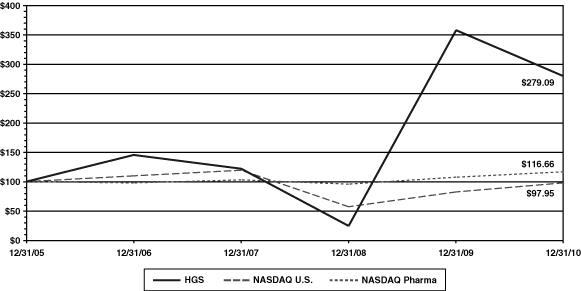
UNITED
STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C.
20549
Form 10-K
ANNUAL REPORT PURSUANT TO
SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31,
2010
Commission File Number
001-14169
HUMAN GENOME SCIENCES,
INC.
(Exact name of
registrant)
| |
|
|
Delaware
(State of
organization)
|
|
22-3178468
(I.R.S. employer
identification number)
|
14200 Shady Grove Road, Rockville, Maryland
20850-7464
(address of principal
executive offices and zip code)
(301) 309-8504
(Registrant’s telephone number)
Securities registered pursuant to Section 12(b) of the
Act:
| |
|
|
|
Title of Each Class
|
|
Name of Each Exchange on Which Registered
|
|
|
|
Common stock, par value $0.01 per share
|
|
The NASDAQ Stock Market LLC
|
Securities registered pursuant to Section 12(g) of the
Act:
None
Indicate by check mark if the registrant is a well-known
seasoned issuer, as defined in Rule 405 of the Securities
Act. Yes þ No
o
Indicate by check mark if the registrant is not required to file
reports pursuant to Section 13 or 15(d) of the Exchange
Act. Yes o No
þ
Indicate by check mark whether the registrant (1) has filed
all reports required to be filed by Section 13 or 15(d) of
the Securities Exchange Act of 1934 during the preceding
12 months (or for such shorter period that the registrant
was required to file such reports) and (2) has been subject
to such filing requirements for the past
90 days. Yes þ No o
Indicate by check mark whether the registrant has submitted
electronically and posted on its corporate Web site, if any,
every Interactive Data File required to be submitted and posted
pursuant to Rule 405 of
Regulation S-T
during the preceding 12 months (or for such shorter period
that the registrant was required to submit and post such
files). Yes o No o
Indicate by check mark if disclosure of delinquent filers
pursuant to Item 405 of
Regulation S-K
is not contained herein and will not be contained, to the best
of the registrant’s knowledge, in definitive proxy or
information statements incorporated by reference in
Part III of this
Form 10-K
or any amendment to this
Form 10-K. o
Indicate by check mark whether the registrant is a large
accelerated filer, an accelerated filer, a non-accelerated
filer, or a smaller reporting company. See the definitions of
“large accelerated filer,” “accelerated
filer” and “smaller reporting company” in
Rule 12b-2
of the Exchange Act. (Check one):
| |
|
|
|
|
|
|
|
Large accelerated
filer þ
|
|
Accelerated
filer o
|
|
Non-accelerated
filer o
(Do not check if a smaller reporting company)
|
|
Smaller reporting
company o
|
Indicate by check mark whether the registrant is a shell company
(as defined in
Rule 12b-2
of the Exchange
Act). Yes o No þ
The number of shares of the registrant’s common stock
outstanding on January 31, 2011 was 189,007,876. As of
June 30, 2010, the aggregate market value of the common
stock held by non-affiliates of the registrant based on the
closing price reported on the National Association of Securities
Dealers Automated Quotations System was approximately
$3,386,315,952.*
DOCUMENTS INCORPORATED BY REFERENCE
Portions of Human Genome Sciences, Inc.’s definitive proxy
statement for the 2011 annual meeting of stockholders are
incorporated by reference into Part III of this Annual
Report.
|
|
| * |
Excludes 38,706,406 shares of common stock deemed to be
held by officers and directors and stockholders whose ownership
exceeds five percent of the shares outstanding at June 30,
2010. Exclusion of shares held by any person should not be
construed to indicate that such person possesses the power,
direct or indirect, to direct or cause the direction of the
management or policies of the registrant, or that such person is
controlled by or under common control with the registrant.
|
TABLE OF CONTENTS
PART I
This annual report on
Form 10-K
contains forward-looking statements, within the meaning of the
Securities Exchange Act of 1934 and the Securities Act of 1933,
that involve risks and uncertainties. In some cases,
forward-looking statements are identified by words such as
“believe,” “anticipate,” “expect,”
“intend,” “plan,” “will,”
“may” and similar expressions. You should not place
undue reliance on these forward-looking statements, which speak
only as of the date of this report. All of these forward-looking
statements are based on information available to us at this
time, and we assume no obligation to update any of these
statements. Actual results could differ from those projected in
these forward-looking statements as a result of many factors,
including those identified in “Risk Factors,”
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and elsewhere. We urge
you to review and consider the various disclosures made by us in
this report, and those detailed from time to time in our filings
with the Securities and Exchange Commission, that attempt to
advise you of the risks and factors that may affect our future
results.
Overview
Human Genome Sciences, Inc. (“HGS”, the
“Company”, “we”, “our” and
“us”) is a biopharmaceutical company that exists to
place new therapies into the hands of those battling serious
disease. Our lead products in development are
BENLYSTA®
(belimumab) for systemic lupus erythematosus (“SLE”)
and raxibacumab for inhalation anthrax.
BENLYSTA continues to progress toward commercialization. On
November 16, 2010, the U.S. Food and Drug
Administration (“FDA”) Arthritis Advisory Committee
voted 13-2
to recommend that the FDA approve BENLYSTA for the treatment of
autoantibody-positive patients with active SLE who are receiving
standard therapy. The Advisory Committee provides non-binding
recommendations for consideration by the FDA, which then makes
the final decision on approval. In December 2010, the FDA
extended the Prescription Drug User Fee Act (“PDUFA”)
target date for its priority review of BENLYSTA from
December 9, 2010 to March 10, 2011.
In June 2010, GlaxoSmithKline (“GSK”), our partner in
the co-development and commercialization of BENLYSTA, submitted
a Marketing Authorization Application (“MAA”) to the
European Medicines Agency (“EMA”) for approval to
market BENLYSTA in Europe. We expect to have the EMA decision in
the second half of 2011. GSK has also submitted regulatory
applications in Canada, Australia, Switzerland, Russia, Brazil
and The Philippines. Regulatory submissions in other
countries are planned in 2011.
In preparation for the anticipated launch and commercialization
of BENLYSTA, we have hired and trained a specialized commercial
team including sales, marketing and medical affairs. We are also
building our own HGS commercial team to work alongside GSK in
Europe. We believe we have produced sufficient BENLYSTA
inventory to meet the anticipated global market need for at
least one year, and are increasing our capacity to meet
long-term demand.
We continue to deliver raxibacumab to the U.S. Strategic
National Stockpile (“SNS”) for emergency use in
treating inhalation anthrax. In 2009, we completed the delivery
of 20,001 doses of raxibacumab to the SNS under an initial order
and as a result recognized $162.5 million in product sales
and manufacturing and development services revenue. In July
2009, the U.S. Government (“USG”) exercised its
option to purchase 45,000 additional doses of raxibacumab, with
delivery to be completed over a three-year period. HGS expects
to receive approximately $142.0 million from this second
order as deliveries are completed, $64.9 million of which
has been recognized as revenue through December 31, 2010.
In May 2009, we submitted a Biologics License Application
(“BLA”) to the FDA for raxibacumab for the treatment
of inhalation anthrax. We received a Complete Response Letter in
November 2009, and we will continue to work closely with the FDA
to obtain approval. HGS will receive approximately
$20.0 million from the USG upon FDA licensure of
raxibacumab.
In addition to these products in our internal pipeline, we have
substantial financial rights to two novel drugs that GSK has
advanced to late-stage development. The first of these is
darapladib, which was discovered by GSK using HGS technology. In
two pivotal Phase 3 trials, GSK is currently evaluating whether
darapladib can reduce the
1
risk of adverse cardiovascular events such as heart attack or
stroke in patients with chronic coronary heart disease and acute
coronary syndrome, respectively. With a planned enrollment of
more than 27,000 patients in the two trials, the Phase 3
clinical program for darapladib is among the largest ever
conducted to evaluate the safety and efficacy of any
cardiovascular medication. The second is albiglutide. GSK has
eight Phase 3 trials in progress to evaluate the long-term
efficacy, safety and tolerability of albiglutide as monotherapy
and add-on therapy for patients with type 2 diabetes mellitus.
Albiglutide was created by HGS using its proprietary
albumin-fusion technology, and the product was licensed to GSK
in 2004.
We are also working to expand and advance our mid- and
early-stage pipeline beyond potential additional indications for
BENLYSTA, with a primary focus on immunology and oncology. We
continue to develop our oncology portfolio around our expertise
in the apoptosis, or programmed cell death, pathway and we plan
to initiate Phase 2 development of our human monoclonal antibody
to the CCR5 receptor for the treatment of ulcerative colitis.
Strategic partnerships are an important driver of our commercial
success. We have a co-development and commercialization
agreement with GSK for BENLYSTA and raxibacumab is being
developed under a contract with the Biomedical Advanced Research
and Development Authority (“BARDA”) of the Office of
the Assistant Secretary for Preparedness and Response
(“ASPR”), U.S. Department of Health and Human
Services (“HHS”). Our strategic partnerships with
leading pharmaceutical and biotechnology companies allow us to
leverage our strengths and gain access to sales and marketing
infrastructure, as well as complementary technologies. Some of
these partnerships provide us with licensing or other fees,
clinical development cost-sharing, milestone payments and rights
to royalty payments as products are developed and
commercialized. In some cases, we are entitled to certain
commercialization, co-promotion, revenue-sharing and other
product rights.
As of December 31, 2010, we had $933.4 million in cash
and investments. With a strong cash position, a management team
experienced in bringing products to market, an experienced drug
development organization and significant capabilities in
biologicals manufacturing, we believe HGS has the resources and
capabilities necessary to achieve near-term commercial success
while sustaining a viable pipeline that supports the long-term
growth of the Company.
We are a Delaware corporation established in 1992, headquartered
at 14200 Shady Grove Road, Rockville, Maryland,
20850-7464.
Our telephone number is
(301) 309-8504.
Our website address is www.hgsi.com. Information contained on
our website is not a part of, and is not incorporated into, this
annual report on
Form 10-K.
Our filings with the SEC are available without charge on our
website as soon as reasonably practicable after filing. HGS,
Human Genome Sciences, BENLYSTA, and ZALBIN are trademarks of
Human Genome Sciences, Inc. Other trademarks referenced are the
property of their respective owners.
Strategy
Over the last few years, we have made strategic decisions that
have transformed HGS on multiple levels and contributed to our
progress toward becoming a fully commercial biopharmaceutical
company capable of generating sustainable revenues and
sustainable growth into the future. Key strategies to help us
achieve this goal include:
Execute
the successful launch and commercialization of BENLYSTA assuming
regulatory approval.
Over the last year, we have taken steps to prepare for the
approval and successful launch of BENLYSTA. We have hired and
trained a specialized commercial team, including sales,
marketing and medical affairs, that understands how to
communicate effectively with rheumatologists and other
stakeholders. The combined HGS and GSK team, including a
U.S. sales force of approximately 150, will work closely
together following approval to communicate effectively and
appropriately with rheumatologists and other stakeholders and
work to ensure that appropriate patients with systemic lupus who
need BENLYSTA will have access to it. HGS and GSK also plan to
support physicians and patients with reimbursement and access
programs.
Under the terms of the 2006 co-development and
co-commercialization agreement with GSK, and assuming regulatory
approval, HGS will have responsibility for the global supply of
BENLYSTA. We believe we have produced sufficient BENLYSTA
inventory to meet anticipated global market needs for at least
one year, and we
2
believe our large-scale manufacturing facility has sufficient
capacity to provide worldwide supply for the first two to three
years following launch. However, HGS and GSK also anticipate
that additional capacity will be required. In July 2010, HGS
announced an agreement with Lonza that will provide for the
future manufacture of BENLYSTA, eventually tripling capacity
with production starting in 2012.
Make
BENLYSTA globally available so patients with need will have
access.
HGS and GSK expect a decision on the European approval of
BENLYSTA in the second half of 2011. GSK has also submitted
regulatory applications in Canada, Australia, Switzerland,
Russia, Brazil and The Philippines. Regulatory submissions
in other countries are planned in 2011. HGS is building its own
commercialization team to work alongside GSK in Europe, with HGS
headquarters in Switzerland and local organizations in Germany,
France and Spain. Elsewhere, GSK will lead local implementation,
with HGS sharing costs and profits equally with GSK.
Work to
achieve the full therapeutic and commercial potential of
BENLYSTA by developing it for other
B-cell
mediated diseases.
Where patients with B-cell mediated autoimmune or other diseases
need new treatment options and we have scientific evidence
supporting the therapeutic potential and commercial viability,
we are committed to continuing the development of BENLYSTA. In
2011, we are considering initiating Phase 2 trials of BENLYSTA
in vasculitis and post-renal transplant. We also plan to
initiate a Phase 3 trial of BENLYSTA in a subcutaneous
formulation that may improve convenience for some patients. In
addition, a number of exploratory studies are currently planned
in Sjögren’s syndrome, Waldenstrom’s disease,
pre-transplant, idiopathic thrombocytopenic purpura (ITP) and
others.
Find and
develop new therapies for our late-stage
pipeline.
We will pursue external opportunities to acquire high-potential
innovative products, with a priority given to products that have
the potential to be
first-in-class
or best-in class, and address large markets or markets on the
verge of escalating growth. We will maintain our primary
therapeutic focus on oncology and immunology —
indications in which we have special expertise and which can be
commercialized with small specialty sales forces. We will be
open to both biologics and small molecule opportunities. We also
look forward to GSK’s continuing progress with late-stage
products to which we have substantial financial rights, such as
darapladib for cardiovascular disease.
Continue
to build a robust mid-and early-stage pipeline.
Strength in our pipeline is important to our long-term future
growth. In 2011, we plan to initiate Phase 2 trials of BENLYSTA
in additional indications, and our CCR5 monoclonal antibody in
ulcerative colitis. In addition, we plan to begin randomization
for our Phase 2 trial of mapatumumab in hepatocellular cancer.
All of these products are investigational and were developed
based on HGS discoveries. We also plan to initiate Phase 1b
combination trials of our in-licensed small-molecule oncology
drug, HGS1029. We are expanding our new targets program to
increase the number of new Investigational New Drugs per year,
and have multiple research collaborations ongoing to complement
our internal efforts.
Remain
strategically opportunistic and vigilant.
Strategically appropriate partnerships and collaborations will
continue to play an important role in our future. We will
continue our partnership with GSK as we explore the full
potential of BENLYSTA. As we bring more products forward in the
future that address large and international markets, other
partnerships with leading companies may allow us to bring new
therapies more quickly to patients who need them on a global
basis. We will also pursue strategic acquisitions and
collaborations to expand our portfolio of new drug candidates,
provide access to complementary technologies, or reduce the cost
and speed the early development of HGS-discovered targets.
Capitalize
on our intellectual property portfolio.
We pursue patents to protect our intellectual property and have
developed a significant intellectual property portfolio, with
hundreds of issued patents covering genes, proteins, antibodies
and proprietary technologies.
3
We have also filed U.S. patent applications covering many
additional discoveries and inventions. We will seek
opportunities to monetize intellectual property assets that we
do not plan to develop internally.
Build
upon our core competencies in manufacturing and process
development.
Our world-class manufacturing and process development
capabilities represent a strategic advantage and allow us to
control quality while maintaining flexibility.
Maintain
a strong financial position.
A strong cash position is essential to our ability to
commercialize new approved drugs, advance promising late-stage
investigational therapies through large and expensive Phase 3
trials, and continue to invest in our earlier-stage pipeline. We
view a strong financial position as a strategic imperative.
Maintaining it requires sound financial controls and the ability
to raise funds on the public or private markets. It also
requires a willingness to advance creative approaches such as
our manufacturing alliance program, through which we have
capitalized on our process development and manufacturing
capability to generate revenues — or raxibacumab,
which is sold to one customer, the U.S. Government, and
generates approximately $50.0 million a year of what
amounts to non-dilutive financing.
Products
HGS has two late stage products: BENLYSTA for systemic lupus and
raxibacumab for inhalation anthrax. We also have financial
rights to certain products in the GSK clinical pipeline. GSK has
advanced two of these products to Phase 3 clinical trials:
darapladib for cardiovascular disease and albiglutide for type
2 diabetes mellitus. In addition, we have a portfolio of
novel drugs in earlier stages of development, led by our TRAIL
receptor antibody mapatumumab, which is in mid-stage development
for cancer.
Clinical
Programs
Late-Stage
Products
BENLYSTA for systemic lupus has the potential to receive
regulatory approval in the United States in March 2011 and in
Europe in the second half of 2011. No new drug for lupus has
been approved by regulatory authorities in more than
50 years. Raxibacumab for inhalation anthrax continues to
generate revenue under our contract with the
U.S. Government as we complete deliveries to the Strategic
National Stockpile, and we continue to work closely with the FDA
to achieve licensure.
BENLYSTA
(belimumab)
BENLYSTA is a human monoclonal antibody and the first in a new
class of drugs called BLYS-specific inhibitors. BENLYSTA
inhibits the biological activity of a naturally occurring
protein known as B-lymphocyte stimulator (BLyS), which was first
discovered by HGS in 1996. In lupus and certain other autoimmune
diseases, elevated levels of BLyS are believed to contribute to
the production of autoantibodies — antibodies that
attack and destroy the body’s own healthy tissues. BENLYSTA
is being developed by HGS and GSK as a potential treatment for
systemic lupus under a co-development and commercialization
agreement entered into in 2006 (described below under “Lead
Commercial Collaborations”).
On November 16, 2010, the FDA Arthritis Advisory Committee
voted 13-2
to recommend that the FDA approve BENLYSTA for the treatment of
autoantibody-positive patients with active SLE who are receiving
standard therapy. In December 2010, the FDA extended the PDUFA
target date for the priority review of BENLYSTA to
March 10, 2011. In June 2010, GSK submitted an application
to EMA seeking approval to market BENLYSTA in Europe. We expect
to have the EMA decision in the second half of 2011. GSK has
also submitted regulatory applications in Canada, Australia,
Switzerland, Russia, Brazil and The Philippines. Regulatory
submissions in other countries are planned in 2011.
We and GSK are examining a range of potential additional
indications for BENLYSTA that might be explored in new clinical
trials. In 2011, we are considering initiating Phase 2 trials of
BENLYSTA in vasculitis and post-renal transplant. We also plan
to initiate a Phase 3 trial of BENLYSTA in a subcutaneous
formulation that may improve
4
convenience for some patients. In addition, a number of
exploratory studies are currently planned in Sjögren’s
syndrome, Waldenstrom’s disease, pre-transplant, idiopathic
thrombocytopenic purpura (ITP) and others.
Raxibacumab
Raxibacumab is a human monoclonal antibody that targets and
blocks Bacillus anthracis protective antigen, which
research has shown to be the key facilitator of the deadly
toxicity of anthrax infection. Raxibacumab represents a new way
to address the anthrax threat. While antibiotics can kill the
anthrax bacteria, they are not effective against the deadly
toxins the bacteria produce. Raxibacumab targets anthrax toxins
after they are released by the bacteria into the blood and
tissues. In an inhalation anthrax attack, people may not know
they are infected with anthrax until the toxins already are
circulating in their blood, and it may be too late for
antibiotics alone to be effective.
We are developing raxibacumab under a contract entered into in
2006 with BARDA. The U.S. Government is currently our only
customer for raxibacumab and has the right to terminate our
contract for convenience at any time. In 2010, HGS continued
delivery of raxibacumab to the U.S. Strategic National
Stockpile. In July 2009, the U.S. Government exercised its
option to purchase 45,000 additional doses of raxibacumab for
the Stockpile for emergency use in treating inhalation anthrax,
with delivery to be completed over a three-year period. HGS
expects to receive approximately $142.0 million from this
second order as deliveries are completed. In 2010, we recognized
$47.2 million in raxibacumab product sales revenue. Also
under our contract, HGS submitted a BLA to the FDA for
raxibacumab for the treatment of inhalation anthrax in May 2009.
We received a Complete Response Letter in November 2009, and we
will continue to work closely with the FDA to obtain approval.
HGS will receive approximately $20.0 million from the
U.S. Government upon FDA licensure of raxibacumab.
Raxibacumab revenue accounted for 31% and 65% of our total
revenue for 2010 and 2009, respectively.
ZALBINtm
(albinterferon alfa-2b)
In April 2010, we announced that Novartis withdrew its MAA
seeking authorization to market ZALBIN in Europe for the
treatment of chronic hepatitis C. The decision was based on
feedback from European regulatory authorities including
questions regarding whether the therapeutic benefit of
JOULFERON®
(known in the U.S. as ZALBIN) was sufficient relative to
risk. In October 2010, as expected, HGS received a Complete
Response Letter from the FDA regarding the Company’s BLA
for 900-mcg ZALBIN. HGS and Novartis have decided not to develop
ZALBIN further.
Mid- and
Early-Stage Pipeline
We are investing strategically to expand and advance our mid-
and early-stage pipeline beyond potential additional indications
for BENLYSTA, with a primary focus on immunology and oncology.
Mapatumumab
HGS has pioneered the development of highly targeted agonistic
antibody therapies for cancer based on the TRAIL receptor
apoptotic pathway. Mapatumumab is a human monoclonal antibody
that specifically binds to TRAIL receptor 1 and causes it to
induce apoptosis in cancer cells. The safety lead-in to a
randomized Phase 2 trial of mapatumumab in combination with
Nexavar (sorafenib) is ongoing in patients with advanced
hepatocellular cancer, which accounts for 80%-90% of all liver
cancers. We expect to initiate the randomization stage of this
study in 2011.
The results of two Phase 2 trials of mapatumumab in combination
with other therapeutic agents were announced in 2010. In March
2010, we announced the results of a randomized Phase 2 trial of
mapatumumab in combination with paclitaxel and carboplatin as
first-line therapy in advanced non-small cell lung cancer. In
June 2010, we announced the results of a randomized Phase 2
trial of mapatumumab in combination with bortezomib (Velcade) in
patients with advanced multiple myeloma. In both studies, the
results showed no difference in disease response or
progression-free survival for patients treated with the
combination that included mapatumumab vs. the control group.
Mapatumumab was well tolerated in both studies.
5
IAP
Inhibitors
The IAP inhibitors are a novel class of compounds that block the
activity of IAP (inhibitor of apoptosis) proteins, allowing
apoptosis to proceed and causing the cancer cells to die. When
IAP proteins are over-expressed in cancer cells, they can help
cancer cells resist apoptosis and resume growth and
proliferation. Preclinical studies of HGS1029 in combination
with mapatumumab demonstrated dramatic synergistic activity
against a number of cancer types, including prostate, breast,
esophageal, colorectal and non-small cell lung. HGS1029 has also
shown significant anti-tumor activity alone and in combination
with other agents in a broad range of cancers. We plan to
develop mapatumumab and our IAP inhibitors in combination with
one another and in combination with other therapeutic agents.
In November 2009, HGS announced the initiation of a Phase 1
study of our lead IAP inhibitor, HGS1029, as monotherapy in
patients with advanced lymphoid tumors. The study’s primary
objectives include evaluation of safety and tolerability, and
dose selection for Phase 2 trials. HGS1029 as monotherapy is
also being studied in an ongoing Phase 1 study in patients with
advanced solid tumors. In 2011, we plan to initiate Phase 1b
studies of HGS1029 in combination with mapatumumab and other
agents; we also plan to complete the Phase 1 studies of HGS1029
as monotherapy.
CCR5
mAb
We plan to initiate a Phase 2 trial of our human monoclonal
antibody to the CCR5 receptor for the treatment of ulcerative
colitis in 2011. This receptor has been shown to play a key role
in the immune system and the inflammation cascade associated
with autoimmune and other diseases.
Products
in the GSK Pipeline
HGS has financial rights to certain products in the GSK clinical
development pipeline (described below under “Lead
Commercial Collaborations”). GSK has advanced two of these
products, darapladib and albiglutide, to Phase 3
development.
Darapladib
Darapladib was discovered by GSK based on our technology. It is
a small-molecule inhibitor of lipoprotein-associated
phospholipase-A2
(Lp-PLA2),
an enzyme associated with the formation of atherosclerotic
plaques and identified in clinical trials as an independent risk
factor for coronary heart disease and ischemic stroke. In two
pivotal Phase 3 trials, GSK is evaluating whether darapladib can
reduce the risk of adverse cardiovascular events, such as heart
attack or stroke, in patients with chronic coronary heart
disease and acute coronary syndrome, respectively. With a
planned enrollment of more than 27,000 patients in the two
trials, the Phase 3 clinical program for darapladib is among the
largest ever conducted to evaluate the safety and efficacy of
any cardiovascular medication. Darapladib was discovered by GSK
based on HGS technology. HGS will receive 10% royalties on
worldwide sales if darapladib is commercialized, and has a 20%
co-promotion option in North America and Europe.
Albiglutide
(formerly
Syncria®)
Albiglutide is a biological product generated from the fusion of
human albumin and modified human GLP-1 peptide, and is designed
to act throughout the body to help maintain normal blood-sugar
levels and to control appetite. GSK currently has eight Phase 3
trials in progress to evaluate the long-term efficacy, safety
and tolerability of albiglutide as monotherapy and add-on
therapy for patients with type 2 diabetes mellitus. Albiglutide
was created by HGS using its proprietary albumin-fusion
technology, and the product was licensed to GSK in 2004. We are
entitled to fees and milestone payments that could amount to as
much as $183.0 million — including
$33.0 million received to date — in addition to
single-digit royalties on worldwide sales if albiglutide is
commercialized.
Research
and Development
HGS has developed core competencies in the discovery and
understanding of human genes and their biological functions, and
in the discovery and development of human protein and antibody
drugs. Research and development
6
expenses were $196.4 million, $173.7 million and
$243.3 million for 2010, 2009, and 2008, respectively.
These expenses are net of amounts reimbursed by our
collaboration partners.
Human
Antibody Technology
We have acquired rights to a variety of human antibody
technologies, have integrated these technologies into our
research and development program, and continue to collaborate
with certain antibody companies. Many medical conditions are the
result of an excess of a specific protein in the body, and some
antibody drugs can inactivate such proteins and bring
therapeutic benefits to patients. These drugs are known as
antagonistic antibodies. For example, BENLYSTA is an
antagonistic human monoclonal antibody.
In certain medical conditions, it may be desirable to stimulate
a specific biological activity. Antibodies that stimulate
biological activity are known as agonistic antibodies.
Mapatumumab is an agonistic antibody that binds to TRAIL
receptor 1 and triggers programmed cell death in cancer cells.
We believe it was the first human agonistic monoclonal antibody
to enter clinical trials.
Albumin-Fusion
Technology
Our albumin-fusion technology allows us to create long-acting
forms of protein drugs by fusing the gene that expresses human
albumin to the gene that expresses a therapeutically active
protein. Our partners are actively pursuing the development of
albumin-fusion drugs based on known therapeutic proteins. For
example, albiglutide was created through the fusion of human
albumin and glucagon-like peptide-1 (GLP-1).
Drug
Development
We have built a drug development organization that has the
expertise necessary to design and implement well-focused,
high-quality clinical trials of multiple compounds. We seek to
gather, document and analyze clinical trial data in such a way
that they can be submitted to regulatory authorities and used to
support BLAs at the appropriate time. We have assembled
experienced teams in key strategic areas of development,
including:
|
|
|
| |
•
|
Clinical Research and Biostatistics. The
clinical research and biostatistics groups are responsible for
the design, planning and analysis of clinical trials.
|
| |
| |
•
|
Clinical Operations. The clinical operations
group executes clinical trials and is responsible for managing
clinical trial sites and ensuring that all proper procedures are
followed during the collection of clinical data. The group
includes our data management team.
|
| |
| |
•
|
Project Management. Our project management
team oversees the process of development of a drug from the
earliest stages of research through the conduct of clinical
development and regulatory filings.
|
| |
| |
•
|
Regulatory Affairs. The regulatory affairs
group manages communications with and submissions to regulatory
authorities.
|
| |
| |
•
|
Drug Safety. As our products advance in
clinical testing, our drug safety group collects and analyzes
information on drug experience and safety, and ensures that
accurate medical information is distributed.
|
| |
| |
•
|
Quality Assurance. The quality assurance group
ensures compliance with all regulatory requirements for the
clinical development and manufacture of our products.
|
| |
| |
•
|
Bioanalytical Sciences. The bioanalytical
sciences group develops and performs highly specialized assays
that are used during monitoring of preclinical tests and
clinical trials. Other assays help to ensure the quality and
consistency of our products.
|
| |
| |
•
|
Biopharmaceutical Development. The
biopharmaceutical development group develops robust
manufacturing processes and product formulations to support
clinical studies and future commercial supply.
|
7
Strategic
Collaborations
Strategic collaborations are a key aspect of our business
strategy. We have co-development and commercialization
agreements with GSK for BENLYSTA, and raxibacumab is being
developed under a contract with the U.S. Government.
Strategic collaborations are an important source of revenues and
drug development cost-sharing. They also allow us to leverage
our strengths and gain access to sales and marketing
infrastructure, international distribution and complementary
technologies.
Other potential collaborations may provide sources of new
product opportunities for in-licensing. In addition, we have
assets that may be a better fit for another company than for
HGS, and therefore could be out-licensed. Each of these
collaborative models is of interest to HGS, and we are committed
to remaining alert to new opportunities.
Lead
Commercial Collaborations
GlaxoSmithKline
BENLYSTA. In 2006, we entered into an
agreement with GSK for the co-development and commercialization
of BENLYSTA. GSK is a world leader that brings global
pharmaceutical development and marketing capabilities to the
BENLYSTA program. Under the BENLYSTA agreement, we and GSK share
Phase 3 and 4 development costs, sales and marketing
expenses, and profits equally. HGS has primary responsibility
for bulk manufacturing. We have received an execution fee of
$24.0 million under this agreement and have recognized this
payment ratably over the development period. We recognized
revenues of $3.4 million in 2010 and $4.7 million in
2009. The BENLYSTA agreement includes cost-sharing provisions
under which we and GSK share clinical development costs. We
recorded cost reimbursement from GSK under this provision of
$62.0 million in 2010 and $43.1 million in 2009, which
was reflected as a reduction in expenses. This agreement will
expire three years after the later of (i) the expiration
date of certain patent rights related to BENLYSTA and
(ii) a period of 10 years after the first commercial
sale of BENLYSTA. These certain patent rights are expected to
expire by 2023, with the potential for later expiration that may
result from any issuance of additional patent
and/or
patent term extensions. GSK may terminate the agreement if
(i) upon the basis of competent scientific evidence or data
regarding commercial potential, GSK determines BENLYSTA does not
merit incurring additional development or marketing expenses or
(ii) BENLYSTA is not approved by the FDA or EMA. In
addition, either party may terminate if the other party commits
a material breach of the agreement or if the other party is
bankrupt or insolvent.
Darapladib. In December 2008, GSK
initiated Phase 3 development of darapladib, a small-molecule
Lp-PLA2
inhibitor discovered by GSK based on HGS technology. We will
receive a 10% royalty on worldwide sales of darapladib if it is
commercialized, and we have a 20% co-promotion option in North
America and Europe. We are also entitled to receive a milestone
payment if darapladib moves through clinical development into
registration.
Albiglutide. In February 2009, GSK
initiated a Phase 3 clinical trial program to evaluate the
efficacy, safety and tolerability of albiglutide in the
long-term treatment of type 2 diabetes mellitus. Albiglutide was
created by HGS using its proprietary albumin-fusion technology,
and the product was licensed to GSK in 2004. HGS is entitled to
fees and milestone payments that could amount to as much as
$183.0 million — including $33.0 million
received to date. We are also entitled to single-digit royalties
on worldwide sales if albiglutide is commercialized.
United
States Government
Raxibacumab. In September 2005, we
entered into a two-phase contract with BARDA to supply
raxibacumab for inhalation anthrax. HHS is the lead agency for
public health and medical response to man-made or natural
disasters, including acts of bioterrorism. Under the first phase
of the contract, we supplied 10 grams of raxibacumab to HHS for
comparative in vitro and in vivo testing. In
June 2006, under the second phase of the contract, the USG
exercised its option to purchase raxibacumab and we agreed to
manufacture and deliver 20,001 doses to the SNS. In 2009, we
achieved our company’s first product sales by completing
these deliveries and recognized $162.5 million in product
sales and manufacturing and development services revenue. In
July 2009, the USG exercised its option to purchase 45,000
additional doses of raxibacumab for the SNS for emergency use in
treating inhalation anthrax, with delivery to be completed over
a three-year period. HGS expects to receive
8
approximately $142.0 million from this second order as
deliveries are completed, including approximately
$47.2 million and $17.7 million recognized as product
sales revenue in 2010 and 2009, respectively. Also under our
contract, HGS submitted a BLA to the FDA for raxibacumab for the
treatment of inhalation anthrax in May 2009. We received a
Complete Response Letter in November 2009, and we will continue
to work closely with the FDA to obtain approval. HGS will
receive approximately $20.0 million from the USG upon FDA
licensure of raxibacumab. Our raxibacumab agreement can be
terminated by the USG if it determines that a termination is in
its interest.
Novartis
ZALBIN. In 2006, we entered into an
exclusive worldwide agreement for the co-development and
commercialization of albinterferon alfa-2b with Novartis, a
global leader in the pharmaceutical industry. In October 2010,
as expected, HGS received a Complete Response Letter from the
FDA regarding the Company’s BLA for 900-mcg ZALBIN
(albinterferon alfa-2b, known in Europe as JOULFERON) dosed
every two weeks for the treatment of chronic hepatitis C.
HGS and Novartis decided not to develop ZALBIN further. Based on
this decision, in September 2010, HGS recognized all remaining
deferred revenue relating to payments previously received under
the ZALBIN agreement with Novartis, an amount totaling
$34.1 million. There will, however, be certain additional
costs incurred by both parties in the near future as activities
are concluded.
Product
Collaborations and Agreements
Aegera Therapeutics. In December 2007,
we and Aegera Therapeutics, Inc. (“Aegera”) completed
a licensing and collaboration agreement providing us with
exclusive worldwide rights (excluding Japan) to develop and
commercialize HGS1029 and other small-molecule inhibitors of IAP
(inhibitor of apoptosis) proteins in oncology. Under the
agreement, we made an upfront payment to Aegera of
$20.0 million as a licensing fee and for an equity
investment. Aegera is entitled to receive up to
$295.0 million in additional development and commercial
milestone payments, including a $5.0 million milestone paid
in 2008 upon FDA clearance of an Investigational New Drug
Application (“IND”). Aegera will receive low
double-digit royalties on net sales in the HGS territory. In
North America, Aegera will have the option to co-promote,
under which it will share certain expenses and profits (30%) in
lieu of its royalties. Aegera retains the non-oncology rights to
its IAP inhibitors that are not selected for development under
this agreement.
Research
and Technology Collaborations
HGS has a rich heritage of scientific discovery that has
produced a substantial intellectual property estate and a
library of thousands of therapeutic and diagnostic targets.
After careful review, we have selected targets for further
research and potential development, with the goal of filing new
INDs to develop the selected targets through co-development or
research collaborations, as well as through our own internal
research, including the application of antibody development
technology from various collaborators.
Process
Development and Manufacturing Alliances
Protein and antibody process development and manufacturing are
core HGS competencies. We currently develop and produce several
protein and antibody drugs in three
state-of-the-art
current good manufacturing practices
(“cGMP”)-compliant process development and
manufacturing facilities — totaling approximately
520,000 square feet. We are leveraging these capabilities
to produce near-term revenue by entering into strategically
appropriate process development and manufacturing alliances.
Patents
and Proprietary Rights
We seek U.S. and foreign patent protection for the genes,
proteins and antibodies that we discover, as well as patents on
therapeutic and diagnostic products and processes, screening and
manufacturing technologies, and other inventions based on genes,
proteins and antibodies. We also seek patent protection or rely
upon trade secret rights to protect certain technologies which
may be used to discover and characterize genes, proteins and
antibodies and which may be used to develop novel therapeutic
and diagnostic products and processes. We believe that in the
9
aggregate, our patent applications, patents and licenses under
patents owned by third parties are of material importance to our
operations.
Important legal issues remain to be resolved as to the extent
and scope of available patent protection for biotechnology
products and processes in the U.S. and other important
markets outside the U.S. We expect that litigation or
administrative proceedings will likely be necessary to determine
the validity and scope of certain of our and others’
proprietary rights. We are currently involved in a number of
administrative proceedings and litigations relating to the scope
of protection of our patents and those of others, and are likely
to be involved in additional proceedings that may affect
directly or indirectly patents and patent applications related
to our products or the products of our partners. For example, we
have been involved in contested proceedings related to products
based on BLyS (such as BENLYSTA). Any such lawsuit or proceeding
may result in a significant commitment of resources in the
future. In addition, changes in, or different interpretations
of, patent laws in the U.S. and other countries may result
in patent laws that allow others to use our discoveries or
develop and commercialize our products. We cannot assure you
that the patents we obtain or the unpatented technology we hold
will afford us significant commercial protection.
