Attached files
| file | filename |
|---|---|
| EX-10.17 - AMENDED AND RESTATED AMERCIAN BRIVISION (HOLDING) CORPORATION 2016 EQUITY INCENT - ABVC BIOPHARMA, INC. | f10k2020ex10-17_americanbriv.htm |
| EX-32.2 - CERTIFICATION - ABVC BIOPHARMA, INC. | f10k2020ex32-2_americanbriv.htm |
| EX-32.1 - CERTIFICATION - ABVC BIOPHARMA, INC. | f10k2020ex32-1_americanbriv.htm |
| EX-31.2 - CERTIFICATION - ABVC BIOPHARMA, INC. | f10k2020ex31-2_americanbriv.htm |
| EX-31.1 - CERTIFICATION - ABVC BIOPHARMA, INC. | f10k2020ex31-1_americanbriv.htm |
| EX-23.1 - CONSENT OF KCCW ACCOUNTANCY CORP - ABVC BIOPHARMA, INC. | f10k2020ex23-1_americanbriv.htm |
| EX-4.2 - DESCRIPTION OF SECURITIES REGISTERED UNDER SECTION 12 OF THE EXCHANGE ACT - ABVC BIOPHARMA, INC. | f10k2020ex4-2_americanbriv.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
☒ ANNUAL REPORT UNDER SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended: December 31, 2020
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission file number: 333-91436
AMERICAN BRIVISION (HOLDING) CORPORATION
(Exact name of Company in its charter)
| Nevada | 26-0014658 | |
| (State or other jurisdiction
of incorporation or organization) |
(I.R.S. Employer
Identification) |
44370 Old Warm Springs Blvd.
Fremont, CA 94538
(Address of principal executive offices, including zip code)
Registrant’s Telephone number, including area code: (510)-668-0881
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act during the preceding 12 months (or such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for at least the part 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (section 232.406 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | |
| Non-accelerated filer | ☒ | Smaller Reporting Company | ☒ | |
| Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates as of March 15, 2021 was $73,261,578, based on the closing price of $3.00 on June 30, 2020.
As of March 15, 2021, the registrant had 24,420,526 shares of common stock outstanding and 0 shares of convertible preferred stock outstanding.
AMERICAN BRIVISION (HOLDING) CORPORATION
Form 10-K
For the Fiscal Year Ended December 31, 2020
Table of Contents
i
As used in this Report, the terms “we”, “us”, “our”, and “our Company” and “the Company” refer to American BriVision (Holding) Corporation and its subsidiaries, unless otherwise indicated.
Except for statements of historical fact, the information presented herein constitutes forward-looking statements. These forward-looking statements generally can be identified by phrases such as “anticipates,” “believes,” “estimates,” “expects,” “forecasts,” “foresees,” “intends,” “plans,” or other words of similar import. Similarly, statements herein that describe our business strategy, outlook, objectives, plans, intentions or goals also are forward-looking statements. Such forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by such forward-looking statements. Such factors include, but are not limited to, our ability to: successfully commercialize our technology; generate revenues and achieve profitability in an intensely competitive industry; compete in products and prices with substantially larger and better capitalized competitors; secure, maintain and enforce a strong intellectual property portfolio; attract additional capital sufficient to finance our working capital requirements, as well as any investment of plant, property and equipment; develop a sales and marketing infrastructure; identify and maintain relationships with third party suppliers who can provide us a reliable source of raw materials; acquire, develop, or identify for our own use, a manufacturing capability; attract and retain talented individuals; continue operations during periods of uncertain general economic or market conditions, and; other events, factors and risks previously and from time to time disclosed in our filings with the Securities and Exchange Commission.
Although we believe the expectations reflected in our forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. Except as required by law, we do not undertake to update or revise any forward-looking statement, whether as a result of new information, future events or otherwise.
ITEM 1. DESCRIPTION OF BUSINESS
Industry Overview
The biotechnology industry focuses on developing breakthrough products and technologies to combat various types of diseases through efficient industrial manufacturing process. Biotechnology is an important business sector in the world’s economies and plays a key role in human health. Companies engaged in biotechnology generally require large amounts of capital investment for their research & development activities and it may take up to tens of years to develop and commercialize a new drug or a new medical device. ABVC (“we” or the “Company”) is an early stage biotechnology company with a pipeline of six new drugs and one medical device under development, all of which are licensed from related parties of the Company.
Our Mission
We devote our resources to building a sophisticated biotech company and becoming a pioneer in the biopharmaceutical industry in the U.S. and Taiwan with a global vision. Dr. Howard Doong, our Chief Executive Officer, and Dr. Tsung-Shann Jiang, the founder and majority shareholder of the Company, understand the challenges and opportunities of the biotech industry in Taiwan and the U.S. ABVC’s mission is to provide therapeutic solutions to significant unmet medical needs and to improve health and quality of human life by developing innovative botanical drugs to treat central nervous system (“CNS”), oncology/ hematology and eye diseases.
1
Business Overview
The Company develops its pipeline by carefully tracking new medical discoveries or medical device technologies in research institutions in the Asia-Pacific region. Pre-clinical, disease animal model and Phase I safety studies are examined closely by the Company’s scientists and other specialists known to the Company to identify drugs or medical devices that it believes demonstrate efficacy and safety based on the Company’s internal qualifications. Once a drug or medical device is shown to be a good candidate for further development and ultimately commercialization, ABVC licenses the drug or medical device from the original researchers and begins to introduce the drugs or medical devices clinical plan to highly respected principal investigators in the United States, Australia and Taiwan. In almost all cases, we have found that research institutions in each of those countries are eager to work with the Company to move forward with Phase II clinical trials.
Currently, institutions conducting phase II clinical trials in partnership with ABVC include:
| ● | Medical Device: ABV-1701, Vitargus® in vitrectomy surgery, Pivotal Study in Australia, Principal Investigator: Andrew Chang, MD, Ph.D., Sydney Eye Hospital, Australia |
| ● | Drug: ABV-1504, Major Depressive Disorder (MDD), Phase II, NCE drug Principal Investigators: Charles DeBattista M.D. and Alan F. Schatzberg, MD, Stanford University Medical Center, Cheng-Ta Li, MD, Ph.D – Taipei Veterans General Hospital |
| ● | Drug: ABV-1505, Adult Attention-Deficit Hyperactivity Disorder (ADHD), Phase II, NCE drug Principal Investigators: Keith McBurnett, Ph.D. and Linda Pfiffner, Ph.D., University of California San Francisco (UCSF), School of Medicine |
| ● | Drug: ABV-1601, Major Depression in Cancer Patients, Phase I/II, NCE drug Principal Investigator: Scott Irwin, MD, Ph.D. – Cedars Sinai Medical Center (CSMC) |
| ● | Drug: ABV-1703, Advanced Inoperable or Metastatic Pancreatic Cancer, Phase II, NCE drug Principal Investigator: Andrew E. Hendifar, MD – Cedars Sinai Medical Center (CSMC) |
| ● | Drug: ABV-1501, A Phase I/II, Open Label Study to Evaluate the Safety and Efficacy of BLEX 404 Oral Liquid Combined with Docetaxel Monotherapy in Patients with Stage IV or Recurrent Breast Cancer Patients |
| ● | Medical Device: ABV-2002, Class I/II through 510K for market launch, Corneal Storage Media, Technology Licensing in progress |
2
Upon successful completion of a Phase II trial, ABVC will seek a partner – a large pharmaceutical company – to complete a Phase III study, submit the New Drug Application (NDA), and commercialize the drug or medical device upon approval by the US FDA, Taiwan TFDA and other country regulatory authorities. We expect to seek our first commercialization partner in 2021 for Vitargus, our vitreous substitute that helps to maintain a round shape and retinal location during vitrectomy surgery.
Another part of our business is conducted by BioKey, a wholly owned subsidiary. BioKey provides a wide range of services, including, API characterization, pre-formulation studies, formulation development, analytical method development, stability studies, IND/NDA/ANDA/510K submissions, and manufacturing clinical trial materials (phase I through phase III) and commercial manufacturing.
Common Stock Reverse Split
On March 12, 2019, the Board by unanimous written consent in lieu of a meeting approved to i) effect a stock reverse split at the ratio of 1-for-18 (the “Reverse Split”) of both the authorized common stock of the Company and the issued and outstanding common stock and ii) to amend the articles of incorporation of the Company to reflect the Reverse Split. The Board approved and authorized the Reverse Split without obtaining approval of the Company’s shareholders pursuant to Section 78.207 of Nevada Revised Statutes.
On May 3, 2019, the Company filed a certificate of amendment to the Company’s articles of incorporation (the “Amendment”) to effect the Reverse Split with the Secretary of State of the State of Nevada. The Reverse Split took effect on May 8, 2019.
Increasing the Authorized Shares
As disclosed on a current report on Form 8-K filed with the SEC on April 7, 2020, on March 12, 2020, our board of directors approved and adopted an amendment to the Company’s Articles of Incorporation, to increase the authorized shares of the common stock, par value $0.001 per share, from 20,000,000 to 100,000,000, such that, after including the previously authorized 20,000,000 shares of preferred stock, par value $0.001 per share, the aggregate number of shares of stock that the Company has authority to issue is 120,000,000 shares.
Name Change and Cusip Number
The Company’s shareholders approved an amendment to the Company’s Articles of Incorporation to change the Company’s corporate name to “ABVC BioPharma, Inc.” and approved and adopted the Certificate of Amendment to affect same at the 2020 annual meeting of shareholders (the “Annual Meeting”). Nevada’s Secretary of State approved the name change on March 8, 2021, but since are still in the FINRA approval process for such name change, the new name is not yet effective. Stock certificates will remain valid and stockholders are not required to submit their stock certificates for exchange as a result of the name change. New stock certificates issued by the Company will be printed with the Company’s new name, ABVC BioPharma, Inc.; existing stock certificates will remain valid.
The Company’s new cusip number is 0091F106. The Company’s stock symbol remains ABVC.
Entry into Securities Purchase Agreements
On April 2, 2020, the Certificates of Amendment to effectuate such amendment became effective after the Certificate of Amendment being filed with the Secretary of State of the State of Nevada.
On January 21, 2020, the Company entered into three note agreements with existing note investors who executed the agreements in 2018. These three investors are Guoliang Yu and Yingfei Wei Family Trust, Keypoint Technology Ltd., and Yoshinobu Odaira. The new agreements bear the same term as other notes investors who executed the contract in 2019. On April 5, 2020, the Company entered into exchange agreements with such note holders. Pursuant to the exchange agreements, the Holders agreed to deliver the Notes to the Company for cancellation, of which the aggregate principal amount plus accrued interest expenses are $931,584, and the Company agreed to issue to the Holders an aggregate of 506,297 shares of the Company’s common stock, and warrants to purchase 506,297 shares of the Company’s common stock.
On April 20, 2020, the Company entered into certain exchange agreements separately with Kuo, Li Shen, Chang, Ping Shan, Lin, Shan Tyan, and Liu, Ching Hsuan in connection with the convertible promissory notes issued by the Company on August 28 and September 4, 2019. Pursuant to the Exchange Agreements, the Holders agreed to deliver the Notes to the Company for cancellation, of which the aggregate principal amount plus accrued interest expenses are $515,196, and the Company agreed to issue to the Holders an aggregate of 289,438 shares of the Company’s common stock, and warrants to purchase 289,438 shares of the Company’s common stock, at an exercise price of $1.78 per share.
In May 2020, the Company received capital contributions of approximately $1,602,040 in cash from 40 investors through private placements with the term of $2.25 per share and a free warrant attaches with each Common stock that was purchased. The exercise price of the warrant will be at $6.00 with a mandatory exercise price of $9.00.
On November 11, 2020, the Company conducted a closing with regard to certain securities purchase agreements (the “SPAs”) dated October 23, 2020, separately with two non-U.S. investors (the “Investors”). Each of the Investors agreed to purchase and the Company agreed to sell to each of the Investors 1,111,112 shares of the Company’s common stock, and warrants to purchase 1,111,112 shares of common stock, for a purchase price of $2,500,000, making the aggregate net proceeds to the Company $5,000,000. The warrants are exercisable upon issuance and expires three years from the date of issuance. The initial exercise price of the warrants is $6.00, subject to stock, splits, stock dividend and other similar events. In addition, when the closing price of the common stock equals or exceeds $9.00 per share for twenty Trading Days (as defined in the exchange agreements) during any thirty-day period, the Company shall have the right to require the investors to exercise all or any portion of the warrants for a cash exercise. The Company and the investors further agreed to amend the terms of the SPA to permit the closing of the offering to occur on a rolling basis.
3
Our Pipeline

| I. | ABV- 1501 Triple Negative Breast Cancer - Combination therapy for Triple Negative Breast Cancer (“TNBC”) |
| ● | ABV- 1501 is developed from BLI-1401-2 whose active pharmaceutical ingredient is Yukiguni Maitake Extract 404. Memorial Sloan Kettering Cancer Center (“MSKCC”) conducted the Phase I clinical trial of a polysaccharide extract from Grifola frondosa (Maitake mushroom), which is very similar to Yukiguni Maitake Extract 404. The Phase I trial focused on Grifola frondosa extract’s immunological effects on breast cancer patients. The results of the Phase I trial showed that oral administration of a polysaccharide extract from Maitake mushroom is associated with both immunologically stimulatory and inhibitory measurable effects in peripheral blood. | |
| ● | Our Investigational New Drug (“IND”) application of ABV-1501 for the Phase II clinical trials referenced with MSKCC Maitake and such Phase II IND was approved in March 2016 by the U.S. FDA. | |
| ● | We are currently collaborating with BHK and in the process to file clinical trial application to the Taiwan FDA (“TFDA”) for conducting this combination therapy trial in Taiwan. | |
| ● | As an alternative route to approval, we are also working towards setting up a clinical site in the U.S. to obtain IRB approval of our IND application, which based on a recent adopted policy in Taiwan, the TFDA will then automatically approve. |
| II. | ABV-1504 Major Depressive Disorder (“MDD”) |
We are developing and researching ABV-1504, a botanical reuptake inhibitor that targets norepinephrine. Prior to clinical trials, we conducted radioligand-binding assay tests on ABV-1504. Radioligand-binding assays are used to characterize the binding effects of a drug to its target receptor. In the case of ABV-1504, the receptors of radioligand-binding assays are norepinephrine, dopamine and serotonin. The radioligand-binding assay test on norepinephrine was conducted from May 3 to May 8, 2007 and the radioligand-binding assay test on dopamine and serotonin was administered from November 26 to December 5, 2007. The result of radioligand-binding assay to norepinephrine of ABV-1504was 2.102 μg/ml of IC50, which indicated ABV-1504’s high inhibitory efficiency on norepinephrine. The results of radioligand-binding assay to dopamine and serotonin were not as good as to norepinephrine, which indicated lower inhibitory efficiency. Because research has shown that norepinephrine inhibitors can alleviate the level of depression, our research team saw ABV-1504’s potential to treat depression and decided to commence the clinical trial process of ABV-1504.
4
In 2013, ABVC successfully completed the Phase I clinical trial of ABV-1504. The primary objective of the Phase I study was to assess the safety profile of ABV-1504. The safety endpoint was assessed based on the results of physical examinations, vital signs, laboratory data, electrocardiograms (“ECG”), Columbia-Suicide Severity Rating Scale evaluation and a number of adverse events during the study period. We began recruiting healthy people as subjects for the Phase I trial in Taiwan on October 30, 2012. For the Phase I trial, we screened 85 healthy volunteers at the Taipei Veterans General Hospital and eventually enrolled 30 people as trial subjects. We divided the subjects into four cohort groups and administered ABV-1504oral capsules of 380 mg, 1140 mg, 2280 mg, and 3800 mg to the subjects in each cohort group, respectively. BioLite visited the first subject the first time on November 13, 2012 and the last subject the last time on July 5, 2013. During the said period, no subject had a serious adverse event nor discontinued the trial due to any adverse events. ABVC did not observe any clinically significant findings in physical examinations, vital signs, electrocardiogram, laboratory measurements, and C-SSRS throughout the treatment period. However, ABVC observed the following mild adverse events: two subjects with flatulence and one subject with constipation in the single-dose 380mg cohort of seven subjects; one subject with somnolence and one subject with stomatitis ulcer in the single-dose 2,280 mg cohort. Comparatively, two subjects with somnolence and one subject with stomatitis ulcer were observed in the placebo group of seven subjects. ABVC did not observe any suicidal ideation or behavior throughout the trial period. ABV-1504’s Phase I clinical trial results reflected that the oral administration of ABV-1504 to healthy volunteers was safe and well-tolerated at the dose levels of from 380 mg to 3,800 mg.
ABVC received an IND approval to proceed with the Phase II clinical trial of ABV-1504 from the F.D.A. in March 2014 and an IND approval of its Phase II trial from the Taiwan F.D.A. in June 2014. For the Phase II trial, BioLite administered oral capsules to 72 MDD patients (the trial subjects) in a randomized, double-blind study with a placebo control group to assess ABV-1504’s efficacy and safety profile, primarily in accordance with the Montgomery-Åsberg Depression Rating Scale (“MADRS”). ABVC via BioLite began recruiting Phase II subjects in March 2015 at the following study sites, Taipei Veterans General Hospital, Linkou Chang Gung Memorial Hospital, Taipei City Hospital-Songde Branch, Tri-Service General Hospital, Wan Fang Hospital and started recruiting MDD patients at Stanford Depression Research Clinic. The first five sites are in Taiwan and the last one is in the United States. The primary endpoint of the Phase II trial is to see changes of the subjects’ MADRS total scores from the baseline scores of the placebo subjects within the first six weeks. The secondary objectives of the Phase II trial are to evaluate the efficacy and safety profile of ABV-1504 on other rating scales with secondary endpoints of (i) demonstrating changes in MADRS total scores from baseline scores within the second to seventh weeks and (ii) showing changes in the total scores on Hamilton Rating Scale for Depression (HAM-D-17), Hamilton Rating Scale for Anxiety (HAM-A), Depression and Somatic Symptoms Scale (DSSS), Clinical Global Impression Scale (CGI) from the baseline scores in the second, fourth, sixth and seventh week. ABVC plans to measure the percentages of partial responders (subjects with a 25% to 50% decrease of total MADRS scores from the baseline score) and responders (subjects with 50% or more decrease of total MADRS scores from the baseline score) by the second, fourth, sixth and seventh week. Additionally, ABVC intends to monitor the subjects’ performance in accordance with the Safety Assessments and Columbia-Suicide Severity Rating Scale from the screening stage to each subject’s last visit as well as to analyze the differences in the mean changes of MADRS, HAM-D-17, HAM-A, DSSS, CGI and Columbia-Suicide Severity Rating Scale scores of the subjects administered with ABV-1504 and the placebo group in the second, fourth, sixth and seventh week.
On May 23, 2019, the Company announced the Phase II clinical study results of ABV-1504. The clinical study results showed that PDC-1421, the active pharmaceutical ingredient of ABV-1504, met the pre-specified primary endpoint of the Phase II clinical trial and significantly improved the symptoms of MDD. The Phase II clinical study was a randomized, double-blind, placebo-controlled, multi-center trial, in which sixty (60) adult patients with confirmed moderate to severe MDD were treated with PDC-1421 in either low dose (380 mg) or high dose (2 x 380 mg) compared with placebo administration, three times a day for six weeks. PDC-1421 high dose (2 x 380 mg) met the pre-specified primary endpoint by demonstrating a highly significant 13.2-point reduction in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score by Intention-To-Treat (ITT) analysis, averaged over the 6-week treatment period (overall treatment effect) from baseline, as compared to 9.2-point reduction of the placebo group. By Per-Protocol (PP) analysis, PDC-1421 showed a dose dependent efficacy toward MDD in which high dose (2 x 380 mg) gave 13.4-point reduction in MADRS total score from baseline and low dose (380 mg) gave 10.4-point reduction as compared to a 8.6-point in the placebo group. Based on the trial results as set forth above, the Company has decided to use the high dose formula for ABV-1504’s Phase III clinical trial.
5
| III. | ABV-1505 Attention Deficit Hyperactivity Disorder (“ADHD”) |
We developed the ADHD indication from the same API of ABV-1504. Also ABV-1505 shares similar pharmaceutical mechanism of action as ABV-1504 in as much as ABV-1505 shows the potential of increasing the level of norepinephrine in the human’s nervous system by inhibiting its reabsorption. Because of ABV-1505’s sufficient similarity with ABV-1504, in January 2016 the FDA approved our IND application to conduct ABV-1505’s Phase II clinical trial based on its preclinical research and the Phase I trial results of ABV-1504.
