Attached files
| file | filename |
|---|---|
| EX-23.2 - RenovoRx, Inc. | ex23-2.htm |
| EX-5.1 - RenovoRx, Inc. | ex5-1.htm |
| EX-4.2 - RenovoRx, Inc. | ex4-2.htm |
| EX-1.1 - RenovoRx, Inc. | ex1-1.htm |
As filed with the Securities and Exchange Commission on August 11, 2021.
No. 333-258071
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Amendment No. 2
To
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
RenovoRx, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 2834 | 27-1448452 | ||
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
4546 El Camino Real, Suite B1
Los Altos, CA 94022
(650)-284-4433
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Shaun Bagai
Chief Executive Officer
RenovoRx, Inc.
4546 El Camino Real, Suite B1
Los Altos, CA 94022
(650)-284-4433
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies of all communications to:
Jeffrey J. Fessler Sheppard Mullin Richter & Hampton LLP 30 Rockefeller Plaza New York, New York 10112 (212) 653-8700 |
Robert Charron Charles Phillips Ellenoff Grossman & Schole LLP 1345 Avenue of the Americas New York, New York 10105 (212) 370-1300 |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. [X]
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act of 1933, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. [ ]
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
| Large accelerated filer | [ ] | Accelerated filer | [ ] | |||
| Non-accelerated filer | [X] | Smaller reporting company | [X] | |||
| Emerging growth company | [X] | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. [ ]
CALCULATION OF REGISTRATION FEE
| Proposed
Maximum Aggregate Offering Price(1)(2) | Amount
of Registration Fee | |||||||
| Units, each Unit consisting of one share of common stock, $0.0001 par value per share, and three-quarter Warrant to purchase one share of common stock (3) | $ | 27,657,500 | $ | 3,017.43 | ||||
| (i) Common stock included in the Units (4) | ||||||||
| (ii) Warrants included in the Units (4) | ||||||||
| Pre-funded Units, each Pre-funded Unit consisting of one Pre-funded Warrant to purchase one share of common stock and three-quarter Warrant to purchase one share of common stock (3) | ||||||||
| (i) Pre-funded Warrants included in the Pre-funded Units (4) | ||||||||
| (ii) Warrants included in the Pre-funded Units (3)(4) | ||||||||
| Shares of common stock underlying Pre-funded Warrants included in the Pre-funded Units (4) | ||||||||
| Shares of common stock underlying Warrants included in the Units and the Pre-funded Units (5) | $ | 24,891,750 | $ | 2,715.69 | ||||
| Underwriter’s warrant (6)(7) | ||||||||
| Common stock underlying underwriter’s warrant (6) | $ | 2,904,038 | $ | 316.83 | ||||
| Total | $ | 55,453,288 | $ | 6,049.95 | (8) | |||
(1) Pursuant to Rule 416, the securities being registered hereunder include such indeterminate number of additional securities as may be issuable to prevent dilution resulting from stock splits, stock dividends or similar transactions.
(2) Estimated solely for the purpose of calculating the registration fee in accordance with Rule 457(o) under the Securities Act of 1933, as amended (the “Securities Act”). Includes shares of our common stock and Warrants that the underwriters have the option to purchase to cover over-allotments, if any, of the Units offered hereby.
(3) The proposed maximum aggregate offering price of the Units proposed to be sold in the offering will be reduced on a dollar-for-dollar basis based on the offering price of any Pre-funded Units offered and sold in the offering, and the proposed maximum aggregate offering price of the Pre-funded Units to be sold in the offering will be reduced on a dollar-for-dollar basis based on the offering price of any Units sold in the offering. Accordingly, the proposed maximum aggregate offering price of the Units and Pre-funded Units, if any, is $27,657,500 (including the underwriter’s over-allotment option).
(4) Filing fee included with the Units or Pre-funded Units, as applicable.
(5) Pursuant to Staff Compliance and Disclosure Interpretation 240.06, equals the aggregate exercise price of the Warrants.
(6) The registrant has agreed to issue, upon the closing of this offering, an underwriter’s warrant to Roth Capital Partners, LLC entitling it to purchase a number of shares of common stock equal to 5% of the aggregate shares of common stock (including shares of common stock underlying the Warrants and any Pre-funded Warrants) sold in this offering. The exercise price of the underwriter’s warrant will be equal to 120% of the public offering price of the Units offered hereby.
(7) No additional registration fee is payable pursuant to Rule 457(g) under the Securities Act of 1933, as amended.
(8) $4,255.77 was previously paid.
The registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This preliminary prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED AUGUST 11, 2021
PRELIMINARY PROSPECTUS
![]()
1,850,000 Units, Each Consisting of One Share of Common Stock and Three-Quarter Warrant to Purchase One Share of Common Stock
1,850,000 Pre-funded Units, Each Consisting of a Pre-funded Warrant to Purchase One Share of Common Stock and Three-Quarter Warrant to Purchase One Share of Common Stock
This is our initial public offering. We are offering up to 1,850,000 of units of securities (the “Units”) pursuant to this prospectus. Prior to this offering, there has been no public market for our securities. We anticipate that the initial public offering price of our Units will be between $11.00 and $13.00.
Each Unit consists of (a) one share of our common stock and (b) three-quarter warrant (the “Warrants”) to purchase one share of our common stock at an exercise price equal to $14.40 (120% of initial public offering price per Unit) per share, assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) exercisable until the fifth anniversary of the issuance date, and subject to certain adjustment and cashless exercise provisions as described herein.
We are also offering to those purchasers, if any, whose purchase of Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if the purchaser so chooses, pre-funded units (each a “Pre-funded Unit”) in lieu of Units that would otherwise result in the purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock. Each Pre-funded Unit will consist of a pre-funded warrant to purchase one share of our common stock at an exercise price of $0.0001 per share (each a “Pre-funded Warrant”) and a Warrant. The purchase price of each Pre-funded Unit is equal to the price per Unit being sold to the public in this offering, minus $0.0001. The Pre-funded Warrants will be immediately exercisable and may be exercised at any time until all of the Pre-funded Warrants are exercised in full.
We are offering up to 1,850,000 Units and up to 1,850,000 Pre-funded Units, in the aggregate not exceeding more than a total of 1,850,000 units. For each Pre-funded Unit we sell, the number of Units we are offering will be decreased on a one-for-one basis. Units and Pre-funded Units will not be issued or certificated. The shares of common stock or Pre-funded Warrants, as the case may be, and the Warrants included in the Units or the Pre-funded Units, can only be purchased together in this offering, but the securities contained in the Units or Pre-funded Units will be issued separately and will be immediately separable upon issuance.
We have been approved to have our common stock listed on the Nasdaq Capital Market under the symbol “RNXT” upon our satisfaction of the exchange’s initial listing criteria, including the completion of this offering. If we do not meet all of Nasdaq’s initial listing criteria, we will not complete this offering. We do not intend to apply for any listing of the Warrants or Pre-funded Warrants on the Nasdaq Capital Market or any other securities exchange or nationally recognized trading system, and we do not expect a market to develop for the Warrants or Pre-funded Warrants.
We are an “emerging growth company” under the federal securities laws and have elected to comply with certain reduced public company reporting requirements.
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 9.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
Per Unit | Per Pre-funded Unit | Total | ||||||||||
| Initial public offering price(1) | $ | $ | $ | |||||||||
| Underwriting discounts and commissions(2) | $ | $ | $ | |||||||||
| Proceeds to us, before expenses | $ | $ | $ | |||||||||
(1) The assumed public offering price corresponds to (x) in respect of a Unit, (i) a public offering price per share of common stock of $ and (ii) a public offering price per Warrant of $0.01 and (y) in respect of a Pre-funded Unit (i) a public offering price per Pre-funded Warrant of $ and (ii) a public offering price per Warrant of $0.01.
(2) We refer you to “Underwriting” beginning on page 113 for additional information regarding underwriters’ compensation.
We have granted Roth Capital Partners, LLC, as representative of the underwriters, an option, to purchase an additional 277,500 shares of our common stock and/or Warrants to purchase 208,125 additional shares of common stock from us (being up to 15% of the shares of common stock, including shares of common stock underlying the Pre-funded Warrants, and/or up to 15% of the Warrants sold in this offering), in any combination thereof, for forty-five (45) days from the date of this prospectus, at prices described herein to cover over-allotments, if any, of the Units and the Pre-funded Units offered hereby. See “Underwriting.”
The underwriters expect to deliver the securities to purchasers on or about , 2021.
Sole Book-Running Manager
Roth Capital Partners
Lead Manager
Maxim Group LLC
The date of this prospectus is , 2021
TABLE OF CONTENTS
Neither we nor the underwriters have authorized anyone to provide you with information other than that contained in this prospectus. We and the underwriters take no responsibility for, and can provide no assurance as to the reliability of, any other information that others may give you. We and the underwriters are offering to sell, and seeking offers to buy, the securities offered hereby only in jurisdictions where offers and sales are permitted. The information contained in this prospectus is accurate only as of the date on the front cover page of this prospectus, or other earlier date stated in this prospectus, regardless of the time of delivery of this prospectus or of any sale of our securities.
No action is being taken in any jurisdiction outside the United States to permit a public offering of our securities or possession or distribution of this prospectus in that jurisdiction. Persons who come into possession of this prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this prospectus applicable to that jurisdiction.
This prospectus includes industry data and forecasts that we have obtained from industry publications and surveys, public filings and internal company sources. Industry publications and surveys and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable, but there can be no assurance as to the accuracy or completeness of the included information. Statements as to our market position and market estimates are based on independent industry publications, government publications, third party forecasts, management’s estimates and assumptions about our markets and our internal research. While we are not aware of any misstatements regarding the market, industry or similar data presented herein, such data involve risks and uncertainties and are subject to change based on various factors, including those discussed under the headings “Risk Factors” and “Cautionary Statement Concerning Forward-Looking Statements” in this prospectus.
| -2- |
The following summary highlights information contained elsewhere in this prospectus. It does not contain all of the information you need to consider in making your investment decision. Before making an investment decision, you should read this entire prospectus carefully and you should consider, among other things, the matters set forth under “Risk Factors” and our financial statements and related notes thereto appearing elsewhere in this prospectus. In this prospectus, except as otherwise indicated, “RenovoRx,” the “Company,” “we,” “our,” and “us” refer to RenovoRx, Inc., a Delaware corporation, and its subsidiaries.
Overview
We are a clinical-stage biopharmaceutical company focused on developing therapies for the local treatment of solid tumors and conducting a Phase 3 registrational trial for our lead product candidate RenovoGem™. Our therapy platform, RenovoRx Trans-Arterial Micro-Perfusion, or RenovoTAMP™ utilizes approved chemotherapeutics with validated mechanisms of action and well-established safety and side effect profiles, with the goal of increasing their efficacy, improving their safety, and widening their therapeutic window. RenovoTAMP combines our patented U.S. Food and Drug Administration, or FDA, cleared delivery system, RenovoCath®, with small molecule chemotherapeutic agents that can be forced across the vessel wall using pressure, targeting these anti-cancer drugs locally to the solid tumors. While we anticipate investigating other chemotherapeutic agents for intra-arterial delivery via RenovoTAMP, our clinical work to date has focused on gemcitabine, which is a generic drug. Our first product candidate, RenovoGem, is a drug and device combination consisting of intra-arterial gemcitabine and RenovoCath. FDA has determined that RenovoGem will be regulated as, and if approved we expect will be reimbursed as, a new oncology drug product. We have secured FDA Orphan Drug Designation for RenovoGem in our first two indications: pancreatic cancer and cholangiocarcinoma (bile duct cancer, or CCA). We have completed our RR1 Phase 1/2 and RR2 observational registry studies, with 20 and 25 patients respectively, in locally advanced pancreatic cancer, or LAPC. These studies demonstrated a median overall survival of 27.9 months in patients treated with RenovoGem and radiation. Based on previous large randomized clinical trials, the expected survival of LAPC patients is 12-15 months in patients receiving only intravenous (IV) systemic chemotherapy or IV chemotherapy plus radiation (which are both considered standard of care). Unlike the randomized trials that established these standard-of-care results, our RR1 and RR2 clinical trials did not prospectively control the standard of care therapy received prior to RenovoTAMP. Based on FDA safety review of our Phase 1/2 study the FDA allowed us to proceed to evaluate RenovoGem within our Phase 3 registration Investigational New Drug, or IND, clinical trial. Our Phase 3 trial is over 40% enrolled as of July 15, 2021 and we expect to report data from a planned interim data readout in the second half of 2022. We intend to evaluate RenovoGem in a second indication in a Phase 2/3 trial in hilar CCA (cancer that occurs in the bile ducts that lead out of the liver and join with the gallbladder, also called extrahepatic cholangiocarcinoma, or HCCA). We plan to propose the trial to the FDA and potentially launch in the first half of 2022. In addition, we may evaluate RenovoGem in other indications, potentially including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma (an aggressive type of cancer that can occur in the brain or spinal cord). To date, we have used gemcitabine, but in the future we may develop other chemotherapeutic agents for intra-arterial delivery via RenovoCath.
Our RenovoTAMP therapy platform is focused on optimizing drug concentration in solid tumors using approved small molecule chemotherapeutics that enable physicians to isolate segments of the vascular anatomy closest to tumors and force chemotherapy across the blood vessel wall to bathe these difficult-to-reach tumors in chemotherapy. More specifically, our patented approach combines local delivery via our patented RenovoCath delivery system utilizing pressure to force small molecule chemotherapy into the tumor tissue with pre-treatment of the local blood vessels and tissue with standard-of-care radiation therapy to decrease chemotherapy washout. We believe there are many advantages to our approach:
| ● | Application of Approved Small Molecule Chemotherapeutic Agents: We use approved small molecule chemotherapeutic agents such as gemcitabine. | |
| ● | Targeted Approach: With our approach, we have demonstrated in our clinical studies up to 100 times higher local drug concentration compared to systemic chemotherapy. We believe our approach decreases systemic exposure and improves patient outcomes. | |
| ● | Delivery Method Independent of Tumor Vascularity: We invented a novel combination platform and delivery system to deliver small molecule chemotherapeutic agents in solid tumors resistant to systemic chemotherapy due to lack of tumor blood vessels or tumor feeders. | |
| ● | Broad Application for Solid Tumor Indications: Our platform is not restricted to a single small molecule chemotherapeutic agent or solid tumor type. As such, our platform and delivery system may be applied for use in additional solid tumor indications, including in solid tumors without identifiable tumor feeders. |
| -3- |
Our lead product candidate, RenovoGem, is a combination of gemcitabine and our patented delivery system, RenovoCath, and is regulated by the FDA as a novel oncology drug product. Our RenovoTAMP platform therapy utilizes pressure mediated delivery of gemcitabine across the arterial wall to bathe the pancreatic tumor tissue in 120mL of saline with 1,000mg/m2 of the drug over a 20-minute delivery time (approximately a total of 1,500-2,000mg of drug dependent upon patient Body Surface Area). RenovoCath is an adjustable double balloon catheter designed to isolate the proximal and distal vessel and adjust the distance between the balloons to exclude any branching blood vessel offshoots.
While the field of oncology has seen progress in treating a handful of deadly cancers over the last few decades, there is a common limitation in chemotherapy: enhanced dosing of the drug to impact the tumor while minimizing systemic toxicity. The characteristics of the vasculature, within and surrounding the tumor, can be a limiting step in this goal. For example, LAPC and HCCA are more difficult to treat due to the lack of blood vessels that feed these tumors, making it difficult to expose tumors to chemotherapy, which is typically delivered intravenously. Trans-arterial chemoembolization (TACE) is an established first line therapy for certain solid tumors. A key component of this approach is to identify and isolate vessels feeding the tumor, known as tumor feeders. However, in patients with pancreatic cancer, no tumor feeder vessels are visible during angiography. In the absence of visible tumor feeders, we can introduce drugs directly across the arterial wall into the surrounding tissue via pressurized diffusion.
We are currently evaluating RenovoGem in patients with LAPC in a Phase 3 trial entitled “Targeted Intra-arterial Gemcitabine vs. Continuation of IV (intravenous) Gemcitabine Plus Nab-Paclitaxel Following Induction With Sequential IV Gemcitabine Plus Nab-Paclitaxel and Radiotherapy for Locally Advanced Pancreatic Cancer”, or TIGeR-PaC, trial at 28 US and Belgian sites. The trial is designed to enroll 340 subjects. 145 patients were enrolled as of July 15, 2021. A planned interim data readout is expected during the second half of 2022. We have secured Orphan Drug Designation for RenovoGem, which would provide us with seven years of exclusivity to market intra-arterial use of gemcitabine for LAPC upon New Drug Application, or NDA, approval.
In addition, we intend to evaluate RenovoGem in patients with HCCA, and we have secured FDA Orphan Drug Designation for this indication. We intend to potentially pursue additional indications including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma.
For our initial indication, LAPC, we have completed two studies. We launched RR1, our first-in-human, dose escalation, Phase 1/2 safety study in May 2015 to evaluate our RenovoTAMP platform by delivering intra-arterial gemcitabine via our patented RenovoCath delivery system. In this safety study, 20 patients with a diagnosis of Stage 3 pancreatic cancer were enrolled. We established the maximum tolerated dose of 1000mg/m2 of intra-arterial gemcitabine delivered via RenovoCath. The most common serious adverse events were neutropenia followed by sepsis. After completion of enrollment and demonstration of an early survival efficacy signal in this study, we launched our RR2 observational registry study in June 2016. The key inclusion criteria was a diagnosis of advanced or borderline resectable pancreatic adenocarcinoma confirmed by histology or cytology. The primary endpoints were survival, tumor response and performance of RenovoCath. A combination analysis of these two studies demonstrated that survival in “all comers” (n=31) receiving at least one cycle (two treatments over one month) was 29% at two years. Looking at the prior-radiation therapy subset (n=10), 24-month survival was 60% with a median overall survival (mOS) of 27.9 months. This compares favorably both to IV chemotherapy alone, with 24-month survival of 12%, and to chemotherapy + radiation with 24-month survival of 5% and mOS of 12-15 months as demonstrated in historical studies.
We intend to submit our proposed Phase 2/3 clinical trial to evaluate RenovoGem in HCCA, BENEFICIAL, to the FDA as part of a pre-IND submission in the fourth quarter of 2021 and to launch the clinical trial in the first half of 2022. Intra-venous (IV), or systemic, delivery of gemcitabine has been considered standard of care for several solid tumors, and the drug’s anti-cancer tumor effects are well profiled. We intend to explore the application of our RenovoTAMP platform in additional indications including locally advanced lung cancer, locally advanced uterine cancer, and glioblastoma. We have completed and presented data on a lung cancer application in pre-clinical studies, and additional pre-clinical experiments in lung cancer may be conducted. Beyond our initial anti-cancer product candidate, RenovoGem, multiple small molecule therapeutics could be incorporated into our RenovoTAMP platform, and we will opportunistically look to develop other potential product candidates.
Our management team, Board of Directors, and Scientific Advisors provide us with expertise across multiple sectors to drive success through clinical development and subsequent commercialization of our novel therapy platform. Our Chief Executive Officer, Shaun Bagai, has extensive experience running clinical trials as well as launching, creating, and developing new markets for novel therapies at Trans Vascular, Medtronic, Ardian, and HeartFlow. Dr. Ramtin Agah, our Co-Founder and Chief Medical Officer, is a practicing cardiovascular specialist who has 20 years of research experience in vascular biology and disease in both academia and industry. Our Board of Directors includes a wide range of public and private company management and Board experience including drug/device combination and oncology experience. Clinical advisors include experts in surgical oncology, interventional radiology, radiation oncology, and medical oncology. Dr. Daniel Von Hoff, a medical oncologist, was instrumental as the Principal Investigator who brought to market standard of care therapies for pancreatic cancer. Dr. Mike Pishvaian, also a medical oncologist, has extensive experience in running oncology studies and is an Associate sociate Professor, and Department of Oncology Director of the Gastrointestinal, Developmental Therapeutics, and Clinical Research Programs at the NCR Kimmel Cancer Center at Sibley Memorial Hospital Johns Hopkins University School of Medicine. Dr. Pishvaian is the Principal Investigator/Global Study Chair of our TIGeR-PaC Phase 3 study. Dr. Peter Muscarella is a surgical oncologist and the Director of Pancreatic Surgery, General Surgery Site Director, Weiler Hospital Associate Program Director, and General Surgery Residency Training Program at Montefiore Medical Center, Bronx, NY. Dr. Karyn Goodman serves as our Radiation Monitor for our TIGeR-PaC Phase 3 study and Professor and Vice Chair of Clinical Research, Department of Radiation Oncology at the Icahn School of Medicine at Mount Sinai, and Associate Director of Clinical Research at the Tisch Cancer Institute at Mount Sinai. We have two interventional radiology scientific advisors: Dr. Reza Malek, Neurointerventional Radiologist at Minimally Invasive Surgical Solutions and Dr. Jacob Cynamon, Professor of Clinical Radiology of the Albert Einstein College of Medicine, Chief of the Division of Vascular and Interventional Radiology, and Program Director of the Vascular and Interventional Radiology Fellowship Program at the Montefiore Medical Center, Bronx, NY.
| -4- |
Summary Risk Factors
Our prospects should be considered in light of the risks, uncertainties, expenses and difficulties frequently encountered by similar companies. Our ability to realize our business objectives and execute our strategies is subject to risks and uncertainties, including, among others, the following:
| ● | We are a clinical stage company and may never earn a profit. |
| ● | We will need to raise substantial additional capital to develop and commercialize RenovoGem, and our failure to obtain funding when needed may force us to delay, reduce or eliminate our product development programs or collaboration efforts. As a result, there is substantial doubt about our ability to operate as a going concern. |
| ● | Our product candidates’ commercial viability remains subject to current and future preclinical studies, clinical trials, regulatory approvals, and the risks generally inherent in the development of a pharmaceutical product candidate. If we are unable to successfully advance or develop our product candidates, our business will be materially harmed. |
| ● | Our product candidates may exhibit undesirable side effects when used alone or in combination with other approved pharmaceutical products or investigational new drugs, which may delay or preclude further development or regulatory approval or limit their use if approved. |
| ● | If the results of preclinical studies or clinical trials for our product candidates are negative, we could be delayed or precluded from the further development or commercialization of our product candidates, which could materially harm our business. |
| ● | If we are unable to satisfy regulatory requirements, we may not be able to commercialize our product candidates. |
| ● | If our product candidates are unable to compete effectively with marketed drugs targeting similar indications as our product candidates, our commercial opportunity will be reduced or eliminated. We may delay or terminate the development of our product candidates at any time if we believe the perceived market or commercial opportunity does not justify further investment, which could materially harm our business. |
| ● | Our future success depends on our ability to retain our key personnel and to attract, retain, and motivate qualified personnel. |
| ● | If we are unable to protect our intellectual property effectively, we may be unable to prevent third parties from using our technologies, which would impair our competitive advantage. |
| ● | The patents issued to us may not be broad enough to provide any meaningful protection, one or more of our competitors may develop more effective technologies, designs, or methods without infringing our intellectual property rights and one or more of our competitors may design around our proprietary technologies. |
| -5- |
| ● | The market price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock in this offering. |
| ● | We have broad discretion in the use of our cash and cash equivalents, including the net proceeds we receive in this offering, and may not use them effectively. | |
| ● | Holders of our Warrants will have no rights as shareholders until they acquire shares of our common stock, if ever, except as set forth in the Warrants. |
In addition, we face other risks and uncertainties that may materially affect our business prospects, financial condition, and results of operations. You should consider the risks discussed in “Risk Factors” and elsewhere in this prospectus before investing in our securities.
Corporate Information
We were incorporated in the State of Delaware on December 17, 2012. Our principal executive offices are located at 4546 El Camino Real, Suite B1, Los Altos, CA 94022. Our telephone number is (650) 284-4433. Our website address is https://renovorx.com. Information contained in our website does not constitute any part of, and is not incorporated into, this prospectus.
Implications of Being an Emerging Growth Company
Upon the completion of this offering, we will qualify as an “emerging growth company” under Jumpstart Our Business Act of 2012, as amended, or the JOBS Act. As a result, we will be permitted to, and intend to, rely on exemptions from certain disclosure requirements. For so long as we are an emerging growth company, we will not be required to:
| ● | have an auditor report on our internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act; | |
| ● | comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (i.e., an auditor discussion and analysis); | |
| ● | submit certain executive compensation matters to stockholder advisory votes, such as “say-on-pay” and “say-on-frequency;” and | |
| ● | disclose certain executive compensation related items such as the correlation between executive compensation and performance and comparisons of the chief executive officer’s compensation to median employee compensation. |
In addition, Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. In other words, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to take advantage of the benefits of this extended transition period. Our financial statements may therefore not be comparable to those of companies that comply with such new or revised accounting standards.
We will remain an emerging growth company for up to five years from the date of the first sale of equity securities pursuant to an effective registration statement, or until the earliest of (i) the last day of the first fiscal year in which our total annual gross revenues exceed $1.07 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three year period.
| -6- |
| Units offered by us | Each Unit consists of (a) one share of our common stock, par value $0.0001 per share and (b) three-quarter Warrant to purchase one share of our common stock at an exercise price equal to $14.40 (120% of initial public offering price per Unit) per share, assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) exercisable until the fifth anniversary of the issuance date. | |
| ||
| Pre-funded Units offered by us | We are also offering to those purchasers, if any, whose purchase of Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if the purchaser so chooses, pre-funded units (each a “Pre-funded Unit”) in lieu of Units that would otherwise result in the purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock. Each Pre-funded Unit will consist of a pre-funded warrant to purchase one share of our common stock at an exercise price of $0.0001 per share (each a “Pre-funded Warrant”) and a Warrant. The purchase price of each Pre-funded Unit is equal to the price per Unit being sold to the public in this offering, minus $0.0001. The Pre-funded Warrants will be immediately exercisable and may be exercised at any time until all of the Pre-funded Warrants are exercised in full. We are offering up to 1,850,000 Units and up to 1,850,000 Pre-funded Units, in the aggregate not exceeding more than a total of 1,850,000 units. For each Pre-funded Unit we sell, the number of Units we are offering will be decreased on a one-for-one basis. Because we will issue one Warrant as part of each Unit or Pre-funded Unit, the number of Warrants sold in this offering will not change as a result of a change in the mix of the Units and Pre-funded Units sold. | |
| Over-allotment option | We have granted the representative an option to purchase an additional 277,500 shares of our common stock and/or Warrants to purchase 208,125 additional shares of common stock from us (being up to 15% of the shares of common stock, including shares of common stock underlying the Pre-funded Warrants, and/or up to 15% of the Warrants sold in this offering), in any combination thereof, for forty-five (45) days from the date of this prospectus, at prices described herein to cover over-allotments, if any, of the Units and the Pre-funded Units offered hereby. See “Underwriting.” | |
| Shares of common stock outstanding before this offering(1) | 2,291,557 shares of common stock. | |
| Shares outstanding after this offering | 8,207,572 shares of common stock (or 8,485,072 shares of common stock if the representative exercises its over-allotment option in full), after the sale of 1,850,000 Units in this offering and after the Preferred Stock Conversions and the Note Conversions (as defined below) and assuming no Pre-funded Warrants are sold in this offering. | |
| Use of proceeds | We estimate that we will receive net proceeds of approximately $19.9 million (or approximately $23.0 million if the representative exercises its over-allotment option in full) from the sale of Units by us in this offering assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus). We plan to use the net proceeds of this offering primarily for completion of our currently ongoing Phase 3 trial for the locally advanced pancreatic cancer indication for RenovoGem, the launch of our Phase 2/3 trial for our second indication of hilar cholangiocarcinoma for RenovoGem, and working capital and general corporate purposes. See “Use of Proceeds.” | |
| Risk factors | Investing in our securities involves a high degree of risk and purchasers of our securities may lose part or all of their investment. See “Risk Factors” for a discussion of factors you should carefully consider before deciding to invest in our securities. | |
| Proposed trading market and symbol | We
have been approved to list our common stock for trading on the Nasdaq Capital Market under the symbol “RNXT” upon
our satisfaction of the exchange’s initial listing criteria, including the completion of this offering.
We do not intend to apply for any listing of the Warrants or Pre-funded Warrants on the Nasdaq Capital Market or any other securities exchange or nationally recognized trading system, and we do not expect a market for the Warrants or Pre-funded Warrants to develop. |
(1) The number of shares outstanding is based on shares outstanding as of June 30, 2021 and excludes the following:
| ● | 992,150 shares of our common stock issuable upon the exercise of outstanding options with a weighted-average exercise price of $0.53 per share; | |
| ● | 708,533 shares of Series A-1 Preferred Stock, 709,219 shares of Series A-2 Preferred Stock, 532,046 shares of Series A-3 Preferred Stock and 1,585,671 shares of Series B Preferred Stock which will convert into 708,533, 709,219, 532,046 and 1,585,671 shares of common stock, respectively, upon the closing of this offering (the “Preferred Stock Conversions”); | |
| ● | 530,546 shares of common stock and 397,901 shares of common stock issuable upon exercise of warrants with an exercise price of $14.40 per share assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) to be issued upon conversion of the 2020 Convertible Notes and 2021 Convertible Notes upon the closing of this offering (the “Note Conversions”);
| |
| ● | up to an additional 10,368 shares of our common stock issuable under our 2013 Equity Incentive Plan; and | |
| ● | 161,875 shares of our common stock underlying the underwriter’s purchase option to be issued to the representative of the underwriters in connection with this offering. |
Except as otherwise indicated herein, all information in this prospectus assumes:
| ● | no exercise by the representative of its option to purchase additional shares of our common stock and/or Warrants from us at prices described herein to cover over-allotments, if any, of the Units offered hereby. | |
| ● | no exercise of the Warrants being offered by this prospectus. | |
| ● | no exercise of the underwriter’s warrant. | |
| ● | no sale of Pre-funded Units in this offering. |
| ● | a 1-to-5 reverse stock split of our common stock pursuant to which (i) every 5 shares of outstanding common stock were decreased to one share of common stock, (ii) the number of shares of common stock for which each outstanding warrant or option to purchase common stock is exercisable was proportionally decreased on a 1-to-5 basis, and (iii) the exercise price of each outstanding warrant or option to purchase common stock was proportionately increased on a 1-to-5 basis, (the “Reverse Stock Split”). |
| -7- |
SUMMARY FINANCIAL INFORMATION
The following tables set forth our summary financial data for the periods and as of the dates indicated. The summary statements of operations data for the years ended December 31, 2019 and 2020 have been derived from our audited financial statements included elsewhere in this prospectus. The summary statements of operations data for the three months ended March 31, 2020 and 2021 and the summary balance sheet data as of March 31, 2021 have been derived from our unaudited interim condensed financial statements included elsewhere in this prospectus. Our unaudited interim condensed financial statements have been prepared on a basis consistent with our audited financial statements and, in the opinion of management, reflect all adjustments, consisting only of normal recurring adjustments, necessary for a fair presentation of the financial information set forth in those statements. Our historical results are not necessarily indicative of the results that may be expected for any other period in the future and our interim results are not necessarily indicative of our expected results for the year ending December 31, 2021. You should read the following summary financial data set forth below in conjunction with our financial statements and the related notes included elsewhere in this prospectus and the information in the section titled Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained elsewhere in this prospectus.
| Year Ended December 31, | Three Months Ended March 31, | |||||||||||||||
| 2019 | 2020 | 2020 | 2021 | |||||||||||||
| (unaudited) | ||||||||||||||||
| (in thousands, except per share data) | ||||||||||||||||
| Statements of Operations Data: | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 2,997 | $ | 2,386 | $ | 805 | $ | 635 | ||||||||
| General and administrative | 899 | 818 | 248 | 418 | ||||||||||||
| Total operating expenses | 3,896 | 3,204 | 1,053 | 1,053 | ||||||||||||
| Loss from operations | (3,896 | ) | (3,204 | ) | (1,053 | ) | (1,053 | ) | ||||||||
| Interest income (expense), net | 63 | (587 | ) | 2 | (230 | ) | ||||||||||
| Other income (expense), net | 2 | (7 | ) | - | (5 | ) | ||||||||||
| Loss on change in fair value of warrant liability | (8 | ) | - | - | - | |||||||||||
| Gain on loan extinguishment | - | - | - | 140 | ||||||||||||
| Total other income (expense), net | 57 | (594 | ) | 2 | (95 | ) | ||||||||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | $ | (1,051 | ) | $ | (1,148 | ) | ||||
| Net loss per share – basic and diluted | $ | (1.76 | ) | $ | (1.72 | ) | $ | (0.48 | ) | $ | (0.51 | ) | ||||
| Weighted average shares used to compute net loss per share – basic and diluted | 2,177 | 2,214 | 2,195 | 2,242 | ||||||||||||
| Pro forma net loss per share – basic and diluted | $ | (0.68 | ) | $ | (0.53 | ) | $ | (0.18 | ) | $ | (0.19 | ) | ||||
| Weighted average shares used to compute pro forma net loss per share – basic and diluted | 5,686 | 6,066 | 5,865 | 6,094 | ||||||||||||
| (1) | The unaudited pro forma net loss per share for the years ended December 31, 2019 and 2020 and the three months ended March 31, 2020 and 2021 was computed using the weighted-average number of shares of common stock outstanding, including the pro forma effect of the conversion of all outstanding shares of convertible preferred stock and convertible notes into shares of common stock, as if such conversion had occurred at the beginning of the period, or their issuance dates, if later. |
| As of March 31, 2021 | ||||||||||||
| Actual | Pro forma (1) | Pro forma as adjusted (2) | ||||||||||
| (in thousands) | ||||||||||||
| Balance Sheet Data: | ||||||||||||
| Cash and cash equivalents | $ | 837 | $ | 837 | $ | 20,733 | ||||||
| Working (deficit) capital | (3,636 | ) | 206 | 20,102 | ||||||||
| Total assets | 1,278 | 1,278 | 21,174 | |||||||||
| Convertible note, net | 2,843 | - | - | |||||||||
| Total liabilities | 4,589 | 747 | 747 | |||||||||
| Convertible preferred stock | 12,451 | - | - | |||||||||
| Stockholders’ (deficit) equity | (15,762 | ) | 531 | 20,427 | ||||||||
| (1) | The pro forma balance sheet data gives effect to the Preferred Stock Conversions and the Notes Conversions into 4,066,015 shares of common stock and warrants to purchase 397,901 shares of common stock immediately prior to the closing of this offering. | |
| (2) | The pro forma as adjusted balance sheet data gives effect to the issuance and sale of 1,850,000 Units in this offering at an assumed initial public offering price of $12.00 per Unit, which is the midpoint of the estimated price range set forth on the cover of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. The pro forma as adjusted information is illustrative only and will depend on the actual initial public offering price and other terms of this offering determined at pricing. |
| -8- |
An investment in our securities involve a high degree of risk. You should carefully consider the risks described below, together with the financial and other information contained in this prospectus, before you decide to purchase our securities. If any of the following risks actually occurs, our business, financial condition, results of operations, cash flows and prospects could be materially and adversely affected. In that event, the trading price of our common stock and the market value of the securities offered hereby could decline, and you may lose all or part of your investment.
Risks Related to Our Business
We are a clinical stage company and may never earn a profit.
We are a clinical stage company and have incurred losses since our formation. As of March 31, 2021, we have an accumulated total deficit of approximately $16.1 million. For both three-month periods ended March 31, 2020 and 2021, we had a net loss of approximately $1.1 million. To date, we have experienced negative cash flow from development of our product candidate, RenovoGem, our platform technology, Renovo Trans-Arterial Micro-Perfusion, or RenovoTAMP, and our RenovoCath delivery system. We have not generated any revenue from operations, and we expect to incur substantial net losses for the foreseeable future as we seek to further develop and commercialize RenovoGem. We cannot predict the extent of these future net losses, or when we may attain profitability, if ever. If we are unable to generate significant revenue from RenovoGem or attain profitability, we will not be able to sustain operations.
Because of the numerous risks and uncertainties associated with developing and commercializing RenovoGem, we are unable to predict the extent of any future losses or when we will attain profitability, if ever. We may never become profitable and you may never receive a return on an investment in our common stock. An investor in our common stock must carefully consider the substantial challenges, risks and uncertainties inherent in the attempted development and commercialization of RenovoGem. We may never successfully commercialize RenovoGem, and our business may not be successful.
We will need to raise substantial additional capital to develop and commercialize RenovoGem, and our failure to obtain funding when needed may force us to delay, reduce or eliminate our product development programs or collaboration efforts. If we do not obtain adequate and timely funding, we may not be able to continue as a going concern.
As of March 31, 2021, our cash and cash equivalents were approximately $0.8 million, and our working capital deficit was approximately $3.6 million. Due to our recurring losses from operations and the expectation that we will continue to incur losses in the future, we will be required to raise additional capital to complete the development and commercialization of our product candidates. We have historically relied upon private sales of our equity as well as debt financings to fund our operations. In order to raise additional capital, we may seek to sell additional equity and/or debt securities, obtain a credit facility or other loan or enter into collaborations, licenses or other similar arrangements, which we may not be able to do on favorable terms, or at all. Our ability to obtain additional financing will be subject to a number of factors, including market conditions, our operating performance and investor sentiment. If we are unable to raise additional capital when required or on acceptable terms, we may have to significantly delay, scale back or discontinue the development and/or commercialization of our product candidate, restrict our operations or obtain funds by entering into agreements on unfavorable terms. Failure to obtain additional capital at acceptable terms would result in a material and adverse impact on our operations. As a result, there is substantial doubt about our ability to operate as a going concern.
| -9- |
Our financial statements have been prepared on a going concern basis and do not include any adjustments that may result from the outcome of this uncertainty. If we fail to raise additional working capital, or do so on commercially unfavorable terms, it would materially and adversely affect our business, prospects, financial condition and results of operations, and we may be unable to continue as a going concern. If we seek additional financing to fund our business activities in the future and there remains substantial doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding to us on commercially reasonable terms, if at all. If we are unable to continue as a going concern, we might have to liquidate our assets and the value we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements, and our shareholders may lose their entire investment in our ordinary shares.
Our product candidates’ commercial viability remains subject to current and future preclinical studies, clinical trials, regulatory approvals, and the risks generally inherent in the development of a pharmaceutical product candidate. If we are unable to successfully advance or develop our product candidate, our business will be materially harmed.
In the near-term, failure to successfully advance the development of our product candidate may have a material adverse effect on us. To date, we have not successfully developed or commercially marketed, distributed, or sold any product candidate. The success of our business depends primarily upon our ability to successfully advance the development of our current and future product candidates through preclinical studies and clinical trials, have the product candidates approved for sale by the FDA or regulatory authorities in other countries, and ultimately have the product candidates successfully commercialized by us or a commercial partner. We cannot assure you that the results of our ongoing preclinical studies or clinical trials will support or justify the continued development of our product candidate, or that we will receive approval from the FDA, or similar regulatory authorities in other countries, to advance the development of our product candidates.
Our product candidates must satisfy rigorous regulatory standards of safety and efficacy before we can advance or complete their clinical development, or they can be approved for sale. To satisfy these standards, we must engage in expensive and lengthy preclinical studies and clinical trials, develop acceptable manufacturing processes, and obtain regulatory approval. Despite these efforts, our product candidates may not:
| ● | offer therapeutic or other medical benefits over existing drugs or other product candidates in development to treat the same patient population; |
| ● | be proven to be safe and effective in current and future preclinical studies or clinical trials; |
| ● | have the desired effects; |
| ● | be free from undesirable or unexpected effects; |
| ● | meet applicable regulatory standards; |
| ● | be capable of being formulated and manufactured in commercially suitable quantities and at an acceptable cost; or |
| ● | be successfully commercialized by us or by collaborators. |
We cannot assure you that the results of late-stage clinical trials will be favorable enough to support the continued development of our product candidates. A number of companies in the pharmaceutical and biopharmaceutical industries have experienced significant delays, setbacks and failures in all stages of development, including late-stage clinical trials, even after achieving promising results in preclinical testing or early-stage clinical trials. Accordingly, results from completed preclinical studies and early-stage clinical trials of our product candidates may not be predictive of the results we may obtain in later-stage trials. Furthermore, even if the data collected from preclinical studies and clinical trials involving our product candidates demonstrate a favorable safety and efficacy profile, such results may not be sufficient to support the submission of an NDA to obtain regulatory approval from the FDA in the U.S., or other similar regulatory agencies in other jurisdictions, which is required to market and sell the product.
| -10- |
Our product candidates will require significant additional research and development efforts, the commitment of substantial financial resources, and regulatory approvals prior to advancing into further clinical development or being commercialized by us or collaborators. We cannot assure you that our product candidates will successfully progress through the drug development process or will result in commercially viable products. We do not expect our product candidates to be commercialized by us or collaborators for at least several years.
Our product candidates may exhibit undesirable side effects when used alone or in combination with other approved pharmaceutical products or investigational new drugs, which may delay or preclude further development or regulatory approval or limit their use if approved.
Throughout the drug development process, we must continually demonstrate the safety and tolerability of our product candidates to obtain regulatory approval to further advance clinical development or to market them. Even if our product candidates demonstrate clinical efficacy, any unacceptable adverse side effects or toxicities, when administered alone or in the presence of other pharmaceutical products, which can arise at any stage of development, may outweigh potential benefits. In preclinical studies and clinical trials we have conducted to date, our product candidate’s tolerability profile is based on studies and trials that have involved a small number of subjects or patients over a limited period of time. We may observe adverse or significant adverse events or drug-drug interactions in future preclinical studies or clinical trial candidates, which could result in the delay or termination of development, prevent regulatory approval, or limit market acceptance if ultimately approved.
Raising additional capital may cause dilution to our existing stockholders, including purchasers of common stock in this offering, restrict our operations or require us to relinquish rights to our product candidates on unfavorable terms to us.
We may seek additional capital through a variety of means, including through public or private equity, debt financings or other sources, including up-front payments and milestone payments from strategic collaborations. To the extent that we raise additional capital through the sale of equity or convertible debt or equity securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Such financing may result in dilution to stockholders, imposition of debt covenants, increased fixed payment obligations or other restrictions that may affect our business. If we raise additional funds through up-front payments or milestone payments pursuant to strategic collaborations with third parties, we may have to relinquish valuable rights to our product candidates or grant licenses on terms that are not favorable to us. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
If the results of preclinical studies or clinical trials for our product candidates, including those that are subject to existing or future license or collaboration agreements, are unfavorable or delayed, we could be delayed or precluded from the further development or commercialization of our product candidates, which could materially harm our business.
In order to further advance the development of, and ultimately receive regulatory approval to sell, our product candidates, we must conduct extensive preclinical studies and clinical trials to demonstrate their safety and efficacy to the satisfaction of the FDA or similar regulatory authorities in other countries, as the case may be. Preclinical studies and clinical trials are expensive, complex, can take many years to complete, and have highly uncertain outcomes. Delays, setbacks, or failures can occur at any time, or in any phase of preclinical or clinical testing, and can result from concerns about safety or toxicity, a lack of demonstrated efficacy or superior efficacy over other similar products that have been approved for sale or are in more advanced stages of development, poor study or trial design, and issues related to the formulation or manufacturing process of the materials used to conduct the trials. The results of prior preclinical studies or clinical trials are not necessarily predictive of the results we may observe in later stage clinical trials. In many cases, product candidates in clinical development may fail to show desired safety and efficacy characteristics despite having favorably demonstrated such characteristics in preclinical studies or earlier stage clinical trials.
| -11- |
In addition, we may experience numerous unforeseen events during, or as a result of, preclinical studies and the clinical trial process, which could delay or impede our ability to advance the development of, receive regulatory approval for, or commercialize our product candidate, including, but not limited to:
| ● | communications with the FDA, or similar regulatory authorities in different countries, regarding the scope or design of a trial or trials; |
| ● | regulatory authorities, including an Institutional Review Board (“IRB”) or Ethical Committee (“EC”), not authorizing us to commence or conduct a clinical trial at a prospective trial site; |
| ● | enrollment in our clinical trials being delayed, or proceeding at a slower pace than we expected, because we have difficulty recruiting patients or participants dropping out of our clinical trials at a higher rate than we anticipated; |
| ● | our third-party contractors, upon whom we rely for conducting preclinical studies, clinical trials and manufacturing of our trial materials, may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| ● | having to suspend or ultimately terminate our clinical trials if participants are being exposed to unacceptable health or safety risks; |
| ● | IRBs, ECs, or regulators requiring that we hold, suspend or terminate our preclinical studies and clinical trials for various reasons, including non-compliance with regulatory requirements; and |
| ● | the supply or quality of drug material or the supply of our RenovoCath device necessary to conduct our preclinical studies or clinical trials being insufficient, inadequate, or unavailable. |
Even if the data collected from preclinical studies or clinical trials involving our product candidates demonstrate a favorable safety and efficacy profile, such results may not be sufficient to support the submission of an NDA to obtain regulatory approval from the FDA in the U.S., or other similar foreign regulatory authorities in foreign jurisdictions, which is required to market and sell the product.
If third party vendors upon whom we intend to rely on to conduct our preclinical studies or clinical trials do not perform or fail to comply with strict regulations, these studies or trials of our product candidate may be delayed, terminated, or fail, or we could incur significant additional expenses, which could materially harm our business.
We have limited resources dedicated to designing, conducting, and managing preclinical studies and clinical trials. We intend to rely on third parties, including clinical research organizations, consultants, and principal investigators, to assist us in designing, managing, monitoring and conducting our preclinical studies and clinical trials. We intend to rely on these vendors and individuals to perform many facets of the drug development process, including certain preclinical studies, the recruitment of sites and patients for participation in our clinical trials, maintenance of good relations with the clinical sites, and ensuring that these sites are conducting our trials in compliance with the trial protocol, including safety monitoring and applicable regulations. If these third parties fail to perform satisfactorily, or do not adequately fulfill their obligations under the terms of our agreements with them, we may not be able to enter into alternative arrangements without undue delay or additional expenditures, and therefore the preclinical studies and clinical trials of our product candidate may be delayed or prove unsuccessful. Further, the FDA, or other similar foreign regulatory authorities, may inspect some of the clinical sites participating in our clinical trials in the U.S., or our third-party vendors’ sites, to determine if our clinical trials are being conducted according to Good Clinical Practices. If we or the FDA determine that our third-party vendors are not in compliance with, or have not conducted our clinical trials according to, applicable regulations we may be forced to delay, repeat, or terminate such clinical trials.
| -12- |
We have limited capacity for recruiting and managing clinical trials, which could impair our timing to initiate or complete clinical trials of our product candidate and materially harm our business.
We have limited capacity to recruit and manage the clinical trials necessary to obtain FDA approval or approval by other regulatory authorities. By contrast, larger pharmaceutical and bio-pharmaceutical companies often have substantial staff with extensive experience in conducting clinical trials with multiple product candidates across multiple indications. In addition, they may have greater financial resources to compete for the same clinical investigators and patients that we are attempting to recruit for our clinical trials. If potential competitors are successful in completing drug development for their product candidates and obtain approval from the FDA, they could limit the demand to participate in clinical trials of our product candidates.
As a result, we may be at a competitive disadvantage that could delay the initiation, recruitment, timing, completion of our clinical trials and obtaining regulatory approvals, if at all, for our product candidates.
We, and our collaborators, if any, must comply with extensive government regulations in order to advance our product candidates through the development process and ultimately obtain and maintain marketing approval for our products in the U.S. and abroad.
The product candidates that we, or our collaborators, are developing or may develop require regulatory approval to advance through clinical development and to ultimately be marketed and sold and are subject to extensive and rigorous domestic and foreign government regulation. In the U.S., the FDA regulates, among other things, the development, testing, manufacture, safety, efficacy, record-keeping, labeling, storage, approval, advertising, promotion, sale, and distribution of pharmaceutical and biopharmaceutical products. Our product candidates are also subject to similar regulation by foreign governments to the extent we seek to develop or market them in those countries. We, or our collaborators, must provide the FDA and foreign regulatory authorities, if applicable, with preclinical and clinical data, as well as data supporting an acceptable manufacturing process, that appropriately demonstrate our product candidate’s safety and efficacy before it can be approved for the targeted indications. Our product candidates have not been approved for sale in the U.S. or any foreign market, and we cannot predict whether we or our collaborators will obtain regulatory approval for any product candidates we are developing or plan to develop. The regulatory review and approval process can take many years, is dependent upon the type, complexity, novelty of, and medical need for the product candidate, requires the expenditure of substantial resources, and involves post-marketing surveillance and vigilance and potentially post-marketing studies or Phase 4 clinical trials. In addition, we or our collaborators may encounter delays in, or fail to gain, regulatory approval for our product candidate based upon additional governmental regulation resulting from future legislative, administrative action or changes in FDA’s or other similar foreign regulatory authorities’ policy or interpretation during the period of product development. Delays or failures in obtaining regulatory approval to advance our product candidate through clinical development, and ultimately commercialize them, may:
| ● | adversely impact our ability to raise sufficient capital to fund the development of our product candidates; |
| ● | adversely affect our ability to further develop or commercialize our product candidates; |
| ● | diminish any competitive advantages that we or our collaborators may have or attain; and |
| -13- |
| ● | adversely affect the receipt of potential milestone payments and royalties from collaborators, if any, from the sale of our products or product revenues in the future. |
Furthermore, any regulatory approvals, if granted, may later be withdrawn. If we or our collaborators fail to comply with applicable regulatory requirements at any time, or if post-approval safety concerns arise, we or our collaborators may be subject to restrictions or a number of actions, including:
| ● | delays, suspension, or termination of clinical trials related to our products; |
| ● | refusal by regulatory authorities to review pending applications or supplements to approved applications; |
| ● | product recalls or seizures; |
| ● | suspension of manufacturing; |
| ● | withdrawals of previously approved marketing applications; and |
| ● | fines, civil penalties, and criminal prosecutions. |
Additionally, at any time we or our collaborators may voluntarily suspend or terminate the preclinical or clinical development of a product candidate, or withdraw any approved product from the market if we believe that it may pose an unacceptable safety risk to patients, or if the product candidate or approved product no longer meets our business objectives. The ability to develop or market a pharmaceutical product outside of the U.S. is contingent upon receiving appropriate authorization from the respective foreign regulatory authorities. Foreign regulatory approval processes typically include many, if not all, of the risks and requirements associated with the FDA regulatory process for drug development and may include additional risks.
Clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Our product candidates may not prove to be safe and efficacious in clinical trials and may not meet all the applicable regulatory requirements needed to receive regulatory approval. In order to receive regulatory approval for the commercialization of our product candidates, we must conduct, at our own expense, extensive preclinical testing and clinical trials to demonstrate safety and efficacy of our product candidate for the intended indication of use. Clinical testing is expensive, can take many years to complete, if at all, and its outcome is uncertain. Failure can occur at any time during the clinical trial process.
The results of preclinical studies and early clinical trials of new drugs do not necessarily predict the results of later-stage clinical trials. The design of our clinical trials is based on many assumptions about the expected effects of our product candidate, and if those assumptions are incorrect, they may not produce statistically significant results. Preliminary results may not be confirmed on full analysis of the detailed results of a clinical trial. Product candidates in later stages of clinical development may fail to show safety and efficacy sufficient to support intended use claims despite having progressed through earlier clinical testing. The data collected from clinical trials of our product candidate may not be sufficient to support the filing of an NDA or to obtain regulatory approval in the United States or elsewhere. Because of the uncertainties associated with drug development and regulatory approval, we cannot determine if or when we will have an approved product for commercialization or achieve sales or profits.
| -14- |
Delays in clinical testing could result in increased costs to us and delay our ability to generate revenue.
We may experience delays in clinical testing of our product candidate. We do not know whether planned clinical trials will begin on time, will need to be redesigned or will be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including pandemics, delays in obtaining regulatory approval to commence a clinical trial, in securing clinical trial agreements with prospective sites with acceptable terms, in obtaining IRB approval to conduct a clinical trial at a prospective site, in recruiting patients to participate in a clinical trial or in obtaining sufficient supplies of clinical trial materials, including RenovoCath. Many factors affect patient enrollment, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the clinical trial, the existing body of safety and efficacy data with respect to the study drug, competing clinical trials, new drugs approved for the conditions we are investigating and health epidemics such as the COVID-19 pandemic. Clinical investigators will need to decide whether to offer their patients enrollment in clinical trials of our product candidate versus treating these patients with commercially available drugs that have established safety and efficacy profiles. Any delays in completing our clinical trials will increase our costs, slow down our product development, timeliness and approval process and delay our ability to generate revenue.
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
The time required to obtain approval by the FDA and comparable foreign authorities is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that our existing product candidate or any product candidate we may seek to develop in the future will ever obtain regulatory approval may fail to receive regulatory approval.
Our product candidate could fail to receive regulatory approval for many reasons, including the following:
| ● | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication; |
| ● | the results of clinical trials may not meet the level of statistical significance required for approval by the FDA or comparable foreign regulatory authorities; |
| ● | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| ● | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| ● | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
| -15- |
This lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market our product candidates, which would significantly harm our business, results of operations and prospects.
In addition, even if we were to obtain approval, regulatory authorities may approve our product candidates for fewer or more limited indications than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our product candidates.
We have not previously submitted an NDA to the FDA, nor similar drug approval filings to comparable foreign authorities, for our product candidates, and we cannot be certain that our product candidates will be successful in clinical trials or receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations. Even if we successfully obtain regulatory approvals to market one or more of our product candidates, our revenues will be dependent on many factors including the size of the markets in the territories for which we gain regulatory approval and have commercial rights. If the markets for patients that we are targeting for our product candidates are not as significant as we estimate, we may not generate significant revenues from sales of such products, if approved.
We plan to seek regulatory approval and to commercialize our product candidates, directly or with collaborators in the United States, the European Union, and other foreign countries which we have not yet identified. While the scope of regulatory approval is similar in other countries, to obtain separate regulatory approval in many other countries we must comply with numerous and varying regulatory requirements of such countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing, and distribution of our product candidates, and we cannot predict success in these jurisdictions.
We may be required to suspend or discontinue clinical trials due to unexpected side effects or other safety risks that could preclude approval of our product candidates.
Our clinical trials may be suspended at any time for a number of reasons. For example, we may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to the clinical trial patients. In addition, the FDA or other regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the clinical trial patients.
Administering our product candidates to humans may produce undesirable side effects. These side effects could interrupt, delay or halt clinical trials of our product candidates and could result in the FDA or other regulatory authorities denying further development or approval of our product candidate for any or all targeted indications. Ultimately, our product candidates may prove to be unsafe for human use. Moreover, we could be subject to significant liability if any volunteer or patient suffers, or appears to suffer, adverse health effects as a result of participating in our clinical trials.
If we fail to comply with healthcare regulations, we could face substantial enforcement actions, including civil and criminal penalties and our business, operations and financial condition could be adversely affected.
As a developer of pharmaceuticals, certain federal and state healthcare laws and regulations pertaining to fraud and abuse, false claims and patients’ privacy rights are and will be applicable to our business. We could be subject to healthcare fraud and abuse laws and patient privacy laws of both the federal government and the states in which we conduct our business. The laws include:
| ● | the federal healthcare program anti-kickback law, which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
| -16- |
| ● | federal false claims laws which prohibit, among other things, individuals, or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent, and which may apply to entities like us which provide coding and billing information to customers; | |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; | |
| ● | the Federal Food, Drug, and Cosmetic Act, which among other things, strictly regulates drug manufacturing and product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples; and | |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payor, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
If our operations are found to be in violation of any of the laws described above or any governmental regulations that apply to us, we may be subject to penalties, including civil and criminal penalties, damages, fines, and the curtailment or restructuring of our operations. Any penalties, damages, fines, curtailment or restructuring of our operations could adversely affect our ability to operate our business and our financial results. Although compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, the risks cannot be entirely eliminated. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert management’s attention from the operation of our business. Moreover, achieving and sustaining compliance with applicable federal and state privacy, security and fraud laws may prove costly.
If we are unable to satisfy regulatory requirements, we may not be able to commercialize our product candidate.
We need FDA approval prior to marketing our product candidates in the United States. If we fail to obtain FDA approval to market our product candidates, we will be unable to sell our product candidates in the United States and we will not generate any revenue.
The FDA’s review and approval process, including among other things, evaluation of preclinical studies and clinical trials of a product candidate as well as the manufacturing process and facility, is lengthy, expensive, and uncertain. To receive approval, we must, among other things, demonstrate with substantial evidence from well-designed and well-controlled pre-clinical testing and clinical trials that the product candidates are both safe and effective for each indication for which approval is sought. Satisfaction of these requirements typically takes several years, and the time needed to satisfy them may vary substantially, based on the type, complexity and novelty of the pharmaceutical product. We cannot predict if or when we will submit an NDA for approval for our product candidate currently under development. Any approvals we may obtain may not cover all of the clinical indications for which we are seeking approval or may contain significant limitations on the conditions of use.
The FDA has substantial discretion in the NDA review process and may either refuse to file our NDA for substantive review or may decide that our data is insufficient to support approval of our product candidates for the claimed intended uses. Following any regulatory approval of our product candidates, we will be subject to continuing regulatory obligations such as safety reporting, required and additional post marketing obligations, and regulatory oversight of promotion and marketing. Even if we receive regulatory approvals, the FDA may subsequently seek to withdraw approval of our NDA if we determine that new data or a reevaluation of existing data show the product is unsafe for use under the conditions of use upon the basis of which the NDA was approved, or based on new evidence of adverse effects or adverse clinical experience, or upon other new information. If the FDA does not file or approve our NDA or withdraws approval of our NDA, the FDA may require that we conduct additional clinical trials, preclinical or manufacturing studies and submit that data before it will reconsider our application. Depending on the extent of these or any other requested studies, approval of any applications that we submit may be delayed by several years, may require us to expend more resources than we have available, or may never be obtained at all. In addition, we have obtained FDA clearance for our RenovoCath delivery system. In the event adverse events arise with respect to the RenovoCath delivery system, the FDA could revoke its clearance which would have a material adverse effect on our business.
| -17- |
We will also be subject to a wide variety of foreign regulations governing the development, manufacture, and marketing of our products to the extent we seek regulatory approval to develop and market our product candidates in a foreign jurisdiction. As of the date hereof we have not identified any foreign jurisdictions which we intend to seek approval from. Whether or not FDA approval has been obtained, approval of a product candidate by the comparable regulatory authorities of foreign countries must still be obtained prior to marketing the product candidate in those countries. The approval process varies, and the time needed to secure approval in any region such as the European Union or in a country with an independent review procedure may be longer or shorter than that required for FDA approval. We cannot assure you that clinical trials conducted in one country will be accepted by other countries or that an approval in one country or region will result in approval elsewhere.
If our product candidates are unable to compete effectively with marketed drugs targeting similar indications as our product candidates, our commercial opportunity will be reduced or eliminated.
We face competition generally from established pharmaceutical and biotechnology companies, as well as from academic institutions, government agencies and private and public research institutions. Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Small or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large, established companies. We are aware of a number of companies in Phase 3 clinical trials for the treatment of LAPC including Angiodynamics, Bausch Health, Fibrogen, NovoCure, and SynCore Biotechnology. In addition, we are aware of a number of companies in Phase 1 and Phase 2 clinical trials for the treatment of LAPC including one interventional company, TriSalus Lifesciences. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize any products that are safer, more effective, have fewer side effects or are less expensive than our product candidates. These potential competitors compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites, and patient enrollment for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
If approved and commercialized, RenovoGem would compete with several currently approved prescription therapies for the treatment of LAPC and hilar cholangiocarcinoma. To our knowledge, other potential competitors are in earlier stages of development. If potential competitors are successful in completing drug development for their product candidates and obtain approval from the FDA, they could limit the demand for RenovoGem.
We expect that our ability to compete effectively will depend upon our ability to:
| ● | successfully identify and develop key points of product differentiation from currently available therapies; |
| ● | successfully and timely complete clinical trials and submit for and obtain all requisite regulatory approvals in a cost-effective manner; |
| ● | maintain a proprietary position for our products and manufacturing processes and other related product technology; |
| -18- |
| ● | attract and retain key personnel; |
| ● | develop relationships with physicians prescribing these products; and |
| ● | build an adequate sales and marketing infrastructure for our products, if approved. |
Because we will be competing against significantly larger companies with established track records, we will have to demonstrate that, based on experience, clinical data, side-effect profiles and other factors, our products, if approved, are competitive with other products. If we are unable to compete effectively and differentiate our products from other marketed drugs, we may never generate meaningful revenue.
We may expend our limited resources to pursue one or more product candidates or indications within our product development strategy, which has and may continue to change over time, and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we intend to focus on developing product candidates for specific indications that we identify as most likely to succeed, in terms of their potential both to gain regulatory approval and to achieve commercialization. As a result, we may forego or delay pursuit of opportunities with other product candidates or in other indications with greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable product candidates. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing, or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to the product candidate.
If the manufacturers upon whom we rely fail to produce our product candidates, in the volumes that we require on a timely basis, or fail to comply with stringent regulations applicable to pharmaceutical drug manufacturers, we may face delays in the development and commercialization of our product candidates.
We do not currently possess internal manufacturing capacity. We plan to utilize the services of GMP, FDA inspected contract manufacturers to manufacture our clinical supplies. Any curtailment in the availability of gemcitabine, however, could result in production or other delays with consequent adverse effects on us. In addition, because regulatory authorities must generally approve raw material sources for pharmaceutical products, changes in raw material suppliers may result in production delays or higher raw material costs.
We obtain our RenovoCath delivery system from a single source. Gemcitabine is supplied from our clinical sites own pharmacies and used off-label for intra-arterial use within our clinical study. We continue to pursue supply agreements for gemcitabine and our RenovoCath delivery system. We may be required to agree to minimum volume requirements, exclusivity arrangements or other restrictions with the contract manufacturers. We may not be able to enter into long-term agreements on commercially reasonable terms, or at all. If we change or add manufacturers, the FDA and comparable foreign regulators may require approval of the changes. Approval of these changes could require new testing by the manufacturer and compliance inspections to ensure the manufacturer is conforming to all applicable laws and regulations and GMP.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products may encounter difficulties in production, particularly in scaling up production. These problems include difficulties with production costs and yields, quality control, including stability of the product and quality assurance testing, shortages of qualified personnel, as well as compliance with federal, state, and foreign regulations. In addition, any delay or interruption in the supply of clinical trial supplies could delay the completion of our clinical trials, increase the costs associated with conducting our clinical trials and, depending upon the period of delay, require us to commence new clinical trials at significant additional expense or to terminate a clinical trial.
| -19- |
We will be responsible for ensuring that our future contract manufacturers comply with the GMP requirements of the FDA and other regulatory authorities from which we seek to obtain product approval. These requirements include, among other things, quality control, quality assurance and the maintenance of records and documentation. The approval process for NDAs includes an inspection of the manufacturer’s compliance with GMP requirements. We will be responsible for regularly assessing a contract manufacturer’s compliance with GMP requirements through record reviews and periodic audits and for ensuring that the contract manufacturer takes responsibility and corrective action for any identified deviations. Manufacturers of our product candidates may be unable to comply with these GMP requirements and with other FDA and foreign regulatory requirements, if any.
While we will oversee compliance of our contract manufacturers, ultimately, we will not have control over our manufacturers’ compliance with these regulations and standards. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval. If the safety of our product candidates is compromised due to a manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to obtain regulatory approval for or successfully commercialize our product candidates, and we may be held liable for any injuries sustained as a result. Any of these factors could cause a delay of clinical trials, regulatory submissions, approvals, or commercialization of RenovoGem or other product candidates, entail higher costs or result in us being unable to effectively commercialize our product candidates. Furthermore, if our manufacturers fail to deliver the required commercial quantities on a timely basis and at commercially reasonable prices, we may be unable to meet demand for any approved products and would lose potential revenues.
We may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates.
To date, our product candidates have been manufactured in small quantities for preclinical studies and clinical trials. If our product candidates are approved by the FDA or comparable regulatory authorities in other countries for commercial sale, we will need to manufacture such product candidates in larger quantities. We may not be able to successfully increase the manufacturing capacity for our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to successfully increase the manufacturing capacity for a product candidate, the clinical trials as well as the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in supply. Our product candidates require precise, high quality manufacturing. Our failure to achieve and maintain these high-quality manufacturing standards in collaboration with our third-party manufacturers, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could harm our business, financial condition and results of operations.
Our product candidates, if approved for sale, may not gain acceptance among physicians, patients, and the medical community, thereby limiting our potential to generate revenues.
If our product candidates are approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product by physicians, healthcare professionals and third-party payors and our profitability and growth will depend on a number of factors, including:
| ● | demonstration of safety and efficacy; |
| ● | changes in the practice guidelines and the standard of care for the targeted indication; |
| -20- |
| ● | relative convenience and ease of administration; |
| ● | the prevalence and severity of any adverse side effects; |
| ● | budget impact of adoption of our product on relevant drug formularies and the availability, cost, and potential advantages of alternative treatments, including less expensive generic drugs; |
| ● | pricing, reimbursement, and cost effectiveness, which may be subject to regulatory control; |
| ● | effectiveness of our or any of our or our partners’ sales and marketing strategies; |
| ● | the product labeling or product insert required by the FDA or regulatory authority in other countries; and |
| ● | the availability of adequate third-party insurance coverage or reimbursement. |
If any product candidate that we develop does not provide a treatment regimen that is as beneficial as, or is perceived as being as beneficial as, the current standard of care or otherwise does not provide patient benefit, that product candidate, if approved for commercial sale by the FDA or other regulatory authorities, likely will not achieve market acceptance. Our ability to effectively promote and sell any approved products will also depend on pricing and cost-effectiveness, including our ability to produce a product at a competitive price and our ability to obtain sufficient third-party coverage or reimbursement. If any product candidate is approved but does not achieve an adequate level of acceptance by physicians, patients and third-party payors, our ability to generate revenues from that product would be substantially reduced. In addition, our efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources, may be constrained by FDA rules and policies on product promotion, and may never be successful.
