Attached files
UNITED
STATES SECURITIES AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
10-K
|X|
ANNUAL REPORT UNDER SECTION 13 OR 15(d) OF THE SECURITIES
EXCHANGE
ACT OF
1934
FOR THE FISCAL YEAR
ENDED JUNE 30, 2009
OR
£
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES
EXCHANGE ACT OF 1934
FOR THE
TRANSITION PERIOD FROM _____ TO __________
Commission
File Number: 000-52321
Cellceutix
Corporation
(Exact
name of registrant as specified in its charter)
|
Nevada
|
13-4303398
|
|||
|
(State
or other jurisdiction
|
(IRS
Employer
|
|||
|
of
incorporation)
|
Identification
No.)
|
100
Cummings Center, Suite 151-B
Beverly,
MA 01915
(Address
of principal executive offices and zip code)
(978)-633-3623
(Registrant’s
telephone number, including area code)
SECURITIES
REGISTERED UNDER SECTION 12(b) OF THE EXCHANGE ACT: NONE SECURITIES REGISTERED
UNDER SECTION 12(g) OF THE EXCHANGE ACT: COMMON STOCK, PAR VALUE $0.0001 PER
SHARE
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in
Rule 405 of the Securities Act. Yes |_| No X
Indicate
by check mark if the registrant is not required to file reports pursuant to
Section 13 or 15(d) of the Act.
Yes
|_| No X
Indicate
by check mark whether the registrant (1) has filed all reports required to be
filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the
past 12 months (or for such shorter period that the registrant was required to
file such reports), and (2) has been subject to such filing requirements for the
past 90 days: Yes |X| No |_|
Indicate by check mark whether the
registrant has submitted electronically and posted on its corporate Web site, if
any, every Interactive Data File required to be submitted and posted pursuant to
Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12
months (or for such shorter period that the registrant was required to submit
and post such
files).
Yes No
|X|.
Indicate
by check mark if disclosure of delinquent filers in response to Item 405 of
Regulation S-K is not contained herein, and will not be contained, to the best
of Registrant's knowledge, in definitive proxy or information statements
incorporated by reference in Part III of this Form 10-K or any amendment to this
Form 10-K |.
Yes No
|X|.
Indicate
by check mark whether the registrant is a large accelerated filer, a
non-accelerated filer, or a smaller reporting company. See definitions of
"accelerated filer and large accelerated filer" and "smaller reporting company"
in Rule 12b-2 of the Exchange Act.
|
Accelerated
filer
|
|
|
Non-accelerated
filer
|
Smaller
reporting company |X|.
|
Indicate
by check mark whether the registrant is a shell company (as defined in Rule
12b-2 of the Exchange Act) YES NO|X|.
Issuer's
revenues for its most recent fiscal year were $0.00
The
aggregate market value of voting stock held by non-affiliates of the Registrant
at June 30, 2009 was $16,696,485 computed by reference to the last traded sale
price as reported on the Over the Counter market on such date.
The
number of shares outstanding of the Registrant's common stock as of September
30, 2009 was 91,861,000 shares.
CELLCEUTIX
CORPORATION
FORM 10-K
INDEX
|
PAGE
NO
|
||
|
PART I
|
||
|
ITEM 1
|
BUSINESS
|
2
|
|
ITEM 1A
|
RISK
FACTORS
|
15
|
|
ITEM 2
|
DESCRIPTION
OF PROPERTIES
|
29
|
|
ITEM 3
|
LEGAL
PROCEEDINGS
|
29
|
|
ITEM 4
|
SUBMISSION
OF MATTERS TO A VOTE OF SECURITY HOLDERS
|
29
|
|
PART II
|
||
|
ITEM 5
|
MARKET
FOR REGISTRANT'S COMMON EQUITY AND RELATED STOCKHOLDER
MATTERS
|
29
|
|
ITEM 6
|
SELECTED
FINANCIAL DATA
|
32
|
|
ITEM 7
|
MANAGEMENT'S
DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
|
32
|
|
ITEM 7A
|
QUANTITATIVE
AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
|
37
|
|
ITEM 8
|
FINANCIAL
STATEMENTS AND SUPPLEMENTARY DATA
|
37
|
|
ITEM 9
|
CHANGES
IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
|
51
|
|
ITEM 9A(T)
|
CONTROLS
AND PROCEDURES
|
51
|
|
ITEM 9B
|
OTHER
INFORMATION
|
52
|
|
PART III
|
||
|
ITEM 10
|
DIRECTORS ,
EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
|
52
|
|
ITEM 11
|
EXECUTIVE
COMPENSATION
|
54
|
|
ITEM 12
|
SECURITY
OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED
STOCKHOLDER MATTERS
|
56
|
|
ITEM 13
|
CERTAIN
RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
|
57
|
|
ITEM 14
|
PRINCIPAL
ACCOUNTANT FEES AND SERVICES
|
58
|
|
PART IV
|
||
|
ITEM 15
|
EXHIBITS
AND FINANCIAL STATEMENT SCHEDULES
|
58
|
|
SIGNATURES
CERTIFICATIONS
|
59
|
|
1
ITEM 1. DESCRIPTION
OF BUSINESS
FORWARD-LOOKING
STATEMENTS
This report
contains forward-looking statements within the meaning of Section 21E of the
Securities Exchange Act of 1934, and Section 27A of the Securities Act of 1933.
Any statements contained in this report that are not statements of historical
fact may be forward-looking statements. When we use the words “intends,” “estimates,” “predicts,”
“potential,” “continues,” “anticipates,”
“plans,” “expects,” “believes,” “should,” “could,” “may,” “will” or the negative of these terms or
other comparable terminology, we are
identifying forward-looking statements. Forward-looking statements involve risks
and uncertainties, which may cause our actual results, performance or
achievements to be materially different from those expressed or implied by
forward-looking statements. These factors include our; research and
development activities, distributor channel; compliance with regulatory
impositions; and our capital needs. Although we believe that the expectations
reflected in the forward-looking statements are reasonable, we cannot guarantee
future results, levels of activity, performance or
achievements.
Except as may be
required by applicable law, we do not undertake or intend to update or revise
our forward-looking statements, and we assume no obligation to update any
forward-looking statements contained in this report as a result of new
information or future events or developments. Thus, you should not assume that
our silence over time means that actual events are bearing out as expressed or
implied in such forward-looking statements. You should carefully review and
consider the various disclosures we make in this report and our other reports
filed with the Securities and Exchange Commission that attempt to advise
interested parties of the risks, uncertainties and other factors that may affect
our business.
For further information about these and
other risks, uncertainties and factors, please review the disclosure included in
this report under “Part I, Item 1A, Description of Business - Risk
Factors.”
GENERAL
Acquisition of Cellceutix Pharma,
Inc.
On December 6, 2007, Cellceutix
Corporation, formerly known as EconoShare, Inc., (the “Company” or the
“Registrant”) acquired Cellceutix Pharma, Inc., a privately owned Delaware
corporation pursuant to an Agreement and Plan of Share Exchange (the
“Exchange”). Cellceutix Pharma, Inc. was incorporated under the laws of
the State of Delaware on June 20, 2007. Its assets consisted of rights
assigned to it for six early stage pharmaceutical compounds by three different
scientists. Upon consummation of the Exchange, the Company adopted the business
plan of Cellceutix Pharma, Inc.
Pursuant to the terms of the Exchange,
the Company acquired Cellceutix Pharma, Inc. in exchange for an aggregate
of 82,000,000 newly issued shares of the Company’s common stock, par value
$0.0001 per share (the “Common Stock”), resulting in an aggregate of 91,791,000
shares of the Company’s common stock issued and outstanding. As
a result of the Exchange, Cellceutix Pharma, Inc. became a wholly-owned
subsidiary of the Company. The Company’s shares were issued to the
Cellceutix Pharma, Inc. shareholders on a pro rata basis, on the basis of 82
shares of Common Stock for each share of Cellceutix Pharma, Inc. common stock
held by such Cellceutix Pharma, Inc. shareholder at the time of the
Exchange.
At the
effective time of the Exchange, our Board of Directors was reconstituted by the
resignation of Mr. Hyman Schwartz and Jacob Werczberger from their role as
directors, and the appointment of George W. Evans and Krishna Menon as directors
(both of whom were directors of Cellceutix Pharma, Inc. immediately prior to the
Exchange). Our executive management team also was reconstituted following the
resignation of Hyman Schwartz as Company president, and new officers were
appointed in place of our former officers (See Item 10 - Directors, Executive
Officers and Corporate Governance).
The former holders of Cellceutix
Pharma, Inc. Common Stock, upon Exchange, owned approximately 89% of the
outstanding shares of the Company’s Common Stock. Accordingly, the Exchange
represents a change in control. As of the date of this report, there are
91,836,000 shares of Common Stock issued and outstanding. For
financial accounting purposes, the acquisition was a reverse acquisition of the
Company by Cellceutix Pharma, Inc., under the purchase method of accounting, and
was treated as a recapitalization with Cellceutix Pharma, Inc. as the legal
acquirer. Upon consummation of the Exchange, the Company adopted the business
plan of Cellceutix Pharma, Inc.
2
On January 14, 2008, a majority of the
shareholders of EconoShare, Inc. approved an amendment to the Registrant’s
articles of incorporation to change the name of the Registrant to Cellceutix
Corporation. Upon the filing of a Definitive Information Statement and
effectiveness of the name change on February 1, 2008, the Company applied to the
National Association of Security Dealers (NASD) to change its stock symbol on
the Over the Counter Bulletin Board which resulted in the Company’s stock symbol
being changed to CTIX.
OVERVIEW
EconoShare, Inc. was incorporated on
August 1, 2005 in the State of Nevada and was organized for the
purpose of developing a B2B (Business to Business) website for an Asset Sharing
market place and transaction system. As a result of the Exchange with
Cellceutix Pharma, Inc., we have adopted the business plan of Cellceutix Pharma,
Inc. and now we are an early stage developmental biopharmaceutical
company. The Company has no customers, products or revenues to date, and
may never achieve revenues or profitable operations.
We have acquired exclusive rights to
seven (7) different pharmaceutical compound candidates that are designed for
treatment of diseases which exist, or may exist in the future. The Company
will initially spend most of its efforts and resources on its anti-cancer
compound, Kevetrin™, for the treatment of certain cancers. This compound
is furthest along in in-vivo studies in small animals. Based on the
results, the Company has decided to advance Kevetrin along the
regulatory and clinical pathway.
We anticipate using our expertise to
manage and perform what we believe are the most critical aspects of the product
development process which includes: (i) the design and oversight of non-clinical
and clinical trials; (ii) the development and execution of strategies for the
protection and maintenance of intellectual property rights; and (ii) the
interaction with regulatory authorities worldwide.
We expect to concentrate on product
development and engage in a limited way in product discovery,
avoiding the significant investment of time and financial resources that is
generally required before a new compound is identified and brought into clinical
trials. In addition, we are currently engaged in pre-clinical testing of two of
our product candidates and intend to out-source clinical trials, pre-clinical
testing and the manufacture of clinical materials to third parties.
Our current portfolio of product
candidates in pre-clinical development includes: (i) two (2) anti-cancer agents
targeting multiple tumors; (ii) one (1) candidate targeting psoriasis; (iii) one
(1) candidate targeting rheumatoid arthritis; (iv) one (1) candidate with
potential for indications of osteo-arthritis/asthma; (v) one (1) candidate
with a potential for indications of neurological disorders for the
treatment of Multiple Sclerosis, Lou Gehrig Disease, and/or Parkinson's Disease;
and (vi) a small molecule with potential for development to treat hypertensive
emergency.
On May 7,
2008, the Company issued convertible debentures, at 9% per annum, for a total
amount of $400,000 (the “2007
Convertible Debentures”). The principle and related accrued
interest are due December 1, 2009, and are secured by the Company’s
assets. The 2007 Convertible Debentures and any accrued and unpaid
interest are convertible into the Company’s common stock, at the holder’s
request, at a conversion price of $1.50. The current principal balance of the
2007 Convertible Debentures is $400,000 as of June 30, 2009 and remains unpaid
as of September 30, 2009. The debenture holders have a first priority security
interest in all of the assets of the Company.
The Company has incurred significant
operating losses since its inception resulting in an accumulated deficit of
$2,005,133 at June 30, 2009. For the year ended June 30, 2009, the
Company had a net loss of $1,485,331. Such losses are expected to
continue for the foreseeable future and until such time, if ever, as the Company
is able to attain sales levels sufficient to support its
operations.
The accompanying financial statements
on pages of this Form 10-K have been prepared assuming that the Company will
continue as a going concern which assumes the Company will be able to continue
to realize assets and satisfy liabilities in the normal course of business.
Accordingly, they do not include any adjustments relating to the realization of
the carrying value of assets or the amounts and classification of liabilities
that might be necessary should the company be unable to continue as a going
concern. These factors raise substantial doubt about the Company's ability to
continue as a going concern.
3
Glossary of
Terms -definitions of
certain technical terms used in this report that are commonly used in the
pharmaceutical and biotechnology industries
Adenocarcinoma: A cancer that
originates in glandular tissue
AKT: Also known as AKT1 or
protein kinase B (PKB) is an important molecule in mammalian cellular
signaling.
Alkylation agent: A compound that
interferes with the cell's DNA and inhibits cancer cell
growth.
Angiogenesis: is a physiological
process involving the growth of new blood vessels from pre-existing
vessels.
Carcinomas: A type of cancer that
arises from the lining cells of the body, called epithelial cells. Epithelial
cells form the outer layer of the skin, and the membranes lining the digestive
tract, bladder and uterus, as well as the tubes and ducts that run through the
body's organs.
Cisplatin: is a platinum-based
chemotherapy drug used to treat various types of
cancers.
Cytotoxicity: is the quality of being
toxic to cells.
Epifluorescence microscope - A
fluorescence microscope uses a much higher intensity light to illuminate the
sample. This light excites fluorescence species in the sample, which then emit
light of a longer wavelength. A fluorescent microscope also produces a magnified
image of the sample, but the image is based on the second light source -- the
light emanating from the fluorescent species -- rather than from the light
originally used to illuminate and excite the sample.
Folate: A B-complex vitamin that
is being studied as a cancer prevention agent.
Immunocytochemistry: A
method of detecting cancer in tissues. Monoclonal antibodies are used to stain
the tissues and cells before examination under a
microscope.
Immunolocalisation: The
immunoresponse noticed locally.
In-vitro: refers to the technique of
performing a given experiment in a test tube, or, generally, in a controlled
environment outside a living organism.
In-vivo: refers that which takes place
inside an organism. In science, in vivo refers to experimentation done in or on
the living tissue of a whole, living organism as opposed to a partial or dead
one. Animal testing and clinical trials are forms of in-vivo
research.
Isotoxic: Compounds that show toxicity
levels equally at given doses.
LTB4; Leukotriene B4 (LTB4) is a
notable participant in inflammation and chemotaxis.
Lysates: are a variety of cell and
tissue used as positive controls for our antibodies.
P53, also known as protein 53: A
tumor suppressor gene that is mutated in many human cancers and results in the
loss of a cell’s ability to check for DNA damage.
4
Small Molecule Drug: A medicinal
drug compound having a molecular weight of less than 1,000 Daltons, and
typically between 300 and 700 Daltons.
Western Blot Analysis: A technique used
to identify and locate proteins based on their ability to bind to specific
antibodies.
Xenograft:
The cells of one species transplanted to another species.
The Company’s
Pipeline Consists of the Following Compounds:
|
Disease
|
Development
Stage
|
|
|
Kevetrin
|
Cancer
|
Preclinical
|
|
KM
277
|
Arthritis
|
Preclinical
|
|
KM
278
|
Arthritis/Asthma
|
Preclinical
|
|
KM
133
|
Psoriasis
|
Preclinical
|
|
KM
362
|
Cancer
|
Early
R&D
|
|
KM-3174
|
MS/ALS/Parkinsons
|
Early
R&D
|
|
KM-732
|
Hypertensive
emergency
|
Early
R&D
|
Compound:
Kevetrin
Disease: Cancer
Our lead product candidate, Kevetrin,
was discovered by Dr. Krishna Menon, a Company founder. The Company acquired
exclusive rights to Kevetrin in August 2007. (See Item 13.
Certain Relationships and Related Transactions)
Kevetrin
is initially being developed to treat certain cancers. There is a potential for
use of Kevetrin in multiple tumor types. The Company has conducted pre-clinical
studies in lung cancer, head and neck cancer, colon cancer, breast cancer and
prostate cancer. Several of these studies have involved cell lines that
are resistant to standard drugs.
Kevetrin
is being developed as an intravenous (IV) infusion therapy (the giving of the
drug directly into the vein).
Information about these cancers is
available at http://www.cancer.gov .
Kevetrin
Studies To-Date
Prior to our acquisition of exclusive
rights to Kevetrin, a small molecule now proprietary to the Company, the
molecule had been subjected to extensive initial in-vitro and in-vivo studies.
Kevetrin acts as an AKT inhibitor with the potential to work through other
pathways, as well. Kevetrin has shown to have potent activity against
various cancer cell lines both in-vitro and in-vivo. Summary results are
presented below:
Growth Inhibition
Assays
The cytotoxicity was determined by the
MTT assay. Briefly, cells were seeded in 24-well tissue culture
plates at 10,000-15,000 cells/well and incubated overnight. The exponentially
growing cells were then exposed to different drug concentrations for three to
four generation times. Cellular viability was determined by exposing cells to
the MTT tetrazolium salt for 4 h at 37°C, and the formation of Formazan was
measured at 560 nm by a microplate reader. The concentration inhibiting cell
growth by 50% compared with untreated controls was determined from the curves
plotting survival as a function of dose. All values are average of at least
three independent experiments each done in duplicates. The results showed
that Kevetrin has potent cytotoxic activity. Therefore, Kevetrin was selected
for further development.
5
Immunolocalisation
of p53
To determine the localization of p53,
immunocytochemistry was carried out. Briefly, HCT-116 cells were attached to
glass slides overnight and exposed to isotoxic concentrations of Kevetrin (300
µg/ml), or cisplatin (11 µg/ml) for 6 h. After drug exposure, cells were
fixed with 3.7% formaldehyde, permeabilized with 0.25% Triton X-100, and blocked
with 1% BSA. Cells were then incubated for 1 h with anti-p53 polyclonal
antibodies (Sc-6243; Santa Cruz Biotechnology) followed by secondary anti-rabbit
FITC-conjugated antibodies (Amersham Life Sciences). Coverslips were mounted in
Vectashield (Vector Laboratories) and analyzed with an epifluorescence
microscope Axiovert 100M equipped with appropriate filters and laser confocal
scanning system LSM 510 by using a plan Apochromat x63 objective (Zeiss). ). p53
is a very important protein in the development of colorectal cancers. This
experiment demonstrated that Kevetrin has potent activity in cancers that
contain p53 proteins. This activity is another reason that we selected Kevetrin
for further development.
Western Blot
Analysis
Western blot analysis was performed.
Whole cell lysates were prepared from cells treated with isotoxic concentrations
of Kevetrin (300 µg/ml), or Cisplatin (11 µg/ml) for 6 h. Proteins (50 µg/lane)
were separated on a 4-12% polyacrylamide SDS gel and transferred to
PolyScreen
membranes. The presence of p53, p21, and Я-actin was revealed by anti-p53
antibodies (Sc-6243; Santa Cruz Biotechnology), anti-p21 antibodies (Sc-3976;
Santa Cruz Biotechnology), and anti-actin antibodies (Sc-1616; Santa Cruz
Biotechnology), respectively, followed by incubation with
peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) and
detection by enhanced chemiluminescence (New England Nuclear). The
proteins p53, p21 and B-actin are very important in the development of cancers.
Suppressing these proteins is vital in the treatment of cancer, and the enhanced
suppression of such proteins was shown in the experiment. Therefore Kevetrin was
selected for development.
Influence of
Kevetrin on the Viability of Human Tumor Cell Lines
The influence of Kevetrin on the
viability of 10 different types of human tumor cells, including carcinomas of
the lung, colon, breast, ovary, prostate, sarcomas, gliomas, and leukemias, was
determined after continuous exposure to Kevetrin for three doubling times. The
cytotoxicity differs >120-fold between the different cell lines ranging from
6 ng/ml for Colo 205 (colon carcinoma cells) to 640 ng/ml for U2-Os
(osteosarcoma cells). Generally, Kevetrin has potent activity against human
tumor cells of epithelial origin. The cytotoxic effect of Kevetrin was most
pronounced toward non-small cell lung, colon and ovarian carcinomas with IC50s
ranging from 11 to 68 ng/ml.
Time Dependence of
Kevetrin Cytotoxicity
To
determine the influence of exposure time on the cytotoxic effects of Kevetrin,
DU-145, HCT-116, and HT-29 carcinoma cells were exposed to different
concentrations of Kevetrin for 5, 10, 20, 30, or 45 minutes or 1, 2, 6, 24, or
120 hours. Clear time-dependent cytotoxic effects of Kevetrin were observed for
all three cell lines with longer exposure times being associated with
increased cytotoxicity . The time dependence was particularly dramatic for
exposure times [See figure 1] 30-45 minutes. In contrast, extending the drug
exposure time beyond 24 hours had no influence on the cytotoxicity.
See Figure 1-Time Dependence of Kevetrin
Cytotoxicity
Comparison of the activity spectra of
Cisplatin and Kevetrin toward 10 different types of human tumor cells
demonstrates clear differences between the two drugs. The activity of Kevetrin
was more marked than that of Cisplatin toward lung, head and neck, breast,
ovary, colon, and hepatic cell lines. Interestingly, Kevetrin showed activity
toward all head and neck (n = 3), non-small cell lung (n = 3), ovary (n = 6),
colon (n = 5), and glioma (n = 2) cell lines tested in contrast to Cisplatin,
which generally exhibited a more heterogeneous response within a given tumor
cell type. The difference between Kevetrin and the Cisplatin was particularly
striking for the three head and neck cancer cell lines. Kevetrin showed activity
toward all of the cell lines, whereas Cisplatin was active toward only one of
the three cell lines. Surprisingly, Kevetrin has only very limited activity
toward leukemias, which is unusual for alkylating agents, and different from
what is observed for Cisplatin.
6
See
Figure 2-Activity Spectra for Kevetrin & Cisplatin
The influence of Kevetrin and Cisplatin
on the viability of the indicated tumor cell lines was measured using the MTT
assay after continuous exposure to Kevetrin for three doubling times. The
indicated values are calculated as follows: log (IC50 individual cell line -
IC50 average). Negative values indicate that the cell line is more sensitive
than the average, whereas positive values indicate that the cell line is more
resistant than the average. The average IC50s for all cell lines tested were 4.9
x 10-7 M for Kevetrin and 2.1 x 10-6 M for
Cisplatin.
Activity toward
Cisplatin-resistant Cells
The development of resistance to
Cisplatin is associated with treatment failure and disease progression in
several tumor types, such as ovarian cancer. We therefore compared the
activities of Kevetrin toward two well-characterized, Cisplatin-resistant
ovarian carcinoma cell lines of A2780. The results show that A2780 resistant
cell lines are not resistant to Kevetrin compared with the 7-fold resistance to
Cisplatin.
Influence of p53 and
p21 Status
Increasing evidence suggests that loss
of p53 function is accompanied by increased resistance to alkylating agents,
such as cisplatin. We have compared the influence of Kevetrin toward parental
HCT-116 human colon adenocarcinoma cells and the HCT-116 p53 -/- subline, where
the p53 gene has been deleted by homogenous recombination. The results show that
loss of p53 function has only marginal effect on the cytotoxicity of
Kevetrin. In contrast, p53 deficiency is associated with [See figure 2]
4-fold resistance to Cisplatin compared with parental cells expressing
p53. P53 is a transcription factor that is expressed at low levels in
the absence of cellular stress, and its expression is induced by a variety of
stimuli, usually including DNA damage. Therefore, the independence of p53 status
with respect to Kevetrin-mediated cytotoxicity could be caused by lack of p53
induction. Alternatively, p53 might be induced by Kevetrin but not playing an
important role in Kevetrin-mediated cell death. To distinguish between these two
possibilities, HCT-116 cells were treated with an isotoxic dose of Cisplatin, or
Kevetrin, followed by immunocytochemistry with a p53-directed antibody. The
results show that not only Cisplatin but Kevetrin also was able to induce the
accumulation of nuclear p53. The induction of p53 was further confirmed by
Western blot analysis. Among the many p53 target genes, the cyclin-dependent
kinase inhibitor p21cip-1/waf-1 is the most universally expressed in tumor cell
lines. Western blot analysis of p21 expression in cells treated with an isotoxic
dose of Kevetrin and Cisplatin shows that both these agents were able to induce
p21, thus suggesting that the drug-induced p53 is transcriptionally active. It
should be noted that untreated HCT-116 control cells express constitutive levels
of p21, which may explain the relatively modest induction of p21 after drug
treatment.
