Attached files
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2009
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 000-51470

AtriCure, Inc.
(Exact name of registrant as specified in its charter)
| DELAWARE | 34-1940305 | |
| (State or other jurisdiction of incorporation or organization) |
(I.R.S. Employer Identification Number) | |
| 6217 Centre Park Drive, West Chester, OH | 45069 | |
| (Address of principal executive offices) | (Zip Code) | |
Registrant’s telephone number including area code: (513) 755-4100
Securities Registered Pursuant to Section 12(b) of the Act:
| Title of each class |
Name of each exchange on which registered | |
| Common Stock, $.001 Par Value Per Share | NASDAQ Global Market |
Securities Registered Pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ¨ No ¨.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer ¨ | Accelerated Filer ¨ | Non-Accelerated Filer ¨ (Do not check if smaller |
Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting Common Stock held by non-affiliates of the registrant, based upon the closing sale price of the Common Stock on June 30, 2009, as reported on the NASDAQ Global Market, was $34.8 million.
As of March 1, 2010 there were 15,451,788 shares of Common Stock, $.001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Items 10, 11, 12, 13 and 14 of Part III of this Form 10-K incorporate information by reference from the registrant’s definitive proxy statement to be filed with the Securities and Exchange Commission within 120 days after the close of the fiscal year covered by this Form 10-K.
Table of Contents
Table of Contents
This Form 10-K, including the sections titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Risk Factors,” contains forward-looking statements regarding our future performance. All forward-looking information is inherently uncertain and actual results may differ materially from assumptions, estimates or expectations reflected or contained in the forward-looking statements as a result of various factors, including those set forth under “Risk Factors” and elsewhere in this Form 10-K. Forward-looking statements convey our current expectations or forecasts of future events. All statements contained in this Form 10-K other than statements of historical fact are forward-looking statements. Forward-looking statements include statements regarding our future financial position, business strategy, budgets, projected costs, plans and objectives of management for future operations. The words “may,” “continue,” “estimate,” “intend,” “plan,” “will,” “believe,” “project,” “expect,” “anticipate” and similar expressions may identify forward-looking statements, but the absence of these words does not necessarily mean that a statement is not forward-looking. With respect to the forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995. These forward-looking statements speak only as of the date of this Form 10-K. Unless required by law, we undertake no obligation to publicly update or revise any forward-looking statements to reflect new information or future events or otherwise.
| ITEM 1. | BUSINESS |
Overview
We are a medical device company and a leader in developing, manufacturing and selling innovative cardiac surgical ablation systems designed to create precise lesions, or scars, in cardiac, or heart, tissue. We have three primary product lines. Our primary product line, which accounts for a majority of our revenues, is our Isolator® Synergy bipolar ablation clamp system, or Isolator system, and related radiofrequency ablation devices. Additionally, we offer a cryoablation product line, which features reusable and disposable cryoablation devices. During the fourth quarter of 2009 we initiated a European launch of our AtriClip™ Gillinov-Cosgrove Left Atrial Appendage System, or AtriClip system, which is designed to safely and effectively exclude the left atrial appendage.
Cardiothoracic surgeons have adopted our Isolator system to treat atrial fibrillation, or AF, in an estimated 70,000 patients since January 2003 and we believe that we are currently the market leader in the surgical treatment of AF. Our products are utilized by cardiothoracic surgeons in open-heart, or open, concomitant surgical procedures and also during sole-therapy minimally invasive cardiac ablation procedures. During an open procedure, the surgeon ablates cardiac tissue and/or treats the left atrial appendage, secondary, or concomitant, to a primary cardiac procedure such as a valve or coronary bypass. Medical journals have described the adoption by leading cardiothoracic surgeons of our Isolator system as a treatment alternative during open-heart surgical procedures to create lesions in cardiac tissue to block the abnormal electrical impulses that cause AF a rapid, irregular quivering of the upper chambers of the heart. Additionally, leading cardiothoracic surgeons and publications in medical journals have described our Isolator system as a standard treatment alternative for AF patients who may be candidates for sole-therapy minimally invasive surgical procedures. To date, none of our products has been approved or cleared by the Food and Drug Administration, or FDA, for the treatment of AF. We anticipate that substantially all of our revenues for the foreseeable future will relate to products we currently sell or are in the process of developing, which surgeons generally use to ablate cardiac tissue for the treatment of AF or for the exclusion of the left atrial appendage.
AF affects approximately 1% of the population in the United States. It is the most common cardiac arrhythmia, or irregular heartbeat, encountered in clinical practice and accounts for more doctor visits and hospital days than any other cardiac arrhythmia. AF is a condition wherein abnormal electrical impulses cause the atria, or upper chambers of the heart, to fibrillate, or quiver, at rapid rates of 400 to 600 beats per minute. As
1
Table of Contents
a result of this quivering, blood in the atria may become static, creating an increased risk that a blood clot will form and cause a stroke or other serious complications. If AF persists, patients often progress from experiencing AF intermittently to having AF continuously, a condition that is more difficult to treat. Symptoms of AF may include heart palpitations, dizziness, fatigue and shortness of breath, and these symptoms may be debilitating and life threatening in some cases. Although there is often no specific cause of AF, the condition is often associated with high blood pressure and other forms of heart disease. In most cases, AF is associated with cardiovascular disease, in particular hypertension, congestive heart failure, left ventricular dysfunction, coronary artery disease and valvular disease.
In the United States we primarily sell our products to medical centers through our direct sales force. AtriCure Europe, B.V., our wholly-owned subsidiary incorporated and based in the Netherlands, markets and sells our products throughout Europe, the Middle East and Africa, or EMEA, primarily through distributors, while in Germany, Switzerland and Austria we sell directly to medical centers. Additionally, we sell our products to other international distributors, primarily in Asia, South America and Canada. Our business is primarily transacted in U.S. dollars with the exception of transactions with our European subsidiary which are primarily transacted in Euros. Our sales outside of the United States represented 19% of our 2009 revenues and 15% of our 2008 revenues.
We were incorporated in the State of Delaware as AtriCure, Inc. on October 31, 2000 in connection with a spin-off transaction from Enable Medical Corporation, in which shares of our common stock were distributed to the Enable shareholders. The spin-off was intended to allow us to focus on the development of products designed to treat AF and to raise capital for that purpose, while Enable continued its broader research and manufacturing activities. On August 5, 2005, we completed an initial public offering of our common stock. On August 10, 2005 we acquired Enable Medical Corporation, the manufacturer of our Isolator clamps, which are an essential part of our Isolator system. Additionally, in December 2005, we formed AtriCure Europe, B.V.
Market Overview
AF is the most commonly diagnosed sustained cardiac arrhythmia, and affects more than 5 million people worldwide, including more than 2.5 million in the United States, where approximately 160,000 new cases of AF are diagnosed each year. According to data from the Framingham Heart Study, a study originally undertaken by the National Heart Institute (now known as the National Heart, Lung and Blood Institute), it is estimated that the incidence of AF doubles with each decade of an adult’s life. At age 40, remaining lifetime risk for AF is 26% for men and 23% for women. AF is an under-diagnosed condition due in large part to the fact that patients with AF often have mild or no symptoms and their AF is only diagnosed when they seek treatment for an associated condition, such as a stroke or heart disease. We believe that increasing awareness of AF and improved diagnostic screening will result in an increased number of patients diagnosed with AF. Also, since the prevalence of AF increases with age, there will likely be an increase in the number of diagnosed AF patients in the United States as the population ages.
According to the American Heart Association, people with AF are about five times more likely to have a stroke and AF is thought to be responsible for approximately 15% to 20% of the estimated 700,000 strokes that occur annually in the United States. Further, 35% of AF patients will have a stroke in their lifetime and AF-related strokes tend to be severe. Studies suggest that 25% of people who have an AF-related stroke die within the first thirty days following their stroke and over 40% are permanently bedridden. AF accounts for $6.7 billion in hospitalization-related costs in the United States each year and an estimated 5 million office visits annually. Additional costs include the cost of drugs and indirect costs, such as the management of AF-related strokes, the costs of which are believed to be significant.
AF is a condition that doctors often find difficult to treat and, historically, there has been no widely accepted long-term cure for AF. Doctors typically begin treating AF with drugs, which are often ineffective, not well-
2
Table of Contents
tolerated and may be associated with serious side effects. Patients who cannot effectively be treated with drugs may be candidates to undergo catheter-based procedures to treat their AF. To perform a catheter ablation, an electrophysiologist performs the ablation from the inside of the heart using a flexible catheter. The heart is reached via a blood vessel, most commonly through the femoral vein. Catheter-based procedures are often technically challenging, can be associated with serious complications, are generally not indicated for a certain population of AF patients and have been known to yield inconsistent results. Implantable devices, such as pacemakers and defibrillators, are sometimes used to reduce the frequency and symptoms of AF although they are not designed to treat the underlying disease. In the past, an open-heart surgical procedure known as the “cut and sew Maze” was used to treat AF, but this procedure has not been widely adopted because it is technically challenging, highly invasive and involves long recovery times.
Of the patients undergoing open-heart surgery in the United States, we estimate that approximately 80,000 of these patients are potential candidates for surgical ablation using our products. Of the United States population diagnosed with AF, approximately 12%, or 300,000, of these patients are symptomatic and do not respond to drug therapy or are intolerant to the drugs used to treat AF. For these patients, the cut and sew Maze procedure is typically too invasive and catheter ablation is often not indicated. Accordingly, we believe that there is a large population of under-treated patients who would potentially benefit from minimally invasive AF treatment using our Isolator system and related products, and that these patients comprise our largest growth opportunity.
It is estimated that 15% to 20% of all strokes are attributable to AF and that a majority of cardiac clots in patients with AF form in the left atrial appendage, which some physicians believe is associated with AF-related strokes. We believe that the surgical practice of excluding the left atrial appendage has become a growing trend in procedures performed to treat AF and current practice guidelines indicate that the left atrial appendage should be removed, when possible, during cardiac surgery in patients at risk of developing postoperative AF. We also believe that our AtriClip system is potentially safer, more effective and easier to use when permanently excluding the left atrial appendage, than current products and techniques. The AtriClip system received CE Mark approval on September 24, 2009 and we began an initial commercial launch in Europe during the fourth quarter of 2009. The AtriClip system has not been approved or cleared by the FDA and, pending approval or clearance by the FDA, we anticipate a commercial release of the AtriClip system in the United States during the second half of 2010. We believe the market for the AtriClip system is large and represents a significant new growth opportunity for us.
The AtriCure Solution and Products
We believe that traditional surgical and catheter-based ablation devices are not ideal for safely, rapidly and reliably creating the transmural lesions required to block the abnormal electrical impulses that cause AF, particularly for patients with more chronic forms of AF. Reports of preliminary clinical studies conducted by doctors at prominent medical centers suggest that our products, including our Isolator system, enable cardiac surgeons to simplify the cut and sew Maze procedure with a faster, less invasive and less technically challenging approach that appears to have comparable effectiveness. Over eighty medical centers in the United States are currently using our Isolator system as a sole-therapy minimally invasive treatment for AF and revenues from our minimally invasive products exceeded $16.5 million in 2009.
Our clinical studies for the use of our products to treat AF are ongoing. Leading cardiothoracic surgeons and electrophysiologists, including those who serve or who have served as consultants to us, have published results of initial clinical studies utilizing our Isolator system. These studies have been conducted at prominent medical centers, including the Cleveland Clinic (Cleveland, Ohio), Washington University (St. Louis, Missouri), the Cardiopulmonary Research Science and Technology Institute (Dallas, Texas), Medical College of Virginia (Richmond, Virginia), Oregon Heart and Vascular Institute (Eugene, Oregon), University of Zurich (Zurich, Switzerland), Maastricht University (Maastricht, the Netherlands), Anzhen Hospital (Beijing, China), Uppsala University Hospital (Uppsala, Sweden) and the Nebraska Heart Hospital (Lincoln, Nebraska). The results of these studies are promising in terms of efficacy, ease of use and safety.
3
Table of Contents
We have three primary product lines:
| • | Isolator System and Related Radio-Frequency Ablation Devices. Our Isolator system and related radio-frequency, or RF, devices, such as our multifunctional pen and Coolrail™ device, represent our primary product line and currently generate a substantial majority of our revenues. Our Isolator system and related RF devices are used in both open and minimally invasive procedures and primarily consist of the following products: |
| • | Isolator Synergy Bipolar Radio-Frequency Ablation Clamps. We sell multiple configurations of our Isolator Synergy clamps. One design is for ablation during open-heart procedures and one design is for ablation during minimally invasive procedures. Our Isolator Synergy clamps are single-use disposables and have jaws that close in a parallel fashion. The parallel closure compresses the tissues and evacuates the blood and fluids from the energy pathway in order to make the ablation more effective. During the fourth quarter of 2009, we introduced and commercially released in Europe a new open-heart configuration of our Isolator Synergy clamps, known as the Isolator Synergy Access clamp. The Isolator Synergy Access clamp features a pivoting clamp head which promotes easier access to challenging anatomy. The Isolator Synergy Access clamp has not yet been cleared for use in the United States. |
| • | Ablation and Sensing Unit, or ASU. Our ASU is a compact power generator that uses our proprietary software and delivers bipolar radio-frequency, or RF, energy. The ASU provides the RF energy necessary for our clamps, multifunctional pens and Coolrail linear ablation device. We generally lend our ASU, free of charge, to our direct customers and sell it to our distributors. |
| • | AtriCure Switch Box, or ASB. Our ASB is a compact switch box which provides the technology needed for the dual pulsing electrodes in our Isolator Synergy clamps as well as the ability to connect and toggle between our multiple RF devices. We generally lend our ASB, free of charge, to our direct customers and sell it to our distributors. |
| • | Isolator Multifunctional Pen. Our Isolator multifunctional pen is a disposable RF device that enables surgeons to evaluate cardiac arrhythmias, perform temporary pacing, stimulation, sensing and ablate cardiac tissue with the same device. When the multifunctional pen is used with our ASB, surgeons are able to toggle back and forth between temporary pacing, sensing, stimulation and ablation. Because of its broad range of capabilities, we believe surgeons are generally using this device in combination with our Isolator clamps during minimally invasive procedures and have also adopted it for use during open-heart procedures. |
| • | Coolrail Linear Ablation Device. During the second quarter of 2008 we released our Coolrail linear ablation device, which is a disposable linear RF ablation device designed to allow physicians to create an expanded cardiac ablation lesion set during minimally invasive procedures. We believe physicians are using our Coolrail device during minimally invasive procedures in order to improve long-term results for patients who have persistent and long-standing persistent AF. |
| • | Cryoablation System. Our cryoablation offering consists of a variety of reusable and disposable devices which use cryothermy, or extreme cold, to ablate cardiac tissue. In August 2007 we acquired the Frigitronics® CCS-200 product line for cardiac ablation, which included a console and a variety of reusable cardiac ablation probes. During the first half of 2009, we launched our Cryo1™ cryoablation device, a disposable device which is used with the CCS-200 console and is being adopted by physicians for AF ablation treatment during certain open-heart procedures, for which physicians prefer cryoablation over RF ablation. We believe our Cryo1 device provides us with a superior competitive product offering and also positions us as the only company to offer both RF and cryo surgical cardiac ablation devices. |
| • | AtriClip System. Our AtriClip system is designed to exclude the left atrial appendage by implanting the device during open or minimally invasive surgical procedures from the outside of the heart, avoiding contact with the circulating blood pool while eliminating blood flow between the left atrial appendage and the atria. We believe that our AtriClip system is potentially safer, more effective and easier to use |
4
Table of Contents
| when permanently excluding the left atrial appendage than current products and techniques. The AtriClip system received CE Mark approval on September 24, 2009 and we began an initial commercial launch in Europe during the fourth quarter of 2009. The AtriClip system has not been approved or cleared by the FDA and, pending approval or clearance by the the FDA, we anticipate a commercial release of the AtriClip system in the United States during the second half of 2010. |
In addition to the above product lines we also sell enabling technologies including our Lumitip™ dissector and MicroPace ORLab™ system. The Lumitip dissector is used by surgeons to gently separate tissues to provide access to key anatomical structures that are targeted for ablation. The Lumitip dissector consists of a shaft with an articulating index finger-shaped tip that illuminates, allowing surgeons to more easily determine the movement, direction and position of the device during procedures. The Lumitip dissector is cleared by the FDA for the dissection of soft tissues during general, thoracic and certain other surgical procedures and was designed in conjunction with Dr. Randall Wolf who is a leader in the field of minimally invasive cardiothoracic surgery. Our ORLab system is a stimulating, mapping and recording system which we believe, when used with a mapping probe, enables physicians to effectively confirm that the ablation lines being created are forming electrical barriers or lines of block.
Current AF Treatment Alternatives
Doctors usually begin treating AF patients with a variety of drugs intended to prevent blood clots, control heart rate or restore the heart to normal sinus rhythm. If a patient’s AF cannot be adequately controlled with drug therapy, doctors may perform one of several procedures that vary depending on the severity of the AF symptoms and whether or not the patient suffers from other forms of heart disease. During 2007 the Heart Rhythm Society published an expert consensus statement on catheter and surgical ablation for the treatment of AF. The expert consensus concluded that the current indications for the surgical treatment of AF are the following:
| • | Symptomatic AF patients undergoing other cardiac surgery; |
| • | Selected asymptomatic AF patients undergoing cardiac surgery in whom the ablation can be performed with minimal risk; or |
| • | Stand-alone (or sole-therapy) AF surgery should be considered for symptomatic AF patients who prefer a surgical approach, have failed one or more attempts at catheter ablation or are not candidates for catheter ablation. |
Other treatment alternatives include:
| • | Drugs. Currently available drugs are often ineffective, not well-tolerated and may be associated with severe side effects. For these reasons, drug therapy for AF fails for as many as 50% of patients within one year. Of those who initially respond to drug therapy, only approximately 25% of patients can continue to be managed with drugs after five years. |
| • | Implantable Devices. Implantable devices, such as defibrillators and pacemakers, can be effective in reducing the symptoms and frequency of AF episodes, but neither device is intended to treat AF. Patients may continue to experience the adverse effects of AF as well as some of the symptoms and complications, including dizziness, fatigue, palpitations and stroke, because the AF continues. |
| • | Catheter-Based Treatment. Catheter ablation is an ablation procedure that is typically performed by an electrophysiologist. The ablations are made from the inside of the heart using a flexible catheter. The heart is reached via a blood vessel, most commonly through the femoral vein. Catheter-based AF treatments are often technically challenging, can be associated with serious complications and have been known to yield inconsistent results. In proportion to the prevalence of AF, only a small number of catheter-based AF treatments are performed each year in the United States. |
| • | Cut and Sew Maze. The cut and sew Maze procedure is a highly invasive open-heart surgical procedure that involves the use of a heart-lung bypass machine and cutting and sewing back together sections of |
5
Table of Contents
| the heart in order to block the abnormal electrical impulses causing AF. Although this procedure is highly effective at treating AF, it is rarely performed because it requires extensive open-heart surgery, is technically challenging and is typically associated with long recovery times. For these reasons, only a limited number of these procedures have been performed by a small number of cardiothoracic surgeons. |
Surgeons have adopted our products for use in open-heart and minimally invasive procedures for the treatment of AF. During elective open-heart surgical procedures, such as bypass or valve surgery, cardiothoracic surgeons use our Isolator or cryoablation system to treat patients with a pre-existing history of AF. Surgeons report that ablation using our products generally adds approximately 20 minutes to an open-heart surgical procedure. Surgeons use our products to perform cardiac procedures that may vary depending on the length of time a patient has been diagnosed with AF and whether the patient’s AF is intermittent, known as paroxysmal, or more continuous, known as persistent, long-standing persistent or permanent AF. Patients who have been diagnosed with AF for a longer duration and have more continuous AF generally receive more extensive ablation procedures than patients who have been diagnosed with AF for a shorter duration or who have paroxysmal AF. Surgeons using our Isolator system or cryoablation system and related products during an open-heart surgical procedure typically perform the following steps:
Pulmonary Vein Isolation. Regardless of the duration or type of AF, surgeons will create lesions in the heart tissue surrounding the pulmonary veins to create an electrical barrier between the pulmonary veins and the atrium, or upper chambers of the heart. In patients with intermittent AF, those lesions are often the extent of the treatment performed and, in some cases, doctors may also use our multifunctional pen to sense, pace, stimulate or ablate cardiac tissue.
Additional Lesions. For those patients who have more continuous AF, doctors may determine that additional lesions are required to treat their AF. In cases where patients require such additional lesions, surgeons may use our Isolator clamps or Cryo1 probe during open-heart or concomitant surgical procedures to create lesions in the atrium that are intended to reproduce similar electrical barriers to those created by surgeons during the cut and sew Maze procedure. In some cases, doctors may also use our multifunctional pen to sense, pace, stimulate or ablate cardiac tissues. Additionally, our reusable cryoablation probes are sometimes used to ablate cardiac tissue near the heart valves.
For those patients with AF who do not require a concomitant open-heart surgical procedure, surgeons have used our Isolator system and other products for minimally invasive AF treatment procedures. These procedures have generally been performed through minimally invasive incisions without the need to place patients on a heart-lung bypass machine. Surgeons have reported that the procedure takes approximately two to three hours and that the average hospitalization period has been two to five days. Similar to the open-heart surgical procedure, patients who have more continuous AF generally require an expanded lesion set that mimics the cut and sew Maze procedure. Our Coolrail device and multifunctional pen are often used during these procedures to enable physicians to perform additional ablations.
Physicians have recently developed and are exploring an emerging minimally invasive stand-alone procedure which combines epicardial ablation (ablation on the outside of the heart) with endocardial ablation and mapping techniques (from the inside of the heart). Physicians are reporting that they are performing this emerging procedure, also known as a hybrid procedure, utilizing our Isolator system and related products in combination with catheter ablation and mapping techniques to primarily treat patients who have more continuous forms of AF. We are in the process of working with the FDA to finalize a clinical trial, DEEP AF, which explores the safety and effectiveness of this procedure when utilizing our Isolator system and related products in combination with a commercially available catheter for the treatment of persistent and long-standing persistent AF. We anticipate trial approval and initial patient enrollment during 2010.
6
Table of Contents
Product Development
Our product development team develops product enhancements and new products to address unmet procedural and market needs with the goal of increasing revenues and optimizing procedural outcomes. Our current product development activity includes projects extending and improving our existing products, the creation of new enabling devices and research into new technologies.
The Cleveland Clinic Foundation and Case Western Reserve University and collaborating businesses, including us, received publicly announced grants from the State of Ohio for, among other things, the creation of the Atrial Fibrillation Innovation Center. Pursuant to the terms of the agreement, effective June 2005, we supplied personnel and materials to accomplish certain research-related activities in connection with the grant and, over a three and one-half-year period ended during 2008, we received a total of $0.9 million for personnel and materials in matching dollars associated with our required $7.7 million spend for research and development-related operating expenses. Further, The Cleveland Clinic acquired $2.4 million in capital equipment for our use in support of our performance of the amended agreement. We earned the $2.4 million in matching capital equipment under the grant by spending $4.8 million in qualifying capital expenditures through the period ended December 31, 2009. Our expenditures represented ordinary course expenditures that we would have otherwise anticipated making.
Business Strategy
Our mission is to expand the treatment options for those patients who suffer from AF through the continued development of our technologies and expansion of our product offerings. The key elements of our strategy include:
New Product Innovation. We plan to continue to develop new and innovative products, including those that allow us to enter new market opportunities or expand our growth in existing markets. During 2009 we launched our Cryo1 probe which expanded our open-heart cardiac ablation product offering. Additionally, the development and commercialization of the AtriClip system provides a new growth platform and allows entrance into a new market. Our product development and growth plans include continued innovation to expand on both new and existing market opportunities.
Provide Product Education. We have recruited and trained sales professionals who have strong backgrounds in the medical device industry to effectively communicate to doctors the unique features and benefits of our technology as they relate to their cleared indications. Our highly trained sales professionals meet with doctors at leading institutions to provide education and technical training on the technical features and benefits of our products. In addition to our sales activities, we provide medical information on our products in response to information requests from physicians, and we have provided educational grants to institutions that have facilitated the education of doctors concerning the treatment of AF, including the use of our products as an AF treatment alternative. As a result of the educational process, we believe that awareness of our technology is growing and will result in the increased use of our products.
Expansion of International Markets and Entrance into New Markets. Our international business represented 19% of our total revenues for 2009. Most of the international markets in which we currently do business are underpenetrated markets which present high growth opportunities for our products. Further, we plan to expand into new geographic markets such as Saudi Arabia, Russia and Australia.
Form Investigational Relationships with Key Opinion Leaders at Leading Institutions. We have formed investigational relationships with key opinion leaders at several leading medical centers including the Cleveland Clinic, Washington University, Medical College of Virginia, the Cardiopulmonary Research Science and Technology Institute, Oregon Heart and Vascular Institute, Nebraska Heart Hospital, the University of Oklahoma, the University of Zurich, the University Hospital Maastricht and Indiana Heart Hospital. These key opinion leaders and others have worked with us as consultants to evaluate and develop our products.
7
Table of Contents
Additionally, several key opinion leaders at these institutions have published peer-reviewed data that describes the use of our products as a treatment alternative for AF. These opinion leaders have assisted and continue to assist us with the design and/or evaluation of our products. To date, there have been over 40 peer-reviewed publications that describe our Isolator systems’ ability to create transmural lesions or the use of our Isolator system as an AF treatment alternative. Key publications and presentations have highlighted promising results utilizing our products to treat patients with AF during sole-therapy minimally invasive surgical procedures. Further, initial presentations and publications have described our AtriClip system as a safe and effective means of excluding the left atrial appendage. We believe that these publications and presentations have contributed to and, we expect, will contribute to the expanded adoption of our products.
Expand Adoption of Our Minimally Invasive Products. We believe that the catalysts for expanded adoption of our minimally invasive products include the publication of peer-reviewed articles, which we believe will help validate the successful, long-term use of our products for patients with AF, new product introductions, and procedural advancements such as the hybrid procedure. Our consultants, or individuals who have acted as consultants to us, have received grant monies to support certain research activities and have presented and published their results of an initial series of studies relating to the use of our minimally invasive products. As results of these peer-reviewed studies are accepted, we believe that this will increase the demand for our minimally invasive products. We believe our consultants are continuing their efforts to investigate, present and publish results from the use of our products to perform minimally invasive procedures and that the results from these research activities will continue to demonstrate that our products can be used to offer certain AF patients an improved treatment alternative. We also believe new procedural techniques, such as the hybrid procedure, will increase the demand for our minimally invasive products. We believe that these ongoing research activities, new procedural techniques, and anticipated presentations and publications will create an increased demand for our minimally invasive products.
Clinical Trials
During 2007 we worked with the FDA and leading cardiothoracic surgeons to design our pivotal clinical trial, ABLATE, which was approved by the FDA for patients with permanent AF (as defined in the trial’s protocol) undergoing concomitant cardiac surgical procedures. The primary efficiency endpoints of the trial are an estimated minimum of 70% of patients treated being free of AF and off of antiarrhythmic drugs at their six-month follow-up. A 24-hour holter monitor was used to determine the rhythm status six months following surgery. We believe that the ABLATE clinical trial has completed enrollment and preliminary follow-up at 55 patients during 2009. We believe that the preliminary results from the trial met or exceeded the defined endpoints and we are in the process of summarizing the clinical data from the trial which we plan to submit to the FDA during the first half of 2010 in support of a PMA approval. This approval would allow us to market our Isolator system for the treatment of patients with permanent AF during open- heart procedures. During February 2010, we submitted to the FDA a continued access protocol for the ABLATE trial, which would provide for enrollment up to an additional 50 patients. We would anticipate that the continued access trial data will be used as additional supporting clinical evidence during the PMA process and at the anticipated panel review/clinical advisory meeting.
In 2007 we initiated a clinical trial in Zurich, Switzerland for our AtrClip system. During 2009 enrollment was completed and 40 patients participated in the trial. The clinical data for this trial was utilized to support the CE Mark approval in Europe which was obtained in the third quarter of 2009.
In 2008 we received FDA approval for our EXCLUDE clinical trial for our AtriClip system, which was used in support of our 510(k) filing. The primary endpoint for the trial is safe and effective exclusion of the left atrial appendage, which was evaluated at three months for all patients and at six months for 30 patients. The clinical trial has been completed with 70 patients treated. 61 patients were followed through the primary endpoint at three months post procedure and 45 patients were followed through six months post procedure. The clinical data was submitted to the FDA in December 2009 and is currently under review.
8
Table of Contents
During the first quarter of 2010 we submitted a protocol to the FDA for a feasibility trial, DEEP AF, which would evaluate the safety and efficacy of our minimally invasive products with catheter ablation and mapping technologies for persistent and long-standing persistent patients. The protocol is currently being reviewed by the FDA.
During 2009 we closed a FDA approved trial which was a second arm of the ABLATE trial designed for patients with persistent AF. We also have a trial, RESTORE SRIIB, which is a feasibility trial designed to demonstrate the safety and efficacy of our Isolator Synergy system during minimally invasive sole-therapy procedures for the treatment of patients with persistent or permanent AF. As of the date of this Form 10-K, we were not actively enrolling patients in RESTORE SRIIB.
Sales, Marketing and Medical Education
Our United States sales and marketing efforts focus on educating doctors concerning our unique technologies and their technical benefits. It is our policy not to market or promote our products for the treatment of AF. Our sales personnel visit cardiac surgeons, electrophysiologists and other doctors to discuss the general attributes of our products and promote them for their FDA cleared indications. We train our sales force on the use of our products to treat AF so that they are able to respond to unsolicited requests from doctors for information on the use of our products for the treatment of AF. In addition, medically trained clinical application specialists attend surgical procedures to discuss the use of our products and to respond in a non-promotional manner to unsolicited requests for information on the use of our Isolator system and other products for the treatment of AF.
We have entered into consulting agreements with leading scientists, cardiothoracic surgeons and electrophysiologists who assist us with the design, clinical testing and evaluation of our products, education of doctors on the use of our technologies and provide advice concerning regulatory submissions. We work closely with these thought leaders to understand unmet needs and emerging applications related to the treatment of AF. We also provide educational grants to several leading medical centers. These institutions have used these grants to sponsor activities evaluating the effectiveness of our Isolator system and our other products and technologies which have increased the number of peer-reviewed publications that cite the use of our Isolator system. These grants have also been used by these institutions to sponsor independent educational programs relating to AF, including programs which focus on the surgical treatment of AF using our products. We provide some guidance to physicians and medical institutions regarding those physicians who are available and qualified for training other physicians in the use of our Isolator system in the treatment of AF.
We have formed a healthcare compliance committee in support of our ongoing compliance efforts with applicable federal and state healthcare laws and regulations. This committee has instituted standard operating procedures relating to our marketing and promotional activities, grant review and funding procedures and the training and education of our sales force. Our training and educational programs include training on federal and state requirements for marketing medical devices and we maintain continuous oversight of our grant application and funding procedures and requirements. During 2010, we entered into a five-year Corporate Integrity Agreement with the Office of Inspector General of the Department of Health and Human Services. The Agreement provides for increased training, monitoring and compliance activities with respect to our healthcare compliance activities.
Our sales team in the United States is led by a Vice President of Sales and has 55 employees supporting approximately 30 sales territories. We select our sales personnel based on their expertise, sales experience and reputation in the medical device industry and their knowledge of our products and technologies. We believe at this time that our sales organization is appropriately sized and do not anticipate significant changes in the foreseeable future.
We market and sell our products in selected markets outside of the United States through independent distributors and, in EMEA markets, through our European subsidiary which includes a combination of
9
Table of Contents
independent distributors and direct sales personnel. During 2009 and 2008, sales outside of the United States accounted for 19% and 15% of our total revenues, respectively. We have a network of distributors outside of the United States who currently market and sell our products and are located primarily in Europe, Asia, South America and Canada. Our international sales team is led by a Vice President of Sales and has a direct sales representative who sells to customers in Germany, Switzerland and Austria. We continue to expand our presence in markets outside of the United States.
Competition
Our industry is highly competitive, subject to change and significantly affected by new product introductions and other activities of industry participants. Many of our competitors have significantly greater financial and human resources than we do and have established reputations with our target customers, as well as worldwide distribution channels that are more established and developed than ours. Our primary competitors include Medtronic, Inc., St. Jude Medical, Inc., ATS Medical, Inc., nContact Surgical, Inc. and Endoscopic Technologies, Inc. We and our competitors provide products that have been adopted by doctors for the off-label treatment of AF. As of December 31, 2009, no company had received FDA approval or clearance to market a surgical ablation system for the treatment of AF. Some of our competitors offer catheter-based treatments, including but not limited to Biosense Webster, Inc. (a subsidiary of Johnson & Johnson), St. Jude Medical, Inc., and Medtronic, Inc. These companies sell products that are used by doctors to treat the population of patients that have AF but are not candidates for open-heart surgery. However, catheter-based treatments often do not effectively treat patients with more continuous forms of AF, which we believe is a segment of the AF patient population that would benefit from minimally invasive AF procedures.
