Attached files
| file | filename |
|---|---|
| EX-99.1 - EXHIBIT 99.1 - Chemomab Therapeutics Ltd. | tm2112883d1_ex99-1.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
Amendment No. 2
FORM 8-K/A
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of Report (Date of earliest event reported): April 14, 2021 (March 17, 2021)
Chemomab Therapeutics Ltd.
(Exact name of Registrant as Specified in Its Charter)
| State of Israel | 001-38807 | 81-3676773 |
| (State or Other Jurisdiction | (Commission | (IRS Employer |
| of Incorporation) | File Number) | Identification No.) |
| Kiryat Atidim, Building 7 | |
| Tel Aviv, Israel | 6158002 |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: +972-77-331-0156
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| ¨ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ¨ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ¨ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ¨ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) |
Name of each exchange on which registered | ||
| American Depositary Shares, each representing twenty (20) ordinary shares, no par value per share | CMMB | Nasdaq Capital Market | ||
| Ordinary shares, no par value per share | N/A | Nasdaq Capital Market* |
* Not for trading; only in connection with the registration of American Depositary Shares.
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§ 230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§ 240.12b-2 of this chapter).
Emerging growth company x
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
- 2 -
EXPLANATORY NOTE
On March 17, 2021, Chemomab Therapeutics Ltd., formally known as Anchiano Therapeutics Ltd. (the “Company” or the “Registrant”) filed a Current Report on Form 8-K (the “Initial Filing”) disclosing the closing of the merger transaction, which closed on March 16, 2021 (the “Closing Date”) pursuant to the Agreement and Plan of Merger by and among the Registrant, Chemomab Ltd. (“Chemomab”) and CMB Acquisition Ltd., an Israeli limited company and wholly-owned subsidiary of the Company (the “Merger”).
On March 19, 2021, the Company filed Amendment No. 1 on Form 8-K/A to amend the Initial Filing to include the required historical financial statements of Chemomab. This Amendment No. 2 on Form 8-K/A (“Amendment No. 2”) amends the Initial Filing to include the required pro forma financial statements as of and for the twelve months ended December 31, 2020 (the “Pro Forma Financial Information”), giving effect to the Merger as required under Item 9.01(b) under cover of Form 8-K/A.
Prior to the Closing Date, and on March 9, 2021, the Registrant filed its Annual Report on Form 10-K (the “Annual Report”) with the Securities and Exchange Commission (the “SEC”), which included disclosures pertinent to Anchiano, which was the operating company of the Registrant prior to the Closing Date. In parallel with the filing of this Amendment No. 2, the Registrant is filing a Registration Statement on Form S-3 to cover the resale of certain issued securities. In addition to filing the Pro Forma Financial Information, the Registrant is filing this Amendment No. 2 in order to update certain disclosures included in the Annual Report, such that the disclosures will relate to Chemomab, which is the current operating company of the Registrant.
Unless the context otherwise requires, all references in this section to “we,” “us,” “our”, the “Registrant” or the “Company” refer to Chemomab and its subsidiaries.
BUSINESS
Overview
Chemomab is a clinical-stage biotech company discovering and developing innovative therapeutics for conditions with high unmet medical need that involve inflammation and fibrosis. CM-101, the company’s lead clinical product candidate, is a first-in-class humanized monoclonal antibody which hinders the fundamental function of the soluble chemokine CCL24, also known as eotaxin-2, as a regulator of major inflammatory and fibrotic pathways. Chemomab has shown that CM-101 interferes with the underlying biology of inflammation and fibrosis using a novel and differentiated mechanism of action and is actively advancing CM-101 into a staggered Phase 2 clinical studies to treat patients with liver, skin, and lung fibrosis. Chemomab has completed two Phase 1a clinical studies at varying doses using different administration methods, as well as a Phase 1b safety, tolerability and proof-of-mechanism clinical study of CM-101 in non-alcoholic fatty liver disease, or NAFLD, patients. Chemomab is currently conducting a Phase 2a clinical study in primary sclerosing cholangitis, or PSC, a rare obstructive and cholestatic liver disease, in Europe and Israel and Chemomab is planning a Phase 2 study in systemic sclerosis, or SSc, a rare autoimmune rheumatic disease characterized by accumulation of collagen, or fibrosis, by the end of this year. Although the primary focus of Chemomab relates to these two rare indications, an additional Phase 2a clinical study expanding the understanding of CM-101 in non-alcoholic steatohepatitis, or NASH, has been initiated and will provide important safety and PK data designed to support the development of CM-101 subcutaneous formulation.
Fibrosis is the abnormal and excessive accumulation of collagen and extracellular matrix, the non-cellular component in all tissues and organs, consisting of macromolecules such as collagen, that provide structural and biochemical support to surrounding cells, leading to scarring and thickening of connective tissues, affecting tissue properties and potentially leading to organ failure. Fibrosis can occur in many different tissues, including lung, liver, kidney, muscle, skin, and the gastrointestinal tract, resulting in a growing number of progressive fibrotic conditions. A healthy inflammatory response is necessary for efficient tissue repair. However, fibrosis and inflammation are intrinsically linked and a prolonged inflammatory response can contribute to the pathogenesis of fibrosis.
- 3 -
Chemomab has pioneered the therapeutic targeting of CCL24, a chemokine that promotes various types of cellular processes that regulate inflammatory and fibrotic activities through the CCR3 receptor. The chemokine is expressed in monocytes, macrophages, activated T cells, fibroblasts, endothelial cells, and epithelial cells, including the bile duct epithelial cells called cholangiocytes. Chemomab has developed a novel CCL24 inhibiting product candidate with dual anti-fibrotic and anti-inflammatory activity allowing it to challenge the complex interplays of both of these inflammatory and fibrotic mechanisms that drive fibrotic indications. This innovative approach is being developed for difficult to treat rare diseases, also known as orphan indications or diseases, such as PSC and SSc, for which patients have no established standard of care treatment options.
Pipeline

Chemomab’s lead product candidate, CM-101, is a first-in-class humanized monoclonal antibody targeting CCL24 that is being advanced in two orphan indications: PSC and SSc. CCL24 has been extensively studied in airway inflammation and, more recently, Chemomab has demonstrated in pre-clinical studies and early clinical studies that it plays an important role in additional indication areas, including inflammation and fibrosis of the liver, skin and lung. Although found in low levels in blood or tissue samples taken from healthy volunteers, elevated levels of both CCL24 and its receptor CCR3, have been found in patients with PSC, SSc and NASH. CCL24 levels have even been correlated to different phases of disease. Chemomab expects that neutralizing CCL24 with an antibody will exert anti-fibrotic and anti-inflammatory effects in patients. CM-101 has been granted orphan drug designation by both the FDA and the EMA in its primary indications of PSC and SSc based on extensive preclinical and non-clinical data. This designation provides multiple benefits, including exclusive marketing and development rights for a period of time for these indications.
PSC is a rare, chronic cholestatic liver disease characterized by progressive inflammation, fibrosis, and destruction of the intrahepatic and extra-hepatic bile ducts with no identifiable cause. Cholestasis is a symptom of liver injury and is characterized as the interruption of bile flow from hepatocytes to the intestine, which leads to bile acid accumulation in the liver, resulting in oxidative stress, inflammation, apoptosis, and fibrosis. PSC affects approximately 30,000-45,000 patients in the United States and is commonly associated with inflammatory bowel disease. Median survival is between 10-12 years. Fibrosis and inflammatory responses induce a progressive spread of the fibrotic condition. No treatment aside from a liver transplant has been associated with change of the disease course or significant long term improvement in the clinical outcome. PSC is a clear serious unmet medical need with no FDA-approved therapeutics and for which the current standard of care is inadequate.
SSc is a connective tissue disease characterized by excessive fibrosis and extracellular matrix accumulation in the skin, lung, and other visceral organs. The disease initiates with an early inflammatory phase involving the immune cell network, as well as endothelial cells. As the disease progresses, the inflammation increase, fibroblasts and myofibroblasts generate tissue fibrosis and endothelial cells promote vascular injury, which leads to skin fibrosis, interstitial lung disease, myocardial insufficiency, vascular obliteration, distal ulcerations, and gangrene. SSC affects approximately 75,000-100,000 patients in the United States. SSc has the highest mortality rate among the systemic rheumatic diseases with high unmet need, as current treatments managing only disease manifestations and there is no disease modifying drug available.
- 4 -
Chemomab is primarily focused on the orphan indications PSC and SSc, and believes that it has additional opportunities in neighboring fibrotic-inflammatory disease areas such as idiopathic pulmonary fibrosis, or IPF and Nonalcoholic steatohepatitis, or NASH. CM-101 has shown promising anti-fibrotic and anti-inflammatory effects in preclinical studies of liver fibrosis and PSC, with significant reductions in fibrotic genes, liver enzymes, bile acid and cholangiocyte proliferation, all reflecting an improvement in disease status. In preclinical studies of SSc, CM-101 reduces inflammatory and fibrotic injury resulting in reductions in dermal thickness, collagen concentration in the skin and the lung and immune cell infiltration in the lung.
Chemomab has completed two Phase 1a single ascending dose studies with intravenous, or IV, and subcutaneous, or SC, administrations of CM-101 in 40 healthy volunteers. The drug was shown to be safe and well-tolerated, with a PK profile supporting dosing once every 2-4 weeks. The company also recently completed a Phase 1b multiple administrations ascending dose study in 16 NAFLD patients, expanding its safety, tolerability, and pharmacodynamics database into patients where an initial confirmation of an anti fibrotic effect was already seen.
Chemomab is currently recruiting patients, in Europe and Israel, for a Phase 2a randomized, double-blind, placebo-controlled study in PSC patients. The company plans to enroll 45 patients over a 15-week treatment window. Two independent-primary endpoints for the clinical study will be changes in alkaline phosphatase, or ALP, liver enzyme levels and enhanced liver fibrosis, or ELF, score, both established parameters for the evaluation of liver health and fibrosis in PSC patients. Secondary and exploratory endpoints will include other liver enzymes, fibrotic and inflammatory markers, and liver stiffness measured by elastography (FibroScanTM). Chemomab intends to initiate a global randomized, double-blind, placebo-controlled Phase 2 clinical study in SSc by the end of this year. The company will evaluate the Combined Response Index in diffuse cutaneous Systemic Sclerosis, or ACR CRISS, score as a primary endpoint. Safety and tolerability, as well as lung function, skin thickness and fibrotic biomarkers, will be secondary and exploratory endpoints. Chemomab may also explore CM-101 in other indications, where the dual activity of CM-101 acting on both inflammation and fibrosis could provide new avenues for treating conditions like IPF and NASH.
Chemomab was founded in 2011, based on a novel discovery originating from the Sourasky Medical Center in Tel-Aviv, Israel, where Professor Jacob George first identified CCL24 as a key regulator of unstable plaque formation in atherosclerotic patients. In its early years, Chemomab focused on research directed at clarifying the role and effectiveness of a CCL24 blockade. In 2015, Chemomab selected its proprietary lead product candidate, CM-101, and started product development directed towards human testing.
Chemomab has assembled an executive team with highly relevant experience in inflammation and fibrosis, and biologics drug discovery and clinical development. Adi Mor, Ph.D., Chemomab’s Chief Executive Officer and Co-founder has 15 years of experience in Immunology and has led the CM-101 program from discovery stage into Phase 2 clinical studies. Arnon Aharon, M.D., Chemomab’s Chief Medical Officer, brings deep experience in clinical development. He previously served as the Chief Medical Officer to BiolineRx where he directed the oncology and immunology pipeline. Dr. Aharon’s professional experience also includes multiple senior management positions at private and publicly traded biotechnology companies, such as Pharmos Ltd. and Thrombotech Ltd. Michal Segal Salto, Ph.D, our VP of R&D, has over 15 years of experience in cell biology, molecular biology and biochemistry and has successfully led the R&D team from early preclinical stages through clinical translation. Sharon Elkobi serves as our VP of Business Development and holds 15 years of experience in prior business development positions, both at Neurim Pharmaceuticals and Teva.
- 5 -
Company strategy
Chemomab aims to become a world-leading company for the treatment of fibrosis, developing novel therapies across a wide range of fibrotic indications. To achieve this, the company is focused on the following key strategies:
• Advance Chemomab’s lead product, CM-101, for the treatment of PSC and SSc, through clinical development to approval
Chemomab is developing CM-101 as a novel therapy for PSC and SSc, two orphan indications with high unmet medical need. Chemomab has completed the Phase 1 clinical studies (single and multiple administration safety and tolerability studies). Chemomab is currently conducting a Phase 2a study in PSC and it intends to commence a Phase 2 study in SSc this year. Following completion of the Phase 2 studies, Chemomab will engage with the FDA and comparable foreign authorities to discuss the requirements from the pivotal study(ies) needed to obtain approval. CM-101 was granted orphan designation for both PSC and SSc from Food and Drug Administration, or FDA and European Medicine Agency, or EMA and, based on the Phase 2 results, will seek expedited regulatory approval such as the Fast Track, Breakthrough Therapy, and Priority Review designations. Chemomab expects that in order to obtain regulatory approval for the use of CM-101 to treat these indications, it will be required to conduct only one Phase 3 pivotal study for each such orphan indication.
• Expand Chemomab’s next generation pipeline
Based on the know-how, knowledge and experience it has gathered in fibrosis and fibrotic diseases, Chemomab intends to expand its pipeline with next generation products developed against new targets. Chemomab will also explore targeting CCL24 with additional, complementary fibrotic or inflammatory mechanisms, including acquiring or in-licensing innovative product candidates.
• Selectively evaluate partnership opportunities
Chemomab continuously explores partnership opportunities to advance CM-101 development in PSC and SSc, identifying companies with drugs (either approved or in development) that could possibly be combined with CM-101, extending the development of CM-101 to new indications beyond PSC and SSc, and seeking additional significant commercial or drug development capabilities that may accelerate CM-101’s time to market.
• Explore opportunities for CM-101 in additional fibrotic indications
Chemomab continuously evaluates the potential benefit of CM-101 outside of its two lead indications, PSC and SSc, in order to maximize the product’s potential. CM-101 has shown anti-fibrotic activity in animal models and human tissue studies of IPF and NASH. The dual activity of CM-101 will continue to drive Chemomab into new disease areas and to build new collaborations with global medical researchers.
• Strengthen Chemomab’s intellectual property portfolio
Chemomab believes that it has developed a strong intellectual property portfolio and will continue to seek, maintain, and defend patent rights, whether developed internally or licensed to protect and enhance the proprietary technology, inventions, and improvements that are commercially important to the development of its business proprietary position in the field of inflammation and fibrosis.
Fibrosis and inflammation
Tissue damage activates a repair process that includes acute inflammation followed by either successful complete repair or tissue replacement by fibrosis. However, persistent and repeated damage results in continuous activation of the repair process leading to chronic inflammation, progressive tissue fibrosis and sclerosis.
Fibrosis is an accumulation of non-functional tissue and can occur in many different tissues, including lung, liver, kidney, muscle, skin and the gastrointestinal tract, resulting in a growing number of chronic fibrotic conditions. Liver fibrosis is the process of excessive accumulation of extracellular matrix proteins, predominantly collagen, which occurs as results of liver injury. In cases of acute temporary damage, these changes are transient and liver fibrosis may resolve. In chronic cases, however, the liver damage persists and chronic inflammation and accumulation of the extracellular matrix eventually leads to cirrhosis. The various fibrotic manifestations in conditions like SSc are still not well understood. Disease progression is characterized by an early inflammatory onset followed by tissue fibrosis, vascular injury and organ damage. Fibrosis, and specifically lung fibrosis, is the main cause of disease progression and mortality.
- 6 -
Fibrosis and inflammation are intrinsically linked; a healthy inflammatory response is necessary for efficient wound healing, however, a prolonged response can contribute to the pathogenesis of fibrosis. The inflammatory response during chronic liver injury is a dynamic process with intrahepatic accumulation of diverse immune cells. Recruitment and infiltration of these cells to the liver and their localization is mainly determined by chemokines and cytokines that are produced by hepatocytes, immune cells, biliary epithelial cells, and endothelial cells. Notably, activated liver fibroblasts, the hepatic stellate cells, or HSCs, secrete various chemokines, thereby contributing to the ongoing immune response during fibrotic liver diseases. Similarly, for SSc, the early inflammatory phase leading to fibrosis in multiple organs of the body include activation of the immune cell network of lymphocytes, eosinophils, and monocytes, as well as endothelial and endothelial progenitor cells. In the advanced SSc phase, fibroblasts and myofibroblasts take the lead to generate tissue fibrosis.
Chemokine involvement in inflammation and fibrosis
Chemokines are a group of small signaling proteins thought to be involved in the etiology, or causation, of multiple inflammatory diseases. They are not only implicated in immune cell recruitment during inflammation, but also contribute to immune surveillance, direct cells to target organs in homeostasis, and exert pleiotropic, or diverse, effects on nonimmune cells, for instance, directly influencing the functionality of fibrogenic cells. Chemokines and their corresponding chemokine receptors are key players in orchestrating the sequential influx of immune cells into damaged or disease organs, driving inflammatory responses to specific triggers.
In the liver, chemokines have a key role in the development of inflammation and wound healing responses, which can lead to either resolution of liver injury or promote, if ongoing, maladaptive responses with chronic inflammation, fibrosis, and development of clinically manifest liver disease. Although the pathophysiology underlying PSC has not yet been fully clarified, animal models of PSC have provided contributions in dissecting the molecular basis of this disease and focusing on the role of cytokines and chemokines as important pathogenetic mediators of liver inflammation and fibrosis. Recently published studies demonstrated that in most of the processes suggested for the onset and the development of PSC, chemokines and chemokine receptors play a key role. HSCs may be the main producers of cytokines and play the initial role in the progression of liver fibrosis by attracting different types of immune cells, resulting in further production of cytokines and liver injury in a vicious disease cycle. Extensive proliferation, transdifferentiation and activation of HSCs results in ongoing chronic tissue remodeling and severe fibrosis. In addition, chemokines are also involved in promoting polarization of the recruited immune cells. Therefore, chemokines may participate in PSC by either promoting migration of inflammatory and fibrotic cells, by activating inflammatory and fibrotic cells locally, or by inducing cytokines that promote collagen and matrix deposition.
Likewise, in SSc pathogenesis, chemokines foster migration and activation of inflammatory and fibrotic cells, inducing the secretion of cytokines that promote collagen and matrix deposition in affected organs. Indeed, patients with SSc exhibit increased systemic levels of proinflammatory chemokines and some have also been shown to correlate with limited or diffuse cutaneous disease phenotype and/or to organ-specific pathology as lung disease or skin vascular inflammation.
The role of CCL24
CCL24 is a chemokine that promotes various types of cellular processes that regulate inflammatory and fibrotic activities through the CCR3 receptor. This chemokine is known to be expressed by activated T cells, monocytes, epithelial cells and endothelial cells, as well as by activated fibroblasts. CCL24 induces chemotaxis and activation of CCR3-expressing cells, including immune cells and fibroblasts.
- 7 -
Chemomab has been the driving force in establishing the role of CCL24 in the pathogenesis of PSC and SSc, however, others have proven its contribution in other indications. For example, published work has shown that both CCL24 and CCR3 are involved in lung and skin inflammation and fibrosis. CCR3 is robustly expressed on eosinophils and recent data has suggested that eosinophilic inflammation may be involved in the pathogenesis and progression of SSc. For example, in SSc patients, eosinophil counts, but not total leukocytes, were significantly higher than in patients with other connective autoimmune diseases. Eosinophil counts correlated positively with both interstitial lung disease severity and the modified Rodnan skin thickness score, or mRSS. Notably, CCR3 was demonstrated to be expressed on oral and dermal fibroblasts where it modulates wound healing and tissue remodeling processes. A recent academic study also demonstrated overexpression of CCR3 on monocyte populations isolated from SSc patients. CCL24 was shown to be involved in proinflammatory reactions, specifically contributing to the type 2 immune reaction involving Th2 lymphocytes and M2 macrophages that were shown to be present in skin lesions of SSc patients. Accordingly, CCL24 was found to play a dominant role in inducing profibrotic effects and to be overexpressed in fibrotic lungs and bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis (IPF), a disease sharing similar lung dysfunction features with SSc. Furthermore, CCL24 was shown to promote collagen production in human lung fibroblasts and to be constitutively expressed by dermal fibroblasts.
Prior studies support the role of CCL24/CCR3 signaling in the pathogenesis of SSc and these findings have been further explored by Chemomab in SSc and for the first time, in PSC.

CCL24 is a critical mediator promoting inflammation and fibrosis
Challenges to drug development in fibrosis and inflammation
Successful treatment of fibrotic disorders has in large part remained elusive, primarily due to incomplete understanding of the complexity and multi-mechanism contributions to disease progression. This has complicated preclinical investigations for new products and new targets, with animal models having limited resemblance to human disease. As such, preclinical animal data is often of short treatment duration and does not capture the effects of treating chronic fibrotic indications. This is particularly applicable to complex, orphan indications like SSc, where there is still no approved standard of care or proven target mechanism. Most drug approvals in this space have been focused on fibrosis of the lungs i.e., idiopathic pulmonary fibrosis, or IPF, interstitial lung disease, or ILD, and pulmonary arterial hypertension, or PAH.
Most approved anti-fibrotic products target extracellular components, given their biological accessibility, and inhibition of receptors and ligands preventing downstream signaling is considered to be an effective choice to alleviate fibrosis. PDGF and TGF-β are commonly studied targets in fibrosis and there are two approved products that target these pathways, pirfenidone and nintedanib. Both pirfenidone and nintedanib are approved for the treatment of IPF, with nintedanib also recently approved for treatment of systemic sclerosis associated interstitial lung disease and chronic fibrosing interstitial lung diseases. Due to the strong associations between inflammation and fibrosis, companies have devoted efforts to anti-inflammatory drugs with the hope that reduction in inflammation will attenuate fibrosis. For example, companies have targeted TNF-α, a commonly explored anti-inflammatory mechanism in fibrotic indications. This has yet to result in any product approvals in fibrosis. Despite the success of targeting cytokines, inflammatory factors and immune cells in pure inflammatory autoimmune diseases, like blocking TNF and IL-6, these results have not been reproduced in studies targeting inflammatory fibrotic indications. Treatments that inhibit certain pure anti fibrotic pathways, such as nintedanib and pirfenidone, have resulted in limited clinical benefit. Chemomab believes that these results highlight the importance of a dual mechanism that, with adequate selectivity, will target inflammatory processes and will directly prevent fibrosis resulting in blockage of multiple disease-contributing mechanisms.
- 8 -
Notwithstanding challenges in the field of fibrosis and inflammation, there is still significant industry interest given the associated unmet medical needs and the open field to identifying optimal therapeutic targets. For example, in 2019 Novartis completed two transactions related to the treatment of NASH, a liver metabolic fibrotic disease. It acquired IFM Tre for NLRP3 antagonists for a $310 million upfront payment and total potential consideration of $1.5 billion and licensed an integrin inhibitor from Pliant Therapeutics for an $80 million upfront payment. Additionally, Gilead Sciences licensed two preclinical programs, one in NASH for a $15 million upfront payment (total potential consideration of $785 million) and the other for TGF-β inhibitors in fibrosis for an $80 million upfront payment and total potential consideration of $1.4 billion. In 2020, Roche acquired Promedior Inc. for a $390 upront payment and total potential consideration of $1 billion in milestones for its Phase 2 product in pulmonary fibrosis and Bayer partnered with Recursion Pharmaceuticals to develop and commercialize preclinical-stage small molecule treatments for fibrotic conditions for a $30 million upfront payment and total potential consideration of $1 billion. Boehringer Ingelheim also acquired Enleofen Bio in a deal worth $1 billion for its NASH and ILD anti-IL11 platform.
Targeting chemokines as a treatment for fibrotic indications
Chemomab believes that its approach, selectively targeting fibrotic conditions by attenuating both inflammation and fibrosis, may be an optimal approach for both effectiveness and reduction of toxicity. As central regulators of initiation and progression of fibrotic disorders, chemokines are an ideal target to impact both inflammation and fibrosis. Some chemokines are also disease-specific, allowing for potential selectivity.
Chemokine receptors, or CCRs, have been more extensively studied as drug targets in fibrotic conditions compared to chemokine ligands, however, the therapeutic effects of CCR inhibitors have generally fallen short in the clinic. Pharmaceutical companies have previously explored the CCL24 ligand receptor, CCR3, and its other ligands CCL7 and CCL11, with small or large molecule inhibitors. These programs were directed at inhibiting eosinophilic trafficking in respiratory and allergic inflammation, however, despite promising preclinical data, most programs were discontinued largely due to poor safety profiles and limited efficacy of the antagonist used. To Chemomab’s knowledge, only Alkahest has an active program that explores CCR3 inhibition, which is under license from Boehringer Ingelheim and is being developed as a treatment for wet AMD. In contrast, Chemomab believes CCL24 presents a more promising opportunity. Unlike other CCR3 ligands, CCL24 binds only to the CCR3 receptor and is also organ/disease-specific which together could provide enhanced selectivity and tolerability. For example, CCL24 is elevated in the liver and cholangiocytes (bile duct epithelia) in PSC patients and is specifically related to fibrosis-related inflammation and not generalized eosinophilic inflammation as its receptor. Likewise, elevation of CCL24 has been shown in fibrotic lungs and bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis, or IPF, a disease sharing similar lung dysfunction features with SSc and which recently was correlated, by Chemomab, with disease severity and lung involvement in a cohort of SSc patients from the United Kingdom. Furthermore, CCL24 is constitutively expressed by skin and dermal fibroblasts. The use of an antibody in targeting this chemokine is a novel approach to targeting fibrosis.
Chemomab’s expertise and approach to drug discovery
Chemomab is a clinical stage biotechnology company focused on the discovery and development of novel drugs to address fibrotic indications with unmet medical needs. CCL24 is a key target promoting fibrosis as it regulates the two main processes that drive fibrosis: fibroblast activation and immune cell migration and activation. Using Chemomab’s expertise in monoclonal antibody, or mAb, development and deep knowledge of chemokines biology. Chemomab is developing CM-101, a proprietary, first-in-class, fully humanized mAb that through research and studies to date, is found to neutralize CCL24 and by so doing inhibits its disease-related functions in both inflammation and fibrosis. This represents an innovative approach to anti-fibrotic drug discovery and a key differentiator for Chemomab. The ability of CM-101 to directly attenuate fibroblast activation and concurrently attenuate recruitment of immune cells is novel and could address a wide-range of hard-to-treat fibrotic diseases.
- 9 -
Chemomab’s ongoing collaborations are complementary in both preclinical and clinical aspects of research and development. Chemomab has created an extensive panel of in vitro, ex vivo and in vivo assays which it has used to further the understanding of fibrotic processes together with the role of CCL24 in various diseases and the effects of its neutralization with CM-101. These assays have allowed Chemomab to sequentially explore target validation and proof of mechanism in disease relevant human and animal samples which continues to de-risk the translation of CM-101 into the clinic.
Target expression and engagement
Chemomab regularly collaborates with leading academic centers around the world to investigate the role of CCL24 and CM-101 in various indications. For example, Chemomab works with The Royal Free Hospital, or RFH, in London, United Kingdom to access liver biopsy and serum samples from patients with PSC. Using immunohistochemistry and florescence microscopy to stain CCL24 and CCR3 it explores the expression patterns of these targets in disease relevant human samples and compares them to healthy volunteers. Similarly, Chemomab has tested biopsies of SSc patients through a collaboration with Florence University in Italy.
Proof of mechanism
Chemomab explores fibroblast activation and immune cell recruitment in response to CM-101 treatment through in house ex vivo and in vitro assays. Chemomab executed a multitude of validated genetic and treatment-based disease models in fibrotic and inflammatory indications in which it has investigated CM-101’s effects. Additionally, as part of a collaboration with Nordic Biosciences, Copenhagen, Denmark, Chemomab has gained access to proprietary tools and expertise to explore the effects of CM-101 on key fibrogenesis and fibrolysis biomarkers. Nordic Biosciences is a world-leading extracellular matrix specialist and continues to contribute as Chemomab analyses its clinical samples.

Chemomab has created a landscape of biological assays to explore CCL24 and CM-101
Chemomab plans to explore next generation biologic products, and, based on its wide database of patient samples and extensive knowledge and experience in fibrosis, aims to identify targets that could complement CCL24 inhibition. Next generation assets may therefore be dual targeting and will be screened through the panel of assays available at Chemomab evaluating target expression in fibrotic tissues as well as the anti-fibrotic activity of potential candidates. Similar to CM-101, this process will establish proof-of-biological-mechanism in both animal models and human tissue prior to commencing product development and initiating clinical studies.
- 10 -
The Chemomab pipeline
CM-101 in PSC and SSc
Chemomab’s lead product, CM-101, is a first-in-class humanized monoclonal antibody targeting CCL24 and being developed initially for treatment of PSC and SSc, with potential future opportunity in other fibrotic-inflammatory indications. Chemomab has completed two Phase 1a studies of CM-101 in healthy volunteers as well as a Phase 1b safety, tolerability and proof-of-mechanism study in NAFLD patients. A Phase 2a study in PSC is now ongoing in Europe and Israel and a Phase 2 study in SSc will follow this year. Although the primary focus of Chemomab is these two orphan indications, a Phase 2a study expanding the safety, tolerability and mechanistic dataset for CM-101 is also planned in NASH patients.
Primary Sclerosing Cholangitis
PSC is a progressive, rare, and chronic cholestatic liver disorder that is characterized by thickening, inflammation, and fibrosis of the bile ducts in which both intra- and extra-hepatic bile ducts are affected. This generally leads to cholestasis, liver damage, cirrhosis, and eventually to liver failure. The exact cause of PSC remains mostly known; however, immune system dysregulation, genes, viruses, and bacteria may be involved. PSC is commonly associated with inflammatory bowel disease, or IBD. Approximately three in every four individuals with PSC also have ulcerative colitis. Most individuals affected with PSC are adults with an average age of diagnosis being 40 years; however, it may also occur in children. Disease progression, symptoms, and severity may vary greatly between individuals. Patients in the initial stages of PSC are generally asymptomatic or have only mild symptoms.
