Attached files
| file | filename |
|---|---|
| EX-31.2 - EX-31.2 - STRATA Skin Sciences, Inc. | d444442dex312.htm |
| EX-31.1 - EX-31.1 - STRATA Skin Sciences, Inc. | d444442dex311.htm |
| EX-32.1 - EX-32.1 - STRATA Skin Sciences, Inc. | d444442dex321.htm |
| EX-23.1 - EX-23.1 - STRATA Skin Sciences, Inc. | d444442dex231.htm |
| EXCEL - IDEA: XBRL DOCUMENT - STRATA Skin Sciences, Inc. | Financial_Report.xls |
Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2012
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number 000-51481
MELA SCIENCES, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 13-3986004 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification No.) |
50 South Buckhout Street, Suite 1
Irvington, New York 10533
(Address, including zip code, of registrant’s principal executive offices)
(914) 591-3783
Registrant’s telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common stock, $0.001 par value | The NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Security Act. Yes ¨ No þ
Indicate by check mark whether the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes þ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ¨ | Accelerated filer þ | Non-accelerated filer ¨ | Smaller reporting company ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No þ
The aggregate market value of the 30,138,724 shares of common stock held by non-affiliates of the registrant as of June 29, 2012 was $98,252,240 based on the last reported sale price of $3.26 per share on the Nasdaq Capital Market on June 29, 2012. (For this computation, the registrant excluded the market value of all the shares of its common stock held by Directors and Officers of the registrant holding approximately 0.6% of the registrant’s outstanding shares; such exclusion shall not be deemed to constitute an admission that any such person is an “affiliate” of the registrant. There were no shareholders holding at least 10% of the Company’s common stock). The number of shares outstanding of the registrant’s common stock as of February 28, 2013 was 43,037,144 shares.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for the 2013 Annual Meeting of Stockholders, which is to be filed subsequent to the date hereof, are incorporated by reference into Part III of this Form 10-K.
Table of Contents
MELA SCIENCES, INC.
2012 FORM 10-K ANNUAL REPORT
Table of Contents
This Annual Report on Form 10-K, including the sections labeled Management’s Discussion and Analysis of Financial Condition and Results of Operations, contains forward-looking statements that you should read in conjunction with the financial statements and notes to financial statements that we have included elsewhere in this report. These statements are based on our current expectations, assumptions, estimates and projections about our business and our industry, and involve known and unknown risks, uncertainties, and other factors that may cause our or our industry’s results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied in, or contemplated by, the forward-looking statements. We generally identify these statements by words or phrases that contain words such as “believe,” “anticipate,” “assuming,” “expect,” “intend,” “plan,” “will,” “may,” “should,” “estimate,” “predict,” “potential,” “continue,” “contemplate”, or the negative of such terms or other similar expressions. Our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements, and you should not place undue reliance on these statements. Factors that might cause such a difference include those discussed below under the section “Risk Factors,” as well as those discussed elsewhere in this Annual Report on Form 10-K. We disclaim any intent or obligation to update any forward-looking statements as a result of developments occurring after the period covered by this report or otherwise.
Overview
We are a medical device company focused on the commercialization of our flagship product, MelaFind®, and the further design and development of MelaFind® and our technology. MelaFind® is a non-invasive, point-of-care (i.e. in the doctor’s office) instrument to aid in the detection of melanoma. MelaFind®, features a hand-held component that emits light of multiple wavelengths to capture digital data from clinically atypical pigmented skin lesions. The data are then analyzed utilizing sophisticated classification algorithms, ‘trained’ on our proprietary database of melanomas and benign lesions, to provide information to assist in the management of the patient’s disease, including information useful in the decision whether to biopsy the lesion.
The components of the MelaFind® system include:
| • | a hand-held component, which employs high precision optics and multi-spectral illumination (multiple colors of light including near infra-red); |
| • | our proprietary database of pigmented skin lesions, which we believe to be the largest in the U.S.; and |
| • | our lesion classifiers, which are sophisticated mathematical algorithms that extract lesion feature information and classify lesions. |
In November 2011, the Company received written approval from the U.S. Food and Drug Administration (“FDA”) for the MelaFind® Pre-Market Approval (“PMA”) application and in September 2011 received Conformite Europeenne (“CE”) Mark approval for MelaFind®. On March 7, 2012, the Company installed the first commercial MelaFind® systems, and proceeded with the first phase of the commercial launch of its breakthrough product for melanoma detection.
In 2012 the Company evolved from a research and development company to a commercial enterprise. The first commercial MelaFind® systems were placed in working office settings to obtain critical market feedback from dermatologists and their patients. The Company utilized this information to adapt the MelaFind® interface and to make system enhancements to optimize the user experience, accordingly. The market feedback from the initial installations was also used to determine the optimal medical messaging to dermatologists and patient-directed materials in order to effectively communicate the features and benefits of MelaFind®.
The training program for dermatologists, part of the U.S. Food and Drug Administration (“FDA”) approval of MelaFind®, was put into practice at launch. The training of the dermatologists and their staffs includes the
1
Table of Contents
appropriate use of MelaFind® and how best to integrate MelaFind® into their practices, protocols and algorithms for melanoma detection.
During 2012, the Company put in place and developed several critical capabilities to support its commercialization goals. Included among these added capabilities were sales and marketing leadership and staff; technical support and field installation resources; implementation of CRM (Customer Resource Management) and ERP (Enterprise Resource Planning) business systems, and expanded and optimized manufacturing processes with the Company’s critical business partners.
The initial launch of MelaFind® was controlled and deliberate, focused primarily on the U.S. Northeast and several select cities in Germany. Midway through 2012, the scope was widened beyond these select cities and at December 31, 2012, we had over 100 signed user agreements with our customers throughout the U.S. and Germany. Management estimates that this phase of the commercial launch will be complete when approximately 275 user agreements are signed (roughly 200 in the US and 75 in Germany). The second phase of the MelaFind® launch will focus primarily on driving MelaFind® utilization.
Prior to the commercial launch of MelaFind® commencing in the first quarter of 2012, the Company had not generated any revenues from MelaFind®.
Skin cancer is the most common form of cancer in the U.S. More than 3.5 million skin cancers in over two million people are diagnosed annually. Each year there are more new cases of skin cancer than the combined incidence of cancers of the breast, prostate, lung and colon. Melanoma is responsible for approximately 75% of skin cancer fatalities and is the deadliest of all skin cancers as there currently is no cure for advanced stage melanoma. However, detection of early melanoma can lead to virtually a 100% cure rate. Advanced stage melanoma is costly to treat and is responsible for approximately 90% of the total spending on melanoma treatment in the US, costing up to $160,000 per patient. If diagnosed early, however, early melanoma is almost always cured by simple resection at a cost of approximately $4,500 per patient. The cost of treating a Stage IV melanoma is estimated to be more than 22 times the cost of treating a melanoma at the melanoma in situ stage.
Because detection of early melanoma is critical to survival, the American Cancer Society recommends that all Americans over the age of 20 undergo complete skin examinations during their periodic health check-ups. Individuals with dysplastic nevi, a type of pigmented skin lesion associated with an increased risk of melanoma, warrant more frequent observation.
Melanomas are mainly diagnosed by dermatologists and/or primary care physicians using visual clinical evaluation. Physicians assess pigmented skin lesions using the “ABCDEPRU” criteria; Asymmetry, Border irregularity, Color variegation, Diameter greater than 6 mm, Evolving — change in “ABCD” over time, Patients concern, Regression and Ugly duckling. This assessment is subjective and results in missed melanomas as well as a highly variable ratio of benign lesions biopsied to melanomas detected. This biopsy ratio is as high as 50 to 1 for dermatologists and up to 80 to 1 for primary care physicians.
We designed MelaFind® to aid in the evaluation of clinically atypical pigmented skin lesions, when a dermatologist chooses to obtain additional information before making a final decision to biopsy to rule out melanoma. MelaFind® acquires and displays multi-spectral (from blue to near infrared) and dermoscopic Red Green Blue (“RGB”) digital data from pigmented skin lesions. It uses automatic data analysis and statistical pattern recognition to help identify lesions to be considered for biopsy to rule out melanoma, the deadliest form of skin cancer.
To date, MelaFind® has been developed, trained and tested on a proprietary database of over 10,000 skin lesions from more than 7,000 patients at over 40 clinics. The Company believes this is the largest such database in the U.S. and a substantial barrier to competition. The landmark MelaFind® pivotal trial, one of the largest prospective clinical studies ever conducted in melanoma detection, achieved sensitivity of greater than 98% (95% lower confidence bound) and specificity statistically significantly higher than that of study clinicians.
2
Table of Contents
We believe that with the assistance provided by MelaFind®, dermatologists could diagnose more melanomas at the most curable stages with fewer false positive biopsies, which would reduce both treatment costs and the number of biopsies of benign moles, and improve quality of life. Our goal is for MelaFind® to become an integral part of the standard of care in melanoma detection.
The Market Opportunity
Cancer of the skin (non-melanoma and melanoma skin cancers combined) is the most common of all cancers, with over 3.5 million skin cancers in over 2 million people diagnosed annually, and is estimated to account for almost 50% of all cancers. Melanoma is the deadliest form of skin cancer. It is estimated that more than 135,000 new cases of melanoma will be diagnosed in the U.S. in 2013 — more than 61,300 non-invasive (in situ) and more than 76,690 invasive. Melanoma causes one death every hour of every day of the year in the U.S. There are three significant forms of skin cancer: basal cell, accounting for approximately 75% of skin cancer cases; squamous cell, totaling approximately 20% of skin cancer cases; and melanoma, which accounts for an estimated 4% of skin cancer cases, but is responsible for approximately 75% of all deaths from skin cancer. The American Cancer Society projects that 9,480 of the more than 12,000 skin cancer deaths in 2013 will be from melanoma. Because approximately 62% of melanomas and 45% of melanoma deaths occur prior to age 65, melanoma places significant burdens on the healthcare system well beyond Medicare.
Melanoma can be fatal if left untreated. If diagnosed and removed early in its evolution, when confined to the outermost skin layer and deemed to be “in situ,” it has a survival rate of almost 100%. Invasive melanomas that are thin and extend into the uppermost regions of the second skin layer still have excellent cure rates (greater than 90%). However, once the cancer advances into the deeper layers of skin, the risk of metastasis (spreading to other parts of the body) increases. Metastases can occur when the tumor enters into lymphatic channels and newly formed blood vessels, potentially resulting in significant morbidity (illness) and mortality (death). Once the cancer has advanced and metastasized to other parts of the body, it is difficult to treat. At this advanced stage, the five-year survival rate is about 15% to 20%. Moreover, survival prospects for those with advanced melanoma have not significantly improved over the past three decades.
Melanoma is currently the fastest growing cancer and the subject of significant attention in the medical community. The incidence rate of melanoma has doubled since 1973. While there has been a 20% decline in cancer deaths overall since 1991, melanoma is one of three cancers facing increasing rates. According to a study from the Mayo Clinic, the incidence of melanoma increased eightfold among women under 40 and fourfold among men under 40 from 1970 to 2009. Unlike many other common cancers, melanoma has a wide age distribution. In fact, it is one of the more common cancers in people younger than 30, the most common cancer in adults aged 25 to 29, the leading cause of cancer death in women ages 25 to 30 and second only to breast cancer in women ages 30 to 34.
Melanoma is virtually 100% curable if caught early, though no cure is currently available for advanced-stage melanoma. In early 2013, the Company sponsored a national survey with 2109 participants conducted by Harris Interactive that found that only 24% of Americans have had a skin cancer screening with a dermatologist. The Company believes this finding communicates an urgent need to educate people about the importance of early detection.
Our Strategy
Our objective is for MelaFind® to become an integral part of the standard of care in melanoma detection. To achieve this objective, we are pursuing the following strategy:
| • | Establish MelaFind® as the leading technology for assisting in the detection of melanoma. We have invested considerable capital and expertise into developing our core technology platform, which is protected by sixteen U.S. and three Australian patents. We will continue to refine and optimize this technology in order to position MelaFind® as the leading system for assisting in the detection of melanoma. |
3
Table of Contents
| • | Commercialize MelaFind® using multiple sales and marketing strategies. We commenced commercialization in March 2012 and continued with a controlled launch of MelaFind® in selected U.S. and European markets. Our marketing efforts focus on high volume, integrated dermatology practices and skin cancer specialists, in key regions of the U.S. and Europe. To enter the larger general dermatology markets in the U.S. and internationally, we may establish partnerships with pharmaceutical and/or diagnostic/device companies that have an established presence in these markets. The Company anticipates that in addition to the continued placement of MelaFind systems, its strategy will also focus on driving MelaFind® utilization. Our plan is for dermatologists to offer their patients examinations with MelaFind® on a self-pay per patient basis. Once there is sufficient evidence to support favorable coding and coverage decisions and to obtain appropriate payment levels, we may pursue national coverage decisions from the Centers for Medicare and Medicaid Services (“CMS”) and private payers for third-party reimbursement. |
| • | Publication and education strategy. We are executing a publication strategy of providing information for continuing medical education efforts in order to communicate the potential of MelaFind® to improve patient care. The Company committed to conduct a Post-Approval Study (“PAS”) of MelaFind® as a condition of its approval by the FDA. In addition to the PAS, we have sponsored clinical trials in order to evaluate MelaFind® in the clinical setting. We anticipate that the results of these studies will also be published in peer-reviewed journals and presented at scientific and medical meetings and that these studies will help to demonstrate the potential of MelaFind® to improve patient care. We anticipate that its adoption by physicians and ultimate reimbursement by payers will be facilitated by medical and scientific evidence published in peer-reviewed journals and presentations at scientific and medical meetings. |
Additionally, our strategy may include the potential acquisition of complementary products and technologies in the dermatological arena.
Limitations of Current Melanoma Diagnosis
Melanoma is mainly diagnosed by dermatologists and primary care physicians using visual clinical evaluation. This subjective interpretation relies on physician experience and skill. In contrast, MelaFind® delivers an objective assessment based on numerical scores assigned to the clinically atypical skin lesion under evaluation. Further, clinical examination is limited to the surface appearance of the clinically atypical pigmented skin lesion, whereas MelaFind® utilizes information derived from up to 2.5 mm below the skin surface.
Dermatologists who specialize in the management of pigmented skin lesions may also use dermoscopy, a method of viewing lesions under magnification. Although dermoscopy provides more information than unaided visual examination, mastery of the technique necessitates many years of training and experience. Proper use of dermoscopy can reduce the number of biopsies of benign lesions, but even experts in dermoscopy biopsy 3-10 benign lesions for every melanoma detected. While many primary care physicians immediately refer patients with clinically atypical pigmented skin lesions to a specialist, an increasing number perform biopsies on skin lesions themselves. This results in a ratio of benign lesions biopsied to confirmed melanomas of up to 80 to 1.
MelaFind® Product Description
MelaFind® is a non-invasive system to aid in the detection of melanoma. The MelaFind® system produces a report at the-point-of-care to assist in the diagnostic process. MelaFind® employs light of multiple wavelengths to obtain data from clinically atypical lesions; and then the data are analyzed against our proprietary database of melanomas and benign lesions using our sophisticated algorithms. The MelaFind® report contains objective information about the lesion that may not be otherwise available, including information useful in making the decision whether to biopsy the lesion. The key components of the MelaFind® system are:
A hand-held component, which is comprised of several components:
| • | an illuminator that shines light of 10 different specific wavelengths, including near infra-red bands; |
4
Table of Contents
| • | a lens system composed of nine elements that focuses the light reflected from the lesions; |
| • | a photon (light) sensor; and |
| • | a processor employing proprietary algorithms to extract many discrete characteristics or features from the lesions. |
Our proprietary database of pigmented skin lesions, which includes in vivo MelaFind® data and corresponding histological results of over 10,000 biopsied skin lesions from over 7,000 patients, which we believe to be the largest such database in the U.S. and a substantial barrier to competition.
Our lesion classifiers are sophisticated mathematical algorithms. The “brain” of the MelaFind® system, the lesion classifier, distinguishes melanoma from non-melanoma using the lesion features extracted and measured by the hand-held component. The mathematical formulas and algorithms used by the lesion classifiers are devised and optimized through the process of “classifier training” using lesions from our proprietary database. Lesion classifier development and training is an iterative process involving: (1) selection of the lesion features that provide for optimal lesion discrimination; (2) optimization of the mathematical formulas to differentiate benign lesions from melanoma; and (3) expansion of the size and diversity of our proprietary lesion database. The performance of future lesion classifiers is directly related to the size of the database used in classifier development, as well as the degree to which the training database is representative of the lesions that will be evaluated by MelaFind® in a practice setting.
As with many diagnostic systems, the diagnostic performance of MelaFind® is characterized using two measures: (1) sensitivity — the ability to detect disease when it is present; and (2) specificity — the ability to exclude disease when it is not present. Since sensitivity and specificity are typically trade-offs, meaning that as one parameter increases the other decreases, the MelaFind® lesion classifier is developed and trained with the intention that MelaFind® will detect all melanomas in the training data set with the highest possible specificity.
Reliable functioning of the MelaFind® system is critical to its utility and success in the marketplace. Automated self-calibration tests are performed by the hand-held device to ensure proper functionality.
History of MelaFind®
MelaFind® Pivotal Clinical Trial
The MelaFind® Pre-Market Approval (“PMA”) application was submitted to the FDA in June 2009. A pivotal clinical trial was conducted at seven centers across the U.S. and included 1,831 pigmented skin lesions from 1,383 patients. A binding Protocol Agreement with the FDA stipulated the sensitivity and specificity endpoints that should be used to determine the safety and effectiveness of MelaFind®. MelaFind® detected 112 of 114 (98% measured sensitivity; lower confidence bound of 95%) melanomas that were eligible and evaluable for primary sensitivity endpoint analysis, and 125 of 127 (98% measured sensitivity; lower confidence bound greater than 95%) melanomas overall. Importantly, MelaFind® detected 172/175 melanomas and “high grade lesions” (98% sensitivity; lower confidence bound greater than 95%). The Protocol Agreement called for sensitivity endpoints of greater than 95% lower confidence bound (a lower confidence bound of greater than 95% indicates that if the study were repeated, there would be less than a 5% chance that the sensitivity would be below 95%). MelaFind®’s specificity (9.5%), the ability to accurately rule out disease, was significantly superior to that of the study dermatologists (3.7%), who are skin cancer experts (p-value less than 0.02). The Protocol Agreement called for MelaFind® to be more specific than the study physicians at a p-value of less than 0.05 (a p-value of less than 0.05 indicates a less than 5% probability that the observed difference was due to chance).
In order to generate a comparison with physicians’ ability to accurately detect melanomas, the Company conducted an online reader study in which 155 physicians participated including 110 dermatologists. Using images and clinical histories for 65 randomly selected melanomas from the pivotal study, this group of
5
Table of Contents
dermatologists, on average, missed (i.e., would not have elected to biopsy) 28% of the melanomas. The biopsy sensitivity of MelaFind was 97% (p < 0.0001 versus dermatologists). In addition, the kappa score of dermatologists was 0.29, indicating only “fair agreement”.
In November 2011, the Company received written approval from the FDA for the MelaFind® PMA. The Company committed to conduct a PAS of MelaFind® as a condition of PMA approval. Agreement with the FDA on the study protocol was reached and the study was initiated during 2012. The Company anticipates that the PAS could be costly and time consuming to complete.
In September 2011, the Company received Conformite Europeenne (“CE”) Mark approval for MelaFind®.
Hardware and Software History
ASKION GmbH (“ASKION”), located in Germany, which specializes in precision optics, has become an integral member of our MelaFind® development team and we expect to continue to work with ASKION for the foreseeable future. ASKION produced the MelaFind® hand-held components used in our pivotal clinical trials and is continuing to build those units for commercial placement. ASKION also performs other developmental activities with respect to MelaFind®.
The Company, primarily through ASKION, engages Carl Zeiss Jena GmbH (“Zeiss”) to build the lenses and assemblies, as well as provide certain technical consulting, for the MelaFind® units used in the Company’s pivotal clinical trial and in the units now being manufactured for placement. This work is expected to continue for MelaFind® units through 2013.
The Company has obtained Underwriters’ Laboratories (“UL”) certification and Certification Bodies’ Scheme (“CB”) test certification for MelaFind®. The Company has received ISO 13485 certification of the Company’s comprehensive management system for the design and manufacture of medical devices.
MelaFind® Sales and Marketing
The Company offers MelaFind® to dermatologists as a non-invasive, point-of-care service that provides additional information about specific moles during a skin exam. Typically, customers pay a fee for placement of the system in their offices and additional fees based on usage. This approach is intended to provide us with recurring revenue corresponding to the number of patients examined and to provide the dermatologist with access to our technology without a significant capital investment. We have established a direct sales force to place MelaFind® in the U.S. and Europe (initially in Germany), focused on introducing it at high-volume dermatologists’ offices and training their staffs in its use. As we continue to increase the number of MelaFind® placements in dermatology practices, we may continue to increase our sales force or possibly establish partnerships to accelerate the product introduction and to maximize the breadth of the commercial opportunity. While we are exploring potential partnership opportunities we have not yet established any such arrangements.
We have, however, recently added a new division to our sales team called, System Operation Specialists. This team strategically works alongside our sales force to train office staff on the proper use of the MelaFind® system and to help implement flow and efficiency models to seamlessly integrate the system into normal office procedures.
In addition to continuing to drive the placements of MelaFind®, we have also developed key strategies that we believe will increase the everyday use of the system, as we strive to have MelaFind® become the new standard of care in melanoma detection. We have engaged a national public relations (“PR”) agency to generate awareness for MelaFind® in the health/beauty/wellness arena, targeting all of our primary demographics. The
6
Table of Contents
intent of this PR campaign is to position MelaFind® as the leader/expert in melanoma detection among print, broadcast and web media tapping into a broader platform around skin cancer prevention. The Company will continue to work to build awareness and education around the various stages of melanoma and how MelaFind® can help dermatologists in their efforts to detect melanoma at its most curable stage.
The MelaFind® Value Proposition for the Healthcare System
We offer MelaFind® on a self-pay (non-reimbursed) basis. Based on market research with physicians and patients, we believe that a self-pay model could support significant utilization of MelaFind® in the dermatologists’ offices.
Following the self-pay introduction of MelaFind®, we will look to build a sufficient body of medical evidence to support favorable coding and coverage decisions at appropriate payment levels by third-party payers. This strategy is consistent with the approach that has been used to support positive coverage decisions by the Centers for Medicare and Medicaid Services (“CMS”) and private payers for other products. The value drivers include the diagnosis of melanoma at the most curable stages, as opposed to advanced stages, allowing for both a greater opportunity to cure and a reduction in treatment costs.
Our Reimbursement Strategy
We may pursue Current Procedural Terminology (“CPT”) code and private insurance coverage, as appropriate, following our initial commercialization efforts, which are on a self-pay basis. We are aware of no CPT code that is specifically applicable to the use of MelaFind®. We have engaged the services of expert consultants with extensive experience in the CPT, coverage and payment decision processes to assist us in our strategy.
In the U.S., healthcare providers that utilize medical systems such as MelaFind® generally rely on third-party payers, including Medicare, Medicaid, private health insurance carriers, and managed care organizations, to reimburse part, but not necessarily all, of the costs and fees associated with the procedures performed using these devices. Public and professional concern about the cost of medical care and new technologies has evoked a variety of remedies. Third-party payers are increasingly challenging the pricing of medical products and procedures. Guidelines have been established that recognize the need for clinical strategies to assess the cost-effectiveness of new diagnostic tools or procedures (Evidence-Based Medicine), in the hope of reducing the variations in diagnostic and treatment protocols and reducing healthcare expenditures. Insurers are also attempting to curb over utilization by applying a rational analysis of the costs versus benefits of new technologies.
It is critical to build a sufficient body of evidence to support favorable coding and coverage decisions and to secure appropriate levels of payment from third-party payers. Following initial commercialization of MelaFind ®, we will consider submitting an application for a new CPT code to the American Medical Association (“AMA”) CPT Editorial Panel pursuant to the establishment of significant clinical evidence to support favorable coding and coverage decisions. If the CPT Editorial Panel concurs that a new CPT code is needed and appropriate, and we are able to demonstrate that MelaFind ® is reasonable and necessary for the Medicare population, we would expect that the new code would be referred to the AMA’s Relative Value Scale Update Committee (“RUC”) to determine the appropriate level of Medicare Part B reimbursement for the procedure, relative to other physician services. This analysis would include a survey of physicians utilizing MelaFind ® in the practice setting. In setting Medicare reimbursement rates, CMS is generally guided, though not bound, by the recommendation of the RUC. Medicare coverage and payment policies significantly influence the practices and policies of private payers, managed care organizations, and state Medicaid agencies. We would expect to commence efforts to obtain positive coverage decisions from private payers, managed care organizations, Medicaid agencies, and state Medicare administrative contractors pursuant to the establishment of significant clinical evidence to support favorable coding and coverage decisions and secure appropriate payment levels at a future date. We believe it is likely that initially the private payers, managed care organizations, and state Medicare administrative contractors will desire to establish pilot programs of MelaFind ® to determine the impact of the product in their systems.
7
Table of Contents
One of the keys to securing reimbursement is the desire of physicians to use a new technology in order to enhance their diagnostic acumen and improve the standard of care. Likewise, we believe that once patients become aware of the availability of MelaFind ®, they may request that their physicians utilize MelaFind ®. We believe that MelaFind ® will represent an improvement in the standard of care for the detection of melanoma. As such, we anticipate that its adoption by physicians and reimbursement by payers will be facilitated by medical and scientific evidence published in peer-reviewed journals and presentations at scientific and medical meetings including the American Academy of Dermatology’s annual and regional meetings. We plan to execute a publication strategy and to provide information for continuing medical education efforts in order to communicate the potential of MelaFind ® to improve patient care. In addition to the PAS, we also plan to sponsor clinical trials in order to evaluate MelaFind® in the clinical setting. We anticipate that the results of these studies will also be published in peer-reviewed journals and presented at scientific and medical meetings and that these studies will help to demonstrate the potential of MelaFind ® to improve patient care.