We believe that we compete favorably against companies that have products used for the surgical treatment of AF during both open-heart and sole-therapy minimally invasive procedures, although we cannot assume that we will be able to continue to do so in the future or that new devices that perform better than our Isolator system and related products will not be introduced. We also believe that our Isolator system competes favorably when compared to catheter-based treatments. Further, we believe our AtriClip system provides an improved treatment alternative for the exclusion of the left atrial appendage.
Due to the size of the AF market and the unmet need for an AF cure, competitors have dedicated and will continue to dedicate significant resources to aggressively develop and market their products. New product developments that could compete with us more effectively are likely because the surgical AF treatment market is characterized by extensive research efforts and technological progress. Further, recent publications and industry events are expanding knowledge of the market and treatment alternatives and have identified the surgical treatment of AF as a treatment alternative for AF patients.
Existing or new competitors may develop technologies and products that are safer, more effective, easier to use or less expensive than our products. To compete effectively, we have to demonstrate that our products are an attractive alternative to other treatments by differentiating our products on the basis of safety, efficacy, performance, ease of use, brand and name recognition, reputation, service and price. We have encountered and expect to continue to encounter potential customers who, due to existing relationships with our competitors, are committed to or prefer the products offered by competitors. Competitive pressures may result in price reductions and reduced gross profit margins for our products over time. Technological advances developed by one or more of our competitors may render our products obsolete or uneconomical.
Third-Party Reimbursement
Payment for patient care in the United States is generally made by third-party payors. These payors include private insurers and government insurance programs, such as Medicare and Medicaid. The Medicare program, the largest single payor in the United States, is a federal health benefit program administered by the Centers for Medicare and Medicaid Services (CMS), and covers certain medical care items and services for eligible
10
Table of Contents
beneficiaries, such as individuals over 65 years old, as well as chronically disabled individuals. Reimbursement under Part A of the Medicare program includes hospitals and other institutional services, while Medicare Part B covers physician services. Because Medicare beneficiaries comprise a large percentage of the populations for which our products are used, and private insurers may follow the coverage and payment policies for Medicare, Medicare’s coding, coverage and payment policies for cardiothoracic surgical procedures are significant to our business.
Medicare’s Part A program pays hospitals for inpatient services under the Inpatient Prospective Payment System, which provides a predetermined payment based on the patient’s discharge diagnosis. Discharge diagnoses are grouped into Medicare Severity Diagnosis Related Groupings (MS-DRGs). There are several cardiac surgery MS-DRGs associated with the surgical treatment of AF, with and without a concomitant open-heart procedure. When an ablation device is used during a concomitant open-heart procedure, reimbursement is based upon the primary surgical procedure. Reimbursement for sole-therapy minimally invasive AF treatment is also influenced by the patient’s severity of illness before the appropriate MS-DRG is assigned. Currently, we believe hospital reimbursement rates for sole therapy and concomitant therapy cardiac tissue ablation are adequate to cover the cost of our Isolator system and other products. Medicare’s coding, coverage, and payment polices are subject to change. As a result, the continuance of current coverage, coding or payment determinations cannot be guaranteed, and any change may have an adverse impact on our business. Currently, there is a proposal by CMS to modify the MS-DRGs associated with hospital reimbursement for sole-therapy minimally invasive AF treatment that would be effective October 1, 2010. If the proposal were to become effective, it could result in a reduction of current reimbursement levels.
Doctors are reimbursed for their services separately under the Medicare Part B physician fee schedule. When surgically performing a cardiac ablation with and without a concomitant open-heart procedure, surgeons report Current Procedural Terminology, or CPT, codes to receive a professional fee. Surgeons have a choice of CPT codes to report sole-therapy and concomitant therapy cardiac tissue ablation.
In addition to the Medicare program, many private payors look to CMS policies as a guideline in setting their coverage policies and payment amounts. The current coverage policies of these private payors may differ from the Medicare program, and payment rates may be higher, lower, or the same as the Medicare program. If CMS or other agencies decrease or limit reimbursement payments to doctors and hospitals, this may negatively impact our business. Additionally, some private payors do not follow the Medicare guidelines and those payors may reimburse only a portion of the cost of cardiac ablation, or not at all.
The FDA generally does not regulate the practice of medicine. Doctors may use our Isolator system and other products in circumstances where they deem it medically appropriate, such as for the treatment of AF, even though the FDA has not approved or cleared our products for that indication. In these circumstances, some government or private payors, including some Medicare carriers, may make coverage and payment determinations on a case-by-case basis. Additionally, some government or private payors may deem the treatment of AF using our products for indications not approved or cleared by the FDA to be experimental or not medically necessary and, as such, may not provide coverage or payment.
Government Regulation
Our products are medical devices and are subject to regulation by the FDA, as well as other federal and state regulatory bodies in the United States and comparable authorities in other countries. We currently market our Isolator system and Coolrail device in the United States under a 510(k) clearance for the ablation of cardiac tissue. Our multifunctional pen is marketed in the United States under a 510(k) clearance for temporary pacing, sensing, stimulating and recording during the evaluation of cardiac arrhythmias and for the ablation of cardiac tissue. We currently market the Lumitip dissector in the United States under a 510(k) clearance for use in the dissection of soft tissues during general, ear, nose and throat, thoracic, urological and gynecological surgical
11
Table of Contents
procedures. We have filed a 510(k) with the FDA seeking clearance to market our AtriClip system for exclusion of the left atrial appendage. Our products may not be marketed for the treatment of AF without obtaining additional approvals from the FDA.
The FDA requires that premarket approval, or PMA, be obtained for a device before it can be marketed for the treatment of AF. During 2007 we worked with the FDA and leading cardiothoracic surgeons to design our clinical trial, ABLATE, which was approved by the FDA for patients with permanent AF undergoing concomitant open-heart surgical ablation procedures. We anticipate filing the final module of our PMA during the first half of 2010 which, if approved by the FDA, would allow us to market our Isolator system for the treatment of patients with permanent AF (pursuant to the study protocol) during open-heart procedures. Since the filing of our ABLATE trial, guidelines with respect to the classification of AF patients has changed. As a result, the approval we may obtain for the type of AF is likely not going to be permanent AF and is unknown at this time. We cannot be certain that we will successfully complete ABLATE, receive approval for any additional clinical trials or submit and obtain approval for any of our products for use in treating AF. Submission of a PMA is a much more demanding process than the 510(k) notification process. Further, both 510(k)s and PMAs must now be submitted with a potentially substantial user fee payment to the FDA, although certain exemptions and waivers of the user fee can apply, including certain exemptions and waivers for small businesses.
FDA regulations govern nearly all of the activities that we perform, or that are performed on our behalf, to ensure that medical products distributed domestically or exported internationally are safe and effective for their intended uses. The activities that the FDA regulates include the following:
| • | product design, development and manufacture; |
| • | product safety, testing, labeling and storage; |
| • | pre-clinical testing in animals and in the laboratory; |
| • | clinical investigations in humans; |
| • | premarketing clearance or approval; |
| • | record keeping and document retention procedures; |
| • | advertising and promotion; |
| • | the import and export of products; |
| • | product marketing, sales and distribution; |
| • | post-marketing surveillance and medical device reporting, including reporting of deaths, serious injuries, device malfunctions or other adverse events; and |
| • | corrective actions, removals and recalls. |
FDA’s Premarket Clearance and Approval Requirements. Unless an exemption applies, each medical device distributed commercially in the United States will require either prior 510(k) clearance or approval of a PMA from the FDA. Medical devices are classified into one of three classes—Class I, Class II, or Class III—depending on the degree of risk and the level of control necessary to assure the safety and effectiveness of each medical device. Devices deemed to pose lower risks are placed in either Class I or II, which requires the manufacturer to submit to the FDA a 510(k) notification requesting clearance to commercially distribute the device. Some low risk devices are exempted from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, or predicate device, are generally placed in Class III, requiring submission of a PMA supported by clinical trial data.
510(k) Clearance Pathway. When 510(k) clearance is required, we must submit a notification to the FDA demonstrating that our proposed device is substantially equivalent to a predicate device, previously cleared and
12
Table of Contents
legally marketed 510(k) device or a device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a PMA. The FDA is required to respond to a 510(k) notification within 90 days of submission, but the response may be a request for additional information or data, including clinical data. As a practical matter, 510(k) clearance often takes significantly longer than 90 days and may take up to a year or more. If the FDA determines that the device, or its intended use, is not substantially equivalent to a previously cleared device or use, the device is automatically placed into Class III, requiring the submission of a PMA. Any modification to a 510(k)-cleared device that would constitute a major change in its intended use, design or manufacture, requires a new 510(k) clearance or, possibly, in connection with safety and effectiveness, approval of a PMA. The FDA requires every manufacturer to make the determination regarding a new 510(k) submission in the first instance, but the FDA may review any manufacturer’s decision. We have made modifications to elements of our products which we believe did not require us to seek additional 510(k) clearance. We have recently been in communication with the FDA regarding our decision not to file a new 510(k) related to a change in indication for our Isolator Synergy clamps. At the time our Isolator clamps received 510(k) clearance for the ablation of cardiac tissue, through our internal and external regulatory review process, we determined that a new 510(k) was not needed for our Isolator Synergy clamps to change their intended use from the ablation of soft tissue to the ablation of cardiac tissue. The FDA has reviewed this decision and has indicated that a 510(k) was required to be filed for us to market our Isolator Synergy clamps for cardiac tissue ablation instead of soft tissue ablation. We are working with the FDA to provide them additional information in support of our decision and to evaluate potential corrective actions.
Premarket Approval Pathway. A PMA must be submitted to the FDA if the device cannot be cleared through the 510(k) process and is not otherwise exempt. A PMA must be supported by extensive data, including but not limited to technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction, the safety and effectiveness of the device.
After a PMA is submitted and the FDA has determined that the application is sufficiently complete to permit a substantive review, the FDA will accept the application for filing. The FDA has 180 days to review an “accepted” PMA, although the review of an application generally occurs over a significantly longer period of time and can take up to several years. During this review period, the FDA may request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a preapproval inspection of the manufacturing facility to ensure compliance with quality system regulations. Any approvals we receive may be limited in scope or may be contingent upon further post-approval study commitments or other conditions. New PMAs or PMA supplements are required for significant modification to the device, including indicated use, manufacturing process, labeling and design of a device that is approved through the premarket approval process. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require as extensive clinical data or the convening of an advisory panel.
Clinical Trials. Clinical trials are required to support a PMA and are sometimes required for 510(k) clearance. In the United States, clinical trials for a significant risk device require the prior submission of an application for an Investigational Device Exemption, or IDE, to the FDA for approval. An IDE amendment must be submitted before initiating a new clinical study. Some trials require a feasibility study followed by a pivotal trial. An IDE supplement is utilized as a means of obtaining approval to initiate a pivotal trial following the conclusion of a feasibility trial. IDE applications must be supported by appropriate data, such as animal and laboratory testing results, and any available data on human clinical experience, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The animal and laboratory testing must meet the FDA’s good laboratory practice requirements.
The IDE and any IDE supplement for a new trial must be approved in advance by the FDA. Clinical trials for significant risk devices may not begin until the IDE application is approved by the FDA and each center’s Institutional Review Board (IRB) overseeing the welfare of the research subjects and responsible for that
13
Table of Contents
particular clinical trial. If the product is considered a non-significant risk device under FDA regulations, only the center’s IRB approval is required. Under its regulations, the agency responds to an IDE application (amendment or supplement) for a new trial within 30 days. The FDA may approve the IDE unconditionally, grant an approval with certain conditions, or identify deficiencies that must be addressed prior to the approval of the study. It is common for the FDA to require additional information before approving an IDE, and thus final FDA approval on a submission commonly extends beyond the initial 30 days. The FDA may also require that a small-scale feasibility study be conducted before a pivotal trial may commence. In a feasibility trial, the FDA limits the number of patients and centers that may participate. Feasibility trials are typically structured to obtain information on safety and to evaluate the clinical efficacy to determine the number of subjects required to demonstrate statistical significance in a pivotal trial.
Clinical trials are subject to extensive recordkeeping and reporting requirements. Our clinical trials must be conducted under the oversight of an IRB for the relevant clinical trial sites and must comply with FDA regulations, including but not limited to those relating to good clinical practices. We are also required to obtain the patients’ written informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the IRB may suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the United States. Similarly, in Europe, the clinical study must be approved by a local ethics committee and, in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
Educational Grants. The FDA permits a device manufacturer to provide financial support, including support by way of grants, to third-parties for the purpose of conducting medical educational activities. If these funded activities are considered by the FDA to be independent of the manufacturer, then the activities fall outside the restrictions on promotion to which the manufacturer is subject.
The FDA considers several factors in determining whether an educational event or activity is independent from the substantive influence of the device manufacturer and therefore nonpromotional, including the following:
| • | whether the intent of the funded activity is to present clearly defined educational content, free from commercial influence or bias; |
| • | whether the third-party grant recipient and not the manufacturer has maintained control over selecting the faculty, speakers, audience, activity content and materials; |
| • | whether the program focuses on a single product of the manufacturer without a discussion of other relevant existing competitive products or treatment options; |
| • | whether there was meaningful disclosure to the audience, at the time of the program, regarding the manufacturer’s funding of the program, any significant relationships between the provider, presenters, or speakers and the supporting manufacturer and whether any unapproved uses will be discussed; and |
| • | whether there are legal, business, or other relationships between the supporting manufacturer and the provider or its employees that could permit the supporting manufacturer to exert influence over the content of the program. |
We seek to ensure that the activities we support pursuant to our educational grants program are in accordance with these criteria for independent educational activities. However, we cannot provide an assurance that the FDA or other government authorities would view the programs we have supported as being independent.
Pervasive and Continuing Regulation. There are numerous regulatory requirements that apply after a product is cleared or approved. These include:
| • | the FDA’s Quality System Regulation, or QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
14
Table of Contents
| • | labeling regulations and FDA prohibitions against the false or misleading promotion or the promotion of products for uncleared, unapproved or off-label use or indication; |
| • | requirements to obtain clearance or approval of product modifications that could significantly affect safety or efficacy or that would constitute a major change in intended use; |
| • | medical device reporting, or MDR, regulations which require that manufacturers comply with reporting requirements of the FDA and report if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
| • | post-approval restrictions or conditions, including post-approval study commitments; |
| • | post-market surveillance regulations which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; and |
| • | requirements to issue notices of correction or removal, or conduct market withdrawals or recalls where quality or other issues arise. |
From January 1, 2009 through February 28, 2010 we submitted to the FDA ten MDRs related to complications during procedures utilizing our products. Of these MDRs, six related to our Isolator clamps, two related to our multifunctional pen, one related to our Coolrail device and one related to our Lumitip dissector. Included in the above MDR filings were two patient deaths, which we included in our MDR filings; however, neither were categorized as outcomes related to the failure of our devices. Additionally, there may have been other incidents, including patient deaths, which have occurred using our products, although we are not aware of any such incidents during the period noted above.
The advertising and promotion of medical devices are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Recently, some promotional activities for FDA-regulated products have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the Federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims.
We have registered with the FDA as a medical device manufacturer. The FDA has broad post-market and regulatory enforcement powers. We are subject to unannounced inspections by the FDA to determine our compliance with the QSR and other regulations, and these inspections may include the manufacturing facilities of our suppliers.
Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other federal or state authorities, which may include any of the following sanctions:
| • | warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | customer notifications, repair, replacement, refunds, recall or seizure of our products; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | refusing our requests for 510(k) clearance or premarket approval of new products, new intended uses or modifications to existing products; |
| • | withdrawing 510(k) clearance or premarket approvals that have already been granted; and |
| • | criminal prosecution. |
Fraud, Abuse and False Claims. We are directly and indirectly subject to various federal and state laws governing our relationship with healthcare providers and pertaining to healthcare fraud and abuse, including anti-kickback laws. In particular, the federal healthcare program Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in
15
Table of Contents
exchange for or to induce either the referral of an individual, or the furnishing, arranging for or recommending a good or service for which payment may be made in whole or part under federal healthcare programs, such as the Medicare and Medicaid programs. Penalties for violations include criminal penalties and civil sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal healthcare programs. The Anti-Kickback Statute is broad and prohibits many arrangements and practices that are lawful in businesses outside of the healthcare industry. In implementing the statute, the Office of Inspector General of the U.S. Department of Health and Human Services, or OIG, has issued a series of regulations, known as the “safe harbors.” These safe harbors set forth provisions that, if all their applicable requirements are met, will assure healthcare providers and other parties that they will not be prosecuted under the Anti-Kickback Statute. The failure of a transaction or arrangement to fit precisely within one or more safe harbors does not necessarily mean that it is illegal or that prosecution will be pursued. However, conduct and business arrangements that do not fully satisfy each applicable element of a safe harbor may result in increased scrutiny by government enforcement authorities, such as the OIG.
The Federal False Claims Act, or FCA, imposes civil liability on any person or entity that submits, or causes the submission of, a false or fraudulent claim to the United States Government. Damages under the FCA can be significant and consist of the imposition of fines and penalties. The FCA also allows a private individual or entity with knowledge of past or present fraud against the federal government to sue on behalf of the government to recover the civil penalties and treble damages. The U.S. Department of Justice, or DOJ, on behalf of the government, has previously alleged that the marketing and promotional practices of pharmaceutical and medical device manufacturers included the off-label promotion of products or the payment of prohibited kickbacks to doctors violated the FCA resulting in the submission of improper claims to federal and state healthcare entitlement programs such as Medicaid. In certain cases, manufacturers have entered into criminal and civil settlements with the federal government under which they entered into plea agreements, paid substantial monetary amounts and entered into corporate integrity agreements that require, among other things, substantial reporting and remedial actions going forward.
As previously reported, in late 2008 we received a letter from the DOJ informing us that they were conducting an investigation for potential FCA and common law violations relating to our surgical ablation devices for the period beginning January 1, 2005. Other manufacturers of medical devices adopted for the treatment of AF reported receiving similar letters. Specifically, the letter stated that the DOJ was investigating our marketing practices utilized in connection with our surgical ablation system to treat AF, a specific use outside the FDA’s 510(k) clearance, and was also investigating whether we instructed hospitals to bill Medicare for cardiac surgical ablation using incorrect billing codes. On February 2, 2010, we entered into a settlement agreement with the DOJ, the Office of Inspector General of the Department of Health and Human Services, or OIG, and Elaine Bennett (also know as Elaine George), the relator in the related qui tam complaint (the “Relator”), which definitively resolved all claims related to the DOJ investigation and qui tam complaint, which has been dismissed. We did not and will not admit wrongdoing in connection with the settlement. Additionally, we entered into a five-year corporate integrity agreement with the OIG. For a discussion of the terms of the settlement, see “Item 3. Legal Proceedings.”
AdvaMed is one of the primary voluntary United States trade associations for medical device manufacturers. This association has established guidelines and protocols for medical device manufacturers in their relationships with healthcare professionals on matters including research and development, product training and education, grants and charitable contributions, support of third-party educational conferences, and consulting arrangements. Adoption of the AdvaMed Code by a medical device manufacturer is voluntary, and while the OIG and other federal and state healthcare regulatory agencies encourage its adoption and may look to the AdvaMed Code, they do not view adoption of the AdvaMed Code as proof of compliance with applicable laws. We have adopted the AdvaMed Code and incorporated its principles in our standard operating procedures, sales force training programs, and relationships with doctors. Key to the underlying principles of the AdvaMed Code is the need to focus the relationships between manufacturers and healthcare professionals on matters of training, education and scientific research, and limit payments between manufacturers and healthcare professionals to fair
16
Table of Contents
market value for legitimate services provided and payment of modest meal, travel and other expenses for a healthcare professional under limited circumstances. We have incorporated these principles into our relationships with healthcare professionals under our consulting agreements, payment of travel and lodging expenses, grant making procedures and sponsorship of third-party conferences. In addition, we have conducted training sessions on these principles. However, we cannot provide any assurance that regulatory or enforcement authorities will view these arrangements as being in compliance with applicable laws.
Regulation Outside of the United States. Sales of medical devices outside of the United States are subject to foreign governmental regulations which vary substantially from country to country. The time required to obtain certification or approval by a foreign country may be longer or shorter than that required for FDA clearance or approval and the requirements may be different.
The primary regulatory body in Europe is that of the European Union, which has adopted numerous directives and promulgated voluntary standards regulating the design, manufacture and labeling of and clinical trials and adverse event reporting for medical devices. Devices that comply with the requirements of a relevant directive will be entitled to bear CE conformity marking, indicating that the device conforms to the essential requirements of the applicable directives and, accordingly, can be commercially distributed throughout the member states of the European Union and other countries that comply with or mirror these directives. The method for assessing conformity varies depending on the type and class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a notified body, an independent and neutral institution appointed by a country to conduct the conformity assessment. This third-party assessment may consist of an audit of the manufacturer’s quality system and specific testing of the manufacturer’s device. Such an assessment is required for a manufacturer to commercially distribute the product throughout these countries. We are compliant with the International Organization for Standardization, (ISO) 13485:2003 Quality Management System. Compliance establishes the presumption of conformity with the essential requirements for a CE Marking. We have the authorization to affix the CE Mark to our Isolator clamps and to commercialize our Isolator clamps in the European Union for the treatment of cardiac arrhythmias, including atrial fibrillation.
Intellectual Property
Protection of our intellectual property is a strategic priority for our business and we rely on a combination of patent, copyright, trademark and trade secret laws to protect our interests. Our ability to protect and use our intellectual property rights in the continued development and commercialization of our technologies and products, operate without infringing the proprietary rights of others, and prevent others from infringing our proprietary rights is crucial to our continued success. We will be able to protect our products and technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, trademarks or copyrights or are effectively maintained as trade secrets, know-how or other proprietary information.
We seek patent protection relating to our Isolator system and other important technologies we develop in both the United States and in selected foreign countries. While we own much of our intellectual property, including patents, patent applications, trademarks, trade secrets, know-how and proprietary information, we also license patents and related technology of importance to the commercialization of our products. For example, to continue developing and commercializing our current and future products, we may license intellectual property from commercial or academic entities to obtain the rights to technology that is required for our research, development and commercialization activities.
All of our employees and technical consultants are required to execute confidentiality agreements in connection with their employment and consulting relationships with us. We also generally require them to agree to disclose and assign to us all inventions conceived in connection with their relationship with us. We cannot provide any assurance that employees and consultants will abide by the confidentiality or assignment terms of
17
Table of Contents
these agreements. Despite measures taken to protect our intellectual property, unauthorized parties might copy aspects of our products or obtain and use information that we regard as proprietary. We devote significant resources to obtaining patents and other intellectual property and protecting our other proprietary information. We have already obtained patents or filed patent applications on a number of our technologies, including patents and patent applications relating to our Isolator system. If valid and enforceable, these patents may give us a means of blocking competitors from using infringing technology to compete directly with our products. We also have certain proprietary trade secrets that may not be patentable or for which we have chosen to maintain secrecy rather than file for patent protection. With respect to proprietary know-how that is not patentable, we have chosen to rely on trade secret protection and confidentiality agreements to protect our interests.
As of December 31, 2009, we had the following portfolio of patents or patent applications covering our proprietary technologies and products:
| • | 36 issued or approved United States patents expiring between 2015 and 2027; |
| • | 28 United States non-provisional patent applications; |
| • | 4 United States provisional patent applications; |
| • | 5 issued foreign patents; and |
| • | 14 pending foreign patent applications that are in various national stages of prosecution. |
Additionally, as of December 31, 2009, we had eight registered and three pending trademark registrations covering our product branding.
Manufacturing
We manufacture a substantial majority of the disposable products we sell and generally purchase items that would be deemed capital equipment, including our ASU, ASB and ORLab. We inspect, assemble, test and package our products in West Chester, Ohio and our products are sterilized by third-party outside sterilizers at their facilities. Purchased components are generally available from more than one supplier. However some products, such as our ASU and ASB, are critical components of our Isolator system, and there are relatively few alternative sources of supply available. We generally carry a six month supply of these products, however, obtaining a replacement supplier for the ASU and ASB, if required, may not be accomplished quickly or at all and could involve significant additional costs. Generally, our suppliers have no contractual obligations to supply us with, and we are not contractually obligated to purchase from them, any of our supplies. During 2007, we entered into a development, manufacturing and supply agreement with MicroPace Pty Ltd of Australia to develop, manufacture and supply our ORLab system. Under the terms of the agreement, we are obligated to certain minimum purchase commitments through 2010 in order to retain exclusive distribution rights.
Order quantities and lead times for components purchased from outside suppliers are based on our forecasts derived from historical demand and anticipated future demand. Lead times may vary significantly depending on the size of the order, time required to fabricate and test the components, specific supplier requirements and current market demand for the components and subassemblies. To date, we have not experienced significant delays in obtaining any of our components. There are no unique or proprietary processes required in manufacturing our components. We generally do not have contractual obligations that preclude us from developing products or sourcing components from new suppliers.
We and our component suppliers are required to manufacture our products in compliance with the FDA’s QSR. The QSR regulates extensively the methods and documentation of the design, testing, control, manufacturing, labeling, quality assurance, packaging, storage and shipping of our products. The FDA enforces the QSR through periodic inspections that may be announced or unannounced and may include the manufacturing facilities of our suppliers. Our failure or the failure of our suppliers to maintain compliance with
18
Table of Contents
the QSR requirements could result in the shutdown of our manufacturing operations or the recall of our products, which would have a material adverse effect on our business. In the event that one of our suppliers fails to maintain compliance with our quality requirements, we may have to qualify a new supplier and could experience manufacturing delays as a result. We also could be subject to injunctions, product seizures, or civil or criminal penalties.
We regularly audit our suppliers for compliance with QSR and applicable ISO standards. We have been a FDA-registered medical device manufacturer since November 2002. We obtained our CE Mark in June of 2002 and our quality systems and facility practices are certified to ISO 13485:2003; MDD 93/42/EEC, or CE Mark, and CMDCAS, or Canadian regulations. We believe that we are currently in good standing with the FDA and are subject to pre-announced inspections. Our current quality system is developed to comply with QSR and ISO standards.
We are subject to numerous federal, state and local laws relating to such matters as laboratory practices, the experimental use of animals, the use and disposal of hazardous or potentially hazardous substances, safe working conditions, manufacturing practices, environmental protection and fire hazard control. We may incur significant costs to comply with those laws and regulations now or in the future, but, as we currently believe we are in compliance with such laws and regulations we do not expect that continued compliance will have a material impact on our business.
Consulting Relationships
We have developed consulting relationships with a number of leading scientists and doctors to give our research and development team additional technical and creative breadth. We work closely with these thought leaders to understand unmet needs and emerging applications for the treatment of AF. We typically enter into a written agreement with the consultant pursuant to which the consultant is obligated to provide services such as advising us regarding the design and development of our products, educating doctors on the FDA-cleared or approved use of our technologies, conducting clinical trials, and providing supporting data for clinical trials and providing advice concerning grants and regulatory submissions. These agreements are generally for a term of one year and may generally be terminated by us or by the consultant upon written notice. Generally, we own the rights to any inventions or ideas made or conceived by our consultants during the performance of the consulting services.
Most of our consulting agreements provide for payment of compensation in cash only and on a per diem basis (in addition to travel and other expenses), upon determination by us that services have been provided to our satisfaction. In addition, under agreements entered into prior to the fourth quarter of 2005, some of our consultants were entitled to receive stock options. We do not expect or require the consultant to utilize or promote our products, and consultants are required to disclose their relationship with us as appropriate, such as when publishing an article in which one of our products is discussed. See “Risk Factors—Risks Relating To Our Business—We may be subject to fines, penalties, injunctions and other sanctions if we are deemed to be promoting the use of our product for non-FDA-approved, or off-label, uses.”
We entered into a consulting agreement, dated as of January 1, 2007, with Michael D. Hooven, our co-founder and also one of our directors. Under the terms of the agreement, Mr. Hooven provided consulting services and advice to us with respect to the creation and development of new products and product platforms relating to cardiac arrhythmias and the prevention or reduction of strokes using cardiac devices. As consideration for his services and for assigning the rights to certain intellectual property as provided for in the agreement, Mr. Hooven was paid $12,000 per month. The term of the agreement was one year, with the exception of certain non-compete and non-solicitation provisions which expired on December 31, 2009.
During February 2009 we entered into a six-month consulting agreement with Enable Medical Technologies, an entity founded and owned by Michael D. Hooven. Under the terms of the agreement, Enable Medical Technologies provided research and development consulting services related to product and procedural
19
Table of Contents
development activities. Under the agreement, Enable Medical Technologies received $216,000 as a development fee and, upon completion of certain milestones, earned an additional $15,000.
Royalty Agreements
We have certain royalty agreements in place with terms that include payment of royalties based on product revenues from sales of current products, certain other inventions, improvements or ideas. As of January 1, 2010, royalty rates are 5% of product revenues related to our AtriClip system and Lumitip dissector. The agreement for the Lumitip dissector also calls for minimum royalty payments and limits the maximum aggregate in royalties during the term of each agreement. Parties to royalty agreements each have the right at any time to terminate the agreement immediately for cause. Royalty expense for each of the years ended December 31, 2009, 2008 and 2007 was $0.2 million.
Employees
As of December 31, 2009, we had approximately 200 full-time employees. None of the employees was represented by a labor union or was covered by a collective bargaining agreement. We have never experienced any employment-related work stoppages and consider our employee relations to be good although we cannot provide any assurance that we will not experience such work stoppages in the future.
Available Information
We are subject to the reporting requirements under the Securities Exchange Act of 1934. Consequently, we are required to file reports and information with the Securities and Exchange Commission, or SEC, including reports on the following forms: Form 10-K, Form 10-Q, Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. These reports and other information concerning us may be accessed through the SEC’s website at http://www.sec.gov. You may also find, free-of-charge, on our website at http://www.atricure.com electronic copies of our Form 10-Ks, Form 10-Qs, Form 8-Ks, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. Such filings are placed on our website as soon as reasonably possible after they are filed with the SEC. Our charters for our Audit, Compensation and Nominating and Corporate Governance Committees and our Code of Ethics are available on our website. In the event that we grant a waiver under our Code of Ethics to any of our officers and directors, we will publish it on our website. Information contained in any of our websites is not deemed to be a part of this Form 10-K.
| ITEM 1A. | RISK FACTORS |
Risks Relating To Our Business
If our products do not achieve widespread market acceptance in the United States, our operating results will be harmed and we may not achieve profitability.
Our success will depend, in large part, on the medical community’s acceptance of our principal products in the United States, which is the largest revenue market in the world for medical devices. The U.S. medical community’s acceptance of our products will depend upon our ability to demonstrate the safety and efficacy, advantages, long-term clinical performance and cost-effectiveness of our products as compared to other products. In addition, acceptance of products for the treatment of AF is dependent upon, among other factors, the level of screening for AF and the awareness and education of the medical community about the surgical treatment of AF, in general, and the existence, effectiveness and, in particular, the safety of our products. Market acceptance and adoption of our products for the treatment of AF also depends on the level of reimbursement to doctors and hospitals for use of our products.
We cannot predict whether the U.S. medical community will accept our products or, if accepted, the extent of their use. Negative publicity resulting from isolated incidents involving our products or other products related
20
Table of Contents
to those we sell could have a significant adverse effect on the overall acceptance of our products. If we encounter difficulties developing a market for our products in the United States, we may not be able to increase our revenue enough to achieve profitability, and our business and operating results will be seriously harmed.
We rely on the Isolator system and related products as our primary sources of revenue. If we are not successful in selling these products, our operating results will be harmed.
Currently, our Isolator system and related RF products generate a large majority of our revenues. We expect that sales of our Isolator system and related products will account for a majority of our revenues for the foreseeable future and that our future revenues will depend on the increasing acceptance by the medical community of our Isolator system and related products as a standard treatment alternative for the surgical treatment of AF during open-heart surgical procedures and as a sole-therapy minimally invasive procedure. We may not be able to maintain or increase market acceptance of our Isolator system and related products for a number of additional reasons, including those set forth elsewhere in this “Risk Factors” section. Since we believe that doctors are using our Isolator system only for the surgical treatment of AF, if doctors do not use our Isolator system and other products to treat AF, we would lose substantially all of our revenues.
Current worldwide economic conditions may have reduced demand for procedures using our products or otherwise resulted in adverse implications on our business, operating results and financial condition.
