Attached files
Table of Contents
As filed with the Securities and Exchange Commission on February 18, 2011
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
ACCENTIA BIOPHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| Florida | 2834 | 04-3639490 | ||
| (State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
324 South Hyde Park Avenue, Suite 350
Tampa, Florida 33606
(813) 864-2554
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Samuel S. Duffey, Esq.
President and General Counsel
Accentia Biopharmaceuticals, Inc.
324 South Hyde Park Avenue, Suite 350
Tampa, Florida 33606
Phone: (813) 864-2554
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
Curt P. Creely, Esq.
Foley & Lardner LLP
100 North Tampa Street, Suite 2700
Tampa, Florida 33602
(813) 229-2300
(813) 221-4210—Fax
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ¨ Accelerated filer ¨
Non-accelerated filer ¨ Smaller reporting company x
CALCULATION OF REGISTRATION FEE
| Title of Each Class of Securities to Be Registered |
Proposed Maximum Aggregate Offering Price(1) |
Amount of Registration Fee | ||
| Units, each consisting of one share of Common Stock, par value $0.001 per share, and a Warrant to purchase 0.5 shares of Common Stock | $5,000,000 | $581 | ||
| Common Stock underlying the Units |
(3) | |||
| Warrants underlying the Units |
(3) | |||
| Common Stock issuable upon exercise of the Warrants underlying the Units(2) |
||||
| Placement Agent Warrants |
||||
| Common Stock issuable upon exercise of the Placement Agent Warrants(2) |
(3) | |||
| Total |
$5,000,000 | $581 | ||
| (1) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act of 1933. |
| (2) | Pursuant to Rule 416, the securities being registered hereunder include such indeterminate number of additional shares of common stock as may be issuable upon the exercise of the warrants registered hereunder as a result of stock splits, stock dividends, or similar transactions. |
| (3) | No fee required pursuant to Rule 457(g). |
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the registration statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED FEBRUARY 18, 2011

Units, Each Consisting of
One Share of Common Stock and
0.5 of a Warrant to Purchase One Share of Common Stock
We are offering up to units, each unit consisting of one share of our common stock, par value $0.001 per share, and 0.5 of a warrant to purchase one share of our common stock. Each full warrant entitles its holder to purchase one share of our common stock at an exercise price of $ per share. The units will separate immediately, the common stock and the warrants will be issued separately, and the common stock will trade separately. For a more detailed description of our units, our common stock, and our warrants, see the section entitled “DESCRIPTION OF SECURITIES” beginning on page 130 of this prospectus.
We are not required to sell any specific dollar amount or number of units, but we will use our best efforts to sell all of the units being offered.
Our common stock is currently quoted on the OTCQB under the symbol “ABPI”. We do not intend to apply to list the warrants on any securities exchange or market. On February 17, 2011, the last reported sale price of our common stock on the OTCQB was $0.71 per share.
Investing in the offered securities involves risks. See “RISK FACTORS” beginning on page 7 of this prospectus.
| Per Unit | Total | |||
| Price to Public |
5,000,000 | |||
| Placement Agent’s Fees |
||||
| Proceeds, Before Expenses, to Accentia Biopharmaceuticals |
has agreed to act as our exclusive placement agent in connection with this offering. may engage one or more sub placement agents or selected dealers. The placement agent is not purchasing the securities offered by us, and is not required to sell any specific number or dollar amount of units, but will assist us in this offering on a “best efforts” basis. We have agreed to pay the placement agent a cash fee equal to % of the gross proceeds of the offering of units by us, as well as “Placement Agent Warrants” to purchase shares of our common stock equal to % of the aggregate number of units sold in the offering. The Placement Agent Warrants will be substantially on the same terms as the warrants offered hereby, except as required by the Financial Industry Regulatory Authority, Inc. We estimate the total expenses of this offering, excluding the placement agent fees, will be approximately $ . Because there is no minimum offering amount required as a condition to closing in this offering, the actual public offering amount, placement agent fees, and proceeds to us, if any, are not presently determinable and may be substantially less than the total maximum offering amounts set forth above. See “PLAN OF DISTRIBUTION” beginning on page 135 of this prospectus for more information regarding this offering and the placement agent arrangements. All costs associated with the offering will be borne by us.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Brokers or dealers effecting transactions in these securities should confirm that the securities are registered under the applicable state law or that an exemption from registration is available.
The date of this prospectus is , 2011.
Table of Contents
| 2 | ||||
| 7 | ||||
| 33 | ||||
| 35 | ||||
| 35 | ||||
| MARKET PRICE OF COMMON STOCK AND RELATED STOCKHOLDER MATTERS |
35 | |||
| 38 | ||||
| MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
41 | |||
| 63 | ||||
| 112 | ||||
| 121 | ||||
| SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT |
127 | |||
| 130 | ||||
| 135 | ||||
| 140 | ||||
| 140 | ||||
| 140 | ||||
| F-1 | ||||
| F-61 | ||||
| S-1 |
You should rely only on the information contained in this prospectus. We have not authorized anyone to provide you with information different from the information contained in this prospectus. We are not making an offer to sell securities in any state where offers and sales are not permitted. The information contained in this prospectus is accurate only as of the date of this prospectus, regardless of when this prospectus is delivered or when any sale of our securities occurs.
FOR INVESTORS OUTSIDE THE UNITED STATES: We have not done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about, and to observe any restrictions relating to, this offering and the distribution of this prospectus.
1
Table of Contents
This summary highlights information that we present more fully in the rest of this prospectus and does not contain all of the information you should consider before investing in our securities. This summary contains forward-looking statements that involve risks and uncertainties, such as statements about our plans, objectives, expectations, assumptions or future events. These statements involve estimates, assumptions, known and unknown risks, uncertainties and other factors that could cause actual results to differ materially from any future results, performances or achievements expressed or implied by the forward-looking statements. You should read the entire prospectus carefully, including the “Risk Factors” section and our consolidated financial statements and related notes.
Overview
Unless the context requires otherwise, as used in this prospectus, the terms “Accentia,” “we,” “us,” “our,” “the Company,” “our company,” and similar references refer to Accentia Biopharmaceuticals, Inc. and its subsidiaries.
Headquartered in Tampa, Florida, Accentia Biopharmaceuticals, Inc. is a biotechnology company that is developing Revimmune™ as a comprehensive system of care for the treatment of autoimmune diseases. Additionally, through our majority-owned subsidiary, Biovest International, Inc. (“Biovest”), we are developing BiovaxID® as a therapeutic cancer vaccine for treatment of follicular non-Hodgkin’s lymphoma and mantle cell lymphoma. Through our wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), we conduct a health economics research and consulting business which we market to the pharmaceutical and biotechnology industries, using our operating cash flow to support our corporate administration and product development activities.
REVIMMUNE
Revimmune™ is being developed as a treatment for various autoimmune diseases. The approximately 80 known autoimmune diseases generally arise from an overactive immune response against substances and/or tissue normally present in the body. As a system of care, Revimmune consists of administering high, pulsed doses of an FDA-approved drug, cyclophosphamide, over a four-day interval as part of an integrated risk management system including a panel of preventive tests, monitoring and medications which are intended to minimize risks while seeking to maximize the clinical benefit.
We have developed a multi-faceted strategy to maintain and protect our proprietary interests in Revimmune, involving various forms of intellectual property, including patent and non-patent exclusivity. We hold the exclusive world-wide rights to commercialize high-dose pulsed cyclophosphamide to treat MS and certain other autoimmune diseases through a sublicense (the “Revimmune Sublicense”) from Revimmune, LLC, which is affiliated with one of our directors and shareholders, which holds the exclusive license for the technology from Johns Hopkins University (the “JHU License”).
To extend our proprietary rights to Revimmune, we have entered into an agreement with Baxter Healthcare Corporation, which we believe is the only cGMP (“current Good Manufacturing Practice” enforced in the U. S. by the FDA) manufacturer of injectable cyclophosphamide in the U.S. This agreement grants us the exclusive right to use Baxter’s regulatory file and drug history (“Drug Master File”) for cyclophosphamide which we believe will facilitate our planned clinical trials and anticipated dealings with the FDA. Additionally, the agreement grants to us the exclusive right to purchase Baxter’s cyclophosphamide for the treatment of various autoimmune diseases including Autoimmune Hemolytic Anemia, Multiple Sclerosis, and Scleroderma.
2
Table of Contents
BIOVAXID
Through a collaboration with the National Cancer Institute (“NCI”), Biovest has developed a patient-specific cancer vaccine, BiovaxID®, which has demonstrated statistically significant Phase 3 clinical benefit by prolonging disease-free survival in follicular non-Hodgkin’s lymphoma (“FL”) patients treated with BiovaxID.
BiovaxID is characterized as an “active immunotherapy”. Active immunotherapies attempt to stimulate the patient’s immune system to respond to a disease. “Specific active immunotherapies” such as BiovaxID specifically seek to induce cellular and/or humoral immune responses focused on specific antigens present on a diseased cell (such as a tumor cell). As a specific, active immunotherapy, BiovaxID targets only the cancerous B-cells while sparing healthy B-cells. Accordingly, BiovaxID is highly targeted. BiovaxID is manufactured specifically and entirely for each patient and is considered to be a highly “personalized therapy”. If approved, BiovaxID will represent the only specific active immunotherapeutic approved for the treatment of FL and therefore will represent a new class of drugs that provide a new therapeutic option for patients with lymphoma.
CONSULTING SERVICES
Through Analytica, we conduct a global research and strategy consulting business that provides professional services to the pharmaceutical and biotechnology industries. Since 1997, Analytica has expertly directed research studies and projects, including traditional health economic modeling projects, database studies, structured reviews, health technology assessments, reimbursement analyses, and value dossiers.
We utilize Analytica’s services for our own product development efforts in order to, among other things, evaluate and analyze the market and potential pricing of our product candidates. We also use Analytica’s services to evaluate the payor reimbursement prospects of our products and to develop reimbursement strategies.
INSTRUMENTS AND DISPOSABLES
Biovest sells hollow-fiber perfusion instruments used for the production of significant quantities of cell culture products. This product line includes: (a) AutovaxID™, a fully automated, reusable instrument that employs a fully disposable, closed-system cell-growth chamber incorporating a hollow-fiber cell-growth cartridge, (b) the Primer HF®, a low cost hollow-fiber cell culture system capable of producing small quantities of monoclonal antibody, (c) the miniMax®, an automated cell culture system that provides the flexibility and technology needed to support optimization studies and research scale production of mammalian cell secreted proteins, (d) the Maximizer®, an automated cell culture system that provides maximum flexibility to support optimization studies and pilot scale production of mammalian cell secreted proteins, and (e) the Xcellerator™, a self-standing floor system containing an incubator and refrigerator section, control fixtures and pump panel.
3
Table of Contents
CELL CULTURE PRODUCTS AND SERVICES
Biovest also manufactures mammalian cell culture products, such as whole cells, recombinant and secreted proteins, and monoclonal antibodies. Additionally, Biovest provides related services as a contract resource to assist customers in developing cell production process protocols, cell line optimization, cell culture production optimization, media evaluation and other related services. This segment of our business represented approximately $1.2 million (approximately 22%) and $1.5 million (approximately 41%) in revenues for the fiscal years ended September 30, 2010, and 2009 respectively.
Chapter 11 Reorganization
On November 10, 2008, we, along with all of our subsidiaries, filed a voluntary petition for reorganization under Chapter 11 in the United States Bankruptcy Court for the Middle District of Florida, Tampa Division (Case No. 8:08-bk-17795-KRM). On August 16, 2010, we filed our First Amended Joint Plan of Reorganization, and, on October 25, 2010, we filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. We emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
Corporate Information
Our principal executive offices are located at 324 South Hyde Park Avenue, Suite 350, Tampa, Florida 33606, and our telephone number is (813) 864-2554. Our website is www.accentia.net. Information contained on our website is not incorporated by reference into this prospectus, and such information should not be considered to be part of this prospectus.
4
Table of Contents
The Offering
| Securities Offered | Up to units. Each unit will consist of one share of our common stock and 0.5 of a warrant to purchase one share of our common stock.
| |
| Offering Price | $ per unit.
| |
| Description of Warrants | The warrants will be exercisable at any time during the period commencing on the date of closing and ending on the fifth anniversary of the closing date at an exercise price of $ per share. See “DESCRIPTION OF SECURITIES” for additional information.
| |
| Common Stock Outstanding Prior to the Offering | 68,632,612 shares.
| |
| Common Stock Outstanding After the Offering | shares.
| |
| OTCQB Symbol | “ABPI”
| |
| Use of Proceeds | We expect to use the net proceeds of this offering for working capital and other general corporate purposes and to reduce the principal and interest on our indebtedness to our senior lender by $ (which represents 30% of the net proceeds of the offering). See “USE OF PROCEEDS” for additional information.
| |
| Risk Factors | See “RISK FACTORS” and other information included in this prospectus for a discussion of factors you should carefully consider before deciding to invest in the units offered by this prospectus.
|
The number of shares of our common stock outstanding after the offering is based on 68,632,612 shares outstanding as of January 31, 2011 and excludes:
| • | 15,067,364 shares of common stock issuable upon the exercise of warrants outstanding as of January 31, 2011, at a weighted average exercise price of $2.45 per share; |
| • | 25,013,433 shares of common stock issuable upon the exercise of options outstanding as of January 31, 2011 at a weighted average exercise price of $0.81 per share; |
| • | 9,215,000 shares of common stock reserved for future grant or issuance as of January 31, 2011 under our 2010 Equity Incentive Option Plan; |
5
Table of Contents
| • | shares of common stock underlying the warrants sold in this offering; and |
| • | shares of common stock underlying the warrants issued to the placement agent in connection with this offering. |
Unless otherwise indicated, all information in this prospectus assumes:
| — | no person will exercise any outstanding options; and |
| — | the sale of units at an assumed offering price of $ per unit. |
6
Table of Contents
An investment in our securities involves a high degree of risk and many uncertainties. You should carefully consider the specific factors listed below together with the other information included in this prospectus before purchasing our securities in this offering. If any of the possibilities described as risks below actually occurs, our operating results and financial condition would likely suffer and the trading price of our securities could fall, causing you to lose some or all of your investment in the securities we are offering. The following is a description of what we consider the key challenges and material risks to our business and an investment in our securities.
Risks Related to Our Business
We have a history of operating losses and expect to incur further losses.
We have never been profitable and we have incurred significant losses and cash flow deficits. For the fiscal years ended September 30, 2010 and 2009, we reported net losses of $47.8 million and $5.3 million, respectively, and negative cash flow from operating activities of $0.4 million and $1.9 million, respectively. As of September 30, 2010, we had an aggregate accumulated deficit of $326.0 million. We anticipate that operations may continue to show losses and negative cash flow, particularly with the anticipated expenses associated with the initial clinical trial of Revimmune™ and Biovest’s efforts to seek regulatory approval for BiovaxID®. There is no assurance that the additional required funds can be obtained on terms acceptable or favorable to us, if at all. The audit opinion issued by our independent auditors with respect to our financial statements for the 2010 fiscal year indicated that there is substantial doubt about our ability to continue as a going concern. The financial statements do not include any adjustments that might result from the outcome of this uncertainty. We expect to receive a similar audit opinion from our independent auditors with respect to our financial statements for the 2011 fiscal year.
Our ability to achieve and sustain profitability is to a large degree dependent on the success of our development efforts with regard to Revimmune and the development and commercialization efforts of our majority owned subsidiary with regard to BiovaxID. We may not be successful in our efforts and even if successful we may not be able to profitably commercialize any of our drug products candidates.
Our independent registered public accountants have expressed substantial doubt as to our ability to continue as a going concern.
As of September 30, 2010, we had a working capital deficit of $147.6 million, including those liabilities subject to compromise through our Chapter 11 bankruptcy proceedings. The working capital deficit at January 31, 2011 was $38.0 million, including approximately $8.0 million of claims yet to be settled under our Chapter 11 bankruptcy proceedings. We expect to continue to incur substantial net operating losses for the foreseeable future. Continued operating losses would impair our ability to continue operations. We may not be able to generate sufficient product revenue to become profitable on a sustained basis, or at all. We have operating and liquidity concerns due to our significant net losses and negative cash flows from operations. As a result of these and other factors, our independent registered certified public accountants, Cherry, Bekaert, and Holland, L.L.P., have indicated, in their report to our 2010 financial statements, that there is substantial doubt about our ability to continue as a going concern. Our ability to continue as a going concern is dependent upon generating sufficient cash flow to conduct operations and obtaining additional capital and financing. Any financing activity is likely to result in significant dilution to current shareholders.
7
Table of Contents
Our financial statements have been prepared assuming we will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty. We incurred net losses of $47.8 million and $5.3 million in 2010 and 2009, respectively, and $7.4 million for the quarter ended December 31, 2010. We have also experienced negative cash flows from operations for the past three fiscal years. In addition, our projected cash receipts from operations for fiscal 2011 are anticipated to be insufficient to finance operations without funding from other sources. Historically, we have had difficulty in meeting our cash requirements. There can be no assurances that we will obtain the necessary funding, reduce the level of historical losses and achieve successful commercialization of any of our drug product candidates. Continuation as a going concern is ultimately dependent upon achieving profitable operations and positive operating cash flows sufficient to pay all obligations as they come due.
We will need substantial additional financing but our access to capital funding is uncertain.
We have limited cash or other liquid assets on hand. During prior years, we met our cash requirements through the use of cash on hand, revenue, the sale of common stock, and loans. We have significant outstanding indebtedness, which, as of December 31, 2010, aggregated approximately $51.2 million of which $25.5 million will be automatically converted into stock at various intervals and approximately $10.1million is voluntarily convertible into stock. Certain of our debt is secured by our assets and may make procuring additional debt or equity financing more difficult or uncertain. Furthermore, our secured creditors may be able to foreclose on our assets if we are unable to meet our obligations as they become due. We are required by certain loan covenants to allocate 30% of the net proceeds from the sale of our capital stock under certain circumstances to re-pay certain indebtedness which may make future capital raises more difficult.
Our ability to continue present operations is dependent upon our ability to obtain significant external funding when required. Additional sources of funding have not been established; however, we anticipate that in the future we will seek additional financing from a number of sources, including, but not limited to, the sale of equity or debt securities, strategic collaborations, recognized research funding programs and domestic and/or foreign licensing. There can be no assurance that we will be successful in securing needed financing at acceptable terms, if at all. If adequate funds are not available, or if we determine it to otherwise be in our best interests, we may consider additional strategic financing options, including sales of assets or business units that are non-essential to the ongoing development or future commercialization of our drug candidates in development or we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or curtail some of our commercialization efforts.
If we are successful in procuring additional financing when required, it will most likely result in our issuing additional shares and/or rights to acquire shares of our capital stock or in the alternative it may result in our selling shares of our majority-owned subsidiary, Biovest, which we currently own. Accordingly, our access to additional financing when needed is anticipated to be dilutive to existing shareholders.
We are largely dependent on the success of BiovaxID® and Revimmune™ and we may not be able to successfully commercialize these therapies.
We have expended and will continue to expend significant time, money, and effort on the development of BiovaxID, which is owned by our majority owned subsidiary, and/or Revimmune. We will incur significant costs and may never generate significant revenues from commercial sales of these products, if approved. None of these products is approved for marketing in any jurisdiction, and they may never be commercialized. Before we can market and sell these products, we will need to demonstrate in clinical trials that these products are safe and effective and will also need to obtain necessary approvals from the FDA, and similar foreign regulatory agencies.
8
Table of Contents
If we fail to successfully commercialize any or all of these product candidates, we may be unable to generate sufficient revenue to sustain and grow our business, and our business, financial condition, and results of operations will be adversely affected.
If we fail to obtain FDA approval of BiovaxID®, Revimmune™ or any of our future product candidates, we will be unable to commercialize these products.
Development, testing, manufacturing and marketing of pharmaceutical products are subject to extensive regulation by numerous governmental authorities in the U.S. and other countries. The process of obtaining FDA approval of pharmaceutical products is costly and time consuming. Any new pharmaceutical product must undergo rigorous preclinical and clinical testing and an extensive regulatory approval process mandated by the FDA. Such regulatory review includes the determination of manufacturing capability and product performance.
In addition to seeking approval from the FDA for BiovaxID and Revimmune, we intend to seek the governmental approval required to market our products in the EU, and potentially additional countries and territories. We anticipate commencing the applications required in some or all of these countries following approval by the FDA; however, we may determine to file applications in advance of the FDA approval if we determine such filings to be both time and cost effective. Marketing of our products in these countries, and in most other countries, is not permitted until we have obtained required approvals or exemptions in each individual country.
In addition, patient-specific active immunotherapies such as BiovaxID are complex, and regulatory agencies have limited experience with them. To date, the FDA has only approved for marketing one active immunotherapy for treating cancer. This limited precedent and experience may lengthen the regulatory review process and impede our ability to obtain timely FDA approval for BiovaxID, if at all. Even if BiovaxID is approved by the FDA, the FDA’s limited precedent and experience with respect to a patient-specific active idiotype vaccine may increase our development costs and otherwise delay or prevent commercialization.
Further, we have not commenced any clinical trials in Revimmune and the development of our product candidates based on Revimmune should be considered to be in an early stage. The structure, size, design, cost and/or length of future trials in Revimmune have not been established and remain uncertain.
There can be no assurance that the products currently in development, or those products acquired or in-licensed by us, will be approved by the FDA. In addition, there can be no assurance that all necessary approvals will be granted for future products or that FDA review or actions will not involve delays caused by the FDA’s request for additional information or testing that could adversely affect the time to market and sale of the products.
Any delay in any approval or any failure to obtain approval of a product could delay or impair our ability to commercialize that product and to generate revenue as well as increase costs for that product.
9
Table of Contents
Before regulatory approval can be sought for BiovaxID® or Revimmune™, we may need to successfully complete clinical trials, outcomes of which are uncertain.
Conducting clinical trials is a lengthy, time-consuming, and expensive process, and the results of these trials are inherently uncertain. The time required to complete necessary clinical trials is often difficult, if not impossible, to predict. Our commencement and rate of completion of clinical trials may be delayed by many factors, including:
| • | ineffectiveness of our product candidate or perceptions by physicians that the product candidate is not safe or effective for a particular indication; |
| • | inability to manufacture sufficient quantities of the product candidate for use in clinical trials; |
| • | delay or failure in obtaining approval of our clinical trial protocols from the FDA or institutional review boards; |
| • | slower than expected rate of patient recruitment and enrollment; |
| • | inability to adequately follow and monitor patients after treatment; |
| • | difficulty in managing multiple clinical sites; |
| • | unforeseen safety issues; |
| • | government or regulatory delays; and |
| • | clinical trial costs that are greater than we currently anticipate. |
Even if we achieve positive interim results in clinical trials, these results do not necessarily predict final results, and positive results in early trials may not be indicative of success in later trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials. Negative or inconclusive results or adverse medical events during a clinical trial could cause us to repeat or terminate a clinical trial or require us to conduct additional trials. We do not know whether our existing or any future clinical trials will demonstrate safety and efficacy sufficiently to result in marketable products. Our clinical trials may be suspended at any time for a variety of reasons, including if the FDA or we believe the patients participating in our trials are exposed to unacceptable health risks or if the FDA finds deficiencies in the conduct of these trials.
Failures or perceived failures in our clinical trials will directly delay our product development and regulatory approval process, damage our business prospects, make it difficult for us to establish collaboration and partnership relationships, and negatively affect our reputation and competitive position in the pharmaceutical community.
10
Table of Contents
We have incurred significant costs in our development efforts to date and may never generate significant revenue from commercial sales of our product candidates, if approved.
We expended significant time and funds to conduct a Phase 3 clinical trial in SinuNase™, a drug candidate to treat a sinus condition. The clinical trial was not considered to be successful and we are not continuing to develop this drug. Nevertheless, we expended significant time and funds in the development of SinuNase. In addition, we will continue to expend significant sums in the development efforts for Revimmune and BiovaxID. We expect to continue to incur significant operating expenses and capital expenditures as we:
| • | conduct clinical trials; |
| • | conduct research and development on existing and new product candidates; |
| • | seek regulatory approvals for our product candidates; |
| • | commercialize our product candidates, if approved; |
| • | hire additional clinical, scientific, sales and marketing and management personnel; and |
| • | identify and license additional product candidates. |
If product candidates fail in clinical trials or do not gain regulatory approval or gain regulatory approval for more restricted indications than we have anticipated, we may not generate significant revenues from any of our product candidates. In addition, we may continue to experience net losses for the foreseeable future, in which case our accumulated deficit will continue to increase, and we may exhaust our resources and be unable to complete the development of our product candidates. If we are unable to fund the continuing development of our product candidates or if we fail to generate significant revenues from any of our product candidates, you could lose all or part of your investment.
We anticipate that we will need substantial additional funding in the future, and if we are unable to raise capital when needed, we would be forced to delay, reduce, or eliminate our product development programs or commercialization efforts.
Developing biopharmaceutical products, conducting clinical trials, establishing manufacturing capabilities, and marketing developed products is expensive. We anticipate that we will need to raise substantial additional capital in the future in order to complete the commercialization of our product candidates, to continue to pursue the submission of new drug applications (“NDAs”) for our product candidates and to fund the development and commercialization of our specialty pharmaceutical product candidates. Further, we anticipate that Biovest will need to raise substantial additional capital from other sources in order to continue the clinical trials for BiovaxID. Additional sources of funding have not been established by us or Biovest; however, additional financing is currently being sought from a number of sources, including the sale of Biovest equity or debt securities, strategic collaborations, recognized research funding programs, as well as domestic and/or foreign licensing of Biovest’s vaccine.
Based on our current operating plans, we expect that our existing capital and cash flow from operations, together with proceeds of one or more anticipated equity financing transactions, will be sufficient to fund our operations and development activities for a limited period, assuming Biovest receives its own funding. We have received a report from our independent registered public accounting firm on our consolidated financial statements for our fiscal years ended September 30, 2010 and 2009, in which our auditors have included explanatory paragraphs indicating that our significant net losses and working capital deficiency cause substantial doubt about our ability to continue as a going concern.
11
Table of Contents
We expect to seek additional financing from a number of sources, including but not limited to public or private equity offerings, debt financings, or corporate collaboration and licensing arrangements. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional dilution, and debt financing, if available, may involve restrictive covenants. If we sell shares of Biovest stock which are currently owned by us, we will reduce our ownership interest in Biovest. As part of prior financings and our Plan of Reorganization, we have granted warrants to purchase an aggregate of up to 14.4 million shares of Biovest owned by us which if fully exercised would provide an aggregate of approximately $21.6 million in additional financing for us. If our Biovest subsidiary raises funds through the issuance of equity securities, our equity interest in Biovest could be substantially diminished. If our Biovest subsidiary raises additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or grant licenses on terms that are not favorable to us. We cannot be certain that additional funding will be available on acceptable terms, or at all. If adequate funds are not available from the foregoing sources, we may consider additional strategic financing options, including sales of assets or business units that are non-essential to the ongoing development or future commercialization of BiovaxID or Revimmune, or we may be required to delay, reduce the scope of, or eliminate one or more of our research or development programs or curtail some of our commercialization efforts. We may seek to access the public or private equity markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at this time.
There is a high risk of failure because we are trying to develop both a novel autoimmune disease treatment and a new anti-cancer vaccine.
We are pursuing novel therapeutic treatments, including a new autoimmune disease treatment approach and a patient specific cancer therapy. Commercialization requires governmental approval, establishment of cost effective production capability, distribution capability and market acceptance. Our Revimmune treatment and our BiovaxID vaccine are subject to all of the risks of failure that are inherent in developing products based on new technologies and the risks associated with drug development generally. These risks include the possibility that:
| • | our technology or the product based on our technology will be ineffective or toxic, or otherwise fail to receive necessary regulatory approvals; |
| • | future products based on our technology will be difficult to manufacture on a large scale or at all or will prove to be uneconomical to produce or market; |
| • | proprietary rights of third parties will prevent us or our collaborators from marketing products; |
| • | third parties will market superior or equivalent products; |
| • | technology advances will render our technology or product to be outdated or less attractive; and |
| • | the products will not attain market acceptance. |
Drug development, including clinical trials required for governmental approval, is expensive and new drugs have a high risk of failure. Based on results at any stage of development, including later-stage clinical trials and our inability to bear the related costs associated with product development or product production or marketing, we may decide to discontinue development or clinical trial at any time.
12
Table of Contents
Preparing for and processing the Biologics License Application (“BLA”) for BiovaxID and conducting a clinical trial for Revimmune will be expensive and time consuming. The FDA response to any application is uncertain and the FDA may reject or deny the application, or may impose additional requirements. Such additional requirements may be expensive and/or time consuming, and meeting these additional requirements may be difficult or impossible.
We might be unable to manufacture our drug candidates on a commercial scale even if approved.
Assuming approval of Revimmune and/or BiovaxID, manufacturing, supply and quality control problems could arise as we, either alone or with subcontractors, attempt to scale-up manufacturing capabilities for products under development. We might be unable to scale-up in a timely manner or at a commercially reasonable cost. Problems could lead to delays or pose a threat to the ultimate commercialization of our product and cause us to fail.
Manufacturing facilities, and those of any future contract manufacturers, are or will be subject to periodic regulatory inspections by the FDA and other federal and state regulatory agencies and these facilities are subject to Quality System Regulation, or “QSR,” requirements of the FDA. If we or our third-party manufacturers fail to maintain facilities in accordance with QSR regulations, other international quality standards, or other regulatory requirements, then the manufacture process could be suspended or terminated, which would harm us.
Because the product development and the regulatory approval process for Revimmune™ and BiovaxID® will be expensive and its outcome is uncertain, we must incur substantial expenses that might not result in any viable product and the process could take longer than expected.
Regulatory approval of a pharmaceutical product is a lengthy, time-consuming and expensive process. Before obtaining regulatory approvals for the commercial sale of either Revimmune and/or the BiovaxID vaccine, we must demonstrate to the satisfaction of applicable regulatory agencies that Revimmune and/or BiovaxID are safe and effective for use in humans. This is expected to result in substantial expense and require significant time.
Historically, the results from pre-clinical testing and early clinical trials often have not been predictive of results obtained in later clinical trials. A number of new drugs have shown promising results in clinical trials, but subsequently failed to establish sufficient safety and efficacy data to obtain necessary regulatory approvals. Data obtained from pre-clinical and clinical activities are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. In addition, regulatory delays or rejections could be encountered as a result of many factors, including changes in regulatory policy during the period of product development.
Clinical trials conducted by us or by third parties on our behalf might not demonstrate sufficient safety, efficacy or statistical significance to obtain the requisite regulatory approval for our products. Regulatory authorities might not permit us to undertake any additional clinical trials for our product candidates.
13
Table of Contents
We may experience difficulties in manufacturing Revimmune™ and/or BiovaxID® or in obtaining approval of the change in manufacturing site or process from the FDA or international regulatory agencies, which could prevent or delay us in the commercialization.
Manufacturing an approved drug is complex and requires coordination internally among our employees, as well as, externally with physicians, hospitals and third-party suppliers and carriers. This process involves several risks that may lead to failures or delays in manufacturing BiovaxID, including:
| • | difficulties in obtaining adequate tumor samples from physicians; |
| • | difficulties in timely shipping of tumor samples to us or in the shipping of BiovaxID to the treating physicians due to errors by third-party carriers, transportation restrictions or other reasons; |
| • | destruction of, or damage to, tumor samples or BiovaxID during the shipping process due to the improper handling by third-party carriers, hospitals, physicians or us; |
| • | destruction of, or damage to, tumor samples or BiovaxID during storage at our facility; and |
| • | difficulties in ensuring the availability, quality, and consistency of materials provided by our suppliers. |
If we experience any difficulties in manufacturing Revimmune and/or BiovaxID, commercialization may be delayed, resulting in delays in generating revenue and increased costs.
In addition, because Biovest has relocated the site of the manufacturing process for BiovaxID to Biovest’s Minneapolis facility following the clinical trials, we are required to demonstrate to the FDA that the product developed under new conditions is comparable to the product that was the subject of earlier clinical testing. This requirement applies to the relocation at the Minneapolis facility as well as future expansions of the manufacturing facilities, such as the possible expansion to additional facilities that may be required for successful regulatory approvals or commercialization of the vaccine, resulting in increased costs. There is also a requirement for validation of the manufacturing process for BiovaxID utilizing our AutovaxID™ instrument.
A showing of comparability requires data demonstrating that the product is consistent with that produced for the clinical trial and continues to be safe, pure, and potent and may be based on chemical, physical, and biological assays and, in some cases, other non-clinical data. If we demonstrate comparability, additional clinical safety and/or efficacy trials with the new product may not be needed. The FDA will determine if comparability data are sufficient to demonstrate that additional clinical studies are unnecessary. If the FDA requires additional clinical safety or efficacy trials to demonstrate comparability, our clinical trials or FDA approval of BiovaxID may be delayed, which would cause delays in generating revenue and increased costs.
14
Table of Contents
Because we have limited experience, we might be unsuccessful in our efforts to develop, obtain approval for, commercially produce or successfully market Revimmune™ and/or BiovaxID®.
The extent to which we develop and commercialize Revimmune and BiovaxID will depend on our ability to:
| • | complete required clinical trials; |
| • | obtain necessary regulatory approvals; |
| • | establish, or contract for, required manufacturing capacity; and |
| • | establish, or contract for, sales and marketing resources. |
We have limited experience with these activities and might not be successful in the trials, product development or commercialization.
Competing technologies may adversely affect us.
Biotechnology has experienced, and is expected to continue to experience, rapid and significant change. The use of monoclonal antibodies as initial or induction therapy, and increasingly for maintenance therapy, has become well-established and generally accepted. Products that are well-established or accepted, including monoclonal antibodies such as Rituxan®, including its anticipated pending expanded approval as a NHL maintenance therapy, may constitute significant barriers to market penetration and regulatory approval which may be expensive, difficult or even impossible to overcome. New developments in biotechnological processes are expected to continue at a rapid pace in both industry and academia, and these developments are likely to result in commercial applications competitive with our proposed vaccine. We expect to encounter intense competition from a number of companies that offer products in our targeted application area. We anticipate that our competitors in these areas will consist of both well-established and development-stage companies and will include:
| • | healthcare companies; |
| • | chemical and biotechnology companies; |
| • | biopharmaceutical companies; and |
| • | companies developing drug discovery technologies. |
The cell culture production and technology business is also intensely competitive and is, in many areas, dominated by large service providers. In many instances, our competitors have substantially greater financial, technical, research and other resources and larger, more established marketing, sales, distribution and service organizations than us. Moreover, these competitors may offer broader product lines and have greater name recognition than us and may offer discounts as a competitive tactic.
Competitors might succeed in developing, marketing, or obtaining FDA approval for technologies, products, or services that are more effective or commercially attractive than those we offer or are developing, or that render our products or services obsolete. As these companies develop their technologies, they might develop proprietary positions, which might prevent us from successfully commercializing products. Also, we might not have the financial resources, technical expertise or marketing, distribution or support capabilities to compete successfully in the future.
15
Table of Contents
Risks related to BiovaxID® (which is being developed by our majority owned subsidiary, Biovest)
Biovest’s clinical trials for BiovaxID® may not be regarded by the FDA or other regulatory authorities as conclusive, and Biovest may decide, or regulators may require Biovest, to conduct additional clinical testing for this product candidate or cease its trials.
In April 2008, Biovest closed its only Phase 3 clinical trial of BiovaxID® with 234 patients enrolled instead of the 563 patients anticipated to be enrolled pursuant to Biovest’s clinical trial protocol. Biovest announced clinical results based upon a relatively smaller number of patients. Additionally, Biovest’s only Phase 3 clinical trial enrolled or randomized patients before their treatment with BiovaxID or control resulting in a significant number of patients being randomized but not treated with either BiovaxID or control because of failure to maintain compliance with protocol requirements including maintenance of complete remission. Biovest does not know if the size or design of its Phase 3 clinical trial or an evaluation, which is limited to only patients that received at least one dose of BiovaxID or control, will be acceptable to the FDA for demonstrating sufficient safety and efficacy required to obtain marketing approval.
While the Phase 3 clinical trial reported clinical benefit for all patients treated with BiovaxID or control that was statistically significant (p-value of 0.045) in patients receiving at least one dose of BiovaxID or control, the results did not achieve “high” statistical significance which is generally considered to be a p-value of 0.01 or better. While it is not required, it is not uncommon for the FDA to require a second confirming Phase 3 clinical trial, when “high” statistical significance has not been demonstrated. The statistical significance of the reported clinical benefit for patients whose tumor and vaccine had an IgM isotype as opposed to an IgG isotype was “highly significant” (p-value of 0.002); however, we do not know how this analysis will impact the FDA’s overall review process. Biovest does not know whether its existing or future clinical trials will be considered by the FDA and/or other regulatory authorities to sufficiently demonstrate safety and efficacy to result in marketing approval. Because Biovest’s clinical trials for BiovaxID may be considered to be inconclusive, Biovest may decide, or regulators may require it, to conduct additional clinical and/or preclinical testing for this product candidate or cease Biovest’s clinical trials. If this happens, Biovest may not be able to obtain approval for this product or the anticipated time to market for this product may be substantially delayed, and Biovest may also experience significant additional development costs. Biovest may also be required to undertake additional clinical testing if Biovest changes or expands the indications for its product candidate.
The ongoing detailed analysis being performed in Biovest’s clinical trials for BiovaxID® may produce negative or inconclusive results and may delay Biovest’s efforts to obtain approval for this product.
Biovest is currently engaged in performing detailed analyses of the safety and efficacy data generated by Biovest’s Phase 3 clinical trial of BiovaxID in follicular non-Hodgkin’s lymphoma (“FL”) and Biovest’s Phase 2 clinical trial in mantle cell lymphoma (“MCL”). Biovest cannot predict with certainty the results of the analyses, and if the results are negative or inconclusive Biovest may decide, or regulators may require it, to conduct additional clinical and/or preclinical testing for this product candidate or cease Biovest’s clinical trials. If this happens, Biovest may not be able to obtain approval for BiovaxID for FL, MCL or both, or the anticipated time to market for this product may be substantially delayed, and Biovest may also experience significant additional development costs.
16
Table of Contents
The clinical trials for BiovaxID® have demonstrated that certain side effects may be associated with this treatment and ongoing or future clinical trials may reveal additional unexpected or unanticipated side effects.
Biovest cannot guarantee that its current or future trials for BiovaxID will not demonstrate additional adverse side effects that may delay or even preclude regulatory approval. Even if BiovaxID receives regulatory approval, if Biovest or others identify previously unknown side effects following approval, regulatory approval could be withdrawn and sales of BiovaxID could be significantly reduced.
Inability to obtain regulatory approval for Biovest’s BiovaxID® manufacturing facility or to manufacture on a commercial scale may delay or disrupt our commercialization efforts.
Before Biovest can obtain FDA approval for any new drug, the manufacturing facility for the drug must be inspected and approved by the FDA. Biovest plans to establish a dedicated BiovaxID manufacturing facility within its existing Minnesota leasehold space. Therefore, before Biovest can obtain the FDA approval necessary to allow it to begin commercially manufacturing BiovaxID, Biovest must pass a pre-approval inspection of its BiovaxID manufacturing facility by the FDA. In order to obtain approval, Biovest will need to ensure that all of its processes, methods, and equipment are compliant with the current Good Manufacturing Practices, or “cGMP,” and perform extensive audits of vendors, contract laboratories, and suppliers. The cGMP requirements govern quality control of the manufacturing process and documentation policies and procedures. Biovest has undertaken steps towards achieving compliance with these regulatory requirements required for commercialization. In complying with cGMP, Biovest will be obligated to expend time, money, and effort in production, record keeping, and quality control to assure that the product meets applicable specifications and other requirements. If Biovest fails to comply with these requirements, Biovest could experience product liability claims from patients receiving its vaccines, Biovest might be subject to possible regulatory action and Biovest may be limited in the jurisdictions in which Biovest is permitted to sell BiovaxID.
In order to commercialize BiovaxID, Biovest will need to develop and qualify one or more additional manufacturing facilities. Preparing a facility for commercial manufacturing may involve unanticipated delays, and the costs of complying with state, local, and FDA regulations may be higher than Biovest anticipated. In addition, any material changes Biovest makes to the manufacturing process may require approval by the FDA and state or foreign regulatory authorities. Obtaining these approvals is a lengthy, involved process, and Biovest may experience delays. Such delays could increase costs and adversely affect its business. In general, the FDA views cGMP standards as being more rigorously applied as products move forward in development and commercialization. In seeking to comply with these standards, Biovest may encounter problems with, among other things, controlling costs and quality control and assurance. It may be difficult to maintain compliance with cGMP standards as the development and commercialization of BiovaxID progresses, if it progresses.
Biovest and its products are subject to comprehensive regulation by the FDA in the U.S. and by comparable authorities in other countries. These national agencies and other federal, state and local entities regulate, among other things, the preclinical and clinical testing, safety, approval, manufacture, labeling, marketing, export, storage, record keeping, advertising and promotion of Biovest’s products. If Biovest violates regulatory requirements at any stage, whether before or after marketing approval is obtained, Biovest may be subject to forced removal of a product from the market, product seizure, civil and criminal penalties and other adverse consequences.
17
Table of Contents
The NCI is not precluded from working with other companies on developing products that are competitive with BiovaxID®.
Biovest’s BiovaxID vaccine is based on research and studies conducted at Stanford University (“Stanford”) and the National Cancer Institute (“NCI”). The concept of producing a patient-specific anti-cancer vaccine through the hybridoma method from a patient’s own cancer cells has been discussed in a variety of publications over a period of many years, and, accordingly, the general method and concept of such a vaccine is not eligible to be patented by us, the NCI, or any other party. Until November 2006, Biovest was a party to a Cooperative Research and Development Agreement, or “CRADA,” with the NCI for the development of a hybridoma-based patient-specific idiotypic vaccine for the treatment of indolent FL. Biovest gave notice of termination of the CRADA in September 2006. Although the NCI transferred sponsorship of the investigational new drug application (“IND”) for BiovaxID to Biovest in 2004, and although there are certain confidentiality protections for information generated pursuant to the CRADA, the CRADA does not prevent the NCI from working with other companies on other hybridoma-based idiotypic vaccines for indolent FL or other forms of cancer, and the NCI or its future partners may be able to utilize certain technology developed under Biovest’s prior CRADA. If the NCI chooses to work with other companies in connection with the development of such a vaccine, such other companies may also develop technology and know-how that may ultimately enable such companies to develop products that compete with BiovaxID. Additionally, through their partnership with the NCI, these companies could develop immunotherapies for other forms of cancer that may serve as barriers to any future products that we may develop for such indications.
Biovest is not able to prevent third parties, including potential competitors, from developing and selling an anti-cancer vaccine for NHL having the same composition of matter as BiovaxID®.
Biovest’s BiovaxID vaccine is based on research and studies conducted at Stanford and the NCI. As a result of published studies, the concept of the vaccine and its composition of matter are in the public domain and cannot be patented by Biovest, the NCI, or any other party. Biovest has filed a Patent Cooperation Treaty (“PCT”) patent application on the type of cell media that is used to grow cell cultures in the production of its vaccine, and Biovest has filed a PCT patent application on certain features of the integrated production and purification system used to produce and purify the vaccine in an automated closed system. Biovest has obtained an exclusive world-wide license for use of a proprietary cell line which it uses to manufacture BiovaxID, but Biovest cannot be certain that it will be successful in preventing others from utilizing this cell line or will be able to maintain and enforce the exclusive license in all jurisdictions. Biovest cannot prevent other companies using different manufacturing processes from developing active immunotherapies that directly compete with BiovaxID. For example, Bayer Innovation GmbH has commenced a Phase 1 clinical trial for its autologous anti-idiotype lymphoma vaccine produced in a tobacco plant cell line.
Several companies, such as Genentech, Inc., Corixa Corporation, Biogen Idec, and Immunomedics, Inc., are involved in the development of passive immunotherapies for NHL. These passive immunotherapies include Rituxan®, a monoclonal antibody, and Zevalin® and Bexxar®, which are passive radioimmunotherapy products. Passive immunotherapies, particularly the monoclonal antibody Rituxan, are well accepted and established in the treatment of NHL and as such will impact both regulatory considerations and market penetration.
18
Table of Contents
Risks Related to Our Industry
If we acquire other complementary technologies or companies, our financial performance could suffer, and such acquisitions involve a number of risks.
We may from time to time actively seek to identify and acquire companies, technologies, or pharmaceutical products with attributes complementary to our products and services. Acquisitions that we make may involve numerous risks, including:
| • | diverting management’s attention from other business concerns; |
| • | being unable to maintain uniform standards, controls, procedures, and policies; |
| • | entering markets in which we have no direct prior experience; |
| • | improperly evaluating new services and technologies or otherwise being unable to fully exploit the anticipated opportunity; and |
| • | being unable to successfully integrate the acquisition. |
Any of the factors listed above would adversely affect our results of operations.
Our proprietary rights may not adequately protect our technologies and product candidates.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of our technologies and product candidates as well as successfully defending these patents against third-party challenges. We will only be able to protect our technologies and product candidates from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them. Furthermore, the degree of future protection of our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage.
For Revimmune, we have published patent applications as well as additional patent protection. For BiovaxID, Biovest holds the patent relating to the method of producing BiovaxID and Biovest has filed additional patent applications for BiovaxID. There are filed patent applications for AutovaxID as well.
The patent positions of life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in such companies’ patents has emerged to date in the U.S. The patent situation outside the U.S. is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. or other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents. For example:
| • | we or our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| • | we or our licensors might not have been the first to file patent applications for these inventions; |
19
Table of Contents
| • | others may independently develop similar or alternative technologies or duplicate any of our technologies; |
| • | it is possible that none of our pending patent applications or the pending patent applications of our licensors will result in issued patents; |
| • | our issued patents and issued patents of our licensors may not provide a basis for commercially viable products, or may not provide us with any competitive advantages, or may be challenged and invalidated by third parties; |
| • | we may not develop additional proprietary technologies or product candidates that are patentable; or |
| • | the patents of others may have an adverse effect on our business. |
We also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. While we use reasonable efforts to protect our trade secrets, our or our strategic partners’ employees, consultants, contractors, or scientific and other advisors may unintentionally or willfully disclose our information to competitors. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time consuming, and the outcome would be unpredictable. In addition, courts outside the United States are sometimes less willing to protect trade secrets. Moreover, if our competitors independently develop equivalent knowledge, methods, and know-how, it will be more difficult for us to enforce our patent rights and our business could be harmed.
If we are not able to defend the patent or trade secret protection position of our technologies and product candidates, then we will not be able to exclude competitors from developing or marketing competing products, and we may not generate enough revenue from product sales to justify the cost of development of our products and to achieve or maintain profitability.
If we are sued for infringing intellectual property rights of third parties, such litigation will be costly and time consuming, and an unfavorable outcome would have a significant adverse effect on our business.
Our ability to commercialize our products depends on our ability to sell such products without infringing the patents or other proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending applications, which are owned by third parties, exist in the various areas in which we have products or are seeking to create products, including patents relating to specific antifungal formulations and methods of using the formulations to treat infections, as well as patents relating to serum-based vaccines and methods for detection of lymphoma. The interpretation of patent claims is complex and uncertain. The legal standards governing claim interpretations are evolving and changing. Thus, any significant changes in the legal standards would impact the way that we interpret the claims of third-party patents in our product areas. In addition, because patent applications can take several years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our product candidates may infringe. There could also be existing patents of which we are not aware that our product candidates may inadvertently infringe.
20
Table of Contents
If a third party claims that we infringe on their patents or other proprietary rights, we could face a number of issues that could seriously harm our competitive position, including:
| • | infringement and other intellectual property claims which, with or without merit, can be costly and time consuming to litigate and can delay the regulatory approval process and divert management’s attention from our core business strategy; |
| • | substantial damages for past infringement which we may have to pay if a court determines that our products or technologies infringe upon a competitor’s patent or other proprietary rights; |
| • | a court prohibiting us from selling or licensing our products or technologies unless the holder licenses the patent or other proprietary rights to us, which it is not required to do; |
| • | if a license is available from a holder, we may have to pay substantial royalties or grant cross licenses to our patents or other proprietary rights; and |
| • | redesigning our process so that it does not infringe, which may not be possible or may require substantial time and expense. |
Such actions could harm our competitive position and our ability to generate revenue and could result in increased costs.
We currently depend on sole-source suppliers for cyclophosphamide, the primary agent utilized in Revimmune™ treatment, and for KLH, a critical raw material used in the manufacture of BiovaxID, and physicians who administer BiovaxID depend on a sole-source supplier for GM-CSF, an immune system stimulant administered with BiovaxID.
We currently depend on single source suppliers for critical raw materials used in Revimmune, BiovaxID and other components used in the manufacturing process and required for the administration of BiovaxID. In particular, the Revimmune treatment consists of high-dose cyclophosphamide. Baxter International, Inc. (“Baxter”) is the exclusive supplier of injectable cyclophosphamide in the U.S. We have entered into an agreement making Baxter our exclusive source of cyclophosphamide under an agreed-upon price structure (the “Baxter Agreement”). The Baxter Agreement provides us with the exclusive, worldwide right to purchase Baxter’s cyclophosphamide, which is marketed under the brand name Cytoxan®, for the treatment of certain designated autoimmune diseases, including but not limited to autoimmune hemolytic anemia, systemic sclerosis and multiple sclerosis. In addition, the manufacturing of BiovaxID requires keyhole limpet hemocyanin, or “KLH,” a foreign carrier protein. Biovest purchases KLH from BioSyn Arzneimittel GmbH (“BioSyn”), a single source supplier. Biovest has entered into a supply agreement with BioSyn, pursuant to which BioSyn has agreed to supply us with KLH. The supply agreement has an initial term of three years and is renewable for indefinite additional terms of five years each at our discretion, so long as we are not in default of our obligations pursuant to this agreement. Either party may terminate the supply agreement earlier upon a breach that is not cured within 60 days or other events relating to insolvency or bankruptcy. Under this agreement BioSyn is not contractually obligated to supply Biovest with the amounts of KLH required for commercialization. Biovest has become aware of additional suppliers who now market KLH, but Biovest has not yet established a relationship with these suppliers and has not performed testing to ensure that the KLH supplied by them is suitable for use in the BiovaxID production process.
21
Table of Contents
When BiovaxID is administered, the administering physician uses a cytokine to enhance the patient’s immune response, and this cytokine is administered concurrently with BiovaxID. The cytokine used by physicians for this purpose is Leukine® sargramostim, a commercially available recombinant human granulocyte-macrophage colony stimulating factor known as GM-CSF. This cytokine is a substance that is purchased by the administering physician and is administered with an antigen to enhance or increase the immune response to that antigen. The physicians who administer BiovaxID will rely on Berlex Inc., or “Berlex,” as a supplier of GM-CSF, and these physicians will generally not have the benefit of a long-term supply contract with Berlex. GM-CSF is not commercially available from other sources in the U.S. or Canada.
Establishing additional or replacement suppliers for these materials or components may take a substantial amount of time. In addition, we may have difficulty obtaining similar components from other suppliers that are acceptable to the FDA. If Biovest has to switch to a replacement supplier, Biovest may face additional regulatory delays and the manufacture and delivery of BiovaxID, or any other immunotherapies that Biovest may develop, could be interrupted for an extended period of time, which may delay completion of Biovest’s commercialization of BiovaxID, or any other immunotherapies that we may develop. If we are unable to obtain adequate amounts of these components, our commercialization will be delayed. In addition, we will be required to obtain regulatory clearance from the FDA to use different components that may not be as safe or as effective. As a result, regulatory approval of BiovaxID may not be received at all. All these delays could cause delays in commercialization of BiovaxID, delays in Biovest’s ability to generate revenue from BiovaxID, and increased costs.
The market may not be receptive to our products upon their introduction.
The biopharmaceutical products that we may develop may not achieve market acceptance among physicians, patients, health care payors, and the medical community. The degree of market acceptance will depend upon a number of factors, including:
| • | the receipt of regulatory approvals; |
| • | limited indications of regulatory approvals; |
| • | the establishment and demonstration in the medical community of the clinical efficacy and safety of our products and their potential advantages over existing treatment methods; |
| • | the prices of such products; |
| • | reimbursement policies of government and third-party payors; |
| • | market acceptance of patient-specific active immunotherapies, in the case of BiovaxID; |
| • | the prevalence and severity of any side effects; |
| • | potential advantages over alternative treatments; |
| • | ability to produce our products at a competitive price; |
| • | stocking and distribution; |
| • | relative convenience and ease of administration; |
| • | the strength of marketing and distribution support; and |
22
Table of Contents
| • | sufficient third-party coverage or reimbursement. |
The failure of our product pipeline to gain market acceptance could impair our ability to generate revenue, which could have a material adverse effect on our future business, financial condition and results of operations.
Our competitors may develop products that are less expensive, safer, or more effective, which may diminish or eliminate the commercial success of any future products that we may commercialize.
We compete with several biopharmaceutical companies, and our competitors may:
| • | develop product candidates and market products that are less expensive or more effective than our future products; |
| • | commercialize competing products before we or our partners can launch any products developed from our product candidates; |
| • | initiate or withstand substantial price competition more successfully than we can; |
| • | have greater success in recruiting skilled scientific workers from the limited pool of available talent; |
| • | more effectively negotiate third-party licenses and strategic relationships; and |
| • | take advantage of acquisition or other opportunities more readily than we can. |
We will compete for market share against large pharmaceutical and biotechnology companies and smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their partners, may develop new product candidates that will compete with ours, and these competitors may, and in certain cases do, operate larger research and development programs or have substantially greater financial resources than we do.
If our competitors market products that are less expensive, safer or more effective than our potential products, or that reach the market sooner than our potential products, we may not achieve commercial success. In addition, the life sciences industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change we may be unable to compete effectively. Our competitors may render our technologies obsolete by advances in existing technological approaches or the development of new or different approaches, potentially eliminating the advantages in our drug discovery process that we believe we derive from our research approach and proprietary technologies.
If we fail to comply with extensive regulations enforced by the FDA, EMEA, and other agencies, the sale of our product candidates would be prevented or delayed.
Research, pre-clinical development, clinical trials, manufacturing, and marketing of our products are subject to extensive regulation by various government authorities. We have not received marketing approval for BiovaxID or Revimmune. The process of obtaining FDA, EMEA, and other required regulatory approvals is lengthy and expensive, and the time required for such approvals is uncertain. The approval process is affected by factors such as:
| • | the severity of the disease; |
23
Table of Contents
| • | the quality of submission; |
| • | the clinical efficacy and safety; |
| • | the strength of the chemistry and manufacturing control of the process; |
| • | the manufacturing facility compliance; |
| • | the availability of alternative treatments; |
| • | the risks and benefits demonstrated in clinical trials; and |
| • | the patent status and marketing exclusivity rights of certain innovative products. |
Any regulatory approvals that we or our partners receive for our product candidates may also be subject to limitations on the indicated uses for which the drug may be marketed or contain requirements for potentially costly post-marketing follow-up studies. The subsequent discovery of previously unknown problems with the drug, including adverse events of unanticipated severity or frequency, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
Our U.S. manufacturing, labeling, storage, and distribution activities also are subject to strict regulation and licensing by the FDA. Our biopharmaceutical manufacturing facilities are subject to periodic inspection by the FDA, the EMEA, and other regulatory authorities and from time to time, we may receive notices of deficiencies from these agencies as a result of such inspections. Our failure, or the failure of our biopharmaceutical manufacturing facilities, to continue to meet regulatory standards or to remedy any deficiencies could result in corrective action by the FDA or these other authorities, including the interruption or prevention of marketing, closure of our biopharmaceutical manufacturing facilities and fines or penalties.
Regulatory authorities also will require post-marketing surveillance to monitor and report to the FDA potential adverse effects of our products or product candidates. Congress or the FDA in specific situations can modify the regulatory process. Once approved, a product’s failure to comply with applicable regulatory requirements could, among other things, result in warning letters, fines, suspension or revocation of regulatory approvals, product recalls or seizures, operating restrictions, injunctions, and criminal prosecutions.
The FDA’s policies may change and additional government regulations may be enacted that could prevent or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we might not be permitted to market our products and our business could suffer.
24
Table of Contents
Distribution of our products outside the U.S. is subject to extensive government regulation. These regulations, including the requirements for approvals or clearance to market, the time required for regulatory review and the sanctions imposed for violations, vary from country to country. There can be no assurance that we will obtain regulatory approvals in such countries or that we will not be required to incur significant costs in obtaining or maintaining these regulatory approvals. In addition, the export by us of certain of our products that have not yet been cleared for domestic commercial distribution may be subject to FDA export restrictions. Failure to obtain necessary regulatory approvals, the restriction, suspension or revocation of existing approvals or any other failure to comply with regulatory requirements would impair our ability to generate revenue, increase our compliance costs, and have a material adverse effect on our future business, financial condition, and results of operations.
The insurance coverage and reimbursement status of newly approved products is uncertain and failure to obtain or maintain adequate coverage and reimbursement for new or current products could limit our ability to market those products and decrease our ability to generate revenue.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. The commercial success of our potential products in both domestic and international markets is substantially dependent on whether third-party coverage and reimbursement is available for the ordering of our potential products by the medical profession for use by their patients. Medicare, Medicaid, health maintenance organizations, and other third-party payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement of new products, and, as a result, they may not cover or provide adequate payment for our potential products. Our product candidates could, if approved, face declining revenues if competitor products are perceived as providing a substantially equivalent therapeutic effect at a lower cost to the payor. They may not view our products as cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our products to be marketed on a competitive basis. Likewise, legislative or regulatory efforts to control or reduce healthcare costs or reform government healthcare programs could result in lower prices or rejection of our products. Changes in coverage and reimbursement policies or healthcare cost containment initiatives that limit or restrict reimbursement for our products may cause our revenue to decline.
We may not be able to maintain sufficient product liability insurance to cover claims against us.
Product liability insurance for the biopharmaceutical industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to maintain such insurance on acceptable terms or that we will be able to secure increased coverage if the commercialization of our products progresses, or that existing, or future claims against us will be covered by our product liability insurance. Moreover, there can be no assurance that the existing coverage of our insurance policy and/or any rights of indemnification and contribution that we may have will offset existing or future claims. We maintain product liability insurance of $1.0 million per occurrence and in the aggregate. We believe that this coverage is currently adequate based on current and projected business activities and the associated risk exposure, although we expect to increase this coverage as our business activities and associated risk grow. A successful claim against us with respect to uninsured liabilities or in excess of insurance coverage and not subject to any indemnification or contribution could have a material adverse effect on our future business, financial condition, and results of operations.
25
Table of Contents
We could be negatively impacted by the application or enforcement of federal and state fraud and abuse laws, including anti-kickback laws and other federal and state anti-referral laws.
We are subject to various federal and state laws pertaining to healthcare fraud and abuse, including anti-kickback laws and physician self-referral laws. Violations of these laws are punishable by criminal and civil sanctions, including, in some instances, imprisonment and exclusion from participation in federal and state healthcare programs, including the Medicare, Medicaid and Veterans Administration health programs. Because of the far-reaching nature of these laws, we may be required to alter or discontinue one or more of our practices to be in compliance with these laws. Healthcare fraud and abuse regulations are complex, and even minor irregularities can potentially give rise to claims that a statute or prohibition has been violated. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our business, financial condition and results of operations. If there is a change in law, regulation or administrative or judicial interpretations, we may have to change or discontinue our business practices or our existing business practices could be challenged as unlawful, which could have a material adverse effect on our business, financial condition and results of operations. In addition, we could become subject to false claims litigation under federal statutes, which can lead to treble damages based on the reimbursements by federal health care programs, civil money penalties (including penalties levied on a per false claim basis), restitution, criminal fines and imprisonment, and exclusion from participation in Medicare, Medicaid and other federal and state healthcare programs. These false claims statutes include the False Claims Act, which allows any person to bring suit on behalf of the federal government alleging the submission of false or fraudulent claims, or causing to present such false or fraudulent claims, under federal programs or contracts claims or other violations of the statute and to share in any amounts paid by the entity to the government in fines or settlement. These suits against biotechnology companies have increased significantly in recent years and have increased the risk that a healthcare company will have to defend a false claim action, pay fines or restitution, or be excluded from the Medicare, Medicaid or other federal and state healthcare programs as a result of an investigation arising out of such action. We cannot assure you that we will not become subject to such litigation or, if we are not successful in defending against such actions, that such actions will not have a material adverse effect on our business, financial condition and results of operations. In addition, we cannot assure you that the costs of defending claims or allegations under the False Claims Act will not have a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Operations
The failure to attract and retain skilled personnel could impair our product development and commercialization efforts.
Our performance is substantially dependent on the performance of our senior management and key scientific and technical personnel. The loss of the services of any member of our senior management, scientific, or technical staff may significantly delay or prevent the achievement of product development and other business objectives by diverting management’s attention to transition matters and identification of suitable replacements, and could have a material adverse effect on our business, operating results, and financial condition. We do not maintain key man life insurance for any of our executive management personnel. We are not aware of any plans by our key personnel to retire or leave us in the near future.
We also rely on consultants and advisors to assist us in formulating our research and development strategy. All of our consultants and advisors are either self-employed or employed by other organizations, and they may have conflicts of interest or other commitments, such as consulting or advisory contracts with other organizations, that may affect their ability to contribute to us.
In addition, we believe that we will need to recruit additional executive management and scientific and technical personnel. There is currently intense competition for skilled executives and employees with relevant scientific and technical expertise, and this competition is likely to continue. The inability to attract and retain sufficient scientific, technical, and managerial personnel could limit or delay our product development efforts, which would adversely affect the development of our product candidates and commercialization of our potential products and growth of our business.
26
Table of Contents
We expect to expand our development, clinical research, and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
We expect to have significant growth in expenditures, the number of our employees and the scope of our operations, in particular with respect to those product candidates that we elect to commercialize independently or together with a partner. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational, and financial systems, expand our facilities, and continue to recruit and train additional qualified personnel. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
We have a limited operating history and financial results are uncertain.
We have a limited history as a consolidated company and face many of the risks of a new business. As a result of our limited operating history, it is difficult to accurately forecast our potential revenue. Our revenue and income potential is unproven and our business model is still emerging. Therefore, we cannot assure you that we will provide a return on investment in the future. An investor in our common stock must consider the challenges, risks, and uncertainties frequently encountered in the establishment of new technologies and products in emerging markets and evolving industries. These challenges include our ability to:
| • | execute our business model; |
| • | create brand recognition; |
| • | manage growth in our operations; |
| • | create a customer base cost-effectively; |
| • | retain customers; |
| • | access additional capital when required; and |
| • | attract and retain key personnel. |
We cannot be certain that our business model will be successful or that it will successfully address these and other challenges, risks, and uncertainties.
The existence of minority shareholders in our Biovest subsidiary creates potential for conflicts of interest.
We own a majority of the outstanding shares of common stock of Biovest and the remaining Biovest stock is owned by approximately 450 shareholders. To the extent that our officers and directors are also officers or directors of Biovest, matters may arise that place the fiduciary duties of these individuals in conflicting positions. Although, we intend that such conflicts will be resolved by independent directors of Biovest, if this occurs, matters important to us could be delayed. Francis E. O’Donnell, Jr., M.D., our Chairman and Chief Executive Officer, is also Chairman and Chief Executive Officer of Biovest; Samuel S. Duffey, Esq., our President and General Counsel, is also President and General Counsel of Biovest; and Alan M. Pearce, our former Chief Financial Officer, was also Chief Financial Officer of Biovest.
27
Table of Contents
Shares of the Biovest stock held by us are transferable under debentures and warrants issued by us.
In September 2006, we entered into a private placement in which we issued to investors an aggregate of $25.0 million of 8% secured convertible debentures together with common stock purchase warrants. On the Effective Date, these debentures and warrants were replaced by new notes and warrants issued pursuant to our Plan of Reorganization (the “Class 5 Debentures” and the “Class 5 Warrants,” respectively). The Class 5 Debentures issued by us are convertible at the option of the holder into shares of our common stock or exchangeable for shares of Biovest stock held by us, and the Class 5 Warrants are exercisable for our common stock or shares of Biovest stock held by us. In connection with the Class 5 Debentures and the Class 5 Warrants, we have pledged into an escrow account 14.4 million shares of the 91.0 million shares of Biovest common stock held by us to be available to holders for exchange as well as to secure the repayment of the Class 5 Debentures. The total number of shares of Biovest common stock transferable by us to the investors in the Class 5 Debentures, whether pursuant to the exchange or exercise of the Class 5 Debentures and Class 5 Warrants or the exercise of rights under the pledge agreement, may not exceed 14.4 million shares in the aggregate. Accordingly, it is possible that our ownership of Biovest common stock could decrease by up to 14.4 million shares as a result of the Class 5 Debentures and/or the Class 5 Warrants. We have also pledged a total of approximately 42 million shares of Biovest common stock owned by us to secure debts owed to three secured creditors. In addition, Biovest may issue shares of its common stock to third parties in connection with debt and/or equity financing transactions. In light of the foregoing, it is possible that we could cease to be the majority shareholder of Biovest.
We occasionally become subject to commercial disputes that could harm our business by distracting our management from the operation of our business, by increasing our expenses and, if we do not prevail, by subjecting us to potential monetary damages and other remedies.
From time to time we are engaged in disputes regarding our commercial transactions. These disputes could result in monetary damages or other remedies that could adversely impact our financial position or operations. Even if we prevail in these disputes, they may distract our management from operating our business and the cost of defending these disputes would reduce our operating results. If we do not prevail in these litigation matters or if we are required to expend a significant amount of resources defending such claims, our operating results, financial position, and cash flows could be adversely impacted.
Risks Related to Our Common Stock
Evolving regulation of corporate governance and public disclosure may result in additional expenses and continuing uncertainty.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002 and new SEC regulations, are creating uncertainty for public companies. As a result of these new rules, we will incur additional costs associated with our public company reporting requirements. In addition, these new rules could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and this could make it difficult for us to attract and retain qualified persons to serve on our board of directors.
28
Table of Contents
We are presently evaluating and monitoring developments with respect to new and proposed rules and cannot predict or estimate the amount of the additional costs we may incur or the timing of such costs. These new or changed laws, regulations, and standards are subject to varying interpretations, in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
We are committed to maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest resources to comply with evolving laws, regulations, and standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new or changed laws, regulations, and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us and we may be harmed.
Our common stock is quoted on the Over-the-Counter Market.
As opposed to a larger or better accepted market, our common stock is quoted on the OTCQB, which is the middle tier of the OTC Market, reserved for companies that are registered and reporting with the SEC or a U.S. banking regulator. Thus, an investor might find it more difficult than it would be on a national exchange to dispose of, or to obtain accurate quotations as to the market value of, our securities. We cannot be certain that an active trading market will develop or, if developed, be sustained. We also cannot be certain that purchasers of our common stock will be able to resell their common stock at prices equal to or greater than their purchase price. The development of a public market having the desirable characteristics of depth, liquidity and orderliness depends upon the presence in the marketplace of a sufficient number of willing buyers and sellers at any given time. We do not have any control over whether there will be sufficient numbers of buyers and sellers. Accordingly, we cannot be certain that an established and liquid market for our common stock will develop or be maintained. The market price of our common stock could experience significant fluctuations in response to our operating results and other factors. In addition, the stock market in recent years has experienced extreme price and volume fluctuations that often have been unrelated or disproportionate to the operating performance of individual companies. These fluctuations, and general economic and market conditions, may hurt the market price of our common stock.
We are also subject to a Securities and Exchange Commission rule that, if we fail to meet certain criteria set forth in such rule, the rule imposes various sales practice requirements on broker-dealers who sell securities governed by the rule to persons other than established customers and accredited investors. For these types of transactions, the broker-dealer must make a special suitability determination for the purchaser and have received the purchaser’s written consent to the transaction prior to sale. Consequently, the rule may have an adverse effect on the ability of broker-dealers to sell our securities and may affect the ability of our stockholders to buy and sell our securities in the secondary market. The additional burdens imposed upon broker-dealers may discourage broker-dealers from effecting transactions in penny stocks, which could reduce the liquidity of our common stock and have a material adverse effect on the trading market for our securities.
We believe we are eligible to upgrade our stock listing to the OTCQX, which is the top tier of the OTC market. The OTCQX is reserved exclusively for companies that meet the highest financial standards and undergo a qualitative review, as determined by the Pink OTC Markets, Inc. However, there can be no assurance that an OTCQX application, if submitted, will be approved.
29
Table of Contents
The price of our stock may be highly volatile.
The market price for our common stock is likely to fluctuate due to factors unique to us and along with the highly volatile market prices of securities of biotechnology companies. You may not be able to resell shares of our common stock following periods of volatility. In addition, you may not be able to resell shares at or above your purchase price.
Our stock price may be affected by many factors, many of which are outside of our control, which may include:
| • | actual or anticipated variations in quarterly operating results; |
| • | the results of clinical trials and preclinical studies involving our products or those of our competitors; |
| • | changes in the status of any of our drug development programs, including delays in clinical trials or program terminations; |
| • | developments in our relationships with other collaborative partners; |
| • | developments in patent or other proprietary rights; |
| • | governmental regulation; |
| • | public concern as to the safety and efficacy of products developed by us, our collaborators or our competitors; |
| • | our ability to fund on-going operations; |
| • | announcements of technological innovations or new products or services by us or our competitors; |
| • | changes in financial estimates by securities analysts; |
| • | conditions or trends in the biotechnology industry; and |
| • | changes in the economic performance or market valuations of other biotechnology companies. |
We may be unable to obtain necessary additional financing.
The capital requirements for our operations have been and will continue to be significant. Our ability to generate cash from operations is dependent upon, among other things, increased demand for our products and services and the successful development of direct marketing and product distribution capabilities. There can be no assurance that we will have sufficient capital resources to implement our business plan and we may need additional external debt and/or equity financing to fund our future operations.
30
Table of Contents
There may not be a market for our common stock.
Our shares are quoted on the OTCQB. The OTCQB is the middle tier of the OTC market. OTCQB companies are registered and reporting with the SEC or a U.S. banking regulator, making it easier for investors to identify companies that are current in their reporting obligations. There are no financial or qualitative standards to be in this tier. The OTCQB™ marketplace was launched in April 2010, by Pink OTC Markets, Inc. to better distinguish OTC securities that are registered and reporting with U.S. regulators. However, no assurance can be given that a holder of our common stock will be able to sell such securities in the future or as to the price at which such securities might trade. The liquidity of the market for such securities and the prices at which such securities trade will depend upon the number of holders thereof, the interest of securities dealers in maintaining a market in such securities and other factors beyond our control.
Penny stock regulations may impose certain restrictions on the marketability of our securities.
The Securities and Exchange Commission (the “SEC”) has adopted regulations which generally define a “penny stock” to be any equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. As a result, our common stock is subject to rules that impose additional sales practice requirements on broker dealers who sell such securities to persons other than established customers and accredited investors (generally those with assets in excess of $1.0 million or annual income exceeding $0.2 million, or $0.3 million together with their spouse). For transactions covered by such rules, the broker dealer must make a special suitability determination for the purchase of such securities and have received the purchaser’s written consent to the transaction prior to the purchase. Additionally, for any transaction involving a penny stock, unless exempt, the rules require the delivery, prior to the transaction, of a risk disclosure document mandated by the SEC relating to the penny stock market. The broker dealer must also disclose the commission payable to both the broker dealer and the registered representative, current quotations for the securities and, if the broker dealer is the sole market maker, the broker dealer must disclose this fact and the broker dealer’s presumed control over the market. Finally, monthly statements must be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. Broker-dealers must wait two business days after providing buyers with disclosure materials regarding a security before effecting a transaction in such security. Consequently, the “penny stock” rules restrict the ability of broker dealers to sell our securities and affect the ability of investors to sell our securities in the secondary market and the price at which such purchasers can sell any such securities, thereby affecting the liquidity of the market for our common stock.
Stockholders should be aware that, according to the SEC, the market for penny stocks has suffered in recent years from patterns of fraud and abuse. Such patterns include:
| • | control of the market for the security by one or more broker-dealers that are often related to the promoter or issuer; |
| • | manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases; |
| • | “boiler room” practices involving high pressure sales tactics and unrealistic price projections by inexperienced sales persons; |
| • | excessive and undisclosed bid-ask differentials and markups by selling broker-dealers; and |
31
Table of Contents
| • | the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, along with the inevitable collapse of those prices with consequent investor losses. |
Our management is aware of the abuses that have occurred historically in the penny stock market.
We have not been the subject of an independent valuation.
No investment banker or underwriter has been retained to value our common stock. We have not attempted to make any estimate of the prices at which our common stock may trade in the market. Moreover, there can be no assurance given as to the market prices that will prevail.
Our shareholders could suffer significant dilution upon the issuance of additional shares.
It is possible that we will need to issue additional shares of common stock or securities or warrants convertible into such shares in order to raise additional equity. In such event, our stockholders could suffer significant dilution.
Risks Related to the Offering
We will have immediate and broad discretion over the use of most of the net proceeds from this offering.
There is no minimum offering amount required as a condition to closing this offering and, therefore, subject to the requirement that we use 30% of the net proceeds of the offering to reduce the principal and interest on our indebtedness to our senior lender, the net proceeds from this offering will be immediately available to us to use at our discretion. We intend to use the net proceeds for working capital and other general corporate purposes and to reduce the principal and interest on our indebtedness to our senior lender by $ (which represents 30% of the net proceeds of the offering). Our judgment may not result in positive returns on your investment and you will not have an opportunity to evaluate the economic, financial, or other information upon which we base our decisions.
You will experience immediate and substantial dilution as a result of this offering.
You will incur immediate and substantial dilution as a result of this offering. Assuming the sale by us of the units offered in this offering at an assumed offering price of $ per unit (based upon the last reported sale price of our common stock on , 2011), and after deducting the placement agent fees and estimated offering expenses payable by us, investors in this offering can expect an immediate dilution of $ per share, assuming no exercise of the warrants. Investors exercising their warrants will experience additional dilution.
32
Table of Contents
Shares eligible for future sale may adversely affect the market.
From time to time, certain of our stockholders may be eligible to sell some or all of their shares of our common stock by means of ordinary brokerage transactions in the open market pursuant to Rule 144, promulgated under the Securities Act of 1933, as amended, subject to certain limitations. In general, pursuant to Rule 144, a stockholder (or stockholders whose shares are aggregated) who has satisfied a six-month holding period may, under certain circumstances, sell within any three month period a number of securities which does not exceed the greater of 1% of the then outstanding shares of common stock or the average weekly trading volume of the class during the four calendar weeks prior to such sale. Rule 144 also permits, under certain circumstances, the sale of securities, without any limitation, by our stockholders that are non-affiliates that have satisfied a one-year holding period. Any substantial sale of our common stock pursuant to Rule 144 or pursuant to any resale prospectus may have a material adverse effect on the market price of our common stock.
This prospectus contains forward-looking statements that involve risks and uncertainties, such as statements about our plans, objectives, expectations, assumptions, or future events. In some cases, you can identify forward-looking statements by terminology such as “anticipate,” “estimate,” “plan,” “project,” “continuing,” “ongoing,” “expect,” “believe,” “intend,” “may,” “will,” “should,” “could,” and similar expressions. Examples of forward-looking statements include, without limitation:
| • | statements regarding our strategies, results of operations or liquidity; |
| • | statements concerning projections, predictions, expectations, estimates or forecasts as to our business, financial and operational results and future economic performance; |
| • | statements of management’s goals and objectives; |
| • | projections of revenue, earnings, capital structure and other financial items; |
| • | assumptions underlying statements regarding us or our business; and |
| • | other similar expressions concerning matters that are not historical facts. |
Forward-looking statements should not be read as a guarantee of future performance or results, and will not necessarily be accurate indications of the times at, or by, which such performance or results will be achieved. Forward-looking statements are based on information available at the time those statements are made or management’s good faith belief as of that time with respect to future events, and are subject to risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to, factors discussed under the headings “RISK FACTORS,” “MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS,” and “BUSINESS.”
33
Table of Contents
Forward-looking statements involve estimates, assumptions, known and unknown risks, uncertainties and other factors that could cause actual results to differ materially from any future results, performances, or achievements expressed or implied by the forward-looking statements. These risks include, but are not limited to, those listed below and those discussed in greater detail under the heading “RISK FACTORS” above:
| • | our history of operating losses; |
| • | our ability to generate sufficient revenue to become profitable; |
| • | our ability to continue as a going concern; |
| • | our ability to obtain additional funding when required; |
| • | our dependence on the success of products that may not be successfully commercialized and for which we have not obtained FDA approval; |
| • | difficulties in manufacturing our products or in obtaining regulatory approval of our products; |
| • | competing technologies may adversely affect us; |
| • | our proprietary rights may not adequately protect our technologies and product candidates; |
| • | our dependence on certain sole source suppliers; |
| • | the market may not be receptive to our products; |
| • | our ability to comply with extensive regulations; |
| • | the potential negative impact associated with the application or enforcement of federal and state regulations; |
| • | the volatility of our stock price; and |
| • | the unpredictability of the market for our common stock. |
Additional risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, financial condition or operating results.
The forward-looking statements speak only as of the date on which they are made, and, except as required by law, we undertake no obligation to update any forward-looking statement to reflect events or circumstances after the date on which the statement is made or to reflect the occurrence of unanticipated events. In addition, we cannot assess the impact of each factor on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. Consequently, you should not place undue reliance on forward-looking statements.
34
Table of Contents
We estimate that we will receive up to $ in net proceeds from the sale of units in this offering if all of the units offered hereby are sold, based on an assumed price of $ per unit (based upon the closing sale price of our common stock on the OTCQB on , 2011) and after deducting estimated placement agent fees and estimated offering expenses payable by us.
If a warrant holder elects to pay the exercise price, rather than exercising the warrants on a “cashless” basis, we may also receive proceeds from the exercise of warrants. We cannot predict when or if the warrants will be exercised. It is possible that the warrants may expire and may never be exercised.
We intend to use the net proceeds of this offering for working capital and other general corporate purposes and to reduce the principal and interest on our indebtedness to our senior lender by $ (which represents 30% of the net proceeds of the offering). Our indebtedness to our senior lender currently accrues interest at a rate of 8.5% per annum and is due and payable in full on November 17, 2012.
Other than the 30% to be paid to our senior lender, we have not yet determined with certainty the manner in which we will allocate the net proceeds. Accordingly, our management will have broad discretion in the application of the net proceeds, and investors will be relying on the judgment of our management regarding the application of the net proceeds of this offering. The amount and timing of these expenditures will vary depending upon a number of factors, including FDA and foreign regulatory approval for our products, the success of our product development efforts, the timing and success of initiating the commercialization of our products, our future sales growth, the cash generated from future operations and actual expenses to operate our business.
Pending the uses described above, we intend to invest the net proceeds in United States government securities and other short-term, investment-grade, interest-bearing instruments.
We have never declared or paid any cash dividends and do not anticipate paying cash dividends in the foreseeable future. We anticipate that all future earnings will be retained to fund the development and expansion of our business. Any future determination to pay dividends will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in future financing instruments, and other factors.
MARKET PRICE OF COMMON STOCK AND RELATED STOCKHOLDER MATTERS
Market for Our Common Stock
Prior to August 19, 2010, our common stock was quoted on the Pink Sheets under the symbol “ABPIQ”. On August 19, 2010, our common stock opened for trading on the OTCQB™ Market under the symbol, “ABPIQ”. On November 18, 2010, our common stock opened for trading on the OTCQB™ Market under the symbol, “ABPI”.
Number of Common Shareholders
As of January 31, 2011, we had approximately 68,632,612 shares of common stock outstanding, which were held by approximately 150 stockholders of record.
35
Table of Contents
Quarterly High/Low Company Stock Price – ABPIQ/ABPI
The following table sets forth the range of high and low bid quotations for our common stock for each of the quarterly periods indicated as reported by the Pink Sheets from October 1, 2008 to August 18, 2010 and thereafter, as reported by the OTCQB™ Market. Bid quotations reflect inter-dealer prices, without retail mark-up, mark-down or commission, and may not represent actual transactions.
|
High |
Low |
|||||||
| Year Ended September 30, 2009: | ||||||||
| First Quarter |
$ | 0.60 | $ | 0.08 | ||||
|
Second Quarter |
$ | 0.44 | $ | 0.12 | ||||
| Third Quarter |
$ | 0.90 | $ | 0.20 | ||||
|
Fourth Quarter |
$ | 0.34 | $ | 0.18 | ||||
|
Year Ended September 30, 2010: |
||||||||
| First Quarter |
$ | 0.35 | $ | 0.15 | ||||
|
Second Quarter |
$ | 1.10 | $ | 0.26 | ||||
| Third Quarter |
$ | 1.84 | $ | 0.77 | ||||
|
Fourth Quarter |
$ | 1.30 | $ | 0.71 | ||||
|
Year Ending September 30, 2011: |
||||||||
| First Quarter |
$ | 1.55 | $ | 0.46 | ||||
| From January 1, 2011 through January 31, 2011 |
$ | 0.79 | $ | 0.54 | ||||
As of January 31, 2011, our closing sale price for our common stock was $0.65.