Guidelines and recommendations published by various organizations can impact the use of our products.
Government agencies promulgate regulations and guidelines directly applicable to us and to our product. In addition, professional societies, practice management groups, private health and science foundations and organizations involved in various diseases from time to time may also publish guidelines or recommendations to the healthcare and patient communities. Recommendations of government agencies or these other groups or organizations may relate to such matters as usage, dosage, route of administration and use of concomitant therapies. Recommendations or guidelines suggesting the reduced use of our products or the use of competitive or alternative products that are followed by patients and healthcare providers could result in decreased use of our proposed products.
If third-party contract manufacturers upon whom we rely to formulate and manufacture our product candidates do not perform, fail to manufacture according to our specifications or fail to comply with strict regulations, our preclinical studies or clinical trials could be adversely affected and the development of our product candidate could be delayed or terminated or we could incur significant additional expenses.
We do not own or operate any manufacturing facilities. We intend to rely on GMP, FDA inspected third-party contractors, at least for the foreseeable future, to formulate and manufacture these preclinical and clinical materials. Our reliance on third-party contract manufacturers exposes us to a number of risks, any of which could delay or prevent the completion of our preclinical studies or clinical trials, or the regulatory approval or commercialization of our product candidate, result in higher costs, or deprive us of potential product revenues. Some of these risks include:
| ● | our third-party contractors failing to develop an acceptable formulation to support later-stage clinical trials for, or the commercialization of, our product candidates; |
| -21- |
| ● | our contract manufacturers failing to manufacture our product candidates according to their own standards, our specifications or Current Good Manufacturing Practice (“cGMP”), or otherwise manufacturing material that we or the FDA may deem to be unsuitable in our clinical trials; |
| ● | our contract manufacturers being unable to increase the scale of, increase the capacity for, or reformulate the form of our product candidate. We may experience a shortage in supply, or the cost to manufacture our products may increase to the point where it adversely affects the cost of our product candidates. We cannot assure you that our contract manufacturers will be able to manufacture our product candidates at a suitable scale, or we will be able to find alternative manufacturers acceptable to us that can do so; |
| ● | our contract manufacturers placing a priority on the manufacture of their own products, or other customers’ products; |
| ● | our contract manufacturers failing to perform as agreed or not remaining in the contract manufacturing business; and |
| ● | our contract manufacturers’ plants being closed as a result of regulatory sanctions or a natural disaster. |
In the event that we need to change our third-party contract manufacturers, our preclinical studies, clinical trials or the commercialization of our product candidate could be delayed, adversely affected or terminated, or such a change may result in significantly higher costs.
Due to regulatory restrictions inherent in an IND or NDA, or for economic reasons, various steps in the manufacture of our product candidate may need to be sole-sourced. We currently obtain our RenovoCath delivery system from a single supplier. In accordance with cGMP regulations, changing manufacturers may require the re-validation of manufacturing processes and procedures, and may require further preclinical studies or clinical trials to show comparability between the materials produced by different manufacturers. Changing our current or future contract manufacturers may be difficult for us and could be costly, which could result in our inability to manufacture our product candidate for an extended period of time and therefore a delay in the development of our product candidate. Further, in order to maintain our development time lines in the event of a change in our third-party contract manufacturer, we may incur significantly higher costs to manufacture our product candidate.
We currently do not have any internal drug discovery capabilities, and therefore we are dependent on identifying drugs that are off patent or on in-licensing or acquiring development programs from third parties in order to obtain additional product candidates.
If in the future we decide to further expand our pipeline, we will be dependent on identifying drugs that are off patent or on in-licensing or acquiring product candidates as we do not have significant internal discovery capabilities at this time. We may face substantial competition from other biotechnology and pharmaceutical companies, many of which may have greater resources then we have, in obtaining in-licensing, sponsored research or acquisition opportunities. In-licensing or acquisition opportunities may not be available to us on terms we find acceptable, if at all. In-licensed compounds that appear promising in research or in preclinical studies may fail to progress into further preclinical studies or clinical trials.
| -22- |
If a product liability claim is successfully brought against us for uninsured liabilities, or such claim exceeds our insurance coverage, we could be forced to pay substantial damage awards that could materially harm our business.
The use of any of our existing or future product candidates in clinical trials and the sale of any approved pharmaceutical products may expose us to significant product liability claims. We have product liability insurance coverage for our proposed clinical trials; however, such insurance coverage may not protect us against any or all of the product liability claims that may be brought against us now or in the future. We may not be able to acquire or maintain adequate product liability insurance coverage at a commercially reasonable cost or in sufficient amounts or scope to protect us against potential losses. In the event a product liability claim is brought against us, we may be required to pay legal and other expenses to defend the claim, as well as uncovered damage awards resulting from a claim brought successfully against us. In the event our product candidate is approved for sale by the FDA and commercialized, we may need to substantially increase the amount of our product liability coverage. Defending any product liability claim or claims could require us to expend significant financial and managerial resources, which could have an adverse effect on our business.
We may delay or terminate the development of our product candidates at any time if we believe the perceived market or commercial opportunity does not justify further investment, which could materially harm our business.
Even though the results of preclinical studies and clinical trials that have been conducted or may be conducted in the future may support further development of our product candidates, we may delay, suspend or terminate the future development of a product candidate at any time for strategic, business, financial or other reasons, including the determination or belief that the emerging profile of the product candidate is such that it may not receive FDA approval, gain meaningful market acceptance, generate a significant return to shareholders, or otherwise provide any competitive advantages in its intended indication or market.
Our future success depends on our ability to retain our key personnel and to attract, retain and motivate qualified personnel.
We are highly dependent on the development, regulatory, commercialization, and business development expertise of Shaun Bagai, our Chief Executive Officer, as well as the other principal members of our management, scientific and clinical teams. Although we have employment agreements, offer letters or consulting agreements with our executive officers, these agreements do not prevent them from terminating their services at any time.
If we lose one or more of our executive officers or key employees, our ability to implement our business strategy successfully could be seriously harmed. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop product candidates, gain regulatory approval, and commercialize new products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be engaged by entities other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us. If we are unable to continue to attract and retain highly qualified personnel, our ability to develop and commercialize product candidates will be limited.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
We are a small company with 7 employees as of July 15, 2021. Future growth of our company will impose significant additional responsibilities on members of management, including the need to identify, attract, retain, motivate and integrate highly skilled personnel. We may increase the number of employees in the future depending on the progress of our development and commercialization of our product candidates. Our future financial performance and our ability to commercialize our product candidate and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to:
| ● | manage our clinical studies effectively; |
| -23- |
| ● | integrate additional management, administrative, manufacturing, and regulatory personnel; |
| ● | maintain sufficient administrative, accounting and management information systems and controls; and |
| ● | hire and train additional qualified personnel. |
There is no guarantee that we will be able to accomplish these tasks, and our failure to accomplish any of them could materially adversely affect our business, prospects, and financial condition.
Business disruptions could seriously harm future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party manufacturers, contract research organizations, or CROs, and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses.
Our corporate headquarters are located in Silicon Valley, California, an area prone to wildfires and earthquakes. These and other natural disasters could severely disrupt our operations, and have a material adverse effect on our business, results of operations, financial condition and prospects. If a natural disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Any disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material adverse effect on our business.
Security threats to our information technology infrastructure and/or our physical buildings could expose us to liability and damage our reputation and business.
It is essential to our business strategy that our and our vendors, partners, clinical trial sites, and third-party providers’ technology and network infrastructure and physical buildings remain secure and are perceived by our customers and corporate partners to be secure. Despite security measures, however, any network infrastructure may be vulnerable to cyber-attacks by hackers and other security threats. We may face cyber-attacks that attempt to penetrate our network security, sabotage, or otherwise disable our research and development activities, products and services, misappropriate our or our customers’ and partners’ proprietary information, which may include personally identifiable information, or cause interruptions of our internal systems and services. Despite security measures, we also cannot guarantee security of our physical buildings. Physical building penetration or any cyber-attacks could negatively affect our reputation, damage our network infrastructure and our ability to deploy our products and services, harm our relationship with customers and partners that are affected, and expose us to financial liability.
| -24- |
Additionally, there are a number of state, federal, and international laws protecting the privacy and security of health information and personal data. For example, the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) imposes limitations on the use and disclosure of an individual’s healthcare information by healthcare providers, healthcare clearinghouses, and health insurance plans, or, collectively, covered entities, and also grants individuals rights with respect to their health information. HIPAA also imposes compliance obligations and corresponding penalties for non-compliance on individuals and entities that provide services to healthcare providers and other covered entities. As part of the American Recovery and Reinvestment Act of 2009 (“ARRA”), the privacy and security provisions of HIPAA were amended. ARRA also made significant increases in the penalties for improper use or disclosure of an individual’s health information under HIPAA and extended enforcement authority to state attorneys general. As amended by ARRA and subsequently by the final omnibus rule adopted in 2013, HIPAA also imposes notification requirements on covered entities in the event that certain health information has been inappropriately accessed or disclosed: notification requirements to individuals, federal regulators, and in some cases, notification to local and national media. Notification is not required under HIPAA if the health information that is improperly used or disclosed is deemed secured in accordance with encryption or other standards developed by the U.S. Department of Health and Human Services. Most states have laws requiring notification of affected individuals and/or state regulators in the event of a breach of personal information, which is a broader class of information than the health information protected by HIPAA. Many state laws impose significant data security requirements, such as encryption or mandatory contractual terms, to ensure ongoing protection of personal information. Activities outside of the U.S. implicate local and national data protection standards, impose additional compliance requirements, and generate additional risks of enforcement for non-compliance. We may be required to expend significant capital and other resources to ensure ongoing compliance with applicable privacy and data security laws, to protect against security breaches and hackers or to alleviate problems caused by such breaches.
We and our third-party contract manufacturers must comply with environmental, health and safety laws and regulations, and failure to comply with these laws and regulations could expose us to significant costs or liabilities.
We and our third-party manufacturers are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the use, generation, manufacture, distribution, storage, handling, treatment, remediation and disposal of hazardous materials and wastes. Hazardous chemicals, including flammable and biological materials, are involved in certain aspects of our business, and we cannot eliminate the risk of injury or contamination from the use, generation, manufacture, distribution, storage, handling, treatment or disposal of hazardous materials and wastes. In the event of contamination or injury, or failure to comply with environmental, health and safety laws and regulations, we could be held liable for any resulting damages and any such liability could exceed our assets and resources. We could also incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials.
Environmental, health and safety laws and regulations are becoming increasingly more stringent. We may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Our failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
Further, with respect to the operations of our third-party contract manufacturers, it is possible that if they fail to operate in compliance with applicable environmental, health and safety laws and regulations or properly dispose of wastes associated with our products, we could be held liable for any resulting damages, suffer reputational harm or experience a disruption in the manufacture and supply of our product candidates or products.
A variety of risks associated with operating internationally could materially adversely affect our business.
Doing business internationally involves a number of risks, including but not limited to:
| ● | multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses; |
| -25- |
| ● | failure by us to obtain and maintain regulatory approvals for the use of our products in various countries; |
| ● | additional potentially relevant third-party patent rights; |
| ● | complexities and difficulties in obtaining protection and enforcing our intellectual property; |
| ● | difficulties in staffing and managing foreign operations; |
| ● | complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems; |
| ● | limits in our ability to penetrate international markets; |
| ● | financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for our products and exposure to foreign currency exchange rate fluctuations; |
| ● | natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; |
| ● | certain expenses including, among others, expenses for travel, translation, and insurance; and |
| ● | regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the U.S. Foreign Corrupt Practices Act, its books and records provisions, or its anti-bribery provisions. |
Any of these factors could significantly harm any current or future international operations and, consequently, our results of operations.
General economic or business conditions may have a negative impact on our business.
Continuing concerns over U.S. healthcare reform legislation and energy costs, geopolitical issues, the availability and cost of credit and government stimulus programs in the U.S. and other countries have contributed to increased volatility. If the economic climate deteriorates or is poor, our business, as well as the financial condition of our suppliers and our third-party payors, could be negatively impacted, which could materially adversely affect our business, prospects and financial condition.
Healthcare reform measures could adversely affect our business.
In the United States and foreign jurisdictions, there have been, and continue to be, a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs. In 2010, the Patient Protection and Affordable Care Act (the “PPACA”) was enacted, which includes measures to significantly change the way healthcare is financed by both governmental and private insurers. Among the provisions of the PPACA of greatest importance to the pharmaceutical and biotechnology industry are the following:
| ● | an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| ● | implementation of the federal physician payment transparency requirements, sometimes referred to as the “Physician Payments Sunshine Act”; |
| -26- |
| ● | a licensure framework for follow-on biologic products; |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
| ● | establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program, to 23.1% and 13% of the average manufacturer price for most branded and generic drugs, respectively and capped the total rebate amount for innovator drugs at 100% of the AMP; |
| ● | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for certain drugs and biologics, including our product candidates, that are inhaled, infused, instilled, implanted or injected; |
| ● | extension of manufacturers’ Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| ● | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability; |
| ● | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and |
| ● | expansion of the entities eligible for discounts under the Public Health program. |
Some of the provisions of the PPACA have yet to be implemented, and there have been legal and political challenges to certain aspects of the PPACA. During President Trump’s administration, he signed two executive orders and other directives designed to delay, circumvent, or loosen certain requirements mandated by the PPACA. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the PPACA. While Congress has not passed repeal legislation, the PPACA includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the PPACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Congress may consider other legislation to repeal or replace elements of the PPACA.
Many of the details regarding the implementation of the PPACA are yet to be determined, and at this time, the full effect that the PPACA would have on our business remains unclear.
Individual states have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access, and marketing cost disclosure and transparency measures, and to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
| -27- |
In addition, given recent federal and state government initiatives directed at lowering the total cost of healthcare, Congress and state legislatures will likely continue to focus on healthcare reform, the cost of prescription drugs and biologics and the reform of the Medicare and Medicaid programs. While we cannot predict the full outcome of any such legislation, it may result in decreased reimbursement for drugs and biologics, which may further exacerbate industry-wide pressure to reduce prescription drug prices. This could harm our ability to generate revenues. Increases in importation or re-importation of pharmaceutical products from foreign countries into the United States could put competitive pressure on our ability to profitably price our products, which, in turn, could adversely affect our business, results of operations, financial condition and prospects. We might elect not to seek approval for or market our products in foreign jurisdictions in order to minimize the risk of re-importation, which could also reduce the revenue we generate from product sales. It is also possible that other legislative proposals having similar effects will be adopted.
Furthermore, regulatory authorities’ assessment of the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of new information, including on other products, changing policies and agency funding, staffing and leadership. We cannot be sure whether future changes to the regulatory environment will be favorable or unfavorable to our business prospects. For example, average review times at the FDA for marketing approval applications can be affected by a variety of factors, including budget and funding levels and statutory, regulatory and policy changes.
The outbreak of the novel coronavirus disease, COVID-19, could materially adversely impact our business, results of operations and financial condition, including our clinical trials.
In January 2020, the World Health Organization declared the outbreak of COVID-19 as a “Public Health Emergency of International Concern,” which continues to spread throughout the world and has adversely impacted global commercial activity and contributed to significant volatility in financial markets. The COVID-19 outbreak and government responses are creating disruption in global supply chains and adversely impacting many industries. The outbreak could have a continued material adverse impact on economic and market conditions. We continue to monitor the impact of the COVID-19 outbreak closely. The extent to which the COVID-19 outbreak will impact our operations or financial results is uncertain.
The outbreak and government measures taken in response have also had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; activity at facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. While the extent of the impact of the COVID-19 pandemic on our business and financial results is uncertain, a continued and prolonged public health crisis such as the COVID-19 pandemic could have a material adverse effect on our business, financial condition and results of operations. As a result of the COVID-19 pandemic, we may experience disruptions that could severely impact our business and clinical trials, including:
| ● | delays or difficulties in enrolling and retaining patients in our clinical trials; |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures (such as endoscopies that are deemed non-essential), which may impact the integrity of subject data and clinical study endpoints; |
| -28- |
| ● | interruption or delays in the operations of the FDA or other regulatory authorities, which may impact review and approval timelines; |
| ● | interruption of, or delays in receiving, supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; |
| ● | limitations on employee and consulting resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees, consultants or their families or the desire of employees or consultants to avoid contact with large groups of people; |
| ● | interruption or delays to our outsourced clinical activities; and |
| ● | changes in clinical site procedures and requirements as well as regulatory requirements for conducting clinical trials during the pandemic. |
We may be required to develop and implement additional clinical trial policies and procedures designed to help protect subjects from the COVID-19 virus. For example, in March 2020, the FDA issued guidance, which FDA subsequently updated, on conducting clinical trials during the pandemic, which describes a number of considerations for sponsors of clinical trials impacted by the pandemic, including the requirement to include in the clinical trial report contingency measures implemented to manage the trial, and any disruption of the trial as a result of the COVID-19 pandemic; a list of all subjects affected by the COVID-19 pandemic related study disruption by unique subject identifier and by investigational site and a description of how the individual’s participation was altered; and analyses and corresponding discussions that address the impact of implemented contingency measures (e.g., participant discontinuation from investigational product and/or study, alternative procedures used to collect critical safety and/or efficacy data) on the safety and efficacy results reported for the trial.
The COVID-19 pandemic continues to evolve rapidly, with the status of operations and government restrictions evolving weekly. The extent to which the outbreak impacts our business and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease.
The ultimate impact of the COVID-19 pandemic on our business operations is highly uncertain and subject to change and will depend on future developments, which cannot be accurately predicted, including the duration of the pandemic, the ultimate geographic spread of the disease, additional or modified government actions, new information that will emerge concerning the severity and impact of COVID-19 and the actions taken to contain COVID-19, including vaccination efforts, or address its impact in the short and long term, among others. We do not yet know the full extent of potential delays or impacts on our business, our clinical trials, our research programs, healthcare systems or the global economy. We will continue to monitor the situation closely.
In addition, our business could be materially adversely affected by other business disruptions to us or our third-party providers that could materially adversely affect our potential future revenue and financial condition and increase our costs and expenses. Our operations, and those of our CROs, third party manufacturers, and other contractors, consultants and third parties could be subject to other global pandemics, earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, medical epidemics and other natural or man-made disasters or business interruptions, for which we are predominantly self-insured. The occurrence of any of these business disruptions could materially adversely affect our operations and financial condition and increase our costs and expenses. We rely on third-party manufacturers to produce and process our product candidate. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption.
| -29- |
Risks Related to Intellectual Property
If we are unable to protect our intellectual property effectively, we may be unable to prevent third parties from using our technologies, which would impair our competitive advantage.
We rely on patent protection as well as a combination of trademark, copyright and trade secret protection, and other contractual restrictions, to protect our proprietary technologies, all of which provide limited protection and may not adequately protect our rights or permit us to gain or keep any competitive advantage. We may not be successful in defending challenges made in connection with our patents and patent applications. If we fail to protect our intellectual property, we will be unable to prevent third parties from using our technologies and they will be able to compete more effectively against us.
In addition to our patents, we rely on contractual restrictions to protect our proprietary technology. We require our employees and third parties to sign confidentiality agreements and our employees are also required to sign agreements assigning to us all intellectual property arising from their work for us. Nevertheless, we cannot guarantee that these measures will be effective in protecting our intellectual property rights. Any failure to protect our intellectual property rights could materially adversely affect our business, prospects and financial condition.
Our currently pending or future patent applications may not result in issued patents and any patents issued to us may be challenged, invalidated, or held unenforceable. Furthermore, we cannot be certain that we were the first to make the invention claimed in our issued patents or pending patent applications in the U.S., or that we were the first to file for protection of the inventions claimed in our foreign issued patents or pending patent applications. In addition, there are numerous recent changes to the patent laws and proposed changes to the rules of the USPTO, which may have a significant impact on our ability to protect our technology and enforce our intellectual property rights. For example, in September 2011, the U.S. enacted sweeping changes to the U.S. patent system under the Leahy-Smith America Invents Act, including changes that transitioned the U.S. from a “first-to-invent” system to a “first-to-file” system and alter the processes for challenging issued patents. These changes could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. In addition, we may become subject to interference proceedings conducted in the patent and trademark offices of various countries to determine our entitlement to patents, and these proceedings may conclude that other patents or patent applications have priority over our patents or patent applications. It is also possible that a competitor may successfully challenge our patents through various proceedings and those challenges may result in the elimination or narrowing of our patents, and therefore reduce our patent protection. Accordingly, rights under any of our issued patents, patent applications or future patents may not provide us with commercially meaningful protection for our products or afford us a commercial advantage against our competitors or their competitive products or processes.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our unregistered trademarks or trade names may be challenged, infringed, circumvented, or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential collaborators or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our unregistered trademarks or trade names. Over the long term, if we are unable to successfully register our trademarks and trade names and establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business may be adversely affected. Our efforts to enforce or protect our proprietary rights related to trademarks, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely impact our financial condition or results of operations.
| -30- |
The patents issued to us may not be broad enough to provide any meaningful protection, one or more of our competitors may develop more effective technologies, designs or methods without infringing our intellectual property rights and one or more of our competitors may design around our proprietary technologies.
If we are not able to protect our proprietary technology, trade secrets and know-how, our competitors may use our inventions to develop competing products. Our patents may not protect us against our competitors, and patent litigation is very expensive. We may not have sufficient cash available to pursue any patent litigation to its conclusion because we currently do not generate revenues other than licensing, milestone and royalty income.
We cannot rely solely on our current patents to be successful. The standards that the USPTO and foreign patent offices use to grant patents, and the standards that U.S. and foreign courts use to interpret patents, are not the same, are not always applied predictably or uniformly and can change, particularly as new technologies develop. As such, the degree of patent protection obtained in the U.S. may differ substantially from that obtained in various foreign countries.
We cannot be certain of the level of protection, if any, that will be provided by our patents if they are challenged in court, where our competitors may raise defenses such as invalidity, unenforceability, or possession of a valid license. In addition, the type and extent of any patent claims that may be issued to us in the future are uncertain. Any patents that are issued may not contain claims that will permit us to stop competitors from using similar technology.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights.
Third parties may challenge the validity, inventorship or ownership of our patents and other intellectual property rights, resulting in costly litigation or other time-consuming and expensive proceedings, which could deprive us of valuable rights. If we become involved in any intellectual property litigation, interference or other judicial or administrative proceedings, we will incur substantial expenses and the attention of our technical and management personnel will be diverted. An adverse determination may subject us to significant liabilities or require us to seek licenses that may not be available from third parties on commercially favorable terms, if at all. Further, if such claims are proven valid, through litigation or otherwise, we may be required to pay substantial monetary damages, which can be tripled if the infringement is deemed willful, or be required to discontinue or significantly delay development, marketing, selling and licensing of the affected products and intellectual property rights.
Our competitors may have filed, and may in the future file, patent applications covering technology similar to ours. Any such patent application may have priority over our patent applications and could further require us to obtain rights to issued patents covering such technologies. There may be third-party patents, patent applications and other intellectual property relevant to our potential products that may block or compete with our potential products or processes. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the USPTO to determine priority of invention in the U.S. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our U.S. patent position with respect to such inventions. In addition, we cannot assure you that we would prevail in any of these suits or that the damages or other remedies that we are ordered to pay, if any, would not be substantial. Claims of intellectual property infringement, misappropriation or other violations against us may require us to enter into royalty or license agreements with third parties that may not be available on acceptable terms, if at all. We may also be subject to injunctions against the further development and use of our technology, which could materially adversely affect our business, prospects and financial condition.
| -31- |
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could materially adversely affect our ability to raise the funds necessary to continue our operations.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent application process and following the issuance of a patent. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In certain circumstances, even inadvertent noncompliance events may permanently and irrevocably jeopardize patent rights. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
We rely on confidentiality agreements to protect our trade secrets. If these agreements are breached by our employees or other parties, our trade secrets may become known to our competitors.
We rely on trade secrets which we seek to protect through confidentiality agreements with our employees and other parties. If these agreements are breached, our competitors may obtain and use our trade secrets to gain a competitive advantage over us. We may not have any remedies against our competitors and any remedies that may be available to us may not be adequate to protect our business or compensate us for the damaging disclosure. In addition, we may have to expend resources to protect our interests from possible infringement by others.
Risks Related to this Offering and Our Common Stock
Our 2020 Convertible Notes and 2021 Convertible Notes automatically convert at the closing of this offering at a discount to the public offering price of the Units, all of which could negatively impact trading in our securities.
In March 2020, we completed the offering of $3.0 million principal amount of 2020 Convertible Notes that provided for the automatic conversion into shares of our common stock and warrants at the closing of this offering at a 20% discount to the public offering price of the Units. In April 2021, we completed the offering of $2.0 million of 2021 Convertible Notes that provided for the automatic conversion into shares of our common stock and warrants at the closing of this offering at a 12.5% discount to the public offering price of the Units. As a result, the investors in this offering will experience immediate dilution when the 2020 Convertible Notes and 2021 Convertible Notes are automatically converted into shares of our common stock and warrants in this offering. We estimate that approximately 336,769 shares and warrants to purchase 252,571 shares will be issuable upon conversion of the 2020 Convertible Notes based on a $9.60 conversion price (assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus)) and taking into account accrued interest through August 15, 2021. We estimate that approximately 193,977 shares and warrants to purchase 145,330 shares will be issuable upon conversion of the 2021 Convertible Notes based on a $10.50 conversion price (assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus)) and taking into account interest earned through August 15, 2021. The automatic conversion of the 2020 Convertible Notes and 2021 Convertible Notes into shares of our common stock and warrants in this offering will be dilutive to our holders and could negatively impact the trading market and price of our common stock following the offering.
No active trading market for our common stock currently exists, and an active trading market may not develop.
Prior to this offering, there has not been an active trading market for our common stock. If an active trading market for our common stock does not develop following this offering, you may not be able to sell your shares quickly or at the market price. Our ability to raise capital to continue to fund operations by selling shares of our common stock and our ability to acquire other companies or technologies by using shares of our common stock as consideration may also be impaired. The initial public offering price of our common stock will be determined by negotiations between us and the underwriters and may not be indicative of the market prices of our common stock that will prevail in the trading market.
The market price of our common stock may be volatile and fluctuate substantially, which could result in substantial losses for purchasers of our common stock in this offering.
The market price of our common stock is likely to be highly volatile and may be subject to wide fluctuations in response to a variety of factors, including the following:
| ● | any delay in the commencement, enrollment and ultimate completion of our clinical trials; |
| ● | any delay in submitting an NDA and any adverse development or perceived adverse development with respect to the FDA’s review of that NDA; |
| ● | failure to successfully develop and commercialize RenovoGem; |
| ● | inability to obtain additional funding; |
| -32- |
| ● | regulatory or legal developments in the United States and other countries applicable to RenovoGem or any other product candidate; |
| ● | adverse regulatory decisions; |
| ● | changes in the structure of healthcare payment systems; |
| ● | inability to obtain adequate product supply for RenovoGem, RenovoCath or any other product candidate, or the inability to do so at acceptable prices; |
| ● | introduction of new products, services or technologies by our competitors; |
| ● | failure to meet or exceed financial projections we provide to the public; |
| ● | failure to meet or exceed the estimates and projections of the investment community; |
| ● | changes in the market valuations of companies similar to ours; |
| ● | market conditions in the pharmaceutical and biotechnology sectors, and the issuance of new or changed securities analysts’ reports or recommendations; |
| ● | announcements of significant acquisitions, strategic collaborations, joint ventures or capital commitments by us or our competitors; |
| ● | significant lawsuits, including patent or stockholder litigation, and disputes or other developments relating to our proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| ● | additions or departures of key scientific or management personnel; |
| ● | sales of our common stock by us or our stockholders in the future; |
| ● | trading volume of our common stock; |
| ● | general economic, industry and market conditions; |
| ● | health epidemics and outbreaks, including the COVID-19 pandemic, or other natural or manmade disasters which could significantly disrupt our preclinical studies and clinical trials, and therefore our receipt of necessary regulatory approvals could be delayed or prevented; and |
| ● | the other factors described in this “Risk Factors” section. |
In addition, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors, as well as general economic, political, regulatory and market conditions, may negatively affect the market price of our common stock, regardless of our actual operating performance. The market price of our common stock may decline below the initial public offering price, and you may lose some or all of your investment. In particular, stock markets have experienced extreme volatility due to the ongoing COVID-19 pandemic and investor concerns and uncertainty related to the impact of the pandemic on the economies of countries worldwide.
We have broad discretion in the use of our cash and cash equivalents, including the net proceeds we receive in this offering, and may not use them effectively.
Our management has broad discretion to use our cash and cash equivalents, including the net proceeds we receive in this offering, to fund our operations and could spend these funds in ways that do not improve our results of operations or enhance the value of our common stock, and you will not have the opportunity as part of your investment decision to assess whether the net proceeds are being used appropriately. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the price of our common stock to decline. Pending their use to fund our operations, we may invest our cash and cash equivalents, including the net proceeds from this offering, in a manner that does not produce income or that loses value.
| -33- |
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against companies following a decline in the market price of their securities. This risk is especially relevant for us because biotechnology companies have experienced significant share price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, the market price for the shares and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. If research analysts do not establish and maintain adequate research coverage or if one or more of the analysts who covers us downgrades our common stock or publishes inaccurate or unfavorable research about our business, the market price for our common stock would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, we could lose visibility in the financial markets, which, in turn, could cause the market price or trading volume for our common stock to decline.
We do not expect to pay dividends in the foreseeable future after this offering, and you must rely on price appreciation of your shares for return on your investment.
We have paid no cash dividends on any class of our stock to date and we do not anticipate paying cash dividends in the near term. For the foreseeable future, we intend to retain any earnings to finance the development and expansion of our business, and we do not anticipate paying any cash dividends on our stock. Accordingly, investors must be prepared to rely on sales of their shares after price appreciation to earn an investment return, which may never occur. Investors seeking cash dividends should not purchase our shares. Any determination to pay dividends in the future will be made at the discretion of our board of directors and will depend on our results of operations, financial condition, contractual restrictions, restrictions imposed by applicable law and other factors our board deems relevant.
As our initial public offering price is substantially higher than our net tangible book value per share, you will experience immediate and substantial dilution.
If you purchase Units in this offering, you will pay more for your shares of common stock than the amount paid by our existing stockholders for their shares on a per share basis. As a result, you will experience immediate and substantial dilution in net tangible book value per share in relation to the price that you paid for your shares. We expect the dilution as a result of the offering and the automatic conversion of all outstanding shares of our preferred stock and the automatic conversion of our outstanding convertible notes immediately prior to the closing of this offering, to be $9.51 per share to new investors purchasing our shares in this offering. In addition, you will experience further dilution to the extent that our shares are issued upon the exercise of any warrants or exercise of stock options under any stock incentive plans. See “Dilution” for a more complete description of how the value of your investment in our shares will be diluted upon completion of this offering.
We will incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, and particularly after we no longer qualify as an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur previously. The Sarbanes-Oxley Act of 2002 (“SOX”), the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of Nasdaq, and other applicable securities rules and regulations impose various requirements on U.S. reporting public companies, including the establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that these rules and regulations may make it more expensive for us to obtain director and officer liability insurance, which in turn could make it more difficult for us to attract and retain qualified senior management personnel or members for our board of directors. In addition, these rules and regulations are often subject to varying interpretations, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. Pursuant to Section 404 of SOX (“Section 404”), we will be required to furnish a report by our senior management on our internal control over financial reporting.
| -34- |
While we remain an emerging growth company, we will not be required to include an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To prepare for eventual compliance with Section 404, once we no longer qualify as an emerging growth company, we will be engaged in a process to document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we will need to continue to dedicate internal resources, potentially engage outside consultants and adopt a detailed work plan to assess and document the adequacy of internal control over financial reporting, continue steps to improve control processes as appropriate, validate through testing that controls are functioning as documented and implement a continuous reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that we will not be able to conclude, within the prescribed timeframe or at all, that our internal control over financial reporting is effective as required by Section 404.
We have identified material weaknesses in our internal control over financial reporting. Failure to maintain effective internal controls could cause our investors to lose confidence in us and adversely affect the market price of our common stock. If our internal controls are not effective, we may not be able to accurately report our financial results or prevent fraud.
Effective internal control over financial reporting is necessary for us to provide reliable financial reports in a timely manner. In connection with the audit of our financial statements as of and for the years ended December 31, 2019 and 2020 and the review of our financial statements for the three month periods ended March 31, 2020 and 2021, we identified material weaknesses in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. Specifically, we have determined that we lack a sufficient number of qualified accounting and financial reporting personnel with an appropriate level of knowledge, training and experience to address complex accounting issues, sufficient written policies and procedures for accounting and financial reporting in accordance with GAAP, and adequate management review controls. In addition, we have determined that our financial statement close process includes significant control gaps mainly driven by the small size of our accounting and finance staff and, as a result, a significant lack of appropriate segregation of duties.
The above material weaknesses could result in a misstatement of our account balances or disclosures that would result in a material misstatement of our annual or interim financial statements that would not be prevented or detected. To address the material weaknesses, we have implemented, and are continuing to implement, measures designed to improve internal control over financial reporting, including expanding our accounting and finance team to add additional qualified accounting and finance resources, which may include third party consultants, and new financial processes. We intend to continue to take steps to remediate the material weaknesses through the hiring or engagement of additional experienced accounting and financial reporting personnel, formalizing documentation of policies and procedures and further evolving the accounting processes, including implementing appropriate segregation of duties. We expect to incur additional costs to remediate these weaknesses, including personnel, consulting and other costs.
We may not be successful in implementing these changes or in developing other internal controls, which may undermine our ability to provide accurate, timely and reliable reports on our financial and operating results. Further, we will not be able to fully assess whether the steps we are taking will remediate the material weakness in our internal control over financial reporting until we have completed our implementation efforts and sufficient time passes in order to evaluate their effectiveness. In addition, until we remediate these weaknesses, or if we identify additional material weaknesses in our internal control over financial reporting, we may not detect errors on a timely basis and our financial statements may be materially misstated. Moreover, in the future we may engage in business transactions, such as acquisitions, reorganizations or implementation of new information systems that could negatively affect our internal control over financial reporting and result in material weaknesses.
| -35- |
If we identify new material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to assert that our internal control over financial reporting is effective, we may be late with the filing of our periodic reports, investors may lose confidence in the accuracy and completeness of our financial reports, and the market price of our common stock could be negatively affected. As a result of such failures, we could also become subject to investigations by the stock exchange on which our securities are listed, the SEC, or other regulatory authorities, and become subject to litigation from investors and stockholders, which could harm our reputation, financial condition or divert financial and management resources from our core business.
Holders of our Pre-funded Warrant and the Warrants will have no rights as shareholders until they acquire shares of our common stock, if ever, except as set forth in the Pre-funded Warrants or the Warrants.
If you acquire the Pre-funded Warrants or Warrants to purchase shares of our common stock in this offering, you will have no rights with respect to our common stock until you acquire shares of such common stock upon exercise of your Pre-funded Warrants or Warrants, except as set forth in the Pre-funded Warrants or the Warrants, respectively. Upon exercise of your Pre-funded Warrant or the Warrants, you will be entitled to exercise the rights of a holder of common stock only as to matters for which the record date occurs after the exercise date.
There is no public market for the Pre-funded Warrants or the Warrants being offered by us in this offering and an active trading market for the same is not expected to develop.
There is no established public trading market for the Pre-funded Warrants or the Warrants being offered in this offering, and we do not expect a market to develop. In addition, we do not intend to apply for any listing of the Pre-funded Warrants or Warrants offered hereby on the Nasdaq Capital Market or any other securities exchange or nationally recognized trading system. Without an active market, the liquidity of the Pre-funded Warrants and the Warrants will be severely limited.
We are an “emerging growth company,” and the reduced reporting requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act (“the JOBS Act”). For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including exemption from compliance with the auditor attestation requirements of Section 404, reduced disclosure obligations regarding executive compensation and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the closing of this offering, (b) in which we have total annual gross revenue of at least $1.07 billion or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock held by non-affiliates exceeds $700 million as of the end of our prior second fiscal quarter, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
In addition, under the JOBS Act, emerging growth companies may delay adopting new or revised accounting standards until such time as those standards apply to private companies. We may elect not to avail ourselves of this exemption from new or revised accounting standards and, therefore, may be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our share price may be more volatile.
Anti-takeover provisions contained in our certificate of incorporation and bylaws to be adopted upon the closing of this offering, as well as provisions of Delaware law, could impair a takeover attempt.
Our certificate of incorporation, bylaws and Delaware law contain or will contain provisions which could have the effect of rendering more difficult, delaying or preventing an acquisition deemed undesirable by our board of directors. Our corporate governance documents include or will include provisions:
| ● | classifying our board of directors into three classes; |
| ● | authorizing “blank check” preferred stock, which could be issued by our board of directors without stockholder approval and may contain voting, liquidation, dividend, and other rights superior to our common stock; |
| ● | limiting the liability of, and providing indemnification to, our directors and officers; |
| ● | limiting the ability of our stockholders to call and bring business before special meetings; |
| ● | requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our board of directors; |
| ● | controlling the procedures for the conduct and scheduling of board of directors and stockholder meetings; and |
| ● | providing our board of directors with the express power to postpone previously scheduled annual meetings and to cancel previously scheduled special meetings. |
These provisions, alone or together, could delay or prevent hostile takeovers and changes in control or changes in our management.
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation law, which prevents some stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of substantially all of our outstanding common stock.
Any provision of our certificate of incorporation, bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
Our certificate of incorporation, as amended, designates the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our certificate of incorporation requires that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will, to the fullest extent permitted by law, be the sole and exclusive forum for each of the following:
| ● | any derivative action or proceeding brought on our behalf; |
| ● | any action asserting a claim for breach of any fiduciary duty owed by any director, officer or other employee of ours to the Company or our stockholders, creditors or other constituents; |
| ● | any action asserting a claim against us or any director or officer of ours arising pursuant to, or a claim against us or any of our directors or officers, with respect to the interpretation or application of any provision of, the DGCL, our certificate of incorporation or bylaws; or |
| ● | any action asserting a claim governed by the internal affairs doctrine; |
provided, that, if and only if the Court of Chancery of the State of Delaware dismisses any of the foregoing actions for lack of subject matter jurisdiction, any such action or actions may be brought in another state court sitting in the State of Delaware.
The exclusive forum provision is limited to the extent permitted by law, and it will not apply to claims arising under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or for any other federal securities laws which provide for exclusive federal jurisdiction.
Furthermore, Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all such Securities Act actions. Accordingly, both state and federal courts have jurisdiction to entertain such claims. To prevent having to litigate claims in multiple jurisdictions and the threat of inconsistent or contrary rulings by different courts, among other considerations, our second amended and restated certificate of incorporation provides that the federal district courts of the United States of America will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. While the Delaware courts have determined that such choice of forum provisions are facially valid, a stockholder may nevertheless seek to bring such a claim arising under the Securities Act against us, our directors, officers, or other employees in a venue other than in the federal district courts of the United States of America. In such instance, we would expect to vigorously assert the validity and enforceability of the exclusive forum provisions of our second amended and restated certificate of incorporation.
Although we believe this provision benefits us by providing increased consistency in the application of Delaware law in the types of lawsuits to which it applies, this provision may limit or discourage a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find the choice of forum provision contained in our certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
We note that there is uncertainty as to whether a court would enforce the provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Although we believe this provision benefits us by providing increased consistency in the application of Delaware law in the types of lawsuits to which it applies, the provision may have the effect of discouraging lawsuits against our directors and officers.
| -36- |
CAUTIONARY STATEMENT CONCERNING FORWARD-LOOKING STATEMENTS
This prospectus contains certain “forward-looking statements” with respect to our business, financial condition, liquidity and results of operations. Words such as “anticipates,” “expects,” “intends,” “plans,” “predicts,” “believes,” “seeks,” “estimates,” “could,” “would,” “will,” “may,” “can,” “continue,” “potential,” “should,” and the negative of these terms or other comparable terminology often identify forward-looking statements. These forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties that could cause actual results to differ materially from the results contemplated by the forward-looking statements. See “Risk Factors” beginning on page 9.
| ● | our estimates regarding sufficiency of our cash resources, anticipated capital requirements and our need for additional financing; |
| ● | the commencement of clinical trials and the results and timing of those clinical trials; |
| ● | our ability to successfully commercialize our product candidates and generate revenue; |
| ● | submission and timing of applications for regulatory approval and approval thereof; |
| ● | our ability to successfully negotiate and enter into agreements with distribution, strategic and corporate partners; and |
| ● | our estimates of potential market opportunities and our ability to successfully realize these opportunities. |
Many of the important factors that will determine these results are beyond our ability to control or predict. You are cautioned not to put undue reliance on any forward-looking statements, which speak only as of the date of this prospectus. Except as otherwise required by law, we do not assume any obligation to publicly update or release any revisions to these forward-looking statements to reflect events or circumstances after such applicable date or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks we describe in the “Risk Factors” section hereof beginning on page 9 and in reports we will file from time to time with the SEC after the date of this prospectus.
We obtained the industry, statistical and market data included in this prospectus from our own internal estimates and research as well as from industry and general publications and research, surveys and studies conducted by third parties. All of the market data used in this prospectus involve a number of assumptions and limitations, and the sources of such data cannot guarantee the accuracy or completeness of such information. While we are not aware of any misstatements regarding the third-party information and we believe that each of these studies and publications is reliable, the industry in which we operate is subject to a high degree of uncertainty and risk due to a variety of important factors, including those described in the section titled “Risk Factors.” These and other factors could cause results to differ materially from those expressed in the estimates made by third parties and by us.
| -37- |
TRADEMARKS, SERVICE MARKS AND TRADE NAMES
We own or have rights to use a number of registered and common law trademarks, service marks and/or trade names in connection with our business in the United States and/or in certain foreign jurisdictions.
Solely for convenience, the trademarks, service marks, logos and trade names referred to in this prospectus are without the ® and ™ symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensors to these trademarks, service marks and trade names. This prospectus contains additional trademarks, service marks and trade names of others, which are the property of their respective owners. All trademarks, service marks and trade names appearing in this prospectus are, to our knowledge, the property of their respective owners. We do not intend our use or display of other companies’ trademarks, service marks, copyrights or trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
We have trademarks for the names RENOVORX, RENOVOGEM, RENOVOCATH, TAMP and DELIVERING THERAPY WHERE IT MATTERS. We have a trademark pending for RenovoTAMP.
| -38- |
We estimate that the net proceeds from this offering will be approximately $19.9 million ($23.0 million if the representative exercises its over-allotment option in full), after deducting the underwriting discount and estimated offering expenses payable by us, based on an assumed initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus).
We intend to use:
| ● | approximately $8.4 million to fund our ongoing TIGeR-PaC Phase 3 clinical trial of RenovoGem in LAPC patients through mid 2023, including our planned interim analysis in our TIGeR-PaC Phase 3 clinical trial which we expect will take place in the second half of 2022; | |
| ● | approximately $3.6 million to fund the launch of our planned BENEFICIAL Phase 2/3 clinical trial of RenovoGem in HCCA patients, which we expect to commence in the first half of 2022, through mid 2023; and | |
| ● | the balance for working capital and general corporate purposes, including the costs of operating as a public company. |
We estimate that our current capital resources, along with the net proceeds from this offering, will be sufficient to fund our operating expenses and capital expenditure requirements through mid 2023. However, the net proceeds from this offering, together with our current cash, will not be sufficient for us to fund the development of RenovoGem through regulatory approval, and we will need to raise additional capital to complete the development and commercialization of RenovoGem. At this time, we cannot predict with certainty the amount of capital needed to complete the development and commercialization of RenovoGem, but we anticipate seeking additional capital in the future to fund such capital needs through further equity offerings and/or debt borrowings. We cannot guarantee that we will be able to raise additional capital on reasonable terms or at all.
We may also use a portion of the net proceeds of this offering to acquire or invest in complementary businesses, products, or technologies, or to obtain the right to use such complementary technologies. We have no commitments with respect to any acquisition or investment, and we are not currently involved in any negotiations with respect to any such transaction.
As of the date of this prospectus, we cannot specify with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering. The amounts and timing of our actual expenditures will depend on numerous factors, including the status of our product development efforts, sales and marketing activities, technological advances, amount of cash generated or used by our operations and competition. Accordingly, our management will have broad discretion in the application of the net proceeds and investors will be relying on the judgment of our management regarding the application of the proceeds of this offering. Pending such use, we intend to invest the net proceeds in interest-bearing investment-grade securities or government securities.
| -39- |
We have never declared or paid cash dividends on our common stock. We currently intend to retain all available funds and any future earnings for use in the operation of our business and do not anticipate paying any cash dividends on our common stock in the near future. We may enter into credit agreements or other borrowing arrangements in the future that will restrict our ability to declare or pay cash dividends on our common stock.
Any future determination to declare dividends will be made at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements, contractual restrictions, general business conditions and other factors that our board of directors may deem relevant.
| -40- |
The following table sets forth our total capitalization as of March 31, 2021:
| ● | on an actual basis; | |
| ● | on a pro forma basis giving effect to the Preferred Stock Conversions and the Notes Conversions into 4,066,015 shares of common stock and warrants to purchase 397,901 shares of common stock immediately prior to the closing of this offering; | |
| ● | on a pro forma as adjusted basis giving further effect to the sale and issuance by us of 1,850,000 Units in this offering at the assumed initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. |
You should read this table together with our financial statements, the related notes included elsewhere in this prospectus and the information under “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| March 31, 2021 | ||||||||||||
| Actual | Pro forma | Pro forma as adjusted | ||||||||||
| (in thousands, except for share and per share data) | ||||||||||||
| Cash and cash equivalents | $ | 837 | $ | 837 | $ | 20,733 | ||||||
| Convertible note | $ | 2,843 | $ | - | - | |||||||
| Convertible preferred stock: | ||||||||||||
| Series A-1 Preferred Stock, $0.0001 par value, 3,542,669 shares authorized and 708,533 shares issued and outstanding, actual; no shares authorized or issued and outstanding, pro forma and pro forma as adjusted | $ | 639 | $ | - | - | |||||||
| Series A-2 Preferred Stock, $0.0001 par value, 3,546,095 shares authorized and 709,219 shares issued and outstanding, actual; no shares authorized or issued and outstanding, pro forma and pro forma as adjusted | 1,099 | - | - | |||||||||
| Series A-3 Preferred Stock, $0.0001 par value, 2,660,230 shares authorized and 532,046 shares issued and outstanding, actual; no shares authorized or issued and outstanding, pro forma and pro forma as adjusted | 2,166 | - | - | |||||||||
| Series B Preferred Stock, $0.0001 par value, 12,611,461 shares authorized and 1,585,671 shares issued and outstanding, actual; no shares authorized or issued and outstanding, pro forma and pro forma as adjusted | 8,547 | - | - | |||||||||
| Stockholders’ (deficit) equity: | ||||||||||||
Common stock, $0.0001 par value, 42,000,000 shares authorized actual, pro forma and pro forma as adjusted; 2,283,197, 6,308,404, and 8,199,212 shares issued and outstanding actual, pro forma and pro forma as adjusted, respectively |
1 | 3 | 2 | |||||||||
| Additional paid-in capital | 345 | 16,636 | 35,534 | |||||||||
| Accumulated deficit | (16,108 | ) | (16,108 | ) | (15,109 | ) | ||||||
| Total stockholders’ (deficit) equity | (15,762 | ) | 531 | 20,427 | ||||||||
| Total capitalization | $ | (3,311 | ) | $ | 531 | $ | 21,174 | |||||
Each $1.00 increase (decrease) in the assumed initial public offering price of $12.00 per Unit, the midpoint of the price range listed on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents and total stockholders’ equity by approximately $1.7 million, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and estimated offering expenses payable by us. Similarly, each increase (decrease) of 100,000 Units in the number of Units offered by us at the assumed initial public offering price of $12.00 per Unit, the midpoint of the price range listed on the cover page of this prospectus, would increase (decrease) the pro forma as adjusted amount of each of cash and cash equivalents, working capital, total assets and total stockholders’ equity by approximately $1.1 million.
| -41- |
If you purchase Units in this offering, your interest will be diluted to the extent of the difference between the initial public offering price per share and our net tangible book value per share after this offering. Dilution results from the fact that the assumed initial public offering price per share is substantially in excess of the net tangible book value per share attributable to the existing stockholders for our presently outstanding common stock.
Our net tangible book value (deficit) was approximately $(3.3) million or $(1.45) per share, as of March 31, 2021. Our net tangible book value represents the amount of our total tangible assets (which is calculated by subtracting net intangible assets, deferred tax assets, and prepaid offering expenses from our total assets), less the amount of our total liabilities.
Our pro forma net tangible book value as of March 31, 2021, was $0.5 million, or $0.08 per share of our common stock. Pro forma net tangible book value represents the amount of our total tangible assets less our total liabilities. Pro forma net tangible book value per share represents pro forma net tangible book value divided by the total number of shares outstanding as of March 31, 2021, after giving effect to the automatic conversion of all outstanding shares of our preferred stock and the automatic conversion of our outstanding convertible notes into 4,066,015 shares of common stock and warrants to purchase 397,901 shares of common stock immediately prior to the closing of this offering as if such conversions had occurred on March 31, 2021.
After giving further effect to the sale and issuance by us of 1,850,000 Units in this offering at the assumed initial public offering price of $12.00 per Unit, (the midpoint of the price range as set forth on the cover page of this prospectus), after deducting underwriting discounts and commissions and estimated offering expenses payable by us, assuming no sale of any Pre-funded Units and no exercise of the Warrants being sold in this offering and the warrants issuable upon the automatic conversion of our outstanding convertible notes, our pro forma as adjusted net tangible book value as of March 31, 2021 would have been $20.4 million or $2.49 per share. This represents an immediate increase in pro forma net tangible book value of $2.41 per share to our existing stockholders, and an immediate dilution in pro forma as adjusted net tangible book value of $9.51 per share to new investors. The following table illustrates this per share dilution:
| Assumed initial public offering price per share | $ | 12.00 | ||||||
| Pro forma net tangible book value per share as of March 31, 2021 | $ | 0.08 | ||||||
| Increase in pro forma as adjusted net tangible book value per share attributable to this offering | $ | 2.41 | ||||||
| Pro forma as adjusted net tangible book value per share, after this offering | 2.49 | |||||||
| Dilution per share to new investors in this offering | $ | 9.51 | ||||||
If the representative’s over-allotment option is exercised in full, our pro forma as adjusted net tangible book value per share after this offering would be $2.78 and dilution per share to new investors purchasing Units in this offering would be $9.22 at the assumed initial public offering price of $12.00 per Unit, (the midpoint of the price range as set forth on the cover page of this prospectus) and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
The pro forma as adjusted information discussed above is illustrative only. Our pro forma as adjusted net tangible book value following the completion of this offering is subject to adjustment based on the actual initial public offering price of our Units and other terms of this offering determined at pricing.
The following tables summarize the differences between our existing stockholders and the investors purchasing Units in this offering with respect to the number of Units purchased from us, the total consideration paid and the average price per share paid, at the assumed initial public offering price of $12.00 per Unit, (the midpoint of the price range as set forth on the cover page of this prospectus) before deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
| -42- |
| Shares Purchased | Total Consideration | Average Price | ||||||||||||||||||
| Number | Percent | Amount | Percent | per Share | ||||||||||||||||
| Common Stock | 2,283,197 | 29.8 | $ | 45,000 | 0.1 | $ | - | |||||||||||||
| Series A-1 Preferred Stock | 708,533 | 9.2 | 660,000 | 1.9 | $ | 0.95 | ||||||||||||||
| Series A-2 Preferred Stock | 709,219 | 9.2 | 1,150,000 | 3.3 | $ | 1.60 | ||||||||||||||
| Series A-3 Preferred Stock | 532,046 | 6.9 | 2,100,000 | 6.1 | $ | 3.95 | ||||||||||||||
| Series B Preferred Stock | 1,585,671 | 20.7 | 8,308,000 | 24.1 | $ | 5.25 | ||||||||||||||
| New investors | 1,850,000 | 24.2 | 22,200,000 | 64.5 | $ | 12.00 | ||||||||||||||
| 7,668,666 | 100.0 | $ | 34,463,000 | 100.0 | ||||||||||||||||
The table above assumes no exercise of the representative’s over-allotment option. If the representative’s over-allotment option is exercised in full, upon completion of this offering, the percentage of common stock held by existing holders would be reduced to 73.2%, and the percentage of common stock held by new investors purchasing common stock in this offering would be increased to 26.8%.
The number of shares outstanding is based on shares outstanding as of March 31, 2021 and except as noted above excludes the following:
| ● | 951,133 shares of our common stock issuable upon the exercise of outstanding options with a weighted-average exercise price of $0.40 per share; | |
| ● | 708,533 shares of Series A-1 Preferred Stock, 709,219 shares of Series A-2 Preferred Stock, 532,046 shares of Series A-3 Preferred Stock and 1,585,671 shares of Series B Preferred Stock which will convert into 708,533, 709,219, 532,046 and 1,585,671 shares of common stock, respectively, upon the closing of this offering (the “Preferred Stock Conversions”); | |
| ● | 530,546 shares of common stock and 397,901 shares issuable upon exercise of warrants with an exercise price of $14.40 per share assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) to be issued upon conversion of the 2020 Convertible Notes and 2021 Convertible Notes upon the closing of this offering (the “Note Conversions”);
| |
| ● | up to an additional 10,368 shares of our common stock issuable under our 2013 Equity Incentive Plan; and | |
| ● | 161,875 shares of our common stock underlying the underwriter’s warrant to be issued to the representative of the underwriters in connection with this offering. |
| -43- |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Unless the context otherwise requires, all references in this section to the “Company,” “we,” “us, or “our” refer to the business of RenovoRx, Inc. You should read the following discussion and analysis of our financial condition and results of operations together with the “Selected Financial Data” section of this prospectus and our financial statements and the related notes appearing elsewhere in this prospectus. This discussion contains forward-looking statements that reflect our plans, estimates, and beliefs that involve risks and uncertainties. As a result of many factors, such as those set forth under the “Risk Factors” and “Special Note Regarding Forward-Looking Statements” sections and elsewhere in this proxy statement/prospectus, our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
We are a clinical-stage biopharmaceutical company focused on developing therapies for the local treatment of solid tumors and conducting a Phase 3 registrational trial for our lead product candidate RenovoGem™. Our therapy platform, RenovoRx Trans-Arterial Micro-Perfusion, or RenovoTAMP™ utilizes approved chemotherapeutics with validated mechanisms of action and well-established safety and side effect profiles, with the goal of increasing their efficacy, improving their safety, and widening their therapeutic window. RenovoTAMP combines our patented FDA cleared delivery system, RenovoCath®, with small molecule chemotherapeutic agents that can be forced across the vessel wall using pressure, targeting these anti-cancer drugs locally to the solid tumors. While we anticipate investigating other chemotherapeutic agents for intra-arterial delivery via RenovoTAMP, our clinical work to date has focused on gemcitabine, which is a generic drug. Our first product candidate, RenovoGem, is a drug and device combination consisting of intra-arterial gemcitabine and RenovoCath. FDA has determined that RenovoGem will be regulated as, and if approved we expect will be reimbursed as, a new oncology drug product. We have secured FDA Orphan Drug Designation for RenovoGem in our first two indications: pancreatic cancer and cholangiocarcinoma (bile duct cancer, or CCA). We have completed our RR1 Phase 1/2 and RR2 observational registry studies, with 20 and 25 patients respectively, in locally advanced pancreatic cancer, or LAPC. These studies demonstrated a median overall survival of 27.9 months in patients treated with RenovoGem and radiation. Based on previous large randomized clinical trials, the expected survival of LAPC patients is 12-15 months in patients receiving only intravenous (IV) systemic chemotherapy or IV chemotherapy plus radiation (which are both considered standard of care). Unlike the randomized trials that established these standard-of-care results, our RR1 and RR2 clinical trials did not prospectively control the standard of care therapy received prior to RenovoTAMP. Based on FDA safety review of our Phase 1/2 study the FDA allowed us to proceed to evaluate RenovoGem within our Phase 3 registration Investigational New Drug, or IND, clinical trial. Our Phase 3 trial is over 40% enrolled as of July 15, 2021 and we expect to report data from a planned interim data readout in the second half of 2022. We intend to evaluate RenovoGem in a second indication in a Phase 2/3 trial in hilar CCA (cancer that occurs in the bile ducts that lead out of the liver and join with the gallbladder, also called extrahepatic cholangiocarcinoma, or HCCA). We plan to propose the trial to the FDA and potentially launch in the first half of 2022. In addition, we may evaluate RenovoGem in other indications, potentially including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma (an aggressive type of cancer that can occur in the brain or spinal cord). To date, we have used gemcitabine, but in the future we may develop other chemotherapeutic agents for intra-arterial delivery via RenovoCath.
Since our inception, we have devoted substantially all of our efforts to developing our cancer therapy platform and product candidates, raising capital and organizing and staffing our company. To date, we have funded our operations with proceeds from the issuance of convertible preferred stock and convertible notes. Through March 31, 2021, we received total net proceeds of $15.0 million of which $11.8 million was from the issuance of convertible preferred stock, $3.0 million from convertible notes and $140,000 from a loan under the Paycheck Protection Program, or PPP. In April 2021, the Company entered into a series of convertible note agreements with certain current and new investors to provide an aggregate $2.0 million in cash proceeds.
We have incurred significant operating losses and generated negative cash flows from operations since our inception. As of March 31, 2021, we had cash and cash equivalents of $837,000. We also had net losses of $1.1 million in each of the three months ended March 31, 2020 and March 31, 2021. As of March 31, 2021, we had an accumulated deficit of $16.1 million. We expect to continue to incur significant expenses, increasing operating losses and negative cash flows from operations in 2021 and for the foreseeable future. We do not expect to generate revenues from product sales unless and until we successfully complete development and obtain regulatory approval for one or more product candidates. We expect that our expenses will increase substantially in connection with our ongoing research and development activities, particularly as we:
| ● | Advance clinical development of RenovoGem and our platform technology by continuing to enroll patients in our ongoing TIGeR-PaC Phase 3 clinical trial, expanding the number of clinical trials including our planned clinical trial in HCCA, and advancing RenovoGem through preclinical and clinical development in additional indications; | |
| ● | Hire additional research, development, and engineering personnel; | |
| ● | Maintain, expand, enforce, defend, and protect our intellectual property portfolio; and | |
| ● | Expand our operational, financial and management systems and increase personnel, including personnel to support our clinical development, manufacturing and commercialization efforts and our operations as a public company. |
In addition to the variables described above, if and when any of our product candidates successfully complete development, we will incur substantial additional costs associated with establishing a sales, marketing, medical affairs and distribution infrastructure to commercialize products for which we may obtain marketing approval, regulatory filings, marketing approval, and post-marketing requirements, in addition to other commercial costs. We cannot reasonably estimate these costs at this time.
As a result, we will need significant additional funding to support our continuing operations. Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through equity issuances, debt financings and collaborations, licenses or other similar arrangements. We currently have no credit facility or committed sources of capital. To the extent that we raise additional capital through the future sale of equity or debt, the ownership interests of our stockholders will be diluted and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our existing common stockholders. If we raise additional funds through the issuance of debt securities, these securities could contain covenants that would restrict our operations. We may require additional capital beyond our currently anticipated amounts and additional capital may not be available on reasonable terms, or at all. If we raise additional funds through collaboration arrangements or other strategic transactions in the future, we may have to relinquish valuable rights to our technologies or future revenue streams or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate development or future commercialization efforts.
Without giving effect to the anticipated net proceeds from this offering, we expect that our existing cash on hand will be insufficient to fund our operating expenses and capital expenditures for at least one year from the date of our financial statements. We will need to raise additional capital to finance our operations, which cannot be assured. We have concluded that this circumstance raises substantial doubt about our ability to continue as a going concern for at least one year from the date of issuance date of our financial statements. See Note 2 to our audited financial statements and Note 2 to our unaudited interim condensed financial statements, each included elsewhere in this prospectus, for additional information on our assessment. Similarly, our independent registered public accounting firm included an explanatory paragraph in its report on our audited financial statements as of and for the year ended December 31, 2020, describing the existence of substantial doubt about our ability to continue as a going concern.
| -44- |
Impact of COVID-19
In December 2019, a novel strain of coronavirus, which causes the disease known as COVID-19, was reported to have surfaced in Wuhan, China. Since then, COVID-19 has spread globally. In March 2020, the World Health Organization declared the COVID-19 outbreak a pandemic. The ongoing COVID-19 global and national health emergency has caused significant disruption in the international and U.S. economies and financial markets. The spread of COVID-19 has caused illness, quarantines, cancellation of events and travel, business and school shutdowns, reduction in business activity and financial transactions, labor shortages, supply chain interruptions and overall economic and financial market instability.
In response to public health directives and orders and to help minimize the risk of the virus to employees, we have taken precautionary measures, including implementing work-from home policies for certain employees. The COVID-19 global pandemic also has negatively affected, and we expect will continue to negatively affect, our clinical studies. For example, we have faced challenges in conducting our clinical trials, including recruiting subjects and accommodating patient visits. Additionally, our service providers and their operations may be disrupted, temporarily closed or experience worker or supply shortages, which could result in additional disruptions or delays in shipments of purchased materials or the continued development of our product candidates. To date, we have not suffered material supply chain disruptions.
We are not able to estimate the duration of the pandemic and the potential impact on our business. As the global pandemic of COVID-19 continues to evolve, it could result in significant long-term disruption of global financial markets, reducing our ability to raise additional capital when needed and on acceptable terms, if at all, which could negatively affect our liquidity. The extent to which the COVID-19 pandemic impacts our clinical development and regulatory efforts will depend on future developments that are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the outbreak, travel restrictions, quarantines and social distancing requirements in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the virus. We will continue to monitor the COVID-19 situation closely.
In April 2020, we received $140,000 in funding as a promissory note under the PPP. This promissory note was subsequently forgiven in February 2021. See Note 6 to the unaudited condensed interim financial statements appearing at the end of this prospectus for additional information.
Components of Our Results of Operations
Revenue
We have not generated any revenue from product sales and do not expect to generate any revenue from the sale of products for several years, if at all. If our development efforts for our current or future product candidates are successful and result in marketing approval or collaboration or license agreements with third parties, we may generate revenue in the future from a combination of product sales or payments from collaboration or license agreements that we may enter into with third parties.
| -45- |
Operating Expenses
Research and Development
Research and development expenses consist of costs related to the research and development of our platform technology. Clinical trial costs are a significant component of research and development expenses and include costs associated with third-party contractors. We outsource a substantial portion of our clinical trial activities, utilizing the service of third-party clinical trial sites and contract research organizations to assist us with the execution of our clinical trials. In addition, we have FDA 510(k) clearance for the RenovoCath delivery device, which comprises part of the RenovoGem product. Accordingly, we were able to charge our clinical trial sites for the RenovoCath delivery device. To date, proceeds from clinical trial sites have been adequate to cover our direct costs of manufacturing the RenovoCath delivery devices for which they have paid. Any proceeds we receive from the clinical trial sites for their purchase of the RenovoCath delivery device are offset against our research and development expenses. We expect our research and development expenses to increase for the foreseeable future as we continue the development of our product candidates and enroll subjects in our ongoing clinical trials, initiate future clinical trials and pursue regulatory approval of our product candidates. It is difficult to predict with any certainty the duration and completion costs of our current or future clinical trials of our product candidates or if, when or to what extent we will achieve regulatory approval and generate revenue from the commercialization and sale of our product candidates. The duration, costs and timing of clinical trials and other development of our product candidates will depend on a variety of factors, including uncertainties in clinical trial enrollment, timing and extent of future clinical trials, development of new product candidates and significant and changing government regulation. We may never succeed in achieving regulatory approval for any of our product candidates.
Our research and development expenses include:
| ● | expenses incurred under agreements with clinical trial sites, contract research organizations, and consultants that conduct our clinical trials, | |
| ● | costs of acquiring and developing clinical trial materials, | |
| ● | personnel costs, including salaries, benefits, bonuses, and stock-based compensation for employees engaged in preclinical and clinical research and development, | |
| ● | costs related to compliance with regulatory requirements, | |
| ● | travel expenses, and | |
| ● | facilities, insurance, and other allocated expenses which include direct and allocated expenses for rent, insurance and other general overhead costs. |
Research and development costs are expensed as incurred. Costs for certain development activities, such as clinical trials and preclinical studies, are recognized based on evaluation of progress to completion of specific tasks using data such as subject enrollment, clinical site activations or information provided to us by third party vendors.
Due to the impact of the COVID-19 pandemic and work-from-home policies and other operational limitations mandated by federal, state, and local governments as a result of the pandemic, certain of our research and development activities have been delayed and may be further delayed until such operational limitations are lifted.
General and Administrative
General and administrative expenses consist of salaries, benefits, and stock-based compensation for personnel in executive, finance and administrative functions, professional services and associated costs related to accounting, tax, audit, legal, intellectual property and other matters, consulting costs, conferences, travel and allocated expenses for rent, insurance and other general overhead costs. Following the listing of our common stock on Nasdaq, we expect to continue to incur additional expenses as a result of operating as a public company, including costs to comply with the rules and regulations of the Securities and Exchange Commission, or SEC, and Nasdaq listing standards and increased expenses in the areas of insurance, professional services and investor relations. As a result, we expect our general and administrative expenses to increase for the foreseeable future. General and administrative expenses are expensed as incurred.
| -46- |
Other Income (Expense), Net
Interest Income (Expense) Net
Interest income (expense), net consists of interest income earned from our cash and cash equivalents net of interest expense. Interest expense consists of charges relating to the amortization of the debt discount and debt issuance costs as well as interest on amounts outstanding on our convertible notes.
