Attached files
| file | filename |
|---|---|
| EX-31.2 - EXHIBIT 31.2 - CUMBERLAND PHARMACEUTICALS INC | d294938dex312.htm |
| EX-32.1 - EXHIBIT 32.1 - CUMBERLAND PHARMACEUTICALS INC | d294938dex321.htm |
| EX-23.1 - EXHIBIT 23.1 - CUMBERLAND PHARMACEUTICALS INC | d294938dex231.htm |
| EX-10.11 - EXHIBIT 10.11 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1011.htm |
| EX-10.13 - EXHIBIT 10.13 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1013.htm |
| EX-10.27 - EXHIBIT 10.27 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1027.htm |
| EX-10.29 - EXHIBIT 10.29 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1029.htm |
| EX-10.26 - EXHIBIT 10.26 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1026.htm |
| EX-10.12 - EXHIBIT 10.12 - CUMBERLAND PHARMACEUTICALS INC | d294938dex1012.htm |
| EXCEL - IDEA: XBRL DOCUMENT - CUMBERLAND PHARMACEUTICALS INC | Financial_Report.xls |
| EX-31.1 - EXHIBIT 31.1 - CUMBERLAND PHARMACEUTICALS INC | d294938dex311.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
(Mark One)
| x | Annual Report Pursuant To Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the Fiscal Year Ended December 31, 2011
| ¨ | Transition Report Pursuant To Section 13 or 15(d) of the Securities Exchange Act of 1934 |
Commission File No. 001-33637
Cumberland Pharmaceuticals Inc.
(Exact name of registrant as specified in its charter)
| Tennessee | 62-1765329 | |
| State or other jurisdiction of | (I.R.S. Employer | |
| Incorporation or organization | Identification No.) |
2525 West End Avenue, Suite 950,
Nashville, Tennessee 37203
(Address of principal executive offices)(Zip Code)
(615) 255-0068
(Registrant’s telephone number, Including area code)
Securities Registered Pursuant to Section 12(b) of the Act
| Title of each class |
Name of each exchange on which registered | |
| Common stock, no par value | Nasdaq Global Select Market |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter time that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ¨ | Accelerated filer | x | |||
| Non-accelerated filer | ¨ (Do not check if smaller reporting company) | Smaller reporting company | ¨ | |||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act.)
Yes ¨ No x
The aggregate market value of common stock held by non-affiliates as of June 30, 2011 was $74,531,644. The number of shares of the registrant’s Common Stock, no par value, outstanding as of March 1, 2012 was 19,987,511.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required in Part III of Form 10-K is incorporated by reference from the registrant’s Proxy Statement for its 2012 annual meeting of shareholders.
Table of Contents
CUMBERLAND PHARMACEUTICALS INC.
Table of Contents
THE COMPANY
Cumberland Pharmaceuticals Inc. (“Cumberland,” the “Company,” or as used in the context of “we,” “us” or “our”), is a growing specialty pharmaceutical company focused on the acquisition, development and commercialization of branded prescription products. Our primary target markets are hospital acute care and gastroenterology, which are characterized by relatively concentrated physician prescriber bases that we believe can be penetrated effectively by relatively small, targeted sales forces. We are dedicated to providing innovative products that improve quality of care for patients and address poorly met medical needs.
Our product portfolio includes Acetadote® (acetylcysteine) Injection for the treatment of acetaminophen poisoning, Caldolor® (ibuprofen) Injection, the first injectable treatment for pain and fever approved in the United States, Kristalose® (lactulose) for Oral Solution, a prescription laxative, and Hepatoren™ (ifetroban) injection, a Phase II candidate for the treatment of critically ill hospitalized patients suffering from hepatorenal syndrome (HRS). We market and sell our products through our dedicated hospital and gastroenterology sales forces in the United States, which together comprised more than 100 sales representatives and managers as of March 1, 2012.
We have both product development and commercial capabilities, and believe we can leverage our existing infrastructure to support our expected growth. Our management team consists of pharmaceutical industry veterans experienced in business development, product development, commercialization and finance. Our business development team identifies, evaluates and negotiates product acquisition, in-licensing and out-licensing opportunities. Our product development team develops proprietary product formulations, manages our clinical trials, prepares all regulatory submissions and manages our medical call center. Our quality and manufacturing professionals oversee the manufacture of our products. Our marketing and sales professionals are responsible for our commercial activities, and we work closely with our third party distribution partner to ensure availability and delivery of our products.
The following table sets forth our total net revenues, net income attributable to common shareholders and earnings per share (basic and diluted) for the periods presented:
| For the Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (in millions, except per share data) | ||||||||||||
| Total revenues, net |
$ | 51.1 | $ | 45.9 | $ | 43.5 | ||||||
| Research and development expense |
5.0 | 4.3 | 5.0 | |||||||||
| Net income attributable to common shareholders |
5.7 | 2.5 | 3.1 | |||||||||
| Earnings per share, basic |
$ | 0.28 | $ | 0.12 | $ | 0.22 | ||||||
| Earnings per share, diluted |
$ | 0.28 | $ | 0.12 | $ | 0.17 | ||||||
We have been profitable since 2004, generating sufficient cash flows to fund our development and marketing programs. In 2009, we completed an initial public offering of our common stock to help further facilitate our growth. Our strategy includes maximizing the potential of our existing products and continuing to expand our portfolio of differentiated products. Our current products are approved for sale in the United States, and we are working with overseas partners to bring them to international markets. We also look for opportunities to expand into additional patient populations through new product indications, whether through our own clinical studies or by supporting investigator-initiated studies at reputable research institutions. We actively pursue opportunities to acquire additional late-stage development product candidates as well as marketed products in our target medical specialties. Further, we are supplementing these growth strategies with the early-stage drug development activities of Cumberland Emerging Technologies, or CET, our 85% owned subsidiary. CET partners with universities and other research organizations to develop promising, early-stage product candidates, which we have the opportunity to commercialize.
We were incorporated in 1999 and have been headquartered in Nashville, Tennessee since inception. Our website address is www.cumberlandpharma.com. We make available, free of charge through our website our press releases, Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports as soon as reasonably practicable after we file them or furnish them to the U.S. Securities and Exchange Commission, or SEC. These filings are also available to the public at www.sec.gov.
1
Table of Contents
PRODUCTS
Our key products include:
| Products |
Indication |
Status | ||
| Acetadote | Acetaminophen Poisoning | Marketed: Approved by the FDA and launched in 2004; new formulation FDA approved in 2011. | ||
| Caldolor | Pain and Fever | Marketed: Approved in 2009. | ||
| Kristalose | Chronic and Acute Constipation | Marketed: Approved in 2006. | ||
| Hepatoren | Hepatorenal Syndrome | In Phase II clinical development. | ||
Acetadote
Acetadote is an intravenous formulation of N-acetylcysteine, or NAC, indicated for the treatment of acetaminophen poisoning. Acetadote, which has been available in the United States since our introduction of the product in 2004, is currently used in hospital emergency departments to prevent or lessen potential liver damage resulting from an overdose of acetaminophen, a common ingredient in many over-the-counter pain relief and fever-reducing products. Acetaminophen overdose continues to be the leading cause of poisonings reported by hospital emergency rooms in the United States, and Acetadote has become a standard of care for treating this potentially life-threatening condition.
Originally approved in January 2004, Acetadote received U.S. Food and Drug Administration, or FDA, approval as an orphan drug, which provided seven years of marketing exclusivity from date of approval. In connection with the FDA’s approval of Acetadote, we committed to certain post-marketing activities for the product. Our first Phase IV commitment (pediatric) was completed in 2004 and resulted in the FDA’s 2006 approval of expanded labeling for Acetadote for use in pediatric patients. Our second Phase IV commitment (clinical) was completed in 2006 and resulted in further revised labeling for the product with FDA approval of additional safety data in 2008. We completed our third and final Phase IV commitment (manufacturing) for Acetadote in 2010, which culminated in the approval and launch of a new, next generation formulation of the product.
In October 2010, we submitted a supplemental new drug application (sNDA) to the FDA for approval of a new formulation of Acetadote designed to replace the original formulation. The new formulation, which is the result of the aforementioned Phase IV commitment made to the FDA, addresses the FDA’s safety concerns and contains no Ethylene diamine tetracetic acid or other stabilization and chelating agents and is preservative-free. In January 2011, we received FDA approval and commenced U.S. launch activities for this new Acetadote formulation. The original formulation has been removed from FDA reference materials and we no longer manufacture it. We filed a patent application with the U.S. Patent and Trademark Office, or USPTO, to protect the proprietary new formulation in 2011. In February 2012, we received a Notification of Allowance from the USPTO for the new formulation of Acetadote. This Notice of Allowance is a composition of matter patent that enables us to protect the product and its formula as we continue to grow the brand. Upon issuance, the patent will expire in August 2025.
In March 2010, we submitted another sNDA to the FDA for the use of Acetadote in patients with non-acetaminophen acute liver failure. The sNDA included data from a clinical trial led by investigators at the University of Texas Southwestern Medical Center indicating that acute liver failure patients treated with Acetadote have a significantly improved chance of survival without a transplant. The study showed that these patients can also survive a significant number of days longer without transplant, which would provide patients requiring transplant increased time for a donor organ to become available.
Acute liver failure is associated with a high mortality rate and frequent need for liver transplantation. Approximately half of acute liver failure cases are caused by acetaminophen poisoning while the other half result from a variety of causes including hepatitis and alcohol. Currently, transplantation of the liver is the only treatment for patients with liver failure not caused by acetaminophen overdose.
2
Table of Contents
In May 2010, the FDA officially accepted the sNDA and granted a priority review with a response expected in September 2010. In August 2010, we announced that the FDA extended its review of the sNDA by three months, resulting in a new Prescription Drug User Fee Act (PDUFA) goal date in December 2010. In December 2010, we received a Complete Response Letter from the FDA indicating that the agency had completed its review of the application and had identified additional items that must be addressed prior to approving the new indication. We are in discussions with the FDA to gain clarity on a pathway to approval for this indication to treat a critically ill patient population with few treatment alternatives. In addition to expanded labeling for Acetadote, we have requested additional exclusivity for the product in association with the potential new indication.
We are also supporting a number of investigator-initiated studies to explore other potential indications for Acetadote.
Market for Acetadote
Acetaminophen is one of the most widely used drugs for oral treatment of pain and fever in the U.S. and can be found in many common over-the-counter, or OTC, products and prescription narcotics. Though safe at recommended doses, the drug can cause liver damage with excessive use. According to the American Association of Poison Control Centers’ National Poison Data System, acetaminophen poisoning was the leading cause of toxic drug ingestions reported to U.S. poison control centers in 2009. In a study published in 2005 that examined acute liver failure, researchers concluded that acetaminophen poisoning was responsible for acute liver failure in over half the patients examined in 2003, up from 28% in 1998. While an estimated 48% of cases were due to the accidental use over several days, causing chronic liver failure, an estimated 44% of the cases were intentional overdoses, causing acute liver failure. According to the FDA, four grams of acetaminophen is the daily maximum dosage recommended for adults. Ingesting just eight grams of acetaminophen a day can cause serious complications, especially in people whose livers are stressed by virus, medication or alcohol. When used in conjunction with opiates, acetaminophen can offer effective pain relief after surgery or injury; however, patients taking acetaminophen/opiate combination drugs on a chronic basis often eventually require increasing amounts to achieve the same level of pain relief, which can also lead to liver failure. In January 2011, the FDA initiated a campaign to heighten awareness of the potential toxicity associated with acetaminophen and announced that it is asking manufacturers of prescription acetaminophen combination products to limit the maximum amount of acetaminophen in these products to 325 mg per tablet in an effort to reduce adverse events.
NAC is widely accepted as the standard of care for acetaminophen overdose. According to The Medical Letter on Drugs and Therapeutics, NAC is virtually 100% effective in preventing severe liver damage, renal failure and death if administered within eight to ten hours of the overdose. Throughout Europe and much of the rest of the world, NAC has been available in an injectable formulation for over 25 years. Until the 2004 approval of Acetadote, however, the only FDA-approved form of NAC available in the U.S. was an oral preparation. Many U.S. hospitals prepared an off-label, IV form of NAC from the oral solution to treat patients suffering from acetaminophen poisoning. For a number of these patients, an IV product is the only reasonable route of administration due to nausea and vomiting associated with oral administration. Given this market dynamic, we concluded that a medical need existed for an FDA-approved, injectable formulation of NAC for the U.S. market.
Competitive Advantages
We believe Acetadote offers clinical benefits relative to oral NAC including ease of administration, minimizing nausea and vomiting associated with oral NAC, accurate dosage control, shorter treatment protocol and reduction in overall cost of acetaminophen overdose management. Acetadote makes NAC administration easier to tolerate for patients and easier to administer for medical providers.
Acetadote also offers a significant cost benefit to both patient and hospital by reducing treatment regimen, usually from three days to one day. An independently conducted study of Acetadote as a cost-saving treatment for acetaminophen poisoning was published in the December 2009 issue of the peer-reviewed Journal of Medical Economics. The study concludes that Acetadote is a less costly treatment regimen than oral NAC in all evaluated scenarios. The cost differential between the use of oral NAC and Acetadote was shown to range between $881 and $2,259, and was primarily attributable to the time required to complete recommended treatment. Under approved therapeutic protocols, the oral product requires 72 hours to administer compared to 21 hours for Acetadote. Consequently, the use of Acetadote results in shorter hospital stays and substantial cost disparity between the treatments.
3
Table of Contents
New Formulation
In January 2011, the FDA approved our sNDA for our new formulation of Acetadote, which was the result of a Phase IV commitment we made to the FDA upon receipt of initial marketing approval of the product. The new formulation does not contain Ethylene diamine tetracetic acid or any other stabilization and chelating agents and is free of preservatives. We launched the next generation product, which replaced the previously marketed formulation, in the first quarter of 2011 and continued to support the transition to this new product during the second and third quarters of 2011.
In July 2011, we filed a response with the USPTO for a patent to protect our proprietary discoveries related to the new Acetadote formulation. This formulation patent was allowed and issued in China in April 2011. We also filed a second U.S. patent application related to the safety profile of the new formulation.
Acetadote was issued a patent for market use in Australia in late 2011 through Phebra Pharmaceuticals, our Australian commercial partner.
Caldolor
Caldolor, our intravenous formulation of ibuprofen, was the first injectable product approved in the United States for the treatment of both pain and fever. The FDA approved Caldolor for marketing in the United States in June 2009 following a priority review. The product is indicated for use in adults for the management of mild to moderate pain, for the management of moderate to severe pain as an adjunct to opioid analgesics, and for the reduction of fever.
In September 2009, we successfully implemented the U.S. launch of Caldolor, with more than 100 experienced sales professionals promoting the product across the country. Caldolor is stocked at the major wholesalers serving hospitals nationwide, and is available in 800mg vials. In early 2010, we focused on securing formulary approval and stocking nationally for Caldolor. Our sales group worked with members of hospital pharmacy and therapeutic committees to secure placement on committee agendas to continue growing formulary approval.
Later in 2011, we began reaching out to a wider audience within hospitals to drive pull-through sales of Caldolor in facilities that have added the product to formulary. Our sales professionals are equipped with marketing documents which highlight key differentiating factors including the product’s ability to be safely dosed not only post-operatively but also at induction of anesthesia. We supported the publication of Caldolor clinical data in 2011, with results from those trials appearing in peer-reviewed journals. In December 2011, we announced the decision to phase out the 400mg vial of Caldolor. This decision was based on our ongoing refinement of the Caldolor strategy.
We are currently enrolling patients in four clinical studies designed to support marketing of Caldolor. Two of these clinical trials are designed to support pediatric use, including a pediatric fever study to evaluate safety, efficacy and pharmacokinetics of Caldolor in hospitalized children as well as a pediatric pain study. Two registry studies with Caldolor are also underway and are designed to gather additional safety and efficacy data on use of the product in adults. The first of these studies is evaluating Caldolor in treating pain and fever in a wide range of hospitalized patients and the second evaluates the product for management of pain in surgical patients.
We have worldwide commercial rights to Caldolor. We market Caldolor in the United States through our existing hospital sales force, and have partnered with institutions to reach outside the United States.
The Market for Caldolor
Therapeutic agents used to treat pain are known as analgesics. Physicians prescribe injectable analgesics for hospitalized patients who have high levels of pain, require rapid pain relief or cannot take oral analgesics. According to IMS, the U.S. market for injectable analgesics exceeded $331 million, or 599 million units, in 2011. This market consists principally of generic opioids and the NSAID ketorolac.
Injectable opioids such as morphine, meperidine, hydromorphone and fentanyl accounted for approximately 622 million units sold in 2009. While opioids are widely used for acute pain management, they are associated with a variety of side effects including sedation, nausea, vomiting, constipation, headache, cognitive impairment,
4
Table of Contents
reduced gastro intestinal motility and respiratory depression. Respiratory depression, if not monitored closely, can be deadly. Opioid-related side effects can warrant dosing limitations, which may reduce overall effectiveness of pain relief. Side effects from opioids can cause a need for further medication or treatment, and can increase lengths of stay in post-anesthesia care units as well as overall hospital stay, which can lead to increased costs for hospitals and patients.
Despite a poor safety profile, use of ketorolac, the only non-opioid injectable analgesic available in the U.S., has grown from approximately 38 million units in 2004, or 5% of the market, to approximately 40 million units in 2011, or 7% of the market, according to IMS Health. The FDA warns that ketorolac should not be used in various patient populations that are at-risk for bleeding, as a prophylactic analgesic prior to major surgery or for intra-operative administration when stoppage of bleeding is critical.
Caldolor is one of only two U.S. approved injectable treatments for fever, with the other being an injectable acetaminophen product. Significant fever, generally defined as a temperature of greater than 102 degrees Fahrenheit, can cause hallucinations, confusion, convulsions and death. Hospitalized patients are subject to increased risk for developing fever, especially from exposure to infectious agents. Patients with endotracheal intubation, sedation, reduced gastric motility, nausea or recent surgery are frequently unable to ingest, digest, absorb, or tolerate oral products to reduce fever. Treatment for these patients ranges from rectal delivery of medication to physical cooling measures such as tepid baths, ice packs and cooling blankets.
Kristalose
Kristalose is a prescription laxative administered orally for the treatment of constipation. An innovative, dry powder crystalline formulation of lactulose, Kristalose is designed to enhance patient compliance and acceptance. We acquired exclusive U.S. commercialization rights to Kristalose in 2006 from Inalco S.p.A., assembled a dedicated field sales force and re-launched the product in September 2006 under the Cumberland brand. We direct our sales efforts to physicians who are the most prolific writers of prescription laxatives, including gastroenterologists, internists and colon and rectal surgeons.
Effective November 15, 2011 through a series of transactions, we have entered into an agreement with Mylan Inc. to obtain certain assets associated with the Kristalose brand including the Kristalose trademark and the FDA registration. We have also entered into a long-term supply agreement for the product.
As a result of these agreements, we have terminated our license agreement and supply agreement with Inalco S.p.A. By entering into these transactions, we now have streamlined the supply chain for the product and expect to further develop the brand.
Market for Kristalose
Constipation is a common condition in the U.S., affecting approximately 20% of the population each year. While many occurrences are non-recurring, a significant number are chronic in nature and require some treatment to control or resolve. Constipation treatments are sold in both the OTC and prescription segments. The prescription laxative market has historically consisted of a few highly promoted brands including MiraLax® (polyethylene glycol 3350), which is now being sold as an OTC product, and Amitiza®, as well as several generic forms of liquid lactulose. According to data from IMS Health, the prescription laxative market had sales of approximately $689 million in 2011.
Competitive Advantages
Kristalose is the only prescription-strength laxative available in pre-measured powder packets, making it very portable. The drug dissolves quickly in four ounces of water, offering patients a virtually tasteless, grit-free and calorie-free alternative to liquid lactulose treatments. We believe that Kristalose has competitive advantages over competing prescription laxatives, such as fewer potential side effects and contraindications as well as lower cost. There are no age limitations or length of use restrictions for Kristalose, and it is the only osmotic prescription laxative still sampled to physicians.
In 2009, we completed a crossover patient preference study evaluating Kristalose compared to similar products in liquid forms. Patient preference was measured through survey responses collected at the end of the study. Overall, more patients preferred Kristalose, noting portability as a key differentiating feature. More patients also preferred the taste of Kristalose as well as the consistency compared to the syrup formulations. We are also exploring opportunities to expand into new indications with Kristalose.
5
Table of Contents
OUR PRODUCT PIPELINE
Our pre-clinical product candidates are being developed through CET. We negotiate rights to develop and commercialize CET product candidates, and in conjunction with research institutions have obtained nearly $1 million in grant funding from the National Institutes of Health to support the development of these programs.
Hepatoren
In April 2011, we entered into an agreement to acquire the rights to ifetroban, a new Phase II product candidate. We have initiated clinical development under the brand name Hepatoren (ifetroban) Injection and are evaluating this candidate for the treatment of critically ill hospitalized patients suffering from HRS, a life-threatening condition involving progressive kidney failure for which there is no U.S. approved pharmaceutical treatment.
Our acquisition of the rights to the ifetroban program includes an extensive clinical database and non-clinical data package as well as manufacturing processes, know-how and intellectual property. Ifetroban was initially developed by Bristol-Myers Squibb, or BMS, for significant cardiovascular indications. BMS conducted extensive preclinical and clinical studies for its own target indications and eventually donated the entire program to Vanderbilt University. Researchers at Vanderbilt identified ifetroban as a potentially valuable compound in treating patients for several niche indications. We acquired the rights to the ifetroban program from Vanderbilt through CET and intend to develop it for several potential indications, including as an Orphan Drug for HRS for which we will pursue seven years of marketing exclusivity.
The FDA has cleared our Investigational New Drug Application, or IND, for this product candidate and we have initiated a Phase II dose escalation clinical study to evaluate Hepatoren for the treatment of HRS. We have commenced manufacturing and have filed patent applications to protect intellectual property related to the new indication. We believe this product candidate is an excellent strategic fit for us given our established presence in the hospital acute care market.
OUR STRATEGY
Maximize sales of Acetadote and Kristalose
Since its launch in June 2004, we have consistently grown product sales for Acetadote, our injectable treatment for acetaminophen poisoning. Net revenue from Acetadote sales grew from $18.8 million in 2007 to $42.5 million in 2011, a compound annual growth rate of 23%. In 2009, we expanded our hospital sales force in preparation for the launch of Caldolor, and are also leveraging this expansion to support Acetadote sales. In early 2011, we received FDA approval for a new formulation of Acetadote and have subsequently launched that new product. The Acetadote patent was approved by the USPTO in February 2012 and, upon issuance, will expire in August 2025, allowing us to protect the product and its formula as we continue to grow the product.
Kristalose competes in the high growth U.S. prescription laxatives market which, based on data from IMS Health, had sales of approximately $689 million in 2011. After acquiring exclusive U.S. rights to Kristalose in April 2006, we assembled an experienced, dedicated sales force and designed a new marketing program, re-launching the product in September 2006. We inherited this product on a downtrend and have been successful in halting that decline and moving toward growth by enhancing brand awareness and highlighting the product’s many positive, competitive attributes.
Successfully commercialize Caldolor
We believe Caldolor, injectable ibuprofen, currently represents our most significant product opportunity based on the large potential markets for intravenous treatment of pain and fever, as well as clinical results for the product to date. In September 2009, we began marketing the product in the U.S. through our expanded hospital sales force. During 2010, we focused on obtaining formulary approval and stocking of the product at U.S. hospitals and other medical facilities. Beginning in the first quarter of 2011, we began working to increase that stocking as well as drive use of the product in those facilities. We hold international patent rights for Caldolor and, in connection with certain current and potential future international partners, are working to seek regulatory approval for and market Caldolor outside of the U.S.
6
Table of Contents
Continue to build a high-performance sales organization to address our target markets
We believe that continuing to build our sales infrastructure will help drive prescription volume and product sales. We currently utilize two distinct sales teams to address our primary target markets: a hospital sales force for the acute care market and a field sales force for the gastroenterology market.
Hospital market: We promote Acetadote and Caldolor through our dedicated hospital sales team. This team addresses hospitals across the U.S., and is comprised of sales professionals with substantial experience in the hospital market. According to IMS Health, U.S. hospitals accounted for approximately $28 billion, or 9%, of U.S. pharmaceutical sales in 2011. However, IMS also reports that only 2% of approximately $23 billion total pharmaceutical industry promotional spending was focused on hospital-use drugs in 2011. The majority of promotional spending is directed toward large, outpatient markets on drugs intended for chronic use rather than short-term, hospital use. We believe the hospital market is underserved and highly concentrated, and that it can be penetrated effectively by a small, dedicated sales force without large-scale promotional activity.
Gastroenterology market: We promote Kristalose through a dedicated field sales force addressing a targeted group of physicians who are responsible for a majority of total retail Kristalose prescriptions nationally. By investing in our marketing program, we believe that we will be able to increase market share for Kristalose and that we will be equipped to promote any further gastroenterology product additions as well. Because the market for gastrointestinal diseases is broad in patient scope, yet relatively narrow in physician base, we believe it provides product opportunities but can be penetrated with a modest sales force.
Expand our product portfolio by acquiring rights to additional products and late-stage product candidates
In addition to our product development activities, we are also seeking to acquire products or late-stage development product candidates to continue to build a portfolio of complementary products. We focus on under-promoted, FDA-approved drugs as well as late-stage development products that address poorly met medical needs, which we believe helps mitigate our exposure to risk, cost and time associated with drug discovery and research. We plan to continue to target products that are competitively differentiated, have valuable trademarks or other intellectual property, and allow us to leverage our existing infrastructure. We also plan to explore opportunities to seek approval for new uses of existing pharmaceutical products.
Develop a pipeline of early-stage products through CET
In order to build our product pipeline, we are supplementing our acquisition and late-stage development activities with the early-stage drug development activities of CET. CET partners with universities and other research organizations to develop promising, early-stage product candidates, and we have the opportunity to negotiate rights to further develop and commercialize them.
CLINICAL DEVELOPMENT OVERVIEW
Two registry studies with Caldolor are underway and are designed to gather additional safety and efficacy data on use of the product in adults.
The first of two registry studies is a Phase IV multi-center, open-label, single-dose surveillance clinical study to assess the safety and efficacy of ibuprofen administered intravenously over five to ten minutes to adult patients in the hospital setting with fever (temperature >101ºF) and/or pain ( visual analog scale (VAS) assessment >3). Eligible patients will be enrolled to receive one of two dose strengths (400mg for treatment of fever, 800 mg for treatment of pain) of intravenous ibuprofen. One hundred fifty patients will be enrolled in this study.
