Attached files
| file | filename |
|---|---|
| EX-21.01 - LIST OF SUBSIDIARIES - DEXCOM INC | dex2101.htm |
| EX-23.01 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - DEXCOM INC | dex2301.htm |
| EX-31.02 - CERTIFICATION OF CFO - DEXCOM INC | dex3102.htm |
| EX-32.02 - CERTIFICATION OF CFO - DEXCOM INC | dex3202.htm |
| EX-32.01 - CERTIFICATION OF CEO - DEXCOM INC | dex3201.htm |
| EX-31.01 - CERTIFICATION OF CEO - DEXCOM INC | dex3101.htm |
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2010
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 000-51222
DEXCOM, INC.
(Exact name of Registrant as Specified in its Charter)
| Delaware | 33-0857544 | |
| (State or Other Jurisdiction of Incorporation or Organization) |
(I.R.S. Employer Identification No.) | |
| 6340 Sequence Drive San Diego, California |
92121 | |
| (Address of Principal Executive offices) | (Zip Code) | |
Registrant’s Telephone Number, including area code: (858) 200-0200
Securities registered pursuant to Section 12(b) of the Exchange Act:
| Title of Each Class |
Name of Each Exchange on Which Registered | |
| Common Stock, $0.001 Par Value Per Share | The NASDAQ Stock Market LLC (Nasdaq Global Market) | |
| Preferred Stock Purchase Rights | The NASDAQ Stock Market LLC (Nasdaq Global Market) |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes x No ¨
Indicate by check mark if the Registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act.
Yes ¨ No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes ¨ No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Rule 405 of Regulation S-K is not contained herein, and will not be contained, to the best of Registrant’s knowledge, in definite proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “Smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one)
Large accelerated Filer ¨ Accelerated Filer x Non-accelerated Filer ¨ Smaller reporting company ¨
Indicate by check mark whether the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ¨ No x
As of June 30, 2010, the aggregate market value of the registrant’s common stock held by non-affiliates of the registrant was approximately $657,285,311 based on the closing sales price as reported on the NASDAQ Global Market.
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date.
| Class |
Outstanding at February 28, 2011 | |
| Common stock, $0.001 par value per share | 62,208,159 shares |
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the documents listed below have been incorporated by reference into the indicated parts of this report, as specified in the responses to the item numbers involved.
Designated portions of the Proxy Statement relating to the 2011 Annual Meeting of the Stockholders (the “Proxy Statement”): Part III (Items 9, 10, 11, 12, and 13). Except with respect to information specifically incorporated by reference in the Form 10-K, the Proxy Statement is not deemed to be filed as part hereof.
Table of Contents
Table of Contents
| Page Number |
||||||
| PART I |
||||||
| ITEM 1. |
1 | |||||
| ITEM 1A. |
23 | |||||
| ITEM 1B. |
43 | |||||
| ITEM 2. |
43 | |||||
| ITEM 3. |
43 | |||||
| PART II |
||||||
| ITEM 5. |
45 | |||||
| ITEM 6. |
46 | |||||
| ITEM 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
47 | ||||
| ITEM 7A. |
59 | |||||
| ITEM 8. |
60 | |||||
| ITEM 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
61 | ||||
| ITEM 9A. |
61 | |||||
| ITEM 9B. |
62 | |||||
| PART III |
||||||
| ITEM 10. |
63 | |||||
| ITEM 11. |
63 | |||||
| ITEM 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholders Matters |
63 | ||||
| ITEM 13. |
Certain Relationships and Related Transactions, and Director Independence |
63 | ||||
| ITEM 14. |
63 | |||||
| PART IV |
||||||
| ITEM 15. |
64 | |||||
Table of Contents
PART I
Except for historical financial information contained herein, the matters discussed in this Form 10-K may be considered forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, and subject to the safe harbor created by the Securities Litigation Reform Act of 1995. Such statements include declarations regarding our intent, belief, or current expectations and those of our management. Prospective investors are cautioned that any such forward-looking statements are not guarantees of future performance and involve a number of risks, uncertainties and other factors, some of which are beyond our control; actual results could differ materially from those indicated by such forward-looking statements. Important factors that could cause actual results to differ materially from those indicated by such forward-looking statements include, but are not limited to: (i) that the information is of a preliminary nature and may be subject to further adjustment; (ii) those risks and uncertainties identified under “Risk Factors;” and (iii) the other risks detailed from time-to-time in our reports and registration statements filed with the Securities and Exchange Commission, or SEC. Except as required by law, we undertake no obligation to revise or update publicly any forward-looking statements, whether as a result of new information, future events or otherwise.
| ITEM 1. | BUSINESS |
Overview
We are a medical device company focused on the design, development and commercialization of continuous glucose monitoring systems for ambulatory use by people with diabetes and for use by healthcare providers in the hospital for the treatment of both diabetic and non-diabetic patients.
Ambulatory Product Line: SEVEN® PLUS
We received approval from the Food and Drug Administration, or FDA, and commercialized our first product in 2006. In 2007, we received approval and began commercializing our second generation system, the SEVEN, and on February 13, 2009, we received approval for our third generation system, the SEVEN PLUS, which is designed for up to seven days of continuous use, and we began commercializing this product in the first quarter of 2009. We no longer market or provide support for the SEVEN system. There are various differences between the SEVEN and the SEVEN PLUS. As compared to the SEVEN, the SEVEN PLUS incorporates additional user interface and algorithm enhancements that are intended to make its glucose monitoring function more accurate and customizable. The approval of the SEVEN PLUS by the FDA allows for the use of the SEVEN PLUS by adults with diabetes to detect trends and track glucose patterns, to aid in the detection of hypoglycemia and hyperglycemia and to facilitate acute and long-term therapy adjustments.
In-Hospital Product Line: GlucoClear®
To address the in-hospital patient population, we entered into an exclusive agreement with Edwards Lifesciences LLC, or Edwards, to develop jointly and market a specific product platform for the in-hospital glucose monitoring market, with an initial focus on the development of an intravenous sensor specifically for the critical care market. On October 30, 2009, we received CE Mark approval for our first generation blood-based in-vivo automated glucose monitoring system, which we have branded the GlucoClear, for use by healthcare providers in the hospital, and are continuing to seek approval for this system from the FDA. In partnership with Edwards, we initiated a very limited launch of the GlucoClear system in Europe in 2009.
Background
From inception to 2006, we devoted substantially all of our resources to start-up activities, raising capital and research and development, including product design, testing, manufacturing and clinical trials. Since 2006,
1
Table of Contents
we have devoted considerable resources to the commercialization of our ambulatory continuous glucose monitoring systems, including the SEVEN PLUS, as well as the continued research and clinical development of our technology platform.
The International Diabetes Federation, or IDF, estimates that 285 million people around the world have diabetes, and the Centers for Disease Control, or CDC, estimates that diabetes affects 25.8 million people in the United States. IDF estimates that by 2030, the worldwide incidence of people suffering from diabetes will reach 438.0 million. The increased prevalence of diabetes is believed to be the result of an aging population, unhealthy diets and increasingly sedentary lifestyles. According to the CDC, diabetes was the seventh leading cause of death by disease in the United States during 2007, and complications related to diabetes include heart disease, limb amputations, loss of kidney function and blindness.
According to a CDC spokesman cited in a New York Times article, one in every three children born in the United States in 2001 was expected to become diabetic in their lifetimes, and every day in the United States, on average, there would be 4,100 people diagnosed with diabetes, 230 people undergoing amputations as a result of diabetes, 120 people who enter end-stage kidney disease programs and 55 people who lose their vision.
According to the American Diabetes Association, or ADA, one in every ten health care dollars was spent on treating diabetes in 2007, and the direct medical costs and indirect expenditures attributable to diabetes in the United States were an estimated $174 billion, an increase of $42 billion since 2002. Of the $174 billion in overall expenses, the ADA estimated that approximately $89 billion were costs associated with chronic complications and excess general medical costs, $27 billion were costs associated with diabetes care and $58 billion were indirect medical costs. The ADA also found that average medical expenditures among people with diagnosed diabetes were 2.3 times higher than for people without diabetes.
We believe continuous glucose monitoring has the potential to enable more people with diabetes to achieve and sustain tight glycemic control. The Diabetes Control and Complications Trial (DCCT) demonstrated that improving blood glucose control lowers the risk of developing diabetes related complications by up to 50%. The study also demonstrated that people with Type 1 diabetes achieved sustained benefits with intensive management. Yet, according to an article published in the Journal of the American Medical Association (JAMA) in 2004, less than 50% of diabetes patients were meeting ADA standards for glucose control (A1c), and only 37% of people with diabetes were achieving their glycemic targets. The CDC estimated that as of 2006, 63.4% of all adults with diabetes were monitoring their blood glucose levels on a daily basis, and that 86.7% of insulin-requiring patients with diabetes monitored daily.
Various clinical studies also demonstrate the benefits of continuous glucose monitoring and that continuous glucose monitoring is equally effective in patients who administer insulin through multiple daily injections or through use of continuous subcutaneous insulin infusion pumps. Results of a Juvenile Diabetes Research Foundation (JDRF) study published in the New England Journal of Medicine in 2008, and the extension phase of the study, published in Diabetes Care in 2009, demonstrated that continuous glucose monitoring improved A1c levels and reduced incidence of hypoglycemia for patients over the age of 25 and for all patients of all ages who utilized continuous glucose monitoring regularly.
Our initial target market in the United States consists of an estimated 30% of people with Type 1 diabetes who utilize insulin pump therapy and an estimated 50% of people with Type 1 diabetes who utilize multiple daily insulin injections. Our broader target market in the United States consists of our initial target market plus an estimated 20% of people with Type 1 diabetes using conventional insulin therapy and the 27% of people with Type 2 diabetes who require insulin. Although our initial focus is within the United States, our CE Mark approval also enables us to commercialize our system in those European, Asian and Latin American countries that recognize the CE Mark.
2
Table of Contents
Close Concerns, Inc., a healthcare information firm exclusively focused on diabetes and obesity, founded dQ&A Market Research Inc., a market research business with over 3,000 panel members that participate in diabetes related surveys. A dQ&A Panel Summary Report from February 2011 estimated that our share of the continuous glucose monitoring system market in the United States was at 48%. The report analyzed responses from 382 panel members who were asked what brand and model of continuous glucose monitoring system they used.
We have built a direct sales organization to call on endocrinologists, physicians and diabetes educators who can educate and influence patient adoption of continuous glucose monitoring. We believe that focusing efforts on these participants is important given the instrumental role they each play in the decision-making process for diabetes therapy. To complement our direct sales efforts, we also employ clinical specialists who educate and provide clinical support in the field, and we have entered into a limited number of distribution arrangements that allow distributors to sell our products. Although we plan to modestly increase the size of the sales organization in 2011, we believe our direct, highly-specialized and focused sales organization is sufficient for us to support our sales efforts.
We are leveraging our technology platform to enhance the capabilities of our current products and to develop additional continuous glucose monitoring products. In January 2008, we entered into two separate development agreements, one with Animas Corporation, or Animas, a subsidiary of Johnson & Johnson, and one with Insulet Corporation, or Insulet, to integrate our technology into the insulin pump product offerings of the respective partner, enabling the partner’s insulin pump to receive glucose readings from our transmitter and display this information on the pump’s screen. In addition, we are continuing to seek approval for our next generation ambulatory system, and are responding to FDA’s requests for additional data in support of that application. We expect our next generation system will further improve sensor reliability, stability and accuracy over the useful life of the sensor, and will be suited for large scale manufacturing. We also intend to seek approval for a pediatric indication (patients under 18 years of age) and a pregnancy indication (patients who develop gestational diabetes) for our product platform in the future. Further, as described above, we are developing in collaboration with Edwards the GlucoClear, which is a blood-based in-vivo automated glucose monitoring system for use by healthcare providers in the hospital. Our development timelines are highly dependent on our ability to achieve clinical endpoints and regulatory requirements and to overcome technology challenges, and our development timelines may be delayed due to extended regulatory approval timelines, scheduling issues with patients and investigators, requests from institutional review boards, sensor performance and manufacturing supply constraints, among other factors. In addition, support of these clinical trials requires significant resources from employees involved in the production of our products, including research and development, manufacturing, quality assurance, and clinical and regulatory personnel. Even if our development and clinical trial efforts are successful, the FDA may not approve our products, and if approved, we may not achieve acceptance in the marketplace by physicians and patients.
As a medical device company, reimbursement from Medicare and private third-party healthcare payors is an important element of our success. Although the Centers for Medicare and Medicaid, or CMS, released 2008 Alpha-Numeric Healthcare Common Procedure Coding System (“HCPCS”) codes applicable to each of the three components of our continuous glucose monitoring systems, to date, our approved products are not reimbursed by virtue of a national coverage decision by Medicare. It is not known when, if ever, Medicare will adopt a national coverage decision with respect to continuous glucose monitoring devices. Until any such coverage decision is adopted by Medicare, reimbursement of our products will generally be limited to those patients covered by third-party payors that have adopted coverage policies for continuous glucose monitoring devices. As of March 2011, the seven largest private third-party payors, in terms of the number of covered lives, have issued coverage policies for the category of continuous glucose monitoring devices. In addition, we have negotiated contracted rates with six of those third-party payors for the purchase of our SEVEN PLUS system by their members. Many of these coverage policies are restrictive in nature and require the patient to comply with extensive documentation and other requirements to demonstrate medical necessity under the policy. In addition, patients
3
Table of Contents
who are insured by payors that do not offer coverage for our devices will have to bear the financial cost of the products. We currently employ in-house reimbursement expertise to assist patients in obtaining reimbursement from private third-party payors. We also maintain a field-based reimbursement team charged with calling on third-party private payors to obtain coverage decisions and contracts. We have had formal meetings and have increased our efforts to create and liberalize coverage policies with third-party payors and expect to continue to do so in 2011. However, unless government and other third-party payors provide adequate coverage and reimbursement for our products, patients may not use them on a widespread basis.
We plan to develop future generations of technologies focused on improved performance and convenience and that will enable intelligent insulin administration. Our next generation of technologies are not yet FDA approved, but in the near term, we are seeking regulatory approval for a next generation sensor platform using advanced manufacturing processes that are more scalable and reliable. Over the longer term, we plan to develop networked platforms with open architecture, connectivity and transmitters capable of communicating with other devices.
Market Opportunity
Diabetes
Diabetes is a chronic, life-threatening disease for which there is no known cure. The disease is caused by the body’s inability to produce or effectively utilize the hormone insulin. This inability prevents the body from adequately regulating blood glucose levels. Glucose, the primary source of energy for cells, must be maintained at certain concentrations in the blood in order to permit optimal cell function and health. Normally, the pancreas provides control of blood glucose levels by secreting the hormone insulin to decrease blood glucose levels when concentrations are too high. In people with diabetes, the body does not produce sufficient levels of insulin, or fails to utilize insulin effectively, causing blood glucose levels to rise above normal. This condition is called hyperglycemia and often results in chronic long-term complications such as heart disease, limb amputations, loss of kidney function and blindness. When blood glucose levels are high, patients often administer insulin in an effort to decrease blood glucose levels. Unfortunately, insulin administration can drive blood glucose levels below the normal range, resulting in hypoglycemia. In cases of severe hypoglycemia, diabetes patients risk acute complications, such as loss of consciousness or death. Due to the drastic nature of acute complications associated with hypoglycemia, many patients are reluctant to reduce blood glucose levels. Consequently, these patients often remain in a hyperglycemic state, increasing their odds of developing long-term chronic complications.
Diabetes is typically classified into two major groups: Type 1 and Type 2. We estimate that there are approximately 1.3 million Type 1 diabetes patients in the United States. Type 1 diabetes usually develops during childhood and is characterized by an absence of insulin, resulting from destruction of the insulin producing cells of the pancreas. Individuals with Type 1 diabetes must rely on frequent insulin injections in order to regulate and maintain blood glucose levels. We estimate that there are approximately 24.5 million people with Type 2 diabetes in the United States, which results when the body is unable to produce sufficient amounts of insulin or becomes insulin resistant. Depending on the severity of Type 2 diabetes, individuals may require diet and nutrition management, exercise, oral medications or insulin injections to regulate blood glucose levels. We estimate that approximately 3.6 million Type 2 patients must use insulin to manage their diabetes.
There are various subgroups of diabetic patients, including in-hospital patients, who present significant management challenges. According to the ADA, diabetes related hospitalizations totaled 24.3 million days in 2007, an increase of 7.4 million days from 2002. Additionally, studies show that many non-diabetic hospital patients suffer episodes of hyperglycemia. According to a Diabetes Care article, as of 1998, as many as 1.5 million hospitalized patients had significant hyperglycemia without a history of diabetes. A November 2001 article in the New England Journal of Medicine summarized a study of over 1,500 hospitalized patients, of which only 13% were diabetic, which concluded that intensive insulin therapy to maintain blood glucose levels within a target range reduced mortality among critically ill patients in the surgical intensive care unit and improved patient outcomes.
4
Table of Contents
According to the National Diabetes Education Program, about 75% of all newly diagnosed cases of Type 1 diabetes in the United States occur in juveniles younger than 18 years of age. In addition, Type 2 diabetes is occurring with increasing frequency in young people. The increase in prevalence is related to an increase in obesity amongst children. As of 2002, approximately 16% of children and teens were overweight, about double the number two decades before.
Importance of Glucose Monitoring
Blood glucose levels can be affected by many factors, including the carbohydrate and fat content of meals, exercise, stress, illness or impending illness, hormonal releases, variability in insulin absorption and changes in the effects of insulin in the body. Given the many factors that affect blood glucose levels, maintaining glucose within a normal range is difficult, resulting in frequent and unpredictable excursions above or below normal blood glucose levels. Patients manage their blood glucose levels by administering insulin or ingesting carbohydrates throughout the day in order to maintain blood glucose within normal ranges. Patients frequently overcorrect and fluctuate between hyperglycemic and hypoglycemic states, often multiple times during the same day. As a result, many patients with diabetes are routinely outside the normal blood glucose range. Patients are often unaware that their glucose levels are either too high or too low, and their inability to completely control blood glucose levels and the associated serious complications can be frustrating and, at times, overwhelming.
In an attempt to maintain blood glucose levels within the normal range, patients with diabetes must first measure their blood glucose levels. Often after measuring their blood glucose levels, patients make therapeutic adjustments. As adjustments are made, additional blood glucose measurements may be necessary to gauge the individual’s response to the adjustments. More frequent testing of blood glucose levels provides patients with information that can be used to better understand and manage their diabetes. The ADA recommends that patients test their blood glucose levels at least three or four times per day.
Clinical outcomes data support the notion that an important component of effective diabetes management is frequent monitoring of blood glucose levels. The landmark 1993 Diabetes Control and Complications Trial, or DCCT, consisting of patients with Type 1 diabetes, and the 1998 UK Prospective Diabetes Study, consisting of patients with Type 2 diabetes, demonstrated that patients who intensely managed blood glucose levels delayed the onset and slowed the progression of diabetes-related complications. In the DCCT, a major component of intensive management was monitoring blood glucose levels at least four times per day using conventional single-point blood glucose meters. The DCCT demonstrated that intensive management reduced the risk of complications by 76% for eye disease, 60% for nerve disease and 50% for kidney disease. However, the DCCT also found that intensive management led to a three-fold increase in the frequency of hypoglycemic events. In the December 2005 edition of the New England Journal of Medicine, the authors of a peer-reviewed study concluded that intensive diabetes therapy has long-term beneficial effects on the risk of cardiovascular disease in patients with Type 1 diabetes. The study showed that intensive diabetes therapy reduced the risk of cardiovascular disease by 42% and the risk of non-fatal heart attack, stroke or death from cardiovascular disease by 57%.
Limitations of Existing Glucose Monitoring Products
Single-point finger stick devices are the most prevalent devices for glucose monitoring. These devices require taking a blood sample with a finger stick, placing a drop of blood on a test strip and inserting the strip into a glucose meter that yields a single point in time blood glucose measurement. We believe that these devices suffer from several limitations, including:
| • | Limited Information. Even if patients test several times each day, each measurement represents a single blood glucose value at a single point in time. Given the many factors that can affect blood glucose levels, excursions above and below the normal range often occur between these discrete measurement points in time. Because patients only have single-point data, they do not gain sufficient information to indicate the direction or rate of change in their blood glucose levels. Without the ability |
5
Table of Contents
| to determine whether their blood glucose level is rising, falling or holding constant, and the rate at which their blood glucose level is changing, the patient’s ability to effectively manage and maintain blood glucose levels within normal ranges is severely limited. In addition, patients cannot test themselves during sleep, when the risk of hypoglycemia is significantly increased. |
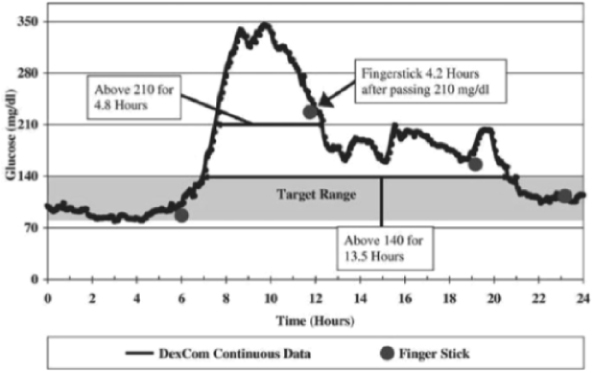
The following graph shows the limited information provided by four single-point measurements during a single day using a traditional single-point finger stick device, compared to the data provided by our continuous sensor. The data presented in the graph is from a clinical trial we completed in 2003 with a continuous glucose monitoring system, where the patient was blinded to the continuous glucose data. The continuous data indicates that, even with four finger sticks in one day, the patient’s blood glucose levels were above the target range of 80-140 mg/dl, or milligrams per deciliter, for a period of 13.5 hours.
Single Day Continuous Data
| • | Inconvenience. The process of measuring blood glucose levels with single-point finger stick devices can cause significant disruption in the daily activities of people with diabetes and their families. Patients using single-point finger stick devices must stop whatever they are doing several times per day, self-inflict a painful prick and draw blood to measure blood glucose levels. To do so, patients must always carry a fully-supplied kit that may include a spring-loaded needle, or lancet, disposable test strips, cleansing wipes, and the meter, and then safely dispose of the used supplies. This process is inconvenient and may cause uneasiness in social situations. |
| • | Difficulty of Use. To obtain a sample with single-point finger stick devices, patients generally prick one of their fingertips or, occasionally, a forearm with a lancet. Patients then squeeze the area to produce the blood sample and another prick may be required if a sufficient volume of blood is not obtained the first time. The blood sample is then placed on a disposable test strip that is inserted into a blood glucose meter. This task can be difficult for patients with decreased tactile sensation and visual acuity, which are common complications of diabetes. |
6
Table of Contents
| • | Pain. Although the fingertips are rich in blood flow and provide a good site to obtain a blood sample, they are also densely populated with highly sensitive nerve endings. This makes the lancing and subsequent manipulation of the finger to draw blood painful. The pain and discomfort are compounded by the fact that fingers offer limited surface area, so tests are often performed on areas that are sore from prior tests. Patients may also suffer pain when the finger prick site is disturbed during regular activities. |
We believe a market opportunity exists for a glucose monitoring system that provides continuous glucose information, including trends, and that is convenient and easy to use. Several companies have attempted to address the limitations of single-point finger stick devices by developing continuous glucose monitoring systems. To date, in addition to DexCom, we are aware of three other companies, Cygnus, Medtronic and Abbott, that have received approval from the FDA to market continuous glucose monitors. We believe that one of the products, originally developed and marketed by Cygnus, is no longer actively marketed. In addition, we believe others are developing invasive and non-invasive continuous glucose monitoring systems, including Bayer Corporation. Except for our SEVEN and SEVEN PLUS, we believe that none of the products that have received FDA approval are labeled for more than five days of use. We also believe that none of the products that have received FDA approval are labeled for use as a replacement for single-point finger stick devices.
The DexCom Solution
Our SEVEN PLUS system offers the following advantages to patients with diabetes:
| • | Improved Outcomes. Data published in a peer-reviewed article based on our approval support trial for our first system demonstrated that patients using the system showed statistically significant improvements in maintaining their glucose levels within the target range when compared to patients relying solely on single-point finger stick measurements. Additional peer-review published data from our approval support trial for the SEVEN demonstrated that patients with access to seven days of continuous glucose data statistically improved glucose control by further increasing their time spent with glucose levels in the target range, thereby reducing time spent in both hyperglycemic and hypoglycemic ranges. Peer-review published data from our repeated use trial demonstrated a statistically significant reduction in hemoglobin A1c levels, a measure of the average amount of glucose in the blood over the prior three months, in patients using our system compared to patients relying solely on single-point finger stick measurements. Finally, results of a major multicenter clinical trial funded by the Juvenile Diabetes Research Foundation demonstrated that patients with Type 1 diabetes who used continuous glucose monitoring devices to help manage their disease experienced significant improvements in glucose control. |
| • | Access to Real-Time Values, Trend Information and Alerts. At the push of a button, patients can view their current glucose value, along with a graphical display of one-, three-, six-, twelve- or twenty-four-hour trend information. Without continuous monitoring, the patient is often unaware if his or her glucose is rising, declining or remaining constant. Access to continuous real-time glucose measurements provides patients with information that may aid in attaining better glucose control. Additionally, our SEVEN PLUS alerts patients when their glucose levels approach inappropriately high or low levels so that they may intervene. |
| • | Intuitive Patient Interface. We have developed a patient interface that we believe is intuitive and easy to use. Our receiver’s ergonomic design includes user-friendly buttons, an easy-to-read display, simple navigation tools, audible alerts and graphical display of trend information. |
| • | Convenience and Comfort. Our SEVEN PLUS provides patients with the benefits of continuous monitoring, without having to perform finger stick tests for every measurement. Additionally, the disposable sensor electrode that is inserted under the skin is a very thin wire, minimizing potential discomfort associated with inserting or wearing the disposable sensor. The external portion of the sensor, including the transmitter, is small, has a low profile and is designed to be easily worn under |
7
Table of Contents
| clothing. The wireless receiver is the size of a small cell phone and can be carried discreetly in a pocket or purse. We believe that convenience is an important factor in achieving widespread adoption of a continuous glucose monitoring system. |
While we believe the SEVEN PLUS offers these advantages, patients may not perceive the benefits of continuous glucose monitoring and may be unwilling to change their current treatment regimens. Furthermore, we do not expect that our SEVEN PLUS will appeal to all types of patients with diabetes. The SEVEN PLUS prompts a patient to insert a disposable sensor electrode under their skin at least every seven days, although we are aware of reports from the field that some patients have been able to use sensors for periods longer than seven days. Patients could find this process to be uncomfortable or inconvenient. Patients may be unwilling to insert a disposable sensor in their body, especially if their current diabetes management involves no more than two finger sticks per day. Additionally, the SEVEN PLUS is not approved as a replacement device for single-point finger stick devices, must be calibrated initially using measurements from two single-point finger stick tests, and thereafter at least every 12 hours using single-point finger stick tests, and may be more costly to use.
Our Strategy
Our objective is to become the leading provider of continuous glucose monitoring systems and related products to enable people with diabetes to more effectively and conveniently manage their disease. We also seek to develop and commercialize products that integrate our continuous glucose monitoring technologies into the insulin pump delivery systems of Animas and Insulet, respectively. In addition, we seek to design, develop and commercialize, in collaboration with Edwards, the GlucoClear, which is a blood-based in-vivo automated glucose monitoring system for use by healthcare providers in the hospital for the treatment of both diabetic and non-diabetic patients. To achieve these objectives, we are pursuing the following business strategies:
| • | Establish our technology platform as the leading approach to continuous glucose monitoring and leverage our development expertise to rapidly bring products to market. We have developed proprietary core technology and expertise that provides a broad platform for the development of innovative products for continuous glucose monitoring. We received approval from the FDA and commercialized our first product in 2006. In 2007, we received approval and began commercializing our second generation system, the SEVEN, and on February 13, 2009, we received approval for our third generation system, the SEVEN PLUS, which is designed for up to seven days of continuous use, and we began commercializing this product in the first quarter of 2009. We plan to continue to invest in the development of our technology platform and to obtain additional FDA approvals for our continuous glucose monitoring systems for both the ambulatory and in-hospital markets as well as for our integrated insulin pump delivery systems. We expect to continue to provide performance improvements and introduce new products to establish and maintain a leadership position in the market. In the future, we may develop our technology to support applications beyond glucose sensing. |
| • | Drive the adoption of our ambulatory products through a direct sales and marketing effort. We have a small direct field sales force, which we plan to modestly expand in 2011, to call on endocrinologists, physicians and diabetes educators who can educate and influence patient adoption of continuous glucose monitoring. We believe that focusing efforts on these participants is important given the instrumental role they each play in the decision-making process for diabetes therapy. To complement our sales efforts, we employ clinical specialists who will educate and provide clinical support to patients, and have entered into distribution arrangements that allow distributors to sell our SEVEN PLUS. We currently sell the SEVEN PLUS only in the United States and in portions of Europe and Israel, but plan to expand our sales elsewhere in the future. |
| • | Drive additional adoption through technology integration partnerships. We have development agreements with Animas and Insulet to develop products that will integrate our ambulatory product technology into the Animas conventional insulin pump and the Insulet OmniPod System PDM, as applicable, enabling the partner’s insulin pump to receive glucose readings from our transmitter and |
8
Table of Contents
| display this information on the pump’s screen. We believe patients who have adopted continuous subcutaneous insulin infusion, or CSII, are patients who more aggressively manage their diabetes and may be more inclined to utilize our continuous glucose monitoring systems. |
| • | Seek broad coverage policies and reimbursement for our products. Our approved products are not reimbursed by virtue of a national coverage decision by Medicare. As of March 2011, the seven largest private third-party payors, in terms of the number of covered lives, have issued coverage policies for the category of continuous glucose monitoring devices. Many of these coverage policies, however, are restrictive in nature and require the patient to comply with extensive documentation and other requirements to demonstrate medical necessity under the policy. We have negotiated contracted rates with six of those third-party payors for the purchase of our products by their members. We currently employ in-house reimbursement expertise to assist patients in obtaining reimbursement from private third-party healthcare payors. We also maintain a field-based reimbursement team charged with calling on third-party private payors to obtain coverage decisions and contracts. |
| • | Expand the use of our products to other patient care settings and patient demographics. Our ambulatory products are approved for use at home and in health care facilities by adults (18 years and older) with diabetes. We believe our sensor technology may be beneficial to pediatric diabetes patients and intend to seek approval for use in patients under the age of 18 in the future. We also believe there is an unmet medical need for continuous glucose monitoring in the hospital setting. According to the ADA, diabetes related hospitalizations totaled 24.3 million days in 2007, an increase of 7.4 million days from 2002. In addition, studies show that many non-diabetic hospital patients suffer episodes of hyperglycemia. As of 1998, as many as 1.5 million hospitalized patients in the United States had significant hyperglycemia without a history of diabetes. A study of over 1,500 hospitalized patients, of which only 13% had a history of diabetes, concluded that intensive insulin therapy to maintain blood glucose levels reduced mortality among critically ill patients in the surgical intensive care unit and improved patient outcomes. To address this patient population, we entered into an exclusive agreement with Edwards to develop jointly and market a specific product platform for the in-hospital glucose monitoring market, with an initial focus on the development of an intravenous sensor specifically for the critical care market. |
| • | Provide a high level of customer support, service and education. We support our sales and marketing efforts with a customer service program that includes customer training and support. We provide direct technical support by telephone 24 hours a day to patients, endocrinologists, physicians and diabetes educators to promote safe and successful use of our products. |
| • | Pursue the highest safety and quality levels for our products. We have established an organization that is highly focused on product quality and patient safety. We have developed in-house engineering, quality assurance, clinical and regulatory expertise, and data analysis capabilities. Additionally, we seek to continue to establish credible and open relationships with regulatory bodies, physician opinion leaders and scientific experts. These capabilities and relationships will assist us in designing products that we believe will meet or exceed expectations for reliable, safe performance. |
Our Technology Platform
The development of a continuous glucose monitor requires successful coordination and execution of a wide variety of technology disciplines, including biomaterials, membrane systems, electrochemistry, low power microelectronics, telemetry, software, algorithms, implant tools and sealed protective housings. We have developed in-house expertise in each of these disciplines. We believe we have a broad technology platform that will support the development of multiple products for glucose monitoring.