Other Income (Expense), Net
Other income (expense), net comprise primarily investment expenses and foreign currency exchange gains and losses. We expect our foreign currency exchange gains and losses to continue to fluctuate in the future due to changes in foreign currency exchange rates.
Gain on Loan Extinguishment
Gain on loan extinguishment resulted from the forgiveness and cancellation of the Company’s PPP loan.
Income Tax Expense
We account for income taxes using the asset and liability method. Under this method, deferred income tax assets and liabilities are recorded based on the estimated future tax effects of differences between the financial statement and income tax basis of existing assets and liabilities. Deferred income tax assets and liabilities are recorded net and classified as noncurrent on the balance sheets. A valuation allowance is provided against our deferred income tax assets when their realization is not reasonably assured.
We are subject to income taxes in the federal and state jurisdictions. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. In accordance with the authoritative guidance on accounting for uncertainty in income taxes, we recognize tax liabilities for uncertain tax positions when it is more likely than not that a tax position will not be sustained upon examination and settlement with various taxing authorities. Liabilities for uncertain tax positions are measured based upon the largest amount of benefit that is more-likely-than-not (greater than 50%) of being realized upon settlement. Our policy is to recognize interest and/or penalties related to income tax matters in income tax expense.
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, was enacted. The CARES Act includes several significant provisions for corporations, including the usage of net operating losses, interest deductions and payroll benefits. Corporate taxpayers may carryback net operating losses, or NOLs, originating during 2018 through 2020 for up to five years.
Results of Operations
The following table summarizes the significant components of our results of operations for the periods presented (in thousands):
| Years Ended December 31, | Three Months Ended March 31, | |||||||||||||||
| 2019 | 2020 | 2020 | 2021 | |||||||||||||
| Statements of Operations Data: | (unaudited) | |||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 2,997 | $ | 2,386 | $ | 805 | $ | 635 | ||||||||
| General and administrative | 899 | 818 | 248 | 418 | ||||||||||||
| Total operating expenses | 3,896 | 3,204 | 1,053 | 1,053 | ||||||||||||
| Loss from operations | (3,896 | ) | (3,204 | ) | (1,053 | ) | (1,053 | ) | ||||||||
| Other income (expense), net | ||||||||||||||||
| Interest income (expense), net | 63 | (587 | ) | 2 | (230 | ) | ||||||||||
| Other income (expense), net | 2 | (7 | ) | - | (5 | ) | ||||||||||
| Loss on change in fair value of warrant liability | (8 | ) | - | - | - | |||||||||||
| Gain on loan extinguishment | - | - | - | 140 | ||||||||||||
| Total other income (expense), net | 57 | (594 | ) | 2 | (95 | ) | ||||||||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | $ | (1,051 | ) | $ | (1,148 | ) | ||||
| -47- |
Comparison of the Three Months Ended March 31, 2020 and 2021
The following table summarizes our results of operations for the three months ended March 31, 2020 and 2021 (in thousands, except percentages):
| Three Months Ended March 31, | Change | |||||||||||||||
| 2020 | 2021 | $ | % | |||||||||||||
| (unaudited) | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 805 | $ | 635 | $ | (170 | ) | (21 | )% | |||||||
| General and administrative | 248 | 418 | 170 | 69 | % | |||||||||||
| Total operating expenses | 1,053 | 1,053 | - | - | ||||||||||||
| Loss from operations | (1,053 | ) | (1,053 | ) | - | - | ||||||||||
| Other income (expense), net | ||||||||||||||||
| Interest income (expense), net | 2 | (230 | ) | (232 | ) | (116 | )% | |||||||||
| Other expense, net | - | (5 | ) | (5 | ) | 100 | % | |||||||||
| Gain on loan extinguishment | - | 140 | 140 | 100 | % | |||||||||||
| Total other income (expense), net | 2 | (95 | ) | (97 | ) | (4,850 | )% | |||||||||
| Net loss | $ | (1,051 | ) | $ | (1,148 | ) | $ | 97 | 9 | % | ||||||
Research and Development
Research and development expenses decreased $170,000, or 21%, in the three months ended March 31, 2021 compared to the three months ended March 31, 2020. This decrease was due a reduction of $50,000 in clinical development spending related to travel and off-site meetings due to the COVID-19 pandemic, a $42,000 reduction in expenses for preclinical studies due to the completion of sample analysis activities, and a $12,000 reduction in regulatory expenses due to lower support for European filings and vendor audits. Also contributing to the decrease in research and development expense was an increase of $45,000 in offsetting clinical supply usage and corresponding vendor payments from clinical trial sites. Vendor payments for delivery devices represents the cash payment made by vendors for the RenovoCath delivery devices used in clinical trials.
General and Administrative Expenses
General and administrative expenses increased $170,000, or 69%, in the three months ended March 31, 2021 compared to the three months ended March 31, 2020. This increase was due to an increase in professional expenses of $285,000 primarily as a result of preparing for this offering offset by a reduction in spending on legal costs of $68,000 due to reduced spending on intellectual property and a decrease of $37,000 in executive-level clinical support costs.
Interest Income (Expense), Net
The interest expense for the three-month period ended March 31, 2021 comprises both the stated interest on the note of 5% per annum, or $37,000, as well as the amortization of the discount and debt issuance costs associated with the 2020 Convertible Notes of $193,000. Interest income for the three months ended March 31, 2020 was de minimis.
Other Expense, Net
Other expense, net for the three months ended March 31, 2021 was de minimis. There was no other expense for the three months ended March 31, 2020.
On March 1, 2021, the Company entered into an amendment to the 2020 Convertible Notes which extends the maturity date of the 2020 Convertible Notes from March 31, 2021 to October 30, 2021 and provides for the conversion of the 2020 Convertible Notes into shares of the Company’s common stock upon a Qualified Financing. No other terms to the 2020 Convertible Notes were amended. This amendment was accounted for as a troubled debt restructuring pursuant to FASB ASC Topic 470-60, “Troubled Debt Restructurings by Debtors”. As the future undiscounted cash flows of the 2020 Convertible Notes were greater than their carrying amount, the carrying amount was not adjusted and no gain was recognized as a result of the modification of terms.
Gain on Loan Extinguishment
The gain on loan extinguishment of $140,000 for the three months ended March 31, 2021 resulted from the forgiveness and cancellation of the Company’s PPP loan.
| -48- |
Comparison of the Years Ended December 31, 2019 and 2020
| Year Ended December 31, | Change | |||||||||||||||
| 2019 | 2020 | $ | % | |||||||||||||
| (in thousands, except percentages) | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 2,997 | $ | 2,386 | $ | (611 | ) | (20 | )% | |||||||
| General and administrative | 899 | 818 | (81 | ) | (9 | )% | ||||||||||
| Total operating expenses | 3,896 | 3,204 | (692 | ) | (18 | )% | ||||||||||
| Loss from operations | (3,896 | ) | (3,204 | ) | 692 | 18 | % | |||||||||
| Other income (expense), net | ||||||||||||||||
| Interest income (expense), net | 63 | (587 | ) | (650 | ) | (1,032 | )% | |||||||||
| Other income (expense), net | 2 | (7 | ) | (9 | ) | (450 | )% | |||||||||
| Loss on change in fair value of warrant liability | (8 | ) | - | 8 | (100 | )% | ||||||||||
| Total other income (expense), net | 57 | (594 | ) | (651 | ) | (1,142 | )% | |||||||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | $ | 41 | 1 | % | ||||||
Research and Development
The following table summarizes our research and development expenses, personnel and our outsourced spending by functional area (in thousands):
| Year Ended December 31, | Increase/ | |||||||||||
| 2019 | 2020 | (Decrease) | ||||||||||
| Clinical development | $ | 2,021 | $ | 1,518 | $ | (503 | ) | |||||
| Vendor payments for delivery devices | (113 | ) | (241 | ) | (128 | ) | ||||||
| Preclinical research and development | 416 | 238 | (178 | ) | ||||||||
| Regulatory | 247 | 339 | 92 | |||||||||
| Personnel | 426 | 532 | 106 | |||||||||
| Total research and development expenses | $ | 2,997 | $ | 2,386 | $ | (611 | ) | |||||
Research and development expenses were $3.0 million for the year ended December 31, 2019 compared to $2.4 million for the year ended December 31, 2020, a net decrease of $0.6 million. This net decrease can be attributed to a number of factors. The decrease in clinical development costs of $0.5 million for the year ended December 31, 2020 is primarily due to the initiation of the European portion of our clinical trial and startup of clinical trial sites in the U.S. in 2019 that did not recur in 2020. Vendor payments for delivery devices represents the cash payment made by vendors for the RenovoCath delivery devices used in clinical trials. To date, proceeds from clinical trial sites have been adequate to cover our direct costs of manufacturing the RenovoCath delivery devices for which they have paid. Preclinical research and development decreased by $0.2 million for the year ended December 31, 2020 due to higher costs associated with early-stage product development in 2019. Regulatory expenses increased by approximately $0.1 million for the year ended December 31, 2020 due to a higher level of support required for communications and filings with the FDA. Our personnel-related expenses increased by $0.1 million in the year ended December 31, 2020 driven primarily by increased staffing costs to support our ongoing clinical trials.
General and Administrative Expenses
The following table summarizes our general and administrative expenses (in thousands):
| Year Ended December 31, | Increase/ | |||||||||||
| 2019 | 2020 | (Decrease) | ||||||||||
| Personnel | $ | 560 | $ | 576 | $ | 16 | ||||||
| Legal fees | 201 | 170 | (31 | ) | ||||||||
| Professional services and other | 138 | 72 | (66 | ) | ||||||||
| Total general and administrative | ||||||||||||
| expenses | $ | 899 | $ | 818 | $ | (81 | ) | |||||
| -49- |
General and administrative expenses were $0.9 million for the year ended December 31, 2019 compared to $0.8 million for the year ended December 31, 2020, a decrease of $0.1 million. The decrease in general and administrative expenses was primarily attributable to reductions in expenses related to legal, professional services, consulting, recruiting, medical conferences and communications.
Interest Income (Expense), Net (in thousands)
| Year Ended December 31, | Increase/ | |||||||||||
| 2019 | 2020 | (Decrease) | ||||||||||
| Interest income | $ | 63 | $ | 3 | $ | (60 | ) | |||||
| Interest expense | - | (590 | ) | (590 | ) | |||||||
| Interest income (expense), net | $ | 63 | (587 | ) | $ | (650 | ) | |||||
Interest income (expense), net decreased by $0.7 million from the year ended December 31, 2019 to the year ended December 31, 2020. The decrease in interest income is due to interest earned on lower cash balances and lower interest rates throughout 2020 compared to 2019. Interest expense increased by $0.6 million during the year ended December 31, 2020 primarily as a result of the 2020 Convertible Notes we issued in 2020. Interest expense comprises both the stated interest on the note of 5% per annum or $0.1 million as well as the amortization of the discount and debt issuance costs associated with the 2020 Convertible Notes of $0.5 million.
Other Expense, Net
Other expense, net increased by $9,000 from the year ended December 31, 2019 to the year ended December 31, 2020, a de minimis amount
Loss on Change in Fair Value of Warrant Liability
Loss on change in fair value of warrant liability decreased by $8,000 from the year ended December 31, 2019 to the year ended December 31, 2020, a de minimis amount.
Liquidity and Capital Resources
For the years ended December 31, 2019 and December 31, 2020, our net losses were $3.8 million in each of those years, and for the three months ended March 31, 2020 and March 31, 2021 we incurred net losses of $1.1 million in each of those periods. As of March 31, 2021, we had an accumulated deficit of $16.1 million. We expect to incur additional losses and increased operating expenses in future periods. Since our inception, our primary sources of liquidity have been the issuance of convertible preferred stock and convertible notes.
As of December 31, 2020 and March 31, 2021, we had $1.8 million and $837,000 in cash and cash equivalents, respectively. During the three months ended March 31, 2021, we used $1.0 million of cash in operations. Our primary requirements for liquidity have been to fund our clinical trial activity and general corporate and working capital needs. Our cash and cash equivalents at these dates was primarily from $3.0 million in net proceeds from convertible notes and a promissory note with Silicon Valley Bank for proceeds of $140,000 pursuant to the PPP under the CARES Act. In February 2021, we received notification and confirmation from Silicon Valley Bank that our PPP loan including related accrued interest has been forgiven in its entirety by the U.S. Small Business Administration and automatically cancelled.
| -50- |
Based on our planned operations, we expect that the proceeds from this offering plus our current cash and cash equivalents will be sufficient to fund our operations for at least 12 months after the date of this prospectus. After this offering, we intend to raise additional capital through equity offerings and/or debt financings. Adequate funding may not be available to us on acceptable terms, or at all. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our clinical trials or other operations. If any of these events occur, our ability to achieve our operational goals would be adversely affected. Our future capital requirements and the adequacy of available funds will depend on many factors, including those described in “Risk Factors.” Depending on the severity and direct impact of these factors on us, we may be unable to secure additional financing to meet our operating requirements on terms favorable to us, or at all.
Sources of Liquidity
Since our inception, we have not generated any revenue from product sales and we have incurred significant operating losses. We do not have any products that have achieved regulatory marketing approval and we do not expect to generate revenue from sales of any product candidates for several years, if ever.
To date, we have funded our operations primarily through the issuance and sale of convertible preferred stock and debt. From our inception through March 31, 2021, we had raised net cash proceeds of $14.9 million from the issuance and sale of our convertible preferred stock and convertible notes. We also received $140,000 from a loan under the PPP which was forgiven in February 2021. As of March 31, 2021, we had cash and cash equivalents of $837,000 and an accumulated deficit of $16.1 million. In April 2021, we entered into a series of convertible note agreements with certain existing and new investors to provide an aggregate $2.0 million in cash proceeds.
Cash Flows
Our primary uses of cash are to fund our operations including research and development and general and administrative expenses. We will continue to incur operating losses in the future and expect that our research and development and general and administrative expenses will continue to increase as we continue our research and development efforts with respect to clinical development of our product candidates and further develop our platform. We expect that we will use a substantial portion of the net proceeds of this offering, in combination with our existing cash and cash equivalents, for these purposes and for the increased expenses associated with being a public company. Cash used to fund operating expenses is impacted by the timing of when we pay expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.
| -51- |
The following table summarizes our cash flows for the periods indicated (in thousands):
| Years Ended December 31, | Three Months Ended March 31, | |||||||||||||||
| 2019 | 2020 | 2020 | 2021 | |||||||||||||
| (unaudited) | ||||||||||||||||
| Net cash provided by (used in): | ||||||||||||||||
| Operating activities | $ | (3,350 | ) | $ | (3,528 | ) | $ | (1,039 | ) | $ | (992 | ) | ||||
| Investing activities | (1 | ) | - | - | - | |||||||||||
| Financing activities | - | 3,199 | 1,312 | 34 | ||||||||||||
| (Decrease) increase in cash and cash equivalents | $ | (3,351 | ) | $ | (329 | ) | $ | 273 | $ | (958 | ) | |||||
Cash Used in Operating Activities
Net cash used in operating activities for the three months ended March 31, 2020 and 2021 was approximately $1.0 million in each of the periods, consisting primarily of our net losses in both periods of $1.1 million.
Net cash used in operating activities for the year ended December 31, 2019 of $3.4 million was primarily attributable to a $3.8 million net loss, partially offset by a net increase in operating assets and liabilities of $0.4 million.
Net cash used in operating activities for the year ended December 31, 2020 of $3.5 million was primarily attributable to a $3.8 million net loss and a decrease in operating assets and liabilities of $0.2 million, partially offset by amortization of debt issuance costs and amortization of debt discount of $0.5 million.
Cash Used in Investing Activities
There were no investing activities for the three months ended March 31, 2020 and March 31, 2021.
Net cash used in investing activities during the year ended December 31, 2019 was insignificant. There were no investing activities during the year ended December 31, 2020.
Cash Provided by Financing Activities
Net cash provided by financing activities in the three months ended March 31, 2020 was $1.3 million, consisting primarily of $1.3 million in proceeds from the issuance of convertible notes.
Net cash provided by financing in the three months ended March 31, 2021 was $34,000 and consisted of proceeds from the exercise of stock options.
We had no financing activities in 2019.
Net cash provided by financing activities during the year ended December 31, 2020 of $3.2 million was primarily attributable to $3.0 million in proceeds from the issuance of the 2020 Convertible Notes and $140,000 in proceeds from the PPP loan.
Contractual Obligations and Other Commitments
The following table summarizes our contractual obligations as of December 31, 2020 (in thousands):
| Payments due by period | ||||||||||||||||
| Total | Less than 1 year | 1 to 3 years | 3 to 5 years | |||||||||||||
| 2020 Convertible Notes | $ | 3,038 | $ | 3,038 | $ | - | $ | - | ||||||||
| PPP loan | 140 | 117 | 23 | - | ||||||||||||
| Total | $ | 3,178 | $ | 3,155 | $ | 23 | $ | - | ||||||||
| -52- |
On February 6, 2021, the Company received notification and confirmation from Silicon Valley Bank that its PPP loan totaling $140,000 has been forgiven in its entirety by the U.S. Small Business Administration and automatically cancelled. There have been no other significant changes in our contractual obligations as of March 31, 2021 compared to December 31, 2020.
Critical Accounting Policies and Significant Judgments and Estimates
The accompanying management’s discussion and analysis of our financial condition and results of operations are based upon our financial statements and the related disclosures, which have been prepared in accordance with accounting principles generally accepted in the United States or GAAP. The preparation of these financial statements requires us to make estimates, assumptions and judgments that affect the reported amounts in our financial statements and accompanying notes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. To the extent that there are material differences between these estimates and actual results, our future financial statement presentation, financial condition, results of operations and cash flows will be affected. While our significant accounting policies are described in the notes to our financial statements included elsewhere in this prospectus, we believe that the following critical accounting policies are most important to understanding and evaluating our reported financial results.
A critical accounting policy is defined as one that is both material to the presentation of our financial statements and requires management to make difficult, subjective, or complex judgments that could have a material effect on our financial condition and results of operations. Specifically, critical accounting estimates have the following attributes: (i) we are required to make assumptions about matters that are highly uncertain at the time of the estimate; and (ii) different estimates we could reasonably have used, or changes in the estimate that are reasonably likely to occur, would have a material effect on our financial condition or results of operations.
We believe the following policies to be the most critical to an understanding of our financial condition and results of operations because they require us to make estimates, assumptions and judgments about matters that are inherently uncertain.
Clinical Trial Expenses
We make payments in connection with clinical trials under contracts with clinical trial sites and contract research organizations that support conducting and managing clinical trials. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee, unit price or on a time and materials basis. A portion of the obligation to make payments under these contracts depends on factors such as the successful enrollment or treatment of patients or the completion of other clinical trial milestones.
Expenses related to clinical trials are accrued based on estimates and/or representations from service providers regarding work performed, including actual level of patient enrollment, completion of patient studies and progress of the clinical trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts we are obligated to pay under clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), the accruals are adjusted accordingly. Revisions to contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain.
Stock-Based Compensation
We calculate the fair value of stock options using the Black-Scholes option pricing model, which incorporates various assumptions including assumptions including the fair value of our common stock, volatility, expected life, and risk-free interest rate. Compensation related to service-based awards is recognized starting on the grant date on a straight-line basis over the vesting period, which is generally four years.
| -53- |
Determining the grant date fair value of options using the Black-Scholes option pricing model requires management to make assumptions and judgments. If any of the assumptions used in the Black-Scholes model change significantly, stock-based compensation for future awards may differ materially compared with the awards granted previously. The assumptions and estimates are as follows:
Fair Value of Common Stock—Given the absence of a public trading market, our Board of Directors considered numerous objective and subjective factors to determine the fair value of our common stock at each grant date. These factors included, but were not limited to: (i) contemporaneous third-party valuations of common stock; (ii) the prices for preferred stock sold to outside investors; (iii) the rights and preferences of preferred stock relative to common stock; (iv) the lack of marketability of our common stock; (v) developments in the business; and (vi) the likelihood of achieving a liquidity event, such as an IPO or sale of the business, given prevailing market conditions. The methodology to determine the fair value of our common stock included estimating the fair value of the enterprise using the “backsolve” method, which is a market approach that assigns an implied enterprise value by accounting for all share class rights and preferences based on the latest round of financing. The total equity value implied was then applied in the context of an option pricing model to determine the value of each class of our shares.
Following this offering, we will rely on the closing price of our common stock as reported on the date of grant to determine the fair value of our common stock, as shares of our common stock will be traded in the public market.
Expected Term—The expected term represents the period that the stock-based awards are expected to be outstanding. We determine the expected term using the simplified method. The simplified method deems the term to be the average of the time-to-vesting and the contractual life of the options. For stock options granted to non-employees, the expected term equals the remaining contractual term of the option from the vesting date.
Expected Volatility—Given the absence of a public trading market, the expected volatility was estimated by taking the average historic price volatility for industry peers, consisting of several public companies in our industry that are either similar in size, stage, or financial leverage, over a period equivalent to the expected term of the awards.
Risk-Free Interest Rate—The risk-free interest rate is calculated using the average of the published interest rates of U.S. Treasury zero-coupon issues with maturities that are commensurate with the expected term.
Dividend Rate—The dividend yield assumption is zero as we have no plans to make dividend payments.
Convertible Instruments and Embedded Derivatives
We evaluate all of our agreements to determine whether such instruments have derivatives or contain features that qualify as embedded derivatives. We account for certain redemption features that are associated with the terms of convertible notes as liabilities at fair value and adjusts the instruments to their fair value at the end of each reporting period. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in other income (expense), net in the statements of operations. Derivative instrument liabilities are classified in the balance sheets as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. As of December 31, 2020 and March 31, 2021, our only derivative financial instrument was related to the 2020 Convertible Notes, which contained certain redemptive features.
| -54- |
Emerging Growth Company and Smaller Reporting Company Status
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012, as amended, or the JOBS Act. Under the JOBS Act, companies have extended transition periods available for complying with new or revised accounting standards. We have elected this exemption to delay adopting new or revised accounting standards. We will remain an emerging growth company until the earlier of (1) December 31, 2026, (2) the last day of the fiscal year in which we have total annual gross revenues of at least $1.07 billion, (3) the date on which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, or (4) the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period. An emerging growth company may take advantage of specified reduced reporting requirements and is relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an emerging growth company,
| ● | we may present only two years of audited financial statements, plus unaudited condensed financial statements for any interim period, and related Management’s Discussion and Analysis of Financial Condition and Results of Operations in this prospectus; | |
| ● | we may avail ourselves of the exemption from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act; | |
| ● | we may provide reduced disclosure about our executive compensation arrangements; and | |
| ● | we do not require stockholder non-binding advisory votes on executive compensation or golden parachute arrangements. |
We have elected to take advantage of certain of the reduced disclosure obligations in the registration statement of which this prospectus is a part and may elect to take advantage of other reduced reporting requirements in future filings. As a result, the information that we provide to our stockholders may be different than you might receive from other public reporting companies in which you hold equity interests.
We are also a “smaller reporting company,” meaning that the market value of our stock held by non-affiliates plus the proposed aggregate amount of gross proceeds to us as a result of this offering is less than $700.0 million and our annual revenue is less than $100.0 million during the most recently completed fiscal year. We may continue to be a smaller reporting company after this offering if either (1) the market value of our stock held by nonaffiliates is less than $250.0 million or (2) our annual revenue is less than $100.0 million during the most recently completed fiscal year and the market value of our stock held by non-affiliates is less than $700.0 million. If we are a smaller reporting company at the time we cease to be an emerging growth company, we may continue to rely on exemptions from certain disclosure requirements that are available to smaller reporting companies. Specifically, as a smaller reporting company we may choose to present only the two most recent fiscal years of audited financial statements in our Annual Report on Form 10-K and, similar to emerging growth companies, smaller reporting companies have reduced disclosure obligations regarding executive compensation
Recently Issued and Adopted Accounting Pronouncements
See Note 2 to our audited financial statements included elsewhere in this prospectus for more information.
Off-Balance Sheet Arrangements
During the periods presented, we did not have any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Quantitative and Qualitative Disclosures about Market Risk
We are exposed to market risks in the ordinary course of our business. These risks primarily include interest rate risks, foreign currency exchange, risks and inflation risks. Periodically, we maintain deposits in accredited financial institutions in excess of federally insured limits. We deposit our cash in financial institutions that we believe have high credit quality and have not experienced any losses on such accounts and do not believe we are exposed to any unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
| -55- |
Interest Rate Risk
Our cash and cash equivalents consisted primarily of cash on hand at December 31, 2020 and March 31, 2021. The fair value of our cash and cash equivalents would not be significantly affected by either an increase or decrease in interest rates.
Our exposure to risks related to interest rates is minimal. The interest rate for our 2020 Convertible Note is a fixed rate.
Foreign Currency Exchange Risk
We are not currently exposed to significant market risk related to changes in foreign currency exchange rates. However, we have contracted with and may continue to contract with vendors such as contract research organizations and clinical trial sites that are located in Europe. We may be subject to fluctuations in foreign currency rates in connection with certain of these agreements. Transactions denominated in currencies other than the U.S. dollar are recorded based on exchange rates at the time such transactions arise. While we have not engaged in hedging our foreign currency transactions to date, we may evaluate the costs and benefits of initiating such a program and may in the future, hedge selected significant transactions denominated in currencies other than the U.S. dollar as we expand our clinical trial sites globally.
Inflation Risk
Inflation generally affects us by increasing our labor and clinical trial costs. We do not believe that inflation had a material effect on our business, financial condition or results of operations during the year ended December 31, 2020 and the three-month period ended March 31, 2021.
Internal Control Over Financial Reporting
Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with generally accepted accounting principles in the United States, or GAAP. Under standards established by the Public Company Accounting Oversight Board, or PCAOB, a deficiency in internal control over financial reporting exists when the design or operation of a control does not allow management or personnel, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. The PCAOB defines a material weakness as a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of annual or interim financial statements will not be prevented or detected on a timely basis.
In preparation for our initial public offering, we identified material weaknesses in our internal control over financial reporting related to our control environment. Specifically, we have determined that we lack a sufficient number of qualified accounting and financial reporting personnel with an appropriate level of knowledge, training and experience to address complex accounting issues, sufficient written policies and procedures for accounting and financial reporting in accordance with GAAP, and adequate management review controls. In addition, we have determined that our financial statement close process includes significant control gaps mainly driven by the small size of our accounting and finance staff and, as a result, a significant lack of appropriate segregation of duties.
To address these material weaknesses, we have implemented, and are continuing to implement, measures designed to improve internal controls over financial reporting, including expanding our accounting and finance team to add additional qualified accounting and finance resources, which may include third party consultants, and have implemented new financial processes. We intend to continue to take steps to remediate the material weaknesses through the hiring or engagement of additional experienced accounting and financial reporting personnel, formalizing documentation of policies and procedures and further evolving the accounting processes, including implementing appropriate segregation of duties.
The process of designing and implementing an effective accounting and financial reporting system is a continuous effort that requires us to anticipate and react to changes in our business and the economic and regulatory environments and to expend significant resources to maintain an accounting and financial reporting system that is adequate to satisfy our reporting obligations. As we continue to evaluate and take actions to improve our internal control over financial reporting, we may determine to take additional actions to address control deficiencies or determine to modify certain of the remediation measures described above. We cannot assure you that the measures we have taken to date, or any measures we may take in the future, will be sufficient to remediate the material weakness we have identified or avoid potential future material weaknesses.
| -56- |
Overview
We are a clinical-stage biopharmaceutical company focused on developing therapies for the local treatment of solid tumors and conducting a Phase 3 registrational trial for our lead product candidate RenovoGem™. Our therapy platform, RenovoRx Trans-Arterial Micro-Perfusion, or RenovoTAMP™ utilizes approved chemotherapeutics with validated mechanisms of action and well-established safety and side effect profiles, with the goal of increasing their efficacy, improving their safety, and widening their therapeutic window. RenovoTAMP combines our patented FDA cleared delivery system, RenovoCath®, with small molecule chemotherapeutic agents that can be forced across the vessel wall using pressure, targeting these anti-cancer drugs locally to the solid tumors. While we anticipate investigating other chemotherapeutic agents for intra-arterial delivery via RenovoTAMP, our clinical work to date has focused on gemcitabine, which is a generic drug. Our first product candidate, RenovoGem, is a drug and device combination consisting of intra-arterial gemcitabine and RenovoCath. FDA has determined that RenovoGem will be regulated as, and if approved we expect will be reimbursed as, a new oncology drug product. We have secured FDA Orphan Drug Designation for RenovoGem in our first two indications: pancreatic cancer and cholangiocarcinoma (bile duct cancer, or CCA). We have completed our RR1 Phase 1/2 and RR2 observational registry studies, with 20 and 25 patients respectively, in locally advanced pancreatic cancer, or LAPC. These studies demonstrated a median overall survival of 27.9 months in patients treated with RenovoGem and radiation. Based on previous large randomized clinical trials, the expected survival of LAPC patients is 12-15 months in patients receiving only intravenous (IV) systemic chemotherapy or IV chemotherapy plus radiation (which are both considered standard of care). Unlike the randomized trials that established these standard-of-care results, our RR1 and RR2 clinical trials did not prospectively control the standard of care therapy received prior to RenovoTAMP. Based on FDA safety review of our Phase 1/2 study the FDA allowed us to proceed to evaluate RenovoGem within our Phase 3 registration Investigational New Drug, or IND, clinical trial. Our Phase 3 trial is over 40% enrolled as of July 15, 2021 and we expect to report data from a planned interim data readout in the second half of 2022. We intend to evaluate RenovoGem in a second indication in a Phase 2/3 trial in hilar CCA (cancer that occurs in the bile ducts that lead out of the liver and join with the gallbladder, also called extrahepatic cholangiocarcinoma, or HCCA). We plan to propose the trial to the FDA and potentially launch in the first half of 2022. In addition, we may evaluate RenovoGem in other indications, potentially including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma (an aggressive type of cancer that can occur in the brain or spinal cord). To date, we have used gemcitabine, but in the future we may develop other chemotherapeutic agents for intra-arterial delivery via RenovoCath.
Our RenovoTAMP therapy platform is focused on optimizing drug concentration in solid tumors using approved small molecule chemotherapeutics that enable physicians to isolate segments of the vascular anatomy closest to tumors and force chemotherapy across the blood vessel wall to bathe these difficult-to-reach tumors in chemotherapy. More specifically, our patented approach combines local delivery via our patented RenovoCath delivery system utilizing pressure to force small molecule chemotherapy into the tumor tissue with pre-treatment of the local blood vessels and tissue with standard-of-care radiation therapy to decrease chemotherapy washout. We believe there are many advantages to our approach:
| ● | Application of Approved Small Molecule Chemotherapeutic Agents: We use approved small molecule chemotherapeutic agents such as gemcitabine. | |
| ● | Targeted Approach: With our approach, we have demonstrated in our clinical studies up to 100 times higher local drug concentration compared to systemic chemotherapy. We believe our approach decreases systemic exposure and improves patient outcomes. | |
| ● | Delivery Method Independent of Tumor Vascularity: We invented a novel combination platform and delivery system to deliver small molecule chemotherapeutic agents in solid tumors resistant to systemic chemotherapy due to lack of tumor blood vessels or tumor feeders. | |
| ● | Broad Application for Solid Tumor Indications: Our platform is not restricted to a single small molecule chemotherapeutic agent or solid tumor type. As such, our platform and delivery system may be applied for use in additional solid tumor indications, including in solid tumors without identifiable tumor feeders. |
Our lead product candidate, RenovoGem, is a combination of gemcitabine and our patented delivery system, RenovoCath, and is regulated by the FDA as a novel oncology drug product. Our RenovoTAMP platform therapy utilizes pressure mediated delivery of the small molecule gemcitabine across the arterial wall to bathe the pancreatic tumor tissue in 120mL of saline with 1,000mg/m2 of the drug over a 20-minute delivery time (approximately a total of 1,500-2,000mg of drug dependent upon patient Body Surface Area). RenovoCath is an adjustable double balloon catheter designed to isolate the proximal and distal vessel and adjust the distance between the balloons to exclude any branching blood vessel offshoots.
| -57- |
While the field of oncology has seen progress in treating a handful of deadly cancers over the last few decades, there is a common limitation in chemotherapy: enhanced dosing of the drug to impact the tumor while minimizing systemic toxicity. The characteristics of the vasculature, within and surrounding the tumor, can be a limiting step in this goal. For example, LAPC and HCCA are more difficult to treat due to the lack of blood vessels that feed these tumors, making it difficult to expose tumors to chemotherapy, which is typically delivered intravenously. Trans-arterial chemoembolization (TACE) is an established first line therapy for solid tumors. A key component of this approach is to identify and isolate vessels feeding the tumor, known as tumor feeders. However, in patients with pancreatic cancer, no tumor feeder vessels are visible during angiography. In the absence of visible tumor feeders, we can introduce drugs directly across the arterial wall into the surrounding tissue via pressurized diffusion.
We are currently evaluating RenovoGem in patients with LAPC in our TIGeR-PaC Phase 3 trial at 28 US and Belgian sites. The trial is designed to enroll 340 subjects. As of July 15, 2021, 145 patients were enrolled, accounting for over 40% of expected total enrollment. A planned interim data readout is expected during the second half of 2022. We have secured Orphan Drug Designation for RenovoGem; which would provide us with seven years of exclusivity to market intra-arterial use of gemcitabine for LAPC upon NDA approval.
In addition, we intend to evaluate RenovoGem in patients with HCCA, and we have secured FDA Orphan Drug Designation for the broader CCA indication. We intend to potentially pursue additional indications including locally advanced lung cancer, locally advanced uterine tumors and glioblastoma.
For our initial indication, LAPC, we have completed two studies. We launched RR1, our first-in-human, dose escalation, Phase 1/2 safety study in May 2015 to evaluate our RenovoTAMP platform by delivering intra-arterial gemcitabine via our patented RenovoCath delivery system. In this safety study, 20 patients with a diagnosis of Stage 3 pancreatic cancer were enrolled. After completion of enrollment and demonstration of an early survival efficacy signal in this study, we launched our RR2 observational registry study in June 2016 to examine the tolerability and initial efficacy of the RenovoTAMP procedure. A combination analysis of these two studies demonstrated that survival in “all comers” (n=31) receiving at least one cycle (two treatments over one month) was 29% at two years. Looking at the prior-radiation therapy subset (n=10), 24-month survival was 60% with a median overall survival (mOS) of 27.9 months. This compares favorably to IV chemotherapy, with 24-month survival of 12%, and to chemotherapy + radiation with 24-month survival of 5% and mOS of 12-15 months as demonstrated in historical studies.
We intend to submit our proposed Phase 2/3 clinical trial to evaluate RenovoGem in HCCA, BENEFICIAL, to the FDA as part of a pre-IND submission in the fourth quarter of 2021 and to launch the clinical trial in the first half of 2022. Gemcitabine has been considered standard of care for several solid tumors, and the drug’s anti-cancer tumor effects are well profiled. We intend to explore applications of our RenovoTAMP platform in additional indications including locally advanced lung cancer, locally advanced uterine cancer, and glioblastoma. We have completed and presented data on a lung cancer application in pre-clinical studies, and additional pre-clinical experiments in lung cancer may be conducted.
We are using gemcitabine in our initial anti-cancer product candidate, RenovoGem, however, multiple small molecule therapeutics are compatible with our RenovoTAMP platform. We intend to opportunistically develop additional anti-cancer product candidates using small molecule therapeutics.
Our management team, Board of Directors, and Scientific Advisors provide us with expertise across multiple sectors to drive success through clinical development and subsequent commercialization of our novel therapy platform. Our Chief Executive Officer, Shaun Bagai, has extensive experience running clinical trials and launching, creating, and developing new markets for novel therapies at Trans Vascular, Medtronic, Ardian, and HeartFlow. Dr. Ramtin Agah, our Co-Founder and Chief Medical Officer, is a practicing cardiovascular specialist who has 20 years of research experience in vascular biology and disease in both academia and industry. Our Board of Directors includes a wide range of public and private company management and Board experience including drug/device combination and oncology experience. Clinical advisors include experts in surgical oncology, interventional radiology, radiation oncology, and medical oncology. Dr. Daniel Von Hoff, a medical oncologist, was instrumental as the Principal Investigator who brought to market standard of care therapies for pancreatic cancer. Dr. Mike Pishvaian, also a medical oncologist, has extensive experience in running oncology studies and is an Associate sociate Professor, and Department of Oncology Director of the Gastrointestinal, Developmental Therapeutics, and Clinical Research Programs at the NCR Kimmel Cancer Center at Sibley Memorial Hospital Johns Hopkins University School of Medicine. Dr. Pishvaian is the Principal Investigator/Global Study Chair of our TIGeR-PaC Phase 3 study. Dr. Peter Muscarella is a surgical oncologist and the Director of Pancreatic Surgery, General Surgery Site Director, Weiler Hospital Associate Program Director, and General Surgery Residency Training Program at Montefiore Medical Center, Bronx, NY. Dr. Karyn Goodman serves as our Radiation Monitor for our TIGeR-PaC Phase 3 study and Professor and Vice Chair of Clinical Research, Department of Radiation Oncology at the Icahn School of Medicine at Mount Sinai, and Associate Director of Clinical Research at the Tisch Cancer Institute at Mount Sinai. We have two interventional radiology scientific advisors: Dr. Reza Malek, Neurointerventional Radiologist at Minimally Invasive Surgical Solutions and Dr. Jacob Cynamon, Professor of Clinical Radiology of the Albert Einstein College of Medicine, Chief of the Division of Vascular and Interventional Radiology, and Program Director of the Vascular and Interventional Radiology Fellowship Program at the Montefiore Medical Center, Bronx, NY.
| -58- |
Research and Development Pipeline
Our portfolio of cancer therapies is based on our RenovoTAMP therapy platform. Our current pipeline is summarized below:
RenovoGem Product Pipeline Addresses Multiple Indications
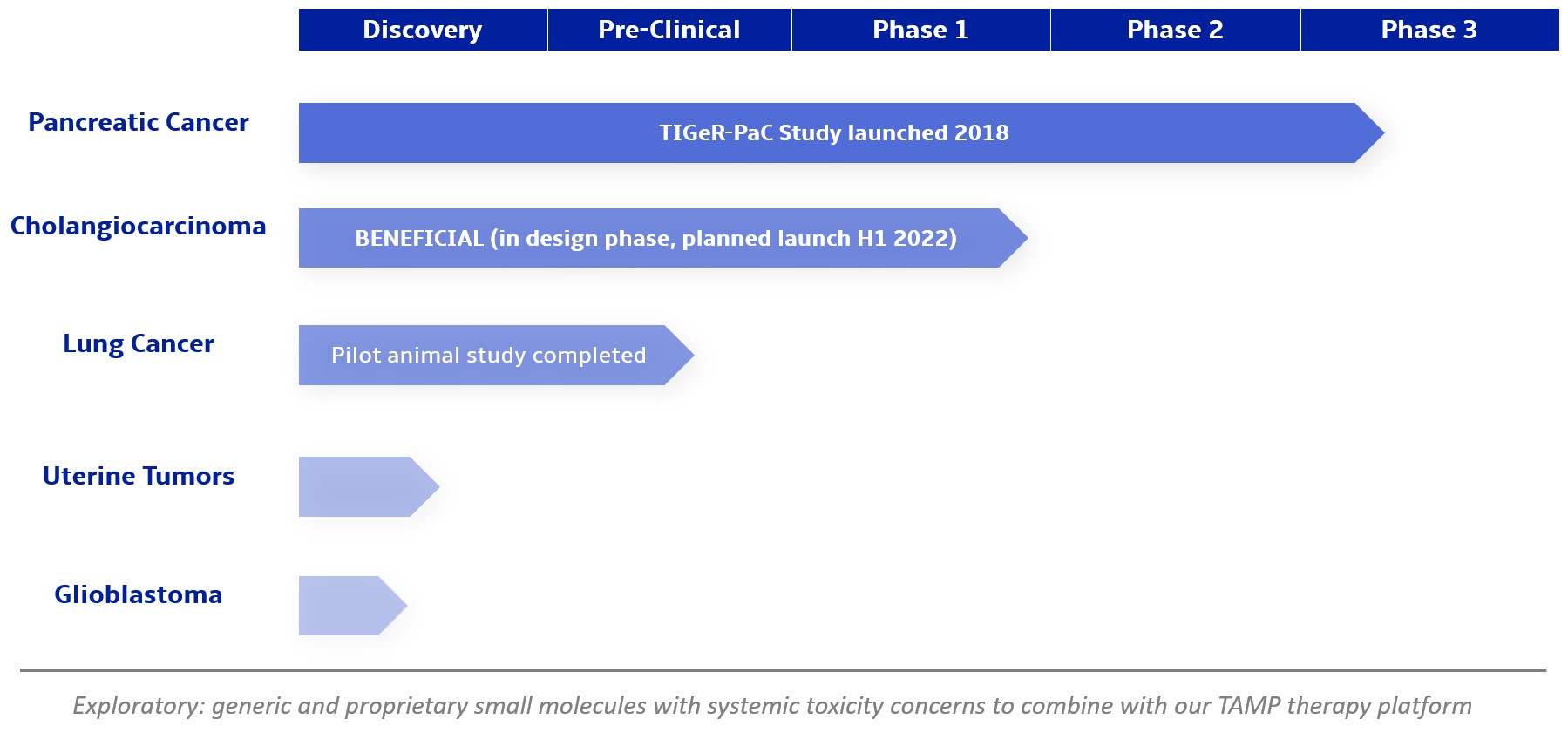
Figure 1 RenovoGem Clinical Pipeline detailing our potential portfolio of cancer therapies based on our RenovoTAMP therapy platform.
Strategy
RenovoGem is a combination of intra-arterial gemcitabine and our patented delivery system, RenovoCath, and is regulated by the FDA as a novel oncology drug product. Our near-term goal is to develop RenovoGem utilizing our RenovoTAMP platform to address the unmet medical needs of LAPC and HCCA patients. We intend to broaden application of our platform, by exploring additional cancer indications including locally advanced lung cancer, locally advanced uterine cancer, and glioblastoma. Our long-term goal is to expand applications of our RenovoTAMP platform beyond RenovoGem by acquiring or licensing other small molecule therapies to continue to address unmet medical needs of cancer patients. To achieve our near-term and long-term goals, we intend to pursue the following strategies:
| ● | Advance our lead product candidate, RenovoGem for use in our first indication, LAPC. In our Phase 1/2 study, we demonstrated a median survival of approximately 28 months from diagnosis which compares favorably to 12-15 months in historical controls in locally advanced pancreatic cancer patients. We are currently conducting our TIGeR-PaC Phase 3 clinical trial with a target enrollment of 340 patients. As of July 15, 2021, we have enrolled 145 patients. We expect to receive an interim data readout on the primary endpoint of overall survival by the second half of 2022. |
| -59- |
| ● | Advance RenovoGem for use in our second indication, HCCA. We have secured Orphan Drug Designation for the broader CCA indication and intend to launch a Phase 2/3 trial for the treatment of HCCA in the first half of 2022. | |
| ● | Expand RenovoGem for use in additional solid tumors indications. We plan to potentially launch IND studies to explore the application of RenovoGem for the treatment of locally advanced lung cancer and other solid tumor indications with data on the use of systemic or intravenously administered gemcitabine such as locally advanced uterine tumors and glioblastoma. | |
| ● | Use RenovoTAMP with different chemotherapeutic agents. Our delivery system, RenovoCath, can be used to deliver almost any small molecule therapeutic agent to solid tumors. RenovoTAMP has the potential to overcome limitations of systemic toxicity by local delivery of small molecule therapeutic agents to the tumor. We may to use our platform to develop products with drugs that are available generically or we may enter strategic collaborations to access other companies’ proprietary drugs. | |
| ● | Explore collaborations with biotechnology and pharmaceutical companies. We have exclusive global development and commercialization rights for RenovoGem and RenovoCath including issued patents on methods of RenovoTAMP for all indications that we may pursue. While we may develop these products independently, we may also enter strategic relationships with biotechnology or pharmaceutical companies to advance our product candidates. |
Our Strengths
| ● | Solid tumor targeting via local therapy. Our platform has the potential to efficiently target locally advanced solid tumors. Many solid tumors cannot be surgically removed and are difficult to treat. Our innovative therapy platform delivers anti-cancer drugs directly to the tumor and does not rely on the existence of extensive vasculature also known as tumor feeders. We believe that RenovoTAMP, which locally delivers directly to the tumor a drug that is standard of care in systemic administration, is a promising approach to improve outcomes in locally advanced solid tumors. | |
| ● | Preliminary data indicate that our approach is feasible and well-tolerated with promising survival results. Our Phase 1/2 data demonstrated that RenovoGem via the RenovoTAMP therapy platform is well tolerated with multiple survival signals. | |
| ● | Pipeline with broad utility. We believe that the flexibility of our platform combined with our exclusive global development and commercialization rights, gives us the ability to grow our product pipeline by targeting a broad range of solid tumor indications and by using additional chemotherapeutic agents. Furthermore, our platform has been used throughout the duration of our clinical trials with demonstrated adoption of the RenovoTAMP technique by physicians. |
Current Treatments and Limitations of Approaches
Currently, solid tumors are typically treated using one or a combination of treatment modalities: surgery, radiation, and pharmacological therapies (chemotherapy). For solid tumors, when possible, surgical resection of the tumor is the most frequently employed treatment approach. If the tumor is detected at an early stage and is localized to the affected organ, surgery may be an effective and potentially curative treatment of the entire tumor is removed. In most cases, surgery is initially completed prior to commencing additional treatment approaches. However, multiple solid tumor types, including LAPC and HCCA are diagnosed at stages that preclude surgery as a treatment approach. In many of these circumstances, the tumor has grown into adjacent anatomical structures making the surgery difficult or impossible.
| -60- |
Intra-venous (IV), or systemic chemotherapy, is considered standard of care for most solid tumors, but limitations include less than acceptable efficacy, systemic toxicities, and side effects.
For the treatment of localized solid tumors, TACE is an established first line therapy. Many companies have used this approach to treat tumors of the liver, uterus, and prostate. Many solid tumors have a dedicated blood supply; small blood vessels, called tumor feeders, that branch off of larger native arteries and terminate in the tumors to provide nutrition to the tumors. A key aspect of TACE is to identify and isolate these tumor feeders during x-ray angiography and then deliver the desired therapy including chemotherapy and embolic agents. In patients LAPC, no tumor feeder vessels are visible during angiography due to the avascular (lack of blood vessels) nature of these tumors. This limitation has rendered TACE ineffective in the treatment of patients with LAPC, HCCA, and with a subset of other solid tumors. The limitations of TACE translate to low survival rates in these tumor subtypes despite attempts with novel therapies including targeted therapies that address a specific molecule on one or more tumor types and immuno-oncology treatment approaches, which harness the body’s immune system to treat cancer. For example, early studies targeting immunotherapies in pancreatic cancer have limited success due to the inability of immune cells to penetrate the tumor tissue.
Our Platform: RenovoTAMP
In Figure 2 below, the panel on the left depicts visualization of the actual tumor, hepatocellular carcinoma, or primary liver cancer, under x-ray angiography as dye injected through the arteries reaches the tumor itself. Further, visible tumor feeders can be reached by simple end-hole catheters to deliver targeted therapy to these liver tumors. The panel on the right demonstrates the typical lack of tumor feeders to the pancreatic tumor. Given the lack of tumor feeders, the dye does not reach the tumor, rendering the tumor “invisible” under x-ray angiography. Rather one can see the native/large blood vessel being “pinched” by the tumor.
Tumors in Liver are Different from Hypovascular Tumors in the Pancreas
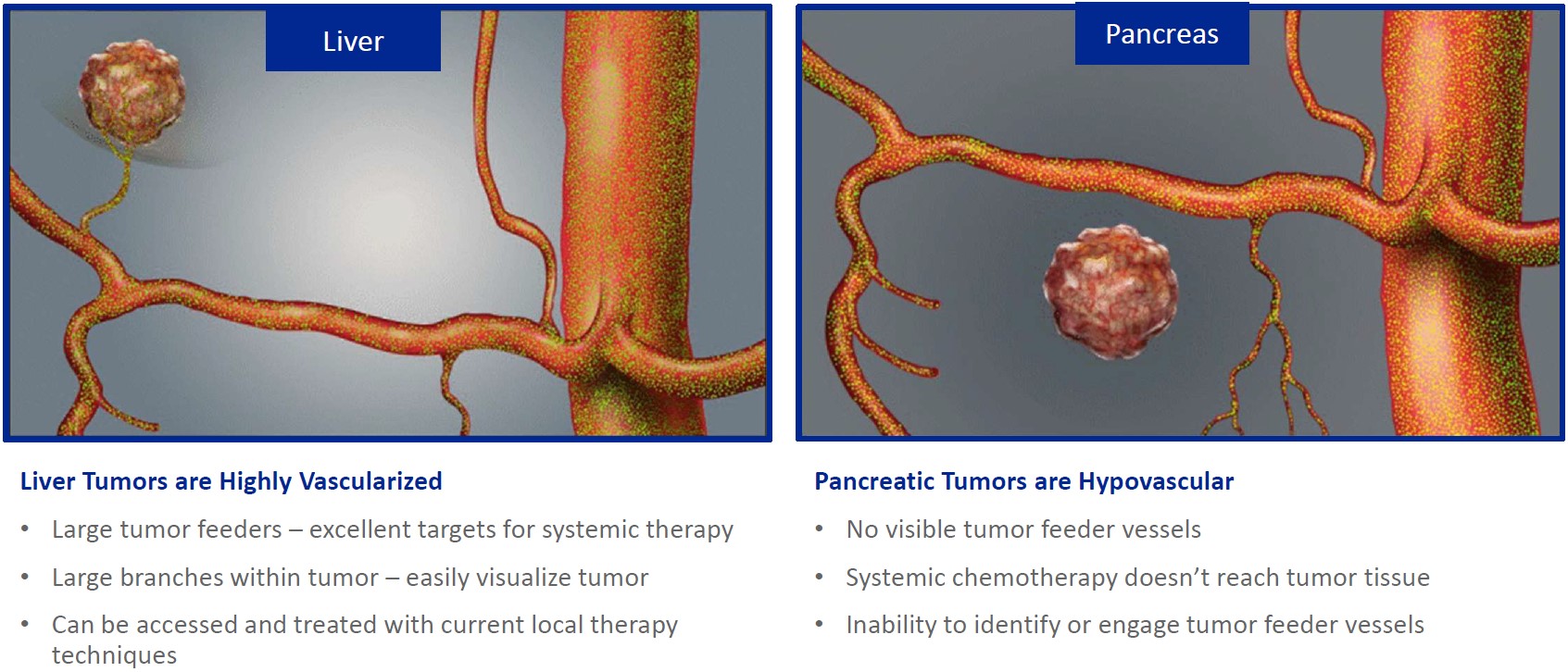
Figure 2 Liver tumors are highly vascularized versus pancreas tumors that are avascular, therefore systemic chemotherapy and local TACE approaches can more easily deliver chemotherapy to liver tumors than to pancreatic tumors. The panel on the left depicts tumor feeders originating from the native liver arteries. Under x-ray angiography, dye injected in the native liver arteries drain into the tumor feeders and clearly define the tumor. The panel on the right depicts the “invisible” pancreatic tumor. From a dye injection through the arteries near the pancreas, one can only visualize a narrowing of the artery due to the tumor pinching on the artery, but tumor feeders, and thus, the tumor itself are not visible.
| -61- |
In 2009 our founder Dr. Ramtin Agah, an experienced interventional cardiologist with a degree in biomedical engineering, invented the concept for RenovoTAMP as a way to deliver chemotherapy locally to treat poorly vascularized tumors. He joined forces with Kamran Najmabadi, who brought significant medical device engineering experience, to found RenovoRx in 2009. We contracted with a contract manufacturer to prototype and manufacture its RenovoCath delivery devices. We received our first FDA 510(k) clearance for RenovoCath in 2014, a second clearance to use the RenovoCath for infusion of chemotherapy agents in 2017, and a further clearance for use with a power-injector in 2019. We are evaluating RenovoGem under an IND and expect that RenovoGem, if approved, will be regulated and reimbursed as a drug.
To overcome the limitation of lack of tumor feeder vessels, we explored a different approach to locally deliver anti-cancer drugs. By isolating a section of the blood vessel and then increasing the intravascular pressure in the isolated segment we can introduce chemotherapy directly across the arterial wall into the surrounding tissue via pressurized diffusion or Trans-Arterial Micro-Perfusion (TAMP®). To isolate the vessel and create this pressure gradient, we developed a patented adjustable double balloon catheter to occlude the proximal and distal part of the vessel (RenovoCath). Using this technique with the RenovoCath delivery system we were able to validate our hypothesis by demonstrating >99% gemcitabine pressurized diffusion in explanted pig aorta and iliac arteries across the arterial wall in the absence of feeder vessels. This mechanism of action was further supported via exploratory acute animal studies measuring the pressure gradient within the artery during double balloon occlusion. Figure 3 demonstrates the change in intra-arterial pressure over time from catheter introduction to balloon inflation, start of infusion, and pressure plateau when chemotherapy is forced out of vessel. These changes in pressure are a result of pressure declining as the first balloon blocks blood inflow and then rising as the drug is administered and fills up the space between the balloons.
RenovoCath Pressurizes Isolated Vessel Segment, Allowing RenovoTAMP
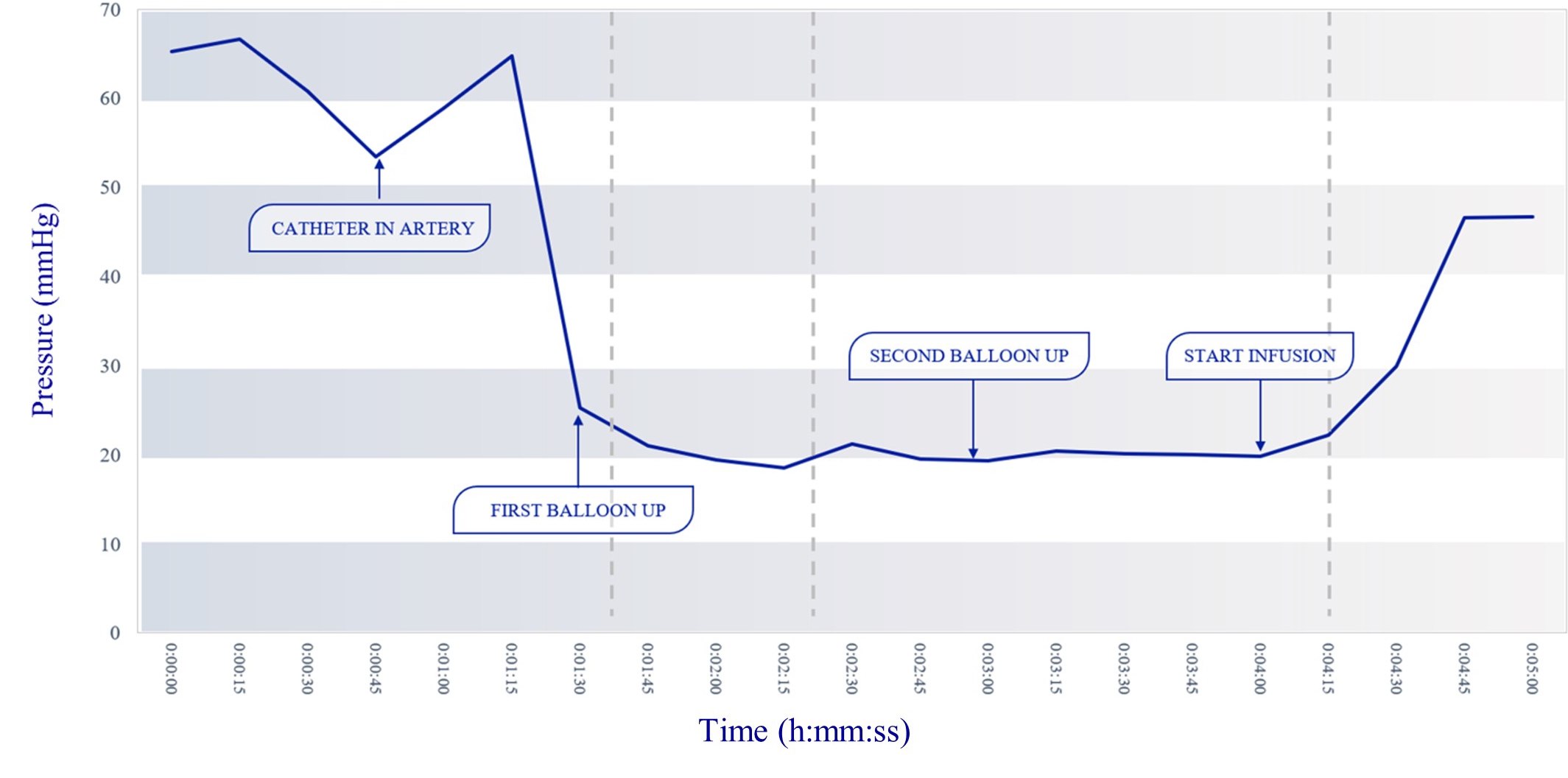
Figure 3 Occluding the vessel with the RenovoCath while changing the balloon-to-balloon distance to exclude all branches established an intravascular interstitial pressure in the isolated segment of approximately 20mmHg. With subsequent infusion of fluids between the balloons at 6mls/minute, the intravascular pressure increases until above 45mmHg, trans-arterially forcing the small molecule drug across the arterial wall via diffusion (RenovoTAMP).
Our RenovoTAMP platform therapy utilizes pressure mediated delivery of the small molecule gemcitabine across the arterial wall to bathe the pancreatic tumor tissue in 120mL of saline with 1,000mg/m2 of the drug over a 20-minute delivery time (approximately a total of 1,500-2,000mg of drug dependent upon patient Body Surface Area). This blanketing approach of large fluid volume delivery over time may enable the drug to approach these difficult-to-reach tumors. Some advantages of RenovoTAMP include:
| ● | Ideal for solid tumors where resection is not possible due to proximity/impingement of tumor on blood vessels, nerves, or other key structures | |
| ● | No need for identifying tumor feeder vessels to deliver the drug. These generally do not exist in avascular or hypovascular tumors such as LAPC and HCCA | |
| ● | In solid tumors without identifiable feeder vessels, technically easier than direct cannulation of small tumor feeders | |
| ● | High local concentration of drug into the tumor tissue | |
| ● | Potential for decreased systemic exposure of drug due to local metabolism prior to systemic exposure |
| -62- |
By isolating the vessel adjacent to the tumor and creating a pressure gradient across the arterial wall between the isolated vessel segment and the surrounding tissue or tumor, we are able to force the small molecule chemotherapy across the vessel directly into surrounding tissue or tumor. To accomplish this, we needed a minimally invasive technique to isolate the blood vessel next to the tumor, exclude any branches that can cause washout of chemotherapy away from the target, and then infuse the chemotherapy into the isolated segment to achieve pressure mediated diffusion through the vessel wall and into the tumor tissue. We accomplished this with our patented RenovoCath delivery system. RenovoCath is a double balloon catheter with the ability to adjust the balloon-to-balloon distance to tailor the treatment zone to each patient’s unique anatomy. The RenovoCath delivery system is inserted into the body through the femoral artery and positioned in the artery closest to the tumor using standard interventional technique, by an interventional radiologist. Once the balloons are inflated and the position is confirmed, chemotherapy is delivered through the handle, exiting the device between the balloons and forced through the vessel wall into the tissue over 20 minutes, depicted below in Figure 4.
RenovoCath Delivers Therapeutic Agent Between Two Balloons
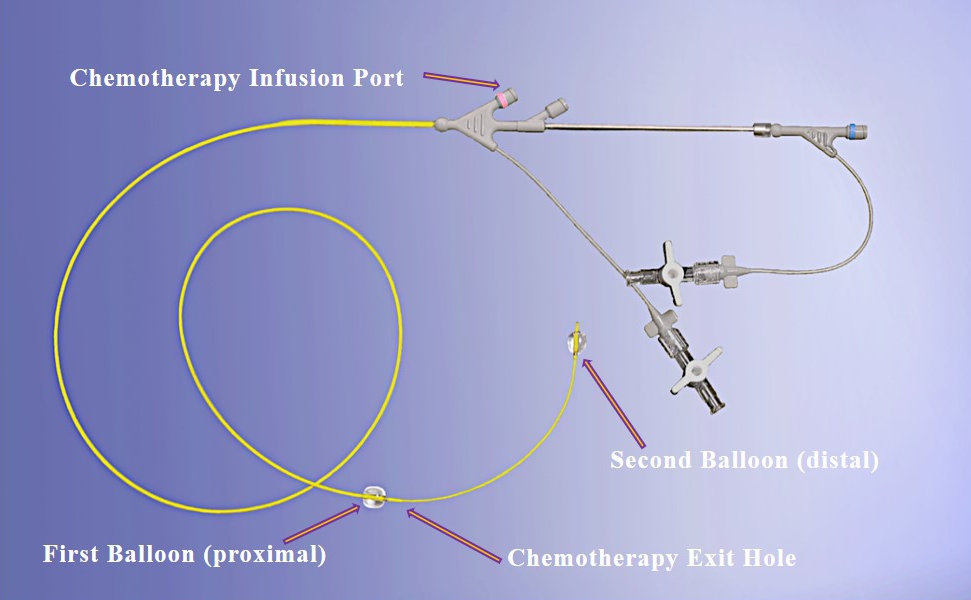
Figure 4 RenovoCath delivery system illustrating two balloon configuration to isolate the target vessel segment, and Chemotherapy delivery port/exit hole to infuse target vessel segment.
| -63- |
After the procedure is complete, RenovoCath is discarded, and the patient is generally discharged the same day. The average procedure is 90 minutes, and the procedure is repeated every two weeks for as long as local chemotherapy is warranted. Interventional Radiologists using the device are typically proctored for their first 2-3 cases only. In addition, platform training for our primary indication should transfer to other indications with ease.
RenovoGem for LAPC
Disease Overview
Pancreatic cancer is one of the deadliest cancers in the U.S. with very poor outcomes. In 2021, it is estimated that 60,430 Americans will be diagnosed with pancreatic cancer in the US and more than 48,220 will die of the disease. Pancreatic cancer currently has a 5-year overall survival rate of 5-10% (Stages I-IV) and is expected to quickly become the second leading cause of cancer-related deaths.
Current Treatment Landscape and Limitations
Pancreatic cancer has limited treatment options including one or a combination of surgery, radiation, chemotherapy, and/or some targeted therapies. Only a small subset of pancreatic cancer patients is eligible for surgery at the time of presentation (Stage I-II: 15-20%); the rest are distributed between having tumors with unresectable LAPC (Stage III: 30%) and metastatic pancreatic cancer (Stage IV: 50%). The curative prognosis for these patients is poor with a 5-year survival of only 7%.
Chemotherapy, which can be used in the neoadjuvant setting (before surgery) to attempt to decrease tumor size in the borderline resectable or resectable patients, in the adjuvant setting (after surgery), or first line in the metastatic/advanced setting, is the forefront of systemic therapy. Specifically, gemcitabine is a nucleoside metabolic inhibitor that exhibits antitumor activity by blocking the synthesis of new DNA, which results in cell death. Gemcitabine administered as an intravenous (IV) infusion has an established role in the treatment of both unresectable LAPC and metastatic pancreatic cancer, or metastatic PC, since its introduction in the US as Gemzar® (gemcitabine for injection) in 1996 with an FDA approved indication as such and remains in the guidelines as standard of care. It has been demonstrated to provide clinical benefit for subjects (decreased pain and improved performance status) as well as to improve the time to tumor progression and survival for subjects with metastatic PC and LAPC. However, major improvement in the survival curve of all pancreatic cancer subjects has been a clinical challenge, with an average median survival time for LAPC stalled at 12-15 months from time of diagnosis.
| -64- |
A key limitation of conventional chemotherapy in these tumors can be attributed to their avascular nature and desmoplasia (fibrosis or the growth of scar tissue) that impedes drug delivery. Pancreatic tumor cells have a thick and poorly perfused stroma, or connective tissue, and high interstitial pressure. This can potentially constrict blood vessels leading to an avascular or hypovascular environment that impedes chemotherapy from reaching tumor cells in high enough volume rendering them relatively chemo resistant.
In patients with metastatic disease, two chemotherapy combination regimens have shown superiority to gemcitabine, albeit with increased toxicity. First, the combination of oxaliplatin, irinotecan, fluorouracil, and leucovorin (FOLFIRINOX) in a relatively young cohort of metastatic pancreatic cancer patients appears superior to gemcitabine by improving survival from 6.8 to 11.1 months. Second, in the Metastatic Pancreatic Adenocarcinoma Clinical Trial (MPACT) trial, the combination of gemcitabine plus nab-paclitaxel (Abraxane) demonstrated an OS benefit of 9 weeks versus gemcitabine alone at the cost of increased toxicity. Despite these modest advances, there is room for improvement.
A major focus of clinicians is to determine the most optimal method to treat patients with LAPC, patients with localized disease who are not surgical candidates, roughly 30% of all pancreatic cancer patients. IV, or systemic, administration of chemotherapy has yielded unsatisfactory results in these patients. Various localized treatments have included high dose local radiation, attempts at local injection of drugs directly, and use of adenoviral vector to deliver toxic agents. The aforementioned treatment options demonstrated limited success in the treatment of LAPC. The lack of successful treatment options represents a recognized unmet medical need for these patients.
Standard of care chemotherapy for the treatment of pancreatic cancer has historically shifted a couple of times with the addition of erlotinib to gemcitabine 15 years ago resulting in a 14-day survival benefit. More recently, the addition of Abraxane to gemcitabine was approved in 2013 with immediate deep market penetration based on an 8-week survival benefit.