The second of two registry studies is a Phase IV multi-center, open-label, single or multiple-dose surveillance clinical study will assess the safety of ibuprofen administered intravenously over five to ten minutes to adult hospitalized patients undergoing surgical procedures. Eligible patients will enroll to receive 800 mg intravenous ibuprofen administered at induction of anesthesia. Three hundred patients will be enrolled in this study.
7
Table of Contents
Phase IV Required Pediatric Assessment
The required pediatric assessment for the Caldolor new drug application, or NDA, was deferred for the treatment of fever and for the management of pain. Two clinical studies are currently underway to address the Phase IV requirements.
The first of two pediatric studies is a multi-center, randomized, open-label, parallel, active comparator, study in pediatric patients less than or equal to 16 years of age with fever greater than or equal to 101.0ºF (38.3ºC) to assess the efficacy, safety and pharmacokinetics of intravenous ibuprofen. Two hundred patients will be enrolled in this study.
The second of two pediatric studies is a multi-center, randomized, double-blind placebo-controlled, single-dose study conducted in pediatric patients 6 to 17 years of age undergoing tonsillectomy to assess the safety and efficacy of intravenous ibuprofen. One hundred sixty patients will be enrolled in this study.
No additional Phase IV commitments were assigned by the FDA.
Safety Summary
Extensive use and worldwide literature support the strong safety profile of oral ibuprofen. Building on the oral safety profile, we have assembled an integrated IV ibuprofen safety database combining data from our clinical trials as well as previously published study data. We used this data to support our NDA filing and will continue to use and update the data as a part of our ongoing safety evaluation. In addition, this data will be used by our sales force and in our marketing materials to promote Caldolor.
In clinical trials supporting our proposed indications, no serious adverse events have been directly attributed to Caldolor. The number and percentage of all patients in pivotal studies who reported treatment emergent adverse events was comparable between IV ibuprofen and placebo treatment groups. Additionally, there have been no safety related differences between Caldolor and placebo involving side effects sometimes observed with oral NSAIDs, such as changes in renal function, bleeding events or gastrointestinal disorders.
BUSINESS DEVELOPMENT
Since inception, we have had an active business development program focused on acquiring rights to marketed products and product candidates that fit our strategy and target markets. We source our business development leads through our senior executives and our international network of pharmaceutical and medical industry insiders. These opportunities are reviewed and considered on a regular basis by a multi-disciplinary team of our managers against a list of selection criteria. We have historically focused on product opportunities with relatively low acquisition, development and commercialization costs, employing a variety of deal structures.
We intend to continue to build a portfolio of complementary, niche products largely through product acquisitions and late-stage product development. Our primary targets are under-promoted, FDA-approved drugs with existing brand recognition and late-stage development product candidates that address unmet medical needs in the hospital acute care and gastroenterology markets. We believe that by focusing mainly on approved or late-stage products, we can minimize the significant risk, cost and time associated with drug development.
Through CET, we are collaborating with a growing list of research institutions. Our business development team is responsible for identifying appropriate CET product candidates and negotiating with our university partners to secure rights to these candidates. Although we believe that these collaborations may be important to our business in the future, they are not material to our business at this time.
CET entered into a new collaboration agreement with Washington University in St. Louis to co-develop promising biomedical technologies. Washington University is a national leader in medical research and ranks among the top U.S. institutions in funding by the National Institutes of Health. This collaboration represents the fourth major university partnership for CET, which has similar arrangements with Vanderbilt University, the University of Tennessee and the University of Mississippi.
These agreements allow us to play an important role in fostering and shaping early-stage biomedical research to improve patient care and provide CET and us with access to promising pipeline candidates such as Hepatoren.
8
Table of Contents
CLINICAL AND REGULATORY AFFAIRS
We have in-house capabilities for the management of our clinical, professional and regulatory affairs. Our team develops and manages our clinical trials, prepares regulatory submissions, manages ongoing product-related regulatory responsibilities and manages our medical information call center. Team members have been responsible for devising the regulatory and clinical strategies and obtaining FDA approvals for Acetadote and Caldolor.
Clinical development
Our clinical development personnel are responsible for:
| • | creating clinical development strategies; |
| • | designing, implementing and monitoring our clinical trials; and |
| • | creating case report forms and other study-related documents. |
Regulatory and quality affairs
Our internal regulatory and quality affairs team is responsible for:
| • | preparing and submitting INDs for clearance to begin patient studies; |
| • | preparing and submitting NDAs and fulfilling post-approval marketing commitments; |
| • | maintaining investigational and marketing applications through the submission of appropriate reports; |
| • | submitting supplemental applications for additional label indications, product line extensions and manufacturing improvements; |
| • | evaluating regulatory risk profiles for product acquisition candidates, including compliance with manufacturing, labeling, distribution and marketing regulations; |
| • | monitoring applicable third-party service providers for quality and compliance with current Good Manufacturing Practices, Good Laboratory Practices, and Good Clinical Practices, and performing periodic audits of such vendors; and |
| • | maintaining systems for document control, product and process change control, customer complaint handling, product stability studies and annual drug product reviews. |
Professional and medical affairs
Our medical team provides in-house, medical information support for our marketed products. This includes interacting directly with healthcare professionals to address any product or medical inquiries through our medical information call center. Prior to the launch of Caldolor, we expanded our medical affairs staff to support inquiries from medical professionals regarding the appropriate use of Caldolor as well as to support the efforts of our expanded hospital sales force. In addition to coordinating the call center, our clinical/regulatory group generates medical information letters, provides informational memos to our sales forces and assists with ongoing training for the sales forces.
SALES AND MARKETING
Our sales and marketing team has broad industry experience in selling branded pharmaceuticals. Our sales and marketing professionals manage our dedicated hospital and gastroenterology sales forces, including more than 100 sales representatives and district managers, direct our national marketing campaigns and maintain key national account relationships. In January 2007, we converted our hospital sales force, which had previously been contracted to us by Cardinal Health Inc., or Cardinal, to Cumberland employees through our wholly-owned subsidiary, Cumberland Pharma Sales Corp.
Our gastroenterology-focused team was formed in September 2006 with our re-launch of Kristalose and is a field sales force addressing high prescribers of laxatives. This gastroenterology sales force was previously contracted to us by Ventiv Commercial Services, LLC, or Inventiv. In September 2010, we converted the field sales force to Cumberland employees as we had previously done with our hospital force.
9
Table of Contents
Our sales and marketing executives conduct ongoing market analyses to evaluate marketing campaigns and promotional programs. The evaluations include development of product profiles, testing of the profiles against the needs of the market, determining what additional product information or development work is needed to effectively market the products and preparing financial forecasts. We utilize professional branding and packaging as well as promotional items to support our products, including direct mail, sales brochures, journal advertising, educational and reminder leave-behinds, patient educational pieces and product sampling. We also regularly attend targeted trade shows to promote broad awareness of our products. Our National Accounts group is responsible for key large buyers and related marketing programs. This group supports sales and marketing efforts by maintaining relationships with our wholesaler customers as well as with third-party payors such as Group Purchasing Organizations, Pharmacy Benefit Managers, Hospital Buying Groups, state and federal government purchasers and influencers and health insurance companies.
International Sales and Marketing
We have licensed to third parties the right to distribute certain products outside the U.S. We have granted Alveda Pharmaceuticals Inc., or Alveda, an exclusive license to distribute Caldolor in Canada subject to receipt of regulatory approval. Alveda is obligated to make payments to us upon Caldolor’s achieving specified regulatory milestones in Canada and to pay us a royalty based on Canadian sales of Caldolor. This license terminates five years after regulatory approval is obtained in Canada for the later of the fever or pain indications.
In December 2009, we announced that we entered into an exclusive partnership with DB Pharm Korea Co. Ltd., a Korean-based pharmaceutical company, for the commercialization of Caldolor in South Korea. Under the terms of the agreement, DB Pharm Korea is responsible for obtaining any regulatory approval for the product and handling ongoing regulatory requirements, product marketing, distribution and sales in Korea. We maintain responsibility for product formulation, development and manufacturing. Under the agreement, we received an upfront payment and will receive milestone payments and a transfer price upon sale of the product to our partner. We will also receive royalties on any future sales of Caldolor in South Korea.
In October 2009, we announced that we entered into an exclusive partnership with Phebra Pty Ltd., or Phebra, an Australian-based specialty pharmaceutical company, for the commercialization of Caldolor in Australia and New Zealand. Phebra has responsibility for obtaining any regulatory approval for the product, and for handling all ongoing regulatory requirements, product marketing, distribution and sales in the territories. We will maintain responsibility for product formulation, development and manufacturing. Under the terms of the agreement, we received an upfront payment and will receive milestone payments and a transfer price upon sale of the product to our partner. We will also receive royalties on any future sales of Caldolor in those territories.
We also granted Phebra an exclusive license to market and distribute Acetadote in Australia, New Zealand, and Southeast Asia, subject to the receipt of regulatory approval. Phebra is obligated to make payments to us upon Phebra’s achieving specified milestones as well as royalty payments. In April 2010, the Therapeutic Goods Administration granted approval for the commercialization of Acetadote in Australia and in October 2010, Phebra commenced with the Australian launch of the product. This introduction of Acetadote in Australia marked the introduction of our products into international markets. In addition to Australia, Phebra has exclusive marketing rights to Acetadote for New Zealand and has obtained marketing approval in that country.
In June 2011, we reached an agreement with Harvest & Health Co, LTD in Taiwan and Insanbakti in Malaysia to market Caldolor and Acetadote. Al-Nabil International became our commercial partner of Caldolor and Acetadote in the U.A.E.in late 2011.
The application for regulatory approval of Caldolor in Canada was submitted by our partner Alveda Pharma and approved in December 2011. Review of the application for approval of Caldolor in Australia submitted by our partner Phebra Pty Ltd is under review by the Australian regulatory authorities. We are also currently working to identify appropriate arrangements for the registration and commercialization of our products in other markets.
Net revenues from non-U.S. customers were approximately $0.1 million for each of the years ended December 31, 2011 and 2010, and approximately $0.7 million for the year ended December 31, 2009.
10
Table of Contents
MANUFACTURING AND DISTRIBUTION
We partner certain non-core, capital-intensive functions, including manufacturing and distribution. Our executives are experienced in these areas and manage these third-party relationships with a focus on quality assurance.
Manufacturing
We have entered into manufacturing agreements for all of our products. For Kristalose, we purchase and maintain an inventory of the active pharmaceutical ingredient, or API, used in production. The API is produced by a single supplier based in Italy, for which we are currently negotiating a long-term supply arrangement. All suppliers of APIs must be approved by the FDA prior to utilizing them. We continuously monitor the production capacity of our supplier and their ability to continue to supply our needs.
Our key manufacturing relationships include:
| • | In July 2000, we established an international manufacturing alliance with a predecessor to Hospira Australia Pty. Ltd., or Hospira. Hospira sources active pharmaceutical ingredients, or APIs, and manufactures Caldolor for us under an agreement that expires in June 2014, subject to early termination upon 45 days prior notice in the event of uncured material breach by us or Hospira. The agreement will automatically renew for successive three-year terms unless Hospira or we provide at least 12 months prior written notice of non-renewal. Under the agreement, we pay Hospira a transfer price per unit of Caldolor supplied. In addition, we reimburse Hospira for agreed-upon development, regulatory and inspection and audit costs. |
| • | Bioniche Teoranta, or Bioniche, sources APIs and has manufactured our Acetadote product for sale in the U.S. at its FDA-approved manufacturing facility in Ireland. Our relationship with Bioniche began in January 2002. Bioniche manufactures and packages Acetadote for us, and we purchase Acetadote from Bioniche pursuant to an agreement that we are currently renegotiating. |
| • | We entered into an agreement with Bayer Healthcare, LLC, or Bayer, in February 2008 for the manufacture of Caldolor and Acetadote. The agreement expires in February 2013, subject to early termination upon 30 days prior written notice in the event of uncured material breach by us or Bayer. The agreement will automatically renew for successive one-year terms unless Bayer or we provide at least six months prior written notice of non-renewal. Under the agreement, we pay Bayer a transfer price per each unit of Caldolor or Acetadote supplied. In addition, we pay Bayer for agreed upon development costs. |
Distribution
Like many other pharmaceutical companies, we employ an outside third-party logistics contractor to facilitate our distribution efforts. Since August 2002, Specialty Pharmaceutical Services, or SPS, (formerly CORD Logistics, Inc.) has exclusively handled all aspects of our product logistics efforts, including warehousing, shipping, customer billing and collections. SPS is a division of Cardinal. SPS’s main facility is located outside of Nashville, Tennessee, with more than 325,000 square feet of space and a well-established infrastructure. In 2008, SPS opened a second, distribution-only facility in Reno, Nevada, with an additional 88,000 square feet of space. We began utilizing this facility for distribution to certain locations in the second half of 2008. We maintain ownership of our finished products until sale to our customers.
TRADEMARKS, PATENTS AND PROPRIETARY RIGHTS
We seek to protect our products from competition through a combination of patents, trademarks, trade secrets, FDA exclusivity and contractual restrictions on disclosure. Proprietary rights, including patents, are an important element of our business. We seek to protect our proprietary information by requiring our employees, consultants, contractors and other advisors to execute agreements providing for protection of our confidential information upon commencement of their employment or engagement. We also require confidentiality agreements from entities that receive our confidential data or materials.
11
Table of Contents
Acetadote
Acetadote was approved by the FDA in January 2004 as an orphan drug for the intravenous treatment of acetaminophen overdose. As an orphan drug, we were entitled to seven years of marketing exclusivity for the treatment of this approved indication, which expired in January 2011. In January 2011, we received FDA approval for our next generation, new formulation of Acetadote, for which we have applied for patent protection through U.S. patent application No. 11/209,804, as well as through international application No. PCT/US06/20691, both of which are directed to acetylcysteine compositions, methods of making the same and methods of using the same. In addition, we have an exclusive, worldwide license to NAC clinical data from Newcastle Master Misercordiae Hospital in Australia. We have no expected outstanding payment obligations pursuant to this contract. In 2011, we also applied for a patent for a second indication for Acute Liver Failure, or ALF, and this is pending approval.
Caldolor
We are the owner of U.S. Patent No. 6,727,286, which is directed to ibuprofen solution formulations, methods of making the same, and methods of using the same, and which expires in 2021. This U.S. patent is associated with our completed international application No. PCT/US01/42894. We have filed for international patent protection in association with this PCT application in various countries, some of which have been allowed and some of which remain pending.
In 2009, we also filed the first of several new patent applications for Caldolor. Part of an ongoing initiative to protect the value of our intellectual property, the new applications address our proprietary method of dosing intravenous ibuprofen.
We have an exclusive, worldwide license to clinical data for intravenous ibuprofen from Vanderbilt University, in consideration for royalty and other payment obligations related to Caldolor.
In addition, we received three years marketing exclusivity upon receipt of FDA approval for Caldolor. We intend to seek further exclusivity from the FDA upon completion of successful pediatric clinical trials for the product.
COMPETITION
The pharmaceutical industry is characterized by intense competition and rapid innovation. Our continued success in developing and commercializing pharmaceutical products will depend, in part, upon our ability to compete against existing and future products in our target markets. Competitive factors directly affecting our markets include but are not limited to:
| • | product attributes such as efficacy, safety, ease-of-use and cost-effectiveness; |
| • | brand awareness and recognition driven by sales and marketing and distribution capabilities; |
| • | intellectual property and other exclusivity rights; |
| • | availability of resources to build and maintain developmental and commercial capabilities; |
| • | successful business development activities; |
| • | extent of third-party reimbursements; and |
| • | establishment of advantageous collaborations to conduct development, manufacturing or commercialization efforts. |
A number of our competitors possess research and development and sales and marketing capabilities as well as financial resources greater than ours. These competitors, in addition to emerging companies and academic research institutions, may be developing, or in the future could develop, new technologies that could compete with our current and future products or render our products obsolete.
Acetadote
Acetadote is our injectable formulation of NAC for the treatment of acetaminophen overdose. NAC is accepted worldwide as the standard of care for acetaminophen overdose. Despite the availability of injectable NAC outside the United States, Acetadote, to our knowledge, is the only injectable NAC product approved in the U.S. to treat acetaminophen overdose. Our competitors in the acetaminophen overdose market are those companies selling orally administered NAC including, but not limited to, Geneva Pharmaceuticals, Inc., Bedford Laboratories division of Ben Venue Laboratories, Inc., Roxane Laboratories, Inc. and Hospira Inc.
12
Table of Contents
Caldolor
Caldolor is marketed for the treatment of pain and fever, primarily in a hospital setting. A variety of other products address the acute pain market, including but not limited to:
| • | Morphine, the most commonly used product for the treatment of acute, post-operative pain, is manufactured and distributed by several generic pharmaceutical companies. |
| • | DepoDur® is an extended release injectable formulation of morphine that is marketed by EKR Therapeutics, Inc. |
| • | Other generic injectable opioids, including fentanyl, meperidine and hydromorphone, address this market. |
| • | Ketorolac (brand name Toradol®), an injectable NSAID, is also manufactured and distributed by several generic pharmaceutical companies. |
| • | Ofirmev®, an injectable acetaminophen product, was approved by the FDA in 2010. |
We are aware of other product candidates in development to treat acute pain including injectable NSAIDs, novel opioids, new formulations of existing therapies and extended release anesthetics. We believe non-narcotic analgesics for the treatment of post-surgical pain are the primary potential competitors to Caldolor.
In addition to the injectable analgesic products above, many companies are developing analgesics for specific indications such as migraine and neuropathic pain, oral extended-release forms of existing narcotic and non-narcotic products, and products with new methods of delivery such as transdermal. We are not aware of any approved injectable products indicated for the treatment of fever in the U.S. other than Caldolor and Ofirmev. There are, however, numerous drugs available to physicians to reduce fevers in hospital settings via oral administration to the patient, including ibuprofen, acetaminophen, and aspirin. These drugs are manufactured by numerous pharmaceutical companies.
Kristalose
Kristalose is a dry powder crystalline prescription formulation of lactulose indicated for the treatment of constipation. The U.S. constipation therapy market includes various prescription and OTC products. The prescription products which we believe are our primary competitors are Amitiza® and liquid lactuloses. Amitiza is indicated for the treatment of chronic idiopathic constipation in adults and is marketed by Sucampo Pharmaceuticals Inc. and Takeda Pharmaceutical Company Limited. Liquid lactulose products are marketed by a number of pharmaceutical companies.
There are several hundred OTC products used to treat constipation marketed by numerous pharmaceutical and consumer health companies. MiraLax® (polyethylene glycol 3350), previously a prescription product, was indicated for the treatment of constipation and manufactured and marketed by Braintree Laboratories, Inc. Under an agreement with Braintree, Schering-Plough introduced MiraLax as an OTC product in February 2007.
GOVERNMENT REGULATION
Pharmaceutical companies are subject to extensive regulation by national, state, and local agencies in the U.S. and additional regulations in other countries in which they do business. The manufacture, distribution, marketing and sale of pharmaceutical products is subject to government regulation in the U.S. and various foreign countries. Additionally, in the U.S., we must follow rules and regulations established by the FDA requiring the presentation of data indicating that our products are safe and efficacious and are manufactured in accordance with cGMP regulations. If we do not comply with applicable requirements, we may be fined, the government may refuse to approve our marketing applications or allow us to manufacture or market our products, and we may be criminally prosecuted. We and our manufacturers and clinical research organizations may also be subject to regulations under other federal, state and local laws, including, but not limited to, the Occupational Safety and Health Act, the Resource Conservation and Recovery Act, the Clean Air Act and import, export and customs regulations as well as the laws and regulations of other countries.
13
Table of Contents
FDA Approval Process
The steps required to be taken before a new prescription drug may be marketed in the U.S. generally include:
| • | completion of pre-clinical laboratory and animal testing; |
| • | the submission to the FDA of an IND, which must be evaluated and found acceptable by the FDA before human clinical trials may commence; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use; and |
| • | submission and approval of an NDA. |
The sponsor of the drug typically conducts human clinical trials in three sequential phases, but the phases may overlap. In Phase I clinical trials, the product is tested in a small number of patients or healthy volunteers, primarily for safety at one or more dosages. In Phase II clinical trials, in addition to safety, the sponsor evaluates the efficacy of the product on targeted indications, and identifies possible adverse effects and safety risks in a patient population. Phase III clinical trials typically involve testing for safety and clinical efficacy in an expanded population at geographically-dispersed test sites.
The FDA requires that clinical trials be conducted in accordance with the FDA’s good clinical practices (GCP) requirements. The FDA may order the partial, temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The institutional review board (IRB), or ethics committee (outside of the U.S.), of each clinical site generally must approve the clinical trial design and patient informed consent and may also require the clinical trial at that site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
The results of the pre-clinical and clinical trials, together with, among other things, detailed information on the manufacture and composition of the product and proposed labeling, are submitted to the FDA in the form of an NDA for marketing approval. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has ten months in which to complete its initial review of a standard NDA and respond to the applicant. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months of the PDUFA goal date. If the FDA’s evaluations of the NDA and the clinical and manufacturing procedures and facilities are favorable, the FDA may issue an approval letter. The FDA may also issue an approvable letter setting forth further conditions that must be met in order to secure final approval of the NDA. If and when those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter. An approval letter authorizes commercial marketing of the drug for certain indications. According to the FDA, the median total approval time for NDAs approved during calendar year 2004 was approximately 13 months for standard applications. If the FDA’s evaluations of the NDA submission and the clinical and manufacturing procedures and facilities are not favorable, it may refuse to approve the NDA and issue a not-approvable letter. The time and cost of completing these steps and obtaining FDA approval can vary dramatically depending on the drug. However, to complete these steps for a novel drug can take many years and cost millions of dollars.
Section 505(b)(2) New Drug Applications
As an alternate path for FDA approval of new indications or new formulations of previously-approved products, a company may file a Section 505(b)(2) NDA, instead of a “stand-alone” or “full” NDA. Section 505(b)(2) of the Food, Drug, and Cosmetic Act, or FDC, was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. Some examples of products that may be allowed to follow a 505(b)(2) path to approval are drugs that have a new dosage form, strength, route of administration, formulation or indication.
14
Table of Contents
We successfully secured FDA approvals for Acetadote in January 2004 and for Caldolor in June 2009 pursuant to the 505(b)(2) pathway. Upon approval of a “full” or 505(b)(2) NDA, a drug may be marketed only for the FDA-approved indications in the approved dosage forms. Further clinical trials are necessary to gain approval for the use of the product for any additional indications or dosage forms. The FDA may also require post-market reporting and may require surveillance programs to monitor the side effects of the drug, which may result in withdrawal of approval after marketing begins.
Special Protocol Assessment Process
The special protocol assessment, or SPA, process generally involves FDA evaluation of a proposed Phase III clinical trial protocol and a commitment from the FDA that the design and analysis of the trial are adequate to support approval of an NDA, if the trial is performed according to the SPA and meets its endpoints. The FDA’s guidance on the SPA process indicates that SPAs are designed to evaluate individual clinical trial protocols primarily in response to specific questions posed by the sponsors. In practice, the sponsor of a product candidate may request an SPA for proposed Phase III trial objectives, designs, clinical endpoints and analyses. A request for an SPA is submitted in the form of a separate amendment to an IND, and the FDA’s evaluation generally will be completed within a 45-day review period under applicable PDUFA goals, provided that the trials have been the subject of discussion at an end-of-Phase II and pre-Phase III meeting with the FDA, or in other limited cases.
On June 14, 2004, we submitted a request for SPA of our Caldolor Phase III clinical study. During a meeting with the FDA on September 29, 2004, the FDA confirmed that the efficacy data from our study of post-operative pain with a positive outcome was considered sufficient to support a 505(b)(2) application for the pain indication. Final determinations by the FDA with respect to a product candidate, including as to the scope of its “labeling,” are made after a complete review of the applicable NDA and are based on the entire data in the application.
Orphan Drug Designation
The Orphan Drug Act of 1983, or Orphan Drug Act, encourages manufacturers to seek approval of products intended to treat “rare diseases and conditions” with a prevalence of fewer than 200,000 patients in the U.S. or for which there is no reasonable expectation of recovering the development costs for the product. For products that receive orphan drug designation by the FDA, the Orphan Drug Act provides tax credits for clinical research, FDA assistance with protocol design, eligibility for FDA grants to fund clinical studies, waiver of the FDA application fee, and a period of seven years of marketing exclusivity for the product following FDA marketing approval. Acetadote received Orphan Drug designation in October 2001 and was approved by the FDA for the intravenous treatment of moderate to severe acetaminophen overdose in January 2004. As an orphan drug, Acetadote was entitled to marketing exclusivity until January 2011 for the treatment of this approved indication, and we intend to seek additional exclusivity for this product through new potential indications. However, even if granted, this exclusivity would not prevent a product with a different formulation from competing with Acetadote.
The Hatch-Waxman Act
Among other things, the Hatch-Waxman Act provides three years of marketing exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) NDAs in certain situations, including new indications, dosages or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application. It is under this provision that we received three years marketing exclusivity for Caldolor upon receipt of FDA approval in June 2009.
Healthcare Patient Protection and Affordable Care Act
On March 23, 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Healthcare and Education Reconciliation Act of 2010, or collectively the PPACA. As enacted, PPACA represents a significant change to the healthcare industry, including the way in which pharmaceuticals are covered and reimbursed. The following highlights certain provisions of PPACA, as enacted, that may affect us.
Pharmaceutical Industry Fee: Beginning in calendar-year 2011, an annual fee is imposed on pharmaceutical manufacturers and importers that sell branded prescription drugs to specified government programs (e.g., Medicare Part D, Medicare Part B, Medicaid, Department of Veterans Affairs programs,
15
Table of Contents
Department of Defense programs and TRICARE). The annual fee is allocated to companies based on their previous calendar-year market share using sales data provided by the government agencies that purchase the pharmaceuticals to the Treasury Department. Although we participate in governmental programs that would subject us to this fee, our sales volume in such programs is less than $10 million, with the first $5 million of sales being exempt from the fee. We do not anticipate this fee will have a material impact on our results of operations.