We recognize that a favorable reimbursement environment could have a significant impact on MelaFind®’s adoption and commercial success. Even if a procedure is eligible for reimbursement, the level of reimbursement may not be adequate. In addition, third-party payers may deny reimbursement if they determine that the device used in the treatment was not cost-effective or was used for a non-approved indication. We have anticipated this need and we plan to employ an active strategy to obtain medical coverage, identify appropriate coding and establish adequate payment.
Competition
We are not aware of any system which is directly competitive to MelaFind®. A number of systems for visualization and assessment of pigmented skin lesions are in use or in development. These include clinical (naked eye) examination, whole body mole mapping systems, dermoscopes (also known as “dermatoscopes”), digital dermoscopes, spectrophotometric intercutaneous analysis (analysis of skin structures through measurement of how they absorb light of different wavelengths), confocal microscopy, spectrophotometric (color) analysis and several newly identified light-based approaches. These systems rely on physician experience and expertise in recognizing patterns that are associated with melanoma and non-melanoma in order to render an interpretation and diagnosis.
The current primary method for detecting melanoma relies on physicians to interpret whether a pigmented skin lesion is suspicious for melanoma (thereby requiring biopsy) based on their ability to recognize patterns using clinical examination. Physicians use the “ABCDE” criteria: Asymmetry, Border irregularity, Color variation, Diameter greater than 6 mm, and Evolving-change in “ABCD”, in their assessment. Recently the letters “PRU” have been added to the criteria; Patient concern, Regression and Ugly duckling. Physicians also use whole body mole mapping which consists of periodic photography of patients, typically those at high risk for developing melanoma. The pictures are reviewed clinically. This service is provided at some diagnostic imaging centers and dermatology offices. DigitalDerm, Inc. offers MoleMapCD ® a computerized system for acquisition, storage, and review of the pictures and Melanoscan, Inc. offers a similar sequential photography system. MoleSafe also offers whole body mapping in clinics across the U.S.
Dermoscopy, or epiluminescence microscopy, allows for non-invasive visualization of colors and microstructures of the epidermis, the dermal-epidermal junction, and the papillary dermis not visible to the naked eye. Manufacturers of dermoscopes include (but are not limited to) Welch Allyn, Inc. (U.S.), Heine Optotechnik (Germany), Riester Medical (Germany) 3Gen, LLC (U.S.), and others. Several manufacturers, such as FotoFinder Systems, GmbH (Germany), are selling apps and hardware that allow an Apple iPhone to be used as a dermoscope.
Dermoscopy is a tool used by approximately 25% of dermatologists in the U.S. and is associated with a long learning curve. Physicians experienced in the use of dermoscopy have been shown to have an increased diagnostic accuracy of 10% to 20% over clinical examination. Although some digital dermoscopes provide information regarding the probability that a lesion may be melanoma compared to a database of lesions, no system, to our knowledge, is under PMA development for objective interpretation.
8
Table of Contents
Digital dermoscopes allow for dermoscopic images to be visualized on a computer screen at a larger magnification. In addition, images may be stored and compared to images taken previously. Manufacturers of digital dermoscopes include (but are not limited to) Derma Medical Systems, Inc. (Austria), manufacturers of the Molemax™ family of digital systems, Biomips Engineering (Italy) and FotoFinder Systems, GmbH the manufacturers of the FotoFinder group of instruments.
ViseoMed, AG a German company, has developed microDerm, a digital dermoscopy system that documents images and analyzes lesions for early recognition of skin cancer including melanoma. The microDerm is comprised of one camera which takes both microscopic images and clinical images in High Definition quality. Using the company’s DANAOS software the system is designed to calculate a classification value of a particular lesion designed to support the physician.
Spectrophotometric intercutaneous analysis is a technique of visualizing collagen, blood, and pigment. MedX Health Corp.’s product, SIMSYS MoleMate, uses this method. The system, also called SIAscopy, integrates non-harmful light and digital imaging to evaluate lesions in five distinct views. This software produces a rating “score” for scanned lesions.
Caliber Imaging & Diagnostics, Inc. (formerly Lucid, Inc.) (US) markets cellular imaging and diagnosis products which utilize confocal microscopy. Their product line, VivaScope, is used for non-invasive visualization of skin structures at the cellular level. Researchers at Vanderbilt University are developing technology called Confocal Raman Micro-Spectroscopy’ which uses a reflective laser to produce a molecular fingerprint of the underlying tissue to indicate the presence or absence of disease. In addition, Verisante Technology, Inc. has received a CE Mark and approval from Health Canada for technology it has licensed from The British Columbia Cancer Agency, an agency of the Provincial Health Services Authority, for skin cancer detection. The company’s product, the Aura, uses NIR Raman Spectroscopy and autofluorescence spectroscopy to identify spectral changes associated with the biochemistry of skin cancer cells in less than a second.
We also compete with other imaging modalities, including molecular imaging in which tagged antibodies search for cancer cell antigens, and with molecular and genetic screening tests. Molecular-based approaches are also being investigated; for example Dermtech, Inc. (U.S.) is exploring Messenger RNA analysis of surface cells. Dermtech’s core technologies are 1) the patented, non-invasive EGIR™ (Epidermal Genetic Information Retrieval) technique that uses an adhesive to painlessly collect cells from the upper layer of the skin, and 2) multi-gene biomarkers that are generated using microarray analysis. The ribonucleic acid (“RNA”) from these cells is then isolated, amplified, and analyzed using molecular biology tools. To date, Dermtech has patented three biomarkers that can be used to identify and correlate changes in the gene expression profile of RNA obtained from the skin with the presence of certain diseases.
Scibase AB is developing electrical impedance technology for melanoma detection. The method is called electrical impedance spectroscopy (EIS). It is based on a technology that uses the varying electrical properties of human tissue to categorize the cell structures and thereby detect malignancies. On August 1, 2012 Scibase announced top line results from its clinical trial called “SIMPS” (SciBase International Melanoma Pivotal Study).
Balter Medical (Norway) uses ‘Optical Transfer Diagnosis’ to detect anomalies in human tissue to support the diagnosis of melanomas. The technology measures how much light is absorbed in healthy versus diseased tissue to determine whether cancer is present. The company is currently conducting a clinical trial in the United Kingdom.
DatInf, GmbH a German company, has two products for mole mapping and analyzing moles, MoleExpert macro and MoleExpert micro. MoleExpert macro utilizes a high resolution digital camera as a mole mapping and screening system for the detection of new or changed lesions. The system automatically detects moles in the first and in follow-up images and extracts features, such as size, shape and color of the moles and uses the two images
9
Table of Contents
for comparison. MoleExpert micro provides a measurement or score of features obtained from microscopic images of the skin (such as the ABCD features) which may support diagnosis of lesions.
Raytheon Corporation has partnered with Arizona Cancer Center, to utilize satellite-based remote imaging technology in detecting skin changes that could indicate the presence of cancer.
Researchers at Ben Gurion University in Israel announced a new device that detects cancerous skin tumors not visible to the naked eye. The Optical Spectro-Polarimetric Imaging (OSPI) instrument reportedly diagnosed 73 types of lesions, some of them cancerous, in initial testing.
Michelson Diagnostics Ltd, a UK based developer and manufacturer of Optical Coherence Tomography (OCT) products, has U.S. FDA 510(k) clearance for its VivoSight™ OCT scanning product for non-melanoma skin cancers. VivoSight™ is a Multi-Beam OCT system indicated for use in the two-dimensional, cross-sectional, real-time imaging of external tissues of the human body. VivoSight™ is a Fourier-Domain OCT scanner that provides sub-surface images of tissue at far higher resolution than is possible with existing technologies such as ultrasound, CT or MRI, in 2D and 3D and in real time, using an easy-to-use lightweight hand-held probe.
Cascade Technologies Corp through its wholly owned subsidiary Spectral Molecular Imaging (a development stage company), is using what it calls “hyperspectral-optical” technology to advance early diagnosis of cancer and pre-cancer conditions. The company’s SkinSpect™ device is being developed for non-invasive diagnosis of and screening for skin cancer.
Agfa Healthcare announced in September 2012 that it was launching its SKINTELL a high-definition optical coherence tomography system. This device allows the clinician to have three viewing modes of the lesion of interest. The operator can choose slice view, 3D view, and a unique parallel to the skin perspective. SKINTELL is not available in the U.S. or Canada.
The broad market for precision optical imaging devices used for medical diagnosis is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. Since MelaFind® is approved for marketing, we will potentially be subject to competition from major optical imaging companies, such as Raytheon Corporation, General Electric Co., Siemens AG, Bayer AG, Olympus Corporation, Carl Zeiss AG Deutschland and others, each of which manufactures and markets precision optical imaging products for the medical market and could decide to develop or acquire a product to compete with MelaFind®.
Manufacturing
We are currently focusing the manufacturing efforts of our contract manufacturers on building MelaFind® systems and in optimizing efficiency and larger-scale manufacturing. To support our commercial marketing of MelaFind®, we have contracted with ASKION in Germany, an ISO 9001 and ISO 13485 certified manufacturer, which specializes in precision optics. Through ASKION, we have contracted with Zeiss, an international optics house, to supply lenses and lens assemblies to be used in the hand-held components. ASKION will provide the system integration for MelaFind® systems to be placed in Europe.
In addition, we are utilizing Nexcore Technology Inc., an FDA GMP compliant and ISO 9001 and ISO 13485 certified original equipment manufacturer of medical devices in New Jersey, to provide the assembled MelaFind® carts and tested MelaFind® systems incorporating the hand-held devices along with the processing computer, software and operator controls for MelaFind® systems to be placed in the U.S.
Research and Development Efforts
We continue to develop refinements and improvements to the hardware and software, including lesion classification algorithms, some of which are likely to require approval of a PMA supplement. Our research and
10
Table of Contents
development (“R&D”) plan also includes further improvements such as faster and easier software downloads for future versions.
We have performed feasibility studies of a MelaFind® software add-on feature called MelaMetertm , an enhancement to MelaFind® that provides information regarding the depth of penetration of a pigmented skin lesion. This information may be useful to physicians in determining the necessary depth and breadth of a biopsy of a pigmented skin lesion. Initial clinical studies of MelaMetertm demonstrate the ability of MelaMetertm to non-invasively estimate the Breslow thickness (the thickness of a cutaneous malignant melanoma measured from the epidermis to the deepest malignant cells present) comparably to histological examination of excised lesions. We plan to continue the development of MelaMetertm and seek FDA approval for it, as appropriate.
We further intend to explore and evaluate the potential use of our light based computer vision platform in other applications, including the non-invasive detection of basal cell carcinoma, the most common skin cancer, as well as squamous cell carcinoma of the skin. New hardware systems for the data collection from blood and blood vessel patterns are needed since the majority of these cancers are not pigmented and, accordingly, the MelaFind® system as currently developed is not appropriate for these uses. However, we believe many software programs and algorithms used in the MelaFind® system will be applicable with some modification.
The Company spent approximately $11,497, $9,656 and $6,792 in 2010, 2011 and 2012, respectively, on R&D. With the commercial launch of MelaFind®, certain costs previously included under R&D, such as costs associated with the PAS, will in the future be recorded as general and administrative costs. R&D efforts going forward will focus on refinements to MelaFind® as well as new, complementary technology.
Intellectual Property
Our policy is to protect our intellectual property by obtaining U.S. and foreign patents to protect technology, inventions and improvements important to the development of our business. Currently, we have twenty issued U.S. patents in force, plus two that are projected to issue in 2013, and these patents have numerous foreign counterparts issued and pending. Of those issued, sixteen U.S. patents and three Australian patents relate to various aspects of MelaFind® technology. Two of the U.S. patents are design patents, while all others are utility patents. In addition, we have ten more U.S. utility patents currently pending, all of which relate to MelaFind®. Of the many pending foreign patent applications that relate to MelaFind ® , ten are currently in the European regional phase (with the European Patent Office), ten are pending in Australia, nine in Canada, three in Japan, and four in Hong Kong. Also, we have obtained non-exclusive licenses from several of our suppliers for critical components of MelaFind®. We have not granted any significant licenses with respect to our intellectual property other than licenses granted in connection with our DIFOTI product, which was discontinued in 2005.
We cannot be certain that our patents will not be challenged or circumvented by competitors. Whether a patent is infringed and is valid, or whether a patent application should be granted, are all complex matters of science and law, and therefore we cannot be certain that, if challenged, our patents, patent applications and/or other intellectual property rights would be upheld. If one or more of those patents, patent applications or other intellectual property rights are invalidated, rejected or found unenforceable, that could reduce or eliminate any competitive advantage that we derive from that intellectual property.
We also rely on trade secrets and technical know-how in the manufacture and marketing of MelaFind®. We require our employees, consultants and contractors to execute confidentiality agreements with respect to our proprietary information.
We have active U.S. trademark registrations for the word marks: MELA, MELA SCIENCES, MELAFIND and MELARECORD, as well as for our “MelaFind®” word-plus-design (logo) mark. The “MELA,” “MELA SCIENCES,” “MELAFIND” and “MELARECORD” word marks are currently registered in the European Union and Australia, and “MELAFIND” also in New Zealand. (Any combinations of upper- and lower-case letters in
11
Table of Contents
any style or color, are covered by “standard character” word mark registrations, indicated here by upper-case lettering.) For the “MELAFIND” word mark and “MelaFind” logo, the description of goods in International Class 10 covered by the U.S. registration is: “medical devices, namely, electro-optical devices incorporating hardware for obtaining digital data in different spectral bands and software for analyzing the digital data for use in analyzing skin lesions and determining the existence of melanoma.” In Europe, besides International Class 10, the “MELAFIND” mark is also registered in International Classes 16 (for printed reports) and 44 (as a service mark). The “MELARECORD” word mark is registered in classes 9, 10 and 16 in the US, the European Union, and Australia. The registration in class 9 covers the “Electronic MelaRecord®” Patient Card, while the registration in class 16 covers printed “MelaRecord® Reports” for patients, physicians or pathologists, for example. The registration in class 10 covers the “MelaRecord® auxiliary card reader,” for example. A Statement of Use has been filed and accepted by the U.S. Patent and Trademark Office for the “MELAFINDER” word mark in International Class 42, for “Providing a website featuring a search engine for locating dermatologists using specialized melanoma detection equipment.” Several additional trademark registrations are pending in the U.S. in International Classes 10 and 16, for which Notices of Allowance have been received but for which no Statements of Use have yet been filed. Those additional marks include the “MELA Sciences” corporate logo and the “MELAMETER” word mark, for example.
We also have registered the internet domain names: www.melasciences.com, www.eosciences.com, www.melafind.com, www.mela.us.com, www.melafind.info, www.melafind.net, www.melafind.org, www.melafind.us, melafinder.com, www.melainc.com, and www.skinsurf.com.
The following table lists our U.S. patents and patent applications relating to melanoma detection:
U.S. Patents Relating to MelaFind®
| Patent # |
Title |
Issued |
Expiration | |||||||
| 6,081,612 | Systems and Methods for the Multispectral Imaging and Characterization of Skin Tissue |
06/27/00 | 02/27/18 | |||||||
| 6,208,749 | Systems and Methods for the Multispectral Imaging and Characterization of Skin Tissue |
03/27/01 | 02/27/18 | * | ||||||
| 6,307,957 | Multispectral Imaging and Characterization of Biological Tissue |
10/23/01 | 06/27/20 | |||||||
| 6,626,558 | Apparatus for Uniform Illumination of an Object |
09/30/03 | 08/31/21 | |||||||
| 6,657,798 | Method for Optimizing the Number of Good Assemblies Manufacturable From a Number of Parts |
12/02/03 | 02/10/23 | |||||||
| 6,710,947 | Method for Assembling Lens Elements |
03/23/04 | 02/27/23 | |||||||
| 7,102,672 | Integrated CMOS Imaging Array & Dark Current Monitor |
09/05/06 | 01/10/24 | |||||||
| 7,127,094 | Method of Controlling Data Gathered at Remote Locations |
10/24/06 | 03/11/25 | |||||||
| D613,866 | Medical Cart |
04/13/10 | 04/13/24 | |||||||
| D613,867 | Table Structure of a Medical Cart |
04/13/10 | 04/13/24 | |||||||
| 7,813,586 | Reducing Noise in Digital Images |
10/12/10 | 05/31/27 | |||||||
| 7,894,651 | Quantitative Analysis of Skin Characteristics |
02/22/11 | 03/19/29 | |||||||
| 8,160,386 | Reducing Noise in Digital Images (CIP) |
04/17/12 | 10/06/30 | |||||||
| 8,208,698 | Characterizing a Texture of an Image |
06/26/12 | 04/25/31 | |||||||
| 8,286,977 | Medical Cart |
10/16/12 | 10/25/30 | |||||||
| 8,381,987 | An Insertable Storage Card Containing a Portable Memory Card Having a Connection Interface |
02/26/13 | 06/29/30 | |||||||
| * | A post-PMA request for patent term extension under the Hatch-Waxman Act was filed 12/19/11. |
12
Table of Contents
Pending Non-Provisional U.S. Patent Applications Relating to MelaFind®
| Published Pat Appl Ser # |
Title |
Filed | ||||
| US2008/0312952A1 |
Regulating Use of a Device to Perform a Procedure on a Subject |
06/12/07 | ||||
| US2009/0060304A1 |
Obtaining Dermatology Information |
09/04/08 | ||||
| US2012/0033863A1 |
Assessing Features for Classification |
08/06/10 | ||||
| US2011/0064287A1 |
Characterizing a Texture of an Image (CIP) |
09/07/10 | ||||
| US2011/0103660A1 |
Showing Skin Lesion Information* |
11/01/10 | ||||
| US2011/0210984A1 |
Showing Skin Lesion Information (CIP) |
05/03/11 | ||||
| US2012/0126503A1 |
Medical Cart (CIP) |
01/27/12 | ||||
| US2012/0118981A1 |
Storage Cart (CIP) |
01/27/12 | ||||
| US2012/0162487A1 |
Reducing Noise in Digital Images (CIP) |
03/01/12 | ||||
| US2012/0224753A1 |
Characterizing a Texture of an Image (CIP) |
05/15/12 | ||||
Note: CIP denotes a Continuation-in-Part patent application.
| * | Notice of Allowance issued 11/30/12; Request for Continued Examination filed 1/30/13 |
Patent No. 6,081,612 relates to the MelaFind® system and methods employed in building MelaFind® classification algorithms involving the use of novel multi-spectral lesion features by means of wavelet maxima representations. Wavelet maxima representations use specific types of mathematical transformations called wavelets to represent a signal, such as digital data of a lesion taken by the MelaFind® system, at different detail levels. The wavelet maxima representation retains information of potential diagnostic value. This information is quantified in the form of statistical features used for automatic classification. Patent No. 6,208,749 relates to methods employed for automatic segmentation of the lesion digital data, and in building the MelaFind ® classification algorithms involving the use of novel features of multispectral lesion data that do not involve the use of wavelet transformations to determine whether the lesion is or is not a melanoma. We believe the inclusion of the described wavelets and non-wavelets features improves significantly the sensitivity and specificity of the melanoma classifiers. Patent No. 6,307,957 extends the use of the novel features of the MelaFind® system to endoscopy (examination of gastro-intestinal tissues using fiber-optic probes). We have no present plans to develop endoscopy applications of our technology.
Patent No. 6,626,558 covers the construction of the array of numerous light-emitting diodes (“LED’s”) that are used in the MelaFind® hand-held device to provide uniform illumination of lesions in multiple spectral bands of illumination. Patent No. 6,657,798 involves the use of a computer algorithm to optimize the number of lens assemblies possible from a given number of sets of lens elements. Patent No. 6,710,947 describes a method that we may employ for the economical assembly of the nine elements of the MelaFind® hand-held device’s optical lens module.
Patent No. 7,102,672 is a process that we may employ to compensate for the effect of temperature-dependent dark current on the data acquired by the MelaFind® hand-held probe, and Patent No. 7,127,094 is a series of methods for central control of the acquisition and processing of the data acquired by MelaFind® probes located at remotes sites. Patent No. 7,813,586 covers a novel method for reducing noise in digital data, which was invented and has been implemented as part of the calibration of all MelaFind® image digital data. Six additional claims are covered in its Continuation-in-Part Patent No. 8,160,386. The two design patents describe novel design aspects of the MelaFind® medical cart. Patent No. 8,286,977 protects certain innovative functional aspects of the medical cart, while the Continuation-in-Part patent application for the Medical Cart filed January 27, 2012 seeks to protect certain additional functional aspects. Patent No. 8,381,987 covers 20 claims for “An Insertable Storage Card Containing a Portable Memory Card Having a Connection Interface,” based on an application filed on July 30, 2010. It protects certain aspects of the MelaRecord® Patient Card used with MelaFind® while a still-pending Continuation-in-Part “Storage Card” application filed January 27, 2012 seeks to protect certain other aspects.
13
Table of Contents
Patent No. 7,894,651 protects devices and methods for quantitative analysis of skin characteristics to identify lesions that require further evaluation by physicians to rule out melanoma. Our June 12, 2007 patent filing relates to innovative ways to control use of our MelaFind® system. Patent No. 8,208,698 and its September 7, 2010 and May 15, 2012 Continuation-in-Part applications relate to new methods for characterizing the “lacunarity” texture of an image. Our September 4, 2008 patent filing concerns certain dermatology information associated with MelaFind® and claims priority to a provisional application filed a year earlier.
A Notice of Allowance was issued November 30, 2012 for 46 claims relating to the MelaFind® user interface as described in our “Showing Skin Lesion Information” patent filed November 1, 2010. Subsequently discovered prior-art references required us to file a Request for Continued Examination on January 30, 2013, delaying issuance of the patent to permit examination of those references. A Continuation-in-Part application filed May 3, 2011, which is still pending, seeks to cover additional claims regarding the user interface.
The patent application for “Assessing Features for Classification” filed August 6, 2010 seeks to protect a novel method of generating lesion classifiers such as those employed for the MelaFind® device.
We also have developed trade secret calibration methods, classifier programs, and search engines. These programs have been developed over many years and incorporate decades of experience in optical computer vision. In addition, our proprietary MelaFind® database of over 10,000 lesions has been compiled over a number of years and would be difficult to replicate.
We believe that our patented methods and apparatus, together with proprietary trade-secret technology, give us a competitive advantage; however, we cannot be certain that, if challenged, our patented methods and apparatus and/or trade-secret technology would be upheld. If one or more of our patented methods, patented apparatus or trade-secret technology rights are invalidated, rejected or found unenforceable, that could reduce or eliminate any competitive advantage we might otherwise have had.
FDA Regulation
Our product, MelaFind®, is regulated as a medical device and is subject to extensive regulation by the FDA and other regulatory authorities in the U.S. The Food, Drug, and Cosmetic Act (“FD&C Act”) and other federal and state statutes and regulations govern the research, design, development, preclinical and clinical testing, manufacturing, safety, approval or clearance, labeling, packaging, storage, record keeping, servicing, promotion, import and export, and distribution of medical devices.
Unless an exemption applies, each medical device we wish to commercially distribute in the U.S. will require prior pre-market notification, 510(k) clearance, or PMA approval from the FDA. The FDA classifies medical devices into one of three classes. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are subject to general controls such as labeling, pre-market notification, and adherence to the FDA’s Quality System Regulation (a set of current good manufacturing practice requirements put forth by the FDA which govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation and servicing of finished devices) (“QSR”). Class II devices are subject to special controls such as performance standards, post-market surveillance, FDA guidelines, as well as general controls. Some Class I and Class II devices are exempted by regulation from the premarket notification, or 510(k), clearance requirement or the requirement of compliance with certain provisions of the QSR. Devices are placed in Class III, which requires approval of a PMA application, if insufficient information exists to determine that the application of general controls or special controls are sufficient to provide reasonable assurance of safety and effectiveness, or they are life-sustaining, life-supporting or implantable devices, or the FDA deems these devices to be “not substantially equivalent” either to a previously 510(k) cleared device or to a “pre-amendment” Class III device in commercial distribution before May 28, 1976, for which PMA applications have not been required. The FDA classifies MelaFind® as a Class III device, requiring PMA approval.
14
Table of Contents
A PMA application must be supported by valid scientific evidence, which typically requires extensive data, including technical, pre-clinical, clinical, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. A PMA application must include, among other things, a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling. A PMA application also must be accompanied by a user fee, unless exempt. For example, the FDA does not require the submission of a user fee for a small business’ first PMA.
New PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling and device specifications, materials or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel.
Clinical trials are almost always required to support a PMA application, and are sometimes required for a 510(k) clearance. These trials generally require submission of an application for an Investigational Device Exemption (“IDE”) to the FDA. An IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for more abbreviated IDE requirements. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent form are approved by appropriate institutional review boards (“IRBs”) at the clinical trial sites. The FDA’s approval of an IDE allows clinical testing to go forward, but does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and effectiveness, even if the trial meets its intended success criteria.
All clinical trials must be conducted in accordance with the FDA’s IDE regulations that govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators.
The clinical studies of MelaFind® are considered by the FDA as Non-significant Risk (“NSR”) studies. Consequently, the trials were conducted under the auspices of an abbreviated IDE. Clinical trials must further comply with the FDA’s regulations for IRB approval and for informed consent. Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended safety and effectiveness success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product.
The withdrawal of previously received approvals or failure to comply with existing or future regulatory requirements would have a material adverse effect on our business, financial condition and results of operations.