Abdominal discomfort, fatigue, and pruritus, or itching, are common initial symptoms of PSC which can be severe and debilitating. The initial step in diagnosing PSC is to evaluate liver enzyme levels through blood tests. Physicians will then confirm a diagnosis with cholangiography ultrasound and, in rare cases, a liver biopsy. As the disease progresses, bile flow from the liver is obstructed and is subsequently absorbed into the bloodstream leading to the yellowing of the mucous membranes, whites of the eyes, and skin. Furthermore, individuals may also experience abdominal pain, malaise, light-colored stools, nausea, dark urine, weight loss, and/or hepatomegaly or splenomegaly. PSC patients have a 40-fold increased risk of liver cancer and a 400-fold increased risk of cholangiocarcinoma, and the disease may lead to other conditions including osteoporosis, bacterial cholangitis, portal hypertension, bleeding, as well as vitamin deficiencies.
There are currently no specific medical therapies that can alter or cure the course of the disease; instead, available treatments are directed towards slowing the progression of PSC and treating symptoms. In certain individuals, endoscopic surgery may be performed to enlarge the narrowed bile ducts and to remove blockages. Complications due to vitamin deficiencies can be prevented with the help of vitamin supplements, while infections and inflammation can be controlled by using antibiotics. Cholestyramine and UCDA can be effective in managing itching and can be used with or without antihistamines. Patients with advanced symptoms such as end-stage liver disease, recurent bacterial cholangitis and intractable pruritus, will undergo liver transplantation, however, in 30% of cases, PSC will recur even after liver transplantation. The median survival is 10-12 years without intervention.
Systemic Sclerosis
SSc is an autoimmune inflammatory condition which results in widespread fibrosis and vascular abnormalities affecting the skin, lungs, gastrointestinal tract, heart and kidneys. Other key features of SSc include thickening and hardening of the skin, autoantibody production and abnormal nail fold capillaries. The underlying mechanisms that cause SSc are complex and for the most part unknown but most likely involve a combination of factors including the immune system, genetics, and environmental triggers. Various pathways are involved in the pathogenesis of SSc including cytokines that injure blood vessels, growth factors that stimulate collagen, integrin signaling, morphogen pathways, and co-stimulatory pathways. SSc is generally diagnosed between the age of 30 and 50 years and is most prevalent in women.
- 11 -
Given that SSc can affect many different parts of the body there are a multitude of different symptoms of the disease. The most widely observed symptoms include fatigue, arthralgia, and myalgia. However, the earliest sign is often the Raynaud phenomenon in which the body’s normal response to cold or emotional stress is exaggerated, resulting in abnormal spasms in arterioles. Cutaneous features include sclerosis of the skin, particularly the face and hands. Gastrointestinal symptoms of the upper tract include acid reflux and of the lower tract include bloating, nausea and incontinence. Cardiopulmonary presentations include interstitial lung disease, pulmonary arterial hypertension and cardiac scleroderma. Renal and ocular symptoms can also present and 20% of SSc patients have an overlapping diagnosis with other connective tissue diseases and can develop arthritis, lupus or myositis. SSc is subdivided into two main types related to the distribution of skin involvement: diffuse cutaneous (two-thirds of cases) and limited cutaneous. Diffuse SSc, or dcSSc, is rapidly progressive with more significant organ involvement.
There is no cure for SSc. Established treatments can help with symptoms and may only modify the disease outcome if given early in the disease course. Prescribed medications, used off-label, primarily focus on suppressing inflammation with NSAIDs and dilating abnormal or constricted blood vessels with losartan, sildenafil, iloprost and SSRIs, as well as treatments to manage individual organ involvement. The only two drugs that are approved for the treatment of SSc symptoms are Bosentan by Actelion Pharmaceuticals, approved in Europe for the prevention of digital ulcer development, and nintedanib by Boehringer Ingelheim, recently approved in the United States, Europe and Japan for the treatment of SSc associated interstitial lung disease. The clinical course of SSc is determined by the extent of vascular and fibrosis complications and has the highest mortality rate among the systemic rheumatic diseases as 40% of patients die within 10 years from disease onset, with pulmonary involvement being the leading cause of death.
Chemomab’s solution is CM-101
The dual anti-fibrotic and anti-inflammatory activity of CM-101 enables the targeting of a wide range of pathogenic mechanisms and affords patients a new treatment that may have a more impactful effect on disease progression.

Targeting CCL24 offers a dual activity approach
In order to understand CCL24’s role in disease pathophysiology, Chemomab has collected data on CCL24 levels from patient with multiple fibrotic-inflammatory indications, including those with PSC, SSc and NASH. PSC patients’ liver biopsies and SSc skin samples were stained for CCL24 and its receptor, CCR3. Blood samples taken from PSC and SSc patients were used to further evaluate the role of the CCL24-CCR3 axis exploring levels of circulating CCL24 and CCR3. To explore the influence of CCL24 on disease status, CCL24 serum levels were correlated with fibrotic biomarkers and disease severity markers.
- 12 -
CCL24 levels in liver biopsies from PSC patients
PSC pathology generally initiates with bile duct damage leading to cholestasis, bile duct inflammation and fibrosis and finally to substantial liver damage. Chemomab assessed the accumulation and cellular localization of CCL24 in livers of PSC patients focusing on CCL24 levels in the periductal damaged zone that is most relevant to disease pathology. CCL24 was mainly found in inflammatory cells in the liver of PSC patients and due to the robust liver inflammatory insult in PSC, reflected by massive accumulation of resident and recruited immune cells in the periductal space, CCL24 positive staining was extensive. Specific and robust CCL24 staining was also shown in cholangiocytes, the epithelial cells of the bile ducts. Activated myofibroblasts that surround the bile ducts, whether they originate from hepatic stellate cells or portal fibroblasts, are the main drivers of the excess extracellular matrix accumulation in this area, comprising the unique “onion ring” shape seen in PSC liver sections. The collective expression pattern shows high CCL24 levels in areas that are most affected in PSC and highlights its central role in PSC related liver pathology.

Elevated CCL24 staining in liver biopsies from PSC patients
CCR3 levels in liver biopsies from PSC patients
To evaluate the levels of CCR3, the receptor of CCL24, and identify the cells that can potentially respond to CCL24 secretion, biopsies were stained for CCR3 As seen for CCL24, specific CCR3 staining was evident in cholangiocytes, surrounding immune cells and fibroblasts.

Elevated CCR3 staining in liver biopsies from PSC patients
CCL24 levels in serum and correlation to a fibrotic biomarker in PSC patients
Together with RFH, Chemomab analyzed serum levels of CCL24 in PSC patients at various stages of disease. CCL24 levels showed a positive correlation to the liver fibrosis biomarker ELF score, which is a commercially available test that reflects liver fibrosis stage based on serum concentrations of several fibrosis-related proteins. When dividing this cohort of PSC serum samples by ALP levels, a circulating parameter used for monitoring PSC activity, there was a stronger relation of the fibrotic biomarker and CCL24 with increased ALP reflected.
- 13 -

CCL24 levels correlate with ELF score
CCR3 levels in circulating PBMCs in PSC patients
Chronic liver inflammation is driven in most hepatic injuries by several different immune cell populations originating from either resident hepatic immune cells or recruited cells from the circulation to the damaged site. In collaboration with the Kaplan Medical Center, Israel, Chemomab explored systemic changes of CCR3, given that this could impact cell recruitment to the PSC damaged liver. PBMCs from ten PSC patients and healthy controls were stained for expression of CCR3 and demonstrated that levels were significantly higher in PSC patient samples compared to healthy donors.
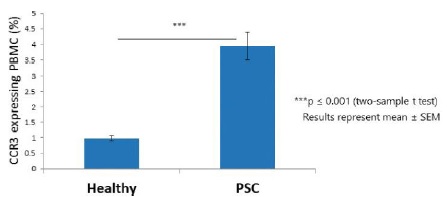
PSC patients showed significantly higher expression of CCR3 on PBMCs
CCL24 and CCR3 levels in skin biopsies from SSc patients
Chemomab analyzed skin samples from diffuse SSc patients and healthy volunteers and the SSc samples showed elevations in CCL24 and CCR3. Specifically, higher accumulation of CCL24 on immune cells skin infiltration was shown in the SSc samples and CCR3 was evident in skin fibroblasts, immune cells and endothelial cells. These elevations led to a CCL24 mediated robust activation of CCR3 expressing cells which enhances the recruitment of immune cells and fibroblasts to the diseased organ.
- 14 -

SSc patients showed elevated levels of CCL24 in skin tissue

SSc patients showed elevated levels of CCR3 in skin tissue
CCL24 levels in serum samples from SSc patients and correlation with fibrotic biomarkers
Chemomab analyzed SSc serum samples which showed that CCL24 levels were significantly increased in SSc patients compared with healthy individuals. Notably, in diffuse SSc patients, CCL24 levels were fourfold higher than in healthy control patients, while in limited SSc patients, a threefold elevation was found compared with healthy controls. Additionally, the levels of CCL24 were correlated with a biomarker of SSc severity, anti-topoisomerase, an autoantibody seen in diffuse SSc patients.
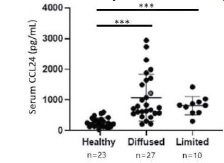
SSc patients showed elevated levels of CCL24 in serum samples
Clinical Development of CM-101
Completed studies
The CM-101 Phase 1 program included two Phase 1a single administration, or SAD, studies, using IV and SC administration with doses ranging from 0.75-10 mg/kg, in healthy volunteers and a Phase 1b multiple administration (MAD) study (5 administrations) in NAFLD patients with normal liver function, testing 2.5 mg/kg IV and 5 mg/kg SC. To date 42 subjects have received at least one CM-101 dose, the majority by IV infusion (12/42 subjects received SC).
- 15 -
Safety
The first Phase 1a study, which was a single-center, randomized double-blind, placebo-controlled, single-dose, dose-escalation study, included four escalating dose groups of eight subjects each. In each dose group subjects were randomized in a 3:1 ratio to receive a single IV infusion of either CM-101 (n=6) or placebo (n=2). A total of 24 subjects were enrolled into the study and randomized to the treatment groups (0.75 mg/ kg, 2.5 mg/kg, 5.0 mg/kg, 10 mg/kg) and eight subjects received a placebo. All 32 subjects completed the study as planned. Single, IV doses of CM-101 were safe and well tolerated up to the highest dose level (10 mg/ kg) in healthy subjects. No severe or serious adverse events, or AEs, occurred during the study and all CM-101 related AEs were mild, with one moderate AE reported in the placebo group (myalgia).
The second Phase 1a study was also a single-center, randomized double-blind, placebo-controlled, single-dose study, but evaluated only one dose group. Subjects were randomized in a 3:1 ratio to receive a single SC injection of either CM-101 5 mg/kg (n=6) or matching placebo (n=2). A total of eight subjects were enrolled into the study and randomized; all eight subjects completed the study as planned. Single, SC administration of 5 mg/kg of CM-101 was safe and well tolerated with no severe or serious AEs occurring during the study. A total of 6 AEs were reported in two subjects treated with CM-101; only one AE was classified as related to CM-101 (change in diastolic blood pressure) and that AE was classified as mild in intensity.
In both Phase 1a studies, all AEs reported were resolved; no subjects discontinued the study prematurely due to AEs, and no concomitant medications were required for treatment of any drug-related AEs. No clinically significant changes in laboratory tests (hematology, chemistry or urinalysis), vital signs, ECG, physical examination or infusion site examination were observed. In the first Phase 1a study with CM-101 delivered by IV administration, the effect on cytokine secretion was tested pre-treatment and one hour, eight hours and 24 hours post drug administration. Serum levels of a panel of cytokines including IL-6, IFNγ, GM-CSF, TNF-α, IL-2, IL-4, IL-8 and IL-10 showed no significant change at all tested CM-101 doses and timepoints. These findings suggest that single CM-101 administration does not cause immune activation nor cytokine secretion. Additionally, none of the subjects in either of the Phase 1a studies tested positive for anti-drug antibodies (ADA).
The multiple administrations randomized, placebo-controlled, Phase 1b study in NAFLD patients with normal liver function tests evaluated two dose levels. The first dose level of 2.5 mg/kg CM-101 was administered as IV infusions and the second dose level of 5 mg/kg was administered as SC injections. Both dose levels involved five drug administrations over 12 weeks (Q3W) providing 15 weeks of treatment coverage. At both dose levels, subjects were randomized in a 3:1 ratio to receive either CM-101 (n=6 per cohort) (2.5 mg/kg IV or 5 mg/kg SC) or matching placebo (n=2 per cohort). Five repeated IV and SC CM-101 administrations were safe and well tolerated and there were no deaths, or severe or serious drug related AEs reported throughout the study. Only mild to moderate AEs were reported in the CM-101 treatment groups of which only two AEs were classified as possibly related to CM-101. No injection site reactions or clinically significant trends in laboratory tests (hematology, chemistry, or urinalysis), vital signs, ECG or physical examination were observed. One patient experienced a non-drug-related SAE. This patient was a 61-year-old female that was subsequently diagnosed with a non-treatment related meningioma. The tumor was treated surgically, and the patient was discontinued from the study.
Pharmacokinetics with single-dose administration
PK analysis was conducted for the Phase 1 studies and the quantification of CM-101 in plasma samples was performed using a validated ELISA-based assay by Eurofins (UK). Following IV infusion in healthy volunteers, CM-101 exhibited a biphasic serum concentration vs. time curve (rapid distribution phase and slow elimination phase) which is typical for monoclonal antibodies. Target-mediated drug disposition (TMDD), or presence of ADAs, was not evident in the analyzed concentration vs. time curves of CM-101, which exhibited linear terminal slope without apparent TMDD kinetics or other concentration-dependent changes of the elimination kinetics. Comparison of the PK data of 5 mg/kg CM-101 using IV administration against SC administration indicates consistent distribution and elimination behavior of CM-101.
At either IV or SC administration, the values of the PK parameters obtained in the non-compartmental and compartmental analysis of CM-101 concentration vs. time data appear to be typical for monoclonal antibodies that undergo FcRn-mediated recycling. The terminal half-life of CM-101 was long for both SC and IV formulations which supports administration of CM-101 at a frequency of once every 2-4 weeks.
- 16 -
Pharmacokinetics with multiple-dose administration
PK analysis of the data from the Phase 1b study was conducted to evaluate CM-101 following multiple IV infusion of 2.5 mg/kg or 5 mg/kg SC injections of CM-101 in NAFLD patients. Following repeated IV infusions (2.5 mg/kg Q3W) and SC injection (5 mg/kg Q3W), CM-101 exhibited a long terminal half-life, similar to the terminal half life seen in the single dose studies. CM-101 accumulated over time, resulting in significant systemic exposure over time and potentially reaching a steady state.
Overall, CM-101 reached steady state conditions more slowly following SC injection, as compared to IV infusion. The inter-patient variability in CM-101 serum concentrations was higher for SC dosing injection, as compared to IV. The trough CM-101 serum concentrations after repeated 5 mg/kg SC injections were proportionally higher than those after 2.5 mg/kg IV infusions, considering the difference in administration modes. Comparison of the PK data of CM-101 in the Phase 1b to the Phase 1a studies indicates a consistency in PK behavior of CM-101.
Pharmacodynamics and target engagement of CM-101
Serum was taken from patients in all three Phase 1 studies at different times and the levels of both CCL24 and CM-101 were measured. Total CCL24 levels represent CM-101’s engagement to its target. Total CCL24 levels were increased following administration of the drug, which indicates that CM-101 is effective in target engagement, as the higher levels of CCL24 correlated significantly with greater doses of CM-101, and such levels decreased gradually from the peak of CM-101 administration. These findings demonstrate that CM-101 effectively binds to CCL24 in the circulation, which reflects a strong drug-target interaction.
In the Phase 1b study, CM-101 treatment of 2.5mg/kg IV attained the highest levels of total CCL24 by the third administration, maintaining such levels until the end of treatment. CM-101 5mg/kg administered by SC injection reached the highest levels of CCL24 by the fourth treatment, and maintained such levels until the end of treatment. The matching placebo did not have any effect on CCL24 levels.
As exemplified in the in-vitro studies, binding of CCL24 by CM-101 attenuates the binding of CCL24 to its cognate CCR3 receptor, thereby reducing its downstream activation. Altogether, CCL24 levels following treatment with CM-101 provide strong evidence for target engagement and pharmacodynamic response of CM-101 in healthy volunteers and patients.
Phase 1b exploratory endpoints- preliminary results
Fibrotic biomarkers were analyzed as part of the Phase 1b study in NAFLD patients with normal liver function. Circulating fibrotic biomarkers were tested in serum pre- and post-treatment. The analysis included data from patients that presented with a more active disease, reflected by baseline elastography (FibroScanTM) score >4 kPa. Tissue inhibitor of metalloproteinases-1 (TIMP-1) and tissue inhibitor of metalloproteinases-2 (TIMP-2), considered well established fibrotic biomarkers, were evaluated, and showed that CM-101 treatment led to reductions of both markers by week 15. The growth factor PDGF-AA, known as a pro-fibrotic secreted factor, was also reduced in CM-101 treated patients. Conversely, in the placebo group TIMP-1, TIMP-2 and PDGF-AA all increased.
Evaluation of the fibrogenesis and fibrolysis/inflammatory biomarkers, Pro-C3, Pro-C4 and C3M measured in serum, conducted by Nordic Bioscience, Copenhagen, Denmark, were also used as sensitive indicators of the liver’s fibrotic state. In accordance with reduced liver stiffness, Pro-C3, Pro-C4 and C3M were all reduced in the CM-101 treated groups. No reductions were identified in the placebo control group.
Changes in liver stiffness, a measurement of liver fibrosis, were also evaluated using FibroScanTM measurements taken at screening and end of treatment (EoT) following 15 weeks of treatment coverage. 80% of CM-101 treated patients had significant decreases in FibroScanTM measurements, unlike placebo patients where there was no significant change from baseline
Overall, these encouraging results provide initial support for CM-101 anti-fibrotic and anti-inflammatory mechanism in humans and support further testing of CM-101 in PSC and SSc patients.
- 17 -
Current and planned clinical studies for PSC and SSc
Chemomab is currently recruiting for a Phase 2a study at multiple sites in Europe and Israel for the study of CM-101 in patients with PSC and is also planning to conduct a Phase 2 study this year across 25 sites in the United States and Europe in SSc.
The ongoing Phase 2a study in PSC is a randomized, double-blind, placebo-controlled, study aimed to evaluate the safety and efficacy of CM-101 in adult subjects with PSC. Recruited subjects will have a serum alkaline phosphatase,or ALP, level of at least 1.5 times the upper limit of normal (x 1.5 ULN). Subjects with concomitant IBD are eligible for recruitment if their disease is stable and there is an absence of high-grade dysplasia in colonic biopsies within 18 months of randomization. Up to 45 subjects will be randomized to receive 10 mg/kg CM-101 IV, or placebo, in a 2:1 ratio. Randomization will be stratified based on ongoing Ursodeoxycholic Acid, or UDCA, treatment and concomitant IBD diagnosis. Subjects will receive a dose of investigational product once every three weeks for a total of five administrations resulting in a total coverage of 15 weeks.
The primary endpoints for the study are changes from baseline in serum alkaline phosphatase, or ALP, levels and the fibrotic marker enhanced liver function, or ELF, score at week 15. ALP is a liver enzyme that is elevated in cholestasis and the ELF score is a biochemical test panel made up of serum markers that are indicators of the extracellular matrix. Secondary endpoints include evaluation of safety and tolerability, changes from baseline in other liver enzymes and additional fibrotic markers, to include AST, ALT, Pro-C3 and Pro-C5. PK, PD and ADA parameters will also be collected.
The measurement of ALP as a primary endpoint is common in other PSC studies, for example the Phase 2 and Phase 3 studies for norUrsodeoxycholic acid (Dr. Falk Pharma GmbH, Freiburg, Germany). However, some studies have shown that improvements in ALP do not always correlate with improved outcomes. Accordingly, Chemomab has included the ELF score as an additional-primary endpoint, a measurement which has also shown successful reductions in other Phase 2 PSC clinical studies, such as one being conducted by NGM Biopharmaceuticals in connection with its development of NGM 282.
The planned Phase 2 study in SSc will be a global randomized, double-blind, placebo-controlled study aimed to evaluate the safety and efficacy of CM-101 in adult subjects with diffuse SSc. Recruited subjects will have had a disease duration of ≤ five years from the first non-Raynaud’s phenomenon manifestation and a modified Rodnan skin score (mRSS) of 15 to 35 units at baseline. Subjects will be randomized to receive either 5 mg/kg or 10 mg/kg CM-101 IV, or placebo, in a 1:1 ratio. Subjects will receive a dose of investigational product once every three weeks for a total duration of 24-52 weeks.
The study plans evaluate the safety and tolerability of multiple doses of CM-101 and the change from baseline in the ACR CRISS composite score. The CRISS score includes core items that assess changes in two common and prominent manifestations of early SSc (skin and lung damage), functional disability (as assessed by the HAQ DI), and patient and physician global assessments. In addition, the ACR CRISS score captures clinically meaningful worsening of internal organ involvement requiring treatment, that classifies the patient as having not improved. ACR CRISS is a commonly used endpoint in clinical studies of SSc and has previously shown meaningful reductions. Chemomab will also include secondary endpoints for change from baseline in the mRSS, a score of skin thickness in 17 body sites, lung function tests, inflammatory and fibrotic markers, PK, ADA and target engagement.
- 18 -
Other clinical development plans for CM-101
To further explore CM-101’s mechanistic effect in liver fibrosis, and given the string of effects seen preclinically, Chemomab is conducting a Phase 2a study in NASH patients to further explore the safety, tolerability and effects of CM-101 on relevant fibrosis related biomarkers using the SC formulation. The study will be initiated early this year and will be conducted across multiple sites in Israel and aims to recruit 40 patients with confirmed NASH and fibrosis without cirrhosis, liver fat content > 10% and at least one associated risk factor. Patients will have a total of eight administrations of CM-101, every two weeks, given at two mg/kg by SC injection.
Primary endpoints for the study will be safety and tolerability, however, Chemomab will explore multiple efficacy markers in its secondary endpoints, namely changes in liver enzymes, fibrotic markers, liver stiffness, liver fat content, PK, ADA, target engagement and PD.
Preclinical safety and toxicology of CM-101
Pre-clinical safety evaluation of CM-101 included tissue cross reactivity, assessment of the effect of CM-101 on pro-inflammatory cytokine secretion ex-vivo, and in vivo GLP toxicology studies in mice and non-human primates. No safety concerns were observed in these pre clinical assessments.
Immunogenicity may be triggered following administration of humanized monoclonal antibodies, an effect that is frequently seen with approved mAbs. Chemomab conducted an immunogenicity analysis and identified few potential non germline residues in CM-101. The company also completed a T-cell proliferation assay, to investigate these findings in vitro. Chemomab observed that T cell proliferation was not induced using the whole antibody (CM-101) while few specific fragments of the mAb were found to induce T cell proliferation. Overall, the fact that no ADA were identified three clinical studies performed to date supports a preliminary conclusion of low immunogenic potential of CM-101.
As summarized below, there were no safety concerns related to CM-101 in any of the other safety experiments.
Summary of key preclinical safety experiments
| Preclinical findings | | | Observation |
| Ex vivo | | | |
| Antibody dependent cell-cytotoxic (ADCC) and complement dependent cell-cytotoxic (CDC) activity was tested in PBMCs from healthy volunteers | | | CM-101 did not have Fc-related effector functions such as ADCC and CDC |
| Cytokine release was assessed in human whole blood from healthy volunteers. | | | CM-101 did not induce pro-inflammatory cytokine secretion |
| Tissue cross reactivity was evaluated from healthy human tissues. | | | CM-101 does not bind non-specifically to healthy tissues, and therefore is expected to only bind to its target, circulating CCL24 |
| In vivo | | | |
| GLP repeated dose 4-week toxicity study of CM-101 (IV) in mice | | |
1. No obvious treatment related adverse reactions 2. No gross or microscopic pathological findings 3. No cases of treatment related mortality were observed 4. No significant elevation was seen in IL1β, IL2, IL4, IL5, IL10, GM-CSF, IFN and TNFα |
- 19 -
| GLP repeated dose (up to 50 mg/kg) 6-month toxicity study of CM-101 (SC) in Cynomolgus Monkey | | |
1. No obvious treatment related adverse reactions 2. No clinical signs or injection site reactions 3. No cases of treatment related mortality were observed 4. Blood and urine tests were found to be within normal ranges for monkeys 5. No treatment-related organ weight changes and no treatment-related necropsy findings 6. No treatment-related histopathology findings 7. Three samples from treated animals were confirmed ADA positive and there was no obvious correlation between positive ADA results and CM-101 serum concentrations or systemic exposure |
Preclinical proof of mechanism studies for CM-101
Chemomab conducted a series of in vitro and in vivo studies to demonstrate the proposed mechanism of action and provide proof-of-concept for administering CM-101 in the clinic for target indications.
Affinity, selectivity, and binding kinetics
Chemomab evaluated the kinetic binding parameters of CM-101 to human CCL24, as well as the specificity of CM-101 binding to other chemokines using commercial binding assays. CM-101 demonstrated a strong and stable, high affinity, binding to CCL24.
CM-101 reduced CCL24 dependent CCR3 activation
In an in vitro assay, CM-101 was shown to robustly attenuate the ability of CCL24 to induce activation of the CCR3 receptor following pre-incubation of CCL24 with CM-101.

CM-101 reduces CCL24 dependent CCR3 activation dose dependently
- 20 -
Pre-clinical Efficacy of CM-101 in models of PSC
| Preclinical experiments in models of PSC |
| Human hepatic stellate cells demonstrated reduced transition to myofibroblasts following incubation of CM-101 with CCL24. |
| Human hepatic stellate cells showed reduced motility towards CCL24 following treatment with CM-101 |
| CM-101 demonstrated in vivo activity on liver fibrosis and cholangiocytes proliferation induced by bile duct ligation in Sprague Dawley rat model. |
| CM-101 (D8) inhibits the progression of liver fibrosis and bile duct damage in a chronic cholangitis cholestasis model using the hepatobiliary toxin ANIT. |
| CM-101 (D8) reduces liver enzymes, bile acid and circulating inflammatory monocytes in an experimental cholangitis model in MDR2 knock out mice. |
| CM-101 reduces liver enzymes, fibrosis, collagen, and fibrotic gene expression in a TAA-induced liver fibrosis model in rats. |
| CM-101 (D8) prevented fibrosis and inflammation in a TAA-induced liver fibrosis model in mice. |
Of the experiments performed above, results from the multi drug resistant 2, or MDR2, knock out mouse model that reflects sclerosing cholangitis and the thioacetamide (TAA) rat model reflecting liver fibrosis are described below.
CM-101 demonstrates anti-cholestatic and anti-fibrotic activity in MDR2 knock out mouse model in vivo
Mice with targeted disruption of the MDR2, transporter gene develop chronic and progressive hepatic sclerosing cholangitis that closely resembles PSC and therefore this model has been extensively used to study the pathogenesis and progression of PSC. Using MDR2 knockout mice (six weeks of age), Chemomab tested the ability of CM-101 (D8) (murine surrogate of CM-101) to attenuate PSC related symptoms. Mice (n=15/group) received either vehicle control, or CM-101 5 mg/kg SC twice weekly during weeks 6-12 following established disease and were sacrificed at the end of week 12. In this study mice were tested for changes in alkaline phosphatase, orALP, bile acid levels, collagen deposition (histology, Sirius red) and circulating inflammatory populations. Chemomab observed a significant decrease in serum collagen deposition, ALP and bile acid levels after CM-101 (D8) treatment compared to non-active treatment.
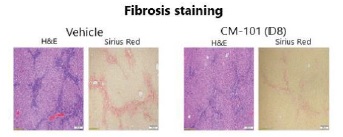
CM-101 reduces liver fibrosis in an MDR2 knockout model
In MDR2 knockout mice during chronic inflammation circulating Ly6Chi monocytes were shown to infiltrate the liver and have a significant role in hepatic damage. In these mice, circulating CD11b+ Ly6Chi monocytes were increased in numbers when compared to healthy mice and CM-101 (D8) treatment was able to normalize the number of inflammatory Ly6Chi monocytes back to normal levels seen in the healthy mice.
- 21 -

CM-101 reduces circulating inflammatory monocytes in MDR2 knockout model
CM-101 demonstrates in vivo activity in a thioacetamide induced liver fibrosis model in rats
To assess potential efficacy of CM-101 on liver fibrosis, Chemomab used the TAA-induced liver fibrosis model. Liver fibrosis was induced by intraperitoneal administration of TAA at a dose of 250 mg/kg twice weekly for eight weeks. Rats (n=10/group) received either vehicle control or CM-101 2.5 mg/kg IV twice weekly during weeks four-eight following established fibrosis and were sacrificed at week eight. After eight weeks of TAA treatment, all vehicle-treated animals had developed liver fibrosis, as confirmed by Sirius-red-stained liver histology.

CM-101 reduces fibrosis in rat livers
Plasma ALP, ALT, and AST levels decreased in the CM-101 study arm. Liver collagen content and fibrotic areas were significantly reduced in the CM-101 treated group compared to non-active treatment. CM-101 was also shown to reduce fibrotic markers in the TAA treated rats.
Efficacy CM-101 in models of SSc
| Preclinical experiments in models of SSc |
| CM-101 reduces SSc serum-induced dermal fibroblast activation and transition to myofibroblasts and interferes with endothelial cell activation. |
| CM-101 treatment attenuated skin fibrotic remodelling in the bleomycin (BLM)-induced dermal fibrosis mouse model. |
| CM-101 attenuated lung fibrosis and inflammation in the bleomycin (BLM)-induced pulmonary fibrosis mouse model. |
Of the experiments listed above, results from the bleomycin (BLM)-induced dermal and lung fibrosis mouse models are set forth below in more detail.