Medicaid Rebate Rate: We currently provide rebates for Kristalose sold to Medicaid beneficiaries. Effective January 1, 2010, the rebate for non-innovator drugs increased from eleven percent to thirteen percent of the average manufacturer price. Our sales of Kristalose under the Medicaid program have been increasing. The increased rebate percentage did not have a material impact on our results of operations for the year ended December 31, 2011.
OTHER LAWS AND REGULATORY REQUIREMENTS
Regulations continue to apply to pharmaceutical products after FDA approval occurs. Post-marketing safety surveillance is required in order to continue to market an approved product. The FDA also may, in its discretion, require post-marketing testing and surveillance to monitor the effects of approved products or place conditions on any approvals that could restrict the commercial applications of these products.
If we seek to make certain changes to an FDA-approved product, such as promoting or labeling a product for a new indication, making certain manufacturing changes or product enhancements or adding labeling claims, we will need FDA review and approval before the change can be implemented. While physicians may use products for indications that have not been approved by the FDA, we may not label or promote the product for an indication that has not been approved. Securing FDA approval for new indications or product enhancements and, in some cases, for manufacturing and labeling claims, is generally a time-consuming and expensive process that may require us to conduct clinical trials under the FDA’s IND regulations. Even if such studies are conducted, the FDA may not approve any change in a timely fashion, or at all. In addition, adverse experiences associated with use of the products must be reported to the FDA, and FDA rules govern how we can label, advertise or otherwise commercialize our products. In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs.
Federal False Claims Act
The federal False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product.
Outside of the U.S., our ability to market our products will also depend on receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country.
ENVIRONMENTAL MATTERS
We are subject to federal, state, and local environmental laws and regulations and we believe that our operations comply with such regulations. We anticipate that the effects of compliance with federal, state and local laws and regulations relating to the discharge of materials into the environment will not have any material effect on our capital expenditures, earnings or competitive position.
16
Table of Contents
SEASONALITY
There are no significant seasonal aspects to our business.
BACKLOG
Due to the relatively short lead-time required to fill orders for our products, backlog of orders is not considered material to our business.
EMPLOYEES
As of March 1, 2012, we had 138 full-time employees. In addition, we believe that utilizing experienced, independent contractors and consultants is a cost-efficient and effective way to accomplish our goals and a number of individuals have provided or are currently providing services to us pursuant to agreements between the individuals or their employers and us. None of our employees are represented by a collective bargaining unit. We believe that we have positive relationships with our employees.
In February 2011, Tan Cheow Choon was appointed Director of International Business. Based in Singapore, Choon is responsible for executing our initiative to expand our product markets across Asia.
Effective October of 2011, we named Rick S. Greene as Vice President and Chief Financial Officer. He had previously been serving as the interim Vice President of Finance and Accounting since April 2011. Mr. Greene has over 20 years of experience in financial management and reporting. Prior to joining us, he supported the accounting activities associated with our initial public offering and the ongoing preparation of our quarterly financial information following out stock exchange listing.
You should carefully consider the risk factors described below and throughout this report, which could materially affect our business. There are also risks that are not presently known or not presently material, as well as the other information set forth in this report that could materially affect our business. In addition, in our periodic filings with the SEC, press releases and other statements, we discuss estimates and projections regarding our future performance and business outlook. By their nature, such “forward-looking statements” involve known and unknown risks, uncertainties and other factors that in some cases are out of our control. For a further discussion of forward-looking statements, please refer to the section entitled “Special Note Regarding Forward-Looking Statements.” These factors could cause our actual results to differ materially from our historical results or our present expectations and projections. These risk factors and uncertainties include, but are not limited to the following:
RISKS RELATED TO OUR BUSINESS
An adverse development regarding our products could have a material and adverse impact on our future revenues and profitability.
A number of factors may impact the effectiveness of our marketing and sales activities and the demand for our products, including:
| • | The prices of our products relative to other drugs or competing treatments; |
| • | Any unfavorable publicity concerning us, our products, or the markets for these products such as information concerning product contamination or other safety issues in any of our product markets, whether or not directly involving our products; |
| • | Perception by physicians and other members of the healthcare community of the safety or efficacy of our products or competing products; |
17
Table of Contents
| • | Regulatory developments related to our marketing and promotional practices or the manufacture or continued use of our products; |
| • | Changes in intellectual property protection available for our products or competing treatments; |
| • | The availability and level of third-party reimbursement for sales of our products; and |
| • | The continued availability of adequate supplies of our products to meet demand. |
If demand for our products weaken, our revenues and profitability will likely decline. Known adverse effects of our marketed products are documented in product labeling, including the product package inserts, medical information disclosed to medical professionals and all marketing-related materials. At this time, no unforeseen or serious adverse effects outside of those specified in current product labeling have been directly attributed to our approved products.
We currently market and sell three products: Caldolor, Acetadote and Kristalose. A product contamination or other safety or regulatory issues, such as a failure to meet certain U.S. Food and Drug Administration, or FDA, reporting requirements involving our products could negatively impact us and possibly lead to a product recall. In addition, changes impacting any of our products in areas such as competition, lack of market acceptance or demand, government regulation, intellectual property, reimbursement and manufacturing could have an adverse impact on our future revenues and profitability.
In 2011, the FDA issued a press announcement asking manufacturers of prescription combination products that contain acetaminophen to limit the amount of acetaminophen to no more than 325 milligrams (mg) in each tablet or capsule. The FDA also is requiring manufacturers to update labels of all prescription combination acetaminophen products to warn of the potential risk for severe liver injury. The actions the FDA is taking for prescription acetaminophen combination products do not affect over-the-counter acetaminophen products. The FDA’s regulation of acetaminophen in prescription combination products and over-the-counter products may reduce the number of acetaminophen overdoses which could result in a lower demand for Acetadote. If the demand for Acetadote decreases, it could have an adverse impact on our future revenues and profitability.
Caldolor was approved by the FDA in June 2009, and we started commercializing Caldolor in the United States in September 2009. The commercial success of Caldolor is dependent on many third-parties, including physicians, pharmacists, hospital pharmacy and therapeutics committees, or P&T committees, suppliers and distributors, all of whom we have little or no control over. We expect Caldolor to be administered primarily to hospitalized patients who are unable to receive oral therapies for the treatment of pain or fever. Before we can distribute Caldolor to any new hospital customers, Caldolor must be approved for addition to the hospitals’ formulary lists by their P&T committees. A hospital’s P&T committee generally governs all matters pertaining to the use of medications within the institution, including review of medication formulary data and recommendations of drugs to the medical staff. We cannot guarantee that we will be successful in getting the approvals we need from enough P&T committees to be able to optimize hospital sales of Caldolor. Even if we obtain hospital approval for Caldolor, we must still convince individual hospital physicians to prescribe Caldolor repeatedly. Because Caldolor is a new drug, any mistakes made in the timely supply of Caldolor, education about how to properly administer Caldolor or any unexpected side effects that develop from use of the drug, may lead physicians to not accept Caldolor as a viable treatment alternative. The commercial success of Caldolor also depends on our ability to coordinate supply, distribution, marketing, sales and education efforts. We have set a price for Caldolor that we believe hospitals and other purchasers are willing to pay, but that will also generate sufficient profits. If we have set a price for Caldolor that hospitals consider too high, we may need to subsequently reduce the price for Caldolor. As with our other products, if the price for Caldolor is not accepted in the marketplace, it could have an adverse impact on our future revenues and profitability.
If any manufacturer we rely upon fails to produce our products in the amounts we require on a timely basis, or fails to comply with stringent regulations applicable to pharmaceutical drug manufacturers, we may be unable to meet demand for our products and may lose potential revenues.
We do not manufacture any of our products, and we do not currently plan to develop any capacity to do so. Our dependence upon third parties for the manufacture of products could adversely affect our profit margins or our ability to develop and deliver products on a timely and competitive basis. If for any reason we are unable to obtain
18
Table of Contents
or retain third-party manufacturers on commercially acceptable terms, we may not be able to sell our products as planned. Furthermore, if we encounter delays or difficulties with contract manufacturers in producing our products, the distribution, marketing and subsequent sales of these products could be adversely affected.
Caldolor is manufactured at Hospira Australia Pty. Ltd.’s facility in Australia. Acetadote is manufactured primarily at Bayer’s facility in Kansas and Bioniche’s manufacturing plant in Ireland is an alternative manufacturing source for Acetadote. The active pharmaceutical ingredient for Kristalose is manufactured at a single facility in Italy. If any one of these facilities is damaged or destroyed, or if local conditions result in a work stoppage, we could suffer an inability to meet demand for our products. Kristalose is manufactured through a complex process. It would be particularly difficult to find a new manufacturer of Kristalose on an expedited basis. As a result of these factors, our ability to manufacture Kristalose may be substantially impaired if the manufacturer is unable or unwilling to supply sufficient quantities of the product.
In addition, all manufacturers of our products and product candidates must comply with current good manufacturing practices, referred to as cGMP, enforced by the FDA through its facilities inspection program. These requirements include quality control, quality assurance and the maintenance of records and documentation. Manufacturers of our products may be unable to comply with cGMP requirements and with other FDA, state and foreign regulatory requirements.
We have no control over our manufacturers’ compliance with these regulations and standards. If our third-party manufacturers do not comply with these requirements, we could be subject to:
| • | fines and civil penalties; |
| • | suspension of production or distribution; |
| • | suspension or delay in product approval; |
| • | product seizure or recall; and |
| • | withdrawal of product approval. |
We are dependent on a variety of other third parties. If these third parties fail to perform as we expect, our operations could be disrupted and our financial results could suffer.
We have a relatively small internal infrastructure. We rely on a variety of third parties, other than our third-party manufacturers, to help us operate our business. Other third parties on which we rely include:
| • | Cardinal Health Specialty Pharmaceutical Services, a logistics and fulfillment company and business unit of Cardinal, which warehouses and ships our marketed products and |
| • | Vanderbilt University and the Tennessee Technology Development Corporation, co-owners with us of CET, and the universities that collaborate with us in connection with CET’s research and development programs. |
If these third parties do not continue to provide services to us, or collaborate with us, we might not be able to obtain others who can serve these functions. This could disrupt our business operations, increase our operating expenses or otherwise adversely affect our operating results.
Competitive pressures could reduce our revenues and profits.
The pharmaceutical industry is intensely competitive. Our strategy is to target differentiated products in specialized markets. However, this strategy does not relieve us from competitive pressures and can entail distinct competitive risks. Certain of our competitors do not aggressively promote their products in our markets. An increase in promotional activity in our markets could result in large shifts in market share, adversely affecting us.
Our competitors may sell or develop drugs that are more effective and useful or less costly than ours, and they may be more successful in manufacturing and marketing their products. Many of our competitors have significantly greater financial and marketing resources than we do. Additional competitors may enter our markets.
19
Table of Contents
The pharmaceutical industry is characterized by constant and significant investment in new product development, which can result in rapid technological change. The introduction of new products could substantially reduce our market share or render our products obsolete. The selling prices of pharmaceutical products tend to decline as competition increases, through new product introduction or otherwise, which could reduce our revenues and profitability.
Governmental and private healthcare payors emphasize substitution of branded pharmaceuticals with less expensive generic equivalents. An increase in the sales of generic pharmaceutical products could result in a decrease in revenues of our branded pharmaceuticals.
Any attempt by us to expand the potential market for any of our products is subject to limitations.
Expansion of the market for our products may be subject to certain limitations. For example, in its June 2009 Caldolor approval letter, the FDA required us to conduct two additional Phase IV pediatric studies by 2011 and 2012, respectively. If the results of these Phase IV clinical studies are not favorable, we may not be able to expand the market for Caldolor to children ages 1-16. We may also experience delays associated with these required Phase IV clinical studies potentially resulting from, among other factors, difficulty enrolling pediatric patients. Such delays could impact our ability to obtain an additional six months of FDA exclusivity.
In addition, we have only obtained regulatory approval to market our products in the United States. In foreign jurisdictions, we have licensed the right to market some of our products to third parties. These third parties are responsible for seeking regulatory approval for the products in their respective jurisdictions. We have no control over these third parties and cannot be sure that marketing approval for our products will be obtained outside the United States.
Our future growth depends on our ability to identify and acquire rights to products. If we do not successfully identify and acquire rights to products and successfully integrate them into our operations, our growth opportunities may be limited.
We acquired rights to Caldolor, Acetadote, Kristalose and Hepatoren. Our business strategy is to continue to acquire rights to FDA-approved products as well as pharmaceutical product candidates in the late stages of development. We do not plan to conduct basic research or pre-clinical product development, except to the extent of our investment in CET. As compared to large multi-national pharmaceutical companies, we have limited resources to acquire third-party products, businesses and technologies and integrate them into our current infrastructure. Many acquisition opportunities involve competition among several potential purchasers including large multi-national pharmaceutical companies and other competitors that have access to greater financial resources than we do. With future acquisitions, we may face financial and operational risks and uncertainties. We may not be able to engage in future product acquisitions, and those we do complete may not be beneficial to us in the long term.
Furthermore, other products in development may encounter unforeseen issues during their clinical trials. Any unforeseen issues or lack of FDA approval will negatively affect marketing and development plans for those products.
Our Hepatoren product candidate has not been approved for sale and may never be successfully commercialized.
We anticipate that a portion of our future growth will come from sales of our Hepatoren product candidate. Hepatoren, which is injectable ifetroban, is a drug used to treat hepatorenal syndrome, or HRS. However, Hepatoren has not been approved by the FDA for marketing, and it is still subject to risks associated with its development.
The FDA has cleared our Investigational New Drug application for this product candidate and we have initiated a Phase II dose escalation clinical study to evaluate Hepatoren for the treatment of HRS. We have commenced manufacturing and have filed patent applications to protect intellectual property related to the new indication. Delays in the completion of the clinical study could significantly delay commercial launch and affect our product development costs. Moreover, results from the clinical study may not be favorable.
20
Table of Contents
Even if Hepatoren is eventually successfully developed and approved by the FDA, it may never gain significant acceptance in the marketplace and therefore never generate substantial revenue or profits for us. Physicians may determine that existing drugs are adequate to address patients’ needs. The extent to which Hepatoren will be reimbursed by the U.S. government or third-party payors is also currently unknown.
As a result of the foregoing and other factors, we do not know the extent to which Hepatoren will contribute to our future growth.
If we are unable to maintain, train and build an effective sales and marketing infrastructure, we will not be able to commercialize and grow our products and product candidates successfully.
As we grow, we may not be able to secure sales personnel or organizations that are adequate in number or expertise to successfully market and sell our products. This risk would be accentuated if we acquire products in areas outside of hospital acute care and gastroenterology since our sales forces specialize in these areas. If we are unable to expand our sales and marketing capability, train our sales force effectively or provide any other capabilities necessary to commercialize our products and product candidates, we will need to contract with third parties to market and sell our products. We must train our employees on proper regulatory compliance, including, but not limited to, “fair balance” promotion of our products and anti-kickback laws. If we are unable to establish and maintain compliant and adequate sales and marketing capabilities, we may not be able to increase our product revenue, may generate increased expenses, may have regulatory compliance issues and may not continue to be profitable.
If governmental or third-party payors do not provide adequate reimbursement for our products, our revenue and prospects for continued profitability may be limited.
Our financial success depends, in part, on the availability of adequate reimbursement from third-party healthcare payors. Such third-party payors include governmental health programs such as Medicare and Medicaid, managed care providers and private health insurers. Third-party payors are increasingly challenging the pricing of medical products and services, while governments continue to propose and pass legislation designed to reduce the cost of healthcare. Adoption of such legislation could further limit reimbursement for pharmaceuticals.
In March 2010, the U.S. government passed into law and enacted the Patient Protection and Affordable Care Act, as amended by the Healthcare and Education Affordability Reconciliation Act, or collectively the Healthcare Reform Act. Among other provisions, the Healthcare Reform Act calls for an increase in certain Medicare drug rebates paid by pharmaceutical manufacturers and an industry fee imposed on pharmaceutical manufacturers according to the individual manufacturer’s relative percentage of total industry sales to specified government programs. At this time no assurances can be given that these measures, or any other measures included in the Healthcare Reform Act, will not have an adverse effect on our revenues in the future. Furthermore, future cost control initiatives, legislation and regulations could decrease the price that we would receive for any products, which would limit our revenue and profitability.
Also, reimbursement practices of third-party payors might preclude us from achieving market acceptance for our products or maintaining price levels sufficient to realize an appropriate return on our investment in product acquisition and development. If we cannot obtain adequate reimbursement levels, our business, financial condition and results of operations would be materially and adversely affected.
Our employees have been trained to submit accurate and correct pricing information to payors. If, despite the training, our employees provide incorrect or fraudulent information, then we will be subject to various administrative and judicial investigations and litigation.
“Formulary” practices of third-party payors could adversely affect our competitive position.
Many managed healthcare organizations are now controlling the pharmaceutical products listed on their formulary lists. Having products listed on these formulary lists creates competition among pharmaceutical companies which, in turn, has created a trend of downward pricing pressure in our industry. In addition, many managed care organizations are pursuing various ways to reduce pharmaceutical costs and are considering formulary contracts primarily with those pharmaceutical companies that can offer a full line of products for a given therapy sector or disease state. Our products might not be included on the formulary lists of managed care organizations, and downward pricing pressure in our industry generally could negatively impact our operations.
21
Table of Contents
Continued consolidation of distributor networks in the pharmaceutical industry as well as increases in retailer concentration may limit our ability to profitably sell our products.
We sell most of our products to large pharmaceutical wholesalers, who in turn sell to, thereby supplying, hospitals and retail pharmacies. The distribution network for pharmaceutical products has become increasingly consolidated in recent years. Further consolidation or financial difficulties could also cause our customers to reduce the amounts of our products that they purchase, which would materially and adversely affect our business, financial condition and results of operations.
Our CET joint initiative may not result in our gaining access to commercially viable products.
Our CET joint initiative with Vanderbilt University and Tennessee Technology Development Corporation is designed to help us investigate, in a cost-effective manner, early-stage products and technologies. However, we may never gain access to commercially viable products from CET for a variety of reasons, including:
| • | CET investigates early-stage products, which have the greatest risk of failure prior to FDA approval and commercialization; |
| • | In some programs, we do not have pre-set rights to product candidates developed by CET. We would need to agree with CET and its collaborators on the terms of any product licensed to, or acquired by, us; |
| • | We rely principally on government grants to fund CET’s research and development programs. If these grants were no longer available, we or our co-owners might be unable or unwilling to fund CET operations at current levels or at all; |
| • | We may become involved in disputes with our co-owners regarding CET policy or operations, such as how best to deploy CET assets or which product opportunities to pursue. Disagreement could disrupt or halt product development; and |
| • | CET may disagree with one of the various universities with which CET is collaborating on research. A disagreement could disrupt or halt product development. |
We depend on our key personnel, the loss of whom would adversely affect our operations. If we fail to attract and retain the talent required for our business, our business will be materially harmed.
We are a relatively small company, and we depend to a great extent on principal members of our management and scientific staff. If we lose the services of any key personnel, in particular, A.J. Kazimi, our Chief Executive Officer, it could have a material adverse effect on our business prospects. Mr. Kazimi, in particular, plays a key role in several operational and strategic decisions such that any loss of his services due to death or disability would adversely effect our day-to-day operations. We currently have a key man life insurance policy covering the life of Mr. Kazimi. We have entered into agreements with each of our employees that contain restrictive covenants relating to non-competition and non-solicitation of our customers and suppliers for one year after termination of employment. Nevertheless, each of our officers and key employees may terminate his or her employment at any time without notice and without cause or good reason, and so as a practical matter these agreements do not guarantee the continued service of these employees. Our success depends on our ability to attract and retain highly qualified scientific, technical and managerial personnel and research partners. Competition among pharmaceutical companies for qualified employees is intense, and we may not be able to retain existing personnel or attract and retain qualified staff in the future. If we experience difficulties in hiring and retaining personnel in key positions, we could suffer from delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect operating results.
The size of our organization and our potential growth may lead to difficulties in managing operations.
As of March 1, 2012, we had 138 full-time employees. We may need to continue to expand our managerial, operational, financial and other resources in order to increase our marketing efforts with regard to our currently marketed products, continue our business development and product development activities and commercialize our product candidates. We have experienced, and may continue to experience, growth and increased expenses in the scope of our operations in connection with the continued marketing and development of our products. Our financial performance will depend, in part, on our ability to manage any such growth and expenses of the current organization effectively.
22
Table of Contents
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product or product candidate and may have to limit its commercialization.
We face an inherent risk of product liability lawsuits related to the testing of our product candidates and the commercial sale of our products. An individual may bring a liability claim against us if one of our product candidates or products causes, or appears to have caused, an injury. If we cannot successfully defend ourselves against the product liability claim, we may incur substantial liabilities. Liability claims may result in:
| • | decreased demand for our products; |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants; |
| • | significant litigation costs; |
| • | substantial monetary awards to or costly settlement with patients; |
| • | product recalls; |
| • | loss of revenue; and |
| • | the inability to commercialize our product candidates. |
We are highly dependent upon medical and patient perceptions of us and the safety and quality of our products. We could be adversely affected if we or our products are subject to negative publicity. We could also be adversely affected if any of our products or any similar products sold by other companies prove to be, or are asserted to be, harmful to patients. Also, because of our dependence upon medical and patient perceptions, any adverse publicity associated with illness or other adverse effects resulting from the use or misuse of our products or any similar products sold by other companies could have a material adverse impact on our results of operations.
We have product liability insurance that covers our clinical trials and the marketing and sale of our products up to a $10 million annual aggregate limit, subject to specified deductibles. Our current or future insurance coverage may prove insufficient to cover any liability claims brought against us.
Because of the increasing costs of insurance coverage, we may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy any liability that may arise.
Regulatory approval for any approved product is limited by the FDA to those specific indications and conditions for which clinical safety and efficacy have been demonstrated.
Any regulatory approval is limited to those specific diseases and indications for which a product is deemed to be safe and effective by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. If we are not able to obtain FDA approval for any desired future indications for our products, our ability to effectively market and sell our products may be reduced and our business may be adversely affected.
While physicians may choose to prescribe drugs for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical studies and approved by the regulatory authorities, our ability to promote the products is limited to those indications that are specifically approved by the FDA. These “off-label” uses are common across medical specialties and may constitute an appropriate treatment for some patients in varied circumstances. Regulatory authorities in the U.S. generally do not regulate the behavior of physicians in their choice of treatments. Regulatory authorities do, however, restrict communications by pharmaceutical companies on the subject of off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to suspend or withdraw an approved product from the market, require a recall or institute fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our business.
23
Table of Contents
Our business and operations would suffer in the event of system failures or adverse events at our corporate headquarters.
Despite the implementation of security measures, our internal computer systems, including those at our corporate headquarters, are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. In the event that our corporate headquarters and/or our computer systems are disabled or materially damaged, it would have a substantial and material negative effect on our operations. Furthermore, any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our drug development programs. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability and the further development of our products or product candidates may be delayed.
RISKS RELATING TO GOVERNMENT REGULATION
We are subject to stringent government regulation. All of our products face regulatory challenges.
Virtually all aspects of our business activities are regulated by government agencies. The manufacturing, processing, formulation, packaging, labeling, distribution, promotion and sampling, advertising of our products, and disposal of waste products arising from such activities are subject to governmental regulation. These activities are regulated by one or more of the FDA, the Federal Trade Commission, or the FTC, the Consumer Product Safety Commission, the U.S. Department of Agriculture and the U.S. Environmental Protection Agency, or the EPA, as well as by comparable agencies in foreign countries. These activities are also regulated by various agencies of the states and localities in which our products are sold. For more information, see “Business—Government Regulation.”
Like all pharmaceutical manufacturers, we are subject to regulation by the FDA under the authority of the Federal Food, Drug and Cosmetic Act, or the FDC Act. All “new drugs” must be the subject of an FDA-approved new drug application, or NDA, before they may be marketed in the United States. The FDA has the authority to withdraw existing NDA approvals and to review the regulatory status of products marketed under the enforcement policy. The FDA may require an approved NDA for any drug product marketed under the enforcement policy if new information reveals questions about the drug’s safety and effectiveness. All drugs must be manufactured in conformity with cGMP, and drug products subject to an approved NDA must be manufactured, processed, packaged, held and labeled in accordance with information contained in the NDA. Since we rely on third parties to manufacture our products, cGMP requirements directly affect our third party manufacturers and indirectly affect us. The manufacturing facilities of our third-party manufacturers are continually subject to inspection by such governmental agencies, and manufacturing operations could be interrupted or halted in any such facilities if such inspections prove unsatisfactory. Our third-party manufacturers are subject to periodic inspection by the FDA to assure such compliance.
Pharmaceutical products must be distributed, sampled and promoted in accordance with FDA requirements. We must train our employees on proper regulatory compliance, including, but not limited to, “fair balance” promotion of our products and anti-kickback laws. The FDA also regulates the advertising of prescription drugs. The FDA has the authority to request post-approval commitments that can be time-consuming and expensive.
Under the FDC Act, the federal government has extensive enforcement powers over the activities of pharmaceutical manufacturers to ensure compliance with FDA regulations. Those powers include, but are not limited to, the authority to initiate court action to seize unapproved or non-complying products, to enjoin non-complying activities, to halt manufacturing operations that are not in compliance with cGMP, and to seek civil monetary and criminal penalties. The initiation of any of these enforcement activities, including the restriction or prohibition on sales of our products, could materially adversely affect our business, financial condition and results of operations.
Any change in the FDA’s enforcement policy could have a material adverse effect on our business, financial condition and results of operations.
24
Table of Contents
We cannot determine what effect changes in regulations or statutes or legal interpretation, when and if promulgated or enacted, may have on our business in the future. Such changes, or new legislation, could have a material adverse effect on our business, financial condition and results of operations.
Proposed legislation may permit re-importation of drugs from other countries into the U.S., including foreign countries where the drugs are sold at lower prices than in the U.S., which could materially adversely affect our operating results and our overall financial condition.
Legislation has been introduced in Congress that, if enacted, would permit more widespread re-importation of drugs from foreign countries into the U.S., which may include re-importation from foreign countries where the drugs are sold at lower prices than in the U.S. Such legislation, or similar regulatory changes, could decrease the price we receive for any approved products which, in turn, could materially adversely affect our operating results and our overall financial condition.
RISKS RELATING TO INTELLECTUAL PROPERTY
Our strategy to secure and extend marketing exclusivity or patent rights may provide only limited protection from competition.