After a device is approved or cleared and placed in commercial distribution, numerous regulatory requirements apply. These include:
| • | establishment registration and device listing; |
| • | QSR, which requires manufacturers to follow design, testing, control, documentation and other quality assurance procedures; |
| • | labeling regulations, which prohibit the promotion of products for unapproved or “off-label” uses and impose other restrictions on labeling; |
| • | medical device reporting regulations, which require that manufacturers report to the FDA if a device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; and |
15
Table of Contents
| • | corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FD&C Act that may present a risk to health. |
The FDA enforces regulatory requirements by conducting periodic, unannounced inspections and market surveillance. Inspections may include the manufacturing facilities of our subcontractors. Thus, we must continue to spend time, money, and effort to maintain compliance.
Failure to comply with applicable regulatory requirements may result in enforcement action by the FDA, which may lead to any of the following sanctions:
| • | warning letters; |
| • | fines and civil penalties; |
| • | unanticipated expenditures; |
| • | delays in approving or refusal to approve our applications, including supplements; |
| • | withdrawal of FDA approval; |
| • | product recall or seizure; |
| • | interruption of production; |
| • | operating restrictions; |
| • | injunctions; and |
| • | criminal prosecution. |
Our contract manufacturers, specification developers, and some suppliers of components are also required to manufacture our products in compliance with current Good Manufacturing Practices (“cGMP”) requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and includes extensive requirements with respect to quality management and organization, device design, equipment, purchase and handling of components, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing, and record keeping. The FDA enforces the QSR through periodic unannounced inspections that may include the manufacturing facilities of our subcontractors. We expect that our subcontractors’ manufacturing facilities will be subject to domestic and international regulatory inspection and review. If the FDA believes any of our contract manufacturers or regulated suppliers are not in compliance with these requirements, it can shut down the manufacturing operations of our contract manufacturers, require recall of our products, refuse to approve new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations, or assess civil and criminal penalties against us or our officers or other employees. Any such action by the FDA would have a material adverse effect on our business. We cannot assure you that we will be able to comply with all applicable FDA regulations.
Non-FDA Government Regulation
The advertising of our MelaFind® product is subject to both FDA and Federal Trade Commission regulations. In addition, the sale and marketing of MelaFind® is subject to a complex system of federal and state laws and regulations intended to deter, detect, and respond to fraud and abuse in the healthcare system. These laws and regulations restrict and may prohibit pricing, discounting, commissions and other commercial practices that may be typical outside of the healthcare business. In particular, anti-kickback and self-referral laws and regulations will limit our flexibility in crafting promotional programs and other financial arrangements in connection with the sale of our products and related services, especially with respect to physicians seeking reimbursement through Medicare or Medicaid. These federal laws include, by way of example, the following:
| • | the anti-kickback statute prohibits certain business practices and relationships that might affect the provision and cost of healthcare services reimbursable under Medicare, Medicaid and other federal |
16
Table of Contents
| healthcare programs, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs; |
| • | the physician self-referral prohibition, commonly referred to as the Stark Law, which prohibits referrals by physicians of Medicare or Medicaid patients to providers of a broad range of designated healthcare services in which the physicians or their immediate family members have ownership interests or with which they have certain other financial arrangements; |
| • | the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; |
| • | the Civil False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs; and |
| • | the Civil Monetary Penalties Law, which authorizes the U.S. Department of Health and Human Services (“HHS”) to impose civil penalties administratively for fraudulent or abusive acts. |
Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. These laws also impose an affirmative duty on those receiving Medicare or Medicaid funding to ensure that they do not employ or contract with persons excluded from the Medicare and other government programs.
Many states have adopted or are considering legislative proposals similar to the federal fraud and abuse laws, some of which extend beyond the Medicare and Medicaid programs to prohibit the payment or receipt of remuneration for the referral of patients and physician self-referrals regardless of whether the service was reimbursed by Medicare or Medicaid. Many states have also adopted or are considering legislative proposals to increase patient protections, such as limiting the use and disclosure of patient-specific health information. These state laws typically impose criminal and civil penalties similar to the federal laws.
In the ordinary course of their business, medical device manufacturers and suppliers have been and are subject regularly to inquiries, investigations and audits by federal and state agencies that oversee these laws and regulations. Federal and state legislation has increased funding for investigations and enforcement actions, which have increased dramatically over the past several years. This trend is expected to continue. Private enforcement of healthcare fraud also has increased, due in large part to amendments to the Civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. These whistleblower suits by private persons, known as qui tam relaters, may be filed by almost anyone, including physicians and their employees and patients, our employees, and even competitors. The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), in addition to its privacy provisions, created a series of new healthcare-related crimes.
Environmental Regulation
Our research and development and clinical processes involve the handling of potentially harmful biological materials as well as hazardous materials. We and our investigators and vendors are subject to federal, state and local laws and regulations governing the use, handling, storage and disposal of hazardous and biological materials and we incur expenses relating to compliance with these laws and regulations. If violations of environmental, health and safety laws occur, we could be held liable for damages, penalties and costs of remedial actions. These expenses or this liability could have a significant negative impact on our financial condition. We may violate environmental, health and safety laws in the future as a result of human error, equipment failure or other causes. Environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. We are subject to potentially conflicting and changing
17
Table of Contents
regulatory agendas of political, business and environmental groups. Changes to or restrictions on permitting requirements or processes, hazardous or biological material storage or handling might require an unplanned capital investment or relocation. Failure to comply with new or existing laws or regulations could harm our business, financial condition and results of operations.
International Regulation
The medical device regulatory process for international distribution is subject to government regulations that may vary by country from those having few or no regulations to those having pre-market controls and pre-market acceptance. In the EU, medical devices require a Conformite Europeenne (“CE”) Mark in order to be placed in the market. The CE Mark certifies that a product has met EU consumer safety, health and environmental requirements. CE marking requires meeting the conditions of the European Directive to which the medical device applies. The directives regulate the design, manufacture, clinical trials, labeling, and post-market surveillance reporting activities for medical devices.
During the process of achieving CE Mark approval in 2011, the Company successfully completed the International Organization for Standardization (“ISO”) 13485 certification inspection of the Company’s comprehensive management system for the design and manufacture of medical devices. In September 2011, after review and approval of the MelaFind® technical file, the Company received CE Mark approval for MelaFind®.
We have also begun evaluation and planning for product distribution in Australia and Brazil. The Australian regulatory process for medical devices is similar to the EU regulatory process. In Australia, devices must comply with the Therapeutic Goods (Medical Devices) Regulations of 2002 or, if the device has a European CE Mark, the CE certificates can be used as “Manufacturer’s Evidence” for compliance when submitted to the Therapeutic Goods Administration (“TGA”). If the device does not have a European CE Mark, the manufacturer is required to submit a Technical File or Design Dossier to TGA. To achieve the required QMS compliance, most manufacturers comply with the ISO 13485 standard.
The Brazilian regulatory process for medical devices is also similar to the EU regulatory process. Medical devices in Brazil are regulated by the Agência Nacional de Vigilância Sanitária (“ANVISA”). A Technical File is needed for registration with ANVISA. The Brazilian quality system requirements are very similar to US FDA 21 CFR Part 820, Quality System Regulations.
Product Liability and Insurance
Our business exposes us to the risk of product liability claims that is inherent in the testing, manufacturing and marketing of medical devices, including those which may arise from the misuse or malfunction of, or design flaws in, our products. We may be subject to product liability claims if MelaFind® causes, or merely appears to have caused, an injury. Claims may be made by patients, healthcare providers or others involved with MelaFind®. We have both general liability insurance and product liability insurance for MelaFind®, which is and will be subject to deductibles and coverage limitations. We have also obtained clinical trial liability insurance in the U.S. and in certain European countries where required by statute or clinical site policy. Our future product liability insurance needs may not be available to us in amounts and on acceptable terms, if at all, and, if available, the coverage may not be adequate to protect us against any future product liability claims. If we are unable to obtain insurance at an acceptable cost or on acceptable terms with adequate coverage or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may harm our business.
Employees
As of December 31, 2012, we had 71 full-time and 3 part-time employees, of whom 11 were engaged in research and development, 25 in production (including clinical, regulatory affairs, document control and quality assurance) and 38 in marketing, sales and administrative activities. We believe that our relationship with our employees is good.
18
Table of Contents
Other
The Company’s Internet address is www.melasciences.com. Our annual report on Form 10-K, quarterly reports on Forms 10-Q, current reports on Forms 8-K, and amendments to those reports are available, without charge, on our website www.melasciences.com as soon as reasonably practical after they are filed electronically with the Securities and Exchange Commission (“SEC”). Copies are also available, without charge, from MELA Sciences, Inc., 50 South Buckhout Street, Suite 1, Irvington, New York, 10533, Attention: Secretary.
You should carefully consider the following risk factors, as well as the other information contained in this report. If any of the following risks actually occur, our business, financial condition and results of operations would likely suffer. In that case, the trading price of our common stock would likely decline.
Risks Relating to Our Business
We are required to conduct a Post-Approval Study of MelaFind®. If the results from this study are negative or we fail to meet the requirements of this condition of approval, we may not be able to maintain the approval of MelaFind®.
As a condition of approval of our PMA, we must conduct a Post-Approval Study evaluating the sensitivity of physicians in diagnosing melanomas and high-grade lesions and the false positive rate after using MelaFind®. Preparing for and conducting this Post-Approval Study will be costly and time consuming.
We are required to submit to the FDA progress reports on this study for every six months during the first two years and annually thereafter. The first progress report was submitted to the FDA in February 2013. If the FDA has questions on the data provided in a progress report, or believes the data are incomplete or insufficient, the agency may request additional information, including through a deficiency letter. The FDA may seek the advice of advisory panels of outside experts when considering the initiation or progress of post-approval studies. If we have not met the study milestones or timeline specified in the study protocol, we must provide a rationale to the FDA in our progress reports. If a change in the study milestones or timeline could significantly affect the outcome of the Post-Approval Study, we will need to submit that revision for the agency’s review and approval. We will need to update MelaFind®’s labeling with the results from this study, including any positive or negative results.
We may be unable to complete our Post-Approval Study if, for example, we institute a recall of MelaFind® from the market. The FDA can terminate our study if we have not fulfilled or cannot fulfill the Post-Approval Study condition of approval; for example, if MelaFind® is not being sold because the device technology is obsolete, study questions are no longer relevant, we withdraw the PMA, or the study cannot answer the Post-Approval Study question. If the FDA determines the study cannot be completed as designed or because of study data inadequacies, but the study objectives remain important, the FDA may terminate the original study and discuss establishing a new post-approval study commitment and schedule. In appropriate circumstances, the FDA may order additional post-market surveillance.
The FDA may initiate withdrawal of approval of the PMA if the agency concludes we have not met the Post-Approval Study condition of approval and have not provided a valid scientific justification for doing so. The FDA also may withdraw the approval of the PMA (1) based on negative results from the Post-Approval Study that indicate the device is unsafe or ineffective under the approved labeling or (2) if we fail to conduct the study in accordance with the FDA’s regulations, including those related to institutional review board and informed consent. If the PMA approval is withdrawn, we would be unable to continue marketing the device without violating the Federal Food, Drug, and Cosmetic Act. The sites involved in our Post-Approval Study and we as sponsor of the study can be inspected by the FDA at any time to assess compliance with the Post-Approval Study agreement, protocol adherence, human subject protection, and data integrity.
19
Table of Contents
The FDA posts information about the status of post-approval studies on its website. These website postings could undermine the credibility of the Company or MelaFind®, or have other collateral effects. For example, the agency will identify the study status as “Progress Inadequate” if the study progress is inconsistent with the protocol, such as if the study is not meeting the enrollment schedule or study timeline, missing timepoint evaluations, if there are poor follow-up rates, or if not all the endpoints are evaluated.
We have incurred losses for over ten years, and anticipate that we will incur continued losses for the foreseeable future.
Since 1999, we have primarily financed our operations through the sale of our equity securities and have devoted substantially all of our resources to research and development relating to MelaFind®. Our net loss for the year ended December 31, 2012 was approximately $22.7 million, and as of December 31, 2012, we had an accumulated deficit of approximately $142.2 million. Our research and development expenses may increase in connection with our continued development activities related to MelaFind®. We expect to incur significant sales, marketing, contract-manufacturing and inventory build-up expenses which will require additional funding. As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity.
We may be unable to continue commercialization and continue development of MelaFind® enhancements or other products without additional funding and we will not be able to achieve significant commercialization without additional funding.
As of December 31, 2012 we had approximately $7.9 million in cash and cash equivalents. Our operations have consumed substantial amounts of cash for each of the last eleven years and we expect that our cash used by operations will increase significantly in each of the next several years. Subsequent to year end, we raised approximately $15.8 million in net proceeds comprised of approximately $8.5 million under our “at-the-market” equity offering program and approximately $7.3 million from a public offering of our common stock. In addition, in February 2013 the Company signed a term sheet to borrow up to $10 million, of which $6 million will be immediately available to the Company upon closing. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described. We believe that our year end cash and cash equivalents, combined with our financing activities subsequent to year end and anticipated revenues, will be sufficient to fund our anticipated levels of operations for at least the next twelve months. In the event that the Company is unable to raise additional funds, the Company has the ability and intent to reduce certain discretionary expenditures. However, we will need substantial funds to broaden the commercial expansion of MelaFind®, including further development of a direct sales force and expansion of our contract-manufacturing capacity. We also expect to continue to spend funds on research and development and product enhancements. Our business or operations may change in a manner that would consume available resources more rapidly than we anticipate. The amount of funding we will need will depend on many factors, including:
| • | the cost of commercialization activities, including product marketing and continuing to build a domestic direct sales force and conducting activities in Germany and ultimately throughout the European Union (“EU”); |
| • | the amount of direct payments we are able to obtain from physicians utilizing MelaFind®; |
| • | the costs of maintaining regulatory approval ; |
| • | reimbursement amounts for the use of MelaFind® that physicians are able to obtain from Medicare and third party payers; |
| • | the success of our research and development efforts in product creation and enhancement, and meeting competitive services and technologies; |
| • | the schedule, costs and results of any clinical trials and studies; |
| • | the costs of maintaining inventory and other manufacturing expenses; |
20
Table of Contents
| • | our ability to establish and maintain any collaborative, licensing or other arrangements, and the terms and timing of any such arrangements; |
| • | the costs involved in defending any patent infringement actions or other litigation claims brought against us by third parties; and |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other rights. |
Additional financing may not be available to us when we need it, or it may not be available on favorable terms. If we are unable to obtain adequate financing on a timely basis, we may be required to significantly curtail or cease one or more of our development and marketing programs. We also may have to reduce marketing, customer support and other resources devoted to our products. We could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own, or that may require us to grant a security interest in our assets. If we raise additional funds by issuing equity securities, our then-existing stockholders will experience ownership dilution, could experience declines in our share price and the terms of any new equity securities may have preferences over our common stock.
If we raise money through debt, we may be required to make monthly payments of principal and interest and the terms of the loan agreement may contain significant restrictions.
In February 2013, we signed a non-binding term sheet with a venture capital lender for a $10 million loan. It is anticipated that the loan will mature 42 months from the initial closing and bear interest at the rate of 10.45% per year. The term sheet contemplates that during the first 12 months of the loan, only interest will be paid to the lender and after that we will make 30 equal payments of principal and interest until maturity. The loan would be secured by a general lien against all of our assets, other than our intellectual property assets. In addition, the lender will have a security interest in the proceeds of the sale of any of our intellectual property assets. In connection with the loan, the lender will receive a warrant to purchase that number of shares of common stock equal to 775,000 divided by the to-be-determined exercise price of the warrant. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described.
MelaFind® may not be widely accepted by the dermatological community.
The success of MelaFind® will depend upon the level of acceptance by dermatologists who perform skin examinations and treat skin disorders, including industry opinion leaders, that the evaluation information provided by MelaFind® is medically useful and reliable. We will be subject to intense scrutiny before physicians will be comfortable incorporating MelaFind® in their diagnostic approaches. We believe that recommendations by respected physicians will be essential for the development and successful marketing of MelaFind®; however, there can be no assurance that a significant number of such recommendations will be obtained. To date, the medical community outside of our customer base has had little exposure to us and MelaFind®. Because the medical community is often skeptical of new companies and new technologies, we may be unable to gain access to potential customers in order to demonstrate the operation and effectiveness of MelaFind®. Even if we gain access to potential customers, no assurance can be given that members of the dermatological medical community will perceive a need for or accept MelaFind®. In particular, given the potentially fatal consequences of failing to detect melanoma at the early, curable stages, dermatologists may remain reluctant to use MelaFind®. Any of the foregoing factors, or other currently unforeseen factors, could limit or detract from market acceptance of MelaFind® by the dermatological community. Insufficient market acceptance of MelaFind® would have a material adverse effect on our business, financial condition and results of operations.
MelaFind® may not achieve general market acceptance at a level that will make us profitable.
Our future growth and profitability will depend, in large part, on speed and level of market acceptance for the use of MelaFind® among physicians, government and third party payers, and regulators.
21
Table of Contents
Physicians tend to be slow to change their diagnostic and medical treatment practices because of perceived liability risks arising from the use of new products and the uncertainty of third party reimbursement. Physicians may not begin to use MelaFind® until there is long-term clinical evidence to convince them to alter their existing methods of diagnosing or evaluating clinically atypical lesions and there are independent demonstrations that MelaFind® is effective. We cannot predict the speed at which physicians may adopt the use of MelaFind®. By limiting the initial cash outlay of MelaFind® to the physician, we believe we will accelerate its adoption and usage. However, by charging on a per patient basis we will increase the initial capital burden on the Company.
The degree of market acceptance of MelaFind® will depend on a number of factors, including:
| • | perceived effectiveness of MelaFind®; |
| • | convenience and cost of use; |
| • | availability and adequacy of third-party coverage or reimbursement; |
| • | publicity concerning MelaFind® or competitive products; |
| • | potential advantages over alternative diagnostic methodologies; |
| • | introduction and acceptance of competing products or technologies; and |
| • | extent and success of our sales, marketing and distribution efforts. |
If MelaFind® does not achieve an adequate level of acceptance by patients, physicians, healthcare payers and regulators, we may not generate significant product revenue and we may not become profitable.
MelaFind® may not be commercially viable if we fail to obtain an adequate level of reimbursement by Medicare, Medicaid and other third party payers.
The availability of medical insurance coverage and reimbursement for newly approved medical devices is uncertain. In the U.S., physicians and other healthcare providers performing biopsies for clinically atypical skin lesions are generally reimbursed for all or part of the cost of the diagnosis and biopsy by Medicare, Medicaid, or other third party payers. Significant commercial success of MelaFind® in both domestic and international markets may depend on whether third-party coverage and reimbursement are available for services involving MelaFind®.
In the U.S., Medicare, Medicaid, health maintenance organizations and other third-party payers are increasingly attempting to contain healthcare costs by limiting both the scope of coverage and the level of reimbursement of new medical devices, and as a result, they may not cover or provide adequate payment for the use of MelaFind®. In order to obtain satisfactory reimbursement arrangements, we may have to agree to a fee or sales price lower than the fee or sales price we might otherwise charge. Even if Medicare and other third-party payers decide to cover procedures involving our product, we cannot be certain that the reimbursement levels will be adequate. Accordingly, unless government and other third-party payers provide adequate coverage and reimbursement for our products, some physicians may be discouraged from using them, and our sales would suffer.
Medicare reimburses for medical devices in a variety of ways, depending on where and how the device is used. However, Medicare only provides reimbursement if the Centers for Medicare & Medicaid Services, the federal agency that administers Medicare (“CMS”), determines that the device should be covered and that the use of the device is consistent with the coverage criteria. A coverage determination can be made at the local level by the Medicare administrative contractor, a private contractor that processes and pays claims on behalf of CMS for the geographic area where the services were rendered, or at the national level by CMS through a national coverage determination. There are statutory provisions intended to facilitate coverage determinations for new technologies. Coverage presupposes that the device has been cleared or approved by the FDA and further, that the coverage will be no broader than the approved intended uses of the device as approved or cleared by the
22
Table of Contents
FDA, but coverage can be narrower. A coverage determination may be so limited that relatively few patients will qualify for a covered use of the device. Should a very narrow coverage determination be made for MelaFind®, it may undermine the commercial viability of MelaFind®.
Germany is the only country in the world with a national skin screening program. Based on this program, public insurance (90% of the population) covers a visual examination only conducted by a General Practitioner or dermatologists — they do not yet cover imaging technologies/diagnostics devices. For coverage of imaging technologies/diagnostic devices, patients must be privately insured, have supplemental insurance or pay out-of-pocket. Private insurance (10% of the population) and/or supplemental insurance coverage reimbursement varies by policy, but ranges from $65 to $195 for imaging technologies. We cannot be certain that all private German insurers will reimburse us or that the reimbursement we do obtain will be adequate for us to maintain our business in Germany.
Even if MelaFind® is approved for reimbursement by Medicare, Medicaid and/or other third party payers, we anticipate there will be significant pressures on pricing.
We expect to experience pricing pressures in connection with the commercialization of MelaFind® due to efforts by private and government-funded payers to reduce or limit the growth of healthcare costs, the increasing influence of health maintenance organizations, and additional legislative proposals to reduce or limit increases in public funding for healthcare services. Private payers, including managed care payers, increasingly are demanding discounted fee structures and the assumption by healthcare providers of all or a portion of the financial risk. Efforts to impose greater discounts and more stringent cost controls upon healthcare providers by private and public payers are expected to continue. Payers frequently review their coverage policies for existing and new diagnostic tools and can, sometimes without advance notice, deny or change their coverage policies. Significant limits on the scope of services covered or on reimbursement rates and fees on those services that are covered could have a material adverse effect on our ability to successfully commercialize MelaFind® and therefore, on our liquidity, margins and our business, financial condition, and results of operations.
Obtaining a coverage determination by Medicare or Medicaid is a time-consuming, expensive and highly uncertain proposition.
Obtaining a coverage determination, whether local or national, is a time-consuming, expensive and highly uncertain proposition, especially for a new technology, and inconsistent local determinations are possible. On average, according to an industry report, Medicare coverage determinations for medical devices lag 15 months to five years or more behind FDA approval for that device. The Medicare statutory framework is also subject to administrative rulings, interpretations and discretion that affect the amount and timing of reimbursement made under Medicare. Medicaid coverage determinations and reimbursement levels are determined on a state by state basis, because Medicaid, unlike Medicare, is administered by the states under a state plan filed with the Secretary of the U.S. Department of Health and Human Services (“HHS”). Medicaid generally reimburses at lower levels than Medicare. Moreover, Medicaid programs and private insurers are frequently influenced by Medicare coverage determinations. The length of time it takes for us to obtain a coverage determination may affect the ability of MelaFind® to become commercially viable.
We depend on clinical investigators and clinical sites and other third parties to manage our clinical trials and to perform related data collection and analysis, and, as a result, we may face costs and delays that are outside of our control.
We have and will continue to rely on clinical investigators and clinical sites, some of which are private practices, and some of which are research, university or government affiliated, to enroll patients in any future clinical trials which we may conduct, as well as our FDA mandated post-approval studies. We have and will continue to rely on: pathologists and pathology laboratories; a contract research organization to assist in monitoring, collecting data, and ensuring FDA Good Clinical Practices (“GCP”) are observed at our sites; a consultant biostatistician; and other third parties to manage trials and to perform related data collection and
23
Table of Contents
analysis. However, we may not be able to control the amount and timing of resources that clinical sites and other third parties devote to our clinical trials or studies. Our agreements with clinical investigators and clinical sites for clinical testing generally place substantial responsibilities on these parties and, if these parties fail to perform as expected, our trials or studies could be delayed or terminated. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain are compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials or studies may be extended, delayed or terminated, and we may be unable to complete our studies or obtain regulatory approval for any other products which may be developed from our core technology. If these clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or studies, or if the clinical sites fail to comply adequately with the clinical protocols, we will be unable to complete any such trials or studies, which could prevent us from obtaining regulatory approvals for the products being developed.
In addition to the foregoing, any future clinical trials may be delayed or halted for numerous other reasons, including, but not limited to, the following:
| • | the FDA, an Institutional Review Board (“IRB”) or other regulatory authorities place our clinical trial on hold; |
| • | patients do not enroll in clinical trials at the rate we expect; |
| • | patient follow-up is not at the rate we expect; |
| • | IRBs and third-party clinical investigators delay or reject our trial protocol; |
| • | third-party organizations do not perform data collection and analysis in a timely or accurate manner; |
| • | regulatory inspections of our clinical trials or facilities manufacturing our products, among other things, require us to undertake corrective action or suspend or terminate our clinical trials, or invalidate our clinical trials; |
| • | changes in governmental regulations or administrative actions; and |
| • | the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness. |
Technological breakthroughs in the diagnosis or treatment of melanoma could render MelaFind® obsolete.
The precision optical imaging field is subject to rapid technological change and product innovation. MelaFind® is based on our proprietary technology, but a number of companies and medical researchers are pursuing new technologies such as confocal microscopy, an approach for non-invasive visualization of skin structures at the cellular level; and confocal Raman Micro-Spectroscopy which uses a reflective laser to produce a molecular fingerprint of the underlying tissue to indicate the presence or absence of disease. Other imaging modalities being developed include molecular imaging, in which tagged antibodies search for cancer cell antigens.
Also being developed is an electrical impedance technology for melanoma detection. The method is based on a technology that uses the varying electrical properties of human tissue to categorize the cell structures and thereby detect malignancies. Furthermore, several additional light based imaging approaches have recently been identified, including:
| • | a technology that measures how much light is absorbed in healthy versus diseased tissue to determine whether cancer is present; |
| • | a satellite-based remote imaging technology for use in detecting skin changes which could indicate the presence of cancer; |
| • | a scanner that provides real-time sub-surface images of tissue at far higher resolution than is possible with existing technologies such as ultrasound, CT or MRI, in 2D and 3D; |
24
Table of Contents
| • | a device that currently uses reflected visual light to analyze non-melanoma lesions; and |
| • | a device for non-invasive diagnosis of and screening for skin cancer; and a method for computer-aided analysis of photographs of skin lesions to detect the cancer which uses a traditional RGB (Red Green Blue) image as its computer source. |
The commercial development, market acceptance and reimbursement approval of any of these new technologies could result in a technological breakthrough in the diagnosis and/or treatment of melanoma, which could render MelaFind® less accepted or obsolete.