Sensor Technology
The key enabling technologies for our sensors include biomaterials, membrane systems, electrochemistry and low power microelectronics. Our membrane technology consists of multiple polymer layers configured to
9
Table of Contents
selectively allow the appropriate mix of glucose and oxygen to travel through the membrane and react with a glucose specific enzyme to create an extremely low level electrical signal, measured in pico-amperes. This electrical signal is then translated into glucose values. We believe that the capability to measure very low levels of an electrical signal and to accurately translate those measurements into glucose values is also a unique and distinguishing feature of our technology. We have also developed technology to allow sensitive electronics to be packaged in a small, fully-contained, lightweight sealed unit which minimizes inconvenience and discomfort for the patient.
Receiver and Transmitter Technology
Our ambulatory glucose monitoring systems use radiofrequency telemetry to wirelessly transmit information from the transmitter, which sits in a pod atop the sensor, to our receiver. We have developed the technology for reliable transmission and reception and have consistently demonstrated a high rate of successful transmissions from sensor to receiver in our clinical trials. Our receiver then processes and displays real-time and trended glucose values, and provides alerts. We have used our extensive database of continuous glucose data from our clinical trials to create software and algorithms for the display of data to patients.
In March 2009, the Federal Communications Commission, or FCC, established a bifurcated Medical Implant Communications System, or MICS, band which requires device manufacturers whose products will operate in the main MICS band to either manufacture their devices using listen-before-transmit technology, or to transmit on a side band outside the main MICS band at lower power. Although the SEVEN PLUS does not comply with existing MICS band listen-before-transmit requirements, the FCC granted a waiver to allow us to continue marketing and operating our SEVEN PLUS through March 2013, which we believe will provide adequate time to design an alternative method of wireless communication.
Other Technology Applications
We have gained our technology expertise by learning to design implants that can withstand the rigors of functioning within the human body for extended periods of time. In addition to the foreign body response, we have overcome other problems related to operating within the human body, such as device sealing, miniaturization, durability and sensor geometry. We believe that, over time, the expertise gained in overcoming these problems may support the development of additional products beyond glucose monitoring.
Our Products
Ambulatory Product Line: SEVEN PLUS
We received approval from the FDA and commercialized our first product in 2006. In 2007, we received approval and began commercializing our second generation system, the SEVEN, and on February 13, 2009, we received approval for our third generation system, the SEVEN PLUS, which is designed for up to seven days of continuous use, and we began commercializing this product in the first quarter of 2009. We no longer market or provide support for the SEVEN system. There are various differences between the SEVEN and the SEVEN PLUS. As compared to the SEVEN, the SEVEN PLUS incorporates additional user interface and algorithm enhancements that are intended to make its glucose monitoring function more accurate and customizable. The approval of the SEVEN PLUS by the FDA allows for the use of the SEVEN PLUS by adults with diabetes to detect trends and track glucose patterns, to aid in the detection of hypoglycemia and hyperglycemia and to facilitate acute and long-term therapy adjustments. The SEVEN PLUS must be prescribed by a physician and includes a disposable sensor, a transmitter and a small handheld receiver. The SEVEN PLUS is indicated for use as an adjunctive device to complement, not replace, information obtained from standard home blood glucose monitoring devices and must be calibrated periodically using a standard home blood glucose monitor. The sensor is inserted by the patient and is intended to be used continuously for up to seven days after which it is removed by the patient and may be replaced by a new sensor. Our transmitter and receiver are reusable. In 2008, we
10
Table of Contents
received CE Mark approval for the SEVEN system, and on September 30, 2009, we received CE Mark approval for the SEVEN PLUS, enabling commercialization of the SEVEN PLUS system in the European Union and the countries in Asia and Latin America that recognize the CE Mark. We initiated a limited commercial launch in the European Union and Israel in 2008 and 2009 and have focused our international sales efforts on a portion of European countries.
In-Hospital Product Line: GlucoClear
To address the in-hospital patient population, we entered into an exclusive agreement with Edwards to develop jointly and market a specific product platform for the in-hospital glucose monitoring market, with an initial focus on the development of an intravenous sensor specifically for the critical care market. On October 30, 2009, we received CE Mark approval for our first generation GlucoClear, a blood-based in-vivo automated glucose monitoring system for use by healthcare providers in the hospital, and are continuing to seek approval for this system from the FDA. In partnership with Edwards, we initiated a very limited launch of the GlucoClear in Europe in 2009.
Products in Development
We are leveraging our technology platform to enhance the capabilities of our current products and to develop additional continuous glucose monitoring products. We are continuing to seek approval for our next generation ambulatory system, and are responding to FDA’s requests for additional data in support of that application. We expect our next generation system will further improve sensor reliability, comfort, stability and accuracy over the useful life of the sensor, and will be suited for large scale manufacturing. We also intend to seek approval for a pediatric indication (patients under 18 years of age) in the future.
In 2008, we entered into two separate development agreements, one with Animas, a subsidiary of Johnson & Johnson, and one with Insulet, to integrate our technology into the insulin pump product offerings of the respective partner, enabling the partner’s insulin pump to receive glucose readings from our transmitter and display this information on the pump’s screen.
Continuous Glucose Monitoring Disposable Sensor & Reusable Transmitter
Our sensor includes a tiny wire-like electrode coated with our sensing membrane system. This disposable sensor comes packaged with an integrated insertion device and is contained in a small plastic housing platform, or pod. The base of the pod has adhesive that attaches it to the skin. The sensor is intended to be easily and reliably inserted by the patient by exposing the adhesive, placing the pod against the surface of the skin of the abdomen and pushing down on the insertion device. The insertion device first extends a narrow gauge needle containing the sensor into the subcutaneous tissue and then retracts the needle, leaving behind the sensor in the tissue and the pod adhered to the skin. The patient then disposes of the insertion device and snaps the reusable transmitter to the pod. After a stabilization period of a few hours, the patient is required to calibrate the receiver with two measurements from a single-point finger stick device and the disposable sensor begins wirelessly transmitting the continuous glucose data at specific intervals to the handheld receiver. Patients are prompted by the receiver to calibrate the system twice per day with finger stick measurements throughout the seven day usage period to ensure reliable operation, which calibration may be accomplished by using any FDA approved blood glucose meter. Currently, the SEVEN PLUS is indicated for use as an adjunctive device to complement, not replace, information obtained from standard home blood glucose monitoring devices, although in the future we may seek replacement claim labeling from the FDA for the use of a future generation sensor as the sole basis for making therapeutic adjustments.
The disposable sensor contained in the SEVEN PLUS is intended to function for up to seven days after which it may be replaced. After seven days, the patient simply removes the pod and attached sensor from the skin
11
Table of Contents
and discards them while retaining the reusable transmitter. A new sensor and pod can then be inserted and used with the same receiver and transmitter for a subsequent seven day period. We are aware of reports from the field, however, that patients have been able to use sensors for periods longer than seven days.
Handheld Receiver
Our small handheld receiver is carried by the patient and wirelessly receives continuous glucose values from the sensor. Proprietary algorithms and software, developed from our extensive database of continuous glucose data from clinical trials, are programmed into the receiver to process the glucose data from the sensor and display it on a user-friendly graphical user interface. With a push of a button, the patient can access their current glucose value and one-, three-, six-, twelve- and twenty-four-hour trended data. Additionally, when glucose values are inappropriately high or low, the receiver provides an audible alert or vibrates. The receiver is a self-contained, durable unit with a rechargeable battery.
Sales and Marketing
We have built a direct sales organization to call on endocrinologists, physicians and diabetes educators who can educate and influence patient adoption of continuous glucose monitoring. We believe that focusing efforts on these participants is important given the instrumental role they each play in the decision-making process for diabetes therapy. To complement our direct sales efforts, we employ clinical specialists who help to educate patients on the benefits of continuous glucose monitoring and provide clinical support to endocrinologists, physicians and diabetes educators who prescribe our products. As of December 31, 2010, we employed approximately 56 direct sales personnel and clinical account specialists. We continue to improve our sales and marketing organization as necessary to support the commercialization of our products, and plan to modestly expand the size of the field sales force during 2011. We believe that referrals by physicians and diabetes educators, together with self-referrals by patients, have driven and will continue to drive adoption of our SEVEN PLUS. We directly market our products in the United States primarily to endocrinologists, physicians and diabetes educators. Although the number of diabetes patients is significant, the number of physicians and educators influencing these patients is relatively small. As of 2008, there were an estimated 4,000 clinical endocrinologists in the United States. As a result, we believe our direct, highly-specialized and focused sales organization is sufficient for us to support our sales efforts for the foreseeable future.
We use a variety of marketing tools to drive adoption, ensure continued usage and establish brand loyalty for our continuous glucose monitoring systems by:
| • | creating awareness of the benefits of continuous glucose monitoring and the advantages of our technology with endocrinologists, physicians, diabetes educators and patients; |
| • | providing strong and simple educational and training programs to healthcare providers and patients to ensure easy, safe and effective use of our systems; and |
| • | maintaining a readily-accessible telephone and web-based technical and customer support infrastructure, which includes clinicians, diabetes educators and reimbursement specialists, to help referring physicians, diabetes educators and patients as necessary. |
Our sales organization competes with the experienced and well-funded marketing and sales operations of our competitors. We have relatively limited experience developing and managing a direct sales organization and we may be unsuccessful in our attempt to manage and expand the sales force. Developing a direct sales organization is a difficult, expensive and time consuming process. To be successful we must:
| • | recruit and retain adequate numbers of effective sales personnel; |
| • | effectively train our sales personnel in the benefits of our products; |
12
Table of Contents
| • | establish and maintain successful sales, marketing, training and education programs that encourage endocrinologists, physicians and diabetes educators to recommend our products to their patients; and |
| • | manage geographically disbursed operations. |
Competition
The market for blood glucose monitoring devices is intensely competitive, subject to rapid change and significantly affected by new product introductions. Four companies, Roche Disetronic, a division of Roche Diagnostics; LifeScan, Inc., a division of Johnson & Johnson; the MediSense and TheraSense divisions of Abbott Laboratories; and Bayer Corporation, currently account for substantially all of the worldwide sales of self-monitored glucose testing systems. These competitors’ products use a meter and disposable test strips to test blood obtained by pricking the finger or, in some cases, the forearm. In addition, other companies are developing or marketing minimally invasive or noninvasive glucose testing devices and technologies that could compete with our devices. There are also a number of academic and other institutions involved in various phases of our industry’s technology development.
Several companies have attempted to address the limitations of single-point finger stick devices by developing continuous glucose monitoring systems. To date, in addition to DexCom, we are aware that three other companies, Cygnus, Medtronic, and Abbott, have received approval from the FDA for continuous glucose monitors. We believe that one of the products, originally developed and marketed by Cygnus, is no longer actively marketed. Except for our SEVEN and SEVEN PLUS, we believe that none of the products that have received FDA approval are labeled for more than five days of use. We also believe that none of the FDA approved products are labeled for use as a replacement for single-point finger stick devices.
A number of companies, including Bayer, are developing next generation real-time continuous glucose monitoring or sensing devices and technologies as well as several other companies that are developing non-invasive continuous glucose monitoring products to measure the patient’s glucose level. The majority of these non-invasive technologies do not pierce the skin, but instead typically analyze signatures reflected back from energy that has been directed into the patient’s skin, tissue or bodily fluids.
Many of our competitors are either publicly traded or are divisions of publicly-traded companies, and they enjoy several competitive advantages, including:
| • | significantly greater name recognition; |
| • | established relations with healthcare professionals, customers and third-party payors; |
| • | established distribution networks; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer higher discounts or incentives to gain a competitive advantage; |
| • | greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products and marketing approved products; and |
| • | greater financial and human resources for product development, sales and marketing, and patent litigation. |
As a result, we cannot assure you that we will be able to compete effectively against these companies or their products.
We believe that the principal competitive factors in our market include:
| • | safe, reliable and high quality performance of products; |
13
Table of Contents
| • | cost of products and eligibility for reimbursement; |
| • | comfort and ease of use; |
| • | effective sales, marketing and distribution; |
| • | brand awareness and strong acceptance by healthcare professionals and patients; |
| • | customer service and support and comprehensive education for patients and diabetes care providers; |
| • | speed of product innovation and time to market; |
| • | regulatory expertise; and |
| • | technological leadership and superiority. |
Manufacturing
We currently manufacture our devices at our headquarters in San Diego, California. These facilities have more than 8,000 square feet of laboratory space and approximately 10,000 square feet of controlled environment rooms. In February 2010, our facility was subject to a post-approval inspection by the FDA. After the close of the inspection, the FDA investigator issued a Form 483 identifying several inspectional observations. Subsequent to the inspection, we also received a warning letter from the FDA requiring us to file medical device reports (MDRs) in accordance with the MDR regulations for complaints involving sensor wire fractures underneath a patient’s skin. The warning letter also recommended that we add certain warnings and precautions statements to the labeling, patient education brochures, and our company website regarding the appropriate use of the SEVEN PLUS system, including that they are not approved for use in children under age 18, pregnant women, or persons on dialysis. In response to the warning letter and the Form 483 inspectional observations, we have taken corrective action to address the observations to achieve substantial compliance with the FDA regulatory requirements applicable to a commercial medical device manufacturer. In October 2010, we were subject to a follow-up site inspection by the FDA, and upon completion of that inspection, we were notified by the inspector that there were no 483 inspectional observations. We also received written notification dated November 1, 2010 from the FDA that we adequately addressed all issues cited in the warning letter.
There are technical challenges to increasing manufacturing capacity, including equipment design and automation, material procurement, problems with production yields, and quality control and assurance. We have focused significant effort on continual improvement programs in our manufacturing operations intended to improve quality, yields and throughput. We have made progress in manufacturing to enable us to supply adequate amounts of product to support our commercialization efforts, however there can be no assurances that supply will not be constrained going forward. Additionally, the production of our continuous glucose monitoring systems must occur in a highly controlled and clean environment to minimize particles and other yield- and quality-limiting contaminants. Developing commercial-scale manufacturing facilities has and will continue to require the investment of substantial additional funds and the hiring and retaining of additional management, quality assurance, quality control and technical personnel who have the necessary manufacturing experience. Manufacturing is subject to numerous risks and uncertainties described in detail in “Risk Factors” below.
We manufacture our SEVEN PLUS with components supplied by outside vendors and with parts manufactured by us internally. Key components that we manufacture internally include our wire-based sensors for our SEVEN PLUS. The remaining components and assemblies are purchased from outside vendors. We then assemble, test, package and ship the finished SEVEN PLUS systems, which includes a reusable transmitter, a receiver, and disposable sensors.
We purchase certain components and materials from single sources due to quality considerations, costs or constraints resulting from regulatory requirements. Currently, those single sources are On Semiconductor Corp, which produces the application specific integrated circuits used in our transmitters; DSM PTG, Inc., which manufactures certain polymers used to synthesize our polymeric membranes for our sensors; Flextronics
14
Table of Contents
International Ltd., which assembles the printed circuit boards for our transmitters and receivers; and The Tech Group, which produces injection molded components. In some cases, agreements with these and other suppliers can be terminated by either party upon short notice. We may not be able to quickly establish additional or replacement suppliers for our single-source components, especially after our products are commercialized, in part because of the FDA approval process and because of the custom nature of the parts we designed. Any supply interruption from our vendors or failure to obtain alternate vendors for any of the components would limit our ability to manufacture our systems, and could have a material adverse effect on our business.
Third Party Reimbursement
As a medical device company, reimbursement from Medicare and private third-party healthcare payors is an important element of our success. Although CMS released 2008 Alpha-Numeric HCPCS codes applicable to each of the three components of our continuous glucose monitoring systems, to date, our approved products are not reimbursed by virtue of a national coverage decision by Medicare. As of March 2011, the seven largest private third-party payors, in terms of the number of covered lives, have issued coverage policies for the category of continuous glucose monitoring devices. In addition, we have negotiated contracted rates with six of those third-party payors for the purchase of our products by their members. Many of these coverage policies are restrictive in nature and require the patient to comply with documentation and other requirements to demonstrate medical necessity under the policy. In addition, patients who are insured by payors that do not offer coverage for our devices will have to bear the financial cost of the products. We currently employ in-house reimbursement expertise to assist patients in obtaining reimbursement from private third-party payors. We also maintain a field-based reimbursement team charged with calling on third-party private payors to obtain coverage decisions and contracts. We have had formal meetings and have increased our efforts to create coverage policies with third-party payors during 2010 and expect to continue to do so in 2011. However, unless government and other third-party payors provide adequate coverage and reimbursement for our products, patients may not use them.
Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medical devices, and, as a result, their coverage policies may be restrictive, or they may not cover or provide adequate payment for our products. In order to obtain reimbursement arrangements, we may have to agree to a net sales price lower than the net sales price we might charge in other sales channels. The continuing efforts of government and third-party payors to contain or reduce the costs of healthcare may limit our revenue. Our initial dependence on the commercial success of our SEVEN PLUS makes us particularly susceptible to any cost containment or reduction efforts. Accordingly, unless government and other third-party payors provide adequate coverage and reimbursement for our products, our financial performance may be harmed.
In some foreign markets, pricing and profitability of medical devices are subject to government control. In the United States, we expect that there will continue to be federal and state proposals for similar controls. Also, the trends toward managed healthcare in the United States and proposed legislation intended to reduce the cost of government insurance programs could significantly influence the purchase of healthcare services and products and may result in lower prices for our products or the exclusion of our products from reimbursement programs.
Intellectual Property
Protection of our intellectual property is a strategic priority for our business. We rely on a combination of patent, copyright and other intellectual property laws, trade secrets, nondisclosure agreements and other measures to protect our proprietary rights. As of February 2011, we had obtained 57 issued U.S. patents, and had 255 additional U.S. patent applications pending. We believe it will take up to five years, and possibly longer, for these pending U.S. patent applications to result in issued patents. As of February 2011, we had 18 international applications filed under the Patent Cooperation Treaty, 3 granted European patents, 49 European patent applications pending, 10 Japanese patent applications pending, 12 registered U.S. trademarks, 6 pending U.S. trademark applications, 9 registered European trademarks, 2 pending European trademark applications, and 3 registered Japanese trademarks. Our patents begin expiring in 2017.
15
Table of Contents
Together, our patents and patent applications seek to protect aspects of our core membrane and sensor technologies, and our product concepts for continuous glucose monitoring. We believe that our patent position provides us with sufficient rights to develop, sell and protect our current and proposed commercial products. However, our patent applications may not result in issued patents, and any patents that have issued or might issue may not protect our intellectual property rights. Furthermore, our patents may not be upheld. Any patents issued to us may be challenged by third parties as being invalid or unenforceable, or third parties may independently develop similar or competing technology that avoids our patents. The steps we have taken may not prevent the misappropriation of our intellectual property, particularly in foreign countries where the laws may not protect our proprietary rights as fully as in the United States.
The medical device industry in general, and the glucose testing sector of this industry in particular, are characterized by the existence of a large number of patents and frequent litigation based on assertions of patent infringement. We are aware of numerous patents issued to third parties that may relate to aspects of our business, including the design and manufacture of continuous glucose monitoring sensors and membranes, as well as methods for continuous glucose monitoring. The owners of each of these patents could assert that the manufacture, use or sale of our continuous glucose monitoring systems infringes one or more claims of their patents. Each of these patents contains multiple claims, any one of which may be independently asserted against us. There may be patents of which we are presently unaware that may relate to aspects of our technology that could materially and adversely affect our business. In addition, because patent applications can take many years to issue, there may be currently pending applications that are unknown to us, which may later result in issued patents that may materially and adversely affect our business.
We are currently engaged in patent litigation with Abbott Diabetes Care, Inc., or Abbott, as further described in “Item 3. Legal Proceedings” of this Annual Report. In connection with this litigation each of Abbott’s seven patents that are the subject of the litigation have one or more associated reexamination requests in various stages at the U.S. Patent and Trademark Office, or the Patent Office. The court has granted a stay of litigation pending completion of the reexamination process. The Board of Patent Appeals and Interferences within the Patent Office has recently rendered decisions on the appeals related to the reexaminations of two of the patents. We believe these decisions are favorable to us; however, Abbott may still seek judicial review of the decisions. Four patents are currently undergoing reexamination at the Patent Office. Reexamination of another patent was completed in 2010 and we recently submitted a subsequent reexamination request.
In addition, since 2008, Abbott has copied claims from certain of our applications, and stated that it may seek to provoke an interference with certain of our pending applications in the Patent Office. If interference is declared and Abbott prevails in the interference, we would lose certain patent rights to the subject matter defined in the interference. Also since 2008, Abbott has filed reexamination requests seeking to invalidate fifteen of our patents. The fifteen reexamination requests are in various stages at the Patent Office. We have filed responses with the Patent Office seeking claim construction to differentiate certain claims from the prior art presented in the reexaminations, seeking to amend certain claims to overcome the prior art presented in the reexaminations, canceling claims and/or seeking to add new claims. It is possible that the Patent Office may determine that some or all of the claims of our patents subject to the reexamination are invalid.
Although it is our position that Abbott’s assertions of infringement have no merit, and that the potential interference and reexamination requests have no merit, the outcome of the litigation and interference or reexamination requests cannot be assessed currently with certainty. We may not successfully defend ourselves against the claims made by Abbott, and we may not prevail in the litigation. If Abbott were to successfully seek an injunction, it could force us to stop making, using, selling or offering to sell our products. The technology at issue in our litigation with Abbott is currently used in our products, including our SEVEN PLUS, which is our only ambulatory product that is approved for commercial sale, and the GlucoClear, our blood-based in-vivo automated glucose monitoring system for in-hospital use. If we were forced to stop selling these products either as a result of an unfavorable outcome in the litigation or in connection with the grant of an injunction, our business and prospects would suffer. In addition, defending against this action, including any injunction action,
16
Table of Contents
could have a number of harmful effects on our business regardless of the final outcome of such litigation. For example, we have incurred, and expect to continue to incur significant costs in defending the action.
Any adverse determination in litigation or interference proceedings to which we are or may become a party relating to patents could subject us to significant liabilities to third parties or require us to seek licenses from other third parties. Furthermore, if we are found to willfully infringe third-party patents, we could, in addition to other penalties, be required to pay treble damages and/or attorney fees for the prevailing party. Although patent and intellectual property disputes in the medical device area have often been settled through licensing or similar arrangements, costs associated with such arrangements may be substantial and would likely require ongoing royalties. We may be unable to obtain necessary licenses on satisfactory terms, if at all. If we do not obtain necessary licenses, we may not be able to redesign our products to avoid infringement and any redesign may not receive FDA approval in a timely manner. Adverse determinations in a judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from manufacturing and selling our products, which would have a significant adverse impact on our business. We also rely on trade secrets, technical know-how and continuing innovation to develop and maintain our competitive position. We seek to protect our proprietary information and other intellectual property by generally requiring our employees, consultants, contractors, outside scientific collaborators and other advisors to execute non-disclosure and assignment of invention agreements on commencement of their employment or engagement. Agreements with our employees also forbid them from bringing the proprietary rights of third parties to us. We also generally require confidentiality or material transfer agreements from third parties that receive our confidential data or materials. We cannot provide any assurance that employees and third parties will abide by the confidentiality or assignment terms of these agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our products or obtain and use information that we regard as proprietary.
Government Regulation
Our products are medical devices subject to extensive and ongoing regulation by the FDA and regulatory bodies in other countries. The Federal Food, Drug and Cosmetic Act, or FDCA, and the FDA’s implementing regulations govern product design and development, pre-clinical and clinical testing, pre-market clearance or approval, establishment registration and product listing, product manufacturing, product labeling, product storage, advertising and promotion, product sales, distribution, recalls and field actions, servicing and post-market clinical surveillance.
FDA Regulation
Unless an exemption applies, each medical device we wish to commercially distribute in the United States will require either prior 510(k) clearance or prior approval from the FDA through the premarket approval (PMA) process. Our SEVEN PLUS system is classified by the FDA as a PMA medical device. The FDA classifies medical devices into one of three classes. Devices requiring fewer controls because they are deemed to pose lower risk are placed in Class I or II. Class I devices are subject to general controls such as labeling, pre-market notification, and adherence to the FDA’s Quality System Regulation, or QSR. Class II devices are subject to special controls such as performance standards, post-market surveillance, FDA guidelines, or particularized labeling, as well as general controls. Some Class I and Class II devices are exempted by regulation from the pre-market notification (i.e., 510(k) clearance) requirement, and/or the requirement of compliance with substantially all of FDA’s manufacturing requirements, known as the QSR. Some devices are placed in Class III, which requires approval of a PMA application, if they are deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or certain implantable devices, or to be “not substantially equivalent” either to a previously 510(k) cleared device or to a “preamendment” Class III device in commercial distribution before May 28, 1976 for which PMA applications have not been required.
Our SEVEN PLUS has been classified as a device requiring PMA approval. A PMA application must be supported by valid scientific evidence, which typically requires extensive data, including technical, pre-clinical,
17
Table of Contents
clinical, manufacturing and labeling data, to demonstrate to the FDA’s satisfaction the safety and efficacy of the device. A PMA application also must include a complete description of the device and its components, a detailed description of the methods, facilities and controls used to manufacture the device, and proposed labeling. After a PMA application is submitted and found to be sufficiently complete, the FDA begins an in-depth review of the submitted information. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA. In addition, the FDA generally will conduct a pre-approval inspection of the manufacturing facility to evaluate compliance with QSR, which requires manufacturers to implement and follow design, testing, control, documentation and other quality assurance procedures. In February 2010, our facility was subject to a post-approval inspection by FDA. After the close of the inspection, the FDA investigator issued a Form 483 identifying several inspectional observations. Subsequent to the inspection, we also received a warning letter from the FDA requiring us to file medical device reports (MDRs) in accordance with the MDR regulations for complaints involving sensor wire fractures underneath a patient’s skin. The warning letter also recommended that we add certain warnings and precautions statements to the labeling, patient education brochures, and our company website regarding the appropriate use of the SEVEN PLUS system, including that they are not approved for use in children under age 18, pregnant women, or persons on dialysis. In response to the warning letter and the Form 483 inspectional observations, we have taken corrective action to address the observations to achieve substantial compliance with the FDA regulatory requirements applicable to a commercial medical device manufacturer. In October 2010, we were subject to a follow-up site inspection by the FDA, and upon completion of that inspection, we were notified by the inspector that there were no 483 inspectional observations. We also received written notification dated November 1, 2010 from the FDA that we adequately addressed all issues cited in the warning letter.
FDA review of a PMA application generally takes between one and three years, but may take significantly longer. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
| • | our systems may not be safe or effective to the FDA’s satisfaction; |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support approval; |
| • | the manufacturing process or facilities we use may not meet applicable requirements; and |
| • | changes in FDA approval policies or adoption of new regulations may require additional data. |
If an FDA evaluation of a PMA application or manufacturing facilities is favorable, the FDA will either issue an approval letter, or approvable letter, which usually contains a number of conditions which must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of a device, subject to the conditions of approval and the limitations established in the approval letter. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA may also determine that additional trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA. The PMA process can be expensive, uncertain and lengthy and a number of devices for which FDA approval has been sought by other companies have never been approved by the FDA for marketing.
New PMA applications or PMA supplements may be required for modifications to the manufacturing process, labeling, device specifications, materials or design of a device that is approved through the PMA process. PMA supplements often require submission of the same type of information as an initial PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the approved PMA application and may or may not require as extensive clinical data or the convening of an advisory panel.
Clinical trials are almost always required to support a PMA application and are sometimes required for a 510(k) clearance. These trials generally require submission of an application for an investigational device
18
Table of Contents
exemption, or IDE, to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and eligible for abbreviated IDE requirements. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate institutional review boards at the clinical trial sites. The FDA’s approval of an IDE allows clinical testing to go forward, but does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and efficacy, even if the trial meets its intended success criteria. All clinical trials must be conducted in accordance with the FDA’s IDE regulations, which govern investigational device labeling, prohibit promotion, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials must further comply with the FDA’s regulations for institutional review board approval and for informed consent and other human subject protections. Required records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product. The commencement or completion of any of our clinical trials may be delayed or halted, or be inadequate to support approval of a PMA application, for numerous reasons, including, but not limited to, the following:
| • | the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| • | patients do not enroll in clinical trials at the rate we expect; |
| • | patients do not comply with trial protocols; |
| • | patient follow-up is not at the rate we expect; |
| • | patients experience adverse side effects; |
| • | patients die during a clinical trial, even though their death may not be related to our products; |
| • | institutional review boards and third-party clinical investigators may delay or reject our trial protocol; |
| • | third-party clinical investigators decline to participate in a trial or do not perform a trial on our anticipated schedule or consistent with the clinical trial protocol, good clinical practices or other FDA requirements; |
| • | the company or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans; |
| • | third-party clinical investigators have significant financial interests related to the company or study that FDA deems to make the study results unreliable, or the company or investigators fail to disclose such interests; |
| • | regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; |
| • | changes in governmental regulations or administrative actions; |
| • | the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or efficacy; and |
| • | the FDA concludes that our trial design is inadequate to demonstrate safety and efficacy. |
After a device is approved and placed in commercial distribution, numerous regulatory requirements apply. These include:
| • | establishment registration and device listing; |
19
Table of Contents
| • | QSR, which requires manufacturers to follow design, testing, control, storage, supplier/contractor selection, complaint handling, documentation and other quality assurance procedures; |
| • | labeling regulations, which prohibit the promotion of products for unapproved or off-label uses or indications and impose other restrictions on labeling, advertising and promotion; |
| • | medical device reporting regulations, which require that manufacturers report to the FDA if a device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| • | voluntary and mandatory device recalls to address problems when a device is defective and/or could be a risk to health; and |
| • | corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health. |
Also, the FDA may require us to conduct post-market surveillance studies or order us to establish and maintain a system for tracking our products through the chain of distribution to the patient level. The FDA and the Food and Drug Branch of the California Department of Health Services enforce regulatory requirements by conducting periodic, unannounced inspections and market surveillance. Inspections may include the manufacturing facilities of our subcontractors.
Failure to comply with applicable regulatory requirements, including those applicable to the conduct of our clinical trials, can result in enforcement action by the FDA, which may lead to any of the following sanctions:
| • | warning letters or untitled letters that require corrective action; |
| • | fines and civil penalties; |
| • | unanticipated expenditures; |
| • | delays in approving or refusal to approve our future continuous glucose monitoring systems or other products; |
| • | FDA refusal to issue certificates to foreign governments needed to export our products for sale in other countries; |
| • | suspension or withdrawal of FDA approval; |
| • | product recall or seizure; |
| • | interruption of production; |
| • | operating restrictions; |
| • | injunctions; and |
| • | criminal prosecution. |
We and our contract manufacturers, specification developers, and some suppliers of components or device accessories, are also required to manufacture our products in compliance with current Good Manufacturing Practice, or GMP, requirements set forth in the QSR. The QSR requires a quality system for the design, manufacture, packaging, labeling, storage, installation and servicing of marketed devices, and includes extensive requirements with respect to quality management and organization, device design, buildings, equipment, purchase and handling of components or services, production and process controls, packaging and labeling controls, device evaluation, distribution, installation, complaint handling, servicing, and record keeping. The FDA evaluates compliance with the QSR through periodic unannounced inspections that may include the manufacturing facilities of our subcontractors. If the FDA believes we or any of our contract manufacturers or regulated suppliers are not in compliance with these requirements, it can shut down our manufacturing
20
Table of Contents
operations, require recall of our products, refuse to approve new marketing applications, institute legal proceedings to detain or seize products, enjoin future violations, or assess civil and criminal penalties against us or our officers or other employees. Any such action by the FDA would have a material adverse effect on our business. We cannot assure you that we will be able to comply with all applicable FDA regulations.
Fraud and Abuse Laws
The healthcare industry is subject to various federal and state laws pertaining to healthcare fraud and abuse. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, exclusion from participation in federal and state healthcare programs, including Medicare and Medicaid.