We seek to secure and extend marketing exclusivity for our products through a variety of means, including FDA exclusivity and patent rights. Acetadote is indicated to prevent or lessen hepatic (liver) injury when administered intravenously within eight to ten hours after ingesting quantities of acetaminophen that are potentially toxic to the liver. Although a patent application and other applications relating to the uses of Acetadote are in prosecution, they have not yet been issued. However, on February 8, 2012, we received a Notice of Allowance from the United States Patent and Trademark Office, or the USPTO, relating to our new formulation of Acetadote. Once issued, this composition of matter patent will expire in 2025. Barriers to competitive market entry include the time and cost associated with the development, regulatory approval and manufacturing of a similar formulation.
We do not have “composition of matter” or “use” patents for our marketed products. As mentioned above, we have received a Notice of Allowance for the Acetadote patent that has not yet been issued. We do have a U.S. patent, No. 6,727,286 for Caldolor, and some related international patents, which are directed to ibuprofen solution formulations, methods of making the same, and methods of using the same, and which are related to our formulation and manufacture of Caldolor. Additionally, the active ingredient in Caldolor—ibuprofen—is in the public domain, and if a competitor were to develop a sufficiently distinct formulation, it could develop and seek FDA approval for another ibuprofen product that competes with Caldolor. Upon receipt of FDA approval in June 2009, we received three years of marketing exclusivity for Caldolor. Upon the expiration of our marketing exclusivity, a competitor with a generic form of injectable ibuprofen could enter the market.
In November 2011, we acquired the rights to the trademark and abbreviated new drug application, or ANDA, related to the manufacture of Kristalose. This patent is not directed to the composition or use of Kristalose and does not prevent a competitor from developing a formulation and developing and seeking FDA approval for a product that competes with Kristalose.
While we consider patent protection when evaluating product acquisition opportunities, any products we acquire in the future may not have significant patent protection. Neither the USPTO nor the courts have a consistent policy regarding the breadth of claims allowed or the degree of protection afforded under many pharmaceutical patents. Patent applications in the U.S. and many foreign jurisdictions are typically not published until 18 months following the filing date of the first related application, and in some cases not at all. In addition, publication of discoveries in scientific literature often lags significantly behind actual discoveries. Therefore, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in these patent applications. In addition, changes in either patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property or narrow the scope of our patent protection. Furthermore, our competitors may independently develop similar technologies or duplicate technology developed by us in a manner that does not infringe our patents or other intellectual property. As a result of these factors, our patent rights may not provide any commercially valuable protection from competing products.
25
Table of Contents
If we are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patents, we rely upon trade secrets, unpatented proprietary know-how and continuing technological innovation where we do not believe patent protection is appropriate or attainable. For example, the manufacturing process for Kristalose involves substantial trade secrets and proprietary know-how. We have entered into confidentiality agreements with certain key employees and consultants pursuant to which such employees and consultants must assign to us any inventions relating to our business if made by them while they are our employees, as well as certain confidentiality agreements relating to the acquisition of rights to products. Confidentiality agreements can be breached, though, and we might not have adequate remedies for any breach. Also, others could acquire or independently develop similar technology.
We depend on our licensors for the maintenance and enforcement of our intellectual property and have limited, if any, control over the amount or timing of resources that our licensors devote on our behalf.
When we license products, we often depend on our licensors to protect the proprietary rights covering those products. We have limited, if any, control over the amount or timing of resources that our licensors devote on our behalf or the priority they place on maintaining patent or other rights and prosecuting patent applications to our advantage. While any such licensor is expected to be under contractual obligations to us to diligently prosecute its patent applications and allow us the opportunity to consult, review and comment on patent office communications, we cannot be sure that it will perform as required. If a licensor does not perform and if we do not assume the maintenance of the licensed patents in sufficient time to make required payments or filings with the appropriate governmental agencies, we risk losing the benefit of all or some of those patent rights.
If the use of our technology conflicts with the intellectual property rights of third parties, we may incur substantial liabilities, and we may be unable to commercialize products based on this technology in a profitable manner or at all.
If our products conflict with the intellectual property rights of others, they could bring legal action against us or our licensors, licensees, manufacturers, customers or collaborators. If we were found to be infringing a patent or other intellectual property rights held by a third party, we could be forced to seek a license to use the patented or otherwise protected technology. We might not be able to obtain such a license on terms acceptable to us or at all. If an infringement or misappropriation legal action were to be brought against us or our licensors, we would incur substantial costs in defending the action. If such a dispute were to be resolved against us, we could be subject to significant damages, and the manufacturing or sale of one or more of our products could be enjoined.
We may be involved in lawsuits to protect or enforce our patents or the patents of our collaborators or licensors, which could be expensive and time consuming.
Competitors may infringe our patents or the patents of our collaborators or licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Interference proceedings brought by the USPTO may be necessary to determine the priority of inventions with respect to our patent applications or those of our collaborators or licensors. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management. We may not be able, alone or with our collaborators and licensors, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, some of our confidential information could be disclosed during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
26
Table of Contents
If we breach any of the agreements under which we license rights to our products and product candidates from others, we could lose the ability to continue commercialization of our products and development and commercialization of our product candidates.
We have exclusive licenses for the marketing and sale of certain products and may acquire additional licenses. Such licenses may terminate prior to expiration if we breach our obligations under the license agreement related to these pharmaceutical products. For example, the licenses may terminate if we fail to meet specified quality control standards, including cGMP with respect to the products, or commit a material breach of other terms and conditions of the licenses. Such early termination could have a material adverse effect on our business, financial condition and results of operations.
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
RISKS RELATED TO OUR FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Our operating results are likely to fluctuate from period to period.
We are a relatively new company seeking to capture significant growth. While our revenues and operating income have increased over time, we anticipate that there may be fluctuations in our future operating results. We may not be able to maintain or improve our current levels of revenue or income. Potential causes of future fluctuations in our operating results may include:
| • | new product launches, which could increase revenues but also increase sales and marketing expenses; |
| • | acquisition activity and other charges (such as for inventory expiration); |
| • | increases in research and development expenses resulting from the acquisition of a product candidate that requires significant additional development; |
| • | changes in the competitive, regulatory or reimbursement environment, which could drive down revenues or drive up sales and marketing or compliance costs; and |
| • | unexpected product liability or intellectual property claims and lawsuits. |
See also “Management’s discussion and analysis of financial condition and results of operations—Liquidity and capital resources.” Fluctuation in operating results, particularly if not anticipated by investors and other members of the financial community, could add to volatility in our stock price.
Our focus on acquisitions as a growth strategy has created a large amount of intangible assets whose amortization could negatively affect our results of operations.
Our total assets include intangible assets related to our acquisitions. As of December 31, 2011, intangible assets relating to product and data acquisitions represented approximately 7% of our total assets. We may never realize the value of these assets. Generally accepted accounting principles require that we evaluate on a regular basis whether events and circumstances have occurred that indicate that all or a portion of the carrying amount of the asset may no longer be recoverable, in which case we would write down the value of the asset and take a corresponding charge to earnings. Any determination requiring the write-off of a significant portion of unamortized intangible assets would adversely affect our results of operations.
27
Table of Contents
We may need additional funding and may be unable to raise capital when needed, which could force us to delay, reduce or eliminate our product development or commercialization and marketing efforts.
We may need to raise additional funds in order to meet the capital requirements of running our business and acquiring and developing new pharmaceutical products. If we require additional funding, we may seek to sell common stock or other equity or equity-linked securities, which could result in dilution to our shareholders. We may also seek to raise capital through a debt financing, which would result in ongoing debt-service payments and increased interest expense. Any financings would also likely involve operational and financial restrictions being imposed on us. We might also seek to sell assets or rights in one or more commercial products or product development programs. Additional capital might not be available to us when we need it on acceptable terms or at all. In addition, the recent downgrade of the U.S. credit rating and the ongoing European debt crisis have contributed to the instability in global credit markets. We are unable to predict the impact of these events, and if economic conditions deteriorate, our business, results of operations and ability to raise needed capital could be materially and adversely affected. If we are unable to raise additional capital when needed due to the reasons listed above and lack of creditworthiness, bank failures, or price decline in market investments, we could be forced to scale back our operations to conserve cash.
If we are unable to establish appropriate internal financial reporting controls and procedures, it could cause us to fail to meet our reporting obligations, result in the restatement of our financial statements, harm our operating results, subject us to regulatory scrutiny and sanction, cause investors to lose confidence in our reported financial information and have a negative effect on the market price for shares of our common stock.
Effective internal controls are necessary for us to provide reliable financial reports and mitigate the risk of fraud. We maintain a system of internal control over financial reporting, which is defined as a process designed by, or under the supervision of, our principal executive officer and principal financial officer, or persons performing similar functions, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the U.S., or GAAP.
We cannot assure you that we will not, in the future, identify areas requiring improvement in our internal control over financial reporting. We cannot assure you that the measures we will take to remediate any areas in need of improvement will be successful or that we will implement and maintain adequate controls over our financial processes and reporting in the future as we continue our growth. If we are unable to establish appropriate internal financial reporting controls and procedures, it could cause us to fail to meet our reporting obligations, result in the restatement of our financial statements, harm our operating results, subject us to regulatory scrutiny and sanction, cause investors to lose confidence in our reported financial information and have a negative effect on the market price for shares of our common stock.
In addition, we maintain a system of internal controls and provide training to employees designed to provide reasonable assurance that unlawful and fraudulent activity, including misappropriation of assets, fraudulent financial reporting, and unauthorized access to sensitive or confidential data is either prevented or timely detected. However in the event that our employees engage in such fraudulent behavior, we could suffer material adverse consequences.
Changes in, or interpretations of, accounting principles and tax laws could have a significant impact on our financial position and results of operations.
We prepare our consolidated financial statements in accordance with GAAP. These principles are subject to interpretation by the SEC and various bodies formed to interpret and create appropriate accounting principles. A change in these principles can have a significant effect on our reported results and may even retroactively affect previously reported transactions.
For example, the U.S.-based Financial Accounting Standards Board, or FASB, is currently working together with the International Accounting Standards Board, or IASB, on several projects to further align accounting principles and facilitate more comparable financial reporting between companies who are required to follow GAAP under SEC regulations and those who are required to follow International Financial Reporting Standards, or IFRS, outside of the U.S. These efforts by the FASB and IASB may result in different accounting principles under GAAP that may result in materially different financial results for us in areas including, but not limited to principles for recognizing revenue and lease accounting.
28
Table of Contents
RISKS RELATED TO OWNING OUR STOCK
The market price of our common stock may fluctuate substantially.
The price for the shares of our common stock sold in our initial public offering was determined by negotiation between the representatives of the underwriters and us. This price may not have reflected the market price of our common stock following our initial public offering. Through March 1, 2012, the closing price of our common stock since our initial public offering has ranged from a low of $4.70 to a high of $17.05 per share. Moreover, the market price of our common stock might decline below current levels. In addition, the market price of our common stock is likely to be highly volatile and may fluctuate substantially. Sales of a substantial number of shares of our common stock in the public market or the perception that these sales may occur could cause the market price of our common stock to decline.
The realization of any of the risks described in these “Risk Factors” could have a dramatic and material adverse impact on the market price of our common stock. In addition, securities class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Any such securities litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could negatively impact our business, operating results and financial condition. Sales of a substantial number of shares of our common stock in the public market or the perception that these sales may occur could cause the market price of our common stock to decline.
Unstable market conditions may have serious adverse consequences on our business.
The economic downturn and market instability has made the business climate more volatile and more costly. Our general business strategy may be adversely affected by unpredictable and unstable market conditions. While we believe we have adequate capital resources to meet current working capital and capital expenditure requirements, a radical economic downturn or increase in our expenses could require additional financing on less than attractive rates or on terms that are dilutive to existing shareholders. Failure to secure any necessary financing in a timely manner and on favorable terms could have a material adverse effect on our growth strategy, financial performance and stock price and could require us to delay or abandon clinical developments plans. There is a risk that one or more of our current service providers, manufacturers and other partners may encounter difficulties during challenging economic times, which would directly affect our ability to attain our operating goals on schedule and on budget.
We are experiencing increased costs and regulatory risk as a result of operating as a public company, and our management will be required to devote additional time to new compliance initiatives.
We have and will continue to incur increased costs as a result of operating as a public company, and our management is required to devote additional time to new compliance initiatives. As a public company, we have and will continue to incur legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002, or Sarbanes-Oxley Act, Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010, and other rules and regulations subsequently implemented by the SEC and Nasdaq, have imposed various requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. These rules and regulations have and will continue to increase our legal and financial compliance costs and will render some activities more time-consuming and costly. Despite the internal controls and procedure put in place to maintain compliance with securities laws and regulations, our employees may still fail to comply with all SEC disclosure and reporting requirements. Such failure could lead to administrative and civil penalties, criminal penalties, and private litigation with shareholders. The consequences could have a significant material effect on our ability to operate and market out products.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. In particular, we must perform system and process evaluation and testing of our internal controls over financial reporting to allow management and our independent registered public accounting firm to report on the effectiveness of our internal controls over financial reporting. Our testing, or the subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses.
29
Table of Contents
Our compliance with Section 404 requires that we incur substantial accounting expense and expend significant management efforts. Moreover, if we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses, the market price of our stock could decline and we could be subject to sanctions or investigations by Nasdaq, the SEC or other regulatory authorities, which would require additional financial and management resources.
We must comply with the Foreign Corrupt Practices Act.
We are required to comply with the United States Foreign Corrupt Practices Act, which prohibits U.S. companies from engaging in bribery or other prohibited payments to foreign officials for the purpose of obtaining or retaining business. Foreign companies, including some of our competitors, are not subject to these prohibitions. If our competitors engage in these practices, they may receive preferential treatment from personnel of some companies, giving our competitors an advantage in securing business or from government officials who might give them priority in obtaining new licenses, which would put us at a disadvantage. We have established formal policies or procedures for prohibiting or monitoring this conduct, but we cannot assure you that our employees or other agents will not engage in such conduct for which we might be held responsible. If our employees or other agents are found to have engaged in such practices, we could suffer severe penalties.
Some provisions of our third amended and restated charter, bylaws, credit facility and Tennessee law may inhibit potential acquisition bids that you may consider favorable.
Our corporate documents contain provisions that may enable our board of directors to resist a change in control of our company even if a change in control were to be considered favorable by you and other shareholders. These provisions include:
| • | the authorization of undesignated preferred stock, the terms of which may be established and shares of which may be issued without shareholder approval; |
| • | advance notice procedures required for shareholders to nominate candidates for election as directors or to bring matters before an annual meeting of shareholders; |
| • | limitations on persons authorized to call a special meeting of shareholders; |
| • | a staggered board of directors; |
| • | a restriction prohibiting shareholders from removing directors without cause; |
| • | a requirement that vacancies in directorships are to be filled by a majority of the directors then in office and the number of directors is to be fixed by the board of directors; and |
| • | no cumulative voting. |
These and other provisions contained in our third amended and restated charter and bylaws could delay or discourage transactions involving an actual or potential change in control of us or our management, including transactions in which our shareholders might otherwise receive a premium for their shares over then current prices, and may limit the ability of shareholders to remove our current management or approve transactions that our shareholders may deem to be in their best interests and, therefore, could adversely affect the price of our common stock.
Under our bank credit agreement, it is an event of default if any person or entity obtains ownership or control, in one or a series of transactions, of more than 30% of our common stock or 30% of the voting power entitled to vote in the election of members of our board of directors.
In addition, we are subject to control share acquisitions provisions and affiliated transaction provisions of the Tennessee Business Corporation Act, the applications of which may have the effect of delaying or preventing a merger, takeover or other change in control of us and therefore could discourage attempts to acquire our company.
30
Table of Contents
We have never paid cash dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have never paid cash dividends on our capital stock. We do not anticipate paying cash dividends to our shareholders in the foreseeable future. The availability of funds for distributions to shareholders will depend substantially on our earnings. Even if we become able to pay dividends in the future, we expect that we would retain such earnings to enhance capital and/or reduce long-term debt.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements in this Annual Report on Form 10-K that are not historical factual statements are “forward-looking statements.” Forward-looking statements include, among other things, statements regarding our intent, belief or expectations, and can be identified by the use of terminology such as “may,” “will,” “expect,” “believe,” “intend,” “plan,” “estimate,” “should,” “seek,” “anticipate” and other comparable terms or the negative thereof. In addition, we, through our senior management, from time to time make forward-looking oral and written public statements concerning our expected future operations and other developments. While forward-looking statements reflect our good-faith beliefs and best judgment based upon current information, they are not guarantees of future performance and are subject to known and unknown risks and uncertainties, including those mentioned in Item 1A, “Risk Factors,” Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere in this Form 10-K. Actual results may differ materially from the expectations contained in the forward-looking statements as a result of various factors. Such factors include, without limitation:
| • | legislative, regulatory or other changes in the healthcare industry at the local, state or federal level which increase the costs of, or otherwise affect our operations; |
| • | changes in reimbursement available to us by government or private payers, including changes in Medicare and Medicaid payment levels and availability of third-party insurance coverage; |
| • | competition; and |
| • | changes in national or regional economic conditions, including changes in interest rates and availability and cost of capital to us. |
Item 1B: Unresolved Staff Comments
None
Item 2: Properties
As of December 31, 2011, we leased approximately 25,500 square feet of office space in Nashville, Tennessee for our corporate headquarters. The lease expires in October 2016. Of the 25,500 square feet of leased office space, we have subleased to others approximately 9,900 square feet. We believe these facilities are adequate to meet our current needs for office space. We currently do not plan to purchase or lease facilities for manufacturing, packaging or warehousing, as such services are provided to us by third-party contract groups.
Under an agreement expiring in July 2016, CET leases approximately 6,900 square feet of office and wet laboratory space in Nashville, Tennessee. CET uses this space to operate the CET Life Sciences Center for product development work to be carried out in collaboration with universities, research institutions and entrepreneurs. The CET Life Sciences Center provides laboratory and office space, equipment and infrastructure to early-stage life sciences companies and university spin-outs.
We are not currently engaged in any legal proceedings.
31
Table of Contents
| Item 5: | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock, no par value, has been traded on the Nasdaq Global Select Market since August 11, 2009 under the symbol “CPIX.” Prior to that time, there was no public market for our common stock. As of March 1, 2012, there were 118 shareholders of record, which excludes shareholders whose shares are held in nominee or street name by brokers. The closing price of our common stock on the Nasdaq Global Select Market on March 1, 2012 was $7.71 per share. The following table sets forth the high and low trading sales prices for our common stock as reported on the Nasdaq Global Select Market for the full quarterly periods since the completion of our initial public offering and through December 31, 2011:
| High | Low | |||||||
| Fiscal year ended December 31, 2010: |
||||||||
| First quarter |
$ | 14.52 | $ | 10.26 | ||||
| Second quarter |
11.11 | 6.16 | ||||||
| Third quarter |
6.90 | 4.70 | ||||||
| Fourth quarter |
8.18 | 5.63 | ||||||
| Fiscal year ended December 31, 2011: |
||||||||
| First quarter |
7.49 | 4.90 | ||||||
| Second quarter |
5.98 | 4.80 | ||||||
| Third quarter |
6.63 | 5.00 | ||||||
| Fourth quarter |
6.31 | 5.22 | ||||||
Dividend Policy
We have not declared or paid any cash dividends on our common stock nor do we anticipate paying dividends for the foreseeable future. We currently intend to retain any future earnings for use in the operation of our business and to fund future growth. The payment of dividends by us on our common stock is limited by our loan agreement. Any future decision to declare or pay dividends will be at the sole discretion of our Board of Directors.
Performance Graph
The following stock performance graph illustrates a comparison of the total cumulative stockholder return on our common stock since August 10, 2009, which is the date of our initial public offering on the Nasdaq Global Select Market, to the Nasdaq Composite and Nasdaq Pharmaceutical Stocks. The graph assumes an initial investment of $100 on August 10, 2009, and that all dividends were reinvested.
32
Table of Contents
Comparison of Cumulative Total Return
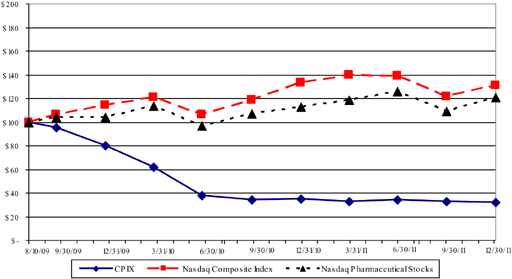
Purchases of Equity Securities
On May 13, 2010, we announced a share repurchase program to purchase up to $10 million of our common stock pursuant to Rule 10b-18 of the Securities Act. In January 2011, our Board of Directors modified the existing repurchase program to provide for the repurchase of $10 million of our outstanding common stock, in addition to the amount repurchased in 2010. The following table summarizes the activity, by month, during the fourth quarter of 2011:
| Period |
Total Number of Shares (or Units) Purchased |
Average Price Paid per Share (or Unit) |
Total Number of Shares (or Units) Purchased as Part of Publicly Announced Plans or Programs |
Maximum Number (or Approximate Dollar Value) of Shares (or Units) that May Yet Be Purchased Under the Plans or Programs |
||||||||||||
| October 1 – October 31 |
46,371 | $ | 5.66 | 46,371 | $ | 7,611,749 | ||||||||||
| November 1 – November 30 |
135,823 | (1) | 5.82 | 43,109 | 7,356,746 | |||||||||||
| December 1 – December 31 |
80,048 | 5.85 | 80,048 | 6,888,368 | ||||||||||||
|
|
|
|||||||||||||||
| Total |
262,242 | |||||||||||||||
|
|
|
|||||||||||||||
| (1) | Includes shares tendered by option holders upon exercise of stock options and other private purchases at the then-current fair market value of common stock. |
33
Table of Contents
Item 6: Selected Financial Data
The selected consolidated financial data set forth below should be read in conjunction with the audited consolidated financial statements and related notes and Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and other financial information appearing elsewhere in this Form 10-K. The historical results are not necessarily indicative of the results to be expected for any future periods.
| Years Ended December 31, | ||||||||||||||||||||
| Statement of income data: |
2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Net revenues |
$ | 51,143 | $ | 45,876 | $ | 43,537 | $ | 35,075 | $ | 28,064 | ||||||||||
| Costs and expenses |
41,293 | 39,375 | 37,761 | 27,793 | 21,338 | |||||||||||||||
| Operating income |
9,849 | 6,502 | 5,777 | 7,282 | 6,725 | |||||||||||||||
| Net income attributable to common shareholders |
5,658 | 2,457 | 3,091 | 4,766 | 4,044 | |||||||||||||||
| Earnings per share – basic |
$ | 0.28 | $ | 0.12 | $ | 0.22 | $ | 0.47 | $ | 0.40 | ||||||||||
| Earnings per share – diluted |
$ | 0.28 | $ | 0.12 | $ | 0.17 | $ | 0.29 | $ | 0.24 | ||||||||||
| As of December 31, | ||||||||||||||||||||
| Balance sheet data: |
2011 | 2010 | 2009 | 2008 | 2007 | |||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Cash and cash equivalents |
$ | 70,599 | $ | 65,894 | $ | 78,702 | $ | 11,830 | $ | 10,815 | ||||||||||
| Working capital |
80,708 | 71,811 | 74,549 | 10,104 | 6,669 | |||||||||||||||
| Total assets |
95,518 | 92,054 | 103,724 | 31,119 | 28,919 | |||||||||||||||
| Total long-term debt and other long-term obligations (including current portion) |
5,485 | 7,802 | 20,155 | 7,666 | 7,623 | |||||||||||||||
| Convertible preferred stock |
— | — | — | 2,604 | 2,743 | |||||||||||||||
| Retained earnings (accumulated deficit) |
12,657 | 6,999 | 4,542 | 1,451 | (3,316 | ) | ||||||||||||||
| Total equity |
82,835 | 77,715 | 72,221 | 17,555 | 16,746 | |||||||||||||||
Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial position and results of operations should be read together with our audited consolidated financial statements and related notes appearing elsewhere in this Form 10-K. This discussion and analysis may contain forward-looking statements that involve risks and uncertainties – please refer to the section entitled, “Special Note Regarding Forward-Looking Statements,” Contained in Part I, Item 1A, “Risk Factors,” of this Form 10-K. You should review the “Risk Factors” section of this Form 10-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements described in the following discussion and analysis.
EXECUTIVE SUMMARY
We are a profitable and growing specialty pharmaceutical company focused on the acquisition, development and commercialization of branded prescription products. Our primary target markets are hospital acute care and gastroenterology, which are characterized by concentrated physician bases that we believe can be penetrated effectively by relatively small, targeted sales forces. We are dedicated to providing innovative products that improve quality of care for patients.
We operate in a single operating segment of specialty pharmaceuticals products. Our marketed product portfolio includes Acetadote® (acetylcysteine) Injection for the treatment of acetaminophen poisoning, Caldolor® (ibuprofen) Injection, the first injectable treatment for pain and fever approved in the United States, and Kristalose® (lactulose) for Oral Solution, a prescription laxative. In early 2011, we acquired rights to a late-stage product candidate that we intend to develop under the brand name Hepatoren™ (ifetroban) Injection for the treatment of hepatorenal syndrome. We market and sell our approved products through our hospital and field sales forces in the United States and are working with partners to reach international markets.