We operate in a highly competitive market, we may face competition from large, well-established medical device manufacturers with significant resources, and we may not be able to compete effectively.
We do not know of any product possessing the diagnostic assistance capabilities of MelaFind®. While several companies including Verisante, Scibase and Caliber Imaging and Diagnostic, Inc. (formerly Lucid, Inc.) have technologies that may be used to assist the dermatologist none of these companies’ products have undergone the rigors of FDA PMA review and subsequent approval. We believe that other products that enhance the visualization and analysis of potential melanomas have been approved or are under development by: Welch Allyn, Inc.; Heine Optotechnik; 3Gen, LLC; Derma Medical Systems, Inc.; MedX Health; Biomips Engineering, Michelson Diagnostics, Riester, ViseoMed, AG and others. In addition, several companies have developed various dermatological apps for use with an Apple iPhone. The broader market for precision optical imaging devices used for medical diagnosis is intensely competitive, subject to rapid change, and significantly affected by new product introductions and other market activities of industry participants. We will potentially be subject to competition from major optical imaging companies, such as: Raytheon Corporation, General Electric Co.; Siemens AG; Bayer AG; Olympus Corporation; Carl Zeiss AG Deutschland; and others, each of which manufactures and markets precision optical imaging products for the medical market, and could decide to develop or acquire a product to compete with MelaFind®. These companies enjoy numerous competitive advantages, including:
| • | significantly greater name recognition; |
| • | established relations with healthcare professionals, customers and third-party payers; |
| • | established distribution networks; |
| • | additional lines of products, and the ability to offer rebates, higher discounts or incentives to gain a competitive advantage; |
| • | greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products, and marketing approved products; and |
| • | greater financial and human resources for product development, sales and marketing, and patent litigation. |
As a result, we may not be able to compete effectively against these companies or their products.
If we are unable to establish adequate sales, marketing and distribution capabilities or enter into and maintain arrangements with third parties to sell, market and distribute MelaFind®, our business may be harmed.
We have a limited sales organization, and have limited experience in the marketing and distribution of MelaFind® or similar devices. To achieve commercial success for MelaFind®, we must develop a full sales and marketing force or enter into arrangements with others to market and sell our products. We have established a small direct sales force to market MelaFind® in the U.S. and Europe (initially in Germany), focused on introducing it at high volume dermatologists’ offices and training their staffs in its use. We anticipate that we will
25
Table of Contents
need additional funds in order to fully implement this marketing plan. In addition to being expensive, developing such a sales force is time consuming and could delay or limit the success of any product launch. We may not be able to develop this capacity on a timely basis or at all. Qualified direct sales personnel with experience in the medical device market are in high demand, and there is no assurance that we will be able to hire or retain an effective direct sales team. Similarly, qualified, independent medical device representatives both within and outside the U.S. are in high demand, and we may not be able to build an effective network for the distribution of our product through representatives. We can give no assurance that we will be able to enter into contracts with representatives on terms acceptable or reasonable to us. Similarly, there is no assurance that we will be able to build an alternate distribution framework, should we attempt to do so.
We will need to contract with third parties in order to sell and install our products in larger markets, including non-specialist dermatologists. To the extent that we enter into arrangements with third parties to perform marketing and distribution services in the U.S. and Europe, our product revenue could be lower and our costs higher than if we directly marketed MelaFind®. Furthermore, to the extent that we enter into co-promotion or other marketing and sales arrangements with other companies, any revenue received will depend on the skills and efforts of others, and we do not know whether these efforts will be successful. If we are unable to establish and maintain adequate sales, marketing and distribution capabilities, independently or with others, we will not be able to generate product revenue, and may not become profitable.
We have limited manufacturing capabilities and if we are not able to procure an adequate supply of MelaFind®, our growth could be limited and our business could be harmed.
We have limited experience in manufacturing MelaFind® for commercial distribution. Since we utilize contract manufacturers for our product, we do not have in-house resources and facilities to commercially manufacture MelaFind®. In order to produce MelaFind® in the quantities we anticipate necessary to meet market demand, we will need to increase our third-party manufacturing capacity. There are technical challenges to increasing manufacturing capacity, including equipment design and automation, material procurement, problems with production yields, and quality control and assurance. Developing commercial-scale manufacturing facilities that meet FDA requirements would require the investment of substantial additional funds and the hiring and retaining of additional management and technical personnel who have the necessary manufacturing experience.
We currently outsource production to contract manufacturers. Any difficulties in the ability of third-party manufacturers to supply devices of the quality, at the times, and in the quantities we need, could have a material adverse effect on our business, financial condition, and results of operations. Similarly, when we enter into contracts for the third-party manufacture of our devices, the quality of the devices will depend on the skills and efforts of others, and we do not know whether these efforts will be successful. Manufacturers often encounter difficulties in scaling up production of new products, including problems involving product yields, controlling and anticipating product costs, quality control and assurance, component supply, and shortages of qualified personnel. We cannot assure you that the third-party contract manufacturers with whom we have developed or are developing relationships will have or sustain the ability to produce the quantities of MelaFind® needed for development or commercial sales, or will be willing to do so at prices that allow MelaFind® to compete successfully in the market.
The failure by us or our suppliers to produce a sufficient number of MelaFind® devices that can operate according to our specifications could delay the full commercialization of MelaFind®, and would adversely affect both our ability to successfully commercialize MelaFind® and our business, financial condition and results of operations.
Our manufacturing operations for MelaFind® are dependent upon third-party suppliers, making us vulnerable to supply problems and price fluctuations, which could harm our business.
For manufacturing MelaFind® we rely on several vendors for critical components and materials such as: ON Semi, Carl Zeiss Jena GmbH (“Zeiss”), AB Electronics, AmeriCad and Canvys Electronics. Additionally, we are
26
Table of Contents
currently working with ASKION in Germany for the provision of the hand-held components and tested MelaFind® systems. We are utilizing Nexcore Technology Inc., an FDA regulated and ISO certified contract manufacturer of medical devices in New Jersey, to provide the assembled MelaFind® carts and tested MelaFind® systems.
There can be no assurance that these third parties will meet their obligations. Each of these suppliers is a sole-source supplier. Our contract suppliers also rely on sole-source suppliers to manufacture some of the components used in our products. Our manufacturers and suppliers may encounter problems during manufacturing due to a variety of reasons, including failure to procure their raw material on time, failure to follow specific protocols and procedures, failure to comply with applicable regulations, equipment malfunction and environmental factors, any of which could delay or impede their ability to meet our demand. Our reliance on these outside manufacturers and suppliers also subjects us to other risks that could harm our business, including:
| • | suppliers may make errors in manufacturing components that could negatively impact the effectiveness or safety of our products, or cause delays in shipment of our products; |
| • | we may not be able to obtain adequate supply in a timely manner or on commercially reasonable terms; |
| • | we may have difficulty locating and qualifying alternative suppliers for our sole-source suppliers; |
| • | switching components may require product redesign and submission to the FDA of a PMA supplement or possibly a separate PMA, either of which could significantly delay production; |
| • | our suppliers manufacture products for a range of customers, and fluctuations in demand for the products these suppliers manufacture for others may affect their ability to deliver components to us in a timely manner; and |
| • | our suppliers may encounter financial hardships unrelated to our demand for components, which could inhibit their ability to fulfill our orders and meet our requirements. |
Any interruption or delay in the supply of components or materials, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders, which would adversely affect both our ability to successfully commercialize MelaFind® and our business, financial condition and results of operations.
We have entered into an agreement with ASKION to continue developmental engineering, production and testing of our hand-held component, and to also assemble and test the integrated finished MelaFind® system, including the cart, for units to be sold within the European Union. Failure to maintain such an agreement with ASKION on mutually acceptable terms would require us to expand our own manufacturing facilities or obtain such services elsewhere. Similarly, through ASKION we have entered into a production agreement with Zeiss for lenses and lens objective assemblies. The manufacturing agreement with ASKION includes the integration of the Zeiss lenses in the hand-held components. Our planned reliance upon an outside provider for assembly and production services subjects us to the risk of adverse consequences from delays and defects caused by the failure of such outside supplier to meet its contractual obligations, including confidentiality obligations in the case of Zeiss, which is an affiliate of Carl Zeiss AG, a potential competitor.
Significant purchases of MelaFind® systems from our contract manufacturers will result in greater amounts of property and equipment on our balance sheet which will lead, to among other things, increased depreciation expenses on our income statement and may affect our return on capital investment.
From time to time we plan to issue significant purchase orders, to be paid and delivered over time, under master production agreements and similar arrangements with our contract manufacturers in order to satisfy the projected placements of MelaFind® systems. Certain purchase orders may be for very substantial amounts, some of which may be in excess of $1 million, and represent firm commitments to purchase MelaFind® systems (or components thereof). The timing and size of these purchase orders will depend on our market forecasts and our
27
Table of Contents
available capital resources. As we purchase larger numbers of MelaFind® systems, the amount of property and equipment on our balance sheet will correspondingly increase. The increased amount of property and equipment on our balance sheet will result in larger amounts of depreciation expense which will impact the amount of net income (loss) reported on our income statement. If we are unable to place purchased MelaFind® systems we may experience expenses for obsolescence. The destruction or loss of any of the placed MelaFind® systems may also result in loss above and beyond the amounts for which they are insured.
Our current business model entails an immediate upfront expense to the Company in connection with the purchase of a MelaFind® system, the cost of which is then offset by an initial installation fee and a future revenue stream to the Company every time the system is used. It may take several years before the anticipated income from a placed MelaFind® system is sufficient to recover the unit’s original cost.
MelaFind ® is complex and may contain undetected design defects and errors, which could have a material adverse impact on our business, financial condition and results of operations.
MelaFind® is complex and may contain undetected design defects and errors when first introduced, or errors that may be introduced when enhancements are released. Such defects and errors may occur despite our testing, and may not be discovered until after our devices have been shipped to and used by our customers. The existence of these defects and errors could result in costly repairs, returns of devices, diversion of development resources and damage to our reputation in the marketplace. In addition, when we contract with third-party manufacturers for the production of our products, these manufacturers may inadvertently produce devices that vary from devices we have produced in unpredictable ways that cause adverse consequences. Any of these conditions could have a material adverse impact on our business, financial condition and results of operations.
We are subject to the risks of international trade, including possible import/export restrictions and fluctuations in foreign currency exchange rates.
Many significant components of the MelaFind® system are manufactured by foreign suppliers and we also market MelaFind® internationally. We may be subject to various import duties applicable to materials manufactured in foreign countries and, in addition, may be affected by various other import and export restrictions, as well as other considerations or developments impacting upon international trade, including economic or political instability, shipping delays and product quotas. These international trade factors may have an adverse impact on the cost of components and the prices we can charge for the MelaFind® system. To the extent that transactions relating to the purchase of components and materials or the sale of products involve currencies other than U.S. dollars, our operating results will be affected by fluctuations in foreign currency exchange rates.
We may be required to purchase obsolete parts for MelaFind®.
Our MelaFind® design incorporates certain unique components, some of which have become obsolete and others of which may, from time-to-time, become obsolete. We will eventually be unable to replace obsolete components and an extensive re-design and re-approval process for MelaFind® will be required in order to utilize substitute components. If we are unable to provide component availability during that re-design and regulatory approval process, or are unable to effectively re-design and complete the re-approval process for MelaFind®, our ability to produce MelaFind® will become impaired, we may incur substantial additional costs and it would have a material adverse effect on our business, financial condition and results of operations. Furthermore, our actions in building inventory of parts that may become obsolete and have to be disposed of at a loss or written-down may add expenses to our operations and reduce our margins.
28
Table of Contents
We will not be able to sell MelaFind® unless its design verification and validation are maintained in accordance with current good manufacturing practices as set forth in the U.S. medical device Quality System Regulation (“QSR”) and ISO 13485 certification.
Prior to the installation of the first commercial MelaFind® system in March of 2012, we completed all the steps necessary to verify and validate the design of the MelaFind® system that were required to be performed prior to commercialization. If we are unable to maintain design verification and validation successfully, we will not be able to sell MelaFind®, and we will not be able to meet our plans for the full commercialization of MelaFind®. Later discovery of previously unknown problems with MelaFind®, including manufacturing problems, or failure to comply with regulatory requirements such as the FDA QSR and ISO 13485, may result in restrictions on MelaFind® or its manufacturing processes, withdrawal of MelaFind® from the market, patient or physician notification, voluntary or mandatory recalls, fines, withdrawal of regulatory approvals, refusal to approve pending applications or supplements to approved applications, refusal to permit the import or export of our products, product seizures, injunctions or the imposition of civil or criminal penalties. Should any of these enforcement actions occur, our business, financial condition and results of operations could be materially and adversely affected.
If we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with MelaFind®, it could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data and promotional activities for such product, will be subject to continuous review and periodic inspections by the FDA and other regulatory bodies, including Germany’s Federal Institute for Drugs and Medical Devices. In particular, we and our suppliers are required to comply with the QSR, ISO 13485 and other U.S. and European regulations which cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, storage, promotion, distribution, and shipping of MelaFind®. We also will be subject to ongoing U.S. and foreign regulatory requirements, including required submissions of safety and other post-market information and reports and registration and listing requirements. Furthermore, our third-party contract manufacturers will be required to adhere to current cGMP requirements enforced by the FDA as part of QSR, or similar regulations required by regulatory agencies in other countries. The manufacturing facilities of our contract manufacturers must be in full compliance with cGMP requirements before approval for marketing. The FDA enforces the QSR and other regulatory requirements through unannounced inspections.
If we are found to be deficient in cGMP or QSR (or any applicable foreign rules and regulations), we could be subject to regulatory action of a type described below, which could negatively affect our ability to successfully commercialize MelaFind®. There can be no assurance that the future interpretations of legal requirements made by the FDA or other U.S. or foreign regulatory bodies with possible retroactive effect, or the adoption of new requirements or policies, will not adversely affect us. We may be slow to adapt, or may not be able to adapt, to these changes or new requirements. Failure by us or one of our suppliers to comply with statutes and regulations administered by the FDA and other U.S. or foreign regulatory bodies, or failure to take adequate response to any observations, could result in, among other things, any of the following actions:
| • | warning letters; |
| • | fines and civil penalties; |
| • | unanticipated expenditures; |
| • | withdrawal of approval by the FDA or other regulatory bodies; |
| • | product recall or seizure; |
| • | interruption of production; |
| • | operating restrictions; |
| • | injunctions; and |
| • | criminal prosecution. |
29
Table of Contents
If any of these actions were to occur, it would harm our reputation and cause our product sales and profitability to suffer.
We are involved in a heavily regulated sector, and our ability to remain viable will depend on favorable government decisions at various points by various agencies.
Healthcare is heavily regulated by national and regional governments, both in the U.S. and other countries. The laws and regulations affecting healthcare change constantly, thereby increasing the uncertainty and risk associated with any healthcare related venture, including our business and MelaFind®.
For example, from time to time, legislation is introduced in the U.S. Congress that could significantly change the statutory provisions governing the approval, manufacture and marketing of a medical device. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance, or interpretations changed, and what the impact of such changes, if any, may be.
The U.S. federal government regulates healthcare through various agencies, including but not limited to the following: (i) the FDA, which administers the Federal Food, Drug, and Cosmetic Act, as well as other relevant laws; (ii) CMS, which administers the Medicare and Medicaid programs; (iii) the Office of Inspector General (“OIG”) which enforces various laws aimed at curtailing fraudulent or abusive practices, including by way of example, the Anti-Kickback Law, the Physician Self-Referral Law, commonly referred to as the Stark Law, the Anti-Inducement Law, the Civil Money Penalty Law, and the laws that authorize the OIG to exclude healthcare providers and others from participating in federal healthcare programs; and (iv) the Office of Civil Rights, which administers the privacy aspects of the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”). Healthcare is also provided or regulated, as the case may be, by the Department of Defense through its TriCare program, the Public Health Service within HHS under the Public Health Service Act, the Department of Justice through the Federal False Claims Act and various criminal statutes, and state governments under Medicaid and other state sponsored or funded programs and their internal laws regulating all healthcare activities.
In addition to regulation by the FDA as a medical device manufacturer, we are subject to general healthcare industry regulations. The healthcare industry is subject to extensive international, federal, state and local laws and regulations relating to:
| • | billing for services; |
| • | quality of medical equipment and services; |
| • | confidentiality, maintenance and security issues associated with medical records and individually identifiable health information; |
| • | false claims; and |
| • | labeling products. |
These laws and regulations are extremely complex and, in some cases, still evolving. In many instances, the industry does not have the benefit of significant regulatory or judicial interpretation of these laws and regulations. If our operations are found to be in violation of any of the international, federal, state or local laws and regulations that govern our activities, we may be subject to the applicable penalty associated with the violation, including civil and criminal penalties, damages, fines or curtailment of our operations. The risk of being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s time and attention from the operation of our business.
30
Table of Contents
Legislation relating to medical devices may have a material adverse effect on us.
On March 23, 2010, President Obama signed the Patient Protection and Affordable Care Act. The legislation imposes significant new excise taxes on medical device transactions. Under the legislation, the total cost to the medical device industry is estimated to be approximately $20 billion over ten years. In January 2013, a 2.3% excise tax on medical devices went into effect as a component of the Patient Protection and Affordable Care Act. This tax along with the others in the Act will result in a significant increase in the tax burden on our industry, which could have a material, negative impact on our results of operations and our cash flows. Other elements of this legislation such as comparative effectiveness research, an independent payment advisory board, payment system reforms including shared savings pilots and other provisions could meaningfully change the way healthcare is developed and delivered, and may materially impact numerous aspects of our business.
We must comply with complex statutes prohibiting fraud and abuse, and both we and physicians utilizing MelaFind® could be subject to significant penalties for noncompliance.
There are extensive federal and state laws and regulations prohibiting fraud and abuse in the healthcare industry that can result in significant criminal and civil penalties. These federal laws include: the anti-kickback statute which prohibits certain business practices and relationships, including the payment or receipt of remuneration for the referral of patients whose care will be paid by Medicare or other federal healthcare programs; the physician self-referral prohibition, commonly referred to as the Stark Law; the anti-inducement law, which prohibits providers from offering anything to a Medicare or Medicaid beneficiary to induce that beneficiary to use items or services covered by either program; the Civil False Claims Act, which prohibits any person from knowingly presenting or causing to be presented false or fraudulent claims for payment by the federal government, including the Medicare and Medicaid programs and; the Civil Monetary Penalties Law, which authorizes HHS to impose civil penalties administratively for fraudulent or abusive acts. Sanctions for violating these federal laws include criminal and civil penalties that range from punitive sanctions, damage assessments, money penalties, imprisonment, denial of Medicare and Medicaid payments, or exclusion from the Medicare and Medicaid programs, or both. As federal and state budget pressures continue, federal and state administrative agencies may also continue to escalate investigation and enforcement efforts to root out waste and to control fraud and abuse in governmental healthcare programs. Private enforcement of healthcare fraud has also increased, due in large part to amendments to the Civil False Claims Act in 1986 that were designed to encourage private persons to sue on behalf of the government. A violation of any of these federal and state fraud and abuse laws and regulations could have a material adverse effect on our liquidity and financial condition. An investigation into the use of MelaFind® by physicians may dissuade physicians from either purchasing or using MelaFind® and could have a material adverse effect on our ability to successfully commercialize MelaFind®.
The application of the privacy provisions of HIPAA is uncertain.
HIPAA, among other things, protects the privacy and security of individually identifiable health information by limiting its use and disclosure. HIPAA directly regulates “covered entities” (insurers, clearinghouses, and most healthcare providers) and indirectly regulates “business associates” with respect to the privacy of patients’ medical information. Certain entities that receive and process protected health information are required to adopt certain procedures to safeguard the security of that information. It is uncertain whether we would be deemed to be a covered entity under HIPAA, and it is unlikely that based on our current business model, we would be a business associate. Nevertheless, we may be contractually required to physically safeguard the integrity and security of the patient information that we or our physician customers receive, store, create or transmit. If we fail to adhere to our contractual commitments, then our physician customers may be subject to civil monetary penalties, and this could adversely affect our ability to market MelaFind®. We also may be liable under state laws governing the privacy of health information.
31
Table of Contents
We may become subject to claims of infringement or misappropriation of the intellectual property rights of others, which could prohibit us from shipping affected products, require us to obtain licenses from third parties or to develop non-infringing alternatives, and subject us to substantial monetary damages and injunctive relief. Our patents may also be subject to challenge on validity grounds, and our patent applications may be rejected.
Third parties could, in the future, assert infringement or misappropriation claims against us with respect to our current or future products. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Therefore, we cannot be certain that we have not infringed the intellectual property rights of such third parties. Our potential competitors may assert that some aspect of MelaFind® infringes their patents. There also may be existing patents of which we are unaware that one or more components of our MelaFind® system may inadvertently infringe.
Any infringement or misappropriation claim could cause us to incur significant costs, could place significant strain on our financial resources, divert management’s attention from our business and harm our reputation. If the relevant patents were upheld as valid and enforceable and we were found to infringe, we could be prohibited from selling our product unless we could obtain licenses to use the technology covered by the patent or are able to design around the patent. We may be unable to obtain a license on terms acceptable to us, if at all, and we may not be able to redesign MelaFind® to avoid infringement.
A court could order us to pay compensatory damages for such infringement, plus prejudgment interest and could, in addition, treble the compensatory damages and award attorney fees. These damages could be substantial and could harm our reputation, business, financial condition and operating results. A court also could enter orders that temporarily, preliminarily or permanently enjoin us and our customers from making, using, selling, offering to sell or importing MelaFind®, and/or could enter an order mandating that we undertake certain remedial activities. Depending on the nature of the relief ordered by the court, we could become liable for additional damages to third parties.
We rely on our patents, patent applications and other intellectual property rights to give us a competitive advantage. Whether a patent is valid, or whether a patent application should be granted, is a complex matter of science and law. Therefore we cannot be certain that, if challenged, our patents, patent applications and/or other intellectual property rights would be upheld. If one or more of those patents, patent applications and other intellectual property rights are invalidated, rejected or found unenforceable, those outcomes could reduce or eliminate any competitive advantage we might otherwise have had.
New product development in the medical device industry is both costly and labor intensive with very low success rates for successful commercialization; if we cannot successfully develop or obtain future products, our growth, beyond the growth related to MelaFind®, would be delayed.
Our long-term success is dependent, in large part, on the successful commercialization of MelaFind® and the further design and development of MelaFind® and our technology. The product development process is time-consuming, unpredictable and costly. There can be no assurance that we will be able to develop or acquire new products, successfully complete any related clinical trials, obtain the necessary regulatory clearances or approvals required from the FDA on a timely basis, or at all, manufacture our potential products in compliance with regulatory requirements or in commercial volumes, or that, even if approved and manufactured, such potential products will achieve market acceptance. In addition, changes in regulatory policy for product approval during the period of product development, and regulatory agency review of each submitted new application, may cause delays or rejections. It may be necessary for us to enter into licensing arrangements in order to market effectively any new products or new indications for existing products. There can be no assurance that we will be successful in entering into such licensing arrangements on terms favorable to us or at all. Failure to develop, obtain necessary regulatory clearances or approvals for, or successfully market potential new products could have a material adverse effect on our business, financial condition and results of operations.
32
Table of Contents
We face the risk of product liability claims and may not be able to obtain or maintain adequate insurance.
Our business exposes us to the risk of product liability claims that is inherent in the testing, manufacturing and marketing of medical devices, including those which may arise from the misuse or malfunction of, or design flaws in, our products. We may be subject to product liability claims if MelaFind® causes, or merely appears to have caused, an injury or if a patient alleges that MelaFind® failed to provide appropriate evaluation information on a lesion where melanoma was subsequently found to be present. Claims may be made by patients, healthcare providers or others involved with MelaFind®. Our coverage may not be adequate to protect us against any future product liability claims. If our insurance proves to be inadequate, we may not be protected against potential product liability claims and we will be exposed to significant liabilities which may harm our business. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could result in significant costs and significant harm to our business.
We may be subject to claims against us even if the apparent injury is due to the actions of others. For example, we rely on the expertise of dermatologists and other associated medical personnel to operate MelaFind®. If these medical personnel are not properly trained or are negligent, we may be subjected to claims and ultimately liability. These liabilities could prevent or interfere with our product commercialization efforts. Defending a suit, regardless of merit, could be costly, could divert management attention and might result in adverse publicity, which could result in reduced acceptance of MelaFind® in the market.
Insurance and surety companies have reassessed many aspects of their business and, as a result, may take actions that could negatively affect our business. These actions could include increasing insurance premiums, requiring higher self-insured retentions and deductibles, reducing limits, restricting coverage, imposing exclusions, and refusing to underwrite certain risks and classes of business. Any of these actions may adversely affect our ability to obtain appropriate insurance coverage at reasonable costs, which could have a material adverse effect on our business, financial condition and results of operations.
We may be adversely affected by a data center failure.
The success of MelaFind® is dependent upon our ability to protect our data center against damage from fire, power loss, telecommunications failure, natural disaster, sabotage or a similar catastrophic event. Substantially all of our computer equipment and data operations are located in a single facility. Our prospective failure to maintain off-site copies of information contained in our MelaFind® database, or our inability to use alternative sites in the event we experience a natural disaster, hardware or software malfunction or other interruption of our data center could adversely impact our business, financial condition and results of operations. While the Company does provide off-site back-up for its critical data, which we believe to be sufficient to meet our needs, there can be no assurance that our current plan can anticipate every possible eventuality.