36
Table of Contents
Equity Compensation Plan Information
Securities authorized for issuance under equity compensation plans as of September 30, 2010:
| Plan Category |
Number
of securities to be issued upon exercise of outstanding options, warrants, and rights (a)
|
Weighted- average exercise price of outstanding options, warrants, and rights (b)
|
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) (c)
|
|||||||||
| Equity compensation plans approved by stockholders |
24,255,580 | $ | 0.81 | 3,987,950 | ||||||||
| Total |
24,255,580 | $ | 0.81 | 3,987,950 | ||||||||
37
Table of Contents
The following table sets forth our capitalization as of January 31, 2011:
| • | on an actual basis, and |
| • | on an as adjusted basis to reflect our sale of units offered by us at a price of $ per unit (based on the last reported sale price of our common stock on , 2011), less the placement agent fees and estimated offering expenses payable by us. |
You should read the information in this table together with “MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS” and our financial statements and the notes thereto.
| As of January 31, 2011 | ||||||||||||
| Actual | As Adjusted | |||||||||||
| (Unaudited) | (Unaudited) | |||||||||||
| Total common shares outstanding |
68,375,259 | |||||||||||
| Warrants |
15,067,364 | |||||||||||
| Options |
25,013,433 | 25,013,433 | ||||||||||
| Class 3 Plan Note – Ryll note(1) |
3,922,877 | 3,922,877 | ||||||||||
| Class 5 Plan Debentures – 2006 Debentures(2) |
2,278,575 | 2,278,575 | ||||||||||
| Class 6 Plan Debentures – 2008 Debentures(3) |
6,235,989 | 6,235,989 | ||||||||||
| Class 9 Plan Debentures – 2007 Debentures(4) |
16,720,844 | 16,720,844 | ||||||||||
| Class 13 Plan Notes – Preferred transaction(5) |
4,290,686 | 4,290,686 | ||||||||||
| Options, warrants, and convertible debt |
73,529,768 | |||||||||||
| Fully Diluted |
142,162,380 | |||||||||||
| (1) | Effective as of November 17, 2010, the effective date of our Bankruptcy Plan (the “Effective Date”), we issued a new promissory note in favor of Dennis Ryll, the holder by assignment of our secured note to Southwest Bank, as payment of our obligation to Southwest Bank prior to the Effective Date of the Plan (the “Class 3 Plan Note”), in an original principal amount of $4,483,284. We are not obligated to pay the Class 3 Plan Note in cash, but rather through seven (7) quarterly conversions into shares of our common stock or, subject to certain conditions, by exchanging the Class 3 Plan Note into shares of Biovest common stock owned by us. The Class 3 Plan Note matures on November 17, 2012. On the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 3 Automatic Conversion Date”), provided that the average of the trading price of our common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 3 Plan Note plus the Class 3 Interest as of the Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the Class 3 Automatic Conversion Price per share of our common stock. The Class 3 Plan Note is secured by a lien on 15 million shares of Biovest’s common stock owned by the Company (the “Class 3 Pledged Shares”), subject to the incremental release of a designated portion of such security upon each quarterly payment under the Class 3 Plan Note. If, on any Class 3 Automatic Conversion Date, the Class 3 Automatic Conversion Price is less than $1.00 per share, Dr. Ryll may, at his election, either: (a) convert the Class 3 Automatic Conversion Amount into shares of our common stock at a conversion rate equal to $1.00 per share of our common stock; or (b) liquidate that number of the Class 3 Pledged Shares which equals the Class 3 Automatic Conversion Amount using a conversion rate for the Class 3 Pledged Shares equal to the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date. |
38
Table of Contents
| (2) | Effective as of November 17, 2010, we issued, in satisfaction of the September 2006 Debentures outstanding prior to the Effective Date, new debentures (the “Class 5 Plan Debentures”) in the original principal amount of $3,109,880. We are not obligated to pay the Class 5 Plan Debentures in cash, but rather through the conversion by the holders into shares of our common stock or, subject to certain conditions, by exchanging the Class 5 Plan Debentures into shares of our Biovest common stock during the term of the Class 5 Plan Debentures which mature on May 17, 2012 (provided, however, in the event that the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 5 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding such maturity date is below $0.75, then the maturity date will automatically be extended for an additional twelve (12) months), and the outstanding principal together with all accrued but unpaid interest is due on such maturity date. At the option of a holder of Class 5 Plan Debentures, all or any portion of the then outstanding balance of such holder’s Class 5 Plan Debentures may be converted into shares of our common stock or exchanged for shares of Biovest’s common stock at the applicable conversion or exchange rate set forth in such holder’s Class 5 Plan Debenture. |
| (3) | Effective as of November 17, 2010, we issued, in satisfaction of the 2008 Secured Debentures outstanding prior to the Effective Date of the Plan, new debentures (the “Class 6 Plan Debentures”) in the original principal amount of $9,730,459. The Class 6 Plan Debentures mature on November 17, 2013 , and the outstanding principal together with all accrued but unpaid interest is due in cash on such date. At the option of a holder of Class 6 Plan Debentures, such holder may elect to convert all of the then outstanding balance of its Class 6 Plan Debentures into shares of our common stock at a conversion rate equal to $1.10 per share of our common stock. Also, commencing on May 15, 2011, if the trading price of our common stock (as determined in accordance wit the Class 6 Plan Debentures and the Plan) is at least 150% of $1.10 per share for any ten (10) consecutive trading days, we, at our option, may convert the then outstanding balance of all of the Class 6 Plan Debentures into shares of our common stock at a conversion rate equal to $1.10 per share of our common stock. |
| (4) | Effective as of November 17, 2010, we issued, in satisfaction of the 2007 Debentures outstanding prior to the Effective Date of the Plan, new debentures (the “Class 9 Plan Debentures”) in the original principal amount of $19,109,554. The Class 9 Plan Debentures mature on November 17, 2012 and no interest will accrue on the outstanding principal balance of the Class 9 Plan Debentures. On the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 9 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 9 Automatic Conversion Date (the “Class 9 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 9 Plan Debentures (the “Class 9 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the lesser of $1.25 per share or the Class 9 Automatic Conversion Price per share. |
39
Table of Contents
| (5) | Effective as of November 17, 2010, we issued, in satisfaction of the Convertible Preferred Stock outstanding prior to the Effective Date of the Plan, new promissory notes (the “Class 13 Plan Notes”) in the original principal amount of $4,903,644. The Class 13 Plan Notes mature on November 17, 2012 and no interest will accrue on the outstanding principal balance of the Class 13 Plan Notes. On the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 13 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 13 Automatic Conversion Date (the “Class 13 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original balance of the Class 13 Plan Notes (the “Class 13 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the Class 13 Automatic Conversion Price per share. On the Effective Date, a total of approximately $0.6 million of the outstanding principal amount of the Class 13 Plan Notes were converted into common stock at a conversion price equal to $1.36 per share, resulting in the issuance of 450,708 shares of our common stock. |
40
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
When you read this section of this prospectus, it is important that you also read our selected consolidated financial data, the consolidated financial statements and related notes included elsewhere in this prospectus. This section of this prospectus contains forward-looking statements that involve risks and uncertainties, such as statements of our plans, objectives, expectations, and intentions. Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons set forth herein, including the factors described below and in “RISK FACTORS.”
Overview
Headquartered in Tampa, Florida, Accentia Biopharmaceuticals, Inc. (Other OTC: “ABPI”) is a biotechnology company that is developing Revimmune™ as a comprehensive system of care for the treatment of autoimmune diseases. Additionally, through our majority-owned subsidiary, Biovest International, Inc., we are developing BiovaxID® as a therapeutic cancer vaccine for treatment of follicular non-Hodgkin’s lymphoma (“FL”) and mantle cell lymphoma (“MCL”). Through our wholly-owned subsidiary, Analytica International, Inc., we conduct a health economics research and consulting business which we market to the pharmaceutical and biotechnology industries, using our operating cash flow to support our corporate administration and product development activities.
Revimmune™ is being developed as a treatment for various autoimmune diseases. The approximately 80 known autoimmune diseases generally arise from an overactive immune response against substances and/or tissue normally present in the body. As a system of care, Revimmune consists of administering high, pulsed doses of an FDA-approved drug, cyclophosphamide, over a four-day interval as part of an integrated risk management system including a panel of preventive tests, monitoring and medications which are intended to minimize risks while seeking to maximize the clinical benefit.
Through a collaboration with the National Cancer Institute (“NCI”), our majority-owned subsidiary, Biovest International, Inc. (OTCQB: “BVTI”) (“Biovest”), has developed a patient-specific cancer vaccine, BiovaxID®, which has demonstrated statistically significant Phase 3 clinical benefit by prolonging disease-free survival in follicular non-Hodgkin’s lymphoma patients treated with BiovaxID.
Additionally, through our wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), based in New York City, we conduct a global research and strategy consulting business that provides professional services to the pharmaceutical and biotechnology industries. Since 1997, Analytica has expertly directed research studies and projects, including traditional health economic modeling projects, database studies, structured reviews, health technology assessments, reimbursement analyses, and value dossiers.
Corporate History and Structure
We were organized in 2002 to develop and commercialize biopharmaceutical products.
We commenced business in April 2002 with the acquisition of Analytica, a provider of analytical and consulting services to the biopharmaceuticals industry, including clinical trial services, pricing and market assessment and outcomes research. We acquired Analytica in a merger transaction for $3.7 million cash, $1.2 million of convertible promissory notes, and the issuance of 8.1 million shares of Series B preferred stock. Analytica, which was founded in 1997, has offices in New York and Germany.
41
Table of Contents
In June 2003, we acquired an 81% interest of Biovest, for an investment of $20.0 million in Biovest pursuant to an investment agreement. Biovest is a biologics company that is developing our BiovaxID patient-specific vaccine for the treatment of FL. As of January 31, 2011, we owned of record approximately 65% of the common stock of Biovest, with the minority interest being held by approximately 450 shareholders of record. Biovest’s common stock is registered under Section 12(g) of the Securities Exchange Act of 1934, and Biovest therefore files periodic and other reports with the SEC.
On November 10, 2008, we, along with all of our subsidiaries, filed a voluntary petition for reorganization under Case No. 8:08-bk-17795-KRM (“Chapter 11”). On August 16, 2010, we filed our First Amended Joint Plan of Reorganization, and, on October 25, 2010, we filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. We emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”). The accompanying audited consolidated financial statements have been prepared assuming we will continue as a going concern as we emerge from Chapter 11.
Financial Measures
REVENUE RECOGNITION
We recognize revenue as follows:
Products
Net sales of cell culture instruments and disposables are recognized in the period in which the risk and rewards of ownership have passed (at point of shipment) to the buyer. We do not provide our customers with a right of return; however, deposits made by customers must be returned to customers in the event of non-performance by us.
Actual product returns, chargebacks, and other sales allowances incurred are dependent upon future events and may be different than our estimates. We continually monitor the factors that influence sales allowance estimates and make adjustments to these provisions when management believes that actual product returns, chargebacks, and other sales allowances may differ from established allowances.
Services
Service revenue is generated primarily by fixed-price contracts for cell culture production and consulting services. Such revenue is recognized over the contract term in accordance with the percentage-of-completion method based on the percentage of service cost incurred during the period compared to the total estimated service cost to be incurred over the entire contract. The nature and scope of our contracts often require us to make judgments and estimates in recognizing revenues.
42
Table of Contents
Estimates of total contract revenues and costs are continuously monitored during the term of the contract, and recorded revenues and costs are subject to revision as each contract progresses. Such revisions may result in increases or decreases to revenues and income and are reflected in the consolidated financial statements in the periods in which they are first identified. Each month we accumulate costs on each contract and compare them to the total current estimated costs to determine the percentage of completion. We then apply this percentage to the total contract value to determine the amount of revenue that can be recognized. Each month we review the total current estimated costs on each contract to determine if these estimates are still accurate and, if necessary, we adjust the total estimated costs for each contract. As the work progresses, we might decide that original estimates were incorrect due to, among other things, revisions in the scope of work, and a contract modification might be negotiated with the customer to cover additional costs. If a contract modification is not agreed to, we could bear the risk of cost overruns. Losses on contracts are recognized during the period in which the loss first becomes probable and reasonably estimable. Reimbursements of contract-related costs are included in revenues. An equivalent amount of these reimbursable costs is included in cost of sales. Because of the inherent uncertainties in estimating costs, it is at least reasonably possible that the estimates used will change within the near term.
Service costs related to cell culture production include all direct materials and subcontract and labor costs and those indirect costs related to contract performance, such as indirect labor, insurance, supplies, and tools. We believe that actual cost incurred in contract cell production services is the best indicator of the performance of the contractual obligations, because the costs relate primarily to the amount of labor incurred to perform such services. The deliverables inherent in each of our cell culture production contracts are not output driven, but rather driven by a pre-determined production run. The duration of our cell culture production contracts range typically from 2 to 14 months.
Service costs relating to our consulting services, consists primarily of internal labor expended in the fulfillment of our consulting projects and, to a lesser extent, outsourced research services. Service costs on a specific project may also consist of a combination of both internal labor and outsourced research service. Our consulting projects are priced and performed in phases, and the projects are managed by phase. As part of the contract bidding process, we develop an estimate of the total number of hours of internal labor required to generate each phase of the customer deliverable (for example, a manuscript or database), and the labor cost is then computed by multiplying the hours dedicated to each phase by a standard hourly labor rate. We also determine whether we need services from an outside research or data collection firm and include those estimated outsourced costs in our total contract cost for the phase. At the end of each month, we collect the cumulative total hours worked on each contract and apply a standard labor cost rate to arrive at the total labor cost incurred to date. This amount is divided by the total estimated contract cost to arrive at the percentage of completion, which is then applied to the total estimated contract revenues to determine the revenue to be recognized through the end of the month. Accordingly, as hours are accumulated against a project and the related service costs are incurred, we concurrently fulfill our contract obligations. The duration of our consulting service contracts range typically from 1 to 12 months. Certain other professional service revenues, such as revenues from maintenance services on cell culture equipment, are recognized as the services are performed.
In our financial statements, unbilled receivables represents revenue that is recognizable under the percentage-of-completion method due to the performance of services for which billings have not been generated as of the balance sheet date. In general, amounts become billable pursuant to contractual milestones or in accordance with predetermined payment schedules. Under our consulting services contracts, the customer is required to pay for contract hours worked by us (based on the standard hourly rate used to calculate the contract price) even if the customer cancels the contract and elects not to proceed to completion of the project. Unearned revenues represent customer payments in excess of revenue earned under the percentage-of-completion method. Such payments are made in accordance with predetermined payment schedules set forth in the contract.
43
Table of Contents
Inventories
Inventories are recorded at the lower of cost or market. We periodically review inventory quantities of raw materials, instrumentation components and disposables on hand, and we record write-downs of inventories to market value based upon contractual provisions and obsolescence, as well as assumptions about future demand and market conditions. If assumptions about future demand change and/or actual market conditions are less favorable than those projected by management, additional write-downs of inventories may be required.
Valuation of Goodwill and Intangible Assets
Our intangible assets include goodwill, trademarks, product rights, non-compete agreements, technology rights, purchased customer relationships, and patents, all of which are accounted for based on Accounting Standard Codification (“ASC”) Topic 350 Intangibles-Goodwill and Other. As described below, goodwill and intangible assets that have indefinite useful lives are not amortized but are tested at least annually for impairment or more frequently if events or changes in circumstances indicate that the asset might be impaired. Intangible assets with limited useful lives are amortized using the straight-line method over their estimated period of benefit, ranging from two to eighteen and one-half years. Goodwill is tested for impairment by comparing the carrying amount to the estimated fair value, in accordance with generally accepted accounting principles. Impairment exists if the carrying amount is less than its estimated fair value, resulting in a write-down equal to the difference between the carrying amount and the estimated fair value. Our carrying value of goodwill at September 30, 2010 and 2009 was $0.9 million and $1.2 million, respectively. The values recorded for goodwill and other intangible assets represent fair values calculated by accepted valuation methods. Such valuations require critical estimates and assumptions derived from and which include, but are not limited to: (i) information included in our business plan, (ii) estimated cash flows, (iii) discount rates, (iv) patent expiration information, (v) terms of license agreements, and (vi) expected timelines and costs to complete any in-process research and development projects to commercialize our products under development.
We capitalized goodwill in connection with our acquisition of Analytica in April 2002 and in connection with the IMOR acquisition in December 2003, based on the fair value of the acquired assets net of assumed liabilities.
We recorded amortization of intangible assets of $0.3 million and $0.2 million in the years ended September 30, 2010 and 2009, respectively. We amortize intangibles based on their expected useful lives and look to a number of factors for such estimations, including the longevity of our license agreements and the remaining life of patents on products currently being marketed. We recognized impairment losses of $0.4 million and $1.5 million during the years ended September 30, 2010 and September 30, 2009, respectively, in connection with certain intangible assets. Our carrying value of intangible assets at September 30, 2010 and 2009 was $1.1 million and $2.6 million, respectively, net of accumulated amortization. We begin amortizing capitalized intangibles on their date of acquisition.
Impairment Testing
Our goodwill impairment testing is calculated at the reporting unit level. Our annual impairment test has two steps. The first identifies potential impairments by comparing the fair value of the reporting unit with its carrying value. If the fair value exceeds the carrying amount, goodwill is not impaired and the second step is not necessary. If the carrying value exceeds the fair value, the second step calculates the possible impairment loss by comparing the implied fair value of goodwill with the carrying amount. If the implied fair value of goodwill is less than the carrying amount, a write-down is recorded.
44
Table of Contents
The impairment test for the other intangible assets is performed by comparing the carrying amount of the intangible assets to the sum of the undiscounted expected future cash flows. In accordance with generally accepted accounting principles, which relates to impairment of long-lived assets other than goodwill, impairment exists if the sum of the future undiscounted cash flows is less than the carrying amount of the intangible asset or to its related group of assets.
We predominately use a discounted cash flow model derived from internal budgets in assessing fair values for our impairment testing. Factors that could change the result of our impairment test include, but are not limited to, different assumptions used to forecast future net sales, expenses, capital expenditures, and working capital requirements used in our cash flow models. In addition, selection of a risk-adjusted discount rate on the estimated undiscounted cash flows is susceptible to future changes in market conditions, and when unfavorable, can adversely affect our original estimates of fair values. In the event that our management determines that the value of intangible assets have become impaired using this approach, we will record an accounting charge for the amount of the impairment.
Share-Based Compensation
We account for stock-based compensation based on ASC Topic 718-Stock Compensation which requires expensing of stock options and other share-based payments based on the fair value of each option awarded. The fair value of each option is estimated on the date of grant using the Black-Scholes valuation model. This model requires management to estimate the expected volatility, expected dividends, and expected term as inputs to the valuation model.
In applying the Black-Scholes-Merton options-pricing model during fiscal 2010, we assumed no dividend yield, risk-free interest rates ranging from 2.35% to 3.11%, expected option terms ranging from 5.0 to 6.0 years, a volatility factor of 140.08%, a share price of $0.44, and option exercise price of $0.44.
We recorded stock-based compensation of approximately $0.5 million and $1.71 million in the twelve months ended September 30, 2010 and 2009, respectively. Stock based compensation for the quarter ended December 31, 2010 was $10.8 million. For both periods, stock-based compensation is classified in general and administrative expense.
Derivative instruments
We generally do not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, we and our consolidated subsidiaries have entered into certain other financial instruments and contracts, such as debt financing arrangements and freestanding warrants with features that are either not afforded equity classification, embody risks not clearly and closely related to host contracts, or may be net-cash settled by the counterparty. Except as provided in ASC Topic 815 Embedded Derivatives, these instruments are required to be carried as derivative liabilities, at fair value, in our consolidated financial statements. In instances where we elect not to bifurcate embedded derivative features, the entire hybrid instrument is carried at fair value in the consolidated financial statements.
45
Table of Contents
We estimate fair values of derivative financial instruments using various techniques (and combinations thereof) that are considered to be consistent with the objective of measuring fair values. In selecting the appropriate technique(s), management considers, among other factors, the nature of the instrument, the market risks that it embodies and the expected means of settlement. For less complex derivative instruments, such as free-standing warrants, we generally use the Black-Scholes-Merton option valuation technique because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. For forward contracts that contingently require net-cash settlement as the principal means of settlement, management projects and discounts future cash flows applying probability-weighted averages to multiple possible outcomes. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in our trading market price which has high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair values, our income will reflect the volatility in these estimate and assumption changes.
Variable interest entities
We evaluate all significant arrangements and relationships for indications of variable interest entities pursuant to generally accepted accounting principles. During April 2006 and December 2006, we entered into financing arrangements with Biovest that involved entities that met the definition of variable interest entities. As a result, our Company and Biovest were required to consolidate these entities and reflect the non-controlling interest in the consolidated financial statements as of and for the years ended September 30, 2010 and 2009. The 2010 and 2009 consolidated financial statements include the variable interest entities as follows: Biovest Investment, LLC, Telesis CDE Two LLC, AutovaxID Investment LLC, St. Louis New Market Tax Credit Fund II LLC and Revimmune, LLC. Biovest has ceased all activities under the New Market Tax Credit transactions and has liquidated all associated subsidiaries. Only Revimmune, LLC remains as a variable interest entity in the December 31, 2010 consolidated financial statements.
Income Taxes
We incurred net operating losses for the years ended September 30, 2010 and 2009 and the quarter ended December 31, 2010, and consequently will not be required to pay federal or foreign income taxes, but we did pay nominal state taxes in several states where we have operations. We have a federal net operating loss (“NOL”) carryover of approximately $212.3 million as of September 30, 2010, which expires incrementally through 2027. Of this amount, approximately $70.7 million is attributable to Biovest and will no longer be available to offset income generated by the other members of the group.
Under Sections 382 and 383 of the Internal Revenue Code, if an ownership change occurs with respect to a “loss corporation” as defined, there are annual limitations on the amount of the net operating loss and other deductions which are available to us. Due to the acquisition transactions in which we have engaged in recent years, we believe that the use of these net operating losses will be significantly limited. In addition, the utilization of our net operating loss carryforwards may be further limited if we experience a change in ownership of more than 50% subsequent to the last change in ownership of September 30, 2003. Accordingly, our net operating loss carryforward available to offset future federal taxable income arising before such ownership changes may be further limited.
We currently have limitations on at least $30.0 million of the NOLs based upon ownership changes through September 30, 2003. Of those losses subject to the limitations, $11.3 million is expected to expire before the losses can be utilized. Of the remaining amounts, the limitation is approximately $1.8 million per year through approximately the year ended September 30, 2012. After that, the annual limitation will decrease to approximately $0.2 million through September 30, 2024.
46
Table of Contents
Our ability to realize our deferred tax assets depends on our future taxable income as well as the limitations on usage discussed above. For financial reporting purposes, a deferred tax asset must be reduced by a valuation allowance if it is more likely than not that some portion or all of the deferred tax asset will not be realized prior to its expiration. Because we believe the realization of our deferred tax assets is uncertain, we have recorded a valuation allowance to fully offset them.
Additionally, since Biovest is no longer part of the consolidated group for income tax purposes, we could in the future have a net loss but we or Biovest could be subject to tax on our income since the losses may not be available to offset the income of the other entity.
Results of Operations
THREE MONTHS ENDED DECEMBER 31, 2010 COMPARED TO THE THREE MONTHS ENDED DECEMBER 31, 2009
Consolidated Results of Operations
Net Sales. Our net sales for the three months ended December 31, 2010 were approximately $2.1 million compared to net sales of $3.7 million for the three months ended December 31, 2009, a decrease of approximately 44%. Sales from both Biovest’s instrumentation segment and Biovest’s cell culture contract segment have decreased year over year. In September 2009, Biovest entered into a $1.5 million contract with the U.S. Department of Defense Naval Health Research Center (“NHRC”) to supply AutovaxID™ bioreactors to evaluate the instrument’s suitability to produce cell-culture based anti-viral vaccines. As a result of this contract, $0.3 million in revenue was recorded in the quarter ended December 31, 2009. Analytica’s sales decreased 49% to $1.1 million compared to the same quarter in the previous fiscal year primarily due to the cessation of the clinical trial services business of our German subsidiary which accounted for $0.9 million of revenue for the three months ended December 31, 2009. Our grant revenue for the quarter ended December 31, 2010 relating to the Qualifying Therapeutic Discovery Program was $0.3 million. There was no grant revenue for the quarter ended December 31, 2009.
Research and Development Expenses. Our research and development costs were approximately $0.3 million for the three months ended December 31, 2010 and December 31, 2009. Research and development costs consist primarily of salaries and research facility costs.
General and Administrative Expenses. Our general and administrative expenses were approximately $12.4 million for the three months ended December 31, 2010; an increase of $10.9 million over the three months ended December 31, 2009. This increase is primarily due to an increase in share-based compensation of $10.4 million. This increase is attributable to the expense related to options granted during our bankruptcy with vesting contingent upon our emergence from bankruptcy in addition to the market value of 1.5 million shares issued to executives on the Effective Date. The remainder of the increase is primarily due to an increase in professional fees in conjunction with our resumption of financial statement audits associated with our filings with the Securities and Exchange Commission (“SEC”).
Derivative loss. Derivative loss was approximately $4.1 million for the three months ended December 31, 2010 as compared to a loss of $0.03 million for the three months ended December 31, 2009. This increase was primarily related to derivative instruments issued in conjunction with our various financings, and was offset by the decrease in our common stock price and Biovest’s common stock price, on which the derivative liabilities are based, during the quarter ended December 31, 2010.
47
Table of Contents
Gain on reorganization. The gain on reorganization was recognized on November 17, 2010, the Effective Date of our Joint Plan of Reorganization. The gain is primarily a result of settling claims with cash or stock at amounts less than the recorded value. Additional gain or loss may be recognized depending on the outcome of the final disposition of certain unresolved claims.
YEAR ENDED SEPTEMBER 30, 2010 COMPARED TO THE YEAR ENDED SEPTEMBER 30, 2009
Consolidated Results of Operations
Net Sales. Our net sales for the year ended September 30, 2010 were $10.5 million compared to $10.6 million for the year ended September 30, 2009. Sales from Biovest’s instrumentation segment experienced an increase of $2.0 million year over year. In September 2009, Biovest entered into a $1.5 million contract with the U.S. Department of Defense Naval Health Research Center (“NHRC”) to supply AutovaxID™ bioreactors and to evaluate the instrument’s suitability to produce cell-culture based anti-viral vaccines. As a result of this contract, Biovest recorded $1.2 million in instrumentation revenue in the year ended September 30, 2010, with no comparable revenue in the prior year. Service revenue decreased $0.3 million compared to the year ended September 30, 2009 due to three significant cell culture contracts that were ongoing in fiscal 2009. These projects were run to completion during the fiscal year ended September 30, 2010. Analytica’s consulting revenue decreased approximately $1.8 million primarily due to the insolvency filing of the German operations.
Research and Development Expenses. Our research and development costs were $1.3 million for the year ended September 30, 2010; a decrease of approximately $20,000 over the year ended September 30, 2009. Prior to the commencement of fiscal 2010, Biovest completed the initial Phase 3 clinical trial in the cancer vaccine, BiovaxID. We have not yet commenced our planned clinical trials for Revimmune and Biovest is not currently conducting a clinical trial and the research and development effort is largely focused on analyzing data available from our completed trial and preparing for anticipated discussions with regulatory agencies regarding next steps in the regulatory process. Accordingly, research and development spending levels have declined over the prior year even though Biovest increased the number of technical and scientific employees in fiscal 2010.
General and Administrative Expenses. Our general and administrative expenses were $5.3 million for the year ended September 30, 2010; a decrease of $2.8 million, or 35%, over the year ended September 30, 2009, due primarily to the reduction in stock compensation to $0.5 million compared to $1.7 million during the twelve months ended September 30, 2009. Business insurance expense decreased $0.2 million due to a drop in premiums from 2009 to 2010.
Impairment of Intangible Assets. Impairment of intangible assets was $0.4 million for the year ended September 30, 2010 as compared to $1.5 million for the year ended September 30, 2009. The impairment in 2010 relates to the insolvency filing for IMOR, our German subsidiary of Analytica. The 2009 expense related primarily to certain acquired product rights upon the closure of our TEAMM subsidiary in November 2008.
Interest Expense. In the year ended September 30, 2010, our interest expense was $16.7 million, an increase of $8.3 million over the year ended September 30, 2009. The increase was due primarily to the adjustment to fair market value of the September 2006 convertible debentures which fluctuates with the market price of our stock.
48
Table of Contents
Derivative gain (loss). Derivative loss was $28.8 million for the year ended September 30, 2010 as compared to a gain of $14.4 million for the year ended September 30, 2009. This difference of approximately $43.2 million is primarily related to the derivative instruments associated with the convertible debt and warrants. The loss was primarily due to an increase in our stock price during the year ended September 30, 2010.
Income (loss) from discontinued operations. Loss from discontinued operations was approximately $12,000 for the year ended September 30, 2010 compared to a gain of $1.7 million for the year ended September 30, 2009. Accentia Pharmaceuticals was in operation from October 1, 2008 through November 10, 2008 during the twelve months ended September 30, 2009. The income in 2009 relates primarily to the adjustment of reserves related to future returns and rebates.
Other. In a letter filed by us with the Securities and Exchange Commission on February 15, 2011, we indicated that in our consolidated statements of operations filed for periods after December 31, 2010, we would include in our reported net loss any loss or income from non-controlled interests in variable interest entities and would separately itemize preferred stock dividends in our net income (loss) per share calculations.
Liquidity and Capital Resources
Sources of Liquidity
Since our inception, we have funded our operations primarily through public and private placements of our capital stock, debt financing, conversions of debt to equity, and financing transactions with our strategic partners. These transactions are described throughout the following pages.
We have historically had significant losses from operations. On December 31, 2010, we had an accumulated deficit of approximately $327 million and a working capital deficit of approximately $38 million. Cash and cash equivalents at December 31, 2010, was approximately $3.8 million, of which $3.5 million was attributable to Biovest. We intend to attempt to meet our cash requirements through proceeds from our cell culture and instrument manufacturing activities from our Biovest subsidiary, income from our Analytica subsidiary, and the use of cash on hand, trade-vendor credit, and short-term borrowings. Additionally, we may seek public or private equity investment, short or long term debt financing or strategic relationships such as investments or licenses. Our ability to continue present operations and to continue our product development efforts are dependent upon our ability to successfully execute the obligations under our Plan and to obtain significant external funding, which raises substantial doubt about our ability to continue as a going concern. The need for funds is expected to grow as we continue our efforts to commercialize Revimmune™, BiovaxID®, and AutovaxID™.
Capital Raised through Equity Issuances
We have received funding from our initial public offering, private placements of our common and preferred stock, the issuance of convertible debentures and the exercise of warrants and options to purchase capital stock.
Corps Real, LLC-Biovest
On November 17, 2010, the effective date of the Biovest Plan (the “Effective Date”), in accordance with the terms of Biovest’s First Amended Plan of Reorganization, Biovest executed and delivered in favor of Corps Real, LLC, an Illinois limited liability company (“Corps Real”), the members of which are directors of, or are affiliated with directors of, Biovest and our company, a secured convertible promissory note (the “DIP Lender Plan Note”) in an original principal amount of $2,291,560. The DIP Lender Plan Note amends and restates the $3.0 million secured line of credit promissory note dated December 22, 2008, previously executed by Biovest in favor of Corps Real. The DIP Lender Plan Note matures on November 17, 2012, and all principal and accrued but unpaid interest is due on such date. Interest accrues and is payable on the outstanding principal amount of the DIP Lender Plan Note at a fixed rate of sixteen percent (16%) per annum, with interest in the amount of ten percent (10%) to be paid monthly and interest in the amount of six percent (6%) to accrue and be paid on the maturity date. Biovest may prepay the DIP Lender Plan Note in full, without penalty, at any time, and Corp Real may convert all or a portion of the outstanding balance of the DIP Lender Plan Note into shares of Biovest’s common stock at a conversion rate of $0.75 per share of Biovest’s common stock. The DIP Lender Plan Note is secured by a first priority lien on all of Biovest’s assets. As of December 31, 2010, the outstanding principal amount of the DIP Lender Plan Note, as issued, on the Effective Date has remained unchanged.
49
Table of Contents
Exit Financing-Biovest
On October 19, 2010, Biovest completed a financing as part of its Plan (the “Exit Financing”). Pursuant to the Exit Financing, Biovest issued an aggregate of $7.0 million in principal amount of Debtor-In-Possession Secured Convertible Notes (the “Initial Notes”) and warrants to purchase shares of Biovest’s common stock (the “Initial Warrants”) to a total of twelve accredited investors (the “Buyers”). In connection with the Exit Financing, Biovest and the Buyers entered into a Securities Purchase Agreement, dated October 19, 2010 (the “Purchase Agreement”), pursuant to which Biovest sold and issued the Initial Notes and the Initial Warrants to the Buyers. Pursuant to the Purchase Agreement, Biovest issued two separate Initial Warrants to the Buyers, Series A Warrants (the “Initial Series A Warrants”) and Series B Warrants (the “Initial Series B Warrants”).
On the Effective Date: (a) the Initial Notes were exchanged pursuant to the terms of the Plan for new unsecured notes (the “Exchange Notes”) in the aggregate principal amount of $7.04 million, (b) the Initial Series A Warrants were exchanged pursuant to the terms of the Plan for new warrants to purchase a like number of shares of Biovest common stock (the “Series A Exchange Warrants”), and (c) the Initial Series B Warrants were exchanged pursuant to the terms of the Biovest Plan for new warrants to purchase a like number of shares of Biovest common stock (the “Series B Exchange Warrants”).
The following are the material terms and conditions of the Exchange Notes:
| • | the Exchange Notes mature on November 17, 2012, and all principal and accrued but unpaid interest is due on such date; |
| • | interest accrues and is payable on the outstanding principal amount of the Exchange Notes at a fixed rate of seven percent (7%) per annum (with a fifteen percent (15%) per annum default rate), and is payable monthly in arrears; the first date for an interest payment was December 1, 2010; |
| • | interest payments are payable at Biovest’s election in either cash or subject to certain specified conditions, in shares of Biovest’s common stock; |
| • | Biovest may from time to time, subject to certain conditions, redeem all or any portion of the outstanding principal amount of the Exchange Notes for an amount, in cash, equal to 110% of the sum of the principal amount being redeemed and certain make-whole interest payments; |
| • | the holders of the Exchange Notes may convert all or a portion of the outstanding balance of the Exchange Notes into shares of Biovest’s common stock at a conversion rate of $0.91 per share of Biovest’s common stock, subject to anti-dilution adjustments in certain circumstances; and |
50
Table of Contents
| • | in the event that the average of the daily volume weighted average price of Biovest’s common stock is at least 150% of the then-effective conversion price for any ten (10) consecutive trading days, Biovest, at its option, may upon written notice to the holders of the Exchange Notes, convert the then outstanding balance of the Exchange Notes into shares of Biovest’s common stock at the conversion price then in effect under the Exchange Notes. |
The following are the material terms and conditions of the Series A Exchange Warrants:
| • | the Series A Exchange Warrants give the Buyers the right to purchase 8,733,096 shares of Biovest common stock (the “Series A Warrant Shares”); |
| • | the Series A Exchange Warrants have an exercise price of $1.20 per share and expire on November 17, 2017; and |
| • | if Biovest issues or sells any options or convertible securities after the issuance of the Series A Exchange Warrants that are convertible into or exchangeable or exercisable for shares of common stock at a price which varies or may vary with the market price of the shares of common stock, including by way of one or more reset(s) to a fixed price, the investors have the right to substitute any of the applicable variable price formulation for the exercise price upon exercise of the warrants held. |
On December 22, 2010, the Series B Exchange Warrants were exercised by a cashless exercise and 1,075,622 shares of Biovest’s common stock were issued to the Buyers.
As of December 31, 2010, a total of $4.2 million in principal on the Exchange Notes had been converted into shares of Biovest common stock, resulting in the issuance to the Buyers of 5.0 million shares of Biovest’s common stock. The remaining principal balance outstanding on the Exchange Notes was $2.8 million as of December 31, 2010.
Debt Financing
Laurus Master Fund, Ltd. and the Valens Funds - Accentia
Effective as of November 17, 2010, the effective date of our Plan (the “Effective Date”), we issued to Laurus Master Fund, Ltd. (in liquidation) (“Laurus”), PSource Structured Debt Limited (“PSource”), Valens Offshore SPV I, Ltd., Valens Offshore SPV II, Corp., Valens U.S. SPV I, LLC, and LV Administrative Services, Inc. (collectively, “Laurus/Valens”), security agreements and term notes in the original principal amount of $8.8 million (the “Laurus/Valens Term Notes”) in satisfaction of allowed claims prior to the Effective Date of the Plan. The following are the material terms and conditions of the Laurus/Valens Term Notes:
| • | the Laurus/Valens Term Notes mature on November 17, 2012 and may be prepaid at any time without penalty; |
| • | interest accrues on the Laurus/Valens Term Notes at the rate of eight and one-half percent (8.5%) per annum (with a twelve and one-half percent (12.5%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
51
Table of Contents
| • | we are required to make mandatory prepayments under the Laurus/Valens Term Notes as follows: |
| • | on May 17, 2012, a payment of principal, in cash, in the amount of $4.4 million, less the amount of any prior optional prepayments of principal by us and the amount of any other mandatory prepayments of principal under the Laurus/Valens Term Notes; and |
| • | a prepayment equal to thirty percent (30%) of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by us) of certain capital raising transactions (with certain exclusions); |
| • | the Laurus/Valens Term Notes are secured by: |
| • | a first lien on all of our assets, junior only to the liens granted under the Plan to holders of the Class 6 Plan Debentures, in the original aggregate principal amount of $8,906,098 and certain permitted liens; |
| • | a pledge by us to Laurus/Valens of (a) all of our equity interests in Analytica, and (b) 20,115,818 shares of common stock of Biovest owned by us; and |
| • | all of the assets of Analytica, which secure a guaranty of Analytica as to the entire indebtedness under the Laurus/Valens Term Notes; and |
| • | with the prior written consent of Laurus/Valens, we may convert all or any portion of the outstanding principal and accrued interest under the Laurus/Valens Term Notes into a number of shares of our common stock equal to (a) the aggregate portion of the principal and accrued but unpaid interest outstanding under the Laurus/Valens Term Note being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported for our common stock for the ten (10) trading days immediately preceding the date of the notice of conversion. |
On the Effective Date, 2,236,848 shares of our common stock were issued to Laurus/Valens for payment of Laurus/Valens’ Class 5 and Class 13 claims under the Plan aggregating approximately $6.0 million at a conversion rate equal to $2.67 per share. As of December 31, 2010, the outstanding principal amounts of the Laurus/Valens Term Notes, as issued, on the Effective Date, have remained unchanged.
Laurus Master Fund, Ltd. and the Valens Funds – Biovest
On the Effective Date, Biovest issued two new notes (the “Laurus/Valens Term A Notes” and “Laurus/Valens Term B Notes) in the aggregate original principal amount of $29.06 million to Laurus, PSource, Valens Offshore SPV I, Ltd. (“Valens I”), Valens Offshore SPV II, Corp. (“Valens II”), Valens U.S. SPV I, LLC (“Valens US” and together with Valens I and Valens II, “Valens”), and LV Administrative Services, Inc., as administrative and collateral agent for Laurus, PSource, and Valens (“LV” and together with Laurus, PSource, Valens, and each of their respective affiliates, “Laurus/Valens”) in compromise and satisfaction of secured claims prior to the Effective Date. The following are the material terms and conditions of the Laurus/Valens Term A Notes:
| • | the original principal amount of the Laurus/Valens Term A Notes was $24.9 million; |
| • | the Laurus/Valens Term A Notes mature on November 17, 2012; |
| • | interest accrues on the Laurus/Valens Term A Notes at the rate of eight percent (8%) per annum (with a twelve percent (12%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
52
Table of Contents
| • | Biovest may prepay the Laurus/Valens Term A Notes, without penalty, at any time; |
| • | Biovest is required to make mandatory prepayments under the Laurus/Valens Term A Notes as follows: |
| • | a prepayment equal to thirty percent (30%) of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by Biovest) of certain capital raising transactions (with certain exclusions), but only up to the then outstanding principal and accrued interest under the Laurus/Valens Term A Notes; |
| • | from any intercompany funding by us to Biovest (with certain exceptions and conditions); and |
| • | a prepayment equal to fifty percent (50%) of the positive net cash flow of Biovest for each fiscal quarter after November 17, 2010, less the amount of certain capital expenditures on certain biopharmaceutical products of Biovest made during such fiscal quarter or during any prior fiscal quarter ending after the Effective Date. |
On November 18, 2010, Biovest prepaid the Laurus/Valens Term A Notes in an amount equal to $1.4 million from the proceeds received in the Biovest Exit Financing. Accordingly, as of December 31, 2010, Biovest’s indebtedness under the Laurus/Valens Term A Notes was $23.5 million.
The following are the material terms and conditions of the Laurus/Valens Term B Notes:
| • | the original principal amount of the Laurus/Valens Term B Notes was $4.16 million; |
| • | the Laurus/Valens Term B Notes mature on November 17, 2013; |
| • | interest accrues on the Laurus/Valens Term B Notes at the rate of eight percent (8%) per annum (with a twelve percent (12%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
| • | Biovest may prepay the Laurus/Valens Term B Notes, without penalty, at any time; and |
| • | provided that the Laurus/Valens Term A Notes have been paid in full, Biovest is required to make mandatory prepayments under the Laurus/Valens Term B Notes from any intercompany funding by us to Biovest (with certain exceptions and conditions), but only up to the outstanding principal and accrued interest under the Laurus/Valens Term B Notes. |
As of December 31, 2010, the outstanding principal amounts of the Laurus/Valens Term B Notes, as issued on the Effective Date, have remained unchanged.
With the prior written consent of Laurus/Valens, Biovest may convert all or any portion of the outstanding principal and accrued interest under either the Laurus/Valens Term A Notes or the Laurus/Valens Term B Notes into shares of Biovest’s common stock. The number of shares of Biovest’s common stock issuable on such a conversion (the “Laurus/Valens Conversion Shares”) is equal to (a) an amount equal to the aggregate portion of the principal and accrued and unpaid interest thereon outstanding under the applicable Laurus/Valens Term A Notes or the Laurus/Valens Term B Notes being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported (or reported by Pink Sheets, LLC) for Biovest’s common stock for the ten (10) trading days immediately preceding the date of the notice of conversion.
53
Table of Contents
The Laurus/Valens Term A Notes and the Laurus/Valens Term B Notes are secured by a first lien on all of the assets of Biovest and its subsidiaries, junior only to the lien granted to Corp Real and to certain permitted liens. The Laurus/Valens Term A Notes and the Laurus/Valens Term B Notes will also be guaranteed by us (the “Accentia Guaranty”), up to a maximum amount of $4,991,360.00. The Accentia Guaranty will be secured by our pledge of 20,115,818 shares of Biovest’s common stock owned by us and by the assets of our subsidiary, Analytica.
McKesson Corporation
On November 17, 2010, we issued, a new promissory note in the original amount of $4,342,771 to McKesson Corporation (“McKesson”) (the “Class 4 Plan Note”) in satisfaction of McKesson’s approved pre-Effective Date secured claims. The Class 4 Plan Note is payable in cash in one installment on March 17, 2014 (unless earlier accelerated) and the outstanding principal together with all accrued but unpaid interest is due on such date. As of December 31, 2010, the outstanding principal amount of the Class 4 Plan Note, as issued on the Effective Date, has remained unchanged.
The following are the material terms and conditions of the Class 4 Plan Note:
| • | interest accrues and is payable on the outstanding principal amount under the Class 4 Plan Note at a fixed rate of five percent (5%) per annum (with a ten percent (10%) per annum default rate); |
| • | we may prepay all or any portion of the Class 4 Plan Note, without penalty, at any time; and |
| • | the Class 4 Plan Note is secured by a lien on 6,102,408 shares of Biovest’s common stock owned by us. |
Credit Facility with Southwest Bank of St. Louis f/k/a Missouri State Bank
On November 17, 2010, we issued, a new promissory note to Dennis Ryll, the holder by assignment of our previously-issued secured note to Southwest Bank, as payment of our obligation to Southwest Bank prior to the Effective Date of our Plan (the “Class 3 Plan Note”), in an original principal amount of $4,483,284. We are not obligated to pay the Class 3 Plan Note in cash, but rather through quarterly conversions into shares of our common stock or, subject to certain conditions, by exchanging the quarterly conversion amounts into shares of Biovest common stock owned by us. The following are the material terms and conditions of the Class 3 Plan Note:
| • | the Class 3 Plan Note matures on November 17, 2012; |
| • | interest accrues and is payable on the outstanding principal balance of the Class 3 Plan Note from time to time (the “Class 3 Interest”) at a fixed rate of six percent (6%) per annum; |
| • | on November 17, 2010 and on each of the following seven (7) quarterly anniversaries thereof (each, a “Class 3 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 3 Plan Note) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 3 Plan Note plus the Class 3 Interest as of the Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the Class 3 Automatic Conversion Price per share of our common stock. On November 17, 2010, the first quarterly installment in the amount of approximately $560,000 was converted into shares of our common stock at a conversion rate equal to $1.36 per share, resulting in the issuance of 412,067 shares of our common stock; |
54
Table of Contents
| • | the Class 3 Plan Note is secured by a lien on 15.0 million shares of Biovest’s common stock owned by us (the “Class 3 Pledged Shares”), subject to the incremental release of a designated portion of such security upon each quarterly payment under the Class 3 Plan Note; and |
| • | if, on any Class 3 Automatic Conversion Date, the Class 3 Automatic Conversion Price is less than $1.00 per share, Dennis Ryll may, at his election, either: |
| • | convert the Class 3 Automatic Conversion Amount into shares of our common stock at a conversion rate equal to $1.00 per share of our common stock; or |
| • | liquidate that number of the Class 3 Pledged Shares which equals the Class 3 Automatic Conversion Amount using a conversion rate for the Class 3 Pledged Shares equal to the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date. |
As of December 31, 2010, the outstanding principal amount of the Class 3 Plan Note was $3,922,873.
September 2006 Debentures and Warrants (Class 5)
Effective as of November 17, 2010, we issued, in satisfaction of the September 2006 Secured Debentures outstanding prior to the Effective Date of the Plan, new debentures (the “Class 5 Plan Debentures”) in the original principal amount of $3,109,880. We are not obligated to pay the Class 5 Plan Debentures in cash, but rather through the conversion by the holders into shares of our common stock or, subject to certain conditions, by exchanging the Class 5 Plan Debentures into shares of Biovest common stock owned by us during the term of the Class 5 Plan Debentures which mature on May 17, 2012.
The following are the material terms and conditions of the Class 5 Plan Debentures:
| • | the Class 5 Plan Debentures mature on May 17, 2012 (provided, however, in the event that the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 5 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding such maturity date is below $0.75, then the maturity date will automatically be extended for an additional twelve (12) months), and the outstanding principal together with all accrued but unpaid interest is due on such maturity date; |
| • | interest accrues and is payable on the outstanding principal amount under the Class 5 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum; |
| • | each of the Class 5 Plan Debentures is secured by a lien on certain shares of Biovest’s common stock owned by us; |
| • | at the option of a holder of Class 5 Plan Debentures, all or any portion of the then outstanding balance of such holder’s Class 5 Plan Debentures may be converted into shares of our common stock or exchanged for shares of Biovest’s common stock owned by us at the applicable conversion or exchange rate set forth in such Class 5 Plan Debentures; |
55
Table of Contents
| • | commencing on August 17, 2011, if the trading price of our common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least 150% of the fixed conversion price for a holder of Class 5 Plan Debentures for any ten (10) consecutive trading days (in the case of a conversion into our common stock), or the trading price of Biovest’s common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least $1.25 for any ten (10) consecutive trading days (in the case of an exchange into Biovest’s common stock), at our option, we may (a) convert the then outstanding balance of all of the Class 5 Plan Debentures into shares of our common stock at a conversion rate equal to the fixed conversion price for each holder of Class 5 Plan Debentures, or (b) exchange the then outstanding balance of all of the Class 5 Plan Debentures into shares of Biovest’s common stock owned by us at a rate equal to $0.75 per share of Biovest’s common stock (with certain exceptions set forth in the Class 5 Plan Debentures and the Plan); and |
| • | in the event a holder of Class 5 Plan Debentures elects to receive shares of Biovest’s common stock, then such holder will be subject to certain restrictions set forth in the Class 5 Plan Debentures regarding Biovest’s common stock from November 17, 2010 through February 17, 2011. |
As of December 31, 2010, the aggregate outstanding principal amount of the Class 5 Plan Debentures, as issued on the Effective Date, has remained unchanged.
Effective as of November 17, 2010, we executed and delivered warrants (the “Class 5 Plan Warrants”) to purchase up to 2,508,960 shares of our common stock or up to 14,400,000 shares of Biovest’s common stock owned by us. The Class 5 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from November 17, 2010, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 5 Plan Warrants.
February 2007 Notes and Warrants (Class 9)
Effective as of November 17, 2010, we issued, in satisfaction of the 2007 Debentures outstanding prior to the Effective Date, new debentures (the “Class 9 Plan Debentures”) in the original principal amount of $19,109,554. We are not obligated to pay the Class 9 Plan Debentures in cash, but rather through the conversion by the holders into shares of our common stock. The Class 9 Plan Debentures mature on November 17, 2012 (the “Class 9 Plan Debenture Maturity Date”) and no interest will accrue on the outstanding principal balance of the Class 9 Plan Debentures.
The following are the material terms and conditions of the Class 9 Plan Debentures:
| • | on November 17, 2010 and on each of the following seven (7) quarterly anniversaries thereof (each, a “Class 9 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 9 Automatic Conversion Date (the “Class 9 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 9 Plan Debentures (the “Class 9 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the lesser of $1.25 per share or the Class 9 Automatic Conversion Price per share. On November 17, 2010, a total of approximately $2.4 million of the original principal amount of the Class 9 Plan Debentures was converted into shares of our common stock at a conversion price equal to $1.25 per share, resulting in the issuance of 1,910,963 shares of our common stock; |
56
Table of Contents
| • | if, on any Class 9 Automatic Conversion Date, the Class 9 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 9 Plan Debentures may elect to convert the Class 9 Automatic Conversion Amount into shares of our common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 9 Plan Debentures at the Class 9 Plan Debenture Maturity Date will be due and payable in full, at our election, in either cash or in shares of our common stock at a conversion rate equal to the average trading price of our common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 9 Plan Debenture Maturity Date (provided that the average trading price for such period is at least $.50 per share); |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of our common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.50 per share for ten (10) consecutive trading days, a holder of the Class 9 Debentures, at its option, may convert any or all of the then outstanding principal balance of its Class 9 Plan Debenture into shares of our common stock at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on November 17, 2010 but not to exceed $1.25 per share; and |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of our common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.88 per share for thirty (30) consecutive trading days, at our option, we may require the conversion of up to $5.0 million of the then aggregate outstanding principal balance of the Class 9 Plan Debentures at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on November 17, 2010 but not to exceed $1.25 per share. |
As of December 31, 2010, the aggregate outstanding principal amount of the Class 9 Plan Debentures was $16,720,844.
On November 17, 2010, we executed and delivered warrants (the “Class 9 Plan Warrants”) to purchase up to 3,154,612 shares of our common stock. The Class 9 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from the Effective Date, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 9 Plan Warrants and the Plan.
January 2008 Notes and Warrants (Class 13)
Effective as of November 17, 2010, we issued, in satisfaction of the Convertible Preferred Stock outstanding prior to the Effective Date of the Plan, new promissory notes (the “Class 13 Plan Notes”) in the original principal amount of $4,903,644. The Class 13 Plan Notes mature on November 17, 2012 (the “Class 13 Plan Notes Maturity Date”), and no interest will accrue on the outstanding principal balance of the Class 13 Plan Notes. We have no obligation to pay the Class 13 Plan Notes in cash at maturity, but rather through the conversions by the holders into shares of our common stock.
57
Table of Contents
The following are the material terms and conditions of the Class 13 Plan Notes:
| • | on November 17, 2010 and on each of the following seven (7) quarterly anniversaries thereof (each, a “Class 13 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 13 Automatic Conversion Date (the “Class 13 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original balance of the Class 13 Plan Notes (the “Class 13 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the Class 13 Automatic Conversion Price per share. On November 17, 2010, a total of approximately $0.6 million of the outstanding principal amount of the Class 13 Plan Notes was converted into shares of our common stock at a conversion price equal to $1.36 per share, resulting in the issuance of 450,708 shares of our common stock; |
| • | if, on any Class 13 Automatic Conversion Date, the Class 13 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 13 Plan Notes may elect to convert the Class 13 Automatic Conversion Amount into shares of our common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 13 Plan Notes at the Class 13 Plan Notes Maturity Date will be due and payable in full, at our election, in either cash or in shares of our common stock at a conversion rate equal to the greater of the average of the trading price of our common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 13 Plan Notes Maturity Date or $1.00; |
| • | if, at any time during the term of the Class 13 Plan Notes, the trading price of our common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 125% of $1.25 per share for ten (10) consecutive trading days, a holder of Class 13 Plan Notes, at its option, may convert any or all of the then outstanding principal balance of its Class 13 Plan Notes into shares of our common stock at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on November 17, 2010; and |
| • | if, at any time during the term of the Class 13 Plan Notes, the trading price of our common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 150% of $1.25 per share for thirty (30) consecutive trading days, at our option, we may require the conversion of all or any portion of the then aggregate outstanding principal balance of the Class 13 Plan Notes at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on November 17, 2010. |
As of December 31, 2010, the aggregate outstanding principal amount of the Class 13 Plan Notes, as issued on the Effective Date, has remained unchanged.
Effective as of November 17, 2010, we executed and delivered warrants (the “Class 13 Plan Warrants”) to purchase up to 1,072,840 shares of our common stock. The Class 13 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from November 17, 2010, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 13 Plan Warrants and the Plan.
58
Table of Contents
June 2008 Debentures and Warrants (Class 6)
Effective as of November 17, 2010, we issued, in satisfaction of the 2008 Secured Debentures outstanding prior to the Effective Date, new debentures (the “Class 6 Plan Debentures”) in the original principal amount of $9,730,459. The Class 6 Plan Debentures mature on November 17, 2013, and the outstanding principal together with all accrued but unpaid interest is due in cash on such date.
The following are the material terms and conditions of the Class 6 Plan Debentures:
| • | interest accrues and is payable on the outstanding principal under the Class 6 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum and each of the Class 6 Plan Debentures is secured by a lien on certain of our assets; |
| • | at the option of a holder of Class 6 Plan Debentures, such holder may elect to convert all of the then outstanding balance of its Class 6 Plan Debentures into shares of our common stock at a conversion rate equal to $1.10 per share of our common stock; and |
| • | commencing on May 15, 2011, if the trading price of our common stock (as determined in accordance with the Class 6 Plan Debentures and the Plan) is at least 150% of $1.10 per share for any ten (10) consecutive trading days, at our option, we may convert the then outstanding balance of all of the Class 6 Plan Debentures into shares of our common stock at a conversion rate equal to $1.10 per share of our common stock. |
On November 17, 2010, we executed and delivered warrants (the “Class 6 Plan Warrants”) to purchase up to 2,979,496 shares of our common stock. The Class 6 Plan Warrants (a) have an exercise price of $1.50 per share and an expiration date of November 17, 2013, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 6 Plan Warrants and the Plan.
During the period ended December 31, 2010, $2,772,548 in Class 6 Plan Debentures were converted into our common stock at a conversion price equal to $1.10 per share, resulting in the issuance of 2,520,496 shares of our common stock. As of December 31, 2010, the aggregate total outstanding principal amount of the Class 6 Plan Debentures was $6,957,912.
Accentia Class 10 Plan Distributions
Effective as of November 17, 2010, we became obligated to pay approximately $2.4 million in cash, on March 17, 2014, to unsecured creditors holding Class 10 claims under the Plan (the “Class 10 Plan Distributions”).
The Class 10 Plan Distributions mature on March 17, 2014, and the outstanding principal together with all accrued but unpaid interest is due on such date. Interest accrues and is payable on the outstanding principal amount under the Class 10 Plan Distributions at a fixed rate of five percent (5%) per annum.
Unsecured creditors holding an aggregate total of $3,287,695 in Class 10 claims elected to convert those Class 10 claims into our common stock valued at the average market price for our common stock over the ten trading days preceding the Effective Date. On the Effective Date, we issued 2,417,431 shares of our common stock to these Class 10 unsecured creditors at a conversion price equal to $1.36 per share.
59
Table of Contents
As of December 31, 2010, the aggregate outstanding principal amount of the Class 10 Plan Distributions has remained unchanged since the Effective Date.
Accentia Promissory Demand Notes
Effective as of November 17, 2010, under Biovest’s Plan, our entire pre-petition claim due from Biovest was converted into shares of Biovest common stock at a conversion rate equal to $0.75 per share, resulting in the issuance of 17,925,720 shares of Biovest common stock.
Biovest Class 8 Option A Promissory Note
Effective as of November 17, 2010, under Biovest’s Plan, Biovest became obligated to pay to the Biovest unsecured creditors approximately $2.7 million in cash, together with interest at five percent (5%) per annum, in one installment on March 27, 2014 (the “Option A Promissory Notes”). These unsecured claims include, but are not limited to the Pulaski Bank notes, the Southwest Bank note, and the guarantor indemnities. As of December 31, 2010, the aggregate outstanding principal amount of the Option A Promissory Notes, as issued on the Effective Date, has remained unchanged.
Biovest Class 8 Option C Promissory Note
Effective as of November 17, 2010, under Biovest’s Plan, Biovest became obligated to its unsecured creditors in the principal amount of approximately $2.0 million (the “Option C Notes”). These creditors received an amount equal to 100% of their allowed Class 8 unsecured claims (including post-petition interest under Biovest’s Plan at a rate of three percent (3%) per annum) in combination of debt and equity resulting in the issuance of a total of $1.8 million in new notes, as well as, 0.2 million shares of Biovest common stock, using an effective conversion rate equal to $1.66 per share. The Option C Notes bear interest at seven percent (7%) and are convertible into shares of Biovest common stock in seven quarterly installments beginning on February 17, 2011 as follows:
| • | provided that the average of the volume weighted average prices for Biovest’s common stock for the ten consecutive trading days immediately preceding each quarterly conversion date (“Ten Day VWAP”) is at least $1.00 per share, one-eighth (1/8th) of the Option C Notes plus accrued interest will be automatically converted into shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP; |
| • | if the Ten Day VWAP is less than $1.00 per share, the Option C Notes will not automatically convert into shares of Biovest common stock, but will instead become payable at maturity (August 17, 2012), unless a holder of an Option C Note elects to convert one-eighth (1/8th) of such holder’s Option C Note plus accrued interest into shares of Biovest common stock at a conversion rate equal to $1.00 per share; |
| • | any portion of the Option C Notes and any Option C interest that are outstanding at maturity (August 17, 2012) will be due and payable in full, at the election of Biovest, either in cash or in shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP; |
60
Table of Contents
| • | if, at any time prior to August 17, 2012, the Ten Day VWAP is at least $1.50 per share, an Option C Note holder, at its option, may convert any or all of its Option C Notes, plus the then accrued and unpaid interest, into shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP used for the initial conversion on the Effective Date ($1.66 per share); and |
| • | if, at any time prior to August 17, 2012, the volume weighted average price for Biovest common stock is at least $1.88 per share for thirty (30) consecutive trading days, Biovest, at its option, may require the conversion of the then aggregate outstanding balance of the Option C Notes plus the then accrued and unpaid Option C interest at a conversion rate equal to the Ten Day VWAP used for the initial conversion on the Effective Date ($1.66 per share). |
April 2006 NMTC Transaction
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “Worcester Restructuring Agreement”) with Telesis CDE Corporation and Telesis CDE Two, LLC (collectively, “Telesis”), contingent upon submission to and approval by the Bankruptcy Court. The Worcester Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to the April 2006 NMTC Transaction, in consideration of retention by Telesis of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $0.3 million along with a settlement payment in the amount of $85,000 to defray certain legal and administrative expenses incurred by Telesis. The Worcester Restructuring Agreement and the compromise of the outstanding claims against Biovest and its affiliates in connection with the April 2006 NMTC Transaction was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, our guaranty, Biovest’s guaranty, and all of Biovest’s subsidiary guaranties from affiliates and third parties and all other obligations of all parties to Transaction I were terminated. Biovest has ceased all activities under the April 2006 NMTC Transaction and Biovest has liquidated the subsidiaries created specifically to conduct activities under the April 2006 NMTC transaction.
December 2006 NMTC Transaction
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “St. Louis Restructuring Agreement”) with St. Louis Development Corporation and Saint Louis New Markets Tax Credit Fund II, LLC (collectively “SLDC”), contingent upon submission to and approval by the Bankruptcy Court. The St. Louis Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to the December 2006 NMTC Transaction, in consideration of retention by SLDC of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $160,000 along with a settlement payment in the amount of $62,000, to defray certain legal and administrative expenses incurred by SLDC. This St. Louis Restructuring Agreement and the compromise of the outstanding claims against Biovest in connection with the December 2006 NMTC Transaction was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, our guaranty, Biovest’s guaranty, and all of the subsidiary guaranties from affiliates and third parties and all other obligations of all parties to the December 2006 NMTC Transaction were terminated. Biovest has ceased all activities under the December 2006 NMTC Transaction and Biovest has liquidated the subsidiaries created specifically to conduct activities under the December 2006 NMTC Transaction.
61
Table of Contents
Cash Resources
Cash Flows for the Three Months Ended December 31, 2010
For the three months ended December 31, 2010, our cash increased by $3.2 million. Net cash flow from operating activities was $1.3 million. The use for operating activities included a net loss of $7.5 million and additions of $10.8 million for share-based compensation and $4.1 million for derivative loss. There was also an adjustment of $11.4 million for the gain on reorganization.
Net cash outflow from investing activities for the three months ended December 31, 2010 was approximately $0.1 million for the acquisition of property, plant and equipment.
We had net cash flows from financing activities of $4.6 million for the three months ended December 31, 2010. Proceeds from notes payable, related party were approximately $0.3 million, in addition to the $7.0 million proceeds from Biovest’s convertible note.
Cash Flows for the Year Ended September 30, 2010
Our cash and cash equivalents were $0.6 million at September 30, 2010 compared with $0.3 million at September 30, 2009.
For the year ended September 30, 2010, we used $0.4 million in cash to fund our operating activities. The use for operating activities included a net loss of $48.2 million, adjustments to net income of $6.2 for the accretion of debt discounts, $28.8 million for derivative losses, and $3.0 million for the change in fair value of convertible debentures.
There were minimal net cash flows from investing activities.
We had net cash flows from financing activities of $0.7 million in the year ended September 30, 2010, primarily due to the debtor-in-possession financing proceeds of $0.6 million.
Off-Balance Sheet Arrangements
We do not maintain any off-balance sheet financing arrangements.
Fluctuations in Operating Results
We anticipate that our results of operations will fluctuate from quarter to quarter for several reasons, including:
| • | the timing and extent of our development activities and clinical trials for BiovaxID, Revimmune, and any biopharmaceutical products that we may develop in the future; |
| • | the timing and outcome of our applications for regulatory approval for our product candidates; |
| • | the timing and extent of our adding new employees and infrastructure; and |
| • | the timing of any milestone payments, license fees, or royalty payments that we may be required to make. |
62
Table of Contents
General
Headquartered in Tampa, Florida, Accentia Biopharmaceuticals, Inc. (Other OTC: “ABPI”) is a biotechnology company that is developing Revimmune™ as a comprehensive system of care for the treatment of autoimmune diseases. Additionally, through our majority-owned subsidiary, Biovest International, Inc., we are developing BiovaxID® as a therapeutic cancer vaccine for treatment of follicular non-Hodgkin’s lymphoma (“FL”) and mantle cell lymphoma (“MCL”). Through our wholly-owned subsidiary, Analytica International, Inc., we conduct a health economics research and consulting business which we market to the pharmaceutical and biotechnology industries, using our operating cash flow to support our corporate administration and product development activities.
Revimmune™ is being developed as a treatment for various autoimmune diseases. The approximately 80 known autoimmune diseases generally arise from an overactive immune response against substances and/or tissue normally present in the body. As a system of care, Revimmune consists of administering high, pulsed doses of an FDA-approved drug, cyclophosphamide, over a four-day interval as part of an integrated risk management system including a panel of preventive tests, monitoring and medications which are intended to minimize risks while seeking to maximize the clinical benefit.
Through a collaboration with the National Cancer Institute (“NCI”), our majority-owned subsidiary, Biovest International, Inc. (OTCQB: “BVTI”) (“Biovest”), has developed a patient-specific cancer vaccine, BiovaxID®, which has demonstrated statistically significant Phase 3 clinical benefit by prolonging disease-free survival in follicular non-Hodgkin’s lymphoma patients treated with BiovaxID.
Additionally, through our wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), based in New York City, we conduct a global research and strategy consulting business that provides professional services to the pharmaceutical and biotechnology industries. Since 1997, Analytica has expertly directed research studies and projects, including traditional health economic modeling projects, database studies, structured reviews, health technology assessments, reimbursement analyses, and value dossiers.
Corporate Overview
We were organized in 2002 to develop and commercialize biopharmaceutical products.
We commenced business in April 2002 with the acquisition of Analytica, a provider of analytical and consulting services to the biopharmaceuticals industry, including clinical trial services, pricing and market assessment and outcomes research. We acquired Analytica in a merger transaction for $3.7 million cash, $1.2 million of convertible promissory notes, and the issuance of 8.1 million shares of Series B preferred stock. Analytica, which was founded in 1997, has offices in New York and Germany.
In June 2003, we acquired an 81% interest of Biovest, for an investment of $20.0 million in Biovest pursuant to an investment agreement. Biovest is a biologics company that is developing our BiovaxID patient-specific vaccine for the treatment of FL. As of September 30, 2010, we owned of record approximately 75% of the common stock of Biovest, with the minority interest being held by approximately 400 shareholders of record. Biovest’s common stock is registered under Section 12(g) of the Securities Exchange Act of 1934, and Biovest therefore files periodic and other reports with the SEC.
63
Table of Contents
Chapter 11 Reorganization
On November 10, 2008, we, along with all of our subsidiaries, filed a voluntary petition for reorganization under Case No. 8:08-bk-17795-KRM (“Chapter 11”). On August 16, 2010, we filed our First Amended Joint Plan of Reorganization, and, on October 25, 2010, we filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. We emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
Our Products and Services
REVIMMUNE™
We are developing Revimmune as a comprehensive system of care for the treatment of various autoimmune diseases.
The Immune System and Autoimmunity
The immune system is the body’s natural defense mechanism for identifying and killing or eliminating disease-causing pathogens (such as bacteria, viruses, or other foreign microorganisms) and tumor cells. In humans, the primary disease fighting function of the immune system is carried out by white blood cells (leukocytes), which mediate two types of immune responses: innate immunity and adaptive immunity. Innate immunity refers to the broad first-line immune defense that recognizes and eliminates certain pathogens prior to the initiation of a more specific adaptive immune response. While the cells of the innate immune system provide a first line of defense, they cannot always eliminate or recognize infectious organisms. In some cases, new infections may not always be recognized or detected by the innate immune system. In these cases, the adaptive immune response has evolved to provide a highly specific and versatile means of defense which also provides long-lasting protection (immune memory) against subsequent re-infection by the same pathogen.
Autoimmune diseases are the result of white blood cells in the body recognizing and injuring or destroying normal (self) organs or tissues. In affected patients, the adaptive immune response (normally targeted against foreign antigens) becomes aberrantly targeted against self-tissues, leading to tissue damage, and chronic inflammation of affected organs. In severe cases patients may experience loss of function leading to disability or death. Autoimmune diseases are generally considered manageable in their early stages with immunosuppressive therapies or immunomodulating therapies. These therapies, however, are rarely considered “curative” and with modern standards of care, patients may suffer from chronic disease progression.
Autoimmune diseases pose a major burden to society. At least ten million Americans suffer from the more than eighty illnesses caused by autoimmunity. Many autoimmune diseases occur among young to middle-aged adults, leading to life-long disease and often life-changing disability. These conditions therefore also pose a disproportionate economic burden to healthcare systems in the industrialized world. Women are especially susceptible and comprise approximately 75% of diagnosed cases. Autoimmune diseases are among the ten leading causes of death among women in all age groups up to age 65.
64
Table of Contents
Although many of these diseases can be treated clinically by currently available conventional immunosuppressive regimens, important problems remain: some patients are refractory to standard immunotherapy, and others respond only partially. In many cases, immunosuppressive therapies or therapies to control the symptoms of the disease must be continued indefinitely, maintaining an impaired immune system, and often resulting in increased risk of infections and other serious health problems.
Revimmune™ for the Treatment of Autoimmune Disease
In contrast to currently approved therapies available to treat autoimmune disease, Revimmune seeks to eliminate virtually all circulating white blood cells, including those driving autoimmunity, while seeking to spare the patient’s stem cells. As the patient’s eliminated white blood cells are replenished with new white blood cells derived from these stem cells, the patient’s immune system becomes effectively replaced or “rebooted”.
Revimmune consists of an approved active drug (cyclophosphamide) administered as part of an integrated risk management system designed to assure its consistent use and minimize the risks of treatment. Cyclophosphamide has been approved by the FDA to treat disorders other than autoimmune disease including various forms of cancer.
To facilitate our development and commercialization of Revimmune, we have entered into an agreement with Baxter Healthcare Corporation, which we believe is the only cGMP (“current Good Manufacturing Practice” enforced in the U. S. by the FDA) manufacturer of injectable/infusion cyclophosphamide used in the U.S. as referenced in the FDA Orange Book. This agreement grants us the exclusive right to use Baxter’s regulatory file and drug history (Drug Master File) for cyclophosphamide, which we believe will advance our planned clinical trials and anticipated communications with the FDA. Additionally, the agreement grants to us the exclusive right to purchase Baxter’s cyclophosphamide for the treatment of various autoimmune diseases including autoimmune hemolytic anemia, multiple sclerosis, and systemic sclerosis.
Cyclophosphamide is a nitrogen mustard alkylating agent (it destroys target cells by binding to DNA and interfering with cell division and function) which is converted by the liver into an active chemotherapeutic agent. Cyclophosphamide’s effects are dose-dependent. As used in Revimmune, cyclophosphamide is planned to be administered at a dose of 50 mg/kg which is a unit of measurement where drug dosage is measured in milligrams based on the patient’s body weight measured in kilograms delivered in a series of 4 daily infusions (which is generally referred to as “Pulsed”) corresponding to a total dose of 200 mg/kg. This dose represents an ultra-high dose of cyclophosphamide, which is generally administered to cancer patients in total doses ranging from approximately 40 to 50 mg/kg or less over a period of 2 to 5 days. We refer to the dosing schedule of cyclophosphamide as used in Revimmune as “High-Dose Pulsed Cyclophosphamide”.
Revimmune includes a comprehensive risk management system to restrict the use of High-Dose Pulsed Cyclophosphamide to those patients anticipated to most benefit from the drug and to exclude patients for whom the drug is contraindicated. The risk management system includes pre- and post-treatment drugs combined with careful monitoring during and after administration of cyclophosphamide to avoid or minimize infections and other adverse side effects which may result from the therapy. This computerized central risk management system, which we refer to as “Revimmune Bolstering Outcomes Of Therapy” or “REBOOT™”, is intended to assist physicians and other Revimmune care providers to maintain a consistent risk management approach when administering Revimmune. We anticipate that this system will incorporate a number of safety questionnaires, protocols, and other educational materials for patients and providers; in addition, we anticipate the system will provide a series of computer-verified diagnostic assays and compliance checks integrated with patients’ treatment. The REBOOT system may also include a patient and physician registry to enable long-term follow-up and research to improve outcomes and minimize risk in treated patients. Furthermore, we expect to enhance patient safety by packaging, labeling and distributing Revimmune therapy only as part of the REBOOT system.
65
Table of Contents
We anticipate that our REBOOT system will be at the core of a formal risk evaluation and mitigation strategy subject to approval by the FDA. As such, it forms a critical part of our planned clinical trial(s) and investigational new drug application(s) (“IND”). Such risk evaluation and mitigation strategies are generally referred to as REMS, which were authorized by the 2007 amendment to the Food and Drug Act. REMS are frequently and increasingly included as part of new drug applications, approvals and/or labels to assure safety and to maximize benefit. REMS are subject to enforcement by the FDA through civil penalties.
Previous Clinical Studies in Cyclophosphamide to Treat Autoimmune Disease
A number of small, single-arm, open label, uncontrolled trials that have been conducted at various institutions suggest the effect of cyclophosphamide on the disease course of certain autoimmune diseases. These prior studies form the basis of our belief that Revimmune, which incorporates High-Dose Pulsed Cyclophosphamide, may potentially represent a new treatment option for certain autoimmune diseases and support our decision to pursue the development of Revimmune. These prior studies are considered to be preliminary and we believe that significant additional development is required before High-Dose Pulsed Cyclophosphamide can be widely accepted as a therapeutic option for autoimmune disease. These additional developments, which are part of our Revimmune development plan, include: (i) conducting a definitive controlled and randomized trial for each specific autoimmune disease sufficient to demonstrate safety and efficacy to the satisfaction of the FDA as required to support potential marketing approval and (ii) developing a REMS for High-Dose Pulsed Cyclophosphamide to assure that the therapy properly balances risks and benefits in a manner which is sufficient to obtain the approval of the FDA under the Food and Drug Administration Amendments Act of 2007.
| Autoimmune Disease |
Study Site |
Number of |
||||
| Aplastic Anemia | JHU1, Hahnemann2, Wayne State | 82 | ||||
| Systemic Sclerosis (diffuse cutaneous) | JHU | 6 | ||||
| Systemic Lupus Erythematosus | JHU, Hahnemann | 43 | ||||
| Multiple Sclerosis | JHU, Hahnemann, Stony Brook3 | 47 | ||||
| Myasthenia Gravis | JHU, Stanford University | 13 | ||||
| Autoimmune Hemolytic Anemia (AIHA) | JHU, Hahnemann, University of Milan | 12 | ||||
| CIDP | Hahnemann | 5 | ||||
| Rheumatoid Arthritis | JHU, Queen Elizabeth Hospital4 | 5 | ||||
| Pemphigus | JHU, University of Miami | 4 | ||||
| TOTAL | 217 | |||||
| 1 | Johns Hopkins University |
| 2 | Medical College of Pennsylvania-Hahnemann Hospital at Drexel University |
| 3 | State University of New York at Stony Brook |
| 4 | Queen Elizabeth Hospital, Adelaide, South Australia |
Table 1. Published preliminary studies investigating High-Dose Pulsed Cyclophosphamide for treatment of Autoimmune Diseases listed by disease.
66
Table of Contents
Revimmune Development Plan
Rationale
Notwithstanding the prior reports of High-Dose Pulsed Cyclophosphamide as a potential therapy for certain autoimmune diseases, cyclophosphamide is not currently FDA-approved for the treatment of any autoimmune diseases. These prior studies of High-Dose Pulsed Cyclophosphamide in the United States have been conducted at a limited number of large academic research hospitals and have featured non-uniform inclusion criteria and/or administration schedules. It is generally recognized that there may be significant potential risks of infection or other side effects when High-Dose Pulsed Cyclophosphamide is not administered by highly-qualified personnel in a controlled and regulated clinical setting. While the previous studies are important to our Revimmune development plan, they are not considered sufficient to support regulatory approval. At the core of our Revimmune development plan is a recognition that controlled and randomized clinical trials will be necessary to demonstrate the efficacy of High-Dose Pulsed Cyclophosphamide to the satisfaction of the FDA and that safety will be an important regulatory and clinical concern which we believe will require an FDA approved formal REMS.
There are approximately 80 recognized autoimmune diseases. We previously planned to conduct our first randomized and controlled trial of Revimmune in multiple sclerosis (“MS”), because of the potentially significant impact of such a study and the number of MS patients previously treated with High Dose Cyclophosphamide (Table 1). However, due to our limited capital and resources, we have modified our Revimmune development plan so as to initially focus on two less prevalent autoimmune diseases where we perceive a significant unmet medical need: diffuse systemic sclerosis and autoimmune hemolytic anemia. Subject to our capital availability, we plan to undertake clinical trials of Revimmune for the treatment of these two autoimmune diseases in a parallel manner. Diffuse systemic sclerosis and autoimmune hemolytic anemia represent diseases for which we believe significant unmet medical needs exist. There is no standard treatment for systemic sclerosis, and while the standard treatment for autoimmune hemolytic anemia is effective for a subset of patients, patients for which this treatment option is not effective, referred to as refractory patients, have very limited options. We believe that the significant unmet medical need, the shorter and severe disease course and the smaller prevalence of these autoimmune diseases make them appropriate for our initial focus. Notwithstanding the initial focus of our current Revimmune development plan, we continue to believe that new treatment options are needed for MS and we remain interested in ultimately conducting studies with Revimmune in MS.
In September 2007, we conducted a meeting with the FDA regarding our proposed design of a clinical trial for Revimmune to treat MS. We considered the FDA meeting to have been constructive. However, with the change in our Revimmune development plan, we are currently preparing for an additional meeting with the FDA to discuss possible studies in diffuse scleroderma and/or autoimmune hemolytoic anemia. Based on FDA input, we anticipate filing an Investigational New Drug Application(s) (IND) under which we hope to conduct our planned clinical trials. Further, we plan to discuss with the FDA our plans for a REMS to be developed under the Food and Drug Administration Amendments Act of 2007.
67
Table of Contents
Provided that our planned clinical trials, once completed, demonstrate clinical benefit and safety, we would anticipate discussing next steps with the FDA which could include the suitability of seeking conditional accelerated approval and/or the appropriateness and design of additional trial(s).
Diffuse Cutaneous Systemic Sclerosis
Disease Background
Systemic sclerosis is a chronic, multisystem autoimmune disease characterized by hardening of the skin and affected tissues, blood vessel (vascular) alterations, and the presence of autoimmune antibodies (autoantibodies) in the blood. Patients with systemic sclerosis often experience Raynaud’s phenomenon, a condition associated with discoloration of fingers and/or toes when exposed to changes in temperature or emotional events. Systemic sclerosis is most common in females who are 30 to 50 years of age.
The underlying disease etiology, or cause, of systemic sclerosis remains largely unknown but in affected patients, prolonged activation of the immune system and inflammation results in extensive tissue damage and repair. Continuous remodeling and repair and an inability to terminate the reparative processes leads to persistent connective tissue remodeling, scarring, and fibrosis. Some patients may have only a few affected areas, but others suffer from progressive skin involvement which becomes the source of much affliction as the condition worsens.
Systemic sclerosis can be divided into two primary forms, limited cutaneous systemic sclerosis (“lcSSc”) and diffuse cutaneous systemic sclerosis (“dcSSc”). Approximately 60% of scleroderma patients have limited cutaneous systemic sclerosis, while approximately 35% have the diffuse cutaneous form. lcSSc typically presents as skin manifestations affecting the hands, arms and face. Additionally, pulmonary arterial hypertension may occur in up to one third of patients and is the most serious complication for this form of systemic sclerosis. Diffuse cutaneous systemic sclerosis comprises a very severe form of systemic sclerosis which progresses rapidly and affects a large area of the skin and one or more internal organs, frequently the kidneys, esophagus, heart and lungs. dcSSc leads to substantial disability and/or death. We plan our clinical trials to study diffuse cutaneous systemic sclerosis, which has an estimated U.S. prevalence of approximately 30,000 cases with an estimated incidence of approximately 2100 annually.
Unmet Medical Need
dsSSc is a severe disease with the highest case-specific mortality of any rheumatic disorder, with 50% of patients dying or developing major organ complications within 3 years of diagnosis (Denton and Black 2005). Currently there is no treatment that has been proven to prevent progression of disease, underscoring a huge unmet medical need in this disease.
68
Table of Contents
Autologous stem cell transplantation can significantly improve the skin score, a measure of disease progression in dcSSc. However, reported transplant-related mortality ranges from 8.7%-17%. Other immune modulatory agents have been studied with varying results; either the trials were too small or uncontrolled to definitively determine the efficacy of the treatment or randomized, controlled trials were conducted without demonstrating a clinically significant benefit.
High-Dose Pulsed Cyclophosphamide Experience in Diffuse Scleroderma
An open-label trial of High-Dose Pulsed Cyclophosphamide in patients with clinically active diffuse systemic sclerosis patients was conducted at Johns Hopkins University (Tehlirian et.al. published these results in 2008). This study forms the basis of our planned clinical trial of Revimmune.
In this study, six patients with diffuse cutaneous systemic sclerosis were treated. One patient died of a pulmonary infection (7 weeks after treatment) that was acquired after the absolute neutrophil count had normalized, indicating that the infection was not acquired during the immune ablation period. This patient experienced acute respiratory distress syndrome associated with a fungal infection and a decline in kidney function. This patient had extensive lung disease at the time of entry into the study. This highlights the need for a robust system of care during High Dose Cyclophosphamide treatment and the requirement for highly selective inclusion criteria, especially in patients with organ involvement and damage as a result of their autoimmune disease.