34
Table of Contents
The following is a summary of our 2011 highlights and recent developments. For more information, please see Part I, Item I, Business, of this Form 10-K.
| • | The FDA approved our sNDA for our new formulation of Acetadote, which was the result of a Phase IV commitment we made to the FDA upon receipt of initial marketing approval of the product. |
| • | We filed a response with the USPTO for a patent to protect our proprietary discoveries related to the new Acetadote formulation. This formulation patent was allowed and issued in China in April 2011. We also recently filed a second U.S. patent application related to the safety profile of the new formulation. In February 2012, we received a Notice of Allowance for the composition of matter related to our new formulation of Acetadote. Once issued, the patent will expire in August 2025. |
| • | We implemented a pull-through sales strategy for Caldolor, with an emphasis on activities required to build volume and use in centers that have already stocked the product. |
| • | We acquired the rights to Hepatoren, an injectable form of ifetroban for the treatment of HRS. |
| • | We continued our efforts in our clinical studies, with four studies designed to support Caldolor, one study for an additional indication for Kristalose and one Phase II dose escalation clinical study to evaluate Hepatoren for the treatment of HRS. |
| • | We acquired the rights to certain assets of Kristalose, including the Kristalose trademark and FDA registration. The acquisition allows us better control of the supply chain, and allows us to pursue additional indications to increase the marketability of Kristalose. |
| • | At CET, we continued to expand our collaboration partners by entering into an agreement with Washington University in St. Louis to co-develop promising biomedical technologies. We continue to collaborate with Vanderbilt University, the University of Tennessee and the University of Mississippi. |
| • | We continued to expand our international presence by entering into marketing agreements with Harvest & Health Co, Ltd in Taiwan and Insanbakti in Malaysia to market Caldolor and Acetadote. In addition, Al-Nabil International became our commercial partner of Caldolor and Acetadote in the U.A.E.in late 2011. |
| • | In addition to the above highlights, we added key individuals to our management team. In February 2011, Tan Cheow Choon was appointed Director of International Business. Based in Singapore, Choon is responsible for executing our initiative to expand our product markets across Asia. In October 2011, we named Rick S. Greene as Vice President and Chief Financial Officer . He had previously been serving as the interim Vice President of Finance and Accounting since April 2011. |
In 2011, we evaluated our financial position and elected to pay in full our outstanding term debt balance, resulting in the elimination of interest costs associated with this debt. We also modified our line of credit facility with Bank of America to provide for up to $10 million of available funds, with an option to increase it to $20 million upon the satisfaction of certain conditions. We reduced the cost of the loan, extended the maturity date to December 2014 and modified certain financial and restrictive covenants more favorable to us.
We continue to repurchase shares under a Rule 10(b)-5 plan that was authorized by our Board of Directors in May 2010 and modified in January 2011. The plan provides for the repurchase of up to $10 million, plus the amount repurchased in 2010, of our outstanding stock from time-to-time in the open market. The timing and amount of purchases are determined by us based on evaluation of market conditions, stock price and other factors.
CRITICAL ACCOUNTING POLICIES AND SIGNIFICANT JUDGMENTS AND ESTIMATES
Accounting Estimates and Judgments
The preparation of the consolidated financial statements in conformity with GAAP requires management to make estimates, judgments and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the period. We base our estimates on past experience and on other factors we deem reasonable given the circumstances. Past results help form the basis of our judgments about the carrying value of assets and liabilities that are not determined from other sources. Actual results could differ from these estimates. These estimates, judgments and assumptions are most critical with respect to our accounting for revenue recognition, inventory, provision for income taxes, stock-based compensation, research and development accounting, and intangible assets.
35
Table of Contents
Revenue Recognition
We recognize revenue in accordance with the SEC’s Staff Accounting Bulletin (SAB) No. 101, Revenue Recognition in Financial Statements, as amended by SAB No. 104 (together, SAB 101), and Topic 605-15 of the Accounting Standards Codification.
Our revenue is derived primarily from the product sales of Acetadote, Caldolor and Kristalose. Revenue is recognized when persuasive evidence of an arrangement exists, delivery has occurred, the fee is fixed and determinable and collectability is probable. Delivery is considered to have occurred upon either shipment of the product or arrival at its destination based on the shipping terms of the transaction. When these conditions are satisfied, we recognize gross product revenue, which is the price we charge generally to our wholesalers for a particular product. Other income, which is a component of net revenues, includes rental and grant income. Other income was less than one percent of net revenues in 2011 and 2009, and less than three percent in 2010.
Our net product revenue reflects the reduction of gross product revenue at the time of initial sales recognition for estimated accounts receivable allowances for chargebacks, discounts and damaged product as well as provisions for sales related accruals of rebates, product returns and administrative fees and fee for services. Our financial statements reflect accounts receivable allowances of $0.2 million at December 31, 2011 and 2010, and $0.1 million at December 31, 2009, respectively, for chargebacks, discounts and allowances for product damaged in shipment.
The following table reflects our sales-related accrual activity for the periods indicated below:
| 2011 | 2010 | 2009 | ||||||||||
| Balance at January 1 |
$ | 2,626,313 | $ | 1,863,012 | $ | 1,040,203 | ||||||
| Current Provision |
4,719,231 | 4,933,553 | 3,436,208 | |||||||||
| Current Provision for Prior Period Sales |
380,235 | 306,706 | 75,589 | |||||||||
| Actual Returns/Credits |
(4,509,157 | ) | (4,476,958 | ) | (2,688,988 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Balance at December 31 |
$ | 3,216,622 | $ | 2,626,313 | $ | 1,863,012 | ||||||
|
|
|
|
|
|
|
|||||||
The allowances for chargebacks, discounts, and damaged products and sales related accruals for rebates and product returns are determined on a product-by-product basis and are established by management as our best estimate at the time of sale based on each product’s historical experience, adjusted to reflect known changes in the factors that impact such allowances and accruals. Additionally, these allowances and accruals are established based on the following:
| • | the contractual terms with customers; |
| • | analysis of historical levels of discounts, returns, chargebacks and rebates; |
| • | communications with customers; |
| • | purchased information about the rate of prescriptions being written and the level of inventory remaining in the distribution channel, if known; and |
| • | expectations about the market for each product, including any anticipated introduction of competitive products. |
The allowances for chargebacks and accruals for rebates and product returns are the most significant estimates used in the recognition of our revenue from product sales. Of the accounts receivable allowances and our sales related accruals, our accrual for fee for services and product returns represents the majority of the balance. Sales related accrued liabilities for rebates, product returns, service fees, and administrative fees totaled $3.2 million, $2.6 million and $1.9 million as of December 31, 2011, 2010 and 2009, respectively. Of these amounts, our estimated liability for fee for services represented $1.0 million, $0.8 million and $0.7 million, respectively, while our accrual for product returns totaled $1.8 million, $1.4 million and $1.0 million, respectively. If the actual amount of cash discounts, chargebacks, rebates, and product returns differs from the amounts estimated by management, material differences may result from the amount of our revenue recognized from product sales. A change in our rebate estimate of one percentage point would have impacted net sales by approximately $0.1 million in each of
36
Table of Contents
the three years ended December 31, 2011. A change in our product return estimate of one percentage point would have impacted net sales by $0.6 million for the year ended December 31, 2011 and $0.5 million for the years ended December 31, 2010 and 2009, respectively. Any expired product return would be from a prior period, given the shelf-life of the products.
As a general rule, we do not allow customers to purchase additional product prior to a scheduled price increase. We occasionally make an exception to this policy when we offer odd-lot quantities at a slightly reduced price or when a customer opens a new facility and requests special terms on its initial purchase. To date, we believe these types of transactions have not been material. Moreover, when we offer special terms, we review the transaction against our revenue recognition policy for proper treatment. If we determine such transactions have become material, we will disclose the impact in the notes to our financial statements.
While we do not have regular access to our customers’ inventory levels, we review each order from all of our customers. To the extent that an order reflects more than a normal purchasing pattern, management discusses the order with the customer prior to agreeing to process the order.
Inventories
We provide valuation reserves for estimated obsolescence or unmarketable inventory in an amount equal to the difference between the cost of inventory and the estimated market value based upon assumptions about remaining shelf life, future demand and market conditions. The reserve for estimated inventory obsolescence was calculated based upon specific review of the inventory expiration dates and the quantity on-hand at December 31, 2011 in comparison to our expected inventory usage. The amount of actual inventory obsolescence and unmarketable inventory could differ (either higher or lower) in the near term from the amounts accrued. Changes in our estimates would be recorded in the income statement in the period of the change.
Income Taxes
We provide for deferred taxes using the asset and liability approach. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to operating loss and tax credit carry-forwards and differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Our principal differences are related to the timing of deductibility of certain items such as depreciation, amortization and expense for options issued to nonemployees. Deferred tax assets and liabilities are measured using management’s estimate of tax rates expected to apply to taxable income in the years in which management believes those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in our results of operations in the period that includes the enactment date.
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment.
The tax benefit associated with the exercise of nonqualified stock options is recognized when the benefit is used to offset income taxes payable. As of December 31, 2011, we have unrecognized federal net operating loss carryforwards associated with the exercise of nonqualified options of $57.6 million.
Stock-Based Compensation
We recognize compensation expense for all share-based payments based on the fair value of the award on the date of grant. In addition, incremental compensation expense is recognized upon the modification, cancellation or repurchase of equity awards. The fair value of stock options and warrants are calculated using the Black-Scholes option-pricing model on the date of grant. We estimate volatility in accordance with SAB No. 107, as amended by SAB No. 110. As there was no public market for our common stock prior to our initial public offering and, therefore, a lack of company-specific historical or implied volatility data, we have determined the share-price volatility based on an analysis of certain publicly-traded companies that we consider to be our peers. The comparable peer companies used for our estimated volatility are publicly-traded companies with operations which we believe to be similar to ours. When identifying companies as peers, we consider such characteristics as the
37
Table of Contents
type of industry, size and/or type of product(s), research and/or product development capabilities, and stock-based transactions. We intend to continue to consistently estimate our volatility to value stock options in this manner until sufficient historical information regarding the volatility of our own shares becomes available, or circumstances change such that the identified entities are no longer similar to us. In this latter case, we would utilize other similar entities whose share prices are publicly available. We estimate the expected life of employee share options based on the simplified method allowed by SAB No. 107, as amended by SAB No. 110. Under this approach, the expected term is presumed to be the average between the weighted-average vesting period and the contractual term. The expected term for options granted to nonemployees is generally the contractual term of the option. The risk-free interest rate is based on the U.S. Treasury Note, Stripped Principal, on the date of grant with a term substantially equal to the corresponding option’s expected term. We have never declared or paid any cash dividends nor do we plan to pay cash dividends in the foreseeable future.
The following assumptions were used in calculating the fair value of employee options granted during 2010 and 2009:
| 2010 | 2009 | |||
| Dividend yield |
– % | – % | ||
| Expected term (in years) |
2.5 – 6.0 | 3.7 – 6.2 | ||
| Expected volatility |
49% – 53% | 50% – 52% | ||
| Risk-free interest rate |
0.8% – 2.8% | 1.4% – 2.7% |
The following assumptions were used in calculating the fair value of nonemployee options granted during 2010 and 2009:
| 2010 | 2009 | |||
| Dividend yield |
– % | – % | ||
| Expected term (in years) |
5.0 | 2.3 – 10.0 | ||
| Expected volatility |
52% – 53% | 51% – 67% | ||
| Risk-free interest rate |
2.2% – 2.4% | 1.1% – 2.7% |
During 2011, we began issuing restricted stock awards at no cost in lieu of stock options to employees, directors and consultants. Compensation expense for restricted stock granted to employees and directors is generally equal to the fair market value of the underlying common stock on the date of grant. If a sufficient disincentive for nonperformance does not exist at the date of grant, the compensation cost is remeasured at each reporting date at the then-current fair market value of the underlying common stock until the award vests.
Research and Development
We account for research and development costs and accrue expenses based on estimates of work performed, patient enrollment or fixed-fee-for-services. As work is performed and/or invoices are received, we adjust our estimates and accruals. To date, our accruals have been within our estimates. Total research and development costs are a function of studies being conducted and will increase or decrease based on the level of activity in any particular year.
Intangible Assets
Intangible assets include license agreements, product rights and other identifiable intangible assets. We assess the impairment of identifiable intangible assets whenever events or changes in circumstances indicate the carrying value may not be recoverable. In determining the recoverability of our intangible assets, we must make assumptions regarding estimated future cash flows and other factors. If the estimated undiscounted future cash flows do not exceed the carrying value of the intangible assets, we must determine the fair value of the intangible assets. If the fair value of the intangible assets is less than the carrying value, an impairment loss will be recognized in an amount equal to the difference. Fair value is determined through various valuation techniques including quoted market prices, third-party independent appraisals and discounted cash flow models, as considered necessary.
38
Table of Contents
RESULTS OF OPERATIONS
Year ended December 31, 2011 compared to year ended December 31, 2010
Net product revenues. Net product revenue increased $6.2 million, or 14%, in 2011 as compared to 2010. Net product revenue increased $7.4 million for Acetadote, which was partially offset be a decreases in Kristalose revenue of $1.0 million and Caldolor of $0.2 million.
An increase in volume of approximately 13% and an increase in the average selling price contributed to the increase in Acetadote revenue. In early 2011, we introduced the new formulation of Acetadote that is free of Ethylene diamine tetracetic acid or other stabilization and chelating agents and is preservative-free. The new formulation of Acetadote has been well-received in the market, and continues to be the treatment of choice for acetaminophen overdose. Additionally, Acetadote revenue was positively impacted by the shortage of the oral form of n-acetylcysteine due to manufacturing delays. The shortage is expected to continue into 2012. Management is unable to determine the degree to which the shortage has contributed to Acetadote revenue or the impact on future revenue when the shortage problem is corrected.
The decrease in Kristalose net revenue was primarily due to a decrease in volume, partially offset by an increase in the average selling price. During 2011, we experienced manufacturing delays caused by a change in ownership of the plant that produces Kristalose. As previously noted, we acquired the full rights to Kristalose in 2011 and believe this will help alleviate some of the manufacturing delays we experienced in 2011.
Gross product revenue for Caldolor increased $0.2 million in 2011 as compared to 2010. The increase in gross revenue was primarily due to increased volume as we continue to penetrate our target market. Additionally, during the first quarter of 2011, we initiated a shift in focus and began transitioning part of our sales and marketing resources to driving pull-through use of Caldolor in facilities stocking the product. In the fourth quarter of 2011, we notified our wholesalers that we will discontinue the 400mg offering of Caldolor and concentrate our sales efforts on 800mg. As a result, we recognized a reserve for potential returns related to the 400mg product, of which a majority was related to sales in prior years. The result of this reserve plus the normal sales allowances resulted in a decrease in net product revenue of $0.2 million.
Other revenue. Other revenue decreased $0.9 million in 2011 as compared to 2010. The decrease was primarily due to the recognition of $0.9 million of federal grant funding from the Qualifying Therapeutic Discovery Project, a component of the healthcare reform legislation enacted in 2010. This program was not available in 2011.
Cost of products sold. Cost of products sold as a percentage of net revenues increased from 7.8% in 2010 to 10.5% in 2011. The increase was primarily due to the recognition of $2.0 million of inventory write-downs during 2011 for potentially obsolete inventory. Excluding this charge, cost of products sold as a percentage of net revenues would have been 6.6%. This decrease was primarily due to the change in our sales mix in the periods.
Selling and marketing. Selling and marketing expense totaled approximately $20.9 million in 2011, representing a decrease of $1.7 million, or 8%, over 2010. The decrease was primarily due a decrease in (1) royalty expense as a result of the Acetadote royalty agreement expiring in January 2011 and (2) sales force and related expenses due to a reduction in headcount and savings associated with bringing the field sales force in-house, rather than outsourcing. These decreases were partially offset by increased marketing and advertising expense due to significant investments made in marketing the new formulation of Acetadote and enhancing the brand message.
Research and development. Research and development expense totaled $5.0 million in 2011, representing an increase of $0.7 million, or 16%, over 2010. The increase was primarily due to (1) increased personnel costs as we expanded our research and development team and (2) increased annual product and establishment fees from the FDA for our products. We are currently performing clinical trials on each of our products and product candidates, and expect research and development costs to increase in 2012 as these studies progress.
General and administrative. General and administrative expense totaled $9.2 million in 2011, representing an increase of $1.2 million, or 15%, over 2010. The increase was primarily due to increased (1) charitable contributions as we donated inventory for humanitarian needs, (2) increased travel expenses as we continue to expand our products world-wide and (3) increased consulting and personnel costs.
39
Table of Contents
Interest expense. Interest expense totaled $0.4 million in 2011, representing a decrease of $1.1 million, or 75%, over 2010. The decrease in interest expense was primarily due to (1) the payoff of our term debt in July 2011 and (2) lower interest rates as a result of modifying our line of credit agreement. In August 2011, we amended our debt agreement to provide up to $10 million of availability under our line of credit, with the option of increasing it to $20 million upon the satisfaction of certain conditions. As a result of amending our debt agreement, we were able to reduce the interest cost through the term of the line of credit facility (December 31, 2014).
Income tax expense. As a percentage of income before income taxes, the tax rate decreased from 54% in 2010 to 42% in 2011. The decrease was primarily due to (1) an increase in pre-tax income without a corresponding increase in the nature and amount of permanent differences and (2) the effects of the Therapeutic Discovery Tax Credit in 2010. As previously noted, we received $0.9 million of federal tax grants in 2010, which represented 50% of the eligible expenses. The full amount of the eligible expenses was not deductible for federal income purposes. The Therapeutic Discovery Tax Credit was not available in 2011.
Year ended December 31, 2010 compared to year ended December 31, 2009
Net revenues. Net revenues for 2010 totaled approximately $45.9 million, representing an increase of approximately $2.3 million, or 5%, over the same period in 2009. Net revenue increased $4.9 million for Acetadote and decreased $0.2 million and $3.2 million for Kristalose and Caldolor, respectively. The increase in Acetadote revenue was positively impacted by a 4% increase in volume and an increase in the average selling price, offset by an increase in fee-for-service deductions due to additional arrangements with our wholesalers. While Kristalose gross revenue increased, net revenue was impacted by an increase in the gross-to-net revenue deductions primarily associated with rebates and expired product returns. Additionally, in the third quarter of 2009, we completed the commercial launch of Caldolor, and recognized $3.3 million of net revenue in 2009. Our sales forces continued to maintain a consistent level of focus on Acetadote and Kristalose while they progressed the promotion of Caldolor.
In 2010, we focused our sales and marketing efforts primarily on securing formulary approval and stocking nationally for Caldolor.
In 2010, we recognized approximately $0.9 million in federal grant funding from the Qualifying Therapeutic Discovery Project, a component of the healthcare reform legislation enacted in 2010.
Cost of products sold. Cost of products sold as a percentage of net revenues decreased from 9.5% for 2009 to 7.8% for the same period in 2010. This decrease was primarily due to the sales mix in the periods.
Selling and marketing. Selling and marketing expense for 2010 totaled approximately $22.7 million, representing an increase of approximately $2.5 million, or 12%, over the same period in 2009. The increase was primarily due to the expansion of our hospital sales force during the third quarter of 2009, and the resulting increases in payroll and related taxes, travel, meals and promotional activities. These increases were offset by a decrease in marketing, advertising and hiring expenses related to Caldolor in 2010 as compared to the significant investment made in 2009 related to the launch.
Research and development. Research and development expense for 2010 totaled approximately $4.3 million, representing a decrease of approximately $0.7 million, or 13%, over the same period in 2009. The decrease was primarily due to the inclusion in 2009 of approximately $2.0 million of milestone expenses incurred upon the FDA approval of Caldolor in June 2009. This decrease was offset by additional costs incurred in 2010 related to annual FDA product and establishment fees, increased salary and related expenses resulting from an increase in personnel and increased costs related to furthering our development efforts for our products and product candidates.
General and administrative. General and administrative expense for 2010 totaled approximately $8.0 million, representing an increase of approximately $0.3 million, or 5%, over the same period in 2009. The increase is primarily due to additional expenses associated with being an SEC registrant, including legal, accounting and insurance costs.
Interest income. Interest income for 2010 totaled approximately $0.2 million, representing an increase of approximately $0.1 million, or 152%, over the same period in 2009. The increase was primarily due to the higher cash balances maintained in 2010 as a result of the proceeds received from the initial public offering in the third quarter of 2009.
40
Table of Contents
Interest expense. Interest expense for 2010 totaled approximately $1.4 million, representing an increase of approximately $0.7 million as compared to the same period in 2009. The increase is primarily attributable to (1) an average higher outstanding debt balance in 2010 as compared to 2009 and (2) the inclusion of approximately $0.1 million of deferred financing costs and approximately $0.2 million of prepayment fees associated with the early extinguishment and amendment of our term debt facility in September 2010.
Income tax expense. Income tax expense for 2010 totaled approximately $2.9 million, representing an increase of approximately $0.8 million, over the same period in 2009. As a percentage of income before income taxes, income tax expense increased from 39.8% for the year ended December 31, 2009 to 54.0% for the same period in 2010. The increase in the percentage was primarily due to (1) research and development expenses utilized in the Therapeutic Discovery Tax Credit not being deductible for federal income tax purposes, (2) an increase in stock compensation expense that is not deductible for income tax purposes and (3) an increase in nondeductible meals and entertainment expenses associated with the expansion of our sales force.
LIQUIDITY AND CAPITAL RESOURCES
Our primary sources of liquidity are cash flows provided by our operations, our borrowings and the cash proceeds from our initial public offering of common stock. We believe that our internally generated cash flows and amounts available under our line of credit agreement will be adequate to service existing debt, finance internal growth and fund capital expenditures. As of December 31, 2011 and 2010, our cash and cash equivalents was $70.6 million and $65.9 million, respectively; working capital (current assets minus current liabilities) was $80.7 million and $71.8 million, respectively; and our current ratio (current assets to current liabilities) was 13.2x and 8.8x, respectively. As of December 31, 2011, we also had the ability to make additional draws of up to approximately $5.1 million on our line of credit.
The information included in Note 6 to the consolidated financial statements included in this annual report on Form 10-K is hereby incorporated by reference into this Item.
The following table summarizes our net changes in cash and cash equivalents for the years ended December 31, 2011, 2010 and 2009:
| Years Ended December 31, | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| (in thousands) | ||||||||||||
| Cash provided by (used in): |
||||||||||||
| Operating activities |
$ | 8,722 | $ | 347 | $ | 405 | ||||||
| Investing activities |
(438 | ) | (769 | ) | (712 | ) | ||||||
| Financing activities |
(3,579 | ) | (12,386 | ) | 67,180 | |||||||
|
|
|
|
|
|
|
|||||||
| Net (decrease) increase in cash and cash equivalents (1) |
$ | 4,705 | $ | (12,808 | ) | $ | 66,872 | |||||
|
|
|
|
|
|
|
|||||||
| (1) | The sum of the individual amounts may not agree due to rounding. |
The net increase in cash and cash equivalents of $4.7 million for the year ended December 31, 2011 was primarily due to cash generated from our operating activities. Our net income increased from $2.4 million in 2010 to $6.0 million in 2011. The increase in cash and cash equivalents from operating activities was offset by increased purchases of fixed assets and intangibles of $0.4 million and cash used in financing activities of $3.6 million. During 2011, we paid in full our term debt facility of $5.3 million. In connection with the termination of the term debt facility, we increased our borrowings under our line of credit by $3.0 million. In addition, our financing activities included the repurchase of common stock of $4.4 million in connection with our share repurchase program discussed in Part II, Item 5, Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities, of this Form 10-K. During 2011, we recognized approximately $2.4 million of excess tax benefits. The excess tax benefit represents the income taxes that would have been paid if not for the tax deductions created upon the exercise of nonqualified stock options. We expect to pay minimal income taxes in 2012 due to the continued usage of the unrecognized tax benefit related to the excess tax deduction described in Note 7 to the consolidated financial statements included in this annual report on Form 10-K.
41
Table of Contents
The net decrease in cash and cash equivalents of $12.8 million for year ended December 31, 2010 was primarily due to cash used in financing activities, which included principal payments on our term debt of $12.7 million and the repurchase of common stock of approximately $4.8 million. These expenditures were offset by proceeds from the exercise of stock options of approximately $1.4 million and the excess tax benefit derived from the exercise of nonqualified options of approximately $3.9 million. Cash provided by operating activities for the year ended December 31, 2010 was primarily due to net income for the period and the collection of accounts receivable offset by (1) the purchase of inventory, (2) the decrease in accounts payable and (3) the excess tax benefit derived from the exercise of stock options.
In July 2009, we amended our debt agreement (the Fourth Amended and Restated Loan Agreement) to provide for $18.0 million in term debt and a $4.0 million revolving credit facility, both with an interest rate of LIBOR plus an applicable margin based on the our Leverage Ratio, as defined in the agreement. The interest rate at December 31, 2009 was 5.73% per annum. In addition, we were required to pay a commitment fee of 0.75% per annum on the unused portion of the commitment. The term debt was payable in quarterly installments of $1.5 million beginning on March 31, 2010 and continuing until December 31, 2012. The revolving credit facility was due on December 31, 2012. The proceeds from the term debt were restricted for the payment, in part, of the minimum statutory tax withholding requirements of approximately $24.6 million due from option holders who exercised options to purchase shares of our common stock at the pricing of our initial public offering. The Fourth Amended and Restated Loan Agreement required us to make an additional principal payment within 120 days after the end of the fiscal year in an amount equal to its Excess Cash Flow, as defined in the agreement. The additional principal payment of $3.1 million was paid during the first quarter of 2010. The Fourth Amended and Restated Loan Agreement contained restrictive covenants, which we were in compliance with during 2010 and 2009.
In September 2010, we further amended our loan agreement (the Agreement) that provided for an increase in the availability under the existing line of credit from $4.0 million to $6.0 million, with interest payable monthly at LIBOR plus an Applicable Margin, as defined in the Agreement (4.76% at December 31, 2010). In addition, the term debt was reduced to $6.0 million, with quarterly payments under the term debt reduced to $666,667, plus interest at the same rate as the line of credit, beginning December 31, 2010. Concurrent with the amendment, we elected to prepay approximately $5.9 million of the outstanding term debt, incurring a prepayment penalty of approximately $0.2 million. At December 31, 2010, the outstanding term loan and line of credit balances were $5.3 million and $1.8 million, respectively.
The Agreement included certain financial and restrictive covenants, all of which we were in compliance with at December 31, 2010. As a condition of the Agreement, we were required to maintain deposits at amounts equal to at least the sum of (a) the maximum amount of the line of credit plus (b) the aggregate principal amount then outstanding under the term debt.
In July 2011, we paid in full the outstanding term debt balance. In addition, we amended our line of credit agreement with them (the Amendment) to provide a line of credit of up to $10 million, with the option to increase the availability to $20 million upon the satisfaction of certain events. The line of credit expires on December 31, 2014, and bears interest at LIBOR plus an applicable margin, as defined in the Amendment (2.29% at December 31, 2011). We reduced the commitment fee from one-half of one percent (0.50%) to one-quarter of one percent (0.25%) per annum on the unused line of credit. Borrowings under the line of credit are collateralized by substantially all of our assets. We are no longer required to maintain minimum deposits with the lender. The Amendment includes certain financial and restricted covenants, all of which we were in compliance with during the period.
Our manufacturing and supply agreement with one manufacturer, which expires in 2014, contains a minimum annual purchase obligation. We expect our normal inventory purchasing levels to be above the required minimum amounts. As of December 31, 2011, we had met our purchase obligations for 2011 under this agreement.
