Attached files
| file | filename |
|---|---|
| EX-10.1(A) - EXHIBIT 10.1(A) - Brooklyn ImmunoTherapeutics, Inc. | brhc10024430_ex10-1a.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): May 6, 2021
BROOKLYN IMMUNOTHERAPEUTICS, INC.
(Exact Name of Registrant as Specified in its Charter)
|
Delaware
|
001-11460
|
31-1103425
|
|
(State or Other Jurisdiction of Incorporation)
|
(Commission File Number)
|
(IRS Employer Identification No.)
|
|
140 58th Street, Building A, Suite 2100
|
||
|
Brooklyn, New York
|
11220
|
|
|
(Address of Principal Executive Offices)
|
(Zip Code)
|
Registrant’s telephone number, including area code: (212) 582-1199
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the
following provisions:
| ☐ |
Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425)
|
| ☐ |
Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12)
|
|
☐
|
Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b))
|
|
☐
|
Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c))
|
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Trading symbol
|
Name of each exchange on which registered
|
||
|
Common Stock, par value $0.005 per share
|
BTX
|
NYSE American
|
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 or Rule 12b-2 of the Securities Exchange Act of 1934:
Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards
provided pursuant to Section 13(a) of the Exchange Act. ☐
| Item 5.02. |
Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.
|
Appointment of Erich Mohr as Director
On May 6, 2021, the board of directors elected Erich Mohr, Ph.D, to serve as a member of the board, with a term effective on May 7, 2021 and continuing
until our 2021 annual meeting of stockholders. It is expected that Dr. Mohr will be appointed to the board’s compensation committee.
Dr. Mohr serves as the Chairman of Oak Bay Biosciences Inc., a developmental biotechnology company focused on treatments for Stargardt disease. Since 2006
Dr. Mohr has been the founder, Chairman and Chief Executive Officer of MedGenesis Therapeutix Inc., a biopharmaceutical company that commercializes treatments for Parkinson’s disease. From 2002 to 2005, Dr. Mohr served as the Executive Vice
President and Chief Scientific Officer of PRA International, a clinical research organization that provided drug development services to pharmaceutical and biotechnology companies. From 1995 to 2002, Dr. Mohr was the founder, Chairman and Chief
Executive Officer of CroMedica International, which later merged with PRA International. Dr. Mohr received a MSc and Ph.D from University of Victoria, British Columbia, Canada, and a BA and BSc from University of the Pacific. He is 67 years old.
There are no family relationships between Dr. Mohr and any of our existing directors or our executive officers, and Dr. Mohr has not had any direct or
indirect material interest in any transaction required to be disclosed pursuant to Item 404(a) of Regulation S-K under the Securities Exchange Act of 1934.
Resignation of Yiannis Monovoukas as Director
On May 6, 2021, Yiannis Monovoukas notified the board of directors of his intention to resign as a director effective in connection with the board’s
identification and election of an additional new director. Mr. Monovoukas’s resignation as a director was effective as of May 7, 2021, upon the appointment of Erich Mohr to the board.
The resignation of Mr. Monovoukas was not the result of any disagreement relating to our operations, policies or practices.
2
| Item 8.01 |
Other Events.
|
In the discussions below, “Brooklyn Inc.” refers to Brooklyn ImmunoTherapeutics, Inc. (formerly known as NTN Buzztime, Inc.) and “Brooklyn LLC” refers to Brooklyn ImmunoTherapeutics LLC, a wholly
owned subsidiary of Brooklyn Inc. All references to “our company,” “we,” “us” or “our” mean Brooklyn Inc. and its subsidiaries, including Brooklyn LLC, unless stated otherwise or the context otherwise requires.
On March 25, 2021, BIT Merger Sub, Inc., a wholly owned subsidiary of Brooklyn Inc. (then known as NTN Buzztime, Inc.) merged with and into Brooklyn LLC, with Brooklyn LLC surviving as a wholly
owned subsidiary of Brooklyn Inc.. This transaction, which we refer to as the Merger, was completed in accordance with the terms of an agreement and plan of merger and reorganization dated August 12, 2020, or the Merger Agreement, among Brooklyn
Inc. (then known as NTN Buzztime, Inc.), BIT Merger Sub, Inc. and Brooklyn LLC.
On March 26, 2021, Brooklyn Inc. sold its rights, title and interest in and to the assets relating to the business it operated prior to the Merger, which it had operated under the name “NTN
Buzztime, Inc.,” to eGames.com Holdings LLC, or eGames.com, in exchange for eGames.com’s payment of a purchase price of $2.0 million and assumption of specified liabilities relating to such pre-Merger business. This transaction, which we refer to
as the Asset Sale, was completed in accordance with the terms of an asset purchase agreement dated September 18, 2020, as amended, or the Asset Purchase Agreement, between Brooklyn Inc. and eGames.com.
Following the completion of the Merger and the Asset Sale, our business consists exclusively of the business conducted by our wholly owned subsidiary Brooklyn LLC.
Business of Brooklyn LLC
We are a clinical-stage biopharmaceutical company focused on exploring the role that cytokine-based therapy can have on the immune system in treating patients with cancer,
both as a single agent and in combination with other anti-cancer therapies. We are seeking to develop IRX‑2, a novel cytokine-based therapy, to treat patients with cancer. IRX‑2 active constituents, namely Interleukin-2, or IL‑2, and other key
cytokines, are postulated to signal, enhance and restore immune function suppressed by the tumor, thus enabling the immune system to attack cancer cells, unlike existing cancer therapies, which rely on targeting the cancer directly. We also are
exploring opportunities to advance oncology, blood disorder, and monogenic disease therapies using gene-editing cell‑therapy technology through a license with Factor Biosciences Limited and Novellus Therapeutics Limited, which we refer to
collectively as the Licensor.
The development of our product candidates could be disrupted and materially adversely affected by the continuing COVID-19 pandemic. As a result of measures imposed by the
governments in affected regions, businesses and schools have been suspended due to quarantines intended to contain this outbreak. The spread of SARS CoV‑2 from China to other countries resulted in the Director General of the World Health
Organization declaring COVID-19 a pandemic in March 2020. While the constraints of the pandemic are slowly being lifted, we are still assessing the longer term impact of the COVID-19 pandemic on our development plans, and on the ability to conduct
our clinical trials There can be no assurance that this analysis will enable us to avoid or remediate part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally. The extent to
which the COVID-19 pandemic and global efforts to contain its spread will impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the
pandemic and the actions taken to contain or treat the COVID-19 pandemic.
The patients in our clinical trials have conditions that make them especially vulnerable to COVID-19, and as a result we have seen slowdowns in enrollment in our clinical
trials. While our Phase 2b clinical study in patients with squamous cell carcinoma of the oral cavity, known as the INSPIRE study, is fully populated, our other clinical studies are likely to continue to encounter delays as a result of the
pandemic. Further, with respect to the INSPIRE study, we anticipate that the COVID-19 pandemic will slow our ability to close out trial sites and report trial data.
IRX‑2 is a therapy based on IL‑2, a type of cytokine-signaling molecule in the immune system. While many of the mechanisms of action of COVID-19 are still unknown, there is
evidence that for some patients, severe COVID-19 patients may result in “cytokine storm syndrome,” in which the body releases cytokines into the body too quickly, which can create symptoms such as high fever, inflammation, severe fatigue and nausea
and can lead to severe or life-threatening symptoms.
3
In June 2020 the Journal of Medical Virology published a letter submitted by Wen Luo, Jia-Wen Zhang, Wei Zhang, Yuan-Long Lin and Qi
Wang, supported by grants from the State Key Laboratory of Veterinary Technology, Harbin Veterinary Research Institute, stating that, based on a review of 25 patients admitted to intensive care units with a confirmed infection of COVID-19, cytokine
storm of a number of interleukins, including IL‑2, was absent. The letter therefore suggested that the severity of COVID-19 symptoms is not directly associated with circulating levels of IL‑2. There can be no assurance, however, that further study
will bear this out or that patients treated with IRX‑2, who are already at higher risk for COVID-19 due to their underlying diagnosis, will not be adversely affected.
IRX‑2 is a primary human cell-derived biological medicinal product containing multiple active cytokine components acting as immunomodulators. It is prepared from the
supernatant of pooled allogeneic peripheral blood mononuclear cells, known as PBMNCs, that have been stimulated using a proprietary process employing a specific population of cells and a specific mitogen.
While IRX‑2 is a cytokine mixture, one of its principal active components is IL‑2, a cytokine-signaling molecule in the immune system. IL‑2 is a protein that regulates the
activities of white blood cells (leukocytes and often lymphocytes) that are responsible for immunity. IL‑2 is part of the body’s natural response to microbial infection, and in discriminating between foreign (“non-self”) and “self,” IL‑2 mediates
its effects by binding to IL‑2 receptors, which are expressed by lymphocytes. The major sources of IL‑2 are activated CD4+ T cells and activated CD8+ T cells.
Unlike existing recombinant IL‑2 therapies, IRX‑2 is naturally derived from human blood cells. This potentially may promote better tolerance, broader targeting and a natural
molecular conformation leading to greater activity, and may permit low physiologic dosing, rather than the high doses needed in other existing IL‑2 therapies. Our ongoing development program is specifically investigating use of IRX‑2 in neoadjuvant
(pre-surgical) and adjuvant (post-operative) treatment for advanced head and neck squamous cell carcinoma, or HNSCC. IRX‑2 has received both fast track designation and orphan drug designation from the U.S. Food and Drug Administration, or FDA, for
this indication. Potential use of our product candidate in other cancer indications is also being evaluated in several investigator-sponsored trials. Finally, we are currently modifying our manufacturing process to allow us to develop additional
drugs with a variety of cytokine mixtures to expand our product offerings.
Our product candidate IRX‑2 currently remains under development and has not yet been approved for marketing authorization in any jurisdiction. The ongoing development program
is investigating use of IRX‑2 as an immunotherapeutic neoadjuvant (pre-surgical) and adjuvant (post-operative) treatment for advanced HNSCC and other indications.
The HNSCC development program is being conducted under FDA Investigational New Drug, or IND, #11,137 filed on June 30, 2003 and is ongoing. Potential use of IRX‑2 in other
cancer indications is also being conducted by independent clinical researchers as investigator‑initiated trials.
The HNSCC program has received fast track designation, approved November 7, 2003, and orphan drug designation, conferred on July 7, 2005, from the FDA. We have not submitted a
request for orphan drug designation in the European Union, or EU, though we may seek such designation in the future.
Our findings to date from nonclinical studies of IRX‑2 include murine acute toxicology as well as acute and chronic primate studies. These studies detected circulating
associated cytokines yet were associated with benign toxicological findings. A further murine study demonstrated PD/PDL‑1 synergy when additively administered with IRX‑2.
Clinical studies in humans involving IRX‑2 show immune marker activation in patients treated with IRX‑2. In a prior clinical trial, a correlation was shown between marker
activation and disease-free survival in head and neck cancer.
4
Our clinical pipeline of therapeutic studies focused on oncology indications of high unmet medical need includes:
| • |
Monotherapy studies:
|
| • |
INSPIRE, a Phase 2B study involving 105 patients with HNSCC. Details of this trial can be found at clinicaltrials.gov (NCT02609386).
|
| • |
BR-101 - A study involving 16 patients with neoadjuvant breast cancer performed at the Providence Portland Medical Center. Details of this trial can be found at clinicaltrials.gov (NCT02950259).
|
| • |
CIN-201 - An open label single arm Phase 2 trial of the IRX‑2 regimen in women with cervical squamous intraepithelial neoplasia 3 or squamous vulvar intraepithelial neoplasia 3. Details of this trial can be found at clinicaltrials.gov (NCT03267680).
|
| • |
Combination studies:
|
| • |
BAS-104 - A basket study originally intended to enroll 100 patients with metastatic bladder, renal, non-small cell lung cancer (NSCLC), melanoma, and head and neck cancer being held at the Moffitt Cancer Center, using IRX‑2 in
conjunction with Opdivo (Nivolumab), an immunotherapy cancer treatment marketed by Bristol-Myers Squibb Company. Details of this trial can be found on clinicaltrials.gov (NCT03758781).
|
| • |
HCC-107 - A study involving 28 patients with metastatic hepatocellular carcinoma, HCC, being held at HonorHealth Research Institute, City of Hope Medical Center and Texas Oncology at Baylor Charles A. Simmons Cancer Center using IRX‑2 in
conjunction with Opdivo, a cancer treatment marketed by Bristol-Myers Squibb Company. Details of this trial can be found at clinicaltrials.gov (NCT03655002).
|
| • |
GI-106 - A study involving 20 patients with metastatic gastric and gastroesophageal junction cancers (GI) being held at HonorHealth Research Institute, City of Hope Medical Center and Texas Oncology at Baylor Charles A. Simmons Cancer
Center using IRX‑2 in conjunction with Keytruda (Pembrolizumab), an immunotherapy cancer treatment marketed by Merck. Details of this trial can be found at clinicaltrials.gov (NCT03918499).
|
| • |
MHN-102 - A study involving 15 patients with metastatic head and neck cancer being held at the H. Lee Moffitt Cancer Center and Research Institute and University of Michigan Health System using IRX‑2 in conjunction with Imfinzi
(Durvalumab), a cancer treatment marketed by AstraZeneca plc. Details of this trial can be found at clinicaltrials.gov (NCT03381183).
|
| • |
BR-202 - A study involving 30 patients with neoadjuvant triple negative breast cancer, held at the Providence Portland Medical Center using IRX‑2 in conjunction with a programmed cell death protein 1 (PD1) and chemotherapy treatments.
Details of this trial can be found at clinicaltrials.gov (NCT04373031).
|
Other than the INSPIRE study, each of the studies described above is an investigator-sponsored study for which we are providing IRX‑2 as study drug and financial support to
conduct the trial.
Our goal is twofold: to rapidly advance our IRX‑2 platform to become a leader in immunologic therapy for various types of cancer as both a first-line therapy and in combination with other cancer
treatments, and to commercialize the gene editing technology licensed from the Licensors:
| • |
Pursue commercialization of gene-editing technology. Develop analog mRNA based technology and proprietary delivery system licensed from the Licensors for gene therapy, and cellular engineering in
the treatment of indications of high unmet medical need in oncology and other conditions.
|
5
| • |
Advance combination trials with checkpoint inhibitors. Once INSPIRE trial are released, we plan to use those results as a catalyst in addition to six other clinical trials with multiple data
read-outs anticipated in 2022 and later.
|
| • |
Pursue partnerships to advance our clinical program. We are pursuing partnering opportunities with leading biopharmaceutical companies for the development and commercialization of IRX‑2.
|
| • |
Opportunistically in-license/acquire complementary programs. We may seek additional products to license or acquire in order to expand our product pipeline. This includes products that we may seek
to develop if we exercise our option to exclusively license certain additional technology from the Licensors.
|
| • |
Regulatory Strategy. We believe that our assets may be deemed to be unique and to represent potential breakthroughs in cancer treatment. We will endeavor to seek breakthrough therapy designation
with regulatory agencies for IRX‑2 for one or more indications and for any other product we may acquire or in‑license that could potentially lead to accelerated clinical development timelines. We cannot, however, assure you that we will
receive breakthrough therapy designation for any indications or that any breakthrough therapy designation we do receive will necessarily lead to a faster approval time.
|
| • |
Intellectual Property. We continue to pursue additional intellectual property based on data from IRX clinical studies.
|
IRX‑2 Technology
IRX‑2 is an allogeneic, reproducible, primary, cell-derived biologic with multiple active cytokine components that act on various parts of the immune system to activate the
tumor microenvironment. IRX‑2 contains multiple human cytokines that promote or enhance an immune response. IRX‑2 is administered as a subcutaneous injection in proximity to lymph node beds harboring metastatic cancer.
IRX‑2 is produced under current good manufacturing practices, or cGMP, following stimulation of a specific population of human peripheral blood mononuclear cells, or PMBC,
using a specified mitogen. These cells consist of lymphocytes (T cells, B cells, NK cells) and monocytes. Cytokine production induced by the employed mitogen mimics that observed after brisk stimulation of human immune cells by an immunogenic
pathogen or an infection. PBMCs are obtained from FDA-licensed blood banks meeting all criteria for further human use.
Early immunotherapeutic approaches to cancer tended to oversimplify the immune system, often based on the hope that a single target or receptor might restore cellular immune
responses. Today we know that the immune system represents a complex interaction of cells. These include the cells that interact to create immunization, antigen‑presenting cells called dendritic cells, or DCs, and different types of T cells
required for an anticancer immune response. It is now known that defects in these cells exist in cancer and that these defects must be reversed to generate effective cellular immune responses. In addition, because tumors induce immune suppression
through multiple mechanisms, next-generation active immunotherapies must also effectively counteract these mechanisms of tumor-induced immune suppression. For these reasons, we believe the multiple cytokines present in IRX‑2 may be demonstrated to
be particularly effective in reversing the multiple immune deficits in cancer patients.
Preclinical data from animal and in vitro studies, as well as biologic data from patients in clinical trials, demonstrate that IRX‑2 acts in multiple ways to augment the
immune response. The following illustration shows how IRX‑2 is believed to use its multiple active cytokine components to enhance the immune response:
6
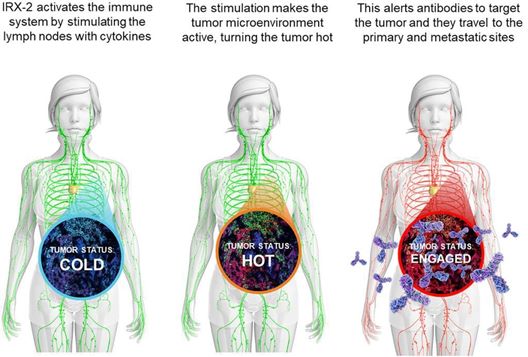
While the sample size in these studies was not statistically significant, data collected to date suggest that IRX‑2 reduces the immune suppression that is often seen in the
cancer tumor microenvironment held to be associated with reduced immune recognition of solid tumors mediated by surface proteins. This immunomodulatory activity appears to occur through the restoration of immune function and activation of a
coordinated immune response against the tumor. IRX‑2 contains numerous active cytokine components, which is believed to restore and activate multiple immune cell types including T cells, dendritic cells, and natural killer cells to recognize and
destroy tumors.
These data are derived from the following three studies of IRX‑2:
| • |
The Phase 2a study described below in “Historical Background of the INSPIRE Study,” the results of which were published in December 2011 in the journal Head and Neck. The article reported that
IRX‑2 showed an immunologically mediated antitumor effect, suggested by pronounced lymphocytic infiltration seen in some tumors and by the tumor reductions observed at the end of the 21-day regimen in 11 patients, and by means of a 75%
reduction of glycolytic activity in the tumor and lymph nodes on post-treatment PET scans in one patient.
|
| • |
The BR-101 early stage breast cancer study described below, the results of which were published in April 2020 in the journal Clinical Cancer Research. The article reported that IRX‑2 showed
potentially favorable immunomodulatory changes, supporting further study of IRX‑2 in early-stage breast cancer and other malignancies.
|
| • |
The INSPIRE study described below in “INSPIRE Phase 2B Study Details,” the results of which were published in December 2020 in the journal Oral Oncology. Paired biopsy and resection specimens
from 62 patients were available for creation of tissue microarray, or TMA, and multiplex immunohistology, or IMH, analysis. In this protocol, tumor infiltrating lymphocyte, or TIL, counts by TMA were performed manually under 200x
magnification by a technician blinded to clinical outcome. Additional details of the methodology of TMA and IMH are provided in the publication. Treatment with IRX‑2 was associated with significant increases in CD8+ infiltrates
(p=0.01) compared to the control arm, as shown in the graph below.
|
7
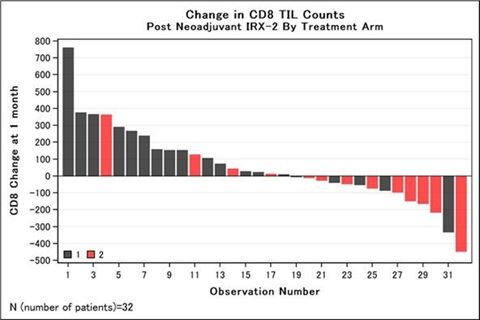
Increases in CD8+ TIL infiltrate scores of at least 10 cells/mm2 were used to characterize immune responders based on prior findings that each 10-cell
increase in CD8 cell density was associated with a significant decrease in multivariable hazard risk of death from study of biopsy specimens obtained from over 200 oral cavity patients treated surgically (Ref 1, 2 below). Immune responders were
more frequent in the IRX arm than the control arm (74% vs 31%, p=0.01), as shown in the table below.
Change in Mean ( ± SEM) Immune Cell Counts1 in Primary Tumor
Before and After Neoadjuvant Immunotherapy with (Regimen 1) or without (Regimen 2) IRX‑2
Before and After Neoadjuvant Immunotherapy with (Regimen 1) or without (Regimen 2) IRX‑2
|
Overall
n= 32 |
Regimen 1
n= 19 |
Regimen 2
n= 13 |
p16+
n=6 |
p16-
n= 26 |
|||||||||||||||||||||||||
|
Mean
|
SEM
|
Mean
|
SEM
|
Mean
|
SEM
|
Mean
|
SEM
|
Mean
|
SEM
|
||||||||||||||||||||
|
CD4
|
14.4
|
20.1
|
29.3
|
31.2
|
-7.4
|
19.2
|
-2.6
|
57.1
|
18.3
|
21.5
|
|||||||||||||||||||
|
CD8
|
56.8
|
40.7
|
131.3
|
52.8
|
-52.2
|
52.4
|
-75.8
|
61
|
87.3
|
46.4
|
|||||||||||||||||||
|
FOXP3
|
37.8
|
43.7
|
59.5
|
42.3
|
6.2
|
89.8
|
-68
|
45.1
|
62.3
|
51.9
|
|||||||||||||||||||
|
CD20
|
-8.4
|
29.1
|
-5.3
|
48.6
|
-12.8
|
14.8
|
-111.3
|
128.9
|
15.4
|
20.2
|
|||||||||||||||||||
|
CD68
|
24.4
|
19.7
|
-7
|
20.1
|
70.2
|
35.8
|
-21.2
|
27.7
|
34.9
|
23.1
|
|||||||||||||||||||
|
TILws2
|
36.2
|
29.5
|
74
|
35.4
|
-19
|
48.6
|
-47.8
|
48
|
55.7
|
33.8
|
|||||||||||||||||||
|
1
|
Cell counts are per mm2.
|
|
2
|
TIL weighted score.
|
|
1-
|
http://refhub.elsevier.com/S1368-8375(20)30364-X/h0155
|
|
2-
|
[33] Spector ME, Bellile E, Amlani L, Zarins K, Smith J, Brenner JC, Rozek L, Nguyen A, Thomas D, McHugh JB, Taylor JMG, Wolf GT, the University of Michigan Head and Neck SPORE Program5. Tumor infiltrating lymphocytes are prognostic
in HNSCC. JAMA Otolaryngol Head Neck Surg. 2019 Sep 5. doi: 10.1001/jamaoto.2019.2427. [Epub ahead of print]. PMID: 31486841.
|
The data from the INSPIRE Phase 2B trial described above are interim data available under the trial protocol in August 2019 (approximately 1.5 years after all patients
received their neoadjuvant regimen and had surgery completed). The primary endpoint of the INSPIRE trial is event-free survival of study subjects who received treatment for their cancer which included IRX‑2, compared to subjects whose treatment for
their cancer did not include IRX‑2. The final trial results on all primary, secondary and exploratory endpoints will be assessed four years after randomization of the last patient (which occurred in February 2018) and is expected to be reported in
the first half of 2022.
8
About Interleukin 2
One of a group of related proteins made by leukocytes (white blood cells) and other cells in the body. IL-2 is made by a type of T lymphocyte. It increases the growth and
activity of other T lymphocytes and B lymphocytes and affects the development of the immune system.
IL‑2 is classified as a “biologic response modifier,” or BRM, or “biologic therapy.” BRMs modify the body’s response to cancer cells. IL-2 is part of a family of proteins
called cytokines. Cytokines act primarily by communicating between the various cells of the body’s immune system.
IL‑2 has essential roles in key functions of the immune system, tolerance and immunity, primarily via its direct effects on T cells. In the thymus, where T cells mature, it
prevents autoimmune diseases by promoting the differentiation of certain immature T cells into regulatory T cells, which suppress other T cells that are otherwise primed to attack normal healthy cells in the body, arguably mitigating off target
effects associated with immune system activation . IL‑2 enhances activation-induced cell death, or AICD. IL‑2 also promotes the differentiation of T cells into effector T cells and into memory T cells when the initial T cell is also stimulated by
an antigen, thus helping the body fight off infections. Together with other polarizing cytokines, IL‑2 stimulates naive CD4+ T cell differentiation into Th1 and Th2 lymphocytes while it impedes differentiation into Th17 and follicular Th
lymphocytes.
IL‑2’s expression and secretion are tightly regulated and function as part of both transient positive and negative feedback loops in mounting and dampening immune responses.
Through its role in the development of T cell immunologic memory, which depends upon the expansion of the number and function of antigen-selected T cell clones, IL‑2 plays a key role in enduring cell-mediated immunity.