Our Solution
We believe that our product candidate, RenovoGem, has the potential to address the recognized unmet medical need. Utilizing our patented RenovoTAMP therapy platform, we believe RenovoGem can enhance local drug concentration, thereby increasing efficacy and decreasing systemic exposure and toxicity to improve patient outcomes. RenovoGem is a drug and device combination therapy of intra-arterial gemcitabine and our proprietary RenovoCath delivery system which forces anti-cancer drug into the tissue. RenovoGem is regulated by the FDA as a novel oncology drug product. We do not intend to sell RenovoCath alone. Instead, we intend to sell RenovoCath only in combination with intra-arterial gemcitabine or potentially with other therapeutic agents.
Based on third-party primary research/market analysis of the U.S. market, we believe that over 5,000 patients per year would be excellent candidates and undergo RenovoGem treatment once approved in the U.S. The independent oncologists interviewed stated their dissatisfaction with current standard of care and the strong desire for a therapy like ours to extend potential survival while maintaining quality of life. Further, the analysis suggests, based on analogous oncology drugs with only a modest efficacy benefit, a novel drug can expect 50-80%+ penetration in a first line setting. The results of the Key Opinion Leader, or KOL interviews revealed that a majority of oncologists would refer 90%+ of their LAPC patients who are eligible for the procedure for RenovoTAMP if the current Phase 3 trial demonstrates at least a 4-month survival benefit over systemic chemotherapy.
| -65- |
RenovoTAMP Therapy Platform and First Product Candidate, RenovoGem

Figure 5 We invented a new therapy platform, RenovoTAMP, that uses pressure to force small molecule chemotherapeutics across the vessel wall into the tissue using our patented RenovoCath delivery system. Our first product candidate, RenovoGem, is a drug-device combination of intra-arterial gemcitabine and the RenovoCath delivery system and is in development for LAPC and HCCA with Orphan Drug Designation secured for both indications.
Clinical Development of RenovoGem in LAPC
Pre-Clinical Studies and Data
Once RenovoCath is introduced via standard interventional technique to the arterial vessel segment next to the targeted tissue, both balloons are inflated and the vessel segment is isolated from the rest of the circulatory system. With inflation of balloons, the pressure is observed to drop within the vessel. However, with infusion of fluids between the balloons, the intravascular pressure increases beyond 45mmHg until plateauing, generating a gradient and trans-arterially forcing the infusate across the arterial wall via diffusion or Trans-Arterial Micro-Perfusion (TAMP). A key aspect of this approach is to adjust the distance between the balloons to exclude any side branches in the isolated segment to allow the increase in pressure gradient, rather than drug washout via the side branches. Figure 6 shows a comparison between proper balloon positioning with no side branches allowing drug across the arterial wall versus improper balloon positioning with side branches washing out drug via the side branches in an animal study.
| -66- |
Infusion Pressure Achieved When Side Branches Are Excluded
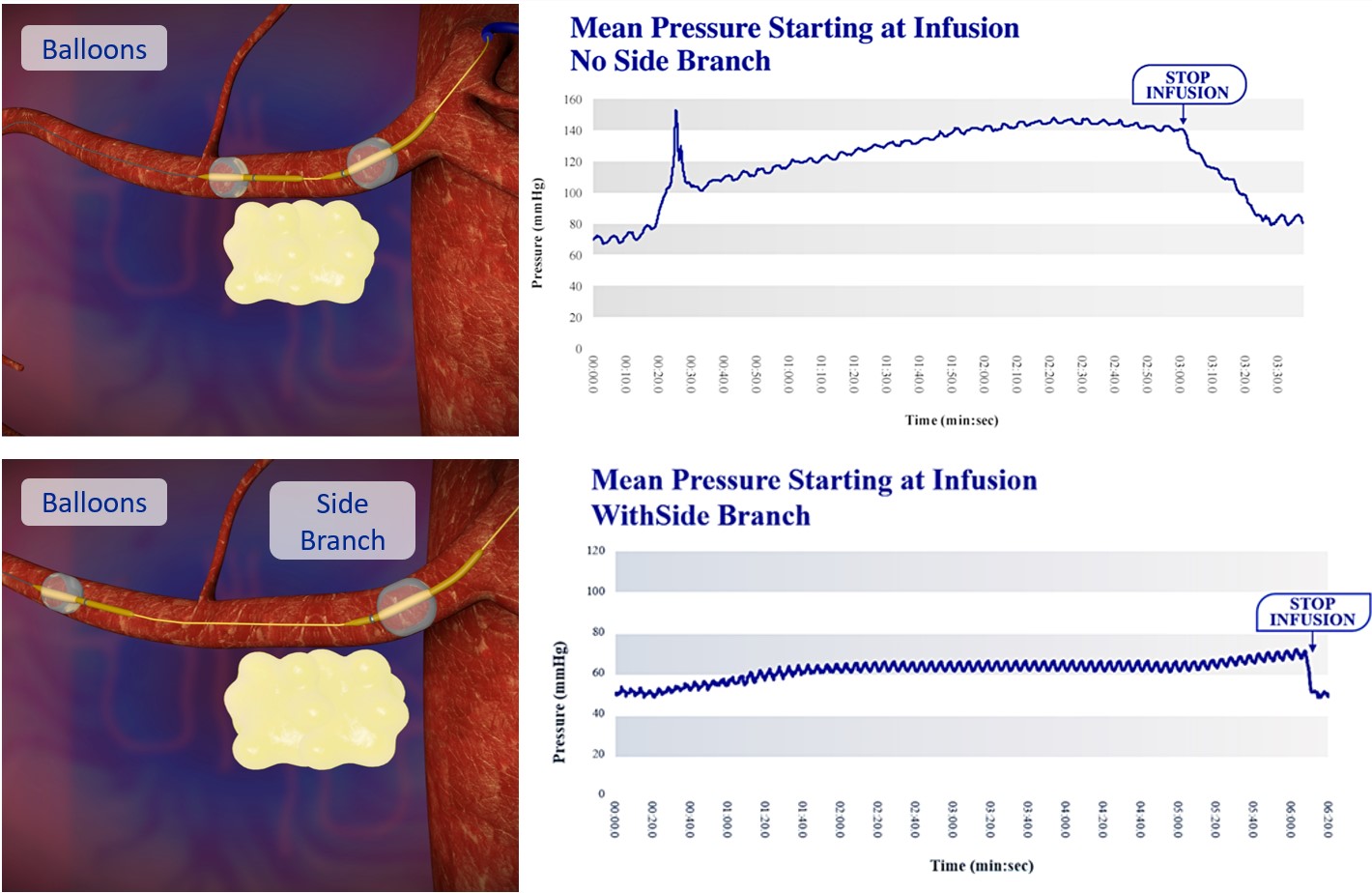
Figure 6 Top panel demonstrates proper balloon positioning with no side branch. Pressure increases with infusion and reaches plateau of approximately 75mmHg higher than initial pressure. Bottom panel demonstrates improper balloon positioning with side branch between the balloons. Pressure increases with infusion and reaches plateau of approximately only 15mmHg higher than initial pressure.
With diffusion of fluids across the arterial wall in RenovoTAMP, we expected to be able to deliver small molecules into the surrounding tissue. We performed following studies to validate this hypothesis:
| 1) | Gemcitabine can cross the arterial wall via RenovoTAMP, with 99% crossing the arterial wall into the tissue. |
In explanted (dissected out of the animal and used separately in a saline water bath) pig iliac and aortic artery, with introduction of RenovoCath and infusion of gemcitabine in the isolated vessel segment, we were able to measure (in a time dependent fashion) the amount of gemcitabine crossing the arterial wall into the surrounding fluid. We isolated the arterial vessel segment using RenovoCath and then delivered 60mg/minute of gemcitabine into the isolated area over 20 minutes. By the end of the infusion, we measured 1188 mg of gemcitabine in the surrounding fluid around the vessel and 9 mg in the analyzed tissue of the vessel. This demonstrated that 99% of the drug crosses the arterial wall and only 0.75% is retained in the arterial tissue (Figure 7).
99% of Chemotherapy Crosses Arterial Wall with RenovoTAMP Delivery
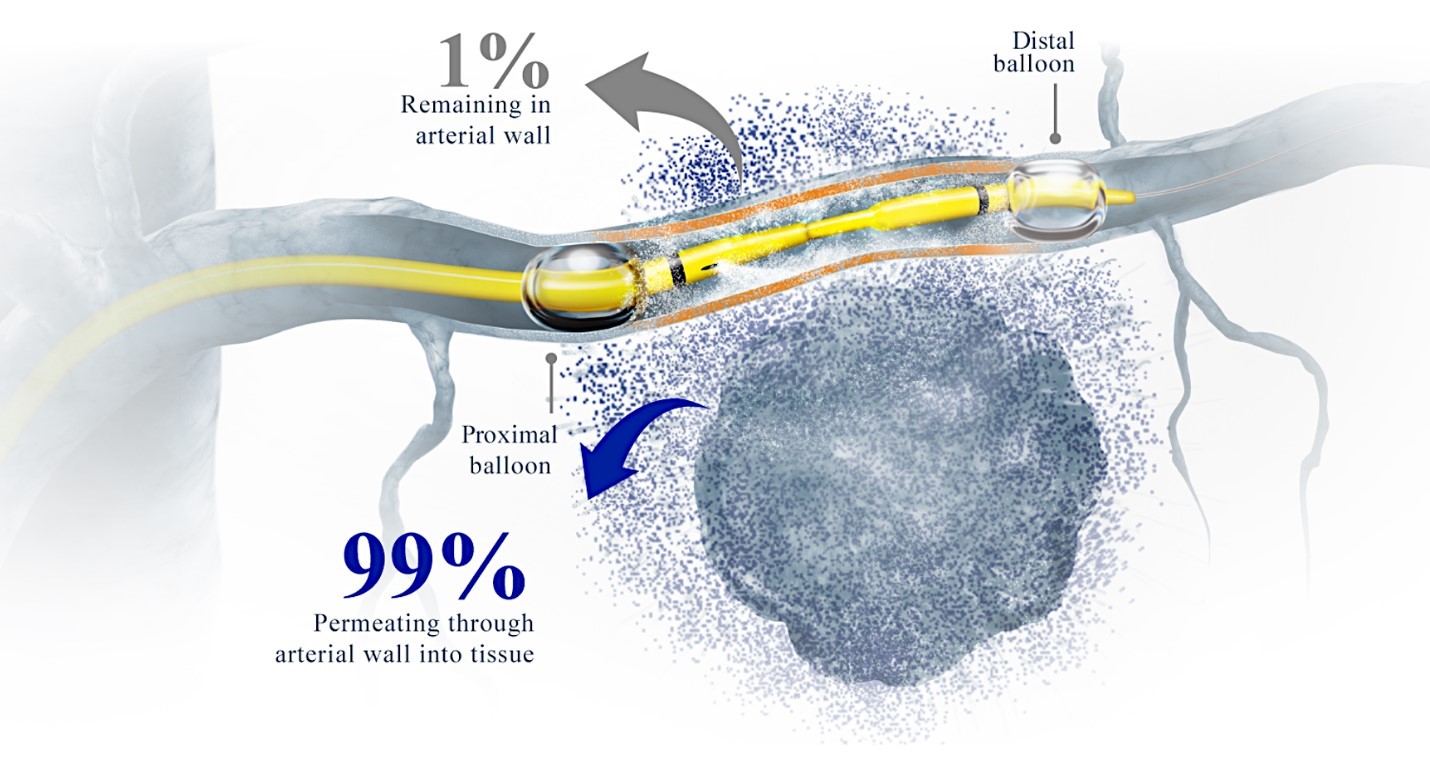
Figure 7 RenovoTAMP: delivery of chemotherapy through the RenovoCath and into the tissue to bathe the tumor in chemotherapy. With gemcitabine, 99% of the drug crosses the arterial wall and less than 0.75% is retained in the arterial tissue.
| 2) | Infusion of gemcitabine via RenovoTAMP has demonstrated vascular safety with acceptable toxicity in the pig model and does not cause loss of vessel integrity or inflammation. |
| -67- |
Six pigs were treated with gemcitabine via RenovoTAMP (6mL/min for 20 minutes). Target vessels included selection of the superficial femoral artery (SFA) and splenic arteries from each animal (either test or saline control). A total of 6 vessels (3 SFA and 3 splenic arteries) were treated with an equal number of control vessels. All animals survived the 7-day in-life period although two of the animals with gemcitabine treatment in the splenic artery experienced atypical pain during the post-operative phase and required additional pain management with eventual complete recovery.
Analysis of the vessels demonstrated preserved vessel shape with intact endothelial cells (cells on the inside of the vessels). Minimal to no inflammation was observed. The only vessel toxicity observed was a reduction of smooth muscles cells in the vessel wall, primarily close to the inside of the vessel.
| 3) | RenovoTAMP can achieve targeted local drug (dye) delivery |
| I. | Targeted small molecule delivery (dye) into pancreatic tissue |
We further validated our approach for tissue drug delivery using acute animal experiments. Using both dye and gemcitabine infusion via the RenovoTAMP therapy, we were able to demonstrate that fully isolating a segment of a vessel (by blocking inflow and outflow in the target vessel with the RenovoCath double balloons as well as side branches) can lead to dye penetration greater than 4.0 cm from the vessel wall and drug tissue concentration (gemcitabine) up to 100-fold greater than systemic administration.
In an acute pig experiment, RenovoCath was introduced into the gastro-duodenal artery (GDA), a side branch was excluded (using small implants that block the artery, coils), and then dye was introduced at 6mls/minute over 2 minutes. Analysis demonstrated that the blue dye diffused covered approximately 10.56 cm2 (2.2 cm x 4.8 cm) of the pancreas.
Dye Demonstrates RenovoTAMP Delivery of Agent into Pancreatic Tissue
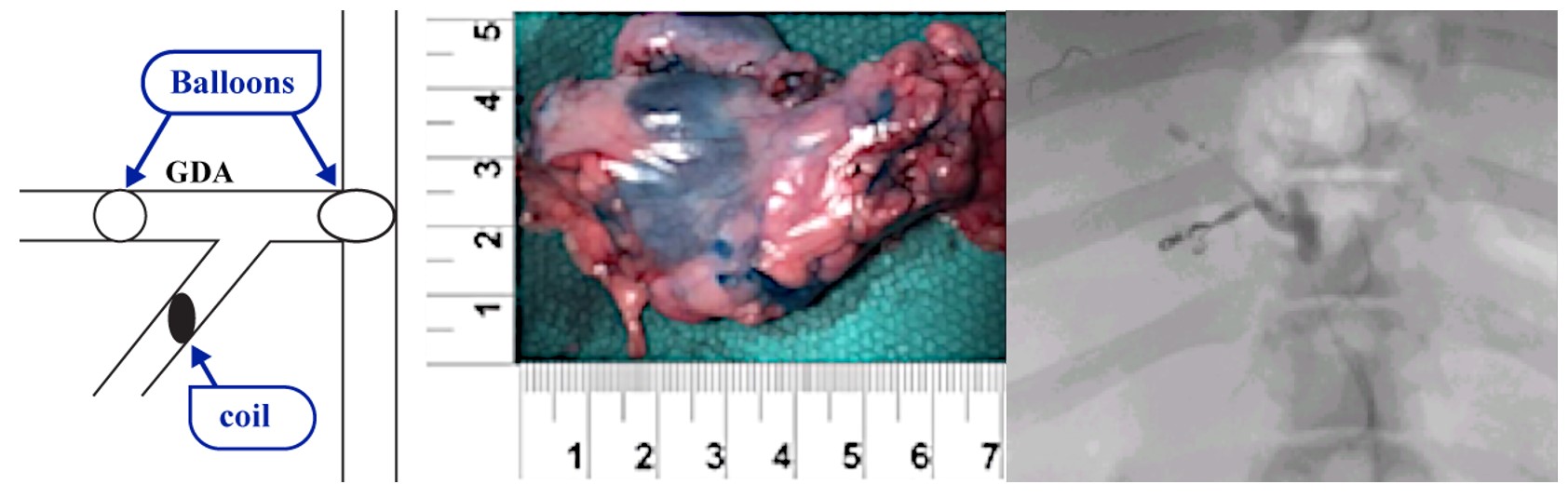
Figure 8 RenovoCath was introduced into the GDA and a side branch was excluded by coiling. This test was conducted in an acute porcine model and demonstrated a dye coverage area of approximately 10.56 cm2 for a 2-minute dye infusion. All dimensions in above figure are in cm.
The study was repeated in 6 other vessel targets to validate the impact of vessel isolation on dye penetration into the surrounding tissue with similar results.
| II. | Small molecule delivery (dye and gemcitabine) locally into lung tissue |
In another set of acute animal experiments, the pulmonary artery was isolated via access through the internal jugular vein. Six ml of methylene blue dye was injected over 1 min and gemcitabine was subsequently delivered locally at rate of 6mls/minute for 20 minutes to the lung tissue using the RenovoTAMP procedure
| -68- |
Dense dye staining localized to the area of the isolated vessel segment was observed. Again, analysis established penetration into surrounding tissue (4cm). Furthermore, RenovoTAMP achieved greater than 100-fold tissue concentration of gemcitabine versus the tissue level achieved by IV (systemic) delivery of gemcitabine at the same infusion rate.
Dye Staining Demonstrates RenovoTAMP Delivery of Agent to Lung Tissue
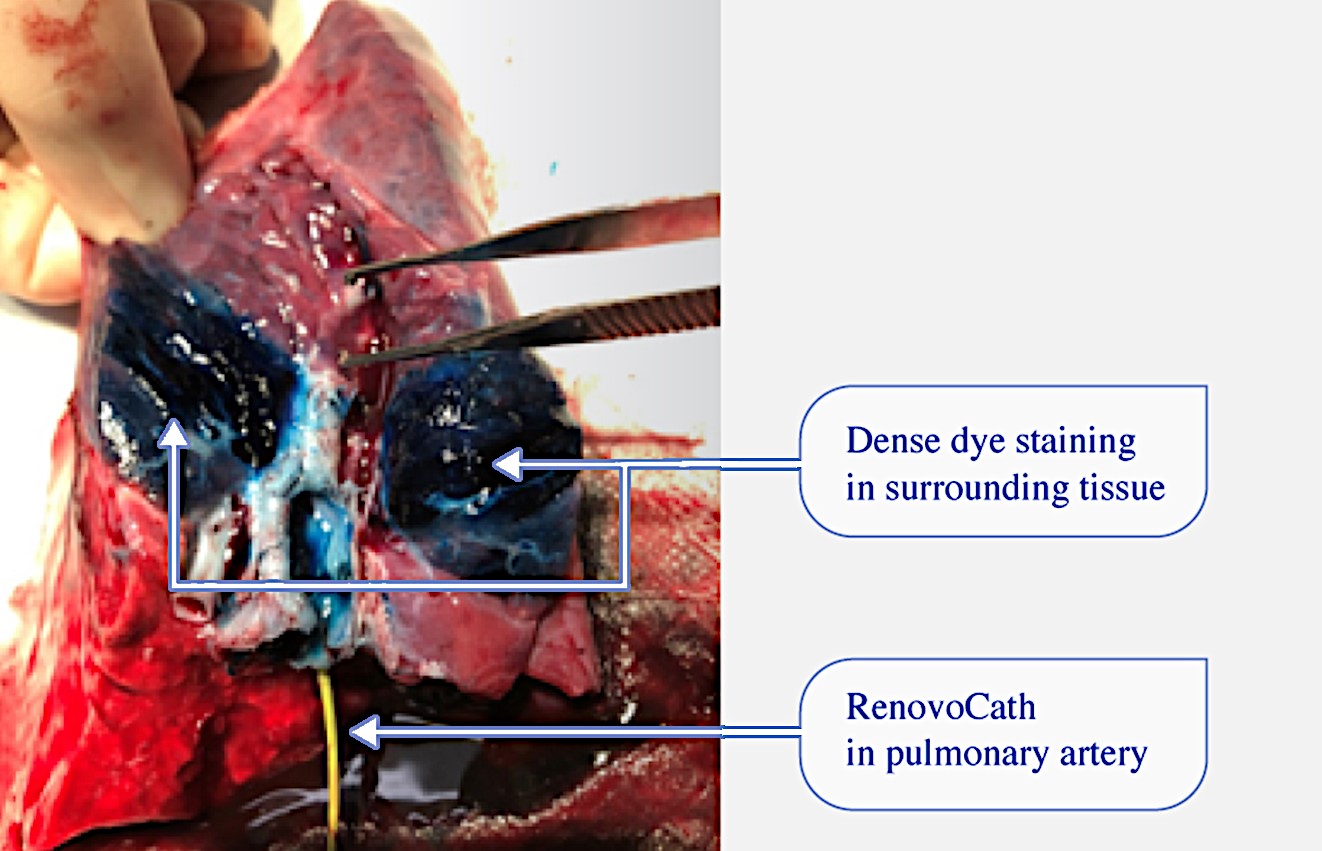
Figure 9 Dense dye staining localized to the area of the isolated pulmonary artery segment and penetrating 4cm into surrounding tissue following 1 minute dye infusion. In addition, gemcitabine was delivered via RenovoTAMP for 20 minutes demonstrating 100-fold increase in tissue concentration of gemcitabine compared to IV delivery of gemcitabine at the same infusion rate.
We concluded that RenovoTAMP can achieve drug penetration into the surrounding tissue and can achieve high dose of local tissue concentration. The tissue concentration with intravenous infusion and/or distant from RenovoTAMP site (likely after recirculation through systemic system) are two orders of magnitudes lower than tissue levels achieved with RenovoTAMP (p<0.02).
RenovoTAMP Increases Local Tissue Concentration of Gemcitabine Compared to IV Infusion.
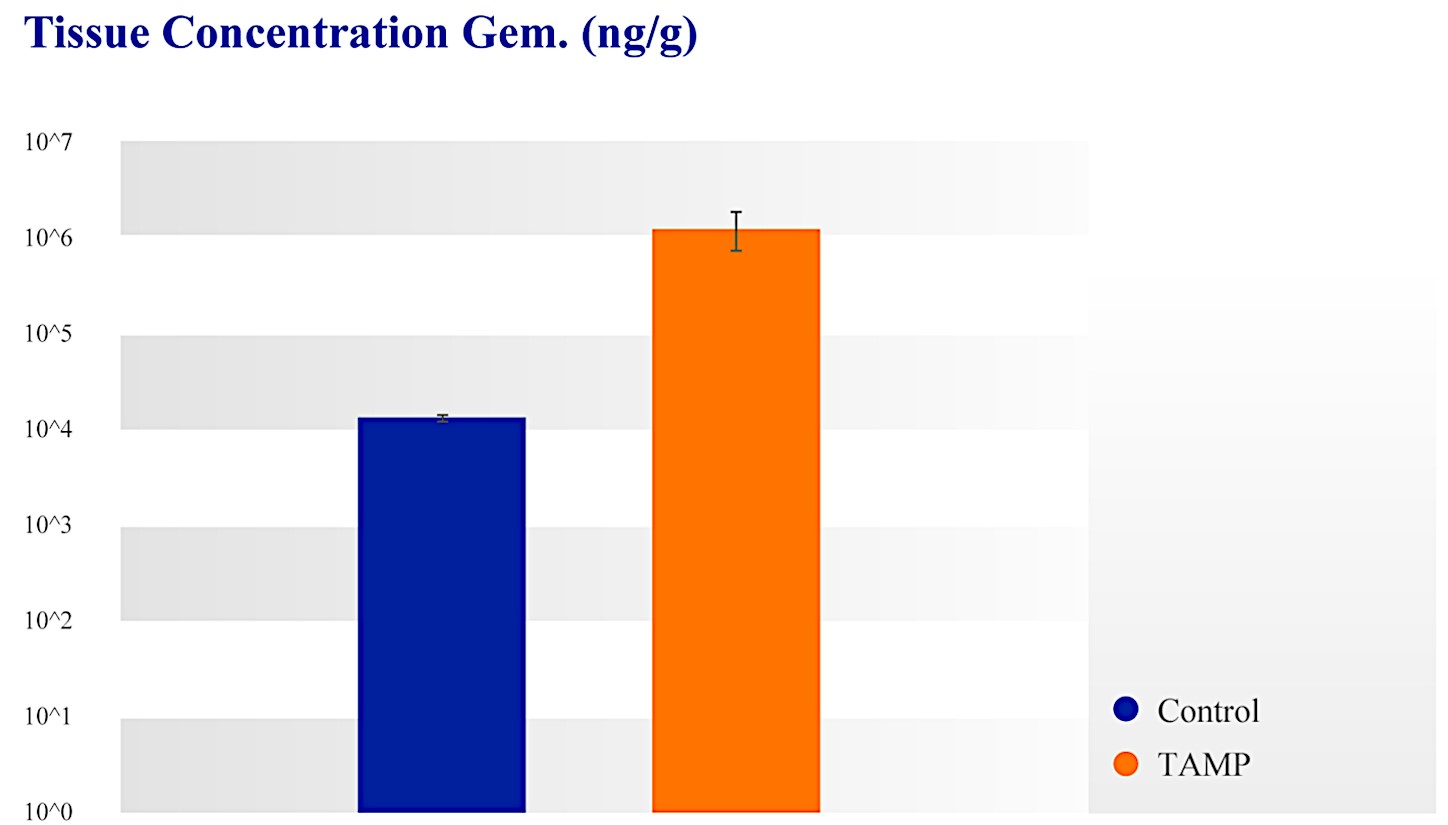
Figure 10 Local tissue concentration of gemcitabine. control (Blue): IV infusion versus RenovoTAMP (Orange): RenovoTAMP: intra-arterial infusion. The tissue concentration with intravenous infusion and/or distant from RenovoTAMP site (likely after recirculation through systemic system) are 100-fold lower than tissue levels achieved with RenovoTAMP.
| -69- |
This animal lung study successfully validated the efficacy and ability for RenovoCath to deliver small molecules locally to lung tissue with the RenovoTAMP therapy.
| III. | Increase in local tissue delivery of gemcitabine in LAPC should enhance tumor reduction and therapeutic response |
In relevant mouse models of pancreatic tumors, it has been demonstrated that targeted intra-arterial (IA) infusion of gemcitabine into the pancreas after surgical isolation of arterial blood flow, has a superior therapeutic effect with greater reduction in tumor volume compared to the same concentration administered by conventional systemic (IV) injection. To achieve a comparable reduction in tumor growth as seen with IA treatment, gemcitabine had to be given IV at over 300 times the dose which was associated with some toxicity.
RenovoTAMP and Radiation
Traditionally the goal of radiation includes a) debulking the tumor and/or b) acting as a chemo-sensitizer. In our RR1 dose escalation safety study and RR2 observational registry study, the benefit of RenovoTAMP seems to be enhanced in patients with prior radiation. As we were observing this effect months after radiation and several randomized studies have not demonstrated a benefit of chemotherapy + radiation versus chemotherapy alone, we hypothesized that a direct effect of radiation on the vasculature may be enhancing the effect of RenovoTAMP. One of the side effects of radiation is a decrease in the micro-vasculature in the irradiated tissue including the small blood vessels that exist in the vessel walls themselves. Therefore, we postulated that by eliminating microvasculature in and around the vessel wall, radiation may enhance drug penetration into the tissue via RenovoTAMP (Figure 11). As such a possible enhancing effect of radiation on RenovoTAMP may involve decreasing washout of the drug as it crosses the arterial wall by preventing draining into the surrounding microvasculature.
We completed a pig study where we observed the impact of RenovoTAMP in recruiting the vasa vasorum (small blood vessels within the larger blood vessel walls) around the vessel during drug/dye infusion. It was discovered that the dye drained into the vasa vasorum and other small vessels in the adjacent tissue (Figure 11); as these vessels can directly connect to the adjacent venous system, the microvascular networks can serve as an “escape route” for drugs. Ultimately this direct washout can reduce the amount of drug concentration in the tissue. Radiation pretreatment may enhance the impact of RenovoTAMP by attenuating this escape route.
RenovoTAMP Combined with Radiation Reduces Venous Outflow by Decreasing the Microvasculature
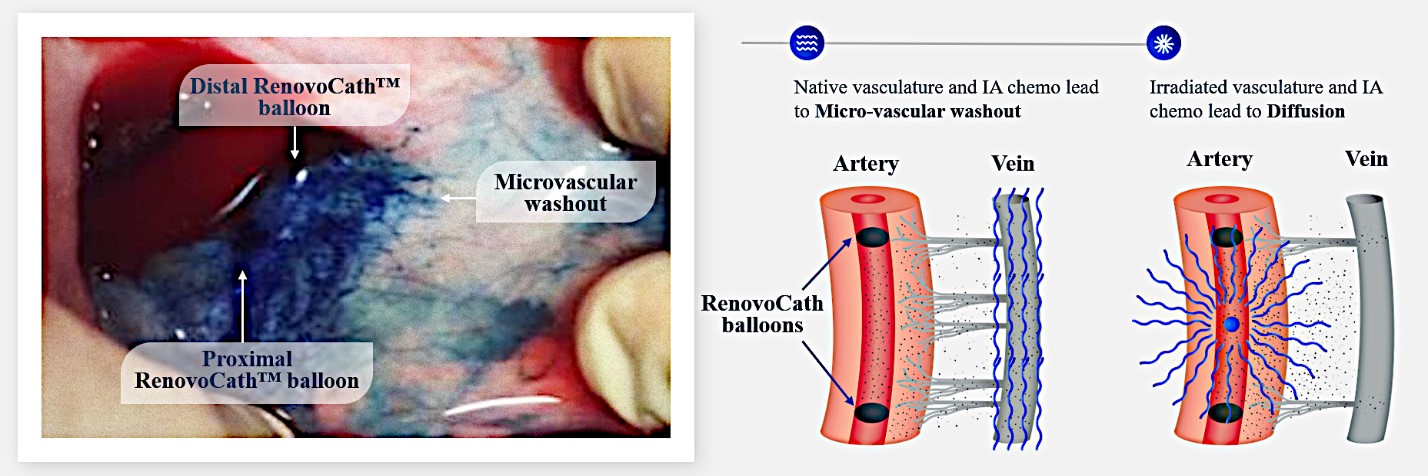
Figure 11 Mechanism of RenovoTAMP and radiation reduces venous outflow by decreasing the microvasculature networks that could act as an “escape route” for the drugs. The photo on the left illustrates this effect in a dye infusion study in the porcine animal model. The panel on the right demonstrates venous chemotherapy washout without radiation versus less venous escape routes for chemotherapy following radiation.
| -70- |
We further advanced this theory by conducting a pig study to directly test whether radiation can enhance tissue uptake by RenovoTAMP. In a single-animal study, we examined the use of Stereotactic Body Radiation Therapy (SBRT) pre-treatment on one leg followed by RenovoTAMP versus RenovoTAMP without prior radiation therapy on the opposite leg. The leg of the animal that was pre-treated with radiation demonstrated more pronounced tissue staining with methylene blue dye and increased gemcitabine concentration via punch biopsy. Based on these findings, we believe that the benefit of prior radiation on clinical outcomes with RenovoTAMP may be improved by the effect of radiation on microvasculature between the vessel wall and the tumor.
Dye test Demonstrates that RenovoTAMP Plus Radiation Increases Concentration of Gemcitabine
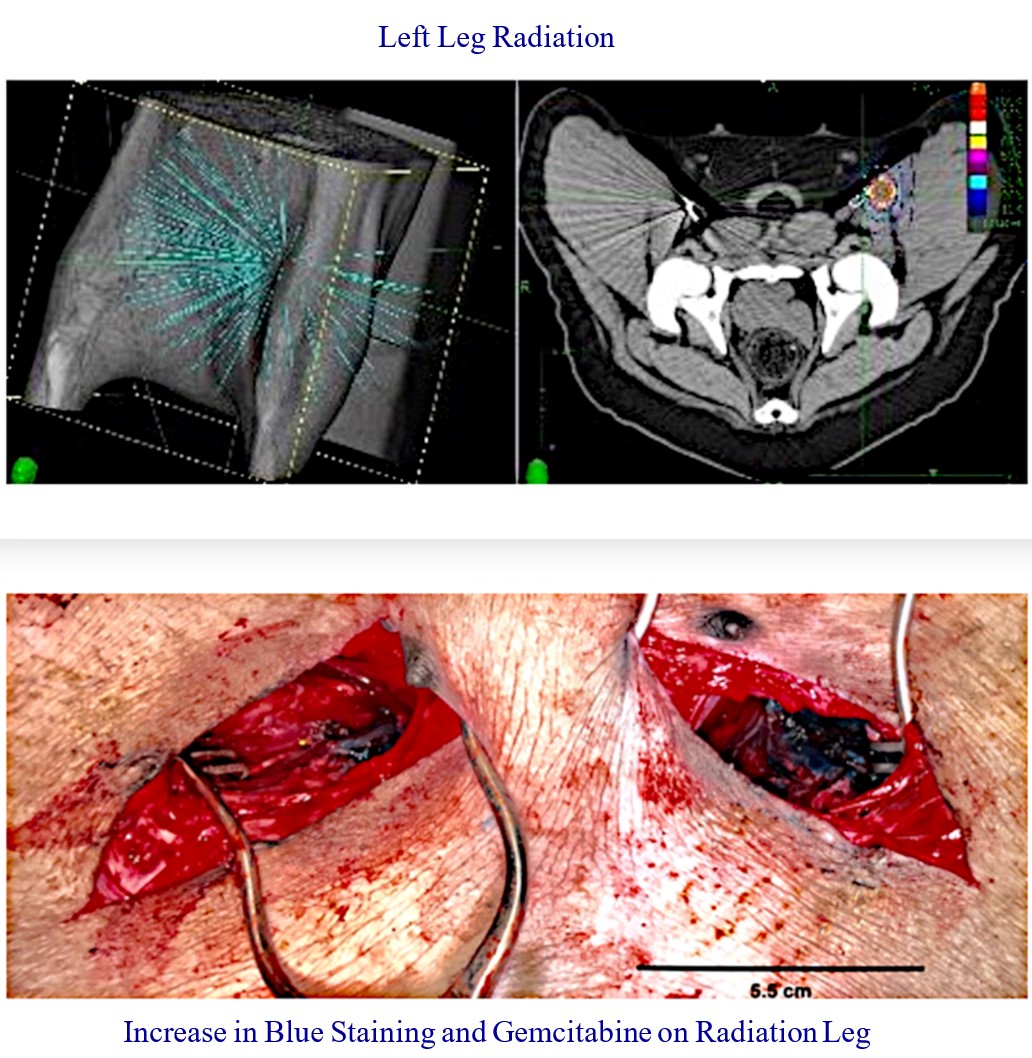
Figure 12 To demonstrate the effect of radiation pre-treatment, we delivered radiation therapy to the left leg of a pig. After waiting for one month for the vascular effect to take place, we performed RenovoTAMP on the left and right leg arteries with blue dye and gemcitabine. Dissection revealed better dye penetration into the tissue on the left (irradiated) leg, and punch biopsy demonstrated higher gemcitabine concentration in the left leg.
We have demonstrated that the RenovoTAMP therapy allows targeted small molecule drug delivery into the tissue surrounding the vessel wall, without need to identify tumor feeders. The mechanism of action is the exclusion of distal (downstream) and side branch vessels in the isolated segment and subsequently achieving a pressure gradient by infusing the drug over time. The pressure gradient results in a diffusion-mediated delivery of drug into the surrounding tissue. With the use of gemcitabine, the procedure appears safe in terms of local toxicity in the vasculature. Using this approach, we can achieve increased drug delivery into the surrounding tissue in the range of 4cm-tissue penetration and concentration orders of magnitude larger than what can be achieved with IV infusion. Lastly, RenovoTAMP appears to be enhanced by prior radiation of tissue, possibly through its effect on decreasing the microvasculature and subsequent potential chemotherapy washout.
| -71- |
LAPC Clinical Development
RenovoTAMP has been studied in a phase 1/2 dose-ranging study of 20 subjects with locally advanced pancreatic cancer (RR1) and in an observational study that enrolled 25 additional subjects with pancreatic cancer (RR2); two subjects from the RR1 safety study continued to receive treatment in the RR2 observational registry study. We subsequently launched a Phase 3 registration trial (TIGeR-PaC) and as of July 15, 2021 we have enrolled 145 patients in this study.
Phase 1/2 Dose-Ranging Study: RR1
Study Design
A phase 1/2 safety study of our RenovoTAMP therapy has been completed in subjects with LAPC (Phase 1/2 RenovoCath/Gem RR1). This multicenter, prospective, open label, interventional, nonrandomized, intra-subject dose escalation study evaluated IA gemcitabine delivered locally to the pancreas using the RenovoCath in 20 subjects with locally advanced pancreatic cancer. The primary objectives of the study were (1) to establish the maximum tolerated dose (MTD) and (2) to study the safety and tolerability of intra-arterial (IA) gemcitabine administered by RenovoCath at doses ranging from 250 mg/m2 to 1000 mg /m2. Secondary endpoints included overall survival, CA 19-9 marker change, change in tumor size based on RECIST 1.1 (Response Evaluation Criteria in Solid Tumors) criteria, and pain scores and narcotic use. Adverse events were collected from the first IA gemcitabine infusion until 3 months following the final IA gemcitabine infusion. Subjects were followed for survival.
Treatment constituted introducing RenovoCath to target vessel (adjacent to tumor) via catheterization, occluding the targeted segments via the RenovoCath balloons, and infusing gemcitabine in the occluded segment. To minimize ischemia (damage due to cessation of blood flow) the infusion was limited to 20 minutes and anticoagulants (heparin) was given during the procedure. Tissue markers were followed post procedure to ensure lack of local tissue damage-toxicity (AST, ALT, Lipase and Amylase).
Treatment was administered in four 28-day cycles, each of which consisted of two IA doses of gemcitabine, one on day 1 and one on day 15, with a two-week rest period between cycles. The first six subjects received a starting does of 250mg/m2, and doses increased by 250 mg/m2 in each subsequent cycle culminating with the full dose of 1,000 mg/m2. After the initial six subjects, the starting dose increased to 500 mg/m2 for one cycle, after which dosing increased to 750 mg/m2 for the second cycle, and then the full 1,000 mg/m2 dose for the remaining 2 cycles. Each subject underwent CT scanning prior to the first procedure for the selection of the optimal target vessel most proximal to the tumor.
Study Subjects and RenovoGem Exposure
The median age of subjects was 66.7 years with a gender distribution of 9 men and 11 women. Prior treatment included chemotherapy and radiation therapy in 6 (30%), chemotherapy alone in 5 (25%) and no prior therapy in 9 (45%) subjects. Collectively the 20-subject cohort received 101 IA treatments. Importantly, 9 of the 20 subjects had a biliary stent or drain in place before the first IA procedure.
Trial Results
Safety
There was no evidence of local tissue toxicity in any patients post procedure as measured by liver and pancreatic enzymes. Adverse Events were reported in 11 subjects, including catheterization/procedure-related events with arterial dissections at treatment sites (3), pseudoaneurysm in a visceral artery (1) away from the treatment site and site complications (2) out of 101 procedures.
Serious adverse events were reported in 9 subjects during the study. Overall survival (including deaths that occurred following disease progression) was followed in all study subjects. The number of subjects with serious adverse events is shown in Table 1 below.
| -72- |
Table 1 Summary of Serious Adverse Events for 9 subjects in RR1 Dose Ranging Study
| Serious Adverse Event | N=20 | |
| Cardiac Arrest | 1/20 (5%) | |
| Dehydration | 1/20 (5%) | |
| Duodenal obstruction | 1/20 (5%) | |
| Gastritis | 1/20 (5%) | |
| Infection | 1/20 (5%) | |
| Intraoperative arterial injury-dissection | 3/20 (15%) | |
| Intraoperative arterial injury-lower extremity | 1/20 (5%) | |
| Pain-Abdominal NOS | 1/20 (5%) | |
| Respiratory failure | 1/20 (5%) | |
| Sepsis | 3/20 (15%) | |
| Neutropenia | 4/20 (20%) |
This table shows serious adverse events reported in 9 of the 20 subjects during the study. Several subjects had more than one serious adverse event.
Efficacy
The principal evaluation of efficacy was survival. All subjects were followed for survival after the end of IA gemcitabine treatment. All subjects have died, with the longest having an overall survival of 35.9 months.
Subjects Who Received More Cycles Survived Longer
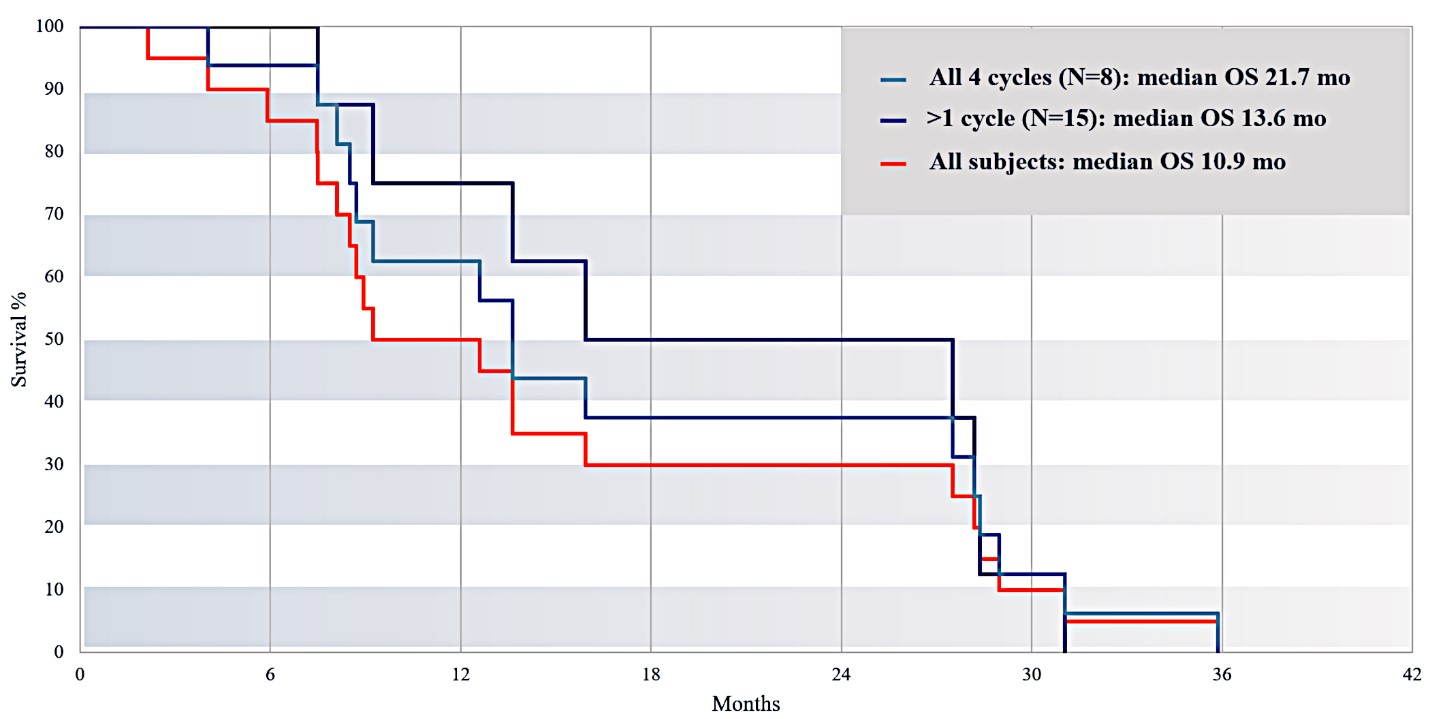
Figure 13 This chart shows survival as a function of total number of IA treatment cycles received. Subjects receiving all 4 cycles (n=8) had a median survival time of 21.7 months, compared to a median survival time of 10.9 months for all subjects (n=20).
| -73- |
Subjects Who Received Greater Cumulative Exposure Survived Longer
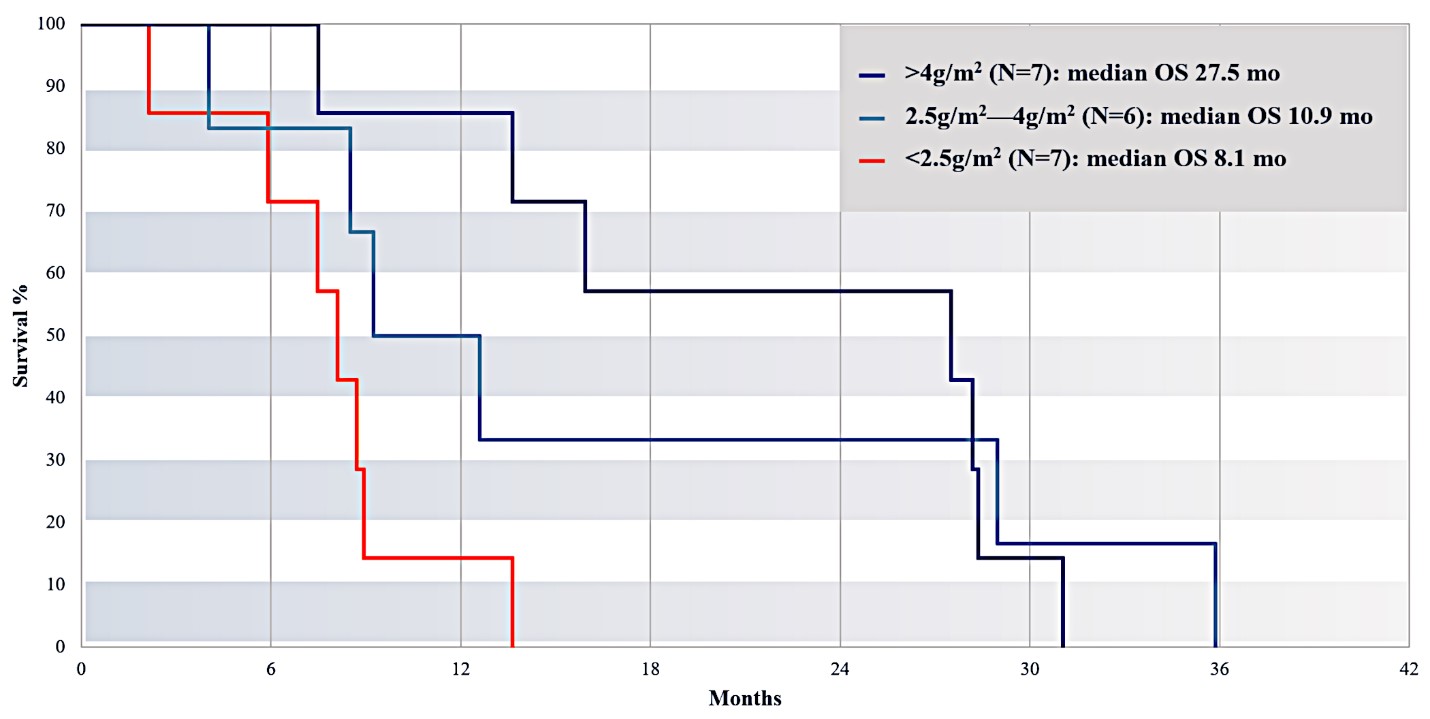
Figure 14 Splitting the entire cohort into equal tertiles based on total dose received, patients receiving the lowest total dose (<2.5g/m2; n=7) demonstrated the lowest median overall survival (8.1 months) compared to patients in the group receiving the next higher total dose (>2.5g/m2, but <4g/m2; n=6; median OS=10.9 months), and patients in the group receiving the highest total dose (>4g/m2; n=7; median OS=27.5 months).
| -74- |
Fifteen subjects received more than 1 cycle of intra-arterial gemcitabine treatment. The blue line depicts subjects without any prior treatment or received prior chemotherapy only (n=10; median OS=13.6 months). The green line depicts subjects who received prior chemoradiation (n=5; median OS=28.2 months). P < 0.05 for survival between the two subsets. Survival appeared to be longer in subjects who had prior chemoradiation as shown below.
Subjects Who Had Prior Radiation Exposure Survived Longer Than Those Who Did Not
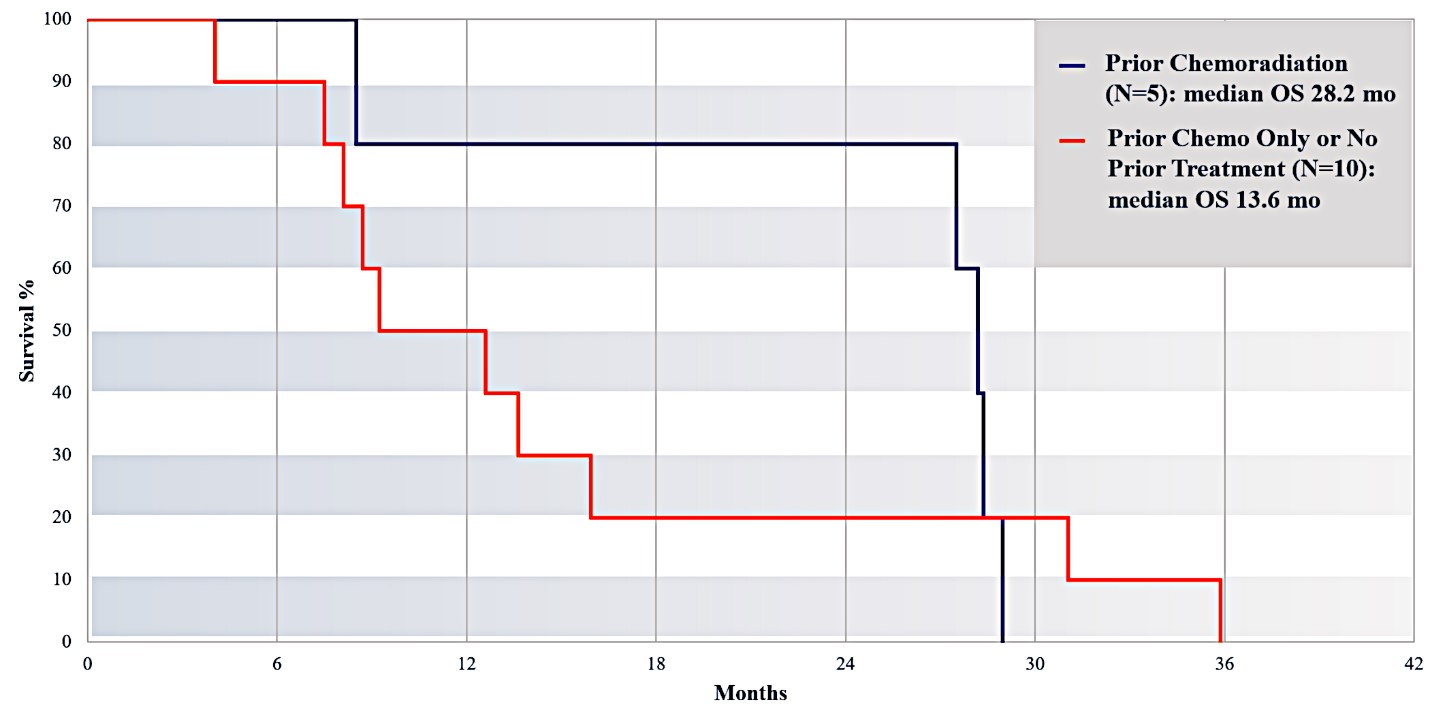
Figure 15 Survival of subjects with or without Prior Chemoradiation. Subjects with prior chemoradiation (n=5) had a median survival time of 28.2 months, compared with a median survival time of 13.6 months for subjects without prior chemoradiation (n=10).
Disease progression based on RECIST 1.1 Criteria
The RECIST 1.1 criteria was used to compare the baseline and follow up CT images submitted by sites. Follow up CT scans obtained 5 months after initiation of IA gemcitabine therapy were submitted for 17 of the 20 subjects and were compared to the baseline images. Six of the 17 subjects (35.3%) experienced tumor progression, 1 (5.9%) had a partial response, and 10 (58.8%) demonstrated stable disease 5 months post treatment initiation. Two of the 6 subjects with tumor progression received less than 1 cycle (only 1 treatment) of IA gemcitabine. Among 15 subjects who received more than 1 cycle (2 treatments), 26.7% had disease progression, 6.7% had partial response and 66.7% had stable disease 5 months post IA therapy.
CA 19-9 Tumor Marker Change
CA19-9 is a protein that can be detected in serum and is a biomarker of pancreatic cancer; its levels can be used to assess tumor response to therapy. Twelve of 20 subjects had measurable CA 19-9 tumor markers. The final CA 19-9 tumor marker levels were lower in 7 of 12 (58%) and greater in 5 of 12 (42%) subjects. It is notable that, final tumor marker levels were lower in 4 of 5 subjects with prior chemoradiation and higher in 5 of 7 subjects without prior chemoradiation.
Observational Registry Study RR2
We launched the RR2 observational registry study in January 2016 to further explore the clinical utility of the RenovoTAMP procedure. The key inclusion criteria included patients with locally advanced or borderline resectable pancreatic adenocarcinoma confirmed by histology or cytology. This was an observational patient registry study with endpoints of safety and survival following intra-arterial gemcitabine treatment with RenovoCath. The study was conducted at 7 sites in the US and subsequently closed on August 2019 except for one US site (that did not participate in the Phase 3 study). This last site the study was officially closed in September 2020. Over the 3 years that the trial was open, we enrolled 25 subjects with LAPC. Two of the subjects participated in our Phase 1/2 RenovoCath/Gem RR1 trial, in which each received 8 IA gemcitabine infusions prior to enrollment in the RR2 study for observation of additional IA gemcitabine infusions. A summary of data updated through January 2021 is presented below.
| -75- |
The study initially enrolled LAPC subjects without regard to prior radiation or chemotherapy; after the observation of longer survival of subjects with prior chemoradiation versus subjects who had not had prior radiotherapy, entry into the registry was restricted to subjects with LAPC who had received prior radiation beginning April 2017. Of note, one subject who had prior pancreatic cancer surgery (Whipple procedure) would not normally have been enrolled in the study but was included for safety observations as her physician had previously planned IA gemcitabine therapy.
Investigators in the study reported all Serious Adverse Events from the first IA gemcitabine infusion to at least 60 days post-last procedure but reporting of non-serious adverse events was optional. All subjects received gemcitabine 1000 mg/m2 every two weeks, except one who received 500 mg/m2, typically for a total of 8 doses. Subjects were followed post treatment for survival.
Study Subjects and RenovoGem Exposure
Twenty-five subjects were enrolled at 7 sites. The study enrolled 15 women (60%) and 10 men (40%); with a mean and median age of 73. Of the 25, 10 (40%) had no prior therapy, 8 (32%) had radiotherapy and chemotherapy, 6 had chemotherapy alone and 1 (4.5%) had surgery (Whipple procedure).
Two subjects were continuations from the previous Phase 1/2 RenovoCath/Gem RR1 study, and as a result, received more than eight treatments (total in both studies). The treatment received summary is shown in Table 2:
Table 2 Dosing Treatments Tally for RR2 Observational Registry Study, for 25 Subjects Enrolled at 7 Sites
Number of Dosing Treatments | N=25 | |
| 1 | 5/25 (20%) | |
| 2 | 3/25 (12%) | |
| 3 | 4/25 (16%) | |
| 4 | 5/25 (20%) | |
| 6 | 2/25 (8%) | |
| 7 | 2/25 (8%) | |
| 8 | 2/25 (8%) | |
| >8 | 2/25 (8%) |
Twenty-five subjects, 15 women (60%) and 10 men (40%); with a mean and median age of 73 were enrolled at 7 sites. Of 25, 10 (40%) had no prior therapy, 8 (32%) had radiotherapy and chemotherapy, 6 had chemotherapy alone and 1 (4.5%) had surgery (Whipple procedure).
Trial Results
Safety
There were number of adverse events reported. The most common were nausea (36%), vomiting (28%), abdominal pain (32%), followed by vascular access complications (16%). The less common adverse events reported (< 5%) included rash, allergic reaction, retroperitoneal hemorrhage, sepsis, ischemic bowel, arterial spasm, atrial fibrillation, chest pain, back pain, hypoglycemia, pruritis, and other GI issues. No deaths were noted in the immediate post-treatment period. No deaths were considered related to study treatment. Survival is summarized as an efficacy evaluation.
| -76- |
Summary of Key Safety Observations
| ● | Neither pancreatitis nor local tissue toxicity was reported in LAPC subjects without prior surgery. | |
| ● | There was no instance of arterial dissection in this study. | |
| ● | The incidence of sepsis was lower in this study (1/25 subjects receiving 94 infusions) compared with the incidence in Phase 1/2 RenovoCath/Gem RR1 (3/20 subjects receiving 101 infusions). The subject with sepsis did not have a biliary stent or drain and the source of the sepsis was not identified. No sepsis events were noted after 51 infusions in 12 subjects with biliary stents, who received peri-procedure antibiotics. The incidence of sepsis in the RR2 observational registry is similar to that of pancreatic cancer subjects receiving myelosuppressive chemotherapeutic regiment in other studies. |
Efficacy
Excluding the subject with prior or post pancreatic cancer surgery, median survival (n=22) from the time of first IA gemcitabine treatment was 5.43 months, as illustrated in and median overall survival was 13.0 months.
Survival of all Subjects from first IA Gemcitabine Treatment (Median 5.43 Months)
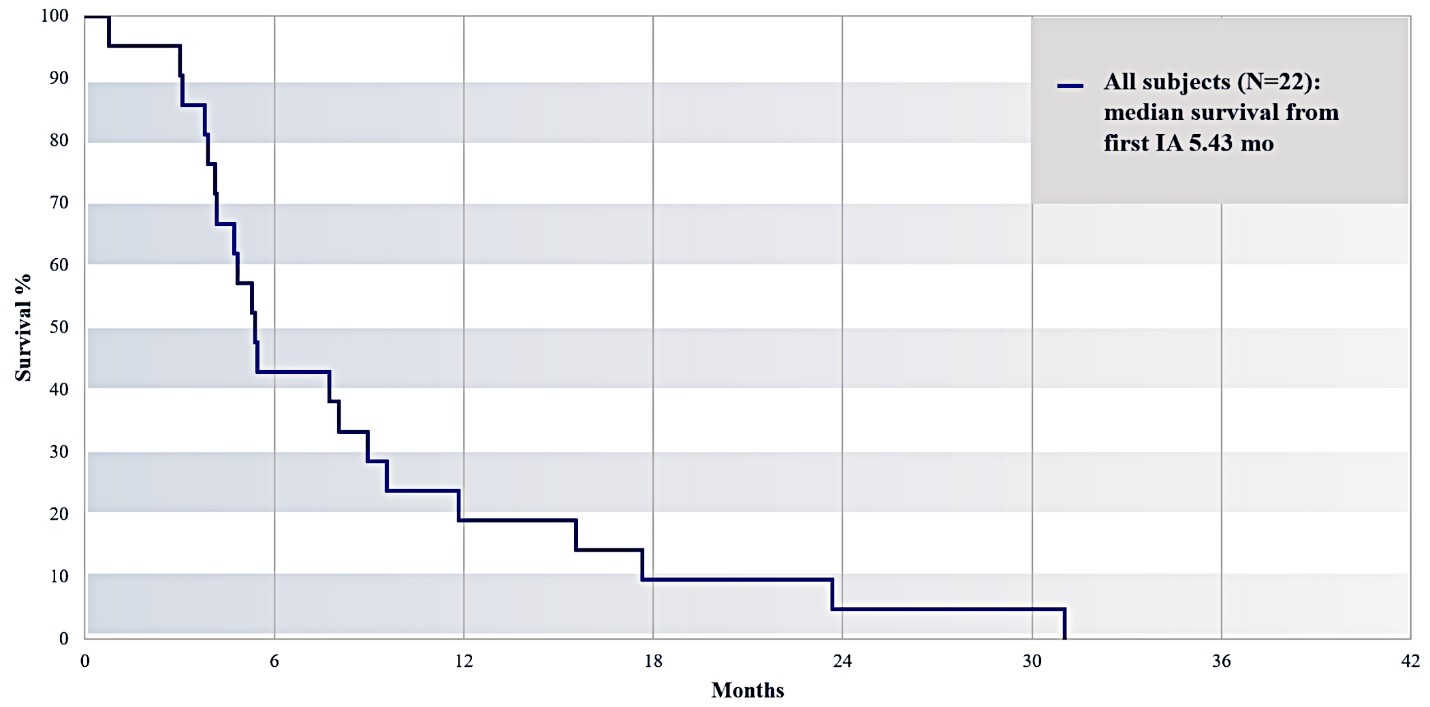
Figure 16 Overall RR2 observational registry study cohort (N=22) survival from first IA treatment until date of death.
| -77- |
Overall Survival of All Subjects (Median Overall Survival 13 Months)
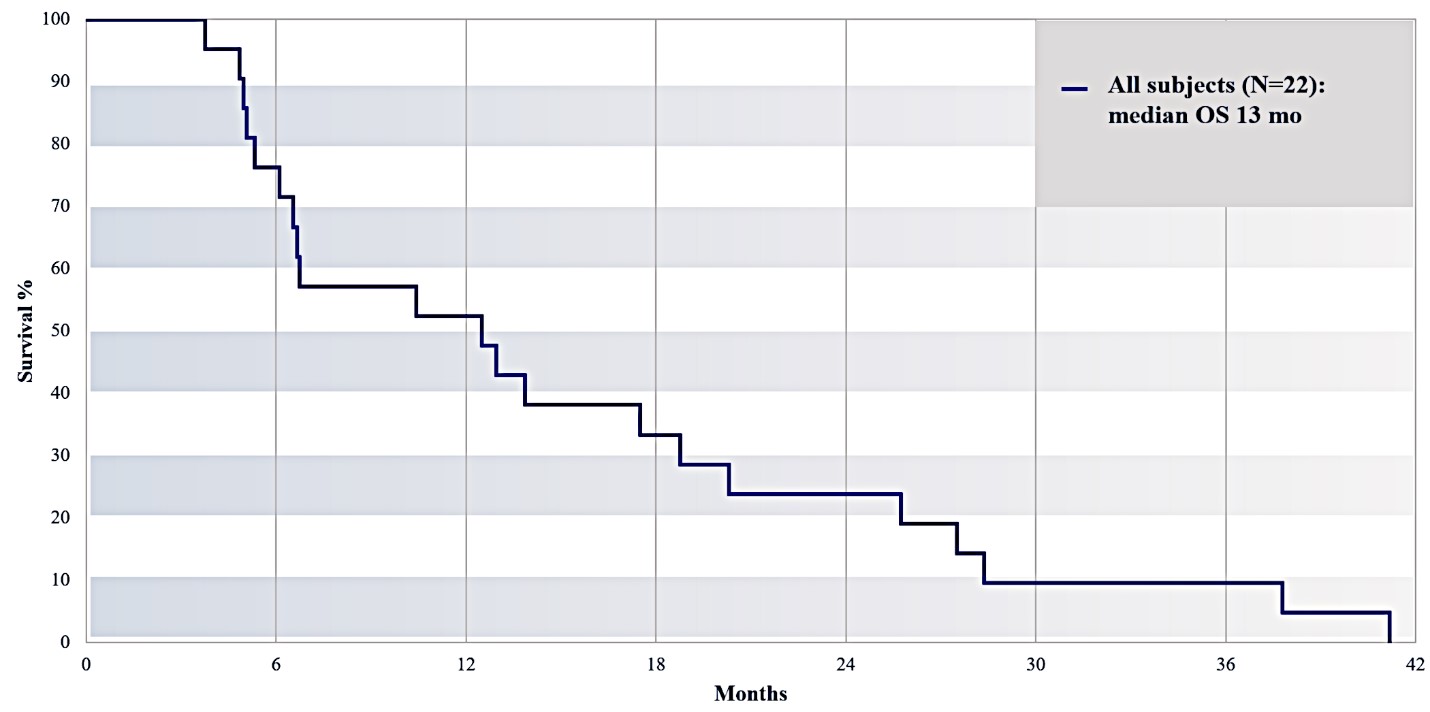
Figure 17 Overall RR2 observational registry study cohort (N=22) overall survival from date of diagnosis.
As in Phase 1/2 RenovoCath/Gem RR1, subjects with prior radiation and chemotherapy demonstrated longer survival than other subjects.