Anti-kickback Laws. The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, receiving, offering or providing remuneration directly or indirectly to induce either the referral of an individual, or the furnishing, recommending, or arranging of a good or service, for which payment may be made under a federal healthcare program such as Medicare and Medicaid. The definition of “remuneration” has been broadly interpreted to include anything of value, including such items as gifts, discounts, the furnishing of supplies or equipment, credit arrangements, waiver of payments, and providing anything at less than its fair market value. The Department of Health and Human Services (“HHS”) has issued regulations, commonly known as safe harbors, that set forth certain provisions which, if fully met, will assure healthcare providers and other parties that they will not be prosecuted under the federal Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable safe harbor may result in increased scrutiny by government enforcement authorities such as the HHS Office of Inspector General.
The penalties for violating the federal Anti-Kickback Statute include imprisonment for up to five years, fines of up to $25,000 per violation and possible exclusion from federal healthcare programs such as Medicare and Medicaid. Many states have adopted prohibitions similar to the federal Anti-Kickback Statute, some of which apply to the referral of patients for healthcare services reimbursed by any source, not only by the Medicare and Medicaid programs.
Federal False Claims Act. The federal False Claims Act prohibits the knowing filing of a false claim or the knowing use of false statements to obtain payment from the federal government. When an entity is determined to have violated the False Claims Act, it must pay three times the actual damages sustained by the government, plus mandatory civil penalties of between $5,500 and $11,000 for each separate false claim. Suits filed under the False Claims Act, known as “qui tam” actions, can be brought by any individual on behalf of the government and such individuals (known as “relators” or, more commonly, as “whistleblowers”) may share in any amounts paid by the entity to the government in fines or settlement. In addition, certain states have enacted laws modeled after the federal False Claims Act. Qui tam actions have increased significantly in recent years, causing greater numbers of healthcare companies to have to defend a false claim action, pay fines or be excluded from Medicare, Medicaid or other federal or state healthcare programs as a result of an investigation arising out of such action.
HIPAA. The Health Insurance Portability and Accountability Act of 1996, or HIPAA, created two new federal crimes: healthcare fraud and false statements relating to healthcare matters. The healthcare fraud statute prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private payors. A violation of this statute is a felony and may result in fines, imprisonment or exclusion from government sponsored programs. The false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. A violation of this statute is a felony and may result in fines or imprisonment.
Additionally, the U.S. Foreign Corrupt Practices Act (“FCPA”) prohibits U.S. corporations and their representatives from offering, promising, authorizing or making payments to any foreign government official,
21
Table of Contents
government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA would include interactions with certain healthcare professionals in many countries. Our present and future business has been and will continue to be subject to various other U.S. and foreign laws, rules and/or regulations.
International Regulation
International sales of medical devices are subject to foreign government regulations, which may vary substantially from country to country. The time required to obtain approval in a foreign country may be longer or shorter than that required for FDA approval, and the requirements may differ. There is a trend towards harmonization of quality system standards among the European Union, United States, Canada and various other industrialized countries.
The primary regulatory environment in Europe is that of the European Union, which includes most of the major countries in Europe. Other countries, such as Switzerland, have voluntarily adopted laws and regulations that mirror those of the European Union with respect to medical devices. The European Union has adopted numerous directives and standards regulating the design, manufacture, clinical trials, labeling and adverse event reporting for medical devices. Devices that comply with the requirements of a relevant directive will be entitled to bear the CE conformity marking, indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout Europe. The method of assessing conformity varies depending on the class of the product, but normally involves a combination of self-assessment by the manufacturer and a third party assessment by a “Notified Body.” This third party assessment may consist of an audit of the manufacturer’s quality system and specific testing of the manufacturer’s product. An assessment by a Notified Body of one country within the European Union is required in order for a manufacturer to commercially distribute the product throughout the European Union. Outside of the European Union, regulatory approval needs to be sought on a country-by-country basis in order for us to market our products.
Environmental Regulation
Our research and development, clinical and manufacturing processes involve the handling of potentially harmful biological materials as well as hazardous materials. We are subject to federal, state and local laws and regulations governing the use, handling, storage and disposal of hazardous and biological materials and we incur expenses relating to compliance with these laws and regulations. If violations of environmental, health and safety laws occur, we could be held liable for damages, penalties and costs of remedial actions. These expenses or this liability could have a significant negative impact on our financial condition. We may violate environmental, health and safety laws in the future as a result of human error, equipment failure or other causes. Environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. We are subject to potentially conflicting and changing regulatory agendas of political, business and environmental groups. Changes to or restrictions on permitting requirements or processes, hazardous or biological material storage or handling might require an unplanned capital investment or relocation. Failure to comply with new or existing laws or regulations could harm our business, financial condition and results of operations.
Advisory Boards and Consultants
We have relied upon the advice of experts in the development and commercialization of our products. Since 2005, we have used experts in various disciplines on a consulting basis as needed to solve problems or accelerate development pathways. We will continue to engage advisors from the academic, consultancy, governmental or other areas to assist us as necessary. We meet with our clinical advisory board on an annual basis.
22
Table of Contents
Employees
As of December 31, 2010, we had 405 full-time employees and 115 contract and temporary employees. Approximately 95 employees are engaged in research and development, clinical, regulatory and quality assurance, 129 in manufacturing and 181 in selling, general and administrative functions. None of our employees are represented by a labor union or covered by a collective bargaining agreement. We have never experienced any employment-related work stoppages and consider our employee relations to be good.
Available Information
Our Internet website address is www.dexcom.com. We provide free access to various reports that we file with or furnish to the SEC through our website, as soon as reasonably practicable after they have been filed or furnished. These reports include, but are not limited to, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to those reports. Our SEC reports can be accessed through the investor relations section of our website, or through www.sec.gov. Also available on our website are printable versions of DexCom’s Audit Committee charter, Compensation Committee charter, Nominating and Corporate Governance Committee charter, and Business Code of Conduct and Ethics. Information on our website does not constitute part of this annual report on Form 10-K or other report we file or furnish with the SEC. Stockholders may request copies of these documents from:
DexCom, Inc.
6340 Sequence Drive
San Diego, CA 92121
(858) 200-0200
| ITEM 1A. | RISK FACTORS |
Factors that May Affect our Financial Condition and Results of Operations
We have a limited operating history and our products may never achieve market acceptance.
We expect that sales of our SEVEN PLUS, which consists of a handheld receiver, reusable transmitter and disposable sensor, will account for substantially all of our product revenue for the foreseeable future. From inception through December 31, 2010, product revenues total approximately $73.1 million. We have relatively limited experience in selling our products and we might be unable to successfully expand the commercialization of our products on a wide scale for a number of reasons, including:
| • | widespread market acceptance of our products by physicians and patients will largely depend on our ability to demonstrate their relative safety, efficacy, reliability, cost-effectiveness and ease of use; |
| • | the limited size of our sales force and our relative inexperience in marketing, selling and distributing our products; |
| • | we may not have sufficient financial or other resources to adequately expand the commercialization efforts for our products; |
| • | our FDA and other regulatory submissions may be delayed, or approved with limited product labeling; |
| • | we may not be able to manufacture our products in commercial quantities or at an acceptable cost; |
| • | patients with diabetes do not generally receive broad reimbursement from third-party payors for their purchase of our products since many payors require that a patient meet specific medical criteria to qualify for reimbursement, which may reduce widespread use of our products; |
| • | the uncertainties associated with establishing and qualifying new manufacturing facilities; |
| • | our SEVEN PLUS is not labeled as a replacement for the information that is obtained from single-point finger stick devices; |
23
Table of Contents
| • | patients will need to incur the costs of our SEVEN PLUS in addition to single-point finger stick devices; |
| • | the relative immaturity of the continuous glucose monitoring market internationally, and the general absence of international reimbursement of continuous glucose monitoring devices by third party payors and government healthcare providers outside the United States; |
| • | the introduction and market acceptance of competing products and technologies; |
| • | our inability to obtain sufficient quantities of supplies at appropriate quality levels from our sole source and other key suppliers; |
| • | our inability to manufacture products that perform in accordance with expectations of consumers; and |
| • | rapid technological change may make our technology and our products obsolete. |
Our SEVEN PLUS is more invasive than current self-monitored glucose testing systems, including single-point finger stick devices, and patients may be unwilling to insert a sensor in their body, especially if their current diabetes management involves no more than two finger sticks per day. Moreover, patients may not perceive the benefits of continuous glucose monitoring and may be unwilling to change their current treatment regimens. In addition, physicians tend to be slow to change their medical treatment practices because of perceived liability risks arising from the use of new products. Physicians may not recommend or prescribe our products until (i) there is more long-term clinical evidence to convince them to alter their existing treatment methods, (ii) there are additional recommendations from prominent physicians that our products are effective in monitoring glucose levels and (iii) reimbursement or insurance coverage is more widely available. We cannot predict when, if ever, physicians and patients may adopt more widespread use of the SEVEN PLUS. If the SEVEN PLUS does not achieve an adequate level of acceptance by patients, physicians and healthcare payors, we may not generate significant product revenue and we may not become profitable.
We have incurred losses since inception and anticipate that we will incur continued losses for the foreseeable future.
We have incurred net losses in each year since our inception in May 1999, including a net loss of $55.2 million for the twelve months ended December 31, 2010. As of December 31, 2010, we had an accumulated deficit of $346.4 million. We have financed our operations primarily through private placements of our equity and debt securities and our public offerings, and have devoted a substantial portion of our resources to research and development relating to our continuous glucose monitoring systems, including our in-hospital product development, and more recently, we have incurred significant sales and marketing and manufacturing expenses associated with the commercialization of the SEVEN PLUS. In addition, we expect our research and development expenses to increase in connection with our clinical trials and other development activities related to our products, including our next generation sensor and the GlucoClear. We also expect that our general and administrative expenses will continue to increase due to the additional operational and regulatory burdens applicable to public healthcare and medical device companies. As a result, we expect to continue to incur significant operating losses for the foreseeable future. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity.
Current uncertainty in global economic conditions makes it particularly difficult to predict product demand and other related matters and makes it more likely that our actual results could differ materially from expectations.
Our operations and performance depend on worldwide economic conditions, which have been adversely impacted by the global macroeconomic downturn. These conditions have and may continue to make it difficult for our customers and potential customers to afford our products, and could cause our customers to stop using our products or to use them less frequently. If that were to occur, we would experience a decrease in revenue and
24
Table of Contents
our performance would be negatively impacted. Furthermore, during economic uncertainty, our customers have experienced job losses and may continue to experience issues gaining timely access to sufficient health insurance or credit, which could result in their unwillingness to purchase products or an impairment of their ability to make timely payments to us. We cannot predict the reoccurrence of any economic slowdown or the strength or sustainability of the economic recovery, worldwide, in the United States, or in our industry. These and other economic factors could have a material adverse affect on our financial condition and operating results.
We may require additional funding to continue the commercialization of our SEVEN PLUS or the development and commercialization of our next generation and other continuous glucose monitoring systems, including the GlucoClear and our systems to be integrated with Animas and Insulet’s insulin pump delivery systems.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts on commercializing our products, including further modest expansion of our direct sales force and growth of our manufacturing capacity, and on research and development, including conducting clinical trials for our GlucoClear in-hospital system as well as our next generation ambulatory continuous glucose monitoring sensors and systems. For the twelve months ended December 31, 2010, our net cash used in operating activities was $42.7 million, compared to $39.4 million for the same period in 2009, and as of December 31, 2010, we had working capital of $50.9 million comprised of $48.8 million in cash, cash equivalents and short-term marketable securities, and includes $1.7 million in restricted cash. We expect that our cash used by operations will increase significantly in each of the next several years, and, although we recently completed a follow-on public offering of 3,277,500 shares of our common stock for net proceeds to the company of approximately $33.0 million, we may need additional funds to continue the commercialization of our products and for the development and commercialization of our next generation sensors and systems. Additional financing may not be available on a timely basis on terms acceptable to us, or at all. Any additional financing may be dilutive to stockholders or may require us to grant a lender a security interest in our assets. The amount of funding we will need will depend on many factors, including:
| • | the revenue generated by sales of our products and other future products; |
| • | the costs, timing and risks of delay of additional regulatory approvals; |
| • | the expenses we incur in manufacturing, developing, selling and marketing our products; |
| • | our ability to scale our manufacturing operations to meet demand for our current and any future products; |
| • | the costs to produce our continuous glucose monitoring systems; |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; |
| • | the rate of progress and cost of our clinical trials and other development activities; |
| • | the success of our research and development efforts; |
| • | the emergence of competing or complementary technological developments; |
| • | the terms and timing of any collaborative, licensing and other arrangements that we may establish; and |
| • | the acquisition of businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
If adequate funds are not available, we may not be able to commercialize our products at the rate we desire and we may have to delay development or commercialization of our other products or license to third parties the rights to commercialize products or technologies that we would otherwise seek to commercialize. We also may have to reduce sales, marketing, customer support or other resources devoted to our products. Any of these factors could harm our financial condition.
25
Table of Contents
If we are unable to continue the development of an adequate sales and marketing organization, or if our direct sales organization is not successful, we may have difficulty achieving market awareness and selling our products.
To achieve commercial success for the SEVEN PLUS and our future products, we must continue to develop and grow our sales and marketing organization and enter into partnerships or other arrangements to market and sell our products. We currently employ a small direct sales force to market our products in the United States. In the United States, our sales force calls directly on healthcare providers and patients throughout the country to initiate sales of our products. Our sales organization competes with the experienced, larger and well-funded marketing and sales operations of our competitors. We plan to modestly increase the size of our sales force during 2011, and may not be able to successfully manage our increasingly dispersed sales force, or increase our product sales in the new territories. We have also entered into distribution arrangements to leverage existing distributors already engaged in the diabetes marketplace. Our U.S. distribution partnerships are focused on accessing underrepresented regions and, in some instances, third-party payors that contract exclusively with distributors. Our European distribution partners call directly on healthcare providers to market and sell our products in Europe. Because of the competition for their services, we may be unable to partner with or retain additional qualified distributors. Further, we may not be able to enter into agreements with distributors on commercially reasonable terms, if at all.
Additionally, to aid our efforts to obtain timely and comprehensive reimbursement of our products for our customers, we must continue to improve our customer service processes and scale our information technology systems.
Developing and managing a direct sales organization is a difficult, expensive and time consuming process. To be successful we must:
| • | recruit and retain adequate numbers of effective and experienced sales personnel; |
| • | effectively train our sales personnel in the benefits and risks of our products; |
| • | establish and maintain successful sales and marketing and education programs that educate endocrinologists, physicians and diabetes educators so they can appropriately inform their patients about our products; and |
| • | manage geographically disbursed sales and marketing operations. |
If we are unable to establish adequate sales, marketing and distribution capabilities or enter into and maintain arrangements with third parties to sell, market and distribute our products, our business may be harmed.
We have entered into distribution arrangements to leverage existing distributors already engaged in the diabetes marketplace. We have entered into a distribution agreement with RGH Enterprises, Inc., or “Edgepark,” pursuant to which we generated approximately 20% of our revenue during 2010. There can be no assurances that this relationship will continue or that we will be able to maintain this volume of sales from this relationship in the future. A substantial decrease or loss of these sales could have a material adverse effect on our operating performance. Additionally, to the extent that we enter into additional arrangements with third parties to perform sales, marketing, distribution and billing services in the United States or Europe, our product margins could be lower than if we directly marketed and sold our products. Furthermore, to the extent that we enter into co-promotion or other marketing and sales arrangements with other companies, any revenue received will depend on the skills and efforts of others, and we cannot predict whether these efforts will be successful. In addition, market acceptance of our products by physicians and patients in Europe will largely depend on our ability to demonstrate their relative safety, efficacy, reliability, cost-effectiveness and ease of use. If we are unable to do so, we may not be able to generate product revenue from our sales efforts in Europe. Finally, if we are unable to establish and maintain adequate sales, marketing and distribution capabilities, independently or with others, we may not be able to generate adequate product revenue and may not become profitable.
26
Table of Contents
Although many third party payors have adopted some form of coverage policy on continuous glucose monitoring devices, our products do not yet have broad-based contractual coverage with third party payors and we frequently experience administrative challenges in obtaining reimbursement for our customers. If we are unable to obtain adequately broad reimbursement at acceptable prices for our products or any future products from third party payors, we will be unable to generate significant revenue.
As a medical device company, reimbursement from Medicare and private third-party healthcare payors is an important element of our success. To date, our products are not reimbursed by virtue of a national coverage decision by Medicare. Although CMS released 2008 Alpha-Numeric HCPCS codes applicable to each of the three components of our continuous glucose monitoring systems, to date, our approved products are not reimbursed by virtue of a national coverage decision by Medicare. It is not known when, if ever, Medicare will adopt a national coverage decision with respect to continuous glucose monitoring devices. Until any such coverage decision is adopted by Medicare, reimbursement of our products will generally be limited to those patients covered by third-party payors that have adopted coverage policies for continuous glucose monitoring devices. As of March 2011, the seven largest private third-party payors, in terms of the number of covered lives, have issued coverage policies for the category of continuous glucose monitoring devices. In addition, we have negotiated contracted rates with six of those third-party payors for the purchase of our products by their members. However, patients without insurance that covers our products will have to bear the financial cost of them. In the United States, patients using existing single-point finger stick devices are generally reimbursed all or part of the product cost by Medicare or other third-party payors. The commercial success of our products in both domestic and international markets will be substantially dependent on whether third-party reimbursement is widely available for patients that use them. While many third party payors have adopted some form of coverage policy on continuous glucose monitoring devices, those coverage policies frequently require significant medical documentation in order for patients to obtain reimbursement, and as a result, we have experienced difficulty in improving the efficiency of our customer service group. In addition, Medicare, Medicaid, health maintenance organizations and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new medical devices, and, as a result, they may not cover or provide adequate payment for our products. In order to obtain additional reimbursement arrangements, we may have to agree to a net sales price lower than the net sales price we might charge in other sales channels. The continuing efforts of government and third-party payors to contain or reduce the costs of healthcare may limit our revenue. Furthermore, we are unable to predict what effect the current or any future healthcare reform will have on our business, or the effect these matters will have on our customers. Our initial dependence on the commercial success of the SEVEN PLUS makes us particularly susceptible to any cost containment or reduction efforts. Accordingly, unless government and other third-party payors provide adequate coverage and reimbursement for the SEVEN PLUS, patients may not use our products.
In some foreign markets, pricing and profitability of medical devices are subject to government control. In the United States, we expect that there will continue to be federal and state proposals for similar controls. Also, the trends toward managed healthcare in the United States and proposed legislation intended to reduce the cost of government insurance programs could significantly influence the purchase of healthcare services and products and may result in lower prices for our products or the exclusion of our products from reimbursement programs.
We may never receive FDA approval or clearance to market our next generation SEVEN PLUS ambulatory system, or the GlucoClear, our blood-based in-vivo automated glucose monitoring system, or any other continuous glucose monitoring system under development.
Our SEVEN PLUS systems are classified by the FDA as PMA medical devices. We are continuing to seek approval for the next generation of our SEVEN PLUS ambulatory system, and are responding to FDA’s requests for additional data in support of that application. The PMA process requires us to prove the safety and efficacy of our ambulatory system to the FDA’s satisfaction. This process can be expensive, prolonged and uncertain, requires detailed and comprehensive scientific and human clinical data, and may never result in the FDA granting a PMA. We cannot predict when, if ever, the next generation of our SEVEN PLUS ambulatory system will obtain FDA approval.
27
Table of Contents
We also intend to seek approvals for the products that integrate our continuous glucose monitoring technology into the insulin delivery systems of Animas and Insulet, respectively, but cannot predict when, if ever, those products will be approved.
In addition, we are continuing to seek 510(k) clearance from the FDA for the GlucoClear, and are working with Edwards to formulate the next step for this submission. The 510(k) process would require us to establish (including through pre-clinical testing, bench testing, and/or potentially clinical data) that our GlucoClear system are substantially equivalent in terms of indication, technological characteristics, and performance to one or more legally marketed devices eligible to be cited as predicates in the 510(k) process. We cannot predict whether FDA will classify the GlucoClear as a 510(k) or PMA product, nor can we predict when, if ever, the GlucoClear will obtain FDA clearance or approval.
The FDA can refuse to grant us 510(k) clearance or delay, limit or deny approval of a PMA application for many reasons, including:
| • | our systems may not be deemed by the FDA to be substantially equivalent to appropriate predicate devices; |
| • | our systems may not satisfy the FDA’s safety or efficacy requirements; |
| • | the data from our pre-clinical studies and clinical trials may be insufficient to support approval; |
| • | the manufacturing process or facilities we use may not meet applicable requirements; and |
| • | changes in FDA approval policies or adoption of new regulations may require additional data. |
Even if approved or cleared by the FDA, the next generation of our SEVEN PLUS ambulatory system, the GlucoClear, or any other continuous glucose monitoring system under development, may not be approved or cleared for the indications that are necessary or desirable for successful commercialization. We may not obtain the necessary regulatory approvals or clearances to market these continuous glucose monitoring systems in the United States. Any delay in, or failure to receive or maintain, approval or clearance for the next generation of our SEVEN PLUS ambulatory system or the GlucoClear, could prevent us from generating revenue from these products or achieving profitability.
If we are unable to successfully complete the pre-clinical studies or clinical trials necessary to support additional PMA or 510(k) applications, we may be unable to commercialize our continuous glucose monitoring systems under development, including our next generation ambulatory system, our GlucoClear system or our systems being developed in collaboration with Animas and Insulet, which could impair our financial position.
We are continuing to seek approval for our next generation ambulatory system, and have been requested to provide additional data in support of that application. We also intend to seek approvals for the products that integrate our continuous glucose monitoring technology into the insulin delivery systems of Animas and Insulet, respectively. In addition, we have submitted to the FDA a 510(k) application for clearance for the GlucoClear, which application is pending before the FDA. The GlucoClear may ultimately be classified by the FDA as either a 510(k) or PMA product, and we may consequently be requested to provide additional data in support of the GlucoClear application.
To support these and any future additional PMA or 510(k) applications we must successfully complete pre-clinical studies, bench-testing, and clinical trials that we believe will demonstrate that the product is safe and effective. Product development, including pre-clinical studies and clinical trials, is a long, expensive and uncertain process and is subject to delays and failure at any stage. Furthermore, the data obtained from the studies and trial may be inadequate to support approval of a PMA or 510(k) application and the FDA may request additional clinical data in support of those applications, which may result in significant additional clinical
28
Table of Contents
expenses and may delay product approvals. While we have in the past obtained, and may in the future obtain, an Investigational Device Exemption, or IDE, prior to commencing clinical trials for our continuous glucose monitoring systems, FDA approval of an IDE application permitting us to conduct testing does not mean that the FDA will consider the data gathered in the trial to be sufficient to support approval of a PMA or 510(k) application, even if the trial’s intended safety and efficacy endpoints are achieved. Additionally, since 2009, the FDA has significantly increased the scrutiny applied to its oversight of companies subject to its regulations, including 510(k) submissions, by hiring new investigators and increasing the frequency and scope of its inspections of manufacturing facilities. The FDA has recently also significantly increased the number of warning letters issued to companies. Unexpected changes to the FDA or foreign regulatory approval processes could also delay or prevent the approval of our products submitted for review. The data drawn from our clinical trials may not be sufficient to support approval of our products or additional or expanded indications. Medical device stock prices have declined significantly in certain circumstances where companies have failed to meet expectations in regards to the timing of regulatory approval. If the FDA’s response causes product approval delays, or is not favorable for any of our products, our stock price could decline substantially.
The commencement or completion of any of our clinical trials may be delayed or halted, or be inadequate to support approval of a PMA or 510(k) application, for numerous reasons, including, but not limited to, the following:
| • | the FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; |
| • | patients do not enroll in clinical trials at the rate we expect; |
| • | patients do not comply with trial protocols; |
| • | patient follow-up does not occur at the rate we expect; |
| • | patients experience adverse side effects; |
| • | patients die during a clinical trial, even though their death may not be related to our products; |
| • | institutional review boards, or IRBs, and third-party clinical investigators may delay or reject our trial protocol; |
| • | third-party clinical investigators decline to participate in a trial or do not perform a trial on our anticipated schedule or consistent with the investigator agreements, clinical trial protocol, good clinical practices or other FDA or IRB requirements; |
| • | the company or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans; |
| • | third-party clinical investigators have significant financial interests related to the company or study that FDA deems to make the study results unreliable, or the company or investigators fail to disclose such interests; |
| • | regulatory inspections of our clinical trials or manufacturing facilities may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; |
| • | changes in governmental regulations, policies or administrative actions; |
| • | the interim or final results of the clinical trial are inconclusive or unfavorable as to safety or efficacy; and |
| • | the FDA concludes that our trial design is inadequate to demonstrate safety and efficacy. |
The results of pre-clinical studies do not necessarily predict future clinical trial results, and prior clinical trial results might not be repeated in subsequent clinical trials. Additionally, the FDA may disagree with our
29
Table of Contents
interpretation of the data from our pre-clinical studies and clinical trials, or may find the clinical trial design, conduct or results inadequate to prove safety or efficacy, and may require us to pursue additional pre-clinical studies or clinical trials, which could further delay the approval of our products. If we are unable to demonstrate the safety and efficacy of our products in our clinical trials to the FDA’s satisfaction, we will be unable to obtain regulatory approval to market our products in the United States. In addition, the data we collect from our current clinical trials, our pre-clinical studies and other clinical trials may not be sufficient to support FDA approval, even if our endpoints are met.
We depend on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trials and to perform related data collection and analysis, and, as a result, we may face costs and delays that are outside of our control.
We rely on clinical investigators and clinical sites to enroll patients in our clinical trials and other third parties to manage the trial and to perform related data collection and analysis. However, we may not be able to control the amount and timing of resources that clinical sites may devote to our clinical trials. If these clinical investigators and clinical sites fail to enroll a sufficient number of patients in our clinical trials or fail to ensure compliance by patients with clinical protocols or fail to comply with regulatory requirements, we will be unable to complete these trials, which could prevent us from obtaining regulatory approvals for our products. Our agreements with clinical investigators and clinical sites for clinical testing place substantial responsibilities on these parties and, if these parties fail to perform as expected, our trials could be delayed or terminated. If these clinical investigators, clinical sites or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated, or the clinical data may be rejected by the FDA, and we may be unable to obtain regulatory approval for, or successfully commercialize, our products.
Healthcare reforms, changes in healthcare policies and changes to third-party reimbursements for our products may affect demand for our products.
Comprehensive healthcare legislation, signed into law in March 2010, imposes stringent compliance, recordkeeping, and reporting requirements on companies in various sectors of the life sciences industry, with which we may need to comply, and enhanced penalties for non-compliance with the new healthcare regulations. The U.S. government may in the future consider healthcare policies and proposals intended to curb rising healthcare costs, including those that could significantly affect reimbursement for healthcare products such as the SEVEN PLUS. These policies have included, and may in the future include: basing reimbursement policies and rates on clinical outcomes, the comparative effectiveness and costs of different treatment technologies and modalities; imposing price controls and taxes on medical device providers; and other measures. Future significant changes in the healthcare systems in the United States or elsewhere could also have a negative impact on the demand for our current and future products. These include changes that may reduce reimbursement rates for our products and changes that may be proposed or implemented by the current U.S. Presidential administration or Congress.
In addition, the comprehensive healthcare reform legislation recently adopted by Congress and subsequently signed into law includes an annual excise tax on the sale of medical devices equal to 2.3% of the price of the device starting on January 1, 2013, which would likely include our SEVEN PLUS and GlucoClear systems. The exact impact of this excise tax, including whether our products would be considered “medical devices” and how such a tax would be assessed, is not currently clear. As a result of such tax, our future operating results could be harmed, which in turn could cause the price of our stock to decline, Additionally, because of the uncertainty surrounding these issues, the impact of this tax has not been reflected in our forward guidance.
30
Table of Contents
We conduct business in a heavily regulated industry and if we fail to comply with these laws and government regulations, we could suffer penalties or be required to make significant changes to our operations.
The healthcare industry is subject to extensive federal, state and local laws and regulations, including those relating to:
| • | billing for services; |
| • | financial relationships with physicians and other referral sources; |
| • | inducements and courtesies given to physicians and other health care providers and patients; |
| • | labeling products; |
| • | quality of medical equipment and services; |
| • | confidentiality, maintenance and security issues associated with medical records and individually identifiable health information; |
| • | medical device reporting; |
| • | false claims; and |
| • | professional licensure. |
These laws and regulations are extremely complex and, in some cases, still evolving. In many instances, the industry does not have the benefit of significant regulatory or judicial interpretation of these laws and regulations. If our operations are found to be in violation of any of the federal, state or local laws and regulations which govern our activities, we may be subject to the applicable penalty associated with the violation, including civil and criminal penalties, damages, fines or curtailment of our operations. The risk of being found in violation of these laws and regulations is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Any action against us for violation of these laws or regulations, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s time and attention from the operation of our business.
In addition, healthcare laws and regulations may change significantly in the future. Any new healthcare laws or regulations may adversely affect our business. A review of our business by courts or regulatory authorities may result in a determination that could adversely affect our operations. Also, the healthcare regulatory environment may change in a way that restricts or adversely impacts our operations.
We are not aware of any governmental healthcare investigations involving our executives or us. However, any future healthcare investigations of our executives, our managers or us could result in significant liabilities or penalties to us, as well as adverse publicity.
We have limited manufacturing capabilities and manufacturing personnel, and if our manufacturing capabilities are insufficient to produce an adequate supply of product at appropriate quality levels, our growth could be limited and our business could be harmed.
We currently have limited resources, facilities and experience in commercially manufacturing sufficient quantities of product to meet expected demand. In the past, we have had difficulty scaling our manufacturing operations to provide a sufficient supply of product to support our commercialization efforts. From time to time, we have also experienced brief periods of backorder and, at times, have had to limit the efforts of our sales force to introduce our products to new customers. We have focused significant effort on continual improvement programs in our manufacturing operations intended to improve quality, yields and throughput. We have made progress in manufacturing to enable us to supply adequate amounts of product to support our commercialization
31
Table of Contents
efforts, however, there can be no assurances that supply will not be constrained in the future. In order to produce our products in the quantities we anticipate will be necessary to meet market demand, we will need to increase our manufacturing capacity by a significant factor over the current level. In addition, we will have to modify our manufacturing design and process if and when our next generation sensor technologies are approved and commercialized. There are technical challenges to increasing manufacturing capacity, including equipment design and automation, materials procurement, manufacturing site expansion, problems with production yields and quality control and assurance. Developing commercial-scale manufacturing facilities will require the investment of substantial additional funds and the hiring and retention of additional management, quality assurance, quality control and technical personnel who have the necessary manufacturing experience. Also, the scaling of manufacturing capacity is subject to numerous risks and uncertainties, and may lead to variability in product quality or reliability, increased construction timelines, as well as resources required to design, install and maintain manufacturing equipment, among others, all of which can lead to unexpected delays in manufacturing output. In addition, any changes to our manufacturing processes may require FDA submission and approval and our facilities may have to undergo additional inspections by the FDA and corresponding state agencies. We cannot assure you that we will be able to develop and expand our manufacturing process and operations or obtain FDA and state agency approval of our facilities in a timely manner or at all. If we are unable to manufacture a sufficient supply of our current products or any future products for which we may receive approval, maintain control over expenses or otherwise adapt to anticipated growth, or if we underestimate growth, we may not have the capability to satisfy market demand and our business will suffer.
Additionally, the production of our products must occur in a highly controlled and clean environment to minimize particles and other yield-and quality-limiting contaminants. Weaknesses in process control or minute impurities in materials may cause a substantial percentage of defective products. If we are not able to maintain stringent quality controls, or if contamination problems arise, our clinical development and commercialization efforts could be delayed, which would harm our business and our results of operations.
Since our commercial launch in 2006, we have experienced periodic field failures, including reports of broken sensors or sensors that become lodged beneath a patient’s skin, as well as reports that a sensor fails to provide glucose values for a full seven days. We do not believe these failures necessitated device explant, other procedures, or non-standard clinical treatment or intervention. To comply with the FDA’s medical device reporting requirements, we have filed reports of all such broken or lodged sensors. Although we believe we have taken and are taking appropriate actions aimed at reducing or eliminating field failures, there can be no assurances that we will not experience additional failures going forward.