Traditional high dose, recombinant IL‑2 therapies have demonstrated severe side effects, including:
| • |
flu-like symptoms (fever, headache, muscle and joint pain, fatigue);
|
| • |
nausea/vomiting;
|
| • |
dry, itchy skin or rash;
|
| • |
weakness or shortness of breath;
|
| • |
diarrhea;
|
| • |
low blood pressure;
|
| • |
drowsiness or confusion; and
|
| • |
loss of appetite.
|
More serious and dangerous side effects sometimes are seen, such as breathing problems, serious infections, seizures, allergic reactions, heart problems, kidney failure or a
variety of other possible complications. The most common adverse effect of high-dose IL‑2 therapy is vascular leak syndrome (VLS; also termed capillary leak syndrome). It is caused by lung endothelial cells expressing high-affinity IL‑2R. These
cells, as a result of IL‑2 binding, causes increased vascular permeability. Thus, intravascular fluid extravasates into organs, predominantly lungs, which leads to life-threatening pulmonary or brain edema. Other drawbacks of traditional
recombinant IL‑2 cancer immunotherapy are its short half-life in circulation and its potential to predominantly expand regulatory T cells at high doses.
Unlike traditional drugs based on IL‑2, IRX‑2 is derived from human blood cells, or hu IL‑2. This difference confers several distinct advantages:
| • |
IRX‑2 has been well tolerated at doses that have been used in clinical trials to date.
|
| • |
IRX‑2 takes advantage of multiple cytokines, while traditional IL‑2 only utilizes a single cytokine.
|
| • |
IRX‑2 is designed to allow for natural folding believed to lead to greater activity, while traditional IL‑2 has shown abnormal folding impacting functionality.
|
| • |
IRX‑2 is administered subcutaneously at physiologic doses, while traditional IL‑2 utilizes high doses which potentially contributes to the severe side effects noted above. Physiologic dosing allows for the
traditional dose to be reduced to levels that allow for the replacement of normal IL‑2 levels in the body.
|
9
The INSPIRE Study
Historical Background of the INSPIRE Study
The IRX‑2 regimen has been studied in patients with HNSCC in two previous open-label, multi-center studies. Also, a phase 1 trial evaluated the IRX‑2 regimen as a therapy for
advanced disease, which reported that the IRX‑2 regimen was well tolerated.
In 2011, results were reported for a Phase 2a trial of IRX‑2. This trial was an open-label study involving 27 patients, 26 of whom completed the study. The primary endpoint of
the study was to further evaluate the safety and efficacy of the immunotherapy regimen including IRX‑2 in the neoadjuvant setting in previously untreated patients with advanced (Stage II to IVa) HNSCC. The primary study objective was to demonstrate
the safety of this immunotherapy regimen based on adverse events (AEs), changes in clinical laboratory measures (hematology, chemistry, and urinalysis), vital signs, and physical examinations. Secondary objectives were clinical, pathologic, and
radiographic tumor response; and patient disease-free survival (DFS) and overall survival (OS).
Most recombinant cytokines, such as IL‑2, are tested in the same manner as traditional oncology drugs, where the maximum tolerated dose is sought. Typical cytokine therapies
in cancer treatment use extremely high doses, in the millions of units per administration. Thus, AEs such as fever, hypotension, malaise, anemia, leukopenia and hepatic and renal dysfunction are commonly reported, and often lead to discontinuation
of the treatment. IV administration of cytokines is frequently associated with an acute phase reaction characterized by rigors, fever, an increase in neutrophils, a decrease in lymphocytes and changes in hormone levels. By contrast, the IRX‑2
regimen, which contains physiologic quantities of cytokines, showed greatly improved tolerability over typical recombinant cytokine therapies.
The results of the Phase 2a study were published in December 2011 in the journal Head and Neck. The article reported that IRX‑2
showed an immunologically mediated antitumor effect, suggested by pronounced lymphocytic infiltration. Lymphocytic infiltration, or LI, was measured by a 100-mm Visual Analog Scale, or VAS, score, in which 100 mm signified lymphocyte infiltration
of the entire primary tumor section and 0 mm signified no lymphocyte infiltration in the tumor specimen. The mean VAS score for all 24 patients was 22.6 mm on the samples obtained at surgery. Patients were grouped into low VAS score (below the
overall mean) and high VAS score (above the overall mean) cohorts. There were 14 patients in the low VAS score cohort with scores between 2 and 21 (median of 9.5), and 10 patients in the high VAS score cohort with scores between 27 and 66 (median
of 37.0). Patients in the high-LI group included fewer oral cavity patients (50% in high LI vs 60% in low LI) but were similar with respect to tumor sites. Seventy percent of high-LI patients were stage IV, whereas only 60% of low LI were stage IV.
The LI score was used to determine whether the degree of LI correlated with survival. Patients with a high-LI score had an improved survival trend compared to those with low LI, and superior to the survival rate for the combined overall group. (See
below.)

10
Interestingly, LI in resected tumor specimens was considered high in 40% of the patients. The 10 patients with a high-LI score showed an improved survival trend in comparison
to the low-LI group (n = 15) and to the entire study population (n = 26). It is difficult to directly compare these subgroups, because there was some imbalance, with a slightly higher percentage of oral cavity patients in the low-LI group. In the
absence of a randomized control, however, it is impossible to directly attribute the LI to the immuno-therapy regimen. In addition, tumor reductions were observed at the end of the 21-day regimen in 11 patients, and a 75% reduction of glycolytic
activity in the tumor and lymph nodes on post-treatment PET scans was observed in one patient.
With regard to the primary endpoint, eight serious adverse events, or SAEs, were reported during treatment and the 30-day post-operative period in 7 patients, including 3
patients with aspiration pneumonia, 1 patient with asthma exacerbation secondary to upper respiratory infection, 1 patient with a post-operative wound infection, 1 patient with a neck abscess, and 1 patient with an episode of alcohol withdrawal.
Only 1 case of aspiration pneumonia was deemed life threatening (grade 4). None of the SAEs was considered related to treatment except for the post-operative wound infection, which was considered possibly related. Other minor (grade 1 or 2) adverse
events included headache (30%), injection-site pain (22%), nausea (22%), constipation (15%), dizziness (15%), fatigue (11%) and myalgia (7%).
After over more than 36 months of follow-up, 11 of the 27 patients enrolled in the Phase 2a study had experienced tumor relapse (n =
1) or death (n = 10). The pattern of first HNSCC relapse included 3 patients with primary site recurrence, 2 with recurrences in the neck, and 2 with distant metastases. Of the 10 patients who died, 6 died
of cancer (1 from a new primary) and 4 died of other causes. The 1-year, 2-year, and 3-year DFS probabilities after surgery were 72%, 64%, and 62%, respectively. Of the 26 patients whose primary tumor was resected surgically, 2 patients died during
the first year and 5 patients died during the second year after surgery. The probability of surviving after surgery was 92% the first year, 73% the second year, and 69% the third year, which was considered to be an encouraging survival rate
compared to historical norms in patients with HNSCC.
A second finding of the study was that some tumors showed some decrease in overall size after the immunotherapy regimen. Overall tumor shrinkage was modest, although in 4
patients, independent, objective imaging documented a greater than 10% decrease in tumor size. This was unexpected and encouraging after only 3 weeks of presurgical neoadjuvant immunotherapy. No patient achieved a true partial response by modified
Response Evaluation Criteria in Solid Tumors, or RECIST, criteria. Increases in tumor measurements were also seen in some patients, but most patients showed negligible change in tumor dimensions. We believe that these findings suggest the safety of
the neoadjuvant regimen, although final decisions on whether a drug product is safe and effective can only be made by the FDA.
The phase 2a trial did not include a randomized control cohort. The manuscript published in Head and Neck in December 2011 stated the
authors’ belief, however, that the safety results and feasibility of this immunotherapy regimen were intriguing enough to warrant further study and appropriate comparison in a randomized trial.
INSPIRE Phase 2B Study Details.

11
The INSPIRE study is intended to further evaluate neoadjuvant therapy with the IRX‑2 regimen in a randomized trial. In addition, the immunotherapy regimen will be enhanced by
adding four adjuvant booster courses of a shorter IRX‑2 regimen during the first post-operative year.
The INSPIRE study is an open label, randomized, multi-center, multi-national Phase 2b clinical trial intended for patients with Stage II, III or IVA untreated SCC of the oral cavity who are
candidates for resection with curative intent. Subjects were randomized 2:1 to either Regimen 1 or Regimen 2 and treated for 21 days prior to surgery and then post-operatively with a booster regimen given every 3 months for 1 year (a total of 4
times.)
| • |
Regimen 1: IRX‑2 Regimen with cyclophosphamide, indomethacin, zinc-containing multivitamins, omeprazole and IRX‑2 as neoadjuvant and adjuvant therapy.
|
| • |
Regimen 2: Regimen 1 with cyclophosphamide, indomethacin, zinc-containing multivitamins, omeprazole but without IRX‑2 as neoadjuvant and adjuvant therapy.
|
Treatments were allocated to study subjects using minimization with a stochastic algorithm based on the range method. Minimization will account for the major prognostic
factors for SCC of the oral cavity (T and N stage) and study center to avoid imbalances in treatment allocation within centers.
Postoperatively, subjects first received standard adjuvant radiation or chemoradiation therapy as determined by the investigators per National Comprehensive Cancer Network
Clinical Practice Guidelines in Oncology, referred to as NCCN guidelines, and then also received Booster Regimen 1 or 2 as determined in the prior randomization.
Subjects will be followed for the Primary, Secondary and Exploratory endpoints. Protocol mandated follow-up will end 4 years after randomization of the last patient.
The Neoadjuvant IRX‑2 Regimen is a 21-day pre-operative regimen of cyclophosphamide on Day 1, indomethacin, zinc-containing multivitamins and omeprazole on Days 1-21, and
subcutaneous IRX‑2 injections in bilateral mastoid insertion regions for 10 days between Days 4 and 21, as shown in the table below:
|
Agent
|
Dose
|
Route of Administration
|
Treatment Days
|
|||
|
Cyclophosphamide
|
300 mg/m2
|
IV
|
1
|
|||
|
IRX‑2
|
230 units daily (Bilateral injections of 115 units)
|
Subcutaneous at or near the mastoid insertion of both sternocleidomastoid muscles
|
Any 10 days between Days 4 and 21
|
|||
|
Indomethacin
|
25 mg TID
|
Oral
|
1-21
|
|||
|
Zinc-Containing Multivitamins
|
1 tablet containing 15-30 mg of zinc
|
Oral
|
1-21
|
|||
|
Omeprazole
|
20 mg
|
Oral
|
1-21
|
12
The Booster IRX‑2 Regimen is given at 3, 6, 9 and 12 months (-14 to +28 days) after surgical resection. It is a 10-day post-operative regimen of cyclophosphamide on Day 1,
indomethacin, zinc-containing multivitamins and omeprazole on Days 1-10 and subcutaneous IRX‑2 injections in bilateral deltoid regions for 5 days between Days 4 and 10 as shown in the table below:
|
Agent
|
Dose
|
Route of Administration
|
Treatment Days
|
|||
|
Cyclophosphamide
|
300 mg/m2
|
IV
|
1
|
|||
|
IRX‑2
|
230 units daily (Bilateral injections of 115 units)
|
Subcutaneous into bilateral deltoid regions
|
Every 3 months.
Any 5 days between Days 4 and 10 |
|||
|
Indomethacin
|
25 mg TID
|
Oral
|
Every 3 months.
Days 1-10 |
|||
|
Zinc-Containing Multivitamins
|
1 tablet containing 15-30 mg of zinc
|
Oral
|
Every 3 months.
Days 1-10 |
|||
|
Omeprazole
|
20 mg daily
|
Oral
|
Every 3 months.
Days 1-10 |
Regimen 2, the control arm of the study, is identical, except that subjects will not receive IRX‑2.
The primary objective of the study is to determine if the event-free survival (EFS) of subjects treated with Regimen 1 is longer than for subjects treated with Regimen 2. The
secondary objections of the study are to (i) determine if OS of subjects treated with Regimen 1 is longer than for subjects treated with Regimen 2, (ii) compare the safety of each Regimen, and (iii) compare the feasibility of each booster regimen.
BR-101 – An open label single arm Phase 1 trial of the IRX‑2 Regimen in early-stage breast cancer. Sixteen subjects with stage I-III
early stage-breast cancer (any histology type) indicated for surgical lumpectomy or mastectomy received 10 days of 230 international units, or IUs, IRX‑2 prior to surgery. The objectives of the study were feasibility, safety, and to assess changes
in stromal tumor-infiltrating lymphocyte score. Preoperative locoregional cytokine administration was feasible in 100% (n = 16/16) of subjects and associated with increases in stromal tumor–infiltrating lymphocytes (P < 0.001). Programmed death
ligand 1 (CD274) was upregulated at the RNA (P < 0.01) and protein level by Ventana PD-L1 (SP142) and immunofluorescence, showing statistical significance. Other immunomodulatory effects included upregulation of RNA signatures of T-cell
activation and recruitment and cyclophosphamide-related peripheral T-regulatory cell depletion. The study concluded that IRX‑2 is well tolerated in early-stage breast cancer and, while Phase 1 trials are generally designed only to test safety and
are not powered to assess efficacy, potentially favorable immunomodulatory changes were observed in this trial, supporting further study of IRX‑2 in early-stage breast cancer and other malignancies.
The primary endpoint of this trial was the number of surgeries delayed due to adverse events from the IRX‑2 regimen, and the secondary endpoint was the change in TIL score as
measured by hematoxylin and eosin TIL count, according to Salgado criteria (a criteria for evaluating TIL set forth by Dr. Roberto Salgado published in the February 2015 edition of the journal Annals of Oncology)
from pre-surgical biopsy to resected tumor specimen. Further information about these endpoints is available on clinicaltrials.gov (NCT02950259).
CIN-201 - An open label single arm Phase 2 trial of the IRX‑2 Regimen in women with cervical intraepithelial neoplasia 3 or squamous
vulvar intraepithelial neoplasia 3. Ten subjects will be treated in the Cervical Interstitial Neoplasia, or CIN arm, if 2 or fewer responses are observed, enrollment will be stopped. If 3 or more responses are seen enrollment, will continue to 22
subjects. Five subjects will be treated in the Vulvar Interstitial Neoplasia, or VIN arm, if there is no response observed, enrollment will be stopped. If one or more responses are seen, enrollment will continue to 10 subjects. Patients will be
treated with 4 days of 230 IU IRX‑2 for a total of 2 cycles (each cycle is 6 weeks). Objective responses will be assessed at Week 25 to determine the percentage of patients that achieve pathologic Complete Response or Partial Response and Human
Papillomavirus, or HPV, status will be assessed at 3 months post-surgical incision.
13
The primary endpoint of this trial is to compare the proportion of subjects who achieve a pathologic complete response, or CR, or partial response, or PR, in regimen 1 versus
regimen 2 at week 25, based on the resected surgical specimen. The secondary endpoints are (i) the evaluation of the toxicity and feasibility of the administration of IRX‑2 in subjects with confirmed CIN or VIN and (ii) the evaluation of multiple
parameters to assess the activity of the IRX‑2 regimen for the treatment of CIN or VIN, including (a) the occurrence of clinical CRs or PRs at weeks 6, 13 and 25; (b) the frequency of elimination of HPV in cervical or vulvar tissue using a
commercial HPV genotyping assay and viral load determination by quantitative polymerase chain reaction, or PCR, (c) analysis of the immune infiltrates in the resected surgical specimens, (d) immunophenotypic analysis of peripheral blood
lymphocytes, (e) frequency of serum antibodies to HPV E6, E7, and L1 proteins by enzyme-linked immunosorbent assay, and (f) ribonucleic acid, or RNA, expression profiling of immune-inflammatory markers from post-treatment resected surgical
specimens. Further information about these endpoints is available on clinicaltrials.gov (NCT03267680)
BAS-104 – A Phase 1b open label trial to evaluate the safety, determine recommended Phase 2 dose, and investigate biologic and
clinical activity in recurrent/metastatic lung, renal, bladder, melanoma, and head and neck cancers. Twenty patients were intended to be enrolled into each of the 5 tumor types (for a total of 100 subjects), 10 which were to be naïve to PD1/PDL1
treatment and 10 of which were to have previously received PD1/PDL1 treatment. Patients will be treated with Nivolumab (on label per approved guidelines) in combination with IRX‑2 for a total of 6 cycles (each cycle is 12 weeks and begins with a 21
day cycle of IRX‑2 treatment). The safety phase of the study consisted of 12 patients, of which 6 will be dosed at 230IU of IRX‑2 and followed for 3 weeks for safety assessment before the next cohort of 6 subjects is enrolled and dosed at 460IU of
IRX‑2 and followed for 3 weeks for safety assessment. If no significant safety issues are noted, the remaining subjects in the trial will be dosed at the 460IU dose of IRX‑2. Objective responses are measured by RECIST 1.1 and iRECIST criteria every
six weeks.
The primary endpoint of this trial is the number of participants who experience dose-limiting toxicities through Day 28. The secondary endpoints are (i) the objective response
rate, determined using RECIST 1.1 and iRECIST criteria, and (ii) progression-free survival as defined by RECIST 1.1 to be the time from Day 1 of treatment to the evidence of progression. Further information about these endpoints is available on clinicaltrials.gov (NCT03758781).
This study was closed to enrollment in the second quarter of 2021 due to strategic realignment within BTX and reimbursement issues at the institutional sponsor after enrolling
11 subjects in the trial. The data are currently being collected and biomarker samples are being analyzed for reporting on the subjects treated in the trial.
HCC-107 – A Phase 1b open label trial of IRX‑2 in combination with Nivolumb (on label per approved guidelines) in patients with
advanced hepatocellular cancer. Twenty eight patients will be enrolled into the study. Patients who were previously treated with PD1/PDL1 agents are not excluded from the trial. Patients will be treated with Nivolumb in combination with IRX‑2 for a
total of 18 cycles (each cycle is 1 month). The study is a standard 3+3 design, where three subjects are enrolled and treated at the 230IU of IRX‑2 and followed for 4 weeks for safety assessment before the next cohort of 3 subjects are enrolled and
dosed at 460IU of IRX‑2 and followed for 4 weeks for safety assessment. If no significant safety issues are noted, the remaining subjects in the trial will be dosed at the 460IU dose of IRX‑2. Objective responses are measured by RECIST 1.1 and
iRECIST criteria every 8-12 weeks and progression free survival, progression free survival at 6 months, and overall survival will be assessed.
The primary endpoint of this trial is to determine the safety profile of combination IRX‑2 regimen and Nivolumab in anti-PD1/PDL1 naïve patients who have failed or not
tolerated at least one line of treatment. The secondary endpoints are (i) to evaluate the overall response rate of the IRX‑2 regimen combined with Nivolumab using RECIST 1.1 and iRECIST criteria, (ii) to evaluate the rate of 6-month
progression-free survival in patients treated with combination IRX‑2 regimen and Nivolumab, and (iii) to evaluate median progression-free survival and overall survival. Further information about these endpoints is available on clinicaltrials.gov
(NCT03655002).
MHN-102 – A Phase 1b open label trial of IRX‑2 in combination with Durvalumab (on label per approved guidelines) in patients with
incurable HNSCC. Fifteen evaluable patients will be enrolled into the study. Patients who were previously treated with PD1/PDL1 agents are not excluded from the trial; however. given current treatment paradigms. it is anticipated that the majority
of subjects in this trial will have received prior PD1/PDL1 treatment. Patients will be treated with Durvalumab in combination with IRX‑2 for a total of 12 cycles (each cycle is 4 weeks). The safety phase of the study will consist of 12 patients. 6
that will be dosed at 230IU of IRX‑2 and followed for 4 weeks for safety assessment before the next cohort of 6 subjects are enrolled and dosed at 460IU of IRX‑2 and followed for 4 weeks for safety assessment. If no significant safety issues are
noted, the remaining subjects in the trial will be dosed at the 460IU dose of IRX‑2. Biopsies will be collected pre and post treatment to assess changes in immune activity. Objective responses are measured by RECIST 1.1 and iRECIST criteria every
2-3 months and progression free survival and progression free survival at 6 months.
14
The primary endpoint of this trial is to determine the safety profile of combination IRX‑2 regimen and Durvalumab. The secondary endpoints are (i) to evaluate the overall
response rate of the IRX‑2 regimen combined with pembrolizumab using RECIST 1.1 and iRECIST criteria, and (ii) to evaluate initial median progression-free and overall survival in patients treated with combination IRX‑2 regimen and pembrolizumab.
Further information about these endpoints is available on clinicaltrials.gov (NCT03918499).
This study was originally designed to treat 24 subjects in the trial, but was downsized to 15 evaluable subjects due to strategic realignment within BTX and the partner
company.
GI-106 – A Phase 1b/2 open label trial of IRX‑2 in combination with Pembrolizumab in patients with advanced gastric and
gastroesophageal junction adenocarcinoma. Twenty six patients will be enrolled into the study. Patients who were previously treated with PD1/PDL1 are excluded from the study. Patients will be treated with Pembrolizumab in combination with IRX‑2 for
a total of 35 cycles (each cycle is 21 days). The study is a standard 3+3 design, where three subjects are enrolled and treated at the 230IU of IRX‑2 and followed for 3 weeks for safety assessment before the next cohort of 3 subjects are enrolled
and dosed at 460IU of IRX‑2 and followed for 3 weeks for safety assessment. If no significant safety issues are noted, the remaining subjects in the trial will be dosed at the 460IU dose of IRX‑2. Objective responses are measured by RECIST 1.1 and
iRECIST criteria every 6-9 weeks and response rate and median progression free survival will be assessed.
The primary endpoint of this trial is to determine the maximum tolerated dose of the combination of the IRX‑2 regimen and Pembrolizumab as outlined in the treatment arm. The
secondary endpoints are (i) objective response to the combination of the IRX‑2 regimen and Pembrolizumab, which will be documented using standard RECIST criteria, (ii) progression-free survival of IRX‑2/ Pembrolizumab treatment participants at six
months post-treatment, as defined by RECIST 1.1 to be the time from Day 1 of treatment to evidence of progression, (iii) median progress free survival of IRX‑2/Pembrolizumab treatment participants, defined as the time from day 1 of treatment to
evidence of progression as defined by RECIST 1.1, and (iv) median overall survival of IRX‑2/Pembrolizumab treatment participants, defined as the length of time from the start of treatment that patients are still alive. Further information about
these endpoints is available on clinicaltrials.gov (NCT03381183).
BR-202 – A randomized, controlled, Phase 2 trial of induction lRX-2 immunotherapy to promote immunologic priming and enhanced response
to neoadjuvant Pembrolizumab plus chemotherapy in triple negative breast cancer. The trial just recently commenced. Thirty patients will be enrolled into the study (15 into the control arm and 15 into the experimental arm). Patients in the control
arm will receive Pembrolizumab plus paclitaxel plus doxorubicin plus cyclophosphamide. Patients in the experimental arm will receive the same as the control arm plus lRX-2. The primary endpoint of this trial is to assess the pathological complete
response rate of Pembrolizumab plus chemotherapy and Pembrolizumab plus IRX‑2 plus chemotherapy. Secondary endpoints include assessment of rate of response based on residual cancer burden index and to evaluate stromal tumor infiltrating lymphocyte
quantity. Further information about these endpoints is available on clinicaltrials.gov (NCT04373031).
The status of each of these trials is set forth below.
|
Protocol
|
Status/number of patients
|
Date of commencement
|
Estimated final trial data availability*
|
|||
|
BR-101
|
Complete/ 16
|
Second quarter 2016
|
N/A (completed and published)
|
|||
|
MHN-102
|
Ongoing/16
|
Third quarter 2019
|
Second quarter 2022
|
|||
|
BAS-104
|
Ongoing/11
|
First quarter 2019
|
Fourth quarter 2023
|
|||
|
GI-106
|
Ongoing/9
|
Second quarter 2019
|
Third quarter 2022
|
|||
|
HCC-107
|
Ongoing/8
|
First quarter 2019
|
Fourth quarter 2022
|
|||
|
BR-202
|
Ongoing/5
|
Fourth quarter 2020
|
Second quarter 2024
|
|||
|
CIN-201
|
Ongoing/10
|
First quarter 2018
|
Third quarter 2023
|
* Interim data may be available sooner.
As we are not the sponsors of these trials, we do not have access to a full listing of all reported AEs for each of the trials. However, to date no SAEs have been reported to
us which were determined to be related to IRX‑2.
15
License and Royalty Agreements
IRX‑2
Unless otherwise stated below, each royalty to be paid under these license and royalty agreements is payable until the last patent for IRX‑2 expires and
runs in perpetuity unless earlier terminated pursuant to the terms described below. There are no milestone payments due under any of these agreements.