Pooled Analysis of
Kevetrin Efficacy in Nude Mice with SCC-15 Head and Neck
Cancer
Three experiments were conducted using
the same protocol at Kard Scientific, Wilmington, MA, Toxicone in Hyderabad,
India and at the University of British Columbia. In each experiment there
were four arms, control, Kevetrin alone, radiation alone, and Kevetrin
administered sequentially with radiation. Each arm included ten nude mice
with head and neck cancer SCC-15, a squamous cell carcinoma. Tumor
reduction was measured every other day and calculated, until it reached 1,500
mg, starting from approximately 100 mg. The results of the experiments
were pooled and means and standard deviations were calculated. Statistical
evaluations of active arms compared to control were performed using the Student
‘s t-test.
7
The
improvements in the active arms compared to controls were also statistically
significant in the individual experiments.
In this analysis, Kevetrin alone,
radiation alone and Kevetrin administered sequentially with radiation were
effective in reducing tumor size. There appeared to be a synergistic
effect with Kevetrin administered sequentially with radiation. Based on
this analysis, Kevetrin appears to be a viable candidate for development for the
treatment of head and neck cancer.
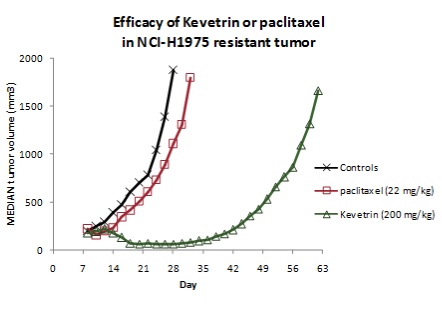
Kevetrin
Efficacy in NCI-H1975 Lung Cancer Cell Line
Summary
Kevetrin
is effective in mouse models of human lung cancer: NCI-H1975
|
·
|
Kevetrin
(200 mg/kg IP x 3 doses)
|
|
o
|
142%
to 156% tumor growth delay compared to
controls
|
|
o
|
44%
to 107% tumor growth delay compared to paclitaxel (22 mg/kg IV x 4
doses)
|
|
·
|
No
decrease in animal weight
|
Details
Nude mice
were implanted with NCI-H1975, a multi-drug resistant human lung non-small cell
lung carcinoma (NSCLC) cell line, subcutaneously in the right flank. Once
tumors reached, on average, ~120 to 200 mm3, the
mice were grouped according to similar tumor size ranges. Mice were treated
intraperitoneally with 200 mg/kg Kevetrin every other day for 3 doses. For
comparison, another group of mice were treated with 22 mg/kg paclitaxel IV every
other day for 4 doses. Another group of mice remained untreated to serve
as controls. Tumors were measured three times per week. During treatments,
mice were observed daily for any adverse affects and mouse body weights were
measured.
The
results of the initial experiment, presented as median tumor volumes over time,
are shown in:
Figure
3
(NCI-H1975 tumor volumes in nude mice
treated with either 200 mg/kg Kevetrin IP qod or 22 mg/kg paclitaxel IV
qod).
The
growth of NCI-H1975 human lung adenocarcinoma tumors was significantly delayed
(p<0.01) following treatment with Kevetrin compared to controls, whereas
paclitaxel had little efficacy in these tumors compared to controls. Tumor
growth delay with Kevetrin was also significantly greater than with paclitaxel
(p<0.01). On measurements of tumor volume at day 28, Kevetrin was
significantly more effective than controls or paclitaxel
(p<0.01).
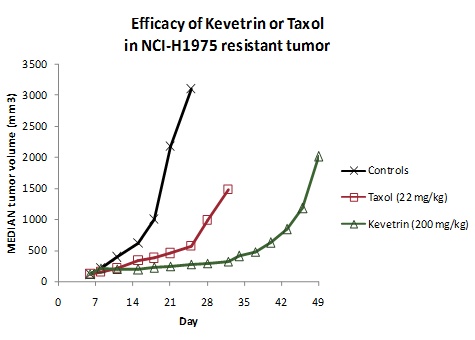
The
results of the repeat experiment are shown in:
Figure
4
( NCI-H1975 tumor volumes in nude
mice treated with either 200 mg/kg Kevetrin IP qod or 22 mg/kg Taxol IV
qod).
8
In this
experiment, the growth of NCI-H1975 human lung adenocarcinoma tumors was
significantly delayed (p<0.01) following treatment with Kevetrin compared to
controls, whereas paclitaxel had only moderate efficacy in these tumors
producing a tumor growth delay of compared to controls. Tumor growth delay with
Kevetrin was also significantly greater than with paclitaxel (p<0.01).
On measurements of tumor volume at day 25, Kevetrin was significantly more
effective than controls or paclitaxel (p<0.01). A significant
therapeutic index was achieved since no weight loss occurred during treatment
with Kevetrin.
These
results demonstrate that Kevetrin, but not paclitaxel, has potent anti-tumor
activity against a human lung adenocarcinoma xenograft tumor model, NCI-H1975,
at a dose and schedule that is well-tolerated as indicated by no weight loss
during treatment. These studies support the development of Kevetrin in lung
carcinoma indications, particularly in cases where tumors have become resistant
to standard chemotherapy.
Kevetrin Efficacy in A-549 Lung
Cancer Cell Line
Summary
Kevetrin
is effective in mouse models of human lung cancer: A549
|
·
|
Kevetrin
(200 mg/kg IP x 3 doses)
|
|
o
|
33%
to 111% tumor growth delay compared to
controls
|
|
o
|
33%
to 100% tumor growth delay compared to paclitaxel (22 mg/kg IV x 4
doses)
|
|
·
|
Only
3% to 4% decrease in animal
weight
|
Details
Kevetrin
was shown to be effective in animal model experiments (mice) with of human lung
cancer cell lines. Nude mice were implanted with A-549, a multi-drug
resistant human lung non-small cell lung carcinoma (NSCLC) cell line,
subcutaneously in the right flank. Once tumors reached, on average, ~120
mm3, the
mice were grouped according to similar tumor size ranges. Mice were treated
intraperitoneally with 200 mg/kg Kevetrin every other day for 3 doses. For
comparison, another group of mice were treated with 22 mg/kg paclitaxel IV every
other day for 4 doses. Another group of mice remained untreated to serve
as controls. Tumors were measured three times per week. During treatments,
mice were observed daily for any adverse affects and mouse body weights were
measured.
The
results of the initial experiment, presented as median tumor volumes over time,
are shown in:
Figure
5
(A549 tumor volumes in nude mice
treated with either 200 mg/kg Kevetrin IP qod or 22 mg/kg paclitaxel
IV qod).
The
growth of A549 human lung adenocarcinoma tumors was significantly delayed (p
< 0. 01) following treatment with Kevetrin, whereas paclitaxel had little
efficacy in these tumors producing a no tumor growth delay.
The
results of the repeat experiment are shown in:
Figure
6
6 (A549 tumor volumes in nude mice
treated with either 200 mg/kg Kevetrin IP qod or 22 mg/kg Taxol IV
qod).
In this
experiment, the growth of A549 human lung adenocarcinoma tumors was
significantly delayed (p < 0. 01) following treatment with Kevetrin, whereas
paclitaxel had little efficacy in these tumors producing a tumor growth delay of
only 11%. A significant therapeutic index was achieved since during
treatment, Kevetrin resulted in only a 3% to 4%, but transient, decrease in
average animal weight.
These
results demonstrate that Kevetrin, but not paclitaxel, has potent anti-tumor
activity against a human lung adenocarcinoma xenograft tumor model, A549, at a
dose and schedule that is well-tolerated as indicated by a small transient
weight loss during treatment. These studies support the development of Kevetrin
in lung carcinoma indications, particularly in cases where tumors have become
resistant to standard chemotherapy.
Acute
Toxicity Studies
The
Company has completed two acute toxicity studies, one in mice and one in
rats.
In the
acute toxicity mouse study, animals were sorted into 4 groups of twenty animals,
each comprised of ten male and ten female mice. One group served as the
controls and the other three groups were administered a single dose of 150
mg/kg, 200 mg/kg or 300mg/kg of Kevetrin. Animals were observed for 15
days. Data was periodically collected measuring food consumption, body
weight and various blood chemistry parameters. At the conclusion of the 15
day trial, post-mortem examinations were made of all the mice. In this
study there were no significant indications of toxicity at expected therapeutic
doses in the mice that had been dosed with Kevetrin.
In the
acute toxicity rat study, animals were sorted into 4 groups of ten animals, each
comprised of five male and five female rats. One group served as the
controls and the other three groups were administered a single dose of 160
mg/kg, 200 mg/kg or 240 mg/kg of Kevetrin. Animals were followed for 15
days. Data was periodically collected measuring food consumption, body
weight and blood chemistry parameters. At the conclusion of the 15 day
trial, post-mortem examinations were made of all
the rats. In this study there were no significant indications of toxicity
at expected therapeutic doses in the rats that had been dosed with
Kevetrin.




9
FIGURES
Figure
1: Time dependence
for cytotoxicity. A) HT-29, B) HCT-116, C) DU-145 carcinoma
cells were exposed to different
concentrations of Kevetrin for 5, 10, 20, 30, or 45 min or 1, 2, 6,
24, or 120 h. Cellular viability, as measured using the MTT assay, expressed as
IC50 was plotted verses time of Kevetrin exposure.
Kevetrin
Cisplatin
Figure
2 : The
influence of Kevetrin and cisplatin on the viability of the indicated tumor cell
lines was measured using the MTT assay after continuous exposure to Kevetrin for
three doubling times. The indicated values are calculated as follows: log
(IC50 individual cell line -
IC50 average). Negative values indicate
that the cell line is more sensitive than the average, whereas positive values
indicate that the cell line is more resistant than the average. The average
IC50s for all cell lines tested were 4.9
x 10-7 M for Kevetrin and 2.1 x
10-6 M for
cisplatin.
Figure 3. Median NCI-H1975 tumor volumes in
nude mice treated with either 200 mg/kg Kevetrin IP qod on days 7-11 or 22 mg/kg
paclitaxel IV qod on day 7-13 after tumor implantation.
10
Figure 4. Median NCI-H1975 tumor volumes in
nude mice treated with either 200 mg/kg Kevetrin IP qod on days 7-11 or 22 mg/kg
Taxol IV qod on day 7-13 after tumor implantation.
Figure 5. Median A549 tumor volumes in nude
mice treated with either 200 mg/kg Kevetrin IP qod on days 7-11 or 22 mg/kg
paclitaxel IV qod on day 7-13 after tumor implantation.
[
Figure 6. Median A549 tumor volumes in nude
mice treated with either 200 mg/kg Kevetrin IP qod on days 7-11 or 22 mg/kg
Taxol IV qod on day 7-13 after tumor implantation.
 ]
]11
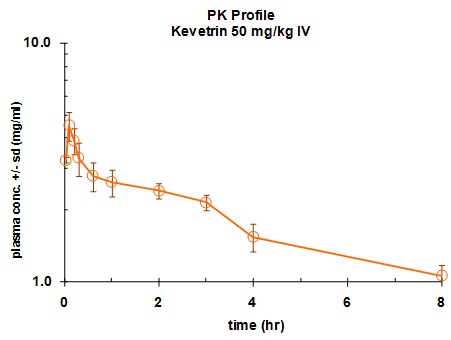
Figure
7. Pharmacokinetics
profile of Kevetrin . Time vs. concentration curve of plasma from
mice treated with 50 mg/kg of Kevetrin
Compound: KM
277
Disease:
Arthritis
In-vivo studies on over 1,000 animal
subjects have been conducted to check for potential efficacy of KM 277 in
rheumatoid arthritis. These studies, as well as related in vitro assays, have
led the Company’s management to believe that the compound has developmental
potential for the treatment of the disease.
Compound: KM
278
Disease:
Arthritis/Asthma
KM 278 showed potential efficacy in
both Osteoarthritis animal models and Asthma animal models. KM 278 was
tested against standard treatment for asthma and Osteoarthritis. These studies
have led the Company’s management to believe that the compound has developmental
potential for the treatment of asthma and
Osteoarthritis.
12
Compound:
KM-133
Disease:
Psoriasis
KM 133 is
a small molecule compound, acting on the principles of folate mechanism, is in
very early stage development and may have potential efficacy against
Psoriasis. After a series of chemical optimization exercises, the
Company is now completing an animal study in a xenograft model of
psoriasis. If the study is successful, the Company will begin to advance
the compound along the regulatory pathway.
Compound:
KM-362
Disease:
Cancer
This is a very early stage
developmental compound in the treatment of cancer. After additional in vitro and
in vivo studies the Company will determine whether to advance this compound for
further development.
Disease:
MS/ALS/Parkinsons
Amyelination (absence of the myelin
sheath on a nerve), is a characteristic in most neurological diseases such as
Lou Gherig Disease, Parkinson’s Disease and Multiple Sclerosis. Initial
studies suggest that this compound aids in demyelination (the loss of nerve
fiber "insulation" due to trauma or disease, which reduces the ability of nerves
to conduct impulses) along with strengthening the functions of the nerves,
spinal chord and the brain tissue. These studies led the Company’s management to
believe that the compound has developmental potential for the treatment of these
diseases.
Compound:
KM-732
Disease:
Hypertensive Emergency
This is a
very early-stage developmental compound with potential for the treatment of
hypertensive emergency. After additional in vitro and in vivo studies, the
Company will determine whether to advance this compound for further
development.
INTELLECTUAL
PROPERTY
The
Company has been assigned all right title and interest to the following seven
pharmaceutical compounds: Kevetrin, KM 277, KM 278, KM 133 KM 362, KM 3174, and
KM 732 by their inventors. The Company agreed to pay the inventors 5% of net
sales of the compounds in countries where composition of matter patents have
been issued and 3% of net sales in other countries. Kevetrin, KM 277, KM 278 and
KM 362 were acquired by the Company from Dr. Krishna Menon, the Company’s
Director, President, and principal shareholder. With
respect to KM 732, the Company has agreed to pay an individual a fixed payment
if the compound is approved for sale in the U.S.
On May
20, 2009, the Company filed a U.S. patent application covering pharmaceutical
formulations of Kevetrin and many novel compounds having similar structures to
Kevetrin that may have potential as drug development candidates. The application
also covers the use of Kevetrin and the other related compounds in various
areas, including cancers. The Company plans to file in other countries
within a year of the U.S. filing.
The
Company intends to file patent applications for the other compounds as funds
become available.
13
OUR
STRATEGY
We are in the business of developing
innovative small molecule therapies to treat diseases with significant medical
need, particularly in the areas of cancer and inflammatory disease. Our
strategy is to use our business and scientific expertise to maximize the value
of our pipeline. We will do this by focusing initially on our lead
compound, Kevetrin, and advancing it as quickly as possible along the regulatory
pathway. We will develop the highest quality data and broadest
intellectual property to support Kevetrin.
We currently own all development and
marketing rights to our products. In order to successfully develop and
market our products, we may have to partner with other companies.
Prospective partners may require that we grant them significant development
and/or commercialization rights in return for agreeing to share the risk of
development and/or commercialization.
MANUFACTURING
The
Company's compounds are still in preclinical development and at this time the
Company does not have, and does not intend to establish, manufacturing
facilities to produce its product candidates in the near or mid-term. In June
2009, the Company signed an agreement with Girindus America, Inc., for the
manufacture of Kevetrin active pharmaceutical ingredient. The Company
may continue to use the services of Kard, Inc. (See Item 13. Certain
Relationships and Related Transactions; and Director Independence)
GOVERNMENT
REGULATION
Our operations and activities are
subject to extensive regulation by numerous government authorities in the United
States and other countries. In the United States, drugs are subject to rigorous
regulation by the United States Food and Drug Administration (“FDA”). The
Federal Food, Drug and Cosmetic Act, and other federal and state statutes and
regulations, govern the testing, manufacture, safety, effectiveness, labeling,
storage, record keeping, approval, advertising and promotion of our products. As
a result of these regulations, product development and the product approval
process is a very expensive and time consuming process. The FDA must approve a
drug before it can be sold in the United States. The general process for FDA
approval of a drug is as follows:
Preclinical
Testing
Before we can test a drug candidate in
humans, we must study the drug in laboratory experiments and in animals to
generate data to support the drug's potential safety and benefits. We submit
this data to the FDA in an investigational new drug application (IND) seeking
their approval to test the compound in humans.
Clinical
Trials
If the FDA accepts the IND, we study
the drug in human clinical trials to determine if the drug is safe and
effective. These clinical trials involve three separate phases that often
overlap, can take many years to compile, and are very expensive. These three
phases, which are themselves subject to considerable regulation, are as
follows:
Phase 1. The drug is given to a small
number of healthy human subjects (patients) to test for safety, dose tolerance,
pharmacokinetics, metabolism, distribution and
excretion.
Phase 2. The drug is given to a limited
patient population to determine the effect of the drug in treating the disease,
the best dose of the drug, and the possible side effects and safety risks of the
drug.
Phase 3. If a compound appears to be
effective and safe in Phase 2 clinical trials, Phase 3 clinical trials are
commenced to confirm those results. Phase 3 clinical trials are long-term,
involve a significantly larger population of patients, are conducted at numerous
sites in different geographic regions, and are carefully designed to provide
reliable and conclusive data regarding the safety and benefits of a
drug. It is not uncommon for a drug that appears promising in Phase 2
clinical trials to fail in the more rigorous and reliable Phase 3 clinical
trials.
FDA Approval
Process
If we believe that the data from the
Phase 3 clinical trials show an adequate level of safety and effectiveness, we
will file a new drug application (NDA) with the FDA seeking approval to sell the
drug for a particular use. The FDA will review the NDA and often will hold a
public hearing where an independent advisory committee of expert advisors asks
additional questions regarding the drug. This committee makes a recommendation
to the FDA, which is not binding on the FDA, but is generally followed. If the
FDA agrees that the compound has met the required level of safety and
effectiveness for a particular use, it will allow the Company to sell the drug
in the United States for that use. It is not unusual, however, for the FDA to
reject an application because it believes that the drug is not safe enough, or
effective enough, or because it does not believe that the data submitted is
reliable or conclusive.
At any point in this process, the
development of a drug could be stopped for a number of reasons including safety
concerns and lack of treatment benefit. We cannot be certain that any clinical
trials that we are currently conducting or any that we conduct in the future,
will be completed successfully or within any specified time period. We may
choose, or the FDA may require us, to delay or suspend our clinical trials at
any time if it appears that the patients are being exposed to an unacceptable
health risk or if the drug candidate does not appear to have sufficient
treatment benefit.
14
The FDA may also require us to: (i)
complete additional testing; (ii) provide additional data or information; (iii),
improve our manufacturing processes, procedures or facilities; and (iv) require
extensive post-marketing testing and surveillance to monitor the safety or
benefits of our product candidates if it determines that our new drug
application does not contain adequate evidence of the safety and benefits of the
drug. In addition, even if the FDA approves a drug, it could limit the uses of
the drug. The FDA can withdraw approvals if it does not believe that a company
is complying with regulatory standards or if problems are uncovered
or occur after approval.
In addition to obtaining FDA approval
for each drug, we must obtain FDA approval of the manufacturing facilities for
any drug we sell, including those of companies who manufacture our drugs for
us.. The FDA must also approve foreign establishments that manufacture products
to be sold in the United States and these facilities are subject to periodic
regulatory inspection.
Should our products be approved for
marketing, we would also be subject to various other State and Federal laws
concerning the marketing and cost reimbursement of our
products.
Other major countries or groups of
countries, such as the European Union, Japan and Canada, have similarly rigorous
regulatory processes. They may also require studies not required by the
FDA, which can add to the cost and risk of development. Products approved
by the FDA might not be approved by these countries. After review by the
health authorities, pricing and cost reimbursement are also subject to separate
approvals in many of these countries.
COMPETITION
Competition in the pharmaceutical and
biotechnology industries is intense. The drugs that we are developing will have
to compete with existing therapies. In addition, a large number of companies are
pursuing the development of pharmaceuticals that target the same diseases and
conditions that we are targeting. Many pharmaceutical or biotechnology companies
have products on the market and are actively engaged in the research and
development of products that are competitive with our potential products. Many
of these companies and institutions, either alone or together with their
collaborative partners, have substantially greater financial, manufacturing,
sales, distribution and technical resources and more experience in research and
development, clinical trials and regulatory matters, than we do. In addition,
our competitors may succeed in developing technologies and drugs that are more
effective, better tolerated or less costly than any which are being developed by
us or which would render our technology or potential drugs obsolete or
noncompetitive.
With respect to Kevetrin, our lead
compound for cancer, there are many drugs approved to treat various cancers and
many more in the publicly disclosed development pipeline. Our success
depends on our ability to identify tumor types where Kevetrin has an advantage
over existing therapies and those in the publicly disclosed development
pipeline.
EMPLOYEES
As of June 30, 2009 the Company had
three (3) employees, Mr. George Evans, Dr. Krishna Menon, and Mr. Leo
Ehrlich. Messrs. Krishna Menon and George Evans executed employment
agreements with the Company on December 7, 2007. The Company expects to
conduct its operations using contractors and consultants for the short term. The
Company may hire additional full-time employees to be engaged in administration,
research and development should the Company have adequate
funds.
CORPORATE
INFORMATION
The Company's corporate headquarters
are located at 100 Cummings Center, Suite 151-B, Beverly, MA, 01915. The
Company's telephone number is (978) 633-3623.
ITEM
1A. RISK FACTORS
Investing in the Company's common stock
involves a high degree of risk. Prospective investors should carefully consider
the risks described below, together with all of the other information included
or referred to in this Annual Report on Form 10-K, before purchasing shares of
the Company's common stock. There are numerous and varied risks, known and
unknown, that may prevent the Company from achieving its goals. The risks
described below are not the only ones the Company will face. If any of these
risks actually occur, the Company's business, financial condition or results of
operation may be materially adversely affected. In such case, the trading price
of the Company's common stock could decline and investors in the Company's
common stock could lose all or part of their investment.
Risks Specific to
Us
Our
company is a development stage company that has no products approved for
commercial sale, has never generated any revenues, and may never achieve
revenues or profitability.
15
We are a development stage
biopharmaceutical company. Currently, we have no products approved for
commercial sale and, to date, we have not generated any revenues. Our ability to
generate revenue depends heavily on:
|
successful
development and demonstration in pre-clinical trials that our
leading drug candidate, Kevetrin, may be studied in human clinical
trials;
|
|
|
•
|
successful
demonstration in human clinical trials that Kevetrin is safe and
effective;
|
|
•
|
our
ability to seek and obtain regulatory approvals, including with respect to
the indications we are seeking;
|
|
•
|
the
successful commercialization of our product candidates;
and
|
|
•
|
market
acceptance of our products.
|
All of our existing product candidates
are in early stages of development. If we do not successfully develop and
commercialize these products, we will not achieve revenues or profitability in
the foreseeable future, if at all. If we are unable to generate revenues or
achieve profitability, we may be unable to continue our
operations.
We
are a development stage company with a limited operating history, making it
difficult for you to evaluate our business and your investment.
We are in the development stage and our
operations and the development of our proposed products are subject to all of
the risks inherent in the establishment of a new business enterprise, including
but not limited to:
• the absence of an operating
history;
• the lack of commercialized
products;
• insufficient capital;
• expected substantial and continual
losses for the foreseeable future;
• limited experience in dealing with
regulatory issues;
• the lack of manufacturing experience
and limited marketing experience;
• possible reliance on third parties
for the development and commercialization of our proposed
products;
• a competitive environment
characterized by numerous, well-established and well capitalized competitors;
and
• reliance on key
personnel.