We have filed U.S. patent applications with respect to many
human genes and their corresponding proteins. We also have filed
U.S. patent applications with respect to all or portions of
the genomes of several infectious and non-infectious
microorganisms. We have hundreds of U.S. patents covering
genes, proteins, antibodies and proprietary technologies. Our
remaining applications may not result in the issuance of any
patents. Our applications may not be sufficient to meet the
statutory requirements for patentability in all cases. In
certain instances, we will be dependent upon our collaborators
to file and prosecute patent applications.
Other companies or institutions have filed, and may in the
future file, patent applications that attempt to patent genes
similar to those covered in our patent applications, including
applications based on our potential products. Any patent
application filed by a third party may prevail over our patent
applications, in which event the third party may require us to
stop pursuing a potential product or to negotiate a royalty
arrangement to pursue the potential product.
We also are aware that others, including universities and
companies working in the biotechnology and pharmaceutical
fields, have filed patent applications and have been granted
patents in the U.S. and in other countries that cover
subject matter potentially useful or necessary to our business.
Some of these patents and patent applications claim only
specific products or methods of making products, while others
claim more general processes or techniques useful in the
discovery and manufacture of a variety of products. The risk of
additional patents and patent applications will continue to
increase as the biotechnology industry progresses. We cannot
predict the ultimate scope and validity of existing patents and
patents that have been or may be granted to third parties, nor
can we predict the extent to which we may wish or be required to
obtain licenses to such patents, or the availability and cost of
acquiring such licenses. To the extent licenses are required,
the owners of the patents could bring legal actions against us
to claim damages or to stop our manufacturing and marketing of
the affected products.
Issued patents may not provide commercially meaningful
protection against competitors and may not provide us with
competitive advantages. Other parties may challenge our patents
or design around our issued patents or develop products
providing effects similar to our products. Furthermore, patents
are issued for a limited time period and may expire before the
useful life of the covered product. In addition, others may
discover uses for genes, proteins or antibodies other than those
uses covered in our patents, and these other uses may be
separately patentable. The holder of a patent covering the use
of a gene, protein or antibody for which we have a patent claim
could exclude us from selling a product for a use covered by its
patent.
We rely on trade secret protection to protect our confidential
and proprietary information. We believe we have developed
proprietary procedures for making libraries of DNA sequences and
genes. We have not sought patent protection for these
procedures. We have developed a substantial database concerning
genes we have identified. We have taken security measures to
protect our data and continue to explore ways to further enhance
the security for our data. However, we may not be able to
meaningfully protect our trade secrets. While we have entered
into confidentiality agreements with employees and
collaborators, we may not be able to prevent their disclosure of
these data or materials. Others may independently develop
substantially equivalent information and techniques.
10
Competition
General. We face intense competition from a
wide range of pharmaceutical, biotechnology and diagnostic
companies, as well as academic and research institutions and
government agencies. Some of these competitors have
substantially greater financial, marketing, research and
development and human resources. Most large pharmaceutical
companies have considerably more experience in undertaking
clinical trials and in obtaining regulatory approval to market
pharmaceutical products.
Basis of Competition. Principal competitive
factors in our industry include:
|
|
|
| |
•
|
the quality and breadth of an organization’s technology;
|
| |
| |
•
|
the skill of an organization’s employees and ability to
recruit and retain skilled employees;
|
| |
| |
•
|
an organization’s intellectual property estate;
|
| |
| |
•
|
the range of capabilities, from target identification and
validation to drug discovery and development to manufacturing
and marketing; and
|
| |
| |
•
|
the availability of substantial capital resources to fund
discovery, development, manufacturing and commercialization
activities.
|
We believe that the quality and breadth of our technology
platform, the skill of our employees and our ability to recruit
and retain skilled employees, our patent portfolio, our
capabilities for research and drug development, and our capital
resources are competitive strengths. However, many large
pharmaceutical and biotechnology companies have significantly
larger intellectual property estates than we do, more
substantial capital resources than we have, and greater
capabilities and experience than we do in preclinical and
clinical development, sales, marketing, manufacturing and
regulatory affairs.
Products. We are aware of products in research
or development by our competitors that address all of the
diseases we are targeting. Any of these products may compete
with our product candidates. Our competitors may succeed in
developing their products before we do, obtaining approvals from
the FDA or other regulatory agencies for their products more
rapidly than we do, or developing products that are more
effective than our products. These products or technologies
might render our technology obsolete or noncompetitive. In
addition, our albumin fusion protein products are designed to be
long-acting versions of existing products. While we believe our
albumin fusion protein products may be a more attractive
alternative to the existing products, the existing product in
some cases has an established market that may make the
introduction of our product more difficult. Competition is based
primarily on product efficacy, safety, timing and scope of
regulatory approvals, availability of supply, marketing and
sales capability, reimbursement coverage, price and patent
position.
Government
Regulation
Regulations in the U.S. and other countries have a
significant impact on our research, product development and
manufacturing activities and will be a significant factor in the
marketing of our products. All of our products require
regulatory approval prior to commercialization. In particular,
our products are subject to rigorous preclinical and clinical
testing and other premarket approval requirements by the FDA and
similar regulatory authorities in other regions. Various
statutes and regulations also govern or influence the
manufacturing, safety, labeling, storage, record keeping and
marketing of our products. The lengthy process of seeking these
approvals, and the subsequent compliance with applicable
statutes and regulations, require the expenditure of substantial
resources. Any failure by us to obtain, or any delay in
obtaining, regulatory approvals could materially adversely
affect our ability to commercialize our products in a timely
manner, or at all.
Preclinical Testing. Before a drug may be
clinically tested in the U.S., it must be the subject of
rigorous preclinical testing. Preclinical tests include
laboratory evaluation of product chemistry and animal studies to
assess the potential safety and efficacy of the product and its
formulations. The results of these studies must be submitted to
the FDA as part of an IND, which is reviewed by the FDA before
clinical testing in humans can begin.
11
Clinical Testing. Typically, clinical testing
involves a three-phase process, which generally lasts four to
seven years, and sometimes longer:
|
|
|
| |
•
|
Phase 1 clinical trials are conducted with a small number of
subjects to determine the early safety profile and the pattern
of drug distribution and metabolism.
|
| |
| |
•
|
Phase 2 clinical trials are conducted with groups of patients
afflicted with a specified disease in order to provide enough
data to evaluate preliminary efficacy and optimal dosages
statistically and to expand evidence of safety.
|
| |
| |
•
|
Phase 3 clinical trials are large-scale, multi-center,
comparative trials, which are designed to gather additional
information for proper dosage and labeling of the drug and to
demonstrate its overall safety and efficacy.
|
The FDA monitors the progress of each phase of testing, and may
require the modification, suspension or termination of a trial
if it is determined to present excessive risks to patients. The
clinical trial process may be accompanied by substantial delay
and expense and there can be no assurance that the data
generated in these studies will ultimately be sufficient for
marketing approval by the FDA.
Marketing Approvals. Before a product can be
marketed and sold, the results of the preclinical and clinical
testing must be submitted to the FDA for approval. This
submission will be either a new drug application or a biologics
license application, depending on the type of drug. In
responding to a new drug application or a biologics license
application, the FDA may grant marketing approval, request
additional information or deny the application if it determines
that the application does not provide an adequate basis for
approval. We cannot assure you that any approval required by the
FDA will be obtained on a timely basis, or at all.
In addition, the FDA may condition marketing approval on the
conduct of specific post-marketing studies to further evaluate
safety and efficacy (such as Phase 4 trials). Rigorous and
extensive FDA regulation of pharmaceutical products continues
after approval, particularly with respect to compliance with
cGMPs, reporting of adverse effects, advertising, promotion and
marketing. Discovery of previously unknown problems or failure
to comply with the applicable regulatory requirements may result
in restrictions on the marketing of a product or withdrawal of
the product from the market as well as possible civil or
criminal sanctions, any of which could materially adversely
affect our business.
Other Regulation. We are also subject to
various laws and regulations relating to safe working
conditions, laboratory and manufacturing practices, the
experimental use of animals and the use and disposal of
hazardous or potentially hazardous substances used in connection
with our research, including radioactive compounds and
infectious disease agents. We also cannot accurately predict the
extent of regulations that might result from any future
legislative or administrative action.
In addition, ethical, social and legal concerns about genetic
testing and genetic research could result in additional
regulations restricting or prohibiting the processes we or our
suppliers may use. Federal and state agencies, congressional
committees and foreign governments have expressed interest in
further regulating biotechnology. More restrictive regulations
or claims that our products are unsafe or pose a hazard could
prevent us from commercializing our products.
Foreign Regulation. We must obtain regulatory
approval by governmental agencies in other countries prior to
commercialization of our products in those countries. Foreign
regulatory systems may be just as rigorous, costly and uncertain
as in the U.S.
Possible Pricing Restrictions. The levels of
revenues and profitability of biopharmaceutical companies like
ours may be affected by the continuing efforts of government and
third-party payers to contain or reduce the costs of health care
through various means. For example, in certain foreign markets,
pricing or profitability of therapeutic and other pharmaceutical
products is subject to governmental control. In the
U.S. there have been, and we expect that there will
continue to be, a number of federal and state proposals to
implement similar governmental control. While we cannot predict
whether any legislative or regulatory proposals will be adopted,
the adoption of such proposals could have a material adverse
effect on our business, financial condition and profitability.
In addition, in the U.S. and elsewhere, sales of
therapeutic and other pharmaceutical products depend in part on
the availability of
12
reimbursement to the consumer from third-party payers, such as
government and private insurance plans. Third-party payers are
increasingly challenging the prices charged for medical products
and services. We cannot assure you that any of our products will
be considered cost effective or that reimbursement to the
consumer will be available or will be sufficient to allow us to
sell our products on a competitive and profitable basis.
Sources
of Supply
Most raw materials and other supplies required in our business
are generally available from various suppliers in quantities
adequate to meet our needs. Certain materials and other supplies
required for manufacturing are currently available only from
single sources. As we prepare for commercialization of our
products, we intend to identify alternative sources of supply
wherever possible.
Manufacturing
We are able to manufacture multiple protein and antibody drugs
for use in research, clinical and commercial activities. We
produce and purify these protein and antibody drugs in three
process development and manufacturing facilities that total
approximately 520,000 square feet and offer both
small-scale and large-scale manufacturing capabilities. We do
not currently manufacture any products approved for commercial
use. We are, however, manufacturing raxibacumab for supply to
the Strategic National Stockpile. We believe that we have
sufficient manufacturing capacity to launch BENLYSTA, if it is
approved by the FDA. We believe our HGS large-scale
manufacturing facility has ample capacity to provide worldwide
supply of BENLYSTA, if approved, for the first two or three
years following launch. We currently have at least a year’s
worth of BENLYSTA inventory on hand. In July 2010, HGS entered
into an agreement with Lonza, a leader in biologics
manufacturing, for the future commercial supply of BENLYSTA.
This agreement will eventually triple our manufacturing capacity
for BENLYSTA with production expected to come on-line for
U.S. production by mid-2012.
Currently each of our lead products, BENLYSTA and raxibacumab,
is produced at a single manufacturing site. BENLYSTA is produced
at our large-scale manufacturing facility in Rockville,
Maryland, and raxibacumab is produced in a separate part of our
small-scale manufacturing facility, also in Rockville, Maryland.
Each of these facilities is the sole source for these products.
We cannot guarantee that one or more of these manufacturing
plants will not encounter problems, including but not limited to
loss of power, equipment failure or viral or microbial
contamination, which could impact our ability to deliver
adequate supply of one or more of these products to the market.
We cannot assure you that we will be able in the future to
consistently manufacture our products economically or in
compliance with cGMPs and other regulatory requirements.
In the future, we may contract with additional third-party
manufacturers or develop products with partners and use the
partners’ manufacturing capabilities. When we use others to
manufacture our products, we depend on those parties to comply
with cGMPs and other regulatory requirements, and to deliver
materials on a timely basis. These parties may not perform
adequately. Any failures by these third parties may delay our
development of products or the submission of these products for
regulatory approval.
Marketing
In preparation for the launch and commercialization of BENLYSTA
assuming regulatory approval, HGS has hired and trained a
specialized commercial team including sales, marketing and
medical affairs. The combined HGS and GSK team, including a
U.S. sales force of approximately 150, will work closely
together following approval to communicate effectively and
appropriately with rheumatologists and other stakeholders and
work to ensure that appropriate patients with systemic lupus who
need BENLYSTA will have access to it. HGS and GSK also plan to
support patients with reimbursement and access programs.
HGS and GSK expect a decision on the European approval of
BENLYSTA in the second half of 2011. GSK has also submitted
regulatory applications in Canada, Australia, Switzerland,
Russia, Brazil and The Philippines. Regulatory submissions
in other countries are planned in 2011. HGS is building its own
commercialization team to work alongside GSK in Europe, with HGS
headquarters in Switzerland and local organizations in Germany,
France and Spain. Elsewhere, GSK will lead local implementation,
with HGS sharing costs and profits equally with GSK.
13
GSK and others also have co-marketing rights with respect to
certain of our other products. For any products that we market
together with partners, we will rely, in whole or in part, on
the marketing capabilities of those parties. We may also
contract with third parties to market certain of our products.
Ultimately, we and our partners may not be successful in
marketing our products. The prices for our products may be
impacted by various factors, including economic analyses of the
burden of the applicable disease, the perceived value of the
product and third-party reimbursement policies.
Employees
As of February 1, 2011, we had approximately
1,100 full-time employees. None of our U.S. employees
is subject to collective bargaining agreements. Our employees
located in certain
non-U.S. countries
are covered by collective bargaining agreements that are
prescribed by local labor law. Additionally, substantially all
of our employees in Europe are covered by national labor laws
which establish the rights of employees, including the amount of
wages and benefits paid and, in certain cases, severance and
similar benefits. We consider relations with our employees to be
good.
14
There are a number of risk factors that could cause our
actual results to differ materially from those that are
indicated by forward-looking statements. Those factors include,
without limitation, those listed below in addition to the other
information in this Annual Report on
Form 10-K.
You should carefully consider these risk factors in evaluating
our business because these risk factors may have a significant
impact on our business, financial condition and results of
operations. The risks described below are not the only risks we
may face. Additional risks and uncertainties not presently
apparent to us, or risks that we currently consider immaterial,
could also negatively affect our business, financial condition
and results of operations.
COMMERCIAL
RISKS
Our near-term prospects are highly dependent on BENLYSTA, our
lead product candidate. If we fail to obtain FDA
and/or EMA
approval for BENLYSTA or fail to successfully commercialize
BENLYSTA, our results of operations and business will be
materially and adversely affected and our stock price would
likely decline.
Our most advanced product candidate is BENLYSTA. The development
program for BENLYSTA involved two large-scale, multi-center
Phase 3 clinical trials, in addition to earlier Phase 1 and
Phase 2 clinical trials. In July 2009, we reported the results
from the first of our two Phase 3 clinical trials for BENLYSTA.
This trial was called BLISS-52. In BLISS-52, BENLYSTA met its
primary efficacy endpoint. In November 2009, we reported the
52 week data from the second Phase 3 clinical trial for
BENLYSTA. This trial was called BLISS-76. In BLISS-76, BENLYSTA
at a dose of 10 mg/kg also met its primary efficacy
endpoint. Although the primary efficacy endpoint of the BLISS-76
study was assessed after 52 weeks, we continued to collect
additional data from this trial for an additional 24 weeks.
In April 2010, we reported the results of the BLISS-76 trial for
these additional weeks. At week 76 in the BLISS-76 trial,
BENLYSTA plus standard of care showed higher response rates
compared with placebo plus standard of care, but this difference
was not statistically significant. We do not know what, if any,
effect these 76-week results may have on our ability to obtain
regulatory approval for BENLYSTA.
In June 2010, we filed a BLA for BENLYSTA in the United States
and our collaboration partner GSK submitted an MAA for BENLYSTA
with the EMA. The Arthritis Advisory Committee of the
U.S. FDA met on November 16, 2010 to consider the
BENLYSTA BLA and voted
13-2 to
recommend FDA approval. On December 3, 2010, the FDA
extended the PDUFA target date for its priority review of the
BLA from December 9, 2010 to March 10, 2011 and
requested some additional information. We currently expect a
decision by the EMA in the second half of 2011. Despite our
determination that the results from the two BENLYSTA trials were
positive and the affirmative vote for approval by the FDA
Advisory Committee, FDA and EMA approval of BENLYSTA depends,
among other things, on regulatory authorities finding the data
from our clinical trials, our manufacturing protocol and
controls and the composition of the product sufficient to
support approval. We cannot offer any assurances or predict with
any certainty that the FDA or EMA will grant marketing approval
for BENLYSTA, or on the expected timeframes. Furthermore,
regulatory approvals, even if obtained, may be limited to
specific indications, limit the type of patients in which the
drug may be used, or otherwise require specific warning or
labeling language, any of which might reduce the commercial
potential of BENLYSTA. Regulatory authorities are likely to
condition BENLYSTA marketing approval on the conduct of specific
post-marketing studies to further evaluate safety and efficacy,
in either particular patient populations or general patient
populations or both. The results of these studies, the discovery
of previously unknown issues involving safety or efficacy or the
failure to comply with post-approval regulatory requirements,
including requirements with respect to manufacturing practices,
reporting of adverse effects, advertising, promotion and
marketing, may result in restrictions on the marketing of
BENLYSTA or the withdrawal of BENLYSTA from the market.
Even if we receive FDA or EMA approval, we might not be
successful in commercializing BENLYSTA. Should we fail to obtain
FDA and/or
EMA approval for BENLYSTA or fail to successfully commercialize
BENLYSTA upon approval, our business, financial condition and
results of operations would be materially adversely affected and
the price of our common stock would likely decline.
15
If we are unable to commercialize BENLYSTA or other product
candidates we develop, we may not be able to recover our
investment in our research, product development, manufacturing
and marketing efforts.
We have made and are continuing to make substantial expenditures
in advance of commercializing BENLYSTA and our other product
candidates. We have invested significant time and resources to
isolate and study genes and determine their functions. We devote
substantial resources to developing proteins, antibodies and
small molecules for the treatment of human disease. Before we
can commercialize a product, we must rigorously test the product
in the laboratory and complete extensive animal and human
clinical studies. We are also devoting substantial resources to
enhancing our manufacturing capabilities and to building our
commercial supply inventories of BENLYSTA to support potential
commercialization. We have and expect to continue to devote
substantial resources to establish and maintain a marketing
capability for BENLYSTA and any of our other products that are
approved by the FDA or other regulatory authorities. These costs
have increased as we near the potential launch of BENLYSTA. If
BENLYSTA or other products we develop are not approved for
commercial sale, we may be unable to recover the large
investment we have made in research, development, manufacturing
and marketing efforts, and our business and financial condition
could be materially adversely affected.
We may be unable to successfully establish commercial
manufacturing capability and may be unable to obtain required
quantities of our product candidates for commercial use.
We have not yet manufactured any products that have been
approved for commercial use. Except for raxibacumab and
quantities of BENLYSTA manufactured to build our commercial
supply inventory of BENLYSTA in support of commercialization, we
have limited experience in manufacturing materials suitable for
commercial use. The FDA must determine that our facilities
comply with cGMPrequirements for commercial production and
license them accordingly. We may not successfully establish
sufficient manufacturing capabilities or manufacture our
products economically or in compliance with cGMPs and other
regulatory requirements. For example, we believe that we have
sufficient manufacturing capacity to launch BENLYSTA if it is
approved by the FDA
and/or EMA
and to supply commercial quantities of BENLYSTA for the first
two or three years following launch. In June 2010, we entered
into a manufacturing agreement with Lonza Sales AG
(“Lonza”) pursuant to which Lonza will manufacture
additional commercial quantities of BENLYSTA. However, this
additional manufacturing capacity may not be available for
18 months or longer, if ever, due, in part, to the time
required to obtain regulatory approvals for the manufacture of
BENLYSTA in Lonza’s facility. If Lonza’s facility
fails to obtain regulatory approval in a timely manner or at
all, we may not be able to build or procure additional capacity
in the required timeframe to meet commercial demand, and our
revenues may accordingly be limited from BENLYSTA. Our revenues
from BENLYSTA also will be limited if the demand for BENLYSTA
exceeds our capacity to supply BENLYSTA to patients.
BENLYSTA is solely produced at our large-scale manufacturing
facility in Rockville, Maryland and raxibacumab is solely
produced at our small-scale manufacturing facility, which is
also in Rockville, Maryland. Furthermore, the filling, finishing
and packaging for both BENLYSTA and raxibacumab are solely
performed by a single third party manufacturer. We cannot
guarantee that one or more of these plants will not encounter
problems, including but not limited to loss of power, equipment
failure or viral or microbial contamination, which could
adversely affect our ability to deliver adequate supply of one
or more of these products to patients or customers.
If we or our third-party manufacturers fail to comply in a
significant way with regulatory requirements imposed on our
manufacturing activities, it could have a material adverse
effect on our business, financial condition and results of
operations.
We have previously engaged third-party manufacturers or utilized
our collaboration partners’ manufacturing capabilities to
manufacture product candidates and we expect to continue to do
so in the future. If we use others to manufacture our products,
we will depend on those parties to comply with cGMPs and other
regulatory requirements and to deliver materials on a timely
basis. In addition, because regulatory approval to manufacture a
drug is generally site-specific, the FDA and other regulatory
authorities will repeatedly inspect our manufacturing facilities
and our current and future third-party manufacturers’
facilities for compliance with cGMPs. If we or our third-party
manufacturers fail to comply with applicable regulatory
requirements, a regulatory agency may: issue warning letters;
suspend or withdraw our regulatory approval for approved or
in-market products; seize or detain products or recommend a
product recall; refuse to approve pending applications or
supplements to approved applications filed
16
by us; suspend any of our ongoing clinical trials; impose
restrictions or obligations on our operations, including costly
new manufacturing requirements; close our facilities or those of
our contract manufacturers; revoke previously granted drug
approvals under certain circumstances; or impose civil or
criminal penalties. Any of these actions could delay our
development of products, the submission of these products for
regulatory approval or result in insufficient product quantity
to support commercial demand. As a result, our business,
financial condition and results of operations could be seriously
harmed.
Because we currently have only a limited marketing capability
and in light of various factors, we may be unable to price or
sell any of our products effectively.
We can provide no assurance as to the price at which we may be
able to sell any of our products, or that we will be able to
price any of our products at a level that is consistent with
other similar products. The prices for our products may be
affected by various factors that could adversely affect our
sales and profit margins, including economic analyses of the
burden of the applicable disease, the perceived value of the
product and third party reimbursement policies.
We currently do not have any marketed products, although we have
sold raxibacumab to the U.S. Government. If we receive
approval for products that can be marketed, we intend to market
the products either independently or together with collaborators
or strategic partners. GSK and others have co-commercialization
rights with respect to certain of our products. If we decide to
market any products, either independently or together with
partners, we will incur significant additional expenditures and
commit significant additional management resources to establish
sales forces. For any products that we market together with
partners, we will rely, in whole or in part, on the marketing
capabilities of those parties. We may also contract with third
parties to market certain of our products. In building a sales
force in anticipation of the approval and commercial launch of
BENLYSTA, we may be unable to retain an adequate number of
qualified sales representatives and may encounter difficulties
in retaining third parties to provide sales, marketing or
distribution resources. Ultimately, we and our partners may not
be successful in marketing our products.
If any product candidate for which we receive regulatory
approval does not achieve broad market acceptance (including as
a result of failing to differentiate our products from
competitor products or as a result of failing to obtain
reimbursement rates for our products that are competitive from
the healthcare provider’s perspective), the revenues we
generate from sales will be limited and our business may not be
profitable.
Our success will depend in substantial part on the extent to
which our products for which we obtain marketing approval from
the FDA and comparable foreign regulatory authorities are
accepted by the medical community and reimbursed by third-party
payors, including government payors. The degree of market
acceptance will depend upon a number of factors, including,
among other things:
|
|
|
| |
•
|
our product’s perceived advantages over existing treatment
methods (including relative convenience and ease of
administration and prevalence and severity of any adverse
events, including any unexpected adverse events of which we
become aware after marketing approval);
|
| |
| |
•
|
claims, limitations, warnings or other information in our
product’s approved labeling;
|
| |
| |
•
|
reimbursement and coverage policies of government and other
third-party payors;
|
| |
| |
•
|
pricing and cost-effectiveness;
|
| |
| |
•
|
in the United States, the ability of group purchasing
organizations, or GPOs (including distributors and other network
providers), to sell our products to their constituencies;
|
| |
| |
•
|
the establishment and demonstration in the medical community of
the safety and efficacy of our products and our ability to
provide acceptable evidence of safety and efficacy;
|
| |
| |
•
|
availability of alternative treatments; and
|
| |
| |
•
|
the prevalence of off-label substitution of biologically
equivalent products.
|
17
We cannot predict whether physicians, patients, healthcare
insurers or maintenance organizations, or the medical community
in general, will accept or utilize any of our products. Our
efforts to educate the medical community and third-party payors
regarding the benefits of our products may require significant
resources and may not be successful in achieving our objectives.
If our products are approved but do not achieve an adequate
level of acceptance by these parties, we may not generate
sufficient revenues from these products to become or remain
profitable.
Changes in the health care system or reimbursement policies
may result in a decline in our potential sales and a reduction
in our expected revenue from our potential products.
The levels of revenues and profitability of biopharmaceutical
companies like ours may be affected by the continuing efforts of
government and third-party payers to contain or reduce the costs
of health care through various means. For example, in certain
foreign markets pricing or profitability of therapeutic and
other pharmaceutical products is subject to governmental
control. In the United States, there have been, and we expect
that there will continue to be, a number of federal and state
proposals to implement similar governmental control. Recently
enacted United States legislation, including implementing rules
and regulations, instituted significant changes to the United
States healthcare system that could have a material adverse
effect on our business, financial condition and profitability.
We cannot predict what effects, if any, this legislation might
have on our company and our products as this legislation is
implemented over the next few years, nor can we predict whether
additional legislative or regulatory proposals may be adopted in
the future.
In addition, in the United States and elsewhere, sales of
therapeutic and other pharmaceutical products depend in part on
the availability of reimbursement to the consumer from
third-party payers, such as government and private insurance
plans. Third-party payers are increasingly challenging the
prices charged for medical products and services. Any
reimbursement granted may not be maintained or limits on
reimbursement available from third parties may reduce the demand
for or negatively affect the price and profitability of those
products. This would likely have a material adverse effect on
our business, financial condition and results of operations. Our
ability to successfully commercialize our products and product
candidates and the demand for our products depend, in part, on
the extent to which reimbursement is available from such
third-party payers.
Uncertainty exists about the reimbursement status of newly
approved biopharmaceutical products. Healthcare providers and
third-party payers use coding systems to identify diagnoses,
procedures, services, and treatments. Proper coding is an
integral component to receiving appropriate reimbursement for
the administration of BENLYSTA and related services. Most payors
recognize the Healthcare Common Procedure Coding System
(“HCPCS”) Level II national codes to identify and
report drugs and the American Medical Association Current
Procedural Terminology, or CPT, codes to report professional
services (including drug administration). As a new drug,
BENLYSTA has not been assigned a permanent, unique HCPCS code
and there is a lack of clarity as to the appropriate CPT Code
that should be used for its administration. We have applied for
a permanent HCPCS code and intend to obtain guidance from The
Centers for Medicare & Medicaid Services
(“CMS”) regarding CPT Code usage. Until a permanent
HCPCS code is assigned for BENLYSTA, physicians may bill using
an unclassified (miscellaneous) code. Use of miscellaneous codes
typically causes claims processing delays and may lead to lower
payments to physicians or other providers and may cause
physicians or other providers to delay use of BENLYSTA until a
permanent code has been assigned.
Under Medicare Part B, reimbursement for BENLYSTA will
currently be computed based on manufacturer’s Average Sales
Price (“ASP”). The Patient Protection and Affordable
Care Act (“PPACA”) made changes to the statutory
definition of ASP and CMS is in the process of interpreting and
implementing those changes, which could result in lower payments
for physician-administered drugs. If government and other
third-party payers do not provide adequate coverage and
reimbursement levels for BENLYSTA, its market acceptance may be
materially adversely affected.
18
Because raxibacumab is a product whose current sole purchaser
is the U.S. Government, the sale of raxibacumab faces risks
in addition to the risks generally associated with the sale of
biopharmaceutical products, including political considerations,
government contracting requirements and government spending
policies.
Raxibacumab, a human monoclonal antibody developed for use in
the treatment of anthrax disease, presents risks in addition to
those associated with our other products. Numerous other
companies and governmental agencies are known to be developing
biodefense pharmaceuticals and related products to combat
anthrax disease. These competitors may have financial or other
resources greater than ours, they may have easier or preferred
access to the likely distribution channels for biodefense
products or they may develop products judged to have greater
efficacy for biodefense. In addition, since the primary
purchaser of biodefense products is the U.S. Government and
its agencies, the success of raxibacumab will depend on
government spending priorities, policies and pricing
restrictions. In the case of the U.S. Government, executive
or legislative action could attempt to impose production and
pricing requirements on us. In the event of extreme urgency, the
government might seek to compel us or we might ourselves choose
to reallocate our production in ways that may not be
economically beneficial to the company.
We have entered into a two-phase contract to supply raxibacumab
to the U.S. Government, which may be terminated by the
U.S. Government at any time. Under the first phase of the
contract, we supplied ten grams of raxibacumab to the HHS for
comparative in vitro and in vivo testing. Under the second
phase of the contract, the U.S. Government ordered 20,001
doses of raxibacumab for the U.S. Strategic National
Stockpile (“SNS”) for use in the treatment of anthrax
disease. We completed delivery of these doses and the
U.S. Government accepted our deliveries. In July 2009, the
U.S. Government agreed to purchase 45,000 additional doses.
As of December 31, 2010, we have delivered approximately
20,000 doses of this second order. We, therefore, have future
deliveries to make and ongoing obligations under the contract,
including the obligation to seek FDA approval.
In November 2009, we received a Complete Response Letter from
the FDA related to our BLA for raxibacumab. In this letter, the
FDA determined that it would not approve our BLA for raxibacumab
in its present form and requested additional studies and data.
Although the government has accepted shipment of raxibacumab
subsequent to the receipt of the FDA’s Complete Response
Letter, we cannot assure you that the government will continue
to accept future shipments or place additional orders.
We will continue to face risks related to the requirements of
the contract. If we are unable to meet our obligations
associated with this contract, the U.S. Government will not
be required to make future payments related to that order.
Although we have received U.S. Government approval for two
orders of raxibacumab, we cannot assure you we will receive
additional orders.
COMPETITIVE
RISKS
Our competitors may develop and market products that are, or
are perceived as, less expensive, more effective, safer, easier
to administer or reach the market sooner. This may diminish or
eliminate the commercial success of any products we may
commercialize.
The development and commercialization of biopharmaceutical
products is highly competitive and subject to rapid
technological advances. We will face competition with respect to
all products we may develop or commercialize in the future from
pharmaceutical and biotechnology companies worldwide. We also
expect to face increasing competition from governments,
universities and other non-profit research organizations. These
institutions carry out a significant amount of research and
development in the field of biologic technologies, and they are
increasingly aware of the commercial value of their findings. As
a result, they are demanding greater patent and other
proprietary rights, as well as licensing and future royalty
revenues.
Many of our competitors have significantly greater financial,
research and development, intellectual property estates,
regulatory, manufacturing, marketing, sales and other resources
than we do. As a result, our competitors may succeed in
developing their products before we do and obtaining approvals
from the FDA or other regulatory agencies for their products
more rapidly than we do. They may be able to devote greater
resources to the development, manufacture, marketing and sale of
their products, initiate or withstand substantial price
competition
19
or otherwise more successfully market their products. These
competing products or technologies might render our technology
or product candidates under development noncompetitive,
uneconomical or obsolete.
Key factors affecting the success of any approved product
include its efficacy, safety profile, drug interactions, method
and frequency of administration, pricing, reimbursement and
level of promotional activity relative to those of competing
products. Competing products may provide greater therapeutic
convenience or clinical or other benefits for a specific
indication than our products, or may offer comparable
performance at a lower cost. The introduction of more
efficacious, safer, cheaper, or more convenient alternatives to
our products could reduce our revenues and the value of our
product development efforts.
We are aware of existing products and products in research or
development by others that address the diseases we are
targeting. Any of these products may compete with our product
candidates. For example, a number of pharmaceutical and
biotechnology companies are currently developing products
targeting the same types of indications that we are targeting
with BENLYSTA, and some of these competitors’ products have
entered clinical trials.
If we successfully develop products but those products do not
achieve and maintain market acceptance, our business will not be
successful. Any reduction in demand for our products as a result
of a competing product could lead to reduced revenues, reduced
margins, reduced levels of profitability, and loss of market
share for our products. These competitive pressures could
adversely affect our business and operating results.
If our products are approved and marketed, we may also face
risks to our profitability and financial condition from generic
drug or biosimilar manufacturers.
The United States has recently enacted legislation establishing
a regulatory pathway for follow-on biologics, also known as
biosimilars. This and similar regulatory and legislative
activity in other countries may make it easier for generic drug
manufacturers to manufacture and sell biological drugs similar
or identical to BENLYSTA and raxibacumab which might affect the
profitability or commercial viability of our products. An
accelerated route to market for generic versions of small
molecule drugs was established in the United States with the
passage of the Hatch-Waxman Amendments in 1984, which also
provides five years of exclusivity for small molecule drugs with
additional exclusivity under certain circumstances. The passage
of the Biologics Price Competition and Innovation Act
(“BPCIA”) in March 2010 established a similar pathway
for FDA approval of a follow-on biologic that provides twelve
years of exclusivity for the original biologic and an additional
six month exclusivity period if certain pediatric studies are
conducted. The European Medicines Agency has issued guidelines
for approving products through an abbreviated pathway under
which more than ten biosimilars have been approved. European
legislation provides ten years of exclusivity for original
drugs, including biologics, and an additional one year of
exclusivity for obtaining marketing approval for certain
additional indications. If a generic or biosimilar version of
one of our products were approved, it could have a material
adverse effect on the sales and gross profits of the product and
adversely affect our business and operating results.
PRODUCT
DEVELOPMENT RISKS
Our product development efforts depend on new technologies,
which may not prove successful.
Our development of new products depends on the use of
cutting-edge, recently discovered technologies that may never
have been successfully utilized in existing commercial products.
As a result, our product development efforts involve risks of
failure inherent in the use of innovative and unproven
technologies and the risks associated with drug development
generally. These risks include the possibility that:
|
|
|
| |
•
|
these technologies or any or all of the molecules based on these
technologies may be ineffective or toxic and, therefore fail to
receive or retain necessary regulatory clearances;
|
| |
| |
•
|
the products, even if safe and effective, may be difficult to
manufacture on a large scale or uneconomical to market;
|
| |
| |
•
|
proprietary rights of third parties may prevent us or our
collaborators from exploiting technologies or marketing
products; and
|
| |
| |
•
|
third parties may market superior or equivalent products.
|
20
We have limited experience in developing and commercializing
products, and we may be unsuccessful in our efforts to do so.
Although we are conducting human studies with respect to a
number of products, we may not be successful in developing or
commercializing these or other products. Our ability to develop
and commercialize products based on proteins, antibodies and
small molecules will depend on our abilities to:
|
|
|
| |
•
|
successfully complete laboratory testing and human studies;
|
| |
| |
•
|
obtain and maintain necessary intellectual property rights to
our products;
|
| |
| |
•
|
obtain and maintain necessary regulatory approvals related to
the efficacy and safety of our products;
|
| |
| |
•
|
maintain production facilities meeting all regulatory
requirements or enter into arrangements with third parties to
manufacture our products on our behalf; and
|
| |
| |
•
|
deploy sales and marketing resources appropriately, efficiently
and effectively or enter into arrangements with third parties to
provide these functions.
|
We are currently a late-stage development company, and we
cannot assure you that we will be able to develop products that
will become commercially successful or that we will achieve
profitability from such products.
We will continue to incur substantial expenditures relating to
research, development, clinical studies and manufacturing
efforts. Depending on the stage of development, our products may
require significant further research, development, testing and
regulatory approvals. Even if regulatory approval is obtained
for the commercial sale of a product, it could take considerable
time following approval, if ever, before we are likely to
receive continuing revenue from product sales or substantial
royalty payments. Until such time, we expect to continue to
incur losses and we cannot assure you that we will ever become
profitable on a sustainable basis.
We are continually evaluating our business strategy and may
modify this strategy in light of developments in our business
and other factors.
Our ability to discover and develop new products will depend on
our internal research capabilities and our ability to acquire
products. Although we continue to conduct research and
development activities on products and have increased our
activities in this area, our limited resources may not be
sufficient to discover and develop new drug candidates.
We continue to evaluate our business strategy and, as a result,
may modify our strategy in the future. In this regard, we may,
from time to time, focus our product development efforts on
different products or may delay or halt the development of
various products, as we did with ZALBIN in 2010. In addition, as
a result of changes in our strategy, we may also change or
refocus our existing drug discovery, development,
commercialization and manufacturing activities. This could
require changes in our facilities and personnel and the
restructuring of various financial arrangements. We cannot
assure you that any product development changes that we
implement will be successful.
Clinical trials for our products are expensive and protracted
and their outcome is uncertain, and we must invest substantial
amounts of time and money that may not yield viable products.
Conducting clinical trials is a lengthy, time-consuming and
expensive process. Before obtaining regulatory approvals for the
commercial sale of any product we must demonstrate through
laboratory, animal and human studies that the product is both
effective and safe for use in humans. We will incur substantial
expense and devote a significant amount of time to conducting
ongoing trials and initiating new trials.