General worldwide economic conditions deteriorated beginning in late 2007 due to the effects of the subprime lending crisis, general credit market crisis, collateral effects on the finance and banking industries, concerns about inflation, slower economic activity, decreased consumer confidence, reduced corporate profits and capital spending, adverse business conditions and liquidity concerns. Although there are signs of an improving economic environment, the deteriorated economic environment continues. Because many procedures using our products are elective, they can be deferred by patients. In addition, patients may not be as willing under current economic conditions, to take time off from work or spend their money on deductibles and co-payments often required in connection with the procedures that use our products.
Beyond patient demand, the worldwide economic crisis, including in particular its effect on the credit and capital markets, may have other adverse implications for our business. For example, our customers’ ability to borrow money from their existing lenders or to obtain credit from other sources to purchase our products may be impaired resulting in a decrease in sales. Although we maintain allowances for estimated losses resulting from the inability of our customers to make required payments, we cannot guarantee that we will accurately predict the loss rates we will experience, especially given the current turmoil in the worldwide economy. A significant change in the liquidity or financial condition of our customers could cause unfavorable trends in our receivable collections and additional allowances may be required, which could adversely affect our operating results.
Healthcare costs have risen significantly over the past decade. There have been and may continue to be proposals by legislators, regulators and third-party payors to keep, contain or reduce healthcare costs.
The continuing efforts of governments, insurance companies and other payors of healthcare costs to contain or reduce these costs, combined with closer scrutiny of such costs, could lead to patients being unable to obtain approval for payment from these third-party payors. The cost containment measures that healthcare providers are instituting both in the U.S. and internationally could harm our business. Some health care providers in the U.S. have adopted or are considering a managed care system in which the providers contract to provide comprehensive health care for a fixed cost per person. Health care providers may attempt to control costs by authorizing fewer elective surgical procedures or by requiring the use of the least expensive devices possible, which could adversely affect the demand for our products or the price at which we can sell our products.
21
Table of Contents
We face significant uncertainty in the industry due to government healthcare reform.
The recently-enacted Patient Protection and Affordable Care Act (the “Patient Act”) as well as other healthcare reform legislation being contemplated by Congress and certain state legislatures may have a significant impact on our business. While we are currently evaluating the effects of the Patient Act on our business, the impact on the health care industry is extensive and includes, among other things, having the federal government assume a larger role in the health care system, expanding healthcare coverage of United States citizens and mandating basic healthcare benefits. In addition, any health care reforms enacted in the future may, like the Patient Act, be phased in over a number of years but, if enacted, could reduce our revenues, increase our costs, or require us to revise the ways in which we conduct business or put us at risk for loss of business. In addition, our results of operations, our financial position and cash flows could be materially adversely affected by changes under the Patient Act and change under any federal or state legislation adopted in the future.
Our quarterly financial results are likely to fluctuate significantly because our sales prospects are uncertain.
Due to current worldwide economic conditions and other factors discussed in this “Risk Factors” section which may impact our sales results, our quarterly operating results are difficult to predict and may fluctuate significantly from quarter to quarter or from prior year to current year periods, particularly because our sales prospects are uncertain. These fluctuations may also affect our annual operating results and may cause those results to fluctuate unexpectedly from year to year.
Restrictions in our ability to train doctors in the use of our products could reduce the market acceptance of our products or result in injuries to patients or other adverse events that lead to litigation that could harm us or could reduce our revenues.
It is critical to the success of our sales efforts to ensure that there are a sufficient number of doctors familiar with, trained on and proficient in the use of our products. While we educate and train doctors as to the skills involved in the proper use of our products, it is not our policy to educate or train them to use any products for the surgical treatment of AF. Doctors learn to use our products for the treatment of AF through independent training programs sponsored by hospitals and universities and through independent peer-to-peer training among doctors. We cannot assure you that a sufficient number of doctors will become aware of training programs, or that doctors will dedicate the time, funds and energy necessary to obtain training for themselves or to train others in the use of our products. In addition, our inability to directly train doctors in off-label use exposes us to a risk that our products may not be used correctly and may also expose us to a greater risk of product liability for injuries sustained during procedures utilizing our products.
Unless and until we obtain FDA approval for our products, we will not be able to promote our products to treat AF, and our ability to maintain and grow our business could be harmed.
We have not received FDA clearance or approval to promote any of our products for the treatment of AF. See “Business—Government Regulation.” Unless and until we obtain FDA clearance or approval for the use of our products for the treatment of AF we, and others acting on our behalf, may not promote our products for such uses, make any claim that our system is safe and effective for such uses, or proactively discuss or provide information on the use of our system in connection with such uses. We cannot assure you that future clearances or approvals of our products will be granted or that current or future approvals will not be withdrawn. Failure to obtain a clearance or approval or loss of an existing clearance or approval, could hurt our ability to maintain and grow our business.
22
Table of Contents
Unless we are able to complete the clinical trials required to support future submissions to the FDA, and unless the data generated by such trials supports the use of our Isolator system and other products as safe and effective for the treatment of AF, we may not be able to secure additional FDA clearances or approvals and our ability to maintain and grow our business could be harmed.
In order to obtain FDA approvals to promote our products for the treatment of AF, we will need to demonstrate in clinical trials that our products are safe and effective for such use. We cannot assure you that any of our clinical trials will be completed in a timely manner or successfully or that the results obtained will be acceptable to the FDA. In addition, if the results obtained from our clinical trials, any other clinical studies, or clinical or commercial experience indicate that any of our products are not safe or effective, or not as safe or effective as other treatment options, the FDA may not approve our products for the treatment of AF, adoption of the use of our products for the treatment of AF may suffer and our business would be harmed.
We have experienced and may continue to experience unfavorable publicity relating to our business and our industry. This publicity has had and may continue to have a negative impact on our ability to attract and retain customers, our sales, clinical studies involving our products, our reputation and our stock price.
We believe that we experienced a negative impact on our business from newspaper articles relating to, among other things, concerns of conflicts of interest between the Cleveland Clinic and us, our compliance with FDA regulations for medical device reporting, concerns that certain of our consultants who are involved with clinical studies and the publication of articles concerning our products failed to adequately disclose their financial relationships with us and our recently settled Department of Justice investigation. We believe that this publicity has had and may continue to have a negative impact on our clinical studies, business, results of operations and financial condition. We also believe that future unfavorable publicity could cause other adverse effects, including a decline in the price of our stock.
We may be subject to fines, penalties, injunctions and other sanctions if we are deemed to be promoting the use of our products for non-FDA approved, or off-label, uses.
Our business and future growth depend on the continued use of our products for the treatment of AF, which is considered an off-label use of our products. Under the Federal Food, Drug, and Cosmetic Act and other laws, we are prohibited from promoting our products for off-label uses. We may not make claims about the safety or effectiveness of our products for the treatment of AF and may not proactively discuss or provide information on the use of our products for the treatment of AF, except in certain limited scientific and other settings.
These limitations present a material risk that the FDA or other federal or state law enforcement authorities could determine that the nature and scope of our sales, marketing and/or product support activities, though designed to comply with all FDA requirements, constitute the promotion of our products for a non-FDA approved use in violation of the law. We also face the risk that the FDA or other governmental authorities might pursue enforcement based on past activities that we have discontinued or changed, including sales activities, arrangements with institutions and doctors, educational and training programs and other activities. Investigations concerning the promotion of off-label uses and related issues, including our completed DOJ investigation that was finalized subsequent to year end (see further discussion in Item 3, “Legal Proceedings” of this Form 10-K), are typically expensive, disruptive and burdensome and generate negative publicity. If our promotional activities are found to be in violation of the law, we may face significant fines and penalties and may be required to substantially change our sales, promotion, grant and educational activities. There is also a possibility that we could be enjoined from selling some or all of our products for any non-FDA approved use, which effectively would bar all sales of our products in the United States until we receive FDA clearances or approval, if ever. In addition, as a result of an enforcement action against us or our executive officers, we could be excluded from participation in government healthcare programs such as Medicare and Medicaid. Also, our failure to comply with the terms of the settlement agreement with the DOJ or the related corporate integrity agreement could result in additional action by the DOJ or the OIG, in fines or penalties or in restrictions on our sales, promotion, grant or educational activities.
23
Table of Contents
The use of products we sell may result in injuries or other adverse events that lead to product liability suits, which could be costly to our business or our customers’ businesses.
The use of products we sell may result in a variety of serious complications, including damage to the heart, internal bleeding, death or other adverse events, potentially leading to product liability claims. Serious complications, including death, have been encountered in connection with the surgical treatment of AF, including in connection with a limited number of sole-therapy minimally invasive procedures in which our Isolator system and other products were used. If products we sell are defectively designed, manufactured or labeled, contain inadequate warnings, contain defective components or are misused, we may become subject to costly litigation by our customers or their patients. We carry product liability insurance that is limited in scope and amount and may not be adequate to fully protect us against product liability claims. We could be required to pay damages that exceed our insurance coverage. Any product liability claim, with or without merit, could result in an increase in our product insurance rates or our inability to secure coverage on reasonable terms, if at all. Even in the absence of a claim, our insurance rates may rise in the future. Any product liability claim, even a meritless or unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, withdrawal of clinical trial participants, injury to our reputation and loss of revenue. Any of these events could negatively affect our earnings and financial condition.
Competition from existing and new products and procedures may decrease our market share and cause our revenues to decline.
The medical device industry, including the market for the treatment of AF, is highly competitive, subject to rapid technological change and significantly affected by new product introductions and promotional activities of other participants. We cannot assure you that our products will compete effectively against drugs, catheter-based ablation, implantable devices, other ablation systems, other products or techniques to exclude the left atrial appendage, or other surgical AF treatments, which may be more well-established among doctors and hospitals. We anticipate that new or existing competitors may develop competing products, procedures or clinical solutions. There are few barriers to prevent new entrants or existing competitors from developing products to compete directly with ours. Some companies also compete with us to attract qualified scientific and technical personnel as well as funding. Some of our competitors have greater financial, manufacturing, marketing and research and development capabilities than we have or may obtain FDA approval for the use of their products in the treatment of AF before we obtain approval for any of our products. The introduction of new products, procedures, clinical solutions or our competitors obtaining an AF approval may result in price reductions, reduced margins or loss of market share and may render our products obsolete, which could adversely affect our net revenues and future profitability.
Our intellectual property rights may not provide meaningful commercial protection for our products, which could enable third-parties to use our technology or methods, or very similar technology or methods, and could reduce our ability to compete.
Our success depends significantly on our ability to protect our proprietary rights to the technologies used in our products. We rely on patent protection, as well as a combination of copyright, trade secret and trademark laws and nondisclosure, confidentiality and other contractual restrictions to protect our proprietary technology. However, these legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. Our patent applications may not issue as patents at all or in a form that will be advantageous to us. Our issued patents and those that may be issued in the future may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products. Although we have taken steps to protect our intellectual property and proprietary technology, we cannot assure you that third parties will not be able to design around our patents or, if they do infringe upon our technology, that we will be successful in or will have sufficient resources to pursue a claim of infringement against those
24
Table of Contents
third parties. We believe that third parties may have developed or are developing products that could infringe upon our patent rights. Any pursuit of an infringement claim by us may involve substantial expense or diversion of management attention. In addition, although we have generally entered into confidentiality agreements and intellectual property assignment agreements with our employees, consultants, investigators and advisors, such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements.
Furthermore, the laws of foreign countries may not protect our intellectual property rights to the same extent as the laws of the United States. Foreign countries generally do not allow patents to cover methods for performing surgical procedures. If our intellectual property does not provide significant protection against foreign or domestic competition, our competitors could compete more directly with us, which could result in a decrease in our market share. All of these factors may harm our competitive position.
The medical device industry is characterized by extensive litigation and administrative proceedings over patent and other intellectual property rights and any litigation or claim against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our business and harm our reputation.
Whether a product infringes a patent involves complex legal and factual issues, the determination of which is often uncertain. Any patent dispute, even one without merit or an unsuccessful one, would be time-consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, the disruption of development and marketing efforts, injury to our reputation and loss of revenue. Any of these events could negatively affect our earnings and financial condition.
In the event of a patent dispute, if a third-party’s patents were upheld as valid and enforceable and we were found to be infringing, we could be prevented from selling our products unless we were able to obtain a license to use technology or ideas covered by such patent or are able to redesign our system to avoid infringement. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our products to avoid any infringement. Modification of our products or development of new products could require us to conduct additional clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. If we are not successful in obtaining a license or redesigning our products, we may be unable to sell our products and our business could suffer.
The increase in cost of medical malpractice premiums to doctors and hospitals or the lack of malpractice insurance coverage due to the use of our products by doctors for an off-label indication may cause certain doctors or hospitals to decide not to use our products and may damage our ability to grow and maintain the market for our system.
Insurance carriers have been raising premiums charged for medical malpractice insurance due, at least in part, to increased risks associated with off-label procedures, including higher damage awards for successful plaintiffs. Insurance carriers may continue to raise premiums or they may deny malpractice coverage for procedures performed using products such as ours on an off-label basis. If this trend continues or worsens, our revenue may fall as doctors or hospitals decide against purchasing our products due to the cost or unavailability of insurance coverage.
We have a history of net losses and we may never become profitable.
We have incurred net losses each year since our inception, including net losses of $16.5 million in 2009, $10.2 million in 2008, $11.3 million in 2007, $13.7 million in 2006, $12.7 million in 2005, $9.5 million in 2004 and $7.1 million in 2003. As of December 31, 2009, we had an accumulated deficit of $94.0 million.
Our net losses have resulted principally from costs and expenses relating to sales and promotional efforts, research and development, seeking regulatory clearances and approvals, goodwill impairment, litigation and
25
Table of Contents
settlement costs associated with the DOJ investigation and general operating expenses. We expect to continue to make substantial expenditures and to incur additional operating losses in the future as we further develop and commercialize our products, including completing clinical trials and seeking regulatory clearances and approvals. If sales of our products do not continue to grow as we anticipate, we will not be able to achieve profitability. Our expansion efforts may prove more expensive than we currently anticipate, and we may not succeed in increasing our revenues sufficiently to offset these higher expenses. Our losses have had, and are expected to continue to have, an adverse impact on our working capital, total assets and stockholders’ deficit and we may never become profitable.
Our federal tax net operating loss and general business credit carryforwards generated prior to the initial public offering of our common stock will be limited or may expire, which could result in greater future income tax expense and adversely impact future cash flows because we experienced an ownership change of more than 50 percentage points upon the initial public offering of our common stock.
In connection with our initial public offering in August 2005, we experienced an ownership change as defined by Section 382 of the Internal Revenue Code of 1986. Section 382 imposes limitations (“Section 382 limitation”) on a company’s ability to use net operating loss and general business credit carryforwards if a company experiences a more-than-50-percent ownership change over a three-year testing period. The Section 382 limitation could limit the availability of our net operating loss and general business credit carryforwards to offset any future taxable income, which may increase our future income tax expense and adversely impact future cash flows. We had federal income tax net operating loss and general business credit carryforwards at August 5, 2005 that, if not utilized to reduce our taxable income, will begin to expire in 2021. In addition, if the company were to experience a second ownership change of more than 50 percentage points in a future period, the company’s NOL carryforward at the date of the original ownership change would be subject to a second Section 382 limitation. In addition, the company’s NOLs generated subsequent to the original ownership change would be subject to the second Section 382 limitation. Since December 31, 2005 the company has generated additional net operating loss and general business credit carryforwards of $29 million and $1.6 million, respectively, that, if not utilized to reduce our taxable income, will begin to expire in 2026.
Our capital needs after the next 12 months are uncertain and we may need to raise additional funds in the future and such funds may not be available on acceptable terms, if at all.
We believe that our current cash, cash equivalents and investments will be sufficient to meet our projected capital requirements for at least the next 12 months. Our current loan agreement (the “Agreement”) with Silicon Valley Bank (the “Bank”) includes a term loan and a revolving credit facility under which we can borrow a maximum of $10.0 million. We have borrowed the maximum amount of $6.5 million under the term loan. We can borrow up to $10.0 million under the revolving loan facility with the availability subject to a borrowing base formula. The Agreement matures on April 30, 2012 and is secured by all of our assets, including intellectual property. Interest on the term loan accrues at a rate of 10.0% per year, and interest on the revolving loan will accrue at a fluctuating rate equal to the Bank’s announced prime rate of interest, subject to a floor of 4.0%, plus between 1.0% and 2.0%, depending on our Adjusted Quick Ratio (as defined in the Agreement).
Our Agreement contains covenants that include, among others, covenants that limit our ability to dispose of assets, enter into mergers or acquisitions, incur indebtedness, incur liens, pay dividends or make distributions on our capital stock, make investments or loans, and enter into certain affiliate transactions, in each case subject to customary exceptions for a credit facility of this size and type. Additional covenants apply when we have outstanding borrowings under the revolving loan facility or when we achieve specific covenant milestones. The occurrence of an event of default could result in an increase to the applicable interest rate by 3.0%, an acceleration of all obligations under the Agreement, an obligation to repay all obligations in full, and a right by the Bank to exercise all remedies available to it under the Agreement and related agreements including the Guaranty and Security Agreement.
26
Table of Contents
If we need to raise additional funds, we cannot be certain that such funds will be available to us on acceptable terms, if at all. Furthermore, if we issue equity securities to raise additional funds, our existing stockholders may experience dilution, and if we issue equity or debt securities, such securities may have rights, preferences and privileges senior to those of our existing stockholders. In addition, if we raise additional funds through collaboration, licensing or other similar arrangements, it may be necessary to relinquish potentially valuable rights to our future products or proprietary technologies, or grant licenses on terms that are not favorable to us. If we cannot raise funds on acceptable terms, we may not be able to expand our operations, develop new products, take advantage of future opportunities or respond to competitive pressures or unanticipated customer requirements.
We depend upon single and limited source third-party suppliers, making us vulnerable to supply problems and price fluctuations which could harm our business.
We currently rely on single and limited source third-party vendors for the manufacture of many of the components used in our products. For example, we rely on one vendor to manufacture our ASU and ASB. It would be a time consuming and lengthy process to secure these products from an alternative supplier. In addition, in some cases there are relatively few, or no, alternative sources of supply for certain other components that are critical to our products.
Our reliance on these outside manufacturers and suppliers also subjects us to risks that could harm our business, including:
| • | we may not be able to obtain adequate supply in a timely manner or on commercially reasonable terms; |
| • | we may have difficulty locating and qualifying alternative suppliers; |
| • | switching components may require product redesign and new submissions to the FDA which could significantly delay production or, if the FDA refuses to approve the changes, completely eliminate our ability to manufacture or sell our products; |
| • | our suppliers manufacture products for a range of customers, and fluctuations in demand for the products those suppliers manufacture for others may affect their ability to deliver components to us in a timely manner; and |
| • | our suppliers may encounter financial hardships unrelated to our demand for components, which could inhibit their ability to fulfill our orders and meet our requirements. |
Identifying and qualifying additional or replacement suppliers for any of the components used in our products, if required, may not be accomplished quickly or at all and could involve significant additional costs. Any interruption or delay in the supply of components or materials, or our inability to obtain components or materials from alternate sources at acceptable prices in a timely manner, could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competitive products and could therefore have a material adverse effect on our business, financial condition and results of operations.
An inability to forecast future revenues or estimate life cycles of products may result in inventory-related charges that would negatively affect our gross margins and results of operations.
To mitigate the risk of supply interruptions, we may choose to maintain excess inventory of our products or component parts. Managing our inventory levels is important to our cash position and results of operations and is more challenging in the current economic environment. As we grow and expand our product offerings, managing our inventory levels becomes more difficult, particularly as we expand into new product areas and bring product enhancements to market. An excessive amount of inventory reduces our cash available for operations and may result in excess or obsolete materials. Conversely, inadequate inventory levels may make it difficult for us to meet customer product demand, resulting in decreased revenues. An inability to forecast future revenues or estimated life cycles of products may result in inventory-related charges that would negatively affect our gross margins and results of operations.
27
Table of Contents
If we or our third-party vendors fail to comply with extensive FDA regulations relating to the manufacturing of our products or any component part, we may be subject to fines, injunctions and penalties, and our ability to commercially distribute and sell our products may be hurt.
Our manufacturing facility and the manufacturing facility of any of our third-party component manufacturers, critical suppliers or third-party sterilization facility are required to comply with the FDA’s quality systems regulations, or QSR, which sets forth minimum standards for the procedures, execution and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our Isolator system and other products we sell. The FDA may enforce its QSR, among other ways, through periodic unannounced inspections. If our manufacturing facility or the manufacturing facility of any of our third-party component manufacturers, critical suppliers or third-party sterilization facility, fails a QSR inspection, our and their operations could be disrupted, and manufacturing interrupted. Failure to take adequate and timely corrective action in response to an adverse QSR inspection could force a shutdown of our manufacturing operations or a recall of our products. Adverse QSR inspections could delay FDA approval of our products and could have an adverse effect on our production, sales and profitability. We and any of our third-party vendors may also encounter other problems during manufacturing including failure to follow specific protocols and procedures, equipment malfunction and environmental factors, any of which could delay or impede our ability to meet demand. The manufacture of our product also subjects us to risks that could harm our business, including problems relating to the sterilization of our products or facilities and errors in manufacturing components that could negatively affect the efficacy or safety of our products or cause delays in shipment of our products. Any interruption or delay in the manufacture of the product or any of its components could impair our ability to meet the demand of our customers and cause them to cancel orders or switch to competitive products, and could therefore have a material adverse effect on our business, financial condition and results of operations.
If we fail to comply with the extensive FDA regulations relating to our business, we may be subject to fines, injunctions and penalties and our ability to commercially distribute and promote our products may be hurt.
Our products are classified by the FDA as medical devices and as such are subject to extensive regulation in the United States by the FDA and numerous other federal, state and foreign governmental authorities. FDA regulations, guidance, notices and other issuances specific to medical devices are broad and regulate, among other things:
| • | product design, development, manufacturing and labeling; |
| • | product testing, including electrical testing, transportation testing and sterility testing; |
| • | pre-clinical laboratory and animal testing; |
| • | clinical trials in humans; |
| • | product safety, effectiveness and quality; |
| • | product manufacturing, storage and distribution; |
| • | pre-market clearance or approval; |
| • | record keeping and document retention procedures; |
| • | product advertising, sales and promotion; |
| • | post-market surveillance and medical device reporting of events where our device caused or contributed to a death or other serious injury, or malfunctioned in such a way that if it were to recur would likely cause or contribute to a death or serious injury; |
| • | product corrective actions, removals and recalls; and |
| • | product import and export. |
28
Table of Contents
Compliance with FDA, state and other regulations can be complex, expensive and time-consuming. The FDA and other authorities have broad enforcement powers. Furthermore, changes in the applicable governmental regulations could prevent further commercialization of our products and technologies and could materially harm our business.
If a serious failure to comply with applicable regulatory requirements were determined, it could result in enforcement action by the FDA or other state or federal agencies, including the DOJ, which may include any of the following sanctions:
| • | warning letters, fines, injunctions, consent decrees and civil penalties; |
| • | repair, replacement, refunds, recall or seizure of our products; |
| • | operating restrictions, partial suspension or total shutdown of production; |
| • | refusing or delaying our pending requests for 510(k) clearance or PMAs, new intended uses or modifications to existing products; |
| • | withdrawing 510(k) clearance or PMAs that have already been granted; and |
| • | criminal prosecution. |
If any of these events were to occur, we could lose customers and our production, product sales, business, results of operations and financial condition would be harmed.
We are also subject to medical device reporting regulations that require us to file reports with the FDA if our products reasonably are the cause of or contribute to an adverse event, death, serious injury or, in the event of product malfunction, that if it were to recur, would likely cause or contribute to a death or serious injury. We have a history of submitting medical device reports to the FDA involving our products, including patient deaths, which were categorized as outcomes based on physician judgment, not on the failure of our devices. There have also been other incidents, including patient deaths, which have occurred during procedures using our products that we have not, and believe were not required to be, reported to the FDA because we and our physician consultants determined that our products did not cause or contribute to the outcomes in these incidents. If the FDA disagrees with us, however, and determines that we should have submitted reports for these adverse events, we could be subject to significant regulatory fines or other penalties. In addition, the number of medical device reports we make, or the magnitude of the problems reported, could cause the FDA or us to terminate or modify our clinical trials or recall or cease the sale of our products, and could hurt commercial acceptance of our products.
Modifications to our products may require new clearances or approvals or require us to cease promoting or to recall the modified products until such clearances or approvals are obtained and the FDA may not agree with our conclusions regarding whether new clearances or approvals were required.
Any modification to a 510(k)-cleared device that would constitute a change in its intended use, design or manufacture, could require a new 510(k) clearance or, possibly, submission and FDA approval of a PMA. The FDA requires every medical device company to make the determination as to whether a new 510(k) is to be filed, but the FDA may review any medical device company’s decision. We have made modifications to our products but do not believe such modifications required us to submit an additional 510(k). The FDA may not agree with our decisions regarding whether new clearances or approvals were required. We have recently been in communication with the FDA regarding our decision not to file a new 510(k) related to a change in indication for our Isolator Synergy clamps. At the time our Isolator clamps received 510(k) clearance for the ablation of cardiac tissue, through our internal and external regulatory review process, we determined that a new 510(k) was not needed for our Isolator Synergy clamps to change their intended use from the ablation of soft tissue to the ablation of cardiac tissue. The FDA has reviewed this decision and has indicated that a 510(k) was required to be filed for us to market our Isolator Synergy clamps for cardiac tissue ablation instead of soft tissue ablation. We are working with the FDA to provide them additional information in support of our decision and to evaluate corrective actions.
29
Table of Contents
If the FDA disagrees with us and requires us to submit a new 510(k) or PMA for then existing modifications, we may be required to cease promoting or to recall the modified product until we obtain clearance or approval. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our products could be subject to recall if the FDA determines, for any reason, that our products are not safe or effective or that appropriate regulatory submissions were not made. Delays in receipt or failure to receive clearances or approvals, the loss of previously received clearances or approvals, or the failure to comply with existing or future regulatory requirements, could reduce our sales, profitability and future growth prospects.
We will spend considerable time and money complying with federal, state and foreign regulations in addition to FDA regulations, and, if we are unable to fully comply with such regulations, we could face substantial penalties.
We are subject to extensive regulation by the federal government and the states and foreign countries in which we conduct our business. The laws that affect our ability to operate our business in addition to the Federal Food, Drug, and Cosmetic Act and FDA regulations include, but are not limited to, the following:
| • | state food and drug laws, including laws regulating the manufacture, promotion and distribution of medical devices; |
| • | state consumer protection, fraud and business practice laws; |
| • | the Federal Anti-Kickback Statute, which prohibits persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce either the referral of an individual, or furnishing or arranging for a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid Programs; |
| • | the Federal False Claims Act, which prohibits submitting a false claim or causing of the submission of a false claim to the government; |
| • | Medicare laws and regulations that prescribe the requirements for coverage and payment, including the amount of such payment, and laws prohibiting false claims for reimbursement under Medicare and Medicaid; |
| • | the federal doctor self-referral prohibition, commonly known as the Stark Law, which, in the absence of a statutory or regulatory exception, prohibits the referral of Medicare patients by a doctor to an entity for the provision of certain designated healthcare services including inpatient and outpatient hospital services, if the doctor or a member of the doctor’s immediate family has a direct or indirect financial relationship, including an ownership interest in, or a compensation arrangement with, the entity and also prohibits that entity from submitting a bill to a federal payor for services rendered pursuant to a prohibited referral; |
| • | state laws that prohibit the practice of medicine by non-doctors and by doctors not licensed in a particular state, and fee-splitting arrangements between doctors and non-doctors, as well as state law equivalents to the Anti-Kickback Statute and the Stark Law, which may not be limited to government-reimbursed items; |
| • | federal and state healthcare fraud and abuse laws or laws protecting the privacy of patient medical information, including the Health Insurance Portability and Accountability Act, or HIPAA; |
| • | the Federal Trade Commission Act and similar laws regulating advertising and consumer protection; and |
| • | similar and other regulations outside the United States. |
Certain federal and state laws regarding Medicare, Medicaid and physician self-referrals are broad and we may be required to change one or more of our practices to be in compliance with these laws. Healthcare fraud and abuse regulations are complex and even minor, inadvertent irregularities in submissions can potentially give rise to claims that a statute has been violated. Any violations of these laws could result in a material adverse effect on our business, financial condition and results of operations. For example, if we were found to be in
30
Table of Contents
violation of the Federal False Claims Act, we would likely face significant fines and penalties and would likely be required to change substantially our sales, promotion, grant and educational activities. There is also a possibility that we could face an injunction that would prohibit in whole or in part our current business activities, and, as a result of enforcement actions against us or our senior officers, we could be excluded from participation in government healthcare programs such as Medicare and Medicaid. If there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which could have a material adverse effect on our business, financial condition and results of operations. Recently, the DOJ conducted an investigation of our marketing and promotional practices. Although we admitted to no wrongdoing and believe there was no wrongdoing on the part us or our, our employees, this investigation resulted in a financial settlement of $4.4 million (which includes interest based on payment terms). Additionally, we incurred substantial legal costs through the investigation and settlement process.
If our past or present operations are found to be in violation of any of the laws described above or the other governmental regulations to which we, our distributors or our customers are subject, we may be subject to the applicable penalty associated with the violation, including civil and criminal penalties, damages, fines, exclusion from Medicare, Medicaid and other government programs and the curtailment or restructuring of our operations. If we are required to obtain permits or licensure under these laws that we do not already possess, we may become subject to substantial additional regulation or incur significant expense. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business and our financial results. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully or clearly interpreted by the regulatory authorities or the courts, and their provisions are subject to a variety of interpretations and additional legal or regulatory change. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
Adverse changes in payors’ policies toward coverage and reimbursement for surgical AF treatment would harm our ability to promote and sell our Isolator system and other products.
Third-party payors are increasingly exerting pressure on medical device companies to reduce their prices. Even to the extent that the treatment of AF using our products is reimbursed by private payors and governmental payors, adverse changes in payors’ policies toward coverage and reimbursement for surgical AF treatment would also harm our ability to promote and sell our products. Payors continue to review their policies and can, without notice, deny coverage for treatments that include the use of our products. Because each third-party payor individually approves coverage and reimbursement, obtaining these approvals may be time-consuming and costly. In addition, third-party payors may require us to provide scientific and clinical support for the use of our products. Alternatively, government or private payors may deem the treatment of AF utilizing our products experimental or not medically necessary and, as such, not provide coverage. Adverse changes in coverage and reimbursement for surgical AF treatment could harm our business and reduce our revenues.
We have limited long-term clinical data regarding the safety and efficacy of our products. Any long-term data that is generated may not be positive or consistent with our limited short-term data, which would affect the rate at which our products are adopted by the medical community.
Important factors upon which the efficacy of our products will be measured include long-term data on the number of patients that continue to experience AF following treatment with our products and the number of patients that have serious complications resulting from AF treatment using our products. Our clinical trials may produce limited data regarding the efficacy of our products for the treatment of AF or may identify unexpected safety issues. We cannot provide any assurance that the data collected during our clinical trials will be compelling to the medical community because it may not be scientifically meaningful and may not demonstrate that procedures utilizing our products are an attractive procedure when compared against data from alternative procedures and products. In addition, the long-term effects of ablation system procedures are not known. Negative long-term data would affect the use of our products and harm our business and prospects.
31
Table of Contents
We sell our products outside of the United States and we are subject to various regulatory and other risks relating to international operations, which could harm our international revenues and profitability.
Doing business outside of the United States exposes us to risks distinct from those we face in our domestic operations. For example, our operations outside of the United States are subject to different regulatory laws and requirements in each jurisdiction where we operate or have sales. Our failure, or the failure of our distributors, to comply with current or future foreign regulatory requirements, or the assertion by foreign authorities that we or they have failed to comply, could result in adverse consequences, including enforcement actions, fines and penalties, recalls, cessation of sales, civil and criminal prosecution, and the consequences could be disproportionate to the relative contribution of our international operations to our results of operations. Moreover, if political or economic conditions deteriorate in these countries, our ability to conduct our international operations could be limited and the costs could be increased, which could negatively affect our operating results. Engaging in business outside of the United States inherently involves a number of other difficulties and risks, including:
| • | export restrictions and controls relating to technology; |
| • | pricing pressure that we may experience internationally; |
| • | difficulties in enforcing agreements and collecting receivables through certain foreign legal systems; |
| • | political and economic instability; |
| • | potentially adverse tax consequences, tariffs and other trade barriers; |
| • | the need to hire additional personnel to promote our Isolator system outside of the United States; |
| • | international terrorism and anti-American sentiment; |
| • | fluctuations in exchange rates for future sales denominated in foreign currency, which represent a majority of our sales outside of the United States; and |
| • | difficulty in obtaining and enforcing intellectual property rights. |
In addition, our business practices in foreign countries comply with U.S. law, including the Foreign Corrupt Practices Act (“FCPA”). We have a compliance program in place designed to reduce the likelihood of potential violations of the FCPA and other U.S. laws. If violations were to occur, they could subject us to fines and other penalties as well as increased compliance costs.