We may be adversely affected by breaches of online security.
Our MelaFind® lesion database does not contain any information that allows us to identify specific patients. However, we must identify certain data as belonging to or as derived from specific patients for regulatory, quality assurance and billing purposes. To the extent that our activities involve the storage and transmission of confidential information, security breaches could damage our reputation and expose us to a risk of loss, or to litigation and possible liability. Our business may be materially adversely affected if our security measures do not prevent security breaches. In addition, such information may be subject to HIPAA privacy and security regulations, the potential violation of which may trigger concerns by healthcare providers, which may adversely impact our business, financial condition and results of operations.
We are dependent upon telecommunications and the internet.
We use the internet to inform the public about the availability of our products and to market to and communicate with physicians who are potential or actual customers. Our success will therefore depend in part on the continued growth and use of the internet. If our ability to use the internet fails, it may materially adversely affect our business.
33
Table of Contents
All of our operations are conducted at a single location. Any disruption at our facility could increase our expenses.
Substantially all of our operations are conducted at a single building in Irvington New York. We take precautions to safeguard our facility, including insurance, health and safety protocols, contracted off-site engineering services, and storage of computer data. However, a natural disaster, such as a fire, flood or earthquake, could cause substantial delays in our operations or cause us to incur additional expenses. The insurance we maintain against fires, floods, earthquakes and other natural disasters may not be adequate to cover our losses in any particular case.
We may be liable for contamination or other harm caused by materials that we handle, and changes in environmental regulations could cause us to incur additional expense.
Our research and development and clinical processes do not generally involve the handling of potentially harmful biological materials or hazardous materials, but they may occasionally do so. We are subject to federal, state and local laws and regulations governing the use, handling, storage and disposal of hazardous and biological materials. If violations of environmental, health and safety laws occur, we could be held liable for damages, penalties and costs of remedial actions. These expenses or this liability could have a significant negative impact on our business, financial condition and results of operations. We may violate environmental, health and safety laws in the future as a result of human error, equipment failure or other causes. Environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. Changes to or restrictions on permitting requirements or processes, hazardous or biological material storage or handling might require an unplanned capital investment or relocation. Failure to comply with new or existing laws or regulations could harm our business, financial condition and results of operations.
Our success will depend on our ability to attract and retain our personnel.
We are highly dependent on our senior management, especially Joseph V. Gulfo, M.D., our Chairman, President and Chief Executive Officer and Dina Gutkowicz-Krusin, Ph.D., our Director of Clinical Research and Principal Scientist. Our success will depend on our ability to retain our current senior management and to attract and retain qualified personnel in the future, including scientists, clinicians, engineers and other highly skilled personnel.
Competition for senior management personnel, as well as scientists, clinicians, engineers, and experienced sales and marketing individuals, is intense, and we may not be able to retain our personnel. The loss of the services of members of our senior management, scientists, clinicians or engineers could prevent the implementation and completion of our objectives, including the successful commercialization of MelaFind®. The loss of a member of our senior management or our professional staff would require the remaining executive officers to divert immediate and substantial attention to seeking a replacement.
We expect to expand our operations and grow our research and development, product development, administrative and marketing operations. This expansion is expected to place a significant strain on our management, and will require hiring a significant number of qualified personnel. Accordingly, recruiting and retaining such personnel in the future will be critical to our success. There is competition from other companies and research and academic institutions for qualified personnel in the areas of our activities. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our development and commercialization activities.
Climate control policy changes, including regulations issued by the Environmental Protection Agency and negotiated international treaties, could have an impact on our Company.
We cannot predict whether climate control legislation will be enacted and treaties ratified, the final form any legislation or treaties might take, or the effects of such legislation or treaties. If climate control legislation and/or regulations are enacted or treaties ratified, our operations or the operations of our suppliers could be adversely impacted affecting our ability to successfully commercialize MelaFind® in the U.S. marketplace.
34
Table of Contents
Results could be impacted by the effects of, and changes in, world-wide economic and capital market conditions.
Our business may be adversely affected by factors in the United States and other countries, such as Germany and the other member states of the European Union, that are beyond our control, such as disruptions in the financial markets or downturns in economic activity. The current world-wide economic conditions could have an adverse impact on the availability and cost of capital, interest rates, tax rates, or regulations.
Risks Relating to our Common Stock
If we fail to maintain the adequacy of our internal controls, our ability to provide accurate financial statements could be impaired and any failure to maintain our internal controls could have an adverse effect on our stock price.
The Sarbanes-Oxley Act of 2002 (“SOX”), as well as rules implemented by the SEC, the Public Company Accounting Oversight Board and the NASDAQ Stock Market, have required changes in the corporate governance practices of public companies. Monitoring compliance with the existing rules and implementing changes required by these rules may increase our legal and financial compliance costs, divert management attention from operations and strategic opportunities, and make legal, accounting and administrative activities more time-consuming and costly. Since 2008, we have retained a consultant experienced in SOX that assists us in the process of instituting changes to our internal procedures to satisfy the requirements of the SOX. We have evaluated our internal control systems in order to allow us to report on, and our independent registered public accounting firm to attest to, our internal controls, as required by Section 404 of the SOX. As a small company with limited capital and human resources, we may need to divert management’s time and attention away from our business in order to ensure continued compliance with these regulatory requirements. We may require new information technologies systems, the auditing of our internal controls, and compliance training for our directors, officers and personnel. Such efforts may entail a significant expense. If we fail to maintain the adequacy of our internal controls as such standards are modified, supplemented or amended from time to time, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal control over financial reporting in accordance with Section 404 of the SOX. Any failure to maintain the adequacy of our internal controls could have an adverse effect on timely and accurate financial reporting and the trading price of our common stock.
An active trading market for our common stock may not be sustained.
An active public market for our common stock may not be sustained. Further, we cannot be certain that the market price of our common stock will not decline below the amount required by NASDAQ to maintain a listing on its Capital Market. Should we fail to meet the minimum standards established by NASDAQ for its Capital Market, we could be de-listed which could have an adverse effect on our stockholders ability to sell their stock.
If our common stock is delisted from The NASDAQ Capital Market, we may be subject to the risks relating to penny stocks.
If our common stock were to be delisted from trading on The NASDAQ Capital Market and the trading price of the common stock were below $5.00 per share on the date the common stock were delisted, trading in our common stock would also be subject to the requirements of certain rules promulgated under the Exchange Act. These rules require additional disclosure by broker-dealers in connection with any trades involving a stock defined as a “penny stock” and impose various sales practice requirements on broker-dealers who sell penny stocks to persons other than established customers and accredited investors, generally institutions. These additional requirements may discourage broker-dealers from effecting transactions in securities that are classified as penny stocks, which could severely limit the market price and liquidity of such securities and the ability of purchasers to sell such securities in the secondary market. A penny stock is defined generally as any non-exchange listed equity security that has a market price of less than $5.00 per share, subject to certain exceptions.
35
Table of Contents
Our ability to issue additional shares of common stock is severely limited.
We are restricted from issuing additional shares of our common stock, or from issuing securities that are convertible into or exchangeable for, or that represent the right to receive, our common stock. Our total number of authorized shares of common stock is 45 million and as of February 28, 2013, we had an aggregate of 43,037,144 shares of common stock issued and outstanding. In addition we had approximately 2.6 million shares of our common stock subject to issuance under outstanding options and warrants, which includes 900,000 shares underlying an outstanding option which is subject to a forbearance agreement precluding exercise until such time as the underlying shares become available. We are planning to take the steps necessary to increase the number of our authorized shares of common stock. However there can be no assurances that we will be able to do so.
Our stock price may be volatile, meaning purchasers of our common stock could incur substantial losses.
Our stock price has been and is likely to continue to be volatile. The stock market in general and the market for medical technology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The following factors, in addition to other risk factors described in this section and general market and economic conditions, may have a significant impact on the market price of our common stock:
| • | the timing of regulatory approval for our future products; |
| • | failure of any of our products to achieve commercial success; |
| • | results of our research and development efforts and our clinical trials; |
| • | the announcement of new products or product enhancements by us or our competitors; |
| • | regulatory developments in the U.S. and foreign countries; |
| • | our ability to manufacture our products to commercial standards; |
| • | developments concerning our clinical collaborators, suppliers or marketing partners; |
| • | changes in financial estimates or recommendations by securities analysts; |
| • | public concern over our products; |
| • | developments or disputes concerning patents or other intellectual property rights; |
| • | product liability claims and litigation against us or our competitors; |
| • | the departure of key personnel; |
| • | the strength of our balance sheet; |
| • | variations in our financial results or those of companies that are perceived to be similar to us; |
| • | changes in the structure of third-party reimbursement in the U.S. and other countries; |
| • | changes in accounting principles or practices; |
| • | general economic, industry and market conditions; and |
| • | future sales of our common stock. |
A decline in the market price of our common stock could cause you to lose some or all of your investment, limit your ability to sell your shares of stock and may adversely impact our ability to attract and retain employees and raise capital. In addition, stockholders have, and may in the future, initiate securities class action lawsuits if the market price of our stock drops significantly. Whether or not meritorious, litigation brought against us could result in substantial costs and could divert the time and attention of our management. Our insurance to cover claims of this sort may not be adequate.
36
Table of Contents
Our charter documents and Delaware law may inhibit a takeover that stockholders consider favorable and could also limit the market price of our stock.
Provisions of our restated certificate of incorporation and bylaws and applicable provisions of Delaware law may make it more difficult for or prevent a third party from acquiring control of us without the approval of our board of directors. These provisions:
| • | set limitations on the removal of directors; |
| • | limit who may call a special meeting of stockholders; |
| • | establish advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted upon at stockholder meetings; |
| • | do not permit cumulative voting in the election of our directors, which would otherwise permit less than a majority of stockholders to elect directors; |
| • | prohibit stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; and |
| • | provide our board of directors the ability to designate the terms of and issue a new series of preferred stock without stockholder approval. |
In addition, Section 203 of the Delaware General Corporation Law generally limits our ability to engage in any business combination with certain persons who own 15% or more of our outstanding voting stock or any of our associates or affiliates who at any time in the past three years have owned 15% or more of our outstanding voting stock.
These provisions may have the effect of entrenching our management team and may deprive you of the opportunity to sell your shares to potential acquirers at a premium over prevailing prices. This potential inability to obtain a control premium could reduce the price of our common stock.
Item 1B. Unresolved Staff Comments
Not applicable.
We lease approximately 21,700 square feet of office, laboratory, and assembly space in a building with the street address of 50 South Buckhout Street, Suite 1, Irvington, New York 10533. The lease expires in December 2016. We believe that this facility is adequate to meet our current and reasonably foreseeable requirements. We believe that we will be able to obtain additional space, if required, on commercially reasonable terms. Manufacturing agreements with our contract manufacturers allow for the inclusion of charges for finished goods warehousing services as a component of their overhead charges.
On November 19, 2010, a purported securities class action complaint was filed in the U.S. District Court for the Southern District of New York, naming as defendants the Company and certain of its officers and directors, entitled Randall J. Pederson, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo, Richard I. Steinhart, and Breaux Castleman, No. 7:10-cv-08774-JFM. Two similar complaints were also filed, one on December 2, 2010 and the other on January 20, 2011, in the same District Court, entitled Amy Steigman, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo, Richard I. Steinhart, and Breaux Castleman, No. 7:10-cv-09024-JFM; and Martin Slove and Linda Slove, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo,
37
Table of Contents
Richard I. Steinhart, and Breaux Castleman, No. 1:11 cv-00429-JFM. These three securities class actions were consolidated into one action on February 15, 2011, entitled In re MELA Sciences, Inc. Securities Litigation, No. 7:10-Cv-08774-JFM (“securities class action”). The securities class action plaintiffs asserted violations of the Securities Exchange Act of 1934, alleging, among other things, that defendants made misstatements and omissions regarding the Company’s product, MelaFind®, and its prospects for FDA approval, on behalf of stockholders who purchased the Company’s common stock during the period from February 13, 2009 through November 16, 2010, and sought unspecified damages. On May 2, 2011, the securities class action plaintiffs filed their amended consolidated complaint, alleging similar claims to their prior complaints. On July 29, 2011, defendants filed a motion to dismiss the consolidated amended complaint in its entirety. Plaintiff’s opposition to the motion to dismiss was filed on September 23, 2011. In light of the Company’s receipt of the Approvable Letter from the FDA for the MelaFind® PMA Application on September 22, 2011, plaintiffs filed a motion for leave to amend the consolidated amended complaint on November 18, 2011, which defendants opposed. On September 19, 2012, the court denied plaintiffs’ motion for leave to amend the consolidated amended complaint. On September 28, 2012, the court reinstated and granted defendants’ motion to dismiss the consolidated amended complaint. On October 22, 2012, plaintiffs filed a notice of appeal from the Judgments denying Lead Plaintiffs’ motion to amend the consolidated amended complaint and granting Defendants’ motion to dismiss the consolidated amended complaint. On December 7, 2012, plaintiffs withdrew the appeal with prejudice and without costs pursuant to a stipulation between the parties. On December 10, 2012, the United States Court of Appeals for the Second Circuit granted the stipulation and dismissed the appeal.
From time to time, we may be a party to certain legal proceedings, incidental to the normal course of our business. These may include controversies relating to contract claims and employment related matters, some of which claims may be material, in which case, we will make separate disclosure as required.
Item 4. Mine Safety Disclosures
Not applicable
PART II
| Item 5. Market | for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock has been traded on the NASDAQ Capital Market since October 28, 2005 under the symbol MELA. Prior to such time, there was no public market for our common stock. The following table sets forth the range of the high and low intraday prices for the period of January 1, 2011 through December 31, 2012 as reported by the NASDAQ Capital Market:
| High | Low | |||||||
| Year Ended December 31, 2012 |
||||||||
| October 1 — December 31, 2012 |
$ | 3.35 | $ | 1.71 | ||||
| July 1 — September 30, 2012 |
$ | 4.37 | $ | 3.01 | ||||
| April 1 — June 30, 2012 |
$ | 4.87 | $ | 2.50 | ||||
| January 1 — March 31, 2012 |
$ | 5.13 | $ | 3.19 | ||||
| Year Ended December 31, 2011 |
||||||||
| October 1 — December 31, 2011 |
$ | 6.96 | $ | 3.31 | ||||
| July 1 — September 30, 2011 |
$ | 6.20 | $ | 1.75 | ||||
| April 1 — June 30, 2011 |
$ | 3.97 | $ | 2.22 | ||||
| January 1 — March 31, 2011 |
$ | 3.87 | $ | 2.44 | ||||
38
Table of Contents
As of January 31, 2012, there were approximately 119 holders of record of our common stock. This number does not include the number of persons whose shares are in nominee or in “street name” accounts through brokers.
Dividend Policy
We have never declared or paid cash dividends on our common stock. We currently intend to retain our cash for the development of our business. We do not intend to pay cash dividends to our stockholders in the foreseeable future.
Any future determination relating to our dividend policy will be made at the discretion of our board of directors and will depend on then existing conditions, including our earnings, financial condition, results of operations, level of indebtedness, contractual restrictions, capital requirements, business prospects and other factors our board of directors may deem relevant. Our board of directors’ ability to declare a dividend is also subject to limits imposed by Delaware law.
Securities Authorized For Issuance Under Equity Compensation Plans
| Plan Category at 12/31/2012 |
Number of securities to be issued upon exercise of outstanding options |
Weighted-average exercise price of outstanding options |
Number of securities remaining available under equity compensation plans (excluding securities reflected in the first column) |
|||||||||
| Equity compensation plans approved by stockholders |
2,426,533 | $ | 4.01 | 1,222,739 | ||||||||
| Equity compensation plans not approved by stockholders |
— | — | — | |||||||||
| Total |
2,426,533 | $ | 4.01 | 1,222,739 | ||||||||
39
Table of Contents
Item 6. Selected Financial Data
The following table sets forth selected financial data. The financial information for the years ended December 31, 2010, 2011, and 2012 and as of December 31, 2011 and 2012 has been derived from our audited financial statements and related notes appearing in Part II Item 8 of this report and should be read together with such financial statements and the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section appearing in Part II Item 7 of this report. The financial information for the years ended December 31, 2008 and 2009 and as of December 31, 2008, 2009, and 2010 have been derived from our audited financial statements not included in this report. The historical results are not necessarily indicative of results of any future periods.
| Year Ended December 31, | ||||||||||||||||||||
| 2008 | 2009 | 2010 | 2011 | 2012 | ||||||||||||||||
| (In thousands, except share and per share data) | ||||||||||||||||||||
| Statements of Operations Data: |
||||||||||||||||||||
| Revenue |
$ | 278 | ||||||||||||||||||
| Cost of revenue |
2,042 | |||||||||||||||||||
|
|
|
|||||||||||||||||||
| (1,764 | ) | |||||||||||||||||||
|
|
|
|||||||||||||||||||
| Research and development expenses |
$ | 12,508 | $ | 10,950 | $ | 11,497 | $ | 9,656 | 6,792 | |||||||||||
| Selling, general and administrative expenses |
5,766 | 7,631 | 8,738 | 10,806 | 14,169 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Operating loss from continuing operations |
(18,274 | ) | (18,581 | ) | (20,235 | ) | (20,462 | ) | (22,725 | ) | ||||||||||
| Interest income |
(468 | ) | (45 | ) | (32 | ) | (54 | ) | (32 | ) | ||||||||||
| Other income, net |
(201 | ) | (83 | ) | (280 | ) | (23 | ) | (20 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Loss from continuing operations |
(17,605 | ) | (18,453 | ) | (19,923 | ) | (20,385 | ) | (22,673 | ) | ||||||||||
| Gain from discontinued operations |
— | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net loss |
(17,605 | ) | (18,453 | ) | (19,923 | ) | (20,385 | ) | (22,673 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Net income ( loss) per share, basic and diluted: |
||||||||||||||||||||
| Continuing operations |
$ | (1.08 | ) | $ | (.96 | ) | $ | (.83 | ) | $ | (.80 | ) | $ | (.74 | ) | |||||
| Discontinued operations |
— | — | — | — | — | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted net loss per common share |
$ | (1.08 | ) | $ | (.96 | ) | $ | (.83 | ) | $ | (.80 | ) | $ | (.74 | ) | |||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Basic and diluted weighted average number of common shares outstanding |
16,282,176 | 19,293,761 | 24,043,135 | 25,415,880 | 30,762,610 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| As of December 31, | ||||||||||||||||||||
| 2008 | 2009 | 2010 | 2011 | 2012 | ||||||||||||||||
| (In thousands) | ||||||||||||||||||||
| Balance Sheet Data: |
||||||||||||||||||||
| Total current assets |
$ | 15,836 | $ | 30,338 | $ | 31,044 | $ | 29,058 | $ | 9,683 | ||||||||||
| Total assets |
16,620 | 32,127 | 33,589 | 31,393 | 17,270 | |||||||||||||||
| Total current liabilities |
1,530 | 1,811 | 1,686 | 1,448 | 3,019 | |||||||||||||||
| Total liabilities |
1,530 | 1,811 | 1,790 | 1,586 | 3,295 | |||||||||||||||
| Accumulated deficit |
(60,767 | ) | (79,220 | ) | (99,143 | ) | (119,527 | ) | (142,200 | ) | ||||||||||
| Total stockholders’ equity |
15,089 | 30,316 | 31,799 | 29,807 | 13,975 | |||||||||||||||
40
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion contains forward-looking statements, which involve risks and uncertainties. Our actual results could differ from those anticipated in these forward-looking statements as a result of various factors, including those set forth above under the caption “Risk Factors”. You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements for the year ended December 31, 2012 and the related notes appearing in Part II Item 8 of this report.
Overview
We are a medical device company focused on the commercialization of our flagship product, MelaFind®, and the further design and development of MelaFind® and our technology. MelaFind® is a non-invasive, point-of-care instrument to aid in the detection of melanoma. MelaFind®, features a hand-held component that emits multiple wavelengths of light to capture digital data from clinically atypical pigmented skin lesions. Prior to the commercial launch of MelaFind® commencing in the first quarter of 2012, the Company had not generated any revenues from MelaFind®. All of our historical revenues have come from activities and products that have since been discontinued.
In November 2011, the Company received written approval from the FDA for the MelaFind® PMA application and in September 2011 received CE Mark approval for MelaFind®. On March 7, 2012, the Company installed the first commercial MelaFind® systems, and proceeded with the first phase of the commercial launch of its breakthrough product for melanoma detection.
In 2012 the Company evolved from a research and development company to a commercial enterprise. The first commercial MelaFind® systems were placed in working office settings to obtain critical market feedback from dermatologists and their patients. The Company utilized this information to adapt the MelaFind® interface and to make system enhancements to optimize the user experience, accordingly. The market feedback from the initial installations was also used to determine the optimal medical messaging to dermatologists and in patient-directed materials in order to effectively communicate the features and benefits of MelaFind®.
The training program for dermatologists, required as part of the FDA approval of MelaFind®, was put into practice at launch. The training of the dermatologists and their staffs includes the appropriate use of MelaFind® and how best to integrate MelaFind® into their practices, protocols and algorithms for melanoma detection.
During 2012, the Company put in place and developed several critical capabilities to support its commercialization goals. Included among these added capabilities were sales and marketing leadership and staff; technical support and field installation resources; implementation of CRM (Customer Resource Management) and ERP (Enterprise Resource Planning) business systems, and expanded and optimized manufacturing processes with the Company’s critical business partners.
The initial launch of MelaFind® was controlled and deliberate, focused primarily on the U.S. Northeast, and several select cities in Germany. Midway through 2012, the scope was widened beyond these select cities and at December 31, 2012, we had over 100 signed user agreements with our customers throughout the U.S. and Germany. Management estimates that this phase of the commercial launch will be complete when approximately 275 user agreements are signed (roughly 200 in the US and 75 in Germany). The second phase of the MelaFind® launch will focus primarily on driving MelaFind® utilization.
Our revenue for the foreseeable future will depend on the success of commercialization of MelaFind®, and may vary substantially from year to year and quarter to quarter. Our operating expenses may also vary substantially from year to year and quarter to quarter in support of the commercialization of MelaFind®. We believe that period-to-period comparisons of our results of operations may not be meaningful and should not be relied on as indicative of our future performance.
41
Table of Contents
We commenced operations in December 1989 as a New York corporation and re-incorporated as a Delaware corporation in September 1997. Since our inception, we have generated significant losses. As of December 31, 2012, we had an accumulated deficit of approximately $142.2 million. We expect to continue to spend significant amounts on the commercialization of MelaFind®.
Subsequent to year end, we raised approximately $15.8 million in net proceeds comprised of approximately$ 8.5 million under our “at-the-market” equity offering program and approximately $7.3 million from a public offering of our common stock. In addition, in February 2013 the Company signed a term sheet to borrow up to $10 million, of which $6 million will be immediately available to the Company upon closing. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described. We believe that our year end cash and cash equivalents, combined with our financing activities subsequent to year end and anticipated revenues, will be sufficient to fund our anticipated levels of operations for at least the next twelve months. However, should the Company experience unforeseen expenses, or if anticipated revenues are not realized, the effect could negatively impact management’s estimated operating results over the next twelve months. The timing and amount of any additional funding the Company may require to broaden the commercialization of Melafind® will be affected by the commercial success of the product. The funding could be in the form of either additional equity or debt financing. In the event that the Company is unable to raise additional funds, the Company has the ability and intent to reduce certain discretionary expenditures.
Most of our expenditures to date have been for research and development activities and general and administrative expenses. Research and development expenses represent costs incurred for product development, clinical trials and activities relating to regulatory filings and manufacturing development efforts. We expense all of our research and development costs as they are incurred.
Our research and development expenses incurred for the year ended December 31, 2012 were related primarily to the development of MelaFind®. We expect to continue to incur certain additional research and development expenses relating to MelaFind®. Additional research and development charges may be incurred for complementary technologies. These additional expenses could exceed our estimated amounts, possibly materially.
Selling, general and administrative expenses consist primarily of salaries and related human resources expenses, legal expenses, general corporate activities and costs associated with our efforts to expand our commercial infrastructure to market and sell MelaFind®. We expect selling, general and administrative expenses to increase as we continue to build our sales and marketing capabilities to support placing MelaFind® systems in selected markets inside and outside the U.S.
At December 31, 2012, we had available income tax benefit from net operating loss carryforwards for federal income tax reporting purposes of approximately $66 million. The net operating loss carryforwards may be available to offset future taxable income expiring at various dates through the year 2032. The Company’s ability to utilize its net operating losses may be significantly limited due to future changes in the Company’s ownership as defined by federal income tax regulations.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements as well as the reported revenues and expenses during the reporting periods. On an
42
Table of Contents
ongoing basis, we evaluate our judgments related to accounting estimates. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 1 to our financial statements included in this annual report, we believe that the following accounting policies and significant judgments and estimates relating to revenue recognition, stock-based compensation charges, and accrued expenses are most critical to aid you in fully understanding and evaluating our reported financial results.
Revenue Recognition
The Company considers revenue to be earned when all of the following criteria are met: persuasive evidence a sales arrangement exists; delivery has occurred or services have been rendered; the price is fixed or determinable; and collectability is reasonably assured. The Company’s agreements with dermatologists regarding the MelaFind® system combine the elements noted above with a future service obligation. While the Company is required to place the MelaFind® systems with dermatologists for their exclusive use, ownership of the MelaFind® systems remains with the Company.