For the Phase II trial, ABVC plans to recruit a maximum number of 105 ADHD patients as trial subjects in the United States and Taiwan, to whom ABVC intends to administer ABV-1505 oral capsules. ABVC together with its CROs designed a randomized, double-blind dose escalation study with a placebo-controlled group to assess the efficacy and safety profile of ABV-1505, primarily against the ADHD Rating Scale-IV (“ADHD-RS-IV”). The primary endpoint of the Phase II trial is a 40% or higher improvement on the ADHD-RS-IV from the respective baseline scores within a period of up to eight weeks. The secondary objective is to determine the efficacy and safety profile of ABV-1505 on other rating scales with secondary endpoints of (i) improvements of the total ADHD symptom scores from the respective baseline scores on the Conners’ Adult ADHD Rating Scale-Self Report: Short Version (“CAARS-S:S”) 18-Item for a treatment period of eight weeks at maximum; and (ii) achievement of scores of two or lower on both the Clinical Global Impression-ADHD- Severity (“CGI-ADHD-S”) and Clinical Global Impression-ADHD-Improvement (“CGI-ADHD-I”) from the subjects’ respective baseline scores. As of the date of this prospectus, ABVC was engaging with the University of California San Francisco (“UCSF”) for conducting the Phase II trial which consists of Part I and Part II. Part I clinical protocol, entitled “A Phase II Tolerability and Efficacy Study of PDC-1421 Treatment in Adult Patients with Attention-Deficit Hyperactivity Disorder (ADHD), Part I”, was initiated on January 14, 2020. Part I was a single center, open label, dose escalation evaluation with two dosage levels in six subjects. Six subjects were initially evaluated for safety and efficacy assessments at low-dose (1 capsule of PDC-1421, three times a day (TID)) for 28 days. A safety checkpoint was evaluated at day-28 for entering the high-dose (2 capsules TID). The subjects who passed the checkpoint were evaluated for safety and efficacy assessments at high-dose (2 capsules of PDC-1421 TID) for 28 days. On July 15, 2020, the last patient last visit (LPLV) marked the final step toward the completion of the ABV-1505 Phase II Part I clinical trial for the treatment of adult ADHD. On October 24, 2020, a full clinical study report (CSR) of ABV-1505 Phase II Part I clinical trial was issued. The study results showed that the PDC-1421 Capsule was safe, well tolerated and efficacious during its treatment and the follow-up period with six adult patients. For the primary endpoints, the percentages of improvement in ADHD-RS-IV score from baseline to 8 weeks treatment were 83.3% (N=5) in the ITT population and 80.0% (N=4) in the PP population. Both low and high doses of PDC-1421 Capsule met the primary end points by passing the required 40% population in ADHD-RS-IV test scores. Overall, the results from this study, which demonstrate the therapeutic value of PDC-1421, support further Phase II Part II clinical development of ABV-1505 for the treatment of adult ADHD.
6
| IV. | ABV-1702 to treat Myelodysplastic syndromes (“MDS”) |
ABVC started the preparation for ABV-1702’s Phase II clinical trials after receiving its IND approval from the FDA in July 2016. ABVC plans to recruit fifty-two subjects in the United States who are diagnosed with either IPSS int-1, IPSS int-2 or high risk MDS or CMML and may take azacitidine as part of the subjects’ prescription. Azacitidine is an FDA-approved drug used to treat MDS. ABVC intends to administer ABV-1702 in the oral liquid form along with azacitidine. The Phase II trial is divided into two parts, where Part 1 is to determine the safety and recommended dose level (“RDL”) of ABV-1702 in combination with azacitidine and Part 2 is to determine whether ABV-1702 under the established RDL reduces bactericidal and fungicidal infection in the subjects’ respiratory systems. The primary endpoint of Part 1 Phase II trial is to assess the safety and RDL profile of ABV-1702 administered with azacitidine by measuring ABV-1702’s prohibited toxicity. The secondary endpoints of Phase II Part 1 are to determine the safety, time-to-first infection after first dose (Day 1) of the first azacitidine treatment cycle, reduction in treatment requirements and duration of infections, enhancement of immune responses, improvements of response rates, progression, and survival rates of the subjects under such ABV-1702 - azacitidine combination treatment. The primary endpoint of Part 2 of Phase II is to determine whether ABV-1702 under the established RDL reduces bactericidal and fungicidal infection risks in the subjects’ respiratory systems in combination with azacitidine as compared to the control group with incidence of infections and incidence/frequency of inpatient hospitalization due to infections. The secondary endpoints of Part 2 of Phase II are to determine the safety, time-to-first infection after first dose (Day 1) of the first azacitidine treatment cycle, reduction in required dosage and duration of infection, enhancement of immune responses, improvement of response rate, progression, and survival rates of the subjects under the trial conditions. In April 2016, BioLite submitted a letter to the FDA in response to its queries with additional information about the proposed Phase II trial.
As of the date of this prospectus, ABVC intends to commence the Phase II clinical trials of ABV-1702 in the fourth quarter of 2021 although neither BioLite nor ABVC can guaranty that the Phase II trial will be initiated as planned. Due to the scarcity of MDS cases, BioLite applied for the orphan drug designation for ABV-1702 or BLI-1301.
| V. | ABV-1703 Pancreatic Cancer |
ABVC developed a new indication for Pancreatic Cancer from Maitake Extract, which is named as ABV-1703 and out licensed it to Rgene for the preparation of its IND application with the FDA. On August 25, 2017, ABV-1703’s Phase II trial was approved by FDA. Pursuant to the ABVC-Rgene Co-development Agreement, ABVC is responsible for coordinating and conducting the clinical trials of ABV-1703 globally and Rgene is responsible for preparing the related FDA applications. As of the date of this prospectus, we are engaging Cedars-Sinai Medical Center in the U.S. to conduct the Phase II clinical trial and plan to initiate the Phase II trial in the third quarter of 2021. We plan to submit ABV-1703’s Phase II clinical trial IND to the Taiwan FDA after we commence the clinical trials in the United States. However, there is no guaranty that we would be able to launch the Phase II trials of ABV-1703 as planned in either the U.S. or Taiwan.
| VI. | ABV-1601 Treating Depression in Cancer Patients |
We developed the treatment of depression in cancer patient indication from the same API as ABV-1504. In addition, ABV-1601 shares the similar pharmaceutical mechanism of action of ABV-1504 in as much as ABV-1601 shows the potential of increasing the level of norepinephrine in human’s nervous system by inhibiting its reabsorption. Because of ABV-1601’s sufficient similarity with ABV-1504, the FDA approved our ABV-1601-001 clinical protocol under IND 112567 (the same IND as for ABV-1504) in December 2018.
For the Phase II trial, ABVC plans to recruit a maximum number of 54 cancer patients with depression, to whom ABVC intends to administer ABV-1601 oral capsules. ABVC is engaging the Principal Investigator at Cedars-Sinai Medical Center in the U.S. which designed a randomized, double-blind dose escalation study with a comparator-controlled group to assess the efficacy and safety profile of ABV-1601, primarily against Montgomery-Åsberg Depression Rating Scale (MADRS) total score. The primary endpoint of the Phase II trial is change in MADRS, Hospital Anxiety and Depression Scale (HADS), and subscales (HADS-A and HADS-D), and Clinical Global Impression Scale (CGI) total scores from baseline in patients taking PDC-1421 compared to the comparator. As of the date of this prospectus, the Part I of Phase II clinical protocol, which is an open trial, has been approved by Cedars-Sinai Medical Center IRB Committee. This study was initiated in the first quarter of 2021.
7
| VII. | ABV-1701 Vitreous Substitute for Vitrectomy and Collaboration Agreement with BioFirst |
On July 24, 2017, BriVision, one of our wholly-owned subsidiaries entered into a collaboration agreement (the “BioFirst Agreement”) with BioFirst, pursuant to which BioFirst granted BriVision the global license to co-develop BFC-1401 Vitreous Substitute for Vitrectom (“BFC-1401”) for medical purposes. BioFirst is a related party to the Company because BioFirst and YuanGene Corporation (“YuanGene”), the Company’s controlling shareholder, are under common control, being both controlled by the controlling beneficiary shareholder of YuanGene.
According to the BioFirst Agreement, we are to co-develop and commercialize BFC-1401 or ABV-1701 with BioFirst and are obligated to pay BioFirst $3,000,000 (the “Total Payment”) in cash or common stock of BriVision on or before September 30, 2018 in two installments. An upfront payment of $300,000, representing 10% of the Total Payment due under the Collaboration Agreement, was to be paid upon execution of the BioFirst Agreement. BriVision is entitled to receive 50% of the future net licensing income or net sales profit when ABV-1701 is sublicensed or commercialized. On June 30, 2019, the Company and BioFirst entered into a Stock Purchase Agreement (the “Purchase Agreement”), pursuant to which the Company will issue 428,571 shares of the Company’s common stock to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst in connection with the BioFirst Collaborative Agreement. For more information about the BioFirst Agreement and Purchase Agreement, please refer to the current reports on Form 8-K filed on July 24, 2017 and July 12, 2019.
On November 7, 2016, the application of Phase I clinical trial prepared and submitted by BioFirst was approved by the Human Research Ethics Committee, Australia (“HREC”), and on November 14, 2016, it was approved by the Therapeutic Goods Administration, Australia (“TGA”).
We successfully finished the Phase I clinical trial of ABV-1701 at Sydney Retina Clinic and Day Surgery, a clinic located in Sydney, Australia. This was the only site for this Phase I clinical trial. The trial started on November 17, 2016, and was completed with positive results in July 2018. The Protocol Title is “A Phase I, single center, safety and tolerability study of Vitargus in the treatment of Retinal Detachment.”
The primary endpoint of this Phase I clinical trial was to evaluate the safety and tolerability of a single intravitreal dose of Vitargus in patients as a vitreous substitute during vitrectomy surgery for retinal detachment. Intravitreal is a route of administration of a drug or other substance, in which the substance is delivered into the eyes. The secondary endpoint of this Phase I clinical trial is to assess retinal attachment and Virtagus degradation at day 90 and to assess best corrected visual acuity (“BVCA”) after vitrectomy surgery. BVCA refers to the best possible vision a person can achieve. The primary and second endpoints are required by HREC for the purpose of evaluation of our Phase I clinical trial application. We enrolled an aggregate number of 10 patient subjects in this trial. On November 17, 2016, we received the approval from the Data and Safety Monitoring Board for the first subject, and nine more subjects were enrolled thereafter. In this trial, Vitargus was injected into the vitreous cavity of vitrectomised eyes, whose vitreous gel was removed from the vitreous cavity after a vitrectomy surgery. On August 24, 2020, a full clinical study report (CSR) of ABV-1701 Phase I clinical trial was issued. The study results showed that ABV-1701 (Vitargus) was well-tolerated as a vitreous substitute without any apparent toxicity to ocular tissues. Further, there was no indication of an increased overall safety risk with Vitargus. For efficacy, participants showed significant improvement in visual acuity. The optical properties of Vitargus allowed the patients to see well and facilitated visualisation of the fundus immediately following surgery. In addition, since Vitargus set as a stable semisolid gel adhering to the retina, it maintained its position without requiring the patient to remain face-down following surgery.
We are currently planning the pivotal study for ABV-1701 and the necessary step to obtain the Premarket Approval for this medical device. The pivotal study for ABV-1701 is designed to be a multi-nation and multi-site clinical trial involving several countries, including Australia, the U.S.A., Japan, Thailand, Taiwan, and the People’s Republic of China. The pivotal study will be initiated in Australia in the third quarter of 2021.
Co-development Agreement with Rgene
On May 26, 2017, American BriVision Corporation entered into a co-development agreement (the “Co-Dev Agreement”) with Rgene Corporation (the “Rgene”), a related party under common control by controlling beneficiary shareholder of YuanGene Corporation and the Company. Pursuant to Co-Dev Agreement, BriVision and Rgene agreed to co-develop and commercialize ABV-1507 HER2/neu Positive Breast Cancer Combination Therapy, ABV-17 Pancreatic Cancer Combination Therapy and ABV-1527 Ovary Cancer Combination Therapy. Under the terms of the Co-Dev Agreement, Rgene is required to pay the Company $3,000,000 in cash or stock of Rgene with equivalent value by August 15, 2017. The payment is for the compensation of BriVision’s past research efforts and contributions made by BriVision before the Co-Dev Agreement was signed and it does not relate to any future commitments made by BriVision and Rgene in this Co-Dev Agreement. In addition to the $3,000,000, the Company is entitled to receive 50% of the future net licensing income or net sales profit earned by Rgene, if any, and any development costs shall be equally shared by both BriVision and Rgene.
8
By June 1, 2017, the Company had delivered all research, technical, data and development data to Rgene. Since both Rgene and the Company are related parties and under common control by a controlling beneficiary shareholder of YuanGene Corporation and the Company, the Company has recorded the full amount of $3,000,000 in connection with the Co-Dev Agreement as additional paid-in capital during the year ended September 30, 2017. During the year ended December 31, 2017, the Company received $450,000 in cash. On December 24, 2018, the Company received the remaining balance of $2,550,000 in the form of newly issued shares of Rgene’s Common Stock, at the price of NT$50 (approximately equivalent to $1.60 per share), for an aggregate number of 1,530,000 shares, which accounted for equity method long-term investment as of December 31, 2018. During the year ended December 31, 2018, the Company has recognized investment loss of $549. On December 31, 2018, the Company determined to fully write off this investment based on the Company’s assessment of the severity and duration of the impairment, and qualitative and quantitative analysis of the operating performance of the investee, adverse changes in market conditions and the regulatory or economic environment, changes in operating structure of Rgene, additional funding requirements, and Rgene’s ability to remain in business. All projects that have been initiated will be managed and supported by the Company and Rgene.
The Company and Rgene signed an amendment to the Co-Dev Agreement on November 10, 2020, pursuant to which both parties agreed to delete AB-1507 HER2/neu Positive Breast Cancer Combination Therapy and AB 1527 Ovary Cancer Combination Therapy and add ABV-1519 EGFR Positive Non-Small Cell Lung Cancer Combination Therapy and ABV-1526 Large Intestine / Colon / Rectal Cancer Combination Therapy to the products to be co-developed and commercialized. Other provisions of the Co-Dev Agreement remain in full force and effect.
BioKey
BioKey provides a variety of regulatory services tailored to the needs of its customers, which include proofreading and regulatory review of submission documents related to formulation development, clinical trials, marketed products, generics, nutraceuticals and OTC products and training presentations. In addition to supporting ABVC’s new drug development, ABVC’s CDMO SBU submits INDs, NDAs, ANDAs, and DMFs to the FDA, on behalf of clients, in compliance with new electronic submission guidelines of the FDA. ABVC provides regulatory consulting services for the entire lifecycle of its clients’ drug development projects.
GMP Manufacturing
ABVC owns a certified GMP manufacturing facility, through BioKey, that is qualified to deliver small quantities of drugs for use by its clients in clinical trials from Phase I to Phase III. The GMP facility can manufacture direct API or blend fill-in capsules, manual and automated encapsulation, wet granulation or tray drying process, tablet compression and coating process, packaging solid dosage forms for ANDA and IND submission.
ABVC manufacturing facility consists of the GMP suite, product development area, analytical laboratory, food processing area, caged GMP storage area, receiving area and two warehouses. The facility was established in December 2008 and received its first drug manufacturing license in June 2009. ABVC’s current drug manufacturing license allows it to manufacture drug products under IND for human clinical trials thereon until the expiration of such license on December 2, 2021.
Market Distribution Strategy
We focus primarily on developing botanical drugs, which are intended for use in the diagnosis, cure, mitigation or treatment of disease in humans. Together with our strategic partners, we plan to market, distribute and sell our drug products internationally once those drug candidates comply with the local authorities regulating drugs and foods. Currently, many countries follow the International Council for Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (the “ICH”) guidelines that are published by European Medicines to provide guidance on quality and safety of pharmaceutical development and new drug commercialization in Japan, the United States and Europe. All of our drug candidates first go through the United States FDA process for new drug development first and then seek regulatory approval from regulators equivalent to the FDA in the jurisdictions where we plan to distribute those candidates.
9
Intellectual Property
The new drug candidates are dependent on or are the subject of the following patents and patent applications.
|
No. |
Status | Patent No. | Patent
Starting Date |
Patent
|
Patent Name | Territory | Patent Owner(1)(2) | |||||||
| 1 | granted | 6911222 | 6/28/2005 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract, Part 1 | The U.S. | MPITDC | |||||||
| 2 | granted | 7175861 | 2/13/2007 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract, Part 2 | The U.S. | MPITDC | |||||||
| 3 | granted | 7179496 | 2/20/2007 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract, Part 3 | The U.S. | MPITDC | |||||||
| 4 | granted | 7223425 | 5/29/2007 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract, Part 4 | The U.S. | MPITDC | |||||||
| 5 | granted | 0001337647 | 1/31/2007 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | Italy | MPITDC | |||||||
| 6 | granted | CH693499 | 9/15/2003 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | Switzerland | MPITDC | |||||||
| 7 | granted | 10220149 | 4/26/2007 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | Germany | MPITDC | |||||||
| 8 | granted | GB2383951 | 6/7/2006 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | United Kingdom | MPITDC | |||||||
| 9 | granted | 4109907 | 6/6/2002 | 6/5/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | Japan | MPITDC | |||||||
| 10 | granted | FR2834643 | 7/18/2003 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | France | MPITDC | |||||||
| 11 | granted | I295576 | 4/11/2008 | 1/10/2022 | Anti-depression Pharmaceutical Composition Containing Polygala Extract | Taiwan | MPITDC | |||||||
| 12 | granted | DE202007003503 U1 | 8/23/2007 | 9/20/2026 | Novel Polygalatenosides and use thereof as an antidepressant agent | Germany | MPITDC | |||||||
| 13 | granted | 7531519 | 5/12/2009 | 9/20/2026 | Novel Polygalatenosides and use thereof as an antidepressant agent | The U.S. | MPITDC | |||||||
| 14 | granted | 4620652 | 11/20/2006 | 11/19/2026 | Novel Polygalatenosides and use thereof as an antidepressant agent | Japan | MPITDC | |||||||
| 15 | granted | I 314453 | 9/21/2006 | 9/20/2026 | Novel Polygalatenosides and use thereof as an antidepressant agent | Taiwan | MPITDC | |||||||
| 16 | granted | I389713 | 3/21/2013 | 10/13/2030 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute (3) | Taiwan | NHRI | |||||||
| 17 | granted | US 8197849 B2 | 6/12/2012 | 8/30/2030 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | The U.S. | NHRI |
10
| 18 | granted | AU 2011/215775 B2 | 4/17/2014 | 2/9/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | Australia | NHRI | |||||||
| 19 | granted | KR 10-1428898 | 8/4/2014 | 2/9/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | Korea | NHRI | |||||||
| 20 | granted | CA 2786911 (C) | 10/6/2015 | 2/10/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | Canada | NHRI | |||||||
| 21 | granted | WO2011100469 A1 | N/A(4) | N/A(4) | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | PCT | NHRI | |||||||
| 22 | granted | EP 2534200 | 4/8/2015 | 2/9/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | European Union (Germany, United Kingdom, France, Switzerland, Spain, Italy) | NHRI | |||||||
| 23 | granted | 特許第 5885349號 | 2/9/2011 | 2/9/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute | Japan | NHRI | |||||||
| 24 | granted | ZL 201180005494.7 | 12/24/2014 | 2/9/2031 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute(3) | China | NHRI | |||||||
| 25 | granted | HK1178188 | 3/6/2015 | 6/21/2030 | Cross-linked oxidized hyaluronic acid for use as a vitreous substitute(3) | Hong Kong (5) | NHRI | |||||||
| 26 | applied | US 16/936,032 | 9/4/2020 | 9/4/2040 | Polygala extract for the treatment of major depresive disorder | US | ABVC | |||||||
| 27 | applied | TW 109130285 | 9/4/2020 | 9/4/2040 | Polygala extract for the treatment of major depresive disorder | Taiwan | ABVC | |||||||
| 28 | applied | US17/120,965 | 12/20/2020 | 12/20/2040 | Polygala Extract for the Treatment of Attention Deficit Hyperactive Disorder | U.S. | ABVC | |||||||
| 29 | applied | TW 110106546 | 2/24/2021 | 2/24/2041 | Polygala Extract for the Treatment of Attention Deficit Hyperactive Disorder | Taiwan | ABVC |
| (1) | “MPITDC” stands for Medical and Pharmaceutical Industry Technology and Development Center, Taiwan. |
| (2) | “NHRI” stands for National Health Research Institutes, Taiwan. |
| (3) | The patent name is translated into English and the original patent name is written as “交联氧化透明质酸作为眼球玻璃体之替代物.” |
| (4) | The starting date and expiration date of patents under PTC are subject to the laws of the specific participating jurisdiction where the patent application is filed. We have subsequently submitted such patent to the jurisdictions listed in No.22 herein above. |
| (5) | NHRI has obtained standard patent in Hong Kong based on the registration of the patent (listed as No.24 herein) granted by the State Intellectual Property Office, People’s Republic of China. |
11
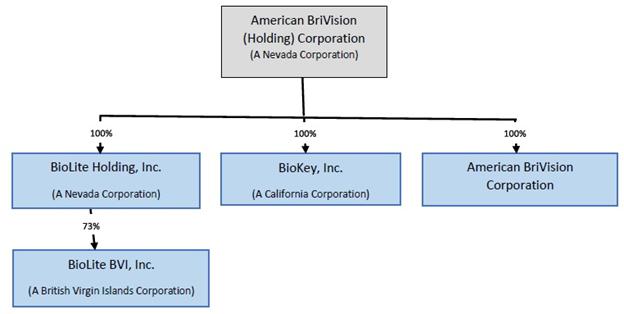
Corporate History and Structure
ABVC was incorporated under the laws of the State of Nevada on February 6, 2002 and has three wholly-owned Subsidiaries: BriVision, BioLite Holding, Inc. and BioKey, Inc. BriVision was incorporated in July 2015 in the State of Delaware and is in the business of developing pharmaceutical products in North America.