License Agreement with the University of South Florida Research Association
On June 28, 2000, IRX Therapeutics, a predecessor to Brooklyn LLC, entered into a series of License Agreements, which we refer to collectively and as amended as the USF
License Agreement, with the University of South Florida Research Association, Inc., or the Research Association. Pursuant to the USF License Agreement, the Research Association licensed the exclusive worldwide rights to certain patents on IRX‑2 in
exchange for royalties equal to 7% of the gross product sales of IRX‑2 (as defined in the USF License Agreement). The USF License Agreement was assigned to Brooklyn LLC in connection with the sale of the assets of IRX Therapeutics to Brooklyn LLC
in November 2018. The Research Association has the right to terminate (i) upon Brooklyn LLC’s entering into bankruptcy or insolvency on a voluntary or involuntary basis, (ii) upon the failure to pay royalties due and payable upon thirty days’
notice, or (iii) upon a material breach or default of the Agreement by Brooklyn LLC, unless such breach or default is cured within a thirty day notice period. Brooklyn LLC may terminate the USF License Agreement for any reason upon six months’
notice to the Research Association.
Royalty Agreement with certain former IRX Therapeutics investors
On May 1, 2012, IRX Therapeutics entered into a royalty agreement, which we refer as the IRX Investor Royalty Agreement, with certain investors who participated in a financing
transaction. The IRX Investor Royalty Agreement was assigned to Brooklyn LLC in November 2018 when Brooklyn LLC acquired the assets of IRX Therapeutics. Pursuant to the IRX Investor Royalty Agreement, when Brooklyn LLC becomes obligated to pay
royalties to the Research Association under the USF License Agreement, it will pay an additional royalty of 1% of gross sales to an entity organized by the investors who participated in such financing transaction. There are no termination
provisions in the IRX Investor Royalty Agreement.
Collaborator License Agreement
Effective June 28, 2018, IRX Therapeutics terminated its Research, Development and Option Facilitation Agreement and its Options Agreement with a collaborative partner, which
we refer to as the Collaborator, pursuant to a Termination Agreement. In connection with the Termination Agreement, all of the rights granted to the Collaborator under the Research, Development and Option Facilitation and the Option Agreement were
terminated, and IRX Therapeutics had no obligation to refund any payments received from the Collaborator. The Termination Agreement was assigned to Brooklyn LLC in connection with the sale of the assets of IRX Therapeutics to Brooklyn LLC in
November 2018.
As consideration for entering into the Termination Agreement, the Collaborator will receive a royalty equal to 6% of revenues from the sale of IRX‑2 for the period of time
beginning with the first sale of IRX‑2 through the later of (i) the twelfth anniversary of the first sale of IRX‑2 and (ii) the expiration of the last IRX patent or other exclusivity of IRX‑2, all as more particularly set forth in the Termination
Agreement. Each party under the Termination Agreement may terminate the agreement upon (i) a material breach of the Termination Agreement by the other party that is not cured within sixty days (or thirty days if such breach is due to Brooklyn LLC’s
non-payment of royalties) or (ii) the other party entering into bankruptcy on a voluntary or involuntary basis where such petition is not dismissed, discharged, bonded or stayed within ninety days.
Investor Royalty Agreement
On March 22, 2021, Brooklyn LLC entered into an Amended and Restated Royalty Agreement and Distribution Agreement, or the Royalty Agreement, with Brooklyn ImmunoTherapeutics
Investors GP LLC, or GP, Brooklyn ImmunoTherapeutics Investors LP, or LP, and certain beneficial holders of GP and LP. Pursuant to the Royalty Agreement, among other things, we are required to pay compensatory royalties equal to 4% of net revenues
of IRX‑2 on an annual basis, of which 3% is to be paid to certain beneficial holders of LP and 1% is to be paid to certain beneficial holders of GP. The royalty continues in perpetuity. The Royalty Agreement amends and restates a royalty agreement
Brooklyn LLC entered into with GP and LP on November 6, 2018.
16
License Agreements with the Licensor
On April 26, 2021 , Brooklyn LLC entered into an exclusive license agreement, or the License Agreement, with the Licensor to license the Licensor’s IP and mRNA cell
reprogramming and gene editing technology for use in the development of certain cell-based therapies to be evaluated and developed for treating human diseases, including certain types of cancer, sickle cell disease, and beta thalassemia. Through
the License Agreement, Brooklyn LLC acquired an exclusive worldwide license to develop and commercialize certain cell-based therapies to treat cancer and rare blood disorders, including sickle cell disease, based on patented technology and know-how
of Novellus Therapeutics Limited.
The License Agreement provides that Brooklyn LLC is obligated to pay the Licensor a total of $4,000,000 in connection with the execution of the License Agreement, of which
$2,500,000 has been paid and the remaining $1,500,000 is expected to be paid in July 2021. Brooklyn LLC is obligated to pay to the Licensor additional fees of $5,000,000 in October 2021 and $7,000,000 in October 2022.
Under the terms of the License Agreement, Brooklyn LLC is required to use commercially reasonably efforts to achieve certain delineated milestones, including specified
clinical development and regulatory milestones and specified commercialization milestones. In general, upon its achievement of these milestones, Brooklyn LLC will be obligated, in the case of development and regulatory milestones, to make milestone
payments to Licensor in specified amounts and, in the case of commercialization milestones, to specified royalties with respect to product sales, sublicense fees or sales of pediatric review vouchers. In the event Brooklyn LLC fails to timely
achieve certain delineated milestones, the Licensor may have the right to terminate the rights of Brooklyn LLC under provisions of the License Agreement relating to those milestones.
The Licensor is responsible for preparing, filing, prosecuting and maintaining all patent applications and patents under the License Agreement. If, however, the Licensor
determines not to maintain a particular licensed patent or not to prepare, file and prosecute a licensed patent, Brooklyn LLC will have the right, but not the obligation, to assume those responsibilities in the territory at its expense.
Novellus is a pre-clinical development, manufacturing, and technology licensing entity focused on engineered cellular medicines. Novellus has created, developed, and patented
mRNA-based cell reprogramming and gene editing technologies to create engineered cellular medicines. The synthetic mRNA developed by Novellus is non-immunogenic—it is capable of successfully evading the immune system while being recognized by
cellular processes. The synthetic mRNA is then capable of expressing high levels of proteins for cell reprogramming and gene editing. The mRNA may be formulated for injection into target tissues for cellular uptake and therapeutic treatment.
The synthetic mRNA technology may be used to edit gene mutations through mRNA chemistry or expressed gene-editing proteins to treat genetic and rare diseases. It may also be
used to reprogram human non-pluripotent cells and induce human pluripotent stem cells, or IPSCs. The IPSCs may then be differentiated into pure populations of varying therapeutic cell types. The reprogramming technology offers a rapid,
cost-effective and patient specific therapy using the engineered stem cells created from IPSCs.
Novellus has over 45 granted patents throughout the world covering synthetic mRNA, RNA-based gene editing, and RNA-based cell reprogramming, in addition to specific patents
covering methods for treating specific diseases. There are also greater than 50 pending patent applications throughout the world focused on these and other aspects of the technology. The patent coverage includes granted patents and pending patent
applications in the United States, Europe, and Japan along with other major life sciences markets.
There can be no assurance that Brooklyn LLC can successfully develop and commercialize the technology licensed under the License Agreement.
17
Our Patent Portfolio
As of January 15, 2021, we owned or controlled approximately 8 patent families filed in the United States and other major markets worldwide, including 99 granted, 10 pending
and 11 published patent applications, directed to novel compounds, formulations, methods of treatments and platform technologies. Patent protection for IRX‑2 includes:
|
Summary Description of
Patent or Patent Application |
Jurisdiction
|
Earliest Effective Date
of Patent Application |
|||
|
IRX‑2 Modified Manufacturing Process
|
Granted: US (No. 8,470,562), EP (BE, CH, DE, DK, ES, FI, FR, GB, IT, LI, NL, SW), AU, CA, JP, MX, TR
|
US: 4/14/2009
EP: 4/14/2009
|
|||
|
Method of Reversing Immune Suppression of Langerhans Cells
|
Granted: US (Nos. 9,333,238 and 9,931,378), EP (BE, CH, DE, DK, ES, FI, FR, GB, LI, NL), AU, CA
Published: CN, HK
|
US: 6/8/2012 (No. 9,333,238), 4/13/2016 (No. 9,931,378)
EP: 12/8/2010
|
|||
|
Method of Increasing Immunological Effect
|
Granted: US (Nos. 7,993,660 and 8,591,956), EP (BE, CH, DE, DK, ES, FI, FR, GB, IT, LI, NL), AU, CA, JP
Published: HK
|
US: 8/9/2011 (No. 7,993,660), 11/26/2013 (No. 8,591,956)
EP: 11/26/2008
|
|||
|
Vaccine Immunotherapy
|
Granted: US (Nos. 6,162,778, 9,492,517, 9,492,519, 9,539,320 and 9,566,331), EP (BE, CH, DE, DK, ES, FI, FR, GB, IT, LI, NL), AU, CA, HK, JP
|
US: 7/24/2007 (No. 6,162,778), 10/8/2009 (No. 9,492,517), 11/15/2011 (No. 9,539,230), 2/20/2013 (No. 9,566,331), 7/12/2013 (No. 9,492,519)
EP: 5/17/2010
|
|||
|
Vaccine Immunotherapy for Immune Suppressed Patients
|
Granted: US (Patent Nos. 6,977,072, 7,153,499, 8,784,796, 9,789,172 and 9,789,173), CA, JP
|
US: 10/26/2001 (No. 6,977,072), 5/5/2003 (No. 7,153,499), 7/12/2013 (No. 8,784,796), 6/4/2014 (Nos. 9,789,172 and 9,789,173
|
|||
|
Immunotherapy for Immune Suppressed Patients
|
Granted: EP (BE, CH, DE, ES, FR, GB, IT, NL, LI)
|
EP: 3/9/2007
|
|||
|
Composition for the Treatment of Advanced Prostate Cancer
|
Granted: CA
Published: EP, HK
|
||||
|
Uses of PD-1/PD-L1 Inhibitors and/or CTLA-4 Inhibitors with a Biologic Containing Multiple Cytokine Components |
Pending: AU, CA, EP, IL, JP, KR, NZ, PH, SG, US
Published: BR, CN, EA, IN, MX, ZA
|
||||
US – United States of America
EP – European Patent Convention
BE – Belgium
CH – Switzerland
DE – Germany
DK – Denmark
ES – Spain
FI – Finland
GB – Great Britain
IT – Italy
LI – Lichtenstein
NL – Netherlands
SW – Sweden
AU – Australia
BR - Brazil
CA – Canada
CN – Peoples’ Republic of China
EA – Eurasian Patent Organization
HK – Hong Kong
IL – Israel
IN - India
JP – Japan
KR – Republic of Korea (South Korea)
MX – Mexico
PH – Philippines
SG - Singapore
TR – Turkey
ZA – South Africa
18
Patent Families
Descriptions of our patent families with issued patents in the United States or EU are as follows:
| • |
IRX‑2 Modified Manufacturing Process - A method of making a primary cell derived biologic, including the steps of: (a) removing contaminating cells from mononuclear cells (MNCs) by loading leukocytes onto lymphocyte separation medium
(LSM), and washing and centrifuging the medium with an automated cell processing and washing system; (b) storing the MNCs overnight in a closed sterile bag system; (c) stimulating the MNCs with a mitogen and ciprofloxacin in a disposable
cell culture system to produce cytokines; (d) removing the mitogen from the mononuclear cells by filtering; (e) incubating the filtered MNCs in a culture medium; (f) producing a clarified supernatant by filtering the MNCs from the culture
medium; (g) producing a chromatographed supernatant by removing DNA from the clarified supernatant by anion exchange chromatography; and (h) removing viruses from the chromatographed supernatant by filtering with dual 15 nanometer filters
in series, thereby producing a primary cell derived biologic, wherein the primary cell derived biologic comprises IL-1.beta., IL‑2, and IFN-.gamma.
|
| • |
Method of Reversing Immune Suppression of Langerhans Cells - A method of treating human papillomavirus (HPV), by administering a therapeutically effective amount of a primary cell-derived biologic to a patient infected with HPV and
inducing an immune response to HPV. A method of overcoming HPV-induced immune suppression of Langerhans cells (LC), by administering a therapeutically effective amount of a primary cell-derived biologic to a patient infected with HPV and
activating LC. A method of increasing LC migration towards lymph nodes, by administering a therapeutically effective amount of a primary cell-derived biologic to a patient infected with HPV, activating LC, and inducing LC migration towards
lymph nodes. A method of generating immunity against HPV, by administering an effective amount of a primary cell derived biologic to a patient infected with HPV, generating immunity against HPV, and preventing new lesions from developing.
|
| • |
Method of Increasing Immunological Effect - A method of increasing immunological effect in a patient by administering an effective amount of a primary cell derived biologic to the patient, inducing immune production, blocking immune
destruction, and increasing immunological effect in the patient. Methods of treating an immune target, treating a tumor, immune prophylaxis, and preventing tumor escape.
|
| • |
Vaccine Immunotherapy/Composition for the Treatment of Advanced Prostate Cancer – A method providing compositions and methods of immunotherapy to treat cancer or other antigen-producing diseases or lesions. According to one embodiment of
the invention, a composition is provided for eliciting an immune response to at least one antigen in a patient having an antigen-producing disease or lesion, the composition comprising an effective amount of a cytokine mixture, preferably
comprising IL-1, IL‑2, IL-6, IL-8, IFN-.gamma. (gamma) and TNF-. alpha. (alpha). The cytokine mixture acts as an adjuvant with the antigen associated with the antigen-producing disease or lesion to enhance the immune response of the patient
to the antigen. Methods are therefore also provided for eliciting an immune response to at least one antigen in a patient having an antigen-producing disease or lesion utilizing the cytokine mixture of the invention. The compositions and
methods are useful in the treatment of antigen-producing diseases such as cancer, infectious diseases or persistent lesions.
|
| • |
Vaccine Immunotherapy for Immune Suppressed Patients - A method for overcoming mild to moderate immune suppression includes the steps of inducing production of naive T-cells and restoring T-cell immunity. A method of vaccine
immunotherapy includes the steps of inducing production of naive T-cells and exposing the naive T-cells to endogenous or exogenous antigens at an appropriate site. Additionally, a method for unblocking immunization at a regional lymph node
includes the steps of promoting differentiation and maturation of immature dendritic cells at a regional lymph node and allowing presentation of processed peptides by resulting mature dendritic cells, thus, for example, exposing tumor
peptides to T-cells to gain immunization of the T-cells. Further, a method of treating cancer and other persistent lesions includes the steps of administering an effective amount of a natural cytokine mixture as an adjuvant to endogenous or
exogenous administered antigen to the cancer or other persistent lesions.
|
| • |
Immunotherapy for Immune Suppressed Patients – A method providing compositions of a natural cytokine mixture (NCM) for treating a cellular immunodeficiency characterized by T lymphocytopenia, one or more
dendritic cell functional defects such as those associated with lymph node sinus histiocytosis, and/or one or more monocyte functional defects such as those associated with a negative skin test to NCM. The invention includes methods of
treating these cellular immunodeficiences using the NCM of the invention. The compositions and methods are useful in the treatment of diseases associated with cellular immunodeficiencies such as cancer. Also provided are compositions and
methods for reversing tumor-induced immune suppression comprising a chemical inhibitor and a non-steroidal anti-inflammatory drug (NSAID). The invention also provides a diagnostic skin test comprising NCM for predicting treatment outcome in
cancer patients.
|
19
Patent Term and Term Extensions
Individual patents have terms for varying periods depending on the date of filing of the patent application or the date of patent issuance and the legal term of patents in the
countries in which they are obtained. Generally, utility patents issued for applications filed in the United States and the EU are granted a term of 20 years from the earliest effective filing date of a non-provisional patent application. In
addition, in certain instances, a patent term can be extended to recapture a portion of the U.S. Patent and Trademark Office, or USPTO, delay in issuing the patent as well as a portion of the term effectively lost as a result of the FDA regulatory
review period. However, as to the FDA component, the restoration period cannot be longer than five years and the restoration period cannot extend the patent term beyond 14 years from FDA approval. The duration of foreign patents varies in
accordance with provisions of applicable local law, but typically are also 20 years from the earliest effective filing date. All taxes or annuities for a patent, as required by the USPTO and various foreign jurisdictions, must be timely paid in
order for the patent to remain in force during this period of time.
The actual protection afforded by a patent may vary on a product-by-product basis, from country to country, and can depend upon many factors, including the type of patent, the
scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Our patents and patent applications may be subject to procedural or legal challenges by others. We may be unable to obtain, maintain and protect the intellectual property
rights necessary to conduct our business, and we may be subject to claims that we infringe or otherwise violate the intellectual property rights of others, which could materially harm our business. For more information, see the section titled “Risk
Factors-Risks Related to Our Intellectual Property.”
Litigation
For purposes of the following descriptions, the “Pre-Merger Company” refers to Brooklyn Inc. while it operated as NTN Buzztime, Inc. prior to the Merger and prior to changing
its name to “Brooklyn ImmunoTherapeutics, Inc.”
Transaction between the Pre-Merger Company and Brooklyn LLC / Related Shareholder Litigation
The Pre-Merger Company and its former directors have been named as defendants in ten substantially similar actions brought by purported stockholders of the Pre-Merger Company
arising out of the Merger, which we refer to collectively as the Stockholder Actions:
| • |
Henson v. NTN Buzztime, Inc., et al., No. 1:20-cv-08663-LGS (S.D.N.Y.);
|
| • |
Monsour v. NTN Buzztime, Inc., et al., No. 1:20-cv-08755-LGS (S.D.N.Y.);
|
| • |
Amanfo v. NTN Buzztime, Inc., et al., No. 1:20-cv-08747-LGS (S.D.N.Y.);
|
| • |
Carlson v. NTN Buzztime, Inc., et al., No. 1:21-cv-00047-LGS (S.D.N.Y.);
|
| • |
Finger v. NTN Buzztime, Inc., et al., No. 1:21-cv-00728-LGS (S.D.N.Y.);
|
| • |
Falikman v. NTN Buzztime, Inc., et al., No. 1:20-cv-05106-EK-SJB (E.D.N.Y.);
|
| • |
Haas v. NTN Buzztime, Inc., et al., No. 3:20-cv-02123-BAS-JLB (S.D. Cal.);
|
| • |
Gallo v. NTN Buzztime, Inc., et al., No. 3:21-cv-00157-WQH-AGS (S.D. Cal.);
|
| • |
Chinta v. NTN Buzztime, Inc., et al., No. 1:20-cv-01401-CFC (D. Del.); and
|
| • |
Nicosia v. NTN Buzztime, Inc., et al., No. 1:21-cv-00125-CFC (D. Del.).
|
The Chinta and Nicosia cases also name Brooklyn LLC as a defendant.
These actions assert claims alleging violations of Sections 14(a) and 20(a) of the U.S. Securities Exchange Act of 1934 and Rule 14a-9 promulgated thereunder by the U.S. Securities and Exchange
Commission, or SEC, and in both the Chinta and Nicosia cases alleged that Brooklyn LLC was a controlling person of the Pre-Merger Company. The complaints generally allege that the defendants failed to disclose allegedly material information in a
Registration Statement on Form S-4 filed with the SEC by the Pre-Merger Company on October 2, 2020, including: (1) certain details regarding any projections or forecasts of the Pre-Merger Company or Brooklyn LLC may have made, and the analyses
performed by Newbridge Securities Corporation, the financial advisor to the Pre-Merger Company in connection with the Merger; (2) conflicts concerning the sales process; and (3) disclosures regarding whether or not
20
The Pre-Merger Company entered into any confidentiality agreements with standstill and/or “don’t ask, don’t waive” provisions. The complaints generally allege that these purported failures to
disclose rendered the Form S-4 false and misleading. The complaints request: preliminary and permanent injunction of the Merger; rescission of the Merger and/or rescissory damages in unspecified amounts; direction to the individual directors to
disseminate a compliant Registration Statement; an accounting by the Pre-Merger Company for all alleged damages suffered; a declaration that certain federal securities laws have been violated; and costs, including attorneys’ and expert fees and
expenses. On or about February 26, 2021, in order to moot certain of the disclosure claims asserted in the Stockholder Actions, to avoid nuisance, potential expense, and delay, and to provide additional information to its stockholders, the
Pre‑Merger Company determined to voluntarily supplement the Proxy Statement with certain additional disclosures. In exchange for those disclosures, the Plaintiffs in each of the ten actions agreed to voluntarily dismiss their claims. All ten
actions have now been dismissed. The parties are presently attempting to resolve a request of plaintiffs’ counsel for an award of attorneys’ fee and expenses based on the purported benefit they contend was conferred on the Pre-Merger Company’s
stockholders as a result of the supplemental disclosures. If agreement cannot be reached, plaintiffs’ counsel have reserved their right to seek a fee and defendants have reserved their right to challenge any such fee application.
Dhesh Govender v. Brooklyn ImmunoTherapeutics, LLC, et al., Index No. 650847/2021 (N.Y. Sup. Ct. N.Y. Cty. 2021)
On or about February 5, 2021, Dhesh Govender, a former short-term consultant of Brooklyn LLC, filed a complaint against Brooklyn LLC and certain individuals who plaintiff
alleges were directors of Brooklyn LLC. Plaintiff purports to state claims against Brooklyn LLC and the individual defendants under the New York State Executive Law, New York State Administrative Code, and other statutory and common law claims for
alleged unlawful and discriminatory conduct based on race, national origin and hostile work environment. Plaintiff also asserts various breach of contract, fraud and quantum meruit claims based on an alleged oral agreement under which he alleges
Brooklyn LLC agreed to hire him as an executive once Brooklyn LLC went public. In particular, plaintiff alleges that in exchange for transferring an opportunity to obtain an agreement to acquire a license from Novellus for its mRNA-based gene
editing and cell reprogramming technology to Brooklyn LLC, he was promised a $500,000 salary and 7% of the equity of Brooklyn LLC. Based on these and other allegations, plaintiff seeks damages of not less than $10 million, a permanent injunction
enjoining Brooklyn LLC from exercising the option to acquire a license from Novellus or completing the Merger. On or about February 19, 2021, an amended complaint was filed asserting the same causes of action but withdrawing the request for
injunctive relief. On or about April 26, 2021, the parties entered into a stipulation whereby the defendants agreed to accept service of the Amended Complaint without waiver of any defenses, including jurisdictional defenses, except for improper
service, and the plaintiff agreed to extend Defendants time to respond to the complaint to June 6, 2021.
Carlson v. Allen Wolff, Michael Gottlieb, Richard Simtob, Susan Miller, and NTN Buzztime, Inc., C.A. No. 2021-0193-KSJM (Del. Ch. Ct.)
On or about March 12, 2021, Douglas Carlson, a purported stockholder of the Pre-Merger Company, filed a verified class action complaint against the Pre-Merger Company and its
then-serving directors for allegedly breaching their fiduciary duties and violating Section 211(c) of the Delaware General Corporation Law. In particular, plaintiff seeks to compel the defendants to hold an annual stockholder meeting. Plaintiff
also moved for summary judgment at the same time that he filed his complaint. In order to moot the claim addressed in the complaint, Brooklyn LLC has agreed to hold its annual meeting on June 29, 2021. The parties have agreed to extend defendants’
time to respond to the complaint by thirty days and are presently negotiating an extended briefing schedule on the motion for summary judgment.
From time to time we may become involved in legal proceedings arising in the ordinary course of business. Except as described above, we do not believe
there is any litigation pending that could have, individually or in the aggregate, a material adverse effect on our results of operations, financial condition or cash flows.
21
Employees
As of April 30, 2021, we had 11 employees. None of our employees are represented by a labor union, and we consider our employee relations to be good.
We lease approximately 8,000 square feet of office and laboratory space in the borough of Brooklyn in New York, New York, at a cost of approximately $32,000 per month. We
believe the facilities that we occupy are adequate for the purposes for which they are currently used and are well-maintained.
We have considerable experience in manufacturing products, particularly the active pharmaceutical ingredient, or API, for clinical or, ultimately, commercial purposes. We
currently have internal API manufacturing capabilities. We have established long-standing finish and fill contract manufacturing relationships with a single manufacturer for packaging of the clinical supplies of IRX‑2, As with any supply program,
obtaining raw materials of the correct quality, and the performance of our single qualified contract fill and finish site cannot be guaranteed and we cannot ensure that we will be successful in the continuation of this endeavor.
At the time of commercial approval, if not prior, and to the extent possible and commercially practicable, we would seek to engage a back-up supplier for IRX‑2. Until such
time, we expect that we will rely on internal API manufacture, and a single contract manufacturer to finish IRX‑2 under cGMP regulations. Our third-party manufacturers have a limited number of facilities in which IRX‑2 can be produced and will have
a single site for manufacturing API for IRX‑2 in quantities sufficient for commercialization. Our third-party manufacturers will have start-up costs, as well as other clients and may have other priorities that could affect their ability to perform
the work satisfactorily and/or on a timely basis. Both of these occurrences would be beyond our control.
We expect to similarly rely on contract manufacturing relationships for any products that we may in-license or acquire in the future. However, there can be no assurance that
we will be able to successfully contract with such manufacturers on terms acceptable to us, or at all.