In the U.S., the Company generates revenue generally from the sale of electronic patient record cards. These cards activate the MelaFind® system, capture data and store the data for each patient visit. In January 2013, the Company began offering two additional billing options. Customers now choose among three billing options: 1) purchase of single-use electronic cards 2) use of high capacity activation cards which allow for billing electronically per session or per lesion, or 3) a fixed monthly rental fee. Additionally, the Company typically charges an initial installation fee for each MelaFind® system, which covers training, delivery, supplies, maintenance and the right to use MelaFind®. In accordance with the accounting guidance regarding multiple-element arrangements, the Company allocates total contract consideration to each element based upon the relative standalone selling prices of each element, and recognizes the associated revenue for each element as delivery occurs or over the related service period, generally expected to be two years.
Costs of revenue are associated with the placement of the MelaFind® system in the doctor’s office, the cost of consumables delivered at installation, the cost of the electronic record cards, technical support costs and costs of the MelaFind® system placed with the customer, which remains the property of the Company. Certain product quality and manufacturing overhead costs associated with supporting the contract manufacturers of MelaFind® are allocated to costs of goods sold and MelaFind® systems on hand.
In Germany, the typical contract with dermatologists calls for an installation fee and/or fixed monthly fee and a per patient usage charge. Revenue generated from German contracts is recognized when earned.
Prior to the installation of the first commercial MelaFind® system in the first quarter of 2012, the Company had no revenues from products since 2005 when it discontinued its DIFOTI operations.
Stock-Based Compensation
We account for non-employee stock-based awards in which goods or services are the consideration received for the equity instruments issued based on the fair value of the equity instruments issued in accordance with FASB ASC 505-50, “Equity Based Payments to Non-Employees.”
We record compensation expense associated with stock options and other forms of equity compensation in accordance with FASB ASC 718, Compensation-Stock Compensation, as interpreted by SEC Staff Accounting Bulletins No. 107 and No. 110. A compensation charge is recorded, when it is probable that performance conditions will be satisfied, over the period estimated to satisfy the performance condition. The probability of vesting is updated at each reporting period and compensation is adjusted prospectively.
43
Table of Contents
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for such service where we have not been invoiced or otherwise notified of the actual cost. Examples of estimated accrued expenses include:
| • | professional service fees; |
| • | contract clinical and regulatory related service fees; |
| • | fees paid to contract manufacturers in conjunction with the production of MelaFind® components or materials; and |
| • | fees paid to third party data collection organizations and investigators in conjunction with the clinical trials and FDA and other regulatory review. |
In connection with such service fees, our estimates are most affected by our projections of the timing of services provided relative to the actual level of services provided by such service providers. The majority of our service providers invoice us monthly in arrears for services performed. In the event that we do not identify certain costs that have begun to be incurred or we under or over estimate the level of services performed or the costs of such services, our actual expenses could differ from such estimates. The date on which certain services commence, the level of services performed on or before a given date, and the cost of such services are often subjective determinations. We make these judgments based upon the facts and circumstances known to us and accrue for such costs in accordance with accounting principles generally accepted in the US. This is done as of each balance sheet date in our financial statements.
Results of Operations (in thousands)
Year Ended December 31, 2012 Compared to Year Ended December 31, 2011
In the year ended December 31, 2012, the Company evolved from being exclusively a research and development company prior to the MelaFind® launch to a commercial company with the controlled installation of MelaFind® systems in the U.S. and Germany. Additions have been made to the direct sales force and field technical support capabilities of the Company commensurate with these additional installations and the increasing demand for MelaFind®. The production levels of our contract manufacturers have been increased to address the growing demand for MelaFind® systems. For the first two months of 2012 the Company continued to record all transactions as an R&D company, as it had in 2011. Subsequent to the commercial launch of MelaFind®, certain costs previously classified as research and development expenses are now classified as cost of revenue, inventory or selling, general and administrative expenses. Sales and marketing efforts were increased in 2012 prior and subsequent to the commercial launch of MelaFind®.
Revenue
Invoicing in the year ended December 31, 2012 totaled $582, with revenue of $278 and deferred revenue of $304 recorded as the Company continued its controlled commercial launch of the MelaFind® product, which commenced during March 2012. Prior to the commercial launch of MelaFind®, the Company had not recorded any product revenue or deferred revenue since the discontinuance of its Difoti product in 2005. In general, the Company signs a user agreement with its customers that includes an installation fee for the placement of the MelaFind® system and provides for the sale of its electronic patient record cards and consumables, which are needed to operate the system, or a fixed monthly rental fee. The Company is addressing unique aspects of the European marketplace through variations of the user agreement. Deferred revenue primarily reflects the timed recognition of the installation fee revenue over the term of the user agreement, which is generally two years.
44
Table of Contents
Cost of Revenue
Costs of $2,042 were recorded as associated with the realization of MelaFind® revenue during the year ended December 31, 2012. These costs were made up of direct costs associated with the placement of the MelaFind® system in the doctor’s office, the cost of consumables delivered at installation, the cost of the electronic record cards, technical support costs and depreciation expense of the MelaFind® system placed with the customer, which remains the property of the Company. Certain product quality and manufacturing overhead costs associated with supporting the contract manufacturers of MelaFind® are allocated to costs of goods sold and product inventory. The Company had not recorded any cost of revenue prior to the commercial launch of MelaFind®.
Research and Development Expense
Research and development (“R&D”) expenses decreased $2,864 or 30% for the year ended December 31, 2012 as compared to the comparable period a year earlier. In 2012, $870 of regulatory expenses and $909 of clinical expenses, which prior to commercialization would have been recorded to R&D, were recorded to selling, general and administrative expense. Also, $662 of technical support expenses, $305 of certain quality expenses, and $236 of certain operating expenses, which prior to commercialization would have been recorded to R&D, were recorded to cost of revenue or inventory. Exclusive of these changes, R&D expenses increased $118 over the comparable period of 2011. This increase was principally made up of the increases in Software development costs of $578 and in U.S. Development expenses of $743 offset by a decrease in Askion Development costs of $1,081in 2012 compared to 2011.
Selling, General and Administrative Expense
Selling, general and administrative (“SG&A”) expenses increased $3,363 or 31% for the year ended December 31, 2012 as compared to the comparable period a year earlier. Other than the costs which were R&D prior to commercialization, SG&A expenses showed a year-to-year increase of approximately $1,584. In 2012, Marketing expenses in the U.S. and Germany increased $2,570 and Information Technology costs increased $639 from the level of the comparable period a year earlier. In addition, share-based compensation expense decreased $1,585 in the year ended December 31, 2012 below the comparable period a year earlier. Approval of the MelaFind PMA in 2011had resulted in the vesting of a significant number of performance-based options and the related charges to share-based compensation.
Interest (Income)/Expense
Interest income for the year ended December 31, 2012 decreased to $32 from $54 for the comparable period of 2011. Interest income decreased primarily as a result of smaller cash balances during the period in 2012.
Other Income, net
In accordance with the terms of our DIFOTI sale and licensing agreement, KaVo will pay us an annual royalty based on the number of DIFOTI related systems sold per calendar year following their commercial re-launch. Other income for both of the years ended December 31, 2012 and 2011, is primarily the $20 royalty annual minimum.
Year Ended December 31, 2011 Compared to Year Ended December 31, 2010
Research and Development Expense
Research and development expense decreased by $1,841 to $9,656 for the year ended December 31, 2011 from $11,497 for the year ended December 31, 2010. R&D costs at Askion decreased by $1,434 primarily in labor and product improvement materials while U.S. development costs decreased by $717, which was primarily related to decreased product improvement, software development and subcontract design activities. Clinical
45
Table of Contents
spending decreased $283, Technical Support decreased $87, and Quality/Regulatory decreased $103. Non-cash share-based compensation expense in R&D, resulting from vesting associated with the achievement of performance and time-based milestones and from the granting of options, increased by $783.
General and Administrative Expense
General and administrative expense increased by $2,068 to $10,806 for the year ended December 31, 2011 from $8,738 for the year ending December 31, 2010. $1,845 of the increase was in non-cash, share-based compensation resulting from vesting associated with the achievement of performance and time-based milestones and from the granting of options and stock awards. G&A costs increased $354 in legal and professional fees while overall G&A salaries experienced a decrease of $125.
Interest (Income)/Expense
Interest income for the year ended December 31, 2011 was $54 compared to $32 for the year ended December 31, 2010. The increase reflected significantly higher interest rates available on accounts in 2011.
Other Income, net
Other income for the year ended December 31, 2011 decreased from the comparable period in 2010 by $258. In 2010 the Company had received a R&D grant from the federal government for $245. The Company did not receive a grant in 2011.
In accordance with the terms of our DIFOTI sale and licensing agreement, KaVo will pay us an annual royalty based on the number of DIFOTI related systems sold per calendar year following commercial re-launch. As KaVo has not re-launched DIFOTI as of 2011 year end, the Company earned the minimum annual royalty of $20 in both 2011 and 2010.
Liquidity and Capital Resources (in thousands)
From inception, we have financed our operations primarily through the use of working capital from the sale of equity securities. As of December 31, 2012, the Company had not borrowed (other than by issuing convertible notes, all of which have been converted into equity) or financed our operations through equipment leases, financing loans or other debt instruments. Subsequent to year end 2012, the Company signed a term sheet for a debt instrument in the amount of $10 million, $6 million of which is available immediately upon closing and $4 million of which will be available based on certain future milestones. As of December 31, 2012, we had $7,862 in cash and cash equivalents as compared to $27,997 at December 31, 2011. The $20,135 decrease in 2012 from 2011 reflects the $5,247 of net cash provided by 2012 financing activities offset by $19,224 of net cash used in operating activities and $6,158 of net cash used in investing activities which was principally for the purchase of MelaFind® systems. Our cash and cash equivalents at December 31, 2012 are liquid investments in cash with two commercial banks and money market accounts held in accounts that substantially exceed FDIC limits.
On May 7, 2009, the Company entered into a committed equity financing facility (“CEFF”) with Kingsbridge Capital Limited, pursuant to which Kingsbridge committed to purchase from time to time at the Company’s sole discretion, up to the lesser of $45 million or 3,327,000 shares of the Company’s common stock, prior to May 25, 2012 subject to various conditions for individual sales, including dollar, timing, and trading volume limitations, a minimum market per share price, and other contractual and regulatory requirements. In connection with this CEFF, the Company issued a 5-year warrant, exercisable as of November 7, 2009, to Kingsbridge to purchase up to 200,000 shares of our common stock at an exercise price of $11.35 per share with a Black Scholes Fair Value of $678,000. The issuance of this warrant was deemed to be a cost of the offering.
46
Table of Contents
Under the CEFF, during 2009, the Company sold 1,824,941 shares of common stock to Kingsbridge Capital Limited, at an average per share price of approximately $9.24, for gross proceeds of approximately $16.9 million, and during 2010, the Company sold 406,744 shares of common stock to Kingsbridge Capital Limited, at an average per share price of approximately $9.22, for gross proceeds of approximately $3.75 million. A proportionate share of the CEFF originating expenses was allocated to each of these sales from deferred offering costs. Net of expenses, proceeds from these sales were approximately $16.8 million in 2009 and $3.727 million in 2010. In May 2012 the CEFF terminated with 1,095,315 shares of common stock remaining unsold. Legal, accounting, and other costs associated with this agreement approximating $62 were charged to operations in the quarter ended June 30, 2012 as the CEFF expired. The 200,000 warrants held by Kingsbridge remain outstanding and, if not exercised, will expire in May 2014.
In May 2010, the Company filed a Form S-3 shelf registration statement for an indeterminate number of shares of common stock, warrants to purchase shares of common stock and units consisting of a combination thereof having an aggregate initial offering price not to exceed $75 million. The registration statement was declared effective by the SEC on June 1, 2010 (File No. 333-167113). On June 30, 2010, the Company entered into an underwriting agreement, relating to the public offering of 2,200,000 shares of the Company’s common stock, at a price to the public of $7.50 per share less underwriting discounts and commissions. The common stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the Securities and Exchange Commission (the “SEC”) on June 30, 2010, in connection with a takedown from the Company’s effective shelf registration statement. The gross proceeds to the Company from the sale of the Common Stock totaled $16.5 million. After deducting the underwriters’ discounts and commissions and other offering expenses payable by the Company, net proceeds were approximately $15.2 million. This offering closed on July 6, 2010.
On December 15, 2011, the Company entered into an underwriting agreement, relating to the public offering of 5,000,000 shares of the Company’s common stock, at a price to the public of $3.25 per share, except for an officer and a director of the Company who collectively purchased 42,915 shares at the closing market price of $3.97, less underwriting discounts and commissions. The common stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the Securities and Exchange Commission (the “SEC”) on December 16, 2011, in connection with a takedown from the Company’s effective shelf registration statement. The gross proceeds to the Company from the sale of the common stock totaled approximately $16.3 million. After deducting the underwriters’ discounts and commissions and other offering expenses payable by the Company, net proceeds were approximately $15 million. This offering closed on December 21, 2011
On June 15, 2012, the Company entered into a sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) to sell shares of its common stock with aggregate gross proceeds of up to $20 million, from time to time, through an “at-the-market” equity offering program (“ATM Program”) under which Cowen will act as sales agent. The common stock was offered and will be sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the SEC on June 15, 2012, in connection with a takedown from the Company’s effective shelf registration statement, leaving $22.2 million available under the shelf registration, assuming full utilization of the ATM Program. Through December 31, 2012, 1,863,830 shares of the Company’s common stock were sold through the ATM Program for gross proceeds of approximately $5.6 million and net proceeds of approximately $5.4 million. Subsequent to year end 2012 and through termination of the Sales Agreement and the ATM Program on February 15, 2013, the Company sold 4,714,365 shares for additional gross proceeds of approximately $8.8 million.
On February 5, 2013, the Company signed a non-binding term sheet with a venture capital lender for a $10 million loan. Of the $10 million, it is anticipated that $6 million will be funded in March 2013 and we will have the option to draw down the remaining $4 million through March 17, 2014, subject to meeting certain sales and revenue targets. It is anticipated that the loan will mature 42 months from the initial closing and bear interest at the rate of 10.45% per year. The term sheet contemplates that during the first 12 months of the loan, only
47
Table of Contents
interest will be paid to the lender and after that we will make 30 equal payments of principal and interest until maturity. The loan would be secured by a general lien against all of the Company’s assets, other than its intellectual property assets. In addition, the lender will have a security interest in the proceeds of the sale of any of the Company’s intellectual property assets. In connection with the loan, the lender will receive a warrant to purchase that number of shares of common stock equal to 775,000 divided by the to-be-determined exercise price of the warrant. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents and was conditioned upon the Company having raised $12.5 million in proceeds since January 1, 2013, which has been satisfied with the proceeds from the ATM Program and the February 2013 public offering (see below). There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described.
On February 12, 2013 the Company entered into an underwriting agreement, relating to the public offering of 6,100,000 shares of the Company’s common stock, at a price to the public of $1.30 per share less underwriting discounts and commissions. The gross proceeds to the Company from the sale of the common Stock totaled $7.9 million. After deducting the Underwriters’ discounts and commissions and other estimated offering expenses payable by the Company, net proceeds were approximately $7.3 million. The offering closed on February 15, 2013. The common Stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the Securities and Exchange Commission (the “SEC”) on February 12, 2013, in connection with a takedown from the Company’s effective shelf registration statement on Form S-3 (File No. 333-167113) declared effective by the SEC on June 1, 2010.
Cash Flows from Operating Activities
Net cash used in operations was $19,224 for the year ended December 31, 2012. For the year ended December 31, 2011, the net cash used in operations was $17,458. For both periods, cash used in operations was attributable primarily to net losses after adjustment for non-cash charges related to non-cash compensation, depreciation and other changes in operating assets and liabilities.
Cash Flows from Investing Activities
Net cash used in our investing activities was $6,158 for the year ended December 31, 2012 principally relating to the purchase of fixed assets generally MelaFind® systems. For the year ended December 31, 2011 net cash used by investing activities was $104 principally relating to the purchase of fixed assets.
Cash Flows from Financing Activities
Net cash provided by financing activities was $5,247 for the year ended December 31, 2012 and reflects the net proceeds received from our ATM Program sale of common stock and proceeds from the exercise of common stock options. For the year ended December 31, 2011, the net cash flows provided by financing activities was $15,038, which reflects the net proceeds received from our December 15, 2011 public offering of common stock and proceeds from the exercise of common stock options.
Operating Capital and Capital Expenditure Requirements
We face certain risks and uncertainties, which are present in many emerging medical device companies. At December 31, 2012, we had an accumulated deficit of $142.2 million. We anticipate that we will continue to incur net losses for the foreseeable future as we proceed with the MelaFind® commercialization process and expand our corporate infrastructure. We do not expect to generate significant product revenue until after we successfully complete a controlled product launch of MelaFind®. However, we will need substantial funds to broaden the commercialization of MelaFind®, including development of a direct sales force and expansion of our contract manufacturing capacity. The timing and amount of any additional funding the Company may require will be affected by the commercial success of the product. The funding could be in the form of either additional equity or debt financing. Subsequent to year end, we raised approximately $15.8 million in net proceeds from a
48
Table of Contents
public offering of our common stock and from sales under our “at-the-market” equity offering program and signed a term sheet to borrow up to $10 million, of which $6 million will be immediately available to the Company upon closing. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described. We believe that our year end cash and cash equivalents, combined with our financing activities subsequent to year end and anticipated revenues, will be sufficient to fund our anticipated levels of operations for at least the next twelve months. However, should the Company experience unforeseen expenses, or if anticipated revenues are not realized, the effect could negatively impact management’s estimated operating results over the next twelve months. If our existing cash is insufficient to satisfy our liquidity requirements, or if we develop additional products, we may seek to sell additional equity or debt securities or obtain a credit facility, which will be even more difficult due to the lack of available capital as a result of the ongoing effects of the recent global economic crisis. If additional funds are raised through the issuance of debt securities, these securities would have rights senior to those associated with our common stock and could contain covenants that would restrict our operations. Any additional financing may not be available in amounts or on terms acceptable to us, or at all. If we are unable to obtain this additional financing, we may be required to reduce the scope of, delay or eliminate some or all of planned product research development and commercialization activities, which could harm our business.
Because of the numerous risks and uncertainties associated with the development and commercialization of medical devices such as MelaFind® and operating our Company, we are unable to estimate the exact amounts of capital outlays and operating expenditures. Our future funding requirements will depend on many factors, including, but not limited to:
| • | the cost of commercialization activities, including product marketing and building a domestic direct sales force and conducting activities in Germany and ultimately in other countries; |
| • | the amount of direct payments we are able to obtain from physicians utilizing MelaFind®; |
| • | the costs of maintaining regulatory approval; |
| • | reimbursement amounts for the use of MelaFind® that we are able to obtain from Medicare and third party payers; |
| • | the success of our research and development efforts in product creation and enhancement, and meeting competitive services and technologies; |
| • | the schedule, costs, and results of our clinical trials; |
| • | the costs of maintaining or potentially building our inventory and other manufacturing expenses; |
| • | our ability to establish and maintain any collaborative, licensing or other arrangements, and the terms and timing of any such arrangements. |
| • | the costs involved in defending any patent infringement actions or other litigation claims brought against us by third parties; and |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims or other rights. |
Contractual Obligations (in thousands)
The following table summarizes our outstanding contractual obligations as of December 31, 2012 and the effect those obligations are expected to have on our liquidity and cash flows in future periods:
Payments Due by Period:
| Contractual Obligations | ||||||||||||||||
| Total | Less Than 1 Year |
1-3 Years | 3-5 Years | |||||||||||||
| (Dollars in thousands) | ||||||||||||||||
| Operating Leases |
$ | 1,895 | $ | 462 | $ | 955 | $ | 478 | ||||||||
49
Table of Contents
Our long-term obligations represent a non-cancelable operating lease for our laboratory, assembly, and office space. The lease on approximately 21,700 square feet of office space expires in December 2016.
Related Party Transactions (in thousands)
Consulting Agreement with Gerald Wagner, Ph.D.
In January 2007, Dr. Wagner, a former Director on the Company’s Board of Directors, entered into an amended and restated consulting contract with the Company. Under the terms of the amended contract, Dr. Wagner is paid a monthly retainer of $2.5 and will be paid $2.5 for each additional consulting day. This amended agreement will end at the option of Dr. Wagner or the Company at any time, by providing fifteen days prior written notice, or immediately upon the mutual agreement of the Company and Dr. Wagner. The amounts paid to Dr. Wagner amounted to $30 in each of 2010, 2011 and 2012. Dr. Wagner resigned from the Company’s Board of Directors in December 2011 with the consulting contract remaining in effect until December 2012, when it was cancelled.
Consulting Agreement with Anne Egger
In March 2009, the Company entered into a consulting agreement with Anne Egger for certain consulting services primarily focusing on physician advocacy. The agreement was for an initial term of three months, was subsequently extended to run through September 2012, when it was not renewed. Under the terms of the agreement, Ms. Egger was entitled to receive a consulting fee of $1.6 per day. Ms. Egger was appointed to the Company’s Board of Directors as of June 10, 2009. During the years ended December 31, 2010, 2011 and 2012, Ms. Egger was paid $60, $8 and $0, respectively, under this agreement.
Off-Balance Sheet Arrangements
We do not currently have, nor have we ever had, any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. As such, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships.
Recently Adopted Accounting Pronouncements
The Financial Accounting Standards Board has issued a number of new accounting standards that require future adoption. Based on the Company’s initial review of these new standards, none are expected to have a material impact on the Company’s financial statements.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Our exposure to market risk at December 31, 2012 is confined to our cash and cash equivalents. We invest in cash and money market accounts with commercial banks. We currently do not hedge interest rate exposure. While declines in interest rates do impact the amount of interest income that our cash and cash equivalents will earn, we do not believe that we have any material exposure to interest rate risk arising from our investments, due to the nature of our accounts.
50
Table of Contents
Item 8. Financial Statements and Supplementary Data
51
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
MELA Sciences, Inc.
We have audited the accompanying balance sheets of MELA Sciences, Inc. as of December 31, 2011 and 2012, and the related statements of operations, stockholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2012. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of MELA Sciences, Inc., as of December 31, 2011 and 2012, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2012, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), MELA Sciences, Inc.’s internal control over financial reporting as of December 31, 2012, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”), and our report dated March 4, 2013 expressed an unqualified opinion thereon.