Before clinical testing in humans can begin, a drug must be
subject to rigorous preclinical testing and documentation. This
documentation must be reviewed by the FDA as part of an
Investigational New Drug Application (“IND”). Data
obtained from tests are susceptible to varying interpretations
which may delay, limit or prevent regulatory approval.
Furthermore, regulatory authorities may refuse or delay approval
as a result of many factors, including changes in regulatory
policy during the period of product development. Even if an IND
is
21
approved, preclinical studies do not predict clinical success.
Many potential drugs have shown promising results in early
testing but subsequently failed to obtain necessary regulatory
approvals.
Completion of clinical trials may take many years. The time
required varies substantially according to the type, complexity,
novelty and intended use of the product candidate. The progress
of clinical trials is monitored by both the FDA and independent
data monitoring committees which may require the modification,
suspension or termination of a trial if it is determined to
present excessive risks to patients. Our rate of commencement
and completion of clinical trials may be delayed by many
factors, including:
|
|
|
| |
•
|
our inability to manufacture sufficient quantities of materials
for use in clinical trials;
|
| |
| |
•
|
unavailability or variability in the number and types of
patients for each study;
|
| |
| |
•
|
difficulty in maintaining contact with patients after treatment,
resulting in incomplete data;
|
| |
| |
•
|
safety issues or side effects;
|
| |
| |
•
|
ineffectiveness of products during the clinical trials; or
|
| |
| |
•
|
government or regulatory delays.
|
Data obtained from our clinical trials may not be sufficient
to support an application for regulatory approval without
further studies.
Studies conducted by us or by third parties on our behalf may
not demonstrate sufficient effectiveness and safety to obtain
the requisite regulatory approvals for these or any other
potential products. For example, we have submitted BLAs to the
FDA for raxibacumab, ZALBIN and BENLYSTA, but the studies we
have conducted to date may not be sufficient to obtain FDA
approval. For example, we had been developing ZALBIN for many
years. In 2010, we and our collaboration partner Novartis
decided to end further development of ZALBIN in anticipation of
a Complete Response Letter from the FDA in which we expected the
FDA to conclude that our existing ZALBIN data would not support
approval of our BLA. Additionally, in November 2009, we received
a Complete Response Letter from the FDA related to our BLA for
raxibacumab. In this letter, the FDA determined that it could
not approve the BLA in its present form and requested additional
studies and data that would be needed prior to the FDA making a
decision as to whether or not to approve the raxibacumab BLA.
For raxibacumab, we may not be able to complete the requested
studies or to generate the required data in a timely manner, if
at all. If we do not complete the additional studies and
generate the additional data within the time required by the
FDA, we may be required to withdraw our existing BLA and
resubmit our BLA after completion of such studies. This will
start a new review cycle. Even if we can complete such studies
and generate such data, the studies and data may not be
sufficient for FDA approval.
For BENLYSTA, we have completed two large-scale, multi-center
Phase 3 clinical trials, in addition to earlier Phase 1 and
Phase 2 clinical trials. See our first risk factor under the
heading “Commercial Risks” for additional discussion
regarding the data from these trials. In June 2010, we filed a
BLA in the United States and our collaboration partner GSK
submitted a Marketing Authorization Application with the
European Medicines Agency. The FDA has assigned a PDUFA target
date of March 10, 2011 and requested some additional
information. We currently expect a decision by the EMA in the
second half of 2011. We may not be able to obtain FDA, EMA or
other regulatory approval of BENLYSTA. Even if FDA, EMA or other
regulatory approval is obtained, it may be limited to specific
indications, limit the type of patients in which the drug may be
used, or otherwise require specific warnings or labeling
language, any of which might reduce the commercial potential of
BENLYSTA.
We depend on third parties to conduct many of our clinical
trials, and we may encounter delays in or lose some control over
our efforts to develop products.
We are dependent on third-party research organizations to enroll
qualified patients and conduct, supervise and monitor many of
our clinical trials. Our reliance on these service providers
does not relieve us of our regulatory responsibilities,
including ensuring that our clinical trials are conducted in
accordance with good clinical practice regulations and the plan
and protocols contained in the relevant regulatory application.
In addition, these
22
organizations may not complete activities on schedule or may not
conduct our preclinical studies or clinical trials in accordance
with regulatory requirements or our trial design. If we are
unable to obtain any necessary services on acceptable terms or
if these service providers do not successfully carry out their
contractual duties or meet expected deadlines, our efforts to
obtain regulatory approvals for our product candidates may be
delayed or prevented.
RISK FROM
COLLABORATION RELATIONSHIPS AND STRATEGIC ACQUISITIONS
Our plan to use collaborations to leverage our capabilities
may not be successful if we are unable to integrate our
partners’ capabilities with our operations or if our
partners’ capabilities do not meet our expectations.
As part of our strategy, we intend to continue to evaluate
strategic partnership opportunities. In order for our future
collaboration efforts to be successful, we must first identify
partners whose capabilities complement and integrate well with
ours. Technologies to which we gain access may prove ineffective
or unsafe. Our current agreements that grant us access to such
technology may expire and may not be renewable or could be
terminated if we or our partners do not meet our obligations.
These agreements are subject to differing interpretations and we
and our partners may not agree on the appropriate interpretation
of specific requirements. In addition, our partners, among other
things, may prove difficult to work with, less skilled than we
originally expected or unable to satisfy their financial
commitments to us. Past collaborative successes are no assurance
of potential future success.
We currently depend on our collaboration partners for
substantial revenue. We may not become profitable on a
sustainable basis if we cannot increase the revenue from our
collaboration partners or other sources.
We have received substantial revenue from payments made under
collaboration agreements with Novartis and GSK, and to a lesser
extent, other agreements. Our agreement with Novartis has been
terminated. The research term of our initial GSK collaboration
agreement and many of our other collaboration agreements expired
in 2001. None of the research terms of these collaboration
agreements was renewed and we may not be able to enter into
additional collaboration agreements. While our partners under
our initial GSK collaboration agreement have informed us that
they have been pursuing research programs involving different
genes for the creation of small molecule, protein and antibody
drugs, we cannot assure you that any of these programs will be
continued or will result in any approved drugs. If our partners
are unsuccessful in such research and development efforts, we
will not receive any revenue from the development or
commercialization of these assets.
Under our present collaboration agreements, we are entitled to
certain development and commercialization payments based on our
development of the applicable product or certain milestone and
royalty payments based on our partners’ development of the
applicable product. We may not receive payments under these
agreements if we or our collaborators fail to:
|
|
|
| |
•
|
develop marketable products;
|
| |
| |
•
|
obtain regulatory approvals for products; or
|
| |
| |
•
|
successfully market products.
|
Further, circumstances could arise under which one or more of
our collaboration partners may allege that we breached our
agreement with them and, accordingly, seek to terminate our
relationship with them. Our collaboration partners may also
terminate these agreements without cause or if competent
scientific evidence or safety risks do not justify moving the
applicable product forward. If any one of these agreements
terminates, this could adversely affect our ability to
commercialize our products and harm our business.
If one of our collaborators pursues a product that competes
with our products, there could be a conflict of interest and we
may not receive expected milestone or royalty payments.
Each of our collaborators is developing a variety of products,
some with other partners. Our collaborators may pursue existing
or alternative technologies to develop drugs targeted at the
same diseases instead of using our licensed technology to
develop products in collaboration with us. Our collaborators may
also develop products that
23
are similar to or compete with products they are developing in
collaboration with us. If our collaborators pursue these other
products instead of our products, we may not receive milestone
or royalty payments.
Our efforts to acquire other biotechnology companies or
in-license and develop additional product candidates may not be
successful, which could have a material adverse effect on our
business, financial condition and results of operations.
Our business strategy includes the acquisition of other
biotechnology companies or in-license of additional product
candidates to complement and supplement our existing product
pipeline, and we may acquire such companies and in-license
additional product candidates that have demonstrated positive
pre-clinical
and/or
clinical data. We have certain criteria by which we assess any
acquisition or in-license and we may not be successful in
identifying, effectively evaluating, acquiring or in-licensing,
and developing additional product candidates, or on acceptable
terms. In addition, product in-licensing involves inherent
risks, including uncertainties due to matters that may affect
the successful development or commercialization of the
in-licensed product as well as the possibility of contractual
disagreements with regard to terms such as patent rights,
license scope or termination rights. Competition for attractive
product opportunities is intense and may require us to devote
substantial resources, both managerial and financial, to an
opportunity that may not result in a successfully developed, or
commercialized, product. Moreover, the cost of acquiring other
companies or in-licensing product candidates could be
substantial and in order to acquire companies or new products we
may need to raise additional financing, which if it involves the
issuance of additional shares of our common stock would dilute
existing stockholders. If we are unsuccessful in our efforts to
acquire other companies or in-license and develop additional
product candidates, or if we acquire or license unproductive
assets, it could have a material adverse effect on the growth of
our business.
In order to achieve the anticipated benefits of an acquisition,
we must integrate the acquired company’s business,
technology and employees in an efficient and effective manner.
The successful combination of companies in a rapidly changing
biotechnology industry may be more difficult to accomplish than
in other industries. The combination of two companies requires,
among other things, integration of the companies’
respective technologies and research and development efforts. We
cannot assure you that this integration will be accomplished
smoothly or successfully. The difficulties of integration may be
increased by any need to coordinate geographically separated
organizations and address differences in corporate cultures and
management philosophies. The integration of certain operations
will require the dedication of management resources and,
accordingly, may temporarily distract attention from the
day-to-day
operations of the combined companies. The business of the
combined companies may also be disrupted by employee retention
uncertainty and lack of focus during integration. The inability
of management to integrate successfully the operations of the
two companies, in particular, the integration and retention of
key personnel, or the inability to integrate successfully two
technology platforms, could have a material adverse effect on
our business, results of operations and financial condition.
REGULATORY
RISKS
Because we are subject to extensive and changing government
regulatory requirements, we may not be able to obtain regulatory
approval of our products in a timely manner, if at all.
Regulations in the United States and other countries have a
significant impact on our research, product development and
manufacturing activities and will be a significant factor in the
marketing of our products. All of our products require
regulatory approval prior to commercialization. In particular,
our products are subject to rigorous preclinical and clinical
testing and other premarket approval requirements by the FDA and
similar regulatory authorities in other regions, such as Europe
and Asia. Various statutes and regulations also govern or
influence the manufacturing, safety, labeling, storage, record
keeping and marketing of our products. The lengthy process of
seeking these approvals, and the subsequent compliance with
applicable statutes and regulations, require the expenditure of
substantial resources. Any failure by us to obtain, or any delay
in obtaining, regulatory approvals could materially adversely
affect our ability to commercialize our products in a timely
manner, or at all.
United States Regulatory Approval. Before a
product can be marketed in the United States, the results of the
preclinical and clinical testing must be submitted to the FDA
for approval. This submission will be either a new drug
application (“NDA”) or a biologics license application
(“BLA”), depending on the type of drug. In responding
to an application, the FDA may grant marketing approval, request
additional information or deny the application if it
24
determines that the application does not provide an adequate
basis for approval. We cannot assure you that any approval
required by the FDA will be obtained on a timely basis, or at
all.
In June 2010 we filed a BLA with the FDA for BENLYSTA. The
Arthritis Advisory Committee of the FDA met on November 16,
2010 to consider the BENLYSTA BLA and voted
13-2 to
recommend FDA approval. On December 3, 2010, the FDA
extended the PDUFA target date for its priority review of the
BLA from December 9, 2010 to March 10, 2011 and
requested some additional information. Even with the positive
recommendation of the Advisory Committee, the FDA may determine
that our BLA is insufficient to support marketing approval or
may deny our BLA, either of which would materially adversely
affect our results of operations and business. Furthermore,
regulatory approvals, even if obtained, may be limited to
specific indications, limit the type of patients in which the
drug may be used, or otherwise require specific warning or
labeling language, any of which might reduce the commercial
potential of BENLYSTA and materially adversely affect our
results of operations and business.
The FDA may condition marketing approval on the conduct of
specific post-marketing studies to further evaluate safety and
efficacy, including in particular patient populations. Rigorous
and extensive FDA regulation of pharmaceutical products
continues after approval, particularly with respect to
compliance with cGMPs, reporting of adverse effects,
advertising, promotion and marketing. Discovery of previously
unknown problems or failure to comply with the applicable
regulatory requirements may result in restrictions on the
marketing of a product or withdrawal of the product from the
market as well as possible civil or criminal sanctions, any of
which could materially adversely affect our business. In
addition, such post-marketing studies may be expensive,
time-consuming and difficult to complete in a timely fashion,
any of which may limit our ability to develop other indications
of existing products as well as indications for new products.
Foreign Regulatory Approvals. We must obtain
regulatory approval by governmental agencies in other countries
prior to commercialization of our products in those countries.
Foreign regulatory systems may be rigorous, costly and
uncertain. In June 2010, our collaboration partner GSK submitted
an MAA for BENLYSTA with the European Medicines Agency. We
currently expect a decision by the EMA in the second half of
2011, but can provide no assurance that the EMA will grant
regulatory approval for BENLYSTA on such timeframe, if at all.
In addition, regulatory approval, even if obtained, may be
limited to specific indications, limit the type of patients in
which the drug may be used, or otherwise require specific
warning or labeling language, any of which might reduce the
commercial potential of BENLYSTA.
To date, neither we nor any of our collaboration partners
have received marketing approval for any product candidate
resulting from our research and development efforts. Because we
may never be able to obtain any such approval, it is possible
that we may not be able to generate any product revenue other
than with respect to raxibacumab.
Although we have active BLAs for two of our product candidates
(raxibacumab and BENLYSTA), we cannot assure you that these
products will receive marketing approval. It is possible that we
will not receive FDA marketing approval for any of our products.
All products being developed by our collaboration partners will
also require additional research and development, preclinical
studies and extensive clinical trials and regulatory approval
prior to any commercial sales. In some cases, the length of time
that it takes for our collaboration partners to achieve various
regulatory approval milestones may affect the payments that we
are eligible to receive under our collaboration agreements. We
and our collaboration partners may need to successfully address
a number of technical challenges in order to complete
development of our products. Moreover, these products may not
prove effective in treating any disease or may prove to have
undesirable or unintended side effects, toxicities or other
characteristics that may preclude obtaining regulatory approval
or prevent or limit commercial use.
If we receive marketing approval for BENLYSTA but are unable
to expand label usage of BENLYSTA, we may not recognize the full
value of the product and there may be adverse effects on our
expected financial and operating results.
BENLYSTA is a human monoclonal antibody that recognizes and
inhibits the biological activity of
B-lymphocyte
stimulator, or BLyS, and is being developed as a potential
treatment for SLE. If the FDA or other regulatory agencies
approve BENLYSTA for the treatment of SLE, we and our partner
intend to conduct new
25
clinical trials for additional approved, or labeled uses, of
BENLYSTA, such as vasculitis, post-renal transplant and other
autoimmune indications and seek expansion of the labeled uses in
the U.S. and in other countries. However, we may be unable
to obtain approval for such label expansion, in full or in part.
If we are not able to obtain approval for expansion of the
labeled uses for BENLYSTA, sales of BENLYSTA may be limited.
We are subject to environmental, health and safety laws that
may restrict us from conducting our business in the most
economically advantageous manner.
We are subject to various laws and regulations relating to safe
working conditions, laboratory and manufacturing practices, the
experimental use of animals, emissions and wastewater
discharges, and the use and disposal of hazardous or potentially
hazardous substances used in connection with our research,
including radioactive compounds and infectious disease agents.
We also cannot accurately predict the extent that regulations
that might result from any future legislative or administrative
action might affect our business. Any of these laws or
regulations could cause us to incur additional expense or
restrict our operations.
INTELLECTUAL
PROPERTY RISKS
If our patent applications do not result in issued patents or
if patent laws or the interpretation of patent laws change, our
competitors may be able to obtain rights to and commercialize
our discoveries.
Our pending patent applications, including those covering
full-length genes and their corresponding proteins, may not
result in the issuance of any patents. Our applications may not
be sufficient to meet the statutory requirements for
patentability in all cases or may be subject to challenge if
they do issue. Important legal issues remain to be resolved as
to the extent and scope of available patent protection for
biotechnology products and processes in the United States and
other important markets outside the United States, such as
Europe and Japan. For example, a recent U.S. district court
decision involving Myriad Genetics expressed concerns regarding
the patentability of isolated human genes and gene-based
diagnostic methods; this case is on appeal to the
U.S. Court of Appeals for the Federal Circuit. In addition,
the United States Congress is considering significant changes to
U.S. intellectual property laws that could affect the
extent and scope of existing protections for biotechnology
products and processes. Foreign markets may not provide the same
level of patent protection as provided under the
U.S. patent system. We expect that litigation or
administrative proceedings will likely be necessary to determine
the validity and scope of certain of our and others’
proprietary rights. We are currently involved in a number of
litigation and administrative proceedings relating to the scope
of protection of our patents and those of others in the United
States and the rest of the world.
On January 25, 2011, HGS filed suit against Genentech,
Inc., and City of Hope in the United States District Court for
the District of Delaware seeking a ruling that US Patent
No. 6,331,415 (the “Cabilly II Patent”) is
invalid, unenforceable, and not infringed by BENLYSTA. In
addition, in a paper filed with the United States District Court
for the Central District of California on January 28, 2011,
Genentech sought leave to add BENLYSTA to an ongoing litigation
between GSK and Genentech in that court related to GSK’s
product
ARZERRAtm
(ofatumumab).
We have also been involved in a number of interference
proceedings brought by the United States Patent and Trademark
Office (“PTO”) and may be involved in additional
interference proceedings in the future. These proceedings
determine the priority of inventions and, thus, the right to a
patent for technology in the U.S.
We are also involved in proceedings in connection with foreign
patent filings, including opposition and revocation proceedings,
and may be involved in other such proceedings in the future. For
example, we are involved in European opposition proceedings
regarding an issued patent of Biogen Idec that HGS and GSK have
licensed. In this opposition, the European Patent Office
(“EPO”) found Biogen Idec’s claims to a method of
treating autoimmune diseases using an antibody to BLyS (such as
BENLYSTA), to be valid. Merck Serono SA has appealed this
decision to an EPO Technical Board of Appeal. A hearing is
expected in late 2011. We are also involved in a revocation
proceeding brought by Eli Lilly and Company with respect to our
United Kingdom (“UK”) patent related to BLyS
compositions, including antibodies. Although an EPO Technical
Board of Appeal held that the corresponding European patent was
valid, the UK Court of Appeal disagreed and upheld a lower UK
court’s ruling that the UK patent was invalid. The UK
Supreme Court granted HGS permission to appeal this decision.
The appeal is expected to be heard in July 2011.
26
In addition, Genentech, Inc. opposed our European patent related
to products based on TRAIL Receptor 1 (such as HGS-ETR1). On
October 5, 2010, the Opposition Division of the EPO upheld
our patent; no appeal was filed by the final deadline.
We cannot assure you that we will be successful in any of these
proceedings. Moreover, any such litigation or proceeding may
result in a significant commitment of resources in the future
and could force us to do one or more of the following: cease
selling or using any of our products that incorporate the
challenged intellectual property, which would adversely affect
our revenue; obtain a license from the holder of the
intellectual property right alleged to have been infringed,
which license may not be available on reasonable terms, if at
all; and redesign our products to avoid infringing the
intellectual property rights of third parties, which may be
time-consuming or impossible to do. In addition, such litigation
or proceeding may allow others to use our discoveries or develop
or commercialize our products. Changes in, or different
interpretations of, patent laws in the United States and other
countries may result in patent laws that allow others to use our
discoveries or develop and commercialize our products or prevent
us from using or commercializing our discoveries and products.
We cannot assure you that the patents we obtain or the
unpatented technology we hold will afford us significant
commercial protection.
If others file patent applications or obtain patents similar
to ours, then the United States Patent and Trademark Office may
deny our patent applications, or others may restrict the use of
our discoveries.
We are aware that others, including universities, government
agencies and companies working in the biotechnology and
pharmaceutical fields, have filed patent applications and have
been granted patents in the United States and in other countries
that cover subject matter potentially useful or necessary to our
business. Some of these patents and patent applications claim
only specific products or methods of making products, while
others claim more general processes or techniques useful in the
discovery and manufacture of a variety of products. The risk of
third parties obtaining additional patents and filing patent
applications will continue to increase as the biotechnology
industry expands. We cannot predict the ultimate scope and
validity of existing patents and patents that may be granted to
third parties, nor can we predict the extent to which we may
wish or be required to obtain licenses to such patents, or the
availability and cost of acquiring such licenses. To the extent
that licenses are required, the owners of the patents could
bring legal actions against us to claim damages or to stop our
manufacturing and marketing of the affected products. We believe
that there will continue to be significant litigation in our
industry regarding patent and other intellectual property
rights. Such litigation could consume a substantial portion of
our resources.
Because issued patents may not fully protect our discoveries,
our competitors may be able to commercialize products similar to
those covered by our issued patents.
Issued patents may not provide commercially meaningful
protection against competitors and may not provide us with
competitive advantages. Other parties may challenge our patents
or design around our issued patents or develop products
providing effects similar to our products. In addition, others
may discover uses for genes, proteins or antibodies other than
those uses covered in our patents, and these other uses may be
separately patentable. The holder of a patent covering the use
of a gene, protein or antibody for which we have a patent claim
could exclude us from selling a product for a use covered by its
patent.
We rely on our collaboration partners to seek patent
protection for the products they develop based on our
research.
A significant portion of our future revenue may be derived from
royalty payments from our collaboration partners. These partners
face patent protection issues similar to those that we and other
biotechnology or pharmaceutical companies face. As a result, we
cannot assure you that any product developed by our
collaboration partners will be patentable, and therefore,
revenue from any such product may be limited, which would reduce
the amount of any royalty payments. We also rely on our
collaboration partners to effectively prosecute their patent
applications. Their failure to obtain or protect necessary
patents could also result in a loss of royalty revenue to us.
27
If we are unable to protect our trade secrets, others may be
able to use our secrets to compete more effectively.
We may not be able to meaningfully protect our trade secrets. We
rely on trade secret protection to protect our confidential and
proprietary information. We believe we have acquired or
developed proprietary procedures and materials for the
production of proteins and antibodies. We have not sought patent
protection for these procedures. While we have entered into
confidentiality agreements with employees and collaborators, we
may not be able to prevent their disclosure of these data or
materials. Others may independently develop substantially
equivalent information and processes.
Other parties may seek to cancel or revoke our trademarks
and/or
restrict the use of our trademarks.
Our trademarks, including BENLYSTA, are important to us and are
generally covered by trademark applications or registrations in
the United States and in other countries. Trademark protection
varies in accordance with local law, and continues in some
countries for as long as the mark is used and in other countries
for as long as the mark is registered. Trademark registrations
are generally for fixed but renewable terms.
Our trademark applications may not be sufficient to meet the
statutory requirements for registration in all cases or may be
subject to challenge if they are registered. Other parties may
seek to cancel or revoke our trademarks
and/or
restrict the use of our trademarks through litigation or
administrative proceedings in both the United States and in the
rest of the world. We cannot assure you that we will be
successful in any such proceedings. Moreover, any such
litigation or proceeding may require us to modify our trademarks
or rebrand our products to avoid infringing the trademark rights
of third parties This may be time-consuming and could adversely
affect our revenue.
FINANCIAL
AND MARKET RISKS
Because of our substantial indebtedness and lease
obligations, we may be unable to adjust our strategy to meet
changing conditions in the future.
As of December 31, 2010, we had convertible subordinated
debt of $372.9 million ($403.8 million on a face value
basis) and a long-term lease financing for our large-scale
manufacturing facility of $250.5 million. During the fiscal
year ended December 31, 2010 we made cash interest payments
on our convertible subordinated debt of $9.1 million.
During the fiscal year ended December 31, 2010 we made cash
payments on our long-term lease financing of $24.5 million.
In addition, we have operating leases, primarily our long-term
operating lease for our headquarters, for which we made cash
payments of $20.9 million during the fiscal year ended
December 31, 2010. Our substantial debt and long-term lease
obligations will have several important consequences for our
future operations. For instance:
|
|
|
| |
•
|
payments of interest on, and principal of, our indebtedness and
our long-term lease obligations will be substantial and may
exceed then current income and available cash;
|
| |
| |
•
|
we may be unable to obtain additional future financing for
marketing efforts, continued clinical trials, capital
expenditures, acquisitions or general corporate purposes;
|
| |
| |
•
|
we may be unable to withstand changing competitive pressures,
economic conditions and governmental regulations; and
|
| |
| |
•
|
we may be unable to make acquisitions or otherwise take
advantage of significant business opportunities that may arise.
|
We may not have adequate financial resources available to
repay our Convertible Subordinated Notes due 2011 (“2011
Notes”) and our Convertible Subordinated Notes due 2012
(“2012 Notes”) at maturity.
As of December 31, 2010, we had $403.8 million in face
value of convertible subordinated debt outstanding, with
$197.1 million and $206.7 million due in 2011 and
2012, respectively. Those notes are convertible into our common
stock at conversion prices of approximately $15.55 and $17.78
per share, respectively. If our stock price does not exceed the
applicable conversion price of those notes, upon maturity, we
may need to pay the note holders in cash or restructure some or
all of the debt. Our recent stock price has been above the
conversion price and we believe we currently have sufficient
unrestricted cash should note holders seek cash payment upon
maturity.
28
However, since there are other potential uses for these funds
and it may be one or more years, if ever, before we are likely
to generate significant positive cash flow from operations, we
may not have enough cash, cash equivalents, short-term
investments and marketable securities available to repay our
debt upon maturity.
To become a successful biopharmaceutical company, we may need
additional funding. If we do not obtain this funding on
acceptable terms, we may not be able to generate sufficient
revenue to repay our debt, to continue our research and
development efforts or to launch and successfully market our
products.
We continue to expend substantial funds on our research and
development programs and human studies on current and future
product candidates. We also expect to expend significant funds
to support pre-launch and commercial marketing activities, to
acquire other biotechnology companies or in-license and develop
additional product candidates, and to enhance our manufacturing
capacity. We may need additional financing to fund these
activities. We may not be able to obtain additional financing on
acceptable terms, if at all. If we raise additional funds by
issuing equity securities, equity-linked securities or debt
securities, the new equity securities may dilute the interests
of our existing stockholders and the new debt securities may
contain restrictive financial covenants.
Our need for additional funding will depend on many factors,
including, without limitation:
|
|
|
| |
•
|
the amount of revenue or cost sharing, if any, that we are able
to obtain from our collaborations, any approved products, and
the time and costs required to achieve those revenues;
|
| |
| |
•
|
the timing, scope and results of preclinical studies and
clinical trials;
|
| |
| |
•
|
the size and complexity of our development programs;
|
| |
| |
•
|
the timing and costs involved in obtaining regulatory approvals;
|
| |
| |
•
|
the timing and costs of increasing our manufacturing capacity;
|
| |
| |
•
|
the costs of commercializing our products, including marketing,
promotional and sales costs;
|
| |
| |
•
|
the commercial success of our products;
|
| |
| |
•
|
our stock price;
|
| |
| |
•
|
our ability to establish and maintain collaboration partnerships;
|
| |
| |
•
|
competing technological and market developments;
|
| |
| |
•
|
the costs involved in filing, prosecuting, enforcing and
defending patent claims;
|
| |
| |
•
|
the costs involved in bringing and defending against
litigation; and
|
| |
| |
•
|
scientific progress in our research and development programs.
|
If we are unable to raise additional funds, we may, among other
things:
|
|
|
| |
•
|
delay, scale back or eliminate some or all of our research and
development programs;
|
| |
| |
•
|
delay, scale back or eliminate some or all of our
commercialization activities;
|
| |
| |
•
|
lose rights under existing licenses;
|
| |
| |
•
|
relinquish more of, or all of, our rights to product candidates
on less favorable terms than we would otherwise seek; and
|
| |
| |
•
|
be unable to operate as a going concern.
|
Our short-term investments, marketable securities and
restricted investments are subject to certain risks that could
materially adversely affect our overall financial position.
We invest our cash in accordance with an established internal
policy and customarily in instruments which historically have
been highly liquid and carried relatively low risk. However, the
capital and credit markets have experienced extreme volatility
and disruption. Over the past several years, the volatility and
disruption reached unprecedented levels. We maintain a
significant portfolio of investments in short-term investments,
marketable debt
29
securities and restricted investments, which are recorded at
fair value. Certain of these transactions expose us to credit
risk in the event of default by the issuer. To seek to minimize
our exposure to credit risk, we invest in securities with strong
credit ratings and have established guidelines relative to
diversification and maturity with the objective of maintaining
safety of principal and liquidity. We do not invest in
derivative financial instruments or auction rate securities, and
we generally hold our investments in debt securities until
maturity. In recent years, certain financial instruments,
including some of the securities in which we have invested, have
sustained downgrades in credit ratings and some high quality
short-term investment securities have suffered illiquidity or
events of default. Deterioration in the credit market may have
an adverse effect on the fair value of our investment portfolio.
Should any of our short-term investments, marketable securities
or restricted investments lose significant value or have their
liquidity impaired, it could materially and adversely affect our
overall financial position by imperiling our ability to fund our
operations and forcing us to seek additional financing sooner
than we would otherwise. Such financing may not be available on
commercially attractive terms, or at all.
We have only a limited amount of insurance that protects us
only from certain business risks, which could leave us exposed
to significant, uninsured liabilities.
We do not carry insurance for all categories of risk that our
business may encounter. We currently maintain general liability,
property, auto, workers’ compensation, product liability,
fiduciary and directors’ and officers’ insurance
policies. Potential risks and liabilities may not be covered by
our insurance policies, insurers may dispute coverage, or the
amount of insurance may not be enough to cover claims and
liabilities. We may be unable to maintain existing insurance,
obtain new coverage, or increase limits in the future, and may
be unable to do so on favorable terms. Any significant uninsured
liability may require us to pay substantial amounts, which would
adversely affect our cash position and results of operations.
We may be subject to product liability or other litigation
that could result in a diminution or inefficient allocation of
our critical resources, delay the implementation of our business
strategy, affect our reputation and, if successful, materially
and adversely harm our business and financial condition.
We face an inherent risk of product liability claims in the
event that the use of our products caused, or is alleged to have
caused, adverse side effects (including death) or drug
interactions. It may be that we will not understand these risks
until the drug has been administered to patients for some time.
We may be subject to product liability claims that can be costly
to defend and may result in large judgments or settlements
against us. Any insurance we obtain may not provide adequate
coverage against any such asserted claims and we may therefore
be exposed to significant litigation costs and liabilities,
which could exceed our total assets and our ability to pay. In
addition, negative publicity associated with any claims,
regardless of their merit, may decrease the future demand for
our products.
We may from time to time become involved in various lawsuits and
legal proceedings that arise in the ordinary course of our
business. Any litigation to which we are subject could require
significant involvement of our senior management and may divert
management’s attention from our business and operations.
Litigation costs or an adverse result in any litigation that may
arise from time to time may materially adversely impact our
business, operating results and financial condition.
OTHER
BUSINESS RISKS
Our success is dependent on our continued ability to attract,
motivate and retain key personnel. If we lose or are unable to
attract key management or other personnel, it could have a
material adverse effect on our business, financial condition and
results of operations.
Much of our progress to date has resulted from the particular
scientific, technical and management skills of personnel
available to us. Competition for qualified employees is intense
among pharmaceutical and biotechnology companies. Part of being
able to attract, motivate and retain key personnel is our
ability to offer a competitive compensation package, including
cash bonus and equity incentive awards. Our ability to offer
attractive equity incentive awards in the future may be limited
or nonexistent if we are unable to increase the number of shares
available under our stock incentive plan. If we are unable to
provide a compensation package that is competitive within our
industry, or otherwise successfully compete to attract, motivate
and retain qualified management and
30
other highly skilled employees, this could materially adversely
affect the implementation of our business strategy, delay the
commercialization of our products or prevent us from becoming
profitable.
We may be unable to fulfill the terms of our contract
manufacturing agreements with our customers for manufacturing
process development and supply of selected biopharmaceutical
products.
To more fully utilize our existing manufacturing capacity, we
have entered into agreements with customers pursuant to which we
have agreed to develop manufacturing processes for, and
manufacture clinical and commercial supplies of, certain
biopharmaceutical products, and may enter into similar
agreements with other potential customers in the future. Our
receipt of revenue under these agreements is dependent on our
ability to successfully manufacture such products. If we are
unable to develop a validated manufacturing process for such
products or are otherwise unable to deliver product that meets
the manufacturing specifications, we may not receive any
additional payments under such agreements. Even if successful,
we may not be able to enter into additional agreements with
other customers. Any current or future customers may decide to
discontinue the products contemplated under these agreements,
and therefore we may not receive revenue from these agreements.
RISKS
RELATED TO OWNERSHIP OF OUR COMMON STOCK
The market price of our common stock may be lower or more
volatile than you expected.
Our stock price, like the stock prices of many other
biotechnology companies, has been highly volatile. During the
preceding twelve months, the closing price of our common stock
has been as low as $21.84 per share and as high as $33.30 per
share. These broad market fluctuations may cause the market
price of our common stock to be lower or more volatile than you
expected.
The price and volume fluctuations in our stock may often be
unrelated to our operating performance. The market price of our
common stock could fluctuate widely because of:
|
|
|
| |
•
|
future announcements about our company or our competitors,
including the results of testing, clinical trials, technological
innovations or new commercial products;
|
| |
| |
•
|
regulatory actions with respect to our potential products or
regulatory approvals with respect to our competitors’
products;
|
| |
| |
•
|
changes in government regulations;
|
| |
| |
•
|
developments in our relationships with our collaboration
partners;
|
| |
| |
•
|
developments affecting our collaboration partners;
|
| |
| |
•
|
announcements relating to health care reform and reimbursement
levels for new drugs;
|
| |
| |
•
|
our failure to acquire or maintain proprietary rights to the
gene sequences we discover or the products we develop;
|
| |
| |
•
|
litigation;
|
| |
| |
•
|
public concern as to the safety of our products; and
|
| |
| |
•
|
political and economic factors affecting market prices generally
or our market segment or our company particularly.
|
The issuance and sale of shares underlying our outstanding
convertible debt securities and options, as well as the sale of
additional equity or equity-linked securities would dilute the
holdings of our existing stockholders and may materially and
adversely affect the price of our common stock.
Sales of substantial amounts of shares of our common stock or
securities convertible into or exchangeable for our common stock
in the public market, or the perception that those sales may
occur, could cause the market price of our common stock to
decline. We have used and may continue to use our common stock
or securities convertible into or exchangeable for our common
stock to acquire technology, product rights or businesses, or
for other purposes. Our authorized capital stock consists of
400,000,000 shares of common stock, par value $0.01 per
share.
31
As of December 31, 2010, we had 188,980,748 shares of
common stock outstanding. In addition, an aggregate of
approximately 24,302,742 shares of our common stock are
issuable upon conversion of our outstanding 2011 Notes and
outstanding 2012 Notes at an applicable conversion price of
$15.55 and $17.78 per share, respectively;
24,028,688 shares of our common stock are issuable upon the
exercise of options outstanding as of December 31, 2010,
having a weighted-average exercise price of $14.55 per share,
including 5,309,761 stock options granted during the fiscal year
ended December 31, 2010 with a weighted-average grant date
fair value of $17.31 per share; and 203,782 shares of our
common stock are issuable upon the vesting of restricted stock
unit awards outstanding as of December 31, 2010. If we
issue additional equity securities, including in exchange for
our outstanding convertible debt or in connection with the
exercise or vesting of equity awards, the price of our common
stock may be materially and adversely affected and the holdings
of our existing stockholders would be diluted.
Our certificate of incorporation and bylaws could discourage
acquisition proposals, delay a change in control or prevent
transactions that may be in your best interests.
Provisions of our certificate of incorporation and bylaws, as
well as Section 203 of the Delaware General Corporation
Law, may discourage, delay or prevent a change in control of our
company that you as a stockholder may consider favorable and may
be in your best interest. Our certificate of incorporation and
bylaws contain provisions that:
|
|
|
| |
•
|
authorize the issuance of up to 20,000,000 shares of
“blank check” preferred stock that could be issued by
our board of directors to increase the number of outstanding
shares and discourage a takeover attempt;
|
| |
| |
•
|
limit who may call special meetings of stockholders; and
|
| |
| |
•
|
establish advance notice requirements for nomination of
candidates for election to the board of directors or for
proposing matters that can be acted upon by stockholders at
stockholders’ meetings.
|
32
|
|
|
ITEM 1B.
|
UNRESOLVED
STAFF COMMENTS
|
None.
We currently lease and occupy approximately
1,020,000 square feet of laboratory, manufacturing and
office space in Rockville, Maryland. Our space includes
approximately 200,000 square feet of laboratory space,
approximately 520,000 square feet of manufacturing and
manufacturing support space and approximately
300,000 square feet of office space. This excludes a
portion of our headquarters facility under lease which is not
being utilized.