BioLite Holding was incorporated under the laws of the State of Nevada on July 27, 2016, with 500,000,000 shares authorized, par value $0.0001. Its key Subsidiaries include BioLite BVI, Inc. (“BioLite BVI”) that was incorporated in the British Virgin Islands on September 13, 2016 and BioLite Inc. (“BioLite Taiwan”), a Taiwanese corporation that was founded in February 2006. BioLite Taiwan has been in the business of developing new drugs for over twelve years. Certain shareholders of BioLite Taiwan exchanged approximately 73% of equity securities in BioLite Taiwan for the Common Stock in BioLite Holding in accordance with a share purchase/ exchange agreement (the “Share Purchase/ Exchange Agreement”). As a result, BioLite Holding owns via BioLite BVI approximately 73% of BioLite Taiwan. The other shareholders who did not enter this Share Purchase/ Exchange Agreement retain their equity ownership in BioLite Taiwan.
Incorporated in California on November 20, 2000, BioKey has chosen to initially focus on developing generic drugs to ride the opportunity of the booming industry.
Upon closing of the Mergers on February 8, 2019, BioLite and BioKey became two wholly-owned subsidiaries of ABVC.
The following chart illustrates the corporate structure of ABVC:
12
Competition
The healthcare industry is highly competitive and subject to significant and rapid technological change as researchers learn more about diseases and develop new technologies and treatments. Significant competitive factors in our industry include product efficacy and safety; quality and breadth of an organization’s technology; skill of an organization’s employees and its ability to recruit and retain key employees; timing and scope of regulatory approvals; the average selling price of products; the availability of raw materials and qualified manufacturing capacity; manufacturing costs; intellectual property and patent rights and their protection; and our capabilities of securing competent collaborators. Market acceptance of our current products and product candidates will depend on a number of factors, including: (i) potential advantages over existing or alternative therapies or tests, (ii) the actual or perceived safety of similar classes of products, (iii) the effectiveness of sales, marketing, and distribution capabilities, and (iv) the scope of any approval provided by the FDA or foreign regulatory authorities.
Since we are a small biopharmaceutical company compared to other companies that we may compete against, it is our intention to license our products to much larger pharmaceutical, specialty pharmaceutical and generic drug companies with the financial, technical and human resources to compete effectively in the markets we address.
We anticipate that our license partners will face intense and increasing competition when and as our new drug candidates enter the markets, as advanced technologies become available and as generic forms of currently branded products become available. Finally, the development of new treatment methods for the diseases we are targeting could render our products non-competitive or obsolete. There can be no assurance that any of our new drug candidates will be clinically superior or scientifically preferable to products developed or introduced by our competitors.
The following chart lists some, not all, of the biopharmaceutical companies that research, develop, commercialize, distribute or sell drugs that are in competition with our drug candidates.
|
Disease |
Drug Name | Pharmaceutical
Companies |
Headquarters | |||
| Major Depressive Disorder | Cymbalta oral | Eli Lilly and Co., Inc. | IN | |||
| Lexapro oral | Forest Laboratories, Inc. | NJ | ||||
| Pfizer Pharmaceuticals, Inc. | CT | |||||
| Attention-Deficit | Adderall XR | Shire Development LLC | MA | |||
| Hyperactivity Disease | Ritalin | Novartis Pharmaceuticals Corporation | NJ | |||
| Dexedrine | Amedra Pharmaceuticals LLC | PA | ||||
| Myelodysplastic | Vidaza | Celgene Corporation | NJ | |||
| Syndromes | Dacogen | Astex Pharmaceuticals, Inc. | CA | |||
| Triple Negative Breast Cancer | Avastin | Genentech, Inc. | CA | |||
| Erbitux (Cetuximab) | ImClone Systems Incorporated | NY | ||||
| Pancreatic Cancer | Abraxane, Abraxis BioScience LLC | Los Angeles | CA | |||
| Novartis Pharma Stein AG | Stein | Switzerland | ||||
| Vitargus for the treatments | Alcon Laboratories, Inc. | Fort Worth | TX | |||
of Retinal Detachment or Vitreous Hemorrhage |
Arcadophta | Toulouse | France |
13
Government Regulations
Currently, we are focusing on the research and development of six therapeutic candidates in the fields of CNS, oncology/hematology and autoimmune, for which regulatory approval must be received before we can commence marketing. In addition, our cGMP facility is subject to review by the FDA. Regulatory approval processes and FDA regulations for ABVC’s current and any future product candidates are discussed below.
Approval Process for Pharmaceutical Products
FDA Approval Process for Pharmaceutical Products
In the U.S., pharmaceutical products are subject to extensive regulation by the FDA. The Federal Food, Drug and Cosmetic Act (the “FDC Act”), and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending NDAs, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution. Pharmaceutical product development in the U.S. typically involves the performance of satisfactory nonclinical, also referred to as pre-clinical, laboratory and animal studies under the FDA’s Good Laboratory Practice, or GLP, regulation, the development and demonstration of manufacturing processes, which conform to FDA mandated current good manufacturing requirements, or cGMPs, including a quality system regulating manufacturing, the submission and acceptance of an IND application, which must become effective before human clinical trials may begin in the U.S., obtaining the approval of Institutional Review Boards, or IRBs, at each site where we plan to conduct a clinical trial to protect the welfare and rights of human subjects in clinical trials, adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought, and the submission to the FDA for review and approval of an NDA. Satisfaction of FDA requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Pre-clinical tests generally include laboratory evaluation of a product candidate, its chemistry, formulation, stability and toxicity, as well as certain animal studies to assess its potential safety and efficacy. Results of these pre-clinical tests, together with chemistry, manufacturing controls and analytical data and the clinical trial protocol, which details the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated, along with other requirements must be submitted to the FDA as part of an IND, which must become effective before human clinical trials can begin. The entire clinical trial and its protocol must be in compliance with what are referred to as good clinical practice, or GCP, requirements. The term, GCP, is used to refer to various FDA laws and regulations, as well as international scientific standards intended to protect the rights, health and safety of patients, define the roles of clinical trial sponsors and assure the integrity of clinical trial data.
14
An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the intended conduct of the trials and imposes what is referred to as a clinical hold. Pre-clinical studies generally take several years to complete, and there is no guarantee that an IND based on those studies will become effective, allowing clinical testing to begin. In addition to FDA review of an IND, each medical site that desires to participate in a proposed clinical trial must have the protocol reviewed and approved by an independent IRB or Ethics Committee, or EC. The IRB considers, among other things, ethical factors, and the selection and safety of human subjects. Clinical trials must be conducted in accordance with the FDA’s GCP requirements. The FDA and/or IRB may order the temporary, or permanent, discontinuation of a clinical trial or that a specific clinical trial site be halted at any time, or impose other sanctions for failure to comply with requirements under the appropriate entity jurisdiction.
Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap.
In Phase I clinical trials, a product candidate is typically introduced either into healthy human subjects or patients with the medical condition for which the new drug is intended to be used. The main purpose of the trial is to assess a product candidate’s safety and the ability of the human body to tolerate the product candidate. Phase I clinical trials generally include less than 50 subjects or patients.
During Phase 2 trials, a product candidate is studied in an exploratory trial or trials in a limited number of patients with the disease or medical condition for which it is intended to be used in order to: (i) further identify any possible adverse side effects and safety risks, (ii) assess the preliminary or potential efficacy of the product candidate for specific target diseases or medical conditions, and (iii) assess dosage tolerance and determine the optimal dose for Phase III trials.
Phase III trials are generally undertaken to demonstrate clinical efficacy and to further test for safety in an expanded patient population with the goal of evaluating the overall risk-benefit relationship of the product candidate. Phase III trials are generally designed to reach a specific goal or endpoint, the achievement of which is intended to demonstrate the candidate product’s clinical efficacy and adequate information for labeling of the approved drug.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the FDA’s threshold determination that it is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA has agreed to certain performance goals in the review of NDAs. Most applications for standard review drug products are reviewed within ten months; most applications for priority review drugs are reviewed within six months. Priority review can be applied to drugs that the FDA determines offer major advances in treatment, or provide a treatment where no adequate therapy exists. The review process for both standard and priority review may be extended by the FDA for three additional months to consider certain late-submitted information, or information intended to clarify information already provided in the submission. The FDA may also refer applications for novel drug products, or drug products which present difficult questions of safety or efficacy, to an advisory committee — typically a panel that includes clinicians and other experts — for review, evaluation, and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug is manufactured. The FDA will not approve the product unless compliance with cGMPs is satisfactory and the NDA contains data that provide substantial evidence that the drug is safe and effective in the indication studied.
After the FDA evaluates the NDA and the manufacturing facilities, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy, or REMS, to help ensure that the benefits of the drug outweigh the potential risks.
15
REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. The requirement for a REMS can materially affect the potential market and profitability of the drug. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing.
Post-Approval Regulations
Even if a product candidate receives regulatory approval, the approval is typically limited to specific clinical indications. Further, even after regulatory approval is obtained, subsequent discovery of previously unknown problems with a product may result in restrictions on its use or even complete withdrawal of the product from the market. Any FDA-approved products manufactured or distributed by us are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse events or experiences. Further, drug manufacturers and their subcontractors are required to register their establishments with the FDA and state agencies, and are subject to periodic inspections by the FDA and state agencies for compliance with cGMPs, which impose rigorous procedural and documentation requirements upon us and our contract manufacturers. ABVC cannot be certain that ABVC or its present or future contract manufacturers or suppliers will be able to comply with cGMPs regulations and other FDA regulatory requirements. Failure to comply with these requirements may result in, among other things, total or partial suspension of production activities, failure of the FDA to grant approval for marketing, and withdrawal, suspension, or revocation of marketing approvals.
If the FDA approves one or more of our product candidates, ABVC must provide certain updated safety and efficacy information. Product changes, as well as certain changes in the manufacturing process or facilities where the manufacturing occurs or other post-approval changes may necessitate additional FDA review and approval. The labeling, advertising, promotion, marketing and distribution of a drug must be in compliance with FDA and Federal Trade Commission, or FTC, requirements which include, among others, standards and regulations for direct-to-consumer advertising, off-label promotion, industry sponsored scientific and educational activities, and promotional activities involving the Internet. The FDA and FTC have very broad enforcement authority, and failure to abide by these regulations can result in penalties, including the issuance of a warning letter directing us to correct deviations from regulatory standards and enforcement actions that can include seizures, fines, injunctions and criminal prosecution.
Foreign Regulatory Approval
Outside of the U.S., ABVC’s ability to market our product candidates will be contingent also upon its receiving marketing authorizations from the appropriate foreign regulatory authorities, whether or not FDA approval has been obtained. The foreign regulatory approval process in most industrialized countries generally encompasses risks similar to those ABVC will encounter in the FDA approval process. The requirements governing conduct of clinical trials and marketing authorizations, and the time required to obtain requisite approvals, may vary widely from country to country and differ from those required for FDA approval.
ABVC will be subject to additional regulations in other countries in which we market, sell and import our products, including Canada. ABVC or its distributors must receive all necessary approvals or clearance prior to marketing and/or importing our products in those markets.
16
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the U.S., the Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety &Health Administration, the Environmental Protection Agency and state and local governments. In the U.S., sales, marketing and scientific/educational programs must also comply with state and federal fraud and abuse laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in the Health Care Reform Law, as amended by the Health Care and Education Affordability Reconciliation Act, or ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive recordkeeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines, imprisonment or other penalties, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
Employees
As of March 15, 2021, we, including the subsidiaries, have 34 employees, 30 of which are full-time, located in the U.S. and Taiwan.
17
Investing in our securities includes a high degree of risk. Prior to making a decision about investing in our securities, you should consider carefully the specific factors discussed below, together with all of the other information contained in this prospectus. If any of the following risks actually occurs, our business, financial condition, results of operations and future prospects would likely be materially and adversely affected. This could cause the market price of our Common Stock to decline and could cause you to lose all or part of your investment.
Risks Related to the Company’s Business
Unfavorable global economic conditions, including as a result of health and safety concerns, could adversely affect our business, financial condition or results of operations.
Our results of operations could be adversely affected by general conditions in the global economy, including conditions that are outside of our control, such as the impact of health and safety concerns from the current outbreak of the COVID-19 coronavirus (“COVID-19”). The spread of the COVID-19, which was declared a pandemic by the World Health Organization in March 2020, has caused different countries and cities to mandate curfews, including “shelter-in-place” and closures of most non-essential businesses as well as other measures to mitigate the spread of the virus.
The negative impact of COVID-19 on our operations is ongoing and the extent of which remains uncertain and potentially wide-spread, including:
| ● | our ability to successfully execute our long-term growth strategy during these uncertain times; |
| ● | our ability to recruit the necessary number of patients to complete future clinical trials; | |
| ● | supply chain disruptions in projects ABV-1504, ABV-1505 and ABV-1601, resulting from reduced workforces, scarcity of raw materials, and scrutiny or embargoing of goods produced in infected areas; |
| ● | our ability to perform on-site due-diligence for project ABV-1505 (MDD Phase II completed new drug candidate) and ABV-1701 (Vitargus FIH completed medical device) with our potential partners/collaborators in US, Mainland China, and Japan; |
| ● | our ability to access capital sources, as well as the ability of our key customers, suppliers, and vendors to do the same in regard to their own obligations; and |
| ● | diversion of management and employee attention and resources from key business activities and risk management outside of COVID-19 response efforts, including maintenance of internal controls. |
The COVID-19 pandemic remains highly volatile and continues to evolve on a daily basis and therefore, despite our efforts and developments to combat the virus, there can be no assurance that these measures will prove successful. The extent to which COVID-19 continues to impact the Company’s business, sales, and results of operations will depend on future developments, which are highly uncertain and cannot be predicted.
18
The Company is a development stage biopharmaceutical company and is thus subject to the risks associated with new businesses in that industry.
The Company acquired the sole licensing rights to develop and commercialize for therapeutic purposes six compounds from BioLite and the right to co-develop with BioFirst a medical device (collectively the “ABVC Pipeline Products”). As such, the Company is a clinical stage biopharmaceutical company with operations that generate unsubstantial revenues. The Company is establishing and implementing many important functions necessary to operate a business, including the clinical research and development of the ABVC Pipeline Products, further establishment of the Company’s managerial and administrative structure, accounting systems and internal financial controls
BioLite and BioKey are expected to continue to have limited revenue and remain unprofitable for an indefinite period of time.
Accordingly, you should consider the Company’s prospects in light of the risks and uncertainties that a pharmaceutical company with a limited operating history and revenue faces. In particular, potential investors should consider that there are significant risks that the Company will not be able to:
| ● | implement or execute its current business plan, or generate profits; |
| ● | attract and maintain a skillful management team; |
| ● | raise sufficient funds in the capital markets or otherwise to effectuate its business plan; |
| ● | determine that the processes and technologies that it has developed are commercially viable; and/or |
| ● | enter into contracts with commercial partners, such as licensors and suppliers. |
If any of the above risks occurs, the Company’s business may fail, in which case you may lose the entire amount of your investment in the Company. The Company cannot assure that any of its efforts in business operations will be successful or result in the timely development of new products, or ultimately produce any material revenue and profits.
As a pre-profit biopharmaceutical company, the Company needs to transition from a company with a research and development focus to a company capable of supporting commercial activities. The Company may not be able to reach such transition point or make such a transition, which would have affect our business, financial condition, results of operations and prospects.
If the Company fails to raise additional capital, its ability to implement its business model and strategy could be compromised.
The Company has limited capital resources and operations. The CDMO Unit generates limited amount of revenue that could partially support the operations of the Company. To date, its operations have been funded partially from the proceeds from financings or loans from its shareholders and management. From time to time, we may seek additional financing to provide the capital required to expand research and development (“R&D”) initiatives and/or working capital, as well as to repay outstanding loans if cash flow from operations is insufficient to do so. We cannot predict with certainty the timing or amount of any such capital requirements.
19
If the Company does not raise sufficient capital to fund its ongoing development activities, it is likely that it will be unable to carry out its business plans, including R&D development and expansion of production facilities. Even if the Company obtains financing for near term operations and product development, the Company may require additional capital beyond the near term. Furthermore, additional capital may not be available in sufficient amounts or on reasonable terms, if at all, and our ability to raise additional capital may be adversely impacted by potential worsening global economic conditions and the recent disruptions to and volatility in the credit and financial markets in the United States and worldwide resulting from the ongoing COVID-19 pandemic. If the Company is unable to raise capital when needed, its business, financial condition and results of operations would be materially adversely affected, and it could be forced to reduce or discontinue our operations.
The Company has no history in obtaining regulatory approval for, or commercializing, any new drug candidate.
With limited operating history, the Company has never obtained regulatory approval for, or commercialized, any new drug candidate. It is possible that the FDA may refuse to accept our planned New Drug Application (or “NDA”) for any of the six drug products for substantive review, or may conclude after review of our data that our application is insufficient to obtain regulatory approval of the new drug candidates or the medical device. Although our CDMO strategic business department has experience in obtaining abbreviated new drug application (or “ANDA”) approvals, the processes and timelines of obtaining an NDA approval and ANDA approval can differentiate substantially. If the FDA does not accept or approve our planned NDA for our product candidates, it may require that we conduct additional clinical, preclinical or manufacturing validation studies, which may be costly. Depending on the FDA required studies, approval of any NDA or application that we submit may be significantly delayed, possibly for several years, or may require us to expend more resources than we have. Any delay in obtaining, or inability to obtain, regulatory approvals of any of our drug candidate will prevent us from sublicensing such product. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the FDA. If any of these outcomes occurs, we may be forced to abandon our planned NDA for such drug candidate, which materially adversely affects our business and could potentially cause us to cease operations. We face similar regulatory risks in a foreign jurisdiction.
Our growth is dependent on our ability to successfully develop, acquire or license new drugs.
Our growth is supported by continuous investment in time, resources and capital to identify and develop new products or new formulations for the market and market penetration. If we are unable to either develop new products on our own or acquire licenses for new products from other parties, our ability to grow revenues and market share will be adversely affected. In addition, we may not be able to recover our investment in the development of new drugs and medical devices, given that projects may be interrupted, unsuccessful, not as profitable as initially contemplated or we may not be able to obtain necessary financing for such development. Similarly, there is no assurance that we can successfully secure such rights from third parties on an economically feasible basis.
20
Our current products have certain side effects. If the side effects associated with our current or future products are not identified prior to their marketing and sale, we may be required to withdraw such products from the market, perform lengthy additional clinical trials or change the labeling of our products, any of which could adversely impact our growth.
The Company researches and develops the following six drug products and one medical device: ABV-1501, ABV-1504, ABV-1505, ABV-1701, ABV-1702, ABV-1601 and ABV-1703. Each of these seven products may cause serious adverse effects to their users. For example, the API of ABV-1501, ABV-1702 and ABV-1703 is Maitake mushroom extract. Side effects, or adverse events, associated with Maitake mushroom extract include blood bilirubin increase, lymphocyte count decrease, neutrophil count decrease, platelet count decrease, white blood cell decrease, headache, and hyperglycemia. Serious adverse events (collectively, the “SAE”) associated with this compound include leukocytosis, platelet count decrease, eye disorders, abdominal pain, gastrointestinal disorders, aphonia, lung infection, muscle weakness right-sided, confusion, edema cerebral, stroke, dyspnea, wheezing, and pruritus.
ABV-1504 and ABV-1505 have the same API, “Radix Polygala”, which is known as Polygala tenuifolia Willd or PDC-1421 Capsule (“Polygala tenuifolia Willd”). Side effects, or adverse events, associated with ABV-1504 and ABV-1505, coming from administration of the trial medicine or examination procedure such as the procedure of taking blood (fainting, pain and/or bruising), may lead to gastrointestinal disorders (abdominal fullness and constipation), nervous system disorders (drowsiness, sleepiness, and oral ulcer). In addition, long-term use may cause miscarriages.
The safety and preliminary efficacy findings from this study, combined with the unique properties of ABV-1701, are supportive of further investigation for its use following vitrectomy surgery in patients requiring vitreous replacement. However, new serious side effects of ABV-1701 may be uncovered as the clinical trials continue.
The occurrence of any of those adverse events would harm our future sales of these medicines and substantially increase the costs and expenses of marketing these medicines, which in turn could cause our revenues and net income to decline. In addition, the reputation and sales of our future medicines could be adversely affected due to the severe side effects discovered.
We may be subject to product liability claims in the future, which could divert our resources, cause us to incur substantial liabilities and limit commercialization of any products that we may develop.
We face an inherent business risk of exposure to product liability claims in the event that the uses of our products are alleged to have caused adverse side effects. Side effects or marketing or manufacturing problems pertaining to any of our products could result in product liability claims or adverse publicity. These risks will exist for those products in clinical development and with respect to those products that receive regulatory approval for commercial sale. Furthermore, although we have not historically experienced any problems associated with claims by users of our products, we do not currently maintain product liability insurance and there could be no assurance that we are able to acquire product liability insurance with terms that are commercially feasible.
