Attached files
| file | filename |
|---|---|
| EX-31.1 - CERTIFICATION - NOVADEL PHARMA INC | exhibit31-1.htm |
| EX-23.1 - CONSENT - NOVADEL PHARMA INC | exhibit23-1.htm |
| EX-32.1 - CERTIFICATION - NOVADEL PHARMA INC | exhibit32-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
[ X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the year ended December 31, 2009
OR
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE ACT OF 1934
COMMISSION FILE NO. 001-32177
NOVADEL PHARMA INC.
(Exact Name of registrant as specified in its charter)
|
Delaware
|
22-2407152
|
|||
|
(State or other jurisdiction
of incorporation or organization)
|
(I.R.S. Employer
Identification No.)
|
1200 ROUTE 22 EAST, SUITE 2000, BRIDGEWATER, NEW JERSEY 08807
(Address of principal executive offices) (Zip Code)
(908) 203-4640
Registrant’s telephone number, including area code
Securities registered pursuant to Section 12(b) of the Exchange Act:
|
Title of each class
|
Name of each exchange on which registered
|
|
Common Stock, par value $0.001 per share
|
OTCBB
|
Securities registered pursuant to Section 12(g) of
the Exchange Act:
None
Indicate by check mark if the registrant is a well-know seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes o No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer o
|
Accelerated filer o
|
|
Non-accelerated filer ¨ (Do not check if a smaller reporting company)
|
Smaller reporting company x
|
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
As of June 30, 2009, the aggregate market value of the voting and non-voting common equity of the issuer held by non-affiliates of the registrant was approximately $17 million based upon the closing sale price of $0.31 for the Registrant’s common stock, $0.001 par value, as reported by the NYSE Amex LLC, formerly known as the American Stock Exchange on that date. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
As of March 24, 2010, the issuer had 89,283,000 shares of common stock, $0.001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the definitive proxy statement to be filed pursuant to Regulation 14A within 120 days of the end of the fiscal year (December 31, 2009) are incorporated by reference into Part III of this Annual Report on Form 10-K.
NOVADEL PHARMA INC.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2009
|
PART I
|
|
|
PART II
|
|
|
PART III
|
|
|
PART IV
|
|
Unless the context otherwise requires, all references to “we,” “us,” “our,” and the “Company” include NovaDel Pharma Inc. (NovaDel).
SAFE HARBOR STATEMENTS UNDER THE PRIVATE SECURITIES LITIGATION REFORM ACT OF 1995
This Annual Report on Form 10-K includes “forward-looking statements,” including statements regarding NovaDel Pharma Inc.’s (the “Company,” “we,” “us” or “NovaDel”) expectations, beliefs, intentions or strategies for the future and the Company’s internal controls and procedures and outstanding financial reporting obligations and other accounting issues. The Company intends that all forward-looking statements be subject to the safe-harbor provisions of the Private Securities Litigation Reform Act of 1995. These forward-looking statements are only predictions and reflect the Company’s views as of the date they are made with respect to future events and financial performance. In particular, the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section in Part II, Item 7 of this Annual Report includes forward-looking statements that reflect the Company’s current views with respect to future events and financial performance. The Company uses words such as “expect,” “anticipate,” “believe,” “intend” and similar expressions to identify forward-looking statements. You can also identify forward-looking statements by the fact that they do not relate strictly to historical or current facts. A number of important risks and uncertainties could, individually or in the aggregate, cause actual results to differ materially from those expressed or implied in any forward-looking statements.
Examples of the risks and uncertainties include, but are not limited to: the inherent risks and uncertainties in developing products of the type the Company is developing (independently and through collaborative arrangements); the inherent risks and uncertainties in completing the pilot pharmacokinetic feasibility studies being conducted by the Company; possible changes in the Company’s financial condition; the progress of the Company’s research and development; clinical trials require adequate supplies of drug substance and drug product, which may be difficult or uneconomical to procure or manufacture; timely obtaining sufficient patient enrollment in the Company’s clinical trials; the impact of development of competing therapies and/or technologies by other companies; the Company’s ability to obtain additional required financing to fund its research programs; the Company’s ability to enter into agreements with collaborators and the failure of collaborators to perform under their agreements with the Company; the progress of the U.S. Food and Drug Administration, or FDA, approvals in connection with the conduct of the Company’s clinical trials and the marketing of the Company’s products; the additional costs and delays which may result from requirements imposed by the FDA in connection with obtaining the required approvals; acceptance for filing by the FDA does not mean that the New Drug Application, or NDA, has been or will be approved, nor does it represent an evaluation of the adequacy of the data submitted; the risks related to the Company’s internal controls and procedures; and the risks identified under the section entitled “Risk Factors” included as Item 1A in Part I of this Annual Report on Form 10-K and other reports, including this report and other filings filed with the Securities and Exchange Commission from time to time.
PART I
GENERAL
NovaDel Pharma Inc., a Delaware corporation, referred to herein as “we”, “us” and “our”, is a specialty pharmaceutical company developing oral spray formulations for a broad range of marketed pharmaceuticals. Our proprietary technology offers, in comparison to conventional oral dosage forms, the potential for faster absorption of drugs into the bloodstream leading to quicker onset of therapeutic effects and possibly lower doses. Oral sprays eliminate the requirement for water or the need to swallow, potentially improving patient convenience and compliance. Our oral spray technology is focused on addressing unmet medical needs for a broad array of existing and future pharmaceutical products. Our most advanced oral spray candidates target angina, insomnia, erectile dysfunction, migraine headaches, nausea and disorders of the central nervous system. We plan to develop these and other products independently and through collaborative arrangements with pharmaceutical and biotechnology companies. Currently, we have nine patents which have been issued in the U.S. and 69 patents which have been issued outside of the U.S. Additionally, we have over 65 patents pending around the world. We look for drug compounds that are off patent or are coming off patent in the near future, and we formulate these compounds in conjunction with our proprietary drug delivery method. Once formulated, we file for new patent applications on these formulated compounds that comprise our product candidates. Our patent portfolio includes patents and patent applications with claims directed to the pharmaceutical formulations, methods of use and methods of manufacturing for our product candidates.
Our goal is to become a leading specialty pharmaceutical company that develops and commercializes improved formulations of existing drugs using our patented oral spray technology. We believe that our technology has application to a broad number of therapeutic areas and product categories. Our strategy is to concentrate our product development activities primarily on pharmaceutical products which meet the following characteristics:
|
|
·
|
Significant prescription sales already exist;
|
|
|
·
|
Our proprietary novel drug delivery technology enhances the performance of the active ingredient of the target compound, potentially addressing unmet patient needs; and
|
|
|
·
|
Applicability of an efficient regulatory pathway to approval using the 505(b)(2) pathway.
|
In today’s environment of escalating drug development costs and time to market, we believe that the ability to bring products with some degree of differentiation and competitive advantage to the marketplace in a timely and cost-effective manner is a viable strategy.
Since inception, substantially all of our revenues have been derived from consulting activities, primarily in connection with product development for various pharmaceutical companies. More recently, we have begun to derive revenues from license fees and milestone payments stemming from our partnership agreements. Our future growth and profitability will be principally dependent upon our ability to successfully develop our product candidates and to market and distribute the final products either internally or with the assistance of strategic partners.
We have a history of recurring losses, giving rise to an accumulated deficit as of December 31, 2009 of $82,766,000, as compared to $74,829,000 as of December 31, 2008. Additionally, we have had negative cash flow from operating activities of $1,578,000 for the year ended December 31, 2009, $5,533,000 for the year ended December 31, 2008, and $15,240,000 for the year ended December 31, 2007. As of December 31, 2009, we had negative working capital of $495,000 as compared to $47,000 as of December 31, 2008, representing a net decrease in working capital of approximately $542,000.
Throughout 2009, our reduced clinical development activities were limited to expenditures required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation development activities for certain other products. We will seek to raise additional capital in 2010 to fund our operations and future development activities through new strategic partnerships or collaborations, the sale of common stock or other equity securities or the issuance of debt. In the event we do not enter into a license agreement or other strategic transaction in which we receive an upfront fee or payment, or we do not undertake a financing of debt or equity securities, we may not have sufficient cash on hand to fund operations. We can give no assurances that we will be able to enter into a strategic transaction or raise any additional capital or, if we do, that such additional capital will be sufficient to meet our needs, or on terms favorable to us. If we are unable to raise additional capital, and we do not use our existing working capital to fund our development plans, we will have sufficient cash on hand to fund operating costs through the fourth quarter 2010. We will need additional financing thereafter until we achieve profitability.
Our audited financial statements for the year ended December 31, 2009 were prepared under the assumption that we will continue our operations as a going concern. We were incorporated in 1982, and have a history of losses. As a result, our independent registered public accounting firm in their audit report has expressed substantial doubt about our ability to continue as a going concern. We believe that the cash inflows that have been generated from our financing transactions and our licensing transactions and any additional potential cash inflows that may be received during 2010 and 2011will improve our ability to continue our operations as a going concern. Continued operations are dependent on our ability to complete equity or debt formation activities or to generate profitable operations. Such capital formation activities may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty.
As previously disclosed on May 14, 2008, we received notice from the NYSE Amex LLC (formally known as the American Stock Exchange) indicating that we are not in compliance with certain of the NYSE Amex LLC continued listing standards. We submitted a plan of compliance with NYSE Amex LLC, which was accepted, and attempted to regain compliance with such plan during fiscal year 2009. On December 2, 2009, we formally notified NYSE Amex LLC of our intent to voluntarily withdraw our listing and registration due to our failure to regain compliance with the continued listing standards and our plan. On December 14, 2009, we filed a Form 25 voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009. On December 24, 2009, our common stock began trading on the Over-the-Counter Bulletin Board, or OTCBB, under its new ticker symbol on OTCBB as NVDL.OB.
Highlights for the year ended December 31, 2009, and additionally through the date of filing of this Annual Report on Form 10-K, include the following product development and business achievements:
Product Pipeline
|
·
|
After careful review of our portfolio of product opportunities we have selected Sildenafil Citrate (Viagra®) as our next product to develop. Our development plans anticipate that our oral spray formulation, Duromist™, will be available for launch in the first half of 2012.
|
Intellectual Property
|
·
|
Announced that we received a Notice of Allowance from the United States Patent and Trademark Office, or USPTO, for claims under U.S. Patent Application No. 10/671,715, entitled “Buccal, Polar and Non-polar Spray Containing Zolpidem,” which covers a method of treating insomnia by administering zolpidem to humans utilizing NovaMist™ Oral Spray technology. Once issued, this patent will expire in 2018.
|
|
·
|
Announced that we received an Issue Notification from the USPTO for a new U.S. Patent, No. 7632517, entitled “Buccal, Polar and Non-polar Spray Containing Zolpidem,” which covers a method of treating insomnia by administering zolpidem to humans utilizing NovaDel's oral spray technology.
|
Other
|
·
|
Announced that Michael E. Spicer resigned as Chief Financial Officer and Corporate Secretary, effective April 1, 2009. Our Board of Directors appointed Deni M. Zodda, Ph.D., our Chief Business Officer, to serve as Interim Chief Financial Officer, Principal Financial Officer and Corporate Secretary, effective April 1, 2009. We also hired Joseph M. Warusz as a consultant to serve as Principal Accounting Officer, effective April 1, 2009.
|
|
·
|
Announced that Deni M. Zodda, Ph.D., Chief Business Officer, Interim Chief Financial Officer and Corporate Secretary, agreed to leave the Company resulting from a reorganization of the executive team. Mr. Zodda has entered into a Separation, Consulting and General Release Agreement under which he received a one-time fee of $137,500 to provide us with certain consulting services through October 31, 2009. Steven B. Ratoff, our Chairman, President and Chief Executive Officer, has been appointed our Interim Chief Financial Officer.
|
|
·
|
Announced the Board of Directors appointed Steven B. Ratoff as President and Chief Executive Officer effective January 1, 2010. Mr. Ratoff will continue to serve as Interim Chief Financial Officer.
|
|
·
|
Announced we executed a lease amendment modifying certain terms to the lease for the property in Flemington, New Jersey. The amendment converted the lease term to month to month commencing on July 1, 2009 with a provision that either party may terminate the lease upon thirty days written notice. We have released the lease escrow of $226,000 to the landlord in order to satisfy rent payments through June 30, 2009. This lease was terminated in December 2009.
|
|
·
|
Effective February 1, 2010, we entered into a one (1) year lease agreement with Regus Management Group LLC for approximately 5,000 square feet of office space in Bridgewater, New Jersey.
|
|
·
|
Announced that we entered into an agreement with Seaside 88, LP, or Seaside. Under the terms of the agreement and subject to the approval of the NYSE Amex LLC, Seaside has committed to purchase up to 13.0 million NovaDel common shares, in a series of closings every two weeks in the amount of 500,000 shares each for a total of up to 26 purchases. We had received approval from NYSE Amex LLC to issue up to 12.0 million shares over twelve (12) months. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP.
|
|
·
|
Announced we entered into an agreement with Arthur W. Wood Company, Inc., or AWW, pursuant to which AWW agreed to assist us as a non-exclusive financial advisor for the purposes of seeking capital related to the Seaside offering, referred to herein as the Placement. In consideration of AWW’s services, we agreed to pay AWW upon closing of a capital-raising transaction, a fee equal to three percent (3%) of the aggregate value of the proceeds paid or payable in the Placement.
|
|
·
|
Announced we received a milestone payment of approximately $150,000 from Velcera, Inc., or Velcera, relating to its License and Development Agreement with Velcera, dated June 22, 2004.
|
|
·
|
Announced we entered into a license and distribution agreement with privately-held Mist Acquisition, LLC to manufacture and commercialize the NitroMist™ lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris, in the United States, Canada and Mexico. Under the terms of the agreement, we received a $1,000,000 licensing fee upon execution of the agreement, and will receive milestone payments totaling an additional $1,000,000 over the next twelve months and ongoing performance payments of up to seventeen percent (17%) of net sales, subject to the terms of the agreement.
|
|
·
|
Announced we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ product in the United States and Canada. ZolpiMist™ is our oral spray formulation of zolpidem tartrate, which was approved by the FDA in December of 2008. Under the terms of the agreement, we received $3,000,000 upon the execution of the agreement and ECR will pay us ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products, subject to the terms of the agreement.
|
|
·
|
Announced that we notified NYSE Amex LLC of our intent to voluntarily delist our common stock from the Exchange. We filed Form 25 on December 14, 2009, voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009. Our common stock began trading on the OTCBB on December 24, 2009. Our new ticker symbol on OTCBB is NVDL.OB.
|
|
·
|
Announced we entered into an amendment agreement with ProQuest Investments L.P. and its affiliates, referred to herein as ProQuest, to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued but unpaid interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of our common stock as of December 31, 2009.
|
PRODUCT DEVELOPMENT
Drug development in the U.S. and most countries throughout the world is a process that includes several steps defined by the U.S. Food and Drug Administration, or FDA, or comparable regulatory authorities in foreign countries. The FDA approval processes relating to new drugs differ, depending on the nature of the particular drug for which approval is sought. With respect to any drug product with active ingredients not previously approved by the FDA, a prospective drug manufacturer is required to submit a New Drug Application, or NDA, which includes complete reports of pre-clinical, clinical and laboratory studies to prove such product’s safety and efficacy. Prior to submission of the NDA, it is necessary to submit an Investigational New Drug, or IND, to obtain permission to begin clinical testing of the new drug. Given that our current product candidates are based on a new technology for formulation and delivery of active pharmaceutical ingredients that have been previously approved and that have been shown to be safe and effective in previous clinical trials, we believe that we will be eligible to submit what is known as a 505(b)(2) NDA. We estimate that the development of new formulations of our pharmaceutical product candidates, including formulation, testing and submission of an NDA, will require significantly less time and lower investments in direct research and development expenditures than is the case for the discovery and development of new chemical entities. However, our estimates may prove to be inaccurate; or pre-marketing approval relating to our proposed products may not be obtained on a timely basis, if at all, and research and development expenditures may significantly exceed management’s expectations.
It is not anticipated that we will generate any revenues from royalties or sales of our product candidates until regulatory approvals are obtained and marketing activities begin. We anticipate generating revenues in 2010 from our existing licensed products, Zolpimist™ and NitroMist™. Any one or more of our product candidates may not prove to be commercially viable, or if viable, may not reach the marketplace on a basis consistent with our desired timetables, if at all. The failure or the delay of any one or more of our proposed products to achieve commercial viability would have a material adverse effect on us.
The successful development of our product candidates is highly uncertain. Estimates of the nature, timing and estimated expenses of the efforts necessary to complete the development of, and the period in which material net cash inflows are expected to commence from, any of our product candidates are subject to numerous risks and uncertainties, including:
|
|
·
|
the scope, rate of progress and expense of our clinical trials and other research and development activities;
|
|
|
·
|
results of future clinical trials;
|
|
|
·
|
the expense of clinical trials for additional indications;
|
|
|
·
|
the terms and timing of any collaborative, licensing and other arrangements that we may establish;
|
|
|
·
|
the expense and timing of regulatory approvals or changes in the regulatory approval process;
|
|
|
·
|
the expense of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
|
|
|
·
|
the effect of competing technologies and market developments; and
|
|
|
·
|
the expense of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights.
|
We expect to spend significant amounts on the development of certain of our product candidates and we expect our costs to increase if we restart certain programs to develop and ultimately commercialize our product candidates. The following table summarizes our product candidates:
|
Active Ingredient or Class of Molecule
|
Indications
|
Stage of Development
|
Partner
|
|
|
Approved Products
|
||||
|
NitroMist™
|
nitroglycerin
|
Angina Pectoris
|
FDA Approved
|
Mist Acquisition, LLC
|
|
Zolpimist™
|
zolpidem
|
Insomnia
|
FDA Approved
|
ECR Pharmaceuticals Company
|
|
Product Candidates
|
||||
|
Duromist™
|
sildenafil
|
Erectile Dysfunction
|
Preclinical development
|
-
|
|
Zensana™
|
ondansetron
|
Nausea/Vomiting
|
Clinical development
|
Hana Biosciences/Par Pharmaceutical, Inc./BioAlliance Pharma S.A.
|
|
NVD-201
|
sumatriptan
|
Migraines
|
Pilot Efficacy study complete
|
-
|
|
NVD-301
|
midazolam
|
Pre-Procedure Anxiety
|
Preclinical development
|
-
|
NitroMist™ (nitroglycerin lingual aerosol). This product is indicated for acute relief of an attack or acute prophylaxis of angina pectoris due to coronary artery disease, and was approved by the FDA in November 2006. Previously, this product was partnered with Par Pharmaceutical, Inc., or Par; however, on August 1, 2007, we announced that Par returned the rights to NitroMist™ to us as part of Par's strategy to concentrate its resources on supportive care in AIDS and oncology markets. Our former contract manufacturer for NitroMist™, INyX Pharma, filed for protection under the Chapter 11 bankruptcy laws in 2007, and ceased operations at its facility in Puerto Rico where our product was to be manufactured during 2008. As a result, we selected an alternative contract manufacturer, DPT Laboratories. On October 27, 2009, we entered into a licensing and distribution agreement with privately-held Mist Acquisition, LLC, or Mist, to manufacture and commercialize the NitroMist™ in the United States, Canada and Mexico. Under the terms of the agreement, Mist paid us a $1,000,000 licensing fee upon execution of the agreement, and will pay us milestone payments totaling an additional $1,000,000 over the next twelve months if certain milestones are met and ongoing performance payments of up to seventeen percent (17%) of net sales. In addition, Mist will assume the activities and costs necessary for the completion of the product transfer to DPT Laboratories.
Zolpimist™ (zolpidem oral spray). Zolpidem is the active ingredient in Ambien®, the leading hypnotic for insomnia marketed by Sanofi-Aventis. Our oral spray formulation of zolpidem was approved for the short-term treatment of insomnia by the FDA in December 2008. In October 2009, we received a Notice of Allowance from the United States Patent and Trademark Office, or USPTO for claims which cover a method of treating insomnia by administering zolpidem to humans utilizing NovaMist™ Oral Spray technology. On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ in the United States and Canada. Under the terms of the agreement, we received a $3,000,000 licensing fee and will receive ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products.
Duromist™ (Sildenafil oral spray). Duromist™ contains sildenafil, the leading PDE-5 inhibitor for the treatment of erectile dysfunction marketed under the brand name Viagra®. We believe that an oral spray of sildenafil has the potential of a faster onset of action and a lower dose compared to tablets.
Erectile dysfunction occurs in approximately 18% of the male population with prevalence of over 50% in men over 65 years of age. PDE-5 inhibitors are effective in approximately 75% of the erectile dysfunction population. Sildenafil is the most popular molecule with over 50% market share in a erectile dysfunction market of over $3 billion.
Development is in progress for a formulation to be used in future clinical trials to begin in 2010, with a development plan that would deliver a FDA approved product available for launch in the second quarter of 2012.
Zensana™ (ondansetron oral spray). Ondansetron is the active ingredient in Zofran®, the leading anti-emetic marketed by GlaxoSmithKline, or GSK. Through July 31, 2007, this product candidate was licensed to Hana Biosciences, who was overseeing all clinical development and regulatory approval activities for this product in the U.S. and Canada. On July 31, 2007, we entered into a Product Development and Commercialization Sublicense Agreement with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a sublicense to Par to develop and commercialize Zensana™. Par is responsible for all development, regulatory, manufacturing and commercialization activities of Zensana™ in the United States and Canada, including the development and re-filing of the NDA in the United States. In addition, we entered into an Amended and Restated License Agreement with Hana Biosciences, pursuant to which Hana Biosciences relinquished its right to pay reduced royalty rates to us until such time as Hana Biosciences had recovered one-half of its costs and expenses incurred in developing Zensana™ from sales of Zensana™ and we agreed to surrender for cancellation all 73,121 shares of the Hana Biosciences common stock we acquired in connection with execution of the original license agreement with Hana Biosciences. Par had previously announced that it expected to complete clinical development on the revised formulation of Zensana™ during 2008, and expected to submit a new NDA for Zensana™ by the end of 2008. However, in November 2008, Par announced that it had completed bioequivalency studies on Zensana™ with mixed results, with bioequivalence to reference drug (Zofran® tablets) achieved in some of the studies and not achieved in others. We have notified Hana and Par that under the terms of our agreement, they are required to return the product to us.
On May 19, 2008, we entered into an agreement with BioAlliance Pharma S.A., whereby BioAlliance acquired the European rights for our ondansetron oral spray. Under the terms of the agreement, BioAlliance paid us a license fee of $3,000,000 upon closing. We are eligible for additional milestone payments totaling approximately $24 million (an approval milestone of $5,000,000 and sales-related milestone payments of approximately $19 million) as well as a royalty on net sales. We anticipate that their development activities will not be initiated until development is completed in the United States.
Sumatriptan oral spray (NVD-201). Sumatriptan is the active ingredient in Imitrex® which is the largest selling migraine remedy marketed by GSK. A pilot PK study of NVD-201 with 9 healthy subjects, completed in the second half of calendar 2004, suggested that the formulation achieved plasma concentrations of sumatriptan in the therapeutic range. In September 2006 we announced positive results from an additional pilot pharmacokinetic study, with NVD-201 which demonstrated that NVD-201 achieves a statistically significant increase in absorption rate as compared with Imitrex® tablets. The rate of drug absorption is believed to be the most important predictor of the degree and speed of migraine relief. NVD-201 was evaluated in a four-arm, crossover pharmacokinetic study comparing 50mg Imitrex® tablets to 20mg and 30mg of the NVD-201 in 10 healthy male volunteers under fasting conditions. At least 90% of subjects receiving NVD-201 had detectable drug levels at three minutes post-dosing, while at the same timepoint, only 10% of subjects receiving 50mg Imitrex® tablets had detectable drug levels. These differences are statistically significant. At 3 to 6 minutes post dosing, all NVD-201 groups had statistically significantly higher mean concentration levels compared to 50mg Imitrex® tablets. Using published data for the currently marketed Imitrex® nasal spray as a proxy for therapeutic blood levels, we observed that by 6 minutes post-dosing, 100% of the 20mg NVD-201 users achieved these critical plasma concentration levels while none of the subjects from the Imitrex® tablet group did so by this timepoint. This result was also statistically significant. Furthermore, the study indicates up to a 50% increase in relative bioavailability of NVD-201 in comparison to the Imitrex® tablet. Additionally, the pharmacokinetics of 20mg NVD-201 after a meal were evaluated. NVD-201 was well tolerated.
While Imitrex® nasal spray was not included in this clinical study, the following represents a discussion of the results of our clinical study as compared to published data for Imitrex® nasal spray. Time to the first peak plasma concentration of sumatriptan -- which represents drug absorbed directly across the oral mucosa -- was approximately 70% faster with the 20mg NVD-201 than what has been reported in the literature for the same dose of the Imitrex® nasal spray (6 min. vs. 20 min.). The mean concentration level achieved during this critical first phase of absorption is approximately 30% greater for the NVD-201 than what was observed in published studies of the nasal spray (10.9 ng/mL vs. 8.5 ng/mL). Relative bioavailability after administration of 20mg NVD-201 appears to be greater than published estimates for the same dose of the Imitrex® nasal spray.
In September 2008, we announced the results from a pilot efficacy study for NVD-201. This was a multi-center, active control, open-label, dose-ranging, efficacy and safety study. Subjects received up to 5 treatments, comprising single doses of the following: Imigran® 50-mg tablets, Imigran® 100-mg tablets, NVD-201 20-mg, NVD-201 30-mg, and NVD-201 40-mg. Their response to Imigran® 50-mg tablets determined whether they were eligible to receive the other four treatments. Patients recorded the severity of each migraine attack on the same 4-point scale immediately before dosing and at 15, 30, 60, 90, 120, and 240 minutes, and at 24 hours post-dosing. Associated symptoms (nausea, vomiting, photophobia, and phonophobia) were also recorded immediately before dosing and at 30, 60, 90 and 120 minutes post-dosing. All dosing was done on an outpatient basis and patients returned to the clinic between migraine attacks.
In the primary analysis of efficacy, the percentage of patients responding to treatment at or before 60 minutes post-dosing, there was a statistically significant greater percentage of subjects receiving the 30- and 40-mg doses of NVD-201 with a reduction in headache pain compared to those receiving the 50-mg s Imigran® tablet (42% and 46%, respectively, vs 12%; P<0.011), and was comparable to the percentage who responded to the higher (100 mg) dose of the tablet formulation (42%). Significantly more patients had responded to all three doses of NVD-201 than to 50-mg Imigran® tablet by 90 minutes post-dosing (57% to 70.0% vs 32%; P<0.028) and all three oral spray doses were comparable to the 100-mg tablet. There were no treatment differences by 2 hours after dosing, when 68% to 77% of patients had responded irrespective of treatment.
Compared to 50-mg Imigran® tablet, at least one dose of NVD-201 also significantly increased percentage of patients who were pain free by 1 to 2 hours post-dosing, with the response ratio indicating significantly faster complete pain relief for the 40-mg dose, and significantly more patients had complete pain relief without use of rescue medication after receiving any dose of NVD-201. In addition, after one or more doses of NVD-201, the percentage of patients who were asymptomatic was significantly increased, and the percentages who experienced nausea, photophobia, or phonophobia were significantly decreased. NVD-201 was comparable to the 100-mg tablet on all the above measures.
We believe NVD-201 may provide clinical benefits to migraine sufferers including, possibly, faster relief than Imitrex® tablets as well as greater tolerability than triptan nasal sprays. Further, if proven to be safe and effective, we believe NVD-201 may be attractive to patients who have trouble taking oral medications due to nausea and vomiting caused by the migraine attack. Previously, we were targeting an NDA submission for our sumatriptan product candidate in the first half of calendar 2008; however, due to funding constraints and other higher priorities associated with our current product pipeline, we have not progressed our development efforts.
We will continue to evaluate this program when sufficient additional funding becomes available.
Midazolam oral spray (NVD-301). NVD-301 contains midazolam which is the leading benzodiazepine used for sedation during diagnostic, therapeutic and endoscopic procedures. We believe that NVD-301 has the potential to be an easy-to use, rapid onset product useful to relieve the pre-procedure anxiety suffered by many patients prior to undergoing a wide variety of procedures performed in hospitals, imaging centers, ambulatory surgery centers and dental offices.
Annually, there are approximately 40 million invasive procedures performed in the ambulatory surgical setting, > 25 million MRI/CT scans and over 90 million pediatric dental procedures performed. Pre- procedure anxiety occurs in approximately 60% of children undergoing surgery and is associated with an increase in post-surgical complications including delirium, pain and sleep disorders, as well as higher levels of use of post-surgical medications. Anxiety interferes with approximately 30% of MRI scans with 5-10% of scans not completed due to anxiety. Pre-procedure anxiety is the number one reason for the use of sedation in dental procedures.
As of the current date, we have not yet secured sufficient financing to resume our clinical development activity. There can be no assurances that we will be able to secure additional capital, and as a result, there can be no assurances as to whether, and when, we will be able to resume our clinical development activities.
Propofol oral spray. Propofol is the active ingredient in Diprivan®, a leading intravenous anesthetic marketed by AstraZeneca. On July 10, 2007, Manhattan Pharmaceuticals, our licensee, announced its intention to pursue appropriate sub-licensing opportunities for this product candidate.
Veterinary. Our veterinary initiatives are being carried out largely by our partner, Velcera, Inc., or Velcera. In June 2007, Velcera announced that it had entered into a global license and development agreement with Novartis Animal Health. The agreement calls for Novartis Animal Health to develop, register and commercialize a novel canine product utilizing Velcera’s Promist™ platform, which is based on our patented oral spray technology. On March 5, 2008, Velcera announced that it had received notice from Novartis that it was terminating the agreement without cause. On August 24, 2009, we issued a press release to announce that we received a milestone payment of approximately $150,000 from Velcera, Inc. relating to our License and Development agreement dated June 22, 2004. This milestone payment resulted from Velcera’s recently announced global licensing agreement for the first canine pain management product delivered in a transmucosal mist form.
As discussed above, certain of our product candidates are in early stages of clinical development and some are in preclinical testing. These product candidates are continuously evaluated and assessed and are often subject to changes in formulation and technology. As a result, these product candidates are subject to a more difficult, time-consuming and expensive regulatory path in order to commence and complete the preclinical and clinical testing of these product candidates as compared to other product candidates in later stages of development.
BUSINESS DEVELOPMENT
To date, we have entered into license agreements with (i) Mist Acquisition, LLC to manufacture and commercialize the NitroMist® lingual spray version of nitroglycerine, (ii) ECR Pharmaceuticals Company, Inc., to commercialize and manufacture ZolpiMist™ in the United States and Canada, (iii) Hana Biosciences, for the development and marketing rights in the U.S. and Canada for our ondansetron oral spray, (iv) Par, for the marketing rights in the U.S. and Canada for NitroMist™, (v) Manhattan Pharmaceuticals, in connection with propofol, (vi) Velcera, in connection with veterinary applications for currently marketed veterinary drugs and (vii) BioAlliance Pharma SA, for the European rights for ondansetron oral spray. In addition, we have entered into a sub-license agreement with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a sublicense to Par to develop and commercialize Zensana™.
We intend to enter into additional license agreements and strategic alliances, including additional marketing partners and strategic alliances as may be appropriate for the remaining present and future products in our development pipeline.
AGREEMENT WITH PAR PHARMACEUTICAL, INC. AND HANA BIOSCIENCES, INC.
In October 2004, we entered into a license and development agreement pursuant to which we granted to Hana Biosciences an exclusive license to develop and market Zensana™, our oral spray version of ondansetron in the U.S. and Canada. Pursuant to the terms of the agreement, in exchange for $1,000,000, Hana Biosciences purchased 400,000 shares of our common stock at a per share price equal to $2.50, a premium of $0.91 per share or $364,000 over the then market value of our common stock. The Company accounted for this premium as deferred revenue related to the license. In connection with the agreement, Hana Biosciences issued to us $500,000 worth of common stock of Hana Biosciences (73,121 shares based on a market value of $6.84 per share). The fair value of the common stock received from Hana Biosciences was included in deferred revenue and was being recognized over the 20-year term of the agreement.
In July 2007, we entered into a Product Development and Commercialization Sublicense Agreement, or the Sublicense Agreement, with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a non-transferable, non-sublicenseable, royalty-bearing, exclusive sublicense to Par to develop and commercialize Zensana™. In connection therewith, Hana Biosciences amended and restated their existing License and Development Agreement, as amended, with us relating to the development and commercialization of Zensana™, referred to herein as the Amended and Restated License Agreement, to coordinate certain of the terms of the Sublicense Agreement. Under the terms of the Sublicense Agreement, Par is responsible for all development, regulatory, manufacturing and commercialization activities of Zensana™ in the United States and Canada. We retain our rights to Zensana™ outside of the United States and Canada.
In addition, under the terms of the Amended and Restated License Agreement, Hana Biosciences relinquished its right to pay reduced royalty rates to us until such time as Hana Biosciences had recovered one-half of its costs and expenses incurred in developing Zensana™ from sales of Zensana™ and we agreed to surrender for cancellation all 73,121 shares of the Hana Biosciences common stock, with a fair value of $140,000, that had been acquired by us in connection with execution of the original License Agreement (See Note 9).
During the three months ended March 31, 2007, we recorded a $360,000 impairment charge to the statement of operations, the only component of other loss, to establish a new cost basis of $140,000 for the investment as of March 31, 2007. The remaining investment balance was written off in the quarter ended September 30, 2007, to reflect the surrender of our 73,121 shares to Hana in connection with the Amended and Restated License Agreement (See Note 9). We may receive additional milestone payments and royalties over the term of the agreement.
LICENSE AND SUPPLY AGREEMENT WITH PAR PHARMACEUTICAL, INC.
In July 2004, we entered into a 10-year license and supply agreement with Par, a wholly owned subsidiary of Par Pharmaceutical Companies, Inc., whereby Par has the exclusive rights to market, sell and distribute our nitroglycerin lingual spray in the U.S. and Canada. The terms of the agreement call for an upfront license fee which was paid to us in July 2004, a milestone payment made to us upon the FDA’s acceptance of an NDA for our nitroglycerin lingual spray for review in September 2004, another potential milestone payment if and when the NDA is approved for marketing in the U.S., and double-digit percentage royalties on net sales of the product in the U.S. and Canada. We are responsible for obtaining regulatory approval for the product and for supplying the product to Par. In July 2007, we and Par agreed to terminate the agreement relating to NitroMist™.
AGREEMENT WITH MANHATTAN PHARMACEUTICALS, INC.
In April 2003, we entered into a license and development agreement with Manhattan Pharmaceuticals for the worldwide, exclusive rights to our proprietary oral spray technology to deliver propofol for pre-procedural sedation. The terms of the agreement call for certain license, milestone and other payments, the first $125,000 of which was received in June 2003. In November 2003, we received $375,000 from Manhattan Pharmaceuticals for license fees. We have included these license fees in deferred revenue and are recognizing these license fees over the 20-year term of the license. In July 2007, Manhattan Pharmaceuticals, our partner for its propofol oral spray product candidate, announced that as part of its change in strategic focus it intends to pursue appropriate sub-licensing opportunities for this product candidate.
AGREEMENT WITH VELCERA PHARMACEUTICALS, INC. (FORMERLY VETCO)
In June 2004, we entered into a 20-year worldwide exclusive license agreement with Velcera, a veterinary company. The license agreement is for the exclusive rights to our propriety oral spray technology in animals. In September 2004, we received $1,500,000 from Velcera as an upfront payment in connection with the commercialization agreement. The upfront payment has been included in deferred revenue and is being recognized in income over the 20-year term of the agreement. In addition, we received an equity stake of 529,500 shares of common stock in Velcera which did not have a material value. Such investment continues to be carried at its cost basis of $0 as of December 31, 2009. In February 2007, Velcera merged with Denali Sciences, Inc., a publicly reporting Delaware corporation. In June 2007, Velcera announced that it had entered into a global license and development agreement with Novartis Animal Health. The agreement called for Novartis Animal Health to develop, register and commercialize a novel canine product utilizing Velcera’s Promist™ platform, which is based on its patented oral spray technology. We may receive additional milestone payments and royalty payments over the 20-year term of the agreement. In November 2007, the common stock of the merged companies began trading on the OTC bulletin board. On March 5, 2008, Velcera announced that it had received notice from Novartis Animal Health that it was terminating the agreement, without cause. On August 24, 2009, we issued a press release to announce that we received a milestone payment of approximately $150,000 from Velcera, Inc. relating to its license agreement. This milestone payment resulted from Velcera’s recently announced global licensing agreement for the first canine pain management product delivered in a transmucosal mist form.
AGREEMENT WITH BIOALLIANCE PHARMA SA
On May 19, 2008, we and BioAlliance Pharma SA or BioAlliance, entered into an agreement where BioAlliance acquired the European rights for our Ondansetron oral spray. Under the terms of the agreement, BioAlliance paid us a license fee of $3,000,000 upon closing. We are eligible for additional milestone payments totaling approximately $24 million (an approval milestone of $5,000,000 and sales-related milestone payments of approximately $19 million) as well as a royalty on net sales. BioAlliance and us anticipate collaborating in the completion of development activities for Europe, with BioAlliance responsible for regulatory and pricing approvals and then commercialization throughout Europe. We will be responsible for supplying the product. The upfront payment has been included in deferred revenue and is being recognized in income over the term of the agreement (nineteen and one half-years). During the three and twelve months ended December 31, 2009, we recognized $38,000 and $154,000 of income related to this contract, respectively.
AGREEMENT WITH MIST ACQUISITION, LLC
On October 27, 2009, we and privately-held Mist Acquisition, LLC, entered into a licensing agreement to manufacture and commercialize the NitroMist® lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris, in the United States, Canada and Mexico. Under terms of the agreement, Mist paid us a $1,000,000 licensing fee upon execution of the agreement, and will pay milestone payments totaling an additional $1,000,000 over twelve months if certain conditions are met, and ongoing performance payments of seventeen percent (17%) of net sales, subject to the terms of the agreement.