Contract manufacturers are subject to ongoing periodic and unannounced inspections by the FDA, the U.S. Drug Enforcement Administration and corresponding state agencies to
ensure strict compliance with cGMP and other state and federal regulations. Our contractors, if any, in Europe will face similar challenges from the numerous EU and member state regulatory agencies and authorized bodies. We do not have control over
third-party manufacturers’ compliance with these regulations and standards, other than through contractual obligations. If they are deemed out of compliance with cGMPs, product recalls could result, inventory could be destroyed, production could be
stopped, and supplies could be delayed or otherwise disrupted.
If we need to change manufacturers after commercialization, the FDA and corresponding foreign regulatory agencies must approve these new manufacturers in advance, which will
involve testing and additional inspections and associated regulatory prior approval submissions to ensure compliance with FDA regulations and standards. This may precipitate significant lead times and delay. Furthermore, switching manufacturers may
be difficult because the number of potential manufacturers is limited. It may be difficult or impossible for us to find a replacement manufacturer quickly or on terms acceptable to us, or at all.
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development,
testing, manufacture, labeling, record-keeping, promotion, storage, advertising, distribution, marketing, and export and import of products such as those we are developing. Drugs must be approved by the FDA through the New Drug Application, or NDA,
process or the Biologic License Application, or BLA, process before they may be legally marketed in the United States.
22
There are two Centers within the FDA that are responsible for the review and approval of drug marketing applications and general regulatory oversight: the Center for Drug
Evaluation and Research, or CDER, and the Center for Biologics Evaluation and Research, or CBER. While all conventional drug products are regulated by CDER, biological products can be regulated by either CDER or CBER, depending on the product’s
classification.
While the majority of BLA submissions are assigned to CBER, BLAs for certain biological product categories are reviewed by CDER instead. These product categories include
monoclonal antibodies for in vivo use, most proteins for therapeutic use, and categories such as cytokines, enzymes and other novel proteins. Based on this, it is likely that a BLA submission for IRX‑2 would fall under CDER. Regardless of the
category, NDAs for all drug products fall under the jurisdiction of CDER.
In the United States, drugs are subject to rigorous regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations, as well as other federal and state
statutes. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to
comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal
to approve pending applications, license suspension or revocation, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil
penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • |
completion of pre-clinical laboratory tests, animal studies and formulation studies according to the FDA’s good laboratory practice, or GLP, regulations;
|
| • |
submission of an investigational new drug application, or IND, which must become effective before human clinical trials may begin and which must include approval by an institutional review board, or IRB, at each clinical site before the
trials are initiated;
|
| • |
performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use conducted in compliance with federal regulations and good clinical practice, or GCP, an
international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators, and monitors;
|
| • |
submission to, and acceptance by, the FDA of an NDA or BLA;
|
| • |
satisfactory completion of an FDA inspection of our manufacturing facility or other facilities at which the drug is produced to assess compliance with current good manufacturing practice, or cGMP, regulations to assure that the
facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity;
|
| • |
potential FDA audit of the non-clinical and clinical trial sites that generated the data in support of the NDA or BLA; and
|
| • |
FDA review and approval of the NDA or BLA.
|
The testing and approval process require substantial time, effort and financial resources, and the receipt and timing of any approval is uncertain.
U.S. Drug Development Process
Once a pharmaceutical candidate is identified for development it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations of product chemistry,
toxicity and formulation, as well as animal studies. Prior to beginning human clinical trials, an IND sponsor must submit an IND to the FDA. The IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information
and analytical data, to the FDA as part of the IND. Some pre-clinical or non-clinical testing may continue even after the IND is submitted. In addition to including the results of the pre-clinical studies, the IND will also include a protocol
detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated, if the trial lends itself to an efficacy evaluation. The IND automatically becomes
effective 30 days after receipt by the FDA, unless the FDA, within the 30 day time period, raises concerns or questions about the conduct of the trial. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the
clinical trial can begin. The FDA may, at any time, impose a clinical hold on ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized
by the FDA.
23
Clinical trials involve the administration of the investigational new drug to healthy volunteers or patients under the supervision of one or more qualified investigators in
accordance with federal regulations and GCP.
Clinical trials must be conducted under protocols detailing the objectives of the trial and the safety and effectiveness criteria to be evaluated. Each protocol must be
submitted to the FDA as part of the IND. Further, an Institutional Review Board, or IRB, at each institution participating in the clinical trial must review and approve each protocol before any clinical trial commences at that institution. All
research subjects must provide informed consent, and informed consent information must be submitted to the IRB for approval prior to initiation of the trial. Progress reports detailing the results of the clinical trials must be submitted at least
annually to the FDA and more frequently if adverse events or other certain types of other changes occur.
Human clinical trials are typically conducted in three phases. A fourth, or post-approval, phase may include additional clinical studies. These phases generally include the following, and may be
sequential, or may overlap or be combined:
| • |
Phase 1 clinical trials involve the initial introduction of the drug into human subjects. These studies are designed to determine the safety of usually single doses of the compound and determine any dose limiting intolerance, as well as
evidence of the metabolism and pharmacokinetics of the drug in humans.
|
| • |
Phase 2 clinical trials usually involve studies in a limited patient population to evaluate the safety and efficacy of the drug for specific, targeted indications, to determine dosage tolerance and optimal dosage, and to identify
possible adverse effects and safety risks.
|
| • |
In Phase 3, if a compound is found to be potentially effective and to have an acceptable safety profile in Phase 2 (or occasionally Phase 1) studies, the Phase 3 studies will be conducted to further confirm clinical efficacy, optimal
dosage and safety within an expanded population which may involve geographically diverse clinical trial sites.
|
| • |
Generally, but not always, two adequate and well-controlled Phase 3 clinical trials are required by the FDA for approval of an NDA.
|
| • |
Phase 4 clinical trials are studies required of or agreed to by a sponsor that are conducted after the FDA has approved a product for marketing. These studies are used to gain additional experience from the
treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of drugs approved under accelerated approval regulations. If the FDA approves a product while a company has ongoing clinical trials
that were not necessary for approval, a company may be able to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement. Failure to promptly conduct Phase 4 clinical trials where necessary could
result in withdrawal of approval for products approved under accelerated approval regulations.
|
While Phase 1, Phase 2, and Phase 3 tests are generally required for approval of an NDA, certain drugs may not require one or more steps in the process depending on other
testing and the situation involved. Additionally, the FDA, an IRB, or the sponsor may stop testing at any time if results show patients being exposed to unnecessary health risks or overly dangerous side effects.
In addition, the manufacturer of an investigational drug in a Phase 2 or Phase 3 clinical trial for a serious or life-threatening disease is required to make available, such
as by posting on its website, its policy on evaluating and responding to requests for expanded access to such investigational drug.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical
characteristics of the drug and finalize a process for manufacturing the product in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other
requirements, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate
that the drug candidate does not undergo unacceptable deterioration over its shelf life.
24
U.S. Drug Review and Approval Process
FDA approval of an NDA or BLA is required before marketing of the product may begin in the United States. The NDA must include the results of product development, pre-clinical
studies and clinical studies, together with other detailed information, including information on the chemistry, manufacture and composition of the product. In addition, a BLA must also demonstrate purity, specifically in terms of showing that the
final product does not contain extraneous material. The FDA has 60 days from its receipt of the NDA or BLA to review the application to ensure that it is sufficiently complete for substantive review before accepting it for filing. The FDA may
request additional information rather than accept an NDA or BLA for filing. In this event, the NDA or BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for
filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The submission of an NDA or BLA is also subject to the payment of a substantial application fee (for FDA fiscal year 2021, this fee is $2,875,842
when clinical data are required), although a waiver of such fee may be obtained under certain limited circumstances, including when the drug that is subject of the application has received Orphan Drug Designation for the indication sought. Further,
the sponsor of an approved NDA or BLA is subject to an annual program fee, which for FDA fiscal year 2021 is $336,432 per prescription drug product. User fees typically increase annually. The approval process is lengthy and difficult, and the FDA
may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the
NDA or BLA does not satisfy the criteria for approval. The FDA may also refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes
clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee. The FDA reviews an NDA to determine, among other
things, whether a product is safe and effective for its intended use. Before approving an NDA or BLA, the FDA will inspect the facility or facilities where the product is manufactured to determine whether its manufacturing is cGMP–compliant to
assure and preserve the product’s identity, strength, quality, purity and stability.
If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA will issue a complete response letter. The complete response letter
outlines the deficiencies in the submission and often requires additional testing or information in order for the FDA to reconsider the application. Even after submitting this additional information, the FDA ultimately may decide that the
application does not satisfy the regulatory criteria for approval. With limited exceptions, the FDA may withhold approval of an NDA regardless of prior advice it may have provided or commitments it may have made to the sponsor.
Once an NDA or BLA is approved, changes to the conditions of approval, including additional indications, are made by the submission of a supplement to the NDA or BLA. The
supplemental NDA, or sNDA, or the supplemental BLA, or sBLA must contain all of the information necessary to support the change. In the case of a new indication, that information usually consists of at least one clinical trial, and often more. Like
an NDA, FDA determines whether the sNDA or sBLA is sufficiently complete to permit review before it files the sNDA. FDA then reviews the sNDA or sBLA. Like an NDA, FDA can either approve the sNDA or issue a complete response letter outlining the
deficiencies in the sNDA or sBLA.
Manufacturing
As part of the approval process, the FDA must inspect and approve each manufacturing facility. Among the conditions of approval is the requirement that a manufacturer’s
quality control and manufacturing procedures conform to cGMP. Manufacturers must expend significant time, money and effort to ensure continued compliance, and the FDA conducts periodic inspections to certify compliance. It may be difficult for our
contract manufacturers or us to comply with the applicable cGMP, as interpreted by the FDA, and other FDA regulatory requirements. If we, or our contract manufacturers, fail to comply, then the FDA may not allow us to market products that have been
affected by the failure.
25
If the FDA grants approval, the approval will be limited to those conditions and patient populations for which the product is safe and effective, as demonstrated through
clinical studies. Further, a product may be marketed only in those dosage forms and for those indications approved in the NDA or BLA. Certain changes to an approved NDA or BLA, including, with certain exceptions, any significant changes to
labeling, require approval of a supplemental application before the drug may be marketed as changed. Any products that we manufacture or distribute pursuant to FDA approvals are subject to continuing monitoring and regulation by the FDA, including
compliance with cGMP and the reporting of adverse experiences with the drugs. The nature of marketing claims that the FDA will permit us to make in the labeling and advertising of our products will generally be limited to those specified in FDA
approved labeling, and the advertising of our products will be subject to comprehensive monitoring and regulation by the FDA. Drugs whose review was accelerated may carry additional restrictions on marketing activities, including the requirement
that all promotional materials are pre-submitted to the FDA. Claims exceeding those contained in approved labeling will constitute a violation of the FDCA. Violations of the FDCA or regulatory requirements at any time during the product development
process, approval process, or marketing and sale following approval may result in agency enforcement actions, including corrective advertising, cessation of violative promotion, withdrawal of approval, recall, seizure of products, warning letters,
injunctions, fines and/or civil or criminal penalties. Any agency enforcement action could have a material adverse effect on our business.
Failure to comply with applicable federal, state and foreign laws and regulations would likely have a material adverse effect on our business. In addition, federal, state and
foreign laws and regulations regarding the manufacture and sale of new drugs are subject to future changes.
Post-Approval Requirements and consideration
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of
drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the internet. As a condition of NDA approval, the FDA
may also require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for the healthcare professionals, and other
Elements to Assure Safe Use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries.
The requirement for a REMS can materially affect the potential market and profitability of the drug.
Drugs may be marketed only for the approved indications and in accordance with the provisions of the approved labeling. Changes to some of the conditions established in an
approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or NDA supplement before the change can be implemented. An NDA supplement for a new
indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing NDAs.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post-marketing testing, known as Phase 4
testing, and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control as well as drug manufacture, packaging, and labeling
procedures must continue to conform to cGMPs after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced
inspections by the FDA during which the agency inspects manufacturing facilities to assess compliance with cGMPs. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain
compliance with cGMPs. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized
problems are subsequently discovered.
Foreign Regulatory Requirements
In addition to regulation by the FDA and certain state regulatory agencies, we are also subject to a variety of foreign regulations governing clinical trials and the marketing
of other products. Outside of the United States, our ability to market a product depends upon receiving a marketing authorization from the appropriate regulatory agencies. The requirements governing the conduct of clinical trials, marketing
authorization, pricing and reimbursement vary widely from country to country. In any country, however, we will only be permitted to commercialize our products if the appropriate regulatory agency is satisfied that we have presented adequate
evidence of safety, quality and efficacy. Whether or not FDA approval has been obtained, approval of a product by the comparable regulatory authorities of foreign countries must be obtained prior to the commencement of marketing of the product in
those countries. The regulatory approval and oversight process in other countries includes all of the risks associated with regulation by the FDA and certain state regulatory agencies as described above.
26
Under the EU regulatory system, applications for drug approval may be submitted either in a centralized or decentralized manner. Under the centralized procedure, a single
application to the European Medicines Agency leads to an approval granted by the European Commission which permits marketing of the product throughout the EU. The decentralized procedure provides for mutual recognition of nationally approved
decisions and is used for products that do not comply with requirements for the centralized procedure. Under the decentralized procedure, the holders of national marketing authorization in one of the countries within the EU may submit further
applications to other countries within the EU, who will be requested to recognize the original authorization based on an assessment report provided by the country in which marketing authorization is held.
Pharmaceutical Pricing and Reimbursement
In both U.S. and foreign markets, our ability to commercialize our products successfully, and to attract commercialization partners for our products, depends in significant
part on the availability of adequate financial coverage and reimbursement from third-party payors, including, in the United States, governmental payors such as Medicare and Medicaid, managed care organizations, private commercial health insurers
and pharmacy benefit managers, or PBMs. Third party payors are increasingly challenging the prices charged for medicines and examining their cost effectiveness, in addition to their safety and efficacy. We may need to conduct expensive
pharmacoeconomic or other studies in order to further demonstrate the value of our products. Even with the availability of such studies, our products may be considered less safe, less effective or less cost-effective than alternative products, and
third-party payors may not provide coverage and reimbursement for our drug candidates, in whole or in part.
Political, economic and regulatory influences are subjecting the health care industry in the United States to fundamental changes. There have been, and we expect there will
continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business, including the Patient Protection and Affordable Care Act of 2010, or the Affordable Care Act. There continue
to be efforts in Congress to repeal the Affordable Care Act and replace it with another law. As a result, there is great uncertainty as to what changes will be made to U.S. healthcare laws and there can be no assurance how changes to those laws may
affect our business.
We anticipate that in the United States, Congress, state legislatures, and private sector entities will continue to consider and may adopt healthcare policies intended to curb rising healthcare
costs. These cost containment measures could include:
| • |
controls on government-funded reimbursement for drugs;
|
| • |
mandatory rebates or additional charges to manufacturers for their products to be covered on Medicare Part D formularies;
|
| • |
controls on healthcare providers;
|
| • |
controls on pricing of pharmaceutical products, including the possible reference of the pricing of U.S. drugs to non-U.S. drug pricing for the same product;
|
| • |
challenges to the pricing of drugs or limits or prohibitions on reimbursement for specific products through other means;
|
| • |
reform of drug importation laws;
|
| • |
entering into contractual agreements with payors; and
|
| • |
expansion of use of managed-care systems in which healthcare providers contract to provide comprehensive healthcare for a fixed cost per person.
|
We are unable to predict what additional legislation, regulations or policies, if any, relating to the healthcare industry or third-party coverage and reimbursement may be
enacted in the future or what effect such legislation, regulations or policies would have on our business. Any cost containment measures, including those listed above, or other healthcare system reforms that are adopted may have a material adverse
effect on our business prospects.
27
Further, the pricing of pharmaceutical products generally, and particularly the pricing of orphan drugs, has recently received scrutiny from the press, from members of
Congress in both parties, and from President Trump. Some members of the medical community have also made statements in the press on the pricing of orphan drugs. The impact of this scrutiny on the pricing of orphan drugs and other pharmaceutical
products generally cannot be determined with any certainty at this time.
The Biologics Price Competition and Innovation Act of 2009, which was included in the Patient Protection and Affordable Care Act, or PPACA, authorized the FDA to approve
similar versions of innovative biologics, commonly known as biosimilars. Under the PPACA, a manufacturer may submit an application for licensure of a biologic product that is “biosimilar to” or “interchangeable with” a previously approved biologic
product or “reference product.” Manufacturers may not submit an application for a biosimilar to the FDA until four years following approval of the reference product, and the FDA may not approve a biosimilar product until 12 years from the date on
which the reference product was approved. Even if IRX‑2 or any other biological product we may acquire or in-license, if approved, are deemed to be reference products eligible for exclusivity, another company could market a competing version of
that product if the FDA approves a full BLA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. Additionally, from
time to time, there are proposals to repeal or modify the PPACA, including proposals that could significantly shorten the exclusivity period for biologics.
Orphan Drug Exclusivity
Some jurisdictions, including the United States and Europe, may designate drugs or biologic products for relatively small patient populations as orphan drugs. Under the Orphan
Drug Act of 1983 (ODA), the FDA may grant orphan drug designation to drugs or biologic products intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the
United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. In
the United States, orphan drug designation must be requested before submitting an application for marketing approval. An orphan drug designation does not shorten the duration of the regulatory review and approval process. The grant of an orphan
drug designation request does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and efficacy of a product candidate must be established through adequate and well-controlled studies. If a product
which has been granted orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to an orphan drug exclusivity period, which means the FDA may not approve any
other application to market the same drug for the same disease or condition for a period of seven years, except in limited circumstances, such as where an alternative product demonstrates clinical superiority to the product with orphan exclusivity.
In addition, holders of exclusivity for orphan drugs are expected to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for
the drug.
The orphan drug exclusivity contained in the ODA has been the subject of recent scrutiny from the press, from some members of Congress and from some in the medical community.
There can be no assurance that the exclusivity granted in ODA to orphan drugs approved by the FDA will not be modified in the future, and as to how any such change might affect our products.
The European Orphan Drug Regulation is considered for drugs intended to diagnose, prevent or treat a life-threatening or very serious condition afflicting five or fewer per
10,000 people in the EU, including compounds that for serious and chronic conditions would likely not be marketed without incentives due to low market return on the sponsor’s development investment. The medicinal product considered should be of
significant benefit to those affected by the condition. Benefits of being granted Orphan Medicinal Product Designation are significant, including eight years of data exclusivity, two years of marketing exclusivity and a potential one-year extension
of both. The EU Community and Member States may not accept or grant for ten years a new marketing authorization or application for another drug for the same therapeutic indication as the orphan drug, although the ten-year period can be reduced to
six years if, after the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of the marketing exclusivity. A supplementary protection certificate may extend the protection six
months beyond patent expiration if that is later than the orphan drug exclusivity period. To apply for the supplementary protection, a pediatric investigation plan, or PIP, must be included in the market application. In Europe all drugs now seeking
marketing authorization need to have a PIP agreed with the European Medicines Agency (EMA) before it can be approved, even if it is a drug being developed specifically for a pediatric indication. If a product is developed solely for use in the
pediatric population, then a Pediatric Use Marketing Authorization, or PUMA, may provide eight years of data exclusivity and ten years of marketing exclusivity.
28
Fast Track Designation and Accelerated Approval
The FDA is required to facilitate the development, and expedite the review, of drugs that are intended for the treatment of a serious or life-threatening disease or condition
for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the fast track program, the sponsor of a new drug candidate may request that FDA designate the drug candidate for
a specific indication as a fast track drug concurrent with, or after, the filing of the IND for the drug candidate. FDA must determine if the drug candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
Under the fast track program and FDA’s accelerated approval regulations, FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic
benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably
likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
In clinical trials, a surrogate endpoint is a measurement of laboratory or clinical signs of a disease or condition that substitutes for a direct measurement of how a patient
feels, functions, or survives. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A drug candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the
completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, will allow FDA to withdraw the
drug from the market on an expedited basis. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by FDA.
In addition to other benefits such as the ability to use surrogate endpoints and engage in more frequent interactions with FDA, FDA may initiate review of sections of a fast
track drug’s NDA before the application is complete. This rolling review is available if the applicant provides, and FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However,
FDA’s time period goal for reviewing an application does not begin until the last section of the NDA or BLA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer
supported by data emerging in the clinical trial process.
Priority Review
Under FDA policies, a drug candidate is eligible for priority review, or review within a six to eight-month time frame from the time a complete NDA or BLA is submitted, if the
drug candidate is intended for the treatment, diagnosis, or prevention of a serious or life-threatening condition, demonstrates the potential to address an unmet medical need, or provides a significant improvement compared to marketed drugs.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs, are required to register and disclose certain clinical trial information. Information related to the
product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical
trials after completion. Disclosure of results of these trials can be delayed in certain circumstances for up to two years after the date of completion of the clinical trial. Competitors may use this publicly-available information to gain knowledge
regarding the progress of development programs. Finally, there can be no assurance that fast track designation will result in a faster review process.
29
Gene Editing Products
Gene editing product candidates we may develop based on the License Agreement with the Licensor are based on new technology, which makes it difficult to predict the time and
cost of development and of subsequently obtaining regulatory approval, if we are able to obtain such approval.
Anti-Kickback, False Claims Laws and the Prescription Drug Marketing Act
In addition to FDA restrictions on marketing of pharmaceutical products, other state and federal laws have been applied to restrict certain marketing practices in the
pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal healthcare program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting
or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare
programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and patients, prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are
punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common
activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they
do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making,
or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in
turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain
marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services
reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payer.
The U.S. Centers for Medicare & Medicaid Services, or CMS, has issued a rule that requires manufacturers of approved prescription drugs to collect and report information
on payments or transfers of value to physicians and teaching hospitals, as well as investment interests held by physicians and their immediate family members. The information reported each year is made publicly available on a searchable website.
Failure to submit required information may result in civil monetary penalties.
In addition, several states now require prescription drug companies to report expenses relating to the marketing and promotion of drug products and to report gifts and
payments to individual physicians in these states. Other states prohibit various other marketing-related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, California,
Connecticut, Massachusetts and Nevada require pharmaceutical companies to implement compliance programs and/or marketing codes. Several additional states are considering similar proposals. Compliance with these laws is difficult and time consuming,
and companies that do not comply with these state laws face civil penalties.
Prescription drug advertising is subject to federal, state and foreign regulations. In the United States, the FDA regulates prescription drug promotion, including
direct-to-consumer advertising. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S.
Prescription Drug Marketing Act, a part of the FDCA. In addition, Title II of the Federal Drug Quality and Security Act of 2013, known as the Drug Supply Chain Security Act or DSCSA, has imposed new “track and trace” requirements on the
distribution of prescription drug products by manufacturers, distributors, and other entities in the drug supply chain. The DSCSA requires product identifiers (i.e., serialization) on prescription drug products in order to eventually establish an
electronic interoperable prescription product to system to identify and trace certain prescription drugs distributed in the United States and preempts existing state drug pedigree laws and regulations on this topic. The DSCSA also establishes new
requirements for the licensing of wholesale distributors and third-party logistic providers, although FDA regulations addressing wholesale distributors and third party logistics providers have not yet been promulgated. We will serialize our product
at both the package and homogeneous case level, pass serialization and required transaction information to customers, and believe that we comply with all such requirements.
30
Risk Factors
Risk Factor Summary
Our business is subject to a number of risks, including risks that may prevent us
from achieving our business objectives or may adversely affect our business, financial condition, results of operations, cash flows and prospects. The risks are discussed more fully below and include, but are not limited to, the risks summarized
below.
Risks Related to Our Business
| • |
the impact of the COVID-19 pandemic on our business plan;
|
| • |
our limited operating history and lack of product revenue;
|
| • |
the expense, uncertainty and time consuming nature of the clinical studies required for our product candidates;
|
| • |
that we are subject to extensive and costly government regulation;
|
| • |
that we do not have, and may never obtain, regulatory approvals required to marked our product candidates;
|
| • |
our product may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives;
|
| • |
that we may be unable to scale up manufacturing of IRX-2 in the required quality and quantity to commercialize our product;
|
| • |
that we will require substantial additional capital to fund our operations;
|
| • |
our ability to identify and acquire or in-license additional product candidates.
|
Risks Related to the Discovery, Development and Regulatory Approval of our Drug Candidates
| • |
our dependence on the success of our internal development programs;
|
| • |
difficulty in enrolling patients in our clinical trials, which could delay or prevent us from proceeding with clinical trials for our product candidates;
|
| • |
the potential for undesirable side effects in our current or future product candidates, which could halt their development or prevent their approval;
|
| • |
that we may expend resources on a product candidate that we fail to capitalize;
|
| • |
our ability to maintain orphan drug exclusivity for IRX-2 or for a future drug candidate, which would adversely affect our revenues;
|
| • |
our ability to comply with environmental health and safety regulations.
|
Risks Related to the Commercialization of our Product Candidates and Other Regulatory Compliance Matters
| • |
our ability to obtain marketing approval for our product candidates;
|
| • |
our current lack of marketing, sales or distribution infrastructure with respect to our product candidates;
|
| • |
the impact on ongoing regulatory obligations and continued regulatory review of our product candidates, which may result in significant additional expenses;
|
| • |
that our competitors may develop and market products that are more effective, safer or less expensive than our own product candidates;
|
| • |
our dependence on the market acceptance of our products by physicians, patients, payers and others in the medical community;
|
| • |
the uncertainty of insurance coverage and reimbursement status of our product candidates, if approved, which could limit our ability to market those products or
generate revenue;
|
| • |
that IRX-2 or any other biological product we may acquire or in-license and for which we intend to seek approval may face competition sooner than anticipated;
|
| • |
the impact of approved generic versions of our product candidates on our sales;
|
| • |
that healthcare legislative reform measures and contains on national budget social security systems may have a material adverse effect on our business and results
of operations.
|
Risks Related to our Dependence on Third Parties
| • |
our reliance on third parties to conduct our clinical studies, which third parties may not perform satisfactorily;
|
31
| • |
our ability to enter into collaborations with third parties for development and commercialization of our product candidates, the failure of which may impact our
ability to capitalize on the market potential of IRX-2 or any other product we may acquire or in-license;
|
Risks Related to our Intellectual Property
| • |
our ability to obtain and maintain patent and other intellectual property protection for our products and product candidates;
|
| • |
that our patents could be found invalid or unenforceable if challenged in court or administrative proceedings;
|
| • |
changes in patent law in the U.S. and other jurisdictions could diminish the value of our patens in general, impairing our ability to protect our products or
product candidates;
|
| • |
our ability to maintain license rights that our important to our business.
|
Risks Related to our Business
We face business disruption and related risks resulting from the recent pandemic of the novel coronavirus (COVID-19), which could have a material adverse
effect on our business plan.