Our manufacturing operations are dependent upon third-party suppliers, making us vulnerable to supply problems and price fluctuations, which could harm our business.
We rely on Flextronics International, Ltd. to manufacture and supply circuit boards for our receiver; we rely on On Semiconductor Corp. to manufacture and supply the application specific integrated circuit, or ASIC, that is incorporated into the transmitter; we rely on DSM PTG, Inc. to manufacture certain polymers used to synthesize our polymeric biointerface membranes for our products; and we rely on The Tech Group to supply our injection molded components. Each of these suppliers is a single-source supplier. In some cases, our agreements with these and our other suppliers can be terminated by either party upon short notice. Our contract manufacturers also rely on single-source suppliers to manufacture some of the components used in our products. Our manufacturers and suppliers may encounter problems during manufacturing for a variety of reasons, including failure to follow specific protocols and procedures, failure to comply with applicable regulations, equipment malfunction and environmental factors, any of which could delay or impede their ability to meet our demand. Some of our single source suppliers, including Flextronics, are shifting their manufacturing and assembly sites to China and other international locations, which sites may require additional FDA approval and inspection. Should any such FDA approval be delayed, or such inspection require corrective action, our supply of critical components may be constrained or unavailable. Our reliance on these outside manufacturers and suppliers also subjects us to other risks that could harm our business, including:
| • | we may not be able to obtain adequate supply in a timely manner or on commercially reasonable terms; |
32
Table of Contents
| • | our products are technologically complex and it is difficult to develop alternative supply sources; |
| • | we are not a major customer of many of our suppliers, and these suppliers may therefore give other customers’ needs higher priority than ours; |
| • | our suppliers may make errors in manufacturing components that could negatively affect the efficacy or safety of our products or cause delays in shipment of our products; |
| • | we may have difficulty locating and qualifying alternative suppliers for our single-source supplies; |
| • | switching components may require product redesign and submission to the FDA of a PMA supplement or possibly a separate PMA, either of which could significantly delay production; |
| • | our suppliers manufacture products for a range of customers, and fluctuations in demand for the products these suppliers manufacture for others may affect their ability to deliver components to us in a timely manner; |
| • | our suppliers may make obsolete components that are critical to our products; and |
| • | our suppliers may encounter financial hardships unrelated to our demand for components, including those related to changes in global economic conditions, which could inhibit their ability to fulfill our orders and meet our requirements. |
We may not be able to quickly establish additional or replacement suppliers, particularly for our single-source components, in part because of the FDA approval process and because of the custom nature of various parts we design. Any interruption or delay in the supply of components or materials, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competitive products.
Potential long-term complications from our products or other continuous glucose monitoring systems under development may not be revealed by our clinical experience to date.
Based on our experience, complications from use of our SEVEN PLUS system may include broken or lodged sensors or skin irritation under the adhesive dressing of the sensor. Inflammation or redness, swelling, minor infection, and minor bleeding at the sensor insertion site are also possible risks with a patient’s use of the device. However, if unanticipated long-term side-effects result from the use of our products or other glucose monitoring systems under development, we could be subject to liability and our systems would not be widely adopted. With respect to our SEVEN PLUS, our clinical trials have been limited to seven days of continuous use. Additionally, we have limited clinical experience with repeated use of our products in the same patient. We cannot assure you that long-term use would not result in unanticipated complications. Furthermore, the interim results from our current pre-clinical studies and clinical trials may not be indicative of the clinical results obtained when we examine the patients at later dates. It is possible that repeated use of our products may result in unanticipated adverse effects, potentially even after the device is removed.
If we or our suppliers fail to comply with ongoing regulatory requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain marketing approval will be subject to continual review and periodic inspections by the FDA and other regulatory bodies, which may include inspection of our manufacturing processes, post-approval clinical data and promotional activities for such product. The FDA’s medical device reporting, or MDR, regulations require that we report to the FDA any incident in which our product may have caused or contributed to a death or serious injury, or in which our product malfunctioned and, if the malfunction were to recur, it would likely cause or contribute to a death or serious injury. We and our suppliers are also required to comply with the FDA’s Quality System Regulation, or QSR, and other regulations, which cover the methods and documentation of the design, testing, production, control, selection and oversight of suppliers or
33
Table of Contents
contractors, quality assurance, labeling, packaging, storage, complaint handling, shipping and servicing of our products. The FDA enforces the QSR through unannounced inspections. We currently manufacture our devices at our headquarters facilities in San Diego, California. In these facilities we have more than 8,000 square feet of laboratory space and approximately 10,000 square feet of controlled environment rooms. In February 2010, our facility was subject to a post-approval inspection by the FDA. After the close of the inspection, the FDA investigator issued a Form 483 identifying several inspectional observations. Subsequent to the inspection, we also received a warning letter from the FDA requiring us to file MDRs in accordance with the MDR regulations for complaints involving sensor wire fractures underneath a patient’s skin. The warning letter also recommended that we add certain warnings and precautions statements to the labeling, patient education brochures, and our company website regarding the appropriate use of the SEVEN PLUS system, including that they are not approved for use in children under age 18, pregnant women, or persons on dialysis. In response to the warning letter and the Form 483 inspectional observations, we have taken corrective action to address the observations to achieve substantial compliance with the FDA regulatory requirements applicable to a commercial medical device manufacturer. In October 2010, we were subject to a follow-up site inspection by the FDA, and upon completion of that inspection, we were notified by the inspector that there were no 483 inspectional observations. We also received written notification dated November 1, 2010 from the FDA that we adequately addressed all issues cited in the warning letter.
In March 2009, the FCC established a bifurcated MICS band which requires device manufacturers whose products will operate in the main MICS band to either manufacture their devices using listen-before-transmit technology, or to transmit on a side band outside the main MICS band at lower power. Although the SEVEN PLUS does not comply with existing MICS band listen-before-transmit requirements, the FCC granted a waiver to allow us to continue marketing and operating our SEVEN PLUS through March 2013, which we believe will provide adequate time to design an alternative method of wireless communication.
Compliance with ongoing regulatory requirements can be complex, expensive and time-consuming. Failure by us or one of our suppliers to comply with statutes and regulations administered by the FDA and other regulatory bodies, or failure to take adequate response to any observations, could result in, among other things, any of the following actions:
| • | warning letters or untitled letters that require corrective action; |
| • | delays in approving or refusal to approve our continuous glucose monitoring systems; |
| • | fines and civil penalties; |
| • | unanticipated expenditures; |
| • | FDA refusal to issue certificates to foreign governments needed to export our products for sale in other countries; |
| • | suspension or withdrawal of approval by the FDA or other regulatory bodies; |
| • | product recall or seizure; |
| • | interruption of production; |
| • | operating restrictions; |
| • | injunctions; and |
| • | criminal prosecution. |
If any of these actions were to occur, it would harm our reputation and cause our product sales and profitability to suffer. In addition, we believe events that could be classified as reportable events pursuant to MDR regulations are generally underreported by physicians and users, and any underlying problems could be of a larger magnitude than suggested by the number or types of MDRs filed by us. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with applicable regulatory requirements.
34
Table of Contents
Even if regulatory approval or clearance of a product is granted, the approval or clearance may be subject to limitations on the indicated uses for which the product may be marketed or contain requirements for costly post-marketing testing or surveillance to monitor the safety or efficacy of the product. Later discovery of previously unknown problems with our products, including software bugs, unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as the QSR, MDR reporting, or other postmarket requirements may result in restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties.
Abbott Diabetes Care, Inc. has filed a patent infringement lawsuit against us. If we are not successful in defending against its claims, our business could be materially impaired.
As further described in Item 3 “Legal Proceedings” of this Annual Report, Abbott Diabetes Care, Inc., or Abbott, has filed a patent infringement lawsuit against us, claiming that our continuous glucose monitor infringes certain patents held by Abbott. We have requested, and the Patent Office has granted, reexamination of each of the patents cited in this lawsuit. On September 30, 2007, the court granted our motion to stay the case pending conclusion of the reexamination proceedings in the Patent Office relating to all seven patents asserted against us.
Each of the seven patents described above has one or more associated reexamination requests in various stages at the Patent Office. Two of the patents are in the appeal process and the other five patents have been issued one or more Certificates of Reexamination. With regard to the two patents in the appeal process, the Board of Patent Appeals and Interferences within the Patent Office has recently rendered decisions. We believe these decisions are favorable to us; however, Abbott may seek judicial review of the decisions. Four of the remaining five patents are undergoing reexamination at the Patent Office. Reexamination of one of the remaining five patents was completed in 2010 and we subsequently submitted a reexamination request on this patent.
Since 2008, Abbott has copied claims from certain of our applications, and stated that it may seek to provoke an interference with certain of our pending applications in the Patent Office. If an interference is declared and Abbott prevails in the interference, we would lose certain patent rights to the subject matter defined in the interference. Also since 2008, Abbott has filed reexamination requests seeking to invalidate fifteen of our patents in the Patent Office. The fifteen reexamination requests are in various stages at the Patent Office. If the Patent Office were to determine in the reexamination that some or all of the claims of our patents are invalid, it could have a significant impact on our ability to protect aspects of our technology.
Although it is our position that Abbott’s assertions of infringement have no merit, and that the potential interference and reexamination requests have no merit, the outcome of the litigation and interference or reexamination requests cannot be assessed currently with any certainty. We may not successfully defend ourselves against the claims made by Abbott or prevail in the litigation. Subject to the stay of litigation, if Abbott were to seek and obtain a preliminary or permanent injunction, it could force us to stop making, using, selling or offering to sell our products. The technology at issue in our litigation with Abbott is currently used in our products, including SEVEN PLUS, our only current ambulatory product that is approved for commercial sale, and our GlucoClear system for in-hospital use. If we were forced to stop selling these products, our business and prospects would suffer. In addition, defending against this action could have a number of harmful effects on our business, including those discussed in the following risk factor, regardless of the final outcome of such litigation. For example, we have incurred, and expect to continue to incur, significant costs in defending the action.
Any adverse determination in litigation or interference proceedings to which we are or may become a party relating to patents could subject us to significant liabilities to third parties or require us to seek licenses from other third parties. Furthermore, if we are found to willfully infringe third-party patents, we could, in addition to other penalties, be required to pay treble damages and/or attorneys fees for the prevailing party. Although patent and intellectual property disputes in the medical device area have often been settled through licensing or similar arrangements, costs associated with such arrangements may be substantial and would likely include ongoing
35
Table of Contents
royalties. We may be unable to obtain necessary licenses on satisfactory terms. If we do not obtain necessary licenses, we may not be able to redesign our products to avoid infringement and any redesign may not receive FDA approval in a timely manner if at all. Adverse determinations in a judicial or administrative proceeding or failure to obtain necessary licenses could prevent us from manufacturing and selling our products, which would have a significant adverse impact on our business.
We are subject to claims of infringement or misappropriation of the intellectual property rights of others, which could prohibit us from shipping affected products, require us to obtain licenses from third parties or to develop non-infringing alternatives, and subject us to substantial monetary damages and injunctive relief. We may also be subject to other claims or suits.
Other companies, including Abbott, could, in the future, assert infringement or misappropriation claims against us with respect to our current or future products. Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Therefore, we cannot be certain that we have not infringed the intellectual property rights of such third parties or others. Our competitors may assert that our continuous glucose monitoring systems or the methods we employ in the use of our systems are covered by U.S. or foreign patents held by them. This risk is exacerbated by the fact that there are numerous issued patents and pending patent applications relating to self-monitored glucose testing systems in the medical technology field. Because patent applications may take years to issue, there may be applications now pending of which we are unaware that may later result in issued patents that our products infringe. There could also be existing patents of which we are unaware that one or more components of our system may inadvertently infringe. As the number of competitors in the market for continuous glucose monitoring systems grows, the possibility of inadvertent patent infringement by us or a patent infringement claim against us increases.
Any infringement or misappropriation claim, including the claim brought by Abbott, could cause us to incur significant costs, could place significant strain on our financial resources, divert management’s attention from our business and harm our reputation. If the relevant patents were upheld as valid and enforceable and we were found to infringe, we could be prohibited from selling our product that is found to infringe unless we could obtain licenses to use the technology covered by the patent or are able to design around the patent. We may be unable to obtain a license on terms acceptable to us, if at all, and we may not be able to redesign our products to avoid infringement. Even if we are able to redesign our products to avoid an infringement claim, we may not receive FDA approval for such changes in a timely manner or at all. A court could also order us to pay compensatory damages for such infringement, plus prejudgment interest and could, in addition, treble the compensatory damages and award attorney fees. These damages could be substantial and could harm our reputation, business, financial condition and operating results. A court also could enter orders that temporarily, preliminarily or permanently enjoin us and our customers from making, using, selling or offering to sell one or more of our products, or could enter an order mandating that we undertake certain remedial activities. Depending on the nature of the relief ordered by the court, we could become liable for additional damages to third parties.
In addition, from time to time, we are subject to various claims and suits arising out of the ordinary course of business, including commercial or employment related matters. Although individually we do not expect these claims or suits to have a material adverse effect on the Company, in the aggregate they may divert significant time and resources from the Company and our staff.
Our inability to adequately protect our intellectual property could allow our competitors and others to produce products based on our technology, which could substantially impair our ability to compete.
Our success and our ability to compete are dependent, in part, upon our ability to maintain the proprietary nature of our technologies. We rely on a combination of patent, copyright and trademark law, and trade secrets and nondisclosure agreements to protect our intellectual property. However, such methods may not be adequate to protect us or permit us to gain or maintain a competitive advantage. Our patent applications may not issue as patents in a form that will be advantageous to us, or at all. Our issued patents, and those that may issue in the
36
Table of Contents
future, may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products. In addition, proposed regulations may limit our ability to file continuing patent applications and pursue patent claims in the Patent Office.
To protect our proprietary rights, we may in the future need to assert claims of infringement against third parties. The outcome of litigation to enforce our intellectual property rights in patents, copyrights, trade secrets or trademarks is highly unpredictable, could result in substantial costs and diversion of resources, and could have a material adverse effect on our financial condition and results of operations regardless of the final outcome of such litigation. In the event of an adverse judgment, a court could hold that some or all of our asserted intellectual property rights are not infringed, invalid or unenforceable, and could award attorney fees.
Despite our efforts to safeguard our unpatented and unregistered intellectual property rights, we may not be successful in doing so or the steps taken by us in this regard may not be adequate to detect or deter misappropriation of our technology or to prevent an unauthorized third party from copying or otherwise obtaining and using our products, technology or other information that we regard as proprietary. Additionally, third parties may be able to design around our patents. Furthermore, the laws of foreign countries may not protect our proprietary rights to the same extent as the laws of the United States.
We operate in a highly competitive market and face competition from large, well-established medical device manufacturers with significant resources, and, as a result, we may not be able to compete effectively.
The market for glucose monitoring devices is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. In selling the SEVEN PLUS, we compete directly with Roche Diabetes Care, a division of Roche Diagnostics; LifeScan, Inc., a division of Johnson & Johnson; the MediSense and TheraSense divisions of Abbott Laboratories; and Bayer Corporation, each of which manufactures and markets products for the single-point finger stick device market. Collectively, these companies currently account for substantially all of the worldwide sales of self-monitored glucose testing systems. Several companies are developing or marketing short-term continuous glucose monitoring products that will compete directly with our products. To date, in addition to us, three other companies, Cygnus, Medtronic and Abbott, have received approval from the FDA to market continuous glucose monitors. We believe that one of the products, originally developed and marketed by Cygnus, is no longer actively marketed. In addition, we believe that others, including Bayer, are developing invasive and non-invasive continuous glucose monitoring systems. Most of the companies developing or marketing competing devices are publicly traded or divisions of publicly-traded companies, and these companies enjoy several competitive advantages, including:
| • | significantly greater name recognition; |
| • | established relations with healthcare professionals, customers and third-party payors; |
| • | established distribution networks; |
| • | additional lines of products, and the ability to offer rebates or bundle products to offer higher discounts or incentives to gain a competitive advantage; |
| • | greater experience in conducting research and development, manufacturing, clinical trials, obtaining regulatory approval for products and marketing approved products; and |
| • | greater financial and human resources for product development, sales and marketing, and patent litigation. |
As a result, we may not be able to compete effectively against these companies or their products.
37
Table of Contents
We have entered into a Collaboration Agreement with Edwards to develop jointly an in-hospital automated blood glucose monitoring device, branded as the GlucoClear, that may not result in the development of a commercially viable product or generation of any future revenues.
On November 10, 2008, we entered into a Collaboration Agreement with Edwards pursuant to which we have agreed to develop jointly and to market the GlucoClear, a blood-based in-vivo automated glucose monitoring system for use by healthcare providers in the hospital. Under the Collaboration Agreement, we expect to receive payments for various milestones related to regulatory approvals and commercial readiness of the product. In addition, we also expect to receive either a profit-sharing payment of 10% of commercial sales of the product, or a royalty of 6% of commercial sales of the product. The Collaboration Agreement provides Edwards with an exclusive license to our intellectual property that relates to blood-based glucose sensors in the critical care sector of the hospital market. However, this collaboration may not result in the development of products that achieve regulatory approval in the United States or commercial success, which would result in various penalties to us under the Collaboration Agreement, up to and including delay or loss of some or all of our milestone payments and rights to any profit-sharing or royalties. On October 30, 2009, we received CE Mark approval for the first generation GlucoClear that we developed in collaboration with Edwards. Although Edwards commenced market evaluations during 2009, this product did not generate significant revenue during 2010 and we do not expect this product to generate significant revenue during 2011. We are continuing to seek 510(k) clearance from the FDA for the GlucoClear, and are working with Edwards to formulate the next step for this submission. We cannot predict whether FDA will classify the GlucoClear as a 510(k) or PMA product, nor can we predict when, if ever, the GlucoClear will obtain FDA clearance or approval.
We enter into collaborations with third parties related to our SEVEN PLUS that may not result in the development of commercially viable products or the generation of significant future revenues.
In the ordinary course of our business, we enter into collaborative arrangements to develop new products and to pursue new markets, such as our agreements with Animas and Insulet, to integrate our continuous glucose monitoring technology into their respective insulin delivery systems. We have also entered into an OUS Commercialization Agreement, as amended, with Animas pursuant to which Animas retains the exclusive right to develop and market outside the United States an ambulatory insulin pump that is combined with our continuous glucose monitoring technology. These collaborations may not result in the development of products that achieve commercial success and could be terminated prior to developing any products. Accordingly, we cannot assure you that any of our collaborations will result in the successful development of a commercially viable product or result in significant additional future revenues. In addition, our development timelines are highly dependent on our ability to achieve clinical endpoints and regulatory requirements and to overcome technology challenges, and may be delayed due to scheduling issues with patients and investigators, requests from institutional review boards, product performance and manufacturing supply constraints, among other factors. In addition, support of these clinical trials requires significant resources from employees involved in the production of our products, including research and development, manufacturing, quality assurance, and clinical and regulatory personnel. Even if our development and clinical trial efforts are successful, the FDA may not approve the combined products or may require additional product testing and clinical trials before approving the combined products, which would result in product launch delays and additional expense. If approved by the FDA, the combined products may not achieve acceptance in the marketplace by physicians and patients.
To date, no continuous glucose monitoring system, including our SEVEN PLUS, has received FDA clearance as a replacement for single-point finger stick devices, and our SEVEN PLUS and future generations may never be approved for that indication.
The SEVEN PLUS does not eliminate the need for single-point finger stick devices and our future products may not be approved for that indication. No precedent for FDA approval of continuous glucose monitoring systems as a replacement for single-point finger stick devices has been established. Accordingly, there is no established study design or agreement regarding performance requirements or measurements in clinical trials for continuous glucose monitoring systems. We have not yet filed for FDA approval for therapeutic or replacement
38
Table of Contents
claim labeling and we cannot assure you that we will not experience delays if we do file. If any of our competitors were to obtain replacement claim labeling for a continuous glucose monitoring system, our products may not be able to compete effectively against that system and our business would suffer.
Technological breakthroughs in the glucose monitoring market could render our products obsolete.
The glucose monitoring market is subject to rapid technological change and product innovation. Our products are based on our proprietary technology, but a number of companies and medical researchers are pursuing new technologies for the monitoring of glucose levels. FDA approval of a commercially viable continuous glucose monitor or sensor produced by one of our competitors could significantly reduce market acceptance of our systems. Several of our competitors, including Bayer, are in various stages of developing continuous glucose monitors or sensors, including non-invasive and invasive devices, and the FDA has approved several of these competing products. In addition, the National Institutes of Health and other supporters of diabetes research are continually seeking ways to prevent, cure or improve treatment of diabetes. Therefore, our products may be rendered obsolete by technological breakthroughs in diabetes monitoring, treatment, prevention or cure.
We face the risk of product liability claims and may not be able to maintain or obtain insurance.
Our business exposes us to the risk of product liability claims that is inherent in the testing, manufacturing and marketing of medical devices, including those which may arise from the misuse or malfunction of, or design flaws in, our products. We may be subject to product liability claims if our products cause, or merely appear to have caused, an injury. Claims may be made by patients, healthcare providers or others selling our products.
Although we have product liability and clinical trial liability insurance that we believe is appropriate, this insurance is subject to deductibles and coverage limitations. Our current product liability insurance may not continue to be available to us on acceptable terms, if at all, and, if available, the coverage may not be adequate to protect us against any future product liability claims. Further, if additional products are approved for marketing, we may seek additional insurance coverage. If we are unable to obtain insurance at an acceptable cost or on acceptable terms with adequate coverage or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which may harm our business. A product liability claim, recall or other claim with respect to uninsured liabilities or for amounts in excess of insured liabilities could result in significant costs and significant harm to our business.
We may be subject to claims against us even if the apparent injury is due to the actions of others or misuse of the device. Our customers, either on their own or following the advice of their physicians, may use our products in a manner not described in the products’ labeling and that differs from the manner in which it was used in clinical studies and approved by the FDA. For example, our SEVEN PLUS is designed to be used by a patient continuously for up to seven days, but the patient might be able to circumvent the safeguards designed into the SEVEN PLUS and use the product for longer than seven days. Off-label use of products by patients is common, and any such off-label use of our products could subject us to additional liability. These liabilities could prevent or interfere with our product commercialization efforts. Defending a suit, regardless of merit, could be costly, could divert management attention and might result in adverse publicity, which could result in the withdrawal of, or inability to recruit, clinical trial volunteers or result in reduced acceptance of our products in the market.
We may be subject to fines, penalties and injunctions if we are determined to be promoting the use of our products for unapproved off-label uses.
Although we believe our promotional materials and training methods are conducted in compliance with FDA and other regulations, if the FDA determines that our promotional materials or training constitutes promotion of an unapproved use, the FDA could request that we modify our training or promotional materials or subject us to regulatory enforcement actions, including the issuance of a warning letter, injunction, seizure, civil
39
Table of Contents
fine and criminal penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
The majority of our operations are conducted at two facilities in San Diego, California. Any disruption at these facilities could increase our expenses.
We take precautions to safeguard our facilities, including insurance, health and safety protocols, and off-site storage of computer data. However, a natural disaster, such as a fire, flood or earthquake, could cause substantial delays in our operations, damage or destroy our manufacturing equipment or inventory, and cause us to incur additional expenses. The insurance we maintain against fires, floods, earthquakes and other natural disasters may not be adequate to cover our losses in any particular case.
We may be liable for contamination or other harm caused by materials that we handle, and changes in environmental regulations could cause us to incur additional expense.
Our research and development and clinical processes involve the handling of potentially harmful biological materials as well as hazardous materials. We are subject to federal, state and local laws and regulations governing the use, handling, storage and disposal of hazardous and biological materials and we incur expenses relating to compliance with these laws and regulations. If violations of environmental, health and safety laws occur, we could be held liable for damages, penalties and costs of remedial actions. These expenses or this liability could have a significant negative impact on our financial condition. We may violate environmental, health and safety laws in the future as a result of human error, equipment failure or other causes. Environmental laws could become more stringent over time, imposing greater compliance costs and increasing risks and penalties associated with violations. We are subject to potentially conflicting and changing regulatory agendas of political, business and environmental groups. Changes to or restrictions on permitting requirements or processes, hazardous or biological material storage or handling might require an unplanned capital investment or relocation. Failure to comply with new or existing laws or regulations could harm our business, financial condition and results of operations.
Failure to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products abroad.
We have begun limited commercial and marketing efforts in Europe and Israel with respect to our SEVEN PLUS and may seek to market our products in other regions in the future. Outside the United States, we can market a product only if we receive a marketing authorization and, in some cases, pricing approval, from the appropriate regulatory authorities. The approval procedure varies among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval in addition to other risks. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States on a timely basis, or at all.
Our success will depend on our ability to attract and retain our personnel.
We are highly dependent on our senior management, especially Terrance H. Gregg, our President and Chief Executive Officer, Steven R. Pacelli, our Chief Operating Officer, Jorge Valdes, our Chief Technical Officer, and Andrew K. Balo, our Senior Vice President of Clinical and Regulatory Affairs. Our success will depend on our ability to retain our current management and to attract and retain qualified personnel in the future, including sales
40
Table of Contents
persons, scientists, clinicians, engineers and other highly skilled personnel. Competition for senior management personnel, as well as sales persons, scientists, clinicians and engineers, is intense and we may not be able to retain our personnel. The loss of the services of members of our senior management, scientists, clinicians or engineers could prevent the implementation and completion of our objectives, including the commercialization of our current products and the development and introduction of additional products. The loss of a member of our senior management or our professional staff would require the remaining executive officers to divert immediate and substantial attention to seeking a replacement. Each of our officers may terminate their employment at any time without notice and without cause or good reason. Additionally, volatility or a lack of positive performance in our stock price may adversely affect our ability to retain key employees.
We expect to continue to expand our operations and grow our research and development, manufacturing, sales and marketing, product development and administrative operations. This expansion is expected to place a significant strain on our management and will require hiring a significant number of qualified personnel. Accordingly, recruiting and retaining such personnel in the future will be critical to our success. There is intense competition from other companies and research and academic institutions for qualified personnel in the areas of our activities. If we fail to identify, attract, retain and motivate these highly skilled personnel, we may be unable to continue our development and commercialization activities.
We have incurred and will incur increased costs as a result of recently enacted and proposed changes in laws and regulations relating to corporate governance matters.
The laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002 and rules adopted or proposed by the SEC will result in increased costs to us as we evaluate the implications of any new rules and regulations and respond to new requirements under such rules and regulations. We are required to comply with many of these rules and regulations, and will be required to comply with additional rules and regulations in the future. Furthermore, on July 21, 2010, President Obama signed into law the Dodd-Frank Wall Street Reform and Consumer Protection Act of 2010. This new law makes significant changes to corporate governance and executive compensation rules for public companies across all industries. As an early commercialization stage company with limited capital and human resources, we will need to divert management’s time and attention away from our business in order to ensure compliance with these regulatory requirements.
Valuation of share-based payments, which we are required to perform for purposes of recording compensation expense under authoritative guidance for share-based payment, involves significant assumptions that are subject to change and difficult to predict.
We record compensation expense in the consolidated statement of operations for share-based payments, such as employee stock options, using the fair value method. The requirements of the authoritative guidance for share-based payment have and will continue to have a material effect on our future financial results reported under Generally Accepted Accounting Principles, or GAAP, and make it difficult for us to accurately predict the impact our future financial results.
For instance, estimating the fair value of share-based payments is highly dependent on assumptions regarding the future exercise behavior of our employees and changes in our stock price. Our share-based payments have characteristics significantly different from those of freely traded options, and changes to the subjective input assumptions of our share-based payment valuation models can materially change our estimates of the fair values of our share-based payments. In addition, the actual values realized upon the exercise, expiration, early termination or forfeiture of share-based payments might be significantly different that our estimates of the fair values of those awards as determined at the date of grant. Moreover, we rely on third parties that supply us with information or help us perform certain calculations that we employ to estimate the fair value of share-based payments. If any of these parties do not perform as expected or make errors, we may inaccurately calculate actual or estimated compensation expense for share-based payments.
41
Table of Contents
The authoritative guidance for share-based payment could also adversely impact our ability to provide accurate guidance on our future financial results as assumptions that are used to estimate the fair value of share-based payments are based on estimates and judgments that may differ from period to period. We may also be unable to accurately predict the amount and timing of the recognition of tax benefits associated with share-based payments as they are highly dependent on the exercise behavior of our employees and the price of our stock relative to the exercise price of each outstanding stock option.
For those reasons, among others, the authoritative guidance for share-based payment may create variability and uncertainty in the share-based compensation expense we will record in future periods, which could adversely impact our stock price and increase our expected stock price volatility as compared to prior periods.
Changes in financial accounting standards or practices or existing taxation rules or practices may cause adverse unexpected revenue and/or expense fluctuations and affect our reported results of operations.
A change in accounting standards or practices or a change in existing taxation rules or practices can have a significant effect on our reported results and may even affect our reporting of transactions completed before the change is effective. New accounting pronouncements and taxation rules and varying interpretations of accounting pronouncements and taxation practice have occurred and may occur in the future. The method in which we market and sell our products may have an impact on the manner in which we recognize revenue. In addition, changes to existing rules or the questioning of current practices may adversely affect our reported financial results or the way we conduct our business. For example, as a result of changes approved by the Financial Accounting Standards Board, or FASB, on January 1, 2006 we began recording compensation expense in our statements of operations for equity compensation instruments, including employee stock options, using the fair value method. Our reported financial results beginning for the first quarter of 2006 and for all foreseeable future periods will be negatively and materially impacted by this accounting change. Other potential changes in existing taxation rules related to stock options and other forms of equity compensation could also have a significant negative effect on our reported results.
Our loan and security agreement contains restrictions that may limit our operating flexibility.
In March 2006, we entered into our Loan Agreement that provided for a loan to finance various equipment and leasehold improvement expenses. In January 2008, we amended our Loan Agreement to enable us to draw an additional $3.0 million. We are required to repay this additional amount at intervals through July 2011. As of December 31, 2010, we had a total outstanding loan balance under the Loan Agreement of $525,000. The Loan Agreement requires us to maintain a minimum cash balance with Square 1 Bank, and also imposes certain limitations on us, including, among others, limitations on our ability to:
| • | transfer all or any part of our businesses or properties, other than transfers done in the ordinary course of business; |
| • | engage in any business other than the businesses in which we are currently engaged; |
| • | relocate our chief executive offices or state of incorporation; |
| • | change our legal name or fiscal year; |
| • | replace our chief executive officer or chief financial officer; |
| • | merge or consolidate with or into any other business organizations, with certain exceptions; |
| • | permit any person to beneficially own a sufficient number of shares entitling such person to elect a majority of our board of directors; |
| • | incur additional indebtedness, with certain exceptions; |
| • | incur liens with respect to any of our properties, with certain exceptions; or |
42
Table of Contents
| • | store any equipment or inventory in which the lender has any interest with any bailee, warehousemen or similar third party unless the third party has been notified of the lender’s security interest. |
Complying with these covenants may make it more difficult for us to successfully execute our business strategy and compete against companies who are not subject to such restrictions.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
| ITEM 2. | PROPERTIES |
We maintain our headquarters in San Diego, California in two leased facilities of approximately 102,844 square feet, which includes our laboratory, research and development, manufacturing and general administration functions. The lease for these facilities expires in 2016. We have the right to extend the term of this lease for one period of five years. During 2010, we also maintained a third facility in San Diego, California, which was located at our former headquarters. The lease for this facility expires in 2011. We have not entered into a sublease agreement for our former headquarters facility. In February 2010, our facility was subject to a post-approval inspection by the FDA. After the close of the inspection, the FDA inspector issued a Form 483 identifying several inspectional observations. Subsequent to the inspection, we also received a warning letter from the FDA requiring us to file MDRs in accordance with the MDR regulations for complaints involving sensor wire fractures underneath a patient’s skin. The warning letter also recommended that we add certain warnings and precautions statements to the labeling, patient education brochures, and our company website regarding the appropriate use of the SEVEN and SEVEN PLUS Systems, including that they are not approved for use in children under age 18, pregnant women, or persons on dialysis. In response to the warning letter and the Form 483 inspectional observations, we have taken corrective action to address the observations to achieve substantial compliance with the FDA regulatory requirements applicable to a commercial medical device manufacturer. In October 2010, we were subject to a follow-up site inspection by the FDA, and upon completion of that inspection, we were notified by the inspector that there were no 483 inspectional observations. We also received written notification dated November 1, 2010 from the FDA that we adequately addressed all issues cited in the warning letter. We previously leased a smaller facility of approximately 7,000 square feet near our former headquarters. We entered into a sublease agreement with an unaffiliated third-party to lease this facility from us through the balance of the lease term. We believe that our existing facilities are adequate to meet our needs for the foreseeable future, and that suitable additional space will be available in the future on commercially reasonably terms as needed.
| ITEM 3. | LEGAL PROCEEDINGS |
On August 11, 2005, Abbott Diabetes Care, Inc., or Abbott, filed a patent infringement lawsuit against us in the United States District Court for the District of Delaware, seeking a declaratory judgment that our continuous glucose monitor infringes certain patents held by Abbott. In August 2005, we moved to dismiss these claims and filed requests for reexamination of the Abbott patents with the United States Patent and Trademark Office, or the Patent Office, and by March 2006, the Patent Office ordered reexamination of each of the four patents originally asserted against us in the litigation. On June 27, 2006, Abbott amended its complaint to include three additional patents owned or licensed by Abbott which are allegedly infringed by our continuous glucose monitor. On August 18, 2006, the court granted our motion to stay the lawsuit pending reexamination by the Patent Office of each of the four patents originally asserted by Abbott, and the court dismissed one significant infringement claim. In approving the stay, the court also granted our motion to strike, or disallow, Abbott’s amended complaint in which Abbott had sought to add three additional patents to the litigation. Subsequent to the court’s August 18, 2006 order striking Abbott’s amended complaint, Abbott filed a separate action in the U.S. District Court for the District of Delaware alleging patent infringement of the three additional patents it had sought to include in the litigation discussed above. On September 7, 2006, we filed a motion to strike Abbott’s new complaint on the
43
Table of Contents
grounds that it was redundant of claims Abbott already improperly attempted to inject into the original case, and because the original case has been stayed, Abbott must wait until the court lifts that stay before it could properly ask the court to consider these claims. Alternatively, we asked the court to consolidate the new case with the original case and thereby stay the entirety of the case pending conclusion of the reexamination proceedings in the Patent Office. In February 2007, the Patent Office ordered reexamination of each of the three patents cited in this new lawsuit. On September 30, 2007, the court granted our motion to consolidate the cases and stay the entirety of the case pending conclusion of the reexamination proceedings in the Patent Office relating to all seven patents asserted against us.