Our exposure to each of these risks may increase our costs and require significant management attention. We cannot assure you that one or more of these factors will not harm our business.
We primarily rely on independent distributors to market and sell our products outside of the United States, and a failure of our independent distributors to successfully market our products in these markets may adversely impact our sales.
We primarily depend on third-party distributors to sell our products outside of the United States and if these distributors do not perform, we may be unable to increase or maintain our level of international revenues. Over the long term, we intend to continue to grow our business outside of the United States, and to do so we will need to attract additional distributors or hire direct sales personnel to expand the territories in which we sell our products. Independent distributors may terminate their relationship with us or devote insufficient sales efforts to our products. We are not able to control our independent distributors, and they may not be successful in implementing our marketing plans. In addition, many of our independent distributors outside of the United States initially obtain and maintain foreign regulatory approval for sale of our products in their respective countries. Our failure to maintain our relationships with our independent distributors outside of the United States, or our failure to recruit and retain additional skilled independent distributors in these locations, could have an adverse effect on our operations. Turnover among our independent distributors, even if replaced, may adversely affect
32
Table of Contents
our short-term financial results while we transition to new independent distributors or direct personnel. Fluctuations in foreign currency exchange rates including, in particular, any strengthening of the U.S. dollar may cause our independent sales distributors to seek longer payment terms to offset the higher prices they are paying in local currency for our products. In addition, in light of the worldwide economic crisis, the ability of our distributors to borrow money from their existing lenders or to obtain credit from other sources to purchase our products may be impaired or our distributors could experience a significant change in their liquidity or financial condition, all of which could impair their ability to distribute our products and eventually lead to distributor turnover.
If coverage and adequate levels of reimbursement from governmental and third-party payors outside of the United States are not attained and maintained, sales of our products outside of the United States may decrease and we may fail to achieve or maintain significant sales outside of the United States.
Our revenues generated from sales outside of the United States are also dependent upon the availability of coverage and reimbursement within prevailing foreign healthcare payment systems. In general, foreign healthcare payors do not provide reimbursement for sole-therapy minimally invasive procedures utilizing ablation devices such as our Isolator system and related products. In addition, healthcare cost containment efforts similar to those we face in the United States are prevalent in many of the other countries in which we sell our products, and these efforts are expected to continue. To the extent that the use of an ablation device such as our Isolator clamp has historically received reimbursement under a foreign healthcare payment system, if any, such reimbursement, if any, has typically been significantly less than the reimbursement provided in the United States. If coverage and adequate levels of reimbursement from governmental and third-party payors outside of the United States are not attained and maintained, sales of our products outside of the United States may decrease and we may fail to achieve or maintain significant sales outside of the United States.
The outcome of litigation in which we have been named as a defendant, including two class action shareholder lawsuits, is unpredictable and an adverse decision in any such matter could have a material adverse affect on our financial position and results of operations.
We, along with certain of our current and former officers, are named defendants in two purported securities class action lawsuits. These proceedings have resulted, and are expected to continue to result, in a diversion of management’s attention and resources and in significant professional fees. These professional fees have increased and, in the near term, may continue to increase our cash needs.
We have certain obligations to indemnify our officers and directors and to advance expenses to such officers and directors. Although we have purchased liability insurance for our directors and officers, if our insurance carriers should deny coverage for all or a portion of the amount to be paid, or if the indemnification costs exceed the insurance coverage, we may be forced to bear some or all of these indemnification costs directly, which could be substantial and may have an adverse effect on our business, financial condition, results of operations and cash flows.
While we have recorded a liability of $2.0 million, representing our estimate of the potential defense and/or settlement costs in connection with the Levine v. AtriCure, Inc. class action, we cannot assure you that we will be able to settle the case without incurring additional liability. We are not able to estimate the amount of damages or internal efforts associated with defending ourselves in our other class action lawsuit. A failure to resolve the Levine case as we currently estimate or a failure to successfully defend ourselves in our other class action lawsuit may adversely affect our business, financial condition, results of operations and cash flows as a result of the damages that we would be required to pay. It is possible that our insurance policies either may not cover potential claims of this type or may not be adequate to indemnify us from all liability that may be imposed. While we believe that the allegations and claims made in these lawsuits are wholly without merit and intend to defend against these actions vigorously, we cannot be certain that we will be successful in any or all of these actions.
33
Table of Contents
An adverse resolution of any lawsuits could have a material adverse affect on our financial position and results of operations.
We depend on our officers and other skilled and experienced personnel to operate our business effectively. If we are not able to retain our current employees or recruit additional qualified personnel, our business will suffer and our future revenue and profitability will be impaired.
We are highly dependent on the skills and experience of our President and Chief Executive Officer, David J. Drachman, and certain other officers and key employees. We do not have any insurance in the event of the death or disability of our key personnel other than Mr. Drachman. Our officers and key employees, with the exception of our Chief Executive Officer and Chief Financial Officer, do not have employment agreements and they may terminate their employment and work elsewhere without notice and without cause or good reason. Currently we have non-compete agreements with our officers and other employees. Due to the specialized knowledge that each of our officers possesses with respect to our products and our operations and the limited pool of people with relevant experience in the medical device field, the loss of service of one or more of these individuals could significantly affect our ability to operate and manage our business. The announcement of the loss of one or more of our key personnel could negatively affect our stock price.
We depend on our scientific and technical personnel for successful product development and innovation, which are critical to the success of our business. In addition, to succeed in the implementation of our business strategy, our management team must rapidly execute our sales strategy, obtain expanded FDA clearances and approvals, achieve market acceptance for our products and further develop products, while managing anticipated growth by implementing effective planning, manufacturing and operating processes. Managing this growth will require us to attract and retain additional management and technical personnel. Our offices are located in West Chester, Ohio where it can be difficult to attract and retain employees with experience in the medical device industry. We rely primarily on direct sales employees to sell our products in the United States and failure to adequately train them in the use and benefits of our products will prevent us from achieving our market share and revenue growth goals. We have key relationships with doctors that involve procedure, product, market and clinical development. If any of these doctors end their relationship with us, our business could be negatively impacted. We cannot assure you that we will be able to attract and retain the personnel and doctor relationships necessary to grow and expand our business and operations. If we fail to identify, attract, retain and motivate these highly skilled personnel and doctors, we may be unable to continue our development and sales activities.
Compliance with environmental laws and regulations may be expensive. Failure to comply with environmental laws and regulations could subject us to significant liability.
Our manufacturing operations and research and development activities involve the use of biological materials and hazardous substances and are subject to a variety of federal, state and local environmental laws and regulations relating to the storage, use, discharge, disposal, remediation of, and human exposure to, hazardous substances. Our research and development and manufacturing operations may produce biological waste materials, such as animal tissues and certain chemical waste. These operations are permitted by regulatory authorities and the resultant waste materials are disposed of in material compliance with environmental laws and regulations. Compliance with these laws and regulations may be expensive and non-compliance could result in substantial liabilities. In addition, we cannot completely eliminate the risk of accidental contamination or injury to third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, we could be held liable for any resulting damages, and any liability could exceed any applicable insurance coverage we may have. In addition, our manufacturing operations may result in the release, discharge, emission or disposal of hazardous substances that could cause us to incur substantial liabilities, including costs for investigation and remediation.
34
Table of Contents
Risks Relating To Our Common Stock
The price and trading volume of our common stock may experience extreme fluctuations and you could lose some or all of your investment.
Because we operate within the medical device segment of the healthcare industry, our stock price is likely to be volatile. The market price of our common stock may have and has had a history of substantial fluctuation due to a variety of factors, including:
| • | doctor and patient acceptance of the surgical treatment of AF using our products; |
| • | adverse regulatory developments with respect to our products, such as recalls, new regulatory requirements, changes in regulatory requirements or guidance and timing of regulatory clearances and approvals for new products; |
| • | coverage and reimbursement determinations for our products and the related procedures; |
| • | the timing of orders received; |
| • | delays or interruptions in manufacturing or shipping of our products; |
| • | pricing of our products; |
| • | media reports, publications and announcements about products or new innovations that could compete with our products or about the medical device product segment in general; |
| • | investigations, claims or allegations by regulatory agencies, such as the DOJ; |
| • | market conditions or trends related to the medical device and healthcare industries or the market in general; |
| • | additions to or departures of our key personnel; |
| • | disputes, litigation or other developments relating to proprietary rights, including patents, and our ability to obtain patent protection for our technologies; |
| • | changes in financial estimates, investors’ perceptions or recommendations by securities analysts; |
| • | variations in our quarterly financial and operating results; |
| • | failure to achieve or maintain an effective healthcare compliance environment; |
| • | changes in accounting principles; and |
| • | failure to achieve and maintain an effective internal control environment. |
These factors, some of which are not within our control, may cause the price of our stock to fluctuate substantially. If our quarterly or annual operating results fail to meet or exceed the expectations of securities analysts or investors, our stock price could drop suddenly and significantly. We believe the quarterly and annual comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
The market prices of the securities of medical device companies, particularly companies like ours without consistent product revenues and earnings, have been highly volatile and are likely to remain highly volatile in the future. This volatility has often been unrelated to the operating performance of particular companies. These market prices generally are not sustainable and are highly volatile. In the past, companies that experience volatility in the market price of their securities have often faced securities class action litigation. Whether or not meritorious, litigation brought against us could result in substantial costs, divert our management’s attention and resources and harm our ability to grow our business.
35
Table of Contents
The ownership of our common stock is highly concentrated. Your interests may conflict with the interests of our existing stockholders and sales of a significant number of shares may cause our stock price to decline.
Our executive officers and directors and their affiliates, together with our current significant stockholders, beneficially owned approximately 30% of our outstanding common stock as of December 31, 2009. Accordingly, these stockholders have significant influence over the outcome of corporate actions requiring stockholder approval. The interests of these stockholders may be different than the interests of other stockholders on these matters. This concentration of ownership could also have the effect of delaying or preventing a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which in turn could reduce the price of our common stock. In addition, the holders of up to 3.9 million shares of our common stock have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. If our common stockholders sell substantial amounts of our common stock in the public market, through a registration statement or otherwise, or the market perceives that such sales may occur, the market price of our common stock could decline.
Sales of common stock by us in a capital raising transaction may dilute your ownership of common stock and cause a decline in the market price of our common stock.
We may need to raise capital in the future to fund our operations. If we raise funds by issuing equity securities, our stock price may decline and our existing shareholders may experience significant dilution. Furthermore, we may enter into financing transactions at prices that represent a substantial discount to market price. A negative reaction by investors and securities analysts to any sale of our equity securities could result in a decline in the trading price of our common stock.
Anti-takeover provisions in our amended and restated certificate of incorporation and amended and restated bylaws and under Delaware law could inhibit a change in control or a change in management that you consider favorable.
Provisions in our certificate of incorporation and bylaws could delay or prevent a change of control or change in management that would provide you with a premium to the market price of your common stock. These provisions include those:
| • | authorizing the issuance without further approval of “blank check” preferred stock that could be issued by our board of directors to increase the number of outstanding shares and thwart a takeover attempt; |
| • | prohibiting cumulative voting in the election of directors, which would otherwise allow less than a majority of stockholders to elect director candidates; |
| • | limiting the ability to remove directors; |
| • | limiting the ability of stockholders to call special meetings of stockholders; |
| • | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of stockholders; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon by stockholders at stockholder meetings. |
In addition, Section 203 of the Delaware General Corporation Law limits business combination transactions with 15% stockholders that have not been approved by our board of directors. These provisions and others could make it difficult for a third party to acquire us, or for members of our board of directors to be replaced, even if doing so would be beneficial to our stockholders. Because our board of directors is responsible for appointing the members of our management team, these provisions could, in turn, affect any attempt to replace the current management team. If a change of control or change in management is delayed or prevented, you may lose an opportunity to realize a premium on your shares of common stock or the market price of our common stock could decline.
36
Table of Contents
We do not expect to pay dividends in the foreseeable future. As a result, you must rely on stock appreciation for any return on your investment.
We do not anticipate paying cash dividends on our common stock in the foreseeable future. Any payment of cash dividends will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our board of directors. Accordingly, you will have to rely on capital appreciation, if any, to earn a return on your investment in our common stock. Furthermore, pursuant to our credit facility, we are currently subject to restrictions on our ability to pay dividends and we may in the future become subject to other contractual restrictions on, or prohibitions against, the payment of dividends.
The requirements of being a public company may strain our resources and distract management.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act. These requirements may place a strain on our systems and resources. The Exchange Act requires that we file annual, quarterly and current reports with respect to our business and financial condition. The Sarbanes-Oxley Act requires that we maintain effective disclosure controls and procedures and internal controls over financial reporting. In order to maintain and improve the effectiveness of our disclosure controls and procedures and internal control over financial reporting, significant resources and management oversight will be required. This may divert management’s attention from other business concerns, which could have a material adverse effect on our business, financial condition, results of operations and cash flows.
| ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
| ITEM 2. | PROPERTIES |
We maintain our headquarters in West Chester, Ohio in a facility of approximately 29,100 square feet, which contains office space in addition to other technical and support functions and manufacturing. We currently pay monthly rent of approximately $20,200. In addition, we have two leases in West Chester, Ohio totaling 17,400 square feet of manufacturing, office and distribution space. We currently pay monthly rent for these properties of approximately $9,300. All West Chester leases will expire in August 2013. Internationally, we maintain office space in the Netherlands. Our monthly rent for this lease is approximately $2,700. We believe that our existing facilities are adequate to meet our immediate needs and that suitable additional space will be available in the future on commercially reasonable terms as needed.
| ITEM 3. | LEGAL PROCEEDINGS |
We are not party to any material pending or threatened litigation, except as described below:
Class Action Lawsuits
AtriCure, Inc. and certain of its current and former officers were named as defendants in a purported securities class action lawsuit filed in the United States District Court for the Southern District of New York (Levine v. AtriCure, Inc., Case No. 06 CV 14324 (United States District Court for the Southern District of New York)). The suit alleges violations of the federal securities laws and seeks damages on behalf of purchasers of our common stock during the period from our initial public offering in August 2005 through February 16, 2006. We filed a motion to dismiss the lawsuit for lack of subject matter jurisdiction. This motion was denied in September 2007, and a motion for reconsideration of that denial was denied in January 2009. Although we admit no wrongdoing, as of December 31, 2009, we have recorded a liability of $2.0 million, which represents an estimate of the potential defense and/or settlement costs. In addition, we recorded a related receivable of $2.0 million from our insurance carrier for the potential defense and/or settlement costs, as recovery is deemed probable.
37
Table of Contents
On December 12, 2008 AtriCure, Inc. and certain of its current executive officers were named in a putative class action lawsuit which is now captioned In re AtriCure, Inc. Securities Litigation, filed in the U.S. District Court for the Southern District of Ohio, Western Division. The plaintiffs allege violations of Sections10(b) and 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder and seek unspecified damages against AtriCure, Inc. and certain of its current executive officers. The plaintiffs allege, among other things, that the defendants issued materially false and misleading statements that failed to disclose that we improperly promoted certain products to physicians and caused the filing of false claims for reimbursement. The class period alleged ran from May 10, 2007 through October 31, 2008. In July 2009 we filed a motion to dismiss and, in September 2009, the plaintiffs filed their memorandum in opposition to our motion to dismiss to which we responded on November 9, 2009. On March 29, 2010, the court granted in part and denied in part our motion to dismiss and, in particular, dismissed the claim that we caused the filing of false claims for reimbursement. We intend to continue vigorously defending this lawsuit. Our liability, if any, resulting from this legal proceeding cannot be estimated and as such we have not recorded a liability within the Consolidated Financial Statements in relation to this matter.
Department of Justice Investigation
We received a letter on October 27, 2008 from the DOJ, informing us that they were conducting an investigation for potential False Claims Act and common law violations relating to our surgical ablation devices for the period beginning January 1, 2005. Specifically, the letter stated that the DOJ was investigating our marketing practices utilized in connection with our surgical ablation system to treat AF, a specific use outside the FDA’s 510(k) clearance. The letter also stated that they were investigating whether we instructed hospitals to bill Medicare for cardiac surgical ablation using incorrect billing codes. We cooperated with the investigation and operated our business in the ordinary course during the investigation. As of December 31, 2009, we reached a tentative settlement with the DOJ to resolve the investigation and recorded a liability and charged operating expenses for a total of $3,955,405, which represented the net present value of the proposed settlement amount to be paid to the DOJ, the relator, and relator’s counsel (total payments based on the settlement inclusive of interest were estimated to be $4,350,000, payable over five years).
On February 2, 2010 the settlement was finalized pursuant to the preliminary terms and we entered into a settlement agreement with the DOJ, the Office of Inspector General of the Department of Health and Human Services, or OIG, and the relator in the qui tam complaint discussed below. The settlement agreement definitively resolved all claims related to the DOJ investigation. We have not admitted nor will we admit any wrongdoing in connection with the settlement.
As part of the resolution, we also entered into a five year Corporate Integrity Agreement with the OIG. This agreement acknowledges the existence of our corporate compliance program and provides for certain other compliance-related activities during the five year term of the agreement. Those activities include specific written standards, monitoring, training, education, independent review, disclosure and reporting requirements.
Qui Tam Complaint
A copy of a qui tam complaint against us was unsealed on July 10, 2009. The qui tam complaint, filed in the U.S. District Court for the Southern District of Texas, was originally filed by the relator in August 2007. The complaint, which was related to the DOJ investigation, alleged a cause of action under the FCA relating to our alleged marketing practices in connection with our surgical cardiac ablation devices. In August 2009, the DOJ declined to intervene in the qui tam complaint. Nonetheless, the relator continued to pursue the litigation on behalf of the federal government. Additionally, upon a showing of good cause, the government had the right to intervene in the action at a later time. The qui tam complaint was settled in February 2010 in accordance with the DOJ settlement agreement.
The Company may from time to time become a party to additional legal proceedings.
| ITEM 4. | RESERVED |
38
Table of Contents
| ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Common Stock Market Price
Our common stock is traded on the NASDAQ Global Market under the symbol “ATRC”. The following table sets forth the high and low closing sales price of our common stock for 2009 and 2008:
| Price Range | ||||||
| High | Low | |||||
| 2009 |
||||||
| First Quarter |
$ | 2.20 | $ | 1.21 | ||
| Second Quarter |
$ | 3.90 | $ | 1.07 | ||
| Third Quarter |
$ | 5.15 | $ | 2.90 | ||
| Fourth Quarter |
$ | 6.05 | $ | 4.00 | ||
| Price Range | ||||||
| High | Low | |||||
| 2008 |
||||||
| First Quarter |
$ | 14.05 | $ | 10.85 | ||
| Second Quarter |
$ | 13.57 | $ | 9.72 | ||
| Third Quarter |
$ | 11.23 | $ | 9.65 | ||
| Fourth Quarter |
$ | 9.53 | $ | 2.00 | ||
As of March 1, 2010, the closing price of our common stock on the NASDAQ Global Market was $5.22 per share, and the number of stockholders of record was 71.
Dividend Policy
Since our incorporation, we have never declared or paid any dividends on our capital stock. Furthermore, pursuant to our credit facility, we are currently subject to certain restrictions on our ability to pay dividends. We currently expect to retain future earnings, if any, for use in the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future.
39
Table of Contents
Performance Graph
The following graph compares the cumulative total stockholder return on our common stock with the cumulative total return of the NASDAQ Composite and the NASDAQ Medical Equipment Index for the period beginning on August 5, 2005, our first day of trading after our initial public offering, and ending on December 31, 2009.
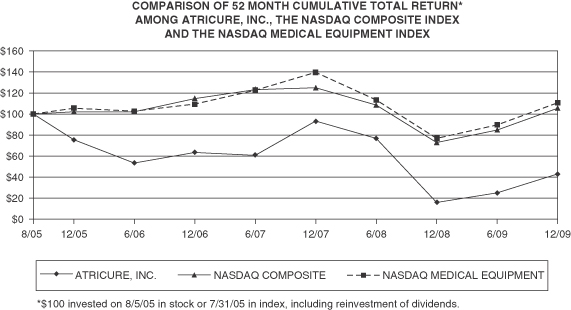
| * | This graph assumes that $100.00 was invested on August 5, 2005 in our common stock, the NASDAQ Composite Index and the NASDAQ Medical Equipment Index, and that all dividends are reinvested. No dividends have been declared or paid on our common stock. Stock performance shown in the above chart for our common stock is historical and should not be considered indicative of future price performance. |
| 8/05 | 12/05 | 6/06 | 12/06 | 6/07 | 12/07 | 6/08 | 12/08 | 6/09 | 12/09 | |||||||||||
| AtriCure, Inc. |
100.00 | 75.64 | 53.41 | 63.49 | 60.65 | 93.11 | 76.70 | 15.77 | 24.86 | 42.90 | ||||||||||
| NASDAQ Composite |
100.00 | 102.20 | 102.02 | 114.54 | 123.17 | 125.06 | 108.57 | 72.86 | 84.83 | 105.28 | ||||||||||
| NASDAQ Medical |
100.00 | 105.42 | 102.78 | 109.37 | 122.59 | 139.52 | 113.47 | 76.85 | 89.54 | 110.69 |
40
Table of Contents
| ITEM 6. | SELECTED FINANCIAL DATA |
The following table reflects selected financial data derived from our Consolidated Financial Statements for each of the last five years. The statement of operations data for the years ended December 31, 2009, 2008 and 2007, and the balance sheet data as of December 31, 2009 and 2008 are derived from our audited financial statements included in this Form 10-K. The statement of operations data for the years ended December 31, 2006 and 2005 and the balance sheet data as of December 31, 2007, 2006, and 2005 are derived from our audited financial statements not included in this Form 10-K and include the operations of Enable Medical Corporation since its acquisition on August 10, 2005. Historical results are not necessarily indicative of future results. The selected financial data set forth below should be read in conjunction with our financial statements, the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Form 10-K.
| Year Ended December 31, | ||||||||||||||||||||
| 2009(3) | 2008 | 2007 | 2006(2) | 2005(1) | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||||||
| Operating Results: |
||||||||||||||||||||
| Revenues |
$ | 54,534 | $ | 55,257 | $ | 48,309 | $ | 38,243 | $ | 30,957 | ||||||||||
| Cost of revenues |
12,751 | 13,225 | 10,137 | 7,626 | 8,057 | |||||||||||||||
| Gross profit |
41,783 | 42,033 | 38,172 | 30,617 | 22,900 | |||||||||||||||
| Gross margin |
76.6 | % | 76.1 | % | 79.0 | % | 80.1 | % | 74.0 | % | ||||||||||
| Operating expenses |
57,295 | 53,031 | 50,740 | 45,386 | 33,750 | |||||||||||||||
| Other (expense) income |
(1,042 | ) | 774 | 1,315 | 1,052 | (1,833 | ) | |||||||||||||
| Income tax benefit |
(59 | ) | (57 | ) | — | — | — | |||||||||||||
| Net loss |
(16,495 | ) | (10,167 | ) | (11,253 | ) | (13,717 | ) | (12,683 | ) | ||||||||||
| Basic and diluted net loss per share |
(1.13 | ) | (0.72 | ) | (0.84 | ) | (1.13 | ) | (2.10 | ) | ||||||||||
| Weighted average shares outstanding |
14,564 | 14,191 | 13,382 | 12,137 | 6,025 | |||||||||||||||
| Financial Position: |
||||||||||||||||||||
| Cash, cash equivalents and short-term investments |
$ | 15,722 | $ | 11,448 | $ | 20,007 | $ | 19,488 | $ | 33,802 | ||||||||||
| Restricted cash and cash equivalents |
— | 6,000 | — | — | — | |||||||||||||||
| Working capital |
19,545 | 17,997 | 24,624 | 23,031 | 35,903 | |||||||||||||||
| Total assets |
34,982 | 43,369 | 46,071 | 39,128 | 50,040 | |||||||||||||||
| Long-term debt and capital leases |
2,670 | 6,037 | 282 | 693 | 1,084 | |||||||||||||||
| Accumulated deficit |
(93,970 | ) | (77,475 | ) | (67,308 | ) | (56,055 | ) | (42,337 | ) | ||||||||||
| Stockholders’ equity |
17,090 | 29,119 | 36,237 | 30,694 | 43,183 | |||||||||||||||
| (1) | On August 10, 2005 we acquired Enable Medical Corporation. |
| (2) | Effective January 1, 2006, we adopted new guidance under FASB ASC 718, “Compensation—Stock Compensation” (“ASC 718”), which requires the measurement and recognition of compensation cost at fair value for all share-based payments. We adopted the new guidance using the modified prospective transition method and, as a result, did not retroactively adjust results from prior periods. For further discussion, see the section entitled “Share-Based Employee Compensation” in Note 1, “Description of Business and Summary of Significant Accounting Policies,” to our Consolidated Financial Statements. |
| (3) | As a result of a reduction in our market capitalization during the first quarter of 2009, we believed an indication of impairment existed and as such, performed an interim analysis of our goodwill as of March 31, 2009 as required by FASB ASC 350, “Goodwill and Other Intangible Assets” (“ASC 350”). The analysis concluded that the carrying value of our goodwill exceeded the estimated fair value, and as such, we recognized a full impairment loss of $6.8 million during 2009. See Note 5, “Goodwill and Intangible Assets” to our Consolidated Financial Statements. Also during 2009, we recorded $4.0 million in expense related to a settlement with the DOJ. See Note 10, “Commitments and Contingencies,” to our Consolidated Financial Statements. |
41
Table of Contents
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the accompanying financial statements and notes thereto contained in Item 8, “Financial Statements and Supplementary Data,” to provide an understanding of our results of operations, financial condition and cash flows. This discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. The actual results may differ from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those set forth under Item 1A “Risk Factors”, the cautionary statement regarding forward-looking statements at the beginning of Part I and elsewhere in this Form 10-K.
Results of Operations
Year Ended December 31, 2009 compared to December 31, 2008
The following table sets forth, for the periods indicated, our results of operations expressed as dollar amounts and as percentages of total revenues:
| Year Ended December 31, | ||||||||||||||
| 2009 | 2008 | |||||||||||||
| % of | % of | |||||||||||||
| Amount | Revenues | Amount | Revenues | |||||||||||
| (dollars in thousands) | ||||||||||||||
| Revenues |
$ | 54,534 | 100.0 | % | $ | 55,257 | 100.0 | % | ||||||
| Cost of revenues |
12,751 | 23.4 | % | 13,225 | 23.9 | % | ||||||||
| Gross profit |
41,783 | 76.6 | % | 42,033 | 76.1 | % | ||||||||
| Operating expenses: |
||||||||||||||
| Research and development expenses |
11,415 | 20.9 | % | 10,609 | 19.2 | % | ||||||||
| Selling, general and administrative expenses |
35,113 | 64.4 | % | 42,422 | 76.8 | % | ||||||||
| Goodwill impairment |
6,812 | 12.5 | % | — | — | |||||||||
| Settlement reserve |
3,955 | 7.3 | % | — | — | |||||||||
| Total operating expenses |
57,295 | 105.1 | % | 53,031 | 96.0 | % | ||||||||
| Loss from operations |
(15,512 | ) | -28.4 | % | (10,998 | ) | -19.9 | % | ||||||
| Other (expense) income: |
||||||||||||||
| Interest expense |
(812 | ) | -1.5 | % | (364 | ) | -0.7 | % | ||||||
| Interest income |
51 | 0.1 | % | 382 | 0.7 | % | ||||||||
| Other |
(281 | ) | -0.5 | % | 756 | 1.4 | % | |||||||
| Other (expense) income |
(1,042 | ) | -1.9 | % | 774 | 1.4 | % | |||||||
| Loss before income tax benefit |
(16,554 | ) | -30.4 | % | (10,225 | ) | -18.5 | % | ||||||
| Income tax benefit |
(59 | ) | -0.1 | % | (57 | ) | -0.1 | % | ||||||
| Net loss |
$ | (16,495 | ) | -30.2 | % | $ | (10,167 | ) | -18.6 | % | ||||
Revenues. Total revenues decreased $0.7 million, or 1.3%, from $55.3 million in 2008 to $54.5 million in 2009. Revenues from domestic sales decreased $2.8 million or 6.0% and revenues from international sales increased $2.1 million or 24.9%. The decrease in domestic revenues was due primarily to a decrease in revenues from the sale of minimally invasive products. During 2008, we introduced and sold our ORLab systems to a large majority of customers who utilize our minimally invasive products, which is the targeted customer for our ORLab system. Neutralizing the impact of foreign currency exchange rate fluctuations, total revenues decreased 0.8% as compared to the reported 1.3%, and international revenues grew 28.2% as compared to the reported 24.9%.
42
Table of Contents
Cost of revenues. Cost of revenues decreased $0.5 million, from $13.2 million in 2008 to $12.8 million in 2009. The decrease in cost of revenues was primarily due to a reduction in total revenues and a reduction in revenues from the sale of capital equipment (including our ORLab system), which carry a higher cost of revenues than our disposable products. As a percentage of revenues, cost of revenues decreased from 23.9% for the year ended December 31, 2008 to 23.4% for the year ended December 31, 2009. The decrease in cost of revenues as a percentage of revenues was due to a reduction in revenues from capital equipment, partially offset by an increased mix of international sales, which carry a lower average selling price than domestic sales and the introduction of new products, which generally carry a higher product cost initially.
Research and development expenses. Research and development expenses increased $0.8 million, from $10.6 million in 2008 to $11.4 million in 2009. As a percentage of revenues, research and development expenses increased from 19.2% in 2008 to 20.9% in 2009. The increase was primarily attributable to a $1.2 million increase in consulting expenses to support clinical trial activities, an increase in clinical trial expense of $0.2 million and an increase in share-based compensation of $0.4 million, partially offset by a decrease in product development project costs of $0.8 million.
Selling, general and administrative expenses. Selling, general and administrative expenses decreased $7.3 million, from $42.4 million in 2008 to $35.1 million in 2009. The decrease was primarily due to lower headcount-related and travel expenses of $6.1 million, primarily the result of a reduction in our sales force which occurred during the fourth quarter of 2008, and a $0.7 million decrease in marketing expenses due primarily to reduced spending in support of tradeshow activities. These reductions in expenses were partially offset by an increase in legal expense of $0.5 million, related primarily to our DOJ investigation, and an increase in share-based compensation expense of $0.7 million, due primarily to the conversion of 2008 executive and management bonuses to be paid in restricted stock. As a percentage of total revenues, selling, general and administrative expenses decreased from 76.8% in 2008 to 64.4% in 2009.
Goodwill impairment. As a result of a reduction in our market capitalization during the first quarter of 2009, we believed an indication of impairment existed and we performed an interim analysis of our goodwill as of March 31, 2009. The analysis concluded that the carrying value of our goodwill exceeded the estimated fair value and we recognized a full impairment loss of $6.8 million as of March 31, 2009.
Settlement reserve. During 2009, in conjunction with the DOJ investigation and related qui tam complaint we recorded a settlement reserve of $4.0 million, which represents the net present value of the settlement amount. See Note 10, “Commitments and Contingencies” to our Consolidated Financial Statements.
Net interest income (expense). Net interest income (expense) decreased $0.8 million from income of $18,214 in 2008 to expense of $0.8 million in 2009. The decrease in income was primarily due to the write-off of deferred financing costs of $0.1 million in connection with the termination of our credit facility with PNC Bank (formerly National City Bank), increased interest expense associated with borrowings under the term loan component of our new credit facility of $0.2 million (driven by a higher effective interest rate and an increase in average borrowings outstanding) and $0.2 million related to the amortization of the discount on long-term debt for the warrant issued in conjunction with our new credit facility.
Other (expense) income. Other (expense) income consists of foreign currency transaction (losses) gains, grant income and non-employee option (expense) income related to the fair market value change for fully vested options outstanding for consultants which are accounted for as free-standing derivatives. In 2009, other expense of $0.3 million included $0.2 million related to foreign currency transaction losses associated with partial settlements of intercompany balances and $0.1 million of certain non-employee option expense due to an increase in the fair market value of the options. Other income of $0.8 million in 2008 included income of $0.5 million associated with a reduction in fair market value of certain non-employee stock options and $0.3 million in grant income related to our grant agreement with the Cleveland Clinic Foundation, partially offset by $0.1 million related to foreign currency transaction losses associated with the partial settlement of intercompany balances.