We face an inherent risk of product liability claims as a result of the clinical testing of our products and potentially commercially selling any products that we may develop. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidate. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our product candidates or products that we may develop; | |
| ● | injury to our reputation and significant negative media attention; |
21
| ● | withdrawal of clinical trial participants; |
| ● | significant costs to defend resulting litigation; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | loss of revenue; | |
| ● | reduced resources of our management to pursue our business strategy; and | |
| ● | the inability to commercialize any products that we may develop. |
We currently have insurance policies to cover liabilities under the clinic trials but do not maintain general liability insurance; and even if we have a general liability insurance in the future, this insurance may not fully cover potential liabilities that we may incur. The cost of any product liability litigation or other proceeding, even if resolved in our favor, could be substantial. We would need to increase our insurance coverage if and when we begin selling any product candidate that receives marketing approval. In addition, insurance coverage is becoming increasingly expensive. If we are unable to obtain or maintain sufficient insurance coverage at an acceptable cost or to otherwise protect against potential product liability claims, it could prevent or inhibit the development and commercial production and sale of our product candidate, which could adversely affect our business, financial condition, results of operations and prospects.
We have conducted, and may in the future conduct, clinical trials for certain of our product candidates at sites outside the United States, and the FDA may not accept data from trials conducted in such locations.
We have conducted and may in the future choose to conduct one or more of our clinical trials outside the United States. Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted and performed by qualified investigators in accordance with ethical principles. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. In addition, while these clinical trials are subject to the applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials also complied with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept the data from any of our clinical trials that we determine to conduct outside the United States, it would likely result in the need for additional trials, which would be costly and time-consuming and delay or permanently halt our development of the product candidate.
In addition, the conduct of clinical trials outside the United States could have a significant impact on us. Risks inherent in conducting international clinical trials include:
| ● | foreign regulatory requirements that could restrict or limit our ability to conduct our clinical trials; | |
| ● | administrative burdens of conducting clinical trials under multiple foreign regulatory schema; | |
| ● | foreign exchange fluctuations; and | |
| ● | diminished protection of intellectual property in some countries. |
If clinical trials of our product candidates fail to demonstrate safety and efficacy to the satisfaction of the FDA and comparable non-U.S. regulators, we may incur additional costs or experience delays in completing, or ultimately be unable to complete the development and commercialization of our product candidates.
We are not permitted to commercialize, market, promote or sell any product candidate in the United States without obtaining marketing approval from the FDA. Comparable non-U.S. regulatory authorities impose similar restrictions. We may never receive such approvals. We must complete extensive preclinical development and clinical trials to demonstrate the safety and efficacy of our product candidate in humans before we will be able to obtain these approvals.
22
Clinical testing is expensive, difficult to design and implement, can take many years to complete and is inherently uncertain as to outcome. Any inability to successfully complete preclinical and clinical development could result in additional costs to us and impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties. In addition, if (1) we are required to conduct additional clinical trials or other testing of our product candidate beyond the trials and testing that we contemplate, (2) we are unable to successfully complete clinical trials of our product candidate or other testing, (3) the results of these trials or tests are unfavorable, uncertain or are only modestly favorable, or (4) there are unacceptable safety concerns associated with our product candidate, we, in addition to incurring additional costs, may:
| ● | be delayed in obtaining marketing approval for our product candidates; |
| ● | not obtain marketing approval at all; |
| ● | obtain approval for indications or patient populations that are not as broad as we intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings; |
| ● | be subject to additional post-marketing testing or other requirements; or |
| ● | be required to remove the product from the market after obtaining marketing approval. |
Even if any of our product candidates receives marketing approval, it may fail to achieve the degree of market acceptance by physicians, patients, third party payors and others in the medical community necessary for commercial success and the market opportunity for the product candidate may be smaller than we estimate.
We have never completed a new drug or new medical device FDA application process from Phase I to FDA approval and commercialization. Even if our products are approved by the appropriate regulatory authorities for marketing and sale, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third party payors and others in the medical community. For example, physicians are often reluctant to switch their patients from existing therapies even when new and potentially more effective or convenient treatments enter the market. Further, patients often acclimate to the therapy that they are currently taking and do not want to switch unless their physicians recommend switching products or they are required to switch therapies due to lack of reimbursement for existing therapies.
The potential market opportunities for our products are difficult to estimate precisely. Our estimates of the potential market opportunities are predicated on many assumptions, including industry knowledge and publications, third party research reports and other surveys. While we believe that our internal assumptions are reasonable, these assumptions involve the exercise of significant judgment on the part of our management, are inherently uncertain and the reasonableness of these assumptions has not been assessed by an independent source. If any of the assumptions proves to be inaccurate, the actual markets for our products could be smaller than our estimates of the potential market opportunities.
We may seek to enter into collaborations with third parties for the development and commercialization of our product candidates. If we fail to enter into such collaborations, or such collaborations are not successful, we may not be able to capitalize on the market potential of our product candidates.
We may seek third-party collaborators for development and commercialization of our products. Our likely collaborators for any marketing, distribution, development, licensing or broader collaboration arrangements include large and mid-size pharmaceutical companies, regional and national pharmaceutical companies, non-profit organizations, government agencies, and biotechnology companies. Our ability to generate revenues from these arrangements will depend on our collaborators’ abilities to successfully perform the functions assigned to them in these arrangements.
23
Collaborations involving our products will pose the following risks to us:
| ● | collaborators may have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| ● | collaborators may not pursue development and commercialization of our product candidate or may elect not to continue or renew development or commercialization programs based on preclinical or clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors such as an acquisition that diverts resources or creates competing priorities; |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or require a new formulation of a product candidate for clinical testing; |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our product candidate if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
| ● | collaborators with marketing and distribution rights to one or more products may not commit sufficient resources to the marketing and distribution of such product or products; |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
| ● | collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; |
| ● | disputes may arise between the collaborators and us that result in the delay or termination of the research, development or commercialization of our product candidate or that result in costly litigation or arbitration that diverts management attention and resources; and |
| ● | collaborations may be terminated and, if terminated, may result in a need for additional capital to pursue further development or commercialization of the applicable product candidates. |
Collaborative agreements may not lead to development or commercialization of our product candidate in the most efficient manner or at all. If a collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program could be delayed, diminished or terminated.
ABVC, through BioLite, may not be able to receive the full amounts available under the collaboration agreement by and between BioLite, Inc. and BioHopeKing, which could increase its burden to seek additional capital to fund the business operations.
In February and December 2015, BioLite, Inc., a subsidiary of BioLite, entered into a total of three collaboration agreements with BioHopeKing to jointly develop ABV-1501 for TNBC (or BLI-1401-2 as used by BioLite internally) and ABV-1504 for MDD (or BLI-1005 as used by BioLite internally) in most Asian countries and BLI-1006, which has been later replaced with BLI-1008 for ADHD in Asia, excluding Japan. ABVC and BioLite are co-developing ABV-1501 for TNBC and ABV-1504 for MDD pursuant to the Collaboration Agreement and its Addendum entered by and between BriVision and BioLite Taiwan where ABVC and BriVision are responsible for the clinical trials of such two new drug candidates. In accordance with the terms of the BioHopeKing Collaboration Agreement for ABV-1501 or BLI-1401-2 and the Addendum thereto, BioLite shall receive payments of a total of $10 million in cash and equity of BioHopeKing or equity securities owned by it at various stages on a schedule dictated by BioLite’s achievements of certain milestones and twelve per cent (12%) of net sales of the drug products when ABV-1501 or BLI-1401-2 is approved for sale in the licensed territories. If BioLite fails to reach any of the milestones in a timely manner, it may not receive the rest of the payments from BioHopeKing. As a result of BioLite’s potential inability to receive the full payments under those collaboration agreements with BioHopeKing, ABVC may have to seek other sources of financing to fund its operation activities.
24
ABVC and its Subsidiaries may not be successful in establishing and maintaining additional strategic partnerships, which could adversely affect ABVC’s ability to develop and commercialize products, negatively impacting its operating results.
In addition to ABVC’s current collaboration with BioHopeKing for selected Asian markets, a part of its strategy is to evaluate and, as deemed appropriate, enter into additional partnerships in the future with major biotechnology or pharmaceutical companies. ABVC’s products may prove to be difficult to effectively license out as planned. Various regulatory, commercial and manufacturing factors may impact ABVC’s ability to seek co-developers of or grow revenues from licensing out any of the six products in the pipeline, none of which has been fully licensed out. Specifically, ABVC may encounter difficulty by virtue of:
| ● | its inability to effectively identify and align with commercial partners in the U.S. to collaborate the development of ABV-1504 for the treatment of Major Depressive Disorder, ABV-1505 to treat Attention-Deficit Hyperactivity Disease, ABV-1501 for the treatment of Triple Negative Breast Cancer, ABV-1703 to the treatment of Pancreatic Cancer, ABV-1601 to treat Depression in Cancer Patients and ABV-1702 to treat Myelodysplastic syndromes and ABV-1701 Vitargus for the treatments of Retinal Detachment or Vitreous Hemorrhage; |
| ● | its inability to secure appropriate contract research organizations (“CRO”s) to conduct data analysis, lab research and FDA communication; and |
| ● | its inability to effectively continue clinical studies on and secure positive research results of all of our investigational new drugs to attract additional commercial collaborators outside the U.S. |
ABVC faces significant competition in seeking appropriate partners for its therapeutic candidates, and the negotiation process is time-consuming and complex. In order for ABVC to successfully partner its autoimmune, CNS and hematology therapeutic candidates, as well as Vitargus, its medical device, potential partners must view these medicinal candidates as economically valuable in markets they determine to be attractive in light of the terms that ABVC is seeking and compared to other available products for licensing by other companies. Even if ABVC is successful in its efforts to establish new strategic partnerships, the terms that ABVC agrees upon may not be favorable, and it may not be able to maintain such strategic partnerships if, for example, development or approval of an autoimmune therapeutic is delayed or sales of an approved product are disappointing. Any delay in entering into new strategic partnership agreements related to any of ABVC’s therapeutic candidates could delay the development and commercialization of such candidates and reduce its competitiveness even if it reaches the market.
If ABVC fails to establish and maintain additional strategic partnerships or collaboration related to its therapeutic candidates that have not been fully licensed, it will bear all of the risk and costs related to the development of any such drug candidate, and it may need to seek additional financing, hire additional employees and otherwise develop expertise for which it has not budgeted. This could negatively affect the development of any incompletely partnered new drug candidates.
ABVC’s licensors may choose to terminate any of the license agreements with ABVC. As a result, ABVC’s research and development of the new drug candidate which contains the underlying API may be terminated abruptly.
If ABVC’s Subsidiary BioLite materially breaches any license agreements it has with Yukiguni Maitake Co. (“Yukiguni”), Medical and Pharmaceutical Industry Technology and Development Center (“MPITDC”) or Industrial Technology Research Institute (“ITRI”), or any of such license agreement terminates unexpectedly, BioLite may not be able to continue its research and development of the new drug candidate which contains the underlying API whose license has been terminated. Pursuant to the Yukiguni License Agreement, if BioLite fails to meet the milestone sales requirement or submit certain applications to the appropriate health authorities on a schedule prescribed therein, Yukiguni shall have the right to terminate the Yukiguni License Agreement. If the Yukiguni License Agreement is terminated involuntarily, BioLite will be forced to discontinue its new drug development of ABV-1702, ABV-1502 and ABV-1501 and terminate the collaboration agreements relating to the three new drug candidates. The termination of the right to use the underlying API will materially disrupt the operations of ABVC. Pursuant to the license agreement between BioLite Taiwan and ITRI, if BioLite Taiwan fails to complete the research submission milestones according to the schedule set forth therein without reasons or with reasons unstatisfied with ITRI, ITRI shall have the right to terminate the license agreement with BioLite Taiwan without refund to BioLite Taiwan. BioLite Taiwan and BioLite have submitted the IND for PDC-1421 and subsequently conducted Phase II clinical trials of two drug candidiates developed from PDC-1421 according to the schedule listed in the license agreement between BioLite Taiwan and MPITDC.
25
ABVC’s Subsidiary BioLite depends on one supplier for the API of ABV-1702, ABV-1502 and ABV-1501 and any failure of such supplier to deliver sufficient quantities of the API that meets its quality standard could have a material adverse effect on its research of these three drug candidates.
Currently BioLite relies primarily on Yukiguni, a Japanese supplier, to provide Yukiguni Maitake Extract 404, the API which is contained in ABV-1702, ABV-1502 and ABV-1501, three of the six drug candidates in BioLite’s oncology/hematology portfolio. It has entered into the Yukiguni License Agreement, among other things, for the delivery of Yukiguni Maitake Extract 404, which is patented in Japan and China. BioLite agrees to fulfill its demand of the Yukiguni Maitake Extract 404 by purchasing first from Yukiguni respecting the therapeutic products and Yukiguni represents that it will provide sufficient quantities of such API that meets cGMP standards. If the supplies of Yukiguni Maitake Extract 404 were interrupted for any reason, BioLite’s research and development activities of these three drug candidates could be delayed. These delays could be extensive and expensive, especially in situations where a substitution is not readily available.
Although BioLite may negotiate with other vendors that could provide Yukiguni Maitake Extract 404, it cannot guarantee that it will be able to find such vendors. Failure to obtain adequate supplies of high quality Yukiguni Maitake Extract 404 in a timely manner could have a disruptive effect on ABVC and BioLite’s research and development activities of ABV-1702, ABV-1502 and ABV-1501, resulting in a material adverse effect on the Company’s business, financial condition and results of operations.
ABVC may use hazardous chemicals and biological materials in its business. Any claims relating to improper handling, storage or disposal of these materials could be time consuming and costly.
ABVC’s research and development may involve the controlled use of hazardous materials, including chemicals and biological materials. ABVC cannot eliminate the risk of accidental contamination or discharge and any resulting injury from these materials. ABVC may be sued for any injury or contamination that results from its use or the use by third parties of these materials, and its liability may exceed any insurance coverage and its total assets. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these hazardous materials and specified waste products, as well as the discharge of pollutants into the environment and human health and safety matters. Although ABVC makes its best efforts to comply with environmental laws and regulations despite the associated high costs and inconvenience, ABVC cannot guarantee that it will not mishandle any hazardous materials in the future. If it fails to comply with these requirements or any improper handling of hazardous materials occurs, it could incur substantial costs, including civil or criminal fines and penalties, clean-up costs or capital expenditures for control equipment or operational changes necessary to achieve and maintain compliance. In addition, ABVC cannot predict the impact on its business of new or amended environmental laws or regulations or any changes in the way existing and future laws and regulations are interpreted and enforced.
The facilities where the samples of drug candidates are manufactured need to be maintained and monitored in compliance with the good manufacturing practice standards, the failure of such maintenance could contaminate the results of our clinical trials and adversely affect our operations.
ABVC’s Subsidiary BioKey operates a laboratory facility that is a certified good manufacturing practice facility (“cGMP”) and some of its contract clinical trial service providers use cGMP facilities to conduct clinical studies. ABVC cannot be certain that ABVC or its present or future contract manufacturers or suppliers will be able to comply with cGMPs regulations and other FDA regulatory requirements. Failure to comply with these requirements may result in, among other things, total or partial suspension of production activities, failure of the FDA to grant approval for marketing, and withdrawal, suspension, or revocation of marketing approvals.
26
Risks Related to Intellectual Property
Pharmaceutical patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to the Company, could negatively impact its respective licensors’ patent position and interrupt its research activities.
The patent positions of pharmaceutical companies and research institutions can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering pharmaceutical compositions may be uncertain and difficult to determine, and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the U.S. Patent and Trademark Office, or USPTO, are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to re-examination proceedings, post-grant review and/or inter parties review in the USPTO. Foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, re-examination, post-grant review, inter parties review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide the Company with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the U.S. and foreign countries may permit others to use discoveries of the Company or to develop and commercialize their new drug candidates without providing any compensation thereto, or may limit the number of patents or claims the Company can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending the intellectual property rights of the Company.
If the Company fails to obtain and maintain patent protection and trade secret protection of its respective products, the Company could lose their competitive advantages and competition it faces would increase, reducing any potential revenues and adversely affecting its ability to attain or maintain profitability.
Developments in patent law could have a negative impact on the Company’s Licensors’ patent positions and the Company’s business.
From time to time, the U.S. Supreme Court, other federal courts, the U.S. Congress or the USPTO may change the standards of patentability and any such changes could have a negative impact on the Company’s business.
In addition, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes the way issued patents are challenged, and changes the way patent applications are disputed during the examination process. These changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013. Substantive changes to patent law associated with the America Invents Act may affect the Company, BioLite and BioKey’s ability to obtain patents, and if obtained, to enforce or defend them. Accordingly, it is not clear what, if any, impact the America Invents Act will ultimately have on the cost of prosecuting the Company’s patent applications, its ability to obtain patents based on its discoveries and its ability to enforce or defend its patents.
27
If the Company is unable to protect the confidentiality of its trade secrets, its business and competitive position would be harmed, respectively.
In addition to patent protection, because the Company operates in the highly technical field of discovery and development of therapies, it relies in part on trade secret protection in order to protect its proprietary technology and processes. However, trade secrets are difficult to protect. The Company has entered into confidentiality and non-disclosure agreements with its employees, consultants, outside scientific and commercial collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties any confidential information developed by the party or made known to the party by the Company during the course of the party’s relationship therewith. These agreements also generally provide that inventions conceived by the party in the course of rendering services to the Company will be ABVC’s exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to the Company.
In addition to contractual measures, the Company tries to protect the confidential nature of its proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for the Company. The Company’s security measures may not prevent an employee or consultant from misappropriating its trade secrets and providing them to a competitor, and recourse it takes against such misconduct may not provide an adequate remedy to protect the Company’s interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the U.S. may be less willing to protect trade secrets. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by the Company. If the Company’s confidential or proprietary information, such as the trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, its competitive position could be harmed.
Third parties may assert that the Company’s employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
The Company might employ individuals who were previously employed at universities or other biopharmaceutical companies, including its competitors or potential competitors. Although through certain non-disclosure covenants and employment agreements with its officers and employees, the Company tries to ensure that its employees and consultants do not use the proprietary information or know-how of others in the work for the Company, the Company may be subject to claims that it or its employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If the Company fails in defending any such claims, in addition to paying monetary damages, the Company may lose valuable intellectual property rights or personnel. Even if the Company is successful in defending against such claims, litigation could result in substantial costs and be a distraction to the Company’s management and other employees.
28
ABVC’s ability to compete may decline if it does not adequately protect its proprietary rights or if it is barred by the intellectual property rights of others.
ABVC’s commercial success depends on obtaining and maintaining proprietary rights to its drug candidates as well as successfully defending these rights against third-party challenges. ABVC obtains its rights to use and research certain proprietary information to further develop the drug candidates primarily from three institutions, MPITDC, ITRI and Yukiguni (collectively the “Licensors”). These three institutions own the intellectual property rights in the products that have been licensed to us and may prosecute new patents of the drug candidates that are invented or discovered within the licensed scope of use under the respective license agreements. ABVC will only be able to protect its new drug candidates from unauthorized use by third parties to the extent that its valid and enforceable patents, or effectively protected trade secrets and know-how, cover them.
ABVC’s ability to obtain new patent protection for its new drug candidates is uncertain due to a number of factors, including that:
| ● | ABVC may not have been the first to make the inventions covered by pending patent applications or issued patents; |
| ● | ABVC may not have been the first to file patent applications for its new drug candidates; |
| ● | others may independently develop identical, similar or alternative products or compositions and uses thereof; |
| ● | ABVC’s disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| ● | any or all of ABVC’s pending patent applications may not result in issued patents; |
| ● | ABVC may not seek or obtain patent protection in countries that may eventually provide a significant business opportunity; |
| ● | any patents issued to ABVC may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties; |
| ● | ABVC’s methods may not be patentable; |
| ● | ABVC’s licensors may successfully challenge that ABVC’s new patent application fall outside the licensed use of the products; or |
| ● | others may design around ABVC’s patent claims to produce competitive products which fall outside of the scope of its patents. |
Even if ABVC has or obtains new patents covering its new drug candidates, ABVC may still be barred from making, using and selling them because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering products that are similar or identical to ABVC. There are many issued U.S. and foreign patents relating to therapeutic products and some of these relate to ABVC’s new drug candidates. These could materially affect ABVC’s ability to develop its drug candidates. Because patent applications can take many years to issue, there may be currently pending applications unknown to ABVC that may later result in issued patents that its new drug candidates may infringe. These patent applications may have priority over patent applications filed by ABVC.
The Company and its respective licensors may not be able to enforce their intellectual property rights throughout the world.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the U.S. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to pharmaceuticals and medical devices. This could make it difficult for the Company and its respective licensors to stop the infringement of some of the Licensors’ patents, or the misappropriation of their other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, the Company and its licensors have chosen in the past and may choose in the future not to seek patent protection in certain countries, and as a result the Company will not have the benefit of patent protection in such countries. Moreover, the Company may choose in the future not to seek patent protection in certain countries, and as a result it will not have the benefit of patent protection in such countries.
29
Proceedings to enforce the Company’s and its licensors’ patent rights in foreign jurisdictions could result in substantial costs and divert its efforts and attention from other aspects of the businesses. Accordingly, the efforts to protect the Company’s intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in the U.S. and foreign countries may affect the Company’s ability to obtain adequate protection for its technology and the enforcement of intellectual property.
Regulatory Risks Relating to Biopharmaceutical Business
The Company is subject to various government regulations.