42
Table of Contents
The following table sets forth a summary of our contractual cash obligations as of December 31, 2011:
| Payments Due by Year | ||||||||||||||||||||||||
| Contractual obligations(1) |
Total (2) | 2012 | 2013 | 2014 | 2015 | 2016+ | ||||||||||||||||||
| (in thousands) | ||||||||||||||||||||||||
| Amounts reflected in the balance sheet: |
||||||||||||||||||||||||
| Line of credit |
$ | 4,860 | $ | — | $ | — | $ | 4,860 | $ | — | $ | — | ||||||||||||
| Estimated interest on debt (3) |
334 | 111 | 111 | 111 | — | — | ||||||||||||||||||
| Other cash obligations not reflected on the balance sheet: |
||||||||||||||||||||||||
| Operating leases |
4,369 | 860 | 886 | 913 | 940 | 769 | ||||||||||||||||||
| Purchase obligations (4) |
2,559 | 975 | 975 | 609 | — | — | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total (1) |
$ | 12,122 | $ | 1,947 | $ | 1,972 | $ | 6,493 | $ | 940 | $ | 769 | ||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| (1) | The table of contractual obligations excludes amounts due under the Kristalose purchase agreement as these amounts can not be determined until sales of the product have occurred. As consideration for the purchase of certain Kristalose assets in November 2011, we agreed to pay the seller a percentage of net sales for a seven-year period beginning November 15, 2011. Payments are due quarterly, in arrears. |
| (2) | The sum of the individual amounts may not agree due to rounding. |
| (3) | Represents the estimated interest payments on our line of credit based on the December 31, 2011 interest rate of LIBOR plus an applicable margin, or 2.29%. Interest payments are due and payable quarterly in arrears. The line of credit becomes due and payable in December 2014. Estimated interest for the line of credit is based on the assumption of a consistent outstanding balance. |
| (4) | Represents minimum purchase obligations under our manufacturing agreements. |
OFF-BALANCE SHEET ARRANGEMENTS
During 2011, 2010 and 2009, we did not engage in any off-balance sheet arrangements.
RECENTLY ISSUED BUT NOT YET ADOPTED ACCOUNTING PRONOUNCEMENTS
There are no recently issued but not yet adopted accounting pronouncements that would materially impact our financial condition or results of operations.
Item 7A: Quantitative and Qualitative Disclosures About Market Risk
Interest Rate Risk
We are exposed to market risk related to changes in interest rates on our cash on deposit in highly-liquid money market accounts and revolving credit facility. We do not utilize derivative financial instruments or other market risk-sensitive instruments to manage exposure to interest rate changes. The main objective of our cash investment activities is to preserve principal while maximizing interest income through low-risk investments. Our investment policy focuses on principal preservation and liquidity.
We believe that our interest rate risk related to our cash and cash equivalents is not material. The risk related to interest rates for these accounts would produce less income than expected if market interest rates fall. Based on current interest rates, we do not believe we are exposed to significant downside risk related to a change in interest on our money market accounts.
In the first quarter of 2012, we analyzed our return on our investments and determined investing in variable rate demand notes and a portfolio of government backed securities (including U.S. Treasuries, government sponsored enterprise debentures and government sponsored adjustable rate mortgage backed securities), would yield a higher return with minimal additional risk. The variable rate demand notes, or VRDNs, are generally issued by municipal governments and are backed by a financial institution letter of credit. We hold a put right on the VRDN, which allows us to liquidate the investment relatively quickly (less than one week). The government backed securities have an active secondary market that generally provides for liquidity in less than one week. The risk related to interest rates for these accounts will produce less income than expected if market interest rates fall.
43
Table of Contents
Based on current interest rates, we do not believe we are exposed to significant downside risk related to change in interest on our investment accounts.
The interest rate risk related to borrowings under our line of credit is a variable rate of LIBOR plus an applicable margin, as defined in the loan agreement (2.29% at December 31, 2011). As of December 31, 2011, we had outstanding borrowings of $4.9 million under our line of credit. If interest rates increased by 1.0%, the impact on interest expense in future periods would be less than $0.1 million. We have sufficient cash balances to pay down the line of credit to minimize our interest rate exposure.
Exchange Rate Risk
While we operate primarily in the U.S., we are exposed to foreign currency risk. A portion of our research and development is performed abroad. As of December 31, 2011, our outstanding payables denominated in a foreign currency totaled less than $0.1 million.
Currently, we do not utilize financial instruments to hedge exposure to foreign currency fluctuations. We believe our exposure to foreign currency fluctuation is minimal as our purchases in foreign currency have a maximum exposure of 90 days based on invoice terms with a portion of the exposure being limited to 30 days based on the due date of the invoice. Foreign currency exchange losses were immaterial for 2011 and 2010. Neither a 5% increase nor decrease from current exchange rates would have had a material effect on our operating results or financial condition.
Item 8: Financial Statements and Supplementary Data
See consolidated financial statements, including the report of the independent registered public accounting firm, starting on page F-1, which is incorporated herein by reference.
Item 9: Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A: Controls and Procedures
Our Chief Executive Officer and Chief Financial Officer have evaluated the effectiveness of the design and operation of our disclosure controls and procedures as of December 31, 2011. Based on that evaluation, they have concluded that our disclosure controls and procedures were effective as of December 31, 2011 to ensure that material information relating to us and our consolidated subsidiaries is made known to officers within these entities in order to allow for timely decisions regarding required disclosure.
Management’s report on internal control over financial reporting and the related attestation report of KPMG LLP, our independent registered public accounting firm, are included on page F-1 and F-3, respectively, of this annual report on Form 10-K, and incorporated herein by reference.
During our fourth quarter of 2011, there were no changes in our internal control over financial reporting (as defined in Rule 13a-15(f) or 15d-15(f)).
On March 5, 2012, Cumberland Pharmaceuticals Inc., Mylan Institutional Inc., and Mylan Pharmaceuticals Inc. (Mylan Institutional Inc. and Mylan Pharmaceuticals are collectively referred to as “Mylan”) executed an agreement, which is effective on November 1, 2011, whereby Mylan will package Cumberland’s lactulose crystal product marketed under the name Kristalose®, hereafter referred to as the “Packaging Agreement.”
Under the Packaging Agreement, Cumberland will be responsible for delivering the active pharmaceutical ingredient for Kristalose to Mylan and will compensate Mylan for packaging Kristalose® at an amount determined by the number of pouches. The price per pouch is determined by a packaging volume requirement. The agreement has an initial term of five years, and the agreement is automatically renewed for subsequent one year terms after the initial term.
The Packaging Agreement is attached hereto as Exhibit 10.29, and the foregoing summary is qualified in its entirety by such agreement.
The information called for by Part III of Form 10-K (Item 10 – Directors, Executive Officers and Corporate Governance, Item 11 – Executive Compensation, Item 12 – Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters, Item 13 – Certain Relationships and Related Transactions, and Director Independence, Item 14 – Principal Accounting Fees and Services), is incorporated by reference from our proxy statement related to our 2012 annual meeting of shareholders, which is expected to be filed with the SEC on or around March 9, 2012.
44
Table of Contents
Item 15: Exhibits, Financial Statement Schedules
| (a) | Documents filed as part of this report: |
| (1) | Financial Statements |
| (b) | Exhibits |
| Exhibit Number |
Description | |
| 3.1 | Third Amended and Restated Charter of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 19 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 17, 2009 | |
| 3.2 | Second Amended and Restated Bylaws of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 19 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 17, 2009 | |
| 4.1 | Specimen Common Stock Certificate of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 5 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on August 6, 2007 | |
| 4.2 | Warrant to Purchase Common Stock of Cumberland Pharmaceuticals Inc., issued to Bank of America, N.A. on October 21, 2003, incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 4.3 | Stock Purchase Warrant, issued to S.C.O.U.T. Healthcare Fund L.P. on April 15, 2004, incorporated herein by reference to the corresponding exhibit to Amendment No. 1 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on June 22, 2007 | |
| 4.4 | Warrant to Purchase Common Stock of Cumberland Pharmaceuticals Inc., issued to Bank of America, N.A. on April 6, 2006, incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 4.5# | Form of Option Agreement under 1999 Stock Option Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 4.6.1# | Form of Incentive Stock Option Agreement under 2007 Long-Term Incentive Compensation Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on May 17, 2010 | |
45
Table of Contents
| Exhibit Number |
Description | |
| 4.6.2# | Form of Nonstatutory Stock Option Agreement under 2007 Long-Term Incentive Compensation Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on May 17, 2010 | |
| 4 .7# | Form of Nonstatutory Stock Option Agreement under 2007 Directors’ Compensation Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on May 17, 2010 | |
| 4.8 | Warrant to Purchase Common Stock of Cumberland Pharmaceuticals Inc., issued to Bank of America, N.A. on July 22, 2009, incorporated herein by reference to the corresponding exhibit to the Registrant’s Annual Report on Form 10-K (File No. 001-33637) as filed with the SEC on March 19, 2010 | |
| 10.1† | Manufacturing and Supply Agreement for N-Acetylcysteine, dated January 15, 2002, by and between Bioniche Life Sciences, Inc. and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 5 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on August 6, 2007 | |
| 10.2 | Novation Agreement, dated January 27, 2006, by and among Bioniche Life Sciences, Inc., Bioniche Pharma Group Ltd., and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 10.3† | First Amendment to Manufacturing and Supply Agreement for N-Acetylcysteine, dated November 16, 2006, by and between Bioniche Teoranta and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 3 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 11, 2007 | |
| 10.3.1† | Second Amendment to Manufacturing and Supply Agreement for N-Acetylcysteine, dated March 25, 2008, by and between Bioniche Teoranta and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 10 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 21, 2008 | |
| 10.3.2† | Third Amendment to Manufacturing and Supply Agreement for N-Acetylcysteine, effective April 25, 2011, by and between Bioniche Teoranta and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Current Report on Form 8-K (File No. 001-33637) as filed with the SEC on June 24, 2011 | |
| 10.7† | Exclusive Distribution Agreement, effective as of July 1, 2010, by and between Cardinal Health 105, Inc. and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit of the Registrant’s Current Report on Form 8-K (File No. 001-33637) as filed with the SEC on August 13, 2010 | |
| 10.8† | Strategic Alliance Agreement, dated July 21, 2000, by and between F.H. Faulding & Co. Limited and Cumberland Pharmaceuticals Inc., including notification of assignment from F.H. Faulding & Co. Limited to Mayne Pharma Pty Ltd., dated April 16, 2002, incorporated herein by reference to the corresponding exhibit to Amendment No. 4 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 23, 2007 | |
| 10.10† | License Agreement, dated May 28, 1999, by and between Vanderbilt University and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 3 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 11, 2007 | |
| 10.11# | Employment Agreement dated March 2, 2012, effective as of January 1, 2012, by and between A.J. Kazimi and Cumberland Pharmaceuticals Inc. | |
| 10 .12# | Employment Agreement dated March 2, 2012, effective as of January 1, 2012, by and between Jean W. Marstiller and Cumberland Pharmaceuticals Inc. | |
| 10.13# | Employment Agreement dated March 2, 2012, effective as of January 1, 2012, by and between Leo Pavliv and Cumberland Pharmaceuticals Inc. | |
46
Table of Contents
| Exhibit Number |
Description | |
| 10.16† | Fifth Amended and Restated Loan Agreement by and between Cumberland Pharmaceuticals Inc. and Bank of America, N.A., dated August 2, 2011, incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on August 8, 2011 | |
| 10.17# | 1999 Stock Option Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 10.18# | 2007 Long-Term Incentive Compensation Plan of Cumberland Pharmaceuticals Inc., as amended on November 4, 2010, incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on November 15, 2010 | |
| 10.19# | 2007 Directors’ Compensation Plan of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on May 17, 2010 | |
| 10 .20 | Form of Indemnification Agreement between Cumberland Pharmaceuticals Inc. and all members of its Board of Directors, incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 10.21† | Lease Agreement, dated September 10, 2005, by and between Nashville Hines Development, LLC and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 3 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on July 11, 2007 | |
| 10.21.1† | First Amendment to Office Lease Agreement, dated April 25, 2008, by and between 2525 West End, LLC (successor in interest to Nashville Hines Development LLC) and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 10 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 21, 2008 | |
| 10.21.2† | Second Amendment to Office Lease Agreement, dated March 2, 2010, by and between 2525 West End, LLC (successor in interest to Nashville Hines Development LLC) and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Quarterly Report on Form 10-Q (File No. 001-33637) as filed with the SEC on May 17, 2010 | |
| 10.23† | Amended and Restated Lease Agreement, dated November 11, 2004, by and between The Gateway to Nashville LLC and Cumberland Emerging Technologies, Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 10.24 | First Amendment to Amended and Restated Lease Agreement, dated August 23, 2005, by and between The Gateway to Nashville LLC and Cumberland Emerging Technologies, Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 10.24.1 | Second Amendment to Amended and Restated Lease Agreement, dated January 9, 2006, by and between The Gateway to Nashville LLC and Cumberland Emerging Technologies, Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 10 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 21, 2008 | |
| 10.25† | Manufacturing Agreement, dated February 6, 2008, by and between Bayer HealthCare, LLC, and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to Amendment No. 12 of the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on June 20, 2008 | |
| 10.26# | Employment Agreement dated March 2, 2012, effective as of January 1, 2012, by and between Martin E. Cearnal and Cumberland Pharmaceuticals Inc. | |
| 10.27# | Employment Agreement dated March 2, 2012, effective as of January 1, 2012, by and between Rick S. Greene and Cumberland Pharmaceuticals Inc. | |
| 10.28† | Asset Purchase and Royalty Agreement for Kristalose dated November 15, 2011 by and between Mylan Inc. and Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit of the Registrant’s Current Report on Form 8-K (File No. 001-33637) as filed with the SEC on November 22, 2011 | |
47
Table of Contents
| Exhibit Number |
Description | |
| 10.29†† | Packaging Agreement effective November 1, 2011 by and among Mylan Institutional Inc., Mylan Pharmaceuticals Inc. and Cumberland Pharmaceuticals Inc. | |
| 21 | Subsidiaries of Cumberland Pharmaceuticals Inc., incorporated herein by reference to the corresponding exhibit to the Registrant’s Registration Statement on Form S-1 (File No. 333-142535) as filed with the SEC on May 1, 2007 | |
| 23.1 | Consent of KPMG LLP | |
| 31.1 | Certification of Chief Executive Officer Pursuant to Rule 13-14(a) of the Securities Exchange Act of 1934 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Certification of Chief Financial Officer Pursuant to Rule 13-14(a) of the Securities Exchange Act of 1934 as Adopted Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification of Chief Executive Officer and Chief Financial Officer Pursuant to 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| # | Indicates a management contract or compensatory plan. |
| † | Confidential treatment has been granted for portions of this exhibit. These portions have been omitted from the Registration Statement and submitted separately to the Securities and Exchange Commission. |
| †† | Confidential treatment has been requested for portions of this exhibit. These portions have been omitted from the Registration Statement and submitted separately to the Securities and Exchange Commission. |
48
Table of Contents
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on the 7th day of March 2012.
| CUMBERLAND PHARMACEUTICALS INC. | ||
| By: |
/s/ A. J. Kazimi | |
| A. J. Kazimi | ||
| Chief Executive Officer | ||
| (Principal Executive Officer) | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature |
Title |
Date | ||
| /s/ A. J. Kazimi |
Chairman and CEO | March 7, 2012 | ||
| A.J. Kazimi |
(Principal Executive Officer and | |||
| Director) | ||||
| /s/ Rick S. Greene |
Vice President and CFO | March 7, 2012 | ||
| Rick S. Greene |
(Principal Financial and Accounting Officer |
|||
| /s/ Robert G. Edwards |
Director | March 7, 2012 | ||
| Robert G. Edwards |
||||
| /s/ Thomas R. Lawrence |
Director | March 7, 2012 | ||
| Thomas R. Lawrence |
||||
| /s/ Lawrence W. Greer |
Director | March 7, 2012 | ||
| Lawrence W. Greer |
||||
| /s/ Martin E. Cearnal |
Director | March 7, 2012 | ||
| Martin E. Cearnal |
||||
| /s/ Gordon Bernard |
Director | March 7, 2012 | ||
| Gordon Bernard |
||||
| /s/ Jonathan Griggs |
Director | March 7, 2012 | ||
| Jonathan Griggs |
||||
| /s/ James Jones |
Director | March 7, 2012 | ||
| James Jones |
||||
| /s/ Joey Jacobs |
Director | March 7, 2012 | ||
| Joey Jacobs |
||||
49
Table of Contents
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
The management of Cumberland Pharmaceuticals Inc. is responsible for establishing and maintaining adequate internal control over financial reporting. Cumberland Pharmaceuticals Inc.’s internal control system was designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements. All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Cumberland Pharmaceuticals Inc.’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2011. In making this assessment, it used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control – Integrated Framework.
Based on its assessment, management has concluded that, as of December 31, 2011, the Company’s internal control over financial reporting was effective based on those criteria.
Cumberland Pharmaceuticals Inc.’s independent registered public accounting firm has issued an audit report on the effectiveness of Cumberland Pharmaceuticals Inc.’s internal control over financial reporting. This report appears on page F-3 of this annual report on Form 10-K.
| /s/ A. J. Kazimi |
| A. J. Kazimi |
| Chief Executive Officer |
| March 7, 2012 |
| /s/ Rick S. Greene |
| Rick S. Greene |
| Chief Financial Officer |
| March 7, 2012 |
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors
Cumberland Pharmaceuticals Inc.:
We have audited the accompanying consolidated balance sheets of Cumberland Pharmaceuticals Inc. and subsidiaries (the Company) as of December 31, 2011 and 2010, and the related consolidated statements of income, cash flows, and equity and comprehensive income for each of the years in the three-year period ended December 31, 2011. In connection with our audits of the consolidated financial statements, we have also audited the financial statement Schedule II – Valuation and Qualifying Accounts for each of the years in the three-year period ended December 31, 2011. These consolidated financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Cumberland Pharmaceuticals Inc. and subsidiaries as of December 31, 2011 and 2010, and the results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2011, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth herein.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the Company’s internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated March 7, 2012 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
/s/ KPMG LLP
Nashville, Tennessee
March 7, 2012
F-2
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors
Cumberland Pharmaceuticals Inc.:
We have audited Cumberland Pharmaceuticals Inc.’s (the Company) internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2011, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Cumberland Pharmaceuticals Inc. and subsidiaries as of December 31, 2011 and 2010, and the related consolidated statements of income, cash flows, and equity and comprehensive income for each of the years in the three-year period ended December 31, 2011, and our report dated March 7, 2012 expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Nashville, Tennessee
March 7, 2012
F-3
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
December 31, 2011 and 2010
| 2011 | 2010 | |||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 70,599,146 | $ | 65,893,970 | ||||
| Accounts receivable, net of allowances |
7,082,890 | 5,145,494 | ||||||
| Inventories |
5,774,694 | 7,683,842 | ||||||
| Prepaid and other current assets |
1,627,455 | 1,336,765 | ||||||
| Deferred tax assets |
2,223,882 | 978,771 | ||||||
|
|
|
|
|
|||||
| Total current assets |
87,308,067 | 81,038,842 | ||||||
| Property and equipment, net |
1,119,339 | 1,220,010 | ||||||
| Intangible assets, net |
7,023,064 | 7,427,223 | ||||||
| Deferred tax assets |
— | 2,265,192 | ||||||
| Other assets |
67,846 | 102,787 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 95,518,316 | $ | 92,054,054 | ||||
|
|
|
|
|
|||||
| LIABILITIES AND EQUITY |
||||||||
| Current liabilities: |
||||||||
| Current portion of long-term debt |
$ | — | $ | 2,666,668 | ||||
| Accounts payable |
1,513,548 | 2,124,654 | ||||||
| Other accrued liabilities |
5,086,400 | 4,436,298 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
6,599,948 | 9,227,620 | ||||||
| Revolving line of credit |
4,859,951 | 1,825,951 | ||||||
| Long-term debt, excluding current portion |
— | 2,666,665 | ||||||
| Deferred tax liability |
645,029 | — | ||||||
| Other long-term obligations, excluding current portion |
578,119 | 618,343 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
12,683,047 | 14,338,579 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies |
||||||||
| Equity: |
||||||||
| Shareholders’ equity: |
||||||||
| Common stock – no par value; 100,000,000 shares authorized; 20,020,535 and 20,338,461 shares issued and outstanding as of December 31, 2011 and 2010, respectively |
70,272,155 | 70,778,874 | ||||||
| Retained earnings |
12,656,662 | 6,998,806 | ||||||
|
|
|
|
|
|||||
| Total shareholders’ equity |
82,928,817 | 77,777,680 | ||||||
| Noncontrolling interests |
(93,548 | ) | (62,205 | ) | ||||
|
|
|
|
|
|||||
| Total equity |
82,835,269 | 77,715,475 | ||||||
|
|
|
|
|
|||||
| Total liabilities and equity |
$ | 95,518,316 | $ | 92,054,054 | ||||
|
|
|
|
|
|||||
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Consolidated Statements of Income
Years ended December 31, 2011, 2010 and 2009
| 2011 | 2010 | 2009 | ||||||||||
| Revenues: |
||||||||||||
| Net product revenue |
$ | 50,893,794 | $ | 44,704,570 | $ | 43,142,350 | ||||||
| Other revenue |
248,982 | 1,171,801 | 394,928 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net revenues |
51,142,776 | 45,876,371 | 43,537,278 | |||||||||
|
|
|
|
|
|
|
|||||||
| Costs and expenses: |
||||||||||||
| Cost of products sold |
5,362,554 | 3,586,646 | 4,136,541 | |||||||||
| Selling and marketing |
20,940,060 | 22,674,505 | 20,194,074 | |||||||||
| Research and development |
5,028,072 | 4,327,485 | 4,993,278 | |||||||||
| General and administrative |
9,197,955 | 7,990,222 | 7,643,070 | |||||||||
| Amortization |
655,302 | 686,911 | 686,904 | |||||||||
| Other |
109,346 | 108,855 | 106,776 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total costs and expenses |
41,293,289 | 39,374,624 | 37,760,643 | |||||||||
|
|
|
|
|
|
|
|||||||
| Operating income |
9,849,487 | 6,501,747 | 5,776,635 | |||||||||
| Interest income |
210,727 | 200,207 | 79,363 | |||||||||
| Interest expense |
(353,497 | ) | (1,423,523 | ) | (772,927 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Income before income taxes |
9,706,717 | 5,278,431 | 5,083,071 | |||||||||
| Income tax expense |
(4,080,204 | ) | (2,851,420 | ) | (2,024,192 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net income |
5,626,513 | 2,427,011 | 3,058,879 | |||||||||
| Net loss at subsidiary attributable to noncontrolling interests |
31,343 | 29,669 | 32,536 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net income attributable to common shareholders |
$ | 5,657,856 | $ | 2,456,680 | $ | 3,091,415 | ||||||
|
|
|
|
|
|
|
|||||||
| Earnings per share attributable to common shareholders |
||||||||||||
| - Basic |
$ | 0.28 | $ | 0.12 | $ | 0.22 | ||||||
| - Diluted |
$ | 0.28 | $ | 0.12 | $ | 0.17 | ||||||
| Weighted-average shares outstanding |
||||||||||||
| - Basic |
20,342,913 | 20,333,932 | 14,199,479 | |||||||||
| - Diluted |
20,572,132 | 21,058,577 | 18,234,171 | |||||||||
See accompanying notes to consolidated financial statements.
F-5
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
Years ended December 31, 2011, 2010 and 2009
| 2011 | 2010 | 2009 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net income |
$ | 5,626,513 | $ | 2,427,011 | $ | 3,058,879 | ||||||
| Adjustments to reconcile net income to net cash flows provided by operating activities: |
||||||||||||
| Depreciation and amortization expense |
1,040,407 | 978,398 | 816,499 | |||||||||
| Deferred tax expense (benefit) |
1,665,110 | (332,349 | ) | (525,467 | ) | |||||||
| Stock-based compensation — nonemployees |
149,719 | 80,222 | 1,056,401 | |||||||||
| Stock-based compensation — employees |
629,586 | 688,408 | 606,395 | |||||||||
| Excess tax benefit derived from exercise of stock options |
(2,355,345 | ) | (3,874,966 | ) | (3,968,894 | ) | ||||||
| Noncash interest expense |
137,487 | 352,484 | 128,800 | |||||||||
| Net changes in assets and liabilities affecting operating activities: |
||||||||||||
| Accounts receivable |
(1,937,396 | ) | 1,031,091 | (3,047,238 | ) | |||||||
| Inventory |
1,909,148 | (2,860,969 | ) | (3,060,097 | ) | |||||||
| Prepaid, other current assets and other assets |
(399,393 | ) | 1,342,032 | (721,464 | ) | |||||||
| Accounts payable and other accrued liabilities |
2,296,535 | 201,725 | 6,572,098 | |||||||||
| Other long-term obligations |
(40,224 | ) | 313,575 | (510,942 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by operating activities |
8,722,147 | 346,662 | 404,970 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Additions to property and equipment |
(257,502 | ) | (577,159 | ) | (601,802 | ) | ||||||
| Additions to trademarks and patents |
(180,269 | ) | (191,483 | ) | (110,541 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(437,771 | ) | (768,642 | ) | (712,343 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activities: |
||||||||||||
| Proceeds from initial public offering of common stock |
— | — | 85,000,000 | |||||||||
| Costs of initial public offering |
— | — | (7,479,011 | ) | ||||||||
| Proceeds from borrowings on long-term debt |
— | — | 18,000,000 | |||||||||
| Principal payments on note payable |
(5,333,333 | ) | (12,666,667 | ) | (5,000,000 | ) | ||||||
| Net borrowings on line of credit |
3,034,000 | — | — | |||||||||
| Costs of financing for long-term debt and credit facility |
(17,637 | ) | (110,000 | ) | (189,660 | ) | ||||||
| Payments made in connection with repurchase of common shares |
(4,247,440 | ) | (4,846,791 | ) | (27,295,808 | ) | ||||||
| Proceeds from exercise of stock options |
629,865 | 1,362,760 | 175,089 | |||||||||
| Excess tax benefit derived from exercise of stock options |
2,355,345 | 3,874,966 | 3,968,894 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net cash (used in) provided by financing activities |
(3,579,200 | ) | (12,385,732 | ) | 67,179,504 | |||||||
|
|
|
|
|
|
|
|||||||
| Net increase (decrease) in cash and cash equivalents |
4,705,176 | (12,807,712 | ) | 66,872,131 | ||||||||
| Cash and cash equivalents, beginning of year |
65,893,970 | 78,701,682 | 11,829,551 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents, end of year |
$ | 70,599,146 | $ | 65,893,970 | $ | 78,701,682 | ||||||
|
|
|
|
|
|
|
|||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid during the year for: |
||||||||||||
| Interest |
$ | 191,410 | $ | 814,373 | $ | 677,387 | ||||||
| Income taxes |
304,480 | 52,136 | 196,187 | |||||||||
| Noncash investing and financing activities: |
||||||||||||
| Change in unpaid invoices for purchases of intangibles |
97,806 | — | — | |||||||||
| Reclass of redeemable common stock to (from) equity |
— | 1,930,000 | (1,930,000 | ) | ||||||||
| Deferred financing costs |
— | — | 335,075 | |||||||||
See accompanying notes to consolidated financial statements.