We anticipate that existing commercial real estate or the
available land located at our laboratory and office campus will
enable us to continue to expand our operations in close
proximity to one another. We believe that our properties are
generally in good condition, well maintained, suitable and
adequate to carry on our business.
|
|
|
ITEM 3.
|
LEGAL
PROCEEDINGS
|
In the ordinary course of business, we are involved in various
legal proceedings, including, among others, patent oppositions,
patent infringement litigation and other matters incidental to
our business. While it is not possible to accurately predict or
determine the eventual outcome of these matters or estimate a
range of loss, one or more of these matters currently pending
could have a material adverse effect on our financial condition,
results of operations or cash flows.
|
|
|
ITEM 4.
|
(REMOVED
AND RESERVED)
|
33
PART II
|
|
|
ITEM 5.
|
MARKET
FOR REGISTRANT’S COMMON EQUITY AND RELATED STOCKHOLDER
MATTERS
|
Our common stock is traded on the NASDAQ Global Market under the
symbol HGSI. The following table presents the quarterly high and
low closing prices as quoted by NASDAQ.
| |
|
|
|
|
|
|
|
|
|
|
|
High
|
|
Low
|
|
|
|
2010
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
32.68
|
|
|
$
|
25.76
|
|
|
Second Quarter
|
|
$
|
33.30
|
|
|
$
|
22.46
|
|
|
Third Quarter
|
|
$
|
30.18
|
|
|
$
|
21.84
|
|
|
Fourth Quarter
|
|
$
|
29.79
|
|
|
$
|
23.60
|
|
|
2009
|
|
|
|
|
|
|
|
|
|
First Quarter
|
|
$
|
2.78
|
|
|
$
|
0.48
|
|
|
Second Quarter
|
|
$
|
3.19
|
|
|
$
|
0.82
|
|
|
Third Quarter
|
|
$
|
20.50
|
|
|
$
|
2.39
|
|
|
Fourth Quarter
|
|
$
|
31.15
|
|
|
$
|
17.96
|
|
As of January 31, 2011, there were 592 holders of record of
our common stock. We have never declared or paid any cash
dividends. We do not anticipate declaring or paying cash
dividends for the foreseeable future, in part because existing
contractual agreements prohibit such dividends. Instead, we will
retain our earnings, if any, for the future operation and
expansion of our business.
The following graph compares the performance of our Common Stock
for the periods indicated with the performance of the NASDAQ
U.S. Stock Market Total Return Index (the “TRI”)
and the NASDAQ Pharmaceutical Index (the “NPI”). The
comparison assumes $100 was invested on December 31, 2005
in our Common Stock and in each of the foregoing indices and
assumes the reinvestment of dividends, if any.
Comparison
of 5 Year Cumulative Total Return
The performance graph and related information shall not be
deemed “soliciting material” or be “filed”
with the Securities and Exchange Commission, nor shall such
information be incorporated by reference into any future filing
under the Securities Act or the Exchange Act, except to the
extent that the Company specifically incorporates it by
reference into such filing.
34
|
|
|
ITEM 6.
|
SELECTED
CONSOLIDATED FINANCIAL DATA
|
We present below our selected consolidated financial data for
the years ended December 31, 2010, 2009 and 2008, and as of
December 31, 2010 and 2009, which have been derived from
the audited consolidated financial statements included elsewhere
herein and should be read in conjunction with such consolidated
financial statements and the accompanying notes. We present
below our selected financial data for the years ended
December 31, 2007 and 2006, and as of December 31,
2008, 2007 and 2006, which have been derived from audited
financial statements not included herein. The results of
operations of prior periods are not necessarily indicative of
results that may be expected for any other period. See
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations” and
“Business.”
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
2007
|
|
|
2006
|
|
|
|
|
(In thousands, except per share and ratio data)
|
|
|
|
|
Statement of Operations Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales
|
|
$
|
47,159
|
|
|
$
|
154,074
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Manufacturing and development services
|
|
|
22,695
|
|
|
|
50,653
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Research and development collaborative agreements
|
|
|
87,497
|
|
|
|
71,022
|
|
|
|
48,422
|
|
|
|
41,851
|
|
|
|
25,755
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue
|
|
|
157,351
|
|
|
|
275,749
|
|
|
|
48,422
|
|
|
|
41,851
|
|
|
|
25,755
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs and expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales
|
|
|
29,941
|
|
|
|
15,805
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Cost of manufacturing and development services
|
|
|
15,016
|
|
|
|
18,215
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Research and development expenses
|
|
|
196,370
|
|
|
|
173,709
|
|
|
|
243,257
|
|
|
|
246,293
|
|
|
|
209,515
|
|
|
General and administrative expenses
|
|
|
106,797
|
|
|
|
61,073
|
|
|
|
60,865
|
|
|
|
55,874
|
|
|
|
53,101
|
|
|
Facility-related exit charges (credits)
|
|
|
—
|
|
|
|
759
|
|
|
|
—
|
|
|
|
(3,673
|
)
|
|
|
29,510
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total costs and expenses
|
|
|
348,124
|
|
|
|
269,561
|
|
|
|
304,122
|
|
|
|
298,494
|
|
|
|
292,126
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income (loss) from operations
|
|
|
(190,773
|
)
|
|
|
6,188
|
|
|
|
(255,700
|
)
|
|
|
(256,643
|
)
|
|
|
(266,371
|
)
|
|
Investment income
|
|
|
16,966
|
|
|
|
12,727
|
|
|
|
23,487
|
|
|
|
32,988
|
|
|
|
27,131
|
|
|
Interest expense
|
|
|
(59,500
|
)
|
|
|
(58,424
|
)
|
|
|
(62,912
|
)
|
|
|
(60,716
|
)
|
|
|
(39,606
|
)
|
|
Gain on extinguishment of debt
|
|
|
—
|
|
|
|
38,873
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Gain on sale of long-term equity investment
|
|
|
—
|
|
|
|
5,259
|
|
|
|
32,518
|
|
|
|
—
|
|
|
|
14,759
|
|
|
Other income (expense)
|
|
|
76
|
|
|
|
(238
|
)
|
|
|
(6,284
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income (loss) before taxes
|
|
|
(233,231
|
)
|
|
|
4,385
|
|
|
|
(268,891
|
)
|
|
|
(284,371
|
)
|
|
|
(264,087
|
)
|
|
Income tax benefit
|
|
|
—
|
|
|
|
1,274
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss)
|
|
$
|
(233,231
|
)
|
|
$
|
5,659
|
|
|
$
|
(268,891
|
)
|
|
$
|
(284,371
|
)
|
|
$
|
(264,087
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) per share, basic and diluted
|
|
$
|
(1.24
|
)
|
|
$
|
0.04
|
|
|
$
|
(1.99
|
)
|
|
$
|
(2.12
|
)
|
|
$
|
(2.00
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ratio of earnings to fixed charges
|
|
|
—
|
|
|
|
1.06
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Coverage deficiency
|
|
$
|
(233,231
|
)
|
|
$
|
—
|
|
|
$
|
(268,891
|
)
|
|
$
|
(284,371
|
)
|
|
$
|
(264,087
|
)
|
35
|
|
|
ITEM 6.
|
SELECTED
CONSOLIDATED FINANCIAL DATA, CONTINUED
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
2007
|
|
2006
|
|
|
|
(In thousands)
|
|
|
|
Balance Sheet Data:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents, short-term investments, marketable
securities and restricted
investments(1)
|
|
$
|
933,382
|
|
|
$
|
1,191,660
|
|
|
$
|
372,939
|
|
|
$
|
603,840
|
|
|
$
|
763,084
|
|
|
Total
assets(1)
|
|
|
1,315,029
|
|
|
|
1,530,630
|
|
|
|
686,832
|
|
|
|
961,566
|
|
|
|
1,161,922
|
|
|
Total debt and lease
financing(2)
|
|
|
623,367
|
|
|
|
598,435
|
|
|
|
664,074
|
|
|
|
637,513
|
|
|
|
612,811
|
|
|
Accumulated deficit
|
|
|
(2,419,897
|
)
|
|
|
(2,186,666
|
)
|
|
|
(2,192,325
|
)
|
|
|
(1,923,434
|
)
|
|
|
(1,639,063
|
)
|
|
Total stockholders’ equity (deficit)
|
|
|
585,763
|
|
|
|
755,415
|
|
|
|
(136,304
|
)
|
|
|
117,145
|
|
|
|
364,892
|
|
|
|
|
|
(1) |
|
“Cash, cash equivalents, short-term investments, marketable
securities and restricted investments” and “Total
assets” for 2010, 2009, 2008, 2007 and 2006 include
$79,510, $88,437, $69,360, $70,931 and $61,165 respectively, of
restricted investments relating to certain leases. |
| |
|
(2) |
|
“Total debt and lease financing” for 2010, 2009, 2008,
2007 and 2006 does not include any operating lease obligations
under various facility and equipment lease arrangements. See
“Management’s Discussion and Analysis of Financial
Condition and Results of Operations — Liquidity and
Capital Resources” for additional discussion. |
36
|
|
|
ITEM 7.
|
MANAGEMENT’S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
Overview
Human Genome Sciences, Inc. (“HGS”) is a
biopharmaceutical company that exists to place new therapies
into the hands of those battling serious disease. Our lead
products in development are
BENLYSTA®
(belimumab) for systemic lupus erythematosus (“SLE”)
and raxibacumab for inhalation anthrax.
BENLYSTA continues to progress toward commercialization. On
November 16, 2010, the U.S. Food and Drug
Administration (“FDA”) Arthritis Advisory Committee
voted 13-2
to recommend that the FDA approve BENLYSTA for the treatment of
autoantibody-positive patients with active SLE who are receiving
standard therapy. The Advisory Committee provides non-binding
recommendations for consideration by the FDA, which then makes
the final decision on approval. In December 2010, the FDA
extended the Prescription Drug User Fee Act (“PDUFA”)
target date for its priority review of BENLYSTA from
December 9, 2010 to March 10, 2011.
In June 2010, GlaxoSmithKline (“GSK”), our partner in
the co-development and commercialization of BENLYSTA, submitted
a Marketing Authorization Application (“MAA”) to the
European Medicines Agency (“EMA”) for approval to
market BENLYSTA in Europe. We expect to have the EMA decision in
the second half of 2011. GSK has also submitted regulatory
applications in Canada, Australia, Switzerland, Russia, Brazil
and The Philippines. Regulatory submissions in other
countries are planned in 2011.
In preparation for the anticipated launch and commercialization
of BENLYSTA, we have hired and trained a specialized commercial
team including sales, marketing and medical affairs. We are also
building our own HGS commercial team to work alongside GSK in
Europe. We believe we have produced sufficient BENLYSTA
inventory to meet the anticipated global market need for at
least one year, and are increasing our capacity to meet
long-term demand.
We continue to deliver raxibacumab to the U.S. Strategic
National Stockpile (“SNS”) for emergency use in
treating inhalation anthrax. In 2009, we completed the delivery
of 20,001 doses of raxibacumab to the SNS under an initial order
and as a result recognized $162.5 million in product sales
and manufacturing and development services revenue. In July
2009, the U.S. Government (“USG”) exercised its
option to purchase 45,000 additional doses of raxibacumab, with
delivery to be completed over a three-year period. HGS expects
to receive approximately $142.0 million from this second
order as deliveries are completed, $64.9 million of which
has been recognized as revenue through December 31, 2010.
In May 2009, we submitted a Biologics License Application
(“BLA”) to the FDA for raxibacumab for the treatment
of inhalation anthrax. We received a Complete Response Letter in
November 2009, and we will continue to work closely with the FDA
to obtain approval. HGS will receive approximately
$20.0 million from the USG upon FDA licensure of
raxibacumab.
In addition to these products in our internal pipeline, we have
substantial financial rights to two novel drugs that GSK has
advanced to late-stage development. The first of these is
darapladib, which was discovered by GSK using HGS technology. In
two pivotal Phase 3 trials, GSK is currently evaluating whether
darapladib can reduce the risk of adverse cardiovascular events
such as heart attack or stroke in patients with chronic coronary
heart disease and acute coronary syndrome, respectively. With a
planned enrollment of more than 27,000 patients in the two
trials, the Phase 3 clinical program for darapladib is among the
largest ever conducted to evaluate the safety and efficacy of
any cardiovascular medication. The second is albiglutide. GSK
currently has eight Phase 3 trials in progress to evaluate the
long-term efficacy, safety and tolerability of albiglutide as
monotherapy and add-on therapy for patients with type 2 diabetes
mellitus. Albiglutide was created by HGS using its proprietary
albumin-fusion technology, and the product was licensed to GSK
in 2004.
We are also working to expand and advance our mid- and
early-stage pipeline beyond potential additional indications for
BENLYSTA, with a primary focus on immunology and oncology. We
continue to develop our oncology portfolio around our expertise
in the apoptosis, or programmed cell death, pathway and we plan
to initiate Phase 2 development of our human monoclonal antibody
to the CCR5 receptor for the treatment of ulcerative colitis.
37
Overview
(continued)
We and Novartis were developing
ZALBINtm
for the treatment of patients with chronic hepatitis C. In
2010 the FDA expressed concerns regarding the risk-benefit
assessment of ZALBIN dosed at 900-mcg every two weeks. Based on
the FDA and similar EMA feedback, we and Novartis decided to end
further development of ZALBIN in 2010.
Strategic partnerships are an important driver of our commercial
success. We have a co-development and commercialization
agreement with GSK for BENLYSTA, and raxibacumab is being
developed under a contract with the Biomedical Advanced Research
and Development Authority of the Office of the Assistant
Secretary for Preparedness and Response, U.S. Department of
Health and Human Services. Our strategic partnerships with
leading pharmaceutical and biotechnology companies allow us to
leverage our strengths and gain access to sales and marketing
infrastructure, as well as complementary technologies. Some of
these partnerships provide us with licensing or other fees,
clinical development cost-sharing, milestone payments and rights
to royalty payments as products are developed and
commercialized. In some cases, we are entitled to certain
commercialization, co-promotion, revenue-sharing and other
product rights.
Critical
Accounting Policies and the Use of Estimates
A “critical accounting policy” is one that is both
important to the portrayal of our financial condition and
results of operations and that requires management’s most
difficult, subjective or complex judgments. Such judgments are
often the result of a need to make estimates about the effect of
matters that are inherently uncertain. The preparation of our
financial statements in conformity with accounting principles
generally accepted in the United States requires management to
make estimates and assumptions that affect the reported amounts
of assets and liabilities and disclosure of contingent assets
and liabilities at the date of the financial statements and the
reported amounts of revenues and expenses during the reporting
period. Actual results could differ materially from those
estimates. See Note B, Summary of Significant Accounting
Policies, of the Notes to the Consolidated Financial Statements
for further discussion.
We currently believe the following accounting policies to be
critical:
Investments. We account for investments in
accordance with the Financial Accounting Standards Board
(“FASB”) Accounting Standards Codification (“FASB
ASC”) Topic 320, Investments — Debt and Equity
Securities. We carry our investments at their respective
fair values. We periodically evaluate the fair values of our
investments to determine whether any declines in the fair value
of investments represent an
other-than-temporary
impairment. This evaluation consists of a review of several
factors, including but not limited to the length of time and
extent that a security has been in an unrealized loss position,
the existence of an event that would impair the issuer’s
future repayment potential, the near term prospects for recovery
of the market value of a security and our intent to hold the
security until the market value recovers, which may be maturity.
We also evaluate whether it is more likely than not that we will
be required to sell the security before its anticipated
recovery. If management determines that such an impairment
exists we would recognize an impairment charge. Because we may
determine that market or business conditions may lead us to sell
a short-term investment or marketable security prior to
maturity, we classify our short-term investments and marketable
securities as
“available-for-sale.”
Investments in securities that are classified as
available-for-sale
and have readily determinable fair values are measured at fair
market value in the balance sheets, and unrealized holding gains
and losses for these investments are reported as a separate
component of stockholders’ equity until realized, or an
other-than-temporary
impairment is recorded. We classify those marketable securities
that may be used in operations within one year as short-term
investments. Those marketable securities in which we have both
the ability to hold until maturity and have a maturity date
beyond one year from our most recent consolidated balance sheet
date are classified as non-current marketable securities.
For investments carried at fair value, we disclose the level
within the fair value hierarchy as prescribed by FASB ASC Topic
820, Fair Value Measurements and Disclosures. We evaluate
the types of securities in our investment portfolio to determine
the proper classification in the fair value hierarchy based on
trading activity and the observability of market inputs. We
generally obtain a single quote or price per instrument from
independent third parties to help us determine the fair value of
securities in Level 1 and Level 2 of the fair value
hierarchy.
38
Critical
Accounting Policies and the Use of Estimates
(continued)
Inventories. Inventories, which are recorded
at the lower of cost or market, include materials, labor and
other direct and indirect costs and are valued using the
first-in,
first-out method. The Company capitalizes inventories produced
in preparation for product launches when the related product
candidates are considered likely to receive regulatory approval
and it is probable that the related costs will be recoverable
through the commercialization of the product.
Leases. We lease various real properties under
operating leases that generally require us to pay taxes,
insurance and maintenance. During 2006, we entered into a
20-year
lease agreement with BioMed Realty Trust, Inc.
(“BioMed”) for our Traville facility. We account for
the Traville lease with BioMed as an operating lease.
During 2006 and as described further in Note F, Long-Term
Debt, of the Notes to the Consolidated Financial Statements, we
sold our large-scale manufacturing facility (“LSM”)
and headquarters land to BioMed, and simultaneously agreed to
lease such assets back over 20 years. We recorded the sale
and leaseback of these assets as a financing transaction and
accordingly recorded the allocated sale proceeds as outstanding
debt on our balance sheet. We account for lease payments under
the related lease agreements as principal and interest payments
on this debt.
Impairments of long-lived assets. Long-lived
assets to be held and used are reviewed for impairment whenever
events or changes in circumstances indicate that the carrying
amount of the assets might not be recoverable. Conditions that
would necessitate an impairment assessment include a significant
decline in the observable market value of an asset, a
significant change in the extent or manner in which an asset is
used, or a significant adverse change that would indicate that
the carrying amount of an asset or group of assets is not
recoverable. Determination of recoverability is based on an
estimate of undiscounted cash flows resulting from the use of
the asset and its eventual disposition. In the event that such
cash flows are not expected to be sufficient to recover the
carrying amount the assets, the assets are written down to their
estimated fair values. Long-lived assets to be sold are carried
at fair value less costs to sell.
Product sales. Revenue from product sales is
recognized when persuasive evidence of an arrangement exists,
title to product and associated risk of loss has passed to the
customer, the price is fixed or determinable, collection from
the customer is reasonably assured, and we have no further
performance obligations.
Manufacturing and development services. We
have entered into agreements for manufacturing process
development, clinical and commercial supply of certain
biopharmaceutical products. Revenue under these agreements is
recognized as services are performed or products delivered,
depending on the nature of the work contracted, using a
proportional performance method of accounting. Performance is
assessed using output measures such as
units-of-work
performed to date as compared to total
units-of-work
contracted. Advance payments received in excess of amounts
earned are classified as deferred revenue until earned.
Research and development collaborative
agreements. Our revenue recognition policies for
all non-refundable up-front license fees and milestone
arrangements are in accordance with the guidance provided in
FASB ASC Topic 605, Revenue Recognition. FASB ASC Topic
605 provides guidance on when an arrangement that involves
multiple revenue-generating activities or deliverables should be
divided into separate units of accounting for revenue
recognition purposes, and if this division is required, how the
arrangement consideration should be allocated among the separate
units of accounting. If the deliverables in a revenue
arrangement constitute separate units of accounting according to
FASB ASC Topic 605’s separation criteria, the revenue
recognition policy must be determined for each identified unit.
If the arrangement is a single unit of accounting, the revenue
recognition policy must be determined for the entire
arrangement. Under arrangements where the license fees and
research and development activities cannot be accounted for as
separate units of accounting, non-refundable up-front license
fees are deferred and recognized as revenue on a straight-line
basis over the expected term of our continued involvement in the
research and development process. Revenues from the achievement
of research and development milestones, if deemed substantive,
are recognized as revenue when the milestones are achieved, and
the milestone payments are due and collectible. If not deemed
substantive, we would recognize such milestones as revenue on a
straight-line basis over the remaining expected term of
continued involvement in the research and development process.
Milestones are considered substantive if all of the following
conditions are met: (1) the milestone is non-refundable;
39
Critical
Accounting Policies and the Use of Estimates
(continued)
(2) achievement of the milestone was not reasonably assured
at the inception of the arrangement; (3) substantive effort
is involved to achieve the milestone; and, (4) the amount
of the milestone appears reasonable in relation to the effort
expended, the other milestones in the arrangement and the
related risk associated with the achievement of the milestone
and any ongoing research and development or other services are
priced at fair value. Payments received in advance of work
performed are recorded as deferred revenue.
The up-front license fee and expense reimbursement received in
2006 from Novartis in connection with ZALBIN along with
subsequently received milestones were recognized ratably over
the estimated four-year clinical development period ending in
2010. Our initial payment from GSK in connection with BENLYSTA
was recognized ratably over the estimated four-year clinical
development period ending in 2010. Our up-front license fee with
GSK in connection with albiglutide is being recognized ratably
over the estimated eight-year clinical development period ending
in 2012. Our revenues from other research and development
collaborative agreements in 2010, 2009 and 2008 have generally
been recognized in full upon receipt, as we have met the
criteria for recognition.
Research and Development. Research and
development expenses primarily include related salaries, outside
services, materials and supplies and allocated facility costs.
Such costs are charged to research and development expense as
incurred. Our drug development expenses include accruals for
clinical site and clinical research organization
(“CRO”) costs. Estimates of the incurred to date but
not yet received invoices must be made for clinical site and CRO
costs in determining the accrued balance in any accounting
period. Reimbursement of research and development expenses
received in connection with collaborative cost-sharing
agreements is recorded as a reduction of such expenses.
Stock Compensation. We have a stock incentive
plan (the “Incentive Plan”) under which options to
purchase shares of our common stock may be granted to employees,
consultants and directors at a price no less than the quoted
market value on the date of grant. The Incentive Plan also
provides for awards in the form of stock appreciation rights,
restricted (non-vested) or unrestricted stock awards,
stock-equivalent units or performance-based stock awards.
We account for share-based awards to employees and non-employee
directors pursuant to FASB ASC Topic 718,
Compensation — Stock Compensation, which
requires that compensation cost resulting from share-based
payment transactions be recognized in the financial statements
at fair value over the service period. The amount of
compensation expense recognized using the fair value method
requires us to exercise judgment and make assumptions relating
to the factors that determine the fair value of our stock option
grants. We use the Black-Scholes-Merton model to estimate the
fair value of our option grants. The fair value calculated by
this model is a function of several factors, including grant
price, the risk-free interest rate, the estimated term of the
option and the estimated future volatility of the option. The
estimated term and estimated future volatility of the options
require our judgment.
Exit Accruals. In 2006, we exited certain
facilities, including certain headquarters space (“Wing
C”), which required us to make significant estimates in
several areas including the realizable values of assets deemed
redundant or excess and the ability to generate sublease income.
We evaluate these estimates on a quarterly basis and adjust the
accrued exit expenses if there is a material change in the
estimates used or if there is a material change in the facts and
circumstances relating to the accrual.
Income taxes. Deferred tax assets and
liabilities are determined based on temporary differences
between the financial reporting basis and the tax basis of
assets and liabilities. These deferred tax assets and
liabilities are measured using the enacted tax rates and laws
that will be in effect when such amounts are expected to reverse
or be utilized. The realization of total deferred tax assets is
contingent upon the generation of future taxable income. A
valuation allowance is provided to reduce such deferred tax
assets to amounts more likely than not to be ultimately realized.
In determining the effective income tax rate, we analyze various
factors, including projections of our annual earnings and taxing
jurisdictions in which the earnings will be generated, the
impact of state and local and foreign income taxes and our
ability to use tax incentives. We file income tax returns in
U.S. federal, state and foreign
40
Critical
Accounting Policies and the Use of Estimates
(continued)
jurisdictions. Our income taxes have not been subject to
examination by any tax jurisdictions since its inception.
Accordingly, all our filed income tax returns are subject to
examination by the taxing jurisdictions.
Results
of Operations
Years Ended December 31, 2010 and 2009
Revenues. We had revenues of
$157.4 million and $275.7 million for 2010 and 2009,
respectively. Revenues for 2010 consisted primarily of
$82.8 million recognized from Novartis primarily relating
to recognition of all remaining unrecognized up-front license
fees and milestones due to the decision to end further
development of ZALBIN, as well as $47.2 million in
raxibacumab product sales and $21.3 million from contract
manufacturing services. Revenues for 2009 consisted primarily of
$154.1 million in raxibacumab product sales,
$54.2 million recognized from Novartis related to
straight-line recognition of up-front license fees and
milestones reached for ZALBIN, $26.1 million related to
raxibacumab development services and $24.4 million from
contract manufacturing services.
Cost of sales. Cost of sales includes both
cost of product sales of $29.9 million and cost of
manufacturing and development services of $15.0 million for
2010. Cost of sales includes both cost of product sales of
$15.8 million and cost of manufacturing and development
services of $18.2 million for 2009. Cost of product sales
for 2010 includes the cost of manufacturing raxibacumab and
royalties whereas the cost of product sales for 2009 included
only royalties, as the manufacturing costs had been previously
expensed. Cost of product sales for 2010 also includes amounts
expensed related to rejected or terminated production batches.
Our manufacturing and development services costs include costs
associated with contract manufacturing services and raxibacumab
development services costs. The decrease in manufacturing and
development services costs is primarily due to reduced contract
manufacturing activities.
Expenses. Research and development expenses
were $196.4 million for 2010 as compared to
$173.7 million for 2009. Our research and development
expenses for 2010 are net of $61.6 million of costs
primarily reimbursed by GSK and Novartis. Our research and
development expenses for 2009 are net of $44.0 million of
costs reimbursed by GSK and Novartis.
We track our research and development expenditures by type of
cost incurred — research, pharmaceutical sciences,
manufacturing and clinical development costs.
Our research costs amounted to $23.2 million for 2010 as
compared to $19.0 million for 2009. This increase is
primarily due to activity related to HGS1029 and new target
development. Our research costs for 2010 and 2009 are net of
$2.9 million and $3.2 million, respectively, of cost
reimbursement primarily from GSK and Novartis under cost sharing
provisions in our collaboration agreements.
Our pharmaceutical sciences costs, where we focus on improving
formulation, process development and production methods,
decreased to $27.8 million for 2010 from $31.4 million
for 2009. This decrease is primarily due to decreased activity
related to contract manufacturing services and ZALBIN, partially
offset by increased BENLYSTA activity. Pharmaceutical sciences
costs for 2010 include $0.6 million of net costs incurred
by GSK and Novartis. Pharmaceutical sciences costs for 2009 are
net of $0.5 million of cost reimbursement from GSK and
Novartis under cost sharing provisions in our collaboration
agreements.
Our manufacturing costs increased to $80.2 million for 2010
from $49.0 million for 2009. This increase is primarily due
to increased BENLYSTA production partially offset by decreased
ZALBIN activity and capitalization of raxibacumab manufacturing
costs in 2010. Our manufacturing costs for 2010 and 2009 are net
of $43.4 million and $9.7 million, respectively, of
anticipated cost reimbursement from GSK and Novartis under the
cost sharing provisions in our collaboration agreements.
Manufacturing costs incurred to produce BENLYSTA prior to the
FDA Advisory Committee vote to recommend approval were expensed
as incurred.
Our clinical development costs decreased to $65.2 million
for 2010 from $74.3 million for 2009. The decrease is
primarily due to the completion of our first BENLYSTA Phase 3
clinical trial in 2009, completion of our second Phase 3
BENLYSTA clinical trial in March 2010 and decreased raxibacumab
development activities. Our clinical
41
Results
of Operations (continued)
Years Ended December 31, 2010 and 2009 (continued)
development costs for 2010 and 2009 are net of
$15.9 million and $30.5 million, respectively, of cost
reimbursement from GSK and Novartis under cost sharing
provisions in our collaboration agreements.
The research and development expenditures noted above are
categorized by functional area. We evaluate and prioritize our
activities according to functional area, rather than on a
per-project basis. For this reason, we do not maintain a formal
accounting system that captures or allocates all costs, both
direct and indirect, on a per-project basis. Therefore, we do
not believe that our available
project-by-project
information would form a reasonable basis for disclosure to
investors.
General and administrative expenses increased to
$106.8 million for 2010 from $61.1 million for 2009.
This increase is primarily due to increased commercial readiness
activities, such as market research and additional personnel
including our sales force, which was hired in late 2010. General
and administrative expenses in future periods are likely to
increase as the level of commercial activities rises.
Facility-related exit charges of $0.8 million for 2009
relate to an adjustment to our exit reserve for Wing C. See
Note J, Facility-Related Exit Charges, of the Notes to the
Consolidated Financial Statements for additional discussion.
Investment income increased to $17.0 million for 2010 from
$12.7 million for 2009. The increase is primarily due to
higher average investment balances partially offset by lower
yields on our portfolio. Investment income also includes
realized net losses on our short-term investments, marketable
securities and restricted investments of $0.3 million for
2010 as compared to net losses of $0.8 million for 2009.
The yield on our investments was approximately 1.6% for 2010, as
compared to approximately 2.5% for 2009. A general decline in
interest rates may adversely affect the interest earned from our
portfolio as securities mature and may be replaced with
securities having a lower interest rate.
Interest expense increased to $59.5 million for 2010
compared to $58.4 million for 2009. Interest expense
includes non-cash interest expense related to amortization of
debt discount of $23.1 million and $21.9 million for
2010 and 2009, respectively, as a result of the adoption of FASB
ASC 470 which requires that the liability and equity
components of convertible debt instruments that may be settled
in cash upon conversion (including partial cash settlement) be
separately accounted for in a manner that reflects an
issuer’s non-convertible debt borrowing rate.
The gain on extinguishment of debt of $38.9 million for
2009 relates to the repurchase of convertible subordinated debt
due in 2011 and 2012 with a face value of approximately
$106.2 million for an aggregate cost of approximately
$50.0 million plus accrued interest. The gain on
extinguishment of debt is net of write-offs of related debt
discount of $16.4 million and deferred financing charges of
$0.9 million.
The gain on sale of long-term equity investment for 2009 of
$5.3 million relates to the 2008 sale of our investment in
CoGenesys, Inc. (“CoGenesys”). The agreement between
CoGenesys and Teva Pharmaceutical Industries Ltd.
(“Teva”) provided for an escrow of a portion of the
purchase price. We received the final payment for our equity
investment during 2009 and recorded a gain of $5.3 million
with respect to this escrow.
We recorded other income of $0.1 million for 2010, and
other expense of $0.2 million for 2009.
Income Tax Benefit. Income tax benefit of
$1.3 million for 2009 represents a credit received in 2009
of $0.5 million for 2008 and an accrued income tax benefit
of $0.8 million for 2009. We elected to accelerate
recognition of research and development tax credits by electing
out of bonus depreciation pursuant to regulations passed in 2008.
Net Income (Loss). We recorded a net loss of
$233.2 million, or $1.24 per basic and diluted share, for
2010, compared to net income of $5.7 million, or $0.04 per
share, for 2009. The change from net income to net loss is
primarily due to lower product sales and manufacturing and
development services revenue in 2010 versus 2009 and
42
Results
of Operations (continued)
Years Ended December 31, 2010 and 2009 (continued)
the gain on extinguishment of debt in 2009, partially offset by
increased revenue recognized from the Novartis agreement in 2010.
Years
Ended December 31, 2009 and 2008
Revenues. We had revenues of
$275.7 million and $48.4 million for 2009 and 2008,
respectively. Revenues for 2009 consisted primarily of
$154.1 million in raxibacumab product sales,
$26.1 million related to raxibacumab development services,
$24.4 million from contract manufacturing services and
$54.2 million recognized from Novartis related to
straight-line recognition of up-front license fees and
milestones reached for ZALBIN. Revenues for 2008 consisted
primarily of $35.4 million recognized from Novartis related
to straight-line recognition of
up-front
license fees and milestones reached for ZALBIN and
$6.5 million recognized from GSK related to straight-line
recognition of an up-front fee for BENLYSTA. Revenue recognized
from Novartis increased in 2009 due to receipt of a
$75.0 million milestone which was being recognized over the
remaining development period, ending in the fall of 2010.
Cost of sales. Cost of sales includes both
cost of product sales of $15.8 million and cost of
manufacturing and development services of $18.2 million for
2009. No cost of sales were incurred in 2008 as we had no
revenue from product sales or manufacturing and development
services. With respect to the initial 2006 order for
raxibacumab, we incurred substantially all of the product and
service costs prior to 2009, and expensed these costs as
incurred. We incurred royalty costs associated with the initial
order during 2009, which are included in cost of product sales.
In addition, we have recorded as cost of product sales the
expenses associated with manufacturing additional raxibacumab
incurred prior to receiving the follow-on order in July 2009.
Our manufacturing and development service costs include
raxibacumab development service costs incurred in 2009 and costs
associated with contract manufacturing services. After approval
of a product, inventoriable costs are capitalized into inventory
and will be expensed as the inventory is sold.
Expenses. Research and development expenses
were $173.7 million for 2009 as compared to
$243.3 million for 2008. Our research and development
expenses for 2009 are net of $0.9 million and
$43.1 million of costs reimbursed by Novartis and GSK,
respectively. Our research and development expenses for 2008 are
net of $36.1 million and $51.8 million of costs
reimbursed by Novartis and GSK, respectively.
We track our research and development expenditures by type of
cost incurred — research, pharmaceutical sciences,
manufacturing and clinical development costs.
Our research costs amounted to $19.0 million for 2009 as
compared to $25.6 million for 2008. This decrease is
primarily due to the conclusion of animal studies being
conducted for raxibacumab in 2008, and a $5.0 million
milestone payment made to Aegera Therapeutics, Inc.
(“Aegera”) in 2008. Our research costs for 2009 and
2008 are net of $3.2 million and $2.4 million,
respectively, of cost reimbursement from Novartis and GSK under
cost sharing provisions in our collaboration agreements.
Our pharmaceutical sciences costs, where we focus on improving
formulation, process development and production methods,
decreased to $31.4 million for 2009 from $35.9 million
for 2008. This decrease is primarily due to decreased activity
related to raxibacumab and ZALBIN, partially offset by increased
activity related to contract manufacturing services.
Pharmaceutical sciences costs for 2009 and 2008 are net of
$0.5 million and $1.2 million, respectively, of cost
reimbursement from Novartis and GSK under cost sharing
provisions in our collaboration agreements.
Our manufacturing costs decreased to $49.0 million for 2009
from $77.1 million for 2008. This decrease is primarily due
to capitalizing raxibacumab production costs beginning in 2009
and decreased production of raxibacumab, ZALBIN and BENLYSTA,
partially offset by increased manufacturing services activities.
Our manufacturing costs for 2009 and 2008 are net of
$9.7 million and $19.9 million, respectively, of
anticipated cost reimbursement from Novartis and GSK under the
commercial cost sharing provisions in our collaboration
agreements.
43
Results
of Operations (continued)
Years Ended December 31, 2009 and 2008 (continued)
Our clinical development costs decreased to $74.3 million
for 2009 from $104.7 million for 2008. This decrease is
primarily due to the substantial completion of our ZALBIN Phase
3 clinical trials in late 2008, completion of the first Phase 3
BENLYSTA clinical trial and wind down of the second Phase 3
BENLYSTA clinical trial during 2009 and decreased HGS-ETR1
clinical trial costs. Our clinical development costs for 2009
and 2008 are net of $30.5 million and $64.4 million,
respectively, of cost reimbursement from Novartis and GSK under
cost sharing provisions in our collaboration agreements.
The research and development expenditures noted above are
categorized by functional area. We evaluate and prioritize our
activities according to functional area, rather than on a
per-project basis. For this reason, we do not maintain a formal
accounting system that captures or allocates all costs, both
direct and indirect, on a per-project basis. Therefore, we do
not believe that our available
project-by-project
information would form a reasonable basis for disclosure to
investors.
General and administrative expenses increased to
$61.1 million for 2009 from $60.9 million for 2008.
This increase is primarily due to increased pre-commercial
launch activities, partially offset by decreased legal expenses
associated with our patents.
Facility-related exit charges of $0.8 million for 2009
relate to an adjustment to our exit reserve for Wing C. Our Wing
C subtenant terminated its lease during 2009, which resulted in
an exit charge of $11.4 million during the second quarter
of 2009. During the fourth quarter of 2009, we decided to resume
production activities in most of Wing C and accordingly,
reversed $10.6 million of the $11.4 million charge,
resulting in a net exit charge of $0.8 million for the full
year. During 2008, we did not incur any facility-related exit
charges. See Note J, Facility-Related Exit Charges, of the
Notes to the Consolidated Financial Statements for additional
discussion.
Investment income decreased to $12.7 million for 2009 from
$23.5 million for 2008. The decrease is primarily due to
lower yields in 2009 as compared to 2008, partially offset by
higher average investment balances. Investment income also
includes realized net losses on our short-term investments,
marketable securities and restricted investments of
$0.8 million for 2009 as compared to net gains of
$0.9 million for 2008. The yield on our investments was
approximately 2.5% for 2009, as compared to approximately 4.6%
for 2008.
Interest expense decreased to $58.4 million for 2009
compared to $62.9 million for 2008. Interest expense
includes non-cash interest expense related to amortization of
debt discount of $21.9 million and $24.2 million for
2009 and 2008, respectively, as a result of the adoption of FASB
ASC Topic 470 which requires that the liability and equity
components of convertible debt instruments that may be settled
in cash upon conversion (including partial cash settlement) be
separately accounted for in a manner that reflects an
issuer’s non-convertible debt borrowing rate. The decrease
in interest expense is primarily due to the February 2009
repurchase of convertible subordinated debt due in 2011 and 2012.