The manufacture and sale of human therapeutic and diagnostic products in the U.S. and foreign jurisdictions are governed by a variety of statutes and regulations. These laws require approval of manufacturing facilities, controlled research and testing of products and government review and approval of a submission containing manufacturing, preclinical and clinical data in order to obtain marketing approval based on establishing the safety and efficacy of the product for each use sought, including adherence to current PIC/S Guide to Good Manufacturing Practice for Medicinal products during production and storage, and control of marketing activities, including advertising and labeling.
The products the Company is currently developing will require significant development, preclinical and clinical testing and investment of substantial funds prior to its commercialization. The process of obtaining required approvals can be costly and time-consuming, and there can be no assurance that future products will be successfully developed and will prove to be safe and effective in clinical trials or receive applicable regulatory approvals. Markets other than the U.S. have similar restrictions. Potential investors and shareholders should be aware of the risks, problems, delays, expenses and difficulties which we may encounter in view of the extensive regulatory environment which controls our business.
The Company cannot be certain that it will be able to obtain regulatory approval for, or successfully commercialize, any of its current or future product candidates.
The Company may not be able to develop any current or future product candidates. The Company’s new drug candidates will require substantial additional clinical development, testing, and regulatory approval before the commencement of commercialization. The clinical trials of the Company’s drug candidates are, and the manufacturing and marketing of our new drug candidates will be subject to extensive and rigorous review and regulation by numerous government authorities in the U.S. and in other countries where the Company intend to test and, if approved, market any new drug candidate. Before obtaining regulatory approvals for the commercial sale of any product candidate, the Company must demonstrate through pre-clinical testing and clinical trials that the product candidate is safe and effective for use in each target indication. This process can take many years and may include post-marketing studies and surveillance, which will require the expenditure of substantial resources. Of the large number of drugs in development in the U.S., only a small percentage successfully completes the FDA regulatory approval process and is commercialized. Accordingly, even if the Company is able to obtain the requisite financing to continue to fund its development and clinical programs, it cannot assure the investors that any of the product candidates will be successfully developed or commercialized.
The Company is not permitted to market a therapeutic product in the U.S. until it receives approval of an NDA or ANDA, for that product from the FDA, or in any foreign countries until they receive the requisite approval from such countries. Obtaining approval of an NDA is a complex, lengthy, expensive and uncertain process, and the FDA may delay, limit or deny approval of any product candidate for many reasons, including, among others:
| ● | Unable to demonstrate that a product candidate is safe and effective to the satisfaction of the FDA; |
| ● | the results of the Company’s clinical trials may not meet the level of statistical or clinical significance required by the FDA for marketing approval; |
30
| ● | the FDA may not approve the formulation of any product candidate; |
| ● | the CROs, that BioLite or the Company retains to conduct its clinical trials may take actions outside of its control that materially adversely impact its clinical trials; |
| ● | delays in patient enrollment, variability in the number and types of patients available for clinical trials, and lower-than anticipated retention rates for patients in clinical trials; |
| ● | the FDA may find the data from pre-clinical studies and clinical trials insufficient to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks, such as the risk of drug abuse by patients or the public in general; |
| ● | the FDA may disagree with the interpretation of data from the Company’s pre-clinical studies and clinical trials; |
| ● | the FDA may not accept data generated at the Company’s clinical trial sites; |
| ● | if an NDA, if and when submitted, is reviewed by an advisory committee, the FDA may have difficulties scheduling an advisory committee meeting in a timely manner or the advisory committee may recommend against approval of our application or may recommend that the FDA require, as a condition of approval, additional pre-clinical studies or clinical trials, limitations on approved labeling or distribution and use restrictions; |
| ● | the FDA may require development of a Risk Evaluation and Mitigation Strategy, or REMS, as a condition of approval or post-approval; or |
| ● | the FDA may change its approval policies or adopt new regulations. |
These same risks apply to applicable foreign regulatory agencies from which the Company, through BioLite, may seek approval for any of our new drug candidates.
Any of these factors, many of which are beyond the Company’s control, could jeopardize its ability to obtain regulatory approval for and successfully market any new drug candidate. As a result, any such setback in the Company’s pursuit of initial or additional regulatory approval would have a material adverse effect on its business and prospects.
If the Company does not successfully complete pre-clinical and Phase I and II clinical development, it will be unable to receive full payments under their respective collaboration agreements, find future collaborators or partners to take the drug candidates to Phase III clinical trials. Even if the Company successfully completes all Phase I and II clinical trials, those results are not necessarily predictive of results of additional trials that may be needed before an NDA for Phase III trials may be submitted to the FDA. Although there are a large number of drugs in development in the U.S. and other countries, only a very small percentage result in commercialization, and even fewer achieve widespread physician and consumer acceptance following the regulatory approval.
In addition, the Company may encounter delays or drug candidate rejections based on new governmental regulations, future legislative or administrative actions, or changes in FDA policy or interpretation during the period of product development. If the Company obtains required regulatory approvals, such approvals may later be withdrawn. Delays or failures in obtaining regulatory approvals may result in:
| ● | varying interpretations of data and commitments by the FDA and similar foreign regulatory agencies; and |
| ● | diminishment of any competitive advantages that such drug candidates may have or attain. |
31
Furthermore, if the Company fails to comply with applicable FDA and other regulatory requirements at any stage during this regulatory process, the Company may encounter or be subject to:
| ● | delays or termination in clinical trials or commercialization; |
| ● | refusal by the FDA or similar foreign regulatory agencies to review pending applications or supplements to approved applications; |
| ● | product recalls or seizures; |
| ● | suspension of manufacturing; |
| ● | withdrawals of previously approved marketing applications; and |
| ● | fines, civil penalties, and criminal prosecutions. |
The Company faces substantial competition from companies with considerably more resources and experience than the Company has, which may result in others discovering, developing, receiving approval for, or commercializing products before or more successfully than the Company.
The Company competes with companies that research, develop, manufacture and market already-existing and new pharmaceutical products in the fields of CNS, hematology/oncology and autoimmune. The Company anticipates that it will face increased competition in the future as new companies enter the market with new drugs and/or technologies and/or their competitors improve their current products. One or more of their competitors may offer new drugs superior to the Company’s and render the Company’s drugs uneconomical. A lot of the Company’s current competitors, as well as many of its respective potential competitors, have greater name recognition, more substantial intellectual property portfolios, longer operating histories, significantly greater resources to invest in new drug development, more substantial experience in product marketing and new product development, greater regulatory expertise, more extensive manufacturing capabilities and the distribution channels to deliver products to customers. If the Company is not able to compete successfully, it may not generate sufficient revenue to become profitable. The Company’s ability to compete successfully will depend largely on its ability to:
| ● | successfully commercialize its drug candidates with commercial partners; |
| ● | discover and develop new drug candidates that are superior to other products in the market; |
| ● | with its collaborators, obtain required regulatory approvals; |
| ● | attract and retain qualified personnel; and |
| ● | obtain patent and/or other proprietary protection for its product candidates. |
Established pharmaceutical companies devote significant financial resources to discovering, developing or licensing novel compounds that could make the Company’s products and product candidates obsolete. Our competitors may obtain patent protection, receive FDA approval, and commercialize medicines before we do. Other companies are or may become engaged in the discovery of compounds or botanical materials that may compete with the drug candidates the Company is developing.
The Company competes with a large number of well-established pharmaceutical companies that may have more resources than the Company does in developing therapeutics in the fields of CNS, oncology/hematology and ophthalmology.
32
Any new drug candidate the Company is developing or commercializing that competes with a currently-approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and/or safety in order to address price competition and be commercially successful. If the Company is not able to compete effectively against its current and future competitors, its business will not grow and its financial condition and operations will suffer.
Risks Relating to Doing Business Outside the United States
Because part of ABVC’s pharmaceutical research and development is conducted outside of the U.S., the Company is subject to the risks of doing business internationally, including periodic foreign economic downturns and political instability, which may adversely affect the Company’s revenue and cost of doing business in Taiwan.
ABVC collaborates with partners whose primary place of business is in Taiwan, Republic of China and the Company has certain key employees in Taiwan. Foreign economic downturns may affect our results of operations in the future. Additionally, other facts relating to the operation of the Company’s business outside of the U.S. may have a material adverse effect on the Company’s business, financial condition and results of operations, including:
| ● | international economic and political changes; |
| ● | the imposition of governmental controls or changes in government regulations, including tax laws, regulations and treaties; |
| ● | changes in, or impositions of, legislative or regulatory requirements regarding the pharmaceutical industry; |
| ● | compliance with U.S. and international laws involving international operations, including the Foreign Corrupt Practices Act and export control laws; |
| ● | difficulties in achieving headcount reductions due to unionized labor and works councils; |
| ● | restrictions on transfers of funds and assets between jurisdictions; and |
| ● | China- Taiwan geo-political instability. |
As the Company continues to operate its business globally, its success will depend in part, on its ability to anticipate and effectively manage these risks. The impact of any one or more of these factors could materially adversely affect the Company’s business, financial condition and results of operations.
The Company may be exposed to liabilities under the U.S. Foreign Corrupt Practices Act (“FCPA”) and Chinese anti-corruption law.
The Company is subject to the FCPA, and other laws that prohibit improper payments or offers of payments to foreign governments, foreign government officials and political parties by U.S. persons as defined by the statute for purposes of obtaining or retaining businesses. The Company may have agreements with third parties who may make sales in mainland China and the U.S., during the process of which the Company may be exposed to corruption. Activities in Taiwan create the risk of unauthorized payments or offers of payments by an employee, consultant or agent of the Company, because these parties are not always subject to the Company’s control.
Although the Company believes to date it has complied in all material aspects with the provisions of the FCPA and Chinese anti-corruption law, the existing safeguards and any future improvements may prove to be less than effective and any of the Company’s employees, consultants or agents may engage in corruptive conduct for which the Company might be held responsible. Violations of the FCPA or Chinese anti-corruption law may result in severe criminal or civil sanctions against the Company and individuals and therefore could negatively affect the Company’s business, operating results and financial condition. In addition, the Taiwanese government may seek to hold the Company liable as a successor for FCPA violations committed by companies in which the Company invests or acquires.
33
International operations expose the Company to currency exchange and repatriation risks, and the Company cannot predict the effect of future exchange rate fluctuations on its business and operating results.
The Company has business operations in Taiwan and collaborative activities in the U.S. and Japan. Substantial amounts of revenues are received and expenses are incurred in New Taiwan Dollars and U.S. dollars. Thus, the Company has exposure to currency fluctuations. The Company cannot assure you that the effect of currency exchange fluctuations will not materially affect its revenues and net income in the future.
We conduct our operations internationally and the effect of business, legal and political risks associated with international operations may seriously harm our business.
Sales to customers outside the United States accounted for 0% for the year ended December 31, 2020 and 2019. Our international sales and operations are subject to a wide range of risks, which may vary from country to country or region to region. These risks include the following:
| ● | export and import duties, changes to import and export regulations, and restrictions on the transfer of funds; |
| ● | political and economic instability; |
| ● | issues arising from cultural or language differences and labor unrest; |
| ● | longer payment cycles and greater difficulty in collecting accounts receivable; |
| ● | compliance with trade and technical standards in a variety of jurisdictions; |
| ● | difficulties in staffing and managing international operations, including the risks associated with fraud, theft and other illegal conduct; |
| ● | compliance with laws and regulations, including environmental, employment and tax laws, which vary from country to country and over time, increasing the costs of compliance and potential risks of non-compliance; |
| ● | difficulties enforcing our contractual and intellectual property rights, especially in those foreign countries that do not respect and protect intellectual property rights to the same extent as the United States and European countries; |
| ● | operations may be affected by political tensions, trade disputes and similar matters, particularly between China and Taiwan or between China and the United States; |
| ● | United States and foreign trade restrictions, including those that may limit the importation of technology or components to or from various countries or impose tariffs or quotas; and |
| ● | imposition of currency exchange controls or taxes that make it impracticable or costly to repatriate funds from foreign countries. |
We cannot assure you that risks relating to our international operations will not seriously harm our business.
If the Company becomes directly subject to the recent scrutiny, criticism and negative publicity involving U.S.-listed Chinese companies, we may have to expend significant resources to investigate and resolve the matters. Any unfavorable results from the investigations could harm our business operations, this offering and our reputation.
Recently, U.S. public companies that have substantially all of their operations in China, have been subjects of intense scrutiny, criticism and negative publicity by investors, financial commentators and regulatory agencies, such as the SEC. Much of the scrutiny, criticism and negative publicity has centered on financial and accounting irregularities, lack of effective internal control over financial accountings, inadequate corporate governance and ineffective implementation thereof and, in many cases, allegations of fraud. As a result of enhanced scrutiny, criticism and negative publicity, the publicly traded stocks of many U.S. listed Chinese companies have sharply decreased in value and, in some cases, have become virtually worthless or illiquid. Many of these companies are now subject to shareholder lawsuits and SEC enforcement actions and are conducting internal and external investigations into the allegations. It is not clear what effects the sector-wide investigations will have on the Company. If the Company becomes a subject of any unfavorable allegations, whether such allegations are proven to be true or untrue, the Company will have to expend significant resources to investigate such allegations and defend the Company. If such allegations were not proven to be baseless, the Company would be severely hampered and the price of the stock of the Company could decline substantially. If such allegations were proven to be groundless, the investigation might have significantly distracted the attention of the Company’s management.
34
Risks Related to the Company’s Financial Condition
Our existing indebtedness may adversely affect our ability to obtain additional funds and may increase our vulnerability to economic or business downturns.
We are subject to a number of risks associated with our indebtedness, including: 1) we must dedicate a portion of our cash flows from operations to pay debt service costs, and therefore we have less funds available for operations and other purposes; 2) it may be more difficult and expensive to obtain additional funds through financings, if available at all; 3) we are more vulnerable to economic downturns and fluctuations in interest rates, less able to withstand competitive pressures and less flexible in reacting to changes in our industry and general economic conditions; and 4) if we default under any of our existing credit facilities or if our creditors demand payment of a portion or all of our indebtedness, we may not have sufficient funds to make such payments. As of December 31, 2020, our outstanding current liabilities were approximately $4.8 million, which consisted primarily of short-term bank loans and accrued expenses. On April 5 and 20, 2020, we entered into certain exchange agreements separately with certain U.S. and non-U.S. holders of certain convertible promissory notes in the aggregate amount of $1,446,780; pursuant to the exchange agreements, we issued to the Holders an aggregate of 795,735 shares of Common Stock and warrants to purchase 795,735 shares of Common Stock. On November 9, 2020, we entered into an exchange agreement with a certain non-U.S. holder of certain convertible promissory notes in the amount of $270,272; pursuant to the exchange agreements, we will issue to the holder an aggregate of 120,121 shares of Common Stock and warrants to purchase 120,121 shares of Common Stock. On October 23, 2020, we entered into a securities purchase agreement with a certain accredited investor. Pursuant to such securities purchase agreement, we issued a convertible promissory note in principal amount of $2,500,000 with a maturity date of twenty-four (24) month anniversary from the issuance date. The convertible promissory note bears an interest rate of ten percent (10%) per annum and may be convertible into our shares of common stock at a fixed conversion price of $2.25 per share. We also agreed to issue an aggregate of 545,182 options of common stock to some of our employees in lieu of their deferred salaries in an aggregate amount of $1,090,360.
Our disclosure controls and procedures were not effective as of December 31, 2020 and as a result of such we do not expect that our disclosure controls and procedures will prevent all errors and all instances of fraud. The ineffective disclosure controls and procedures may lead to restatement of our financial statements, harm our operating results, subject us to regulatory scrutiny and sanction, cause investors to lose confidence in our reported financial information and have a negative effect on the market prices for our Common Stock.
Effective internal controls are necessary for us to provide reliable financial reports and effectively prevent fraud. We maintain a system of internal control over financial reporting, which is defined as a process designed by, or under the supervision of, our principal executive officer and principal financial officer, or persons performing similar functions, and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
We maintain disclosure controls and procedures designed to provide reasonable assurance that material information required to be disclosed by us in the reports we file or submit under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that the information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure. We performed an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered by this prospectus. Based on their evaluation, our management, including our Chief Executive Officer and Chief Financial Officer, concluded that our disclosure controls and procedures were not effective as of December 31, 2020.
35
We do not expect that our disclosure controls and procedures will prevent all errors and all instances of fraud. Disclosure controls and procedures, no matter how well conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the disclosure controls and procedures are met. Further, the design of disclosure controls and procedures must reflect the fact that there are resource constraints, and the benefits must be considered relative to their costs. Because of the inherent limitations in all disclosure controls and procedures, no evaluation of disclosure controls and procedures can provide absolute assurance that we have detected all our control deficiencies and instances of fraud, if any. The design of disclosure controls and procedures is also based partly on certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions.
Our articles of incorporation allow for our board to create new series of preferred stock without further approval by our shareholders, which could adversely affect the rights of the holders of our Common Stock.
Our Board of Directors has the authority to fix and determine the relative rights and preferences of preferred stock without shareholder approval. As a result, our Board of Directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation, the right to receive dividend payments before dividends are distributed to the holders of Common Stock and the right to the redemption of the shares, together with a premium, prior to the redemption of our Common Stock. In addition, our Board of Directors could authorize the issuance of a series of preferred stock that has greater voting power than our Common Stock or that is convertible into our Common Stock, which could decrease the relative voting power of our Common Stock or result in dilution to our existing shareholders.
We may create any additional series of preferred stock and issue such shares in the future although we do not have any present intention of doing so.
We may not be able to secure financing needed for future operating needs on acceptable terms, or on any terms at all.
From time to time, we may seek additional financing to provide the capital required to expand our production facilities, Research and development (“R&D”) initiatives and/or working capital, as well as to repay outstanding loans if cash flow from operations is insufficient to do so. We cannot predict with certainty the timing or amount of any such capital requirements. If such financing is not available on satisfactory terms, we may be unable to expand our business or to develop new business at the rate desired. If we are able to incur debt, we may be subject to certain restrictions imposed by the terms of the debt and the repayment of such debt may limit our cash flow and growth. If we are unable to incur debt, we may be forced to issue additional equity, which could have a dilutive effect on our current shareholders.
Our internal computer systems, or those of our third-party contractors or consultants, may fail or suffer security breaches, which could result in a material disruption of our product development programs.
Despite the implementation of security measures, our internal computer systems and those of our third-party contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we do not believe that we have experienced any such system failure, accident, or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a loss of clinical trial data for our new drug candidates which could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications or other data or applications relating to our technology or new drug candidates, or inappropriate disclosure of confidential or proprietary information, we could incur liabilities and the further development of our product candidates could be delayed.
36
The elimination of personal liability against our directors and officers under Nevada law and the existence of indemnification rights held by our directors, officers and employees may result in substantial expenses.
ABVC Bylaws eliminate the personal liability of our directors and officers to us and our shareholders for damages for breach of fiduciary duty as a director or officer to the extent permissible under Nevada law. Further, our Bylaws provide that we are obligated to indemnify each of our directors or officers to the fullest extent authorized by Nevada law and, subject to certain conditions, advance the expenses incurred by any director or officer in defending any action, suit or proceeding prior to its final disposition. Those indemnification obligations could expose us to substantial expenditures to cover the cost of settlement or damage awards against our directors or officers, which we may be unable to afford. Further, those provisions and resulting costs may discourage us or our shareholders from bringing a lawsuit against any of our current or former directors or officers for breaches of their fiduciary duties, even if such actions might otherwise benefit our shareholders.
Risks Related to the Company’s Common Stock
The share price of our Common Stock is volatile and may be influenced by numerous factors, some of which are beyond our control.
There is currently only a limited public market for our Common Stock, which is listed on the OTCQB Market, and there can be no assurance that a trading market will develop further or be maintained for our Common Stock in the future. The trading price of our Common Stock is likely to be highly volatile, and could be subject to wide fluctuations in response to various factors, some of which are beyond our control. In addition to the factors discussed in this “Risk Factors” section and elsewhere in this prospectus, these factors include:
| ● | the new drug candidates we acquire for commercialization; |
| ● | the product candidates we seek to pursue, and our ability to obtain rights to develop those product candidates; |
| ● | our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial; |
| ● | actual or anticipated adverse results or delays in our pre-clinical studies and clinical trials; |
| ● | our failure to get any of our new drug candidates approved; |
| ● | unanticipated serious safety and environmental concerns related to the use and research activities of any of our new drug candidates; |
| ● | overall performance of the equity markets and other factors that may be unrelated to our operating performance or the operating performance of our competitors, including changes in market valuations of similar companies; |
| ● | conditions or trends in the healthcare, biotechnology and pharmaceutical industries; |
| ● | introduction of new products offered by us or our competitors; |
| ● | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors; |
| ● | our ability to maintain an adequate rate of growth and manage such growth; |
| ● | issuances of debt or equity securities by us; |
| ● | sales of our securities by us or our shareholders in the future, or the perception that such sales could occur; |
| ● | trading volume of our Common Stock; |
| ● | ineffectiveness of our internal control over financial reporting or disclosure controls and procedures; |
| ● | general political and economic conditions in U.S. and other countries and territories where we conduct our business; |
| ● | effects of natural or man-made catastrophic events; and |
37
| ● | adverse regulatory decisions; |
| ● | additions or departures of key scientific or management personnel; |
| ● | changes in laws or regulations applicable to our product candidates, including without limitation clinical trial requirements for approvals; |
| ● | disputes or other developments relating to patents and other proprietary rights and our ability to obtain protection for our products; |
| ● | our dependence on third parties, including CROs and scientific and medical advisors; |
| ● | failure to meet or exceed any financial guidance or expectations regarding development milestones that we may provide to the public; |
| ● | actual or anticipated variations in quarterly operating results; |
| ● | failure to meet or exceed the estimates and projections of the investment community; |
| ● | other events or factors, many of which are beyond our control. |
In addition, the stock market in general, and the stocks of small-cap healthcare, biotechnology and pharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our Common Stock, regardless of our actual operating performance. The realization of any of the above risks or any of a broad range of other risks, including those described in these “Risk Factors,” could have a dramatic and material adverse impact on the market price of our Common Stock.