The development of our product candidates could be disrupted and materially adversely affected by the recent outbreak of COVID-19. As a result of measures imposed by the governments in affected regions, businesses
and schools have been suspended due to quarantines intended to contain this outbreak. The spread of SARS CoV‑2 from China to other countries resulted in the Director General of the World Health Organization declaring COVID-19 a pandemic in March
2020. We are still assessing our business plans and the impact COVID-19 may have on our ability to conduct or the timeline of our clinical trials and the clinical trials being undertaken by investigators, but there can be no assurance that this
analysis will enable us to avoid part or all of any impact from the spread of COVID-19 or its consequences, including downturns in business sentiment generally. The extent to which the COVID-19 pandemic and global efforts to contain its spread will
impact our operations will depend on future developments, which are highly uncertain and cannot be predicted at this time, and include the duration, severity and scope of the pandemic and the actions taken to contain or treat the COVID-19 pandemic.
The patients in our clinical trials have conditions that make them especially vulnerable to COVID-19, and as a result we have seen slowdowns in enrollment in our clinical trials. While the INSPIRE study is fully
populated, our other clinical studies may see significant delays as a result of the pandemic.
IRX‑2 is a therapy based on IL‑2, a type of cytokine signaling molecule in the immune system. While many of the mechanisms of action of COVID-19 are still unknown, there is evidence that for some patients, severe
COVID-19 may result in “cytokine storm syndrome,” in which the body releases cytokines into the body too quickly, creating symptoms such as high fever, inflammation, severe fatigue and nausea, and can lead to severe or life-threatening symptoms.
There can be no assurance that further study will bear this out or that patients treated with IRX‑2, who are already at higher risk for COVID-19 due to their underlying diagnosis, will not be adversely affected.
We have a limited operating history and have never generated any product revenue.
We are a clinical-stage biopharmaceutical company with a limited operating history. Brooklyn ImmunoTherapeutics LLC was formed on September 27, 2018, for the purpose of consummating a business combination with IRX
Therapeutics, Inc., which business combination was consummated on November 6, 2018. Since inception, we have incurred significant net losses. As of September 30, 2020, it had an accumulated deficit of approximately $16.0 million. Since inception,
it financed its operations with approximately $23 million in capital contributions from the beneficial holders of its Class A membership interests.
Our ability to generate product revenue and become profitable depends upon our ability to successfully complete the development of, and obtain the necessary regulatory approvals for, our product candidates in
development, including IRX‑2. We have never been profitable, have no products approved for commercial sale, and have not generated any product revenue.
Even if we receive regulatory approval for any of IRX‑2 or any future product candidates (including any products that may be developed from the licensed technology if Brooklyn exercises its
option to exclusively license certain technology from Novellus), we do not know when or if such product candidate will generate product revenue. Our ability to generate product revenue depends on a number of factors, including, but not limited to,
our ability to:
| • |
successfully complete pre-clinical studies and clinical trials deemed adequate by regulatory authorities and obtain and maintain regulatory approval for the marketing of our product candidates;
|
| • |
add operational, financial and management information systems personnel, including personnel to support its clinical, manufacturing and planned future commercialization efforts and operations as a public company;
|
| • |
conduct internal manufacturing meeting specifications and in compliance with applicable good clinical manufacturing practices (“cGMP”)
|
32
| • |
establish or maintain collaborations, licensing or other arrangements;
|
| • |
initiate and continue relationships with third-party suppliers and manufacturers or continue to further develop our own manufacturing capabilities and have commercial quantities of our product candidates manufactured at acceptable cost
and quality levels and in compliance with the FDA and other regulatory requirements;
|
| • |
launch commercial sales of our products, whether alone or in collaboration with others, including establishing sales, marketing and distribution systems for its product candidates;
|
| • |
set an acceptable price for any approved product candidates and obtain coverage and adequate reimbursement from third-party payors;
|
| • |
achieve market acceptance of our products in the medical community and with third-party payors and consumers; and
|
| • |
maintain, expand and protect our intellectual property portfolio.
|
Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses, or when or if, it will be able to achieve or maintain
profitability. Neither will be able to predict the state of market competition which would adversely affect our potential revenues from product sales. Our expenses could increase beyond expectations if we are required by the FDA or comparable
non-U.S. regulatory authorities to perform studies or clinical trials in addition to those that we currently anticipate. Even if any of our product candidates is approved for commercial sale, we anticipate incurring significant costs associated
with their commercial launch. If we cannot successfully execute any one of the foregoing, our business may not succeed, and your investment will be negatively impacted.
Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront capital expenditures and significant risk that a product candidate will fail to
gain regulatory approval or fail to become commercially viable. We have never generated any product revenue, and we cannot estimate with precision the extent of our future losses. We do not currently have any products that are available for
commercial sale and we may never generate product revenue or achieve profitability.
We expect to continue to incur substantial and increasing losses through the commercialization of IRX‑2 or any other product candidate we may acquire or in-license, if approved. None of our product candidates have
been approved for marketing anywhere in the world, and we may never receive such approval. As a result, we are uncertain when or if we will achieve profitability and, if so, whether we will be able to sustain it. Our ability to generate product
revenue and achieve profitability is dependent on our ability to complete the development of our product candidates, obtain necessary regulatory approvals for such product candidates, and manufacture and successfully market our product candidates
alone or in collaboration with others. There can be no assurance that we will be profitable even if we successfully commercialize IRX‑2 or any other product candidate we may acquire or in-license (including any products that may be developed from
the licensed technology if Brooklyn exercises its option to exclusively license certain technology from Novellus). If we do successfully obtain regulatory approval to market any of our product candidates, our revenue will be dependent upon, in part
and among other things, the size of the markets in the territories for which we gain regulatory approval, the number of competitors in such markets, the accepted price for any such product candidate and whether we own the commercial rights for
those territories. If the indication approved by regulatory authorities is narrower than we expect, or the treatment population is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales
of any of our product candidates, even if approved. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Failure to become and remain profitable may adversely impact the market
price of the common stock and our ability to raise capital and continue operations.
We expect that our research and development expenses in connection with our development programs for product candidate will continue to be significant. In addition, as we prepare for and if we obtain regulatory
approval for any of our product candidates, we expect to incur increased sales, marketing and manufacturing expenses. As a result, we expect to continue to incur significant and increasing operating losses and negative cash flows for the
foreseeable future. These losses have harmed and will continue to harm our results of operations, financial position and working capital.
33
Our business is heavily dependent on the successful development, regulatory approval and commercialization of our
product candidate.
We currently have no products that are approved for commercial sale and may never be able to develop marketable products. We expect that a substantial portion of our efforts and expenditures will be devoted to the
continued clinical evaluation of our lead product candidate, IRX‑2, and the monotherapy and combination studies related thereto and the commercialization of such product candidates following regulatory approval, if received, as well as the
continued clinical and preclinical evaluation of any IRX‑2 and any other product we may acquire or in-license (including any products that may be developed from the licensed technology if Brooklyn exercises its option to exclusively license certain
technology from Novellus). Accordingly, our business currently depends heavily on the successful completion of our clinical trials for its product candidates and subsequent regulatory approval and commercialization of such product candidate.
We cannot be certain that IRX‑2 or any other product candidate we may acquire or in-license (including any products that may be developed from the licensed technology if Brooklyn exercises its option to exclusively
license certain technology from Novellus) will receive regulatory approval or be successfully commercialized even if it receives regulatory approval. The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of
products are, and will remain, subject to extensive regulation by the FDA and other regulatory authorities in the United States and in other countries that each have differing regulations. We are not permitted to market IRX‑2 or any other product
candidate we may acquire or in-license in the United States (including any products that may be developed from the licensed technology if Brooklyn exercises its option to exclusively license certain technology from Novellus) until we receive
approval of a new drug application, or NDA, or Biologic License Application, or BLA or in any foreign country until it receives the requisite approvals from the appropriate authorities in such countries for marketing authorization. In addition, we
have not yet demonstrated our ability to complete later-stage or pivotal clinical trials for any of our product candidates.
We have not submitted an NDA or BLA for IRX‑2 to the FDA or any comparable application to any other regulatory authority. Obtaining approval of an NDA or BLA or similar regulatory approval is an
extensive, lengthy, expensive and inherently uncertain process, and the FDA or other foreign regulatory authorities may delay, limit or deny approval of any of our product candidates for many reasons, including:
| • |
We may not be able to demonstrate that any IRX‑2 is safe or effective as a treatment for any of our currently targeted indications to the satisfaction of the FDA or other relevant regulatory authorities;
|
| • |
the relevant regulatory authorities may require additional pre-approval studies or clinical trials which would increase our costs and prolong its development timelines or could grant conditional approval with a requirement to perform
additional studies at a significant cost;
|
| • |
the results of our clinical trials may not meet the level of statistical or clinical significance required by the FDA or other relevant regulatory authorities for marketing approval;
|
| • |
the FDA or other relevant regulatory authorities may disagree with the number, design, size, conduct or implementation of our clinical trials, including the design of its future pivotal Phase 3 clinical trials;
|
| • |
CROs that we may retain to conduct clinical trials may take actions outside of our control, or otherwise commit errors or violations of protocols, that adversely impact our clinical trials and ability to obtain market approvals;
|
| • |
the FDA or other relevant regulatory authorities may not find the data from nonclinical studies or clinical trials sufficient to demonstrate that the clinical and other benefits of these products outweigh their safety risks;
|
| • |
the FDA or other relevant regulatory authorities may disagree with our interpretation of data or significance of results from the nonclinical studies and clinical trials of IRX‑2 or any other product candidate we may acquire or
in-license or may require that it conduct additional studies;
|
| • |
the FDA or other relevant regulatory authorities may not accept data generated from our clinical trial sites;
|
| • |
if our NDA or other foreign application is reviewed by an advisory committee, the FDA or other relevant regulatory authority, as the case may be, may have difficulties scheduling an advisory committee meeting in a timely manner or the
advisory committee may recommend against approval of such application or may recommend that the FDA or other relevant regulatory authority, as the case may be, require, as a condition of approval, additional nonclinical studies or clinical
trials, limitations on approved labeling or distribution and use restrictions;
|
34
| • |
the FDA or other relevant regulatory authorities may require development of a Risk Evaluation and Mitigation Strategy (REMS) , or its equivalent, as a condition of approval;
|
| • |
the FDA or other relevant regulatory authorities may require additional post-marketing studies and/or a patient registry, which would be costly;
|
| • |
the FDA or other relevant regulatory authorities may find the chemistry, manufacturing and controls data insufficient to support the quality of IRX - 2 or any other product candidate we may acquire or in-license;
|
| • |
the FDA or other relevant regulatory authorities may identify deficiencies in the manufacturing processes or facilities of our third-party manufacturer or our own manufacturing processes or facilities; or
|
| • |
the FDA or other relevant regulatory authorities may change their approval policies or adopt new regulations.
|
Even if we do receive regulatory approval to market any product candidate, any such approval may be subject to limitations on the indicated uses or patient populations for
which it may market the product. Accordingly, even if we are able to obtain the requisite financing to continue to fund its development programs, we cannot assure you that IRX - 2 or any other product candidate we may acquire or in-license
(including any products that may be developed from the licensed technology if Brooklyn exercises its option to exclusively license certain technology from Novellus) will be successfully developed or commercialized.
In addition, because IRX‑2 targets one or more indications in the medical oncology field, if IRX‑2 or any other product candidate we may develop or in-license, encounter
safety or efficacy problems, developmental delays, regulatory issues, supply issues, or other problems, our development plans for the affected product candidate and some or all of our other product candidates could be significantly harmed, which
would harm its business. Further, competitors who are developing products in the oncology field or that target the same indications as us with products that have a similar mechanism of action may experience problems with its products that could
identify problems that would potentially harm our business.
Clinical studies required for our product candidates are expensive and time-consuming, and their outcome is
uncertain.
In order to obtain FDA approval to market a new pharmaceutical product, we must demonstrate proof of safety and efficacy of the product in humans. To meet these requirements,
we must conduct “adequate and well controlled” clinical studies. Conducting clinical studies is a lengthy, time-consuming, and expensive process. The length of time may vary substantially according to the type, complexity, novelty, and intended use
of the product candidate, and often can be several years or more per study. Delays associated with products for which we are directly conducting clinical studies may cause them to incur additional operating expenses. The commencement and rate of
completion of clinical studies may be delayed by many factors, including, for example: inability to manufacture sufficient quantities of stable and qualified materials under cGMP, for use in clinical studies; slower than expected rates of patient
recruitment; failure to recruit a sufficient number of patients; modification of clinical study protocols; changes in regulatory requirements for clinical studies; the lack of effectiveness during clinical studies; the emergence of unforeseen
safety issues; delays, suspension, or termination of the clinical studies due to the IRB responsible for overseeing the study at a particular study site; and government or regulatory delays or “clinical holds” requiring suspension or termination of
the studies.
The results from early clinical studies are not necessarily predictive of results obtained in later clinical studies. Accordingly, even if we obtain positive results from
early clinical studies, future clinical studies may not have the same positive results. Clinical studies may not demonstrate sufficient safety and effectiveness to obtain the requisite regulatory approvals for product candidates.
35
In some cases, IRX‑2 and any other product we may acquire or in-license (including any products that may be developed from the licensed technology if Brooklyn exercises its
option to exclusively license certain technology from Novellus) may be expected to be used in combination with approved therapies that may have significant adverse event profiles. During the course of treatment, these patients could suffer adverse
medical events or die for reasons that may or may not be related to our product candidates. We cannot ensure that safety issues will not arise with respect to our product candidates in clinical development. In addition, several of our studies
involving IRX‑2 involve IRX‑2 being studied in conjunction with current preferred treatments for certain types of cancer, including head and neck cancers. If the drugs being studied in conjunction with IRX‑2 are found to be faulty in any way, or if
they fall out of favor as a preferred treatment, our clinical studies may be adversely effected, as we may have to restart such studies with the newer, preferred treatment in conjunction with IRX‑2.
The failure of clinical studies to demonstrate safety and effectiveness for the desired indications could harm the development of that product candidate and other product
candidates. This failure could cause us to abandon a product candidate and could delay development of other product candidates. Any delay in, or termination of, our clinical studies would delay the filing of its NDAs with the FDA and, ultimately,
our ability to commercialize its product candidates and generate product revenues. Any change in, or termination of, our clinical studies could materially harm our business, financial condition, and results of operations.
We are subject to extensive and costly government regulation.
Product candidates employing medical technology are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of
the U.S. Department of Health and Human Services, the U.S. Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and nonclinical testing and clinical
studies, manufacture, safety, effectiveness, record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of biopharmaceutical products. Each new chemical entity presented to the FDA must
first be preceded by an Investigational New Drug Application, or IND. The FDA regulates small molecule chemical entities as drugs, subject to an NDA under the FDCA. The FDA applies the same standard for biologics, requiring a BLA prior to
licensure. Other products, such as vaccines, are also regulated under the Public Health Service Act. FDA has conflated the standards for approval of NDAs and BLAs so that it requires the same types of information on safety, effectiveness, and
chemistry, manufacturing, and controls, or CMCs. If products employing our technologies are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval for a given
product and its uses. Such foreign regulation may be equally or more demanding than corresponding U.S. regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our products. The regulatory review and approval process, which includes preclinical and
nonclinical testing and clinical studies of each product candidate, is lengthy, expensive, and uncertain. we or our collaborators must obtain and maintain regulatory authorization to conduct clinical studies. We or our collaborators must obtain
regulatory approval for each product it intends to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires the submission of extensive preclinical,
nonclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product’s safety and efficacy, and in the case of biologics also potency and purity, for each intended use. The
development and approval process takes many years, requires substantial resources, and may never lead to the approval of a product.
Even if we are able to obtain regulatory approval for a particular product, the approval may limit the indicated medical uses for the product, may otherwise limit our ability to promote, sell, and distribute the
product, may require that we conduct costly post-marketing surveillance, and/or may require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling,
may require further regulatory review and approval. Once obtained, any approvals may be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators, or our contract manufacturing organizations, or CMOs, fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in,
among other things delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; warning
letters; fines; import and/or export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the
FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
36
We do not have, and may never obtain, the regulatory approvals we need to market our product candidates.
Following completion of clinical studies, the results are evaluated and, depending on the outcome, submitted to the FDA in the form of an NDA or BLA in order to obtain FDA approval of the product and authorization to
commence commercial marketing. In responding to an NDA, the FDA may require additional testing or information, may require that the product labeling be modified, may impose post-approval study and other commitments or reporting requirements or
other restrictions on product distribution, or may deny the application. The FDA has established performance goals for review of NDAs or BLAs: six months for priority applications and ten months for standard applications. However, the FDA is not
required to complete its review within these time periods. The timing of final FDA review and action varies greatly but can take years in some cases and may involve the input of an FDA advisory committee of outside experts. Product sales in the
United States may commence only when an NDA or BLA is approved.
It is possible that none of our product candidates will be approved for marketing. Failure to obtain regulatory approvals, or delays in obtaining regulatory approvals, may adversely affect the successful
commercialization of any drugs or biologics that Brooklyn or its partners develop, may impose additional costs on us or our collaborators, may diminish any competitive advantages that us or our partners may attain, and/or may adversely affect our
receipt of revenues or royalties.
Any fast track designation or grant of priority review status by the FDA may not actually lead to a faster
development or regulatory review or approval process, nor will it assure FDA approval of our product candidates. Additionally, our product candidates may treat indications that do not qualify for priority review vouchers.
Our drug candidate, IRX‑2, has received fast track designation in head and neck cancers. If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to
address unmet medical needs for this condition, the drug sponsor may apply for FDA fast track designation. If a product candidate offers major advances in treatment, the FDA may designate it eligible for a priority review. The FDA has broad
discretion whether or not to grant these designations, so even if we believe a particular product candidate is eligible for these designations, we cannot assure you that the FDA would decide to grant them. We may also seek fast track designation
for IRX‑2 in the other indications or for future products we may seek to develop. Even if we are granted fast track designation or priority review, we may not experience a faster development process, review or approval compared to conventional FDA
procedures. The FDA may also withdraw fast track designation if it believes that the designation is no longer supported by data from Brooklyn’s clinical development program.
Even if we are able to commercialize any product candidate that we may develop, the product may become subject to
unfavorable pricing regulations, third-party payor reimbursement practices or healthcare reform initiatives that could harm our business.
The commercial success of our current or future product candidates will depend substantially, both domestically and abroad, on the extent to which the costs of its product candidates will be paid by health
maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities (such as Medicare and Medicaid), private health coverage insurers and other third-party
payors. If reimbursement is not available, or is available only to limited levels, Brooklyn may not be able to successfully commercialize its products. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow
us to establish and maintain pricing sufficient to realize a meaningful return on its investment.
There is significant uncertainty related to third-party payor coverage and reimbursement of newly approved drugs. Marketing approvals, pricing and reimbursement for new drug products vary widely from country to
country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some non-U.S. markets, prescription
pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then be subject to price regulations that
delay commercial launch of the product, possibly for lengthy time periods, which may negatively impact the revenues it is able to generate from the sale of the product in that country. Adverse pricing limitations may hinder our ability to recoup
our investment in one or more product candidates, even if our product candidates obtain marketing approval.
37
Our ability to commercialize our product candidate will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from government health
administration authorities, private health insurers and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will cover and establish
reimbursement levels. The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of
reimbursement for particular medications, which could affect our ability to sell our product candidates profitably. These payors may not view our products, if any, as cost-effective, and coverage and reimbursement may not be available to our
customers, or may not be sufficient to allow our products, if any, to be marketed on a competitive basis. Cost-control initiatives could cause us to decrease the price we might establish for products, which could result in lower than anticipated
product revenues. If the prices for our products, if any, decrease or if governmental and other third-party payors do not provide adequate coverage or reimbursement, our prospects for revenue and profitability will suffer.
There may also be delays in obtaining coverage and reimbursement for newly approved drugs, and coverage may be more limited than the indications for which the drug is approved by the FDA or comparable non-U.S.
regulatory authorities. Moreover, eligibility for reimbursement does not imply that any drug will be paid for in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Reimbursement rates
may vary, by way of example, according to the use of the drug and the clinical setting in which it is used. Reimbursement rates may also be based on reimbursement levels already set for lower cost drugs or may be incorporated into existing payments
for other services.
In addition, increasingly, third-party payors are requiring higher levels of evidence of the benefits and clinical outcomes of new technologies and are challenging the prices charged. We cannot be sure that coverage
will be available for any product candidate that we may commercialize and, if available, that the reimbursement rates will be adequate. Further, the net reimbursement for drug products may be subject to additional reductions if there are changes to
laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for
any of our product candidates for which we obtain marketing approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Gene editing product candidates we may develop based on our option agreements with Novellus are based on new
technology, which makes it difficult to predict the time and cost of development and of subsequently obtaining regulatory approval, if we are able to obtain such approval.
Gene editing technology is relatively new, and only a few products based on such technology have been approved in the United States or the EU to date, and only a limited number of clinical trials of product
candidates based on gene-editing technologies have been commenced. As such, it is difficult to accurately predict the developmental challenges we may incur if we exclusively license the technology from Novellus, as they proceed through product
discovery or identification, preclinical studies and clinical trials. There may be long-term effects from treatment with any such product candidates that we may develop that we cannot predict at this time. Any product candidates we may develop may
interact with genetic material (RNA/DNA) and because animal genetic materials differ from human genetic material, past testing of any such product candidates in animal models may not be predictive of results in human clinical trials for safety or
efficacy. As a result of these factors, it is more difficult to predict the time and cost of such product candidate development, and we cannot predict whether the application of gene editing technology, or other similar or competitive gene editing
technologies, will result in the identification, development and regulatory approval of any products. There can be no assurance that any development problems we may experience related to such gene editing technology or any of our research programs
that use such technology will not cause significant delays or anticipated costs, or that such development problems can be solved. Any of these factors may prevent us from completing preclinical studies or clinical trials based on such technology or
from commercializing any such product candidates on a timely or profitable basis, if at all.
38
The clinical trial requirements of the FDA, the EMA and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product candidate vary substantially according to
the type, complexity, novelty and intended use and market of the product candidate. Only a few products based on gene-editing technologies have been approved by regulators to date. As a result, the regulatory approval process for product candidates
using such technology is uncertain and may be more expensive and take longer than the approval process for product candidates based on other, better known or more extensively studied technologies. It is difficult to determine how long it will take
or how much it will cost to obtain regulatory approvals for product candidates using this technology in either the United States or the EU or how long it will take to commercialize any product candidates. Delay or failure to obtain, or unexpected
costs in obtaining, the regulatory approval necessary to bring a potential product candidate to market could decrease our ability to generate sufficient product revenue, and our business, financial condition, results of operations and prospects may
be harmed.
The FDA, NIH and EMA have demonstrated caution in their regulation of gene therapy treatments, and ethical and
legal concerns about gene therapy and genetic testing may result in additional regulations or restrictions on the development and commercialization of such products, which may be difficult to predict.
The FDA, NIH and EMA have each expressed interest in further regulating biotechnology, including gene therapy and genetic testing. For example, the EMA advocates a risk-based approach to the development of a gene
therapy product. Agencies at both the federal and state level in the United States, as well as the U.S. congressional committees and other governments or governing agencies, have also expressed interest in further regulating the biotechnology
industry. Such action may delay or prevent commercialization any such product candidates we may seek to develop if we exercise our option agreements with Novellus.