Because we are subject to these risks,
you may have a difficult time evaluating our business and your investment in our
company.
Our ability to become profitable
depends primarily on the following factors:
●
our ability to develop
drugs, obtain approval for such drugs, and if approved, to successfully
commercialize our drugs;
●
our R&D efforts,
including the timing and cost of clinical trials; and
16
●
our ability to enter into
favorable alliances with third-parties who can provide substantial capabilities
in clinical development, regulatory affairs, sales, marketing and
distribution.
Even if we successfully develop and
market our drug candidates, we may not generate sufficient or sustainable
revenue to achieve or sustain profitability.
If
the Company is unable to repay the balance of the debentures, the holders of the
debentures may initiate foreclosure proceedings against the assets of the
Company.
On May 7,
2008, the Company issued convertible debentures, at 9% per annum, for a total
amount of $400,000 (the “2007
Convertible Debentures”). The principle and related accrued
interest are due December 01, 2009, and are secured by the Company’s
assets. The 2007 Convertible Debentures and any accrued and unpaid
interest are convertible into the Company’s common stock, at the holder’s
request, at a conversion price of $1.50. The current principal balance of the
2007 Convertible Debentures is $400,000 as of June 30, 2009 and remains unpaid
as of September 30, 2009. The debenture holders have a first priority security
interest in all of the assets of the Company. At the present time the
Company does not have sufficient cash available to repay the debenture
holders.
The
report of our independent registered public accounting firm includes a going
concern opinion, and we may not be profitable in the future, if
ever.
As of June 30, 2009 we have $140,380
cash available to support operations or our business plan. Our operating
cash needs, cash consumption, and doubt as to whether we will ever become
profitable, are factors which raise substantial doubt as to our ability to
continue as a going concern. Consequently, our independent registered public
accounting firm has included a going concern opinion in its report which is
included elsewhere in this Form 10-K. It is uncertain at this time how the going
concern opinion by our independent registered public accounting firm will affect
our ability to raise capital. If we are unable to achieve revenues or obtain
financing on terms and
conditions acceptable to the Company, then we may not be able to commence
revenue-generating operations or continue as an on-going
concern.
We
will need to raise substantial additional capital in the future to fund our
operations and we may be unable to raise such funds when needed and on
acceptable terms.
We currently do not have resources to
complete the development and commercialization of any of our proposed products.
We expect to incur costs of approximately nine million dollars ($9,000,000) in
the upcoming twelve (12) months to operate our business in accordance with our
business plans and budgets. We may not be able to secure this amount of
financing on terms and conditions acceptable to the Company. In the
event that we cannot obtain acceptable financing, we would be unable to complete
preclinical development and file an IND application with the FDA for our
anti-cancer drug, Kevetrin. This will delay:
• research and development
programs;
• preclinical studies and clinical
trials; material characterization studies, regulatory processes;
• establishment of our own laboratory
or a search for third party marketing partners to market our products for
us.
The amount of capital we may require
will depend on many factors, including the:
• progress, timing and scope of our
research and development programs;
• progress, timing and scope of our
preclinical studies and clinical trials;
• time and cost necessary to obtain
regulatory approvals;
• time and cost necessary to establish
our own marketing capabilities or to seek marketing partners;
• time and cost necessary to respond to
technological and market developments;
17
• changes made or new developments in
our existing collaborative, licensing and
• other commercial relationships;
and
• new collaborative, licensing and
other commercial relationships that we may
establish.
Our fixed expenses, such as rent and
other contractual commitments, may increase in the future, as we
may:
• enter into leases for new facilities
and capital equipment;
• enter into additional licenses and
collaborative agreements; and
• incur additional expenses associated
with being a public company.
We
have limited experience in drug development and may not be able to successfully
develop any drugs.
We have limited experience in drug
development and may not be able to successfully develop any drugs. Our ability
to achieve revenues and profitability in our business will depend, among other
things, on our ability to:
• develop products internally or obtain
rights to them from others on favorable terms;
• complete laboratory testing and human
studies;
• obtain and maintain necessary
intellectual property rights to our products;
• successfully complete regulatory
review to obtain requisite governmental agency
approvals
• enter into arrangements with third
parties to manufacture our products on our behalf; and
• enter into arrangements with third
parties to provide sales and marketing functions.
Development
of pharmaceutical products is a time-consuming process, subject to a number of
factors, many of which are outside of our control. Consequently, we can provide
no assurance of the successful and timely development of new
drugs.
Our drug candidates are in early
developmental stage. Further development and extensive testing will be required
to determine their technical feasibility and commercial viability. Our success
will depend on our ability to achieve scientific and technological advances and
to translate such advances into reliable, commercially competitive drugs on a
timely basis. Drugs that we may develop are not likely to be commercially
available for several years, if ever. The proposed development schedules for our
drug candidates may be affected by a variety of factors, including technological
difficulties, proprietary technology of others, and changes in government
regulation, many of which will not be within our control. Any delay in the
development, introduction or marketing of our drug candidates could result
either in such drugs being marketed at a time when their cost and performance
characteristics would not be competitive in the marketplace or in the shortening
of their commercial lives. In light of the long-term nature of our projects, the
unproven technology involved and the other factors described elsewhere in “Risk
Factors”, we may not be able to complete successfully the development or
marketing of any of our drug candidates.
We may fail to successfully develop and
commercialize our drug candidates because they:
18
• are found to be unsafe or ineffective
in clinical trials;
• do not receive necessary approval
from the FDA or foreign regulatory agencies;
• fail to conform to a changing
standard of care for the diseases they seek to treat;
or
• are less effective or more expensive
than current or alternative treatment methods.
Drug development failure can occur at
any stage of clinical trials and as a result of many factors and there can be no
assurance that we or our collaborators will reach our anticipated clinical
targets. Even if we or our collaborators complete our clinical trials, we do not
know what the long-term effects of exposure to our drug candidates will be.
Furthermore, our drug candidates may be used in combination with other
treatments and there can be no assurance that such use will not lead to unique
safety issues. Failure to complete clinical trials or to prove that our drug
candidates are safe and effective would have a material adverse effect on our
ability to generate revenue and could require us to reduce the scope of or
discontinue our operations.
We
must comply with significant and complex government regulations, compliance with
which may delay or prevent the commercialization of our drug
candidates.
The R&D, manufacture and marketing
of drug candidates are subject to regulation, primarily by the FDA in the United
States, and by comparable authorities in other countries. These national
agencies and other federal, state, local and foreign entities regulate, among
other things, R&D activities (including testing in animals and in humans)
and the testing, manufacturing, handling, labeling, storage, record keeping,
approval, advertising and promotion of the products that we are developing.
Noncompliance with applicable requirements can result in various adverse
consequences, including approval delays or refusals to approve drug licenses or
other applications, suspension or termination of clinical investigations,
revocation of approvals previously granted, fines, criminal prosecution, recalls
or seizures of products, injunctions against shipping drugs and total or partial
suspension of production and/or refusal to allow a company to enter into
governmental supply contracts.
The process of obtaining FDA approval
for a drug has historically been costly and time consuming. Current FDA
requirements for a new human drug or biological product to be marketed in the
United States include: (i) the successful conclusion of pre-clinical laboratory
and animal tests, if appropriate, to gain preliminary information on the
product's safety; (ii) filing with the FDA of an IND application to conduct
human clinical trials for drugs or biologics; (iii) the successful completion of
adequate and well-controlled human clinical investigations to establish the
safety and efficacy of the product for its recommended use; and (iv) filing by a
company and acceptance and approval by the FDA of a New Drug Application
(“NDA”), for a drug product or a biological license application (“BLA”), for a
biological product to allow commercial distribution of the drug or biologic. A
delay in one or more of the procedural steps outlined above could be harmful to
the Company in terms of getting our drug candidates through clinical testing and
to market.
The FDA reviews the results of the
clinical trials and may order the temporary or permanent discontinuation of
clinical trials at any time if it believes the drug candidate exposes clinical
subjects to an unacceptable health risk. Investigational drugs used in clinical
studies must be produced in compliance with current good manufacturing practice
(“cGMP”) rules pursuant to FDA regulations.
Sales outside the United States of
products that we develop will also be subject to additional regulatory
requirements governing human clinical trials and marketing for drugs and
biological products and devices. The requirements vary widely from country to
country, but typically the registration and approval process takes several years
and requires significant resources.
We also are subject to the following
risks and obligations, related to the approval of our products:
• The FDA or foreign regulators may
interpret data from pre-clinical testing and clinical trials in different ways
than we interpret them.
19
• If regulatory approval of a product
is granted, the approval may be limited to specific indications or limited with
respect to its distribution. In addition, many foreign countries control pricing
and coverage under their respective national social security
systems.
• The FDA
or foreign regulators may not approve our manufacturing processes or
manufacturing facilities.
• The FDA or foreign regulators may
change their approval policies or adopt new
regulations.
• Even if regulatory approval for any
of our product is obtained, the corresponding marketing license will be subject
to continual review, and newly discovered or developed safety or effectiveness
data may result in suspension or revocation of the marketing license.
• If regulatory approval of the product
candidate is granted, the marketing of that product would be subject to adverse
event reporting requirements and a general prohibition against promoting
products for unapproved uses.
• In some foreign countries, we may be
subject to official release requirements that require each batch of the product
we produce to be officially released by regulatory authorities prior to its
distribution by us.
• We will be subject to continual
regulatory review and periodic inspection and approval of manufacturing
modifications, including compliance with cGMP regulations.
We
can provide no assurance that our drug candidates will obtain regulatory
approval or that the results of clinical studies will be
favorable.
The work-plan we have developed for the
next twelve (12) months should enable us to file an Investigational New Drug
(“IND”) application for Kevetrin, our anti-cancer drug in our 2009-2010 fiscal
year. We need to be able to undertake further studies in animal models to obtain
necessary data regarding the pharmaco-kinetic and pharmaco-dynamic profiles of
our drug candidates as well as toxicity profiles. The data will then be used to
file an IND application, an important first step towards the goal of obtaining
FDA approval for testing the drug in human patients.
The testing, marketing and
manufacturing of any product for use in the United States will require approval
from the FDA. We cannot predict with any certainty the amount of time necessary
to obtain such FDA approval and whether any such approval will ultimately be
granted. Preclinical and clinical trials may reveal that one or more products
are ineffective or unsafe, in which event further development of such products
could be seriously delayed or terminated. Moreover, obtaining approval for
certain products may require testing on human subjects of substances whose
effects on humans are not fully understood or documented. Delays in obtaining
FDA or any other necessary regulatory approvals of any proposed drugs, and
failure to receive such approvals, would have an adverse effect on the drug's
potential commercial success and on our business, prospects, financial condition
and results of operations. In addition, it is possible that a proposed drug may
be found to be ineffective or unsafe due to conditions or facts that arise after
development has been completed and regulatory approvals have been obtained. In
this event, we may be required to withdraw such proposed drug from the market.
To the extent that our success will depend on any regulatory approvals from
government authorities outside of the United States that perform roles similar
to that of the FDA, uncertainties similar to those stated above will also
exist.
Even
if we obtain regulatory approvals, our marketed drug candidates will be subject
to ongoing regulatory review. If we fail to comply with continuing U.S. and
foreign regulations, we could lose our approvals to market these drugs and our
business would be seriously harmed.
Following any initial regulatory
approval of any drugs we may develop, we will also be subject to continuing
regulatory review, including the review of adverse experiences and clinical
results that are reported after our drug candidates are made commercially
available. This would include results from any post-marketing tests or vigilance
required as a condition of approval. The manufacturer and manufacturing
facilities we use to make any of our drug candidates will also be subject to
periodic review and inspection by the FDA. The discovery of any previously
unknown problems with the drug, manufacturer or facility may result in
restrictions on the drug or manufacturer or facility, including withdrawal of
the drug from the market. If we are required to withdraw all or more of our
drugs from the market, we may be unable to continue revenue generating
operations. We do not have, and currently do not intend to develop, the ability
to manufacture material for our clinical trials or on a commercial scale.
Reliance on third-party manufacturers entails risks to which we would not be
subject if we manufactured drugs ourselves, including reliance on the
third-party manufacturer for regulatory compliance. Our drug promotion and
advertising is also subject to regulatory requirements and continuing FDA
review.
20
We
have no experience in conducting or supervising clinical trials and must
outsource all clinical trials.
We have no experience in conducting or
supervising clinical trials that must be performed to obtain data to submit in
concert with applications for approval by the FDA. The regulatory process to
obtain approval for drugs for commercial sale involves numerous steps. Drugs are
subjected to clinical trials that allow development of case studies to examine
safety, efficacy, and other issues to ensure that sale of drugs meets the
requirements set forth by various governmental agencies, including the FDA. In
the event that our protocols do not meet standards set forth by the FDA, or that
our data is not sufficient to allow such trials to validate our drugs in the
face of such examination, we might not be able to meet the requirements that
allow our drugs to be approved for sale.
Because we have no experience in
conducting or supervising clinical trials, we must outsource our clinical trials
to third parties. We have no control over their compliance with procedures and
protocols used to complete clinical trails in accordance with standards required
by the agencies that approve drugs for sale. If these subcontractors fail to
meet these standards, the validation of our drugs would be adversely affected,
causing a delay in our ability to engage in revenue-generating operations
We
are subject to risks inherent in conducting clinical trials. The risk of non
compliance with FDA-approved good clinical practices by clinical investigators,
clinical sites, or data management services could delay or prevent us from
developing or ever commercializing our drug
candidates.
Agreements with clinical investigators
and medical institutions for clinical testing and with other third parties for
data management services place substantial responsibilities on these parties,
which could result in delays in, or termination of, our clinical trials if these
parties fail to perform as expected. For example, if any of our clinical trial
sites fail to comply with FDA-approved good clinical practices, we may be unable
to use the data gathered at those sites. If these clinical investigators,
medical institutions or other third parties do not carry out their contractual
duties or obligations or fail to meet expected deadlines, or if the quality or
accuracy of the clinical data they obtain is compromised due to their failure to
adhere to our clinical protocols or for other reasons, our clinical trials may
be extended, delayed or terminated, and we may be unable to obtain regulatory
approval for or successfully commercialize our drug
candidates.
We or regulators may suspend or
terminate our clinical trials for a number of reasons. We may voluntarily
suspend or terminate our clinical trials if at any time we believe that they
present an unacceptable risk to the patients enrolled in our clinical trials. In
addition, regulatory agencies may order the temporary or permanent
discontinuation of our clinical trials at any time if they believe that the
clinical trials are not being conducted in accordance with applicable regulatory
requirements or that they present an unacceptable safety risk to the patients
enrolled in our clinical trials. In addition, clinical trials may have
independent monitoring boards composed of experts in the field. These
boards may also have the authority to suspend or terminate clinical
trials.
Our clinical trial operations will be
subject to regulatory inspections at any time. If regulatory inspectors conclude
that we or our clinical trial sites are not in compliance with applicable
regulatory requirements for conducting clinical trials, we may receive reports
of observations or warning letters detailing deficiencies, and we will be
required to implement corrective actions. If regulatory agencies deem our
responses to be inadequate, or are dissatisfied with the corrective actions that
we or our clinical trial sites have implemented, our clinical trials may be
temporarily or permanently discontinued, we may be fined, we or our
investigators may be precluded from conducting any ongoing or any future
clinical trials, the government may refuse to approve our marketing applications
or allow us to manufacture or market our drug candidates or we may be criminally
prosecuted. If we are unable to complete clinical trials and have our products
approved due to our failure to comply with regulatory requirements, we will be
unable to commence revenue generating operations.
21
The
Company is exposed to product liability, clinical and preclinical liability
risks which could place a substantial financial burden upon the Company should
it be sued, because the Company does not currently have product liability
insurance.
The Company does not currently have
products in clinical trials nor does it have marketed products. Should its
products advance to the clinical trial or marketing stage, however, it could be
exposed to potential product liability and other liability risks that are
inherent in the testing, manufacturing and marketing of pharmaceutical products.
The Company cannot assure that such potential claims will not be asserted
against it. In addition, the use in the Company's clinical trials of
pharmaceutical products that it may develop and the subsequent sale of these
products by the Company or its potential collaborators may cause the Company to
bear a portion of or all product liability risks. A successful liability claim
or series of claims brought against the Company could have a material adverse
effect on its business, financial condition and results of operations.
The Company does not currently have
product liability insurance or other liability insurance relating to clinical
trials or to the marketing of any products or compounds. The Company cannot
assure that it will be able to obtain or maintain adequate product liability
insurance on acceptable terms, if at all, or that such insurance will provide
adequate coverage against the Company's potential liabilities. Furthermore, the
Company's current and potential partners with whom the Company has collaborative
agreements with or its future licensees may not be willing to indemnify the
Company against these types of liabilities and may not themselves be
sufficiently insured or have a net worth sufficient to satisfy any product
liability claims. Claims or losses in excess of any product liability insurance
coverage that may be obtained by the Company could have a material adverse
effect on its business, financial condition and results of operations.
Confidentiality
agreements with employees and others may not adequately prevent disclosure of
trade secrets and other proprietary information. Disclosure of our trade secrets
or proprietary information could compromise any competitive advantage that we
have.
We depend upon confidentiality
agreements with our officers, employees, consultants, and subcontractors to
maintain the proprietary nature of the technology. These measures may not afford
us sufficient or complete protection, and may not afford an adequate remedy in
the event of an unauthorized disclosure of confidential information. In
addition, others may independently develop technology similar to ours, otherwise
avoiding the confidentiality agreements, or produce patents that would
materially and adversely affect our business, prospects, financial condition,
and results of operations.
We
may be unable to obtain or protect intellectual property rights relating to our
products, and we may be liable for infringing upon the intellectual property
rights of others.
Our ability to compete effectively will
depend on our ability to maintain the proprietary nature of our compounds and
the proprietary compounds of others with which we have entered into
licensing agreements. We have filed one patent application and expect to file a
number of additional patent applications in the coming years. There can be
no assurance that any of these patent applications will ultimately result in the
issuance of a patent with respect to the proprietary compounds owned by us or
licensed to us. The patent position of pharmaceutical or biotechnology
companies, including ours, is generally uncertain and involves complex legal and
factual considerations. The standards that the United States Patent and
Trademark Office use to grant patents are not always applied predictably or
uniformly and can change. There is also no uniform, worldwide policy regarding
the subject matter and scope of claims granted or allowable in pharmaceutical or
biotechnology patents. Accordingly, we do not know the degree of future
protection for our proprietary rights or the breadth of claims that will be
allowed in any patents issued to us or to others. Further, we rely on a
combination of trade secrets, know-how, technology and nondisclosure, and other
contractual agreements and technical measures to protect our rights in the
proprietary compounds. If any trade secret, know-how or other proprietary
information and/or compounds not protected by a patent were to be disclosed to
or independently developed by a competitor, our business and financial condition
could be materially adversely affected.
We do not believe that any of the drug
candidates we are currently developing infringe upon the rights of any third
parties nor are they infringed upon by third parties; however, there can be no
assurance that our proprietary compounds will not be found in the future to
infringe upon the rights of others or be infringed upon by others. In such a
case, others may assert infringement claims against us, and should we be found
to infringe upon their patents, or otherwise impermissibly utilize their
intellectual property, we might be forced to pay damages, potentially including
treble damages, if we are found to have willfully infringed on such parties'
patent rights. In addition to any damages we might have to pay, we may be
required to obtain licenses from the holders of this intellectual property,
enter into royalty agreements, or redesign our drug candidates so as not to
utilize this intellectual property, each of which may prove to be uneconomical
or otherwise impossible. Conversely, we may not always be able to successfully
pursue our claims against others that infringe upon our proprietary
compounds. Thus, the proprietary nature of our technology or
technology licensed by us may not provide adequate protection against
competitors.
22
Moreover, the cost to us of any
litigation or other proceeding relating to our patents and other intellectual
property rights, even if resolved in our favor, could be substantial, and the
litigation would divert our management's efforts. Uncertainties resulting from
the initiation and continuation of any litigation could limit our ability to
continue our operations.
We
have limited manufacturing experience
The Company has never manufactured
products in the highly regulated environment of pharmaceutical manufacturing.
There are numerous regulations and requirements that must be maintained to
obtain licensure and permitting required to commence manufacturing, as well as
additional requirements to continue manufacturing pharmaceutical products. We do
not own or lease facilities currently that could be used to manufacture any
products that might be developed by the Company, nor do we have the resources at
this time to acquire or lease suitable facilities.
We
have no sales and marketing personnel.
We are an early stage development
Company with limited resources. We do not currently have any products available
for sale, so have not secured sales and marketing staff at this early stage of
operations. We cannot generate sales without sales or marketing staff and must
rely on officers to provide any sales or marketing services until such staff are
secured, if ever.
Even
if we were to successfully develop approvable drugs, we will not be able to sell
these drugs if we or our third party manufacturers fail to comply with
manufacturing regulations.
If we were to successfully develop
approvable drugs, before we can begin selling these drugs, we must obtain
regulatory approval of our cGMP manufacturing facility and process or the cGMP
manufacturing facility and process of the third party or parties with whom we
may outsource our manufacturing activities. The cGMP regulations govern quality
control and documentation policies and procedures. Our manufacturing facilities,
if any in the future, and the manufacturing facilities of our third party
manufacturers will be continually subject to inspection by the FDA and other
state, local and foreign regulatory authorities, before and after product
approval. We cannot guarantee that we, or any potential third party manufacturer
of our products, will be able to comply with the cGMP regulations or other
applicable manufacturing regulations.
Our
potential collaborative relationships with third parties could cause us to
expend significant resources and incur substantial business risk with no
assurance of financial return.
We may have to rely substantially upon
strategic collaborations for marketing and the commercialization of our drug
candidates, and we may rely even more on strategic collaborations for R&D of
our other drug candidates. Our business will depend on our ability to sell drugs
to both government agencies and to the general pharmaceutical market. We may
have to sell our drugs through strategic partnerships with pharmaceutical
companies. If we are unable to establish or manage such strategic collaborations
on terms favorable to us in the future, our revenue and drug development may be
limited. To date, we have not entered into any strategic collaborations with
third parties capable of providing these services. In addition, we have not yet
marketed or sold any of our drug candidates or entered into successful
collaborations for these services in order to ultimately commercialize our drug
candidates.
If we determine to enter into R&D
collaborations during the early phases of drug development, our success will in
part depend on the performance of our research collaborators. We will not
directly control the amount or timing of resources devoted by our research
collaborators to activities related to our drug candidates. Our research
collaborators may not commit sufficient resources to our programs. If any
research collaborator fails to commit sufficient resources, our preclinical or
clinical development programs related to this collaboration could be delayed or
terminated. Also, our collaborators may pursue existing or other
development-stage products or alternative technologies in preference to those
being developed in collaboration with us. Finally, if we fail to make required
milestone or royalty payments to our collaborators, or to observe other
obligations in our agreements with them, our collaborators may have the right to
terminate those agreements.
Establishing strategic collaborations
is difficult and time-consuming. Our discussion with potential collaborators may
not lead to the establishment of collaborations on favorable terms, if at all.
Potential collaborators may reject collaborations based upon their assessment of
our financial, regulatory or intellectual property position. Even if we
successfully establish new collaborations, these relationships may never result
in the successful development or commercialization of our drug candidates or the
generation of sales revenue. To the extent that we enter into collaborative
arrangements, our drug revenues are likely to be lower than if we directly
marketed and sold any drugs that we may develop.
Management of our relationships with
our collaborators will require:
• significant time and effort from our
management team;
• coordination of our marketing and
R&D programs with the marketing and R&D priorities of our collaborators;
and
• effective allocation of our resources
to multiple projects.
We
may not be able to attract and retain highly skilled
personnel.
Our ability to attract and retain
highly skilled personnel is critical to our operations and expansion. We face
competition for these types of personnel from other pharmaceutical companies and
more established organizations, many of which have significantly larger
operations and greater financial, technical, human and other resources than us.