- 22 -
CM-101 treatment attenuates skin fibrotic remodeling in the bleomycin (BLM)-induced dermal fibrosis mouse model
The activity of CM-101 (D8) (murine surrogate of CM-101) in SSc was tested in the dermal bleomycin model. Treatment started after the onset of fibrotic signs, eight days following the first BLM injection. Histological assessment of skin lesions stained with H&E and Masson’s trichrome revealed significant elevation of dermal thickness and collagen deposition following 21 days of BLM administration. This elevation was significantly reduced when mice were treated with 2.5 mg/kg CM-101 with significant reductions in both skin thickness and collagen deposition compared with the mouse group treated with BLM alone.

CM-101 treatment attenuates skin fibrotic remodeling in the bleomycin-induced dermal fibrosis mouse model
Another feature that characterizes the BLM model and is representative of human SSc is the development of bronchoalveolar inflammation. To evaluate the effect of CM-101 on lung inflammation, Chemomab collected bronchoalveolar lavage, or, BAL, fluid, and assessed the number of white blood cells, or WBC, and mononuclear cells. Treatment with BLM for 21 days significantly increased WBC and mononuclear cells in BAL fluid and the number of WBC and mononuclear cells was decreased significantly following CM-101 treatment compared with the group that was administered only with BLM. This data supports the anti inflammatory effect of CM-101 in SSc.
CM-101 inhibits lung fibrosis in the BLM-induced pulmonary fibrosis mouse model
Chemomab also tested CM-101 in the experimental lung SSc model where mice were given a single intratracheal administration of BLM followed by either CM-101, non-active treatments (PBS or control immunoglobulin G (IgG)) or the approved anti-fibrosis drugs, pirfenidone and nintedanib. CM-101 had a significant anti-fibrotic and anti-inflammatory effect in the experimental BLM-induced lung fibrosis model as compared with non-active tratments treated animals. BLM animals treated with non-active treatments showed massive immune cell infiltration, extensive fibrosis and severe tissue injury. CM-101-treated mice exhibited significantly reduced levels of lung fibrosis down to similar levels in healthy animals and showed superior effects compared to approved fibrosis drugs pirfenidone and nintedanib.

CM-101 attenuates lung fibrosis and collagen deposition in the bleomycin (BLM)-induced pulmonary fibrosis mouse model
- 23 -
Competition
The development and commercialization of new drug products is highly competitive across major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. Chemomab faces competition with respect to its current product and expects to face competition with respect to any product candidates that it may develop or commercialize in the future. Specifically, there are a number of companies developing treatments for fibrotic/inflammatory diseases, including multiple major pharmaceutical and biotechnology companies with substantially greater resources than Chemomab. Chemomab is a small biotech company with limited resources compared to the major pharmaceutical companies, however, Chemomab believes that the unique CM-101 platform together with its knowledge and experience in inflammatory fibrotic research provides the company with competitive advantages.
Therapeutic options for PSC and SSc are limited and despite significant biopharmaceutical industry investment, the FDA has not approved any disease modifying therapies for the treatment of PSC or SSc. Liver transplant is currently the only treatment shown to improve clinical outcomes for PSC patients while SSc patients are being treated with drugs that were approved for different manifestations of the disease like interstitial lung disease (Nintedanib, Boehringer Ingelheim and Tocilizumab, Hoffmann-La Roche).
Chemomab is advancing CM-101, a first in class monoclonal antibody that interferes directly with both inflammation and fibrosis, into clinical development for the treatment of PSC and SSc. There are a number of large biopharmaceutical and biotechnology companies that are currently pursuing the development of products for the treatment of fibrotic indications like PSC and SSc such as Gilead Sciences, Inc., Mitsubishi Tanabe Pharma Corporation, Horizon Therapeutics plc, Pliant Therapeutics, Inc., Kadmon Holdings, Inc and others. However, Chemomab knows of no other companies currently in clinical development with a monoclonal antibody that targets CCL24.
Although the approach is novel with respect to targeting both inflammation and fibrosis, Chemomab will need to compete with products further advanced in the pipeline towards market approval. Investigational products in the leading disease areas, include:
• PSC
There are currently no FDA-approved therapies for the treatment of PSC. Companies currently developing product candidates in Phase 3 clinical studies include Gilead and Dr. Falk Pharma, targeting cholestasis and liver metabolism (Gilead; Cilofexor, Dr. Falk; norUrso). Additional companies with clinical candidates in earlier stages of development include HighTide Biopharmaceutical, Mirum Pharmaceuticals and Pliant Inc.
• SSc
There are currently two FDA approved products for the treatment of clinical manifestations of SSc, Nintedanib, marketed by Boehringer Ingelheim GmbH and Tocilizumab, marketed by Hoffmann-La Roche for the treatment of interstitial lung disease. Companies currently developing product candidates in SSc in early clinical stage include Kadmon Holdings, Inc., Horizon, BMS, GSK, Vicore, Sanofi and Emerald Health Sciences Inc.
The availability of reimbursement from government and other third-party payors will affect the pricing and competitiveness of CM-101 and any future products. More advanced competitors also may obtain regulatory approval for their products more rapidly than Chemomab, which could result in competitors establishing a strong market position.
Intellectual Property
Overview
Chemomab strives to protect and enhance the proprietary technology, inventions, and improvements that are commercially important to the development of its business, including seeking, maintaining, and defending patent rights, whether developed internally or licensed from third parties. Chemomab also relies on trade secrets relating to its proprietary technology platform and know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen, and maintain its proprietary position in the field of inflammation and fibrosis that may present areas of opportunity for the development of its business. Chemomab may also rely on regulatory protection afforded through data exclusivity, market exclusivity, and patent term extensions, where available.
- 24 -
Chemomab’s commercial success may depend in part on its ability to: obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to its business; defend and enforce its patents; preserve the confidentiality of its trade secrets; and operate without infringing the valid enforceable patents and proprietary rights of third parties. Chemomab’s ability to prevent third parties from making, using, selling, offering to sell, or importing Chemomab’s products may depend on the extent to which it has rights under valid and enforceable licenses, patents, or trade secrets that cover these activities. In certain cases, enforcement of these rights may depend on third party licensors. With respect to both licensed and company-owned intellectual property rights, Chemomab cannot be sure that patents will be granted with respect to any of its pending patent applications or with respect to any patent applications that may be filed by Chemomab in the future, nor can Chemomab be sure that any of its existing patents or any patents that may be granted to it in the future will be commercially useful in protecting its commercial products and methods of manufacturing the same.
As of March 31st, 2021, Chemomab owned or licensed three pending or issued US patents and patent applications as well as patents and patent applications in other jurisdictions. The first patent family has been issued in each of the United States, Europe (validated in France, Germany and the United Kingdom) and Israel to the Tel Aviv Souraski Medical Center, which rights have been licensed to Chemomab on an exclusive basis. To date, two additional patent families were filed by Chemomab, including a composition of matter patent that was issued in United States and certain corresponding foreign jurisdictions and a patent family concerning the use of anti CCL24 antibodies in specific fibrotic indications. Finally, two provisional US patent applications were filed during 2020 concerning dosing regimens, routes of administration, and additional indications. Chemomab will seek United States and foreign patent protection for a variety of additional technologies, including: research compounds and methods, candidate compounds and antibodies for modulating the activity of CCL24, methods for treating diseases of interest, and methods for treating its products. Chemomab will seek additional protection, in part, through confidentiality and proprietary information agreements.
Company Owned Intellectual Property
Chemomab owns multiple families of patent applications that are pertain to anti CCL24 monoclonal antibodies compositions capable of blocking CCL24 activity and methods for treating or preventing diseases associated with inflammation and fibrosis. Certain applications in these families relate to Chemomab’s CM-101 antibody, backup variants, various unit dosages, dosing regimens, and other routes of administration. Patents that are or will be issued from these submissions will expire between the years 2035 to 2041, subject to possible patent term adjustments and/or extensions.
In addition to the above, Chemomab has established expertise and development capabilities focused in the areas of preclinical research and development, manufacturing and manufacturing process development, quality control, quality assurance, regulatory affairs, and clinical study design and implementation. Chemomab believes that its focus and expertise will help Chemomab develop products based on its proprietary intellectual property.
Licensed IP
As mentioned above, Chemomab has obtained an exclusive license from the Tel Aviv Souraski Medical Center for one patent, which is expected to expire in 2029. This patent was issued in each of the United States, Europe and Israel, which patent and pertains to anti CCL24 inhibitors and methods of using such inhibitors for treating inflammatory, autoimmune and cardiovascular diseases.
Trade Secret Protection
Chemomab may rely, in some circumstances, on trade secrets to protect its technology. Chemomab seeks to protect its proprietary technology and processes, in part, by entering into confidentiality agreements with its employees, consultants, scientific advisors, and contractors. Chemomab also seeks to preserve the integrity and confidentiality of its data and trade secrets by maintaining physical security of its premises and physical and electronic security of its information technology systems.
- 25 -
Material Agreements
Tel-Aviv Souraski Medical Center (TASMC) License Agreement
In December 2011, Chemomab entered into a license agreement, or the TASMC Agreement, with the Medical Research, Infrastructure, Health Services Fund of the Tel Aviv Souraski Medical Center., or TASMC, for the research, development and commercialization of the CCL24 platform and CCR3 blockade platform (CM-101), which license includes patent rights covering the foregoing platforms and related know how and products. Under the terms of the TASMC Agreement, Chemomab is responsible for the research, development, manufacturing and commercialization of CM-101. This license was granted on an exclusive basis and Chemomab was also granted rights to sublicense the instant license to third parties pursuant to certain terms described therein.
In accordance with the TASMC Agreement, Chemomab paid TASMC a non-refundable and non-creditable payment in four milestone installments, related to TASMC’s past patent maintenance and prosecution costs.
Certain additional terms of the TASMC Agreement include:
• Chemomab will be required to pay TASMC non-refundable and non-creditable milestone payments of up to (i) $300,000 upon the submission of an NDA, BLA or equivalent for each of the licensed products to the FDA and to equivalent European and Asian foreign regulatory agencies, and (ii) $600,000 upon the grant by the FDA or equivalent European and/or Asian regulatory agencies of their marketing approval for each licensed product;
• In the event of an “exit,” as such term is defined therein, Chemomab must pay TASMC an exit fee of 1% of the transaction consideration (which shall be capped at $3 million);
• In the event Chemomab sublicenses a licensed product, Chemomab must pay TASMC a sublicense fee of 10% of all attributed income, in addition to a low-single digit percentage tiered royalty payment of our earned royalties.
Unless terminated earlier, the TASMC Agreement will expire upon the later of the expiration of the last-to-expire valid patent claim and any extension granted prior thereto. The termination of the TASMC Agreement will not preclude TASMC from receiving sublicense payments or royalties. In addition to the foregoing, the TASMC Agreement includes customary termination provisions.
CMC Collaboration Agreement
In June 2015, Chemomab entered into a collaboration agreement, or the CMC Agreement, with CMC ICOS Biologics, Inc. (acquired by AGC Biologics in 2018), or CMC, which, under the terms thereof, granted Chemomab certain licenses to use proprietary rights, materials and know how of CMC for purposes of research and development of CM-101 as well as commercialization thereof. Pursuant to the terms of the CMC Agreement, Chemomab received (i) a worldwide, non-exclusive, non-transferable, non-sublicensable license for research purposes, or the Research License, and (ii) an option, or the Option License, to a worldwide, non-exclusive, non-transferable, sublicensable license for commercialization purposes, subject to a fee schedule in addition to that described below.
In accordance with the terms of the CMC Agreement, Chemomab agreed to pay in exchange for the foregoing license payments to CMC upon the achievement of certain pre-determined clinical and regulatory events, an amount stipulated in the CMC Agreement, aggregating a six-digit number. Additionally, for any product that is commercialized pursuant to the CMC Agreement, Chemomab is required to pay CMC a royalty payment based on annual aggregate worldwide net sales thresholds for such products. In the event CMC exclusively manufactures Chemomab’s products, CMC agrees to waive the foregoing royalty.
Unless terminated earlier pursuant to the customary termination provisions set forth in the CMC Agreement, the Research License will expire upon the conclusion of the term as defined therein, and the Option License will expire upon the later of (a) the tenth anniversary following Chemomab’s obtainment of regulatory approval, or (b) the last to expire of the patent rights and country-by-country basis.
- 26 -
Manufacturing
Chemomab’s product candidate, CM-101, is a monoclonal antibody amenable to standard formulation technologies. Chemomab has developed the biological process and manufactured kilogram quantities through processes similar to the manufacturing processes that will be required to provide drug product for our anticipated Phase 2 clinical studies. The manufacturing process of the drug substance used for such product candidates is robust, well established and requires the use of readily available starting materials. The biological route is amenable to large-scale production and does not require unconventional equipment or handling during the manufacturing process. Chemomab has obtained an adequate supply chain of the drug substance for CM-101 from Chemomab’s first European contract manufacturing organization, or CMO, to satisfy both Chemomab’s clinical and preclinical requirements this year. Chemomab relies on a sole supplier for the manufacture of CM-101. Chemomab’s manufacturer has the capabilities to support late stage clinical studies as well as product launch and marketing.
Chemomab does not own or operate facilities for clinical drug manufacturing, storage, distribution or quality testing. Currently, all of Chemomab’s clinical manufacturing is outsourced to third-party manufacturers. As Chemomab’s development programs expand and it builds new process efficiencies, Chemomab expects to continually evaluate this strategy with the objective of satisfying demand for its clinical studies and, if approved, the manufacture, sale and distribution of commercial products.
Commercialization
Chemomab intends to develop and, if approved by the FDA, to commercialize its product candidates alone or in collaboration with others. Chemomab may work in combination with one or more large pharmaceutical partners for certain indications, where specialist capabilities are needed. Chemomab intends to enter into distribution or licensing arrangements for global or regional commercialization rights. Chemomab will, however, continuously review its partnering strategy in the light of new clinical data and market understanding.
Regulatory Matters
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs, such as those Chemomab is developing. These agencies and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of Chemomab’s product candidates.
United States government regulation of drug products
In the United States, the FDA regulates drugs under the FDCA and its implementing regulations. The FDA also regulates biological products under the FDCA and the Public Health Service Act, or PHSA. If Chemomab advances clinical development of a biologic candidate in the future, these development activities will be subject to additional regulatory requirements specific to biologics. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending New Drug Applications, or NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
- 27 -
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
• Completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations;
• Submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical studies may begin;
• Approval by an IRB at each clinical site before each study may be initiated;
• Performance of adequate and well-controlled human clinical studies in accordance with Good Clinical Practice, or GCP requirements to establish the safety and efficacy of the proposed drug product for each indication;
• Completion of all manufacturing requirements to ensure robust manufacturing process, and product quality and safety as per Good Manufacturing Practice, or cGMP guidelines;
• Completion of non-clinical reproductive studies, as applicable, prior to late stage clinical studies and New Drug Application, or NDA and Biologics License Application, or BLA submission.
• Development of an appropriate pediatric plan for clinical testing or exclusion, pre or post approval, as applicable.
• Submission to the FDA of an NDA;
• Satisfactory completion of an FDA advisory committee review, if applicable;
• Satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practice, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity;
• Satisfactory completion of FDA audits of clinical study sites to assure compliance with GCPs and the integrity of the clinical data;
• Payment of user fees and securing FDA approval of the NDA; and
• Compliance with any post-approval requirements, including the potential requirement to implement a REMS and the potential requirement to conduct post-approval studies.
Preclinical studies
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical studies and places the clinical study on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin. As a result, submission of an IND may not result in the FDA allowing clinical studies to initiate.
Clinical studies
Clinical studies involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical study. Clinical studies are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical study and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical study must review and approve the plan for any clinical study before it initiates at that institution. Information about certain clinical studies must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
- 28 -
Human clinical studies are typically conducted in three sequential phases, which may overlap or be combined:
• Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness.
• Phase 2: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
• Phase 3: The drug is administered to an expanded patient population, generally at geographically dispersed clinical study sites, in well-controlled clinical studies to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical studies must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 studies may not be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA or BLA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA, for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to FDA because the FDA has approximately two months to make a “filing” decision.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. An Agreed Initial Pediatric Study Plan requesting a waiver from the requirement to conduct clinical studies has been submitted to the FDA.
The FDA also may require submission of a REMS plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans, and/or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
- 29 -
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity.
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, which reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical study sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical study sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical studies, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Orphan drug designation and exclusivity
Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition (generally meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product). A company must request orphan product designation before submitting an NDA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process. As of the current date, Chemomab has obtained orphan drug designation for three indications, PSC, SSc and IPF.
If a product with orphan status receives the first FDA approval for the disease or condition for which it has such designation or for a select indication or use within the rare disease or condition for which it was designated, the product generally will be receiving orphan product exclusivity. Orphan product exclusivity means that the FDA may not approve any other applications for the same product for the same indication for seven years, except in certain limited circumstances. If a drug or drug product designated as an orphan product ultimately receives marketing approval for an indication broader than what was designated in its orphan product application, it may not be entitled to exclusivity. Orphan exclusivity will not bar approval of another product under certain circumstances, including if a subsequent product with the same active ingredient for the same indication is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care, or if the company with orphan drug exclusivity is not able to meet market demand. Further, the FDA may approve more than one product for the same orphan indication or disease as long as the products contain different active ingredients. Moreover, competitors may receive approval of different products for the indication for which the orphan product has exclusivity or obtain approval for the same product but for a different indication for which the orphan product has exclusivity.
- 30 -
United States marketing exclusivity
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an Abbreviated New Drug Application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for the original non-modified version of the drug. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical studies necessary to demonstrate safety and effectiveness.
Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing regulatory exclusivity periods. This six-month exclusivity may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
Post-approval requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There are continuing, annual user fee requirements for any marketed products and the establishments where such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical studies, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies, and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
- 31 -
Once an approval of a drug is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
• Restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls;
• Fines, warning letters or holds on post-approval clinical studies;
• Refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals;
• Product seizure or detention, or refusal to permit the import or export of products; and
• Injunctions or the imposition of civil or criminal penalties.
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted by a manufacturer and any third parties acting on behalf of a manufacturer only for the approved indications and in a manner consistent with the approved label for the product. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. On May 10, 2019, the Centers for Medicare and Medicaid Services announced a new pricing transparency rule, which was set to take effect on July 9, 2019. The rule would have required direct-to-consumer television advertisements for prescription drugs and biological products for which reimbursement is available, directly or indirectly, through or under Medicare or Medicaid to include the list price of that product, except for a prescription drug or biological product that has a list price of less than $35 per month for a 30-day supply or typical course of treatment. The final rule was vacated by the D.C. District Court prior to taking effect. Several states have adopted price transparency requirements and those as well as any future federal price transparency requirements that may be implemented in the future could have a negative effect on Chemomab’s business.
Other healthcare laws
Healthcare providers, physicians, and third party payors play a primary role in the recommendation and prescription of drug products for which Chemomab obtains marketing approval. Arrangements with third party payors, healthcare providers and physicians, in connection with the clinical research, sales, marketing and promotion of products, once approved, and related activities, may expose a pharmaceutical manufacturer to broadly applicable fraud and abuse and other healthcare laws and regulations. In the United States, these laws include, without limitation, state and federal anti-kickback, false claims, physician transparency, and patient data privacy and security laws and regulations, including but not limited to those described below:
• the federal Anti-Kickback Statute, or AKS, which makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer or pay any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, that is intended to induce or reward, referrals including the purchase recommendation, order or prescription of a particular drug for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act, or FCA;
- 32 -
• the federal civil and criminal false claims laws, including the FCA, which can be enforced through “qui tam” or “whistleblower” actions, and civil monetary penalty laws, which impose criminal and civil penalties against individuals or entities for, among other things, knowingly presenting, or causing to be presented, claims for payment or approval from Medicare, Medicaid, or other federal health care programs that are false or fraudulent; knowingly making or causing a false statement material to a false or fraudulent claim or an obligation to pay or transmit money or property to the federal government; or knowingly concealing or knowingly and improperly avoiding or decreasing such an obligation. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation.
• the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters;
• HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose requirements on certain covered healthcare providers, health plans, and healthcare clearinghouses as well as their respective business associates that perform services for them that involve the creation, use, receipt, maintenance or disclosure of individually identifiable health information, relating to the privacy, security and transmission of individually identifiable health information;
• the federal Physician Payments Sunshine Act, created under Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the ACA, and its implementing regulations, which require manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to the Centers for Medicare and Medicaid Services, or CMS, under the Open Payments Program, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Effective January 1, 2022, these reporting obligations will extend to include transfers of value made during the previous year to certain non-physician providers such as physician assistants and nurse practitioners; and
• analogous state and foreign laws and regulations, such as state and foreign anti-kickback, false claims, consumer protection and unfair competition laws which may apply to pharmaceutical business practices, including but not limited to, research, distribution, sales and marketing arrangements as well as submitting claims involving healthcare items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government that otherwise restricts payments that may be made to healthcare providers and other potential referral sources; state laws that require drug manufacturers to file reports with states regarding pricing and marketing information, such as the tracking and reporting of gifts, compensations and other remuneration and items of value provided to healthcare professionals and entities; state and local laws requiring the registration of pharmaceutical sales representatives; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
- 33 -
Because of the breadth of these laws and the narrowness of the statutory exceptions and regulatory safe harbors available, it is possible that some of a pharmaceutical manufacturer’s business activities could be subject to challenge under one or more of such laws. Efforts to ensure that business arrangements comply with applicable healthcare laws involve substantial costs. It is possible that governmental and enforcement authorities will conclude that a pharmaceutical manufacturer’s business practices do not comply with current or future statutes, regulations or case law interpreting applicable fraud and abuse or other healthcare laws and regulations. If any such actions are instituted against a pharmaceutical manufacturer, and it is not successful in defending itself or asserting its rights, those actions could have a significant impact on its business, including the imposition of significant civil, criminal and administrative penalties, damages, disgorgement, imprisonment, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, reporting obligations and oversight if Chemomab becomes subject to integrity and oversight agreements to resolve allegations of non-compliance, contractual damages, reputational harm, diminished profits and future earnings, and curtailment of operations, any of which could adversely affect a pharmaceutical manufacturer’s ability to operate its business and the results of operations. In addition, commercialization of any drug product outside the United States will also likely be subject to foreign equivalents of the healthcare laws mentioned above, among other foreign laws.
In the United States, numerous federal and state laws and regulations, including state data breach notification laws, state health information privacy laws, and federal and state consumer protection laws, govern the collection, use, disclosure, and protection of health-related and other personal information. For example, in June 2018, the State of California enacted the California Consumer Privacy Act of 2018, or the CCPA, which came into effect on January 1, 2020 and provides new data privacy rights for consumers and new operational requirements for companies, which may increase Chemomab’s compliance costs and potential liability. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. While there is currently an exception for protected health information that is subject to HIPAA and clinical study regulations, as currently written, the CCPA may impact certain of Chemomab’s business activities. The CCPA could mark the beginning of a trend toward more stringent state privacy legislation in the United States, which could increase Chemomab’s potential liability and adversely affect its business.
In the event Chemomab decides to conduct clinical studies or continue to enroll subjects in its ongoing or future clinical studies, Chemomab may be subject to additional privacy restrictions. The collection, use, storage, disclosure, transfer, or other processing of personal data regarding individuals in the European Economic Area, or EEA, including personal health data, is subject to the EU General Data Protection Regulation, or GDPR, which became effective on May 25, 2018. The GDPR is wide-ranging in scope and imposes numerous requirements on companies that process personal data, including requirements relating to processing health and other sensitive data, obtaining consent of the individuals to whom the personal data relates, providing information to individuals regarding data processing activities, implementing safeguards to protect the security and confidentiality of personal data, providing notification of data breaches, and taking certain measures when engaging third-party processors. The GDPR also imposes strict rules on the transfer of personal data to countries outside the EEA, including the United States, and permits data protection authorities to impose large penalties for violations of the GDPR, including potential fines of up to €20 million or 4% of annual global revenues, whichever is greater. The GDPR also confers a private right of action on data subjects and consumer associations to lodge complaints with supervisory authorities, seek judicial remedies, and obtain compensation for damages resulting from violations of the GDPR. In addition, the GDPR includes restrictions on cross-border data transfers. The GDPR may increase Chemomab’s responsibility and liability with respect to personal data that Chemomab processes where such processing is subject to the GDPR, and it may be required to put in place additional mechanisms to ensure compliance with the GDPR, including as implemented by individual countries. Compliance with the GDPR will be a rigorous and time-intensive process that may increase Chemomab’s cost of doing business or require it to change its business practices, and despite those efforts, there is a risk that Chemomab may be subject to fines and penalties, litigation, and reputational harm in connection with its European activities. Further, the United Kingdom’s decision to leave the EU, often referred to as Brexit, has created uncertainty with regard to data protection regulation in the United Kingdom. In particular, it is unclear how data transfers to and from the United Kingdom will be regulated now that the United Kingdom has left the EU.
Current and future healthcare reform legislation
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory changes to the health care system. In particular, in 2010 the ACA was enacted, which, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program, extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations, subjected manufacturers to new annual fees and taxes for certain branded prescription drugs, and provided incentives to programs that increase the federal government’s comparative effectiveness research.
- 34 -
There remain judicial, administrative, executive, and legislative challenges to certain aspects of the ACA, and Chemomab expects there will be additional challenges and amendments to the ACA in the future. On December 14, 2018, a Texas U.S. District Court Judge ruled that the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress as part of the Tax Act. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari to review this case, and has allotted one hour for oral arguments, which are expected to occur in the fall. In addition, the Trump Administration has issued various Executive Orders which eliminated cost sharing subsidies and various provisions that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. Additionally, Congress has introduced several pieces of legislation aimed at significantly revising or repealing the ACA. It is unclear whether the ACA will be overturned, repealed, replaced, or further amended. Chemomab cannot predict what effect further changes to the ACA would have on its business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. In August 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect in 2013, and, due to subsequent legislative amendments, will remain in effect through 2030 unless additional Congressional action is taken. However, the Medicare sequester reductions under the Budget Control Act of 2011 will be suspended from May 1, 2020 through December 31, 2020 due to the COVID-19 pandemic. The American Taxpayer Relief Act of 2012 further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. The Bipartisan Budget Act of 2018, also amended the ACA, effective January 1, 2019, by increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and closing the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”.
Additionally, there has been heightened governmental scrutiny in the United States of pharmaceutical pricing practices in light of the rising cost of prescription drugs and biologics. At the federal level, the U.S. Presidential administration’s budget proposal for the fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Moreover, the U.S. Presidential administration previously released a “Blueprint” to lower drug prices and reduce out of pocket costs of drugs that contained proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. Although a number of these and other measures may require additional authorization to become effective, Congress and the U.S. Presidential administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. Any reduction in reimbursement from Medicare and other government programs may result in a similar reduction in payments from private payers. In addition, individual states in the United States have also increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
- 35 -
Legislative and regulatory proposals, and enactment of laws, at the foreign, federal and state levels, directed at containing or lowering the cost of healthcare, will continue into the future.
Rest of World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing product development, the conduct of clinical studies, manufacturing, distribution, marketing approval, product licensing, pricing and reimbursement vary from country to country. Additionally, clinical studies must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If Chemomab fails to comply with applicable foreign regulatory requirements, Chemomab may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Additionally, to the extent that any of Chemomab’s product candidates, once approved, are sold in a foreign country, it may be subject to applicable post-marketing requirements, including safety surveillance, anti-fraud and abuse laws and implementation of corporate compliance programs and reporting of payments or other transfers of value to healthcare professionals.
Coverage and reimbursement
Successful commercialization of new drug products depends in part on the extent to which reimbursement for those drug products will be available from government health administration authorities, private health insurers, and other organizations. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drug products they will pay for and establish reimbursement levels. The availability and extent of reimbursement by governmental and private payors is essential for most patients to be able to afford a drug product. Sales of drug products depend substantially, both domestically and abroad, on the extent to which the costs of drugs products are paid for by health maintenance, managed care, pharmacy benefit and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers and other third-party payors.
A primary trend in the United States healthcare industry and elsewhere is cost containment. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular drug products. In many countries, the prices of drug products are subject to varying price control mechanisms as part of national health systems. In general, the prices of drug products under such systems are substantially lower than in the United States. Other countries allow companies to fix their own prices for drug products, but monitor and control company profits. Accordingly, in markets outside the United States, the reimbursement for drug products may be reduced compared with the United States.
In the United States, the principal decisions about reimbursement for new drug products are typically made by CMS, an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new drug product will be covered and reimbursed under Medicare, and private payors tend to follow CMS to a substantial degree. However, no uniform policy of coverage and reimbursement for drug products exists among third-party payors and coverage and reimbursement levels for drug products can differ significantly from payor to payor.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. While all Medicare drug plans must give at least a standard level of coverage set by Medicare, Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each Part D prescription drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for drugs for which Chemomab obtains marketing approval. Any negotiated prices for any of Chemomab’s products covered by a Part D prescription drug plan will likely be lower than the prices it might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
- 36 -
For a drug product to receive federal reimbursement under the Medicaid or Medicare Part B programs or to be sold directly to United States government agencies, the manufacturer must extend discounts to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the average manufacturer price, or AMP, and Medicaid rebate amounts reported by the manufacturer. As of 2010, the ACA expanded the types of entities eligible to receive discounted 340B pricing, although under the current state of the law these newly eligible entities (with the exception of children’s hospitals) will not be eligible to receive discounted 340B pricing on orphan drugs. As 340B drug pricing is determined based on AMP and Medicaid rebate data, the revisions to the Medicaid rebate formula and AMP definition described above could cause the required 340B discount to increase. The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. The plan for the research was published in 2012 by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures are made to Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of Chemomab’s product candidates, if any such drug or the condition that they are intended to treat are the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s drug could adversely affect the sales of Chemomab’s product candidate. If third-party payors do not consider Chemomab’s drugs to be cost-effective compared to other available therapies, they may not cover Chemomab’s drugs after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow Chemomab to sell its drugs on a profitable basis.