Through a separate license agreement with Mist, Akrimax Pharmaceuticals, LLC will receive the exclusive right to manufacture, distribute, market and sell NitroMist® in North America. NitroMist® provides acute relief of an attack or acute prophylaxis of angina pectoris due to coronary artery disease. The lingual spray form of the drug is conveniently administered and is rapidly absorbed into the bloodstream via the oral mucosa, providing patients a fast and tolerable treatment option for the prevention or relief of pain associated with such attacks.
AGREEMENT WITH ECR PHARMACEUTICALS COMPANY, INC.
On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. (a wholly-owned subsidiary of Hi-Tech Pharmacal Co., Inc.) to commercialize and manufacture ZolpiMist™ in the United States and Canada. ZolpiMist™ is the Company’s oral spray formulation of zolpidem tartrate approved by the FDA in December of 2008.
Under the terms of the agreement, we received a $3,000,000 licensing fee from ECR upon execution of the agreement. ECR will assume responsibility for manufacturing and marketing the product in the United States and Canada. In addition, ECR will pay royalties of up to 15% on net sales of ZolpiMist™ as well as an additional milestone payment if sales reach a specified level.
BUSINESS STRATEGY
Strategy
Our goal is to become a leading specialty pharmaceutical company that develops and commercializes improved formulations of existing drugs using our patented oral spray technology. We believe that our technology has application to a broad number of therapeutic areas and product categories. Our strategy is to concentrate our product development activities primarily on pharmaceutical products which meet the following characteristics:
|
·
|
Significant prescription sales already exist;
|
|
·
|
Our proprietary novel drug delivery technology enhances the performance of the active ingredient of the target compound, potentially addressing unmet patient needs; and
|
|
·
|
Applicability of an efficient regulatory pathway to approval using the 505(b)(2) pathway.
|
In today’s environment of escalating drug development costs and time to market, we believe that the ability to bring products with some degree of differentiation and competitive advantage to the marketplace in a timely and cost-effective manner is a viable strategy.
Products
We currently have four product candidates in our pipeline. Our NitroMist™ and Zolpimist™ products are approved and currently licensed to Mist Acquisition, LLC and ECR Pharmaceuticals Company, Inc., respectively. Zensana™, is currently licensed to a marketing partner who we expect to commercialize this product candidate, with us receiving milestone and royalty income from revenue upon product approval. For the remainder of our pipeline, we expect to secure marketing partners for these product candidates after we have generated sufficient clinical data to demonstrate the effectiveness of these product candidates. We anticipate that such marketing partners for both our approved and our development products would provide us with milestone payments and royalties based on revenues.
In addition to our existing product candidates, we intend to continue to identify and pursue additional product candidates for development.
PATENTED AND PATENT PENDING DELIVERY SYSTEMS
We have certain patents and pending patent applications for our oral spray delivery system. FDA approval is not a prerequisite for patent approval. The expected year of marketability of a given product candidate will vary depending upon the specific drug product with which the delivery system will be utilized. Each individual use of the delivery system will require registration with and/or approval by the FDA or other relevant health authority prior to marketability, and the amount of regulatory oversight required by the FDA or other regulatory agencies will also depend on the specific type of drug product for which the delivery system is implemented. Our aerosol and pump spray formulations release drugs in the form of a fine mist into the buccal portion of the mouth for rapid absorption into the bloodstream via the mucosal membranes. Our proprietary technology offers, in comparison to conventional oral dosage forms, the potential for faster absorption of drugs into the bloodstream leading to quicker onset of therapeutic effects and possibly reduced first pass liver metabolism, which may result in lower doses. Oral sprays eliminate the requirement for water or the need to swallow, potentially improving patient convenience and adherence. Our oral spray technology is focused on addressing unmet medical needs for a broad array of existing and future pharmaceutical products.
MARKETING AND DISTRIBUTION
To date, we have chosen to license products developed with our technology to other drug companies. We intend to pursue additional strategic alliances, as well as to consider fully developing and commercializing product candidates internally.
We anticipate that promotion of our product candidates, whether conducted by us or by a strategic partner, will be characterized by an emphasis on their distinguishing characteristics, such as dosage form and packaging, as well as possible therapeutic advantages of such product candidates. We intend to position our product candidates as alternatives or as line extensions to brand-name products. We believe that to the extent our formulated products are patent-protected, such formulations may offer brand-name manufacturers the opportunity to expand their product lines. Alternatively, products which are not patented may be offered to brand-name manufacturers as improved substitute products after patent protection on existing products expire.
In as much as we do not currently have the financial or other resources to undertake extensive marketing activities, we generally intend to seek to enter into marketing arrangements, including possible joint ventures or license or distribution arrangements, with third parties. We believe that such third-party arrangements will permit us to maximize the promotion and distribution of pharmaceutical products while minimizing our direct marketing and distribution costs. If we are unable to enter into additional agreements, we may not be able to successfully market our product candidates.
We have not yet determined strategies relating to marketing of our other proposed formulated products; these will be formulated in advance of anticipated completion of development activities relating to the particular formulated product. As a company, we have no experience in marketing or distribution of our product candidates, and our ability to fund such marketing activities will require us to raise additional funds and/or consummate a strategic alliance or combination with a well-funded business partner.
MANUFACTURING
We intend to contract out the manufacturing of our product candidates. The manufacture of our pharmaceutical product candidates is subject to cGMP prescribed by the FDA and pre-approval inspections by the FDA and foreign authorities prior to the commercial manufacture of any such products. See Item 1, Business-“Raw Materials and Suppliers” and “Government Regulation.”
In February 2008, we entered into a Master Services Agreement with Rechon Life Sciences (Malmo, Sweden), whereby Rechon will provide services related to the manufacturing development and the manufacture of clinical supplies for certain of our products. Rechon provides these services on a fee-for-service basis.
On December 28, 2009, DPT Laboratories became our contract manufacturer for Duromist™, sildenafil citrate oral spray.
RAW MATERIALS AND SUPPLIERS
We believe that the active ingredients used in the manufacture of our product candidates are presently available from numerous suppliers located in the U.S., Europe and Japan and can be delivered to our manufacturing facility by such suppliers. We intend to enter into arrangements with such third-party suppliers for supplies of active and inactive pharmaceutical ingredients and packaging materials used in the manufacture of our product candidates. Accordingly, we may be subject to various import duties applicable to both finished products and raw materials and may be affected by various other import and export restrictions as well as other developments impacting upon international trade. These international trade factors will, under certain circumstances, have an impact on the manufacturing costs (which will, in turn, have an impact on the cost of our product candidates). To the extent that transactions relating to the purchase of raw materials involve currencies other than U.S. dollars, our operating results will be affected by fluctuations in foreign currency exchange rates.
Generally, certain raw materials, including inactive ingredients, are available from a limited number of suppliers and certain packaging materials intended for use in connection with our product candidates may be available only from sole source suppliers. Although we believe that we will not encounter difficulties in obtaining the inactive ingredients or packaging materials necessary for the manufacture of our products, we may not be able to enter into satisfactory agreements or arrangements for the purchase of commercial quantities of such materials. A failure to enter into agreements or otherwise arrange for adequate or timely supplies of principal raw materials and the possible inability to secure alternative sources of raw material supplies could have a material adverse effect on our ability to manufacture formulated products.
Development and regulatory approval of our product candidates are dependent upon our ability to procure active ingredients and certain packaging materials from FDA-approved sources. Since the FDA approval process requires manufacturers to specify their proposed suppliers of active ingredients and certain packaging materials in their applications, FDA approval of a supplemental application to use a new supplier would be required if active ingredients or such packaging materials were no longer available from the specified supplier, which could result in manufacturing delays. Accordingly, we intend to locate alternative FDA approved suppliers.
GOVERNMENT REGULATION
FDA approval process
In the United States, pharmaceutical products are subject to extensive regulation by the U.S. Food and Drug Administration, or the FDA. The Federal Food, Drug, and Cosmetic Act, or the FDC Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending new drug applications or NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Pharmaceutical product development in the U.S. typically involves preclinical laboratory and animal tests, the submission to the FDA of a notice of claimed investigational exemption or an investigational new drug application or IND, which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements including good laboratory practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long term preclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not commented on or questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin.
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations, good clinical practices or GCP, as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial subjects. The study protocol and informed consent information for subjects in clinical trials must also be submitted to an institutional review board, or IRB, for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to assess metabolism, pharmacokinetics, pharmacological actions, side effects associated with increasing doses and, if possible, early evidence of effectiveness. Phase 2 usually involves trials in a limited patient population, to determine the effectiveness of the drug for a particular indication or indications, dosage tolerance and optimum dosage, and identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain additional information about clinical efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug.
Under the Pediatric Research Equity Act of 2003, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers.
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing of the product may begin in the U.S. The NDA must include the results of all preclinical, clinical and other testing and a compilation of data relating to the product's pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial. Under federal law, the submission of most NDAs is additionally subject to a substantial application user fee, currently $1,178,000, and the manufacturer and/or sponsor under an approved new drug application are also subject to annual product and establishment user fees, currently $65,030 per product and $392,700 per establishment. These fees are typically increased annually.
The FDA has 60 days from its receipt of a NDA to determine whether the application will be accepted for filing based on the agency's threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of new drug applications. Most such applications for non-priority drug products are reviewed within ten months. The review process may be extended by FDA for three additional months to consider certain new information or clarification regarding information already provided in the submission. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with good clinical practices, or GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. FDA will not approve the product unless compliance with current good manufacturing practices is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication proposed for marketing.
After FDA evaluates the NDA and the manufacturing facilities, it issues an approval letter, an approvable letter or a not-approvable letter. Both approvable and not-approvable letters generally outline the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA's satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. FDA has committed to reviewing such resubmissions in 2 or 6 months depending on the type of information included.
An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require substantial post-approval testing and surveillance to monitor the drug's safety or efficacy and may impose other conditions, including labeling restrictions which can materially affect the potential market and profitability of the drug. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
The Hatch-Waxman Act
In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s product. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated new drug application, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredients in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. ANDA applicants are not required to conduct or submit results of pre-clinical or clinical tests to prove the safety or effectiveness of their drug product, other than the requirement for bioequivalence testing. Drugs approved in this way are commonly referred to as "generic equivalents" to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid is called a Paragraph IV certification. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the ANDA applicant.
The ANDA also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. Federal law provides a period of five years following approval of a drug containing no previously approved active ingredients, during which ANDAs for generic versions of those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original product approval. Federal law provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was required to be supported by new clinical trials conducted by or for the sponsor, during which FDA cannot grant effective approval of an ANDA based on that listed drug.
Section 505(b)(2) New Drug Applications
Most drug products obtain FDA marketing approval pursuant to an NDA or an ANDA. A third alternative is a special type of NDA, commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the safety and efficacy data of an existing product, or published literature, in support of its application.
505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant may rely upon certain preclinical or clinical studies conducted for an approved product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the label indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on studies conducted for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the Orange Book to the same extent that an ANDA applicant would. Thus, approval of a 505(b)(2) NDA can be stalled until all the listed patents claiming the referenced product have expired, until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired, and, in the case of a Paragraph IV certification and subsequent patent infringement suit, until the earlier of 30 months, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant.
We expect that the majority of our product candidates in development will require the filing of 505(b)(2) NDAs because, although such products contain previously approved chemical entities, we or our licensees may seek to make new claims regarding therapeutic effects or lessened side effects, or both.
Other Regulatory Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, FDA closely regulates the marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet.
Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4 testing, risk minimization action plans, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as drug manufacture, packaging, and labeling procedures must continue to conform to current good manufacturing practices, or cGMPs, after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies, and are subject to periodic inspections by the FDA during which the agency inspects manufacturing facilities to access compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
|
Anti-Kickback, False Claims Laws & The Prescription Drug Marketing Act
|
In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Physician Drug Samples
As part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. The Prescription Drug Marketing Act, or the PDMA, imposes requirements and limitations upon the provision of drug samples to physicians, as well as prohibits states from licensing distributors of prescription drugs unless the state licensing program meets certain federal guidelines that include minimum standards for storage, handling and record keeping. In addition, the PDMA sets forth civil and criminal penalties for violations.
COMPETITION
The markets which we intend to enter are characterized by intense competition, often from organizations which are larger and/or better capitalized than us. We will be competing against established pharmaceutical companies which currently market products which are equivalent or functionally similar to those we intend to market. Prices of drug products are significantly affected by competitive factors and tend to decline as competition increases. In addition, numerous companies are developing or may, in the future, engage in the development of products competitive with our proposed products. We expect that technological developments will occur at a rapid rate and that competition is likely to intensify as enhanced delivery system technologies gain greater acceptance. Additionally, the markets for formulated products which we have targeted for development are intensely competitive, involving numerous competitors and products. We intend to enhance our competitive position by focusing our efforts on our novel dosage forms.
We also face, and will continue to face, competition from colleges, universities, governmental agencies and other public and private research organizations. These competitors are becoming more active in seeking patent protection and licensing arrangements to collect royalties for use of technology that they have developed. Some of these technologies may compete directly with the technologies that we are developing. These institutions will also compete with us in recruiting highly qualified scientific personnel. We expect that developments in the areas in which we are active may occur at a rapid rate and that competition will intensify as advances in this field are made. As a result, we need to continue to devote substantial resources and efforts to research and development activities.
PATENTS AND PROTECTION OF PROPRIETARY INFORMATION
We have applied for U.S. and foreign patent protection for our buccal spray delivery systems which are the primary focus of our development activities. Currently, we have nine patents which have been issued in the U.S. and 69 patents which have been issued outside of the U.S. Additionally, we have over 65 patents pending around the world. Additional patent applications may not be granted, or, if granted, may not provide adequate protection to us. We also intend to rely on whatever protection the law affords to trade secrets, including unpatented know-how. Other companies, however, may independently develop equivalent or superior technologies or processes and may obtain patents or similar rights with respect thereto.
Although we believe that we have developed our technology independently and have not infringed, and do not infringe, on the patents of others, third parties may make claims, however, that our technology does infringe on their patents or other intellectual property. In the event of infringement, we may, under certain circumstances, be required to modify our infringing product or process or obtain a license. We may not be able to do either of those things in a timely manner if at all, and failure to do so could have a material adverse effect on our business. In addition, we may not have the financial or other resources necessary to enforce a patent infringement or proprietary rights violation action or to defend ourselves against such actions brought by others. If any of the products we develop infringe upon the patent or proprietary rights of others, we could, under certain circumstances, be enjoined or become liable for damages, which would have a material adverse effect on our business.
We also rely on confidentiality and nondisclosure agreements with our licensees and potential development candidates to protect our technology, intellectual property and other proprietary property. Pursuant to the foregoing and for other reasons, we face the risk that our competitors may acquire information which we consider to be proprietary, that such parties may breach such agreements or that such agreements will be inadequate or unenforceable.
Buccal Nonpolar Sprays. On April 12, 1996, we filed an application with the U.S. Patent and Trademark Office, or the USPTO, with claims directed to our buccal spray composition containing certain amounts of propellant, a non-polar solvent, and certain classes of drugs, as well as specific drugs within those classes. The application also included claims directed to soft-bite gelatin capsules containing these drugs. On September 1, 1998, the USPTO allowed the claims directed to buccal spray propellant compositions, but rejected the claims directed to the capsules. In November 1998, we deleted the capsule claims from this application to pursue issuance of a patent with claims directed to the buccal non-polar spray compositions and methods of administering the class of drugs using the buccal spray compositions. On September 21, 1999, U.S. Patent No. 5,955,098 was issued to us with claims directed to the above-described buccal non-polar spray propellant compositions and methods. This patent expires on April 12, 2016.
On February 21, 1997, we filed an application under the Patent Cooperation Treaty, or the PCT, (PCT Publication No. WO 97/38663) for the above-subject matter. The International Preliminary Examination Authority issued an International Preliminary Examination Report alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
With respect to the above PCT application, in October and November 1998, we entered the national phase in Canada and Europe, with claims directed to the above subject matter. On April 16, 2003, European Patent No. EP 0 904 055 was granted to us with claims directed to propellant containing buccal non-polar spray compositions containing similar drugs (i.e., anti-histamines, steroid hormones, non-steroidal anti-inflammatories, benzodiazepines, anti-depressants and nicotine) to those in the corresponding issued U.S. patent. This European patent has been validated in the UK, Germany, France, Italy, Belgium, Switzerland/Liechtenstein, Austria, Sweden, Denmark, Finland, Luxembourg, the Netherlands, Spain, Greece, Monaco, Portugal and Ireland so that there is patent protection in these countries. We have filed a divisional application based on this European patent. On April 17, 2007 this application issued to us as European Patent No. 1 275 374 with claims directed to a buccal spray composition containing a propellant, a non-polar solvent and an active compound selected from alkaloids and analgesics This European patent has been validated in the U.K., Germany, France, Italy, Belgium, Switzerland/Lichtenstein, Sweden, the Netherlands, Spain, and Greece, so that there is patent protection in these countries. No opposition has been filed to this application and the time for filing any opposition has expired.
With respect to the Canadian application, we filed a request for examination with the Canadian Patent Office on February 7, 2002. We received an Office Action from the Canadian Patent Office dated April 13, 2004, pursuant to which we were requested to elect for prosecution either claims directed to buccal spray compositions or claims to the soft-bite gelatin capsules. We elected to prosecute the claims directed to buccal spray compositions. The Canadian Patent Office granted the application on December 27, 2005 as Canadian Patent No. 2,252,050. The allowed claims are similar to those granted by the European Patent Office.
Buccal Polar Sprays. On April 12, 1996, we filed an application with the USPTO with claims directed to propellant free buccal polar spray compositions containing certain amounts of a polar solvent and certain classes of drugs (i.e., non-steroidal anti-inflammatories, anti-histamines, steroid hormones, benzodiazepams, and anti-depressants), as well as specific drugs within those classes. The application also contained claims to soft-bite gelatin capsules containing such drugs. A continuation-in-part, or CIP, application was filed directed to this subject matter before the original application was allowed to go abandoned. The USPTO initially rejected the claims in the CIP application. We deleted the claims from this application (including the soft-bite capsule claims) and replaced them with claims directed to methods of using the above-described propellant free buccal polar spray compositions to administer the drugs. On August 29, 2000, U.S. Patent No. 6,110,486 was issued to us with claims directed to the above-described methods of administering the drugs. This patent expires on April 12, 2016.
On February 21, 1997, we filed an application under the PCT (PCT Publication No. WO 97/38662) for the above-described subject matter. The International Preliminary Examination Authority issued an International Preliminary Examination Report alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
With respect to the above PCT application, in October and November 1998, we entered the national phase in Canada and Europe, respectively, with claims directed to the above subject matter.
On February 2, 2005, European Patent No. 0 910 339 was granted to us with claims directed to use of polar solvent containing pump sprays containing similar drugs to those in the corresponding issued U.S. patent. This European patent has been validated in the UK, Germany, France, Italy, Belgium, Switzerland/Liechtenstein, Austria, Sweden, Denmark, Finland, Luxembourg, the Netherlands, Spain, Greece, Monaco, Portugal and Ireland so that there was patent protection in these countries. In November 2005, Akzo Nobel N.V. filed a successful opposition against this patent in the European Patent Office alleging “lack of inventive step.” We have decided not to file any appeal in connection with this opposition. As a result, the European Patent is no longer in force.
With respect to the Canadian application, we filed a request for examination with the Canadian Patent Office on February 7, 2002. We received an Office Action from the Canadian Patent Office dated April 13, 2004, pursuant to which we were requested to elect for prosecution either claims directed to buccal spray compositions or claims to the soft-bite gelatin capsules. We elected to prosecute the claims directed to buccal spray compositions. On February 10, 2006, the Canadian Patent Office issued a Notice of Allowance for this application. On October 10, 2006, Canadian Patent No. 2,252,038 was granted to us with claims directed to the use of a pharmacologically active compound selected from the group consisting of non-steroidal anti-inflammatories, anti-histamines, steroid hormones, benzodiazepines, and anti-depressants for the preparation of a buccal aerosol pump spray composition for being absorbed through the oral mucosa.
Buccal Nonpolar Spray for Nitroglycerin. On April 12, 1996, we filed an application with the USPTO with claims directed to a buccal spray containing certain amounts of nitroglycerin, a non-polar solvent, and a propellant. The claims were allowed and on February 9, 1999, the USPTO issued U.S. Patent No. 5,869,082 to us for said nitroglycerin buccal spray. This patent expires on April 12, 2016.
On February 21, 1997, we filed a PCT application (PCT Publication No. WO 97/38687) directed to the above-described subject matter. The International Preliminary Examination Authority issued an International Preliminary Examination Report alleging that the subject matter of the invention lacks an inventive step. This opinion, with which we disagree, is not dispositive.
In October 1998, we entered the national phase in Canada. We filed a request for examination on February 7, 2002. The Canadian Patent Office issued a second office action to us dated July 11, 2005. We responded to the office action on January 11, 2006. As a result, Canadian Patent No. 2,251,564 was granted to us on January 9, 2007, with claims directed to a buccal spray containing certain amounts of nitroglycerin, a non-polar solvent and a propellant.
In November 1998, we entered the national phase in Europe. European Patent No. 0 927 032 was granted to us on April 16, 2003, with claims directed to a buccal spray containing certain amounts of nitroglycerin, a non-polar solvent and a propellant. This European patent has been validated in the UK, Germany, France, Italy, Belgium, Switzerland/Liechtenstein, Austria, Sweden, Denmark, Finland, Luxembourg, the Netherlands, Spain, Greece, Monaco, Portugal and Ireland so that there is patent protection in these countries.
Buccal Polar/Nonpolar Sprays or Capsules. On October 1, 1997, we filed a PCT application (PCT Publication No. WO 99/16417) designating a large number of countries including the U.S., directed to the buccal sprays and soft-bite capsules. The application included claims directed to: (A) a buccal spray composition containing either (1) a polar solvent with certain classes of drugs, as well as specific drugs in those classes with or without a propellant or (2) a non-polar solvent with or without a propellant with certain classes of drugs, as well as specific drugs in those classes; (B) buccal spray composition containing a non-polar solvent, a flavoring agent and certain classes of drugs; and (C) methods of administering these drugs using the buccal spray compositions. The application also contained claims to soft-bite gelatin capsules containing such drugs. This application differs from the first three applications, discussed above, in that the claimed compositions include different classes of drugs from those described in the first three applications. The International Preliminary Examination Authority issued an International Preliminary Examination Report alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
On March 29, 2000, we entered the national phase in the U.S. by filing a CIP of the above-identified PCT application with the USPTO. The CIP application included claims directed to propellant free buccal spray compositions containing certain amounts of polar or non-polar solvents, and certain classes of drugs, as well as specific drugs in those classes; buccal spray compositions containing certain amounts of a propellant, a polar or non-polar solvent and certain classes of drugs, as well as specific drugs in those classes; and methods of administering said drugs using these types of buccal spray compositions. The application is currently being prosecuted with claims directed to the propellant free buccal spray compositions and methods of administering said drugs using these types of buccal spray compositions.
Subsequently, we filed two divisional applications claiming priority to the CIP. The first divisional application was issued to us as U.S. Patent No. 6,998,110 with claims directed to methods of administering a biologically active peptides, central nervous system active amines, sulfonyl ureas, antibiotics, antifungals, sleep inducers, antiasthmatics, antiemetics, antivirals, histamine H-2 receptor antagonists, barbiturates, prostaglandins, or bronchial dilators using the buccal spray compositions containing certain amounts of a propellant, a polar or non-polar solvent and certain classes of drugs. This patent expires on October 1, 2017. Another application has been filed directed to additional formulations relating to U.S. Patent No. 6,998,110. The second divisional application was issued to us as U.S. Patent No. 6,676,931. This patent expires on October 1, 2017. The claims of this patent are directed to a propellant free pump spray composition containing certain amounts of a polar solvent, certain amounts of a flavoring agent and certain amounts of cyclosporin or ondansetron hydrochloride. Another application has been filed directed to the additional classes of drugs and specific drugs and formulations that were not included in the claims of U.S. Patent No. 6,676,931.
Based on the above-identified PCT application, we entered the national phase in Canada on March 29, 2000. We filed a request for examination in Canada on August 29, 2002. An office action has been received from the Canadian Patent Office and we have responded to that office action.
Based on the above-identified PCT application, we also entered the national phase in Japan on April 3, 2000. An office action rejecting the pending claims has been received from the Japanese Patent Office. We have demanded a trial in response to that office action. In addition, we are in the process of filing a divisional application in Japan claiming priority to this application.
Based on the above-identified PCT application, we also entered the national phase in Europe in April 2000. The European application includes claims directed to propellant free buccal spray compositions containing certain amounts of a polar solvent and certain classes of drugs, as well as specific drugs in those classes and the use thereof to prepare a medicament for use as a buccal spray for transmucosal administration. We have filed three applications related to this application in Europe. The first application included claims directed to buccal spray compositions containing certain amounts of a non-polar solvent, a propellant and certain classes of drugs as well as specific drugs in those classes and the use thereof to prepare a medicament for use as a buccal spray for transmucosal administration. This application was granted to us on April 18, 2007, as European Patent No. 1 295 536 with claims directed to a buccal spray composition including a propellant, a non-polar solvent, and one of the following active compounds: biologically active peptides, central nervous system active amines, sulfonyl ureas, antibiotics, antifungals, antivirals, sleep inducers, antihistamines, antiemetics, histamine H-2 receptor antagonists, barbiturates, prostoglandins, and bronchial dilators selected from the group consisting of terbutaline, and theophylline. A divisional application has been filed claiming priority from this patent. The second application included claims directed to propellant free buccal spray compositions containing certain amounts of a non-polar solvent and certain classes of drugs, as well as specific drugs in those classes. The third application included claims directed to a buccal spray composition containing certain amounts of a polar solvent, a propellant and certain classes of drugs, as well as specific drugs in those classes. Each of the above-identified European applications is currently being prosecuted.
Furthermore, in August 2002, we filed a number of U.S. patent applications directed to buccal spray compositions containing certain classes of drugs as well as specific drugs for treating particular types of disorders. In August 2003, we filed PCT applications related to these U.S. applications. We have subsequently filed corresponding applications in Europe, Japan and Canada for the subject matter for a majority of these CIP applications.
From these U.S. patent applications, we have been granted U.S. Patent No. 6,969,508 with claims directed to methods for administering an effective amount of anti-opioid agents, anti-migraine agents, pain control agents, anesthetics, and mixtures thereof using a buccal spray composition containing a polar solvent and a propellant. We have also been granted U.S. Patent No. 6,977,070 with claims directed to methods for administering an effective amount of a pharmacologically active compound to a mammal to provide transmucosal absorption of a pharmacologically effective amount of acetylcholinesterase inhibitors, nerve impulse inhibitors, anti-cholinergics, anti-convulsants, anti-psychotics, anxiolytic agents, dopamine metabolism inhibitors, agents to treat post stroke sequelae, neuroprotectants, agents to treat Alzheimer's disease, neurotransmitters, neurotransmitter agonists, sedatives, agents for treating attention deficit disorder, agents for treating narcolepsy, central adregenic antagonists, anti-depression agents, agents for treating Parkinson's disease, benzodiazepine antagonists, stimulants, neurotransmitter antagonists, tranquilizers, and mixtures there of using a buccal spray containing a polar solvent and a propellant.
In addition, in September 2003, we filed a number of U.S. patent applications directed to buccal spray compositions containing specific drugs. We have subsequently filed corresponding applications in Europe, Japan, Canada, Israel and Korea for the subject matter a majority of these CIP applications.
Stable Hydroalcoholic Oral Spray Formulations and Methods. On April 19, 2007, we filed an application with the USPTO with claims directed to hydroalcoholic spray compositions and methods. The application was published on October 25, 2007, and is currently pending. Substantive examination of the application by the USPTO has not yet begun.
On April 19, 2007 we also filed a corresponding PCT application (PCT Publication No. WO 2007/123955) to the above noted subject matter. On October 30, 2008, the International Bureau issued an International Preliminary Report on Patentability alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
Based on the above-identified PCT application, we entered the national phase in Canada, Europe and Japan in October 2008.
Anti-Migraine Oral Spray Formulations and Methods. On July 27, 2007 we filed an application with the USPTO with claims directed to compositions comprising a selective 5-hydroxytryptamine receptor subtype agonist and methods of treatment. The application was published on February 7, 2008, and is currently pending. Substantive examination of the application by the USPTO has not yet begun.
On July 27, 2007 we also filed a corresponding PCT application (PCT Publication No. W0 2008/013929) to the above noted subject matter. On April 25, 2008, the International Searching Authority issued a Written Opinion alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
Based on the above-identified PCT application, we entered the national phase in Canada, Europe and Japan in January 2009.
Stable Anti-Nausea Oral Spray Formulations and Methods. On December 21, 2007 we filed an application with the USPTO with claims directed to formulations containing a selective 5-hydroxytryptamine receptor antagonist and methods of treatment. The application was published on July 17, 2008, and is currently pending. Substantive examination of the application by the USPTO has not yet begun.
On December 21, 2007 we also filed a corresponding PCT application (PCT Publication No. W0 2008/079295) to the above noted subject matter. On May 1, 2008, the International Searching Authority issued a Written Opinion alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
Anti-Insomnia Compositions and Methods. On May 12, 2008 we filed an application with the USPTO with claims directed to administering an anti-insomnia composition by buccal spray for transmucosal absorption to a patient. The application was published on November 13, 2008, and is currently pending.
On May 12, 2008 we also filed a corresponding PCT application (PCT Publication No. W0 2008/141264) to the above noted subject matter. On July 30, 2008, the International Searching Authority issued a Written Opinion alleging that the subject matter of the invention lacked novelty and/or lacked an inventive step. This opinion, with which we disagree, is not dispositive.
Antihistamine Syrup and Ointment. On November 10, 1997, we filed an application with the USPTO with claims directed to a spray composition for topical administration containing an antihistamine and a polar solvent or an antihistamine, a non-polar solvent and a propellant. In October 1998, the PTO rejected the claims. The claims were deleted and replaced with a claim directed to a method of controlling the occurrence of delayed contact dermatitis by applying a lotion composition containing certain amounts of certain antihistamines in certain amounts of a polar or non-polar solvent. On May 21, 2002, U.S. Patent No. 6,391,282 was issued to us for the above-described method. This patent expires on November 10, 2017.
General Comment with Respect to Entering the National Phase for Each of the Foregoing PCT Applications. In addition to our patents and patent applications in the U.S., we are interested in entering the national phase and obtaining patent protection in Europe, Japan and Canada. At the present time, it is not possible to accurately predict the expenses involved in pursuing the foregoing applications in Canada, Japan and Europe. For example, we anticipate that, in the case of the European applications, it may become necessary to file appeals with the Board of Appeals in Munich. Expenses may exceed $100,000 (in the aggregate) before a final disposition is obtained. We expect that this process may take between two and four years.
EMPLOYEES
As of March 24, 2010, we had 3 employees, all of whom were full-time employees.
The names and ages of our Directors and Executive Officers as of the date of filing this Annual Report on Form 10-K are set out below. All Directors are elected annually, to serve until the next annual meeting of stockholders and until their successors are duly elected and qualified. Executive Officers are elected annually by the Board of Directors and serve at the Board of Directors’ pleasure. The Board of Directors has determined that the following individuals are the Executive Officers of the Company: Mr. Ratoff, Dr. Bergstrom and Mr. Warusz.
|
NAME
|
AGE
|
POSITION WITH THE COMPANY
|
|
Mark J. Baric
|
51
|
Director
|
|
Thomas E. Bonney
|
45
|
Director
|
|
Charles Nemeroff, M.D., Ph.D.
|
60
|
Director
|
|
Steven B. Ratoff
|
67
|
Chairman of the Board of Directors, President, Chief Executive Officer and Interim Chief Financial Officer
|
|
David H. Bergstrom, Ph.D.
|
55
|
Senior Vice President and Chief Operating Officer
|
|
Joseph M. Warusz
|
53
|
Principal Accounting Officer
|
On March 19, 2009, Michael E. Spicer notified our Board of Directors of his intention to resign as Chief Financial Officer and Corporate Secretary, effective April 1, 2009. There is no disagreement between us and Mr. Spicer concerning our policies, procedures and operations. Effective April 2009, Deni Zodda, Ph.D. served as Interim Chief Financial Officer and Corporate Secretary and Joseph M. Warusz, a consultant, shall serve as Principal Accounting Officer. On April 28, 2009, Steven Ratoff was appointed Interim Chief Financial Officer and Corporate Secretary concurrent with the resignation of Deni Zodda.
Mark J. Baric, Director, 51. Mr. Baric was elected to the Board in February 2007. Since 2005, Mr. Baric has been the President and co-founder of CeNeRx BioPharma, Inc., a privately-held development company with a therapeutic focus on diseases of the central nervous system. In 2001 he co-founded and served, until 2005, as Chief Executive Officer and Chairman of 2ThumbZ Entertainment Inc., a privately-held company which develops and markets entertainment applications for users of handheld wireless devices and networks. From 1996 to 2001, Mr. Baric was Chairman and Chief Executive Officer of Virtus Entertainment Corporation, an emerging company in the fast-growing interactive entertainment industry. From 1990 to 1996, Mr. Baric held various leadership positions, including Chief Operating Officer and Chief Financial and Administrative Officer of Seer Technologies Inc. (now known as Cicero, Inc.), a provider of business integration software. Prior to 1990, Mr. Baric held various leadership positions at several firms, including CS First Boston and Coopers and Lybrand. Mr. Baric serves on the boards of CeNeRx BioPharma, Inc. and 2ThumbZ Entertainment Inc. Mr. Baric received an M.B.A. from the Wharton School of the University of Pennsylvania and a B.S. from Clarion University. He is our chair of our Corporate Governance and Nominating Committee, and a member of our Audit and Compensation Committees.
Thomas E. Bonney, CPA, Director, 45. Mr. Bonney was elected to the Board in March 2005. From 2002 to the present, Mr. Bonney has been Managing Director of CMF Associates, LLC, a financial and management consulting firm. Since December 2006, Mr. Bonney has been a General Partner in West Place LLC, and West Place Restaurant Group, LLC, privately-held companies that invest in and manage hotels and real estate. Since June 2005, Mr. Bonney has been a Director of Leblon Holdings LLC, a privately-held beverage supplier and from June 2005 through July 2007 was the Chief Financial Officer of Leblon Holdings, LLC. From 2001 to 2002, he was Chief Financial Officer of Akcelerant Holdings, Inc., a technology holding company. From 1995 to 2001, Mr. Bonney was President and a Director of Polaris Consulting & Information Technologies, a technology solutions provider. Mr. Bonney was at Deloitte & Touche from 1987 to 1995 in various positions including Senior Manager. Mr. Bonney received his B.S. in Accounting at the Pennsylvania State University and is a member of the Pennsylvania Institute of Certified Public Accountants. He is our lead director, chair of our Audit Committee and a member of our Compensation and Corporate Governance and Nominating Committees.
Charles Nemeroff, M.D., Ph.D., Director, 60. Dr. Nemeroff was elected to the Board in September 2003. Dr. Nemeroff is the Leonard M. Miller Professor and Chairman of the Department of Psychiatry and Behavioral Sciences at the University of Miami Leonard M. Miller School of Medicine in Miami, Florida since 2009. Previously, he served as the Reunette W. Harris Professor and Chairman of the Department of Psychiatry and Behavioral Sciences at Emory University School of Medicine in Atlanta, Georgia. Dr. Nemeroff has served on the Scientific Advisory Board of numerous publicly-traded pharmaceutical companies, including Astra-Zeneca Pharmaceuticals and Forest Laboratories. In 2002, he was elected to the Institute of Medicine of the National Academy of Sciences. Dr. Nemeroff received his B.S. from the City College of New York, his M.S. from Northeastern University, and his M.D., Ph.D. and post doctoral training from the University of North Carolina. Dr. Nemeroff is chair of our Scientific Advisory Board. He is also chair of our Compensation Committee and a member of our Audit and Corporate Governance and Nominating Committees.
Steven B. Ratoff, Chairman of the Board, President, Chief Executive Officer, Interim Chief Financial Officer and Secretary, 67. Mr. Ratoff was elected to the Board in January 2006 and was elected Chairman of the Board on September 15, 2006. He was appointed as Interim President and Chief Executive Officer of NovaDel on July 23, 2007. On December 31, 2009, he was appointed President and Chief Executive Officer. Mr. Ratoff is a private investor and since December 2004 has served as a venture partner with ProQuest, a health care venture capital firm. Mr. Ratoff served as director, since May 2005, and was Chairman of the Board, from September 2005 to October 2006, of Torrey Pines Therapeutics Inc. (formerly Axonyx Inc.), a NASDAQ development stage pharmaceutical company which has recently merged with Raptor. Mr. Ratoff served as a director of Inkine Pharmaceuticals, Inc. from February 1998 to its sale to Salix, Inc. in September 2005. He also served as a board member since March 1995 and as Chairman of the Board and Interim Chief Executive Officer of CIMA Labs, Inc. from May 2003 to its sale to Cephalon, Inc. in August 2004. Mr. Ratoff also served as a director, since 1998 and as President and Chief Executive Officer of MacroMed, Inc. from February to December 2001. From December 1994 to February 2001, Mr. Ratoff served as Executive Vice President and Chief Financial Officer of Brown-Forman Corporation, a publicly-traded manufacturer and marketer of alcoholic beverages. Mr. Ratoff also was employed by Bristol Myers Squibb from 1975 to 1991, serving in a number of executive positions, the last of which was as Senior Vice President and Chief Financial Officer of the Pharmaceutical Group. Mr. Ratoff received his B.S. in Business Administration from Boston University and an M.B.A. with Distinction from the University of Michigan.