F-6
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Consolidated Statements of Equity
Years ended December 31, 2011, 2010 and 2009
| Cumberland Pharmaceuticals Inc. Shareholders | ||||||||||||||||||||||||||||
| Non- | ||||||||||||||||||||||||||||
| Preferred stock | Common stock | Retained | controlling | Total | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | earnings | interests | equity | ||||||||||||||||||||||
| Balance, December 31, 2008 |
812,749 | $ | 2,604,070 | 9,903,047 | $ | 13,500,034 | $ | 1,450,711 | $ | — | $ | 17,554,815 | ||||||||||||||||
| Initial public offering of common stock, net of offering costs |
— | — | 5,000,000 | 74,801,596 | — | — | 74,801,596 | |||||||||||||||||||||
| Stock-based compensation – employees |
— | — | — | 606,395 | — | — | 606,395 | |||||||||||||||||||||
| Issuance of common stock for services received |
— | — | 20,250 | 338,240 | — | — | 338,240 | |||||||||||||||||||||
| Stock-based compensation – nonemployees |
— | — | — | 845,661 | — | — | 845,661 | |||||||||||||||||||||
| Conversion of preferred stock into common stock |
(812,749 | ) | (2,604,070 | ) | 1,625,498 | 2,604,070 | — | — | — | |||||||||||||||||||
| Repurchase of common shares |
— | — | (4,018 | ) | (52,234 | ) | — | — | (52,234 | ) | ||||||||||||||||||
| Issuance of common stock warrants |
— | — | — | 97,575 | — | — | 97,575 | |||||||||||||||||||||
| Exercise of options and related tax benefit |
— | — | 3,635,709 | (23,099,591 | ) | — | — | (23,099,591 | ) | |||||||||||||||||||
| Net income |
— | — | — | — | 3,091,415 | (32,536 | ) | 3,058,879 | ||||||||||||||||||||
| Reclass of redeemable common stock |
— | — | — | (1,930,000 | ) | — | — | (1,930,000 | ) | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, December 31, 2009 |
— | — | 20,180,486 | 67,711,746 | 4,542,126 | (32,536 | ) | 72,221,336 | ||||||||||||||||||||
| Stock-based compensation – employees |
— | — | — | 688,408 | — | — | 688,408 | |||||||||||||||||||||
| Issuance of common stock for services received |
— | — | 5,636 | 55,140 | — | — | 55,140 | |||||||||||||||||||||
| Stock-based compensation – nonemployees |
— | — | — | 43,101 | — | — | 43,101 | |||||||||||||||||||||
| Repurchase of common shares |
— | — | (615,455 | ) | (4,887,247 | ) | — | — | (4,887,247 | ) | ||||||||||||||||||
| Exercise of options and related tax benefit |
— | — | 767,794 | 5,237,726 | — | — | 5,237,726 | |||||||||||||||||||||
| Net income |
— | — | — | — | 2,456,680 | (29,669 | ) | 2,427,011 | ||||||||||||||||||||
| Reclass of redeemable common stock |
— | — | — | 1,930,000 | — | — | 1,930,000 | |||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, December 31, 2010 |
— | — | 20,338,461 | 70,778,874 | 6,998,806 | (62,205 | ) | 77,715,475 | ||||||||||||||||||||
| Stock-based compensation – employees |
— | — | — | 627,353 | — | — | 627,353 | |||||||||||||||||||||
| Stock-based compensation – nonemployees |
— | — | 10,144 | 128,158 | — | — | 128,158 | |||||||||||||||||||||
| Repurchase of common shares |
— | — | (743,073 | ) | (4,247,440 | ) | — | — | (4,247,440 | ) | ||||||||||||||||||
| Exercise of options and related tax benefit |
— | — | 415,003 | 2,985,210 | — | — | 2,985,210 | |||||||||||||||||||||
| Net income |
— | — | — | — | 5,657,856 | (31,343 | ) | 5,626,513 | ||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Balance, December 31, 2011 |
— | $ | — | 20,020,535 | $ | 70,272,155 | $ | 12,656,662 | $ | (93,548 | ) | $ | 82,835,269 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes to consolidated financial statements.
F-7
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
| (1) | Organization |
Cumberland Pharmaceuticals Inc. and its subsidiaries (the Company or Cumberland) is a specialty pharmaceutical company incorporated in Tennessee on January 6, 1999. The Company’s mission is to provide high-quality products to address underserved medical needs. Cumberland is focused on acquiring rights to, developing and commercializing branded prescription products for the hospital acute care and gastroenterology markets.
Cumberland focuses its resources on maximizing the commercial potential of its products, as well as developing new product candidates, and has both internal development and commercial capabilities. The Company’s products are manufactured by third parties, which are overseen by Cumberland’s quality control and manufacturing professionals. The Company works closely with its third-party distribution partner to make its products available in the United States.
In order to create access to a pipeline of early-stage product candidates, the Company formed a subsidiary, Cumberland Emerging Technologies, Inc. (CET), which assists universities and other research organizations to help bring biomedical projects from the laboratory to the marketplace. The Company’s ownership in CET is 85%. The remaining interest is owned by Vanderbilt University and the Tennessee Technology Development Corporation. The operating results of CET are allocated to the noncontrolling interests in the consolidated statements of income, and totaled approximately $31,000, $30,000 and $33,000 for the years ended December 31, 2011, 2010 and 2009, respectively.
Effective January 1, 2007, the Company formed a wholly-owned subsidiary, Cumberland Pharma Sales Corp. (CPSC), for the purpose of employing the hospital sales force that promotes the Company’s products, Acetadote® and Caldolor®, in the acute care market. In September 2010, the Company converted its field sales force, which promotes Caldolor and Kristalose®, to Cumberland employees. Previously, these sales forces were contracted through third-party contract sales organizations.
The Company operates in a single operating segment of specialty pharmaceutical products. All of the Company’s assets are located in the United States. Total revenues are primarily attributable to U.S. customers. Net revenues from non-U.S. customers were approximately $0.1 million, $0.1 million and $0.7 million for the years ended December 31, 2011, 2010 and 2009, respectively.
| (2) | Significant Accounting Policies |
| (a) | Principles of Consolidation |
These consolidated financial statements are stated in U.S. dollars and are prepared under U.S. generally accepted accounting principles. The consolidated financial statements include the accounts of the Company and its majority-owned subsidiaries. All significant intercompany transactions and accounts have been eliminated.
| (b) | Cash and Cash Equivalents |
Cash and cash equivalents include highly liquid investments with an original maturity of three months or less when purchased.
F-8
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (c) | Accounts Receivable |
Trade accounts receivable are recorded at the invoiced amount and do not bear interest. The Company records allowances for uncollectible amounts, cash discounts, chargebacks and credits to be taken by customers for product damaged in shipment based on historical experience. The Company reviews each customer balance for collectibility.
Discounts are reductions to invoiced amounts offered to customers for payment within a specified period of time from the date of the invoice.
The majority of the Company’s products are distributed through independent pharmaceutical wholesalers. Net product revenue and accounts receivable take into account the sale of the product at the wholesale acquisition cost, and an accrual is recorded to reflect the difference between the wholesale acquisition cost and the estimated average end-user contract price. This accrual is calculated on a product-specific basis and is based on the estimated number of outstanding units sold to wholesalers that will ultimately be sold under end-user contracts. When the wholesaler sells the product to the end-user at the agreed upon end-user contract price, the wholesaler charges the Company for the difference between the wholesale acquisition price and the end-user contract price and that chargeback is offset against the initial accrual balance.
The Company’s estimate of the allowance for damaged product is based upon historical experience of claims made for damaged product. At the time the transaction is recognized as a sale, the Company records a reduction in revenue for the estimate of product damaged in shipment.
| (d) | Inventories |
The Company works closely with third parties to manufacture and package finished goods for sale, takes title to the finished goods at the time of shipment from the manufacturer and warehouses such goods until distribution and sale. Inventories are stated at the lower of cost or market with cost determined using the first-in, first-out method.
The Company continually evaluates inventory for potential losses due to excess, obsolete or slow-moving inventory by comparing sales history and sales projections to the inventory on hand. When evidence indicates the carrying value may not be recoverable, a charge is taken to reduce the inventory to the net realizable value.
During 2009 and 2010, the Company built inventory in preparation for the Caldolor product launch. Caldolor inventory represented the majority of net inventory on hand at December 31, 2011 and 2010, respectively, and has varying expiration dates through January 2015. At December 31, 2011 and 2010, the Company has recognized a reserve for potential obsolescence and discontinuance primarily for Caldolor of approximately $2.1 million and $0.1 million, respectively. If actual sales in future periods are less than projected sales, the Company could incur additional obsolescence losses.
In the fourth quarter of 2010, the Company purchased certain packaging materials related to the manufacture of Caldolor. As these materials are consumed as part of the manufacturing process, the costs associated with these materials will be used to offset the finished goods price from the packager.
In connection with the purchase of certain Kristalose assets in 2011 as discussed in Note 4, the Company purchases the active pharmaceutical ingredient for Kristalose, and maintains the inventory at the third-party manufacturer. As the ingredients are consumed in production, the value of the ingredients is transferred from raw materials to finished goods.
F-9
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
As of December 31, 2011 and 2010, inventory was comprised of the following:
| December 31, | ||||||||
| 2011 | 2010 | |||||||
| Raw materials |
$ | 774,637 | $ | 356,676 | ||||
| Finished goods |
5,000,057 | 7,327,166 | ||||||
|
|
|
|
|
|||||
| Total |
$ | 5,774,694 | $ | 7,683,842 | ||||
|
|
|
|
|
|||||
| (e) | Prepaids and Other Current Assets |
Prepaid and other current assets consist of unamortized deferred financing costs, prepaid insurance premiums, prepaid consulting services and annual fees to the U.S. Food and Drug Administration (FDA). The Company expenses all prepaid amounts as used or over the period of benefit primarily on a straight-line basis, as applicable.
| (f) | Property and Equipment |
Property and equipment, including leasehold improvements, are stated at cost. Depreciation is provided using the straight-line method over the estimated useful lives of the assets. Leasehold improvements are amortized over the shorter of the initial lease term plus its renewal options, if reasonably assured, or the remaining useful life of the asset. Upon retirement or disposal of assets, the asset and accumulated depreciation or amortization accounts are adjusted accordingly, and any gain or loss is reflected as a component of operating income in the consolidated statement of income. Repairs and maintenance costs are expensed as incurred. Improvements that extend an asset’s useful life are capitalized.
| (g) | Intangible Assets |
The Company’s intangible assets consist of costs incurred related to product rights, licenses, trademarks and patents.
The cost of acquiring product rights and licenses of products that are approved for commercial use are capitalized based on the fair value and amortized ratably over the estimated economic life of the products. At the time of acquisition, the economic life is estimated based upon the term of the license agreement, patent life or market exclusivity of the products and our assessment of future sales and profitability of the product. We assess this estimate regularly during the amortization period and adjust the asset value or useful life when appropriate.
Patents consist of outside legal costs associated with obtaining patents for products that have already been approved for marketing by the FDA. If it becomes probable that a patent will not be issued, related costs associated with the patent application will be expensed at the time such determination is made. All costs associated with obtaining patents for products that have not been approved for marketing by the FDA are expensed as incurred.
Amortization expense is recognized on a straight-line basis over the following periods:
| Product rights | 15 years | |
| License rights | Term of license agreement | |
| Trademarks | 10 years | |
| Patents | Life of patent |
F-10
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (h) | Impairment of Long-Lived Assets |
Long-lived assets, such as property and equipment and purchased intangible assets subject to amortization, are reviewed for impairment whenever events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. If circumstances require a long-lived asset to be tested for possible impairment, the Company first compares undiscounted cash flows expected to be generated by an asset to the carrying value of the asset. If the carrying amount of the long-lived asset is not recoverable on an undiscounted cash flow basis, an impairment charge is recognized to the extent that the carrying value exceeds its fair value. Fair value is determined through various valuation techniques including quoted market prices, third-party independent appraisals and discounted cash flow models, as considered necessary. Assets to be disposed of would be separately presented in the consolidated balance sheet and reported at the lower of the carrying amount or fair value less costs to sell, and would no longer be depreciated. The assets and liabilities of a disposed group classified as held-for-sale would be presented separately in the appropriate asset and liability sections of the consolidated balance sheet. The Company recorded no impairment charges during the three-year period ended December 31, 2011.
| (i) | Revenue Recognition |
Revenue is realized or realizable and earned when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the seller’s price to the buyer is fixed and determinable; and (4) collectibility is reasonably assured. Delivery is considered to have occurred upon either shipment of the product or arrival at its destination, depending upon the shipping terms of the transaction.
The Company is a party to several licensing arrangements that allows the licensee access to our FDA registration file. In addition, the licensee is required to purchase product from the Company. Typical arrangements require an up-front payment in exchange for access to the FDA registration file, royalties and milestone payments upon the achievement of specific sales levels. Generally, the amounts received for access to the FDA registration file are recognized as revenue over the term of the arrangement, and royalties and milestones are recognized as income when earned.
The Company’s net product revenue reflects reduction from gross product revenue for estimated allowances for chargebacks, discounts, and damaged goods and for accruals for rebates, product returns, certain administrative fees and fee for services. Allowances of $0.2 million as of December 31, 2011 and 2010 for chargebacks, discounts and product damaged in shipment are recorded as a reduction of accounts receivable, and liabilities of $3.2 million and $2.6 million as of December 31, 2011 and 2010, respectively, for rebates, product returns, administrative fees and fee for services are included in other accrued liabilities.
As discussed in Note 2(c) above, the allowances for chargebacks, discounts and damaged goods are determined on a product-by-product basis, and are established by management as the Company’s best estimate at the time of sale based on each product’s historical experience adjusted to reflect known changes in the factors that impact such allowances. These allowances are established based on the contractual terms with direct and indirect customers and analyses of historical levels of chargebacks, discounts and credits claimed for damaged product.
Other organizations, such as managed care providers, pharmacy benefit management companies and government agencies, may receive rebates from the Company based on either negotiated contracts to carry the Company’s products or reimbursements for filled prescriptions. These entities represent indirect customers of the Company. In addition, the Company may provide rebates to the end-user. In conjunction with recognizing a sale to a wholesaler, sales revenues are reduced and accrued liabilities are increased by the Company’s estimates of the rebates that will be owed.
F-11
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
Consistent with industry practice, the Company maintains a return policy that allows customers to return product within a specified period prior to and subsequent to the expiration date. The Company’s estimate of the provision for returns is based upon historical experience. Any changes in the assumptions used to estimate the provision for returns are recognized in the period those assumptions were changed.
The Company has agreements with certain key wholesalers that include fee for service costs. These costs have been netted against product revenues.
The Company’s net product revenue (loss) consisted of the following as of December 31:
| Net product revenue | ||||||||||||
| 2011 | 2010 | 2009 | ||||||||||
| Acetadote |
$ | 42,454,055 | $ | 35,092,796 | $ | 30,176,981 | ||||||
| Kristalose |
8,517,873 | 9,510,275 | 9,688,998 | |||||||||
| Caldolor |
(78,134 | ) | 101,499 | 3,276,371 | ||||||||
|
|
|
|
|
|
|
|||||||
| $ | 50,893,794 | $ | 44,704,570 | $ | 43,142,350 | |||||||
|
|
|
|
|
|
|
|||||||
The Company obtained FDA approval for Caldolor in June 2009 and launched the product in September 2009. In December 2011, the Company discontinued sales of the 400mg offering and focused on the 800mg offering. Gross product revenue for Caldolor was approximately $0.3 million for the year ended December 31, 2011. Gross product revenue for Caldolor was approximately $0.1 million and $3.6 million for the years ended December 31, 2010 and 2009, respectively. The Company recognized approximately $0.4 million of sales allowances in the fourth quarter of 2011 for estimated returns of discontinued product.
Other revenue is comprised of revenue generated by CET through grant funding from federal Small Business (SBIR/STTR) grant programs, lease income generated by CET’s Life Sciences Center and contract services. The Life Sciences Center is a research center that provides scientists with access to flexible lab space and other resources to develop biomedical products. Revenue related to grants is recognized when all conditions related to such grants have been met. Grant revenue from SBIR/STTR programs totaled approximately $0.1 million for the years ended December 31, 2011 and 2010, and $0.2 million for the year ended December 31, 2009, respectively.
In addition to the items identified above, other revenue in 2010 includes approximately $0.9 million of federal grants associated with the Therapeutic Discovery Project Credit, a component of the U.S. health care reform act enacted in March 2010. The Therapeutic Discovery Project Credit allowed entities to apply for funding based on qualified research activities. Funds were then granted to entities based on their qualified research expenses. Revenue was recognized after the application was approved and as qualified research expenses were incurred.
| (j) | Income Taxes |
The Company provides for deferred taxes using the asset and liability approach. Under this method, deferred tax assets and liabilities are recognized for future tax consequences attributable to operating loss and tax credit carryforwards, as well as differences between the carrying amounts of existing assets and liabilities and their respective tax bases. The Company’s principal differences are related to the timing of deductibility of certain items, such as depreciation, amortization and expense for nonqualified stock options. Deferred tax assets and liabilities are measured using enacted tax rates that are expected to apply to taxable income in the years such temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in
F-12
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
tax rates is recognized in income in the period of enactment. The Company only recognizes income tax benefits associated with an income tax position where it is “more likely than not” that the position would be sustained upon examination by the taxing authorities.
In assessing the realizability of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income and tax planning strategies in making this assessment. Excluding the alternative minimum tax (AMT) tax credits, the Company will need to generate future taxable income of approximately $4.0 million in order to fully realize the deferred tax assets. Taxable income, excluding tax deductions generated by the exercise of nonqualified options, for the years ended December 31, 2011, 2010 and 2009 was approximately $5.7 million, $7.3 million and $7.0 million, respectively. Based upon the level of taxable income over the last three years and projections for future taxable income over the periods in which the deferred tax assets are deductible, management believes it is more likely than not that the Company will realize the benefits of these deductible differences, net of the existing valuation allowances at December 31, 2011. The amount of the deferred tax assets considered realizable, however, could be reduced in the near term if estimates of future taxable income during the carryforward period are reduced.
The tax benefit associated with the exercise of nonqualified stock options is recognized when the benefit is used to offset income taxes payable.
The Company’s accounting policy with respect to interest and penalties arising from income tax settlements is to recognize them as part of the provision for income taxes.
| (k) | Share-Based Payments |
The Company recognizes compensation cost for all share-based payments issued, modified, repurchased or cancelled. The cost of stock options is measured based on the grant-date fair value using the Black-Scholes option-pricing model, and the expense is recognized over the employee’s requisite service period. Depending on the nature of the vesting provisions, restricted stock awards are measured using either the fair value on the grant date or the fair value of common stock on the date the vesting provisions lapse. Prior to the lapse for those options not valued on the grant date, the fair value is measured on the last day of the reporting period.
| (l) | Research and Development |
Research and development costs are expensed in the period incurred. Research and development costs are comprised mainly of clinical trial expenses, salary and wages and other related costs such as materials and supplies. Development expense includes activities performed by third-party providers participating in the Company’s clinical studies. The Company accounts for these costs based on estimates of work performed, patients enrolled or fixed fees for services.
| (m) | Advertising Costs |
Advertising costs are expensed as incurred and amounted to $0.9 million, $0.8 million and $1.4 million in 2011, 2010 and 2009, respectively, and are included as a component of selling and marketing expenses in the consolidated statements of income.
| (n) | Selling and Marketing Expense |
Selling and marketing expense consists primarily of expense relating to the promotion, distribution and sale of products, including royalty expense, salaries and related costs.
F-13
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (o) | Distribution Costs |
The Company expenses distribution costs as incurred. Distribution costs included in selling and marketing expenses amounted to $1.2 million in 2011 and 2010, and $1.1 million in 2009.
| (p) | Cost of Products Sold |
Cost of products sold consists principally of the cost to acquire each unit of product sold, including in-bound freight expense. Cost of products sold also includes expenses associated with the write-down of slow-moving or expired product.
| (q) | Earnings per Share |
Basic earnings per share is calculated by dividing net income attributable to common shareholders by the weighted-average number of shares outstanding. Except where the result would be antidilutive to income from continuing operations, diluted earnings per share is calculated by assuming the vesting of unvested restricted stock and the exercise of stock options and warrants, unrecognized compensation costs, as well as their related income tax benefits. The following table reconciles the numerator and the denominator used to calculate diluted earnings per share:
| 2011 | 2010 | 2009 | ||||||||||
| Numerator: |
||||||||||||
| Net income attributable to common shareholders |
$ | 5,657,856 | $ | 2,456,680 | $ | 3,091,415 | ||||||
|
|
|
|
|
|
|
|||||||
| Denominator: |
||||||||||||
| Weighted-average shares outstanding – basic |
20,342,913 | 20,333,932 | 14,199,479 | |||||||||
| Convertible preferred stock shares |
— | — | 986,840 | |||||||||
| Dilutive effect of other securities |
229,219 | 724,645 | 3,047,852 | |||||||||
|
|
|
|
|
|
|
|||||||
| Weighted-average shares outstanding – diluted |
20,572,132 | 21,058,577 | 18,234,171 | |||||||||
|
|
|
|
|
|
|
|||||||
| Antidilutive restricted shares and options outstanding not included above |
1,079,904 | 640,718 | 246,332 | |||||||||
|
|
|
|
|
|
|
|||||||
| (r) | Comprehensive Income |
Total comprehensive income was comprised solely of net income for all periods presented.
| (s) | Use of Estimates |
The preparation of the consolidated financial statements in conformity with U.S. generally accepted accounting principles requires management of the Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the period. Significant items subject to estimates and assumptions include those related to chargebacks, rebates, discounts, credits for damaged product and returns, the valuation and determination of useful lives of intangible assets and the rate such assets are amortized, the realization of deferred tax assets, inventory reserves and stock-based compensation. Actual results could differ from those estimates.
F-14
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (t) | Fair Value of Financial Instruments |
The Company’s financial instruments include cash and cash equivalents, accounts receivable, accounts payable, accrued liabilities, revolving line of credit and long-term debt. The carrying values for cash and cash equivalents, accounts receivable, accounts payable and accrued liabilities approximate fair value due to their short-term nature. The terms of the revolving line of credit and term debt include variable interest rates, which approximate current market rates. The term debt was paid in full during 2011.
| (3) | Property and Equipment |
Property and equipment consisted of the following at December 31:
| Range of | ||||||||||
| useful lives | 2011 | 2010 | ||||||||
| Computer hardware and software |
3 – 5 years | $ | 536,702 | $ | 417,681 | |||||
| Office equipment |
3 – 15 years | 116,502 | 108,140 | |||||||
| Furniture and fixtures |
5 – 15 years | 598,510 | 488,982 | |||||||
| Leasehold improvements |
3 – 15 years, or remaining lease term |
951,688 | 931,097 | |||||||
|
|
|
|
|
|||||||
| 2,203,402 | 1,945,900 | |||||||||
| Less accumulated depreciation and amortization |
(1,084,063 | ) | (725,890 | ) | ||||||
|
|
|
|
|
|||||||
| $ | 1,119,339 | $ | 1,220,010 | |||||||
|
|
|
|
|
|||||||
Depreciation expense, including amortization expense related to leasehold improvements, during 2011, 2010 and 2009 was approximately $0.4 million, $0.3 million and $0.1 million, respectively, and is included in general and administrative expense in the consolidated statements of income.
| (4) | Intangible Assets |
Intangible assets consisted of the following at December 31:
| 2011 | 2010 | |||||||
| Product rights |
$ | 6,518,798 | $ | — | ||||
| Less accumulated amortization |
(54,259 | ) | — | |||||
|
|
|
|
|
|||||
| Total product rights |
6,464,539 | — | ||||||
|
|
|
|
|
|||||
| Trademarks |
9,020 | 9,020 | ||||||
| Less accumulated amortization |
(8,667 | ) | (8,123 | ) | ||||
|
|
|
|
|
|||||
| Total trademarks |
353 | 897 | ||||||
|
|
|
|
|
|||||
| License |
— | 10,303,595 | ||||||
| Less accumulated amortization |
— | (3,262,805 | ) | |||||
|
|
|
|
|
|||||
| Total license |
— | 7,040,790 | ||||||
|
|
|
|
|
|||||
| Patents |
608,561 | 409,536 | ||||||
| Less accumulated amortization |
(50,389 | ) | (24,000 | ) | ||||
|
|
|
|
|
|||||
| Total patents |
558,172 | 385,536 | ||||||
|
|
|
|
|
|||||
| $ | 7,023,064 | $ | 7,427,223 | |||||
|
|
|
|
|
|||||
In April 2006, the Company acquired the exclusive U.S. commercialization rights for Kristalose from Inalco Biochemicals, Inc. and Inalco S.p.A. (collectively Inalco) for $10,303,595. In November 2011, the Company completed the acquisition of the remaining rights associated with the Kristalose brand, including the FDA
F-15
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
registration and trademark. In connection with the acquisition, the unamortized balance of the license rights was reclassified to product rights. The agreement requires the Company to make future quarterly payments over a seven-year period equal to a percentage of Kristalose net sales. The payments are being treated as consideration for the assets acquired, and are being capitalized and amortized over the remaining expected useful life of the acquired asset, generally 15 years.