The primary endpoint was the modified Rodnan skin score (“mRSS”), a typical efficacy endpoint for systemic sclerosis trials. The mRSS measures skin thickness and collagen content of skin. In dcSSc, increasing skin thickness is associated with increased disease activity, and a reduction in mRSS is associated with a more favorable outcome, with a reduction of 25% considered clinically significant. The study reported that in 5 evaluable patients, mRSS was reduced by 60%, 55%, 41%, 31%, and 0%. The study further reported that four patients out of five responded initially, while one responded but relapsed at 6 months.
The physician’s global assessment (“PGA”) was reported to have improved for four of five patients 47%, 69%, 56%, and 59% within the first month after treatment and for one of five within the first 3 months (80%). The forced vital capacity (“FVC”), a measure of lung function, also stabilized in four of six patients. One patient’s FVC worsened due to an infection, then returned to baseline. Five of five patients improved on the Health Assessment Questionnaire Disability Index.
Planned Clinical Trial of Revimmune in dcSSc
Similar to the prior clinical trial conducted using High-Dose Pulsed Cyclophosphamide to treat diffuse cutaneous systemic sclerosis, we intend to enroll patients meeting established criteria for this diagnosis and who have evidence of moderately severe organ damage and clinical evidence of active disease.
69
Table of Contents
Autoimmune Hemolytic Anemia (AIHA)
Disease Background
We plan to study autoimmune hemolytic anemia (“AIHA”), a disease in which the body’s immune system attacks its own red blood cells (“RBCs”), leading to their destruction, or hemolysis. While studies of prevalence of AIHA in the U.S. are few, we estimate the U.S. prevalence of AIHA at approximately 50,000 cases based on epidemiological studies in other countries. The U.S. incidence of AIHA is approximately 2,400 per year.
In affected patients, a process of increased destruction of RBCs takes place mediated by the formation of anti-RBC autoantibodies (an autoantibody is an antibody produced by the body in reaction to the body’s own cells). AIHA can occur at any age and affects women more often than men. About half of the time, the cause of autoimmune hemolytic anemia cannot be determined (idiopathic autoimmune hemolytic anemia). Autoimmune hemolytic anemia can also be caused by or occur with another disorder, such as systemic lupus erythematosus (lupus), and rarely it follows the use of certain drugs, such as penicillin.
AIHA frequently commences as an acute, sometimes life-threatening disease often requiring hospitalization, but is considered a chronic disease. In some people, the destruction may stop after a period of time. In other people, red blood cell destruction persists and becomes chronic. There are two main types of autoimmune hemolytic anemia: warm antibody hemolytic anemia (“WAIHA”) and cold antibody hemolytic anemia (“CAIHA”). In the warm antibody type, the autoantibodies attach to and destroy red blood cells at temperatures equal to or in excess of normal body temperature. In the cold antibody type, the autoantibodies become most active and attack red blood cells only at temperatures well below normal body temperature. Rarely, patients have both cold- and warm-reactive autoantibodies and are classified as mixed-type AIHA.
Autoimmune hemolytic anemia as the cause of hemolysis is confirmed when blood tests detect increased amounts of certain antibodies, either attached to red blood cells (direct antiglobulin or Coombs’ test) or in the liquid portion of the blood (indirect antiglobulin or Coombs’ test). Other tests sometimes help determine the cause of the autoimmune reaction that is destroying red blood cells.
If symptoms are mild or if destruction of red blood cells seems to be slowing on its own, no treatment is needed. If red blood cell destruction is increasing, a corticosteroid such as prednisone is usually the first choice for treatment for WAIHA. High doses are used at first, followed by a gradual reduction of the dose over many weeks or months. When WAIHA patients do not respond to corticosteroids or when the corticosteroid causes intolerable side effects, surgery to remove the spleen (splenectomy) is often the next treatment. The spleen is removed because it is one of the places where antibody-coated red blood cells are destroyed. When destruction of red blood cells persists after removal of the spleen or when surgery cannot be done, immunosuppressive drugs may be used.
When red blood cell destruction is severe, blood transfusions are sometimes needed, but they do not treat the cause of the anemia and provide only temporary relief.
70
Table of Contents
Unmet Medical Need
We believe that there is a need for additional therapeutic options to treat both warm and cold AIHA. Only 15-20% of patients achieve complete remission with standard first line therapy, corticosteroids. Adverse events related to corticosteroid therapy include excessive weight gain, skin flushing, neuropsychiatric disorders, sleep disturbances, increased risk of cardiovascular events, cataracts, and myopathy. Co-morbidities exacerbated by corticosteroids include diabetes, hypertension, hyperlipidemia, heart failure, glaucoma, and peptic ulcer disease.
Usually splenectomy, the surgical removal of the spleen, is used as a second-line therapy, which elicits a 50% response rate. Options for patients unresponsive or refractory to corticosteroids and splenectomy are limited. In WAIHA, the chance of spontaneous or drug-induced remission or cure is extremely low. An urgent need exists for new, better treatments of WAIHA, especially for steroid-refractory or unresponsive patients. CAIHA does not respond to either steroids or splenectomy and therefore remains a significant clinical challenge.
High-Dose Pulsed Cyclophosphamide Experience with AIHA
Following studies in other severe autoimmune disorders, High-Dose Pulsed Cyclophosphamide was studied in patients with severe AIHA that was refractory to standard therapies (Moyo, Smith et al. 2002). Nine patients were treated at Hahnemann University, Medical College of Pennsylvania and Johns Hopkins University. Seven patients displayed the warm type (WAIHA), one the cold-type (CAIHA), and one patient was mixed warm/cold. These patients had previously failed at least 2 standard therapies (primary AIHA) or one standard therapy (secondary AIHA), and were steroid dependent.
High-Dose Pulsed Cyclophosphamide was well-tolerated in these patients. All patients were reported to have experienced transfusion independence following treatment. The study reported that six patients underwent a complete response, and the remaining 3 achieved a partial remission. At last follow-up (median follow-up duration 15 months, ranging from 4-29 months), only one patient in CR was still receiving doses of corticosteroids (although these were tapering) and one patient in PR was receiving a low dose of corticosteroids. All others discontinued steroids and no patient experienced relapse at a median of 15 months after treatment.
Three other patients with AIHA have been treated with High Dose Cyclophosphamide, in one small study (Brodsky 1998) and one case report (Panceri 2002). Of the two patients in the former study, one had Evan’s syndrome (a combination of AIHA and another disease). The AIHA patient experienced a complete recovery for 16 months but then came down with immune thrombocytopenic purpura, another autoimmune disease and subsequently died. The other patient experienced no symptomatic manifestations of disease after treatment and was independent of transfusion for more than 10 months. The case report (Panceri 2002) described the striking sudden improvement in a child with severe AIHA who was unresponsive to four other treatments. However, due to the absence of defined inclusion criteria as used in the Moyo study, these three patients have extremely heterogeneous characteristics and may not be comparable to the larger Moyo study in which patients were more similar.
Therefore, the small Moyo study forms the basis of our planned clinical trial of Revimmune in AIHA.
71
Table of Contents
Planned Clinical Trial of Revimmune in AIHA
Similar to the prior clinical trial conducted using High-Dose Pulsed Cyclophosphamide to treat AIHA we intend to enroll patients with a diagnosis of severe autoimmune hemolytic anemia. We expect that these patients will have had a failure of at least 2 standard treatment approaches (e.g., prednisone therapy, splenectomy, intravenous immunoglobulin, or other immunosuppressants.), or an inability to taper prednisone dose to less than 10 mg/day. We plan to enroll both patients with both the warm and cold-types of AIHA and plan to structure the trial analyses to control for the differences between these types of AIHA. For example, if Revimmune is effective for only WAIHA, then the analysis plan will still be sufficient to determine efficacy in only the warm type.
Proprietary Rights to Revimmune
We have developed a multi-faceted strategy to maintain and protect our proprietary interests in Revimmune, involving various forms of intellectual property, including patent and non-patent exclusivity. We hold the exclusive world-wide rights to commercialize High-Dose Pulsed Cyclophosphamide to treat MS and certain other autoimmune diseases through a sublicense (the “Revimmune Sublicense”) from Revimmune, LLC, which is affiliated with one of our directors and shareholders, which holds the exclusive license for the technology from JHU (the “JHU License”). Under the Revimmune Sublicense, we are obligated to pay a royalty of 8% of net sales of Revimmune which is equally split between JHU and Revimmune, LLC until the later of the expiration of the last to expire patent under the JHU License on a country by country basis, or 10 years following the first commercial sale of a regulatory approved product regardless of the issuance of any such patents.
We believe that our JHU License creates an important relationship between us, JHU, and the inventors who are JHU faculty and pioneers in the study and development of High-Dose Pulsed Cyclophosphamide. JHU has filed patent applications in the U.S. and several foreign countries with claims pertaining to the use of High-Dose Pulsed Cyclophosphamide to treat MS and certain other autoimmune diseases which are covered by our JHU License. Some of these licensed patent applications are undergoing examination and are subject to pending patent office objections and/or rejections. Additionally, we have filed patent applications for our REBOOT system and certain screening protocols that maximize the safety and effectiveness of cyclophosphamide treatment regimens. Further, we anticipate that our computer software program being developed to implement our REBOOT system will be proprietary and protected through trademark and copyright filings.
To extend our proprietary rights to Revimmune, effective on November 29, 2010, we entered into an agreement with Baxter Healthcare Corporation (“Baxter”), which we believe is the only cGMP (“current Good Manufacturing Practice” enforced in the U. S. by the FDA) manufacturer of injectable cyclophosphamide in the U.S. This agreement makes Baxter our exclusive source of cyclophosphamide under an agreed-upon price structure. The Agreement requires us to make defined royalty payments to Baxter in connection with net sales of cyclophosphamide for the designated indications, including without limitation any sales by subdistributors. The Agreement also grants us the exclusive right to use Baxter’s regulatory file and drug history (Drug Master File) for cyclophosphamide which we believe will facilitate our planned clinical trials and anticipated dealings with the FDA. Additionally, the agreement grants to us the exclusive right to purchase Baxter’s cyclophosphamide for the treatment of various autoimmune diseases including Autoimmune Hemolytic Anemia, Multiple Sclerosis, and Scleroderma.
72
Table of Contents
BIOVAXID® - THERAPEUTIC CANCER VACCINE
Our majority-owned subsidiary, Biovest International, Inc. (“Biovest”), is developing BiovaxID as a cancer vaccine to treat follicular lymphoma (“FL”) and mantle cell lymphoma (“MCL”).
The Human Immune System
The immune system functions as the body’s natural defense mechanism for identifying and killing or eliminating disease-causing pathogens, such as bacteria, viruses, or other foreign microorganisms. However, with regard to cancer, including lymphomas, the immune system’s natural defense mechanism is believed to be largely thwarted by natural immune system mechanisms which seek to protect “self-cells” from attack. In humans, the primary disease fighting function of the immune system is carried out by white blood cells (leukocytes), which mediate two types of immune responses: innate immunity and adaptive immunity. Innate immunity refers to the broad first-line immune defense that recognizes and eliminates certain pathogens prior to the initiation of a more specific adaptive immune response. While the cells of the innate immune system provide a first line of defense, they cannot always eliminate or recognize infectious organisms. In some cases, new infections may not always be recognized or detected by the innate immune system. In these cases, the adaptive immune response has evolved to provide a highly-specific and versatile means of defense which also provides long-lasting protection (immune memory) against subsequent re-infection by the same pathogen. This adaptive immune response facilitates the use of preventative vaccines that protect against viral and bacterial infections such as measles, polio, diphtheria, and tetanus. Biovest believes that BiovaxID creates an adaptive immune response to cancerous B-cells.
Adaptive immunity is mediated by a subset of white blood cells called lymphocytes, which are divided into two types: B-cells and T-cells. In the bloodstream, B-cells and T-cells recognize antigens, which are molecules that are capable of triggering a response in the immune system. Antigens are molecules from bacterial, viral, or fungal origin, foreign (non-self) proteins, and in some cases, tumor-derived proteins that can stimulate an immune response. The human body makes millions of different types of B-cells that circulate in the blood and lymphatic systems and perform immune surveillance. Each B-cell has a unique receptor protein (immunoglobulin) on its surface that binds to one particular antigen. Once a B-cell recognizes its specific antigen and receives additional signals from a T-helper cell, it can proliferate and become activated in order to secrete antibodies (immunoglobulins; Ig) which can neutralize the antigen and target it for destruction. T-cells may also recognize antigens on foreign cells, whereby they can promote the activation of other white blood cells or initiate destruction of the targeted cells directly. A person’s B-cells and T-cells can collectively recognize a wide variety of antigens, but each individual B-cell or T-cell will recognize only one specific antigen. Consequently, in each person’s bloodstream, only a relatively few lymphocytes will recognize the same antigen.
Since B-cell cancers such as non-Hodgkin’s lymphoma (“NHL”) are tumors arising from a single malignant transformed B-cell, the tumor cells in NHL maintain on their surface the original malignant B-cell’s immunoglobulin (collectively referred to as, the “tumor idiotype”) that is distinct from those found on normal B cells. The idiotype maintained on the surface of each B-cell lymphoma serves as the tumor-specific antigen for the BiovaxID cancer vaccine.
In many cases, including in NHL, cancer cells produce molecules known as tumor-associated antigens, which may or may not be present in normal cells but may be over-produced in cancer cells. T-cells and B-cells have receptors on their surfaces that enable them to recognize the tumor associated antigens. While cancer cells may naturally trigger a B- or T-cell-based immune response during the initial appearance of the disease, this response may be only weakly specific or attenuated in such a way that it does not fully eradicate all tumor cells. Subsequently, tumor cells gradually evolve and escape from this weak immune response and are able to grow into larger tumors. In addition, because cancer cells arise from normal tissue cells, they are often able to exploit or increase existing immune tolerance mechanisms to suppress the body’s immune response which would normally destroy them. In other cases, chemotherapy or other treatment regimens used to treat the cancer may themselves weaken the immune response and render it unable to reject and kill tumor cells. Even with an activated immune system; however, the number and size of tumors can often overwhelm the immune system.
73
Table of Contents
In the case of cancer and other diseases, immunotherapies are designed to activate a person’s immune system in an attempt to combat the disease. There are two forms of immunotherapy used to treat diseases: passive and active. Passive immunotherapy is exemplified by the intravenous infusion into a patient of antibodies specific to the particular antigen. While passive immunotherapies have shown clinical benefits in some cancers, they require repeated infusions and can cause the destruction of normal cells in addition to cancer cells. An example of passive immunotherapy to treat lymphoma is monoclonal antibodies such as rituximab. An active immunotherapy, on the other hand, seeks to generate a durable adaptive immune response by introducing an antigen into a patient, often in combination with other components that can enhance an immune response to the antigen. BiovaxID is an example of active specific immunotherapy. Although active immunotherapies have been successful in preventing many infectious diseases, their ability to combat cancers of various types has been limited by a variety of factors, including the inability of tumor antigens to elicit an effective immune response, difficulty in identifying suitable target tumor antigens, inability to manufacture tumor antigens in sufficiently pure form, and inability to manufacture sufficient quantities of tumor antigens.
Nevertheless, in 2010 one active immunotherapy, Provenge®, developed by Dendreon Corporation, received marketing approval from the FDA. This represents the first case of an active immunotherapy to successfully gain marketing approval in the U.S. In addition to BiovaxID, there are a number of other active immunotherapeutics for cancer in various stages of clinical trials that have demonstrated promising results.
A number of features of the non-Hodgkin’s lymphomas make these tumors particularly suitable for treatment with a therapeutic cancer vaccine. The malignant B-cell lymphocytes of NHL express a unique, identifiable tumor-specific antigen which is not expressed by other (healthy) cells in the body. In contrast, the majority of human cancers typically lack strong ubiquitous expression of tumor-specific antigens to distinguish them from normal cells, or they express a potentially widely-varying mix of antigens which can be difficult to identify and formulate into a successful therapeutic vaccine.
Non-Hodgkin’s Lymphoma (“NHL”)
NHL is a heterogeneous group of malignancies of the lymphatic system with differing clinical behaviors and responses to treatment. BiovaxID has been studied in two distinct forms of non-Hodgkin’s lymphoma, namely, FL and MCL. NHL is the fifth most common type of cancer in the U.S., with an estimated prevalence of 438,325 cases in 2007 in the U.S. NHL accounts for 5% of all cancer deaths in the U.S. NHL is one of the few malignancies in which there continues to be a rise in incidence. Since the early 1970’s, incidence rates for NHL have nearly doubled. Moreover, in spite of recent advances in the standard of care, the overall five-year survival rate remains at approximately 63%. According to the NCI, in 2010 it was estimated that approximately 66,000 new cases of NHL would be diagnosed and approximately 19,000 Americans would die from the disease, with a comparable number estimated in Europe.
74
Table of Contents
NHL is usually classified for clinical purposes as being either “indolent” or “aggressive,” depending on how quickly the cancer cells are likely to grow and spread. The indolent, or slow-growing, form of NHL has a very slow growth rate and may need little or no treatment for months or possibly years. Aggressive, or fast-growing, NHL tends to grow and spread quickly and cause severe symptoms, and patients with aggressive NHL have shorter overall survival.
Follicular Lymphoma
Indolent (slow growing) and aggressive NHL each constitute approximately half of all newly diagnosed B-cell NHL, and roughly half of the indolent B-cell NHL is FL. Accordingly, approximately 22% of new cases of NHL fall into the category of disease known as indolent FL. The American Cancer Society’s most recent estimates for 2010 anticipated that there would be approximately 66,000 new cases of NHL in the U.S. This translates into approximately 15,000 new cases of FL diagnosed in 2010. According to the most recent data published by SEER (Surveillance, Epidemiology, and End Results), on January 1, 2007, there were approximately 438,325 patients alive who had a history of NHL. This figure translates into approximately 97,000 patients with FL. Biovest has conducted a Phase 2 clinical trial followed by a Phase 3 clinical trial in FL under Biovest’s IND. FL is a form of NHL that is derived from a type of cell known as a follicle center cell. Despite its slow progression, FL is almost invariably fatal. The median survival reported for FL patients ranges between 8 and 10 years, although these figures may have become slightly higher within the last decade as a result of improvements in the standard of care for FL.
The current standard of care for treatment of advanced, bulky follicular lymphoma (bulky Stage II, Stage III-IV) as specified by the National Comprehensive Cancer Network includes initial treatment of newly-diagnosed patients with rituximab-containing chemotherapy. Rituximab is a monoclonal antibody (an immune protein capable of selectively recognizing and binding to a molecule) which targets a protein primarily found on the surface of both healthy and cancerous B cells, known as CD20. Accordingly, rituximab seeks to bind and destroy all B-cells, including healthy B-cells, as a means of controlling the progression of FL in treated patients.
Rituximab and other biologics currently approved for lymphoma are characterized as “passive immunotherapies”. Following administration, rituximab exerts its effects primarily through an unselective and near total destruction of a patient’s B-cells, including malignant as well as healthy B-cells. Rituximab and other passive immunotherapies are often administered in sequential, repeated doses to achieve their effect, and following cessation of administration are over time eliminated from the patient’s circulation by normal bodily functions. Rituximab is characterized as a targeted therapy since it targets CD20, which is present on both healthy and tumor cells. Rituximab is manufactured in bulk and is not considered to be a personalized therapy.
By comparison, BiovaxID is characterized as an “active immunotherapy”. Active immunotherapies attempt to stimulate the patient’s immune system to respond to a disease. “Specific active immunotherapies” such as BiovaxID specifically seek to induce cellular and/or humoral immune responses focused on specific antigens present on a diseased cell (such as a tumor cell). As a specific, active immunotherapy, BiovaxID targets only the cancerous B-cells while sparing healthy B-cells. Accordingly, BiovaxID is highly targeted. BiovaxID is manufactured specifically and entirely for each patient and is considered to be a highly “personalized therapy”. If approved, BiovaxID will represent the only specific active immunotherapeutic approved for the treatment of FL and therefore will represent a new class of drugs that provide a new therapeutic option for patients with lymphoma.
75
Table of Contents
The EMEA has recently approved the use of two years of rituximab maintenance therapy in FL patients responding to first line chemo-immunotherapy. In this context, patients receive rituximab repeatedly over a period of two years, during which the agent seeks to deplete all B-cells in the body in an attempt to extend tumor remission. BiovaxID, if approved, would likewise be considered to be a “maintenance” or remission consolidation therapy. However, BiovaxID differs from rituximab maintenance therapy in that BiovaxID is administered for a 6-month period during which it seeks to establish a targeted and persistent immune response to the patient’s tumor. BiovaxID seeks to establish an adaptive immune response which be can come into play and destroy the tumor cells every time the patient’s trained immune system encounters the specific tumor target.
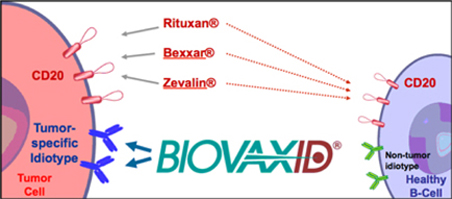
Figure 1. BiovaxID targets tumor-specific idiotype, a protein unique to the tumor and not found on healthy (non-malignant) B-cells. In contrast, current monoclonal antibody-based therapies for NHL, including rituximab (Rituxan®), tositumomab (Bexxar®), and ibritumomab tiuxetan (Zevalin®) target CD20, a cell-surface protein expressed by both tumor and healthy B-cells. As such, through its unique mode of action, BiovaxID represents a new therapeutic approach to treating FL.
Mantle Cell Lymphoma (“MCL”)
MCL is a rare, aggressive subtype of NHL characterized by short remissions and rapid progression similar to aggressive lymphomas and successive relapses, reflecting incurability similar to indolent lymphomas. The median overall survival for MCL has been cited as 3 to 5 years and the disease currently lacks a consensus standard of care. MCL represents approximately 6% of all NHL cases and worldwide there are approximately 7,800 new cases each year of which approximately one half are in the U.S.
The majority of MCL patients have disseminated disease and bone marrow involvement at diagnosis. Patients’ clinical outcomes from currently available therapies are poor. Although many therapeutic regimens are capable of rendering high initial response rates, these responses are of short duration (i.e., about 20 months) and the relative survival rates of MCL patients are among the lowest compared to other types of NHL. The prognostic after the first relapse is very poor, with an expected median overall survival of about 1-2 years. No currently available therapeutic regimens are curative.
While several therapeutic regimens are available to treat MCL patients, there currently exists no consensus standard of care for treatment of first-line relapsed MCL. As such, MCL remains incurable and it is generally considered that additional treatment options are required given this significant unmet medical need.
76
Table of Contents
Currently upon first diagnosis MCL patients are often evaluated for eligibility for autologous stem cell transplantation (“autoSCT”). Stem cell transplantation, an aggressive treatment protocol consisting of high-dose chemotherapy, immunotherapy and full-body radiation, aims to treat the patient’s tumor and purge the bone marrow of lymphoma cells. MCL patients who are eligible for autoSCT receive either R-CHOP (rituximab, cyclophophamide, doxorubicin, vincristine, prednisone) followed by autoSCT or R-HyperCVAD (rituximab, cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with rituximab plus high dose methotrexate and cytarabine) followed by observation. Although these therapeutic approaches do yield high response rates, they are associated with high rates of treatment discontinuation, high risk of myelodysplastic syndrome, and high mortality rates. Consequently, the considerable toxicity associated with these regimens largely limits these options primarily to a select subset of the MCL patients who are younger and better fit to tolerate these high-intensity treatments. However, even this subset ultimately gains only modest benefits from existing treatment options. Moreover, the use of these more aggressive regimens appears not to result in superior overall survival as compared to standard therapies. Given that the median age for newly diagnosed mantle cell lymphoma patients is 60 years, less aggressive therapeutic approaches are needed.
Development Status of BiovaxID
Introduction
Preliminary studies demonstrated that treatment of patients with NHL with an active immunotherapy could allow a patient’s immune system to produce B-cells and/or T-cells that recognized numerous portions of their tumor antigen and generate clinically significant immune responses. These studies provided the rationale for large-scale trials of active specific immunotherapy of this disease. These studies have been published in The New England Journal of Medicine (October 1992), Blood (May 1997), and Nature Medicine (October 1999). In the treatment of cancer, residual tumor cells remaining in the patient after completion of surgery or anti-tumor therapy are often the cause of tumor relapse. These residual tumor cells cannot always be detected by standard imaging techniques but their destruction may be feasible by active immunotherapy. The use of such vaccines differs from traditional cancer treatment in that the ultimate mechanism of action against the tumor is indirect: the anti-tumor immunity induced by vaccination, rather than the vaccine itself, is ultimately responsible for treatment benefit.
In 1994, the NCI filed for initiation of an IND for the purpose of conducting clinical trial(s) investigating the use of BiovaxID in NHL. Under this IND, the NCI began in 1994 a Phase 2 clinical trial in FL; in 1999, the Phase 3 clinical trial in FL; and in 2000 a Phase 2 clinical trial in MCL. The NCI selected Biovest to produce the vaccine for the initial Phase 2 clinical trial in FL. In 2001, Biovest entered into a formal cooperative research and development agreement (“CRADA”) with the NCI which formalized Biovest’s collaboration with the NCI. The IND filed by the NCI was formally transferred to Biovest in April 2004, which made Biovest the exclusive sponsor of the IND with full rights to complete the NCI-initiated Phase 3 clinical trial in FL and the NCI-initiated Phase 2 clinical trial in MCL, to communicate and negotiate with the FDA relating to marketing approval for BiovaxID and to conduct other clinical studies in NHL under the IND.
77
Table of Contents
BiovaxID Clinical Trials
Phase 2 Clinical Trial of BiovaxID for Treatment of Follicular Lymphoma (FL)
In 1994, the Phase 2 clinical trial was commenced by the NCI to evaluate the ability of BiovaxID to eradicate residual lymphoma cells in 20 patients with FL who were in chemotherapy-induced first clinical complete remission. All 11 patients with a detectable lymphoma gene sequence (translocation) in their primary tumors had cells from the malignant clone detectable in their blood by DNA polymerase chain reaction (PCR) analysis both at diagnosis and after chemotherapy, despite being in complete remission. In this clinical trial, molecular remission was defined as patients lacking any detectable residual cancer cells bearing the translocation as determined by a very sensitive PCR technique. After vaccination, 8 of 11 patients converted to lacking cells in their blood from the malignant lymphoma clone detectable by PCR. Anti-tumor T-cell responses were found in the vast majority of the patients (19 of 20 patients), whereas anti-tumor antibodies were detected, but apparently were not required for molecular remission. Vaccination was thus associated with clearance of residual tumor cells from the blood and long-term disease-free survival. The demonstration of molecular remissions and uniform, specific T-cell responses against lymphoma tumor targets, as well as the addition of granulocyte–monocyte colony-stimulating factor (“GM-CSF”) to the vaccine formulation provided the rationale for the initiation of a larger Phase 3 clinical trial at the NCI in 2000. These results were published in Nature Medicine (October 1999).
Phase 2 Clinical Trial of BiovaxID for Treatment of Mantle Cell Lymphoma (MCL)
In 2000, the NCI initiated a Phase 2 open-label clinical trial (NCT00020215) of BiovaxID for the treatment of MCL. This Phase 2 clinical trial was based upon the NCI’s Phase 2 clinical trial in FL. The primary objective of this Phase 2 clinical trial was to study BiovaxID in treatment-naïve patients with MCL and to determine the safety and efficacy of BiovaxID following a rituximab-based immunotherapy. Twenty-six patients with untreated, mostly (92%) stage IV MCL, were enrolled. All patients received six cycles of EPOCH-R (which is an chemo-immunotherapy consisting of etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab); 92% of the patients achieved complete response (“CR”) and 8% achieved partial response (“PR”). All but 3 patients (i.e., due to disease progression or inability to manufacture the vaccine) received BiovaxID together with KLH on day 1, along with GM-CSF (100 µg/m2/day) on days 1-4 at 1, 2, 3, 4, and 6 months starting at least 3 months post-chemotherapy.
The results of NCI’s MCL Phase 2 clinical trial were reported in Nature Medicine (August 2005). As reported in Nature Medicine, after a median follow-up of 46 months, the OS was 89%, the median event-free survival (“EFS”) was 22 months, and five patients remained in continuous first CR. Antibody responses to immunization were detected in 30% of the patients, following a delayed pattern (i.e., detected mostly after the 4-5th vaccination) which paralleled the peripheral blood B-cell recovery. Most importantly, specific CD4+ and CD8+ T-cell responses were detected in 87% of patients post-vaccine, and in 7 of 9 patients tested these responses were detected after the 3rd vaccination when peripheral B-cells were by and large undetectable. The detected cytokine release response included GM-CSF, INF-g, and TNF-a (type I). In this study, BiovaxID induced both humoral and cellular immune responses following almost complete depletion of B-cells following rituximab-containing chemotherapy. The adverse events observed in this trial were minimal and comparable to the toxicities observed in the FL studies. These were limited mostly to injection site reactions, similar to those reported in the Phase 2 and Phase 3 FL clinical trials.
78
Table of Contents
Phase 3 Clinical Trial of BiovaxID for Treatment of Follicular Lymphoma (FL)
Overview and Objectives. In January 2000, the Phase 3 clinical trial in FL was initiated by the NCI. The Phase 3 clinical trial was a multi-center, double-blind, randomized, controlled clinical trial that was designed to confirm the results reported in the NCI’s Phase 2 clinical trial.
As studied in the Phase 3 clinical trial, BiovaxID consisted of the patient-specific idiotype protein (“Id”) derived from the patient’s cancer cells conjugated or combined with keyhole limpet hemocyanin (KLH immunogenic carrier protein) and administered with granulocyte-monocyte colony stimulating factor (which is known as GM-CSF and which is a biological response enhancer). The comparator studied in the Phase 3 clinical trial was a control vaccine consisting of keyhole limpet hemocyanin (“KLH”) and administered with granulocyte-monocyte colony stimulating factor (“GM-CSF”; a biological response enhancer). Accordingly, the only difference between BiovaxID and the control vaccine was the inclusion of the idiotype protein from the patient’s own tumor in BiovaxID. BiovaxID or control vaccine were administered following chemotherapy (also referred to as induction therapy) with a drug combination referred to as PACE. Induction therapy represents the “first-line” treatment for FL patients and attempts to induce complete tumor remission as defined by radiological evidence (CT scans). In FL, patients treated with the current standard of care often achieve complete remission but these remissions almost always are of limited duration and most treated patients must eventually be re-treated for their disease. In the majority of cases, however, even with re-treatment, the disease often relapses and develops resistance to therapy, leading to a need for bone marrow transplant and eventually resulting in the death of the patient. In the Phase 3 clinical trial, patients who achieved complete response following induction therapy were assigned to a limited waiting period prior to vaccination to allow for immune reconstitution following the induction chemotherapy. Patients who relapsed during this immune reconstitution period did not receive either BiovaxID or control treatment. Patients who maintained their complete remission following this immune recovery period received either BiovaxID or control administered as five subcutaneous injections monthly over a six month period (one month was skipped).
The primary objectives of the Phase 3 clinical trial were to confirm the safety and efficacy of BiovaxID in two predefined groups:
| (1) | All Randomized Patients: all randomized patients (the “Randomized Patients”) including patients who completed initial chemotherapy but relapsed and did not receive either BiovaxID or control. |
| (2) | All Treated Patients: the Randomized Patients who were disease-free at the time of vaccination and consequently received at least one dose of BiovaxID or control. |
The secondary objectives of the Phase 3 clinical trial included: (1) to determine the ability of BiovaxID to produce a molecular complete response in subjects in clinical complete response, but with polymerase chain reaction (“PCR”) evidence of residual disease after standard chemotherapy; (2) to determine the impact of BiovaxID on molecular remission in FL patients; (3) to evaluate the ability of BiovaxID to generate an immune response against autologous tumor; (4) to determine and compare the overall survival (“OS”) of subjects randomized to receive either treatment assignment; and (5) to evaluate the safety of BiovaxID administered with GM-CSF.
79
Table of Contents
Biopsy, Chemotherapy, and Immune Recovery. Prior to chemotherapy, a small tumor biopsy was performed to obtain tissue for tumor classification and characterization, and to provide starting material necessary to manufacture BiovaxID. Following this biopsy patients were initially treated with PACE chemotherapy (a combination of prednisone, doxorubicin, cyclophosphamide, etoposide) in order to induce a complete response (“CR”) or a complete response unconfirmed (“Cru”) as measured by CT radiological scans.
The trial protocol stipulated that for all patients, an Immune Recovery Period of approximately 6 months following completion of chemotherapy was required to be completed without relapse before vaccination. The Immune Recovery Period was required in order to maximize the potential for immune response to vaccine and to avoid confounding factors from any potential lingering immunosuppressive effects of chemotherapy.
Randomization to Immune Recovery Followed by BiovaxID or Control. When the NCI designed the Phase 3 clinical trial protocol, a decision was made to randomize patients immediately after completion of chemotherapy and to not wait for the completion of the Immune Recovery Period in an effort to avoid expending NCI resources to manufacture patient-specific vaccines for patients who were not anticipated to receive the vaccine (e.g., control patients). In the Phase 3 clinical trial, of 234 patients initially enrolled into the clinical trial, 177 patients completed chemotherapy successfully and were randomized.
As per the design of the study, patients who relapsed during the Immune Recovery Period were excluded from treatment with BiovaxID or control notwithstanding the fact that they had been randomized. In the clinical trial, of the 177 initially randomized patients, 117 remained eligible to be treated with either BiovaxID (76 patients) or control (41 patients) at the end of the Immune Recovery Period. Sixty patients of the 177 randomized patients relapsed during the Immune Recovery Period and were not treated with either BiovaxID or control (Figure 2).
80
Table of Contents
Figure 2. Flow Diagram of Phase 3, double-blind, randomized clinical trial of patient-specific vaccination with BiovaxID + GM-CSF in first complete remission. 234 patients were enrolled at 14 centers and assessed for eligibility. Of those enrolled, 57 were excluded from randomization for reasons indicated. 177 patients were randomized (ITT population), of which 118 were allocated to the BiovaxID (Id-KLH + GM-CSF) arm (treatment) and 59 were allocated to the KLH + GM-CSF arm (control). Patients that failed to remain in CR/CRu (60 total) did not receive either vaccine. As a result, 76 patients were vaccinated with Id-KLH + GM-CSF and 41 were vaccinated with KLH + GM-CSF, comprising the modified ITT (mITT) population. Patients receiving less than 5 immunizations either withdrew from the study or relapsed before completion.
Trial Enrollment and the Use of Rituximab-Containing Induction Chemotherapy. During the course of the Phase 3 clinical trial, the standard of care for induction chemotherapy in FL changed to include rituximab, which reduced the ability to recruit and enroll patients into the study. In order to facilitate enrollment in the clinical trial, Biovest amended the study protocol in 2007 to permit the use of a rituximab-containing chemotherapy regimen (CHOP-R), as induction therapy. However, the FDA requested that Biovest abstain from vaccinating any patients who received CHOP-R and Biovest did not vaccinate any of the patients who received CHOP-R chemotherapy under the clinical trial protocol.
81
Table of Contents
Due to the protracted enrollment, the clinical trial’s Independent Data Monitoring Committee (“DMC”, a committee responsible for reviewing the available unblinded clinical trial data in the study and responsible for recommendations to the sponsor and the FDA) recommended an interim analysis of the clinical trial’s endpoints and overall safety profile which resulted in the termination and halting of the trial in 2008.
As of April 15, 2008, when the clinical trial was officially closed, a total of 234 subjects had been enrolled and 177 subjects had been randomized in the Phase 3 clinical trial, which was less than the original clinical trial plan which called for 629 subjects to be enrolled and 540 to be randomized. While the termination of the clinical trial before completion of the planned accrual resulted in a smaller sample size than was originally intended, Biovest believes that the randomized nature of Biovest’s clinical trial yields a valid conclusion because the baseline characteristics of the patients in the two groups were balanced, the allocation to treatment arms was concealed, and the study was double-blinded.
Results of Phase 3 Clinical Trial
As reported at the plenary session of the Annual Meeting of the American Society of Clinical Oncology (ASCO 2009, Orlando, FL), the patient cohort of the 177 All Randomized patients (which included 117 (66%) Treated Patients and 60 (35%) patients who were not treated) did not demonstrate statistically significant difference in median DFS from randomization between treatment and control arms.
82
Table of Contents
Figure 3. Disease-free survival (DFS) according to study group for all randomized patients (N = 177). Kaplan-Meier actuarial curves for DFS for all randomized patients are shown according to their study group of Id-KLH+GM-CSF (N = 118) or KLH+GM-CSF (N = 59). The number of events, median, and 95% confidence intervals for each group are also presented.
At ASCO, Biovest further reported the median DFS data for the patients who received at least one vaccination either with BiovaxID or control. In this cohort of 117 patients, which represents a modified intent-to-treat population, median DFS was 13.6 months longer in patients who received BiovaxID compared to patients who received control. This analysis reflects the prospectively defined primary clinical trial objective. Accordingly, there were 60 patients who were randomized but who did not receive either BiovaxID or control and who are not included in this analysis. Of these 117 treated patients, 76 patients received at least one dose of BiovaxID (referred to as the “BiovaxID Arm”) and 41 patients received at least one dose of control (referred to as the “Control Arm”). No serious adverse events were reported in either the BiovaxID Arm or the Control Arm. At the median follow-up of 56.6 months (range 12.6-89.3 months), a statistically significant improvement of 13.6 months was observed in DFS between patients in the BiovaxID Arm (44.2 months), versus the Control Arm (30.6 months) (log-rank p-value = 0.045; HR = 1.6). Using a Cox proportional-hazard model, a statistically significant hazard ratio (“HR”) of 0.62 was achieved (p=0.048; 95% CI: 0.39, 0.99). This means that patients receiving BiovaxID experienced an approximately 61% (1/0.62) lower risk of cancer recurrence compared to patients who received the control vaccine. The Phase 3 clinical trial’s secondary endpoint of overall survival (“OS”) has not yet been reached for either group due to the length of follow-up to date.
83
Table of Contents
Figure 4. Disease-free survival (DFS) according to study group for the randomized patients who received blinded vaccinations (N = 117). Kaplan-Meier actuarial curves for DFS for the randomized patients who received at least one dose of the Id-KLH+GM-CSF (N = 76) or KLH+GM-CSF (N = 41) are shown. The number of events, median, and 95% confidence intervals for each group are also presented.
84
Table of Contents
Analysis of Patients by Isotype. A typical antibody (immunoglobulin), including the lymphoma idiotype expressed on the surface of each cancerous lymphoma cell, is composed of protein “heavy chains” and “light chains”. In humans, the heavy chains are classified as IgG, IgM, IgA, IgD and IgE, and the light chains are classified as either kappa or lambda. The Id protein expressed on the surface of FL cells is an immunoglobulin protein characteristic of the single B-cell from which the tumor arose. The immunoglobulin protein contains a region known as the “heavy chain” and a region known as the “light chain” (Figure 5). Almost always in FL, the heavy chain region is characterized as either an IgM-isotype or an IgG-isotype. Figure 5 below illustrates the dramatic differences in the structure of immunoglobulin protein characterized as an IgM-isotype as opposed that characterized as an IgG-isotype. Accordingly, an antibody may be referred to as IgG-isotype or IgM-isotype depending on its heavy-chain classification. In the normal immune response, antibody isotypes may have different roles and may help direct the appropriate immune response. The small region at the tip of the antibody is known as the “variable region”, or antibody binding site, and the balance of the isotype is known as the “constant region”. When Biovest manufactures BiovaxID, Biovest screens each patient’s tumor cells obtained by biopsy for the isotype. Approximately 60% of patients with FL are diagnosed with tumors expressing an IgM isotype and approximately 40% of patients bear tumors expressing an IgG isotype. In rare cases (<1%), patients are diagnosed with another isotype (e.g. IgA). Infrequently, the patient’s tumor also contains cells with one or more isotype (a heterogenous or “mixed” isotype); in these patients we select either an IgG or IgM isotype for manufacture of BiovaxID. Each patient’s tumor isotype can be readily determined by standard analytical techniques (flow cytometry) at the time of the patient’s tumor biopsy. In both the Phase 2 and Phase 3 clinical trials, the determination of tumor heavy-chain isotype determined the specific manufacturing and purification process used to make that patient’s vaccine. For patients who have tumors expressing an IgG (or an IgG-containing “mixed” isotype), we manufacture an IgG isotype vaccine and for patients determined to have tumors expressing an IgM (or an IgM-containing “mixed” isotype), we manufacture an IgM vaccine. Due to Biovest’s manufacturing process (rescue fusion hybridoma), the isotype (IgG or IgM) of the tumor is directly reproduced in each patient’s vaccine so that each patient’s BiovaxID vaccine matches the patient’s original tumor isotype (IgG or IgM).
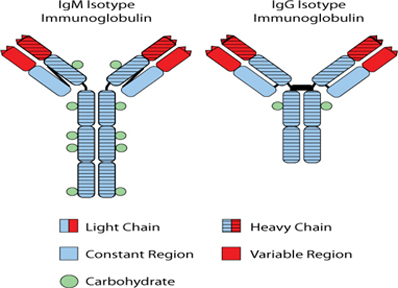
Figure 5. The Id protein expressed on the surface of FL cells is an immunoglobulin protein characteristic of the single B-cell from which the tumor arose. The immunoglobulin protein contains a region known as the “heavy chain” and a region known as the “light chain”. Almost always in FL, the heavy chain region is characterized as either an IgM-isotype or an IgG-isotype. The figure illustrates the dramatic differences in the structure of immunoglobulin protein characterized as an IgM-isotype as opposed that characterized as an IgG-isotype.
85
Table of Contents
Preclinical data indicates that the ability to develop an immune response differs between IgM-isotype and IgG-isotype idiotypes. The IgG-isotype idiotype was reported to be tolerogenic, meaning that the immune response against the specific tumor target is suppressed. On the other hand, the IgM-isotype idiotype was reported to be highly immunogenic, meaning that it induces an ample, persistent immune response against the specific tumor target. The unique feature of Biovest’s trial—the manufacturing and administration of tumor-matched isotype idiotype vaccines—allowed Biovest to investigate whether these preclinical data translate into differential clinical efficacy of the two isotype vaccines in Biovest’s clinical trial.
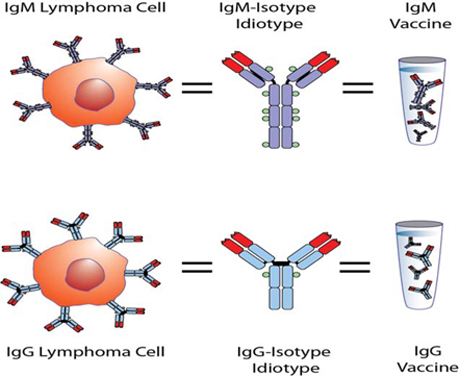
Figure 6. The vaccines produced for each individual patient consist of the tumor idiotype with the same isotype as the tumor cells from which the vaccine was produced. Therefore, patients with IgM isotype tumors received IgM vaccine, and patients with IgG isotype tumors received IgG vaccine manufactured from their own tumor cells.
Biovest analyzed differences in median DFS in vaccinated patients in our Phase 3 clinical trial separately by tumor isotype. There were 35 IgM isotype patients who received BiovaxID and 25 IgM isotype patients who received control. There were 40 IgG isotype patients who received BiovaxID and 15 IgG isotype patients who received control. Two patients had mixed IgM/IgG biopsy isotypes and were excluded from this analysis. The baseline characteristics of the patients who received either IgM or IgG vaccine were balanced between each respective BiovaxID and control group.
86
Table of Contents
In the IgM isotype group, Biovest observed that patients who were treated with isotype-matched BiovaxID had significantly longer DFS (52.9 months, versus 28.7 months) than patients with IgM isotype tumors who received control vaccine (Figure 7). In contrast, in the IgG isotype group, there was no difference in median DFS between the patients who received isotype-matched BiovaxID and the patients with IgG isotype tumors who received control vaccine (Figure 8). Although a separate manufacturing process is prescribed for the IgM isotype and for the IgG isotype, the clinical trial protocol did not include planned analyses to address a subset efficacy analysis by isotype.
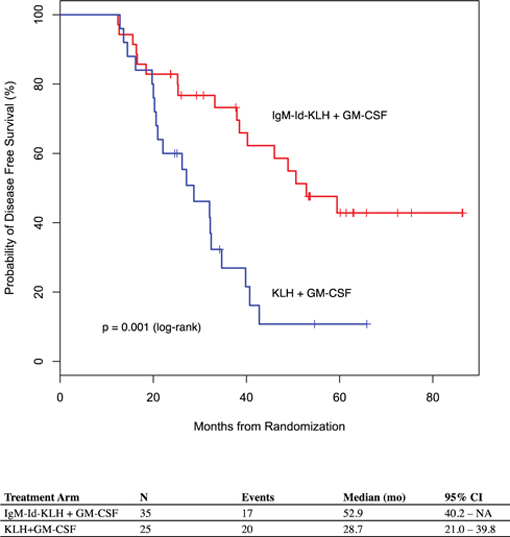
Figure 7. Disease-free survival (DFS) for the randomized patients with tumor IgM heavy chain isotype who received blinded vaccinations. Kaplan-Meier actuarial curves for DFS for the Id vaccinated [igM-id-KLH + GM-CSF] and control [KLH+GM-CSF] groups for the IgM isotype are shown. The number of events, median, and 95% confidence intervals for each group are also presented.
87
Table of Contents
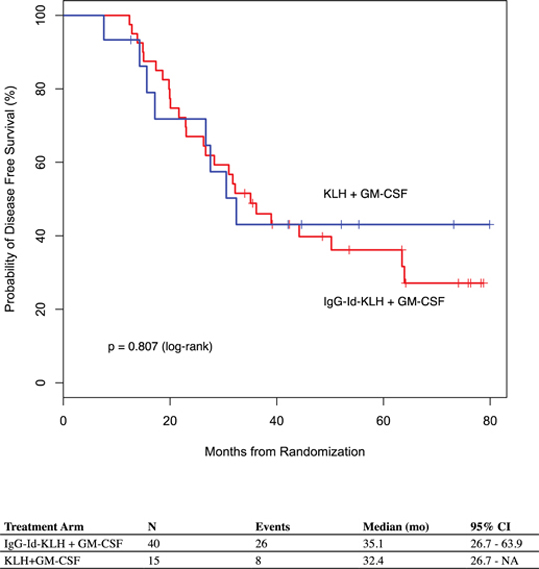
Figure 8. Disease-free survival (DFS) for the randomized patients with tumor IgG heavy chain isotype who received blinded vaccinations. Kaplan-Meier actuarial curves for DFS for the Id vaccinated [igM-id-KLH + GM-CSF] and control [KLH+GM-CSF] groups for the IgG isotype are shown. The number of events, median, and 95% confidence intervals for each group are also presented.
88
Table of Contents
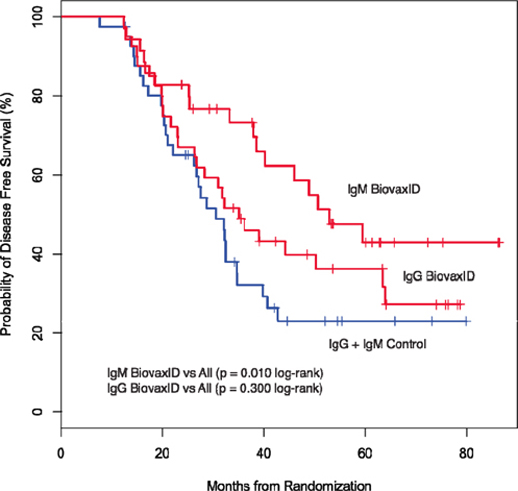
Figure 9. Shown is a standard statistical analysis (Kaplan-Meier survival curves) used to measure the fraction of patients free of disease for a certain amount of time after treatment. In this Figure, IgM-isotype patients who receive an IgM-isotype vaccine are compared with IgG-isotype patients who received an IgG-isotype vaccine compared with all control patients (patients with both IgM-isotype and IgG-isotype).
Biovest’s clinical trial has two unique manufacturing features, where (1) the vaccine consists of the full structure of the idiotype protein (that is, both the variable and the constant regions of the immunoglobulin), and (2) the idiotype of the vaccine matches the idiotype of the patient’s own tumor. These unique features allowed us to be the first to investigate the clinical efficacy implications of the two tumor isotypes. The prior Phase 3 clinical trials of FL idiotype vaccines conducted by Genitope Corporation and Favrille, Inc. used a manufacturing process known as recombinant manufacturing that universally linked the patient’s variable region of the idiotype into an IgG isotype without regard to the actual isotype of each patient’s tumor. Biovest believes that the use of an IgG isotype was due to the comparative ease of manufacture and purification of IgG proteins as well as to their relatively long half-life. There are two implications of the manufacturing processes used by these prior trials: (1) clinical efficacy cannot be compared by isotype group and (2) the lack of clinical efficacy observed in these trials may be due to the tolerogenic effect of the universal IgG isotype used in the vaccine manufacturing. As such, Biovest believes that its analysis by tumor isotype may provide profound insight into the efficacy of BiovaxID and may also suggest methods by which cancer vaccines in general could be developed in the future.
89
Table of Contents
BiovaxID ® Regulatory Status
Biovest is currently preparing to discuss with the FDA and potentially international regulatory agencies the next steps required to achieve potential marketing approval of BiovaxID. In preparation for these anticipated discussions, Biovest is continuing to analyze data available from the Phase 3 clinical trial so that Biovest can have as comprehensive as possible discussions regarding the safety and efficacy results of Biovest’s Phase 3 clinical trial. In addition, Biovest continues to advance its efforts to comply with various regulatory validation and comparability requirements related to its manufacturing process and facility. Biovest anticipates conducting additional regulatory discussions with the FDA in 2011.
Proprietary Rights to BiovaxID®
As a result of Orphan Drug designation for the treatment of FL and MCL, Biovest has seven (7) years of market exclusivity in the U.S. from the date of FDA marketing approval. Biovest has ten (10) years of market exclusivity in Europe as a result of Orphan Medicinal Product designation for the treatment of FL by the EMEA.
In addition to market exclusivity based on governmental regulation, Biovest relies on proprietary rights provided by a combination of an exclusive world-wide license to the cell line that is used in the production of BiovaxID, patent protection, trade secret protection, and Biovest’s ongoing innovation. Although the composition of matter of the BiovaxID vaccine is not patentable, Biovest has filed an international patent application (“PCT”) relating to methods of treatment using Biovest’s vaccine. In addition, Biovest has filed U.S. and foreign patent applications relating to certain features of the AutovaxID instrument used in the production of the vaccine. Biovest’s proprietary production system will use fully enclosed and disposable components for each patient’s vaccine. Biovest believes that, without the availability of an automated production system, the methods used to produce a patient-specific immunotherapy are time-consuming and labor-intensive, resulting in a very expensive process that would be difficult to scale up. Following the finds related to the apparent role of the IgM isotype in clinical benefit from vaccine, Biovest filed a broad range of patent applications covering various aspects of this finding. Biovest has been granted the registration of the trademark BiovaxID. BiovaxID is manufactured with a proprietary cell line, which Biovest has licensed on a world-wide exclusive basis from Stanford University (“Stanford”). This may be significant, because we believe that the use of any cell line other than Biovest’s exclusively licensed cell line, in the production of a similar idiotype vaccine would require filing a separate IND application and undergoing clinical testing evaluation by the FDA.
BiovaxID® Manufacturing Process and Facility
Manufacturing Process
The BiovaxID manufacturing production process begins when a sample of the patient’s tumor is extracted by a biopsy and the sample is shipped refrigerated to Biovest’s facility in Minneapolis, Minnesota. At Biovest’s facility, Biovest identifies the idiotype that is expressed on the surface of the patient’s tumor cells through laboratory analysis. Additionally, Biovest identifies whether the isotype is IgM or IgG. In NHL, the tumor B-cells bear the surface idiotype (immunoglobulin or antibody) derived from the original transformed malignant B-cell, but do not typically secrete it in an amount suitable for vaccine production. In order to make sufficient quantities of idiotype for vaccination, the patient’s tumor cells are then fused with an exclusively licensed cell line (mouse/human heterohybridoma cell line K6H6) from Stanford to create a hybridoma or hybrid cell.
90
Table of Contents
After the creation of the hybridoma, we determine which hybridoma cells display the same antigen idiotype as the patient’s tumor cells, and those cells are selected to produce the vaccine. The selected hybridoma cells are then seeded into our proprietary hollow-fiber bioreactors, where they are cultured and where they secrete or produce idiotype antigen. The secreted idiotype is then collected from the cells growing in the hollow-fiber reactor. After a sufficient amount of idiotype is collected for the production of an appropriate amount of the vaccine, the patient’s idiotype is purified using multi-step purification processes.
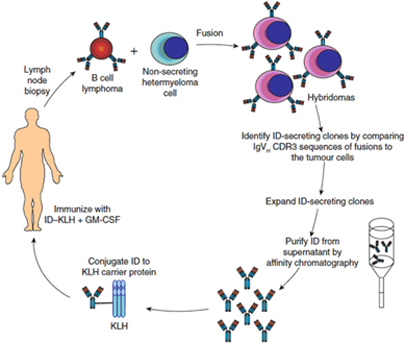
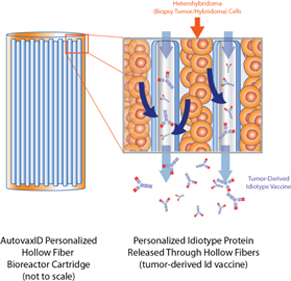
91
Table of Contents
Figure 10. Individualized Manufacturing Process for BiovaxID immunotherapy: (Clockwise) Beginning with an excisional (>2cm) lymph node biopsy, tumor cells are fused with our proprietary mouse/human heterohybridoma in order to induce secretion of normally surface-bound tumor immunoglobulin (idiotype). Id-secreting clones are identified by comparing their unique idiotype sequence to the tumor’s after which they are cultured (expanded) in a proprietary hollow-fiber bioreactor system (not shown). During culture, supernatant (containing idiotype) is collected until sufficient amounts have been produced to yield adequate dosage of vaccine. This supernatant is purified by affinity chromatography and conjugated (bonded) to KLH carrier protein, resulting in a finished vaccine that can be shipped and administered to patients. In the Phase 3 clinical trial, manufacturing success was approximately 95% of treated patients. (Fig. reprinted from Neelapu, et al. Exp. Opin Biol Ther 2007).
Biovest uses a method known as “hollow-fiber perfusion” to produce the cell cultures used in the manufacture of BiovaxID. Hollow-fiber perfusion, as compared to other cell culture methods, seeks to grow cells to higher densities more closely approaching the density of cells naturally occurring in body tissue. The hollow-fiber perfusion method involves using hair-like plastic fibers with hollow centers which are intended to simulate human capillaries. Thousands of these fibers are inserted in a cartridge, which Biovest refers to as a bioreactor. The cells are grown on the outside of the hollow fibers while nutrient media used to support cell growth is delivered through the hollow centers of the fibers. The fiber walls have small pores, allowing nutrients to pass from the hollow center to the cells. The fibers act as filters and yield concentrated secreted products. Because the cells are immobilized in the bioreactor, the concentrated product can be harvested during the ongoing cell growth process. Biovest believes that hollow-fiber technology permits the harvest of cell culture products with generally higher purities than stirred-tank fermentation, a common alternative cell culture method, thereby reducing the cost of purification as compared to stirred tank fermentation. Additionally, the technology associated with the hollow-fiber process generally minimizes the amount of costly nutrient media required for cell growth as opposed to other cell culturing techniques.
After manufacture and purification, the resulting purified idiotype is then conjugated, or joined together, with KLH, to create the vaccine. KLH is a foreign carrier protein that is used to improve the immunogenicity, or ability to evoke an immune response, of the tumor-specific idiotype. The vaccine is then frozen and shipped to the treating physician. At the treating physician’s office, the vaccine is thawed and injected into the patient.
The vaccine is administered in conjunction with granulocyte macrophage colony- stimulating factor (“GM-CSF”), a natural immune system growth factor that is administered with the idiotype vaccine to stimulate the immune system and increase the response to the idiotype vaccine. In the clinical trials patients were administered five monthly BiovaxID injections in the amount of 0.5 milligram of idiotype per injection, with the injections being given over a six-month period of time in which the fifth month is skipped. Through this process, the patient-specific idiotype is used to stimulate the patient’s immune system into targeting and destroying malignant B-cells bearing the same idiotype.
Biovest estimates that an average of three (3) months is required to manufacture each vaccine, which for most patients may overlap the time period when induction chemotherapy is being administered. While the manufacturing process for the BiovaxID vaccine is highly personalized to each patient Biovest considers it to be highly controlled and predictable. During the Phase 3 clinical trial, Biovest experienced approximately 95% success rate in manufacturing vaccines. The most common reason for a failure to successfully produce a patient’s vaccine were the presence of rare idiotype variants as opposed to the failure of a step in the manufacturing process.
92
Table of Contents
Manufacturing Facility
BiovaxID is a personalized medicine which is produced separately for each individual patient through a laboratory process based on the patient’s own tumor cells derived by biopsy. Following U.S. regulatory approval of BiovaxID, Biovest plans to initially produce BiovaxID in Biovest’s existing leasehold space located in Minneapolis, Minnesota. I n order to facilitate the regulatory process, Biovest is preparing a dedicated suite of laboratory clean rooms especially designed to produce BiovaxID. As the regulatory process advances toward completion, Biovest anticipates expanding its U.S. production capability, potentially by expanding its leasehold space and the dedicated manufacturing facility as required to meet anticipated commercial requirements. If BiovaxID receives regulatory approval in non-U.S. jurisdictions, Biovest anticipates establishing manufacturing capability, including appropriate laboratory space and Biovest’s proprietary AutovaxID instruments, which will be dedicated to serving lymphoma patients in that geographic area. Such facilities are anticipated to be operated either by Biovest or by contract parties. During the Phase 3 clinical trial, BiovaxID was produced at Biovest’s facility in Worcester, Massachusetts. Because Biovest has relocated the site of the manufacturing process to its Minneapolis facility following the clinical trials and because Biovest is expanding that facility, Biovest is currently in the process of attempting to demonstrate to the FDA that the product under these new conditions is comparable to the product that was the subject of earlier clinical testing. This requirement will also apply to future expansions of the manufacturing facility, such as the possible expansion to additional facilities that may be required for successful commercialization of the vaccine. There is also a requirement for validation of the manufacturing process for BiovaxID utilizing our AutovaxIDTM instrument. A showing of comparability requires data demonstrating that the product continues to be safe, pure, and potent and may be based on chemical, physical, and biological assays and, in some cases, other non-clinical data.
CONSULTING SERVICES - ANALYTICA INTERNATIONAL, INC.
Our wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), provides a broad range of consulting services to companies and institutions in the pharmaceutical, biotechnology, and medical markets, including some of the world’s largest pharmaceutical companies. Analytica provides these services to clients throughout the world, and we also utilize these services for our own product development efforts in order to, among other things, evaluate and analyze the market and potential pricing of our product candidates. Analytica’s development and commercialization services include outcomes research on the economic profiles of pharmaceuticals and biologics, pricing and market assessment on these products, and various services designed to expedite clinical trials. We also use these services to evaluate the payor reimbursement prospects of our products and to develop reimbursement strategies.
Analytica provides its commercialization and development services through a team of employees who are based in offices in New York and Germany. This team includes research professionals at the Master’s and Doctoral level in the fields of medicine, epidemiology, biochemistry, statistics, engineering, public health, pharmacy, health economics, and business administration.
93
Table of Contents
INSTRUMENTS AND DISPOSABLES
Our majority owned subsidiary, Biovest, sells hollow-fiber perfusion instruments used for the production of significant quantities of cell culture products. This product line includes:
AutovaxID™
AutovaxID is a fully automated, reusable instrument that employs a fully disposable, closed-system cell-growth chamber incorporating a hollow-fiber cell-growth cartridge. Since it is fully enclosed, computer controlled and automated, AutovaxID requires limited supervision and manpower to operate compared to manual instruments. AutovaxID is suitable for growing antibody-secreting cell lines, including hybridomas and Chinese hamster ovary (“CHO”) cells which are among the leading kinds of cell lines used for commercial therapeutic protein manufacture. We plan to utilize the AutovaxID technology to streamline commercial manufacture of our proprietary anti-cancer vaccine, BiovaxID. AutovaxID is the first cell culture system that enables production of personalized cell-based treatments economically and in compliance with U.S. Food and Drug Administration Good Manufacturing Practices (“GMPs”). In September 2009, Biovest entered into a contract with the U.S. Department of Defense (“DoD”) to further develop and modify the AutovaxID instrument including for purposes of applying this technology to applications of potential military interest, including research and production of antiviral vaccines such as those targeting influenzas.
Primer HF®
The Primer HF is a low cost hollow-fiber cell culture system capable of producing small quantities of monoclonal antibody. This system also provides a relatively inexpensive option to evaluate the efficacy of new cell lines in perfusion technology.
Mini MAX®
The miniMax provides the flexibility and technology needed to support optimization studies and research scale production of mammalian cell secreted proteins. This automated cell culture system is a table-top unit complete with microprocessor controller, self-contained incubator, and pump panel. The miniMax is an economical tool for researching scale-up processes and producing small quantities of protein of up to 10 grams per month.
Maximizer®
The Maximizer provides maximum flexibility to support optimization studies and pilot scale production of mammalian cell secreted proteins. This automated cell culture system is a table-top unit complete with validated microprocessor controller, self-contained incubator, and pump panel. With production rates up to one gram a day, the Maximizer is a tool for process development and production.
XCellerator™
The XCellerator is a self-standing floor system containing an incubator and refrigerator section, control fixtures and pump panel. Each Xcellerator supports two independent flowpaths, is controlled by a process control computer and has the capability of remote monitoring. The combined features of the XCellerator support production of 60-500 grams of protein per month, per XCellerator unit.
94
Table of Contents
In addition to instruments sales, Biovest has recurring revenue from the sale of hollow-fiber bioreactors, cultureware, tubing sets and other disposable products and supplies for use with Biovest’s instruments. Revenues from such disposable products represented approximately 36% and 45% of our total revenue from this business segment for the fiscal years ended September 30, 2010 and 2009 respectively.
Currently Biovest assembles, validates and packages the instruments and disposables which it sells. Customers for Biovest’s instruments and disposables are the same potential customers targeted for Biovest’s contract production services which include biopharmaceutical and biotech companies, medical schools, universities, research facilities, hospitals and public and private laboratories.
CELL CULTURE PRODUCTS AND SERVICES
Biovest manufactures mammalian cell culture products such as, whole cells, recombinant and secreted proteins, and monoclonal antibodies. Additionally, Biovest provides related services as a contract resource to assist customers in developing cell production process protocols, cell line optimization, cell culture production optimization, media evaluation and other related services. This segment of our business represented approximately $1.2 million (approximately 22%) and $1.5 million (approximately 41%) in revenues for the fiscal years ended September 30, 2010, and 2009 respectively.
Customers include biopharmaceutical and biotech companies, medical schools, universities, research facilities, hospitals and public and private laboratories. Biovest generally produce cell culture products pursuant to contracts which specify the customer’s requirements for the cell culture products to be produced or the services to be performed.
There are various processes commonly used to produce mammalian cells generally used in the production of antibodies. These may include hollow-fiber bioreactor perfusion, stirred tank fermentation, roller bottle and other processes. Biovest primarily uses hollow-fiber bioreactor technology to expand customer provided cell lines and produce the respective monoclonal antibodies. This technology grows cells to higher densities which more closely mimics mammalian physiology. Biovest has significant expertise with in vitro (outside the living body) cell culture methods for a wide variety of mammalian cells. Mammalian cells are complicated and dynamic, with constantly changing needs. A primary component of hollow-fiber bioreactors is fibers made of plastic polymers. The fibers are hair-like with hollow centers which simulate human capillaries. Thousands of these fibers are inserted in a cartridge, which we refer to as a bioreactor. The cells are grown on the outside of the hollow fibers while nutrient media used to support cell growth is perfused through the lumen of the fibers. The fiber walls have small pores, allowing nutrients to pass from the hollow center to the cells. The fibers act as filters and yield concentrated secreted products. Because the cells are immobilized in the bioreactor, the concentrated product can be harvested during the on-going cell growth process. Hollow-fiber technology permits harvests of cell culture products with generally higher purities thereby reducing the cost of downstream purification processes. This technology generally minimizes the amount of costly nutrient media required for cell growth.
95
Table of Contents
The most generally used process for mammalian cell production is stirred tank fermentation. Hollow-fiber bioreactor technology can be contrasted with the competitive stirred tank fermentation process which takes place in tanks of various sizes. Cells are grown inside the tanks in culture medium which is maintained under controlled conditions and continuously stirred to stimulate growth. At the end of the growing process, as opposed to incrementally during the growth process, cells are separated from the medium and the protein of interest is isolated through a series of complex purification processes. The size of the tanks generally result in stirred tank fermentation facilities requiring significantly more start-up costs, space and infrastructure than comparable production facilities using hollow-fiber technology. While stirred tank fermentation and hollow fiber technology are both used for cell production of various quantities, Biovest believes that the stirred tank fermentation process is currently more commonly used for larger scale commercial production requirements. Biovest believes that hollow-fiber technology has advantages in scalability, start-up time and cost in the early development of antibody production. In the expanding field of personalized medicine where patient specific drugs and therapeutics are frequently envisioned, such as the therapeutic vaccine which Biovest is developing, Biovest believes that hollow-fiber technology may be the appropriate cell culture production technology.
Competition
Biotechnology has experienced, and is expected to continue to experience, rapid and significant change. The use of monoclonal antibodies as initial or induction therapy, and increasingly for maintenance therapy, has become well-established and generally accepted. Products that are well-established or accepted, including monoclonal antibodies such as Rituxan®, may constitute significant barriers to market penetration and regulatory approval which may be expensive, difficult or even impossible to overcome. New developments in biotechnological processes are expected to continue at a rapid pace in both industry and academia, and these developments are likely to result in commercial applications competitive with our proposed vaccine. We expect to encounter intense competition from a number of companies that offer products in our targeted application area. We anticipate that our competitors in these areas will consist of both well-established and development-stage companies and will include:
| • | healthcare companies; |
| • | chemical and biotechnology companies; |
| • | biopharmaceutical companies; and |
| • | companies developing drug discovery technologies. |
We expect to compete on, among other things, the safety and efficacy of our products and more desirable treatment regimens, combined with the effectiveness of our experienced management team. Competing successfully will depend on our continued ability to attract and retain skilled and experienced personnel, to identify and secure the rights to and develop pharmaceutical products and compounds and to exploit these products and compounds commercially before others are able to develop competitive products.
COMPETITION FOR BIOVAXID
If approved, BiovaxID® will be required to compete with currently approved therapies, as well as therapies which may be approved in the future. One such approved therapy is rituximab, which is a monoclonal antibody manufactured by XX under the brand name XX. This therapy, which is considered part of the standard of care, is discussed under the section captioned “BIOVAXID-THERAPEUTIC CANCER VACCINE- Non-Hodgkin’s Lymphoma (“NHL”)-Follicular Lymphoma”. There are currently no approved active immunotherapeutic drugs which seek to induce an adaptive, specific and durable immune response to identify and eradicate the residual lymphoma cells remaining after a patient achieves remission in an effort to extend that remission or avoid relapse. BiovaxID is a therapy designed to be administered to lymphoma patients who have achieved complete remission after initial chemotherapy treatment. If approved, BiovaxID would represent a new class of drugs available to treat FL potentially offering a new treatment option for FL patients.
96
Table of Contents
BiovaxID is the only personalized cancer vaccine for treatment of FL that has demonstrated significant clinical benefit in a Phase 3 clinical trial. Two other vaccines (MyVaxTM developed by Genitope Corporation and Specifid™ developed by Favrille, Inc.) which were studied in Phase 3 trials in FL patients did not report statistically significant clinical benefit and Biovest believes are no longer under development. There are fundamental structural differences between BiovaxID and the personalized cancer vaccines developed by Genitope Corporation and Favrille, Inc.; Genitope and Favrille manufactured their respective vaccines with IgG isotypes without regard to the patient’s actual isotype and the clinical trial designs under which the clinical efficacy of these vaccines were tested were different, which Biovest believes explain why BiovaxID achieved significant clinical benefit while the other vaccines did not.
Chemotherapy and monoclonal antibodies are widely used for the treatment of FL. Although chemotherapy and monoclonal antibodies can substantially reduce the tumor mass and in most instances achieve clinical remission, the remission is generally of limited duration. FL patients generally relapse and the cancer usually becomes increasingly resistant to further chemotherapy treatments. The patient’s response to therapy becomes briefer and weaker with each additional course of therapy, that eventually further chemotherapy would offer no clinical benefit.
A number of passive immunotherapies, such as rituximab and radioimmunotherapeutic agents (radioisotopes linked to monoclonal antibodies), are approved by the FDA for the treatment of FL. A monoclonal antibody is a type of antibody produced in large quantity that is specific to an antigen that is expressed by tumor cells but may also be expressed by at least some normal cells. These therapies have been used as primary treatment and also as part of combination induction therapy including chemotherapy and rituximab based therapy is considered to be the standard of care to treat FL. In an effort to prolong the duration of the clinical remission monoclonal antibodies have increasingly been used as maintenance therapies; however, such treatments are “off-label” and no maintenance therapy is currently FDA approved to treat FL. Supplemental marketing applications have been filed with the FDA and EMEA in 2010 to expand the indication for rituximab to include its use as a maintenance therapy for NHL, and it is anticipated that such approvals will be granted. However, the prolonged use of rituximab results in a substantial proportion of patients becoming non-responsive to rituximab-based therapy over time.
If approved to treat FL, BiovaxID will face competition from the then current standard of care recognized to treat FL. Today, the standard of care to treat FL generally includes therapies that contain rituximab. Penetrating a market and achieving usage by physicians and patients in the face of an established standard of care is anticipated to represent a significant challenge.
If BiovaxID is approved for treatment of MCL, it will be required to compete with other approved and/or development therapies for the treatment of MCL. There are currently no FDA-approved therapies for the first line treatment of MCL and the only approved therapy, bortezomib (Velcade®) is indicated for patients in relapse.
97
Table of Contents
Government Regulation
Government authorities in the U.S. at the federal, state, and local levels and foreign countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing, and import and export of pharmaceutical products, biologics, and medical devices. All of our products in development will require regulatory approval by government agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical trials and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal, state, local, and foreign statutes and regulations also govern testing, manufacturing, safety, labeling, storage, and record-keeping related to such products and their marketing. The process of obtaining these approvals and the subsequent process of maintaining substantial compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. In addition, statutes, rules, regulations, and policies may change and new legislation or regulations may be issued that could delay such approvals.
PHARMACEUTICAL PRODUCT REGULATION
In the U.S., the FDA regulates drugs and well-characterized biologics under the Federal Food, Drug, and Cosmetic Act (“FDCA”), and implementing regulations that are adopted under the FDCA. I n the case of biologics, the FDA regulates such products under the Public Health Service Act. If we fail, to comply with the applicable requirements under these laws and regulations at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of its operations, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us. The FDA also administers certain controls over the export of drugs and biologics from the U.S.
Under the U.S. regulatory scheme, the development process for new pharmaceutical products can be divided into three distinct phases:
| • | Preclinical Phase. The preclinical phase involves the discovery, characterization, product formulation and animal testing necessary to prepare an IND, for submission to the FDA. The IND must be accepted by the FDA before the drug can be tested in humans. |
| • | Clinical Phase. The clinical phase of development follows a successful IND submission and involves the activities necessary to demonstrate the safety, tolerability, efficacy, and dosage of the substance in humans, as well as the ability to produce the substance in accordance with the FDA’s current Good Manufacturing Processes (“cGMP”) requirements. Data from these activities are compiled in a New Drug Application (“NDA”) or for biologic products a Biologics License Application (“BLA”), for submission to the FDA requesting approval to market the drug. |
98
Table of Contents
| • | Post-Approval Phase. The post-approval phase follows FDA approval of the NDA or BLA, and involves the production and continued analytical and clinical monitoring of the product. The post- approval phase may also involve the development and regulatory approval of product modifications and line extensions, including improved dosage forms, of the approved product, as well as for generic versions of the approved drug, as the product approaches expiration of patent or other exclusivity protection. |
Each of these three phases is discussed further below.
Preclinical Phase
The development of a new pharmaceutical agent begins with the discovery or synthesis of a new molecule or well-characterized biologic. These agents are screened for pharmacological activity using various animal and tissue models, with the goal of selecting a lead agent for further development. Additional studies are conducted to confirm pharmacological activity, to generate safety data, and to evaluate prototype dosage forms for appropriate release and activity characteristics. Once the pharmaceutically active molecule is fully characterized, an initial purity profile of the agent is established. During this and subsequent stages of development, the agent is analyzed to confirm the integrity and quality of material produced. In addition, development and optimization of the initial dosage forms to be used in clinical trials are completed, together with analytical models to determine product stability and degradation. A bulk supply of the active ingredient to support the necessary dosing in initial clinical trials must be secured. Upon successful completion of preclinical safety and efficacy studies in animals, an IND submission is prepared and provided to the FDA for review prior to commencement of human clinical trials. The IND consists of the initial chemistry, analytical, formulation, and animal testing data generated during the preclinical phase. In general, the review period for an IND submission is 30 days, after which, if no comments are made by the FDA, the product candidate can be studied in Phase 1 clinical trials.
The process for the development of biologic products, such as our BiovaxID® product, parallels the process outlined above. Biologics, in contrast to drugs that are chemically synthesized, are derived from living sources, such as humans, animals, and microorganisms. Most biologics are complex mixtures that are not easily identified or characterized and have activity that is different from the activity of small, organic molecules normally found in drugs. Because of the diversity of the nature of biologic products and their substantial molecular size (usually hundreds of times larger than small, organic molecules associated with drugs), special technology is often required for their production and subsequent analysis. Biologic products, especially proteins, may be produced with living cells. Purity testing of biologics can be complex since living cells may harbor viruses and other agents. The potential presence of these agents and the requirement to establish degradation profiles and identify impurities associated with production and purification, further require establishing, validating, and conducting specialized tests and analyses. Formulation development in this area is often more complex than for small, organic drug substances. For example, molecules produced using recombinant DNA technology, are inherently less stable than their organic counterparts because structural integrity must be maintained through administration and distribution of the product. Accordingly, certain aspects of the development process for biologic products may be more challenging than similar aspects encountered in the development of drugs.
99
Table of Contents
Clinical Phase
Following successful submission of an IND, the sponsor is permitted to conduct clinical trials involving the administration of the investigational product candidate to human subjects under the supervision of qualified investigators in accordance with good clinical practice. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study and the parameters to be used in assessing the safety and the efficacy of the drug. Each protocol must be submitted to the FDA as part of the IND prior to beginning the trial. Each trial must be reviewed, approved and conducted under the auspices of an independent Institutional Review Board, and each trial, with limited exceptions, must include the patient’s informed consent. Typically, clinical evaluation involves the following time-consuming and costly three-phase sequential process:
| • | Phase 1. Phase 1 human clinical trials are conducted in a limited number of healthy individuals to determine the drug’s safety and tolerability and includes biological analyses to determine the availability and metabolization of the active ingredient following administration. The total number of subjects and patients included in Phase 1 clinical trials varies, but is generally in the range of 20 to 80 people. |
| • | Phase 2. Phase 2 clinical trials involve administering the drug to individuals who suffer from the target disease or condition to determine the drug’s potential efficacy and ideal dose. These clinical trials are typically well controlled, closely monitored, and conducted in a relatively small number of patients, usually involving no more than several hundred subjects. These trials require scale up for manufacture of increasingly larger batches of bulk chemical. These batches require validation analysis to confirm the consistent composition of the product. |
| • | Phase 3. Phase 3 clinical trials are performed after preliminary evidence suggesting effectiveness of a drug has been obtained and safety (toxicity), tolerability, and an ideal dosing regimen have been established. Phase 3 clinical trials are intended to gather additional information about the effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug and to complete the information needed to provide adequate instructions for the use of the drug, also referred to as the Official Product Information. Phase 3 trials usually include from several hundred to several thousand subjects |
Throughout the clinical phase, samples of the product made in different batches are tested for stability to establish shelf life constraints. In addition, large-scale production protocols and written standard operating procedures for each aspect of commercial manufacture and testing must be developed.
Phase 1, 2, and 3 testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request additional clinical trials be conducted as a condition to product approval. Additionally, new government requirements may be established that could delay or prevent regulatory approval of our products under development. Furthermore, institutional review boards, which are independent entities constituted to protect human subjects in the institutions in which clinical trials are being conducted, have the authority to suspend clinical trials at any time for a variety of reasons, including safety issues.
100
Table of Contents
New Drug Application (“NDA”) or Biologics License Application (“BLA”)
After the successful completion of Phase 3 clinical trials, the sponsor of the new drug submits an NDA or BLA in the case of biologics, to the FDA requesting approval to market the product for one or more indications. An NDA, or BLA, is a comprehensive, multi-volume application that includes, among other things, the results of all preclinical and clinical studies, information about the drug’s composition, and the sponsor’s plans for producing, packaging, and labeling the drug. Under the Pediatric Research Equity Act of 2003, an application also is required to include an assessment, generally based on clinical study data, on the safety and efficacy of drugs for all relevant pediatric populations before the NDA is submitted. The statute provides for waivers or deferrals in certain situations. I n most cases, the NDA or BLA must be accompanied by a substantial user fee. In return, the FDA assigns a goal of 10 months from acceptance of the application to return of a first “complete response,” in which the FDA may approve the product or request additional information.
The submission of the application is no guarantee that the FDA will find it complete and accept it for filing. The FDA reviews all NDAs and BLAs submitted before it accepts them for filing. It may refuse to file the application and request additional information rather than accept the application for filing, in which case, the application must be resubmitted with the supplemental information. After application is deemed filed by the FDA, the FDA reviews an NDA or BLA to determine, among other things, whether a product is safe and effective for its intended use. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA or BLA. Drugs that successfully complete NDA or BLA review may be marketed in the U.S., subject to all conditions imposed by the FDA. Prior to granting approval, the FDA generally conducts an inspection of the facilities, including outsourced facilities, which will be involved in the manufacture, production, packaging, testing and control of the drug product for cGMP compliance. The FDA will not approve the application unless cGMP compliance is satisfactory. If the FDA determines that the marketing application, manufacturing process, or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the marketing application does not satisfy the regulatory criteria for approval and refuse to approve the application by issuing a “not approvable” letter.
The length of the FDA’s review ranges from a few months to many years.
Post-Approval Phase
If the FDA approves the NDA, BLA, or abbreviated new drug application (“ANDA”) application, as applicable, the pharmaceutical product becomes available for physicians to prescribe in the U.S. After approval, we are still subject to continuing regulation by FDA, including record keeping requirements, submitting periodic reports to the FDA, reporting of any adverse experiences with the product, and complying with drug sampling and distribution requirements. In addition, we are required to maintain and provide updated safety and efficacy information to the FDA. We are also required to comply with requirements concerning advertising and promotional labeling. In that regard, our advertising and promotional materials must be truthful and not misleading. We are also prohibited from promoting any non-FDA approved or “off-label” indications of products. Failure to comply with those requirements could result in significant enforcement action by the FDA, including warning letters, orders to pull the promotional materials, and substantial fines. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval.
101
Table of Contents
Drug and biologics manufacturers and their subcontractors are required to register their facilities and products manufactured annually with FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA to assess compliance with cGMP regulations. Facilities may also be subject to inspections by other federal, foreign, state, or local agencies. In addition, approved biological drug products may be subject to lot-by-lot release testing by the FDA before these products can be commercially distributed. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. We use, and will continue to use, third-party manufacturers, to produce certain of our products in clinical and commercial quantities, and future FDA inspections may identify compliance issues at its facilities or at the facilities of its contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
In addition, following FDA approval of a product, discovery of problems with a product or the failure to comply with requirements may result in restrictions on a product, manufacturer, or holder of an approved marketing application, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications. Also, the FDA may require post-market testing and surveillance to monitor the product’s safety or efficacy, including additional clinical studies, known as Phase 4 clinical trials, to evaluate long-term effects.
Orphan Drug Designation and Exclusivity
Some jurisdictions, including the U.S. and the EU, designate drugs intended for relatively small patient populations as “orphan drugs.” The FDA, for example, grants orphan drug designation to drugs intended to treat rare diseases or conditions that affect fewer than 200,000 individuals in the U.S. or drugs for which there is no reasonable expectation that the cost of developing and making the drugs available in the U.S. will be recovered. In the U.S. orphan drug designation must be requested before submitting an application for approval of the product.
Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to a marketing exclusivity. For seven (7) years, the FDA may not approve any other application, including NDAs or ANDAs, to market the “same drug” for the same indication. The only exceptions are (i) where the second product is shown to be “clinically superior” to the product with orphan drug exclusivity, as that phrase is defined by the FDA and (ii) if there is an inadequate supply.