David H. Bergstrom, Ph.D., Senior Vice President and Chief Operating Officer, 55. Dr. Bergstrom joined NovaDel in December 2006 as Senior Vice President and Chief Operating Officer. From 1999 to November 2006, Dr. Bergstrom served in several capacities at Cardinal Health, Inc., including Vice President, Research & Development and Senior Vice President and General Manager. From 1998 to 1999, Dr. Bergstrom was Vice President of Pharmaceutical & Chemical Development at Guilford Pharmaceuticals Inc. Dr. Bergstrom was employed by Hoechst Marion Roussel, Inc. as the Director of Pharmaceutical and Analytical Sciences from 1996 to 1998. Dr. Bergstrom served as Director of Pharmaceutical and Analytical Development for the predecessor company, Hoechst-Roussel Pharmaceuticals Inc., from 1991 to 1996, and Group Manager, Formulations, Pharmaceutical Research from 1990 to 1991. Prior thereto, Dr. Bergstrom held various positions at Ciba-Geigy Corporation. Dr. Bergstrom received his Ph.D. in Pharmaceutics at the University of Utah in 1985. In addition, he received his M.S. in Pharmaceutical Chemistry at the University of Michigan in 1982 and his B.S. degree in Pharmacy in 1978 at Ferris State University.
Joseph M. Warusz, Principal Accounting Officer, 53. Mr. Warusz joined NovaDel as a consultant in April 2009, serving as Principal Accounting Officer. Since March 2006, Mr. Warusz has been providing consulting services to a broad range of clients in the life sciences sector. From August 2005 to March 2006, Mr. Warusz was Vice President, Finance, of Orchid Cellmark Inc. (formerly known as Orchid Biosciences, Inc.). Mr. Warusz is a Certified Public Accountant and holds an undergraduate degree in accounting and an MBA from Drexel University.
AVAILABLE INFORMATION
We file annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission, or the Commission. You may read and copy any document we file with the Commission at the Commission’s public reference rooms at 100 F Street, N.E., Washington, D.C. 20549. Please call the Commission at 1-800-SEC-0330 for further information on the public reference room. Our Commission filings are also available to the public from the Commission’s Website at “http://www.sec.gov.” We make available free of charge our annual, quarterly and current reports, proxy statements and other information upon request. To request such materials, please send an e-mail to jwarusz@novadel.com or contact Joseph Warusz, our Principal Accounting Officer, at 1200 Route 22 East, Suite 2000, Bridgewater, New Jersey, 08807or at 908-203-4643.
We maintain a website at “http://www.novadel.com” (this is not a hyperlink; you must visit this website through an Internet browser). Our website and the information contained therein or connected thereto are not incorporated into this Annual Report on Form 10-K.
This report contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those discussed in this report. Factors that could cause or contribute to these differences include, but are not limited to, those discussed below, elsewhere in this report, and in any documents incorporated in this report by reference.
RISKS RELATED TO OUR BUSINESS
OUR AUDITORS HAVE EXPRESSED SUBSTANTIAL DOUBT ABOUT OUR ABILITY TO CONTINUE AS A GOING CONCERN.
Our audited financial statements for the year ended December 31, 2009, were prepared under the assumption that we will continue our operations as a going concern. We were incorporated in 1982, and have a history of losses. As a result, our independent registered public accounting firm in their audit report on our 2009 Financial Statements has expressed substantial doubt about our ability to continue as a going concern. Continued operations are dependent on our ability to complete equity or debt formation activities or to generate profitable operations. Given the recent downturn in the economy, such capital formation activities may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we cannot continue as a viable entity, our stockholders may lose some or all of their investment in us.
WE WILL REQUIRE SIGNIFICANT ADDITIONAL CAPITAL TO FUND OUR OPERATIONS.
Our operations to date have required significant cash expenditures. Our future capital requirements will depend on the results of our research and development activities, and preclinical studies.
We have significantly reduced clinical development activities on our product candidate pipeline since the fourth quarter 2007 and continuing throughout 2009, limiting our expenditures primarily to those required to support our two approved products NitroMist™ and Zolpimist™. We have initiated product development of Duromist™, an oral spray of sildenafil citrate, for the treatment of erectile dysfunction. We will need to obtain more funding in the future through collaborations or other arrangements with research institutions and corporate partners or public and private offerings of our securities, including debt or equity financing, to complete the development of this product and other products in our product development pipeline.
On October 27, 2009, we entered into a licensing agreement with privately-held Mist Acquisition, LLC to manufacture and commercialize the NitroMist™ lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris. Under the terms of the agreement, we received a $1,000,000 licensing fee upon execution of the agreement, and we will receive milestone payments totaling an additional $1,000,000 over the next twelve months and ongoing performance payments of up to seventeen percent (17%) of net sales.
On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ in the United States and Canada. Under the terms of the agreement, we received a $3,000,000 licensing fee and will receive ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products.
In addition, on December 31, 2009, we entered into an amendment agreement with ProQuest Investments L.P. and its affiliates, referred to herein as ProQuest, to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued but unpaid interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of our common stock as of December 31, 2009.
We have entered into a common stock purchase agreement with Seaside 88, LP, whereby Seaside 88, LP will purchase 500,000 shares of common stock in a series of closings occurring every two weeks for a total of up to 26 closings, provided that the 3 day volume weighed average price prior to the scheduled closing is greater than or equal to the stated floor price of $0.25 per share. We have received $1,055,000 in gross proceeds for the closings that have occurred through December 31, 2009. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP as of such date.
We may not be able to obtain adequate funds for our operations from these sources when needed or on acceptable terms. Future collaborations or similar arrangements may require us to license valuable intellectual property to, or to share substantial economic benefits with, our collaborators. If we raise additional capital by issuing additional equity or securities convertible into equity, our stockholders may experience dilution and our share price may decline. Any debt financing may result in restrictions on our spending.
If we are unable to raise additional funds, we will need to do one or more of the following:
|
|
·
|
further delay, scale-back or eliminate some or all of our research and product development programs;
|
|
|
·
|
license third parties to develop and commercialize products or technologies that we would otherwise seek to develop and commercialize ourselves;
|
|
|
·
|
attempt to sell our company;
|
|
|
·
|
cease operations; or
|
|
|
·
|
declare bankruptcy.
|
We are seeking to raise additional capital in 2010 to fund our operations and future development. A capital raise could include the securing of funds through new strategic partnerships or collaborations, the sale of common stock or other equity securities or the issuance of debt. In the event we do not enter into a license agreement or other strategic transaction in which we receive an upfront fee or payment, or we do not undertake a financing of debt or equity securities, we may not have sufficient cash on hand to fund operations. We can give no assurances that we will be able to enter into a strategic transaction or raise any additional capital or if we do, that such additional capital will be sufficient to meet our needs, or on terms favorable to us.
If we are unable to raise additional capital, and we do not use our existing working capital to fund our development plans, we will have sufficient cash on hand to fund operating costs through the fourth quarter 2010.
WE WILL REQUIRE SIGNIFICANT CAPITAL FOR PRODUCT DEVELOPMENT AND COMMERCIALIZATION IN THE NEAR TERM.
The research, development, testing and approval of our product candidates involve significant expenditures, and, accordingly, we require significant capital to fund such expenditures. Due to our small revenue base, negative working capital and, until recently, our relative inability to increase the number of development agreements with pharmaceutical companies, we have been unable to pursue aggressively our product development strategy. Until and unless our operations generate significant revenues and cash flow, we will attempt to continue to fund operations from cash on hand, license agreements and sale of equity securities. Our long-term liquidity is contingent upon achieving sales and positive cash flows from operating activities, and/or obtaining additional financing. The most likely sources of financing include private placements of our equity or debt securities or bridge loans to us from third-party lenders, license payments from current and future partners, and royalty payments from sales of approved product candidates by partners. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs, or on terms favorable to us. Since the fourth quarter 2007 and continuing throughout 2009, we have significantly reduced clinical development activities on our product candidate pipeline, such that we have limited our expenditures primarily to those required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation development activities for certain other products, as we did not believe that we had sufficient cash to sustain such activities.
WE ARE A PRE-COMMERCIALIZATION COMPANY, HAVE A LIMITED OPERATING HISTORY AND HAVE NOT GENERATED ANY REVENUES FROM THE SALE OF PRODUCTS TO DATE.
We are a pre-commercialization specialty pharmaceutical company developing oral spray formulations of a broad range of marketed treatments. There are many uncertainties and complexities with respect to such companies. We have not generated any revenue from the commercial sale of our proposed products, however our licensees for Nitromist and Zolpimist are expected to commercially launch these products in the second half of 2010. This limited history may not be adequate to enable one to fully assess our ability to develop our technologies and proposed products, obtain U.S. Food and Drug Administration, or FDA, approval and achieve market acceptance of our proposed products and respond to competition. The filing of a New Drug Application, or NDA, with the FDA is an important step in the approval process in the U.S. Acceptance for filing by the FDA does not mean that the NDA has been or will be approved, nor does it represent an evaluation of the adequacy of the data submitted. We cannot be certain as to when to anticipate commercializing and marketing any of our product candidates in development, if at all, and do not expect to generate sufficient revenues from proposed product sales to cover our expenses or achieve profitability in the near future.
We had an accumulated deficit as of December 31, 2009 of approximately $82,766,000. We incurred losses in each of our last ten fiscal years, including net losses of approximately $7,577,000 for the year ended December 31, 2009, $9,586,000 for the year ended December 31, 2008, and $16,963,000 for the year ended December 31, 2007. Additionally, we have reported negative cash flows from operations of approximately $1,578,000 for the year ended December 31, 2009, $5,533,000 for the year ended December 31, 2008, and $15,240,000 for the year ended December 31, 2007. We anticipate that, even with our limited research and development activities, we could incur substantial operating expenses in connection with continued research and development, clinical trials, testing and approval of our proposed products, and expect these expenses will result in continuing and, perhaps, significant operating losses until such time, if ever, that we are able to achieve adequate product sales levels. Our ability to generate revenue and achieve profitability depends upon our ability, alone or with others, to complete the development of our product candidates, obtain the required regulatory approvals and manufacture, market and sell our product candidates.
OUR ADDITIONAL FINANCING REQUIREMENTS COULD RESULT IN DILUTION TO EXISTING STOCKHOLDERS.
The additional financings we require may be obtained through one or more transactions which effectively dilute the ownership interests of our existing stockholders. Given the recent downturn in the economy, we may not be able to secure such additional financing on terms acceptable to us, if at all. We have the authority to issue additional shares of our common stock, as well as additional classes or series of ownership interests or debt obligations which may be convertible into any one or more classes or series of ownership interests. We are authorized to issue a total of 200,000,000 shares of common stock and 1,000,000 shares of preferred stock. Such securities may be issued without the approval or other consent of our stockholders.
OUR TECHNOLOGY PLATFORM IS BASED SOLELY ON OUR PROPRIETARY DRUG DELIVERY TECHNOLOGY. OUR ONGOING CLINICAL TRIALS FOR CERTAIN OF OUR PRODUCT CANDIDATES MAY BE DELAYED, OR FAIL, WHICH WILL HARM OUR BUSINESS.
Our strategy is to concentrate our product development activities primarily on pharmaceutical products for which there already are significant prescription sales, where the use of our proprietary, novel drug delivery technology could potentially enhance speed of onset of therapeutic effect, could potentially reduce side effects through a reduction of the amount of active drug substance required to produce a given therapeutic effect and improve patient convenience or compliance.
Companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier trials. Data obtained from tests are susceptible to varying interpretations which may delay, limit or prevent regulatory approval. In addition, companies may be unable to enroll patients quickly enough to meet expectations for completing clinical trials. The timing and completion of current and planned clinical trials of our product candidates depend on, among other factors, the rate at which patients are enrolled, which is a function of many factors, including:
|
|
·
|
the number of clinical sites;
|
|
|
·
|
the size of the patient population;
|
|
|
·
|
the proximity of patients to the clinical sites;
|
|
|
·
|
the eligibility criteria for the study;
|
|
|
·
|
the existence of competing clinical trials; and
|
|
|
·
|
the existence of alternative available products.
|
Delays in patient enrollment in clinical trials may occur, which would likely result in increased costs, program delays or both.
THERE ARE CERTAIN INTERLOCKING RELATIONSHIPS AND POTENTIAL CONFLICTS OF INTEREST.
As of March 24, 2010, ProQuest, a significant stockholder, directly and indirectly, of us, beneficially owns approximately 40.6% of our outstanding common stock (assuming full exercise of certain warrants held by ProQuest). As such, ProQuest may be deemed to be our affiliate. Mr. Steven B. Ratoff, our Chairman, President, Chief Executive Officer and Interim Chief Financial Officer, has served as a venture partner with ProQuest since December 2004, although he has no authority for investment decisions by ProQuest.
OUR BUSINESS AND REVENUE IS DEPENDENT ON THE SUCCESSFUL DEVELOPMENT OF OUR PRODUCTS.
Revenue received from our product development efforts consists of payments by pharmaceutical companies for research and bioavailability studies, pilot clinical trials and similar milestone-related payments. Our future growth and profitability will be dependent upon our ability to successfully raise additional funds to complete the development of, obtain regulatory approvals for and license out or market our product candidates. Accordingly, our prospects must be considered in light of the risks, expenses and difficulties frequently encountered in connection with the establishment of a new business in a highly competitive industry, characterized by frequent new product introductions. We anticipate that we will incur substantial operating expenses in connection with the development, testing and approval of our product candidates and expect these expenses to result in continuing and significant operating losses until such time, if ever, that we are able to achieve adequate levels of sales or license revenues. We may not be able to raise additional financing, increase revenues significantly, or achieve profitable operations.
SOME OF OUR PRODUCT CANDIDATES ARE IN EARLY STAGES OF CLINICAL DEVELOPMENT AND SOME ARE IN PRECLINICAL TESTING, WHICH MAY AFFECT OUR ABILITY OR THE TIME WE REQUIRE TO OBTAIN NECESSARY REGULATORY APPROVALS.
Some of our product candidates are in early stages of clinical development and some are in preclinical testing. These product candidates are continuously evaluated and assessed and are often subject to changes in formulation and technology. The regulatory requirements governing these types of products may be less well defined or more rigorous than for conventional products. As a result, we may experience delays with our preclinical and clinical testing, and a longer and more expensive regulatory process in connection with obtaining regulatory approvals of these types of product candidates as compared to others in our pipeline at later stages of development. These delays may negatively affect our business and operations.
WE DO NOT HAVE COMMERCIALLY AVAILABLE PRODUCTS.
Our principal efforts are the development of obtaining regulatory approvals for and licensing our product candidates. We anticipate that marketing activities by our licensees for our two approved products, will not begin until the second half of 2010.
There can be no assurances that our licensees will successfully market out two approved product candidates, or that such product candidates will become commercially available.
WE HAVE NOT COMPLETED PRODUCT DEVELOPMENT.
We have not completed the development of our product candidates and we will be required to devote considerable effort and expenditures to complete such development. In addition to obtaining adequate financing, satisfactory completion of development, testing, government approval and sufficient production levels of such product candidates must be obtained before the product candidates will become available for commercial sale. We have recently obtained strategic partners for both NitroMist™ and Zolpimist™. Other potential products remain in the conceptual or very early development stage and remain subject to all the risks inherent in the development of pharmaceutical products, including unanticipated development problems and possible lack of funds to undertake or continue development. These factors could result in abandonment or substantial change in the development of a specific formulated product. We may not be able to successfully develop any one or more of our product candidates or develop such product candidates on a timely basis. Further, such product candidates may not be commercially accepted if developed. The inability to successfully complete development, or a determination by us, for financial or other reasons, not to undertake to complete development of any product candidates, particularly in instances in which we have made significant capital expenditures, could have a material adverse effect on our business and operations.
WE DO NOT HAVE DIRECT CONSUMER MARKETING EXPERIENCE.
We have no experience in marketing or distribution at the consumer level of our product candidates. Moreover, we do not have the financial or other resources to undertake extensive marketing and advertising activities. Accordingly, we intend generally to rely on marketing arrangements, including possible joint ventures or license or distribution arrangements with third-parties. Except for our agreements with Mist, ECR, BioAlliance, Par, Manhattan Pharmaceuticals, Velcera and Hana Biosciences, we have not entered into any significant agreements or arrangements with respect to the marketing of our product candidates. We may not be able to enter into any such agreements or similar arrangements in the future and we may not be able to successfully market our products. If we fail to enter into these agreements or if we or the third parties do not perform under such agreements, it could impair our ability to commercialize our products.
We have stated our intention to possibly market our own products in the future, although we have no such experience to date. Substantial investment will be required in order to build infrastructure and provide resources in support of marketing our own products, particularly the establishment of a marketing force. If we do not develop a marketing force of our own, then we will depend on arrangements with corporate partners or other entities for the marketing and sale of our remaining products. The establishment of our own marketing force, or a strategy to rely on third party marketing arrangements, could adversely affect our profit margins.
WE MUST COMPLY WITH GOOD MANUFACTURING PRACTICES.
The manufacture of our pharmaceutical products under development will be subject to current Good Manufacturing Practices, or cGMP, prescribed by the FDA, pre-approval inspections by the FDA or comparable foreign authorities, or both, before commercial manufacture of any such products and periodic cGMP compliance inspections thereafter by the FDA. We, or any of our third party manufacturers, may not be able to comply with cGMP or satisfy pre- or post-approval inspections by the FDA or comparable foreign authorities in connection with the manufacture of our product candidates. Failure or delay by us or any such manufacturer to comply with cGMP or satisfy pre- or post-approval inspections would have a material adverse effect on our business and operations.
WE ARE DEPENDENT ON OUR SUPPLIERS.
We believe that the active ingredients used in the manufacture of our product candidates are presently available from numerous suppliers located in the U.S., Europe, India and Japan. We believe that certain raw materials, including inactive ingredients, are available from a limited number of suppliers and that certain packaging materials intended for use in connection with our spray products currently are available only from sole source suppliers. Although we do not believe we will encounter difficulties in obtaining the inactive ingredients or packaging materials necessary for the manufacture of our product candidates, we may not be able to enter into satisfactory agreements or arrangements for the purchase of commercial quantities of such materials.
In February 2008, we entered into a Master Services Agreement with Rechon Life Sciences (Malmo, Sweden), whereby Rechon will provide services related to the manufacturing development and the manufacture of clinical supplies for our products. Rechon provides these services on a fee-for-service basis.
On December 28, 2009, DPT Laboratories became our contract manufacturer for Duromist, sildenafil citrate oral spray.
With respect to other suppliers, we operate primarily on a purchase order basis beyond which there is no contract memorializing our purchasing arrangements. The inability to enter into agreements or otherwise arrange for adequate or timely supplies of principal raw materials and the possible inability to secure alternative sources of raw material supplies, or the failure of DPT Laboratories, or Rechon Life Sciences to comply with their supply obligations to us, could have a material adverse effect on our ability to arrange for the manufacture of formulated products. In addition, development and regulatory approval of our products are dependent upon our ability to procure active ingredients and certain packaging materials from FDA-approved sources. Since the FDA approval process requires manufacturers to specify their proposed suppliers of active ingredients and certain packaging materials in their applications, FDA approval of a supplemental application to use a new supplier would be required if active ingredients or such packaging materials were no longer available from the originally specified supplier, which may result in manufacturing delays. If we do not maintain important manufacturing relationships, we may fail to find a replacement manufacturer or to develop our own manufacturing capabilities. If we cannot do so, it could delay or impair our ability to obtain regulatory approval for our products and substantially increase our costs or deplete any profit margins. If we do find replacement manufacturers, we may not be able to enter into agreements with them on terms and conditions favorable to us and, there could be a substantial delay before a new facility could be qualified and registered with the FDA and foreign regulatory authorities.
FAILURE TO ACHIEVE AND MAINTAIN EFFECTIVE INTERNAL CONTROLS IN ACCORDANCE WITH SECTION 404 OF THE SARBANES-OXLEY ACT OF 2002 COULD HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS AND OPERATING RESULTS. IN ADDITION, CURRENT AND POTENTIAL STOCKHOLDERS COULD LOSE CONFIDENCE IN OUR FINANCIAL REPORTING, WHICH COULD HAVE A MATERIAL ADVERSE EFFECT ON OUR STOCK PRICE.
Effective internal controls are necessary for us to provide reliable financial reports and effectively prevent fraud. If we cannot provide reliable financial reports or prevent fraud, our operating results and financial condition could be harmed.
We are required to document and test our internal control procedures in order to satisfy the requirements of Section 404 of the Sarbanes-Oxley Act of 2002, which requires annual management assessments of the effectiveness of our internal controls over financial reporting. During the course of our testing we may identify deficiencies which we may not be able to remediate in time to meet the deadline imposed by the Sarbanes-Oxley Act of 2002 for compliance with the requirements of Section 404. In addition, if we fail to maintain the adequacy of our internal controls, as such standards are modified, supplemented or amended from time to time, we may not be able to ensure that we can conclude on an ongoing basis that we have effective internal controls over financial reporting in accordance with Section 404 of the Sarbanes-Oxley Act of 2002. Failure to achieve and maintain an effective internal control environment could also cause investors to lose confidence in our reported financial information, which could have a material adverse effect on the price of our common stock.
COMPLIANCE WITH CHANGING REGULATION OF CORPORATE GOVERNANCE AND PUBLIC DISCLOSURE MAY RESULT IN ADDITIONAL EXPENSES.
Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new regulations promulgated by the Securities and Exchange Commission, or SEC, and NYSE Amex, or NYSE Amex rules, are creating uncertainty for companies such as ours. These new or changed laws, regulations and standards are subject to varying interpretations in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies, which could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, our efforts to comply with evolving laws, regulations and standards have resulted in, and are likely to continue to result in, increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. In particular, our recent efforts to comply with Section 404 of the Sarbanes-Oxley Act of 2002 and the related regulations regarding our required assessment of our internal controls over financial reporting and our independent registered public accounting firm’s audit of that assessment requires the commitment of significant financial and managerial resources. In addition, it has become more difficult and more expensive for us to obtain director and officer liability insurance. We expect these efforts to require the continued commitment of significant resources. Further, our Board members, Chief Executive Officer and Chief Financial Officer could face an increased risk of personal liability in connection with the performance of their duties. As a result, we may have difficulty attracting and retaining qualified board members and executive officers, which could harm our business. If our efforts to comply with new or changed laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, our reputation may be harmed.
WE FACE INTENSE COMPETITION.
The markets which we intend to enter are characterized by intense competition. We, or our licensees, may be competing against established, larger and/or better capitalized pharmaceutical companies with currently marketed products which are equivalent or functionally similar to those we intend to market. Prices of drug products are significantly affected by competitive factors and tend to decline as competition increases. In addition, numerous companies are developing or may, in the future, engage in the development of products competitive with our product candidates. We expect that technological developments will occur at a rapid rate and that competition is likely to intensify as enhanced dosage from technologies gain greater acceptance. Additionally, the markets for formulated products which we have targeted for development are intensely competitive, involving numerous competitors and products. Most of our prospective competitors possess substantially greater financial, technical and other resources than we do. Moreover, many of these companies possess greater marketing capabilities than we do, including the resources necessary to enable them to implement extensive advertising campaigns. We may not be able to compete successfully with such competitors.
Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA or comparable foreign approval or commercializing products before us. If we commence commercial product sales, we will compete against companies with greater marketing and manufacturing capabilities who may successfully develop and commercialize products that are more effective or less expensive than ours. Our competitors may be more successful in receiving third party reimbursements from government agencies and others for their commercialized products which are similar to our products. If we cannot receive third party reimbursement for our products, we may not be able to commercialize our products. These are areas in which, as yet, we have limited or no experience. In addition, developments by our competitors may render our product candidates obsolete or noncompetitive.
We also face, and will continue to face, competition from colleges, universities, governmental agencies and other public and private research organizations. These competitors are becoming more active in seeking patent protection and licensing arrangements to collect royalties for use of technology that they have developed. Some of these technologies may compete directly with the technologies that we are developing. These institutions will also compete with us in recruiting highly qualified scientific personnel. We expect that developments in the areas in which we are active may occur at a rapid rate and that competition will intensify as advances in this field are made. As a result, we need to continue to devote substantial resources and efforts to research and development activities.
LIMITED PRODUCT LIABILITY INSURANCE COVERAGE MAY AFFECT OUR BUSINESS.
We may be exposed to potential product liability claims by end-users of our products. Although we obtain product liability insurance per contractual obligations, before the commercialization of any of our product candidates, we cannot guarantee such insurance will be sufficient to cover all possible liabilities to which we may be exposed. Any product liability claim, even one that was not in excess of our insurance coverage or one that is meritless and/or unsuccessful, could adversely affect our cash available for other purposes, such as research and development. In addition, the existence of a product liability claim could affect the market price of our common stock. In addition, certain food and drug retailers require minimum product liability insurance coverage as a condition precedent to purchasing or accepting products for retail distribution. Product liability insurance coverage includes various deductibles, limitations and exclusions from coverage, and in any event might not fully cover any potential claims. Failure to satisfy such insurance requirements could impede the ability of us or our distributors to achieve broad retail distribution of our product candidates, which could have a material adverse effect on us.
EXTENSIVE GOVERNMENT REGULATION MAY AFFECT OUR BUSINESS.
The development, manufacture and commercialization of pharmaceutical products is generally subject to extensive regulation by various federal and state governmental entities. The FDA, which is the principal U.S. regulatory authority over pharmaceutical products, has the power to seize adulterated or misbranded products and unapproved new drugs, to request their recall from the market, to enjoin further manufacture or sale, to publicize certain facts concerning a product and to initiate criminal proceedings. As a result of federal statutes and FDA regulations pursuant to which new pharmaceuticals are required to undergo extensive and rigorous testing, obtaining pre-market regulatory approval requires extensive time and expenditures. Under the Federal Food, Drug, and Cosmetic Act, or FFDCA, as amended (21 U.S.C. 301 et. seq.), a new drug may not be commercialized or otherwise distributed in the U.S. without the prior approval of the FDA or pursuant to an applicable exemption from the FFDCA. The FDA approval processes relating to new drugs differ, depending on the nature of the particular drug for which approval is sought. With respect to any drug product with active ingredients not previously approved by the FDA, a prospective drug manufacturer is required to submit an NDA, which includes complete reports of pre-clinical, clinical and laboratory studies to prove such product’s safety and efficacy. Prior to submission of the NDA, it is necessary to submit an Investigational New Drug, or IND, to obtain permission to begin clinical testing of the new drug. Such clinical trials are required to meet good clinical practices under the FFDCA. Given that our current product candidates are based on a new technology for formulation and delivery of active pharmaceutical ingredients that have been previously approved and that have been shown to be safe and effective in previous clinical trials, we believe that we will be eligible to submit what is known as a 505(b)(2). We estimate that the development of new formulations of pharmaceutical products, including formulation, testing and NDA submission, generally takes two to three years under the 505(b)(2) NDA process. Our determinations may prove to be inaccurate or pre-marketing approval relating to our proposed products may not be obtained on a timely basis, if at all. The failure by us to obtain necessary regulatory approvals, whether on a timely basis or at all, would have a material adverse effect on our business. The filing of an NDA with the FDA is an important step in the approval process in the U.S. Acceptance for filing by the FDA does not mean that the NDA has been or will be approved, nor does it represent an evaluation of the adequacy of the data submitted.
THE CLINICAL TRIAL AND REGULATORY APPROVAL PROCESS FOR OUR PRODUCTS IS EXPENSIVE AND TIME CONSUMING, AND THE OUTCOME IS UNCERTAIN.
In order to sell our proposed products, we must receive separate regulatory approvals for each product. The FDA and comparable agencies in foreign countries extensively and rigorously regulate the testing, manufacture, distribution, advertising, pricing and marketing of drug products like our products. This approval process for an NDA includes preclinical studies and clinical trials of each pharmaceutical compound to establish its safety and effectiveness and confirmation by the FDA and comparable agencies in foreign countries that the manufacturer maintains good laboratory and manufacturing practices during testing and manufacturing. Clinical trials generally take two to five years or more to complete. Even if favorable testing data is generated by clinical trials of drug products, the FDA may not accept an NDA submitted by a pharmaceutical or biotechnology company for such drug product for filing, or if accepted for filing, may not approve such NDA.
The approval process is lengthy, expensive and uncertain. It is also possible that the FDA or comparable foreign regulatory authorities could interrupt, delay or halt any one or more of our clinical trials. If we, or any regulatory authorities, believe that trial participants face unacceptable health risks, any one or more of our trials could be suspended or terminated. We also may fail to reach agreement with the FDA and/or comparable foreign agencies on the design of any one or more of the clinical studies necessary for approval. Conditions imposed by the FDA and comparable agencies in foreign countries on our clinical trials could significantly increase the time required for completion of such clinical trials and the costs of conducting the clinical trials. Data obtained from clinical trials are susceptible to varying interpretations which may delay, limit or prevent regulatory approval.
Delays and terminations of the clinical trials we conduct could result from insufficient patient enrollment. Patient enrollment is a function of several factors, including the size of the patient population, stringent enrollment criteria, the proximity of the patients to the trial sites, having to compete with other clinical trials for eligible patients, geographical and geopolitical considerations and others. Delays in patient enrollment can result in greater costs and longer trial timeframes. Patients may also suffer adverse medical events or side effects.
The FDA and comparable foreign agencies may withdraw any approvals we obtain. Further, if there is a later discovery of unknown problems or if we fail to comply with other applicable regulatory requirements at any stage in the regulatory process, the FDA may restrict or delay our marketing of a product or force us to make product recalls. In addition, the FDA could impose other sanctions such as fines, injunctions, civil penalties or criminal prosecutions. To market our products outside the U.S., we also need to comply with foreign regulatory requirements governing human clinical trials and marketing approval for pharmaceutical products. Other than the approval of NitroMist™ and ZolpiMist™, the FDA and foreign regulators have not yet approved any of our products under development for marketing in the U.S. or elsewhere. If the FDA and other regulators do not approve any one or more of our products under development, we will not be able to market such products.
WE EXPECT TO FACE UNCERTAINTY OVER REIMBURSEMENT AND HEALTHCARE REFORM.
In both the U.S. and other countries, sales of our products will depend in part upon the availability of reimbursement from third-party payers, which include government health administration authorities, managed care providers and private health insurers. Third-party payers are increasingly challenging the price and examining the cost effectiveness of medical products and services.
OUR STRATEGY INCLUDES ENTERING INTO COLLABORATION AGREEMENTS WITH THIRD PARTIES FOR CERTAIN OF OUR PRODUCT CANDIDATES AND WE MAY REQUIRE ADDITIONAL COLLABORATION AGREEMENTS. IF WE FAIL TO ENTER INTO THESE AGREEMENTS OR IF WE OR THE THIRD PARTIES DO NOT PERFORM UNDER SUCH AGREEMENTS, IT COULD IMPAIR OUR ABILITY TO COMMERCIALIZE OUR PROPOSED PRODUCTS.
Our strategy for the completion of the required development and clinical testing of certain of our product candidates and for the manufacturing, marketing and commercialization of such product candidates includes entering into collaboration arrangements with pharmaceutical companies to market, commercialize and distribute the products.
Through December 31, 2008, we entered into strategic license agreements with: (i) Hana Biosciences, for the development and marketing rights in the U.S. and Canada for our ondansetron oral spray, (ii) Par, for the marketing rights in the U.S. and Canada for NitroMist™, (iii) Manhattan Pharmaceuticals, in connection with propofol, (iv) Velcera, in connection with veterinary applications for currently marketed veterinary drugs and (v) BioAlliance Pharma SA, for the European rights for Ondansetron oral spray. Subsequent to December 31, 2008, the following events occurred with respect our strategic license agreements:
On October 27, 2009, we entered into a license and distribution agreement with privately-held Mist Acquisition, LLC to manufacture and commercialize the NitroMist™ lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris, in the United States, Canada and Mexico. Under the terms of the agreement, we received a $1,000,000 licensing fee upon execution of the agreement, and will receive milestone payments totaling an additional $1,000,000 over the next twelve months and ongoing performance payments of up to seventeen percent (17%) of net sales subject to potential reduction, subject to the terms of the agreement.
On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture the Company’s ZolpiMist™ in the United States and Canada. ZolpiMist™ is our oral spray formulation of zolpidem tartrate, which was approved by the FDA in December of 2008. Under the terms of the agreement, ECR paid us $3,000,000 upon the execution of the agreement and will pay ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products, subject to the terms of the agreement.
Our success depends upon obtaining additional collaboration partners and maintaining our relationships with our current partners. In addition, we may depend on our partners’ expertise and dedication of sufficient resources to develop and commercialize proposed products. We may, in the future, grant to collaboration partners, rights to license and commercialize pharmaceutical products developed under collaboration agreements. Under these arrangements, our collaboration partners may control key decisions relating to the development of the products. The rights of our collaboration partners could limit our flexibility in considering alternatives for the commercialization of such product candidates. If we fail to successfully develop these relationships or if our collaboration partners fail to successfully develop or commercialize such product candidates, it may delay or prevent us from developing or commercializing our proposed products in a competitive and timely manner and would have a material adverse effect on our business.
IF WE CANNOT PROTECT OUR INTELLECTUAL PROPERTY, OTHER COMPANIES COULD USE OUR TECHNOLOGY IN COMPETITIVE PRODUCTS. IF WE INFRINGE THE INTELLECTUAL PROPERTY RIGHTS OF OTHERS, OTHER COMPANIES COULD PREVENT US FROM DEVELOPING OR MARKETING OUR PRODUCTS.
We seek patent protection for our technology so as to prevent others from commercializing equivalent products in substantially less time and at substantially lower expense. The pharmaceutical industry places considerable importance on obtaining patent and trade secret protection for new technologies, products and processes. Our success will depend in part on our ability and that of parties from whom we license technology to:
|
|
·
|
defend our patents and otherwise prevent others from infringing on our proprietary rights;
|
|
|
·
|
protect our trade secrets; and
|
|
|
·
|
operate without infringing upon the proprietary rights of others, both in the U.S. and in other countries.
|
The patent position of firms relying upon biotechnology is highly uncertain and involves complex legal and factual questions for which important legal principles are unresolved. To date, the U.S. Patent and Trademark Office, or USPTO, has not adopted a consistent policy regarding the breadth of claims that the USPTO allows in biotechnology patents or the degree of protection that these types of patents afford. As a result, there are risks that we may not develop or obtain rights to products or processes that are or may seem to be patentable.
Section 505(b)(2) of the FFDCA was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. For example, the Hatch-Waxman Act permits an applicant to rely upon the FDA’s findings of safety and effectiveness for an approved product. The FDA may also require companies to perform one or more additional studies or measurements to support the change from the approved product. The FDA may then approve the new formulation for all or some of the label indications for which the referenced product has been approved, or a new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s findings for an already-approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (1) the required patent information has not been filed (paragraph I certification); (2) the listed patent has expired (paragraph II certification); (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration (paragraph III certification); or (4) the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new product (paragraph IV certification). If the applicant does not challenge the listed patents, the Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced product have expired, and once any pediatric exclusivity expires. The Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired.
If the applicant has provided a paragraph IV certification to the FDA, the applicant must also send notice of the paragraph IV certification to the NDA holder and patent owner once the NDA has been accepted for filing by the FDA. The NDA holder and patent owner may then initiate a legal challenge to the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of their receipt of a paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA until the earliest of 30 months, expiration of the patent, settlement of the lawsuit or a decision in an infringement case that is favorable to the Section 505(b)(2) applicant. Thus, a Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its products only to be subject to significant delay and patent litigation before its products may be commercialized. Alternatively, if the NDA holder or patent owner does not file a patent infringement lawsuit within the required 45-day period, the applicant’s NDA will not be subject to the 30-month stay.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA changes its interpretation of Section 505(b)(2), this could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
EVEN IF WE OBTAIN PATENTS TO PROTECT OUR PRODUCTS, THOSE PATENTS MAY NOT BE SUFFICIENTLY BROAD AND OTHERS COULD COMPETE WITH US.
We, and the parties licensing technologies to us, have filed various U.S. and foreign patent applications with respect to the products and technologies under our development, and the USPTO and foreign patent offices have issued patents with respect to our products and technologies. These patent applications include international applications filed under the Patent Cooperation Treaty. Currently, we have nine patents which have been issued in the U.S. and 69 patents which have been issued outside of the U.S. Additionally, we have over 65 patents pending around the world. Our pending patent applications, those we may file in the future and those we may license from third parties, may not result in the USPTO or any foreign patent office issuing patents. Also, if patent rights covering our products are not sufficiently broad, they may not provide us with sufficient proprietary protection or competitive advantages against competitors with similar products and technologies. Furthermore, if the USPTO or foreign patent offices issue patents to us or our licensors, others may challenge the patents or circumvent the patents, or the patent office or the courts may invalidate the patents. Thus, any patents we own or license from or to third parties may not provide any protection against competitors.
Furthermore, the life of our patents is limited. Such patents, which include relevant foreign patents, expire on various dates. We have filed, and when possible and appropriate, will file, other patent applications with respect to our product candidates and processes in the U.S. and in foreign countries. We may not be able to develop additional products or processes that will be patentable or additional patents may not be issued to us. See also “Risk Factors - If We Cannot Meet Requirements Under our License Agreements, We Could Lose the Rights to our Products.”
INTELLECTUAL PROPERTY RIGHTS OF THIRD PARTIES COULD LIMIT OUR ABILITY TO MARKET OUR PRODUCTS.