/s/ EisnerAmper LLP
New York, New York
March 4, 2013
52
Table of Contents
BALANCE SHEETS
| December 31, 2011 |
December 31, 2012 |
|||||||
| ASSETS | ||||||||
| Current Assets: |
||||||||
| Cash and cash equivalents |
$ | 27,996,871 | $ | 7,861,524 | ||||
| Accounts receivable |
— | 179,956 | ||||||
| Inventory |
— | 675,602 | ||||||
| Prepaid expenses and other current assets |
1,061,550 | 965,624 | ||||||
|
|
|
|
|
|||||
| Total Current Assets |
29,058,421 | 9,682,706 | ||||||
| Property and equipment, net |
1,626,791 | 7,349,531 | ||||||
| Patents and trademarks, net |
59,208 | 47,308 | ||||||
| Deferred financing costs |
62,391 | 106,141 | ||||||
| Other assets |
586,498 | 84,127 | ||||||
|
|
|
|
|
|||||
| Total Assets |
$ | 31,393,309 | $ | 17,269,813 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current Liabilities: |
||||||||
| Accounts payable (includes related parties of $36,027 and $59,689 as of December 31, 2011 and December 31, 2012, respectively) |
$ | 670,950 | $ | 1,850,102 | ||||
| Accrued expenses |
745,754 | 956,541 | ||||||
| Deferred revenue |
— | 171,726 | ||||||
| Other current liabilities |
30,993 | 40,811 | ||||||
|
|
|
|
|
|||||
| Total Current Liabilities |
1,447,697 | 3,019,180 | ||||||
|
|
|
|
|
|||||
| Long Term Liabilities: |
||||||||
| Deferred rent |
138,216 | 143,772 | ||||||
| Deferred revenue |
— | 131,651 | ||||||
|
|
|
|
|
|||||
| Total Long Term Liabilities |
138,216 | 275,423 | ||||||
|
|
|
|
|
|||||
| Total Liabilities |
1,585,913 | 3,294,603 | ||||||
|
|
|
|
|
|||||
| COMMITMENTS, CONTINGENCIES and LITIGATION (Note 4) |
||||||||
| Stockholders’ Equity |
||||||||
| Preferred stock — $.10 par value; authorized 10,000,000 shares; issued and outstanding: none |
||||||||
| Common stock — $.001 par value; authorized 45,000,000 shares; issued and outstanding 30,307,538 shares at December 31, 2011 and 32,204,720 at December 31, 2012 |
30,308 | 32,205 | ||||||
| Additional paid-in capital |
149,304,424 | 156,142,873 | ||||||
| Accumulated deficit |
(119,527,336 | ) | (142,199,868 | ) | ||||
|
|
|
|
|
|||||
| Stockholders’ Equity |
29,807,396 | 13,975,210 | ||||||
|
|
|
|
|
|||||
| Total Liabilities and Stockholders’ Equity |
$ | 31,393,309 | $ | 17,269,813 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements
53
Table of Contents
STATEMENTS OF OPERATIONS
| Year Ended | ||||||||||||
| December 31, 2010 |
December 31, 2011 |
December 31, 2012 |
||||||||||
| Revenue |
$ | — | $ | — | $ | 278,461 | ||||||
| Cost of revenue |
— | — | 2,042,333 | |||||||||
|
|
|
|
|
|
|
|||||||
| — | — | (1,763,872 | ) | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating expenses: |
||||||||||||
| Research and development |
11,496,634 | 9,656,003 | 6,791,940 | |||||||||
| Selling, general, and administrative, |
8,738,203 | 10,806,228 | 14,168,754 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating loss |
(20,234,837 | ) | (20,462,231 | ) | (22,724,566 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Interest income |
(31,582 | ) | (54,371 | ) | (32,034 | ) | ||||||
| Other income, net |
(280,740 | ) | (23,145 | ) | (20,000 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| (312,322 | ) | (77,516 | ) | (52,034 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (19,922,515 | ) | $ | (20,384,715 | ) | $ | (22,672,532 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted net loss per common share |
$ | (.83 | ) | $ | (.80 | ) | $ | (.74 | ) | |||
|
|
|
|
|
|
|
|||||||
| Basic and diluted weighted average number of common shares outstanding |
24,043,135 | 25,415,880 | 30,762,610 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements
54
Table of Contents
STATEMENTS OF STOCKHOLDERS’ EQUITY
Years Ended December 31, 2010, 2011 and 2012
| Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
Total Stockholders’ Equity |
|||||||||||||||||
| Shares | Amount | |||||||||||||||||||
| Balance at January 1, 2010 |
22,354,317 | $ | 22,354 | $ | 109,513,582 | $ | (79,220,106 | ) | $ | 30,315,830 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Exercise of options |
12,944 | 13 | 33,075 | 33,088 | ||||||||||||||||
| Cashless exercise of options |
16,262 | 16 | (16 | ) | — | |||||||||||||||
| Exercise of warrants |
239,723 | 240 | 1,691,394 | 1,691,634 | ||||||||||||||||
| Cashless exercise of warrants |
32,548 | 33 | (33 | ) | — | |||||||||||||||
| Issuance of shares of common stock in connection with a public offering (net of expenses) |
2,200,000 | 2,200 | 15,231,471 | 15,233,671 | ||||||||||||||||
| Issuance of shares of common stock in connection with a Committed Equity Financing Facility (CEFF) (net of expenses) |
406,744 | 407 | 3,719,697 | 3,720,104 | ||||||||||||||||
| Share-based compensation expense |
727,156 | 727,156 | ||||||||||||||||||
| Net loss |
(19,922,515 | ) | (19,922,515 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2010 |
25,262,538 | 25,263 | 130,916,326 | (99,142,621 | ) | 31,798,968 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Exercise of options |
5,000 | 5 | 13,345 | 13,350 | ||||||||||||||||
| Issuance of common stock award |
40,000 | 40 | 171,960 | 172,000 | ||||||||||||||||
| Issuance of shares of common stock in connection with a public offering (net of expenses) |
5,000,000 | 5,000 | 15,019,662 | 15,024,662 | ||||||||||||||||
| Share-based compensation expense |
3,183,131 | 3,183,131 | ||||||||||||||||||
| Net loss |
(20,384,715 | ) | (20,384,715 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2011 |
30,307,538 | 30,308 | 149,304,424 | (119,527,336 | ) | 29,807,396 | ||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Cashless exercise of options |
11,868 | 12 | (12 | ) | — | |||||||||||||||
| Exercise of options |
21,484 | 21 | 44,738 | 44,759 | ||||||||||||||||
| Issuance of shares of common stock in connection with an ATM public offering (net of expenses) |
1,863,830 | 1,864 | 5,306,173 | 5,308,037 | ||||||||||||||||
| Share-based compensation expense |
1,487,550 | 1,487,550 | ||||||||||||||||||
| Net loss |
(22,672,532 | ) | (22,672,532 | ) | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| Balance at December 31, 2012 |
32,204,720 | $ | 32,205 | $ | 156,142,873 | $ | (142,199,868 | ) | $ | 13,975,210 | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
The accompanying notes are an integral part of these financial statements
55
Table of Contents
STATEMENTS OF CASH FLOWS
| Year Ended | ||||||||||||
| December 31, 2010 |
December 31, 2011 |
December 31, 2012 |
||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (19,922,515 | ) | $ | (20,384,715 | ) | $ | (22,672,532 | ) | |||
|
|
|
|
|
|
|
|||||||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Gain on sale of fixed assets |
(8,811 | ) | — | — | ||||||||
| Depreciation and amortization |
552,860 | 562,803 | 969,500 | |||||||||
| Issuance of common stock award |
— | 172,000 | — | |||||||||
| Write off of unamortized financing costs |
— | — | 62,391 | |||||||||
| Noncash compensation |
727,156 | 3,183,131 | 1,487,550 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Increase in accounts receivable |
— | — | (179,956 | ) | ||||||||
| Increase in inventory |
— | — | (675,602 | ) | ||||||||
| Decrease (increase) in prepaid expenses and other current assets |
141,290 | (537,878 | ) | 95,926 | ||||||||
| (Decrease) increase in accounts payable and accrued expenses |
(121,321 | ) | (239,776 | ) | 1,389,939 | |||||||
| (Decrease) increase in other current liabilities |
(3,747 | ) | 1,455 | 9,818 | ||||||||
| Increase in other assets |
(289,705 | ) | (248,793 | ) | (19,643 | ) | ||||||
| Increase in deferred rent |
104,304 | 33,912 | 5,556 | |||||||||
| Increase in deferred revenue |
— | — | 303,377 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(18,820,489 | ) | (17,457,861 | ) | (19,223,676 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Purchases of property and equipment |
(1,044,079 | ) | (104,092 | ) | (6,158,326 | ) | ||||||
| Proceeds from disposal of fixed assets |
10,284 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(1,033,795 | ) | (104,092 | ) | (6,158,326 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Net proceeds from private placements/public offerings |
15,233,671 | 15,024,662 | 5,201,896 | |||||||||
| Net proceeds from Committed Equity Financing Facility |
3,743,283 | — | — | |||||||||
| Proceeds from exercise of stock options |
33,088 | 13,350 | 44,759 | |||||||||
| Proceeds from exercise of stock warrants |
1,691,634 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
20,701,676 | 15,038,012 | 5,246,655 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
847,392 | (2,523,941 | ) | (20,135,347 | ) | |||||||
| Cash and cash equivalents at beginning of year |
29,673,420 | 30,520,812 | 27,996,871 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents at end of year |
$ | 30,520,812 | $ | 27,996,871 | $ | 7,861,524 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental Schedule of Noncash Investing and Financing Activities: |
||||||||||||
| Amortization of deferred financing costs |
$ | 23,179 | — | $ | 41,179 | |||||||
| Reclassification of MelaFind® components from other assets to property and equipment |
— | — | $ | 522,014 | ||||||||
The accompanying notes are an integral part of these financial statements
56
Table of Contents
Notes to Financial Statements
(In thousands, except for share and per share data and unless stated otherwise)
| 1. | Principal Business Activities and Summary of Significant Accounting Policies: |
Organization and Business
MELA Sciences, Inc., a Delaware corporation (the “Company”), is a medical device company focused on the commercialization of its flagship product, MelaFind®, and the further design and development of MelaFind® and its technology. MelaFind® is a non-invasive, point-of-care (in the doctor’s office) instrument to aid in the detection of melanoma. MelaFind® features a hand-held component that emits light of multiple wavelengths to capture digital data from clinically atypical pigmented skin lesions. The data are then analyzed utilizing sophisticated classification algorithms, ‘trained’ on our proprietary database of melanomas and benign lesions, to provide information to assist in the management of the patient’s disease, including information useful in the decision of whether to biopsy the lesion.
On March 7, 2012, the Company installed the first commercial MelaFind® systems, and proceeded with a controlled launch of MelaFind ® in selected U.S. and European markets throughout 2012. Prior to the commercial launch of MelaFind® commencing in the first quarter of 2012, the Company had not generated any revenues from MelaFind®.
The Company anticipates that it will continue to incur net losses for the foreseeable future in the commercialization of the MelaFind® device, the conduct of a Post Approval Study (“PAS”) evaluating the sensitivity of physicians in diagnosing melanomas and high-grade lesions and the false positive rate after using MelaFind®, the further development of MelaFind® and the Company’s technology and the expansion of its corporate infrastructure. From inception, the Company has financed operations initially through the sale of convertible preferred stock prior to becoming a public company in 2005 and subsequently through the sale of common stock.
On June 15, 2012, the Company entered into a sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) to sell shares of its common stock with aggregate gross proceeds of up to $20 million, from time to time, through an “at-the-market” equity offering program (“ATM Program”) under which Cowen will act as sales agent. As of December 31, 2012, there were 1,863,830 shares of the Company’s common stock sold through the ATM Program for gross proceeds of approximately $5.6 million and net proceeds of approximately $5.4 million. At December 31, 2012, approximately $14.4 million remains available under the Company’s ATM Program.
The Company faces certain risks and uncertainties which are present in many emerging medical device companies regarding future profitability, ability to obtain future capital, protection of patents and intellectual property rights, competition, rapid technological change, government regulations including levying of a medical device excise tax, changing health care marketplace, recruiting and retaining key personnel, and reliance on third party manufacturing organizations.
As of December 31, 2012, the Company’s total of cash and cash equivalents was approximately $7.9 million. Subsequent to year end, the Company raised approximately $15.8 million in net proceeds comprised of approximately$ 8.5 million under the Company’s “at-the-market” equity offering program and approximately $7.3 million from a public offering of the Company’s common stock. In addition, in February 2013 the Company signed a term sheet to borrow up to $10 million, of which $6 million will be immediately available to the Company upon closing. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described. Management believes that the year end cash and cash equivalents, combined with the Company’s financing activities subsequent to year end and anticipated revenues, will be
sufficient to fund the Company’s anticipated levels of operations for at least the next twelve months. However,
57
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
the Company will need substantial funds to broaden the commercialization of MelaFind®, including further development of a direct sales force and expansion of the Company’s contract manufacturing capacity. Should the Company experience unforeseen expenses or if anticipated revenues are not realized, the effect could negatively impact Management’s estimated operating results over the next twelve months.
Working capital at December 31, 2012 and the additional proceeds received from the ATM Program and the February 2013 public offering are intended to be used to continue the commercial launch of MelaFind® in the U.S. and the European Union, for continued research & development activities and for general corporate purposes. The Company may need to raise additional financing in the future and there can be no assurances that the Company will be able to obtain such financing on acceptable terms if at all. Any additional funding that the Company may obtain in the future could be in the form of debt or equity that could be dilutive to common stockholders and could provide new investors with rights and preferences senior to common stockholders. In the event that the Company is unable to raise additional funds, the Company has the ability and intent to reduce certain discretionary expenditures.
Foreign Exchange
The Company’s operations in Germany use the U.S. dollar as its functional currency and from time to time conducts business in Euros. For all periods presented, aggregate foreign exchange transaction gains and losses were not material.
Revenue recognition
The Company considers revenue to be earned when all of the following criteria are met: persuasive evidence a sales arrangement exists; delivery has occurred or services have been rendered; the price is fixed or determinable; and collectability is reasonably assured. The Company’s agreements with dermatologists regarding the MelaFind® system combine the elements noted above with a future service obligation. While the Company is required to place the MelaFind® systems with dermatologists for their exclusive use, ownership of the MelaFind® systems remains with the Company.
In the U.S., the Company generates revenue from the sale of single-use electronic patient record cards. These cards activate the MelaFind® system, capture data and store the data for each patient visit. Additionally, the Company typically charges an initial installation fee for each MelaFind® system which covers training, delivery, supplies, maintenance and the right to use MelaFind®. In accordance with the accounting guidance regarding multiple-element arrangements, the Company allocates total contract consideration to each element based upon the relative standalone selling prices of each element, and recognizes the associated revenue for each element as delivery occurs or over the related service period, generally expected to be two years. Revenues associated with undelivered elements are deferred until delivery occurs or services are rendered.
Costs of revenue are associated with; the placement of the MelaFind® system in the doctor’s office, the cost of consumables delivered at installation, the cost of the electronic record cards, technical support costs and depreciation expense of the MelaFind® system placed with the customer which remains the property of the Company. Certain product quality and manufacturing overhead costs associated with supporting the contract manufacturers of MelaFind® are allocated to costs of revenue and product inventory.
In Germany, the typical contract with dermatologists calls for an installation or fixed monthly fee and a per patient usage charge. Revenue generated from German contracts is recognized when earned.
58
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
Inventories
Inventories consist of finished products that are stated at the lower of cost (first-in, first-out) or market value.
Business Segments
The Company’s operations are confined to one business segment: the design, development and commercialization of MelaFind®.
Cash and Cash Equivalents
The Company’s cash is held in nationally-chartered banks and the amount the Company currently maintains with these banks exceeds the current federal insurance limits provided by the Federal Deposit Insurance Company. The Company has not experienced any loss of its cash or interest income. Cash equivalents are highly liquid debt instruments with an original maturity of three months or less at the date of acquisition. The carrying value of these instruments approximates fair value.
Property and Equipment
Depreciation of property and equipment is provided for by the straight-line method over the estimated useful lives of the related assets. Leasehold improvements are amortized over the lesser of the assets’ useful lives or the remaining term of the lease.
Patents
Patents are carried at cost less accumulated amortization which is calculated on a straight-line basis over a period of 15 years.
Litigation
Legal fees incurred in connection with contingent claims made against the Company are expensed as incurred.
Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires the use of estimates and assumptions by management that affect reported amounts of assets and liabilities and disclosures of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The most significant estimates relate to stock-based compensation arrangements, use of estimates to determine the elements of our revenue and deferred revenue, and accrued expenses. Actual results could differ from these estimates.
Long-lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An asset is considered to be impaired when the sum of the undiscounted future net cash flows expected to result from the use of the asset and its eventual disposition exceeds its carrying amount. The amount of impairment loss, if any, is measured as the difference between the net book value of the asset and its estimated fair value.
59
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
Research and Development
Research and development costs are expensed as incurred.
Stock-Based Compensation
The Company records compensation expense associated with stock options and other forms of equity compensation.
The Company grants options to employees that vest over either a defined service period, generally four years, or upon the achievement of defined performance milestones. The fair value of an option award granted to an employee is measured at grant date using the Black-Scholes Model and expensed over the service period or, in the case of performance milestones, when such awards are probable of vesting. The probability of performance milestones vesting is updated each reporting period and compensation expense is adjusted in the period for any change in estimate.
The Company also grants fully vested stock options to non-employees. The fair value of an option award granted to a non-employee is measured at the vesting date using the Black-Scholes Model and expensed at the time of grant.
Financial Instruments
The Company’s financial instruments consist principally of cash and cash equivalents, accounts receivable and accounts payable. The Company believes the financial instruments’ recorded values approximate current values because of their nature and respective durations.
Net Loss per Common Share
Basic net loss per share excludes dilution for potentially dilutive securities and is computed by dividing loss attributable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net loss per share gives effect to dilutive options, warrants and other potential common shares outstanding during the period. Diluted net loss per common share is equal to basic net loss per common share since all potentially dilutive securities are anti-dilutive for each of the periods presented. Potential common stock equivalents excluded consist of stock options and warrants which are summarized as follows:
| Year Ended December 31, | ||||||||||||
| 2010 | 2011 | 2012 | ||||||||||
| Common stock options |
2,132,879 | 2,057,104 | 2,426,533 | |||||||||
| Warrants |
546,781 | 546,781 | 200,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
2,679,660 | 2,603,885 | 2,626,533 | |||||||||
|
|
|
|
|
|
|
|||||||
Comprehensive loss
For all periods presented, the Company had no comprehensive income items and accordingly there is no difference between the reported net loss, and per share amounts per the Statement of Operations and comprehensive net loss and related per share amounts.
60
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
Recently Issued Accounting Standards
The Financial Accounting Standards Board has issued a number of new accounting standards that require future adoption. Based on the Company’s initial review of these new standards, none are expected to have a material impact on the Company’s financial statements.
| 2. | Property and Equipment: |
Property and equipment, at cost, consists of the following:
| December 31, | Estimated Useful Life |
|||||||||||
| 2011 | 2012 | |||||||||||
| Leasehold improvements |
$ | 786 | $ | 906 | Lease Term | |||||||
| Laboratory and research equipment |
975 | 1,084 | 3-5 years | |||||||||
| Office furniture and equipment |
1,878 | 2,023 | 3-5 years | |||||||||
| MelaFind Systems |
— | 6,306 | 3 years | |||||||||
|
|
|
|
|
|
|
|||||||
| 3,639 | 10,319 | |||||||||||
| Accumulated depreciation and amortization |
(2,012 | ) | (2,969 | ) | ||||||||
|
|
|
|
|
|||||||||
| $ | 1,627 | $ | 7,350 | |||||||||
|
|
|
|
|
|||||||||
Depreciation expense amounted to approximately $541, $551 and $957 for the years ended December 31, 2010, 2011 and 2012, respectively.
| 3. | Patents and trademarks: |
Patents and trademarks as shown in the accompanying balance sheets are net of accumulated amortization of $215 and $227 at December 31, 2011 and 2012, respectively. Amortization expense related to all patents was approximately $12, $12 and $12 for the years ended December 31, 2010, 2011 and 2012, respectively. Amortization expense of currently held patents is expected to amount to $5 for each of the years ending December 31, 2013 through 2017, respectively.
| 4. | Commitments, Contingencies and Litigation: |
The Company is obligated under a non-cancelable operating lease for office, lab, and manufacturing space expiring December 2016. The lease is subject to escalations for increases in operating expenses. For the years ended December 31, the approximate aggregate minimum future payments due under this lease are as follows:
| 2013 |
$ | 462 | ||
| 2014 |
477 | |||
| 2015 |
478 | |||
| 2016 |
478 | |||
|
|
|
|||
| $ | 1,895 | |||
|
|
|
Rent expense charged to operations amounted to approximately $416, $414 and $446 for the years ended December 31, 2010, 2011, and 2012, respectively.
61
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
ASKION GmbH (“ASKION”), located in Gera Germany, which specializes in precision optics, has become an integral member of the MelaFind® development team and the Company expects to continue to work with ASKION for the foreseeable future. ASKION produced the MelaFind® hand-held components used in our pivotal clinical trials and is currently under contract to build additional units and perform additional developmental activities.
Beginning in August 2006, the Company, primarily through ASKION, engaged Carl Zeiss Jena GmbH (“Zeiss”) to build the lenses and assemblies, as well as provide certain technical consulting, for the MelaFind® units which have been used in the Company’s pivotal clinical trials. This work was performed from 2006 through 2012 and is expected to continue on commercial MelaFind® units throughout 2013.
On November 19, 2010, a purported securities class action complaint was filed in the U.S. District Court for the Southern District of New York, naming as defendants the Company and certain of its officers and directors, entitled Randall J. Pederson, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo, Richard I. Steinhart, and Breaux Castleman, No. 7:10-cv-08774-JFM. Two similar complaints were also filed, one on December 2, 2010 and the other on January 20, 2011, in the same District Court, entitled Amy Steigman, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo, Richard I. Steinhart, and Breaux Castleman, No. 7:10-cv-09024-JFM; and Martin Slove and Linda Slove, Individually and on Behalf of All Others Similarly Situated v. MELA Sciences, Inc., Joseph V. Gulfo, Richard I. Steinhart, and Breaux Castleman, No. 1:11 cv-00429-JFM. These three securities class actions were consolidated into one action on February 15, 2011, entitled In re MELA Sciences, Inc. Securities Litigation, No. 7:10-Cv-08774-JFM (“securities class action”). The securities class action plaintiffs asserted violations of the Securities Exchange Act of 1934, alleging, among other things, that defendants made misstatements and omissions regarding the Company’s product, MelaFind®, and its prospects for FDA approval, on behalf of stockholders who purchased the Company’s common stock during the period from February 13, 2009 through November 16, 2010, and sought unspecified damages. On May 2, 2011, the securities class action plaintiffs filed their amended consolidated complaint, alleging similar claims to their prior complaints. On July 29, 2011, defendants filed a motion to dismiss the consolidated amended complaint in its entirety. Plaintiff’s opposition to the motion to dismiss was filed on September 23, 2011. In light of the Company’s receipt of the Approvable Letter from the FDA for the MelaFind® PMA Application on September 22, 2011, plaintiffs filed a motion for leave to amend the consolidated amended complaint on November 18, 2011, which defendants opposed. On September 19, 2012, the court denied plaintiffs’ motion for leave to amend the consolidated amended complaint. On September 28, 2012, the court reinstated and granted defendants’ motion to dismiss the consolidated amended complaint. On October 22, 2012, plaintiffs filed a notice of appeal from the Judgments denying Lead Plaintiffs’ motion to amend the consolidated amended complaint and granting Defendants’ motion to dismiss the consolidated amended complaint. On December 7, 2012, plaintiffs withdrew the appeal with prejudice and without costs pursuant to a stipulation between the parties. On December 10, 2012, the United States Court of Appeals for the Second Circuit granted the stipulation and dismissed the appeal.
From time to time, we may be a party to certain legal proceedings, incidental to the normal course of our business. These may include controversies relating to contract claims and employment related matters, some of which claims may be material in which case we will make separate disclosure as required.
| 5. | Employee Benefit Plan: |
The Company has a SIMPLE IRA defined contribution plan covering all qualified employees. An officer of the Company serves as trustee of the plan. The Company provides a matching contribution of up to 3% of each employee’s salary. Company contributions to this plan amounted to approximately $114, $106 and $151 for the years ended December 31, 2010, 2011 and 2012, respectively.
62
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
| 6. | Stockholders’ Equity |
In May 2009, the Company entered into a committed equity financing facility (“CEFF”) with Kingsbridge Capital Limited, pursuant to which Kingsbridge committed to purchase from time to time at the Company’s sole discretion, up to the lesser of $45 million or 3,327,000 shares of the Company’s common stock, prior to May 25, 2012. In connection with this CEFF, the Company issued a 5 year warrant, exercisable as of November 7, 2009, to Kingsbridge to purchase up to 200,000 shares of the Company’s common stock at an exercise price of $11.35 per share.
During 2009 and 2010, 1,824,941 shares and 406,744 shares respectively were issued in accordance with the CEFF and in May 2012 the CEFF terminated with 1,095,315 shares of common stock remaining unsold. Deferred financing costs, comprised of legal, accounting, and other costs associated with this agreement totaling approximating $62, were charged to operations in the quarter ended June 30, 2012 as the CEFF expired. The 200,000 warrants held by Kingsbridge remain outstanding and, if not exercised, will expire in May 2014.
In May 2010, the Company filed a Form S-3 shelf registration statement for an indeterminate number of shares of common stock, warrants to purchase shares of common stock and units consisting of a combination thereof having an aggregate initial offering price not to exceed $75 million. The registration statement was declared effective by the SEC on June 1, 2010. On June 30, 2010, the Company entered into an underwriting agreement, relating to the public offering of 2,200,000 shares of the Company’s common stock, at a price to the public of $7.50 per share less underwriting discounts and commissions. The common stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the SEC on June 30, 2010, in connection with a takedown from the Company’s effective shelf registration statement.
On December 15, 2011, the Company entered into an underwriting agreement, relating to the public offering of 5,000,000 shares of the Company’s common stock, at a price to the public of $3.25 per share less underwriting discounts and commissions. The common stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the SEC on December 16, 2011, in connection with a takedown from the Company’s effective shelf registration statement. The gross proceeds to the Company from the sale of the common stock totaled approximately $16.3 million. After deducting the underwriters’ discounts and commissions and other offering expenses payable by the Company, net proceeds were approximately $15 million. This offering closed on December 21, 2011.
On June 15, 2012, the Company entered into a sales agreement (the “Sales Agreement”) with Cowen and Company, LLC (“Cowen”) to sell shares of its common stock with aggregate gross proceeds of up to $20 million, from time to time, through an “at-the-market” equity offering program (“ATM Program”) under which Cowen will act as sales agent. The common stock was offered and will be sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the SEC on June 15, 2012, in connection with a takedown from the Company’s effective shelf registration statement, leaving $22.2 million available under the shelf registration, assuming full utilization of the ATM Program. Through December 31, 2012, 1,863,830 shares of the Company’s common stock were sold through the ATM Program for gross proceeds of approximately $5.6 million and net proceeds of approximately $5.4 million. The ATM Program was terminated during February 2013. See note 12.
On February 12, 2013 the Company entered into an underwriting agreement, relating to the public offering of 6,100,000 shares of the Company’s common stock, at a price to the public of $1.30 per share less underwriting discounts and commissions. The gross proceeds to the Company from the sale of the Common Stock totaled $7.9 million. After deducting the Underwriters’ discounts and commissions and other estimated offering expenses payable by the Company, net proceeds were approximately $7.3 million. The Offering closed on
63
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
February 15, 2013. The Common Stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the Securities and Exchange Commission (the “SEC”) on February 11, 2013, in connection with a takedown from the Company’s effective shelf registration statement on Form S-3 (File No. 333-167113) declared effective by the SEC on June 1, 2010. At February 15, 2013, the Company had $19.9 million available under its shelf registration, including $5.6 million previously allocated to the ATM Program and remaining unused upon termination of the ATM Program.
As of December 31, 2012, the Company had 10,000,000 shares of $0.10 par value preferred stock authorized with no preferred shares issued and outstanding. The Company’s Board of Directors has the authority to issue preferred stock in series that may contain rights and preference senior to common stockholders.
During the four year period ended December 31, 2009, the Company issued approximately 1.2 million warrants to various financial advisors, including 200,000 warrants issued to Kingsbridge Capital in 2009, as discussed above in connection with the equity offerings of the Company’s common stock. Cumulative through December 31, 2012 approximately 1 million warrants were exercised or expired. As of December 31, 2012, the only outstanding warrants are the 200,000 warrants held by Kingsbridge Capital that are exercisable at $11.35 per share and expire in May 2014. In February 2013, we signed a non-binding term sheet with a venture capital lender for a $10 million loan. In connection with the loan, the lender will receive a warrant to purchase that number of shares of our common stock equal to 775,000 divided by the to-be-determined exercise price of the warrant.
| 7. | Stock-Based Compensation: |
Stock Options
The Company has one stock option plan, the 2005 Stock Incentive Plan (“2005 Plan”), under which the board of directors may currently grant incentives to employees, directors, consultants and collaborating scientists in the form of incentive stock options, nonqualified stock options and restricted stock awards. The Company also has two other stock-based compensation plans pursuant to which stock options are outstanding but no new grants may be made.