We may not be successful in attracting and retaining qualified personnel on a
timely basis, on competitive terms, or at all. If we are not successful in
attracting and retaining these personnel, our business, prospects, financial
condition and results of operations will be materially adversely
affected.
We
depend upon our senior management and their loss or unavailability could put us
at a competitive disadvantage.
We currently depend upon the efforts
and abilities of our management team. The loss or unavailability of the services
of any of these individuals for any significant period of time could have a
material adverse effect on our business, prospects, financial condition and
results of operations. We have not obtained, do not own, nor are we the
beneficiary of key-person life insurance.
The Company believes the following
persons are critical to the success of the Company as well as the terms of the
employment agreements between them and the Company:
On December 7, 2007, the Company
entered into employment agreements with its two executive officers, Mr. George
Evans, Chief Executive Officer, and Dr. Krishna Menon, Chief Scientific Officer.
Both agreements provide for a three year term with minimum annual base salaries
of $200,000 in the first year, $300,000 in the second year and $400,000 in the
third year. In addition, the agreements provide for bonuses according to
the following schedule:
Upon receiving IND: $250,000 if
received within 10 months; $150,000 if received within 12 months; $100,000 if
received within 16 months; and no bonus if received thereafter or not
received.
23
Completion of Phase 1with clinical
results that would have Kevetrin proceed to Phase 2/3: $450,000 if received
within 18 months; $350,000 if received within 24 months; $150,000 if received
within 28 months; and no bonus if received thereafter or not
received.
Start Phase 2/3: $500,000 if within 36
months; $350,000 if within 42 months; $150,000 if within 48 months; and no bonus
if received thereafter or not received.
The bonus obligations under the
agreements do not commence until the Company receives a financing commitment in
an amount of at least $4,000,000.
The agreement with Mr. Evans also
provides a grant of options to purchase a number of shares of the Company's
stock equal to one percent of the issued and outstanding common stock following
the first anniversary of the commencement of the
agreement. Thereafter Mr. Evans shall be issued an additional grant
of options following the successive anniversaries of the commencement date of a
number of shares of common stock equal to one percent of the issued and
outstanding common stock at a purchase price equal to the average closing bid
price of the common stock on its primary exchange for the fifteen successive
trading days immediately prior to the date of the
grant.
There
are conflicts of interest among our officers, directors and
stockholders.
Certain of our executive officers and
directors and their affiliates are engaged in other activities and have
interests in other entities on their own behalf or on behalf of other persons.
Neither we nor any of our stockholders will have any rights in these ventures or
their income or profits. In particular:
• Our executive officers or directors
or their affiliates may have an economic interest in, or other business
relationship with, partner companies that invest in us or are engaged in
competing drug development.
• Our executive officers or directors or
their affiliates have interests in entities that provide products or services to
us. Presently, Kard Scientific, a company controlled by Dr. Krishna Menon,
President and Director, provides preclinical and manufacturing services to the
Company and leases space to the Company.
In any of these cases:
• Our executive officers or directors
may have a conflict between our current interests and their personal financial
and other interests in another business venture.
• Our executive officers or directors
may have conflicting fiduciary duties to us and the other
entity.
• The terms of transactions with the
other entity may not be subject to arm's length negotiations and therefore may
be on terms less favorable to us than those that could be procured through arm's
length negotiations.
While the Company is not aware of any
conflict that has arisen to date, Dr. Menon may have conflicting fiduciary
duties between the Company and Kard Scientific. Currently, the Company does not
have any policy in place to deal with such should such a conflict
arise.
Risks Related to the
Biotechnology/Biopharmaceutical Industry
The
biotechnology and biopharmaceutical industries are characterized by rapid
technological developments and a high degree of competition. We may be unable to
compete with enterprises equipped with more substantial resources than
us.
24
The biotechnology and biopharmaceutical
industries are characterized by rapid technological developments and a high
degree of competition based primarily on scientific and technological factors.
These factors include the availability of patent and other protection for
technology and products, the ability to commercialize technological developments
and the ability to obtain government approval for testing, manufacturing and
marketing.
We compete with biopharmaceutical firms
in the United States, Europe and elsewhere, as well as a growing number of large
pharmaceutical companies that are applying biotechnology to their operations.
Many biopharmaceutical companies have focused their development efforts in the
human therapeutics area, including cancer. Many major pharmaceutical companies
have developed or acquired internal biotechnology capabilities or made
commercial arrangements with other biopharmaceutical companies. These companies,
as well as academic institutions, government agencies and private research
organizations, also compete with us in recruiting and retaining highly qualified
scientific personnel and consultants. Our ability to compete successfully with
other companies in the pharmaceutical field will also depend to a considerable
degree on the continuing availability of capital on terms and conditions
acceptable to us.
We are aware of numerous products under
development or manufactured by competitors that are used for the prevention or
treatment of certain diseases we have targeted for drug development. Various
companies are developing biopharmaceutical products that potentially directly
compete with our drug candidates even though their approach to such treatment is
different.
With respect to Kevetrin, our lead
compound for cancer, there are many drugs approved to treat various cancers and
many more in the publicly disclosed pipeline. Our success depends on our
ability to identify tumor types where Kevetrin has an advantage over existing
therapies and those in the publicly disclosed
pipeline.
Our competition will be determined in
part by the potential indications for which drugs are developed and ultimately
approved by regulatory authorities. Additionally, the timing of the market
introduction of some of our potential drugs or of competitors' products may be
an important competitive factor. Accordingly, the relative speed with which we
can develop drugs, complete pre-clinical testing, clinical trials, approval
processes and supply commercial quantities to market are important competitive
factors. We expect that competition among drugs approved for sale will be based
on various factors, including product efficacy, safety, reliability,
availability, price and patent protection.
The successful development of
biopharmaceuticals is highly uncertain. A variety of factors including,
pre-clinical study results or regulatory approvals, could cause us to abandon
development of our drug candidates.
Successful
development of biopharmaceuticals is highly uncertain and is dependent on
numerous factors, many of which are beyond our control. Products that appear
promising in the early phases of development may fail to reach the market for
several reasons including:
• pre-clinical study results that may
show the product to be less effective than desired (e.g., the study failed to
meet its primary objectives) or to have harmful or problematic side
effects;
• failure to receive the necessary
regulatory approvals or a delay in receiving such approvals. Among other things,
such delays may be caused by slow enrollment in clinical studies, length of time
to achieve study endpoints, additional time requirements for data analysis or a
IND and later NDA, preparation, discussions with the FDA, an FDA request for
additional pre-clinical or clinical data or unexpected safety or manufacturing
issues;
• manufacturing costs, pricing or
reimbursement issues, or other factors that make the product not economical;
and
• the proprietary rights of others and
their competing products and technologies that may prevent the product from
being commercialized.
25
Success in pre-clinical and early
clinical studies does not ensure that large-scale clinical studies will be
successful. Clinical results are frequently susceptible to varying
interpretations that may delay, limit or prevent regulatory approvals. The
length of time necessary to complete clinical studies and to submit an
application for marketing approval for a final decision by a regulatory
authority varies significantly from one product to the next, and may be
difficult to predict.
Risks Related to the
Securities Markets and Investments in Our Common
Stock
Because
our common stock is quoted on the "OTCBB," your ability to sell your shares in
the secondary trading market may be limited.
Our common stock is currently quoted on
the over-the-counter market on the OTC Electronic Bulletin Board. Consequently,
the liquidity of our common stock is impaired, not only in the number of shares
that are bought and sold, but also through delays in the timing of transactions,
and coverage by security analysts and the news media, if any, of our company. As
a result, prices for shares of our common stock may be lower than might
otherwise prevail if our common stock was quoted and traded on NASDAQ or a
national securities exchange.
Because
our shares are "penny stocks," you may have difficulty selling them in the
secondary trading market.
Federal regulations under the
Securities Exchange Act of 1934 regulate the trading of so-called "penny
stocks," which are generally defined as any security not listed on a national
securities exchange or NASDAQ, priced at less than $5.00 per share and offered
by an issuer with limited net tangible assets and revenues. Since our common
stock currently is quoted on the "OTCBB" at less than $5.00 per share, our
shares are "penny stocks" and may not be traded unless a disclosure schedule
explaining the penny stock market and the risks associated therewith is
delivered to a potential purchaser prior to any
trade.
In addition, because our common stock
is not listed on NASDAQ or any national securities exchange and currently is
quoted at and trades at less than $5.00 per share, trading in our common stock
is subject to Rule 15g-9 under the Securities Exchange Act. Under this rule,
broker-dealers must take certain steps prior to selling a "penny stock," which
steps include:
• obtaining financial and investment
information from the investor;
• obtaining a written suitability
questionnaire and purchase agreement signed by the investor;
and
• providing the investor a written
identification of the shares being offered and the quantity of the
shares.
If these penny stock rules are not
followed by the broker-dealer, the investor has no obligation to purchase the
shares. The application of these comprehensive rules will make it more difficult
for broker-dealers to sell our common stock and our shareholders, therefore, may
have difficulty in selling their shares in the secondary trading
market.
Our
stock price may be volatile and your investment in our common stock could suffer
a decline in value.
As of June 30, 2009, the last trade
price of our common stock, as quoted on the OTC Bulletin Board, was
$0.38. The price may fluctuate significantly in response to a number
of factors, many of which are beyond our control. These factors
include:
• progress of our products through the
regulatory process;
• results of preclinical studies and
clinical trials;
26
• announcements of technological
innovations or new products by us or our competitors;
• government regulatory action
affecting our products or our competitors' products in both the United States
and foreign countries;
• developments or disputes concerning
patent or proprietary rights;
• general market conditions for
emerging growth and pharmaceutical companies;
• economic conditions in the United
States or abroad;
• actual or anticipated fluctuations in
our operating results;
• broad market fluctuations;
and
• changes in financial estimates by
securities analysts.
A
registration of a significant amount of our outstanding restricted stock may
have a negative effect on the trading price of our
stock.
At June 30, 2009,
shareholders of the
Company had approximately 56,391,000 shares of restricted stock, or 61% of the
outstanding common stock. If we were to file a registration statement including
all of these shares, and the registration is allowed by the SEC, these shares
would be freely tradable upon the effectiveness of the planned registration
statement. If investors holding a significant number of freely tradable shares
decide to sell them in a short period of time following the effectiveness of a
registration statement, such sales could contribute to significant downward
pressure on the price of our stock.
Our
directors and executive officers own or control a sufficient number of shares of
our common stock to control our company, which could discourage or prevent a
takeover, even if an acquisition would be beneficial to our
shareholders.
At June 30, 2009, our directors and
executive officers own or control approximately 48% of our outstanding voting
power. Accordingly, these shareholders, individually and as a group, may be able
to influence the outcome of shareholder votes, involving votes concerning the
election of directors, the adoption or amendment of provisions in our articles
of incorporation and bylaws and the approval of certain mergers or other similar
transactions, such as sales of substantially all of our assets. Such control by
existing shareholders could have the effect of delaying, deferring or preventing
a change in control of our company.
We
do not intend to pay any cash dividends in the foreseeable future and,
therefore, any return on your investment in our capital stock must come from
increases in the fair market value and trading price of the capital
stock.
We have not paid any cash dividends on
our common stock and do not intend to pay cash dividends on our common stock in
the foreseeable future. We intend to retain future earnings, if any, for
reinvestment in the development and expansion of our business. Any credit
agreements, which we may enter into with institutional lenders, may restrict our
ability to pay dividends. Whether we pay cash dividends in the future will be at
the discretion of our board of directors and will be dependent upon our
financial condition, results of operations, capital requirements and any other
factors that the board of directors decides is relevant. Therefore, any return
on your investment in our capital stock must come from increases in the fair
market value and trading price of the capital
stock.
We
may issue additional equity shares to fund the Company's operational
requirements which would dilute your share
ownership.
27
The Company's continued viability
depends on its ability to raise capital. Changes in economic, regulatory or
competitive conditions may lead to cost increases. Management may also determine
that it is in the best interest of the Company to develop new services or
products. In any such case additional financing is required for the Company to
meet its operational requirements. There can be no assurances that the Company
will be able to obtain such financing on terms acceptable to the Company and at
times required by the Company, if at all. In such event, the Company may be
required to materially alter its business plan or curtail all or a part of its
operational plans as detailed further in Management's Discussion and Analysis in
this Form 10-K. While the Company currently has no offers to sell it securities
to obtain financing, sale or the proposed sale of substantial amounts of our
common stock in the public markets may adversely affect the market price of our
common stock and our stock price may decline substantially. In the event that
the Company is unable to raise or borrow additional funds, the Company may be
required to curtail significantly its operational plans as further detailed in
Requirements for Additional Capital in the Management Discussion and Analysis of
this Form 10-K.
The Company is authorized to issue up
to 300,000,000 total shares of Common Stock without additional approval by
shareholders. As of June 30, 2009 we had 91,836,000 shares of common stock
outstanding, and warrants and options convertible to 5,297,487 shares of common
stock outstanding.
Because
our common stock is quoted only on the OTCBB, your ability to sell your shares
in the secondary trading market may be limited.
Our common stock is quoted only on the
OTCBB. Consequently, the liquidity of our common stock is impaired, not only in
the number of shares that are bought and sold, but also through delays in the
timing of transactions, and coverage by security analysts and the news media, if
any, of our company. As a result, prices for shares of our common stock may be
different than might otherwise prevail if our common stock was quoted or traded
on a national securities exchange such as the New York Stock Exchange.
Large
amounts of our common stock will be eligible for resale under Rule 144.
As of June 30, 2009, approximately
56,391,000 of the 91,836,000 issued and outstanding shares of the Company's
common stock are restricted securities as defined under Rule 144 of the
Securities Act of 1933, as amended (the “Act”) and under certain circumstances
may be resold without registration pursuant to Rule 144.
Approximately 8,500,000 shares of our
restricted shares of common stock are held by non-affiliates who may avail
themselves of the public information requirements and sell their shares in
accordance with Rule 144. As a result, some or all of these shares may be sold
in accordance with Rule 144 potentially causing the price of the Company's
shares to decline.
In general, under Rule 144, a person
(or persons whose shares are aggregated) who has satisfied a six month holding
period may, under certain circumstances, sell within any three-month period a
number of securities which does not exceed the greater of 1% of the then
outstanding shares of common stock or the average weekly trading volume of the
class during the four calendar weeks prior to such sale. Rule 144 also permits,
under certain circumstances, the sale of securities, without any limitation, by
a person who is not an Affiliate, as such term is defined in Rule 144(a)(1), of
the Company and who has satisfied a one-year holding period. Any substantial
sale of the Company's common stock pursuant to Rule 144 may have an adverse
effect on the market price of the Company's shares. This filing will satisfy
certain public information requirements necessary for such shares to be sold
under Rule 144.
The
requirements of complying with the Sarbanes-Oxley act may strain our resources
and distract management
We are subject to the reporting
requirements of the Securities Exchange Act of 1934, as amended (the “Exchange
Act”), and the Sarbanes-Oxley Act of 2002. The costs associated with these
requirements may place a strain on our systems and resources. The Exchange Act
requires that we file annual, quarterly and current reports with respect to our
business and financial condition. The Sarbanes-Oxley Act requires that we
maintain effective disclosure controls and procedures and internal controls over
financial reporting. Historically, as a private company we have maintained a
small accounting staff, but in order to maintain and improve the effectiveness
of our disclosure controls and procedures and internal control over financial
reporting, significant additional resources and management oversight will be
required. This includes, among other things, retaining independent public
accountants. This effort may divert management's attention from other business
concerns, which could have a material adverse effect on our business, financial
condition, results of operations and cash flows. In addition, we may need to
hire additional accounting and financial persons with appropriate public company
experience and technical accounting knowledge, and we cannot assure you that we
will be able to do so in a timely fashion.
28
ITEM 2. DESCRIPTION OF PROPERTY
Our principal offices are located at 100
Cummings Center, Suite 151-B, Beverly, MA 01915. Dr. Menon, the
Company’s principal shareholder, President and Director also serves as the COO
and Director of Kard Scientific. On December 7, 2007, Cellceutix Pharma began
renting 200 square feet of office space from Kard Scientific, on a month to
month basis for $900 per month. In September 2008, when Kard moved to its
present location in Beverly, MA, the Company moved along with them. The
Company's telephone number is (978) 633-3623.
We
subcontract the laboratory research and development work to Kard Scientific
which occupies 10,000 square foot in the same building. In September
of 2007, the Company engaged Kard Scientific to conduct specified pre-clinical
studies necessary for the Company to prepare an IND submission to the FDA.
We do not have an exclusive arrangement with KARD. All work performed by
Kard must have prior approval of the executive officers of the Company; and we
retain all intellectual property resulting from the services by KARD. To date we
have incurred charges from KARD of $390,587.
Management believes that the property
arrangement satisfies the Company’s current needs and is sufficient for the
Company to monitor the developmental progress at its
subcontractors.
ITEM
3. LEGAL PROCEEDINGS
ITEM 4. SUBMISSION OF MATTERS TO A
VOTE OF SECURITY HOLDERS
None,
ITEM 5. MARKET FOR COMMON EQUITY AND
RELATED STOCKHOLDER MATTERS
On January 14, 2008 shareholders
approved an amendment to the articles of incorporation to change the name of the
Company to Cellceutix Corporation. Upon the filing on February 1, 2008 of
a Definitive Information Statement, and effectiveness of the name change, the
Company applied to the National Association of Security Dealers to change its
stock symbol from “ECSR”. This resulted in the issuance of the
Company’s new stock symbol of “CTIX”.
The Company’s Common Stock is quoted on
the Over the Counter Bulletin Board (OTCBB). The table below sets forth the high
and low prices for the Company’s Common Stock. Quotations reflect inter-dealer
prices, without retail mark-up, mark-down commission, and may not represent
actual transactions. Since the Company's common stock trades sporadically, there
is not an established active public market for its common stock. No assurance
can be given that an active market will exist for the Company's common stock and
the Company does not expect to declare dividends in the foreseeable future since
the Company intends to utilize its earnings, if any, to finance its future
growth, including possible acquisitions.
Quarter
ended Low price High
price
|
June
30, 2009
|
$0.20
|
$1.00
|
|
March
31, 2009
|
$0.20
|
$0.20
|
|
December
31, 2008
|
$0.15
|
$0.30
|
|
September
30, 2008
|
$0.38
|
$0.92
|
|
June
30, 2008
|
$0.57
|
$1.01
|
|
March
31, 2008
|
$1.90
|
$1.90
|
29
December
11 to December 31, 2007
(1) No Trading
Occurred
(1) Effective date of reverse merger,
December 11, 2007
Number
of Shareholders
As of
June 30, 2009, a total of 91,836,000 of the Company’s common stock (shares) are
outstanding and held by approximately 100 shareholders of record. Of this
amount, 35,445,030 shares are unrestricted 8,493,118 shares are restricted
securities held by non-affiliates, and the remaining 47,897,882 shares are
restricted securities held by affiliates. These shares may only be sold in
accordance with Rule 144. As of June 30, 2009, there were 2,964,000 warrants and
2,066,820 stock options to purchase the Company’s Common Stock
outstanding.
Dividends.
The Company has not paid any cash
dividends since its inception. The Company currently intends to retain any
earnings for use in its business, and therefore does not anticipate paying
dividends in the foreseeable future.
Long-Term Incentive
Plans Awards In Last Fiscal Year
None
Recent
Cancellation of Unregistered Securities
On April
15, 2008 the Company signed a service agreement with a consultant to provide
public relations and strategic communications advice and services to the Company
for one year beginning April 28, 2008. In October, the Company was notified of
the death of the consultant and the agreement was terminated in accordance with
its terms. The 100,000 shares of the Company’s common stock
previously issued to the consultant were returned to the Company and
cancelled.
Recent
Sales of Unregistered Securities
From
August 2005 until September 27, 2006 we sold 741,000 shares of Common Stock
to investors as part of the Series A Units. The Units consisted of Common
Stock and Common Stock Purchase Warrants. The Units itself will
not be tradable and are not being registered. We are registering the
components of the Units which
include Common Stock and Common Shares underlying the
Common Stock Purchase Warrants. Each Unit was sold to investors at $0.05 per
Unit and consisted of one
(1) Share of Common Stock $.0001 par value and
four series of Warrants. In conjunction with the sale of
these Units, we issued 741,000 shares of Common Stock and Warrants to
purchase 2,964,000 shares of Common Stock underlying four
(4) Series of Warrants.
On
December 6, 2007, in accordance with the Share Exchange Agreement with
Cellceutix Pharma, Inc., we issued 82,000,000 newly issued shares of Common
Stock to the shareholders of Cellceutix Pharma.
On
December 7, 2007, we entered into an employment agreement with George Evans
which provides a grant of options to purchase a number of shares of our common
stock equal to one percent of the issued and outstanding common stock following
the first anniversary of the commencement of the agreement, at a purchase
price of $0.15 per share. Thereafter Mr. Evans shall be issued an additional
grant of options following the successive anniversaries of the commencement date
of a number of shares of common stock equal to one percent of the issued and
outstanding common stock at a purchase price equal to the average closing bid
price of the common stock on its primary exchange for the fifteen successive
trading days immediately prior to the date of the grant.
30
On May 7,
2008 the Company issued Convertible Debentures, at 9% per annum, for a total
amount of $400,000. The principle and related accrued interest are
due December 2009, and are secured by the Company’s assets. The
Debentures and any accrued and unpaid interest are convertible into the
Company’s common stock, at the holder’s request, at a conversion price of
$1.50.
On March
2, 2009, the Company entered into an agreement with a patent attorney to prepare
patent applications for Cellceutix products. The Company expects to file
multiple patent applications during the term of this agreement. The
agreement provides that the Company will grant 40,000 options to purchase shares
of the Company’s common stock as compensation for each application filed in lieu
of cash compensation. The options will be granted on the day that the work on
the application begins. As of June 30, 2009, the attorney had been issued 80,000
options for the preparation of two patents, valued at $11,843 and recorded in
the accompanying Statements of Operations as patent expense.
On April
1, 2009, the Company entered into a three month
agreement with a Consultant to assist the Company's Chief Scientific Officer to
organize, manage and display data from animal studies as well as information
relating to Active Pharmaceutical Ingredients and formulations of the Company's
products. The Consultant was compensated at the rate of $4,000 per month payable
on the last day of each month. In addition, at the end of each month of services
provided, the Consultant was granted options to purchase 10,000 shares of
Company's common stock. For the year ended June 30, 2009, the
Consultant was awarded a total of 30,000 options to purchase common stock valued
at $7,507 to be vested over one year. For the year ended June 30,
2009, the Company has expensed $600 to professional fees expense for the
amortization of those options. As of June 30, 2009, the Company has
accrued $4,000 for the June 30, 2009 payment.
On May 6,
2009, the Company entered into a one year agreement with a Consultant to provide
advice on financial and administrative matters as specifically requested by the
Company. In exchange for these services, the Company granted the
Consultant an option to purchase 100,000 shares of common stock and the option
expires on May 6, 2012. The option is valued at $36,085 and vests
over a one year period. For the year ended June 30, 2009, the Company
has expensed $6,015 to professional fees expense.
On June
9, 2009, the Company entered into an agreement
with a Consultant to assist the Company by providing advice on FDA regulatory
matters as specifically requested by Officers of the Company and it is expected
that the primary focus of the services will be to prepare for and arrange a
pre-IND meeting for Kevetrin. The Consultant will be compensated at
the rate of $100 per hour for partial days and $1,000 per day for full days,
plus reasonable and documented expenses. In addition, the Consultant
was granted options to purchase 20,000 shares common stock valued at $5,065 and
vests over a one year period. The options are exercisable for three
years at an exercise price of $0.15 per share. For the year ended
June 30, 2009, the Company has expensed $422 to professional fees
expense.
On June
11, 2009, the Company determined that they would award the patent attorney
additional compensation of 20,000 shares of common stock, valued at
$7,600. This amount has been recorded in the accompanying Statements of
Operations as patent expense.