Insiders have substantial control over us, and they could delay or prevent a change in our corporate control even if our other shareholders wanted it to occur.
Our executive officers, directors, and principal shareholders own, in the aggregate, approximately 62.9% of our outstanding Common Stock. As a result of their stockholdings, these shareholders are able to assert substantial control over matters requiring shareholder approval, including the election of directors and approval of significant corporate transactions. This could delay or prevent an outside party from acquiring or merging with us even if our other shareholders wanted it to occur.
The market price of our Common Stock may be volatile and there may not be sufficient liquidity in the market for our securities in order for investors to sell their securities.
The market price of our Common Stock has been and will likely continue to be highly volatile, as is the stock market in general. Factors that may materially affect the market price of our Common Stock are beyond our control, these factors may materially adversely affect the market price of our Common Stock, regardless of our performance. In addition, the public stock markets have experienced extreme price and trading volume volatility. These broad market fluctuations may influence the market price of our Common Stock. There is currently only a limited public market for our Common Stock, which is listed on the OTCQB Market, and there can be no assurance that a trading market will develop further or be maintained in the future.
The stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies, including very recently in connection with the ongoing COVID-19 pandemic, which has resulted in decreased stock prices for many companies notwithstanding the lack of a fundamental change in their underlying business models or prospects. These fluctuations have often been unrelated or disproportionate to the operating performance of those companies. Broad market and industry factors, including potentially worsening economic conditions and other adverse effects or developments relating to the ongoing COVID-19 pandemic, political, regulatory and other market conditions, may negatively affect the market price of shares of our common stock, regardless of our actual operating performance. The market price of shares of our common stock may decline and you may lose some or all of your investment.
38
We have not paid dividends in the past and do not expect to pay dividends in the future, and any return on investment may be limited to the value of our shares.
We have never paid any cash dividends on our Common Stock and do not anticipate paying any cash dividends in the foreseeable future, and any return on investment may be limited to the value of our Common Stock. We plan to retain any future earnings to finance growth.
Under applicable Nevada law, we, as a Nevada corporation, generally may not make a distribution if i) we would not be able to pay our debts as they become due in the usual course of business, or ii) our total assets would be less than the sum of our total liabilities plus the amount that would be needed, if we were to be dissolved at the time of distribution, to satisfy the preferential rights upon dissolution of shareholders whose preferential rights are superior to those receiving the distribution.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and any trading volume could decline.
Any trading market for our Common Stock that may develop will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on us or our business. If no securities or industry analysts commence coverage of our company, the trading prices for our Common Stock could be negatively affected. If securities or industry analysts initiate coverage, and one or more of those analysts downgrade our stock or publish inaccurate or unfavorable research about our business, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our Common Stock could decrease, which might cause our stock price and any trading volume to decline.
Future sales and issuances of our Common Stock or rights to purchase Common Stock, including pursuant to our equity incentive plan or otherwise, could result in dilution of the percentage ownership of our shareholders and could cause our stock price to fall.
We expect that we will need significant additional capital in the future to continue our planned operations. To raise capital, we may sell Common Stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell Common Stock, convertible securities or other equity securities in more than one transaction, including issuance of equity securities pursuant to any future stock incentive plan to our officers, directors, employees and non-employee consultants for their services to us, investors in a prior transaction may be materially diluted by subsequent sales. Additionally, any such sales may result in material dilution to our existing shareholders, and new investors could gain rights, preferences and privileges senior to those of holders of our Common Stock. Further, any future sales of our Common Stock by us or resales of our Common Stock by our existing shareholders could cause the market price of our Common Stock to decline. Any future grants of options, warrants or other securities exercisable or convertible into our Common Stock, or the exercise or conversion of such shares, and any sales of such shares in the market, could have an adverse effect on the market price of our Common Stock.
Our Common Stock may be subject to the “penny stock” rules of the Securities and Exchange Commission, which may make it more difficult for shareholders to sell our Common Stock.
The SEC has adopted Rule 15g-9 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, the rules require that a broker or dealer approve a person’s account for transactions in penny stocks, and the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased.
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must obtain financial information and investment experience objectives of the person, and make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
39
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form sets forth the basis on which the broker or dealer made the suitability determination, and that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of the Company’s Common Stock if and when such shares are eligible for sale and may cause a decline in the market value of its stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stock.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable to us since we are not an accelerated filer, a large accelerated filer or a well-known seasoned issuer under SEC rules.
Our Subsidiary BioLite has its laboratories located in Hsinchu Biomedical Science Park, with an address of 20, Sec. 2, Shengyi Rd., 2nd Floor, Zhubei City, Hsinchu County 302, Taiwan (R.O.C.). On January 1, 2015, BioLite Taiwan entered into a lease agreement with the National Science Park Administrative Office (Hsinchu City) under which it rents two dormitory buildings in Hsinchu County, Taiwan for a period of five years. On January 1, 2020, BioLite Taiwan extended the contract for another five years. The new expiration date is on Dec 31, 2024. The rent increases by a small percentage each year during the term of the lease agreement. During the fiscal years of 2019 and 2018, BioLite paid approximately $50,038 and $43,619, respectively for the rent. In addition, BioLite leases two spaces as its laboratories in Hsinchu County, Taiwan. BioLite Taiwan and the National Science Park Administrative Office (Hsinchu City) entered into two five-year term leases which each commenced respectively on January 1, 2020. The aggregate leasing area amounts to approximately 678 square meters (equivalent to approximately 7,298 square feet) on the second floor of the building. The leased space counts for approximately 1.9% of the total space of the building. In the fiscal year of 2019 and 2018, BioLite incurred rental expenses relating the laboratory spaces in the amount of approximately $9,000 per month. BioLite paid $64,506 and $9,912 for the years ended December 31, 2020 and 2019, respectively.
Another of our Subsidiary BioKey is headquartered in Fremont, California. BioKey’s office lease will end on February 28, 2026 and the office occupies approximately 28,186 square feet. BioKey’s space consists of offices, research and production laboratories, and manufacturing facilities, which are GMP certified. BioKey has an option to extend the lease for its offices in Fremont for a period of five years commencing February 28, 2026, and BioKey may exercise this option for 5 more years. The total BioKey’s rental expenses were $403,776 and $306,876 for the years ended December 31, 2020 and 2019, respectively.
Unless disclosed otherwise, we are currently not a party to any material legal or administrative proceedings and are not aware of any pending legal or administrative proceedings against us. We may from time to time become a party to various legal or administrative proceedings arising in the ordinary course of our business.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable
40
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information. Our common stock, par value $.001 per share (the “Common Stock”), is currently quoted on the OTCQB under the symbol “ABVC”.
The following table sets forth the range of high and low bid quotations for our common stock. The quotations represent inter-dealer prices without retail markup, markdown or commission, and may not necessarily represent actual transactions.
| Quarter Ended | High Bid* | Low Bid* | ||||||
| 12/31/20 | 5.50 | 2.75 | ||||||
| 9/30/20 | 4.00 | 2.60 | ||||||
| 6/30/20 | 3.00 | 1.05 | ||||||
| 3/31/20 | 5.05 | 1.80 | ||||||
| 12/31/19 | 7.18 | 2.00 | ||||||
| 9/30/19 | 13.00 | 5.12 | ||||||
| 6/30/19 | 27.00 | 11.95 | ||||||
| 3/31/19 | 37.80 | 26.10 | ||||||
* as adjusted for the 1-for-18 reverse stock split effective on May 8, 2019.
Holders. As of March 15, 2021, we had approximately 711 shareholders of record of our common stock.
Dividends. Holders of our common stock are entitled to receive such dividends as may be declared by our board of directors. No dividends on our common stock have ever been paid, and we do not anticipate that dividends will be paid on our common stock in the foreseeable future.
Securities authorized for issuance under equity incentive plans.
Equity Compensation Plan Information
The following table discloses information as of December 31, 2020, with respect to compensation plans (including individual compensation arrangements) under which our equity securities are authorized for issuance, aggregated as follows:
| Plan category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted-average exercise price of outstanding options, warrants and rights | Shares of common stock remaining available for future issuance under equity compensation plans | |||||||||
| Equity compensation plans approved by security holders | 545,182 | $ | 2.00 | 2,812,949 | ||||||||
| Equity compensation plans not approved by security holders | - | - | - | |||||||||
| Total | 545,182 | $ | 2.00 | 2,812,949 | ||||||||
See, Item 11, Executive Compensation for additional details about our option plan.
41
Recent Sales of Unregistered Securities.
During the period covered by this report, the Company has not issued unregistered securities to any person, except as described below. None of these transactions involved any underwriters, underwriting discounts or commissions, except as specified below, or any public offering, and, unless otherwise indicated below, the Registrant believes that each transaction was exempt from the registration requirements of the Securities Act by virtue of Section 4(a)(2) thereof and/or Rule 506 of Regulation D promulgated thereunder, and/or Regulation S promulgated thereunder regarding offshore offers and sales. All recipients had adequate access, though their relationships with the Registrant, to information about the Registrant.
Between January 1, 2018 and August 6, 2019, the Company issued convertible notes of an aggregate amount of $800,000 to three non-U.S. investors for the Company’s general working capital purposes in reliance on an exemption from registration set forth in section 4(2) of the Securities Act of 1933, as amended.
On February 8, 2019, after the Merger, the Company issued 74,997,546 shares to the shareholders of BioLite and 29,561,231 shares to the shareholders of BioKey.
On June 30, 2019, the Company entered into a Stock Purchase Agreement with BioFirst, pursuant to which the Company agreed to issue 428,571 shares of the Company’s common stock to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst. These common shares have been issued during the year ended December 31, 2019.
On August 1, 2019, the Company entered into certain agreements to convert certain related party debts in an aggregate amount of $4,872,340 into shares of our common stock at a conversion price of $7.00 per share, which are being issued as of the date of this prospectus.
On April 5, 2020 and April 20, 2020, the Company entered into certain exchange agreements separately with certain U.S. and non-U.S. holders who are holders of certain convertible promissory notes issued by the Company in the aggregate amount of $1,446,780. Pursuant to the exchange agreements, the Company agreed to issue to the Holders an aggregate of 795,735 shares of the Company’s common stock, and warrants to purchase 795,735 shares of common stock.
Each warrant is exercisable upon issuance and expires three years from the date of issuance. The initial exercise price of the warrant is $5.00, subject to stock, splits, stock dividend and other similar events. In addition, when the closing price of the common stock equals or exceeds $9.00 per share for twenty Trading Days (as defined in the exchange agreements) during any thirty-day period, the Company shall have the right to require the holders to exercise all or any portion of the note holders’ warrants for a cash exercise. On September 30, 2020, the Company has issued such note holders’ shares warrants to the holders and closed the transactions contemplated by the Exchange Agreements. The note holder’s shares and warrants were exempt from the registration requirements of the Securities Act, pursuant to Regulation S and Regulation D promulgated thereunder.
In May 2020, the Company received capital contributions of approximately $1,602,040 in cash from 40 investors through private placements of the sale of certain number of Common Stocks for the purchase price of $2.25 per share of Common Stock and a free warrant attaches with each Common stock that was purchased. The exercise price of the warrant is at $6.00 per common stock with a mandatory redemption at $9.00 per common stock pursuant to the terms and conditions of the warrants.
On July 8, 2020, the Company entered an agreement with View Trade Securities Inc. (“ViewTrade”) to engage ViewTrade as the placement agent and the Company’s advisor with respect to its ongoing capital events. Pursuant to the agreement, the Company agreed to pay View Trade (“ViewTrade Securities”) 60,000 restricted common shares of the Company and 60,000 warrants to purchase common shares of the Company at an exercise price of $6 per share for a period of 5 years with cashless exercise provision.
As of December 31, 2020, the Company has issued 60,000 shares of common stock to ViewTrade for the advisory services with an estimated value of $135,000. The warrants were never issued and the parties mutually agreed to terminate the agreement on November 19, 2020. As a termination fee, the Company agreed to issue ViewTrade 50,000 restricted common shares of the Company.
Also on November 19, 2020, the Company and ViewTrade agreed to a new Advisory agreement under which ViewTrade was engaged to provide advisory services only. In addition to a retainer fee, the Company agreed to issue 200,000 warrants, with an exercise price of $2.25, an industry standard cashless exercise provision, and a term of 5 years from November 19, 2020.
42
On September 30, 2020, the Company also issued to Ever Adventure inv. (Formosa) Consultant Co., Ltd. (or its designee), Jinwei International Co., Ltd. (or its designee), and Thalia Media Ltd. (or its designee) (the “Consultants”) 120,000 shares, 180,000 shares and 120,000 shares of common stock (collectively, “Consultants’ Shares”), respectively, as their compensation as the Company’s investor relations and business development advisors. Each Consultant has entered into certain consulting agreement with the Company.
On September 30, 2020, the Company issued an aggregate of 795,735 shares of Common Stock to five previous note holders, who had converted their outstanding principals and accrued and unpaid interests during the nine months ended September 30, 2020.
On November 8, 2020, the Company entered into an exchange agreement with a holder of convertible promissory notes issued by the Company in the aggregate amount of $270,272. Pursuant to the exchange agreements, the Company agreed to issue to the Holder an aggregate of 120,121 shares of the Company’s common stock, and warrants to purchase 120,121 shares of common stock. On December 31, 2021, the Company issued an aggregated of 120,121 shares of Common Stock to the note holder.
On November 11, 2020, the Company conducted a closing with regard to certain securities purchase agreements (the “SPAs”) dated October 23, 2020, separately with two non-U.S. investors (the “Investors”). Each of the Investors agreed to purchase and the Company agreed to sell to each of the Investors 1,111,112 shares of the Company’s common stock, and warrants to purchase 1,111,112 shares of common stock, for a purchase price of $2,500,000. The warrants are exercisable upon issuance and expires three years from the date of issuance. The initial exercise price of the warrants is $6.00, subject to stock, splits, stock dividend and other similar events. In addition, when the closing price of the common stock equals or exceeds $9.00 per share for twenty Trading Days (as defined in the exchange agreements) during any thirty-day period, the Company shall have the right to require the investors to exercise all or any portion of the warrants for a cash exercise. The aggregate net proceeds of the Offering were $5,000,000. The Company and the investors further agreed to amend the terms of the SPA to permit the closing of the offering to occur on a rolling basis.
During the year ended December 31, 2020, the Company entered into consulting agreements with four service providers for consulting and advisory services, pursuant to which the Company agreed to pay the service fee by issuing 521,887 shares of unrestricted common shares, valued at the closing price from $2 to $3.68 per share on the grant date. As of December 31, 2020, these shares have been issued.
During the year ended December 31, 2019, the Company entered into service agreements with Euro-Asia Investment & Finance Corp Ltd. (a related party), Ever Adventure inv. (Formosa) Consultant Co., Ltd., New Eastern Asia (a related party),and Kimho Consultants Co., Ltd. (a related party) for the maintenance of the listing in the U.S. stock exchange market, investor relations, and business development. Pursuant to the agreements, the Company issued 644,972 shares of the Company’s common stock for the consulting service from July 2019 to July 2024 for the service fee of $4,514,800 in aggregate, and recorded as stock subscription receivable. As of December 31, 2020 and 2019, stock subscription receivable was $3,160,360 and $4,063,320, respectively.
The Company paid the following fees to a FINRA member firm in connection with the Offering: (i) a cash success fee of $175,000 and (ii) warrants to purchase a number of shares of Common Stock equal to 7% of the number of shares of Common Stock sold in this offering, at an exercise price per share equal to $6.00 subject to adjustment (the “Comp Warrants”). The Comp Warrants are exercisable on a cashless basis, at the holder’s discretion
ITEM 6. SELECTED FINANCIAL DATA
Not applicable since we are a smaller reporting company as defined under the applicable SEC rules.
43
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Caution Regarding Forward-Looking Information
The following discussion and analysis of our financial condition and result of operations should be read in conjunction with our audited consolidated financial statements and the notes to those financial statements appearing elsewhere in this Form 10-K. This discussion contains forward-looking statements and involves numerous risks and uncertainties contained in this report and the other reports we file with the Securities and Exchange Commission. Our actual results may differ materially from those contained in any forward-looking statements.
Overview
From its inception, the Company has not generated substantial revenue from its medical device and new drug development. For the twelve months ended December 31, 2020, the Company generated $483,045 in revenue, mainly from the sale of Contract Development & Manufacturing Organization (“CDMO”) services.
Business Overview
American BriVision Corporation, which was incorporated in July 2015 in the State of Delaware, is a clinical stage biopharmaceutical company focused on development of new drugs and medical devices, all of which are derived from plants.
Medicines derived from plants have a long history of relieving or preventing many diseases and, typically, have exhibited fewer side effects than drugs developed from animals or chemical ingredients. Perhaps the most famous example is aspirin, which evolved from a compound found in the bark and leaves of the willow tree and was later marketed by Bayer starting in 1899. Aspirin has very few serious side effects and has proven to be one of the most successful drugs in medical history. Some 50 years later, scientists identified anticancer compounds in the rosy periwinkle, which Eli Lilly subsequently produced for the treatment of leukemia and Hodgkins disease. Other well-known examples of successful botanical drugs include the cancer-fighting Taxol, isolated from the Pacific yew tree.
The Company develops its pipeline by carefully tracking new medical discoveries or medical device technologies in research institutions in the Asia-Pacific region. Pre-clinical, disease animal model and Phase I safety studies are examined closely by the Company’s scientists and other specialists known to the Company to identify drugs that it believes demonstrate efficacy and safety based on the Company’s internal qualifications. Once a drug is shown to be a good candidate for further development and ultimately commercialization, BriVision licenses the drug or medical device from the original researchers and begins to introduce the drugs clinical plan to highly respected principal investigators in the United States, Australia and Taiwan. In almost all cases, we have found that research institutions in each of those countries are eager to work with the Company to move forward with Phase II clinical trials.
44
Currently, institutions conducting phase II clinical trials in partnership with ABVC include:
| ● | Medical Device: ABV-1701, Vitargus® in vitrectomy surgery, Pivotal Study in Australia, Principal Investigator: Andrew Chang, MD, Ph.D., Sydney Eye Hospital, Australia |
| ● | Drug: ABV-1504, Major Depressive Disorder (MDD), Phase II, NCE drug Principal Investigators: Charles DeBattista M.D. and Alan F. Schatzberg, MD, Stanford University Medical Center, Cheng-Ta Li, MD, Ph.D – Taipei Veterans General Hospital |
| ● | Drug: ABV-1505, Adult Attention-Deficit Hyperactivity Disorder (ADHD), Phase II, NCE drug Principal Investigators: Keith McBurnett, Ph.D. and Linda Pfiffner, Ph.D., University of California San Francisco (UCSF), School of Medicine |
| ● | Drug: ABV-1601, Major Depression in Cancer Patients, Phase I/II, NCE drug Principal Investigator: Scott Irwin, MD, Ph.D. – Cedars Sinai Medical Center (CSMC) |
| ● | Drug: ABV-1703, Advanced Inoperable or Metastatic Pancreatic Cancer, Phase II, NCE drug Principal Investigator: Andrew E. Hendifar, MD – Cedars Sinai Medical Center (CSMC) |
| ● | Drug: ABV-1501, A Phase I/II, Open Label Study to Evaluate the Safety and Efficacy of BLEX 404 Oral Liquid Combined with Docetaxel Monotherapy in Patients with Stage IV or Recurrent Breast Cancer Patients |
| ● | Medical Device: ABV-2002, Class I/II through 510K for market launch, Corneal Storage Media, Technology Licensing in progress |
Upon successful completion of the Phase II trial, the Company will seek a partner – a large pharmaceutical company – to complete a Phase III study, submit the New Drug Application (NDA), and commercialize the drug upon approval by the FDA and Taiwan FDAs. The Company expects to seek its first commercialization partner in 2021 for Vitargus, its vitreous substitute that helps to maintain a round shape and retinal location during vitrectomy surgery.
Another part of the Company’s business is conducted by BioKey, a wholly owned subsidiary, that is engaged in a wide range of services, including, API characterization, pre-formulation studies, formulation development, analytical method development, stability studies, IND/NDA/ANDA/510K submissions, and manufacturing clinical trial materials (phase I through phase III) and commercial manufacturing.