The gain on extinguishment of debt of $38.9 million for
2009 relates to the repurchase of convertible subordinated debt
due in 2011 and 2012 with a face value of approximately
$106.2 million for an aggregate cost of approximately
$50.0 million plus accrued interest. The gain on
extinguishment of debt is net of write-offs of related debt
discount of $16.4 million and deferred financing charges of
$0.9 million.
The gain on sale of long-term equity investment for 2009 and
2008 of $5.3 million and $32.5 million respectively,
relates to the 2008 sale of our investment in CoGenesys. We
received initial proceeds in 2008 of $47.3 million. Our
cost basis in this investment was $14.8 million, resulting
in a gain of $32.5 million. The agreement between CoGenesys
and Teva provided for an escrow of a portion of the purchase
price. We received the final payment for our equity investment
during 2009 and recorded an additional gain of $5.3 million.
Other expense of $0.2 million for 2009 primarily represents
unrealized, non-cash foreign currency translation losses related
to our investment in Aegera, which is denominated in Canadian
dollars. Other expense of $6.3 million for 2008 was due to
an
other-than-temporary
impairment of our investment in debt securities issued by Lehman
44
Results
of Operations (continued)
Years Ended December 31, 2009 and 2008 (continued)
Brothers Holdings, Inc. (“LBHI”). During 2008, LBHI
experienced a significant deterioration in its credit worthiness
and filed a petition under Chapter 11 of the
U.S. Bankruptcy Code.
Income Tax Benefit. Income tax benefit of
$1.3 million represents a credit received in 2009 of
$0.5 million for 2008 and an accrued income tax benefit of
$0.8 million for 2009. We elected to accelerate recognition
of research and development tax credits by electing out of bonus
depreciation pursuant to regulations passed in 2008.
Net Income (Loss). We recorded net income of
$5.7 million, or $0.04 per basic and diluted share, for
2009, compared to a net loss of $268.9 million, or $1.99
per share, for 2008. The improvement to net income from a net
loss is primarily due to 2009 activity including revenue from
raxibacumab, gain on extinguishment of debt and reduced research
and development expenses.
Liquidity
and Capital Resources
We had working capital of $250.2 million as of
December 31, 2010 compared to working capital of
$616.6 million as of December 31, 2009. The decrease
in our working capital is primarily due to the use of working
capital to fund our operations and the classification of
$188.6 million of our
21/4% Convertible
Subordinated Notes due 2011 as a current liability as of
December 31, 2010 compared to non-current as of
December 31, 2009.
We expect to continue to incur substantial expenses relating to
our research and development efforts, as we focus on clinical
trials and manufacturing required for the development of our
active product candidates. We will also continue to incur costs
related to our pre- and post-commercial launch activities, and
will incur significant sales and marketing costs if BENLYSTA is
approved. In the event our working capital needs exceed our
available working capital, we can utilize our non-current
marketable securities, which are classified as
“available-for-sale”.
In 2009, the USG agreed to purchase 45,000 additional doses of
raxibacumab for the SNS, to be delivered over a three-year
period, which began in 2009. We expect to receive a total of
approximately $142.0 million from this order as deliveries
are completed, $64.9 million of which was recognized as
revenue through 2010. We may also receive payments under
collaboration agreements, to the extent milestones are met,
which would further improve our working capital position. We
continue to evaluate our working capital position on an ongoing
basis.
To minimize our exposure to credit risk, we invest in securities
with strong credit ratings and have established guidelines
relative to diversification and maturity with the objectives of
maintaining safety of principal and liquidity. We do not invest
in derivative financial instruments or auction rate securities,
and we generally hold our investments in debt securities until
maturity.
The amounts of expenditures that will be needed to carry out our
business plan are subject to numerous uncertainties, which may
adversely affect our liquidity and capital resources. We have
completed our fourth Phase 3 trial and have several ongoing
Phase 1 and Phase 2 trials and expect to initiate additional
trials in the future. The duration and cost of our clinical
trials are a function of numerous factors such as the number of
patients to be enrolled in the trial, the amount of time it
takes to enroll them, the length of time they must be treated
and observed, and the number of clinical sites and countries for
the trial. However, the duration of these phases may vary
considerably according to the type, complexity, novelty and
intended use of the drug candidate. Some trials may take
considerably longer to complete.
45
Liquidity
and Capital Resources (continued)
Our clinical development expenses are impacted by the clinical
phase of our drug candidates. Our expenses increase as our drug
candidates move to later phases of clinical development. The
status of our clinical projects is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Clinical Trial Status
|
|
|
|
|
|
as of
December 31,(2)
|
|
Product Candidate
(1)
|
|
Indication
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
BENLYSTA
|
|
Systemic Lupus Erythematosus
|
|
Phase 3(3)
|
|
Phase 3
|
|
Phase 3
|
|
BENLYSTA
|
|
Rheumatoid Arthritis
|
|
(4)
|
|
Phase 2
|
|
Phase 2
|
|
Raxibacumab
|
|
Anthrax
|
|
(5)
|
|
(5)
|
|
(5)
|
|
HGS1029
|
|
Cancer
|
|
Phase 1
|
|
Phase 1
|
|
Phase 1
|
|
HGS-ETR1
|
|
Cancer
|
|
Phase 2
|
|
Phase 2
|
|
Phase 2
|
|
ZALBIN
|
|
Hepatitis C
|
|
(6)
|
|
Phase 3
|
|
Phase 3
|
|
HGS-ETR2
|
|
Cancer
|
|
(7)
|
|
(7)
|
|
Phase 1
|
|
|
|
|
(1) |
|
Includes only those candidates for which an Investigational New
Drug Application (“IND”) has been filed with the FDA. |
| |
|
(2) |
|
Clinical Trial Status defined as when patients are being dosed. |
| |
|
(3) |
|
Results from two Phase 3 clinical trials reported; MAA and BLA
filed in June 2010; BLA PDUFA target date of March 10, 2011. |
| |
|
(4) |
|
Phase 2 trial completed; treatment IND ongoing. |
| |
|
(5) |
|
BLA filed in 2009; Complete Response Letter received from FDA;
additional work ongoing. |
| |
|
(6) |
|
Further development discontinued. |
| |
|
(7) |
|
Ongoing Phase 1 trial by National Institutes of Health; further
development not anticipated. |
We identify our drug candidates by conducting numerous
preclinical studies. We may conduct multiple clinical trials to
cover a variety of indications for each drug candidate. Based
upon the results from our trials, we may elect to discontinue
clinical trials for certain indications or certain drugs in
order to concentrate our resources on more promising drug
candidates.
We are advancing a number of drug candidates, including
antibodies, and a small molecule, in part to diversify the risks
associated with our research and development spending. In
addition, our manufacturing plants have been designed to enable
multi-product manufacturing capability.
We must receive regulatory clearance to advance each of our
products into and through each phase of clinical testing.
Moreover, we must receive regulatory approval to launch any of
our products commercially. In order to receive such approval,
the appropriate regulatory agency must conclude that our
clinical data establish safety and efficacy and that our
products and the manufacturing facilities meet all applicable
regulatory requirements. We cannot be certain that we will
establish sufficient safety and efficacy data to receive
regulatory approval for any of our drugs or that our drugs and
the manufacturing facilities will meet all applicable regulatory
requirements.
Part of our business plan includes collaborating with others.
For example, we entered into a collaboration agreement with GSK
in 2006 with respect to BENLYSTA and received a payment of
$24.0 million. We and GSK share Phase 3 and 4 development
costs, and share sales and marketing expenses and profits of any
product that is commercialized in accordance with the
collaboration agreement. We also entered into a collaboration
agreement with Novartis in 2006. During 2010, we recorded
approximately $61.6 million due primarily from GSK and
Novartis with respect to our cost sharing agreements as a
reduction of research and development expenses. We recognized
the up-front fees and milestones received from GSK and Novartis
as revenue ratably over the estimated remaining development
periods. Based on the decision in September 2010 to end further
development of ZALBIN, all remaining deferred revenue related to
the payments received from Novartis was recognized. Furthermore,
we will not be receiving any of the remaining
$300.0 million in milestone payments from Novartis with
respect to ZALBIN.
46
Liquidity
and Capital Resources (continued)
We have collaborators who have sole responsibility for product
development. For example, GSK is developing other products under
separate agreements as part of our overall relationship with
them. We have no control over the progress of GSK’s
development plans. We cannot forecast with any degree of
certainty whether any of our current or future collaborations
will affect our drug development.
Because of the uncertainties discussed above, the costs to
advance our research and development projects are difficult to
estimate and may vary significantly. Our long-term strategy
includes the acquisition of other biotechnology companies or
in-license of additional product candidates to complement and
supplement our existing product pipeline, and we may acquire
such companies and in-license additional product candidates that
have demonstrated positive pre-clinical
and/or
clinical data. Any such acquisition or in-license agreement
could include consideration in the form of cash, which would
have an adverse effect on our liquidity. We expect that our
existing funds, payments received under the raxibacumab contract
and other agreements and investment income will be sufficient to
fund our operations for at least the next twelve months.
Our future capital requirements and the adequacy of our
available funds will depend on many factors, primarily including
the scope and costs of our clinical development programs, the
scope and costs of our manufacturing and process development
activities, the magnitude of our discovery and preclinical
development programs and the level of our pre- and
post-commercial launch activities. There can be no assurance
that any additional financing required in the future will be
available on acceptable terms, if at all.
Depending upon market and interest rate conditions, we may take
actions to strengthen further our financial position. We may
undertake financings and may repurchase or restructure some or
all of our outstanding convertible debt instruments in the
future depending upon market and other conditions. During 2009
we repurchased approximately $106.2 million of our
convertible subordinated debt due in 2011 and 2012 at a cost of
approximately $50.0 million plus accrued interest. In
August and December 2009 we completed public offerings of our
common stock, resulting in net cash proceeds of approximately
$812.9 million.
We have certain contractual obligations that may have a future
effect on our financial condition, changes in financial
condition, results of operations, liquidity, capital
expenditures or capital resources. Our operating leases, along
with our unconditional purchase obligations, are not recorded on
our balance sheets. Debt associated with the 2006 sale of our
LSM to BioMed and accompanying leaseback is recorded on our
balance sheet as of December 31, 2010 and 2009. We have an
option to purchase the Traville facility in 2016 for
$303.0 million. This is not reflected in the contractual
obligations table below because we are not obligated to exercise
this option.
47
Liquidity
and Capital Resources (continued)
Our contractual obligations as of December 31, 2010 are
summarized as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by Period
|
|
|
|
|
|
|
|
One Year
|
|
|
Two to
|
|
|
Four to
|
|
|
After
|
|
|
|
|
Total
|
|
|
or Less
|
|
|
Three Years
|
|
|
Five Years
|
|
|
Five Years
|
|
|
|
|
(dollars in millions)
|
|
|
|
|
Contractual Obligations
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Long-term debt — convertible
notes(1)
|
|
$
|
417.6
|
|
|
$
|
206.2
|
|
|
$
|
211.4
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
Long-term lease commitment —
BioMed(2)
|
|
|
446.5
|
|
|
|
25.0
|
|
|
|
51.5
|
|
|
|
53.6
|
|
|
|
316.4
|
|
|
Operating
leases(3)
|
|
|
346.6
|
|
|
|
20.9
|
|
|
|
42.8
|
|
|
|
44.4
|
|
|
|
238.5
|
|
|
Manufacturing source of
supply(4)
|
|
|
25.9
|
|
|
|
25.9
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Raxibacumab
royalties(5)
|
|
|
4.8
|
|
|
|
2.9
|
|
|
|
1.9
|
|
|
|
—
|
|
|
|
—
|
|
|
Other long-term liabilities reflected on our balance
sheets(6)
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total contractual cash
obligations(7)
|
|
$
|
1,241.4
|
|
|
$
|
280.9
|
|
|
$
|
307.6
|
|
|
$
|
98.0
|
|
|
$
|
554.9
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Contractual interest obligations related to our convertible
subordinated notes included above total $13.7 million as of
December 31, 2010. Contractual interest obligations of
$9.1 million, $4.6 million are due in one year or less
and two to three years, respectively. |
| |
|
(2) |
|
Contractual interest obligations related to BioMed are included
above and aggregate $393.5 million as of December 31,
2010. Contractual interest obligations of $25.0 million,
$51.5 million, $53.6 million and $263.4 million
are due in one year or less, two to three years, four to five
years and after five years, respectively. |
| |
|
(3) |
|
Includes Traville headquarters operating lease with BioMed with
aggregate payments of $325.5 million. Lease payments of
$18.2 million, $37.6 million, $39.1 million and
$230.6 million are due in one year or less, two to three
years, four to five years and after five years, respectively. |
| |
|
(4) |
|
Amounts relate to supply commitments with respect to BENLYSTA.
Under the terms of the contract we could be obligated to pay at
least an additional $88.0 million, however the contract
includes termination clauses and therefore these amounts have
been excluded from the table above. |
| |
|
(5) |
|
Includes royalties associated with the delivery of raxibacumab
to the U.S. Strategic National Stockpile. |
| |
|
(6) |
|
In the event we reach certain development milestones for
BENLYSTA or raxibacumab such as regulatory approval, we would be
obligated to make payments of up to $9.0 million over the
next five years. In the event we reach certain development
milestones related to HGS1029, we would be obligated to pay up
to $204.0 million. Our other products are in either Phase 1
or Phase 2 and would also obligate us to make certain milestone
payments should they reach Phase 3 or regulatory approval. These
other payments could result in aggregate milestone payments of
$14.5 million. Because we cannot forecast with any degree
of certainty whether any of these products will reach these
milestones, we have excluded these amounts and any royalty
payments, except for those royalty payments related to
raxibacumab sales under the current order from the USG, from the
above table. |
| |
|
(7) |
|
For additional discussion of our debt obligations and lease
commitments, see Note F, Long-Term Debt and Note G,
Commitments and Other Matters, of the Notes to the Consolidated
Financial Statements. |
As of December 31, 2010, we had net operating loss
carryforwards (“NOLs”) for federal income tax purposes
of approximately $1.8 billion, excluding approximately
$0.3 billion of stock-based compensation NOLs, which
expire, if unused, through December 31, 2030. We also have
available research and development tax credit and other tax
credit carryforwards of approximately $35.0 million, the
majority of which will expire, if unused, through
December 31, 2030.
Our unrestricted and restricted funds may be invested in
U.S. Treasury securities, government agency obligations,
high grade corporate debt securities and various money market
instruments rated “A−” or better.
48
Liquidity
and Capital Resources (continued)
Such investments reflect our policy regarding the investment of
liquid assets, which is to seek a reasonable rate of return
consistent with an emphasis on safety, liquidity and
preservation of capital.
Off-Balance
Sheet Arrangements
During 1997 and 1999, we entered into two long-term leases with
the Maryland Economic Development Corporation
(“MEDCO”) expiring January 1, 2019 for a
small-scale manufacturing facility aggregating
127,000 square feet and built to our specifications. We
have accounted for these leases as operating leases. The
facility was financed primarily through a combination of bonds
issued by MEDCO (“MEDCO Bonds”) and loans issued to
MEDCO by certain State of Maryland agencies. We have no equity
interest in MEDCO.
Rent is based upon MEDCO’s debt service obligations and
annual base rent under the leases is currently approximately
$2.6 million. The MEDCO Bonds are secured by letters of
credit issued for the account of MEDCO which were renewed in
December 2009. We are required to have restricted investments of
approximately $34.3 million which serve as security for the
MEDCO letters of credit reimbursement obligation. Upon default
or early lease termination, the MEDCO Bond indenture trustee can
draw upon the letters of credit to pay the MEDCO Bonds as they
are tendered. In such an event, we could lose part or all of our
restricted investments and could record a charge to earnings for
a corresponding amount. Alternatively, we have an option through
the end of the lease term to purchase this facility for an
aggregate amount that declines from approximately
$36.0 million in 2011 to approximately $21.0 million
in 2019.
49
Safe
Harbor Statement under the Private Securities Litigation Reform
Act of 1995
Certain statements contained in “Management’s
Discussion and Analysis of Financial Condition and Results of
Operations” are forward-looking statements within the
meaning of Section 27A of the Securities Act of 1933, as
amended, and Section 21E of the Securities Exchange Act of
1934, as amended. The forward-looking statements are based on
our current intent, belief and expectations. These statements
are not guarantees of future performance and are subject to
certain risks and uncertainties that are difficult to predict.
Actual results may differ materially from these forward-looking
statements because of our unproven business model, our
dependence on new technologies, the uncertainty and timing of
clinical trials, our ability to develop and commercialize
products, our dependence on collaborators for services and
revenue, our substantial indebtedness and lease obligations, our
changing requirements and costs associated with facilities,
intense competition, the uncertainty of patent and intellectual
property protection, our dependence on key management and key
suppliers, the uncertainty of regulation of products, the impact
of future alliances or transactions and other risks described in
this filing and our other filings with the Securities and
Exchange Commission. Existing and prospective investors are
cautioned not to place undue reliance on these forward-looking
statements, which speak only as of today’s date. We
undertake no obligation to update or revise the information
contained in this Annual Report on
Form 10-K
whether as a result of new information, future events or
circumstances or otherwise.
50
|
|
|
ITEM 7A.
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
We do not currently have operations of a material nature that
are subject to risks of foreign currency fluctuations. We do not
use derivative financial instruments in our operations or
investment portfolio. Our investment portfolio may be comprised
of low-risk U.S. Treasuries, government-sponsored
enterprise securities, high-grade debt having at least an
“A−” rating at time of purchase and various
money market instruments. The short-term nature of these
securities, which currently have an average term of
approximately 13 months, decreases the risk of a material
loss caused by a market change related to interest rates.
We believe that a hypothetical 100 basis point adverse move
(increase) in interest rates along the entire interest rate
yield curve would adversely affect the fair value of our cash,
cash equivalents, short-term investments, marketable securities
and restricted investments by approximately $10.2 million,
or approximately 1.1% of the aggregate fair value of
$933.4 million, as of December 31, 2010. For these
reasons, and because these securities are generally held to
maturity, we believe we do not have significant exposure to
market risks associated with changes in interest rates related
to our debt securities held as of December 31, 2010. We
believe that any interest rate change related to our investment
securities held as of December 31, 2010 is not material to
our consolidated financial statements. As of December 31,
2010, the yield on comparable one-year investments was
approximately 0.3%, as compared to our current portfolio yield
of approximately 1.7%. However, given the short-term nature of
these securities, a general decline in interest rates may
adversely affect the interest earned from our portfolio as
securities mature and may be replaced with securities having a
lower interest rate.
To minimize our exposure to credit risk, we invest in securities
with strong credit ratings and have established guidelines
relative to diversification and maturity with the objectives of
maintaining safety of principal and liquidity. We do not invest
in derivative financial instruments, auction rate securities,
loans held for sale or mortgage-backed securities backed by
sub-prime or
Alt-A collateral, and we generally hold our investments in debt
securities until maturity. However, adverse changes in the
credit markets relating to credit risks would adversely affect
the fair value of our cash, cash equivalents, short-term
investments, marketable securities and restricted investments.
Our facility leases for our Traville headquarters and LSM
require us to maintain minimum levels of restricted investments
of approximately $39.8 million, or $39.5 million if in
the form of cash, as collateral for these facilities. Together
with the requirement to maintain approximately
$34.3 million in restricted investments with respect to our
small-scale manufacturing facility leases, our overall level of
restricted investments is currently required to be approximately
$74.1 million. Although the market value for these
investments may rise or fall as a result of changes in interest
rates, we will be required to maintain this level of restricted
investments in either a rising or declining interest rate
environment.
Our convertible subordinated notes bear interest at fixed rates.
As a result, our interest expense on these notes is not affected
by changes in interest rates.
Our wholly-owned subsidiary, Human Genome Sciences Europe GmbH
(“HGS Europe”) assists in our clinical trials and
clinical research collaborations in European countries. Although
HGS Europe’s activities are denominated primarily in euros,
we believe the foreign currency fluctuation risks to be
immaterial to our operations as a whole. Our wholly-owned
subsidiary, Human Genome Sciences Pacific Pty Ltd. (“HGS
Pacific”) sponsors some of our clinical trials in the
Asia/Pacific region. We currently do not anticipate HGS Pacific
to have any operational activity and therefore we do not believe
we will have any foreign currency fluctuation risks with respect
to HGS Pacific. During 2010 we formed additional European
subsidiaries in preparation for commercial activity in that
region. The formation of these subsidiaries slightly increases
our exposure to foreign currency fluctuation risks, as they are
predominantly denominated in euros. We do not believe the risk
of foreign currency gains or losses to be material to our
overall operations.
51
|
|
|
ITEM 8.
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
The information required by this item is set forth on pages
F-1 — F-37.
|
|
|
ITEM 9.
|
CHANGES
AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
|
None.
|
|
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES
|
Disclosure
Controls and Procedures
Our management, including our principal executive and principal
financial officers, has evaluated the effectiveness of our
disclosure controls and procedures as of December 31, 2010.
Our disclosure controls and procedures are designed to provide
reasonable assurance that the information required to be
disclosed in this annual report on
Form 10-K
has been appropriately recorded, processed, summarized and
reported within the time periods specified in the Securities and
Exchange Commission’s rules and forms, and that such
information is accumulated and communicated to our management,
including our principal executive and principal financial
officers, to allow timely decisions regarding required
disclosure. Based on that evaluation, our principal executive
and principal financial officers have concluded that our
disclosure controls and procedures are effective at the
reasonable assurance level.
Changes
in Internal Control
Our management, including our principal executive and principal
financial officers, has evaluated any changes in our internal
control over financial reporting that occurred during the
quarterly period ended December 31, 2010, and has concluded
that there was no change that occurred during the quarterly
period ended December 31, 2010 that has materially
affected, or is reasonably likely to materially affect, our
internal control over financial reporting.
Management
Report on Internal Control over Financial Reporting
The management of the Company is responsible for establishing
and maintaining adequate internal control over financial
reporting. Internal control over financial reporting is defined
in
Rule 13a-15(f)
or 15d-15(f)
promulgated under the Securities Exchange Act of 1934, as
amended, as a process designed by, or under the supervision of,
the Company’s principal executive and principal financial
officers and effected by the Company’s board of directors,
management and other personnel, to provide reasonable assurance
regarding the reliability of financial reporting and the
preparation of financial statements for external purposes in
accordance with generally accepted accounting principles and
includes those policies and procedures that:
|
|
|
| |
•
|
pertain to the management of records that in reasonable detail
accurately and fairly reflect the transactions and dispositions
of the assets of the Company;
|
| |
| |
•
|
provide reasonable assurance that transactions are recorded as
necessary to permit preparation of financial statements in
accordance with generally accepted accounting principles, and
that receipts and expenditures of the Company are being made
only in accordance with authorizations of management and
directors of the Company; and
|
| |
| |
•
|
provide reasonable assurance regarding prevention or timely
detection of unauthorized acquisition, use or disposition of the
Company’s assets that could have a material effect on the
financial statements.
|
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Projections of any evaluation of effectiveness to future periods
are subject to the risk that controls may become inadequate
because of changes in conditions, or that the degree of
compliance with the policies or procedures may deteriorate. All
internal control systems, no matter how well designed, have
inherent limitations. Therefore, even those systems determined
to be effective can provide only reasonable assurance with
respect to financial statement preparation and presentation.
The Company’s management assessed the effectiveness of the
Company’s internal control over financial reporting as of
December 31, 2010. In making this assessment, the
Company’s management used the criteria set
52
|
|
|
ITEM 9A.
|
CONTROLS
AND PROCEDURES (continued)
|
forth by the Committee of Sponsoring Organizations of the
Treadway Commission (COSO) in Internal Control-Integrated
Framework.
Based on our assessment, management believes that, as of
December 31, 2010, the Company’s internal control over
financial reporting is effective based on those criteria.
The Company’s independent auditors have issued an audit
report on internal control over financial reporting which
follows herein.
|
|
|
ITEM 9B.
|
OTHER
INFORMATION
|
None.
53
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Board of Directors and Stockholders of
Human Genome Sciences, Inc.
Rockville, Maryland
We have audited Human Genome Sciences Inc.’s internal
control over financial reporting as of December 31, 2010,
based on criteria established in Internal Control —
Integrated Framework issued by the Committee of Sponsoring
Organizations of the Treadway Commission (the COSO criteria).
Human Genome Sciences, Inc.’s management is responsible for
maintaining effective internal control over financial reporting,
and for its assessment of the effectiveness of internal control
over financial reporting included in the accompanying
“Management Report on Internal Control over Financial
Reporting.” Our responsibility is to express an opinion on
the company’s internal control over financial reporting
based on our audit.
We conducted our audit in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether effective internal control
over financial reporting was maintained in all material
respects. Our audit included obtaining an understanding of
internal control over financial reporting, assessing the risk
that a material weakness exists, testing and evaluating the
design and operating effectiveness of internal control based on
the assessed risk, and performing such other procedures as we
considered necessary in the circumstances. We believe that our
audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a
process designed to provide reasonable assurance regarding the
reliability of financial reporting and the preparation of
financial statements for external purposes in accordance with
generally accepted accounting principles. A company’s
internal control over financial reporting includes those
policies and procedures that (1) pertain to the maintenance
of records that, in reasonable detail, accurately and fairly
reflect the transactions and dispositions of the assets of the
company; (2) provide reasonable assurance that transactions
are recorded as necessary to permit preparation of financial
statements in accordance with generally accepted accounting
principles, and that receipts and expenditures of the company
are being made only in accordance with authorizations of
management and directors of the company; and (3) provide
reasonable assurance regarding prevention or timely detection of
unauthorized acquisition, use, or disposition of the
company’s assets that could have a material effect on the
financial statements.
Because of its inherent limitations, internal control over
financial reporting may not prevent or detect misstatements.
Also, projections of any evaluation of effectiveness to future
periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree
of compliance with the policies or procedures may deteriorate.
In our opinion, Human Genome Sciences, Inc. maintained, in all
material respects, effective internal control over financial
reporting as of December 31, 2010, based on the COSO
criteria.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), the
consolidated balance sheets of Human Genome Sciences, Inc. as of
December 31, 2010 and 2009, and the related consolidated
statements of operations, stockholders’ equity (deficit),
and cash flows for each of the three years in the period ended
December 31, 2010 and our report dated February 24,
2011 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Baltimore, Maryland
February 24, 2011
54
PART III
|
|
|
ITEM 10.
|
DIRECTORS,
EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
We incorporate herein by reference the relevant information
concerning directors, executive officers and corporate
governance to be included in our definitive proxy statement for
the 2011 annual meeting of stockholders (the “2011 Proxy
Statement”).
|
|
|
ITEM 11.
|
EXECUTIVE
COMPENSATION
|
We incorporate herein by reference the relevant information
concerning executive compensation to be included in the 2011
Proxy Statement.
|
|
|
ITEM 12.
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS
|
We incorporate herein by reference the relevant information
concerning security ownership of certain beneficial owners and
management to be included in the 2011 Proxy Statement.
|
|
|
ITEM 13.
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR
INDEPENDENCE
|
We incorporate herein by reference the relevant information
concerning certain other relationships and related transactions
to be included in the 2011 Proxy Statement.
|
|
|
ITEM 14.
|
PRINCIPAL
ACCOUNTING FEES AND SERVICES
|
We incorporate herein by reference the relevant information
concerning principal accounting fees and services to be included
in the 2011 Proxy Statement.
55
PART IV
|
|
|
ITEM 15.
|
EXHIBITS AND
FINANCIAL STATEMENT SCHEDULES
|
(a) The following documents are filed as part of this
Annual Report:
(1) Index to Consolidated Financial Statements
| |
|
|
|
|
|
|
|
Page
|
|
|
|
Number
|
|
|
|
|
|
|
F-2
|
|
|
|
|
|
F-3
|
|
|
|
|
|
F-4
|
|
|
|
|
|
F-5
|
|
|
|
|
|
F-6
|
|
|
|
|
|
F-8
|
|
(2) Financial Statement Schedules
Financial statement schedules are omitted because they are not
required.
(3) Exhibits
| |
|
|
|
Exhibit No.
|
|
Description
|
|
|
|
3.1
|
|
Certificate of Incorporation (incorporated by reference to
Exhibit 3.1 to the Company’s Annual Report on
Form 10-K,
filed on March 31, 1994; Exhibit 3.3 to the
Company’s Annual Report on
Form 10-K/A,
filed on April 3, 1998; Exhibit 3.1 to the
Company’s Current Report on
Form 8-K,
filed on December 16, 1999; Exhibit 3.1 to the
Company’s Quarterly Report on
Form 10-Q,
filed on July 31, 2001; Exhibit 3.1 to the
Company’s Current Report on
Form 8-K,
filed on May 8, 2008; and Exhibit 3.2 to the
Company’s Current Report on
Form 8-K,
filed on May 24, 2010).
|
|
3.2
|
|
Amended and Restated By-laws (incorporated by reference to
Exhibit 3 to the Company’s Current Report on
Form 8-K,
filed on October 6, 2010).
|
|
4.1
|
|
Form of Common Stock Certificate (incorporated by reference to
Exhibit 4.2 to the Company’s Registration Statement on
Form S-3
(Registration
No. 333-45272),
filed on September 6, 2000).
|
|
4.2
|
|
Indenture, dated as of October 4, 2004, between the Company
and The Bank of New York, as trustee, including the form of
21/4% Convertible
Subordinated Note due 2011 (incorporated by reference to
Exhibit 4.1 to the Company’s Current Report on
Form 8-K,
filed on October 4, 2004).
|
|
4.3
|
|
Indenture, dated as of August 9, 2005, between the Company
and The Bank of New York, as trustee, including the form of
21/4% Convertible
Subordinated Note due 2012 (incorporated by reference to
Exhibit 4.1 to the Company’s Current Report on
Form 8-K,
filed on August 9, 2005).
|
|
10.1†
|
|
Co-development and Commercialization Agreement, dated as of
August 1, 2006, between Glaxo Group Limited and the Company
(incorporated by reference to Exhibit 10.25 to the
Company’s Annual Report on
Form 10-K,
filed on February 26, 2009).
|
|
10.2†*
|
|
Amendment No. 1, dated as of November 25, 2009, to
Co-development and Commercialization Agreement between Glaxo
Group Limited and the Company.
|
|
10.3†*
|
|
Amendment No. 2, dated as of October 5, 2010, to
Co-development and Commercialization Agreement between Glaxo
Group Limited and the Company.
|
|
10.4
|
|
Lease Agreement, dated as of December 1, 1997, between
Maryland Economic Development Corporation and the Company
(incorporated by reference to Exhibit 10.67 to the
Company’s Annual Report on
Form 10-K,
filed on March 31, 1998).
|
|
10.5
|
|
Amendment No. 1, dated as of December 1, 2009, to
Lease Agreement between Maryland Economic Development
Corporation and the Company (incorporated by reference to
Exhibit 10.10 to the Company’s Annual Report on
Form 10-K,
filed on March 2, 2010).
|
56
| |
|
|
|
Exhibit No.
|
|
Description
|
|
|
|
10.6
|
|
Amended and Restated Lease Agreement, dated as of
December 1, 2009, between Maryland Economic Development
Corporation and the Company (incorporated by reference to
Exhibit 10.12 to the Company’s Annual Report on
Form 10-K,
filed on March 2, 2010).
|
|
10.7
|
|
Purchase and Sale Agreement, dated as of May 2, 2006,
between BioMed Realty, L.P. and the Company (incorporated by
reference to Exhibit 10.2 to the Company’s Quarterly
Report on
Form 10-Q,
filed on August 9, 2006).
|
|
10.8
|
|
Lease Agreement, dated as of May 24, 2006, between
BMR-Belward Campus Drive LSM LLC and the Company (incorporated
by reference to Exhibit 10.3 to the Company’s
Quarterly Report on
Form 10-Q,
filed on August 9, 2006).
|
|
10.9
|
|
Lease Agreement, dated as of May 24, 2006, between
BMR-Shady Grove Road HQ LLC and the Company (incorporated by
reference to Exhibit 10.4 to the Company’s Quarterly
Report on
Form 10-Q,
filed on August 9, 2006).
|
|
10.10
|
|
Collateral Pledge Agreement, dated as of December 1, 2009,
among the Company, as Pledgor, Manufacturers and Traders
Trust Company, as Pledgee, and Manufacturers and Traders
Trust Company, as the Collateral Agent (incorporated by
reference to Exhibit 10.14 to the Company’s Annual
Report on
Form 10-K,
filed on March 2, 2010).
|
|
10.11
|
|
Reimbursement Agreement, dated as of December 1, 2009,
between the Company and Manufacturers and Traders
Trust Company, relating to 1997 Series Revenue Bonds
(incorporated by reference to Exhibit 10.11 to the
Company’s Annual Report on
Form 10-K,
filed on March 2, 2010).
|
|
10.12
|
|
Reimbursement Agreement, dated as of December 1, 2009,
between the Company and Manufacturers and Traders
Trust Company, relating to Series 1999 A and B Revenue
Bonds (incorporated by reference to Exhibit 10.13 to the
Company’s Annual Report on
Form 10-K,
filed on March 2, 2010).
|
|
10.13†
|
|
Solicitation (as amended) and Modification of Contract, dated
June 24, 2006, awarded by the U.S. Department of Health and
Human Services, Biomedical Advanced Research and Development
Authority to the Company (incorporated by reference to
Exhibit 10.5 to the Company’s Quarterly Report on
Form 10-Q/A,
filed on September 27, 2007).
|
|
10.14†
|
|
Amendment of Solicitation/Modification of Contract, dated
July 17, 2009, awarded by the U.S. Department of Health and
Human Services, Biomedical Advanced Research and Development
Authority to the Company (incorporated by reference to
Exhibit 10.1 to the Company’s
Form 10-Q,
filed on October 29, 2009).
|
|
C 10.15
|
|
Form of Indemnification Agreement entered into with directors
and officers (incorporated by reference to Exhibit 10.1 to
the Company’s Quarterly Report on
Form 10-Q,
filed on July 22, 2010).
|
|
C 10.16
|
|
Employment Agreement, dated as of November 21, 2004,
between the Company and H. Thomas Watkins (incorporated by
reference to Exhibit 10.1 to the Company’s Current
Report on
Form 8-K,
filed on November 23, 2004).
|
|
C 10.17
|
|
First Amendment, dated as of January 1, 2008, to Employment
Agreement between the Company and H. Thomas Watkins
(incorporated by reference to Exhibit 10.1 to the
Company’s Current Report on
Form 8-K,
filed on December 20, 2007).
|
|
C 10.18
|
|
Second Amendment, dated as of January 1, 2009, to
Employment Agreement between the Company and H. Thomas Watkins
(incorporated by reference to Exhibit 10.26 to the
Company’s Annual Report on
Form 10-K,
filed on February 26, 2009).
|
|
C 10.19
|
|
Form of Executive Agreement (incorporated by reference to
Exhibit 10.3 to the Company’s Current Report on
Form 8-K,
filed on December 20, 2007).
|
|
C 10.20
|
|
Form of First Amendment to Executive Agreement (incorporated by
reference to Exhibit 10.27 to the Company’s Annual
Report on
Form 10-K,
filed on February 26, 2009).
|
|
C 10.21
|
|
Second Amended and Restated Key Executive Severance Plan
(incorporated by reference to Exhibit 10.2 to the
Company’s Current Report on
Form 8-K,
filed on December 20, 2007).
|
|
C 10. 22
|
|
Form of First Amendment to Second Amended and Restated Key
Executive Severance Plan (incorporated by reference to
Exhibit 10.28 to the Company’s Annual Report on
Form 10-K,
filed on February 26, 2009).
|
57
| |
|
|
|
Exhibit No.
|
|
Description
|
|
|
|
C 10.23
|
|
Human Genome Sciences, Inc. Discretionary Bonus Policy
(incorporated by reference to Exhibit 10.4 to the
Company’s Current Report on
Form 8-K,
filed on December 20, 2007).
|
|
C 10.24
|
|
Human Genome Sciences, Inc. Amended and Restated Stock Incentive
Plan (incorporated by reference to Annex A to the
Company’s Definitive Proxy Statement on Schedule 14A,
filed on March 24, 2009).
|
|
C 10.25
|
|
Form of Restricted Stock Agreement (incorporated by reference to
Exhibit 10.1 to the Company’s Quarterly Report on
Form 10-Q,
filed on August 1, 2005).
|
|
C 10.26
|
|
Form of Stock Option Agreement (incorporated by reference to
Exhibit 10.2 to the Company’s Current Report on
Form 8-K,
filed on September 20, 2004).
|
|
C 10.27
|
|
Form of Stock Unit Grant Agreement under the Non-Employee
Director Equity Compensation Plan (incorporated by reference to
Exhibit 10.5 to the Company’s Current Report on
Form 8-K,
filed on December 20, 2007).
|
|
12.1*
|
|
Ratio of Earnings to Fixed Charges.
|
|
21.1*
|
|
Subsidiaries.
|
|
23.1*
|
|
Consent of Ernst & Young LLP, Independent Registered
Public Accounting Firm.
|
|
31.1*
|
|
Rule 13a-14(a)
Certification of Principal Executive Officer.
|
|
31.2*
|
|
Rule 13a-14(a)
Certification of Principal Financial Officer.
|
|
32.1*
|
|
Section 1350 Certification of Chief Executive Officer.
|
|
32.2*
|
|
Section 1350 Certification of Chief Financial Officer.
|
|
|
|
|
* |
|
Filed herewith. |
| |
|
† |
|
Confidential treatment requested for certain portions of this
Exhibit pursuant to
Rule 24b-2
under the Securities Exchange Act of 1934, as amended, which
portions are omitted and filed separately with the Securities
and Exchange Commission. |
| |
|
C |
|
Management contract or compensatory plan or arrangement. |
58
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the
Securities Exchange Act of 1934, the registrant has duly caused
this report to be signed on its behalf by the undersigned
thereunto duly authorized.