As of the date of this Report, each of Abbott’s seven patents that are the subject of the litigation has one or more associated reexamination requests in various stages at the Patent Office. Abbott has filed responses with the Patent Office seeking claim construction to differentiate certain claims from the prior art we have presented, seeking to amend certain claims to overcome the prior art we have presented, canceling claims and/or seeking to add new claims. The Board of Patent Appeals and Interferences within the Patent Office has recently rendered decisions on the appeals related to the reexaminations of two of the patents. We believe these decisions are favorable to us; however, Abbott may still seek judicial review of the decisions. Four patents are currently undergoing reexamination at the Patent Office. Reexamination of another patent was completed in 2010 and we recently submitted a subsequent reexamination request.
In addition, since 2008, Abbott has copied claims from certain of our applications, and stated that it may seek to provoke an interference with certain of our pending applications in the Patent Office. If interference is declared and Abbott prevails in the interference, we would lose certain patent rights to the subject matter defined in the interference. Also since 2008, Abbott has filed reexamination requests seeking to invalidate fifteen of our patents. The fifteen reexamination requests are in various stages at the Patent Office. We have filed responses with the Patent Office seeking claim construction to differentiate certain claims from the prior art presented in the reexaminations, seeking to amend certain claims to overcome the prior art presented in the reexaminations, canceling claims and/or seeking to add new claims. It is possible that the Patent Office may determine that some or all of the claims of our patents subject to the reexamination are invalid.
Although it is our position that Abbott’s assertions of infringement in the litigation and requests for interference and reexamination proceedings with the Patent Office have no merit, the outcome of the litigation and interference and reexamination requests cannot be assessed currently with any certainty. Rulings against us in the litigation or in any potential interference or reexamination proceeding could have a material adverse impact on our business and prospects.
44
Table of Contents
PART II
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
DexCom’s common stock is traded on the NASDAQ Global Market under the symbol “DXCM.” As of February 28, 2011, there were approximately 91 stockholders of record, excluding stockholders whose shares were held in nominee or street name by brokers. We have not paid any cash dividends and do not currently have plans to do so in the foreseeable future. Additionally, our loan agreement prohibits us from paying cash dividends without the lender’s prior written consent.
The following table sets forth the high and low intraday sales price per share for DexCom’s common stock for the periods indicated:
| High | Low | |||||||
| Year Ended December 31, 2010 |
||||||||
| First Quarter |
$ | 10.95 | $ | 8.21 | ||||
| Second Quarter |
$ | 12.02 | $ | 8.92 | ||||
| Third Quarter |
$ | 14.18 | $ | 10.68 | ||||
| Fourth Quarter |
$ | 14.47 | $ | 9.91 | ||||
| High | Low | |||||||
| Year Ended December 31, 2009 |
||||||||
| First Quarter |
$ | 4.72 | $ | 2.62 | ||||
| Second Quarter |
$ | 7.05 | $ | 3.53 | ||||
| Third Quarter |
$ | 8.96 | $ | 5.25 | ||||
| Fourth Quarter |
$ | 8.26 | $ | 6.53 | ||||
Neither we nor any affiliated purchaser repurchased any of our equity securities in fiscal year 2010.
45
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA |
The consolidated statements of operations data for the years ended December 31, 2010, 2009, and 2008 and the consolidated balance sheet data as of December 31, 2010 and 2009 have been derived from our audited consolidated financial statements included elsewhere in this annual report. The statements of operations data for the years ended December 31, 2007 and 2006 and the balance sheet data as of December 31, 2008, 2007 and 2006 have been derived from our audited financial statements not included in this annual report. The following selected financial data should be read in conjunction with our “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and consolidated financial statements and related notes to those statements included elsewhere in this annual report.
| Years Ended December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Consolidated Statements of Operations Data: |
||||||||||||||||||||
| Product revenue |
$ | 40,175 | $ | 18,036 | $ | 8,108 | $ | 4,627 | $ | 2,170 | ||||||||||
| Development grant and other revenue |
8,456 | 11,657 | 1,730 | — | — | |||||||||||||||
| Total revenue |
48,631 | 29,693 | 9,838 | 4,627 | 2,170 | |||||||||||||||
| Product cost of sales |
26,104 | 18,216 | 13,383 | 12,736 | 10,959 | |||||||||||||||
| Development and other cost of sales |
4,084 | 7,816 | 1,984 | — | — | |||||||||||||||
| Total cost of sales |
30,188 | 26,032 | 15,367 | 12,736 | 10,959 | |||||||||||||||
| Gross margin (deficit) |
18,443 | 3,661 | (5,529 | ) | (8,109 | ) | (8,789 | ) | ||||||||||||
| Operating expenses: |
||||||||||||||||||||
| Research and development |
23,227 | 14,294 | 19,629 | 16,131 | 19,419 | |||||||||||||||
| Selling, general and administrative |
40,506 | 35,200 | 27,669 | 22,436 | 21,111 | |||||||||||||||
| Total operating expenses |
63,733 | 49,494 | 47,298 | 38,567 | 40,530 | |||||||||||||||
| Operating loss |
(45,290 | ) | (45,833 | ) | (52,827 | ) | (46,676 | ) | (49,319 | ) | ||||||||||
| Other income |
63 | — | 34 | — | — | |||||||||||||||
| Interest income |
95 | 354 | 1,220 | 3,782 | 2,815 | |||||||||||||||
| Interest expense |
(1,548 | ) | (8,045 | ) | (7,283 | ) | (5,560 | ) | (95 | ) | ||||||||||
| Loss on debt extinguishment upon conversion of convertible debt |
(8,490 | ) | — | — | — | — | ||||||||||||||
| Net loss |
$ | (55,170 | ) | $ | (53,524 | ) | $ | (58,856 | ) | $ | (48,454 | ) | $ | (46,599 | ) | |||||
| Basic and diluted net loss per share attributable to common stockholders(1) |
$ | (0.97 | ) | $ | (1.21 | ) | $ | (2.00 | ) | $ | (1.71 | ) | $ | (1.71 | ) | |||||
| Shares used to compute basic and diluted net loss per share attributable to common stockholders(1) |
56,881 | 44,347 | 29,487 | 28,313 | 27,236 | |||||||||||||||
| As of December 31, | ||||||||||||||||||||
| 2010 | 2009 | 2008 | 2007 | 2006 | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Consolidated Balance Sheet Data: |
||||||||||||||||||||
| Cash, cash equivalents, and marketable securities |
$ | 47,113 | $ | 28,016 | $ | 27,068 | $ | 64,323 | $ | 54,508 | ||||||||||
| Working capital |
50,896 | 18,124 | 17,062 | 58,844 | 52,126 | |||||||||||||||
| Total assets |
77,164 | 46,948 | 43,882 | 76,388 | 64,553 | |||||||||||||||
| Long term obligations |
1,042 | 46,597 | 48,354 | 38,009 | 2,118 | |||||||||||||||
| Total stockholders’ equity (deficit) |
60,993 | (18,445 | ) | (19,468 | ) | 29,932 | 56,828 | |||||||||||||
| (1) | See Note 2 of the notes to our consolidated financial statements for a description of the method used to compute basic and diluted net loss per share attributable to common stockholders. |
46
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This document, including the following Management’s Discussion and Analysis of Financial Condition and Results of Operations, contains forward-looking statements that are based upon current expectations. These forward-looking statements fall within the meaning of the federal securities laws that relate to future events or our future financial performance. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “intend,” “potential” or “continue” or the negative of these terms or other comparable terminology. Forward-looking statements involve risks and uncertainties. Our actual results and the timing of events could differ materially from those anticipated in our forward-looking statements as a result of many factors, including product performance, a lack of acceptance in the marketplace by physicians and patients, the inability to manufacture products in commercial quantities at an acceptable cost, possible delays in our research and development programs, the inability of patients to receive reimbursements from third-party payors, inadequate financial and other resources, global economic conditions, and the other risks set forth below under “Risk Factors” and elsewhere in this report. We assume no obligation to update any of the forward-looking statements after the date of this report or to conform these forward-looking statements to actual results.
Overview
We are a medical device company focused on the design, development and commercialization of continuous glucose monitoring systems for ambulatory use by people with diabetes and for use by healthcare providers in the hospital for the treatment of both diabetic and non-diabetic patients. The majority of our product revenue comes from sales of our SEVEN PLUS ambulatory continuous glucose monitoring system, which we began commercializing in the first quarter of 2009. We also have received CE Mark approval for the GlucoClear in-hospital system, and in partnership with Edwards, we initiated a very limited launch of the GlucoClear system in Europe in 2009.
From inception to 2006, we devoted substantially all of our resources to start-up activities, raising capital and research and development, including product design, testing, manufacturing and clinical trials. Since 2006, we have devoted considerable resources to the commercialization of our ambulatory continuous glucose monitoring systems, including the SEVEN PLUS, as well as the continued research and clinical development of our technology platform.
From inception through December 31, 2010, we generated $95.0 million of product and development grant and other (non-product) revenue, and we have incurred net losses in each year since our inception in May 1999. From inception through December 31, 2010, we had an accumulated deficit of $346.4 million. We expect our losses to continue as we proceed with our commercialization and research and development activities. We have financed our operations primarily through offerings of equity securities and convertible debt. In April 2005, we completed our initial public offering in which we sold 4,700,000 shares of common stock for net proceeds of $50.5 million. In March 2006, we entered into a Loan Agreement, which was subsequently amended in January 2008. As of December 31, 2010, we had an outstanding balance of $525,000 under the Loan Agreement. In May 2006, we completed a follow-on public offering of 2,117,375 shares of our common stock for net proceeds of $47.0 million. In March 2007, we issued an aggregate principal amount of $60.0 million of 4.75% convertible senior notes due in 2027, all of which convertible senior notes have converted into shares of our common stock. In February 2009, we completed a follow-on public offering of 15,994,000 shares of our common stock for net proceeds of approximately $45.6 million. In January 2010, we completed a follow-on public offering of 4,025,000 shares of our common stock for net proceeds of approximately $33.0 million. In November 2010, we completed a follow-on public offering of 3,277,500 shares of our common stock for net proceeds of approximately $33.0 million.
47
Table of Contents
Financial Operations
Revenue
From inception through December 31, 2010, we generated $73.1 million in product revenue from the sale of our continuous glucose monitoring systems. We expect that revenues we generate from the sales of our products will fluctuate from quarter to quarter. During the first quarter of 2008, we entered into a joint development agreement with Animas and we recognize development grant and other revenue received pursuant to that agreement ratably over the term of the development period. During the fourth quarter of 2008, we entered into a collaboration agreement with Edwards and we recognize development grant and other revenue received pursuant to that agreement ratably over the term of the development period. From inception through December 31, 2010, we recognized $21.8 million in development grant and other revenue, which includes milestones and services.
Cost of Sales
Product cost of sales includes direct labor and materials costs related to each product sold or produced, including assembly, test labor and scrap, as well as factory overhead supporting our manufacturing operations. Factory overhead includes facilities, material procurement and control, manufacturing engineering, quality assurance, supervision and management. These costs are primarily salary, fringe benefits, share-based compensation, facility expense, supplies and purchased services. The majority of our costs are currently fixed due to our relatively low production volumes compared to our potential capacity. All of our manufacturing costs are included in product cost of sales. Development and other cost of sales consists primarily of salaries, fringe, facilities, and supplies directly attributable to our development contracts.
Research and Development
Our research and development expenses primarily consist of engineering and research expenses related to our continuous glucose monitoring technology, clinical trials, regulatory expenses, quality assurance programs, materials and products for clinical trials. Research and development expenses are primarily related to employee compensation, including salary, fringe benefits, share-based compensation, and temporary employee expenses. We also incur significant expenses to operate our clinical trials including clinical site reimbursement, clinical trial product and associated travel expenses. Our research and development expenses also include fees for design services, contractors and development materials.
Selling, General and Administrative
Our selling, general and administrative expenses primarily consist of salary, fringe benefits and share-based compensation for our executive, financial, sales, marketing and administrative functions. Other significant expenses include trade show expenses, sales samples, insurance, professional fees for our outside legal counsel and independent auditors, litigation expenses and consulting expenses.
Results of Operations
Fiscal year ended December 31, 2010 Compared to December 31, 2009
Revenue, Cost of Sales and Gross Margin
Product revenues increased $22.1 million to $40.2 million for the twelve months ended December 31, 2010 compared to $18.0 million for the twelve months ended December 31, 2009 based primarily on increased sales volume of our durable systems and disposable sensors, and higher average per unit selling prices. Product cost of sales increased $7.9 million to $26.1 million for the twelve months ended December 31, 2010 compared to $18.2 million for the twelve months ended December 31, 2009. The increased product cost of sales associated with additional product sales was offset primarily by increased manufacturing absorption for the twelve months ended December 31, 2010 as compared to the same period in 2009. The product gross margin of $14.1 million for the
48
Table of Contents
twelve months ended December 31, 2010 increased $14.3 million compared to a loss of $180,000 for the same period in 2009, primarily due to increased revenue and improved direct labor utilization. During the twelve months ended December 31, 2010, we increased overall inventory levels to meet sales forecast requirements and in anticipation of Flextronics’ relocation of its production site from California to China. Flextronics, which manufactures our ambulatory hardware components, relocated its production lines during the first calendar quarter of 2011. The increase in our inventory levels resulted in additional absorption of manufacturing costs and a corresponding nominal improvement in product gross margin.
Development grant and other revenues decreased $3.2 million to $8.5 million for the twelve months ended December 31, 2010 compared to $11.7 million for the twelve months ended December 31, 2009. Development and other cost of sales decreased $3.7 million to $4.1 million for the twelve months ended December 31, 2010 compared to $7.8 million for the twelve months ended December 31, 2009. The decrease in development grant and other revenues during the twelve months ended December 31, 2010 was based on longer than expected development and regulatory review timelines under our collaboration arrangements with Edwards and Animas. The decrease in costs associated with development was primarily due to fewer development obligations during the year with respect to our collaboration arrangements.
Research and Development. Research and development expense increased $8.9 million to $23.2 million for the twelve months ended December 31, 2010, compared to $14.3 million for the twelve months ended December 31, 2009. The increase in research and development expense was primarily due to increased development efforts for our future generation ambulatory products and by decreased activity with respect to our development and collaboration agreements. Major elements of increased research and development costs include $4.4 million in additional salaries, bonus, and payroll related costs, $1.2 million in additional share-based compensation, and $764,000 in additional facilities costs.
Selling, General and Administrative. Selling, general and administrative expense increased $5.3 million to $40.5 million for the twelve months ended December 31, 2010, compared to $35.2 million for the twelve months ended December 31, 2009. The increase was primarily due to higher selling, information technology, and international development costs to support revenue growth and the continued commercialization of our products. Major elements of increased selling, general, and administrative expenses include $4.0 million in higher salaries, bonus, and payroll related costs, $590,000 in higher depreciation expense, and $420,000 in higher commissions.
Interest Income. Interest income decreased $259,000 to $95,000 for the twelve months ended December 31, 2010, compared to $354,000 for the twelve months ended December 31, 2009. The decrease in interest income was primarily due to lower average interest bearing cash and marketable securities balances and lower yields earned on those balances during the twelve months ended December 31, 2010 as compared to the same period of 2009.
Interest Expense. Interest expense decreased $6.5 million to $1.5 million for the twelve months ended December 31, 2010, compared to $8.0 million for the twelve months ended December 31, 2009. The decrease in interest expense was primarily due to lower non-cash interest expense relating to the accretion of the debt discount for the 4.75% senior convertible notes (the “Notes”) issued in March 2007, and lower coupon interest expense relating to the Notes outstanding as a result of the conversions of the Notes that occurred during the twelve months ended December 31, 2010.
Loss on Debt Extinguishment upon Conversion of Convertible Debt
For the twelve months ended December 31, 2010, we completed exchanges with prior holders of our issued and outstanding Notes, under which we issued an aggregate of approximately 7.9 million shares of our common stock, par value $0.001 per share, in exchange for $60.0 million in aggregate principal amount of the Notes previously held by the exchanging holders. We incurred a loss on the extinguishment of the Notes in the amount
49
Table of Contents
of $8.5 million for the twelve months ended December 31, 2010, which includes the difference between the carrying value and the fair value of the Notes on the conversion date, other consideration given to note holders to induce early conversion and transaction costs incurred with third parties, other than the investors, to settle the conversion of the Notes.
Fiscal year ended December 31, 2009 Compared to December 31, 2008
Revenue, Cost of Sales and Gross Margin
Product revenues increased $9.9 million to $18.0 million for the twelve months ended December 31, 2009 compared to $8.1 million for the twelve months ended December 31, 2008 based primarily on increased sales volume and higher average per unit selling prices. Product cost of sales increased $4.8 million to $18.2 million for the twelve months ended December 31, 2009 compared to $13.4 million for the twelve months ended December 31, 2008. The increased product cost of sales associated with additional product sales was offset primarily by increased manufacturing absorption for the twelve months ended December 31, 2009 as compared to the same period in 2008. The product gross margin loss of $180,000 for the twelve months ended December 31, 2009 decreased $5.1 million compared to $5.3 million for the same period in 2008, primarily due to increased revenue and better direct labor utilization.
Development grant and other revenues increased $10.0 million to $11.7 million for the twelve months ended December 31, 2009 compared to $1.7 million for the twelve months ended December 31, 2008. Development and other cost of sales increased $5.8 million to $7.8 million for the twelve months ended December 31, 2009 compared to $2.0 million for the twelve months ended December 31, 2008. The increase in both revenues and costs associated with development was primarily due to our continuing performance obligations under a joint development agreement with Animas originally signed in the first quarter of 2008 and our continuing performance obligations under a collaboration agreement with Edwards originally signed in the fourth quarter of 2008.
Research and Development. Research and development expense decreased $5.3 million to $14.3 million for the twelve months ended December 31, 2009, compared to $19.6 million for the twelve months ended December 31, 2008. The decrease in research and development expense was primarily due to the joint development and collaboration agreements originally entered into with Animas and Edwards in 2008, and the corresponding allocation of expenses to development cost of sales for the ongoing delivery of these agreements. Changes in research and development expense include $3.8 million in lower development costs and $1.6 million in lower clinical and regulatory and quality assurance costs. Major elements of decreased research and development costs include $2.3 million in lower salaries and payroll related costs, $1.1 million in lower facilities costs, and $509,000 in lower consulting costs.
Selling, General and Administrative. Selling, general and administrative expense increased $7.5 million to $35.2 million for the twelve months ended December 31, 2009, compared to $27.7 million for the twelve months ended December 31, 2008. The increase was primarily due to higher selling, marketing, and international development costs to support revenue growth and the continued commercialization of our products. Major elements of increased selling, general, and administrative expenses include $4.3 million in higher salaries, bonus, and payroll related costs, $839,000 in higher commissions, and $722,000 in higher share-based compensation.
Interest Income. Interest income decreased $866,000 to $354,000 for the twelve months ended December 31, 2009, compared to $1.2 million for the twelve months ended December 31, 2008. The decrease in interest income was primarily due to lower average interest bearing cash and marketable securities balances and lower yields earned on those balances during the twelve months ended December 31, 2009 as compared to the same period of 2008.
50
Table of Contents
Interest Expense. Interest expense increased $762,000 to $8.0 million for the twelve months ended December 31, 2009, compared to $7.3 million for the twelve months ended December 31, 2008. The increase in interest expense was primarily due to additional non-cash interest expense relating to the 4.75% convertible notes issued in March of 2007 and the accretion of the debt discount to interest expense over the instrument’s expected life using the effective interest method.
Liquidity and Capital Resources
We are in the early commercialization stage and have incurred losses since our inception in May 1999. As of December 31, 2010, we had an accumulated deficit of $346.4 million and had working capital of $50.9 million. Our cash, cash equivalents and short-term marketable securities totaled $47.1 million, excluding $1.7 million in restricted cash. We have funded our operations primarily from the sale of equity and debt securities and our bank line. As of December 31, 2010 we had a total of $525,000 outstanding under our amended bank equipment loan that we are required to repay through July 2011. In January 2010, we completed a follow-on public offering of 4,025,000 shares of our common stock for net proceeds of approximately $33.0 million. In November 2010, we completed a public follow-on offering of 3,277,500 shares of our common stock for net proceeds of approximately $33.0 million.
Net Cash Used in Operating Activities. Net cash used in operating activities increased $3.3 million to $42.7 million for the twelve months ended December 31, 2010, compared to $39.4 million for the same period in 2009. The increase in cash used in operating activities was primarily due to $7.6 million in additional changes in operating assets and liabilities and $1.6 million in higher net loss, offset by $5.9 million additional non-cash charges primarily comprised of loss on the extinguishment of debt upon conversion of our Notes. The additional changes in operating assets and liabilities was primarily due to $5.3 million in increased inventory levels to meet sales forecast requirements and in anticipation of our ambulatory hardware manufacturer, Flextronics, relocating its production site from California to China.
Net Cash Provided By Investing Activities. Net cash used in investing activities was $25.6 million for the twelve months ended December 31, 2010, compared to $14.1 million provided for the same period of 2009. The increase in cash used in investing activities was primarily due to $8.2 million increase in cash used to purchase available-for-sale marketable securities and by $3.9 million increase in cash used to purchase property and equipment for the twelve months ended December 31, 2010 as compared to the same period in 2009. For the twelve months ended December 31, 2010, we invested $6.9 million in equipment to support manufacturing improvements compared to $3.0 million during the same period in 2009.
Net Cash Provided by Financing Activities. Net cash provided by financing activities increased $25.3 million to $69.6 million for the twelve months ended December 31, 2010, compared to $44.4 million for the same period of 2009. The increase was primarily due to the $33.0 million in net proceeds generated by the sale of common stock in the follow on public offering completed in January 2010 and the $33.0 million in net proceeds generated by the sale of common stock in the follow on public offering completed in November 2010 for the twelve months ending December 31, 2010 compared to $45.6 million in the same period of 2009.
Operating Capital and Capital Expenditure Requirements
We anticipate that we will continue to incur net losses for the foreseeable future as we incur expenses to continue expand the commercialization of our approved products, develop additional continuous glucose monitoring products, and expand our marketing, manufacturing and corporate infrastructure.
We believe that our cash, cash equivalents, short-term marketable securities balances, and projected cash contributions from existing partnership arrangements will be sufficient to meet our anticipated cash requirements with respect to the continued scale-up of our commercialization activities, research and development activities, including clinical trials, the expansion of our marketing, manufacturing and corporate infrastructure, and to meet
51
Table of Contents
our other anticipated cash through at least December 31, 2011. If our available cash, cash equivalents and short-term marketable securities are insufficient to satisfy our liquidity requirements, or if we develop additional products, we may seek to sell additional equity or debt securities or obtain an additional credit facility. The sale of additional equity and debt securities may result in additional dilution to our stockholders. If we raise additional funds through the issuance of debt securities or preferred stock, these securities could have rights senior to those of our common stock and could contain covenants that would restrict our operations. We may require additional capital beyond our currently forecasted amounts. Any such required additional capital may not be available on reasonable terms, if at all. Additionally, there can be no assurance that we will be successful in obtaining additional cash contributions from future partnership arrangements. Our ability to transition to attaining profitable operations is dependent upon achieving a level of revenues adequate to support our cost structure. If events or circumstances occur such that we do not meet our operating plan as expected, or if we are unable to obtain additional financing, we may be required to reduce planned increases in compensation related expenses or other operating expenses related to research, development, and commercialization activities, which could have an adverse impact on our ability to achieve our intended business objectives.
Because of the numerous risks and uncertainties associated with the development of continuous glucose monitoring technologies, we are unable to estimate the exact amounts of capital outlays and operating expenditures associated with our current and anticipated clinical trials. Our future funding requirements will depend on many factors, including, but not limited to:
| • | the revenue generated by sales of our approved products and other future products; |
| • | the expenses we incur in manufacturing, developing, selling and marketing our products; |
| • | the quality levels of our products and services; |
| • | the third party reimbursement of our products for our customers; |
| • | our ability to efficiently scale our manufacturing operations to meet demand for our current and any future products; |
| • | the costs, timing and risks of delays of additional regulatory approvals; |
| • | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights, including, but not limited to, defending the patent infringement lawsuit filed against us by Abbott; |
| • | the rate of progress and cost of our clinical trials and other development activities; |
| • | the success of our research and development efforts; |
| • | the emergence of competing or complementary technological developments; |
| • | the terms and timing of any collaborative, licensing and other arrangements that we may establish; and |
| • | the acquisition of businesses, products and technologies, although we currently have no commitments or agreements relating to any of these types of transactions. |
Contractual Obligations
On January 31, 2008, we amended our bank equipment loan to enable us to draw an additional $3.0 million. Beginning April 2008, this additional amount requires monthly amortized payments through the maturity date of July 2011. As of December 31, 2010, we had an outstanding balance of $525,000 due on this bank equipment loan.
In April 2006, we entered into an office lease agreement for approximately 66,400 square feet of additional facilities located in San Diego, California. In connection with the lease, we entered into a $664,000 letter of credit to secure future payments under the lease and paid a security deposit in the amount of $89,640 in April
52
Table of Contents
2006. In August 2010, we entered into a First Amendment to Office Lease (the “Agreement”) with respect to facilities in the buildings at 6340 Sequence Drive and 6310 Sequence Drive, each in San Diego, California (the “Buildings”). Under the Agreement, we have leased approximately 102,844 square feet of space in the Buildings, and retain the right and obligation to lease an additional 25,971 square feet in the Buildings. The lease term for the Buildings extends through November 2016 and we have a five-year option to renew the lease upon the expiration of the initial term. Excluding real estate taxes and operating costs, we are required to make total future monthly payments for all of our real estate obligations for the period from January 2011 through November 2016 totaling $14.4 million.
We are party to various purchase arrangements related to components used in production and research and development activities. As of December 31, 2010, we had purchase commitments with certain vendors totaling approximately $3.3 million due within one year. There are no material purchase commitments due beyond one year.
The following table summarizes our outstanding contractual obligations as of December 31, 2010 and the effect those obligations are expected to have on our liquidity and cash flows in future periods (in thousands):
| Contractual Obligations |
Total | Less than 1 Year |
1-3 Years |
3-5 Years |
More than 5 Years |
|||||||||||||||
| Equipment lines |
$ | 525 | $ | 525 | $ | — | $ | — | $ | — | ||||||||||
| Operating leases |
14,410 | 2,027 | 7,207 | 5,176 | — | |||||||||||||||
| Purchase commitments |
3,348 | 3,348 | — | — | — | |||||||||||||||
| Total |
$ | 18,283 | $ | 5,900 | $ | 7,207 | $ | 5,176 | $ | — | ||||||||||
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet activities.
Related Party Transactions
Our former Chairman of the Board is Donald L. Lucas, who retired from the Board on May 19, 2010. Mr. Lucas is a director of Oracle Corporation. We incurred costs totaling $284,000, $94,000, and $105,000 relating to an Oracle ERP system for the years ended December 31, 2010, 2009 and 2008, respectively. Mr. Lucas was not involved in the selection of our ERP system. We believe that the aforementioned arrangement was at no less favorable rates to us than those that could have been obtained from unrelated third parties based on review of price quotations with third parties.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which we have prepared in accordance with generally accepted accounting principles. The preparation of these consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements as well as the reported revenue and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
53
Table of Contents
While our significant accounting policies are more fully described in Note 1 to our consolidated financial statements included in our annual report on Form 10-K, we believe that the following accounting policies and estimates are most critical to a full understanding and evaluation of our reported financial results.
Revenue Recognition
We sell durable systems and disposable units through a direct sales force in the United States as well as through distribution arrangements in the United States, in portions of Europe, and Israel. Components are individually priced and can be purchased separately or together. The SEVEN PLUS durable system includes a transmitter, a receiver, a power cord, data management software and a USB cable. Disposable sensors for use with the SEVEN PLUS system are sold separately in packages of four. The initial SEVEN PLUS durable system price is not dependent upon the purchase of any amount of disposable sensors. We discontinued sales of our SEVEN system in the United States in the first quarter of 2009, although we continue to sell disposable sensors for use with both the SEVEN and SEVEN PLUS durable systems.
Revenue is recognized when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed or determinable, and collectability is reasonably assured. Revenue on product sales is recognized upon shipment, which is when title and the risk of loss have been transferred to the customer and there are no other post-shipment obligations. With respect to customers who directly pay for the products, the products are generally paid for at the time of shipment using a customer’s credit card and do not include customer acceptance provisions. We recognize revenue from contracted insurance payors based on the contracted rate. For non-contracted insurance payors, we obtain a prior authorization from the payor and recognize revenue based on the agreed upon price, estimated collectible amount and historical experience. We also receive a prescription or statement of medical necessity and, for insurance reimbursement customers, an assignment of benefits prior to shipment.
We provide a “30-day money back guarantee” program whereby customers who purchase the SEVEN PLUS durable system and a package of four disposable sensors may return the SEVEN PLUS durable system for any reason within thirty days of purchase and receive a full refund of their purchase price. At December 31, 2010, we maintained a reserve balance of $36,000 relating to this program. We accrue for estimated returns and/or refunds by reducing revenues and establishing a liability account at the time of shipment based on historical experience.
During 2008 and 2009, we entered into distribution agreements with RGH Enterprises, Inc., or “Edgepark,” and other distributors that allow the distributors to sell our durable systems and disposable units. Revenue on product sales to distributors is recognized at the time of shipment, which is when title and risk of loss have been transferred to the distributor and there are no other post-shipment obligations. Revenue is recognized based on contracted prices and invoices are either paid by check following the issuance of a purchase order or letter of credit, or they are paid by wire at the time of placing the order. Terms of distributor orders are FOB shipping point (FCA shipping point for international orders). Distributors do not have rights of return per their distribution agreement outside of our standard warranty. We accrue for estimated returns, refunds and rebates by reducing revenues and establishing a liability account at the time of shipment based on historical experience. Our distributors typically have a limited time frame to notify us of any missing, damaged, defective or non-conforming products. For any such products, we shall either, at our option, replace the portion of defective or non-conforming product at no additional cost to the distributor or cancel the order and refund any portion of the price paid to us at that time for the sale in question. We have no intention of refunding or unwinding a prior sale and view any potential product non-conformity solely as a warranty issue.