MANUFACTURING
With regard to Biovest’s development of BiovaxID, Biovest is currently undertaking the construction of a new manufacturing suite for BiovaxID to be located within Biovest’s existing facility in Minneapolis, Minnesota. If it receives FDA approval of the vaccine, Biovest may continue to manufacture the vaccine at its facility in Minnesota, although Biovest will likely need to expand this existing facility and/or develop additional facilities to fully support commercial production for the U.S. markets. Changes to the manufacturing process or site during or following the completion of clinical trials requires sponsors to demonstrate to the FDA that the product under new conditions is comparable to the product that was the subject of earlier clinical testing. This requirement applies to relocations or expansions of manufacturing facilities, such as Biovest’s relocation of the BiovaxID production process and planned expansion of such facilities or additional facilities that may be required upon successful commercialization of the vaccine. A showing of comparability requires data demonstrating that the product continues to be safe, pure, and potent and may be based on chemical, physical, and biological assays and, in some cases, other non-clinical data. If Biovest demonstrates comparability, additional clinical safety and/or efficacy trials with the new product may not be needed. If the FDA requires additional clinical safety or efficacy trials to demonstrate comparability, its clinical trials or the FDA approval of BiovaxID may be delayed. Additionally, if BiovaxID receives regulatory approval in any non-U.S. jurisdictions, Biovest anticipates that it would be required to establish manufacturing capability, including appropriate laboratory space incorporating Biovest’s proprietary AutovaxID instruments, that would be dedicated to serving lymphoma patients in that geographic area. Such facilities, which could potentially be operated by Biovest or by contract parties, are anticipated to require approval from the applicable jurisdiction as part of the approval process for BiovaxID and would be regulated by that jurisdiction.
102
Table of Contents
We anticipate that sufficient supplies of Cyclophosphamide to be used in our planned Revimmune product will be manufactured by Baxter Healthcare Corporation pursuant to our exclusive agreement.
MEDICAL DEVICE REGULATION
New medical devices are also subject to FDA approval and extensive regulation under the FDCA. Under the FDCA, medical devices are classified into one of three classes: Class I, Class II, or Class III. The classification of a device into one of these three classes generally depends on the degree of risk associated with the medical device and the extent of control needed to ensure safety and effectiveness.
Class I devices are those for which safety and effectiveness can be assured by adherence to a set of general controls. These general controls include compliance with the applicable portions of the FDA’s Quality System Regulation, which sets forth good manufacturing practice requirements; facility registration and product reporting of adverse medical events listing; truthful and non-misleading labeling; and promotion of the device only for its cleared or approved intended uses. Class II devices are also subject to these general controls, and any other special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. Review and clearance by the FDA for these devices is typically accomplished through the so-called 510(k) pre-market notification procedure.
When 510(k) clearance is sought, a sponsor must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to a previously approved device. If the FDA agrees that the proposed device is substantially equivalent to the predicate device, then 510(k) clearance to market will be granted. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require pre-market approval. Our instruments and disposables used for the production of cell cultures are generally regulated as Class I devices exempt from the 510(k) clearance process.
Clinical trials are almost always required to support a PMA application and are sometimes required for a 510(k) pre-market notification. These trials generally require submission of an application for an investigational device exemption, or IDE. An IDE must be supported by pre-clinical data, such as animal and laboratory testing results, which show that the device is safe to test in humans and that the study protocols are scientifically sound. The IDE must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and is eligible for more abbreviated investigational device exemption requirements.
103
Table of Contents
Both before and after a medical device is commercially distributed, manufacturers and marketers of the device have ongoing responsibilities under FDA regulations. The FDA reviews design and manufacturing practices, labeling and record keeping, and manufacturers’ required reports of adverse experiences and other information to identify potential problems with marketed medical devices. Device manufacturers are subject to periodic and unannounced inspection by the FDA for compliance with the Quality System Regulation, current good manufacturing practice requirements that govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, servicing, labeling, storage, installation, and distribution of all finished medical devices intended for human use.
If the FDA finds that a manufacturer has failed to comply or that a medical device is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, ranging from a public warning letter to more severe actions such as:
| • | fines, injunctions, and civil penalties; |
| • | recall or seizure of products; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | refusing requests for 510(k) clearance or PMA approval of new products; |
| • | withdrawing 510(k) clearance or PMA approvals already granted; and |
| • | criminal prosecution. |
The FDA also has the authority to require repair, replacement or refund of the cost of any medical device.
The FDA also administers certain controls over the export of medical devices from the U.S., as international sales of medical devices that have not received FDA approval are subject to FDA export requirements. Additionally, each foreign country subjects such medical devices to its own regulatory requirements. In the EU, a single regulatory approval process has been created, and approval is represented by the CE Mark.
OTHER REGULATION IN THE UNITED STATES
The Biologics Price Competition and Innovation Act (2010)
The Biologics Price Competition and Innovation Act (2010) establishes an abbreviated approval pathway for “biosimilar” biological products. Among the provisions potentially applicable to the Company’s products are: (1) innovator manufacturers of reference biological products (such as BiovaxID) are granted 12 years of exclusive use before biosimilars can be approved for marketing in the U.S.; and (2) an application for a biosimilar product may not be submitted to the FDA until 4 years after the date on which the BLA for the reference product was first approved. The FDA is still early in the process of developing regulations to implement the provisions of this legislation.
104
Table of Contents
Toxic Substances Control Act
The Environmental Protection Agency, or “EPA,” has promulgated regulations under Section 5 of the Toxic Substances Control Act, or “TSCA,” which require notification procedures for review of certain so-called Intergeneric microorganisms before they are introduced into commerce. Intergeneric microorganisms are those formed by deliberate combinations of genetic material from organisms classified in different taxonomic genera, which are types of animal or plant groups. The regulations provide exemptions from the reporting requirements for new microorganisms used for research and development when the researcher or institution is in mandatory compliance with the National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules, or “NIH Guidelines.” Those researchers voluntarily following the NIH Guidelines can, by documenting their use of the NIH Guidelines, satisfy EPA’s requirements for testing in contained structures. The EPA may enforce the TSCA through enforcement actions such as seizing noncompliant substances, seeking injunctive relief, and assessing civil or criminal penalties. We believe that its research and development activities involving intergeneric microorganisms comply with the TSCA, but there can be no assurance that restrictions, fines or penalties will not be imposed on us in the future.
Health Care Coverage and Reimbursement
Commercial success in marketing and selling our products depends, in part, on the availability of adequate coverage and reimbursement from third-party health care payers, such as government and private health insurers and managed care organizations. Third-party payers are increasingly challenging the pricing of medical products and services. Government and private sector initiatives to limit the growth of health care costs, including price regulation, competitive pricing, coverage and payment policies, and managed-care arrangements, are continuing in many countries where we do business, including the U.S. These changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective medical products.
Government programs, including Medicare and Medicaid, private health care insurance and managed-care plans have attempted to control costs by limiting the amount of reimbursement they will pay for particular procedures or treatments. This has created an increasing level of price sensitivity among customers for our products. Examples of how limits on drug coverage and reimbursement in the United States may cause drug price sensitivity include the growth of managed care, changing Medicare reimbursement methodologies, and drug rebates and price controls. Some third-party payors must also approve coverage for new or innovative devices or therapies before they will reimburse health care providers who use the medical devices or therapies. Even though a new medical product may have been cleared for commercial distribution, we may find limited demand for the product until reimbursement approval has been obtained from governmental and private third-party payors.
Anti-Kickback Laws
In the United States, there are federal and state anti-kickback laws that prohibit the payment or receipt of kickbacks, bribes or other remuneration to induce the purchase, order or recommendation of health care products and services. These laws constrain the sales, marketing and other promotional activities of pharmaceutical companies, such as us, by limiting the kinds of financial arrangements (including sales programs). We may have with prescribers, purchasers, dispensers and users of drugs and biologics. The HHS Office of Inspector General (“OIG”) has issued Compliance Guidance for pharmaceutical manufacturers which, among other things, identifies manufacturer practices implicating the federal anti-kickback law (42 U.S.C. § 1320a-7b(b)) and describes elements of an effective compliance program. The OIG Compliance Guidance is voluntary, and we have not adopted a formal compliance program modeled after the one described in the OIG guidance. Although none of our practices have been subject to challenge under any anti-kickback laws, due to the breadth of the statutory provisions of some of these laws, it is possible that some of our practices might be challenged under one or more of these laws in the future. Violations of these laws can lead to civil and criminal penalties, including imprisonment, fines and exclusion from participation in federal health care programs. Any such violations could have a material adverse effect on our business, financial condition, results of operations or cash flows.
105
Table of Contents
Health Information Privacy and Security
Individually identifiable health information is subject to an array of federal and state regulation. Federal rules promulgated pursuant to the Health Information Portability and Accountability Act of 1996 (“HIPAA”) regulate the use and disclosure of health information by “covered entities” (which includes individual and institutional providers from which we may receive individually identifiable health information). These regulations govern, among other things, the use and disclosure of health information for research purposes, and require the covered entity to obtain the written authorization of the individual before using or disclosing health information for research. Failure of the covered entity to obtain such authorization (absent obtaining a waiver of the authorization requirement from an Institutional Review Board) could subject the covered entity to civil and criminal penalties. As the implementation of this regulation is still in its early phases, we may experience delays and complex negotiations as we deal with each entity’s differing interpretation of the regulations and what is required for compliance. Further, HIPAA’s criminal provisions are not limited in their applicability to “covered persons,” but apply to any “person” that knowingly and in violation of the statute obtains or discloses individually identifiable health information. Also, where its customers or contractors are covered entities, including hospitals, universities, physicians or clinics, we may be required by the HIPAA regulations to enter into “business associate” agreements that subject us to certain privacy and security requirements, including making its books and records available for audit and inspection by HHS and implementing certain health information privacy and security safeguards. In addition, many states have laws that apply to the use and disclosure of health information, and these laws could also affect the manner in which we conduct its research and other aspects of its business. Such state laws are not preempted by the federal privacy law where they afford greater privacy protection to the individual. While activities to assure compliance with health information privacy laws are a routine business practice, we are unable to predict the extent to which its resources may be diverted in the event of an investigation or enforcement action with respect to such laws.
FOREIGN REGULATION
Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement also vary greatly from country to country. Although governed by the applicable country, clinical trials conducted outside of the United States typically are administered under a three-phase sequential process similar to that discussed above for pharmaceutical products. Clinical trials conducted in the EU must comply with the EU Clinical Trials Directive.
106
Table of Contents
Under EU regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure for most products. The centralized procedure, which is available for medicines produced by biotechnology or which are highly innovative, provides for the grant of a single marketing authorization that is valid for all EU member states. Under European Commission Regulation 726/2004, the centralized authorization procedure is required for all biotechnology-derived medicinal products developed through recombinant DNA technology, controlled expression of genes coding for biologically active proteins, and hybridoma and monoclonal antibody methods. It is also required for designated orphan medicinal products and all new active substances indicated for the treatment of AIDS, cancer, neurodegenerative disorder, or diabetes. This authorization is a marketing authorization approval, or “MAA.” The decentralized procedure provides for mutual recognition of national regulatory authority approval decisions. Under this procedure, the holder of a national marketing authorization granted by one member state may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval.
This procedure is referred to as the mutual recognition procedure, or “MRP.”
In addition, regulatory approval of prices is required in most countries other than the U.S. We face the risk that the prices which result from the regulatory approval process would be insufficient to generate an acceptable return to us or our collaborators.
Intellectual Property
We are pursuing a number of methods to establish and maintain market exclusivity for our product candidates to the greatest extent possible, including seeking patent protection, the use of statutory market exclusivity provisions, and otherwise protecting our intellectual property.
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology, and know-how; to operate without infringing the proprietary rights of others; and to prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications when possible relating to our proprietary technology, inventions, and improvements that are important to our business. We also rely on trade secrets, know-how, continuing technological innovation, and in-licensing opportunities to develop and maintain our proprietary position.
The following is information regarding Biovest’s owned and licensed patents and patent applications that we consider material to its business.
Biovest owns several patents covering various aspects of its hollow fiber perfusion process, instruments and proprietary cell culturing methods. The patents also cover aspects of Biovest’s therapeutic vaccine production process. Biovest plans to continue pursuing patent and other proprietary protection for its cancer vaccine technology and instrumentation. Currently, Biovest has four (4) issued U.S. patents. Additionally, Biovest has filed several U.S. and foreign patent applications that are pending. The expiration dates of Biovest’s presently issued United States patents range from October 2011 to November 2017.
Biovest has recently filed a number of provisional patent applications asserting claims largely based on or related to various aspects of Biovest’s analysis of clinical benefit based on isotype.
107
Table of Contents
Biovest also possesses licensed intellectual property used in the development and manufacture of BiovaxID. BiovaxID is manufactured with a proprietary cell line, which we have licensed on a world-wide exclusive basis from Stanford University. This is significant, because Biovest believes that the use of any cell line other than Biovest’s exclusively licensed cell line, in the production of a similar idiotype vaccine, would require filing a separate IND application and undergoing clinical testing evaluation by the FDA.
Additionally, Biovest considers trademarks to be important to its business. Biovest has established trademarks covering various aspects of its hollow fiber perfusion process, instruments and proprietary cell culturing methods. Biovest has registered the trademark BiovaxID® in connection with its therapeutic cancer vaccine. Biovest plans to continue aggressively pursuing trademark and other proprietary protection for Biovest’s therapeutic vaccine technology and instrumentation, including seeking protection of its trademarks internationally.
A list of published U.S. and foreign patent applications within the Revimmune portfolio, which are licensed or wholly or jointly owned by us are as follows:
| Publication No.
|
Title and Inventor(s)
|
Filing Date/Publication
|
Countries/Regions
| |||
| US2007/0202077 | USE OF HIGH-DOSE OXAZAPHOSPHORINE DRUGS FOR TREATING IMMUNE DISORDERS by Robert A. Brodsky et al. | Dec. 2, 2006/Aug. 30, 2007 | United States | |||
| WO2007/065167 | USE OF HIGH-DOSE OXAZAPHOSPHORINE DRUGS FOR TREATING IMMUNE DISORDERS by Robert A. Brodsky et al. | Dec. 2, 2006/June 7, 2007 | Australia, Canada, Europe, Mexico | |||
| WO2008/034071 | METHOD OF IDENTIFYING PATIENTS SUITALE FOR HIGH-DOSE CYCLOPHOSPHAMIDE TREATMENT by Robert A. Brodsky et al. | Sept. 14, 2007/Mar. 20, 2008 | United States | |||
| WO2008/034074 | USE OF HIGH-DOSE CYCLOPHOSPHAMIDE IN COMBINATION WITH ANTI-IDIOTYPIC VACCINES IN ANTI-CANCER THERAPY by Robert A. Brodsky et al. | Nov. 14, 1997/Dec 14, 1999 | United States | |||
| WO2008/156494 | USE OF HIGH-DOSE OXAZAPHOSPHORINE DRUGS IN COMBINATION WITH MONOCLONAL ANTIBODIES FOR TREATING IMMUNE DISORDERS by Robert A. Brodsky et al. | Sept. 14, 2007/Mar. 20, 2008 | United States | |||
| WO2009/067690 | METHODS FOR SAFE AND EFFECTIVE TREATMENT USING OXAZAPHOSPHORINE DRUGS by Francis E. O’Donnell, Jr. et al. | Nov. 21, 2008/May 28, 2009 | United States | |||
| WO2009/067699 | METHODS FOR PROVIDING A SYSTEM OF CARE FOR AN OXAZAPHOSPHORINE DRUG REGIMEN by Francis E. O’Donnell, Jr. et al. | Sept. 14, 2007/Mar. 20, 2008 | United States | |||
| WO2009/094456 | USE OF HIGH-DOSE, POST-TRANSPLANTATION OXAZAPHOSPHORINE DRUGS FOR REDUCTION OF TRANSPLANT REJECTION by Ephraim Fuchs et al. | Jan. 22. 2009/July 30, 2009 | International | |||
108
Table of Contents
Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated, or circumvented, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our products. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies or duplicate any technology developed by us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We rely in some circumstances on trade secrets to protect our technology, particularly with respect to certain aspects of Biovest’s BiovaxID manufacturing process. However, trade secrets are difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors, and other contractors. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Third-Party Reimbursement and Pricing Controls
In the U.S. and elsewhere, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. It will be time-consuming and expensive for us to go through the process of seeking reimbursement from Medicare and private payors. Our products may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis. The passage of the Medicare Prescription Drug and Modernization Act of 2003 imposes new requirements for the distribution and pricing of prescription drugs which may affect the marketing of our products.
In many foreign markets, including the countries in the EU, pricing of pharmaceutical products is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
109
Table of Contents
Insurance
We may be exposed to potential product liability claims by users of our products. We presently maintain product liability insurance coverage, in connection with our systems and other products and services, in amounts which we believe to be adequate and on acceptable terms.
Although, we believe that our current level of coverage is adequate to protect our business from foreseeable product liability and clinical trial claims, we may seek to increase our insurance coverage in the future in the event that we significantly increase our level of contract production services. There can be no assurance; however, that we will be able to maintain our existing coverage or obtain additional coverage on acceptable terms, or that such insurance will provide adequate coverage against all potential claims to which we may be exposed. A successful partially or completely uninsured claim against us could have a material adverse effect on our operations. Biovest’s cell culture production services may expose us to potential risk of liability. We seek to obtain agreements from contract production customers to mitigate such potential liability and to indemnify us under certain circumstances. There can be no assurance, however, that we will be successful in obtaining such agreements or that such indemnification, if obtained, will adequately protect us against potential claims.
The terms and conditions of our sales and instruments include provisions which are intended to limit our liability for indirect, special, incidental or consequential damages.
Employees
As of January 31, 2011, we had 60 employees. None of our employees is represented by labor unions or covered by collective bargaining agreements. We supplement our staff with temporary employees and consultants as required. We believe that our relations with employees are satisfactory.
Our ability to continue to develop and improve marketable products and to establish and maintain our competitive position in light of technological developments will depend, in part, upon our ability to attract and retain qualified technical personnel.
Properties
We lease approximately 7,400 square feet of office space in Tampa, Florida, which is our principal executive office and administrative office. The lease will expire on September 30, 2011. We are in the process of negotiating a potential extension of the lease with our landlord.
Our wholly-owned subsidiary, Analytica, leases approximately 4,000 square feet of office space, located at 24 West 40th Street, New York, NY 10018, and office space located at Meeraner Platz 1, 79539 Lorrach, Germany, which is occupied by Analytica’s employees in Germany (the “Lorrach Lease”). The Lorrach Lease will expire on November 1, 2011.
Our majority-owned subsidiary, Biovest, leases approximately 35,000 square feet in Minneapolis, Minnesota, which Biovest uses for offices, a laboratory, manufacturing, and warehousing areas to support the production of perfusion cell culture equipment, and contract cell culture services. On December 2, 2010, Biovest entered into a new long-term lease for this facility, wherein its landlord (in conjunction with the City of Coon Rapids and the State of Minnesota) has agreed to fund and amortize improvements to the facility to provide a dedicated laboratory space for the production of BiovaxID and potential future expansion to the facility to permit additional BiovaxID production capacity when required.
110
Table of Contents
We plan to continue to evaluate our requirements for facilities during fiscal 2011. We anticipate that as our development of Revimmune and/or BiovaxID advances and as we prepare for the future commercialization of these products, our facilities requirements will continue to change on an ongoing basis.
Legal Proceedings
BANKRUPTCY PROCEEDINGS
On November 10, 2008, we, along with our subsidiaries, filed a voluntary petition for reorganization under Chapter 11 of the United States Bankruptcy Code (“Chapter 11”) in the U.S. Bankruptcy Court for the Middle District of Florida, Tampa Division (the “Bankruptcy Court”). During the pendency of the Chapter 11 proceedings, we operated our business as a debtor-in-possession in accordance with the provisions of Chapter 11 and subject to the jurisdiction of the Bankruptcy Court. On August 16, 2010, we filed our First Amended Joint Plan of Reorganization, and, on October 25, 2010, we filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. We emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
Notwithstanding the effectiveness of our Plan, the Bankruptcy Court retains jurisdiction to adjudicate any remaining issues regarding, inter alia, the validity, amount, and method of payment of claims filed in connection with our Chapter 11 proceeding. Accordingly, we anticipate that there may be ongoing proceedings before the Bankruptcy Court to resolve any filed objections or disputes as to claims filed in the Chapter 11 proceeding.
BIOVEST LITIGATION
On August 4, 2008, our majority-owned subsidiary, Biovest, was served with a summons and complaint filed in California Superior Court on behalf of Clinstar LLC for breach of contract for non-payment of certain fees for clinical trial studies and pass-through expenses in the amount of $385,000. Biovest intends to seek the dismissal of this litigation and plans to defend these claims vigorously. Upon the filing of Biovest’s Chapter 11 petition on November 10, 2008, this litigation was automatically stayed pursuant to provisions of federal bankruptcy law. Biovest anticipates that the claims involved in this litigation will be contested and resolved by the Bankruptcy Court as part of an objection to claim which Biovest expects to file shortly.
OTHER PROCEEDINGS
Except for the foregoing, we are not party to any material legal proceedings, and management is not aware of any threatened legal proceedings that could cause a material adverse impact on our business, assets, or results of operations.
111
Table of Contents
Executive Officers and Directors
The following table provides information with respect to our directors and officers as of January 31, 2011:
| Name
|
Age | Position | ||
| Francis E. O’Donnell, Jr., M.D. | 60 | Chief Executive Officer (“CEO”), Chairman and Director
| ||
| Samuel S. Duffey, Esq. | 65 | President and General Counsel
| ||
| Garrison J. Hasara | 41 | Acting Chief Financial Officer, Controller
| ||
| Douglas M. Calder | 43 | Vice President of Strategic Planning and Capital Markets
| ||
| Carlos F. Santos | 33 | Senior Vice President of Product Development and Scientific/Regulatory Affairs
| ||
| Edmund C. King | 75 | Director
| ||
| David M. Schubert | 44 | Director
| ||
| Christopher C. Chapman, M.D. | 58 | Director
| ||
| William S. Poole | 64 |
Director
|
Francis E. O’Donnell, Jr., M.D., age 60, has served as our Chairman of the Board since the Company’s inception in 2002 and has served as our CEO since 2003. Dr. O’Donnell also served as our President from September 2003 through November 2004. Since February 2009, Dr. O’Donnell has been Chief Executive Officer and Chairman of our majority-owned subsidiary, Biovest International, Inc. (“Biovest”). Since 2003, Dr. O’Donnell had been a Director and Vice-Chairman (non-executive) of Biovest. He is also the Chairman and a Director of BioDelivery Sciences International, Inc. (“BDSI”). Dr. O’Donnell was the founder and for more than the last five years has served as manager of The Hopkins Capital Group (“HCG”), an affiliation of limited liability companies which engage in business development of disruptive healthcare technologies. HCG entities are significant stockholders of Accentia and BDSI. Dr. O’Donnell is a 1975, summa cum laude graduate of the Johns Hopkins School of Medicine. He received his specialty training at the Wilmer Ophthalmological Institute, Johns Hopkins Hospital. He is the former Professor and Chairman, Department of Ophthalmology, St Louis University School of Medicine. Dr. O’Donnell has published over 30 peer-reviewed scientific articles and he has been awarded over 34 U.S. Patents. He is the recipient of the 2000 Jules Stein Award from Retinitis Pigmentosa International. He is a Trustee for St. Louis University. We believe that Dr. O’Donnell’s experience and skills make him a qualified and valuable member of our Board of Directors.
112
Table of Contents
Samuel S. Duffey, age 65, was appointed our President in December 2008 and continues to serve as our General Counsel. Mr. Duffey served a director of our Company from 2003 to 2005. Since February 2009, Mr. Duffey was appointed President and General Counsel of Biovest. Prior to that, Mr. Duffey practiced business law with Duffey and Dolan P.A. beginning in 1992. From February 2000 to September 2003, Mr. Duffey served as the non-executive chairman and as a member of the Board of Directors of Invisa, Inc., a small publicly held safety company, and from October 2001 to May 2004, Mr. Duffey also served as the non-executive chairman and as a member of the Board of Directors of FlashPoint International, Inc., a publicly held automotive parts company which is currently named Navitrak International Corporation. Mr. Duffey received his B.A. and J.D. degrees from Drake University. We believe that Mr. Duffey’s experience and skills make him a qualified and valuable member of our management team. Mr. Duffey has been instrumental in facilitating our capital raises and was instrumental in managing our company through the very complex Chapter 11 process.
Garrison J. Hasara, age 41, was appointed as our Acting Chief Financial Officer on January 15, 2011. Mr. Hasara has also served as our Controller since June 2005. From November 2003 to June 2005, Mr. Hasara was our Compliance Specialist. Mr. Hasara has been a licensed Certified Public Accountant since 1993 and received his B.S. from the University of South Florida in 1991.
Douglas M. Calder, age 43, was appointed as our Vice President of Strategic Planning and Capital Markets on January 15, 2011. Also, on January 15, 2011, Mr. Calder was appointed to serve as Biovest’s Vice President of Strategic Planning and Capital Markets. From December 2007 until his recent appointment, Mr. Calder was the Director of Investment Relations for both Accentia and Biovest. Prior to joining Accentia and Biovest, Mr. Calder served as the Director of Communications for the publicly-traded biotechnology company, Viragen, Inc., from June 2000 through October 2007. Viragen was engaged in the research and development of natural human alpha interferon, avian transgenic technology and monoclonal antibodies. Mr. Calder previously served on the Communications Committee and Animal Biotechnology Committee for the Biotechnology Industry Organization (“BIO”). BIO is the world’s largest biotechnology organization, providing advocacy, business development and communications services to its members. Mr. Calder has more than 18-years of life science executive experience as a financial portfolio manager and investor relations professional in managing corporate communications, business development, media strategies and capital markets strategies. Mr. Calder previously served as a registered financial portfolio manager with a biotechnology focus working for the New York Stock Exchange Member Firms: Dean Witter Reynolds, Inc.; Gruntal & Co.; and Moors & Cabot, Inc. Mr. Calder received his B.A. in English from Florida State University, Tallahassee, Florida.
Carlos F. Santos, Ph.D., age 33, was appointed as our Senior Vice President of Product Development and Scientific/Regulatory Affairs on January 15, 2011. Dr. Santos joined our company in July 2003 and, from March 2005 until his recent appointment, Dr. Santos was our Chief Science Officer. Dr. Santos joined Biovest in January 2007 and served as its Vice President of Regulatory Affairs. Also, on January 15, 2011, Dr. Santos was appointed to serve as Biovest’s Senior Vice President of Product Development and Scientific/Regulatory Affairs. Since 1998, Dr. Santos has served as a scientist and advisor with the Hopkins Capital Group (“HCG”) (St. Louis, MO) of companies. HCG is an affiliation of limited liability companies which engage in business development of disruptive healthcare technologies. HCG entities are significant stockholders of our company. Dr. Santos obtained a B.Sc. (Computer Science) from Washington University in St. Louis (St. Louis, MO), an M.Sc. (Bioinformatics) from the University of Michigan (Ann Arbor, MI), and a Ph.D. (Bioinformatics) from the University of Michigan (Ann Arbor, MI). At the University of Michigan, Dr. Santos led the development of a large-scale automated system for biomolecular pathway integration and search engine at the University of Michigan’s Center for Computational Medicine and Bioinformatics and the National Center for Integrative Biomedical Informatics. Dr. Santos has authored numerous peer-reviewed publications and is listed as co-inventor on pending BiovaxID and Revimmune-related patents.
113
Table of Contents
Edmund C. King, age 75, has served as a director since October 2006. He has also served as a director of our subsidiary, Biovest, since June 2010. Mr. King was a partner in Ernst & Young, an international accounting and consulting firm. While at Ernst & Young, Mr. King was that firm’s southern California senior healthcare partner and prior to that directed the southern California healthcare practice for Arthur Young & Company, one of the predecessor firms of Ernst & Young. During his 30 years with Ernst & Young, Mr. King counseled clients in structuring acquisitions and divestitures; advised on the development of strategic plans; directed the preparation of feasibility studies; assisted with operational and financial restructuring; directed and supervised audits of client financial statements; and provided expert witness testimony and technical SEC consultation. Commencing in 1999, Mr. King became a financial consultant to SmartGate, L.C. Mr. King has served as Chief Financial Officer and Director of Invisa, Inc. since November 2000 and Acting President since November 2007. Mr. King is also a member of the Board of Directors of LTC Properties, Inc., a NYSE listed real estate investment trust. Mr. King is a graduate of Brigham Young University having served on the National Advisory Council of that school’s Marriott School of Management, and has completed a Harvard University management course sponsored by Ernst & Young. Mr. King also has served as Chairman of the MPMA’s Long-Term Care Committee (Los Angeles Chapter) and is a past member of the National Association of Corporate Directors. We believe that Mr. King’s experience and background make him a qualified and valuable member of our Board of Directors. Specifically, Mr. King’s background in accounting and finance make him a valuable resource on our Board.
David M. Schubert, age 44, has served as a director since October 2005. Mr. Schubert is currently the Chief Business Officer of Accelerator Corporation, a venture capital-backed biotechnology investment company located in Seattle, Washington. Prior to joining Accelerator in 2005, Mr. Schubert served as President and founder of Cellexsys, Inc., a privately held biotechnology company that he founded in January 2001 that was acquired by Chromos Molecular Systems in July 2004. Following the sale of Cellexsys, Mr. Schubert has worked as an independent consultant providing advisory services to biotechnology companies. Prior to founding Cellexsys, Mr. Schubert worked for Targeted Genetics Corporation, a publicly held developer of gene-based treatments, as Senior Director, Strategic Initiatives from April 2000 to December 2000 and as Senior Director, Communications and Strategic Relations from November 1997 through March 2000. Mr. Schubert’s prior work experience also includes serving as a Senior Market Manager- Immunotherapy for Baxter Healthcare Corporation. Mr. Schubert is a graduate of Eastern Nazarene College with Bachelor’s degrees in Biology and Psychology, Utah State University with a Master’s degree in Biology, and The Pennsylvania State University with an MBA. We believe that Mr. Schubert’s experience and background make him a qualified and valuable member of our Board of Directors. Specifically, Mr. Schubert’s background in biotechnology and healthcare make him a valuable resource on our Board.
Christopher C. Chapman, M.D., age 58, has served as a director of our company since April 2008. He has been a director of our subsidiary, Biovest, since March 2004. From 2004 to the present, Dr. Chapman has been manager of Chapman Pharmaceutical Consulting, Inc., a consulting organization that provides support on clinical and regulatory issues for pharmaceutical and biotech companies in North America, Europe, Japan, India and Africa. Dr. Chapman received his M.D. degree from Georgetown University in Washington, D.C. in 1987 where he completed his internship in Internal Medicine. He completed a residency in Anesthesiology and a fellowship in Cardiovascular and Obstetric Anesthesiology at Georgetown University. Since 1995, Dr. Chapman has been a critical care physician on the staff at Doctor’s Community Hospital, Lanham, Maryland. In 2009, Dr. Chapman joined Takeda Pharmaceuticals, Inc. and manages pharmacovigilance for an ongoing Phase 3 clinical trial. Dr. Chapman also is a consultant manager for Middle Brook Pharmaceuticals and a manager for staff at Enzon Pharmaceuticals. Dr. Chapman was the Executive Vice President of Medical and Regulatory Affairs and Director of New Business Development (pharmaceuticals) for BDSI on a part time basis from October 2000 to November 2004. From 1995 to April 2000, Dr. Chapman was Executive Director, Medical Affairs, Quintiles Consulting and a founding Co-Director of Quintiles BRI (QBRI) Medical Affairs, Drug Safety and Medical Writing Departments. We believe that Dr. Chapman’s experience and skills make him a qualified and valuable member of our Board of Directors. Specifically, Dr. Chapman’s background in drug development consultation and clinical trials make him a valuable resource on our Board.
114
Table of Contents
William S. Poole, age 64, has served as a director of our company in June 2007. In recent years, Mr. Poole has acted as a private consultant. Mr. Poole was President and CEO of Spherics, Inc. from January 2007 to July 2008, a biotechnology company focusing on unique mechanisms of certain drugs for the treatment of CNS disease. In addition, Mr. Poole is a board member of BDSI and is Chairman of its Compensation Committee. From 1972 to early 1996, Mr. Poole worked for Lederle Laboratories, a Division of American Cyanamid Company. During his 24-year career at Cyanamid, Mr. Poole held positions of increasing responsibility and held the position of World-Wide Division President of the Medical Device Division when Wyeth acquired Cyanamid in 1995. He later served as President, North American Pharmaceuticals, of Novo Nordisk Pharmaceuticals, and also as President of Biovail Pharmaceuticals. In both of these companies, Mr. Poole was instrumental in growing revenue, building a solid management team and improving profitability. As President of these firms, Mr. Poole had total profit and loss responsibility and directly oversaw vice presidents in charge of manufacturing, research and development, sales, legal, marketing, finance, regulatory and human resources functions. We believe that Mr. Poole’s experience and background make him a qualified and valuable member of our Board of Directors. Specifically, Mr. Poole’s background in biopharmaceutical and medical device industry make him a valuable resource on our Board.
Director Independence
Currently, our board of directors consists of five members – Francis E. O’Donnell, Jr., M.D., Edmund C. King, David M. Schubert, Christopher C. Chapman, M.D., and William S. Poole. Our board has determined that four of its members are “independent directors” as defined under the applicable rules of The NASDAQ Stock Market and the Securities Exchanged Commission. These four “independent directors” are Edmund C. King, David M. Schubert, Christopher C. Chapman, M.D., and William Poole. In making their determinations of independence, our Governance and Nominating Committee and our board of directors considered questionnaires completed by each director and all ordinary course transactions between our company and all entities with which each director is employed. With regard to Mr. Poole, our board considered Mr. Poole’s relationship as a director of BioDelivery Sciences International, Inc., which has relationships with the Company. With regard to Dr. Chapman and Mr. King, our board considered their relationship as directors of Biovest International, Inc., the Company’s majority-owned subsidiary. Additionally, our board determined that Francis E. O’Donnell, Jr., M.D. and Alan M. Pearce, who was a director of our company from August 2004 until January 19, 2011, are not independent.
115
Table of Contents
Compensation of Executive Officers
SUMMARY COMPENSATION TABLE
| Name and principal position |
Year
|
Salary ($)
|
Option Awards ($)
|
All Other Compensation ($)
|
Total ($)
|
|||||||||||||||
| Francis E, O’Donnell, Jr., M.D., CEO (10) |
2010 | 1 | (1) | 1,415,750 | 16,863 | (4) | 1,432,614 | |||||||||||||
| 2009 | 1 | (1) | 92,360 | (2) | 19,936 | (5) | 112,297 | (2) | ||||||||||||
| Alan M. Pearce, CFO* (10) |
2010 | 129,500 | 1,019,250 | 17,507 | (6) | 1,166,257 | ||||||||||||||
| 2009 | 131,250 | 140,700 | (3) | 9,307 | (7) | 281,257 | (3) | |||||||||||||
| Samuel S. Duffey, Esq., General Counsel/ President (10) |
2010 | 175,672 | 1,732,725 | 23,313 | (8) | 1,931,710 | ||||||||||||||
| 2009 | 176,683 | 262,300 | 8,576 | (9) | 447,559 | |||||||||||||||
| *Effective | December 31, 2010, Mr. Pearce retired from the Company. |
| (1) | Pursuant to his employment agreement, Dr. O’Donnell was paid one dollar. |
| (2) | Includes director compensation in the amount of $51,750 for Fiscal 2009. |
| (3) | Includes director compensation in the amount of $41,400 for Fiscal 2009. |
| (4) | In Fiscal 2010, Dr. O’Donnell was paid $16,863 in other compensation which related to medical, dental and life insurance and long and short term disability. |
| (5) | In Fiscal 2009, Dr. O’Donnell was paid $19,936 in other compensation which consisted of payments related to medical, dental and life insurance and long and short term disability. |
| (6) | In Fiscal 2010, Mr. Pearce was paid $17,507 in other compensation which consisted of the following payments: $11,507 related to medical, dental and life insurance and long and short term disability and $6,000 related to auto allowances. |
| (7) | In Fiscal 2009, Mr. Pearce was paid $9,307 in other compensation which consisted of the following payments: $6,307 related to medical, dental and life insurance and long and short term disability and $3,000 related to auto allowances. |
| (8) | In Fiscal 2010, Mr. Duffey was paid $23,313 in other compensation which consisted of the following payments: $17,313 related to medical, dental and life insurance and long and short term disability and $6,000 related to auto allowances. |
| (9) | In Fiscal 2009, Mr. Duffey was paid $8,576 in other compensation which consisted of the following payments: $5,576 related to medical, dental and life insurance and long and short term disability and $3,000 related to auto allowances. |
| (10) | In Fiscal 2009 and 2010, Messrs. O’Donnell, Pearce and Duffey received additional compensation from our Biovest subsidiary. See Biovest’s Annual Report on Form 10-K for Fiscal 2010. |
Employment Agreements with Executives. On January 1, 2005, Messrs. O’Donnell, Pearce and Duffey all entered into employment agreements, each with an initial term of five years. On January 1, 2010, these employment agreements expired and Messrs. O’Donnell, Pearce and Duffey each continued (and Messrs. O’Donnell and Duffey still continue) on an “at-will” employment basis. The employment agreements provide that Messrs. O’Donnell, Pearce and Duffey may not solicit customers or engage in or own any business that is competitive with us during the time of their employment and for a period ending two years from the termination of their respective agreements.
116
Table of Contents
Base Salaries. The base salaries reflected above were established by employment agreements existing prior to fiscal 2010, which are described in more detail above. During fiscal 2010, we did not establish specific performance objectives for these executives and our decisions were based on their overall performances. Of particular note is our CEO’s employment agreement which since 2005 has fixed his base salary at one dollar per year. Notwithstanding this nominal salary, Dr. O’Donnell serves as our CEO on a full-time basis. Additionally, Dr. O’Donnell receives no salary from our subsidiaries. This nominal base salary reflects our CEO’s preference for equity compensation. We believe that this form of compensation paid to our CEO in fiscal 2010 most closely aligns the interest of our CEO and our shareholders.
Cash Bonuses and Incentives. In fiscal 2010, no cash bonuses were paid or accrued to our executive officers.
Option Awards. The amounts in the “Option Awards” column include the aggregate grant date fair value computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our stock on the date of exercise. Assumptions used in the calculation of these amounts are included in Note 1 to our audited financial statements.
Stock Options Plans. We have four option plans – the 2003 Stock Option Plan (“2003 Plan”), the 2005 Equity Incentive Plan (“2005 Plan”), the Amended and Restated 2008 Equity Incentive Plan (the “2008 Plan”), and the 2010 Equity Incentive Plan (the “2010 Plan”) (collectively, the “Stock Option Plans”). The Stock Option Plans provide for the issuance of qualified and non-qualified options as those terms are defined by the Internal Revenue Code. The 2003 Plan, as amended, provides for the issuance of 3.5 million shares of common stock. As of January 15, 2011, there were approximately 1.7 million options available for issuance under the 2003 Plan. The 2005 Plan provides for the issuance of 3.0 million shares of common stock. As of January 15, 2011, there were approximately 1.6 million options available for issuance under the 2005 Plan. The 2008 Plan provides for the issuance of 23.0 million shares of common stock. As of January 15, 2011, there were approximately 0.8 million options available for issuance under the 2008 Plan. On November 17, 2010, our board of directors approved the 2010 Plan and, on January 19, 2011, our shareholders approved the 2010 Plan. The 2010 Plan provides for the issuance of 10.0 million shares of common stock. As of January 15, 2011, there were approximately, 9.2 million options available for issuance under the 2010 Plan. The Stock Option Plans cannot have a term greater than ten years.
Perquisites. Consistent with our philosophy to preserve cash, we have sought to limit perquisites. Perquisites paid to our named executive officers are discussed in the footnotes to the Summary Compensation Table above. Our policy for paying medical and dental insurance is to pay 75% of the insurance premium. Our policy for paying life insurance, long-term and short-term disability insurances and accidental death and dismemberment insurance is to pay 100% of the insurance premiums. Our policy with regard to unused vacation for our executive group is to pay at the base salary rate for vacation not used in the prior fiscal year.
Change in Control Severance Policy. Each of our named executive officers entered into a written employment agreement with us prior to our IPO; each of their employment agreements expired according to its respective terms in January 2010, and these named executive officers are now employed on an “at-will” basis. None of our named executive officers have change in control severance provisions in their employment agreements.
117
Table of Contents
OUTSTANDING EQUITY AWARDS AT 2010 FISCAL YEAR END
| Name |
Number of Securities Underlying Unexercised Options (#) - Exercisable
|
Number of Securities Underlying Unexercised Options (#) - Unexercisable
|
Option Exercise Price ($)
|
Option Expiration Date
|
||||||||||||
| Francis E, O’Donnell, Jr., M.D., CEO |
2,252 | 2.22 | 04/10/2013 (a) | |||||||||||||
| 12,500 | 25,000 | 2.61 | 03/01/2018 (a) | |||||||||||||
| 60,000 | 0.18 | 02/24/2019 (a) | ||||||||||||||
| 1,000,000 | 0.44 | 02/12/2020 (a) | ||||||||||||||
| 2,500,000 | 0.44 | 02/12/2020 (a) | ||||||||||||||
| 20,000 | 0.50 | 03/13/2014 (b) | ||||||||||||||
| 20,000 | 0.50 | 03/10/2015 (b) | ||||||||||||||
| 60,000 | 1.13 | 10/19/2016 (b) | ||||||||||||||
| 75,000 | 0.66 | 04/11/2018 (b) | ||||||||||||||
| 1,000,000 | 0.06 | 02/24/2019 (b) | ||||||||||||||
| 125,000 | 0.07 | 02/24/2019 (b) | ||||||||||||||
| 1,750,000 | 0.69 | 02/22/2020 (b) | ||||||||||||||
| 200,000 | 0.69 | 02/22/2020 (b) | ||||||||||||||
| Alan M. Pearce, CFO |
84,856 | — | 7.59 | 02/09/2016 (a) | ||||||||||||
| 37,425 | — | 3.70 | 12/15/2016 (a) | |||||||||||||
| 250,000 | — | 3.56 | 12/20/2016 (a) | |||||||||||||
| 275,000 | — | 2.69 | 01/07/2018 (a) | |||||||||||||
| 20,000 | 10,000 | 2.61 | 03/01/2018 (a) | |||||||||||||
| 1,000,000 | 0.18 | 02/24/2019 (a) | ||||||||||||||
| 50,000 | 0.18 | 02/24/2019 (a) | ||||||||||||||
| 2,500,000 | 0.44 | 02/12/2020 (a) | ||||||||||||||
| 150,000 | — | 0.60 | 04/11/2018 (b) | |||||||||||||
| 1,000,000 | 0.06 | 02/24/2019 (b) | ||||||||||||||
| 1,750,000 | 0.69 | 02/22/2020 (b) | ||||||||||||||
| Samuel S. Duffey, Esq., President/General Counsel |
118,765 | — | 2.11 | 11/07/2013 (a) | ||||||||||||
| 41,168 | — | 7.59 | 02/09/2016 (a) | |||||||||||||
| 391,168 | — | 3.70 | 12/15/2016 (a) | |||||||||||||
| 275,000 | — | 2.69 | 01/07/2018 (a) | |||||||||||||
| — | 2,000,000 | 0.18 | 11/13/2018 (a) | |||||||||||||
| 4,250,000 | 0.44 | 02/12/2020 (a) | ||||||||||||||
| 500,000 | — | 0.50 | 11/11/2013 (b) | |||||||||||||
| 500,000 | — | 0.72 | 02/10/2016 (b) | |||||||||||||
| 150,000 | — | 0.60 | 04/11/2018 (b) | |||||||||||||
| 1,000,000 | 0.06 | 02/24/2019 (b) | ||||||||||||||
| 1,750,000 | 0.69 | 02/22/2020 (b) | ||||||||||||||
| (a) | Accentia Biopharmaceuticals, Inc. options |
118
Table of Contents
| (b) | Biovest International, Inc. options |
Option Grants. In fiscal 2010, we granted the following Accentia options under our Amended and Restated 2008 Equity Incentive Plan.
| • | 3,500,000 to Dr. O’Donnell; |
| • | 2,500,000 to Mr. Pearce; and |
| • | 4,250,000 to Mr. Duffey. |
The option grants had an exercise price that was equal to 100% of the closing market price for our common stock on the date of the option grant. The unexercisable option awards outstanding in the table above will vest as follows:
| • | With regard to Dr. O’Donnell’s option awards with an expiration date of February 12, 2020, 333,334 vested on November 17, 2010, 1/3rd will vest on February 17, 2011 (333,333), and 1/3rd will vest on May 17, 2011 2,833,333; |
| • | Mr. Pearce’s options awards with an expiration date of February 12, 2020 will vest on May 17, 2011; and |
| • | Mr. Duffey’s options awards with an expiration date of February 12, 2020 will vest on May 17, 2011. |
Our decision to grant options was based primarily on the recommendation of our Compensation Committee and our desire to retain and motivate our employees.
DIRECTOR COMPENSATION FOR FISCAL YEAR ENDED SEPTEMBER 30, 2010
| Name |
Fees Earned or Paid in Cash ($) |
Option Awards ($) |
Total ($) | |||||||||
| Francis E. O’Donnell, Jr., M.D., Chairman |
— | — | — | |||||||||
| Edmund C. King(1) |
— | 101,925 | 101,925 | |||||||||
| Christopher C. Chapman, M.D.(2) |
— | 81,540 | 81,540 | |||||||||
| David M. Schubert(3) |
— | 81,540 | 81,540 | |||||||||
| Alan M. Pearce |
— | — | — | |||||||||
| William S. Poole(4) |
— | 81,540 | 81,540 | |||||||||
| (1) | As of September 30, 2010, the aggregate number of outstanding option awards held by Mr. King was 485,000. |
| (2) | As of September 30, 2010, the aggregate number of outstanding option awards held by Dr. Chapman was 382,500. |
119
Table of Contents
| (3) | As of September 30, 2010, the aggregate number of outstanding option awards held by Mr. Schubert was 467,500. |
| (4) | As of September 30, 2010, the aggregate number of outstanding option awards held by Mr. Poole was 385,000. |
See the above Outstanding Equity Awards table for Dr. O’Donnell and Mr. Pearce’s aggregate number of outstanding option awards.
The amounts in the “Option Awards” column include the aggregate grant date fair value computed in accordance with Financial Accounting Standards Board Accounting Standards Codification Topic 718. The fair value of the stock options will likely vary from the actual value the holder receives because the actual value depends on the number of options exercised and the market price of our stock on the date of exercise. The assumptions used in the calculation of these amounts are included in Note 1 to our audited financial statements in our Annual Report on Form 10-K for the year ended September 30, 2010.
Directors receive the following compensation for serving as members of the Board of Directors or as members or chairmen of various committees of the Board of Directors: (i) all directors receive reimbursement of expenses, and (ii) non-employee directors receive an annual grant of non-qualified options to purchase shares of common stock at 100% of the closing market price on the day of the option grant. The director compensation stock options issued for fiscal 2010 are set forth above. Mr. King is the Chairman of the Audit Committee.
During fiscal 2010, although each non-employee director was entitled to an annual cash fee in the amount of $18,000, the non-employee directors opted to receive no annual cash compensation until a vote to reinstate such compensation. Our directors received a stock option grant equal to 200,000 shares of our common stock for serving on the board. The grant of options to directors for fiscal 2010 was made on February 12, 2010. The exercise price of the options granted on February 12, 2010 was set at the close of market on February 12, 2010, which was $0.44 per share. The next option grant to non-employee directors and to employee directors will made under the 2010 Equity Incentive Plan, which we anticipate to be in February 2011.
120
Table of Contents
During our fiscal years ended September 30, 2010 and 2009, we were a party to the following transactions with certain of our executive officers, directors, holders of more than 5% of our voting securities, and their respective affiliates. We believe that the terms of these transactions were no less favorable to us than the terms that could have been obtained from unaffiliated third parties. In some instance transactions which occurred in prior years are also described in order to provide proper background.
Relationship with Biovest International, Inc.
In June 2003, we purchased 81% of the outstanding capital stock of Biovest for $20.0 million pursuant to an Investment Agreement with Biovest.
Dr. O’Donnell, our Chairman and CEO, is also the Chairman and a director of Biovest. Mr. Pearce, our former CFO, was also the CFO of Biovest. Mr. Duffey, our President and General Counsel, is also President and General Counsel of Biovest.
On February 5, 2008, we were granted an option by Biovest permitting us, in our discretion, to convert part or all of the principal and interest due to us on the date of the conversion under the intercompany debt owed by Biovest into shares of Biovest common stock at a conversion price of $1.10 per share (the “Conversion Price”) subject to adjustment in the event of certain recapitalizations or in the event of Biovest’s sale of its stock at prices below the Conversion Price. Biovest granted demand and piggyback registration rights to us for the shares underlying this conversion option. On May 9, 2008, as the result of additional Biovest financing transactions, the Conversion Price was reset to $0.32 per share.
On February 5, 2008, we entered into an amendment to the existing royalty agreement with Biovest on sales of BiovaxID to amend and clarify the calculation of royalty payments from any sublicense to specify that in all cases the royalty amounts will be calculated as a percentage of sales to customers/patients.
On November 10, 2008, we, along with Biovest, filed a voluntary petition for reorganization under Chapter 11 of the United States Bankruptcy Code in the United States Bankruptcy Court for the Middle District of Florida. We continued to operate as “debtors-in-possession” under the jurisdiction of the Bankruptcy Court and in accordance with the applicable provisions of the Bankruptcy Code and orders of the Bankruptcy Court, until the effective date of our respective First Amended Plans of Reorganization, which occurred on November 17, 2010. On November 17, 2010, all of our then-outstanding indebtedness of Biovest was eliminated by conversion into common stock of Biovest at a conversion price of $0.75 per share, and our existing royalty agreement between was terminated, thereby eliminating any of our royalty interest in Biovest’s biologic products.
Relationship with BioDelivery Sciences International, Inc.
We have entered into various transactions with BioDelivery Sciences International, Inc. (“BDSI”), a publicly traded drug delivery technology company. Dr. O’Donnell also is BDSI’s principal shareholder and Chairman of the Board. Previously, Dr. O’Donnell also served as the President and Chief Executive Officer of BDSI. The Hopkins Capital Group, LLC (“Hopkins”) and MOAB Investments, LP (“MOAB”) are principal shareholders of BDSI. Also, two employees are shared between BDSI and our company.
121
Table of Contents
In August 2004, BDSI acquired Arius Pharmaceuticals, Inc. (“Arius”), a pharmaceutical development company that is our development partner for our Emezine product. In March 2004, prior to the acquisition of Arius by BDSI, we obtained exclusive U.S. distribution rights to Emezine under a distribution agreement that we entered into with Arius.
On February 17, 2010, the Bankrtupcy Court entered an Order approving the Emezine Settlement Agreement (the “Settlement Agreement”) with BDSI, and entered into as of December 30, 2009. Parties to the Settlement Agreement are us and our wholly-owned subsidiary, TEAMM, BDSI, and BDSI’s wholly-owned subsidiary, Arius.
The purpose of the Settlement Agreement is to memorialize the terms and conditions of a settlement, between our company and BDSI (the “Settlement”) regarding claims by our company relating to allegations of breach of the distribution agreement dated March 12, 2004. Following the issuance in February 2006 by the U.S. Food and Drug Administration (“FDA”) of a non-approvable letter with respect to the New Drug Application (“NDA”) for Emezine, BDSI ceased its Emezine related development efforts and on December 17, 2008, the distribution agreement was terminated. The Settlement Agreement resolves our claims against BDSI under the terminated distribution agreement.
The Settlement Agreement provided that our company and BDSI mutually release all claims that either may have against each other and, in connection therewith, we:
(a) received $2.5 million from BDSI; and
(b) received the following royalty rights (the “Product Rights”) from BDSI with respect to BDSI’s BEMA Granisetron product candidate (“BEMA Granisetron”) (or in the event it is not BEMA Granisetron, the third BDSI product candidate, excluding BEMA Bupremorphine, as to which BDSI files an NDA, which, together with BEMA Granisetron, shall be referred to hereinafter as the “Product”):
(i) 70/30 split (BDSI/Company) of royalty received if a third party sells the Product and 85/15 split on net sales if BDSI sells the Product; and
(ii) BDSI will, from the sale of the Product, fully recover amounts equal to (1) all internal and external worldwide development costs of the Product (“Costs”) plus interest (measured on weighted average prime interest rate from first dollar spent until Product launch) and (2) the $2.5 Million Payment plus interest (measured on weighted average prime interest rate from the time of payment until Product launch) before our company begins to receive its split as described in (b)(i) above.
(c) issued to BDSI a warrant (“Warrant”) to purchase two (2) million shares of our Biovest common stock, with an exercise price of $0.84 per share and an expiration date of March 4, 2017. During the initial two (2) year exercise period, any exercise of the Warrant by BDSI will be subject to approval by Biovest.
In the event of a proposed sale of BDSI or its assets, BDSI has the right to terminate its Product Right payment obligations to us under the Settlement Agreement upon the payment to us of an amount equal to the greater of: (i) Four Million Five Hundred Thousand Dollars ($4.5 million), or (ii) the fair market value of the Product Rights as determined by an independent third party appraiser. Further, if the Product Right is terminated, the Warrant will be terminated if not already exercised, and, if exercised, an amount equal to the strike price will, in addition to the amount in (i) or (ii) above, be paid to us.
122
Table of Contents
Relationship to Revimmune, LLC
On February 27, 2007, we entered into a sublicense agreement (the “Accentia Sublicense”) with Revimmune, LLC under which we were granted the exclusive worldwide right to Revimmune™. The perpetual sublicense allows us to develop and market a patent pending treatment for autoimmune diseases. The Accentia Sublicense covers the potential treatment of all autoimmune diseases including but not limited to multiple sclerosis. Revimmune, LLC’s manager is a director of our company.
Other material terms and conditions of the Accentia Sublicense are as follows:
| • | We assumed certain future development, milestone and minimum royalty obligations of Revimmune, LLC under its license with Johns Hopkins University (“JHU”). In connection with the Accentia Sublicense, we did not pay an upfront fee or reimbursement of expenses. We also agreed to pay to Revimmune, LLC a royalty of 4% on net sales, and in the event of a sublicense, to pay 10% of net proceeds received from any such sublicense to Revimmune, LLC. |
| • | Upon the approval of the sublicensed treatment in the U.S. for each autoimmune disease, we are required to issue to Revimmune, LLC vested warrants to purchase 0.8 million shares of our common stock. The warrant which will be granted at the approval of the first sublicensed product will have an exercise price of $8 per share and any subsequent warrant to be issued will have an exercise price equal to the average of the volume weighted average closing prices of our common stock during the ten (10) trading days immediately prior to the grant of such warrant. |
| • | We will be responsible, at Revimmune’s sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the licensed products. |
| • | We have assumed the cost and responsibility for patent prosecution as provided in the license between Revimmune, LLC and JHU to the extent that the claims actually and directly relate to sublicensed products. |
On January 16, 2008, Biovest entered into a sublicense agreement (the “Biovest Sublicense”) with Revimmune, LLC under which Biovest was granted the exclusive worldwide rights to Revimmune™. The Biovest Sublicense allows Biovest to develop and market a patent-pending pharmaceutical treatment in late-stage development for the treatment of and prevention of transplant rejection including rejection following a bone marrow transplant.
Other material terms and conditions of the Biovest Sublicense are as follows:
| • | Biovest is obligated to pay to Revimmune, LLC a royalty of 6% on net sales, and in the event of a sublicense by Biovest, to pay 20% of sublicense consideration received. Biovest did not pay an upfront fee in connection with the Biovest Sublicense but upon the approval of the sublicensed treatment in the U.S. for each sublicensed indication, Biovest is required to issue to Revimmune, LLC vested warrants to purchase 2.0 million shares of Biovest’s common stock. Each such warrant which will be granted at the approval of each successive sublicensed product will have an exercise price of $1.10 per share or, at the discretion of Biovest, at a price equal to the fair market value of Biovest’s common stock on the date of the grant of such warrant. |
123
Table of Contents
| • | Biovest assumed certain obligations under Revimmune, LLC’s license with JHU related to the sublicensed technology, including the payment of all royalty obligations due JHU for the sublicensed products which includes a 4% royalty on licensed products and services and a 20% royalty on sublicense consideration. |
| • | Biovest will be responsible, at its sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the sublicensed products. |
Relationships with Affiliates
Pursuant to our Plan of Reorganization, effective as of November 17, 2010, we entered into the following transactions involving related parties.
TRANSACTIONS WITH LAURUS MASTER FUND, LTD. AND ITS AFFILIATES
On the Effective Date, we issued to Laurus/Valens term notes in the original principal amount of $8.8 million (the “Laurus/Valens Term Notes”) in satisfaction of allowed claims prior to the Effective Date of the Plan. The following are the material terms and conditions of the Laurus/Valens Term Notes:
| • | the Laurus/Valens Term Notes mature on November 17, 2012 and may be prepaid at any time without penalty; |
| • | interest accrues on the Laurus/Valens Term Notes at the rate of eight and one-half percent (8.5%) per annum (with a twelve and one-half percent (12.5%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
| • | we are required to make mandatory prepayments under the Laurus/Valens Term Notes as follows: |
| • | on May 17, 2012, a payment of principal, in cash, in the amount of $4.4 million, less the amount of any prior optional prepayments of principal by us and the amount of any other mandatory prepayments of principal under the Laurus/Valens Term Notes; and |
| • | a prepayment equal to thirty percent (30%) of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by us) of certain capital raising transactions (with certain exclusions); |
| • | the Laurus/Valens Term Notes are secured by: |
| • | a first lien on all of our assets, junior only to the liens granted under the Plan to holders of the 8% Original Issue Discount Secured Convertible Debentures Due June 19, 2011, issued by us in June 2008, in the original aggregate principal amount of $8,906,098 and certain permitted liens; |
| • | a pledge by us to Laurus/Valens of (a) all of our equity interests in Analytica, and (b) 20,115,818 shares of common stock of Biovest owned by us; and |
| • | all of the assets of Analytica, which secure a guaranty of Analytica as to the entire indebtedness under the Laurus/Valens Term Notes; and |
124
Table of Contents
| • | with the prior written consent of Laurus/Valens, we may convert all or any portion of the outstanding principal and accrued interest under the Laurus/Valens Term Notes into a number of shares of our common stock equal to (a) the aggregate portion of the principal and accrued but unpaid interest outstanding under the Laurus/Valens Term Note being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported for our common stock for the ten (10) trading days immediately preceding the date of the notice of conversion. |
On the Effective Date, 2,236,848 shares of our common stock were issued to Laurus/Valens for payment of Laurus/Valens’ Class 5 and Class 13 claims under the Plan aggregating approximately $6.0 million at a conversion rate equal to $2.67 per share.
On November 17, 2010, all of the following warrants were terminated and cancelled pursuant to the Plan:
| • | the common stock purchase warrant dated August 16, 2005, issued by us to Laurus Master Fund, Ltd. (in liquidation) (“Laurus”), for the purchase of up to 1,000,000 shares of our common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated September 29, 2006, issued by us to Laurus, for the purchase of up to 627,240 shares of our common stock at an exercise price of $2.75 per share; |
| • | the common stock purchase warrant dated October 31, 2007, issued by us to Laurus, for the purchase of up to 4,024,398 shares of our common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated January 18, 2008, issued by us to Valens Offshore SPV I, Ltd., for the purchase of up to 365,169 shares of our common stock at an exercise price of $2.67 per share; and |
| • | the common stock purchase warrant dated January 18, 2008, issued by us to Valens U.S. SPV I, LLC, for the purchase of up to 196,629 shares of our common stock at an exercise price of $2.67 per share. |
TRANSACTIONS WITH DENNIS RYLL, M.D.
On November 17, 2010, we issued, a new promissory note to Dennis Ryll, M.D., a shareholder and an individual affiliated Hopkins Capital II, LLC, who is the holder by assignment of our secured note to Southwest Bank, as payment of our obligation to Southwest Bank prior to the Effective Date of our Plan (the “Class 3 Plan Note”), in an original principal amount of $4,483,284. We are not obligated to pay the Class 3 Plan Note in cash, but rather through quarterly conversions into shares of our common stock or, subject to certain conditions, by exchanging the quarterly conversion amounts into shares of Biovest common stock owned by us. The following are the material terms and conditions of the Class 3 Plan Note:
| • | the Class 3 Plan Note matures on November 17, 2012; |
125
Table of Contents
| • | interest accrues and is payable on the outstanding principal balance of the Class 3 Plan Note from time to time (the “Class 3 Interest”) at a fixed rate of six percent (6%) per annum; |
| • | on November 17, 2010 and on each of the following seven (7) quarterly anniversaries of that date (each, a “Class 3 Automatic Conversion Date”), and provided that the average of the trading price of our common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 3 Plan Note plus the Class 3 Interest as of the Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Amount”) will be automatically converted into shares of our common stock at a conversion rate equal to the Class 3 Automatic Conversion Price per share of our common stock. On November 17, 2010, the first quarterly installment in the amount of approximately $560,000 was converted into shares of our common stock at a conversion rate equal to $1.36 per share, resulting in the issuance of 412,067 shares of our common stock; |
| • | the Class 3 Plan Note is secured by a lien on 15.0 million shares of our Biovest common stock (the “Class 3 Pledged Shares”), subject to the incremental release of a designated portion of such security upon each quarterly payment under the Class 3 Plan Note; and |
| • | if, on any Class 3 Automatic Conversion Date, the Class 3 Automatic Conversion Price is less than $1.00 per share, Dennis Ryll may, at his election, either: |
| • | convert the Class 3 Automatic Conversion Amount into shares of our common stock (also, the “Class 3 Plan Shares”) at a conversion rate equal to $1.00 per share of our common stock; or |
| • | liquidate that number of the Class 3 Pledged Shares which equals the Class 3 Automatic Conversion Amount using a conversion rate for the Class 3 Pledged Shares equal to the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date. |
126
Table of Contents
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth the beneficial ownership of shares of our common stock as of January 31, 2011 for:
| • | each person (or group of affiliated persons) known by us to beneficially own more than 5% of our common stock; |
| • | each of our directors; |
| • | each of our named executive officers; and |
| • | all of our directors and executive officers as a group. |
Information with respect to beneficial ownership has been furnished by each director, officer or beneficial owner of more than 5% of our common stock. Beneficial ownership is determined in accordance with the rules of the SEC and generally requires that such person have voting or investment power with respect to securities. In computing the number of shares beneficially owned by a person listed below and the percentage ownership of such person, shares of common stock underlying options, warrants or convertible securities held by each such person that are exercisable or convertible within 60 days of January 31, 2011 are deemed outstanding, but are not deemed outstanding for computing the percentage ownership of any other person.
Except as otherwise noted below, and subject to applicable community property laws, the persons named have sole voting and investment power with respect to all shares of common stock shown as beneficially owned by them.
| Name of Beneficial Owner |
Number of Shares of Common Stock Beneficially Owned
|
Percentage of Shares Beneficially Owned(1)
| ||
| 5% Shareholders |
||||
| The Hopkins Capital Group, LLC(2) 865 Longboat Club Road Long Boat Key, FL 34228 |
6,524,592 | 9.09% | ||
| Timothy D. Ryll(3) 930 W. Sheridan, #2SE Chicago, IL 60613 |
4,188,737 | 5.97% | ||
| Pharmaceutical Product Development, Inc.(4) 3151 South 17th Street Wilmington, NC 28412 |
4,270,323 | 6.11% | ||
| Named Executive Officers and Directors |
||||
| Francis E. O’Donnell, Jr., M.D.(5) |
7,853,844 | 11.11% | ||
| Samuel S. Duffey, Esq.(6) |
2,344,620 | 3.24% | ||
| Alan M. Pearce(7) |
2,839,803 | 3.98% | ||
| Garrison J. Hasara(8) |
201,309 | * | ||
| Douglas M. Calder(9) |
65,000 | * | ||
| Carlos F. Santos, Ph.D.(10) |
174,015 | * | ||
127
Table of Contents
| Name of Beneficial Owner |
Number of Shares of Stock Beneficially Owned
|
Percentage
| ||
| David M. Schubert(11) |
320,834 | * | ||
| Edmund King(12) |
313,334 | * | ||
| William S. Poole(13) |
245,000 | * | ||
| Christopher C. Chapman, M.D.(14) |
221,667 | * | ||
| Executive Officers and Directors as Group (10 persons) |
14,579,426 | 19.20% | ||
| * | Less than 1.0% |
| (1) | These percentages were calculated using the 68,632,612 shares of our common stock issued and outstanding on January 31, 2011. Please note that, although we have given instructions to our transfer agent to issue our common stock; not all of the shareholders to whom these shares have been issued have electronically claimed/picked-up the shares of common stock issued to them. |
| (2) | Includes 4,349,592 shares of common stock held by The Hopkins Capital Group, LLC (“Hopkins”) and 2,175,000 shares of common stock issuable pursuant to warrants held by Hopkins. Voting and investment power over the shares held by Hopkins is exercised by its manager, Francis E. O’Donnell, Jr., M.D., our Chairman and Chief Executive Officer. |
| (3) | Includes: |
| (a) | 3,442,471 shares of common stock held by MOAB Investments, LP (“MOAB”) and 428,573 shares of our common stock held by MOAB-II Investments, LP (“MOAB-II” and, together with MOAB, the “MOAB Entities”); |
| (b) | 200,000 shares of common stock issuable pursuant to warrants held by MOAB; and |
| (c) | 117,693 shares of common stock held by Timothy D. Ryll, as the Trustee of three different trusts (the “Timothy Ryll Trusts”). |
Timothy Ryll is the sole shareholder and sole director of MOAB Management Company, Inc., which is the sole general partner of each of the MOAB Entities. Mr. Ryll is the trustee of the Timothy Ryll Trusts.
| (4) | Pharmaceutical Product Development International Holdings, Inc., or “PPD International,” is a wholly owned subsidiary of Pharmaceutical Product Development, Inc. (“PPD”), a publicly held corporation. PPD exercises voting and investment control over PPD International. In providing this information, we relied on PPD’s previous Schedule 13(g) filing. |
| (5) | Includes: |
| (a) | 750,000 shares of common stock held by Dr. O’Donnell, 491,018 shares of common stock held by Kathleen M. O’Donnell, Trustee of the Francis O’Donnell, Jr. Descendent Trust (“Descendent Trust”), 4,349,592 shares of common stock held by The Hopkins Capital Groups, LLC (“Hopkins”), 638,846 shares of common stock and 2,175,000 warrants held by The Hopkins Capital Group II, LLC (“Hopkins II”) and 875,000 shares of common stock held by Hopkins Capital Partners, Inc. and |
| (b) | 748,918 shares of common stock issuable pursuant to options held by Dr. O’Donnell that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
Dr. O’Donnell holds voting and investment power over shares held by each of Hopkins and Hopkins II as its Manager and as President of Hopkins Capital Partners, Inc. and disclaims beneficial ownership of the reported securities except to the extent of his pecuniary interest therein. Dr. O’Donnell does not have any voting and investment power of the shares held by Descendent Trust and disclaims beneficial ownership of the reported securities except to the extent of his pecuniary interest therein.
128
Table of Contents
| (6) | Consists of 2,344,620 shares of common stock issuable pursuant to options held by Mr. Duffey that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (7) | Includes: |
| (a) | 987,507 shares of common stock held by Mr. Pearce, 380,011 shares of common stock held jointly by Mr. Pearce and his wife, and 95,003 shares of common stock held by The Pearce Family Limited Partnership; and |
| (b) | 1,377,282 shares of common stock issuable pursuant to options held by Mr. Pearce that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
As a general partner of The Pearce Family Limited Partnership, Mr. Pearce exercises voting and investment power over The Pearce Family Limited Partnership. Effective as of December 31, 2010, Mr. Pearce retired from his position as our Chief Financial Officer, and, effective as of January 19, 2011, Mr. Pearce was no longer a member of our board of directors.
| (8) | Includes 201,309 shares of common stock issuable pursuant to options held by Mr. Hasara that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (9) | Includes 65,000 shares of common stock issuable pursuant to options held by Mr. Calder that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (10) | Includes 174,015 shares of common stock issuable pursuant to options held by Dr. Santos that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (11) | Includes 320,834 shares of common stock issuable pursuant to options held by Mr. Schubert that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (12) | Includes 313,334 shares of common stock issuable pursuant to options held by Mr. King that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (13) | Consists of 245,000 shares of common stock issuable pursuant to options held by Mr. Poole that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
| (14) | Consists of 221,667 shares of common stock issuable pursuant to options held by Dr. Chapman that are currently exercisable or that are exercisable within 60 days of January 31, 2011. |
129
Table of Contents
General
We are authorized to issue up to 300,000,000 shares of common stock, par value $0.001 per share, of which approximately 68,632,612 shares were issued and outstanding as of January 31, 2011. We are also authorized to issue up to 50,000,000 shares of preferred stock having no par value, of which no shares were issued and outstanding as of January 31, 2011.
Common Stock
Holders of our common stock are entitled to one vote per share on all matters to be voted upon by stockholders. In accordance with Florida law, if a quorum exists, action on a matter (other than the election of directors) by a voting group is approved if the votes cast within the voting group favoring the action exceed the votes cast opposing the action.
Shares of our common stock have no preemptive rights, no redemption or sinking fund provisions, and are not liable for further call or assessment. The holders of such common stock are entitled to receive dividends when and as declared by our board of directors out of funds legally available for dividends. Our board of directors has never declared or paid any cash dividends (except for limited dividends on our Series E preferred stock), and our board of directors does not currently anticipate paying any cash dividends in the foreseeable future on our common stock.
Upon a liquidation of our company, our creditors and any holders of our preferred stock with preferential liquidation rights will be paid before any distribution to holders of common stock. The holders of common stock would be entitled to receive a pro rata distribution per share of any excess amount. The rights, preferences, and privileges of holders of common stock are subject to, and may be adversely affected by, the rights of the holders of shares of any series of preferred stock which we may designate and issue in the future.
Units
In this offering, we are offering units, consisting in the aggregate of shares of our common stock and warrants to purchase up to shares of our common stock. Each unit consists of one share of our common stock and 0.5 of a warrant to purchase one share of our common stock. The units will separate immediately and the common stock and the warrants will be issued separately. There will be no market for the units. This prospectus also relates to the offering of shares of our common stock upon exercise, if any, of the warrants.
Warrants
In connection with this offering, we will issue warrants to purchase up to shares of common stock. Each full warrant entitles the holder to purchase one share of common stock at an exercise price of $ per share. The warrants will be exercisable at any time during the period commencing on the date of closing and ending on the fifth anniversary of the closing date. After the expiration of the exercise period, warrant holders will have no further rights to exercise such warrants.
130
Table of Contents
The warrants may be exercised only for full shares of common stock, and may be exercised on a cashless basis only if the registration statement covering the shares issuable upon exercise of the warrants underlying the units is no longer effective. If the registration statement covering the shares issuable upon exercise of the warrants underlying the units is no longer effective, the warrants will be issued with restrictive legends unless such shares are eligible for sale under Rule 144. We will not issue fractional shares of common stock or cash in lieu of fractional shares of common stock. Warrant holders do not have any voting or other rights as a stockholder of our company. The exercise price and the number of shares of common stock purchasable upon the exercise of each warrant are subject to adjustment upon the happening of certain events, such as stock dividends, distributions, and splits.
In addition, we have agreed to issue to the placement agent placement agent warrants on substantially the same terms as the warrants offered in this offering as part of their compensation in connection with the offering, except that these placement agent warrants will comply with FINRA Rule 5110(g)(1) in that for a period of 180 days after the issuance date of the placement agent warrants (which shall not be earlier than the applicable closing date of this offering), neither the placement agent warrants nor any shares of our common stock issued upon exercise of the placement agent warrants shall be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of such securities by any person for a period of 180 days immediately following the date of effectiveness or commencement of sales of the offering pursuant to which the placement agent warrants are being issued, except the transfer of any security:
| • | by operation of law or by reason of reorganization of our company; |
| • | to any FINRA member firm participating in this offering and the officers or partners thereof, if all securities so transferred remain subject to the lock-up restriction described above for the remainder of the time period; |
| • | if the aggregate amount of our securities held by either the placement agent or related persons do not exceed 1% of the securities being offered; |
| • | that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund, and participating members in the aggregate do not own more than 10% of the equity in the fund; or |
| • | the exercise or conversion of any security, if all securities received remain subject to the lock-up restriction set forth above for the remainder of the time period. |
Indemnification of Directors and Executive Officers and Limitation of Liability
The Florida Business Corporation Act, or “FBCA,” permits a Florida corporation to indemnify any person who may be a party to any third party proceeding by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee, or agent of another entity, against liability incurred in connection with such proceeding (including any appeal thereof) if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
131
Table of Contents
The FBCA permits a Florida corporation to indemnify any person who may be a party to a derivative action if such person acted in any of the capacities set forth in the preceding paragraph, against expenses and amounts paid in settlement not exceeding, in the judgment of the board of directors, the estimated expenses of litigating the proceeding to conclusion, actually and reasonably incurred in connection with the defense or settlement of such proceeding (including appeals), provided that the person acted under the standards set forth in the preceding paragraph. However, no indemnification shall be made for any claim, issue, or matter for which such person is found to be liable unless, and only to the extent that, the court determines that, despite the adjudication of liability, but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnification for such expenses which the court deems proper.
The FBCA provides that any indemnification made under the above provisions, unless pursuant to a court determination, may be made only after a determination that the person to be indemnified has met the standard of conduct described above. This determination is to be made by a majority vote of a quorum consisting of the disinterested directors of the board of directors, by duly selected independent legal counsel, or by a majority vote of the disinterested stockholders. The board of directors also may designate a special committee of disinterested directors to make this determination. Notwithstanding the foregoing, the FBCA provides that a Florida corporation must indemnify any director, officer, employee or agent of a corporation who has been successful in the defense of any proceeding referred to above.
Notwithstanding the foregoing, the FBCA provides, in general, that no director shall be personally liable for monetary damages to our company or any other person for any statement, vote, decision, or failure to act, regarding corporate management or policy, unless: (a) the director breached or failed to perform his duties as a director; and (b) the director’s breach of, or failure to perform, those duties constitutes (i) a violation of criminal law, unless the director had reasonable cause to believe his conduct was lawful or had no reasonable cause to believe his conduct was unlawful, (ii) a transaction from which the director derived an improper personal benefit, either directly or indirectly, (iii) an approval of an unlawful distribution, (iv) with respect to a proceeding by or in the right of the company to procure a judgment in its favor or by or in the right of a stockholder, conscious disregard for the best interest of the company, or willful misconduct, or (v) with respect to a proceeding by or in the right of someone other than the company or a stockholder, recklessness or an act or omission which was committed in bad faith or with malicious purpose or in a manner exhibiting wanton and willful disregard of human rights, safety, or property. The term “recklessness,” as used above, means the action, or omission to act, in conscious disregard of a risk: (a) known, or so obvious that it should have been known, to the director; and (b) known to the director, or so obvious that it should have been known, to be so great as to make it highly probable that harm would follow from such action or omission.
The FBCA further provides that the indemnification and advancement of payment provisions contained therein are not exclusive and it specifically empowers a corporation to make any other further indemnification or advancement of expenses of any of its directors, officers, employees or agents under any bylaw, agreement, vote of stockholders or disinterested directors, or otherwise, both for actions taken in an official capacity and for actions taken in other capacities while holding such office. However, a corporation cannot indemnify or advance expenses if a judgment or other final adjudication establishes that the actions of the director, officer, employee, or agent were material to the adjudicated cause of action and the director, officer, employee, or agent (a) violated criminal law, unless the director, officer, employee, or agent had reasonable cause to believe his or her conduct was unlawful, (b) derived an improper personal benefit from a transaction, (c) was or is a director in a circumstance where the liability for unlawful distributions applies, or (d) engaged in willful misconduct or conscious disregard for the best interests of the corporation in a proceeding by or in right of the corporation to procure a judgment in its favor or in a proceeding by or in right of a stockholder.
132
Table of Contents
We have adopted provisions in our articles of incorporation and bylaws providing that our directors and officers and our former directors and officers shall be indemnified to the fullest extent permitted by applicable law.
There is no pending litigation or proceeding involving any of our directors, officers, employees or other agents as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director, officer, employee or other agent.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is therefore unenforceable.
Anti-Takeover Provisions – Florida Law
We are subject to several anti-takeover provisions under Florida law that apply to public corporations organized under Florida law, unless the corporation has elected to opt out of those provisions in its articles of incorporation or bylaws. We have not elected to opt out of those provisions. The FBCA prohibits the voting of shares in a publicly-held Florida corporation that are acquired in a “control share acquisition” unless the holders of a majority of the corporation’s voting shares (exclusive of shares held by officers of the corporation, inside directors, or the acquiring party) approve the granting of voting rights as to the shares acquired in the control share acquisition. A “control share acquisition” is defined in the FBCA as an acquisition that immediately thereafter entitles the acquiring party to vote in the election of directors within any of the following ranges of voting power: one-fifth or more but less than one-third of all voting power, one-third or more but less than a majority of all voting power, and a majority or more of all voting power. However, an acquisition of a publicly-held Florida corporation’s shares is not deemed to be a control-share acquisition if it is either (i) approved by such corporation’s board of directors before the acquisition, or (ii) made pursuant to a merger agreement to which such Florida corporation is a party.
The FBCA also contains an “affiliated transaction” provision that prohibits a publicly-held Florida corporation from engaging in a broad range of business combinations or other extraordinary corporate transactions with any person who, together with affiliates and associates, beneficially owns more than 10% of the corporation’s outstanding voting shares, otherwise referred to as an “interested stockholder,” unless:
| • | the transaction is approved by a majority of disinterested directors before the person becomes an interested stockholder, |
| • | the interested stockholder has owned at least 80% of the corporation’s outstanding voting shares for at least five years, or |
| • | the transaction is approved by the holders of two-thirds of the corporation’s voting shares other than those owned by the interested stockholder. |
Anti-Takeover Provisions – Articles of Incorporation and Bylaws
Our amended and restated articles of incorporation and amended and restated bylaws include a number of provisions that may have the effect of deterring hostile takeovers or delaying or preventing changes in our control or our management, including, but not limited to, the following:
| • | Our board of directors can issue up to 50,000,000 shares of preferred stock, with such rights, preferences, privileges, and restrictions as are fixed by the board of directors (which could include the right to approve or not approve an acquisition or other change in control). |
133
Table of Contents
| • | Our amended and restated bylaws provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide timely notice in writing and also specify requirements as to the form and content of a stockholder’s notice. These provisions may delay or preclude stockholders from bringing matters before a meeting of stockholders or from making nominations for directors at a meeting of stockholders, which could delay or deter takeover attempts or changes in management. |
| • | Our amended and restated bylaws provide that special meetings of the stockholders may be called only by the board of directors or by the Secretary upon the written request of the stockholders who together own of record 25% of the outstanding stock of all classes entitled to vote at such meeting. |
| • | Our amended and restated bylaws provide that vacancies, including newly created directorships, may be filled by the affirmative vote of a majority of the directors then in office, even if less than a quorum; provided, however, that the stockholders have the right to fill any vacancy created by removal of a director by action of the stockholders. In addition, our amended and restated bylaws provide that our board of directors may establish the number of directors by a vote of the directors then in office; provided, however, that the board may not have more than nine directors or less than one director. |
| • | Our amended and restated articles of incorporation do not provide for cumulative voting for directors. The absence of cumulative voting may make it more difficult for stockholders who own an aggregate of less than a majority of our stock to elect any directors to our board. |
These and other provisions contained in our amended and restated articles of incorporation and amended and restated bylaws could delay or discourage transactions involving an actual or potential change in control of us or our management, including transactions in which our stockholders might otherwise receive a premium for their shares over then current prices, and may limit the ability of stockholders to remove our current management or approve transactions that our stockholders may deem to be in their best interests and, therefore, could adversely affect the price of our common stock.
Trading on the OTCQB
Our common stock is currently quoted on the OTCQB under the symbol “ABPI”.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is StockTrans, Inc. The transfer agent’s address is 44 West Lancaster Avenue, Ardmore, PA 19003, and its telephone number is (610) 649-7300.
134
Table of Contents
, which we refer to as the placement agent, has agreed to act as the exclusive placement agent in connection with this offering subject to the terms and conditions of a placement agency agreement, dated , 2011. The placement agent may engage selected dealers to assist in the placement of the units. The placement agent is not purchasing or selling any units offered by this prospectus, nor is it required to arrange the purchase or sale of any specific number or dollar amount of the units, but has agreed to use its commercially reasonable efforts to arrange for the sale of all of the units offered hereby. We will enter into purchase agreements directly with investors in connection with this offering and we may not sell the entire amount of units offered pursuant to this prospectus. The price per unit has been determined based upon arm’s-length negotiations between the purchasers and us.
The placement agent proposes to arrange for the sale to one or more purchasers of the units offered pursuant to this prospectus through direct purchase agreements between the purchasers and us.
Commissions and Expenses
We have agreed to pay the placement agent an aggregate cash placement fee equal to percent of the gross proceeds in this offering.