Our commercial success also significantly depends on our ability to operate without infringing the patents or violating the proprietary rights of others. The USPTO keeps U.S. patent applications confidential while the applications are pending. As a result, we cannot determine which inventions third parties claim in pending patent applications that they have filed. We may need to engage in litigation to defend or enforce our patent and license rights or to determine the scope and validity of the proprietary rights of others. It will be expensive and time consuming to defend and enforce patent claims. Thus, even in those instances in which the outcome is favorable to us, the proceedings can result in the diversion of substantial resources from our other activities. An adverse determination may subject us to significant liabilities or require us to seek licenses that third parties may not grant to us or may only grant at rates that diminish or deplete the profitability of the products to us. An adverse determination could also require us to alter our products or processes or cease altogether any related research and development activities or product sales.
IF WE CANNOT MEET REQUIREMENTS UNDER OUR LICENSE AGREEMENTS, WE COULD LOSE THE RIGHTS TO OUR PRODUCTS.
We depend, in part, on licensing arrangements with third parties to maintain the intellectual property rights to our products under development. These agreements may require us to make payments and/or satisfy performance obligations in order to maintain our rights under these licensing arrangements. All of these agreements last either throughout the life of the patents, or with respect to other licensed technology, for a number of years after the first commercial sale of the relevant product.
In addition, we are responsible for the cost of filing and prosecuting certain patent applications and maintaining certain issued patents licensed to us. If we do not meet our obligations under our license agreements in a timely manner, we could lose the rights to our proprietary technology.
In addition, we may be required to obtain licenses to patents or other proprietary rights of third parties in connection with the development and use of our products and technologies. Licenses required under any such patents or proprietary rights might not be made available on terms acceptable to us, if at all.
WE RELY ON CONFIDENTIALITY AGREEMENTS THAT COULD BE BREACHED AND MAY BE DIFFICULT TO ENFORCE.
Although we believe that we take reasonable steps to protect our intellectual property, including the use of agreements relating to the non-disclosure of confidential information to third parties, as well as agreements that purport to require the disclosure and assignment to us of the rights to the ideas, developments, discoveries and inventions of our employees and consultants while we employ them, the agreements can be difficult and costly to enforce. Although we seek to obtain these types of agreements from our consultants, advisors and research collaborators, to the extent that they apply or independently develop intellectual property in connection with any of our projects, disputes may arise as to the proprietary rights to this type of information. If a dispute arises, a court may determine that the right belongs to a third party, and enforcement of our rights can be costly and unpredictable. In addition, we will rely on trade secrets and proprietary know-how that we will seek to protect in part by confidentiality agreements with our employees, consultants, advisors or others. Despite the protective measures we employ, we still face the risk that:
|
|
·
|
they will breach these agreements;
|
|
|
·
|
any agreements we obtain will not provide adequate remedies for this type of breach or that our trade secrets or proprietary know-how will otherwise become known or competitors will independently develop similar technology; and
|
|
|
·
|
our competitors will independently discover our proprietary information and trade secrets.
|
WE ARE DEPENDENT ON EXISTING MANAGEMENT AND BOARD MEMBERS.
Our success is substantially dependent on the efforts and abilities of the principal members of our management team and our directors. Decisions concerning our business and our management are and will continue to be made or significantly influenced by these individuals. The loss or interruption of their continued services could have a materially adverse effect on our business operations and prospects. Although our employment agreements with members of management generally provide for severance payments that are contingent upon the applicable officer’s refraining from competition with us, the loss of any of these persons’ services could adversely affect our ability to develop and market our products and obtain necessary regulatory approvals, and the applicable noncompetition provisions can be difficult and costly to monitor and enforce. Further, we do not maintain key-man life insurance.
Our future success also will depend in part on the continued service of our key scientific and management personnel and our ability to identify hire and retain additional personnel, including scientific, development and manufacturing staff.
RISKS RELATED TO OUR COMMON STOCK
BECAUSE OUR COMMON STOCK IS LISTED ON THE OVER-THE-COUNTER BULLETIN BOARD, THE LIQUIDITY OF OUR COMMON STOCK MAY BE IMPAIRED.
On December 24, 2009, we announced that our common stock began trading on the Over-the-Counter Bulletin Board, or OTCBB. Our new ticker symbol on OTCBB is NVDL.OB. We filed Form 25 on December 14, 2009, voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009.
Because our common stock is listed on the OTCBB, the liquidity of the common stock is impaired, not only in the
number of shares that are bought and sold, but also through delays in the timing of transactions, and limited coverage by security analysts and the news media. As a result, prices for shares of our common stock may be lower than might otherwise prevail if our common stock was traded on NYSE Amex LLC or another national securities exchange.
As of December 31, 2009, our net worth position was a deficit of $4,341,000 and as of December 31, 2008, our net worth position was a deficit of $2,741,000.
WE ARE INFLUENCED BY CURRENT STOCKHOLDERS, OFFICERS AND DIRECTORS.
Our directors, executive officers and principal stockholders and certain of our affiliates have the ability to influence the election of our directors and most other stockholder actions. As of March 24, 2010, management and our affiliates currently beneficially own, including shares they have the right to acquire, approximately 42.1% of the common stock on a fully-diluted basis. This determination of affiliate status is not necessarily a conclusive determination for other purposes. Specifically, ProQuest has the ability to exert significant influence over matters submitted to our stockholders for approval. Such positions may discourage or prevent any proposed takeover of us, including transactions in which our stockholders might otherwise receive a premium for their shares over the then current market prices. Our directors, executive officers and principal stockholders may influence corporate actions, including influencing elections of directors and significant corporate events.
THE MARKET PRICE OF OUR STOCK AND OUR EARNINGS MAY BE ADVERSELY AFFECTED BY MARKET VOLATILITY.
The market price of our common stock, like that of many other development stage pharmaceutical or biotechnology companies, has been and is likely to continue to be volatile. In addition to general economic, political and market conditions, the price and trading volume of our common stock could fluctuate widely in response to many factors, including:
|
|
·
|
announcements of the results of clinical trials by us or our competitors;
|
|
|
·
|
adverse reactions to products;
|
|
|
·
|
governmental approvals, delays in expected governmental approvals or withdrawals of any prior governmental approvals or public or regulatory agency concerns regarding the safety or effectiveness of our products;
|
|
|
·
|
changes in the U.S. or foreign regulatory policy during the period of product development;
|
|
|
·
|
developments in patent or other proprietary rights, including any third party challenges of our intellectual property rights;
|
|
|
·
|
announcements of technological innovations by us or our competitors;
|
|
|
·
|
announcements of new products or new contracts by us or our competitors;
|
|
|
·
|
actual or anticipated variations in our operating results due to the level of development expenses and other factors;
|
|
|
·
|
changes in financial estimates by securities analysts and whether our earnings meet or exceed the estimates;
|
|
|
·
|
conditions and trends in the pharmaceutical and other industries;
|
|
|
·
|
new accounting standards; and
|
|
|
·
|
the occurrence of any of the risks set forth in these Risk Factors and other reports, including this prospectus and other filings filed with the Securities and Exchange Commission from time to time.
|
Our common stock is currently listed for trading on the OTCBB under the symbol “NVDL.OB” and was previously traded on the NYSE Amex LLC from May 11, 2004 to December 23, 2009. During the twelve-month period ended December 31, 2009, the closing price of our common stock has ranged from $0.12 to $0.52. We expect the price of our common stock to remain volatile. The average daily trading volume in our common stock varies significantly. For the twelve-month period ended December 31, 2009, the average daily trading volume in our common stock was approximately 506,125 shares. Our relatively low average volume and low average number of transactions per day may affect the ability of our stockholders to sell their shares in the public market at prevailing prices and a more active market may never develop.
In the past, following periods of volatility in the market price of the securities of companies in our industry, securities class action litigation has often been instituted against companies in our industry. If we face securities litigation in the future, even if without merit or unsuccessful, it would result in substantial costs and a diversion of management attention and resources, which would negatively impact our business.
BECAUSE THE AVERAGE DAILY TRADING VOLUME OF OUR COMMON STOCK IS LOW, THE ABILITY TO SELL OUR SHARES IN THE SECONDARY TRADING MARKET MAY BE LIMITED.
Because the average daily trading volume of our common stock is low, the liquidity of our common stock may be impaired. As a result, prices for shares of our common stock may be lower than might otherwise prevail if the average daily trading volume of our common stock was higher. The average daily trading volume of our common stock may be low relative to the stocks of exchange-listed companies, which could limit investors’ ability to sell shares in the secondary trading market.
WE LIKELY WILL ISSUE ADDITIONAL EQUITY SECURITIES, WHICH WILL DILUTE CURRENT STOCKHOLDERS’ SHARE OWNERSHIP.
We likely will issue additional equity securities to raise capital and through the exercise of options and warrants that are outstanding or may be outstanding. These additional issuances will dilute current stockholders’ share ownership.
PENNY STOCK REGULATIONS MAY IMPOSE CERTAIN RESTRICTIONS ON MARKETABILITY OF OUR SECURITIES.
The SEC has adopted regulations which generally define a “penny stock” to be any equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to certain exceptions. As a result, our common stock is subject to rules that impose additional sales practice requirements on broker dealers who sell such securities to persons other than established customers and accredited investors (generally those with assets in excess of $1,000,000 or annual income exceeding $200,000, or $300,000 together with their spouse). For transactions covered by such rules, the broker dealer must make a special suitability determination for the purchase of such securities and have received the purchaser’s written consent to the transaction prior to the purchase. Additionally, for any transaction involving a penny stock, unless exempt, the rules require the delivery, prior to the transaction, of a risk disclosure document mandated by the SEC relating to the penny stock market. The broker dealer must also disclose the commission payable to both the broker dealer and the registered representative, current quotations for the securities and, if the broker dealer is the sole market maker, the broker dealer must disclose this fact and the broker dealer’s presumed control over the market. Finally, monthly statements must be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks. Broker-dealers must wait two business days after providing buyers with disclosure materials regarding a security before effecting a transaction in such security. Consequently, the “penny stock” rules restrict the ability of broker dealers to sell our securities and affect the ability of investors to sell our securities in the secondary market and the price at which such purchasers can sell any such securities, thereby affecting the liquidity of the market for our common stock.
Stockholders should be aware that, according to the SEC, the market for penny stocks has suffered in recent years from patterns of fraud and abuse. Such patterns include:
|
|
·
|
control of the market for the security by one or more broker-dealers that are often related to the promoter or issuer;
|
|
|
·
|
manipulation of prices through prearranged matching of purchases and sales and false and misleading press releases;
|
|
|
·
|
“boiler room” practices involving high pressure sales tactics and unrealistic price projections by inexperienced sales persons;
|
|
|
·
|
excessive and undisclosed bid-ask differentials and markups by selling broker-dealers; and
|
|
|
·
|
the wholesale dumping of the same securities by promoters and broker-dealers after prices have been manipulated to a desired level, along with the inevitable collapse of those prices with consequent investor losses.
|
Our management is aware of the abuses that have occurred historically in the penny stock market.
ADDITIONAL AUTHORIZED SHARES OF OUR COMMON STOCK AND PREFERRED STOCK AVAILABLE FOR ISSUANCE MAY ADVERSELY AFFECT THE MARKET.
We are authorized to issue a total of 200,000,000 shares of common stock and 1,000,000 shares of preferred stock. Such securities may be issued without the approval or other consent of our stockholders. As of March 24, 2010, there were 89,283,000 shares of common stock issued and outstanding. However, the total number of shares of our common stock issued and outstanding does not include shares reserved in anticipation of the exercise of options or warrants. As of March 24, 2010, we had outstanding stock options and warrants to purchase approximately 28.1 million shares of common stock, the exercise prices of which range between $0.17 per share and $3.18 per share, and we have reserved shares of our common stock for issuance in connection with the potential exercise thereof.
On July 16, 2009, we received approval from the NYSE Amex LLC to issue up to 12,000,000 shares over the next twelve (12) months. We have entered into a common stock purchase agreement with Seaside 88, LP, whereby Seaside 88, LP will purchase 500,000 shares of common stock in a series of closings occurring every two weeks for a total of up to 26 closings, provided that the 3 day volume weighed average price prior to the scheduled closing is greater than or equal to the stated floor price of $0.25 per share. We have received $1,055,000 in gross proceeds for the closings that have occurred through December 31, 2009. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP as of such date.
The following table provides an overview of our stock options and corresponding plans, as of December 31, 2009:
|
Plan
|
Shares Authorized
|
Options Outstanding at December 31, 2009
|
Remaining Shares Available for Issuance
|
Comments
|
||||
|
1992 Stock Option Plan
|
500,000
|
40,000
|
—
|
Plan Closed
|
||||
|
1997 Stock Option Plan
|
500,000
|
50,000
|
—
|
Plan Closed
|
||||
|
1998 Stock Option Plan
|
3,400,000
|
2,414,000
|
691,000
|
—
|
||||
|
2006 Equity Incentive Plan
|
6,000,000
|
5,195,000
|
180,000
|
—
|
||||
|
Non-Plan
|
n/a
|
581,000
|
—
|
|||||
|
Total
|
10,400,000
|
8,280, 000
|
871,000
|
As of March 24, 2010, there are 2,414,000 and 5,195,000 options outstanding under the 1998 Stock Option Plan and the 2006 Equity Incentive Plan, respectively. As a result, as of March 24, 2010, 691,000 and 180,000 shares remain available for issuance under the 1998 Stock Option Plan and the 2006 Equity Incentive Plan, respectively.
To the extent such options or warrants are exercised, the holders of our common stock will experience further dilution.
In addition, in the event that any future financing should be in the form of, be convertible into or exchangeable for, equity securities, and upon the exercise of options and warrants, investors may experience additional dilution.
See “Risk Factors - Our Additional Financing Requirements Could Result In Dilution To Existing Stockholders” included herein. The exercise of the outstanding derivative securities will reduce the percentage of common stock held by our stockholders in relation to our aggregate outstanding capital stock. Further, the terms on which we could obtain additional capital during the life of the derivative securities may be adversely affected, and it should be expected that the holders of the derivative securities would exercise them at a time when we would be able to obtain equity capital on terms more favorable than those provided for by such derivative securities. As a result, any issuance of additional shares of our common stock may cause our current stockholders to suffer significant dilution which may adversely affect the market.
In addition to the above referenced shares of our common stock which may be issued without stockholder approval, we have 1,000,000 shares of authorized preferred stock, the terms of which may be fixed by our Board. We presently have no issued and outstanding shares of preferred stock and while we have no present plans to issue any shares of preferred stock, our Board has the authority, without stockholder approval, to create and issue one or more series of such preferred stock and to determine the voting, dividend and other rights of holders of such preferred stock. The issuance of any of such series of preferred stock may have an adverse effect on the holders of our common stock.
SHARES ELIGIBLE FOR FUTURE SALE MAY ADVERSELY AFFECT THE MARKET.
From time to time, certain of our stockholders may be eligible to sell all or some of their shares of our common stock by means of ordinary brokerage transactions in the open market pursuant to Rule 144, promulgated under the Securities Act of 1933, as amended, subject to certain limitations. In general, pursuant to Rule 144, a stockholder (or stockholders whose shares are aggregated) who has satisfied a six-month holding period may, under certain circumstances, sell within any three month period a number of securities which does not exceed the greater of 1% of the then outstanding shares of common stock or the average weekly trading volume of the class during the four calendar weeks prior to such sale. Rule 144 also permits, under certain circumstances, the sale of securities, without any limitation, by our stockholders that are non-affiliates that have satisfied a one-year holding period. Any substantial sale of our common stock pursuant to Rule 144 or pursuant to any resale prospectus may have a material adverse effect on the market price of our common stock.
LIMITATION ON DIRECTOR/OFFICER LIABILITY.
As permitted by Delaware law, our certificate of incorporation limits the liability of our directors for monetary damages for breach of a director’s fiduciary duty except for liability in certain instances. As a result of our charter provision and Delaware law, stockholders may have limited rights to recover against directors for breach of fiduciary duty. In addition, our certificate of incorporation provides that we shall indemnify our directors and officers to the fullest extent permitted by law.
WE HAVE NO HISTORY OF PAYING DIVIDENDS ON OUR COMMON STOCK.
We have never paid any cash dividends on our common stock and do not anticipate paying any cash dividends on our common stock in the foreseeable future. We plan to retain any future earnings to finance growth. If we decide to pay dividends to the holders of our common stock, such dividends may not be paid on a timely basis.
PROVISIONS OF OUR CERTIFICATE OF INCORPORATION AND DELAWARE LAW COULD DETER A CHANGE OF OUR MANAGEMENT WHICH COULD DISCOURAGE OR DELAY OFFERS TO ACQUIRE US.
Provisions of our certificate of incorporation and Delaware law may make it more difficult for someone to acquire control of us or for our stockholders to remove existing management, and might discourage a third party from offering to acquire us, even if a change in control or in management would be beneficial to our stockholders. For example, our certificate of incorporation allows us to issue shares of preferred stock without any vote or further action by our stockholders. Our Board has the authority to fix and determine the relative rights and preferences of preferred stock. Our Board also has the authority to issue preferred stock without further stockholder approval, including large blocks of preferred stock. As a result, our Board could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of our common stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our common stock.
SALES OF LARGE QUANTITIES OF OUR COMMON STOCK, INCLUDING THOSE SHARES ISSUABLE IN CONNECTION WITH PRIVATE PLACEMENT TRANSACTIONS, COULD REDUCE THE PRICE OF OUR COMMON STOCK.
On July 16, 2009, we received approval from the NYSE Amex LLC to issue up to 12,000,000 shares over the next twelve (12) months. We have entered into a common stock purchase agreement with Seaside 88, LP, whereby Seaside 88, LP will purchase 500,000 shares of common stock in a series of closings occurring every two weeks for a total of up to 26 closings, provided that the 3 day volume weighed average price prior to the scheduled closing is greater than or equal to the stated floor price of $0.25 per share. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP as of such date.
In October 2008, we sold securities in the subsequent closing of the 2008 Financing, resulting in the issuance of notes convertible into 10,744,681 shares of our common stock, and warrants to purchase 6,446,809 shares of our common stock. The sale of the notes and warrants resulted in gross proceeds to us of $2,525,000, before deducting certain fees and expenses.
In May 2008, we sold securities in the initial closing of the 2008 Financing, resulting in the issuance of notes convertible into 5,000,000 shares of our common stock, and warrants to purchase 3,000,000 shares of our common stock. The sale of the notes and warrants resulted in gross proceeds to us of $1,475,000, before deducting certain fees and expenses.
In December 2006, we sold securities in a private placement transaction resulting in the issuance of 9,823,983 shares of our common stock, and warrants to purchase 4,383,952 shares of our common stock. The sale of the shares of common stock and warrants resulted in gross proceeds to us of approximately $14.2 million, prior to offering expenses.
On July 20, 2006, we filed a shelf registration statement on Form S-3 registering for sale by us of up to 14,000,000 shares of our common stock. Such shelf registration statement was declared effective by the SEC on August 2, 2006. We may offer and sell such shares from time to time, in one or more offerings in amounts and at prices, and on terms determined at the time of the offering. Such offerings of our common stock may be made through agents we select or through underwriters and dealers we select. If we use agents, underwriters or dealers, we will name them and describe their compensation at the time of the offering. As of the filing date of this prospectus, such shelf registration statement is no longer effective.
In April 2006, we sold securities in a private placement transaction resulting in the issuance of 8,092,796 shares of our common stock, and warrants to purchase 2,896,168 shares of our common stock. The sale of the shares of common stock and warrants resulted in gross proceeds to us of approximately $11.8 million, prior to offering expenses.
In May 2005, we sold securities in a private placement transaction resulting in the issuance of 6,733,024 shares of our common stock, and certain warrants to purchase 2,693,210 shares of our common stock. The sales of the shares of common stock and warrants resulted in gross proceeds to us of approximately $7.1 million, prior to offering expenses.
The offering of, and/or resale of our common stock and the exercise of the warrants described immediately above in this risk factor are subject to currently effective registration statements filed by us on Forms S-3. There can be no assurance as to the prices at which our common stock will trade in the future, although they may continue to fluctuate significantly. Prices for our common stock will be determined in the marketplace and may be influenced by many factors, including the following:
|
|
·
|
The depth and liquidity of the markets for our common stock;
|
|
|
·
|
Investor perception of us and the industry in which we participate; and
|
|
|
·
|
General economic and market conditions.
|
Any sales of large quantities of our common stock could reduce the price of our common stock. The holders of the shares may sell such shares at any price and at any time, as determined by such holders in their sole discretion without limitation. If any such holders sell such shares in large quantities, our common stock price may decrease and the public market for our common stock may otherwise be adversely affected because of the additional shares available in the market.
As of March 24, 2010, we have 89,283,000 shares of common stock issued and outstanding and approximately 28.1 million shares of common stock issuable upon the exercise of outstanding stock options and warrants. In the event we wish to offer and sell shares of our common stock in excess of the 200,000,000 shares of common stock currently authorized by our certificate of incorporation, we will first need to receive stockholder approval. Such stockholder approval has the potential to adversely affect the timing of any potential transactions.
THE SECURITIES ISSUED IN OUR PRIVATE PLACEMENTS ARE RESTRICTED SECURITIES.
At the time of the offer and sale of the common stock and the shares of common stock underlying the convertible notes and the warrants, as applicable, in our December 2006 private placement and 2008 private placement, the common stock was not registered under the Securities Act or the securities laws of any state. Accordingly, these securities may not be sold or otherwise transferred unless such sale or transfer is subsequently registered under the Securities Act and applicable state securities laws or unless exemptions from such registration are available. The registration statements covering the December 2006 private placement and the 2008 private placement were declared effective by the SEC on January 26, 2007, and July 16, 2008 and May 5, 2009, respectively. Notwithstanding our registration obligations regarding these securities, investors may be required to hold these securities for an indefinite period of time. All investors who purchase these securities are required to make representations that it will not sell, transfer, pledge or otherwise dispose of any of the securities in the absence of an effective registration statement covering such transaction under the Securities Act and applicable state securities laws, or the receipt by us of an opinion of counsel to the effect that registration is not required.
WE HAVE BROAD DISCRETION AS TO THE USE OF THE PROCEEDS FROM OUR FINANCINGS AND MAY USE THE PROCEEDS IN A MANNER WITH WHICH YOU DISAGREE.
Our Board and management has broad discretion over the use of the net proceeds from our past financings, and will have broad discretion over the use of the net proceeds from any future financings. Stockholders may disagree with the judgment of the Board and management regarding the application of the proceeds. We cannot predict that investments of the proceeds will yield a favorable, or any, return.
WE MAY INCUR SIGNIFICANT COSTS FROM CLASS ACTION LITIGATION DUE TO OUR EXPECTED STOCK VOLATILITY.
In the past, following periods of large price declines in the public market price of a company’s stock, holders of that stock occasionally have instituted securities class action litigation against the company that issued the stock. If any of our stockholders were to bring this type of lawsuit against us, even if the lawsuit is without merit, we could incur substantial costs defending the lawsuit. The lawsuit also could divert the time and attention of our management, which would hurt our business. Any adverse determination in litigation could also subject us to significant liabilities.
THE UNCERTAINTY CREATED BY CURRENT ECONOMIC CONDITIONS AND POSSIBLE TERRORIST ATTACKS AND MILITARY RESPONSES THERETO COULD MATERIALLY ADVERSELY AFFECT OUR ABILITY TO SELL OUR PRODUCTS, AND PROCURE NEEDED FINANCING.
Current conditions in the domestic and global economies continue to present challenges. We expect that the future direction of the overall domestic and global economies will have a significant impact on our overall performance. Fiscal, monetary and regulatory policies worldwide will continue to influence the business climate in which we operate. If these actions are not successful in spurring continued economic growth, we expect that our business will be negatively impacted, as customers will be less likely to buy our products, if and when we commercialize our products. In addition, the potential for future terrorist attacks or war as a result thereof has created worldwide uncertainties that make it very difficult to estimate how the world economy will perform going forward.
OUR INABILITY TO MANAGE THE FUTURE GROWTH THAT WE ARE ATTEMPTING TO ACHIEVE COULD SEVERELY HARM OUR BUSINESS.
We believe that, given the right business opportunities, we may expand our operations rapidly and significantly. If rapid growth were to occur, it could place a significant strain on our management, operational and financial resources. To manage any significant growth of our operations, we will be required to undertake the following successfully:
|
|
·
|
We will need to improve our operational and financial systems, procedures and controls to support our expected growth and any inability to do so will adversely impact our ability to grow our business. Our current and planned systems, procedures and controls may not be adequate to support our future operations and expected growth. Delays or problems associated with any improvement or expansion of our operational systems and controls could adversely impact our relationships with customers and harm our reputation and brand.
|
|
|
·
|
We will need to attract and retain qualified personnel, and any failure to do so may impair our ability to offer new products or grow our business. Our success will depend on our ability to attract, retain and motivate managerial, technical, marketing, and administrative personnel. Competition for such employees is intense, and we may be unable to successfully attract, integrate or retain sufficiently qualified personnel.
|
If we are unable to hire, train, retain or manage the necessary personnel, we may be unable to successfully introduce new products or otherwise implement our business strategy. If we are unable to manage growth effectively, our business, results of operations and financial condition could be materially adversely affected.
WE MAY BE OBLIGATED, UNDER CERTAIN CIRCUMSTANCES, TO PAY LIQUIDATED DAMAGES TO HOLDERS OF OUR COMMON STOCK.
We have entered into agreements with the holders of our common stock that requires us to continuously maintain as effective, a registration statement covering the underlying shares of common stock. Such registration statements were declared effective on January 26, 2007, May 30, 2006 and July 28, 2005 and must continuously remain effective for a specified term. If we fail to continuously maintain such a registration statement as effective throughout the specified term, we may be subject to liability to pay liquidated damages.
None.
As of February 1, 2010, our executive offices are located at 1200 Route 22 East, Suite 2000, Bridgewater, New Jersey 08807. We no longer maintain laboratory and warehousing space. Before February 1, 2010, our executive offices, laboratory, and warehousing space was located at 25 Minneakoning Road, Flemington, New Jersey, known as the Facility. The Facility, constituting approximately 31,800 square feet, was occupied under a 10-year lease, expiring in August 2013. During 2009, we only occupied a portion of our space in the Facility. During the years ended December 31, 2007, 2008 and 2009, we paid rent for the Facility of approximately $443,000, $453,000 and $257,000, respectively. We have contracted out manufacturing for our product candidates. The manufacture of our product candidates is subject to current Good Manufacturing Practices, or cGMP, prescribed by the Food & Drug Administration, or FDA, and pre-approval inspections by the FDA and foreign authorities prior to the commercial manufacture of any such products. See Item 1, “Business- Raw Materials and Suppliers” and “Business-Government Regulations.”
We are not a named party in any material legal proceedings.
PART II
Our common stock is traded on the Over-the-Counter Bulletin Board (OTCBB) under the ticker symbol “NVDL.OB” since December 24, 2009. It was previously listed on the NYSE Amex LLC since May 11, 2004. The following table sets forth the range of high and low closing sales prices of our common stock as reported by the NYSE Amex LLC and OTCBB during the year ended December 31, 2008 and December 31, 2009.
|
CLOSING SALE PRICES
|
||
| ($) | ||
|
YEAR ENDED DECEMBER 31, 2008
|
||
|
First Quarter (January 1, 2008 through March 31, 2008)
|
0.51
|
0.28
|
|
Second Quarter (April 1, 2008 through June 30, 2008)
|
0.35
|
0.22
|
|
Third Quarter (July 1, 2008 through September 30, 2008)
|
0.30
|
0.17
|
|
Fourth Quarter (October 1, 2008 through December 31, 2008)
|
0.46
|
0.06
|
|
HIGH
|
LOW
|
|
|
YEAR ENDED DECEMBER 31, 2009
|
||
|
First Quarter (January 1, 2009 through March 31, 2009)
|
0.40
|
0.20
|
|
Second Quarter (April 1, 2009 through June 30, 2009)
|
0.42
|
0.20
|
|
Third Quarter (July 1, 2009 through September 30, 2009)
|
0.32
|
0.23
|
|
Fourth Quarter (October 1, 2009 through December 31, 2009)
|
0.32
|
0.13
|
We have never declared or paid a dividend on our common stock and management expects that all or a substantial portion of our future earnings will be retained for expansion or development of our business. The decision to pay dividends, if any, in the future is within the discretion of our Board of Directors and will depend upon our earnings, capital requirements, financial condition and other relevant factors such as contractual obligations. Management does not anticipate that we will pay dividends on our common stock in the foreseeable future. Moreover, we may never issue dividends in the future.
EQUITY COMPENSATION PLANS
The following table provides information with respect to our compensation plans under which equity compensation is authorized as of December 31, 2009.
|
Plan Category
|
Number of securities to be issued upon exercise of outstanding options, warrants and rights
|
Weighted-average exercise price of outstanding options, warrants and rights
|
Number of securities remaining available for future issuance under equity compensation plans
|
||||||||
|
Equity compensation plans approved by security holders
|
7,698,000 | $ | 0.69 | 871,000 | |||||||
|
Equity compensation plans not approved by security holders
|
581,000 | 2.38 | — | ||||||||
|
Total
|
8,279,000 | $ | 0.81 | 871,000 | |||||||
PERFORMANCE GRAPH
The graph below compares changes in the cumulative total stockholder return (change in stock price plus reinvested dividends) for the period from July 31, 2004 through December 31, 2009 of an initial investment of $100 invested in (a) NovaDel Pharma Inc.’s common stock, (b) the Total Return Index for the AMEX Composite and (c) the Research Data Group (RDG) Microcap Pharmaceutical Index. Total Return Index values are prepared by the Research Data Group. The stock price performance is not included to forecast or indicate future price performance.
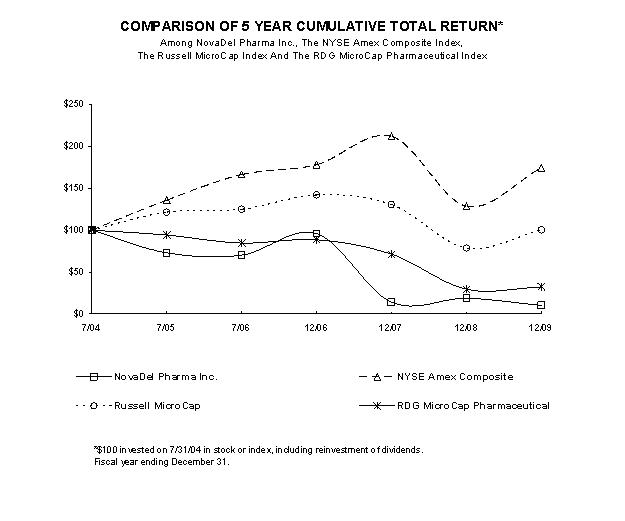
| 7/04 | 7/05 | 7/06 | 12/06 | 12/07 | 12/08 | 12/09 | |||||||||||||||||||||
|
NovaDel Pharma Inc.
|
$ | 100.00 | $ | 73.10 | $ | 70.18 | $ | 95.91 | $ | 14.04 | $ | 18.71 | $ | 10.06 | |||||||||||||
|
NYSE AMEX Composite
|
$ | 100.00 | $ | 135.85 | $ | 166.48 | $ | 178.23 | $ | 212.06 | $ | 128.64 | $ | 174.52 | |||||||||||||
|
Russell MicroCap
|
$ | 100.00 | $ | 121.64 | $ | 125.03 | $ | 142.12 | $ | 130.75 | $ | 78.74 | $ | 100.38 | |||||||||||||
|
RDG MicroCap Pharmaceutical
|
$ | 100.00 | $ | 94.25 | $ | 84.72 | $ | 88.58 | $ | 71.34 | $ | 29.55 | $ | 32.30 |
The following Selected Financial Data should be read in conjunction with our Financial Statements and the related Notes thereto, Management’s Discussion and Analysis of Financial Condition and Results of Operations and other financial information included elsewhere in this Annual Report on Form 10-K. The data set forth below with respect to our Statements of Operations for the years ended December 31, 2009, 2008 and 2007, the five months ended December 31, 2006 and for the fiscal year ended July 31, 2006, and the Balance Sheet data as of December 31, 2009, 2008 and 2007 are derived from our Financial Statements which are included elsewhere in this Annual Report on Form 10-K and are qualified by reference to such Financial Statements and related Notes thereto. The data set forth below for the year ended December 31, 2006 and for the five months ended December 31, 2005 are unaudited. There are no seasonal or other significant factors which affect comparability. The data set forth below with respect to our Statements of Operations for the fiscal years ended July 31, 2005 and the Balance Sheet data as of July 31, 2006 and July 31, 2005 are derived from our Financial Statements, which are not included elsewhere in this Annual Report on Form 10-K. Our historical results are not necessarily indicative of future results of operations.
|
Years Ended December 31,
|
Five Months Ended December 31,
|
Years Ended July 31,
|
||||||||||||||||||||||||||||||
|
Statement of Operations Data:
|
2009
|
2008
|
2007
|
2006
|
2006
|
2005
|
2006
|
2005
|
||||||||||||||||||||||||
|
(unaudited)
|
(unaudited)
|
|||||||||||||||||||||||||||||||
|
Total Revenues
|
$ | 422,000 | $ | 361,000 | $ | 469,000 | $ | 3,280,000 | $ | 2,067,000 | $ | 677,000 | $ | 1,890,000 | $ | 439,000 | ||||||||||||||||
|
Total Expenses
|
6,517,000 | 8,951,000 | 18,656,000 | 13,544,000 | 6,519,000 | 5,429,000 | 12,454,000 | 10,217,000 | ||||||||||||||||||||||||
|
Loss from Operations
|
(6,095,000 | ) | (8,590,000 | ) | (18,187,000 | ) | (10,264,000 | ) | (4,452,000 | ) | (4,752,000 | ) | (10,564,000 | ) | (9,778,000 | ) | ||||||||||||||||
|
Other, net
|
(385,000 | ) | — | (66,000 | ) | — | — | — | — | — | ||||||||||||||||||||||
|
Interest Expense
|
2,160,000 | 1,868,000 | — | — | — | — | — | — | ||||||||||||||||||||||||
|
Interest Income
|
6,000 | 137,000 | 632,000 | 337,000 | 180,000 | 67,000 | 224,000 | 87,000 | ||||||||||||||||||||||||
|
Income Tax Benefit
|
(1,057,000 | ) | (735,000 | ) | (658,000 | ) | (467,000 | ) | (467,000 | ) | (256,000 | ) | (256,000 | ) | (241,000 | ) | ||||||||||||||||
|
Net Loss
|
$ | (7,577,000 | ) | $ | (9,586,000 | ) | $ | (16,963,000 | ) | $ | (9,460,000 | ) | (3,805,000 | ) | $ | (4,429,000 | ) | $ | (10,084,000 | ) | $ | (9,450,000 | ) | |||||||||
|
Basic and Diluted Loss Per Common Share
|
$ | (0.12 | ) | $ | (0.16 | ) | $ | (0.29 | ) | $ | (0.20 | ) | $ | (0.08 | ) | $ | (0.11 | ) | $ | (0.23 | ) | $ | (0.27 | ) | ||||||||
|
Weighted Average Number of Shares of Common Stock Used in Computation of Basic and Diluted Loss Per Share
|
61,345,671 | 59,592,000 | 59,497,000 | 46,732,000 | 49,522,000 | 40,619,000 | 43,000,000 | 34,808,000 | ||||||||||||||||||||||||
|
December 31,
|
July 31,
|
|||||||||||||||||||||||
|
BALANCE SHEET DATA:
|
2009
|
2008
|
2007
|
2006
|
2006
|
2005
|
||||||||||||||||||
|
Cash, cash equivalents, and short-term investments
|
$ | 2,663,000 | $ | 4,328,000 | $ | 6,384,000 | $ | 20,276,000 | $ | 10,138,000 | $ | 8,223,000 | ||||||||||||
|
Total Assets
|
4,453,000 | 7,316,000 | 10,363,000 | 24,316,000 | 14,822,000 | 13,028,000 | ||||||||||||||||||
|
Total Current Liabilities
|
4,588,000 | 5,563,000 | 4,211,000 | 3,146,000 | 2,200,000 | 2,405,000 | ||||||||||||||||||
|
Total Liabilities
|
8,794,000 | 10,057,000 | 6,189,000 | 5,718,000 | 4,777,000 | 5,079,000 | ||||||||||||||||||
|
Accumulated deficit
|
(82,766,000 | ) | (74,829,000 | ) | (65,243,000 | ) | (48,280,000 | ) | (44,475,000 | ) | (34,391,000 | ) | ||||||||||||
|
Total Stockholders’ Equity (Deficiency)
|
$ | (4,341,000 | ) | $ | (2,741,000 | ) | $ | 4,174,000 | $ | 18,598,000 | $ | 10,045,000 | $ | 7,949,000 | ||||||||||
The following discussion of our financial condition and result of operations should be read in conjunction with the financial statements and the notes to those statements included elsewhere in this Annual Report on Form 10-K. The discussion includes forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth in Item 1A “Risk Factors” of this Annual Report on Form 10-K, our actual results may differ materially from those anticipated in these forward-looking statements.
GENERAL
NovaDel Pharma Inc. is a specialty pharmaceutical company developing oral spray formulations for a broad range of marketed drugs. Our proprietary technology offers, in comparison to conventional oral dosage forms, the potential for faster absorption of drugs into the bloodstream leading to quicker onset of therapeutic effects and possibly lower doses. Oral sprays eliminate the requirement for water or the need to swallow, potentially improving patient convenience and compliance. Our oral spray technology is focused on addressing unmet medical needs for a broad array of existing and future pharmaceutical products. Our most advanced oral spray candidates target angina, nausea, insomnia, migraine headaches and disorders of the central nervous system. We plan to develop these and other products independently and through collaborative arrangements with pharmaceutical and biotechnology companies. Currently, we have nine patents which have been issued in the U.S. and 69 patents which have been issued outside of the U.S. Additionally, we have over 65 patents pending around the world. We look for drug compounds that are off patent or are coming off patent in the near future, and we formulate these compounds in conjunction with our proprietary drug delivery method. Once formulated, we file for new patent applications on these formulated compounds that comprise our product candidates. Our patent portfolio includes patents and patent applications with claims directed to the pharmaceutical formulations, methods of use and methods of manufacturing for our product candidates.