Regulatory requirements in the United States and in other jurisdictions governing gene therapy products have changed frequently and may continue to change in the future. In January 2020, the FDA issued several new
guidance documents on gene therapy products. The FDA established the Office of Tissues and Advanced Therapies within its Center for Biologics Evaluation and Research to consolidate the review of gene therapy and related products, and established
the Cellular, Tissue and Gene Therapies Advisory Committee to advise this review. In addition to the government regulators, the IBC and IRB of each institution at which we conduct clinical trials of our product candidates, or a central IRB if
appropriate, would need to review the proposed clinical trial to assess the safety of the trial. In addition, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA or other oversight bodies to change
the requirements for approval of any of our product candidates. Similarly, the EMA governs the development of gene therapies in the EU and may issue new guidelines concerning the development and marketing authorization for gene therapy products and
require that we comply with these new guidelines. These regulatory review agencies and committees and the new requirements or guidelines they promulgate may lengthen the regulatory review process, require us to perform additional studies or trials,
increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of any product candidates we may develop if we exercise our option agreements with Novellus or lead to
significant post-approval limitations or restrictions. As we advance any such product candidates, we will be required to consult with these regulatory agencies and committees and comply with applicable requirements and guidelines. If we fail to do
so, we may be required to delay or discontinue development of such product candidates. These additional processes may result in a review and approval process that is longer than we otherwise would have expected. Delays as a result of an increased
or lengthier regulatory approval process or further restrictions on the development of our product candidates can be costly and could negatively impact our or our collaborators’ ability to complete clinical trials and commercialize our current and
future product candidates in a timely manner, if at all.
We may be unable to successfully scale up manufacturing of IRX‑2 in sufficient quality and quantity, which may
delay or prevent us from commercializing the product even if approved for marketing by the FDA or other regulatory agencies.
In order to commercialize IRX‑2 or any other product candidate we may acquire or in-license (including any products that may be developed from the licensed technology if Brooklyn exercises its option to exclusively
license certain technology from Novellus), we will need to manufacture them in large quantities. Currently, we manufacture the active pharmaceutical ingredient for IRX‑2 before it is sent to a finishing manufacturer. We are planning to increase the
capacity of our proprietary facility to produce commercial quantities of IRX‑2, if approved, and we are currently exploring options for implementing scale-up activities in anticipation of study completion and submitting applications for marketing
approval, if supported by study data. However, we may be unable to successfully increase manufacturing capability in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities.
39
Further, in order to release product and demonstrate stability of product candidates for future commercial use, our analytical methods must be validated in accordance with regulatory guidelines. We may not be able to
successfully validate, or maintain validation of, its analytical methods during scale-up or demonstrate adequate purity, stability or comparability of the biological product candidates in a timely or cost-effective manner, or at all. Even if we
believe our manufacturing processes meet all of the regulatory manufacturing requirements, the FDA will review those processes and the manufacturing facility as part of the review of any future NDA or BLA for IRX‑2, if submitted after completion of
the INSPIRE trial. If we are unable to successfully scale up the manufacture of IRX‑2 in sufficient quality and quantity, or if we encounter validation issues, the development, testing, and clinical trials of future product candidates, may be
delayed or infeasible, and regulatory approval or commercial launch of any resulting product, including IRX‑2, may be delayed or may not be successfully achieved.
We will require substantial additional capital to fund our operations, and if we fail to obtain necessary
financing, we may not be able to complete the development and commercialization of any of our product candidates.
Brooklyn expects to spend substantial capital to complete the development of, seek regulatory approvals for and commercialize IRX‑2 as well as any of other product candidate it may develop. We
will require additional capital to complete the development and potential commercialization of our product candidates. Because the length of time and activities associated with successful development of our product candidates are highly uncertain,
we are unable to estimate with certainty the actual funds we will require for development and any approved marketing and commercialization activities. Our future funding requirements, both near- and long-term, will depend on many factors,
including, but not limited to:
| • |
the timing, progress, costs and results of our Phase 2b INSPIRE clinical monotherapy study for the treatment of squamous cell cancer of the head and neck;
|
| • |
the costs of any other clinical trials we may initiate, including the costs to conduct the study and to produce the supply of IRX‑2 that may be required for our own studies or for investigator-sponsored studies;
|
| • |
the outcome, timing and cost of meeting regulatory requirements established by the FDA and other comparable foreign regulatory authorities;
|
| • |
the cost of filing, prosecuting, defending and enforcing its patent claims and other intellectual property rights;
|
| • |
the cost of defending potential intellectual property disputes, including patent infringement actions brought by third parties against it or any of its current or future product candidates;
|
| • |
the effect of competing market developments;
|
| • |
the cost and timing of completion of commercial-scale manufacturing activities;
|
| • |
the cost of establishing sales, marketing and distribution capabilities for our products in regions where we choose to commercialize our products on our own; and
|
| • |
the initiation, progress, timing and results of the commercialization of our product candidates, if approved for commercial sale.
|
Based on currently available information and our ongoing operations, we believe that our existing cash, inclusive of the funding committed by certain beneficial holders of Brooklyn’s Class A membership interests will
be sufficient for us to fund our operating expenses and capital expenditure requirements through at least the first quarter of 2022. This estimate is based on assumptions that may prove to be incorrect, and we could use our available capital
resources sooner than we currently expect. We cannot be certain that additional capital will be available on acceptable terms, or at all. If we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we may have
to significantly delay, scale back or discontinue the development or commercialization of any product candidate, or potentially discontinue operations altogether. In addition, attempting to secure additional capital may divert the time and
attention of our management from day-to-day activities and harm its product candidate development efforts. Because of the numerous risks and uncertainties associated with the development and potential commercialization of its product candidates, we
are unable to estimate the amounts of increased capital outlays, operating expenditures and capital requirements associated with its current product development programs.
40
Raising additional funds by issuing equity securities may cause dilution to existing holders, raising additional
funds through debt financings may involve restrictive covenants, and raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.
We expect that significant additional capital will be needed in the future to continue our planned operations. Until such time, if ever, that we can generate substantial product revenue, we expect to finance our cash
needs through a combination of equity offerings, debt financings, strategic alliances and license and development agreements or other collaborations. To the extent that we raise additional capital by issuing equity securities, existing stockholder
ownership may experience substantial dilution, and the securities may include preferred shares with liquidation or other preferences that could harm the rights of a common stockholder.
If we raise additional funds through collaborations, strategic alliances or marketing, distribution or licensing arrangements with third parties, it may have to relinquish valuable rights to its technologies, future
revenue streams, research programs or product candidates, or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds when needed, we may be required to delay, limit, reduce or terminate its product
development or future commercialization efforts, or grant rights to develop and market product candidates that we would otherwise develop and market themselves.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully
implement our business strategy.
Our ability to compete in the highly competitive pharmaceuticals industry depends in large part upon its ability to attract highly qualified managerial, scientific and medical personnel. In order to induce valuable
employees to remain with us, we intend to provide employees with stock options that vest over time. The value to employees of stock options that vest over time will be significantly affected by movements in the price of the common stock that it
will not be able to control and may at any time be insufficient to counteract more lucrative offers from other companies.
Our management team has expertise in many different aspects of drug development. However, we will need to hire additional personnel as we continue to develop our drug candidates. Competition for skilled personnel in
the pharmaceutical industry is intense and competition for experienced scientists may limit our ability to hire and retain highly qualified personnel on acceptable terms. Despite our efforts to retain valuable employees, members of our management,
scientific and medical teams may terminate their employment with us on short notice. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level, and senior managers as well as junior,
mid-level, and senior scientific and medical personnel.
Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than we do. Other pharmaceutical
companies also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what Brooklyn has to offer. If we are unable to continue to attract
and retain high-quality personnel, the rate and success at which we can develop and commercialize product candidates would be limited.
We or our affiliates’ employees, independent contractors, principal investigators, consultants, commercial
collaborators, service providers and other vendors or potential collaborators may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could harm our results of operations.
We are exposed to the risk that its or its affiliates’ employees and contractors, including any prospective or current principal investigators, CROs, consultants, commercial collaborators, service providers and other
vendors may engage in misconduct or other illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or other unauthorized activities that violate the laws and regulations of the FDA or other similar
regulatory bodies, including those laws that require the reporting of true, complete and accurate information to such regulatory bodies; manufacturing and the FDA’s Good Clinical Practice, or GCP, or current Good Manufacturing Practice, or cGMP,
standards; federal, state and foreign healthcare fraud and abuse laws and data privacy; or laws that require the true, complete and accurate reporting of financial information or data. In particular, sales, marketing and other business arrangements
in the healthcare industry are subject to extensive laws intended to prevent fraud, kickbacks, self-dealing, bribery, corruption, antitrust violations and other abusive practices. These laws may restrict or prohibit a wide range of business
activities, including research, manufacturing, distribution, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements.
41
Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials, creating fraudulent data in our nonclinical studies or clinical trials
or illegal misappropriation of drug product, which could result in data being eliminated from the final analysis of studies, regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter employee or
third-party misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting them from governmental investigations or other actions or lawsuits
stemming from a failure to comply with such laws or regulations. Additionally, we are subject to the risk that a person, including any person who may have engaged in any fraud or misconduct, or government agency could allege such fraud or other
misconduct, even if none occurred. Furthermore, we rely on our CROs and clinical trial sites to adequately report data from its ongoing clinical trials. For example, any failure by such parties to adequately report safety events to us in a
timely manner from any such trials may also affect the approvability of our product candidates or cause delays and disruptions for the approval of any of our product candidates, if at all. If we or our affiliates’ employees, independent
contractors, principal investigators, consultants, commercial collaborators, service providers or other vendors are alleged or found to be in violation of any such regulatory standards or requirements, or become subject to a corporate integrity
agreement or similar agreement and curtailment of our operations, it could have a significant impact on our business and financial results, including the imposition of significant civil, criminal and administrative penalties, damages, monetary
fines, suspension or delay in Brooklyn’s clinical trials, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs or formulary and pharmacy benefit management programs, FDA debarment, contractual
damages, reputational harm, diminished profits and future earnings, and additional reporting requirements and oversight, any of which could harm our ability to operate our business and results of operations.
We may not be successful in our efforts to identify and acquire or in-license additional product candidates, or
to enter into collaborations or strategic alliances for the development and commercialization of any such future product candidates.
We may seek to identify and acquire or in-license novel product candidates in the oncology field. The process by which we identify product candidates may fail to yield product candidates for
clinical development for a number of reasons, including those discussed in these risk factors and also:
| • |
potential product candidates may, upon further study, be shown to have harmful side effects or other characteristics that indicate that they are unlikely to be products that will receive marketing approval and achieve market
acceptance;
|
| • |
potential product candidates may not be effective in treating their targeted diseases; or
|
| • |
the acquisition or in-licensing transactions can entail numerous operational and functional risks, including exposure to unknown liabilities, disruption of our business, or incurrence of substantial debt
or dilutive issuances of equity securities to pay transaction consideration or costs, higher than expected acquisition or integration costs.
|
We may choose to focus our efforts and resources on a potential product candidate that ultimately proves to be unsuccessful. We also cannot be certain that, following an acquisition or in-licensing transaction,
we will achieve the revenue or specific net income that justifies such transaction. Further, time and resources spent identifying, acquiring and developing potential product candidates may distract management’s attention from our primary
business or other development programs. If we are unable to identify and acquire suitable product candidates for clinical development, this would adversely impact our business strategy, financial position and share price.
In the future, we may also decide to collaborate with other pharmaceutical companies for the development and potential commercialization of our product candidates in the United States or other countries or
territories of the world. We will face significant competition in seeking appropriate collaborators. We may not be successful in our efforts to establish a strategic partnership or other alternative arrangements for its product candidates
because such product candidates may be deemed to be at too early of a stage of development for collaborative effort and third parties may not view our product candidates as having the requisite potential to demonstrate safety and efficacy. If
and when we collaborate with a third party for development and commercialization of a product candidate, we can expect to relinquish some or all of the control over the future success of that product candidate to the third party. Our ability to
reach a definitive agreement for a collaboration will depend, among other things, upon its assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s
evaluation of a number of factors.
42
International expansion of our business exposes us to business, legal, regulatory, political, operational,
financial and economic risks associated with conducting business outside of the United States.
Part of our business strategy involves potential expansion internationally with third-party collaborators to seek regulatory approval for our product candidates outside the United States.
Doing business internationally involves a number of risks, including but not limited to:
| • |
multiple conflicting and changing laws and regulations such as tax laws, export and import restrictions, employment laws, anti-bribery and anti-corruption laws, regulatory requirements and other governmental approvals, permits and
licenses;
|
| • |
failure by us or our collaborators to obtain appropriate licenses or regulatory approvals for the sale or use of IRX‑2 or any other product candidate we may acquire or in license. if approved, due to differing regulations promulgated
by diverse countries;
|
| • |
difficulties in managing foreign operations;
|
| • |
complexities associated with managing multiple payor-reimbursement regimes or self-pay systems;
|
| • |
financial risks, such as longer payment cycles, difficulty enforcing contracts and collecting accounts receivable and exposure to foreign currency exchange rate fluctuations;
|
| • |
reduced protection for intellectual property rights;
|
| • |
natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions; and
|
| • |
failure to comply with the U.S. Foreign Corrupt Practices Act, or FCPA, including its books and records provisions and its anti-bribery provisions, the United Kingdom Bribery Act 2010, or U.K. Bribery
Act, and similar anti-bribery and anti-corruption laws in other jurisdictions, for example by failing to maintain accurate information and control over sales or distributors’ activities.
|
Any of these risks, if encountered, could significantly harm our future international expansion and operations and, consequently, negatively impact its financial condition, results of operations and cash flows.
Our business and operations would suffer in the event of system failures, cyber-attacks or a deficiency in our
cyber-security.
Our computer systems, as well as those of various third parties on which it relies, may sustain damage from computer viruses, unauthorized access, data breaches, phishing attacks, cybercriminals, natural
disasters (including hurricanes and earthquakes), terrorism, war and telecommunication and electrical failures. We rely on our third-party providers to implement effective security measures and identify and correct for any such failures,
deficiencies or breaches. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments and cyber terrorists, has generally increased as the number,
intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of its drug development
programs. For example, the loss of nonclinical or clinical trial data from completed, ongoing or planned trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To
the extent that any disruption or security breach were to result in a loss of or damage to our data or applications, or inappropriate disclosure of personal, confidential or proprietary information, we could incur liability and the further
development of any product candidate could be delayed.
43
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required
to limit commercialization of our drug candidates.
We face a potential risk of product liability as a result of the clinical testing of our drug candidates and will face an even greater risk if we commercialize our drug candidates. For
example, we may be sued if any product we develop or any materials that we use in our products allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability
claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer
protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our drug candidates. Even a successful defense would require
significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| • |
decreased demand for our drug candidates;
|
| • |
injury to our reputation;
|
| • |
withdrawal of clinical trial participants;
|
| • |
costs to defend the related litigation;
|
| • |
a diversion of management’s time and its resources;
|
| • |
substantial monetary awards to trial participants or patients;
|
| • |
product recalls, withdrawals or labeling, marketing or promotional restrictions;
|
| • |
the inability to commercialize our drug candidates; and
|
| • |
a decline in the value of the common stock.
|
Any inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we
develop. We intend to obtain product liability insurance covering our clinical trials. Although we will maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not
covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage.
We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed its coverage limitations or that are not covered by its insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We may acquire businesses, assets or products, or form strategic alliances, in the future, and we may not
realize the benefits of such acquisitions.
We may acquire additional businesses, assets or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire
businesses with promising markets or technologies, it may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter
numerous difficulties in developing, manufacturing and marketing any new delay or prevent it from realizing its expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected
synergies to justify the transaction.
Risks Related to the Discovery, Development and Regulatory Approval of our Drug Candidates
We are substantially dependent on the success of our internal development programs and our product pipeline
candidates may not successfully complete clinical trials, receive regulatory approval or be successfully commercialized.
Our future success will depend heavily on the success of our development of IRX‑2 and any other product we may acquire, license or develop (including any products that may be developed from
the licensed technology if Brooklyn exercises its option to license certain technology from Novellus). Our ability to successfully commercialize IRX‑2 and any other product we may acquire, license or develop, will depend on, among other things,
our ability to:
44
| • |
successfully complete preclinical studies and clinical trials;
|
| • |
receive regulatory approvals from the FDA, the EMA and other similar regulatory authorities;
|
| • |
establish and maintain collaborations with third parties for the development and/or commercialization of our product candidates, or otherwise build and maintain strong development, sales, distribution and marketing capabilities that
are sufficient to develop products and launch commercial sales of any approved products;
|
| • |
obtain coverage and adequate reimbursement from payors such as government health care systems, pharmacy benefit managers, and insurance companies and achieve commercially attractive levels of pricing;
|
| • |
secure acceptance of our product candidates from physicians, health care payors, patients and the medical community;
|
| • |
produce, through a validated process, in manufacturing facilities inspected and approved by regulatory authorities, including the FDA, sufficiently large quantities of our product candidates to permit successful commercialization;
|
| • |
manage our spending as expenses increase due to government mandated post-approval clinical trials and commercialization; and
|
| • |
obtain and enforce sufficient intellectual property rights for any approved products and product candidates.
|
Of the large number of drugs in development in the pharmaceutical industry, only a small percentage result in the submission of a new drug application, or NDA, or biologics licensing application, or BLA, to the
FDA and even fewer are approved for commercialization. Furthermore, even if we do receive regulatory approval to market our product candidates, any such approval may be subject to limitations on the indicated uses or patient populations for
which we may market the product. Accordingly, even if we are able to obtain the requisite financing to continue to fund our development programs, we cannot assure you that our product candidates will be successfully developed or commercialized.
If we are unable to develop, or obtain regulatory approval for, or, if approved, to successfully commercialize our product candidates, we may not be able to generate sufficient revenue to continue our business.
Our product candidates and those of any collaborators will need to undergo preclinical and clinical trials that
are time-consuming and expensive, the outcomes of which are unpredictable, and for which there is a high risk of failure. If preclinical or clinical trials of our or their product candidates fail to satisfactorily demonstrate safety and
efficacy to the FDA, the EMA and any other comparable regulatory authority, additional costs may be incurred or delays experienced in completing, the development of these product candidates, or their development may be abandoned.
The FDA in the United States, the EMA in the EU and the European Economic Area, and other comparable regulatory authorities in other jurisdictions must approve new product candidates before they can be marketed,
promoted or sold in those territories. While we have submitted an IND in the United States and comparable drug approval filings with certain foreign regulatory authorities, the timing of the filing of an NDA in the United States or comparable
filing in other nations is unknown. We must provide these regulatory authorities with data from preclinical studies and clinical trials that demonstrate that our product candidates are safe and effective for a specific indication before they
can be approved for commercial distribution. We cannot be certain that our clinical trials for our product candidates will be successful or that any of our product candidates will receive approval from the FDA, the EMA or any other comparable
regulatory authority.
Preclinical studies and clinical trials are long, expensive and unpredictable processes that can be subject to extensive delays. We cannot guarantee that any clinical trials will be conducted as planned or
completed on schedule, if at all. It may take several years and require significant expenditures to complete the preclinical studies and clinical trials necessary to commercialize a product candidate, and delays or failure are inherently
unpredictable and can occur at any stage. We may also be required to conduct additional clinical trials or other testing of our product candidates beyond the trials and testing that we contemplate, which may lead to us incurring additional
unplanned costs or result in delays in clinical development. In addition, we may be required to redesign or otherwise modify our plans with respect to an ongoing or planned clinical trial and changing the design of a clinical trial can be
expensive and time consuming. An unfavorable outcome in one or more trials would be a major setback for our product candidates and for us. An unfavorable outcome in one or more trials may require us to delay, reduce the scope of or eliminate
one or more product development programs, which could have a material adverse effect on our business, financial position, results of operations and future growth prospects.
45
Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of marketing approval for our product candidates. The FDA, EMA or
any other comparable regulatory authority may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for
our clinical trials.
In connection with clinical trials of our product candidates, we face a number of risks, including risks that:
| • |
a product candidate is ineffective or inferior to existing approved products for the same indications;
|
| • |
a product candidate causes or is associated with unacceptable toxicity or has unacceptable side effects;
|
| • |
patients may die or suffer adverse effects for reasons that may or may not be related to the product candidate being tested;
|
| • |
the results may not confirm the positive results of earlier trials;
|
| • |
the results may not meet the level of statistical significance required by the FDA, the EMA or other relevant regulatory agencies to establish the safety and efficacy of our product candidates for continued trial or marketing
approval; and
|
| • |
our collaborators may be unable or unwilling to perform under their contracts.
|
Furthermore, we sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include our
expectations regarding the commencement or completion of scientific studies, clinical trials, the submission of regulatory filings or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these
milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, the receipt of marketing approval or a commercial launch of a product. The achievement of many of these milestones may be outside of our
control. All of these milestones are based on a variety of assumptions, which may cause the timing of achievement of the milestones to vary considerably from our estimates. If we fail to achieve milestones in the timeframes we expect, the
commercialization of our product candidates may be delayed, we may not be entitled to receive certain contractual payments, which could have a material adverse effect on our business, financial position, results of operations and future growth
prospects.
We may find it difficult to enroll patients in our clinical trials, which could delay or prevent us from
proceeding with clinical trials of our product candidates.
Identifying and qualifying patients to participate in clinical trials of our product candidates is critical to our success. The timing of our clinical trials depends on our ability to recruit patients to
participate as well as the completion of required follow-up periods. Patients may be unwilling to participate in our clinical trials because of negative publicity from adverse events related to novel therapeutic approaches, competitive clinical
trials for similar patient populations, the existence of current treatments or for other reasons. Enrollment risks are heightened with respect to certain indications that we may target for one or more of our product candidates that may be rare
diseases, which may limit the pool of patients that may be enrolled in our planned clinical trials. The timeline for recruiting patients, conducting trials and obtaining regulatory approval of our product candidates may be delayed, which could
result in increased costs, delays in advancing our product candidates, delays in testing the effectiveness of our product candidates or termination of the clinical trials altogether.
We may not be able to identify, recruit and enroll a sufficient number of patients, or those with the required or desired characteristics, to complete our clinical trials in a timely manner.
For example, due to the nature of the indications that we are initially targeting, patients with advanced disease progression may not be suitable candidates for treatment with our product candidates and may be ineligible for enrollment in our
clinical trials. Therefore, early diagnosis in patients with our target diseases is critical to our success. Patient enrollment and trial completion is affected by factors including the:
| • |
size of the patient population and process for identifying subjects;
|
46
| • |
design of the trial protocol;
|
| • |
eligibility and exclusion criteria;
|
| • |
safety profile, to date, of the product candidate under study;
|
| • |
perceived risks and benefits of the product candidate under study;
|
| • |
perceived risks and benefits of our approach to treatment of diseases;
|
| • |
availability of competing therapies and clinical trials;
|
| • |
severity of the disease under investigation;
|
| • |
degree of progression of the subject’s disease at the time of enrollment;
|
| • |
proximity and availability of clinical trial sites for prospective subjects;
|
| • |
ability to obtain and maintain subject consent;
|
| • |
risks that enrolled patients will drop out before completion of the trial;
|
| • |
patient referral services of physicians; and
|
| • |
ability to monitor subjects adequately during and after treatment.
|
If we have difficulty enrolling a sufficient number of patients to conduct our clinical trials as planned, we may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an
adverse effect on our business, financial condition, results of operations and prospects.
Results of preclinical studies and early clinical trials may not be predictive of results of future clinical
trials.
The outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results of clinical trials do not necessarily predict success in the results
of completed clinical trials. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in earlier development, and we could face similar
setbacks. For example, results from the Phase 2b INSPIRE trial of IRX‑2 may not be positive or replicated at clinical trial sites in a later stage clinical trial conducted by us or our collaborators. The design of a clinical trial can determine
whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We may be unable to design and execute a clinical trial to support marketing
approval.
Preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical
trials have nonetheless failed to obtain marketing approval for the product candidates. Even if we, or any collaborators, believe that the results of clinical trials for our product candidates warrant marketing approval, the FDA or comparable
foreign regulatory authorities may disagree and may not grant marketing approval of our product candidates.
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures
set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen and other clinical trial protocols and the rate of dropout among clinical trial participants. If we fail to
receive positive results in clinical trials of our product candidates, the development timeline and regulatory approval and commercialization prospects for our most advanced product candidates, and, correspondingly, our business and financial
prospects would be negatively impacted.
Our current or future product candidates may cause undesirable side effects or have other properties when used
alone or in combination with other approved products or investigational new drugs that could halt their clinical development, prevent their marketing approval, limit their commercial potential or result in significant negative consequences.
Undesirable or clinically unmanageable side effects could occur and cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or
denial of marketing approval by the FDA or comparable foreign regulatory authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics.
47
If unacceptable side effects arise in the development of our product candidates, we, the FDA or comparable foreign regulatory authorities, the Institutional Review Boards, or IRBs, or independent ethics
committees at the institutions in which our studies are conducted, or the Data Safety Monitoring Board, or DSMB, could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory authorities could order us to cease
clinical trials or deny approval of our product candidates for any or all targeted indications. Treatment-related side effects could also affect patient recruitment or the ability of enrolled subjects to complete the trial, or result in
potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We may be required to train medical personnel using our product candidates to understand the side
effect profiles for our clinical trials and upon any commercialization of any of our product candidates. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury or
death. Any of these occurrences may prevent us from achieving or maintaining market acceptance of the affected product candidate and may harm our business, financial condition and prospects significantly.