HUMAN GENOME SCIENCES, INC.
|
|
|
| |
|
BY: /s/ H.
Thomas Watkins
|
H. Thomas Watkins
President and Chief Executive Officer
Dated: February 24, 2011
Pursuant to the requirements of the Securities Exchange Act of
1934, this report has been signed below by the following persons
on behalf of the registrant and in the capacities and the dates
indicated:
| |
|
|
|
|
|
|
|
Signature
|
|
Title
|
|
Date
|
|
|
|
|
|
|
|
|
|
/s/ H.
Thomas Watkins
H.
Thomas Watkins
|
|
President, Chief Executive Officer and Director
(Principal Executive Officer)
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ David
P. Southwell
David
P. Southwell
|
|
Executive Vice President and
Chief Financial Officer
(Principal Financial Officer and Principal Accounting Officer)
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Argeris
N. Karabelas, Ph.D.
Argeris
N. Karabelas, Ph.D.
|
|
Chairman of the Board
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Richard
J. Danzig
Richard
J. Danzig
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Jürgen
Drews, M.D.
Jürgen
Drews, M.D.
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Colin
Goddard, Ph.D.
Colin
Goddard, Ph.D.
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Maxine
Gowen, Ph.D.
Maxine
Gowen, Ph.D.
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Tuan
Ha-Ngoc
Tuan
Ha-Ngoc
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ John
LaMattina, Ph.D.
John
LaMattina, Ph.D.
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Augustine
Lawlor
Augustine
Lawlor
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Gregory
Norden
Gregory
Norden
|
|
Director
|
|
February 24, 2011
|
|
|
|
|
|
|
|
/s/ Robert
C. Young, M.D.
Robert
C. Young, M.D.
|
|
Director
|
|
February 24, 2011
|
59
INDEX TO
CONSOLIDATED FINANCIAL STATEMENTS
| |
|
|
|
|
|
|
|
Page
|
|
|
|
Number
|
|
|
|
|
|
|
F-2
|
|
|
|
|
|
F-3
|
|
|
|
|
|
F-4
|
|
|
|
|
|
F-5
|
|
|
|
|
|
F-6
|
|
|
|
|
|
F-8
|
|
F-1
REPORT OF
INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Stockholders of
Human Genome Sciences, Inc.
Rockville, Maryland
We have audited the accompanying consolidated balance sheets of
Human Genome Sciences, Inc. as of December 31, 2010 and
2009, and the related consolidated statements of operations,
stockholders’ equity (deficit) and cash flows for each of
the three years in the period ended December 31, 2010.
These financial statements are the responsibility of the
Company’s management. Our responsibility is to express an
opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the
Public Company Accounting Oversight Board (United States). Those
standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are
free of material misstatement. An audit includes examining, on a
test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the
accounting principles used and significant estimates made by
management, as well as evaluating the overall financial
statement presentation. We believe that our audits provide a
reasonable basis for our opinion.
In our opinion, the financial statements referred to above
present fairly, in all material respects, the consolidated
financial position of Human Genome Sciences, Inc. at
December 31, 2010 and 2009, and the consolidated results of
its operations and its cash flows for each of the three years in
the period ended December 31, 2010, in conformity with
U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the
Public Company Accounting Oversight Board (United States), Human
Genome Sciences, Inc.’s internal control over financial
reporting as of December 31, 2010, based on criteria
established in Internal Control — Integrated Framework
issued by the Committee of Sponsoring Organizations of the
Treadway Commission and our report dated February 24, 2011
expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Baltimore, Maryland
February 24, 2011
F-2
HUMAN
GENOME SCIENCES, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
(in thousands)
|
|
|
|
|
ASSETS
|
|
|
|
|
|
|
|
|
|
Current assets:
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents
|
|
$
|
155,691
|
|
|
$
|
567,667
|
|
|
Short-term investments
|
|
|
282,016
|
|
|
|
151,528
|
|
|
Accounts receivable
|
|
|
25,958
|
|
|
|
23,892
|
|
|
Collaboration receivables
|
|
|
18,856
|
|
|
|
10,356
|
|
|
Inventory
|
|
|
43,091
|
|
|
|
20,149
|
|
|
Prepaid expenses and other current assets
|
|
|
5,569
|
|
|
|
7,176
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current assets
|
|
|
531,181
|
|
|
|
780,768
|
|
|
Marketable securities
|
|
|
416,165
|
|
|
|
384,028
|
|
|
Property, plant and equipment (net of accumulated depreciation)
|
|
|
253,122
|
|
|
|
263,123
|
|
|
Restricted investments
|
|
|
79,510
|
|
|
|
88,437
|
|
|
Collaboration receivables, non-current
|
|
|
29,225
|
|
|
|
6,920
|
|
|
Long-term equity investments
|
|
|
3,241
|
|
|
|
3,016
|
|
|
Other assets
|
|
|
2,585
|
|
|
|
4,338
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL ASSETS
|
|
$
|
1,315,029
|
|
|
$
|
1,530,630
|
|
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
|
|
|
|
|
|
|
|
Current liabilities:
|
|
|
|
|
|
|
|
|
|
Accounts payable and accrued expenses
|
|
$
|
41,798
|
|
|
$
|
38,369
|
|
|
Accrued payroll and related taxes
|
|
|
30,157
|
|
|
|
30,997
|
|
|
Convertible subordinated debt
|
|
|
188,620
|
|
|
|
—
|
|
|
Collaboration payable
|
|
|
12,984
|
|
|
|
4,000
|
|
|
Deferred revenues
|
|
|
5,134
|
|
|
|
88,565
|
|
|
Accrued exit expenses
|
|
|
2,251
|
|
|
|
2,227
|
|
|
|
|
|
|
|
|
|
|
|
|
Total current liabilities
|
|
|
280,944
|
|
|
|
164,158
|
|
|
Convertible subordinated debt, non-current
|
|
|
184,231
|
|
|
|
349,807
|
|
|
Lease financing
|
|
|
250,516
|
|
|
|
248,628
|
|
|
Deferred rent
|
|
|
10,358
|
|
|
|
8,665
|
|
|
Deferred revenues, non-current
|
|
|
2,517
|
|
|
|
1,978
|
|
|
Accrued exit expenses, non-current
|
|
|
700
|
|
|
|
1,979
|
|
|
|
|
|
|
|
|
|
|
|
|
Total liabilities
|
|
|
729,266
|
|
|
|
775,215
|
|
|
|
|
|
|
|
|
|
|
|
|
Stockholders’ equity:
|
|
|
|
|
|
|
|
|
|
Preferred stock — $0.01 par value;
20,000,000 shares authorized; none outstanding
|
|
|
—
|
|
|
|
—
|
|
|
Common stock — $0.01 par value;
400,000,000 shares authorized; 188,980,748 and
185,254,660 shares issued and oustanding at
December 31, 2010 and 2009, respectively
|
|
|
1,890
|
|
|
|
1,853
|
|
|
Additional paid-in capital
|
|
|
2,996,645
|
|
|
|
2,932,863
|
|
|
Accumulated other comprehensive income
|
|
|
7,125
|
|
|
|
7,365
|
|
|
Accumulated deficit
|
|
|
(2,419,897
|
)
|
|
|
(2,186,666
|
)
|
|
|
|
|
|
|
|
|
|
|
|
Total stockholders’ equity
|
|
|
585,763
|
|
|
|
755,415
|
|
|
|
|
|
|
|
|
|
|
|
|
TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY
|
|
$
|
1,315,029
|
|
|
$
|
1,530,630
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated Financial Statements are
an integral part hereof.
F-3
HUMAN
GENOME SCIENCES, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(in thousands, except share and per share amounts)
|
|
|
|
|
Revenue:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Product sales
|
|
$
|
47,159
|
|
|
$
|
154,074
|
|
|
$
|
—
|
|
|
Manufacturing and development services
|
|
|
22,695
|
|
|
|
50,653
|
|
|
|
—
|
|
|
Research and development collaborative agreements
|
|
|
87,497
|
|
|
|
71,022
|
|
|
|
48,422
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total revenue
|
|
|
157,351
|
|
|
|
275,749
|
|
|
|
48,422
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs and expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of product sales
|
|
|
29,941
|
|
|
|
15,805
|
|
|
|
—
|
|
|
Cost of manufacturing and development services
|
|
|
15,016
|
|
|
|
18,215
|
|
|
|
—
|
|
|
Research and development expenses
|
|
|
196,370
|
|
|
|
173,709
|
|
|
|
243,257
|
|
|
General and administrative expenses
|
|
|
106,797
|
|
|
|
61,073
|
|
|
|
60,865
|
|
|
Facility-related exit charges
|
|
|
—
|
|
|
|
759
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total costs and expenses
|
|
|
348,124
|
|
|
|
269,561
|
|
|
|
304,122
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income (loss) from operations
|
|
|
(190,773
|
)
|
|
|
6,188
|
|
|
|
(255,700
|
)
|
|
Investment income
|
|
|
16,966
|
|
|
|
12,727
|
|
|
|
23,487
|
|
|
Interest expense
|
|
|
(59,500
|
)
|
|
|
(58,424
|
)
|
|
|
(62,912
|
)
|
|
Gain on extinguishment of debt
|
|
|
—
|
|
|
|
38,873
|
|
|
|
—
|
|
|
Gain on sale of long-term equity investment
|
|
|
—
|
|
|
|
5,259
|
|
|
|
32,518
|
|
|
Other income (expense)
|
|
|
76
|
|
|
|
(238
|
)
|
|
|
(6,284
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Income (loss) before taxes
|
|
|
(233,231
|
)
|
|
|
4,385
|
|
|
|
(268,891
|
)
|
|
Income tax benefit
|
|
|
—
|
|
|
|
1,274
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss)
|
|
$
|
(233,231
|
)
|
|
$
|
5,659
|
|
|
$
|
(268,891
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic net income (loss) per share
|
|
$
|
(1.24
|
)
|
|
$
|
0.04
|
|
|
$
|
(1.99
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diluted net income (loss) per share
|
|
$
|
(1.24
|
)
|
|
$
|
0.04
|
|
|
$
|
(1.99
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding, basic
|
|
|
187,791,437
|
|
|
|
149,334,426
|
|
|
|
135,406,642
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding, diluted
|
|
|
187,791,437
|
|
|
|
155,053,473
|
|
|
|
135,406,642
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated Financial Statements are
an integral part hereof.
F-4
HUMAN
GENOME SCIENCES, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional
|
|
|
Other
|
|
|
|
|
|
|
|
|
|
|
Common Stock
|
|
|
Paid-In
|
|
|
Comprehensive
|
|
|
Accumulated
|
|
|
|
|
|
|
|
Shares
|
|
|
Amount
|
|
|
Capital
|
|
|
Income (Loss)
|
|
|
Deficit
|
|
|
Total
|
|
|
|
|
(in thousands, except share amounts)
|
|
|
|
|
Balance — December 31, 2007
|
|
|
134,936,512
|
|
|
$
|
1,349
|
|
|
$
|
2,036,078
|
|
|
$
|
3,152
|
|
|
$
|
(1,923,434
|
)
|
|
$
|
117,145
|
|
|
Comprehensive income (loss):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(268,891
|
)
|
|
|
(268,891
|
)
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(7,052
|
)
|
|
|
—
|
|
|
|
(7,052
|
)
|
|
Cumulative translation adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(590
|
)
|
|
|
—
|
|
|
|
(590
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(276,533
|
)
|
|
Shares of common stock issued pursuant to stock-based
compensation plans
|
|
|
803,466
|
|
|
|
8
|
|
|
|
4,589
|
|
|
|
—
|
|
|
|
—
|
|
|
|
4,597
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
18,487
|
|
|
|
—
|
|
|
|
—
|
|
|
|
18,487
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance — December 31, 2008
|
|
|
135,739,978
|
|
|
|
1,357
|
|
|
|
2,059,154
|
|
|
|
(4,490
|
)
|
|
|
(2,192,325
|
)
|
|
|
(136,304
|
)
|
|
Comprehensive income (loss):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
5,659
|
|
|
|
5,659
|
|
|
Unrealized gain on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
11,264
|
|
|
|
—
|
|
|
|
11,264
|
|
|
Cumulative translation adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
591
|
|
|
|
—
|
|
|
|
591
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive income
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
17,514
|
|
|
Issuance of common stock pursuant to public offerings
|
|
|
44,522,250
|
|
|
|
446
|
|
|
|
812,423
|
|
|
|
—
|
|
|
|
—
|
|
|
|
812,869
|
|
|
Shares of common stock issued pursuant to stock-based
compensation plans
|
|
|
4,992,432
|
|
|
|
50
|
|
|
|
48,762
|
|
|
|
—
|
|
|
|
—
|
|
|
|
48,812
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
12,524
|
|
|
|
—
|
|
|
|
—
|
|
|
|
12,524
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance — December 31, 2009
|
|
|
185,254,660
|
|
|
|
1,853
|
|
|
|
2,932,863
|
|
|
|
7,365
|
|
|
|
(2,186,666
|
)
|
|
|
755,415
|
|
|
Comprehensive income (loss):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(233,231
|
)
|
|
|
(233,231
|
)
|
|
Unrealized loss on investments
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(212
|
)
|
|
|
—
|
|
|
|
(212
|
)
|
|
Cumulative translation adjustment
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
(28
|
)
|
|
|
—
|
|
|
|
(28
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Comprehensive loss
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(233,471
|
)
|
|
Issuance of common stock pursuant to bond redemption
|
|
|
562
|
|
|
|
—
|
|
|
|
8
|
|
|
|
—
|
|
|
|
—
|
|
|
|
8
|
|
|
Shares of common stock issued pursuant to stock-based
compensation plans
|
|
|
3,725,526
|
|
|
|
37
|
|
|
|
38,344
|
|
|
|
—
|
|
|
|
—
|
|
|
|
38,381
|
|
|
Stock-based compensation expense
|
|
|
—
|
|
|
|
—
|
|
|
|
25,430
|
|
|
|
—
|
|
|
|
—
|
|
|
|
25,430
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance — December 31, 2010
|
|
|
188,980,748
|
|
|
$
|
1,890
|
|
|
$
|
2,996,645
|
|
|
$
|
7,125
|
|
|
$
|
(2,419,897
|
)
|
|
$
|
585,763
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying Notes to Consolidated Financial Statements are
an integral part hereof.
F-5
HUMAN
GENOME SCIENCES, INC.
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
(in thousands)
|
|
|
|
|
Cash flows from operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss)
|
|
$
|
(233,231
|
)
|
|
$
|
5,659
|
|
|
$
|
(268,891
|
)
|
|
Adjustments to reconcile net income (loss) to net cash used in
operating activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense
|
|
|
25,430
|
|
|
|
12,524
|
|
|
|
18,593
|
|
|
Depreciation and amortization
|
|
|
21,305
|
|
|
|
21,255
|
|
|
|
21,143
|
|
|
Amortization of debt discount
|
|
|
23,052
|
|
|
|
21,936
|
|
|
|
24,183
|
|
|
Facility-related exit charges
|
|
|
—
|
|
|
|
759
|
|
|
|
—
|
|
|
Accrued interest on short-term investments, marketable
securities and restricted investments
|
|
|
5,552
|
|
|
|
(493
|
)
|
|
|
581
|
|
|
Gain on extinguishment of long-term debt
|
|
|
—
|
|
|
|
(38,873
|
)
|
|
|
—
|
|
|
Gain on sale of long-term equity investment
|
|
|
—
|
|
|
|
(5,259
|
)
|
|
|
(32,518
|
)
|
|
Non-cash expenses and other
|
|
|
2,177
|
|
|
|
3,759
|
|
|
|
8,265
|
|
|
Changes in operating assets and liabilities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable
|
|
|
(2,066
|
)
|
|
|
(21,021
|
)
|
|
|
—
|
|
|
Collaboration receivables
|
|
|
(30,805
|
)
|
|
|
4,800
|
|
|
|
13,792
|
|
|
Inventory
|
|
|
(22,942
|
)
|
|
|
(20,149
|
)
|
|
|
—
|
|
|
Prepaid expenses and other assets
|
|
|
2,110
|
|
|
|
(1,640
|
)
|
|
|
2,760
|
|
|
Accounts payable and accrued expenses
|
|
|
3,444
|
|
|
|
(9,152
|
)
|
|
|
(7,247
|
)
|
|
Accrued payroll and related taxes
|
|
|
(840
|
)
|
|
|
12,423
|
|
|
|
4,126
|
|
|
Collaboration payable
|
|
|
8,984
|
|
|
|
(4,000
|
)
|
|
|
—
|
|
|
Deferred revenues
|
|
|
(82,892
|
)
|
|
|
17,234
|
|
|
|
(44,959
|
)
|
|
Accrued exit expenses
|
|
|
(1,533
|
)
|
|
|
(1,953
|
)
|
|
|
(2,083
|
)
|
|
Deferred rent
|
|
|
1,566
|
|
|
|
1,859
|
|
|
|
1,992
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash used in operating activities
|
|
|
(280,689
|
)
|
|
|
(332
|
)
|
|
|
(260,263
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from investing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of short-term investments and marketable securities
|
|
|
(840,145
|
)
|
|
|
(625,041
|
)
|
|
|
(15,065
|
)
|
|
Proceeds from sale and maturities of short-term investments and
marketable securities
|
|
|
681,687
|
|
|
|
388,277
|
|
|
|
211,722
|
|
|
Capital expenditures — property, plant, and equipment
|
|
|
(9,782
|
)
|
|
|
(10,019
|
)
|
|
|
(9,724
|
)
|
|
Proceeds from sale of long-term equity investment
|
|
|
—
|
|
|
|
5,259
|
|
|
|
47,336
|
|
|
Release of restricted investments
|
|
|
300
|
|
|
|
3,291
|
|
|
|
4,877
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by (used in) investing activities
|
|
|
(167,940
|
)
|
|
|
(238,233
|
)
|
|
|
239,146
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash flows from financing activities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of restricted investments
|
|
|
(31,672
|
)
|
|
|
(47,002
|
)
|
|
|
(28,897
|
)
|
|
Proceeds from sale and maturities of restricted investments
|
|
|
30,081
|
|
|
|
26,426
|
|
|
|
26,120
|
|
|
Proceeds from issuance of common stock
|
|
|
39,269
|
|
|
|
861,573
|
|
|
|
4,432
|
|
|
Purchase of treasury stock
|
|
|
(1,025
|
)
|
|
|
(15
|
)
|
|
|
(105
|
)
|
|
Extinguishment of long-term debt
|
|
|
—
|
|
|
|
(49,998
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash provided by financing activities
|
|
|
36,653
|
|
|
|
790,984
|
|
|
|
1,550
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net increase (decrease) in cash and cash equivalents
|
|
|
(411,976
|
)
|
|
|
552,419
|
|
|
|
(19,567
|
)
|
|
Cash and cash equivalents — beginning of period
|
|
|
567,667
|
|
|
|
15,248
|
|
|
|
34,815
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents — end of period
|
|
$
|
155,691
|
|
|
$
|
567,667
|
|
|
$
|
15,248
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-6
SUPPLEMENTAL
DISCLOSURES OF CASH FLOW INFORMATION, NON-CASH OPERATING,
INVESTING AND FINANCING ACTIVITIES
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
(in thousands)
|
|
|
|
Cash paid (received) during the period for:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest
|
|
$
|
33,169
|
|
|
$
|
33,609
|
|
|
$
|
34,729
|
|
|
Income taxes
|
|
$
|
(1,948
|
)
|
|
$
|
809
|
|
|
$
|
—
|
|
During 2010, the Company was able to reduce certain of its
lease-related collateral obligations and transferred restricted
investments of $9,014 into short-term investments.
During 2010, 2009 and 2008, the Company recorded non-cash
accretion of $277, $1,384 and $466, respectively, related to its
exit accrual for certain exited space.
During 2010, 2009 and 2008, lease financing increased as a
result of non-cash accretion with respect to the Company’s
2006 leases with BioMed Realty Trust, Inc. (“BioMed”)
by $1,889, $2,151 and $2,378 respectively. Because the lease
payments are less than the amount of calculated interest expense
for the first nine years of the leases, the lease balance will
increase during this period.
The accompanying Notes to Consolidated Financial Statements are
an integral part hereof.
F-7
HUMAN
GENOME SCIENCES, INC.
(dollars in thousands, except per share data)
(NOTE A) —
The Company
Human Genome Sciences, Inc. (the “Company”) is a
biopharmaceutical company that exists to place new therapies
into the hands of those battling serious disease. Its lead
products in development are
BENLYSTA®
(belimumab) for systemic lupus erythematosus (“SLE”)
and raxibacumab for inhalation anthrax.
BENLYSTA continues to progress toward commercialization. The
Company is awaiting the U.S. Food and Drug Administration
(“FDA”) decision, which is currently expected by
March 10, 2011, regarding marketing approval of BENLYSTA.
GlaxoSmithKline (“GSK”), the Company’s BENLYSTA
partner, submitted a Marketing Authorization Application
(“MAA”) to the European Medicines Agency
(“EMA”) for approval to market BENLYSTA in Europe with
an EMA decision expected in the second half of 2011. In
preparation for the anticipated launch and commercialization of
BENLYSTA, the Company has hired and trained a specialized
commercial team including sales, marketing and medical affairs.
The Company is also building its own commercial team to work
alongside GSK in Europe.
The Company, which is located primarily in the United States,
operates in a single business segment.
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies
|
Use of
Estimates
The preparation of financial statements in conformity with
U.S. generally accepted accounting principles requires
management to make estimates and assumptions that affect the
reported amounts of assets and liabilities and disclosure of
contingent assets and liabilities at the date of the financial
statements and the reported amounts of revenues and expenses
during the reporting period. Such estimates are based on
historical experience and on various assumptions that the
Company believes to be reasonable under the circumstances.
Actual results could differ from these estimates under different
assumptions or conditions.
Principles
of Consolidation
The consolidated financial statements include the accounts of
Human Genome Sciences, Inc. and its subsidiaries, all of which
are wholly-owned. All significant intercompany accounts and
transactions have been eliminated.
Cash
Equivalents, Short-term Investments, Marketable Securities,
Long-term Equity Investments and Restricted
Investments
The Company considers all highly liquid investment instruments
purchased with a maturity of three months or less to be cash
equivalents.
The Company classifies its short-term investments, marketable
securities and long-term equity investments with readily
determinable fair values as
“available-for-sale.”
Investments in securities that are classified as
available-for-sale
are measured at fair market value in the balance sheets, and
unrealized holding gains and losses on investments are reported
as a separate component of stockholders’ equity until
realized. Investments of less than 20% of privately-held
companies are accounted for as cost-method investments. The
Company reviews the carrying value of such investments on a
periodic basis for indicators of impairment. Additionally,
certain of the Company’s investments are held as restricted
investments. Restricted investments with maturities less than
three months are not classified as cash in the Company’s
consolidated balance sheets. See Note C, Investments, for
additional information.
F-8
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies (continued)
|
Investment
Risk
The Company has invested its cash in obligations of the
U.S. Government, government agencies and in high-grade debt
securities and various money market instruments. The
Company’s investment policy limits investments to certain
types of instruments issued by institutions with credit ratings
of “A−” or better, and places restrictions on
maturities and concentrations in certain industries and by
issuer. The Company does not hold auction rate securities, loans
held for sale or mortgage-backed securities backed by
sub-prime or
Alt-A collateral.
Other-Than-Temporary
Impairment of Investments
Periodically, the Company evaluates whether any investments have
incurred an
other-than-temporary
impairment, based on the criteria under Financial Accounting
Standards Board (“FASB”) Accounting Standards
Codification (“FASB ASC”) Topic 320,
Investments — Debt and Equity Securities. This
evaluation consists of a review of several factors, including
but not limited to the length of time and extent that a security
has been in an unrealized loss position, the existence of an
event that would impair the issuer’s future repayment
potential, the near term prospects for recovery of the market
value of a security and the intent of the Company to hold the
security until the market value recovers and whether it is not
more likely than not that the Company will be required to sell
the security. If the Company determines that such impairment
exists, the Company will recognize a charge in the consolidated
statement of operations equal to the amount of such impairment.
See Note C, Investments, for additional discussion.
Inventories
Inventories, which are recorded at the lower of cost or market,
include materials, labor and other direct and indirect costs and
are valued using the
first-in,
first-out method. The Company capitalizes inventories produced
in preparation for product launches when the related product
candidates are considered likely to receive regulatory approval
and it is probable that the related costs will be recoverable
through the commercialization of the product. In the case of
raxibacumab, inventory costs were capitalized after the Company
received a follow-on order from the U.S. Government
(“USG”) in July 2009. BENLYSTA costs were capitalized
after the FDA Advisory Committee vote in November 2010 to
recommend approval of the product. See Note E, Other
Financial Information and Cost of product sales discussion
within this Note B, for additional information.
Depreciation
Depreciation is computed using the straight-line method over the
estimated useful lives of the assets as follows:
| |
|
|
|
Buildings
|
|
30 years
|
|
Land improvements
|
|
lesser of the lease term or the useful life
|
|
Production equipment
|
|
5 - 10 years
|
|
Laboratory equipment
|
|
3 - 10 years
|
|
Computer equipment and software
|
|
3 - 5 years
|
|
Furniture and office equipment
|
|
3 - 5 years
|
|
Leasehold improvements
|
|
lesser of the lease term or the useful life
|
Impairment
of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or
changes in circumstances indicate that the carrying amount of an
asset may not be recoverable based on the criteria for
accounting for the impairment or disposal of long-lived assets
under FASB ASC Topic 360, Property, Plant and Equipment.
F-9
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies (continued)
|
Product
sales
Revenue from product sales is recognized when persuasive
evidence of an arrangement exists, title to product and
associated risk of loss has passed to the customer, the price is
fixed or determinable, collection from the customer is
reasonably assured, and the Company has no further performance
obligations.
Manufacturing
and development services
As part of its raxibacumab contract with the USG and the
Biomedical Advanced Research and Development Authority
(“BARDA”), the Company performed a variety of drug
development services primarily relating to the conduct of animal
and human studies. Upon BARDA’s acceptance of the initial
raxibacumab delivery, the Company billed the USG for the drug
development work previously performed, and recorded this as
manufacturing and development services revenue during 2009. The
Company has been recording additional development revenue as
services are performed. The Company has entered into agreements
with certain commercial parties for manufacturing process
development, clinical and commercial supply of certain
biopharmaceutical products. Revenue under these agreements is
recognized as services are performed or products delivered,
depending on the nature of the work contracted, using the
proportional performance method of accounting. Performance is
assessed using output measures such as
units-of-work
performed to date as compared to total
units-of-work
contracted. Advance payments received in excess of amounts
earned are classified as deferred revenue until earned.
Research
and development collaborative agreements
Collaborative research and development agreements can provide
for one or more of up-front license fees, research payments and
milestone payments. Agreements with multiple components
(“deliverables” or “items”) are evaluated to
determine if the deliverables can be divided into more than one
unit of accounting. An item can generally be considered a
separate unit of accounting if all of the following criteria are
met: (1) the delivered item(s) has value to the customer on
a stand-alone basis; (2) there is objective and reliable
evidence of the fair value of the undelivered item(s); and
(3) if the arrangement includes a general right of return
relative to the delivered item(s), delivery or performance of
the undelivered item(s) is considered probable and substantially
in control of the Company. Items that cannot be divided into
separate units are combined with other units of accounting, as
appropriate. Consideration received is allocated among the
separate units based on their respective fair values or based on
the residual value method and is recognized in full when the
following criteria are met: (1) persuasive evidence of an
arrangement exists; (2) delivery has occurred or services
have been rendered; (3) the sales price is fixed or
determinable; and (4) collectibility is probable. The
Company deems service to have been rendered if no continuing
obligation exists on the part of the Company.
Revenue associated with non-refundable up-front license fees
under arrangements where the license fees and research and
development activities cannot be accounted for as separate units
of accounting are deferred and recognized as revenue on a
straight-line basis over the expected term of the Company’s
continued involvement in the research and development process.
Revenues from the achievement of research and development
milestones, if deemed substantive, are recognized as revenue
when the milestones are achieved, and the milestone payments are
due and collectible. If not deemed substantive, the Company
would recognize such milestone as revenue on a straight-line
basis over the remaining expected term of continued involvement
in the research and development process. Milestones are
considered substantive if all of the following conditions are
met: (1) the milestone is non-refundable;
(2) achievement of the milestone was not reasonably assured
at the inception of the arrangement; (3) substantive effort
is involved to achieve the milestone; and, (4) the amount
of the milestone appears reasonable in relation to the effort
expended, the other milestones in the arrangement and the
related risk associated with the achievement of the milestone
and any ongoing research and development or other services are
priced at fair value. Payments received in advance of work
performed are recorded as deferred revenue.
F-10
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies (continued)
|
Cost of
product sales
The Company capitalizes inventories produced in preparation for
product launches when the related product candidates are
considered likely to receive regulatory approval and it is
probable that the related costs will be recoverable through the
commercialization of the product. Prior to capitalization, the
cost of manufacturing drug product is recognized as research and
development expense in the period that the cost is incurred.
Therefore, manufacturing costs incurred prior to capitalization
are not included in cost of product sales when revenue is
recognized from the sale of that drug product.
Prior to receiving a follow-on order for raxibacumab from the
USG in July 2009, the Company did not capitalize inventory costs
related to this product. Although authorization to ship to the
U.S. Strategic National Stockpile was received in January
2009, there continued to be uncertainty around future product
orders. Beginning in July 2009, the cost of manufacturing
raxibacumab is recognized as a cost of product sales
(capitalized and then expensed when revenue is recognized),
rather than research and development expenses in the period that
the cost is incurred.
Cost of product sales also includes royalties paid or payable to
third parties based on the sales levels of certain products.
Cost of
manufacturing and development services
Cost of manufacturing and development services represents costs
associated with the Company’s contract manufacturing
arrangements and other development services. The costs
associated with work previously performed to conduct animal and
human studies for raxibacumab were recognized as research and
development expenses in the period that the costs were incurred.
Therefore, these pre-acceptance development costs are not
included in cost of manufacturing and development services for
2009. The Company is recording additional raxibacumab
development services costs as incurred.
Research
and Development
Research and development costs are charged to expense as
incurred, unless otherwise capitalized pursuant to FASB ASC
Topic 730, Research and Development. Research and
development costs include salaries and related benefits, outside
services, licensing fees or milestones, materials and supplies,
building costs and allocations of certain support costs.
Research and development direct expenditures were $196,370,
$173,709 and $243,257 for 2010, 2009 and 2008, respectively.
Reimbursement of research and development expenses received in
connection with collaborative cost-sharing agreements is
recorded as a reduction of such expenses.
Leases
The Company accounts for its leases under FASB ASC Topic 840,
Leases. The Company has a number of operating leases and
has entered into sale-leaseback transactions for land and
facilities. See Note G, Commitments and Other Matters, for
additional discussion.
Stock-Based
Compensation
The Company has a stock incentive plan (the “Incentive
Plan”) under which options to purchase shares of the
Company’s common stock may be granted to employees,
consultants and directors with an exercise price no less than
the quoted market value on the date of grant. The Incentive Plan
also provides for the issuance of non-vested common stock
(restricted stock) and other share-based compensation. The
Company recognizes stock-based compensation expense related to
employee stock options under FASB ASC Topic 718,
Compensation — Stock Compensation. For income
tax purposes, the Company follows the “with and
without” method of accounting for the tax effect of excess
tax benefits generated from stock-based compensation. See
Note H, Stockholders’ Equity, for additional
discussion.
F-11
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies (continued)
|
Financing
Costs Related to Long-term Debt
Costs associated with obtaining long-term debt are deferred and
amortized over the term of the related debt on a straight-line
basis which approximates the effective interest method.
Patent
Application Costs
Patent application costs are charged to expense as incurred.
Net
Income (Loss) Per Share
The Company follows the provisions under FASB ASC Topic 260,
Earnings Per Share, which requires the Company to present
basic and diluted earnings per share. The Company’s basic
and diluted income (loss) per share is calculated by dividing
the net income (loss) by the weighted average number of shares
of common stock outstanding during all periods presented. Shares
issuable upon the conversion of the Company’s convertible
subordinated debt are excluded from diluted earnings per share
calculations for 2010, 2009 and 2008 because the effects are
anti-dilutive.
Foreign
Currency
Assets and liabilities of the Company’s international
operations are translated into U.S. dollars at exchange
rates that are in effect as of the balance sheet date, and
equity accounts are translated at historical rates. Revenue and
expenses are translated at average exchange rates that are in
effect during the year. Translation adjustments are accumulated
in other comprehensive income (loss) as a separate component of
stockholders’ equity in the consolidated balance sheets.
Transaction gains and losses are included in other income
(expense) in the consolidated statements of operations.
Comprehensive
Income (Loss)
FASB ASC Topic 220, Comprehensive Income, requires
unrealized gains and losses on the Company’s
available-for-sale
short-term investments, marketable securities and long-term
equity investments and the activity for the cumulative
translation adjustment to be included in other comprehensive
income.
The components of accumulated other comprehensive income are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Net unrealized gains on:
|
|
|
|
|
|
|
|
|
|
Short-term investments and marketable securities
|
|
$
|
6,313
|
|
|
$
|
5,496
|
|
|
Long-term equity investment in VIA Pharmaceuticals
|
|
|
4
|
|
|
|
21
|
|
|
Restricted investments
|
|
|
805
|
|
|
|
1,816
|
|
|
Foreign currency translation
|
|
|
3
|
|
|
|
32
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated other comprehensive income
|
|
$
|
7,125
|
|
|
$
|
7,365
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated other comprehensive income excludes net realized
losses included in net loss of $303 for 2010. Accumulated other
comprehensive income excludes net realized gains included in net
income (loss) of $4,504 and $33,619 for 2009 and 2008,
respectively. The effect of income taxes on items in other
comprehensive income is $0 for all periods presented.
F-12
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE B) —
|
Summary
of Significant Accounting Policies (continued)
|
Sources
of Supply
The Company is currently able to obtain most of its raw
materials, supplies and equipment from various sources, and
generally has no dependence upon a single supplier. However,
certain materials required for manufacturing are currently
available only from single sources. As the Company proceeds with
commercialization of its products, it intends to identify
alternative sources of supply wherever possible.
Recent
Accounting Pronouncements
In October 2009, the FASB issued new revenue recognition
standards for arrangements with multiple deliverables. The new
standards permit entities to initially use management’s
best estimate of selling price to value individual deliverables
when those deliverables do not have objective and reliable
evidence of fair value. Additionally, these new standards modify
the manner in which the transaction consideration is allocated
across the separately identified deliverables. These new
standards are effective for the Company as of January 1,
2011, however early adoption is permitted. The Company does not
expect the adoption of these standards to have a material effect
on its consolidated results of operations, financial position or
liquidity.
In January 2010, the FASB issued Accounting Standards Update
(“ASU”)
2010-06,
Improving Disclosures about Fair Value Measurements
(“ASU
2010-06”).
ASU 2010-06
requires disclosing the amounts of significant transfers in and
out of Level 1 and 2 fair value measurements and to
describe the reasons for the transfers. The disclosures were
effective for reporting periods beginning after
December 15, 2009, and had no material impact on the
Company’s financial statements for 2010. Additionally,
disclosures of the gross purchases, sales, issuances and
settlements activity in Level 3 fair value measurements
will be required for fiscal years beginning after
December 15, 2010. The Company does not expect the
additional provisions of ASU
2010-06 to
have a material effect on its consolidated results of
operations, financial position or liquidity.
In April 2010, the FASB issued ASU
2010-17,
Milestone Method of Revenue Recognition (“ASU
2010-17”),
which provides guidance on defining a milestone and determining
when it may be appropriate to apply the milestone method of
revenue recognition for research or development transactions.
Research or development arrangements frequently include payment
provisions whereby a portion or all of the consideration is
contingent upon milestone events such as successful completion
of phases in a study or achieving a specific result from the
research or development efforts. The amendments in this ASU
provide guidance on the criteria that should be met for
determining whether the milestone method of revenue recognition
is appropriate. ASU
2010-17 is
effective for fiscal years and interim periods within those
years beginning on or after June 15, 2010, with early
adoption permitted. This ASU is effective for the Company on
January 1, 2011. The Company does not expect the adoption
of ASU
2010-17 to
have a material effect on its consolidated results of
operations, financial position or liquidity.
In December 2010, the FASB issued ASU
2010-27,
Fees Paid to the Federal Government by Pharmaceutical
Manufacturers (“ASU
2010-27”),
which specifies that the liability for the new fee mandated by
the Patient Protection and Affordable Care Act of 2010, as
amended by the Health Care and Education Reconciliation Act,
should be estimated and recorded in full upon the first
qualifying sale with a corresponding deferred cost that is
amortized to expense using a straight-line method of allocation
unless another method better allocates the fee over the calendar
year that it is payable. The amendments in this update are
effective for calendar years beginning after December 31,
2010. The Company is currently evaluating the impact ASU
2010-27 will
have on its consolidated results of operations, financial
position and liquidity.