We have had a history of recurring losses, giving rise to an accumulated deficit as of December 31, 2009 of $82,766,000, as compared to $74,829,000 as of December 31, 2008. We have had negative cash flow from operating activities of $1,578,000 and $5,533,000 for the years ended December 31, 2009 and 2008, respectively. As of December 31, 2009, we had negative working capital of $495,000 as compared to working capital of $47,000 as of December 31, 2008, representing a net decrease in working capital of approximately $542,000.
Throughout 2009, our reduced clinical development activities were limited to expenditures required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation development activities for certain other products. We will seek to raise additional capital in 2010 to fund our operations and future development activities through new strategic partnerships or collaborations, the sale of common stock or other equity securities or the issuance of debt. In the event we do not enter into a license agreement or other strategic transaction in which we receive an upfront fee or payment, or we do not undertake a financing of debt or equity securities, we may not have sufficient cash on hand to fund operations. We can give no assurances that we will be able to enter into a strategic transaction or raise any additional capital or, if we do, that such additional capital will be sufficient to meet our needs, or on terms favorable to us. If we are unable to raise additional capital, and we do not use our existing working capital to fund our development plans, we will have sufficient cash on hand to fund operating costs through the fourth quarter 2010. We will need additional financing thereafter until we achieve profitability.
Our audited financial statements for the fiscal year ended December 31, 2009 were prepared under the assumption that we will continue our operations as a going concern. We were incorporated in 1982, and have a history of losses. As a result, our independent registered public accounting firm in their audit report has expressed substantial doubt about our ability to continue as a going concern. We believe that the cash inflows that have been generated from our financing transactions and our licensing transactions and any additional potential cash inflows that may be received during 2010 will improve our ability to continue our operations as a going concern. Continued operations are dependent on our ability to complete equity or debt formation activities or to generate profitable operations. Such capital formation activities may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty.
As previously disclosed on May 14, 2008, we received notice from the NYSE Amex LLC (formally known as the American Stock Exchange) indicating that we are not in compliance with certain of the NYSE Amex LLC continued listing standards. We submitted a plan of compliance with NYSE Amex LLC, which was accepted, and attempted to regain compliance with such plan during fiscal year 2009. On December 2, 2009, we formally notified NYSE Amex LLC of our intent to voluntarily withdraw our listing and registration due to our failure to regain compliance with the continued listing standards and our plan. On December 14, 2009, we filed a Form 25 voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009. On December 24, 2009, our common stock began trading on the Over-the-Counter Bulletin Board, or OTCBB, under its new ticker symbol on OTCBB as NVDL.OB.
Since inception, substantially all of our revenues have been derived from consulting activities, primarily in connection with product development for various pharmaceutical companies. More recently, we have begun to derive revenues from license fees and milestone payments stemming from our partnership agreements. Our future growth and profitability will be principally dependent upon our ability to successfully develop our products and to market and distribute the final products either internally or with the assistance of a strategic partner.
Highlights for the year ended December 31, 2009, and additionally through the date of filing of this Annual Report on Form 10-K, include the following:
Other
|
·
|
Announced that Michael E. Spicer resigned as Chief Financial Officer and Corporate Secretary, effective April 1, 2009. Our Board of Directors appointed Deni M. Zodda, Ph.D., our Chief Business Officer, to serve as Interim Chief Financial Officer, Principal Financial Officer and Corporate Secretary, effective April 1, 2009. We also hired Joseph M. Warusz as a consultant to serve as Principal Accounting Officer, effective April 1, 2009.
|
|
·
|
Announced that Deni M. Zodda, Ph.D., Chief Business Officer, Interim Chief Financial Officer and Corporate Secretary, agreed to leave the Company resulting from a reorganization of the executive team. Mr. Zodda has entered into a Separation, Consulting and General Release Agreement under which he received a one-time fee of $137,500 to provide us with certain consulting services through October 31, 2009. Steven B. Ratoff, our Chairman, President and Chief Executive Officer, has been appointed our Interim Chief Financial Officer.
|
|
·
|
Announced the Board of Directors appointed Steven B. Ratoff as President and Chief Executive Officer effective January 1, 2010. Mr. Ratoff will continue to serve as Interim Chief Financial Officer.
|
|
·
|
Announced we executed a lease amendment modifying certain terms to the lease for the property in Flemington, New Jersey. The amendment converted the lease term to month to month commencing on July 1, 2009 with a provision that either party may terminate the lease upon thirty days written notice. We have released the lease escrow of $226,000 to the landlord in order to satisfy rent payments through June 30, 2009. This lease was terminated in December 2009.
|
|
·
|
Effective February 1, 2010, we entered into a one (1) year lease agreement with Regus Management Group LLC for approximately 5,000 square feet of office space in Bridgewater, New Jersey.
|
|
·
|
Announced that we entered into an agreement with Seaside 88, LP, or Seaside. Under the terms of the agreement and subject to the approval of the NYSE Amex LLC, Seaside has committed to purchase up to 13.0 million NovaDel common shares, in a series of closings every two weeks in the amount of 500,000 shares each for a total of up to 26 purchases. We had received approval from NYSE Amex LLC to issue up to 12.0 million shares over twelve (12) months. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP.
|
|
·
|
Announced we entered into an agreement with Arthur W. Wood Company, Inc., or AWW, pursuant to which AWW agreed to assist us as a non-exclusive financial advisor for the purposes of seeking capital related to the Seaside offering, referred to herein as the Placement. In consideration of AWW’s services, we agreed to pay AWW upon closing of a capital-raising transaction, a fee equal to three percent (3%) of the aggregate value of the proceeds paid or payable in the Placement.
|
|
·
|
Announced we received a milestone payment of approximately $150,000 from Velcera, Inc., or “Velcera,” relating to our License and Development Agreement with Velcera, dated June 22, 2004.
|
|
·
|
Announced we entered into a licensing agreement with privately-held Mist Acquisition, LLC to manufacture and commercialize the NitroMist™ lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris. Under the terms of the agreement, we received a $1,000,000 licensing fee upon execution of the agreement, and will receive milestone payments totaling an additional $1,000,000 over the next twelve months and ongoing performance payments of up to seventeen percent (17%) of net sales subject to potential reduction based upon the terms of the agreement.
|
|
·
|
Announced we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ in the United States and Canada. ZolpiMist™ is our oral spray formulation of zolpidem tartrate, which was approved by the FDA in December of 2008. Under the terms of the Agreement, we received $3,000,000 upon the execution of the agreement and ECR will pay us ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products, subject to the terms of the agreement.
|
|
·
|
Announced that we notified NYSE Amex LLC ("Exchange") of our intent to voluntarily delist our common stock from the Exchange. We filed Form 25 on December 14, 2009, voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009. Our common stock began trading on the OTCBB on December 24, 2009. Our new ticker symbol on OTCBB is NVDL.OB.
|
|
·
|
Announced we entered into an amendment agreement with ProQuest Investments L.P. and its affiliates, referred to herein as ProQuest, to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued but unpaid interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of our common stock as of December 31, 2009.
|
Drug development in the U.S. and most countries throughout the world is a process that includes several steps defined by the U.S. Food and Drug Administration, or FDA, or comparable regulatory authorities in foreign countries. The FDA approval processes relating to new drugs differ, depending on the nature of the particular drug for which approval is sought. With respect to any drug product with active ingredients not previously approved by the FDA, a prospective drug manufacturer is required to submit a New Drug Application, or NDA, which includes complete reports of pre-clinical, clinical and laboratory studies to prove such product’s safety and efficacy. Prior to submission of the NDA, it is necessary to submit an Investigational New Drug, or IND, to obtain permission to begin clinical testing of the new drug. Given that our current product candidates are based on a new technology for formulation and delivery of active pharmaceutical ingredients that have been previously approved and that have been shown to be safe and effective in previous clinical trials, we believe that we will be eligible to submit what is known as a 505(b)(2) NDA. We estimate that the development of new formulations of our pharmaceutical product candidates, including formulation, testing and submission of an NDA, will require significantly less time and lower investments in direct research and development expenditures than is the case for the discovery and development of new chemical entities. However, our estimates may prove to be inaccurate; or pre-marketing approval relating to our proposed products may not be obtained on a timely basis, if at all, and research and development expenditures may significantly exceed management’s expectations.
It is not anticipated that we will generate any revenues from royalties or sales of our product candidates until regulatory approvals are obtained and marketing activities begin. Any one or more of our product candidates may not prove to be commercially viable, or if viable, may not reach the marketplace on a basis consistent with our desired timetables, if at all. The failure or the delay of any one or more of our proposed products to achieve commercial viability would have a material adverse effect on us.
The successful development of our product candidates is highly uncertain. Estimates of the nature, timing and estimated expenses of the efforts necessary to complete the development of, and the period in which material net cash inflows are expected to commence from any of our product candidates are subject to numerous risks and uncertainties, including:
|
|
·
|
the scope, rate of progress and expense of our clinical trials and other research and development activities;
|
|
|
·
|
results of future clinical trials;
|
|
|
·
|
the expense of clinical trials for additional indications;
|
|
|
·
|
the terms and timing of any collaborative, licensing and other arrangements that we may establish;
|
|
|
·
|
the expense and timing of regulatory approvals or changes in the regulatory approval process;
|
|
|
·
|
the expense of establishing clinical and commercial supplies of our product candidates and any products that we may develop;
|
|
|
·
|
the effect of competing technologies and market developments; and
|
|
|
·
|
the expense of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights.
|
We expect to spend significant amounts on the development of our product candidates and we expect our costs to increase if we restart programs to develop and ultimately commercialize our product candidates. The following table summarizes our product candidates:
|
Active Ingredient or Class of Molecule
|
Indications
|
Stage of Development
|
Partner
|
|
|
Approved Products
|
||||
|
NitroMist™
|
nitroglycerin
|
Angina Pectoris
|
FDA Approved
|
Mist Acquisition, LLC
|
|
Zolpimist™
|
zolpidem
|
Insomnia
|
FDA Approved
|
ECR Pharmaceuticals Company
|
|
Product Candidates
|
||||
|
Duromist™
|
sildenafil
|
Erectile Dysfunction
|
Preclinical development
|
-
|
|
Zensana™
|
ondansetron
|
Nausea/Vomiting
|
Clinical development
|
Hana Biosciences/Par Pharmaceutical, Inc./BioAlliance Pharma S.A.
|
|
NVD-201
|
sumatriptan
|
Migraines
|
Pilot Efficacy study complete
|
-
|
|
NVD-301
|
midazolam
|
Pre-Procedure Anxiety
|
Preclinical development
|
-
|
NitroMist™ (nitroglycerin lingual aerosol). This product is indicated for acute relief of an attack or acute prophylaxis of angina pectoris due to coronary artery disease, and was approved by the FDA in November 2006. Previously, this product was partnered with Par Pharmaceutical, Inc., or Par; however, on August 1, 2007, we announced that Par returned the rights to NitroMist™ to us as part of Par's strategy to concentrate its resources on supportive care in AIDS and oncology markets. Our former contract manufacturer for NitroMist™, INyX Pharma, filed for protection under the Chapter 11 bankruptcy laws in 2007, and ceased operations at its facility in Puerto Rico where our product was to be manufactured during 2008. As a result, we selected an alternative contract manufacturer, DPT Laboratories. On October 27, 2009, the Company entered into a licensing and distribution agreement with privately-held Mist Acquisition, LLC, or Mist, to manufacture and commercialize the NitroMist™ in the United States, Canada and Mexico. Under the terms of the agreement, Mist paid us a $1,000,000 licensing fee upon execution of the agreement, and will pay us milestone payments totaling an additional $1,000,000 over the next twelve months if certain milestones are met and ongoing performance payments of up to seventeen percent (17%) of net sales. In addition, Mist will assume the activities and costs necessary for the completion of the product transfer to DPT Laboratories.
Zolpimist™ (zolpidem oral spray). Zolpidem is the active ingredient in Ambien®, the leading hypnotic for insomnia marketed by Sanofi-Aventis. Our oral spray formulation of zolpidem was approved for the short-term treatment of insomnia by the FDA in December 2008. In October 2009, we received a Notice of Allowance from the United States Patent and Trademark Office, or USPTO for claims which cover a method of treating insomnia by administering zolpidem to humans utilizing NovaMist™ Oral Spray technology. On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ in the United States and Canada. Under the terms of the agreement, we received a $3,000,000 licensing fee and will receive ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products.
Duromist™ (Sildenafil oral spray). Duromist contains sildenafil, the leading PDE-5 inhibitor for the treatment of erectile dysfunction marketed under the brand name Viagra®. We believe that an oral spray of sildenafil has the potential of a faster onset of action and a lower dose compared to tablets.
Erectile dysfunction occurs in approximately 18% of the male population with prevalence of over 50% in men over 65 years of age. PDE-5 inhibitors are effective in approximately 75% of the erectile dysfunction population. Sildenafil is the most popular molecule with over 50% market share in a erectile dysfunction market of over $3 billion.
Development is in progress for a formulation to be used in future clinical trials to begin in 2010, with a development plan that would deliver a FDA approved product available for launch in the second quarter of 2012.
Zensana™ (ondansetron oral spray). Ondansetron is the active ingredient in Zofran®, the leading anti-emetic marketed by GlaxoSmithKline, or GSK. Through July 31, 2007, this product candidate was licensed to Hana Biosciences, who was overseeing all clinical development and regulatory approval activities for this product in the U.S. and Canada. On July 31, 2007, we entered into a Product Development and Commercialization Sublicense Agreement with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a sublicense to Par to develop and commercialize Zensana™. Par is responsible for all development, regulatory, manufacturing and commercialization activities of Zensana™ in the United States and Canada, including the development and re-filing of the NDA in the United States. In addition, we entered into an Amended and Restated License Agreement with Hana Biosciences, pursuant to which Hana Biosciences relinquished its right to pay reduced royalty rates to us until such time as Hana Biosciences had recovered one-half of its costs and expenses incurred in developing Zensana™ from sales of Zensana™ and we agreed to surrender for cancellation all 73,121 shares of the Hana Biosciences common stock we acquired in connection with execution of the original license agreement with Hana Biosciences. Par had previously announced that it expected to complete clinical development on the revised formulation of Zensana™ during 2008, and expected to submit a new NDA for Zensana™ by the end of 2008. However, in November 2008, Par announced that it had completed bioequivalency studies on Zensana™ with mixed results, with bioequivalence to reference drug (Zofran® tablets) achieved in some of the studies and not achieved in others. We have notified Hana and Par that, under the terms of our agreement, they are required to return the product to us.
On May 19, 2008, we entered into an agreement with BioAlliance Pharma S.A., whereby BioAlliance acquired the European rights for our ondansetron oral spray. Under the terms of the agreement, BioAlliance paid us a license fee of $3,000,000 upon closing. We are eligible for additional milestone payments totaling approximately $24 million (an approval milestone of $5,000,000 and sales-related milestone payments of approximately $19 million) as well as a royalty on net sales. We anticipate that their development activities will not be initiated until development is completed in the United States.
Sumatriptan oral spray (NVD-201). Sumatriptan is the active ingredient in Imitrex® which is the largest selling migraine remedy marketed by GSK. A pilot PK study of NVD-201 with 9 healthy subjects, completed in the second half of calendar 2004, suggested that the formulation achieved plasma concentrations of sumatriptan in the therapeutic range. In September 2006 we announced positive results from an additional pilot pharmacokinetic study, with NVD-201 which demonstrated that NVD-201 achieves a statistically significant increase in absorption rate as compared with Imitrex® tablets. The rate of drug absorption is believed to be the most important predictor of the degree and speed of migraine relief. NVD-201 was evaluated in a four-arm, crossover pharmacokinetic study comparing 50mg Imitrex® tablets to 20mg and 30mg of the NVD-201 in 10 healthy male volunteers under fasting conditions. At least 90% of subjects receiving NVD-201 had detectable drug levels at three minutes post-dosing, while at the same timepoint, only 10% of subjects receiving 50mg Imitrex® tablets had detectable drug levels. These differences are statistically significant. At 3 to 6 minutes post dosing, all NVD-201 groups had statistically significantly higher mean concentration levels compared to 50mg Imitrex® tablets. Using published data for the currently marketed Imitrex® nasal spray as a proxy for therapeutic blood levels, we observed that by 6 minutes post-dosing, 100% of the 20mg NVD-201 users achieved these critical plasma concentration levels while none of the subjects from the Imitrex® tablet group did so by this timepoint. This result was also statistically significant. Furthermore, the study indicates up to a 50% increase in relative bioavailability of NVD-201 in comparison to the Imitrex® tablet. Additionally, the pharmacokinetics of 20mg NVD-201 after a meal were evaluated. NVD-201 was well tolerated.
While Imitrex® nasal spray was not included in this clinical study, the following represents a discussion of the results of our clinical study as compared to published data for Imitrex® nasal spray. Time to the first peak plasma concentration of sumatriptan -- which represents drug absorbed directly across the oral mucosa -- was approximately 70% faster with the 20mg NVD-201 than what has been reported in the literature for the same dose of the Imitrex® nasal spray (6 min. vs. 20 min.). The mean concentration level achieved during this critical first phase of absorption is approximately 30% greater for the NVD-201 than what was observed in published studies of the nasal spray (10.9 ng/mL vs. 8.5 ng/mL). Relative bioavailability after administration of 20mg NVD-201 appears to be greater than published estimates for the same dose of the Imitrex® nasal spray.
In September 2008, we announced the results from a pilot efficacy study for NVD-201.This was a multi-center, active control, open-label, dose-ranging, efficacy and safety study. Subjects received up to 5 treatments, comprising single doses of the following: Imigran® 50-mg tablets, Imigran® 100-mg tablets, NVD-201 20-mg, NVD-201 30-mg, and NVD-201 40-mg. Their response to Imigran® 50-mg tablets determined whether they were eligible to receive the other four treatments. Patients recorded the severity of each migraine attack on the same 4-point scale immediately before dosing and at 15, 30, 60, 90, 120, and 240 minutes, and at 24 hours post-dosing. Associated symptoms (nausea, vomiting, photophobia, and phonophobia) were also recorded immediately before dosing and at 30, 60, 90 and 120 minutes post-dosing. All dosing was done on an outpatient basis and patients returned to the clinic between migraine attacks.
In the primary analysis of efficacy, the percentage of patients responding to treatment at or before 60 minutes post-dosing, there was a statistically significant greater percentage of subjects receiving the 30- and 40-mg doses of NVD-201 with a reduction in headache pain compared to those receiving the 50-mg s Imigran® tablet (42% and 46%, respectively, vs 12%; P<0.011), and was comparable to the percentage who responded to the higher (100 mg) dose of the tablet formulation (42%). Significantly more patients had responded to all three doses of NVD-201 than to 50-mg Imigran® tablet by 90 minutes post-dosing (57% to 70.0% vs 32%; P<0.028) and all three oral spray doses were comparable to the 100-mg tablet. There were no treatment differences by 2 hours after dosing, when 68% to 77% of patients had responded irrespective of treatment.
Compared to 50-mg Imigran® tablet, at least one dose of NVD-201 also significantly increased percentage of patients who were pain free by 1 to 2 hours post-dosing, with the response ratio indicating significantly faster complete pain relief for the 40-mg dose, and significantly more patients had complete pain relief without use of rescue medication after receiving any dose of NVD-201. In addition, after one or more doses of NVD-201, the percentage of patients who were asymptomatic was significantly increased, and the percentages who experienced nausea, photophobia, or phonophobia were significantly decreased. NVD-201 was comparable to the 100-mg tablet on all the above measures.
We believe NVD-201 may provide clinical benefits to migraine sufferers including, possibly, faster relief than Imitrex® tablets as well as greater tolerability than triptan nasal sprays. Further, if proven to be safe and effective, we believe NVD-201 may be attractive to patients who have trouble taking oral medications due to nausea and vomiting caused by the migraine attack. Previously, we were targeting an NDA submission for our sumatriptan product candidate in the first half of calendar 2008; however, due primarily to funding constraints, at the present time, in view of the other higher priorities associated with our current product pipeline, we do not anticipate further efforts on the project.
We will continue to evaluate this program when sufficient additional funding becomes available.
Midazolam oral spray (NVD-301). NVD-301 contains midazolam which is the leading benzodiazepine used for sedation during diagnostic, therapeutic and endoscopic procedures. We believe that NVD-301 has the potential to be an easy-to use, rapid onset product useful to relieve the pre-procedure anxiety suffered by many patients prior to undergoing a wide variety of procedures performed in hospitals, imaging centers, ambulatory surgery centers and dental offices.
Annually, there are approximately 40 million invasive procedures performed in the ambulatory surgical setting, > 25 million MRI/CT scans and over 90 million pediatric dental procedures performed. Pre- procedure anxiety occurs in approximately 60% of children undergoing surgery and is associated with an increase in post-surgical complications including delirium, pain and sleep disorders, as well as higher levels of use of post-surgical medications. Anxiety interferes with approximately 30% of MRI scans with 5-10% of scans not completed due to anxiety. Pre-procedure anxiety is the number one reason for the use of sedation in dental procedures.
As of the current date, we have not yet secured sufficient additional financing, and have therefore not resumed clinical development activity. There can be no assurances that we will be able to secure additional capital, and as a result, there can be no assurances as to whether, and when, we will be able to resume our clinical development activities.
Propofol oral spray. Propofol is the active ingredient in Diprivan®, a leading intravenous anesthetic marketed by AstraZeneca. We continue to support our partner, Manhattan Pharmaceuticals, Inc., or Manhattan Pharmaceuticals, who will oversee all clinical development and regulatory approval for this product candidate. On July 10, 2007, Manhattan Pharmaceuticals announced its intention to pursue appropriate sub-licensing opportunities for this product candidate.
Veterinary. Our veterinary initiatives are being carried out largely by our partner, Velcera, Inc., or Velcera. In June 2007, Velcera announced that it had entered into a global license and development agreement with Novartis Animal Health. The agreement calls for Novartis Animal Health to develop, register and commercialize a novel canine product utilizing Velcera’s Promist™ platform, which is based on our patented oral spray technology. On March 5, 2008, Velcera announced that it had received notice from Novartis that it was terminating the agreement without cause. On August 24, 2009, we issued a press release to announce that we received a milestone payment of approximately $150,000 from Velcera, Inc. relating to our License and Development agreement dated June 22, 2004. This milestone payment resulted from Velcera’s recently announced global licensing agreement for the first canine pain management product delivered in a transmucosal mist form.
As discussed above, certain of our product candidates are in early stages of clinical development and some are in preclinical testing. These product candidates are continuously evaluated and assessed and are often subject to changes in formulation and technology. As a result, these product candidates are subject to a more difficult, time-consuming and expensive regulatory path in order to commence and complete the preclinical and clinical testing of these product candidates as compared to other product candidates in later stages of development.
CRITICAL ACCOUNTING POLICIES
USE OF ESTIMATES - The accompanying financial statements have been prepared in conformity with accounting principles generally accepted in the U.S. This requires our management to make estimates about the future resolution of existing uncertainties that affect the reported amounts of assets, liabilities, revenues and expenses which in the normal course of business are subsequently adjusted to actual results. Actual results could differ from such estimates. In preparing these financial statements, management has made its best estimates and judgments of the amounts and disclosures included in the financial statements giving due regard to materiality.
CASH AND CASH EQUIVALENTS – Cash equivalents include certificates of deposit and money market instruments with original maturities of three months or less when purchased. We maintain our cash and cash equivalents with several financial institutions. Deposits held with banks may exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed on demand and are maintained with high quality financial institutions, therefore reducing credit risk.
REVENUE RECOGNITION – We receive revenue from license agreements and consulting services. Upfront license agreement payments are initially deferred and subsequently amortized into revenue over the contractual period. Milestone payments related to license agreements are recognized as revenue when earned. Consulting revenues from contract clinical research are recognized in the period in which the services are rendered, provided that collection is reasonably assured.
DEFERRED FINANCING COSTS – We capitalize the costs related to the issuance of our convertible notes, and amortize such deferred costs to interest expense on a straight-line basis over the life of the related notes. We capitalized approximately $238,000 of deferred financing costs associated with the issuance of our convertible notes during 2008. We amortized approximately $25,000 to expense during the year ended December 31, 2009, upon which these costs are fully amortized.
WARRANTS ISSUED WITH CONVERTIBLE NOTES – The value of warrants and the intrinsic value of beneficial conversion rights arising from the issuance of convertible notes are determined by allocating an appropriate portion of the proceeds received from the debt instruments to the debt and warrants based on their relative fair value, which was determined using the Black-Scholes model. We adopted Accounting Standards Codification, or ASC, 815-40-15 on January 1, 2009. ASC 815-40-15 provides guidance in assessing whether an equity-linked financial instrument (or embedded feature) is indexed to an entity’s own stock. The adoption of ASC 815-40-15 resulted in an adjustment to opening accumulated deficit in the amount of $360,000 to reclassify the fair value of certain outstanding warrants from stockholders’ deficiency to liability. These warrants expired during the first quarter of 2009 and, as a result, the fair value of the warrant liability was reduced to zero and the Company recognized Other Income of $360,000 at the end of the reporting period.
VALUATION OF LONG-LIVED ASSETS – We assess the impairment of long-lived assets whenever events or changes in circumstances indicate that the carrying value may not be recoverable. Our long-lived assets as of December 31, 2009 were represented by property and equipment, as we have no intangible assets on our balance sheet. Factors we consider important which could trigger an impairment review include the following:
|
|
·
|
significant underperformance relative to expected historical or projected future operating results;
|
|
|
·
|
significant changes in the manner of our use of the acquired assets or the strategy for our overall business;
|
|
|
·
|
significant negative industry or economic trends; and
|
|
|
·
|
significant decrease in the market value of the assets.
|
The impairment test is based upon a comparison of the estimated undiscounted cash flows to the carrying value of the long-lived assets. If we determine that the carrying value of long-lived assets may not be recoverable based upon the existence of one or more of the above indicators of impairment, we measure any impairment based on projected discounted cash flows. The cash flow estimates used to determine the impairment, if any, contain management’s best estimate using appropriate assumptions and projections at that time. Net long-lived property and equipment as of December 31, 2009 was $324,000. We reviewed our long-lived property and equipment as of December 31, 2009, and have determined that their estimated fair value exceeds the carrying amount of such assets; therefore, we have not recognized an impairment loss for our long-lived property and equipment.
STOCK-BASED COMPENSATION – We calculate the fair value of stock-based compensation using the Black-Scholes method. Stock based compensation costs are recorded as earned for all unvested stock options outstanding. The charge is being recognized in research and development and consulting, selling, general and administrative expenses over the remaining service period after the adoption date based on the original estimate of fair value of the options as of the grant date. We recorded share-based compensation expense of $326,000 for the year ended December 31, 2009, $771,000 for the year ended December 31, 2008, and $910,000 for the year ended December 31, 2007. We will continue to incur share-based compensation charges in future periods. As of December 31, 2009, unamortized share-based compensation expense of $880,000 remains to be recognized, which is comprised of $482,000 related to non-performance based stock options to be recognized over a weighted average period of 0.7 years, $104,000 related to restricted stock to be recognized over a weighted average period of 1.1 years, and $294,000 related to performance-based stock options which vest upon reaching certain milestones. Expenses related to the performance-based stock options will be recognized if and when we determine that it is probable that the milestone will be reached.
We used the following weighted average assumptions in determining fair value under the Black-Scholes model for grants of all stock options in the respective periods:
|
Years Ended
|
|||
|
December 31,
2009
|
December 31,
2008
|
||
|
Expected volatility
|
85%
|
83%
|
|
|
Dividend yield
|
0%
|
0%
|
|
|
Expected term (years)
|
3.7
|
3.7
|
|
|
Risk-free interest rate
|
1.8%
|
2.3%
|
|
The above table represents the weighted-average assumptions for all stock options granted during the twelve months ended December 31, 2009 and 2008.
Expected volatility is based on historical volatility of our common stock. The expected term of options is estimated based on the average of the vesting period and contractual term of the option. The risk-free rate is based on U.S. Treasury yields for securities in effect at the time of grant with terms approximating the expected term until exercise of the option. In addition, the fair value of stock options granted is recognized as expense over the service period, net of estimated forfeitures. We are utilizing a 5% forfeiture rate, which we believe is a reasonable assumption to estimate forfeitures. However, the estimation of forfeitures requires significant judgment, and to the extent actual results or updated estimates differ from our current estimates, the effects of such resulting adjustment will be recorded in the period estimates are revised.
RESEARCH AND DEVELOPMENT EXPENSES - Research and development costs are expensed as incurred.
NEW ACCOUNTING PRONOUNCEMENTS – In June 2009, the FASB issued FASB ASC 105, Generally Accepted Accounting Principles, or GAAP, which establishes the FASB Accounting Standards Codification as the sole source of authoritative generally accepted accounting principles. Pursuant to the provisions of FASB ASC 105, we have updated references to GAAP in our financial statements issued for the year ended December 31, 2009. The adoption of FASB ASC 105 did not impact our financial position or results of operations.
In April 2009, the FASB issued guidance now codified as FASB ASC Topic 825, Financial Instruments, which amends previous Topic 825 guidance to require disclosures about fair value of financial instruments in interim as well as annual financial statements. The adoption did not have a material impact on our results from operations or on our financial condition. Financial instruments include cash and cash equivalents and accounts payable. The amounts reported for financial instruments are considered to be reasonable approximations of their fair values.
RESULTS OF OPERATIONS
YEARS ENDED DECEMBER 31, 2009 AND DECEMBER 31, 2008
License fees and milestone fees earned for the year ended December 31, 2009 were $422,000 as compared to $361,000 for the year ended December 31, 2008.
Research and development expenses for the year ended December 31, 2009 were $2,473,000 as compared to $3,878,000 for the year ended December 31, 2008. Research and development costs consist primarily of salaries and benefits, contractor and consulting fees, clinical drug supplies of preclinical and clinical development programs, consumable research supplies and allocated facility and administrative costs. Below is a summary of our research and development expenses for the years ended December 31, 2009 and December 31, 2008.
|
Fiscal Year Ended
|
|||||||
|
December 31, 2009
|
December 31, 2008
|
||||||
|
NitroMist™
|
$ | 592,000 | $ | 135,000 | |||
|
Zolpimist™
|
322,000 | 893,000 | |||||
|
Sumatriptan
|
170,000 | 369,000 | |||||
|
Zensana™
|
5,000 | 37,000 | |||||
|
Tizanidine
|
- | 41,000 | |||||
|
Other research and development costs
|
210,000 | 242,000 | |||||
|
Internal costs
|
1,174,000 | 2,161,000 | |||||
|
Total research and development expenses
|
$ | 2,473,000 | $ | 3,878,000 | |||
In the preceding table, research and development expenses are set forth in the following categories:
|
|
·
|
NitroMist™, Zolpimist™, Sumatriptan and Tizanidine - third-party direct project expenses relating to the development of the respective product candidates. The majority of our research and development resources were devoted to our zolpidem and sumatriptan product candidates. Although we have significantly reduced clinical development activities on our product candidate pipeline since the fourth quarter 2007 and continuing throughout 2009, such that we have limited our expenditures primarily to those required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation development activities for certain other products, we believe that we will need to obtain more funding in the future through collaborations or other arrangements with research institutions and corporate partners or public and private offerings of our securities, including debt or equity financing. There can be no assurances that we will be able to secure additional capital, and as a result, there can be no assurances as to whether, and when, we will be able to resume our clinical development activities;
|
|
|
·
|
Zensana™ - third-party direct project expenses relating to the development of Zensana™. As our partner for the Zensana™, Par, is overseeing all clinical development and regulatory approval activities, we do not expect to devote a significant amount of resources to this product candidate. In light of Hana Biosciences’ announcements in February 2007 and March 2007 regarding the status of Zensana™, as described above, we devoted resources to this project during the year ended December 31, 2007, including approximately $204,000 in third-party costs;
|
|
|
·
|
Other research and development costs – direct expenses not attributable to a specific product candidate; and
|
|
|
·
|
Internal costs – costs related primarily to personnel and overhead. We do not allocate these expenses to specific product candidates as these costs relate to all research and development activities.
|
Research and development expenses in the year ended December 31, 2009 decreased primarily as a result of the following items:
|
|
·
|
$457,000 increase in costs associated with our NitroMist™ product candidate primarily due to process validation, method transfer activities and lab supplies in the year ended December 31, 2009;
|
|
|
·
|
$571,000 decrease in product development costs for our Zolpimist™ product candidate, as development efforts were substantially completed during 2007, including filing of an NDA. Costs for zolpidem in the year ended December 31, 2009 related to usage and lab supplies;
|
|
|
·
|
$199,000 decrease in product development costs for our Sumatriptan product candidate, due to delayed activity on this project;
|
|
|
·
|
$987,000 decrease in internal costs is due to restructuring activities and substantially reduced efforts on R&D activities.
|
Consulting, selling, general and administrative expenses for the year ended December 31, 2009 were $4,044,000 as compared to $4,722,000 for the year ended December 31, 2008. General and administrative expenses consist primarily of salaries and related expenses for executive, finance, legal and other administrative personnel, recruitment expenses, professional fees and other corporate expenses. The decrease in general and administrative expenses is primarily attributable to our employee-related costs and reduction in stock compensation expense due to decrease in headcount during the year.
Primarily as a result of the factors described above, total expenses for the year ended December 31, 2009 were $6,517,000, as compared to $8,951,000 for the year ended December 31, 2008.
Other income/(expense) for the year ended December 31, 2009 was $(385,000) which relates to the reversal of the warrant liability (upon expiration of the related warrants) initially recorded upon our adoption of ASC 815-40-15 in the amount of $360,000, offset with a loss on disposition of fixed assets in the amount of $745,000.
Interest expense for the year ended December 31, 2009 was $2,160,000 primarily related to the convertible notes that were issued during the year ended December 31, 2008.
Interest income for the year ended December 31, 2009 was $6,000 as compared to $137,000 for the year ended December 31, 2008, due to lower average cash and cash equivalent balances.
The resulting net loss for the year ended December 31, 2009 was $7,577,000 as compared to $9,586,000 for the year ended December 31, 2008.
YEARS ENDED DECEMBER 31, 2008 AND DECEMBER 31, 2007
License fees and milestone fees earned for the year ended December 31, 2008 were $361,000, as compared to $469,000 for the year ended December 31, 2007. The decrease is primarily due to a non-recurring milestone payment received in the year ended December 31, 2007 from our license agreement with Velcera for veterinary products, which more than offset a one-time payment received during 2008 in connection with a product candidate that had been in development several years ago, and was no longer in our active product candidate pipeline.
Research and development expenses for the year ended December 31, 2008 were $3,878,000 as compared to $11,940,000 for the year ended December 31, 2007. Research and development costs consist primarily of salaries and benefits, contractor and consulting fees, clinical drug supplies of preclinical and clinical development programs, consumable research supplies and allocated facility and administrative costs. Below is a summary of our research and development expenses for the years ended December 31, 2008 and 2007:
|
Fiscal Year Ended
|
|||||||
|
December 31, 2008
|
December 31, 2007
|
||||||
|
NitroMist™
|
$ | 135,000 | $ | 558,000 | |||
|
Zolpimist™
|
893,000 | 5,669,000 | |||||
|
Sumatriptan
|
369,000 | 813,000 | |||||
|
Zensana™
|
37,000 | 213,000 | |||||
|
Tizanidine
|
41,000 | 75,000 | |||||
|
Ropinirole
|
— | 3,000 | |||||
|
Other research and development costs
|
242,000 | 1,763,000 | |||||
|
Internal costs
|
2,161,000 | 2,846,000 | |||||
|
Total research and development expenses
|
$ | 3,878,000 | $ | 11,940,000 | |||
In the preceding table, research and development expenses are set forth in the following categories:
|
·
|
NitroMist™, Zolpimist™, Sumatriptan, Tizanidine and Ropinirole - third-party direct project expenses relating to the development of the respective product candidates. The majority of our research and development resources were devoted to our zolpidem and sumatriptan product candidates. Since the fourth quarter 2007 and continuing throughout 2008, we have significantly reduced clinical development activities on our product candidate pipeline, such that we have limited our expenditures primarily to those required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation activities for certain other products, as we did not believe that we had sufficient cash to sustain such activities. As of the current date, we have not yet secured sufficient additional financing, and have therefore not resumed clinical development activity. There can be no assurances that we will be able to secure additional capital, and as a result, there can be no assurances as to whether, and when, we will be able to resume our clinical development activities;
|
|
·
|
Zensana™ - third-party direct project expenses relating to the development of Zensana™. As our partner for the Zensana™, Par, is overseeing all clinical development and regulatory approval activities, we do not expect to devote a significant amount of resources to this product candidate. In light of Hana Biosciences’ announcements in February 2007 and March 2007 regarding the status of Zensana™, as described above, we devoted resources to this project during the year ended December 31, 2007, including approximately $204,000 in third-party costs;
|
|
·
|
Other research and development costs – direct expenses not attributable to a specific product candidate; and
|
|
·
|
Internal costs – costs related primarily to personnel and overhead. We do not allocate these expenses to specific product candidates as these costs relate to all research and development activities.
|
Research and development expenses in the year ended December 31, 2008 decreased primarily as a result of the following items:
|
·
|
$4,776,000 decrease in product development costs for our Zolpimist™ product candidate, as development efforts were substantially completed during the fourth quarter 2007, including filing of an NDA. Development costs for zolpidem in the first quarter 2007 included costs for clinical trials, manufacturing preparedness and other NDA preparatory costs;
|
|
·
|
$176,000 decrease in product development costs related to Zensana™, as noted above;
|
|
·
|
$423,000 decrease in costs associated with our NitroMist™ product candidate primarily due to process validation and method transfer activities in the year ended December 31, 2007, which were substantially lower in the year ended December 31, 2008;
|
|
·
|
$444,000 decrease in product development costs for our Sumatriptan product candidate, as we substantially reduced our development activities on our product candidate pipeline beginning in the fourth quarter 2007; and
|
|
·
|
$1,521,000 decrease in other research and development costs as we substantially reduced our development activities on our product candidate pipeline beginning in the fourth quarter 2007.
|
Consulting, selling, general and administrative expenses for the year ended December 31, 2008 were $4,722,000 as compared to $6,716,000 for the year ended December 31, 2007. General and administrative expenses consist primarily of salaries and related expenses for executive, finance, legal and other administrative personnel, recruitment expenses, professional fees and other corporate expenses. The decrease in general and administrative expenses is primarily attributable to reduced salaries, benefits and other employee-related expenses, and to lower stock compensation charges.