43
Table of Contents
Year Ended December 31, 2008 compared to December 31, 2007
The following table sets forth, for the periods indicated, our results of operations expressed as dollar amounts and as percentages of total revenues:
| Year Ended December 31, | ||||||||||||||
| 2008 | 2007 | |||||||||||||
| % of | % of | |||||||||||||
| Amount | Revenues | Amount | Revenues | |||||||||||
| (dollars in thousands) | ||||||||||||||
| Revenues |
$ | 55,257 | 100.0 | % | $ | 48,309 | 100.0 | % | ||||||
| Cost of revenues |
13,225 | 23.9 | % | 10,137 | 21.0 | % | ||||||||
| Gross profit |
42,033 | 76.1 | % | 38,172 | 79.0 | % | ||||||||
| Operating expenses: |
||||||||||||||
| Research and development expenses |
10,609 | 19.2 | % | 10,987 | 22.7 | % | ||||||||
| Selling, general and administrative expenses |
42,422 | 76.8 | % | 39,753 | 82.3 | % | ||||||||
| Total operating expenses |
53,031 | 96.0 | % | 50,740 | 105.0 | % | ||||||||
| Loss from operations |
(10,998 | ) | -19.9 | % | (12,568 | ) | -26.0 | % | ||||||
| Other income (expense): |
||||||||||||||
| Interest expense |
(364 | ) | -0.7 | % | (213 | ) | -0.4 | % | ||||||
| Interest income |
382 | 0.7 | % | 948 | 2.0 | % | ||||||||
| Other |
756 | 1.4 | % | 580 | 1.2 | % | ||||||||
| Other income |
774 | 1.4 | % | 1,315 | 2.7 | % | ||||||||
| Loss before income tax benefit |
(10,225 | ) | -18.5 | % | (11,253 | ) | -23.3 | % | ||||||
| Income tax benefit |
(57 | ) | -0.1 | % | — | — | % | |||||||
| Net loss |
$ | (10,167 | ) | -18.4 | % | $ | (11,253 | ) | -23.3 | % | ||||
Revenues. Total revenues increased $6.9 million, or 14.4%, from $48.3 million in 2007 to $55.3 million in 2008. The increase in revenues was due primarily to the sale of new products and an increase in unit sales from existing products to international customers.
Cost of revenues. Cost of revenues increased $3.1 million, from $10.1 million in 2007 to $13.2 million in 2008, primarily due to an increase in the total number of units sold and a change in product mix. As a percentage of revenues, cost of revenues increased from 21.0% for the year ended December 31, 2007 to 23.9% for the year ended December 31, 2008. The increase in cost of revenues as a percentage of revenues was primarily due to the introduction and sale of new products, including our ORLab system, which carries a higher cost of revenues than our disposable products and an increased mix of international sales, which have a lower average selling price than sales in the United States.
Research and development expenses. Research and development expenses decreased $0.4 million, from $11.0 million in 2007 to $10.6 million in 2008. As a percentage of revenues, research and development expenses decreased from 22.7% in 2007 to 19.2% in 2008. The decrease was primarily attributable to the redeployment during 2007 of several individuals who previously focused on clinical activities to selling activities of approximately $0.7 million, which are recorded as a component of selling, general and administrative expenses, partially offset by increased expenditures in support of clinical trials.
Selling, general and administrative expenses. Selling, general and administrative expenses increased $2.7 million, from $39.8 million in 2007 to $42.4 million in 2008. The increase was primarily attributable to an increase in personnel related costs of approximately $2.9 million and an increase in non-cash compensation of $0.6 million, partially offset by reductions in overall general administrative expenses. As a percentage of total revenues, selling, general and administrative expenses decreased from 82.3% in 2007 to 76.8% in 2008.
44
Table of Contents
Net interest income. Net interest income decreased $0.7 million, from $0.7 million in 2007, due primarily to a decrease in average net cash, cash equivalents, investments and restricted cash and cash equivalents outstanding, an increase in debt outstanding and a reduced average effective interest rate.
Other income. Other income consists of grant income, net foreign currency transaction gains and non-employee option expense. Grant income decreased $0.3 million, from $0.6 million in 2007 to $0.3 million in 2008 and consisted of income related to expense sharing under a grant for research and development related activities. Foreign currency transaction loss was $0.1 million in 2008 in connection with a partial settlement of our intercompany payable balance with our subsidiary. Non-employee option income was $0.5 million in 2008 compared to expense of $0.2 million in 2007 and is related to the fair market value change for fully vested options outstanding for consultants, which are accounted for as free standing derivatives.
Liquidity and Capital Resources
As of December 31, 2009 we had cash, cash equivalents and short-term investments of $15.7 million and short-term and long-term debt of $4.9 million (net of $0.3 million in warrants), resulting in a net cash position of $10.8 million. We had working capital of $19.5 million and an accumulated deficit of $94.0 million.
On May 30, 2007, we completed a private placement of 1,789,649 shares of common stock and received net proceeds (after deducting transaction-related expenses) of $15.2 million. The purpose of the offering was to raise additional funds for working capital and general purposes, including research and development activities and potential acquisitions or other strategic initiatives.
Cash flows provided by (used in) operating activities. Net cash provided by operating activities was $0.4 million in 2009 compared to net cash used in operating activities of $5.7 million and $8.1 million in 2008 and 2007, respectively. Net cash provided by operating activities in 2009 was primarily attributable to a goodwill impairment charge of $6.8 million, the recording of a settlement reserve related to the DOJ investigation of $4.0 million, non-cash charges related to share-based compensation of $3.9 million, depreciation and amortization of $2.7 million and a decrease in inventory of $1.5 million. These changes in cash were partially offset by a net loss of $16.5 million, a decrease in accounts payable of $1.5 million and an increase in accounts receivable of $0.7 million. Net cash used in operating activities in 2008 was primarily attributable to the net loss of $10.2 million and cash used for an increase in inventory of $1.1 million, due primarily to an expansion of our product offering and a reduction in fourth quarter 2008 sales, and a reduction in accounts receivable due primarily to an improvement in days sales outstanding and a reduction in fourth quarter 2008 sales. These changes in cash were partially offset by depreciation and amortization of $2.8 million and non-cash charges related to stock-based compensation of $2.7 million. Net cash used in operating activities in 2007 was primarily attributable to the net loss of $11.3 million and increases in accounts receivable, inventory and other current assets of $0.6 million, $1.4 million and $0.2 million, respectively, which increased as revenues increased and we expanded our product offering. Those increases were partially offset by adjustments for depreciation and amortization of $2.3 million and non-cash charges related to stock-based compensation of $1.9 million and increases in payables and accrued liabilities of $0.9 million, due primarily to the growth in the business and expansion of our product offering.
Cash flows used in investing activities. Net cash used in investing activities was $2.2 million, $1.2 million and $8.8 million in 2009, 2008 and 2007, respectively. Cash used in investing activities reflected purchases of property and equipment of $1.4 million, $1.7 million and $3.0 million for 2009, 2008 and 2007, respectively, the net purchases and maturities of investments of $6.8 million, ($7.0) million and $2.4 million for 2009, 2008 and 2007, respectively and, in 2008 and 2007, cash paid for an acquisition, net of cash acquired of $0.4 million and $3.3 million, respectively, for the Frigitronics CCS-200 product line. During 2007, the increase in the purchase of property, plant and equipment was primarily due to the introduction of our ASB in accordance with the launch of our Isolator Synergy platform. The ASB is hardware which we generally loan to our direct customers.
45
Table of Contents
Cash flows (used in) provided by financing activities. Net cash used in financing activities was $0.9 million in 2009 compared with net cash provided by financing activities of $5.3 million and $15.0 million in 2008 and 2007, respectively. In 2009, cash flows used in financing activities included $7.5 million in payments on debt and capital leases, partially offset by borrowing under our credit facility with Silicon Valley Bank of $6.5 million. In 2008, cash flows provided by financing activities included $6.0 million related to our credit facility with PNC Bank (formerly National City Bank), partially offset by the repayment of existing debt, capital leases and debt fees of $1.1 million. In 2007, cash flows provided by financing activities included $15.2 million in net proceeds from our May 2007 private placement of 1.8 million shares of our common stock.
Credit facility. On May 1, 2009, we entered into a Loan and Security Agreement (the “Agreement”) with Silicon Valley Bank (the “Bank”) that provides a term loan and a revolving credit facility under which we can borrow a maximum of $10.0 million. We have borrowed the maximum amount of $6.5 million under the term loan. We can borrow up to $10.0 million under the revolving loan facility with the availability subject to a borrowing base formula. The Agreement also includes up to a $1.0 million sublimit for stand-by letters of credit. The Agreement matures on April 30, 2012 and is secured by all of our assets, including intellectual property.
Interest on the term loan accrues at a rate of 10.0% per year, and interest on the revolving loan will accrue at a fluctuating rate equal to the Bank’s announced prime rate of interest, subject to a floor of 4.0%, plus between 1.0% and 2.0%, depending on our Adjusted Quick Ratio (as defined in the Agreement). Principal on the term loan will be amortized over 36 months of equal principal payments, plus applicable interest. In addition, in connection with the term loan under the Agreement, the Bank received a warrant to purchase 371,732 shares of our common stock at $1.224 per share, exercisable for a term of 10 years. On October 6, 2009 the Warrant was exercised via a net share settlement and 276,143 shares were issued.
On November 4, 2009 and effective September 30, 2009, we entered into a Consent, Waiver and First Loan Modification Agreement (“First Amendment”) with Silicon Valley Bank, which amended, among other things, the financial covenants in the Agreement. On March 26, 2010, we entered into a Waiver and Second Loan Modification Agreement (“Second Amendment”) with Silicon Valley Bank, which amended, among other things, the financial covenants in the Agreement and waived a compliance violation which occurred during February 2010.
As of December 31, 2009 we had no borrowings under our revolving credit facility and borrowing availability of $1.1 million. Also as of December 31, 2009, we had borrowings of $5.1 million under the term loan which includes approximately $2.2 million classified as current. We are required to make monthly principal payments on the term loan of $0.2 million plus interest. The warrant associated with our term loan was recorded as discount on long-term debt at its intrinsic value and is being amortized over the term of the loan and is reflected as a reduction of long-term debt. Amortization expense totaled $0.2 million in 2009. The effective interest rate on borrowings under the term loan, including amortization of the warrant and debt issuance costs, is 15.2%. As of December 31, 2009, the Company had an outstanding letter of credit of $250,000 issued to its corporate credit card program provider which expires on July 31, 2010.
On July 1, 2008 we entered into a two-year credit facility with PNC Bank (formerly National City Bank), which was terminated effective May 1, 2009 and the outstanding balance was repaid in full. As of December 31, 2008, $6.0 million was outstanding under the credit facility and $6.0 million was held as restricted cash and cash equivalents and reported as long-term liabilities and assets, respectively. On July 2, 2008, as a condition to entering into the credit facility, we repaid in full our outstanding indebtedness to Lighthouse Capital Partners V.L.P. We paid $0.7 million to Lighthouse, which consisted of outstanding principal, accrued interest and a final payment fee due at maturity.
Unsecured promissory note. Under the terms and conditions of the Bill of Sale and Assignment Agreement with CooperSurgical, Inc. (“Cooper”) we entered into an unsecured promissory note agreement for $0.4 million, which bore interest at 5.0%. The note was paid in full in January 2008 and was recorded as additional cash paid for acquisition in our Consolidated Statement of Cash Flows during 2008.
46
Table of Contents
Uses of liquidity and capital resources. Our future capital requirements depend on a number of factors, including possible acquisitions and joint ventures, the rate of market acceptance of our current and future products, the resources we devote to developing and supporting our products, future expenses to expand and support our sales and marketing efforts, costs relating to changes in regulatory policies or laws that affect our operations and costs of filing, costs associated with clinical trials and securing regulatory approval for new products, and costs associated with prosecuting, defending and enforcing our intellectual property rights. Global economic turmoil may adversely impact our revenue, access to the capital markets or future demand for our products.
We believe that our current cash, cash equivalents and short-term investments, along with the cash we expect we may generate or use for operations or access via our credit facility, will be sufficient to meet our anticipated cash needs for working capital and capital expenditures for at least the next 12 months. If these sources of cash are insufficient to satisfy our liquidity requirements, we may seek to sell additional equity or debt securities or obtain a revised or additional credit facility. The sale of additional equity or convertible debt securities could result in dilution to our stockholders. If additional funds are raised through the issuance of debt securities, these securities could have rights senior to those associated with our common stock and could contain covenants that would restrict our operations. Additional financing may not be available at all, or in amounts or terms acceptable to us. If we are unable to obtain this additional financing, we may be required to reduce the scope of our planned research and development and selling and marketing efforts.
Contractual Obligations and Commitments
DOJ Settlement
On February 2, 2010, we entered into a settlement agreement among the Company, the DOJ, the OIG and the relator in the DOJ investigation and qui tam complaint (“Settlement Agreement”). The Settlement Agreement and dismissal of the qui tam complaint definitively resolve all claims related to the DOJ investigation and qui tam complaint. We have not admitted nor will we admit to any wrongdoing in connection with the settlement.
The Settlement Agreement provides that we will pay a settlement amount of $3,766,623 (total payments based on the settlement inclusive of interest are $4,150,000) and legal fees to counsel for the relator of $200,000. Payment of the settlement amount will be made over a five-year period and $500,000 was paid in February 2010. A majority of the amount payable is payable during the fourth and fifth years. Payment of the relator legal fees will be made in ratable quarterly payments over four years and the first payment was made in February 2010.
As part of the resolution, we also entered into a five-year Corporate Integrity Agreement with OIG. This agreement acknowledges the existence of our corporate compliance program and provides for certain other compliance-related activities during the five-year term of the agreement. Those activities include specific written standards, monitoring, training, education, independent review, disclosure and reporting requirements.
Purchase Agreement
On June 15, 2007 we entered into a purchase agreement with MicroPace Pty Ltd Inc., (“MicroPace”), which was amended in June 2008. Under the amended agreement, MicroPace produced a derivative of one of their products tailored for the cardiac surgical environment, known as the “MicroPace ORLab” for worldwide distribution by us. Pursuant to the terms of the amended agreement, in order for us to retain exclusive distribution rights, we are required to purchase a minimum of 70 units during 2008 and 80 units for each 2009 and 2010. As of December 31, 2009, we had purchased a total of 158 units. Units purchased in excess of yearly minimums in a year reduce future minimum purchase requirements. We have 72 units to purchase by December 31, 2010 in order to retain exclusive distribution rights.
47
Table of Contents
Life Support Technology, LST b.v.
In September 2007 we settled multiple proceedings with Life Support Technology, LST b.v., or L.S.T., a former distributor of AtriCure products in Europe. The settlement agreement provides for us to pay LST €257,360 (euros) in 16 payments of €16,085, with the final payment due January 1, 2011. If the U.S. Dollar to Euro conversion rate on any of the 16 payment due dates set forth in the agreement is less than $1.36 to the Euro, we will owe LST additional compensation, up to a maximum of €28,310, which reduces over time. We have recorded liabilities of $109,755 and $184,632 as of December 31, 2009 and December 31, 2008, respectively.
The following sets forth our approximate aggregate obligations at December 31, 2009 for future payments under contracts and other contingent commitments:
| Contractual Obligations |
Total | Less than 1 year | 1-3 years | 3-5 years | ||||||||
| Long-term debt and capital leases(1) |
$ | 5,819,178 | $ | 2,648,325 | $ | 3,160,135 | $ | 10,718 | ||||
| DOJ Settlement(2) |
4,350,000 | 562,500 | 1,250,000 | 2,537,500 | ||||||||
| Purchase obligations(3) |
2,162,800 | 2,162,800 | — | — | ||||||||
| Operating leases(4) |
1,914,889 | 628,224 | 987,124 | 299,541 | ||||||||
| Royalty obligations(5) |
1,000,000 | 200,000 | 400,000 | 400,000 | ||||||||
| LST settlement agreement |
109,755 | 87,500 | 22,255 | — | ||||||||
| Research grants |
629,960 | 629,960 | — | — | ||||||||
| Physician consulting agreements(6) |
60,000 | 60,000 | — | — | ||||||||
| Total contractual obligations |
$ | 16,046,582 | $ | 6,979,309 | $ | 5,819,514 | $ | 3,247,759 | ||||
| (1) | Long-term debt represents principal repayment related to our term loan that matures in 2012. Interest on the term loan accrues at a rate of 10% per year and is included above. Capital leases consist of principal and interest payments related to computer equipment. |
| (2) | The DOJ settlement provides that we pay a settlement amount of $3,955,405, which represents the net present value of the settlement amount to be paid to the DOJ, the relator, and relator’s counsel (total payments based on the settlement inclusive of interest are $4,350,000 and payable over five years). |
| (3) | Represents estimated minimum number of units to be purchased from MicroPace for the ORLab units in order to maintain exclusive distribution rights. Represents 2010 purchase of an additional 72 units. In addition, includes purchase orders with other vendors. |
| (4) | Represents lease commitments under various operating leases. |
| (5) | Represents minimum payments required under the terms of a royalty agreement, not to exceed in aggregate $2.0 million from January 1, 2010 through December 31, 2015. The table above reflects the minimum amount due under the terms of the agreement. Our other royalty agreement is for a new product offering, and we are unable to estimate the expense associated with the agreement. As such, no estimate of future obligation has been included in this table. See Note 10, “Commitments and Contingencies” to our Consolidated Financial Statements. |
| (6) | Represents estimated minimum payments to various physicians for consulting services. |
Off-Balance-Sheet Arrangements
As of December 31, 2009 we had operating lease agreements not recorded on the Consolidated Balance Sheets. Operating leases are utilized in the normal course of business.
Also as of December 31, 2009, we had an outstanding letter of credit of $250,000, issued to our corporate credit card program provider. The letter of credit expires on July 31, 2010.
Inflation
Inflation has not had a significant impact on our historical operations and we do not expect it to have a significant impact on our results of operations or financial condition in the foreseeable future.
48
Table of Contents
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of financial statements requires management to make estimates and judgments that affect the reported amounts of assets and liabilities, revenues and expenses, and disclosures of contingent assets and liabilities at the date of the financial statements. On a periodic basis, we evaluate our estimates, including those related to sales returns and allowances, accounts receivable, inventories and share-based compensation. We use authoritative pronouncements, historical experience and other assumptions as the basis for making estimates. Actual results could differ from those estimates under different assumptions or conditions.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements.
Share-Based Employee Compensation—We follow FASB ASC 718 “Compensation—Stock Compensation” (“ASC 718”), to record share-based compensation for all share-based awards made to employees and directors, including employee stock options, restricted stock, performance shares and employee stock purchases related to an employee stock purchase plan, based on estimated fair values. Our employee share-based compensation expense recognized under ASC 718 for the years ended December 31, 2009, 2008, and 2007 was $3.9 million, $2.7 million and $1.5 million, respectively on a before and after tax basis.
FASB ASC 718 requires companies to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in our Consolidated Statement of Operations. The expense has been reduced for estimated forfeitures. FASB ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
We estimate the fair value of options on the date of grant using the Black-Scholes option-pricing model (“Black-Scholes model”). Our determination of fair value of share-based payment awards on the date of grant using an option-pricing model is affected by our stock price as well as assumptions regarding a number of highly complex and subjective variables. These variables include but are not limited to our and the peer group’s expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors.
We estimate the fair value of restricted stock and performance share awards based upon the grant date closing market price of our common stock. Our determination of fair value is affected by our stock price as well as assumptions regarding the number of shares expected to be granted and, in the case of performance shares, the likelihood that the performance measures will be achieved.
We also have an employee stock purchase plan (“ESPP”) which is available to all eligible employees as defined by the ESPP plan document. Under the ESPP, shares of our common stock may be purchased at a discount. We estimate the number of shares to be purchased under the Plan and record compensation expense based upon the fair value of the stock at the beginning of the purchase period using the Black-Scholes model.
Certain of our share-based arrangements are outside the scope of FASB ASC 718 and are subject to FASB ASC 815, “Derivatives and Hedging” (“ASC 815”), which requires vested stock options held by certain non-employee consultants to be accounted for as liability awards until these awards are exercised or forfeited. The fair value of these awards is remeasured at each financial statement date until the awards are settled or expire. During the year ended December 31, 2009, $0.1 million of expense was recorded as a result of the remeasurement of the fair value of these awards compared with $0.5 million of income in 2008 and $0.2 million of expense in 2007. As of December 31, 2009 and 2008, respectively, options to acquire 52,359 and 54,660 shares of common stock held by non-employee consultants remained unexercised and a liability of $180,288 and $40,368, respectively, was included in accrued liabilities in the Consolidated Balance Sheets.
49
Table of Contents
Also outside the scope of FASB ASC 718, in connection with our $6.5 million term loan, we issued a warrant to purchase shares of our common stock. The warrant allowed Silicon Valley to purchase 371,732 shares of our common stock at $1.224 per share, exercisable for a term of 10 years. The warrant was immediately exercisable and provided for net share settlement. We determined this arrangement met the requirements of FASB ASC 715, “Compensation-Retirement Benefits” (“ASC 715”), and other relevant literature and, therefore, the warrant was recorded at its grant date intrinsic value and was classified as an equity transaction. The intrinsic value of the warrant was recorded as a reduction of debt and is being amortized over the life of the credit facility. On October 6, 2009, the warrant was exercised through a net share settlement and 276,143 shares were issued.
Revenue Recognition—Revenues are generated primarily from the sale of our disposable surgical devices. Pursuant to our standard terms of sale, revenues are recognized when title to the goods and risk of loss transfers to customers and there are no remaining obligations that will affect the customers’ final acceptance of the sale. Generally, our standard terms of sale define the transfer of title and risk of loss to occur upon shipment to the respective customer. We generally do not maintain any post-shipping obligations to the recipients of the products. Typically, no installation, calibration or testing of this equipment is performed by us subsequent to shipment to the customer in order to render it operational. Product revenues include shipping revenues of $0.7 million, $0.8 million and $0.5 million in 2009, 2008, and 2007, respectively. Cost of freight for shipments made to customers is included in cost of revenues. Sales and other value-added taxes collected from customers and remitted to governmental authorities are excluded from product revenues. We sell our products primarily through our direct sales force and through our wholly-owned subsidiary AtriCure Europe, B.V. Terms of sale are generally consistent for both end-users and distributors and payment terms are generally net 30 days for end-users and net 60 days for distributors.
We account for revenues in accordance with FASB ASC 605, “Revenue Recognition” (“ASC 605”). We determine the timing of revenue recognition based upon factors such as passage of title, installation, payment terms and ability to return products. We recognize revenue when all of the following criteria are met: (i) persuasive evidence that an arrangement exists; (ii) delivery of the products and/or services has occurred; (iii) the selling price is fixed or determinable; and (iv) collectability is reasonably assured.
Allowance for Uncollectible Accounts Receivable—We evaluate the collectability of accounts receivable in order to determine the appropriate reserve for doubtful accounts. In determining the amount of the reserve, we consider aging of account balances, historical credit losses, customer-specific information, and other relevant factors. Increases to the allowance for doubtful accounts results in a corresponding expense. Periodically, we review accounts receivable and adjust the allowance based on current circumstances and charge-off uncollectible receivables against the allowance when all attempts to collect the receivable have failed.
Inventories—Inventories are stated at the lower of cost or market using the first-in, first-out (“FIFO”) cost method and consist of raw materials, work in process, and finished goods. A reserve for inventory is estimated and recorded for excess, slow-moving and obsolete inventory, as well as inventory with a carrying value in excess of its net realizable value. Write-offs are recorded when the product is destroyed. We review inventory on hand at least quarterly and record provisions for excess and obsolete inventory based on several factors including our current assessment of future product demand, anticipated release of new products into the market, historical experience and product expiration. Our industry is characterized by rapid product development and frequent new product introductions. Uncertain timing of product approvals, variability in product launch strategies, product recalls and variation in product utilization all impact the estimates related to excess and obsolete inventory.
Property and Equipment—Property and equipment is stated at cost, less accumulated depreciation. Depreciation is computed on the straight-line method for financial reporting purposes over the estimated useful lives of the assets. The estimated useful life by major asset category is the following: machinery and equipment is three to seven years, computer and other office equipment is three years, furniture and fixtures is three to seven years, and leasehold improvements and leased equipment under a capital lease are the shorter of their useful life or remaining lease term. Maintenance and repair costs are expensed as incurred.
50
Table of Contents
Included in property and equipment are generators and other capital equipment (such as our ASB) that are loaned at no cost to direct customers that use our disposable products. These generators are depreciated over a three year period and such depreciation is included in cost of revenues. The total of such depreciation was $1.1 million, $1.1 million and $0.8 million in 2009, 2008, and 2007, respectively.
Impairment of Long-Lived Assets (Other than Goodwill)—We review property and equipment and definite-lived intangibles for impairment using our best estimates based on reasonable and supportable assumptions and projections in accordance with FASB ASC 360, “Property, Plant and Equipment” (“ASC 360”). We recorded a charge of $5,517 in 2009 for the impairment of fixed assets. We recorded a charge of $0.2 million in 2008 for the impairment of fixed assets and machinery and equipment related to discontinued product lines. In 2007, we recorded a charge of $0.1 million for the impairment of obsolete machinery and equipment and tooling.
Goodwill and Intangible Assets—As of December 31, 2008, we had $6.8 million in goodwill recorded. Goodwill represents the excess of costs over the fair value of the net assets acquired in business combinations. We historically tested goodwill for impairment annually during our fourth quarter, or more frequently if impairment indicators were present or changes in circumstances indicated that the carrying value of the asset exceeded the estimated fair value. ASC 350, “Intangibles—Goodwill and Other” (“ASC 350”) requires a two-step approach to determine any potential goodwill impairment. The first step (Step 1) requires a comparison of the carrying value of the reporting unit to its fair value. Goodwill is considered potentially impaired if the carrying value of the reporting unit is greater than the estimated fair value. If potential impairment exists based upon completion of Step 1, Step 2 must be completed, which compares the implied fair value of a reporting unit’s goodwill to its carrying value. Step 2 involves an analysis allocating the fair value determined in Step 1 (as if it was the purchase price in a business combination). If the calculated fair value of the goodwill resulting from this allocation is lower than the carrying value of the goodwill of the reporting unit, an impairment loss is recorded. We recorded a full impairment loss related to our goodwill during the three months ended March 31, 2009, based on the results of our Step 1 analysis. During the three month period ended June 30, 2009 we performed our Step 2 analysis and concluded that the charge recorded was appropriate. See Note 5, “Goodwill and Intangible Assets” for additional information related to this impairment.
Intangible assets with determinable useful lives are amortized on a straight-line basis over the estimated periods benefited, which range from four to eight years.
Income Taxes—Income taxes are computed using the asset and liability method in accordance with FASB ASC 740, “Income Taxes” (“ASC 740”), under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of our assets and liabilities. Deferred taxes are measured using provisions of currently enacted tax laws. A valuation allowance against deferred tax assets is recorded when it is more likely than not that such assets will not be fully realized. Tax credits are accounted for as a reduction of income taxes in the year in which the credit originates.
Our estimate of the valuation allowance for deferred tax assets requires us to make significant estimates and judgments about our future operating results. Our ability to realize the deferred tax assets depends on our future taxable income as well as limitations on their utilization. A deferred tax asset is reduced by a valuation allowance if it is more likely than not that some portion or all of the deferred tax asset will not be realized prior to its expiration. The projections of our operating results on which the establishment of a valuation allowance is based involve significant estimates regarding future demand for our products, competitive conditions, product development efforts, approvals of regulatory agencies and product cost. If actual results differ from these projections, or if our expectations of future results change, it may be necessary to adjust the valuation allowance.
Recent Accounting Pronouncements
In April 2009, the FASB issued a staff position within ASC 805, “Business Combinations” (“ASC 805”), amending and clarifying the new business combination standard to address application issues associated with initial recognition and measurement, subsequent measurement and accounting, and disclosure of assets and
51
Table of Contents
liabilities arising from contingencies in a business combination. The staff position is effective for assets or liabilities arising from contingencies in business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. The implementation of this standard did not have a material impact on our consolidated financial position and results of operations.
In June 2009, the FASB issued guidance, now codified as ASC 105, which establishes the FASB Accounting Standards Codification™ (the “Codification”) as the source of authoritative accounting principles recognized by the FASB to be applied by nongovernmental entities in the preparation of financial statements in conformity with U.S. GAAP. Use of the new Codification is effective for interim and annual periods ending after September 15, 2009. We adopted and are utilizing the new Codification references in our reporting and such adoption has not impacted our consolidated financial position or our results of operations.
In August 2009, the FASB issued additional guidance in ASC 825, “Financial Instruments” (“ASC 825”), which clarified how to measure the fair value of liabilities in circumstances when a quoted price in an active market for the identical liability is not available. ASC 825 is effective for the first reporting period beginning after the issuance of this standard, which for us was the year ended December 31, 2009. The implementation of this standard did not have a material impact on our consolidated financial position or our results of operations.
In September 2009, the FASB amended ASC 820 which provides amendments for the fair value measurement of investments to create a practical expedient to measure the fair value of an investment in certain entities on the basis of the net asset value per share of the investment (or its equivalent) determined as of the reporting entity’s measurement date. Therefore, certain attributes of the investment, such as restrictions on redemption, and transaction prices from principal-to-principal or brokered transactions, will not be considered in measuring the fair value of the investment if the practical expedient is used. The amendment in this ASU also requires disclosures by major category of investment about the attributes of those investments, such as the nature of any restrictions on the investor’s ability to redeem its investments at measurement date, any unfunded commitments, and the investment strategies of the investees. The amendments in this ASU are effective for interim and annual periods ending after December 15, 2009. The implementation of this standard did not have a material impact on our consolidated financial position or our results of operations.
In October 2009, the FASB issued new guidance in ASU 985, “Software” (“ASU 985”), which amends the accounting and disclosure for revenue recognition. These amendments, effective for fiscal years beginning on or after June 15, 2010 (early adoption is permitted), modify the criteria for recognizing revenue in multiple element arrangements and the scope of what constitutes a non-software deliverable. We are currently assessing the impact of ASU 985 on our consolidated financial position and our results of operations.
52
Table of Contents
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We have financial instruments accounted for as free-standing derivatives related to certain of the Company’s share-based payment arrangements that are outside the scope of FASB ASC 718 and are subject to FASB ASC 815, which requires vested stock options held by certain non-employee consultants to be accounted for as liability awards until these awards are exercised or forfeited. The fair value of these awards is remeasured at each financial statement date until the awards are settled or expire. Income (expense) recorded based on the remeasurement of these options was ($0.1) million, $0.5 million and ($0.2) million for the years ended December 31, 2009, 2008, and 2007, respectively. As of December 31, 2009 stock options to acquire 52,359 shares of common stock held by non-employee consultants remained unexercised and a liability of $0.2 million is included in accrued liabilities in the accompanying Consolidated Balance Sheet. We are exposed to the volatility of the market price of our stock. If the market price of our stock increased by $1 as of December 31, 2009, we would have recorded approximately $41,393 in additional expense related to these awards.
We are exposed to various market risks, which include potential losses arising from adverse changes in market rates and prices, such as foreign exchange fluctuations and changes in interest rates. Borrowings under our term loan with Silicon Valley Bank bears interest at a rate of 10.0% per year, and interest on the revolving loan will accrue at a fluctuating rate equal to the Bank’s announced prime rate of interest, subject to a floor of 4.0%, plus between 1.0% and 2.0%, depending on our Adjusted Quick Ratio (as defined in the Agreement). At December 31, 2009, our effective borrowing rate was 15.2% and the carrying value and fair value of the outstanding balance under the term loan was $5.1 million. Based upon this debt level, a 10% increase in the interest rate would not have resulted in a material impact to our financial results.
For the years ended December 31, 2009 and 2008, products sold by AtriCure Europe, B.V. accounted for 9.8% and 8.1%, respectively, of our total revenues. Since such revenues were primarily denominated in Euros, we have exposure to exchange rate fluctuations between the Euro and the U.S. Dollar. To date, the effect of the foreign exchange rate fluctuations on our financial results has not been significant. In 2009, we recorded foreign currency transaction losses of $140,593 in connection with partial settlements of our intercompany receivable balance with our subsidiary. For revenues denominated in Euros, if there is an increase in the rate at which Euros are exchanged for U.S. Dollars, it will require more Euros to equal a specified amount of U.S. Dollars than before the rate increase. In such cases, and if we price our products in Euros, we will receive less in U.S. Dollars than we did before the rate increase went into effect. If we price our products in U.S. Dollars and competitors price their products in Euros, an increase in the relative strength of the U.S. Dollar could result in our price not being competitive in a market where business is transacted in Euros. The Euro to U.S. dollar conversion rate fluctuations may impact our reported revenues and expenses.