These laws, and state and federal healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices Chemomab may obtain for any product candidates for which it may obtain regulatory approval or the frequency with which any such product candidate is prescribed or used.
Outside of the United States, the pricing of pharmaceutical products and medical devices is subject to governmental control in many countries. For example, in the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost effectiveness of a particular therapy to currently available therapies or so-called health technology assessments, in order to obtain reimbursement or pricing approval. Other countries may allow companies to fix their own prices for products, but monitor and control product volumes and issue guidance to physicians to limit prescriptions. Efforts to control prices and utilization of pharmaceutical products and medical devices will likely continue as countries attempt to manage healthcare expenditures.
Employees and Human Capital Resources
As of March 31, 2021, Chemomab had 12 full-time employees, including five with Ph.D. or M.D. degrees and 10 who are engaged in research and development activities. Chemomab is dependent on its management and scientific personnel, and it is crucial that it continues to attract and retain valuable employees. To facilitate attraction and retention, Chemomab strives to make itself an inclusive and safe workplace, with opportunities for its employees to grow and develop in their careers, supported by strong compensation and benefits programs. None of Chemomab’s employees are represented by labor unions or covered by collective bargaining agreements.
- 37 -
Facilities
Chemomab leases a facility containing 2,486.46 square feet of laboratory and office space, which is located at Kiryat Atidim, Building 7, Tel Aviv, Israel 6158002. The lease expires in June 2023 with an option to extend for an additional 36 months. Chemomab believes that its current facilities are sufficient to meet its current and near-term needs and that, should it be needed, suitable additional space will be available.
Legal Proceedings
As of the date of this Current Report on Form 8-K, Chemomab is not party to any material legal matters or claims. Chemomab may become party to legal matters and claims arising in the ordinary course of business. Chemomab cannot predict the outcome of any such legal matters or claims, and despite the potential outcomes, the existence thereof may have an adverse impact on Chemomab because of defense and settlement costs, diversion of management resources and other factors.
RISK FACTORS
Risks Related to Chemomab’s Business, Research and Development and the Biopharmaceutical Industry
Chemomab has a limited operating history and funding, which may make it difficult to evaluate its prospects and likelihood of success.
Chemomab is a clinical-stage biopharmaceutical company with a limited operating history. Chemomab was incorporated in 2015, has no products approved for commercial sale and has not generated any revenue. Its operations to date have been limited to organizing and staffing the company, business planning, raising capital, establishing its intellectual property portfolio and conducting research and development of its product candidates, technology related to CCL24 and novel therapies for the treatment of inflammation and fibrosis. Chemomab’s approach to the discovery and development of product candidates is unproven, and Chemomab does not know whether it will be able to develop any products of commercial value. In addition, Chemomab’s lead product candidate, CM-101, is in early clinical development for the treatment of PSC and SSc. The clinical programs will require substantial additional development and clinical research, both in time and resources, before Chemomab is in a position to apply for or receive regulatory approvals and begin generating revenue in connection with the sale of such product candidates. Chemomab has not yet demonstrated the ability to successfully complete a large-scale, pivotal clinical trial, obtain marketing approval, manufacture a commercial scale product, or arrange for a third party to do so on its behalf, or conduct sales and marketing activities necessary for successful product commercialization. Consequently, predictions about Chemomab’s future success or viability may not be as accurate as they could be if Chemomab had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
In addition, as a business with a limited operating history, Chemomab may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors and risks frequently experienced by early-stage biopharmaceutical companies in rapidly evolving fields. Consequently, Chemomab has no meaningful history of operations upon which to evaluate its business, and predictions about its future success or viability may not be as accurate as they could be if Chemomab had a longer operating history or a history of successfully developing and commercializing drug products. Chemomab will eventually need to transition from a company with a research and development focus to a company capable of supporting commercial activities. Chemomab may not be successful in such a transition and, as a result, its business may be adversely affected.
As Chemomab continues to build its business, it expects its financial condition and operating results to fluctuate significantly from quarter to quarter and year to year due to a variety of factors, many of which are beyond Chemomab’s control.
- 38 -
Chemomab’s business is highly dependent on the success of its lead product candidate, CM-101, and any other product candidates that it advances into clinical studies. All of Chemomab’s programs will require significant additional clinical development.
Chemomab currently has no products that are approved for commercial sale and may never be able to develop marketable products. Chemomab is very early in its development efforts and has only one product candidate, CM-101, in early clinical development. Because CM-101 is Chemomab’s lead product candidate, if CM-101 encounters safety or efficacy problems, development delays, regulatory issues or other problems, Chemomab’s development plans and business would be significantly harmed. Chemomab has completed a Phase 1a SAD study with healthy volunteers, a Phase 1b MAD study of CM-101 in non-alcoholic fatty liver disease, or NAFLD, and is recruiting volunteers to participate in its in Phase 2a biomarkers study in NASH patients. Chemomab plans to initiate a Phase 2 SSc study by the end of 2021, , however, for certain additional risks described herein, Chemomab cannot guarantee it will reach this clinical milestone or produce desirable results.
Chemomab expects that a substantial portion of its efforts and expenditures over the next few years will be devoted to CM-101, which will require additional clinical development, management of clinical and manufacturing activities, regulatory approval in multiple jurisdictions, obtaining manufacturing supply, building of a commercial organization, substantial investment and significant marketing efforts before it can generate any revenues from any commercial sales. Chemomab cannot be certain that it will be able to successfully complete any of these activities. In addition, if one or more of Chemomab’s product candidates are approved, Chemomab must ensure access to sufficient commercial manufacturing capacity and conduct significant marketing efforts in connection with any commercial launch. These efforts will require substantial investment, and Chemomab may not have the financial resources to continue the development of its product candidates.
Chemomab’s approach in the area of fibrotic diseases is novel and unproven and may not result in marketable products.
Chemomab’s central objective is to design and develop targeted treatments for inflammation and fibrosis with an initial focus on the antagonism of CCL24 signaling, which is known to regulate fibrotic and inflammatory processes. While several studies are currently underway, this mechanism has not yet been definitively proven to successfully treat inflammation and fibrosis. Targeting CCL24 to treat inflammation and fibrosis is a novel approach in a rapidly developing field, and there can be no assurance that Chemomab can avoid unforeseen problems or delays in the development of its product candidates, that such problems or delays will not result in unanticipated costs, or that any such development problems can or will be solved. Chemomab has only tested its lead product candidate, CM-101, in healthy volunteers and NAFLD patients. Therefore, Chemomab may ultimately discover that its approach does not possess properties required for therapeutic effectiveness. As a result, Chemomab may elect to abandon the program or never succeed in developing a marketable product, which would have a significant effect on the success and profitability of its business.
Clinical development involves a lengthy, complex and expensive process, with an uncertain outcome.
Before obtaining the requisite regulatory approvals from the FDA or other comparable foreign regulatory authorities for the sale of any of its product candidates, Chemomab must support its application with clinical studies that prove that such product candidate is safe and effective in humans. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. In particular, the general approach for FDA approval of a new drug requires positive data from two well-controlled Phase 3 clinical studies of the relevant drug in the relevant patient population. Failure can occur at any time during the clinical study process. Chemomab may experience delays in initiating and completing any clinical studies that it is conducting or intends to conduct, including as a result of the COVID-19 pandemic, and Chemomab does not know whether its ongoing or planned clinical studies will begin or progress on schedule, need to be redesigned, enroll patients on time or be completed on schedule, or at all.
- 39 -
Phase 3 clinical studies typically involve hundreds of patients, have significant costs and take years to complete. A product candidate can fail at any stage of testing, even after observing promising signals of activity in earlier preclinical studies or clinical trials. The results of preclinical studies and early clinical trials of Chemomab’s product candidates may not be predictive of the results of later-stage clinical studies. In addition, initial or interim success in clinical studies may not be indicative of results obtained when such studies are completed. There is typically an extremely high rate of attrition from the failure of product candidates proceeding through clinical studies. Product candidates in later stages of clinical studies may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical studies due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier studies. Most product candidates that commence clinical studies are never approved as products and there can be no assurance that any of Chemomab’s future clinical studies will ultimately be successful or support further clinical development of CM-101. Product candidates that appear promising in the early phases of development may fail to reach the market for several reasons, including:
• the FDA or comparable foreign regulatory authorities disagreeing as to the design or implementation of Chemomab’s clinical studies;
• obtaining regulatory authorizations to commence a trial or consensus with regulatory authorities on trial’s design;
• reaching an agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
• obtaining Institutional Review Board, or IRB, approval at each site, or Independent Ethics Committee, or IEC, approval at sites outside the United States;
• imposition of a clinical hold by regulatory authorities, including as a result of unforeseen safety issues or side effects or failure of trial sites to adhere to regulatory requirements or follow trial protocols;
• clinical studies may show the product candidates to be less effective than expected (e.g., a clinical study could fail to meet its primary endpoint(s)) or to have unacceptable side effects or toxicities;
• failure to establish clinical endpoints that applicable regulatory authorities would consider clinically meaningful;
• the occurrence of serious adverse events in trials of the same class of agents conducted by other companies;
• adding a sufficient number of clinical study sites;
• manufacturing sufficient quantities of product candidate with sufficient quality for use in clinical studies;
• having patients complete a trial or return for post-treatment follow-up;
• recruiting suitable patients to participate in a trial in a timely manner and in sufficient numbers;
• a facility manufacturing Chemomab’s product candidates or any of their components being ordered by the FDA or comparable foreign regulatory authorities to temporarily or permanently shut down due to violations of current good manufacturing practice, or cGMP, regulations or other applicable requirements, or infections or cross-contaminations of product candidates in the manufacturing process;
• third-party clinical investigators losing the licenses or permits necessary to perform Chemomab’s clinical studies, not performing its clinical studies on its anticipated schedule or consistent with the clinical study protocol, good clinical practices, or GCP, or other regulatory requirements;
• third-party contractors not performing data collection or analysis in a timely or accurate manner;
• manufacturing costs, formulation issues, pricing or reimbursement issues, or other factors that make a product candidate uneconomical; or
• the proprietary rights of others and their competing products and technologies that may prevent one of Chemomab’s product candidates from being commercialized.
- 40 -
In addition, differences in trial design between early-stage clinical studies and later-stage clinical studies make it difficult to extrapolate the results of earlier clinical studies to later clinical studies. Moreover, clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in clinical studies have nonetheless failed to obtain marketing approval of their products.
In addition, the standards used by the FDA and comparable foreign regulatory authorities when regulating Chemomab require judgment and can change, which makes it difficult to predict with certainty how they will be applied. For more information, see “Risk Factors — Risks Related to Chemomab’s Regulatory Approvals.”
Successful completion of clinical studies is a prerequisite to submitting a marketing application to the FDA and similar marketing applications to comparable foreign regulatory authorities, for each product candidate and, consequently, the ultimate approval and commercial marketing of any product candidates. Chemomab may experience negative or inconclusive results, which may result in it deciding, or it being required by regulators, to conduct additional clinical studies or trials or abandon some or all of its product development programs, which could have a material adverse effect on Chemomab’s business.
Chemomab may incur additional costs or experience delays in completing the development and commercialization of CM-101 or any other product candidates.
Chemomab may experience delays in initiating or completing clinical studies. It also may experience numerous unforeseen events during, or as a result of, any future clinical studies that could delay or prevent its ability to receive marketing approval or commercialize CM-101 or any other product candidates, including:
• regulators, IRBs, or ethics committees may not authorize Chemomab or its investigators to commence a clinical study or conduct a clinical study at a prospective trial site;
• the FDA or other comparable regulatory authorities may disagree with Chemomab’s clinical study design, including with respect to dosing levels administered in its planned clinical studies, which may delay or prevent Chemomab from initiating its clinical studies with its originally intended trial design;
• Chemomab may experience delays in reaching, or fail to reach, agreement on acceptable terms with prospective trial sites and prospective CROs, which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites;
• the number of subjects required for clinical studies of any product candidates may be larger than Chemomab anticipates or subjects may drop out of these clinical studies or fail to return for post-treatment follow-up at a higher rate than it anticipates;
• Chemomab’s third-party contractors may fail to comply with regulatory requirements or meet its contractual obligations to Chemomab in a timely manner, or at all, or may deviate from the clinical study protocol or drop out of the trial, which may require that Chemomab add new clinical study sites or investigators;
• due to the impact of the COVID-19 pandemic. Chemomab has experienced, and may continue to experience, delays and interruptions to clinical studies, it may experience delays or interruptions to its manufacturing supply chain, or it could suffer delays in reaching, or it may fail to reach, agreement on acceptable terms with third-party service providers on whom it relies;
• additional delays and interruptions to Chemomab’s clinical studies could extend the duration of the trials and increase the overall costs to finish the trials as its fixed costs are not substantially reduced during delays;
- 41 -
• Chemomab may elect to, or regulators, IRBs, Data Safety Monitoring Boards or ethics committees may require that it or its investigators, suspend or terminate clinical research or trials for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks;
• Chemomab may not have the financial resources available to begin and complete the planned trials, or the cost of clinical studies of any product candidates may be greater than it anticipates; and
• the supply or quality of Chemomab’s product candidates or other materials necessary to conduct clinical studies of its product candidates may be insufficient or inadequate to initiate or complete a given clinical study.
Chemomab’s product development costs will increase if it experiences additional delays in clinical testing or in obtaining marketing approvals. Chemomab does not know whether any of its clinical studies will begin as planned, will need to be restructured or will be completed on schedule, or at all. If Chemomab does not achieve its product development goals in the time frames it announces and expects, the approval and commercialization of its product candidates may be delayed or prevented entirely. Significant clinical study delays also could shorten any periods during which it may have the exclusive right to commercialize its product candidates and may allow its competitors to bring products to market before Chemomab does, potentially impairing its ability to successfully commercialize its product candidates and harming its business and results of operations. Any delays in Chemomab’s clinical development programs may harm its business, financial condition and results of operations significantly.
Chemomab’s ongoing and future clinical studies may reveal significant adverse events or immunogenicity related responses and may result in a safety profile that could delay or prevent regulatory approval or market acceptance of its product candidate.
Chemomab completed its Phase 1a and Phase 1b clinical studies of its lead product candidate, CM-101, in healthy volunteers and NAFLD patients, and, with the exception of a number of reported minor adverse events (including mild headaches, changes in blood pressure and mild-moderate increases in liver enzymes), CM-101 was observed to be generally well-tolerated across all doses in 42 trial participants. Some potential therapeutics developed in the biopharmaceutical industry that initially showed therapeutic promise in early-stage trials have later been found to cause side effects that prevented their further development and ultimately commercialization. Even if side effects do not preclude the product candidate from obtaining or maintaining marketing approval, undesirable side effects may inhibit market acceptance of the approved product due to its tolerability versus other therapies.
Protein biopharmaceuticals, including, monoclonal antibodies, or mAbs, may be immunogenic and promote immune responses against themselves. In particular, anti-drug antibodies, or ADAs, may be produced by patients following infusion mAbs and may disturb the pharmacokinetics of mAbs, neutralize their therapeutic activities or induce allergic or autoimmune symptoms. Clinical immunogenicity can range from mild, transient antibody responses with no apparent clinical manifestations to loss of therapeutic efficacy and even life-threatening reactions. Several approved therapeutic antibodies have been found to induce neutralizing antibodies, as illustrated by the approved anti-TNFa antibodies infliximab and adalimumab as well as anti IL-17 approved mAb ixekizumab. Chemomab’s product candidate, CM-101, is a humanized antibody that, similar to other humanized approved mAbs, was shown to include several non-germline sequences that may serve as a source for immunogenicity in therapeutic antibodies. In vitro testing was conducted and revealed that while T cell proliferation was not induced using the whole antibody (CM-101), specific fragments of the mAb that contained non-germline residues, induced T cell proliferation. Clinical studies to date have shown a lack of anti-drug antibodies, or ADAs. Additional larger clinical studies will be needed to address the risk of immunogenicity and, if discovered, Chemomab’s business will be materially and adversely affected.
Additionally, if unacceptable side effects, including materialized risks of immunogenicity, do arise in the development of Chemomab’s product candidates, Chemomab, the FDA or the IRBs at the institutions in which its studies are conducted, or the Data Safety Monitoring Board, if constituted for its clinical studies, could recommend a suspension or termination of Chemomab’s clinical studies, or the FDA or comparable foreign regulatory authorities could order Chemomab to cease further development of or deny approval of a product candidate for any or all targeted indications. In addition, drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete a trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. Chemomab expects to have to train medical personnel using its product candidates to understand the side effect profiles for its clinical studies and upon any commercialization of any of its product candidates. Inadequate training in recognizing or managing the potential side effects of its product candidates could result in patient injury or death. Any of these occurrences may harm Chemomab’s business, financial condition and prospects significantly.
- 42 -
Additionally, if one or more of Chemomab’s product candidates receives marketing approval, and Chemomab or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
• regulatory authorities may withdraw approvals of such product;
• regulatory authorities may require additional warnings on the label, such as a “black box” warning or contraindication;
• additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof;
• Chemomab may be required to implement a Risk Evaluation and Mitigation Strategy, or REMS, or create a medication guide outlining the risks of such side effects for distribution to patients;
• Chemomab could be sued and held liable for harm caused to patients;
• the product may become less competitive; and
• Chemomab’s reputation may suffer.
Any of these events could prevent Chemomab from achieving or maintaining market acceptance of a product candidate, if approved, and could significantly harm Chemomab’s business, results of operations and prospects.
If Chemomab encounters difficulties enrolling patients in its clinical studies, including due to COVID-19, its clinical development activities could be delayed or otherwise adversely affected.
Chemomab may experience difficulties in patient enrollment in its clinical studies for a variety of reasons. The timely completion of clinical studies in accordance with its protocols depends, among other things, on Chemomab’s ability to enroll a sufficient number of patients who remain in the trial until its conclusion. The enrollment of patients depends on many factors, including:
• the patient eligibility and exclusion criteria defined in the protocol;
• the size of the patient population required for analysis of the trial’s primary endpoints and the process for identifying patients;
• the willingness or availability (including legality under applicable COVID-19 shelter-in-place regulations) of patients to participate in Chemomab’s trials (including due to fears of contracting COVID-19);
• the proximity of patients to trial sites;
• the design of the trial;
• Chemomab’s ability to recruit clinical study investigators with the appropriate competencies and experience;
- 43 -
• clinicians’ and patients’ perceptions as to the potential advantages and risks of the product candidate being studied with respect to other available therapies, including any new products that may be approved for the indications Chemomab is investigating;
• the availability of competing commercially available therapies and other competing product candidates’ clinical studies;
• Chemomab’s ability to obtain and maintain patient informed consents; and
• the risk that patients enrolled in clinical studies will drop out of the trials before completion.
Further, timely enrollment in clinical studies is reliant on clinical study sites which may be adversely affected by global health matters, including, among other things, pandemics. For example, Chemomab’s clinical study sites have been affected by the COVID-19 pandemic. Commencement of the enrollment of Chemomab’s clinical studies of CM-101 in PSC has been delayed. Further, Chemomab anticipates delays in the enrollment for its CM-101 PSC Phase 2 study, and it could experience slower than expected enrollment. In addition, after enrollment in these trials, if patients contract COVID-19 during participation in Chemomab’s trials or are subject to isolation or shelter-in-place restrictions, this may cause them to drop out of Chemomab’s trials, miss scheduled doses or follow-up visits or otherwise fail to follow trial protocols. If patients are unable to follow the trial protocols or if Chemomab’s trial results are otherwise disputed due to the effects of the COVID-19 pandemic or actions taken to mitigate its spread, the integrity of data from Chemomab’s trials may be compromised or not accepted by the FDA or other regulatory authorities, which would represent a significant setback for the applicable program.
Some factors from the COVID-19 pandemic that Chemomab believes may adversely affect enrollment in its trials include:
• the diversion of healthcare resources away from the conduct of clinical study matters to focus on pandemic concerns, including the attention of infectious disease physicians serving as Chemomab’s clinical study investigators, hospitals serving as Chemomab’s clinical study sites and hospital staff supporting the conduct of its clinical studies;
• the inability of patients to come to hospitals to participate in Chemomab’s trials, which may force Chemomab to conduct its trials in patients’ homes, rendering the trials more difficult and costly to conduct;
• limitations on travel that interrupt key trial activities, such as clinical study site initiations and monitoring; and
• employee furlough days that delay necessary interactions with local regulators, ethics committees and other important agencies and contractors.
These and other factors arising from the COVID-19 pandemic could worsen in countries that are already afflicted with the virus or could continue to spread to additional countries, each of which may further adversely impact Chemomab’s clinical studies. The global outbreak of the COVID-19 pandemic continues to evolve and the conduct of Chemomab’s trials may continue to be adversely affected, despite efforts to mitigate this impact.
The market opportunities for CM-101, if approved, may be smaller than Chemomab anticipates.
Chemomab expects to initially seek approval of CM-101 for the treatment of PSC and SSc. Its projections of the number of PSC and SSc patients is based on its beliefs and estimates. These estimates have been derived from a variety of sources, including scientific literature, patient foundations and publicly available databases, and may prove to be incorrect. Further, new sources may reveal a change in the estimated number of patients, and the number of patients may turn out to be lower than Chemomab expected. The potential addressable patient population for Chemomab’s current programs or future product candidates may be limited. The ultimate market opportunity for Chemomab’s product candidates will depend on, among other things, the final labeling for such product candidates as agreed with the FDA or comparable foreign regulatory authorities, acceptance by the medical community and patient access, potential competition and drug pricing and reimbursement. Even if Chemomab obtains significant market share for any product candidate, if approved, if the potential target populations are small, Chemomab may never achieve profitability without obtaining marketing approval for additional indications.
- 44 -
Chemomab may not be successful in its efforts to identify or discover additional product candidates in the future.
Chemomab’s research programs may initially show promise in identifying potential product candidates, yet may fail to yield product candidates for clinical development for a number of reasons, including:
• Chemomab’s inability to design such product candidates with the pharmacological properties that it desires or attractive pharmacokinetics; or
• potential product candidates may, on further study, be shown to have harmful side effects or other characteristics that indicate that they are unlikely to be medicines that will receive marketing approval and achieve market acceptance.
Research programs to identify new product candidates require substantial technical, financial and human resources. If Chemomab is unable to identify suitable compounds for preclinical and clinical development, it will not be able to obtain product revenue in future periods, which likely would result in significant harm to Chemomab’s financial position and adversely impact the price of its ADSs.
Due to Chemomab’s limited resources and access to capital, it must make decisions on the allocation of resources to certain programs and product candidates; these decisions may prove to be wrong and may adversely affect Chemomab’s business.
Chemomab has limited financial and human resources and intends to initially focus on research programs and product candidates for a limited set of indications. As a result, Chemomab may forgo or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential or a greater likelihood of success.
There can be no assurance that Chemomab will ever be able to identify additional therapeutic opportunities for its product candidates or to develop suitable potential product candidates through internal research programs, which could materially adversely affect its future growth and prospects. Chemomab may focus its efforts and resources on potential product candidates or other potential programs that ultimately prove to be unsuccessful.
If product liability lawsuits are brought against Chemomab, it may incur substantial financial or other liabilities and may be required to limit commercialization of its product candidates.
Chemomab faces an inherent risk of product liability as a result of testing CM-101, and will face an even greater risk if Chemomab commercializes any products. For example, Chemomab may be sued if any of its product candidates cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical studies, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If Chemomab cannot successfully defend itself against product liability claims, it may incur substantial liabilities or be required to limit commercialization of its product candidates. Even successful defense would require significant financial and management resources. Chemomab’s inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products Chemomab develops. Chemomab will need to obtain additional insurance for clinical studies as it continues clinical development of CM-101 and as additional product candidates enter clinical studies. However, Chemomab may be unable to obtain, or may obtain on unfavorable terms, clinical study insurance in amounts adequate to cover any liabilities from any of its clinical studies. Chemomab’s insurance policies may also have various exclusions, and Chemomab may be subject to a product liability claim for which it has no coverage. Chemomab may have to pay any amount awarded by a court or negotiated in a settlement that exceed its coverage limitations or that are not covered by insurance, and Chemomab may not have, or be able to obtain, sufficient capital to pay such amounts. Even if Chemomab’s agreements with any future corporate collaborators entitles Chemomab to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
- 45 -
Chemomab has been granted Orphan Drug Designation for CM-101 in connection with three indications and may seek Orphan Drug Designation for other indications or product candidates, and Chemomab may be unable to maintain the benefits associated with Orphan Drug Designation, including the potential for market exclusivity, and may not receive Orphan Drug Designation for other indications or for its other product candidates.
Regulatory authorities in some jurisdictions, including the United States and Europe, may designate drugs intended for relatively small patient populations as orphan drugs. Under the Orphan Drug Act, the FDA may designate a drug as an orphan drug if it is a drug intended to treat a rare disease or condition, which is generally defined as a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States. In the United States, Orphan Drug Designation entitles a party to financial incentives such as opportunities for grant funding toward clinical study costs, tax advantages and user-fee waivers. In addition, if a product that has Orphan Drug Designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA to market the same product for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or where the manufacturer is unable to assure sufficient product quantity. However, Orphan Drug Designation neither shortens the development time or regulatory review time of a drug nor gives the drug any advantage in the regulatory review or approval process.
The FDA and EMA granted Orphan Drug Designation to CM-101 in its primary indications of PSC, SSc and IPF. Chemomab may seek Orphan Drug Designations for CM-101 in other indications or for other product candidates. There can be no assurance that Chemomab will be able to obtain such designations.
Even if Chemomab obtains Orphan Drug Designation for any product candidate in specific indications, it may not be the first to obtain marketing approval of such product candidate for the orphan-designated indication due to the uncertainties associated with developing pharmaceutical products. In addition, exclusive marketing rights in the United States may be limited if Chemomab seeks approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Further, even if Chemomab obtains orphan drug exclusivity in the United States for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties can be approved for the same condition. Even after an orphan product is approved, the FDA can subsequently approve the same drug with the same active moiety for the same condition if the FDA concludes that the later drug is safer, more effective or makes a major contribution to patient care.
Chemomab will need to expand its organization, and it may experience difficulties in managing this growth, which could disrupt its operations.
As of March 31, 2021, Chemomab had 12 employees. Chemomab expect to experience significant growth in the number of its employees and the scope of its operations, particularly in the areas of product candidate development, regulatory affairs and sales and marketing. Chemomab may have difficulty identifying, hiring and integrating new personnel. Future growth would impose significant additional responsibilities on its management, including the need to identify, recruit, maintain, motivate and integrate additional employees, consultants and contractors. Also, Chemomab’s management may need to divert a disproportionate amount of its attention away from its day-to-day activities and devote a substantial amount of time to managing these growth activities. Chemomab may not be able to effectively manage the expansion of its operations, which may result in weaknesses in its infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Chemomab’s expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of product candidates. If Chemomab’s management is unable to effectively manage its growth, its expenses may increase more than expected, its ability to generate and/or grow revenues could be reduced, and it may not be able to implement its business strategy. Chemomab’s future financial performance and its ability to commercialize its product candidates and compete effectively will depend, in part, on its ability to effectively manage any future growth.
- 46 -
Many of the biopharmaceutical companies that Chemomab competes against for qualified personnel and consultants have greater financial and other resources, different risk profiles and a longer history in the industry than Chemomab does. If Chemomab is unable to continue to attract and retain high-quality personnel and consultants, the rate and success at which it can discover and develop product candidates and operate its business will be limited.
Chemomab has incurred significant operating losses since its inception and anticipates it will incur continued losses for the foreseeable future.
Chemomab has funded its operations to date through proceeds from sales of its equity and grants from the Israel Innovation Authority, or the IIA, which as of December 31, 2020, resulted in gross proceeds of approximately $35 million. As of December 31, 2020, Chemomab’s cash and cash equivalents were $11.7 million. Chemomab has incurred net losses in each year since its inception, and it has an accumulated deficit of $23.7 million as of December 31, 2020.
Substantially all of Chemomab’s operating losses have resulted from general and administrative costs associated with its operations, and costs associated with its research and development programs, including for its preclinical and clinical product candidates. Chemomab expects to incur increasing levels of operating losses over the next several years and for the foreseeable future. Chemomab’s prior losses, combined with expected future losses, have had and will continue to have an adverse effect on its shareholders’ deficit and working capital. In any particular quarter or quarters, Chemomab’s operating results could be below the expectations of securities analysts or investors, which could cause the price of Chemomab’s ADSs to decline.
Chemomab expects its research and development expenses to significantly increase in connection with its clinical studies of its product candidates. In addition, if Chemomab obtains marketing approval for its product candidates, it will incur significant sales and marketing, legal, and outsourced-manufacturing expenses. As a public company, Chemomab expects to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, Chemomab is also unable to predict the extent of any future losses or when it will become profitable, if at all. Even if Chemomab does become profitable, it may not be able to sustain or increase its profitability on a quarterly or annual basis.
The current pandemic of COVID-19 and the future outbreak of other highly infectious or contagious diseases could seriously harm Chemomab’s research, development and potential future commercialization efforts, increase its costs and expenses and have a material adverse effect on its business, financial condition and results of operations.
Broad-based business or economic disruptions have, and could continue to, adversely affect Chemomab’s ongoing or planned research and development activities. For example, to date, the COVID-19 pandemic has caused significant disruptions to the Israeli, United States and global economy and has contributed to significant volatility and negative pressure in financial markets. The global impact of the outbreak is continually evolving and, as additional cases of the virus are identified, many countries, including Israel and the United States, have reacted by instituting quarantines, restrictions on travel and mandatory closures of businesses. Most countries, including where Chemomab or the third parties with whom it engages operate, have also reacted by instituting quarantines, restrictions on travel, “shelter in place” rules, and restrictions on types of business that may continue to operate.