The loss on disposal of assets held for sale was $351,000 for the year ended December 31, 2008.
Primarily as a result of the factors described above, total expenses for the year ended December 31, 2008 were $8,951,000, as compared to $18,656,000 for the year ended December 31, 2007.
Other, net for the year ended December 31, 2007 was $66,000, as further detailed below in the comparison for the years ended December 31, 2007. There was no Other, net for the year ended December 31, 2008.
Interest expense for the year ended December 31, 2008 was $1,868,000, of which $1,837,000 related to the convertible notes that were issued during 2008. This included $1,498,000 related to the amortization of the debt discount related to the beneficial conversion feature and fair value of the warrants, as well as $213,000 related to the amortization of the deferred financing costs.
Interest income for the year ended December 31, 2008 was $137,000 as compared to $632,000 for the year ended December 31, 2007 due to lower average cash and short-term investment balances.
The resulting net loss for the year ended December 31, 2008 was $9,586,000, as compared to $16,963,000 for the year ended December 31, 2007.
LIQUIDITY AND CAPITAL RESOURCES
From our inception, our principal sources of capital have been consulting revenues, private placements and public offerings of our securities, as well as loans and capital contributions from our principal stockholders. We have had a history of recurring losses, giving rise to an accumulated deficit as of December 31, 2009 of $82,766,000, as compared to $74,829,000 as of December 31, 2008. We have had negative cash flows from operating activities of $1,578,000 and $5,533,000 for the years ended December 31, 2009 and December 31, 2008, respectively. As of December 31, 2009, we had a working capital deficiency of $495,000 as compared to working capital of $47,000 as of December 31, 2008, representing a net decrease in working capital of approximately $542,000. Decrease is due to the conversion of convertible notes and associated liquidated damages and accrued interest of $3,700,000 to common stock, increase in other assets relating to receivable of $1,057,000 for net tax benefit offset by increase of deferred revenue of $4,000,000 and our loss for the year ended December 31, 2009 of $7,577,000.
Net cash used in operating activities was $1,578,000 for the year ended December 31, 2009, as compared to $5,533,000 for the year ended December 31, 2008. The $3,955,000 decrease in cash used is primarily due to the following:
|
|
·
|
A reduction in net loss from $9,586,000 for the year ended December 31, 2008 to $7,577,000 for the year ended December 31, 2009, representing an improvement of $2,009,000.
|
|
|
·
|
Non-cash expense of $1,360,000 upon debt conversion to common stock, $428,000 for the amortization of debt discount and deferred financing fees, and $746,000 on the loss on disposal of fixed assets, during the year ended December 31, 2009.
|
|
|
·
|
$3,734,000 increase in deferred revenue for the year ended December 31, 2009 due to non-refundable license fees of $4,000,000 received during the fourth quarter 2009.
|
Net cash provided from financing activities was a decrease of $128,000 due to the following:
|
|
·
|
$1,007,000 proceeds from the issuance of common stock during the year ended December 31, 2009.
|
|
|
·
|
$1,000,000 partial payment of a convertible note made on April 29, 2009.
|
Until and unless our operations generate significant revenues and cash flow, we will attempt to continue to fund operations from cash on hand and through the sources of capital described below. Our long-term liquidity is contingent upon achieving sales and positive cash flows from operating activities, and/or obtaining additional financing. The most likely sources of financing include private placements of our equity or debt securities or bridge loans to us from third-party lenders, license payments from current and future partners, and royalty payments from sales of approved drugs by partners. We can give no assurances that any additional capital that we are able to obtain will be sufficient to meet our needs.
We received $1,475,000 in gross proceeds on May 30, 2008 from the Initial Closing of a convertible note financing with certain funds affiliated with ProQuest and received $2,525,000 in gross proceeds on October 17, 2008 from the Subsequent Closing of such convertible note financing. The convertible notes issued in the Initial Closing matured on November 30, 2008 and, in the Subsequent Closing, on April 17, 2009. On April 29, 2009, we remitted $1,000,000 to ProQuest against the $4,000,000 of convertible notes issued during 2008. On December 31, 2009, we entered into an amendment agreement with ProQuest to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of our common stock.
During the second quarter of 2008, we also entered into a European partnership for our ondansetron oral spray with BioAlliance, as a result of which we received an immediate non-refundable license fee of $3,000,000.
We have entered into a common stock purchase agreement with Seaside 88, LP, whereby Seaside 88, LP will purchase 500,000 shares of common stock in a series of closings occurring every two weeks for a total of up to 26 closings, provided that the 3 day volume weighed average price prior to the scheduled closing is greater than or equal to the stated floor price of $0.25 per share. We have received $1,055,000 in gross proceeds for the closings that have occurred through December 31, 2009. As of March 24, 2010, we have received $200,140 in gross proceeds for 2010. On March 26, 2010, we mutually agreed to terminate the common stock purchase agreement with Seaside 88, LP.
On August 24, 2009, we received a milestone payment of approximately $150,000 from Velcera, Inc. relating to our License and Development agreement dated June 22, 2004. This milestone payment resulted from Velcera’s recently announced global licensing agreement for the first canine pain management product delivered in a transmucosal mist form.
On October 27, 2009, we entered into a licensing and distribution agreement with privately-held Mist Acquisition, LLC to manufacture and commercialize the NitroMist™ lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris. Under the terms of the agreement, we received a $1,000,000 licensing fee upon execution of the agreement, and will receive milestone payments totaling an additional $1,000,000 over the next twelve months and ongoing performance payments of up to seventeen percent (17%) of net sales subject to potential reduction based upon the terms of the Agreement. The Agreement contains customary termination provisions. In addition, the Agreement may be terminated by Mist for any reason upon written notice to us, which will be effective 180 days from the date of receipt of such notice, provided that Mist may not terminate until the second anniversary after the first commercial sale of NitroMist™ by Mist or its affiliates.
Through a separate license agreement with Mist, Akrimax Pharmaceuticals, LLC will receive the exclusive right to manufacture, distribute, market and sell NitroMist™ in the United States, Canada and Mexico. Under the terms of the Agreement, we will receive a percentage of any income received by Mist under any sublicense agreement relating to NitroMist™.
On November 13, 2009, we entered into an exclusive license and distribution agreement with ECR Pharmaceuticals Company, Inc. to commercialize and manufacture our ZolpiMist™ in the United States and Canada. ZolpiMist™ is our oral spray formulation of zolpidem tartrate, which was approved by the FDA in December of 2008. Under the terms of the agreement, ECR paid us $3,000,000 upon the execution of the agreement and ongoing performance payments of up to 15% of net sales on branded products and a lesser percent of net sales on authorized generic products, subject to the terms of the agreement. A performance milestone will be due to us if net sales reach a certain level. We have an opportunity to co-promote zolpidem tartrate oral spray in the United States and Canada with ECR’s consent, and retain commercialization rights for all other territories. ECR will assume responsibility for manufacturing the product for commercialization in the United States and Canada, including any activities required from the date of the agreement. The agreement contains customary termination provisions. In addition, the agreement may be terminated by ECR for any reason upon written notice to us, which will be effective 180 days from the date of receipt of such notice, provided that ECR may not terminate until the second anniversary after the first commercial sale of ZolpiMist™ by ECR or its affiliates.
We will seek to raise additional capital in 2010 to fund our operations and future development activities through new strategic partnerships or collaborations, the sale of common stock or other equity securities or the issuance of debt. In the event we do not enter into a license agreement or other strategic transaction in which we receive an upfront fee or payment, or we do not undertake a financing of debt or equity securities, we may not have sufficient cash on hand to fund operations. We can give no assurances that we will be able to enter into a strategic transaction or raise any additional capital or, if we do, that such additional capital will be sufficient to meet our needs, or on terms favorable to us. If we are unable to raise additional capital, and we do not use our existing working capital to fund our development plans, we will have sufficient cash on hand to fund operating costs through the fourth quarter 2010.
Our audited financial statements for the fiscal year ended December 31, 2009 were prepared under the assumption that we will continue our operations as a going concern. We were incorporated in 1982, and have a history of losses. As a result, our independent registered public accounting firm in their audit report has expressed substantial doubt about our ability to continue as a going concern. We believe that the cash inflows that have been generated from our financing transactions and our licensing transactions and any additional potential cash inflows that may be received during 2010 will improve our ability to continue our operations as a going concern. Continued operations are dependent on our ability to complete equity or debt formation activities or to generate profitable operations. Such capital formation activities may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty.
As previously disclosed on May 14, 2008, we received notice from the NYSE Amex LLC (formally known as the American Stock Exchange) indicating that we are not in compliance with certain of the NYSE Amex LLC continued listing standards. We submitted a plan of compliance with NYSE Amex LLC, which was accepted, and attempted to regain compliance with such plan during fiscal year 2009. On December 2, 2009, we formally notified NYSE Amex LLC of our intent to voluntarily withdraw our listing and registration due to our failure to regain compliance with the continued listing standards and our plan. On December 14, 2009, we filed a Form 25 voluntarily withdrawing our listing and registration from NYSE Amex LLC. The final day of trading on NYSE Amex LLC was December 23, 2009. On December 24, 2009, our common stock began trading on the Over-the-Counter Bulletin Board, or OTCBB, under its new ticker symbol on OTCBB as NVDL.OB.
OFF-BALANCE SHEET ARRANGEMENTS
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, results of operations, liquidity or capital resources.
CONTRACTUAL OBLIGATIONS
The following table sets forth our aggregate contractual cash obligations as of December 31, 2009.
|
Payments Due By Period
|
|||||||||||||||||||
|
Total
|
<1 year
|
1-3 years
|
3-5 years
|
5 years +
|
|||||||||||||||
|
Capital leases
|
$ | 14,000 | $ | 10,000 | $ | 4,000 | $ | — | $ | — | |||||||||
|
Operating leases
|
42,500 | 39,000 | 3,500 | — | — | ||||||||||||||
|
Employment agreements
|
627,000 | 627,000 | — | — | — | ||||||||||||||
|
Total contractual cash obligations
|
$ | 683,500 | $ | 676,000 | $ | 7,500 | $ | — | $ | — | |||||||||
We invest primarily in short-term, highly-rated investments, including U.S. government securities and certificates of deposit guaranteed by banks. Our market risk exposure consists principally of exposure to changes in interest rates. Because of the short-term maturities of our investments, however, we do not believe that a decrease in interest rates would have a significant negative impact on the value of our investment portfolio.
The financial statements required by this Item are included as a separate section of this report commencing on page F-1.
None.
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures or controls and other procedures that are designed to ensure that the information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act of 1934, or Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the Securities and Exchange Commission, or SEC, rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in the reports that a company files or submits under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding required disclosure.
We carried out an evaluation, under the supervision and with the participation of our chief executive officer and chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rule 13a-15(e) and Rule 15d-15(e) of the Exchange Act) as of December 31, 2009. Based on this evaluation, our chief executive officer and chief financial officer concluded that as of December 31, 2009, our disclosure controls and procedures were effective at providing reasonable assurance that the information required to be disclosed by us in reports filed under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms; and (ii) accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding disclosure.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act and is a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America and includes those policies and procedures that:
|
|
·
|
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
|
|
|
·
|
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of our management and directors; and
|
|
|
·
|
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
|
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2009. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework.
Based on its evaluation, our management has concluded that, as of December 31, 2009, our internal control over financial reporting was effective. This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit us to provide only management’s report in this annual report.
Changes in Internal Controls over Financial Reporting
During the fourth quarter 2009, there were no changes in our internal control over financial reporting (as defined in Rule 13a-15(e) and Rule 15d-15(f) under the Exchange Act) that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
PART III
Certain of the information required to be disclosed by this Item with respect to our executive officers is set forth under the caption “Executive Officers and Directors” contained in Part I, Item 1 of this Annual Report on Form 10-K.
Certain information required to be disclosed by this Item about our board of directors is incorporated in this Annual Report on Form 10-K by reference from the section entitled “Election of Directors,” and “Board of Directors and Committees” contained in our definitive proxy statement for our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
Information required to be disclosed by this Item about the Section 16(a) compliance of our directors and executive officers is incorporated in this Annual Report on Form 10-K by reference from the section entitled “Section 16(a) Beneficial Ownership Reporting Compliance” contained in our definitive proxy statement for our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
Information required to be disclosed by this Item about our board of directors, the audit committee of our board of directors, our audit committee financial expert, our Business Conduct Policy, and other corporate governance matters is incorporated in this Annual Report on Form 10-K by reference from the section entitled “Meetings and Committees of our Board” contained in our definitive proxy statement related to our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
The text of our Business Conduct Policy, which applies to all of our directors, officers and employees is posted in the “Corporate Governance” section of our website, www.novadel.com. A copy of the Business Conduct Policy can be obtained free of charge on our website or can be obtained and will be provided to any person without charge upon written request to our Corporate Secretary at our executive offices, 1200 Route 22 East, Suite 2000, Bridgewater, New Jersey 08807. We intend to disclose on our website any amendments to, or waivers from, our Business Conduct Policy that are required to be disclosed pursuant to the rules of the Securities and Exchange Commission.
Incorporated by reference to “Compensation Discussion and Analysis,” “Compensation Committee Report,” “Summary Compensation Table,” “Grants of Plan-Based Awards,” “Outstanding Equity Awards,” “Option Exercises and Stock Vested,” “Potential Payments Upon Termination” and “Directors Compensation” and “Compensation Committee Interlocks and Insider Participants” contained in our definitive proxy statement related to our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
Incorporated by reference to “Stock Ownership of Directors, Management and Certain Beneficial Owners” contained in our definitive proxy statement related to our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
Incorporated by reference to “Certain Relationships and Related Transactions” and “Independence of Directors” contained in our definitive proxy statement related to our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
Incorporated by reference to “Independent Registered Public Accounting Firm’s Fee Summary” contained in our definitive proxy statement related to our annual meeting of stockholders scheduled to be held in June 2010, which we intend to file within 120 days of the end of our fiscal year (December 31, 2009).
PART IV
|
(a)
|
Financial Statements and Schedules:
|
|
1.
|
Financial Statements
|
The following financial statements and report of independent registered public accounting firm are included herein:
|
Report of Independent Registered Public Accounting Firm
|
F-1
|
|
Balance Sheets
|
F-2
|
|
Statements of Operations
|
F-3
|
|
Statements of Changes in Stockholders’ Equity (Deficiency)
|
F-4
|
|
Statements of Cash Flows
|
F-5
|
|
Notes to Financial Statements
|
F-6
|
|
2.
|
Financial Statement Schedules
|
Not applicable.
|
3.
|
List of Exhibits
|
INDEX TO EXHIBITS
The following exhibits are included with this Annual Report on Form 10-K. All management contracts or compensatory plans or arrangements are marked with an asterisk.
|
EXHIBIT NO.
|
DESCRIPTION
|
METHOD OF FILING
|
||
|
3.1
|
Restated Certificate of Incorporation of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Quarterly Report on Form 10-QSB, as filed with the SEC on June 14, 2004.
|
||
|
3.2
|
Certificate of Amendment to the Certificate of Incorporation of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
3.3
|
Amended and Restated By-laws of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Form 8-K, as filed with the SEC on September 9, 2005.
|
||
|
4.1
|
Form of Class C Warrant for the Purchase of Shares of Common Stock
|
Incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the SEC on January 12, 2004.
|
||
|
4.2
|
Form of Warrant issued to certain accredited investors and placement agents
|
Incorporated by reference to Exhibit 4.1 of the Company’s Form 8-K, as filed with the SEC on April 17, 2006.
|
||
|
4.3
|
Form of Warrant issued to certain accredited investors and the placement agent
|
Incorporated by reference to Exhibit 4.1 of the Company’s Current Report on Form 8-K filed with the SEC on January 4, 2007.
|
||
|
4.4
|
Form of Warrant
|
Incorporated by reference to Exhibit 4.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on June 3, 2008.
|
||
|
10.1*
|
1992 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.2*
|
Form of Incentive Stock Option Agreement under the 1992 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.3*
|
1997 Stock Option Plan
|
Incorporated by reference to Exhibit 10.8 to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.4*
|
Form of Non-Qualified Option Agreement under the 1997 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.5*
|
1998 Stock Option Plan
|
Incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
||
|
10.6*
|
Form of Stock Option Agreement under the 1998 Stock Option Plan
|
Incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
||
|
10.7*
|
Form of Non-Qualified Option Agreement
|
Incorporated by reference to Exhibit 4.3 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
|
10.8
|
Common Stock and Warrant Purchase Agreement, dated December 12, 2001, by and among the Company and certain purchasers
|
Incorporated by reference to Exhibit A to the Schedule 13D as filed by Lindsay A. Rosenwald with the SEC on December 21, 2001.
|
||
|
10.9
|
Amendment No. 1, dated January 6, 2002, to the Common Stock and Warrant Purchase Agreement dated December 12, 2001 between the Company and certain purchasers
|
Incorporated by reference to Exhibit 10.25 to the Company’s Registration Statement of Form SB-2, as filed with the SEC on April 15, 2002 (File No. 333-86262).
|
||
|
10.10
|
License and Development Agreement, effective as of April 4, 2003, by and between the Company and Manhattan Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.31to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on March 11, 2004.
|
||
|
10.11
|
Development, Manufacturing and Supply Agreement, dated July 28, 2004, by and between the Company and Par Pharmaceutical, Inc.
|
Incorporated by reference to Exhibit 10.13 to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on November 15, 2004.
|
||
|
10.12
|
Second Amendment to License and Development Agreement, dated as of June 22, 2004, by and between the Company and the Veterinary Company, Inc.
|
Incorporated by reference to Exhibit 10.14 to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on November 15, 2004.
|
||
|
10.13*
|
Disclosure and Release Agreement Related to the Exchange of Non-Plan Options for Stock Options under the NovaDel Pharma Inc. 1998 Stock Option Plan by and between the Company and Thomas E. Bonney
|
Incorporated by reference to Exhibit 10.3 of the Company’s Form 8-K, as filed with the SEC on August 2, 2005.
|
||
|
10.14*
|
Disclosure and Release Agreement Related to the Exchange of Non-Plan Options for Stock Options under the NovaDel Pharma Inc. 1998 Stock Option Plan by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.4 of the Company’s Form 8-K, as filed with the SEC on August 2, 2005.
|
||
|
10.15*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated July 28, 2005, by and between the Company and Thomas E. Bonney
|
Incorporated by reference to Exhibit 10.25 of the Company’s Annual Report on Form 10-KSB for the period ended July 31, 2005, as filed with the SEC on October 31, 2005.
|
||
|
10.16*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated July 28, 2005, by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.29 of the Company’s Annual Report on Form 10-KSB for the period ended July 31, 2005, as filed with the SEC on October 31, 2005.
|
||
|
10.17
|
Amendment No. 1 to License and Development Agreement dated as of August 8, 2005, by and between the Company and Hana Biosciences Inc.
|
Incorporated by reference to Exhibit 99.1 of the Company’s Form 8-K, as filed with the SEC on August 12, 2005.
|
||
|
10.18*
|
NovaDel Pharma Inc. 2006 Equity Incentive Plan
|
Incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K, as filed with the SEC on January 23, 2006.
|
||
|
10.19*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Thomas Bonney
|
Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.20*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.5 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.21*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Steven Ratoff
|
Incorporated by reference to Exhibit 10.6 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.22*
|
Employment Agreement dated December 4, 2006 by and between the Company and David H. Bergstrom, Ph.D.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.23*
|
Incentive Stock Option Award between the Company and David H. Bergstrom dated December 4, 2006
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.24*
|
Nonqualified Stock Option Award between the Company and David H. Bergstrom, dated December 4, 2006
|
Incorporated by reference to Exhibit 10.3 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.25*
|
Amendment 2007-1 to the NovaDel Pharma Inc. 1998 Stock Option Plan dated March 2, 2007
|
Incorporated by reference to Exhibit 10.45 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
10.26*
|
Amendment 2007-1 to the NovaDel Pharma Inc. 2006 Equity Incentive Plan dated March 2, 2007
|
Incorporated by reference to Exhibit 10.46 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
10.27
|
Amended and Restated License and Development Agreement, dated as of July 31, 2007, by and between NovaDel Pharma Inc. and HANA Biosciences, Inc.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.28
|
Product Development and Commercialization Sublicense Agreement, dated as of July 31, 2007, by and among NovaDel Pharma Inc., HANA Biosciences and PAR Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.29
|
Termination Agreement, dated as of July 31, 2007, by and between NovaDel Pharma Inc. and PAR Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.30+
|
License Agreement, dated May 19, 2008, by and among the Company and BioAlliance Pharma SA.
|
Incorporated by reference to Exhibit 10.4 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on August 7, 2008.
|
||
|
10.31+
|
Supply Agreement, dated July 7, 2008, by and among the Company and BioAlliance Pharma SA.
|
Incorporated by reference to Exhibit 10.5 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on August 7, 2008.
|
||
|
10.32*
|
Separation, Consulting and General Release Agreement, effective as of April 30, 2009, by and between NovaDel Pharma Inc. and Deni M. Zodda
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on May 1, 2009.
|
||
|
10.33
|
Common Stock Purchase Agreement, by and between the Company and Seaside 88, LP, dated June 26, 2009
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on June 30, 2009.
|
||
|
10.34
|
Agreement, by and between the Company and Arthur W. Wood Company, dated June 15, 2009
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on July 20, 2009.
|
||
|
10.35+
|
License and Distribution Agreement, dated October 27, 2009, between NovaDel Pharma Inc. and Mist Acquisition, LLC
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 10, 2009.
|
||
|
10.36+
|
License and Distribution Agreement, dated November 12, 2009, between NovaDel Pharma Inc. and ECR Pharmaceuticals Company, Inc.
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 10, 2009.
|
||
|
10.37
|
Lease Agreement, dated as of December 7, 2009 and effective as of February 1, 2010, by and between Regus Management Group, LLC, as Landlord, and NovaDel Pharma Inc., as Tenant
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on January 14, 2010.
|
||
|
10.38
|
Amendment Agreement, dated December 31, 2009, by and among NovaDel Pharma Inc., ProQuest Investment II, L.P., ProQuest Investment Advisors Fund II, L.P. and ProQuest Investments III, L.P.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on January 7, 2010.
|
||
|
21.1
|
Subsidiaries of the Registrant
|
The registrant has no subsidiaries.
|
||
|
23.1
|
Consent of J.H. Cohn LLP
|
Filed herewith.
|
||
|
31.1
|
Certification of Principal Executive Officer under Rule 13a-14(a)
|
Furnished herewith.
|
||
|
32.1
|
Certifications of the Principal Executive Officer and Principal Financial Officer under 18 USC 1350
|
Furnished herewith.
|
_____________________
* Compensation Related Contract.
+ Confidential Treatment Requested. Confidential Materials omitted and filed separately with the Securities and Exchange Commission.
|
(b)
|
Exhibits.
|
See Item 15(a)(3) above.
|
(c)
|
Financial Statement Schedules.
|
See Item 15(a)(2) above.
In accordance with Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
NovaDel Pharma Inc.
|
||
|
Date: March 31, 2010
|
By:
|
/s/ STEVEN B. RATOFF
|
|
Steven B. Ratoff
|
||
|
President and Chief Executive Officer
|
||
In accordance with the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
SIGNATURES
|
TITLE
|
DATE
|
|
/s/ STEVEN B. RATOFF
|
Chairman, President, Chief Executive Officer, Interim Chief Financial Officer and Secretary
(Principal Executive Officer)
|
March 31, 2010
|
|
Steven B. Ratoff
|
||
|
/S/ MARK J. BARIC
|
Director
|
March 31, 2010
|
|
Mark J. Baric
|
||
|
/S/ THOMAS E. BONNEY
|
Director
|
March 31, 2010
|
|
Thomas E. Bonney
|
||
|
/S/ CHARLES NEMEROFF
|
Director
|
March 31, 2010
|
|
Charles Nemeroff
|
||
INDEX TO FINANCIAL STATEMENTS
The following financial statements are included in Part II, Item 8:
|
Report of Independent Registered Public Accounting Firm
|
F-1
|
|
Balance Sheets
|
F-2
|
|
Statements of Operations
|
F-3
|
|
Statements of Changes in Stockholders’ Equity (Deficiency)
|
F-4
|
|
Statements of Cash Flows
|
F-5
|
|
Notes to Financial Statements
|
F-6
|
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Stockholders and
Board of Directors
NovaDel Pharma Inc.
We have audited the accompanying balance sheets of NovaDel Pharma Inc. as of December 31, 2009 and 2008, and the related statements of operations, changes in stockholders’ equity (deficiency) and cash flows for each of the three years in the period ended December 31, 2009. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of NovaDel Pharma Inc. as of December 31, 2009 and 2008 and its results of operations and cash flows for each of the three years in the period ended December 31, 2009, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has suffered recurring losses from operations and negative cash flows from operating activities that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
|
/s/ J.H. COHN LLP
Roseland, New Jersey
March 31, 2010
|
NOVADEL PHARMA INC.
BALANCE SHEETS
AS OF DECEMBER 31, 2009 AND 2008
|
ASSETS
|
December 31,
2009
|
December 31,
2008
|
||||||
|
Current Assets:
|
||||||||
|
Cash and cash equivalents
|
$ | 2,663,000 | $ | 4,328,000 | ||||
|
Assets held for sale
|
— | 299,000 | ||||||
|
Deferred financing costs, net of accumulated amortization of $238,000 and $213,000, respectively
|
— | 25,000 | ||||||
|
Prepaid expenses and other current assets
|
1,430,000 | 958,000 | ||||||
|
Total Current Assets
|
4,093,000 | 5,610,000 | ||||||
|
Property and equipment, net
|
324,000 | 1,447,000 | ||||||
|
Other assets
|
36,000 | 259,000 | ||||||
|
TOTAL ASSETS
|
$ | 4,453,000 | $ | 7,316,000 | ||||
|
LIABILITIES AND STOCKHOLDERS’DEFICIENCY
|
||||||||
|
Current Liabilities:
|
||||||||
|
Secured convertible notes payable, net of unamortized discount of zero and $403,000, respectively
|
$ | — | $ | 3,597,000 | ||||
|
Accounts payable
|
195,000 | 654,000 | ||||||
|
Accrued expenses and other current liabilities
|
117,000 | 924,000 | ||||||
|
Current portion of deferred revenue
|
4,266,000 | 266,000 | ||||||
|
Current portion of capital lease obligations
|
10,000 | 122,000 | ||||||
|
Total Current Liabilities
|
4,588,000 | 5,563,000 | ||||||
|
Non-current portion of deferred revenue
|
4,202,000 | 4,468,000 | ||||||
|
Non-current portion of capital lease obligations
|
4,000 | 26,000 | ||||||
|
Total Liabilities
|
8,794,000 | 10,057,000 | ||||||
|
COMMITMENTS AND CONTINGENCIES
|
||||||||
|
STOCKHOLDERS’ DEFICIENCY
|
||||||||
|
Preferred stock, $.001 par value:
|
||||||||
|
Authorized 1,000,000 shares, none issued
|
— | — | ||||||
|
Common stock, $.001 par value:
|
||||||||
|
Authorized 200,000,000 shares, Issued 88,343,457 and 60,692,260 at December 31, 2009 and 2008, respectively
|
89,000 | 60,000 | ||||||
|
Additional paid-in capital
|
78,342,000 | 72,034,000 | ||||||
|
Accumulated deficit
|
(82,766,000 | ) | (74,829,000 | ) | ||||
|
Less: Treasury stock, at cost, 3,012 shares
|
(6,000 | ) | (6,000 | ) | ||||
|
Total Stockholders’ Deficiency
|
(4,341,000 | ) | (2,741,000 | ) | ||||
|
TOTAL LIABILITIES AND STOCKHOLDERS’ DEFICIENCY
|
$ | 4,453,000 | $ | 7,316,000 | ||||
See accompanying notes to financial statements.
NOVADEL PHARMA INC.
STATEMENTS OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2009, 2008 AND 2007
|
Year Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
License Fees and Milestone Payments Earned
|
$ | 422,000 | $ | 361,000 | $ | 469,000 | ||||||
|
Research and Development Expenses
|
2,473,000 | 3,878,000 | 11,940,000 | |||||||||
|
Consulting, Selling, General and Administrative Expenses
|
4,044,000 | 4,722,000 | 6,716,000 | |||||||||
|
Loss on Assets Held for Sale
|
— | 351,000 | ||||||||||
|
Total Expenses
|
6,517,000 | 8,951,000 | 18,656,000 | |||||||||
|
Loss From Operations
|
(6,095,000 | ) | (8,590,000 | ) | (18,187,000 | ) | ||||||
|
Other, net
|
(385,000 | ) | — | (66,000 | ) | |||||||
|
Interest Expense
|
(2,160,000 | ) | (1,868,000 | ) | — | |||||||
|
Interest Income
|
6,000 | 137,000 | 632,000 | |||||||||
|
Loss Before Income Tax Benefit
|
(8,634,000 | ) | (10,321,000 | ) | (17,621,000 | ) | ||||||
|
Income Tax Benefit
|
(1,057,000 | ) | (735,000 | ) | (658,000 | ) | ||||||
|
Net Loss
|
$ | (7,577,000 | ) | $ | (9,586,000 | ) | $ | (16,963,000 | ) | |||
|
Basic and Diluted Loss Per Common Share
|
$ | (0.12 | ) | $ | (0.16 | ) | $ | (0.29 | ) | |||
|
Weighted Average Number of Common Shares Used in Computation of Basic and Diluted Loss Per Common Share
|
61,346,000 | 59,592,000 | 59,497,000 | |||||||||
See accompanying notes to financial statements.
NOVADEL PHARMA INC.
STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIENCY)
FOR THE YEARS ENDED DECEMBER 31, 2009, 2008 AND 2007
|
Common Stock
|
||||||||||||||||||||||||||||
|
Shares
|
Amount
|
Additional Paid-In Capital
|
Accumulated
Deficit
|
Accumulated Other Comprehensive Income (Loss)
|
Treasury Stock
|
Total Stockholders’ Equity (Deficiency)
|
||||||||||||||||||||||
|
BALANCE, December 31, 2006
|
58,358,818 | $ | 58,000 | $ | 66,860,000 | $ | (48,280,000 | ) | $ | (34,000 | ) | $ | (6,000 | ) | $ | 18,598,000 | ||||||||||||
|
Share-based compensation expense
|
— | — | 910,000 | — | — | — | 910,000 | |||||||||||||||||||||
|
Stock issued in connection with private placement, net of costs
|
961,914 | 1,000 | 1,394,000 | — | — | — | 1,395,000 | |||||||||||||||||||||
|
Stock issued for options and warrants exercised
|
271,528 | — | 200,000 | — | — | — | 200,000 | |||||||||||||||||||||
|
Comprehensive income (loss): Unrealized loss on investment in marketable equity security
|
— | — | — | — | 34,000 | — | 34,000 | |||||||||||||||||||||
|
Net Loss
|
— | — | — | (16,963,000 | ) | — | — | (16,963,000 | ) | |||||||||||||||||||
|
Total comprehensive loss
|
— | — | — | — | — | — | (16,929,000 | ) | ||||||||||||||||||||
|
BALANCE, December 31, 2007
|
59,592,260 | 59,000 | 69,364,000 | (65,243,000 | ) | — | (6,000 | ) | 4,174,000 | |||||||||||||||||||
|
Share-based compensation expense
|
— | — | 771,000 | — | — | — | 771,000 | |||||||||||||||||||||
|
Restricted stock issued
|
1,100,000 | 1,000 | (1,000 | ) | — | — | — | — | ||||||||||||||||||||
|
Warrants issued to investors and beneficial conversion feature embedded in convertible notes
|
— | — | 1,900,000 | — | — | — | 1,900,000 | |||||||||||||||||||||
|
Net loss
|
— | — | — | (9,586,000 | ) | — | — | (9,586,000 | ) | |||||||||||||||||||
|
BALANCE, December 31, 2008
|
60,692,260 | 60,000 | 72,034,000 | (74,829,000 | ) | — | (6,000 | ) | (2,741,000 | ) | ||||||||||||||||||
|
Share-based compensation expense
|
— | — | 326,000 | — | — | — | 326,000 | |||||||||||||||||||||
|
Cumulative effect for the adoption of ASC 815-40-15 relating to outstanding warrants indexed to the entity’s own stock
|
— | — | — | (360,000 | ) | — | — | (360,000 | ) | |||||||||||||||||||
|
Restricted stock cancelled
|
(575,000 | ) | — | — | — | — | — | — | ||||||||||||||||||||
|
Cashless exercise of warrants
|
489,114 | 1,000 | (1,000 | ) | — | — | — | — | ||||||||||||||||||||
|
Issuance of common stock
|
27,737,083 | 28,000 | 5,983,000 | — | — | — | 6,011,000 | |||||||||||||||||||||
|
Net loss
|
— | — | — | (7,577,000 | ) | — | — | (7,577,000 | ) | |||||||||||||||||||
|
BALANCE, December 31, 2009
|
88,343,457 | $ | 89,000 | $ | 78,342,000 | $ | (82,766,000 | ) | $ | — | $ | (6,000 | ) | $ | (4,341,000 | ) | ||||||||||||
|
See accompanying notes to financial statements.
|
NOVADEL PHARMA INC.
STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, 2009, 2008 AND 2007
|
Year Ended December 31,
|
||||||||||||
|
2009
|
2008
|
2007
|
||||||||||
|
CASH FLOWS FROM OPERATING ACTIVITIES
|
||||||||||||
|
Net loss
|
$ | (7,577,000 | ) | $ | (9,586,000 | ) | $ | (16,963,000 | ) | |||
|
Adjustments to reconcile net loss to net cash used in operating activities:
|
||||||||||||
|
Amortization of discount on short-term investments
|
— | — | (101,000 | ) | ||||||||
|
Debt conversion to common stock expense
|
1,360,000 | — | — | |||||||||
|
Expiration of warrants
|
(360,000 | ) | — | — | ||||||||
|
Share-based compensation expense
|
326,000 | 771,000 | 910,000 | |||||||||
|
Amortization of debt discount and deferred financing fees
|
428,000 | 1,711,000 | — | |||||||||
|
Loss on assets held for sale
|
— | 351,000 | — | |||||||||
|
Loss on disposal of fixed assets
|
746,000 | 19,000 | — | |||||||||
|
Loss from return of investment in marketable security available-for-sale to issuer
|
— | — | 140,000 | |||||||||
|
Other than temporary impairment of investment in marketable equity security available-for-sale
|
— | — | 360,000 | |||||||||
|
Depreciation and amortization
|
369,000 | 506,000 | 685,000 | |||||||||
|
Changes in operating assets and liabilities:
|
||||||||||||
|
Inventories
|
— | — | (99,000 | ) | ||||||||
|
Prepaid expenses and other current assets
|
(472,000 | ) | 151,000 | (574,000 | ) | |||||||
|
Other assets
|
223,000 | 110,000 | (10,000 | ) | ||||||||
|
Accounts payable
|
(459,000 | ) | (978,000 | ) | 834,000 | |||||||
|
Accrued expenses and other current liabilities
|
104,000 | (1,344,000 | ) | 206,000 | ||||||||
|
Deferred revenue
|
3,734,000 | 2,756,000 | (628,000 | ) | ||||||||
|
Net cash used in operating activities
|
(1,578,000 | ) | (5,533,000 | ) | (15,240,000 | ) | ||||||
|
CASH FLOWS FROM INVESTING ACTIVITIES:
|
||||||||||||
|
Purchases of property and equipment
|
— | (121,000 | ) | (179,000 | ) | |||||||
|
Proceeds from sale of fixed assets
|
41,000 | — | — | |||||||||
|
Purchases of short-term and long-term investments
|
— | — | (9,737,000 | ) | ||||||||
|
Maturities of short-term and long-term investments
|
— | — | 13,528,000 | |||||||||
|
Net cash provided by (used in) investing activities
|
41,000 | (121,000 | ) | 3,612,000 | ||||||||
|
CASH FLOWS FROM FINANCING ACTIVITIES:
|
||||||||||||
|
Proceeds from issuance of common stock through private placements
|
1,007,000 | — | 1,395,000 | |||||||||
|
Proceeds from issuance of convertible notes
|
— | 4,000,000 | — | |||||||||
|
Deferred financing costs
|
(238,000 | ) | ||||||||||
|
Proceeds from options and warrants exercised
|
— | — | 200,000 | |||||||||
|
Payments of convertible note obligation
|
(1,000,000 | ) | — | — | ||||||||
|
Payments of capital lease obligations
|
(135,000 | ) | (164,000 | ) | (169,000 | ) | ||||||
|
Net cash provided by (used in) financing activities
|
(128,000 | ) | 3,598,000 | 1,426,000 | ||||||||
|
NET DECREASE IN CASH AND CASH EQUIVALENTS
|
(1,665,000 | ) | (2,056,000 | ) | (10,202,000 | ) | ||||||
|
CASH AND CASH EQUIVALENTS, BEGINNING OF YEAR
|
4,328,000 | 6,384,000 | 16,586,000 | |||||||||
|
CASH AND CASH EQUIVALENTS, END OF YEAR
|
$ | 2,663,000 | $ | 4,328,000 | $ | 6,384,000 | ||||||
|
SUPPLEMENTAL DISCLOSURE OF CASH PAID FOR INTEREST
|
$ | 10,000 | $ | 28,000 | $ | 37,000 | ||||||
|
SUPPLEMENTAL DISCLOSURE OF NONCASH INVESTING AND FINANCING ACTIVITIES:
|
||||||||||||
|
Issuance of common stock for note conversion
|
$ | 3,000,000 | — | — | ||||||||
|
Issuance of common stock to convert penalties and interest
|
$ | 657,000 | — | — | ||||||||
|
Settlement of obligation with transfer of fixed assets
|
$ | 267,000 | ||||||||||
|
Equipment acquired under capital lease obligations
|
$ | — | — | $ | 228,000 | |||||||
See accompanying notes to financial statements.