On June
30, 2009, the Company entered into a one year agreement with a Consultant to
serve as a scientific advisor and to participate as a member of the Company’s
Scientific Advisory Board. In exchange for these services, the
Company has granted the Consultant 25,000 shares of common stock valued at
$9,500. At June 30, 2009, the Company has included $9,500 as a
prepaid expense to be amortized over the Consultant’s one year of
service.
In June
2009, the Company signed an agreement with Girindus America, Inc., for the cGMP
manufacture of Kevetrin active pharmaceutical ingredient. As of June
30, 2009, no payment has been made under this contract, nor has any work been
performed.
Except as
noted above, the sales of the securities identified above were made pursuant to
privately negotiated transactions that did not involve a public offering of
securities and, accordingly, we believe that these transactions were exempt from
the registration requirements of the Securities Act pursuant to Section 4(2)
thereof and rules promulgated there under. Each of the above-referenced
investors in our stock represented to us in connection with their investment
that they were “accredited investors” (as defined by Rule 501 under the
Securities Act) and were acquiring the shares for investment and not
distribution, that they could bear the risks of the investment and could hold
the securities for an indefinite period of time. The investors received written
disclosures that the securities had not been registered under the Securities Act
and that any resale must be made pursuant to a registration or an available
exemption from such registration. All of the foregoing securities are deemed
restricted securities for purposes of the Securities Act.
31
ITEM
6. SELECTED FINANCIAL DATA
Not
applicable for smaller reporting companies.
ITEM 7. MANAGEMENT'S
DISCUSSION AND FINANCIAL CONDITION AND RESULTS OF
OPERATIONS
The following discussion and plan of
operations should read in conjunction with the financial statements and the
notes to those statements included in this Form 10-K. This discussion includes
forward-looking statements that involve risk and uncertainties. As a result of
many factors, such as those set forth under “Risk Factors,” actual results may
differ materially from those anticipated in these forward-looking statements.
Acquisition and
Reorganization
On December 6, 2007, the Acquisition of
Cellceutix Pharma, Inc. was completed, and the business of Cellceutix Pharma,
Inc. was adopted as our business. As such, the following Management Discussion
is focused on the current and historical operations of Cellceutix Pharma, Inc.
and excludes the prior operations of EconoShare, Inc..
Management’s Plan of
Operation
As a result of the Exchange with
Cellceutix Pharma, Inc., we are an early stage developmental biopharmaceutical
company. We have no product sales to date and we will not receive any product
revenue until we receive approval from the FDA or equivalent foreign regulatory
bodies to begin selling our pharmaceutical candidates. Developing pharmaceutical
products, however, is a lengthy and very expensive process. Assuming we do not
encounter any unforeseen safety issues during the course of developing our
product candidates, we do not expect to complete the development of a product
candidate for several years, if ever.
In August and October 2007, we acquired
exclusive rights to a total of six (6) pharmaceutical compound candidates that
are designed for treatment of diseases which may be either existing or diseases
identified in the future. In July, 2009 we acquired the rights to an
additional compound. The Company will initially spend most of its efforts
and resources on its anti-cancer compound, Kevetrin, for the treatment
of certain cancers. Kevetrin is furthest along in in-vivo
studies in small animals. Based on the experimental studies results to
date, the Company has decided to advance Kevetrin along the regulatory and
clinical pathway. We anticipate using our expertise to manage and perform what
we believe are the most critical aspects of the product development process
which include: (i) the design and oversight of clinical trials; (ii) the
development and execution of strategies for the protection and maintenance of
intellectual property rights; and (iii) the interaction with regulatory
authorities internationally. We expect to concentrate on product development and
engage in a limited way in product discovery, avoiding the significant
investment of time and financial resources that is generally required before a
compound is identified and brought into clinical trials. In addition, we are
currently engaged in pre-clinical testing of one of our product candidates and
intend to out-source clinical trials, pre-clinical testing and have currently
outsourced the cGMP manufacturing of Kevetrin to third parties (Girindus
America, Inc.).
We are now engaged in organizational
activities, sourcing compounds, synthesis of experimental quantities, and the
outsourcing of cGMP manufacturing of Kevetrin. We have not obtained
sufficient funding for our drug development business plan, nor have we generated
any revenues, nor do we not expect to generate revenues in the near future. We
may not be successful in developing our drugs and start selling our products
when planned, or that we will become profitable in the future. We have incurred
net losses in each fiscal period since inception of our
operations.
Liquidity and
Capital Resources
As of June 30, 2009 the Company had a
cash balance of $140,380. The Company will need to raise substantial
funds in order to execute its product development plan. Based upon
our expected rate of expenditures, we currently do not have sufficient cash
reserves to meet all of our anticipated obligations through our fiscal year end
of June 30, 2010. The Company may seek to raise capital through an
offering of our common stock or other securities of the Company. However, there
can be no assurance that we will be successful in securing the capital we
require or that we may obtain financing on terms and conditions that are
acceptable to the Company.
32
Subsequent to June 30, 2008, Mr. Ehrlich,
an officer of the Company, converted previous amounts provided in cash to the
Company to a loan (the “Ehrlich Promissory Note A”). The
Ehrlich Promissory Note A is an unsecured, 6% per annum simple interest bearing,
demand note. The principal balance of the Ehrlich Promissory Note A is $32,310
as of June 30, 2009 and 2008 and remains unpaid as of September 30, 2009.
On May 7,
2008, the Company issued convertible debentures, at 9% per annum, for a total
amount of $400,000 (the “2007
Convertible Debentures”). The principle and related accrued
interest are due December 01, 2009, and are secured by the Company’s
assets. The 2007 Convertible Debentures and any accrued and unpaid
interest are convertible into the Company’s common stock, at the holder’s
request, at a conversion price of $1.50. The current principal balance of the
2007 Convertible Debentures is $400,000 as of June 30, 2009 and remains unpaid
as of September 30, 2009.
Subsequent
to June 30, 2009, Mr. Ehrlich, an officer of the Company, provided cash in the
form of a loan to the Company (the “Ehrlich Promissory Note B”). The
Ehrlich Promissory Note B is an unsecured, 6% per annum simple interest bearing,
demand note. The current principal balance of the Ehrlich Promissory Note B is
$85,000 and remains unpaid as of September 30, 2009.
Requirement for
Additional Capital
Research and Development Costs.
The Company has engaged in limited research and development activities. We
currently do not have sufficient funds to meet our planned drug development for
the next twelve (12) months and we may not be able to obtain the necessary
financing on terms and conditions acceptable to the Company. Assuming that we
are successful in raising additional financing, we plan to incur the following
expenses over the next twelve (12) months:
|
Research
and Development of $4,500,000: Includes planned costs for Kevetrin of
$2,500,000 for additional in-vivo, in-vitro, pharmaco-kinetic,
pharmaco-dynamic, and toxicology studies; and cGMP materials, which should
result with the data required to file an investigational new drug
application (“IND”) with the FDA; and $2,000,000 in preclinical
development costs for our other compounds. In August 2009, the Company
signed an agreement with Girindus America, Inc., for the cGMP manufacture
of Kevetrin’s active pharmaceutical ingredient.
|
|
|
2
|
Clinical
trials – We have budgeted $2,000,000 for our Phase 1 trials (assumes
success of Company’s IND filing for Kevetrin)..
|
|
3
|
Corporate
overhead of $1,250,000: Budgeted office salaries, legal, accounting and
other costs expected to be incurred.
|
|
4
|
Capital
costs of $500,000: Estimated cost for equipment and laboratory
improvements.
|
|
5
|
Staffing
costs of $500,000: The Company expects to incur these costs for
the filing of the IND which include additional scientific staff and
consulting firms to assist with FDA compliance, material characterization,
pharmaco-kinetic, pharmaco-dynamic and toxicology
studies.
|
The Company will be unable to proceed
with its full planned drug development program (s), meet its administrative
expense requirements, capital costs, or staffing costs without obtaining
additional financing of approximately $8,750,000 (as per current
management’s budgets). The Company does not have any arrangements at this time
for equity or other financings towards meeting this financing requirement. If we
are unable to obtain additional financing on terms and conditions acceptable to
the Company, our business plan will be significantly
delayed.
Time Schedules,
Milestones and Development Costs
In the event that sufficient funding
can be obtained, we shall endeavor to achieve completion of the following event
within the next twelve months:
33
|
Drug
Development of Kevetrin™ for Cancer
|
|
|
Current
status
|
Kevetrin is currently in
preclinical studies. Kevetrin acts as an AKT inhibitor with possible
activity in other pathways. Our experiments to date suggest that it
is a potent anti-cancer agent. The compound was well tolerated and
showed little toxicity in various animal studies. We are currently
conducting further studies concerning the potential safety of the compound
in order to request permission from FDA to begin clinical
studies.
|
|
Nature,
timing and estimated costs
|
We expect to submit the compound
for an IND submission in 2010 as a drug candidate for the treatment of
certain cancers. The Company has budgeted approximately $2,500,000 for the
material development, production and testing of this drug during this
period. Should management determine the results to be satisfactory, we
will proceed to a Phase I clinical study with the approval of
the FDA which we have presently budgeted
at $5,000,000, of which we
expect $2,000,000 to be incurred over the next 12
months.
|
|
Risks
and uncertainties associated with completing development on schedule, and
the consequences to operations, financial position and liquidity if not
completed timely
|
The
outcome of clinical testing can not be known at this time, and this poses
substantial risk and uncertainty as to whether or when if ever, Kevetrin
will become marketable.
|
The Company intends to commit limited
resources towards the development of compounds other than Kevetrin during the
next twelve (12) months.
The Company has limited experience with
pharmaceutical drug development. As such, these budget estimates may not be
accurate. In addition, the actual work to be performed is not known at this
time, other than a broad outline, as is normal with any scientific work. As
further work is performed, additional work may become necessary or change in
plans or workload may occur. Such changes may have an adverse impact on our
estimated budget. Such changes may also have an adverse impact on our projected
timeline of drug development.
The Company has contracted the
manufacturing of the Kevetrin in June 2009, with Girindus America, Inc., for the
cGMP manufacture of Kevetrin’s active pharmaceutical ingredient( site certified by the FDA) in order
for the Company to produce experimental materials that can be sent to outside
scientists for pharmaco-kinetic, pharmaco-dynamic and toxicology studies. These
three sets of studies must be completed prior to the Company filing an IND with
the FDA to begin the human safety and efficacy trials (Phase I , II and III ).
The
Company believes it can contract the manufacturing of its other compounds at
sites certified by the FDA in order for the Company to produce experimental
materials that can be sent to outside scientists for pharmaco-kinetic,
pharmaco-dynamic and toxicology studies. These three sets of studies must be
completed prior to the Company filing an IND with the FDA to begin the human
safety and efficacy trials of its other compounds (Phase I, II and
III).
The work-plan we have developed for the
next twelve (12) months is expected to enable us to file the IND in our fiscal
year. If we find that we have underestimated the time duration of our
studies, or we have to undertake additional studies, due to various reasons
within or outside of our control, this may significantly impact our development
timelines and/or our financing needs.
Management intends to use capital and
debt financing, as required, to fund the Company's operations. There can be no
assurance that the Company will be able to obtain the additional capital
resources on terms and conditions acceptable to the Company to fund its
anticipated obligations for the next twelve (12)
months.
The Company is considered to be a
development stage company and will continue in the development stage until
generating revenues from the sales of its products or services. As a result, the
report of the independent registered public accounting firm on our financial
statements as of June 30, 2009, contains an explanatory paragraph regarding a
substantial doubt about our ability to continue as a going concern.
34
Off
Balance Sheet Arrangements
There were no off-balance sheet
arrangements as of June 30, 2009.
CRITICAL ACCOUNTING
POLICIES AND ESTIMATES
The preparation of the financial
statements in conformity with accounting principles generally accepted in the
United States of America requires management to make estimates and assumptions
that affect the amounts reported in the financial statements and accompanying
notes. Actual results could differ materially from those
estimates.
We believe that there are several
accounting policies that are critical to understanding our historical and future
performance, as these policies affect the reported amounts of revenue and the
more significant areas involving management’s judgments and estimates. These
significant accounting policies relate to research and development costs,
valuation of inventory, valuation of long-lived assets and income taxes. These
policies, and the related procedures, are described in detail
below.
Research
and Development
Expenditures
for research, development, and engineering of products are expensed as
incurred. For the periods ended June 30, 2009 and 2008, and since
inception, the Company incurred $390,587, $0 and $390,587 of research and
development costs, respectively.
Intangible
Assets – Patents
Costs
incurred to file patent applications are capitalized when the Company believes
that there is a high likelihood that the patent will issue and there will be
future economic benefit associated with the patent. These costs will
be amortized on a straight-line basis over a 17 year life from the date of
patent filing. All costs associated with abandoned patent
applications are expensed. In addition, the Company will review the
carrying value of patents for indicators of impairment on a periodic basis and
if it determines that the carrying value is impaired, it values the patent at
fair value. As of June 30, 2009, the Company has expensed the costs
associated with obtaining patents as the Company has not yet developed products,
which have gained market acceptance. For the years ended June 30,
2009 and 2008 and from inception to June 30, 2009, the Company has recorded
$19,443, $0 and $19,443, respectively.
Long-Lived
Assets
The
Company follows SFAS No. 144, "Accounting for the Impairment or Disposal of
Long-Lived Assets." SFAS No. 144 addresses the financial accounting and
reporting for the impairment of long-lived assets, excluding goodwill and
intangible assets, not subject to amortization, to be held and used or disposed
of. In accordance with SFAS No. 144, the carrying values of
long-lived assets are periodically reviewed by the Company and impairments would
be recognized if the expected future operating non-discounted cash flows derived
from an asset were less than its carrying value and if the carrying value is
more than the fair value of the asset. At June 30, 2009, the Company did not
assign a value to its intangible assets, as they will continue to require
additional development and it has yet to be determined the underlying value of
the assets.
Income
Taxes
Deferred
income tax assets and liabilities arise from temporary differences associated
with differences between the financial statements and tax basis of assets and
liabilities, as measured by the enacted tax rates, which are expected to be in
effect when these differences reverse. Deferred tax assets and
liabilities are classified as current or non-current, depending upon the
classification of the asset or liabilities to which they
relate. Deferred tax assets and liabilities not related to an asset
or liability are classified as current or non-current depending on the periods
in which the temporary differences are expected to reverse. Valuation
allowances are established when necessary to reduce deferred tax assets to the
amount expected to be realized.
35
The
Company has generated net losses since inception and accordingly has not
recorded a provision for income taxes. The deferred tax assets were primarily
comprised of federal and state tax net operating loss, or NOL, carryforwards.
Due to uncertainties surrounding the Company’s ability to generate future
taxable income to realize these tax assets, a full valuation allowance has been
established to offset the deferred tax assets. Additionally, the future
utilization of the NOL carryforwards to offset future taxable income may be
subject to an annual limitation as a result of ownership changes that may have
occurred previously or that could occur in the future. The Company has not yet
determined whether such an ownership change has occurred. If necessary, the
deferred tax assets will be reduced by any carryforwards that expire prior to
utilization as a result of such limitations, with a corresponding reduction of
the valuation allowance.
The
Company has adopted the provisions of FASB Interpretation No. 48, “Accounting
for Uncertainty in Income Taxes” (FIN 48). As of June 30, 2009, the
Company had no unrecognized tax benefits or related interest and
penalties. Under FIN 48, we will include future interest and
penalties associated with any unrecognized benefits within provision for income
taxes on the Statements of Operations. We do not anticipate any
unrecognized benefits in the next 12 months that would result in a material
change to our financial position.
Accounting
for Stock Based Compensation
In
December 2004, the FASB issued SFAS No. 123 (revised 2004), Share-Based Payment
(“SFAS 123R”), which replaces SFAS No. 123; Accounting for Stock-Based
Compensation, (“SFAS 123”) and supersedes APB Opinion No. 25, Accounting for
Stock Issued to Employees, (“APB 25”). SFAS 123R requires all share-based
payments to employees, including grants of employee stock options, to be
recognized in the financial statements based on their fair values. Under SFAS
123R, the Company is required to measure the cost of employee services received
in exchange for stock options and similar awards based on the grant-date fair
value of the award and recognize this cost in the income statement over the
period during which an employee is required to provide service in exchange for
the award. The pro forma disclosures previously permitted under SFAS 123 are no
longer an alternative to financial statement recognition. The Company adopted
SFAS 123R on June 20, 2007 using the modified prospective method, which did not
require the recognition of any non-cash charges, as there were no unvested stock
options on that date.
The fair
value concepts were not changed significantly in FAS 123R; however, in adopting
FAS 123R, companies must choose among alternative valuation models and
amortization assumptions. After assessing alternative valuation models and
amortization assumptions, the Company will continue using the Black-Scholes
valuation model and has elected to use the ratable method to amortize
compensation expense over the vesting period of the grant.
Impairment
of Long-Lived Assets
In accordance with SFAS No. 144
"Accounting for the Impairment or Disposal of Long-Lived Assets" ("SFAS 144"),
our long-lived assets to be held and used in the business are reviewed for
impairment. When impairment is noted, assets are evaluated for impairment at the
lowest level for which there is identifiable cash flows. If the sum of expected
undiscounted future cash flows is less than the carrying amount of the asset, a
loss is recognized based on the amount by which the carrying value exceeds the
fair value of the asset. Fair value is determined primarily using the
anticipated cash flows discounted at a rate commensurate with the risk involved.
Losses on assets to be disposed of are determined in a similar manner, except
that the fair values are reduced for disposal costs. Considerable management
judgment and assumptions are necessary to identify indicators of impairment and
to estimate discounted future cash flows. Accordingly, actual results could vary
significantly from such estimates. At June 30, 2009, the Company did not assign
a value to its intangible assets, as they will continue to require additional
development and it has yet to be determined the underlying value of the
assets.
Contingencies
In the ordinary course of business, we
have entered into various contractual relationships with strategic corporate
partners, customers, distributors, research laboratories and universities,
licensors, licensees, suppliers, vendors and other parties. As such, we could be
subject to litigation, claims or assessments arising from any or all of these
relationships. We account for contingencies such as these in accordance with
Statement of Financial Accounting Standards No. 5, "Accounting for
Contingencies" ("SFAS 5"). SFAS 5 requires us to record an estimated loss
contingency when information available prior to issuance of our financial
statements indicates that it is probable that an asset has been impaired or a
liability has been incurred at the date of the financial statements and the
amount of the loss can be reasonably estimated. Accounting for contingencies
arising from contractual or legal proceedings requires that we use our best
judgment when estimating an accrual related to such contingencies. As additional
information becomes known, our accrual for a loss contingency could fluctuate,
thereby creating variability in our results of operations from period to period.
Likewise, an actual loss arising from a loss contingency which significantly
exceeds the amount accrued for in our financial statements could have a material
adverse impact on our operating results for the period in which such actual loss
becomes known.
Subsequent
Events
On May
28, 2009, FASB issued SFAS No. 165, “Subsequent Events.” The objective of this
statement is to establish general standards of accounting for and disclosure of
events that occur after the balance sheet date but before financial statements
are issued or are available to be issued. SFAS No. 165 has not had a material
impact on the financial reporting of the Company.
In
accordance with SFAS 165, the Company has evaluated subsequent events through
October 8, 2009, the date of issuance of the Financial Statements. During the
period from July 1, 2009 to October 8, 2009, the Company did not have any
material recognizable subsequent events.
36
Not
applicable.
ITEM
8. FINANCIAL STATEMENTS AND SUPPLEMENTARY
DATA
Report
of Independent Registered Public Accounting Firm
Board of
Directors
and
Stockholders
Cellceutix
Corporation
Beverly,
Massachusetts
We have
audited the accompanying balance sheets of Cellceutix Corporation (a development
stage enterprise) as of June 30, 2009 and 2008, and the related statements of
operations, stockholders' equity (deficit) and cash flows for the two years then
ended and the cumulative period from June 20, 2007 (date of inception) through
June 30, 2009. These financial statements are the responsibility of the
Company's management. Our responsibility is to express an opinion on these
financial statements based on our audits.
We
conducted our audits in accordance with the standards of the Public Company
Accounting Oversight Board (United States). Those standards require that we plan
and perform the audits to obtain reasonable assurance about whether the
financial statements are free of material misstatement. The Company is not
required to have, nor were we engaged to perform, an audit of its internal
control over financial reporting. Our audits included consideration of internal
control over financial reporting as a basis for designing audit procedures that
are appropriate in the circumstances, but not for the purpose of expressing an
opinion on the effectiveness of the Company's internal control over financial
reporting. Accordingly, we express no such opinion. An audit also includes
examining, on a test basis, evidence supporting the amounts and disclosures in
the financial statements. An audit also includes assessing the accounting
principles used and significant estimates made by management, as well as,
evaluating the overall financial statement presentation. We believe that our
audits provide a reasonable basis for our opinion.
In our
opinion, the financial statements referred to above present fairly,
in all material respects, the financial position of Cellceutix
Corporation (a development stage enterprise) as of June 30, 2009 and 2008, and
the results of its operations and its cash flows for the two years then ended
and the cumulative period from June 20, 2007 (date of inception) through June
30, 2009, in conformity with accounting principles generally accepted in the
United States of America.
The
accompanying financial statements have been prepared assuming that the Company
will continue as a going concern. As discussed in Note 2 to the financial
statements, the Company has no revenues, has suffered significant operating
losses, and is dependent upon its stockholders to provide sufficient working
capital to meet its obligations and sustain its operations. These circumstances
raise substantial doubt about the Company's ability to continue as a going
concern. Management's plans regarding those matters are also described in Note
2. The accompanying financial statements do not contain any adjustments that
might result from the outcome of these uncertainties.
/s/ Holtz
Rubenstein Reminick LLP
New York,
New York
October 8, 2009
37
Cellceutix
Corporation
(A
Development Stage Enterprise)
Balance
Sheets
|
June
30, 2009
|
June
30, 2008
|
|||||||||
|
Assets
|
||||||||||
|
Current
assets:
|
||||||||||
|
Cash
|
$
|
140,380
|
$
|
351,860
|
||||||
|
Prepaid expenses
|
14,853
|
97,917
|
||||||||
|
Total
current assets
|
155,233
|
449,777
|
||||||||
|
Total
assets
|
$
|
155,233
|
$
|
449,777
|
||||||
|
Liabilities
and Stockholders' Deficit
|
||||||||||
|
Current
liabilities:
|
||||||||||
|
Accounts payable, including related party payables of $327,179 and $0,
respectively
|
$
|
340,013
|
$
|
13,730
|
||||||
|
Accrued expenses, including related party rent accrual of $17,100 and
$6,300, respectively
|
59,100
|
20,349
|
||||||||
|
Accrued officers’ salaries and payroll taxes
|
1,116,869
|
345,378
|
||||||||
|
Note
payable to officer
|
32,310
|
32,310
|
||||||||
|
Convertible
debentures
|
400,000
|
-
|
||||||||
|
Total
current liabilities
|
1,948,292
|
411,767
|
||||||||
|
Long
term liabilities:
|
||||||||||
|
Convertible
debentures
|
-
|
400,000
|
||||||||
|
Total
liabilities
|
1,948,292
|
811,767
|
||||||||
|
Commitments
and contingencies
|
||||||||||
|
Stockholders'
deficit:
|
||||||||||
|
Preferred
stock; $.0001 par value; 10,000,000 shares
|
||||||||||
|
authorized;
0 shares issued and outstanding
|
-
|
-
|
||||||||
|
Common
stock; $.0001 par value; 300,000,000 shares
|
||||||||||
|
authorized;
91,836,000 and 91,891,000 shares issued and outstanding,
respectively
|
9,184
|
9,189
|
||||||||
|
Additional
paid in capital
|
202,890
|
148,623
|
||||||||
|
Deficit
accumulated during development stage
|
(2,005,133
|
)
|
(519,802
|
)
|
||||||
|
Total
stockholders' deficit
|
(1,793,059
|
)
|
(361,990
|
)
|
||||||
|
Total
liabilities and stockholders' deficit
|
$
|
155,233
|
$
|
449,777
|
||||||
The accompanying notes are an
integral part of these financial statements.