We currently invest our cash primarily in money market accounts, U.S. government agencies and securities, corporate notes, corporate bonds, medium term notes, money market securities and commercial paper. Although we believe our cash is invested in a conservative manner, with cash preservation being our primary investment objective, the value of the securities we hold will fluctuate with changes in the financial markets including, among other things, changes in interest rates, credit quality and general volatility. We manage this risk by investing in high quality investment grade securities with very short-term maturities.
Financial instruments that potentially subject us to credit risk consist of cash and cash equivalent balances. Certain of our cash and cash equivalents balances exceed FDIC insured limits or are invested in money market accounts with investment banks that are not FDIC insured. We place our cash and cash equivalents in what we believe to be credit-worthy financial institutions. As of December 31, 2009, $1,488,580 of the cash balance was in excess of the FDIC limits.
53
Table of Contents
| ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
ATRICURE, INC. AND SUBSIDIARY
| Page | ||
| Financial Statements: |
||
| 55 | ||
| 56 | ||
| 57 | ||
| 58 | ||
| 59 | ||
| 60 | ||
| Financial Statement Schedule: |
||
| 83 | ||
54
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
AtriCure, Inc.
West Chester, Ohio
We have audited the accompanying consolidated balance sheets of AtriCure, Inc. and subsidiary (the “Company”) as of December 31, 2009 and 2008, and the related consolidated statements of operations, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2009. Our audits also included the financial statement schedule listed in the Index at Item 15. These financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on the financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. For 2009, the Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our 2009 audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of AtriCure, Inc. and subsidiary at December 31, 2009 and 2008, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2009, in conformity with accounting principles generally accepted in the United States of America. Also, in our opinion, such financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, present fairly, in all material respects, the information set forth therein.
/s/ Deloitte & Touche LLP
Cincinnati, Ohio
March 29, 2010
55
Table of Contents
CONSOLIDATED BALANCE SHEETS
DECEMBER 31, 2009 and 2008
| 2009 | 2008 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 8,905,425 | $ | 11,448,451 | ||||
| Short-term investments |
6,816,673 | — | ||||||
| Accounts receivable, less allowance for doubtful accounts of $24,400 and $40,480, respectively |
7,248,087 | 6,511,594 | ||||||
| Inventories, net |
4,869,708 | 6,361,242 | ||||||
| Other current assets |
3,511,335 | 1,781,825 | ||||||
| Total current assets |
31,351,228 | 26,103,112 | ||||||
| Property and equipment, net |
3,008,699 | 3,682,819 | ||||||
| Intangible assets |
287,653 | 569,153 | ||||||
| Goodwill |
— | 6,812,389 | ||||||
| Restricted cash and cash equivalents |
— | 6,000,000 | ||||||
| Other assets |
334,756 | 201,359 | ||||||
| Total Assets |
$ | 34,982,336 | $ | 43,368,832 | ||||
| Liabilities and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 3,599,943 | $ | 5,150,033 | ||||
| Accrued liabilities |
5,979,176 | 2,922,563 | ||||||
| Current maturities of debt and capital leases |
2,227,431 | 34,004 | ||||||
| Total current liabilities |
11,806,550 | 8,106,600 | ||||||
| Long-term debt and capital leases |
2,669,666 | 6,036,605 | ||||||
| Other liabilities |
3,416,360 | 106,470 | ||||||
| Total Liabilities |
17,892,576 | 14,249,675 | ||||||
| Commitments and contingencies (Note 10) |
— | — | ||||||
| Stockholders’ Equity: |
||||||||
| Common stock, $.001 par value, 90,000,000 shares authorized and 15,353,288 and 14,274,884 issued and outstanding, respectively |
15,353 | 14,275 | ||||||
| Additional paid-in capital |
110,900,087 | 106,636,653 | ||||||
| Accumulated other comprehensive (loss) income |
144,290 | (56,789 | ) | |||||
| Accumulated deficit |
(93,969,970 | ) | (77,474,982 | ) | ||||
| Total Stockholders’ Equity |
17,089,760 | 29,119,157 | ||||||
| Total Liabilities and Stockholders’ Equity |
$ | 34,982,336 | $ | 43,368,832 | ||||
See accompanying notes to consolidated financial statements.
56
Table of Contents
CONSOLIDATED STATEMENTS OF OPERATIONS
YEARS ENDED DECEMBER 31, 2009, 2008 and 2007
| 2009 | 2008 | 2007 | ||||||||||
| Revenues |
$ | 54,533,558 | $ | 55,257,023 | $ | 48,309,063 | ||||||
| Cost of revenues |
12,750,745 | 13,224,504 | 10,136,776 | |||||||||
| Gross profit |
41,782,813 | 42,032,519 | 38,172,287 | |||||||||
| Operating expenses: |
||||||||||||
| Research and development expenses |
11,414,889 | 10,608,668 | 10,987,477 | |||||||||
| Selling, general and administrative expenses |
35,112,006 | 42,422,133 | 39,752,513 | |||||||||
| Goodwill impairment |
6,812,389 | — | — | |||||||||
| Settlement reserve |
3,955,405 | — | — | |||||||||
| Total operating expenses |
57,294,689 | 53,030,801 | 50,739,990 | |||||||||
| Loss from operations |
(15,511,876 | ) | (10,998,282 | ) | (12,567,703 | ) | ||||||
| Other income (expense): |
||||||||||||
| Interest expense |
(812,326 | ) | (364,071 | ) | (213,104 | ) | ||||||
| Interest income |
51,089 | 382,285 | 947,888 | |||||||||
| Other |
(280,514 | ) | 755,564 | 579,853 | ||||||||
| Loss before income tax benefit |
(16,553,627 | ) | (10,224,504 | ) | (11,253,066 | ) | ||||||
| Income tax benefit |
(58,639 | ) | (57,252 | ) | — | |||||||
| Net loss |
$ | (16,494,988 | ) | $ | (10,167,252 | ) | $ | (11,253,066 | ) | |||
| Basic and diluted net loss per share |
$ | (1.13 | ) | $ | (0.72 | ) | $ | (0.84 | ) | |||
| Weighted average shares outstanding—basic and diluted |
14,563,710 | 14,191,000 | 13,381,715 | |||||||||
See accompanying notes to consolidated financial statements.
57
Table of Contents
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
YEARS ENDED DECEMBER 31, 2009, 2008, and 2007
| Accumulated Other Comprehensive Income (Loss) |
Total Stockholders’ Equity |
Comprehensive Loss |
|||||||||||||||||||||||
| Common Stock | Additional Paid-in Capital |
Accumulated Deficit |
|||||||||||||||||||||||
| Shares | Amount | ||||||||||||||||||||||||
| Balance—December 31, 2006 |
12,188,600 | $ | 12,189 | $ | 86,646,064 | $ | (56,054,664 | ) | $ | 90,673 | $ | 30,694,262 | |||||||||||||
| Issuance of common stock under equity incentive plans and warrants |
154,175 | 154 | 174,788 | — | — | 174,942 | |||||||||||||||||||
| Non-employee stock option fair market value adjustment |
— | — | 381,856 | — | — | 381,856 | |||||||||||||||||||
| Share-based employee compensation expense |
— | — | 1,510,361 | — | — | 1,510,361 | |||||||||||||||||||
| Unrealized gains on investments |
— | — | — | — | 7,343 | 7,343 | 7,343 | ||||||||||||||||||
| Foreign currency translation |
— | — | — | — | (92,730 | ) | (92,730 | ) | (92,730 | ) | |||||||||||||||
| Reclassification of non-employee option liability |
— | — | (433,407 | ) | — | — | (433,407 | ) | |||||||||||||||||
| Private placement of common shares |
1,789,649 | 1,789 | 15,245,152 | — | — | 15,246,941 | |||||||||||||||||||
| Net loss |
— | — | — | (11,253,066 | ) | — | (11,253,066 | ) | (11,253,066 | ) | |||||||||||||||
| Comprehensive loss |
$ | (11,338,453 | ) | ||||||||||||||||||||||
| Balance—December 31, 2007 |
14,132,424 | 14,132 | 103,524,814 | (67,307,730 | ) | 5,286 | 36,236,502 | ||||||||||||||||||
| Issuance of common stock under equity incentive plans |
87,537 | 88 | 337,252 | — | — | 337,340 | |||||||||||||||||||
| Issuance of common stock under stock purchase plan |
54,923 | 55 | 103,584 | — | — | 103,639 | |||||||||||||||||||
| Non-employee stock option fair market value adjustment |
— | — | 1,681 | — | — | 1,681 | |||||||||||||||||||
| Share-based employee compensation expense |
— | — | 2,669,322 | — | — | 2,669,322 | |||||||||||||||||||
| Reversal of prior period unrealized gains on investments |
— | — | — | — | (12,129 | ) | (12,129 | ) | (12,129 | ) | |||||||||||||||
| Foreign currency translation |
— | — | — | — | (49,946 | ) | (49,946 | ) | (49,946 | ) | |||||||||||||||
| Net loss |
— | — | — | (10,167,252 | ) | — | (10,167,252 | ) | (10,167,252 | ) | |||||||||||||||
| Comprehensive loss |
$ | (10,229,327 | ) | ||||||||||||||||||||||
| Balance—December 31, 2008 |
14,274,884 | 14,275 | 106,636,653 | (77,474,982 | ) | (56,789 | ) | 29,119,157 | |||||||||||||||||
| Issuance of common stock under equity incentive plans |
684,790 | 685 | (357,801 | ) | — | — | (357,116 | ) | |||||||||||||||||
| Issuance of common stock under stock purchase plan |
117,471 | 117 | 280,918 | — | — | 281,035 | |||||||||||||||||||
| Non-employee stock option fair market value adjustment |
— | — | 19,675 | — | — | 19,675 | |||||||||||||||||||
| Share-based employee compensation expense |
— | — | 3,865,921 | — | — | 3,865,921 | |||||||||||||||||||
| Issuance of common stock under warrants |
276,143 | 276 | 454,721 | — | 454,997 | ||||||||||||||||||||
| Unrealized gain on investments |
2,685 | 2,685 | 2,685 | ||||||||||||||||||||||
| Foreign currency translation |
— | — | — | — | 198,394 | 198,394 | 198,394 | ||||||||||||||||||
| Net loss |
— | — | — | (16,494,988 | ) | (16,494,988 | ) | (16,494,988 | ) | ||||||||||||||||
| Comprehensive loss |
$ | (16,293,909 | ) | ||||||||||||||||||||||
| Balance—December 31, 2009 |
15,353,288 | $ | 15,353 | $ | 110,900,087 | $ | (93,969,970 | ) | $ | 144,290 | $ | 17,089,760 | |||||||||||||
See accompanying notes to consolidated financial statements.
58
Table of Contents
CONSOLIDATED STATEMENTS OF CASH FLOW
YEARS ENDED DECEMBER 31, 2009, 2008 and 2007
| 2009 | 2008 | 2007 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (16,494,988 | ) | $ | (10,167,252 | ) | $ | (11,253,066 | ) | |||
| Adjustments to reconcile net loss to net cash provided by (used in) operating activities: |
||||||||||||
| Depreciation |
2,132,300 | 2,400,704 | 2,030,737 | |||||||||
| Amortization of deferred financing costs |
87,473 | 113,678 | 48,924 | |||||||||
| Write-off of deferred financing costs |
102,485 | — | — | |||||||||
| Amortization of discount on long-term debt |
169,106 | — | — | |||||||||
| Amortization of intangible assets |
281,500 | 281,500 | 242,125 | |||||||||
| Loss on disposal of equipment |
5,517 | 151,518 | 91,396 | |||||||||
| (Benefit from) provision for losses in accounts receivable |
(9,581 | ) | 20,440 | (132,308 | ) | |||||||
| Goodwill impairment |
6,812,389 | — | — | |||||||||
| Settlement reserve |
3,955,405 | — | — | |||||||||
| Share-based compensation expense |
3,885,596 | 2,671,003 | 1,892,217 | |||||||||
| Changes in assets and liabilities, excluding effects of acquired business: |
||||||||||||
| Accounts receivable |
(685,986 | ) | 609,337 | (561,132 | ) | |||||||
| Inventories |
1,504,706 | (1,149,231 | ) | (1,380,956 | ) | |||||||
| Other current assets |
169,163 | (342,710 | ) | (204,052 | ) | |||||||
| Accounts payable |
(1,550,090 | ) | 597,461 | 1,172,689 | ||||||||
| Accrued liabilities |
140,624 | (745,874 | ) | (321,471 | ) | |||||||
| Other non-current assets and non-current liabilities |
(85,671 | ) | (150,514 | ) | 259,269 | |||||||
| Net cash provided by (used in) operating activities |
419,948 | (5,709,940 | ) | (8,115,628 | ) | |||||||
| Cash flows from investing activities: |
||||||||||||
| Purchases of equipment |
(1,360,459 | ) | (1,747,590 | ) | (3,044,546 | ) | ||||||
| Proceeds from sale of equipment |
2,000 | — | — | |||||||||
| Purchases of available-for-sale securities |
(8,015,866 | ) | (1,900,756 | ) | (8,208,668 | ) | ||||||
| Maturities of available-for-sale securities |
1,201,877 | 8,894,670 | 5,808,000 | |||||||||
| Change in restricted cash and cash equivalents |
6,000,000 | (6,000,000 | ) | — | ||||||||
| Cash paid for acquisition |
— | (417,292 | ) | (3,341,349 | ) | |||||||
| Net cash used in investing activities |
(2,172,448 | ) | (1,170,968 | ) | (8,786,563 | ) | ||||||
| Cash flows from financing activities: |
||||||||||||
| Payments on debt and capital leases |
(7,493,269 | ) | (721,917 | ) | (393,675 | ) | ||||||
| Proceeds from borrowings of debt |
6,500,000 | 6,000,000 | — | |||||||||
| Payment of debt fees and premium on retirement of debt |
(235,110 | ) | (340,932 | ) | — | |||||||
| Proceeds from issuance of common stock under employee stock purchase plan |
281,035 | 103,640 | — | |||||||||
| Net proceeds from sale of common stock |
— | — | 15,246,941 | |||||||||
| Proceeds from stock option exercises |
33,335 | 239,873 | 174,942 | |||||||||
| Net cash (used in) provided by financing activities |
(914,009 | ) | 5,280,664 | 15,028,208 | ||||||||
| Effect of exchange rate changes on cash |
123,483 | 48,043 | (15,748 | ) | ||||||||
| Net decrease in cash and cash equivalents |
(2,543,026 | ) | (1,552,201 | ) | (1,889,731 | ) | ||||||
| Cash and cash equivalents—beginning of period |
11,448,451 | 13,000,652 | 14,890,383 | |||||||||
| Cash and cash equivalents—end of period |
$ | 8,905,425 | $ | 11,448,451 | $ | 13,000,652 | ||||||
| Supplemental cash flow information: |
||||||||||||
| Cash paid for interest |
$ | 460,927 | $ | 127,656 | $ | 72,951 | ||||||
| Cash paid for taxes |
$ | 17,300 | $ | 14,000 | $ | 8,500 | ||||||
| Non-cash investing and financing activities: |
||||||||||||
| Purchases of equipment in current liabilities |
$ | 15,746 | $ | 21,036 | $ | 94,179 | ||||||
| Unsecured note payable in connection with acquisition |
$ | — | $ | — | $ | 417,292 | ||||||
| Assets acquired through capital lease |
$ | 105,651 | $ | 102,197 | $ | — | ||||||
| Warrant issued in conjunction with credit facility |
$ | 455,000 | $ | — | $ | — | ||||||
See accompanying notes to consolidated financial statements
59
Table of Contents
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Nature of the Business—AtriCure, Inc. (the “Company” or “AtriCure”) was incorporated in the State of Delaware on October 31, 2000. The Company develops, manufactures and sells devices designed primarily for the surgical ablation of cardiac tissue. The Company sells its medical devices to hospitals and medical centers in the United States and internationally. International sales were $10,414,357, $8,338,932 and $6,591,278 in 2009, 2008, and 2007, respectively.
Principles of Consolidation—The consolidated financial statements include the accounts of the Company and AtriCure Europe, B.V., the Company’s wholly-owned subsidiary incorporated in the Netherlands. All intercompany accounts and transactions have been eliminated in consolidation.
Cash and Cash Equivalents—The Company considers highly liquid investments with maturities of three months or less at the date of acquisition as cash equivalents in the accompanying consolidated financial statements.
Short-Term Investments—The Company places its investments primarily in U.S. Government agencies and securities, corporate bonds and commercial paper. The Company classifies all investments as available-for-sale. Such investments are recorded at fair value, with unrealized gains and losses recorded as a separate component of stockholders’ equity. The Company recognizes gains and losses when these securities are sold using the specific identification method.
Revenue Recognition—Revenues are generated primarily from the sale of the Company’s disposable surgical devices. Pursuant to the Company’s standard terms of sale, revenues are recognized when title to the goods and risk of loss transfers to customers and there are no remaining obligations that will affect the customers’ final acceptance of the sale. Generally, the Company’s standard terms of sale define the transfer of title and risk of loss to occur upon shipment to the respective customer. The Company generally does not maintain any post-shipping obligations to the recipients of the products. Typically, no installation, calibration or testing of this equipment is performed by the Company subsequent to shipment to the customer in order to render it operational. Product revenues include shipping and handling revenues of $669,328, $757,722 and $468,377 in 2009, 2008, and 2007, respectively. Cost of freight for shipments made to customers is included in cost of revenues. Sales and other value-added taxes collected from customers and remitted to governmental authorities are excluded from product revenues. The Company sells its products primarily through a direct sales force and through AtriCure Europe, B.V. Terms of sale are generally consistent for both end-users and distributors and payment terms are generally net 30 days for end-users and net 60 days for distributors.
The Company accounts for revenues in accordance with FASB ASC 605, “Revenue Recognition” (“ASC 605”). The Company determines the timing of revenue recognition based upon factors such as passage of title, installation, payment terms and ability to return products. The Company recognizes revenue when all of the following criteria are met: (i) persuasive evidence that an arrangement exists; (ii) delivery of the products and/or services has occurred; (iii) the selling price is fixed or determinable; and (iv) collectability is reasonably assured.
Sales Returns and Allowances—The Company maintains a provision for sales returns and allowances to account for potential returns of defective or damaged products and price reductions given to customers. The Company’s management estimates such provision based primarily on a specific identification basis. Increases to the provision result in reductions of revenues.
Allowance for Uncollectible Accounts Receivable—The Company evaluates the collectability of accounts receivable in order to determine the appropriate reserve for doubtful accounts. In determining the amount of the
60
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
reserve, the Company considers aging of account balances, historical credit losses, customer-specific information and other relevant factors. An increase to the allowance for doubtful accounts results in a corresponding increase in expense. The Company reviews accounts receivable and adjusts the allowance based on current circumstances and charges off uncollectible receivables against the allowance when all attempts to collect the receivable have failed.
Inventories—Inventories are stated at the lower of cost or market using the first-in, first-out cost method (“FIFO”) and consist of raw materials, work in process, and finished goods. A reserve for inventory is estimated and recorded for excess, slow moving and obsolete inventory as well as inventory with a carrying value in excess of its net realizable value. Write-offs are recorded when a product is destroyed. The Company reviews inventory on hand at least quarterly and records provisions for excess and obsolete inventory based on several factors including current assessment of future product demand, anticipated release of new products into the market, historical experience and product expiration. The Company’s industry is characterized by rapid product development and frequent new product introductions. Uncertain timing of product approvals, variability in product launch strategies, and variation in product utilization all impact the estimates related to excess and obsolete inventory.
Property and Equipment—Property and equipment is stated at cost, less accumulated depreciation. Depreciation is computed on the straight-line method for financial reporting purposes and applied over the estimated useful lives of the assets. The estimated useful life by major asset category is the following: machinery and equipment is three to seven years, computer and other office equipment is three years, furniture and fixtures is three to seven years, and leasehold improvements and equipment leased under a capital lease are the shorter of their useful life or remaining lease term. Maintenance and repair costs are expensed as incurred.
Included in property and equipment are generators and other capital equipment (such as the Company’s ASB) that are loaned at no cost to direct customers that use the Company’s disposable products. These generators are depreciated over a three year period and such depreciation is included in cost of revenues. The total of such depreciation was $1,125,786, $1,069,135 and $801,520 in 2009, 2008, and 2007, respectively.
Impairment of Long-Lived Assets (Other than Goodwill)—The Company reviews property and equipment and definite-lived intangibles for impairment using its best estimates based on reasonable and supportable assumptions and projections in accordance with FASB ASC 360, “Property, Plant and Equipment” (“ASC 360”). The Company recorded a charge within operating expense of $5,517 in 2009 for the impairment of fixed assets. The Company recorded a charge within cost of revenues of $151,518 in 2008 for the impairment of fixed assets and machinery and equipment related to discontinued product lines. In 2007, the Company recorded a charge of $91,396 for the impairment of obsolete machinery and equipment and tooling.
Goodwill and Intangible Assets—As of December 31, 2008 the Company had $6,812,389 in goodwill, which represented the excess of costs over the fair value of the net assets acquired in business combinations. The Company historically tested its goodwill for impairment annually during its fourth quarter, or more frequently if impairment indicators were present or changes in circumstances indicated that carrying value of the asset exceeded the estimated fair value. FASB ASC 350, “Intangibles—Goodwill and Other” (“ASC 350”) requires a two-step approach to determine any potential goodwill impairment. The first step (Step 1) requires a comparison of the carrying value of the reporting unit to its fair value. Goodwill is considered potentially impaired if the carrying value of the reporting unit is greater than the estimated fair value. If potential impairment exists based upon completion of Step 1, Step 2 must be completed, which compares the implied fair value of a reporting unit’s goodwill to its carrying value. Step 2 involves an analysis allocating the fair value determined in Step 1 (as if it was the purchase price in a business combination). If the calculated fair value of the goodwill resulting from this allocation is lower than the carrying value of the goodwill of the reporting unit, an impairment loss is
61
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
recorded. During the first quarter of 2009, the Company’s market capitalization dropped and was less than its recorded net book value, which indicated that a potential impairment existed. The Company recorded a full impairment loss related to its goodwill during the first quarter of 2009, based on the results of its Step 1 analysis. During the second quarter of 2009, the Company performed its Step 2 analysis and concluded that the charge recorded was appropriate.
Intangible assets with determinable useful lives are amortized on a straight-line basis over the estimated periods benefited, which range from four to eight years.
Restricted Cash and Cash Equivalents—As of December 31, 2008, $6,000,000 had been borrowed under a revolving credit facility and in accordance with the terms of the agreement, $6,000,000 was held as restricted cash and cash equivalents. The credit facility was terminated effective May 1, 2009. No restricted cash and cash equivalents were recorded as of December 31, 2009.
Grant Income—Through December 31, 2008, the Company had received research grants, which were recognized as funds earned and not as awarded by awarding agencies. No research grants were earned during year ended December 31, 2009.
Income Taxes—Income taxes are computed using the asset and liability method in accordance with FASB ASC 740 “Income Taxes” (“ASC 740”), under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of the Company’s assets and liabilities. Deferred taxes are measured using provisions of currently enacted tax laws. A valuation allowance against deferred tax assets is recorded when it is more likely than not that such assets will not be fully realized. Tax credits are accounted for as a reduction of income taxes in the year in which the credit originates.
The Company’s estimate of the valuation allowance for deferred tax assets requires it to make significant estimates and judgments about its future operating results. The Company’s ability to realize the deferred tax assets depends on its future taxable income as well as limitations on their utilization. A deferred tax asset is reduced by a valuation allowance if it is more likely than not that some portion or all of the deferred tax asset will not be realized prior to its expiration. The projections of the Company’s operating results on which the establishment of a valuation allowance is based involve significant estimates regarding future demand for the Company’s products, competitive conditions, product development efforts, approvals of regulatory agencies and product cost. If actual results differ from these projections, or if the Company’s expectations of future results change, it may be necessary to adjust the valuation allowance.
Net Loss Per Share—Basic net loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Since the Company has experienced net losses for all periods presented, net loss per share excludes the effect of 2,894,886, 2,791,203 and 2,296,035 options, restricted stock, performance shares and warrants in 2009, 2008, and 2007, respectively, because such options, restricted stock, performance shares and warrants are anti-dilutive. Therefore the number of shares calculated for basic net loss per share is also used for the diluted net loss per share calculation.
62
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Accumulated Other Comprehensive Income (Loss)—Other comprehensive income (loss) consisted of the following:
| Unrealized Gains (Losses) on Short-Term and Long-Term Investments |
Foreign Currency Translation Adjustment |
Other Comprehensive Income (Loss) |
||||||||||
| Balance as of December 31, 2006 |
$ | 4,786 | $ | 85,887 | $ | 90,673 | ||||||
| Current-period change |
7,343 | (92,730 | ) | (85,387 | ) | |||||||
| Balance as of December 31, 2007 |
12,129 | (6,843 | ) | 5,286 | ||||||||
| Current-period change |
(12,129 | ) | (49,946 | ) | (62,075 | ) | ||||||
| Balance as of December 31, 2008 |
— | (56,789 | ) | (56,789 | ) | |||||||
| Current-period change |
2,685 | 198,394 | 201,079 | |||||||||
| Balance as of December 31, 2009 |
$ | 2,685 | $ | 141,605 | $ | 144,290 | ||||||
Foreign Currency Transaction Gain—The Company recorded foreign currency transaction (losses) gains of ($140,593), ($64,176) and $246,562 for the years ended December 31, 2009, 2008 and 2007, respectively, in connection with partial settlements of its intercompany balance with its subsidiary.
Research and Development—Research and development costs are expensed as incurred. These costs include compensation and other internal and external costs associated with the development and research related to new products or concepts, preclinical studies, clinical trials and cost of products used in trials and tests.
Share-Based Employee Compensation—The Company follows FASB ASC 718 “Compensation-Stock Compensation” (“ASC 718”), to record share-based compensation for all share-based payment awards made to employees, including employee stock options, restricted stock, performance shares and employee stock purchases related to an employee stock purchase plan, based on estimated fair values. The Company’s employee share-based compensation expense recognized under ASC 718 for the years ended December 31, 2009, 2008 and 2007 was $3,865,922, $2,669,322 and $1,510,361, respectively on a before and after tax basis.
FASB ASC 718 requires companies to estimate the fair value of share-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as expense over the requisite service periods in the Company’s Consolidated Statement of Operations. The expense has been reduced for estimated forfeitures. FASB ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
The Company estimates the fair value of options on the date of grant using the Black-Scholes option-pricing model (“Black-Scholes model”). The Company’s determination of fair value of share-based payment awards on the date of grant using an option-pricing model is affected by the Company’s stock price, as well as assumptions regarding a number of highly complex and subjective variables. These variables include but are not limited to the Company’s and the peer group’s expected stock price volatility over the term of the awards and actual and projected employee stock option exercise behaviors.
The Company estimates the fair value of restricted stock and performance share awards based upon the grant date closing market price of the Company’s common stock. The Company’s determination of fair value is affected by the Company’s stock price as well as assumptions regarding the number of shares expected to be granted and, in the case of performance shares, the likelihood that the performance measures will be achieved.
63
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The Company also has an employee stock purchase plan (“ESPP” or the “Plan”) which is available to all eligible employees as defined by the Plan. Under the ESPP, shares of the Company’s common stock may be purchased at a discount. The Company estimates the number of shares to be purchased under the Plan and records compensation expense based upon the fair value of the stock at the beginning of the purchase period using the Black-Scholes model.
Certain of the Company’s share-based payment arrangements are outside the scope of ASC 718 and are subject to FASB ASC 815, “Derivatives and Hedging” (“ASC 815”), which requires vested stock options held by certain non-employee consultants to be accounted for as liability awards until these awards are exercised or forfeited. The fair value of these awards is remeasured at each financial statement date until the awards are settled or expire. During the year ended December 31, 2009, $140,620 of expense was recorded as a result of the remeasurement of the fair value of these awards compared with $522,993 of income in 2008 and $227,421 of expense in 2007. As of December 31, 2009 and December 31, 2008, respectively, fully vested options to acquire 52,359 and 54,660 shares of common stock held by non-employee consultants remained unexercised and a liability of $180,288 and $40,368 was included in accrued liabilities in the Consolidated Balance Sheets.
Also outside the scope of ASC 718, in connection with the Company’s $6.5 million term loan, the Company issued a warrant to purchase shares of the Company’s common stock. The warrant allowed the Bank to purchase 371,732 shares of the Company’s common stock at $1.224 per share and was exercisable for a term of 10 years. The warrant was immediately exercisable and provided for net share settlement. The Company determined this arrangement met the requirements of ASC 815 and other relevant literature and, therefore, the warrant was recorded at its grant date intrinsic value and is classified as an equity transaction. On October 6, 2009, the warrant was exercised through a net share settlement and 276,143 shares were issued.
Use of Estimates—The preparation of the financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Fair Value Disclosures—The fair value of the Company’s financial instruments, including cash and cash equivalents, accounts receivable, restricted cash and cash equivalents, short and long-term investments, short and long-term other assets, accounts payable, accrued expenses, other liabilities and fixed and variable interest rate debt, approximate their fair values.
2. RECENT ACCOUNTING PRONOUNCEMENTS
In April 2009, the FASB issued a staff position within ASC 805, “Business Combinations” (“ASC 805”), amending and clarifying the new business combination standard to address application issues associated with initial recognition and measurement, subsequent measurement and accounting, and disclosure of assets and liabilities arising from contingencies in a business combination. The staff position is effective for assets or liabilities arising from contingencies in business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. The implementation of this standard did not have a material impact on the Company’s consolidated financial position or results of operations.
In June 2009, the FASB issued guidance now codified as ASC 105 which establishes the FASB Accounting Standards Codification™ (the “Codification”) as the source of authoritative accounting principles recognized by
64
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
the FASB to be applied by nongovernmental entities in the preparation of financial statements in conformity with U.S. GAAP. Use of the new Codification is effective for interim and annual periods ending after September 15, 2009. The Company adopted and is utilizing the new Codification references in its reporting and such adoption has not impacted its consolidated financial position or results of operations.
In August 2009, the FASB issued additional guidance in ASC 825, “Financial Instruments” (“ASC 825”), which clarified how to measure the fair value of liabilities in circumstances when a quoted price in an active market for the identical liability is not available. ASC 825 is effective for the first reporting period beginning after the issuance of this standard which, for the Company, was its year ended December 31, 2009. The implementation of this standard did not have a material impact on the Company’s consolidated financial position or results of operations.
In September 2009, the FASB amended ASC 820 which provides amendments for the fair value measurement of investments to create a practical expedient to measure the fair value of an investment in certain entities on the basis of the net asset value per share of the investment (or its equivalent) determined as of the reporting entity’s measurement date. Therefore, certain attributes of the investment, such as restrictions on redemption, and transaction prices from principal-to-principal or brokered transactions, will not be considered in measuring the fair value of the investment if the practical expedient is used. The amendment in this ASU also requires disclosures by major category of investment about the attributes of those investments, such as the nature of any restrictions on the investor’s ability to redeem its investments at measurement date, any unfunded commitments, and the investment strategies of the investees. The amendments in this ASU are effective for interim and annual periods ending after December 15, 2009. The implementation of this standard did not have a material impact on the Company’s consolidated financial position or our results of operations.
In October 2009, the FASB issued new guidance in ASU 985, “Software” (“ASU 985”), which amends the accounting and disclosure for revenue recognition. These amendments, effective for fiscal years beginning on or after June 15, 2010 (early adoption is permitted), modify the criteria for recognizing revenue in multiple element arrangements and the scope of what constitutes a non-software deliverable. The Company is currently assessing the impact on its consolidated financial position or results of operations.