We shipped product directly to Edgepark’s customers and recognized $9.6 million and $4.5 million in revenue, which represents 20% and 15% of our revenues for the twelve months ended December 31, 2010 and 2009, respectively. With respect to other distributors which stock inventory of our product and fulfill orders from their inventory, we shipped product to these distributors and recognized $4.7 million and $1.4 million in revenue
54
Table of Contents
from these arrangements for the twelve months ended December 31, 2010 and 2009, respectively. We monitor shipments to, and on-hand inventory levels of, these distributors, and at December 31, 2010 these distributors had limited amounts of our product in their inventory.
During 2008, we entered into collaborative license and development arrangements with strategic partners for the development and commercialization of products utilizing our technologies. The terms of these agreements obligate us to multiple deliverables (for example, license rights, provision of research and development services, and manufacture of clinical materials) in exchange for our right to receive various forms of consideration including non-refundable license fees, funding of research and development activities, payments based upon achievement of development milestones and royalties in the form of a designated percentage of product sales or profits. With the exception of royalties, these types of consideration are classified as development grant and other revenue in our consolidated statements of operations when revenue recognition is appropriate.
Non-refundable license fees are recognized as revenue when we have a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and we have no further performance obligations under the license agreement. Multiple element arrangements, such as license, development and other multiple element service arrangements, are analyzed to determine how the arrangement consideration should be allocated among the separate units of accounting, or whether they must be accounted for as a single unit of accounting.
For transactions containing multiple element arrangements entered into or materially modified after January 1, 2010, we consider deliverables as separate units of accounting and recognize deliverables as revenue upon delivery only if (i) the deliverable has stand-alone value and (ii) if the arrangement includes a general right of return relative to the delivered item(s), delivery of the undelivered item(s) is probable and substantially controlled by us. We allocate consideration to the separate units of accounting using the relative selling price method, in which allocation of consideration is based on vendor-specific objective evidence (“VSOE”) if available, third party evidence (“TPE”), or if VSOE or TPE is not available, management’s best estimate of a stand alone selling price for elements. See Recent Accounting Pronouncements in Note 1 of the notes to the consolidated financial statements for additional information related to our adoption of authoritative guidance for revenue recognition for multiple-deliverable revenue arrangements.
For transactions containing multiple element arrangements entered into prior to January 1, 2010, we considered deliverables as separate units of accounting and recognized deliverables as revenue upon delivery only if (i) the deliverable had stand-alone value, (ii) if the arrangement included a general right of return relative to the delivered item(s), delivery of the undelivered item(s) was probable and substantially controlled by us, and (iii) the fair value of the undelivered performance obligations could be determined. In those instances when objective and reliable evidence of fair value existed for the undelivered items but not for the delivered items, the residual method was used to allocate the arrangement consideration. Under the residual method, the amount of arrangement consideration allocated to the delivered items equaled the total arrangement consideration less the aggregate fair value of the undelivered items. If we were unable to establish stand-alone value for delivered items or when fair value of undelivered items had not been established, revenue was deferred until all elements were delivered and services had been performed, or until fair value could objectively be determined for any remaining undelivered elements.
We use judgment in estimating the value allocable to product revenues or development grant and other revenue based on our estimate of the fair value attributable to the related deliverables. For arrangements that are accounted for as a single unit of accounting, total payments under the arrangement are recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations. We review the estimated period of our performance obligations on a periodic basis and update the recognition period as appropriate. The cumulative amount of revenue earned is limited to the cumulative amount of payments received as of the period ending date.
55
Table of Contents
If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential, then revenue is deferred until we can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance. Deferred revenue amounts are classified as current liabilities to the extent that revenue is expected to be recognized within one year.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement.
During the first quarter of 2008, we entered into a development agreement with Animas, as amended on January 12, 2009 and July 30, 2009, which provided us with a development grant. During the fourth quarter of 2008, we entered into a collaboration agreement with Edwards, as amended on May 5, 2009, which provided us with a development grant. We recognized $8.5 million in development grant and other revenue for the twelve months ended December 31, 2010, respectively. As of December 31, 2010, we had $3.5 million in deferred revenue relating to our development and other agreements.
Share-Based Compensation
We measure and recognize compensation expense for all share-based payment awards made to employees, non-employee directors, and consultants including employee stock options, restricted stock, restricted stock units and employee stock purchases related to the Employee Stock Purchase Plan based on estimated fair values. Share-based compensation expense for the years ended December 31, 2010, 2009 and 2008 was $9.4 million, $8.4 million, and $7.7 million, respectively. As of December 31, 2010, there was $15.9 million of unrecognized compensation cost related to unvested options, restricted stock and restricted stock units that is expected to be recognized as a component of our operating expenses through 2014. Compensation costs will be adjusted for future changes in estimated forfeitures.
We estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods as share-based compensation expense using the straight-line single option method in our consolidated statement of operations. We utilize the Black-Scholes option-pricing model as our method of valuation for stock options granted and we use the grant date fair value of our common stock for valuing restricted stock unit awards. Our determination of the fair value of share-based payment awards on the date of grant using an option-pricing model is affected by our stock price as well as assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors. Option-pricing models were developed for use in estimating the value of traded options that have no vesting or hedging restrictions and are fully transferable. Because our employee stock options have certain characteristics that are significantly different from traded options, and because changes in the subjective assumptions can materially affect the estimated value, the existing valuation models may not provide an accurate measure of the fair value of the our employee stock options. Although the fair value of employee stock options is determined using an option-pricing model, that value may not be indicative of the fair value observed in a willing buyer/willing seller market transaction.
Inventory
Inventories are valued at the lower of cost or market value. We make adjustments to reduce the cost of inventory to its net realizable value, if required, for estimated excess, obsolete and potential scrapped inventories. We estimate excess and obsolete inventories by identifying the amount of on hand and on order materials and comparing those to expected future sales for the next twelve months, taking into account clinical trial and development usage along with new product introductions on a part-by-part basis. Once written down the
56
Table of Contents
adjustments are considered permanent and are not reversed until the related inventory is sold or disposed. We utilize a standard cost system to track inventories on a part-by-part basis that approximates first in, first out. If necessary, adjustments are made to the standard materials, standard labor and standard overhead costs to approximate actual labor and actual overhead costs. The labor and overhead elements of our inventory are based on full utilization of our manufacturing capacity.
Clinical Trial Accounting
We record accruals for estimated clinical study expenses, comprising payments for work performed by contract research organizations, physicians and participating hospitals. These expenses can be a significant component of research and development expenses. We accrue expenses for clinical studies performed by contract research organizations based on estimates of work performed under the contracts. Expenses for setting up clinical trial sites and study initiation are accrued immediately. Clinical expenses related to patient enrollment and ongoing monitoring are accrued as the trials progress.
Warranty Accrual
We accrue for estimated warranty costs at the time of shipment. We estimate warranty accruals by analyzing the timing, cost and amount of returned product. We evaluate assumptions and historical warranty experience on at least a quarterly basis to determine the continued appropriateness of such assumptions.
Bonus Accrual
For the 2010 bonus plan, the Compensation Committee authorized an amount of up to 80% of salary and wages for non sales employees to be awarded from the pool based on the weighted average achievement measured against certain objectives. As various targets were met, bonuses were paid or accrued, as of December 31, 2010, under the 2010 bonus plan totaling $3.0 million.
Foreign Currency
The consolidated financial statements of our non-U.S. subsidiary, whose functional currency is the Swedish Krona, is translated into U.S. dollars for financial reporting purposes. Assets and liabilities are translated at period-end exchange rates, and revenue and expense transactions are translated at average exchange rates for the period. Cumulative translation adjustments are recognized as part of comprehensive income and are included in accumulated other comprehensive income in the consolidated balance sheet. Gains and losses on transactions denominated in other than the functional currency are reflected in operations.
Income Taxes
In July 2006, the FASB issued authoritative guidance for accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement process for recording in the financial statements uncertain tax positions taken or expected to be taken in a tax return. Additionally, the authoritative guidance provides detail on the derecognition, classification, accounting in interim periods and disclosure requirements for uncertain tax positions. Only tax positions that meet the more likely than not recognition threshold at the effective date may be recognized upon adoption of the authoritative guidance.
57
Table of Contents
The following table summarizes the activity related to our gross unrecognized tax benefits (in thousands):
| Balance at January 1, 2008 |
$ | 2,586 | ||
| Increases related to current year tax positions |
491 | |||
| Balance at December 31, 2008 |
3,077 | |||
| Adjustments related to prior year tax positions |
51 | |||
| Increases related to current year tax positions |
604 | |||
| Decreases due to IRC Section 382 limitation |
(837 | ) | ||
| Balance at December 31, 2009 |
2,895 | |||
| Increases related to current year tax positions |
677 | |||
| Balance at December 31, 2010 |
$ | 3,572 | ||
Due to the valuation allowance, $51,000 of the total unrecognized tax benefits as of December 31, 2010, would reduce our annual effective tax rate if recognized. We do not expect our unrecognized tax benefits to change significantly over the next 12 months.
We file income tax returns in the United States and in various state jurisdictions with varying statutes of limitations. Due to net operating losses incurred, our income tax returns from inception to date are subject to examination by taxing authorities. Our policy is to recognize interest expense and penalties related to income tax matters as a component of income tax expense. As of December 31, 2010, we had no interest or penalties accrued for uncertain tax positions.
Fair Value Measurements
The fair value hierarchy is based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value and include the following:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
The book values of cash and cash equivalents, short-term marketable securities, accounts receivable and accounts payable approximate their respective fair values due to the short-term nature of these instruments.
Recent Accounting Pronouncements
In October 2009, the FASB issued authoritative guidance for multiple-deliverable revenue arrangements that provides amendments to the criteria for separating consideration in multiple-deliverable arrangements. As a result of these amendments, multiple-deliverable revenue arrangements will be separated in more circumstances than under existing U.S. GAAP by establishing a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence if available, third-party evidence if vendor-specific objective evidence is not available, or estimated selling price if neither vendor-specific objective evidence nor third-party evidence is available. A vendor will be required to
58
Table of Contents
determine its best estimate of selling price in a manner that is consistent with that used to determine the price to sell the deliverable on a standalone basis. This guidance also eliminates the residual method of allocation and will require that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method, which allocates any discount in the overall arrangement proportionally to each deliverable based on its relative selling price. Expanded disclosures of qualitative and quantitative information regarding application of the multiple-deliverable revenue arrangement guidance are also required under the guidance. The guidance does not apply to arrangements for which industry specific allocation and measurement guidance exists, such as long-term construction contracts and software transactions. This guidance is effective for fiscal years beginning on or after June 15, 2010 and early adoption is permitted. We adopted the provisions of the authoritative guidance as of January 1, 2010 on a prospective basis. Prospective application required us to apply the guidance to new arrangements entered into or arrangements materially modified since the beginning of 2010. The prospective application had no impact on our consolidated financial statements for the twelve months ended December 31, 2010. There would not have been a material difference in amounts recognized for development grant and other revenue for 2009 if the multiple element arrangements entered into or materially modified during 2009 were subject to the measurement requirements of the amendments of this guidance.
In October 2009, the FASB issued authoritative guidance for certain revenue arrangements that include software elements that reduces the types of transactions that fall within the current scope of software revenue recognition guidance. Existing software revenue recognition guidance requires that its provisions be applied to an entire arrangement when the sale of any products or services containing or utilizing software when the software is considered more than incidental to the product or service. As a result of the amendments included in the guidance, many tangible products and services that rely on software will be accounted for under the multiple-element arrangements revenue recognition guidance rather than under the software revenue recognition guidance. Under the guidance, the following components would be excluded from the scope of software revenue recognition guidance: the tangible element of the product, software products bundled with tangible products where the software components and non-software components function together to deliver the product’s essential functionality, and undelivered components that relate to software that is essential to the tangible product’s functionality. The guidance also provides direction on how to allocate transaction consideration when an arrangement contains both deliverables within the scope of software revenue guidance (software deliverables) and deliverables not within the scope of that guidance (non-software deliverables). The guidance is effective for fiscal years beginning on or after June 15, 2010 and early adoption is permitted. We must elect the same transition method for this guidance as that chosen for the guidance for multiple-deliverable revenue arrangements. We adopted the provisions of the authoritative guidance as of January 1, 2010 on a prospective basis. The prospective application had no impact on our consolidated financial statements for the twelve months ended December 31, 2010.
In April 2010, the FASB reached a consensus on the Milestone Method of Revenue Recognition which provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. The updated guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years beginning on or after June 15, 2010, with early adoption permitted. We are currently evaluating the impact of this guidance on our consolidated results of operations and financial condition.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK. |
Interest Rate Risk
The primary objective of our investment activities is to preserve our capital for the purpose of funding operations while at the same time maximizing the income we receive from our investments without significantly increasing risk. To achieve these objectives, our investment policy allows us to maintain a portfolio of cash
59
Table of Contents
equivalents and short-term investments in a variety of securities, including money market funds, U.S. Treasury debt and corporate debt securities. Due to the short-term nature of our investments, we believe that we have no material exposure to interest rate risk.
Foreign Currency Risk
To date we have recorded no product sales in other than U.S. dollars. We have only limited business transactions in foreign currencies. We do not currently engage in hedging or similar transactions to reduce our foreign currency risks. We believe we have no material exposure to risk from changes in foreign currency exchange rates at this time. We will continue to monitor and evaluate our internal processes relating to foreign currency exchange, including the potential use of hedging strategies.
| ITEM 8. | CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
The information required is set forth under “Report of Independent Registered Public Accounting Firm,” “Consolidated Balance Sheets,” “Consolidated Statements of Operations,” “Consolidated Statements of Stockholders’ Equity (Deficit),” “Consolidated Statements of Cash Flows” and “Notes to Consolidated Financial Statements” on pages F-2 to F-28 of this annual report.
60
Table of Contents
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| F-2 | ||||
| F-3 | ||||
| F-4 | ||||
| F-5 | ||||
| F-6 | ||||
| F-7 | ||||
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of DexCom, Inc.:
We have audited the accompanying consolidated balance sheets of DexCom, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2010. Our audits also included the financial statement schedule listed in the index at Item 15(a). These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of DexCom, Inc. at December 31, 2010 and 2009, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2010 in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), DexCom, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 3, 2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 3, 2011
F-2
Table of Contents
CONSOLIDATED BALANCE SHEETS
(In thousands—except par value data)
| As of December 31, | ||||||||
| 2010 | 2009 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 4,889 | $ | 3,577 | ||||
| Short-term marketable securities, available-for-sale |
42,224 | 24,439 | ||||||
| Accounts receivable, net |
6,671 | 3,490 | ||||||
| Inventory |
8,112 | 2,641 | ||||||
| Restricted cash |
1,439 | — | ||||||
| Prepaid and other current assets |
2,690 | 2,773 | ||||||
| Total current assets |
66,025 | 36,920 | ||||||
| Property and equipment, net |
10,763 | 6,422 | ||||||
| Restricted cash |
275 | 2,414 | ||||||
| Other assets |
101 | 1,192 | ||||||
| Total assets |
$ | 77,164 | $ | 46,948 | ||||
| Liabilities and stockholders’ equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable and accrued liabilities |
$ | 5,350 | $ | 5,745 | ||||
| Accrued payroll and related expenses |
5,730 | 4,406 | ||||||
| Current portion of long-term debt |
525 | 900 | ||||||
| Current portion of deferred revenue |
3,524 | 7,745 | ||||||
| Total current liabilities |
15,129 | 18,796 | ||||||
| Other liabilities |
1,042 | 840 | ||||||
| Long-term debt, net of current portion |
— | 45,757 | ||||||
| Total liabilities |
16,171 | 65,393 | ||||||
| Commitments and contingencies (Note 4) |
||||||||
| Stockholders’ equity (deficit): |
||||||||
| Preferred stock, $0.001 par value per share, 5,000 shares authorized; no shares issued and outstanding at December 31, 2010 and December 31, 2009, respectively. |
— | — | ||||||
| Common stock, $0.001 par value per share, 100,000 authorized; 62,360 and 62,078 shares issued and outstanding, respectively, at December 31, 2010, and 46,324 and 46,045 shares issued and outstanding, respectively, at December 31, 2009 |
62 | 46 | ||||||
| Additional paid-in capital |
407,375 | 272,730 | ||||||
| Accumulated other comprehensive loss |
(66 | ) | (13 | ) | ||||
| Accumulated deficit |
(346,378 | ) | (291,208 | ) | ||||
| Total stockholders’ equity (deficit) |
60,993 | (18,445 | ) | |||||
| Total liabilities and stockholders’ equity (deficit) |
$ | 77,164 | $ | 46,948 | ||||
See accompanying notes.
F-3
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
(In thousands—except per share data)
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Product revenue |
$ | 40,175 | $ | 18,036 | $ | 8,108 | ||||||
| Development grant and other revenue |
8,456 | 11,657 | 1,730 | |||||||||
| Total revenue |
48,631 | 29,693 | 9,838 | |||||||||
| Product cost of sales |
26,104 | 18,216 | 13,383 | |||||||||
| Development and other cost of sales |
4,084 | 7,816 | 1,984 | |||||||||
| Total cost of sales |
30,188 | 26,032 | 15,367 | |||||||||
| Gross margin (deficit) |
18,443 | 3,661 | (5,529 | ) | ||||||||
| Operating expenses |
||||||||||||
| Research and development |
23,227 | 14,294 | 19,629 | |||||||||
| Selling, general and administrative |
40,506 | 35,200 | 27,669 | |||||||||
| Total operating expenses |
63,733 | 49,494 | 47,298 | |||||||||
| Operating loss |
(45,290 | ) | (45,833 | ) | (52,827 | ) | ||||||
| Other income |
63 | — | 34 | |||||||||
| Interest income |
95 | 354 | 1,220 | |||||||||
| Interest expense |
(1,548 | ) | (8,045 | ) | (7,283 | ) | ||||||
| Loss on debt extinguishment upon conversion of convertible debt |
(8,490 | ) | — | — | ||||||||
| Net loss |
$ | (55,170 | ) | $ | (53,524 | ) | $ | (58,856 | ) | |||
| Basic and diluted net loss per share attributable to common stockholders |
$ | (0.97 | ) | $ | (1.21 | ) | $ | (2.00 | ) | |||
| Shares used to compute basic and diluted net loss per share attributable to common stockholders |
56,881 | 44,347 | 29,487 | |||||||||
See accompanying notes.
F-4
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
(In thousands—except per share data)
| Common stock | Additional paid-in capital |
Accumulated other comprehensive income (loss) |
Accumulated deficit |
Total stockholders’ equity (deficit) |
||||||||||||||||||||
| Shares | Amount | |||||||||||||||||||||||
| Balance at December 31, 2007 |
28,624 | $ | 29 | $ | 208,718 | $ | 13 | $ | (178,828 | ) | $ | 29,932 | ||||||||||||
| Settlement of the second tranche of the call spread option |
(118 | ) | — | — | — | — | — | |||||||||||||||||
| Issuance of common stock under equity incentive plans |
1,103 | 1 | 1,173 | — | — | 1,174 | ||||||||||||||||||
| Issuance of stock for cash |
92 | — | — | — | — | — | ||||||||||||||||||
| Issuance of common stock for Employee Stock Purchase Plan |
98 | — | 588 | — | — | 588 | ||||||||||||||||||
| Share-based compensation for employee stock options and award grants |
— | — | 7,477 | — | — | 7,477 | ||||||||||||||||||
| Issuance of stock for services |
25 | — | 180 | — | — | 180 | ||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Unrealized gains on available-for-sale investment securities |
— | — | — | 37 | — | 37 | ||||||||||||||||||
| Net loss |
— | — | — | — | (58,856 | ) | (58,856 | ) | ||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (58,819 | ) | |||||||||||||||||
| Balance at December 31, 2008 |
29,824 | 30 | 218,136 | 50 | (237,684 | ) | (19,468 | ) | ||||||||||||||||
| Issuance of stock in follow-on offering in February 2009 at $3.00 per share for cash, net of offering costs of $2,409 |
15,994 | 16 | 45,633 | — | — | 45,649 | ||||||||||||||||||
| Issuance of common stock under equity incentive plans |
34 | — | 120 | — | — | 120 | ||||||||||||||||||
| Issuance of common stock for Employee Stock Purchase Plan |
193 | — | 541 | — | — | 541 | ||||||||||||||||||
| Share-based compensation for employee stock options and award grants |
— | — | 8,300 | — | — | 8,300 | ||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Unrealized loss on available-for-sale investment securities |
— | — | — | (54 | ) | — | (54 | ) | ||||||||||||||||
| Foreign currency translation adjustments |
— | — | — | (9 | ) | — | (9 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (53,524 | ) | (53,524 | ) | ||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (53,587 | ) | |||||||||||||||||
| Balance at December 31, 2009 |
46,045 | 46 | 272,730 | (13 | ) | (291,208 | ) | (18,445 | ) | |||||||||||||||
| Issuance of stock in follow-on offering in January 2010 at $8.75 per share for cash, net of offering costs of $416 |
4,025 | 4 | 32,988 | — | — | 32,992 | ||||||||||||||||||
| Issuance of stock in follow-on offering in November 2010 at $10.60 per share for cash, net of offering costs of $207 |
3,278 | 3 | 32,987 | — | — | 32,990 | ||||||||||||||||||
| Issuance of stock upon conversion of convertible debt |
7,929 | 8 | 54,640 | — | — | 54,648 | ||||||||||||||||||
| Issuance of common stock under equity incentive plans |
594 | 1 | 3,643 | — | — | 3,644 | ||||||||||||||||||
| Issuance of common stock for Employee Stock Purchase Plan |
207 | — | 920 | — | — | 920 | ||||||||||||||||||
| Share-based compensation for employee stock options and award grants |
— | — | 9,467 | — | — | 9,467 | ||||||||||||||||||
| Comprehensive loss: |
||||||||||||||||||||||||
| Unrealized loss on available-for-sale investment securities |
— | — | — | (5 | ) | — | (5 | ) | ||||||||||||||||
| Foreign currency translation adjustments |
— | — | — | (48 | ) | — | (48 | ) | ||||||||||||||||
| Net loss |
— | — | — | — | (55,170 | ) | (55,170 | ) | ||||||||||||||||
| Comprehensive loss |
— | — | — | — | — | (55,223 | ) | |||||||||||||||||
| Balance at December 31, 2010 |
62,078 | $ | 62 | $ | 407,375 | $ | (66 | ) | $ | (346,378 | ) | $ | 60,993 | |||||||||||
See accompanying notes.
F-5
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Operating activities |
||||||||||||
| Net loss |
$ | (55,170 | ) | $ | (53,524 | ) | $ | (58,856 | ) | |||
| Adjustments to reconcile net loss to cash used in operating activities: |
||||||||||||
| Depreciation and amortization |
2,437 | 2,373 | 3,036 | |||||||||
| Share-based compensation |
9,441 | 8,366 | 7,682 | |||||||||
| Non-cash restructuring charge |
— | (362 | ) | 355 | ||||||||
| Accretion and amortization related to investments, net |
721 | 891 | 1 | |||||||||
| Accretion of non-cash interest expense related to convertible notes |
1,059 | 4,861 | 4,059 | |||||||||
| Compensation expense associated with stock options issued to consultants |
— | — | 1 | |||||||||
| Loss on debt extinguishment upon conversion of convertible debt |
8,490 | — | — | |||||||||
| Amortization of debt issuance costs |
36 | 246 | 159 | |||||||||
| Loss on disposal of equipment |
101 | — | — | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Accounts receivable |
(3,181 | ) | (2,372 | ) | (903 | ) | ||||||
| Inventory |
(5,471 | ) | (195 | ) | (1,307 | ) | ||||||
| Prepaid and other assets |
1,288 | (1,004 | ) | 148 | ||||||||
| Restricted cash |
700 | 1,856 | (3,356 | ) | ||||||||
| Accounts payable and accrued liabilities |
(432 | ) | 1,508 | (291 | ) | |||||||
| Accrued payroll and related expenses |
1,324 | 2,291 | (422 | ) | ||||||||
| Deferred revenue |
(4,221 | ) | (4,275 | ) | 12,020 | |||||||
| Deferred rent and other liabilities |
202 | (49 | ) | 223 | ||||||||
| Net cash used in operating activities |
(42,676 | ) | (39,389 | ) | (37,451 | ) | ||||||
| Investing activities |
||||||||||||
| Purchase of available-for-sale marketable securities |
(73,447 | ) | (65,270 | ) | (36,986 | ) | ||||||
| Proceeds from the maturity of available-for-sale marketable securities |
54,691 | 53,856 | 63,802 | |||||||||
| Purchase of property and equipment |
(6,860 | ) | (2,992 | ) | (2,492 | ) | ||||||
| Proceeds from the sale of equipment |
6 | 302 | — | |||||||||
| Net cash (used in) provided by investing activities |
(25,610 | ) | (14,104 | ) | 24,324 | |||||||
| Financing activities |
||||||||||||
| Net proceeds from issuance of common stock |
70,546 | 46,310 | 1,762 | |||||||||
| Proceeds from equipment loan |
— | — | 3,000 | |||||||||
| Repayment of equipment loan |
(900 | ) | (1,931 | ) | (2,050 | ) | ||||||
| Net cash provided by financing activities |
69,646 | 44,379 | 2,712 | |||||||||
| Effect of exchange rate changes on cash and cash equivalents |
(48 | ) | (9 | ) | — | |||||||
| Increase (decrease) in cash and cash equivalents |
1,312 | (9,123 | ) | (10,415 | ) | |||||||
| Cash and cash equivalents, beginning of year |
3,577 | 12,700 | 23,115 | |||||||||
| Cash and cash equivalents, ending of year |
$ | 4,889 | $ | 3,577 | $ | 12,700 | ||||||
| Non-cash investing and financing transactions: |
||||||||||||
| Unrealized gain (loss) on marketable securities |
$ | (5 | ) | $ | (54 | ) | $ | 37 | ||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid during the year for interest |
$ | 357 | $ | 2,932 | $ | 3,074 | ||||||
| Non-cash investing and financing transactions: |
||||||||||||
| Conversion of convertible notes to common stock |
$ | 46,270 | $ | — | $ | — | ||||||
See accompanying notes.
F-6
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2010
1. Organization and Summary of Significant Accounting Policies
Organization and Business
DexCom, Inc. is a medical device company focused on the design, development and commercialization of continuous glucose monitoring systems for ambulatory use by people with diabetes and by healthcare providers in the hospital for the treatment of both diabetic and non-diabetic patients. Unless the context requires otherwise, the terms “we,” “us,” “our,” the “company,” or “DexCom” refer to DexCom, Inc. and its subsidiary.
Basis of Presentation
We have incurred operating losses since our inception and have an accumulated deficit of $346.4 million at December 31, 2010. As of December 31, 2010, we had available cash, cash equivalents and short-term investments totaling $47.1 million, excluding $1.7 million of restricted cash, and working capital of $50.9 million. Our ability to transition to attaining profitable operations is dependent upon achieving a level of revenues adequate to support our cost structure. If events or circumstances occur such that we do not meet our operating plan as expected, we may be required to reduce planned increases in compensation related expenses or other operating expenses which could have an adverse impact on our ability to achieve our intended business objectives. We believe our working capital resources will be sufficient to fund our operations through at least December 31, 2011.
Principles of Consolidation
The consolidated financial statements include the accounts of DexCom and our wholly owned subsidiary. All significant intercompany balances and transactions have been eliminated in consolidation.
Segment Reporting
An operating segment is identified as a component of a business that has discrete financial information available, and one that the chief operating decision maker must decide the level of resource allocation directed to the segment. In addition, the guidance for segment reporting indicates certain quantitative thresholds. We consider our operations and manage our business as one operating segment.
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires us to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from these estimates. Significant estimates include excess or obsolete inventories, warranty accruals, employee bonus, clinical study expenses, trade show expenses, allowances for returned product, allowance for bad debt, and share-based compensation expense. Excess and obsolete inventories are estimated by identifying the amount of on hand and on order materials compared to expected future sales, taking into account clinical trial and development usage along with new product introductions. Employee bonus estimates are based, in part, on the 2010 bonus plan’s authorized target bonus amounts of up to 80%, 70%, 70%, 40%, 30% and 25% of base salary for our Chief Executive Officer, Chief Operating Officer, Chief Technical Officer, our Senior Vice President of Clinical and Regulatory Affairs, our Vice Presidents and the remainder of our non-sales management employees, respectively, to be awarded from the bonus pool based on the weighted average achievement of certain objectives. With respect to the CEO, generally, 75% of any bonus paid under the 2010 Plan was based on achieving certain annual revenue goals and 25% was
F-7
Table of Contents
based on achieving certain operating loss goals. For the remainder of our eligible employees, generally, 60% of any bonus paid under the 2010 Plan was based on achieving certain annual revenue goals, 20% was based on achieving targeted operating loss goals and 20% was based on achieving certain performance milestones. Clinical trial expenses are accrued based on estimates of progress under related contracts and include initial set up costs as well as ongoing monitoring over multiple sites in the U.S. and abroad. An allowance for refunds for returned products is determined by analyzing the timing and amounts of past refund activity.
Cash and Cash Equivalents
We invest our excess cash in bank deposits, money market accounts, and debt securities. We consider all highly liquid investments with an original maturity of 90 days or less at the time of purchase to be cash equivalents.
Short-Term Marketable Securities
We have classified our short-term investments as “available-for-sale” and carry them at fair value with unrealized gains and losses, if any, reported as a separate component of stockholders’ equity and included in comprehensive loss. Realized gains and losses are calculated on the specific identification method and recorded as interest income.
Accounts Receivable
We grant credit to various customers in the normal course of business. We maintain an allowance for doubtful accounts for potential credit losses. Uncollectible accounts are written-off against the allowance after appropriate collection efforts have been exhausted and when it is deemed that a customer account is uncollectible. Generally, receivable balances greater than one year past due are deemed uncollectable.
Fair Value of Financial Instruments
Financial instruments, including cash and cash equivalents, prepaid expenses, accounts payable and accrued liabilities, are carried at cost, which we believe approximates fair value given their short-term nature.
The estimated fair value of our convertible notes was determined by using observable market information and valuation methodologies that correlate fair value with the market price of our common stock. As of December 31, 2010, there were no convertible notes outstanding. The estimated fair value and carrying amounts of our convertible notes at December 31, 2009 was as follows (in thousands):
| December 31, 2009 | ||||||||
| Fair Value | Carrying Value | |||||||
| Convertible notes |
$ | 69,661 | $ | 45,232 | ||||
Letters of Credit
At December 31, 2010 and 2009, we had irrevocable letters of credit outstanding with a commercial bank for approximately $914,000 securing our facility leases. The letters of credit are secured by cash and an equal amount of restricted cash has been separately disclosed in the accompanying consolidated balance sheets.
Concentration of Credit Risk
Financial instruments which potentially subject us to concentrations of credit risk consist primarily of cash, cash equivalents, short-term investment securities, and accounts receivable. We limit our exposure to credit loss by placing our cash with high credit quality financial institutions. We have established guidelines relative to
F-8
Table of Contents
diversification of our cash and investment securities and their maturities that are intended to secure safety and liquidity. These guidelines are periodically reviewed and modified to take advantage of trends in yields and interest rates and changes in our operations and financial position. The following table summarizes customers who accounted for 10% or more of net accounts receivable:
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Customer A |
23 | % | 18 | % | 27 | % | ||||||
| Customer B |
16 | % | 8 | % | — | |||||||
| Customer C |
12 | % | 19 | % | — | |||||||
Property and Equipment
Property and equipment is stated at cost and depreciated over the estimated useful lives of the assets, generally three years for computer equipment, four years for machinery and equipment, and five years for furniture and fixtures, using the straight-line method. Leasehold improvements are stated at cost and amortized over the shorter of the estimated useful lives of the assets or the lease term.