F-13
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
(NOTE C) —
Investments
Available for sale investments, including accrued interest, as
of December 31, 2010 and 2009 were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2010
|
|
|
|
|
|
|
|
Gross
|
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
Unrealized
|
|
|
Fair
|
|
|
|
|
Cost
|
|
|
Gains
|
|
|
Losses
|
|
|
Value
|
|
|
|
|
Government-sponsored enterprise securities
|
|
$
|
140,849
|
|
|
$
|
848
|
|
|
$
|
(13
|
)
|
|
$
|
141,684
|
|
|
Corporate debt securities
|
|
|
138,691
|
|
|
|
1,660
|
|
|
|
(19
|
)
|
|
|
140,332
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Short-term investments
|
|
|
279,540
|
|
|
|
2,508
|
|
|
|
(32
|
)
|
|
|
282,016
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
105,007
|
|
|
|
1,164
|
|
|
|
(140
|
)
|
|
|
106,031
|
|
|
Corporate debt securities
|
|
|
307,322
|
|
|
|
3,695
|
|
|
|
(883
|
)
|
|
|
310,134
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Marketable securities
|
|
|
412,329
|
|
|
|
4,859
|
|
|
|
(1,023
|
)
|
|
|
416,165
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted cash and cash equivalents
|
|
|
7,455
|
|
|
|
—
|
|
|
|
—
|
|
|
|
7,455
|
|
|
U.S. Treasury and agencies
|
|
|
1,302
|
|
|
|
10
|
|
|
|
—
|
|
|
|
1,312
|
|
|
Government-sponsored enterprise securities
|
|
|
19,605
|
|
|
|
200
|
|
|
|
(6
|
)
|
|
|
19,799
|
|
|
Corporate debt securities
|
|
|
50,343
|
|
|
|
630
|
|
|
|
(29
|
)
|
|
|
50,944
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Restricted investments
|
|
|
78,705
|
|
|
|
840
|
|
|
|
(35
|
)
|
|
|
79,510
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
770,574
|
|
|
$
|
8,207
|
|
|
$
|
(1,090
|
)
|
|
$
|
777,691
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
|
|
|
|
|
|
Gross
|
|
|
Gross
|
|
|
|
|
|
|
|
Amortized
|
|
|
Unrealized
|
|
|
Unrealized
|
|
|
Fair
|
|
|
|
|
Cost
|
|
|
Gains
|
|
|
Losses
|
|
|
Value
|
|
|
|
|
U.S. Treasury and agencies
|
|
$
|
7,997
|
|
|
$
|
32
|
|
|
$
|
—
|
|
|
$
|
8,029
|
|
|
Government-sponsored enterprise securities
|
|
|
34,667
|
|
|
|
672
|
|
|
|
(29
|
)
|
|
|
35,310
|
|
|
Corporate debt securities
|
|
|
107,283
|
|
|
|
1,099
|
|
|
|
(193
|
)
|
|
|
108,189
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Short-term investments
|
|
|
149,947
|
|
|
|
1,803
|
|
|
|
(222
|
)
|
|
|
151,528
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
172,191
|
|
|
|
2,009
|
|
|
|
(673
|
)
|
|
|
173,527
|
|
|
Corporate debt securities
|
|
|
208,824
|
|
|
|
2,106
|
|
|
|
(429
|
)
|
|
|
210,501
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Marketable securities
|
|
|
381,015
|
|
|
|
4,115
|
|
|
|
(1,102
|
)
|
|
|
384,028
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted cash and cash equivalents
|
|
|
6,693
|
|
|
|
—
|
|
|
|
—
|
|
|
|
6,693
|
|
|
Government-sponsored enterprise securities
|
|
|
20,316
|
|
|
|
394
|
|
|
|
(17
|
)
|
|
|
20,693
|
|
|
Corporate debt securities
|
|
|
59,612
|
|
|
|
1,470
|
|
|
|
(31
|
)
|
|
|
61,051
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Restricted investments
|
|
|
86,621
|
|
|
|
1,864
|
|
|
|
(48
|
)
|
|
|
88,437
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
617,583
|
|
|
$
|
7,782
|
|
|
$
|
(1,372
|
)
|
|
$
|
623,993
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company’s restricted investments with respect to its
headquarters (“Traville”) lease serve as collateral
for a letter of credit which serves as the security deposit for
the duration of the lease, although the Company has the ability
to reduce the restricted investments that are in the form of
securities by substituting a cash security deposit in the amount
of $19,750 to be maintained with the landlord. Presently, to
secure the security deposit letter of credit, the Company is
required to maintain margin value of the collateral of at least
$19,750.
F-14
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
(NOTE C) —
Investments (continued)
The Company’s restricted investments with respect to its
large-scale manufacturing facility (“LSM”) lease, as
amended, will serve as collateral in favor of the landlord in
lieu of providing the landlord with either a cash deposit or a
standby letter of credit. Under the LSM lease, the Company is
required to pledge to the landlord a minimum of $20,000 in
marketable securities or provide the landlord with a $19,750
cash security deposit.
In addition, the Company is also required to maintain $34,300 in
restricted investments with respect to two leases with the
Maryland Economic Development Corporation (“MEDCO”)
for its small-scale manufacturing facility. The facility was
financed primarily through a combination of bonds issued by
MEDCO (“MEDCO Bonds”) and loans issued to MEDCO by
certain State of Maryland agencies. The MEDCO Bonds are secured
by letters of credit issued for the account of MEDCO which
expire in December 2011. The Company is required to maintain
restricted investments which serve as security for the MEDCO
letters of credit reimbursement obligation.
Short-term
investments, Marketable securities and Restricted
investments — unrealized losses
The Company’s gross unrealized losses and fair value of
investments with unrealized losses were as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2010
|
|
|
|
|
Loss Position
|
|
|
Loss Position
|
|
|
|
|
|
|
|
for Less Than
|
|
|
for Greater Than
|
|
|
|
|
|
|
|
Twelve Months
|
|
|
Twelve Months
|
|
|
Total
|
|
|
|
|
Fair
|
|
|
Unrealized
|
|
|
Fair
|
|
|
Unrealized
|
|
|
Fair
|
|
|
Unrealized
|
|
|
|
|
Value
|
|
|
Losses
|
|
|
Value
|
|
|
Losses
|
|
|
Value
|
|
|
Losses
|
|
|
|
|
Government-sponsored enterprise securities
|
|
$
|
54,718
|
|
|
$
|
(13
|
)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
54,718
|
|
|
$
|
(13
|
)
|
|
Corporate debt securities
|
|
|
38,718
|
|
|
|
(18
|
)
|
|
|
1,536
|
|
|
|
(1
|
)
|
|
|
40,254
|
|
|
|
(19
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Short-term investments
|
|
|
93,436
|
|
|
|
(31
|
)
|
|
|
1,536
|
|
|
|
(1
|
)
|
|
|
94,972
|
|
|
|
(32
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
100,983
|
|
|
|
(140
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
100,983
|
|
|
|
(140
|
)
|
|
Corporate debt securities
|
|
|
65,392
|
|
|
|
(883
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
65,392
|
|
|
|
(883
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Marketable securities
|
|
|
166,375
|
|
|
|
(1,023
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
166,375
|
|
|
|
(1,023
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
1,502
|
|
|
|
(6
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
1,502
|
|
|
|
(6
|
)
|
|
Corporate debt securities
|
|
|
7,110
|
|
|
|
(29
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
7,110
|
|
|
|
(29
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Restricted investments
|
|
|
8,612
|
|
|
|
(35
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
8,612
|
|
|
|
(35
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
268,423
|
|
|
$
|
(1,089
|
)
|
|
$
|
1,536
|
|
|
$
|
(1
|
)
|
|
$
|
269,959
|
|
|
$
|
(1,090
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-15
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
(NOTE C) —
Investments (continued)
Short-term
investments, Marketable securities and Restricted
investments — unrealized losses (continued)
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
|
|
|
Loss Position
|
|
|
Loss Position
|
|
|
|
|
|
|
|
for Less Than
|
|
|
for Greater Than
|
|
|
|
|
|
|
|
Twelve Months
|
|
|
Twelve Months
|
|
|
Total
|
|
|
|
|
Fair
|
|
|
Unrealized
|
|
|
Fair
|
|
|
Unrealized
|
|
|
Fair
|
|
|
Unrealized
|
|
|
|
|
Value
|
|
|
Losses
|
|
|
Value
|
|
|
Losses
|
|
|
Value
|
|
|
Losses
|
|
|
|
|
Government-sponsored enterprise securities
|
|
$
|
23,026
|
|
|
$
|
(29
|
)
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
23,026
|
|
|
$
|
(29
|
)
|
|
Corporate debt securities
|
|
|
24,751
|
|
|
|
(181
|
)
|
|
|
4,474
|
|
|
|
(12
|
)
|
|
|
29,225
|
|
|
|
(193
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Short-term investments
|
|
|
47,777
|
|
|
|
(210
|
)
|
|
|
4,474
|
|
|
|
(12
|
)
|
|
|
52,251
|
|
|
|
(222
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
154,032
|
|
|
|
(673
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
154,032
|
|
|
|
(673
|
)
|
|
Corporate debt securities
|
|
|
76,595
|
|
|
|
(405
|
)
|
|
|
1,231
|
|
|
|
(24
|
)
|
|
|
77,826
|
|
|
|
(429
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Marketable securities
|
|
|
230,627
|
|
|
|
(1,078
|
)
|
|
|
1,231
|
|
|
|
(24
|
)
|
|
|
231,858
|
|
|
|
(1,102
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Government-sponsored enterprise securities
|
|
|
7,317
|
|
|
|
(17
|
)
|
|
|
14
|
|
|
|
—
|
|
|
|
7,331
|
|
|
|
(17
|
)
|
|
Corporate debt securities
|
|
|
6,212
|
|
|
|
(31
|
)
|
|
|
—
|
|
|
|
—
|
|
|
|
6,212
|
|
|
|
(31
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Subtotal — Restricted investments
|
|
|
13,529
|
|
|
|
(48
|
)
|
|
|
14
|
|
|
|
—
|
|
|
|
13,543
|
|
|
|
(48
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
291,933
|
|
|
$
|
(1,336
|
)
|
|
$
|
5,719
|
|
|
$
|
(36
|
)
|
|
$
|
297,652
|
|
|
$
|
(1,372
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company has evaluated its investments and has determined
that no investments have an
other-than-temporary
impairment, as it has no intent to sell securities with
unrealized losses and it is not more likely than not that the
Company will be required to sell any securities with unrealized
losses, given the Company’s current and anticipated
financial position.
The Company owned 322
available-for-sale
U.S Treasury obligations, government-sponsored enterprise
securities and corporate debt securities as of December 31,
2010. Of these 322 securities, 62 had unrealized losses as of
December 31, 2010.
The Company’s equity investments of less than 20% in
privately-held companies are carried at cost and are included in
long-term equity investments on the consolidated balance sheets.
See Note D, Collaborations and U.S. Government
Agreement, for information regarding the Company’s
investment in Aegera Therapeutics, Inc. There were no events or
circumstances during 2010 that would have a significant adverse
effect on the carrying value of these investments.
F-16
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
(NOTE C) —
Investments (continued)
Other
Information
The following table summarizes maturities of the Company’s
short-term investments, marketable securities and restricted
investment securities as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Short-term
|
|
|
Marketable
|
|
|
Restricted
|
|
|
|
|
Investments
|
|
|
Securities
|
|
|
Investments
|
|
|
|
|
Amortized
|
|
|
Fair
|
|
|
Amortized
|
|
|
Fair
|
|
|
Amortized
|
|
|
Fair
|
|
|
|
|
Cost
|
|
|
Value
|
|
|
Cost
|
|
|
Value
|
|
|
Cost
|
|
|
Value
|
|
|
|
|
Less than one year
|
|
$
|
279,540
|
|
|
$
|
282,016
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
$
|
39,842
|
|
|
$
|
40,126
|
|
|
Due in year two through year three
|
|
|
—
|
|
|
|
—
|
|
|
|
323,222
|
|
|
|
326,506
|
|
|
|
36,623
|
|
|
|
37,077
|
|
|
Due in year four through year five
|
|
|
—
|
|
|
|
—
|
|
|
|
70,004
|
|
|
|
70,680
|
|
|
|
1,951
|
|
|
|
2,013
|
|
|
Due after five years
|
|
|
—
|
|
|
|
—
|
|
|
|
19,103
|
|
|
|
18,979
|
|
|
|
289
|
|
|
|
294
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
279,540
|
|
|
$
|
282,016
|
|
|
$
|
412,329
|
|
|
$
|
416,165
|
|
|
$
|
78,705
|
|
|
$
|
79,510
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company’s short-term investments include
mortgage-backed securities with an aggregate cost of $53,944 and
$22,471 as of December 31, 2010 and 2009 respectively, and
an aggregate fair value of $54,718 and $23,026 as of
December 31, 2010 and 2009, respectively. The
Company’s marketable securities include mortgage-backed
securities with an aggregate cost of $79,605 and $58,534 as of
December 31, 2010 and 2009, respectively, and an aggregate
fair value of $80,746 and $59,981 as of December 31, 2010
and 2009, respectively. The Company’s restricted
investments include mortgage-backed securities with an aggregate
cost of $4,748 and $7,483 as of December 31, 2010 and 2009,
respectively, and an aggregate fair value of $4,834 and $7,593
as of December 31, 2010 and 2009, respectively. These
securities have no single maturity date and, accordingly, have
been allocated on a pro rata basis to each maturity range based
on each maturity range’s percentage of the total value.
The Company’s net proceeds, realized gains and realized
losses from its investments are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
Proceeds on sale of investments prior to maturity
|
|
$
|
708,896
|
|
|
$
|
368,560
|
|
|
$
|
237,861
|
|
|
Realized gains
|
|
|
1,673
|
|
|
|
7,026
|
|
|
|
34,113
|
|
|
Realized losses
|
|
|
(1,976
|
)
|
|
|
(2,522
|
)
|
|
|
(479
|
)
|
Realized gains and losses related to the Company’s
short-term investments, marketable securities and restricted
investments are included in investment income in the
consolidated statements of operations. The cost of the
securities sold is based on the specific identification method.
Realized gains shown above also include gains related to the
sale of long-term equity investments, which are shown separately
on the consolidated statements of operations.
During 2010, 2009 and 2008, the Company recognized interest
income of $17,269, $13,506 and $22,406 respectively, in
investment income.
(NOTE D) —
Collaborations and U.S. Government Agreement
Principal
Agreements
Agreements
with GlaxoSmithKline
During 2006, the Company entered into a license agreement with
GSK for the co-development and commercialization of BENLYSTA
arising from an option GSK exercised in 2005, relating to an
earlier collaboration agreement.
F-17
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE D) —
|
Collaborations
and U.S. Government Agreement (continued)
|
Principal
Agreements (continued)
Agreements
with GlaxoSmithKline (continued)
The agreement grants GSK a co-development and
co-commercialization license, under which both companies will
jointly conduct activities related to the development and sale
of products in the United States and abroad. The Company and GSK
share Phase 3 and 4 development costs, and share sales and
marketing expenses and profits of any product commercialized
under the agreement. The Company will have primary
responsibility for bulk manufacturing and for commercial
manufacturing of the finished drug product. During 2010, the
Company completed the second of two Phase 3 clinical trials and
filed a Biologics License Application (“BLA”) with the
FDA for BENLYSTA. In partial consideration of the rights granted
to GSK in this agreement, the Company received a non-refundable
payment of $24,000 during 2006 and recognized this payment as
revenue over the remaining clinical development period, which
ended in 2010. The Company recognized revenue of $3,445, $4,737
and $6,545 in 2010, 2009 and 2008, respectively, relating to
this payment.
The BENLYSTA agreement arises from a 1993 agreement, as amended,
in which the Company entered into a collaboration agreement
providing GSK a first right to develop and market products in
human and animal health care (“GSK Products”), based
upon human genes identified by the Company. In June 1996, this
agreement was substantially amended (the “1996 GSK
Agreement”).
With respect to the Company’s rights under the 1996 GSK
Agreement, the Company is entitled to (1) royalties on the
net sales of certain GSK Products developed pursuant to the
agreement, (2) product development milestones and
(3) the option to co-promote up to 20% of any product
developed by GSK under the collaboration agreement. If the
Company were to exercise its option to co-promote any GSK
Products, it would be entitled to receive additional amounts
from GSK in proportion to its level of co-promotion. The Company
has been informed that GSK is pursuing research programs
involving specific genes for the creation of small molecule
drugs. The Company cannot provide any assurance that any of
these programs will be continued or result in any approved drugs.
In 2004, the Company entered into an agreement with GSK under
which GSK acquired exclusive worldwide rights to develop and
commercialize albiglutide, a drug that had been in late-stage
preclinical development by the Company for potential use in the
treatment of diabetes. In 2004, the Company received an up-front
fee of $6,000 and is recognizing this revenue ratably over the
clinical development period, which is estimated to be eight
years. With respect to this fee, the Company recognized $460 as
revenue in 2010 and $741 as revenue each year in the two year
period ended December 31, 2009. As of December 31,
2010, the Company has also received and recognized development
milestones aggregating $27,000 under the agreement, including
$9,000 received and recognized in 2009.
Agreement
with Novartis
During 2006, the Company entered into an agreement with Novartis
International Pharmaceutical Ltd. (“Novartis”) for the
co-development and commercialization of
ZALBINtm.
In June 2010, the Company announced that it had received
preliminary written feedback from the FDA regarding the BLA it
filed in November 2009 for ZALBIN. The FDA expressed concerns
regarding the risk-benefit assessment of ZALBIN dosed at 900-mcg
every two weeks. In September 2010, the Company and Novartis
decided to end further development of ZALBIN in anticipation of
a Complete Response Letter to be received from the FDA in early
October. In October 2010, the Company received the FDA’s
Complete Response Letter.
Under the agreement, Novartis had paid the Company $207,500,
including a non-refundable up-front license fee and payments
upon the successful attainment of certain milestones. The
Company was recognizing these payments as revenue ratably over
the estimated remaining development period. Based on the
decision to end further
F-18
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE D) —
|
Collaborations
and U.S. Government Agreement (continued)
|
Principal
Agreements (continued)
Agreement
with Novartis (continued)
development of ZALBIN, the Company recognized all remaining
deferred revenue related to these payments in September 2010.
The Company recognized revenue of $82,806, $54,158 and $35,408
in 2010, 2009, and 2008, respectively, under this agreement.
License
Agreement and Manufacturing Services Agreement with Teva
Biopharmaceuticals USA, Inc. (formerly CoGenesys)
In 2008, Teva Pharmaceuticals Industries, Ltd.
(“Teva”) acquired all of the outstanding stock of
CoGenesys and CoGenesys became a wholly-owned subsidiary of Teva
called Teva Biopharmaceuticals USA, Inc. (“Teva Bio”).
The Company had sold its CoGenesys division in 2006 and entered
into a license agreement, as amended, that is now with Teva Bio.
Under the license agreement, as amended, the Company is entitled
to various milestone and royalty rights on certain products, if
they are developed and commercialized. Teva Bio can obtain
additional product rights by extending the initial seven-year
research term upon the payment of additional consideration. In
addition, the Company entered into a three-year manufacturing
services agreement, as amended, which ended during 2009. The
Company allocated, based on estimated fair values, $7,575 of its
consideration received to the product license and manufacturing
services agreement, which was recognized ratably over the term
of the manufacturing services agreement, as amended. The Company
recognized license revenue of $1,052 and $2,525 during 2009 and
2008, respectively, and manufacturing services revenue of $1,035
and $367 during 2009 and 2008, respectively, relating to these
agreements. See Note L, Teva Biopharmaceuticals USA, Inc.
(formerly CoGenesys), for additional discussion.
Collaboration
reimbursements with respect to Novartis and GSK
The Company’s research and development expenses in 2010 of
$196,370 are net of $62,022 of costs reimbursed by GSK and
include $663 of costs incurred by Novartis. Research and
development expenses of $173,709 in 2009 were net of $43,069 and
$851 of costs reimbursed by GSK and Novartis, respectively.
Research and development expenses of $243,257 in 2008 were net
of $51,783 and $36,104 reimbursed by GSK and Novartis,
respectively. The Company shares certain research and
development costs including personnel costs, outside services,
manufacturing, and overhead with Novartis and GSK under cost
sharing provisions in the collaboration agreements.
U.S.
Government Agreement
During 2006, the USG exercised its option under the second phase
of a 2005 contract to purchase 20,001 doses of raxibacumab for
its U.S. Strategic National Stockpile (“SNS”).
Under this two-phase contract, the Company has supplied
raxibacumab, a human monoclonal antibody developed for use in
the treatment of anthrax disease, to the USG. Along with the
cost to manufacture the 20,001 doses, the Company has incurred
the cost to conduct several animal and human studies as part of
this contract. During 2009, the Company received authorization
from BARDA to ship raxibacumab to the SNS and delivered all of
the 20,001 doses. In July 2009, the USG agreed to purchase
45,000 additional doses of raxibacumab for the SNS, to be
delivered over a three-year period beginning in 2009. The
Company expects to receive a total of approximately $142,000
from this order as deliveries are completed The Company
recognized $47,159 and $154,074 in product revenue related to
raxibacumab in 2010 and 2009, respectively. The Company
recognized $1,438 and $26,146 in manufacturing and development
services revenue related to the work to conduct the animal and
human studies and other raxibacumab activities in 2010 and 2009,
respectively. The Company is entitled to receive approximately
$20,000 under the contract with the USG upon FDA licensure of
raxibacumab.
F-19
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE D) —
|
Collaborations
and U.S. Government Agreement (continued)
|
Other
Collaborative and License Agreements
During 2007, the Company entered into a collaboration and
license agreement with Aegera Therapeutics, Inc.
(“Aegera”) of Montreal, Canada under which the Company
acquired exclusive worldwide rights (excluding Japan) to develop
and commercialize certain oncology molecules and related backup
compounds to be chosen during a three-year research period.
During 2010, the Company and Aegera agreed to extend the
research period by one year. Under the agreement, the Company
paid Aegera an aggregate of $20,000 for the license and for an
equity investment in Aegera. The Company allocated $16,852 to
the license fee and $3,148 to the investment. The value per
share assigned to this investment was equal to the value per
share obtained by Aegera through external financing earlier in
2007. Aegera will be entitled to receive up to $295,000 in
future development and commercial milestone payments, including
a $5,000 milestone payment made by the Company during 2008.
Aegera will receive royalties on net sales in the Company’s
territory. In North America, Aegera will have the option to
co-promote with the Company, under which Aegera will share
certain expenses and profits in lieu of its royalties. The
Company incurred and expensed research costs of $2,364, $2,321
and $2,249 related to the Aegera agreement during 2010, 2009 and
2008, respectively.
As of December 31, 2010, the Company values its cost basis
investment in Aegera at $3,237, based on year-end exchange
rates, which is included in long-term equity investments on the
consolidated balance sheets.
In 1999, the Company entered into a collaborative agreement with
Cambridge Antibody Technology Ltd. (assumed by MedImmune LLC
(“MedImmune”) through acquisition) to jointly pursue
the development of fully human monoclonal antibody therapeutics.
MedImmune will receive milestone payments from the Company in
connection with the development of any such antibodies as well
as royalty payments on the Company’s net sales of such
licensed product following regulatory approval. In the event of
the achievement of other milestones or successful product
launch, the Company would be obligated to pay MedImmune
additional compensation. Since 1999, the Company has exercised
one option and made payments to MedImmune totaling $5,000
pursuant to this agreement.
In 2000, the Company entered into a second agreement with
Cambridge Antibody Technology Ltd. The 2000 agreement provides
the Company with rights to use MedImmune technology to develop
and sell an unlimited number of fully human antibodies for
therapeutic and diagnostic purposes. The Company will pay
MedImmune clinical development milestones and royalties based on
product sales. Under this same agreement, the Company paid
MedImmune $12,000 for research support. Since 2000, the Company
has exercised several options and made certain payments. During
2010 and 2009, the Company incurred milestone and royalty
expenses to MedImmune of approximately $2,389 and $8,600,
respectively, associated with the sale of raxibacumab to the
USG, which are included in cost of product sales on the
consolidated statements of operations. No option or milestone
expenses were incurred in 2008.
|
|
|
(NOTE E) —
|
Other
Financial Information
|
Collaboration
Receivables
Collaboration receivables of $18,856 as of December 31,
2010 includes $13,165 due to the Company from GSK for
manufacturing costs incurred to produce pre-launch commercial
product which will be sold within the next year. Collaboration
receivables also includes $5,166 in unbilled receivables from
GSK in connection with the Company’s cost-sharing
agreements and other unbilled receivables. Collaboration
receivables of $10,356 as of December 31, 2009 includes
$9,461 in unbilled receivables from GSK in connection with the
Company’s cost sharing agreements, and other billed and
unbilled receivables.
Collaboration receivables, non-current of $29,225 relates to the
amount due to the Company from GSK for manufacturing costs
incurred to produce pre-launch commercial product which will not
be sold within the next year.
F-20
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE E) —
|
Other
Financial Information (continued)
|
Collaboration
Receivables (continued)
Within the December 31, 2009 consolidated balance sheet,
$6,920 has been reclassified from other assets to collaboration
receivables, non-current, to conform to current year
presentation. Collaboration receivables, non-current, was deemed
immaterial in 2009 for separate disclosure. Due to the
Company’s continued production of pre-launch commercial
product, it has become material and therefore is separately
disclosed. The effect of the reclassification is not material to
the consolidated financial statements.
Inventory
Inventories consist of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Raw materials
|
|
$
|
12,641
|
|
|
$
|
4,293
|
|
|
Work-in-process
|
|
|
23,426
|
|
|
|
9,512
|
|
|
Finished goods
|
|
|
7,024
|
|
|
|
6,344
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
43,091
|
|
|
$
|
20,149
|
|
|
|
|
|
|
|
|
|
|
|
Property,
Plant and Equipment
Property, plant and equipment are stated at cost and are
summarized as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Building (LSM)
|
|
$
|
204,151
|
|
|
$
|
204,151
|
|
|
Laboratory and production equipment
|
|
|
96,462
|
|
|
|
94,644
|
|
|
Computer equipment and software
|
|
|
39,296
|
|
|
|
38,793
|
|
|
Land and improvements
|
|
|
30,521
|
|
|
|
30,521
|
|
|
Leasehold improvements
|
|
|
26,678
|
|
|
|
25,046
|
|
|
Furniture and office equipment
|
|
|
7,274
|
|
|
|
6,629
|
|
|
Construction-in-progress
|
|
|
5,364
|
|
|
|
3,469
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
409,746
|
|
|
|
403,253
|
|
|
Less: accumulated depreciation
|
|
|
(156,624
|
)
|
|
|
(140,130
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
253,122
|
|
|
$
|
263,123
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation expense was $20,055, $19,960 and $19,584 for 2010,
2009 and 2008, respectively.
F-21
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE E) —
|
Other
Financial Information (continued)
|
Accounts
Payable and Accrued Expenses
Accounts payable and accrued expenses are comprised of the
following:
| |
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Accounts payable
|
|
$
|
6,364
|
|
|
$
|
1,036
|
|
|
Accrued clinical trial costs
|
|
|
13,665
|
|
|
|
22,083
|
|
|
Other accrued expenses
|
|
|
21,769
|
|
|
|
15,250
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
41,798
|
|
|
$
|
38,369
|
|
|
|
|
|
|
|
|
|
|
|
Accrued clinical trial costs consist primarily of investigator
fees, contract research organization services and laboratory
costs.
Collaboration
Payable
Collaboration payable of $12,984 as of December 31, 2010
represents cost reimbursements due to GSK and Novartis in
connection with the Company’s cost sharing agreements for
the six months ended December 31, 2010. Collaboration
payable of $4,000 as of December 31, 2009 represents cost
reimbursements due to Novartis in connection with the
Company’s cost sharing agreement for the six months ended
December 31, 2009. Within the December 31, 2009
consolidated balance sheet, $4,000 has been reclassified from
accounts payable and accrued expenses to collaboration payable
to conform to the current year presentation. Collaboration
payable was deemed immaterial in 2009 for separate disclosure.
|
|
|
(NOTE F) —
|
Long-Term
Debt
|
The components of long-term debt are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
Debt
|
|
Interest Rate
|
|
|
Maturities
|
|
|
2010
|
|
|
2009
|
|
|
|
|
21/4% Convertible
Subordinated Notes due 2011
|
|
|
2.25
|
%
|
|
|
October 2011
|
|
|
$
|
188,620
|
|
|
$
|
178,104
|
|
|
21/4% Convertible
Subordinated Notes due 2012
|
|
|
2.25
|
%
|
|
|
August 2012
|
|
|
|
184,231
|
|
|
|
171,703
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
372,851
|
|
|
|
349,807
|
|
|
BioMed lease financing
|
|
|
11.0
|
%
|
|
|
May 2026
|
|
|
|
250,516
|
|
|
|
248,628
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
623,367
|
|
|
|
598,435
|
|
|
Less current portion
|
|
|
|
|
|
|
|
|
|
|
(188,620
|
)
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
434,747
|
|
|
$
|
598,435
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-22
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE F) —
|
Long-Term
Debt (continued)
|
Annual maturities of all long-term debt (representing cash to be
paid) are as follows:
| |
|
|
|
|
|
2011
|
|
$
|
197,100
|
|
|
2012
|
|
|
206,740
|
|
|
2013
|
|
|
—
|
|
|
2014
|
|
|
—
|
|
|
2015
|
|
|
187
|
|
|
2016 and thereafter
|
|
|
52,774
|
|
|
|
|
|
|
|
|
|
|
$
|
456,801
|
|
|
|
|
|
|
|
The difference between total debt of $623,367 and annual
maturities of $456,801 is due to the accounting for the
sale-leaseback of the Company’s headquarters land and LSM
as a financing transaction and the debt discount relating to the
convertible subordinated notes. During 2006, the Company entered
into a purchase and sale agreement with BioMed in connection
with the Company’s Traville headquarters land and LSM. The
Company is accounting for the sale-leaseback as a financing
transaction. Payments due for the BioMed debt resulting from
this financing are based upon an allocation of fair value of the
properties included in the transaction. Aggregate lease
financing payments, including interest, over the remaining
fifteen year period are approximately $446,522 including an
annual lease escalation of 2%. Interest expense associated with
this debt is being calculated at approximately 11%, which
approximated the Company’s incremental borrowing rate at
the time of the agreement. For the first nine years of the
leases, the payments are less than the amount of calculated
interest expense, which results in an increase in the debt
balance during this period, reaching $254,699 in 2015.
Accordingly, the Company has classified the full amount of the
debt outstanding as of December 31, 2010 as long-term.
Beginning in 2015, the payments begin to reduce the debt balance
and are reflected in the annual maturities shown herein. At the
end of the twenty-year leases, the remaining debt will be
approximately $201,738.
During 2004, the Company completed the private placement of
$280,000 of
21/4% Convertible
Subordinated Notes due 2011
(“21/4% Notes
due 2011”), convertible into common stock at approximately
$15.55 per share. Under FASB ASC Topic 470, Debt,
$191,804 of the proceeds from the
21/4% Notes
due 2011 was allocated to long-term debt and $88,196 was
allocated to equity based on the Company’s non-convertible
borrowing rate in effect at the time the notes were issued. Debt
issuance costs for the $191,804 of
21/4% Notes
due 2011 amounted to approximately $5,924, which are being
amortized on a straight-line basis, which approximates the
effective interest method, over the life of the
21/4% Notes
due 2011. Accumulated amortization of the debt issuance costs
for the
21/4% Notes
due 2011 is approximately $5,091 and $4,245 as of
December 31, 2010 and 2009, respectively. As of
December 31, 2010 and 2009, the net balance in deferred
financing costs associated with the
21/4% Notes
due 2011 was $436 and $1,032, respectively. During 2009, the
Company repurchased
21/4% Notes
due 2011 with a face value of $82,900 (as discussed below), and
wrote off the related unamortized debt issuance costs and debt
discount. The
21/4% Notes
due 2011 also contain a provision for a “make-whole”
premium to be paid by the Company to holders of the
21/4% Notes
due 2011 in the event of certain changes in control that could
occur during the life of the
21/4% Notes
due 2011. The premium is payable in the form of cash, the
Company’s common stock, or the same form of consideration
used to pay for the shares of the Company’s common stock in
connection with the transaction constituting the change in
control. The premium declines over time and is based upon the
price of the Company’s stock as of the effective date of
the change in control. As of December 31, 2010, the maximum
premium possible is approximately $21,533, or approximately 11%
of the aggregate face value of
21/4% Notes
due 2011 outstanding, in the event a qualified change in control
occurs with a stock price of $16.00 per share at such date. If
the stock price on the effective date of a change in control is
less than $11.105 per share or greater than $55.00 per share, no
premium will be paid.
During 2005, the Company completed the private placement of
$230,000 of
21/4% Convertible
Subordinated Notes due 2012
(“21/4% Notes
due 2012”), convertible into common stock at approximately
$17.78 per share. Under FASB
F-23
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE F) —
|
Long-Term
Debt (continued)
|
ASC Topic 470, $143,266 of the proceeds from the
21/4% Notes
due 2012 was allocated to long-term debt and $86,734 was
allocated to equity based on the Company’s non-convertible
borrowing rate in effect at the time the notes were issued. Debt
issuance costs for the $143,266 of
21/4% Notes
due 2012 amounted to approximately $4,220, which are being
amortized on a straight-line basis, which approximates the
effective interest method, over the life of the
21/4% Notes
due 2012. Accumulated amortization of the debt issuance costs
for the
21/4% Notes
due 2012 is approximately $3,204 and $2,601 as of
December 31, 2010 and 2009, respectively. As of
December 31, 2010 and 2009, the net balance in deferred
financing costs associated with the
21/4% Notes
due 2012 was $869 and $1,411, respectively. During 2009, the
Company repurchased
21/4% Notes
due 2012 with a face value of $23,250 (as discussed below), and
wrote off the related unamortized debt issuance costs and debt
discount. The
21/4% Notes
due 2012 also contain a provision for a “make-whole”
premium to be paid by the Company to holders of the
21/4% Notes
due 2012 in the event of certain changes in control that could
occur during the life of the
21/4% Notes
due 2012. The premium is payable in the form of the
Company’s common stock by increasing the conversion rate to
the holders of the notes who convert their notes. The premium,
which is expressed as additional shares of common stock per one
thousand dollars principal amount of notes, is based upon the
price of the Company’s stock as of the effective date of
the change in control. The maximum premium possible is
approximately $34,456, or approximately 17% of the aggregate
face value of
21/4% Notes
due 2012 outstanding, in the event a qualified change in control
occurs with a stock price of at least $14.82 per share at such
date. If the stock price on the effective date of a change in
control is less than $14.82 per share or greater than $100.00
per share, no premium will be paid.
During 2009, the Company repurchased
21/4% Notes
due 2011 with a face value of $82,900 and
21/4% Notes
due 2012 with a face value of $23,250 for an aggregate cost of
approximately $50,000 plus accrued interest. The repurchase
resulted in a gain on extinguishment of debt of $38,873, net of
the related debt discount of $16,424 and deferred financing
charges of $855.
The carrying amount and fair value of the Company’s
long-term debt are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
|
|
Carrying
|
|
|
Fair
|
|
|
Carrying
|
|
|
Fair
|
|
|
|
|
Amount
|
|
|
Value
|
|
|
Amount
|
|
|
Value
|
|
|
|
|
21/4% Convertible
Subordinated Notes due 2011
|
|
$
|
188,620
|
|
|
$
|
320,311
|
|
|
$
|
178,104
|
|
|
$
|
402,084
|
|
|
21/4% Convertible
Subordinated Notes due 2012
|
|
|
184,231
|
|
|
|
314,245
|
|
|
|
171,703
|
|
|
|
372,150
|
|
|
BioMed lease financing
|
|
|
250,516
|
|
|
|
266,016
|
|
|
|
248,628
|
|
|
|
265,293
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
623,367
|
|
|
$
|
900,572
|
|
|
$
|
598,435
|
|
|
$
|
1,039,527
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The components of the convertible subordinated debt are as
follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2010
|
|
|
|
|
Face
|
|
|
Unamortized
|
|
|
Carrying
|
|
|
|
|
Value
|
|
|
Debt Discount
|
|
|
Amount
|
|
|
|
|
21/4% Convertible
Subordinated Notes due 2011
|
|
$
|
197,100
|
|
|
$
|
(8,480
|
)
|
|
$
|
188,620
|
|
|
21/4% Convertible
Subordinated Notes due 2012
|
|
|
206,740
|
|
|
|
(22,509
|
)
|
|
|
184,231
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
403,840
|
|
|
$
|
(30,989
|
)
|
|
$
|
372,851
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
F-24
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE F) —
|
Long-Term
Debt (continued)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31, 2009
|
|
|
|
|
Face
|
|
|
Unamortized
|
|
|
Carrying
|
|
|
|
|
Value
|
|
|
Debt Discount
|
|
|
Amount
|
|
|
|
|
21/4% Convertible
Subordinated Notes due 2011
|
|
$
|
197,100
|
|
|
$
|
(18,996
|
)
|
|
$
|
178,104
|
|
|
21/4% Convertible
Subordinated Notes due 2012
|
|
|
206,750
|
|
|
|
(35,047
|
)
|
|
|
171,703
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
403,850
|
|
|
$
|
(54,043
|
)
|
|
$
|
349,807
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
With respect to the Company’s convertible subordinated
notes (the “Notes”), the Notes are unsecured
obligations of the Company and rank junior in right of payment
to the Company’s existing and future senior indebtedness.