3. FAIR VALUE
The FASB ASU 820, “Fair Value Measurements and Disclosures” (“ASU 820”) defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
| • | Level 1—Quoted prices in active markets for identical assets or liabilities. |
| • | Level 2—Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. |
| • | Level 3—Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. |
65
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
In accordance with ASC 820, the following table represents the Company’s fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2009:
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Other Unobservable Inputs (Level 3) |
Total | |||||||||
| Assets: |
||||||||||||
| Money market funds |
$ | — | $ | 7,173,778 | $ | — | $ | 7,173,778 | ||||
| Commercial paper |
— | 2,397,445 | — | 2,397,445 | ||||||||
| U.S. government agencies and securities |
4,018,252 | — | — | 4,018,252 | ||||||||
| Corporate bonds |
400,976 | — | — | 400,976 | ||||||||
| Total assets |
$ | 4,419,228 | $ | 9,571,223 | $ | — | $ | 13,990,451 | ||||
| Liabilities: |
||||||||||||
| Derivative instruments |
$ | — | $ | — | $ | 180,288 | $ | 180,288 | ||||
| Total liabilities |
$ | — | $ | — | $ | 180,288 | $ | 180,288 | ||||
|
In accordance with FASB ASC 820, the following table represents the Company’s fair value hierarchy for its financial assets and liabilities measured at fair value on a recurring basis as of December 31, 2008:
| ||||||||||||
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Other Unobservable Inputs (Level 3) |
Total | |||||||||
| Assets: |
||||||||||||
| Money market funds |
$ | — | $ | 15,570,883 | $ | — | $ | 15,570,883 | ||||
| Total assets |
$ | — | $ | 15,570,883 | $ | — | $ | 15,570,883 | ||||
| Liabilities: |
||||||||||||
| Derivative instruments |
$ | — | $ | — | $ | 40,368 | $ | 40,368 | ||||
| Total liabilities |
$ | — | $ | — | $ | 40,368 | $ | 40,368 | ||||
Certain of the Company’s share-based payment arrangements are outside the scope of ASC 718 and are subject to ASC 815, which requires vested stock options held by certain non-employee consultants to be accounted for as liability awards until these awards are exercised or forfeited. The fair value of these awards is remeasured at each financial statement date until the awards are settled or expire. In calculating the fair value of the options they are estimated on the grant date using the Black-Scholes model subject to change in stock price utilizing assumptions of risk-free interest rate, contractual life of option, expected volatility, weighted average volatility and dividend yield. Due to the lack of certain observable market quotes the Company utilizes valuation models that rely on some Level 3 inputs. Specifically, due to the Company’s limited trading history, the Company uses an equal weighting of both the Company’s implied volatility and the implied volatility of a group of comparable companies in determining the Company’s volatility.
66
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| 2009 | 2008 | |||||||
| Fair Value Measurements Using Significant Other Unobservable Inputs (Level 3) |
Fair Value Measurements Using Significant Other Unobservable Inputs (Level 3) |
|||||||
| Derivative Instruments | Derivative Instruments | |||||||
| Beginning Balance |
$ | 40,368 | $ | 660,827 | ||||
| Total losses (gains) (realized/unrealized) included in earnings |
140,620 | (522,993 | ) | |||||
| Purchases, issuances and settlements |
(700 | ) | (97,466 | ) | ||||
| Ending Balance |
$ | 180,288 | $ | 40,368 | ||||
| The amount of total (losses) gains for the period included in earnings (or changes in net assets) attributable to the change in unrealized (losses) gains relating to assets still held at reporting date |
$ | (140,620 | ) | $ | 522,993 | |||
4. INVESTMENTS
As of December 31, 2009, the Company had no long-term investments. Short-term investments as of December 31, 2009 consisted of the following:
| Cost Basis | Unrealized Gains (losses) |
Fair Value | ||||||||
| U.S. Government agencies and securities |
$ | 4,014,972 | $ | 3,279 | $ | 4,018,251 | ||||
| Commercial paper |
2,397,783 | (337 | ) | 2,397,446 | ||||||
| Corporate bonds |
401,233 | (257 | ) | 400,976 | ||||||
| Total |
$ | 6,813,988 | $ | 2,685 | $ | 6,816,673 | ||||
The Company has not experienced any significant realized gains or losses on its investments in the periods presented in the Consolidated Statements of Operations and no short or long-term investments were outstanding at December 31, 2008.
5. GOODWILL AND INTANGIBLE ASSETS
Intangible assets with definite lives are amortized over their estimated useful lives. The following table provides a summary of the Company’s intangible assets with definite lives:
| Proprietary Manufacturing Technology |
Non-Compete Agreement |
Tradename | Total | |||||||||||||
| Net carrying amount as of December 31, 2006 |
$ | 772,778 | $ | — | $ | — | $ | 772,778 | ||||||||
| Gross carrying amount recorded |
— | 100,000 | 220,000 | 320,000 | ||||||||||||
| Amortization |
(214,000 | ) | (5,208 | ) | (22,917 | ) | (242,125 | ) | ||||||||
| Net carrying amount as of December 31, 2007 |
558,778 | 94,792 | 197,083 | 850,653 | ||||||||||||
| Amortization |
(214,000 | ) | (12,500 | ) | (55,000 | ) | (281,500 | ) | ||||||||
| Net carrying amount as of December 31, 2008 |
344,778 | 82,292 | 142,083 | 569,153 | ||||||||||||
| Amortization |
(214,000 | ) | (12,500 | ) | (55,000 | ) | (281,500 | ) | ||||||||
| Net carrying amount as of December 31, 2009 |
$ | 130,778 | $ | 69,792 | $ | 87,083 | $ | 287,653 | ||||||||
67
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Amortizable intangible assets are being amortized over eight years for a non-compete arrangement, four years for tradename usage and five years for proprietary manufacturing technology. For the years ended December 31, 2009, 2008 and 2007, amortization expense related to intangible assets with definite lives was $281,500, $281,500 and $242,125, respectively.
Future amortization expense related to intangible assets with definite lives is projected as follows:
| Year |
Amortization | ||
| 2010 |
$ | 198,278 | |
| 2011 |
44,583 | ||
| 2012 |
12,500 | ||
| 2013 |
12,500 | ||
| 2014 |
12,500 | ||
| 2015 |
7,292 | ||
| Total |
$ | 287,653 | |
Goodwill represents the excess of costs over the fair value of the net assets acquired in business combinations. The Company historically tested its goodwill for impairment annually during the fourth quarter, or more frequently if impairment indicators were present or changes in circumstances indicated the carrying value of the asset exceeds the estimated fair value. FASB ASC 350 requires a two-step approach to determine any potential goodwill impairment. The first step (Step 1) requires a comparison of the carrying value of the reporting unit to the fair value of the unit. Goodwill is considered potentially impaired if the carrying value of the reporting unit is greater than the estimated fair value. If potential impairment exists based upon completion of Step 1, Step 2 is required, which compares the implied fair value of a reporting unit’s goodwill to its carrying value. Step 2 involves an analysis allocating the fair value determined in Step 1 (as if it was the purchase price in a business combination). If the calculated fair value of the goodwill resulting from this allocation is lower than the carrying value of the goodwill of the reporting unit, an impairment loss is recorded.
As a result of a reduction in the Company’s market capitalization during the first quarter of 2009, the Company believed an indication of impairment existed and performed a Step 1 analysis of its goodwill as of March 31, 2009. The Step 1 process concluded that the carrying value of the Company’s single reporting unit exceeded its estimated fair value.
To estimate the fair value of the reporting unit for Step 1, the Company utilized the market valuation approach. Under the market valuation approach the estimated fair value of the reporting unit is based on the Company’s market capitalization using the closing market price of the Company’s stock and number of shares outstanding as of March 31, 2009. The Company also considered a control premium that represents the estimated amount an investor would pay for a controlling interest in the Company. An income approach was also used to corroborate the results of the Step 1 test. The discounted cash flow method was used to measure the fair value of the Company’s equity under the income approach. Determining the fair value using a discounted cash flow method includes assumptions about future market conditions and operating results. The judgments are based upon historical experience, current market trends and projected estimated future revenues and profit margins. The Company believes that these estimates and assumptions are reasonable and that different estimates and assumptions could result in a different outcome. Determining the control premium to apply to the reporting unit is a subjective process that involves the use of estimates and judgments. The income approach supported the interim Step 1 test result using the market valuation approach in determining that the carrying value of the reporting unit exceeded the fair value.
68
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Step 2 of the goodwill impairment test compares the implied fair value of the reporting unit’s goodwill with the carrying amount of the goodwill. If the carrying amount of the Company’s goodwill exceeds the implied fair value of goodwill, an impairment loss is recognized for an amount equal to that excess. As required, the Company performed Step 2 of the goodwill impairment test during the three month period ended June 30, 2009. Based on the results of this test, the Company concluded its goodwill was fully impaired and that the impairment of $6,812,389 on a before and after tax basis was appropriately recorded as of March 31, 2009. This impairment was recorded as an increase in operating expenses, loss from operations, and net loss in the Consolidated Statement of Operations during the three months ended March 31, 2009.
The changes in the net carrying amount of goodwill for the years ended December 31, 2009 and 2008 are as follows:
| Net carrying amount as of December 31, 2007 |
$ | 6,763,259 | ||
| Goodwill amount recorded |
49,130 | |||
| Net carrying amount as of December 31, 2008 |
6,812,389 | |||
| Goodwill impairment |
(6,812,389 | ) | ||
| Net carrying amount as of December 31, 2009 |
$ | — | ||
The additional goodwill recorded in 2008 relates to an increase in inventory reserves related to the August 7, 2007 Cooper acquisition. The goodwill impairment of $6,812,389 during 2009 represents the cumulative impairment taken since inception.
6. INVENTORIES
Inventories consisted of the following at December 31:
| 2009 | 2008 | |||||||
| Raw material |
$ | 1,839,610 | $ | 2,518,226 | ||||
| Work in process |
411,738 | 425,641 | ||||||
| Finished goods |
2,801,530 | 3,601,270 | ||||||
| Reserve for obsolescence |
(183,170 | ) | (183,895 | ) | ||||
| Inventories, net |
$ | 4,869,708 | $ | 6,361,242 | ||||
7. PROPERTY AND EQUIPMENT
Property and equipment consisted of the following at December 31:
| 2009 | 2008 | |||||||
| Machinery and equipment |
$ | 6,248,020 | $ | 7,064,477 | ||||
| Computer and other office equipment |
1,276,273 | 1,535,249 | ||||||
| Furniture and fixtures |
384,535 | 459,103 | ||||||
| Leasehold improvements |
65,435 | 424,582 | ||||||
| Equipment under capital leases |
207,847 | 102,197 | ||||||
| Construction in progress |
75,806 | 48,507 | ||||||
| Total |
8,257,916 | 9,634,115 | ||||||
| Less accumulated depreciation |
(5,249,217 | ) | (5,951,296 | ) | ||||
| Property and equipment, net |
$ | 3,008,699 | $ | 3,682,819 | ||||
69
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
8. ACCRUED LIABILITIES
Accrued liabilities consisted of the following at December 31:
| 2009 | 2008 | |||||
| Accrued class action settlement reserve |
$ | 2,000,000 | $ | — | ||
| Accrued commissions |
1,251,681 | 847,872 | ||||
| Other accrued liabilities |
883,574 | 850,057 | ||||
| Accrued bonus |
779,949 | 69,525 | ||||
| Accrued settlement reserve (current portion) |
562,500 | — | ||||
| Accrued taxes |
269,491 | 343,455 | ||||
| Accrued vacation |
175,607 | 232,577 | ||||
| Accrued severance |
56,374 | 579,077 | ||||
| Total |
$ | 5,979,176 | $ | 2,922,563 | ||
9. FINANCING ARRANGEMENTS
Long-term debt and capital leases consisted of the following at December 31:
| 2009 | 2008 | |||||
| Credit facility, due 2010 |
$ | — | $ | 6,000,000 | ||
| Credit facility, due 2012 (net of discount on debt) |
4,769,662 | — | ||||
| Capital leases |
127,435 | 70,609 | ||||
| Total debt and capital leases |
4,897,097 | 6,070,609 | ||||
| Less: Current maturities |
2,227,431 | 34,004 | ||||
| Total long-term debt and capital leases |
$ | 2,669,666 | $ | 6,036,605 | ||
On May 1, 2009, the Company and Silicon Valley Bank (the “Bank”) entered into a Loan and Security Agreement (the “Agreement”) that provides a term loan and a revolving credit facility under which the Company can borrow a maximum of $10,000,000. The Company can borrow up to $10,000,000 under the revolving loan facility with the availability subject to a borrowing base formula. On May 1, 2009, the Company borrowed the maximum amount of $6,500,000 under the term loan. In connection with the term loan, the Bank received a warrant to purchase 371,732 shares of the Company’s common stock at $1.224 per share, exercisable for a term of 10 years (the “Warrant”). The Warrant was immediately exercisable and was exercised via a net share settlement exercise on October 6, 2009, resulting in the issuance of 276,143 shares of the Company’s common stock. The Agreement also includes up to a $1,000,000 million sublimit for stand-by letters of credit.
Interest on the term loan accrues at a rate of 10.0% per year, and interest on the revolving loan will accrue at a fluctuating rate equal to the Bank’s announced prime rate of interest, subject to a floor of 4.0%, plus between 1.0% and 2.0%, depending on the Company’s Adjusted Quick Ratio (as defined in the Agreement). Principal on the term loan is being paid over 36 months of equal principal payments, plus applicable interest. The Agreement matures on April 30, 2012 and is secured by all of the Company’s assets, including intellectual property.
The Agreement contains covenants that include, among others, covenants that limit the Company’s and its subsidiaries’ ability to dispose of assets, enter into mergers or acquisitions, incur indebtedness, incur liens, pay dividends or make distributions on the Company’s capital stock, make investments or loans, and enter into certain affiliate transactions, in each case subject to customary exceptions for a credit facility of this size and
70
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
type. Additional covenants apply when the Company has outstanding borrowings under the revolving loan facility or when the Company achieves specific covenant milestones. The occurrence of an event of default could result in an increase to the applicable interest rate by 3.0%, an acceleration of all obligations under the Agreement, an obligation of the Company to repay all obligations in full, and a right by the Bank to exercise all remedies available to it under the Agreement and related agreements including the Guaranty and Security Agreement.
On November 4, 2009, effective September 30, 2009, the Company entered into a Consent, Waiver and First Loan Modification Agreement (“First Amendment”) with Silicon Valley Bank, which amended, among other things, the financial covenants in the Agreement. On March 26, 2010, the Company entered into a Waiver and Second Loan Modification Agreement (“Second Amendment”) with Silicon Valley Bank, which amended, among other things, the financial covenants in the Agreement and waived a compliance violation which occurred during February 2010.
As of December 31, 2009, the Company had no borrowings under its revolving credit facility and borrowing availability of $1,054,144. Also as of December 31, 2009, the Company had $5,055,556 outstanding under its term loan, which includes $2,166,667 classified as current maturities of long-term debt. The Company is required to make monthly principal payments on its term loan of $180,556 plus interest. The Warrant was recorded as discount on long-term debt at its intrinsic value and is being amortized over the term of the loan. For the year ended December 31, 2009, amortization expense related to the Warrant totaled $169,106. The effective interest rate on borrowings under the term loan, including amortization of the Warrant and debt issuance costs, is 15.2%. As of December 31, 2009 the Company had an outstanding letter of credit of $250,000 issued to its corporate credit card program provider. The letter of credit expires on July 31, 2010.
As of December 31, 2009 the Company had capital leases for computer equipment that expire at various terms through 2013. The cost of the assets under lease was $207,847. These assets are depreciated over the estimated useful life of the asset, which equals the term of the lease. Accumulated amortization on the capital leases was $82,218 at December 31, 2009.
Maturities on long-term debt (gross of discount on long-term debt), including capital lease obligations are as follows:
| 2010 |
$ | 2,227,431 | |
| 2011 |
2,193,356 | ||
| 2012 |
751,706 | ||
| 2013 |
10,498 | ||
| Total maturities on long-term debt and capital lease |
$ | 5,182,991 | |
On July 1, 2008, the Company entered into a two-year $10,000,000 credit facility with National City Bank (subsequently merged with PNC Bank). The credit facility was secured by all of the Company’s assets and property, tangible and intangible. As of December 31, 2008, the Company had $6,000,000 outstanding under its credit facility with National City Bank, all of which was held as restricted cash and cash equivalents and reported as long-term liabilities and assets, respectively. On May 1, 2009, the Company terminated its facility with National City Bank.
On July 2, 2008, as a condition to entering into the credit facility with National City Bank, the Company repaid in full its outstanding indebtedness to Lighthouse Capital Partners V, L.P. The Company paid $713,032 to Lighthouse, which consisted of outstanding principal, accrued interest and a final payment fee due at maturity.
71
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
10. COMMITMENTS AND CONTINGENCIES
Operating Leases. The Company leases various types of office, manufacturing and warehouse facilities and equipment under noncancelable operating leases that expire at various terms through 2014. Future minimum lease payments under non-cancelable operating leases are as follows:
| Year |
Amount | ||
| 2010 |
$ | 628,224 | |
| 2011 |
547,109 | ||
| 2012 |
440,015 | ||
| 2013 |
298,893 | ||
| 2014 |
648 | ||
| Total |
$ | 1,914,889 | |
Rent expense was approximately $793,845, $566,132 and $568,574 in 2009, 2008, and 2007, respectively.
Royalty Agreements. The Company has certain royalty agreements in place with terms that include payment of royalties based on product revenues from sales of current products. Effective January 1, 2010, royalty rates are 5% of such revenues and one agreement includes minimum quarterly payments of $50,000 through 2015 and a maximum of $2,000,000 in total royalties from 2010 through 2015. Parties to royalty agreements each have the right at any time to terminate the agreement immediately for cause. Royalty expense was $205,082, $200,000, and $200,000 for the years ended December 31, 2009, 2008 and 2007, respectively.
Purchase Agreement. On June 15, 2007 the Company entered into a purchase agreement with MicroPace Pty Ltd Inc., (“MicroPace”), which was amended in June 2008. Under the amended agreement, MicroPace produced a derivative of one of their products tailored for the cardiac surgical environment, known as the “MicroPace ORLab™” for worldwide distribution by the Company. Pursuant to the terms of the amended agreement, in order for the Company to retain exclusive distribution rights, the Company is required to purchase a minimum of 70 units during 2008 and is required to purchase 80 units each for 2009 and 2010. As of December 31, 2009, a total of 158 units have been purchased by the Company. Units purchased in excess of yearly minimums in a year reduce future minimum purchase requirements. The Company has 72 units remaining to purchase by December 31, 2010 under the commitment in order to retain exclusive distribution rights.
Life Support Technology, LST b.v. In September of 2007, multiple proceedings between the Company and Life Support Technology, LST b.v., or LST., a former distributor of AtriCure products in Europe, were settled. The settlement agreement provides for the Company to pay LST €257,360 (euros) in 16 payments of €16,085, with the final payment due January 1, 2011. If the U.S. Dollar to Euro conversion rate on any of the 16 payment due dates set forth in the agreement is less than $1.36 to the Euro, the Company will owe LST additional compensation, up to a maximum of €28,310, which reduces over time. The Company has recorded liabilities of $109,755 and $184,632 as of December 31, 2009 and 2008, respectively.
Grant Rights and Obligations. On July 18, 2006 the Company entered into an agreement effective as of June 6, 2005 with The Cleveland Clinic relating to the Company’s rights and obligations with respect to the publicly announced grants from the State of Ohio for, among other things, the creation of an Atrial Fibrillation Innovation Center. Pursuant to the terms of the agreement, the Company was required to supply personnel and materials to accomplish certain research-related activities in connection with the grant and, over a four and one-half year period. The Company could receive up to a total of approximately $900,000 for personnel and materials and The Cleveland Clinic would acquire up to approximately $2,400,000 in capital equipment for the
72
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Company’s use in support of its performance of the agreement. Over the period of the agreement, the Company was required to expend up to approximately $7,700,000 for operating expenses and up to approximately $4,800,000 for capital expenses in support of the agreement. The Company believes these amounts represented ordinary course expenditures that it would have otherwise anticipated making.
The terms of the agreement specified the division of ownership of intellectual property developed in the performance of the agreement and provided, among other things, that the Company would own all intellectual property it developed alone and certain intellectual property that jointly developed and had the option to license certain intellectual property that is owned by The Cleveland Clinic and developed in the performance of the agreement. Through December 31, 2009, the Company earned the entire $900,000 in support of operating expenses and $2,400,000 in acquired capital equipment.
Legal. We are not party to any material pending or threatened litigation, except as described below:
Class Action Lawsuits
AtriCure, Inc. and certain of its current and former officers were named as defendants in a purported securities class action lawsuit filed in the United States District Court for the Southern District of New York (Levine v. AtriCure, Inc., Case No. 06 CV 14324 (United States District Court for the Southern District of New York)). The suit alleges violations of the federal securities laws and seeks damages on behalf of purchasers of the Company’s common stock during the period from our initial public offering in August 2005 through February 16, 2006. The Company filed a motion to dismiss the lawsuit for lack of subject matter jurisdiction. This motion was denied in September 2007, and a motion for reconsideration of that denial was denied in January 2009. Although the Company admitted no wrongdoing, as of December 31, 2009, the Company recorded a liability of $2.0 million, which represented an estimate of the potential defense and/or settlement costs. In addition, the Company recorded a related receivable of $2.0 million from its insurance carrier for the potential defense and/or settlement costs, as recovery is deemed probable.
On December 12, 2008 AtriCure, Inc. and certain of its current executive officers were named in a putative class action lawsuit which is now captioned In re AtriCure, Inc. Securities Litigation, filed in the U.S. District Court for the Southern District of Ohio, Western Division. The plaintiffs allege violations of Sections10(b) and 20(a) of the Securities Exchange Act of 1934 and Rule 10b-5 promulgated thereunder and seek unspecified damages against AtriCure, Inc. and certain of its current executive officers. The plaintiffs allege, among other things, that the defendants issued materially false and misleading statements that failed to disclose that the Company improperly promoted certain products to physicians and caused the filing of false claims for reimbursement. The class period alleged ran from May 10, 2007 through October 31, 2008. In July 2009 the Company filed a motion to dismiss, and in September 2009, the plaintiffs filed their memorandum in opposition to the Company’s motion to dismiss to which the Company responded on November 9, 2009. On March 29, 2010, the court granted in part and denied in part the Company’s motion to dismiss and, in particular, dismissed the claim that the Company caused the filing of false claims for reimbursement. The Company intends to continue vigorously defend this lawsuit. The Company’s liability, if any, resulting from this legal proceeding cannot be estimated and as such no liability is recorded within the Consolidated Financial Statements related to this matter.
Department of Justice Investigation
The Company received a letter on October 27, 2008 from DOJ informing the Company that the DOJ was conducting an investigation for potential False Claims Act and common law violations relating to its surgical ablation devices. Specifically, the letter stated that the DOJ was investigating the Company’s marketing practices utilized in connection with its surgical ablation system to treat AF, a specific use outside the FDA’s 510(k)
73
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
clearance. The letter also stated that the DOJ was investigating whether the Company instructed hospitals to bill Medicare for cardiac surgical ablation using incorrect billing codes. The Company cooperated with the investigation and operated its business in the ordinary course during the investigation. As of December 31, 2009, the Company reached a tentative settlement with the DOJ to resolve the investigation and recorded a liability and charged operating expenses for a total of $3,955,405, which represented the net present value of the proposed settlement amount to be paid to the DOJ, the relator, and relator’s counsel (total payments based on the settlement inclusive of interest were estimated to be $4,350,000, payable over five years).
On February 2, 2010, the settlement was finalized pursuant to the preliminary terms and the Company entered into a settlement agreement with the DOJ, the OIG, and the relator in the qui tam complaint discussed below. The settlement agreement definitively resolved all claims related to the DOJ investigation. The Company did not admit nor will it admit to any wrongdoing in connection with the settlement.
As part of the resolution, the Company also entered into a five year Corporate Integrity Agreement with the OIG. This agreement acknowledges the existence of the Company’s corporate compliance program and provides for certain other compliance-related activities during the five year term of the agreement. Those activities include specific written standards, monitoring, training, education, independent review, disclosure and reporting requirements.
Qui Tam Complaint
A copy of a qui tam complaint against the Company was unsealed on July 10, 2009. The qui tam complaint, filed in the U.S. District Court for the Southern District of Texas, was originally filed by the relator in August 2007. The complaint, which was related to the DOJ investigation, alleged a cause of action under the FCA relating to the Company’s alleged marketing practices in connection with its surgical cardiac ablation devices. In August 2009 the DOJ declined to intervene in the qui tam complaint. Nonetheless, the relator continued to pursue the litigation on behalf of the federal government. Additionally, upon a showing of good cause, the government had the right to intervene in the action at a later time. The qui tam complaint was settled in February 2010 in accordance with the DOJ settlement agreement.
The Company may from time to time become a party to additional legal proceedings.
11. INCOME TAXES
The Company files federal, state, and foreign income tax returns in jurisdictions with varying statutes of limitations. Income taxes are computed using the asset and liability method in accordance with FASB ASC 740 under which deferred income taxes are provided for the temporary differences between the financial reporting basis and the tax basis of the Company’s assets and liabilities. Deferred taxes are measured using provisions of currently enacted tax laws. A valuation allowance against deferred tax assets is recorded when it is more likely than not that such assets will not be fully realized. Tax credits are accounted for as a reduction of income taxes in the year in which the credit originates. The Company does not expect any significant unrecognized tax benefits to arise over the next twelve months.
74
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The detail of deferred tax assets and liabilities at December 31 is as follows:
| 2009 | 2008 | |||||||
| Deferred tax assets (liabilities): |
||||||||
| Net operating loss carryforward |
$ | 19,134,000 | $ | 17,307,000 | ||||
| Research and development credit carryforward |
2,923,000 | 2,692,000 | ||||||
| Equity compensation |
2,445,000 | 1,443,000 | ||||||
| Accruals and reserves |
257,000 | 268,000 | ||||||
| Intangible assets |
917,000 | (204,000 | ) | |||||
| Fixed assets |
409,000 | 353,000 | ||||||
| Inventory |
109,000 | 305,000 | ||||||
| Other, net |
— | 2,000 | ||||||
| Subtotal |
26,194,000 | 22,166,000 | ||||||
| Less valuation allowance |
(26,194,000 | ) | (22,166,000 | ) | ||||
| Total |
$ | — | $ | — | ||||
The Company’s provision for income taxes is as follows:
| 2009 | 2008 | 2007 | ||||||||||
| Current income tax benefit |
$ | (58,639 | ) | $ | (57,252 | ) | $ | — | ||||
| Deferred tax benefit |
(4,028,000 | ) | (3,204,000 | ) | (4,325,000 | ) | ||||||
| Increase in valuation allowance |
4,028,000 | 3,204,000 | 4,325,000 | |||||||||
| Total income tax benefit |
$ | (58,639 | ) | $ | (57,252 | ) | $ | — | ||||
The Company has a federal net operating loss carryforward of approximately $51,272,000 which will begin to expire in 2021. The Company also has state net operating loss carryforwards of approximately $23,742,000 which have varying expirations ranging from 5 years to 20 years. The Company also has a foreign net operating loss carryforward of approximately $4,609,000 which will begin to expire in 2016. Additionally, the Company has a research and development credit carryforward of approximately $2,923,000 which will begin to expire in 2022.
The Company’s 2009 and 2008 effective income tax rate differs from the federal statutory rate as follows:
| 2009 | 2008 | |||||||||||||
| Tax at statutory rate |
34.00 | % | $ | (5,628,232 | ) | 34.00 | % | $ | (3,476,331 | ) | ||||
| R&D credit |
1.85 | (307,050 | ) | (2.02 | ) | 206,376 | ||||||||
| Valuation allowance |
(24.33 | ) | 4,027,943 | (31.34 | ) | 3,204,151 | ||||||||
| Goodwill impairment |
(7.89 | ) | 1,305,885 | — | — | |||||||||
| DOJ Settlement |
(3.87 | ) | 640,326 | — | — | |||||||||
| Other |
0.59 | (97,511 | ) | (0.07 | ) | 8,552 | ||||||||
| Effective tax rate |
0.35 | % | $ | (58,639 | ) | 0.57 | % | $ | (57,252 | ) | ||||
The Company’s pretax book loss for AtriCure, Inc. and its subsidiary, Atricure Europe, B.V., was ($15,001,246) and ($1,552,381), respectively for 2009 and ($9,136,096) and ($1,088,406), respectively for 2008.
On January 1, 2007, the Company adopted the provisions of FIN 48. Application of the provisions of FIN 48 did not result in any change to the Company’s tax account balances. The Company has continued to examine
75
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
its tax positions and has concluded that each meets the more-likely-than-not recognition threshold of FIN 48 and is appropriately measured. The Company does not expect any significant unrecognized tax benefits to arise over the next twelve months. The Company currently has not had to accrue interest and penalties related to unrecognized tax benefits, however, when or if the situation occurs, the Company will recognize interest and penalties within the income tax expense (benefit) line in the accompanying Consolidated Statements of Operations and within the related tax liability line in the Consolidated Balance Sheets. The Company files federal, state, and foreign income tax returns in jurisdictions with varying statutes of limitations. All of the Company’s federal, state and foreign income tax returns open under the statutes of limitations remain subject to examination.
12. CONCENTRATIONS
During fiscal 2009, 2008 and 2007, approximately 22.0%, 17.5% and 19.1%, respectively, of the Company’s total net revenues were derived from its top ten customers. During 2009, 2008, and 2007 no customer accounted for more than 10% of the Company’s revenues.
The Company maintains cash balances which at times exceed FDIC limits. As of December 31, 2009, $1,488,580 of the cash balance was in excess of the FDIC limits.
13. RELATED PARTY
During February 2009, the Company entered into a consulting agreement with Enable Medical Technologies, an entity founded and owned by Michael D. Hooven, the Company’s co-founder and also one of its directors. Under the terms of the agreement, Enable Medical Technologies provided research and development consulting services related to product and procedural development activities. Under the agreement, Enable Medical Technologies received $216,000 as a development fee and, upon completion of certain milestones, earned an additional $15,000. The agreement expired in July 2009.
The Company entered into a Consulting Agreement, dated as of January 1, 2007, with Michael D. Hooven. Under the terms of the agreement, Mr. Hooven provided consulting services and advice to the Company with respect to the creation and development of new products and product platforms relating to cardiac arrhythmias and the prevention or reduction of strokes using cardiac devices. As consideration for his services and for assigning the rights to certain intellectual property as provided for in the agreement, Mr. Hooven was paid $12,000 per month. The term of the consulting services portion of the agreement was for one year and expired on December 31, 2007. The agreement also contained certain non-compete and non-solicitation provisions which expired on December 31, 2009.
14. EMPLOYEE BENEFIT PLANS
The Company sponsors the AtriCure, Inc. 401(k) Plan, a defined contribution plan covering substantially all employees of the Company (the “Plan”). Eligible employees may contribute up to 50% of their pre-tax annual compensation (up to 15% prior to January 1, 2007). During 2008 and 2007, the Company made matching contributions of 50% of the first 6% of employee contributions to the Plan. The Plan was amended effective January 1, 2009 primarily to reflect modifications to the definition of compensation and employee eligibility and effective January 1, 2009 employer contributions to the Plan were suspended. The Company’s matching contributions expensed during 2008 and 2007 were approximately $452,887 and $430,910, respectively. Additional amounts may be contributed to the Plan at the discretion of the Company’s board of directors. No such discretionary contributions were made during 2009, 2008 or 2007.
76
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
15. EQUITY COMPENSATION PLANS
The Company has several share-based incentive plans: the 2001 Stock Option Plan (the “2001 Plan”), the 2005 Equity Incentive Plan (the “2005 Plan”) and the 2008 Employee Stock Purchase Plan (the “ESPP”).
During the fourth quarter of 2009, the Company identified a computational error in the calculation of its employee share-based compensation expense for current and prior year periods after upgrading to a new version of the Company’s third-party equity software. The well-known equity accounting software incorrectly calculated share-based compensation expense by inappropriately applying forfeiture rates over the vesting periods of the share awards. The correction of the error during the fourth quarter of 2009 resulted in changes to the timing of share-based compensation expense over the vesting period of the awards during the relevant periods, but did not change the cumulative share-based compensation expense related to those awards. Because share-based compensation expense is a non-cash item, there is no impact to net cash provided by operations in any period. The cumulative impact of the error was $495,629, which was included in operating expenses within the 2009 Consolidated Statement of Operations.
The Company believes the correction of this error is not material to its previously issued historical consolidated financial statements and the Company does not plan to restate prior periods that were impacted by this error.
2001 Plan and 2005 Plan
The 2001 Plan is no longer used for granting incentives. Under the 2005 Plan, the Board of Directors may grant incentive stock options to employees and any parent or subsidiary’s employees, and may grant nonstatutory stock options, restricted stock, stock appreciation rights, performance units or performance shares to employees, directors and consultants of the Company and any parent or subsidiary’s employees, directors and consultants. The administrator (which is made up of the Company’s Board of Directors or a committee of the Board of Directors) has the power to determine the terms of any awards, including the exercise price of options, the number of shares subject to each award, the exercisability of the awards and the form of consideration.
Options granted under the 2001 Plan and the 2005 Plan generally expire 10 years from the date of grant. Options granted from the 2001 Plan are generally exercisable beginning one year from the date of grant in cumulative yearly amounts of 25% of the shares granted. Options granted from the 2005 Plan generally vest at a rate of 25% on the first anniversary date of the grant and ratably each month thereafter. Certain options granted were exercisable at the time of the grant and the underlying unvested shares are subject to the Company’s repurchase rights as stated in the applicable plan agreement.