Subjects with Prior Chemo or Prior Chemoradiation Survive Longer
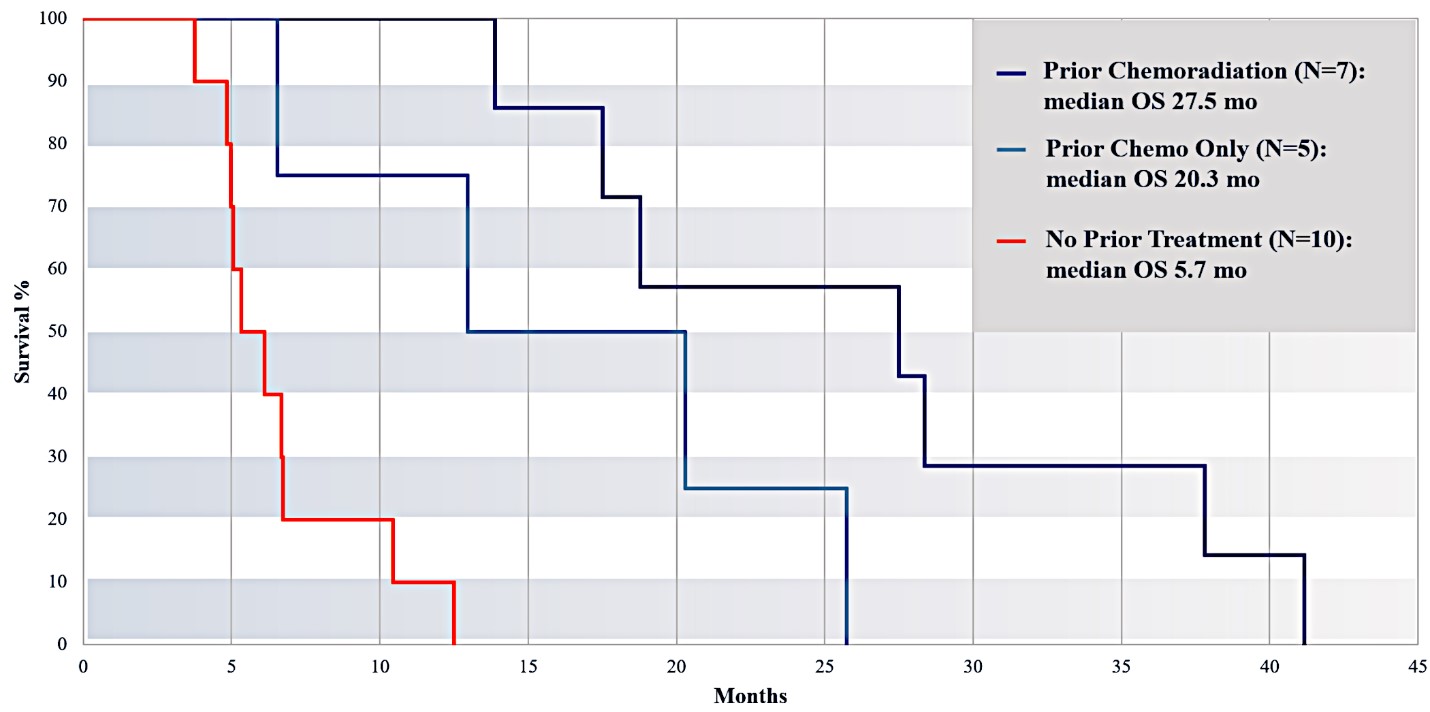
Figure 18 Survival as function of previous treatment received for RR2 registry study subjects. As in RR1 study, subjects with prior radiation and chemotherapy demonstrated longer survival than other subjects.
| -78- |
In summary the results of RR2 observational registry, build on the finding of the Phase 1/2 RenovoCath/Gem RR1 study, where the use of RenovoCath in this patient population can be undertaken with adequate safety, with adequate attention to procedural technique including careful use/manipulation of guide catheter to prevent arterial dissection, and administration of peri-procedure antibiotics in patients with prior biliary stent/drain.
RR1 and RR2 Conclusions
Patients with LAPC treated with RenovoTAMP showed efficacy signals:
| ● | Survival of patients with LAPC following RenovoTAMP was similar to that observed in the previous Phase 1/2 RenovoCath/Gem RR1 study. | |
| ● | Patients with biliary stents or drains who received RenovoTAMP who received prophylactic peri-procedure antibiotics experienced no episodes of sepsis. | |
| ● | LAPC patients who received prior radiation and chemotherapy had longer survival than those without prior radiotherapy. |
Based on the FDA’s safety review of our phase 1/2 study and clinical outcome, the FDA allowed us to proceed to evaluate RenovoGem within our Phase 3 registration clinical trial.
TIGeR-PaC Phase 3 Trial (RR3)
With completion of RR1 and RR2 we obtained FDA approval for Phase 3 IND study in Feb 2018 comparing RenovoTAMP with intra-arterial gemcitabine to standard of care. In the FDA pre-IND meeting, the FDA confirmed the study design and endpoints and indicated that this Phase 3 study should result in New Drug Application approval if successful. Simultaneously in April 2018 we obtained Orphan drug designation for use of RenovoGem in patients with LAPC.
The primary endpoint of the study is overall survival, from time of randomization until death. Secondary endpoints include but not limited to progression free survival and quality of life questionnaire results. The study is a multi-center, open-label, randomized active-controlled study of subjects with locally advanced pancreatic adenocarcinoma which is unresectable according to NCCN guidelines. The study is currently enrolling patients in the US and Belgium.
The study design is follows: all patients receive a four-month induction phase of IV chemotherapy and radiation prior to randomizing to 4 cycles (8 treatments) of RenovoTAMP or 4 cycles of continuation of IV chemotherapy. While RenovoTAMP data versus historical controls predicts a much greater survival benefit, the TIGeR-PaC study is powered to detect a 6-month survival benefit. The TIGeR-PaC SAP includes an interim analysis with the potential of early FDA approval or Breakthrough Status with a strong efficacy signal early in the study. A study flowchart is shown below. Subjects with stable or responding disease after approximately 4 months in induction therapy and who are not surgical candidates will then be randomized 1:1.
| -79- |
TIGeR-PaC Study Flowchart with Chemoradiation Induction Phase
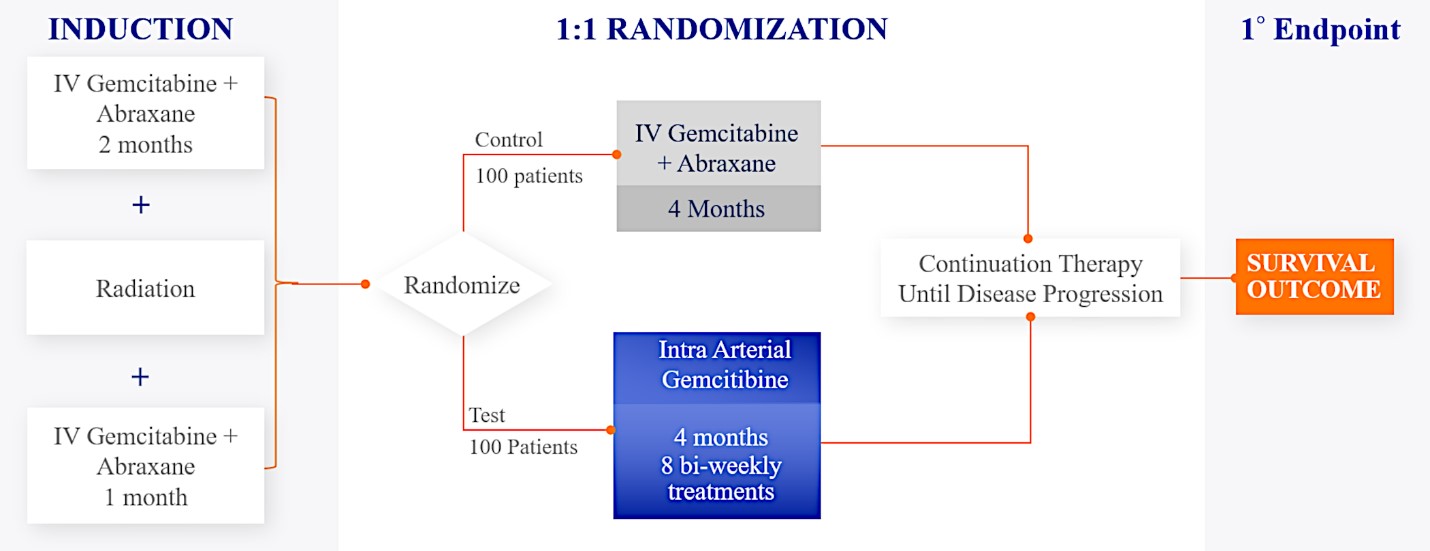
Figure 19 TIGeR-PaC Study Flowchart. All subjects undergo a 4-month induction phase that includes IV gemcitabine + Abraxane and radiation therapy. If the subjects are stable with LAPC post-induction, they are randomized 1:1 into control group (IV gemcitabine + Abraxane) versus treatment group (intra-arterial gemcitabine via RenovoTAMP therapy). Subjects are then administered continuation therapy until disease progression and followed through survival.
The study currently has 28 active clinical sites. As of July 15, 2021, 145 participants have enrolled in the study and ultimately approximately 340 participants are expected to be enrolled in the study with 200 participants randomized in the US and Europe. An interim analysis is planned when a total of 65 deaths (randomized patients) have been observed and is projected to be in the second half of 2022.
It is projected that enrollment and randomization of the entire cohort will be completed in 2023.
Clinical Pharmacokinetic (PK) Data in Patients with LAPC Treated with Gemcitabine via RenovoTAMP
We expect intra-arterial gemcitabine delivered via the RenovoTAMP technique to have distinct PK from IV gemcitabine dosing. Furthermore, with local delivery of gemcitabine into the tissue via RenovoTAMP and drainage into the liver prior to systemic circulation, lower systemic levels of gemcitabine would be anticipated.
We are collecting blood samples for PK analysis in 15 patients from our TIGeR-PaC Phase 3 study (Figure 20, below). Our initial data on the first 5 patients demonstrate a 3-fold decrease in Cmax, or maximum serum concentration of a drug, with RenovoTAMP compared to established levels for IV gemcitabine.
| -80- |
Systemic Levels of Gemcitabine Reduced by 2/3 with RenovoTAMP versus IV Gemcitabine
Figure 20 Initial blood PK data demonstrated a 3-fold decrease in systemic concentration of gemcitabine (Cmax) with RenovoTAMP compared to established levels for IV gemcitabine.
Second Solid Tumor Indication
HCCA Overview
Cholangiocarcinoma is the second most common primary malignant tumor of the liver with over 7,000 new cases diagnosed annually in the US. Cholangiocarcinoma (CCA) develops after malignant transformation of the biliary tract mucosa. The global market of CCA was estimated to be $385 million in 2018 and the US accounted for the largest market size of CCA. Furthermore, the market size for global CCA therapeutics is estimated to grow by $83 million during 2019-2023 with a compound annual growth rate of 6%. Advanced age, male gender, primary sclerosing cholangitis (PSC), inflammatory bowel disease, pancreatitis, and cirrhosis are some predisposing factors for development of CCA.
Based on the tumor location CCA is defined as intra-hepatic, or within the liver, and extra-hepatic, or hilar. The HCCA subset of CCA patients are about 3000 cases per year. HCCA is a disease with an exceptionally poor prognosis.
HCCA Current Treatment Landscape and Limitations
Most patients with HCCA have localized disease with possible extension of the tumor around the bile duct. Based on local extension of the disease, treatment options include surgery, chemotherapy, and radiation therapy. Surgical resection offers the only chance for curative therapy for patients with HCCA; however, the surgery is associated with high mortality and morbidity and most patients are not candidates. Systemic chemotherapy is a primary mode of treatment in these patients as a form of palliation, which is associated with morbidity and limited improvement in survival.
Current standard chemotherapy treatment in these patients is based on the ABC-2 Trial: a randomized trial of 410 patients with unresectable CCA (the study included both intrahepatic, within the liver, and hilar cholangiocarcinoma patients). Patients were treated with gemcitabine plus cisplatin, consisting of a three-week cycle, with treatments on Days 1 and 8 and dosing of gemcitabine at 1000mg/m2 and cisplatin at 25mg/m2. Reported median O.S. for patients on such a regimen (11.7 months) was greater than patients receiving gemcitabine alone (8.1 months). Commonly observed Grade 3-4 toxicity of this standard of care treatment include anemia, leukopenia, neutropenia, thrombocytopenia, lethargy, nausea/vomiting, and anorexia. In the ABC-02 trial the efficacy of gemcitabine/cisplatin combination was not significantly different from that of gemcitabine alone in patients with hilar cholangiocarcinoma. For this reason, a practice standard of care has not been established for hilar cholangiocarcinoma.
| -81- |
RenovoGem for the Treatment of HCCA
Similar to RenovoGem for LAPC, we believe that RenovoGem may overcome the current treatment limitations of HCCA. In this setting, patients with HCCA have several features that make them attractive for treatment via our RenovoTAMP therapy.
| ● | Local disease with possible extension of disease to local vasculature | |
| ● | Avascular nature of the tumor lends itself to our RenovoTAMP approach, overcoming the limitations in drug delivery by targeting the periductal proper hepatic artery or left or right hepatic artery | |
| ● | Gemcitabine, used as a target molecule for this tumor type, has already been demonstrated to be safe in terms of local toxicity targeted via our approach to this vasculature and organ | |
| ● | The bile duct around the hilum is usually within 1-14mm (mean of 3.8 mm) of the hepatic artery: a reasonable target for RenovoTAMP therapy given the potential 4cm tissue penetration of drug |
Clinical Development of RenovoGem in HCCA
BENEFICIAL Study Design and Rationale
The FDA granted us Orphan Drug Designation in June of 2020 for the treatment of CCA with RenovoTAMP. Shortly after the granted designation we began working on the BENEFICIAL study protocol. We intend to meet with the FDA to propose a phase 2/3 trial for approval of RenovoTAMP for HCCA and, if approved by the FDA, to initiate such a study in the first half of 2022.
We expect the study duration will be up to 4.5 years, with an estimated 30 months of enrollment and 2 years of follow-up to have 150 subjects enrolled and 100 subjects randomized. The study is a multi-center, open-label, randomized active-controlled study of subjects with hilar cholangiocarcinoma which is unresectable according to NCCN guidelines. We intend to conduct the study in up to 8 U.S. sites.
The primary endpoint of the study is time to treatment failure (progression or if the subject can no longer tolerate treatment) from randomization. Secondary endpoints include, but not limited to, overall survival, time to progression from randomization to radiologic progression, progression free survival and tumor response by RECIST 1.1 guidelines.
There are four phases to the study after screening: Induction, Randomization Treatment, Continuing Therapy and Survival follow-up. All subjects receive an initial four-month induction phase of IV chemotherapy (gemcitabine + cisplatin) and radiation. Subjects with stable or responding disease after approximately 3 months of induction therapy, and who are not surgical candidates, will then be randomized 1:1 to either 4 cycles (8 treatments) of RenovoTAMP or 5 cycles of IV chemotherapy in addition to the 3 received during induction. Subjects will receive capecitabine during continuing therapy after completion of randomization treatment without disease progression or treatment intolerance. Subjects will be followed for up to three years for survival.
Key inclusion criteria include biopsy proven cholangiocarcinoma within 6 weeks of consent, unresectable cholangiocarcinoma (stage 4A included), measurable disease as per RECIST 1.1 must be present, and ECOG Performance Status score of 0-1. Key exclusion criteria include contraindications to angiography and selective visceral catheterization, location of tumor more than 15mm from targeted IA therapy location as per imaging, cirrhosis (Child-Pugh > Class B7) and clinically evident ascites.
| -82- |
BENEFICIAL Study Flowchart
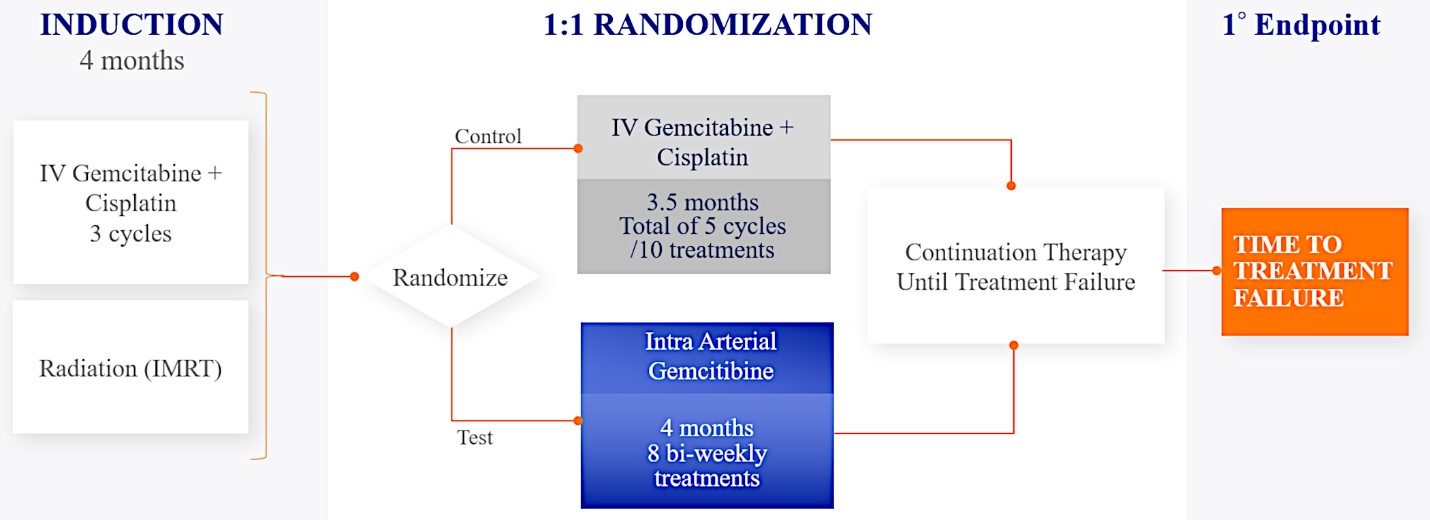
Figure 21 BENEFICIAL study flowchart. All subjects undergo a 4-month induction phase that includes IV gemcitabine + Cisplatin and radiation therapy. If the subjects are stable with LAPC post-induction, they are randomized 1:1 into control group (IV gemcitabine + Cisplatin) versus treatment group (intra-arterial gemcitabine via RenovoTAMP therapy). Subjects are then administered continuation therapy until treatment failure (cessation of continuation therapy, change in continuation therapy, progression, and/or death).
Market Opportunity
We are currently developing RenovoGem for LAPC, intend to develop it for HCCA, and potentially for locally advanced lung cancer, locally advanced uterine cancer and glioblastoma. We estimate that the total annualized addressable market opportunity for RenovoGem for our first market, LAPC, in the United States is approximately $0.5 billion and globally can exceed $1 billion based on a third-party market research analysis. The total cost of care of a patient on the standard of care treatment of gemcitabine + Abraxane is estimated at $67,216, which if applied to 60,000 pancreatic cancer cases per year would total $4 billion per year for the total U.S. pancreatic cancer market.
Beyond our initially targeted subset of LAPC patients, we see potential to evaluate RenovoGem in additional settings where it may help to get more patients to surgery, prolong life, enhance systemic therapy or provide local therapy with fewer side effects than alternative treatments. These may include patients with stage 1 or stage 2 pancreatic cancer receiving neoadjuvant as well as in subpopulations of patients with metastases who also have locally advanced disease. From third-party market research, a number of physicians mentioned a role for local therapy as an adjunct for systemic chemotherapy as well as for patients who decline systemic chemotherapy. Beyond pancreatic cancer, there are additional markets we will be exploring.
Below is published epidemiology data showing the 2021 estimated annual incidence of the following tumor types in the United States to be greater than 350,000 patients in the aggregate.
| -83- |
Market Opportunity for RenovoTAMP
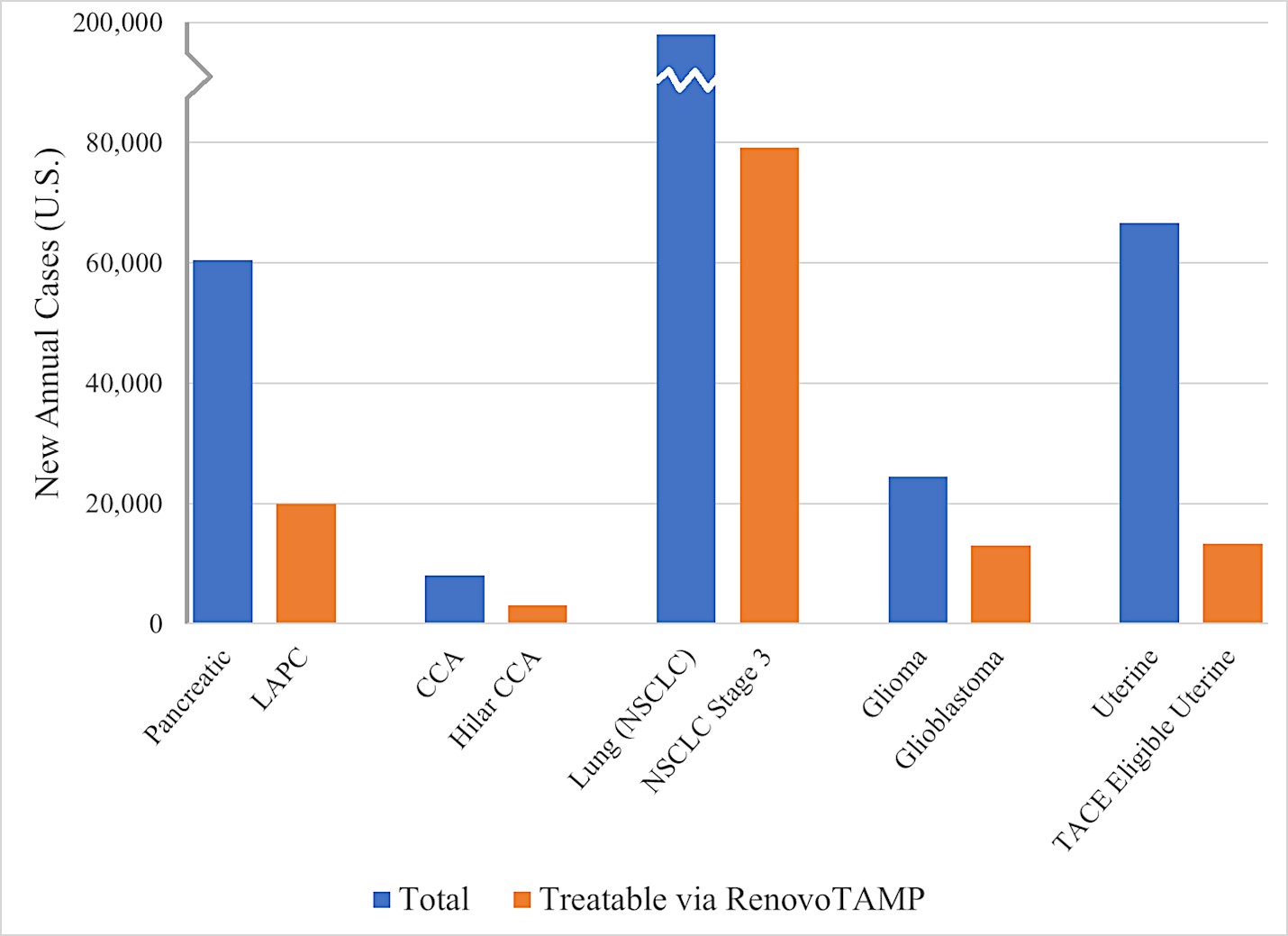
Figure 22 Market Opportunity of target cancers, showing overall incidence in blue, and those treatable via RenovoTAMP in orange.
| ● | All Pancreatic Cancers compared to Locally Advanced Pancreatic Cancers (LAPC) | |
| ● | All Cholangiocarcinoma (CCA) compared to Hilar CCA | |
| ● | All Non-Small Cell Lung Cancers (NSCLC) compared to Stage 3 NSCLC | |
| ● | All Glioma compared to Glioblastoma | |
| ● | All Uterine cancers versus Transcatheter arterial chemoembolization (TACE) eligible uterine cancers |
Intellectual Property
Our success depends in part on our ability to obtain patents and trademarks, maintain trade secret and know-how protection, enforce our proprietary rights against infringers, and operate without infringing on the proprietary rights of third parties. Because of the length of time and expense associated with developing new products and bringing them through the regulatory approval process, the health care industry places considerable emphasis on obtaining patent protection and maintaining trade secret protection for new technologies, products, processes, know-how, and methods.
Our intellectual property protection stems from several issued device and method patents on our RenovoCath delivery system that optimizes delivery of the anti-cancer drug and the RenovoTAMP therapy platform. Our issued patents also provide exclusivity as it relates to utilizing RenovoCath with anti-cancer drugs.
| -84- |
We have 7 US patents issued, 1 European patent issued, and 1 patent pending in each of China, Japan, Europe, and India. In addition, we have three pending US patents. We continue to explore additional opportunities to further bolster our IP position. Table 3 below describes our issued patents, all of which have been assigned to us.
Table 3 RenovoRx has Significant Patent Protection with 8 Issued Patents
Family | App. No. Filing Date | Type of Patent Protection | Patent Focus | Patent # | Estimated Expiration | |||||
| Dual Balloon Methods and Apparatuses | 12/958711
Filed: 12/2/2010 | U.S. Utility patent | Methods: isolating splenic artery with 2 balloons (sliding inner catheter) | U.S. 8,821,476 | January 25, 2033 | |||||
| Dual Balloon Methods and Apparatuses | 14/870833
Filed: 9/30/2015 | U.S. Utility patent | Apparatus: 2 balloons, seal to isolate lumen, and infusion aperture | U.S. 9,463,304 | December 2, 2030 | |||||
| Dual Balloon Methods and Apparatuses | 14/293603
Filed: 6/2/2014 | U.S. Utility patent | Apparatus: 2 balloons (sliding inner catheter), 2 ports for fluid handling | U.S. 9,457,171 | April 16, 2031 | |||||
| Dual Balloon Methods and Apparatuses | 15/351922
File: 11/15/2016 | U.S. Utility patent | Kits for chemotherapy including catheter with 2 balloons, an infusion aperture and 2 ports | U.S. 10,512,761 | April 16, 2031 | |||||
| Dual Balloon Methods and Apparatuses | 10/8351107
Filed: 12/2/2010 | E.U. Utility patent, nationalized in BE, CH, DE, ES, FR, GB, IE, IT and NL | A two occlusion element, adjustable delivery apparatus having inner and outer catheter, seal to isolate lumen | E.U. 2506913 | December 2, 2030 | |||||
| Side Branch Isolation Device and Methods | 14/958428
Filed: 12/3/2015 | U.S. Utility patent | Apparatuses and Methods: 3 balloon catheters for isolating side branches | U.S. 10,099,040 | December 3, 2035 | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | 15/807011
Filed: 11/8/2017 | U.S. Utility patent | Methods delivering radiation to devascularize then TAMP | U.S., 10,695,543 | August 28, 2038 | |||||
| Dual Balloon Methods and Apparatuses | 16/685950
Filed: 11/15/2019 | U.S.
Utility patent (Pending application) | Methods of treating bile duct | PENDING | April 16, 2031* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | 16/685974
Filed: 11/15/2019 | U.S.
Utility patent (Pending application) | Devascularization in conjunction with TAMP | U.S. 11,052,224 | November 8, 2037* | |||||
| Dual Balloon Methods and Apparatuses | IN
1632MUMNP2012 Filed: 12/2/2010 | IN
Utility patent (Pending application) | A two occlusion element, adjustable delivery apparatus having inner and outer catheter, seal to isolate lumen | PENDING | December 2, 2030* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | CN
2018800033529 Filed: 5/18/2018 | CN
Utility patent (Pending application) | Devascularization in conjunction with TAMP | PENDING | November 8, 2037* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | EP
187315908 Filed: 5/18/2018 | EP
Utility patent (Pending application) | Devascularization in conjunction with TAMP | PENDING | November 8, 2037* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | JP
2020514151 Filed: 5/18/2018 | JP
Utility patent (Pending application) | Devascularization in conjunction with TAMP | PENDING | November 8, 2037* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | 17/315220 Filed: 5/7/2021 |
U.S. Utility patent (Pending application) |
Devascularization in conjunction with TAMP | PENDING | November 8, 2037* | |||||
| Trans-Arterial Micro-Perfusion (TAMP) | 17/367046 Filed: 7/2/21 |
U.S. Utility patent (Pending application) |
Devascularization in conjunction with TAMP | PENDING | November 8, 2037* |
* Predicted earliest expiration date. The actual expiration date will depend on factors related to patent prosecution and issuance.
** Assumes all maintenance fees are paid.
We have additional market exclusivity protection with Orphan Drug Designation for seven years post-approval. Gemcitabine is generic; however, we have exclusivity for the intra-arterial route of administration. RenovoGem is regulated by the FDA as a novel oncology drug product. We intend to make intra-arterial gemcitabine and RenovoCath available as a combined product and not to make either component available separately. Once approved, we will have exclusivity over the use of intra-arterial gemcitabine as it will be approved by the FDA in combination with RenovoCath.
When appropriate, we actively pursue protection of our proprietary products, technologies, processes, and methods by filing United States and international patent and trademark applications. We seek to pursue additional patent protection for technology invented through research and development, manufacturing, and clinical use of our technology that will enable us to expand our patent portfolio around advances to our current systems, technology, and methods for our current applications as well as beyond the treatment of cancers in the liver.
There can be no assurance that the pending patent applications will result in the issuance of patents, that patents issued to or licensed by us will not be challenged or circumvented by competitors, or that these patents will be found to be valid or sufficiently broad to protect our technology or provide us with a competitive advantage.
To maintain our proprietary position, we also rely on trade secrets and proprietary technological experience to protect proprietary manufacturing processes, technology, and know-how relating to our business. We rely, in part, on confidentiality agreements with our marketing partners, employees, advisors, vendors and consultants to protect our trade secrets and proprietary technological expertise. In addition, we also seek to maintain our trade secrets through maintenance of the physical security of the premises where our trade secrets are located. There can be no assurance that these agreements will not be breached, that we will have adequate remedies for any breach, that others will not independently develop equivalent proprietary information or that third parties will not otherwise gain access to our trade secrets and proprietary knowledge.
| -85- |
In certain circumstances, United States patent law allows for the extension of a patent’s duration for a period of up to five years after FDA approval. We intend to seek extension for one of our patents after FDA approval if it has not expired prior to the date of approval. In addition to our proprietary protections, the FDA has granted us two orphan drug designations that provide us a seven-year period of exclusive marketing beginning on the date that our NDA is approved by the FDA for the designated orphan drug. While the exclusivity only applies to the indication for which the drug has been approved, we believe that this exclusivity will provide us with added protection once commercialization of an orphan drug designated product begins.
There has been and continues to be substantial litigation regarding patent and other intellectual property rights in the pharmaceutical and medical device areas. If a third party asserts a claim against us, we may be forced to expend significant time and money defending such actions and an adverse determination in any patent litigation could subject us to significant liabilities to third parties, require us to redesign our product, require us to seek licenses from third parties, and, if licenses are not available, prevent us from manufacturing, selling or using our system. Additionally, we plan to enforce our intellectual property rights vigorously and may find it necessary to initiate litigation to enforce our patent rights or to protect our trade secrets or know-how. Patent litigation can be costly and time consuming and there can be no assurance that the outcome will be favorable to us.
Manufacturing and Supply
We rely on a single-source contract manufacturer, Medical Murray, North Barrington, IL, for the device component, RenovoCath, of the drug-device combination and are subject to regulatory requirements of the FDA’s Quality System Regulation (QSR), for medical devices sold in the United States, and the European Medical Device Directive 93/42/EEC and amendments, or MDD, for medical devices marketed in the European Union. We have an agreement in place with Medical Murray to produce the RenovoCath through October 2024 with automatic annual renewal until termination by either party with 12 months’ notice. While we believe Medical Murray has the capabilities to scale RenovoCath production to peak forecasted commercial volumes, manufacturing can be transferred to additional vendors if needed.
The FDA monitors compliance with QSR through periodic inspections of both our facility and the facility of our contact manufacturer. Our European Union Notified Body, British Standards Institute (BSI), monitors compliance with the MDD requirements through both annual scheduled audits and periodic unannounced audits of our facilities as well as our contract manufacturer’s facilities.
Our failure or the failure of our contract manufacturer to maintain acceptable quality requirements could result in the shutdown of our manufacturing operations or the recall of products which could be detrimental to our company. If our contract manufacturer fails to maintain acceptable quality requirements, we may have to qualify a new contract manufacturer and could experience a material adverse effect to manufacturing and manufacturing delays as a result.
We do not own or operate and do not intend to establish our own gemcitabine manufacturing facilities.
Within our TIGeR-PaC Phase 3 trial, hospitals are sourcing generic gemcitabine labeled for IV use from their own respective pharmacies to use in conjunction with the RenovoCath for the RenovoTAMP procedures. In the commercial setting, we expect to rely on contract manufacturing organizations for gemcitabine production, relabeling and co-packaging with the RenovoCath. The formulation of gemcitabine used in the TIGeR-PaC Phase 3 trial and in the commercial setting will be identical, however, the labeling of gemcitabine will be intra-arterial gemcitabine to be used exclusively in conjunction with the RenovoCath.
Government Regulation
Our products are subject to extensive and rigorous government regulation by foreign regulatory agencies and the FDA. Foreign regulatory agencies, the FDA and comparable regulatory agencies in state and local jurisdictions impose extensive requirements upon the clinical development, pre-market clearance and approval, manufacturing, labeling, marketing, advertising and promotion, pricing, storage and distribution of pharmaceutical and medical device products. Failure to comply with applicable foreign regulatory agency or FDA requirements may result in Warning Letters, fines, civil or criminal penalties, suspension or delays in clinical development, recall or seizure of products, partial or total suspension of production or withdrawal of a product from the market.
| -86- |
United States Regulatory Environment
In the US, the FDA regulates drug and device products under the FDCA, and its implementing regulations. RenovoGem is subject to regulation as a combination product, which means it is composed of both a drug component and device component. Each component of a combination product is subject to the requirements established by the FDA for that type of component and if marketed individually, each component would be subject to different regulatory pathways and reviewed by different centers within the FDA. A combination product, however, is assigned to a center that will have primary jurisdiction over its pre-market review and regulation based on a determination of its primary mode of action, which is the single mode of action that provides the most important therapeutic action. In the case of RenovoGem, the primary mode of action is attributable to the drug component of the product, which means that the Center for Drug Evaluation and Research, has primary jurisdiction over its pre-market development and review. Combination products where the drug provides the primary mechanism of action are often referred to as “drug-led combination” products.
The process required by the FDA before drug product candidates, including drug-led combination products, may be marketed in the United States generally involves the following:
| ● | submission to the FDA of an IND, which must become effective before human clinical trials may begin and must be updated periodically, but at least annually; | |
| ● | completion of extensive preclinical laboratory tests and preclinical animal studies, all performed in accordance with the FDA’s Good Laboratory Practice, or GLP, regulations; | |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication; | |
| ● | submission to the FDA of an NDA after completion of all pivotal clinical trials; | |
| ● | a determination by the FDA within 60 days of its receipt of an NDA to file the NDA for review; | |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product is produced and tested to assess compliance with current good manufacturing practice, or cGMP, regulations; and | |
| ● | FDA review and approval of an NDA prior to any commercial marketing or sale of the drug in the United States. |
The development and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product will be granted on a timely basis, if at all.
The results of preclinical tests (which include laboratory evaluation as well as GLP studies to evaluate toxicity in animals) for a particular product candidate, together with related manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. IND submissions may not result in FDA authorization to commence a clinical trial. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development. Further, an independent institutional review board, or IRB, for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the study until completed. The FDA, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive good clinical practice regulations and regulations for informed consent and privacy of individually identifiable information. Similar requirements to the United States IND are required in the EU and other jurisdictions in which we may conduct clinical trials.
| -87- |
Clinical Trials
For purposes of NDA submission and approval, clinical trials are typically conducted in the following sequential phases, which may overlap:
| ● | Phase 1 Clinical Trials. Studies are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, distribution, metabolism and excretion, typically in healthy humans, but in some cases in patients. | |
| ● | Phase 2 Clinical Trials. Studies are generally conducted in a limited patient population to identify possible adverse effects and safety risks, explore the initial efficacy of the product for specific targeted indications and to determine dose range or pharmacodynamics. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. | |
| ● | Phase 3 Clinical Trials. These are commonly referred to as pivotal studies. When Phase 2 evaluations demonstrate that a dose range of the product is effective and has an acceptable safety profile, Phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, provide substantial evidence of clinical efficacy and further test for safety in an expanded and diverse patient population at multiple, geographically dispersed clinical trial centers. | |
| ● | Phase 4 Clinical Trials. The FDA may approve an NDA for a product candidate, but require that the sponsor conduct additional clinical trials to further assess the drug after NDA approval under a post-approval commitment. In addition, a sponsor may decide to conduct additional clinical trials after the FDA has approved an NDA. Post-approval trials are typically referred to as Phase 4 clinical trials. |
Sponsors of clinical trials may submit proposals for the design, execution, and analysis for their pivotal trials under a Special Protocol Assessment, or SPA. A SPA is an evaluation by the FDA of a protocol with the goal of reaching an agreement that the Phase 3 trial protocol design, clinical endpoints, and statistical analyses are acceptable to support regulatory approval of the drug product candidate with respect to effectiveness for the indication studied. Under a SPA, the FDA agrees to not later alter its position with respect to adequacy of the design, execution or analyses of the clinical trial intended to form the primary basis of an effectiveness claim in an NDA, without the sponsor’s agreement, unless the FDA identifies a substantial scientific issue essential to determining the safety or efficacy of the drug after testing begins.
Prior to initiating our currently ongoing Phase 3 clinical trial(s), we submitted a proposal for the design, execution and analysis under a SPA.
New Drug Applications (NDAs)
The results of drug development, preclinical studies and clinical trials are submitted to the FDA as part of an NDA. NDAs also must contain extensive chemistry, manufacturing and control information. An NDA must be accompanied by a significant user fee, which may be waived in certain circumstances. Once the submission has been accepted for filing, the FDA’s goal is to review applications within ten months of submission or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months from submission. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. For new oncology products, the FDA will often solicit an opinion from an Oncologic Drugs Advisory Committee, or ODAC, a panel of expert authorities knowledgeable in the fields of general oncology, pediatric oncology, hematologic oncology, immunologic oncology, biostatistics, and other related professions. The ODAC panel reviews and evaluates data concerning the safety and effectiveness of marketed and investigational human drug products for use in the treatment of cancer, and makes appropriate recommendations to the Commissioner of Food and Drugs. The FDA is not bound by the recommendation of an advisory committee. The FDA may deny approval of an NDA by issuing a Complete Response Letter, or CRL, if the applicable regulatory criteria are not satisfied. A CRL may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Approval may be contingent on a Risk Evaluation and Mitigation Strategy, or REMS, that limits the labeling, distribution or promotion of a drug product. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing, including Phase IV clinical trials, and surveillance programs to monitor the safety effects of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
| -88- |
There are three primary regulatory pathways for an NDA under Section 505 of the FDCA: Section 505 (b)(1), Section 505 (b)(2) and Section 505(j). A Section 505 (b)(1) application is used for approval of a new drug (for clinical use) whose active ingredients have not been previously approved. A Section 505 (b)(2) application is used for a new drug that relies on data not developed by the applicant. Section 505(b)(2) of the FDCA was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, also known as the Hatch-Waxman Act. This statutory provision permits the approval of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The Hatch-Waxman Act permits the applicant to rely in part upon the FDA’s findings of safety and effectiveness for previously approved products. Section 505(j) application, also known as an abbreviated NDA, is used for a generic version of a drug that has already been approved.
The NDA review process for drug-led combination includes a review of the device constituent. In this case, the device constituent for RenovoGem is RenovoCath, which is cleared by the FDA.
Orphan Drug Exclusivity
Some jurisdictions, including the United States, may designate drugs for relatively small patient populations as orphan drugs. Pursuant to the Orphan Drug Act, the FDA grants orphan drug designation to drugs intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States. The orphan designation is granted for a combination of a drug entity and an indication and therefore it can be granted for an existing drug with a new (orphan) indication. Applications are made to the Office of Orphan Products Development at the FDA and a decision or request for more information is rendered in 60 days. NDAs for designated orphan drugs are exempt from user fees, obtain additional clinical protocol assistance, are eligible for tax credits up to 50% of research and development costs, and are granted a seven-year period of exclusivity upon approval. The FDA cannot approve the same drug for the same condition during this period of exclusivity, except in certain circumstances where a new product demonstrates superiority to the original treatment. Exclusivity begins on the date that the marketing application is approved by the FDA for the designated orphan drug, and an orphan designation does not limit the use of that drug in other applications outside the approved designation in either a commercial or investigational setting.
We have received orphan drug designations for RenovoGem for LAPC and for HCCA.
The granting of orphan drug designations does not mean that the FDA has approved a new drug. Companies must still pursue the rigorous development and approval process that requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product will be granted at all, or on a timely basis.
Other Regulatory Requirements
Products manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping, annual product quality review and reporting requirements. Adverse event experiences with the product, including both drug-related and device-related adverse events (including device malfunctions), must be reported to the FDA in a timely fashion and pharmacovigilance programs to proactively look for these adverse events are mandated by the FDA. Drug and device manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMP, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Following such inspections, the FDA may issue notices on Form 483, Untitled Letters or Warning Letters that could cause us or our third-party manufacturers to modify certain activities. A Form 483 Notice, if issued at the conclusion of an FDA inspection, list conditions the FDA investigators believe may have violated cGMP or other FDA regulations or guidelines. In addition to Form 483 Notices and Untitled Letters or Warning Letters, failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as suspension of manufacturing, seizure of product, injunctive action or possible civil penalties. We cannot be certain that we or our present or future third-party manufacturers or suppliers will be able to comply with the cGMP regulations and other ongoing FDA regulatory requirements. If we or our present or future third-party manufacturers or suppliers are not able to comply with these requirements, the FDA may require us to recall our products from distribution or withdraw any potential approvals of an NDA for that product.
| -89- |
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, dissemination of off-label information, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional preclinical studies and clinical trials. Failure to comply with these requirements can result in adverse publicity, Warning Letters, corrective advertising and potential civil and criminal penalties.
Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties, in particular in oncology. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers’ communications regarding off-label use.
Foreign Regulatory Environment
If we seek to market RenovoGem in foreign jurisdictions, we could become subject to a variety of foreign regulations regarding development, approval, commercial sales, and distribution of our products in addition to regulations in the United States. Whether or not we obtain FDA approval for a product, we must obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and can involve additional product testing and additional review periods. The review process may take longer or shorter than that required to obtain FDA approval. The requirements governing, among other things, the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. If we fail to comply with applicable foreign regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, and/or criminal prosecution.
Reimbursement
We are developing a new drug product, RenovoGem, which is intra-arterial gemcitabine delivered via the proprietary RenovoCath delivery system. If the drug is approved, it is expected to be sold together with the catheter used to administer the drug in the National Drug Code (NDC) created when the drug receives FDA approval. The reimbursement pathway involves separate payments for the drug product and for the occlusion procedure to administer it. As to the latter, it is anticipated that the procedure is accurately described by an existing code with existing payment levels. Given the expectation that the drug will be a novel, non-generic drug, a unique code and payment based on pricing information for the product should be established.
For the reasons discussed above, we believe there is a clear path to reimbursement for RenovoGem and its related procedure in both the hospital outpatient and physician office settings (which may include freestanding entities such as catheterization laboratories). As is typical for a product still in clinical development, it is difficult to predict whether there would be any Medicare coverage obstacles, which there usually are not for FDA approved drugs being used for on-label use. We believe the most important step we can take to enhance reimbursement for our products is the development of published, peer-reviewed clinical literature supporting their clinical benefit.
| -90- |
Competition
The oncology biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies and strong competition. While we believe that our knowledge, leadership, experience, scientific resources, intellectual property, regulatory barriers, and the advanced stage of our clinical development provide us with competitive advantages, we may face competition from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies, worldwide. Many potential competitors have substantially greater scientific, research, financial, technical, and/or human resources than we do.
Many companies are active in the oncology market both in terms of commercially marketed products and products in development that could potentially compete with our products and product candidates for the treatment of solid tumors. Any product candidates that we successfully develop and commercialize may compete directly with approved and/or new therapies that may be approved in the future. Our competitors may also obtain FDA or foreign regulatory approval for their products more rapidly than we may obtain approval for our product candidates which could result in our competitors establishing a strong market position prior to us entering the market. Key competitive factors affecting the success of our product candidates, if approved, are likely to be their safety, efficacy, convenience, price, and the availability of reimbursement from government and other third-party payors. Many companies are developing new therapeutics and we cannot predict what the standard of care will be as our product candidate progresses through clinical development.
Currently, there are a handful of companies in Phase 3 clinical trials for the treatment of LAPC including Angiodynamics, Bausch Health, Fibrogen, NovoCure, and SynCore Biotechnology. We are aware of a number of companies in Phase 1 and Phase 2 clinical trials for the treatment of LAPC including one interventional company, TriSalus Lifesciences.
Many of our competitors have substantially greater financial, technological, research and development, marketing and personnel resources. In addition, some of our competitors have considerable experience in conducting clinical trials, regulatory, manufacturing and commercialization capabilities. Our competitors may develop alternative treatment methods, or achieve earlier product development, in which case the likelihood of us achieving meaningful revenues or profitability will be substantially reduced.
Employees
As of July 15, 2021, we had 7 full-time employees. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We believe our relationship with our employees is good. We have 8 key consultants in the areas of quality, regulatory, finance, legal, IT, clinical, and marketing.
Facilities
Our administrative headquarters is located at 4546 El Camino Real, Ste. 223, Los Altos, CA 94019. The office space is approximately 900 square feet, and the rent is $3,600/month. We believe that our facilities are adequate for our operations.
Legal Proceedings
From time to time, we are engaged in various legal actions, claims and proceedings arising in the ordinary course of business, none of which are expected to be material.
| -91- |
MANAGEMENT, EXECUTIVE COMPENSATION AND CORPORATE GOVERNANCE
Directors and Executive Officers
The following sets forth information about our directors, director nominees, and executive officers as of the date of this prospectus:
| Name | Age | Position | ||
| Shaun R. Bagai | 44 | Chief Executive Officer and Director | ||
| Christopher J. Lehman | 60 | Chief Financial Officer Nominee | ||
| Paul Manners | 70 | Vice President, Finance | ||
| Ramtin Agah | 55 | Chief Medical Officer, Founder, Director | ||
| Laurence J. Marton | 77 | Director | ||
| Una S. Ryan | 79 | Director | ||
| Maky Zanganeh | 50 | Director | ||
| Kristen Angela Macfarlane | 56 | Director | ||
| David Diamond | 71 | Director |
Shaun R. Bagai. Mr. Bagai has served as our Chief Executive Officer and director since June 2014. Prior to joining us, Mr. Bagai led Global Market Development for HeartFlow, Inc. from 2011 to 2014, which included directing Japanese market research, regulatory/payer collaboration, and Key Opinion Leader development to create value resulting in a company investment to form HeartFlow-Japan. During his tenure at HeartFlow, he successfully orchestrated their largest clinical trial to date and contracted HeartFlow’s first global customers. In addition, Mr. Bagai has launched new technologies into regional and global marketplaces in both large corporations and growth-phase novel technology companies. He was instrumental in developing the European market for renal denervation for the treatment of hypertension which led to the acquisition of the first renal denervation company, Ardian, Inc. by Medtronic in 2011. Mr. Bagai is a graduate from the University of California, Santa Barbara with a BSc. in Biology/Pre-Med. Mr. Bagai was selected to serve on our board of directors due to his tenure with our company and his industry experience.
Christopher J. Lehman. Since December 2020, Mr. Lehman has been affiliated with LS Associates, Inc. which provides on-demand C-level executives. Pursuant to our contract with LS Associates, Inc., Mr. Lehman has agreed to become our Chief Financial Officer upon consummation of this offering. From December 2019 to August 2020, Mr. Lehman was Chief Financial Officer of Avellino Lab USA, Inc., a precision medicine company. From October 2017 to February 2019, he was affiliated with Eureka Therapeutics, Inc., an immuno-oncology company, as Vice President, Finance from 2017 to 2018 and Chief Financial Officer from 2018 to 2019. From October 2013 to June 2017, Mr. Lehman was Vice President, Financial Strategy of Coherus Biosciences, Inc., a global biosimilars and immuno-oncology company. For over 25 years, Mr. Lehman has worked with venture-backed private and public industry leaders in areas of therapeutics, biosimilars, diagnostics, contract research and industrial biotechnology. He has experience in leveraging his broad industry network with investment banks, venture capital, and strategic pharmaceutical partners to raise dilutive and non-dilutive capital and has experience with investors and strategic partners in the United States, Europe, and Asia
Paul Manners. Mr. Manners has served as our Vice President, Finance since July 2021 and was our Chief Financial Officer from July 2019 to July 2021. In addition, since 2011 he has held several positions for Sandstone Diagnostics, an early stage medical device company, and is currently Senior Director Finance. Previously, Mr. Manners was a VP Finance & Marketing in Chiron/Novartis’ Diagnostic Division from 2003 to 2010 and the Chief Financial Officer, VP Sales & Customer Service for Amira Medical from 1997 to 2002. In addition, he has previously held various accounting and finance positions in various units for Johnson & Johnson from 1973 until 1996. Mr. Manners received an MBA from Rider University and a BBA from Northeastern University.
Ramtin Agah, MD Dr. Agah has served as the Chief Medical Officer and Co-Founder of the Company since December 2009 and has acted as Chairman of the Board of Directors since May 2018. Dr. Agah is currently an Interventional Cardiologist at El Camino Hospital, Mountain View, a role he began in September 2005. He also has acted as a physician consultant for Abbott Vascular since July 2012. Previously, Dr. Agah was an Assistant Professor of Internal Medicine with the Division of Cardiology, University of Utah. Dr. Agah completed a Fellowship in Interventional Cardiology with Cleveland Clinic Foundation, a Residency in Internal Medicine with Baylor College of Medicine, Houston, Texas, and a Fellowship in Cardiology with U.C.S.F. in San Francisco, CA. He is a MD graduate of University of Texas Southwestern Medical School. Dr. Agah was selected to serve on our board of directors due to his tenure with our company and his industry experience.
Laurence J. Marton. Dr. Marton has served as our director since December 2012. Dr. Marton is currently the Executive Chairman of Omniox Pharma, a role he began in July 2020. In addition, in the nonprofit sector, Dr. Marton serves on the Board of Trustees of the American Association for Cancer Research Foundation, and on the Board of Directors of Cancer Commons, Rapid Science, and the Bay Area American Committee for the Weizmann Institute of Science. In the for-profit sector, he serves on the Board of Directors of TOMA Biosciences, Microsonic Systems and Pathologica, is Chair of the Scientific Advisory Board of PharmaJet, and is on the Advisory Boards of Gem Pharmaceuticals and Silicom Ventures. Previously, Dr. Marton was Dean of the University of Wisconsin Medical School and Chaired the Department of Laboratory Medicine at UCSF, where he was a Professor in the Departments of Laboratory Medicine and Neurological Surgery. His research has resulted in more than 195 original publications, 60 scientific reviews and chapters, four books, and numerous patents. Dr. Marton received his MD from the Albert Einstein College of Medicine and his BA from Yeshiva University. Mr. Morton was selected to serve on our board of directors due to his tenure with our company and his industry experience.
| -92- |
Una S. Ryan, PhD, OBE. Dr. Ryan has served as our director since 2013. Dr. Ryan is currently the Managing Director of Golden Seeds (Silicon Valley), an angel investing group focused on women-led companies since 2009. Her board positions include Cortexyme, Inc., Elemental Machines, and Cambridge America. From 2010 until 2013, Dr. Ryan acted as President and CEO of Diagnostics For All, a nonprofit organization developing inexpensive diagnostics for global health and agricultural uses. Previously, from 1993 to 2008, she was President and CEO of AVANT Immunotherapeutics, Inc. a company developing vaccines and immunotherapeutics for cancer, travelers, food safety, and global health, and also a member of its Board of Directors. Dr. Ryan has held a number of positions in academia, including Research Professor of Surgery at Washington University School of Medicine in St. Louis and Professor of Medicine at the University of Miami School of Medicine. She was also Chair of the Massachusetts Biotechnology Council, served on its Board, as well as the Biotechnology Industry Organization, New England Healthcare Institute, Board of Associates of the Whitehead Institute, Strategy & Policy Council of the MIT Center for Biomedical Innovation, Massachusetts Life Sciences Collaborative Leadership Council and the Goddard Council on science, technology, engineering and mathematics education. Dr. Ryan holds a PhD in Cellular and Molecular Biology from Cambridge University and BS degrees in Zoology, Microbiology and Chemistry from Bristol University. In 2007, Dr. Ryan received the Albert Einstein Award for outstanding achievement in the life sciences, and in 2009, she received an Honorary Doctor of Science degree from Bristol University. Dr. Ryan was selected to serve on our board of directors due to her tenure with our company and her industry experience.
Maky Zanganeh, DDS, MBA. Dr, Zanganeh has served as our director since 2018. Dr. Zanganeh has more than 16 years of strategic planning, management, corporate, clinical, business and marketing experience in the pharmaceutical, medical device and technology industries. She has been the Chief Operating Officer of Summit Therapeutics since 2020 and the Founder and Chief Executive Officer of Maky Zanganeh and Associates, Inc., an investment, consulting and management group since 2016. Previously, from 2008 to 2016, Dr. Zanganeh was the Chief Operating Officer at Pharmacyclics, Inc., where she oversaw all clinical, research, commercial and business-related matters. In addition, from 1997 to 2002 Dr. Zanganeh held senior and executive positions including International Product Manager for IRCAD-EITS, General Director Pos De Competitive for Therapeutic Innovation and President Director General (Chief Executive Officer) of Europe, Middle East and Africa and World-Wide Vice President of Training and Education for Computer Motion. Dr. Zanganeh’s current directorships include Human Longevity Inc., Radial Medical and Pulse Biosciences. Dr. Zanganeh earned her Doctor of Dental Surgery and her Master of Business Administration in International Business in France. She is a licensed investment advisor (Series 65) and has her dentistry license. She earned the Fierce Biotech “2013 Top Women in Biotech” Award and the Ernst & Young Entrepreneur of the Year Award in 2013. Dr. Zanganeh was selected to serve on our board of directors due to her industry experience.
Kristen Angela Macfarlane. Ms. Macfarlane has served as our director since September 2018. She currently acts as Chief Executive Officer for Voyant Biotherapeutics, LLC, an early-stage biotech company dedicated to solving age-related macular degeneration since 2018. In addition to serving as CEO of ForSight Labs, LLC an ophthalmic incubator formed in 2005, from 2007 to 2016, from 2008 to 2016, Ms. Macfarlane served as the operating Chief Executive Officer for three ForSight companies, which included acting as a founding Chief Executive Officer through the acquisition of ForSight VISION4, Inc. (acquired by ROCHE), and ForSight VISION5, Inc., (Acquired by Allergan). Previously, Ms. Macfarlane served as Chief Technology Counsel to The Foundry, a medical technology incubator, and Technology Counsel for Thomas J. Fogarty, M.D., a renowned physician/entrepreneur where she participated in formation development of nine companies from 1999 to 2004. She previously served on the senior management teams and counsel at TransVascular, Inc. (acquired by Medtronic), AneuRx, Inc. (acquired by Medtronic), and VidaMed Inc. (through IPO). Ms. Macfarlane is an inventor on 25 U.S. issued patents and received her BA in Business Administration from San Francisco State University, and her J.D. from Golden Gate University School of Law. She currently serves on the board of the Fogarty Institute for Innovation and is a mentor in the Ferolyn Powell Leadership Program. Ms. Macfarlane was selected to serve on our board of directors due to her industry experience.
David Diamond. Mr. Diamond was appointed to the Board on May 9, 2021. Mr. Diamond currently provides strategic guidance and operational oversight to CEOs and boards of directors in the Life Sciences industry. Mr. Diamond has significant experience assisting management teams and boards of directors with capital financing and strategic business planning nationally and internationally and has built strong relationships with prominent investment bankers. He currently serves as the National Life Sciences and Technology Practice Lead at Mayer Hoffman McCann P.C., a national CPA firm since 2015 and has over 30 years of experience in both public accounting and industry. Mr. Diamon also serves as a director of Oncolytics Biotech, Inc. Mr. Diamond previously served as a Board member for Kreston International ($2 billion CPA network), a member of the board of San Diego Venture Group and was a Founding Member of UCSD Connect. He is a Certified Director in Corporate Governance from UCLA Anderson Graduate School of Management and an active CPA, licensed in the United States, Israel and South Africa. Mr. Diamond was selected to serve on our board of directors due to his extensive experience extensive experience as a strategic guide and oversight to CEOs and Boards of companies in the life sciences space.
| -93- |
Family Relationships
There are no other family relationships among any of our officers or directors.
Involvement in Certain Legal Proceedings
To the best of our knowledge, none of our directors or executive officers were involved in any legal proceedings described in Item 401(f) of Regulation S-K in the past ten years.
Corporate Governance
The Board’s Role in Risk Oversight
The board of directors oversees that the assets of our company are properly safeguarded, that the appropriate financial and other controls are maintained, and that our business is conducted wisely and in compliance with applicable laws and regulations and proper governance. Included in these responsibilities is the board’s oversight of the various risks facing our company. In this regard, our board seeks to understand and oversee critical business risks. Our board does not view risk in isolation. Risks are considered in virtually every business decision and as part of our business strategy. Our board recognizes that it is neither possible nor prudent to eliminate all risk. Indeed, purposeful and appropriate risk-taking is essential for our company to be competitive on a global basis and to achieve its objectives.
While the board oversees risk management, company management is charged with managing risk. Management communicates routinely with the board and individual directors on the significant risks identified and how they are being managed. Directors are free to, and indeed often do, communicate directly with senior management.
Our board administers its risk oversight function as a whole by making risk oversight a matter of collective consideration. Once the board establishes committees, it is anticipated that much of the work will be delegated to such committees, which will meet regularly and report back to the full board. It is anticipated that the audit committee will oversee risks related to our financial statements, the financial reporting process, accounting and legal matters, that the compensation committee will evaluate the risks and rewards associated with our compensation philosophy and programs, and that the nominating and corporate governance committee will evaluate risk associated with management decisions and strategic direction.
Director Independence
Our board of directors has undertaken a review of the independence of each director. Based on information provided by each director concerning his background, employment and affiliations, our board of directors has determined that Mr. Marton, Mr. Diamond and Mses. Ryan, Zanganeh and Macfarlane do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the listing standards of the Nasdaq Capital Market. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director, and the transactions involving them described in the section titled “Certain Relationships and Related Person Transactions.”
| -94- |
Committees of the Board of Directors
Our board has established an audit committee, a compensation and nominating and corporate governance committee, each with its own charter that has been approved by the board. Upon completion of this offering, we intend to make each committee’s charter available on our website at www.renovorx.com.
Our board of directors may, from time to time, designate one or more additional committees, which shall have the duties and powers granted to it by our board of directors.
Audit Committee
The members of our audit committee are David Diamond, Una Ryan and Laurence Marton. David Diamond serves as chairperson of the committee. All members of our audit committee meet the independence requirements of Rule 10A-3 under the Exchange Act and the applicable rules of the Nasdaq Capital Market, or the Nasdaq rules. Our board of directors has determined that David Diamond is an audit committee financial expert, as defined by the rules of the SEC, and satisfies the financial sophistication requirements of the Nasdaq rules.
The audit committee will oversee our accounting and financial reporting processes and the audits of the financial statements of our company.
The audit committee will be responsible for, among other things: (i) retaining and overseeing our independent registered public accounting firm; (ii) assisting the board in its oversight of the integrity of our financial statements, the qualifications, independence and performance of our independent registered public accounting firm and our compliance with legal and regulatory requirements; (iii) reviewing and approving the plan and scope of the internal and external audit; (iv) pre-approving any audit and non-audit services provided by our independent registered public accounting firm; (v) approving the fees to be paid to our independent registered public accounting firm; (vi) reviewing with our chief executive officer and chief financial officer and independent registered public accounting firm the adequacy and effectiveness of our internal controls; (vii) reviewing hedging transactions; and (viii) reviewing and assessing annually the audit committee’s performance and the adequacy of its charter.
Compensation Committee
The members of our compensation committee are Maky Zanganeh, Kristen Angela Macfarlane and David Diamond. Maky Zanganeh serves as the chairperson of the committee. The compensation committee will assist the board in reviewing and approving the compensation structure, including all forms of compensation, relating to our directors and executive officers.
The compensation committee will be responsible for, among other things: (i) reviewing and approving the remuneration of our executive officers; (ii) reviewing the compensation of our independent directors; (iii) making recommendations to the board regarding equity-based and incentive compensation plans, policies and programs; and (iv) reviewing and assessing annually the compensation committee’s performance and the adequacy of its charter.
Nominating and Corporate Governance Committee
The members of our nominating and governance committee are Una Ryan, Kristen Angela Macfarlane and Laurence Marton. Una Ryan serves as the chairperson of the committee. The nominating and corporate governance committee will assist the board of directors in selecting individuals qualified to become our directors and in determining the composition of the board and its committees.
The nominating and corporate governance committee will be responsible for, among other things: (i) identifying and evaluating individuals qualified to become members of the board by reviewing nominees for election to the board submitted by stockholders and recommending to the board director nominees for each annual meeting of stockholders and for election to fill any vacancies on the board, (ii) advising the board with respect to board organization, desired qualifications of board members, the membership, function, operation, structure and composition of committees (including any committee authority to delegate to subcommittees), and self-evaluation and policies, (iii) advising on matters relating to corporate governance and monitoring developments in the law and practice of corporate governance, (iv) overseeing compliance with our code of ethics, and (v) approving any related party transactions.
| -95- |
The nominating and corporate governance committee’s methods for identifying candidates for election to our board of directors (other than those proposed by our stockholders, as discussed below) will include the solicitation of ideas for possible candidates from a number of sources—members of our board of directors, our executives, individuals personally known to the members of our board of directors, and other research. The nominating and corporate governance committee may also, from time-to-time, retain one or more third-party search firms to identify suitable candidates.
In making director recommendations, the nominating and corporate governance committee may consider some or all of the following factors: (i) the candidate’s judgment, skill, experience with other organizations of comparable purpose, complexity and size, and subject to similar legal restrictions and oversight; (ii) the interplay of the candidate’s experience with the experience of other board members; (iii) the extent to which the candidate would be a desirable addition to the board and any committee thereof; (iv) whether or not the person has any relationships that might impair his or her independence; and (v) the candidate’s ability to contribute to the effective management of our company, taking into account the needs of our company and such factors as the individual’s experience, perspective, skills and knowledge of the industry in which we operate.
Code of Business Conduct and Ethics
We plan to adopt a code of ethics that applies to all of our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer. Such code of ethics addresses, among other things, honesty and ethical conduct, conflicts of interest, compliance with laws, regulations and policies, including disclosure requirements under the federal securities laws, and reporting of violations of the code. Upon our listing on the Nasdaq Capital Market, our code of business conduct and ethics will be available under the Corporate Governance section of our website.
We will be required to disclose any amendment to, or waiver from, a provision of our code of ethics applicable to our principal executive officer, principal financial officer, principal accounting officer, controller, or persons performing similar functions. We intend to use our website as a method of disseminating this disclosure, as permitted by applicable SEC rules. Any such disclosure will be posted to our website within four business days following the date of any such amendment to, or waiver from, a provision of our code of ethics.
Executive and Director Compensation
As an emerging growth company under the JOBS Act we have opted to comply with the executive compensation disclosure rules applicable to “smaller reporting companies,” which require compensation disclosure for our principal executive officer and the two most highly compensated executive officers (other than our principal executive officer) serving as executive officers at the end of our most recently completed fiscal year (collectively, our “Named Executive Officers”). This section describes the executive compensation program in place for our Named Executive Officers during the year ended December 31, 2020, who are the individuals who served as our principal executive officer and two most highly compensated executive officers.
This section discusses the material components of the executive compensation program for our executive officers who are named in the “Summary Compensation Table” below and the non-employee members of our board of directors. In 2020, our “Named Executive Officers” and their positions were:
| ● | Shaun Bagai, Chief Executive Officer; | |
| ● | Paul Matters, Chief Financial Officer; and | |
| ● | Ramtin Agah, MD, Chief Medical Officer. |
| -96- |
2020 Summary Compensation Table
The following table sets forth information concerning the compensation of our Named Executive Officers for the year ended December 31, 2020 and 2019 (in thousands).
| Name & Principal Position | Year | Salary ($) | Salary/ Bonus ($) | Option Awards ($)(1) | Total ($) | |||||||||||||||
| Shaun Bagai, CEO | 2020 | 253 | $ | 253 | ||||||||||||||||
| 2019 | 240 | 30 | $ | 270 | ||||||||||||||||
| Paul Manners, Vice President, Finance | 2020 | 37 | $ | 37 | ||||||||||||||||
| 2019 | 19 | 20 | $ | 39 | ||||||||||||||||
| Ramtin Agah, MD, CMO | 2020 | 120 | $ | 120 | ||||||||||||||||
| 2019 | 78 | $ | 78 | |||||||||||||||||
| (1) | The amount disclosed represents the aggregate grant date fair value of the award as calculated in accordance with ASC 718. The assumptions used in calculating the grant date fair value of the award disclosed in this column are set forth in the notes to our audited financial statements included elsewhere in this prospectus. This amount does not correspond to the actual value that may be recognized upon vesting of the award. |
Compensation Arrangements with our Executive Officers
Shaun R. Bagai
Shaun R. Bagai has been our Chief Executive Officer since 2014, initially as a consultant. We entered into an offer letter in December 2015 with Mr. Bagai that set forth the terms and conditions of his employment. The letter became effective on January 1, 2016. The offer letter was amended in June 2017 and December 2020. The most recent amended offer letter provides for (i) an annual base salary of $300,000; (ii) a $2,500 monthly stipend for Mr. Bagai’s health insurance expenses, which will continue until we begin offering health insurance as a benefit to all employees; (iii) a $70,000 bonus to be paid in connection with this initial public offering; and (iv) an additional performance bonus upon achievement of certain clinical and company milestones established by the Board in 2021. Upon consummation of the offering, Mr. Bagai’s annual base salary will be $363,000.
Christopher J. Lehman
Christopher J. Lehman has agreed to become our Chief Financial Officer upon consummation of this offering. In July, 2021, we entered into an agreement with LS Associates, Inc., or LS, pursuant to Mr. Lehman is providing consulting services to us and has agreed to become our Chief Financial Officer. The term of the agreement is for two years and we will be billed monthly by LS at a rate of $400 per hour. For the first three months the fees will be capped at $25,000 unless agreed upon by us and Mr. Lehman. After the first three months, LS will adjust its time to meet a monthly fee range of $15,000 per month unless agreed upon by us and Mr. Lehman.