On February 8, 2019, the Company, BioLite Holding, Inc. (“BioLite”), BioKey, Inc. (“BioKey”), BioLite Acquisition Corp., a direct wholly-owned subsidiary of the Company (“Merger Sub 1”), and BioKey Acquisition Corp., a direct wholly-owned subsidiary of the Company (“Merger Sub 2”) (collectively referred to as the “Parties”) completed the business combination pursuant to that certain Agreement and Plan of Merger (the “Merger Agreement”), dated January 31, 2018, pursuant to which the Company acquired BioLite and BioKey via issuing shares of the Company’s Common Stock to the shareholders of BioLite and BioKey. As a result, BioLite and BioKey became two wholly-owned subsidiaries of the Company on February 8, 2019. The Company issued an aggregate of 104,558,777 shares of Common Stock (prior to the reverse stock split in 2019) to the shareholders of both BioLite and BioKey under a registration statement on Form S-4 (file number 333-226285), which became effective by operation of law on or about February 5, 2019.
45
BioLite was incorporated under the laws of the State of Nevada on July 27, 2016, with 500,000,000 shares authorized, par value $0.0001. BioLite’s key subsidiaries include BioLite BVI, Inc. (“BioLite BVI”) that was incorporated in the British Virgin Islands on September 13, 2016 and BioLite, Inc. (“BioLite Taiwan”), a Taiwanese corporation that was founded in February 2006. BioLite Taiwan has been in the business of developing new drugs for over ten years.
BioLite and BioLite BVI are holding companies and have not carried out substantive business operations of their own.
In January 2017, BioLite, BioLite BVI, BioLite Taiwan, and certain shareholders of BioLite Taiwan entered into a share purchase / exchange agreement (the “BioLite Share Purchase / Exchange Agreement”). Pursuant to the BioLite Share Purchase / Exchange Agreement, the shareholder participants to the BioLite Share Purchase / Exchange Agreement sold their equity in BioLite Taiwan and used the proceeds from such sales to purchase shares of Common Stock of BioLite at the same price per share, resulting in their owning the same number of shares of Common Stock as they owned in BioLite Taiwan. Upon closing of the Share Purchase/ Exchange Agreement in August 2017, BioLite owns, via BioLite BVI, approximately 73% of BioLite Taiwan. The other shareholders who did not enter this Share Purchase/ Exchange Agreement retain their equity ownership in BioLite Taiwan.
BioKey was incorporated on August 9, 2000 in the State of California. It is engaged primarily in research and development, manufacturing, and distribution of generic drugs and nutraceuticals with strategic partners. BioKey provides a wide range of services, including, API characterization, pre-formulation studies, formulation development, analytical method development, stability studies, IND/NDA/ANDA/510K submissions, and manufacturing clinical trial materials (phase 1 through phase 3) and commercial manufacturing. It also licenses out its technologies and initiates joint research and development processes with other biotechnology, pharmaceutical, and nutraceutical companies.
Common Stock Reverse Split
On March 12, 2019, the Board by unanimous written consent in lieu of a meeting approved to i) effect a stock reverse split at the ratio of 1-for-18 (the “Reverse Split”) of both the authorized common stock of the Company and the issued and outstanding common stock and ii) to amend the articles of incorporation of the Company to reflect the Reverse Split. The Board approved and authorized the Reverse Split without obtaining approval of the Company’s shareholders pursuant to Section 78.207 of Nevada Revised Statutes.
On May 3, 2019, the Company filed a certificate of amendment to the Company’s articles of incorporation (the “Amendment”) to effect the Reverse Split with the Secretary of State of the State of Nevada. The Reverse Split took effect on May 8, 2019.
Series A Convertible Preferred Stock
On June 28, 2019, the Company filed a certificate of designation (the “Series A COD”) of Series A Convertible Preferred Stock (the “Series A Stock”) with the Secretary of the State of Nevada.
Pursuant to the Series A COD, the Company designated 3,500,000 shares of preferred stock as Series A Stock, par value of $0.001 per share. Subject to the laws of Nevada, the Company will pay cumulative dividends on the Series A Stock on each anniversary from the date of original issue for a period of four calendar years. The Series A Stock will rank senior to the outstanding common stock of the Company, par value $0.001 (the “Common Stock”) with respect to dividend rights, rights upon liquidation, dissolution or winding up in the amount of accrued but unpaid dividend. Holders of the Series A Stock will have the same voting rights as the Company’s Common Stock holders. Each share of Series A Stock is initially convertible at any time at the option of the holder into one share of Common Stock and automatically converts into one share of Common Stock on the four-year anniversary of its issuance.
As of December 31, 2020, no Series A Convertible Preferred Stock has been issued by the Company.
Increasing the Authorized Shares
On March 12, 2020, our board of directors approved and adopted an amendment to the Company’s Articles of Incorporation, to increase the authorized shares of the common stock, par value $0.001 per share, from 20,000,000 to 100,000,000, such that, after including the previously authorized 20,000,000 shares of preferred stock, par value $0.001 per share, the aggregate number of shares of stock that the Company has authority to issue is 120,000,000 shares. The amendment became effective on April 2, 2020.
46
Recent Financing
On November 11, 2020, we conducted a closing with regard to certain securities purchase agreements (the “SPAs”) dated October 23, 2020, separately with two non-U.S. investors (the “Investors”). Each of the Investors agreed to purchase and the Company agreed to sell to each of the Investors 1,111,112 shares of the Company’s common stock, par value $0.001 (“Common Stock”), and warrants (the “Warrants”) to purchase 1,111,112 shares of Common Stock, for a purchase price of $2,500,000 (the “Offering”). The Warrants are exercisable upon issuance and will expire three years from the date of issuance. The initial exercise price of the Warrants is $6.00, subject to stock, splits, stock dividend and other similar events. In addition, when the closing price of the Common Stock equals or exceeds $9.00 per share for twenty Trading Days (as defined in the Exchange Agreements) during any thirty-day period, the Company shall have the right to require the Investors to exercise all or any portion of the Warrants for a cash exercise. The aggregate net proceeds of the Offering were $5,000,000. The Company and the Investors further agreed to amend the terms of the SPA to permit the closing of the offering to occur on a rolling basis.
The Company paid the following fees to a FINRA member firm in connection with the Offering: (i) a cash success fee of $175,000 and (ii) warrants to purchase a number of shares of Common Stock equal to 7% of the number of shares of Common Stock sold in the Offering, at an exercise price per share equal to $6.00 subject to adjustment (the “Comp Warrants”). The Comp Warrants are exercisable on a cashless basis, at the holder’s discretion.
On October 23, 2020, we entered into a Securities Purchase Agreement (the “October SPA”) with one accredited investor. Pursuant to the October SPA, the Company sold and issued a convertible promissory note (the “October Note”) in the principal amount of $2,500,000 to the investor and received the payment from such investor on October 30, 2020.
The October Note was issued on October 23, 2020 and the maturity date of the October Note is the twenty-four (24) month anniversary from the issuance date (the “Maturity Date”). Upon the Maturity Date, the Company shall pay to the holder, in cash, an amount representing all outstanding principal amount and accrued and unpaid interest under the October Note. The October Note bears an interest rate of ten percent (10%) per annum and may be convertible into shares of the Company’s common stock at a fixed conversion price of $2.25 per share. The holder of the October Note may elect to convert part or all of the outstanding balance of the October Note from the issuance date until the Maturity Date. The Company may prepay the outstanding amount at any time, in whole or in part, without any penalty.
In connection with the October Note and pursuant to the terms of an agreement entered into between the Company and a FINRA member firm, such firm shall receive (i) a cash success fee of $78,750 and (ii) upon conversion of the October Note, warrants equal to 7.0% of the number of shares of Common Stock received by the investor at the time of conversion (“Note Warrants”). The warrants are exercisable on a cashless basis, at the holder’s discretion.
The issuance and sale of the Common Stock, the Investor Warrants, Comp Warrants, Note Warrants and the shares of Common Stock underlying the Investor Warrants, the Comp Warrants and the October Note were made in reliance on an exemption from registration contained in either Regulation D or Regulation S of the Securities Act of 1933, as amended (the “Securities Act”).
Entry into Securities Purchase Agreements
On January 21, 2020, the Company entered into three note agreements with existing note investors who executed the agreements in 2018. These three investors are Guoliang Yu and Yingfei Wei Family Trust, Keypoint Technology Ltd., and Yoshinobu Odaira. The new agreements bear the same term as other notes investors who executed the contract in 2019. On April 5, 2020, the Company entered into exchange agreements with such note holders. Pursuant to the exchange agreements, the Holders agreed to deliver the Notes to the Company for cancellation, of which the aggregate principal amount plus accrued interest expenses are $931,584, and the Company agreed to issue to the Holders an aggregate of 506,297 shares of the Company’s common stock, and warrants to purchase 506,297 shares of the Company’s common stock.
On August 28, 2019 and September 4, 2019, the Company issued convertible promissory notes in the aggregate principal amount plus accrued interest expenses are $515,196 to Kuo, Li Shen, Chang, Ping Shan, Lin, Shan Tyan, and Liu, Ching Hsuan. On April 20, 2020, the Company entered into separate exchange agreements with each note holder. Pursuant to the exchange agreements, the note holders agreed to cancel the notes and the Company agreed to issue to the holders an aggregate of 289,438 shares of the Company’s common stock and warrants to purchase 289,438 shares of the Company’s common stock.
In May 2020, the Company received capital contributions of approximately $1,602,040 in cash from 40 investors through private placements with the term of $2.25 per share and a free warrant attaches with each Common stock that was purchased. The exercise price of the warrant will be at $6.00 with a mandatory exercise price of $9.00.
47
Pursuant to the terms of an agreement entered into between the Company and a FINRA member firm, such firm shall receive (i) a cash success fee of $60,831.65 (ii) a warrant to purchase 37,852 shares of Common Stock with an exercise price of $2.25 per share, and (iii) a warrant to purchase 37,852 shares of Common Stock with an exercise price of $6.00 per share.
Recent PPP Loan
On April 14, 2020, the Company received a loan in the amount of $124,400 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from East West Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll. The loan was granted pursuant to a promissory note dated April 14, 2020 issued by the Company, which matures on April 13, 2022 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is two years after the date of the promissory note. On October 23, American BriVision (Holding) Corporation (the “Company”) has started the application with the US Government regarding the loan forgiveness program.
On January 29, 2021, BioKey received a loan in the amount of $132,331 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from East West Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll. The loan was granted pursuant to a promissory note dated January 27, 2021 issued by the Company, which matures on January 28, 2026 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is five years after the date of the promissory note. In addition, the Company will start the application with the US Government regarding the loan forgiveness program as soon as the US Government allows. No collateral or personal guarantees are required.
On February 7, 2021, the Company received a loan in the amount of $104,167 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from Cathay Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll. The loan was granted pursuant to a promissory note dated February 7, 2021 issued by the Company, which matures on February 6, 2026 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is five years after the date of the promissory note. In addition, the Company will start the application with the US Government regarding the loan forgiveness program as soon as the US Government allows. No collateral or personal guarantees are required.
Recent Research Results
On May 23, 2019, the Company announced its internal Phase II clinical study results of ABV-1504 for Major Depression Disorder (“MDD”). The clinical study results showed that PDC-1421, the active pharmaceutical ingredient of ABV-1504, met the pre-specified primary endpoint of the Phase II clinical trial and significantly improved the symptoms of MDD.
The Phase II clinical study was a randomized, double-blind, placebo-controlled, multi-center trial, in which 60 adult patients with confirmed moderate to severe MDD were treated with PDC-1421 in either low dose (380 mg) or high dose (2 x 380 mg) compared with placebo administration, three times a day for six weeks. PDC-1421 high dose (2 x 380 mg) met the pre-specified primary endpoint by demonstrating a highly significant 13.2-point reduction in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score by Intention-To-Treat (ITT) analysis, averaged over the 6-week treatment period (overall treatment effect) from baseline, as compared to 9.2-point reduction of the placebo group. By Per-Protocol (PP) analysis, PDC-1421 showed a dose dependent efficacy toward MDD in which high dose (2 x 380 mg) gave 13.4-point reduction in MADRS total score from baseline and low dose (380 mg) gave 10.4-point reduction as compared to a 8.6-point in the placebo group. The Company has decided to use the high dose formula in the Phase III clinical trial of ABV-1504.
On September 9, 2020 the Company issued a full clinical study report (CSR) of Vitargus® First-in-Human Phase I Clinical Trial. The safety and preliminary efficacy findings from this study, combined with the unique properties of Vitargus® (BFC-1401), are supportive of further development for its use during vitrectomy surgery in patients requiring vitreous replacement.
The study was an open label, Phase I study undertaken at a single study center in Sydney, Australia. A total of 11 participants were enrolled for the study in which each participant had been diagnosed with either (1) a complex or rhegmatogenous retinal detachment or chronic retinal detachment with failure of gas or silicone oil treatment or (2) a vitreous hemorrhage that requires vitrectomy surgery. The study found that Vitargus® was well-tolerated as a vitreous substitute without any apparent toxicity to ocular tissues. Further, there was no indication of an increased overall safety risk with Vitargus®.
48
On November 9, 2020 the Company issued a full clinical study report (CSR) of its ABV-1505 Phase II Part I clinical trial conducted at the University of California, San Francisco (UCSF) for the treatment of adult Attention-Deficit Hyperactivity Disorder (ADHD).
The Phase II Part I clinical study for treating ADHD found that the PDC-1421 Capsule was safe, well tolerated and efficacious during its treatment and the follow-up period with six adult patients. For the primary endpoints, the percentages of improvement in Adult Attention-Deficit/Hyperactivity Disorder Rating Scale-Investigator Rated-IV (ADHD-RS-IV) score from baseline to 8 weeks treatment were 83.3% (N=5) in the Intention-To-Treat (ITT) population and 80.0% (N=4) in the Per-Protocol (PP) population. Both low and high doses of PDC-1421 Capsule met the primary end points by passing the required 40% population in ADHD-RS-IV test scores.
Overall, the results from this study, which demonstrate the therapeutic value of PDC-1421, support further clinical development of ABV-1505 for the treatment of adult ADHD.
On November 4, 2020, we executed an amendment to our collaboration agreement with BioFirst to add BFC-1403 Intraocular Irrigation Solution and BFC-1404 Corneal Storage Solution to our agreement. BFC-1404 is utilized during a corneal transplant procedure to replace a damaged or diseased cornea while BFC-1403 has broader utilization during a variety of ocular procedures.
Initially the Company will focus on BFC-1404, a solution utilized to store a donor cornea prior to either penetrating keratoplasty (full thickness cornea transplant) or endothelial keratoplasty (back layer cornea transplant). Designated ABV-2002 under the Company’s product identification system, the solution is comprised of a specific poly amino acid that protects ocular tissue from damage caused by external osmolarity exposure during pre-surgery storage. The specific polymer in ABV 2002 can adjust osmolarity to maintain a range of 330 to 390 mOsM thereby permitting hydration within the corneal stroma during the storage period. Stromal hydration results in (a) maintaining acceptable corneal transparency and (b) prevents donor cornea swelling. ABV-2002 also contains an abundant phenolic phytochemical found in plant cell walls that provides antioxidant antibacterial properties and neuroprotection.
Early testing by BioFirst indicates that ABV-2002 may be more effective for protecting the cornea and retina during long-term storage than other storage media available today and can be manufactured at lower cost. Categorized as a Class I Medical Device which has the lowest risk to patients, the Company intends to submit a Premarket Notification 510(K) submission to the FDA before the end of 2021 to demonstrate the device is at least as safe and effective as current products on the market.
Strategy
Key elements of our business strategy include:
| ● | Advancing to the pivotal trial phase of ABV-1701 Vitargus for the treatments of Retinal Detachment or Vitreous Hemorrhage, which we expect to generate revenues in the future. |
| ● | Focusing on licensing ABV-1504 for the treatment of major depressive disorder after the successful completion of its Phase II clinical trials. |
| ● | Completing Phase II, Part 2 clinical trial for ABV-1505 for the treatment of attention deficit hyperactivity disorder ADHD. |
| ● | Out licensing drug candidates and medical device candidates to major pharmaceutical companies for phase III and pivotal clinical trials, as applicable, and further marketing if approved by the FDA. |
49
We plan to augment our core research and development capability and assets by conducting Phase I and II clinical trials for investigational new drugs and medical devices in the fields of CNS, Hematology/Oncology and Ophthalmology.
Our management team has extensive experiences across a wide range of new drug and medical device development and we have in-licensed new drug and medical device candidates from large research institutes and universities in both the U.S. and Taiwan. Through an assertive product development approach, we expect that we will build a substantial portfolio of Oncology/ Hematology, CNS and Ophthalmology products. We primarily focus on Phase I and II research of new drug candidates and out license the post-Phase-II products to pharmaceutical companies; we do not expect to devote substantial efforts and resources to building the disease-specific distribution channels.
Business Objectives
The Company is operating its core business based on collaborative activities that can generate current and future revenues through collaborative research, development and/or commercialization agreements. The terms of these agreements typically include payment to the Company related to one or more of the following: nonrefundable upfront license fees, development and commercial milestones, partial or complete reimbursement of research and development costs, and royalties on net sales of licensed products. Each type of payments results in collaborative revenues except for revenues from royalties on net sales of licensed products, which are classified as royalty revenues. To date, we have not received any royalty revenues. Revenue is recognized upon satisfaction of a performance obligation by transferring control of a good or service to the collaboration partners.
As part of the accounting for these arrangements, the Company applies judgment to determine whether the performance obligations are distinct, and develop assumptions in determining the stand-alone selling price for each distinct performance obligation identified in the collaboration agreements. To determine the stand-alone selling price, the Company relies on assumptions which may include forecasted revenues, development timelines, reimbursement rates for R&D personnel costs, discount rates and probabilities of technical and regulatory success.
The Company had multiple deliverables under the collaborative agreements, including deliverables relating to grants of technology licenses, regulatory and clinical development, and marketing activities. Estimation of the performance periods of the Company’s deliverables requires the use of management’s judgment. Significant factors considered in management’s evaluation of the estimated performance periods include, but are not limited to, the Company’s experience in conducting clinical development, regulatory and manufacturing activities. The Company reviews the estimated duration of its performance periods under its collaborative agreements on an annually basis, and makes any appropriate adjustments on a prospective basis. Future changes in estimates of the performance period under its collaborative agreements could impact the timing of future revenue recognition.
(i) Nonrefundable upfront payments
If a license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in an arrangement, the Company recognizes revenue from the related nonrefundable upfront payments based on the relative standalone selling price prescribed to the license compared to the total selling price of the arrangement. The revenue is recognized when the license is transferred to the collaboration partners and the collaboration partners are able to use and benefit from the license. To date, the receipt of nonrefundable upfront fees was solely for the compensation of past research efforts and contributions made by the Company before the collaborative agreements entered into and it does not relate to any future obligations and commitments made between the Company and the collaboration partners in the collaborative agreements.
(ii) Milestone payments
The Company is eligible to receive milestone payments under the collaborative agreement with collaboration partners based on achievement of specified development, regulatory and commercial events. Management evaluated the nature of the events triggering these contingent payments, and concluded that these events fall into two categories: (a) events which involve the performance of the Company’s obligations under the collaborative agreement with collaboration partners, and (b) events which do not involve the performance of the Company’s obligations under the collaborative agreement with collaboration partners.
50
The former category of milestone payments consists of those triggered by development and regulatory activities in the territories specified in the collaborative agreements. Management concluded that each of these payments constitute substantive milestone payments. This conclusion was based primarily on the facts that (i) each triggering event represents a specific outcome that can be achieved only through successful performance by the Company of one or more of its deliverables, (ii) achievement of each triggering event was subject to inherent risk and uncertainty and would result in additional payments becoming due to the Company, (iii) each of the milestone payments is nonrefundable, (iv) substantial effort is required to complete each milestone, (v) the amount of each milestone payment is reasonable in relation to the value created in achieving the milestone, (vi) a substantial amount of time is expected to pass between the upfront payment and the potential milestone payments, and (vii) the milestone payments relate solely to past performance. Based on the foregoing, the Company recognizes any revenue from these milestone payments in the period in which the underlying triggering event occurs.
(iii) Multiple Element Arrangements
The Company evaluates multiple element arrangements to determine (1) the deliverables included in the arrangement and (2) whether the individual deliverables represent separate units of accounting or whether they must be accounted for as a combined unit of accounting. This evaluation involves subjective determinations and requires management to make judgments about the individual deliverables and whether such deliverables are separate from other aspects of the contractual relationship. Deliverables are considered separate units of accounting provided that: (i) the delivered item(s) has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially within its control. In assessing whether an item under a collaboration has standalone value, the Company considers factors such as the research, manufacturing, and commercialization capabilities of the collaboration partner and the availability of the associated expertise in the general marketplace. The Company also considers whether its collaboration partners can use the other deliverable(s) for their intended purpose without the receipt of the remaining element(s), whether the value of the deliverable is dependent on the undelivered item(s), and whether there are other vendors that can provide the undelivered element(s).
The Company recognizes arrangement consideration allocated to each unit of accounting when all of the revenue recognition criteria in ASC 606 are satisfied for that particular unit of accounting. In the event that a deliverable does not represent a separate unit of accounting, the Company recognizes revenue from the combined unit of accounting over the Company’s contractual or estimated performance period for the undelivered elements, which is typically the term of the Company’s research and development obligations. If there is no discernible pattern of performance or objectively measurable performance measures do not exist, then the Company recognizes revenue under the arrangement on a straight-line basis over the period the Company is expected to complete its performance obligations. Conversely, if the pattern of performance in which the service is provided to the customer can be determined and objectively measurable performance measures exist, then the Company recognizes revenue under the arrangement using the proportional performance method. Revenue recognized is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line method or proportional performance method, as applicable, as of the period ending date.