As of December 31, 2009, 4,804,149 shares of common stock had been reserved for issuance under the 2005 Plan. The shares authorized for issuance under the 2005 Plan include (a) shares reserved but unissued under the 2001 Plan as of August 10, 2005, (b) shares returned to the 2001 Plan as the result of termination of options or the repurchase of shares issued under such plan, and (c) annual increases in the number of shares available for issuance on the first day of each year equal to the lesser of:
| • | 3.25% of the outstanding shares of common stock on the first day of the fiscal year; |
| • | 825,000 shares; or |
| • | an amount the Company’s Board of Directors may determine. |
Effective January 1, 2009, an additional 463,934 shares were authorized for issuance under the 2005 Equity Incentive Plan representing 3.25% of the outstanding shares on that date. As of December 31, 2009 there were 681,798 shares available for future grants under the plans.
77
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Activity under the Plans during 2009 was as follows:
| Stock Options |
Number of Shares Outstanding |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value | |||||||
| Outstanding at January 1, 2009 |
2,629,310 | $ | 8.51 | ||||||||
| Granted |
118,500 | $ | 2.62 | ||||||||
| Cancelled or forfeited |
(192,936 | ) | $ | 9.64 | |||||||
| Exercised |
(20,897 | ) | $ | 1.60 | |||||||
| Outstanding at December 31, 2009 |
2,533,977 | $ | 8.20 | 6.03 | $ | 2,937,181 | |||||
| Vested and expected to vest |
2,486,288 | $ | 8.22 | 5.96 | $ | 2,881,439 | |||||
| Exercisable at December 31, 2009 |
1,826,766 | $ | 7.98 | 5.25 | $ | 2,523,284 | |||||
| Restricted Stock |
Number of Shares Outstanding |
Weighted Average Grant Date Fair Value |
|||||||||
| Outstanding at January 1, 2009 |
161,893 | $ | 2.15 | ||||||||
| Granted |
666,320 | $ | 2.48 | ||||||||
| Forfeited |
(28,500 | ) | $ | 2.83 | |||||||
| Released |
(438,804 | ) | $ | 1.68 | |||||||
| Outstanding at December 31, 2009 |
360,909 | $ | 3.26 | ||||||||
Activity under the Plans during 2008 was as follows:
| Stock Options |
Number of Shares Outstanding |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value | |||||||
| Outstanding at January 1, 2008 |
2,296,035 | $ | 8.11 | ||||||||
| Granted |
660,230 | $ | 9.99 | ||||||||
| Cancelled or forfeited |
(239,418 | ) | $ | 10.85 | |||||||
| Exercised |
(87,537 | ) | $ | 2.74 | |||||||
| Outstanding at December 31, 2008 |
2,629,310 | $ | 8.51 | 6.99 | $ | 506,458 | |||||
| Vested and expected to vest |
2,476,174 | $ | 8.41 | 6.88 | $ | 506,458 | |||||
| Exercisable at December 31, 2008 |
1,392,372 | $ | 7.02 | 5.55 | $ | 506,458 | |||||
| Restricted Stock |
Number of Shares Outstanding |
Weighted Average Grant Date Fair Value |
|||||||||
| Outstanding at January 1, 2008 |
— | $ | — | ||||||||
| Granted |
161,893 | $ | 2.15 | ||||||||
| Outstanding at December 31, 2008 |
161,893 | $ | 2.15 | ||||||||
78
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
The total intrinsic value of options exercised during the years ended December 31, 2009, 2008 and 2007 was $65,403, $770,381 and $1,302,202, respectively, and as a result of the Company’s tax position, no tax benefit was recognized related to the stock option exercises. For the years ended December 31, 2009, 2008 and 2007, $33,335, $239,873 and $174,942, respectively, in cash proceeds was included in the Company’s Consolidated Statements of Cash Flows as a result of the exercise of stock options.
The exercise price per share of each option is equal to the fair market value of the underlying share on the date of grant. The Company issues registered shares of common stock to satisfy stock option exercises and restricted stock grants.
The Company recognized expense related to stock options and restricted stock for the years ended December 31, 2009, 2008, and 2007 of $3,577,317, $2,494,369 and $1,510,361, respectively. As of December 31, 2009 there was $4,138,366 of unrecognized compensation costs related to non-vested share-based compensation arrangements ($3,074,713 relating to stock options and $1,063,653 relating to restricted stock). This cost is expected to be recognized over a weighted-average period of 2.0 years for stock options and 1.7 years for restricted stock.
In 2008 the Company issued performance shares to certain employees to incent and reward them for the achievement of specified performance metrics during 2009 and 2010 over a service period ending December 31, 2010. The participant received an award for a specified number of shares of the Company’s common stock at the beginning of the award period, which entitles the participant to the shares at the end of the award period, if achievement of the specified metrics and service requirements occurs. As of December 31, 2009 the Company has the potential to issue 269,750 shares of common stock based upon each participant meeting all of the specified metrics. In accordance with FASB ASC 718, the Company estimates the number of shares to be granted based upon the probability that the performance metric and service period will be achieved. The fair value of the estimated award is expensed over the award period. During the year ended December 31, 2009, the Company recognized expense related to the performance shares of $185,413. The probability of meeting the specified metrics is reviewed quarterly and the estimated expense is adjusted in the current period. As of December 31, 2009, there was $425,566 of unrecognized compensation costs related to non-vested share-based compensation arrangements associated with these performance shares. This cost is expected to be recognized over a weighted-average period of 1.0 years.
Employee Stock Purchase Plan (ESPP)
During the second quarter of 2008, the Company established its 2008 Employee Stock Purchase Plan (“ESPP”) which is available to eligible employees as defined in the plan. Under the ESPP, shares of the Company’s common stock may be purchased at a discount (currently 15%) of the lesser of the closing price of the Company’s common stock on the first trading day or the last trading day of the offering period. The offering period (currently six months) and the offering price are subject to change. Participants may not purchase more than $25,000 of the Company’s common stock in a calendar year and effective January 1, 2009, may not purchase more than 1,500 shares during an offering period. Beginning on January 1, 2009 and on the first day of each fiscal year thereafter during the term of the ESPP, the number of shares available for sale under the ESPP shall be increased by the lesser of (i) two percent (2%) of the Company’s outstanding shares of common stock as of the close of business on the last business day of the prior calendar year, not to exceed 600,000 shares, or (ii) a lesser amount determined by the Board of Directors. At December 31, 2009, there were 413,104 shares available for future issuance under the ESPP, including 285,498 shares approved for issuance by the Company’s Board of Directors effective January 1, 2009. Share-based compensation expense with respect to the ESPP was $103,191 and $33,903 for the years ended December 31, 2009 and 2008, respectively.
79
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Valuation and Expense Information Under FASB ASC 718
The following table summarizes share-based compensation expense related to employee share-based compensation under FASB ASC 715 for 2009, 2008 and 2007. This expense was allocated as follows:
| 2009 | 2008 | 2007 | |||||||
| Cost of revenues |
$ | 284,817 | $ | 151,270 | $ | 85,902 | |||
| Research and development expenses |
757,660 | 346,698 | 243,246 | ||||||
| Selling, general and administrative expenses |
2,823,444 | 2,171,354 | 1,181,213 | ||||||
| Total |
$ | 3,865,921 | $ | 2,669,322 | $ | 1,510,361 | |||
In calculating compensation expense, the fair value of the options is estimated on the grant date using the Black-Scholes model including the following assumptions:
| 2009 | 2008 | 2007 | ||||
| Risk free interest rate |
2.07 - 3.08% | 1.89 - 3.54% | 3.42 - 5.07% | |||
| Expected life of option (years) |
6.0 to 6.25 | 6.0 to 6.25 | 6.0 | |||
| Expected volatility of stock |
53.50 - 62.00% | 43.00 - 51.00% | 42.00 - 45.00% | |||
| Weighted-average volatility |
60.43% | 44.31% | 44.08% | |||
| Dividend yield |
0.00% | 0.00% | 0.00% |
Due to the Company’s limited operating and trading history, volatility is estimated based on an equal weighting of both the Company’s trading history and other companies in the industry. The risk-free interest rate assumption is based upon the U.S. treasury yield curve at the time of grant for the expected option life. The simplified method is utilized in determining the expected life of the option.
Based on the assumptions noted above, the weighted average estimated fair values of the options granted in the years ended December 31, 2009, 2008, and 2007 were $1.53, $4.71, and $5.21, respectively.
Non-Employee Stock Compensation
The Company has issued nonstatutory common stock options to consultants to purchase shares of common stock. Such options vest over a service period ranging from immediately to four years. After January 1, 2006, all stock options were issued with a four year vesting period and vest at a rate of 25% on the first anniversary date of the grant and ratably each month thereafter.
The fair value at the date of grant, which is subject to adjustment at each vesting date based upon the fair value of the Company’s common stock, was determined using the Black-Scholes model. There were no non-employee stock options granted during 2009. The fair value at the date of grant, which is subject to adjustment at each vesting date based upon the fair value of the Company’s common stock, was determined using the Black-Scholes model with the following assumptions:
| 2008 | 2007 | |||||
| Risk free interest rate |
3.45 | % | 4.73 | % | ||
| Expected life of option (years) |
10.0 | 6.0 | ||||
| Expected volatility of stock |
43.00 | % | 45.00 | % | ||
| Weighted-average volatility |
43.00 | % | 45.00 | % | ||
| Dividend yield |
0.00 | % | 0.00 | % |
The values attributable to non-employee options have been amortized over the service period on a graded vesting method and the vested portion of these options was remeasured at each vesting date.
80
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
Stock compensation expense with respect to non-employee stock options totaled $19,675, $1,681 and $381,858 for 2009, 2008 and 2007, respectively.
Certain of the Company’s share-based payment arrangements are outside the scope of FASB ASC 718 and are subject to FASB ASC 815, which requires vested stock awards held by certain non-employee consultants to be accounted for as liability awards until these awards are exercised or forfeited. The fair value of these awards is remeasured at each financial statement date until the awards are settled or expire. The Company recorded (expense) income as a result of the remeasurement of the fair value of these awards during 2009, 2008, and 2007 of ($140,620), $522,992 and ($227,421), respectively. As of December 31, 2009 and 2008, respectively, options to acquire 52,359 and 54,660 shares of common stock held by non-employee consultants remained unexercised and a liability of $180,288 and $40,368 was included in accrued liabilities in the Consolidated Balance Sheets.
16. SEGMENT AND GEOGRAPHIC INFORMATION
The Company considers reporting segments in accordance with FASB ASC 280, “Segment Reporting”. The Company develops, manufactures, and sells devices designed primarily for the surgical ablation of cardiac tissue. These devices are developed and marketed to a broad base of medical centers in the United States and internationally. Management considers all such sales to be part of a single operating segment.
Geographic revenues were as follows:
| Revenues: | 2009 | 2008 | 2007 | ||||||
| United States |
$ | 44,119,201 | $ | 46,918,091 | $ | 41,717,785 | |||
| International |
10,414,357 | 8,338,932 | 6,591,278 | ||||||
| Total |
$ | 54,533,558 | $ | 55,257,023 | $ | 48,309,063 | |||
Substantially all of the Company’s long-lived assets are located in the United States.
17. SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED)
(Dollars in thousands, except per share data)
| For the Three Months Ended | ||||||||||||||||||||||||||||||||
| March 31, | June 30, | September 30, | December 31, | |||||||||||||||||||||||||||||
| 2009(1) | 2008 | 2009 | 2008 | 2009(2) | 2008 | 2009 | 2008 | |||||||||||||||||||||||||
| Operating Results: |
||||||||||||||||||||||||||||||||
| Revenues |
$ | 13,674 | $ | 13,530 | $ | 13,778 | $ | 14,859 | $ | 13,281 | $ | 14,802 | $ | 13,800 | $ | 12,066 | ||||||||||||||||
| Gross profit |
10,729 | 10,299 | 10,670 | 11,364 | 10,003 | 11,406 | 10,380 | 8,964 | ||||||||||||||||||||||||
| Loss from operations |
(7,932 | ) | (3,896 | ) | (1,033 | ) | (1,825 | ) | (4,432 | ) | (1,818 | ) | (2,114 | ) | (3,458 | ) | ||||||||||||||||
| Net loss |
(7,965 | ) | (3,605 | ) | (1,443 | ) | (1,593 | ) | (4,697 | ) | (1,770 | ) | (2,390 | ) | (3,199 | ) | ||||||||||||||||
| Net loss per share (basic and diluted) |
$ | (0.56 | ) | $ | (0.25 | ) | $ | (0.10 | ) | $ | (0.11 | ) | $ | (0.32 | ) | $ | (0.12 | ) | $ | (0.16 | ) | $ | (0.22 | ) | ||||||||
| (1) | As a result of a reduction in our market capitalization during the quarter ended March 31, 2009, we believed an indication of impairment existed and as such, performed an interim analysis of our goodwill as of March 31, 2009 as required by FASB ASC 350, “Goodwill and Other Intangible Assets” (“ASC 350”). The analysis concluded that the carrying value of our goodwill exceeded the estimated fair value, and as such, we recognized a full impairment loss of $6.8 million during 2009. See Note 5, “Goodwill and Intangible Assets” to our Consolidated Financial Statements. |
81
Table of Contents
ATRICURE, INC. AND SUBSIDIARY
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS—(Continued)
| (2) | During the third quarter ended September 30, 2009, we recorded $4.0 million in expense related to a settlement with the DOJ. See Note 10, “Commitments and Contingencies,” to our Consolidated Financial Statements. |
Amounts may not sum to consolidated totals for the full year due to rounding. Basic and diluted net loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly per share amounts will not necessarily equal the total for the year.
18. SUBSEQUENT EVENTS
Subsequent to year end, on February 2, 2010, the Company entered into a settlement agreement among the Company, the DOJ, the OIG and the relator in the DOJ investigation and qui tam complaint (“Settlement Agreement”). The Settlement Agreement definitively resolved all claims related to the DOJ investigation and qui tam complaint. The Company did not admit nor will it admit to any wrongdoing in connection with the settlement.
As part of the settlement, the Company also entered into a five year Corporate Integrity Agreement with the OIG. This agreement acknowledges the existence of the Company’s corporate compliance program and provides for certain other compliance-related activities during the five year term of the agreement. Those activities include specific written standards, monitoring, training, education, independent review, disclosure and reporting requirements.
On March 26, 2010, the Company entered into a Waiver and Second Loan Modification Agreement (“Second Amendment”) with Silicon Valley Bank, which amended, among other things, the financial covenants in the Agreement and waived a compliance violation which occurred during February 2010.
82
Table of Contents
VALUATION AND QUALIFYING ACCOUNTS
| Beginning Balance |
Additions | Deductions | Ending Balance | |||||||||
| Allowance for doubtful accounts receivable |
||||||||||||
| Year ended December 31, 2009 |
$ | 40,480 | $ | — | $ | 16,080 | $ | 24,400 | ||||
| Year ended December 31, 2008 |
26,181 | 20,979 | 6,680 | 40,480 | ||||||||
| Year ended December 31, 2007 |
343,127 | — | 316,946 | 26,181 | ||||||||
| Reserve for sales returns and allowances |
||||||||||||
| Year ended December 31, 2009 |
$ | 71,251 | $ | — | $ | 67,806 | $ | 3,445 | ||||
| Year ended December 31, 2008 |
73,937 | 71,251 | 73,937 | 71,251 | ||||||||
| Year ended December 31, 2007 |
— | 73,937 | — | 73,937 | ||||||||
| Allowance for inventory valuation |
||||||||||||
| Year ended December 31, 2009 |
$ | 183,895 | $ | 25,346 | $ | 26,071 | $ | 183,170 | ||||
| Year ended December 31, 2008 |
116,858 | 121,854 | 54,817 | 183,895 | ||||||||
| Year ended December 31, 2007 |
94,667 | 36,425 | 14,234 | 116,858 | ||||||||
| Valuation allowance for deferred tax assets |
||||||||||||
| Year ended December 31, 2009 |
$ | 22,166,000 | $ | 4,028,000 | $ | — | $ | 26,194,000 | ||||
| Year ended December 31, 2008 |
18,962,000 | 3,204,000 | — | 22,166,000 | ||||||||
| Year ended December 31, 2007 |
14,637,000 | 4,325,000 | — | 18,962,000 | ||||||||
83
Table of Contents
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
ITEM 9A(T). CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
We have evaluated the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rules 13(a)-15(e) and 15(d)-15(e) of the Securities Exchange Act of 1934 (the “Exchange Act”), as of the end of the period covered by this report. Our management, including the Chief Executive Officer and Chief Financial Officer, supervised and participated in the evaluation. Based on the evaluation, we concluded that, as of the end of the period covered by this report, our disclosure controls and procedures were effective in providing reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s forms and rules, and the material information relating to the Company is accumulated and communicated to management, including the Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosures.
Control systems, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that control objectives are met. Because of inherent limitations in all control systems, no evaluation of controls can provide assurance that all control issues and instances of fraud, if any, within a company will be detected. Additionally, controls can be circumvented by individuals, by collusion of two or more people or by management override. Over time, controls can become inadequate because of changes in conditions or the degree of compliance may deteriorate. Further, the design of any system of controls is based in part upon assumptions about the likelihood of future events. There can be no assurance that any design will succeed in achieving its stated goals under all future conditions. Because of the inherent limitations in any cost-effective control system, misstatements due to errors or fraud may occur and not be detected.
Management’s Annual Report on Internal Control Over Financial Reporting
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting. The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. Internal control over financial reporting includes policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with U.S. generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements. The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2009. No matter how well designed, because of inherent limitations in all control systems, internal control over financial reporting may not prevent or detect misstatements should they occur. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the control procedures may deteriorate. In making this assessment, the Company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control- Integrated Framework. Based on such assessment, management has concluded that the Company’s internal control over financial reporting was effective as of December 31, 2009. This annual report does not include an attestation report of the Company’s registered public accounting firm regarding internal control over financial
84
Table of Contents
reporting. Management’s report was not subject to attestation by the Company’s registered public accounting firm pursuant to temporary rules of the Securities and Exchange Commission that permit the Company to provide only management’s report in this annual report.
Changes in Internal Control over Financial Reporting
There were no changes in the Company’s internal control over financial reporting that occurred during the quarter ended December 31, 2009 that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
| ITEM 9B. | OTHER INFORMATION |
None.
| ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this Item is incorporated by reference to the definitive proxy statement for our 2010 Annual Meeting of Stockholders to be filed with the Securities and Exchange Commission within 120 days after the end of 2009 (the “Proxy Statement”).
| ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item is incorporated by reference to the Proxy Statement.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item is incorporated by reference to the Proxy Statement.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE |
The information required by this Item is incorporated by reference to the Proxy Statement.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
The information required by this Item is incorporated by reference to the Proxy Statement.
85
Table of Contents
| ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES |
(1) The financial statements required by Item 15(a) are filed in Item 8 of this Form 10-K.
(2) The financial statement schedules required by Item 15(a) are filed in Item 8 of this Form 10-K.
(3) The following exhibits are included in this Form 10-K or incorporated by reference in this Form 10-K:
| Exhibit No. |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation (incorporated by reference to our Registration Statement on Form S-1 (Registration No. 333-124197) filed on April 20, 2005)). | |
| 3.2 | Second Amended and Restated Bylaws (incorporated by reference to our Registration Statement on Form S-1 (Registration No. 333-124197) filed on April 20, 2005). | |
| 4.1 | Amended and Restated Investors’ Rights Agreement, dated June 6, 2002 between AtriCure, Inc. and each of the signatory Investors (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 4.2 | Amendment No. 1 to Amended and Restated Investors’ Rights Agreement, dated March 8, 2005 between AtriCure, Inc. and each of the signatory Investors (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 4.3 | Specimen common stock certificate (incorporated by reference to Amendment No. 2 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on July 7, 2005). | |
| 4.4 | Registration Rights Agreement, dated May 24, 2007, by and between AtriCure, Inc. and those purchasers executing the Registration Rights (incorporated by reference to our Current Report on Form 8-K, filed on May 25, 2007). | |
| 4.5 | Warrant to purchase AtriCure, Inc. common stock issued to Silicon Valley Bank on May 1, 2009 (incorporated by reference to our Quarterly Report on Form 10-Q, filed on August 10, 2009). | |
| 10.1# | 2001 Stock Option Plan (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 10.2# | 2005 Equity Incentive Plan (incorporated by reference to Amendment No. 2 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on July 7, 2005). | |
| 10.3 | Agreement, dated as of July 18, 2006, by and between AtriCure, Inc. and the Cleveland Clinic (incorporated by reference to our Current Report on Form 8-K, filed on July 20, 2006). | |
| 10.4 | Amendment No. 1, dated as of December 1, 2008, to Agreement dated as of July 18, 2006 by and between AtriCure, Inc. and the Cleveland Clinic (incorporated by reference to our Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.5 | Amendment No. 2, effective as of December 28, 2009, to Agreement dated as of July 18, 2006 by and between AtriCure, Inc. and the Cleveland Clinic. | |
| 10.6# | Consulting Agreement, dated as of January 1, 2007, between AtriCure, Inc. and Michael D. Hooven (incorporated by reference to our Current Report on Form 8-K, filed on January 5, 2007). | |
| 10.7# | Employment Agreement, dated as of January 5, 2007, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on January 9, 2007). | |
| 10.8# | Amendment of Employment Agreement, dated as of April 17, 2007, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on April 20, 2007). | |
86
Table of Contents
| Exhibit No. |
Description | |
| 10.9# | Amendment No. 2 to Employment Agreement, effective as of January 1, 2010, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on March 12, 2010). | |
| 10.10# | Employment Agreement, dated as of February 9, 2007, between AtriCure, Inc. and David J. Drachman (incorporated by reference to our Current Report on Form 8-K, filed on February 14, 2007). | |
| 10.11# | Amendment No. 1 to Employment Agreement, effective as of January 1, 2010, between AtriCure, Inc. and David J. Drachman (incorporated by reference to our Current Report on Form 8-K, filed on March 12, 2010). | |
| 10.12 | Bill of Sale and Assignment Agreement, dated as of August 7, 2007, between CooperSurgical, Inc. and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on August 9, 2007). | |
| 10.13 | Non-Competition Agreement, dated as of August 7, 2007, between CooperSurgical, Inc. and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on August 9, 2007). | |
| 10.14 | 2005 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to the Registrant’s Form S-8 Registration Statement (File No. 333-152014) filed on June 30, 2008). | |
| 10.15 | 2008 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.1 to the Registrant’s Form S-8 Registration Statement (File No. 333-152013) filed on June 30, 2008). | |
| 10.16 | Credit Agreement, dated as of July 1, 2008, among AtriCure, Inc., the lenders party thereto, and National City Bank as administrative agent for the lenders (incorporated by reference to our Current Report on Form 8-K, filed on July 2, 2008). | |
| 10.17# | Form of Performance Share Agreement (incorporated by reference to our Current Report on Form 8-K, filed on October 31, 2008). | |
| 10.18# | Amended Form of Performance Share Agreement (incorporated by reference to our Current Report on Form 8-K, filed on March 30, 2009). | |
| 10.19 | Loan and Security Agreement, dated as of May 1, 2009, between Silicon Valley Bank and AtriCure, Inc. (incorporated by reference to our Quarterly Report on Form 10-Q, filed on August 10, 2009). | |
| 10.20 | Consent, Waiver and First Loan Modification Agreement dated November 4, 2009, between Silicon Valley Bank and AtriCure, Inc. (incorporated by reference to our Quarterly Report on Form 10-Q, filed on November 11, 2010). | |
| 10.21 | Waiver and Second Loan Modification Agreement dated March 26, 2010, between Silicon Valley Bank and AtriCure, Inc. | |
| 10.22 | Settlement Agreement as of February 2, 2010 by and among the United States of America, acting through the United States Department of Justice and on behalf of the Office of Inspector General of the Department of Health and Human Services, the Company and the Relator (incorporated by reference to our Current Report on Form 8-K, filed on February 5, 2010). | |
| 10.23 | Corporate Integrity Agreement between the Office of Inspector General of the Department of Health and Human Services and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on February 5, 2010). | |
| 21 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Deloitte & Touche LLP. | |
| 31.1 | Rule 13a-14(a) Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
87
Table of Contents
| Exhibit No. |
Description | |
| 31.2 | Rule 13a-14(a) Certification of Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350 by the Chief Executive Officer, as adopted, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350 by the Chief Financial Officer, as adopted, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
| † | Portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
| # | Compensatory plan or arrangement. |
88
Table of Contents
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this Form 10-K to be signed on our behalf by the undersigned, thereunto duly authorized.
| AtriCure, Inc. | ||
| (REGISTRANT) | ||
| Date: March 29, 2010 | /s/ David J. Drachman | |
| David J. Drachman President and Chief Executive Officer (Principal Executive Officer) | ||
| Date: March 29, 2010 | /s/ Julie A. Piton | |
| Julie A. Piton Vice President of Finance and Administration (Principal Financial and Accounting Officer) | ||
KNOW ALL MEN AND WOMEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints David J. Drachman, his attorney-in-fact, with the power of substitution, for him in any and all capacities, to sign any and all amendments to this Form 10-K, and to file the same, with exhibits thereto and other documents in connection therewith, with the U.S. Securities and Exchange Commission, granting unto said attorneys-in-fact, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact, and any of them or his substitute or substitutes, may do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Form 10-K has been signed by the following persons on behalf of the registrant and in the capacities indicated on March 29, 2010:
| Signature |
Title(s) | |
| /s/ Richard M. Johnston Richard M. Johnston |
Richard M. Johnston Chairman of the Board | |
| /s/ David J. Drachman David J. Drachman |
David J. Drachman Director, President and Chief Executive Officer (Principal Executive Officer) | |
| /s/ Julie A. Piton Julie A. Piton |
Julie A. Piton Vice President of Finance and Administration and Chief Financial Officer (Principal Financial and Accounting Officer) | |
| /s/ Mark A. Collar Mark A. Collar |
Mark A. Collar Director | |
| /s/ Donald C. Harrison Donald C. Harrison |
Donald C. Harrison Director | |
| /s/ Michael D. Hooven Michael D. Hooven |
Michael D. Hooven Director | |
89
Table of Contents
| Signature |
Title(s) | |
| /s/ Elizabeth D. Krell Elizabeth D. Krell |
Elizabeth D. Krell Director | |
| /s/ Mark R. Lanning Mark R. Lanning |
Mark R. Lanning Director | |
| /s/ Karen P. Robards Karen P. Robards |
Karen P. Robards Director | |
90
Table of Contents
EXHIBIT INDEX
| Exhibit No. |
Description | |
| 3.1 | Amended and Restated Certificate of Incorporation (incorporated by reference to our Registration Statement on Form S-1 (Registration No. 333-124197) filed on April 20, 2005)). | |
| 3.2 | Second Amended and Restated Bylaws (incorporated by reference to our Registration Statement on Form S-1 (Registration No. 333-124197) filed on April 20, 2005). | |
| 4.1 | Amended and Restated Investors’ Rights Agreement, dated June 6, 2002 between AtriCure, Inc. and each of the signatory Investors (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 4.2 | Amendment No. 1 to Amended and Restated Investors’ Rights Agreement, dated March 8, 2005 between AtriCure, Inc. and each of the signatory Investors (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 4.3 | Specimen common stock certificate (incorporated by reference to Amendment No. 2 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on July 7, 2005). | |
| 4.4 | Registration Rights Agreement, dated May 24, 2007, by and between AtriCure, Inc. and those purchasers executing the Registration Rights (incorporated by reference to our Current Report on Form 8-K, filed on May 25, 2007). | |
| 4.5 | Warrant to purchase AtriCure, Inc. common stock issued to Silicon Valley Bank on May 1, 2009 (incorporated by reference to our Quarterly Report on Form 10-Q, filed on August 10, 2009). | |
| 10.1# | 2001 Stock Option Plan (incorporated by reference to Amendment No. 1 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on June 14, 2005). | |
| 10.2# | 2005 Equity Incentive Plan (incorporated by reference to Amendment No. 2 to our Registration Statement on Form S-1 (Registration No. 333-124197), filed on July 7, 2005). | |
| 10.3 | Agreement, dated as of July 18, 2006, by and between AtriCure, Inc. and the Cleveland Clinic (incorporated by reference to our Current Report on Form 8-K, filed on July 20, 2006). | |
| 10.4 | Amendment No. 1, dated as of December 1, 2008, to Agreement dated as of July 18, 2006 by and between AtriCure, Inc. and the Cleveland Clinic (incorporated by reference to our Annual Report on Form 10-K filed on March 16, 2009). | |
| 10.5 | Amendment No. 2, effective as of December 28, 2009, to Agreement dated as of July 18, 2006 by and between AtriCure, Inc. and the Cleveland Clinic. | |
| 10.6# | Consulting Agreement, dated as of January 1, 2007, between AtriCure, Inc. and Michael D. Hooven (incorporated by reference to our Current Report on Form 8-K, filed on January 5, 2007). | |
| 10.7# | Employment Agreement, dated as of January 5, 2007, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on January 9, 2007). | |
| 10.8# | Amendment of Employment Agreement, dated as of April 17, 2007, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on April 20, 2007). | |
| 10.9# | Amendment No. 2 to Employment Agreement, effective as of January 1, 2010, between AtriCure, Inc. and Julie A. Piton (incorporated by reference to our Current Report on Form 8-K, filed on March 12, 2010). | |
| 10.10# | Employment Agreement, dated as of February 9, 2007, between AtriCure, Inc. and David J. Drachman (incorporated by reference to our Current Report on Form 8-K, filed on February 14, 2007). | |
91
Table of Contents
| Exhibit No. |
Description | |
| 10.11# | Amendment No. 1 to Employment Agreement, effective as of January 1, 2010, between AtriCure, Inc. and David J. Drachman (incorporated by reference to our Current Report on Form 8-K, filed on March 12, 2010). | |
| 10.12 | Bill of Sale and Assignment Agreement, dated as of August 7, 2007, between CooperSurgical, Inc. and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on August 9, 2007). | |
| 10.13 | Non-Competition Agreement, dated as of August 7, 2007, between CooperSurgical, Inc. and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on August 9, 2007). | |
| 10.14 | 2005 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 to the Registrant’s Form S-8 Registration Statement (File No. 333-152014) filed on June 30, 2008). | |
| 10.15 | 2008 Employee Stock Purchase Plan (incorporated by reference to Exhibit 10.1 to the Registrant’s Form S-8 Registration Statement (File No. 333-152013) filed on June 30, 2008). | |
| 10.16 | Credit Agreement, dated as of July 1, 2008, among AtriCure, Inc., the lenders party thereto, and National City Bank as administrative agent for the lenders (incorporated by reference to our Current Report on Form 8-K, filed on July 2, 2008). | |
| 10.17# | Form of Performance Share Agreement (incorporated by reference to our Current Report on Form 8-K, filed on October 31, 2008). | |
| 10.18# | Amended Form of Performance Share Agreement (incorporated by reference to our Current Report on Form 8-K, filed on March 30, 2009). | |
| 10.19 | Loan and Security Agreement, dated as of May 1, 2009, between Silicon Valley Bank and AtriCure, Inc. (incorporated by reference to our Quarterly Report on Form 10-Q, filed on August 10, 2009). | |
| 10.20 | Consent, Waiver and First Loan Modification Agreement dated November 4, 2009, between Silicon Valley Bank and AtriCure, Inc. (incorporated by reference to our Quarterly Report on Form 10-Q, filed on November 11, 2010). | |
| 10.21 | Waiver and Second Loan Modification Agreement dated March 26, 2010, between Silicon Valley Bank and AtriCure, Inc. | |
| 10.22 | Settlement Agreement as of February 2, 2010 by and among the United States of America, acting through the United States Department of Justice and on behalf of the Office of Inspector General of the Department of Health and Human Services, the Company and the Relator (incorporated by reference to our Current Report on Form 8-K, filed on February 5, 2010). | |
| 10.23 | Corporate Integrity Agreement between the Office of Inspector General of the Department of Health and Human Services and AtriCure, Inc. (incorporated by reference to our Current Report on Form 8-K, filed on February 5, 2010). | |
| 21 | Subsidiaries of the Registrant. | |
| 23.1 | Consent of Deloitte & Touche LLP. | |
| 31.1 | Rule 13a-14(a) Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 31.2 | Rule 13a-14(a) Certification of Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
| 32.1 | Certification pursuant to 18 U.S.C. Section 1350 by the Chief Executive Officer, as adopted, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | |
92
Table of Contents
| Exhibit No. |
Description | |
| 32.2 | Certification pursuant to 18 U.S.C. Section 1350 by the Chief Financial Officer, as adopted, pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| † | Portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission pursuant to a request for confidential treatment. |
| # | Compensatory plan or arrangement. |
93