During the fourth quarter of 2009, we sold Edwards certain equipment totaling $302,000, which were sold at their net book value. We also sold Edwards supplies totaling $241,000, which were offset against cost of sales and research and development expense.
Impairment of Long-Lived Assets
We will record impairment losses on long-lived assets used in operations when events and circumstances indicate that assets might be impaired and the undiscounted cash flows estimated to be generated by those assets are less than the carrying amount of those assets. We have not experienced any material impairment losses on assets used in operations.
Share-Based Compensation
We measure and recognize compensation expense for all share-based payment awards made to employees, non-employee directors, and consultants including employee stock options, restricted stock, restricted stock units and employee stock purchases related to the Employee Stock Purchase Plan based on estimated fair values. Share-based compensation expense recognized for the years ended December 31, 2010, 2009 and 2008 was $9.4 million, $8.4 million and $7.7 million, respectively.
We estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods as share-based compensation expense using the straight-line single-option method in our consolidated statement of operations. As share-based compensation expense recognized in the consolidated statement of operations during fiscal 2010, 2009, and 2008 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
We utilize the Black-Scholes option-pricing model as our method of valuation for stock options granted and we use the grant date fair value of our common stock for valuing restricted stock unit awards. Our determination of the fair value of share-based payment awards on the date of grant using an option-pricing model is affected by our stock price as well as assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility over the term of the awards, and actual and projected employee stock option exercise behaviors.
F-9
Table of Contents
Revenue Recognition
We sell our durable systems and disposable units through a direct sales force in the United States and through distribution arrangements in the United States and in portions of Europe. Components are individually priced and can be purchased separately or together. We receive payment directly from patients who use our products, as well as from distributors and third party payors. Our durable system includes a reusable transmitter, a receiver, a power cord, data management software and a USB cable. Disposable sensors for use with the durable system are sold separately in packages of four. The initial durable system price is not dependent upon the purchase of any amount of disposable sensors. We discontinued sales of our SEVEN durable system in the United States in the first quarter of 2009, although we continue to sell disposable sensors for use with both the SEVEN and SEVEN PLUS durable systems.
Revenue is recognized when persuasive evidence of an arrangement exists, delivery has occurred or services have been rendered, the price is fixed or determinable, and collectability is reasonably assured. Revenue on product sales is recognized upon shipment, which is when title and the risk of loss have been transferred to the customer and there are no other post shipment obligations. With respect to customers who directly pay for products, the products are generally paid for at the time of shipment using a customer’s credit card and do not include customer acceptance provisions. We recognize revenue from contracted insurance payors based on the contracted rate. For non-contracted insurance payors, we obtain prior authorization from the payor and recognize revenue based on the estimated collectible amount and historical experience. We also receive a prescription or statement of medical necessity and, for insurance reimbursement customers, an assignment of benefits prior to shipment.
We provide a “30-day money back guarantee” program whereby customers who purchase a durable system and a package of four disposable sensors may return the durable system for any reason within thirty days of purchase and receive a full refund of their purchase price. We accrue for estimated returns and/or refunds by reducing revenues and establishing a liability account at the time of shipment based on historical experience.
We have entered into distribution agreements with RGH Enterprises, Inc., or “Edgepark,” and other distributors that allow the distributors to sell our durable systems and disposable units. Revenue on product sales to distributors is recognized at the time of shipment, which is when title and risk of loss have been transferred to the distributor and there are no other post-shipment obligations. Revenue is recognized based on contracted prices and invoices are either paid by check following the issuance of a purchase order or letter of credit, or they are paid by wire at the time of placing the order. Terms of distributor orders are FOB shipping point (FCA shipping point for international orders). Distributors do not have rights of return per their distribution agreement outside of our standard warranty. We accrue for estimated returns, refunds and rebates by reducing revenues and establishing a liability account at the time of shipment based on historical experience. The distributors typically have a limited timeframe to notify us of any missing, damaged, defective or non-conforming products. For any such products, we shall either, at our option, replace the portion of defective or non-conforming product at no additional cost to the distributor or cancel the order and refund any portion of the price paid to us at that time for the sale in question.
We shipped product directly to Edgepark’s customers and recognized $9.6 million and $4.5 million in revenue, which represents 20% and 15% of our total revenues, respectively, for the twelve months ended December 31, 2010 and 2009. With respect to other distributors which stock inventory of our product and fulfill orders from their inventory, we shipped product to these distributors and recognized $4.7 million and $1.4 million in revenue from these arrangements for the twelve months ended December 31, 2010 and 2009, respectively.
During 2008, we entered into collaborative license and development arrangements with strategic partners for the development and commercialization of products utilizing our technologies. The terms of these agreements obligate us to multiple deliverables (for example, license rights, provision of research and development services, and manufacture of clinical materials) in exchange for our right to receive various forms of consideration
F-10
Table of Contents
including non-refundable license fees, funding of research and development activities, payments based upon achievement of development milestones and royalties in the form of a designated percentage of product sales or profits. With the exception of royalties, these types of consideration are classified as development grant and other revenue in our consolidated statements of operations when revenue recognition is appropriate.
Non-refundable license fees are recognized as revenue when we have a contractual right to receive such payment, the contract price is fixed or determinable, the collection of the resulting receivable is reasonably assured and we have no further performance obligations under the license agreement. Multiple element arrangements, such as license, development and other multiple element service arrangements, are analyzed to determine how the arrangement consideration should be allocated among the separate units of accounting, or whether they must be accounted for as a single unit of accounting.
For transactions containing multiple element arrangements entered into or materially modified after January 1, 2010, we consider deliverables as separate units of accounting and recognize deliverables as revenue upon delivery only if (i) the deliverable has stand-alone value and (ii) if the arrangement includes a general right of return relative to the delivered item(s), delivery of the undelivered item(s) is probable and substantially controlled by us. We allocate consideration to the separate units of accounting using the relative selling price method, in which allocation of consideration is based on vendor-specific objective evidence (“VSOE”) if available, third party evidence (“TPE”), or if VSOE or TPE is not available, management’s best estimate of a stand alone selling price for elements. See Recent Accounting Pronouncements in Note 1 of the notes to the consolidated financial statements for additional information related to our adoption of authoritative guidance for revenue recognition for multiple-deliverable revenue arrangements.
For transactions containing multiple element arrangements entered into prior to January 1, 2010, we considered deliverables as separate units of accounting and recognized deliverables as revenue upon delivery only if (i) the deliverable had stand-alone value, (ii) if the arrangement included a general right of return relative to the delivered item(s), delivery of the undelivered item(s) was probable and substantially controlled by us, and (iii) the fair value of the undelivered performance obligations could be determined. In those instances when objective and reliable evidence of fair value existed for the undelivered items but not for the delivered items, the residual method was used to allocate the arrangement consideration. Under the residual method, the amount of arrangement consideration allocated to the delivered items equaled the total arrangement consideration less the aggregate fair value of the undelivered items. If we were unable to establish stand-alone value for delivered items or when fair value of undelivered items had not been established, revenue was deferred until all elements were delivered and services had been performed, or until fair value could objectively be determined for any remaining undelivered elements.
We use judgment in estimating the value allocable to product revenues or development grant and other revenue based on our estimate of the fair value attributable to the related deliverables. For arrangements that are accounted for as a single unit of accounting, total payments under the arrangement are recognized as revenue on a straight-line basis over the period we expect to complete our performance obligations. We review the estimated period of our performance obligations on a periodic basis and update the recognition period as appropriate. The cumulative amount of revenue earned is limited to the cumulative amount of payments received as of the period ending date.
If we cannot reasonably estimate when our performance obligation either ceases or becomes inconsequential, then revenue is deferred until we can reasonably estimate when the performance obligation ceases or becomes inconsequential. Revenue is then recognized over the remaining estimated period of performance. Deferred revenue amounts are classified as current liabilities to the extent that revenue is expected to be recognized within one year.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement.
F-11
Table of Contents
Under the collaboration agreement with Edwards which provided us with a development grant, we recognized $6.9 million in development grant and other revenue, which represents 14% of our total revenues for the twelve months ended December 31, 2010.
Warranty Accrual
Estimated warranty costs are recorded at the time of shipment. We estimate future warranty costs by analyzing the timing, cost and amount of returned product. Assumptions and historical warranty experience are evaluated on at least a quarterly basis to determine the continued appropriateness of such assumptions.
Research and Development
All costs of research and development are expensed as incurred. Research and development expenses primarily include salaries, bonus and payroll related costs, overhead, part components, share-based compensation, and fees paid to consultants.
Foreign Currency
The consolidated financial statements of our non-U.S. subsidiary, whose functional currency is the Swedish Krona, is translated into U.S. dollars for financial reporting purposes. Assets and liabilities are translated at period-end exchange rates, and revenue and expense transactions are translated at average exchange rates for the period. Cumulative translation adjustments are recognized as part of comprehensive income and are included in accumulated other comprehensive income in the consolidated balance sheet. Gains and losses on transactions denominated in other than the functional currency are reflected in operations.
Comprehensive Loss
We report all components of comprehensive income (loss), including net income (loss), in the consolidated financial statements in the period in which they are recognized. Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Net income (loss) and other comprehensive income (loss), including unrealized gains and losses on investments and foreign currency translation adjustments, shall be reported, net of their related tax effect, to arrive at comprehensive income (loss). Our comprehensive loss is as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Net loss |
$ | (55,170 | ) | $ | (53,524 | ) | $ | (58,856 | ) | |||
| Unrealized gain (loss) on available-for-sale marketable securities |
(5 | ) | (54 | ) | 37 | |||||||
| Foreign currency translation loss |
(48 | ) | (9 | ) | — | |||||||
| Comprehensive loss |
$ | (55,223 | ) | $ | (53,587 | ) | $ | (58,819 | ) | |||
Inventory
Inventory is valued at the lower of cost or market value. We make adjustments to reduce the cost of inventory to its net realizable value, if required, for estimated excess, obsolete and potential scrapped inventories. Factors influencing these adjustments include inventories on hand and on order compared to estimated future usage and sales for existing and new products, as well as judgments regarding quality control testing data, and assumptions about the likelihood of scrap and obsolescence. Once written down the adjustments are considered permanent and are not reversed until the related inventory is sold or disposed. We utilize a standard cost system to track inventories on a part-by-part basis that approximates first in, first out. If necessary, adjustments are made to the standard materials, standard labor and standard overhead costs to approximate actual labor and actual overhead costs. The labor and overhead elements of inventory are based on full utilization of our manufacturing capacity.
F-12
Table of Contents
Deferred Rent
Rent expense is recorded on a straight-line basis over the term of the lease. The difference between rent expense accrued and amounts paid under the lease agreement is recorded as deferred rent in the accompanying consolidated balance sheets.
Income Taxes
In July 2006, the FASB issued authoritative guidance for accounting for uncertainty in income taxes, which prescribes a recognition threshold and measurement process for recording in the consolidated financial statements uncertain tax positions taken or expected to be taken in a tax return. Additionally, the accounting standard provides guidance on the derecognition, classification, accounting in interim periods and disclosure requirements for uncertain tax positions.
The following table summarizes the activity related to our gross unrecognized tax benefits (in thousands):
| Balance at January 1, 2008 |
$ | 2,586 | ||
| Increases related to current year tax positions |
491 | |||
| Balance at December 31, 2008 |
3,077 | |||
| Adjustments related to prior year tax positions |
51 | |||
| Increases related to current year tax positions |
604 | |||
| Decreases due to IRC Section 382 limitation |
(837 | ) | ||
| Balance at December 31, 2009 |
2,895 | |||
| Increases related to current year tax positions |
677 | |||
| Balance at December 31, 2010 |
$ | 3,572 | ||
Due to the valuation allowance, $51,000 of the total unrecognized tax benefits as of December 31, 2010 would reduce our annual effective tax rate if recognized.
We file income tax returns in the United States and in various state jurisdictions with varying statutes of limitations. Due to net operating losses incurred, our income tax returns from inception to date are subject to examination by taxing authorities. Our policy is to recognize interest expense and penalties related to income tax matters as a component of income tax expense. As of December 31, 2010, we had no interest or penalties accrued for uncertain tax positions.
Fair Value Measurements
The fair value hierarchy is based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value and include the following:
Level 1—Quoted prices in active markets for identical assets or liabilities.
Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities.
F-13
Table of Contents
The following table represents our fair value hierarchy for our financial assets (cash equivalents and investments) measured at fair value on a recurring basis as of December 31, 2010 (in thousands):
| Fair Value Measurements Using | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Cash equivalents |
$ | 1,000 | — | — | $ | 1,000 | ||||||||||
| Marketable securities, available for sale |
$ | 42,224 | — | — | $ | 42,224 | ||||||||||
| Restricted cash |
$ | 1,714 | — | — | $ | 1,714 | ||||||||||
We have maintained only Level 1 financial assets during the twelve months ended December 31, 2010.
The following table represents our fair value hierarchy for our financial assets (cash equivalents and investments) measured at fair value on a recurring basis as of December 31, 2009 (in thousands):
| Fair Value Measurements Using | ||||||||||||||||
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Cash equivalents |
$ | 1,000 | — | — | $ | 1,000 | ||||||||||
| Marketable securities, available for sale |
$ | 24,439 | — | — | $ | 24,439 | ||||||||||
| Restricted cash |
$ | 2,414 | — | — | $ | 2,414 | ||||||||||
The book values of cash and cash equivalents, short-term marketable securities, accounts receivable and accounts payable approximate their respective fair values due to the short-term nature of these instruments.
Recent Accounting Pronouncements
In October 2009, the FASB issued authoritative guidance for multiple-deliverable revenue arrangements that provides amendments to the criteria for separating consideration in multiple-deliverable arrangements. As a result of these amendments, multiple-deliverable revenue arrangements will be separated in more circumstances than under existing U.S. GAAP by establishing a selling price hierarchy for determining the selling price of a deliverable. The selling price used for each deliverable will be based on vendor-specific objective evidence if available, third-party evidence if vendor-specific objective evidence is not available, or estimated selling price if neither vendor-specific objective evidence nor third-party evidence is available. A vendor will be required to determine its best estimate of selling price in a manner that is consistent with that used to determine the price to sell the deliverable on a standalone basis. This guidance also eliminates the residual method of allocation and will require that arrangement consideration be allocated at the inception of the arrangement to all deliverables using the relative selling price method, which allocates any discount in the overall arrangement proportionally to each deliverable based on its relative selling price. Expanded disclosures of qualitative and quantitative information regarding application of the multiple-deliverable revenue arrangement guidance are also required under the guidance. The guidance does not apply to arrangements for which industry specific allocation and measurement guidance exists, such as long-term construction contracts and software transactions. This guidance is effective for fiscal years beginning on or after June 15, 2010 and early adoption is permitted. We adopted the provisions of the authoritative guidance as of January 1, 2010 on a prospective basis. Prospective application required us to apply the guidance to new arrangements entered into or arrangements materially modified since the beginning of 2010. The prospective application had no impact on our consolidated financial statements for the twelve months ended December 31, 2010. There would not have been a material difference in amounts recognized for development grant and other revenue for 2009 if the multiple element arrangements entered into or materially modified during 2009 were subject to the measurement requirements of the amendments of this guidance.
In October 2009, the FASB issued authoritative guidance for certain revenue arrangements that include software elements that reduces the types of transactions that fall within the current scope of software revenue recognition guidance. Existing software revenue recognition guidance requires that its provisions be applied to an
F-14
Table of Contents
entire arrangement when the sale of any products or services containing or utilizing software when the software is considered more than incidental to the product or service. As a result of the amendments included in the guidance, many tangible products and services that rely on software will be accounted for under the multiple- element arrangements revenue recognition guidance rather than under the software revenue recognition guidance. Under the guidance, the following components would be excluded from the scope of software revenue recognition guidance: the tangible element of the product, software products bundled with tangible products where the software components and non-software components function together to deliver the product’s essential functionality, and undelivered components that relate to software that is essential to the tangible product’s functionality. The guidance also provides direction on how to allocate transaction consideration when an arrangement contains both deliverables within the scope of software revenue guidance (software deliverables) and deliverables not within the scope of that guidance (non-software deliverables). The guidance is effective for fiscal years beginning on or after June 15, 2010 and early adoption is permitted. We must elect the same transition method for this guidance as that chosen for the guidance for multiple-deliverable revenue arrangements. We adopted the provisions of the authoritative guidance as of January 1, 2010 on a prospective basis. The prospective application had no impact on our consolidated financial statements for the twelve months ended December 31, 2010.
In April 2010, the FASB reached a consensus on the Milestone Method of Revenue Recognition which provides guidance on the criteria that should be met for determining whether the milestone method of revenue recognition is appropriate. A vendor can recognize consideration that is contingent upon the achievement of a milestone in its entirety as revenue in the period in which the milestone is achieved only if the milestone meets all criteria to be considered substantive. The updated guidance is effective on a prospective basis for milestones achieved in fiscal years, and interim periods within those years beginning on or after June 15, 2010, with early adoption permitted. We are currently evaluating the impact of this guidance on our consolidated results of operations and financial condition.
2. Net Loss Per Common Share
Basic net loss per share attributable to common stockholders is calculated by dividing the net loss attributable to common stockholders by the weighted-average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share attributable to common stockholders is computed by dividing the net loss attributable to common stockholders by the weighted-average number of common share equivalents outstanding for the period determined using the treasury-stock method. For purposes of this calculation, options, and the conversion of convertible senior notes are considered to be common stock equivalents and are only included in the calculation of diluted net loss per share when their effect is dilutive.
Historical outstanding anti-dilutive securities not included in diluted net loss per share attributable to common stockholders calculation (in thousands):
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Options outstanding to purchase common stock |
8,353 | 7,972 | 7,112 | |||||||||
| Unvested restricted stock and restricted stock units |
504 | 31 | 75 | |||||||||
| Convertible senior notes |
— | 7,692 | 7,692 | |||||||||
| Total |
8,857 | 15,695 | 14,879 | |||||||||
F-15
Table of Contents
3. Financial Statement Details
Short Term Marketable Securities, Available for Sale
Short-term investment securities, consisting solely of debt securities with contractual maturities of less than one year, were as follows (in thousands):
| December 31, 2010 | ||||||||||||||||
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Market Value |
|||||||||||||
| U.S. government agencies |
$ | 32,231 | $ | 2 | $ | (10 | ) | $ | 32,223 | |||||||
| Commercial paper |
4,996 | — | — | 4,996 | ||||||||||||
| Corporate debt |
5,006 | 1 | (2 | ) | 5,005 | |||||||||||
| Total |
$ | 42,233 | $ | 3 | $ | (12 | ) | $ | 42,224 | |||||||
| December 31, 2009 | ||||||||||||||||
| Amortized Cost |
Gross Unrealized Gains |
Gross Unrealized Losses |
Estimated Market Value |
|||||||||||||
| U.S. government agencies |
$ | 16,198 | $ | 7 | $ | (10 | ) | $ | 16,195 | |||||||
| Corporate debt |
8,244 | 1 | (1 | ) | 8,244 | |||||||||||
| Total |
$ | 24,442 | $ | 8 | $ | (11 | ) | $ | 24,439 | |||||||
Accounts Receivable
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accounts receivable |
$ | 7,174 | $ | 3,811 | ||||
| Less allowance for doubtful accounts, sales returns and discounts |
(503 | ) | (321 | ) | ||||
| Total |
$ | 6,671 | $ | 3,490 | ||||
Inventory
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Raw materials |
$ | 5,041 | $ | 1,613 | ||||
| Work in process |
575 | 398 | ||||||
| Finished goods |
2,496 | 630 | ||||||
| Total |
$ | 8,112 | $ | 2,641 | ||||
Property and Equipment
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Furniture and fixtures |
$ | 2,462 | $ | 1,408 | ||||
| Computer equipment |
6,635 | 4,258 | ||||||
| Machinery and equipment |
10,782 | 8,712 | ||||||
| Leasehold improvements |
5,908 | 4,843 | ||||||
| Total |
25,787 | 19,221 | ||||||
| Accumulated depreciation and amortization |
(15,024 | ) | (12,799 | ) | ||||
| Property and equipment, net |
$ | 10,763 | $ | 6,422 | ||||
F-16
Table of Contents
Depreciation and amortization expense for the years ended December 31, 2010, 2009, and 2008 was $2.4 million, $2.4 million, and $3.0 million, respectively.
Accounts Payable and Accrued Liabilities
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accounts payable trade |
$ | 1,758 | $ | 2,600 | ||||
| Accrued tax, audit, and legal fees |
783 | 761 | ||||||
| Clinical trials |
134 | 238 | ||||||
| Accrued interest on convertible debt |
— | 831 | ||||||
| Accrued other including warranty |
2,675 | 1,315 | ||||||
| Total |
$ | 5,350 | $ | 5,745 | ||||
Accrued Payroll and Related Expenses
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Accrued paid time off |
$ | 1,570 | $ | 1,254 | ||||
| Accrued wages, bonus and taxes |
3,596 | 2,779 | ||||||
| Other accrued employee benefits |
564 | 373 | ||||||
| Total |
$ | 5,730 | $ | 4,406 | ||||
Accrued Warranty
| Year Ended December 31, | ||||||||
| 2010 | 2009 | |||||||
| Beginning balance |
$ | 129 | $ | 71 | ||||
| Charges to costs and expenses |
2,178 | 1,223 | ||||||
| Costs incurred |
(1,872 | ) | (1,165 | ) | ||||
| Ending balance |
$ | 435 | $ | 129 | ||||
4. Commitments and contingencies
Equipment Line
In March 2006, we entered into a loan and security agreement (the “Loan Agreement”) that provided for up to $5.0 million to finance various equipment purchases through March 2007. In January 2008, we entered into an amendment to the Loan Agreement to finance additional equipment purchases. The amendment allows us to draw an additional amount of up to $3.0 million under a new and additional Facility B Equipment Line.
At December 31, 2010, we had total borrowings of $525,000 under the Loan Agreement pursuant to the Facility A Equipment Line and Facility B Equipment Line and none was available for future borrowings. The loan bears an interest rate equal to the lender’s prime rate plus 0.25% and at December 31, 2010, the interest rate was 3.5%. Beginning April 2008, terms of the Facility B Equipment Line began to require monthly amortized payments through the maturity date of July 2011. Under the amended Loan Agreement, we continue to grant a security interest in substantially all of our personal property as collateral for the loan and are required to maintain cash balances equal to total outstanding loan balances with the lender.
F-17
Table of Contents
Convertible Senior Notes
In March 2007, we issued $60 million aggregate principal amount of Convertible Senior Notes due 2027 in a private offering. The notes were convertible into shares of common stock based on an initial conversion rate of 128.2051 shares of common stock per $1,000 principal amount of notes, which was equivalent to an initial conversion price of approximately $7.80 per share.
The aggregate underwriting commissions and other debt issuance costs incurred with respect to the issuance of the notes was $2.7 million. These costs have been allocated to the debt and equity components of the convertible debt per the guidance for accounting for convertible debt instruments that may be settled in cash upon conversion (including partial cash settlement), and were capitalized as debt issuance costs on our consolidated balance sheet.
For the twelve months ended December 31, 2010, we completed exchanges with prior holders of our issued and outstanding Notes, under which we issued an aggregate of approximately 7.9 million shares of our common stock, par value $0.001 per share, in exchange for $60.0 million in aggregate principal amount of the Notes previously held by the exchanging holders. We incurred a loss on the extinguishment of the Notes in the amount of $8.5 million for the twelve months ended December 31, 2010, which includes the difference between the carrying value and the fair value of the Notes on the conversion date, other consideration given to note holders to induce early conversion and transaction costs incurred with third parties, other than the investors, to settle the conversion of the Notes. Determining the fair value of the liability component requires the use of accounting estimates and assumptions which are judgmental in nature and could have a significant impact on the valuation. As of September 30, 2010, no Convertible Senior Notes remained outstanding.
Call Spread Option
In March 2007, we entered into hedge transactions to minimize the potential dilution of our common stock upon conversion of the Convertible Senior Notes if our stock price exceeds $7.80 per share through March 2009. We had the right to purchase a number of shares of common stock equal to the number of shares underlying the $60 million principal amount of the notes, at a strike price equal to the conversion price of the notes, or $7.80 per share. The call spread options were structured in four tranches with one tranche expiring in each six-month interval for two years from the date of March 6, 2007. Each of the four options capped the potential benefit to us at market prices ranging from $9.00 for the option which expired in September 2007 to $18.50 for the option which expired in March 2009. The call spread options were separate transactions entered into by us and were not part of the terms of the Convertible Senior Notes.
In accordance with authoritative guidance for accounting for derivative financial instruments indexed to, and potentially settled in, a company’s own stock, we recorded the $11.0 million cost of the call spread transactions as a net reduction in paid in capital in the Balance Sheet for the quarter ended March 31, 2007, and will not recognize subsequent changes in fair value. During September 2007, we received approximately 154,000 shares of our common stock with a value of $1.4 million on the date the shares were returned to us as settlement of the first tranche. During March 2008, we received approximately 118,000 shares of our common stock with a value of $869,000 on the date the shares were returned to us as settlement for the second tranche.
F-18
Table of Contents
Leases
In April 2006, we entered into an office lease agreement for facilities located in San Diego, California. In August 2010, we entered into a First Amendment to Office Lease (the “Agreement”) with respect to facilities in the buildings at 6340 Sequence Drive and 6310 Sequence Drive, each in San Diego, California (the “Buildings”). Under the Agreement, we have leased additional space in the Buildings, and retain the right and obligation to lease additional space in the Buildings. The lease term for the Buildings extends through November 2016 and we have a five-year option to renew the lease upon the expiration of the initial term. We also currently maintain a second lease for our former headquarters facility which expires in 2011. These facility leases have annual rental increases ranging from approximately 2.5% to 4.0%. The difference between the straight-line expense over the term of the lease and actual amounts paid are recorded as deferred rent. In September 2008, our subsidiary in Sweden entered into a three year lease for a small shared office space, which has a quarterly adjustment clause for rent to increase or decrease in proportion to changes in consumer prices. Rental obligations, excluding real estate taxes, operating costs, and tenant improvement allowances, under all lease agreements as of December 31, 2010 were as follows (in thousands):
| Fiscal Year Ending |
||||
| 2011 |
2,027 | |||
| 2012 |
2,117 | |||
| 2013 |
2,503 | |||
| 2014 |
2,587 | |||
| 2015 |
2,665 | |||
| Thereafter |
2,511 | |||
| Total |
$ | 14,410 | ||
Rent expense for the years ended December 31, 2010, 2009, and 2008 was $2.1 million, $1.5 million, and $2.2 million, respectively.
Litigation
On August 11, 2005, Abbott Diabetes Care, Inc., or Abbott, filed a patent infringement lawsuit against us in the United States District Court for the District of Delaware, seeking a declaratory judgment that our continuous glucose monitor infringes certain patents held by Abbott. In August 2005, we moved to dismiss these claims and filed requests for reexamination of the Abbott patents with the United States Patent and Trademark Office, or the Patent Office, and by March 2006, the Patent Office ordered reexamination of each of the four patents originally asserted against us in the litigation. On June 27, 2006, Abbott amended its complaint to include three additional patents owned or licensed by Abbott which are allegedly infringed by our continuous glucose monitor. On August 18, 2006, the court granted our motion to stay the lawsuit pending reexamination by the Patent Office of each of the four patents originally asserted by Abbott, and the court dismissed one significant infringement claim. In approving the stay, the court also granted our motion to strike, or disallow, Abbott’s amended complaint in which Abbott had sought to add three additional patents to the litigation. Subsequent to the court’s August 18, 2006 order striking Abbott’s amended complaint, Abbott filed a separate action in the U.S. District Court for the District of Delaware alleging patent infringement of the three additional patents it had sought to include in the litigation discussed above. On September 7, 2006, we filed a motion to strike Abbott’s new complaint on the grounds that it is redundant of claims Abbott already improperly attempted to inject into the original case, and because the original case is now stayed, Abbott must wait until the court lifts that stay before it can properly ask the court to consider these claims. Alternatively, we asked the court to consolidate the new case with the original case and thereby stay the entirety of the case pending conclusion of the reexamination proceedings in the Patent Office. In February 2007, the Patent Office ordered reexamination of each of the three patents cited in this new lawsuit. On September 30, 2007, the court granted our motion to consolidate the cases and stay the entirety of the case pending conclusion of the reexamination proceedings in the Patent Office relating to all seven patents asserted against us.
F-19
Table of Contents
In connection with this litigation each of Abbott’s seven patents that are the subject of the litigation have one or more associated reexamination requests in various stages at the Patent Office. Abbott has filed responses with the Patent Office seeking claim construction to differentiate certain claims from the prior art we have presented, seeking to amend certain claims to overcome the prior art we have presented, canceling claims and/or seeking to add new claims. The Board of Patent Appeals and Interferences within the Patent Office has recently rendered decisions on the appeals related to the reexaminations of two of the patents. We believe these decisions are favorable to us; however, Abbott may still seek judicial review of the decisions. Four patents are currently undergoing reexamination at the Patent Office. Reexamination of another patent was completed in 2010 and we recently submitted a subsequent reexamination request.
In addition, since 2008, Abbott has copied claims from certain of our applications, and stated that it may seek to provoke an interference with certain of our pending applications in the Patent Office. If interference is declared and Abbott prevails in the interference, we would lose certain patent rights to the subject matter defined in the interference. Also since 2008, Abbott has filed reexamination requests seeking to invalidate fifteen of our patents. The fifteen reexamination requests are in various stages at the Patent Office. We have filed responses with the Patent Office seeking claim construction to differentiate certain claims from the prior art presented in the reexaminations, seeking to amend certain claims to overcome the prior art presented in the reexaminations, canceling claims and/or seeking to add new claims. It is possible that the Patent Office may determine that some or all of the claims of our patents subject to the reexamination are invalid.
Although it is our position that Abbott’s assertions of infringement have no merit, and that the potential interference and reexamination requests have no merit, neither the outcome of the litigation nor the amount and range of potential fees associated with the litigation, potential interference or reexamination requests can be assessed, and as of December 31, 2010, no amounts have been accrued.
Purchase Commitments
We are party to various purchase arrangements related to our development activities including materials used in our glucose monitoring systems. As of December 31, 2010, we had purchase commitments with vendors totaling $3.3 million due within one year. There are no material purchase commitments due beyond one year.
5. Development Agreements
Insulet Corporation
On January 7, 2008, we entered into a development agreement with Insulet to integrate our continuous glucose monitoring technology into Insulet’s wireless, handheld OmniPod System Personal Diabetes Manager. The agreement is non-exclusive and does not impact either party’s existing third party development agreements.
Animas Corporation
On January 10, 2008, as amended on January 12, 2009 and July 30, 2009 (“the Animas Amendments”), we entered into a joint development agreement with Animas to integrate our continuous glucose monitoring technology into Animas insulin pumps. Under the terms of the amended agreement, Animas will contribute up to $1.1 million to DexCom to offset certain development, clinical and regulatory expenses. The agreement is non-exclusive in the United States, but exclusive outside the United States and does not impact either party’s existing third party development agreements. In January 2008 we received $500,000. In January 2009 we received $250,000. We recorded $189,000 in development grant and other revenue for the twelve months ended December 31, 2010, compared to $279,000 for the same period in 2009. Pursuant to the Animas Amendments, we will collaborate with Animas to develop a modified version of our transmitter to support a single, global CGM-enabled insulin pump launch by Animas. We were entitled to receive a one-time $1.0 million milestone payment upon the achievement of performance qualification of a manufacturing line for the modified transmitter,
F-20
Table of Contents
which was earned in December 2010, and we received this $1.0 million milestone payment in January 2011. We are also entitled to receive an additional $4.0 million payment upon the first regulatory body approval outside the United States for the new system.