Amortization expense related to product rights, trademarks and license rights totaled approximately $0.7 million in 2011, 2010 and 2009, and is expected to be approximately $0.5 million in each of the years 2012 through 2016.
| (5) | Other Accrued Liabilities |
Other accrued liabilities consisted of the following at December 31:
| 2011 | 2010 | |||||||
| Rebates, fee for services, and product returns |
$ | 3,216,622 | $ | 2,626,383 | ||||
| Employee wages and benefits |
1,071,691 | 1,078,367 | ||||||
| Other |
798,087 | 731,548 | ||||||
|
|
|
|
|
|||||
| $ | 5,086,400 | $ | 4,436,298 | |||||
|
|
|
|
|
|||||
| (6) | Debt |
In September 2010, the Company amended its debt agreement with its primary lender (the Amendment) to provide for an increase in the line of credit from $4.0 million to $6.0 million and a decrease in the term debt to $6.0 million. Interest on the line of credit and term debt was payable monthly at LIBOR plus an Applicable Margin, as defined in the Amendment (4.76% at December 31, 2010). The quarterly payments under the term debt were reduced to $666,667, plus interest, beginning December 31, 2010. The Company reduced its commitment fee from three-quarters of one percent (0.75%) to one-half of one percent (0.50%) per annum on the unused line of credit. The borrowings were collateralized by a first priority lien on all of the Company’s assets. Concurrent with the amendment of the Agreement, the Company elected to prepay approximately $5.9 million of its term debt, incurring a prepayment penalty of approximately $0.2 million. The prepayment penalty is included as a component of interest expense in the consolidated statement of income for the year ended December 31, 2010.
The Amendment contained restrictive covenants which the Company was in compliance with during 2010.
In July 2011, the outstanding term debt balance of $4.0 million was paid in full. The Company did not incur any prepayment penalties or other fees associated with the payoff. In connection with the repayment, approximately $0.1 million of unamortized debt issue costs associated with the term debt was written off. These costs are included in interest expense in the consolidated statement of income for the year ended December 31, 2011.
In August 2011, the Company entered into a Fifth Amended and Restated Loan Agreement with its primary lender (the Agreement) to provide for an increase in the line of credit to $10 million. The credit facility may be increased up to $20 million upon the satisfaction of certain conditions. The interest rate is the BBA LIBOR Daily Floating Rate plus an Applicable Margin, as those terms are defined in the Agreement (2.29% at December 31, 2011). In addition, a commitment fee of 0.25% per annum is charged on the unused line of credit. The credit facility was extended to expire on December 31, 2014, at which time all principal amounts are due and payable. Interest is payable quarterly. Borrowings are collateralized by substantially all of the Company’s assets.
F-16
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
Under the Agreement, the Company is subject to certain financial covenants including, but not limited to, maintaining a Leverage Ratio and Interest Coverage Ratio, as those terms are defined in the Agreement, that are determined on a quarterly basis, and other restrictive covenants. The Company was in compliance with all covenants at December 31, 2011.
Furthermore, the lender may terminate the Agreement and require the Company to repay all outstanding amounts under certain conditions, as described in the Agreement, including, but not limited to: (1) cross-default on any other credit agreement with an outstanding principal amount in excess of $500,000, (2) material adverse change in our business condition, operations or properties, (3) violation of any covenant or (4) a change in control of the Company.
| (7) | Income Taxes |
In 2009, the Company generated a net operating loss as a result of the exercise of nonqualified options. These benefits will be recognized in the year in which they are able to reduce current income taxes payable. The usage of these net operating losses and related carryforwards resulted in the Company paying minimal income taxes in 2009 through 2011, and expects to pay minimal income taxes in 2012.
Components of the net deferred tax assets at December 31 are as follows:
| 2011 | 2010 | |||||||
| Deferred Tax Assets |
||||||||
| Net operating loss and tax credits |
$ | 1,025,621 | $ | 957,888 | ||||
| Property and equipment and intangibles |
92,470 | 181,156 | ||||||
| Allowance for accounts receivable |
92,977 | 62,951 | ||||||
| Reserve for expired product |
735,992 | 559,492 | ||||||
| Inventory |
1,079,541 | 141,492 | ||||||
| Deferred charges |
563,141 | 507,306 | ||||||
| Cumulative compensation costs incurred on deductible equity awards |
584,212 | 914,540 | ||||||
|
|
|
|
|
|||||
| Total deferred tax assets |
4,173,954 | 3,324,825 | ||||||
| Deferred Tax Liabilities |
||||||||
| Intangibles |
(2,500,642 | ) | — | |||||
|
|
|
|
|
|||||
| 1,673,312 | 3,324,825 | |||||||
| Less deferred tax asset valuation allowance |
(94,459 | ) | (80,862 | ) | ||||
|
|
|
|
|
|||||
| Net deferred tax assets |
$ | 1,578,853 | $ | 3,243,963 | ||||
|
|
|
|
|
|||||
The deferred tax liability was primarily due to the write-off for tax purposes of the net book value of the Kristalose product license rights. For book purposes, the net book value was capitalized as a component of Kristalose product rights and will be amortized over the useful life of the asset.
F-17
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
The following table summarizes the amount and year of expiration of federal and state net operating loss carryforwards as of December 31, 2011:
| Year of expiration |
Federal | State | ||||||
| 2014 |
$ | — | $ | 2,685,129 | ||||
| 2015 - 2017 |
— | 499,034 | ||||||
| 2018 - 2024 |
— | 60,573,367 | ||||||
| 2029 |
57,614,797 | — | ||||||
|
|
|
|
|
|||||
| $ | 57,614,797 | $ | 63,757,530 | |||||
|
|
|
|
|
|||||
The state net operating loss carryforwards above include approximately $2.0 million that is subject to a full valuation allowance at December 31, 2011
Income tax benefit (expense) includes the following components:
| 2011 | 2010 | 2009 | ||||||||||
| Current: |
||||||||||||
| Federal |
$ | (1,992,804 | ) | $ | (2,665,404 | ) | $ | (2,240,827 | ) | |||
| State |
(422,290 | ) | (518,365 | ) | (308,832 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| (2,415,094 | ) | (3,183,769 | ) | (2,549,659 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Deferred: |
||||||||||||
| Federal |
(1,543,261 | ) | 268,563 | 528,602 | ||||||||
| Stat10e |
(121,849 | ) | 63,786 | (3,135 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| (1,665,110 | ) | 332,349 | 525,467 | |||||||||
|
|
|
|
|
|
|
|||||||
| $ | (4,080,204 | ) | $ | (2,851,420 | ) | $ | (2,024,192 | ) | ||||
|
|
|
|
|
|
|
|||||||
The Company’s deferred tax expense in 2011 was primarily due to the write-off for tax purposes of the Kristalose license rights but maintained as a component of products rights for book purposes, and inventory write-downs. The deferred tax benefit for 2010 was primarily due to rent and expired product expenses recognized for book purposes in 2010 that will not be deductible for tax purposes until the future. The deferred tax benefit for 2009 was primarily due to the expense for nonqualified stock options issued to employees.
The deferred income tax benefit (expense) is comprised of the following components for the years ended December 31:
| 2011 | 2010 | 2009 | ||||||||||
| Deferred tax benefit exclusive of components listed below |
$ | 439,744 | $ | 459,575 | $ | 125,367 | ||||||
| Inventory write-downs |
817,840 | 31,228 | — | |||||||||
| Creation (utilization) of operating loss carryforwards |
11,348 | 9,567 | (60,266 | ) | ||||||||
| Creation (utilization) of tax credit carryforwards |
56,395 | (3,115 | ) | 7,172 | ||||||||
| Change in valuation allowance due to changes in net deferred tax asset balances |
(13,597 | ) | (10,750 | ) | (11,342 | ) | ||||||
| Deductible equity awards |
(330,329 | ) | (132,193 | ) | 419,255 | |||||||
| Intangible assets |
(2,646,511 | ) | (21,963 | ) | 45,281 | |||||||
|
|
|
|
|
|
|
|||||||
| Deferred income tax (expense) benefit |
$ | (1,665,110 | ) | $ | 332,349 | $ | 525,467 | |||||
|
|
|
|
|
|
|
|||||||
The valuation allowance at December 31, 2011 and 2010 is primarily related to state tax benefits at CET that will likely not be realized.
F-18
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
The Company’s effective income tax rate for 2011, 2010 and 2009 reconciles with the federal statutory tax rate as follows:
| 2011 | 2010 | 2009 | ||||||||||
| Federal tax expense at statutory rate |
35 | % | 34 | % | 34 | % | ||||||
| State income tax expense (net of federal income tax benefit) |
4 | 6 | 4 | |||||||||
| Permanent differences associated with tax grants |
— | 5 | — | |||||||||
| Permanent differences associated with stock options |
2 | 4 | 2 | |||||||||
| Other permanent differences |
2 | 4 | 1 | |||||||||
| Other |
(1 | ) | 1 | (1 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net income tax expense |
42 | % | 54 | % | 40 | % | ||||||
|
|
|
|
|
|
|
|||||||
During 2010, the Company applied for and received tax-free grants under the Therapeutic Discovery Project. Qualifying expenses certified under this program are nondeductible for federal income tax purposes. Approximately $0.4 million of qualifying expenses related to 2009 for which the Company filed an amended tax return in 2011. The Company also incurred expenses in 2011, 2010 and 2009 associated with the grant of incentive stock options. These expenses are nondeductible for federal income tax purposes.
The Company’s 2009 federal tax return is currently under examination. Federal tax years that remain open to examination are 2010 and 2011. Due to a 2009 net operating loss carryback, federal tax years 2006 – 2008 remain open to the extent of net operating losses utilized in those years. State tax years that remain open to examination are 2006 to 2011.
| (8) | Shareholders’ Equity |
| (a) | Initial Public Offering |
On August 10, 2009, the Company completed its initial public offering of 5,000,000 shares of common stock at a price of $17.00 per share, raising gross proceeds of $85.0 million. After deducting underwriting discounts of approximately $6.0 million and offering costs incurred of approximately $4.2 million, the net proceeds to the Company were approximately $74.8 million. Contemporaneously with the offering, each outstanding share of preferred stock was automatically converted into two shares of common stock.
| (b) | Preferred Stock |
The Company is authorized to issue 20,000,000 shares of preferred stock. The Board of Directors is authorized to divide these shares into classes or series, and to fix and determine the relative rights, preferences, qualifications and limitations of the shares of any class or series so established. At December 31, 2011 and 2010, there was no preferred stock outstanding.
| (c) | Common Stock |
During 2011, 2010 and 2009, the Company issued 10,144, 5,636 and 2,750 shares of common stock, respectively, valued at $59,000, $56,000 and $39,750, respectively, as compensation for services, which is included in general and administrative expenses in the consolidated statements of income. The Company issued 2,924,202 shares of common stock to a key executive and an advisor upon exercise of options in 2009.
The payment of dividends is restricted by the Agreement with the Company’s primary lender.
F-19
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (d) | Warrants |
In 2003, the Company issued warrants to purchase 25,000 shares of common stock at an exercise price of $6.00 per share as partial consideration for a modification to its line of credit. The warrants expire 10 years from the date of issuance. All of these warrants were outstanding and exercisable as of December 31, 2011 and 2010.
In connection with the issuance of shares of common stock to a related party in 2004, the Company issued warrants to purchase 40,000 shares of common stock at $6.00 per share at any time within ten years of issuance. All of these warrants were outstanding and exercisable as of December 31, 2011 and 2010.
In 2006, the Company signed a new line of credit agreement along with a term loan agreement with a financial institution. In conjunction with these agreements, the Company issued warrants to purchase up to 3,958 shares of common stock at $9.00 per share that expire in April 2016, which were outstanding and exercisable as of December 31, 2011 and 2010. In connection with the amendment to the debt agreements in 2009, the Company issued warrants to purchase up to 7,500 shares of common stock at $17.00 per share that expire in July 2019.
| (e) | Share Repurchases |
In February and April 2010, the Company repurchased 163,022 shares of common stock totaling approximately $1.9 million for the settlement of tax liabilities associated with the exercise of certain stock options in 2009. The repurchase amount was based on the fair-market value of common stock on the date of settlement.
In May 2010, the Company announced a share repurchase program to repurchase up to $10.0 million of its outstanding common shares. In January 2011, the Company’s Board of Directors modified the repurchase program to provide for the repurchase of $10.0 million of outstanding common stock, in addition to the amount repurchased in 2010. Pursuant to the plan, the Company repurchased 678,073 and 452,433 shares of common stock for approximately $3.9 million and $3.0 million during the year ended December 31, 2011 and 2010, respectively. In addition, the Company purchased 65,000 outside of the plan during the year ended December 31, 2011 at the then-current fair market value of common stock.
| (9) | Stock-Based Compensation Plans |
The Company has grants outstanding under three equity compensation plans, with two available for future grants of equity compensation awards to employees, consultants and directors. All of the equity plans were approved by shareholders. The 2007 Long-Term Incentive Compensation Plan (the 2007 Plan) and the 2007 Directors’ Incentive Plan (the Directors’ Plan) superseded the 1999 Stock Option Plan. The 2007 Plan and the Directors’ Plan provide for the issuance of stock options, stock appreciation rights and restricted stock. Vesting is determined on a grant-by-grant basis in accordance with the terms of the plans and the related grant agreements. The Company has reserved 2.4 million shares of common stock for issuance under the 2007 Plan and 250,000 shares for issuance under the Directors’ Plan.
The exercise price of stock options is generally 100% of the fair market value of the underlying common stock on the grant date. The exercise price of incentive stock options granted to a shareholder who owns more than 10% of the total combined voting power of all classes of stock must be at least 110% of the fair market value of the underlying common stock on the grant date. The maximum contractual term of stock options is ten years from the date of grant, except for incentive stock options granted to 10% shareholders, which are five years.
F-20
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
During 2011, the Company began issuing shares of restricted stock with no exercise price to employees and directors. Restricted stock issued to employees generally cliff-vests on the fourth anniversary of the date of grant. Restricted stock issued to directors vests on the one-year anniversary of the date of grant.
Stock compensation expense is presented as a component of general and administrative expense in the accompanying consolidated statements of income. At December 31, 2011, there was approximately $1.1 million of unrecognized compensation cost related to share-based payments, which is expected to be recognized over a weighted-average period of 2.6 years. This amount relates primarily to unrecognized compensation cost for employees.
Stock Options
Stock option activity for the year ended December 31, 2011 was as follows:
| Number of shares |
Weighted- average exercise price per share |
Weighted- average remaining contractual term (years) |
Aggregate intrinsic value |
|||||||||||||
| Outstanding, December 31, 2010 |
1,905,470 | $ | 6.45 | |||||||||||||
| Options granted |
— | — | ||||||||||||||
| Options exercised |
(503,411 | ) | 2.19 | |||||||||||||
| Options forfeited/expired |
(125,901 | ) | 10.24 | |||||||||||||
|
|
|
|||||||||||||||
| Outstanding, December 31, 2011 |
1,276,158 | 7.75 | 2.9 | $ | 700,294 | |||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2011 |
1,161,809 | $ | 7.34 | 2.8 | $ | 699,899 | ||||||||||
Information related to the stock option plans during 2011, 2010 and 2009 was as follows:
| 2011 | 2010 | 2009 | ||||||||||
| Intrinsic value of options exercised |
$ | 1,742,103 | $ | 5,519,588 | $ | 86,155,328 | ||||||
| Weighted-average fair value of equity granted |
$ | 2.06 | $ | 4.13 | $ | 6.42 | ||||||
The fair value of employee options granted during 2010 and 2009 was estimated using the Black-Scholes option-pricing model and the following assumptions:
| 2010 | 2009 | |||
| Dividend yield |
— | — | ||
| Expected term (years) |
2.5 – 6.0 | 3.7 – 6.2 | ||
| Expected volatility |
49% – 53% | 50% – 52% | ||
| Risk-free interest rate |
0.8% – 2.8% | 1.4% – 2.7% |
F-21
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
The fair value of nonemployee options granted during 2010 and 2009 were estimated using the Black-Scholes option-pricing model and the following assumptions:
| 2010 | 2009 | |||
| Dividend yield |
— | — | ||
| Expected term (years) |
5 | 2.3 – 10.0 | ||
| Expected volatility |
52% – 53% | 51% – 67% | ||
| Risk-free interest rate |
2.2% – 2.4% | 1.1% –2.7% |
The Company determined the expected life of employee share options based on the simplified method allowed by SEC Staff Accounting Bulletin (SAB) No. 107, as amended by SAB No. 110. Under this approach, the expected term is presumed to be the average between the weighted-average vesting period and the contractual term. The expected term for options granted to nonemployees is generally the contractual term of the option. The expected volatility over the term of the respective option was based on the volatility of similar publicly-traded entities. In evaluating similarity, the Company considered factors such as industry, stage of life cycle, size, and financial leverage. The risk-free interest rate is based on the U.S. Treasury Note, Stripped Principal, on the date of grant with a term substantially equal to the corresponding option’s expected term. The Company has never declared or paid any cash dividends and does not presently plan to pay cash dividends in the foreseeable future.
In the first quarter of 2009, options to purchase 773,556 shares of common stock were exercised with a weighted-average exercise price of $0.11 per share. A portion of the options were exercised using a net-share settlement feature that provided for the option holder to use 204,245 shares acquired upon exercise to settle the minimum statutory tax withholding requirements of approximately $2.7 million. During the third quarter of 2009, options to purchase 4,605,962 shares of common stock were exercised with a weighted-average exercise price of $0.55 per share. A portion of the options was exercised using a net-share settlement feature that provided for the option holder to use 1,445,074 shares acquired upon exercise to settle the minimum statutory tax withholding requirements of approximately $24.6 million. The payment of the exercise price for these options of approximately $2.6 million was settled by cash and the tendering of 140,788 shares of common stock by the optionees. In connection with these exercises, the Company agreed to repurchase up to $1.9 million in common stock during the first quarter of 2010 to provide for the settlement of the remaining tax liabilities associated with the exercise. The repurchase of these shares was completed in 2010.
Restricted Stock Awards
As previously noted, the Company began issuing restricted stock to employees and directors in 2011 under the provisions of the 2007 Plan and the Directors’ Plan. Restricted stock activity during 2011 was as follows:
| Number of shares |
Weighted- average grant-date fair value |
|||||||
| Nonvested, December 31, 2010 |
— | $ | — | |||||
| Shares granted |
149,320 | 5.40 | ||||||
| Shares vested |
(1,000 | ) | 5.28 | |||||
| Shares forfeited |
(12,150 | ) | 5.28 | |||||
|
|
|
|||||||
| Nonvested, December 31, 2011 |
136,170 | 5.41 | ||||||
|
|
|
|||||||
The fair value of restricted stock granted during 2011 was based on the closing market price of the Company’s common stock on the date of grant.
F-22
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
| (10) | Leases |
The Company is obligated under long-term real estate leases for corporate office space expiring in October 2016. In addition, the research lab space at CET is leased through 2016, with an option to extend the lease through July 2021. The Company also subleases a portion of the space under these leases. Rent expense is recognized over the expected term of the lease, including renewal option periods, if applicable, on a straight-line basis. Rent expense for 2011, 2010 and 2009 was approximately $0.8 million, $0.6 million and $0.6 million, respectively, and sublease income was approximately $0.4 million, $0.3 million and $0.2 million. Future minimum sublease income under noncancelable sublease operating leases is approximately $0.8 million through October 2016. Future minimum lease payments under noncancelable operating leases (with initial or remaining lease terms in excess of one year) are:
| Year ending December 31: |
||||
| 2012 |
$ | 860,279 | ||
| 2013 |
886,187 | |||
| 2014 |
912,693 | |||
| 2015 |
940,057 | |||
| 2016 and thereafter |
769,352 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 4,368,568 | ||
|
|
|
| (11) | Manufacturing and Supply Agreements |
The Company utilizes one primary supplier to manufacture each of its respective products and product candidates. In February 2008, the Company entered into an agreement with a second supplier of Acetadote. The agreement for the second supplier expires in February 2013. Although there are a limited number of manufacturers of pharmaceutical products, the Company believes it could utilize other suppliers to manufacture its prescription products on comparable terms. A change in suppliers, any problems with such manufacturing operations or capacity, or contract disputes with the suppliers, however, could cause a delay in manufacturing and a possible loss of sales, which would adversely affect operating results.
| (12) | Commitments and Contingencies |
In connection with the acquisition of certain Kristalose assets during 2011, the Company is required to make quarterly payments based on a percentage of Kristalose net sales through November 2018. The payments are being treated as consideration for the assets acquired, and are being capitalized and amortized over the remaining expected useful life of the acquired asset, generally 15 years.
In connection with its licensing agreements for Caldolor, the Company is required to pay a royalty based on Caldolor net sales over the life of the contracts. Royalty expense is recognized as a component of selling and marketing expense in the period that revenue is recognized.
| (13) | Employment Agreements |
The Company has entered into employment agreements with its full-time and part-time employees. Each employment agreement provides for a salary for services performed, a potential annual bonus and, if applicable, a grant of restricted common shares pursuant to a restricted stock agreement. Four of the employment agreements address expense reimbursements for relevant and applicable licenses and continuing education. Employment agreements are amended each successive one-year period, unless terminated.
| (14) | Market Concentrations |
The Company currently focuses on acquiring, developing, and commercializing branded prescription products for the acute care and gastroenterology markets. The Company’s principal financial instruments subject to potential concentration of credit risk are accounts receivable, which are unsecured, and cash equivalents. The Company’s cash equivalents consist primarily of money market funds. Certain bank deposits may at times be in excess of the Federal Deposit Insurance Corporation (FDIC) insurance limits.
F-23
Table of Contents
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements — (Continued)
The Company’s primary customers are wholesale pharmaceutical distributors in the U.S. Total revenues from customers representing 10% or more of total revenues for the respective years are summarized as follows:
| 2011 | 2010 | 2009 | ||||||||||
| Customer 1 |
36 | % | 35 | % | 37 | % | ||||||
| Customer 2 |
28 | 31 | 29 | |||||||||
| Customer 3 |
31 | 26 | 27 | |||||||||
Additionally, 95% and 80% of the Company’s accounts receivable balances were due from these three customers at December 31, 2011 and 2010, respectively.
| (15) | Employee Benefit Plan |
The Company sponsors an employee benefit plan that was established on January 1, 2006, the Cumberland Pharmaceuticals 401(k) Plan (the Plan), under Section 401(k) of the Internal Revenue Code of 1986, as amended, for the benefit of all employees over the age of 21, having been employed by the Company for at least six months. The Plan provides that participants may contribute up to the maximum amount of their compensation as set forth by the Internal Revenue Service each year. Employee contributions are invested in various investment funds based upon elections made by the employees. During 2011 and 2010, the Company contributed less than $50,000 per year to the Plan as an employer match of participant contributions.
| (16) | Quarterly Financial Information (Unaudited) |
The following table sets forth the unaudited operating results for each fiscal quarter of 2011 and 2010:
| First | Second | Third | Fourth | |||||||||||||||||
| Quarter | Quarter | Quarter | Quarter | Total | ||||||||||||||||
| 2011 |
||||||||||||||||||||
| Net revenues |
$ | 10,666,927 | $ | 14,389,741 | $ | 13,054,278 | $ | 13,031,830 | $ | 51,142,776 | ||||||||||
| Operating income |
1,408,001 | 3,631,857 | 3,098,360 | 1,711,269 | 9,849,487 | |||||||||||||||
| Net income attributableto common shareholders |
721,160 | 2,177,619 | 1,847,412 | 911,665 | 5,657,856 | |||||||||||||||
| Earnings per share attributableto common shareholders (1) |
||||||||||||||||||||
| - Basic |
$ | 0.04 | $ | 0.11 | $ | 0.09 | $ | 0.05 | $ | 0.28 | ||||||||||
| - Diluted |
$ | 0.03 | $ | 0.11 | $ | 0.09 | $ | 0.04 | $ | 0.28 | ||||||||||
| 2010 |
||||||||||||||||||||
| Net revenues |
$ | 10,130,652 | $ | 10,739,935 | $ | 12,190,870 | $ | 12,814,914 | $ | 45,876,371 | ||||||||||
| Operating income |
810,508 | 1,009,860 | 2,443,857 | 2,237,522 | 6,501,747 | |||||||||||||||
| Net income attributableto common shareholders |
323,578 | 287,304 | 1,008,244 | 837,554 | 2,456,680 | |||||||||||||||
| Earnings per share attributableto common shareholders (1) |
||||||||||||||||||||
| - Basic |
$ | 0.02 | $ | 0.01 | $ | 0.05 | $ | 0.04 | $ | 0.12 | ||||||||||
| - Diluted |
$ | 0.02 | $ | 0.01 | $ | 0.05 | $ | 0.04 | $ | 0.12 | ||||||||||
| (1) | Due to the nature of interim earnings per share calculations, the sum of the quarterly earnings per share amounts may not equal the reported earnings per share for the year. |
F-24
Table of Contents
Schedule II
CUMBERLAND PHARMACEUTICALS INC. AND SUBSIDIARIES
Valuation and Qualifying Accounts
Years ended December 31, 2011, 2010 and 2009
| Column A |
Column B | Column C | Column D | Column E | ||||||||||||||||
| Description |
Balance at beginning of period |
Charged to costs and expenses |
Charged to other accounts – describe |
Deductions – describe (1) |
Balance at end of period |
|||||||||||||||
| Allowance for uncollectible amounts, cash discounts, chargebacks, and credits issued for damaged products: |
||||||||||||||||||||
| For the period ended: |
||||||||||||||||||||
| December 31, 2009 |
$ | 147,046 | $ | 1,734,521 | $ | — | $ | (1,646,287 | ) | $ | 235,280 | |||||||||
| December 31, 2010 |
235,280 | 1,494,834 | — | (1,566,366 | ) | 163,748 | ||||||||||||||
| December 31, 2011 |
163,748 | 2,151,890 | — | (2,080,058 | ) | 235,580 | ||||||||||||||
| Valuation allowance for deferred tax assets: |
||||||||||||||||||||
| For the period ended: |
||||||||||||||||||||
| December 31, 2009 |
$ | 58,770 | $ | 11,342 | $ | — | $ | — | $ | 70,112 | ||||||||||
| December 31, 2010 |
70,112 | 10,750 | — | — | 80,862 | |||||||||||||||
| December 31, 2011 |
80,862 | 13,597 | — | — | 94,459 | |||||||||||||||
See accompanying report of independent registered public accounting firm.
F-25