The following table shows the per unit and total cash placement agent’s fees we will pay to the placement agent in connection with the sale of the units offered pursuant to this prospectus assuming the purchase of all of the units offered hereby:
| Per Unit |
$ | |||||||
|
Total |
$ |
In addition, we have agreed to issue to the placement agent, or its designees, warrants exercisable for an aggregate of percent of the unit shares issued in this offering. The placement agent warrants will be exercisable at any time beginning on the date that is six months from the effective date of the registration statement of which this prospectus is a part hereof until 5:00 p.m. (New York time) on the date that is five years following such effective date at an exercise price of $ per share. This prospectus also covers the sale of the placement agent warrants and the shares of common stock issuable upon the exercise of the placement agent warrants. The placement agent warrants will have terms substantially similar to the terms of the warrants included in the units offered hereby, except that, as required by the Financial Industry Regulatory Authority, Inc., or FINRA, neither the placement agent warrants nor any shares of common stock issued upon exercise of the placement agent warrants may be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of such securities by any person for a period of 180 days immediately following the effective date of the registration statement of which this prospectus is a part, except the transfer of any security:
| • | by operation of law or by reason of our reorganization; |
135
Table of Contents
| • | to any FINRA member firm participating in the offering and the officers or partners thereof, if all securities so transferred remain subject to the lock-up restriction described above for the remainder of the time period; |
| • | if the aggregate amount of our securities held by the placement agent or related person do not exceed 1% of the securities being offered; |
| • | that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund, and participating members in the aggregate do not own more than 10% of the equity in the fund; or |
| • | the exercise or conversion of any security, if all securities received remain subject to the lock-up restriction set forth above for the remainder of the time period. |
Because there is no minimum offering amount required as a condition to closing in this offering, the actual total offering commissions, if any, are not presently determinable and may be substantially less than the maximum amount set forth above. We have also agreed to reimburse the placement agent for its out-of-pocket expenses in an aggregate amount not to exceed $ .
Our obligation to issue and sell units to the purchasers is subject to the conditions set forth in the purchase agreements, which may be waived by us at our discretion. A purchaser’s obligation to purchase units is subject to the conditions set forth in his or her purchase agreement as well, which may also be waived.
We currently anticipate that the sale of the units will be completed on or about , 2011. We estimate the total offering expenses of this offering that will be payable by us, excluding the placement agent’s fee, will be approximately $ , which includes legal and printing costs, various other fees and reimbursement of the placements agent’s expenses. At the closing, The Depository Trust Company will credit the shares of common stock to the respective accounts of the investors. We will mail warrants directly to the investors at the respective addresses set forth in their purchase agreements with us.
Indemnification
We have agreed to indemnify the placement agent against liabilities under the Securities Act of 1933, as amended. We have also agreed to contribute to payments the placement agent may be required to make in respect of such liabilities.
Lock-up Agreements
We and our officers and directors have agreed, subject to certain exceptions, for a period of 90 days after the date of this prospectus, not to offer, sell, contract to sell, pledge, grant any option to purchase, make any short sale or otherwise dispose of, directly or indirectly any common shares or any securities convertible into or exchangeable for our common shares either owned as of the date hereof or thereafter acquired without the prior written consent of the placement agent. This 90-day period may be extended if (1) during the last 17 days of the 30-day period, we issue an earnings release or material news or a material event regarding us occurs or (2) prior to the expiration of the 90-day period, we announce that we will release earnings results during the 16-day period beginning on the last day of the 90-day period, then the period of such extension will be 18-days, beginning on the issuance of the earnings release or the occurrence of the material news or material event. If after any announcement described in clause (2) of the preceding sentence, we announce that we will not release earnings results during the 16-day period, the lock-up period shall expire on the later of the expiration of the 90-day period and the end of any extension of such period made pursuant to clause (1) of the preceding sentence. The placement agent may, in its sole discretion and at any time or from time to time before the termination of the lock-up period, without notice, release all or any portion of the securities subject to lock-up agreements.
136
Table of Contents
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the placement agent, or by an affiliate. Other than this prospectus in electronic format, the information on the placement agent’s website and any information contained in any other website maintained by the placement agent is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the placement agent, and should not be relied upon by investors.
The foregoing does not purport to be a complete statement of the terms and conditions of the placement agency agreement and purchase agreements. A copy of the placement agency agreement and the form of purchase agreement with the investors are included as exhibits to our current report on Form 8-K that will be filed with the SEC. See “WHERE YOU CAN FIND MORE INFORMATION” on page 140.
Regulation M Restrictions
The placement agent may be deemed to be an underwriter within the meaning of Section 2(a)(11) of the Securities Act, and any commissions received by it and any profit realized on the resale of the units sold by them while acting as principals might be deemed to be underwriting discounts or commissions under the Securities Act. As an underwriter, the placement agent would be required to comply with the requirements of the Securities Act and the Securities Exchange Act of 1934, as amended (the “Exchange Act”), including, without limitation, Rule 415(a)(4) under the Securities Act and Rule 10b-5 and Regulation M under the Exchange Act. These rules and regulations may limit the timing of purchases and sales of shares by the placement agent acting as a principal. Under these rules and regulations, the placement agent:
| • | must not engage in any stabilization activity in connection with our securities; and |
| • | must not bid for or purchase any of our securities or attempt to induce any person to purchase any of our securities, other than as permitted under the Exchange Act, until it has completed its participation in the distribution. |
Affiliations
The placement agent and its affiliates may provide various investment banking, financial advisory and other services to us and our affiliates for which services they have received, and may in the future receive, customary fees. In the course of their businesses, the placement agent and its affiliates may actively trade our securities or loans for their own account or for the accounts of customers, and, accordingly, the placement agent and its affiliates may at any time hold long or short positions in such securities or loans.
137
Table of Contents
Notice to Investors in the United Kingdom
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member State”), an offer to the public of any securities which are the subject of the offering contemplated by this prospectus may not be made in that Relevant Member State except that an offer to the public in that Relevant Member State of any such securities may be made at any time under the following exemptions under the Prospectus Directive, if they have been implemented in that Relevant Member State:
| (a) | to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| (b) | to any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts; |
| (c) | by the placement agent to fewer than 100 natural or legal persons (other than qualified investors as defined in the Prospectus Directive); or |
| (d) | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of shares shall result in a requirement for the publication by the issuer or the placement agent of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer to the public” in relation to any security in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and any securities to be offered so as to enable an investor to decide to purchase such securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
The placement agent has represented, warranted and agreed that:
| (a) | it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000 (the “FSMA”)) received by it in connection with the issue or sale of any of our securities in circumstances in which section 21(1) of the FSMA does not apply to the issuer; and |
| (b) | it has complied with and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to our securities in, from or otherwise involving the United Kingdom. |
138
Table of Contents
European Economic Area
In particular, this document does not constitute an approved prospectus in accordance with European Commission’s Regulation on Prospectuses no. 809/2004 and no such prospectus is to be prepared and approved in connection with this offering. Accordingly, in relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (being the Directive of the European Parliament and of the Council 2003/71/EC and including any relevant implementing measure in each Relevant Member State) (each, a “Relevant Member State”), with effect from and including the date on which the Prospectus Directive is implemented in that Relevant Member State (the “Relevant Implementation Date”) an offer of securities to the public may not be made in that Relevant Member State prior to the publication of a prospectus in relation to such securities which has been approved by the competent authority in that Relevant Member State or, where appropriate, approved in another Relevant Member State and notified to the competent authority in that Relevant Member State, all in accordance with the Prospectus Directive, except that the placement agent may, with effect from and including the Relative Implementation Date, make an offer of securities to the public in that Relevant Member State at any time:
| • | to legal entities which are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| • | to any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than 43,000,000 euros; and (3) an annual net turnover of more than 50,000,000 euros, as shown in the last annual or consolidated accounts; or |
| • | in any other circumstances which do not require the publication by the issuer of a prospectus pursuant to Article 3 of the Prospectus Directive. |
For the purposes of this provision, the expression an “offer of securities to the public” in relation to any common shares in any Relevant Member State means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe such securities, as the same may be varied in that Member State by any measure implementing the Prospectus Directive in that Member State. For these purposes the units are “securities.”
139
Table of Contents
The validity of the securities offered by this prospectus will be passed upon for us by the law firm of Foley & Lardner LLP, Tampa, Florida. Certain legal matters in connection with this offering will be passed upon for the placement agents by the law firm of .
The consolidated financial statements included in this prospectus have been audited by Cherry, Bekaert & Holland, L.L.P., independent auditors, as stated in their report appearing herein and are included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the Securities and Exchange Commission, under the Securities Act of 1933, a registration statement on Form S-1 relating to the securities offered hereby. This Prospectus does not contain all of the information set forth in the registration statement and the exhibits and schedules thereto. For further information with respect to our company and the securities we are offering by this Prospectus you should refer to the registration statement, including the exhibits and schedules thereto. You may inspect a copy of the registration statement without charge at the Public Reference Room of the Securities and Exchange Commission at 100 F Street, NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the Securities and Exchange Commission at 1-800-SEC-0330. The Securities and Exchange Commission also maintains an internet site that contains reports, proxy and information statements and other information regarding registrants that file electronically with the Securities and Exchange Commission. The Securities and Exchange Commission’s internet address is http://www.sec.gov.
We file periodic reports, proxy statements and other information with the Securities and Exchange Commission in accordance with requirements of the Exchange Act. These periodic reports, proxy statements and other information are available for inspection and copying at the public reference facilities and internet site of the Securities and Exchange Commission referred to above. In addition, you may request a copy of any of our periodic reports filed with the Securities and Exchange Commission at no cost, by writing or telephoning us at the following address:
Accentia Biopharmaceuticals, Inc.
Attn: Investor Relations
324 South Hyde Park Avenue, Suite 350
Tampa, Florida 33606
(813) 864-2554
140
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC.
INDEX TO SEPTEMBER 30, 2010 FINANCIAL STATEMENTS
Accentia Biopharmaceuticals, Inc. and Subsidiaries Consolidated Financial Statements
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
Accentia Biopharmaceuticals, Inc. and Subsidiaries
Tampa, Florida
We have audited the accompanying consolidated balance sheets of Accentia Biopharmaceuticals, Inc. and its subsidiaries as of September 30, 2010 and 2009 and the related consolidated statements of operations, stockholders’ deficit, and cash flows for the years ended September 30, 2010 and 2009. The consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the consolidated financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States of America). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of Accentia Biopharmaceuticals, Inc. and its subsidiaries as of September 30, 2010 and 2009 and the consolidated results of their operations and their cash flows for the years ended September 30, 2010 and 2009 in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. The Company incurred cumulative net losses of approximately $53.2 million during the two years ended September 30, 2010, and had a working capital deficiency of approximately $147.6 million including liabilities subject to compromise. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regards to these matters are described in Note 2. The consolidated financial statements do not include any adjustments with respect to the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that might result from the outcome of this uncertainty.
As discussed in Note 1 to the accompanying consolidated financial statements, Accentia Biopharmaceuticals, Inc. and Subsidiaries changed the presentation of the non-controlling interest in its consolidated financial statements effective October 1, 2009.
As discussed in Note 1 to the accompanying consolidated financial statements, on November 10, 2008, Accentia Biopharmaceuticals, Inc. and its subsidiaries filed voluntary petitions for reorganization under Chapter 11 of the United States Bankruptcy Code.
/s/ CHERRY, BEKAERT & HOLLAND, L.L.P.
Tampa, Florida
December 14, 2010
F-2
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED BALANCE SHEETS
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 558,452 | $ | 325,350 | ||||
| Accounts receivable: |
||||||||
| Trade, net of allowance for doubtful accounts of $10,031 at September 30, 2010 and September 30, 2009 |
1,287,363 | 1,412,549 | ||||||
| Related party |
— | 112,389 | ||||||
| Inventories |
417,087 | 509,287 | ||||||
| Unbilled receivables |
151,303 | 1,503,869 | ||||||
| Deferred finance costs |
16,077 | — | ||||||
| Prepaid expenses and other current assets |
243,998 | 384,841 | ||||||
| Total current assets |
2,674,280 | 4,248,285 | ||||||
| Goodwill |
893,000 | 1,193,437 | ||||||
| Intangible assets |
1,083,962 | 2,633,090 | ||||||
| Furniture, equipment and leasehold improvements, net |
142,276 | 331,853 | ||||||
| Deferred finance costs, less current portion |
— | 1,125,849 | ||||||
| Other assets |
216,791 | 291,648 | ||||||
| Other assets from discontinued operations |
— | 12,159 | ||||||
| $ | 5,010,309 | $ | 9,836,321 | |||||
(Continued)
F-3
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED BALANCE SHEETS
(Continued)
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT |
||||||||
| Liabilities not subject to compromise: |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 1,139,817 | 1,532,019 | |||||
| Accrued expenses |
1,269,395 | 2,210,592 | ||||||
| Unearned revenues |
263,778 | 955,637 | ||||||
| Notes payable, related parties |
2,041,005 | 1,332,420 | ||||||
| Customer deposits |
134,613 | 208,557 | ||||||
| Derivative liabilities |
1,844,200 | — | ||||||
| Total current liabilities |
6,692,808 | 6,239,225 | ||||||
| Total liabilities not subject to compromise |
6,692,808 | 6,239,225 | ||||||
| Liabilities subject to compromise |
143,570,128 | 98,614,732 | ||||||
| Total liabilities |
150,262,936 | 104,853,957 | ||||||
| Commitments and contingencies |
— | — | ||||||
| Series A convertible redeemable preferred stock, $1.00 par value; 8,950 shares authorized; 7,529 shares issued and outstanding at September 30, 2010 and September 30, 2009 |
7,528,640 | 5,196,110 | ||||||
| Stockholders’ deficit: |
||||||||
| Common stock, $0.001 par value; 300,000,000 shares authorized; 58,048,208 shares issued and outstanding at September 30, 2010 and September 30, 2009 |
58,048 | 58,048 | ||||||
| Treasury stock, 194,907 shares, September 30, 2010 and September 30, 2009 |
(170,057 | ) | (170,057 | ) | ||||
| Additional paid-in capital |
203,828,364 | 205,100,477 | ||||||
| Accumulated deficit |
(325,882,720 | ) | (277,838,822 | ) | ||||
| Total stockholders’ deficit attributable to Accentia Biopharmaceuticals, Inc. |
(122,166,365 | ) | (72,850,354 | ) | ||||
| Non-controlling interests |
(30,614,902 | ) | (27,363,392 | ) | ||||
| (152,781,267 | ) | (100,213,746 | ) | |||||
| $ | 5,010,309 | $ | 9,836,321 | |||||
The accompanying footnotes are an integral part of these consolidated financial statements.
F-4
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED STATEMENTS OF OPERATIONS
| 2010 | 2009 | |||||||
| Net Sales: |
||||||||
| Products |
$ | 4,160,095 | $ | 2,202,407 | ||||
| Services |
6,295,418 | 8,353,531 | ||||||
| Total net sales |
10,455,513 | 10,555,938 | ||||||
| Cost of sales: |
||||||||
| Products |
1,959,935 | 1,456,090 | ||||||
| Services |
4,953,798 | 6,381,677 | ||||||
| Total cost of sales (exclusive of amortization of acquired product rights) |
6,913,733 | 7,837,767 | ||||||
| Gross margin |
3,541,780 | 2,718,171 | ||||||
| Operating expenses: |
||||||||
| Research and development |
1,303,638 | 1,320,212 | ||||||
| Royalty |
20,000 | 20,000 | ||||||
| Sales and marketing |
119,619 | 133,977 | ||||||
| General and administrative |
5,318,559 | 8,077,725 | ||||||
| Impairment of intangible assets and goodwill |
394,570 | 1,519,367 | ||||||
| Total operating expenses |
7,156,386 | 11,071,281 | ||||||
| Operating loss |
(3,614,606 | ) | (8,353,110 | ) | ||||
| Other income (expense): |
||||||||
| Interest expense, including change in fair market value of convertible debentures |
(16,659,590 | ) | (8,436,835 | ) | ||||
| Derivative gain (loss) |
(28,783,951 | ) | 14,439,361 | |||||
| Loss on deconsolidation of insolvent subsidiary |
(845,314 | ) | — | |||||
| Other income (expense) |
657,502 | (422,050 | ) | |||||
| Loss before reorganization items, non-controlling interest in losses from variable interest entities, discontinued operations and income taxes |
(49,245,959 | ) | (2,772,634 | ) | ||||
| Reorganization items: |
||||||||
| Professional Fees |
(1,070,276 | ) | (759,000 | ) | ||||
| Gain on reorganization |
59,188 | — | ||||||
| Provision for the rejection of lease (net of $43,553 gain on allowance for leasehold improvements) |
— | 29,553 | ||||||
| Provision for indemnity agreements |
2,061,818 | (3,894,545 | ) | |||||
| 1,050,730 | (4,623,992 | ) | ||||||
| Loss before non-controlling interest in variable interest entities |
(48,195,229 | ) | (7,396,626 | ) | ||||
| Non-controlling interest in losses from variable interest entities |
411,711 | 454,926 | ||||||
| Loss before discontinued operations and income taxes |
(47,783,518 | ) | (6,941,700 | ) | ||||
| (Loss) income from discontinued operations |
(12,159 | ) | 1,683,848 | |||||
| (Loss) income before taxes |
(47,795,677 | ) | (5,257,852 | ) | ||||
| Income taxes |
(3,156 | ) | (4,840 | ) | ||||
| Net Loss |
(47,798,833 | ) | (5,262,692 | ) | ||||
| Preferred stock dividend |
(2,332,505 | ) | (2,799,035 | ) | ||||
(Continued)
F-5
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED STATEMENTS OF OPERATIONS
(Continued)
| 2010 | 2009 | |||||||
| Loss attributable to common shareholders |
$ | (50,131,338 | ) | $ | (8,061,727 | ) | ||
| Weighted average shares outstanding, basic and diluted |
58,048,208 | 58,025,558 | ||||||
| Per share amounts, basic and diluted: |
||||||||
| Continuing operations |
(0.82 | ) | (0.12 | ) | ||||
| Discontinued operations |
0.00 | 0.03 | ||||||
| Loss attributable to common stockholders per common share |
$ | (0.86 | ) | $ | (0.14 | ) | ||
The accompanying footnotes are an integral part of these consolidated financial statements.
F-6
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
YEARS ENDED SEPTEMBER 30, 2010 AND 2009
| Common Stock | ||||||||||||||||||||||||||||
| Shares | Amount | Additional Paid In Capital |
Treasury Stock |
Accumulated Deficit |
Non-Controlling Interest |
Total | ||||||||||||||||||||||
| Balances, October 1, 2008 |
54,103,894 | $ | 54,104 | $ | 200,944,102 | $ | (170,057 | ) | $ | (272,926,155 | ) | $ | (23,759,405 | ) | $ | (95,857,411 | ) | |||||||||||
| Share-based compensation |
— | — | 1,716,854 | — | — | — | 1,716,854 | |||||||||||||||||||||
| Common stock issued upon the conversion of debentures and related interest |
3,353,800 | 3,354 | 1,672,970 | — | — | — | 1,676,324 | |||||||||||||||||||||
| Common stock issued upon the conversion of preferred stock |
590,514 | 590 | 79,334 | — | — | — | 79,924 | |||||||||||||||||||||
| Biovest common stock issued for payment of interest on outstanding debt |
— | — | 242,063 | — | — | — | 242,063 | |||||||||||||||||||||
| Common stock of Biovest issued for extension of debt |
— | — | 32,000 | — | — | — | 32,000 | |||||||||||||||||||||
| Biovest common stock warrants issued for modification of debt |
— | — | 62,325 | — | — | — | 62,325 | |||||||||||||||||||||
| Reclassification of financial instruments from derivative liabilities to equity |
— | — | 350,829 | — | — | — | 350,829 | |||||||||||||||||||||
| Accretion of preferred stock liability |
— | — | — | — | (2,799,035 | ) | (2,799,035 | ) | ||||||||||||||||||||
| Net loss for the year |
— | — | — | — | (2,113,632 | ) | (3,603,987 | ) | (5,717,619 | ) | ||||||||||||||||||
| Balances, September 30, 2009 |
58,048,208 | $ | 58,048 | $ | 205,100,477 | $ | (170,057 | ) | $ | (277,838,822 | ) | $ | (27,363,392 | ) | $ | (100,213,746 | ) | |||||||||||
The accompanying footnotes are an integral part of these consolidated financial statements.
F-7
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ DEFICIT
YEARS ENDED SEPTEMBER 30, 2010 AND 2009
(Continued)
| Common Stock | ||||||||||||||||||||||||||||
| Shares | Amount | Additional Paid
In Capital |
Treasury Stock |
Accumulated Deficit |
Non-Controlling Interest |
Total | ||||||||||||||||||||||
| Balances, October 1, 2009 |
58,048,208 | $ | 58,048 | $ | 205,100,477 | $ | (170,057 | ) | $ | (277,838,822 | ) | $ | (27,363,392 | ) | $ | (100,213,746 | ) | |||||||||||
| Share-based compensation |
— | — | 482,313 | — | — | — | 482,313 | |||||||||||||||||||||
| Cumulative effect of change in accounting principle |
— | — | (2,154,426 | ) | — | (752,359 | ) | — | (2,906,785 | ) | ||||||||||||||||||
| Biovest shares issued in settlement agreement |
— | — | 400,000 | — | — | — | 400,000 | |||||||||||||||||||||
| Accretion of preferred stock liability |
— | — | — | — | (2,332,505 | ) | — | (2,332,505 | ) | |||||||||||||||||||
| Net loss for the year |
— | — | — | — | (44,959,034 | ) | (3,251,510 | ) | (48,210,544 | ) | ||||||||||||||||||
| Balances, September 30, 2010 |
54,103,894 | $ | 58,048 | $ | 203,838,364 | $ | (170,057 | ) | $ | (325,882,720 | ) | $ | (30,614,902 | ) | $ | (152,781,267 | ) | |||||||||||
F-8
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
(DEBTOR-IN-POSSESSION)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| For the Years ended September 30, | ||||||||
| 2010 | 2009 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss before non-controlling interest in variable interest entity and subsidiary |
$ | (48,210,544 | ) | $ | (5,717,619 | ) | ||
| Adjustments to reconcile net loss to net cash flows from operating activities: |
||||||||
| Loss on disposal of assets |
— | 335,000 | ||||||
| Depreciation |
273,870 | 248,666 | ||||||
| Amortization |
318,395 | 233,497 | ||||||
| Impairment of intangibles |
94,133 | 1,519,367 | ||||||
| Impairment of goodwill |
300,437 | — | ||||||
| Share-based compensation |
482,313 | 1,716,854 | ||||||
| Accretion of debt discounts |
6,186,111 | 6,276,204 | ||||||
| Accretion of royalty liability |
617,690 | 557,302 | ||||||
| Accretion of capitalized finance costs |
1,149,773 | 1,474,322 | ||||||
| Derivative gain (loss) |
28,783,951 | (14,439,361 | ) | |||||
| Change in fair market value adjustment of convertible debentures (charged to interest expense) |
2,955,701 | (5,671,361 | ) | |||||
| Issuance of common stock warrants for finance costs |
— | 32,000 | ||||||
| Gain on settlement |
(342,001 | ) | — | |||||
| Proceeds from settlement |
2,500,000 | — | ||||||
| Gain on reorganization |
(59,187 | ) | — | |||||
| Increase (decrease) in cash resulting from changes in: |
||||||||
| Accounts receivable |
125,193 | 921,889 | ||||||
| Inventories |
92,200 | 703,451 | ||||||
| Unbilled receivables |
1,352,566 | 711,401 | ||||||
| Prepaid expenses and other current assets |
140,843 | 194,973 | ||||||
| Other assets |
74,857 | 37,592 | ||||||
| Assets from discontinued operations |
— | (664,144 | ) | |||||
| Accounts payable |
(741,308 | ) | 1,848,507 | |||||
| Accrued expenses |
4,250,576 | 9,764,866 | ||||||
| Unearned revenues |
(691,859 | ) | (262,297 | ) | ||||
| Customer deposits |
(73,944 | ) | 39,218 | |||||
| Liabilities from discontinued operations |
— | (1,792,670 | ) | |||||
| Net cash flows from operating activities |
(420,234 | ) | (1,932,343 | ) | ||||
| Cash flows from investing activities: |
||||||||
| Acquisition of furniture, equipment, and leasehold improvements |
(72,133 | ) | — | |||||
| Net cash flows from investing activities |
$ | (72,133 | ) | $ | — | |||
(Continued)
F-9
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
(Continued)
| For the Years ended September 30, | ||||||||
| 2010 | 2009 | |||||||
| Cash flows from financing activities: |
||||||||
| Payments on notes payable and long-term debt |
$ | — | $ | (50,000 | ) | |||
| Proceeds (payments) made to related party |
125,469 | (17,024 | ) | |||||
| Payment of financing costs |
(40,000 | ) | (64,613 | ) | ||||
| Proceeds (payments) on related party loans including line of credit |
640,000 | 1,250,000 | ||||||
| Net cash flows from financing activities |
725,469 | 1,118,363 | ||||||
| Net change in cash and cash equivalents |
233,102 | (813,980 | ) | |||||
| Cash and cash equivalents at beginning of period |
325,350 | 1,139,330 | ||||||
| Cash and cash equivalents at end of period |
$ | 558,452 | $ | 325,350 | ||||
| Supplemental cash flow information: |
||||||||
| Cash paid for: |
||||||||
| Interest |
$ | — | $ | 16,667 | ||||
| Income taxes |
$ | 3,156 | $ | 3,000 | ||||
Supplemental Disclosure of Non-cash Investing and Financing Activities:
2010:
| • | The cumulative effect of change in accounting principle was approximately $2.9 million. |
| • | Biovest issued 600,000 shares of Biovest common stock in a settlement agreement. |
2009:
| • | The Company issued 2,342,624 shares of common stock upon the conversion of $1.3 million of convertible debentures (face value). |
| • | The Company issued 1,011,176 shares of common stock for $0.5 million of accrued interest payable. |
| • | The Company issued 590,514 shares of common stock upon the conversion of $0.3 million of preferred stock. |
The accompanying footnotes are an integral part of these consolidated financial statements.
F-10
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Description of business and summary of significant accounting policies:
Business and organization
Headquartered in Tampa, Florida, Accentia Biopharmaceuticals, Inc. (Other OTC: “ABPI”) is a biotechnology company that is developing Revimmune™ as a comprehensive system of care for the treatment of autoimmune diseases. Additionally, through the Company’s, majority-owned subsidiary, Biovest International, Inc., the Company is developing BiovaxID® as a therapeutic cancer vaccine for treatment of follicular non-Hodgkin’s lymphoma (“FL”) and mantle cell lymphoma (“MCL”). Through the Company’s wholly-owned subsidiary, Analytica International, Inc., they conduct a health economics research and consulting business which Analytica markets to the pharmaceutical and biotechnology industries, using the Company’s operating cash flow to support the Company’s corporate administration and product development activities.
Revimmune™ is being developed as a treatment for various autoimmune diseases. The approximately 80 known autoimmune diseases generally arise from an overactive immune response against substances and/or tissue normally present in the body. As a system of care, Revimmune consists of administering high, pulsed doses of an FDA-approved drug, cyclophosphamide, over a four-day interval as part of an integrated risk management system including a panel of preventive tests, monitoring and medications which are intended to minimize risks while seeking to maximize the clinical benefit.
Through a collaboration with the National Cancer Institute (“NCI”), the Company’s majority-owned subsidiary, Biovest International, Inc. (OTCQB: “BVTI”) (“Biovest”) has developed a patient-specific cancer vaccine, BiovaxID®, which has demonstrated statistically significant Phase 3 clinical benefit by prolonging disease-free survival in follicular non-Hodgkin’s lymphoma patients treated with BiovaxID.
Additionally, through the Company’s wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), based in New York City, the Company conducts a global research and strategy consulting business that provides professional services to the pharmaceutical and biotechnology industries. Since 1997, Analytica has expertly directed research studies and projects, including traditional health economic modeling projects, database studies, structured reviews, health technology assessments, reimbursement analyses, and value dossiers.
Principles of consolidation
The Company consolidates all entities controlled by ownership of a majority interest and, effective February 27, 2007, has consolidated a variable interest entity of which the Company is the primary beneficiary. The consolidated financial statements include Accentia Biopharmaceuticals, Inc. and its wholly-owned subsidiaries, Analytica International, Inc. (“Analytica”), TEAMM Pharmaceuticals, Inc. d/b/a Accentia Pharmaceuticals (“TEAMM” or “Accentia Pharmaceuticals”), AccentRx, Inc. (“AccentRx”), and Accentia Specialty Pharmacy (“ASP”); its majority owned subsidiary, Biovest (and its consolidated entities), and Revimmune, LLC, an entity in which the Company has a controlling financial interest and has been determined to be the primary beneficiary. All significant intercompany balances and transactions have been eliminated in consolidation.
The consolidated financial statements of Biovest include its wholly owned subsidiaries, Biovax, Inc., AutovaxID, Inc., Biolender, LLC and Biolender II, LLC; and certain variable interest entities of Biovest, Biovax Investment LLC, Telesis CDE Two LLC, AutovaxID Investment LLC, and St. Louis New Markets Tax Credit Fund II LLC.
F-11
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Variable interest entities
The Company evaluates all significant arrangements and relationships for indications of variable interest entities pursuant to generally accepted accounting principles. During April 2006 and December 2006, the Company and Biovest entered into financing arrangements that involved entities that met the definition of variable interest entities. As a result, the Company and Biovest were required to consolidate these entities and reflect the non-controlling interest in the consolidated financial statements. The consolidated financial statements include the variable interest entities as follows: Biovest Investment, LLC, Telesis CDE Two LLC, AutovaxID Investment LLC, St. Louis New Market Tax Credit Fund II LLC and Revimmune, LLC.
Voluntary Petition for Bankruptcy:
On November 10, 2008, the Company and its wholly-owned subsidiaries, Analytica International, Inc. (“Analytica”), TEAMM Pharmaceuticals, Inc. d/b/a Accentia Pharmaceuticals (“TEAMM” or “Accentia Pharmaceuticals”), AccentRx, Inc. (“AccentRx”), and Accentia Specialty Pharmacy (“ASP”) (collectively, the “Debtors”) filed voluntary petitions for reorganization under Chapter 11 in the Bankruptcy Court. During the pendency of the Chapter 11 proceedings, the Company operated its business as a debtor-in-possession in accordance with the provisions of Chapter 11, and was subject to the jurisdiction of the Bankruptcy Court. On August 16, 2010, the Company filed its First Amended Joint Plan of Reorganization, and, on October 25, 2010, the Company filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a Confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. The Company emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”)
ASC Topic 852-Reorganizations, which is applicable to companies in Chapter 11, generally does not change the manner in which financial statements are prepared. However, it does require that the financial statements for periods subsequent to the filing of the Chapter 11 petition distinguish transactions and events that are directly associated with the reorganization from the ongoing operations of the business. Revenues, expenses, realized gains and losses, and provisions for losses that can be directly associated with the reorganization and restructuring of the business must be reported separately as reorganization items in the statements of operations beginning in the quarter ending December 31, 2008. The balance sheet must distinguish prepetition liabilities subject to compromise from both those prepetition liabilities that are not subject to compromise and from post-petition liabilities. Liabilities that may be affected by a plan of reorganization must be reported at the amounts expected to be allowed, even if they may be settled for lesser amounts. In addition, cash provided by reorganization items must be disclosed separately in the statement of cash flows. Accentia became subject to ASC Topic 852 on November 10, 2008, and will segregate those items as outlined above for all reporting periods subsequent to such date.
Use of estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make judgments, assumptions and estimates that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ materially from those estimates.
Cash and cash equivalents
The Company considers all highly-liquid investments purchased with an original maturity of three months or less to be cash equivalents.
F-12
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Accounts receivable, concentrations of credit risk and customer concentrations
Financial instruments that subject the Company to concentrations of credit risk include cash and accounts receivable. The Company places its cash in several high-quality financial institutions. Such amounts are insured by the FDIC up to $0.25 million per institution.
Accounts receivable are customer obligations due under normal trade terms. The Company sells its products and services to retail organizations and drug development companies. The Company performs ongoing credit evaluations of customers’ financial condition and does not require collateral.
Management reviews accounts receivable on a monthly basis to determine collectability. Balances that are determined to be uncollectible are written off to the allowance for doubtful accounts. The allowance for doubtful accounts contains a general accrual for estimated bad debts. Management considers the balance of approximately $.0.01 million adequate as of September 30, 2010; however actual write-offs may exceed the allowance.
Inventories
Inventories consist primarily of supplies and parts used in instrumentation assembly and related materials. Inventories are stated at the lower of cost or market with cost determined using the first-in first-out (“FIFO”) method. In evaluating whether inventory is stated at the lower of cost or market, management considers such factors as the amount of inventory on hand, estimated time required to sell such inventory, and current and expected market conditions, including levels of competition. An allowance is recorded to reduce inventories to their net realizable value, as considered necessary.
Furniture, equipment and leasehold improvements
Furniture, equipment and leasehold improvements are stated at cost, less accumulated depreciation. Depreciation is determined using straight-line and accelerated methods over the estimated useful lives of three to seven years for furniture and equipment. Amortization of leasehold improvements is over the shorter of the improvements’ estimated economic lives or the related lease terms.
Goodwill and intangible assets
Intangible assets include trademarks, product rights, noncompete agreements, technology rights, purchased customer data relationships and patents, and are accounted for based on ASC Topic 350-Intangibles. In that regard, goodwill and intangible assets that have indefinite useful lives, are not amortized but are tested at least annually for impairment or more frequently if events or changes in circumstances indicate that the asset might be impaired. The Company has identified certain trademarks, and purchased customer relationships as intangible assets with indefinite lives and, therefore, these assets are not amortized.
Intangible assets with finite useful lives are amortized over the estimated useful lives from the date of acquisition as follows:
| Customer relationships |
10 years | |||||||
| Trademarks |
3 years | |||||||
| Patents |
3 years |
The Company recognized impairment losses of $0.4 million and $1.5 million related to the impairment of intangible assets for the year ended September 30, 2010 and 2009, respectively.
Deferred finance costs
Deferred finance costs include fees paid in conjunction with obtaining long-term debt, notes payable and lines of credit and are amortized over the contractual term of the related financial instrument. The balance of the deferred finance costs at September 30, 2010 will be amortized during the year ended September 30, 2011.
F-13
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Income taxes
Deferred income tax assets and liabilities are computed annually for differences between the financial statements and income tax bases of assets and liabilities that will result in taxable or deductible amounts in the future, based on enacted tax laws and rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized.
Management has evaluated the effect of a new accounting standard related to accounting for uncertainty in income taxes that became effective in 2009 and has determined the Company had no uncertain income tax positions that could have a significant affect on the consolidated financial statements as of and for the year ended September 30, 2010. The Company’s federal income tax returns for 2007, 2008 and 2009 are subject to examination by the Internal Revenue Service, generally for three years after the federal income tax returns were filed.
Financial instruments
Financial instruments, as defined in ASC Topic 825—Financial Instruments, consist of cash, evidence of ownership in an entity and contracts that both (i) impose on one entity a contractual obligation to deliver cash or another financial instrument to a second entity, or to exchange other financial instruments on potentially unfavorable terms with the second entity, and (ii) conveys to that second entity a contractual right (a) to receive cash or another financial instrument from the first entity or (b) to exchange other financial instruments on potentially favorable terms with the first entity. Accordingly, the Company’s financial instruments consist of cash and cash equivalents, accounts receivable, accounts payable, accrued liabilities, lines of credit, notes payable, long-term debt, royalty liability, other liabilities, related party, derivative financial instruments, and convertible debentures.
The Company carries cash and cash equivalents, accounts receivable, accounts payable, accrued liabilities, and lines of credit at historical costs; their respective estimated fair values approximate carrying values due to their current nature. The Company also carries notes payable and certain convertible debt, included in long-term debt, at historical cost less discounts attributable to the concurrent issuance of detachable warrants, beneficial conversion features and bifurcated embedded derivatives. However, fair values of these debt instruments are estimated for disclosure purposes based upon the present value of the estimated cash flows at market interest rates applicable to similar instruments. The Company carries certain convertible debentures at fair value pursuant to generally accepted accounting principles.
Derivative instruments
Fair Value of Financial Assets and Liabilities
The Company measures the fair value of financial assets and liabilities in accordance with GAAP which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
GAAP defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. GAAP also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. GAAP describes three levels of inputs that may be used to measure fair value:
Level 1 – quoted prices in active markets for identical assets or liabilities
Level 2 – quoted prices for similar assets and liabilities in active markets or inputs that are observable
Level 3 – inputs that are unobservable (for example cash flow modeling inputs based on assumptions)
The Company generally does not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, the Company and its consolidated subsidiaries have entered into certain other financial instruments and contracts, such as debt financing arrangements and freestanding warrants with features that are either (i) not afforded equity classification, (ii) embody risks not clearly and closely related to host contracts, or (iii) may be net-cash settled by the counterparty. These instruments are required to be carried as derivative liabilities, at fair value.
The Company estimates fair values of all derivative instruments, such as free-standing warrants, and embedded conversion features utilizing level 3 inputs. The Company generally uses the Black-Scholes-Merton option valuation technique because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the Company’s trading market price which has high-historical volatility. Since derivative financial instruments are initially and subsequently carried at fair values, the Company’s income will reflect the volatility in these estimates.
F-14
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Foreign currency translation
The Company translates the assets and liabilities of its non-U.S. functional currency subsidiary into dollars at the current rates of exchange in effect at the end of each reporting period, while net sales and expenses are translated using the average exchange rate for each reporting period. Foreign currency translation adjustments were nominal during the year end September 30, 2010 and 2009, and as such, no adjustments have been recognized in the accompanying consolidated financial statements.
Revenue recognition
The Company recognizes revenue as follows:
Services
Service revenue is generated primarily by fixed price contracts for cell culture production and consulting services. Such revenue is recognized over the contract term based on the percentage of services cost incurred during the period compared to the total estimated service cost to be incurred over the entire contract. The nature and scope of the Company’s contracts often require the Company to make judgments and estimates in recognizing revenues. Estimates of total contract revenues and costs are continuously monitored during the term of the contract, and recorded revenues and costs are subject to revision as each contract progresses. Such revisions may result in increases or decreases to revenues and income and are reflected in the consolidated financial statements in the periods in which they are first identified. Losses on contracts are recognized during the period in which the loss first becomes probable and reasonably estimable. Reimbursements of contract-related expenses are included in revenues. An equivalent amount of these reimbursable costs is included in cost of sales. Because of the inherent uncertainties in estimating costs, it is at least reasonably possible that the estimates used will change within the near term.
Contract costs related to cell culture production include all direct material, subcontract and labor costs and those indirect costs related to contract performance, such as indirect labor, insurance, supplies and tools. The Company believes that actual cost incurred in contract cell production services is the best indicator of the performance of the contractual obligations, because the costs relate primarily to the amount of labor incurred to perform such services. The deliverables inherent in each of the Company’s cell culture production contracts are not output driven, but rather driven by a pre-determined production run. The duration of the Company’s cell culture production contracts range typically from 2 to 14 months.
Service costs relating to the Company’s consulting services consist primarily of internal labor expended in the fulfillment of the Company’s consulting projects and, to a lesser extent, outsourced research services. The duration of the Company’s consulting service contracts range typically from 1 to 6 months. Certain other professional service revenues are recognized as the services are performed.
The asset unbilled receivables represents revenue that is recognizable under the percentage of completion method due to the performance of services for which billings have not been generated as of the balance sheet date. In general, amounts become billable pursuant to contractual milestones or in accordance with predetermined billing schedules. Under the Company’s consulting services contracts, the customer is required to pay for contract hours worked by the Company (based on the standard hourly rate used to calculate the contract price) even if the customer cancels the contract and elects not to proceed to completion of the project. Unearned revenues represent customer payments in excess of revenue earned under the percentage of completion method. Such payments are made in accordance with predetermined payment schedules set forth in the contract.
Products
Net sales of instruments and disposables are recognized in the period in which the rewards of ownership have passed (at point of shipment) to the buyer. The Company does not provide its customers of instruments and disposables with a right of return; however, deposits made by customers must be returned to customers in the event of non-performance by the Company.
F-15
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
During 2004, the Company entered into an agreement with Pharmaceutical Product Development, Inc. (“PPD”), a common stockholder. In connection with the agreement, PPD acquired future royalty rights in exchange for $2.5 million received by the Company in September 2004; however, the agreement provides for return of the net purchase price ($2.5 million less royalty payments remitted to date) should royalties received by PPD through December 2009, be less than $2.5 million. In addition, there are certain other default provisions that would require the Company’s return of the net funds received. As of September 30, 2010 and 2009, the liability is approximately $2.2 million, and is included in Liabilities Subject to Compromise in the consolidated balance sheets.
Cost of sales
Cost of sales excludes amortization of acquired product rights of $0.3 million and $0.2 million for each of the years ended September 30, 2010 and 2009.
Shipping and handling costs
Shipping and handling costs are included as a component of cost of sales in the accompanying consolidated statements of operations.
Research and development expense
The Company expenses research and development costs as incurred. Such costs include payroll and related costs, facility costs, consulting and professional fees, equipment rental and maintenance, lab supplies, and certain other indirect cost allocations that are directly related to research and development activities. Research and development expense, related party consists of expenses incurred with a vendor that is a greater than 10% shareholder.
Share-based compensation
The Company follows the guidance of the accounting provisions of ASC Topic 718—Share-based Compensation, which requires the use of the fair value based method to determine compensation for all arrangements under which employees and others receive shares of stock or equity instruments (warrants and options). The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. Expected volatilities are based on weighted averages of the limited historical volatility of the Company’s stock and selected peer group comparable volatilities and other factors estimated over the expected term of the options. The expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for the period of the expected term.
In applying the Black-Scholes options-pricing model, assumptions are as follows:
| 2010 | 2009 | |||||||
| Dividend yield |
$ | 0 | $ | 0 | ||||
| Expected volatility |
140.08 | % | 148.5 | % | ||||
| Risk free interest rate |
2.35% – 3.11 | % | 1.89 – 2.43 | % | ||||
| Expected life |
5.0 to 6.0 years | 5.0 to 6.0 years | ||||||
Loss per common share
The Company had net losses for all periods presented in which potential common shares were in existence. Diluted loss per share assumes conversion of all potentially dilutive outstanding common stock options, warrants, or other convertible financial instruments. Potential common shares outstanding are excluded from the calculation of diluted loss per share if their effect is anti-dilutive. As such, dilutive loss per share is the same as basic loss per share for all periods presented as the effect of all potential common shares outstanding is anti-dilutive.
F-16
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following table sets forth the calculations of basic and diluted loss per share:
| 2010 | 2009 | |||||||
| Numerator: |
||||||||
| Loss applicable to common stockholders |
$ | (50,131,338 | ) | $ | (8,061,727 | ) | ||
| Denominator: |
||||||||
| Weighted average shares—basic loss per share |
58,048,208 | 58,025,558 | ||||||
| Effect of dilutive securities |
— | — | ||||||
| Weighted average shares for dilutive loss per share |
58,048,208 | 58,025,558 | ||||||
| Gain (loss) per shares from discontinued operations |
$ | — | $ | 0.03 | ||||
| Loss per share applicable to common stockholders, basic and diluted |
$ | (0.86 | ) | $ | (0.14 | ) | ||
| Effect on loss per share from preferred dividends |
$ | 0.04 | $ | 0.05 | ||||
The effect of common stock equivalents and common shares indexed to convertible debt securities are not considered in the calculation of diluted loss per share because the effect would be anti-dilutive. They are as follows as of:
| 2010 | 2009 | |||||||
| Options and warrants to purchase common stock |
44,536,380 | 29,553,837 | ||||||
| Convertible debt instruments |
30,454,666 | 28,192,866 | ||||||
| Preferred stock convertible to common stock |
4,906,060 | 4,906,060 | ||||||
| 79,897,106 | 62,652,763 | |||||||
Recent Accounting Pronouncements:
In December 2007, the FASB issued new guidance on accounting for business combinations which establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed, any non-controlling interest in the acquiree and the goodwill acquired. This new guidance also establishes disclosure requirements to enable the evaluation of the nature and financial effects of the business combination. This new guidance is effective for fiscal years beginning after December 15, 2008. Earlier adoption is prohibited. The adoption of this standard is not expected to have a material effect on our results of operations or financial position.
In December 2007, the FASB issued new guidance to establish accounting and reporting standards for the non-controlling interest in a subsidiary and for the deconsolidation of a subsidiary. It clarifies that a non-controlling interest in a subsidiary, which is sometimes referred to as minority interest, is an ownership interest in the consolidated entity that should be reported as equity in the consolidated financial statements. Among other requirements, this statement requires consolidated net income to be reported at amounts that include the amounts attributable to both the parent and the non-controlling interest. It also requires disclosure, on the face of the consolidated income statement, of the amounts of consolidated net income attributable to the parent and to the non-controlling interest. This will require a reclassification of non-controlling interests in variable interest entities on the Company’s consolidated balance sheet to equity and the elimination of non-controlling interest in losses from variable interest entities on the consolidated statement of operations once adopted for our fiscal year beginning October 1, 2010 the adoption of this standard required a reclassification of approximately $25.0 million from accumulated deficit and non-controlling interest in variable interests entities in the liabilities section of the consolidated balance sheet to non-controlling interests in the statement of stockholders’ deficit.
In June 2008, the FASB issued new guidance for determining whether an equity-linked financial instrument (or embedded feature) is indexed to an entity’s own stock. The Company adopted this new guidance effective October 1, 2009. Certain of the Company’s outstanding warrants and convertible debt contain features which fall under the scope of this guidance resulting in a decrease of $2.2 million and $0.8 million to the opening balances of additional paid-in capital and accumulated deficit respectively.
F-17
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In May 2009, the FASB issued new guidance to be used in the evaluation of trends and transactions that may occur and result in recognition or disclosure in the financial statements after the balance sheet date through the date the financial statements are issued. Provisions within this new guidance address the circumstances under which such events or transactions which occur subsequent to the reporting period-end must be recognized in the financial statements. The Company has adopted this new guidance during fiscal year 2009 and it did not have a material effect on its consolidated financial statements.
In June 2009, the FASB issued new guidance amending the existing pronouncement related to the consolidation of variable interest entities. This new guidance requires the reporting entities to evaluate former qualified special purpose entities’ for consolidation, changes the approach to determine a variable interest entity’s primary beneficiary from a quantitative assessment to a qualitative assessment designed to identify a controlling financial interest, and increases the frequency of required assessments to determine whether we are the primary beneficiary of any variable interest entities which we are a party to. This new guidance is not effective for us until October 1, 2010 and earlier adoption is prohibited. We are currently evaluating the impact the adoption of this new guidance will have on our consolidated financial statements.
In October 2009, the FASB issued Accounting Standards Update No. 2009-13, or ASU 2009-13, which addresses the accounting for multiple-deliverable arrangements to enable vendors to account for products or services (deliverables) separately rather than as a combined unit. ASU 2009-13 is effective prospectively for revenue arrangements entered into or materially modified beginning in fiscal years on or after June 15, 2010. Early adoption is permitted. We are currently evaluating the impact that the adoption of this standard will have on our financial statements, if any.
In April 2010, the FASB issued Accounting Standards Update No. 2010-17 (“ASU 2010-17”) which provides guidance on defining a milestone and determining when it may be appropriate to apply the milestone method of revenue recognition for research or development transactions. ASU 2010-17 is effective prospectively for milestones achieved in fiscal years and interim periods within those years, beginning in fiscal years on or after June 15, 2010. The Company has adopted this standard and adjusted its revenue recognition policy to apply the milestone method of revenue recognition for future research and development contracts.
In January 2010, the FASB issued ASU 2010-06- Improving Disclosures about Fair Value Measurements. ASU 2010-06 requires additional disclosures about fair value measurements including transfers in and out of Levels 1 and 2 and a higher level of disaggregation for the different types of financial instruments. For the reconciliation of Level 3 fair value measurements, information about purchases, sales, issuances and settlements should be presented separately. This ASU is effective for annual and interim reporting periods beginning after December 15, 2009 for most of the new disclosures and for periods beginning after December 15, 2010 for the new Level 3 disclosures. Comparative disclosures are not required in the first year the disclosures are required. The Company is currently reviewing the guidance and will update its disclosures when the guidance becomes applicable.
2. Liquidity and management’s plans:
The accompanying consolidated financial statements have been prepared on a going concern basis, which assumes the Company will realize its assets and discharge its liabilities in the normal course of business. As reflected in the accompanying consolidated financial statements, the Company incurred net losses of $47.8 million during the twelve months ended September 30, 2010. The Company’s independent auditors issued a “going concern” uncertainty on the financial statements for the year ended September 30, 2010, citing significant losses and working capital deficits at that date, which raised substantial doubt about the Company’s ability to continue as a going concern.
F-18
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Regulatory strategy and commercialization expenditures:
Notwithstanding the prior reports of High-Dose Pulsed Cyclophosphamide as a potential therapy for certain autoimmune diseases. cyclophosphamide is not currently FDA-approved for the treatment of any autoimmune diseases. These prior studies of High-Dose Pulsed Cyclophosphamide in the United States have been conducted at a limited number of large academic research hospitals and have featured non-uniform inclusion criteria and/or administration schedules. It is generally recognized that there may be significant potential risks of infection or other side effects when High-Dose Pulsed Cyclophosphamide is not administered by highly-qualified personnel in a controlled and regulated clinical setting. While the previous studies are important to the Company’s Revimmune development plan, they are not considered sufficient to support regulatory approval. At the core of the Company’s Revimmune development plan is a recognition that controlled and randomized clinical trials will be necessary to demonstrate the efficacy of High-Dose Pulsed Cyclophosphamide to the satisfaction of the FDA and that safety will be an important regulatory and clinical concern which we believe will require an FDA approved formal REMS.
There are approximately 80 recognized autoimmune diseases. The Company previously planned to conduct its first randomized and controlled trial of Revimmune in multiple sclerosis (MS), because of the potentially significant impact of such a study and the number of MS patients previously treated with High Dose Cyclophosphamide. However, due to the Company’s limited capital and resources, the Company has modified its Revimmune development plan so as to initially focus on two less prevalent autoimmune diseases where the Company perceives a significant unmet medical need: diffuse systemic sclerosis and autoimmune hemolytic anemia. Subject to the Company’s capital availability, it plans to undertake clinical trials of Revimmune for the treatment of these two autoimmune diseases in a parallel manner. Diffuse systemic sclerosis and autoimmune hemolytic anemia represent diseases for which the Company believes significant unmet medical needs exist. There is no standard treatment for systemic sclerosis, and while the standard treatment for autoimmune hemolytic anemia is effective for a subset of patients, patients for which this treatment options is not effective, referred to as refractory patients, have very limited options. The Company believes that the significant unmet medical need, the shorter and severe disease course and the smaller prevalence of these autoimmune diseases make them appropriate for the Company’s initial focus. Notwithstanding the initial focus of the Company’s current Revimmune development plan, it continues to believe that new treatment options are needed for MS and the Company remains interested in ultimately conducting studies with Revimmune in MS.
In September 2007, the Company conducted a meeting with the FDA regarding its proposed design of a clinical trial for Revimmune to treat MS. The Company considered the FDA meeting to have been constructive. However, with the change in the Company’s Revimmune development plan, it is currently preparing for an additional meeting with the FDA to discuss possible studies in diffuse scleroderma and/or autoimmune hemolytoic anemia. Based on FDA input, the Company anticipates filing an Investigational New Drug Application(s) (IND) under which it hopes to conduct its planned clinical trials. Further, the Company plans to discuss with the FDA its plans for a REMS to be developed under the Food and Drug Administration Amendments Act of 2007.
Provided that the Company’s planned clinical trials, once completed, demonstrate clinical benefit and safety, the Company would anticipate discussing next steps with the FDA which could include the suitability of seeking conditional accelerated approval and/or the appropriateness and design of additional trial(s).
Biovest has completed its Phase 3 clinical trial of BiovaxID for the indication of FL. Under Biovest’s current regulatory strategy, the Company is performing in-depth analyses of the available clinical trial data in preparation for submission of the data to the Federal Food and Drug Administration (“FDA”) and European Medicines Agency (“EMEA”). Based upon a recommendation from the independent Data Monitoring Committee (“DMC”) which monitors the safety and efficacy profile of the clinical trial and Biovest’s analysis of the available clinical trial data, Biovest plans to seek accelerated and conditional approval of BiovaxID® with FDA, EMEA, and other international agencies, for the indication of FL. Pending the outcome of these anticipated applications for accelerated and conditional approval, Biovest has ceased enrolling new patients in its Phase 3 clinical trial and has discontinued most clinical trial activities which had the effect of decreasing clinical trial expenses compared to those recorded for prior periods. Accelerated or conditional approval would require Biovest to perform additional clinical studies as a condition to continued marketing of BiovaxID. Accordingly, should Biovest receive accelerated and/or conditional approval, clinical trial activities and related expenses may return to the levels experienced in periods prior to the application for conditional approval until any such clinical trial activity is completed. There can be no assurances Biovest will receive accelerated or conditional approval. Biovest’s ability to timely access required financing will continue to be essential to support the ongoing commercialization efforts. Biovest’s inability to obtain required funds or any substantial delay in obtaining required funds will have a material adverse effect on the ongoing commercialization efforts.
F-19
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Additional expected financing activity:
Management intends to attempt to meet its cash requirements through proceeds from its cell culture and instrument manufacturing activities, the use of cash on hand, trade vendor credit, restructuring of our outstanding debt obligations through the Chapter 11 reorganization proceedings, short-term borrowings, debt and equity financings, and strategic transactions such as collaborations and licensing. The Company’s ability to continue present operations, pay its liabilities as they become due, and meet its obligations for vaccine development is dependent upon the Company’s ability to obtain significant external funding in the very short term. Additional sources of funding have not been established; however, additional financing is currently being sought by the Company from a number of sources, including the sale of equity or debt securities, strategic collaborations, recognized research funding programs, as well as domestic and/or foreign licensing of the Company’s vaccine. Management is currently in the process of exploring these various financing alternatives. There can be no assurance that the Company will be successful in securing such financing at acceptable terms, if at all. Accordingly, our ability to continue present operations, pay our existing liabilities as they become due, and continue our ongoing clinical trial is dependent upon our ability to successfully complete the Chapter 11 reorganization process and to obtain significant external funding from sources other than Accentia in the very near term, which raises substantial doubt about our ability to continue as a going concern. If adequate funds are not available from the foregoing sources in the near term, or if the Company determines it to otherwise be in the Company’s best interest, the Company may consider additional strategic financing options, including sales of assets, or the Company may be required to delay, reduce the scope of, or eliminate one or more of its research or development programs or curtail some or all of its commercialization efforts.
3. Discontinued operations:
Disposition
On November 10, 2008, in conjunction with the Company’s Chapter 11 bankruptcy filing, the Company decided to cease operation their 100% owned subsidiary, TEAMM Pharmaceuticals, Inc., a/k/a Accentia Pharmaceuticals (“TEAMM”)
The operating results for the year ended September 30, 2010 and 2009 are reported as discontinued operations.
The following represents a summary of the Company’s operating results for TEAMM:
| Years Ended September 30, | ||||||||
| 2010 | 2009 | |||||||
| Net sales |
$ | — | $ | 1,254,613 | ||||
| Cost of sales |
— | 311,501 | ||||||
| Gross margin |
— | 943,112 | ||||||
| Operating expenses |
12,159 | 776,731 | ||||||
| Recovery of inventory and sales reserves |
— | (1,517,467 | ) | |||||
| Income (loss) from discontinued operations |
(12,159 | ) | 1,683,848 | |||||
| Non-current assets: |
||||||||
| Furniture, equipment and leasehold improvements, net |
— | 12,159 | ||||||
| Total assets |
— | 12,159 | ||||||
| Current liabilities: |
||||||||
| Accounts payable |
852,587 | 852,587 | ||||||
| Accrued expenses |
425,557 | 425,557 | ||||||
| Total current liabilities (1) |
$ | 1,278,144 | $ | 1,278,144 | ||||
| (1) | 2009 and 2010 current liabilities included in Liabilities Subject to Compromise on the Consolidated Balance Sheet |
F-20
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
4. Inventories:
Inventories consist of the following:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Finished goods, other |
$ | 104,155 | $ | 147,431 | ||||
| Work-in-process |
— | 7,890 | ||||||
| Raw materials |
312,932 | 353,966 | ||||||
| $ | 417,087 | $ | 509,287 | |||||
5. Unbilled receivables and unearned revenues:
Unbilled receivables and unearned revenues are as follows:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Costs incurred on uncompleted service contracts |
$ | 978,628 | $ | 11,371,596 | ||||
| Estimated earnings |
1,253,352 | 4,902,625 | ||||||
| 2,231,980 | 16,274,221 | |||||||
| Less billings to date |
(2,344,455 | ) | (15,725,989 | ) | ||||
| $ | (112,475 | ) | $ | 548,232 | ||||
These amounts are presented in the accompanying consolidated balance sheets as follows:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Unbilled receivables |
$ | 151,303 | $ | 1,503,869 | ||||
| Unearned revenues |
(263,778 | ) | (955,637 | ) | ||||
| $ | (112,475 | ) | $ | 548,232 | ||||
F-21
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
6. Intangible assets:
Intangible assets consist of the following:
| September 30, | Weighted Average Amortization Period |
|||||||||||
| 2010 | 2009 | |||||||||||
| Indefinite-life intangible assets: |
||||||||||||
| Trademarks (a) |
— | $ | 1,176,433 | |||||||||
| Amortizable intangible assets: |
||||||||||||
| Noncompete agreements |
2,104,000 | 2,104,000 | 3.5 years | |||||||||
| Patents |
103,244 | 149,872 | 3.0 years | |||||||||
| Purchased customer relationships |
666,463 | 1,043,813 | 10.0 years | |||||||||
| Product rights |
28,321 | 1,628,321 | 18.4 years | |||||||||
| Software |
438,329 | 498,416 | 3.5 years | |||||||||
| Trademarks |
1,285,960 | 109,527 | 3.0 years | |||||||||
| 4,626,317 | 5,533,949 | |||||||||||
| Less accumulated amortization |
(3,542,355 | ) | (4,077,292 | ) | ||||||||
| 1,083,962 | 1,456,657 | |||||||||||
| Total intangible assets |
$ | 1,083,962 | $ | 2,633,090 | ||||||||
| (a) | During the year ended September 30, 2010, the Company determined certain trademarks had finite lives and began amortizing these assets over their remaining useful lives. |
F-22
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following represents intangible asset additions, amortization, disposals, and impairment of intangible assets for the years ended September 30, 2010, and 2009:
| Indefinite Life Intangibles | Amortizable Intangible Assets | |||||||||||||||||||||||||||||||||||||||||||||||
| Trademarks | Purchased Customer Relationships |
Total | Noncompete Agreements |
Patents | Purchased Customer Relationships |
Software | Trademarks | Product Rights |
Accumulated Amortization |
Total | Grand Total |
|||||||||||||||||||||||||||||||||||||
| Balance, September 30, 2008 |
1,176,433 | 225,137 | 1,401,570 | 2,104,000 | 149,872 | 1,043,813 | 498,416 | 109,527 | 3,706,813 | (4,628,057 | ) | 2,984,384 | 4,385,954 | |||||||||||||||||||||||||||||||||||
| Amortization |
— | — | — | — | — | — | — | (233,497 | ) | (233,497 | ) | (233,497 | ) | |||||||||||||||||||||||||||||||||||
| Impairments (1) |
(225,137 | ) | (225,137 | ) | — | — | — | — | — | (2,078,490 | ) | 784,260 | (1,294,230 | ) | (1,519,367 | ) | ||||||||||||||||||||||||||||||||
| Balance, September 30, 2009 |
1,176,433 | — | 1,176,433 | 2,104,000 | 149,872 | 1,043,813 | 498,416 | 109,527 | 1,628,323 | (4,077,294 | ) | 1,456,657 | 2,633,090 | |||||||||||||||||||||||||||||||||||
| Reclass |
(1,176,433 | ) | — | (1,176,433 | ) | — | — | — | — | 1,176,433 | — | — | 1,176,433 | — | ||||||||||||||||||||||||||||||||||
| Amortization |
— | — | — | — | — | — | — | — | — | (318,395 | ) | (318,395 | ) | (318,395 | ) | |||||||||||||||||||||||||||||||||
| Impairments (2) |
— | — | — | — | — | (377,350 | ) | — | — | — | 283,217 | (94,133 | ) | (94,133 | ) | |||||||||||||||||||||||||||||||||
| Disposals |
— | — | — | — | (46,628 | ) | — | (60,087 | ) | — | (1,600,002 | ) | 570,117 | (1,136,600 | ) | (1,136,600 | ) | |||||||||||||||||||||||||||||||
| Balance, September 30, 2010 |
— | — | — | 2,104,000 | 103,244 | 666,463 | 438,329 | 1,285,960 | 28,321 | (3,542,355 | ) | 1,083,962 | 1,083,962 | |||||||||||||||||||||||||||||||||||
F-23
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
(1) 2009 Impairment
TEAMM intangible impairment
TEAMM’s product rights and purchased customer relationships were impaired during the twelve months ended September 30, 2009 due the discontinuance of TEAMM’s operations on November 10, 2008.
The Company’s intangible assets include goodwill, trademarks, product rights, non-compete agreements, technology rights, purchased customer relationships, and patents, all of which are accounted for based on ASC Topic 350. As described below, goodwill and intangible assets that have indefinite useful lives are not amortized but are tested at least annually for impairment or more frequently if events or changes in circumstances indicate that the asset might be impaired. Intangible assets with limited useful lives are amortized using the straight-line method over their estimated period of benefit, ranging from two to eighteen and one-half years. The Company obtains a valuation of all intangibles “when” purchased and undertakes an annual impairment analysis. Goodwill is tested for impairment by comparing the carrying amount to the estimated fair value, in accordance with ASC Topic 350. Impairment exists if the book value of the entity or operating unit is less than its estimated fair value, resulting in a write-down equal to this difference. The Company made no impairment adjustments to recorded goodwill. The Company’s carrying value of goodwill at September 30, 2009 and 2008 was $1.2 million. The values recorded for goodwill and other intangible assets represent fair values calculated by accepted valuation methods. Such valuations require critical estimates and assumptions derived from and which include, but are not limited to: (i) information included in our business plan, (ii) estimated cash flows, (iii) discount rates, (iv) patent expiration information, (vi) terms of license agreements, and (vii) expected timelines and costs to complete any in-process research and development projects to commercialize our products under development.
(2) 2010 Impairment
IMOR intangible impairment
The Company impaired all intangible assets associated with its IMOR subsidiary during 2010. IMOR filed for insolvency on March 1, 2010 under the provisions of the Insolvency Ordinance administered by the German courts.
Impairment Testing
Goodwill impairment testing is calculated at the reporting unit level. The Company’s annual impairment test has two steps. The first identifies potential impairments by comparing the fair value of the reporting unit with its carrying value. If the fair value exceeds the carrying amount, goodwill is not impaired and the second step is not necessary. If the carrying value exceeds the fair value, the second step calculates the possible impairment loss by comparing the implied fair value of goodwill with the carrying amount. If the implied fair value of goodwill is less than the carrying amount, a write-down is recorded. Impairment tests for the other intangible assets are performed by comparing the carrying amount of the intangible assets to the sum of the undiscounted expected future cash flows. In accordance with generally accepted accounting principles (GAAP), which relates to impairment of long-lived assets other than goodwill, impairment exists if the sum of the future undiscounted cash flows is less than the carrying amount of the intangible asset or to its related group of assets.
The Company predominately uses a discounted cash flow model derived from internal budgets in assessing fair values for our impairment testing. Factors that could change the result of our impairment test include, but are not limited to, different assumptions used to forecast future net sales, expenses, capital expenditures, and working capital requirements used in our cash flow models. In addition, selection of a risk-adjusted discount rate on the estimated undiscounted cash flows is susceptible to future changes in market conditions, and when unfavorable, can adversely affect the original estimates of fair values. In the event that the Company’s management determines that the value of intangible assets have become impaired using this approach, the Company will record an accounting charge for the amount of the impairment.
F-24
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Estimated future amortization of amortizable intangible assets with finite lives is as follows:
| Year ending September 30: |
||||
| 2011 |
458,792 | |||
| 2012 |
427,292 | |||
| 2013 |
197,878 | |||
| $ | 1,083,962 | |||
7. Furniture, equipment and leasehold improvements:
Furniture, equipment and leasehold improvements consist of the following:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Furniture |
$ | 220,319 | $ | 220,319 | ||||
| Office and laboratory equipment |
1,736,411 | 2,007,219 | ||||||
| Leasehold improvements |
113,102 | 403,482 | ||||||
| 2,069,832 | 2,631,020 | |||||||
| Less: accumulated depreciation and amortization |
(1,927,556 | ) | (2,299,167 | ) | ||||
| $ | 142,276 | $ | 331,853 | |||||
8. Note payable, related parties: debtor-in-possession note
On December 22, 2008, Biovest completed the closing of a debtor-in-possession financing transaction (the “DIP Transaction”) with Corps Real, LLC, a Illinois limited liability company, the principal owners of which are a director of, or affiliated with directors of Biovest (the “DIP Lender”). Pursuant to the transaction, the DIP Lender provided to Biovest a secured line of credit in an amount of up to $3.0 million in accordance with an order entered by the Bankruptcy Court (the “Facility”). As of September 30, 2010, the DIP Lender had advanced $1.9 million to Biovest.
The DIP Transaction was memorialized by a Secured Promissory Note (the “DIP Note”) and Security Agreement dated December 22, 2008. The following describes certain material terms of the DIP Transaction:
| • | Principal Amount- Up to $3.0 million |
| • | Priority and Security- The facility is a secured super-priority loan to a Debtor-in-Possession and is secured by all assets of Biovest that is senior to all prior and existing liens of Biovest. |
| • | Advances under the Facility- For all amounts in excess of $1.0 million, Lender and Biovest shall within 30 days of the entry of the Initial Order agree on a list of “milestone” events to be achieved by Biovest through use of these Advances, and Biovest shall be required to make a written request(s) detailing the amount and use and Lender shall in its reasonable discretion approve or reject the written request based upon whether Biovest demonstrates reasonable progress in achieving the agreed milestones. |
| • | Term- All loans outstanding under the Facility (the “Loans”) shall become due and payable on the earlier of: (i) December 31, 2010; (ii) dismissal of the Chapter 11 proceeding; (iii) conversion of the Chapter 11 proceeding to a Chapter 7 proceeding; or (iv) confirmation of Biovest’s Plan of Reorganization. |
| • | Interest- Loans will bear interest at 16% per annum computed on a daily basis based on the principal amount outstanding. Interest shall be paid as follows: (i) 10% interest shall be paid monthly and (ii) 6% interest shall accrue and be paid at maturity. |
| • | Points- At the Closing, Biovest paid in cash 4% of the initial $1.0 million of the Facility ($40,000). At the time that Biovest borrows in excess of $1.0 million, Biovest shall pay in cash 4% of the second $1.0 million of the Facility ($40,000). At the time that Biovest borrows in excess of $2.0 million, Biovest shall pay in cash 4% of the third $1.0 million of the Facility ($40,000). |
| • | Expenses- Biovest issued payment of $25,000 in costs at the Closing. |
F-25
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| • | Prepayment- Loans may be prepaid at any time in an amount of $50,000 or multiples of $50,000 in excess thereof, provided, however, that Biovest’s senior secured lender (Laurus and the Valens Funds) provides its consent to each prepayment via written notification to Biovest and Lender. |
See the Subsequent Events footnote for information on the treatment of this liability in Biovest’s Plan.
9. Derivative Liabilities:
As a result of the Company’s Chapter 11 proceedings, some derivative liabilities listed in the table below became prepetition indebtedness under the Debtors’ plan of reorganization. Accordingly, these obligations have been classified as ‘Liabilities subject to compromise’ in the Company’s condensed consolidated balance sheet. See the Subsequent Events footnote for information on the treatment of this liability in the Company’s Plan.
BDSI/Arius Settlement:
On February 17, 2010, the Bankruptcy Court entered an Order approving an Emezine Settlement Agreement (the “Settlement Agreement”) between the Company and BioDelivery Sciences Inc., a related party (“BDSI”), entered into as of December 30, 2009. Parties to the Settlement Agreement are the Company, our wholly-owned subsidiary, TEAMM, BDSI, and BDSI’s wholly-owned subsidiary, Arius Pharmaceuticals, Inc. (“Arius”).
The purpose of the Settlement Agreement is to memorialize the terms and conditions of a settlement between the Company and BDSI of claims relating to a Distribution Agreement dated March 12, 2004 between Arius and TEAMM (the “Distribution Agreement”) related to the marketing and distribution of Emezine, a product licensed by Arius from Reckitt Benckiser Healthcare (UK) Limited. Following the issuance in February 2006 by the FDA of a non-approvable letter with respect to the NDA for Emezine, BDSI ceased its Emezine related development efforts and on December 17, 2008, the Distribution Agreement was terminated. The Settlement Agreement resolves our claims against BDSI under the terminated Distribution Agreement.
The Settlement Agreement provided that the parties mutually release all claims that either may have against each other and, in connection therewith, we:
| (a) | received $2.5 million from BDSI (the “$2.5 Million Payment”); and |
| (b) | received the following royalty rights (the “Product Rights”) from BDSI with respect to BDSI’s BEMA Granisetron product candidate (“BEMA Granisetron”) (or in the event it is not BEMA Granisetron, the third BDSI product candidate, excluding BEMA Bupremorphine, as to which BDSI files an NDA, which, together with BEMA Granisetron, shall be referred to hereinafter as the “Product”): |
| (i) |
70/30 split (BDSI/Company) of royalty received if a third party sells the Product and 85/15 split on net sales if BDSI sells the Product; and |
| (ii) | BDSI will, from the sale of the Product, fully recover amounts equal to (1) all internal and external worldwide development costs of the Product (“Costs”) plus interest (measured on weighted average prime interest rate from first dollar spent until Product launch) and (2) the $2.5 Million Payment plus interest (measured on weighted average prime interest rate from the time of payment until Product launch) before the Company begins to receive its split as described in (b) (i) above; and |
| (c) | issued to BDSI a warrant (“Warrant”) to purchase two (2) million shares of our majority-owned subsidiary, Biovest, from us, with an exercise price equal to 120% of the closing bid price ($0.84) of Biovest’s common stock as of the date the Bankruptcy Court enters a final order authorizing us to carry out the Settlement Agreement (February 17, 2010), with the issuance of the Warrant to occur upon receipt of the $2.5 Million Payment by us (March 4, 2010). The Warrant will be exercisable immediately and for a period of seven (7) years from the date of issuance. During the initial two (2) year exercise period, any exercise of the Warrant by BDSI will be subject to approval by Biovest. |
In the event that BDSI receives any sublicensing or milestone payments associated with the Product up to and including the NDA approval, BDSI will apply 30% of such payments toward payback of the Costs of the Product plus interest and the $2.5 Million Payment plus the interest.
F-26
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
On March 4, 2010, the $2.5 Million Payment was received by the Company. The transfer of Product Rights became effective on November 17, 2010.
In the event of a proposed sale of BDSI or its assets, BDSI has the right to terminate its Product Right payment obligations to us under the Settlement Agreement upon the payment to us of an amount equal to the greater of: (i) $4.5 million; or (ii) the fair market value of the Product Rights as determined by an independent third party appraiser. Further, if the Product Right is terminated, the Warrant described above will be terminated if not already exercised, and, if exercised, an amount equal to the strike price will, in addition to the amount in (i) or (ii) above, be paid to us.
The fair value of the warrant issued to BDSI was estimated on the date of grant using the Black-Scholes-Merton valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. The value included in derivative liability is adjusted quarterly and the change is recorded to derivative gain or loss. The value on the grant date was approximately $1.0 million. The value at the end of current period was approximately $1.8 million, resulting in a derivative loss for the twelve months ended September 30, 2010 of approximately $0.8 million which is recorded as a post-petition liability as of September 30, 2010 on accompanying balance sheet as shown below.
The following tabular presentation reflects the components of derivative financial instruments as of September 30:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Embedded derivative instruments, bifurcated |
$ | 18,224,001 | $ | 3,766,838 | ||||
| Freestanding derivatives: |
||||||||
| Warrants issued with convertible debt |
10,020,236 | 2,252,294 | ||||||
| Warrants issued with note payable |
7,594,600 | — | ||||||
| Warrants issued with preferred stock |
1,077,118 | 193,762 | ||||||
| Warrants issued with other debt |
467,564 | 384,541 | ||||||
| Warrants issued with settlement(1) |
1,844,200 | — | ||||||
| Default and investment put options, Biovest |
219,700 | 196,930 | ||||||
| Investment put option, Accentia |
2,928,838 | 3,328,943 | ||||||
| $ | 42,376,257 | $ | 10,123,308 | |||||
| (1) | Warrants issued with settlement are not subject to compromise and are presented as Derivative Liabilities on the balance sheet. All other derivatives listed above are subject to compromise. |
The following tabular presentation reflects the number of common shares into which the aforementioned derivatives are indexed as of:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Accentia Common shares indexed: |
||||||||
| Freestanding and embedded derivative |
46,864,748 | 40,913,582 | ||||||
| Investment put |
1,353,229 | 1,353,229 | ||||||
| 48,217,977 | 42,266,811 | |||||||
| Biovest Common shares indexed: |
||||||||
| Freestanding derivatives (warrants to purchase Biovest shares issued to BioDelivery Sciences International, Inc. |
2,000,000 | — | ||||||
F-27
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Derivative gains (losses) in the accompanying statements of operations relate to the individual derivatives as follows:
| For the years ending September 30, | ||||||||
| 2010 | 2009 | |||||||
| Embedded derivative instruments, bifurcated |
$ | (15,917,213 | ) | $ | 10,762,809 | |||
| Freestanding derivatives: |
||||||||
| Warrants issued with convertible debentures |
(6,767,115 | ) | 1,314,818 | |||||
| Warrants issued with term note payable |
(4,687,754 | ) | 3,114,000 | |||||
| Warrants issued with preferred stock |
(883,365 | ) | 334,701 | |||||
| Warrants issued with other debt |
(83,021 | ) | (670,723 | ) | ||||
| Default and investment put options, Biovest |
(22,788 | ) | (20,425 | ) | ||||
| Investment put option, Accentia |
400,105 | (395,819 | ) | |||||
| Warrants issued for settlement |
(822,800 | ) | — | |||||
| $ | (28,783,951 | ) | $ | 14,439,361 | ||||
14. Related party transactions*:
Debtor-in-possession note:
On December 22, 2008, Biovest completed the closing of the DIP Transaction with the DIP Lender. Pursuant to the transaction, the DIP Lender provided to Biovest a secured line of credit in an amount of up to $3.0 million in accordance with an order entered by the Bankruptcy Court (the “Facility”). As of September 30, 2010, the DIP Lender had advanced $1.9 million to Biovest.
The DIP Transaction was memorialized by a Secured Promissory Note (the “DIP Note”) and Security Agreement dated December 22, 2008. The following describes certain material terms of the DIP Transaction:
| • | Principal Amount- Up to $3.0 million |
| • | Priority and Security- The Facility is a secured super-priority loan to a Debtor-in-Possession and is secured by all assets of Biovest that is senior to all prior and existing liens of Biovest. |
| • | Advances under the Facility- For all amounts in excess of $1.0 million, Lender and Biovest shall within 30 days of the entry of the Initial Order agree on a list of “milestone” events to be achieved by Biovest through use of these Advances, and Biovest shall be required to make a written request(s) detailing the amount and use and Lender shall in its reasonable discretion approve or reject the written request based upon whether Biovest demonstrates reasonable progress in achieving the agreed milestones. |
| • | Term- All loans outstanding under the Facility (the “Loans”) shall become due and payable on the earlier of: (i) December 31, 2010; (ii) dismissal of the Chapter 11 Proceeding; (iii) conversion of the Chapter 11 Proceeding to a Chapter 7 Proceeding; or (iv) confirmation of Biovest’s Plan of Reorganization. |
| • | Interest- Loans will bear interest at 16% per annum computed on a daily basis based on the principal amount outstanding. Interest shall be paid as follows: (i) 10% interest shall be paid monthly and (ii) 6% interest shall accrue and be paid at maturity. |
| • | Points- At the Closing, Biovest paid in cash 4% of the initial $1.0 million of the Facility ($40,000). At the time that Biovest borrows in excess of $1.0 million, Biovest shall pay in cash 4% of the second $1.0 million of the Facility ($40,000). At the time that Biovest borrows in excess of $2.0 million, Biovest shall pay in cash 4% of the third $1.0 million of the Facility ($40,000). |
| • | Expenses- Biovest issued payment of $25,000 in costs at the Closing. |
| • | Prepayment- Loans may be prepaid at any time in an amount of $50,000 or multiples of $50,000 in excess thereof, provided, however, that Biovest’s senior secured lender (Laurus and the Valens Funds) provides its consent to each prepayment via written notification to Biovest and Lender. |
| * | See the Subsequent Events footnote for information on the treatment of this liability in the Company’s Plan. |
F-28
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
15. Liabilities subject to compromise*:
As a result of the Company’s Chapter 11 filings, the payment of prepetition indebtedness may be subject to compromise or other treatment under the Debtors’ Plan of Reorganization. Although actions to enforce or otherwise effect payment of prepetition liabilities are stayed, at hearings held in November 2008, the Bankruptcy Court granted approval of the Debtors’ administrative motions, generally designed to stabilize the Debtors’ operations and covering, among other things, human capital obligations, business operations, tax matters, cash management, utilities, case management and retention of professionals. See the Subsequent Events footnote below for information on the treatment of this liability in the Company’s Plan.
The Debtors have been paying and intend to continue to pay undisputed post-petition claims in the ordinary course of business. In addition, the Debtors may reject prepetition executory contracts and unexpired leases with respect to the Debtors’ operations, with the approval of the Bankruptcy Court. Damages resulting from rejection of executory contracts and unexpired leases are treated as general unsecured claims and will be classified as liabilities subject to compromise. On August 16, 2010, the Company filed its First Amended Joint Plan of Reorganization, and, on October 25, 2010, the Company filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a Confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. The Company emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”). See Note 21 - Subsequent Events below for information on the treatment of these liabilities in the Company’s Plan.
ASC Topic 852 requires prepetition liabilities that are subject to compromise to be reported at the amounts expected to be allowed, even if they may be settled for lesser amounts. The amounts currently classified as liabilities subject to compromise may be subject to future adjustments depending on Bankruptcy Court actions, further developments with respect to disputed claims, determinations of the secured status of certain claims, the values of any collateral securing such claims, or other events.
Generally accepted accounting principles require prepetition liabilities that are subject to compromise to be reported at the amounts expected to be allowed, even if they may be settled for lesser amounts. The amounts currently classified as liabilities subject to compromise may be subject to future adjustments depending on Bankruptcy Court actions, further developments with respect to disputed claims, determinations of the secured status of certain claims, the values of any collateral securing such claims, or other events.
| * | See the Subsequent Events footnote for information of this liability in the Company’s Plan. |
F-29
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Liabilities subject to compromise consist of the following:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Accounts payable and accrued expenses(1)(2)(3) |
$ | 15,232,863 | $ | 14,471,456 | ||||
| Hybrid financial instrument |
3,608,674 | 652,971 | ||||||
| Convertible debentures |
28,626,149 | 23,042,458 | ||||||
| Laurus term note |
8,800,000 | 8,800,000 | ||||||
| Valens 15% convertible note, Biovest |
926,300 | 427,237 | ||||||
| Secured promissory notes payable to Laurus and the Valens Funds, Biovest |
28,522,108 | 24,056,608 | ||||||
| Unsecured promissory notes payable to Pulaski Bank and Trust Company(1) |
1,161,900 | 1,078,254 | ||||||
| Unsecured promissory note payable to Southwest Bank of St. Louis |
228,330 | 213,628 | ||||||
| Southwest line of credit |
4,000,000 | 4,000,000 | ||||||
| Notes payable, related parties |
2,313,623 | 1,574,616 | ||||||
| Other notes payable |
610,683 | 518,288 | ||||||
| Minimum royalty due to Laurus on net sales of AutovaxID instrumentation |
6,318,233 | 5,700,543 | ||||||
| Derivative liabilities |
40,532,057 | 10,123,308 | ||||||
| Dividend payable |
479,452 | 479,452 | ||||||
| Other |
2,209,756 | 3,475,913 | ||||||
| $ | 143,570,128 | $ | 98,614,732 | |||||
| (1) | The notes issued to Pulaski Bank and Trust Company (“Pulaski Bank”) are guaranteed by individuals affiliated with Biovest or Accentia. Biovest agreed to indemnify and hold harmless each guarantor should their guarantees be called by Pulaski Bank. In addition, in the event of default by Biovest resulting in a payment to the Lender by the guarantors, Biovest agreed to compensate each affected guarantor by issuance of that number of shares of Biovest’s restricted common stock determined by dividing 700% of the amount guaranteed by $1.10. On December 29, 2008, Pulaski Bank called the guarantees resulting in an aggregate payment of $1.0 million by the guarantors. As a result of their indemnification agreements, the guarantors are allowed an unsecured claim for approximately 6.4 million shares of Biovest common stock. |
| (2) | In an effort to decrease operating expenses, Biovest through its Chapter 11 bankruptcy proceedings rejected its lease in Worcester, Massachusetts on December 8, 2008. Biovest recorded a provision of $644,000 to allow for damages resulting from the rejection of this unexpired lease, which are treated by the bankruptcy code as general unsecured claims and are limited to either 15 percent of the balance of the rent reserved in the lease or the rent reserved for one year from the filing date or the date the premises were surrendered, whichever is earlier. |
| (3) | Includes accounts payable and accrued expenses from discontinued operations. |
16. Preferred stock:
On January 18, 2008, the Company entered into definitive agreements and closed a private placement (the “January 2008 Private Placement”) of $8.7 million of convertible preferred stock (the “Preferred Stock”) (8,950 shares at $1,000 per share stated value). The net proceeds after placement agent fees of approximately $0.6 million, but before other expenses associated with the transaction, were approximately $8.1 million. In connection with the January 2008 Private Placement, the Company also issued to the Placement Agent warrants to purchase an aggregate of 163,464 shares of the Company’s common stock at an exercise price of $2.67 per share.