38
Cellceutix
Corporation
(A
Development Stage Enterprise)
Statements
of Operations
|
For
the Year Ended June 30, 2009
|
For
the year ended June 30, 2008
|
For
the cumulative period from June 20, 2007 (Date of Inception) through June
30, 2009
|
||||||||||
|
Revenues
|
$
|
-
|
$
|
-
|
$
|
-
|
||||||
|
Operating
expenses:
|
||||||||||||
|
General
and administrative
expenses
|
47,459
|
25,099
|
73,088
|
|||||||||
|
Officers’
payroll and payroll tax expense
|
894,773
|
388,911
|
1,283,684
|
|||||||||
|
Research
and development
|
390,587
|
-
|
390,587
|
|||||||||
|
Professional
fees
|
98,713
|
90,183
|
188,896
|
|||||||||
|
Patent
expense
|
19,443
|
-
|
19,443
|
|||||||||
|
Total
operating expenses
|
1,450,975
|
504,193
|
1,955,698
|
|||||||||
|
Loss
from operations
|
(1,450,975
|
)
|
(504,193
|
)
|
(1,955,698
|
)
|
||||||
|
Interest
expense - net
|
(34,356)
|
(6,000)
|
(40,356)
|
|||||||||
|
Net
loss before provision for income taxes
|
(1,485,331
|
)
|
(510,193
|
)
|
(1,996,054)
|
|||||||
|
Provision
for income taxes
|
-
|
-
|
-
|
|||||||||
|
Net
loss
|
$
|
(1,485,331
|
)
|
$
|
(510,193
|
)
|
$
|
(1,996,054
|
)
|
|||
|
Basic
and diluted loss per share
|
$
|
(0.02
|
)
|
$
|
(0.01
|
)
|
||||||
|
Weighted
average number of common shares used in basic and fully diluted per share
calculations
|
91,842,452
|
52,366,221
|
||||||||||
The accompanying notes are an
integral part of these financial statements.
39
Cellceutix
Corporation
(A
Development Stage Enterprise)
Statement
of Changes in Stockholders’ Deficit
For the
cumulative
Period
June 20, 2007 (Date of Inception)
through
June 30, 2009
|
Common
Stock
|
Additional
Paid
|
Deficit
Accumulated
During
Development
|
||||||||||||||||||
|
Shares
|
Par
Value $0.0001
|
In
Capital
|
Stage
|
Total
|
||||||||||||||||
|
Shares
issued June 20, 2007 (Inception)
|
1,000,000
|
$
|
100
|
$
|
-
|
$
|
-
|
$
|
100
|
|||||||||||
|
Net
loss
|
-
|
-
|
-
|
(530
|
)
|
(530
|
)
|
|||||||||||||
|
Balance,
June 30, 2007
|
1,000,000
|
100
|
-
|
(530
|
)
|
(430
|
)
|
|||||||||||||
|
Share
exchange with Cellceutix Pharma, Inc. December 6, 2007
|
(1,000,000
|
)
|
(100
|
)
|
-
|
100
|
-
|
|||||||||||||
|
SharShare
exchange in reverse merger with Cellceutix Pharma, Inc. December 6,
2007
|
82,000,000
|
8,200
|
-
|
(8,200
|
)
|
-
|
||||||||||||||
|
Shares
exchanged in a reverse acquisition of Cellceutix Pharma, December 6,
2007
|
9,791,000
|
979
|
-
|
(979
|
)
|
-
|
||||||||||||||
|
Issuance
of stock options
|
-
|
-
|
43,533
|
-
|
43,533
|
|||||||||||||||
|
Forgiveness
of debt from a
stockholder
|
-
|
-
|
50
|
-
|
50
|
|||||||||||||||
|
Capital
contribution from a stockholder
|
-
|
-
|
50
|
-
|
50
|
|||||||||||||||
|
Shares
issued for services, April 28, 2008 at $1.05
|
100,000
|
10
|
104,990
|
-
|
105,000
|
|||||||||||||||
|
Net
loss
|
-
|
-
|
-
|
(510,193
|
)
|
(510,193
|
)
|
|||||||||||||
|
Balance,
June 30, 2008
|
91,891,000
|
9,189
|
148,623
|
(519,802
|
)
|
(361,990
|
)
|
|||||||||||||
|
Cancellation
of shares issued for services, December 31, 2008
|
(100,000)
|
(10)
|
(104,990)
|
-
|
(105,000)
|
|||||||||||||||
|
Issuance
of stock options
|
-
|
-
|
142,162
|
-
|
142,162
|
|||||||||||||||
|
Shares
issued for services, June 11, 2009 at $0.38
|
20,000
|
2
|
7,598
|
-
|
7,600
|
|||||||||||||||
|
Shares
issued for services, June 30, 2009 at $0.38
|
25,000
|
3
|
9,497
|
9,500
|
||||||||||||||||
|
Net
loss for the year ended
June
30, 2009
|
-
|
-
|
-
|
(1,485,331
|
)
|
(1,485,331
|
)
|
|||||||||||||
|
Balance,
June 30, 2009
|
91,836,000
|
$
|
9,184
|
$
|
202,890
|
$
|
(2,005,133
|
)
|
$
|
(1,793,059
|
)
|
|||||||||
The
accompanying notes are an integral part of these financial
statements.
40
Cellceutix
Corporation
(A
Development Stage Enterprise)
Statements
of Cash Flows
|
For
the Year Ended June 30, 2009
|
For
the Year Ended
June
30, 2008
|
For
the Cumulative Period June 20, 2007 (Date of Inception)
through
June
30, 2009
|
||||||||
|
CASH
FLOWS FROM OPERATING ACTIVITIES:
|
||||||||||
|
Net
loss
|
$
|
(1,485,331
|
)
|
$(510,193)
|
$
|
(1,996,054
|
)
|
|||
|
Adjustments
to reconcile net loss to net cash used in operating
activities:
|
||||||||||
|
Stock
based compensation
|
142,162
|
43,533
|
185,695
|
|||||||
|
Issuance
of common stock for services
|
7,600
|
—
|
7,600
|
|||||||
|
Cancellation
of stock issued for services
|
(17,500)
|
—
|
(17,500)
|
|||||||
|
Amortization
of prepaid expenses
|
—
|
17,500
|
17,500
|
|||||||
|
Changes
in operating assets and liabilities:
|
||||||||||
|
Prepaid
expenses
|
5,064
|
(10,417)
|
(5,353)
|
|||||||
|
Accounts
payable
|
326,283
|
13,350
|
340,063
|
|||||||
|
Accrued
expenses
|
38,751
|
20,349
|
59,100
|
|||||||
|
Accrued
officers’ salaries and payroll taxes
|
771,491
|
345,378
|
1,116,869
|
|||||||
|
Net
cash used in operating activities
|
(211,480
|
)
|
(80,500)
|
(292,080
|
)
|
|||||
|
CASH
FLOWS FROM FINANCING ACTIVITIES:
|
||||||||||
|
Capital
contribution from stockholder
|
—
|
50
|
50
|
|||||||
|
Sale
of common stock
|
—
|
—
|
100
|
|||||||
|
Loan
from officer
|
—
|
32,310
|
32,310
|
|||||||
|
Proceeds
from convertible debentures
|
—
|
400,000
|
400,000
|
|||||||
|
Net
cash provided by financing activities
|
—
|
432,360
|
432,460
|
|||||||
|
NET
(DECREASE) INCREASE IN CASH AND CASH EQUIVALENTS
|
(211,480)
|
351,860
|
140,380
|
|||||||
|
CASH
AND CASH EQUIVALENTS, BEGINNING OF PERIOD
|
351,860
|
—
|
—
|
|||||||
|
CASH
AND CASH EQUIVALENTS, END OF PERIOD
|
$
|
140,380
|
$351,860
|
$
|
140,380
|
|||||
SUPPLEMENTAL
DISCLOSURE OF NON-CASH FLOW FINANCING ACTIVITIES:
|
Common
stock issued for acquisition
|
$
|
—
|
$9,079
|
$
|
9,079
|
|||||
|
Forgiveness
of debt
|
$
|
—
|
$50
|
$
|
50
|
|||||
|
25,000
shares of common stock issued for future services
|
$
|
7,600
|
—
|
$
|
7,600
|
|||||
|
100,000
shares of common stock issued for services
|
$
|
—
|
$105,000
|
$
|
105,000
|
|||||
|
Cancellation
of 100,000 shares of common stock for services
|
$
|
(105,000)
|
—
|
$
|
(105,000)
|
The
accompanying notes are an integral part of these financial
statements.
41
Cellceutix
Corporation
(A
Development Stage Enterprise)
Notes to
Financial Statements
June
30, 2009
1. Background
Information
EconoShare,
Inc. was incorporated on August 1, 2005 in the State of Nevada and was organized
for the purpose of developing a B2B (Business to Business) website for an Asset
Sharing market place and transaction system.
On
December 6, 2007, EconoShare, Inc. acquired Cellceutix Pharma, Inc., a privately
owned Delaware corporation (“Cellceutix Pharma”), pursuant to an Agreement and
Plan of Share Exchange (the “Exchange”), with Cellceutix
Pharma becoming a wholly-owned subsidiary of EconoShare,
Inc. Cellceutix Pharma, Inc. was incorporated under the laws of the
State of Delaware on June 20, 2007. Its assets consisted of rights
assigned to it for six early stage pharmaceutical compounds by three different
scientists. Upon consummation of the Exchange, EconoShare, Inc. adopted the
business plan of Cellceutix Pharma, Inc.
Pursuant
to the terms of the Exchange, EconoShare, Inc. acquired Cellceutix Pharma, Inc.
in exchange for an aggregate of 82,000,000 newly issued shares of EconoShare,
Inc.’s common stock, par value $0.0001 per share (the “Common Stock”), which
resulted at that time in an aggregate of 91,791,000 shares (the “Exchange of
Shares”) of EconoShare, Inc. Common Stock issued and outstanding. As a result of
the Exchange, Cellceutix Pharma, Inc. became a wholly-owned subsidiary of
EconoShare, Inc. The Exchange Shares were issued to the Cellceutix
Pharma, Inc. shareholders on a pro rata basis, on the basis of 82 shares of
Common Stock for each share of Cellceutix Pharma, Inc. common stock held by such
Cellceutix Pharma, Inc. shareholder at the time of the
Exchange.
The
former holders of Cellceutix Pharma Common Stock at the time of the exchange
owned approximately 89% of the outstanding shares of our Common Stock.
Accordingly, the Exchange represented a change in control. As of June 30, 2009,
there are 91,836,000 shares of Common Stock issued and
outstanding. For financial accounting purposes, the acquisition was a
reverse acquisition of EconoShare, Inc. by Cellceutix Pharma, Inc., under the
purchase method of accounting, and was accounted for as a recapitalization as of
June 20, 2007 with Cellceutix Pharma, Inc. as the accounting
acquirer.
On
January 14, 2008, a majority of the shareholders of EconoShare, Inc. approved an
amendment to the Registrant’s articles of incorporation to change the name of
the Registrant to Cellceutix Corporation (“the Company”). Upon the
filing of a Definitive Information Statement and effectiveness of the name
change the Company applied to the National Association of Security Dealers to
change its stock symbol on the Over the Counter Bulletin Board, resulting in the
Company’s new stock symbol of “CTIX”. The Company is considered a development
stage company at this time.
2. Going
Concern
The
accompanying financial statements have been prepared assuming the Company will
continue as a going concern. For the period since June 20, 2007 (date of
inception) through June 30, 2009, the Company has had a cumulative net loss of
$1,996,054, no revenues and a working capital deficit of $1,793,059 at June 30,
2009. As of June 30, 2009, the Company has not emerged from the
development stage. In view of these matters, the ability of the Company to
continue as a going concern is dependent upon the Company’s ability to generate
additional financing. Since inception, the Company has financed its activities
principally from the use of equity securities and debt issuance to pay for its
operations. The Company intends on financing its future development activities
and its working capital needs largely from the issuance of debt and the sale of
equity securities, until such time that funds provided by operations are
sufficient to fund working capital requirements. There can be no assurance that
the Company will be successful at achieving its financing goals at reasonably
commercial terms, if at all.
The
recent economic downturn and market instability has made the business climate
more volatile and more costly. If the current equity and credit
markets deteriorate further or do not improve, it may make necessary debt or
equity financing more difficult, more costly and more
dilutive. Failure to secure any necessary financing in a timely
manner and on favorable terms could have a material adverse effect on the
Company’s growth strategy, financial performance and stock price and could
require the delay of new product development and clinical trial
plans. Currently, the Company has not made any arrangements for
equity or any other type of financing.
42
These
factors raise substantial doubt about the Company's ability to continue as a
going concern. The accompanying financial statements do not include any
adjustments relating to the recoverability of the recorded assets or the
classification of liabilities that may be necessary should the Company be unable
to continue as a going concern.
3. Significant
Accounting Policies
The
significant accounting policies followed are:
Cash
and Cash Equivalents
Cash and
cash equivalents consist of cash and short-term highly liquid investments
purchased with original or remaining maturities of three months or less. There
were no cash equivalents at June 30, 2009 and 2008.
Concentrations
of Credit Risk
All cash
is maintained with a major financial institution in the United
States. Deposits with this bank may exceed the amount of insurance
provided on such deposits. Generally, these deposits may be redeemed
upon demand and, therefore, bear minimal risk.
Financial
Instruments
The
Company’s financial instruments include cash, accounts payable and accrued
liabilities. The carrying amounts of these financial instruments
approximate their fair value, due to the short-term nature of these items and
due to the use of market rates of interest.
Use
of Estimates
The
preparation of financial statements in conformity with accounting principles
generally accepted in the United States of America requires management to make
estimates and assumptions that affect the reported amounts of assets and
liabilities and disclosure of contingent assets and liabilities at the date of
the financial statements and the reported amounts of revenues and expenses
during the reporting period. Actual results could differ from those
estimates.
Research
and Development
Expenditures
for research, development, and engineering of products are expensed as
incurred. For the periods ended June 30, 2009 and 2008, and since
inception, the Company incurred $390,587, $0 and $390,587 of research and
development costs, respectively.
Intangible
Assets – Patents
Costs
incurred to file patent applications are capitalized when the Company believes
that there is a high likelihood that the patent will issue and there will be
future economic benefit associated with the patent. These costs will
be amortized on a straight-line basis over a 17 year life from the date of
patent filing. All costs associated with abandoned patent
applications are expensed. In addition, the Company will review the
carrying value of patents for indicators of impairment on a periodic basis and
if it determines that the carrying value is impaired, it values the patent at
fair value. As of June 30, 2009, the Company has expensed the costs
associated with obtaining patents as the Company has not yet developed products,
which have gained market acceptance. For the years ended June 30,
2009 and 2008 and from inception to June 30, 2009, the Company has recorded
$19,443, $0 and $19,443, respectively.
Long-Lived
Assets
The
Company follows SFAS No. 144, "Accounting for the Impairment or Disposal of
Long-Lived Assets." SFAS No. 144 addresses the financial accounting and
reporting for the impairment of long-lived assets, excluding goodwill and
intangible assets, not subject to amortization, to be held and used or disposed
of. In accordance with SFAS No. 144, the carrying values of
long-lived assets are periodically reviewed by the Company and impairments would
be recognized if the expected future operating non-discounted cash flows derived
from an asset were less than its carrying value and if the carrying value is
more than the fair value of the asset. At June 30, 2009, the Company did not
assign a value to its intangible assets, as they will continue to require
additional development and it has yet to be determined the underlying value of
the assets.
43
Income
Taxes
Deferred
income tax assets and liabilities arise from temporary differences associated
with differences between the financial statements and tax basis of assets and
liabilities, as measured by the enacted tax rates, which are expected to be in
effect when these differences reverse. Deferred tax assets and
liabilities are classified as current or non-current, depending upon the
classification of the asset or liabilities to which they
relate. Deferred tax assets and liabilities not related to an asset
or liability are classified as current or non-current depending on the periods
in which the temporary differences are expected to reverse. Valuation
allowances are established when necessary to reduce deferred tax assets to the
amount expected to be realized.
The
Company has generated net losses since inception and accordingly has not
recorded a provision for income taxes. The deferred tax
assets were primarily comprised of federal and state tax net operating loss, or
NOL, carryforwards. Due to uncertainties surrounding the Company’s ability to
generate future taxable income to realize these tax assets, a full valuation
allowance has been established to offset the deferred tax
assets. Additionally, the future utilization of the NOL carryforwards to offset
future taxable income may be subject to an annual limitation as a result of
ownership changes that could occur in the future. If necessary, the
deferred tax assets will be reduced by any carryforwards
that expire prior to utilization as a result of such limitations, with a
corresponding reduction of the valuation allowance.
The
Company has adopted the provisions of FASB Interpretation No. 48, “Accounting
for Uncertainty in Income Taxes” (FIN 48). As of June 30, 2009, the
Company had no unrecognized tax benefits or related interest and
penalties. Under FIN 48, we will include future interest and
penalties associated with any unrecognized benefits within provision for income
taxes on the Statements of Operations. We do not anticipate any
unrecognized benefits in the next 12 months that would result in a material
change to our financial position.
Basic
Earnings (Loss) per Share
Basic
Earnings (Loss) per Share is calculated in accordance with SFAS No. 128,
“Earnings per Share,” by dividing income or loss attributable to common
stockholders by weighted average common stock outstanding. Diluted
earnings per share is calculated in accordance with SFAS No. 128 by adjusting
weighted average common shares outstanding by assuming conversion of all
potentially dilutive shares. In periods where a net loss is recorded,
no effect is given to potentially dilutive securities, since the effect would be
antidilutive. Total stock options and warrants not included in the
calculation of common shares outstanding (including both exercisable and
nonexercisable) and convertible debentures, for the periods ended June 30, 2009
and 2008 were 5,297,487 and 4,148,577, respectively.
Common
stock equivalents for the all periods presented were anti-dilutive due to the
net losses sustained by the Company during these periods.
Reclassifications
Certain
balances in the prior period’s financial statements have been reclassified to
conform to the presentation adopted in the current year. Such
reclassifications did not affect the net loss.
Accounting
for Stock Based Compensation
In
December 2004, the FASB issued SFAS No. 123 (revised 2004), Share-Based Payment
(“SFAS 123R”), which replaces SFAS No. 123; Accounting for Stock-Based
Compensation, (“SFAS 123”) and supersedes APB Opinion No. 25, Accounting for
Stock Issued to Employees, (“APB 25”). SFAS 123R requires all share-based
payments to employees, including grants of employee stock options, to be
recognized in the financial statements based on their fair values. Under SFAS
123R, the Company is required to measure the cost of employee services received
in exchange for stock options and similar awards based on the grant-date fair
value of the award and recognize this cost in the income statement over the
period during which an employee is required to provide service in exchange for
the award. The pro forma disclosures previously permitted under SFAS 123 are no
longer an alternative to financial statement recognition. The Company adopted
SFAS 123R on June 20, 2007 using the modified prospective method, which did not
require the recognition of any non-cash charges, as there were no unvested stock
options on that date.
44
The fair
value concepts were not changed significantly in FAS 123R; however, in adopting
FAS 123R, companies must choose among alternative valuation models and
amortization assumptions. After assessing alternative valuation models and
amortization assumptions, the Company will continue using the Black-Scholes
valuation model and has elected to use the ratable method to amortize
compensation expense over the vesting period of the grant.
The fair
value of each option for the year ended June 30, 2009 and 2008 was estimated on
the date of grant using the Black Scholes model that uses assumptions noted in
the following table. Expected volatility is based on the monthly trading of a
similar Company’s underlying common stock (as the Company does not have an
adequate trading history for an accurate calculation) and other
factors.
|
2009
|
2008
|
|||||||
|
Expected
term (in years)
|
3 | 3 | ||||||
|
Expected
stock price volatility
|
108.4% - 114.9 | % | 86.4 | % | ||||
|
Risk-free
interest rate
|
1.27%– 1.93 | % | 3.15 | % | ||||
|
Expected
dividend yield
|
0 | % | 0 | % | ||||
|
Estimated
fair value per option granted
|
0.13 - 0.36 | 0.05 | ||||||
The
Company accounts for equity instruments issued to nonemployees in accordance
with the provisions of Emerging Issues Task Force No. 96-18, Accounting for Equity Instruments
That Are Issued to Other Than Employees for Acquiring, or in Conjunction with
Selling, Goods or Services ("EITF 96-18"). The equity instruments, consisting of stock options,
are valued using the Black-Scholes valuation model. The measurement of
stock-based compensation is subject to periodic adjustments as the underlying
equity instruments vest.
The
components of stock based compensation recognized in the Company’s Statement of
Operations for the years ended June 30, 2009 and 2008 and since inception are as
follows:
|
For
the year ended June 30, 2009
|
For
the year ended June 30, 2008
|
For
the cumulative period from June 20, 2007 (Date of Inception) through June
30, 2009
|
||||||||||
|
Officers’
payroll and payroll tax expense
|
$
|
123,282
|
$
|
43,533
|
$
|
166,815
|
||||||
|
Professional
fees
|
7,037
|
-
|
7,037
|
|||||||||
|
Patent
expense
|
19,443
|
-
|
19,443
|
|||||||||
|
$
|
149,762
|
$
|
43,533
|
$
|
193,295
|
|||||||
Recent Accounting
Pronouncements
On May
28, 2009, FASB issued SFAS No. 165, “Subsequent Events.” The
objective of this statement is to establish general standards of accounting for
and disclosure of events that occur after the balance sheet date but before
financial statements are issued or are available to be issued. SFAS No. 165 has
not had a material impact on the financial reporting of the
Company.
In
accordance with SFAS 165, the Company has evaluated subsequent events through
October 8, 2009, the date of issuance of the Financial Statements. During the
period from July 1, 2009 to October 8, 2009, the Company did not have any
material recognizable subsequent events.
In June
2009, the Financial Accounting Standards Board (FASB) issued SFAS No. 168,
“The FASB Accounting Standards Codification and the Hierarchy of Generally
Accepted Accounting Principles—a replacement of FASB Statement No. 162,”
(SFAS 168). SFAS 168 replaces SFAS No. 162, “The Hierarchy of Generally
Accepted Accounting Principles,” and establishes the FASB Accounting Standards
Codification (Codification) as the source of authoritative accounting principles
recognized by the FASB to be applied by nongovernmental entities in the
preparation of financial statements in conformity with GAAP. Rules and
interpretive releases of the Securities and Exchange Commission (SEC) under
authority of federal securities laws are also sources of authoritative GAAP for
SEC registrants. The FASB will no longer issue new standards in the form of
Statements, FASB Staff Positions, or Emerging Issues Task Force Abstracts;
instead the FASB will issue Accounting Standards Updates. Accounting Standards
Updates will not be authoritative in their own right as they will only serve to
update the Codification. The issuance of SFAS 168 and the Codification does not
change GAAP. SFAS 168 becomes effective for the Company for the period ending
September 30, 2009. The adoption of SFAS 168 will not have an impact on the
financial statements.
45
In June
2009, the FASB issued SFAS No. 167, “Amendments to FASB Interpretation
No. 46(R),” (SFAS 167). SFAS 167 amends FASB Interpretation No. 46
(Revised December 2003), “Consolidation of Variable Interest Entities—an
interpretation of ARB No. 51,” (FIN 46(R)) to require an enterprise to
perform an analysis to determine whether the enterprise’s variable interest or
interests give it a controlling financial interest in a variable interest
entity; to require ongoing reassessments of whether an enterprise is the primary
beneficiary of a variable interest entity; to eliminate the quantitative
approach previously required for determining the primary beneficiary of a
variable interest entity; to add an additional reconsideration event for
determining whether an entity is a variable interest entity when any changes in
facts and circumstances occur such that holders of the equity investment at
risk, as a group, lose the power from voting rights or similar rights of those
investments to direct the activities of the entity that most significantly
impact the entity’s economic performance; and to require enhanced disclosures
that will provide users of financial statements with more transparent
information about an enterprise’s involvement in a variable interest entity.
SFAS 167 becomes effective for the Company on July 1, 2010. Management is
currently evaluating the potential impact of SFAS 167 on the financial
statements.
Other
recent accounting pronouncements issued by FASB (including EITF), the AICPA and
the SEC did not or are not believed by management to have a material impact on
the Company’s present financial statements.