NOVADEL PHARMA INC.
NOTES TO FINANCIAL STATEMENTS
NOTE 1 - NATURE OF THE BUSINESS
NovaDel Pharma Inc. (the “Company”) is a specialty pharmaceutical company developing oral spray formulations of a broad range of marketed pharmaceuticals. The Company’s proprietary technology offers, in comparison to conventional oral dosage forms, the potential for faster absorption of drugs into the bloodstream leading to quicker onset of therapeutic effects and possibly reduced first pass liver metabolism, which may result in lower doses. Oral sprays eliminate the requirement for water or the need to swallow, potentially improving patient convenience and compliance. The Company’s oral spray technology is focused on addressing unmet medical needs for a broad array of existing and future pharmaceutical products, with the most advanced oral spray candidates targeting angina, nausea, insomnia, migraine headaches and disorders of the central nervous system.
To date, we have entered into license agreements with (i) Mist Acquisition, LLC to manufacture and commercialize the NitroMist(R) lingual spray version of nitroglycerine, (ii) ECR Pharmaceuticals Company, Inc., to commercialize and manufacture ZolpiMist(TM) in the United States and Canada, (iii) Hana Biosciences, for the development and marketing rights in the U.S. and Canada for our ondansetron oral spray, (iv) Par Pharmaceuticals, Inc., or Par, for the marketing rights in the U.S. and Canada for NitroMist™, (v) Manhattan Pharmaceuticals, in connection with propofol, (vi) Velcera, in connection with veterinary applications for currently marketed veterinary drugs and (vii) BioAlliance Pharma SA, for the European rights for ondansetron oral spray. In addition, we have entered into a sub-license agreement with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a sublicense to Par to develop and commercialize Zensana™.
On November 13, 2009, we and ECR Pharmaceuticals Company, Inc., or ECR, (a wholly-owned subsidiary of Hi-Tech Pharmacal Co., Inc.) entered into an exclusive license and distribution agreement to commercialize and manufacture ZolpiMist™ in the United States and Canada. ZolpiMist™ is the Company’s oral spray formulation of zolpidem tartrate approved by the FDA in December of 2008.
Under the terms of the agreement, ECR paid the Company $3 million upon execution of the agreement. ECR will assume responsibility for manufacturing and marketing the product in the United States and Canada. In addition, ECR will pay royalties of up to 15% on net sales of ZolpiMist™ as well as an additional milestone payment if sales reach a specified level.
On October 27, 2009, we and privately-held Mist Acquisition, LLC, or Mist, entered into a licensing agreement to manufacture and commercialize the NitroMist® lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris. Under terms of the agreement, Mist paid the Company a $1,000,000 licensing fee upon execution of the agreement, and will pay milestone payments totaling an additional $1,000,000 over twelve months, and ongoing performance payments of seventeen percent (17%) of net sales.
Through a separate license agreement with Mist, Akrimax Pharmaceuticals, LLC will receive the exclusive right to manufacture, distribute, market and sell NitroMist® in the United States, Canada and Mexico. NitroMist® provides acute relief of an attack or acute prophylaxis of angina pectoris due to coronary artery disease. The lingual spray form of the drug is conveniently administered and is rapidly absorbed into the bloodstream via the oral mucosa, providing patients a fast and tolerable treatment option for the prevention or relief of pain associated with such attacks.
On May 19, 2008, the Company and BioAlliance Pharma SA or BioAlliance, entered into an agreement where BioAlliance acquired the European rights for NovaDel’s Ondansetron oral spray. Under the terms of the agreement, BioAlliance paid NovaDel a license fee of $3,000,000 upon closing. The Company is eligible for additional milestone payments totaling approximately $24 million (an approval milestone of $5 million and sales-related milestone payments of approximately $19 million) as well as a royalty on net sales. BioAlliance and the Company anticipate collaborating in the completion of development activities for Europe, with BioAlliance responsible for regulatory and pricing approvals and then commercialization throughout Europe. The Company will be responsible for supplying the product. The upfront payment has been included in deferred revenue and is being recognized in income over the term of the agreement (nineteen and one half-years).
In July 2007, the Company entered into a Product Development and Commercialization Sublicense Agreement (the “Sublicense Agreement”) with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a non-transferable, non-sublicenseable, royalty-bearing, exclusive sublicense to Par to develop and commercialize Zensana™. In connection therewith, the Company and Hana Biosciences amended and restated their existing License and Development Agreement, as amended, relating to the development and commercialization of Zensana™ (the “Amended and Restated License Agreement”) to coordinate certain of the terms of the Sublicense Agreement. Under the terms of the Sublicense Agreement, Par is responsible for all development, regulatory, manufacturing and commercialization activities of Zensana™ in the United States and Canada. The Company retains its rights to Zensana™ outside of the United States and Canada. In addition, under the terms of the Amended and Restated License Agreement, Hana Biosciences relinquished its right to pay reduced royalty rates to the Company until such time as Hana Biosciences had recovered one-half of its costs and expenses incurred in developing Zensana™ from sales of Zensana™ and the Company agreed to surrender for cancellation all 73,121 shares of the Hana Biosciences common stock acquired by the Company in connection with execution of the original License Agreement. Also in July 2007, the Company and Par agreed to terminate the agreement relating to NitroMist™.
On November 18, 2004, the Company entered into a manufacturing and supply agreement with INyX whereby INyX manufactures and supplies NitroMist™. For a five-year period that began November 18, 2004, INyX was to be the exclusive provider of the nitroglycerin lingual spray to the Company substantially worldwide. Pursuant to the terms and conditions of the agreement, it would be INyX’s responsibility to manufacture, package and supply NitroMist™ in such territories. Thereafter, INyX would have a non-exclusive right to manufacture such spray for an additional five years. In July 2007, INyX announced it filed for protection under the Chapter 11 bankruptcy laws. The Company was informed by the trustees for INyX in June 2008 that the facility in Puerto Rico where manufacturing operations for NitroMist™ were conducted would be ceasing operations as of the end of July 2008. As a result, the Company selected an alternative contract manufacturing company, DPT Laboratories Inc (“DPT”), and is in the process of transferring manufacturing operations for NitroMist™ to DPT.
NOTE 2 - LIQUIDITY AND BASIS OF PRESENTATION
The Company has reported a net loss of $7,577,000 $9,586,000 and $16,963,000, and negative cash flows from operating activities of $1,578,000, $5,533,000 and $15,240,000, for the years ended December 31, 2009, 2008 and 2007, respectively. As of December 31, 2009, the Company had negative working capital of $495,000 and cash and cash equivalents of $2,663,000. Until and unless the Company’s operations generate significant revenues and cash flow, we will attempt to continue to fund operations from cash on hand and through the sources of capital described below. The Company’s long-term liquidity is contingent upon achieving sales and positive cash flows from operating activities, and/or obtaining additional financing. The most likely sources of financing include private placements of the Company’s equity or debt securities or bridge loans to the Company from third-party lenders, license payments from current and future partners, and royalty payments from sales of approved product candidates by partners. The Company can give no assurances that any additional capital that it is able to obtain will be sufficient to meet its needs, or on terms favorable to it.
Since the fourth quarter 2007 and continuing throughout 2009, we have significantly reduced clinical development activities on our product candidate pipeline, such that we have limited our expenditures primarily to those required to support our two approved products NitroMist™ and Zolpimist™ and minor expenditures to support formulation development activities for certain other products, as we did not believe that we had sufficient cash to sustain such activities. Despite this reduction in expenditures for clinical activities, the Company requires capital to sustain its existing organization until such time as clinical activities can be resumed. The Company received $1,055,000 in gross proceeds for common stock purchased by Seaside 88, LP during the year ended December 31, 2009. During the fourth quarter of 2009, the Company also entered into licensing and distribution agreements with ECR Pharmaceuticals Company, Inc. and Mist Acquisition, LLC, as a result of which it received non-refundable license fees of $4,000,000.
The Company will seek to raise additional capital in 2010 to fund our operations and future development activities through new strategic partnerships or collaborations, the sale of common stock or other equity securities or the issuance of debt. In the event we do not enter into a license agreement or other strategic transaction in which we receive an upfront fee or payment, or we do not undertake a financing of debt or equity securities, we may not have sufficient cash on hand to fund operations. We can give no assurances that we will be able to enter into a strategic transaction or raise any additional capital or, if we do, that such additional capital will be sufficient to meet our needs, or on terms favorable to us. If we are unable to raise additional capital, and we do not use our existing working capital to fund our development plans, we will have sufficient cash on hand to fund operating costs through the fourth quarter 2010.
Our audited financial statements for the year ended December 31, 2009, were prepared under the assumption that we will continue our operations as a going concern. We were incorporated in 1982, and have a history of losses. As a result, our independent registered public accounting firm in their audit report has expressed substantial doubt about our ability to continue as a going concern. Continued operations are dependent on our ability to complete equity or debt formation activities or to generate profitable operations. Such capital formation activities may not be available or may not be available on reasonable terms. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we cannot continue as a viable entity, our stockholders may lose some or all of their investment in the Company.
On May 14, 2008, we received notice from the NYSE Amex LLC (formally known as the American Stock Exchange) indicating that we are not in compliance with certain of the NYSE Amex LLC continued listing standards. Specifically, the NYSE Amex LLC has notified us that we are not in compliance with Section 1003(a)(iii) of the NYSE Amex LLC Company Guide with stockholders’ equity of less than $6,000,000 and losses from continuing operations and net losses in our five most recent fiscal years, and Section 1003(a)(iv) of the NYSE Amex LLC Company Guide in that we have sustained losses which are so substantial in relation to our overall operations or our existing financial resources, or our financial condition has become so impaired that it appears questionable, in the opinion of the NYSE Amex LLC, as to whether we will be able to continue operations and/or meet our obligations as they mature. On December 2, 2009, we formally notified NYSE Amex LLC of our intent to voluntarily withdraw our listing and registration. On December 14, 2009, we filed Form 25 voluntarily withdrawing our listing and registration from NYSE Amex LLC (“AMEX”). The final day of trading on AMEX was December 23, 2009. On December 24, 2009, we announced that our common stock will begin trading on the Over-the-Counter Bulletin Board (“OTCBB”) under its new ticker symbol on OTCBB as NVDL.OB
NOTE 3 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
REVENUE RECOGNITION – The Company receives revenue from license agreements and consulting services. Upfront license agreement payments are initially deferred and subsequently amortized into revenue over the contractual period. Milestone payments related to license agreements are recognized as revenue when earned. Consulting revenues from contract clinical research are recognized in the period in which the services are rendered, provided that collection is reasonably assured.
CASH EQUIVALENTS AND INVESTMENTS - Cash equivalents include certificates of deposit and money market instruments with original maturities of three months or less when purchased. At times, such investments may be in excess of the Federal Deposit Insurance Corporation (FDIC) insurance limit.
FINANCIAL INSTRUMENTS – Financial instruments include cash and cash equivalents and accounts payable. The amounts reported for financial instruments are considered to be reasonable approximations of their fair values.
PROPERTY AND EQUIPMENT - Property and equipment, including leasehold improvements, are stated at cost. The Company provides for depreciation and amortization using the straight-line method, based upon estimated useful lives of five to ten years or the lease term, if shorter.
RESEARCH AND DEVELOPMENT COSTS - Research and development costs are expensed as incurred.
INCOME TAXES - Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Temporary differences between financial statement and income tax reporting result primarily from net operating losses. As a result of these temporary differences, the Company has recorded a deferred tax asset with an offsetting valuation allowance for the same amount. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when it is considered more likely than not that some portion or all of the deferred tax asset will not be realized.
DEFINED CONTRIBUTION RETIREMENT PLANS - During January 2004, the Company established a 401(k) retirement plan that is available to all employees and requires matching contributions by the Company. During the years ended December 31, 2009, 2008 and 2007, the Company contributed approximately $38,000, $69,000 and $95,000, respectively.
VALUATION OF LONG-LIVED ASSETS – We assess the impairment of long-lived assets whenever events or changes in circumstances indicate that the carrying value may not be recoverable. Our long-lived assets as of December 31, 2009 and 2008 were represented by property and equipment, as we have no intangible assets on our balance sheet. Factors we consider important which could trigger an impairment review include the following:
|
|
·
|
significant underperformance relative to expected historical or projected future operating results;
|
|
|
·
|
significant changes in the manner of our use of the acquired assets or the strategy for our overall business;
|
|
|
·
|
significant negative industry or economic trends; and
|
|
|
·
|
significant decrease in the market value of the assets.
|
The impairment test is based upon a comparison of the estimated undiscounted cash flows to the carrying value of the long-lived assets. If we determine that the carrying value of long-lived assets may not be recoverable based upon the existence of one or more of the above indicators of impairment, we measure any impairment based on projected discounted cash flows. The cash flow estimates used to determine the impairment, if any, contain management’s best estimate using appropriate assumptions and projections at that time. Net long-lived property and equipment as of December 31, 2009 and 2008 was $324,000 and $1,447,000, respectively. We reviewed our long-lived property and equipment as of December 31, 2009, and have determined that their estimated fair value exceeds the carrying amount of such assets; therefore, we have not recognized an impairment loss for our long-lived property and equipment.
USE OF ESTIMATES – The accompanying financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America. This requires our management to make estimates about the future resolution of existing uncertainties that affect the reported amounts of assets, liabilities, revenues and expenses which in the normal course of business are subsequently adjusted to actual results. Actual results could differ from such estimates. In preparing these financial statements, management has made its best estimates and judgments of the amounts and disclosures included in the financial statements giving due regard to materiality.
LOSS PER SHARE – The Company’s basic loss per common share is computed as net loss divided by the weighted average number of common shares outstanding for the period. Diluted net loss per common share is the same as basic net loss per common share, since potentially dilutive securities from the assumed exercise of all outstanding options and warrants would have an antidilutive effect because the Company incurred a net loss during each period presented. As of December 31, 2009, 2008 and 2007, there were 28.1 million, 48.0 million, and 35.1 million common shares, respectively, issuable upon exercise of options and warrants, and the vesting of non-vested restricted common stock.
STOCK-BASED COMPENSATION - The Company calculates the fair value of stock-based compensation using the Black-Scholes method. Stock-based compensation costs are recorded as earned for all unvested stock options outstanding. The charge is being recognized in research and development and consulting, general and administrative expenses over the remaining service period after the adoption date based on the original estimate of fair value of the options as of the grant date. The Company recorded share-based compensation of approximately $326,000, $771,000 and $910,000 for the years ended December 31, 2009, 2008 and 2007, respectively. The Company will continue to incur share-based compensation charges in future periods. As of December 31, 2009, unamortized stock-based compensation expense of $880,000 remains to be recognized, which is comprised of $482,000 related to non performance-based stock options to be recognized over a weighted average period of 0.7 years, $104,000 related to restricted stock to be recognized over a weighted average period of 1.1 years, and $294,000 related to performance-based stock options which vest upon reaching certain milestones. Expenses related to the performance-based stock options will be recognized if and when the Company determines that it is probable that the milestone will be reached.
NEW ACCOUNTING PRONOUNCEMENTS – In June 2009, the FASB issued FASB ASC 105, Generally Accepted Accounting Principles (“GAAP”), which establishes the FASB Accounting Standards Codification as the sole source of authoritative generally accepted accounting principles. Pursuant to the provisions of FASB ASC 105, the Company has updated references to GAAP in its financial statements issued for the period ended December 31, 2009. The adoption of FASB ASC 105 did not impact the Company’s financial position or results of operations.
In April 2009, the FASB issued guidance now codified as FASB ASC Topic 825, Financial Instruments, which amends previous Topic 825 guidance to require disclosures about fair value of financial instruments in interim as well as annual financial statements. The adoption did not have a material impact on our results from operations or on our financial condition. Financial instruments include cash and cash equivalents, short-term investments, and accounts payable. The amounts reported for financial instruments are considered to be reasonable approximations of their fair values.
NOTE 4 – CONVERTIBLE NOTES
On May 6, 2008, the Company entered into a binding Securities Purchase Agreement by and among ProQuest Investments II, L.P., ProQuest Investments II Advisors Fund, L.P., and ProQuest Investments III, L.P., referred to herein as the Purchasers, as amended pursuant to Amendment No. 1 to the Securities Purchase Agreement, dated May 28, 2008, by and among the Company and the Purchasers, to sell up to $4,000,000 of secured convertible promissory notes, referred to herein as the convertible notes, and accompanying warrants to such Purchasers, referred to herein as the 2008 Financing. Mr. Steven Ratoff, the Company’s Chairman, Interim President, Chief Executive Officer and Interim Chief Financial Officer, is a private investor in, and since December 2004 has served as a venture partner with, ProQuest Investments.
On May 30, 2008, the Company closed the initial portion of the transaction, referred to herein as the Initial Closing, for $1,475,000, representing no more than 5,000,000 shares of the common stock underlying the convertible notes, upon receipt of approval from the NYSE Amex LLC, and satisfaction of customary closing conditions. The 5,000,000 shares, along with the prior securities owned by the Purchasers, represented 19.8% of the Company’s outstanding common stock upon execution of the Securities Purchase Agreement. At its Annual Stockholders’ Meeting on September 8, 2008, the Company sought and received stockholder approval to fund additional amounts such that the total commitment, inclusive of the amount at the Initial Closing, equaled up to $4,000,000, referred to herein as the Subsequent Closing and together with the Initial Closing, the Closings. On October 17, 2008, the Company closed the Subsequent Closing, for gross proceeds of $2,525,000.
In the Initial Closing, the Company issued the convertible notes, which convert into its common stock at a fixed price of $0.295 per share subject to certain adjustments, and five-year warrants to purchase 3,000,000 shares of its common stock, with an exercise price of $0.369 per share. The maturity date of the convertible notes issued in the Initial Closing was November 30, 2008.
In the Subsequent Closing, the Company issued the convertible notes, which convert into 10,744,681 shares of its common stock at a fixed price of $0.235 per share subject to certain adjustments, and five-year warrants to purchase 6,446,809 shares of its common stock, with an exercise price of $0.294 per share. The maturity date of the convertible notes issued in the Subsequent Closing was April 17, 2009.
The convertible notes accrue interest on their outstanding principal balances at an annual rate of 10%. All unpaid principal, together with any accrued but unpaid interest and other amounts payable under the convertible notes, shall be due and payable upon the earliest to occur of (i) when such amounts are declared due and payable by the Purchasers on or after the date that is 180 days after the date of issuance; or (ii) upon the occurrence of any change of control event. At the option of the Purchasers, interest may be paid in cash or in common stock of the Company. If the Company pays interest in common stock, the stock will be valued at the related conversion price for such convertible note. Therefore, on November 30, 2008, with respect to the Initial Closing and on April 17, 2009, with respect to the Subsequent Closing, the noteholders had the ability to either convert the convertible notes issued in such closing into shares of common stock or demand payment of the outstanding principal balance, plus accrued and unpaid interest at a rate of 10% per annum.
At its option, the Company had the ability to redeem without penalty or premium a portion of, or all of, the principal owed under the convertible notes by providing the Purchasers with at least 5 days’ written notice; provided that the Purchasers shall retain conversion rights in respect of the convertible notes for such period of 5 days after the Company has given such notice. Each prepayment shall be accompanied by the payment of accrued and unpaid interest on the amount being prepaid, through the date of the prepayment. On April 29, 2009, the Company remitted $1.0 million to ProQuest Investments and related entities against the $4.0 million of convertible notes issued during 2008.
On December 31, 2009, the Company announced an amendment agreement with ProQuest Investments L.P. and its affiliates (collectively, "ProQuest") to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of the Company's common stock as of December 31, 2009.
The Company filed an initial registration statement with the Securities Exchange Commission (“SEC”) to register the resale of common stock issuable in connection with the Initial Closing (excluding interest shares), referred to herein as the initial registrable shares, on June 26, 2008, which registration statement became effective as of July 16, 2008. These registration rights will cease once the initial registrable shares are eligible for sale by the Purchasers without restriction under Rule 144. Upon certain events, the Company has agreed to pay as liquidated damages an amount equal to 1.0% of the aggregate purchase price paid by the Purchasers for any convertible notes then held by the Purchasers, but these payments may not exceed 10% of the aggregate purchase price paid by the Purchasers.
The Company has entered into agreements with the holders of our common stock that require us to continuously maintain as effective, a registration statement covering the underlying shares of common stock. Such registration statements were declared effective on January 26, 2007, May 30, 2006 and July 28, 2005 and must continuously remain effective for a specified term. If we fail to continuously maintain such a registration statement as effective throughout the specified term, the Company may be subject to liability to pay liquidated damages.
With respect to the subsequent closing of the 2008 private placement, we agreed to file a registration statement with the SEC to register the resale of 17,978,724 shares of common stock issuable pursuant to the 2008 private placement, referred to herein as the subsequent registrable shares, within 30 days of the related closing. Also, we agreed to respond to all SEC comment letters as promptly as reasonably possible and to use our best efforts to have the registration statement declared effective within 90 days of the related closing. However, we were unable to register 9,044,649 of the subsequent registrable shares in accordance with the rules and regulations of the SEC.
Therefore, we have filed the registration statement with the SEC to register the resale of 8,934,075 subsequent registrable shares issuable pursuant to the 2008 private placement. In connection with our reduction of subsequent registrable shares being registered on the registration statement, in January 2009 we had agreed with the purchasers to pay, as liquidated damages, an amount equal to 1.0% of the aggregate purchase price paid by the purchasers for the shares that were not able to register for resale under the registration statement. Such liquidated damages equaled $12,703 for each 30 day period during which the shares remain unregistered, beginning on February 15, 2009 and ending on the date on which such subsequent registrable shares are registered. However, these payments could not exceed 10% of the aggregate purchase price paid by the purchasers, or $127,030, which the Company had recorded as a liability. The registration statement for the 8,934,075 shares did not become effective until May 5, 2009.
Consequently, the Company renegotiated the registration penalty with the purchasers due to the delay in registering the 8,934,075 shares. As a result, the Company agreed to pay the purchasers a registration penalty for the full amount of shares (17,978,924) for the period beginning on January 19, 2009 and ending on May 5, 2009. This resulted in an increase in the registration penalty of $44,770, for a maximum registration penalty of $171,800. The liquidated damages will be paid in the form of a non-convertible promissory note, which accrues interest at a rate of 10% per annum and all interest and principal will become due and payable upon the earlier to occur of (i) the maturity date, which is twelve months following the date of issuance or (ii) a change of control (as defined in the liquidated damages note). As of December 31, 2009, the Company had issued $172,000 in non-convertible promissory notes to the purchasers. As part of the Company’s December 31, 2009 announcement entering into an amendment agreement with ProQuest to convert the outstanding aggregate principal balance of all convertible notes, the liquidated damages notes were also converted into shares of the Company’s common stock as of December 31, 2009.
The Purchasers represented that they are “accredited investors” and agreed that the securities issued in the 2008 Financing bear a restrictive legend against resale without registration under the Securities Act. The convertible notes and warrants were sold pursuant to the exemption from registration afforded by Section 4(2) of the Securities Act and Regulation D thereunder.
F-11
The value of the warrants issued to the investors was calculated relative to the total amount of the debt offering. The relative fair value of the warrants issued to the investors in the Initial Closing was determined to be $467,000, or 31.7% of the total offering. This was determined using the Black Scholes Model and the following key assumptions were used; a discount rate of 3.41%, volatility of 80.26%, 5 year expected term, and dividend yield of 0%.
The relative fair value of the warrants issued in the Initial Closing (equaling $467,000), along with the effective beneficial conversion feature of the debt in the Initial Closing of $743,000 (calculated as the difference between the conversion price specified in the Securities Purchase Agreement and the calculated intrinsic value of the conversion feature) total $1,210,000 and are not in excess of the face value of the debt. The Company is using the straight-line method to amortize the debt discount and beneficial conversion feature through the maturity dates of the convertible notes, which result does not differ materially from the effective interest rate method. For the year ended December 31, 2009, the Company has recorded additional interest expense of $403,000, related to the amortization of the debt discount for the Initial Closing.
|
The balance of the convertible debt as of December 31, 2009 is summarized as follows:
|
||
|
Face amount
|
$ | 3,000,000 |
|
Total debt discount and beneficial conversion feature
|
1,900,000 | |
|
Amortization of debt discount and beneficial conversion feature
|
1,900,000 | |
|
Net unamortized debt discount and beneficial conversion feature
|
- | |
|
Debt Conversion
|
3,000,000 | |
|
Net debt recorded at December 31, 2009
|
$ | — |
Related to the issuance of the Initial Closing and the Subsequent Closing, the Company paid debt finance costs totaling $238,000, which were capitalized as deferred financing costs. These costs were amortized into interest expense using the straight-line method, which result does not differ materially from the effective interest rate method. For the year ended December 31, 2009, the Company had recorded expense of approximately $25,000 related to the amortization of the deferred financing costs. Related to the amended agreement and conversion of the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued interest, the Company recorded additional interest expense of approximately $1,360,000.
Related to the debt conversion agreement with ProQuest of $3,657,000 to 23,237,083 shares of the Company’s common stock, $0.001 par value per share, the conversion consists of the following:
|
Initial Closing
|
$ | 1,475,000 |
|
Subsequent Closing
|
2,525,000 | |
|
Total Proceeds
|
4,000,000 | |
|
Less principal payment
|
1,000,000 | |
|
Net Amount Outstanding
|
3,000,000 | |
|
Liquidated Damages
|
172,000 | |
|
Accrued Interest
|
485,000 | |
|
Conversion at December 31, 2009
|
$ | 3,657,000 |
In addition to the convertible notes and liquidated damages notes, ProQuest received 9,446,809 warrants with exercise prices of 125% of the related note conversion price. As consideration for converting all of the notes, the Company agreed to set a conversion price of $0.1574 per share for the notes and to reduce the exercise price of the warrants to $0.1888 per share, which resulted in an approximate $170,000 charge to interest expense for the year ended December 31, 2009.
The Company accounted for the conversion of notes in accordance with FASB ASC 470, Debt with Conversion and Other Options. Accordingly, the Company recognized a loss equal to the fair value of all securities and other consideration transferred in the transaction in excess of the fair value of the consideration issuable in accordance with the original conversion terms. The fair value of the consideration in accordance with the original terms was approximately $3,657,000. The Company determined the fair value of the consideration transferred, along with the effective beneficial conversion feature of the debt, was approximately $1,200,000 greater than the fair value of the notes and related interest on December 31, 2009. Thus, the Company recorded this expense as interest expense on the Company’s Statement of Operations for the year ended December 31, 2009.
The Company accounted for this modification in accordance with FASB ASC 718 – Compensation-Stock Compensation. Accordingly, the Company calculated the total fair value of the original warrants to purchase common stock immediately prior to the modification, as well as, the fair value of the warrants to purchase common stock in accordance with the modification and recorded the incremental fair value as expense in the Company’s Statement of Operations for the year ended December 31, 2009. The fair value was determined using the Black-Scholes Model and the following ranges of key assumptions were used:
|
Discount Rate
|
1.7%
|
|
Volatility
|
157 - 163%
|
|
Expected Term
|
0.9 – 1.99 years
|
|
Dividend Yield
|
0%
|
In addition, the Agreement also provides that warrants to purchase 220,726 shares of the Common Stock issued previously to ProQuest in past transactions will be retired and the exercise price of all other warrants held by ProQuest, which consist of warrants to purchase 1,986,536 shares of the Common Stock, will be reduced to $0.1888 per share. The Company accounted for this modification in accordance with FASB ASC 718 – Compensation-Stock Compensation. Accordingly, the Company calculated the total fair value of the original warrants to purchase common stock immediately prior to the modification, as well as, the fair value of the warrants to purchase common stock in accordance with the modification and recorded the incremental fair value as expense in the Company’s Statement of Operations for the year ended December 31, 2009. The fair value was determined using the Black-Scholes Model and the following ranges of key assumptions were used:
|
Discount Rate
|
1.7%
|
|
Volatility
|
134 – 199%
|
|
Expected Term
|
0.9 – 1.99 years
|
|
Dividend Yield
|
0%
|
As a result of these modifications, the Company recorded approximately an additional $1.69 million as interest expense on the Company’s Statement of Operations for the year ended December 31, 2009. Upon conversion of all of the outstanding notes in accordance with the Agreement, the security interest granted to ProQuest in prior securitizations shall extinguish and be of no further force or effect. As a result of the Agreement, ProQuest’s equity ownership in the Company will consist of (i) 29,504,653 shares of the Common Stock and (ii) warrants to purchase 11,433,345 shares of the Common Stock at an exercise price of $0.1888 per share.
NOTE 5 – ASSETS HELD FOR SALE
As of December 31, 2009, the Company reclassified assets with a net book value of $299,000, which on December 31, 2008 had been reported as “assets held for sale” to fixed assets which will be used in the production of its Duromist product line. For the year ended December 31, 2007, the Company owned inventory of $131,000 related to the production of its NitroMist™ product line, which inventory was disposed of during the year ended December 31, 2008. During the year ended December 31, 2008, the Company wrote off $129,000 in inventory and $183,000 of the net property, plant and equipment, as a result of transferring manufacturing operations for NitroMist™ from its contract manufacturer in Manati, Puerto Rico, to its new contract manufacturer in Texas. The total amount of the inventory and equipment disposal, inclusive of $39,000 for the costs of such disposal, was recognized as a loss on disposal of assets held for sale of $351,000. In addition, the Company invested an additional $121,000 during the year ended December 31, 2008 for tooling to support product development activities located at the facility of its contract manufacturer for spray valves in England.
NOTE 6 - PROPERTY AND EQUIPMENT
Property and equipment are summarized as follows:
|
December 31, 2009
|
December 31, 2008
|
||||||
|
Equipment
|
$ | 514,000 | $ | 2,164,000 | |||
|
Furniture and fixtures
|
— | 455,000 | |||||
|
Leasehold improvements
|
— | 1,432,000 | |||||
| 514,000 | 4,051,000 | ||||||
|
Less: Accumulated depreciation and amortization
|
190,000 | 2,604,000 | |||||
| $ | 324,000 | $ | 1,447,000 | ||||
Property and equipment as of December 31, 2009 has been significantly reduced versus December 31, 2008. The net reduction in fixed assets of $1,123,000 is the result of assets being disposed of during the fiscal year due to the Company relocating to much smaller office space. Depreciation expense for December 31, 2009, 2008 and 2007 was $369,000, $506,000 and $685,000, respectively.
As of December 31, 2009 and 2008, the Company had total gross fixed assets of $34,000 and $513,000, with an accumulated depreciation of $17,000 and $93,000, respectively, recorded under a capital lease.
The Company reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. An asset is considered to be impaired when the sum of the undiscounted future net cash flows expected to result from the use of the asset and its eventual disposition is less than its carrying amount. The amount of impairment loss, if any, is measured as the difference between the carrying amount of the asset and its estimated fair value. The Company has reviewed its long-lived property and equipment as of December 31, 2009, and has determined that their estimated fair value exceeds the carrying amount of such assets; therefore, the Company has not recognized an impairment loss for its long-lived property and equipment.
NOTE 7 - RELATED PARTY TRANSACTIONS
PRIVATE PLACEMENTS – On May 6, 2008, the Company entered into a binding Securities Purchase Agreement with ProQuest Investments (see Note 8), as amended, to sell up to $4,000,000 of secured convertible promissory notes and accompanying warrants to ProQuest Investments. On December 31, 2009, the Company announced entering into an amendment agreement with ProQuest Investments L.P. and its affiliates (collectively, "ProQuest") to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of the Company's common stock as of December 31, 2009. As such, ProQuest Investments may be deemed to be our affiliate. Mr. Steven B. Ratoff, our President and Chief Executive Officer, has served as a venture partner with ProQuest Investments since December 2004, although he has no authority for investment decisions by ProQuest Investments.
In September 2006, the Company’s Board of Directors appointed Steven B. Ratoff as Chairman of the Board. In connection with Mr. Ratoff’s appointment as Chairman of the Board, the Board entered into a consulting arrangement to compensate Mr. Ratoff for his efforts. This arrangement is on a month-to-month basis and compensates Mr. Ratoff at a rate of $17,500 per month. Pursuant to this consulting arrangement, the Company paid Mr. Ratoff approximately $210,000 for each of the years ended December 31, 2009 and 2008; and approximately $207,000 for the year ended December 31, 2007. In March 2007, Mr. Ratoff’s monthly compensation was reduced to $10,000 to reflect his decreased day-to-day time involvement at the Company, and in June, 2007, Mr. Ratoff’s monthly compensation was increased to $17,500 per month to reflect his appointment as the Company’s Interim President and Chief Executive Officer. In January 2010, the Company’s Board of Directors appointed Steven B. Ratoff as President and Chief Executive Officer effective January 1, 2010. Mr. Ratoff will continue to serve as Interim Chief Financial Officer.
In September 2007, in connection with his resignation, Dr. Egberts (the Company’s former Chief Executive Officer) and the Company entered into a Separation, Consulting and General Release Agreement (the “Agreement”). Pursuant to the Agreement, the Company paid Dr. Egberts approximately $223,000 and $140,000 for the years ended December 31, 2008 and 2007, respectively.
NOTE 8 - STOCKHOLDERS’ EQUITY (DEFICIENCY)
On May 6, 2008, the Company entered into a binding Securities Purchase Agreement by and among ProQuest Investments II, L.P., ProQuest Investments II Advisors Fund, L.P., and ProQuest Investments III, L.P., referred to herein as the Purchasers, as amended pursuant to Amendment No. 1 to the Securities Purchase Agreement, dated May 28, 2008, by and among the Company and the Purchasers, to sell up to $4,000,000 of secured convertible promissory notes, referred to herein as the convertible notes, and accompanying warrants to such Purchasers, referred to herein as the 2008 Financing. On May 30, 2008, the Company closed the initial portion of the transaction, referred to herein as the Initial Closing, for $1,475,000, representing 5,000,000 shares of the common stock underlying the convertible notes. On October 17, 2008, the Company closed the Subsequent Closing, for gross proceeds of $2,525,000, representing 10,744,681 shares of the common stock underlying the convertible notes (see Note 4).
On December 31, 2009, the Company announced an amendment agreement with ProQuest Investments L.P. and its affiliates (collectively, "ProQuest") to convert the outstanding aggregate principal balance of all convertible notes and all liquidated damages notes, in each case, plus all accrued interest, in an aggregate amount equal to $3,657,000 to 23,237,083 shares of the Company's common stock as of December 31, 2009.
On June 26, 2009, the Company entered into a common stock purchase agreement with Seaside 88, LP. As of December 31, 2009, the Company issued 4,500,000 shares of the common stock and received gross proceeds of $1,055,000.
The Company has entered into registration rights agreements with certain holders of our common stock that require us to continuously maintain an effective registration statement covering the underlying shares of common stock. Such registration statements have been declared effective and must continuously remain effective for a specified term. If we fail to continuously maintain such registration statements as effective throughout the specified terms, the Company may be subject to liability to pay liquidated damages.
PREFERRED STOCK - The Company’s Certificate of Incorporation authorizes the issuance of up to 1,000,000 shares of Preferred Stock. None of the Preferred Stock has been designated or issued through December 31, 2009. The Board is authorized to issue shares of Preferred Stock from time to time in one or more series and to establish and designate any such series and to fix the number of shares and the relative conversion and voting rights, and terms of redemption and liquidation.
NOTE 9 - COMMITMENTS AND CONTINGENCIES
EMPLOYMENT AGREEMENTS - At December 31, 2009, the Company had an employment agreement with one officer of the Company providing for an aggregate salary of $300,000 in each of the years ending December 31, 2009 and 2010, excluding potential Company matching contributions to the officer’s 401(k) plan for 2009. Effective January 1, 2010, the Company matching contributions to the 401(k) plan was discontinued. Effective January 1, 2010, the Company entered into an employment agreement with the newly appointed President and Chief Executive Officer providing an aggregate salary of $350,000 for the year ending December 31, 2010.
The remaining terms of the officer’s employment agreement are outlined below. Generally, in the event an officer is terminated prior to the end of such agreement, the officer is entitled to severance payments equal to the officer’s salary for the shorter of twelve months or the remaining term of the officer’s employment agreement.
The employment agreements with Dr. Bergstrom expire in December 2010 and Mr. Ratoff’s agreement has no defined expiration date.
In September 2007, in connection with his resignation, Dr. Egberts and the Company entered into a Separation, Consulting and General Release Agreement, pursuant to which the Company agreed to pay Dr. Egberts fees for services at a rate of $363,000 per annum through July 25, 2008. At December 31, 2009, the Company had no further obligations to Dr. Egberts under the Agreement.
All of the foregoing employment agreements provide for the potential issuance of bonuses based on certain factors. Such agreements also provide for the grant of options to purchase shares of the Company’s common stock.