Moreover, clinical trials of our product candidates are conducted in carefully defined sets of patients who have agreed to enter into clinical trials. Consequently, it is possible that our
clinical trials may indicate an apparent positive effect of a product candidate that is greater than the actual positive effect, if any, or alternatively fail to identify undesirable side effects. If, following approval of a product candidate,
we, or others, discover that the product is less effective than previously believed or causes undesirable side effects that were not previously identified, any of the following consequences could occur:
| • |
regulatory authorities may withdraw their approval of the product or seize the product;
|
| • |
we, or any collaborators, may need to recall the product, or be required to change the way the product is administered or conduct additional clinical trials;
|
| • |
additional restrictions may be imposed on the marketing of, or the manufacturing processes for, the particular product;
|
| • |
we may be subject to fines, injunctions or the imposition of civil or criminal penalties;
|
| • |
regulatory authorities may require the addition of labeling statements, such as a boxed warning or a contraindication;
|
| • |
we, or any collaborators, may be required to create a medication guide outlining the risks of the previously unidentified side effects for distribution to patients;
|
| • |
we, or any collaborators, could be sued and held liable for harm caused to patients;
|
| • |
the product may become less competitive; and
|
| • |
our reputation may suffer.
|
If any of our current or future product candidates fail to demonstrate safety and efficacy in clinical trials or do not gain marketing approval, we will not be able to generate revenue and our business will be
harmed. Any of these events could harm our business and operations and could negatively impact the price of our common stock.
We may not be successful in our efforts to identify and acquire or in-license additional product candidates.
Although we intend to explore other therapeutic opportunities in addition to the product candidates that we are currently developing, we may fail to identify other product candidates for clinical development
for a number of reasons. For example, our research methodology may not be successful in identifying potential product candidates or those we identify may be shown to have harmful side effects or other characteristics that make them
unmarketable or unlikely to receive regulatory approval. Additional product candidates will require additional, time-consuming development efforts prior to commercial sale, including preclinical studies, clinical trials and approval by the
FDA and/or applicable foreign regulatory authorities. All product candidates are prone to the risks of failure that are inherent in pharmaceutical product development. If we fail to identify and develop additional potential product
candidates, we may be unable to grow our business and our results of operations could be materially harmed.
48
We may expend our limited resources to pursue a particular product candidate or indication and fail to
capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we intend to focus on developing product candidates for specific indications that we identify as most likely to succeed, in terms of both their
potential for marketing approval and commercialization, and there can be no assurance that the products we focus on will succeed. Because we must decide where to focus our resources, we may forego or delay pursuit of opportunities with other
product candidates or for other indications that may later prove to have greater commercial potential.
Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and
product candidates for specific indications may not yield any commercially viable product candidates. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable
rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to the product candidate.
If we fail to maintain orphan drug exclusivity for IRX‑2 or we fail to obtain or maintain such exclusivity for
any future drug candidate we may license, our competitors may sell products to treat the same conditions, and our revenues would be significantly adversely affected.
We have been granted orphan drug designation for IRX‑2 for head or neck cancer by the FDA. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant
funding towards clinical trial costs, tax advantages and user-fee waivers. The company that first obtains FDA approval for a designated orphan drug for a given rare disease receives marketing exclusivity for use of that drug for the stated
disease or condition for a period of seven years, with an additional six months if for a pediatric indication. Orphan drug exclusive marketing rights may be lost if the FDA later determines that the request for designation was materially
defective, a subsequent product is deemed clinically superior, or if the manufacturer is unable to deliver sufficient quantity of the drug.
In the EU, the EMA’s Committee for Orphan Medicinal Products, or COMP, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of
life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU Community and for which no satisfactory method of diagnosis, prevention, or treatment has been authorized (or the product would be
a significant benefit to those affected). Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without
incentives, it is unlikely that sales of the drug in the EU would be sufficient to justify the necessary investment in developing the medicinal product. An EU orphan drug designation entitles a party to financial incentives such as reduction of
fees or fee waivers and 10 years of market exclusivity is granted following medicinal product approval. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the
product is sufficiently profitable not to justify maintenance of market exclusivity. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or
shorten the duration of, the regulatory review and approval process.
Because the extent and scope of patent protection for IRX‑2 may be particularly limited, orphan drug designation is especially important. We plan to rely on the orphan exclusivity period to maintain a competitive
position. However, if we cannot maintain orphan exclusivity for our IRX‑2, our competitors may then sell the same drug to treat the same condition and our revenues will be reduced. Also, without strong patent protection, competitors may sell a
generic version upon the expiration of orphan exclusivity, if our patent position is not upheld.
Even if we obtain orphan drug designation for our future drug candidates, we may not fulfill the criteria for exclusivity, or we may not be the first to obtain marketing approval for any orphan indication.
Further, even if we obtain orphan drug exclusivity for a particular product, that exclusivity may not effectively protect the product from competition because different drugs can be approved for the same condition. Even after an orphan drug is
approved, the FDA can subsequently approve a drug for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care. The FDA can discontinue Orphan Drug exclusivity after it
has been granted if the orphan drug cannot be manufactured in sufficient quantities to meet demand.
49
Orphan Drug Designation and Fast Track Designation may not actually lead to a faster review process.
Under the Prescription Drug User Fee Act, the FDA has a goal of responding to NDAs or BLAs for new molecular entities within 10 months of the date that it is filed for standard review, but the timeframe is also
often extended. We have in the past and we may in the future seek approval of IRX‑2 or any other drug candidate we may acquire or license under programs designed to accelerate the FDA’s review and approval of NDAs or BLAs. For example, fast
track designation is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions that fill an unmet clinical need. The purpose is to get important new drugs to the patient earlier. In our case,
IRX‑2 has been granted fast track designation for the treatment of head and neck cancer. In the future, we may request fast track designation from the FDA for other diseases or for other drug candidates we may acquire or in-license, but we
cannot assure that we will obtain such designations. Further, even if we obtain fast track designation, the designation does not guarantee FDA approval of any NDA or BLA that we file, that the development program or review timeline will
ultimately be shorter than if we had not obtained the designations, or that the FDA will not request additional information, including requesting additional clinical studies (although potentially a post-marketing requirement), during its
review. Any request for additional information or clinical data could delay the FDA’s timely review of any NDA or BLA that we submit.
Obtaining and maintaining marketing approval of our current and future product candidates in one jurisdiction
does not mean that we will be successful in obtaining marketing approval of our current and future product candidates in other jurisdictions.
Obtaining and maintaining marketing approval of our current and future product candidates in one jurisdiction does not guarantee that we will be able to obtain or maintain marketing approval in any other
jurisdiction, while a failure or delay in obtaining marketing approval in one jurisdiction may have a negative effect on the marketing approval process in others. For example, even if the FDA grants marketing approval of a product candidate,
comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and
administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials as clinical studies conducted in one jurisdiction may not be accepted by regulatory
authorities in other jurisdictions. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to
charge for our products is also subject to approval. We do not have experience in obtaining reimbursement or pricing approvals in international markets. Further, pricing obtained in other jurisdictions may impact pricing realized in the United
States and in other jurisdictions where the product is approved.
Obtaining marketing approvals and compliance with regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our products in certain
countries outside of the United States. If we fail to comply with the regulatory requirements in international markets and/or receive applicable marketing approvals, our target market will be reduced and our ability to realize the full market
potential of our product candidates will be harmed.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to
fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and wastes generating in our
manufacturing facility. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from the use of hazardous materials, we could be held liable for any resulting damages, and
the amount of the liability could exceed our resources. We could also incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
Risks Related to the Commercialization of our Product Candidates and Other Regulatory Compliance Matters
Even if we complete the necessary preclinical studies and clinical trials, the marketing approval process is
expensive, time consuming and uncertain and may prevent us or any collaborators from obtaining approvals for the commercialization of some or all of our product candidates. As a result, we cannot predict when or if, and in which territories,
we, or any collaborators, will obtain marketing approval to commercialize a product candidate.
50
The process of obtaining marketing approvals, both in the United States and abroad, is lengthy, expensive and uncertain. It may take many years, if approval is obtained at all, and can vary substantially based
upon a variety of factors, including the type, complexity and novelty of the product candidates involved. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory
authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of
manufacturing facilities by, the regulatory authorities. The FDA or other regulatory authorities may determine that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects,
toxicities or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that
render the approved product not commercially viable.
In addition, changes in marketing approval policies during the development period, changes in or the enactment or promulgation of additional statutes, regulations or guidance or changes in regulatory review for
each submitted product application, may cause delays in the approval or rejection of an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data
are insufficient for approval and require additional preclinical, clinical or other studies. Varying interpretations of the data obtained from preclinical and clinical testing could delay, limit or prevent marketing approval of a product
candidate. We cannot commercialize a product until the appropriate regulatory authorities have reviewed and approved the product candidate. Even if our product candidates demonstrate safety and efficacy in clinical trials, the regulatory
agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or
restrictions on approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product
development, clinical trials and the review process. Any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product commercially unviable.
Moreover, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain
circumstances, we may be required to report some of these relationships to the FDA or other regulatory authority. The FDA or other regulatory authority may conclude that a financial relationship between us and a principal investigator has
created a conflict of interest or otherwise affected interpretation of the study. The FDA or other regulatory authority may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the
clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA or other regulatory authority, as the case may be, and may ultimately lead to the denial of marketing
approval of one or more of our product candidates.
In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our product candidates. For example, regulatory agencies may approve a
product candidate for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. Regulators may approve a product candidate for a smaller patient population, a different drug
formulation or a different manufacturing process, than we are seeking. If we are unable to obtain necessary regulatory approvals, or more limited regulatory approvals than we expect, our business, prospects, financial condition and results of
operations may suffer.
Any delay in obtaining or failure to obtain required approvals could negatively impact our ability to generate revenue from the particular product candidate, which likely would result in significant harm to our
financial position and adversely impact the price of our common stock.
51
We currently have no marketing, sales or distribution infrastructure with respect to our product candidates. If
we are unable to develop our sales, marketing and distribution capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our product candidates.
We currently have no marketing, sales or distribution capabilities and have limited sales or marketing experience within our organization. If one or more of our product candidates is approved, we intend either to
establish a sales and marketing organization with technical expertise and supporting distribution capabilities to commercialize that product candidate, or to outsource this function to a third party. There are risks involved with either
establishing our own sales and marketing capabilities and entering into arrangements with third parties to perform these services.
Recruiting and training an internal commercial organization is expensive and time consuming and could delay any product launch. Some or all of these costs may be incurred in advance of any approval of any of our
product candidates. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these
commercialization expenses. This may be costly and our investment would be lost if we cannot retain or reposition our sales and marketing personnel. In addition, we may not be able to hire a sales force in the United States or other target
market that is sufficient in size or has adequate expertise in the medical markets that we intend to target.
Factors that may inhibit our efforts to commercialize our product candidates on our own include:
| • |
the inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
|
| • |
the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future product that we may develop;
|
| • |
the lack of complementary treatments to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and
|
| • |
unforeseen costs and expenses associated with creating an independent sales and marketing organization.
|
If we enter into arrangements with third parties to perform sales, marketing and distribution services, our product revenue or the profitability to us from these revenue streams is likely to be lower than if we
were to market and sell any product candidates that we develop ourselves. In addition, we may not be successful in entering into arrangements with third parties to sell and market our product candidates or may be unable to do so on terms that
are favorable to us. We likely will have little control over such third parties and any of them may fail to devote the necessary resources and attention to sell and market our product candidates effectively. If we do not establish sales and
marketing capabilities successfully, either on our own or in collaboration with third parties, we may not be successful in commercializing our product candidates.
The market opportunities for any current or future product candidate we develop, if and when approved, may be
limited to those patients who are ineligible for established therapies or for whom prior therapies have failed, and therefore may be small.
Cancer therapies are sometimes characterized as first-line, second-line, or third-line, and the FDA often approves new therapies initially only for third-line use. When cancer is detected early enough, first-line
therapy, usually chemotherapy, hormone therapy, surgery, radiation therapy, immunotherapy or a combination of these, is sometimes adequate to cure the cancer or prolong life without a cure. Second- and third-line therapies are administered to
patients when prior therapy is not effective. We may initially seek approval of IRX‑2 and any other product candidates we develop as a therapy for patients who have received one or more prior treatments. Subsequently, for those products that
prove to be sufficiently beneficial, if any, we would expect to seek approval potentially as a first-line therapy, but there is no guarantee that product candidates we develop, even if approved, would be approved for first-line therapy, and,
prior to any such approvals, we may have to conduct additional clinical trials.
The number of patients who have the cancers we are targeting may turn out to be lower than we expect, or may change as we develop the product candidate (e.g., lower rates of smoking around the globe may lead to
lower incidence levels of head and neck cancer at the time of approval). Additionally, the potentially addressable patient population for our current programs or future product candidates may be limited, if and when approved. Even if we obtain
significant market share for any product candidate, if and when approved, if the potential target populations are small, we may never achieve profitability without obtaining marketing approval for additional indications, including use as first-
or second-line therapy.
52
Even if we receive marketing approval of a product candidate, we will be subject to ongoing regulatory
obligations and continued regulatory review, which may result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products, if
approved.
Any marketing approvals that we receive for any current or future product candidate may be subject to limitations on the approved indicated uses for which the product may be marketed or the
conditions of approval, or contain requirements for potentially costly post-market testing and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a Risk Evaluation and Mitigation Strategy, or
REMS, as a condition of approval of any product candidate, which could include requirements for a medication guide, physician education, development of communication plans or additional elements to ensure safe use, such as restricted
distribution methods, patient registries and other risk minimization tools. If the FDA or a comparable foreign regulatory authority approves a product candidate, the manufacturing processes, labeling, packaging, distribution, adverse event
reporting, storage, advertising, promotion, import and export and record keeping for the product candidate will be subject to extensive and ongoing regulatory requirements. These requirements include, among others, prohibitions on the promotion
of an approved product for uses not included in the product’s approved labeling, submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current Good Manufacturing Practice, or
cGMP, and Good Clinical Practice, or GCP, for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with any approved candidate, including adverse events of unanticipated severity or frequency, or
with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • |
restrictions on the labeling, distribution, marketing or manufacturing of the product, withdrawal of the product from the market, or product recalls;
|
| • |
untitled and warning letters, or holds on clinical trials;
|
| • |
refusal by the FDA to approve pending applications or supplements to approved applications we filed or suspension or revocation of license approvals;
|
| • |
requirements to conduct post-marketing studies or clinical trials;
|
| • |
restrictions on coverage by third-party payors;
|
| • |
fines, restitution or disgorgement of profits or revenues;
|
| • |
suspension or withdrawal of marketing approvals;
|
| • |
product seizure or detention, or refusal to permit the import or export of the product; and
|
| • |
injunctions or the imposition of civil or criminal penalties.
|
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay marketing approval of a product. We cannot predict the
likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption
of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
We face significant competition and if our competitors develop and market products that are more effective,
safer or less expensive than the product candidates we develop, our commercial opportunities will be negatively impacted.
The life sciences industry is highly competitive. We are currently developing therapeutics that will compete, if approved, with other products and therapies that currently exist, are being developed or will in
the future be developed, some of which we may not currently be aware.
53
We have competitors both in the United States and internationally, including major multinational pharmaceutical companies, established biotechnology companies, specialty pharmaceutical companies, universities and
other research institutions. Many of our competitors have significantly greater financial, manufacturing, marketing, product development, technical and human resources than we do. Large pharmaceutical companies, in particular, have extensive
experience in clinical testing, obtaining marketing approvals, recruiting patients and manufacturing pharmaceutical products. These companies also have significantly greater research and marketing capabilities than we do and may also have
products that have been approved or are in late stages of development, and collaborative arrangements in our target markets with leading companies and research institutions. Established pharmaceutical companies may also invest heavily to
accelerate discovery and development of novel compounds or to in-license novel compounds that could make the product candidates that we develop obsolete. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in
even more resources being concentrated among a smaller number of our competitors. As a result of all of these factors, our competitors may succeed in obtaining patent protection and/or marketing approval or discovering, developing and
commercializing products in our field before we do.
There are a large number of companies developing or marketing treatments for cancer, including many major pharmaceutical and biotechnology companies. These treatments consist both of small molecule drug products,
such as traditional chemotherapy, as well as novel immunotherapies. Our commercial opportunities could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe
effects, are more convenient, have a broader label, are marketed more effectively, are reimbursed or are less expensive than any products that we may develop. Our competitors also may obtain FDA, EMA or other marketing approval for their
products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. Even if the product candidate we develop achieve marketing approval,
they may be priced at a significant premium over competitive products if any have been approved by then, resulting in reduced competitiveness.
Smaller and other early stage companies may also prove to be significant competitors. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing
clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. In addition, the biopharmaceutical industry is characterized by rapid technological
change. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our product candidates obsolete, less competitive or not
economical.
The commercial success of any current or future product candidate will depend upon the degree of market
acceptance by physicians, patients, payors and others in the medical community.
We have never commercialized a product, and even if we obtain any regulatory approval for our product candidates, the commercial success of our product candidates will depend in part on the
medical community, patients, and payors accepting our product candidates as effective, safe and cost-effective. Any product that we bring to the market may not gain market acceptance by physicians, patients, payors and others in the medical
community. Physicians are often reluctant to switch their patients from existing therapies even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are
currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of reimbursement for existing therapies.
| • |
The degree of market acceptance of these product candidates, if approved for commercial sale, will depend on a number of factors, including:
|
| • |
the potential efficacy and potential advantages over alternative treatments;
|
| • |
the frequency and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling;
|
| • |
the frequency and severity of any side effects resulting from follow-up requirements for the administration of our product candidates;
|
| • |
the relative convenience and ease of administration;
|
| • |
the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
|
| • |
the strength of marketing and distribution support and timing of market introduction of competitive products;
|
54
| • |
formulary acceptance;
|
| • |
publicity concerning our products or competing products and treatments; and
|
| • |
sufficient third-party insurance coverage and adequate reimbursement.
|
Even if a product candidate displays a favorable efficacy and safety profile in preclinical studies and clinical trials, market acceptance of the product, if approved for commercial sale, will not be known until
after it is launched. Our efforts to educate the medical community and payors on the benefits of our product candidates may require significant resources and may never be successful. Such efforts to educate the marketplace may require more
resources than are required by the conventional technologies marketed by our competitors. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable.
If the market opportunities for our product candidates are smaller than we believe they are, our product
revenues may be adversely affected, and our business may suffer.
We currently focus our research and product development on treatments for oncology indications. Our understanding of both the number of people who have these diseases, as well as the subset of people with these
diseases who have the potential to benefit from treatment with our product candidates, are based on estimates. These estimates may prove to be incorrect and new studies may reduce the estimated incidence or prevalence of these diseases. Patient
identification efforts also influence the ability to address a patient population. If efforts in patient identification are unsuccessful or less impactful than anticipated, we may not address the entirety of the opportunity we are seeking.
The insurance coverage and reimbursement status of newly-approved products is uncertain. Failure to obtain or
maintain adequate coverage and reimbursement for any of our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The availability and extent of reimbursement by governmental and private payors is essential for most patients to be able to afford expensive treatments. Sales of our product candidates will depend substantially,
both domestically and abroad, on the extent to which the costs of our product candidates will be paid by private payors, such as private health coverage insurers, health maintenance, managed care, pharmacy benefit and similar healthcare
management organizations, or reimbursed by government health care programs, such as Medicare and Medicaid. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement. If reimbursement is not
available, or is available only at limited levels, we may not be able to successfully commercialize our product candidates, even if approved. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to
establish or maintain pricing sufficient to realize a sufficient return on our investment.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. In the United States, the principal decisions about coverage and reimbursement for new medicines
are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services, as the CMS decides whether and to what extent a new medicine will be covered and reimbursed under
Medicare. Private payors tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to coverage and reimbursement for novel products such as ours, as there is no body of established practices and
precedents for these new products. Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is: (1) a covered benefit under its health plan; (2)
safe, effective and medically necessary; (3) appropriate for the specific patient; (4) cost-effective; and (5) neither experimental nor investigational. In the United States, no uniform policy of coverage and reimbursement for products exists
among third-party payors. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no
assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Even if we obtain coverage for a given product, the resulting reimbursement payment rates might not be adequate for us to achieve
or sustain profitability or may require co-payments that patients find unacceptably high. Third-party payors may limit coverage to specific drug products on an approved list, also known as a formulary, which might not include all of the
approved drugs for a particular indication.
Additionally, third-party payors may not cover, or provide adequate reimbursement for, long-term follow-up evaluations required following the use of product candidates. Patients are unlikely to use our product
candidates unless coverage is provided, and reimbursement is adequate to cover a significant portion of the cost of our product candidates. There is significant uncertainty related to insurance coverage and reimbursement of newly approved
products. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
55
Moreover, increasing efforts by governmental and third-party payors in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of
reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty
drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of
prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. We expect to experience pricing pressures in connection with the
sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, cost containment initiatives and additional legislative changes.
Outside the United States, certain countries, including a number of member states of the EU, set prices and reimbursement for pharmaceutical products, or medicinal products, as they are commonly referred to in
the EU. These countries have broad discretion in setting prices and we cannot be sure that such prices and reimbursement will be acceptable to us or our collaborators. If the regulatory authorities in these jurisdictions set prices or
reimbursement levels that are not commercially attractive for us or our collaborators, our revenues from sales by us or our collaborators, and the potential profitability of our drug products, in those countries would be negatively affected. An
increasing number of countries are taking initiatives to attempt to reduce large budget deficits by focusing cost-cutting efforts on pharmaceuticals for their state-run health care systems. These international price control efforts have
impacted all regions of the world but have been most drastic in the EU. Additionally, some countries require approval of the sale price of a product before it can be lawfully marketed. In many countries, the pricing review period begins after
marketing or product licensing approval is granted. To obtain reimbursement or pricing approval in some countries, we, or any collaborators, may be required to conduct a clinical trial that compares the cost-effectiveness of our product to
other available therapies. As a result, we might obtain marketing approval for a product in a particular country, but then may experience delays in the reimbursement approval of our product or be subject to price regulations that would delay
our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country. Within the EU, the establishment of lower
pricing by one member-state may establish a pricing ceiling for the entirety of the EU.
Moreover, efforts by governments and payors, in the United States and abroad, to cap or reduce healthcare costs may cause such organizations to limit both coverage and level of reimbursement for new products
approved and, as a result, they may not cover or provide adequate reimbursement for our product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty drug practices.
Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare,
review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. We expect to experience pricing pressures in connection with the sale of any of our product
candidates, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and
other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at
unsatisfactory levels, our business could be harmed.
IRX‑2, and any other biological product we may acquire or in-license and for which we intend to seek approval,
may face competition sooner than anticipated.
Our ability to compete may be affected in many cases by insurers or other third-party payors seeking to encourage the use of biosimilar products. The Biologics Price Competition and Innovation Act of 2009, which
was included in the Patient Protection and Affordable Care Act, or PPACA, authorized the FDA to approve similar versions of innovative biologics, commonly known as biosimilars. Under the PPACA, a manufacturer may submit an application for
licensure of a biologic product that is “biosimilar to” or “interchangeable with” a previously approved biologic product or “reference product.” Manufacturers may not submit an application for a biosimilar to the FDA until four years following
approval of the reference product, and the FDA may not approve a biosimilar product until 12 years from the date on which the reference product was approved. Even if IRX‑2 or any other biological product we may acquire or license, if approved,
are deemed to be reference products eligible for exclusivity, another company could market a competing version of that product if the FDA approves a full BLA or such product containing the sponsor’s own preclinical data and data from adequate
and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. Additionally, from time to time, there are proposals to repeal or modify the PPACA, including proposals that could significantly shorten the
exclusivity period for biologics.
56
If the FDA or comparable foreign regulatory authorities approve generic versions of any of our product
candidates that receive marketing approval, or such authorities do not grant such products appropriate periods of data exclusivity before approving generic versions of such products, the sales of such products could be adversely affected.
In the United States, manufacturers may seek approval of biosimilar versions of biologics approved by the FDA under a BLA through submission of abbreviated biologic license applications, or ABLAs. In support of
an ABLA, a biosimilar manufacturer generally must show to the satisfaction of the FDA that its product is similar to the original biologic product. Biosimilar products may be less costly to bring to market than the original biologic and
companies that produce biosimilar products are sometimes able to offer them at lower prices. Thus, following the introduction of a biosimilar product, a significant percentage of the sales of the original biologic may be lost to the biosimilar
product, and the price of the original biologic product may be lowered.
Competition that our products may face from biosimilar versions of our products could negatively impact our future revenue, profitability and cash flows and substantially limit our ability to obtain a return on
our investments in those product candidates.