Paul Manners
As of July 2021, Mr. Manners became our Vice President, Finance. We entered into a consulting agreement in July 2019 with Paul Manners, then our Chief Financial Officer, that set forth the terms and conditions of the services he was to render. The agreement was amended in December 2020. The amended agreement establishes an hourly consulting rate of one hundred and fifty dollars ($150) per hour. We also agreed to reimburse Mr. Manners for any reasonable out-of-pocket business expenses incurred in connection with the services, provided such expenses are approved in advance by us and fully documented our satisfaction. For the years ended December 31, 2019 and 2020 and the three months ended March 31, 2020 and 2021, we paid Mr. Manners $19,000, $37,000, $12,000 and $54,000, respectively.
Mr. Manners was also granted an option to purchase 28,000 shares of our common stock at the fair market value as determined by the Board as of the grant date, pursuant to our 2013 Equity Incentive Plan. The option fully vested on July 9, 2021.
Ramtin Agah
In January 2013, we entered into a consulting agreement with Dr. Agah, one of our co-founders, whereby he would provide consulting services as our Chief Medical Officer by overseeing Company-sponsored clinical trials. The agreement is for a term of 15 years with automatic one-year renewals. The sole compensation payable to Dr. Agah is the continued vesting of shares of common stock held by Dr. Agah. In July 2018, he was awarded options for the purchase of 40,000 shares of our common stock with 25% vested after one year and the remaining 75% vesting ratably over 36 months. We entered into an amendment to the agreement with Dr. Agah in 2019 providing cash compensation to Dr. Agah of $10,000 per month for additional services. Consulting fees paid to Dr. Agah were $78,000, $120,000, $30,000 and $30,000 for the years ended December 31, 2019 and 2020 and March 31, 2020 and 2021, respectively. Upon consummation of the offering, Dr. Agah’s compensation will be $260,000 per year provided he spends 60% of his time on Company matters. On June 7, 2021 we granted to Dr. Agah options to purchase 20,000 shares of common stock at an exercise price of $2.45 per share which vests in 24 equal consecutive monthly installments commencing on June 14, 2021.
Director Compensation
None of our non-employee directors received option awards during the year ended December 31, 2020. We did not compensate our board during the last fiscal year. Under our non-employee director compensation policy, each non-employee director received the following annual compensation for his or her service:
| Committee membership | ||||||||||||||||
| Annual retainer non-employee board member | Audit | Compensation | Corporate Governance/Nominating | |||||||||||||
| Chair | $ | 36,000 | $ | 15,000 | $ | 10,000 | $ | 10,000 | ||||||||
| Member | $ | 36,000 | $ | 5,000 | $ | 5,000 | $ | 5,000 | ||||||||
| -97- |
Equity compensation
Outstanding equity awards at March 31, 2021
The following table sets forth information concerning the outstanding equity awards held by each of our Named Executive Officers as of March 31, 2021:
| Option Awards(1) | ||||||||||||||||
| Name | Option Grant Date | Number
of Securities Underlying Unexercised Options (#) Exercisable | Number
of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | |||||||||||
| Shaun Bagai | August 6, 2014(2) | 246,057 | 0 | $ | 0.15 | August 5, 2024 | ||||||||||
| May 19, 2017(3) | 56,250 | 3,750 | $ | 0.50 | May 18, 2027 | |||||||||||
| July 11, 2018(4) | 87,500 | 32,500 | $ | 0.65 | July 10, 2028 | |||||||||||
| Paul Manners | February 5, 2020(5) | 0 | 4,666 | $ | 0.70 | February 4, 2030 | ||||||||||
| Ramtin Agah, MD | May 19, 2017(2) | 60,000 | 0 | $ | 0.50 | May 18, 2027 | ||||||||||
| July 11, 2018(6) | 29,166 | 10,833 | $ | 0.65 | July 10, 2028 | |||||||||||
| (1) | Each of the outstanding equity awards was granted pursuant to our 2013 Equity Incentive Plan. | |
| (2) | The shares underlying this option are fully vested and immediately exercisable. | |
| (3) | The shares underlying this option vest, subject to Mr. Bagai’s continued role as a service provider to us, 25% on the one-year anniversary of the June 1, 2017 vesting commencement date and then in 48 equal monthly installments. | |
| (4) | The shares underlying this option vest, subject to Mr. Bagai’s continued role as a service provider to us, 25% on the one-year anniversary of the April, 19, 2018 vesting commencement date and then in 48 equal monthly installments. | |
| (5) | The shares underlying this option vest, subject to Mr. Manner’s continued role as a service provider to us, 25% on the six-month anniversary of the July 9, 2019 vesting commencement date and then in 24 equal monthly installments. On March 19, 2021 Mr. Manners exercised 23,333 options. | |
| (6) | The shares underlying this option vest, subject to Dr. Agah’s continued role as a service provider to us, 25% on the one-year anniversary of the April 19, 2018 vesting commencement date and then in 36 equal monthly installments. |
Incentive plan
Equity Incentive Plans
2021 Omnibus Equity Incentive Plan
On July 19, 2021 our board of directors adopted, and our stockholders approved, the RenovoRx, Inc. 2021 Omnibus Equity Incentive Plan, or the 2021 Plan, which will become effective immediately prior to the closing of this offering. We intend to use the 2021 Plan following the closing of this offering to provide incentives that will assist us to attract, retain, and motivate employees, including officers, consultants, and directors. We may provide these incentives through the grant of stock options, stock appreciation rights, restricted stock, RSUs, performance shares, and units and other cash-based or share-based awards
| -98- |
Shares Available
The maximum number of shares of common stock reserved and available for issuance under the 2021 Plan will be equal to the sum of (i) 2,175,000 shares of common stock; (ii) the number of shares of common stock reserved, but unissued under the 2013 Plan, (iii) the number of shares of common stock underlying forfeited awards under the 2013 Plan and (iv) an annual increase on the first day of each calendar year beginning with the first January 1 following the Effective Date and ending with the last January 1 during the initial ten-year term of the Plan, equal to the lesser of (A) three percent (3%) of the Shares outstanding (on an as-converted basis) on the final day of the immediately preceding calendar year and (B) such lesser number of Shares as determined by the Board; provided that shares of common stock issued under the 2021 Plan with respect to an Exempt Award will not count against the share limit. We use the term “Exempt Award” to mean (i) an award granted in assumption of, or in substitution for, outstanding awards previously granted by another business entity acquired by us or any of our subsidiaries or with which we or any of our subsidiaries merge, or (ii) an award that a participant purchases at fair market value.
Administration
The 2021 Plan is administered by the Board or by one or more committees of directors appointed by the Board (the “Administrator”). The Board may delegate different levels of authority to different committees with administrative and grant authority under the 2021 Plan. Any committee delegated administrative authority under the 2021 Plan may further delegate its authority under the Plan to another committee of directors, and any such delegate shall be deemed to be an Administrator of the 2021 Plan. The Administrator comprised solely of directors may also delegate, to the extent permitted by Section 157 of the Delaware General Corporation Law and any other applicable law, to one or more officers of the Company, its powers under this Plan (a) to designate Eligible Persons who will receive grants of awards under this Plan, and (b) to determine the number of shares subject to, and the other terms and conditions of, such awards. It is anticipated that the Administrator (either generally or with respect to specific transactions) will be constituted so as to comply, as necessary or desirable, with the requirements of Section 162(m)of Internal Revenue Code of 1986, as amended (the “Code”), and Rule 16b-3 promulgated under the Exchange Act.
Eligibility
Awards may be granted pursuant to the 2021 Plan only to persons who are eligible persons. Under the 2021 Plan, “Eligible Person” means any person who is either: (a) an officer (whether or not a director) or employee of the Company or one of its subsidiaries; (b) a director of the Company or one of its subsidiaries; or (c) a consultant who renders bona fide services to the Company or one of its subsidiaries; provided, however, that Incentive Stock Options (“ISOs”) may be granted only to employees.
Awards
The 2021 Plan permits the grant of: (a) stock options, which may be intended as ISOs or as nonqualified stock options (options not meeting the requirements to qualify as ISOs); (b) stock appreciation rights (“SARs”); (c) restricted stock; (d) restricted stock units; (e) cash incentive awards; or (f) other awards, including: (i) stock bonuses, performance stock, performance units, dividend equivalents, or similar rights to purchase or acquire shares, whether at a fixed or variable price or ratio related to the common stock, upon the passage of time, the occurrence of one or more events, or the satisfaction of performance criteria or other conditions, or any combination thereof; or (ii) any similar securities with a value derived from the value of or related to the common stock and/or returns thereon.
| -99- |
Consideration for Awards
The purchase price for any award granted under the 2021 Plan or the common stock to be delivered pursuant to any such award, as applicable, may be paid by means of any lawful consideration as determined by the Administrator, including, without limitation, one or a combination of the following methods:
| ● | services rendered by the recipient of such award; | |
| ● | cash, check payable to the order of the Company, or electronic funds transfer; | |
| ● | notice and third party payment in such manner as may be authorized by the Administrator; | |
| ● | the delivery of previously owned and fully vested shares of common stock; | |
| ● | by a reduction in the number of shares otherwise deliverable pursuant to the award; or | |
| ● | subject to such procedures as the Administrator may adopt, pursuant to a “cashless exercise” with a third party who provides financing for the purposes of (or who otherwise facilitates) the purchase or exercise of awards. |
Certain Federal Tax Consequences
The following summary of the federal income tax consequences of the 2021 Plan transactions is based upon federal income tax laws in effect as of August 5, 2021. This summary does not purport to be a complete description of all applicable rules, and does not discuss state, local or non-U.S. tax consequences.
Nonqualified Stock Options. The grant of a nonqualified stock option under the 2021 Plan will not result in any U.S. federal income taxes to the participant or to the Company. Upon exercise of a nonqualified stock option, the participant will recognize ordinary compensation income equal to the excess of the fair market value of the shares of Common stock at the time of exercise over the option exercise price. If the participant is an employee, this income is subject to withholding for federal income and employment tax purposes. The Company is entitled to an income tax deduction in the amount of the income recognized by the participant, subject to possible limitations imposed by the Code, including Section 162(m) thereof. Any gain or loss on the participant’s subsequent disposition of the shares will be treated as long-term or short-term capital gain or loss, depending on the sales proceeds received and whether the shares are held for more than one year following exercise. The Company does not receive a tax deduction for any subsequent capital gain realized by the participant.
Incentive Options. The grant of an ISO under the 2021 Plan will not result in any U.S. federal income tax consequences to the participant or to the Company. A participant recognizes no federal taxable income upon exercising an ISO (subject to the alternative minimum tax rules discussed below), and the Company receives no deduction at the time of exercise. In the event of a disposition of stock acquired upon exercise of an ISO, the tax consequences depend upon how long the participant has held the shares. If the participant does not dispose of the shares within two years after the ISO was granted, nor within one year after the ISO was exercised, the participant will recognize a long-term capital gain (or loss) equal to the difference between the sale price of the shares and the exercise price. The Company is not entitled to any deduction under these circumstances.
If the participant fails to satisfy either of the foregoing holding periods (referred to as a “disqualifying disposition”), he or she will recognize ordinary compensation income in the year of the disposition. The amount of ordinary compensation income generally is the lesser of (i) the difference between the amount realized on the disposition and the exercise price or (ii) the difference between the fair market value of the stock at the time of exercise and the exercise price. Such amount is not subject to withholding for federal income and employment tax purposes, even if the participant is an employee of the Company. Any gain in excess of the amount taxed as ordinary income will generally be treated as a short-term capital gain. The Company, in the year of the disqualifying disposition, is entitled to a deduction equal to the amount of ordinary compensation income recognized by the participant, subject to possible limitations imposed by the Code, including Section 162(m) thereof.
| -100- |
The “spread” under an ISO — i.e., the difference between the fair market value of the shares at exercise and the exercise price — is classified as an item of adjustment in the year of exercise for purposes of the alternative minimum tax. If a participant’s alternative minimum tax liability exceeds such participant’s regular income tax liability, the participant will owe the alternative minimum tax liability.
Restricted Stock. Restricted stock is generally taxable to the participant as ordinary compensation income on the date that the restrictions lapse (i.e. the date that the stock vests), in an amount equal to the excess of the fair market value of the shares on such date over the amount paid for such stock (if any). If the participant is an employee, this income is subject to withholding for federal income and employment tax purposes. The Company is entitled to an income tax deduction in the amount of the ordinary income recognized by the participant, subject to possible limitations imposed by the Code, including Section 162(m) thereof. Any gain or loss on the participant’s subsequent disposition of the shares will be treated as long-term or short-term capital gain or loss treatment depending on the sales price and how long the stock has been held since the restrictions lapsed. The Company does not receive a tax deduction for any subsequent gain.
Participants receiving restricted stock awards may make an election under Section 83(b) of the Code (“Section 83(b) Election”) to recognize as ordinary compensation income in the year that such restricted stock is granted, the amount equal to the excess of the fair market value on the date of the issuance of the stock over the amount paid for such stock. If such an election is made, the recipient recognizes no further amounts of compensation income upon the lapse of any restrictions and any gain or loss on subsequent disposition will be long-term or short-term capital gain or loss to the recipient. The Section 83(b) Election must be made within 30 days from the time the restricted stock is issued.
Other Awards. Other awards (such as restricted stock units) are generally treated as ordinary compensation income as and when common stock or cash are paid to the participant upon vesting or settlement of such awards. If the participant is an employee, this income is subject to withholding for income and employment tax purposes. The Company is generally entitled to an income tax deduction equal to the amount of ordinary income recognized by the recipient, subject to possible limitations imposed by the Code, including Section 162(m) thereof.
Section 162(m) Limitation. In general, under Section 162(m) of the Code, income tax deductions of publicly-held corporations may be limited to the extent total compensation (including base salary, annual bonus, stock option exercises and non-qualified benefits paid) for certain executive officers exceeds $1 million (less the amount of any “excess parachute payments” as defined in Section 280G of the Code) in any one year. Prior to the Tax Cuts and Jobs Act of 2017 (the “TCJA”), covered employees generally consisted of our Chief Executive Officer and each of the next three highest compensated officers serving at the end of the taxable year other than our Chief Financial Officer, and compensation that qualified as “performance-based” under Section 162(m) was exempt from this $1 million deduction limitation. As part of the TCJA, the ability to rely on this exemption was, with certain limited exceptions, eliminated; in addition, the definition of covered employees was expanded to generally include all named executive officers. Certain awards under the 2013 Plan granted prior to November 2, 2017 (to the extent not materially modified thereafter) may be grandfathered from the changes made by the TCJA under certain limited transition relief, however, for grants after that date and any grants which are not grandfathered, we will no longer be able to take a deduction for any compensation in excess of $1 million that is paid to a covered employee. There is no guarantee that we will be able to take a deduction for any compensation in excess of $1 million that is paid to a covered employee under the 2013 Plan.
Amended and Restated 2013 Equity Incentive Plan
The 2013 Plan was originally adopted by our board of directors and approved by our stockholders in January 23, 2013. The maximum aggregate number of shares of common stock that may be issued under the 2013 Plan is 6,606,504. Upon the closing of this offering, our board of directors will terminate the 2013 Plan and we will not grant any further awards under such plan, but the 2013 Plan will continue to govern outstanding awards granted thereunder. Our compensation committee administers the 2013 Plan and has the authority, among other things, to construe and interpret the terms of the 2013 Plan and awards granted thereunder.
On January 23, 2013, our Board of Directors adopted the 2013 Equity Incentive Plan, or the 2013 Plan. The following summary describes the material terms of the 2013 Plan. This summary is not a complete description of all provisions of the 2013 Plan and is qualified in its entirety by reference to the 2013 Plan, which is filed as an exhibit to the registration statement of which this prospectus is a part.
The purpose of the 2013 Plan is to secure and retain the services of the eligible recipients, to provide incentives for such persons to exert maximum efforts for the success of the Company, and to provide a means by which such eligible recipients may be given an opportunity to benefit from increases in value of the common stock of the Company.
Administration. The plan is administered by the Board. The Board shall have authority in its discretion to determine the eligible persons to whom, and the time or times at which, awards may be granted, the number of shares, units or other rights subject to each award, the exercise, base or purchase price of an award (if any), the time or times at which an award will become vested, exercisable or payable, the performance criteria, performance goals and other conditions of an award, the duration of the award, and all other terms of the award, including the fair market value thereof.
Available shares. The maximum aggregate number of shares of common stock which may be issued under all awards granted to participants under the plan initially shall be 6,346,504 shares. All 6,346,504 of such authorized shares initially available may be issued in respect of incentive stock options.
Eligibility for participation. An incentive stock option may only be granted to an employee of ours or any subsidiary of ours. Awards other than incentive stock options may be granted to employees, directors and consultants.
Types of awards. The 2013 Plan provides for the grant of nonqualified stock options, incentive stock options (“ISOs”), stock appreciation rights (“SARs”), restricted stock and stock units.
● Stock options and SARs. The Board may grant stock options, including ISOs, and SARs. A stock option is a right entitling the holder to acquire our common shares upon payment of the applicable exercise price. A SAR is a right entitling the holder upon exercise to receive an amount (payable in cash or shares of equivalent value) equal to the excess of the fair market value of the shares subject to the right over the base value from which appreciation is measured. The exercise price of each stock option, and the base value of each SAR, granted under the 2013 Plan shall be no less than 100% of the fair market value of a share of common stock on the date of grant. Each stock option and SAR will have a maximum term of not more than ten years from the date of grant.
● Restricted and unrestricted stock and stock units. The administrator of the 2013 Plan may grant awards of shares, stock units, restricted stock and restricted stock units. A stock unit is an unfunded and unsecured promise, denominated in shares, to deliver shares or cash measured by the value of shares in the future, and a restricted stock unit is a stock unit that is subject to the satisfaction of specified performance or other vesting conditions. Restricted stock are shares subject to restrictions requiring that they be redelivered or forfeited to the company if specified conditions are not satisfied.
Change in control. An award under the 2013 Plan may be subject to additional acceleration of vesting and exercisability upon or after a ‘Change in Control’ as may be provided in the applicable award agreement for such stock award or as may be provided in any other written agreement between the Company and the participant, but in the absence of such provision, no such acceleration shall occur.
Stockholder rights. Except as otherwise provided in the applicable award agreement, and with respect to an award of restricted stock, a participant will have no rights as a stockholder with respect to shares of our common stock covered by any award until the participant becomes the record holder of such shares.
Amendments and termination. The Board of Directors may suspend or terminate the 2013 Plan at any time and may amend the 2013 Plan at any time and from time to time in such respects as the Board of Directors may deem advisable or in our best interests; provided, however, that stockholder approval is required for any amendment to the 2013 Plan that (i) increases the number of shares of common stock available for issuance under the 2013 Plan, or (ii) changes the persons or class of persons eligible to receive awards under the 2013 Plan.
| -101- |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND
RELATED STOCKHOLDER MATTERS
Based solely upon information made available to us, the following table sets forth information regarding the beneficial ownership of our common stock as of the date of this prospectus, held by: (i) each director and director nominees; (ii) each of the named executive officers; (iii) all of our directors and executive officers as a group; and (iv) each additional person or group who is known by us to own beneficially more than 5% of our common stock. Except as indicated in the footnotes below, the address of the persons or groups named below is c/o RenovoRx, Inc., 4546 El Camino Real, Suite B1 Los Altos, California 94022.
The percentage of shares beneficially owned is computed on the basis of 2,291,557 shares of our common stock outstanding as of June 30, 2021. Shares of our common stock that a person has the right to acquire within 60 days after June 30, 2021 are deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all directors and executive officers as a group. Common Stock beneficially owned after the offering includes an aggregate 3,535,469 shares of common stock issuable upon the Preferred Stock Conversions and an aggregate 530,546 shares of common stock and warrant to purchase 397,901 shares of common stock issuable upon the Note Conversions.
| Number
of Shares Beneficially Owned | Percentage
of Shares Beneficially Owned | |||||||||||
| Name of Beneficial Owner | Prior
to Offering | Prior
to Offering | After
Offering | |||||||||
| 5% or Greater Stockholders | ||||||||||||
| Kamran Najmabadi(1) | 1,045,000 | 44.4 | 12.6 | |||||||||
| Executive Officer Nominee | ||||||||||||
| Christopher J. Lehman | - | - | - | |||||||||
| Executive Officers and Directors | ||||||||||||
| Shaun R. Bagai(2) | 406,057 | 15.1 | 4.7 | |||||||||
| Paul Manners(3) | 27,999 | 1.2 | * | |||||||||
| Ramtin Agah(4) | 1,070,833 | 44.9 | 12.9 | |||||||||
| Laurence J. Marton(5) | 74,584 | 3.2 | * | |||||||||
| Una S. Ryan(6) | 11,666 | * | * | |||||||||
| Maky Zanganeh(7) | 23,333 | 1.0 | * | |||||||||
| Kristen Angela Macfarlane(8) | 23,333 | 1.0 | * | |||||||||
| David Diamond(9) | 500 | * | * | |||||||||
| Directors and Officers as a Group (8 persons) (10) | 1,638,305 | 55.9 | 18.5 | |||||||||
* Represents less than 1% of the beneficial ownership of the outstanding shares of our common stock.
| (1) | Consists of 741,250 shares held of record by Mr. Najmabadi, our founder and technical engineering advisor, and/or entities controlled by Mr. Najmabadi, 60,000 shares subject to options exercisable within 60 days of June 30, 2021 and 121,875 shares of common stock held by each of The Leili Najmabadi Irrevocable Trust dated April 22, 2021 and The Navid Najmabadi Irrevocable Trust dated April 22, 2021. Mr. Najmabadi and his wife are trustees of the trusts. | |
| (2) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Mr. Bagai. | |
| (3) | Consists of 23,333 shares held of record by Mr. Manners and 4,666 shares subject to options exercisable within 60 days of June 30, 2021. | |
| (4) |
Consists of 975,000 shares held of record by Dr. Agah and 95,833 shares subject to options exercisable within 60 days of June 30, 2021. Also includes 726 shares issuable upon exercise of warrants issued upon the Note Conversion. | |
| (5) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Dr. Marton. | |
| (6) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Dr. Ryan. | |
| (7) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Dr. Zanganeh. | |
| (8) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Ms. Macfarlane. | |
| (9) | Consists of shares subject to options exercisable within 60 days of June 30, 2021 held by Mr. Diamond. | |
| (10) | Includes 639,972 shares subject to options exercisable within 60 days of June 30, 2021. Also includes 726 shares issuable upon exercise of warrants issued upon the Note Conversion. |
| -102- |
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
The following includes a summary of transactions since January 1, 2019 to which we have been a party in which the amount involved exceeded or will exceed the lesser of $120,000 or 1% of the average of our total assets as of December 31, 2020 and 2019, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described under “Executive and Director Compensation.” We also describe below certain other transactions with our directors, executive officers, and stockholder.
In January 2013, we entered into an agreement, or the Agreement, with Dr. Agah, one of our co-founders, whereby he would provide consulting services as our Chief Medical Officer by overseeing Company-sponsored clinical trials. The Agreement is for a term of 15 years with automatic one-year renewals. The sole compensation payable to Dr. Agah was continued vesting of shares of common stock held by Dr. Agah. We entered into an amendment to the Agreement in 2019 providing cash compensation to Dr. Agah of $10,000 per month for additional services. Consulting fees paid to Dr. Agah were $78,000, $120,000 and $30,000 for the years ended December 31, 2019 and 2020 and the three months ended March 31, 2021, respectively. On June 7, 2021 we granted to Dr. Agah options to purchase 20,000 shares of common stock at an exercise price of $2.45 per share which vests in 24 equal consecutive monthly installments commencing on June 14, 2021.
In July 2019, we entered into a consulting agreement, the CFO Agreement, with the Mr. Manners, our Chief Financial Officer. In February 2020, we granted Mr. Manners an option to purchase 28,000 shares of common stock, of which 25% were vested upon grant and the remaining 75% vested ratably over 18 months. We entered into an amendment to the CFO Agreement in December 2020 providing for cash compensation of $150 per hour. Consulting fees paid to Mr. Manners were $19,000, $37,000 and $54,000 for the years ended December 31, 2019 and 2020 and the three months ended March 31, 2021, respectively.
Mr, Kamran Najmabadi, another of our co-founders, has served as our consulting technical engineering advisor on manufacturing and intellectual property matters since January 2020. Previously Mr. Najmabadi was our CEO from inception in December 2009 until January 2013; Chief Technical and Operations Officer from January 2013 until January 2019; and Chief Technology Officer from January 2019 to January 2020. Mr. Najmabadi currently receives cash compensation quarterly of $3,000. He received option grants in 2016 and 2018, which are now fully vested.
Policies and Procedures for Related Person Transactions
Our board of directors intends to adopt a written related person transaction policy, to be effective upon the closing of this offering, setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy will cover, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, where the amount involved exceeds the lesser of $120,000 or 1% of the average of our total assets as of December 31, 2019 and 2020 and a related person had, has or will have a direct or indirect material interest, including without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction. All of the transactions described in this section occurred prior to the adoption of this policy.
| -103- |
General
Upon completion of this offering, our authorized capital stock will consist of 250,000,000 shares of common stock, par value $0.001 per share, and 15,000,000 shares of preferred stock, par value $0.001 per share.
As of June 30, 2021, there were 2,291,557 shares of common stock issued and outstanding. In addition, as of June 30, 2021, the following series of convertible preferred stock was authorized and issued and outstanding, which shares of preferred stock are convertible into an aggregate of 3,535,469 shares of common stock upon closing of this offering.
| Preferred Series | Shares Authorized | Shares Issued and Outstanding | Liquidation Value | |||||||||
| B | 12,611,461 | 1,585,671 | $ | 8,745,000 | ||||||||
| A-3 | 2,660,230 | 532,046 | 2,227,000 | |||||||||
| A-2 | 3,546,095 | 709,219 | 1,150,000 | |||||||||
| A-1 | 3,542,669 | 708,533 | 660,000 | |||||||||
| 22,360,455 | 3,535,469 | $ | 12,782,000 | |||||||||
The following description of our capital stock and provisions of our Sixth Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws to be effective upon the completion of this offering is only a summary. You should also refer to our Sixth Amended and Restated Certificate of Incorporation, a copy of which is filed as an exhibit to the registration statement of which this prospectus is a part, and our Amended and Restated Bylaws, a copy of which is filed as an exhibit to the registration statement of which this prospectus is a part.
We are offering 1,850,000 Units, assuming a public offering price of $12.00 (the midpoint of the price range as set forth on the cover page of this prospectus). Each Unit will consist of one share of our common stock and three-quarter Warrant. We are also offering to those purchasers, if any, whose purchase of Units in this offering would otherwise result in the purchaser, together with its affiliates and certain related parties, beneficially owning more than 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock immediately following the consummation of this offering, the opportunity to purchase, if the purchaser so chooses, Pre-funded Units in lieu of Units that would otherwise result in the purchaser’s beneficial ownership exceeding 4.99% (or, at the election of the purchaser, 9.99%) of our outstanding common stock, each Pre-funded Unit consisting of one Pre-Funded Warrant and three-quarter Warrant. We are offering up to 1,850,000 Units and up to 1,850,000 Pre-funded Units, in the aggregate not exceeding more than a total of 1,850,000 units. For each Pre-funded Unit we sell, the number of Units we are offering will be decreased on a one-for-one basis. Units and Pre-funded Units will not be issued or certificated. The shares of common stock or Pre-funded Warrants, as the case may be, and the Warrants included in the Units or the Pre-funded Units, can only be purchased together in this offering, but the securities contained in the Units or Pre-funded Units will be issued separately and will be immediately separable upon issuance.
Reverse Stock Split
On August 5, 2021 we filed our Second Amendment to the Fifth Amended and Restated Certificate of Incorporation which effectuated a one to five (1:5) reverse stock split (the “Reverse Stock Split”) of our common stock without any change to its par value. No fractional shares will be issued in connection with the Reverse Stock Split as all fractional shares will be rounded down to the next whole share. All references to share and per share amounts of our common stock listed in this prospectus have been adjusted to give effect to the Reverse Stock Split.
Units Offered Hereby
We are offering 1,850,000 Units at a fixed price of $12.00 per Unit, the midpoint of the range set forth on the cover page of this prospectus. Each Unit consists of (a) one share of our common stock and (b) three-quarter warrant to purchase one share of our common stock at an exercise price equal to $14.40 (120% of initial public offering price per Unit) per share, assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) exercisable until the fifth anniversary of the issuance date, and subject to certain adjustment and cashless exercise provisions as described herein.
Common Stock
Voting
Holders of our common stock are entitled to one vote per share on matters to be voted on by stockholders and also are entitled to receive such dividends, if any, as may be declared from time to time by our board of directors in its discretion out of funds legally available therefor. Holders of our common stock have exclusive voting rights for the election of our directors and all other matters requiring stockholder action, except with respect to amendments to our certificate of incorporation that alter or change the powers, preferences, rights or other terms of any outstanding preferred stock if the holders of such affected series of preferred stock are entitled to vote on such an amendment or filling vacancies on the board of directors.
Dividends
Holders of common stock are entitled to share ratably in any dividends declared by our board of directors, subject to any preferential dividend rights of any outstanding preferred stock. Dividends consisting of shares of common stock may be paid to holders of shares of common stock. We do not intend to pay cash dividends in the foreseeable future.
Liquidation and Dissolution
Upon our liquidation or dissolution, the holders of our common stock will be entitled to receive pro rata all assets remaining available for distribution to stockholders after payment of all liabilities and provision for the liquidation of any shares of preferred stock at the time outstanding.
| -104- |
Preferred Stock
Our board of directors will have the authority, without further action by the stockholders, to issue up to 15,000,000 shares of preferred stock in one or more series and to fix the designations, powers, preferences, privileges, and relative participating, optional, or special rights as well as the qualifications, limitations, or restrictions of the preferred stock, including dividend rights, conversion rights, voting rights, terms of redemption, and liquidation preferences, any or all of which may be greater than the rights of the common stock. Our board of directors, without stockholder approval, will be able to issue convertible preferred stock with voting, conversion, or other rights that could adversely affect the voting power and other rights of the holders of common stock. Preferred stock could be issued quickly with terms calculated to delay or prevent a change of control or make removal of management more difficult. Additionally, the issuance of preferred stock may have the effect of decreasing the market price of our common stock, and may adversely affect the voting and other rights of the holders of common stock. At present, we have no plans to issue any shares of preferred stock following this offering.
Warrant Agent
The Warrants will be issued in registered form under a warrant agent agreement (the “Warrant Agent Agreement”) between us and our warrant agent, Philadelphia Stock Transfer, Inc. (the “Warrant Agent”). The material provisions of the warrants are set forth herein and a copy of the Warrant Agent Agreement has been filed as an exhibit to the Registration Statement on Form S-1, of which this prospectus forms a part. The Company and the Warrant Agent may amend or supplement the Warrant Agent Agreement without the consent of any holder for the purpose of curing any ambiguity, or curing, correcting or supplementing any defective provision contained therein or adding or changing any other provisions with respect to matters or questions arising under the Warrant Agent Agreement as the parties thereto may deem necessary or desirable and that the parties determine, in good faith, shall not adversely affect the interest of the Warrant holders. All other amendments and supplements to the Warrant Agent Agreement shall require the vote or written consent of holders of at least 50.1% of the Warrants.
Warrants Offered Hereby
The Warrants entitle the registered holder to purchase one share of our common stock at a price equal to $14.40 (120% of initial public offering price per Unit) per share, assuming an initial public offering price of $12.00 per Unit (the midpoint of the price range set forth on the cover of this prospectus) subject to adjustment as discussed below, terminating at 5:00 p.m., New York City time, on the fifth (5th) anniversary of the date of issuance.
The exercise price and number of shares of common stock issuable upon exercise of the Warrants may be adjusted in certain circumstances, including in the event of a stock dividend, extraordinary dividend on or recapitalization, reorganization, merger or consolidation.
The Warrants may be exercised upon surrender of the warrant certificate on or prior to the expiration date at the offices of the Warrant Agent, with the exercise form attached to the warrant certificate completed and executed as indicated, accompanied by full payment of the exercise price, by certified or official bank check payable to us, for the number of warrants being exercised. The Warrant holders do not have the rights or privileges of holders of common stock or any voting rights until they exercise their Warrants and receive shares of common stock, except as set forth in the Warrants. After the issuance of shares of common stock upon exercise of the warrants, each holder will be entitled to one vote for each share held of record on all matters to be voted on by stockholders.
No Warrants will be exercisable for cash unless at the time of the exercise a prospectus or prospectus relating to common stock issuable upon exercise of the Warrants is current and the common stock has been registered or qualified or deemed to be exempt under the securities laws of the state of residence of the holder of the warrants. Under the terms of the Warrant Agent Agreement, we have agreed to use our best efforts to maintain a current prospectus or prospectus relating to common stock issuable upon exercise of the Warrants until the expiration of the Warrants. Additionally, the market for the Warrants may be limited if the prospectus or prospectus relating to the common stock issuable upon exercise of the Warrants is not current or if the common stock is not qualified or exempt from qualification in the jurisdictions in which the holders of such Warrants reside. In no event will the registered holders of a Warrant be entitled to receive a net-cash settlement in lieu of physical settlement in shares of our common stock.
No fractional shares of common stock will be issued upon exercise of the Warrants. If, upon exercise of the Warrants, a holder would be entitled to receive a fractional interest in a share, we will, upon exercise, round down to the nearest whole number the number of shares of common stock to be issued to the Warrant holder. If multiple Warrants are exercised by the holder at the same time, we will aggregate the number of whole shares issuable upon exercise of all the Warrants.
The price of the Warrants has been arbitrarily established by us and the underwriters after giving consideration to numerous factors, including but not limited to, the pricing of the Units in this offering. No particular weighting was given to any one aspect of those factors considered. We have not performed any valuation of the Warrants.
Pre-Funded Warrants
The following summary of certain terms and provisions of the Pre-funded Warrants included in the Pre-funded Units that are being offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the Pre-funded Warrants, the form of which is filed as an exhibit to the registration statement of which this prospectus forms a part. Prospective investors should carefully review the terms and provisions of the form of Pre-funded Warrant for a complete description of the terms and conditions of the Pre-funded Warrants.
| -105- |
Duration and Exercise Price
Each Pre-funded Warrant offered hereby will have an initial exercise price per share equal to $0.0001. The Pre-funded Warrants will be immediately exercisable and may be exercised at any time until the Pre-funded Warrants are exercised in full. The exercise price and number of shares of common stock issuable upon exercise is subject to appropriate adjustment in the event of stock dividends, stock splits, reorganizations or similar events affecting our common stock and the exercise price. The Pre-funded Warrants will be issued separately from the accompanying Warrants included in the Pre-funded Units.
Exercisability
The Pre-funded Warrants will be exercisable, at the option of each holder, in whole or in part, by delivering to us a duly executed exercise notice accompanied by payment in full for the number of shares of our common stock purchased upon such exercise (except in the case of a cashless exercise as discussed below). A holder (together with its affiliates) may not exercise any portion of a Pre-funded Warrant to the extent that the holder would own more than 4.99% of the outstanding common stock immediately after exercise, except that upon at least 61 days’ prior notice from the holder to us, the holder may increase the amount of ownership of outstanding stock after exercising the holder’s Pre-funded Warrants up to 9.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Pre-funded Warrants. Purchasers of Pre-funded Units in this offering may also elect prior to the issuance of the Pre-funded Units to have the initial exercise limitation set at 9.99% of our outstanding common stock.
Cashless Exercise
If, at the time a holder exercises its Pre-funded Warrants, a registration statement registering the issuance of the shares of common stock underlying the Pre-funded Warrants under the Securities Act of 1933, as amended, or the Securities Act, is not then effective or available for the issuance of such shares, then in lieu of making the cash payment otherwise contemplated to be made to us upon such exercise in payment of the aggregate exercise price, the holder may elect instead to receive upon such exercise (either in whole or in part) the net number of shares of common stock determined according to a formula set forth in the Pre-funded Warrants.
Transferability
Subject to applicable laws, a Pre-funded Warrant may be transferred at the option of the holder upon surrender of the Pre-funded Warrant to us together with the appropriate instruments of transfer.
Fractional Shares
No fractional shares of common stock will be issued upon the exercise of the Pre-funded Warrants. Rather, the number of shares of common stock to be issued will be rounded to the nearest whole number.
Trading Market
There is no trading market available for the Pre-funded Warrants on any securities exchange or nationally recognized trading system.
Right as a Stockholder
Except as otherwise provided in the Pre-funded Warrants or by virtue of such holder’s ownership of shares of our common stock, the holders of the Pre-funded Warrants do not have the rights or privileges of holders of our common stock, including any voting rights, until they exercise their Pre-funded Warrants.
Fundamental Transaction
In the event of a fundamental transaction, as described in the Pre-funded Warrants and generally including any reorganization, recapitalization or reclassification of our common stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, the acquisition of at least 50% of our outstanding common stock, or any person or group becoming the beneficial owner of at least 50% of the voting power represented by our outstanding common stock, any unexercised portion of the Pre-funded Warrants will be automatically exercised on a cashless basis determined according to a formula set forth in the Pre-funded Warrants.
Exclusive Forum
Our Sixth Amended and Restated Certificate of Incorporation to be effective upon completion of this offering provides that unless we consent in writing to the selection of an alternative forum, the State of Delaware is the sole and exclusive forum for: (i) any derivative action or proceeding brought on behalf of us, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee of our Company to us or our stockholders, (iii) any action asserting a claim against us, our directors, officers or employees arising pursuant to any provision of the DGCL or our Sixth Amended and Restated Certificate of Incorporation or our Amended and Restated Bylaws to be effective upon completion of this offering, or (iv) any action asserting a claim against us, our directors, officers, employees or agents governed by the internal affairs doctrine, except for, as to each of (i) through (iv) above, any claim as to which the Court of Chancery determines that there is an indispensable party not subject to the jurisdiction of the Court of Chancery (and the indispensable party does not consent to the personal jurisdiction of the Court of Chancery within ten days following such determination), which is vested in the exclusive jurisdiction of a court or forum other than the Court of Chancery, or for which the Court of Chancery does not have subject matter jurisdiction.
| -106- |
Additionally, our Sixth Amended and Restated Certificate of Incorporation to be effective upon completion of this offering provides that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act or the Securities Exchange Act of 1934, as amended. Any person or entity purchasing or otherwise acquiring any interest in shares of our capital stock are deemed to have notice of and consented to this provision.
Anti-Takeover Effects of Delaware law and Our Sixth Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws
The provisions of Delaware law, our Sixth Amended and Restated Certificate of Incorporation and our Amended and Restated Bylaws to be adopted upon the closing of this offering, described below may have the effect of delaying, deferring or discouraging another party from acquiring control of us.
Section 203 of the Delaware General Corporation Law
We are subject to Section 203 of the Delaware General Corporation Law, which prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years after the date that such stockholder became an interested stockholder, with the following exceptions:
| ● | before such date, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; | |
| ● | upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction began, excluding for purposes of determining the voting stock outstanding (but not the outstanding voting stock owned by the interested stockholder) those shares owned (i) by persons who are directors and also officers and (ii) employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| ● | on or after such date, the business combination is approved by the board of directors and authorized at an annual or special meeting of the stockholder, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines business combination to include the following:
| ● | any merger or consolidation involving the corporation and the interested stockholder; | |
| ● | any sale, transfer, pledge or other disposition of 10% or more of the assets of the corporation involving the interested stockholder; | |
| ● | subject to certain exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; | |
| ● | any transaction involving the corporation that has the effect of increasing the proportionate share of the stock or any class or series of the corporation beneficially owned by the interested stockholder; or | |
| ● | the receipt by the interested stockholder of the benefit of any loss, advances, guarantees, pledges or other financial benefits by or through the corporation. |
In general, Section 203 defines an “interested stockholder” as an entity or person who, together with the person’s affiliates and associates, beneficially owns, or within three years prior to the time of determination of interested stockholder status did own, 15% or more of the outstanding voting stock of the corporation.
Board of Directors Vacancies
Our Sixth Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws authorize only our board of directors to fill vacant directorships. In addition, the number of directors constituting our board of directors may be set only by resolution of the majority of the incumbent directors.
Stockholder Action; Special Meeting of Stockholders
Our Sixth Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws provide that our stockholders may not take action by written consent. Our Sixth Amended and Restated Certificate of Incorporation and Amended and Restated Bylaws further provide that special meetings of our stockholders may be called by a majority of the board of directors, the Chief Executive Officer, or the Chairman of the board of directors.
Advance Notice Requirements for Stockholder Proposals and Director Nominations
Our Amended and Restated Bylaws provide that stockholders seeking to bring business before our annual meeting of stockholders, or to nominate candidates for election as directors at our annual meeting of stockholders, must provide timely notice of their intent in writing. To be timely, a stockholder’s notice must be delivered to the secretary at our principal executive offices not later than the close of business on the 90th day nor earlier than the close of business on the 120th day prior to the first anniversary of the preceding year’s annual meeting; provided, however, that in the event the date of the annual meeting is more than 30 days before or more than 60 days after such anniversary date, or if no annual meeting was held in the preceding year, notice by the stockholder to be timely must be so delivered not earlier than the close of business on the 120th day prior to such annual meeting and not later than the close of business on the later of the 90th day prior to such annual meeting or the 10th day following the day on which a public announcement of the date of such meeting is first made by us. These provisions may preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our annual meeting of stockholders.
Authorized but Unissued Shares
Our authorized but unissued shares of common stock and preferred stock are available for future issuance without stockholder approval and may be utilized for a variety of corporate purposes, including future public offerings to raise additional capital, corporate acquisitions and employee benefit plans. The existence of authorized but unissued and unreserved common stock and preferred stock could render more difficult or discourage an attempt to obtain control of us by means of a proxy contest, tender offer, merger or otherwise. If we issue such shares without stockholder approval and in violation of limitations imposed by the Nasdaq Capital Market or any stock exchange on which our stock may then be trading, our stock could be delisted.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Philadelphia Stock Transfer, Inc.
| -107- |
SHARES ELIGIBLE FOR FUTURE SALE
Upon completion of this offering, we will have 8,207,572 shares of common stock issued and outstanding. All of the shares sold in this offering will be freely transferable without restriction under the Securities Act unless purchased by one of our affiliates as that term is defined in Rule 144 under the Securities Act, which generally includes directors, executive officers and 10% stockholders. Sales of substantial amounts of our shares in the public market could adversely affect prevailing market prices of our shares.
All outstanding shares prior to this offering are “restricted securities” as that term is defined in Rule 144 and may be sold only if they are sold pursuant to an effective registration statement under the Securities Act or an exemption from the registration requirements of the Securities Act such as those provided in Rules 144 and 701 promulgated under the Securities Act, which rules are summarized below. Restricted shares may also be sold outside of the United States in accordance with Regulation S under the Securities Act. This prospectus may not be used in connection with any resale of our shares acquired in this offering by our affiliates.
Rule 144
In general, under Rule 144 of the Securities Act, a person or entity that has beneficially owned our common stock for at least six months and is not our “affiliate” will be entitled to sell our common stock, subject only to the availability of current public information about us, and will be entitled to sell shares held for at least one year without any restriction. A person or entity that is our “affiliate” and has beneficially owned our common stock for at least six months will be able to sell, within a rolling three month period, the number of shares that does not exceed the greater of the following:
(i) 1% of the then outstanding common stock, which immediately after this offering will equal approximately 82,076 shares if the maximum number of shares being offered by us; and
(ii) the average weekly trading volume of our common stock on the Nasdaq Capital Market during the four calendar weeks preceding the date on which notice of the sale is filed with the SEC.
Sales by affiliates under Rule 144 must be made through unsolicited brokers’ transactions. They are also subject to manner of sale provisions, notice requirements and the availability of current public information about us.
Rule 701
In general, under Rule 701 of the Securities Act as currently in effect, each of our employees, directors or consultants who purchases our common stock from us pursuant to a compensatory stock or option plan or other written agreement relating to compensation is eligible to resell such common stock 90 days after we become a reporting company under the Exchange Act in reliance on Rule 144, but without compliance with some of the restrictions, such as the holding period, contained in Rule 144. However, the Rule 701 shares would remain subject to lock-up arrangements and would only become eligible for sale when the lock-up period expires.
| -108- |
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS OF THE COMPANY’S COMMON STOCK, PRE-FUNDED WARRANTS AND WARRANTS
The following is a summary of the material U.S. federal income tax consequences to non-U.S. holders (as defined below) of the ownership and disposition of our common stock, Warrants and Pre-Funded Warrants but does not purport to be a complete analysis of all the potential tax considerations relating thereto. This summary is based upon the provisions of the Internal Revenue Code of 1986, as amended, or the Internal Revenue Code, Treasury regulations promulgated thereunder, administrative rulings and judicial decisions, all as of the date hereof. These authorities may be changed, possibly retroactively, so as to result in U.S. federal income tax consequences different from those set forth below. No ruling on the U.S. federal, state, or local tax considerations relevant to the Company’s operations or to the purchase, ownership or disposition of its shares, has been requested from the IRS or other tax authority. No assurance can be given that the IRS would not assert, or that a court would not sustain, a position contrary to any of the tax consequences described below.
This summary also does not address the tax considerations arising under the laws of any non-U.S., state or local jurisdiction, or under U.S. federal gift and estate tax laws, except to the limited extent set forth below. In addition, this discussion does not address tax considerations applicable to an investor’s particular circumstances or to investors that may be subject to special tax rules, including, without limitation:
| ● | banks, insurance companies or other financial institutions, regulated investment companies or real estate investment trusts; | |
| ● | persons subject to the alternative minimum tax or Medicare contribution tax on net investment income; | |
| ● | tax-exempt organizations or governmental organizations; | |
| ● | controlled foreign corporations, passive foreign investment companies and corporations that accumulate earnings to avoid U.S. federal income tax; | |
| ● | brokers or dealers in securities or currencies; | |
| ● | traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; | |
| ● | persons that own, or are deemed to own, more than five percent of the Company’s capital stock (except to the extent specifically set forth below); | |
| ● | US expatriates and certain former citizens or long-term residents of the United States; | |
| ● | partnerships or entities classified as partnerships for U.S. federal income tax purposes or other pass- through entities (and investors therein); | |
| ● | persons who hold the Company’s common stock as a position in a hedging transaction, “straddle,” “conversion transaction” or other risk reduction transaction or integrated investment; | |
| ● | persons who hold or receive the Company’s common stock pursuant to the exercise of any employee stock option or otherwise as compensation; | |
| ● | persons who do not hold the Company’s common stock as a capital asset within the meaning of Section 1221 of the Internal Revenue Code; or | |
| ● | persons deemed to sell the Company’s common stock under the constructive sale provisions of the Internal Revenue Code. |
| -109- |
In addition, if a partnership or entity classified as a partnership for U.S. federal income tax purposes holds our common stock, Warrants or Pre-funded Warrants, the tax treatment of a partner generally will depend on the status of the partner and upon the activities of the partnership. Accordingly, partnerships that hold our common stock, Warrants or Pre-funded Warrants, and partners in such partnerships, should consult their tax advisors.
Although the characterization of the Pre-funded Warrants for U.S. federal income tax purposes is not entirely clear, because the exercise price of the Pre-funded Warrants is a nominal amount, the Company expects to treat the Pre-funded Warrants as common stock of the Company for U.S. federal income tax purposes. Except where noted, the remainder of this discussion assumes that the Pre-funded Warrants will be so treated. Each non-U.S. holder should consult its own tax advisor regarding the proper characterization of the Pre-funded Warrants for U.S. federal, state and local, and non-U.S. tax purposes, and the consequences to them of such treatment given their individual circumstances. Some portions of the below discussion make reference to potential consequences associated with the purchase, ownership and disposition of the Pre-funded Warrants independent of their potential characterization as common shares
You are urged to consult your tax advisor with respect to the application of the U.S. federal income tax laws to your particular situation, as well as any tax consequences of the purchase, ownership and disposition of our common stock, Warrants or Pre-funded Warrants, arising under the U.S. federal estate or gift tax rules or under the laws of any state, local, non-U.S., or other taxing jurisdiction or under any applicable tax treaty.
Non-U.S. Holder Defined
For purposes of this discussion, you are a non-U.S. holder (other than a partnership) if you are any holder other than:
| ● | an individual citizen or resident of the United States (for U.S. federal income tax purposes); | |
| ● | a corporation or other entity taxable as a corporation created or organized in the United States or under the laws of the United States, any state thereof, or the District of Columbia; | |
| ● | an estate whose income is subject to U.S. federal income tax regardless of its source; or | |
| ● | a trust (x) whose administration is subject to the primary supervision of a U.S. court and which has one or more “U.S. persons” (within the meaning of Section 7701(a)(30) of the Internal Revenue Code) who have the authority to control all substantial decisions of the trust or (y) which has made a valid election to be treated as a U.S. person |
Distributions
As described in “Dividend Policy,” we have never declared or paid cash dividends on our common stock and do not anticipate paying any dividends on our common stock in the foreseeable future. However, if we do make distributions on our common stock, those payments will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent those distributions exceed both our current and accumulated earnings and profits, they will constitute a return of capital and will first reduce your basis in our common stock, but not below zero, and then will be treated as gain from the sale of stock as described below under “—Gain on Disposition of Common Stock.”
Subject to the discussion below on effectively connected income, backup withholding and foreign accounts, any dividend paid to a non-U.S. holder generally will be subject to U.S. withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable income tax treaty. In order to receive a reduced treaty rate, a non-U.S. holder must provide us with an IRS Form W-8BEN, IRS Form W-8BEN-E, or other appropriate version of IRS Form W-8 certifying qualification for the reduced rate. A non-U.S. holder of shares of our common stock eligible for a reduced rate of U.S. withholding tax pursuant to an income tax treaty may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. If the non-U.S. holder holds the stock through a financial institution or other agent acting on the non-U.S. holder’s behalf, the non-U.S. holder will be required to provide appropriate documentation to the agent, which then will be required to provide certification to us or our paying agent, either directly or through other intermediaries.
| -110- |
Dividends received by a non-U.S. holder that are effectively connected with such non-U.S. holder’s conduct of a U.S. trade or business (and, if required by an applicable income tax treaty, attributable to a permanent establishment maintained by a non-U.S. holder in the United States) are generally exempt from the withholding tax described above. In order to obtain this exemption, a non-U.S. holder must provide us with an IRS Form W-8ECI or other applicable IRS Form W-8 properly certifying such exemption. Such effectively connected dividends, although not subject to withholding tax, are taxed at the same graduated rates applicable to U.S. persons, net of certain deductions and credits (if any). In addition, if a non-U.S. holder is a corporate non-U.S. holder, dividends received by such non-U.S. holder that are effectively connected with such non-U.S. holder’s conduct of a U.S. trade or business may also be subject to a branch profits tax at a rate of 30% or such lower rate as may be specified by an applicable income tax treaty. You should consult your tax advisor regarding any applicable tax treaties that may provide for different rules.
Gain on Disposition of Common Stock, Warrants or Pre-funded Warrants
Subject to the discussion below regarding backup withholding and foreign accounts, a non-U.S. holder generally will not be required to pay U.S. federal income tax on any gain realized upon the sale or other disposition of our common stock, Warrants or Pre-funded Warrants unless:
| ● | the gain is effectively connected with the non-U.S. holder’s conduct of a U.S. trade or business (and, if required by an applicable income tax treaty, the gain is attributable to a permanent establishment maintained by such non-U.S. holder in the United States); | |
| ● | the non-U.S. holders are a non-resident alien individual who is present in the United States for a period or periods aggregating 183 days or more during the taxable year in which the sale or disposition occurs and certain other conditions are met; or | |
| ● | our common stock, Warrants and Pre-funded Warrants, constitute a United States real property interest by reason of our status as a “United States real property holding corporation,” or USRPHC, for U.S. federal income tax purposes at any time within the shorter of (i) the five-year period preceding the non-U.S. holder’s disposition of our common stock, Warrants or Pre-funded Warrants, or (ii) the non-U.S. holder’s holding period for our common stock, Warrants or Pre-funded Warrants. |
We believe that we are not currently and will not become a USRPHC for U.S. federal income tax purposes, and the remainder of this discussion so assumes. However, because the determination of whether we are a USRPHC depends on the fair market value of our U.S. real property relative to the fair market value of our other business assets, there can be no assurance that we will not become a USRPHC in the future. Even if we become a USRPHC, however, as long as our common stock is regularly traded on an established securities market, such common stock will be treated as U.S. real property interests only if the non-U.S. holder’s actually or constructively hold more than five percent of such regularly traded common stock at any time during the shorter of (i) the five-year period preceding the non-U.S. holder’s disposition of our common stock, or (ii) the non-U.S. holder’s holding period for our common stock.
Gains described in the first bullet point above, generally will be subject to U.S. federal income tax on a net income basis at the regular graduated U.S. federal income tax rates, and a corporate non-U.S. holder described in the first bullet above also may be subject to the branch profits tax at a 30% rate, or such lower rate as may be specified by an applicable income tax treaty. An individual non-U.S. holder described in the second bullet above, you will be required to pay a 30% tax (or such lower rate specified by an applicable income tax treaty) on the gain derived from the sale, which gain may be offset by U.S. source capital losses for the year (provided the non-U.S. holder has timely filed U.S. federal income tax returns with respect to such losses). You should consult any applicable income tax or other treaties that may provide for different rules.
| -111- |
Exercise of a Warrant
In general, a non-U.S. holder will not recognize gain or loss for U.S. federal income tax purposes upon exercise of a Warrant, except to the extent the non-U.S. holder receives a cash payment for any such fractional share that would otherwise have been issuable upon exercise of the Warrant which will be treated as a sale subject to the rules described under “Gain on Disposition of Common Stock, Warrants or Pre-funded Warrants” above. The non-U.S. holder will take a tax basis in the shares acquired on the exercise of a Warrant equal to the exercise price of the Warrant. The non-U.S. holder’s holding period in the shares of our common stock acquired on exercise of the Warrant will begin on the date of exercise of the Warrant, and will not include any period for which the non-U.S. holder held the Warrant.
Expiration of a Warrant
Expiration of Warrants will be treated as if the non-U.S. holder sold or exchanged the Warrants and recognized a capital loss equal to the non-U.S. holder’s tax basis in the Warrants. However, a non-U.S. holder will not be able to utilize a loss recognized upon expiration of a Warrant against the non-U.S. holder’s U.S. federal income tax liability unless the loss is effectively connected with the non-U.S. holder’s conduct of a trade or business within the United States (and, if an income tax treaty applies, is attributable to a permanent establishment in the United States) or is treated as a U.S.-source loss and the non-U.S. holder is present 183 days or more in the taxable year of disposition and certain other conditions are met.
Certain Adjustments to the Warrants or Pre-funded Warrants and Payments in Respect of the Warrants or Pre-funded Warrants
Under Section 305 of the Code, an adjustment to the number of shares of common stock that will be issued on the exercise of the Warrants or Pre-funded Warrants, or an adjustment to the exercise price of the Warrants or Pre-funded Warrants, may be treated as a constructive distribution to a non-U.S. Holder of the Warrants or Pre-funded Warrants if, and to the extent that, such adjustment has the effect of increasing such non-U.S. Holder’s proportionate interest in our “earnings and profits” or assets, depending on the circumstances of such adjustment (for example, if such adjustment is to compensate for a distribution of cash or other property to our shareholders). Adjustments to the exercise price of Warrants or Pre-funded Warrants made pursuant to a bona fide reasonable adjustment formula that has the effect of preventing dilution of the interest of the non-U.S. Holder of the Warrants or Pre-funded Warrants generally should not be considered to result in a constructive distribution. Such constructive distribution would be treated as a dividend, return of capital or capital gain as described under the heading “Non-U.S. Holders—Distributions on Common Stock” above. Any such constructive distribution would be taxable whether or not there is an actual distribution of cash or other property.
In addition, regulations governing “dividend equivalents” under Section 871(m) of the Code may apply to the Pre-funded Warrants. Under those regulations, an implicit or explicit payment under the Pre-funded Warrants that references a dividend distribution on our common stock (including an adjustment to the amount due on the Pre-funded Warrant to take into account a dividend distribution on our common stock) would be taxable to a non-U.S. Holder as described under the heading “Non-U.S. Holders—Distributions” above. Such dividend equivalent amount would be taxable and subject to withholding whether or not there is actual payment of cash or other property, and the Company may satisfy any withholding obligations it has in respect of the Pre-funded Warrants by withholding from other amounts due to the non-U.S. holder. Non-U.S. holders are encouraged to consult their own tax advisors regarding the application of Section 871(m) of the Code to the Pre-funded Warrants.
Backup Withholding and Information Reporting
Generally, we must report annually to the IRS, regardless of whether any tax was withheld, the amount of dividends paid to a non-U.S. holder, the non-U.S. holder’s name and address and the amount of tax withheld, if any. A similar report will be sent to the non-U.S. holder. Pursuant to applicable income tax treaties or other agreements, the IRS may make these reports available to tax authorities in the non-U.S. holder’s country of residence.
Payments of dividends or of proceeds on the disposition of stock, Warrants or Pre-funded Warrants made to a non-U.S. holder may be subject to information reporting and backup withholding at a current rate of 24% unless such non-U.S. holder establishes an exemption, for example, by properly certifying such non-U.S. holder’s non-U.S. status on an IRS Form W-8BEN, IRS Form W-8BEN-E, or another appropriate version of IRS Form W-8.
Backup withholding is not an additional tax; rather, the U.S. federal income tax liability of persons subject to backup withholding will be reduced by the amount of tax withheld. If withholding results in an overpayment of taxes, a refund or credit may generally be obtained from the IRS, provided that the required information is furnished to the IRS in a timely manner.
Foreign Account Tax Compliance
The Foreign Account Tax Compliance Act, or FATCA, imposes withholding tax at a rate of 30% on dividends on and gross proceeds from the sale or other disposition of our common stock, Warrants or Pre-funded Warrants paid to “foreign financial institutions” (as specially defined under these rules), unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding the U.S. account holders of such institution (which includes certain equity and debt holders of such institution, as well as certain account holders that are foreign entities with U.S. owners) or otherwise establishes an exemption. FATCA also generally imposes a U.S. federal withholding tax of 30% on dividends on and gross proceeds from the sale or other disposition of our common stock paid to a “non-financial foreign entity” (as specially defined for purposes of these rules) unless such entity provides the withholding agent with a certification identifying certain substantial direct and indirect U.S. owners of the entity, certifies that there are none or otherwise establishes an exemption. The withholding provisions under FATCA generally apply to dividends on our common stock, and under current transition rules, are expected to apply with respect to the gross proceeds from the sale or other disposition of our common stock. An intergovernmental agreement between the United States and an applicable foreign country may modify the requirements described in this paragraph. Non-U.S. holders should consult their own tax advisors regarding the possible implications of this legislation on their investment in our common stock.
Each prospective investor should consult its own tax advisor regarding the particular U.S. federal, state and local and non-U.S. tax consequences of purchasing, holding and disposing of our common stock, Warrants or Pre-funded Warrants, including the consequences of any proposed change in applicable laws.
| -112- |
We have entered into an underwriting agreement with Roth Capital Partners, LLC, acting as the representative of several underwriters named below. Subject to the terms and conditions of the underwriting agreement, we have agreed to sell to the underwriters, and the underwriters have agreed to purchase from us, Units and Pre-funded Units. We have been approved to list our common stock on the Nasdaq Capital Market under the symbol “RNXT” upon our satisfaction of the exchange’s initial listing criteria, including the completion of this offering.
Pursuant to the terms and subject to the conditions contained in the underwriting agreement, we have agreed to sell to the underwriters named below, and the underwriters have agreed to purchase from us, the respective number of Units and Pre-funded Units set forth opposite its name below:
| Underwriter | Number of Units | Number of Pre-funded Units | ||||||
| Roth Capital Partners, LLC | ||||||||
| Maxim Group LLC | ||||||||
| Total | ||||||||
The underwriting agreement provides that the obligation of the underwriters to purchase the Units and Pre-funded Units offered by this prospectus is subject to certain conditions. The underwriters are obligated to purchase all of the Units and Pre-funded Units offered hereby if any of the Units or Pre-funded Units are purchased.
We have granted the representative an option, exercisable for 45 days from the date of this prospectus, to purchase up to 277,500 additional shares of common stock at the public offering price per share, less underwriting discounts and commissions, and/or additional Warrants to purchase up to 208,125 shares of common stock at the public offering price per Warrant, less underwriting discounts and commissions, in any combination thereof, to cover over-allotments, if any, in connection with the units offered by this prospectus.
Discounts, Commissions and Expenses
The underwriters propose to offer Units and Pre-funded Units purchased pursuant to the underwriting agreement to the public at the initial public offering price set forth on the cover page of this prospectus and to certain dealers at that price less a concession not in excess of $__ per Unit. After this offering, the initial public offering price and concession may be changed by the underwriters. No such change shall change the amount of proceeds to be received by us as set forth on the cover page of this prospectus.
In connection with the sale of the Units and Pre-funded Units to be purchased by the underwriters, the underwriters will be deemed to have received compensation in the form of underwriting commissions and discounts. The underwriters’ commissions and discounts will be 7.0% of the gross proceeds of this offering, or $ ____ per Unit, based on the public offering price per Unit set forth on the cover page of this prospectus.
We have also agreed to reimburse Roth Capital Partners at closing for legal expenses incurred by it in connection with the offering up to a maximum of $150,000.
The following table shows the underwriting discounts and commissions payable to the underwriters by us in connection with this offering (assuming both the exercise and non-exercise of the over-allotment option to purchase additional Units, Pre-funded Units, shares of our common stock, Pre-funded Warrants and/or Warrants from us at prices described herein to cover over-allotments, if any, of the Units and the Pre-funded Units offered hereby we have granted to the underwriters):
| Per Unit | Per Pre-funded Unit | Total Without Over- allotment | Total With Over- allotment | |||||||||||||
| Initial public offering price | $ | $ | $ | $ | ||||||||||||
| Underwriting discounts and commissions paid by us | $ | $ | $ | $ | ||||||||||||
| -113- |
Underwriter’s Warrant
We will issue to Roth Capital Partners or its designees a warrant to purchase up to 5% of shares of common stock (including shares of common stock underlying the Pre-funded Warrants and the Warrants) sold in this offering. The warrant will have an exercise price equal to 120% of the initial public offering price of the Units set forth on the cover page of this prospectus (or $_____ per share and accompanying warrant), subject to standard anti-dilution adjustments for share splits and similar transactions (the “Underwriter’s Warrant”). The Underwriter’s Warrant will be exercisable, in whole or in part, six months after issuance and will expire on the fifth anniversary of the commencement of sales in this offering in accordance with FINRA Rule 5110(g)(8)(A). The Underwriter’s Warrant will provide for one demand registration right at our expense and an additional demand registration right at the holder’s expense and unlimited piggyback registration rights at our expense for a period of five years following the date of commencement of sales of this offering. Pursuant to FINRA Rule 5110(e), the Purchase Option and any shares of common stock issued upon exercise of the Underwriter’s Warrant shall not be sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put or call transaction that would result in the effective economic disposition of the securities by any person for a period of 180 days immediately following the date of commencement of sales of this offering, except the transfer of any security: (i) by operation of law or by reason of reorganization of the issuer; (ii) to any FINRA member firm participating in the offering and the officers, partners, registered persons or affiliates thereof, if all securities so transferred remain subject to the lock-up restriction set forth above for the remainder of the time period; (iii) if the aggregate amount of our securities held by the Representative or related persons does not exceed 1% of the securities being offered; (iv) that is beneficially owned on a pro-rata basis by all equity owners of an investment fund, provided that no participating member manages or otherwise directs investments by the fund and the participating members in the aggregate do not own more than 10% of the equity in the fund; (v) the exercise or conversion of any security, if all securities remain subject to the lock-up restriction set forth above for the remainder of the time period; (vi) if we meet the registration requirements of Forms S-3, F-3 or F-10; or (vii) back to us in a transaction exempt from registration with the SEC. The Underwriter’s Warrant and the shares of common stock underlying the Underwriter’s Warrant are registered in the registration statement of which this prospectus is a part.
Right of First Refusal
We have granted Roth Capital Partners an 18-month right first refusal to act as the exclusive placement agent or sole book-running manager and sole lead managing underwriter for any private or public offering of equity, equity-linked or debt securities undertaken by us, provided that this offering is consummated.
Indemnification
Pursuant to the underwriting agreement, we have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, or to contribute to payments that the underwriters or such other indemnified parties may be required to make in respect of those liabilities.
Lock-Up Agreements
We and each of our directors, officers and certain stockholders representing approximately 94% of our shares outstanding prior to this offering have agreed not to offer, sell, agree to sell, directly or indirectly, or otherwise dispose of any shares of common stock or any securities convertible into or exchangeable for shares of common stock for a period of 180 days after the closing date of the offering pursuant to the underwriting agreement without the prior written consent of Roth Capital Partners. These lock-up agreements provide for limited exceptions and their restrictions may be waived at any time by Roth Capital Partners.
Electronic Distribution
This prospectus may be made available in electronic format on websites or through other online services maintained by the underwriters or by their affiliates. In those cases, prospective investors may view offering terms online and prospective investors may be allowed to place orders online. Other than this prospectus and the accompanying prospectus in electronic format, the information on the underwriters’ websites or our website and any information contained in any other websites maintained by the underwriters or by us is not part of this prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or the underwriter in its capacity as underwriter, and should not be relied upon by investors.