The Preferred Stock was convertible at any time at the option of the holder into shares of the Company’s common stock at $2.67 per share, subject to adjustment for stock splits, stock dividends, and the like. In the event that the Company issues or grants in the future any rights to purchase shares of the Company’s common stock, or other security convertible into the Company’s common stock, for an effective per share price less than the conversion price then in effect, the conversion price of all unconverted Preferred Stock would decrease to equal such lower price. This adjustment to the conversion price or exchange price for future stock issuances will not apply to certain exempt issuances, including stock issuances pursuant to employee stock option plans and strategic transactions. On June 17, 2008, pursuant to a private placement transaction, the issued Preferred Stock’s conversion price was reset without further anti-dilution protection to $1.25 per share for certain January 2008 Private Placement holders who participated in the June 17, 2008 transaction.
F-30
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The Preferred Stock has liquidation preferences ahead of the holders of common stock and the holders of junior preferred stock in the event of liquidation or bankruptcy up to the amount of the Purchasers’ purchase price for the Preferred Stock. The Company has the right to force the conversion of the Preferred Stock into shares of its common stock if the Company’s common stock trades at 250% of the purchase price for a stated time and certain defined conditions are met. All remaining unconverted Preferred Stock will be redeemed by cash payment at the stated value on the maturity date of March 31, 2011 provided the convertible debentures issued to certain investors in September 2006 and February 2007 (collectively, the “Debentures”) are no longer outstanding. Commencing on January 18, 2009, the Company had the option to redeem some or all of the outstanding Preferred Stock upon notice by payment of a cash redemption price equal to 130% of the then-applicable conversion price and providing the Debentures are then converted, redeemed, or otherwise retired or the consent of the holders is obtained. Upon any liquidation, dissolution or winding-up of the Company, whether voluntary or involuntary, the Holders shall be entitled to receive out of the assets, whether capital or surplus, of the Company an amount equal to the stated value for each share of Preferred Stock before any distribution or payment shall be made to the holders of any junior securities, and if the assets of the Company shall be insufficient to pay in full such amounts, then the entire assets to be distributed to the holders shall be ratably distributed among the holders in accordance with the respective amounts that would be payable on such shares if all amounts payable thereon were paid in full.
As a part of the January 2008 Private Placement, the Company issued short-term warrants (the “Short-Term Warrants”) to the purchasers of the Preferred Stock giving them the right to purchase up to 3,269,283 shares of the Company’s common stock at an exercise price of $2.67 per share. These Short-Term Warrants expired April 23, 2008.
The Company also issued long-term warrants (the “Long-Term Warrants”) to the purchasers of the Preferred Stock giving them the right to purchase up to 1,634,639 shares of the Company’s common stock at an exercise price of $2.67 per share. The Long-Term Warrants will expire on January 17, 2014. On June 17, 2008, pursuant to a private placement transaction, the issued Warrants’ exercise price was reset without further anti-dilution protection to $1.50 per share for certain January 2008 Private Placement holders who participated in the June 17, 2008 transaction.
Other key provisions of the January 2008 Private Placement included:
| • | The Preferred Stock has no voting rights. |
| • | Triggering events that would cause mandatory conversions include the effectiveness of the April 17, 2008 registration statement lapses for a period of more than 20 consecutive Trading Days or 30 non-consecutive Trading Days during any 12 month period and prior to such the end of any such period; fewer than all the conversion shares shall being freely saleable pursuant to Rule 144; the Company shall fail to deliver certificates representing conversion shares issuable upon a conversion; the Company shall fail for any reason to pay in full the amount of cash due pursuant to a buy-in within five calendar days after notice is delivered. |
| • | So long as any shares of Preferred Stock are outstanding, unless the holders of at least 67% of stated value of the then outstanding shares of Preferred Stock shall have otherwise given prior written consent, the Company shall not, and shall not permit any of its subsidiaries (whether or not a subsidiary on the original issue date) to, directly or indirectly amend its certificate of incorporation, bylaws or other charter documents so as to materially and adversely affect any rights of any holder; repay, repurchase or offer to repay, repurchase or otherwise acquire shares of its common stock, common stock equivalents or junior securities, except for the conversion shares to the extent permitted or required under the transaction documents or as otherwise permitted by the transaction documents; enter into any agreement or understanding with respect to any of the foregoing; or pay cash dividends or distributions on junior securities of the Company. |
The Preferred Stock was not recorded as a liability based on generally accepted accounting principles because the security may be converted into an equity instrument at the option of the holder. The Preferred Stock is not considered mandatorily redeemable until the conversion option expires since the conversion option could be exercised prior to the redemption date. As such, the Preferred Stock has been classified in the mezzanine section of the balance sheet in accordance with Securities and Exchange Commission Accounting Series Release 268.
The Preferred Stock embodies certain features, including, an embedded conversion feature, which are defined as derivative financial instruments under ASC 815. Consideration of bifurcation and liability classification of the derivatives was, in part, based upon an overall conclusion that the preferred stock was more akin to a debt instrument, rather than an equity instrument. Accordingly, the conversion feature was evaluated under ASC 815, and the result of that analysis was that the embedded conversion feature does not require bifurcation. In addition, the preferred stock embodied certain cash-settled contingent redemption features (that is, Put Features) and call options that were not clearly and closely related to the host contract and, accordingly, required bifurcation and classification as derivative liabilities.
F-31
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The warrants issued in connection with the January 2008 Private Placement are derivative financial instruments. The warrants issued to the Investors and the Placement Agent did not meet all of the requisite conditions for equity classification, as provided in generally accepted accounting principles because the contracts extended firm registration rights to these investors and placement agents without an economic alternative in the event that the Company is not able to deliver registered shares. Accordingly, the warrants are carried as derivative liabilities, at fair value.
The following table illustrates details of the sale of the Preferred Stock and how the net proceeds arising from the January 2008 Private Placement financing was allocated on the financing inception date:
| Gross cash proceeds |
$ | 8,729,000 | ||
| Financing fees (paid in cash) |
(641,040 | ) | ||
| Net cash proceeds |
8,087,960 | |||
| Allocation of proceeds: |
||||
| Series A-1 Preferred Stock |
— | |||
| Paid-in capital (beneficial conversion feature), net of $223,873 deferred financing fees attributable to the conversion feature |
(2,456,495 | ) | ||
| Derivative liabilities: |
||||
| Investor Warrants liability |
(4,585,503 | ) | ||
| Agent Warrants liability |
(230,362 | ) | ||
| Put liability |
(3,298,330 | ) | ||
| Deferred financing cost |
532,992 | |||
| Retained Earnings (preferred dividend) |
114,537 | |||
| Day-one derivative loss |
1,835,201 | |||
| Total |
$ | (8,087,960 | ) | |
Total financing fees associated with the January 2008 Private Placement financing are as follows:
| Financing fees paid in cash |
$ | 641,040 | ||
| Financing fees (placement agent warrants) |
230,362 | |||
| $ | 871,402 | |||
The following table illustrates the allocation of financing costs associated with the January 2008 Private Placement financing (allocated based upon the relative fair values of the instruments):
| Paid-in capital (attributable to conversion feature) |
$ | 223,873 | ||
| Deferred financing costs (attributable to derivative liabilities) |
532,992 | |||
| Retained earnings (attributable to preferred stock and accounted for as a dividend) |
114,537 | |||
| $ | 871,402 | |||
On June 23, 2008, the Company offered to each January 2008 Private Placement holder that executed and delivered an Acceptance, the right to have all of the shares of Preferred Stock held by such holder be redeemed (“Accepting Preferred Stockholder”) with the following redemption terms:
On July 15, 2008 and on the 15th day of each month thereafter (each such date, a “Monthly Preferred Redemption Date”), the Company shall redeem a number of shares of Preferred Stock held by such Accepting Preferred Stockholder equal to the aggregate number of shares of Preferred Stock held by such Accepting Preferred Stockholder divided by 32, with all shares of Preferred Stock held by such Accepting Preferred Stock Holder being redeemed on or before March 15, 2011, provided, however, any fractional share of Preferred Stock required to be redeemed hereunder shall be rounded up to the next full share of Preferred Stock (the shares of Preferred Stock being redeemed on each Monthly Preferred Redemption Date, the “Redeemed Preferred Stock”). The Company shall redeem the Redeemed Preferred Stock by issuing to each Accepting Preferred Stockholder a stock certificate representing that number of shares of common stock determined by multiplying the stated value by the number of shares of Redeemed Preferred Stock and then
F-32
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
dividing the total by a number equal to ninety percent (90%) of the volume weighted average price (“VWAP”) for the 20 calendar days prior to each Monthly Preferred Redemption Date (such number of shares of common stock, the “Redemption Consideration”), provided, however, that any fractional share of common stock shall be rounded up to the next full share of common stock. The shares of Redeemed Preferred Stock shall be automatically cancelled upon payment by the Company of the Redemption Consideration. An Accepting Preferred Stockholder may, at any time, in its sole and absolute discretion, elect to discontinue the redemption of its Preferred Stock.
Holders representing approximately 94% of the initial $8.7 million investment accepted the redemption offer. As a result of the nature of the Redemption Considerations previously discussed (variable number of common shares dependent upon the Company’s stock price), the Company cannot conclude that it has adequate authorized shares to settle this or other instruments, which are indexed to the Company’s common stock that were previously recorded in equity. As such, the Company reclassified approximately $11.9 million from stockholders’ equity to derivative liabilities and approximately $0.2 million from stockholders’ equity to capitalized finance costs, which is the fair value of three instruments recorded in equity. Of this reclassification, approximately $3.9 million related to the embedded conversion feature of the January 2008 Private Placement, approximately $4.2 million related to the embedded conversion feature of the February 2007 Debenture (originally reclassified to stockholders’ equity at the reset of the conversion price in May 2007), approximately $1.4 million related to the detachable warrants issued with the February 2007 Debenture, and approximately $2.3 million related to other warrants that were previously issued in separate transactions. The fair market value of these instruments was calculated on June 23, 2008 and reclassified from stockholders’ equity to derivative liabilities.
As a result of the Company’s and its subsidiaries’ Chapter 11 filings all actions to enforce or otherwise effect the Company’s and/or its subsidiaries’ obligations and payments under any debt facility were automatically stayed pursuant to provision of federal bankruptcy law. See the Subsequent Events footnote for information on the treatment of this liability in the Company’s Plan.
17. Income taxes
The Company’s deferred tax assets and liabilities consist of the following:
| September 30, | ||||||||
| 2010 | 2009 | |||||||
| Deferred tax assets: |
||||||||
| Accrued expenses deductible in future |
$ | 7,535,000 | $ | 4,187,000 | ||||
| Allowance for doubtful accounts |
3,000 | 84,000 | ||||||
| Basis difference in assets |
3,735,000 | 2,722,000 | ||||||
| Inventory valuation allowance |
490,000 | 345,000 | ||||||
| Share-based compensation |
66,000 | 2,099,000 | ||||||
| Intangible assets |
2,757,000 | 2,315,000 | ||||||
| Net operating loss carryforward |
80,606,000 | 76,544,000 | ||||||
| Debt extinguishment |
5,149,000 | 2,652,000 | ||||||
| Other |
20,859,000 | 5,980,000 | ||||||
| Valuation allowance |
(121,200,000 | ) | (96,928,000 | ) | ||||
| $ | — | $ | — | |||||
F-33
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Income tax (expense) benefit consists of the following:
| 2010 | 2009 | |||||||
| Current |
$ | — | $ | — | ||||
| Deferred |
(25,269,000 | ) | (20,384,000 | ) | ||||
| Benefit of net operating loss carryover |
— | 19,388,000 | ||||||
| Increase in valuation allowance |
25,269,000 | 996,000 | ||||||
| $ | — | $ | — | |||||
| Allocation between continuing and discontinued operations: |
||||||||
| Continuing operations |
$ | — | $ | — | ||||
| Discontinued operations |
— | — | ||||||
| $ | — | $ | — | |||||
The expected income tax benefit at the statutory tax rate differed from income taxes in the accompanying consolidated statements of operations as follows:
| 2010 | 2009 | |||||||
| Statutory tax rate |
34% | 34% | ||||||
| State taxes |
4% | 4% | ||||||
| Permanent items |
16% | — | ||||||
| Other |
(1)% | (5)% | ||||||
| Change in valuation allowance |
(53)% | (33)% | ||||||
| Effective tax rate in accompanying statement of operations |
0% | 0% | ||||||
Deferred tax assets are reduced by a valuation allowance if, based on the weight of available evidence, it is more likely than not (a likelihood of more than 50 percent) that some portion or all of the deferred tax assets will not be realized. The valuation allowance should be sufficient to reduce the deferred tax asset to the amount that is more likely than not to be realized. As a result, the Company recorded a valuation allowance with respect to all of the Company’s deferred tax assets.
Under Section 382 and 383 of the Internal Revenue Code, if an ownership change occurs with respect to a “loss corporation”, as defined, there are annual limitations on the amount of the net operating loss and other deductions, which are available to the Company. Due to the acquisition transactions in which the Company has engaged in recent years, the Company believes that the use of these net operating losses will be significantly limited.
The Company has a federal net operating loss carryover of approximately $212.3 million as of September 30, 2010, which expires through 2027, and of which approximately $30.0 million is subject to various Section 382 limitations based upon ownership changes that occurred through September 30, 2003. Of those losses subject to the limitations, $11.3 million is expected to expire before the losses can be utilized. Of the remaining amounts, the limitation is approximately $1.8 million per year through approximately the year ended September 30, 2012. Subsequent to that date, the annual limitation will decrease to approximately $0.2 million through September 30, 2024. The NOLs incurred after September 30, 2003 may also be subject to further limitations.
F-34
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
18. Stockholders’ equity
Common stock
The Company has one class of common stock with an aggregate authorization of three hundred million shares. Each share of common stock carries equal voting rights, dividend preferences, and a par value of $.001 per share.
Preferred stock
The Company has an aggregate of one hundred fifty million authorized shares of preferred stock, one hundred twenty-five million eight thousand nine hundred fifty of which have been issued and designated in six series (the “preferred stock”), each at a par value of $1.00 per share as follows:
Preferred Shares authorized:
| Series A |
10,000,000 | |||
| Series A-1 |
8,950 | |||
| Series B |
30,000,000 | |||
| Series C |
10,000,000 | |||
| Series D |
15,000,000 | |||
| Series E |
60,000,000 | |||
| 125,008,950 | ||||
The Preferred Stock will be convertible at any time at the option of the holder into shares of the Company’s common stock at $2.67 per share, subject to adjustment for stock splits, stock dividends, and the like. In the event that the Company issues or grants in the future any rights to purchase shares of the Company’s common stock, or other security convertible into the Company’s common stock, for an effective per share price less than the conversion price then in effect, the conversion price of all unconverted Preferred Stock will be decreased to equal such lower price. This adjustment to the conversion price or exchange price for future stock issuances will not apply to certain exempt issuances, including stock issuances pursuant to employee stock option plans and strategic transactions. The Preferred Stock has liquidation preferences ahead of the holders of common stock and the holders of junior preferred stock in the event of liquidation or bankruptcy up to the amount of the Purchasers’ purchase price for the Preferred Stock. The Company has the right to force the conversion of the Preferred Stock into shares of its common stock if the Company’s common stock trades at 250% of the purchase price for a stated time and certain defined conditions are met. All remaining unconverted Preferred Stock will be redeemed by cash payment on the maturity date of March 31, 2011 provided the convertible debentures issued to certain investors in September 2006 and February 2007 (the “Debentures”) are no longer outstanding. Commencing on January 18, 2009, the Company has the option to redeem some or all of the outstanding Preferred Stock upon notice by payment of a cash redemption price equal to 130% of the then-applicable conversion price and providing the Debentures are then converted, redeemed, or otherwise retired or the consent of the holders is obtained. The preferences and features of the Preferred Stock are fully described in a Certificate of Designation and amendment to the Company’s Articles of Incorporation filed on the date of closing of the Private Placement and attached as an exhibit hereto.
Stock options and warrants
The Company provides for three option plans, the 2003 Stock Option Plan (“2003 Plan”) per its second amendment on February 27, 2004, the 2005 Equity Incentive Plan (“2005 Plan”), and the 2008 Equity Incentive Plan (the “2008 Plan”) (collectively, the “Stock Option Plans”). The Stock Option Plans provide for the issuance of qualified and non-qualified options as those terms are defined by the Internal Revenue Code.
The 2003 Plan, as amended, provides for the issuance of 3.5 million shares of common stock, and 762,571 shares of Series D Preferred Stock. At September 30, 2008, all Series D Preferred options have been converted into common share options. All options issued, pursuant to the 2003 Plan, cannot have a term greater than ten years. Options granted under this plan vest over periods established in the option agreement. As of September 30, 2010, there are 1,681,910 options available for issuance under the 2003 Plan.
F-35
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
On February 1, 2005, the Company’s board of directors adopted the Accentia Biopharmaceuticals, Inc. 2005 Equity Incentive Plan. The 2005 Plan provides for the issuance of 3.0 million shares of common stock. All options issued, pursuant to the 2005 Plan, cannot have a term greater than ten years. Options granted under this plan vest over periods established in the option agreement. As of September 30, 2010, there are 1,576,207 options available for issuance under the 2005 Plan. The Company may, at any time, amend or modify the 2005 Plan without limitation.
On November 29, 2007 and effective as of December 31, 2007, the Company’s Board of Directors has adopted the Accentia Biopharmaceuticals, Inc. 2008 Equity Incentive Plan. The 2008 Plan provides for the issuance of 3.0 million shares of common stock. All options issued, pursuant to the 2008 Plan, cannot have a term greater than ten years. Options granted under this plan vest over periods established in the option agreement. As of September 30, 2010, there are 729,833 options available for issuance under the 2008 Plan. The Company may, at any time, amend or modify the Plan without limitation. Subsequent to the period, the Company’s Board of Directors amended the 2008 Plan to increase the number of available options to an aggregate total of 23.0 million.
Stock options and warrants granted, exercised, terminated, and forfeited during the years ended September 30, 2010 and 2009 are as follows:
| Shares | Average Exercise price per share |
|||||||
| Options: |
||||||||
| Outstanding, October 1, 2008 |
4,180,545 | $ | 3.33 | |||||
| Granted |
4,712,000 | 0.18 | ||||||
| Terminated or forfeited |
(619,508 | ) | 2.69 | |||||
| Exercised |
— | |||||||
| Outstanding September 30, 2009 |
8,273,037 | 1.58 | ||||||
| Granted |
16,330,000 | 0.44 | ||||||
| Terminated or forfeited |
(347,457 | ) | 3.20 | |||||
| Exercised |
— | |||||||
| Outstanding September 30, 2010 |
24,255,580 | $ | 0.81 | |||||
| Warrants: |
||||||||
| Outstanding, October 1, 2008 |
23,175,933 | $ | 2.33 | |||||
| Granted |
— | — | ||||||
| Terminated or forfeited |
(1,895,133 | ) | 1.62 | |||||
| Exercised |
— | — | ||||||
| Outstanding September 30, 2009 |
21,280,800 | $ | 2.51 | |||||
| Granted |
— | — | ||||||
| Terminated or forfeited |
(1,000,000 | ) | 2.67 | |||||
| Exercised |
— | — | ||||||
| Outstanding September 30, 2010 |
20,280,800 | $ | 2.50 | |||||
The weighted average grant date fair values of stock options and warrants granted during the years ended September 30, 2010 and 2009 were as follows:
| Weighted Average Grant Date Fair Value |
||||||||
| Options | Warrants | |||||||
| 2010 |
$ | 0.41 | n/a | |||||
| 2009 |
$ | 0.17 | n/a | |||||
F-36
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The following table summarizes information for options outstanding and exercisable at September 30, 2010:
| Outstanding |
Exercisable | |||||||||||||||||||||||||||
| Range of Exercise Prices |
Number | Weighted average remaining life |
Intrinsic Value |
Weighted average exercise price |
Number | Weighted average exercise price |
Intrinsic Value |
|||||||||||||||||||||
| $0.00-1.05 |
20,897,617 | 9.12 years | $ | 0.19 | 74,784 | $ | 0.73 | |||||||||||||||||||||
| $1.06-2.11 |
296,200 | 3.03 years | 2.11 | 296,200 | 2.11 | |||||||||||||||||||||||
| $2.12-2.63 |
335,702 | 6.34 years | 2.58 | 260,702 | 2.55 | |||||||||||||||||||||||
| $2.64-9.00 |
2,726,061 | 6.45 years | 3.87 | 2,726,061 | 3.72 | |||||||||||||||||||||||
| 24,255,580 | $ | 13,084,010 | 3,357,747 | $ | 21,600 | |||||||||||||||||||||||
The following table summarizes information for options outstanding and exercisable at September 30, 2009:
| Outstanding |
Exercisable | |||||||||||||||||||||||||||
| Range of Exercise Prices |
Number | Weighted average remaining life |
Intrinsic Value |
Weighted average exercise price |
Number | Weighted average exercise price |
Intrinsic Value |
|||||||||||||||||||||
| $0.00-1.05 |
4,746,617 | 9.26 years | $ | 0.19 | 43,951 | $ | 0.76 | |||||||||||||||||||||
| $1.06-2.11 |
296,200 | 4.03 years | 2.11 | 296,200 | 2.11 | |||||||||||||||||||||||
| $2.12-2.63 |
374,240 | 7.09 years | 2.58 | 224,240 | 2.55 | |||||||||||||||||||||||
| $2.64-9.00 |
2,855,980 | 7.36 years | 3.87 | 2,431,822 | 3.89 | |||||||||||||||||||||||
| 8,273,037 | $ | 140,130 | 2,996,213 | $ | 0 | |||||||||||||||||||||||
A summary of the status of the Company’s nonvested stock options as of September 30, 2010, and changes during the year then ended, is summarized as follows:
| Nonvested Shares |
Shares | Weighted- Average Grant- Date Fair Value |
Intrinsic Value |
|||||||||
| Nonvested at October 1, 2009 |
5,276,824 | |||||||||||
| Granted |
16,340,000 | |||||||||||
| Vested |
(371,534 | ) | ||||||||||
| Forfeited |
(347,457 | ) | ||||||||||
| Nonvested at September 30, 2010 |
20,897,833 | $ | 0.36 | $ | 13,062,410 | |||||||
19. Employee benefit plan:
The Accentia 401(k) and Profit Sharing Plan (the “Plan”) was established effective July 1, 2004 as a defined contribution plan. The Plan is subject to the provisions of the Employee Retirement Income Security Act of 1974 (ERISA). The Plan includes eligible employees of Accentia and its affiliates including the Company. Employees of the Company who have completed one month of service are eligible to participate in the plan. Participants are permitted to make annual pre-tax salary reduction contributions of up to 50% of their compensation subject to certain limitations. Employer and participant contributions are invested at the direction of the participant in various money market funds or pooled/mutual funds. Employer matching and profit sharing contributions are based upon discretionary amounts and percentages authorized by the Board of Directors of the Company. For the fiscal years ended September 30, 2010 and 2009, the Company made no employer contributions pursuant to the Plan
F-37
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
20. Segment Information:
During 2004, the Company through its subsidiary, Analytica, made an insignificant acquisition of a foreign entity, IMOR, this subsidiary ceased operations during the quarter ended March 31, 2010. Segment information on a geographic basis for the years ended September 30, 2010 and 2009 is as follows:
| Year ended September 30, 2010 | Year ended September 30, 2009 | |||||||||||||||||||||||
| Domestic | International (Europe) |
Total | Domestic | International (Europe) |
Total | |||||||||||||||||||
| Net sales |
$ | 9,512,259 | $ | 943,254 | $ | 10,455,513 | $ | 7,870,417 | $ | 2,685,521 | $ | 10,555,938 | ||||||||||||
| Net loss |
(47,490,854 | ) | (307,979 | ) | (47,798,833 | ) | (4,071,788 | ) | (1,554,904 | ) | (5,626,692 | ) | ||||||||||||
| Total Assets |
5,010,309 | — | 5,010,309 | 7,965,353 | 1,870,968 | 9,836,321 | ||||||||||||||||||
| Goodwill |
893,000 | — | 893,000 | 893,000 | 300,437 | 1,193,437 | ||||||||||||||||||
21. Product rights and obligations:
Product rights and obligations
The Company has entered into certain product development and licensing agreements which provide for the acquisition of product rights and performance payments based on achievement of milestones as it relates to product development. The Company is contractually obligated to pay aggregate acquisition and performance payments of approximately $8.2 million if all milestones are achieved.
Parties to these agreements are as follows:
| • | Mayo Foundation for Medical Education and Research (“MAYO”) |
| • | Arius Pharmaceuticals, Inc. (“Arius”) |
| • | Respirics, Inc. (“Respirics”) |
| • | Mikart, Inc. (“Mikart”) |
| • | Arbor Pharmaceuticals, Inc. (“Arbor”) |
Stanford
In September 2004, Biovest entered into an agreement with Stanford University (“Stanford”) providing for worldwide rights to use two proprietary hybridoma cell lines, that are used in the production of the BiovaxID vaccine. These are the same cell lines that been used by researchers at Stanford and the National Cancer Institute to perform their studies of the hybridoma idiotype vaccine in non-Hodgkins Lymphoma. This agreement gives Biovest exclusivity to this cell line through 2019 in the fields of B-cell and T-cell cancers, and it provides non-exclusive rights in such fields of use at all times thereafter. The agreement also gives Biovest the right to sublicense or transfer the licensed biological materials to collaborators in the licensed fields. Under the agreement with Stanford, Biovest is obligated to pay Stanford an up-front license fee of $15,000 within 30 days following the execution of the agreement, and an annual maintenance fee of $10,000 thereafter. If BiovaxID is approved by the FDA, the agreement provides for a $0.1 million payment to Stanford upon approval, and following approval, Stanford will receive a royalty of the greater of $50.00 per patient or 0.05% of the amount received by us for each BiovaxID patient treated using this cell line. This running royalty will be creditable against the yearly maintenance fee. The agreement with Stanford obligates Biovest to diligently develop, manufacture, market, and sell BiovaxID and to provide progress reports to Stanford regarding these activities. Biovest can terminate this agreement at any time upon 30 days prior written notice, and Stanford can terminate the agreement upon a breach of the agreement by Biovest that remains uncured for 30 days after written notice of the breach from Stanford.
F-38
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
22. Commitments and contingencies:
Operating leases
The Company has operating leases for various facilities, automobiles, machinery, and equipment, which expire at various times through 2012. The annual aggregate rental commitments under non-cancelable leases are as follows:
| Year ending September 30, |
||||
| 2011 |
$ | 401,465 | ||
| 2012 |
236,611 | |||
| 2013 |
243,709 | |||
| 2014 |
165,901 | |||
| $ | 1,047,686 | |||
Rent expense for all operating leases was approximately $0.8 million and $1.3 million, for the year ended September 30, 2010 and 2009 respectively.
Bankruptcy proceedings:
On November 10, 2008, the Company filed a voluntary petition for reorganization under Chapter 11 of the United States Bankruptcy Code (“Chapter 11”) in the U.S. Bankruptcy Court for the Middle District of Florida, Tampa Division (the “Bankruptcy Court”). During the pendency of the Chapter 11 proceedings, the Company operated its business as a debtor-in-possession in accordance with the provisions of Chapter 11 and subject to the jurisdiction of the Bankruptcy Court. On August 16, 2010, the Company filed its First Amended Joint Plan of Reorganization. Subsequent to the filing of this report, on October 25, 2010, the Company filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. The Company emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
Notwithstanding the effectiveness of our Plan, the Bankruptcy Court retains jurisdiction to adjudicate any remaining issues regarding, inter alia, the validity, amount, and method of payment of claims filed in connection with our Chapter 11 proceeding. Accordingly, we anticipate that there may be ongoing proceedings before the Bankruptcy Court to resolve any filed objections or disputes as to claims filed in the Chapter 11 proceeding.
Biovest litigation:
On August 4, 2008, the Company’s majority-owned subsidiary, Biovest was served with a summons and complaint filed in California Superior Court on behalf of Clinstar LLC for breach of contract for non-payment of certain fees for clinical trial studies and pass-through expenses in the amount of $385,000. Biovest intends to seek the dismissal of this litigation and plan to defend these claims vigorously. Upon the filing of Biovest’s Chapter 11 petition on November 10, 2008, this litigation was automatically stayed pursuant to provisions of federal bankruptcy law. The Company anticipates that the claims involved in this litigation will be contested and resolved by the Bankruptcy Court as part of an objection to claim which the Company expects to file shortly.
Analytica litigation:
On December 2, 2005, the Company’s subsidiary, Analytica commenced litigation in the Supreme Court of New York, New York County against a former employee, alleging breach of covenants not to compete, breach of confidentiality agreements and misappropriation of proprietary information. The defendant has filed an Answer containing counterclaims alleging that he was owed contractual bonus and other compensation and seeking damages for wrongful termination against Analytica, the Company and an officer of the Company. Both Analytica and the former employee sought injunctive relief and monetary damages from one another. The Company filed a motion seeking to dismiss all claims naming the Company and the Company’s officer personally, and to dismiss certain
F-39
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
claims against all defendants. In June 2007, the Court granted the Company’s motion and dismissed defendant’s counterclaims against the Company’s officer with prejudice, and dismissed all other counterclaims as well, allowing defendant an opportunity to replead by July 31, 2007. The defendant refiled an Amended Counterclaim in July 2007, and the Company has filed a motion seeking to dismiss virtually all claims in the Amended Counterclaim. The Company is pursuing the Company’s affirmative claims in this matter vigorously and has asserted all available defenses against the counterclaims, which the Company believes are without merit. Upon the filing of the Company’s Chapter 11 petition on November 10, 2008, this litigation was automatically stayed pursuant to provisions of federal bankruptcy law. The Company anticipates that the claims involved in this litigation will be contested and resolved by the Bankruptcy Court as part of an objection to claim which the Company expects to file shortly.
Other proceedings:
Further, from time to time we are subject to various legal proceedings in the normal course of business, some of which are covered by insurance.
Employment agreements
The Company has no employment agreements officers and executives as of September 30, 2010.
Cooperative Research and Development Agreement:
In September 2001, Biovest entered into a definitive Cooperative Research and Development Agreement (“CRADA”) with the National Cancer Institute (“NCI”) for the development and ultimate commercialization of patient-specific vaccines for the treatment of non-Hodgkin’s low-grade follicular lymphoma. The terms of the CRADA, as amended, included, among other things, a requirement to pay $0.5 million quarterly to NCI for expenses incurred in connection with the ongoing Phase 3 clinical trials. Since the transfer to Biovest of the investigational new drug application for development of this vaccine, which occurred in April 2004, these payments to NCI have been reduced to a small fraction of this original obligation (approximately $0.2 million per year). On September 25, 2006, the Company provided written notice to the NCI in accordance with the terms of the CRADA to terminate the CRADA at the end of the sixty day notice period. Under the terms of the CRADA, the Company is obligated to continue to provide vaccine to the NCI at no charge for purposes of the NCI’s studies that are within the scope of the CRADA if the Company were to abandon work on the vaccine. As the Company is actively developing the vaccine for commercialization and intends to do so to its completion, no estimated costs have been accrued as of September 30, 2009.
Food and Drug Administration:
The FDA has extensive regulatory authority over biopharmaceutical products (drugs and biological products), manufacturing protocols and procedures and the facilities in which mammalian proteins will be manufactured. Any new bioproduct intended for use in humans (including, to a somewhat lesser degree, in vivo biodiagnostic products), is subject to rigorous testing requirements imposed by the FDA with respect to product efficacy and safety, possible toxicity and side effects. FDA approval for the use of new bioproducts (which can never be assured) requires several rounds of extensive preclinical testing and clinical investigations conducted by the sponsoring pharmaceutical company prior to sale and use of the product. At each stage, the approvals granted by the FDA include the manufacturing process utilized to produce the product. Accordingly, our cell culture systems used for the production of therapeutic or biotherapeutic products are subject to significant regulation by the FDA under the Federal Food, Drug and Cosmetic Act, as amended (the “FD&C Act”).
Product liability:
The contract production services for therapeutic products offered exposes an inherent risk of liability as the proteins or other substances manufactured, at the request and to the specifications of customers, could foreseeably cause adverse effects. The Company obtains agreements from contract production customers indemnifying and defending the Company from any potential liability arising from such risk. There can be no assurance, however, that the Company will be successful in obtaining such agreements in the future or that such indemnification agreements will adequately protect the Company against potential claims relating to such contract production services. The Company may also be exposed to potential product liability claims by users of its products. A successful partial or completely uninsured claim against the Company could have a material adverse effect on the Company’s operations.
F-40
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Guarantee Indemnifications:
Under the terms of the January and March Pulaski Bank notes payable, several board members and affiliates issued personal guarantees. Biovest agreed to indemnify and hold harmless each guarantor should their guarantees be called by Pulaski Bank by agreeing to compensate each affected guarantor by an issuance of restricted common stock equal to 700% of the amount of their guarantee. The stock was to be issued using a pre-determined value of $1.10 per share. On December 29, 2008, Pulaski Bank called its guaranty resulting in an aggregate payment of $1.0 million by the guarantors. As a result of the indemnification agreements, Biovest originally recorded a provision for approximately 6.4 million shares of Biovest’s common stock valued using the trading price of Biovest’s stock. In March, 2010, Biovest reached an agreement with the guarantors whereby the indemnities will be valued using a price of $0.24 per share, which represents the closing price of Biovest’s stock on November 9, 2008, the day prior to Biovest’s petition for reorganization, and represents the amount that will be allowed as an unsecured claim of the Pulaski guarantors in Biovest’s reorganization proceedings (approximately $1.5 million). See the Subsequent Events footnote below for information on the treatment of these liabilities in the Company’s Plan.
Under the terms of the Southwest Bank note payable issued June 26, 2007, an officer of the Company’s and Biovest issued a personal guarantee. Biovest agreed to indemnify and hold harmless the guarantor should the guarantee be called by Southwest Bank by agreeing to compensate the guarantor by an issuance of restricted common stock equal to 700% of the amount of the guarantee. The stock was to be issued using a pre-determined value of $1.10 per share. In January 2009, Southwest Bank called its guaranty resulting in an aggregate payment of $0.2 million by the guarantor. As a result of the indemnification agreement, Biovest originally recorded a provision for approximately 1.2 million shares of Biovest’s common stock valued using the trading price of Biovest’s stock. In March 2010, Biovest reached an agreement with the guarantor whereby the indemnity will be valued using a price of $0.24 per share, which represents the closing price of Biovest’s stock on November 9, 2008, the day prior to Biovest’s petition for reorganization, and represents the amount that will be allowed as an unsecured claim of the guarantor in Biovest’s reorganization proceedings (approximately $0.3 million).
Distribution agreement:
On June 1, 2007, Biovest entered into a non-exclusive distribution agreement with VWR International, Inc. (“VWR”), to distribute the AutovaxID automated cell culture device in North America. Under the terms of this agreement, Biovest is obligated to pay to VWR 25% of net sales revenue generated from the sales and marketing efforts of VWR on AutovaxID cell culture systems and disposable cultureware used in conjunction with the AutovaxID system. The initial term of this agreement is for 24 months and can be terminated at any time by either party upon 180 days’ prior written notice. Biovest has not incurred any distribution expense to date related to this agreement. Subsequent to the period of this report, effective as of March 21, 2010, Biovest terminated the non-exclusive distribution agreement.
Sublicense agreement with related party
On February 27, 2007, the Company entered into a sublicense agreement (the “Accentia Sublicense”) with Revimmune, LLC under which Accentia was granted the exclusive worldwide right to Revimmune™. The perpetual sublicense allows the Company to develop and market a patent pending treatment for autoimmune diseases. The Accentia Sublicense covers the potential treatment of all autoimmune diseases including but not limited to multiple sclerosis.
Other material terms and conditions of the Accentia Sublicense are as follows:
| • | The Company assumed certain future development, milestone and minimum royalty obligations of Revimmune, LLC under its license with Johns Hopkins University (“JHU”). In connection with the Accentia Sublicense, the Company did not pay an upfront fee or reimbursement of expenses. The Company also agreed to pay to Revimmune, LLC a royalty of 4% on net sales, and in the event of a sublicense, to pay 10% of net proceeds received from any such sublicense to Revimmune, LLC. |
| • | Upon the approval of the sublicensed treatment in the U.S. for each autoimmune disease, the Company is required to issue to Revimmune, LLC vested warrants to purchase 0.8 million shares of the Company’s common stock. The warrant which will be granted at the approval of the first sublicensed product will have an exercise price of $8 per share and any subsequent warrant to be issued will have an exercise price equal to the average of the volume weighted average closing prices of the Company’s common stock during the ten (10) trading days immediately prior to the grant of such warrant. |
| • | The Company will be responsible, at its sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the licensed products. |
F-41
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| • | The Company has assumed the cost and responsibility for patent prosecution as provided in the license between Revimmune, LLC and JHU to the extent that the claims actually and directly relate to sublicensed products. |
| • | Revimmune, LLC’s manager is a director of the Company. |
On January 16, 2008, Biovest entered into a sublicense agreement (the “Biovest Sublicense”) with Revimmune, LLC under which Biovest was granted the exclusive worldwide rights to Revimmune™. The Biovest Sublicense allows Biovest to develop and market, a patent-pending pharmaceutical treatment in late-stage development for the treatment of and prevention of transplant rejection including rejection following a bone marrow transplant.
Other material terms and conditions of the Biovest Sublicense are as follows:
| • | Biovest is obligated to pay to Revimmune, LLC a royalty of 6% on net sales, and in the event of a sublicense by Biovest, to pay 20% of sublicense consideration received. Biovest did not pay an upfront fee in connection with the Biovest Sublicense but upon the approval of the sublicensed treatment in the U.S. for each sublicensed indication, Biovest is required to issue to Revimmune, LLC vested warrants to purchase 2.0 million shares of Biovest’s common stock. Each such warrant which will be granted at the approval of each successive sublicensed product will have an exercise price of $1.10 per share or, at the discretion of Biovest, at a price equal to the fair market value of Biovest’s common stock on the date of the grant of such warrant. |
| • | Biovest assumed certain obligations under Revimmune, LLC’s license with JHU related to the sublicensed technology, including the payment of all royalty obligations due JHU for the sublicensed products which includes a 4% royalty on licensed products and services and a 20% royalty on sublicense consideration. |
| • | Biovest will be responsible, at its sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the sublicensed products. |
23. Variable Interest Entities*:
Accounting for the NMTC financing arrangement and variable interest entities
The Company evaluated the structure of the NMTC financing arrangements and entities so involved under the context of generally accepted accounting principles. This provides a framework for determining whether certain entities should be consolidated (irrespective of equity ownership) based upon a variable interests model. This model determines the control and consolidation based upon potential variability in gains and losses of the entity being evaluated for consolidation. Generally, a variable interest holder that absorbs a majority of the entity’s expected losses, if they occur, receives a majority of the entity’s expected residual return, if they occur, or both is identified as the primary beneficiary for consolidation purposes.
| * | See the Subsequent Events footnote for information on the treatment of this liability in the Company’s Plan. |
F-42
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The Company concluded that Biovax Investment, LLC, Telesis CDE Two, LLC, Autovax Investment, LLC and St. Louis New Market Tax Credit Fund II, LLC met the definition of variable interest entity. However, for the Company to be required to apply the provisions of generally accepted accounting principles, it must have a variable interest in the entity. Variable interests in a variable interest entity are contractual, ownership or other monetary interests in an entity that change with changes in the value of the net assets of the entity. The following tables illustrate the variable interests have been identified in each of the entities considered by the Company and the related holder:
New Market Tax Credit Transaction I:
| Variable Interest Holder |
Variable Interests Biovax Investment, LLC |
Variable Interests Telesis CDE Two, LLC | ||
| Biovest and its Related Parties | Senior beneficial interest Guaranty Agreement Indemnification Agreement Put (VIE Equity) Call (VIE Equity) |
Senior beneficial interest Guarantee Agreement | ||
| Biovax Investment, LLC | VIE Equity (99.9%) | |||
| US Bancorp | VIE Equity (99.9%) | Tax Credit Rights | ||
| Biovax Investment Corp. | VIE Equity (0.01%) | |||
| Telesis CDE, Corp | VIE Equity (0.01%) | |||
New Market Tax Credit Transaction II:
| Variable Interest Holder |
Variable Interests AutovaxID Investment, LLC |
Variable Interests St. Louis NMTC Fund II, LLC | ||
| Biovest and its Related Parties | Senior beneficial interest Guaranty Agreement Indemnification Agreement Put (VIE Equity) Call (VIE Equity) |
Senior beneficial interest Guarantee Agreement | ||
| AutovaxID Investment, LLC | VIE Equity (99.9%) | |||
| US Bancorp | VIE Equity (100%) | Tax Credit Rights | ||
| St. Louis Development Corporation | VIE Equity (0.01%) | |||
The above table illustrates the weight of the variable interests that are held by the Company. In addition, in performing quantitative valuation, the Company afforded significant weight to the guarantee agreements, indemnifications and put features, the preponderance of which limit the equity investor’s risk of loss on the venture. In evaluating both qualitative and quantitative considerations, the Company concluded that its variable interests in the entity absorb most of the variable interest entities’ losses and should, therefore, consolidate the entities under the scope of ASC 810.
F-43
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Assets of $21.5 million and liabilities of $17.9 million of the variable interest entities identified above are limited to the instruments referred to in the description of the NMTC financing arrangement above. In accordance with consolidation principles, these assets and liabilities are eliminated in consolidation leaving the non-controlling interests of Telesis CDE Two, LLC, Biovax Investment, LLC, AutovaxID Investment, LLC, and St. Louis NMTC Fund II, LLC, reflected on the Company’s September 30, 2010 consolidated balance sheet as non-controlling interests in variable interest entities.
Revimmune, LLC
Although, the Company does not have an equity interest in Revimmune, LLC, the Company has the controlling financial interest of Revimmune, LLC, because of the sublicense agreement between the parties and is considered the primary beneficiary, and therefore, the financial statements of Revimmune, LLC have been consolidated with the Company as of February 27, 2007 and through September 30, 2010. As of September 30, 2010, Revimmune, LLC’s assets and equity were approximately $28,000. The Company’s had no non-controlling interest in earnings from Revimmune, LLC for the twelve months ended September 30, 2010.
Non-controlling interest in income (loss)
The Company’s non-controlling interest in (income) loss from variable interest entities on its consolidated statement of operations for the year ended September 30, 2010 consists of the following:
| Biovax Investment LLC |
$ | 455,765 | ||
| Telesis CDE Two, LLC |
(59,247 | ) | ||
| AutovaxID Investment, LLC |
448,000 | |||
| St. Louis NMTC Fund II, LLC |
(432,807 | ) | ||
| Non-controlling interest in earnings from variable interest entities |
$ | 411,711 | ||
24. New Market Tax Credit Transactions:
April 2006 NMTC Transaction:
On April 25, 2006, Biovest through its wholly owned subsidiary, Biovax, Inc. (“Biovax”) closed a financing transaction (“Transaction I”) that was structured in an effort to obtain certain perceived advantages and enhancements from the New Markets Tax Credit (“NMTC”) regulations adopted under the auspices of the United States Department of the Treasury in 2002 to provide incentive for investing in businesses located in “qualifying census tracts,” or areas with a median income below the poverty line. The NMTC was provided for in the Community Renewal Relief Act of 2000 (the “Act”) and permits taxpayers (whether companies or individuals) to claim credits against their Federal income taxes for up to 39% of qualified investments in the equity of Community Development Entities (“CDE”). CDE are privately managed investment institutions that are certified to make Qualified Low-Income Community Investments (“QLICI”). The following parties were involved in Transaction I: the Company, Biolender, LLC (“Biolender”), Biovax Investment Corp., Biovax Investment, LLC (“Fund”), U.S. Bancorp Community Investment Corporation (“U.S. Bancorp”), Telesis CDE Two, LLC (“CDE”), Telesis CDE Corporation, Biovax, and Laurus. Biovax is a qualified, active low-income business and is eligible to receive investment capital under the NMTC regulations.
On March 31, 2006, in contemplation of Transaction I, Biovest closed a financing transaction with Laurus pursuant to which Laurus purchased from Biovest a secured promissory note in the principal amount of $7.799 million (the “Laurus Note”). The Company originally guaranteed 64% of any amounts outstanding under the note. On May 30, 2008, this guaranty was modified to reflect a fixed principal amount of $5.0 million. Under the terms of the Laurus Note, $7.5 million of the principal amount was deposited into a restricted bank account of Biovest pursuant to a restricted account agreement between Biovest and Laurus. The Company, Analytica and Laurus also entered into an Amended and Restated Stock Pledge Agreement pledging the Company’s shares of TEAMM, Analytica, Biovest, and Biolender, who was added as obligor by way of joinder, to secure the obligations owed to Laurus as a result of the Laurus Note.
F-44
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In contemplation of Transaction I, Biovest and the Company formed Biolender, as a Delaware limited liability company. On April 21, 2006, $2.5 million was released from the restricted account created under the Laurus Note. The release of these funds was contingent upon their use in Transaction I. These proceeds were used to purchase a 29.5% equity investment in Biolender for $2.5 million. Biovest used the proceeds of a $6.0 million intraday loan from First Bank to purchase the remaining 70.5% equity interest in Biolender. The $6.0 million loan from First Bank was fully guaranteed by two of the Company’s directors. On April 27, 2006, Biovest redeemed 10 million shares of its common stock owned of record by the Company for a $6.0 million cash payment which equaled the market price of $0.60 per share. The Company used the proceeds of the stock redemption to repay its intraday loan due First Bank. Subsequently, on October 31, 2006, the Company entered into a Purchase Agreement with Biovest whereby Biovest purchased the Company’s 70.5% ownership interest in Biolender. In consideration of the sale of this interest in Biolender, Biovest issued to the Company 10 million shares of common stock, representing the negotiated value of the purchased interest.
In contemplation of Transaction I, Biovax Investment, LLC (the “Fund”) was established. U.S. Bancorp invested $3.6 million for a 99.99% equity interest in the Fund. Biovax Investment Corp., the Fund manager, invested an additional $100 for the remaining 0.01% equity interest. On April 25, 2006, Biolender loaned the Fund $8.5 million pursuant to a 5.18%, annual rate, senior secured, convertible note receivable, due October 27, 2013. Interest on the note is payable as follows: (i) 0.64% interest per annum, non-compounding, shall be payable on the first day of each calendar month until October 27, 2013; and (ii) any remaining accrued and unpaid interest shall be payable in one installment on October 27, 2013. The note is convertible at the option of the fund into shares of the Company’s common stock near the maturity date.
The proceeds received by the Fund from the aforementioned financing transactions were used to make a contemporaneous 99.99% equity investment in Telesis CDE II, LLC ($12.0 million), as well as, make payment for associated management, legal and accounting fees ($0.1 million). The $12.0 million investment by the Fund to the CDE constituted a qualified equity investment (“QEI”) under the NMTC Program authorized by Section 45D of the Internal Revenue Code of 1986, as amended (the “Code”), resulting in $4.7 million in tax credits.
The CDE is a Community Development Entity that is certified through the U.S. Treasury Department to make QLICI, and is managed and partially owned (0.01%) by Telesis CDE Corporation, a private financial institution. Telesis CDE Corporation paid $1,200 in consideration for its 0.01% interest in Telesis. The CDE, upon receipt of its equity funding, contemporaneously issued $11.5 million to Biovax for a 1.0% convertible promissory note payable, due October 27, 2013. The convertible promissory note is convertible into common stock at the option of the CDE within 5 days of the maturity date at a conversion price equaling the then trading market price of the common stock. The overall arrangement provides that in the event the CDE converts the note payable, the aforementioned note receivable is subject to immediate conversion at the same conversion price. Biovest also issued to Telesis CDE Corporation warrants to purchase 1.2 million shares of Biovest’s common stock over a period of nine-years at a fixed price of $1.30. These warrants were reflected as an equity financing cost in stockholders’ equity at a fair value of $517,000 computed using the Black-Scholes-Merton option pricing model. We also issued warrants to Telesis CDE Corporation to purchase 0.2 million shares of the Company’s common stock over a period of seven years at a fixed price of $9.00.
Biovax used the proceeds of the $11.5 million convertible promissory note as follows: $6.0 million was paid to Biovest pursuant to an Asset Purchase and Sale Agreement dated April 18, 2006 and described further below, $1.6 million was issued as a dividend to Biovest and $1.3 million was paid to Biovest for BiovaxID anti-cancer vaccines in various stages of production. The remaining $2.6 million was used to cover ongoing operational expenses.
The transaction was structured so that, upon maturity, Biovax will have paid approximately $12.4 million in principal and interest payments to the CDE. The operating agreement of the CDE stipulates that in the event the QLICI is repaid in the combination of stock and cash, the stock received shall be distributed to the Fund. Furthermore, any distributable cash received by Telesis CDE II, LLC shall be distributed to the Fund in proportion to the Fund’s respective percentage interest in the CDE in an amount sufficient to fully pay the Fund’s note payable to Biolender. Upon maturity, the Fund will have paid approximately $11.9 million in principal and interest payments to Biolender. At maturity, total equity of the Fund is approximated to be $100,000 resulting from the difference of $12.4 million in principal and interest payments received less $11.9 million in principal and interest paid less approximately $0.4 million in estimated operating costs of the Fund over the 7.5 year term of the notes. Biolender
F-45
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
and U.S. Bancorp entered into a put option wherein U.S. Bancorp will have the right to put its investment in the Fund to Biovest near the maturity of the instruments at a price of $180,000. Management has concluded that the fair value of U.S. Bancorp’s investment in the Fund at maturity of both notes (approximately $0.1 million) would be less than the $180,000 U.S. Bancorp would receive upon exercise of the put option and thus it is management’s expectation that this option will be exercised. Thus, prior to maturity of both notes, it is anticipated that the Fund will be 99.99% owned by Biolender. Biovest accounted for this option as a derivative liability that requires recognition at fair value. Biovest utilized a probability-based, discounted cash flow approach to value the put. Accordingly, the valuation technique provided for the recognition of the full put amount ($180,000) at the present value of its cash flows, using market interest rates that would likely be considered by market participants trading similar instruments.
Salient terms and conditions of Transaction I are as follows:
| • | Under an Asset Purchase and Sale Agreement dated as of April 18, 2006, Biovest transferred all or substantially all of the assets of its vaccine manufacturing business situated at 377 Plantation Street, Worcester, Massachusetts (the “Plant” and the assets hereinafter the “Equipment”) and its rights under that certain lease agreement for the Plant and that certain letter of intent with the landlord to potentially lease additional space adjacent to the Plant (collectively the “Leasehold”) to Biovax. As full purchase price for the Equipment, Biovax paid Biovest $1.5 million. In addition, Biovax advanced rental payments for the Leasehold in the amount of $4.5 million. Under the Asset Purchase Agreement, Biovest is required to treat the advance as unrestricted and non-segregated funds provided that Biovest uses the funds to make all required lease payments. Finally, Biovax also hired all of Biovest’s employees related to the vaccine manufacturing business and assumed responsibility for all accrued vacation time and the maintenance of existing health and other benefits. |
| • | The tax credits arising from the transaction were fully assigned to U.S. Bancorp. Biovax entered into an indemnification agreement directly with U.S. Bancorp that provides for indemnification in the event of tax credit recapture from events caused by Biovax. Biovax is contractually required to maintain the following covenants to avoid tax credit recapture: (i) Biovax shall maintain its status as a qualified active low-income business; (ii) At least fifty percent of the use of the tangible property of the Biovax (whether owned or leased) will be within the low-income community as defined by Section 45D(e) of the U.S. Tax Code; (iii) At least fifty percent of the services performed for Biovax by its employees will be within low-income community as defined by Section 45D(e) of the U.S. Tax Code; (iv) Less than five percent of the average of the unadjusted bases of the property of Biovax will be attributable to collectibles (as defined in Section 408(m)(2) of the Code, and which includes any work of art; any rug or antique; any metal or gem; any stamp or coin; and any alcoholic beverage) other than collectibles that are held primarily for sale to customers in the ordinary course of business; (v) Less than five percent of the average of the unadjusted bases of the property of Biovax will be attributable to nonqualified financial property (as defined in Section 1397C(e) of the Code and in Section 1.45D-1(d)(4)(i)(E) of the Regulations, and which includes debt, stock, partnership interests, options, futures contracts, forward contracts, warrants, notional principal contracts, annuities and other similar property); (vi) No part of the business activities of Biovax will consist of the operation of any private or commercial golf course, country club, massage parlor, hot tub facility, or suntan facility, race track or other facility used for gambling, or store the principal business of which is the sale of alcoholic beverages for consumption off premises; (vii) No part of Biovax’s business activities will include the rental to others of residential rental property (the term “residential rental property” is defined in Section 168(e)(2)(A) of the Code as meaning any building or structure if eighty percent or more of the gross rental income from such building or structure for the taxable year is rental income from dwelling units); (viii) The predominant trade or business of Biovax will not include the development or holding of intangibles for sale or license, as provided under Section 1.45D-1(d)(5)(iii) of the Regulations; (ix) Farming (within the meaning of Section 2032A(e)(5)(A) or (B) of the Code) will not be an activity of Biovax; (x) Biovax will generate revenues by the date of April 25, 2009; (xi) Biovax shall not discontinue conducting business, shall not materially change the nature of its business, and shall not materially change the manner in which its business activities are conducted, other than changes in the nature of its business or the manner in which it conducts its business that do not cause the making of the Loan by the Lender to cease to constitute a “qualified low-income community investment” as such term is used in Section 45D of the Code (as determined by the Lender in its good faith judgment and based upon the advice of |
F-46
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| counsel); (xii) Biovax will not be a bank, credit union or other financial institution; (xiii) Biovax will not maintain a qualified low-income building under Section 42 of the Code; (xiv) Biovax will not become a single-member entity treated as disregarded as separate from its owner for federal income tax purposes, nor be liquidated or merged into another entity without the written consent of Telesis CDE II, LLC; and (xv) Biovax and Biovest will operate consistently with the Asset Purchase Agreement between Biovax and Biovest, and will not amend such agreement without prior written consent of Telesis CDE II, LLC. An example of an event that would not cause a recapture is changes in the Internal Revenue Code that results in such recapture. The total indemnification amount could be $4.7 million (representing 39% of the $12.0 million qualified investment). However, in accordance with Financial Interpretation 45 Guarantor’s Accounting and Disclosure Requirements for Guarantees, including Indirect Guarantees of the Indebtedness of Others, the conditions and events that could result in recapture are within the control of Biovax. Therefore, this potential liability is not reflected in the consolidated financial statements. |
| • | The Company, Biovest and certain their directors entered into a guarantee arrangement with the CDE for the debt service of Biovax. Biovest has guaranteed 100% of the debt service while the directors of the Company and have guaranteed up to $6.0 million of the debt service. In addition, the Company has partially guaranteed debt service with limitations established at no greater than $60,000 each year the instrument is outstanding. Biovest issued warrants to purchase 1.0 million shares of common stock to the officers and directors as compensation for their guarantees. The guarantees were treated in a manner similar to contributed service and their fair value of $460,000 was charged to expense upon issuance. Biovest also indemnified the guarantors from any claims of any kind to the extent that the guarantors are called upon to pledge funds, assets or collateral in connection with the guarantee being executed. |
| • | Various legal and accounting fees of $108,000 paid directly by Biovax and involved in structuring this transaction were recorded as a reduction to additional paid-in capital. Various legal and accounting fees of $170,000 paid by entities in which Biovest has a variable interest and involved in structuring this transaction (the Fund and the CDE) were recorded as a reduction to non-controlling interests in variable interest entities on Biovest’s consolidated balance sheet. Professional fees of $360,000 involved in the continuing management of this transaction were recorded as a prepaid expense and will be amortized over a period of seven and one half years, representing the duration of both convertible notes issued by the Fund and Biovax. |
As a result of Biovest’s Chapter 11 filing on November 10, 2008, Biovax discontinued interest payments due to its QLICI promissory note. In July 2010, Biovest and certain of its affiliates entered into an agreement (the “Worcester Restructuring Agreement”) with Telesis CDE Corporation and Telesis CDE Two, LLC (collectively, “Telesis”), contingent upon submission to and approval by the Bankruptcy Court. The Worcester Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to Transaction I, in consideration of retention by Telesis of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $0.3 million along with a settlement payment in the amount of $85,000 to defray certain legal and administrative expenses incurred by Telesis. Upon approval by the Bankruptcy Court, Biovest’s Guaranty, our Guaranty, and all of Biovest’s subsidiary Guaranties from affiliates and third parties and all other obligations to all parties to Transaction I will be terminated.
December 2006 NMTC Transaction:
On December 8, 2006, Biovest through its wholly owned subsidiary, AutovaxID, Inc. (“AutovaxID”) closed a second NMTC financing transaction (“Transaction II”). The following parties were involved in Transaction II: AutovaxID, the Company, Biolender II, LLC (“Biolender II”), St. Louis New Market Tax Credit Fund II, LLC (“CDE II”), St. Louis Development Corp., AutovaxID Investment LLC (“Fund II”), U.S. Bancorp, and Laurus.
On December 8, 2006, the Company loaned to Biovest $3.1 million pursuant to a Secured Promissory Note (the “Accentia Note”). Under the terms of the Accentia Note, interest accrues at a rate equal to prime rate, and is convertible, at the Company’s option, to shares of Biovest’s common stock at a conversion price of $0.32 per share. Upon closing of Transaction II, Biovest repaid the Company $1.1 million. The remaining $2.0 million of principal and all accrued and unpaid interest is included in notes payable, related parties in the accompanying September 30, 2008 consolidated balance sheet, and is due upon 30 days advance written notice by the Company.
F-47
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In contemplation of Transaction II, Biovest formed Biolender II as a Delaware limited liability company. On December 8, 2006, $2.5 million was released from the restricted account created under the Laurus Note. The release of these funds was contingent upon their use in Transaction II. These funds, together with the amount loaned to Biovest under the Accentia Note funded the purchase of a 100% equity interest in Biolender II for the benefit of Biovest. Biovest’s entire equity interest of $5.6 million in Biolender II has been pledged to Laurus as collateral to secure the Laurus Note.
In contemplation of Transaction II, Fund II was established. U.S. Bancorp invested $2.4 million for a 100% equity interest in Fund II. Additionally, Biolender II and Fund II entered into a Loan and Security Agreement pursuant to which Biolender II made a loan to Fund II in the principal amount of $5.6 million (the “Leverage Loan”), evidenced by a promissory note dated December 8, 2006 payable from Fund II to Biolender II (the “Leverage Note”). The Leverage Note becomes due on June 9, 2014, and bears an interest rate of 8%, non-compounding. Payment of interest is due annually on the first calendar day of each year through maturity. The outstanding principal amount on the Leverage Loan and any unpaid interest is due on maturity in cash.
The proceeds received by Fund II from U.S. Bancorp and Biolender II were used to make a contemporaneous 99.99% equity investment in CDE II. The $8.0 million investment by Fund II to CDE II constituted a qualified equity investment (“QEI”) under the NMTC Program authorized by Section 45D of the Internal Revenue Code of 1986, as amended, resulting in $3.12 million in tax credits which were allocated to U.S. Bancorp. All of Fund II interest in CDE II has been pledged to Biolender II as collateral for the Leverage Loan.
CDE II is a Community Development Entity that is certified through the U.S. Treasury Department to make QLICI, and is managed and partially owned (0.01%) by St. Louis Development Corporation, a not-for-profit corporation organized in Missouri. St. Louis Development Corporation paid $1,000 in consideration for its 0.01% interest in CDE II. CDE II, upon receipt of its equity funding, contemporaneously issued a QLICI to AutovaxID, evidenced by a $7.7 million subordinated promissory note dated as of December 8, 2006 which matures on December 8, 2036. Pursuant to a call right, for a period of six months starting on December 8, 2013, the CDE will have the right to call for the repayment of the CDE II Loan in the amount of $5.7 million, in full satisfaction of the principal on the CDE II Loan. Interest on the outstanding principal amount of the CDE II Loan accrues at the rate of 5.82% per annum, non-compounding and is payable in arrears on an annual basis having commenced on January 2, 2007 and continuing until maturity. The CDE II Loan is guaranteed in full by Biovest and also guaranteed up to an amount of $4.5 million by the Company’s officers and directors. Biovest issued warrants to purchase 2.6 million shares of common stock to these officers and directors as compensation for their guarantees. The guarantees were treated in a manner similar to contributed service and their fair value of $1.4 million was charged to expense upon issuance. Biovest also indemnified the guarantors from any claims of any kind to the extent that the guarantors are called upon to pledge funds, assets or collateral in connection with the guarantee being executed.
AutovaxID used the proceeds from the $7.7 million promissory note to pay Biovest $6.1 million pursuant to an Asset Purchase and License Agreement dated December 8, 2006 and described further below. The remaining $1.6 million was used to cover ongoing operational expenses of AutovaxID.
The transaction was structured so that, on June 9, 2014, AutovaxID will have paid approximately $9.1 million in principal and interest payments to CDE II. The operating agreement of CDE II stipulates that any distributable cash received shall be distributed to Fund II in proportion to the respective percentage interest Fund II has in CDE II in an amount sufficient to fully pay the Fund II note payable to Biolender II. On June 9, 2014, Fund II will have paid approximately $9.0 million in principal and interest payments to Biolender II (assuming CDE II exercises its right to call the CDE II loan for $5.7 million on June 9, 2014). At maturity then, total equity of the Fund is approximated to be $100,000, resulting from the difference of $9.1 million in principal and interest payments received less $9.0 million in principal and interest paid over the 7.5 year term of the notes.
Biolender II and U.S. Bancorp entered into a put option wherein U.S. Bancorp will have the right to put its investment in Fund II to the Company near the maturity of the instruments at a price of $120,000. Management has concluded that the fair value of U.S. Bancorp’s investment in Fund II at maturity of both notes (approximately $100,000) would be less than the $120,000 U.S. Bancorp would receive upon exercise of the put option and thus it is management’s expectation that this option will be exercised. Thus, prior to maturity of both notes, it is anticipated that the Fund will be 100% owned by Biolender II. Biovest accounted for this option as a derivative liability that requires recognition at fair value. Biovest utilized a probability-based, discounted cash flow approach to value the
F-48
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
put. Accordingly, the valuation technique provided for the recognition of the full put amount ($120,000) at the present value of its cash flows, using market interest rates that would likely be considered by market participants trading similar instruments.
Salient terms and conditions of Transaction II are as follows:
| • | Under a License and Asset Purchase Agreement dated as of December 8, 2006, Biovest granted a non-exclusive license to the intellectual property enabling AutovaxID to manufacture and sell automated cell culture instruments in the United States, Canada and Mexico (the “License”), which license became exclusive upon the occupancy by AutovaxID of a space located at 1031 Macklind Avenue, St. Louis, Missouri (the “New Plant”) in June 2007. As full purchase price for the License and related business opportunity, AutovaxID paid Biovest $5.6 million. Biovest also agreed to sell AutovaxID certain equipment upon the occupancy by AutovaxID of the New Plant for the fair market value of $0.5 million. |
| • | The CDE II Loan is secured by second lien on all assets of AutovaxID for the benefit of CDE II pursuant to a Second-Lien Security Agreement between AutovaxID and CDE II dated as of December 8, 2006. Laurus has a senior lien on the assets of AutovaxID through the security agreement from Biovest to Laurus, which AutovaxID joined by way of a Joinder Agreement. |
| • | AutovaxID does not have the right to prepay the CDE Loan prior to June 8, 2014. AutovaxID does have the right to prepay the CDE Loan after this date, provided that (i) it prepays the entire CDE Loan amount, (ii) the CDE consents to such prepayment and U.S. Bancorp and the managing member of the CDE agree on the reinvestment of such proceeds in an alternative investment that would qualify as a QLICI and (iii) AutovaxID or the Individual Guarantors (as defined below) under the Tax Credit and Reimbursement and Indemnity Agreement pay to U.S. Bancorp the recapture amount so specified is such agreement. |
| • | All indebtedness owed by AutovaxID and its subsidiaries to CDE II, including its right to receive payments of principal and interest under the CDE II Loan, is expressly subordinate to the extent set forth under the Telesis Subordination Agreement dated as of December 8, 2006 entered into by Laurus, CDE II, U.S. Bancorp, AutovaxID and the Company. |
| • | The tax credits arising from this transaction were fully assigned to U.S. Bancorp. AutovaxID has entered into an indemnification agreement directly with U.S. Bancorp that provides for indemnification in the event of tax credit recapture from events caused by AutovaxID. AutovaxID is contractually required to maintain the following covenants to avoid tax credit recapture: (i) AutovaxID shall maintain its status as a qualified active low-income business; (ii) At least fifty percent of the use of the tangible property of the AutovaxID (whether owned or leased) will be within the low-income community as defined by Section 45D(e) of the U.S. Tax Code; (iii) At least fifty percent of the services performed for AutovaxID by its employees will be within low-income community as defined by Section 45D(e) of the U.S. Tax Code; (iv) Less than five percent of the average of the unadjusted bases of the property of AutovaxID will be attributable to collectibles (as defined in Section 408(m)(2) of the Code, and which includes any work of art; any rug or antique; any metal or gem; any stamp or coin; and any alcoholic beverage) other than collectibles that are held primarily for sale to customers in the ordinary course of business; (v) Less than five percent of the average of the unadjusted bases of the property of AutovaxID will be attributable to nonqualified financial property (as defined in Section 1397C(e) of the Code and in Section 1.45D-1(d)(4)(i)(E) of the Regulations, and which includes debt, stock, partnership interests, options, futures contracts, forward contracts, warrants, notional principal contracts, annuities and other similar property); (vi) No part of the business activities of AutovaxID will consist of the operation of any private or commercial golf course, country club, massage parlor, hot tub facility, or suntan facility, race track or other facility used for gambling, or store the principal business of which is the sale of alcoholic beverages for consumption off premises; (vii) No part of AutovaxID’s business activities will include the rental to others of residential rental property (the term “residential rental property” is defined in Section 168(e)(2)(A) of the Code as meaning any building or structure if eighty percent or more of the gross rental income from such building or structure for the taxable year is rental income from dwelling units); (viii) The predominant trade or business of AutovaxID will not include the development or holding of intangibles for sale or license, as provided under Section 1.45D-1(d)(5)(iii) of the Regulations; (ix) Farming (within the meaning of Section 2032A(e)(5)(A) or (B) of the Code) will not |
F-49
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| be an activity of AutovaxID; (x) AutovaxID will generate revenues by the date of December 8, 2009; (xi)AutovaxID shall not discontinue conducting business, shall not materially change the nature of its business, and shall not materially change the manner in which its business activities are conducted, other than changes in the nature of its business or the manner in which it conducts its business that do not cause the making of the Loan by the Lender to cease to constitute a “qualified low-income community investment” as such term is used in Section 45D of the Code (as determined by the Lender in its good faith judgment and based upon the advice of counsel); (xii) AutovaxID will not be a bank, credit union or other financial institution; (xiii) AutovaxID will not maintain a qualified low-income building under Section 42 of the Code; (xiv) AutovaxID will not become a single-member entity treated as disregarded as separate from its owner for federal income tax purposes, nor be liquidated or merged into another entity without the written consent of CDE II; and (xv) AutovaxID and Biovest will operate consistently with the License between AutovaxID and Biovest, and will not amend such agreement without prior written consent of CDE II. An example of an event that would not cause a recapture is changes in the Internal Revenue Code that results in such recapture. The total indemnification amount could be $3.12 million (representing 39% of the $8.0 million qualified investment). However, in accordance with Financial Interpretation 45 Guarantor’s Accounting and Disclosure Requirements for Guarantees, including Indirect Guarantees of the Indebtedness of Others, the conditions and events that could result in recapture are within AutovaxID’s control. Therefore, this potential liability is not reflected in the consolidated financial statements. |
| • | Under a Reimbursement Agreement dated as of December 8, 2006, Biovest agreed to reimburse St. Louis Development Corp (the 0.01% owner and managing member of CDE II) up to $32,000 annually for expenses incurred by St. Louis Development Corp in connection with its management of CDE II. |
| • | Various legal, accounting, and professional fees of $433,000 paid directly by Biovest and AutovaxID and involved in structuring this transaction were recorded as a reduction to additional paid-in capital. Various legal and accounting fees of $180,000 paid by entities in which Biovest has a variable interest (CDE II, and Fund II) and involved in structuring this transaction were recorded as a reduction to non-controlling interests in variable interest entities on Biovest’s consolidated balance sheet. Professional fees of $115,000 involved in the continuing management of this transaction were recorded as a prepaid expense and will be amortized over a period of seven and one half years, representing the duration of the note issued by Fund II and payable to Biolender II. |
As a result of Biovest’s Chapter 11 filing on November 10, 2008, AutovaxID discontinued interest payments due on its QLICI promissory note. In July 2010, Biovest and certain of its affiliates entered into an agreement (the “St. Louis Restructuring Agreement”) with St. Louis Development Corporation and Saint Louis New Markets Tax Credit Fund II, LLC (collectively “SLDC”), contingent upon submission to and approval by the Bankruptcy Court. The St. Louis Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to Transaction II, in consideration of retention by SLDC of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $160,000 along with a settlement payment in the amount of $62,000 to defray certain legal and administrative expenses incurred by SLDC. Upon approval by the Bankruptcy Court, Biovest’s Guaranty, the Company’s Guaranty, and all of Biovest’s subsidiary Guaranties from affiliates and third parties and all other obligations to all parties to Transaction II will be terminated. See the Subsequent Events footnote for information on the treatment of this liability in the Company’s Plan.
F-50
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
25. Subsequent Events:
Plan of Reorganization:
On August 16, 2010, the Company and its subsidiaries (collectively, the “Debtors”) filed their First Amended Joint Plan of Reorganization, and, on October 25, 2010, the Debtors filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the United States Bankruptcy Court for the Middle District of Florida, Tampa Division (the “Bankruptcy Court”) held a Confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. In connection with the confirmation hearing all creditor classes deemed “impaired” pursuant to the Bankruptcy Code voted to support the Plan. The Debtors emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”). Pursuant to the Plan, the date on which a claim in any Class listed in the Plan became an “allowed claim” by order of the Bankruptcy Court or by agreement between Debtors and the claimant is the “Determination Date”.
Amendments to Articles of Incorporation or Bylaws.
On the Effective Date, the Company amended and restated its articles of incorporation and its bylaws to incorporate provisions required by the Plan, the Confirmation Order, and/or the U.S. Bankruptcy Code. As of the Effective Date, the Company’s second amended and restated articles of incorporation authorize the Company to issue up to 50,000,000 shares of preferred stock and up to 300,000,000 shares of common stock, and each share of the Company’s common stock outstanding immediately before the Effective Date will, under the terms of the Plan, remain outstanding after the Effective Date. The amendments to the articles also included an elimination of the “classified” board of directors and a provision establishing the quorum required for action to be taken at an Annual Meeting of Shareholders at one-third (33.33%) of the number of shares issued and outstanding.
Laurus/Valens Term Notes (Class 2):
Pursuant to the Plan, Laurus Master Fund, Ltd. (in liquidation), PSource Structured Debt Limited, Valens Offshore SPV I, Ltd., Valens Offshore SPV II, Corp., Valens U.S. SPV I, LLC, and LV Administrative Services, Inc. (“Laurus/Valens”), have an allowed secured claim against the Company in the amount of $8.8 million. On the Effective Date, pursuant to a Term Loan and Security Agreement, the Company executed and delivered to Laurus/Valens term notes (the “Laurus/Valens Term Notes”) evidencing such allowed secured claim in the original principal amount of $8.8 million. The following are the material terms and conditions of the Laurus/Valens Term Notes:
| • | the Laurus/Valens Term Notes mature on the second (2nd) anniversary of the Effective Date and may be prepaid at any time without penalty; |
| • | interest accrues on the Laurus/Valens Term Notes at the rate of eight and one-half percent (8.5%) per annum (with a twelve and one-half percent (12.5%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
| • | the Company is required to make mandatory prepayments under the Laurus/Valens Term Notes as follows: |
| • | on that date which is eighteen (18) months following the Effective Date, a payment of principal, in cash, in the amount of $4.4 million, less the amount of any prior optional prepayments of principal by the Company and the amount of any other mandatory prepayments of principal under the Laurus/Valens Term Notes; and |
| • | a prepayment equal to thirty percent (30%) of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by the Company) of certain capital raising transactions (with certain exclusions); |
F-51
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| • | the Laurus/Valens Term Notes are secured by: |
| • | a first lien in all of the assets of the Company, junior only to the liens granted under the Plan to holders of the 8% Original Issue Discount Secured Convertible Debentures Due June 19, 2011, issued by the Company in June 2008, in the original aggregate principal amount of $8,906,098.00 and certain permitted liens; |
| • | a pledge by the Company to Laurus/Valens of (a) all of its equity interests in Analytica, and (b) 20,115,818 shares of common stock of Biovest owned by the Company; and |
| • | all of the assets of Analytica, which secure a guaranty of Analytica as to the entire indebtedness under the Laurus/Valens Term Notes; and |
| • | with the prior written consent of Laurus/Valens, the Company may convert all or any portion of the outstanding principal and accrued interest under the Laurus/Valens Term Notes into a number of shares of the Company’s common stock (the “Laurus/Valens Conversion Shares”) equal to (a) the aggregate portion of the principal and accrued but unpaid interest outstanding under the Laurus/Valens Term Note being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported for the Company’s common stock for the ten (10) trading days immediately preceding the date of the notice of conversion. |
| • | On the Effective Date, 2,236,848 shares of the Company’s common stock were issued to Laurus/Valens for payment of Laurus/Valens Class 5 and Class 13 claims aggregating approximately $6.0 million at a conversion rate equal to $2.67 per share. |
Southwest Bank/Dennis Ryll – (Class 3)
On the Effective Date, the Company executed and delivered in favor of Dennis Ryll, the holder by assignment of the Company’s secured note to Southwest Bank, a promissory note (the “Class 3 Plan Note”) in an original principal amount of $4,483,284. The following are the material terms and conditions of the Class 3 Plan Note:
| • | the Class 3 Plan Note matures on the date that is twenty-four (24) months following the Effective Date; |
| • | interest accrues and is payable on the outstanding principal balance of the Class 3 Plan Note from time to time (the “Class 3 Interest”) at a fixed rate of six percent (6%) per annum; |
| • | on the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 3 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 3 Plan Note plus the Class 3 Interest as of the Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the Class 3 Automatic Conversion Price per share of the Company’s common stock. On the Effective Date, the first quarterly installment in the amount of approximately $0.56 million was converted into the Company’s common stock at a conversion rate equal to $1.36 per share, resulting in the issuance of 412,067 Class 3 Plan Shares; |
| • | the Class 3 Plan Note is secured by a lien on 15 million shares of Biovest’s common stock owned by the Company (the “Class 3 Pledged Shares”), subject to incremental release of a designated portion of such security upon each quarterly payment under the Class 3 Plan Note; and |
| • | if, on any Class 3 Automatic Conversion Date, the Class 3 Automatic Conversion Price is less than $1.00 per share, Dennis Ryll may, at his election, either: |
| • | convert the Class 3 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share of the Company’s common stock; or |
F-52
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| • | liquidate that number of the Class 3 Pledged Shares which equals the Class 3 Automatic Conversion Amount using a conversion rate for the Class 3 Pledged Shares equal to the average of the trading price of the Biovest’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date. |
McKesson Corporation (Class 4):
On the Effective Date, the Company executed and delivered in favor of McKesson Corporation (“McKesson”) a promissory note (the “Class 4 Plan Note”) in an original principal amount of $4,341,767. The following are the material terms and conditions of the Class 4 Plan Note:
| • | the Class 4 Plan Note matures on the date that is forty (40) months following the Effective Date (unless earlier accelerated) and the outstanding principal together with all accrued but unpaid interest is due on such date; |
| • | interest accrues and is payable on the outstanding principal amount under the Class 4 Plan Note at a fixed rate of five percent (5%) per annum (with a ten percent (10%) per annum default rate); |
| • | the Company may prepay all or any portion of the Class 4 Plan Note, without penalty, at any time; and |
| • | the Class 4 Plan Note is secured by a lien on 6,102,408 shares of Biovest’s common stock owned by the Company. |
September 2006 Secured Debentures and Warrants (Class 5):
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered to holders of the Company’s 2006 Secured Debentures $3,109,880 in principal amount of new debentures (the “Class 5 Plan Debentures”):
The following are the material terms and conditions of the Class 5 Plan Debentures:
| • | the Class 5 Plan Debentures mature on the date that is eighteen (18) months following the Effective Date (provided, however, in the event that the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 5 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding such maturity date is below $0.75, then the maturity date will automatically be extended for an additional twelve (12) months), and the outstanding principal together with all accrued but unpaid interest is due on such maturity date; |
| • | interest accrues and is payable on the outstanding principal amount under the Class 5 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum; and |
| • | each of the Class 5 Plan Debentures is secured by a lien on certain shares of Biovest’s common stock owned by the Company; |
| • | at the option of a holder of Class 5 Plan Debentures, all or any portion of the then outstanding balance of such holder’s Class 5 Plan Debentures may be converted into shares of the Company’s common stock or exchanged for shares of Biovest’s common stock at the applicable conversion or exchange rate set forth in such holder’s Class 5 Plan Debenture; |
| • | commencing on the first day that is six (6) months following the Class 5 Lock-Up Period (as defined below), if the trading price of the Company’s common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least 150% of the fixed conversion price for a holder of Class 5 Plan Debentures for any ten (10) consecutive trading days (in the case of a conversion into the Company’s common stock), or the trading price of Biovest’s common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least $1.25 for any ten (10) consecutive trading days (in the case of a exchange into Biovest’s common stock), the Company, at its option, may (a) convert the then outstanding balance of all of the Class 5 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to the fixed conversion price for each holder of |
F-53
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| Class 5 Plan Debentures, or (b) exchange the then outstanding balance of all of the Class 5 Plan Debentures into shares of Biovest’s common stock owned by the Company at a rate equal to $0.75 per share of Biovest’s common stock (with certain exceptions set forth in the Class 5 Plan Debentures and the Plan); and |
| • | in the event a holder of Class 5 Plan Debentures elects to receive shares of Biovest’s common stock, then such holder will be subject to certain restrictions set forth in the Class 5 Plan Debentures and the Plan regarding Biovest’s common stock during the ninety (90) day period following the Effective Date (the “Class 5 Lock-Up Period”). |
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered the following warrants (the “Class 5 Plan Warrants”) to purchase up to 2,508,960 shares of the Company’s common stock or up to 14.4 million shares of Biovest’s common stock owned by the Company:
The following are the material terms and conditions of the Class 5 Plan Warrants:
The Class 5 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from the Effective Date, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 5 Plan Warrants and the Plan.
June 2008 Secured Debentures and Warrants (Class 6):
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered to the holders of the Company’s 2008 Secured Debentures $9,730,459 in principal amount of debentures (the “Class 6 Plan Debentures”):
The following are the material terms and conditions of the Class 6 Plan Debentures:
| • | the Class 6 Plan Debentures mature on the date that is thirty-six (36) months following the Effective Date, and the outstanding principal together with all accrued but unpaid interest is due on such date; |
| • | interest accrues and is payable on the outstanding principal under the Class 6 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum and each of the Class 6 Plan Debentures is secured by a lien on property of the Company; |
| • | at the option of a holder of Class 6 Plan Debentures, such holder may elect to convert all of the then outstanding balance of its Class 6 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to $1.10 per share of the Company’s common stock; and |
| • | commencing on the date six (6) months after the Effective Date, if the trading price of the Company’s common stock (as determined in accordance wit the Class 6 Plan Debentures and the Plan) is at least 150% of $1.10 per share for any ten (10) consecutive trading days, the Company, at its option, may convert the then outstanding balance of all of the Class 6 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to $1.10 per share of the Company’s common stock. |
On the Effective Date, the Company executed and delivered the following warrants (the “Class 6 Plan Warrants”) to purchase up to 2,979,496 shares of the Company’s common stock:
The Class 6 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from the Effective Date, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 6 Plan Warrants and the Plan.
$385,959 in Class 6 Debentures were converted into Company common stock on Effective Date at a conversion price equal to $1.10 per share, resulting in the issuance of 350,872 of the Company’s common stock.