Edwards Lifesciences LLC
On November 10, 2008, and as amended on May 5, 2009, we entered into a Collaboration Agreement (the “Collaboration Agreement”) with Edwards. Pursuant to the Agreement, we and Edwards agreed to develop jointly and to market an in-hospital automatic blood glucose monitoring system. Under the terms of the Collaboration Agreement, as amended, Edwards was obligated to pay us an upfront fee of $13.0 million. In addition, we are entitled to receive up to $22.0 million for product development costs and milestones related to regulatory approvals and manufacturing readiness. We will also receive either a profit-sharing payment of up to 10% of commercial sales of the product, or a royalty of up to 6% of commercial sales of the product. The Collaboration Agreement provides Edwards with an exclusive license under our intellectual property to the critical care sector in the hospital market. Edwards will be responsible for global sales and marketing, and we will initially be responsible for manufacturing. In November 2008 we received $13.0 million. We received an additional $3.0 million during the twelve months ended December 31, 2010. We recorded $6.9 million in development grant and other revenue for the twelve months ended December 31, 2010, compared to $11.1 million for the same period in 2009.
6. Stockholders’ Equity
Follow-on Stock Offering
In January 2010, we completed a follow-on public offering of 4,025,000 shares of our common stock for net proceeds of approximately $33.0 million. In November 2010, we completed a public follow-on stock offering of 3,277,500 shares of our common stock for net proceeds of approximately $33.0 million.
7. Income Taxes
We have recorded a net tax benefit of $31,000 and $15,000 for the years ended December 31, 2010 and 2009, respectively, related to refundable income tax credits and partial offset by foreign income tax expense. The amounts have been included in other income in the consolidated financial statements.
At December 31, 2010, we had federal and state tax net operating loss carryforwards of approximately $237.6 million and $169.4 million, respectively. The federal and state tax loss carryforwards will begin to expire in 2019 and 2013, respectively, unless previously utilized. We also had federal and state research and development tax credit carryforwards of approximately $3.1 million and $5.6 million, respectively. The federal research and development tax credit will begin to expire in 2019, unless previously utilized.
Utilization of net operating losses and credit carryforwards are subject to an annual limitation due to ownership change limitations provided by Section 382 and 383 of the Internal Revenue Code of 1986, as amended, and similar state provisions. An ownership change limitation occurred as a result of the stock offering completed in February 2009. The limitation will likely result in approximately $2.1 million of U.S. income tax credits and approximately $9.2 million of state net operating loss carryforwards that will expire unused. The related deferred tax assets have been removed from the components of our deferred tax assets as summarized below. The tax benefits related to the remaining federal and state net operating losses and tax credit carryforwards may be further limited or lost if future cumulative changes in ownership exceed 50% within any three-year period.
F-21
Table of Contents
Significant components of our deferred tax assets as of December 31, 2010 and 2009 are shown below (in thousands). A valuation allowance of approximately $118.3 million has been established as of December 31, 2010 to offset the deferred tax assets, as realization of such assets is uncertain.
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Net operating loss carryforwards |
$ | 89,069 | $ | 75,325 | ||||
| Capitalized research and development expenses |
15,743 | 16,578 | ||||||
| Tax credits |
3,975 | 3,165 | ||||||
| Share-based compensation |
6,750 | 5,627 | ||||||
| Fixed and intangible assets |
1,397 | 1,358 | ||||||
| Other, net |
1,329 | 815 | ||||||
| Total gross deferred tax assets |
118,263 | 102,868 | ||||||
| Less: valuation allowance |
(118,263 | ) | (97,353 | ) | ||||
| Net deferred tax assets |
— | 5,515 | ||||||
| Deferred tax liabilities |
||||||||
| Convertible debt |
— | (5,515 | ) | |||||
| Net deferred tax assets |
$ | — | $ | — | ||||
We recognize windfall tax benefits associated with the exercise of stock options directly to stockholders’ equity only when realized. Accordingly, deferred tax assets are not recognized for net operating loss carryforwards resulting from windfall tax benefits occurring from January 1, 2006 onward. At December 31, 2010, deferred tax assets do not include $3.6 million of excess tax benefits from share-based compensation.
The reconciliation between our effective tax rate on income (loss) from continuing operations and the statutory rate is as follows:
| December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Income taxes (benefit) at statutory rates |
35.00 | % | 35.00 | % | 35.00 | % | ||||||
| State income tax, net of federal benefit |
3.46 | % | 3.16 | % | 3.06 | % | ||||||
| Permanent items |
(2.68 | )% | (2.20 | )% | (1.58 | )% | ||||||
| Research and development credits |
2.51 | % | 2.32 | % | 1.77 | % | ||||||
| Tax attribute carryover limitation |
— | (4.89 | )% | — | ||||||||
| Other |
(0.36 | )% | 1.09 | % | (1.23 | )% | ||||||
| Change in valuation allowance |
(37.88 | )% | (34.45 | )% | (36.96 | )% | ||||||
| 0.06 | % | 0.03 | % | 0.06 | % | |||||||
8. Related Party Transactions
Our former Chairman is a director of Oracle Corporation. Costs incurred relating to an Oracle ERP system for the years ended December 31, 2010, 2009 and 2008 totaled $284,000, $94,000, and $105,000, respectively.
F-22
Table of Contents
9. Employee Benefit Plans
401(k) Plan
We have a defined contribution 401(k) retirement plan, or the 401(k) Plan, covering substantially all employees that meet certain age requirements. Employees may contribute up to 90% of their compensation per year (subject to a maximum limit by federal tax law). Under the 401(k) Plan, we may elect to match a discretionary percentage of contributions. No such matching contributions have been made to the 401(k) Plan since its inception.
Employee Stock Purchase Plan
The Employee Stock Purchase Plan (“ESPP”) permits our eligible employees to purchase shares of common stock, at semi-annual intervals, through periodic payroll deductions. Payroll deductions may not exceed 10% of the participant’s cash compensation subject to certain limitations, and the purchase price will not be less than 85% of the lower of the fair market value of the stock at either the beginning of the applicable “Offering Period” or the Purchase Date. Each Offering Period is 12 months, with new Offering Periods commencing every six months on the dates of February 1 and August 1 of each year. Each Offering Period consists of two (2) six month purchase periods (each a “Purchase Period”) during which payroll deductions of the participants are accumulated under the ESPP. The last business day of each Purchase Period is referred to as the “Purchase Date.” The First Offering Period ran from April 13, 2005 to July 31, 2006 and included the Purchase Dates of January 31 and July 31 of 2006. Thereafter, Purchase Dates are every six months on the dates of January 31 and July 31. Annually in January of each year, subject to Board discretion and certain limitations, shares reserved for the ESPP will automatically be increased by a number of shares equal to 1% of the total number of issued and outstanding shares of our common stock at the preceding year end. On January 31, 2008, July 31, 2008, January 30, 2009, July 31, 2009, January 29, 2010 and July 30, 2010, we issued 48,584, 49,238, 84,062, 108,629, 146,500 and 60,909 respectively, shares of common stock under the ESPP.
Equity Incentive Plans
In 2005, we adopted the 2005 Equity Incentive Plan (“2005 Plan”) which replaced the 1999 Incentive Stock Plan and provides for the grant of incentive and nonstatutory stock options, restricted stock, stock bonuses, stock appreciation rights, and restricted stock units to employees, directors or consultants of the Company. Shares reserved include all shares that were available under the 1999 Incentive Stock Plan on the day it was terminated. Options generally vest over four years and expire ten years from the date of grant. In addition, incentive stock options may not be granted at a price less than the 100% of the fair market value on the date of grant. The term of the 2005 Plan is scheduled to end in March 2015. Annually in January of each year, subject to Board discretion and certain limitations, shares reserved for the 2005 Plan will automatically be increased by a number of shares equal to 3% of the total number of issued and outstanding shares of our common stock during the preceding year end.
F-23
Table of Contents
A summary of our stock option activity, and related information for the year ended December 31, 2010 is as follows (in thousands except weighted-average exercise price and weighted-average remaining contractual term):
| Number of Shares |
Weighted- Average Exercise Price |
Weighted- Average Remaining Contractual Term (years) |
Aggregate Intrinsic Value |
|||||||||||||
| Outstanding at December 31, 2009 |
7,972 | $ | 7.12 | |||||||||||||
| Granted |
1,145 | $ | 10.40 | |||||||||||||
| Exercised |
(549 | ) | $ | 6.63 | ||||||||||||
| Cancelled |
(215 | ) | $ | 8.52 | ||||||||||||
| Outstanding at December 31, 2010 |
8,353 | $ | 7.56 | 7.3 | $ | 52,996 | ||||||||||
| Exercisable at December 31, 2010 |
5,528 | $ | 7.74 | 6.7 | $ | 34,831 | ||||||||||
The weighted average fair values of options granted was $6.49, $3.42, and $3.00 for the three years ended December 31, 2010, 2009, and 2008, respectively. The total intrinsic value of options exercised was $2.6 million, $110,000, and $7.8 million determined as of the date of exercise during the years ended December 31, 2010, 2009, and 2008, respectively.
The risk-free interest rate assumption is based upon observed interest rates appropriate for the expected life of our employee stock options and stock purchase plan. The dividend yield assumption is based on our history and expectation of dividend payouts.
Due to our limited history as a publicly traded company that began in April 2005, our expected volatility is based on both our historical stock prices and the historical prices of similar companies, as determined by us. Accordingly, we used the simplified method to determine the expected life.
As share-based compensation expense recognized in the consolidated statement of operations for fiscal 2010, 2009 and 2008 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. Authoritative guidance for share-based payment requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Forfeitures were estimated based on historical experience.
The following table summarizes information about stock options outstanding at December 31, 2010 (in thousands except for exercise price and contractual life):
| Options Outstanding |
Options Exercisable | |||||||||||||||||||||||||||
| Range of Exercise Price |
Number of Shares |
Weighted Average Remaining Contractual Life |
Weighted Average Exercise Price |
Aggregate Intrinsic Value |
Number of Shares |
Weighted Average Exercise Price |
Aggregate Intrinsic Value |
|||||||||||||||||||||
| $0.30 – $2.40 |
296 | 3.5 | $ | 1.58 | $ | 3,575 | 293 | $ | 1.58 | $ | 3,533 | |||||||||||||||||
| $3.19 – $4.71 |
1,775 | 8.0 | $ | 3.51 | 18,001 | 958 | $ | 3.62 | 9,616 | |||||||||||||||||||
| $5.05 – $8.06 |
3,440 | 7.2 | $ | 7.03 | 22,769 | 2,536 | $ | 7.07 | 16,679 | |||||||||||||||||||
| $8.26 – $9.56 |
918 | 7.0 | $ | 8.93 | 4,330 | 777 | $ | 8.93 | 3,669 | |||||||||||||||||||
| $9.73 – $13.65 |
1,554 | 8.0 | $ | 10.87 | 4,321 | 608 | $ | 11.46 | 1,334 | |||||||||||||||||||
| $13.86 – $25.80 |
370 | 5.1 | $ | 19.45 | — | 356 | $ | 19.65 | — | |||||||||||||||||||
| $0.30 – $25.80 |
8,353 | 7.3 | $ | 7.56 | $ | 52,996 | 5,528 | $ | 7.74 | $ | 34,831 | |||||||||||||||||
F-24
Table of Contents
We define in-the-money options at December 31, 2010 as options that had exercise prices that were lower than the $13.65 closing market price of our common stock at that date. The aggregate intrinsic value of options outstanding at December 31, 2010 is calculated as the difference between the exercise price of the underlying options and the market price of our common stock for the 8.0 million options that were in-the-money at that date. There were 5.2 million in-the-money options exercisable at December 31, 2010.
The following table sets forth a summary of our nonvested stock options and activity as of and for the year ended December 31, 2010:
| Shares | Weighted Average Grant Date Fair Value |
|||||||
| (in thousands) | ||||||||
| Nonvested at December 31, 2009 |
3,679 | $ | 3.23 | |||||
| Granted |
1,146 | 6.49 | ||||||
| Vested |
(1,786 | ) | 3.76 | |||||
| Forfeited |
(214 | ) | 4.49 | |||||
| Nonvested at December 31, 2010 |
2,825 | $ | 4.21 | |||||
Valuation and expense information
The following table summarizes share-based compensation expense related to employee stock options, restricted stock, restricted stock units and employee stock purchases for the years ended December 31, 2010, 2009 and 2008 were allocated as follows (in thousands):
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Cost of sales |
$ | 746 | $ | 580 | $ | 561 | ||||||
| Research and development |
2,695 | 1,850 | 1,740 | |||||||||
| Selling, general and administrative |
6,000 | 5,936 | 5,382 | |||||||||
| Share-based compensation expense included in operating expenses |
$ | 9,441 | $ | 8,366 | $ | 7,683 | ||||||
We estimated the fair value of each option grant and ESPP purchase rights on the date of grant using the Black-Scholes option pricing model with the below assumptions.
Options:
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Risk free interest rate |
1.8 – 2.8 | % | 2.0 – 2.8 | % | 4.5 | % | ||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Expected volatility of the Company’s stock |
0.67 – 0.68 | 0.63 – 0.67 | 0.48 | |||||||||
| Expected life (in years) |
6.1 | 6.1 | 6.1 | |||||||||
ESPP:
| Years Ended December 31, | ||||||||||||
| 2010 | 2009 | 2008 | ||||||||||
| Risk free interest rate |
0.35 – .38 | % | 0.5 – 1.0 | % | 4.6 | % | ||||||
| Dividend yield |
0 | % | 0 | % | 0 | % | ||||||
| Expected volatility of the Company’s stock |
0.32 – 0.45 | 0.69 – 0.91 | 0.48 | |||||||||
| Expected life (in years) |
1 | 1 | 1 | |||||||||
F-25
Table of Contents
Restricted Stock Awards and Restricted Stock Units (RSUs)
We have periodically granted unvested restricted common stock awards to certain employees. As of December 31, 2010, a total of 151,963 such shares had been granted. The grant awards typically vest 25% annually and are fully vested following the fourth anniversary of the vesting start date. RSU awards typically vest annually over one to three years. Vesting of all restricted common stock awards and RSUs is subject to continued employment and we have the right to repurchase unvested restricted stock award shares at the original issuance price of $0.001 per share subject to certain terms and conditions. The restricted stock and RSUs had a weighted-average fair value of $9.67 per share at date of grant. As of December 31, 2010, there were 4,283 restricted stock shares subject to repurchase with an intrinsic value of $58,000.
The following table sets forth a summary of our RSU activity as of and for the year ended December 31, 2010 (in thousands except weighted average grant date fair value):
| Shares | Weighted Average Grant Date Fair Value |
Aggregate Intrinsic Value |
||||||||||
| Nonvested at December 31, 2009 |
— | $ | — | |||||||||
| Granted |
555 | 9.95 | ||||||||||
| Vested |
(48 | ) | 10.07 | |||||||||
| Forfeited |
(8 | ) | 9.80 | |||||||||
| Nonvested at December 31, 2010 |
499 | $ | 9.94 | $ | 6,814 | |||||||
Reserved Shares
We have reserved shares of common stock for future issuance as follows (in thousands):
| December 31, | ||||||||
| 2010 | 2009 | |||||||
| Stock options and awards under our plans: |
||||||||
| Stock options granted and outstanding |
8,353 | 7,972 | ||||||
| Unvested RSUs |
499 | — | ||||||
| Reserved for future grant |
41 | 136 | ||||||
| Employee Stock Purchase Plan |
1,102 | 849 | ||||||
| Convertible senior notes |
— | 7,692 | ||||||
| Total |
9,995 | 16,649 | ||||||
F-26
Table of Contents
10. Quarterly Financial Information (Unaudited)
The following is a summary of the quarterly results of operations for the years ended December 31, 2010 and 2009 (in thousands except per share data):
| For the Three Months Ended | ||||||||||||||||
| December 31 | September 30 | June 30 | March 31 | |||||||||||||
| Year ended December 31, 2010 |
||||||||||||||||
| Revenues |
$ | 15,636 | $ | 11,664 | $ | 11,786 | $ | 9,545 | ||||||||
| Gross margin |
6,989 | 3,476 | 4,519 | 3,459 | ||||||||||||
| Total operating costs |
16,875 | 16,538 | 15,787 | 14,533 | ||||||||||||
| Net loss attributable to common stockholders |
(9,812 | ) | (13,398 | ) | (11,687 | ) | (20,273 | ) | ||||||||
| Basic and diluted net loss per share attributable to common stockholders |
$ | (0.16 | ) | $ | (0.23 | ) | $ | (0.20 | ) | $ | (0.40 | ) | ||||
| Year ended December 31, 2009 |
||||||||||||||||
| Revenues |
$ | 10,471 | $ | 7,259 | $ | 6,751 | $ | 5,212 | ||||||||
| Gross margin (deficit) |
4,107 | 865 | (1,048 | ) | (263 | ) | ||||||||||
| Total operating costs |
13,569 | 12,444 | 12,407 | 11,074 | ||||||||||||
| Net loss attributable to common stockholders |
(11,529 | ) | (13,523 | ) | (15,330 | ) | (13,142 | ) | ||||||||
| Basic and diluted net loss per share attributable to common stockholders |
$ | (0.25 | ) | $ | (0.29 | ) | $ | (0.33 | ) | $ | (0.33 | ) | ||||
F-27
Table of Contents
SCHEDULE II—VALUATION AND QUALIFYING ACCOUNTS
For the Years Ended December 31, 2010, 2009 and 2008
(in thousands)
| Allowance for doubtful accounts |
||||
| Balance December 31, 2007 |
$ | 25 | ||
| Provision for doubtful accounts |
61 | |||
| Write-off and adjustments |
(19 | ) | ||
| Recoveries |
— | |||
| Balance December 31, 2008 |
$ | 67 | ||
| Allowance for doubtful accounts |
||||
| Balance December 31, 2008 |
$ | 67 | ||
| Provision for doubtful accounts |
356 | |||
| Write-off and adjustments |
(137 | ) | ||
| Recoveries |
— | |||
| Balance December 31, 2009 |
$ | 286 | ||
| Allowance for doubtful accounts |
||||
| Balance December 31, 2009 |
$ | 286 | ||
| Provision for doubtful accounts |
658 | |||
| Write-off and adjustments |
(497 | ) | ||
| Recoveries |
6 | |||
| Balance December 31, 2010 |
$ | 453 | ||
F-28
Table of Contents
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
Not applicable.
| ITEM 9A. | CONTROLS AND PROCEDURES. |
Disclosure Controls and Procedures
Regulations under the Securities Exchange Act of 1934 require public companies to maintain “disclosure controls and procedures,” which are defined to mean a company’s controls and other procedures that are designed to ensure that information required to be disclosed in the reports that it files or submits under the Securities Exchange Act of 1934 is accumulated and timely communicated to management, including our Chief Executive Officer and Chief Financial Officer, recorded, processed, summarized, and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms. Our management, including our Chief Executive Officer and our Chief Financial Officer, conducted an evaluation as of the end of the period covered by this report of the effectiveness of our disclosure controls and procedures. Based on their evaluation, our Chief Executive Officer and our Chief Financial Officer concluded that our disclosure controls and procedures were effective for this purpose.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934. Our internal control over financial reporting is designed to provide reasonable assurance to our management and Board of Directors regarding the preparation and fair presentation of published financial statements.
Our management, with the participation of the Chief Executive and Chief Financial Officers, assessed the effectiveness of our internal control over financial reporting as of December 31, 2010. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control—Integrated Framework. Based on this assessment, our management, with the participation of the Chief Executive and Chief Financial Officers, believes that, as of December 31, 2010, our internal control over financial reporting is effective based on those criteria. The effectiveness of our internal control over financial reporting as of December 31, 2010 has been audited by Ernst & Young LLP an independent Public Registered Accounting firm, as stated in their report which is included herein.
The certifications of our Chief Executive Officer and Chief Financial Officer required under Section 302 of the Sarbanes-Oxley Act have been filed as Exhibits 31.01 and 31.02 to this report.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Limitation on Effectiveness of Controls
It should be noted that any system of controls, however well designed and operated, can provide only reasonable, and not absolute, assurance that the objectives of the system are met. The design of any control system is based, in part, upon the benefits of the control system relative to its costs. Control systems can be circumvented by the individual acts of some persons, by collusion of two or more people, or by management override of the control. In addition, over time, controls may become inadequate because of changes in conditions, or the degree of compliance with the policies or procedures may deteriorate. Because of these and other inherent limitations of control systems, there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote.
61
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders of DexCom, Inc.
We have audited DexCom, Inc.’s internal control over financial reporting as of December 31, 2010, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). DexCom, Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, DexCom, Inc. maintained in all material respects effective internal control over financial reporting as of December 31, 2010 based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of DexCom, Inc. as of December 31, 2010 and 2009, and the related consolidated statements of operations, stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2010 of DexCom, Inc. and our report dated March 3, 2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 3, 2011
| ITEM 9B. | OTHER INFORMATION. |
None.
62
Table of Contents
PART III
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information concerning our directors required by this Item is incorporated by reference to the section in our Proxy Statement entitled “Proposal No. 1—Election of Directors.”
The information concerning our executive officers required by this Item is incorporated by reference to the section in our Proxy Statement entitled “Executive Officers.”
The information concerning compliance with Section 16(a) of the Securities Exchange Act of 1934 required by this Item is incorporated by reference to the section in our Proxy Statement entitled “Section 16(a) Beneficial Ownership Reporting Compliance.”
We have adopted a written code of ethics for financial employees that applies to our principal executive officer, principal financial officer, principal accounting officer, controller and other employees of the finance department designated by our Chief Financial Officer. This code of ethics, titled the “Code of Conduct and Ethics for Chief Executive Officer and Senior Finance Personnel,” is publicly available on our Internet website at http://investor.shareholder.com/dexcom/governance.cfm. The information contained on our Internet website is not incorporated by reference into this Report on Form 10-K.
The information concerning the audit committee of the Board of Directors required by this Item is incorporated by reference to information set forth in the Proxy Statement.
The information concerning material changes to the procedures by which stockholders may recommend nominees to the Board of Directors required by this Item is incorporated by reference to information set forth in the Proxy Statement.
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item concerning executive compensation and our Compensation Committee is incorporated by reference to information set forth in the Proxy Statement.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item is incorporated by reference to information set forth in the Proxy Statement under the headings “Principal Stockholders and Stock Ownership by Management” and “Equity Compensation Plan Information.”
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item with respect to director independence is incorporated by reference to information set forth in the Proxy Statement.
The information concerning certain relationships and related transactions required by the Item is incorporated by reference to the section in our Proxy Statement entitled “Certain Transactions.”
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information concerning principal accountant fees and services required by this Item is incorporated by reference to the section in our Proxy Statement entitled “Ratification of Selection of Independent Registered Public Accounting Firm.”
63
Table of Contents
PART IV
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
| (a) | The following documents are filed as part of this annual report: |
| 1. | Financial Statements. The financial statements in Part II, Item 8 of this annual report are incorporated by reference. |
| 2. | Financial Statement Schedules. |
For the three fiscal years ended December 31, 2010—Schedule II Valuation and Qualifying Accounts, the financial statements in Part II, Item 8 of this annual report are incorporated by reference.
Schedules not listed above have been omitted because information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
| 3. | Exhibits. |
| Exhibit |
Exhibit Description |
Incorporated by Reference | Exhibit Number |
Provided Herewith |
||||||||||||
| Form | File No. | Date of First Filing |
||||||||||||||
| 3.01 | Registrant’s Restated Certificate of Incorporation. | S-1/A | 333-122454 | March 3, 2005 | 3.03 | |||||||||||
| 3.02 | Registrant’s Amended and Restated Bylaws. | 8-K | 000-51222 | May 22, 2009 | 99.01 | |||||||||||
| 4.01 | Form of Specimen Certificate for Registrant’s common stock. | S-1/A | 333-122454 | March 24, 2005 | 4.01 | |||||||||||
| 4.02 | Second Amended and Restated Investors’ Rights Agreement, dated December 30, 2004. | S-1 | 333-122454 | February 1, 2005 | 4.02 | |||||||||||
| 4.03 | Form of Rights Agreement, between DexCom, Inc. and American Stock Transfer & Trust Company, including the Certificate of Designations of Series A Junior Participating Preferred Stock, Summary of Stock Purchase Rights and Forms of Right Certificate attached thereto as Exhibit A, B and C, respectively. | S-1/A | 000-51222 | March 24, 2005 | 4.03 | |||||||||||
| 10.01 | Form of Indemnity Agreement between Registrant and each of its directors and executive officers. | S-1 | 333-122454 | February 1, 2005 | 10.01 | |||||||||||
| 10.02 | 1999 Stock Option Plan and related agreements.* | S-1 | 333-122454 | February 1, 2005 | 10.02 | |||||||||||
| 10.03 | 2005 Equity Incentive Plan and forms of stock option agreement and stock option exercise agreements.* | S-1/A | 000-51222 | March 24, 2005 | 10.03 | |||||||||||
| 10.04 | 2005 Employee Stock Purchase Plan and form of subscription agreement.* | S-1/A | 000-51222 | March 24, 2005 | 10.04 | |||||||||||
64
Table of Contents
| Exhibit |
Exhibit Description |
Incorporated by Reference | Exhibit Number |
Provided Herewith |
||||||||||||
| Form | File No. | Date of First Filing |
||||||||||||||
| 10.05 | Sorrento Valley Business Park Lease dated December 3, 2003 between Hub Properties Trust and DexCom, Inc. | S-1/A | 000-51222 | March 25, 2005 | 10.08 | |||||||||||
| 10.06 | Exclusive Patent License Agreement dated August 17, 2001 between SM Technologies, LLC and DexCom, Inc.** | S-1/A | 000-51222 | April 5, 2005 | 10.09 | |||||||||||
| 10.07 | Agreement Regarding Terms of Sale dated May 23, 2003 between AMI Semiconductor, Inc. and DexCom, Inc.** | S-1/A | 333-122454 | April 5, 2005 | 10.10 | |||||||||||
| 10.08 | Offer letter between DexCom, Inc. and Jorge Valdes dated October 16, 2005.* | 10-K | 000-51222 | February 27, 2006 | 10.14 | |||||||||||
| 10.09 | Loan and Security Agreement between Square 1 Bank and DexCom, Inc., dated March 20, 2006. | 8-K | 000-51222 | March 24, 2006 | 99.01 | |||||||||||
| 10.10 | Office Lease Agreement, dated March 31, 2006, between DexCom, Inc. and Kilroy Realty, L.P. | 8-K | 000-51222 | April 7, 2006 | 99.01 | |||||||||||
| 10.11 | Offer letter between DexCom, Inc. and Steven R. Pacelli dated April 10, 2006.* | 8-K | 000-51222 | April 13, 2006 | 99.01 | |||||||||||
| 10.12 | First Amendment to Loan and Security Agreement, dated January 31, 2008, between DexCom, Inc. and Square 1 Bank. | 8-K | 000-51222 | February 5, 2008 | 99.01 | |||||||||||
| 10.13 | Collaboration Agreement, dated November 10, 2008 between DexCom, Inc. and Edwards Lifesciences LLC.** | 8-K/A | 000-51222 | January 28, 2009 | 10.1 | |||||||||||
| 10.14 | Amended and Restated Joint Development Agreement, dated January 12, 2009, between DexCom, Inc. and Animas Corporation.** | 8-K/A | 000-51222 | January 28, 2009 | 10.1 | |||||||||||
| 10.15 | OUS Commercialization Agreement, dated January 12, 2009, between DexCom, Inc. and Animas Corporation.** | 8-K/A | 000-51222 | January 28, 2009 | 10.2 | |||||||||||
| 10.16 | Form of Amended and Restated Executive Change of Control & Severance Agreement.* | 10-K | 000-51222 | March 5, 2009 | 10.20 | |||||||||||
65
Table of Contents
| Exhibit |
Exhibit Description |
Incorporated by Reference | Exhibit Number |
Provided Herewith |
||||||||||||
| Form | File No. | Date of First Filing |
||||||||||||||
| 10.17 | Amended and Restated Offer Letter Agreement dated December 19, 2008 between DexCom, Inc. and Terrance H. Gregg.* | 10-K | 000-51222 | March 5, 2009 | 10.21 | |||||||||||
| 10.18 | Letter Agreement, between Edwards Lifesciences LLC and DexCom, Inc., dated May 5, 2009. | 10-Q | 000-51222 | August 3, 2009 | 10.22 | |||||||||||
| 10.19 | Non-Exclusive Distribution Agreement, between RGH Enterprises, Inc. and DexCom, Inc., dated April 30, 2008.** | 10-Q | 000-51222 | August 3, 2009 | 10.23 | |||||||||||
| 10.20 | Letter of Amendment of the Amended and Restated Joint Development Agreement, between Animas Corporation and DexCom, Inc., dated July 30, 2009.** | 10-Q | 000-51222 | November 4, 2009 | 10.24 | |||||||||||
| 10.21 | Amendment No. 1 to the Commercialization Agreements, between Animas Corporation and DexCom, Inc., dated July 30, 2009.** | 10-Q | 000-51222 | November 4, 2009 | 10.25 | |||||||||||
| 10.22 | Amended and Restated Development, Manufacturing, Licensing and Supply Agreement, between DSM PTG, Inc. and DexCom, Inc., dated February 19, 2010.** | 10-K | 000-51222 | March 9, 2010 | 10.25 | |||||||||||
| 10.23 | Form of Restricted Stock Unit Award Agreement. | 10-Q | 000-51222 | May 5, 2010 | 10.26 | |||||||||||
| 10.24 | First Amendment to Office Lease between DexCom, Inc. and Kilroy Realty, L.P., dated August 18, 2010. | 10-Q | 000-51222 | November 4, 2010 | 10.27 | |||||||||||
| 21.01 | List of Subsidiaries. | X | ||||||||||||||
| 23.01 | Consent of Independent Registered Public Accounting Firm. | X | ||||||||||||||
| 24.01 | Power of Attorney. (See page 68 of this Form 10-K). | X | ||||||||||||||
| 31.01 | Certification of Chief Executive Officer Pursuant to Securities Exchange Act Rule 13a-14(a). | X | ||||||||||||||
| 31.02 | Certification of Chief Financial Officer Pursuant to Securities Exchange Act Rule 13a-14(a). | X | ||||||||||||||
66
Table of Contents
| Exhibit |
Exhibit Description |
Incorporated by Reference | Exhibit Number |
Provided Herewith |
||||||||||||
| Form | File No. |
Date of First Filing |
||||||||||||||
| 32.01 | Certification of Chief Executive Officer Pursuant to 18 U.S.C. Section 1350 and Securities Exchange Act Rule 13a-14(b).*** | X | ||||||||||||||
| 32.02 | Certification of Chief Financial Officer Pursuant to 18 U.S.C. Section 1350 and Securities Exchange Act Rule 13a-14(b).*** | X | ||||||||||||||
| * | Represents a management contract or compensatory plan. |
| ** | Confidential treatment has been requested for certain portions of this document pursuant to an application for confidential treatment sent to the Securities and Exchange Commission. Such portions are omitted from this filing and were filed separately with the Securities and Exchange Commission. |
| *** | This certification is not deemed “filed” for purposes of Section 18 of the Securities Exchange Act, or otherwise subject to the liability of that section. Such certification will not be deemed to be incorporated by reference into any filing under the Securities Act of 1933 or the Securities Exchange Act of 1934, except to the extent that DexCom specifically incorporates it by reference. |
67
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| DEXCOM, INC. | ||||||
| (Registrant) | ||||||
| Dated: March 3, 2011 |
By: | /s/ JESS ROPER | ||||
| Jess Roper, Chief Financial Officer | ||||||
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Terrance Gregg and Jess Roper, jointly and severally, his attorneys-in-fact, each with the power of substitution, for him in any and all capacities, to sign any amendments to this Report on Form 10-K and to file same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and dates indicated.
| Signature |
Title |
Date | ||
| /s/ TERRANCE GREGG Terrance Gregg |
President, Chief Executive Officer and Director |
March 3, 2011 | ||
| /s/ JESS ROPER Jess Roper |
Chief Financial Officer |
March 3, 2011 | ||
| /s/ JONATHAN LORD Jonathan Lord, M.D. |
Chairman of the Board of Directors |
March 3, 2011 | ||
| /s/ NICHOLAS AUGUSTINOS Nicholas Augustinos |
Director |
March 3, 2011 | ||
| /s/ KEVIN SAYER Kevin Sayer |
Director |
March 3, 2011 | ||
| /s/ JAY SKYLER Jay Skyler, M.D. |
Director |
March 3, 2011 | ||
| /s/ ERIC TOPOL Eric Topol, M.D. |
Director |
March 3, 2011 | ||
68