We may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims
laws, health information privacy and security laws, and other health care laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations will be directly, or indirectly through our
prescribers, customers and purchasers, subject to various federal and state fraud and abuse laws and regulations, including, without limitation, the federal Health Care Program Anti-Kickback Statute, or Anti-Kickback Statute, the federal civil
and criminal False Claims Act and Physician Payments Sunshine Act and regulations. These laws will impact, among other things, our proposed sales, marketing and educational programs. In addition, we may be subject to patient privacy laws by
both the federal government and the states in which we conduct our business. The laws that will affect our operations include, but are not limited to:
| • |
the Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration (including any kickback, bribe or rebate), directly or
indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, either the referral of an individual, or the purchase, lease, order, arrangement, or recommendation of any good, facility, item or service for which
payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. “Remuneration” has been interpreted broadly to include anything of value. A person or entity does not need to have
actual knowledge of the Anti-Kickback Statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the
Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or FCA, or federal civil money penalties. The Anti-Kickback Statute has been interpreted to apply to arrangements between
pharmaceutical manufacturers on the one hand and prescribers, purchasers, and formulary managers on the other. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution;
|
| • |
the federal civil and criminal false claims laws and civil monetary penalty laws, including the FCA, which impose criminal and civil penalties against individuals or entities for, among other things: knowingly presenting, or causing
to be presented, to the federal government, claims for payment that are false or fraudulent; knowingly making, using or causing to be made or used, a false statement of record material to a false or fraudulent claim or obligation to pay
or transmit money or property to the federal government. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or
fraudulent claims. The FCA also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the FCA and to share in any monetary recovery;
|
57
| • |
the beneficiary inducement provisions of the CMP Law, which prohibits, among other things, the offering or giving of remuneration, which includes, without limitation, any transfer of items or services for free or for less than fair
market value (with limited exceptions), to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection of a particular supplier of items or services reimbursable by a
federal or state governmental program;
|
| • |
the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit a person from knowingly and willfully executing, or attempting to execute, a scheme to
defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program,
regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious, or fraudulent statements or
representations in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; similar to the Anti-Kickback Statute, a person or entity does not need to have actual knowledge
of the statute or specific intent to violate it in order to have committed a violation;
|
| • |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and their respective implementing regulations, which impose requirements on certain healthcare providers, health plans, and
healthcare clearinghouses, known as covered entities, as well as their respective business associates, individuals and entities that perform services on their behalf that involve the use or disclosure of individually identifiable health
information, relating to the privacy, security and transmission of individually identifiable health information;
|
| • |
federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; and
|
| • |
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers.
|
Additionally, we are subject to state and foreign equivalents of each of the healthcare laws and regulations described above, among others, some of which may be broader in scope and may apply regardless of the
payer. Many U.S. states have adopted laws similar to the Anti-Kickback Statute and False Claims Act, and may apply to our business practices, including, but not limited to, research, distribution, sales or marketing arrangements and claims
involving healthcare items or services reimbursed by non-governmental payors, including private insurers. In addition, some states have passed laws that require pharmaceutical companies to comply with the April 2003 Office of Inspector General
Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s Code on Interactions with Healthcare Professionals. Several states also impose other marketing restrictions or
require pharmaceutical companies to make marketing or price disclosures to the state. There are ambiguities as to what is required to comply with these state requirements, and if we fail to comply with an applicable state law requirements we
could be subject to penalties. Finally, there are state and foreign laws governing the privacy and security of health information, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating
compliance efforts.
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of our business activities could be subject to challenge under
one or more of such laws. Law enforcement authorities are increasingly focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Efforts to ensure that our current and future
business arrangements with third parties, and our business generally, will comply with applicable healthcare laws and regulations will involve substantial costs. If our operations, including our arrangements with physicians and other healthcare
providers, some of whom receive stock options as compensation for services provided, are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without
limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from
participation in federal and state healthcare programs (such as Medicare and Medicaid), additional reporting requirements and/or oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of
non-compliance with these laws, and individual imprisonment, any of which could adversely affect our ability to operate our business and our financial results. Any action for violation of these laws, even if successfully defended, could cause a
pharmaceutical manufacturer to incur significant legal expenses and divert management’s attention from the operation of the business. Prohibitions or restrictions on sales or withdrawal of future marketed products could materially affect
business in an adverse way.
58
Healthcare legislative reform measures and constraints on national budget social security systems may have a
material adverse effect on our business and results of operations.
Payors, whether domestic or foreign, or governmental or private, are developing increasingly sophisticated methods of controlling healthcare costs and those methods are not always specifically adapted for new
technologies such as those we are developing. In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system that could impact our ability to sell our
products profitably. In particular, in the United States, the ACA was enacted in 2010 which, among other things, subjects biologic products to potential competition by lower-cost biosimilars; addresses a new methodology by which rebates owed by
manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; increases the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program;
extends the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; subjects manufacturers to new annual fees and taxes for certain branded prescription drugs; and provides
incentives to programs that increase the federal government’s comparative effectiveness research.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the current administration to repeal or replace certain aspects of the ACA.
Further, since January 2017, President Trump has signed two Executive Orders and other directives designed to delay the implementation of certain provision of the ACA or otherwise circumvent some of the requirements for health insurance
mandated by the ACA. In addition, CMS recently issued a final rule that will give states greater flexibility, starting in 2020, in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of
relaxing the essential health benefits required under the ACA for plans sold through such marketplaces.
Concurrently, Congress has considered legislation that would repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of
certain taxes under the ACA have been signed into law. The Tax Cuts and Jobs Act of 2017, or TCJA, includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals
who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal
year 2018 that delayed the implementation of certain ACA-mandated fees, including the so-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain high cost employer-sponsored insurance
plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device exercise tax on non-exempt medical devices. Further, the Bipartisan Budget Act of 2018, or BBA, among other things, amends the
ACA, effective January 1, 2019, to increase from 50 percent to 70 percent the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans,
commonly referred to as the “donut hole.” More recently, in July 2018, CMS published a final rule permitting further collections and payments to and from certain ACA qualified health plans and health insurance issuers under the ACA risk
adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. Congress also could consider additional legislation to repeal or replace other elements of the
ACA. Thus, the full impact of the ACA, any law repealing or replacing elements of it, and the political uncertainty surrounding any repeal or replacement legislation on our business remains unclear.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for
spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.5 trillion for the years 2013 through 2021, was unable to reach required goals, thereby
triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in April 2013, and due to subsequent legislative
amendments, including the BBA, will remain in effect through 2027 unless additional Congressional action is taken. In January 2013, the American Taxpayer Relief Act of 2012, was signed into law, which, among other things, further reduced
Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
59
Also, there has been heightened governmental scrutiny recently over the manner in which drug manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and
proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program
reimbursement methodologies for drug products. For example, the current administration released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition,
increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. For example, in November 2018, CMS
issued a proposed rule for comment that would, among other things, provide Medicare prescription drug plans under Part D more transparency in pricing and greater flexibility to negotiate discounts for, and in certain circumstances exclude,
drugs in the six “protected” formulary classes and allow Medicare Advantage plans to use certain drug management tools such as step therapy for physician-administered drugs. Although a number of these, and other proposed measures will require
authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and
containing or lowering the cost of healthcare. We cannot predict the initiatives that may be adopted in the future. The continuing efforts of these governments and other payors to contain or reduce costs of healthcare and/or impose price
controls may adversely affect:
| • |
the demand for our product candidates, if we obtain regulatory approval;
|
| • |
our ability to set a price that we believe is fair for our products;
|
| • |
our ability to generate revenue and achieve or maintain profitability;
|
| • |
the level of taxes that we are required to pay; and
|
| • |
the availability of capital.
|
Any denial in coverage or reduction in reimbursement from Medicare or any other government programs may result in a similar denial or reduction in payments from private payors, which may adversely affect our
future profitability.
Risks Related to our Dependence on Third Parties
We rely, and expect to continue to rely, on third parties to conduct our clinical studies, and those third parties may not perform satisfactorily,
including failing to meet deadlines for the completion of such studies.
Outside of the INSPIRE trial, each of the trials involving IRX‑2 that are currently being performed are investigator-sponsored trials. Outside our providing study drug and financial support for these studies, we
have less involvement and less control of these studies than we would if these were our studies. Negative results from these studies could have material adverse effects on our business despite our lack of control.
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidates. If we fail to enter
into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of IRX‑2 or any other product we may acquire or in-license.
We may seek third-party collaborators for development and commercialization of IRX‑2 or any other product we may acquire or in-license. Our likely collaborators for any marketing, distribution, development,
licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. We are currently
party to a limited number of such arrangements and have limited control over the amount and timing of resources that our collaborators dedicate to the development or commercialization of our product candidates. Our ability to generate revenues
from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
60
Collaborations involving our product candidates currently pose, and will continue to pose, the following risks to it:
| • |
collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations;
|
| • |
collaborators may not pursue development and commercialization of our product candidates or may elect not to continue or renew development or commercialization programs based on preclinical or clinical study results, changes in the
collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities;
|
| • |
collaborators may delay clinical studies, provide insufficient funding for a clinical study program, stop a clinical study or abandon a product candidate, repeat or conduct new clinical studies or require a new formulation of a
product candidate for clinical testing;
|
| • |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidates if the collaborators believe that competitive products are more likely to be
successfully developed or can be commercialized under terms that are more economically attractive than ours;
|
| • |
collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products;
|
| • |
collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate its intellectual property or
proprietary information or expose it to potential litigation;
|
| • |
collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability;
|
| • |
disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidates or that result in costly litigation or arbitration that diverts
management attention and resources; and
|
| • |
collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates.
|
Collaboration agreements may not lead to development or commercialization of our product candidates in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination,
the continued pursuit and emphasis on its product development or commercialization program could be delayed, diminished or terminated.
If we are not able to establish collaborations, we may have to alter our development and commercialization plans.
Our drug development programs, and the potential commercialization of our product candidates will require substantial additional cash to fund expenses. We may decide to collaborate with pharmaceutical and
biotechnology companies for the development and potential commercialization of its product candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for a collaboration will depend, among other things, upon its assessment of the collaborator’s
resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of preclinical studies or clinical studies, the
likelihood of approval by the FDA or similar regulatory authorities outside the United States, the potential market for the subject product candidates, the costs and complexities of manufacturing and delivering such product candidate to
patients, the potential of competing products, the existence of uncertainty with respect to its ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and
market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a collaboration could be more attractive than the
one with us for our product candidates. We may also be restricted under future license agreements from entering into agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and
document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
61
We may not be able to negotiate collaborations on a timely basis, on acceptable terms, or at all. If we are unable to do so, we may have to curtail the development of our product candidates, reduce or delay its
development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to
increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not
be able to further develop out product candidates or bring it to market and generate product revenue.
Risks Related to our Intellectual Property
If we are unable to obtain and maintain patent and other intellectual property protection for our products and product candidates, or if the scope of
the patent and other intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize our products and
product candidates may be adversely affected.
Our ability to compete effectively will depend, in part, on our ability to maintain the proprietary nature of our technology and manufacturing processes. We rely on research, manufacturing and other know-how,
patents, trade secrets, license agreements and contractual provisions to establish our intellectual property rights and protect our products and product candidates. These legal means, however, afford only limited protection and may not
adequately protect our rights. As of January 29, 2021, our intellectual property portfolio includes 14 patents in the United States and an additional 85 patents around the world.
In certain situations and as considered appropriate, we have sought, and we intend to continue to seek to protect our proprietary position by filing patent applications in the United States and, in at least some
cases, one or more countries outside the United States relating to current and future products and product candidates that are important to our business. However, we cannot predict whether the patent applications currently being pursued will
issue as patents, or whether the claims of any resulting patents will provide us with a competitive advantage or whether we will be able to successfully pursue patent applications in the future relating to our current or future products and
product candidates. Moreover, the patent application and approval processes are expensive and time-consuming. We may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner.
Furthermore, we, or any future partners, collaborators, or licensees, may fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on
them. Therefore, we may miss potential opportunities to seek additional patent protection. It is possible that defects of form in the preparation or filing of patent applications may exist, or may arise in the future, for example with respect
to proper priority claims, inventorship, claim scope, or requests for patent term adjustments. If we fail to establish, maintain or protect such patents and other intellectual property rights, such rights may be reduced or eliminated. If there
are material defects in the form, preparation, prosecution or enforcement of our patents or patent applications, such patents may be invalid and/or unenforceable, and such applications may never result in valid, enforceable patents.
Even if they are unchallenged, our patents and patent applications, if issued, may not provide us with any meaningful protection or prevent competitors from designing around our patent claims by developing
similar or alternative technologies or therapeutics in a non-infringing manner. For example, a third party may develop a competitive therapy that provides benefits similar to one or more of our product candidates but that falls outside the
scope of our patent protection. If the patent protection provided by the patents and patent applications we hold or pursue with respect to our product candidates is not sufficiently broad to impede such competition, our ability to successfully
commercialize our product candidates could be negatively affected.
Other parties, many of whom have substantially greater resources and have made significant investments in competing technologies, have developed or may develop technologies that may be related or competitive with
our approach, and may have filed or may file patent applications and may have been issued or may be issued patents with claims that overlap or conflict with our patent applications, either by claiming the same compositions, formulations or
methods or by claiming subject matter that could dominate our patent position. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. As a result, any patents we may obtain in
the future may not provide us with adequate and continuing patent protection sufficient to exclude others from commercializing products similar to our products and product candidates.
62
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain. No consistent policy regarding the breadth of claims allowed in biotechnology and pharmaceutical patents has
emerged to date in the United States or in many foreign jurisdictions. In addition, the determination of patent rights with respect to pharmaceutical compounds commonly involves complex legal and factual questions, which has in recent years
been the subject of much litigation. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our competitors may also seek approval to market their own products similar to or
otherwise competitive with our products. Alternatively, our competitors may seek to market generic versions of any approved products by submitting ANDAs or ABLAs to the FDA in which they claim that our patents are invalid, unenforceable or not
infringed. In these circumstances, we may need to defend or assert our patents, or both, including by filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or other agency with jurisdiction may find our
patents invalid or unenforceable, or that our competitors are competing in a non-infringing manner. Thus, even if we have valid and enforceable patents, these patents still may not provide protection against competing products or processes
sufficient to achieve our business objectives.
In addition to patent protection, we expect to rely heavily on trade secrets, know-how and other unpatented technology, which are difficult to protect. Although we seek such protection in part by entering into
confidentiality agreements with our vendors, employees, consultants and others who may have access to proprietary information, we cannot be certain that these agreements will not be breached, adequate remedies for any breach would be available,
or our trade secrets, know-how and other unpatented proprietary technology will not otherwise become known to or be independently developed by our competitors. If we are unsuccessful in protecting our intellectual property rights, sales of our
products may suffer and our ability to generate revenue could be severely impacted.
Issued patents covering our products and product candidates could be found invalid or unenforceable if challenged in court or in administrative
proceedings. We may not be able to protect our trade secrets in court.
If we initiate legal proceedings against a third-party to enforce a patent covering one of our products or product candidates, should such a patent issue, the defendant could counterclaim that the patent covering
our product or product candidate is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged
failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description or non- enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of
the patent withheld information material to patentability from the USPTO, or made a misleading statement, during prosecution. Third parties also may raise similar claims before administrative bodies in the United States or abroad, even outside
the context of litigation. Such mechanisms include re- examination, post grant review, inter partes review and equivalent proceedings in foreign jurisdictions. An adverse determination in any of the foregoing proceedings could result in the
revocation or cancellation of, or amendment to, our patents in such a way that they no longer cover our products or product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to
the validity question, for example, we cannot be certain that there is no invalidating prior art, of which the patent examiner and we were unaware during prosecution. If a defendant or third party were to prevail on a legal assertion of
invalidity or unenforceability, we could lose at least part, and perhaps all, of the patent protection on one or more of our products and product candidates. Such a loss of patent protection could have a material adverse impact on our business.
In addition, our trade secrets may otherwise become known or be independently discovered by competitors. Competitors and other third parties could purchase our products and product candidates and attempt to
replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe, misappropriate or otherwise violate our intellectual property rights, design around our protected technology or develop their own
competitive technologies that fall outside of our intellectual property rights. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them, or
those to whom they communicate it, from using that technology or information to compete with us. If our trade secrets are not adequately protected or sufficient to provide an advantage over our competitors, our competitive position could be
adversely affected, as could our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating our trade secrets.
63
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements
imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non- compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the USPTO and various governmental patent agencies outside of the
United States in several stages over the lifetime of the patents and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions
during the patent application process and after a patent has issued. There are situations in which non- compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in
the relevant jurisdiction. The terms of one or more licenses that we enter into the future may not provide us with the ability to maintain or prosecute patents in the portfolio, and must therefore rely on third parties to do so.
If we do not obtain patent term extension and data exclusivity for our products and product candidates, our business may be materially harmed.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Various
extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired for a product candidate, we may be open to
competition from competitive products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are
commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
In the future, if we obtain an issued patent covering one of our present or future product candidates, depending upon the timing, duration and specifics of any FDA marketing approval of such product candidates,
such patent may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years
as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended
and only those claims covering the approved drug, a method for using it or a method for manufacturing it may be extended. A patent may only be extended once and only based on a single approved product. However, we may not be granted an
extension because of, for example, failure to obtain a granted patent before approval of a product candidate, failure to exercise due diligence during the testing phase or regulatory review process, failure to apply within applicable deadlines,
failure to apply prior to expiration of relevant patents or otherwise our failure to satisfy applicable requirements. A patent licensed to us by a third party may not be available for patent term extension. Moreover, the applicable time period
or the scope of patent protection afforded could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, our competitors may obtain approval of competing products
following our patent expiration, and our revenue could be reduced, possibly materially.
Changes in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to
protect our products and product candidates.
Changes in either the patent laws or the interpretation of the patent laws in the United States or other jurisdictions could increase the uncertainties and costs surrounding the prosecution of patent applications
and the enforcement or defense of issued patents. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. When implemented, the Leahy-Smith Act included several significant changes to U.S. patent
law that impacted how patent rights could be prosecuted, enforced and defended. In particular, the Leahy-Smith Act also included provisions that switched the United States from a “first-to-invent” system to a “first-to-file” system, allowed
third- party submission of prior art to the USPTO during patent prosecution and set forth additional procedures to attack the validity of a patent by the USPTO administered post grant proceedings. Under a first-to-file system, assuming the
other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO developed
new regulations and procedures governing the administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions, only became effective on
March 16, 2013. It remains unclear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution
of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business.
64
In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. Recent rulings from the U.S. Court of Appeals for the Federal
Circuit and the U.S. Supreme Court have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination of events has created uncertainty with respect
to the validity and enforceability of patents, once obtained. Depending on future actions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could have a
material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, maintaining, defending and enforcing patents on products and product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in
some countries outside the United States could be less extensive than those in the United States. The requirements for patentability may differ in certain countries, particularly in developing countries; thus, even in countries where we do
pursue patent protection, there can be no assurance that any patents will issue with claims that cover our products. There can be no assurance that we will obtain or maintain patent rights in or outside the United States under any future
license agreements. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from
practicing our inventions in all countries outside the United States, even in jurisdictions where we pursue patent protection, or from selling or importing products made using our inventions in and into the United States or other jurisdictions.
Competitors may use our technologies in jurisdictions where we have not pursued and obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection,
but enforcement is not as strong as that in the United States. These products may compete with our products and product candidates and our patents or other intellectual property rights may not be effective or sufficient to prevent them from
competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing
countries, do not favor the enforcement of patents, trade secrets and other intellectual property protection, particularly those relating to biotechnology and pharmaceutical products, which could make it difficult for us to stop the
infringement of our patents or marketing of competing products in violation of our proprietary rights generally. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties.
Proceedings to enforce our patent rights, even if obtained, in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated
or interpreted narrowly and our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may
not be commercially meaningful. While we intend to protect our intellectual property rights in major markets for our products, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may
wish to market our products. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop.
If we are sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or
delay us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates without infringing the intellectual property and other proprietary rights of third parties.
Third parties may have U.S. and non-U.S. issued patents and pending patent applications relating to compounds, methods of manufacturing compounds and/or methods of use for the treatment of the disease indications for which we are developing our
product candidates. If any third-party patents or patent applications are found to cover our product candidates or their methods of use or manufacture, we and our collaborators or sublicensees may not be free to manufacture or market our
product candidates as planned without obtaining a license, which may not be available on commercially reasonable terms, or at all. We may also be required to indemnify our collaborators or sublicensees in such an event.
65
There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party to, or threatened with, litigation or other adversarial proceedings
regarding intellectual property rights with respect to our products candidates, including interference and post-grant proceedings before the USPTO. There may be third-party patents or patent applications with claims to materials, formulations,
methods of manufacture or methods for treatment related to the composition, use or manufacture of our product candidates. We cannot guarantee that any of our patent searches or analyses including, but not limited to, the identification of
relevant patents, the scope of patent claims or the expiration of relevant patents are complete or thorough, nor can we be certain that we have identified each and every patent and pending application in the United States and abroad that is
relevant to or necessary for the commercialization of our product candidates in any jurisdiction. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued
patents that our product candidates may be accused of infringing. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Accordingly, third parties may assert
infringement claims against us based intellectual property rights that exist now or arise in the future. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The
pharmaceutical and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use or manufacture.
The scope of protection afforded by a patent is subject to interpretation by the courts, and the interpretation is not always uniform. If we were sued for patent infringement, we would need to demonstrate that our product candidates, products
or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving
invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our
management and scientific personnel could be diverted in pursuing these proceedings, which could significantly harm our business and operating results. In addition, we may not have sufficient resources to bring these actions to a successful
conclusion.
If we are found to infringe a third party’s intellectual property rights, we could be forced, including by court order, to cease developing, manufacturing or commercializing the infringing product candidate or
product. Alternatively, we may be required to obtain a license from such third party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate or product.
However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to
the same technologies licensed to us; alternatively, or additionally it could include terms that impede or destroy our ability to compete successfully in the commercial marketplace. In addition, we could be found liable for monetary damages,
including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations,
which could harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims by third parties asserting that our employees or we have misappropriated their intellectual property or claiming ownership
of what we regard as our own intellectual property.
Many of our current and former employees, including our senior management, were previously employed at universities or at other biotechnology or pharmaceutical companies, including some which may be competitors
or potential competitors. Some of these employees may be subject to proprietary rights, non-disclosure and non- competition agreements, or similar agreements, in connection with such previous employment. Although we try to ensure that our
employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees have used or disclosed intellectual property, including trade secrets or other proprietary
information, of any such third party. Litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or
sustain damages. Such intellectual property rights could be awarded to a third party, and we could be required to obtain a license from such third party to commercialize our technology or products. Such a license may not be available on
commercially reasonable terms or at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
66
In addition, while we typically require our employees, consultants and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us,
we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related to the ownership of such intellectual property. If
we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims, litigation could result
in substantial costs and be a distraction to our senior management and scientific personnel.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time-consuming
and unsuccessful.
Competitors may infringe our patents, trademarks, copyrights or other intellectual property. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and
time consuming and divert the time and attention of our management and scientific personnel. In addition, our patents may become, involved in inventorship, priority, or validity disputes. To counter or defend against such claims can be
expensive and time-consuming, and our adversaries may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we can. Any claims we assert against perceived infringers could provoke these parties to
assert counterclaims against us alleging that we infringe their patents, in addition to counterclaims asserting that our patents are invalid or unenforceable, or both.
In an infringement proceeding, a court may decide that a patent is invalid or unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover
the technology in question. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating intellectual property rights we own or control. An adverse result in any litigation proceeding
could put one or more of our owned or in-licensed patents at risk of being invalidated or interpreted narrowly. Further, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a
risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Even if resolved in our favor, the court may decide not to grant an injunction against further infringing activity and instead award only monetary damages, which may or may not be an adequate remedy. Litigation
or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results
of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or
proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities.
We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more
effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could
have a material adverse effect on our ability to compete in the marketplace.
If we fail to comply with our obligations under any future intellectual property licenses with third parties, we could lose license rights that are
important to our business.
In connection with our efforts to build our product candidate pipeline, we may enter into license agreements in the future. We expect that such license agreements will impose, various diligence, milestone
payment, royalty, insurance and other obligations on us. If we fail to comply with our obligations under these licenses, our licensors may have the right to terminate these license agreements, in which event we might not be able to market any
product that is covered by these agreements, or our licensors may convert the license to a non-exclusive license, which could negatively impact the value of the product candidate being developed under the license agreement. Termination of these
license agreements or reduction or elimination of our licensed rights may also result in our having to negotiate new or reinstated licenses with less favorable terms.
67
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our marks of interest and our
business may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We rely on both registration and common law protection for our
trademarks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. During
trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many
foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks
may not survive such proceedings. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected.
68
|
Item 9.01
|
Financial Statements and Exhibits.
|
|
(d)
|
Exhibits.
|
|
Exhibit No.
|
Description
|
|
|
Form of Indemnification Agreement (incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed on April 16, 2021)
|
||
|
Schedule identifying agreements substantially identical to the form of Indemnification Agreement filed as Exhibit 10.1
|
† Management contract or compensatory plan.
69
SIGNATURE
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, hereunto duly authorized.
|
BROOKLYN IMMUNOTHERAPEUTICS, INC.
|
||
|
Dated: May 11, 2021
|
By:
|
/s/ Howard J. Federoff
|
|
Howard J. Federoff
|
||
|
Chief Executive Officer and President
|
||
70