F-54
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
February 2007 Debentures and Warrants (Class 9):
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered to holders of the Company’s 2007 Debentures $19,109,554 in principal amount of debentures (the “Class 9 Plan Debentures”):
The following are the material terms and conditions of the Class 9 Plan Debentures:
| • | the Class 9 Plan Debentures mature on the date that is twenty-four (24) months following the Effective Date (the “Class 9 Plan Debenture Maturity Date”) and no interest will accrue on the outstanding principal balance of the Class 9 Plan Debentures; |
| • | on the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 9 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 9 Automatic Conversion Date (the “Class 9 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 9 Plan Debentures (the “Class 9 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the lesser of $1.25 per share or the Class 9 Automatic Conversion Price per share. On the Effective Date, a total of approximately $2.4 million were converted into Company common stock at a conversion price equal to $1.25 per share, resulting in the issuance of 1,910,963 of the Company’s common stock; |
| • | if, on any Class 9 Automatic Conversion Date, the Class 9 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 9 Plan Debentures may elect to convert the Class 9 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 9 Plan Debentures at the Class 9 Plan Debenture Maturity Date will be due and payable in full, at the election of the Company, in either cash or in shares of the Company’s common stock at a conversion rate equal to the average trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 9 Plan Debenture Maturity Date (provided that the average trading price for such period is at least $.50 per share); |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.50 per share for ten (10) consecutive trading days, a holder of the Class 9 Debentures, at its option, may convert any or all of the then outstanding principal balance of its Class 9 Plan Debenture into shares of the Company’s common stock at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on the Effective Date but not to exceed $1.25 per share; and |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.88 per share for thirty (30) consecutive trading days, the Company, at its option, may require the conversion of up to $5.0 million of the then aggregate outstanding principal balance of the Class 9 Plan Debentures at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on the Effective Date but not to exceed $1.25 per share. |
On the Effective Date, the Company executed and delivered the following warrants (the “Class 9 Plan Warrants”) to purchase up to 3,154,612 shares of the Company’s common stock:
The Class 9 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from the Effective Date, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 9 Plan Warrants and the Plan.
F-55
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Plan Distributions to Unsecured Creditors (Class 10):
Effective as of the Effective Date and issued as of the Determination Date, the Company became obligated to make cash distributions in the amount of approximately $2.4 million to unsecured creditors holding Class 10 claims under the Plan (the “Class 10 Plan Distributions”).
The Class 10 Plan Distributions mature on the date that is forty (40) months following the Effective Date and the outstanding principal together with all accrued but unpaid interest is due on such date. Interest accrues and is payable on the outstanding principal amount under the Class 10 Plan Distributions at a fixed rate of five percent (5%) per annum.
Unsecured creditors holding a total of $3,287,695 in Class 10 claims elected to convert those Class 10 claims into Company common stock valued at the average market price for the Company’s common stock over the ten trading days preceding the Effective Date, and the Company issued 2,417,431 of the Company’s common stock to these Class 10 creditors at a conversion price equal to $1.36 per share.
January 2008 Notes and Warrants (Class 13):
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered to holders of the Company’s Convertible Preferred Stock $4,903,644 in principal amount of promissory notes (the “Class 13 Plan Notes”):
The following are the material terms and conditions of the Class 13 Plan Notes:
| • | the Class 13 Plan Notes mature on the date that is twenty-four (24) months following the Effective Date (the “Class 13 Plan Note Maturity Date”) and no interest will accrue on the outstanding principal balance of the Class 13 Plan Notes; |
| • | on the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 13 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 13 Automatic Conversion Date (the “Class 13 Automatic Conversion Price”) is at least $1.00 per share, one-eighth ( 1/8th) of the original balance of the Class 13 Plan Notes (the “Class 13 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the Class 13 Automatic Conversion Price per share. On the Effective Date, a total of approximately $0.6 million of the outstanding principal amount of the Class 13 Plan Notes were converted into Company common stock at a conversion price equal to $1.36 per share, resulting in the issuance of 450,708 of the Company’s common stock; |
| • | if, on any Class 13 Automatic Conversion Date, the Class 13 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 13 Plan Notes may elect to convert the Class 13 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 13 Plan Notes at the Class 13 Plan Note Maturity Date will be due and payable in full, at the election of the Company, in either cash or in shares of the Company’s common stock at a conversion rate equal to the greater of the average of the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 13 Plan Note Maturity Date or $1.00; |
| • | if, at any time during the term of the Class 13 Plan Note, the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 125% of $1.25 per share for ten (10) consecutive trading days, a holder of Class 13 Plan Notes, at its option, may convert any or all of the then outstanding principal balance of its Class 13 Plan Note into shares of the Company’s common stock at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on the Effective Date; and |
F-56
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| • | if, at any time during the term of the Class 13 Plan Notes, the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 150% of $1.25 per share for thirty (30) consecutive trading days, the Company, at its option, may require the conversion of all or any portion of the then aggregate outstanding principal balance of the Class 13 Plan Notes at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on the Effective Date. |
Effective as of the Effective Date and issued as of the Determination Date, the Company executed and delivered the following warrants (the “Class 13 Plan Warrants”) to purchase up to 1,702,840 shares of the Company’s common stock:
The Class 13 Plan Warrants (a) have an exercise price of $1.50 per share and a term of three (3) years from the Effective Date, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 13 Plan Warrants and the Plan.
Equity Interests (Class 15):
Each Company common stockholder on the Effective Date was deemed to receive one (1) share of Reorganized Accentia Common Stock (the “Class 15 Plan Shares”) for each share of Existing Accentia Common Stock held by such stockholder as of the Effective Date. The Company’s Class 15 Plan Shares shall be deemed issued pursuant to Section 1145 of the Bankruptcy Code and shall not have any legend restricting the sale thereof under federal securities laws.
April 2006 NMTC Transaction:
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “Worcester Restructuring Agreement”) with Telesis CDE Corporation and Telesis CDE Two, LLC (collectively, “Telesis”), contingent upon submission to and approval by the Bankruptcy Court. The Worcester Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to Transaction I, in consideration of retention by Telesis of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $0.3 million along with a settlement payment in the amount of $85,000 to defray certain legal and administrative expenses incurred by Telesis. This Worcester Restructuring Agreement and the compromise of the outstanding claims against Biovest and its affiliates in connection with Transaction I was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, the Company’s and Biovest’s Guaranty, and all of Biovest’s subsidiary Guaranties from affiliates and third parties and all other obligations to all parties to Transaction I was terminated.
December 2006 NMTC Transaction:
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “St. Louis Restructuring Agreement”) with St. Louis Development Corporation and Saint Louis New Markets Tax Credit Fund II, LLC (collectively “SLDC”), contingent upon submission to and approval by the Bankruptcy Court. The St. Louis Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to Transaction II, in consideration of retention by SLDC of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $160,000 along with a settlement payment in the amount of $62,000, to defray certain legal and administrative expenses incurred by SLDC. This St. Louis Restructuring Agreement and the compromise of the outstanding claims against Biovest in connection with Transaction II was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, the Company’s and Biovest’s Guaranty, and all of our subsidiary Guaranties from affiliates and third parties and all other obligations to all parties to Transaction II were terminated. On November 17, 2010 pursuant to Biovest’s Plan, the outstanding balance due under the $3.1 million pursuant to a Secured Promissory Note from the Company was converted into Biovest’s common stock issued to the Company at a conversion rate equal to $0.75 per share.
F-57
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Termination of Warrants and Issuance of Shares:
On the Effective Date, all of the following warrants were terminated and cancelled pursuant to the Plan:
| • | the common stock purchase warrant dated August 16, 2005, issued by the Company to Laurus Master Fund, Ltd. (in liquidation), for the purchase of up to 1,000,000 shares of the Company’s common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated September 29, 2006, issued by the Company to Laurus Master Fund, Ltd. (in liquidation), for the purchase of up to 627,240 shares of the Company’s common stock at an exercise price of $2.75 per share; |
| • | the common stock purchase warrant dated October 31, 2007, issued by the Company to Laurus Master Fund, Ltd. (in liquidation), for the purchase of up to 4,024,398 shares of the Company’s common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated January 18, 2008, issued by the Company to Valens Offshore SPV I, Ltd., for the purchase of up to 365,169 shares of the Company’s common stock at an exercise price of $2.67 per share; and |
| • | the common stock purchase warrant dated January 18, 2008, issued by the Company to Valens U.S. SPV I, LLC, for the purchase of up to 196,629 shares of the Company’s common stock at an exercise price of $2.67 per share. |
Termination of Material Agreements:
On the Effective Date all of the following documents were terminated pursuant to the Plan:
| • | all documents evidencing or relating to loans made by Laurus/Valens to the Company prior to the Effective Date; |
| • | that certain revolving credit agreement between Southwest Bank and the Company dated as of December 30, 2005, that certain stock pledge agreement by and between the Company and Southwest Bank dated as of June 16, 2008, and all other documents executed in connection therewith; |
| • | all documents evidencing or relating to loans made by McKesson to the Company prior to the Effective Date (with certain exceptions set forth in the Plan); |
| • | all documents evidencing or relating to the 8% secured convertible debentures due September 29, 2010, issued by the Company in September 2006, in the original aggregate principal amount of $25,000,000.00; |
| • | all documents evidencing or relating to the 8% original issue discount secured convertible debentures due June 19, 2011, issued by the Company in June 2008, in the original aggregate principal amount of $8,906,098.00; |
| • | all documents evidencing or relating to the 8% convertible debentures due February 28, 2011, issued by the Company in February 2007, in the original aggregate principal amount of $24,940,000.00; and |
| • | all documents evidencing or relating to the Company’s Series A-1 convertible preferred stock, par value $1.00 per share. |
Cancellation and Reduction of Royalty Interests.
On the Effective Date, the Company and Biovest entered into a royalty termination agreement (the “Royalty Termination Agreement”), which was acknowledged by Laurus/Valens, and provides for the termination of that certain Royalty Agreement by and between the Company and Biovest, dated as of October 31, 2006, as amended by a letter agreement dated February 5, 2008, as further amended, modified or supplemented thereafter in accordance with its terms.
F-58
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Proforma Balance Sheet:
| September 30, 2010 |
Adjustments | Pro Forma September 30, 2010 (unaudited) |
||||||||||
| ASSETS |
||||||||||||
| Current assets |
||||||||||||
| Cash and cash equivalents |
$ | 558,452 | $ | 5,075,623 | $ | 5,634,075 | ||||||
| Other current assets |
2,115,828 | (63,333 | ) | 2,052,495 | ||||||||
| Total current assets |
2,674,280 | 5,012,290 | 7,686,570 | |||||||||
| Goodwill and intangibles |
1,976,962 | — | 1,976,962 | |||||||||
| Deferred finance costs |
— | 629,825 | 629,825 | |||||||||
| Other assets |
359,067 | — | 359,067 | |||||||||
| $ | 5,010,309 | $ | 5,642,115 | $ | 10,652,424 | |||||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT |
||||||||||||
| Current liabilities |
||||||||||||
| Accounts payable |
$ | 1,139,817 | $ | (143,000 | ) | $ | 996,817 | |||||
| Accrued expenses |
1,269,395 | — | 1,269,395 | |||||||||
| Reserve for unresolved claims |
— | 5,438,338 | 5,438,338 | |||||||||
| Unearned revenues |
263,778 | — | 263,778 | |||||||||
| Convertible promissory notes |
— | 10,686,202 | 10,686,202 | |||||||||
| Notes payable |
— | 1,024,196 | 1,024,196 | |||||||||
| Notes payable, related parties |
2,041,005 | (2,041,005 | ) | — | ||||||||
| Customer deposits |
134,613 | — | 134,613 | |||||||||
| Derivative liabilities |
1,844,200 | 13,811,063 | 15,655,263 | |||||||||
| Total current liabilities |
6,692,808 | 28,775,794 | 35,468,602 | |||||||||
| Liabilities subject to compromise |
143,570,128 | (143,570,128 | ) | — | ||||||||
| Notes payable |
— | 46,829,465 | 46,829,465 | |||||||||
| Convertible notes payable |
— | 21,482,531 | 21,482,531 | |||||||||
| Convertible notes payable, related party |
— | 153,948 | 153,948 | |||||||||
| Total liabilities |
150,262,936 | (46,328,390 | ) | 103,934,546 | ||||||||
| Preferred stock |
7,528,640 | (7,528,640 | ) | — | ||||||||
| Stockholders’ deficit |
||||||||||||
| Common stock |
58,048 | 11,360 | 69,408 | |||||||||
| Treasury stock |
(170,057 | ) | (1,326,164 | ) | (1,496,221 | ) | ||||||
| Additional paid-in capital |
203,828,364 | 59,938,597 | 263,766,961 | |||||||||
| Accumulated deficit |
(325,882,720 | ) | 4,440,886 | (321,441,834 | ) | |||||||
| Total stockholders’ deficit attributable to Accentia Biopharmaceuticals, Inc. |
(122,166,365 | ) | 63,064,679 | (59,101,686 | ) | |||||||
| Non-controlling interests |
(30,614,902 | ) | (3,565,534 | ) | (34,180,436 | ) | ||||||
| (152,781,267 | ) | 59,499,145 | (93,282,122 | ) | ||||||||
| $ | 5,010,309 | $ | 5,642,115 | $ | 10,652,424 | |||||||
F-59
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The Qualifying Therapeutic Discovery Project
On October 31, 2010, the Company received notice from the U.S. Internal Revenue Service (IRS) that it was approved to receive a Federal grant in the amount of approximately $245,000.00 under the Qualifying Therapeutic Discovery Project. The Qualifying Therapeutic Discovery Project tax credit is provided under new section 48D of the Internal Revenue Code (IRC), enacted as part of the Patient Protection and Affordable Care Act of 2010 (P.L. 111-148). The credit is a tax benefit targeted to therapeutic discovery projects that show a reasonable potential to:
| • | Result in new therapies to treat areas of unmet medical need or prevent, detect or treat chronic or acute diseases and conditions, |
| • | Reduce the long-term growth of health care costs in the United States, or |
| • | Significantly advance the goal of curing cancer within 30 years. |
Allocation of the credit will also take into consideration which projects show the greatest potential to create and sustain high-quality, high-paying U.S. jobs and to advance U.S. competitiveness in life, biological and medical sciences. The funds were awarded to support the advancement of Revimmune.
2010 Equity Incentive Plan
On November 15, 2010, the Company’s Board of Directors approved the 2010 Equity Incentive Plan, pursuant to which the Company may grant an aggregate of up to 10.0 million shares of the Company’s common stock for stock option awards, and other equity based awards, to employees, directors, and consultants of the Company and its subsidiaries.
F-60
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC.
INDEX TO UNAUDITED DECEMBER 31, 2010 FINANCIAL STATEMENTS
| Condensed Consolidated Balance Sheets as of December 31, 2010 (unaudited) and September 30, 2010 |
F-62 | |||
| Condensed Consolidated Statements of Operations for the Three Months Ended December 31, 2010 and 2009 (unaudited) |
F-64 | |||
| Condensed Consolidated Statement of Stockholders’ Deficit for the Three Months Ended December 31, 2010 (unaudited) |
F-66 | |||
| Condensed Consolidated Statements of Cash Flows for the Three Months Ended December 31, 2010 and 2009 (unaudited) |
F-67 | |||
| Notes to Condensed Consolidated Financial Statements |
F-69 | |||
F-61
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED BALANCE SHEETS
| December 31, 2010 (Unaudited) |
September 30, 2010 |
|||||||
| ASSETS |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 3,764,396 | $ | 558,452 | ||||
| Accounts receivable: |
||||||||
| Trade, net of allowance for doubtful accounts of $10,031 at December 31, 2010 and September 30, 2010 |
1,139,699 | 1,287,363 | ||||||
| Inventories |
429,348 | 417,087 | ||||||
| Unbilled receivables |
245,173 | 151,303 | ||||||
| Deferred finance costs |
234,124 | 16,077 | ||||||
| Prepaid expenses and other current assets |
1,059,129 | 243,998 | ||||||
| Total current assets |
6,871,869 | 2,674,280 | ||||||
| Goodwill |
893,000 | 893,000 | ||||||
| Intangible assets |
969,263 | 1,083,962 | ||||||
| Furniture, equipment and leasehold improvements, net |
221,022 | 142,276 | ||||||
| Deferred finance costs, less current portion |
195,104 | - | ||||||
| Other assets |
|
37,584
|
|
|
216,791
|
| ||
| TOTAL ASSETS |
$ | 9,187,842 | $ | 5,010,309 | ||||
(Continued)
F-62
Table of Contents
| December 31, 2010 (Unaudited) |
September 30, 2010 | |||||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| Liabilities not subject to compromise: |
||||||||
| Current liabilities: |
||||||||
| Current maturities of long-term debt, convertible |
$ | 22,866,918 | $ | — | ||||
| Accounts payable |
2,485,802 | 1,139,817 | ||||||
| Accrued expenses |
1,541,587 | 1,269,395 | ||||||
| Reserve for unresolved claims |
8,798,070 | — | ||||||
| Due to related party |
24,993 | — | ||||||
| Unearned revenues |
283,024 | 263,778 | ||||||
| Note payable |
149,528 | — | ||||||
| Notes payable, related parties |
— | 2,041,005 | ||||||
| Customer deposits |
107,910 | 134,613 | ||||||
| Derivative liabilities |
8,079,209 | 1,844,200 | ||||||
| Total current liabilities |
44,337,041 | 6,692,808 | ||||||
| Long-term debt, net of current maturities |
||||||||
| Convertible promissory notes |
11,323,177 | — | ||||||
| Convertible promissory notes, related party |
182,043 | — | ||||||
| Other long-term debt |
45,884,429 | |||||||
| Liabilities subject to compromise |
|
—
|
|
|
143,570,128
|
| ||
| Total liabilities |
101,726,690 | 150,262,936 | ||||||
| Commitments and contingencies |
— | — | ||||||
| Series A convertible redeemable preferred stock, $1.00 par value; none as of December 31, 2010, and 8,950 shares authorized; 7,529 shares issued and outstanding at September 30, 2010 |
— | 7,528,640 | ||||||
| Stockholders’ deficit: |
||||||||
| Common stock, $0.001 par value; 300,000,000 shares authorized; 69,768,010 and 58,243,115 shares issued and 68,219,874 and 58,048,208 shares outstanding at December 31, 2010, and September 30, 2010, respectively |
69,768 | 58,048 | ||||||
| Treasury stock, 1,548,136 and 194,907 shares, December 31, 2010 and September 30, 2010, respectively |
(1,496,417) | (170,057) | ||||||
| Additional paid-in capital |
275,142,373 | 203,828,364 | ||||||
| Accumulated deficit |
(327,297,713) | (325,882,720) | ||||||
| Total stockholders’ deficit attributable to Accentia Biopharmaceuticals, Inc. |
(53,581,989) | (122,166,365) | ||||||
| Non-controlling interests |
|
(38,956,859)
|
|
|
(30,614,902)
|
| ||
| Total stockholders’ deficit |
(92,538,848) | (152,781,267) | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIT |
$ | 9,187,842 | $ | 5,010,309 | ||||
The accompanying footnotes are an integral part of these condensed consolidated financial statements.
F-63
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited)
| For the Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| Net Sales: |
||||||||
| Products |
$ | 307,825 | $ | 919,018 | ||||
| Services |
1,434,328 | 2,766,956 | ||||||
| Grant revenue |
319,667 | — | ||||||
| Total net sales |
2,061,820 | 3,685,974 | ||||||
| Cost of sales: |
||||||||
| Products |
245,488 | 412,693 | ||||||
| Services |
1,062,569 | 2,016,517 | ||||||
| Grants |
72,011 | — | ||||||
| Total cost of sales (exclusive of amortization of acquired product rights) |
1,380,068 | 2,429,210 | ||||||
| Gross margin |
681,752 | 1,256,764 | ||||||
| Operating expenses: |
||||||||
| Research and development |
293,799 | 290,704 | ||||||
| Sales and marketing |
25,983 | 28,625 | ||||||
| General and administrative |
12,385,706 | 1,543,794 | ||||||
| Total operating expenses |
12,705,488 | 1,863,123 | ||||||
| Operating loss |
(12,023,736) | (606,359) | ||||||
| Other (expense) income: |
||||||||
| Interest expense, including change in fair market value of convertible debentures |
(2,391,771) | (3,367,235) | ||||||
| Derivative loss |
(4,073,380) | (28,814) | ||||||
| Other income |
10,044 | 40,099 | ||||||
| Loss before reorganization items, non-controlling interest from variable interest entities, discontinued operations and income taxes |
(18,478,843) | (3,962,309) | ||||||
| Reorganization items: |
||||||||
| Gain on reorganization |
11,375,530 | — | ||||||
| Professional fees |
(357,059) | (282,000) | ||||||
| Gain (loss) for provision for indemnity agreements |
— | 687,272 | ||||||
| 11,018,471 | 405,272 | |||||||
| Loss before non-controlling interest |
(7,460,372) | (3,557,037) | ||||||
| Non-controlling interest from variable interest entities and subsidiary |
— | (26,389) | ||||||
| Loss before income taxes |
(7,460,372) | (3,583,426) | ||||||
| Income taxes |
— | (4,556) | ||||||
| Net income (loss) |
(7,460,372) | (3,587,982) | ||||||
| Preferred stock dividend |
$ | — | $ | (699,759) | ||||
(Continued)
F-64
Table of Contents
| For the Three Months Ended December 31, |
||||||||
| 2010 | 2009 | |||||||
| Loss attributable to common shareholders |
$ | (7,460,372) | $ | (4,287,741) | ||||
| Weighted average shares outstanding, basic and diluted |
62,976,851 | 58,048,208 | ||||||
| Per share amounts, basic and diluted: |
||||||||
| Loss attributable to common stockholders per common share |
$ | (0.12) | $ | (0.08) | ||||
The accompanying footnotes are an integral part of these condensed consolidated financial statements.
F-65
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENT OF STOCKHOLDERS’ DEFICIT
(Unaudited)
| Common Stock | ||||||||||||||||||||||||||||
| Shares | Amount | Additional Paid In Capital |
Treasury Stock |
Accumulated Deficit |
Non-Controlling Interests |
Total | ||||||||||||||||||||||
| Balances, October 1, 2010 |
58,048,208 | $ | 58,048 | $ | 203,828,364 | $ | (170,057) | $ | (325,882,720) | $ | (30,614,902) | $ | (152,781,267) | |||||||||||||||
| Reclassification upon dissolution of variable interest entities |
— | — | — | — | 3,565,534 | (3,565,534) | — | |||||||||||||||||||||
| Share-based compensation |
1,500,000 | 1,500 | 10,847,092 | — | — | — | 10,848,592 | |||||||||||||||||||||
| Reclassification of derivative liability to equity |
— | — | 35,093,356 | — | — | — | 35,093,356 | |||||||||||||||||||||
| Accentia shares issued on effective date upon the conversion of debt |
10,072,644 | 10,073 | 12,563,585 | — | — | — | 12,573,658 | |||||||||||||||||||||
| Accentia shares issued upon the conversion of promissory notes |
90,908 | 91 | 75,817 | — | — | — | 75,908 | |||||||||||||||||||||
| Accentia shares issued for services |
56,250 | 56 | 40,444 | — | — | — | 40,500 | |||||||||||||||||||||
| Treasury shares received on effective date |
— | — | — | (1,326,360) | 8 | — | (1,326,352) | |||||||||||||||||||||
| Biovest warrants issued |
— | — | 1,247,582 | — | — | — | 1,247,582 | |||||||||||||||||||||
| Biovest shares issued on effective date for conversion of debt |
— | — | 6,631,156 | — | — | — | 6,631,156 | |||||||||||||||||||||
| Reclassification of Biovest beneficial conversion feature to equity |
— | — | 2,138,789 | — | — | — | 2,138,789 | |||||||||||||||||||||
| Biovest shares issued upon conversion of Empery debenture |
— | — | 60,195 | — | — | — | 60,195 | |||||||||||||||||||||
| Biovest shares issued for interest |
— | — | 313,407 | — | — | — | 313,407 | |||||||||||||||||||||
| Biovest shares issued upon the conversion of employee stock options |
— | — | 6,000 | — | — | — | 6,000 | |||||||||||||||||||||
| Adjustment to non-controlling interests for change in ownership percentage of majority-owned subsidiary |
— | — | 2,296,586 | — | — | (2,296,586) | — | |||||||||||||||||||||
| Net loss for the period |
— | — | — | — | (4,980,535) | (2,479,837) | (7,460,372) | |||||||||||||||||||||
| Balances, December 31, 2010 |
69,798,010 | $ | 69,768 | $ | 275,142,373 | $ | (1,496,417) | $ | (327,297,713) | $ | (38,956,859) | $ | (92,538,848) | |||||||||||||||
The accompanying footnotes are an integral part of these condensed consolidated financial statements.
F-66
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited)
| For the Three Months Ending December 31, |
||||||||
| 2010 | 2009 | |||||||
| Cash flows from operating activities: |
||||||||
| Net loss before non-controlling interest in variable interest entities and subsidiary |
$ | (7,460,372) | $ | (3,561,593) | ||||
| Adjustments to reconcile net loss to net cash flows from operating activities: |
||||||||
| Change in fair market value adjustment of convertible debentures |
— | (432,526) | ||||||
| Depreciation |
19,656 | 42,639 | ||||||
| Amortization |
114,698 | 44,012 | ||||||
| Share-based compensation |
10,848,592 | 379,653 | ||||||
| Accretion of capitalized finance costs |
764,484 | 341,724 | ||||||
| Accretion of debt discounts |
406,873 | 1,958,302 | ||||||
| Accretion of royalty liability |
— | 148,517 | ||||||
| Derivative loss (gain) |
4,073,380 | 28,814 | ||||||
| Gain on the conversion of debt |
(24,902) | — | ||||||
| Issuance of common stock shares for interest expense |
313,407 | — | ||||||
| Issuance of common stock warrants for finance costs |
1,247,582 | — | ||||||
| Issuance of common stock shares for services |
40,500 | — | ||||||
| Increase (decrease) in cash resulting from changes in: |
||||||||
| Accounts receivable |
147,664 | (21,928) | ||||||
| Inventories |
(12,261) | 11,201 | ||||||
| Unbilled receivables |
(93,870) | 566,814 | ||||||
| Prepaid expenses and other current assets |
(815,132) | (38,634) | ||||||
| Other assets |
179,207 | 16,145 | ||||||
| Accounts payable |
521,959 | 318,517 | ||||||
| Accrued expenses |
135,005 | 1,546,520 | ||||||
| Unearned revenues |
19,246 | 54,939 | ||||||
| Customer deposits |
(26,703) | (58,000) | ||||||
| Net cash flows from operating activities before reorganization items |
10,399,013 | 1,345,116 | ||||||
| Reorganization items: |
||||||||
| Gain on reorganization plan |
(11,375,530) | — | ||||||
| Decrease in accrued professional fees |
(325,333) | 79,543 | ||||||
| Decrease in provision for indemnity agreement |
— | (687,000) | ||||||
| Net cash flows from reorganization items |
(11,700,863) | (607,457) | ||||||
| Net cash flows from operating activities |
(1,301,850) | 737,659 | ||||||
| Cash flows from investing activities: |
||||||||
| Acquisition of property, plant and equipment |
$ | (98,403) | — | |||||
| Net cash flows from investing activities |
(98,403) | — | ||||||
(Continued)
F-67
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| For the Three Months Ending December 31, |
||||||||
| 2010 | 2009 | |||||||
| Cash flows from financing activities: |
||||||||
| Proceeds from long-term convertible notes |
$ | 7,000,000 | $ | — | ||||
| Proceeds from the exercise of stock options |
6,000 | — | ||||||
| Payments on notes payable and long-term debt |
(1,466,703) | — | ||||||
| Payment of deferred financing costs |
(1,177,634) | (40,000) | ||||||
| Proceeds from notes payable, related party |
250,000 | 140,000 | ||||||
| Payments made from related party, net |
(5,466) | (6,762) | ||||||
| Net cash flows from financing activities |
4,606,197 | 93,238 | ||||||
| Net change in cash and cash equivalents |
3,205,944 | 830,897 | ||||||
| Cash and cash equivalents at beginning of period |
558,452 | 325,350 | ||||||
| Cash and cash equivalents at end of period |
$ | 3,764,396 | $ | 1,156,247 | ||||
| Supplemental cash flow information: |
||||||||
| Cash paid for: |
||||||||
| Interest |
$ | — | $ | 60,000 | ||||
| Income taxes |
— | — | ||||||
| Supplemental disclosure of non-cash financing activity: |
||||||||
| Reclassification of derivative to equity |
$ | 35,093,356 | $ | 2,906,786 | ||||
| Accentia common stock issued on effective date upon the conversion of debt |
12,573,463 | — | ||||||
| Accentia common stock issued for services |
40,500 | — | ||||||
| Accentia common stock issued upon the conversion of promissory notes |
75,908 | — | ||||||
| Biovest common stock issued on effective date upon the conversion of debt |
6,631,156 | — | ||||||
| Reclassification of Biovest beneficial conversion feature to equity |
2,138,789 | — | ||||||
| Biovest shares for interest |
313,407 | — | ||||||
| Biovest shares issued upon the conversion of Empery debenture |
60,195 | — | ||||||
The accompanying footnotes are an integral part of these condensed consolidated financial statements.
F-68
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
1. Description of the company:
Headquartered in Tampa, Florida, Accentia Biopharmaceuticals, Inc. (OTCQB: “ABPI”) is a biotechnology company that is developing Revimmune™ as a comprehensive system of care for the treatment of autoimmune diseases. Additionally, through the Company’s, majority-owned subsidiary, Biovest International, Inc., the Company is developing BiovaxID® as a therapeutic cancer vaccine for treatment of follicular non-Hodgkin’s lymphoma (“FL”) and mantle cell lymphoma (“MCL”). Through the Company’s wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), they conduct a health economics research and consulting business which Analytica markets to the pharmaceutical and biotechnology industries, using Analytica’s operating cash flow to support the Company’s corporate administration and product development activities.
Revimmune™ is being developed as a treatment for various autoimmune diseases. The approximately 80 known autoimmune diseases generally arise from an overactive immune response against substances and/or tissue normally present in the body. As a system of care, Revimmune consists of administering high, pulsed doses of an FDA-approved drug, cyclophosphamide, over a four-day interval as part of an integrated risk management system including a panel of preventive tests, monitoring and medications which are intended to minimize risks while seeking to maximize the clinical benefit.
Through a collaboration with the National Cancer Institute (“NCI”), the Company’s majority-owned subsidiary, Biovest International, Inc. (OTCQB: “BVTI”) (“Biovest”) has developed a patient-specific cancer vaccine, BiovaxID®, which has demonstrated statistically significant Phase 3 clinical benefit by prolonging disease-free survival in FL patients treated with BiovaxID.
Additionally, through the Company’s wholly-owned subsidiary, Analytica International, Inc. (“Analytica”), based in New York City, the Company conducts a global research and strategy consulting business that provides professional services to the pharmaceutical and biotechnology industries. Since 1997, Analytica has expertly directed research studies and projects, including traditional health economic modeling projects, database studies, structured reviews, health technology assessments, reimbursement analyses, and value dossiers.
2. Chapter 11 Reorganization:
On November 10, 2008, the Company and its wholly-owned subsidiaries, Analytica, TEAMM Pharmaceuticals, Inc. d/b/a Accentia Pharmaceuticals (“TEAMM” or “Accentia Pharmaceuticals”), AccentRx, Inc. (“AccentRx”), and Accentia Specialty Pharmacy (“ASP”) (collectively, the “Debtors”) filed voluntary petitions for reorganization under Chapter 11 in the Bankruptcy Court. During the pendency of the Chapter 11 proceedings, the Company operated its business as a debtor-in-possession in accordance with the provisions of Chapter 11, and was subject to the jurisdiction of the Bankruptcy Court. On August 16, 2010, the Company filed its First Amended Joint Plan of Reorganization, and, on October 25, 2010, the Company filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a Confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. The Company emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
3. Significant accounting policies and consolidation policy:
Basis of presentation
The accompanying condensed consolidated financial statements have been derived from unaudited interim financial information prepared in accordance with the rules and regulations of the Securities and Exchange Commission for quarterly financial statements. Certain information and footnote disclosures normally included in financial statements prepared in accordance with accounting principles generally accepted in the United States of America have been condensed or omitted pursuant to those rules and regulation, although the Company believes that the disclosures made are adequate so that the information presented is not misleading. The condensed consolidated financial statements of the Company, in the opinion of management, include all normal and recurring adjustments necessary for a fair presentation of results as of the dates and for the periods covered by the condensed consolidated financial statements.
Operating results for the three months ended December 31, 2010 are not necessarily indicative of the results that may be expected for the entire fiscal year. For further information, refer to the consolidated financial statements and footnotes thereto included in the Company’s Annual Report on Form 10-K for the fiscal year ended September 30, 2010.
F-69
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Consolidation policy
Principles of consolidation
The Company consolidates all entities controlled by ownership of a majority interest and, effective February 27, 2007, has consolidated a variable interest entity of which the Company is the primary beneficiary. The consolidated financial statements include Accentia Biopharmaceuticals, Inc. and its wholly-owned subsidiaries, Analytica International, Inc. (“Analytica”), TEAMM Pharmaceuticals, Inc. d/b/a Accentia Pharmaceuticals (“TEAMM” or “Accentia Pharmaceuticals”), AccentRx, Inc. (“AccentRx”), and Accentia Specialty Pharmacy (“ASP”); its majority owned subsidiary, Biovest (and its consolidated entities), and Revimmune, LLC, an entity in which the Company has a controlling financial interest and has been determined to be the primary beneficiary. All significant intercompany balances and transactions have been eliminated in consolidation.
Voluntary Petition for Bankruptcy:
ASC Topic 852-Reorganizations, which is applicable to companies in Chapter 11, generally does not change the manner in which financial statements are prepared. However, it does require that the financial statements for periods subsequent to the filing of the Chapter 11 petition distinguish transactions and events that are directly associated with the reorganization from the ongoing operations of the business. Revenues, expenses, realized gains and losses, and provisions for losses that can be directly associated with the reorganization and restructuring of the business must be reported separately as reorganization items in the statements of operations beginning in the quarter ending December 31, 2008. The balance sheet must distinguish prepetition liabilities subject to compromise from both those prepetition liabilities that are not subject to compromise and from post-petition liabilities. Liabilities that may be affected by a plan of reorganization must be reported at the amounts expected to be allowed, even if they may be settled for lesser amounts. In addition, cash provided by reorganization items must be disclosed separately in the statement of cash flows. The Company became subject to ASC Topic 852 on November 10, 2008, and emerged from Chapter 11 protection on November 17, 2010. The Company has segregated those items as outlined above for all reporting periods between such dates.
Financial instruments:
Financial instruments, as defined in ASC Topic 825, consist of cash, evidence of ownership in an entity and contracts that both (i) impose on one entity a contractual obligation to deliver cash or another financial instrument to a second entity, or to exchange other financial instruments on potentially unfavorable terms with the second entity, and (ii) conveys to that second entity a contractual right (a) to receive cash or another financial instrument from the first entity or (b) to exchange other financial instruments on potentially favorable terms with the first entity. Accordingly, the Company’s financial instruments consist of cash, accounts receivable, accounts payable, accrued liabilities, notes payable, long-term debt, royalty liabilities, and derivative financial instruments.
The Company carries cash, accounts receivable, accounts payable, and accrued liabilities at historical costs of which their respective the estimated fair values of these assets and liabilities approximate carrying values due to their current nature. The Company also carries notes payable and long-term debt at historical cost less discounts from warrants issued as loan financing costs; however, fair values of these debt instruments are estimated for disclosure purposes based upon the present value of the estimated cash flows at market interest rates applicable to similar instruments.
Fair Value of Financial Assets and Liabilities:
The Company measures the fair value of financial assets and liabilities in accordance with GAAP which defines fair value, establishes a framework for measuring fair value, and expands disclosures about fair value measurements.
GAAP defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. GAAP also establishes a fair value hierarchy, which requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. GAAP describes three levels of inputs that may be used to measure fair value:
Level 1 – quoted prices in active markets for identical assets or liabilities
Level 2 – quoted prices for similar assets and liabilities in active markets or inputs that are observable
Level 3 – inputs that are unobservable (for example cash flow modeling inputs based on assumptions)
F-70
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
The Company generally does not use derivative financial instruments to hedge exposures to cash-flow, market or foreign-currency risks. However, the Company and its consolidated subsidiaries have entered into certain other financial instruments and contracts, such as debt financing arrangements and freestanding warrants with features that are either (i) not afforded equity classification, (ii) embody risks not clearly and closely related to host contracts, or (iii) may be net-cash settled by the counterparty. These instruments are required to be carried as derivative liabilities, at fair value.
The Company estimates fair values of all derivative instruments, such as free-standing warrants, and embedded conversion features utilizing Level 2 inputs. The Company generally uses the Black-Scholes-Merton option valuation technique because it embodies all of the requisite assumptions (including trading volatility, estimated terms and risk free rates) necessary to fair value these instruments. Estimating fair values of derivative financial instruments requires the development of significant and subjective estimates that may, and are likely to, change over the duration of the instrument with related changes in internal and external market factors. In addition, option-based techniques are highly volatile and sensitive to changes in the Company’s trading market price and the trading market price of various peer companies, which have historically had high volatility. Since derivative financial instruments are initially and subsequently carried at fair value, the Company’s income will reflect the volatility in these estimate and assumption changes.
The Company reports its derivative liabilities at fair value on the accompanying consolidated balance sheets as of December 31, 2010 and September 30, 2010.
Grant revenue:
Grant revenue is the result of the Company and Biovest being awarded the Qualifying Therapeutic Discovery Program Grant from the federal government during 2010. Grant revenue is recognized up to 50% of the reimbursable expenses incurred during 2010 and 2009 for Biovest and 2011 and 2009 for the Company.
Recent accounting pronouncements:
In December 2007, the FASB issued new guidance requiring non-controlling interests to be treated as a separate component of equity, not as a liability or other item outside of permanent equity. This new guidance is applicable to the accounting for non-controlling interests and transactions with non-controlling interest holders in consolidated financial statements and is effective for fiscal years beginning on or after December 15, 2008. Accordingly, we have adopted this new pronouncement as of October 1, 2009 resulting in a change in stockholders’ equity on our opening fiscal year 2010 consolidated statement of financial condition; however, the adoption of this pronouncement did not have a material effect on our results from operations or cash flows.
In June 2008, the FASB issued new guidance for determining whether an equity-linked financial instrument (or embedded feature) is indexed to an entity’s own stock. The Company adopted this new guidance effective October 1, 2009. Certain of the Company’s outstanding warrants and convertible debt contain features which fall under the scope of this guidance resulting in a decrease of $2.2 million and $0.8 million to the October 1, 2009 balances of additional paid-in capital and accumulated deficit respectively.
F-71
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
In June 2009, the FASB issued new guidance amending the existing pronouncement related to the consolidation of variable interest entities. This new guidance requires the reporting entities to evaluate former QSPE’s for consolidation, changes the approach to determine a variable interest entity’s primary beneficiary from a quantitative assessment to a qualitative assessment designed to identify a controlling financial interest, and increases the frequency of required assessments to determine whether the Company is the primary beneficiary of any variable interest entities which it is a party to. This new guidance is not effective for the Company until October 1, 2010 and earlier adoption is prohibited. This new guidance became effective for the Company on October 1, 2010 and did not have a material impact on our consolidated financial statements.
In December 2010, the FASB issued Accounting Standards Update (“ASU”) 2010-27 “Fees Paid to the Federal Government by Pharmaceutical Manufacturers” amending Accounting Standards Codification (“ASC”) 720 “Other Expenses” to address questions concerning how pharmaceutical manufacturers should recognize the annual fees imposed by the Patient Protection and Affordable Care Act for each calendar year beginning January 1, 2011. The ASU requires that the liability related to the annual fee be estimated and recorded in full upon the first qualifying sale with a corresponding deferred cost that is amortized to expense over the calendar year that it is payable. The amendment is effective commencing with the quarter ended March 31, 2011 and is not expected to have a significant impact on the Company’s financial statements.
4. Liquidity and management’s plans:
During the three months ended December 31, 2010, the Company had a net loss of $7.5 million. On December 31, 2010, the Company had an accumulated deficit of approximately $327.3 million and a working capital deficit of approximately $38 million. Cash and cash equivalents at December 31, 2010, was $3.8 million of which $3.5 million was attributable to Biovest. The Company’s independent registered public accounting firm’s report included a “going concern” uncertainty on the financial statements for the year ended September 30, 2010, citing significant losses and working capital deficits at that date, which raised substantial doubt about the Company’s ability to continue as a going concern.
Regulatory strategy and commercialization expenditures:
Notwithstanding the prior reports of High-Dose Pulsed Cyclophosphamide as a potential therapy for certain autoimmune diseases. Cyclophosphamide is not currently FDA-approved for the treatment of any autoimmune diseases. These prior studies of High-Dose Pulsed Cyclophosphamide in the United States have been conducted at a limited number of large academic research hospitals and have featured non-uniform inclusion criteria and/or administration schedules. It is generally recognized that there may be significant potential risks of infection or other side effects when High-Dose Pulsed Cyclophosphamide is not administered by highly-qualified personnel in a controlled and regulated clinical setting. While the previous studies are important to the Company’s Revimmune™ development plan, they are not considered sufficient to support regulatory approval. At the core of the Company’s Revimmune development plan is a recognition that controlled and randomized clinical trials will be necessary to demonstrate the efficacy of High-Dose Pulsed Cyclophosphamide to the satisfaction of the FDA and that safety will be an important regulatory and clinical concern which the Company believes will require an FDA approved formal REMS.
There are approximately 80 recognized autoimmune diseases. The Company previously planned to conduct its first randomized and controlled trial of Revimmune in multiple sclerosis (“MS”) , because of the potentially significant impact of such a study and the number of MS patients previously treated with High Dose Cyclophosphamide. However, due to the Company’s limited capital and resources, the Company has modified its Revimmune development plan so as to initially focus on less prevalent autoimmune diseases where the Company perceives a significant unmet medical need including: diffuse systemic sclerosis and autoimmune hemolytic anemia. Subject to the Company’s capital availability, it plans to undertake clinical trials of Revimmune for the treatment of these two autoimmune diseases in a parallel manner. Diffuse systemic sclerosis and autoimmune hemolytic anemia represent diseases for which the Company believes significant unmet medical needs exist. There is no standard treatment for systemic sclerosis, and while the standard treatment for autoimmune hemolytic anemia is effective for a subset of patients, patients for which this treatment options is not effective, referred to as refractory patients, have very limited options. The Company believes that the significant unmet medical need, the shorter and severe disease course and the smaller prevalence of these autoimmune diseases make them appropriate for the Company’s initial focus. Notwithstanding the initial focus of the Company’s current Revimmune development plan, it continues to believe that new treatment options are needed for MS and the Company remains interested in ultimately conducting studies with Revimmune in MS.
F-72
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
In September 2007, the Company conducted a meeting with the Federal Food and Drug Administration (“FDA”) regarding its proposed design of a clinical trial for Revimmune to treat MS. The Company considered the FDA meeting to have been constructive. However, with the change in the Company’s Revimmune development plan, it is currently preparing for an additional meeting with the FDA to discuss possible studies in diffuse scleroderma and/or autoimmune hemolytoic anemia. Based on FDA input, the Company anticipates filing an Investigational New Drug Application(s) (“IND”) under which it hopes to conduct its planned clinical trials. Further, the Company plans to discuss with the FDA its plans for a REMS to be developed under the Food and Drug Administration Amendments Act of 2007.
Biovest has completed its Phase 3 clinical trial of BiovaxID for the indication of FL. Under Biovest’s current regulatory strategy, the Company is performing in-depth analyses of the available clinical trial data in preparation for submission of the data to the FDA and European Medicines Agency (“EMEA”) to discuss next steps required for approval. Biovest has ceased enrolling new patients in its Phase 3 clinical trial and has discontinued most clinical trial activities which had the effect of decreasing clinical trial expenses compared to those recorded for prior periods. Biovest’s ability to timely access required financing will continue to be essential to support the ongoing commercialization efforts. Biovest’s inability to obtain required funds or any substantial delay in obtaining required funds will have a material adverse effect on the ongoing commercialization efforts.
Additional expected financing activity:
Management intends to attempt to meet its cash requirements through proceeds from its cell culture and instrument manufacturing and development activities as well as its global research and strategy consulting business, the use of cash on hand, trade vendor credit, restructuring of outstanding debt obligations through the Chapter 11 reorganization proceedings, short-term borrowings, debt and equity financings, and strategic transactions such as collaborations and licensing. The Company’s ability to continue present operations, pay its liabilities as they become due, and meet its obligations for product development is dependent upon the Company’s ability to obtain additional funding in the near term. Additional sources of funding have not been established; however, the Company anticipates seeking additional financing potentially from a number of sources, including the sale of equity or debt securities, strategic collaborations and recognized research funding programs. There can be no assurance that the Company will be successful in securing such financing at acceptable terms, if at all. Accordingly, the Company’s ability to continue present operations, pay its existing liabilities as they become due is dependent upon the Company’s ability to obtain significant external funding, which raises substantial doubt about the Company’s ability to continue as a going concern. If adequate funds are not available from the foregoing sources when required, or if the Company determines it to otherwise be in the Company’s best interest, the Company may consider additional strategic financing options, including sales of assets, or the Company may be required to delay, reduce the scope of, or eliminate one or more of its research or development programs or curtail some or all of its commercialization efforts.
5. Inventories:
Inventories consist of the following:
| December 31, 2010 (Unaudited) |
September 30, 2010 |
|||||||
| Finished goods |
$ | 57,509 | $ | 104,155 | ||||
| Work-in-process |
93,961 | — | ||||||
| Raw materials |
277,878 | 312,932 | ||||||
| $ | 429,348 | $ | 417,087 | |||||
F-73
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
6. Intangible assets:
Intangible assets consist of the following:
| December 31, 2010 (Unaudited) |
September 30, 2010 |
Weighted Average Amortization Period |
||||||||||
| Amortizable intangible assets: |
||||||||||||
| Noncompete agreements |
2,104,000 | 2,104,000 | 3.5 years | |||||||||
| Patents |
103,244 | 103,244 | 3.0 years | |||||||||
| Purchased customer relationships |
666,463 | 666,463 | 10.0 years | |||||||||
| Product rights |
28,321 | 28,321 | 18.4 years | |||||||||
| Software |
438,329 | 438,329 | 3.5 years | |||||||||
| Trademarks |
1,285,960 | 1,285,960 | 3.0 years | |||||||||
| 4,626,317 | 4,626,317 | |||||||||||
| Less accumulated amortization |
(3,657,054) | (3,542,355) | ||||||||||
| Total intangible assets |
$ | 969,263 | $ | 1,083,962 | ||||||||
7. Reserve for unresolved claims:
Reserve for unresolved claims consists of disputed amounts in the Company’s Reorganization Plan (Note 2). These claims remain outstanding before the Bankruptcy Court, and the Company anticipates each claim will be resolved during the current fiscal year.
8. Notes payable:
Notes payable of approximately $0.1 million consists primarily of priority and convenience claims to be paid within six months of the Effective Date. All notes payable at September 30, 2010 had been classified as ‘Liabilities subject to compromise’ in the Company’s condensed consolidated balance sheet as of September 30, 2010, as a result of the Company’s Chapter 11 filings on November 10, 2008 (Note 2).
9. Convertible long-term debt:
Convertible promissory notes consist of the following:
| in thousands | ||||||||
| December 31, 2010 | September 30, 2010 | |||||||
| Accentia Class 3 Plan Notes |
3,923 | — | ||||||
| Accentia Class 5 Plan Debentures |
479 | — | ||||||
| Accentia Class 6 Plan Debentures |
6,957 | — | ||||||
| Accentia Class 9 Plan Debentures |
16,721 | — | ||||||
| Accentia Class 13 Plan Notes |
4,291 | — | ||||||
| Biovest Class 8 Option C Promissory Note |
1,773 | — | ||||||
| Biovest Empery Promissory Notes |
46 | — | ||||||
| 34,190 | — | |||||||
| Less current maturities |
(22,867) | — | ||||||
| 11,323 | — | |||||||
F-74
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Accentia Class 3 Plan Note:
Effective as of November 17, 2010, the effective date of the Company’s Plan (the “Effective Date”), the Company issued, a new promissory note to Dennis Ryll, the holder by assignment of the Company’s previously-issued secured note to Southwest Bank, and as payment of the Company’s obligation to Southwest Bank prior to the Effective Date of the Plan (the “Class 3 Plan Note”), in an original principal amount of $4,483,284. The Company is not obligated to pay the Class 3 Plan Note in cash, but rather through quarterly conversions into shares of the Company’s common stock or, subject to certain conditions, by exchanging the quarterly conversion amounts into shares of Biovest common stock owned by the Company. The following are the material terms and conditions of the Class 3 Plan Note:
| • | the Class 3 Plan Note matures on November 17, 2012; |
| • | interest accrues and is payable on the outstanding principal balance of the Class 3 Plan Note from time to time (the “Class 3 Interest”) at a fixed rate of six percent (6%) per annum; |
| • | on November 17, 2010 and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 3 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 3 Plan Note plus the Class 3 Interest as of the Class 3 Automatic Conversion Date (the “Class 3 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the Class 3 Automatic Conversion Price per share of the Company’s common stock. On the Effective Date, the first quarterly installment in the amount of approximately $0.56 million was converted into shares of the Company’s common stock at a conversion rate equal to $1.36 per share, resulting in the issuance of 412,067 shares of the Company’s common stock; |
| • | the Class 3 Plan Note is secured by a lien on 15 million shares of Biovest’s common stock owned by the Company (the “Class 3 Pledged Shares”), subject to the incremental release of a designated portion of such security upon each quarterly payment under the Class 3 Plan Note; and |
| • | if, on any Class 3 Automatic Conversion Date, the Class 3 Automatic Conversion Price is less than $1.00 per share, Dennis Ryll may, at his election, either: |
| • | convert the Class 3 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share of the Company’s common stock; or |
| • | liquidate that number of the Class 3 Pledged Shares which equals the Class 3 Automatic Conversion Amount using a conversion rate for the Class 3 Pledged Shares equal to the average of the trading price of shares of Biovest common stock (as determined in accordance with the Class 3 Plan Note and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 3 Automatic Conversion Date. |
Accentia Class 5 Plan Debenture and Warrants:
Effective as of November 17, 2010, the Company issued, in satisfaction of the 2006 Secured Debentures outstanding prior to the Effective Date, new debentures (the “Class 5 Plan Debentures”) in the original principal amount of $3,109,880. The Company is not obligated to pay the Class 5 Plan Debentures in cash, but rather through the conversion by the holders into shares of the Company’s common stock or, subject to certain conditions, by exchanging the Class 5 Plan Debentures into shares of Biovest common stock owned by the Company during the term of the Class 5 Plan Debentures which mature on May 17, 2012.
The following are the material terms and conditions of the Class 5 Plan Debentures:
| • | the Class 5 Plan Debentures mature on May 17, 2012 (provided, however, in the event that the average of the trading price of Biovest’s common stock (as determined in accordance with the Class 5 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding such maturity date is below $0.75, then the maturity date will automatically be extended for an additional twelve (12) months [May 17, 2013]), and the outstanding principal together with all accrued but unpaid interest is due on such maturity date; |
F-75
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | interest accrues and is payable on the outstanding principal amount under the Class 5 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum; and |
| • | each of the Class 5 Plan Debentures is secured by a lien on certain shares of Biovest’s common stock owned by the Company; |
| • | at the option of a holder of Class 5 Plan Debentures, all or any portion of the then outstanding balance of such holder’s Class 5 Plan Debentures may be converted into shares of the Company’s common stock or exchanged for shares of Biovest’s common stock at the applicable conversion or exchange rate set forth in such holder’s Class 5 Plan Debenture; |
| • | commencing on August 15, 2011, if the trading price of the Company’s common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least 150% of the fixed conversion price for a holder of Class 5 Plan Debentures for any ten (10) consecutive trading days (in the case of a conversion into the Company’s common stock), or the trading price of Biovest’s common stock (determined in accordance with the Class 5 Plan Debentures and the Plan) is at least $1.25 for any ten (10) consecutive trading days (in the case of an exchange into Biovest’s common stock), the Company, at its option, may (a) convert the then outstanding balance of all of the Class 5 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to the fixed conversion price for each holder of Class 5 Plan Debentures, or (b) exchange the then outstanding balance of all of the Class 5 Plan Debentures into shares of Biovest’s common stock owned by the Company at a rate equal to $0.75 per share of Biovest’s common stock (with certain exceptions set forth in the Class 5 Plan Debentures and the Plan); and |
| • | in the event a holder of Class 5 Plan Debentures elects to receive shares of Biovest’s common stock, then such holder will be subject to certain restrictions set forth in the Class 5 Plan Debentures and the Plan regarding Biovest’s common stock from November 17, 2010 through February 15, 2011. |
Effective as of November 17, 2010, the Company executed and delivered the following warrants (the “Class 5 Plan Warrants”) to purchase up to 2,508,960 shares of the Company’s common stock or up to 14.4 million shares of Biovest’s common stock owned by the Company. The Class 5 Plan Warrants (a) have an exercise price of $1.50 per share with a expiration date of November 17, 2013, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 5 Plan Warrants and the Plan.
Accentia Class 6 Plan Debentures and Warrants:
Effective as of November 17, 2010, the Company issued, in satisfaction of the 2008 Secured Debentures outstanding prior to the Effective Date, new debentures (the “Class 6 Plan Debentures”) in the original principal amount of $9,730,459. The Class 6 Plan Debentures mature on November 17, 2013, and the outstanding principal together with all accrued but unpaid interest is due in cash on such date.
The following are the material terms and conditions of the Class 6 Plan Debentures:
| • | interest accrues and is payable on the outstanding principal under the Class 6 Plan Debentures at a fixed rate of eight and one-half percent (8.50%) per annum and each of the Class 6 Plan Debentures is secured by a lien on certain assets of the Company; |
| • | at the option of a holder of Class 6 Plan Debentures, such holder may elect to convert all of the then outstanding balance of its Class 6 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to $1.10 per share of the Company’s common stock; and |
| • | commencing on May 15, 2011, if the trading price of the Company’s common stock (as determined in accordance with the Class 6 Plan Debentures and the Plan) is at least 150% of $1.10 per share for any ten (10) consecutive trading days, the Company, at its option, may convert the then outstanding balance of all of the Class 6 Plan Debentures into shares of the Company’s common stock at a conversion rate equal to $1.10 per share of the Company’s common stock. |
F-76
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
On November 17, 2010, the Company executed and delivered the following warrants (the “Class 6 Plan Warrants”) to purchase up to 2,979,496 shares of the Company’s common stock. The Class 6 Plan Warrants (a) have an exercise price of $1.50 per share with a expiration date of November 17, 2013, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 6 Plan Warrants and the Plan.
On November 17, 2010, $2,672,548 in Class 6 Plan Debentures were converted into the Company’s common stock at a conversion price equal to $1.10 per share, resulting in the issuance of 2,429,588 shares of the Company’s common stock.
Accentia Class 9 Plan Debentures and Warrants:
Effective as of November 17, 2010, , the Company issued, in satisfaction of the 2007 Debentures outstanding prior to the Effective Date, new debentures (the “Class 9 Plan Debentures”) in the original principal amount of $19,109,554. The Company is not obligated to pay the Class 9 Plan Debentures in cash, but rather through the conversion by the holders into shares of the Company’s common stock. The Class 9 Plan Debentures mature on November 17, 2012 (the “Class 9 Plan Debenture Maturity Date”) and no interest will accrue on the outstanding principal balance of the Class 9 Plan Debentures.
The following are the material terms and conditions of the Class 9 Plan Debentures:
| • | on the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 9 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 9 Automatic Conversion Date (the “Class 9 Automatic Conversion Price”) is at least $1.00 per share, one-eighth (1/8th) of the original principal balance of the Class 9 Plan Debentures (the “Class 9 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the lesser of $1.25 per share or the Class 9 Automatic Conversion Price per share. On the Effective Date, a total of approximately $2.4 million of the original principal amount of the Class 9 Plan Debentures was converted into Company common stock at a conversion price equal to $1.25 per share, resulting in the issuance of 1,910,963 shares of the Company’s common stock; |
| • | if, on any Class 9 Automatic Conversion Date, the Class 9 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 9 Plan Debentures may elect to convert the Class 9 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 9 Plan Debentures at the Class 9 Plan Debenture Maturity Date will be due and payable in full, at the election of the Company, in either cash or in shares of the Company’s common stock at a conversion rate equal to the average trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 9 Plan Debenture Maturity Date (provided that the average trading price for such period is at least $.50 per share); |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.50 per share for ten (10) consecutive trading days, a holder of the Class 9 Debentures, at its option, may convert any or all of the then outstanding principal balance of its Class 9 Plan Debenture into shares of the Company’s common stock at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on the Effective Date but not to exceed $1.25 per share; and |
| • | if, at any time during the term of the Class 9 Plan Debentures, the trading price of the Company’s common stock (as determined in accordance with the Class 9 Plan Debentures and the Plan) is at least $1.88 per share for thirty (30) consecutive trading days, the Company, at its option, may require the conversion of up to $5.0 million of the then aggregate outstanding principal balance of the Class 9 Plan Debentures at a conversion rate equal to the Class 9 Automatic Conversion Price used for the initial conversion on the Effective Date but not to exceed $1.25 per share. |
On the Effective Date, the Company executed and delivered the following warrants (the “Class 9 Plan Warrants”) to purchase up to 3,154,612 shares of the Company’s common stock. The Class 9 Plan Warrants (a) have an exercise price of $1.50 per share with a expiration date of November 17, 2013, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 9 Plan Warrants and the Plan.
F-77
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Accentia Class 13 Plan Note and Warrants:
Effective as of November 17, 2010, the Company issued, in satisfaction of the Convertible Preferred Stock outstanding prior to the Effective Date of the Plan, new promissory notes (the “Class 13 Plan Notes”) in the original principal amount of $4,903,644. The Class 13 Plan Notes mature on November 17, 2012 (the “Class 13 Plan Notes Maturity Date”), and no interest will accrue on the outstanding principal balance of the Class 13 Plan Notes. The Company has no obligation to pay the Class 13 Plan Notes in cash at maturity, but rather through the conversions by the holders into shares of the Company’s common stock.
The following are the material terms and conditions of the Class 13 Plan Notes:
| • | on the Effective Date and on each of the following seven (7) quarterly anniversaries of the Effective Date (each, a “Class 13 Automatic Conversion Date”), and provided that the average of the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the then applicable Class 13 Automatic Conversion Date (the “Class 13 Automatic Conversion Price”) is at least $1.00 per share, one-eighth ( 1/8th) of the original balance of the Class 13 Plan Notes (the “Class 13 Automatic Conversion Amount”) will be automatically converted into shares of the Company’s common stock at a conversion rate equal to the Class 13 Automatic Conversion Price per share. On the Effective Date, a total of approximately $0.6 million of the outstanding principal amount of the Class 13 Plan Notes was converted into the Company’s common stock at a conversion price equal to $1.36 per share, resulting in the issuance of 450,708 of the Company’s common stock; |
| • | if, on any Class 13 Automatic Conversion Date, the Class 13 Automatic Conversion Price is less than $1.00 per share and therefore the automatic conversion described above does not occur, a holder of Class 13 Plan Notes may elect to convert the Class 13 Automatic Conversion Amount into shares of the Company’s common stock at a conversion rate equal to $1.00 per share; |
| • | any principal amount outstanding under the Class 13 Plan Notes at the Class 13 Plan Note Maturity Date will be due and payable in full, at the election of the Company, in either cash or in shares of the Company’s common stock at a conversion rate equal to the greater of the average of the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) for the ten (10) consecutive trading days ending on the trading day that is immediately preceding the Class 13 Plan Notes Maturity Date or $1.00; |
| • | if, at any time during the term of the Class 13 Plan Notes, the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 125% of $1.25 per share for ten (10) consecutive trading days, a holder of Class 13 Plan Notes, at its option, may convert any or all of the then outstanding principal balance of its Class 13 Plan Notes into shares of the Company’s common stock at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on the Effective Date; and |
| • | if, at any time during the term of the Class 13 Plan Notes, the trading price of the Company’s common stock (as determined in accordance with the Class 13 Plan Notes and the Plan) is at least 150% of $1.25 per share for thirty (30) consecutive trading days, the Company, at its option, may require the conversion of all or any portion of the then aggregate outstanding principal balance of the Class 13 Plan Notes at a conversion rate equal to the Class 13 Automatic Conversion Price used for the initial conversion on the Effective Date. |
Effective as of November 17, 2010, the Company executed and delivered the following warrants (the “Class 13 Plan Warrants”) to purchase up to 1,072,840 shares of the Company’s common stock. The Class 13 Plan Warrants (a) have an exercise price of $1.50 per share with a expiration date of November 17, 2013, (b) can only be exercised for cash (no cashless exercise), and (c) are subject to certain call provisions set forth in the Class 13 Plan Warrants and the Plan.
F-78
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Biovest Class 8 Option C Note:
Effective as of November 17, 2010, the effective date of Biovest’s Plan (the “Effective Date”), Biovest became obligated to certain of its unsecured creditors in the principal amount of approximately $2.0 million (the “Option C Notes”). The holders received an amount equal to 100% of such claimant’s allowed Class 8 unsecured claim (including post-petition interest under the Biovest’s Plan at the rate of three percent (3%) per annum) in a combination of debt and equity resulting in the issuance of a total of $1.8 million in new notes, as well as, 0.2 million shares of Biovest common stock, using an effective conversion rate equal to $1.66 per share. The Option C Notes bear interest at seven percent (7%) and are convertible into shares of Biovest common stock in seven quarterly installments beginning on February 17, 2011 as follows:
| • | Provided that the average of the volume weighted average prices for Biovest’s common stock for the ten consecutive trading days immediately preceding each quarterly conversion date (“Ten Day VWAP”) is at least $1.00 per share, one-eighth (1/8th) of the Option C Notes plus accrued interest will be automatically converted into shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP. |
| • | If the Ten Day VWAP is less than $1.00 per share, the notes will not automatically convert into shares of Biovest common stock, but will instead become payable at maturity (August 17, 2012), unless the Option C Note holder elects to convert one-eighth (1/8th) of the Option C Notes plus accrued interest into shares of Biovest common stock at a conversion rate equal to $1.00 per share. |
| • | Any portion of the Option C Notes and any Option C interest that are outstanding at maturity (August 17, 2012) will be due and payable in full, at the election of Biovest, in either cash or in shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP. |
| • | If, at any time prior to August 17, 2012, the Ten Day VWAP is at least $1.50 per share, an Option C Notes holder, at its option, may convert any or all of the Option C Notes, plus the then accrued and unpaid interest into shares of Biovest common stock at a conversion rate equal to the Ten Day VWAP used for the initial conversion on the Effective Date ($1.66 per share). |
| • | If, at any time prior to August 17, 2012, the volume weighted average price for Biovest common stock is at least $1.88 per share for thirty (30) consecutive trading days, Biovest, at its option, may require the conversion of the then aggregate outstanding balance of the Option C Notes plus the then accrued and unpaid Option C interest at a conversion rate equal to the Ten Day VWAP used for the initial conversion on the Effective Date ($1.66 per share). |
Biovest Exit Financing and Warrant Transaction:
On October 19, 2010, Biovest completed a financing as part of its Plan (the “Exit Financing”). Pursuant to the Exit Financing, Biovest issued an aggregate of $7.0 million in principal amount of Debtor-In-Possession Secured Convertible Notes (the “Initial Notes”) and warrants to purchase shares of Biovest’s common stock (the “Initial Warrants”) to twelve accredited investors (the “Buyers”). Pursuant to the transaction, Biovest issued two separate Initial Warrants to the Buyers, Series A Warrants (the “Initial Series A Warrants”) and Series B Warrants (the “Initial Series B Warrants”).
On the Effective Date: (a) the Initial Notes were exchanged pursuant to the terms of the Plan for new unsecured notes (the “Exchange Notes”) in the aggregate principal amount of $7.04 million, (b) the Initial Series A Warrants were exchanged pursuant to the terms of the Plan for new warrants to purchase a like number of shares of Company common stock (the “Series A Exchange Warrants”), and (c) the Initial Series B Warrants were exchanged pursuant to the terms of the Plan for new warrants to purchase a like number of shares of Company common stock (the “Series B Exchange Warrants”).
The following are the material terms and conditions of the Exchange Notes:
| • | the Exchange Notes mature on November 17, 2012, and all principal and accrued but unpaid interest is due on such date; |
| • | interest accrues and is payable on the outstanding principal amount of the Exchange Notes at a fixed rate of seven percent (7%) per annum (with a fifteen percent (15%) per annum default rate), and is payable monthly in arrears; the first date for an interest payment was December 1, 2010; |
F-79
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | interest payments are payable at Biovest’s election in either cash or subject to certain specified conditions, in shares of Biovest’s common stock; |
| • | Biovest may from time to time, subject to certain conditions, redeem all or any portion of the outstanding principal amount of the Exchange Notes for an amount, in cash, equal to 110% of the sum of the principal amount being redeemed and certain make-whole interest payments; |
| • | the holders of the Exchange Notes may convert all or a portion of the outstanding balance of the Exchange Notes into shares of Biovest’s common stock at a conversion rate of $0.91 per share, subject to anti-dilution adjustments in certain circumstances; and |
| • | in the event that the average of the daily volume weighted average price of Biovest’s common stock is at least 150% of the then-effective conversion price for any ten (10) consecutive trading days, Biovest, at its option, may upon written notice to the holders of the Exchange Notes, convert the then outstanding balance of the Exchange Notes into shares of Biovest’s common stock at the conversion price then in effect under the Exchange Notes. |
The following are the material terms and conditions of the Series A Exchange Warrants:
| • | the Series A Exchange Warrants give the investors the right to purchase 8,733,096 shares of Biovest’s common stock (the “Series A Warrant Shares”) and; |
| • | the Series A Exchange Warrants have an exercise price of $1.20 per share and expire on November 17, 2017; and |
| • | if Biovest issues or sells any options or convertible securities after the issuance of the Series A Exchange Warrants that are convertible into or exchangeable or exercisable for shares of common stock at a price which varies or may vary with the market price of the shares of common stock, including by way of one or more reset(s) to a fixed price, the investors have the right to substitute any of the applicable variable price formulation for the exercise price upon exercise of the warrants held. |
On December 22, 2010, the Series B Exchange Warrants were exercised by a cashless exercise and 1,075,622 shares of Biovest’s common stock were issued to the Buyers.
As of December 31, 2010, a total of $4.2 million in principal on the Exchange Notes had been converted to Biovest common stock, resulting in the issuance to the Buyers of 5.0 million shares of Biovest’s common stock. The remaining principal balance outstanding on the Exchange Notes is $2.8 million as of December 31, 2010.
The Exchange Notes and Warrants contain conversion and adjustment features required to be classified as derivative instruments and recorded at fair value. As a result, the Exchange Notes have been recorded at a discount which will be amortized to interest expense over two years.
Future maturities of convertible debt are as follows:
| Years ending December 31, |
||||
| 2011 |
15,430 | |||
| 2012 |
17,584 | |||
| 2013 |
6,958 | |||
| Total maturities |
39,972 | |||
| Less unamortized discount: |
(5,782) | |||
| 34,190 | ||||
F-80
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
10. Other long-term debt
Other long-term debt consists of the following:
| in thousands | ||||
|
December 31, 2010 |
September 30, 2010 | |||
| Accentia Class 2 Laurus/Valens Term Note |
8,800 | — | ||
| Accentia Class 4 Promissory Note |
4,343 | — | ||
| Accentia Class 10 Plan Distributions |
2,402 | — | ||
| Biovest Laurus/Valens Term Notes |
27,627 | — | ||
| Biovest Class 8 Plan Distributions |
2,712 | — | ||
| 45,884 | — | |||
| Less current maturities |
- | — | ||
| 45,884 | — | |||
Accentia Class 2 Laurus/Valens Term Note:
Effective as of November 17, 2010, the effective date of the Company’s Plan (the “Effective Date”), the Company issued term notes and security agreements to Laurus Master Fund, Ltd. (in liquidation), PSource Structured Debt Limited, Valens Offshore SPV I, Ltd., Valens Offshore SPV II, Corp., Valens U.S. SPV I, LLC, and LV Administrative Services, Inc. (“Laurus/Valens”), in the original principal amount of $8.8 million (the “Laurus/Valens Term Notes”) in satisfaction of allowed claims prior to the Effective Date. The following are the material terms and conditions of the Laurus/Valens Term Notes:
| • | the Laurus/Valens Term Notes mature on November 17, 2012 and may be prepaid at any time without penalty; |
| • | interest accrues on the Laurus/Valens Term Notes at the rate of eight and one-half percent (8.5%) per annum (with a twelve and one-half percent (12.5%) per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
| • | the Company is required to make mandatory prepayments under the Laurus/Valens Term Notes as follows: |
| • | on May 17, 2012, a payment of principal, in cash, in the amount of $4.4 million, less the amount of any prior optional prepayments of principal by the Company and the amount of any other mandatory prepayments of principal under the Laurus/Valens Term Notes; and |
| • | a prepayment equal to thirty percent (30%) of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by the Company) of certain capital raising transactions (with certain exclusions); |
| • | the Laurus/Valens Term Notes are secured by: |
| • | a first lien on all of the assets of the Company, junior only to the liens granted under the Plan to holders of the Class 6 Plan Debentures, in the original aggregate principal amount of $8,906,098 and certain permitted liens; |
| • | a pledge by the Company to Laurus/Valens of (a) all of the Company’s equity interests in Analytica, and (b) 20,115,818 shares of common stock of Biovest owned by the Company; |
| • | all of the assets of Analytica, which secure a guaranty of Analytica as to the entire indebtedness under the Laurus/Valens Term Notes; and |
F-81
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | with the prior written consent of Laurus/Valens, the Company may convert all or any portion of the outstanding principal and accrued interest under the Laurus/Valens Term Notes into a number of shares of the Company’s common stock (the “Laurus/Valens Conversion Shares”) equal to (a) the aggregate portion of the principal and accrued but unpaid interest outstanding under the Laurus/Valens Term Note being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported for the Company’s common stock for the ten (10) trading days immediately preceding the date of the notice of conversion. |
On the November 17, 2010, 2,236,848 shares of the Company’s common stock were issued to Laurus/Valens for payment of Laurus/Valens Class 5 and Class 13 claims aggregating approximately $6.0 million at a conversion rate equal to $2.67 per share.
Accentia Class 4 Promissory Note:
Effective as of November 17, 2010, the Company issued, a new promissory note in the original principal amount of $4,342,771 to McKesson Corporation (“McKesson”) (the “Class 4 Plan Note”) in satisfaction of McKesson’s approved pre-Effective Date secured claims. The Class 4 Plan Note is payable in cash in one installment on March 17, 2014 (unless earlier accelerated), and the outstanding principal together with all accrued but unpaid interest is due on such date. The following are the material terms and conditions of the Class 4 Plan Note:
| • | interest accrues and is payable on the outstanding principal amount under the Class 4 Plan Note at a fixed rate of five percent (5%) per annum (with a ten percent (10%) per annum default rate); |
| • | the Company may prepay all or any portion of the Class 4 Plan Note, without penalty, at any time; and |
| • | the Class 4 Plan Note is secured by a lien on 6,102,408 shares of Biovest’s common stock owned by the Company. |
Accentia Class 10 Plan Distributions:
Effective as of November 17, 2010, the Company became obligated required to pay approximately $2.4 million in cash on March 17, 2014, to unsecured creditors holding Class 10 claims under the Plan (the “Class 10 Plan Distributions”).
The Class 10 Plan Distributions mature on March 17, 2014, and the outstanding principal together with all accrued but unpaid interest is due on such date. Interest accrues and is payable on the outstanding principal amount under the Class 10 Plan Distributions at a fixed rate of five percent (5%) per annum.
Unsecured creditors holding an aggregate total of $3,287,695 in Class 10 claims elected to convert those Class 10 claims into shares of the Company’s common stock valued at the average market price for the Company’s common stock over the ten trading days preceding the Effective Date. On the Effective Date, the Company issued 2,417,431 shares of the Company’s common stock to these Class 10 unsecured creditors at a conversion price equal to $1.36 per share.
Biovest Laurus/Valens Term Notes:
Under the Biovest Plan, Biovest issued two new notes (the “Laurus/Valens Term A Notes” and “Laurus/Valens Term B Notes”) in the aggregate original principal amount of $29.06 million to Laurus/Valens in compromise, and satisfaction of secured claims prior to the Effective Date.
The following are the material terms and conditions of the Laurus/Valens Term A Notes:
| • | the original principal amount of the Laurus/Valens Term A Notes was $24.9 million; |
| • | the Laurus/Valens Term A Notes are due in one installment of principal and interest due at maturity on November 17, 2012; |
| • | interest accrues at the rate of eight percent per annum (with a twelve percent per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
F-82
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | Biovest may prepay the Laurus/Valens Term A Notes, without penalty, at any time; |
| • | Biovest is required to make mandatory prepayments under the Laurus/Valens Term A Notes as follows: |
| • | a prepayment equal to thirty percent of the net proceeds (i.e., the gross proceeds received less any investment banking or similar fees and commissions and legal costs and expenses incurred by Biovest) of certain capital raising transactions (with certain exclusions), but only up to the then outstanding principal and accrued interest under the Laurus/Valens Term A Notes; |
| • | from any intercompany funding by the Company to Biovest (with certain exceptions and conditions); and |
| • | a prepayment equal to fifty percent (50%) of the positive net cash flow of Biovest for each fiscal quarter after the Effective Date, less the amount of certain capital expenditures on certain biopharmaceutical products of Biovest made during such fiscal quarter or during any prior fiscal quarter ending after November 17, 2010. |
On November 18, 2010, Biovest prepaid the Laurus/Valens Term A Notes in an amount equal to $1.4 million from the proceeds received in the Biovest Exit Financing.
The following are the material terms and conditions of the Laurus/Valens Term B Notes:
| • | The original principal amount of the Laurus/Valens Term B Notes was $4.16 million; |
| • | the Laurus/Valens Term B Notes mature on November 17, 2013; |
| • | interest accrues at the rate of eight percent per annum (with a twelve percent per annum default rate), and is payable at the time of any principal payment or prepayment of principal; |
| • | Biovest may prepay the Laurus/Valens Term B Notes, without penalty, at any time; and |
| • | provided that the Laurus/Valens Term A Notes have been paid in full, Biovest is required to make mandatory prepayments under the Laurus/Valens Term B Notes from any intercompany funding by the Company to Biovest (with certain exceptions and conditions), but only up to the outstanding principal and accrued interest under the Laurus/Valens Term B Notes. |
With the prior written consent of Laurus/Valens, Biovest may convert all or any portion of the outstanding principal and accrued interest under either the Laurus/Valens Term A Notes or the Laurus/Valens Term B Notes into shares of Biovest’s common stock. The number of shares of Biovest’s common stock issuable on such a conversion (the “Laurus/Valens Conversion Shares”) is equal to (a) an amount equal to the aggregate portion of the principal and accrued and unpaid interest thereon outstanding under the applicable Laurus/Valens Term A Notes or the Laurus/Valens Term B Notes being converted, divided by (b) ninety percent (90%) of the average closing price publicly reported (or reported by Pink Sheets, LLC) for shares of Biovest common stock for the ten (10) trading days immediately preceding the date of the notice of conversion.
The Laurus/Valens Term A Notes and the Laurus/Valens Term B Notes are secured by a first lien on all of the assets of Biovest and its subsidiaries, junior only to the lien granted to Corp Real and to certain permitted liens. The Laurus/Valens Term A Notes and the Laurus/Valens Term B Notes are guaranteed by the Company (the “Accentia Guaranty”), up to a maximum amount of $4,991,360. The Accentia Guaranty is secured by a pledge by the Company of 20,115,818 shares of Biovest’s common stock owned by the Company and by the assets of the Company’s subsidiary, Analytica International, Inc.
The Laurus/Valens Term A and B Notes have replaced the following obligations due to Laurus and its affiliates:
| • | $7.799 million note payable to Laurus originated March 2006; |
| • | $0.250 million note payable to Valens Offshore SPV II, Corp originated October 2007; |
| • | $0.245 million note payable to Valens U.S. SPV I, LLC originated October 2007; |
| • | $3.6 million note payable to Valens Offshore SPV II, Corp originated December 2007; |
| • | $4.9 million note payable to Valens U.S. SPV I, LLC originated December 2007; |
F-83
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | $7.5 million minimum royalty due on sales of AutovaxID™ instrumentation originated in April 2007; and |
| • | $4.4 million loan modification fee, originated July 2008, in consideration for modifying the terms of all the then outstanding debt due to Laurus/Valens. |
The fair value of the Laurus/Valens Term A and B Notes was recorded against the combined carrying value of the obligations listed above resulting in a $6.7 million gain on reorganization for the three months ended December 31, 2010.
Biovest Class 8 Option A Obligations:
Effective as of November 17, 2010, under Biovest’s Plan, Biovest became obligated to pay to the Biovest unsecured creditors approximately $2.7 million in cash together with interest at five percent (5%) per annum to be paid in one installment on March 27, 2014. These unsecured claims include, but is not limited to the Pulaski Bank notes, the Southwest Bank note, and the guarantor indemnities. The fair value of the Option A Obligations was recorded against the carrying value of each claim holder electing an Option A distribution as of the Effective Date, resulting in a $0.5 million gain on reorganization for the three months ended December 31, 2010.
Future maturities of other long-term debt are as follows:
| Years ending December 31, |
||||
| 2012 |
27,866 | |||
| 2013 |
8,560 | |||
| 2014 |
9,458 | |||
| 45,884 | ||||
11. Derivative Liabilities
The following tabular presentation reflects the components of derivative financial instruments:
| December 31, 2010 (unaudited) |
September 30, 2010(1) | |||||||
| Embedded derivative instruments, bifurcated |
$ | — | $ | 18,224,001 | ||||
| Freestanding derivatives: |
||||||||
| Warrants issued with convertible debt |
— | 10,020,236 | ||||||
| Warrants issued with note payable |
— | 7,594,600 | ||||||
| Warrants issued with preferred stock |
— | 1,077,118 | ||||||
| Warrants issued with other debt |
— | 467,564 | ||||||
| Warrants issued with settlement |
1,299,031 | 1,844,200 | ||||||
| Default and investment put options, Biovest |
— | 219,700 | ||||||
| Investment put option, Accentia |
— | 2,928,838 | ||||||
| Biovest investor share distribution |
442,750 | — | ||||||
| Biovest warrants issued with convertible debt |
5,675,639 | — | ||||||
| Biovest debt conversion option |
661,789 | — | ||||||
| $ | 8,079,209 | $ | 42,376,257 | |||||
| (1) | As a result of the Company’s Chapter 11 proceedings, some derivative liabilities listed in the table above became prepetition indebtedness under the Plan. Accordingly, these obligations have been classified as ‘Liabilities subject to compromise’ in the Company’s condensed consolidated balance sheet at September 30, 2010. Only warrants issued with settlement were classified as derivative liabilities at September 30, 2010. |
F-84
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Derivative gains (losses) in the accompanying statements of operations relate to the individual derivatives as follows:
| December 31,
2010 (unaudited) |
December 31, 2009 | |||||||
| Embedded derivative instruments, bifurcated |
$ | (3,576,764) | $ | 179,739 | ||||
| Freestanding derivatives: |
||||||||
| Warrants issued with convertible debentures |
289,271 | (772,048) | ||||||
| Warrants issued with term note payable |
(1,583,088) | 615,051 | ||||||
| Warrants issued with preferred stock |
— | (93,009) | ||||||
| Warrants issued with other debt |
— | (75,872) | ||||||
| Default and investment put options, Biovest |
(3,030) | (5,465) | ||||||
| Investment put option, Accentia |
— | 121,790 | ||||||
| Investor share distribution, Biovest |
255,063 | — | ||||||
| Warrants issued for settlement |
545,168 | — | ||||||
| $ | (4,073,380) | $ | (29,814) | |||||
The following table summarizes assets and liabilities measured at fair value on a recurring basis for the periods presented:
| December 31, 2010 (unaudited) |
September 30, 2010 | |||||||||||||||||||||||||||||||
| Fair value measurements: |
Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | Total | ||||||||||||||||||||||||
| Liabilities |
||||||||||||||||||||||||||||||||
| Derivative liabilities |
$ — | $ 8,079,209 | $ — | $ 8,079,209 | $ — | $ 42,376,257 | $ — | $ 42,376,257 | ||||||||||||||||||||||||
12. Related party transactions:
Biovest Claims of Ronald E. Osman:
Effective as of November 17, 2010, the holder of Biovest’s May 9, 2008 promissory note, Ronald E. Osman, a director of Biovest, elected to convert the entire outstanding principal balance under the note (approximately $1.0 million) plus accrued interest into shares of Biovest common stock at a conversion rate equal to $1.66 per share, resulting in the issuance of 608,224 shares of Biovest common stock.