The extent to which COVID-19 may impact Chemomab’s preclinical studies or clinical trial operations will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the duration of the outbreak, the severity of COVID-19, or the effectiveness of actions to contain and treat COVID-19. The continued spread of COVID-19 globally could adversely impact Chemomab’s preclinical studies or clinical study operations in Israel and the United States, including its ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography. COVID-19 may also affect employees of third-party CROs located in affected geographies that Chemomab relies upon to carry out its clinical studies. Any negative impact COVID-19 has on patient enrollment or treatment or the execution of its current product candidates and any future product candidates could cause costly delays to clinical study activities, which could adversely affect Chemomab’s ability to obtain regulatory approval for and to commercialize its current product candidates and any future product candidates, increase its operating expenses, and have a material adverse effect on its financial results.
- 47 -
Chemomab cannot presently predict the scope and severity of any potential business shutdowns or disruptions. If Chemomab or any of the third parties with whom it engages, however, were to experience shutdowns or other business disruptions, Chemomab’s ability to conduct its business in the manner and on the timelines presently planned could be materially and negatively affected, which could have a material adverse impact on its business and its results of operation and financial condition.
Risks Related to Chemomab’s Intellectual Property Rights
If Chemomab is unable to protect its patents or other proprietary rights, or if Chemomab infringes the patents or other proprietary rights of others, its competitiveness and business prospects may be materially damaged.
Patent and other proprietary rights are essential to Chemomab’s business. Chemomab’s success depends to a significant degree on its ability to obtain and enforce patents and licenses to patent rights, both in the United States and in other countries. Chemomab cannot guarantee that pending patent applications will result in issued patents, that patents issued or licensed will not be challenged or circumvented by competitors, that the patents and other intellectual property rights of Chemomab and its business partners will not be found to be invalid or that the intellectual property rights of others will not prevent Chemomab from selling its products or from executing on its strategies.
The patent position of a biopharmaceutical company is often uncertain and involves complex legal and factual questions. Significant litigation concerning patents and products is pervasive in Chemomab’s industry. Patent claims include challenges to the coverage and validity of Chemomab’s patents on products or processes as well as allegations that its products infringe patents held by competitors or other third parties. A loss in any of these types of cases could result in a loss of patent protection or the ability to market products, which could lead to a significant loss of sales, or otherwise materially affect future results of operations. Chemomab also relies on trademarks, copyrights, trade secrets and know-how to develop, maintain and strengthen its competitive positions. Third parties may know, discover or independently develop equivalent proprietary information or techniques, or they may gain access to Chemomab’s trade secrets or disclose such trade secrets to the public.
Although Chemomab’s employees, consultants, parties to collaboration agreements and other business partners are generally subject to confidentiality or similar agreements to protect its confidential and proprietary information, these agreements may be breached, and Chemomab may not have adequate remedies for any breach. In addition, Chemomab’s trade secrets may otherwise become known or be independently discovered by competitors. To the extent that Chemomab’s employees, consultants, parties to collaboration agreements and other business partners use intellectual property owned by others in their work for the company, disputes may arise as to the rights in related or resulting know-how and inventions.
Furthermore, Chemomab’s intellectual property, other proprietary technology and other sensitive company data is potentially vulnerable to loss, damage or misappropriation from system malfunction, computer viruses, unauthorized access to data or misappropriation or misuse thereof by those with permitted access and other events. While Chemomab has invested to protect its intellectual property and other data, and continue to work diligently in this area, there can be no assurance that its precautionary measures will prevent breakdowns, breaches, cyber incidents or other events. Such events could have a material adverse effect on Chemomab’s reputation, business, financial condition or results of operations. Misappropriation or other loss of Chemomab’s intellectual property from any of the foregoing could have a material adverse effect on its competitive position and may cause it to incur substantial litigation costs.
- 48 -
Chemomab may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of a third-party patent, which might adversely affect its ability to develop, manufacture and market its product candidates.
From time to time Chemomab may identify patents or applications in the same general area as its products and product candidates. Chemomab may determine these third-party patents are irrelevant to its business based on various factors, including its interpretation of the scope of the patent claims and its interpretation of when the patent expires. If the patents are asserted against Chemomab, however, a court may disagree with its determinations. Further, while Chemomab may determine that the scope of claims that will issue from a patent application does not present a risk, it is difficult to accurately predict the scope of claims that will issue from a patent application, its determination may be incorrect, and the issuing patent may be asserted against Chemomab. Chemomab cannot guarantee that it will be able to successfully settle or otherwise resolve such infringement claims. If Chemomab fails in any such dispute, in addition to being forced to pay monetary damages, it may be temporarily or permanently prohibited from commercializing its product candidates or be required to obtain a license under such patent, which may not be available on reasonable terms or at all. Chemomab might, if possible, also be forced to redesign its product candidates so that it no longer infringes, misappropriates or otherwise violates the third-party intellectual property rights. Any of these events, even if Chemomab were ultimately to prevail, could require it to divert substantial financial and management resources that it would otherwise be able to devote to its business. Any of the foregoing could have a material adverse effect on Chemomab’s business, financial condition, results of operations, and prospects.
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing Chemomab’s ability to protect its product candidates.
As is the case with other biopharmaceutical and pharmaceutical companies, Chemomab’s success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical and pharmaceutical industries involve both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical and pharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the Leahy-Smith America Invents Act, or the AIA, which was passed in September 2011, resulted in significant changes to the United States patent system.
An important change introduced by the AIA is that, as of March 16, 2013, the United States transitioned from a “first-to-invent” to a “first-to-file” system for deciding which party should be granted a patent when two or more patent applications are filed by different parties claiming the same invention. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to a patent on the invention regardless of whether another inventor had made the invention earlier. A third party that files a patent application in the USPTO after that date but before Chemomab could therefore be awarded a patent covering an invention of ours even if it made the invention before it was made by the third party. This will require Chemomab to be cognizant of the time from invention to filing of a patent application and be diligent in filing patent applications, but circumstances could prevent it from promptly filing patent applications on its inventions.
Among some of the other changes introduced by the AIA are changes that limit where a patentee may file a patent infringement suit and providing opportunities for third parties to challenge any issued patent with the USPTO. This applies to all of Chemomab’s United States patents, even those issued before March 16, 2013. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action.
Accordingly, a third party may attempt to use the USPTO procedures to invalidate Chemomab’s patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. It is not clear what, if any, impact the AIA will have on the operation of its business. However, the AIA and its implementation could increase the uncertainties and costs surrounding the prosecution of Chemomab or its licensors’ patent applications and the enforcement or defense of Chemomab or its licensors’ issued patents.
Chemomab may become involved in opposition, interference, derivation, inter partes review, post-grant review, reexamination or other proceedings challenging Chemomab or its licensors’ patent rights, and the outcome of any proceedings are highly uncertain. An adverse determination in any such proceeding could reduce the scope of, or invalidate, Chemomab’s owned or in-licensed patent rights, in whole or in part, allow third parties to commercialize its technology or products and compete directly with Chemomab, without payment to it, or result in Chemomab’s inability to manufacture or commercialize products without infringing third-party patent rights.
- 49 -
Additionally, the United States Supreme Court has ruled on several patent cases in recent years either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to Chemomab’s ability to obtain patents in the future, this combination of events has created uncertainty with respect to the validity, enforceability and value of patents, once obtained. Depending on decisions by Congress, the federal courts and the USPTO, as well as similar bodies in other jurisdictions, the laws and regulations governing patents could change in unpredictable ways that could weaken Chemomab’s ability to obtain new patents or to enforce its existing patents and patents that it might obtain in the future. Similarly, the complexity and uncertainty of European patent laws have also increased in recent years. In addition, the European patent system is relatively stringent in the type of amendments that are allowed during prosecution. Complying with these laws and regulations could limit Chemomab’s ability to obtain new patents in the future that may be important for its business, and these laws and regulations patents could continue to change in unpredictable ways that could have a material adverse effect on Chemomab’s existing patent rights and its ability to protect and enforce its intellectual property in the future.
Obtaining and maintaining Chemomab’s patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and Chemomab’s patent protection could be reduced or eliminated for non-compliance with these requirements.
The USPTO and European and other patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In addition, periodic maintenance, renewal and annuity fees on any issued patent are due to be paid to the USPTO and European and other patent agencies over the lifetime of a patent. While an inadvertent failure to make payment of such fees or to comply with such provisions can in many cases be cured by additional payment of a late fee or by other means in accordance with the applicable rules, there are situations in which such noncompliance will result in the abandonment or lapse of the patent or patent application, and the partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents within prescribed time limits. If Chemomab or its licensors fails to maintain the patents and patent applications covering its product candidates or if Chemomab or its licensors otherwise allow its patents or patent applications to be abandoned or lapse, its competitors might be able to enter the market, which would hurt Chemomab’s competitive position and could impair its ability to successfully commercialize its product candidates in any indication for which they are approved, which could have a material adverse effect on Chemomab’s business, financial condition, results of operations, and prospects.
Risks Related to Chemomab’s Regulatory Approvals
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if Chemomab is ultimately unable to obtain regulatory approval for CM-101 or any other product candidates, its business will be substantially harmed.
The time required to obtain approval by the FDA and comparable foreign authorities is unpredictable but typically takes many years following the commencement of clinical studiesand depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that Chemomab’s data is insufficient for approval and require additional preclinical, clinical or other data. Even if Chemomab eventually completes clinical testing and receives approval of any regulatory filing for its product candidates, the FDA and other comparable foreign regulatory authorities may approve Chemomab’s product candidates for a more limited indication or a narrower patient population than it originally requested. Chemomab has not obtained regulatory approval for any product candidate and it is possible that it will never obtain regulatory approval for CM-101 or any other product candidate. Chemomab is not permitted to market any of its product candidates in the United States until it receives regulatory approval of an NDA from the FDA.
- 50 -
Prior to obtaining approval to commercialize a product candidate in the United States or abroad, Chemomab must demonstrate with substantial evidence from well-controlled clinical studies, and to the satisfaction of the FDA or foreign regulatory agencies, that such product candidate is safe and effective for its intended use. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if Chemomab believes the preclinical or clinical data for its product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities.
The FDA or any foreign regulatory bodies can delay, limit or deny approval of Chemomab’s product candidates or require it to conduct additional preclinical or clinical testing or abandon a program for many reasons, including:
• the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of Chemomab’s clinical studies;
• Chemomab may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication;
• serious and unexpected drug-related side effects experienced by participants in Chemomab’s clinical studies or by individuals using drugs similar to its product candidates, or other products containing the active ingredient in Chemomab’s product candidates;
• negative or ambiguous results from Chemomab’s clinical studies or results that may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval;
• the population studied in the clinical study may not be sufficiently broad or representative to assure efficacy and safety in the full population for which Chemomab seeks approval;
• Chemomab may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks;
• the FDA or comparable foreign regulatory authorities may disagree with Chemomab’s interpretation of data from preclinical studies or clinical trials;
• the data collected from clinical studies of Chemomab’s product candidates may not be acceptable or sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere, and Chemomab may be required to conduct additional clinical studies;
• the FDA’s or the applicable foreign regulatory agency’s disagreement regarding the formulation, labeling and/or the specifications of Chemomab’s product candidates;
• the FDA or comparable foreign regulatory authorities may fail to approve or find deficiencies with the manufacturing processes or facilities of third-party manufacturers with which Chemomab contracts for clinical and commercial supplies; and
• the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering Chemomab’s clinical data insufficient for approval.
Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless failed to obtain FDA or comparable foreign regulatory authority approval. Chemomab cannot guarantee that the FDA or foreign regulatory authorities will interpret trial results as it does, and more trials could be required before Chemomab is able to submit applications seeking approval of its product candidates. To the extent that the results of the trials are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, Chemomab may be required to expend significant resources, which may not be available to it, to conduct additional trials in support of potential approval of Chemomab’s product candidates. Furthermore, the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering Chemomab’s clinical data insufficient for approval, which may lead to the FDA or comparable foreign regulatory authorities delaying, limiting or denying approval of its product candidates.
- 51 -
Of the large number of drugs in development, only a small percentage successfully complete the regulatory approval processes and are commercialized. This lengthy approval process, as well as the unpredictability of future clinical trial results, may result in Chemomab failing to obtain regulatory approval to market CM-101 or any other product candidate, which would significantly harm Chemomab’s business, results of operations and prospects.
In addition, the FDA or the applicable foreign regulatory agency also may approve a product candidate for a more limited indication or patient population than Chemomab originally requested, and the FDA or applicable foreign regulatory agency may approve a product candidate with a REMS or a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. Regulatory authorities may also grant approval contingent on the performance of costly post-marketing clinical trials. Any of the foregoing scenarios could materially harm the commercial prospects for Chemomab’s product candidates.
Obtaining and maintaining regulatory approval of Chemomab’s product candidates in one jurisdiction does not mean that it will be successful in obtaining regulatory approval of its product candidates in other jurisdictions.
In order to market any product outside of the United States, Chemomab must establish and comply with the numerous and varying safety, efficacy, and other regulatory requirements of other countries. Obtaining and maintaining regulatory approval of its product candidates in one jurisdiction does not guarantee that Chemomab will be able to obtain or maintain regulatory approval in any other jurisdiction, but a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. Chemomab’s product candidates may not receive marketing approval even if they achieve their primary endpoints in future Phase 3 clinical studies or registrational trials. The FDA or comparable foreign regulatory authorities may disagree with Chemomab’s trial designs and its interpretation of data from preclinical studies or clinical trials. In addition, any of these regulatory authorities may change requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal Phase 3 or registrational clinical study. In addition, any of these regulatory authorities may also approve a product candidate for fewer or more limited indications than Chemomab’s request or may grant approval contingent on the performance of costly post-marketing clinical trials. The FDA or comparable foreign regulatory authorities may not approve the labeling claims that Chemomab believes would be necessary or desirable for the successful commercialization of its product candidates, if approved.
Furthermore, even if the FDA or other comparable foreign regulatory authority grants marketing approval of a product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing, and promotion of the product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from those in the United States, including additional preclinical studies or clinical trials as clinical studies conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. The marketing approval processes in other countries may implicate all of the risks detailed above regarding FDA approval in the United States, as well as other risks. In many jurisdictions outside the United States, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that Chemomab intends to charge for its products is also subject to approval.
Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties, and costs for Chemomab and could delay or prevent the introduction of its products in certain countries. Failure to obtain marketing approval in other countries or any delay or other setback in obtaining such approval would impair its ability to market its product candidates in such foreign markets. Any such impairment would reduce the size of its potential market, which could have a material adverse impact on its business, results of operations, and prospects.
- 52 -
Even if Chemomab obtains regulatory approval for CM-101 or any product candidate, it will still face extensive and ongoing regulatory requirements and obligations and any product candidates, if approved, may face future development and regulatory difficulties.
Any product candidate for which Chemomab obtains marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, packaging, distribution, adverse event reporting, storage, recordkeeping, export, import, advertising and promotional activities for such product, among other things, will be subject to extensive and ongoing requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, establishment registration and drug listing requirements, continued compliance with cGMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians and recordkeeping and GCP requirements for any clinical studies that Chemomab conducts post-approval.
Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product candidate may be marketed or to the conditions of approval, including a requirement to implement a REMS. If any of Chemomab’s product candidates receives marketing approval, the accompanying label may limit the approved indicated use of the product candidate, which could limit sales of the product candidate. The FDA may also impose requirements for costly post-marketing studies or clinical trials and surveillance to monitor the safety or efficacy of a product. Violations of the Federal Food, Drug, and Cosmetic Act, or FDCA, relating to the promotion of prescription drugs may lead to FDA enforcement actions and investigations alleging violations of federal and state healthcare fraud and abuse laws, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with Chemomab’s products, manufacturers or manufacturing processes or failure to comply with regulatory requirements, may yield various results, including:
• restrictions on manufacturing such products;
• restrictions on the labeling or marketing of products;
• restrictions on product manufacturing, distribution or use;
• requirements to conduct post-marketing studies or clinical trials;
• warning letters or untitled letters;
• withdrawal of the products from the market;
• refusal to approve pending applications or supplements to approved applications that Chemomab submits;
• recall of products;
• fines, restitution or disgorgement of profits or revenues;
• suspension or withdrawal of marketing approvals;
• refusal to permit the import or export of Chemomab’s products;
• product seizure; or
• injunctions or the imposition of civil or criminal penalties.
Further, the FDA’s policies may change, and additional government regulations may be enacted that could impose extensive and ongoing regulatory requirements and obligations on any product candidate for which Chemomab obtains marketing approval. If Chemomab is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if Chemomab is not able to maintain regulatory compliance, it may lose any marketing approval that it may have obtained, which would adversely affect its business, prospects and ability to achieve or sustain profitability.
- 53 -
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder Chemomab’s ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, approved or commercialized in a timely manner or at all, which could negatively impact its business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would harm Chemomab’s business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the United States government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process Chemomab’s regulatory submissions, which could harm its business.
The COVID-19 pandemic has also resulted in the FDA imposing preventive measures, including postponements of non-United States manufacturing and product inspections. If global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process Chemomab’s regulatory submissions, which could have a material adverse effect on its business.
Risks Related to Commercialization of Chemomab’s Product Candidates
If Chemomab does not achieve its projected development and commercialization goals in the timeframes it announces and expects, the commercialization of its product candidates may be delayed and Chemomab’s business will be harmed.
For planning purposes, Chemomab sometimes estimates the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include Chemomab’s expectations regarding the commencement or completion of scientific studies and clinical trials, the regulatory submissions or commercialization objectives. From time to time, Chemomab may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical study, the initiation of other clinical studies, receipt of regulatory approval or the commercial launch of a product. The achievement of many of these milestones may be outside of Chemomab’s control. All of these milestones are based on a variety of assumptions which may cause the timing of achievement of the milestones to vary considerably from Chemomab’s estimates, including:
• Chemomab’s available capital resources or capital constraints it experiences;
• the rate of progress, costs and results of Chemomab’s clinical studies and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators;
• Chemomab’s ability to identify and enroll patients who meet clinical study eligibility criteria;
• Chemomab’s receipt of authorizations by the FDA and comparable foreign regulatory authorities, and the timing thereof;
• other actions, decisions or rules issued by regulators;
- 54 -
• Chemomab’s ability to access sufficient, reliable and affordable supplies of materials used in the manufacture of its product candidates;
• Chemomab’s ability to manufacture and supply clinical study materials to its clinical sites on a timely basis;
• the severity, duration and impact of the COVID-19 pandemic;
• the efforts of Chemomab’s collaborators with respect to the commercialization of its products, if any; and
• the securing of, costs related to, and timing issues associated with, commercial product manufacturing as well as sales and marketing activities.
If Chemomab fails to achieve announced milestones in the timeframes it expects, the commercialization of any of its product candidates may be delayed, and its business, results of operations, financial condition and prospects may be adversely affected.
Chemomab faces substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than it.
The development and commercialization of new drug products is highly competitive. Chemomab may face competition with respect to any product candidates that it seeks to develop or commercialize in the future from major biopharmaceutical companies, specialty biopharmaceutical companies, and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
There are a number of large biopharmaceutical and biotechnology companies that are currently pursuing the development of products for the treatment of inflammation and fibrosis. Companies that Chemomab is aware of that are targeting the treatment of inflammation and fibrosis include large companies with significant financial resources such as Biogen, Inc., AbbVie Inc., Gilead Sciences, Inc., FibroGen, Inc., Galapagos NV, Bristol Myers Squibb Co., and Novartis AG. However, Chemomab does not know of any other companies currently in clinical development with an anti CCL24 mAb. For additional information regarding Chemomab’s competition, see “Chemomab Business — Competition.”
Many of Chemomab’s current or potential competitors, either alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical studies, obtaining regulatory approvals, and marketing approved products than Chemomab does.
Even if CM-101 or any other product candidate Chemomab develops receives marketing approval, it may fail to achieve market acceptance by physicians, patients, third-party payors or others in the medical community necessary for commercial success.
If CM-101 or any other product candidate Chemomab develops receives marketing approval, it may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community. If it does not achieve an adequate level of acceptance, Chemomab may not generate significant product revenues or become profitable. The degree of market acceptance of Chemomab’s product candidates, if approved, will depend on a number of factors, including but not limited to:
• the efficacy and potential advantages compared to alternative treatments;
• effectiveness of sales and marketing efforts;
• the cost of treatment with respect to alternative treatments, including any similar generic treatments;
• Chemomab’s ability to offer its products for sale at competitive prices;
- 55 -
• the convenience and ease of administration compared to alternative treatments;
• the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies;
• the strength of marketing and distribution support;
• the timing of market introduction of competitive products;
• the availability of third-party coverage and adequate reimbursement;
• product labeling or product insert requirements of the FDA, EMA or other regulatory authorities, including any limitations on warnings contained in a product’s approved labeling;
• the prevalence and severity of any side effects; and
• any restrictions on the use of Chemomab’ product together with other medications.
Because Chemomab expects sales of its product candidates, if approved, to generate substantially all of its revenues for the foreseeable future, the failure of its product candidates to find market acceptance would harm its business and could require it to seek additional financing.
Chemomab relies completely on third-party suppliers to manufacture its clinical drug supplies for its product candidates, and Chemomab intends to rely on third parties to produce preclinical, clinical, and commercial supplies of any future product candidates.
Chemomab does not currently have, nor does Chemomab plan to acquire, the infrastructure or capability to internally manufacture its clinical drug supply of its product candidates, or any future product candidates, for use in the conduct of its preclinical studies and clinical trials.
Chemomab lacks the internal resources and the capabilities to manufacture any product candidates on a clinical or commercial scale. The facilities used by its contract manufacturers to manufacture the active pharmaceutical ingredient and final drug product must complete a pre-approval inspection by the FDA and other comparable foreign regulatory agencies to assess compliance with applicable requirements, including cGMPs, after Chemomab submits its NDA or relevant foreign regulatory market application to the applicable regulatory agency.
Chemomab is responsible for setting the product specifications and approving master batch records, but does not oversee the manufacturing process itself, and is completely dependent on its contract manufacturers to comply with cGMPs for manufacture of both active drug substances and finished drug products. If its contract manufacturers cannot successfully manufacture material that conforms to its specifications and the strict regulatory requirements of the FDA or applicable foreign regulatory agencies, they will not be able to pass a pre-approval inspection or secure and/or maintain regulatory approval for their manufacturing facilities. In addition, Chemomab has no direct control over its contract manufacturers’ ability to maintain adequate quality control, quality assurance, and qualified personnel. Furthermore, all of its contract manufacturers are engaged with other companies to supply and/or manufacture materials or products for such companies, which exposes its manufacturers to regulatory risks for the production of such materials and products. As a result, failure to satisfy the regulatory requirements for the production of those materials and products may affect the regulatory clearance of its contract manufacturers’ facilities generally. If the FDA or an applicable foreign regulatory agency determines now or in the future that these facilities for the manufacture of its product candidates are noncompliant, Chemomab may need to find alternative manufacturing facilities, which would adversely impact its ability to develop, obtain regulatory approval for or market its product candidates. Its reliance on contract manufacturers also exposes Chemomab to the possibility that they, or third parties with access to their facilities, will have access to and may compromise its trade secrets or other proprietary information.
- 56 -
If Chemomab is unable to establish sales, marketing and distribution capabilities either on its own or in collaboration with third parties, it may not be successful in commercializing CM-101, if approved.
Chemomab does not have any infrastructure for the sales, marketing or distribution of CM-101, and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market and successfully commercialize CM-101 or any other product candidate Chemomab develops, if approved, it must build its sales, distribution, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. Chemomab expects to build a focused sales, distribution and marketing infrastructure to market CM-101, if approved. There are significant expenses and risks involved with establishing Chemomab’s own sales, marketing and distribution capabilities, including its ability to hire, retain and appropriately incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of Chemomab’s internal sales, marketing and distribution capabilities could delay any product launch, which would adversely impact the commercialization of that product. Additionally, if the commercial launch of CM-101 for which Chemomab recruits a sales force and establishes marketing capabilities is delayed or does not occur for any reason, it would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and Chemomab’s investment would be lost if it cannot retain or reposition its sales and marketing personnel.
Factors that may inhibit Chemomab’s efforts to commercialize its product candidates on its own include:
• Chemomab’s inability to recruit, train and retain adequate numbers of effective sales and marketing personnel;
• the inability of sales personnel to obtain access to physicians or attain adequate numbers of physicians to prescribe Chemomab’s products; and
• unforeseen costs and expenses associated with creating an independent sales and marketing organization.
Chemomab does not anticipate having the resources in the foreseeable future to allocate to the sales and marketing of its product candidates, if approved, in certain markets overseas. Therefore, Chemomab’s future success will depend, in part, on its ability to enter into and maintain collaborative relationships for such capabilities, the collaborator’s strategic interest in a product and such collaborator’s ability to successfully market and sell the product. Chemomab intends to pursue collaborative arrangements regarding the sale and marketing of CM-101, if approved, for certain markets overseas; however, Chemomab cannot guarantee that it will be able to establish or maintain such collaborative arrangements, or if able to do so, that it will have effective sales forces. To the extent that Chemomab depends on third parties for marketing and distribution, any revenues it receives will depend upon the efforts of such third parties, and there can be no assurance that such efforts will be successful.
If Chemomab is unable to build its own sales force or negotiate a collaborative relationship for the commercialization of CM-101, Chemomab may be forced to delay the potential commercialization of CM-101 or reduce the scope of its sales or marketing activities for CM-101. If Chemomab needs to increase its expenditures to fund commercialization activities for CM-101, it will need to obtain additional capital, which may not be available to it on acceptable terms, or at all. Chemomab may also have to enter into collaborative arrangements for CM-101 at an earlier stage than otherwise would be ideal and it may be required to relinquish rights to CM-101 or otherwise agree to terms unfavorable to it. Any of these occurrences may have an adverse effect on Chemomab’s business, operating results and prospects.
If Chemomab is unable to establish adequate sales, marketing and distribution capabilities, either on its own or in collaboration with third parties, it will not be successful in commercializing its product candidates and may never become profitable. Chemomab will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, it may be unable to compete successfully against these more established companies.
- 57 -
A variety of risks associated with operating internationally could materially adversely affect Chemomab’s business.
Chemomab’s principal executive offices are located in Israel and certain of its product candidates may be manufactured at third-party facilities located in Europe. In addition, Chemomab’s business strategy includes potentially expanding internationally if any of its product candidates receives regulatory approval. Doing business internationally involves a number of risks, including but not limited to:
• multiple, conflicting and changing laws and regulations, such as privacy regulations, tax laws, export and import restrictions, employment laws, regulatory requirements and other governmental approvals, permits and licenses;
• failure by Chemomab to obtain and maintain regulatory approvals for the use of its products in various countries;
• additional potentially relevant third-party patent rights;
• complexities and difficulties in obtaining protection and enforcing Chemomab’s intellectual property;
• difficulties in staffing and managing foreign operations;
• complexities associated with managing multiple payor reimbursement regimes, government payors or patient self-pay systems;
• limits in Chemomab’s ability to penetrate international markets;
• financial risks, such as longer payment cycles, difficulty collecting accounts receivable, the impact of local and regional financial crises on demand and payment for Chemomab’s products and exposure to foreign currency exchange rate fluctuations;
• natural disasters, political and economic instability, including wars, terrorism and political unrest, outbreak of disease, boycotts, curtailment of trade and other business restrictions;
• certain expenses including, among others, expenses for travel, translation and insurance; and
• regulatory and compliance risks that relate to maintaining accurate information and control over sales and activities that may fall within the purview of the United States Foreign Corrupt Practices Act, its books and records provisions, or its anti-bribery provisions.
Any of these factors could significantly harm Chemomab’s international expansion and operations and, consequently, its results of operations.
Risks Related to Chemomab’s Incorporation and Location in Israel
Conditions in Israel could materially and adversely affect Chemomab’s business.
Many of Chemomab’s employees, including certain management members operate from its offices that are located in Tel Aviv, Israel. In addition, a number of Chemomab’s officers and directors are residents of Israel. Accordingly, political, economic, and military conditions in Israel and the surrounding region may directly affect its business and operations. In recent years, Israel has been engaged in sporadic armed conflicts with Hamas, an Islamist terrorist group that controls the Gaza Strip, with Hezbollah, an Islamist terrorist group that controls large portions of southern Lebanon, and with Iranian-backed military forces in Syria. In addition, Iran has threatened to attack Israel and may be developing nuclear weapons. Some of these hostilities were accompanied by missiles being fired from the Gaza Strip against civilian targets in various parts of Israel, including areas in which Chemomab’s employees and some of its consultants are located, and negatively affected business conditions in Israel. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its trading partners could adversely affect Chemomab’s operations and results of operations.
Chemomab’s commercial insurance does not cover losses that may occur as a result of events associated with war and terrorism. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, Chemomab cannot guarantee that this government coverage will be maintained or that it will sufficiently cover its potential damages. Any losses or damages incurred by Chemomab could have a material adverse effect on its business. Any armed conflicts or political instability in the region would likely negatively affect business conditions and could harm Chemomab’s results of operations.
- 58 -
Further, in the past, the State of Israel and Israeli companies have been subjected to economic boycotts. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on Chemomab’s operating results, financial condition or the expansion of its business. A campaign of boycotts, divestment and sanctions has been undertaken against Israel, which could also adversely impact Chemomab’s business.
In addition, many Israeli citizens are obligated to perform several days, and in some cases more, of annual military reserve duty each year until they reach the age of 40 (or older, for reservists who are military officers or who have certain occupations) and, in the event of a military conflict, may be called to active duty. In response to increases in terrorist activity, there have been periods of significant call-ups of military reservists. It is possible that there will be military reserve duty call-ups in the future. Chemomab’s operations could be disrupted by such call-ups, which may include the call-up of members of Chemomab’s management. Such disruption could materially adversely affect Chemomab’s business, prospects, financial condition and results of operations.