LICENSE AND DEVELOPMENT AGREEMENTS - In April 2003, the Company entered into a license and development agreement with Manhattan Pharmaceuticals for the worldwide, exclusive rights to the Company’s proprietary oral spray technology to deliver propofol for pre-procedural sedation. The terms of the agreement call for certain milestone and other payments, the first $125,000 of which was partially received during June 2003. In November 2003, the Company received $375,000 from Manhattan Pharmaceuticals for license fees. The Company has included these license fees in deferred revenue and is recognizing these license fees over the 20-year term of the license.
In June 2004, the Company entered into a 20-year worldwide exclusive license agreement with Velcera, a veterinary company. The license agreement is for the exclusive rights to the Company’s propriety oral spray technology in animals. In September 2004, the Company received $1,500,000 from Velcera as an upfront payment in connection with the commercialization agreement. The upfront payment has been included in deferred revenue and will be recognized in income over the 20-year term of the agreement. In addition, the Company received an equity stake of 529,500 shares of common stock, approximately 15% at the time the shares were issued, in Velcera which did not have a material value. The Company may receive additional milestone payments and royalty payments over the 20-year term of the agreement. During the year ended December 31, 2007, the Company invoiced Velcera $125,000 for contractual milestones that were reached.
In July 2004, the Company entered into a licensing agreement with Par for the exclusive right to market, sell and distribute nitroglycerin lingual spray in the U.S. and Canada. The Company has received $250,000 in upfront and milestone payments and may receive additional fees and royalty payments over the 10-year term of the license. The upfront payment has been included in deferred revenue and will be recognized in income over the 10-year term of the agreement. In July 2007, the Company and Par agreed to terminate the agreement relating to NitroMist™.
In October 2004, the Company entered into a license and development agreement pursuant to which the Company granted to Hana Biosciences an exclusive license to develop and market the Company’s oral spray version of ondansetron in the U.S. and Canada. Pursuant to the terms of the agreement, in exchange for $1,000,000, Hana Biosciences purchased 400,000 shares of the Company’s common stock at a per share price equal to $2.50, a premium of $.91 per share or $364,000 over the then market value of the Company’s common stock. The Company accounted for this premium as deferred revenue related to the license. In connection with the agreement, Hana Biosciences issued to the Company $500,000 worth of common stock of Hana Biosciences (73,121 shares based on a market value of $6.84 per share). The proceeds received from Hana Biosciences attributable to the premium are included in deferred revenue and are being recognized over the 20-year term of the agreement. The Company may receive additional license fees and royalties over the 20-year term of the agreement.
In July 2007, the Company entered into a sublicense agreement with Hana Biosciences and Par, pursuant to which Hana Biosciences granted a non-transferable, non-sublicenseable, royalty-bearing, exclusive sublicense to Par to develop and commercialize Zensana™. In connection therewith, the Company and Hana Biosciences amended and restated their existing license agreement, as amended, relating to the development and commercialization of Zensana™ to coordinate certain of the terms of the sublicense agreement. Under the terms of the sublicense agreement, Par is responsible for all development, regulatory, manufacturing and commercialization activities of Zensana™ in the United States and Canada. The Company retains its rights to Zensana™ outside of the United States and Canada. In addition, under the terms of the Amended and Restated License Agreement, Hana Biosciences relinquished its right to pay reduced royalty rates to the Company until such time as Hana Biosciences had recovered one-half of its costs and expenses incurred in developing Zensana™ from sales of Zensana™ and the Company agreed to surrender for cancellation all 73,121 shares of the Hana Biosciences common stock acquired by the Company in connection with execution of the original License Agreement.
On May 19, 2008, the Company and BioAlliance Pharma SA or BioAlliance, entered into an agreement where BioAlliance acquired the European rights for NovaDel’s Ondansetron oral spray. Under the terms of the agreement, BioAlliance paid NovaDel a license fee of $3,000,000 upon closing. The Company is eligible for additional milestone payments totaling approximately $24 million (an approval milestone of $5,000,000 and sales-related milestone payments of approximately $19 million) as well as a royalty on net sales. BioAlliance and the Company anticipate collaborating in the completion of development activities for Europe, with BioAlliance responsible for regulatory and pricing approvals and then commercialization throughout Europe. The Company will be responsible for supplying the product. The upfront payment has been included in deferred revenue and is being recognized in income over the term of the agreement (nineteen and one half-years).
On November 13, 2009, the Company and ECR (a wholly-owned subsidiary of Hi-Tech Pharmacal Co., Inc.) entered into an exclusive license and distribution agreement to commercialize and manufacture ZolpiMist™ in the United States and Canada. ZolpiMist™ is the Company’s oral spray formulation of zolpidem tartrate approved by the FDA in December 2008.
Under the terms of the agreement, ECR paid the Company $3 million upon execution of the agreement. ECR will assume responsibility for manufacturing and marketing the product in the United States and Canada. In addition, ECR will pay royalties of up to 15% on net sales of ZolpiMist™ as well as an additional milestone payment if sales reach a specified level.
On October 27, 2009, the Company and privately-held Mist entered into a licensing agreement to manufacture and commercialize the NitroMist® lingual spray version of nitroglycerine, a widely-prescribed and leading short-acting nitrate for the treatment of angina pectoris. Under terms of the agreement, Mist paid the Company a $1,000,000 licensing fee upon execution of the agreement, and will pay milestone payments totaling an additional $1,000,000 over twelve months, and ongoing performance payments of seventeen percent (17%) of net sales.
Through a separate license agreement with Mist, Akrimax Pharmaceuticals, LLC will receive the exclusive right to manufacture, distribute, market and sell NitroMist® in North America. NitroMist® provides acute relief of an attack or acute prophylaxis of angina pectoris due to coronary artery disease. The lingual spray form of the drug is conveniently administered and is rapidly absorbed into the bloodstream via the oral mucosa, providing patients a fast and tolerable treatment option for the prevention or relief of pain associated with such attacks.
CAPITAL LEASE OBLIGATIONS – As of December 31, 2009, the Company has aggregate capital lease obligations of $14,000, of which $10,000 and $4,000 are scheduled to be paid in the years ending December 31, 2010 and 2011, respectively.
OPERATING LEASES - In March 2003, the Company entered into a 10-year lease for office, laboratory, manufacturing and warehouse space. During the first five years of the lease, the annual rent was approximately $332,000 plus a proportionate share of real estate taxes and common area charges. Beginning in the sixth year and continuing through the tenth year of the lease, the annual rent was approximately $366,000 plus a proportionate share of real estate taxes and common areas. Effective July 1, 2009, the Company executed a lease amendment modifying certain terms to the existing lease. The amendment converts the lease term to month to month with a provision to terminate upon thirty days written notice. During the years ended December 31, 2009, 2008, and 2007, the Company paid rent of approximately $257,000, $453,000, and $443,000, respectively.
In January 2010, the Company terminated the March 2003 lease effective January 31, 2010 and relocated its corporate office to Bridgewater, NJ upon signing a one-year lease effective February 1, 2010.
Future minimum rental payments subsequent to December 31, 2009 are as follows:
|
Years Ending December 31,
|
||
|
2010
|
$ | 39,000 |
|
2011
|
3,500 | |
| $ | 42,500 | |
NOTE 10 – OTHER, NET
For the year ended December 31, 2007, the Company recorded a charge of $500,000 to account for the return of stock of Hana Biosciences shares. This amount was offset by $434,000 benefit recognized to write off the remaining deferred revenue related to the shares received from Hana Biosciences resulting in a net $66,000 expense for Other, net.
For the year ended December 31, 2009 the Company recorded income relating to the reversal of the warrant liability initially recorded upon the Company’s adoption of ASC 815-40-15 in the amount of $360,000. This amount was offset with a loss on disposition of fixed assets in the amount of $745,000 resulting in a net $385,000 expense for Other, net.
NOTE 11 – ACCRUED EXPENSES AND OTHER CURRENT LIABILITIES
Accrued expenses and other current liabilities are comprised of the following at December 31, 2009 and 2008:
|
December 31,
2009
|
December 31,
2008
|
||||||
|
Professional fees
|
$ | 31,000 | $ | 60,000 | |||
|
Accrued compensation
|
— | 40,000 | |||||
|
Accrued interest expense
|
— | 126,000 | |||||
|
Non-registration penalty
|
— | 127,000 | |||||
|
Product development costs
|
— | 437,000 | |||||
|
Insurance premiums
|
84,000 | 129,000 | |||||
|
Other
|
2,000 | 5,000 | |||||
| $ | 117,000 | $ | 924,000 | ||||
NOTE 12 - INCOME TAXES
The significant components of the Company’s net deferred tax assets are summarized as follows:
|
December 31,
2009
|
December 31,
2008
|
||||||
|
Stock-based compensation
|
$ | 374,000 | $ | 272,000 | |||
|
Net operating loss (“NOL”) carryforwards
|
23,035,000 | 22,542,000 | |||||
|
Deferred revenue
|
3,387,000 | 1,894,000 | |||||
|
Property and equipment
|
(27,000 | ) | (166,000 | ) | |||
|
Research and development credit
|
1,568,000 | 1,350,000 | |||||
|
Capital loss carryforwards
|
200,000 | 200,000 | |||||
|
Accrued expenses and other reserves
|
— | 216,000 | |||||
|
Total gross deferred tax assets
|
28,537,000 | 26,308,000 | |||||
|
Valuation allowance
|
(28,537,000 | ) | (26,308,000 | ) | |||
|
Net deferred tax assets
|
$ | — | $ | — | |||
At December 31, 2009, the Company had Federal and state net operating loss carryforwards for financial reporting and income tax purposes of approximately $ 64.7 million and $16.4 million, respectively, which can be used to offset current and future Federal and state taxable income, if any, through 2029 and 2016, respectively. In addition, the Company has federal and state research and development tax credits of $ 1.0 million and $0.5 million, respectively, which will expire beginning 2020 and 2013, respectively. A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. The Company has provided valuation allowances to offset its deferred tax assets due to the significant uncertainties related to its ability to generate future taxable income. The net increases in the total valuation allowance for the years ended December 31, 2009 and 2008 were $2.2 million and $1.9 million, respectively.
The tax benefits recognized in the years ended December 31, 2009, 2008 and 2007 of $1,057,000, $735,000 and $658,000, respectively, relate solely to the sale of certain of the Company’s state net operating loss carryforwards.
The following is a reconciliation of the income tax benefit computed at the statutory rate to the provision for income taxes:
|
Years Ending
|
|||||
|
December 31,
2009
|
December 31,
2008
|
December 31,
2007
|
|||
|
Federal tax at statutory rate
|
(34.0%)
|
(34.0%)
|
(34.0%)
|
||
|
State income tax
|
3.4%
|
(6.0%)
|
(6.0%)
|
||
|
Other
|
(1.2%)
|
2.0%
|
0.2%
|
||
|
Sale of net operating losses
|
(8.1%)
|
(7.1%)
|
(3.7%)
|
||
|
Amortization of convertible debt discount
|
1.6%
|
5.4%
|
—
|
||
|
Cancelled stock option
|
—
|
5.7%
|
—
|
||
|
Expired Federal NOL
|
0.3%
|
1.1%
|
—
|
||
|
Increase in valuation allowance
|
25.8%
|
25.8%
|
39.8%
|
||
|
Totals
|
(12.2%)
|
(7.10%)
|
(3.7%)
|
||
The Tax Reform Act of 1986 (the “Act”) provides for a limitation on the annual use of NOL carryforwards (following certain ownership changes, as defined by the Act), which could significantly limit the Company’s ability to utilize these carryforwards. The Company has experienced various ownership changes, as defined by the Act, as a result of past financings and may experience others in connection with future financings. Accordingly, the Company’s ability to utilize the aforementioned Federal operating loss carryforwards will be limited. The Company is in the process of determining the impact of ownership changes that have occurred, as defined by the Act. Additionally, because U.S. tax laws limit the time during which these carryforwards may be applied against future taxes, the Company may not be able to take full advantage of these attributes for Federal income tax purposes.
SALE OF NET OPERATING LOSS CARRYFORWARDS: The State of New Jersey has enacted legislation permitting certain corporations located in New Jersey to sell state tax loss carryforwards and state research and development credits, or net operating loss carryforwards, in order to obtain tax benefits. The Company recorded an income tax benefit of $1,057,000, $732,000, and $658,000 for the years ended December 31, 2009, 2008 and 2007, respectively, from the sale of its New Jersey net operating loss carryforwards. If still available under New Jersey law, the Company may attempt to sell its remaining New Jersey net operating loss carryforwards of $16.4 million as of December 31, 2009. The Company cannot estimate, however, what percentage of its saleable net operating loss carryforwards New Jersey will permit it to sell, how much money will be received in connection with the sale, if the Company will be able to find a buyer for its net operating loss carryforwards or if such funds will be available in a timely manner or at all.
The Company files income tax returns in the U.S. Federal jurisdiction and in the State of New Jersey. With certain exceptions, the Company is no longer subject to U.S. Federal and state income tax examinations by tax authorities for years prior to 2004. However, NOL and tax credits generated from those prior years could still be adjusted upon audit. The Company adopted ASC 740-10 (Formerly FIN 48) Accounting for Uncertainty in Income Taxes – an interpretation of ASC 740 (Formerly FASB statement No. 109) on January 1, 2007 with no material impact to the financial statements.
The Company had no unrecognized tax benefits at December 31, 2009 that would affect the annual effective tax rate. Further, the Company is unaware of any positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next twelve months.
NOTE 13 - STOCK OPTIONS AND WARRANTS
At December 31, 2009, the Company had two plans which allow for the issuance of stock options and other awards: the 1998 Stock Option Plan and the 2006 Equity Incentive Plan (the “Plans”). On January 17, 2006, the stockholders of the Company, upon recommendation of the Board of Directors of the Company, approved the NovaDel Pharma Inc. 2006 Equity Incentive Plan (the “2006 Plan”). The 2006 Plan authorizes the grant of several types of stock-based awards, including stock options, stock appreciation rights and stock (including restricted stock). The number of shares of common stock originally reserved for issuance under the 2006 Plan was 6 million shares. These Plans are administered by the Compensation Committee of the Board of Directors. Incentive Stock Options (“ISOs”) may be granted to employees and officers of the Company and non-qualified options may be granted to consultants, directors, employees and officers of the Company. Options to purchase the Company’s common stock may not be granted at a price less than the fair market value of the common stock at the date of grant and will expire not more than 10 years from the date of grant, and vesting is determined by the Compensation Committee of the Board of Directors. ISOs granted to a 10% or more stockholder may not be for less than 110% of fair market value or for a term of more than five years. As of December 31, 2009, there were approximately 0.9 million shares available for issuance under the Plans (see Note 14).
The Company selected the Black-Scholes method of valuation for share-based compensation effective August 1, 2005. Compensation costs are recorded as earned for all unvested stock options outstanding at the beginning of the first quarter of 2005 and for all options granted after the date of adoption. The charge is being recognized in research and development and consulting, selling, general and administrative expenses over the remaining service period after the adoption date based on the original estimate of fair value of the options as of the grant date.
Information with respect to stock option activity for the years ended December 31, 2009, 2008 and 2007 is as follows:
|
Options
|
Shares (000)
|
Weighted-Average Exercise Price
|
Weighted-Average Remaining Contractual Terms (Years)
|
Aggregate Intrinsic Value ($000)
|
|||||||||||
|
Outstanding at January 1, 2007
|
8,775 | $ | 1.68 | — | — | ||||||||||
|
Grants
|
3,239 | 1.67 | — | — | |||||||||||
|
Exercises
|
(268 | ) | .75 | — | — | ||||||||||
|
Forfeitures/Cancellations
|
(3,317 | ) | 1.72 | — | — | ||||||||||
|
Outstanding at December 31, 2007
|
8,429 | $ | 1.69 | 5.9 | $ | — | |||||||||
|
Exercisable at December 31, 2007
|
5,549 | $ | 1.75 | 5.0 | $ | — | |||||||||
|
Outstanding at January 1, 2008
|
8,429 | 1.69 | — | — | |||||||||||
|
Grants
|
338 | .24 | — | — | |||||||||||
|
Exercises
|
— | — | — | — | |||||||||||
|
Forfeitures/Cancellations
|
(3,300 | ) | 1.72 | — | — | ||||||||||
|
Outstanding at December 31, 2008
|
5,467 | $ | 1.59 | 5.4 | $ | — | |||||||||
|
Vested and expected to vest at December 31, 2008
|
5,349 | $ | 1.59 | 5.4 | |||||||||||
|
Exercisable at December 31, 2008
|
3,102 | $ | 1.74 | 4.2 | $ | — | |||||||||
|
Outstanding at January 1, 2009
|
5,467 | 1.59 | — | — | |||||||||||
|
Grants
|
5,103 | .25 | |||||||||||||
|
Exercises
|
— | — | — | — | |||||||||||
|
Forfeitures/Cancellations
|
(2,291 | ) | 1.41 | ||||||||||||
|
Outstanding at December 31, 2009
|
8,279 | $ | 0.81 | 4.1 | — | ||||||||||
|
Vested and expected to vest at December 31, 2009
|
8,073 | $ | 0.82 | 4.1 | — | ||||||||||
|
Exercisable at December 31, 2009
|
4,148 | $ | 1.16 | 3.2 | $ | — | |||||||||
The Company recorded share-based compensation for options using the fair value method of approximately $326,000 for the year ended December 31, 2009, $771,000 for the year ended December 31, 2008, $910,000 for the year ended December 31, 2007, which amounts are included in the Company’s net loss for each period.
|
·
|
On February 6, 2008, the Company’s Board of Directors, upon the recommendation of the Compensation Committee, approved grants of 750,000 shares of restricted common stock to the executive officers of the Company and an additional 350,000 shares of restricted common stock to other employees of the Company. The restricted stock was awarded from the 1998 Stock Option Plan. The restrictions on the restricted stock shall lapse over a three-year period, subject to reduction as follows: (1) in the event of a $5 million non-dilutive financing by the Company on or before December 31, 2008, the three-year restriction shall be accelerated such that the restrictions on the restricted stock shall lapse over a two-and-one-half year period; (2) in the event of an additional $5 million (or $10 million in the aggregate) non-dilutive financing by the Company on or before December 31, 2008, the three-year restriction shall be accelerated such that the restrictions on the restricted stock shall lapse over a two-year period; and (3) in the event of a $20 million (or $20 million in the aggregate) non-dilutive financing by the Company, the restrictions shall immediately lapse. Additionally, the Board, upon the recommendation of the Compensation Committee, agreed that, in the case of the Company’s Chief Executive Officer, an additional 200,000 shares of restricted stock shall be granted as follows: (1) upon achieving a $5 million non-dilutive financing by the Company on or before December 31, 2008, an additional 100,000 shares of restricted stock shall be granted; and (2) upon achieving an additional $5 million (or $10 million in the aggregate) in non-dilutive financing by the Company on or before December 31, 2008, an additional 100,000 shares of restricted stock shall be granted. The restrictions on such additional shares shall lapse over a three-year period. The events triggering the aforementioned issuance of restricted common stock did not occur on or before December 31, 2008.
|
|
·
|
During the year ended December 31, 2009, the Company additionally granted 5,102,500 additional stock options. The exercise price of such options ranged from $0.17 per share to $0.34 per share.
|
The Company used the following weighted average assumptions in determining fair value under the Black-Scholes model for grants in the respective periods:
|
Year Ended
|
|||||
|
|
2009
|
2008
|
2007
|
||
|
Expected volatility
|
85%
|
83%
|
63%
|
||
|
Dividend yield
|
0%
|
0%
|
0%
|
||
|
Expected term until exercise (years)
|
2.6
|
3.7
|
4.9
|
||
|
Risk-free interest rate
|
1.8%
|
2.3%
|
4.8%
|
||
Expected volatility is based on historical volatility of the Company’s common stock. The expected term of options is estimated based on the average of the vesting period and contractual term of the option. The risk-free rate is based on U.S. Treasury yields for securities in effect at the time of grant with terms approximating the expected term until exercise of the option. In addition, the fair value of stock options granted is recognized as expense over the service period, net of estimated forfeitures. The Company is utilizing a 5% forfeiture rate, which it believes is a reasonable assumption to estimate forfeitures. However, the estimation of forfeitures requires significant judgment, and to the extent actual results or updated estimates differ from our current estimates, the effects of such resulting adjustment will be recorded in the period estimates are revised. The weighted average grant date fair value of options granted was $0.25, $0.15, and $0.93 during the years ended December 31, 2009, 2008 and 2007. The total intrinsic value of options exercised was $0, $172,000 and $398,000 during the years ended December 31, 2009, 2008 and 2007, respectively. There were no options exercised during the year ended December 31, 2009.
At December 31, 2009, there were approximately 581,000 non-plan options reserved for issuance.
A summary of the status of the Company’s non-vested restricted common stock as of December 31, 2009 and changes during the twelve months ended December 31, 2009 is presented below:
|
Non-Vested Restricted Common Stock
|
Shares (000)
|
Weighted Average Grant-Date Fair Value
|
|||||
|
January 1, 2009
|
1,133 | $ | 0.51 | ||||
|
Vested
|
(33 | ) | $ | 1.71 | |||
|
Forfeited
|
(575 | ) | $ | 0.47 | |||
|
Granted
|
— | ||||||
|
December 31, 2009
|
525 | $ | 0.47 | ||||
The following table summarizes information related to warrants outstanding at December 31, 2009:
|
Price Range
|
Number of Warrants Outstanding and Exercisable 000’s
|
Remaining Contractual Life (Years)
|
|||||
| $ | 0.01 – 0.99 | 11,854 | 3.1 | ||||
| $ | 1.00 – 1.99 | 7,966 | 1.5 | ||||
|
Totals
|
19,820 | ||||||
NOTE 14 – SUBSEQUENT EVENTS
Seaside Closings
Under the common stock purchase agreement with Seaside 88, LP, the Company has received $200,140 in gross proceeds for the closings that have occurred after December 31, 2009 through March 31, 2010. On March 26, 2010, the Company and Seaside 88, LP mutually agreed to terminate the common stock purchase agreement which was entered into in 2009.
On March 31, 2010, the Company announced it will receive approximately $1.5 million in gross proceeds in a registered direct offering (the "Offering") of 9,100,001 shares of common stock, par value $0.001 per share (the "Common Shares"), at a price of $0.165 per share. The investors will also receive five-year warrants (the "Series A Warrants") to purchase 4,550,001 shares of common stock with an exercise price of $0.25 per share and six-month warrants (the "Series B Warrants," together with the Common Shares and the Series A Warrants, the "Securities") to purchase 3,033,334 shares of common stock at an exercise price of $0.25 per share. The Company expects to receive total proceeds, net of offering expenses and placement agency fees, of approximately $1.4 million. The exercise price of the Series A and Series B Warrants are subject to adjustment as provided by such warrants. The Offering closed on March 31, 2010 and the Company sold the Securities pursuant to an effective registration statement.
NOTE 15 – QUARTERLY RESULTS OF OPERATIONS (UNAUDITED)
Unaudited quarterly financial data for the years ended December 31, 2009, 2008 and 2007 as follows:
| Three Months Ended | Year Ended | |||||||||||||||||||
|
March 31,
2009
|
June 30,
2009
|
September 30,
2009
|
December 31,
2009
|
December 31,
2009
|
||||||||||||||||
|
Total Revenues
|
$ | 66,000 | $ | 67,000 | $ | 223,000 | $ | 66,000 | $ | 422,000 | ||||||||||
|
Total Expenses
|
2,084,000 | 1,560,000 | 1,503,000 | 1,370,000 | 6,517,000 | |||||||||||||||
|
Loss from Operations
|
(2,018,000 | ) | (1,493,000 | ) | (1,280,000 | ) | (1,304,000 | (6,095,000 | ) | |||||||||||
|
Other, net
|
360,000 | (59,000 | ) | — | (686,000 | ) | (385,000 | ) | ||||||||||||
|
Interest Expense
|
486,000 | 150,000 | 81,000 | 1,443,000 | 2,160,000 | |||||||||||||||
|
Interest Income
|
5,000 | 1,000 | — | — | 6,000 | |||||||||||||||
|
Income Tax Benefit
|
— | — | — | 1,057000 | 1,057,000 | |||||||||||||||
|
Net Loss
|
$ | (2,139,000 | ) | $ | (1,701,000 | ) | $ | (1,361,000 | ) | $ | (2,376,000 | ) | $ | (7,577,000 | ) | |||||
|
Basic and Diluted Loss Per Common Share
|
$ | (0.04 | ) | $ | (0.03 | ) | $ | (0.02 | ) | $ | (0.04 | ) | $ | (0.12 | ) | |||||
|
Weighted Average Number of Shares of Common Stock Used in Computation of Basic and Diluted Loss Per Common Share
|
59,892,000 | 60,081,000 | 61,386,000 | 65,282,000 | 61,346,000 | |||||||||||||||
| Three Months Ended | Year Ended | |||||||||||||||||||
|
March 31,
2008
|
June 30,
2008
|
September 30,
2008
|
December 31,
2008
|
December 31,
2008
|
||||||||||||||||
|
Total Revenues
|
$ | 103,000 | $ | 51,000 | $ | 104,000 | $ | 103,000 | $ | 361,000 | ||||||||||
|
Total Expenses
|
2,110,000 | 2,987,000 | 1,878,000 | 1,976,000 | 8,951,000 | |||||||||||||||
|
Loss from Operations
|
(2,007,000 | ) | (2,936,000 | ) | (1,774,000 | ) | (1,873,000 | ) | (8,590,000 | ) | ||||||||||
|
Interest Expense
|
— | 294,000 | 750,000 | 824,000 | 1,868,000 | |||||||||||||||
|
Interest Income
|
35,000 | 28,000 | 21,000 | 53,000 | 137,000 | |||||||||||||||
|
Income Tax Benefit
|
— | — | — | 735,000 | 735,000 | |||||||||||||||
|
Net Loss
|
$ | (1,972,000 | ) | $ | (3,202,000 | ) | $ | (2,503,000 | ) | $ | (1,909,000 | ) | $ | (9,586,000 | ) | |||||
|
Basic and Diluted Loss Per Common Share
|
$ | (0.03 | ) | $ | (0.05 | ) | $ | (0.04 | ) | $ | (0.03 | ) | $ | (0.16 | ) | |||||
|
Weighted Average Number of Shares of Common Stock Used in Computation of Basic and Diluted Loss Per Common Share
|
59,592,000 | 59,592,000 | 59,592,000 | 59,592,000 | 59,592,000 | |||||||||||||||
| Three Months Ended | Year Ended | |||||||||||||||||||
|
March 31,
2007
|
June 30,
2007
|
September 30,
2007
|
December 31,
2007
|
December 31,
2007
|
||||||||||||||||
|
Total Revenues
|
$ | 40,000 | $ | 165,000 | $ | 206,000 | $ | 58,000 | $ | 469,000 | ||||||||||
|
Total Expenses
|
5,334,000 | 5,676,000 | 3,037,000 | 4,609,000 | 18,656,000 | |||||||||||||||
|
Loss from Operations
|
(5,294,000 | ) | (5.511,000 | ) | (2,831,000 | ) | (4,551,000 | ) | (18,187,000 | ) | ||||||||||
|
Other Income/(Loss)
|
(360,000 | ) | — | 294,000 | (66,000 | ) | ||||||||||||||
|
Interest Income
|
230,000 | 187,000 | 127,000 | 88,000 | 632,000 | |||||||||||||||
|
Income Tax Benefit
|
— | — | — | 658,000 | 658,000 | |||||||||||||||
|
Net Loss
|
$ | (5,424,000 | ) | $ | (5,324,000 | ) | $ | (2,410,000 | ) | $ | (3,805,000 | ) | $ | (16,963,000 | ) | |||||
|
Basic and Diluted Loss Per Common Share
|
$ | (0.09 | ) | $ | (0.09 | ) | $ | (0.04 | ) | $ | (0.06 | ) | $ | (0.29 | ) | |||||
|
Weighted Average Number of Shares of Common Stock Used in Computation of Basic and Diluted Loss Per Common Share
|
59,264,000 | 59,537,000 | 59,591,000 | 59,592,000 | 59,497,000 | |||||||||||||||
The sum of the quarters may not equal the full year basic and diluted loss per share since each period is calculated separately.
INDEX TO EXHIBITS
The following exhibits are included with this Annual Report on Form 10-K. All management contracts or compensatory plans or arrangements are marked with an asterisk.
|
EXHIBIT NO.
|
DESCRIPTION
|
METHOD OF FILING
|
||
|
3.1
|
Restated Certificate of Incorporation of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Quarterly Report on Form 10-QSB, as filed with the SEC on June 14, 2004.
|
||
|
3.2
|
Certificate of Amendment to the Certificate of Incorporation of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
3.3
|
Amended and Restated By-laws of the Company
|
Incorporated by reference to Exhibit 3.1 of the Company’s Form 8-K, as filed with the SEC on September 9, 2005.
|
||
|
4.1
|
Form of Class C Warrant for the Purchase of Shares of Common Stock
|
Incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the SEC on January 12, 2004.
|
||
|
4.2
|
Form of Warrant issued to certain accredited investors and placement agents
|
Incorporated by reference to Exhibit 4.1 of the Company’s Form 8-K, as filed with the SEC on April 17, 2006.
|
||
|
4.3
|
Form of Warrant issued to certain accredited investors and the placement agent
|
Incorporated by reference to Exhibit 4.1 of the Company’s Current Report on Form 8-K filed with the SEC on January 4, 2007.
|
||
|
4.4
|
Form of Warrant
|
Incorporated by reference to Exhibit 4.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on June 3, 2008.
|
||
|
10.1*
|
1992 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.2*
|
Form of Incentive Stock Option Agreement under the 1992 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.3*
|
1997 Stock Option Plan
|
Incorporated by reference to Exhibit 10.8 to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.4*
|
Form of Non-Qualified Option Agreement under the 1997 Stock Option Plan
|
Incorporated by reference to the Company’s Registration Statement on Form SB-2, as filed with the SEC on August 8, 1997 (File No. 333-33201).
|
||
|
10.5*
|
1998 Stock Option Plan
|
Incorporated by reference to Exhibit 4.1 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
||
|
10.6*
|
Form of Stock Option Agreement under the 1998 Stock Option Plan
|
Incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
||
|
10.7*
|
Form of Non-Qualified Option Agreement
|
Incorporated by reference to Exhibit 4.3 to the Company’s Registration Statement on Form S-8, as filed with the SEC on June 18, 2004 (File No. 333-116665).
|
||
|
10.8
|
Common Stock and Warrant Purchase Agreement, dated December 12, 2001, by and among the Company and certain purchasers
|
Incorporated by reference to Exhibit A to the Schedule 13D as filed by Lindsay A. Rosenwald with the SEC on December 21, 2001.
|
||
|
10.9
|
Amendment No. 1, dated January 6, 2002, to the Common Stock and Warrant Purchase Agreement dated December 12, 2001 between the Company and certain purchasers
|
Incorporated by reference to Exhibit 10.25 to the Company’s Registration Statement of Form SB-2, as filed with the SEC on April 15, 2002 (File No. 333-86262).
|
||
|
10.10
|
License and Development Agreement, effective as of April 4, 2003, by and between the Company and Manhattan Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.31to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on March 11, 2004.
|
||
|
10.11
|
Development, Manufacturing and Supply Agreement, dated July 28, 2004, by and between the Company and Par Pharmaceutical, Inc.
|
Incorporated by reference to Exhibit 10.13 to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on November 15, 2004.
|
||
|
10.12
|
Second Amendment to License and Development Agreement, dated as of June 22, 2004, by and between the Company and the Veterinary Company, Inc.
|
Incorporated by reference to Exhibit 10.14 to the Company’s Annual Report on Form 10-KSB, as filed with the SEC on November 15, 2004.
|
||
|
10.13*
|
Disclosure and Release Agreement Related to the Exchange of Non-Plan Options for Stock Options under the NovaDel Pharma Inc. 1998 Stock Option Plan by and between the Company and Thomas E. Bonney
|
Incorporated by reference to Exhibit 10.3 of the Company’s Form 8-K, as filed with the SEC on August 2, 2005.
|
||
|
10.14*
|
Disclosure and Release Agreement Related to the Exchange of Non-Plan Options for Stock Options under the NovaDel Pharma Inc. 1998 Stock Option Plan by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.4 of the Company’s Form 8-K, as filed with the SEC on August 2, 2005.
|
||
|
10.15*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated July 28, 2005, by and between the Company and Thomas E. Bonney
|
Incorporated by reference to Exhibit 10.25 of the Company’s Annual Report on Form 10-KSB for the period ended July 31, 2005, as filed with the SEC on October 31, 2005.
|
||
|
10.16*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated July 28, 2005, by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.29 of the Company’s Annual Report on Form 10-KSB for the period ended July 31, 2005, as filed with the SEC on October 31, 2005.
|
||
|
10.17
|
Amendment No. 1 to License and Development Agreement dated as of August 8, 2005, by and between the Company and Hana Biosciences Inc.
|
Incorporated by reference to Exhibit 99.1 of the Company’s Form 8-K, as filed with the SEC on August 12, 2005.
|
||
|
10.18*
|
NovaDel Pharma Inc. 2006 Equity Incentive Plan
|
Incorporated by reference to Exhibit 10.1 of the Company’s Form 8-K, as filed with the SEC on January 23, 2006.
|
||
|
10.19*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Thomas Bonney
|
Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.20*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Charles Nemeroff
|
Incorporated by reference to Exhibit 10.5 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.21*
|
1998 Stock Option Plan Nonqualified Stock Option Agreement dated January 17, 2006, by and between the Company and Steven Ratoff
|
Incorporated by reference to Exhibit 10.6 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on March 15, 2006.
|
||
|
10.22*
|
Employment Agreement dated December 4, 2006 by and between the Company and David H. Bergstrom, Ph.D.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.23*
|
Incentive Stock Option Award between the Company and David H. Bergstrom dated December 4, 2006
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.24*
|
Nonqualified Stock Option Award between the Company and David H. Bergstrom, dated December 4, 2006
|
Incorporated by reference to Exhibit 10.3 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 8, 2006.
|
||
|
10.25*
|
Amendment 2007-1 to the NovaDel Pharma Inc. 1998 Stock Option Plan dated March 2, 2007
|
Incorporated by reference to Exhibit 10.45 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
10.26*
|
Amendment 2007-1 to the NovaDel Pharma Inc. 2006 Equity Incentive Plan dated March 2, 2007
|
Incorporated by reference to Exhibit 10.46 of the Company’s Annual Report on Form 10-K, as filed with the SEC on March 26, 2007.
|
||
|
10.27
|
Amended and Restated License and Development Agreement, dated as of July 31, 2007, by and between NovaDel Pharma Inc. and HANA Biosciences, Inc.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.28
|
Product Development and Commercialization Sublicense Agreement, dated as of July 31, 2007, by and among NovaDel Pharma Inc., HANA Biosciences and PAR Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.2 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.29
|
Termination Agreement, dated as of July 31, 2007, by and between NovaDel Pharma Inc. and PAR Pharmaceuticals, Inc.
|
Incorporated by reference to Exhibit 10.3 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on November 14, 2007.
|
||
|
10.30+
|
License Agreement, dated May 19, 2008, by and among the Company and BioAlliance Pharma SA.
|
Incorporated by reference to Exhibit 10.4 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on August 7, 2008.
|
||
|
10.31+
|
Supply Agreement, dated July 7, 2008, by and among the Company and BioAlliance Pharma SA.
|
Incorporated by reference to Exhibit 10.5 of the Company’s Quarterly Report on Form 10-Q, as filed with the SEC on August 7, 2008.
|
||
|
10.32*
|
Separation, Consulting and General Release Agreement, effective as of April 30, 2009, by and between NovaDel Pharma Inc. and Deni M. Zodda
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on May 1, 2009.
|
||
|
10.33
|
Common Stock Purchase Agreement, by and between the Company and Seaside 88, LP, dated June 26, 2009
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on June 30, 2009.
|
||
|
10.34
|
Agreement, by and between the Company and Arthur W. Wood Company, dated June 15, 2009
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on July 20, 2009.
|
||
|
10.35+
|
License and Distribution Agreement, dated October 27, 2009, between NovaDel Pharma Inc. and Mist Acquisition, LLC
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 10, 2009.
|
||
|
10.36+
|
License and Distribution Agreement, dated November 12, 2009, between NovaDel Pharma Inc. and ECR Pharmaceuticals Company, Inc.
|
Incorporated by reference to Exhibit 10.2 of the Company’s Current Report on Form 8-K, as filed with the SEC on December 10, 2009.
|
||
|
10.37
|
Lease Agreement, dated as of December 7, 2009 and effective as of February 1, 2010, by and between Regus Management Group, LLC, as Landlord, and NovaDel Pharma Inc., as Tenant
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on January 14, 2010.
|
||
|
10.38
|
Amendment Agreement, dated December 31, 2009, by and among NovaDel Pharma Inc., ProQuest Investment II, L.P., ProQuest Investment Advisors Fund II, L.P. and ProQuest Investments III, L.P.
|
Incorporated by reference to Exhibit 10.1 of the Company’s Current Report on Form 8-K, as filed with the SEC on January 7, 2010.
|
||
|
21.1
|
Subsidiaries of the Registrant
|
The registrant has no subsidiaries.
|
||
|
23.1
|
Consent of J.H. Cohn LLP
|
Filed herewith.
|
||
|
31.1
|
Certification of Principal Executive Officer under Rule 13a-14(a)
|
Furnished herewith.
|
||
|
32.1
|
Certifications of the Principal Executive Officer and Principal Financial Officer under 18 USC 1350
|
Furnished herewith.
|
_________________________________
* Compensation Related Contract.
+ Confidential Treatment Requested. Confidential materials omitted and filed separately with the Securities and Exchange Commission