Biovest DIP Lender Plan Note:
Effective as of November 17, 2010, Biovest executed and delivered in favor of Corps Real, LLC, an Illinois limited liability company (“Corps Real”), the members of which are directors of or are affiliated with directors of Biovest and the Company, a secured convertible promissory note (the “DIP Lender Plan Note”), in an original principal amount equal to $2,291,560 and allows Biovest to draw up to an additional $0.9 million on the note. The DIP Lender Plan Note replaces the $3.0 million secured line of credit promissory note dated December 22, 2008, which we previously executed in favor of Corps Real. The DIP Lender Plan Note matures on November 17, 2012 and all principal and accrued but unpaid interest is payable in cash on such date. Interest accrues and is payable on the outstanding principal amount of the DIP Lender Plan Note at a fixed rate of sixteen percent (16%) per annum, with interest in the amount of ten percent (10%) to be paid monthly and interest in the amount of six percent (6%) to accrue and be paid on the maturity date. Biovest may prepay the DIP Lender Plan Note in full, without penalty, at any time, and Corps Real may convert all or a portion of the outstanding balance of the DIP Lender Plan Note into shares of Biovest common stock at a conversion rate of $0.75 per share of Biovest common stock. The DIP Lender Plan Note is secured by a first priority lien on all of Biovest’s assets.
F-85
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
BDSI/Arius Settlement:
On February 17, 2010, the Bankruptcy Court entered an Order approving an Emezine Settlement Agreement (the “Settlement Agreement”) between the Company and BioDelivery Sciences International, Inc. (“BDSI”), and entered into as of December 30, 2009. Parties to the Settlement Agreement are the Company, the Company’s wholly-owned subsidiary, TEAMM, BDSI, and BDSI’s wholly-owned subsidiary, Arius Pharmaceuticals, Inc. (“Arius”).
The purpose of the Settlement Agreement is to memorialize the terms and conditions of a settlement, between the Company and BDSI regarding claims relating to a Distribution Agreement dated March 12, 2004 between Arius and TEAMM (the “Distribution Agreement”) related to the marketing and distribution of Emezine, a product licensed by Arius from Reckitt Benckiser Healthcare (UK) Limited. Following the issuance in February 2006 by the FDA of a non-approvable letter with respect to the NDA for Emezine, BDSI ceased its Emezine related development efforts and on December 17, 2008, the Distribution Agreement was terminated. The Settlement Agreement resolves the Company’s claims against BDSI under the terminated Distribution Agreement.
The Settlement Agreement provides that the parties mutually release all claims that either may have against each other and, in connection therewith, the Company:
| (a) | received $2.5 million from BDSI (the “$2.5 Million Payment”); and |
| (b) | received the following royalty rights (the “Product Rights”) from BDSI with respect to BDSI’s BEMA Granisetron product candidate (“BEMA Granisetron”) (or in the event it is not BEMA Granisetron, the third BDSI product candidate, excluding BEMA Bupremorphine, as to which BDSI files an NDA, which, together with BEMA Granisetron, shall be referred to hereinafter as the “Product”): |
| (i) | 70/30 split (BDSI/Company) of royalty received if a third party sells the Product and 85/15 split on net sales if BDSI sells the Product; and |
| (ii) | BDSI will, from the sale of the Product, fully recover amounts equal to (1) all internal and external worldwide development costs of the Product (“Costs”) plus interest (measured on weighted average prime interest rate from first dollar spent until Product launch) and (2) the $2.5 Million Payment plus interest (measured on weighted average prime interest rate from the time of payment until Product launch) before the Company begins to receive its split as described in (b) (i) above; and |
| (c) | issued to BDSI a warrant (“Warrant”) to purchase two (2) million shares of Biovest common stock held by the Company, with an exercise price of $0.84 and an expiration date of March 4, 2017. During the initial two (2) year exercise period, any exercise of the Warrant by BDSI will be subject to approval by Biovest. |
In the event that BDSI receives any sublicensing or milestone payments associated with the Product up to and including the NDA approval, BDSI will apply 30% of such payments toward payback of the Costs of the Product plus interest and the $2.5 Million Payment plus the interest.
In the event of a proposed sale of BDSI or its assets, BDSI has the right to terminate its Product Right payment obligations to the Company under the Settlement Agreement upon the payment to the Company of an amount equal to the greater of: (i) $4.5 million; or (ii) the fair market value of the Product Rights as determined by an independent third party appraiser. Further, if the Product Right is terminated, the Warrant described above will be terminated if not already exercised, and, if exercised, an amount equal to the strike price will, in addition to the amount in (i) or (ii) above, be paid to the Company.
The fair value of the warrant issued to BDSI was estimated on the date of grant using the Black-Scholes-Merton valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. The value included in derivative liability is adjusted quarterly and the change is recorded to derivative gain or loss. The value on the grant date was approximately $1.0 million. The value at the end of current period was approximately $1.3 million, resulting in a derivative gain for the three months ended December 31, 2010 of approximately $0.5 million which is recorded as a derivative liability as of December 31, 2010 on accompanying balance sheet.
F-86
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
13. Liabilities subject to compromise:
As a result of the Company’s Chapter 11 filings, the payment of prepetition indebtedness was subject to compromise or other treatment under the Plan prior to the Effective Date. ASC Topic 852 requires prepetition liabilities that are subject to compromise to be reported at the amounts expected to be allowed, even if they may be settled for lesser amounts.
The following table reflects liabilities subject to compromise prior to November 17, 2010:
| September 30, 2010 | ||||
| Accounts payable and accrued expenses(1)(2)(3) |
$ 15,232,863 | |||
| Hybrid financial instrument |
3,608,674 | |||
| Convertible debentures |
28,626,149 | |||
| Laurus term note |
8,800,000 | |||
| Valens 15% convertible note, Biovest |
926,300 | |||
| Secured promissory notes payable to Laurus and the Valens Funds, Biovest |
28,522,108 | |||
| Unsecured promissory notes payable to Pulaski Bank and Trust Company(1) |
1,161,900 | |||
| Unsecured promissory note payable to Southwest Bank of St. Louis |
228,330 | |||
| Southwest line of credit |
4,000,000 | |||
| Notes payable, related parties |
2,313,623 | |||
| Other notes payable |
610,683 | |||
| Minimum royalty due to Laurus on net sales of AutovaxID instrumentation |
6,318,233 | |||
| Derivative liabilities |
40,532,057 | |||
| Dividend payable |
479,452 | |||
| Other |
2,209,756 | |||
| $ 143,570,128 | ||||
14. Stockholders’ equity:
Stock options and warrants
The fair value of each option award is estimated on the date of grant using the Black-Scholes-Merton valuation model that uses assumptions for expected volatility, expected dividends, expected term, and the risk-free interest rate. Expected volatilities are based on historical volatility of a peer company’s stock and other factors estimated over the expected term of the options. The expected term of options granted is derived using the “simplified method” which computes expected term as the average of the sum of the vesting term plus the contract term. This method is used because the Company does not currently have adequate historical option exercise or forfeiture information as a basis to determine expected term. The risk-free rate is based on the U.S. Treasury yield curve in effect at the time of grant for the period of the expected term.
F-87
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Stock options and warrants issued, terminated/forfeited and outstanding as of December 31, 2010 are as follows:
| Shares | Average Exercise Price Per Share |
|||||||
| Options: |
||||||||
| Outstanding, October 1, 2010 |
24,255,580 | $ | 0.81 | |||||
| Granted |
— | — | ||||||
| Terminated or forfeited |
(27,147) | 2.89 | ||||||
| Exercised |
— | — | ||||||
| Outstanding December 31, 2010 |
24,228,433 | $ | 0.81 | |||||
| Warrants: |
||||||||
| Outstanding, October 1, 2010 |
20,280,800 | $ | 2.51 | |||||
| Granted |
— | — | ||||||
| Terminated or forfeited |
(5,213,436) | 2.68 | ||||||
| Exercised |
— | — | ||||||
| Outstanding December 31, 2010 |
15,067,364 | $ | 2.45 | |||||
A summary of the status of the Company’s nonvested stock options as of December 31, 2010, and changes during the three months then ended, is summarized as follows:
| Nonvested Shares |
Shares | Weighted- Average Grant- Date Fair Value |
Intrinsic Value |
|||||||||
| Nonvested at September 30, 2010 |
20,897,833 | |||||||||||
| Granted |
— | |||||||||||
| Vested |
(2,584,394) | |||||||||||
| Forfeited |
(27,147) | |||||||||||
| Nonvested at December 31, 2010 |
18,286,292 | $ | 0.40 | $ | 7,417,348 | |||||||
There were no stock options grants during the quarter ended December 31, 2010.
Stock compensation expense was $10.8 million for the quarter ended December 31, 2010 and $0.4 million for the quarter ended December 31, 2009. Approximately $1.8 million in stock compensation expense will be recognized over the next five (5) quarters, as a result of the vesting of shares.
15. Segment information:
During 2004, the Company through its subsidiary, Analytica, made an insignificant acquisition of a foreign entity, IMOR, this subsidiary ceased operations during the quarter ended March 31, 2010. Therefore, segment information on a geographic basis is only presented below for the year quarter ended December 31, 2010:
| Three months ended December 31, 2010 | ||||||||||||
| Domestic | International (Europe) |
Total | ||||||||||
| Net sales |
$ | 2,742,720 | $ | 943,254 | $ | 3,685,974 | ||||||
| Net loss |
(3,280,003 | ) | (307,979 | ) | (3,587,982 | ) | ||||||
| Total Assets |
8,145,716 | 1,600,884 | 9,746,600 | |||||||||
| Goodwill |
893,000 | 300,437 | 1,193,437 | |||||||||
F-88
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
16. Commitments and contingencies:
Legal proceedings:
Bankruptcy proceedings:
On November 10, 2008, the Company filed a voluntary petition for reorganization under Chapter 11 of the United States Bankruptcy Code (“Chapter 11”) in the U.S. Bankruptcy Court for the Middle District of Florida, Tampa Division (the “Bankruptcy Court”). During the pendency of the Chapter 11 proceedings, the Company operated its business as a debtor-in-possession in accordance with the provisions of Chapter 11 and subject to the jurisdiction of the Bankruptcy Court. On August 16, 2010, the Company filed its First Amended Joint Plan of Reorganization. Subsequent to the filing of this report, on October 25, 2010, the Company filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the Bankruptcy Court held a confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. The Company emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”).
Notwithstanding the effectiveness of the Plan, the Bankruptcy Court retains jurisdiction to adjudicate any remaining issues regarding, inter alia, the validity, amount, and method of payment of claims filed in connection with the Company’s Chapter 11 proceeding. Accordingly, the Company anticipates that there may be ongoing proceedings before the Bankruptcy Court to resolve any filed objections or disputes as to claims filed in the Chapter 11 proceeding.
Biovest litigation:
On August 4, 2008, the Company’s majority-owned subsidiary, Biovest was served with a summons and complaint filed in California Superior Court on behalf of Clinstar LLC for breach of contract for non-payment of certain fees for clinical trial studies and pass-through expenses in the amount of $385,000. Biovest intends to seek the dismissal of this litigation and plan to defend these claims vigorously. Upon the filing of Biovest’s Chapter 11 petition on November 10, 2008, this litigation was automatically stayed pursuant to provisions of federal bankruptcy law. The Company anticipates that the claims involved in this litigation will be contested and resolved by the Bankruptcy Court as part of an objection to claim which the Company expects to file shortly.
Other proceedings:
Further, from time to time the Company is subject to various legal proceedings in the normal course of business, some of which are covered by insurance.
Cooperative Research and Development Agreement:
In September 2001, Biovest entered into a definitive Cooperative Research and Development Agreement (“CRADA”) with the NCI for the development and ultimate commercialization of patient-specific vaccines for the treatment of non-Hodgkin’s low-grade follicular lymphoma. The terms of the CRADA, as amended, included, among other things, a requirement to pay $0.5 million quarterly to NCI for expenses incurred in connection with the ongoing Phase 3 clinical trials. Since the transfer to Biovest of the investigational new drug application for development of this vaccine, which occurred in April 2004, these payments to NCI have been reduced to a small fraction of this original obligation (approximately $0.2 million per year). On September 25, 2006, Biovest provided written notice to the NCI in accordance with the terms of the CRADA to terminate the CRADA at the end of the sixty day notice period. Under the terms of the CRADA, Biovest is obligated to continue to provide vaccine to the NCI at no charge for purposes of the NCI’s studies that are within the scope of the CRADA if the Company were to abandon work on the vaccine.
Employment agreements:
The Company has no employment agreements with officers and executives as of December 31, 2010.
F-89
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Food and Drug Administration:
The FDA has extensive regulatory authority over biopharmaceutical products (drugs and biological products), manufacturing protocols and procedures and the facilities in which mammalian proteins will be manufactured. Any new bioproduct intended for use in humans (including, to a somewhat lesser degree, in vivo biodiagnostic products), is subject to rigorous testing requirements imposed by the FDA with respect to product efficacy and safety, possible toxicity and side effects. FDA approval for the use of new bioproducts (which can never be assured) requires several rounds of extensive preclinical testing and clinical investigations conducted by the sponsoring pharmaceutical company prior to sale and use of the product. At each stage, the approvals granted by the FDA include the manufacturing process utilized to produce the product. Accordingly, Biovest’s cell culture systems used for the production of therapeutic or biotherapeutic products are subject to significant regulation by the FDA under the Federal Food, Drug and Cosmetic Act, as amended (the “FD&C Act”).
Product liability:
The contract production services for therapeutic products offered exposes an inherent risk of liability as the proteins or other substances manufactured, at the request and to the specifications of customers, could foreseeably cause adverse effects. The Company obtains agreements from contract production customers indemnifying and defending the Company from any potential liability arising from such risk. There can be no assurance, however, that the Company will be successful in obtaining such agreements in the future or that such indemnification agreements will adequately protect the Company against potential claims relating to such contract production services. The Company may also be exposed to potential product liability claims by users of its products. A successful partial or completely uninsured claim against the Company could have a material adverse effect on the Company’s operations.
Royalty agreements:
| • | Cancellation and Reduction of Royalty Interests: On the Effective Date, the Company, Biovest, and Laurus/Valens entered into several agreements (the “Laurus/Valens Royalty Termination Agreements”) terminating the following royalties pursuant to the Plan. As a result of the foregoing agreements, the aggregate royalty obligation on BiovaxID® and Biovest’s other biologic products was reduced from 35.25% to 6.30%. Additionally, the aggregate royalty obligation on the AutovaxID™ instrument was reduced from 3.0% to no obligation, including the elimination of the $7.5 million minimum royalty obligation. |
Below is the detail of the royalty obligations following the Effective Date:
| • | BiovaxID® and other biologic products |
| • | Accentia. The Royalty Agreement by and between Biovest and the Company, dated as of October 31, 2006, as amended, was terminated, which resulted in the cancellation of all royalty interest of the Company in Biovest’s biologic products. Under the Royalty Agreement, the Company had a 15.5% royalty interest in the gross revenue of Biovest’s biologic products including BiovaxID, as defined in the agreement after allowing for the 4.00% royalty assigned by the Company to Laurus/Valens pursuant to the four (4) separate Assignment of Rights Under Royalty Agreements, each dated as of June 18, 2008, by and among the Company, Biovest, Erato Corp., Valens U.S., Valens I, and PSource, as amended, modified or supplemented thereafter in accordance with their terms; and |
| • | Laurus/Valens. (i) The Royalty Agreement by and between Biovest and Valens II dated October 30, 2007; (ii) the Royalty Agreement by and between Biovest and Valens II dated December 10, 2007, as amended by a letter agreement dated May 30, 2008, and as further amended, modified or supplemented thereafter in accordance with their terms); (iii) the Royalty Agreement by and between Biovest and Valens U.S. dated October 30, 2007; (iv) the Royalty Agreement by and between Biovest and Valens U.S. dated December 10, 2007, and as amended, modified or supplemented thereafter in accordance with their terms), were all terminated. |
F-90
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
| • | AutovaxID™ |
| • | The previously granted royalty equal to three percent (3%) of world-wide net sales (i.e., gross receipts from the world-wide sales of the automated cell and biologic production instrument known as AutovaxID manufactured by Biovest (the “AutovaxID Instruments”) less any rebates, returns and discounts) of AutovaxID Instruments for a period of five (5) years through May 31, 2012, granted to Laurus by Biovest and AutovaxID, Inc. in a letter agreement dated March 19, 2007, was terminated (including the guaranteed minimum royalty in the amount of $7.5 million remaining under such royalty). |
Stanford University agreement:
In September 2004, Biovest entered into an agreement with Stanford University (“Stanford”) allowing worldwide rights to use two proprietary hybridoma cell lines that are used in the production of BiovaxID. Under the agreement with Stanford, Biovest is obligated to pay a yearly maintenance fee of $10,000 per year. The agreement also provides that Biovest will pay Stanford $0.1 million within one year following FDA approval of BiovaxID or five years following the agreement date (whichever occurs first), and following approval Biovest is required to pay Stanford a running royalty of the higher of $50.00 per patient or 0.05% of revenues received by Biovest for each BiovaxID patient treated using this cell line. This running royalty will be creditable against the yearly maintenance fee. Biovest’s agreement with Stanford obligates Biovest to diligently develop, manufacture, market, and sell BiovaxID and to provide progress reports to Stanford regarding these activities. Biovest can terminate this agreement at any time upon 30 days’ prior written notice, and Stanford can terminate the agreement upon a breach of the agreement by Biovest that remains uncured for 30 days after written notice of the breach from Stanford.
Sublicense agreements with related parties
On February 27, 2007, the Company entered into a sublicense agreement (the “Accentia Sublicense”) with Revimmune, LLC under which the Company was granted the exclusive worldwide rights to Revimmune™. Revimmune, LLC’s manager is a director of the Company. The perpetual sublicense allows the Company to develop and market a patent pending treatment for autoimmune diseases. The Accentia Sublicense covers the potential treatment of all autoimmune diseases including but not limited to multiple sclerosis.
Other material terms and conditions of the Accentia Sublicense are as follows:
| • | The Company assumed certain future development, milestone and minimum royalty obligations of Revimmune, LLC under its license with Johns Hopkins University (“JHU”). In connection with the Accentia Sublicense, the Company did not pay an upfront fee or reimbursement of expenses. The Company also agreed to pay to Revimmune, LLC a royalty of 4% on net sales, and in the event of a sublicense, to pay 10% of net proceeds received from any such sublicense to Revimmune, LLC. |
| • | Upon the approval of the sublicensed treatment in the U.S. for each autoimmune disease, the Company is required to issue to Revimmune, LLC vested warrants to purchase 0.8 million shares of the Company’s common stock. The warrant which will be granted at the approval of the first sublicensed product will have an exercise price of $8 per share and any subsequent warrant to be issued will have an exercise price equal to the average of the volume weighted average closing prices of the Company’s common stock during the ten (10) trading days immediately prior to the grant of such warrant. |
| • | The Company will be responsible, at its sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the licensed products. |
| • | The Company has assumed the cost and responsibility for patent prosecution as provided in the license between Revimmune, LLC and JHU to the extent that the claims actually and directly relate to sublicensed products. |
F-91
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
On January 16, 2008, Biovest entered into a sublicense agreement (the “Biovest Sublicense”) with Revimmune, LLC under which Biovest was granted the exclusive worldwide rights to Revimmune™. The Biovest Sublicense allows Biovest to develop and market, a patent-pending pharmaceutical treatment in late-stage development for the treatment of and prevention of transplant rejection including rejection following a bone marrow transplant.
Other material terms and conditions of the Biovest Sublicense are as follows:
| • | Biovest is obligated to pay to Revimmune, LLC a royalty of 6% on net sales, and in the event of a sublicense by Biovest, to pay 20% of sublicense consideration received. Biovest did not pay an upfront fee in connection with the Biovest Sublicense but upon the approval of the sublicensed treatment in the U.S. for each sublicensed indication, Biovest is required to issue to Revimmune, LLC vested warrants to purchase 2.0 million shares of Biovest’s common stock. Each such warrant which will be granted at the approval of each successive sublicensed product will have an exercise price of $1.10 per share or, at the discretion of Biovest, at a price equal to the fair market value of Biovest’s common stock on the date of the grant of such warrant. |
| • | Biovest assumed certain obligations under Revimmune, LLC’s license with JHU related to the sublicensed technology, including the payment of all royalty obligations due JHU for the sublicensed products which includes a 4% royalty on licensed products and services and a 20% royalty on sublicense consideration. |
| • | Biovest will be responsible, at its sole cost and expense, for the development, clinical trial(s), promotion, marketing, sales and commercialization of the sublicensed products. |
17. Variable Interest Entities:
Revimmune, LLC
Although the Company does not have an equity interest in Revimmune, LLC, the Company has the controlling financial interest of Revimmune, LLC, because of the sublicense agreement between the parties and is considered the primary beneficiary, and therefore, the financial statements of Revimmune, LLC has been consolidated with the Company as of February 27, 2007 and through December 31, 2010. As of December 31, 2010, Revimmune, LLC’s assets and equity were approximately $28,321. The Company had no non-controlling interest in earnings from Revimmune, LLC for the three months ended December 31, 2010.
18. Effective Date (November 17, 2010) of Bankruptcy Plan:
Plan of Reorganization:
On August 16, 2010, the Company and its subsidiaries (collectively, the “Debtors”) filed their First Amended Joint Plan of Reorganization, and, on October 25, 2010, the Debtors filed the First Modification to the First Amended Joint Plan of Reorganization (collectively and as amended and supplemented, the “Plan”). On October 27, 2010, the United States Bankruptcy Court for the Middle District of Florida, Tampa Division (the “Bankruptcy Court”) held a Confirmation hearing and confirmed the Plan, and, on November 2, 2010, the Bankruptcy Court entered an Order Confirming Debtors’ First Amended Joint Plan of Reorganization Under Chapter 11 of the Bankruptcy Code (the “Confirmation Order”), which approved and confirmed the Plan, as modified by the Confirmation Order. In connection with the confirmation hearing all creditor classes deemed “impaired” pursuant to the Bankruptcy Code voted to support the Plan. The Debtors emerged from Chapter 11 protection, and the Plan became effective, on November 17, 2010 (the “Effective Date”). Pursuant to the Plan, the date on which a claim in any Class listed in the Plan became an “allowed claim” by order of the Bankruptcy Court or by agreement between Debtors and the claimant is the “Determination Date”.
Equity Interests (Class 15):
Each of the Company’s common stockholder on the Effective Date was deemed to receive one (1) share of Reorganized Accentia Common Stock (the “Class 15 Plan Shares”) for each share of the Company’s existing common stock held by such stockholder as of the Effective Date. The Company’s Class 15 Plan Shares shall be deemed issued pursuant to Section 1145 of the Bankruptcy Code and shall not have any legend restricting the sale thereof under federal securities laws.
F-92
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
April 2006 NMTC Transaction:
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “Worcester Restructuring Agreement”) with Telesis CDE Corporation and Telesis CDE Two, LLC (collectively, “Telesis”), contingent upon submission to and approval by the Bankruptcy Court. The Worcester Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to the April 2006 NMTC Transaction, in consideration of retention by Telesis of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $0.3 million along with a settlement payment in the amount of $85,000 to defray certain legal and administrative expenses incurred by Telesis. The Worcester Restructuring Agreement and the compromise of the outstanding claims against Biovest and its affiliates in connection with the April 2006 NMTC Transaction was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, the Company’s Guaranty, Biovest’s Guaranty, and all of Biovest’s subsidiary Guaranties from affiliates and third parties and all other obligations of all parties to the April 2006 NMTC Transaction were terminated. Biovest has ceased all activities under the April 2006 NMTC Transaction and Biovest has liquidated the subsidiaries created specifically to conduct activities under the April 2006 NMTC transaction.
December 2006 NMTC Transaction:
Biovest and certain of its affiliates entered into an agreement in July 2010 (the “St. Louis Restructuring Agreement”) with St. Louis Development Corporation and Saint Louis New Markets Tax Credit Fund II, LLC (collectively “SLDC”), contingent upon submission to and approval by the Bankruptcy Court. The St. Louis Restructuring Agreement effectively terminates all agreements and obligations of all parties pursuant to the December 2006 NMTC Transaction, in consideration of retention by SLDC of an unsecured claim in Biovest’s Chapter 11 proceeding in the amount of $160,000 along with a settlement payment in the amount of $62,000, to defray certain legal and administrative expenses incurred by SLDC. This St. Louis Restructuring Agreement and the compromise of the outstanding claims against Biovest in connection with the December 2006 NMTC Transaction was approved by the Bankruptcy Court in an Order entered on December 1, 2010. As a result, the Company’s Guaranty, Biovest’s Guaranty, and all of the subsidiary Guaranties from affiliates and third parties and all other obligations of all parties to the December 2006 NMTC Transaction were terminated. Biovest has ceased all activities under the December 2006 NMTC Transaction and Biovest has liquidated the subsidiaries created specifically to conduct activities under the December 2006 NMTC Transaction.
Termination of Warrants:
On the Effective Date, all of the following warrants were terminated and cancelled:
| • | the common stock purchase warrant dated August 16, 2005, issued by the Company to Laurus for the purchase of up to 1,000,000 shares of the Company’s common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated September 29, 2006, issued by the Company to Laurus for the purchase of up to 627,240 shares of the Company’s common stock at an exercise price of $2.75 per share; |
| • | the common stock purchase warrant dated October 31, 2007, issued by the Company to Laurus for the purchase of up to 4,024,398 shares of the Company’s common stock at an exercise price of $2.67 per share; |
| • | the common stock purchase warrant dated January 18, 2008, issued by the Company to Valens Offshore, for the purchase of up to 365,169 shares of the Company’s common stock at an exercise price of $2.67 per share; and |
| • | the common stock purchase warrant dated January 18, 2008, issued by the Company to Valens, for the purchase of up to 196,629 shares of the Company’s common stock at an exercise price of $2.67 per share. |
F-93
Table of Contents
ACCENTIA BIOPHARMACEUTICALS, INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
THREE MONTHS ENDED DECEMBER 31, 2010 AND 2009
Termination of Material Agreements:
On the Effective Date all of the following documents were terminated:
| • | all documents evidencing or relating to loans made by Laurus/Valens to the Company prior to the Effective Date; |
| • | certain revolving credit agreement between Southwest Bank and the Company dated as of December 30, 2005, certain stock pledge agreement by and between the Company and Southwest Bank dated as of June 16, 2008, and all other documents executed in connection therewith; |
| • | all documents evidencing or relating to loans made by McKesson to the Company prior to the Effective Date (with certain exceptions set forth in the Plan); |
| • | all documents evidencing or relating to the 8% secured convertible debentures due September 29, 2010, issued, by the Company in September 2006, in the original aggregate principal amount of $25 million; |
| • | all documents evidencing or relating to the 8% original issue discount secured convertible debentures due June 19, 2011, issued, by the Company in June 2008, in the original aggregate principal amount of $8,906,098.00; |
| • | all documents evidencing or relating to the 8% convertible debentures due February 28, 2011, issued, by the Company in February 2007, in the original aggregate principal amount of $24,940,000.00; and |
| • | all documents evidencing or relating to the Company’s Series A-1 convertible preferred stock, par value $1.00 per share. |
19. Subsequent Events:
Issuance of Common Stock:
On January 10, 2011, pursuant to the Retirement Settlement Agreements with Alan M. Pearce, the Company issued to Mr. Pearce 66,000 shares of the Company’s common stock and Biovest issued to Mr. Pearce 56,000 shares of Biovest’s common stock.
Appointment of Certain Officers:
Effective as of January 15, 2011, Garrison J. Hasara, C.P.A. was appointed to serve in the position of Acting Chief Financial Officer and Controller of the Company. In addition, Mr. Hasara was also designated to serve as the Company’s Principal Financial Officer and Principal Accounting Officer in connection with dealings with the Company’s independent audit firm and filings with the SEC.
Effective as of January 15, 2011, Brian D. Bottjer, C.P.A. was appointed to serve in the position of Acting Chief Financial Officer and Comptroller of Biovest. In addition, Mr. Bottjer was also designated to serve as Biovest’s Principal Financial Officer and Principal Accounting Officer in connection with dealings with Biovest’s independent audit firm and filings with the SEC.
F-94
Table of Contents

Units
PROSPECTUS
, 2011
No dealer, salesperson or other person has been authorized to give any information or to make any representations other than those contained in this Prospectus in connection with the offering made by this Prospectus, and, if given or made, such information or representations must not be relied upon as having been authorized by the Company. This Prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities other than those specifically offered hereby or an offer to sell or a solicitation of an offer to buy any of these securities in any jurisdiction to any person to whom it is unlawful to make such offer or solicitation. Neither the delivery of this Prospectus nor any sale hereunder shall under any circumstances create any implication that there has been no change in the affairs of the Company since the date hereof.
Table of Contents
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
The following table sets forth the costs and expenses, other than placement agent fees and expenses, payable by the registrant in connection with this offering. All amounts are estimates, except for the Securities and Exchange Commission registration fee and the FINRA filing fee. All of these costs and expenses will be borne by the registrant.
| SEC filing fee |
$ | 581 | (1) | |||||
| FINRA filing fee |
$ | |||||||
| Printing and engraving expenses |
$ | |||||||
| Accountants’ fees and expenses |
$ | |||||||
| Legal fees and expenses |
$ | |||||||
| Miscellaneous |
$ | |||||||
| Total |
$ | |||||||
| (1) | Rounded up to nearest whole number. |
| Item 14. | Indemnification of Directors and Officers. |
The Florida Business Corporation Act, or “FBCA,” permits a Florida corporation to indemnify any person who may be a party to any third party proceeding by reason of the fact that such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee, or agent of another entity, against liability incurred in connection with such proceeding (including any appeal thereof) if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
The FBCA permits a Florida corporation to indemnify any person who may be a party to a derivative action if such person acted in any of the capacities set forth in the preceding paragraph, against expenses and amounts paid in settlement not exceeding, in the judgment of the board of directors, the estimated expenses of litigating the proceeding to conclusion, actually and reasonably incurred in connection with the defense or settlement of such proceeding (including appeals), provided that the person acted under the standards set forth in the preceding paragraph. However, no indemnification shall be made for any claim, issue, or matter for which such person is found to be liable unless, and only to the extent that, the court determines that, despite the adjudication of liability, but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnification for such expenses which the court deems proper.
II-1
Table of Contents
The FBCA provides that any indemnification made under the above provisions, unless pursuant to a court determination, may be made only after a determination that the person to be indemnified has met the standard of conduct described above. This determination is to be made by a majority vote of a quorum consisting of the disinterested directors of the board of directors, by duly selected independent legal counsel, or by a majority vote of the disinterested stockholders. The board of directors also may designate a special committee of disinterested directors to make this determination. Notwithstanding the foregoing, the FBCA provides that a Florida corporation must indemnify any director, officer, employee or agent of a corporation who has been successful in the defense of any proceeding referred to above.
Notwithstanding the foregoing, the FBCA provides, in general, that no director shall be personally liable for monetary damages to our company or any other person for any statement, vote, decision, or failure to act, regarding corporate management or policy, unless: (a) the director breached or failed to perform his duties as a director; and (b) the director’s breach of, or failure to perform, those duties constitutes (i) a violation of criminal law, unless the director had reasonable cause to believe his conduct was lawful or had no reasonable cause to believe his conduct was unlawful, (ii) a transaction from which the director derived an improper personal benefit, either directly or indirectly, (iii) an approval of an unlawful distribution, (iv) with respect to a proceeding by or in the right of the company to procure a judgment in its favor or by or in the right of a stockholder, conscious disregard for the best interest of the company, or willful misconduct, or (v) with respect to a proceeding by or in the right of someone other than the company or a stockholder, recklessness or an act or omission which was committed in bad faith or with malicious purpose or in a manner exhibiting wanton and willful disregard of human rights, safety, or property. The term “recklessness,” as used above, means the action, or omission to act, in conscious disregard of a risk: (a) known, or so obvious that it should have been known, to the director; and (b) known to the director, or so obvious that it should have been known, to be so great as to make it highly probable that harm would follow from such action or omission.
The FBCA further provides that the indemnification and advancement of payment provisions contained therein are not exclusive and it specifically empowers a corporation to make any other further indemnification or advancement of expenses of any of its directors, officers, employees or agents under any bylaw, agreement, vote of stockholders or disinterested directors, or otherwise, both for actions taken in an official capacity and for actions taken in other capacities while holding such office. However, a corporation cannot indemnify or advance expenses if a judgment or other final adjudication establishes that the actions of the director, officer, employee, or agent were material to the adjudicated cause of action and the director, officer, employee, or agent (a) violated criminal law, unless the director, officer, employee, or agent had reasonable cause to believe his or her conduct was unlawful, (b) derived an improper personal benefit from a transaction, (c) was or is a director in a circumstance where the liability for unlawful distributions applies, or (d) engaged in willful misconduct or conscious disregard for the best interests of the corporation in a proceeding by or in right of the corporation to procure a judgment in its favor or in a proceeding by or in right of a stockholder.
We have adopted provisions in our articles of incorporation and bylaws providing that our directors and officers and our former directors and officers shall be indemnified to the fullest extent permitted by applicable law.
II-2
Table of Contents
There is no pending litigation or proceeding involving any of our directors, officers, employees or other agents as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director, officer, employee or other agent.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers or persons controlling the registrant pursuant to the foregoing provisions, the registrant has been informed that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is therefore unenforceable.
| Item 15. | Recent Sales of Unregistered Securities |
Since January 2008, we have issued the following securities which were not registered under the Securities Act of 1933, as amended (the “Securities Act”):
1. In January 2008, we issued 233,102 shares of our common stock to various debenture holders in connection with the payment of interest conversion shares pursuant to our February 2007 Convertible Debentures. These shares were issued in lieu of cash interest payments totaling approximately $529,141.54 (or $2.27 per share).
2. In January 2008, we issued a warrant to purchase up to 163,464 shares of our common stock, at an exercise price of $2.67 per share, to Rodman & Renshaw, LLC. These warrants were granted to Rodman & Renshaw, LLC pursuant to the placement agent agreement between Rodman & Renshaw, LLC and our company.
3. In January 2008 through December 2008, we granted stock options to our employees under our stock option plan covering an aggregate of 4,036,000 shares of our common stock (net of expirations and cancellations), at exercises prices ranging from $0.18 to $2.69 per share, with a weighted exercise price of $1.41 per share.
4. In February 2008, we issued 121,952 shares of our common stock to Nixon Peabody, LLP, a law firm, in consideration of legal services provided by Nixon Peabody, LLP to our Company. These shares were issued in lieu of cash payments for legal services totaling approximately $325,611.84 (or $2.67 per share).
5. In February 2008, we issued an aggregate of 6,094 shares of our common stock to Robert Rocke, Jon Sbar, and Robert McLean, attorneys, in consideration of legal services provided by Rocke, Sbar & McLean, P.A. These shares were issued in lieu of cash payments for legal services totaling approximately $16,270.98 (or $2.67 per share).
6. In April 2008, we issued 296,010 shares of our common stock to various debenture holders in connection with the payment of interest conversion shares pursuant to our February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $287,129.70 (or $0.97 per share).
7. In June 2008, we issued 187,266 shares of our common stock to various debenture holders in connection with a settlement payment with regard to the payment of monthly redemption and interest conversion shares pursuant to our February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $500,000.22 (or $2.67 per share).
II-3
Table of Contents
8. In June 2008, we issued 50,000 shares of our common stock to McKesson Corporation in connection with the Amendment to No. 1 to Termination Agreement Re Biologics Distribution Agreement dated June 16, 2008. These shares were issued to guarantee payment to McKesson of approximately $133,500 (or $2.67 per share). Because these shares were issued to guarantee payment, they will be returned to us by McKesson once we pay the guaranteed amounts to McKesson.
9. In June 2008, we issued a warrant to purchase up to 372,437 shares of our common stock, at an exercise price of $1.21 per share, to Rodman & Renshaw, LLC. These warrants were granted to Rodman & Renshaw, LLC pursuant to the placement agent agreement between Rodman & Renshaw, LLC and our company.
10. In July 2008, we issued 2,235,035 shares of our common stock to various debenture holders in connection with the payment of debenture and interest conversion shares pursuant to our September 2006 and February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $2,279,735.70 (or $1.02 per share).
11. In July 2008, we issued 60,000 shares of our common stock to Diamond Opportunity Fund, LLC in connection with the payment of debenture conversion shares pursuant to our February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $75,000 (or $1.25 per share).
12. In July 2008, we issued 275,605 shares of our common stock to various preferred convertible stockholders in connection with the election to redeem the Series A-1 Preferred Convertible Stock under the Preferred Convertible PIPE dated January 18, 2008. These shares were issued in lieu of cash payments totaling approximately $253,556.60 (or $0.92 per share).
13. In August 2008, we issued 1,523,485 shares of our common stock to various debenture holders in connection with the payment of debenture conversion shares pursuant to our September 2006 and February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $1,264,492.55 (or $0.83 per share).
14. In August 2008, we issued 76,923 shares of our common stock to various preferred convertible stockholders in connection with the voluntary election to redeem the Series A-1 Preferred Convertible Stock under the Preferred Convertible PIPE dated January 18, 2008. These shares were issued in lieu of cash payments totaling approximately $96,153.75 (or $1.25 per share).
15. In August 2008, we issued 310,798 shares of our common stock to various preferred convertible stockholders in connection with the election to redeem the Series A-1 Preferred Convertible Stock under the Preferred Convertible PIPE dated January 18, 2008. These shares were issued in lieu of cash payments totaling approximately $279,718.20 (or $0.90 per share).
16. In September 2008, we issued 1,669,092 shares of our common stock to various debenture holders in connection with the payment of debenture conversion shares pursuant to our September 2006 and February 2007 Convertible Debentures. These shares were issued in lieu of cash payments totaling approximately $1,268,509.92 (or $0.76 per share).
17. In September 2008, we issued 377,375 shares of our common stock to various preferred convertible stockholders in connection with the election to redeem the Series A-1 Preferred Convertible Stock under the Preferred Convertible PIPE dated January 18, 2008. These shares were issued in lieu of cash payments totaling approximately $252,841.25 (or $0.67 per share).
II-4
Table of Contents
18. From January 2009 through December 2009, we granted stock options to our employees under our stock option plan covering an aggregate of 2,712,000 shares of our common stock (net of expirations and cancellations), at exercises prices ranging from $0.18 to $0.35 per share, with a weighted exercise price of $0.18 per share.
19. From January 2010 through December 2010, we granted stock options to our employees under our stock option plan covering an aggregate of 16,330,000 shares of our common stock (net of expirations and cancellations), at an exercise price of $0.44 per share, with a weighted exercise price of $0.44 per share.
20. In November 2010, pursuant to our Plan, we issued warrants to various creditors to purchase up to an aggregate of 9,715,908 shares of our common stock at an exercise price of $1.50 per share.
21. From November 2010 through January 2011, pursuant to our Plan with an effective date of November 17, 2010 and Section 1145 of the United States Bankruptcy Code, we issued 10,315,383 shares of our common stock in satisfaction of allowed claims under our Plan, with conversion prices ranging from $1.25 to $2.67 per share.
22. In November 2010, we issued 1,500,000 shares of our common stock to our employees under our stock option plan. These shares were issued for $1.50 per share.
23. In December 2010, we issued 56,250 shares of our common stock to Rocke, Sbar & McLean, P.A., in consideration of legal services provided by Rocke, Sbar & McLean, P.A. These shares were issued in lieu of cash payments for legal services totaling approximately $40,500 (or $0.72 per share).
24. In January 2011, we granted stock options to our employees under our stock option plan covering an aggregate of 785,000 shares of our common stock (net of expirations and cancellations), at an exercise price of $0.74 per share, with a weighted exercise price of $0.74 per share.
25. In January 2011, we issued 66,000 shares of our common stock to Alan M. Pearce in connection with the Resignation Settlement Agreement with an effective date of December 31, 2010. These shares were issued in consideration of the settlement and release of certain claims by Mr. Pearce under the Resignation Settlement Agreement.
We claimed exemption from registration under the Securities Act for the sales and issuances of securities in the transactions described in paragraphs 1, 2, 4-17, 23, and 25 above by virtue of Section 4(2) of the Securities Act in that such sales and issuances did not involve a public offering. The recipients of securities in each of these transactions represented their intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof. Appropriate legends were affixed to the share certificates and instruments issued in all such transactions. All recipients had adequate access, through their relationships with us, to information about us.
We claimed exemption from registration under the Securities Act for the issuances of securities in the transactions described in paragraphs 20 and 21 above by virtue of Section 1145(a) of the United States Bankruptcy Code in that such issuances were made under our Bankruptcy Plan in exchange for claims against, or interests in, our company.
II-5
Table of Contents
We claimed exemption from registration under the Securities Act for the sales and issuances of securities in the transactions described in paragraphs 3, 18, 19, 22, and 24 by virtue of Section 4(2) of the Securities Act and by virtue of Rule 701 promulgated under the Securities Act, in that they were offered and sold either pursuant to written compensatory plans or pursuant to a written contract relating to compensation, as provided by Rule 701. Such sales and issuances did not involve any public offering, were made without general solicitation or advertising, and each purchaser represented its intention to acquire the securities for investment only and not with view to or for sale in connection with any distribution thereof, and appropriate legends were affixed to the share certificates and instruments issued in such transactions. All recipients had adequate access, through their relationships with us, to information about us.
No underwriters were employed in any of the above transactions.
| Item 16. | Exhibits and Financial Statement Schedules. |
| (a) | Exhibits. See Exhibit Index. |
(b) Financial Statement Schedules. All schedules have been omitted because the required information is not present in amounts sufficient to require submission of the schedules, or because the required information is included in the consolidated financial statements or notes thereto.
| Item 17. | Undertakings. |
The undersigned registrant hereby undertakes to provide to the placement agent at the closing specified in the placement agreement certificates in such denominations and registered in such names as required by the placement agent to permit prompt delivery to each purchaser.
The undersigned registrant hereby undertakes:
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933; |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; |
II-6
Table of Contents
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
| (2) | That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b) (1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (5) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
II-7
Table of Contents
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Tampa, State of Florida, on the 18th day of February, 2011.
| ACCENTIA BIOPHARMACEUTICALS, INC. | ||
| By: | /s/ Francis E. O’Donnell, Jr. | |
| Francis E. O’Donnell, Jr., M.D. Chairman and Chief Executive Officer (Principal Executive Officer) | ||
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities and on the dates indicated. Each person whose signature appears below constitutes and appoints Francis E. O’Donnell, Jr. and Garrison J. Hasara and each of them individually, as his or her true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments (including post-effective amendments) to this registration statement and any Rule 462(b) registration statement and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or either or them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
| Signature |
Title |
Date | ||
| /s/ Francis E. O’Donnell, Jr. Francis E. O’Donnell, Jr., M.D. |
Chief Executive Officer; Chairman of the Board; Director (Principal Executive Officer) |
February 18, 2011 | ||
| /s/ Garrison J. Hasara Garrison J. Hasara, CPA |
Acting Chief Financial Officer; Controller (Principal Accounting Officer and Principal Financial Officer) |
February 18, 2011 | ||
| /s/ Edmund C. King Edmund C. King |
Director | February 18, 2011 | ||
| /s/ David M. Schubert David M. Schubert |
Director | February 18, 2011 | ||
| /s/ William S. Poole William S. Poole |
Director | February 18, 2011 | ||
| /s/ Christopher C. Chapman Christopher C. Chapman, M.D. |
Director | February 18, 2011 | ||
S-1
Table of Contents
EXHIBIT INDEX
The following exhibits are filed as part of, or are incorporated by reference into, this Registration Statement on Form S-1:
| Exhibit Number |
Document Description | |
| 1.1* | Form of Placement Agent Agreement. | |
| 3.1 | Amended and Restated Bylaws effective November 17, 2010 (filed as Exhibit 3.1 to Accentia’s Form 8-K filed on November 23, 2010 and incorporated herein by reference). | |
| 3.2 | Amended and Restated Articles of Incorporation effective November 17, 2010 (filed as Exhibit 3.2 to Accentia’s Form 8-K filed on November 23, 2010 and incorporated herein by reference). | |
| 4.1 | Reference is made to Exhibits 3.1 and 3.2. | |
| 4.2 | First Amended Joint Plan of Reorganization of Accentia Biopharmaceuticals, Inc., Analytica International, Inc., TEAMM Pharmaceuticals, Inc., AccentRx, Inc., and Accentia Specialty Pharmacy, Inc. Under Chapter 11 of Title 11, United States Code (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on November 2, 2010 and incorporated herein by reference). | |
| 4.3 | First Modification to First Amended Joint Plan of Reorganization of Accentia Biopharmaceuticals, Inc., Analytica International, Inc., TEAMM Pharmaceuticals, Inc., AccentRx, Inc., and Accentia Specialty Pharmacy, Inc. Under Chapter 11 of Title 11, United States Code (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on November 2, 2010 and incorporated herein by reference). | |
| 4.4* | Form of Warrant. | |
| 5.1* | Opinion of Foley & Lardner LLP. | |
| 10.1 |
License Agreement, dated April 12, 2004, between Accentia and BioDelivery Sciences International, Inc. (“BDSI”), as amended pursuant to an Asset Purchase Agreement dated September 7, 2004 and as further amended by those certain letter agreements dated March 28, 2005 and April 25, 2005 (filed as Exhibit 10.1 to the Registration Statement on Form S-1 (Amendment No. 2) filed on May 16, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.2(b) |
License Agreement, dated February 10, 2004, between Accentia and Mayo Foundation for Medical Education and Research (“MAYO”), as amended on December 12, 2004 (filed as Exhibit 10.2 to the Registration Statement on Form S-1 (Amendment No. 1) filed on April 6, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.3 |
Exclusive Agreement, dated September 17, 2004, between Accentia and The Board of Trustees of the Leland Stanford Junior University (filed as Exhibit 10.3 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
Table of Contents
| 10.4 | Investment Agreement, dated April 10, 2003, between Accentia and Biovest International, Inc. (“Biovest”), as amended (filed as Exhibit 10.4 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.5(b) | Distribution Agreement, dated March 12, 2004, between Accentia and Arius Pharmaceuticals, Inc. (filed as Exhibit 10.6 to the Registration Statement on Form S-1 (Amendment No. 3) filed on June 13, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.6 | Cooperative Research and Development Agreement, dated May 27, 1999, between Accentia and The National Cancer Institute, as amended by that certain amendment dated April 6, 2005 (filed as Exhibit 10.12 to the Registration Statement on Form S-1 (Amendment No. 2) filed on May 16, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.7(a) | 2003 Stock Option Plan, as amended (filed as Exhibit 10.23 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.8(a) | Employment Agreement, dated January 1, 2005, between Accentia and Francis E. O’Donnell, Jr. (filed as Exhibit 10.24 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.9(a) | Employment Agreement, dated January 1, 2005, between Accentia and Alan M. Pearce (filed as Exhibit 10.27 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.10(a) | Employment Agreement, dated January 1, 2005, between Accentia and Samuel S. Duffey (filed as Exhibit 10.28 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.11(a) | Form of Non-Qualified Stock Option Agreement (filed as Exhibit 10.33 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.12(a) | Form of Incentive Stock Option Agreement (filed as Exhibit 10.34 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.13(a) | 2005 Equity Incentive Plan (filed as Exhibit 10.35 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.14 | Form of Director and Officer Indemnity Agreement (filed as Exhibit 10.29 to the Registration Statement on Form S-1 (Amendment No. 3) filed on June 13, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.15 | Form of Warrant for Purchase of Common Stock granted by Accentia to Common Stock Holder (filed as Exhibit 10.32 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
Table of Contents
| 10.16 | Revolving Credit Agreement, dated March 30, 2004, between Missouri State Bank and Trust Company (“MSB”) and Accentia, as amended on March 22, 2005 (filed as Exhibit 10.36 to the Registration Statement on Form S-1 (Amendment No. 1) filed on April 6, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.17 | Accentia Assumption of Debt and Security Agreement, dated December 31, 2003, between Accentia and McKesson, as amended by the First Amendment, dated February 9, 2005, and as modified on May 31, 2005, June 28, 2005, July 8, 2005, August 15, 2005, and September 13, 2005 (filed as Exhibit 10.39 to the Registration Statement on Form S-1 (Amendment No. 8) filed on October 3, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.18 | Warrant Purchase Agreement, dated December 1, 1998, between Accentia and McKesson (filed as Exhibit 10.41 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.19 | Credit Agreement, dated November 30, 1998, between Accentia and McKesson (filed as Exhibit 10.42 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.20 | Security Agreement, dated November 30, 1998, between Accentia and McKesson Corporation (filed as Exhibit 10.43 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.21 | Lease Agreement, dated November 2004, between Accentia and Bay Villa Developers, Inc., as General Partner for Hyde Park Plaza Associates, Ltd. (filed as Exhibit 10.64 to the Registration Statement on Form S-1 (Amendment No. 1) filed on April 6, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.22 | Securities Purchase Agreement, dated April 29, 2005, between Accentia and Laurus Master Fund, Ltd. (“Laurus”) (filed as Exhibit 10.67 to the Registration Statement on Form S-1 (Amendment No. 2) filed on May 16, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.23 | Security Agreement dated April 29, 2005, between Accentia and Laurus (filed as Exhibit 10.68 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.24 | Amended and Restated Secured Convertible Term Note, dated April 29, 2005, of Accentia payable to Laurus (filed as Exhibit 10.69 to the Registration Statement on Form S-1 filed on March 6, 2006 (Registration No. 333-132237) and incorporated by reference). | |
| 10.25 | Amended and Restated Convertible Minimum Borrowing Note dated April 29, 2005, of Accentia and Laurus (filed as Exhibit 10.2 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.26 | Secured Revolving Note dated April 29, 2005, of Accentia and Laurus (filed as Exhibit 10. 7 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.27 | Stock Pledge Agreement and InterCompany Note Pledge Agreement, dated April 29, 2005, between Accentia and Laurus (filed as Exhibit 10.72 to the Registration Statement on Form S-1 (Amendment No. 2) filed on May 16, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.28 | Amended and Restated Common Stock Purchase Warrant, dated August 16, 2005, granted by Accentia to Laurus (filed as Exhibit 10.73 to the Registration Statement on Form S-1 (Amendment No. 7) filed on September 2, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.29 | Subsidiary Guaranty, dated April 29, 2005, between Accentia and Laurus (filed as Exhibit 10.74 to the Registration Statement on Form S-1 (Amendment No. 2) filed on May 16, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.30 | Registration Rights Agreement, dated April 29, 2005, between Accentia and Laurus, as amended (filed as Exhibit 10.75 to the Registration Statement on Form S-1 (Amendment No. 8) filed on October 3, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 10.31 | Amended and Restated Registration Rights Agreement dated February 13, 2006 between Accentia and Laurus (filed as Exhibit 10.5 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.32(b) | Option Agreement, dated December 6, 2005, between Accentia and MAYO (filed as Exhibit 10.83 to Accentia’s Form 10-K filed on December 29, 2005 and incorporated herein by reference). | |
| 10.33(b) | Licensing and Distribution Agreement, dated November 22, 2005, between Accentia and Collegium Pharmaceuticals, Inc. (“Collegium”) (filed as Exhibit 10.84 to Accentia’s Form 10-K filed on December 29, 2005 and incorporated herein by reference). | |
| 10.34 | Promissory Note Dated September 30, 2005 (filed as Exhibit 10.85 to Accentia’s Form 10-K filed on December 29, 2005 and incorporated herein by reference). | |
| 10.35 | Agreement with Collegium (filed as Exhibit 10.84 to Accentia’s Form 10-K filed on December 29, 2005 and incorporated herein by reference). | |
| 10.36 | Overadvance Letter Agreement and Additional Common Stock Purchase Warrant, dated December 29, 2005, between Accentia and Laurus (filed as Exhibit 10.86 to the Registration Statement on Form S-1 filed on March 6, 2006 (Registration No. 333-132237) and incorporated by reference). | |
| 10.37 | Trust Ratification dated February 13, 2006 between Accentia and Laurus (filed as Exhibit 10.1 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.38 | MSB Subordination Agreement dated February 13, 2006 between Accentia and Laurus (filed as Exhibit 10.3 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.39 | Second Omnibus Amendment dated February 13, 2006 between Accentia and Laurus (filed as Exhibit 10.6 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.40 | Joinder Agreement dated February 13, 2006 between TEAMM and Laurus (filed as Exhibit 10.8 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.41 | Revolving Credit Agreement dated December 30, 2005 between Accentia and MSB (filed as Exhibit 10.9 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.42 | Revolving Credit Note dated December 30, 2005 between Accentia and MSB (filed as Exhibit 10.10 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.43 | Security Agreement dated December 30, 2005 between Accentia and MSB (filed as Exhibit 10.11 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.44 | Continuing Contract of Guaranty dated December 30, 2005 between Accentia, MSB, and other parties (filed as Exhibit 10.12 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.45 | Security Agreement dated December 30, 2005 between TEAMM and MSB (filed as Exhibit 10.13 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.46 | Stock Pledge Agreement dated December 30, 2005 between The Francis E. O’Donnell, Jr. Irrevocable Trust No. 1 dated May 25, 1990 and MSB (filed as Exhibit 10.14 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.47 | Securities Pledge and Security Agreement dated December 30, 2005 between Dennis L. Ryll and MSB (filed as Exhibit 10.15 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.48 | Warrant dated February 13, 2006 from Accentia to Laurus (filed as Exhibit 10.19 to Accentia’s Form 10-Q filed on February 14, 2006 and incorporated herein by reference). | |
| 10.49 | Amended and Restated Stock Pledge Agreement, dated as of April 29, 2005 and amended and restated as of April 25, 2006, among Laurus, Accentia, and each other Pledgor party thereto (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.50 | Demand Note, dated April 25, 2006, issued by Biovest to Laurus (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.51 | First Bank Subordination Agreement dated as of April 25, 2006, by and among Laurus, First Bank and Accentia (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.52 | Telesis Subordination Agreement dated as of April 25, 2006, by and among Laurus, Telesis CDE Two, LLC (“Telesis CDE”), Biovax, Inc. (“Biovax”), Biovest and Accentia (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.53 | Promissory Note, dated April 25, 2006, issued by Biovax Investment LLC (“Biovax Leverage Fund”) to Biolender, LLC (“Biolender”) (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.54 | Loan and Security Agreement, dated as of April 25, 2006, between Biovax Leverage Fund and Biolender (filed as Exhibit 10.6 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.55 | Subordinated Convertible Promissory Note, dated April 25, 2006, from Biovax to Telesis CDE (filed as Exhibit 10.7 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.56 | Convertible Loan Agreement dated as of April 25, 2006, by and among Biovax, Telesis CDE and Biovest (filed as Exhibit 10.8 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.57 | Guaranty, dated April 25, 2006, made by Frances E. O’Donnell, Jr., Kathleen M. O’Donnell (as Trustee of the Frances E. O’Donnell, Jr. Irrevocable Trust), Dennis L. Ryll, Ronald Osman, Steven J. Stogel, Donald L. Ferguson, Donald L. Ferguson (as trustee of the Donald L. Ferguson Revocable Trust), Biovest and Accentia in favor of U.S. Bancorp Community Investment Corporation (“USBCIC”) and Telesis CDE (filed as Exhibit 10.9 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.58 | Limited Liability Company Agreement of Biolender, dated April 25, 2006, between Biovest and Accentia (filed as Exhibit 10.10 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.59 | Put Option Agreement dated April 25, 2006, between Biovax IC, Biovax Leverage Fund, USBCIC and Biolender (filed as Exhibit 10.11 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.60 | Purchase Option Agreement dated April 25, 2006, between Biovax IC, Biovax Leverage Fund, USBCIC and Biolender (filed as Exhibit 10.12 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.61 | Common Stock Purchase Warrant, dated April 25, 2006, issued by Biovest to Telesis CDE (filed as Exhibit 10.13 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.62 | Common Stock Purchase Warrant, dated April 25, 2006, issued by Accentia to Telesis CDE (filed as Exhibit 10.14 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.63 | Tax Credit Reimbursement and Indemnity Agreement, dated as of April 25, 2006, between Biovax and USBCIC (filed as Exhibit 10.15 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.64 | Asset Purchase Agreement dated April 18, 2006 between Biovest and Biovax (filed as Exhibit 10.16 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.65 | Vaccine Purchase and Sale Agreement, dated as of April 28, 2006, between Biovax and Biovest (filed as Exhibit 10.17 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.66 | Indemnification Agreement, dated as of April 25, 2006, from Biovest to Dennis L. Ryll (filed as Exhibit 10.18 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.67 | Indemnification Agreement, dated as of April 25, 2006, from Biovest to Steven J. Stogel (filed as Exhibit 10.19 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.68 | Indemnification Agreement, dated as of April 25, 2006, from Biovest to Donald L. Ferguson (filed as Exhibit 10.20 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.69 | Indemnification Agreement, dated as of April 25, 2006, from Biovest to Ronald E. Osman (filed as Exhibit 10.21 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.70 | Indemnification Agreement, dated as of April 25, 2006, from Biovest to Francis E. O’Donnell, Jr. (filed as Exhibit 10.22 to Accentia’s Form 8-K filed on May 2, 2006 and incorporated herein by reference). | |
| 10.71 | Note and Warrant Purchase Agreement, dated March 31, 2006, between Biovest and Laurus (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on April 6, 2006 and incorporated herein by reference). | |
| 10.72 | Secured Promissory Note, dated March 31, 2006, issued by Biovest to Laurus (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on April 6, 2006 and incorporated herein by reference). | |
| 10.73 | Restricted Account Agreement, dated March 31, 2006, among Biovest, Laurus, and North Fork Bank (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on April 6, 2006 and incorporated herein by reference). | |
| 10.74 | Restricted Account Letter Agreement, dated March 31, 2006, between Biovest and Laurus (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on April 6, 2006 and incorporated herein by reference). | |
| 10.75 | Stock Pledge Agreement, dated March 31, 2006, between Laurus and Accentia (filed as Exhibit 10.8 to Accentia’s Form 8-K filed on April 6, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.76 | Securities Purchase Agreement, dated May 15, 2006, among Accentia and the parties identified as “Buyers” therein (“Securities Purchase Agreement”) (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on May 19, 2006 and incorporated herein by reference). | |
| 10.77 | Form of Common Stock Purchase Warrant, dated May 15, 2006, issued by Accentia pursuant to Securities Purchase Agreement (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on May 19, 2006 and incorporated herein by reference). | |
| 10.78 | Registration Rights Agreement, dated May 15, 2006, among Accentia and Buyers under Securities Purchase Agreement (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on May 19, 2006 and incorporated herein by reference). | |
| 10.79 | Overadvance Letter Agreement, dated July 13, 2006, among Laurus, Accentia, The Analytica Group, Inc., and TEAMM (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on July 19, 2006 and incorporated herein by reference). | |
| 10.80 | Amendment to Option Agreement, dated July 20, 2006, between Accentia and MAYO together with Form of Common Stock Purchase Warrant (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on July 24, 2006 and incorporated herein by reference). | |
| 10.81 | Amendment and Consent to Release between Biovest and Laurus dated August 2, 2006 (filed as Exhibit 10.8 to Accentia’s Form 10-Q filed on August 14, 2006 and incorporated herein by reference). | |
| 10.82(b) | Second Amendment to License Agreement, dated August 22, 2006, between Accentia and MAYO (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on August 28, 2006 and incorporated herein by reference). | |
| 10.83 | Common Stock Purchase Warrant dated August 22, 2006, between Accentia and MAYO (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on August 28, 2006 and incorporated herein by reference). | |
| 10.84 | Side Letter dated August 22, 2006 between Accentia and MAYO (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on August 28, 2006 and incorporated herein by reference). | |
| 10.85 | Securities Purchase Agreement, dated as of September 29, 2006, among Accentia and each of the purchasers named therein (including form of Common Stock Purchase Warrant and form of Secured Convertible Debenture issued thereunder) (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on October 2, 2006 and incorporated herein by reference). | |
| 10.86 | Registration Rights Agreement, dated as of September 29, 2006, among Accentia and each of the purchasers of Secured Convertible Debentures (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on October 2, 2006 and incorporated herein by reference). | |
| 10.87 | Registration Rights Agreement, dated as of September 29, 2006, among Biovest, and each of the purchasers of Secured Convertible Debentures (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on October 2, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.88 | Pledge Agreement, dated as of September 29, 2006, among Accentia, each of the purchasers of Secured Convertible Debentures, and American Stock Transfer & Trust Company (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on October 2, 2006 and incorporated herein by reference). | |
| 10.89 | Royalty Agreement between Accentia and Biovest dated October 31, 2006 (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
| 10.90 | Termination Agreement between Accentia and Biovest dated October 31, 2006 (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
| 10.91 | Purchase Agreement of Biolender between Accentia and Biovest dated October 31, 2006 (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
| 10.92 | Consent between Accentia and Laurus dated October 31, 2006 (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
| 10.93 | Common Stock Purchase Warrant, dated October 31, 2006 from Accentia to Laurus (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
| 10.94 | License and Asset Purchase Agreement, dated as of December 8, 2006, between Biovest and AutovaxID, Inc. (“AutovaxID”) (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.95 | License Agreement, dated as of December 8, 2006, between Biovest and AutovaxID (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.96 | Secured Promissory Note, dated as of December 8, 2006, made by Biovest for the benefit of Accentia (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.97 | Loan and Security Agreement, dated as of December 8, 2006, between AutovaxID Investment LLC (“AutovaxID Leverage Fund”) and Biolender II (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.98 | Promissory Note, dated December 8, 2006, issued by AutovaxID Leverage Fund to Biolender II, LLC (“Biolender II”) (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.99 | Subordinated Promissory Note, dated December 8, 2006, from AutovaxID to the St. Louis New Markets Tax Credit Fund-II, LLC (“St. Louis CDE”) (filed as Exhibit 10.6 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.100 | QLICI Loan Agreement dated as of December 8, 2006, by and among AutovaxID, the St. Louis CDE and Biovest (filed as Exhibit 10.7 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.101 | Subordination Agreement dated as of December 8, 2006, by and among Laurus, St. Louis CDE, US Bancorp Community Investment Corporation (“USBCIC”), AutovaxID and Biovest (filed as Exhibit 10.8 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.102 | Second Lien Security Agreement, dated December 8, 2006, from AutovaxID to the CDE (filed as Exhibit 10.9 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.103 | Tax Credit Reimbursement and Indemnity Agreement, dated as of December 8, 2006, between AutovaxID and USBCIC (filed as Exhibit 10.10 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.104 | Guaranty, dated December 8, 2006, made by Hopkins II, Frances E. O’Donnell, Jr., Kathleen M. O’Donnell (as Trustee of the Frances E. O’Donnell, Jr. Irrevocable Trust), Dennis L. Ryll, Ronald E. Osman, Alan M. Pearce, Steven R. Arikian, Steven J. Stogel, Donald L. Ferguson, Donald L. Ferguson (as trustee of the Donald L. Ferguson Revocable Trust) and Biovest in favor of USBCIC and the St. Louis CDE (filed as Exhibit 10.11 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.105 | Limited Liability Company Agreement of Biolender II, dated December 8, 2006 (filed as Exhibit 10.12 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.106 | Put Option Agreement dated December 8, 2006, between AutovaxID, AutovaxID Leverage Fund, USBCIC and Biolender II (filed as Exhibit 10.13 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.107 | Purchase Option Agreement dated December 8, 2006, between AutovaxID, AutovaxID Leverage Fund, USBCIC and Biolender II (filed as Exhibit 10.14 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.108 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Dennis L. Ryll (filed as Exhibit 10.15 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.109 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Steven J. Stogel (filed as Exhibit 10.16 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.110 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Donald L. Ferguson (filed as Exhibit 10.17 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.111 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Ronald E. Osman (filed as Exhibit 10.18 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.112 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Francis E. O’Donnell, Jr. (filed as Exhibit 10.19 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.113 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Alan M. Pearce (filed as Exhibit 10.20 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.114 | Indemnification Agreement, dated as of December 8, 2006, from Biovest to Steven R. Arikian (filed as Exhibit 10.21 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.115 | Subscription Agreement, dated December 8, 2006, between Biovest and St. Louis CDE (filed as Exhibit 10.27 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.116 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Dennis L. Ryll (filed as Exhibit 10.28 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.117 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Steven J. Stogel (filed as Exhibit 10.29 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.118 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Donald L. Ferguson (filed as Exhibit 10.30 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.119 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Ronald E. Osman (filed as Exhibit 10.31 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.120 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Hopkins II (filed as Exhibit 10.32 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.121 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Alan M. Pearce (filed as Exhibit 10.33 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.122 | Common Stock Purchase Warrant dated December 14, 2006 issued by Biovest to Steven R. Arikian (filed as Exhibit 10.34 to Accentia’s Form 8-K filed on December 14, 2006 and incorporated herein by reference). | |
| 10.123 | Notice to Exercise of Option to Terminate Services dated December 15, 2006 from Accentia to Pharmaco Investments, Inc. and Pharmaceutical Product Development, Inc. (filed in Accentia’s Form 10-K filed on December 29, 2006 and incorporated herein by reference). | |
Table of Contents
| 10.124 | Securities Purchase Agreement, dated as of February 28, 2007, among Accentia and each of the purchasers named therein (including exhibits) (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on February 28, 2007 and incorporated herein by reference). | |
| 10.125 | Sublicense Agreement between Accentia and Revimmune, LLC (“Revimmune”) dated February 27, 2007 (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on February 28, 2007 and incorporated herein by reference). | |
| 10.126 | Amendment to Option Agreement dated May 9, 2007 between Accentia and MAYO (filed as Exhibit 10.11 to Accentia’s Form 8-K filed on May 14, 2007 and incorporated herein by reference). | |
| 10.127 | Third Amendment to the License Agreement dated May 10, 2007 between Accentia and MAYO (filed as Exhibit 10.1 to Accentia’s Form 10-Q filed on May 15, 2007 and incorporated herein by reference). | |
| 10.128 | Letter of Understanding between Accentia and Respirics, Inc. dated May 10, 2007 filed as Exhibit 10.2 to Accentia’s Form 10-Q filed on May 15, 2007 and incorporated herein by reference). | |
| 10.129 | Form of Common Stock Purchase Warrant (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on June 19, 2007 and incorporated herein by reference). | |
| 10.130 | Termination of Biologics Agreement dated August 22, 2007 between Accentia and McKesson (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on August 24, 2007 and incorporated herein by reference). | |
| 10.131 | Overadvance Side Letter dated August 29, 2007 between Accentia and Laurus (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on September 5, 2007 and incorporated herein by reference). | |
| 10.132 | Form of the Amendment to Conversion Agreement between Accentia and Midsummer Investment, Ltd. (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on September 5, 2007 and incorporated herein by reference). | |
| 10.133 | Form of Common Stock Purchase Warrant to Debenture Investors (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on October 25, 2007 and incorporated herein by reference). | |
| 10.134 | Amendment No. 1 to Overadvance Side Letter dated as of October 21, 2007 between Accentia and Laurus (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on November 2, 2007 and incorporated herein by reference). | |
| 10.135 | Common Stock Purchase Warrant issued to Laurus (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on November 2, 2007 and incorporated herein by reference). | |
| 10.136 | Subordination Agreement dated October 31, 2007 between Accentia and LV Administrative Services, Inc. as agent to Valens U.S. SPV I, LLC and Valens Offshore SPV II, Corp. (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on November 2, 2007 and incorporated herein by reference). | |
Table of Contents
| 10.137 | Subordination Agreement dated December 10, 2007 between Accentia and LV Administrative Services, Inc. as agent to Valens U.S. SPV I, LLC and Valens Offshore SPV II, Corp. (filed in Accentia’s Form 10-K filed on December 28, 2007 and incorporated herein by reference). | |
| 10.138 | Accentia’s 2008 Equity Incentive Plan effective as of December 31, 2007 (filed in Accentia’s Form 10-K filed on December 28, 2007 and incorporated herein by reference). | |
| 10.139 | Extension Letter dated December 20, 2007 from Southwest Bank to Accentia (filed in Accentia’s Form 10-K filed on December 28, 2007 and incorporated herein by reference). | |
| 10.140 | Securities Purchase Agreement, dated as of January 18, 2008, among Accentia and each of the purchasers named therein (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on January 22, 2008 and incorporated herein by reference). | |
| 10.141 | Registration Rights Agreement, dated as of January 18, 2008, among Accentia and each of the purchasers of Preferred Stock (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on January 22, 2008 and incorporated herein by reference). | |
| 10.142 | Form of Short-Term Common Stock Purchase Warrant (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on January 22, 2008 and incorporated herein by reference). | |
| 10.143 | Form of Long-Term Common Stock Purchase Warrant (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on January 22, 2008 and incorporated herein by reference). | |
| 10.144 | Amendment No. 2 to Overadvance Letter effective January 31, 2008 between Accentia, Analytica International, Inc. (“Analytica”), Teamm, and Laurus (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on February 6, 2008 and incorporated herein by reference). | |
| 10.145 | Letter Amendment to Accentia Warrants effective January 31, 2008 between Accentia and Laurus (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on February 6, 2008 and incorporated herein by reference). | |
| 10.146 | Amendment and clarification of Accentia Royalty Agreement dated February 5, 2008 between Accentia and Biovest (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on February 6, 2008 and incorporated herein by reference). | |
| 10.147 | Conversion Agreement dated February 5, 2008 between Accentia and Biovest (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on February 6, 2008 and incorporated herein by reference). | |
| 10.148 | Revolving Credit Note Modification Agreement dated as of March 31, 2008 between Accentia and Southwest Bank (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on April 4, 2008 and incorporated herein by reference). | |
| 10.149 | Amendment Letter dated May 30, 2008 between Biovest, Valens U.S. SPV I, LLC and Valens Offshore SPV II, Corp. (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on June 5, 2008 and incorporated herein by reference). | |
Table of Contents
| 10.150 | December 10, 2007 Royalty Agreement Amendment dated May 30, 2008 between Biovest and Laurus, Valens U.S. SPV I, LLC, Valens U.S. SPV I, Ltd., Valens Offshore SPV II, Corp., and PSource Structured Debt Limited (“PSource”) (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on June 5, 2008 and incorporated herein by reference). | |
| 10.151 | Reaffirmation and Ratification Agreement dated May 30, 2008 between Biovest and its subsidiaries, Revimmune, LV Administrative Services, Inc., Valens U.S. SPV I, LLC, and Valens Offshore SPV II, Corp. (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on June 5, 2008 and incorporated herein by reference). | |
| 10.152 | Guaranty Side Letter dated May 30, 2008 between Accentia, Laurus, Valens U.S. SPV I, LLC, Valens U.S. SPV I, Ltd., Valens Offshore SPV II, Corp., Valens Offshore SPV II, LLC and PSource (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on June 5, 2008 and incorporated herein by reference). | |
| 10.153 | Securities Purchase Agreement, dated June 17, 2008, among Accentia and each of the purchasers of the Private Placement (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.154 | Registration Rights Agreement, dated June 17, 2008, among Accentia and each of the purchasers of the Private Placement (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.155 | 8% Original Issue Discount Secured Convertible Debenture, dated June 17, 2008, among Accentia and each of the purchasers of the Private Placement (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.156 | Security Agreement, dated June 17, 2008, among Accentia and its subsidiaries, Accentia Specialty Pharmacy, Inc. (“ASP”) and AccentRX and each of the purchasers of the Private Placement (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.157 | Common Stock Purchase Warrant dated June 17, 2008 (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.158 | Subsidiary Guarantee dated June 17, 2008 among ASP and AccentRx and each of the purchasers of the Private Placement (filed as Exhibit 10.6 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.159 | Third Amendment to Revolving Credit Agreement dated June 17, 2008, among Southwest Bank and Accentia (filed as Exhibit 10.7 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.160 | Revolving Credit Note Modification Agreement dated June 17, 2008, among Southwest Bank and Accentia (filed as Exhibit 10.8 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.161 | Stock Pledge Agreement dated June 17, 2008, among Southwest Bank and Accentia (filed as Exhibit 10.9 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
Table of Contents
| 10.162 | Reaffirmation of Guaranty dated June 17, 2008, among Southwest Bank and Accentia (filed as Exhibit 10.10 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.163 | Amendment No. 1 to Termination Agreement Re Biologics Distribution Agreement dated June 17, 2008, among McKesson and Accentia (filed as Exhibit 10.11 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.164 | Stock Pledge Agreement dated June 17, 2008, among McKesson and Accentia (filed as Exhibit 10.12 to Accentia’s Form 8-K filed on June 18, 2008 and incorporated herein by reference). | |
| 10.165 | Payoff Letter and Agreement dated June 18, 2008, between Accentia, Analytica, TEAMM, Laurus, Valens Offshore SPV I, Ltd, Valens US SPV I, LLC and PSource (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.166 | Assignment of Sale Proceeds dated June 18, 2008, between Accentia, Analytica, Valens Offshore SPV I, Ltd, Valens US SPV I, LLC and PSource (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.167 | Assignment of Rights Under Royalty Agreement dated June 18, 2008, between Accentia, Biovest and Erato Corp. (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.168 | Assignment of Rights Under Royalty Agreement dated June 18, 2008, between Accentia, Biovest and Valens Offshore SPV I, Ltd. (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.169 | Assignment of Rights Under Royalty Agreement dated June 18, 2008, between Accentia, Biovest and Valens US SPV I, LLC (filed as Exhibit 10.5 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.170 | Reaffirmation and Ratification Agreement dated June 18, 2008, between Accentia, Analytica, TEAMM, Laurus, Valens Offshore SPV I, Ltd., Valens US SPV I, LLC and PSource (filed as Exhibit 10.6 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.171 | Collateral Assignment dated June 18, 2008 between Accentia and Biovest (filed as Exhibit 10.7 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.172 | Schedules to the Security Purchase Agreement dated June 17, 2008 which was filed as Exhibit 10.1 to Accentia’s Form 8-K dated June 18, 2008 (filed as Exhibit 10.8 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.173 | Schedules to the Security Agreement dated June 17, 2008 which was filed as Exhibit 10.4 to Accentia’s Form 8-K dated June 18, 2008 (filed as Exhibit 10.9 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
| 10.174 | Amendment to Sublicense dated June 16, 2008 between Accentia and Revimmune (filed as Exhibit 10.10 to Accentia’s Form 8-K filed on June 20, 2008 and incorporated herein by reference). | |
Table of Contents
| 10.175 | Promissory Note dated as of June 30, 2008, between Biovest and Pulaski Bank (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on August 1, 2008 and incorporated herein by reference). | |
| 10.176 | Promissory Note dated as of June 30, 2008, between Biovest and Pulaski Bank (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on August 1, 2008 and incorporated herein by reference). | |
| 10.177 | Amendment Agreement dated July 31, 2008 between Biovest and Laurus, Valens Offshore SPV I, Ltd., Valens Offshore SPV II, Corp., Valens U.S. SPV I, LLC and PSource (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on August 1, 2008 and incorporated herein by reference). | |
| 10.178 | Reaffirmation and Ratification Agreement dated July 31, 2008 between Biovest, Biovax, AutovaxID, Biolender, Biolender II, Accentia, Revimmune, and LV Administrative Services, Inc., as Agent, Laurus, Valens U.S. SPV I, LLC, Valens Offshore SPV I, Ltd., Valens Offshore SPV II, Corp., PSource (filed as Exhibit 10.4 to Accentia’s Form 8-K filed on August 1, 2008 and incorporated herein by reference). | |
| 10.179 | Emezine Settlement Agreement between Accentia, TEAMM, BDSI, and Arius dated December 30, 2009 (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on February 22, 2010 and incorporated herein by reference). | |
| 10.180 | Order Confirming First Amended Joint Plan of Reorganization of Accentia Biopharmaceuticals, Inc., Analytica International, Inc., TEAMM Pharmaceuticals, Inc., AccentRx, Inc., and Accentia Specialty Pharmacy, Inc. Under Chapter 11 of Title 11, United States Code dated as of August 16, 2010, as Modified, Pursuant to 11 U.S.C. §1129 (filed as Exhibit 10.3 to Accentia’s Form 8-K filed on November 2, 2010 and incorporated herein by reference). | |
| 10.181 | Notice of Effectiveness Dated November 17, 2010 (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on November 23, 2010 and incorporated herein by reference). | |
| 10.182(b) | Agreement between Accentia Biopharmaceuticals, Inc. and Baxter Healthcare Corporation (filed as Exhibit 10.1 to Accentia’s Form 8-K filed on December 3, 2010 and incorporated herein by reference). | |
| 10.183 | Addendum to Agreement between Accentia Biopharmaceuticals, Inc. and Baxter Healthcare Corporation dated November 29, 2010 (filed as Exhibit 10.2 to Accentia’s Form 8-K filed on December 3, 2010 and incorporated herein by reference). | |
| 10.184 | Accentia’s 2010 Equity Incentive Plan (filed as Exhibit 10.182 to Accentia’s Form 10-K filed on December 14, 2010 and incorporated herein by reference). | |
| 10.185 | Term Loan and Security Agreement, dated November 17, 2010, among LV Administrative Services, Inc., the Lenders and Accentia (filed as Exhibit 10.2 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
Table of Contents
| 10.186 | Secured Term Note, dated November 17, 2010, between Accentia and Erato Corp. (filed as Exhibit 10.3 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.187 | Secured Term Note, dated November 17, 2010, between Accentia and PSource Structured Debt Limited (filed as Exhibit 10.4 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.188 | Secured Term Note, dated November 17, 2010, between Accentia and Valens Offshore SPV II, Corp. (filed as Exhibit 10.5 to Accentia’s Form 10-Q filed on February 10, 2011 and incorporated herein by reference). | |
| 10.189 | Secured Term Note, dated November 17, 2010, between Accentia and Valens U.S. SPV I, LLC (filed as Exhibit 10.6 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.190 | Warrant Termination Agreement, dated November 17, 2010, between Accentia and Laurus Master Fund, Ltd. (in liquidation) (filed as Exhibit 10.7 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.191 | Warrant Termination Agreement, dated November 17, 2010, between Accentia and Valens Offshore SPV I, Ltd. (filed as Exhibit 10.8 to Accentia’s Form 10-Q filed on February 10, 2011 and incorporated herein by reference). | |
| 10.192 | Warrant Termination Agreement, dated November 17, 2010, between Accentia and Valens U.S. SPV I, LLC (filed as Exhibit 10.9 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.193 | Guaranty, dated November 17, 2010, among LV Administrative Services, Inc., the Lenders and Analytica International, Inc. (filed as Exhibit 10.10 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.194 | Stock Pledge Agreement (Biovest Common Stock), dated November 17, 2010, between Accentia and LV Administrative Services, Inc. (filed as Exhibit 10.11 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.195 | Stock Pledge Agreement (Analytica Common Stock), dated November 17, 2010, between Accentia and LV Administrative Services, Inc. (filed as Exhibit 10.12 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.196 | Security Agreement, dated November 17, 2010, between Analytica and LV Administrative Services, Inc. (filed as Exhibit 10.13 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.197 | Grant of Security Interest in Intellectual Property Agreement, dated November 17, 2010, between Analytica and LV Administrative Services, Inc. (filed as Exhibit 10.14 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
Table of Contents
| 10.198 | Grant of Security Interest in Intellectual Property Agreement, dated November 17, 2010, between Accentia and LV Administrative Services, Inc. (filed as Exhibit 10.15 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.199 | Form of 8.5% Secured Convertible Debenture due May 17, 2012, dated November 17, 2010 (Class 5) (filed as Exhibit 10.16 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.200 | Form of Common Stock Purchase Warrant (Class 5), dated November 17, 2010 (filed as Exhibit 10.17 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.201 | Form of Pledge Agreement, dated November 17, 2010 (filed as Exhibit 10.18 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.202 | Form of Subsidiary Guaranty, dated November 17, 2010 (filed as Exhibit 10.19 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.203 | Form of 8.5% Secured Convertible Debenture Due November 17, 2012, dated November 17, 2010 (Class 6) (filed as Exhibit 10.20 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.204 | Form of Common Stock Purchase Warrant (Class 6), dated November 17, 2010 (filed as Exhibit 10.21 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.205 | Form of Security Agreement (Class 6), dated November 17, 2010 (filed as Exhibit 10.22 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.206 | Form of Subsidiary Guarantee (Class 6), dated November 17, 2010 (filed as Exhibit 10.23 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.207 | Form of Convertible Debenture Due November 17, 2012, dated November 17, 2010 (Class 9) (filed as Exhibit 10.24 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.208 | Form of Common Stock Purchase Warrant (Class 6), dated November 17, 2010 (filed as Exhibit 10.25 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.209 | Form of Class 13 Plan Convertible Note, dated November 17, 2010 (filed as Exhibit 10.26 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.210 | Form of Common Stock Purchase Warrant (Class 13), dated November 17, 2010 (filed as Exhibit 10.27 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
| 10.211 | Resignation Settlement, dated December 31, 2010, between Accentia and Alan M. Pearce (filed as Exhibit 10.28 to Accentia’s Form 10-Q filed on February 11, 2011 and incorporated herein by reference). | |
Table of Contents
| 10.212 | Lease Between Biovest International, Inc. and JMS Holdings, LLC dated December 2, 2010. | |
| 21.1 | Subsidiaries of Accentia (filed as Exhibit 21 to the Registration Statement on Form S-1 filed on February 11, 2005 (Registration No. 333-122769) and incorporated herein by reference). | |
| 23.1 | Consent of Cherry, Bekaert & Holland, L.L.P. | |
| 23.2* | Consent of Foley & Lardner LLP (contained in Exhibit 5.1). | |
| 24.1 | Power of Attorney (included on signature pages hereto). | |
* To be filed by amendment.
(a) Indicates management contract or compensatory plan.
(b) Portions of this exhibit have been omitted pursuant to a confidential treatment request. Omitted information has been filed separately with the Securities and Exchange Commission.