4. Commitments
and Contingencies
Consulting
Agreements
On April
15, 2008 the Company signed a service agreement with a consultant to provide
public relations and strategic communications advice and services to the Company
for one year beginning April 28, 2008. In October, the Company was notified of
the death of the consultant and the agreement was terminated in accordance with
its terms. The 100,000 shares of the Company’s common stock
previously issued to the consultant were returned to the Company and
cancelled.
On March
2, 2009, the Company entered into an agreement with a patent attorney to prepare
patent applications for Cellceutix products. The Company expects to file
multiple patent applications during the term of this agreement. The
agreement provides that the Company will grant 40,000 options to purchase shares
of the Company’s common stock as compensation for each application filed in lieu
of cash compensation. The options will be granted on the day that the work on
the application begins. As of June 30, 2009, the attorney had been issued 80,000
options for the preparation of two patents, valued at $11,843 and recorded in
the accompanying Statements of Operations as patent expense.
On April
1, 2009, the Company entered into a three month
agreement with a Consultant to assist the Company's Chief Scientific Officer to
organize, manage and display data from animal studies as well as information
relating to Active Pharmaceutical Ingredients and formulations of the Company's
products. The Consultant was compensated at the rate of $4,000 per month payable
on the last day of each month. In addition, at the end of each month of services
provided, the Consultant was granted options to purchase 10,000 shares of
Company's common stock. For the year ended June 30, 2009, the
Consultant was awarded a total of 30,000 options to purchase common stock valued
at $7,507 to be vested over one year. For the year ended June 30,
2009, the Company has expensed $600 to professional fees expense for the
amortization of those options. As of June 30, 2009, the Company has
accrued $4,000 for the June 30, 2009 payment.
On May 6,
2009, the Company entered into a one year agreement with a Consultant to provide
advice on financial and administrative matters as specifically requested by the
Company. In exchange for these services, the Company granted the
Consultant an option to purchase 100,000 shares of common stock and the option
expires on May 6, 2012. The option is valued at $36,085 and vests
over a one year period. For the year ended June 30, 2009, the Company
has expensed $6,015 to professional fees expense.
46
On June
9, 2009, the Company entered into an agreement
with a Consultant to assist the Company by providing advice on FDA regulatory
matters as specifically requested by Officers of the Company and it is expected
that the primary focus of the services will be to prepare for and arrange a
pre-IND meeting for Kevetrin. The Consultant will be compensated at
the rate of $100 per hour for partial days and $1,000 per day for full days,
plus reasonable and documented expenses. In addition, the Consultant
was granted options to purchase 20,000 shares common stock valued at $5,065 and
vests over a one year period. The options are exercisable for three
years at an exercise price of $0.15 per share. For the year ended
June 30, 2009, the Company has expensed $422 to professional fees
expense.
On June
11, 2009, the Company determined that they would award the patent attorney
additional compensation of 20,000 shares of common stock, valued at
$7,600. This amount has been recorded in the accompanying Statements of
Operations as patent expense.
On June
30, 2009, the Company entered into a one year agreement with a Consultant to
serve as a scientific advisor and to participate as a member of the Company’s
Scientific Advisory Board. In exchange for these services, the
Company has granted the Consultant 25,000 shares of common stock valued at
$9,500. At June 30, 2009, the Company has included $9,500 as a
prepaid expense to be amortized over the Consultant’s one year of
service.
In June
2009, the Company signed an agreement with Girindus America, Inc., for the cGMP
manufacture of Kevetrin active pharmaceutical ingredient. As of June
30, 2009, no payment has been made under this contract, nor has any work been
performed.
Pharmaceutical
Compounds
On August
2, 2007, the Company was assigned all right, title, and interest to three
pharmaceutical compounds; Kevetrin, KM 277 and KM 278, by their inventors. The
Company was assigned all right, title, and interest to an additional three
pharmaceutical compounds on October 17, 2007, KM 133 KM 362 and KM
3174, and in July 2009 was assigned all right, title, and interest to
KM 732. In exchange for these compounds, the Company agreed to pay
the inventors 5% of net sales of the compounds in countries where composition of
matter patents have been issued and 3% of net sales in other countries.
Kevetrin, KM 277, KM 278 and KM 362 were acquired from our President and
director, Dr. Krishna Menon. The Company filed a patent application
for Kevetrin and intends to file patent applications for each of the other six
compounds as funds become available.
The
Company is continuing the research and development of these Compounds and has
therefore, assigned no value to these Compounds.
Employment
Agreements
On
December 7, 2007, the Company entered into employment agreements with its two
executive officers, George Evans, Chief Executive Officer, and Krishna Menon,
Chief Scientific Officer. Both agreements provide for a three year term with
minimum annual base salaries of $200,000 in the first year, $300,000 in the
second year and $400,000 in the third year. In addition, the
agreements provide for bonuses according to the following schedule:
Upon
receiving New Drug Application (“IND”): $250,000 if received within 10
months
$150,000
if received within 12 months
$100,000
if received within 16 months
Completion
of Phase 1with clinical results that would have Kevitrin proceed to Phase
2/3:
$450,000
if received within 18 months
$350,000
if received within 24 months
$150,000
if received within 28 months
Start
Phase 2/3:
$500,000
if within 36 months
$350,000
if within 42 months
$150,000
if within 48 months
The bonus
obligations do not commence until the Company receives a financing commitment in
an amount of at least $4,000,000.
47
The
agreement with Mr. Evans also provides a grant of options to purchase 918,910
and 917,910 shares of the Company's stock with exercise prices of $0.20 and
$0.15 per share and fair values of $123,282 and $43,533, for the years ended
June 30, 2009 and 2008, respectively. The agreement calls for the
issuance options to purchase up to 1% of the common shares outstanding at each
subsequent anniversary year.
As of
June 30, 2009, the Company has recorded accrued officer’s salaries and payroll
taxes are as follows:
George
Evans $375,000
Krisna
Menon
375,000
Leo
Ehrlich
287,500
Accrued
Payroll
Taxes 79,369
$1,116,869
5. Related
Party Transactions
Office
Lease
Dr.
Menon, the Company’s principal shareholder, President, and Director, also serves
as the Chief Operating Officer and Director of Kard Scientific (“KARD”). On
December 7, 2007, the Company began renting office space from KARD, on a month
to month basis for $900 per month. At June 30, 2009 and 2008,
payables of $17,100 and $6,300 to KARD were included in accrued expenses,
respectively. For the years ended June 30, 2009 and 2008, the Company has
included $10,800 and $6,300 in general and administrative expenses,
respectively.
Clinical
Studies
As of
September 28, 2007 the Company engaged KARD to conduct specified pre-clinical
studies necessary for the Company to prepare an IND submission to the
FDA. The Company does not have an exclusive arrangement with
KARD. All work performed by KARD must have prior approval by the
executive officers of the Company, and the Company retains all intellectual
property resulting from the services by KARD. To date the Company has incurred
$390,587 of research and development expenses by KARD, all of which was during
the June 30, 2009 year end, of which $321,505 is included in accounts
payable.
6. Due
To Related Party
Subsequent
to June 30, 2008, Mr. Ehrlich, an officer of the Company, converted previous
amounts provided in cash to the Company to a loan (the “Ehrlich Promissory Note A”). The
Ehrlich Promissory Note A is an unsecured, 6% per annum simple interest bearing,
demand note. The principal balance of the Ehrlich Promissory Note A is $32,310
as of June 30, 2009 and 2008 and remains unpaid as of September 30,
2009.
Subsequent
to June 30, 2009, Mr. Ehrlich, an officer of the Company, provided cash in the
form of a loan to the Company (the “Ehrlich Promissory Note B”). The
Ehrlich Promissory Note B is an unsecured, 6% per annum simple interest bearing,
demand note. The principal balance of the Ehrlich Promissory Note B is for a
maximum of $100,000. Mr. Ehrlich has provided cash of $85,000 and
this amount remains unpaid as of September 30, 2009.
7. Stock
Options and Warrants
On April
5, 2009 the Board of Directors of the Registrant adopted the 2009 Stock Option
Plan ("the Plan"). The Plan permits the grant of 2,000,000 shares of both
Incentive Stock Options ("ISOs"), intended to qualify under section 422 of the
Code, and Non-Qualified Stock Options.
Stock
Options
The
following table summarizes all stock option activity for options granted since
inception:
48
|
Number
of Options
|
Weighted
Average Exercise Price
|
Weighted
Average Remaining Contractual Life (Years)
|
Aggregate
Intrinsic Value
|
||||||||||
|
Outstanding
at June 20, 2007
|
—
|
$
|
—
|
—
|
$
|
—
|
|||||||
|
Granted
|
917,910
|
$
|
0.15
|
2.44
|
$
|
385,522
|
|||||||
|
Exercised
|
—
|
—
|
—
|
—
|
|||||||||
|
Forfeited/expired
|
—
|
—
|
—
|
—
|
|||||||||
|
Outstanding
at June 30, 2008
|
917,910
|
$
|
0.15
|
2.44
|
$
|
385,522
|
|||||||
|
Granted
|
1,148,910
|
$
|
0.19
|
2.51
|
$
|
207,404
|
|||||||
|
Exercised
|
—
|
—
|
—
|
—
|
|||||||||
|
Forfeited/expired
|
—
|
—
|
—
|
—
|
|||||||||
|
Outstanding
at June 30, 2009
|
2,066,820
|
$
|
0.17
|
2.04
|
$
|
418,523
|
|||||||
|
Exercisable
at June 30, 2009
|
1,937,653
|
$
|
0.17
|
1.98
|
$
|
399,481
|
|||||||
The
following table represents our non vested share-based payment activity for the
year ended June 30, 2009:
|
Weighted
Average
|
||||||||
|
Number
of
|
Grant
Date
|
|||||||
|
Options
|
Fair
Value
|
|||||||
|
Nonvested
options – June 30, 2008
|
-
|
-
|
||||||
|
-
|
||||||||
|
Granted
|
1,148,910
|
$
|
0.19
|
|||||
|
Vested
|
(1,019,743
|
)
|
0.19
|
|||||
|
Forfeited
|
-
|
|||||||
|
Nonvested
options – June 30, 2009
|
129,167
|
$
|
0.19
|
|||||
The
following table summarizes information about stock options outstanding and
exercisable at June 30, 2009.
|
Options
Exercisable
|
|||||||||||||||||||||
|
Weighted
|
Weighted
|
Weighted
|
|||||||||||||||||||
|
Average
|
Average
|
Average
|
|||||||||||||||||||
|
Exercise
|
Number
|
Remaining
|
Exercise
|
Number
|
Exercise
|
||||||||||||||||
|
Price
|
of
Shares
|
Life
(Years)
|
Price
|
of
Shares
|
Price
|
||||||||||||||||
|
$
|
0.15
|
927,910
|
1.44
|
$
|
0.15
|
919,577
|
$
|
0.15
|
|||||||||||||
|
0.20
|
918,910
|
2.44
|
0.20
|
918,910
|
0.20
|
||||||||||||||||
|
0.14
|
80,000
|
2.67
|
0.14
|
80,000
|
0.14
|
||||||||||||||||
|
0.18
|
100,000
|
2.85
|
0.18
|
16,667
|
0.18
|
||||||||||||||||
|
0.35
|
10,000
|
2.92
|
0.35
|
833
|
0.35
|
||||||||||||||||
|
0.37
|
20,000
|
2.95
|
0.37
|
1,667
|
0.37
|
||||||||||||||||
|
0.38
|
10,000
|
3.00
|
0.38
|
-
|
0.38
|
||||||||||||||||
|
2,066,820
|
1,937,653
|
||||||||||||||||||||
For the
year ended June 30, 2009 and 2008 and the period from inception to June 30,
2009, there was $142,162, $43,533 and $185,695, respectively, of compensation
cost related to option awards granted and $41,621 unamortized compensation cost
expected to be recognized over the next year.
As of
June 30, 2009 and 2008, there were 2,964,000 warrants issued and
outstanding. The weighted average exercise price was $0.81 and the
options expire in September 2010. There were no new warrants issued
during the years ended June 30, 2009 and 2008.
49
8. Convertible
Debenture
On May 7,
2008, the Company issued convertible debentures, at 9% per annum, for a total
amount of $400,000 (the “2007
Convertible Debentures”). The principle and related accrued
interest are due December 1, 2009, and are secured by the Company’s
assets. The 2007 Convertible Debentures and any accrued and unpaid
interest are convertible into the Company’s common stock, at the holder’s
request, at a conversion price of $1.50. The current principal and accrued
interest balance of the 2007 Convertible Debentures is $400,000 and $42,000,
respectively, as of June 30, 2009 and remains unpaid as of September 30, 2009.
The debenture holders have a first priority security interest in all of the
assets of the Company.
9. Income
Tax
Deferred
income tax assets and liabilities are recognized for the expected future tax
consequences of events that have been reflected in the consolidated financial
statements. Deferred tax assets and liabilities are determined based on the
differences between the book values and the tax bases of particular assets and
liabilities and the tax effects of net operating loss and capital loss carry
forwards. Deferred tax assets and liabilities are measured using enacted tax
rates expected to apply to taxable income in the years in which those temporary
differences are expected to be recovered or settled. The effect on deferred tax
assets and liabilities of a change in the tax rate is recognized as income or
expense in the period that included the enactment date.
The
Company has incurred operating losses since its inception and, therefore, no tax
liabilities have been incurred for the periods presented. The amount of unused
federal tax losses available to carry forward and apply against taxable income
in future years totaled approximately $1,113,000 at June 30, 2009. The loss
carry forwards expire beginning in 2028. Internal Revenue Code Sec. 382 places
limitations on the utilization of net operating losses. Due to the
limitation and the Company’s historical losses, the Company has placed a
full valuation allowance. The valuation allowance increased by $633,300 at
June 30, 2009 and $175,800 at June 30, 2008.
The
income tax provision benefit differs from the amount of tax determined by
applying the Federal statutory rate as follows:
| Years Ended |
June
20, 2007
|
|||||||||||
|
|
|
through
|
||||||||||
|
|
June
30, 2009
|
June
30, 2008
|
June
30, 2009
|
|||||||||
|
Income
tax benefit as statutory rate
|
$ | (219,500 | ) | $ | (158,700 | ) | $ | (269,200 | ) | |||
|
Increase
(decrease) in income tax due to:
|
||||||||||||
|
State
income taxes, net
|
(23,400 | ) | (16,900 | ) | (28,700 | ) | ||||||
|
Valuation
allowance
|
242,900 | 175,600 | 297,900 | |||||||||
| $ | - | $ | - | $ | - | |||||||
There was
no current or deferred provision or benefit for income taxes for the year ended
June 30, 2009 and 2008 and for the period from June 20, 2007 (Date of Inception)
through June 30, 2009. The components of deferred tax assets as of
June 30, 2009 and 2008 are as follows:
|
June
30, 2009
|
June
30, 2008
|
|||||||
|
Deferred
tax (liability) asset:
|
||||||||
|
Accrued
payroll
|
$ | 390,400 | $ | 120,700 | ||||
|
Net
operating loss carryforward
|
418,700 | 55,100 | ||||||
| 809,100 | 175,800 | |||||||
|
Valuation
allowance
|
(809,100 | ) | (175,800 | ) | ||||
|
Total
deferred taxes
|
$ | - | $ | - | ||||
50
ITEM
9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL
DISCLOSURE
There
have been no changes in or disagreements with the Company's accountants on
accounting and financial disclosure.
Evaluation of disclosure
controls and procedures.
The
Company’s Chief Executive Officer and Chief Financial Officer has evaluated the
effectiveness of the Company’s disclosure controls and procedures (as defined in
Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of the fiscal period
ending June 30, 2009 covered by this Annual Report on Form 10-K. Based
upon such evaluation, the Chief Executive Officer and Chief Financial Officer
have concluded that, as of the end of such period, the Company’s disclosure
controls and procedures were not effective as required under Rules 13a-15(e) and
15d-15(e) under the Exchange Act. This conclusion by the Company’s Chief
Executive Officer and Chief Financial Officer does not relate to reporting
periods after June 30, 2009.
Management’s Report on
Internal Control Over Financial Reporting
Management
is responsible for establishing and maintaining adequate internal control over
financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange
Act) of the Company. Internal control over financial reporting is a
process designed to provide reasonable assurance regarding the reliability of
financial reporting and the preparation of financial statements for external
purposes in accordance with accounting principles generally accepted in the
United States of America.
The
Company’s internal control over financial reporting includes those policies and
procedures that (i) pertain to the maintenance of records that, in reasonable
detail, accurately and fairly reflect the transactions and dispositions of the
assets of the company; (ii) provide reasonable assurance that transactions are
recorded as necessary to permit preparation of financial statements in
accordance with accounting principles generally accepted in the United States of
America, and that receipts and expenditures of the Company are being made only
in accordance with authorizations of management and directors of the Company;
and (iii) provide reasonable assurance regarding prevention or timely detection
of unauthorized acquisition, use or disposition of the Company’s assets that
could have a material effect on the financial statements.
Because
of its inherent limitations, internal control over financial reporting may not
prevent or detect misstatements. Also, projections of any evaluation of
effectiveness to future periods are subject to the risk that controls may become
inadequate because of changes in conditions, or that the degree of compliance
with the policies and procedures may deteriorate.
Management,
under the supervision of the Company’s Chief Executive Officer and Chief
Financial Officer, conducted an evaluation of the effectiveness of internal
control over financial reporting based on the framework in Internal Control—Integrated
Framework issued by the Committee of Sponsoring Organizations of the
Treadway Commission. Based on this evaluation, management concluded that
the Company’s internal control over financial reporting was not effective as of
June 30, 2009 under the criteria set forth in the Internal Control—Integrated
Framework.
A
material weakness is a deficiency, or combination of deficiencies, in internal
control over financial reporting such that there is a reasonable possibility
that a material misstatement of the Company’s annual or interim financial
statements will not be prevented or detected on a timely basis. Management
has determined that material weaknesses exist due to the lack of
segregation of duties, i.e. all of the accounting tasks are performed by a
single individual. Currently, the Company’s management reviews all
transactions. Until the Company raises additional capital, it cannot
remediate this weakness.
51
This
annual report does not include an attestation report of the Company’s registered
public accounting firm regarding internal control over financial
reporting. Management’s report was not subject to attestation by our
registered public accounting firm pursuant to temporary rules of the SEC that
permit us to provide only management’s report in this Annual Report on Form
10-K.
Changes in Internal Control
Over Financial Reporting
No change
in the Company’s internal control over financial reporting occurred during the
quarter ended June 30, 2009, that materially affected, or is reasonably likely
to materially affect, the Company’s internal control over financial
reporting.
ITEM
9B. OTHER INFORMATION.
None
PART
III
ITEM 10. DIRECTORS, EXECUTIVE
OFFICERS AND CORPORATE GOVERNANCE; COMPLIANCE WITH SECTION 16(a) OF THE EXCHANGE
ACT.
The
following table sets forth information regarding the members of the Company’s
board of directors and its executive officers. All of the Company’s executive
officers and directors were appointed on December 6, 2007, the effective date of
the Acquisition. All directors hold office until the first annual meeting of the
stockholders of the Company and until the election and qualification of their
successors or their earlier removal or retirement.
Officers
are elected annually by the board of directors and serve at the discretion of
the board.
|
Age
|
Title
|
|
|
George
W. Evans
|
54
|
Chief
Executive Officer; Chairman of the Board of
Directors
|
|
Krishna
Menon
|
61
|
President;
Chief Scientific Officer, Director
|
|
Leo
Ehrlich
|
51
|
Chief
Financial Officer; Director
|
|
Hyman
Schwartz
|
57
|
Former
President and Director (resigned on December 6,
2007)
|
|
Jacob
Werczberger
|
35
|
Former
Secretary and Director (resigned on December 6,
2007)
|
(1)
Each of the persons performs services for the Company on a part-time
basis.
George W. Evans JD, MBA served
as CEO of Cellceutix Pharma since inception in June 2007. Following the
acquisition, he was appointed CEO and Chairman of the board of directors of the
Company. Prior to joining Cellceutix Pharma, Mr. Evans worked for Pfizer Inc.
from 1980 to 2006. Mr. Evans has more than 25 years experience in the
Pharmaceutical industry. He worked in a number of positions for Pfizer Inc,
ending his career there as General Counsel for Pfizer's worldwide prescription
drug unit, and a member of the unit's leadership team. Mr. Evans was a member of
the Editorial Advisory Board Of the Food and Drug Law Journal and the Finance
Committee of the Food and Drug Law Institute and a member of the Board of the
New York City Bar Fund. He is a graduate of Williams College and Columbia
University’s Law and Business schools.
Krishna Menon RCM, PhD, VMD
served as President of Cellceutix Pharma since inception in June 2007.
Following the acquisition, he was appointed President and a director. Dr. Menon,
simultaneously therewith, also serves as the Chief Operating Officer
at Kard Scientific, Inc. Dr. Menon is also the inventor of Kevetrin,
our lead compound. Since June 2005, Dr. Menon is also the Chief
Regulatory Officer (a non- executive officer position) at NanoViricides, Inc.
Dr. Menon has more than 35 years in drug development for academia and industry.
Originally trained as a veterinary surgeon, Menon began his career as Chief
Government Veterinarian for a major Parish in Jamaica. He segued to a three-year
stint as Director of Agriculture for the Cayman Islands, in the British
Caribbean and, in 1982, moved to the Dana Farber Cancer Research Institute,
where he worked under the direction of Nobel Laureate Dr. Tom Frye. Two years
later, he earned his PhD in Pharmacology from Harvard University. Menon's PhD
work focused on anti-folate therapy of various cancers. Menon was Research
Scientist at Dana Farber from 1985 to 1990 and Senior Research Scientist, In
Vivo Research (Cancer), at Bayer Pharmaceuticals (Miles Laboratories) from 1991
to 1993. After a year operating his own veterinary oncology and drug development
consultancy practice, Menon was tapped Group Leader, Cancer In Vivo Research and
Clinical Development, for Eli Lilly (1995-2001), where he played a key role in
lead selection and pre-clinical development of Gemzar and Alimta which in 2008
had approximately $2.9 billion dollars in sales, and co-developed another seven
compounds currently in late-stage clinical development. In 1999, Eli Lilly honored
Menon with the “President's Recognition Award”. (See Item 13. Certain
Relationships and Related Transactions; and Director
Independence)
52
Leo Ehrlich, CPA, served
as Chief Financial Officer (CFO) of Cellceutix Pharma since inception in June
2007. Following the acquisition, he was appointed CFO and a director.
From October 8, 1999 to December 31, 2008, Mr. Ehrlich had been a director
at StatSure Diagnostic Systems, Inc. and has held different executive officer
positions at that company including CEO, President, and CFO. Mr. Ehrlich was
also CFO and a director of NanoViricides, Inc. from June 1, 2005 until May
2007. Mr. Ehrlich is a Certified Public Accountant and received his BBA
from Bernard Baruch College of the City University of New
York.
Hyman Schwartz, resigned on
December 6, 2007. He had been employed at Econoshare, Inc. as President,
Chief Operations Officer, Treasurer, and Chairman of the Board of Directors
since August 1, 2005. Mr. Schwartz is also the president of The Hyett
Group Ltd. since 1996. Through divisions, the Hyett Group Ltd.
provides various business services to its clients, including software
design, mergers & acquisitions, business consulting and operates
a business concept lab. Mr. Schwartz received his degree
in accounting from La Salle University in 1975. From August 1999
until January 2004, Hyman Schwartz was a director of New Medium Enterprises,
Inc. a publicly traded company.
Jacob Werczberger,
resigned on December 6, 2007. He had been employed at EconoShare Inc. as
Corporate Secretary since August 15, 2005. Mr. Werczberger is also
employed at Valley Supplies Ltd. in the capacity of Director of Operations
since June 2003. From November 1997 until June 2003, Jacob
Werczberger was employed at S& K trading International in the capacity
of General Manager. In June 2003, Valley Supplies Ltd. merged with S &K
Trading International. Mr. Werczberger attended Yeshiva Torah Veyirah and
graduated high school. Mr. Werczberger has no prior experience in public
company administration.