Israel’s internal political scene has recently become unstable, with four general elections having been held within the last two years. The one government that was formed following one of the recent elections did not succeed to pass a budget, given strong disagreements among the politicians and political parties that were partners to the government. The COVID-19 pandemic has led to an increase in the national budget deficit, given the economic assistance that was granted to individuals and businesses that were most severely hurt by the pandemic. To the extent the political system does not stabilize soon, that could lead to economic problems that could adversely impact our business operations.
Because a certain portion of Chemomab’s expenses are incurred in currencies other than the U.S. Dollar, its results of operations may be harmed by currency fluctuations and inflation.
Chemomab’s reporting and functional currency is the United States Dollar, but some portion of its clinical studies and operations expenses are in NIS. As a result, Chemomab is exposed to some currency fluctuation risks. Fluctuation in the exchange rates of foreign currency has an influence on the cost of goods sold and Chemomab’s financing revenues and expenses. Chemomab may, in the future, decide to enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the exchange rate of the currencies mentioned above with respect to the U.S. Dollar. These measures, however, may not adequately protect Chemomab from adverse effects.
Chemomab received Israeli government grants for certain of their research and development activities as detailed below. The terms of those grants require us to satisfy specified conditions in order to transfer outside of Israel the manufacture of products based on know-how funded by the Israel Innovation Authority or to transfer outside of Israel the know-how itself. If we fail to comply with the requirements of Israeli law in this regard, we may be required to pay penalties, and it may impair our ability to sell our technology outside of Israel.
Some of Chemomab’s research and development efforts were financed through grants that were received from the Israel Innovation Authority of the Israeli Ministry of Economy and Industry, or the IIA (formerly known as the Office of the Chief Scientist). When know-how is developed using IIA grants, the Encouragement of Research, Development and Technological Innovation in Industry Law 5744-1984, or the Innovation Law, and the regulations thereunder, restrict our ability to transfer outside of Israel either the manufacture of products based on IIA-funded know-how or the know-how itself. Such restrictions continue to apply even after financial obligations to the IIA are paid in full. The consideration available to our shareholders in a future transaction involving the transfer outside of Israel of know-how developed with IIA funding (such as a merger or similar transaction) may be reduced by any amounts that we are required to pay to the IIA.
- 59 -
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis relates to the financial statements for the fiscal year ended December 31, 2020, of the Registrant’s wholly-owned subsidiary, Chemomab Ltd., an Israeli company (the “Subsidiary”). You should read the following discussion and analysis of the Subsidiary’s financial condition and results of operations together with the Subsidiary’s financial statements and the related notes included in our Current Report on Form 8-K/A filed with the SEC on March 19, 2021. Some of the information contained in this discussion and analysis, particularly with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. You should read “Risk Factors” in Item 1A of this Current Report on Form 8-K for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Results of Operations
Comparison of the Years Ended December 31, 2020 and 2019
The following table summarizes the Subsidiary’s results of operations for the years ended December 31, 2020 and 2019:
| Year ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| USD In thousands | ||||||||
| Research and development expenses, net | $ | 4,684 | 5,818 | |||||
| General and administrative expenses | 1,288 | 960 | ||||||
| Operating Loss | 5,972 | 6,778 | ||||||
| Financial (Income) expenses, net | (21 | ) | 2 | |||||
| Loss for the Year | $ | 5,951 | 6,780 | |||||
Research and Development Expenses, net
Research and development expenses, net were $4.7 million for the year ended December 31, 2020, compared to $5.8 million for the year ended December 31, 2019. The decrease of $1.1 million, or 19%, in the year ended December 31, 2020, compared to the prior year is primarily due decrease in costs of subcontractors in the amount of $1.4 million, offset by an increase in salaries and related expenses of $0.3 million, as a result of the Subsidiary increasing research and development headcount.
General and Administrative Expenses
General and administrative expenses were $1.3 million for the year ended December 31, 2020, compared to $1.0 million for the year ended December 31, 2019. The increase of $0.3 million, or 34%, was primarily due to increase in personnel-related costs, as well as an increase in legal and other professional services fees, mainly related to the Merger.
Liquidity and Capital Resources
Since inception, the Subsidiary has not generated any revenue and has incurred significant operating losses and negative cash flows from its operations. The Subsidiary has funded its operations to date primarily with proceeds from the sale of its common and preferred shares. Through December 31, 2020, the Subsidiary had received gross proceeds of approximately $34.3 million from sales of its common and preferred shares and $1.2 million in grants from the Israel Innovation Authority (The "IIA").
- 60 -
Cash in excess of immediate requirements is invested primarily with a view to liquidity and capital preservation.
Cash Flows
The following table summarizes the Subsidiary's cash flows for each of the periods presented (USD in thousands):
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Net cash used in operating activities | $ | (5,242 | ) | $ | (6,626 | ) | ||
| Net cash used in investing activities | (62 | ) | (3 | ) | ||||
| Net cash provided by financing activities | 4,750 | 14,077 | ||||||
| Net increase (decrease) in cash and cash equivalents | $ | (554 | ) | $ | 7,448 | |||
Operating Activities
Net cash used in operating activities for the year ended December 31, 2020 included the Subsidiary’s net loss of $6.0 million, partially offset by net cash provided by changes in the Subsidiary's operating assets and liabilities of $0.6 million and non-cash charges of approximately $20,000, which mainly included share-based compensation expenses of $0.1 million. Net changes in the Subsidiary's operating assets and liabilities for the year ended December 31, 2020 consisted primarily of an increase in accrued expenses and employees and related expenses of $0.4 million and $0.2 million, respectively, offset by increase of $0.1 million in other receivables.
Net cash used in operating activities for the year ended December 31, 2019 included the Subsidiary's net loss of $ 6.8 million, partially offset by net cash provided by non-cash charges of approximately $0.2 million, which mainly included share-based compensation expenses of $0.1 million.
Investing Activities
During the year ended December 31, 2020, net cash used in investing activities was $60,000, mainly reflecting an increase in short-term deposits of $20,000, and purchases of fixed assets in the amount of $40,000.
During the year ended December 31, 2019, net cash used in investing activities was $3,000, which reflected purchases of fixed assets.
Financing Activities
During the year ended December 31, 2020, net cash provided by financing activities was $4.7 million, consisting of $3.5 million of proceeds from the sale of the Subsidiary's Series C preferred shares and $1.2 million from exercise of warrants of the Subsidiary's Series A preferred shares.
During the year ended December 31, 2019, net cash provided by financing activities was $14.1 million, consisting of net proceeds from the sale of the Subsidiary's Series C preferred shares in the amount of $14.0 million, as well as proceeds from exercise of options in the amount of $0.1 million.
- 61 -
Quantitative and Qualitative Disclosures about Market Risks
Interest Rate Risk
As of December 31, 2020, the Subsidiary had cash and cash equivalents, restricted cash and short term bank deposits of $11.7 million, which consisted of cash, restricted cash and short term bank deposits.
Foreign Currency Exchange Risk
The Subsidiary is exposed to foreign exchange rate risk. The Subsidiary’s headquarters are located in Israel, where the majority of its general and administrative expenses costs are incurred in New Israeli Shekels. During each of the years ended December 31, 2020 and 2019, the Subsidiary recognized foreign currency transaction loss of $17,100 and $1,000, respectively. The Subsidiary’s functional currency is the U.S. Dollar. These foreign currency transaction gains and losses were recorded in financial expenses, net in its statements of comprehensive loss. The Subsidiary believes that a 10% change in the exchange rate between the U.S. Dollar and New Israeli Shekel would not have a material impact on its financial position or results of operations.
As the Subsidiary continues to grow its business, its results of operations and cash flows will be subject to fluctuations due to changes in foreign currency exchange rates, which could adversely impact the Subsidiary’s results of operations. To date, it has not entered into any foreign currency hedging contracts to mitigate the Subsidiary’s exposure to foreign currency exchange risk.
Emerging Growth Company Status
The Subsidiary is, and the post-combination company is an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and it may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies. The Subsidiary may take advantage of these exemptions until it is no longer an emerging growth company. Section 107 of the JOBS Act provides that an emerging growth company can take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards. The Subsidiary has irrevocably elected not to avail itself of this extended transition period and, as a result, it will adopt new or revised accounting standards on the relevant dates on which adoption of such standards is required for other public companies. The Subsidiary may take advantage of these exemptions up until the last day of the fiscal year following the fifth anniversary of the Subsidiary’s first registration statement filed under the United States Securities Act of 1933, as amended, or such earlier time that it is no longer an emerging growth company. The Subsidiary would cease to be an emerging growth company if it has more than $1.07 billion in annual revenue, it has more than $700.0 million in market value of its shares held by non-affiliates (and it has been a public company for at least 12 months and have filed one annual report on Form 10-K) or it issues more than $1.0 billion of non-convertible debt securities over a three-year period.
Off-Balance Sheet Arrangements
The Subsidiary did not have, during the periods presented, and it does not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the Securities and Exchange Commission.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact the Subsidiary’s financial position and results of operations is disclosed in Note 2 to the Subsidiary’s audited financial statements.
Contractual Obligations and Commitments
The following table summarizes the Subsidiary’s contractual obligations as of December 31, 2020 and the effects that such obligations are expected to have on the Subsidiary’s liquidity and cash flows in future periods:
| Payments Due by Period | ||||||||||||||||||||
| Total | Less than 1 Year |
1 to 3 Years |
4 to 5 Years |
More than 5 Years |
||||||||||||||||
| (USD in thousands) | ||||||||||||||||||||
| Operating and related lease commitments | $ | 428 | 70 | 219 | 139 | - | ||||||||||||||
| Commitments to sub-contractor | $ | 191 | 133 | 58 | - | |||||||||||||||
| Total | $ | 619 | 203 | 277 | 139 | - | ||||||||||||||
- 62 -
In addition, pursuant to the Subsidiary’s research and license agreements, it is required to make certain milestone and royalty payments to its licensors and collaborators. See “The Subsidiary Ltd.’s Business—Material Agreements—License Agreements” and audited Financial Statements—Note 7—Commitments and Contingent Liabilities for additional details regarding its payment obligations to these licensors.
Those milestone and royalty payments are not included in the above table as they are contingent in nature.
The Subsidiary received grants from the IIA. According to the terms of such grants, it will pay royalties of 3% - 4% of future sales up to the accumulated grant received including annual interest of LIBOR linked to the U.S. Dollar, provided however, that it shall not be obligated to repay such grants if no sales were generated. As of December 31, 2020, no sales were generated. The Subsidiary may be obligated to pay additional royalties upon the occurrence of certain events as determined by the IIA that are within the control of the Subsidiary.
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth information, as of April 12, 2021, regarding beneficial ownership of our ordinary shares (including ordinary shares represented by ADSs):
| · | each person who is known by us to own beneficially more than 5% of our ordinary shares; |
| · | each director; |
| · | each executive officer; and |
| · | all of our directors and executive officers collectively. |
Beneficial ownership is determined in accordance with the rules of the SEC. Under these rules, a person is deemed to be a beneficial owner of a security if that person has or shares voting power, which includes the power to vote or to direct the voting of the security, or investment power, which includes the power to dispose of or to direct the disposition of the security. For purposes of the table below, we deem ordinary shares issuable pursuant to options or warrants that are currently exercisable or exercisable within 60 days of the date of this Current Report on Form 10-K, if any, to be outstanding and to be beneficially owned by the person holding the options or warrants for the purposes of computing the percentage ownership of that person, but we do not treat them as outstanding for the purpose of computing the percentage ownership of any other person.
Unless otherwise noted, the address of each director and current and former executive officer of Chemomab is Kiryat Atidim, Building 7, Tel Aviv, Israel 6158002.
| NAME OF BENEFICIAL OWNER | Total (American Depositary Shares) |
Percentage of Ordinary Shares Beneficially Owned* |
||||||
| 5% and Greater Shareholders | ||||||||
| OrbiMed Israel Partners Limited Partnership(1) | 2,606,991 | 24.3 | % | |||||
| The Centillion Fund(2) | 661,370 | 6.2 | % | |||||
| Rivendell Investments 2017-9(3) | 1,131,563 | 10.6 | % | |||||
| Kobi George(4) | 1,296,540 | 12.0 | % | |||||
| Apeiron group(10) | 914,473 | 8.5 | % | |||||
- 63 -
| NAME OF BENEFICIAL OWNER | Total Beneficial Ownership |
Percentage of Ordinary Shares Beneficially Owned* |
||||||
| Directors and Executive Officers | ||||||||
| Adi Mor(5) | 1,296,540 | 12.0 | % | |||||
| Neil Cohen(6) | 2,409 | 0.02 | % | |||||
| Arnon Aharon(7) | 46,459 | 0.43 | % | |||||
| Stephen Squinto(8) | 109,883 | 1.02 | % | |||||
| Nissim Darvish(9) | 10,123 | 0.09 | % | |||||
| Joel Maryles | — | — | ||||||
| Alan Moses | — | — | ||||||
| Claude Nicaise | — | — | ||||||
| Sigal Fattal | — | — | ||||||
| All current executive officers and directors as a group (9 persons) | 917,194 | 13.40 | % | |||||
* Percentage ownership based on 10,697,997 American Depositary Shares outstanding as of April 12, 2021.
(1) Represents 2,578,174 ADSs, representing 51,563,580 ordinary shares, held by OrbiMed Israel Partners Limited Partnership, or OIP, as reported by OIP on Schedule 13G filed with the SEC on March 26, 2021, and 28,817 ADSs, representing 576,340 Ordinary Shares, issuable upon the exercise of warrants to purchase ADSs. The percentage is calculated based upon 10,697,997 ADSs outstanding, representing 213,959,940 Ordinary Shares, and giving effect to the additional 28,817 ADSs, representing 576,340 Ordinary Shares, that would be outstanding following the exercise of the warrants held by OIP. OIP is the shareholder of record. OrbiMed Israel BioFund GP Limited Partnership, or OrbiMed BioFund, is the general partner of OIP, and OrbiMed Israel GP Ltd., or OrbiMed Israel GP, is the general partner of OrbiMed BioFund. By virtue of such relationships, OrbiMed BioFund and OrbiMed Israel GP may be deemed to have voting and investment power with respect to the shares held directly by OIP and as a result, may be deemed to have beneficial ownership over such securities. OrbiMed Israel GP exercises this investment and voting power through a management committee comprised of Carl Gordon, Jonathan T. Silverstein, Nissim Darvish, Anat Naschitz and Erez Chimovits, each of whom disclaims beneficial ownership of the shares held by OIP. The address of OIP is 89 Medinat HaYehudim St., Build E, 11th Floor, Herzliya 46766 Israel.
(2) The address of Centillion Fund, Inc. is 10 Manoel Street, Castries, Saint Lucia.
(3) Represents 1,108,509 ADSs, representing 22,170,180 ordinary shares, held by Rivendell Investments 2017-9 LLC, or Rivendell, as reported by Rivendell on Schedule 13G filed with the SEC on March 26, 2021, and 23,054 ADSs, representing 461,080 Ordinary Shares, issuable upon the exercise of warrants to purchase ADSs. The percentage is calculated based upon 10,697,975 ADSs outstanding, representing 213,959,500 Ordinary Shares, and giving effect to the additional 23,054 ADSs, representing 461,080 Ordinary Shares, that would be outstanding following the exercise of the warrants held by Rivendell. Rivendell is the shareholder of record. Peter Thiel is the beneficial owner of Rivendell and has sole voting and investment power over the securities held by Rivendell. The address of Rivendell is 1209 Orange Street, Wilmington, Delaware 19801.
(4) Consists of (i) 514,495 ADSs owned directly by Dr. George, (ii) 649,550 ADSs owned by Dr. Adi Mor (Dr. George’s spouse), (iii) 33,725 ADSs of the Registrant issued directly to Dr. George, issuable upon the exercise of options, and (iv) 98,770 ADSs of the Registrant issued to Dr. Mor, (Dr. George’s spouse) issuable upon the exercise of options.
(5) Includes (i) 649,550 ADSs owned directly by Dr. Mor, (ii) 514,495 ADSs owned by Dr. George, (Dr. Mor’s spouse), (iii) 98,770 ADSs of the Registrant issued to Dr. Mor, issuable upon the exercise of options, and (iv) 33,725 ADSs of the Registrant issued to Dr. George, (Dr. Mor’s spouse) upon the exercise of options.
(6) Includes 2,409 ADSs of the Registrant, as reported by Mr. Neil Cohen on Form 4 filed with the SEC on March 26, 2021.
(7) Represents 46,459 ADSs of the Registrant issuable upon the exercise of options, as reported by Mr. Arnon Aharon on Form 3 filed with the SEC on March 25, 2021.
(8) Includes 47,938 ADSs of the Registrant, and 61,945 ADSs of the Registrant issuable upon the exercise of options, as reported by Mr. Stephen Squinto on Form 3 filed with the SEC on March 25, 2021.
- 64 -
(9) Represents 10,123 ADSs of the Registrant issuable upon the exercise of options, as reported by Mr. Nissim Darvish on Form 3 filed with the SEC on March 25, 2021.
(10) The Apeiron Group consists of (i) Apeiron SICAV Ltd. – Presight Capital Fund One, of which owns 438,993 ADSs, (ii) Apeiron Presight Capital Fund II, LP, of which owns 316,987 ADSs, and (iii) Apeiron Investment Group Ltd., of which owns 158,493 ADSs. Each of Fabian Hansen and Christian Angermayer may be deemed to share voting and investment power with respect to the ADSs held by the Apeiron Group.
EXECUTIVE OFFICERS AND DIRECTORS
The following table sets forth the names and ages of the current directors and executive officers:
| Name | Age | Positions Held | ||
| Stephen Squinto† | 64 | Chairman of the Board | ||
| Adi Mor* | 40 | Chief Executive Officer, Chief Scientific Officer and Director | ||
| Nissim Darvish† | 56 | Director | ||
| Joel Maryles† | 61 | Director | ||
| Alan Moses† | 73 | Director | ||
| Claude Nicaise† | 68 | Director | ||
| Neil Cohen† | 57 | Director | ||
| Arnon Aharon* | 52 | Chief Medical Officer | ||
| Sigal Fattal* | 50 | Chief Financial Officer |
|
* † |
Executive Officer Independent Director |
Stephen Squinto, Ph.D. has served on our board of directors as chairman since March 16, 2021. Dr. Squinto is a Partner at OrbiMed Advisors LLC, an investment firm, and has also served as acting Head of Research and Development of SpringWorks Therapeutics, Inc. Dr. Squinto currently serves on the boards of directors of Compass Therapeutics Inc., SpringWorks Therapeutics, Inc., VectivBio Holding AG, and several private companies. Dr. Squinto also previously served on the boards of directors of 89Bio, Inc., Arvinas, Inc., Audentes Therapeutics, Inc., Gemini Therapeutics Inc., and Passage Bio Inc. Previously, Dr. Squinto co-founded Alexion Pharmaceuticals, a biotechnology company, and served as its Executive Vice President and Chief Global Operations Officer from 2012 to January 2015 and as its Global Head of Research and Development from 2007 to 2012. Dr. Squinto holds a Ph.D. in biochemistry and biophysics and a B.A. in chemistry from Loyola University of Chicago. We believe Dr. Squinto is qualified to serve as a director due to his extensive experience as an entrepreneur and investor in the life sciences industry and his service on the boards of other public and private biopharmaceutical and biotechnology companies.
Adi Mor, Ph.D. is a co-founder of Chemomab and has served as Chemomab’s Chief Executive Officer, Chief Scientific Officer and member of Chemomab’s board of directors since its formation in 2011. Dr. Mor has in-depth knowledge in immunology focusing on rare diseases and broad experience in designing, developing and patenting a novel class of monoclonal antibodies to treat inflammatory and fibrotic diseases. Dr. Mor received her Ph.D. in immunology from Tel Aviv University in the Department of Neurobiochemistry in Israel and is the lead author of numerous scientific journal publications in immunology and inflammatory disorders.
Nissim Darvish, M.D., Ph.D. has served on our board of directors since March 16, 2021. Dr. Darvish is a Venture Partner at OrbiMed Israel, an investment firm. Dr. Darvish currently serves as a director of several private companies. Dr. Darvish previously served as a member of the boards of directors of 9 Meters Biopharma Inc. and Medigus Ltd. Previously, Dr. Darvish was employed at Pitango Venture Capital, where he was a General Partner managing life sciences investments. He was also the founder and CEO of Impulse Dynamics, where he culminated in a $250 million realization event. Dr. Darvish obtained his M.D. and Ph.D. in Biophysics and Physiology from the Technion in Israel, and subsequently conducted his post-doctoral research at NIH. He has published over 100 patents and authored over 20 publications. We believe that Dr. Darvish is qualified to serve on our board of directors based on his roles on several public and private boards of directors as well as his extensive experience in investing in healthcare companies.
- 65 -
Joel Maryles has served on our board of directors since March 16, 2021. Mr. Maryles currently serves on the board of directors and as the Chairman of Remuneration Committee of Jefferies International Ltd., since 2016. Mr. Maryles previously served on the board of directors of Radware Ltd. (NASDAQ: RDWR), from 2014 to 2020 and from 2014 to 2016, on the board of directors of EZchip Semiconductor Ltd., which was acquired by Mellanox Technologies in 2016. From 2015 to 2018, Mr. Maryles was a Partner at OurCrowd and from 2007 to 2012, he served as a Portfolio Manager at T-Cubed Investments, which he founded. Prior to that, Mr. Maryles served as a Managing Director in the Investment Banking division of Citigroup, Israel and of Furman Selz. Mr. Maryles holds a Bachelor of Science in Mechanical Engineering from the University of Illinois and an MBA from the University of Chicago, Illinois.
Alan Moses, MD, FACP has served on our board of directors since March 16, 2021. Dr. Moses is board certified by the ABIM with subspecialty certification in Endocrinology and Metabolism and is a Fellow of the American College of Physicians. Dr. Moses currently serves on the board of directors of BioFabUSA, since 2018. Prior to that time, from 2008 to 2018, Dr. Moses served as the Global Chief Medical Officer of Novo Nordisk A/S (CPH: NOVO-B), which he joined in 2004. Dr. Moses served as a Professor of Medicine at Harvard Medical School from 2002 to 2006, and in collaboration with MIT, he co-founded and co-directed the Clinical Investigator Training Program, which focused on training physician-scientists in translational research. Dr. Moses previously served as the Senior Vice President and Chief Medical Officer of the Joslin Diabetes Center, from 1998 to 2004. Dr. Moses holds a BS from Duke University, North Carolina and an MD from Washington University School of Medicine, Missouri.
Claude Nicaise, MD has served on our board of directors since March 16, 2021. Dr. Nicaise is a physician with extensive US and international experience in clinical drug development, strategic management, worldwide regulatory strategy, pharmaceuticals, biotechnology, including clinical cancer research, infectious diseases and neuroscience. Dr. Nicaise is the owner and founder of Clinical Regulatory Services, which provides consulting services to the life science and biotechnology industry in support of all aspects of clinical and regulatory development. Dr. Nicaise currently serves on the board of directors and as the Chairman of the Compensation Committee of Sarepta Therapeutics, Inc. (NASDAQ: SRPT), since 2015, as well as on the board of directors of Mynoryx Therapeutics, since 2017. Prior to that time, from 2008 to 2014, Dr. Nicaise served as the Senior Vice President of Alexion Pharmaceuticals Inc. (NASDAQ: ALXN), and between 1984 and 2008, he held numerous senior management roles at Bristol Myers Squibb (NYSE: BMY). Dr. Nicaise holds an MD and a degree in Internal Medicine, Clinical Oncology, from Brussels University, Belgium.
Neil Cohen has served as a member of Anchiano’s board of directors since April 2020 and as Anchiano’s interim Chief Executive Officer since October 2020. Mr. Cohen has served as the Chairman and Chief Executive Officer of Castel Partners Ltd. since January 2012. In 1994, he co-founded Israel Seed Partners, a leading venture capital firm, and managed the firm until 2019. Mr. Cohen has invested in and served on the boards of directors of many private technology companies, including a large number which were acquired or completed successful initial public offerings, including Compugen (Nasdaq: CGEN), Shopping.com (Nasdaq: SHOP, acquired by EBAY), Broadlight (acquired by Broadcom, Nasdaq: AVGO) and Cyota (acquired by RSA). He is a venture partner at SKY, an Israeli middle-market private equity firm, Hetz Ventures Management Ltd., an early-stage Israeli venture capital fund, and Shavit Capital. Mr. Cohen was previously the Business Editor of The Jerusalem Post and began his career in the private equity group at N M Rothschild & Sons Limited in London. Mr. Cohen received a B.A. and M.A. in Oriental Studies, with first class honors, from Oxford University.
Arnon Aharon, M.D. has served as Chemomab’s Chief Medical Officer since January 2018. Prior to his promotion, Dr. Aharon served as Chemomab’s Head of Clinical Development from December 2016 to December 2017. Prior to joining Chemomab, from January 2014 to November 2016, Dr. Aharon served as Chief Medical Officer at BioLineRx Ltd., a company listed on Nasdaq and The Tel Aviv Stock Exchange, where he directed the oncology and immunology pipeline. Dr. Aharon also served at multiple senior management positions at biotechnology companies, including Pharmos Ltd., a company listed on Nasdaq and The Tel Aviv Stock Exchange, Thrombotech Ltd., and LycoRed Ltd. Dr. Aharon is a member of several industry advisory groups and provides consulting services to biotechnology companies and academic institutions. Dr. Aharon received his M.D. from the University of Tel Aviv in Israel.
- 66 -
Sigal Fattal has served as Chemomab’s interim Chief Financial Officer since October 2020. Prior to joining Chemomab, from March 2017 to December 2019, Ms. Fattal served as Chief Financial Officer at BiomX (NYSE American: PHGE), a clinical stage microbiome product discovery company. Prior to joining BiomX, Ms. Fattal served as Chief Financial Officer at Evogene (Nasdaq and TASE: EVGN), a computational biology company, from 2013 to 2016. Prior to that time, Ms. Fattal served in multiple financial and operational executive roles in various companies. Ms. Fattal also currently serves as co-founder of Simbiz, which was started in September 2020 and which offers one-stop-shop corporate services to startup companies. Ms. Fattal is a certified CPA (Isr.), and holds a BA in Accounting and Economics, and an MBA, both from Tel Aviv University.
Composition of the Board of Directors
The members of the Registrant’s board of directors are appointed to three staggered director classes, which are as follows:
Class I consists of Stephen Squinto, Nissim Darvish and Joel Maryles, each with a term expiring at the 2022 annual meeting of shareholders.
Class II consists of Neil Cohen and Claude Nicaise, each with a term expiring at the 2023 annual meeting of shareholders.
Class III consists of Adi Mor and Alan Moses, each with a term expiring at the 2024 annual meeting of shareholders.
The division of the Registrant’s board of directors into three classes with staggered three-year terms may delay or prevent a change of management or a change of control of the Registrant.
Director Independence
Based upon information requested from and provided by each director concerning their background, employment and affiliations, including family relationships, the Registrant’s board of directors has determined that each of the directors is independent as defined under Nasdaq listing standards, with the exception of Dr. Mor. The Registrant’s board of directors also determined that Nissim Darvish and Neil Cohen, who comprise the compensation committee and Neil Cohen and Joel Maryles, who comprise the corporate governance and nominating committee, all satisfy the independence standards for such committees established by the SEC and Nasdaq listing standards, as applicable. With respect to the Audit Committee, the Registrant’s board of directors has determined that Joel Maryles, Alan Moses and Claude Nicaise satisfy the independence standards for such committee established by Rule 10A-3 under the Exchange Act, the SEC and Nasdaq listing standards, as applicable, and that Joel Maryles is a financial expert under the rules of the SEC. The board of directors considered the relationships between such directors and certain of the investors of the Registrant and determined that such relationships did not affect such directors’ independence under the standards of Nasdaq, or, where applicable, under SEC rules.
Involvement in Certain Legal Proceedings
To the Registrant’s knowledge, the directors and executive officers of the Registrant have not been involved in any of the following events during the past ten years:
| a) | any bankruptcy petition filed by or against such person or any business of which such person was a general partner or executive officer either at the time of the bankruptcy or within two years prior to that time; | |
| b) | any conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
- 67 -
| c) | being subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction, permanently or temporarily enjoining him from or otherwise limiting his involvement in any type of business, securities or banking activities or to be associated with any person practicing in banking or securities activities; | |
| d) | being found by a court of competent jurisdiction in a civil action, the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; | |
| e) | being subject of, or a party to, any federal or state judicial or administrative order, judgment decree, or finding, not subsequently reversed, suspended or vacated, relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or | |
| f) | being subject of or party to any sanction or order, not subsequently reversed, suspended, or vacated, of any self-regulatory organization, any registered entity or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
Board Committees
The Registrant’s board of directors has established three standing committees to assist it in fulfilling its responsibilities to the Registrant and its shareholders: the audit committee, the compensation committee and the corporate governance and nominating committee. Each committee acts pursuant to a written charter, each of which has been posted in the “Investor Relations” section of Chemomab’s website accessible at www.chemomab.com. Each committee reviews its charter on an annual basis. In addition to the three standing committees, the Registrant’s board of directors may approve from time to time the creation of other committees to assist the board in carrying out its duties.
Audit Committee
The functions of the audit committee under the Israeli Companies Law, 5759-1999 (the “Companies Law”) include identifying and addressing flaws in the business management of the Registrant, reviewing and approving related party transactions, establishing whistleblower procedures, overseeing the Registrant’s internal audit system and the performance of its internal auditor, and assessing the scope of the work and recommending the fees of the Registrant’s independent accounting firm. In addition, the audit committee is required to determine whether certain related party actions and transactions are “material” or “extraordinary” for the purpose of the requisite approval procedures under the Companies Law and to establish procedures for considering proposed transactions with a controlling shareholder.\
In accordance with U.S. law and Nasdaq requirements, the Registrant’s audit committee is also responsible for the appointment, compensation and oversight of the work of its independent auditors and for assisting the board of directors in monitoring financial statements, the effectiveness of internal controls and compliance with legal and regulatory requirements.