Price Stabilization, Short Positions and Penalty Bids
In connection with the offering the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Exchange Act:
| -114- |
| ● | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. | |
| ● | Over-allotment involves sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any covered short position by either exercising their over-allotment option and/or purchasing shares in the open market. | |
| ● | Syndicate covering transactions involve purchases of the common stock in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. A naked short position occurs if the underwriters sell more shares than could be covered by the over-allotment option. This position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. | |
| ● | Penalty bids permit the underwriters to reclaim a selling concession from a syndicate member when the common stock originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of the common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. These transactions may be discontinued at any time
Neither we nor the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our shares of common stock. In addition, neither we nor the underwriters make any representation that the underwriter will engage in these transactions or that any transaction, if commenced, will not be discontinued without notice.
Offer restrictions outside the United States
Other than in the United States, no action has been taken by us or the underwriter that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to this offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Australia
This prospectus is not a disclosure document under Chapter 6D of the Australian Corporations Act, has not been lodged with the Australian Securities and Investments Commission and does not purport to include the information required of a disclosure document under Chapter 6D of the Australian Corporations Act. Accordingly, (i) the offer of the securities under this prospectus is only made to persons to whom it is lawful to offer the securities without disclosure under Chapter 6D of the Australian Corporations Act under one or more exemptions set out in section 708 of the Australian Corporations Act, (ii) this prospectus is made available in Australia only to those persons as set forth in clause (i) above, and (iii) the offeree must be sent a notice stating in substance that by accepting this offer, the offeree represents that the offeree is such a person as set forth in clause (i) above, and, unless permitted under the Australian Corporations Act, agrees not to sell or offer for sale within Australia any of the securities sold to the offeree within 12 months after its transfer to the offeree under this prospectus.
| -115- |
Canada
The securities may be sold in Canada only to purchasers purchasing, or deemed to be purchasing, as principal that are accredited investors, as defined in National Instrument 45-106 Prospectus Exemptions or subsection 73.3(1) of the Securities Act (Ontario), and are permitted clients, as defined in National Instrument 31-103 Registration Requirements, Exemptions and Ongoing Registrant Obligations. Any resale of the securities must be made in accordance with an exemption from, or in a transaction not subject to, the prospectus requirements of applicable securities laws. Securities legislation in certain provinces or territories of Canada may provide a purchaser with remedies for rescission or damages if this prospectus (including any amendment thereto) contains a misrepresentation, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser’s province or territory. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser’s province or territory for particulars of these rights or consult with a legal advisor. Pursuant to section 3A.3 of National Instrument 33-105 Underwriting Conflicts (NI 33-105), the underwriter is not required to comply with the disclosure requirements of NI33-105 regarding underwriter conflicts of interest in connection with this offering.
China
The information in this document does not constitute a public offer of the securities, whether by way of sale or subscription, in the People’s Republic of China (excluding, for purposes of this paragraph, Hong Kong Special Administrative Region, Macau Special Administrative Region and Taiwan). The securities may not be offered or sold directly or indirectly in the PRC to legal or natural persons other than directly to “qualified domestic institutional investors.”
European Economic Area — Belgium, Germany, Luxembourg and Netherlands
The information in this document has been prepared on the basis that all offers of securities will be made pursuant to an exemption under the Directive 2003/71/EC (“Prospectus Directive”), as implemented in Member States of the European Economic Area (each, a “Relevant Member State”), from the requirement to produce a prospectus for offers of securities.
An offer to the public of securities has not been made, and may not be made, in a Relevant Member State except pursuant to one of the following exemptions under the Prospectus Directive as implemented in that Relevant Member State:
| ● | to legal entities that are authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; | |
| ● | to any legal entity that has two or more of (i) an average of at least 250 employees during its last fiscal year; (ii) a total balance sheet of more than €43,000,000 (as shown on its last annual unconsolidated or consolidated financial statements) and (iii) an annual net turnover of more than €50,000,000 (as shown on its last annual unconsolidated or consolidated financial statements); | |
| ● | to fewer than 100 natural or legal persons (other than qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive) subject to obtaining our prior consent or any underwriter for any such offer; or | |
| ● | in any other circumstances falling within Article 3(2) of the Prospectus Directive, provided that no such offer of securities shall require us to publish a prospectus pursuant to Article 3 of the Prospectus Directive. |
| -116- |
France
This document is not being distributed in the context of a public offering of financial securities (offre au public de titres financiers) in France within the meaning of Article L.411-1 of the French Monetary and Financial Code (Code monétaire et financier) and Articles 211-1 et seq. of the General Regulation of the French Autorité des marchés financiers (“AMF”). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in France.
This document and any other offering material relating to the securities have not been, and will not be, submitted to the AMF for approval in France and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in France.
Such offers, sales and distributions have been and shall only be made in France to (i) qualified investors (investisseurs qualifiés) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2 and D.411-1 to D.411-3, D. 744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation and/or (ii) a restricted number of non-qualified investors (cercle restreint d’investisseurs) acting for their own account, as defined in and in accordance with Articles L.411-2-II-2° and D.411-4, D.744-1, D.754-1 and D.764-1 of the French Monetary and Financial Code and any implementing regulation.
Pursuant to Article 211-3 of the General Regulation of the AMF, investors in France are informed that the securities cannot be distributed (directly or indirectly) to the public by the investors otherwise than in accordance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 to L.621-8-3 of the French Monetary and Financial Code.
Ireland
The information in this document does not constitute a prospectus under any Irish laws or regulations and this document has not been filed with or approved by any Irish regulatory authority as the information has not been prepared in the context of a public offering of securities in Ireland within the meaning of the Irish Prospectus (Directive 2003/71/EC) Regulations 2005 (the “Prospectus Regulations”). The securities have not been offered or sold, and will not be offered, sold or delivered directly or indirectly in Ireland by way of a public offering, except to (i) qualified investors as defined in Regulation 2(l) of the Prospectus Regulations and (ii) fewer than 100 natural or legal persons who are not qualified investors.
Israel
The securities offered by this prospectus have not been approved or disapproved by the Israeli Securities Authority (the ISA), or ISA, nor have such securities been registered for sale in Israel. The shares may not be offered or sold, directly or indirectly, to the public in Israel, absent the publication of a prospectus. The ISA has not issued permits, approvals or licenses in connection with this offering or publishing the prospectus; nor has it authenticated the details included herein, confirmed their reliability or completeness, or rendered an opinion as to the quality of the securities being offered. Any resale in Israel, directly or indirectly, to the public of the securities offered by this prospectus is subject to restrictions on transferability and must be effected only in compliance with the Israeli securities laws and regulations.
Italy
The offering of the securities in the Republic of Italy has not been authorized by the Italian Securities and Exchange Commission (Commissione Nazionale per le Società e la Borsa, “CONSOB” pursuant to the Italian securities legislation and, accordingly, no offering material relating to the securities may be distributed in Italy and such securities may not be offered or sold in Italy in a public offer within the meaning of Article 1.1(t) of Legislative Decree No. 58 of 24 February 1998 (“Decree No. 58”), other than:
| -117- |
| ● | to Italian qualified investors, as defined in Article 100 of Decree no. 58 by reference to Article 34-ter of CONSOB Regulation no. 11971 of 14 May 1999 (“Regulation no. 1197l”) as amended (“Qualified Investors”); and | |
| ● | in other circumstances that are exempt from the rules on public offer pursuant to Article 100 of Decree No. 58 and Article 34-ter of Regulation No. 11971 as amended. |
Any offer, sale or delivery of the securities or distribution of any offer document relating to the securities in Italy (excluding placements where a Qualified Investor solicits an offer from the issuer) under the paragraphs above must be:
| ● | made by investment firms, banks or financial intermediaries permitted to conduct such activities in Italy in accordance with Legislative Decree No. 385 of 1 September 1993 (as amended), Decree No. 58, CONSOB Regulation No. 16190 of 29 October 2007 and any other applicable laws; and | |
| ● | in compliance with all relevant Italian securities, tax and exchange controls and any other applicable laws. |
Any subsequent distribution of the securities in Italy must be made in compliance with the public offer and prospectus requirement rules provided under Decree No. 58 and the Regulation No. 11971 as amended, unless an exception from those rules applies. Failure to comply with such rules may result in the sale of such securities being declared null and void and in the liability of the entity transferring the securities for any damages suffered by the investors.
Japan
The securities have not been and will not be registered under Article 4, paragraph 1 of the Financial Instruments and Exchange Law of Japan (Law No. 25 of 1948), as amended (the “FIEL”) pursuant to an exemption from the registration requirements applicable to a private placement of securities to Qualified Institutional Investors (as defined in and in accordance with Article 2, paragraph 3 of the FIEL and the regulations promulgated thereunder). Accordingly, the securities may not be offered or sold, directly or indirectly, in Japan or to, or for the benefit of, any resident of Japan other than Qualified Institutional Investors. Any Qualified Institutional Investor who acquires securities may not resell them to any person in Japan that is not a Qualified Institutional Investor, and acquisition by any such person of securities is conditional upon the execution of an agreement to that effect.
New Zealand
The shares of common stock offered hereby have not been offered or sold, and will not be offered or sold, directly or indirectly in New Zealand and no offering materials or advertisements have been or will be distributed in relation to any offer of shares in New Zealand, in each case other than:
| ● | to persons whose principal business is the investment of money or who, in the course of and for the purposes of their business, habitually invest money; | |
| ● | to persons who in all the circumstances can properly be regarded as having been selected otherwise than as members of the public; | |
| ● | to persons who are each required to pay a minimum subscription price of at least NZ$500,000 for the shares before the allotment of those shares (disregarding any amounts payable, or paid, out of money lent by the issuer or any associated person of the issuer); or | |
| ● | in other circumstances where there is no contravention of the Securities Act 1978 of New Zealand (or any statutory modification or reenactment of, or statutory substitution for, the Securities Act 1978 of New Zealand). |
| -118- |
Portugal
This document is not being distributed in the context of a public offer of financial securities (oferta pública de valores mobiliários) in Portugal, within the meaning of Article 109 of the Portuguese Securities Code (Código dos Valores Mobiliários). The securities have not been offered or sold and will not be offered or sold, directly or indirectly, to the public in Portugal. This document and any other offering material relating to the securities have not been, and will not be, submitted to the Portuguese Securities Market Commission (Comissão do Mercado de Valores Mobiliários) for approval in Portugal and, accordingly, may not be distributed or caused to distributed, directly or indirectly, to the public in Portugal, other than under circumstances that are deemed not to qualify as a public offer under the Portuguese Securities Code. Such offers, sales and distributions of securities in Portugal are limited to persons who are “qualified investors” (as defined in the Portuguese Securities Code). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Sweden
This document has not been, and will not be, registered with or approved by Finansinspektionen (the Swedish Financial Supervisory Authority). Accordingly, this document may not be made available, nor may the securities be offered for sale in Sweden, other than under circumstances that are deemed not to require a prospectus under the Swedish Financial Instruments Trading Act (1991:980) (Sw. lag (1991:980) om handel med finansiella instrument). Any offering of securities in Sweden is limited to persons who are “qualified investors” (as defined in the Financial Instruments Trading Act). Only such investors may receive this document and they may not distribute it or the information contained in it to any other person.
Switzerland
The securities may not be publicly offered in Switzerland and will not be listed on the SIX Swiss Exchange (“SIX”) or on any other stock exchange or regulated trading facility in Switzerland. This document has been prepared without regard to the disclosure standards for issuance prospectuses under art. 652a or art. 1156 of the Swiss Code of Obligations or the disclosure standards for listing prospectuses under art. 27 ff. of the SIX Listing Rules or the listing rules of any other stock exchange or regulated trading facility in Switzerland. Neither this document nor any other offering material relating to the securities may be publicly distributed or otherwise made publicly available in Switzerland.
Neither this document nor any other offering material relating to the securities have been or will be filed with or approved by any Swiss regulatory authority. In particular, this document will not be filed with, and the offer of securities will not be supervised by, the Swiss Financial Market Supervisory Authority (FINMA).
This document is personal to the recipient only and not for general circulation in Switzerland.
United Arab Emirates
Neither this document nor the securities have been approved, disapproved or passed on in any way by the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates, nor have we received authorization or licensing from the Central Bank of the United Arab Emirates or any other governmental authority in the United Arab Emirates to market or sell the securities within the United Arab Emirates. This document does not constitute and may not be used for the purpose of an offer or invitation. We may not render services relating to the securities within the United Arab Emirates, including the receipt of applications and/or the allotment or redemption of such shares.
No offer or invitation to subscribe for securities is valid or permitted in the Dubai International Financial Centre.
| -119- |
United Kingdom
Neither the information in this document nor any other document relating to the offer has been delivered for approval to the Financial Services Authority in the United Kingdom and no prospectus (within the meaning of section 85 of the Financial Services and Markets Act 2000, as amended (“FSMA”)) has been published or is intended to be published in respect of the securities. This document is issued on a confidential basis to “qualified investors” (within the meaning of section 86(7) of FSMA) in the United Kingdom, and the securities may not be offered or sold in the United Kingdom by means of this document, any accompanying letter or any other document, except in circumstances which do not require the publication of a prospectus pursuant to section 86(1) FSMA. This document should not be distributed, published or reproduced, in whole or in part, nor may its contents be disclosed by recipients to any other person in the United Kingdom.
Any invitation or inducement to engage in investment activity (within the meaning of section 21 of FSMA) received in connection with the issue or sale of the securities has only been communicated or caused to be communicated and will only be communicated or caused to be communicated in the United Kingdom in circumstances in which section 21(1) of FSMA does not apply us.
In the United Kingdom, this document is being distributed only to, and is directed at, persons (i) who have professional experience in matters relating to investments falling within Article 19(5) (investment professionals) of the Financial Services and Markets Act 2000 (Financial Promotions) Order 2005 (“FPO”), (ii) who fall within the categories of persons referred to in Article 49(2)(a) to (d) (high net worth companies, unincorporated associations, etc.) of the FPO or (iii) to whom it may otherwise be lawfully communicated (together “relevant persons”). The investments to which this document relates are available only to, and any invitation, offer or agreement to purchase will be engaged in only with, relevant persons. Any person who is not a relevant person should not act or rely on this document or any of its contents.
The validity of the securities offered hereby will be passed upon by Sheppard Mullin Richter & Hampton LLP, New York, New York. Ellenoff Grossman & Schole LLP, New York, New York, is acting as counsel for the underwriters in connection with this offering.
The financial statements as of December 31, 2019 and 2020 and for the years then ended included in this prospectus and in the registration statement have been so included in reliance on the report of Frank, Rimerman & Co. LLP, an independent registered public accounting firm, (the report on the financial statements contains an explanatory paragraph regarding the Company’s ability to continue as a going concern) appearing elsewhere herein and in the registration statement, given on the authority of said firm as experts in auditing and accounting.
WHERE TO FIND MORE INFORMATION
This prospectus, which constitutes a part of the registration statement, does not contain all the information set forth in the registration statement, some of which is contained in exhibits to the registration statement as permitted by the rules and regulations of the SEC. For further information with respect to us and our securities, we refer you to the registration statement, including the exhibits filed as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other document are not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, please see the copy of the contract or document that has been filed. Each statement in this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit. You may obtain copies of this information by mail from the Public Reference Section of the SEC, 100 F Street, N.E., Room 1580, Washington, D.C. 20549, at prescribed rates. You may obtain information on the operation of the public reference rooms by calling the SEC at 1-800-SEC-0330. The SEC also maintains an internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov. You may also request a copy of these filings, at no cost, by writing us at:
RenovoRx, Inc.
4546 El Camino Real, Suite B1
Los Altos, California 94022
Attn: Shaun R. Bagai, Chief Executive Officer
E-Mail: info@renovorx.com
Telephone: (650) 284-4433
We are subject to the information and reporting requirements of the Exchange Act and, in accordance with this law, file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information are available for inspection and copying at the SEC’s public reference facilities and the website of the SEC referred to above. We also maintain a website at www.RenovoRx.com. Information contained in, or accessible through, our website is not a part of this prospectus, and the inclusion of our website address in this prospectus is only as an inactive textual reference. You may access these materials free of charge as soon as reasonably practicable after they are electronically filed with, or furnished to, the SEC.
| -120- |
| Page | ||
Audited Financial Statements for the Years Ended December 31, 2019 and 2020 | ||
| Report of Independent Registered Public Accounting Firm | F-2 | |
| Balance Sheets | F-3 | |
| Statements of Operations | F-4 | |
| Statements of Convertible Preferred Stock and Stockholders’ Deficit | F-5 | |
| Statements of Cash Flows | F-6 | |
| Notes to Financial Statements | F-7 | |
Unaudited Condensed Interim Financial Statements for the Three Months Ended March 31, 2020 and 2021 | ||
| Condensed Balance Sheets as of December 31, 2020 and March 31, 2021 | F-28 | |
| Condensed Statements of Operations for the Three Months Ended March 31, 2020 and 2021 (unaudited) | F-29 | |
| Condensed Statements of Convertible Preferred Stock and Stockholders’ Deficit for the Three Months Ended March 31, 2020 and 2021 (unaudited) | F-30 | |
| Condensed Statements of Cash Flows for the Three Months Ended March 31, 2020 and 2021 (unaudited) | F-31 | |
| Notes to Unaudited Condensed Interim Financial Statements | F-32 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and the Board of Directors of
RenovoRx, Inc.
Los Altos, CA
Opinion on the Financial Statements
We have audited the accompanying balance sheets of RenovoRx, Inc. (the “Company”) as of December 31, 2019 and 2020 and the related statements of operations, convertible preferred stock and stockholders’ deficit, and cash flows for each of the years in the two-year period ended December 31, 2020, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2020, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2020, in conformity with accounting principles generally accepted in the United States of America.
Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has incurred recurring losses from operations, has negative cash flows from operating activities and has an accumulated deficit that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (the “PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the U.S. Securities and Exchange Commission (the “SEC”) and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/ Frank, Rimerman +Co. LLP | |
| We have served as the Company’s auditor since 2019. | |
| San Francisco, California | |
| May 12, 2021 (August 5, 2021 as to the effects of the reverse stock split discussed in Note 14) |
| F-2 |
RenovoRx, Inc.
(in thousands, except share and per share data)
| As of December 31, | ||||||||
| 2019 | 2020 | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 2,104 | $ | 1,795 | ||||
| Restricted cash | 20 | - | ||||||
| Prepaid expenses and other current assets | 142 | 115 | ||||||
| Total current assets | 2,266 | 1,910 | ||||||
| Deposits | 4 | 4 | ||||||
| Total assets | $ | 2,270 | $ | 1,914 | ||||
| Liabilities, Convertible Preferred Stock and Stockholders’ Deficit | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 538 | $ | 162 | ||||
| Accrued expenses | 232 | 311 | ||||||
| Promissory note, current portion | - | 117 | ||||||
| Convertible note | - | 2,650 | ||||||
| Derivative liability | - | 856 | ||||||
| Warrant liability | 35 | - | ||||||
| Total current liabilities | 805 | 4,096 | ||||||
| Promissory note, net of current portion | - | 23 | ||||||
| Total liabilities | 805 | 4,119 | ||||||
| Commitments and contingencies (Note 11) | ||||||||
| Convertible preferred stock, $0.0001 par value; 22,360,455 shares authorized; 3,508,631 and 3,535,469 shares issued and outstanding at December 31, 2019 and 2020, respectively (aggregate liquidation preference of $12,757 and $12,782, respectively) | 12,391 | 12,451 | ||||||
| Stockholders’ deficit: | ||||||||
| Common stock, $0.0001 par value; 42,000,000 shares authorized; 2,177,187 and 2,233,139 shares issued and outstanding, respectively | 1 | 1 | ||||||
| Additional paid-in capital | 235 | 303 | ||||||
| Accumulated deficit | (11,162 | ) | (14,960 | ) | ||||
| Total stockholders’ deficit | (10,926 | ) | (14,656 | ) | ||||
| Total liabilities, convertible preferred stock and | ||||||||
| stockholders’ deficit | $ | 2,270 | $ | 1,914 | ||||
See accompanying notes to financial statements.
| F-3 |
RenovoRx, Inc.
(in thousands, except per share data)
| Year Ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 2,997 | $ | 2,386 | ||||
| General and administrative | 899 | 818 | ||||||
| Total operating expenses | 3,896 | 3,204 | ||||||
| Loss from operations | (3,896 | ) | (3,204 | ) | ||||
| Other income (expenses), net: | ||||||||
| Interest income (expense), net | 63 | (587 | ) | |||||
| Other income (expense), net | 2 | (7 | ) | |||||
| Loss on change in fair value of warrant liability | (8 | ) | - | |||||
| Total other income (expense), net | 57 | (594 | ) | |||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | ||
| Net loss per share - basic and diluted | $ | (1.76 | ) | $ | (1.72 | ) | ||
| Weighted average shares of common stock - basic and diluted | 2,177 | 2,214 | ||||||
See accompanying notes to financial statements.
| F-4 |
RenovoRx, Inc.
Statements of Convertible Preferred Stock and Stockholders’ Deficit
(in thousands, except share data)
| Convertible | Additional | Total | ||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In | Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||
| Balance at January 1, 2019 | 3,508,631 | $ | 12,391 | 2,177,187 | $ | - | $ | 198 | $ | (7,323 | ) | $ | (7,125 | ) | ||||||||||||||
| Stock-based compensation expense | - | - | - | - | 38 | - | 38 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (3,839 | ) | (3,839 | ) | |||||||||||||||||||
| Balance at December 31, 2019 | 3,508,631 | 12,391 | 2,177,187 | - | 236 | (11,162 | ) | (10,926 | ) | |||||||||||||||||||
| Issuance of restricted stock award to nonemployee for service | - | - | 24,478 | - | 17 | - | 17 | |||||||||||||||||||||
| Issuance of Series A-1 convertible preferred stock upon exercise of warrant | 26,838 | 25 | - | - | - | - | - | |||||||||||||||||||||
| Issuance of common stock upon exercise of stock options | - | - | 31,474 | - | 18 | - | 18 | |||||||||||||||||||||
| Warrant liability transferred to mezzanine equity | - | 35 | - | - | - | - | - | |||||||||||||||||||||
| Stock-based compensation expense | - | - | - | - | 33 | - | 33 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (3,798 | ) | (3,798 | ) | |||||||||||||||||||
| Balance at December 31, 2020 | 3,535,469 | $ | 12,451 | 2,233,139 | $ | - | $ | 304 | $ | (14,960 | ) | $ | (14,656 | ) | ||||||||||||||
See accompanying notes to financial statements.
| F-5 |
RenovoRx, Inc.
(in thousands)
| Years Ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense | 38 | 33 | ||||||
| Issuance of restricted stock award to nonemployee | - | 17 | ||||||
| Loss on change in fair value of warrant liability | 8 | - | ||||||
| Amortization of debt discount | - | 477 | ||||||
| Amortization of debt issuance cost | - | 12 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (24 | ) | 27 | |||||
| Accounts payable | 343 | (376 | ) | |||||
| Accrued expenses | 124 | (21 | ) | |||||
| Interest accrued on convertible notes | - | 101 | ||||||
| Net cash used in operating activities | (3,350 | ) | (3,528 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Change in deposits | (1 | ) | - | |||||
| Net cash used in investing activities | (1 | ) | - | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from convertible notes | - | 3,038 | ||||||
| Payment of debt issuance costs | - | (22 | ) | |||||
| Proceeds from promissory note | - | 140 | ||||||
| Proceeds from exercise of Series A-1 warrant | - | 25 | ||||||
| Proceeds from exercise of stock options | - | 18 | ||||||
| Net cash provided by financing activities | - | 3,199 | ||||||
| Net decrease in cash, cash equivalents and restricted cash | (3,351 | ) | (329 | ) | ||||
| Cash, cash equivalents and restricted cash, beginning of year | 5,475 | 2,124 | ||||||
| Cash, cash equivalents and restricted cash, end of year | $ | 2,124 | $ | 1,795 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Derivative liability | $ | - | $ | 856 | ||||
| Warrant liability transferred to equity | $ | - | $ | 35 | ||||
See accompanying notes to financial statements.
| F-6 |
Description of the Business
RenovoRx, Inc. (“RenovoRx” or the “Company”) was incorporated in Delaware in December 2012 and operates from its headquarters in Los Altos, California. The Company is a clinical-stage biopharmaceutical company focused on developing therapies for the local treatment of solid tumors and conducting a Phase 3 registrational trial for its lead product candidate RenovoGem™. The Company’s therapy platform, RenovoRx Trans-Arterial Micro-Perfusion, or RenovoTAMP™ utilizes approved chemotherapeutics with validated mechanisms of action and well-established safety and side effect profiles, with the goal of increasing their efficacy, improving their safety, and widening their therapeutic window. RenovoTAMP combines the Company’s patented FDA cleared delivery system, RenovoCath®, with small molecule chemotherapeutic agents that can be forced across the vessel wall using pressure, targeting these anti-cancer drugs locally to the solid tumors. While the Company anticipates investigating other chemotherapeutic agents for intra-arterial delivery via RenovoTAMP, the Company’s clinical work to date has focused on gemcitabine, which is a generic drug. The Company’s first product candidate, RenovoGem, is a drug and device combination consisting of intra-arterial gemcitabine and RenovoCath. FDA has determined that RenovoGem will be regulated as, and if approved the Company expects will be reimbursed as, a new oncology drug product. The Company has secured FDA Orphan Drug Designation for RenovoGem in its first two indications: pancreatic cancer and cholangiocarcinoma (bile duct cancer, or CCA). The Company has completed its RR1 Phase 1/2 and RR2 observational registry studies, with 20 and 25 patients respectively, in locally advanced pancreatic cancer, or LAPC. These studies demonstrated a median overall survival of 27.9 months in patients treated with RenovoGem and radiation versus expected survival of 12-15 months in patients receiving only intravenous (IV) systemic chemotherapy dosed at 1,000mg/m2 based on historical control data. Unlike the historical controls, the Company’s RR1 and RR2 studies were not randomized or controlled for potential confinement. Based on FDA safety review of the Company’s Phase 1/2 study the FDA allowed the Company to proceed to evaluate RenovoGem within its Phase 3 registration Investigational New Drug, or IND, clinical trial. This Phase 3 trial is 40% enrolled as of April 30, 2021 and the Company expects to report data from a planned interim data readout in the second half of 2022. The Company intends to evaluate RenovoGem in a second indication in a Phase 2/3 trial in hilar CCA (cancer that occurs in the bile ducts that lead out of the liver and join with the gallbladder, also called extrahepatic cholangiocarcinoma, or HCCA). The Company plans to propose the trial to the FDA and potentially launch in the first half of 2022. In addition, the Company may evaluate RenovoGem in other indications, potentially including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma (an aggressive type of cancer that can occur in the brain or spinal cord). To date, the Company has used gemcitabine, but in the future it may develop other chemotherapeutic agents for intra-arterial delivery via RenovoCath.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying financial statements and the related disclosures have been prepared in conformity with accounting principles generally accepted in the United States (“GAAP”), applicable rules and regulations of the U.S. Securities and Exchange Commission (“SEC”) and include all adjustments necessary for the fair presentation of the Company’s financial position for the periods presented.
Liquidity and Going Concern
The financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. In order to continue its operations, the Company must raise additional equity or debt financings and achieve profitable operations. Although management has historically been successful in raising capital, there can be no assurance that the Company will be able to obtain additional equity or debt financing on terms acceptable to the Company, or at all. The failure to obtain sufficient funds on acceptable terms when needed could have a material adverse effect on the Company’s business, financial position, results of operations, and future cash flows. The Company is seeking to complete an initial public offering (“IPO”) of its common stock. Upon the closing of an IPO, on specified terms, the Company’s outstanding convertible preferred stock will automatically convert into shares of common stock (see Note 8). In the event the Company does not complete an IPO, the Company expects to seek additional funding through other capital sources including through the sale of equity, debt financings or other capital sources including collaborations with other companies or other strategic transactions. However, the Company may be unable to raise additional funds or enter into such agreements or arrangements when needed on acceptable terms, or at all.
| F-7 |
As a company with no commercial operating history, the Company is subject to all of the risks and expenses associated with a start-up company. The Company must among other things respond to competitive developments, attract, retain and motivate qualified personnel and support ongoing clinical trials for its product candidate. The Company has generated operating losses and negative cash flows from operations in each year since inception. The Company has not generated any revenue from product sales to date and will continue to incur significant research and development and other expenses related to its ongoing operations. The Company has incurred net losses of $3.8 million for each of the years ended December 31, 2019 and 2020, and had an accumulated deficit of $15.0 million at December 31, 2020. The Company has funded its operations primarily through the sale and issuance of convertible preferred stock and convertible notes. The Company has reviewed the relevant conditions and events surrounding its ability to continue as a going concern including among others: historical losses, projected future results, including the effects of the novel coronavirus (“COVID-19”), cash requirements for the upcoming year, terms of the Company’s current debt arrangements, funding capacity, net working capital, total stockholders’ deficit and future access to capital. These factors along with the Company’s cash and cash equivalents, raise substantial doubt about the Company’s ability to continue as a going concern for at least one year from the date the financial statements are issued. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty. If future financing is not achieved, the Company may be required to curtail spending to reduce cash outflows.
Risks and Uncertainties
The Company is subject to a number of risks associated with companies at a similar stage, including the risk associated with the development of products that must receive regulatory approval before market launch, dependence on key individuals, competition from larger and established companies, volatility of the industry, ability to obtain adequate financing to support growth, the ability to attract and retain additional qualified personnel to manage the anticipated growth of the Company and general economic conditions.
In March 2020, the World Health Organization declared COVID-19 a pandemic. The COVID-19 pandemic has negatively impacted the global economy, disrupted global supply chains and created significant volatility and disruption of financial markets. The full extent to which the COVID-19 pandemic will directly or indirectly impact the Company’s business, results of operations and financial condition, including expenses, clinical trials and research and development costs, will depend on future developments that are highly uncertain, including as a result of new information that may emerge concerning COVID-19 and the actions taken to contain or treat COVID-19, as well as the economic impact on local, regional, national and international markets. The Company has made estimates of the impact of COVID-19 within its financial statements and there may be changes to those estimates in future periods. Actual results could materially differ from those estimates.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, income and expenses as well as the disclosure of contingent assets and liabilities, at the date of the financial statements during the reporting periods. Significant estimates and assumptions made in the accompanying financial statements include, but are not limited to, the accrual of certain liabilities, the valuation of financial instruments, the valuation allowance related to deferred income tax assets, the fair value of the Company’s common stock and the fair value of options granted under the Company’s equity incentive plan. Actual results could differ from materially from these estimates.
Concentration of Credit Risk and Significant Suppliers
Financial instruments that potentially subject the Company to concentration of credit risk consist of cash and cash equivalents. At December 31, 2019, the Company maintained approximately $2.1 million in cash, cash equivalents and restricted cash, and at December 31, 2020, the Company maintained $1.8 million in cash and cash equivalents, with one financial institution. The Company is exposed to credit risk in the event of default by the financial institutions to the extent that cash and cash equivalent deposits are in excess of the $250,000 insured by the Federal Deposit Insurance Corporation. These deposits routinely exceed the insurable limit. To date, the Company has not experienced any losses on its cash and cash equivalents.
| F-8 |
The Company is dependent on third-party manufacturers to supply products and services for its research and development activities, including preclinical and clinical development. In particular, the Company relies, and expects to continue to rely, on a small number of third-party manufacturers to manufacture and supply its RenovoCath devices and its product candidates for clinical trials. These activities could be adversely affected by a significant interruption in the supply of these items.
Deferred Offering Costs
The Company capitalizes certain legal, professional, accounting and other third-party fees that are directly associated with in-process financings as deferred offering costs until such financings are consummated. After consummation of the financing, these costs are recorded as a reduction of the proceeds received from the financing. If a planned financing is abandoned, the deferred offering costs are expensed as a charge to operating expenses in the statement of operations.
There were no deferred offering costs on the Company’s balance sheets at December 31, 2019 and 2020.
Net Loss per Share
Basic net loss per common share is calculated by dividing net loss by the weighted-average number of shares of common stock outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is computed by dividing net loss by the weighted-average number of shares of common stock and common stock equivalents of potentially dilutive securities outstanding for the period determined using the treasury stock and if-converted methods. For the years ended December 31, 2019 and 2020, the Company’s potentially dilutive common stock equivalents are comprised of convertible preferred stock, convertible notes, options outstanding under the Company’s stock option plan and the warrant to purchase Series A-1 preferred stock.
Fair Value of Financial Instruments
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability of fair value measures, the following hierarchy prioritizes the inputs to valuation methodologies used to measure fair value.
Level 1 – Valuations based on quoted prices for identical assets and liabilities in active markets.
Level 2 – Valuations based on observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, or other inputs that are observable or can be corroborated by observable market data.
Level 3 – Valuations based on unobservable inputs reflecting the Company’s assumptions, consistent with reasonably available assumptions made by other market participants. These valuations require significant judgment.
The estimated fair value of financial instruments disclosed in the financial statements has been determined by using available market information and appropriate valuation methodologies. In certain cases where there is limited activity or less transparency around inputs to valuation, securities are classified as Level 3.
The carrying value of all remaining current assets and current liabilities approximates fair value because of their short-term nature.
Cash and Cash Equivalents and Restricted Cash
The Company considers all highly liquid investments purchased with a remaining maturity date upon acquisition of three months or less to be cash equivalents. Cash and cash equivalents consist primarily of cash held in checking and money market accounts. As of December 31, 2019, the Company had restricted cash of $20,000, which consisted of reserve funds held at one financial institution to collateralize the Company’s credit cards. The Company held no funds in restricted cash at December 31, 2020.
The following table provides a reconciliation of cash, cash equivalents and restricted cash reported within the balance sheets to the amounts shown in the statements of cash flows (in thousands):
| 2019 | 2020 | |||||||
| Cash and cash equivalents | $ | 2,104 | $ | 1,795 | ||||
| Restricted cash | 20 | - | ||||||
| Total cash and cash equivalents and restricted cash shown in the statements of cash flows | $ | 2,124 | $ | 1,795 | ||||
| F-9 |
Convertible Instruments and Embedded Derivatives
The Company evaluates all of its agreements to determine if such instruments have derivatives or contain features that qualify as embedded derivatives. The Company accounts for certain redemption features that are associated with convertible notes as liabilities at fair value and adjusts the instruments to their fair value at the end of each reporting period. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in other income (expense), net in the statements of operations. Derivative instrument liabilities are classified in the balance sheets as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. The Company had no derivative liability as of December 31, 2019. As of December 31, 2020, the Company’s only derivative financial instrument was related to the 2020 Convertible Notes (defined in Note 5), which contained certain redemptive features (see Note 5).
Research and Development Costs
Research and development costs, which include direct and allocated expenses, are expensed in the period incurred. Research and development costs include primarily clinical development-related expenses, pre-clinical research and development expenses, employee compensation and related benefits, regulatory support and related services, clinical consulting, travel-related expenses and allocated expenses for rent, insurance and other general overhead costs. The Company also receives payments from vendors in performing clinical trials on behalf of the Company for the delivery device used by such vendors in performing such clinical trials. These payments are offset against the Company’s research and development expenses.
Clinical Trial Expenses
The Company makes payments in connection with clinical trials under contracts with clinical trial sites and contract research organizations that support conducting and managing clinical trials. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price or on a time and materials basis. A portion of the obligation to make payments under these contracts depends on factors such as the successful enrollment or treatment of patients or the completion of other clinical trial milestones.
Expenses related to clinical trials are accrued based on estimates and/or representations from service providers regarding work performed, including actual levels of patient enrollment, completion of patient studies and progress of the clinical trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts the Company is obligated to pay under clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), the accruals are adjusted accordingly. Revisions to contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. As noted above, the Company receives payments from vendors performing clinical trials on behalf of the Company for the delivery device used by such vendors in performing such clinical trials.
General and Administrative
General and administrative expenses consist primarily of personnel costs, including employee compensation and related benefits and consulting. Additionally, these expenses include professional fees, including audit, legal, recruiting services and allocated expenses for rent, insurance and other general overhead costs. General and administrative expenses are expensed in the period incurred.
| F-10 |
Convertible Preferred Stock
The Company records preferred stock at fair value on the date of issuance, net of issuance costs. The preferred stock is recorded outside of stockholders’ deficit because the shares contain liquidation features that are not solely within the Company’s control. As a result, the preferred stock is classified as mezzanine equity (temporary equity). The Company has elected not to adjust the carrying value of the preferred stock to the liquidation preferences of such shares because it is uncertain whether or when an event would occur that would obligate the Company to pay the liquidation preferences to holders of shares of preferred stock. Subsequent adjustments to the carrying values to the liquidation preferences will be made only when it becomes probable that such a liquidation event will occur.
Stock-Based Compensation
The Company calculates the fair value of stock options using the Black-Scholes option pricing model, which incorporates various assumptions including volatility, expected life and risk-free interest rate. Compensation related to service-based awards is recognized starting on the grant date on a straight-line basis over the vesting period, which is generally four years.
The determination of the fair value of each stock award using this option-pricing model is affected by the Company’s assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, the fair value of the common stock at the date of grant, the expected term of the awards, the expected stock price volatility over the term of the awards, the risk-free interest rate, and the dividend rate as follows:
Fair Value of Common Stock—Given the absence of a public trading market, the Company’s Board of Directors considered numerous objective and subjective factors to determine the fair value of the Company’s common stock at each grant date. These factors included, but were not limited to: (i) contemporaneous third-party valuations of common stock; (ii) the prices for preferred stock sold to outside investors; (iii) the rights and preferences of preferred stock relative to Common stock; (iv) the lack of marketability of the Company’s common stock; (v) developments in the business; and (vi) the likelihood of achieving a liquidity event, such as an IPO or sale of the business, given prevailing market conditions. The methodology to determine the fair value of the Company’s common stock included estimating the fair value of the enterprise using the “backsolve” method, which is a market approach that assigns an implied enterprise by accounting for all share class rights and preferences based on the latest round of financing. The total equity value implied was then applied in the context of an option pricing model to determine the value of each class of the Company’s shares.
Expected Term—The expected term represents the period that the stock-based awards are expected to be outstanding. The Company determines the expected term using the simplified method. The simplified method deems the term to be the average of the time-to-vesting and the contractual life of the options. For stock options granted to non-employees, the expected term equals the remaining contractual term of the option from the vesting date.
Expected Volatility—Given the absence of a public trading market, the expected volatility was estimated by taking the average historic price volatility for industry peers, consisting of several public companies in the Company’s industry that are either similar in size, stage, or financial leverage, over a period equivalent to the expected term of the awards.
Risk-Free Interest Rate—The risk-free interest rate is calculated using the average of the published interest rates of U.S. Treasury zero-coupon issues with maturities that are commensurate with the expected term.
Dividend Rate—The dividend yield assumption is zero as the Company has no plans to make dividend payments.
The Company generally grants stock options to its employees for a fixed number of shares with an exercise price equal to the fair value of the underlying shares on the date of grant. The Company accounts for all stock option grants using the fair value method and stock-based compensation is recognized as the underlying options vest.
| F-11 |
Preferred Stock Warrant Liability
The Company accounts for its warrants as either equity or liability based upon the characteristics and provisions of each instrument. Warrants classified as derivative liabilities are recorded on the Company’s accompanying balance sheets at their fair value on the date of issuance and are revalued at each subsequent balance sheet date, with fair value changes recognized as increases or reductions to other income (expense), net in the statements of operations.
Income Taxes
The Company accounts for income taxes using the asset and liability method. Under this method, deferred income tax assets and liabilities are recorded based on the estimated future tax effects of differences between the financial statement and income tax basis of existing assets and liabilities. Deferred income tax assets and liabilities are recorded net and classified as noncurrent on the balance sheets. A valuation allowance is provided against the Company’s deferred income tax assets when their realization is not reasonably assured.
The Company is subject to income taxes in the federal and state jurisdictions. Tax regulations within each jurisdiction are subject to the interpretation of the related tax laws and regulations and require significant judgment to apply. In accordance with the authoritative guidance on accounting for uncertainty in income taxes, the Company recognizes tax liabilities for uncertain tax positions when it is more likely than not that a tax position will not be sustained upon examination and settlement with various taxing authorities. Liabilities for uncertain tax positions are measured based upon the largest amount of benefit that is more-likely-than-not (greater than 50%) of being realized upon settlement. The Company’s policy is to recognize interest and/or penalties related to income tax matters in income tax expense.
Segment Reporting
Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) Topic 280, Segment Reporting, requires use of the “management approach” model for segment reporting. The management approach model is based on the way a company’s management organizes segments within the company for making operating decisions and assessing performance.
The Company determined it has one reportable segment, the development of a platform technology to deliver de-risked small molecules for localized treatment of solid cancer tumors. The segment is based on financial information that is utilized by the Company’s Chief Operating Decision Maker who is the Company’s Chief Executive Officer, to assess performance and allocate resources.
Emerging Growth Company Status
From time to time, new accounting pronouncements, or Accounting Standards Updates (“ASU”) are issued by the FASB or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on the Company’s financial position or results of operations upon adoption.
As an “emerging growth company” (“EGC”) under the Jumpstart Our Business Startups Act (“JOBS Act”), the Company may elect to take advantage of certain forms of relief from various reporting requirements that are applicable to public companies. The relief afforded under the JOBS Act includes an extended transition period for the implementation of new or revised accounting standards. The Company has elected to take advantage of this extended transition period and, as a result, the Company’s financial statements may not be comparable to those of companies that implement accounting standards as of the effective dates for public companies. The Company may take advantage of the relief afforded under the JOBS Act up until the last day of the fiscal year following the fifth anniversary of an offering or such earlier time that it is no longer an EGC.
Recently Adopted Accounting Pronouncements
In June 2018, the FASB issued ASU No. 2018-07, Improvements to Nonemployee Share-Based Payment Accounting. The standard simplifies the accounting for share-based payments granted to nonemployees for goods and services and aligns most of the guidance on such payments to the nonemployees with the requirements for share-based payments granted to employees. ASU 2018-07 is effective for the Company for annual reporting periods beginning after December 15, 2019, and interim periods within fiscal years beginning after December 15, 2020 with early adoption permitted. The guidance should be applied to new awards granted after the date of adoption. The Company adopted this new standard on January 1, 2020 and the adoption of this standard did not have an impact on its financial statements.
| F-12 |
In August 2018, the FASB issued ASU 2018-13, Fair Value Measurement (Topic 820), Disclosure Framework — Changes to the Disclosure Requirements for Fair Value Measurement. The standard eliminates, adds and modifies certain disclosure requirements for fair value measurements. Entities will no longer be required to disclose the amount of and reasons for transfers between Level 1 and Level 2 of the fair value hierarchy, but public companies will be required to disclose the range and weighted average of significant unobservable inputs used to develop Level 3 fair value measurements. The standard is effective for annual reporting periods beginning after December 15, 2019, and for interim periods within those periods. The Company adopted this new standard on January 1, 2020, with no material impact on its financial statements.
Effective January 1, 2019, the Company adopted FASB ASU 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash, which requires that a statement of cash flows explain the change during the period in the total of cash, cash equivalents and restricted cash. Therefore, amounts described as restricted cash should be included with cash and cash equivalents when reconciling the beginning of period and end of period amounts shown on the statement of cash flows. The Company adopted this guidance on January 1, 2019. The adoption of ASU 2016-18 did not have an impact on the Company’s financial results, but it did result in a change in the presentation of restricted cash and cash equivalents within the statements of cash flows.
Recent Accounting Pronouncements Not Yet Adopted
In February 2016, the FASB issued ASU No. 2016-02, Leases (Topic 842). The guidance requires lessees to recognize assets and liabilities related to long-term leases on the balance sheet and expands disclosure requirements regarding leasing arrangements. In July 2018, the FASB issued additional guidance, which offers a transition option to entities adopting the new lease standards, and a package of practical expedients an entity can elect to utilize to reduce the level of effort required for adoption. Under the transition option, entities can elect to apply the new guidance using a modified retrospective approach at the beginning of the year in which the new lease standard is adopted, rather than to the earliest comparative period presented in their financial statements. In November 2019, the FASB issued ASU 2019-10 deferring the effective date for private entities for fiscal years beginning after December 15, 2020 and interim periods within fiscal years beginning after December 15, 2021. In June 2020, the FASB issued ASU 2020-05 which further defers the effective date for private entities for fiscal years beginning after December 15, 2021 and interim periods within fiscal years beginning after December 15, 2022. The Company is currently evaluating its contracts to determine whether there will be a significant impact from the adoption of this guidance on its financial statements.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments – Credit Losses, which requires the measurement of expected credit losses for financial instruments carried at amortized cost, such as accounts receivable, held at the reporting date based on historical experience, current conditions and reasonable forecasts. The main objective of this standard is to provide financial statement users with more decision-useful information about the expected credit losses on financial instruments and other commitments to extend credit held by a reporting entity at each reporting date. In November 2018, the FASB issued ASU No. 2018-19, Codification Improvements to Topic 326, Financing Instruments – Credit Losses, which included an amendment of the effective date. The standard is effective for the Company for annual reporting periods beginning after December 15, 2022, and for interim periods within those periods. Early adoption is permitted. The Company plans to adopt this new standard on January 1, 2023 and does not believe that adoption will have a significant impact on its financial statements.
In December 2019, the FASB issued ASU No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, which simplifies the accounting for income taxes. For the Company, ASU 2019-12 is effective on a prospective basis for annual reporting periods beginning after December 15, 2021 and for interim periods within fiscal years beginning after December 15, 2022, with early adoption permitted. The Company plans to adopt this new standard on January 1, 2022 and does not believe that adoption will have a significant impact on its financial statements.
| F-13 |
In August 2020, the FASB issued ASU No. 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity, which simplifies the accounting for certain financial instruments with characteristics of liabilities and equity, including convertible instruments and contracts on an entity’s own equity. ASU 2020-06 is effective on a prospective basis for annual reporting periods beginning after December 15, 2023 and for interim periods within those periods. Early adoption is permitted. The Company has not yet determined the impact that this new standard will have on its financial position and results of operations.
| 3. | Accrued Expenses |
A summary of the components of accrued expenses is as follows (in thousands):
| December 31, | ||||||||
| 2019 | 2020 | |||||||
| Accrued clinical trials | $ | 219 | $ | 171 | ||||
| Accrued interest | - | 101 | ||||||
| Accrued personnel | 13 | 39 | ||||||
| $ | 232 | $ | 311 | |||||
| 4. | Fair Value Measurements |
The following table sets forth by level, within the fair value hierarchy, the financial assets and liabilities that are measured at fair value on a recurring basis at December 31, 2019 and 2020 (in thousands):
| Fair Value Measurements at December 31, 2019 using: | ||||||||||||||||
| Assets: | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Money market funds | $ | 1,087 | $ | - | $ | - | $ | 1,087 | ||||||||
| $ | 1,087 | $ | - | $ | - | $ | 1,087 | |||||||||
| Liabilities: | ||||||||||||||||
| Series A-1 preferred stock warrant liability | $ | $ | - | $ | 35 | $ | 35 | |||||||||
| $ | $ | - | $ | 35 | $ | 35 | ||||||||||
| F-14 |
| Fair Value Measurements at December 31, 2020 using: | ||||||||||||||||
| Assets: | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Money market funds | $ | 1,703 | $ | - | $ | - | $ | 1,703 | ||||||||
| $ | 1,703 | $ | - | $ | - | $ | 1,703 | |||||||||
| Liabilities: | ||||||||||||||||
| Derivative liability – 2020 Convertible Notes | $ | - | $ | - | $ | 856 | $ | 856 | ||||||||
| $ | - | $ | - | $ | 856 | $ | 856 | |||||||||
The change in the fair value of the Series A-1 preferred stock warrant liability is summarized below (in thousands):
| Fair value as of January 1, 2019 | $ | 27 | ||
| Change in fair value recorded in other income (expense), net | 8 | |||
| Fair value as of December 31, 2019 | $ | 35 | ||
| Change in fair value upon warrant exercise in January 2020 recorded in other income (expense), net | - | |||
| Fair value as of January 2020 | $ | 35 | ||
| Transfer of warrant liability to mezzanine equity upon exercise of warrant | (35 | ) | ||
| Fair value as of December 31, 2020 | $ | - |
The Series A-1 preferred stock warrant liability consisted of the fair value of the warrant to purchase Series A-1 convertible preferred stock and was based on significant inputs not observable in the market, which represents a Level 3 measurement within the fair value hierarchy. The fair value of the Series A-1 preferred stock warrant liability was determined using the “backsolve” method to estimate the enterprise value of the Company and the Option Pricing Model to allocate the enterprise value of the Company. The enterprise value was allocated among the various share classes and warrant using the Option Pricing Model. Generally, increases or decreases in the fair value of the underlying convertible preferred stock would result in a directionally similar impact in the fair value measurement of the warrant liability.
There was no derivative liability as of December 31, 2019.
The change in the fair value of the derivative liability for the year ended December 31, 2020 is summarized below (in thousands):
| Fair value as of December 31, 2019 | $ | - | ||
| Derivative liability upon issuance of 2020 Convertible Notes | 856 | |||
| Change in fair value recorded in other income (expense), net | - | |||
| Fair value as of December 31, 2020 | $ | 856 |
The derivative liability in the table above related to the 2020 Convertible Notes and represents the fair value of the redemption-like contingent conversion feature. The Company calculated the fair value of the derivative liability using a probability weighted discounted cash flow analysis. The inputs used to determine the estimated fair value of the derivative were based primarily on the probability of an underlying event occurring that would trigger the embedded derivative and the timing of such event. The Company’s derivative liability is measured at fair value on a recurring basis and is classified as a Level 3 liability. The Company records subsequent adjustments to reflect the increase or decrease in estimated fair value at each reporting date in other income (expense), net in the statements of operations (see Note 5).
| F-15 |
There were no transfers among Level 1, Level 2 or Level 3 categories during any of the periods presented. The Company had no other financial assets or liabilities that were required to be measured at fair value on a recurring basis.
| 5. | Convertible Notes |
In March 2020, the Company entered into a note purchase agreement for the issuance of up to $4.0 million of convertible promissory notes. The Company entered into a series of convertible note payable agreements (the “2020 Convertible Notes”) for aggregate borrowings of approximately $3.0 million. Outstanding borrowings under the 2020 Convertible Notes and accrued interest are due in March 2021, if not previously converted. The 2020 Convertible Notes bear interest at the rate of 5% per annum. The 2020 Convertible Notes cannot be prepaid prior to the maturity date unless approved in writing by the Company and requisite holders.
Pursuant to the 2020 Convertible Notes, the outstanding principal and accrued interest are automatically convertible into equity shares in the next equity financing round with total proceeds of not less than $10.0 million (a “Qualified Financing’), at a conversion price per share equal to 80% of the price per share paid by investors purchasing such equity securities in a Qualified Financing. For purposes of the 2020 Convertible Notes, equity securities mean the Company’s common stock, preferred stock or any securities providing for rights to purchase the Company’s common stock, preferred stock or securities convertible into or exchangeable for the Company’s common stock or preferred stock issued in the Qualified Financing. If the Company consummates a Change of Control prior to a Qualified Financing, the Company will repay each holder in cash an amount equal to the greater of (a) two times (2x) the entire outstanding principal balance of the 2020 Convertible Notes or (b) the amount the holder would receive if the 2020 Convertible Notes had been converted into shares of the Company’s Series B convertible preferred stock immediately prior to the consummation of the Change in Control, at a conversion price equal to the Series B convertible preferred stock Original Issue Price ($1.1030, adjusted for any stock dividends, combinations, splits, and recapitalizations).
The Company evaluated whether the 2020 Convertible Notes contain embedded features that meet the definition of derivatives under FASB ASC Topic 815, Derivatives and Hedging. The Company determined that these redemption features contained rights and obligations for conversion contingent upon a potential future financing event or a change in control. Thus, the embedded redemption features were bifurcated from the face value of the note and accounted for as a derivative liability to be remeasured at the end of each reporting period. The derivative liability had a fair value of approximately $856,000 on the issuance date with the offsetting amount being recorded as a debt discount. The Company also incurred $22,000 of debt issuance costs. The derivative liability is subject to fair value remeasurement at the end of each reporting period. The discount and debt issuance costs are being amortized to interest expense using the effective interest method over the expected term of the 2020 Convertible Notes. The Company recognized approximately $477,000 of amortization of debt discount and $12,000 of amortization of debt issuance costs as interest expense in the statement of operations for the year ended December 31, 2020. The effective interest rate on the 2020 Convertible Notes was 30.8%. During the year ended December 31, 2020, the Company also recognized interest expense in the statement of operations of approximately $101,000 related to the 2020 Convertible Notes. The Company incurred no interest expense during the year ended December 31, 2019.
| 6. | Promissory Note |
On April 22, 2020, the Company entered into a promissory note with Silicon Valley Bank that provided for the receipt by the Company of loan proceeds of $140,000 (the “PPP Loan”) pursuant to the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). Under certain conditions, the loan and accrued interest are forgivable, if the loan proceeds are used for eligible purposes, including payroll, benefits, rent and utilities, and maintaining payroll levels. In October 2020, the Paycheck Protection Program Flexibility Act of 2020 extended the deferral period for borrower payments of principal, interest, and fees on all PPP loans from 6 months to 10 months. As of December 31, 2020, payments were deferred for 10 months. The PPP Loan matures on April 22, 2022 and bears interest at a rate of 1.0% per annum. The PPP Loan contains events of default and other provisions customary for a loan of this type. The Company has recorded the PPP Loan as a promissory note in the December 31, 2020 balance sheet as both a current and non-current liability.
| F-16 |
| 7. | Warrant Liability |
In conjunction with the January 2013 Series A-1 convertible preferred stock financing, the Company issued a warrant to purchase 26,838 shares of the Company’s Series A-1 convertible preferred stock at $0.9315 per share. The warrant was to expire on the earlier of (a) the date that was seven (7) years after the date of the original issuance of the warrant, (b) the date of consummation of an acquisition or (c) the effective date of an IPO. The Company accounts for stock warrants in accordance with FASB ASC Topic 480, Distinguishing Liabilities from Equity, either as derivative liabilities or as equity instruments depending on the specific terms of the warrant agreement. As described in Note 8, all of the Company’s issued and outstanding convertible preferred stock is classified in mezzanine equity. The Company determined that the warrant should be classified as a liability, because it is exercisable for shares of Series A-1 preferred stock that are puttable upon a deemed liquidation event. In 2019, the fair value of the warrant liability of $35,000 was included in current liabilities as the seven-year expiration date was January 23, 2020. The warrant was exercised on January 23, 2020 for proceeds of $25,000. Upon exercise, the warrant liability associated with this warrant was adjusted to its fair value of $35,000. The fair value of $35,000 was subsequently transferred to mezzanine equity as of the date of exercise. The Company also recognized a non-cash loss on settlement of the warrant of $8,000 which was recorded in other income (expense), net in the statement of operations as of December 31, 2020.
| F-17 |
| 8. | Convertible Preferred Stock and Stockholders’ Deficit |
Common Stock
Pursuant to the December 2019 fifth amended and restated certificate of incorporation, the Company is authorized to issue 42,000,000 shares of common stock with a par value of $0.0001 per share. Each holder of common stock is entitled to one vote per share of common stock held. The Company’s common stock reserved for future issuance related to the convertible preferred stock and common stock options as of December 31, 2019 and 2020 are as follows:
| As of December 31, | ||||||||
| 2019 | 2020 | |||||||
| Series A-1 convertible preferred stock | 708,533 | 708,533 | ||||||
| Series A-2 convertible preferred stock | 709,219 | 709,219 | ||||||
| Series A-3 convertible preferred stock | 532,046 | 532,046 | ||||||
| Series B convertible preferred stock | 2,522,292 | 2,522,292 | ||||||
| Common stock options outstanding | 1,003,375 | 997,266 | ||||||
| Common stock options reserved for issuance | 62,738 | 12,893 | ||||||
| Total | 5,538,203 | 5,482,249 | ||||||
The shares that would be issued upon a conversion of the 2020 Convertible Notes have been excluded from the table above as the number of common shares that would be issued is contingent upon the occurrence of a future Qualified Financing and the conversion price per share would be equal to 80% of the price per share paid by the investors in the future Qualified Financing.
Convertible Preferred Stock
As of December 31, 2020, the Company was authorized to issue two classes of stock consisting of common stock and preferred stock.
In December 2019, the Company filed an amendment to its Certificate of Incorporation to re-designate its issuable convertible preferred stock. Series D convertible preferred stock was re-designated as Series B, Series C convertible preferred stock was re-designated as Series A-3 convertible preferred stock (Series A-3), Series B convertible preferred stock was re-designated as Series A-2 convertible preferred stock (Series A-2) and Series A convertible preferred stock was re-designated as Series A-1 convertible preferred stock (Series A-1). The re-designation did not change any of the rights, privileges or preferences of any series of preferred stock. The accompanying financial statements and notes to the financial statements have been updated to reflect these re-designations.
The total number of shares the Company is authorized to issue is 64,360,455 of which 42,000,000 shares shall be common stock and 22,360,455 shares shall be preferred stock with both the common stock and preferred stock having a par value of $0.0001 per share. As of December 31, 2019 and 2020, the Board of Directors designated the convertible preferred stock as follows (in thousands, except shares):
| F-18 |
| December 31, 2019 | ||||||||||||||||
| Preferred Series | Shares
Authorized | Shares Issued and Outstanding | Net Carrying Value | Liquidation Value | ||||||||||||
| Series A-1 | 3,542,669 | 681,695 | $ | 579 | $ | 635 | ||||||||||
| Series A-2 | 3,546,095 | 709,219 | 1,099 | 1,150 | ||||||||||||
| Series A-3 | 2,660,230 | 532,046 | 2,166 | 2,227 | ||||||||||||
| Series B | 12,611,461 | 1,585,671 | 8,547 | 8,745 | ||||||||||||
| 22,360,455 | 3,508,631 | $ | 12,391 | $ | 12,757 | |||||||||||
| December 31, 2020 | ||||||||||||||||
| Preferred Series | Shares
Authorized | Shares Issued and Outstanding | Net Carrying Value | Liquidation Value | ||||||||||||
| Series A-1 | 3,542,669 | 708,533 | $ | 639 | $ | 660 | ||||||||||
| Series A-2 | 3,546,095 | 709,219 | 1,099 | 1,150 | ||||||||||||
| Series A-3 | 2,660,230 | 532,046 | 2,166 | 2,227 | ||||||||||||
| Series B | 12,611,461 | 1,585,671 | 8,547 | 8,745 | ||||||||||||
| 22,360,455 | 3,535,469 | $ | 12,451 | $ | 12,782 | |||||||||||
All classes of convertible preferred stock have a par value of $0.0001 per share and are not redeemable at the option of the holder. Any shares of convertible preferred stock that are redeemed, purchased, converted or exchanged by the Company will be cancelled and retired and will not be reissued or transferred.
Voting
The holders of shares of the convertible preferred stock are entitled to vote, together with the holders of the common stock and not as a separate class, on all matters submitted to stockholders to vote. Each holder of convertible preferred stock is entitled to one vote for each share of common stock into which their shares would convert.
As long as any shares of Series A-2 and Series A-1 remain outstanding, the holders of Series A-2 and Series A-1, voting together as a separate class, are entitled to elect one member of the Company’s Board of Directors. The holders of common stock, voting as a separate class, are entitled to elect two members of the Company’s Board of Directors. The holders of common stock and convertible preferred stock, voting together on an as-if-converted basis, are entitled to elect three members of the Company’s Board of Directors.
Protective Provisions
The holders of convertible preferred stock have certain protective provisions. As long as any shares of preferred stock remain outstanding, the Company cannot, without the approval of a majority of the voting power of preferred stock then outstanding, voting together as a single class on an as-converted basis, take any actions to, among other things: (i) amend the Company’s Certificate of Incorporation or Bylaws; (ii) increase or decrease the total number of authorized shares of common stock or convertible preferred stock; (iii) authorize or designate any new series of stock or any other securities convertible into equity securities; (iv) redeem or repurchase shares of convertible preferred stock or common stock or pay or declare dividends; (v) result in any agreement for merger, consolidation or sale of control (including any liquidation event, asset transfer or acquisition); (vi) create or authorize the issuance of any debt security; or (vii) increase the number of shares available for issuance under the Company’s equity incentive plan.
Conversion Rights
Any shares of the convertible preferred stock may, at the option of the holder, be converted at any time after the date of issuance into fully paid and nonassessable shares of common stock. The number of shares of common stock to which the holder of the convertible preferred stock is entitled upon conversion will be determined by multiplying the conversion rate by the number of shares of Series A-1, A-2, A-3 and B being converted.
| F-19 |
Each share of convertible preferred stock automatically converts into the number of shares of common stock determined in accordance with the then-effective and applicable convertible preferred stock conversion price, (i) at any time upon the affirmative vote or written consent or agreement of the holders of at least a two-thirds of the outstanding shares of Series A-1, A-2, A-3 and B, or (ii) immediately upon the closing of the sale of shares of common stock to the public in a firm-commitment underwritten public offering of common stock resulting in at least $50.0 million of gross cash proceeds to the Company.
The conversion rate is determined by dividing the Original Issue Price for each series of convertible preferred stock of such shares of each series by the original conversion price of the series. The conversion price is equal to the Original Issue Price for the respective series of convertible preferred stock, as adjusted for any stock splits, dividends, reclassifications and the like. As of December 31, 2020, the conversion price for each share of convertible preferred stock is equal to the Original Issue Price.
Dividends
The holders of preferred stock are entitled to receive non-cumulative dividends prior to and in preference to any declaration or payment of dividends on common stock, when and if declared by the Board of Directors. Dividends would be payable at the non-cumulative rates of 6% of the Original Issue Price per share with Original Issue Price per share is defined as follows: $0.9315 for Series A-1, $1.6215 for Series A-2, $4.1850 for Series A-3, and $5.5150 for Series B, as adjusted for any recapitalizations and the like. After payment of the above dividends to holders of convertible preferred stock, any additional dividends will be distributed pro rata amongst the holders of common stock. No dividends have been declared or paid as of December 31, 2019 and 2020.
Liquidation Rights
In the event of any liquidation, dissolution or winding up of the Company, either voluntary or involuntary, the holders of convertible preferred stock then outstanding are entitled to be paid, out of the available funds and assets, and prior and in preference to any payment or distribution of any such funds on any shares of common stock, an amount per share equal to the Original Issue Price for the convertible preferred stock, plus all accrued and declared but unpaid dividends. The holders of convertible preferred stock have liquidation preferences over the common stockholders in the following amounts: $0.9315, $1.6215, $4.1850 and $5.5150 for Series A-1, Series A-2, Series A-3, and Series B, respectively. The liquidation preferences totaled approximately $12.8 million as of December 31, 2019 and 2020. If, upon the occurrence of a liquidation, dissolution or winding up of the Company, the assets and funds to be distributed among the holders of convertible preferred stock are insufficient to permit the payment to such holders, then the entire assets and funds of the Company legally available for distribution will be distributed ratably among the holders of convertible preferred stock in proportion to the preferential amount each such holder is otherwise entitled to receive. After the liquidation preferences of the holders of convertible preferred stock have been satisfied, the remaining assets of the Company will be distributed ratably among the holders of outstanding shares of common stock and convertible preferred stock on an as-if-converted basis.
If at any time after the Series B convertible preferred stock issue date, the Company sells or issues additional shares of common stock for no consideration or at a price below the then-effective convertible preferred stock conversion price, then the existing convertible preferred stock conversion price on the sale or issue date will be reduced.
Mezzanine Equity
The convertible preferred stock does not have a mandatory redemption date. However, while it is not mandatorily redeemable, the convertible preferred stock was reclassified into mezzanine equity because it will become redeemable at the option of the stockholders upon the occurrence of certain deemed liquidation events that are considered not solely within the Company’s control. That is, unless a majority of the holders of the then outstanding convertible preferred stock, on an as-if-converted to common stock basis, elect otherwise, deemed liquidation events include a sale of all or substantially all of the Company’s assets or a sale of at least fifty percent (50%) of the issued and outstanding voting securities, capital stock, or other comparable equity or ownership interest in the Company.
| F-20 |
Upon issuance of the convertible preferred stock, the Company assessed the embedded conversion and liquidation features of the securities. The Company determined that the convertible preferred stock did not require the Company to separately account for the liquidation features. The Company also concluded that no beneficial conversion feature existed upon the issuance date of the convertible preferred stock.
| 9. | Stock-Based Compensation |
In January 2013, the Board of Directors of the Company adopted the 2013 Equity Incentive Plan (the “2013 Plan”). The 2013 Plan provides for the grant of incentive and nonqualified stock options, stock appreciation rights, restricted stock awards and restricted stock units to employees, directors and consultants. The 2013 Plan allows for up to 1,269,300 shares of the Company’s common stock to be issued with respect to awards granted. The exercise price of incentive stock options and nonqualified stock options will be no less than 100% of the fair value per share of the Company’s common stock on the grant date. If an individual owns capital stock representing more than 10% of the voting shares, the exercise price of each share will be at least 110% of the fair market value on the date of grant. Fair value is determined by the Board of Directors. The Company’s stock options generally have 10-year contractual terms (five years for stockholders owning greater than 10% of all classes of stock) and generally vest over a four-year period from the date of grant. The Board of Directors determines the period over which options vest and become exercisable. The Company has a repurchase option for shares issued under unvested options exercisable upon the voluntary or involuntary termination of the purchaser’s employment with, or service to, the Company for any reason.