Stock awards under the Company’s stock option plans have been granted with exercise prices which are no less than the market value of the stock on the date of the grant. Options granted under the 2005 Plan are generally time-based or performance-based options and vesting varies accordingly. Options under this plan expire up to a maximum of ten years from the date of grant. The plan provides for the granting of a maximum number of shares of common stock of 3,724,028 of which 1,222,739 are available for future grant as of December 31, 2012. Compensation expense recognized in the Statement of Operations during 2010, 2011 and 2012 for stock options and restricted stock awards amounted to $727, $3,355 and $1,488, respectively. Cash received from options exercised under all share-based payment arrangements for the years ended December 31, 2010, 2011 and 2012 was $33, $13 and $45, respectively.
The fair value of each option award granted is estimated on the date of grant using the Black-Scholes option valuation and assumptions as noted in the following table:
| For the Year Ended | ||||||||||||
| December 31, 2010 | December 31, 2011 | December 31, 2012 | ||||||||||
| Expected life |
5-10 years | 5-10 years | 5-10 years | |||||||||
| Expected volatility |
60-67 | % | 71-78 | % | 74-80 | % | ||||||
| Risk-free interest rate |
2.26-3.56 | % | 1.30-2.98 | % | 0.91-1.60 | % | ||||||
| Dividend yield |
0 | 0 | 0 | |||||||||
64
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
The expected life of the options is based on the observed and expected time to full-vesting, forfeiture and exercise. Groups of employees that have similar historical exercise behavior are considered separately for valuation purposes. The expected volatility assumptions were determined based upon the historical volatility of the Company’s daily closing stock price. The risk-free interest rate is based on rates provided by the U.S. Treasury with a term equal to the expected life of the option. The expected dividend yield is zero as the Company has never paid dividends and does not currently anticipate paying any in the foreseeable future.
At December 31, 2012, stock options to purchase 2,426,533 shares of common stock at exercise prices ranging from $1.00 to $11.11 per share are outstanding and are exercisable at various dates through 2022. The total number of options exercisable at December 31, 2010, 2011, and 2012 was 942,916, 1,627,329 and 1,620,320 respectively, with weighted average exercise prices of $5.42, $4.08 and $ 3.99, respectively. The aggregate intrinsic value of the options exercisable at December 31, 2012 is $29.
The status of the Company’s stock option plans during the periods indicated is summarized as follows:
| Number of Shares |
Weighted Average Exercise Price per Share |
Weighted Average Remaining Contractual Term in Years |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at January 1, 2010 |
2,031,023 | $ | 5.09 | 5.2 | ||||||||||||
|
|
|
|||||||||||||||
| Granted |
250,300 | 6.12 | 8.6 | |||||||||||||
| Exercised |
(29,206 | ) | 3.92 | |||||||||||||
| Forfeited or expired |
(119,238 | ) | 5.72 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31, 2010 |
2,132,879 | 5.19 | 5.4 | |||||||||||||
|
|
|
|||||||||||||||
| Granted |
554,850 | 3.39 | 9.5 | |||||||||||||
| Exercised |
(5,000 | ) | 2.67 | |||||||||||||
| Forfeited or expired |
(625,625 | ) | 6.38 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31,2011 |
2,057,104 | 4.35 | 6.6 | |||||||||||||
|
|
|
|||||||||||||||
| Granted |
863,202 | 3.35 | 9.5 | |||||||||||||
| Exercised |
(61,796 | ) | 2.91 | |||||||||||||
| Forfeited or expired |
(431,977 | ) | 4.44 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding at December 31,2012 |
2,426,533 | 4.01 | 7.0 | $ | 29 | |||||||||||
|
|
|
|||||||||||||||
| Vested and exercisable at December 31, 2012 |
1,620,320 | 3.99 | 6.2 | $ | 29 | |||||||||||
|
|
|
|||||||||||||||
During the years ended December 31, 2010, 2011 and 2012 the weighted average fair value of options granted, estimated as of the grant date using the Black-Scholes option valuation model, was $4.26 , $ 2.29 and $2.28 per share, respectively. The total intrinsic value of options exercised during the years ended December 31, 2010, 2011 and 2012 was $87, $5 and $86, respectively. The requisite service periods for options granted during 2010, 2011 and 2012 for employees was four years and for directors was one year.
65
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
The following table summarizes information about stock options outstanding at December 31, 2012:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Range of Exercise Prices |
Number Outstanding |
Weighted- Average Remaining Contractual Life |
Weighted- Average Exercise Price |
|||||||||||||||||
| Number Exercisable |
Weighted- Average Exercise Price |
|||||||||||||||||||
| $1.00 |
37,156 | 0.0 years | $ | 1.00 | 37,156 | $ | 1.00 | |||||||||||||
| $1.01-$4.50 |
1,968,902 | 7.4 years | 3.47 | 1,413,839 | 3.60 | |||||||||||||||
| $4.51-$11.11 |
420,475 | 5.8 years | 6.83 | 169,325 | 7.90 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
| $1.00-$11.11 |
2,426,533 | 7.0 years | $ | 4.01 | 1,620,320 | $ | 3.99 | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
As of December 31, 2012, of the total 2,426,533 options outstanding, 806,213 have not vested. Of this total unvested amount, 265,700 will vest upon the attainment of certain milestones, and the balance will vest over the requisite service period. There was $947 of total unrecognized compensation cost related to unvested options, of which approximately $285 will be recognized upon achievement of performance milestones and $662 upon completion of the requisite service period. On February 11, 2013, one of the Company’s employees contractually agreed to not exercise 900,000 fully vested options until such time as the stockholders of the Company approve an increase in the number of authorized shares of the Company’s common stock, or, if earlier, the Company’s written consent.
8. Related Party Agreements
| 8. | Related Party Agreements (see also Note 4): |
The Company has in place the following consulting agreements with related parties.
Consulting Agreement with Gerald Wagner, Ph.D.
In January 2007, Dr. Wagner, a former Director on the Company’s Board of Directors, entered into an amended and restated consulting contract with the Company. Under the terms of the amended contract, Dr. Wagner is paid a monthly retainer of $2.5 and will be paid $2.5 for each additional consulting day. This amended agreement will end at the option of Dr. Wagner or the Company at any time, by providing fifteen days prior written notice, or immediately upon the mutual agreement of the Company and Dr. Wagner. The amounts paid to Dr. Wagner amounted to $30 in each of 2010, 2011 and 2012. Dr. Wagner resigned from the Company’s Board of Directors in December 2011 with the consulting contract remaining in effect until termination on December 31, 2012.
Consulting Agreement with Anne Egger
In March 2009, the Company entered into a consulting agreement with Anne Egger for certain consulting services primarily focusing on physician advocacy. The agreement was for an initial term of three months, was subsequently extended to run through September 2012, when it was not renewed. Under the terms of the agreement, Ms. Egger is entitled to receive a consulting fee of $1.6 per day of consulting services rendered. Ms. Egger was appointed to the Company’s Board of Directors as of June 10, 2009. During the years ended December 31, 2010, 2011 and 2012, Ms. Egger was paid $60, $8 and $0, respectively, under this agreement.
| 9. | Other Income: |
In 2005, the Company discontinued all operations associated with its DIFOTI product. Under an exclusive sale and licensing agreement with KaVo Dental GmbH (“KaVo”) to further develop and commercialize DIFOTI,
66
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
KaVo pays the Company an annual royalty based on the number of DIFOTI related systems sold per calendar year. Other income includes approximately $20 in royalty income in each of the years in the three year period ended December 31, 2012. In addition in 2010, the Company received a $245 research and development grant from the federal government.
| 10. | Income Taxes: |
The Company accounts for income taxes using the asset and liability method for deferred income taxes.
The provision for income taxes includes federal, state and local income taxes currently payable and deferred taxes resulting from temporary differences between the financial statement and tax bases of assets and liabilities. Valuation allowances are recorded to reduce deferred tax assets when it is more likely than not that a tax benefit will not be realized.
With respect to uncertain tax positions, the Company would recognize the tax benefit from an uncertain tax position only if it is more-likely-than-not that the tax position will be sustained upon examination by the taxing authorities, based on the technical merits of the position. The tax benefits to be recognized in the financial statements from such a position would be measured based on the largest benefit that has a greater than fifty percent likelihood of being realized upon ultimate resolution. The Company’s reassessment of its tax positions did not have a material impact on its results of operations and financial position.
The Company has incurred net losses since inception, accordingly, it has not provided for income taxes for the years ended December 31, 2010, 2011 and 2012.
The difference between the actual income tax benefit and that computed by applying the U.S. federal income tax rate of 34% to pretax loss from continuing operations is summarized below:
| Year Ended December 31, | ||||||||||||
| 2010 | 2012 | 2012 | ||||||||||
| Computed expected tax benefit |
$ | (6,774 | ) | $ | (6,931 | ) | $ | (7,709 | ) | |||
| State tax benefit, net of federal effect |
(1,195 | ) | (1,223 | ) | (1,360 | ) | ||||||
| Increase in the valuation allowance |
7,969 | 8,154 | 9,069 | |||||||||
|
|
|
|
|
|
|
|||||||
| Provision for income taxes |
$ | — | $ | — | $ | — | ||||||
|
|
|
|
|
|
|
|||||||
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets and liabilities as of December 31, 2011 and 2012 are as follows:
| December 31, | ||||||||
| 2011 | 2012 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 20,256 | $ | 26,471 | ||||
| Capitalized research and developmental costs |
24,023 | 26,511 | ||||||
| Non-cash compensation |
2,834 | 3,200 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
47,113 | 56,182 | ||||||
| Less valuation allowance |
(47,113 | ) | (56,182 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | — | $ | — | ||||
|
|
|
|
|
|||||
67
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Based on the Company’s historical net losses, management does not believe that it is more likely than not that the Company will realize the benefits of these deferred tax assets and, accordingly, a full valuation allowance has been recorded against the deferred tax assets as of December 31, 2011 and 2012. The Company’s valuation allowance against its deferred tax assets increased by $7,969, $8,154 and $9,069 for the years ended December 31, 2010, 2011 and 2012, respectively.
At December 31, 2012, the Company has net operating loss carryforwards of approximately $66 million to offset future taxable income. The Company has experienced certain ownership changes which, under the provisions of Section 382 of the Internal Revenue Code of 1986, as amended, result in annual limitations on the Company’s ability to utilize its net operating losses in the future. The Company believes that these limitations will not materially impact the Company’s ability to utilize its net operating losses in the future. However, any future equity raise by the Company may limit the use of these net operating loss carryforwards.
FASB ASC 740 “Income Taxes” contains guidance with respect to uncertain tax positions which applies to all tax positions and clarifies the recognition of tax benefits in the financial statements by providing for a two-step approach of recognition and measurement. The first step involves assessing whether the tax position is more likely than not to be sustained upon examination based upon its technical merits. The second step involves measurement of the amount to recognize. Tax positions that meet the more likely than not threshold are measured at the largest amount of tax benefit that is greater than 50% likely of being realized upon ultimate finalization with the taxing authority.
The Company does not have any unrecognized tax benefits which would favorably affect the effective tax rate if recognized in future periods, or accrued penalties and interest. If such matters were to arise, the Company would recognize interest and penalties related to income tax matters in income tax expense. The earliest open tax year subject to examination is 2009.
| 11. | Quarterly Operating Results (Unaudited) |
The following is a summary of operating results by quarter for the years ended December 31, 2011 and 2012:
| Quarter Ended | ||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||
| 2011 |
||||||||||||||||
| Net loss |
$ | (4,942 | ) | $ | (4,810 | ) | $ | (6,111 | ) | $ | (4,522 | ) | ||||
| Basic and diluted net loss per share of common stock |
$ | (0.20 | ) | $ | (0.19 | ) | $ | (0.24 | ) | $ | (0.17 | ) | ||||
| 2012 |
||||||||||||||||
| Net loss |
$ | (5,753 | ) | $ | (5,484 | ) | $ | (5,357 | ) | $ | (6,079 | ) | ||||
| Basic and diluted net loss per share of common stock |
$ | (0.19 | ) | $ | (0.18 | ) | $ | (0.17 | ) | $ | (0.20 | ) | ||||
| 12. | Subsequent Events |
In February 2013, we signed a non-binding term sheet with a venture capital lender for a $10 million loan. Of the $10 million, it is anticipated that $6 million will be funded in March 2013 and we will have the option to draw down the remaining $4 million through March 17, 2014, subject to the satisfaction of meeting certain sales
68
Table of Contents
MELA SCIENCES, INC.
Notes to Financial Statements — (Continued)
and revenue targets. It is anticipated that the loan will mature 42 months from the initial closing and bear interest at the rate of 10.45% per year. The term sheet contemplates that during the first 12 months of the loan, only interest will be paid to the lender and after that we will make 30 equal payments of principal and interest until maturity. The loan would be secured by a general lien against all of our assets, other than our intellectual property assets. In addition, the lender will have a security interest in the proceeds of the sale of any of our intellectual property assets. In connection with the loan, the lender will receive a warrant to purchase that number of shares of our common stock equal to 775,000 divided by the to-be-determined exercise price of the warrant. The loan is subject to satisfactory completion of the lender’s due diligence and the execution of definitive loan documents and was conditioned upon the Company having raised $12.5 million in proceeds since January 1, 2013. This condition was satisfied with the proceeds of the ATM offering and the February 11, 2013 offering. There can be no assurance that the loan will be consummated or that the material terms of the loan will be as described.
On February 12, 2013 the Company entered into an underwriting agreement, relating to the public offering of 6,100,000 shares of the Company’s common stock, at a price to the public of $1.30 per share less underwriting discounts and commissions. The gross proceeds to the Company from the sale of the Common Stock totaled $7.9 million. After deducting the Underwriters’ discounts and commissions and other estimated offering expenses payable by the Company, net proceeds were approximately $7.3 million. The offering closed on February 15, 2013. The Common Stock was offered and sold pursuant to the Company’s Prospectus dated June 1, 2010 and the Company’s Prospectus Supplement filed with the Securities and Exchange Commission (the “SEC”) on February 12, 2013, in connection with a takedown from the Company’s effective shelf registration statement on Form S-3 (File No. 333-167113) declared effective by the SEC on June 1, 2010. On February 11, 2013, an employee of the Company contractually agreed to not exercise 900,000 fully vested options until such time as the stockholders of the Company approve an increase in the number of authorized shares of the Company’s common stock or, if earlier, the Company’s written consent.
In June 2012, the Company entered into a sales agreement with Cowen and Company, LLC, to sell shares of the Company’s common stock through an “at-the-market” equity offering program (the “ATM Program”), which was terminated on February 15, 2013 in conjunction with the public offering described above. Subsequent to year end 2012 and through termination of the agreement, the Company sold approximately 4.7 million shares for gross and net proceeds of approximately $8.8 million and $8.5 million, respectively.
As of February 15, 2013, the Company sold an aggregate of approximately 6.6 million shares of its common stock through the ATM Program for gross proceeds of approximately $14.4 million.
69
Table of Contents
Item 9 Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
Evaluation of disclosure controls and procedures
Our Company’s management, with the participation of our chief executive officer and our chief financial officer, has evaluated the effectiveness of our “disclosure controls and procedures” (as defined in Rule 13a-15(e) under the Securities and Exchange Act of 1934) as of December 31, 2012.
Based on such evaluation, our chief executive officer and our chief financial officer have concluded that, as of December 31, 2012, our disclosure controls and procedures were effective to ensure that the information we are required to disclose in reports that we file or submit to the SEC is (1) recorded, processed, summarized and reported within the time periods specified under the rules and forms of the SEC and (2) accumulated and communicated to our management, including our chief executive officer and our chief financial officer, as appropriate to allow timely decisions regarding required disclosures.
70
Table of Contents
Report of Management on Internal Control Over Financial Reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. Under the rules of the SEC, “internal control over financial reporting procedures” is defined as a process designed to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America.
Internal control over financial reporting includes maintaining records, that in reasonable detail, accurately and fairly reflect our transactions and our dispositions of assets; provide reasonable assurance that transactions are recorded as necessary for preparation of our financial statements in accordance with accounting principles generally accepted in the United States of America; provide reasonable assurance that receipts and expenditures of company assets are made only in accordance with management authorization; and provide reasonable assurance regarding the prevention or the timely detection of the unauthorized acquisition, use or disposition of company assets that could have a material effect on our financial statements. Because of its inherent limitations, internal control over financial reporting may not provide absolute assurance that a misstatement of our financial statements would be prevented or detected.
Management conducted an evaluation of the effectiveness of our internal control over financial reporting using the criteria set forth by COSO in Internal Control — Integrated Framework. Based on this evaluation, management concluded that the company’s internal control over financial reporting was effective as of December 31, 2012.
EisnerAmper LLP, the independent registered public accounting firm, has issued their report on our internal control over financial reporting as of December 31, 2012. Their report is included in this Item 9A.
71
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
MELA Sciences, Inc.
We have audited MELA Sciences, Inc.’s internal control over financial reporting as of December 31, 2012, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Report of Management on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions, and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’ s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, MELA Sciences, Inc., maintained, in all material respects, effective internal control over financial reporting as of December 31, 2012, based on criteria established in Internal Control — Integrated Framework issued by COSO.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of MELA Sciences, Inc. as of December 31, 2011 and December 31, 2012, and the related statements of operations, stockholders’ equity and cash flows for each of the years in the three-year period ended December 31, 2012, and our report dated March 4, 2013 expressed an unqualified opinion thereon.
/s/ EisnerAmper LLP
New York, New York
March 4, 2013
72
Table of Contents
Change in internal control over financial reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2012 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on the effectiveness of controls
Our disclosure controls and procedures are designed to provide reasonable, not absolute, assurance that the objectives of our disclosure control system are met. Because of inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues, if any, within a company have been detected.
Not applicable.
Performance Graph
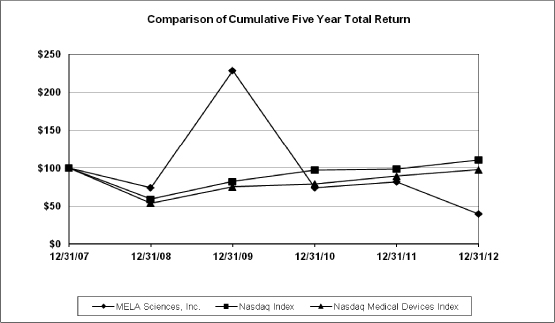
PART III
Item 10. Directors, Executive Officers, and Corporate Governance
The information required by this item will be contained in our definitive proxy statement to be filed with the Securities and Exchange Commission in connection with the Annual Meeting of our Stockholders (the “Proxy Statement”), which is expected to be filed no later than 120 days after the end of our fiscal year ended December 31, 2012, and is incorporated in this report by reference.
Item 11. Executive Compensation
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
73
Table of Contents
| Item 12. Security | Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
Item 14. Principal Accountant Fees and Services
The information required by this item will be set forth in the Proxy Statement and is incorporated in this report by reference.
PART IV
Item 15. Exhibits and Financial Statement Schedules
(a) Exhibits and Financial Statement Schedules:
(1) Financial Statements
See the “Index to Financial Statements” in Part II Item 8 of this report.
(2) Financial Statement Schedules
Not applicable.
(3) Exhibits
A list of exhibits required by Item 601 of Regulation S-K filed or incorporated by reference is found in the Exhibit Index immediately following Part IV of this report.
74
Table of Contents
EXHIBIT INDEX
| Exhibit Number |
Exhibit Title | |
| 3.1 | Fourth Amended and Restated Certificate of Incorporation of the Registrant.(1) | |
| 3.2 | Third Amended and Restated Bylaws of the Registrant.(2) | |
| 4.1 | Specimen Stock Certificate.(2) | |
| 4.2 | Form of Warrant (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on November 1, 2006). | |
| 4.3 | Warrant dated May 7, 2009 issued by Electro-Optical Sciences, Inc. to Kingsbridge Capital Limited (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on May 8, 2009). | |
| 10.1* | Form of Indemnification Agreement for directors and executive officers.(2) | |
| 10.2* | 1996 Stock Option Plan.(3) | |
| 10.3* | 2003 Stock Incentive Plan, as amended.(3) | |
| 10.4* | 2005 Stock Incentive Plan.(2) | |
| 10.5* | Employment Agreement dated as of January 5, 2004 between the Registrant and Joseph V. Gulfo.(3) | |
| 10.6 | Consulting Agreement dated as of May 31, 2005 between the Registrant and Marek Elbaum.(3) | |
| 10.7* | Consulting Agreement dated as of June 20, 2003 between the Registrant and Breaux Castleman, as amended.(1) | |
| 10.8 | Consulting Agreement dated as of June 1, 2005 between the Registrant and Robert Friedman, M.D.(1) | |
| 10.9* | Amended and Restated Consulting Agreement effective as of April 1, 2006 between the Registrant and Gerald Wagner Consulting LLC. (Incorporated by reference to the Registrant’s Annual Report on Form 10-K filed on March 29, 2006). | |
| 10.10* | Employment Offer Letter, dated April 24, 2006, between the Registrant and Richard I. Steinhart. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on April 27, 2006). | |
| 10.11 | Licensing Agreement between the Registrant and KaVo Dental GmbH, dated as of December 5, 2006. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on December 11, 2006). | |
| 10.12* | Amendment No. 1 to Amended and Restated Consulting Agreement dated as of January 30, 2007 by and among the Registrant, Gerald Wagner and Gerald Wagner Consulting LLC. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on January 31, 2007). | |
| 10.13 | Research and Feasibility Agreement between Registrant and L’Oreal S.A. dated as of March 26, 2007. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on March 28, 2007). | |
| 10.14 | Common Stock Purchase Agreement dated as of May 27 between Electro-Optical Sciences, Inc. and Kingsbridge Capital Limited. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on May 8, 2009). | |
| 10.15 | Registration Rights Agreement dated as of May 7, 2009 between Electro-Optical Sciences, Inc. and Kingsbridge Capital Limited.( Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on May 8, 2009). | |
| 10.16 | Agreement of Lease, dated as of July 14, 2009, by and between Stanford Bridge LLC and Electro-Optical Sciences, Inc. (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on July 14, 2009). | |
| 10.17 | Underwriter Agreement dated as of June 30, 2010 among MELA Sciences, Inc., Needham & Company, LLC and Leerink Swann LLC (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on June 30, 2010) | |
75
Table of Contents
| Exhibit Number |
Exhibit Title | |
| 10.18 | Supply Agreement with Arrow Electronics, Inc., dated April 8, 2011 (Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2011 filed on August 5, 2011).+ | |
| 10.19 | Underwriter Agreement dated as of December 15, 2011, among MELA Sciences, Inc., Leerink Swann LLC, Needham & Company, LLC and First Analysis Securities Corporation (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on December 16, 2011). | |
| 10.20 | Production Agreement, dated as of January 6, 2012, by and between MELA Sciences, Inc. and Askion GmbH (Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2012 filed on May 3, 2012).+ | |
| 10.21 | Service Agreement, dated March 21, 2012, by and between MELA Sciences, Inc. and QUINTILES Commercial Germany GmbH (Incorporated by reference to the Registrant’s Quarterly Report on Form 10-Q for the quarterly period ended March 31, 2012 filed on May 3, 2012). | |
| 10.22 | Sales Agreement dated June 15, 2012 between MELA Sciences, Inc. and Cowen and Company, LLC (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on June 15, 2012) | |
| 10.23 | Underwriting Agreement, dated February 12, 2013, by and between MELA Sciences, Inc. and Cowen and Company, LLC (Incorporated by reference to the Registrant’s Current Report on Form 8-K filed on February 12, 2013) | |
| 23.1# | Consent of EisnerAmper LLP | |
| 31.1# | Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2# | Certification of Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1# | Certifications of Chief Executive Officer and Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 101# | The following materials from the Company’s Annual Report on Form 10-K for the year ended December 31, 2012 formatted in Extensible Business Reporting Language (XBRL): (i) the Balance Sheets, (ii) the Statements of Operations, (iii) the Statements of Stockholders’ Equity (iv) the Statements of Cash Flows, and (v) the Notes to Financial Statements. | |
| * | Indicates management compensatory plan, contract or arrangement |
| # | Filed herewith |
| + | Portions of this agreement have been omitted pursuant to a request for confidential treatment. |
| (1) | Incorporated by reference to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-125517), as filed on July 15, 2005. |
| (2) | Incorporated by reference to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-125517), as filed on August 8, 2005. |
| (3) | Incorporated by reference to the Registrant’s Registration Statement on Form S-1, as amended (File No. 333-125517), as filed on June 3, 2005. |
76
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15 (d) of the Securities and Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| MELA SCIENCES, INC. | ||
| By: | /s/ Joseph V. Gulfo, M.D. | |
| Joseph V. Gulfo, M.D. | ||
| Chairman, President and Chief Executive Officer | ||
| (Principal Executive Officer) | ||
Dated: March 5, 2013
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ Joseph V. Gulfo, M.D. Joseph V. Gulfo, M.D. |
Chairman, President and Chief Executive Officer (Principal Executive Officer) |
March 5, 2013 | ||
| /s/ Richard I. Steinhart Richard I. Steinhart |
Sr. Vice President, Finance and Chief Financial Officer (Principal Financial and Accounting Officer) |
March 5, 2013 | ||
| /s/ Robert Coradini Robert Coradini |
Director | March 5, 2013 | ||
| /s/ Anne Egger Anne Egger |
Director | March 5, 2013 | ||
| /s/ Mark Fabiani Mark Fabiani |
Director | March 5, 2013 | ||
| /s/ John Goddard John Goddard |
Director | March 5, 2013 | ||
| /s/ Mindy Meads Mindy Meads |
Director | March 5, 2013 | ||
| /s/ David K. Stone David K. Stone |
Director | March 5, 2013 | ||
| /s/ LuAnn Via LuAnn Via |
Director | March 5, 2013 | ||
77