The members of the audit committee are Joel Maryles, Alan Moses and Claude Nicaise. Joel Maryles is the chairperson of the audit committee and is a financial expert under the rules of the SEC. The Registrant’s board of directors has concluded that the composition of the audit committee meets the requirements for independence under the rules and regulations of Nasdaq and SEC.
Compensation Committee
The functions of the compensation committee under the Companies Law include recommending to the board of directors, for ultimate shareholder approval by a special majority, a policy governing the compensation of directors and officers based on specified criteria, reviewing modifications to and implementing such compensation policy from time to time, and approving the actual compensation terms of directors and officers prior to approval by the board of directors.
- 68 -
The members of the compensation committee are Nissim Darvish and Neil Cohen. Nissim Darvish is the chairperson of the compensation committee. The Registrant’s board of directors has determined that each member of the compensation committee is independent within the meaning of the independent director guidelines of Nasdaq and under Rule 10C-1 under the Exchange Act.
Corporate Governance and Nominating Committee
The corporate governance and nominating committee evaluates and recommends to the board of directors nominees for each election of directors and helps oversee the Registrant’s regulatory and compliance matters.
The members of the corporate governance and nominating committee are Neil Cohen and Joel Maryles. Neil Cohen is the chairperson of the corporate governance and nominating committee. The Registrant’s board of directors has determined that each member of the corporate governance and nominating committee is independent within the meaning of the independent director guidelines of Nasdaq.
EXECUTIVE COMPENSATION
Director Compensation
Chemomab currently pays its independent, non-employee Chairman of the Board annual director fees of $65,000 and grants to him options from time to time.[OTHER BOARD MEMBER?]
Chemomab Executive Compensation
The aggregate amounts of salaries, consulting and director fees, pension, retirement and other similar benefits, and share-based compensation, that were payable to Chemomab’s executive officers and directors and/or to their respective affiliates in respect of employment, consulting and directorship agreements and arrangements (which includes other amounts described in the “Director and Officer Compensation” section of Amendment No. 1 to the Registration Statement on Form S-4, filed with the SEC on February 10, 2021) during the year ended December 31, 2020 is set forth in the below table. The table does not include any amounts that Chemomab paid to reimburse any of such persons for costs incurred in providing it with services during that period.
| (in thousands) | Salary, Fees and Related Benefits | Pension, Retirement and Other Similar Benefits | Share Based Compensation | |||||||||
| All directors and senior management as a group, consisting of 6 persons | $ | 671 | $ | 78 | $ | 91 | ||||||
The Registrant compensates its directors and senior management team in accordance with the recommendation of its compensation committee and, generally, subject to the approval of the Registrant’s board of directors and shareholders. That compensation will generally need to be consistent with the terms of our compensation policy, which will require periodic approval, in accordance with the requirements of the Companies Law.
On March 15, 2021, the shareholders of the Registrant approved an amendment to the compensation terms of its current and future directors, and related amendments to the Registrant’s compensation policy, which went effective on March 16, 2021. These amendments are as follows:
Cash Compensation
The Registrant’s directors are entitled to certain cash fee amounts annually in respect of each of board service and board committee service. The amendment sets maximum director cash fee levels higher under the compensation policy than what the Registrant will pay to the board members in practice, in order to maintain flexibility under the compensation policy in the event that the Registrant desires to increase the fees at a later time. Any such increase in cash fees for the director fees will require shareholder approval pursuant to the requirements of the Companies Law.
- 69 -
| Actual Cash Fees Paid | Cash Fee Upper Limit Under Amendment to Compensation Policy |
||||||||||||
| Annual Cash Fees: | |||||||||||||
| Each board member, other than the Chairman of the Board | $ | 35,000 | $ | 50,000 | |||||||||
| Chairman of the Board | $65,000 (in addition to this amount, the Chairman may receive annual fees for board committee service) |
$100,000 (this maximum cannot be exceeded even after including any additional fees payable for board committee service) |
|||||||||||
| Annual Fees for Board Committee Service: | |||||||||||||
| Committee Chairpersons: | |||||||||||||
| Audit Committee | $ | 15,000 | $ | 15,000 | |||||||||
| Compensation Committee | $ | 10,000 | $ | 10,000 | |||||||||
| Nominating / Governance Committee | $ | 8,000 | $ | 8,000 | |||||||||
| Other Committee Members: | |||||||||||||
| Audit Committee | $ | 7,500 | $ | 7,500 | |||||||||
| Compensation Committee | $ | 5,000 | $ | 5,000 | |||||||||
| Nominating / Governance Committee | $ | 4,000 | $ | 4,000 | |||||||||
Equity Compensation
The amendment to the compensation policy implements terms for actual equity compensation that will be granted to directors on a regular basis. We believe this will better ensure the tying of the performance of the Company to the compensation of its directors. As with the cash fee program for directors, the maximum director equity grant levels higher under the compensation policy than what we will grant to the board members in practice, in order to maintain flexibility under the policy in the event that we desire to increase the amount of the equity grant at a later time. Any such increase in equity grant amounts for the directors will anyway be subject to the shareholder approval requirements under the Companies Law. The following is the equity grant levels for directors:
| Actual Equity Grant Level for Directors | Equity Grant Upper Limit Under Amendment to Compensation Policy | |||||||
| (All equity grants are option grants to purchase the below percentages of the Registrant’s issued and outstanding shares on a fully diluted basis) | ||||||||
| Initial Option Grant (upon initial election to the board of directors): | ||||||||
| Each board member, other than the Chairman of the Board: | 0.1 | % | 0.1 | % | ||||
| Chairman of the Board | 0.2 | % | 0.4 | % | ||||
| Annual Option Grant (first annual grant to be allocated one year following the initial grant): | ||||||||
| Each board member, other than the Chairman of the Board: | 0.05 | % | 0.05 | % | ||||
| Chairman of the Board | 0.1 | % | 0.2 | % | ||||
- 70 -
Additionally, the amendment to the compensation policy included the following changes:
| · | increased the maximum annual value limit for equity grants for each director or for the chairman of the board to 400% of the maximum cash fees (including fees for board committee service) permitted to be paid to that director or the chairman of the board (as applicable) under the compensation policy; and |
| · | modified the vesting terms for director equity grants under the compensation policy as follows: |
| o | Initial grants: Monthly vesting over the course of a three-year period (i.e., 2.77% of grant vests each month during a 36 month vesting period); and |
| o | Annual grants: Vesting occurs at one time (“cliff vesting”) on the one year anniversary of the grant date. |
Employment Agreements
Employment Agreement with Chemomab Chief Executive Officer
Under the employment agreement, dated April 25, 2013, as amended (most recently as of January 1, 2020) that Chemomab entered into with its Chief Executive Officer, Dr. Adi Mor, Dr. Mor is entitled to a gross monthly salary of NIS 52,500 (approximately $16,406). Dr. Mor is also entitled to an annual performance bonus of three monthly salaries, subject to her meeting certain performance milestones, as to be determined by Chemomab’s board of directors on an annual basis. Besides base salary and bonus, Dr. Mor receives under the agreement other benefits that are provided for by Israeli law or that are customary for senior executives in Israel, including reimbursement for reasonable expenses incurred in connection with her services, and the right to use (including certain related fixed and variable costs in respect of) a leased car and a cellular phone. In lieu of a leased car, Dr. Mor may elect to receive a monthly car allowance payment. Dr. Mor is furthermore entitled to company contributions equivalent to 8.33%, 2.5%, and 5% of her gross monthly salary towards certain severance, disability and tax-advantaged savings funds (known as a manager’s insurance policy), respectively. Dr. Mor also contributes 5.5% of her gross monthly salary towards the manager’s insurance policy. The employment engagement is terminable by either party upon 60 days prior written notice, and contains customary provisions regarding noncompetition, confidentiality of information and assignment of inventions. As required under Israeli law, the terms of Dr. Mor’s engagement with Chemomab have been approved by Chemomab’s board of directors and shareholders.
Dr. Mor has been granted, pursuant to her employment agreement, an aggregate of 10,239 options to purchase Chemomab shares, of which 7,679 have vested or will vest within 60 days of April 12, 2021), under Chemomab’s share option and incentive plan.
Employment Agreement with Chemomab Chief Medical Officer
Under the employment agreement, dated March 1, 2019, that Chemomab entered into with its Chief Medical Officer, Dr. Arnon Aharon, Dr. Aharon is entitled to a gross monthly salary of NIS 55,000 (approximately $17,187). Dr. Aharon is also entitled to an annual performance bonus of up to 20% of his annual salary, subject to his meeting certain performance criteria, as to be set forth in a personal bonus policy. Besides base salary and bonus, Dr. Aharon receives under the agreement other benefits that are provided for by Israeli law or that are customary for senior executives in Israel, including reimbursement for reasonable expenses incurred in connection with his services, and the right to a monthly travel allowance of NS 3,000 (approximately $938). Dr. Aharon is furthermore entitled to company contributions equivalent to 8.33%, 2.5%, 7.5% and at least 5% of his gross monthly salary towards certain severance, disability, study fund and tax-advantaged savings funds (known as a manager’s insurance policy or pension), respectively. Dr. Aharon also contributes 6% and 2.5% of his gross monthly salary towards the manager’s insurance policy/pension and study fund, respectively. The employment engagement is terminable immediately by either party upon two months’ prior written notice, and contains customary provisions regarding noncompetition, confidentiality of information and assignment of inventions. As required under Israeli law, the terms of Dr. Aharon’s engagement with Chemomab have been approved by Chemomab’s board of directors.
- 71 -
Dr. Aharon has been granted, pursuant to his employment agreement, an aggregate of 5,849 options to purchase Chemomab shares, of which 3,612 have vested or will vest within 60 days of April 12, 202128), under Chemomab’s share option and incentive plan.
Share Incentive Plans
Chemomab maintains the Chemomab Ltd. 2015 Share Incentive Plan, or the 2015 Plan — which was assumed by Anchiano upon the effectiveness of the Merger. At that time, outstanding options under the 2015 Plan became exercisable for such number of ADSs of Anchiano as was determined based on the exchange ratio in the Merger Agreement, with a reciprocal adjustment to exercise price. As of December 31, 2020, a total of 68,584 Chemomab ordinary shares were reserved for issuance under the 2015 Plan, of which 5,124 shares had been issued pursuant to previous exercises options, and 40644 shares were issuable under outstanding options. Of such outstanding options, options to purchase 22,382 ordinary shares had vested and were exercisable as of that date, with a weighted average exercise price of $16.30 per share.
2015 Share Incentive Plan
In November 2015, Chemomab’s board of directors adopted, and its shareholders subsequently approved, the 2015 Plan. The 2015 Plan provides for the grant of options, restricted shares, restricted share units and other share-based awards to Chemomab’s and its subsidiaries’ and affiliates’ directors, employees, officers, consultants, advisors, and any other person whose services are considered valuable to Chemomab or its affiliates. Any such grants are intended to incentivize the foregoing persons to continue as service providers, to increase their efforts on Chemomab’s behalf or on behalf of its subsidiaries or affiliates, and to promote the success of its business.
The 2015 Plan is administered by Chemomab’s board of directors or by a committee designated by the board of directors, which determines, subject to Israeli law, the grantees of awards and the terms of the grant, including, exercise prices, vesting schedules, acceleration of vesting and the other matters necessary in the administration of the 2015 Plan. The 2015 Plan enables Chemomab to issue awards under various tax regimes, including, without limitation, pursuant to Section 102 of the Israeli Income Tax Ordinance, or the Ordinance, and under Section 3(i) of the Ordinance and Section 422 of the United States Internal Revenue Code of 1986, as amended, or the Code.
The 2015 Plan provides that options granted to Chemomab’s employees, directors and officers who are not controlling shareholders and who are considered Israeli residents are intended to qualify for special tax treatment under the “capital gain track” provisions of Section 102(b) of the Ordinance. Chemomab’s Israeli non-employee service providers and controlling shareholders may only be granted options under Section 3(i) of the Ordinance, which does not provide for similar tax benefits.
Options granted under the 2015 Plan to U.S. residents may qualify as “incentive stock options” within the meaning of Section 422 of the Code, or may be non-qualified. The exercise price for “incentive stock options” must not be less than the fair market value on the date on which an option is granted, or 110% of the fair market value if the option holder holds more than 10% of Chemomab’s share capital.
Options and other awards granted under the 2015 Plan generally vest over four years commencing on the date of grant, such that 25% vests on the first anniversary of the date of grant and an additional 6.25% vests at the end of each subsequent calendar quarter over the course of the next three years, provided that the participant remains continuously employed or engaged by Chemomab.
Options, other than certain incentive share options, that are not exercised within ten years from the grant date expire, unless otherwise determined by Chemomab’s board of directors or its designated committee, as applicable. Share options that qualify as “incentive stock options” and are granted to a person holding more than 10% of Chemomab’s voting power will expire within five years from the date of the grant. In the event of the death of a grantee while employed by or performing service for Chemomab or its subsidiary or within three months after the date of the employee’s termination, or the termination of a grantee’s employment or services for reasons of disability, the grantee, or in the case of death, his or her legal successor, may exercise options or other awards that have vested prior to termination within a period of one year from the date of disability or death. If Chemomab terminates a grantee’s employment or service for cause, all of the grantee’s vested and unvested options or other awards will expire on the date of termination. If a grantee’s employment or service is terminated for any other reason, the grantee may generally exercise his or her vested options or other award within three months of the date of termination. Any expired or unvested options return to the pool and become available for reissuance. From time to time, Chemomab may consider issuing options with slightly different terms or accelerating, extending or otherwise modifying options in accordance with applicable law and regulation and the terms of the 2015 Plan.
- 72 -
In the event of a merger or consolidation of Chemomab, or a sale of all, or substantially all, of Chemomab’s shares or assets or other transaction having a similar effect on Chemomab, then without the consent of the option holder, Chemomab’s board of directors or its designated committee, as applicable, may, but is not required, to (i) cause any outstanding award to be assumed or an equivalent award to be substituted by such successor corporation, or (ii) in case the successor corporation does not assume or substitute the award (a) provide the grantee with the option to exercise the award as to all or part of the shares or (b) cancel the options and pay in cash an amount determined by the board of directors or the committee as fair in the circumstances. Notwithstanding the foregoing, Chemomab’s board of directors or its designated committee may upon such event amend, modify or terminate the terms of any award, including conferring the right to purchase any other security or asset that the board of directors or the committee shall deem, in good faith, appropriate.
Compensation Committee Interlocks and Insider Participation
Each member of the compensation committee of the Registrant is an “outside” director as that term is defined in Section 162(m) of the Internal Revenue Code of 1986, as amended, a “non-employee” director within the meaning of Rule 16b-3 of the rules promulgated under the Exchange Act and independent within the meaning of the independent director guidelines of the Nasdaq. None of the Registrant’s executive officers serves as a member of the board of directors or compensation committee of any entity that has one or more executive officers on the Registrant’s board of directors or compensation committee.
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Described below are any transactions occurring since January 1, 2019, and any currently proposed transactions to which either Anchiano or Chemomab was a party and in which:
• The amounts involved exceeded or will exceed the lesser of (i) $120,000 and (ii) one percent of the average of Anchiano’s or Chemomab’s total assets at year end for the last two completed fiscal years; and
• A director, executive officer, holder of more than 5% of the outstanding share capital of Anchiano or Chemomab, or any member of such person’s immediate family had or will have a direct or indirect material interest.
Chemomab (f/k/a Anchiano) Transactions
The following transactions relate to the Registrant, formerly known as Anchiano Therapeutics Ltd., prior to the consummation of the Merger.
Chemomab entered into employment agreements with each of its executive officers.
Chemomab’s Articles of Association permit it to insure each of its directors and officers to the fullest extent permitted by the Companies Law. Chemomab has obtained directors and officers insurance for its executive officers and directors.
All related party transactions are reviewed and approved by the audit committee of Chemomab, as required by the audit committee charter.
- 73 -
Related Party Transactions
Under the Israel’s Companies Law, 5759-1999, or the Companies Law, a related party transaction in which an “office holder” has a personal interest may be approved only if it is for the benefit of the company. An office holder is defined in the Companies Law as a director, a general manager, chief business manager, deputy general manager, vice general manager, any other person assuming the responsibilities of any of these positions regardless of such person’s title, and any other manager directly subordinate to the general manager. A transaction that is not an extraordinary transaction in which an office holder has a personal interest requires the approval of the board of directors, unless the articles of association of the company provide otherwise. If the transaction is an extraordinary transaction, it must be approved by the audit committee and the board of directors, and, under certain circumstances, by the shareholders of the company. An “extraordinary transaction” is a transaction other than in the ordinary course of business, other than on market terms or that is likely to have a material impact on the company’s profitability, assets or liabilities.
Pursuant to the Companies Law, extraordinary transactions in which a controlling shareholder has a personal interest require the approval of the audit committee, or the compensation committee if the transaction is in connection with employment or service with the company, the board of directors and the shareholders of the company. The shareholder approval must be by a simple majority of all votes cast, provided that (i) such majority includes a simple majority of the votes cast by non-controlling shareholders having no personal interest in the matter or (ii) the total number of votes of shareholders mentioned in clause (i) above who voted against such transaction does not exceed 2% of the total voting rights in the company.
In most cases, the Companies Law prohibits any director who has a personal interest in a transaction from being present for the discussion or voting pertaining to such transaction in the audit committee or board of directors. Nevertheless, a director who has a personal interest may be present at the meeting and vote on the matter if a majority of the directors or members of the audit committee have a personal interest in the approval of such transaction; in this case, however, the transaction also requires shareholder approval.
Director and Officer Compensation
Under the Companies Law, Chemomab is required to approve, at least once every three years, a compensation policy with respect to office holders. Following the recommendation of Chemomab’s compensation committee, the compensation policy must be approved by the board of directors and shareholders. The shareholder approval must be by a simple majority of all votes cast, provided that (i) such majority includes a simple majority of the votes cast by non-controlling shareholders having no personal interest in the matter or (ii) the total number of votes of shareholders mentioned in clause (i) above who voted against such transaction does not exceed 2% of the total voting rights in the company. In general, the terms of compensation of directors, the chief executive officer and any employee or service provider who is considered a controlling shareholder must be approved separately by the compensation committee, the board of directors and the shareholders. The compensation terms of other officers who report directly to the chief executive officer requires the approval of the compensation committee and the board of directors.
Information Rights Agreement
Anchiano entered into an information rights agreement, effective as of December 19, 2018, with one of its principal shareholders, CBI. The information rights agreement provides CBI with rights to receive Anchiano’s annual and quarterly financial statements, auditor consent letters and valuation reports, and other information reasonably required by CBI to enable it to prepare its financial statements. The information rights agreement also requires that Anchiano provide CBI with information material to Anchiano and mandated to be disclosed by the requirements applicable to CBI, as well as certain other material information of Anchiano. The information rights agreement contains customary confidentiality provisions and terminates when CBI, and any company that controls CBI, is no longer required to issue public reports relating to Anchiano pursuant to the Exchange Act.
Chemomab Ltd. Transactions
The following transactions relate to Chemomab Ltd. prior to the consummation of the Merger and therefore do not reflect or give effect to the reverse split of the Registrant’s ordinary shares that took effect on the Closing Date.
- 74 -
Series C Preferred Shares
On September 23, 2019, Chemomab entered into a Series C Preferred Share Purchase Agreement, or the Series C Agreement, pursuant to which it issued and sold to investors 130,831 shares of Chemomab’s Series C preferred shares, nominal value NIS 0.01 per share, at a purchase price of $87.90 per share, for an aggregate consideration of approximately $11.5 million. On November 6, 2019 and May 21, 2020, Chemomab entered into joinder agreements to that Series C Agreement, pursuant to which it issued and sold to investors an additional total 68,260 shares of Chemomab’s Series C preferred shares at a purchase price of $87.90 per share, for an aggregate consideration of approximately $6.0 million.
The following table sets forth the aggregate number of shares of Chemomab’s share capital acquired by beneficial owners of more than 5% of its share capital in the financing transactions described above, which were completed prior to the closing of the Merger.
| Participants | Series C Preferred Stock | |||
| 5% or Greater Stockholders(1) | | |||
| OrbiMed Israel Partners Limited Partnership | 56,883 | |||
| Rivendell Investments 2017-9 LLC | 34,130 | |||
| SBI JI Innovation Fund | 11,377 | |||
| Milestone View Limited | 22,753 | |||
| Apeiron SICAV Ltd. – Presight Capital Fund One | 34,130 | |||
| Boryung Pharmaceuticals Ltd. | 34,130 | |||
Some of our directors are associated with our principal shareholders as indicated in the table below:
| Director | | Principal Shareholder |
| Nissim Darvish | | OrbiMed Israel Partners Limited Partnership |
| Stephen Squinto | | OrbiMed Israel Partners Limited Partnership |
Investors’ Rights Agreement
In connection with the Series C Agreement, Chemomab entered into an Amended and Restated Investors’ Rights Agreement, dated September 23, 2019, with the holders of Chemomab’s preferred shares, including entities with which certain of Chemomab’s directors are related. The agreement provides for certain rights relating to the registration of such holders’ securities, including shares issuable upon conversion of preferred shares. This agreement was terminated prior to closing of the Merger.
Anchiano and Chemomab Transactions
Registration Rights Agreement
On December 14, 2020, Anchiano, Chemomab and certain shareholders of Chemomab, including, among others, Adi Mor, Chemomab’s Chief Executive Officer and Director, and other entities with which certain of Chemomab’s directors are related. The agreement provides for certain rights relating to the registration of such holders’ securities, including shares issuable upon conversion of preferred shares into ADSs of the Registrant.
- 75 -
RECENT SALES OF UNREGISTERED SECURITIES
The description of the Registrant’s sales of unregistered securities that occurred subsequent to the Annual Report is incorporated by reference to Item 3.02 of the Registrant’s Current Report on Form 8-K, filed with the SEC on March 15, 2021.
DESCRIPTION OF SHARE CAPITAL
The following is a description of the material terms of our amended and restated articles of association. The following descriptions of share capital and provisions of our amended and restated articles of association are summaries and are qualified by reference to our amended and restated articles of association, a copy of which is filed with the SEC as an exhibit to the Amendment No. 1 to the Registration Statement on Form S-4, as filed with the SEC on February 10, 2021.
Registration Number and Purposes of the Company
We are registered with the Israeli Registrar of Companies. Our registration number is 51-4694991. Our affairs are governed by our amended and restated articles of association, applicable Israeli law and the Companies Law. Our purpose as set forth in our amended and restated articles of association is to engage in any lawful act or activity.
Voting Rights
All ordinary shares will have identical voting and other rights in all respects.
Transfer of Shares
Our fully paid ordinary shares are issued in registered form and may be freely transferred under our amended and restated articles of association, unless the transfer is restricted or prohibited by another instrument, applicable law or the rules of the Nasdaq Capital Market. The ownership or voting of our ordinary shares by non-residents of Israel is not restricted in any way by our amended and restated articles of association or the laws of the State of Israel, except for ownership by nationals of some countries that are, have been, or will be, in a state of war with Israel.
Election of Directors
Under our amended and restated articles of association, our board of directors must consist of not less than three (3) but no more than 11 (eleven) directors. Pursuant to our amended and restated articles of association, each of our directors will be appointed by a simple majority vote of holders of our ordinary shares, participating and voting at an annual general meeting of our shareholders, provided that (i) in the event of a contested election, the method of calculation of the votes and the manner in which the resolutions will be presented to our shareholders at the general meeting shall be determined by our board of directors in its discretion, and (ii) in the event that our board of directors does not or is unable to make a determination on such matter, then the directors will be elected by a majority of the voting power represented at the general meeting in person or by proxy and voting on the election of directors, provided that if the number of such nominees exceeds the number of directors to be elected, then as among such elected nominees the election shall be by a plurality of the votes cast. In addition, our directors are divided into three classes, one class being elected each year at the annual general meeting of our shareholders, and shall serve on our board of directors until the third annual general meeting following such election or re-election or until they are removed by a vote of 65% of the total voting power of our shareholders at a general meeting of our shareholders or upon the occurrence of certain events, in accordance with the Companies Law and our amended and restated articles of association. In addition, our amended and restated articles of association provide that vacancies on our board of directors may be filled by a vote of a simple majority of directors then in office. Any director so appointed will hold office until the next annual general meeting of our shareholders for the election of the class of directors in respect of which the vacancy was created, or in the case of a vacancy due to the number of directors being less than the maximum number of directors stated in our amended and restated articles of association, until the next annual general meeting of our shareholders for the election of the class of directors to which such director was assigned by our board of directors.
- 76 -
Dividend and Liquidation Rights
We may declare a dividend to be paid to the holders of our ordinary shares in proportion to their respective shareholdings. Under the Companies Law, dividend distributions are determined by the board of directors and do not require the approval of the shareholders of a company unless the company’s articles of association provide otherwise. Our amended and restated articles of association do not require shareholder approval of a dividend distribution and provide that dividend distributions may be determined by our board of directors.
Pursuant to the Companies Law, the distribution amount is limited to the greater of retained earnings or earnings generated over the previous two years, according to our then last reviewed or audited financial statements (less the amount of previously distributed dividends, if not reduced from the earnings), provided that the end of the period to which the financial statements relate is not more than six months prior to the date of the distribution. If we do not meet such criteria, then we may distribute dividends only with court approval. In each case, we are only permitted to distribute a dividend if our board of directors and, if applicable, the court determines that there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of our ordinary shares in proportion to their shareholdings. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
Exchange Controls
There are currently no Israeli currency control restrictions on remittances of dividends on our ordinary shares, proceeds from the sale of the ordinary shares or interest or other payments to non-residents of Israel, except for shareholders who are subjects of countries that are, have been, or will be, in a state of war with Israel.
Shareholder Meetings
Under Israeli law, we are required to hold an annual general meeting of our shareholders once every calendar year and no later than 15 months after the date of the previous annual general meeting. All meetings other than the annual general meeting of shareholders are referred to in our amended and restated articles of association as special general meetings. Our board of directors may call special general meetings of our shareholders whenever it sees fit, at such time and place, within or outside of Israel, as it may determine. In addition, the Companies Law provides that our board of directors is required to convene a special general meeting of our shareholders upon the written request of (i) any two or more of our directors, (ii) one-quarter or more of the serving members of our board of directors or (iii) one or more shareholders holding, in the aggregate, either (a) 5% or more of our outstanding issued shares and 1% or more of our outstanding voting power or (b) 5% or more of our outstanding voting power.
Under Israeli law, one or more shareholders holding at least 1% of the voting rights at the general meeting of the shareholders may request that the board of directors include a matter in the agenda of a general meeting of the shareholders to be convened in the future, provided that it is appropriate to discuss such a matter at the general meeting. Our amended and restated articles of association contain procedural guidelines and disclosure items with respect to the submission of shareholder proposals for general meetings.
Subject to the provisions of the Companies Law and the regulations promulgated thereunder, shareholders entitled to participate and vote at general meetings of shareholders are the shareholders of record on a date to be decided by the board of directors, which, as a company listed on an exchange outside Israel, may be between four and 40 days prior to the date of the meeting. Furthermore, the Companies Law requires that resolutions regarding the following matters must be passed at a general meeting of shareholders:
| · | amendments to our articles of association; |
| · | appointment, terms of service or and termination of service of our auditors; |
| · | appointment of directors, including external directors (if applicable); |
| · | approval of certain related party transactions; |
| · | increases or reductions of our authorized share capital; |
| · | a merger; and |
- 77 -
| · | the exercise of our board of directors’ powers by a general meeting, if our board of directors is unable to exercise its powers and the exercise of any of its powers is required for our proper management. |
The Companies Law requires that a notice of any annual general meeting or special general meeting be provided to shareholders at least 21 days prior to the meeting and if the agenda of the meeting includes, among other things, the appointment or removal of directors, the approval of transactions with office holders or interested or related parties, or an approval of a merger, notice must be provided at least 35 days prior to the meeting. Under the Companies Law and our amended and restated articles of association, shareholders are not permitted to take action by way of written consent in lieu of a meeting.
Quorum
Pursuant to our amended and restated articles of association, holders of our ordinary shares have one vote for each ordinary share held on all matters submitted to a vote before the shareholders at a general meeting of shareholders. The quorum required for our general meetings of shareholders consists of at least two shareholders present in person or by proxy who hold or represent between them at least 33⅓% of the total outstanding voting rights, provided, however, that with respect to any general meeting that was convened pursuant to a resolution adopted by the board of directors and which at the time of such general meeting we qualify as a “foreign private issuer,” the requisite quorum shall consist of two or more shareholders present in person or by proxy who hold or represent between them at least 25% of the total outstanding voting rights. The requisite quorum shall be present within half an hour of the time fixed for the commencement of the general meeting. A general meeting adjourned for lack of a quorum shall be adjourned either to the same day in the next week, at the same time and place, to such day and at such time and place as indicated in the notice to such meeting, or to such day and at such time and place as the chairperson of the meeting shall determine. At the reconvened meeting, any number of shareholders present in person or by proxy shall constitute a quorum, unless a meeting was called pursuant to a request by our shareholders, in which case the quorum required is one or more shareholders, present in person or by proxy and holding the number of shares required to call the meeting as described above.
Vote Requirements
Our amended and restated articles of association provide that all resolutions of our shareholders require a simple majority vote, unless otherwise required by the Companies Law or by our amended and restated articles of association. Under the Companies Law, certain actions require the approval of a special majority, including: (i) an extraordinary transaction with a controlling shareholder or in which the controlling shareholder has a personal interest, (ii) the terms of employment or other engagement of a controlling shareholder of the company or a controlling shareholder’s relative (even if such terms are not extraordinary) and (iii) certain compensation-related matters described above under “Management—Compensation Committee—Compensation Policy under the Companies Law.” Under our amended and restated articles of association, the alteration of the rights, privileges, preferences or obligations of any class of our shares (to the extent there are classes other than ordinary shares) requires the approval of a simple majority of the class so affected (or such other percentage of the relevant class that may be set forth in the governing documents relevant to such class), in addition to a majority of all classes of shares voting together as a single class at a shareholder meeting.