AUDIT COMMITTEE. The Company
intends to establish an audit committee, which will consist of independent
directors. The audit committee's duties would be to recommend to the Company's
board of directors the engagement of independent auditors to audit the Company's
financial statements and to review its accounting and auditing principles. The
audit committee would review the scope, timing and fees for the annual audit and
the results of audit examinations performed by the internal auditors and
independent public accountants, including their recommendations to improve the
system of accounting and internal controls. The audit committee would at all
times be composed exclusively of directors who are, in the opinion of the
Company's board of directors, free from any relationship which would
interfere with the exercise of independent judgment as a committee member and
who possess an understanding of financial statements and generally accepted
accounting principles.
COMPENSATION COMMITTEE. Our
board of directors does not have a standing compensation committee responsible
for determining executive and director compensation. Instead, the entire
board of directors fulfills this function, and each member of the Board
participates in the determination. Given the small size of the Company and
its Board and the Company's limited resources, locating, obtaining and retaining
additional independent directors is extremely difficult. In the absence of
independent directors, the Board does not believe that creating a separate
compensation committee would result in any improvement in the compensation
determination process. Accordingly, the board of directors has concluded
that the Company and its stockholders would be best served by having the entire
Board of Directors act in place of a compensation committee. When acting
in this capacity, the Board does not have a charter.
53
In
considering and determining executive and director compensation, our Board of
Directors reviews compensation that is paid by other similar public companies to
its officers and takes that into consideration in determining the compensation
to be paid to the Company’s officers. The Board of Directors also
determines and approves any non-cash compensation to any employee. The Company
does not engage any compensation consultants to assist in determining or
recommending the compensation to the Company’s officers or employees.
CODE OF
ETHICS
We have
adopted a code of ethics meeting the requirements of Section 406 of the
Sarbanes-Oxley Act of 2002. We believe our code of ethics is reasonably designed
to deter wrongdoing and promote honest and ethical conduct; provide full, fair,
accurate, timely and understandable disclosure in public reports; comply with
applicable laws; ensure prompt internal reporting of violations; and provide
accountability for adherence to the provisions of the code of ethic. Our code of
ethics is filed as an exhibit to this Form 10-K.
COMPLIANCE WITH SECTION 16(A) OF THE
EXCHANGE ACT
Our
executive officers and directors, and persons who own beneficially more than ten
percent of our equity securities, are required under Section 16(a) of the
Securities Exchange Act of 1934 to file reports of ownership and changes in
their ownership of our securities with the Securities and Exchange Commission.
They must also furnish copies of these reports to us. Based solely on a review
of the copies of reports furnished to us and written representations that no
other reports were required, we believe that for fiscal year 2008 our executive
officers, directors and 10% beneficial owners complied with all applicable
Section 16(a) filing requirements, except that a Form 4 was filed late.
ITEM 11. EXECUTIVE
COMPENSATION
Summary Compensation Table
The
following table sets forth all of the compensation awarded to, earned by or paid
to (i) each individual serving as our principal executive officer during our
last completed fiscal year; and (ii) each other individual that served as an
executive officer at the conclusion of the fiscal year ended June 30, 2008 and
who received in excess of $100,000 in the form of salary and bonus during such
fiscal year (collectively, the Named Executives).
|
Year
|
Salary
|
Bonus
|
Option
Awards
|
Non-Equity
Incentive Plan Compensation
|
All
Other Compensation
|
Total
|
||||||||||||||||
|
George
W. Evans (1)(2)(3)(4)
Chief Executive Officer, Chairman of the Board
|
2009
2008
|
$
$
|
258,333
116,667
|
$
$
|
0
0
|
$
$
|
123,282
43,533
|
0
0
|
$
$
|
0
0
|
$
$
|
381,615
160,200
|
||||||||||
|
Dr.
Krishna Menon (3)(5)(6)
President, Chief Scientific Officer, Director
|
2009
2008
|
$
$
|
258,333
116,667
|
$
$
|
0
0
|
$
$
|
0
0
|
0
0
|
$
$
|
0
0
|
$
$
|
258,333
116,667
|
||||||||||
|
Leo
Ehrlich, (7)(8)Chief
Financial Officer,, Director
|
2009
2008
|
$
$
|
200,000
87,500
|
$
$
|
|
0
0
|
$
$
|
0
0
|
0
0
|
$
$
|
0
0
|
$
$
|
200,000
87,500
|
|||||||||
|
(1)
|
Mr.
Evans has served as Chief Executive Officer and Chairman of the Board of
the Company from June 2007 until
present.
|
|
(2)
|
Represents
the amount of Mr. Evans’ base salary paid by the Company. Mr.
Evans’ total base salary for 2008 was $200,000 for the period of December
7, 2007 through December 7, 2008, and $300,000 for the period of December
7, 2008 through December 7, 2009. $116,667 was deemed earned by Mr. Evans
for the period December 7, 2007 through June 30, 2008 and was originally
deferred until June 30, 2009, and remains unpaid as of September 30,
2009. Mr Evans’ salary deemed earned for the period of
July 1, 2008 through June 30, 2009 was $258,333 and was deferred and
remains unpaid as of September 30,
2009.
|
|
(3)
|
Officer
is eligible for bonuses upon the successful achievement of agreed upon
corporate and individual performance based milestones after the Company
receives a financing commitment in amount of at least $4,000,000,
upon achieving the following milestones: FDA IND $250,000 if
received within 10 months $150,000 if received within 12 months
$100,000 if received within 16 months Completion of Phase 1with clinical
results that would have Kevetrin proceed to Phase 2/3: $450,000 if
received within 18 months $350,000 if received within 24 months $150,000
if received within 28 months; start Phase 2/3: $500,000 if within 36
months $350,000 if within 42 months $150,000 if within 48
months.
|
|
(4)
|
Mr.
Evans’ employment agreement provides a grant of options to purchase a
number of shares of the Company's stock equal to one percent of the issued
and outstanding common stock following the first anniversary of the
commencement date of the agreement, at a purchase price of $0.15 per
share. Mr. Evans shall thereafter be issued an additional grant of options
to purchase a number of shares of common stock equal to one percent of the
issued and outstanding common stock at a purchase price equal to the
average closing bid price of the common stock on its primary exchange for
the fifteen successive trading days immediately prior to the date of the
grant.
|
|
(5)
|
Dr.
Menon has served as President, Chief Scientific Officer and a Director of
the Company from June 2007 until
present.
|
|
(6)
|
Represents
the amount of Dr. Menon’s base salary paid by the Company. Dr.
Menon’s total base salary for 2008 was $200,000 for the period of December
7, 2007 through December 7, 2008, and $300,000 for the period of December
7, 2008 through December 7, 2009. $116,667 was deemed earned by
Dr. Menon for the period December 7, 2007 through June 30, 2008 and was
originally deferred until June 30, 2009, and remains unpaid as of
September 30, 2009. Dr Menon’s salary deemed earned for the
period of July 1, 2008 through June 30, 2009 was $258,333 and was deferred
and remains unpaid as of September 30,
2009.
|
|
(7)
|
Mr.
Ehrlich has served as Chief Financial Officer and a Director of the
Company from June 2007 until
present.
|
|
(8)
|
Represents
the amount of Mr. Ehrlich’s base salary paid by the
Company. Mr. Ehrlich’s total base salary for 2008 was $150,000
for the period of December 7, 2007 through December 7, 2008, and $250,000
for the period of December 7, 2008 through December 7,
2009. $87,500 was deemed earned by Mr. Ehrlich for the period
December 7, 2007 through June 30, 2008 and was originally deferred until
June 30, 2009, and remains unpaid as of September 30, 2009. Mr
Ehrlich’s salary deemed earned for the period of July 1, 2008 through June
30, 2009 was $200,000 and was deferred and remains unpaid as of September
30, 2009.
|
54
COMPENSATION
POLICY
Our
Company’s executive compensation plan is based on attracting and retaining
qualified professionals who possess the skills and leadership necessary to
enable our Company to achieve earnings and profitability growth to satisfy our
stockholders. We must, therefore, create incentives for these executives to
achieve both Company and individual performance objectives through the use of
performance-based compensation programs. No single component is considered by
itself, rather all components of the compensation package are considered in
aggregate. Wherever possible, objective measurements will be utilized to
quantify performance, however, many subjective factors still come into play when
determining performance.
Compensation
Components. As an early-stage development company, the main elements of our
compensation package consist of base salary, stock options and
bonus.
Base
Salary. As we continue to grow and financial conditions improve, these base
salaries, bonuses and incentive compensation will be reviewed for possible
adjustments. Base salary adjustments will be based on both individual and
Company performance and will include both objective and subjective criteria
specific to each executive’s role and responsibility with the
Company.
COMPENSATION OF DIRECTORS
At this
time, Directors receive no remuneration for their services as directors of the
Company, nor does the Company reimburse its directors for expenses incurred in
their service to the Board of Directors. The Company does not expect to pay any
fees to its directors for the 2009 fiscal year.
EMPLOYMENT AGREEMENTS, TERMINATION OF
EMPLOYMENT AND CHANGE-IN-CONTROL ARRANGEMENTS
On
December 7, 2007, the Company entered into employment agreements with two (2)
executive officers, George Evans, Chief Executive Officer, and Dr. Krishna
Menon, President. Both agreements provide for a three year term with minimum
annual base salaries of $200,000 in the first year, $300,000 in the second year
and $400,000 in the third year. In addition, the agreements provide for
bonuses according to the following schedule:
Upon
receiving IND: $250,000 if received within 10 months; $150,000 if received
within 12 months; $100,000 if received within 16 months; and no bonus if
received thereafter or not received.
Completion
of Phase 1with clinical results that would have Kevetrin proceed to Phase 2/3:
$450,000 if received within 18 months; $350,000 if received within 24 months;
$150,000 if received within 28 months; and no bonus if received thereafter or
not received.
Start
Phase 2/3: $500,000 if within 36 months; $350,000 if within 42 months; $150,000
if within 48 months; and no bonus if received thereafter or not received.
The bonus
obligations of the Company under the agreements do not commence until the
Company receives a financing commitment in an amount of at least $4,000,000.
The
agreement with Mr. Evans also provides a grant of options to purchase a number
of shares of the Company's stock equal to one percent of the issued and
outstanding common stock following the first anniversary of the commencement of
the agreement, at a purchase price of $0.15 per share.. Thereafter Mr. Evans
shall be issued an additional grant of options following the successive
anniversaries of the commencement date of a number of shares of common stock
equal to one percent of the issued and outstanding common stock at a purchase
price equal to the average closing bid price of the common stock on its primary
exchange for the fifteen successive trading days immediately prior to the date
of the grant.
55
COMPENSATION OF SCIENTIFIC ADVISORY
BOARD
The
Company has established a Scientific Advisory Board consisting
of:
Emil Frei III, MD Dr. Frei is
one of the world’s leading oncologists, a pioneer of chemotherapy and a leader
in medical research, clinical practice and education. His distinguished career
includes 40 years in top leadership positions such as Chief of Medicine at
National Cancer Institute, Associate Scientific Director at M. D. Anderson, and
Director and Physician-in-Chief at the Dana-Farber Cancer Institute. He
continues as Physician-in-Chief, Emeritus, at
Dana-Farber.
Samuel Danishefsy, PhD.
Dr. Danishefsky is the incumbent of a Eugene W. Kettering Chair and a
member of the Molecular Pharmacology and Chemistry Program in the
Sloan-Kettering Institute (SKI). He is also a professor of chemistry at Columbia
University.
Dr.
Danishefsky earned his BS degree from Yeshiva University, his PhD degree from
Harvard University, and conducted postdoctoral research at Columbia with Gilbert
Stork. In 1996, Drs. Danishefsky and Stork shared the Wolf Prize. Dr.
Danishefsky has previously won the American Chemical Society Award for Creative
Work in Chemical Synthesis, the Cope Medal, and the Tetrahedron Prize, among
others, as well as membership in the National Academy of Sciences. He joined
Memorial Sloan-Kettering Cancer Center in 1991 after serving on the faculties of
both the University of Pittsburgh and Yale University.
Members
of the Scientific Advisory Board have each received 25,000 shares of the
Company's common stock as remuneration for their services. The Company has
also agreed to reimburse members of the Scientific Advisory Board for expenses
incurred in their service to the Company.
ITEM 12. SECURITY OWNERSHIP OF
CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER
MATTERS
The
following table summarizes certain information regarding the beneficial
ownership (as such term is defined in Rule 13d-3 under the Securities Exchange
Act of 1934) of outstanding EconoShare Common Stock as of December 11, 2007
(after giving effect to the Exchange) by (i) each person known by us to be the
beneficial owner of more than 5% of the outstanding Cellceutix Common Stock,
(ii) each of our directors, (iii) each of our named executive officers (as
defined in Item 402(a)(3) of Regulation S-B under the Securities Act), and (iv)
all executive officers and directors as a group. Except as indicated in the
footnotes below, the security and stockholders listed below possess sole voting
and investment power with respect to their
shares.
|
Amount
and Nature of Beneficial Owner (1)
|
Percent
of Class (2)
|
|
|
Dr.
Krishna Menon C/O Cellceutix
100
Cumming Ctr., Suite 151-B
Beverley,
MA 01915
|
32,048,286
|
34.9%
|
|
George
W. Evans (3)
C/O Cellceutix
100
Cumming Ctr., Suite 151-B
Beverley,
MA 01915
|
4,602,312
|
5.0%
|
|
Leo
Ehrlich (4)
C/O Cellceutix
100
Cumming Ctr., Suite 151-B
Beverley,
MA 01915
|
11,247,284
|
12.2%
|
|
All
Directors and Executive
Officers
as a Group (3 persons) (5)
|
47,897,882
|
52.1%
|
56
|
1.
|
"Beneficial
Owner" means having or sharing, directly or indirectly (i) voting power,
which includes the power to vote or to direct the voting, or (ii)
investment power, which includes the power to dispose or to direct the
disposition, of shares of the common stock of an issuer. The definition of
beneficial ownership includes shares, underlying options or warrants to
purchase common stock, or other securities convertible into common stock,
that currently are exercisable or convertible or that will become
exercisable or convertible within 60 days. Unless otherwise indicated, the
beneficial owner has sole voting and investment
power.
|
|
2.
|
For
each shareholder, the calculation of percentage of beneficial ownership is
based upon 91,836,000 shares of Common Stock outstanding as of June 30,
2009, and shares of Common Stock subject to options, warrants and/or
conversion rights held by the shareholder that are currently exercisable
or exercisable within 60 days, which are deemed to be outstanding and to
be beneficially owned by the shareholder holding such options, warrants,
or conversion rights. The percentage ownership of any shareholder is
determined by assuming that the shareholder has exercised all options,
warrants and conversion rights to obtain additional securities and that no
other shareholder has exercised such
rights.
|
|
3.
|
George
W. Evans, Chief Executive Officer and Chairman. Includes
2,766,496 shares of EconoShare Inc.’s common stock held by Mr. Evans and
includes 1,835,816 shares of EconoShare, Inc.’s common stock held by the
children of George W.
Evans.
|
|
4.
|
Leo
Ehrlich, Chief Financial Officer and Director. Includes
7,745,002 shares of EconoShare, Inc.’s common stock held by Mr. Ehrlich
and includes 3,502,282 shares of EconoShare, Inc.’s common stock held by
the wife and children of Leo
Ehrlich.
|
|
5.
|
Includes
6,088,070 shares of Common Stock indirectly owned by certain of the
Executive Officers and Directors as a group but excludes vested options to
acquire approximately 1,836,820 additional shares of Common Stock by
Executive Officers and Directors, as a
group.
|
ITEM 13. CERTAIN RELATIONSHIPS
AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Dr.
Krishna Menon
On August
2, 2007 and October 17, 2007, Cellceutix Pharma entered into Compound Assignment
Agreements, with Dr. Krishna Menon, our largest shareholder, covering Kevetrin,
KM 277, KM 278 and KM 362. On June 23, 2009, the Company entered into a
Compound Assignment Agreement with Dr. Menon covering KM-732. In each
Agreement, the Company agreed to pay Dr. Menon 5% of net sales of the compounds
in countries where a composition of matter patent is issued and 3% of net sales
in other countries.
These
transactions have identical terms to transactions we entered into with other
inventors who are unrelated third parties.
KARD
Scientific, Inc.
Dr.
Menon, the Company’s principal shareholder, President and Director also serves
as the COO and Director of Kard Scientific. On December 7, 2007, Cellceutix
Pharma began renting 200 square feet of office space from Kard Scientific, on a
month to month basis for $900 per month. In September of 2007, the Company
engaged Kard Scientific to conduct specified pre-clinical studies necessary for
the Company to prepare an IND submission to the FDA. We do not have an
exclusive arrangement with KARD. All work performed by Kard must have
prior approval of the executive officers of the Company; and we retain all
intellectual property resulting from the services by KARD. To date we
have incurred charges from KARD of $390,587.
Mr.
Leo Ehrlich
Subsequent
to June 30, 2008, Mr. Ehrlich, an officer of the Company, converted previous
amounts provided in cash to the Company to a loan (the “Ehrlich Promissory Note A”). The
Ehrlich Promissory Note A is an unsecured, 6% per annum simple interest bearing,
demand note. The principal balance of the Ehrlich Promissory Note A is $32,310
as of June 30, 2009 and 2008 and remains unpaid as of September 30,
2009.
57
Subsequent
to June 30, 2009, Mr. Ehrlich, an officer of the Company, provided cash in the
form of a loan to the Company (the “Ehrlich Promissory Note B”). The
Ehrlich Promissory Note B is an unsecured, 6% per annum simple interest bearing,
demand note. The principal balance of the Ehrlich Promissory Note B is for a
maximum of $100,000. Mr. Ehrlich has provided cash of $85,000 and
this amount remains unpaid as of September 30, 2009.
Currently,
we have no independent directors on our Board of Directors, and therefore have
no formal procedures in effect for reviewing and pre-approving any transactions
between us, our directors, officers and other affiliates. We will use our best
efforts to insure that all transactions are on terms at least as favorable to
the Company as we would negotiate with unrelated third
parties.
Director
Independence
Our
common stock trades on the Over the Counter Bulletin Board (OTCBB). As such, we
are not currently subject to corporate governance standards of listed companies,
which require, among other things, that the majority of the board of directors
be independent.
Since we
are not currently subject to corporate governance standards relating to the
independence of our directors, we choose to define an “independent” director in
accordance with the NASDAQ Global Market's requirements for independent
directors (NASDAQ Marketplace Rule 4200). We do not currently have an
independent director under the above definition. We do not list that definition
on our Internet website.
ITEM
14. PRINCIPAL ACCOUNTANTS FEES AND SERVICES
Audit
Fees. During the years ended June 30, 2009 and 2008, the aggregate fees billed
by the Company's auditors, Holtz Rubinstein Reminick LLP, for services rendered
for the audit of our annual financial statements and the review of the financial
statements included in our quarterly reports on Form 10-Q and for services
provided in connection with the statutory and regulatory filings or engagements
for those fiscal years was $35,000 and $23,500, respectively.
Audit-Related
Fees. During years ended June 30, 2009 and 2008, our auditors, Holtz Rubinstein
Reminick LLP, did not receive any fees for any audit-related services other than
as set forth in paragraph (a) above.
Tax Fees.
Our auditors, Holtz Rubinstein Reminick LLP, did not provide tax compliance or
tax planning advice during the years ended June 30, 2009 and 2008.
All Other
Fees. There were no fees billed for services rendered by Holtz Rubenstein
Reminick LLP for 2009 and 2008, other than the services described
above.
PART IV
(A)
EXHIBITS INCLUDED HEREIN:
EXHIBIT NO.
DESCRIPTION
2.1
Agreement and Plan of Share Exchange, by and among EconoShare, Inc., Cellceutix
Pharma, Inc., and the Shareholders of Cellceutix Pharma, Inc. dated as of
December 6, 2007 (1)
3.1 Articles
of Incorporation of Cellceutix Pharma, Inc. (1)
3.2 By-laws
of Cellceutix Pharma, Inc. (1)
10.1 Employment
Agreement between EconoShare, Inc. and George W. Evans dated December 7, 2007
(1)
10.2 Employment
Agreement between EconoShare, Inc. and Krishna Menon dated December 7, 2007
(1)
10.3 Assignment
Agreement between Cellceutix Pharma, Inc. and. Krishna Menon dated August 2,
2007(1)
10.4 Assignment
Agreement between Cellceutix Pharma, Inc. and Krishna Menon dated August 2,
2007(1)
10.5 Assignment
Agreement between Cellceutix Pharma, Inc. and Krishna Menon dated August 2,
2007(1)
10.6 Assignment
Agreement between Cellceutix Pharma, Inc. and Geetha Kamburath dated August 21,
2007(1)
10.7 Assignment
Agreement between Cellceutix Pharma, Inc. and Krishna Menon dated October 17,
2007(1)
10.8
Assignment Agreement between Cellceutix Pharma, Inc. and Adam Harris dated
October 17, 2007(1)
|
|
10.9 Confidential
Disclosure Agreement between Kard Scientific, Inc. and Cellceutix Pharma,
Inc. dated September 28, 2007.
(1)
|
10.10 Laboratory
Services Agreement Kard Scientific, Inc. and Cellceutix Pharma, Inc. dated
September 28,2007.
(1)
10.11 Cellceutix
Lease with Kard Scientific, Inc. dated December 7, 2007(1)
10.12
Assignment Agreement between Cellceutix Pharma,
Inc. and Krishna Menon dated June 23, 2009(1)
|
|
10.1.1
Security Agreement, dated as of May 7, 2008,
between Cellceutix Corp. and Putnam Partners, White Star LLC,
and Dahlia Nordlicht, filed with the Company’s Form 10-KSB with the
Securities Commission on September 24,
2008
|
|
|
10.2.2
Convertible Promissory Note dated as of May 7,
2008, between Cellceutix Corp. and Putnam Partners, White Star
LLC, and Dahlia Nordlicht, filed with the Company’s Form 10-KSB with the
Securities Commission on September 24,
2008
|
|
|
10.3.4
Guaranty in favor of Putnam Partners, White Star LLC,
and Dahlia Nordlicht dated as of May 7, 2008, filed with the Company’s
Form 10-KSB with the Securities Commission on September 24,
2008
|
|
|
10.13 Agreement
with Paul Ginsburg, Attorney filed on Form 8-K with the Securities
Commission on April 6, 2009
|
|
|
10.14
Consulting Agreement with Sylvia Holden,
Consultant filed on Form 8-K with the Securities Commission on April
6, 2009
|
|
|
10.15
Cellceutix Corporation 2009 Stock Option
Plan filed on Form 8-K with the Securities Commission on April 6,
2009
|
|
|
10.16 Consulting
Agreement with James DeAngelis filed on Form 8-K with the
Securities Commission on May 21,
2009
|
10.17
Agreement between Cellceutix Pharma, Inc. and
Girindus America dated June 22, 2009*
14.15
Code of Ethics (2)
58
31. 1 Chairman
of the Board and Chief Executive Officer Certifications required under Section
302 of the
Sarbanes Oxley Act of
2002*
31.2 Chief
Financial Officer Certifications required under Section 302 of the Sarbanes
Oxley Act of 2002*
32. Certifications
required under Section 906 of the Sarbanes Oxley Act of 2002*
*
An asterisk (*) indicates the exhibit is identified in this report.
(1)
Incorporated by reference to the Company’s registration statement on Form 8-K,
filed with the Securities Commission on December 11, 2007
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of
1934, the Registrant has duly caused this report to be signed on its behalf by
the undersigned, thereunto duly authorized.
Dated:
October 8, 2009
CELLCEUTIX
CORPORATION
/s/
George W. Evans
--------------------------------------
George W.
Evans
Chairman
of the Board and
Chief
Executive Officer
/s/ Leo
Ehrlich
--------------------------------------
Leo
Ehrlich
Chief Financial Officer and
Secretary
Pursuant
to the requirements of the Securities Exchange Act of 1934, this Annual Report
on Form 10-K has been signed below by the following persons on behalf of the
Registrant and in the capacities indicated on October 8,
2009.
/s/
Krishna Menon
-----------------------------
President
and Director
Krishna
Menon
59