The Notes are not redeemable prior to maturity, but can be
repurchased by the Company on the open market.
The indentures under which the Notes have been issued contain no
financial covenants or any restriction on the payments of
dividends, the incurrence of senior indebtedness, or other
indebtedness, or the Company’s issuance or repurchase of
securities. There are no sinking fund requirements with respect
to the Notes.
The fair value of the BioMed lease financing is determined using
a discounted cash flow analysis and current rates for corporate
debt having similar characteristics and companies with similar
credit worthiness. The Company concluded that its incremental
borrowing rate as of December 31, 2010 as compared to
December 31, 2009 has increased, resulting in an increase
in the fair value of the debt to $266,016.
|
|
|
(NOTE G) —
|
Commitments
and Other Matters
|
Leases
The Company leases office and laboratory premises pursuant to
operating leases expiring at various dates through 2026. Minimum
annual rentals are as follows:
| |
|
|
|
|
|
Year Ending December 31,
|
|
|
|
|
|
|
2011
|
|
$
|
20,876
|
|
|
2012
|
|
|
21,241
|
|
|
2013
|
|
|
21,613
|
|
|
2014
|
|
|
21,993
|
|
|
2015
|
|
|
22,380
|
|
|
2016 and thereafter
|
|
|
238,496
|
|
|
|
|
|
|
|
|
|
|
$
|
346,599
|
|
|
|
|
|
|
|
The operating lease commitment of $346,599 includes lease
payments associated with the Company’s lease with BioMed
for its Traville headquarters. During 2006 the Company entered
into a lease with BioMed for its Traville headquarters following
the termination of the Company’s Traville lease with its
former lessor. Based upon an allocation of fair value, the
initial annual rent for Traville was approximately $16,653. The
aggregate rental payments over the remaining lease term are
approximately $325,479, including an annual escalation of 2%
which is accounted for on a straight-line basis over the lease
term. The Company has an option to purchase the Traville
facility in 2016 for $303,000. There are no financial covenants
with respect to the BioMed lease.
As part of its agreement with BioMed, the Company committed to
exercise purchase options with respect to certain equipment
currently used at the Traville facility at the end of the
applicable equipment lease terms. The equipment was subject to
several operating leases with an unrelated party. During 2008,
the Company exercised the purchase option with regard to certain
leases at a cost of approximately $4,200, and exercised the
purchase option related to the remaining leases in 2009 at a
cost of approximately $5,300. The Company will transfer
ownership of this
F-25
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE G) —
|
Commitments
and Other Matters (continued)
|
Leases
(continued)
facility-related equipment to BioMed at the earlier of the end
of the Traville lease term or at certain other pre-specified
events.
The Company has entered into two long-term leases, as amended,
with MEDCO expiring January 1, 2019 for a small-scale
manufacturing facility aggregating 127,000 square feet and
built to the Company’s specifications. The Company has
accounted for these leases as operating leases. The facility was
financed primarily through a combination of MEDCO Bonds and
loans issued to MEDCO by certain State of Maryland agencies. The
Company has no equity interest in MEDCO.
Rent is based upon MEDCO’s debt service obligations. Annual
base rent under the leases is currently approximately $2,640.
The MEDCO Bonds are secured by letters of credit issued for the
account of MEDCO which expire in December 2011. The Company has
restricted investments of approximately $35,400 and $34,500 as
of December 31, 2010 and 2009, respectively, associated
with these leases which serve as security for the MEDCO letters
of credit reimbursement obligation. Upon default or early lease
termination, the MEDCO Bond indenture trustee can draw upon the
letters of credit to pay the MEDCO Bonds as they are tendered.
In such an event, the Company could lose part or all of its
restricted investments and could record a charge to earnings for
a corresponding amount. Alternatively, the Company has an option
during or at the end of the lease term to purchase this facility
for an aggregate amount that declines from approximately $36,000
in 2011 to approximately $21,000 in 2019. The amended leases
contain no debt covenants with respect to the Company’s
financial condition.
See Note C, Investments, for additional discussion of the
Company’s restricted investments.
The Company’s leases for office and laboratory space
provide for certain rent escalations on each anniversary of the
lease commencement date. For financial reporting purposes, rent
expense is charged to operations on a straight-line basis over
the term of the lease, resulting in a liability for deferred
rent of $10,358 and $8,664 as of December 31, 2010 and
2009, respectively.
The Company had entered into various sale-leaseback transactions
resulting in equipment leases with rental and buy-out payments,
with initial terms ranging from five to seven years. The Company
accounted for these leases as operating leases. Under the
leases, the Company was required to maintain minimum levels of
unrestricted cash, cash equivalents and marketable securities.
During 2009 and 2008, the Company exercised its remaining
purchase options at the end of the initial lease terms and
released the related restricted investments.
Rent expense aggregated $20,949, $22,357 and $27,588 for 2010,
2009 and 2008, respectively. The decrease in rent expense each
year is due to the expiration of certain equipment leases.
401(k)
Plan
The Company has a 401(k) pension plan available to eligible
full-time employees. Participating employees may contribute up
to 100% of their total eligible compensation to the plan,
subject to Internal Revenue Service limitations. The Company
currently matches a portion of the employee contributions. The
Company’s contribution was $2,592, $1,645 and $1,945 for
2010, 2009 and 2008, respectively.
Contingent
Liabilities
In the ordinary course of business, the Company is involved in
various legal proceedings, including, among others, patent
oppositions, patent infringement litigation and other matters
incidental to its business. While it is not possible to
accurately predict or determine the eventual outcome of these
matters or estimate a range of loss, one or more of these
matters currently pending could have a material adverse effect
on the Company’s financial condition, results of operations
or cash flows.
F-26
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE H) —
|
Stockholders’
Equity
|
Public
Offerings of Common Stock
During 2009, the Company completed two public offerings of its
common stock. The Company issued 26,697,250 shares in
August 2009 at a price of $14.00 per share, resulting in net
proceeds of approximately $356,500. The Company also issued
17,825,000 shares in December 2009 at a price of $26.75 per
share, resulting in net proceeds of approximately $456,400.
Stock-based
Compensation Plans
The Company has two stock-based compensation plans as described
below. The following is a summary of the stock-based
compensation expense that has been recorded in the consolidated
statements of operations for the years indicated:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Employee stock option and employee stock purchase plan
|
|
$
|
24,277
|
|
|
$
|
11,935
|
|
|
$
|
17,630
|
|
|
Restricted stock units
|
|
|
1,068
|
|
|
|
589
|
|
|
|
647
|
|
|
Restricted stock awards
|
|
|
85
|
|
|
|
—
|
|
|
|
316
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total
|
|
$
|
25,430
|
|
|
$
|
12,524
|
|
|
$
|
18,593
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
No income tax benefit was recognized in the consolidated
statements of operations for stock-based compensation for the
years presented due to the Company’s net loss position.
Stock
Incentive Plan
Stock Options
The Company has an Incentive Plan under which options to
purchase new shares of the Company’s common stock may be
granted to employees, consultants and directors at an exercise
price no less than the quoted market value on the date of grant.
The Incentive Plan also provides for awards in the form of stock
appreciation rights, restricted (non-vested) or unrestricted
stock awards, stock-equivalent units or performance-based stock
awards. The Company issues both qualified and non-qualified
options under the Incentive Plan. The vesting period of the
options is determined by the Board of Directors and is generally
four years. Upon acquisition by a person, or group of persons,
of more than 50% of the Company’s outstanding common stock,
outstanding options shall immediately vest in full and be
exercisable. The Company recognizes compensation expense for an
award with only service conditions that has a graded vesting
schedule on a straight-line basis over the requisite service
period for the entire award. All options expire after ten years
or earlier from the date of grant.
As of December 31, 2010, the total authorized number of
shares under the Incentive Plan, including prior plans, was
54,809,554. Options available for future grant were 4,706,605 as
of December 31, 2010.
F-27
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE H) —
|
Stockholders’
Equity (continued)
|
Stock-based
Compensation Plans (continued)
Stock
Incentive Plan (continued)
A summary of stock option activity for 2010 is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
|
|
|
|
|
|
|
|
Weighted-Average
|
|
|
Contractual Term
|
|
|
Aggregate Intrinsic
|
|
|
|
|
Shares
|
|
|
Exercise Price
|
|
|
(years)
|
|
|
Value(1)
|
|
|
|
|
Outstanding at January 1, 2010
|
|
|
24,601,174
|
|
|
$
|
13.62
|
|
|
|
5.85
|
|
|
$
|
476,728
|
|
|
Granted
|
|
|
5,309,761
|
|
|
|
30.36
|
|
|
|
|
|
|
|
|
|
|
Exercised
|
|
|
(3,600,641
|
)
|
|
|
10.51
|
|
|
|
|
|
|
|
66,779
|
|
|
Forfeited
|
|
|
(440,817
|
)
|
|
|
9.09
|
|
|
|
|
|
|
|
|
|
|
Expired
|
|
|
(1,840,789
|
)
|
|
|
57.13
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2010
|
|
|
24,028,688
|
|
|
|
14.55
|
|
|
|
6.32
|
|
|
|
275,278
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Vested and expected to vest at December 31, 2010
|
|
|
23,184,862
|
|
|
|
14.47
|
|
|
|
6.24
|
|
|
|
266,731
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exercisable at December 31, 2010
|
|
|
15,715,563
|
|
|
|
12.46
|
|
|
|
5.09
|
|
|
|
200,972
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
|
Aggregate intrinsic value includes only options in which the
exercise price of the option is less than the market value of
the Company’s stock on December 31, 2010, or for
exercised options, the intrinsic value on the exercise date. |
The following table summarizes information about stock options
outstanding as of December 31, 2010:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Options Outstanding
|
|
|
Options Exercisable
|
|
|
|
|
|
|
|
Weighted-
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Average
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Remaining
|
|
|
Weighted-Average
|
|
|
|
|
|
|
|
|
|
|
Number
|
|
|
Contractual
|
|
|
Exercise
|
|
|
Number
|
|
|
Weighted-Average
|
|
|
Range of Exercise Price
|
|
Outstanding
|
|
|
Life (In Years)
|
|
|
Price
|
|
|
Exercisable
|
|
|
Exercise Price
|
|
|
|
|
$0.52 to $10.00
|
|
|
7,414,134
|
|
|
|
7.28
|
|
|
$
|
3.71
|
|
|
|
4,113,051
|
|
|
$
|
4.67
|
|
|
$10.01 to $12.50
|
|
|
7,237,748
|
|
|
|
4.93
|
|
|
|
10.95
|
|
|
|
7,021,482
|
|
|
|
10.96
|
|
|
$12.51 to $15.00
|
|
|
2,534,306
|
|
|
|
3.73
|
|
|
|
12.85
|
|
|
|
2,531,487
|
|
|
|
12.85
|
|
|
$15.01 to $35.00
|
|
|
6,070,839
|
|
|
|
8.60
|
|
|
|
29.08
|
|
|
|
1,277,882
|
|
|
|
26.16
|
|
|
$35.01 to $70.00
|
|
|
771,661
|
|
|
|
0.78
|
|
|
|
43.65
|
|
|
|
771,661
|
|
|
|
43.65
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24,028,688
|
|
|
|
6.32
|
|
|
|
14.55
|
|
|
|
15,715,563
|
|
|
|
12.46
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
During 2010, 2009 and 2008, the Company issued 3,600,641,
4,584,767 and 364,236 shares of common stock, respectively,
in conjunction with stock option exercises. The Company received
cash proceeds from the exercise of these stock options of
approximately $37,841, $48,147 and $3,443, for 2010, 2009 and
2008, respectively.
As of December 31, 2010, total unrecognized compensation
cost related to stock options amounted to $85,447, which is
expected to be recognized over a weighted-average period of
2.6 years as the options vest. There were non-vested stock
options outstanding for 8,329,125 shares as of
December 31, 2010.
The total intrinsic value of stock options exercised during
2010, 2009 and 2008 was approximately $66,779, $51,205 and $720,
respectively. The total fair value of stock options which vested
during 2010, 2009 and 2008 was
F-28
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE H) —
|
Stockholders’
Equity (continued)
|
Stock-based
Compensation Plans (continued)
Stock
Incentive Plan (continued)
approximately $20,286, $12,353 and $17,078, respectively. The
weighted-average grant-date fair value of stock options granted
during 2010, 2009 and 2008 was $17.31, $0.91 and $2.25 per
share, respectively.
The fair values of employee stock options granted during 2010,
2009 and 2008 were determined based on the Black-Scholes-Merton
option-pricing model using the following range of assumptions:
| |
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
2010
|
|
2009
|
|
2008
|
|
|
|
Expected life:
|
|
|
|
|
|
|
|
Stock options
|
|
5.5 years
|
|
5.5 years
|
|
5.4 years
|
|
Employee stock purchase plan rights
|
|
1.0 years
|
|
1.0 years
|
|
1.0 years
|
|
Interest rate
|
|
1.3% - 2.7%
|
|
1.4% - 2.8%
|
|
1.2% - 3.6%
|
|
Volatility
|
|
55.0% - 65.4%
|
|
53.0% - 83.5%
|
|
41.9% - 57.3%
|
|
Dividend yield
|
|
0%
|
|
0%
|
|
0%
|
An explanation of the above assumptions is as follows:
Expected Life of Stock-based Awards — The
expected life of stock-based awards is the period of time for
which the stock-based award is expected to be outstanding. This
estimate is based on historical exercise data.
Interest Rate — The risk-free rate over the
expected life of the option is based on the U.S. Treasury
yield curve in effect at the time of grant.
Volatility — Volatility is a measure of the
amount by which a financial variable such as a share price has
fluctuated (historical volatility) or is expected to fluctuate
(implied volatility) during a period. The Company uses a
combination of historical volatility and the implied volatility
of its traded convertible notes as the basis for its expected
volatility. Because the Company’s traded convertible notes
are nearing maturity, in 2010 the Company added historical
volatility to its overall volatility calculation. The weighted
average volatility used was 63.5%, 55.9% and 43.7% for 2010,
2009 and 2008, respectively.
Dividend Yield — The Company has never
declared or paid dividends and has no plans to do so in the
foreseeable future.
Restricted
Stock
Under the Incentive Plan, the Company has granted both
restricted stock awards and restricted stock units
(“RSUs”). Beginning in 2007, employees of the Company
could elect to receive RSUs in lieu of a portion of their stock
option grants. RSUs have service conditions and vest ratably on
an annual basis over a four-year period. During 2010, the
Company awarded 81,722 RSUs at a weighted-average grant date
fair value of $31.91 per share. The Company incurred $1,153,
$589 and $963 of compensation expense for 2010, 2009 and 2008,
respectively, related to both RSUs and restricted stock awards.
F-29
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE H) —
|
Stockholders’
Equity (continued)
|
Stock-based
Compensation Plans (continued)
Stock
Incentive Plan (continued)
A summary of the status of the Company’s restricted stock
as of December 31, 2010 and changes during 2010, is
presented below:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted-Average
|
|
|
|
|
|
|
|
Grant-Date Fair
|
|
|
|
|
Shares
|
|
|
Value
|
|
|
|
|
Restricted stock at January 1, 2010
|
|
|
205,737
|
|
|
$
|
6.39
|
|
|
Granted
|
|
|
81,722
|
|
|
|
31.91
|
|
|
Vested
|
|
|
(79,982
|
)
|
|
|
7.61
|
|
|
Forfeited
|
|
|
(3,695
|
)
|
|
|
13.63
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted stock at December 31, 2010
|
|
|
203,782
|
|
|
|
16.01
|
|
|
|
|
|
|
|
|
|
|
|
|
Expected to vest at December 31, 2010
|
|
|
177,263
|
|
|
|
16.56
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation expense under FASB ASC Topic 718,
Compensation — Stock Compensation, for 2010, 2009
and 2008 is not necessarily representative of the level of
stock-based compensation expense in future years due to, among
other things, (1) the vesting period of the stock-based
compensation and (2) the number and fair value of
additional stock-based grants in future years.
Employee
Stock Purchase Plan
The Company has an Employee Stock Purchase Plan (the
“Purchase Plan”), as amended, registering
2,000,000 shares of $0.01 par value common stock for
issuance under this plan. Under the Purchase Plan, eligible
employees may purchase shares of common stock on certain dates
and at certain prices as set forth in the plan. The common stock
is purchased under the Purchase Plan at a discounted rate,
currently at 15%, which results in this plan qualifying as
compensatory. The first purchase period for the Purchase Plan
began January 1, 2001. During 2010, the Company issued
71,798 shares of common stock pursuant to the Purchase Plan
and recorded compensation cost of approximately $478. The
weighted-average fair value of the employee stock purchase plan
rights granted during 2010, 2009 and 2008 was $6.65, $0.53 and
$0.84 per share, respectively. Common stock reserved for future
employee purchase under the Purchase Plan aggregated
664,649 shares as of December 31, 2010. There are no
other investment options for participants.
F-30
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
The Company provides for income taxes using the liability
method. The difference between the tax provision and the amount
that would be computed by applying the statutory Federal income
tax rate to income before taxes is attributable to the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Federal income tax provision at 35%
|
|
$
|
(77,112
|
)
|
|
$
|
1,491
|
|
|
$
|
(91,416
|
)
|
|
State taxes, net of federal tax benefit
|
|
|
(7,324
|
)
|
|
|
185
|
|
|
|
(14,161
|
)
|
|
Tax credits, principally for research and development
|
|
|
428
|
|
|
|
(1,813
|
)
|
|
|
(3,500
|
)
|
|
Research and development credit refunds
|
|
|
—
|
|
|
|
(1,274
|
)
|
|
|
—
|
|
|
Other
|
|
|
3,064
|
|
|
|
(3,303
|
)
|
|
|
3,447
|
|
|
Change in valuation allowance on deferred tax asset
|
|
|
80,944
|
|
|
|
3,440
|
|
|
|
105,630
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
(1,274
|
)
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The change in valuation allowance as reported above excludes the
change in valuation allowance associated with the net deferred
tax liability recorded in connection with the net unrealized
gains on investments, as such amounts are recorded as a
component of other comprehensive loss.
Temporary differences and carryforwards that give rise to a
significant portion of deferred tax assets and liabilities are
as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
Current
|
|
|
Long-Term
|
|
|
|
|
Asset
|
|
|
Asset (Liability)
|
|
|
|
|
December 31, 2010
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforward
|
|
$
|
—
|
|
|
$
|
796,645
|
|
|
Research and development and other tax credit carryforwards
|
|
|
—
|
|
|
|
34,854
|
|
|
Deferred revenue
|
|
|
2,182
|
|
|
|
761
|
|
|
Lease termination charges
|
|
|
—
|
|
|
|
3,302
|
|
|
Net unrealized gains on investments
|
|
|
—
|
|
|
|
(2,806
|
)
|
|
Intangible assets
|
|
|
385
|
|
|
|
4,104
|
|
|
Equity-based compensation
|
|
|
—
|
|
|
|
13,664
|
|
|
Depreciation
|
|
|
—
|
|
|
|
18,633
|
|
|
Unamortized debt discount
|
|
|
—
|
|
|
|
(11,922
|
)
|
|
Reserves and accruals
|
|
|
7,453
|
|
|
|
10,968
|
|
|
Deferred tax charge
|
|
|
5,909
|
|
|
|
18,712
|
|
|
Other
|
|
|
—
|
|
|
|
1,437
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
15,929
|
|
|
|
888,352
|
|
|
Less valuation allowance
|
|
|
(15,929
|
)
|
|
|
(888,352
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
F-31
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE I) —
|
Income
Taxes (continued)
|
| |
|
|
|
|
|
|
|
|
|
|
|
Current
|
|
|
Long-Term
|
|
|
|
|
Asset
|
|
|
Asset (Liability)
|
|
|
|
|
December 31, 2009
|
|
|
|
|
|
|
|
|
|
Net operating loss carryforward
|
|
$
|
—
|
|
|
$
|
739,900
|
|
|
Research and development and other tax credit carryforwards
|
|
|
—
|
|
|
|
35,282
|
|
|
Deferred revenue
|
|
|
34,935
|
|
|
|
781
|
|
|
Lease termination charges
|
|
|
—
|
|
|
|
3,957
|
|
|
Net unrealized gains on investments
|
|
|
—
|
|
|
|
(2,885
|
)
|
|
Intangible assets
|
|
|
394
|
|
|
|
4,602
|
|
|
Equity-based compensation
|
|
|
—
|
|
|
|
11,561
|
|
|
Depreciation
|
|
|
—
|
|
|
|
14,154
|
|
|
Unamortized debt discount
|
|
|
—
|
|
|
|
(21,317
|
)
|
|
Reserves and accruals
|
|
|
11,687
|
|
|
|
10,068
|
|
|
Other
|
|
|
—
|
|
|
|
726
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
47,016
|
|
|
|
796,829
|
|
|
Less valuation allowance
|
|
|
(47,016
|
)
|
|
|
(796,829
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
The Company recognized a valuation allowance to the full extent
of its deferred tax assets since the likelihood of realization
of the benefit is not more likely than not. The valuation
allowance increased by $60,435 during 2010 to $904,281 as of
December 31, 2010. The increase is primarily related to the
net operating loss carryforward generated in the current year
and the adjustment to the federal statutory rate.
Provision for income taxes is comprised of the following:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Current:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal
|
|
$
|
—
|
|
|
$
|
(1,274
|
)
|
|
$
|
—
|
|
|
State
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Foreign taxes
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
Deferred
|
|
|
—
|
|
|
|
—
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$
|
—
|
|
|
$
|
(1,274
|
)
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company has available tax credit carryforwards of
approximately $34,616 which expire, if unused, from the year
2011 through the year 2030. The Company has net operating loss
(“NOL”) carryforwards for federal income tax purposes
of approximately $1,827,934, excluding stock-based compensation
NOLs, which expire, if unused, from the year 2011 through the
year 2030. The Company’s ability to utilize these NOLs may
be limited under Internal Revenue Code Section 382
(“Section 382”). In connection with the adoption
of stock-based compensation guidance in 2006, the Company
elected to follow the
with-and-without
approach to determine the sequence in which deductions and NOL
carryforwards are utilized. As of December 31, 2010,
approximately $340,521 of the NOL carryforwards relate to
stock-based compensation for which future tax benefits will be
credited to equity.
Section 382 imposes annual limitations on the utilization
of NOL carryfowards and other tax attributes upon an ownership
change. In general terms, an ownership change may result from
transactions that increase the aggregate
F-32
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE I) —
|
Income
Taxes (continued)
|
ownership of certain stockholders in the Company’s stock by
more than 50 percentage points over a testing period
(generally three years). The Company completed a
Section 382 analysis during 2009. Based on this analysis,
the Company does not believe that its NOLs and other tax
attributes are limited under Section 382. The
Company’s future utilization of all of the Company’s
NOLs and other tax attributes is dependent upon the
Company’s ability to generate sufficient income during the
carryforward periods and no further significant changes in
ownership.
The Company accounts for uncertain tax positions pursuant to the
guidance of FASB ASC Topic 740, Income Taxes. The Company
recognizes interest and penalties related to uncertain tax
positions, if any, in income tax expense. As of
December 31, 2010 and 2009, the Company did not accrue any
interest related to uncertain tax positions. The Company’s
income taxes have not been subject to examination by any tax
jurisdictions since the Company’s inception. Accordingly,
all income tax returns filed by the Company are subject to
examination by the taxing jurisdictions.
A reconciliation of the beginning and ending amount of gross
unrecognized tax benefits is as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Balance as of January 1
|
|
$
|
30,022
|
|
|
$
|
30,282
|
|
|
$
|
29,611
|
|
|
Gross increases related to prior year tax positions
|
|
|
—
|
|
|
|
—
|
|
|
|
62
|
|
|
Gross decreases related to prior year tax positions
|
|
|
(803
|
)
|
|
|
(1,108
|
)
|
|
|
(558
|
)
|
|
Gross increases related to current year tax positions
|
|
|
304
|
|
|
|
848
|
|
|
|
1,167
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance as of December 31
|
|
$
|
29,523
|
|
|
$
|
30,022
|
|
|
$
|
30,282
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company believes that any of its uncertain tax positions
would not result in adjustments to its effective tax rate
because likely corresponding adjustments to deferred tax assets
would be offset by adjustments to recorded valuation allowances.
It is reasonably possible that the balance of uncertain tax
positions will decrease by up to $635 within the next
12 months.
(NOTE J) —
Facility-Related Exit Charges
During 2006, the Company exited certain headquarters space
(“Wing C”) and recorded a charge of $9,156, net of
estimated sublease income, pursuant to FASB ASC Topic 420,
Exit or Disposal Cost Obligations. During the second
quarter of 2009, the Company’s sublessee terminated its
sublease, which resulted in an exit charge of approximately
$11,400, comprised of an increase of $10,585 in accrued exit
expenses and $850 in additional fixed asset impairments. During
the fourth quarter of 2009, the Company decided to resume
production activities in Wing C in order to support increased
contract manufacturing services and increased early-stage
clinical development and reversed approximately $10,675 of the
exit reserve, representing the portion of the Wing C exit
reserve related to the production space. The Company’s exit
reserve is approximately $2,951 as of December 31, 2010.
F-33
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE J) —
|
Facility-Related
Exit Charges (continued)
|
The following table summarizes the activity related to the
reserve for exit charges for 2010, all of which is
facilities-related:
| |
|
|
|
|
|
Balance as of January 1, 2010
|
|
$
|
4,206
|
|
|
Accretion recorded
|
|
|
278
|
|
|
|
|
|
|
|
|
Subtotal
|
|
|
4,484
|
|
|
Cash items
|
|
|
(1,533
|
)
|
|
|
|
|
|
|
|
Balance as of December 31, 2010
|
|
|
2,951
|
|
|
Less current portion
|
|
|
(2,251
|
)
|
|
|
|
|
|
|
|
|
|
$
|
700
|
|
|
|
|
|
|
|
|
|
|
(NOTE K) —
|
Fair
Value Measurements
|
The FASB guidance regarding the fair value of all assets and
liabilities defines fair value, provides guidance for measuring
fair value and requires certain disclosures. This guidance does
not require any new fair value measurements, but rather applies
to all other accounting pronouncements that require or permit
fair value measurements. This guidance does not apply to
measurements related to share-based payments.
The FASB Codification discusses valuation techniques, such as
the market approach (comparable market prices), the income
approach (present value of future income or cash flow), and the
cost approach (cost to replace the service capacity of an asset
or replacement cost). The guidance utilizes a fair value
hierarchy that prioritizes the inputs to valuation techniques
used to measure fair value into three broad levels. The
following is a brief description of those three levels:
| |
|
|
|
Level 1:
|
|
Observable inputs such as quoted prices (unadjusted) in active
markets for identical assets or liabilities.
|
|
Level 2:
|
|
Inputs other than quoted prices that are observable for the
asset or liability, either directly or indirectly. These include
quoted prices for similar assets or liabilities in active
markets and quoted prices for identical or similar assets or
liabilities in markets that are not active.
|
|
Level 3:
|
|
Unobservable inputs that reflect the reporting entity’s own
assumptions.
|
Active markets are those in which transactions occur with
sufficient frequency and volume to provide pricing information
on an ongoing basis. Inactive markets are those in which there
are few transactions for the asset, prices are not current, or
price quotations vary substantially either over time or among
market makers, or in which little information is released
publicly. With regard to the Company’s financial assets
subject to fair value measurements, the Company believes that
all of the assets it holds are actively traded because there is
sufficient frequency and volume to obtain pricing information on
an ongoing basis.
F-34
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE K) —
|
Fair
Value Measurements (continued)
|
The Company’s assets and liabilities subject to fair value
measurements on a recurring basis and the related fair value
hierarchy are as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair Value as of December 31, 2010
|
|
|
|
|
Level 1
|
|
|
Level 2
|
|
|
Level 3
|
|
|
|
|
Residential mortgage-backed securities
|
|
$
|
—
|
|
|
$
|
54,718
|
|
|
$
|
—
|
|
|
Government-sponsored enterprise securities
|
|
|
—
|
|
|
|
86,965
|
|
|
|
—
|
|
|
Asset-backed securities
|
|
|
—
|
|
|
|
12,988
|
|
|
|
—
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
127,345
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Short-term investments
|
|
$
|
—
|
|
|
$
|
282,016
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Residential mortgage-backed securities
|
|
$
|
—
|
|
|
$
|
80,746
|
|
|
$
|
—
|
|
|
Government-sponsored enterprise securities
|
|
|
—
|
|
|
|
25,285
|
|
|
|
—
|
|
|
Asset-backed securities
|
|
|
—
|
|
|
|
58,904
|
|
|
|
—
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
251,230
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Marketable securities
|
|
$
|
—
|
|
|
|
416,165
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Money market funds
|
|
$
|
7,455
|
|
|
$
|
—
|
|
|
$
|
—
|
|
|
U.S. Treasury securities
|
|
|
1,311
|
|
|
|
—
|
|
|
|
—
|
|
|
Residential mortgage-backed securities
|
|
|
—
|
|
|
|
6,496
|
|
|
|
—
|
|
|
Government-sponsored enterprise securities
|
|
|
—
|
|
|
|
13,303
|
|
|
|
—
|
|
|
Asset-backed securities
|
|
|
—
|
|
|
|
4,422
|
|
|
|
—
|
|
|
Corporate debt securities
|
|
|
—
|
|
|
|
46,523
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted investments
|
|
$
|
8,766
|
|
|
$
|
70,744
|
|
|
$
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The Company evaluates the types of securities in its investment
portfolios to determine the proper classification in the fair
value hierarchy based on trading activity and the observability
of market inputs. The Company’s privately-held equity
investment is carried at cost and is not included in the table
above, and is reviewed for impairment at each reporting date.
The Company generally obtains a single quote or price per
instrument from independent third parties to help it determine
the fair value of securities in Level 1 and Level 2 of
the fair value hierarchy. The Company’s Level 1 cash
and money market instruments are valued based on quoted prices
from third parties, and the Company’s Level 1
U.S. Treasury securities are valued based on broker quotes.
The Company’s Level 2 assets are valued using a
multi-dimensional pricing model that includes a variety of
inputs including actual trade data, benchmark yield data,
non-binding broker/dealer quotes, issuer spread data, monthly
payment information, collateral performance and other reference
information. These are all observable inputs. The Company
reviews the values generated by the multi-dimensional pricing
model for reasonableness, which could include reviewing other
publicly available information.
The Company does not hold auction rate securities, loans held
for sale, mortgage-backed securities backed by
sub-prime or
Alt-A collateral or any other investments which require the
Company to determine fair value using a discounted cash flow
approach. Therefore, the Company does not need to adjust its
analysis or change its assumptions specifically to factor
illiquidity in the markets into its fair values.
The fair value of the Company’s collaboration receivables,
other assets, accounts payable, accrued expenses and
collaboration payable approximate their carrying amount due to
the relatively short maturity of these items. The fair value of
the Company’s convertible subordinated debt is based on
quoted market prices. The quoted market price of the
Company’s convertible subordinated debt was approximately
$635,000 as of December 31, 2010. With respect to its lease
financing, the Company evaluated its incremental borrowing rate
as of December 31, 2010, based on the current interest rate
environment an d the Company’s credit risk. The fair value
of the BioMed lease financing was
F-35
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE K) —
|
Fair
Value Measurements (continued)
|
approximately $266,000 as of December 31, 2010 based on a
discounted cash flow analysis, and current rates for corporate
debt having similar characteristics and companies with similar
creditworthiness.
|
|
|
(NOTE L) —
|
Teva
Biopharmaceuticals USA, Inc. (formerly CoGenesys)
|
In 2008, Teva acquired all of the outstanding stock of
CoGenesys, which became Teva Bio. CoGenesys had been a division
of the Company until 2006, when the Company completed the sale
of assets and concurrently entered into a license agreement and
manufacturing services agreement.
As consideration for the assets conveyed, liabilities assumed
and intellectual property licensed, the Company obtained a 14%
equity interest in CoGenesys. As part of this transaction,
$7,575 was allocated to the intellectual property license and
manufacturing services agreement and was recognized ratably over
the term of the manufacturing services agreement, as amended,
which ended in 2009.
Under the license agreement, as amended, the Company is entitled
to various milestone and royalty rights on certain products, if
they are developed and commercialized. Teva Bio can obtain
additional product rights by extending the initial seven-year
research term upon the payment of additional consideration.
As a result of Teva’s acquisition of CoGenesys in 2008, the
Company received $47,336 as partial payment for its equity
investment in CoGenesys and recorded a gain on sale of long-term
equity investment of $32,518 in 2008. The terms of the agreement
between Teva and CoGenesys required an escrow account be
established for 10% of the purchase price as security for
CoGenesys’ representations, warranties, and covenants.
Because the Company had no information concerning the likelihood
of the terms of the escrow agreement being satisfied, the
Company did not include any potential proceeds from escrow in
the calculation of the gain on the sale of its investment in
2008. During 2009, the Company received the final payment for
its equity investment in CoGenesys and recorded a gain of $5,259.
|
|
|
(NOTE M) —
|
Earnings
Per Share
|
Diluted net income (loss) per share was determined as follows:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
December 31,
|
|
|
|
|
2010
|
|
|
2009
|
|
|
2008
|
|
|
|
|
Numerator:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) used for diluted net income (loss) per share
|
|
$
|
(233,231
|
)
|
|
$
|
5,659
|
|
|
$
|
(268,891
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Denominator:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares outstanding
|
|
|
187,791,437
|
|
|
|
149,334,426
|
|
|
|
135,406,642
|
|
|
Effect of dilutive securities:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Employee stock options and restricted stock units
|
|
|
—
|
|
|
|
5,719,047
|
|
|
|
—
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average shares used for diluted net income (loss) per
share
|
|
|
187,791,437
|
|
|
|
155,053,473
|
|
|
|
135,406,642
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Diluted net income (loss) per share
|
|
$
|
(1.24
|
)
|
|
$
|
0.04
|
|
|
$
|
(1.99
|
)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock issued in connection with the Company’s
Purchase Plan and through exercised options granted pursuant to
the Incentive Plan are included in the Company’s weighted
average share balance based upon the issuance date of the
related shares. As of December 31, 2010, 2009 and 2008, the
Company had 24,028,688, 24,601,174 and 28,373,151, respectively,
stock options outstanding. The Company had 24,302,742,
24,303,304 and 30,942,877 shares issuable upon the
conversion of the Company’s convertible subordinated debt
as of December 31,
F-36
HUMAN
GENOME SCIENCES, INC.
NOTES TO
CONSOLIDATED FINANCIAL STATEMENTS
(dollars in thousands, except per share data)
|
|
|
(NOTE M) —
|
Earnings
Per Share (continued)
|
2010, 2009 and 2008, respectively. The shares issuable upon the
conversion of the Company’s convertible subordinated debt
are excluded from the weighted average shares as they are
anti-dilutive.
|
|
|
(NOTE N) —
|
Quarterly
Financial Information (unaudited)
|
Quarterly financial information for 2010 and 2009 is presented
in the following table:
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1st
Quarter
|
|
2nd
Quarter
|
|
3rd
Quarter
|
|
4th
Quarter
|
|
|
|
2010
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
46,514
|
|
|
$
|
38,792
|
|
|
$
|
50,782
|
|
|
$
|
21,263
|
|
|
Loss from operations
|
|
|
(37,773
|
)
|
|
|
(46,992
|
)
|
|
|
(29,826
|
)
|
|
|
(76,182
|
)
|
|
Net loss
|
|
|
(47,877
|
)
|
|
|
(56,863
|
)
|
|
|
(40,859
|
)
|
|
|
(87,632
|
)
|
|
Net loss per share, basic and diluted
|
|
|
(0.26
|
)
|
|
|
(0.30
|
)
|
|
|
(0.22
|
)
|
|
|
(0.46
|
)
|
|
2009
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue
|
|
$
|
177,277
|
|
|
$
|
26,681
|
|
|
$
|
18,834
|
|
|
$
|
52,957
|
|
|
Income (loss) from operations
|
|
|
97,795
|
|
|
|
(53,414
|
)
|
|
|
(37,964
|
)
|
|
|
(229
|
)
|
|
Net income (loss)
|
|
|
129,813
|
|
|
|
(65,411
|
)
|
|
|
(49,003
|
)
|
|
|
(9,740
|
)
|
|
Net income (loss) per share, basic
|
|
|
0.96
|
|
|
|
(0.48
|
)
|
|
|
(0.32
|
)
|
|
|
(0.06
|
)
|
|
Net income (loss) per share, diluted
|
|
|
0.85
|
|
|
|
(0.48
|
)
|
|
|
(0.32
|
)
|
|
|
(0.06
|
)
|
The Company’s results for the third quarter of 2010 include
additional revenue recognized based on the decision to end
future development of ZALBIN of $34,126, or $0.18 per basic and
diluted share.
The Company’s results for the first quarter of 2009 include
a gain on extinguishment of debt of $38,873, or $0.29 per basic
share and $0.24 per diluted share and a gain on the sale of an
equity investment of $5,259, or $0.04 per basic share and $0.03
per diluted share.
F-37