Under our amended and restated articles of association, the approval of the holders of at least 65% of the total voting power of our shareholders is generally required to remove any of our directors from office, to amend the provision requiring the approval of at least 65% of the total voting power of our shareholders to remove any of our directors from office, or certain other provisions regarding our staggered board, shareholder proposals, the size of our board and plurality voting in contested elections. Another exception to the simple majority vote requirement is a resolution for the voluntary winding up, or an approval of a scheme of arrangement or reorganization, of the company pursuant to Section 350 of the Companies Law, which requires the approval of holders holding at least 75% of the voting rights represented at the meeting and voting on the resolution.
- 78 -
Access to Corporate Records
Under the Companies Law, all shareholders generally have the right to review minutes of our general meetings, our shareholder register (including with respect to material shareholders), our articles of association, our financial statements, other documents as provided in the Companies Law and any document we are required by law to file publicly with the Israeli Registrar of Companies or the Israeli Securities Authority. Any shareholder who specifies the purpose of its request may request to review any document in our possession that relates to any action or transaction with a related party which requires shareholder approval under the Companies Law. We may deny a request to review a document if we determine that the request was not made in good faith, that the document contains a trade secret or a patent or that the document’s disclosure may otherwise impair our interests.
Acquisitions under Israeli Law
Full Tender Offer
A person wishing to acquire shares of a public Israeli company who would, as a result, hold over 90% of the target company’s voting rights or the target company’s issued and outstanding share capital (or of a class thereof), is required by the Companies Law to make a tender offer to all of the company’s shareholders for the purchase of all of the issued and outstanding shares of the company (or the applicable class). If (a) the shareholders who do not accept the offer hold less than 5% of the issued and outstanding share capital of the company (or the applicable class) and the shareholders who accept the offer constitute a majority of the offerees that do not have a personal interest in the acceptance of the tender offer or (b) the shareholders who did not accept the tender offer hold less than 2% of the issued and outstanding share capital of the company (or of the applicable class), all of the shares that the acquirer offered to purchase will be transferred to the acquirer by operation of law. A shareholder who had its shares so transferred may petition an Israeli court within six months from the date of acceptance of the full tender offer, regardless of whether such shareholder agreed to the offer, to determine whether the tender offer was for less than fair value and whether the fair value should be paid as determined by the court. However, an offeror may provide in the offer that a shareholder who accepted the offer will not be entitled to petition the court for appraisal rights as described in the preceding sentence, as long as the offeror and the company disclosed the information required by law in connection with the full tender offer. If the full tender offer was not accepted in accordance with any of the above alternatives, the acquirer may not acquire shares of the company that will increase its holdings to more than 90% of the company’s voting rights or the company’s issued and outstanding share capital (or of the applicable class) from shareholders who accepted the tender offer. Shares purchased in contradiction to the full tender offer rules under the Companies Law will have no rights and will become dormant shares.
Special Tender Offer
The Companies Law provides that an acquisition of shares of an Israeli public company must be made by means of a special tender offer if, as a result of the acquisition, the purchaser would become a holder of 25% or more of the voting rights in the company. This requirement does not apply if there is already another holder of 25% or more of the voting rights in the company. Similarly, the Companies Law provides that an acquisition of shares of an Israeli public company must be made by means of a special tender offer if, as a result of the acquisition, the purchaser would become a holder of more than 45% of the voting rights in the company, if there is no other shareholder of the company who holds more than 45% of the voting rights in the company. These requirements do not apply if (i) the acquisition occurs in the context of a private placement by the company that received shareholder approval as a private placement whose purpose is to give the purchaser 25% or more of the voting rights in the company, if there is no person who holds 25% or more of the voting rights in the company or as a private placement whose purpose is to give the purchaser 45% of the voting rights in the company, if there is no person who holds 45% of the voting rights in the company, (ii) the acquisition was from a shareholder holding 25% or more of the voting rights in the company and resulted in the purchaser becoming a holder of 25% or more of the voting rights in the company, or (iii) the acquisition was from a shareholder holding more than 45% of the voting rights in the company and resulted in the purchaser becoming a holder of more than 45% of the voting rights in the company. A special tender offer must be extended to all shareholders of a company. A special tender offer may be consummated only if (i) at least 5% of the voting power attached to the company’s outstanding shares will be acquired by the offeror and (ii) the number of shares tendered in the offer exceeds the number of shares whose holders objected to the offer (excluding the purchaser, its controlling shareholders, holders of 25% or more of the voting rights in the company and any person having a personal interest in the acceptance of the tender offer, or anyone on their behalf, including any such person’s relatives and entities under their control).
In the event that a special tender offer is made, a company’s board of directors is required to express its opinion on the advisability of the offer, or may abstain from expressing any opinion if it is unable to do so, provided that it gives the reasons for its abstention. The board of directors shall also disclose any personal interest that any of the directors has with respect to the special tender offer or in connection therewith. An office holder in a target company who, in his or her capacity as an office holder, performs an action the purpose of which is to cause the failure of an existing or foreseeable special tender offer or to impair the chances of its acceptance, is liable to the potential purchaser and shareholders for damages, unless such office holder acted in good faith and had reasonable grounds to believe he or she was acting for the benefit of the company. However, office holders of the target company may negotiate with the potential purchaser in order to improve the terms of the special tender offer, and may further negotiate with third parties in order to obtain a competing offer.
- 79 -
If a special tender offer is accepted, then shareholders who did not respond to or that had objected the offer may accept the offer within four days of the last day set for the acceptance of the offer and they will be considered to have accepted the offer from the first day it was made.
In the event that a special tender offer is accepted, then the purchaser or any person or entity controlling it or under common control with the purchaser or such controlling person or entity at the time of the offer may not make a subsequent tender offer for the purchase of shares of the target company and may not enter into a merger with the target company for a period of one year from the date of the offer, unless the purchaser or such person or entity undertook to effect such an offer or merger in the initial special tender offer. Shares purchased in contradiction to the special tender offer rules under the Companies Law will have no rights and will become dormant shares.
Merger
The Companies Law permits merger transactions if approved by each party’s board of directors and, unless certain conditions described under the Companies Law are met, a simple majority of the outstanding shares of each party to the merger that are represented and voting on the merger. The board of directors of a merging company is required pursuant to the Companies Law to discuss and determine whether in its opinion there exists a reasonable concern that as a result of a proposed merger, the surviving company will not be able to satisfy its obligations towards its creditors, such determination taking into account the financial status of the merging companies. If the board of directors determines that such a concern exists, it may not approve a proposed merger. Following the approval of the board of directors of each of the merging companies, the boards of directors must jointly prepare a merger proposal for submission to the Israeli Registrar of Companies.
For purposes of the shareholder vote of a merging company whose shares are held by the other merging company, or by a person or entity holding 25% or more of the voting rights at the general meeting of shareholders of the other merging company, or by a person or entity holding the right to appoint 25% or more of the directors of the other merging company, unless a court rules otherwise, the merger will not be deemed approved if a majority of the shares voted on the matter at the general meeting of shareholders (excluding abstentions) that are held by shareholders other than the other party to the merger, or by any person or entity who holds 25% or more of the voting rights of the other party or the right to appoint 25% or more of the directors of the other party, or any one on their behalf including their relatives or corporations controlled by any of them, vote against the merger. In addition, if the non-surviving entity of the merger has more than one class of shares, the merger must be approved by each class of shareholders. If the transaction would have been approved but for the separate approval of each class or the exclusion of the votes of certain shareholders as provided above, a court may still approve the merger upon the request of holders of at least 25% of the voting rights of a company, if the court holds that the merger is fair and reasonable, taking into account the valuation of the merging companies and the consideration offered to the shareholders. If a merger is with a company’s controlling shareholder or if the controlling shareholder has a personal interest in the merger, then the merger is instead subject to the same special majority approval that governs all extraordinary transactions with controlling shareholders.
Under the Companies Law, each merging company must deliver to its secured creditors the merger proposal and inform its unsecured creditors of the merger proposal and its content. Upon the request of a creditor of either party to the proposed merger, the court may delay or prevent the merger if it concludes that there exists a reasonable concern that, as a result of the merger, the surviving company will be unable to satisfy the obligations of the merging company, and may further give instructions to secure the rights of creditors.
In addition, a merger may not be completed unless at least 50 days have passed from the date that a proposal for approval of the merger is filed with the Israeli Registrar of Companies and 30 days from the date that shareholder approval of both merging companies is obtained.
- 80 -
Anti-Takeover Measures
The Companies Law allows us to create and issue shares having rights different from those attached to our ordinary shares, including shares providing certain preferred rights with respect to voting, distributions or other matters and shares having preemptive rights. As of this Current Report on Form 8-K, no preferred shares will be authorized under our amended and restated articles of association. In the future, if we do authorize, create and issue a specific class of preferred shares, such class of shares, depending on the specific rights that may be attached to it, may have the ability to frustrate or prevent a takeover or otherwise prevent our shareholders from realizing a potential premium over the market value of their ordinary shares. The authorization and designation of a class of preferred shares will require an amendment to our amended and restated articles of association, which requires the prior approval of the holders of a majority of the voting power attached to our issued and outstanding shares at a general meeting of our shareholders. The convening of the meeting, the shareholders entitled to participate and the vote required to be obtained at such a meeting will be subject to the requirements set forth in the Companies Law and our amended articles of association, as described above in “—Shareholder Meetings.” In addition, as disclosed under “—Election of Directors,” we have a classified board structure, which effectively limits the ability of any investor or potential investor or group of investors or potential investors to gain control of our board of directors.
Borrowing Powers
Pursuant to the Companies Law and our amended and restated articles of association, our board of directors may exercise all powers and take all actions that are not required under law or under our amended and restated articles of association to be exercised or taken by our shareholders, including the power to borrow money for company purposes.
Changes in Capital
Our amended and restated articles of association enable us to increase or reduce our share capital. Any such changes are subject to Israeli law and must be approved by a resolution duly passed by our shareholders at a general meeting of shareholders. In addition, transactions that have the effect of reducing capital, such as the declaration and payment of dividends in the absence of sufficient retained earnings or profits, require the approval of both our board of directors and an Israeli court.
Exclusive Forum
Our amended and restated articles of association provide that unless we consent in writing to the selection of an alternative forum, the United States District Court for the Southern District of New York shall be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act and/or the Exchange Act. Our amended and restated articles of association also provide that unless we consent in writing to the selection of an alternative forum, the competent courts in Tel Aviv, Israel shall be the exclusive forum for any derivative action or proceeding brought on behalf of the Company, any action asserting a breach of a fiduciary duty owed by any of our directors, officers or other employees to the Company or our shareholders or any action asserting a claim arising pursuant to any provision of the Companies Law or the Israeli Securities Law.
Transfer Agent and Registrar
The transfer agent and registrar for our ordinary shares and ADSs is Computershare. Its address is 1290 Avenue of the Americas, 9th Floor, New York, NY 10104, and its telephone number is (212) 805-7100.
Listing
Our ordinary shares represented by American Depositary Shares are listed on Nasdaq under the symbol “CMMB.”
DESCRIPTION OF AMERICAN DEPOSITARY SHARES
American Depositary Shares
The Bank of New York Mellon, as depositary, will register and deliver the Chemomab’s ADSs. Each ADS will represent ordinary shares (or a right to receive twenty (20) ordinary shares) . Each ADS will also represent any other securities, cash or other property which may be held by the depositary. The depositary’s office at which our ADSs will be administered and its principal executive office are located at 240 Greenwich Street, New York, New York 10286.
- 81 -
You may hold ADSs either (A) directly (i) by having an ADR, which is a certificate evidencing a specific number of ADSs, registered in your name or (ii) by having uncertificated ADSs registered in your name or (B) indirectly by holding a security entitlement in ADSs through your broker or other financial institution that is a direct or indirect participant in DTC. If you hold ADSs directly, you are a registered ADS holder, also referred to as an ADS holder. This description assumes you are an ADS holder. If you hold our ADSs indirectly, you must rely on the procedures of your broker or other financial institution to assert the rights of ADS holders described in this section. You should consult with your broker or financial institution to find out what those procedures are.
Registered holders of uncertificated ADSs will receive statements from the depositary confirming their holdings.
As an ADS holder, we will not treat you as one of our shareholders and you will not have shareholder rights. Israeli law governs shareholder rights. The depositary will be the holder of the ordinary shares underlying your ADSs. As a registered holder of ADSs, you will have ADS holder rights. A deposit agreement among us, the depositary, ADS holders and all other persons indirectly or beneficially holding ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and our ADSs.
The following is a summary of the material provisions of the deposit agreement. For more complete information, you should read the entire deposit agreement and the form of ADR. For directions on how to obtain copies of those documents see “Where You Can Find More Information.”
Dividends and Other Distributions
How will you receive dividends and other distributions on the shares?
The depositary has agreed to pay or distribute to ADS holders the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities, upon payment or deduction of its fees and expenses. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent.
Cash. The depositary will convert any cash dividend or other cash distribution we pay on the ordinary shares into U.S. dollars, if it can do so on a reasonable basis and can transfer the U.S. dollars to the United States. If that is not possible or if any government approval is needed and cannot be obtained, the deposit agreement allows the depositary to distribute the foreign currency only to those ADS holders to whom it is possible to do so. It will hold the foreign currency it cannot convert for the account of our ADS holders who have not been paid. It will not invest the foreign currency and it will not be liable for any interest.
Before making a distribution, any withholding taxes or other governmental charges that must be paid will be deducted. See “Taxation and Government Programs.” It will distribute only whole U.S. dollars and cents and will round fractional cents to the nearest whole cent. If the exchange rates fluctuate during a time when the depositary cannot convert the foreign currency, you may lose some or all of the value of the distribution.
Shares. The depositary may distribute additional ADSs representing any ordinary shares we distribute as a dividend or free distribution. The depositary will only distribute whole ADSs. It will sell ordinary shares which would require it to deliver a fraction of an ADS (or ADSs representing those ordinary shares) and distribute the net proceeds in the same way as it does with cash. If the depositary does not distribute additional ADSs, the outstanding ADSs will also represent the new shares. The depositary may sell a portion of the distributed ordinary shares (or ADSs representing those ordinary shares) sufficient to pay its fees and expenses in connection with that distribution.
Rights to purchase additional shares. If we offer holders of our securities any rights to subscribe for additional ordinary shares or any other rights, the depositary may (i) exercise those rights on behalf of ADS holders, (ii) distribute those rights to ADS holders or (iii) sell those rights and distribute the net proceeds to ADS holders, in each case after deduction or upon payment of its fees and expenses. To the extent the depositary does not do any of those things, it will allow the rights to lapse. In that case, you will receive no value for them. The depositary will exercise or distribute rights only if we ask it to and provide satisfactory assurances to the depositary that it is legal to do so. If the depositary will exercise rights, it will purchase the securities to which the rights relate and distribute those securities or, in the case of ordinary shares, new ADSs representing the new ordinary shares, to subscribing ADS holders, but only if ADS holders have paid the exercise price to the depositary. U.S. securities laws may restrict the ability of the depositary to distribute rights or ADSs or other securities issued on exercise of rights to all or certain ADS holders, and the securities distributed may be subject to restrictions on transfer.
- 82 -
Other Distributions. The depositary will send to ADS holders anything else we distribute on deposited securities by any means it thinks is legal, fair and practical. If it cannot make the distribution in that way, the depositary will have a choice. It may decide to sell what we distributed and distribute the net proceeds, in the same way as it does with cash. Or, it may decide to hold what we distributed, in which case ADSs will also represent the newly distributed property. However, the depositary is not required to distribute any securities (other than ADSs) to ADS holders unless it receives satisfactory evidence from us that it is legal to make that distribution. The depositary may sell a portion of the distributed securities or property sufficient to pay its fees and expenses in connection with that distribution. U.S. securities laws may restrict the ability of the depositary to distribute securities to all or certain ADS holders, and the securities distributed may be subject to restrictions on transfer.
The depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any ADS holders. We have no obligation to register ADSs, ordinary shares, rights or other securities under the Securities Act. We also have no obligation to take any other action to permit the distribution of ADSs, shares, rights or anything else to ADS holders. This means that you may not receive the distributions we make on our ordinary shares or any value for them if it is illegal or impractical for us to make them available to you.
Deposit, Withdrawal and Cancellation
How are ADSs issued?
The depositary will deliver ADSs if you or your broker deposits ordinary shares or evidence of rights to receive ordinary shares with the custodian. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will register the appropriate number of ADSs in the names you request and will deliver our ADSs to or upon the order of the person or persons that made the deposit.
How can ADS holders withdraw the deposited securities?
You may surrender your ADSs for the purpose of withdrawal at the depositary’s office. Upon payment of its fees and expenses and of any taxes or charges, such as stamp taxes or stock transfer taxes or fees, the depositary will deliver the ordinary shares and any other deposited securities underlying our ADSs to the ADS holder or a person the ADS holder designates at the office of the custodian. Or, at your request, risk and expense, the depositary will deliver the deposited securities at its office, if feasible. However, the depositary is not required to accept surrender of ADSs to the extent it would require delivery of a fraction of a deposited share or other securities. The depositary may charge you a fee and its expenses for instructing the custodian regarding delivery of deposited securities.
How do ADS holders interchange between certificated ADSs and uncertificated ADSs?
You may surrender your ADR to the depositary for the purpose of exchanging your ADR for uncertificated ADSs. The depositary will cancel that ADR and will send to the ADS holder a statement confirming that the ADS holder is the registered holder of uncertificated ADSs. Alternatively, upon receipt by the depositary of a proper instruction from a registered holder of uncertificated ADSs requesting the exchange of uncertificated ADSs for certificated ADSs, the depositary will execute and deliver to the ADS holder an ADR evidencing those ADSs.
- 83 -
Voting Rights
How do you vote?
ADS holders may instruct the depositary how to vote the number of deposited ordinary shares their ADSs represent. If we request the depositary to solicit your voting instructions (and we are not required to do so), the depositary will notify you of a shareholders’ meeting and send or make voting materials available to you. Those materials will describe the matters to be voted on and explain how ADS holders may instruct the depositary how to vote. For instructions to be valid, they must reach the depositary by a date set by the depositary. The depositary will try, as far as practical, subject to the laws of Israel and the provisions of our articles of association or similar documents, to vote or to have its agents vote the ordinary shares or other deposited securities as instructed by ADS holders. If we do not request the depositary to solicit your voting instructions, you can still send voting instructions, and, in that case, the depositary may try to vote as you instruct, but it is not required to do so.
Except by instructing the depositary as described above, you won’t be able to exercise voting rights unless you surrender your ADSs and withdraw the ordinary shares. However, you may not know about the meeting enough in advance to withdraw the ordinary shares. In any event, the depositary will not exercise any discretion in voting deposited securities and it will only vote or attempt to vote as instructed.
We cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote your shares. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that you may not be able to exercise your right to vote and there may be nothing you can do if your ordinary shares are not voted as you requested.
In order to give you a reasonable opportunity to instruct the Depositary as to the exercise of voting rights relating to Deposited Securities, if we request the Depositary to act, we agree to give the Depositary notice of any such meeting and details concerning the matters to be voted upon at least 45 days in advance of the meeting date.
Fees and Expenses
| Persons depositing or withdrawing ordinary shares or ADS holders must pay |
| | For | |
| $5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | | | Issuance of ADSs, including issuances resulting from a distribution of ordinary shares or rights or other property Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates | |
| $.05 (or less) per ADS | | | Any cash distribution to ADS holders | |
| A fee equivalent to the fee that would be payable if securities distributed to you had been ordinary shares and the ordinary shares had been deposited for issuance of ADSs | | | Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders | |
| $.05 (or less) per ADS per calendar year | | | Depositary services | |
- 84 -
| Persons depositing or withdrawing ordinary shares or ADS holders must pay |
| | For | |
| Registration or transfer fees | | | Transfer and registration of ordinary shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw ordinary shares | |
| Expenses of the depositary | | |
Cable, telex and facsimile transmissions (when expressly provided in the deposit agreement)
Converting foreign currency to U.S. dollars |
|
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or ordinary shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes | | | As necessary | |
| Any charges incurred by the depositary or its agents for servicing the deposited securities | | | As necessary | |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing ordinary shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of our ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
The depositary may convert foreign currency itself or through any of its affiliates and, in those cases, acts as principal for its own account and not as an agent, fiduciary or broker on behalf of any other person and earns revenue, including, without limitation, fees and spreads that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be most favorable to ADS holders, subject to its obligations under the deposit agreement. The methodology used to determine exchange rates used in currency conversions is available upon request.
Payment of Taxes
You will be responsible for any taxes or other governmental charges payable on your ADSs or on the deposited securities represented by any of your ADSs. The depositary may refuse to register any transfer of your ADSs or allow you to withdraw the deposited securities represented by your ADSs until such taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by your ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to ADS holders any proceeds, or send to ADS holders any property, remaining after it has paid the taxes.
Tender and Exchange Offers; Redemption, Replacement or Cancellation of Deposited Securities
The depositary will not tender deposited securities in any voluntary tender or exchange offer unless instructed to do by an ADS holder surrendering ADSs and subject to any conditions or procedures the depositary may establish.
If deposited securities are redeemed for cash in a transaction that is mandatory for the depositary as a holder of deposited securities, the depositary will call for surrender of a corresponding number of ADSs and distribute the net redemption money to the holders of called ADSs upon surrender of those ADSs.
- 85 -
If there is any change in the deposited securities such as a subdivision, combination or other reclassification, or any merger, consolidation, recapitalization or reorganization affecting the issuer of deposited securities in which the depositary receives new securities in exchange for or in lieu of the old deposited securities, the depositary will hold those replacement securities as deposited securities under the deposit agreement. However, if the depositary decides it would not be lawful and practical to hold the replacement securities because those securities could not be distributed to ADS holders or for any other reason, the depositary may instead sell the replacement securities and distribute the net proceeds upon surrender of our ADSs.
If there is a replacement of the deposited securities and the depositary will continue to hold the replacement securities, the depositary may distribute new ADSs representing the new deposited securities or ask you to surrender your outstanding ADRs in exchange for new ADRs identifying the new deposited securities.
If there are no deposited securities underlying ADSs, including if the deposited securities are cancelled, or if the deposited securities underlying ADSs have become apparently worthless, the depositary may call for surrender or of those ADSs or cancel those ADSs upon notice to the ADS holders.
Amendment and Termination
How may the deposit agreement be amended?
We may agree with the depositary to amend the deposit agreement and the ADRs without your consent for any reason. If an amendment adds or increases fees or charges, except for taxes and other governmental charges or expenses of the depositary for registration fees, facsimile costs, delivery charges or similar items, or prejudices a substantial right of ADS holders, it will not become effective for outstanding ADSs until 30 days after the depositary notifies ADS holders of the amendment. At the time an amendment becomes effective, you are considered, by continuing to hold your ADSs, to agree to the amendment and to be bound by the ADRs and the deposit agreement as amended.
How may the deposit agreement be terminated?
The depositary will initiate termination of the deposit agreement if we instruct it to do so. The depositary may initiate termination of the deposit agreement if:
• 90 days have passed since the depositary told us it wants to resign but a successor depositary has not been appointed and accepted its appointment;
• we delist our ADSs from an exchange on which they were listed and do not list our ADSs on another exchange within a reasonable time;
• we appear to be insolvent or enter insolvency proceedings;
• all or substantially all the value of the deposited securities has been distributed either in cash or in the form of securities;
• there are no deposited securities underlying our ADSs or the underlying deposited securities have become apparently worthless; or
• there has been a replacement of deposited securities.
If the deposit agreement will terminate, the depositary will notify ADS holders at least 90 days before the termination date. At any time after the termination date, the depositary may sell the deposited securities. After that, the depositary will hold the money it received on the sale, as well as any other cash it is holding under the deposit agreement, unsegregated and without liability for interest, for the pro rata benefit of the ADS holders that have not surrendered their ADSs. Normally, the depositary will sell as soon as practicable after the termination date.
- 86 -
After the termination date and before the depositary sells, ADS holders can still surrender their ADSs and receive delivery of deposited securities, except that the depositary may refuse to accept a surrender for the purpose of withdrawing deposited securities or reverse previously accepted surrenders of that kind if it would interfere with the selling process. The depositary may refuse to accept a surrender for the purpose of withdrawing sale proceeds until all the deposited securities have been sold. The depositary will continue to collect distributions on deposited securities, but, after the termination date, the depositary is not required to register any transfer of ADSs or distribute any dividends or other distributions on deposited securities to the ADSs holder (until they surrender their ADSs) or give any notices or perform any other duties under the deposit agreement except as described in this paragraph.
Limitations on Obligations and Liability
Limits on our obligations and the obligations of the depositary; Limits on liability to holders of ADSs
The deposit agreement expressly limits our obligations and the obligations of the depositary. It also limits our liability and the liability of the depositary. We and the depositary:
• are only obligated to take the actions specifically set forth in the deposit agreement without negligence or bad faith and the depositary will not be a fiduciary or have any fiduciary duty to holders of ADSs;
• are not liable if we are or it is prevented or delayed by law or circumstances beyond our or its control from performing our or its obligations under the deposit agreement;
• are not liable if we exercise or it exercises discretion permitted under the deposit agreement;
• are not liable for the inability of any holder of ADSs to benefit from any distribution on deposited securities that is not made available to holders of ADSs under the terms of the deposit agreement, or for any special, consequential or punitive damages for any breach of the terms of the deposit agreement;
• have no obligation to become involved in a lawsuit or other proceeding related to our ADSs or the deposit agreement on your behalf or on behalf of any other person;
• are not liable for the acts or omissions of any securities depository, clearing agency or settlement system;
• may rely upon any documents we believe or it believes in good faith to be genuine and to have been signed or presented by the proper person; and
• the depositary has no duty to make any determinations or provide any information as to our status, or any liability for any tax consequences that may be incurred by ADS holders as a result of owning or holding ADSs or liable for the inability or failure of an ADS holder to obtain the benefit of a foreign tax credit reduced rate of withholdings or refund of amounts withheld in respect of tax or any other tax benefit.
In the deposit agreement, we and the depositary agree to indemnify each other under certain circumstances.
Requirements for Depositary Actions
Before the depositary will deliver or register a transfer of ADSs, make a distribution on ADSs, or permit withdrawal of shares, the depositary may require:
• payment of stock transfer or other taxes or other governmental charges and transfer or registration fees charged by third parties for the transfer of any ordinary shares or other deposited securities;
• satisfactory proof of the identity and genuineness of any signature or other information it deems necessary; and
- 87 -
• compliance with regulations it may establish, from time to time, consistent with the deposit agreement, including presentation of transfer documents.
The depositary may refuse to deliver ADSs or register transfers of ADSs when the transfer books of the depositary or our transfer books are closed or at any time if the depositary or we think it advisable to do so.
Your Right to Receive the Ordinary Shares Underlying your ADSs
ADS holders have the right to cancel their ADSs and withdraw the underlying ordinary shares at any time except:
• when temporary delays arise because: (i) the depositary has closed its transfer books or we have closed our transfer books; (ii) the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting; or (iii) we are paying a dividend on our shares;
• when you owe money to pay fees, taxes and similar charges; or
• when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
This right of withdrawal may not be limited by any other provision of the deposit agreement.
Direct Registration System
In the deposit agreement, all parties to the deposit agreement acknowledge that the Direct Registration System, or DRS, and Profile Modification System, or Profile, will apply to our ADSs. DRS is a system administered by DTC that facilitates interchange between registered holding of uncertificated ADSs and holding of security entitlements in ADSs through DTC and a DTC participant. Profile is a feature of DRS that allows a DTC participant, claiming to act on behalf of a registered holder of uncertificated ADSs, to direct the depositary to register a transfer of those ADSs to DTC or its nominee and to deliver those ADSs to the DTC account of that DTC participant without receipt by the depositary of prior authorization from the ADS holder to register that transfer.
In connection with and in accordance with the arrangements and procedures relating to DRS/Profile, the parties to the deposit agreement understand that the depositary will not determine whether the DTC participant that is claiming to be acting on behalf of an ADS holder in requesting registration of transfer and delivery as described in the paragraph above has the actual authority to act on behalf of the ADS holder (notwithstanding any requirements under the Uniform Commercial Code). In the deposit agreement, the parties agree that the depositary’s reliance on and compliance with instructions received by the depositary through the DRS/Profile system and in accordance with the deposit agreement will not constitute negligence or bad faith on the part of the depositary.
Shareholder Communications; Inspection of Register of Holders of ADSs
The depositary will make available for your inspection at its office all communications that it receives from us as a holder of deposited securities that we make generally available to holders of deposited securities. The depositary will send you copies of those communications or otherwise make those communications available to you if we ask it to. You have a right to inspect the register of holders of ADSs, but not for the purpose of contacting those holders about a matter unrelated to our business or our ADSs.
Jury Trial Waiver
The deposit agreement provides that, to the extent permitted by law, ADS holders waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to our shares, our ADSs or the deposit agreement, including any claim under the U.S. federal securities laws. If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable in the facts and circumstances of that case in accordance with applicable case law.
- 88 -
| Item 9.01 | Financial Statements and Exhibits |
(b) This Current Report on Form 8-K is made to comply with, among other things, Item 9.01(b) by providing the pro forma financial statements of Anchiano Therapeutics Ltd. and Chemomab Ltd.
(d) Exhibits Below is a list of exhibits included with this Current Report on Form 8-K.
| Exhibit No. |
Document | |
| 99.1 | Pro Forma Combined Condensed Financial Statements of Anchiano Therapeutics Ltd. and Chemomab Ltd. |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| Chemomab Therapeutics Ltd. | ||
| By: | /s/ Adi Mor | |
| Name: | Adi Mor | |
| Title: | Chief Executive Officer and Chief Scientific Officer | |
Date: April 14, 2021