Activity under the 2013 Plan for the years ended December 31, 2019 and December 31, 2020 is set forth below (in thousands except shares, per share amounts and years):
| Shares Available for Grant | Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (Years) | Aggregate Intrinsic Value | ||||||||||||||||
| Outstanding at January 1, 2019 | 10,603 | 985,509 | $ | 0.40 | 6.75 | $ | 232 | |||||||||||||
| Authorized | 70,000 | - | - | |||||||||||||||||
| Options granted | (56,700 | ) | 56,700 | 0.65 | ||||||||||||||||
| Options cancelled | 38,834 | (38,834 | ) | 0.50 | ||||||||||||||||
| Outstanding at December 31, 2019 | 62,737 | 1,003,375 | $ | 0.40 | 6.75 | $ | 232 | |||||||||||||
| Options granted | (48,700 | ) | 48,700 | 0.70 | ||||||||||||||||
| Options exercised | - | (31,475 | ) | 0.55 | ||||||||||||||||
| Options cancelled | 23,334 | (23,334 | ) | 0.65 | ||||||||||||||||
| Restricted stock award granted | (24,478 | ) | - | - | ||||||||||||||||
| Outstanding at December 31, 2020 | 12,893 | 997,266 | $ | 0.40 | 5.84 | $ | 276 | |||||||||||||
| Vested and exercisable at December 31, 2020 | 847,613 | $ | 0.40 | 5.47 | $ | 269 | ||||||||||||||
| Vested and expected to vest at December 31, 2020 | 996,891 | $ | 0.40 | 5.84 | $ | 276 | ||||||||||||||
The aggregate intrinsic value of options outstanding, vested and exercisable, and vested and expected to vest were calculated as the difference between the exercise price of the option and the estimated fair value of the Company’s common stock, as determined by the Board of Directors, as of December 31, 2020.
During the year ended December 31, 2019, the Company recognized $38,000 in stock-based compensation expense. During the year ended December 31, 2020, the Company recognized $50,000 in stock-based compensation expense comprised of $33,000 from stock option grants and $17,000 from the issuance of a restricted stock award to a consultant for services rendered. The compensation expense is allocated on a departmental basis, based on the classification of the option holder. No income tax benefits have been recognized in the statements of operations for stock-based compensation arrangements.
The Company uses the Black-Scholes option pricing model to estimate the fair value of each option grant on the date of grant or any other measurement date. The following sets forth the weighted average assumptions used to determine the fair value of stock options during the years ended December 31, 2019 and 2020:
| F-21 |
| Years Ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| Expected term (years) | 6.98 | 5.24 | ||||||
| Risk-free interest rate | 1.88 | % | 1.29 | % | ||||
| Volatility factor | 37 | % | 38 | % | ||||
| Dividend yield | - | - | ||||||
Expected volatility is based on historical volatilities of public companies operating in the Company’s industry. The expected term of the options represents the period of time options are expected to be outstanding and is estimated considering vesting terms and historical exercise and post-vesting employment termination behavior. The risk-free interest rate is based on the U.S. Treasury yield curve in effect at the time of grant. The weighted-average fair value of options granted was $0.25 per share in both 2019 and 2020.
Future stock-based compensation for unvested employee options granted and outstanding as of December 31, 2020 is $38,000, to be recognized over a weighted-average remaining requisite service period of 1.56 years.
As of December 31, 2020, options outstanding had a weighted-average remaining contractual life of 5.8 years. As of December 31, 2020, 4,238,068 options were vested and exercisable with a weighted-average exercise price of $0.08 and a weighted-average remaining contractual life of 5.5 years.
In February 2020, the Company granted 24,478 shares of restricted common stock under the 2013 Plan to a consultant as partial consideration for services rendered, with a deemed fair value of $0.70 per share or $17,000. The fair value of this restricted stock award was expensed on the date of grant as they were fully vested on that date.
Stock-based compensation is included in the statements of operations in general and administrative and research and development, depending on the nature of the services provided. Stock-based compensation expense recorded to operations for stock options was as follows (in thousands):
| Years ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| General and administrative | $ | 23 | $ | 25 | ||||
| Research and development | 15 | 8 | ||||||
| Total | $ | 38 | $ | 33 | ||||
The Company also issued a restricted stock award to a consultant during the year ended December 31, 2020 for which $17,000 in stock-based compensation was included in general and administrative in the statement of operations.
| F-22 |
| 10. | Income Taxes |
For the years ended December 31, 2019 and 2020, the Company’s income tax provision is zero due to a full valuation allowance against the deferred tax assets.
The differences between the statutory tax expense (benefit) rate and the effective tax expense (benefit) rate, were as follows (in thousands):
| 2019 | 2020 | |||||||
| Statutory federal income tax rate | $ | (806 | ) | $ | (799 | ) | ||
| Increase (decrease) resulting from: | ||||||||
| Change in valuation allowance | 999 | 844 | ||||||
| Permanent Items | 8 | 107 | ||||||
| Prior year true ups | (12 | ) | - | |||||
| Tax credits | (184 | ) | (125 | ) | ||||
| State | (5 | ) | (28 | ) | ||||
| Other | - | 1 | ||||||
| Income tax provision/(benefit) | $ | - | $ | - | ||||
The components of our deferred tax assets and liabilities consist of (in thousands):
| 2019 | 2020 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryforwards | $ | 2,516 | $ | 3,146 | ||||
| Tax credit carryforwards | 405 | 607 | ||||||
| Stock based compensation | 18 | 24 | ||||||
| Fixed assets/intangible assets | 65 | 74 | ||||||
| Charitable contributions | 1 | 1 | ||||||
| Accruals and other | 3 | - | ||||||
| 3,008 | 3,852 | |||||||
| Valuation allowance | (3,008 | ) | (3,852 | ) | ||||
| Total deferred tax assets | $ | - | $ | - | ||||
We have established a valuation allowance to offset net deferred tax assets as of December 31, 2019 and 2020 due to the uncertainty of realizing future tax benefits from such assets.
As of December 31, 2020, the Company had U.S. federal and state net operating loss (“NOL”) carryforward amounts of $12.9 million and $6.4 million. The federal NOL carryforward consists of $4.7 million that has a carryforward of 20 years and begin to expire in 2030 and can and can be used to offset 100% of taxable income while $8.2 million can be carried forward indefinitely and may be able to be used against 100% of taxable income through the tax year ending December 31, 2020, as updated for the Coronavirus Aid, Relief, and Economic Security Act (P.L. 116-136), otherwise known as the CARES Act. The state NOL carryforward will begin to expire in 2033.
As of December 31, 2020, we had federal and state tax credit carryforwards of $0.7 million and $0.2 million, respectively. The federal tax credit carryforwards will begin to expire in 2033. The state tax credit carryforwards do not expire.
The Company follows Financial Accounting Standards Board No. 48, Accounting for Uncertainty in Income Taxes – an interpretation of FASB No. 109, as codified in FASB ASC 740-10, Income Taxes. At December 31, 2020, the Company recorded unrecognized tax benefits related to federal and state tax credits of $0.2 million. The Company’s policy is to recognize interest and penalties related to income tax matters in income tax expense. The Company did not have tax-related interest and penalties at December 31, 2020. The Company does not expect significant changes to its unrecognized tax benefits in the next twelve months. If recognized, none of the unrecognized tax benefits would affect the effective tax rate:
| F-23 |
The following summarizes the activity related to the Company’s unrecognized tax benefits for the years ended December 31, 2019 and December 31, 2020 (in thousands):
| Balance at January 1, 2019 | $ | - | ||
| Tax positions related to the current year: | ||||
| Additions | 77 | |||
| Reductions | - | |||
| Tax positions related to the prior year: | ||||
| Additions | 65 | |||
| Reductions | - | |||
| Settlements | - | |||
| Lapses in statute | ||||
| Balance at December 31, 2019 | 142 | |||
| Tax positions related to the current year: | ||||
| Additions | 70 | |||
| Reductions | - | |||
| Tax positions related to the prior year: | ||||
| Additions | - | |||
| Reductions | - | |||
| Settlements | - | |||
| Lapses in Statute | - | |||
| Balance at December 31, 2020 | $ | 212 |
The utilization of NOLs and tax credit carryforwards to offset future taxable income may be subject to an annual limitation as a result of ownership changes that have occurred previously or that may occur in the future. Under Sections 382 and 383 of the Internal Revenue Code (“IRC”) a corporation that undergoes an ownership change may be subject to limitations on its ability to utilize its pre-change NOLs and other tax attributes otherwise available to offset future taxable income and/or tax liability. An ownership change is defined as a cumulative change of 50% or more in the ownership positions of certain stockholders during a rolling three-year period. The Company has not completed a formal study to determine if any ownership changes within the meaning of IRC Section 382 and 383 have occurred. If an ownership change has occurred, the Company’s ability to use its NOLs or tax credit carryforwards may be restricted, which could require the Company to pay federal or state income taxes earlier than would be required if such limitations were not in effect.
| F-24 |
The Company files income tax returns in the U.S. federal jurisdiction and various state jurisdictions. The Company is subject to U.S. federal and state income tax examination for calendar tax years beginning in 2010 due to net operating losses that are being carried forward for tax purposes.
On March 27, 2020, the Coronavirus Aid, Relief and Economic Security Act (“CARES Act”) was enacted and signed into law in response to the COVID-19 pandemic. GAAP requires recognition of the tax effects of new legislation during the reporting period that includes the enactment date. The CARES Act includes changes to the tax provisions that benefits business entities and makes certain technical corrections to the 2017 Tax Cuts and Jobs Act. The tax relief measures for businesses include a five-year net operating loss carryback, suspension of the annual deduction limitation of 80% of taxable income from net operating losses generated in a tax year beginning after December 31, 2017, changes in the deductibility of interest, acceleration of alternative minimum tax credit refunds, payroll tax relief, technical corrections on net operating loss carryforwards for fiscal year taxpayers and allows accelerated deduction qualified improvement property. The CARES Act also provides other non-tax benefits to assist those impacted by the pandemic. The Company filed for and received a PPP loan. We evaluated the impact of the CARES Act and determined that there was no material impact for the year ended December 31, 2020.
On June 29, 2020, California Assembly Bill 85 was signed into law. The legislation suspends the California net operating loss deductions for 2020, 2021, and 2022 for certain taxpayers and imposes a limitation of certain California tax credits for 2020, 2021, and 2022. The legislation disallows the use of California net operating loss deductions if the taxpayer recognizes business income and its adjusted gross income is greater than $1.0 million. The carryover periods for net operating loss deductions disallowed by this provision will be extended. Additionally, any business credit will only offset a maximum of $5.0 million of California tax. Given our loss position in the current year, the new legislation did not impact the current year provision or our financial statements for the year ended December 31, 2020. We will continue to monitor possible California net operating loss and credit limitations in future periods.
On December 27, 2020, the Consolidated Appropriations Act, 2021 was enacted and signed into law to further COVID-19 economic relief and extend certain expiring tax provisions. The relief package includes a tax provision clarifying that businesses with forgiven PPP loans can deduct regular business expenses that are paid for with the loan proceeds. Additional pandemic relief tax measures include an expansion of the employee retention credit, enhanced charitable contribution deductions, and a temporary full deduction for business expenses for food and beverages provided by a restaurant. The provisions did not have a material impact on our financial statements for the year ended December 31, 2020.
| F-25 |
| 11. | Net Loss per Share |
Basic and diluted net loss per common share was calculated as follows (in thousands except per share amounts):
| Year ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| Numerator: | ||||||||
| Net loss | $ | (3,839 | ) | $ | (3,798 | ) | ||
| Denominator: | ||||||||
| Weighted average shares used in computing net loss per share – basic and diluted | 2,177 | 2,214 | ||||||
| Net loss per share – basic and diluted | $ | (1.76 | ) | $ | (1.72 | ) | ||
For the years ended December 31, 2019 and 2020, the Company had a net loss and as such, all outstanding shares of potentially dilutive securities were excluded from the calculation of diluted net loss per share as the inclusion would be anti-dilutive.
Potentially dilutive securities not included in the computation of diluted net loss per share because to do so would be antidilutive are as follows (in common stock equivalent shares):
| Year ended December 31, | ||||||||
| 2019 | 2020 | |||||||
| Convertible preferred stock | 3,508,631 | 3,535,469 | ||||||
| Options to purchase common stock | 1,003,375 | 997,266 | ||||||
| Series A-1 preferred stock warrant | 26,838 | - | ||||||
| 4,538,844 | 4,532,735 | |||||||
The table above omits the potentially dilutive shares into which the 2020 Convertible Notes would convert.
| 12. | Commitments and Contingencies |
Operating Lease
The Company leases its headquarters in Los Altos, California under a non-cancelable operating lease agreement which expired and became month-to-month on July 31, 2019. As a result, there are no future minimum lease payments for 2020. Rent expense under the operating lease was $42,000 and $44,000 in 2019 and 2020, respectively.
Legal Proceedings
From time to time, the Company may become involved in legal proceedings arising in the ordinary course of business. Management is not currently aware of any matters that will have a material adverse effect on the financial position, results of operations or cash flows of the Company.
Guarantees and Indemnifications
In the normal course of business, the Company enters into agreements that contain a variety of representations and provide for general indemnification. The Company’s exposure under these agreements is unknown because it involves claims that may be made against the Company in the future. To date, the Company has not paid any claims or been required to defend any action related to its indemnification obligations. As of December 31, 2020, the Company is not subject to any material indemnification claims whose assertion is probable or reasonably possible. Consequently, the Company has not recorded any related liabilities.
| F-26 |
| 13. | Related Party Transactions |
In January 2013, the Company entered into a consulting agreement (the “Agreement”) with one of the Company’s co-founders whereby the co-founder provides consulting services as the Company’s Chief Medical Officer (“CMO”) by overseeing Company-sponsored clinical trials. The Agreement is for a term of 15 years with automatic one-year renewals. The sole compensation payable to the CMO is the continued vesting of shares of common stock of the Company held by the CMO. In July 2018, the CMO was awarded options for the purchase of 40,000 shares of the Company’s common stock with 25% vested after one year and the remaining 75% vesting ratably over 36 months. The Company and the CMO entered into an amendment to the Agreement in 2019 providing cash compensation to the CMO of $10,000 per month for additional services. Consulting fees paid to the CMO were $78,000 and $120,000 for the years ended December 31, 2019 and 2020, respectively.
In July 2019, the Company entered into a consulting agreement (the “CFO Agreement”) with the Company’s Chief Financial Officer (“CFO”). In February 2020, the Company granted the CFO an option to purchase 28,000 shares of the Company’s common stock, of which 25% were vested at the grant date and the remaining 75% vested ratably over 18 months. The Company entered into an amendment to the Agreement in December 2020 increasing the cash compensation to $150 per hour. Consulting fees paid to the CFO were $19,000 and $37,000 for the years ended December 31, 2019 and 2020, respectively.
Mr. Najmabadi, another co-founder of the Company has served as our consulting technical engineering advisor on manufacturing and intellectual property matters since January 2020. Previously he was CEO of the Company from inception in December 2009 until January 2013; Chief Technical and Operations Officer from January 2013 until January 2019; and Chief Technology Officer from January 2019 to January 2020. He currently receives cash compensation quarterly compensation of $3,000.
| 14. | Subsequent Events |
For the financial statements as of December 31, 2020 and for the year then ended, the Company evaluated subsequent events through August 11, 2021, the date on which those financial statements were issued. The Company has concluded that no events or transactions have occurred that require disclosure in the accompanying financial statements, other than the following:
On February 6, 2021, the Company received notification and confirmation from Silicon Valley Bank that its PPP loan and related accrued interest has been forgiven in its entirety by the U.S. Small Business Administration and automatically cancelled.
On March 1, 2021, the Company entered into an amendment to the 2020 Convertible Notes which provides for the extension of the maturity date of the 2020 Convertible Notes from March 2021 to October 30, 2021 and the conversion of the 2020 Convertible Notes into shares of the Company’s common stock upon a Qualified Financing that is an IPO. No other terms to the 2020 Convertible Notes were amended.
In April 2021, the Company entered into a note purchase agreement and a series of convertible note payable agreements (the “2021 Convertible Notes”) for aggregate borrowings of approximately $2.0 million. Outstanding borrowings under the 2021 Convertible Notes and accrued interest are due in April 2022, if not previously converted. The 2021 Convertible Notes bear interest at the rate of 5% per annum. Pursuant to the 2021 Convertible Notes, the outstanding principal and accrued interest are automatically convertible into equity shares a Qualified Financing at a conversion price per share equal to 87.5% of the price per share paid by investors purchasing such equity securities in a Qualified Financing.
In June 2021, the Company amended the 2013 Plan to increase the number of shares of common stock reserved for issuance by 52,000 and also granted 57,400 stock options with a weighted average exercise price of $2.45 per share and a grant fair value of approximately $56,000.
In connection with preparing for its initial public offering, the Company filed a certificate of amendment to its Fifth Amended and Restated Certificate of Incorporation to effect a 1-for-5 reverse stock split of its issued and outstanding preferred stock and common stock, which became effective on August 5, 2021. Adjustments corresponding to the reverse stock split were made to the ratio at which the Company’s convertible preferred stock will convert into the Company’s common stock. Accordingly, all share and per share amounts related to the common stock, stock options, warrants and restricted stock awards for all periods presented in the accompanying financial statements and notes thereto have been retroactively adjusted, where applicable, to reflect the reverse stock split.
| F-27 |
Condensed Balance Sheets
(in thousands, except share and per share data)
| December 31, | March 31, | |||||||
| 2020 | 2021 | |||||||
| (Note 2) | (Unaudited) | |||||||
| Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 1,795 | $ | 837 | ||||
| Prepaid expenses and other current assets | 115 | 116 | ||||||
| Total current assets | 1,910 | 953 | ||||||
| Other assets | 4 | 325 | ||||||
| Total assets | $ | 1,914 | $ | 1,278 | ||||
| Liabilities, Convertible Preferred Stock and Stockholders’ Deficit | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 162 | $ | 472 | ||||
| Accrued expenses | 311 | 413 | ||||||
| Promissory note, current | 117 | - | ||||||
| Convertible note | 2,650 | 2,843 | ||||||
| Derivative liability | 856 | 861 | ||||||
| Total current liabilities | 4,096 | 4,589 | ||||||
| Promissory note, net of current portion | 23 | - | ||||||
| Total liabilities | 4,119 | 4,589 | ||||||
| Commitments and contingencies | ||||||||
| Convertible preferred stock, $0.0001 par value; 22,360,455 shares authorized; 3,535,469 shares issued and outstanding at December 31, 2020 and March 31, 2021 (aggregate liquidation preference of $12,782 at December31, 2020 and March 31, 2021) | 12,451 | 12,451 | ||||||
| Stockholders’ deficit: | ||||||||
| Common stock, $0.0001 par value; 42,000,000 shares authorized; 2,233,139 and 2,283,197 shares issued and outstanding at December 31, 2020 and March 31, 2021, respectively | 1 | 1 | ||||||
| Additional paid-in capital | 303 | 345 | ||||||
| Accumulated deficit | (14,960 | ) | (16,108 | ) | ||||
| Total stockholders’ deficit | (14,656 | ) | (15,762 | ) | ||||
| Total liabilities, convertible preferred stock and stockholders’ deficit | $ | 1,914 | $ | 1,278 | ||||
The accompanying notes are an integral part of these financial statements.
| F-28 |
Condensed Statements of Operations
(Unaudited)
(in thousands, except per share data)
| Three Months Ended March 31, | ||||||||
| 2020 | 2021 | |||||||
| Operating expenses: | ||||||||
| Research and development | $ | 805 | $ | 635 | ||||
| General and administrative | 248 | 418 | ||||||
| Total operating expenses | 1,053 | 1,053 | ||||||
| Loss from operations | (1,053 | ) | (1,053 | ) | ||||
| Other income (expense), net: | ||||||||
| Interest income (expense), net | 2 | (230 | ) | |||||
| Other expense, net | - | (5 | ) | |||||
| Gain on loan extinguishment | - | 140 | ||||||
| Total other income (expense), net | 2 | (95 | ) | |||||
| Net loss | $ | (1,051 | ) | $ | (1,148 | ) | ||
| Net loss per share - basic and diluted | $ | (0.48 | ) | $ | (0.51 | ) | ||
| Weighted average shares of common stock - basic and diluted | 2,195 | 2,242 | ||||||
The accompanying notes are an integral part of these financial statements.
| F-29 |
Condensed Statements of Convertible Preferred Stock and Stockholders’ Deficit
(Unaudited)
(in thousands, except share amounts)
| Convertible Preferred Stock | Common Stock | Additional Paid-In | Accumulated | Total Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||
| Balance at December 31, 2019 | 3,508,631 | $ | 12,391 | 2,177,187 | $ | - | $ | 236 | $ | (11,162 | ) | $ | (10,926 | ) | ||||||||||||||
| Issuance of restricted stock award to | ||||||||||||||||||||||||||||
| nonemployee for service | - | - | 24,478 | - | 17 | - | 17 | |||||||||||||||||||||
| Issuance of common stock upon | ||||||||||||||||||||||||||||
| exercise of stock options | - | - | 16,666 | - | 11 | - | 11 | |||||||||||||||||||||
| Issuance of Series A-1 convertible | ||||||||||||||||||||||||||||
| preferred stock upon exercise | ||||||||||||||||||||||||||||
| of warrant | 26,838 | 25 | - | - | - | - | - | |||||||||||||||||||||
| Warrant liability transferred to | ||||||||||||||||||||||||||||
| mezzanine equity | - | 35 | - | - | - | - | - | |||||||||||||||||||||
| Stock-based compensation expense | - | - | - | - | 10 | - | 10 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (1,051 | ) | (1,051 | ) | |||||||||||||||||||
| Balance at March 31, 2020 | 3,535,469 | $ | 12,451 | 2,218,331 | $ | - | $ | 274 | $ | (12,213 | ) | $ | (11,939 | ) | ||||||||||||||
| Balance at December 31, 2020 | 3,535,469 | $ | 12,451 | 2,233,139 | $ | - | $ | 304 | $ | (14,960 | ) | $ | (14,656 | ) | ||||||||||||||
| Issuance of common stock upon | ||||||||||||||||||||||||||||
| exercise of stock options | - | - | 50,058 | - | 34 | - | 34 | |||||||||||||||||||||
| Stock-based compensation expense | - | - | - | - | 8 | - | 8 | |||||||||||||||||||||
| Net loss | - | - | - | - | - | (1,148 | ) | (1,148 | ) | |||||||||||||||||||
| Balance at March 31, 2021 | 3,535,469 | $ | 12,451 | 2,283,197 | $ | - | $ | 346 | $ | (16,108 | ) | $ | (15,762 | ) | ||||||||||||||
The accompanying notes are an integral part of these financial statements.
| F-30 |
Condensed Statements of Cash Flows
(Unaudited)
(in thousands)
| Three Months Ended March 31, | ||||||||
| 2020 | 2021 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (1,051 | ) | $ | (1,148 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Stock-based compensation expense | 10 | 8 | ||||||
| Loss on change in fair value of derivative liability | - | 5 | ||||||
| Gain on loan extinguishment | - | (140 | ) | |||||
| Amortization of debt discount | - | 188 | ||||||
| Amortization of debt issuance cost | - | 5 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (66 | ) | (1 | ) | ||||
| Other assets | - | (321 | ) | |||||
| Accounts payable | (132 | ) | 310 | |||||
| Accrued expenses | 200 | 102 | ||||||
| Net cash used in operating activities | (1,039 | ) | (992 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Proceeds from convertible notes | 1,290 | - | ||||||
| Payment of debt issuance costs | (14 | ) | - | |||||
| Proceeds from exercise of Series A-1 warrant | 25 | - | ||||||
| Proceeds from exercise of stock options | 11 | 34 | ||||||
| Net cash provided by financing activities | 1,312 | 34 | ||||||
| Increase (decrease) in cash, cash equivalents and restricted cash | 273 | (958 | ) | |||||
| Cash, cash equivalents and restricted cash, beginning of period | 2,124 | 1,795 | ||||||
| Cash, cash equivalents and restricted cash, end of period | $ | 2,397 | $ | 837 | ||||
| Supplemental disclosure of non-cash investing and financing activities: | ||||||||
| Derivative liability | $ | 363 | $ | 861 | ||||
| Supplemental disclosure of non-cash activity: | ||||||||
| Debt issuance costs - 2021 Convertible Notes | $ | - | $ | 6 | ||||
The accompanying notes are an integral part of these financial statements.
| F-31 |
Description of the Business
RenovoRx, Inc. (“RenovoRx” or the “Company”) was incorporated in Delaware in December 2012 and operates from its headquarters in Los Altos, California. The Company is a clinical-stage biopharmaceutical company focused on developing therapies for the local treatment of solid tumors and conducting a Phase 3 registrational trial for its lead product candidate RenovoGem™. The Company’s therapy platform, RenovoRx Trans-Arterial Micro-Perfusion, or RenovoTAMP™ utilizes approved chemotherapeutics with validated mechanisms of action and well-established safety and side effect profiles, with the goal of increasing their efficacy, improving their safety, and widening their therapeutic window. RenovoTAMP combines the Company’s patented FDA cleared delivery system, RenovoCath®, with small molecule chemotherapeutic agents that can be forced across the vessel wall using pressure, targeting these anti-cancer drugs locally to the solid tumors. While the Company anticipates investigating other chemotherapeutic agents for intra-arterial delivery via RenovoTAMP, to date the Company’s clinical work has focused on gemcitabine, which is a generic drug. The Company’s first product candidate, RenovoGem, is a drug and device combination consisting of intra-arterial gemcitabine and RenovoCath. FDA has determined that RenovoGem will be regulated as, and if approved the Company expects will be reimbursed as, a new oncology drug product. The Company has secured FDA Orphan Drug Designation for RenovoGem in its first two indications: pancreatic cancer and cholangiocarcinoma (bile duct cancer, or CCA). The Company has completed its RR1 Phase 1/2 and RR2 observational registry studies, with 20 and 25 patients respectively, in locally advanced pancreatic cancer, or LAPC. These studies demonstrated a median overall survival of 27.9 months in patients treated with RenovoGem and radiation versus expected survival of 12-15 months in patients receiving only intravenous (IV) systemic chemotherapy dosed at 1,000mg/m2 based on historical control data. Unlike the historical controls, the Company’s RR1 and RR2 studies were not randomized or controlled for potential confinement. Based on FDA safety review of the Company’s Phase 1/2 study the FDA allowed the Company to proceed to evaluate RenovoGem within its Phase 3 registration Investigational New Drug, or IND, clinical trial. This Phase 3 trial is 40% enrolled as of April 30, 2021 and the Company expects to report data from a planned interim data readout in the second half of 2022. The Company intends to evaluate RenovoGem in a second indication in a Phase 2/3 trial in hilar CCA (cancer that occurs in the bile ducts that lead out of the liver and join with the gallbladder, also called extrahepatic cholangiocarcinoma, or HCCA). The Company plans to propose the trial to the FDA and potentially launch in the first half of 2022. In addition, the Company may evaluate RenovoGem in other indications, potentially including locally advanced lung cancer, locally advanced uterine tumors, and glioblastoma (an aggressive type of cancer that can occur in the brain or spinal cord). To date, the Company has used gemcitabine, but in the future it may develop other chemotherapeutic agents for intra-arterial delivery via RenovoCath.
2. Summary of Significant Accounting Policies
Unaudited Financial Statements
The accompanying unaudited condensed interim financial statements should be read in conjunction with the audited financial statements and notes thereto included elsewhere in this prospectus. The unaudited condensed interim balance sheet as of December 31, 2020 included herein was derived from the audited financial statements as of that date. The results of operations for the three months ended March 31, 2021 are not necessarily indicative of the results for the fiscal year ending December 31, 2021 or any future interim period. The financial statement data and the other financial information contained in these notes to the unaudited condensed interim financial statements related to the three-month periods are also unaudited. Certain disclosures have been condensed or omitted from the unaudited condensed interim financial statements.
Basis of Presentation
The accompanying unaudited condensed interim financial statements are presented in U.S. dollars and have been prepared in conformity with accounting principles generally accepted in the United States of America (“GAAP”) and applicable rules and regulations of the U.S. Securities and Exchange Commission (“SEC”) for interim reporting. Any reference in these notes to applicable guidance is meant to refer to the authoritative GAAP as found in the Accounting Standards Codification (“ASC”) and as amended by Accounting Standards Update (“ASU”) of the Financial Accounting Standards Board (“FASB”).
| F-32 |
In the opinion of management, the accompanying unaudited condensed interim financial statements include all normal and recurring adjustments (which consist primarily of accruals, estimates and assumptions that impact the financial statements) considered necessary to present fairly the Company’s financial position as of March 31, 2021 and its results of operations and changes in convertible preferred stock and stockholders’ deficit for the three months ended March 31, 2020 and 2021 and cash flows for the three months ended March 31, 2020 and 2021. Operating results for the three months ended March 31, 2021 are not necessarily indicative of the results that may be expected for the year ending December 31, 2021.
Liquidity and Going Concern
The financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. In order to continue its operations, the Company must raise additional equity or debt financings and achieve profitable operations. Although management has historically been successful in raising capital, there can be no assurance that the Company will be able to obtain additional equity or debt financing on terms acceptable to the Company, or at all. The failure to obtain sufficient funds on acceptable terms when needed could have a material adverse effect on the Company’s business, financial position, results of operations, and future cash flows. The Company is seeking to complete an initial public offering (“IPO”) of its common stock. Upon the closing of an IPO, on specified terms, the Company’s outstanding convertible preferred stock will automatically convert into shares of common stock (see Note 8). In the event the Company does not complete an IPO, the Company expects to seek additional funding through other capital sources including through the sale of equity, debt financings or other capital sources including collaborations with other companies or other strategic transactions. However, the Company may be unable to raise additional funds or enter into such agreements or arrangements when needed on acceptable terms, or at all.
As a company with no commercial operating history, the Company is subject to all of the risks and expenses associated with a start-up company. The Company must among other things respond to competitive developments, attract, retain and motivate qualified personnel and support ongoing clinical trials for its product candidate. The Company has generated operating losses and negative cash flows from operations in each year since inception and anticipates that it will continue to incur net losses into the foreseeable future. The Company has not generated any revenue from product sales to date and will continue to incur significant research and development and other expenses related to its ongoing operations. As of March 31, 2021, the Company had cash and cash equivalents of $837,000 and had an accumulated deficit of $16.1 million. In April 2021, the Company entered into a series of convertible note agreements with several lenders including current investors to provide an aggregate $2.0 million in cash proceeds. The Company has funded its operations primarily through the sale and issuance of convertible preferred stock and convertible notes. The Company has reviewed the relevant conditions and events surrounding its ability to continue as a going concern including among others: historical losses, projected future results, including the effects of the novel coronavirus (“COVID-19”), cash requirements for the upcoming year, terms of the Company’s current debt arrangements, funding capacity, net working capital, total stockholders’ deficit and future access to capital. These factors along with the Company’s cash and cash equivalents raise substantial doubt about the Company’s ability to continue as a going concern for at least one year from the date the financial statements are issued. The financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty. If future financing is not achieved, the Company may be required to curtail spending to reduce cash outflows.
| F-33 |
Risks and Uncertainties
The Company is subject to a number of risks associated with companies at a similar stage, including the risk associated with the development of products that must receive regulatory approval before market launch, dependence on key individuals, competition from larger and established companies, volatility of the industry, ability to obtain adequate financing to support growth, the ability to attract and retain additional qualified personnel to manage the anticipated growth of the Company and general economic conditions.
In March 2020, the World Health Organization declared COVID-19 a pandemic. The COVID-19 pandemic has negatively impacted the global economy, disrupted global supply chains and created significant volatility and disruption of financial markets. The full extent to which the COVID-19 pandemic will directly or indirectly impact the Company’s business, results of operations and financial condition, including expenses, clinical trials and research and development costs, will depend on future developments that are highly uncertain, including as a result of new information that may emerge concerning COVID-19 and the actions taken to contain or treat COVID-19, as well as the economic impact on local, regional, national and international markets. The Company has made estimates of the impact of COVID-19 within its financial statements and there may be changes to those estimates in future periods. Actual results could materially differ from those estimates.
Use of Estimates
The preparation of unaudited condensed interim financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, income and expenses as well as the disclosure of contingent assets and liabilities, at the date of the financial statements during the reporting periods. Significant estimates and assumptions made in the accompanying unaudited condensed interim financial statements include, but are not limited to, the accrual of certain liabilities, the valuation of financial instruments, the fair value of the Company’s common stock and the fair value of options granted under the Company’s equity incentive plan. Actual results could differ from materially from these estimates.
Recent Accounting Pronouncements
From time to time, new accounting pronouncements, or Accounting Standard Updates (“ASU”) are issued by the Financial Accounting Standards Board (“FASB”), or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on the Company’s financial position or results of operations upon adoption.
Concentration of Credit Risk and Significant Suppliers
Financial instruments that potentially subject the Company to concentration of credit risk consist of cash and cash equivalents. At December 31, 2020 and March 31, 2021, the Company maintained $1.8 million and $837,000 in cash and cash equivalents, respectively, with one financial institution. The Company is exposed to credit risk in the event of default by the financial institutions to the extent that cash and cash equivalent deposits are in excess of the $250,000 insured by the Federal Deposit Insurance Corporation. These deposits routinely exceed the insurable limit. To date, the Company has not experienced any losses on its cash and cash equivalents.
The Company is dependent on third-party manufacturers to supply products and services for its research and development activities, including preclinical and clinical development. In particular, the Company relies, and expects to continue to rely, on a small number of third-party manufacturers to manufacture and supply its RenovoCath devices and its product candidates for clinical trials. These activities could be adversely affected by a significant interruption in the supply of these items.
Deferred Offering Costs
The Company capitalizes certain legal, professional, accounting and other third-party fees that are directly associated with in-process financings as deferred offering costs until such financings are consummated. After consummation of the financing, these costs are recorded as a reduction of the proceeds received from the financing. If a planned financing is abandoned, the deferred offering costs are expensed as a charge to operating expenses in the statements of operations.
There were zero and $321,000 in deferred offering costs included in other assets in the Company’s condensed balance sheets at December 31, 2020 and March 31, 2021, respectively.
| F-34 |
Net Loss per Share
Basic net loss per common share is calculated by dividing net loss by the weighted-average number of shares of common stock outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is computed by dividing net loss by the weighted-average number of shares of common stock and common stock equivalents of potentially dilutive securities outstanding for the period determined using the treasury stock and if-converted methods. Potentially dilutive common stock equivalents are comprised of convertible preferred stock, convertible notes and options outstanding under the Company’s stock option plan. For the three months ended March 31, 2020 and 2021, there was no difference in the number of shares used to calculate basic and diluted shares outstanding as the inclusion of the potentially dilutive securities would be anti-dilutive.
Fair Value of Financial Instruments
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. To increase the comparability of fair value measures, the following hierarchy prioritizes the inputs to valuation methodologies used to measure fair value.
Level 1 – Valuations based on quoted prices for identical assets and liabilities in active markets.
Level 2 – Valuations based on observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets, quoted prices for identical or similar assets and liabilities in markets that are not active, or other inputs that are observable or can be corroborated by observable market data.
Level 3 – Valuations based on unobservable inputs reflecting the Company’s assumptions, consistent with reasonably available assumptions made by other market participants. These valuations require significant judgment.
The estimated fair value of financial instruments disclosed in the financial statements has been determined by using available market information and appropriate valuation methodologies. In certain cases where there is limited activity or less transparency around inputs to valuation, securities are classified as Level 3.
The carrying value of all remaining current assets and current liabilities approximates fair value because of their short-term nature.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with a remaining maturity date upon acquisition of three months or less to be cash equivalents. Cash and cash equivalents consist primarily of cash held in checking and money market accounts. As of March 31, 2020, the Company had restricted cash of $20,000, which consisted of reserve funds held at one financial institution to collateralize the Company’s credit cards. The Company held no funds in restricted cash at December 31, 2020 or March 31, 2021.
Convertible Instruments and Embedded Derivatives
The Company evaluates all of its agreements to determine if such instruments have derivatives or contain features that qualify as embedded derivatives. The Company accounts for certain redemption features that are associated with convertible notes as liabilities at fair value and adjusts the instruments to their fair value at the end of each reporting period. For derivative financial instruments that are accounted for as liabilities, the derivative instrument is initially recorded at its fair value and is then re-valued at each reporting date, with changes in the fair value reported in other income (expense), net in the condensed statements of operations. Derivative instrument liabilities are classified in the condensed balance sheets as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date. As of December 31, 2020 and March 31, 2021, the Company’s only derivative financial instrument was related to the 2020 Convertible Notes (defined in Note 5), which contained certain redemptive features (see Note 5).
| F-35 |
Research and Development Costs
Research and development costs, which include direct and allocated expenses, are expensed in the period incurred. Research and development costs include primarily clinical development-related expenses, pre-clinical research and development expenses, employee compensation and related benefits, regulatory support and related services, clinical consulting, travel-related expenses and allocated expenses for rent, insurance and other general overhead costs. The Company also receives payments from vendors in performing clinical trials on behalf of the Company for the delivery device used by such vendors in performing such clinical trials. These payments are offset against the Company’s research and development expenses.
Clinical Trial Expenses
The Company makes payments in connection with clinical trials under contracts with clinical trial sites and contract research organizations that support conducting and managing clinical trials. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price or on a time and materials basis. A portion of the obligation to make payments under these contracts depends on factors such as the successful enrollment or treatment of patients or the completion of other clinical trial milestones.
Expenses related to clinical trials are accrued based on estimates and/or representations from service providers regarding work performed, including actual levels of patient enrollment, completion of patient studies and progress of the clinical trials. Other incidental costs related to patient enrollment or treatment are accrued when reasonably certain. If the amounts the Company is obligated to pay under clinical trial agreements are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), the accruals are adjusted accordingly. Revisions to contractual payment obligations are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. As noted above, the Company receives payments from vendors performing clinical trials on behalf of the Company for the delivery device used by such vendors in performing such clinical trials.
General and Administrative
General and administrative expenses consist primarily of personnel costs, including employee compensation and related benefits and consulting. Additionally, these expenses include professional fees, including audit, legal, recruiting services and allocated expenses for rent, insurance and other general overhead costs. General and administrative expenses are expensed in the period incurred.
Convertible Preferred Stock
The Company records preferred stock at fair value on the date of issuance, net of issuance costs. The preferred stock is recorded outside of stockholders’ deficit because the shares contain liquidation features that are not solely within the Company’s control. As a result, the preferred stock is classified as mezzanine equity (temporary equity). The Company has elected not to adjust the carrying value of the preferred stock to the liquidation preferences of such shares because it is uncertain whether or when an event would occur that would obligate the Company to pay the liquidation preferences to holders of shares of preferred stock. Subsequent adjustments to the carrying values to the liquidation preferences will be made only when it becomes probable that such a liquidation event will occur.
Stock-Based Compensation
The Company calculates the fair value of stock options using the Black-Scholes option pricing model, which incorporates various assumptions including volatility, expected life and risk-free interest rate. Compensation related to service-based awards is recognized starting on the grant date on a straight-line basis over the vesting period, which is generally four years.
| F-36 |
The determination of the fair value of each stock award using this option-pricing model is affected by the Company’s assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, the fair value of the common stock at the date of grant, the expected term of the awards, the expected stock price volatility over the term of the awards, the risk-free interest rate, and the dividend rate as follows:
Fair Value of Common Stock—Given the absence of a public trading market, the Company’s Board of Directors considered numerous objective and subjective factors to determine the fair value of the Company’s common stock at each grant date. These factors included, but were not limited to: (i) contemporaneous third-party valuations of common stock; (ii) the prices for preferred stock sold to outside investors; (iii) the rights and preferences of preferred stock relative to Common stock; (iv) the lack of marketability of the Company’s common stock; (v) developments in the business; and (vi) the likelihood of achieving a liquidity event, such as an IPO or sale of the business, given prevailing market conditions. The methodology to determine the fair value of the Company’s common stock included estimating the fair value of the enterprise using the “backsolve” method, which is a market approach that assigns an implied enterprise by accounting for all share class rights and preferences based on the latest round of financing. The total equity value implied was then applied in the context of an option pricing model to determine the value of each class of the Company’s shares.
Expected Term—The expected term represents the period that the stock-based awards are expected to be outstanding. The Company determines the expected term using the simplified method. The simplified method deems the term to be the average of the time-to-vesting and the contractual life of the options. For stock options granted to non-employees, the expected term equals the remaining contractual term of the option from the vesting date.
Expected Volatility—Given the absence of a public trading market, the expected volatility was estimated by taking the average historic price volatility for industry peers, consisting of several public companies in the Company’s industry that are either similar in size, stage, or financial leverage, over a period equivalent to the expected term of the awards.
Risk-Free Interest Rate—The risk-free interest rate is calculated using the average of the published interest rates of U.S. Treasury zero-coupon issues with maturities that are commensurate with the expected term.
Dividend Rate—The dividend yield assumption is zero as the Company has no plans to make dividend payments.
The Company generally grants stock options to its employees for a fixed number of shares with an exercise price equal to the fair value of the underlying shares at date of grant. The Company accounts for all stock option grants using the fair value method and stock-based compensation is recognized as the underlying options vest.
Income Taxes
In determining quarterly provisions for income taxes, the Company uses the annual estimated effective tax rate applied to the actual year-to-date profit or loss, adjusted for discrete items arising in that quarter. The Company’s annual estimated effective tax rate differs from the U.S. federal statutory rate primarily as a result of state taxes, foreign taxes, and changes in the Company’s valuation allowance against its deferred tax assets. For the three months ended March 31, 2020 and 2021, the Company recorded an immaterial provision for income taxes.
Segment Reporting
Financial Accounting Standards Board (“FASB”) ASC Topic 280, Segment Reporting, requires use of the “management approach” model for segment reporting. The management approach model is based on the way a Company’s management organizes segments within the company for making operating decisions and assessing performance.
| F-37 |
The Company determined it has one reportable segment, the development of a platform technology to deliver de-risked small molecules for localized treatment of solid cancer tumors. The segment is based on financial information that is utilized by the Company’s Chief Operating Decision Maker who is the Company’s Chief Executive Officer, to assess performance and allocate resources.
Emerging Growth Company Status
From time to time, new accounting pronouncements, or Accounting Standards Updates (“ASU”) are issued by the FASB or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on the Company’s financial position or results of operations upon adoption.
As an “emerging growth company” (“EGC”) under the Jumpstart Our Business Startups Act (“JOBS Act”), the Company may elect to take advantage of certain forms of relief from various reporting requirements that are applicable to public companies. The relief afforded under the JOBS Act includes an extended transition period for the implementation of new or revised accounting standards. The Company has elected to take advantage of this extended transition period and, as a result, the Company’s financial statements may not be comparable to those of companies that implement accounting standards as of the effective dates for public companies. The Company may take advantage of the relief afforded under the JOBS Act up until the last day of the fiscal year following the fifth anniversary of an offering or such earlier time that it is no longer an EGC.
3. Accrued Expenses
A summary of the components of accrued expenses is as follows (in thousands):
| December 31, 2020 | March 31, 2021 | |||||||
| Accrued clinical trials | $ | 171 | $ | 254 | ||||
| Accrued interest | 101 | 138 | ||||||
| Accrued personnel | 39 | 21 | ||||||
| $ | 311 | $ | 413 | |||||
4. Fair Value Measurements
The following table sets forth by level, within the fair value hierarchy, the financial assets and liabilities that are measured at fair value on a recurring basis at December 31, 2020 and March 31, 2021 (in thousands):
| Fair Value Measurements at December 31, 2020 using: | ||||||||||||||||
| Assets: | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Money market funds | $ | 1,703 | $ | - | $ | - | $ | 1,703 | ||||||||
| $ | 1,703 | $ | - | $ | - | $ | 1,703 | |||||||||
| Liabilities: | ||||||||||||||||
| Derivative liability – 2020 Convertible Notes | $ | $- | $ | 856 | $ | 856 | ||||||||||
| $ | $- | $ | 856 | $ | 856 | |||||||||||
| F-38 |
| Fair Value Measurements at March 31, 2021 using: | ||||||||||||||||
| Assets: | Level 1 | Level 2 | Level 3 | Total | ||||||||||||
| Money market funds | $ | 803 | $ | - | $ | - | $ | 803 | ||||||||
| $ | 803 | $ | - | $ | - | $ | 803 | |||||||||
| Liabilities: | ||||||||||||||||
| Derivative liability – 2020 Convertible Notes | $ | - | $ | - | $ | 861 | $ | 861 | ||||||||
| $ | - | $ | - | $ | 861 | $ | 861 | |||||||||
The change in the fair value of the derivative liability is summarized below (in thousands):
Three Months Ended March 31 | ||||||||
| 2020 | 2021 | |||||||
| Fair value at beginning of the period | $ | - | $ | 856 | ||||
| Initial fair value of instruments issued | 363 | - | ||||||
| Change in fair value of instruments | - | 5 | ||||||
| Fair value at end of the period | $ | 363 | $ | 861 | ||||
The derivative liability in the table above related to the 2020 Convertible Notes and represents the fair value of the redemption-like contingent conversion feature. The Company calculated the fair value of the derivative liability using a probability weighted discounted cash flow analysis. The inputs used to determine the estimated fair value of the derivative were based primarily on the probability of an underlying event occurring that would trigger the embedded derivative and the timing of such event. The Company’s derivative liability is measured at fair value on a recurring basis and is classified as a Level 3 liability. The Company records subsequent adjustments to reflect the increase or decrease in estimated fair value at each reporting date in other income (expense), net in the condensed statements of operations (see Note 5).
There were no transfers among Level 1, Level 2 or Level 3 categories during any of the periods presented. The Company had no other financial assets or liabilities that were required to be measured at fair value on a recurring basis.
5. Convertible Notes
In March 2020, the Company entered into a note purchase agreement for the issuance of up to $4.0 million of convertible promissory notes. The Company entered into a series of convertible note payable agreements (the “2020 Convertible Notes”) for aggregate borrowings of approximately $3.0 million. Outstanding borrowings under the 2020 Convertible Notes and accrued interest are due in March 2021, if not previously converted. The 2020 Notes bear interest at the rate of 5% per annum. The 2020 Notes cannot be prepaid prior to the maturity date unless approved in writing by the Company and requisite holders.
| F-39 |
The Company evaluated whether the 2020 Convertible Notes contain embedded features that meet the definition of derivatives under FASB ASC 815, Derivatives and Hedging. The Company determined that these redemption features contained rights and obligations for conversion contingent upon a potential future financing event or a change in control. Thus, the embedded redemption features were bifurcated from the face value of the note and accounted for as a derivative liability to be remeasured at the end of each reporting period. The fair value of the derivative liability at December 31, 2020 and March 31, 2021 was $856,000 and $861,000, respectively with the offsetting amount being recorded as a debt discount. Debt issuance costs were $22,000 at December 31, 2020. There were no debt issuance costs during the three months ended March 31, 2021. The derivative liability is subject to fair value remeasurement at the end of each reporting period. The discount and debt issuance costs are being amortized to interest expense using the effective interest method over the expected term of the 2020 Convertible Notes. There was no amortization of debt discount or debt issuance costs for the three months ended March 31, 2020. For the three months ended March 31, 2021, the Company recognized $193,000 for amortization of the debt discount and debt issuance costs, of which $188,000 pertains to the debt discount and $5,000 pertains to debt issuance costs. This amortization expense is recognized as interest expense in the condensed statements of operations for the three months ended March 31, 2021. For the three months ended March 31, 2020 and 2021, the effective interest rate of the 2020 Convertible Notes was 30.8% compared to the stated rate of 5%. As a result, the Company’s reported interest expense was significantly higher than the contractual cash interest payments. During the three months ended March 31, 2020 and 2021, the Company recognized interest expense in the condensed statements of operations of zero and $37,000, respectively, related to the 2020 Convertible Notes.
On March 1, 2021, the Company entered into an amendment to the 2020 Convertible Notes which extends the maturity date of the 2020 Convertible Notes from March 31, 2021 to October 30, 2021 and provides for the conversion of the 2020 Convertible Notes into shares of the Company’s common stock upon a Qualified Financing that is an IPO. No other terms to the 2020 Convertible Notes were amended. This amendment was accounted for as a troubled debt restructuring pursuant to FASB ASC Topic 470-60, “Troubled Debt Restructurings by Debtors”. As the future undiscounted cash flows of the 2020 Convertible Notes were greater than their carrying amount, the carrying amount was not adjusted and no gain was recognized as a result of the modification of terms.
6. Promissory Note
On April 22, 2020, the Company entered into a promissory note with Silicon Valley Bank that provided for the receipt by the Company of loan proceeds of $140,000 (the “PPP Loan”) pursuant to the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). Under certain conditions, the loan and accrued interest are forgivable, if the loan proceeds are used for eligible purposes, including payroll, benefits, rent and utilities, and maintaining payroll levels. In October 2020, the Paycheck Protection Program Flexibility Act of 2020 extended the deferral period for borrower payments of principal, interest, and fees on all PPP loans from 6 months to 10 months. As of December 31 2020, payments were deferred for 10 months. The PPP Loan matures on April 22, 2022 and bears interest at a rate of 1.0% per annum. The PPP Loan contains events of default and other provisions customary for a loan of this type. The Company has recorded the PPP Loan as a promissory note in the December 31, 2020 balance sheet as both a current and non-current liability.
On February 6, 2021, the Company received notification and confirmation from Silicon Valley Bank that its PPP loan and related accrued interest have been forgiven in their entirety by the U.S. Small Business Administration and automatically cancelled. During the three-months ended March 31, 2021, a gain on loan extinguishment of $140,000 was recognized and included in other income (expense), net in the condensed statements of operations.
7. Warrant Liability
In conjunction with the January 2013 Series A-1 convertible preferred stock financing, the Company issued a warrant to purchase 26,838 shares of the Company’s Series A-1 convertible preferred stock at $0.9315 per share. The warrant was to expire on the earlier of (a) the date that was seven (7) years after the date of the original issuance of the warrant, (b) the date of consummation of an acquisition or (c) the effective date of an IPO. The warrant was exercised on January 23, 2020 for proceeds of $25,000. Upon exercise, the warrant liability associated with this warrant was adjusted to its fair value of $35,000 and the fair value was subsequently transferred to mezzanine equity as of the date of exercise.
| F-40 |
8. Common Stock
The Company’s common stock reserved for future issuance related to the convertible preferred stock and common stock options as of March 31, 2020 and 2021 are as follows:
| As of March 31, | ||||||||
| 2020 | 2021 | |||||||
| Series A-1 convertible preferred stock | 708,533 | 708,533 | ||||||
| Series A-2 convertible preferred stock | 709,219 | 709,219 | ||||||
| Series A-3 convertible preferred stock | 532,046 | 532,046 | ||||||
| Series B convertible preferred stock | 2,522,292 | 2,522,292 | ||||||
| Common stock options outstanding | 1,002,575 | 951,133 | ||||||
| Common stock options reserved for issuance | 22,393 | 8,968 | ||||||
| Total | 5,497,058 | 5,432,191 | ||||||
The shares that would be issued upon a conversion of the 2020 Convertible Notes have been excluded from the table above as the number of common shares that would be issued is contingent upon the occurrence of a future Qualified Financing and the conversion price per share would be equal to 80% of the price per share paid by the investors in the future Qualified Financing.
9. Stock-Based Compensation
Activity under the 2013 Plan for the three-month period ended March 31, 2021 is set forth below (in thousands except shares, per share amounts and years):
Shares Available for Grant | Number of Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Life (Years) | Aggregate Intrinsic Value | ||||||||||||||||
| Outstanding at December 31, 2020 | 12,893 | 997,266 | $ | 0.40 | 5.84 | $ | 276 | |||||||||||||
| Options granted | (10,800 | ) | 10,800 | 0.80 | ||||||||||||||||
| Options exercised | - | (50,058 | ) | 0.70 | ||||||||||||||||
| Options forfeited | 6,875 | (6,875 | ) | 0.65 | ||||||||||||||||
| Outstanding at March 31, 2021 | 8,968 | 951,133 | $ | 0.40 | 5.41 | $ | 369 | |||||||||||||
| Vested
and exercisable at March 31, 2021 | 830,028 | $ | 0.40 | 5.05 | $ | 352 | ||||||||||||||
| Vested
and expected to vest at March 31, 2021 | 951,133 | $ | 0.40 | 5.41 | $ | 369 | ||||||||||||||
During the three-month period ended March 31, 2020, the Company recognized $10,000 in stock-based compensation expense from stock option grants. During the three-month period ended March 31, 2021, the Company recognized $8,000 in stock-based compensation expense. The compensation expense is allocated on a departmental basis, based on the classification of the option holder. No income tax benefits have been recognized in the condensed statements of operations for stock-based compensation arrangements.
The following sets forth the weighted average assumptions used to determine the fair value of stock options for the three months ended March 31, 2020 and 2021:
Three Months Ended March 31, | ||||||||
| 2020 | 2021 | |||||||
| Expected term (years) | 4.64 | 5.51 | ||||||
| Risk-free interest rate | 1.60 | % | 0.72 | % | ||||
| Volatility factor | 37 | % | 42 | % | ||||
| Dividend yield | - | - | ||||||
The weighted-average fair value of options granted was $0.25 per share and $0.30 per share in the three-months ended March 31, 2020 and 2021, respectively.
Future stock-based compensation for unvested employee options granted and outstanding as of March 31, 2021 is $30,000, to be recognized over a weighted-average remaining requisite service period of 1.46 years.
10. Income Tax
The Company had no income tax expense for the three months ended March 31, 2020 and 2021. During the three months ended March 31, 2020 and 2021, the Company had a net operating loss (“NOL”) for each period that generated deferred tax assets for NOL carryforwards. Deferred income tax assets and liabilities are recognized for temporary differences between the financial statements and income tax carrying values using tax rates in effect for the years such differences are expected to reverse. Due to uncertainties surrounding our ability to generate future taxable income and consequently realize such deferred income tax assets, the Company has determined that it is more-likely-than-not that these deferred tax assets will not be realized. Accordingly, the Company has established a full valuation allowance against its deferred tax assets as of March 31, 2021.
The Company’s policy is to recognize any interest and penalties related to unrecognized tax benefits as a component of income tax expense. As of the three months ended March 31, 2020 and 2021, the Company had no accrued interest or penalties related to uncertain tax positions.
| F-41 |
11. Net Loss per Share
Basic and diluted net loss per common share was calculated as follows (in thousands except per share amounts):
Three Months Ended March 31, | ||||||||
| 2020 | 2021 | |||||||
| Numerator: | ||||||||
| Net loss | $ | (1,051 | ) | $ | (1,148 | ) | ||
| Denominator: | ||||||||
| Weighted average shares used in computing net loss per share – basic and diluted | 2,195 | 2,242 | ||||||
| Net loss per share – basic and diluted | $ | (0.48 | ) | $ | (0.51 | ) | ||
For the three-month periods ended March 31, 2020 and 2021, the Company had a net loss and as such, all outstanding shares of potentially dilutive securities were excluded from the calculation of diluted net loss per share as the inclusion would be anti-dilutive.
Potentially dilutive securities not included in the computation of diluted net loss per share because to do so would be antidilutive are as follows (in common stock equivalent shares):
Three Months Ended March 31, | ||||||||
| 2020 | 2021 | |||||||
| Convertible preferred stock | 3,535,469 | 3,535,469 | ||||||
| Options to purchase common stock | 1,002,575 | 951,133 | ||||||
| 4,538,044 | 4,486,602 | |||||||
The table above omits the potentially dilutive shares into which the 2020 Convertible Notes would convert.
12. Related Party Transactions
In January 2013, the Company entered into a consulting agreement (the “Agreement”) with one of the Company’s co-founders whereby the co-founder provides consulting services as the Company’s Chief Medical Officer (“CMO”) by overseeing Company-sponsored clinical trials. An amendment to the Agreement was signed in 2019 providing cash compensation to the CMO of $10,000 per month for additional services. Consulting fees paid to the CMO were $30,000 for each of the three-month periods ended March 31, 2020 and 2021.
In July 2019, the Company entered into a consulting agreement (the “CFO Agreement”) with the Company’s Chief Financial Officer (“CFO”) providing for compensation of $50 per hour. In February 2020, the Company granted the CFO an option to purchase 28,000 shares of the Company’s common stock, of which 25% were vested at the grant date and the remaining 75% vested ratably over 18 months. The Company entered into an amendment to the Agreement in December 2020 increasing the cash compensation to $150 per hour. Consulting fees paid to the CFO were $12,000 and $54,000 for the three months ended March 31, 2020 and 2021, respectively.
Another co-founder of the Company has served as our consulting technical engineering advisor on manufacturing and intellectual property matters since January 2020. Previously he was CEO of the Company from inception in December 2009 until January 2013; Chief Technical and Operations Officer from January 2013 until January 2019; and Chief Technology Officer from January 2019 to January 2020. He currently receives cash compensation quarterly compensation of $3,000.
13. Subsequent Events
The Company has evaluated all events occurring through August 11, 2021, the date on which the unaudited condensed interim financial statements were issued.
In April 2021, the Company entered into a note purchase agreement and a series of convertible note payable agreements (the “2021 Convertible Notes”) for aggregate borrowings of approximately $2.0 million. Outstanding borrowings under the 2021 Convertible Notes and accrued interest are due in April 2022, if not previously converted. The 2021 Convertible Notes bear interest at the rate of 5% per annum. Pursuant to the 2021 Convertible Notes, the outstanding principal and accrued interest are automatically convertible into equity shares a Qualified Financing at a conversion price per share equal to 87.5% of the price per share paid by investors purchasing such equity securities in a Qualified Financing.
In June 2021, the Company amended the 2013 Plan to increase the number of shares of common stock reserved for issuance by 52,000 and also granted 57,400 stock options with a weighted average exercise price of $2.45 per share and a grant fair value of approximately $56,000.
In connection with preparing for its initial public offering, the Company filed a certificate of amendment to its Fifth Amended and Restated Certificate of Incorporation to effect a 1-for-5 reverse stock split of its issued and outstanding preferred stock and common stock, which became effective on August 5, 2021. Adjustments corresponding to the reverse stock split were made to the ratio at which the Company’s convertible preferred stock will convert into the Company’s common stock. Accordingly, all share and per share amounts related to the common stock, stock options, warrants and restricted stock awards for all periods presented in the accompanying financial statements and notes thereto have been retroactively adjusted, where applicable, to reflect the reverse stock split.
| F-42 |
![]()
1,850,000 Units, Each Consisting of One Share of Common Stock and
Three-Quarter Warrant to Purchase One Share of Common Stock
1,850,000 Pre-funded Units, Each Consisting of a Pre-funded Warrant to
Purchase One Share
of Common Stock and Three-Quarter Warrant to Purchase
One Share of Common Stock
PROSPECTUS
Sole Book-Running Manager
Roth Capital Partners
Lead Manager
Maxim Group LLC
The date of this prospectus is , 2021
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
The following table sets forth the costs and expenses, other than underwriter fees to be paid by us in connection with the sale of our securities being registered hereby. All amounts are estimates except for the SEC registration fee, the FINRA filing fee and the NASDAQ listing fee. All such expenses will be borne by us.
| SEC registration fee | $ | 6,050 | ||
| FINRA filing fee | 8,818 | |||
| NASDAQ listing fee | 55,000 | |||
| Legal fees and expenses | 250,000 | |||
| Accounting fees and expenses | 250,000 | |||
| Printing and engraving expenses | 10,000 | |||
| Transfer agent and registrar fees and expenses | 5,000 | |||
| Other expenses | 15,132 | |||
| Total | $ | 600,000 |
| Item 14. | Indemnification of Directors and Officers. |
Section 102(b)(7) of Delaware’s General Corporation Law (“DGCL”) allows a corporation to provide in its certificate of incorporation that a director of the corporation will not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, except where the director breached the duty of loyalty, failed to act in good faith, engaged in intentional misconduct or knowingly violated a law, authorized the payment of a dividend or approved a stock repurchase in violation of Delaware corporate law or obtained an improper personal benefit. Our amended and restated certificate of incorporation provides for this limitation of liability.
Section 145 of the DGCL, or Section 145, provides that a Delaware corporation may indemnify any person who was, is or is threatened to be made, party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person is or was an officer, director, employee or agent of such corporation or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who are, were or are a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person is or was a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit, provided such person acted in good faith and in a manner he reasonably believed to be in or not opposed to the corporation’s best interests, provided that no indemnification is permitted without judicial approval if the officer, director, employee or agent is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him against the expenses which such officer or director has actually and reasonably incurred.
| -121- |
Section 145 further authorizes a corporation to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the corporation or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation or enterprise, against any liability asserted against him and incurred by him in any such capacity, or arising out of his or her status as such, whether or not the corporation would otherwise have the power to indemnify him under Section 145.
Our amended and restated bylaws provide that we must indemnify our directors and officers to the fullest extent permitted by the DGCL and must also pay expenses incurred in defending any such proceeding in advance of its final disposition upon delivery of an undertaking, by or on behalf of an indemnified person, to repay all amounts so advanced if it should be determined ultimately that such person is not entitled to be indemnified.
We will enter into indemnification agreements with certain of our executive officers and directors pursuant to which we will agree to indemnify such persons against all expenses and liabilities incurred or paid by such person in connection with any proceeding arising from the fact that such person is or was an officer or director of our company, and to advance expenses as incurred by or on behalf of such person in connection therewith.
The indemnification rights set forth above shall not be exclusive of any other right which an indemnified person may have or hereafter acquire under any statute, provision of our certificate of incorporation, our bylaws, agreement, vote of stockholders or disinterested directors or otherwise.
We maintain standard policies of insurance that provide coverage (1) to our directors and officers against loss rising from claims made by reason of breach of duty or other wrongful act and (2) to us with respect to indemnification payments that we may make to such directors and officers.
| Item 15. | Recent Sales of Unregistered Securities |
During the past three years, we issued the following securities, which were not registered under the Securities Act.
In March 2020, the Company entered into a note purchase agreement for the issuance of up to $4.0 million of convertible promissory notes. The Company entered into a series of convertible note payable agreements (the “2020 Convertible Notes”) for aggregate borrowings of approximately $3.0 million. Outstanding borrowings under the 2020 Convertible Notes and accrued interest are due in March 2021, if not previously converted. The 2020 Convertible Notes bear interest at the rate of 5% per annum. The 2020 Convertible Notes cannot be prepaid prior to the maturity date unless approved in writing by the Company and requisite holders.
On April 22, 2020, the Company entered into a promissory note with Silicon Valley Bank that provided for the receipt by the Company of loan proceeds of $140,000 (the “PPP Loan”) pursuant to the Paycheck Protection Program under the Coronavirus Aid, Relief and Economic Security Act (the “CARES Act”). Under certain conditions, the loan and accrued interest are forgivable, if the loan proceeds are used for eligible purposes, including payroll, benefits, rent and utilities, and maintaining payroll levels. In October 2020, the Paycheck Protection Program Flexibility Act of 2020 extended the deferral period for borrower payments of principal, interest, and fees on all PPP loans from 6 months to 10 months. As of December 31, 2020, payments were deferred for 10 months. The PPP Loan matures on April 22, 2022 and bears interest at a rate of 1.0% per annum.
In April 2021, the Company entered into a note purchase agreement and a series of convertible note payable agreements (the “2021 Convertible Notes”) for aggregate borrowings of approximately $2.0 million. Outstanding borrowings under the 2021 Convertible Notes and accrued interest are due in April 2022, if not previously converted. The 2021 Convertible Notes bear interest at the rate of 5% per annum. Pursuant to the 2021 Convertible Notes, the outstanding principal and accrued interest are automatically convertible into equity shares in a Qualified Financing at a conversion price per share equal to 87.5% of the price per share paid by investors purchasing such equity securities in a Qualified Financing.
We deemed the offers, sales and issuances of the securities described above to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act, including Regulation D and Rule 506 promulgated thereunder, relative to transactions by an issuer not involving a public offering.
| -122- |
| Item 16. | Exhibits and Financial Statement Schedules |
* Previously filed
+ Portions of this exhibit (indicated by asterisks) have been redacted in compliance with Regulation S-K Item 601(b)(10)(iv).
| -123- |
| Item 17. | Undertakings |
(a) The undersigned registrant hereby undertakes as follows:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the Commission pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement;
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser, each prospectus filed pursuant to Rule 424(b) as part of a registration statement relating to an offering, other than registration statements relying on Rule 430B or other than prospectuses filed in reliance on Rule 430A, shall be deemed to be part of and included in the registration statement as of the date it is first used after effectiveness. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such first use, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such date of first use.
(5) That, for the purpose of determining any liability under the Securities Act of 1933 to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant;
| -124- |
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or our securities provided by or on behalf of the undersigned registrant; and
(iv) Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser.
(b) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the undersigned pursuant to the foregoing provisions, or otherwise, the undersigned has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the undersigned of expenses incurred or paid by a director, officer or controlling person of the undersigned in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the undersigned will, unless in the opinion of our counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
| -125- |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, RenovoRx, Inc., a Delaware corporation, has duly caused this Registration Statement on Form S-1 to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Los Altos, state of California, on August 11, 2021.
| RENOVORX, INC. | ||
| By: | /s/ Shaun R. Bagai | |
| Name: | Shaun R. Bagai | |
| Title: | Chief Executive Officer | |
Pursuant to the requirements of the Securities Act of 1933, this Amendment to Registration Statement on Form S-1 has been signed by the following persons in the capacities indicated.
| SIGNATURE | TITLE | DATE | ||
| /s/ Shaun R. Bagai | Chief Executive Officer and Director | |||
| Shaun R. Bagai | (Principal Executive Officer) | August 11, 2021 | ||
* |
Vice President, Finance | |||
| Paul Manners | (Principal Financial Officer) | August 11, 2021 | ||
| * | ||||
| Ramtin Agah | Director | August 11, 2021 | ||
* |
||||
| Laurence J. Marton | Director | August 11, 2021 | ||
* |
||||
| Una S. Ryan | Director | August 11, 2021 | ||
| Maky Zanganeh | Director | August 11, 2021 | ||
* |
||||
| Kristen Angela Macfarlane | Director | August 11, 2021 | ||
* |
||||
| David Diamond | Director | August 11, 2021 |
| *By: | /s/ Shaun Bagai | |
| Attorney-in-Fact |
| -126- |