At the inception of an arrangement that includes milestone payments, the Company evaluates whether each milestone is substantive and at risk to both parties on the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether: (1) the consideration is commensurate with either the Company’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from its performance to achieve the milestone, (2) the consideration relates solely to past performance and (3) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, clinical, regulatory, commercial, and other risks that must be overcome to achieve the particular milestone and the level of effort and investment required to achieve the particular milestone in making this assessment. There is considerable judgment involved in determining whether a milestone satisfies all of the criteria required to conclude that a milestone is substantive. Milestones that are not considered substantive are recognized as earned if there are no remaining performance obligations or over the remaining period of performance, assuming all other revenue recognition criteria are met.
51
(iv) Royalties and Profit Sharing Payments
Under the collaborative agreement with the collaboration partners, the Company is entitled to receive royalties on sales of products, which is at certain percentage of the net sales. The Company recognizes revenue from these events based on the revenue recognition criteria set forth in ASC 606. Based on those criteria, the Company considers these payments to be contingent revenues, and recognizes them as revenue in the period in which the applicable contingency is resolved.
Revenues Derived from Research and Development Activities Services — Revenues related to research and development and regulatory activities are recognized when the related services or activities are performed, in accordance with the contract terms. The Company typically has only one performance obligation at the inception of a contract, which is to perform research and development services. The Company may also provide its customers with an option to request that the Company provides additional goods or services in the future, such as active pharmaceutical ingredient, API, or IND/NDA/ANDA/510K submissions. The Company evaluates whether these options are material rights at the inception of the contract. If the Company determines an option is a material right, the Company will consider the option a separate performance obligation.
If the Company is entitled to reimbursement from its customers for specified research and development expenses, the Company accounts for the related services that it provides as separate performance obligations if it determines that these services represent a material right. The Company also determines whether the reimbursement of research and development expenses should be accounted for as revenues or an offset to research and development expenses in accordance with provisions of gross or net revenue presentation. The Company recognizes the corresponding revenues or records the corresponding offset to research and development expenses as it satisfies the related performance obligations.
The Company then determines the transaction price by reviewing the amount of consideration the Company is eligible to earn under the contracts, including any variable consideration. Under the outstanding contracts, consideration typically includes fixed consideration and variable consideration in the form of potential milestone payments. At the start of an agreement, the Company’s transaction price usually consists of the payments made to or by the Company based on the number of full-time equivalent researchers assigned to the project and the related research and development expenses incurred. The Company does not typically include any payments that the Company may receive in the future in its initial transaction price because the payments are not probable. The Company would reassess the total transaction price at each reporting period to determine if the Company should include additional payments in the transaction price.
The Company receives payments from its customers based on billing schedules established in each contract. Upfront payments and fees may be recorded as advance from customers upon receipt or when due, and may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements. Amounts are recorded as accounts receivable when the right of the Company to consideration is unconditional. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customers and the transfer of the promised goods or services to the customers will be one year or less.
Examples of collaborative agreements the Company has entered into are as follows:
Collaborative agreements with BHK
(i) On February 24, 2015, BioLite Taiwan and BioHopeKing Corporation (the “BHK”) entered into a co-development agreement, (the “BHK Co-Development Agreement”), pursuant to which it is collaborative with BHK to develop and commercialize BLI-1401-2 (Botanical Drug) Triple Negative Breast Cancer (TNBC) Combination Therapy (BLI-1401-2 Products) in Asian countries excluding Japan for all related intellectual property rights, and has developed it for medicinal use in collaboration with outside researchers. The development costs shall be shared 50/50 between BHK and the Company. The BHK Co-Development Agreement will remain in effect for fifteen years from the date of first commercial sale of the Product in Asia excluding Japan.
52
On July 27, 2016, BioLite Taiwan and BHK agreed to amend the payment terms of the milestone payment in an aggregate amount of $10 million based on the following schedule:
| ● | Upon the signing of the BHK Co-Development Agreement: $1 million, or 10% of total payment |
| ● | Upon the first Investigational New Drug (IND) submission and BioLite Taiwan will deliver all data to BHK according to FDA Reviewing requirement: $1 million, or 10% of total payment |
| ● | At the completion of first phase II clinical trial: $1 million, or 10% of total payment |
| ● | At the initiation of phase III of clinical trial research: $3 million, or 30% of total payment |
| ● | Upon the New Drug Application (NDA) submission: $4 million, or 40% of total payment |
In December 2015, BHK has paid a non-refundable upfront cash payment of $1 million, or 10% of $10,000,000, upon the signing of BHK Co-Development Agreement. The Company concluded that the deliverables are considered separate units of accounting as the delivered items have value to the customer on a standalone basis and recognized this cash receipt as collaboration revenue when all research, technical, and development data was delivered to BHK in 2015. The receipt is for the compensation of past research efforts and contributions made by BioLite Taiwan before this collaborative agreement was signed and it does not relate to any future commitments made by BioLite Taiwan and BHK in this collaborative agreement. In August 2016, the Company has received the second milestone payment of NT$31,649,000, approximately equivalent to $1 million, and recognized collaboration revenue for the year ended December 31, 2016. As of the date of this report, the Company has not completed the first phase II clinical trial.
In addition to the milestone payments, BioLite Taiwan is entitled to receive royalty on 12% of BHK’s net sales related to BLI-1401-2 Products. As of December 31, 2020, the Company has not earned royalties under the BHK Co-Development Agreement.
(ii) On December 9, 2015, BioLite Taiwan entered into another two collaborative agreements (the “BHK Collaborative Agreements”), pursuant to which it is collaborative with BHK to co-develop and commercialize BLI-1005 for “Targeting Major Depressive Disorder” (BLI-1005 Products) and BLI-1006 for “Targeting Inflammatory Bowel Disease” (BLI-1006 Products) in Asia excluding Japan for all related intellectual property rights, and has developed it for medicinal use in collaboration with outside researchers. The development costs shall be shared 50/50 between BHK and the Company. The BHK Co-Development Agreement will remain in effect for fifteen years from the date of first commercial sale of the Product in in Asia excluding Japan.
In 2015, the Company recognized the cash receipt in a total of NT$50 million, approximately equivalent to $1.6 million, as collaboration revenue when all research, technical, and development data was delivered to BHK. The Company concluded that the deliverables are considered separate units of accounting as the delivered items have value to the customer on a standalone basis and recognized this payment as collaboration revenue when all research, technical, data and development data was delivered to BHK. The cash receipt is for the compensation of past research efforts and contributions made by BioLite Taiwan before this BHK Collaborative Agreements was signed and it does not relate to any future commitments made by BioLite Taiwan and BHK in this BHK Collaborative Agreements.
In addition to the total of NT$50 million, approximately equivalent to $1.60 million, BioLite Taiwan is entitled to receive 50% of the future net licensing income or net sales profit. As of December 31, 2020, the Company has not earned the royalty under the BHK Collaborative Agreements.
Co-Development agreement with Rgene Corporation, a related party
On May 26, 2017, American BriVision Corporation entered into a co-development agreement (the “Co-Dev Agreement”) with Rgene Corporation (the “Rgene”), a related party under common control by controlling beneficiary shareholder of YuanGene Corporation and the Company (See Note 12). Pursuant to Co-Dev Agreement, BriVision and Rgene agreed to co-develop and commercialize ABV-1507 HER2/neu Positive Breast Cancer Combination Therapy, ABV-1511 Pancreatic Cancer Combination Therapy and ABV-1527 Ovary Cancer Combination Therapy. Under the terms of the Co-Dev Agreement, Rgene is required to pay the Company $3,000,000 in cash or stock of Rgene with equivalent value by August 15, 2017. The payment is for the compensation of BriVision’s past research efforts and contributions made by BriVision before the Co-Dev Agreement was signed and it does not relate to any future commitments made by BriVision and Rgene in this Co-Dev Agreement. In addition to $3,000,000, the Company is entitled to receive 50% of the future net licensing income or net sales profit earned by Rgene, if any, and any development costs shall be equally shared by both BriVision and Rgene.
53
On June 1, 2017, the Company has delivered all research, technical, data and development data to Rgene. Since both Rgene and the Company are related parties and under common control by a controlling beneficiary shareholder of YuanGene Corporation and the Company, the Company has recorded the full amount of $3,000,000 in connection with the Co-Dev Agreement as additional paid-in capital during the year ended September 30, 2017. During the year ended December 31, 2017, the Company has received $450,000 in cash. On December 24, 2018, the Company received the remaining balance of $2,550,000 in the form of newly issued shares of Rgene’s Common Stock, at the price of NT$50 (approximately equivalent to $1.60 per share), for an aggregate number of 1,530,000 shares, which accounted for equity method long-term investment as of December 31, 2018. During the year ended December 31, 2018, the Company has recognized investment loss of $549. On December 31, 2018, the Company determined to fully write off this investment based on the Company’s assessment of the severity and duration of the impairment, and qualitative and quantitative analysis of the operating performance of the investee, adverse changes in market conditions and the regulatory or economic environment, changes in operating structure of Rgene, additional funding requirements, and Rgene’s ability to remain in business. All projects that have been initiated will be managed and supported by the Company and Rgene.
The Company and Rgene signed an amendment to the Co-Dev Agreement on November 10, 2020, pursuant to which both parties agreed to delete AB-1507 HER2/neu Positive Breast Cancer Combination Therapy and AB 1527 Ovary Cancer Combination Therapy and add ABV-1519 EGFR Positive Non-Small Cell Lung Cancer Combination Therapy and ABV-1526 Large Intestine / Colon / Rectal Cancer Combination Therapy to the products to be co-developed and commercialized. Other provisions of the Co-Dev Agreement remain in full force and effect.
Collaborative agreement with BioFirst Corporation, a related party
On July 24, 2017, American BriVision Corporation entered into a collaborative agreement (the “BioFirst Collaborative Agreement”) with BioFirst Corporation, a corporation incorporated under the laws of Taiwan (“BioFirst”), pursuant to which BioFirst granted the Company the global licensing right for medical use of the product (the “Product”): BFC-1401 Vitreous Substitute for Vitrectomy. BioFirst is a related party to the Company because a controlling beneficiary shareholder of YuanGene Corporation and the Company is one of the directors and Common Stock shareholders of BioFirst (See Note 12).
Pursuant to the BioFirst Collaborative Agreement, the Company will co-develop and commercialize the Product with BioFirst and pay BioFirst in a total amount of $3,000,000 in cash or stock of the Company before September 30, 2018. The amount of $3,000,000 is in connection with the compensation for BioFirst’s past research efforts and contributions made by BioFirst before the BioFirst Collaborative Agreement was signed and it does not relate to any future commitments made by BioFirst and BriVision in this BioFirst Collaborative Agreement. In addition, the Company is entitled to receive 50% of the future net licensing income or net sales profit, if any, and any development cost shall be equally shared by both BriVision and BioFirst.
On September 25, 2017, BioFirst has delivered all research, technical, data and development data to BriVision. The Company determined to fully expense the entire amount of $3,000,000 since currently the related licensing rights do not have alternative future uses. According to ASC 730-10-25-1, absent alternative future uses the acquisition of product rights to be used in research and development activities must be charged to research and development expenses immediately. Hence, the entire amount of $3,000,000 is fully expensed as research and development expense during the year ended September 30, 2017.
On June 30, 2019, BriVision entered into a Stock Purchase Agreement (the “Purchase Agreement”) with BioFirst. Pursuant to the Purchase Agreement, the Company issued 428,571 shares of the Company’s common stock (the “Shares”) to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst (the “Total Payment”) in connection with a certain collaborative agreement between the Company and BioFirst dated July 24, 2017 (the “Collaborative Agreement”). Pursuant to the Collaborative Agreement, BioFirst granted the Company the global licensing right to co-develop BFC-1401 or ABV-1701 Vitreous Substitute for Vitrectomy for medical purposes in consideration for the Total Payment.
54
On August 5, 2019, BriVision entered into a second Stock Purchase Agreement (“Purchase Agreement 2”) with BioFirst. Pursuant to Purchase Agreement 2, the Company issued 414,702 shares of the Company’s common stock (the “Shares”) to BioFirst in consideration for $2,902,911 owed by the Company to BioFirst (the “Total Payment”) in connection with a loan provided to BriVision from BioFirst.
On November 4, 2020, the Company executed an amendment to the collaboration agreement with BioFirst to add BFC-1403 Intraocular Irrigation Solution and BFC-1404 Corneal Storage Solution to the agreement. BFC-1404 is utilized during a corneal transplant procedure to replace a damaged or diseased cornea while BFC-1403 has broader utilization during a variety of ocular procedures.
Initially the Company will focus on BFC-1404, a solution utilized to store a donor cornea prior to either penetrating keratoplasty (full thickness cornea transplant) or endothelial keratoplasty (back layer cornea transplant). Designated ABV-2002 under the Company’s product identification system, the solution is comprised of a specific poly amino acid that protects ocular tissue from damage caused by external osmolarity exposure during pre-surgery storage. The specific polymer in ABV 2002 can adjust osmolarity to maintain a range of 330 to 390 mOsM thereby permitting hydration within the corneal stroma during the storage period. Stromal hydration results in (a) maintaining acceptable corneal transparency and (b) prevents donor cornea swelling. ABV-2002 also contains an abundant phenolic phytochemical found in plant cell walls that provides antioxidant antibacterial properties and neuroprotection.
Early testing by BioFirst indicates that ABV-2002 may be more effective for protecting the cornea and retina during long-term storage than other storage media available today and can be manufactured at lower cost. Categorized as a lower risk Class I Medical Device, the Company intends to submit a Premarket Notification 510(K) submission to the FDA before the end of 2021 to demonstrate the device is at least as safe and effective as current products on the market.
BioKey Revenues
In addition to collaborative agreements, ABVC earns revenue through its wholly owned BioKey subsidiary which provides a wide range of Contract Development & Manufacturing Organization (“CDMO”) services including API characterization, pre-formulation studies, formulation development, analytical method development, stability studies, IND/NDA/ANDA/510K submissions, and manufacturing clinical trial materials (from Phase I through Phase III) and commercial manufacturing of pharmaceutical products.
In addition, BioKey provides a variety of regulatory services tailored to the needs of its customers, which include proofreading and regulatory review of submission documents related to formulation development, clinical trials, marketed products, generics, nutraceuticals and OTC products and training presentations. In addition to supporting ABVC’s new drug development, BioKey submits INDs, NDAs, ANDAs, and DMFs to the FDA, on ABVC’s behalf in compliance with new electronic submission guidelines of the FDA.
Impact of COVID-19 Outbreak
On January 30, 2020, the World Health Organization declared the coronavirus outbreak a “Public Health Emergency of International Concern” and on March 10, 2020, declared it to be a pandemic. Actions taken around the world to help mitigate the spread of the coronavirus include restrictions on travel, and quarantines in certain areas, and forced closures for certain types of public places and businesses. The coronavirus and actions taken to mitigate it have had and are expected to continue to have an adverse impact on the economies and financial markets of many countries, including the geographical area in which the Company operates. While the closures and limitations on movement, domestically and internationally, are expected to be temporary, if the outbreak continues on its current trajectory the duration of the supply chain disruption could reduce the availability, or result in delays, of materials or supplies to and from the Company, which in turn could materially interrupt the Company’s business operations. Given the speed and frequency of the continuously evolving developments with respect to this pandemic, the Company cannot reasonably estimate the magnitude of the impact to its consolidated results of operations. We have taken every precaution possible to ensure the safety of our employees.
55
Due to the COVID-19, our revenue for the first half of fiscal 2020 was significantly impacted. As we have not seen a stronger signal to indicate that overall global economic will be back to normal in the third quarter, our business’s overall revenue stream may be impacted further until the restrictions of COVID-19 can be released, then the company can start operating normally. The global pandemic of COVID-19 continues to evolve rapidly, and we will continue to monitor the situation closely, including its potential effect on our plans and timelines.
Additionally, it is reasonably possible that estimates made in the financial statements have been, or will be, materially and adversely impacted in the near term as a result of these conditions, including losses on inventory; impairment losses related to goodwill and other long-lived assets and current obligations.
Summary of Critical Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with the generally accepted accounting principles in the United States of America (the “U.S. GAAP”). All significant intercompany transactions and account balances have been eliminated.
This basis of accounting involves the application of accrual accounting and consequently, revenues and gains are recognized when earned, and expenses and losses are recognized when incurred. The Company’s financial statements are expressed in U.S. dollars.
Fiscal Year
The Company changed its fiscal year from the period beginning on October 1st and ending on September 30th to the period beginning on January 1st and ending on December 31st, beginning January 1, 2018. All references herein to a fiscal year prior to December 31, 2017 refer to the twelve months ended September 30th of such year.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amount of revenues and expenses during the reporting periods. Actual results could differ materially from those results.
Inventory
Inventory consists of raw materials, work-in-process, finished goods, and merchandise. Inventories are stated at the lower of cost or market and valued on a moving weighted average cost basis. Market is determined based on net realizable value. The Company periodically reviews the age and turnover of its inventory to determine whether any inventory has become obsolete or has declined in value, and incurs a charge to operations for known and anticipated inventory obsolescence.
Reclassifications
Certain classifications have been made to the prior year financial statements to conform to the current year presentation. The reclassification had no impact on previously reported net loss or accumulated deficit.
Forward Stock Split
On March 21, 2016, the Board of Directors of the Company approved an amendment to Articles of Incorporation to effect a forward split at a ratio of 1 to 3.141 and increase the number of our authorized shares of Common Stock, par value $0.001 per share, to 360,000,000, which was effective on April 8, 2016.
56
Stock Reverse Split
On March 12, 2019, the Board of Directors of the Company by unanimous written consent in lieu of a meeting approved to i) effect a stock reverse split at the ratio of 1-for-18 (the “Reverse Split”) of both the authorized common stock of the Company (the “Common Stock”) and the issued and outstanding Common Stock and ii) to amend the articles of incorporation of the Company to reflect the Reverse Split. The Board approved and authorized the Reverse Split without obtaining approval of the Company’s shareholders pursuant to Section 78.207 of Nevada Revised Statutes. On May 3, 2019, the Company filed a certificate of amendment to the Company’s articles of incorporation (the “Amendment”) to effect the Reverse Split with the Secretary of State of Nevada. The Financial Industry Regulatory Authority (“FINRA”) informed the Company that the Reverse Split was effective on May 8, 2019. All shares and related financial information in this Form 10-Q reflect this 1-for-18 reverse stock split.
Fair Value Measurements
FASB ASC 820, “Fair Value Measurements” defines fair value for certain financial and nonfinancial assets and liabilities that are recorded at fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. It requires that an entity measure its financial instruments to base fair value on exit price, maximize the use of observable units and minimize the use of unobservable inputs to determine the exit price. It establishes a hierarchy which prioritizes the inputs to valuation techniques used to measure fair value. This hierarchy increases the consistency and comparability of fair value measurements and related disclosures by maximizing the use of observable inputs and minimizing the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that reflect the assumptions market participants would use in pricing the assets or liabilities based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy prioritizes the inputs into three broad levels based on the reliability of the inputs as follows:
| ● | Level 1 - Inputs are quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. Valuation of these instruments does not require a high degree of judgment as the valuations are based on quoted prices in active markets that are readily and regularly available. |
| ● | Level 2 - Inputs other than quoted prices in active markets that are either directly or indirectly observable as of the measurement date, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. | |
| ● | Level 3 - Valuations based on inputs that are unobservable and not corroborated by market data. The fair value for such assets and liabilities is generally determined using pricing models, discounted cash flow methodologies, or similar techniques that incorporate the assumptions a market participant would use in pricing the asset or liability. |
The carrying values of certain assets and liabilities of the Company, such as cash and cash equivalents, restricted cash, accounts receivable, due from related parties, inventory, prepaid expenses and other current assets, accounts payable, accrued liabilities, and due to related parties approximate fair value due to their relatively short maturities. The carrying value of the Company’s short-term bank loan, convertible notes payable, and accrued interest approximates their fair value as the terms of the borrowing are consistent with current market rates and the duration to maturity is short. The carrying value of the Company’s long-term bank loan approximates fair value because the interest rates approximate market rates that the Company could obtain for debt with similar terms and maturities.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities of three months or less, when purchased, to be cash equivalents. As of December 31, 2020 and 2019, the Company’s cash and cash equivalents amounted $4,273,208 and $144,295, respectively. Some of the Company’s cash deposits are held in financial institutions located in Taiwan where there is currently regulation mandated on obligatory insurance of bank accounts. The Company believes this financial institution is of high credit quality.
57
Restricted Cash Equivalents
Restricted cash equivalents primarily consist of cash held in a reserve bank account in Taiwan. As of December 31, 2020 and 2019, the Company’s restricted cash equivalents amounted $728,163 and $16,148, respectively.
Concentration of Credit Risk
The Company’s financial instruments that are exposed to concentrations of credit risk consist primarily of cash and cash equivalents. The Company places its cash and temporary cash investments in high quality credit institutions, but these investments may be in excess of Taiwan Central Deposit Insurance Corporation and the U.S. Federal Deposit Insurance Corporation’s insurance limits. The Company does not enter into financial instruments for hedging, trading or speculative purposes.
Revenue Recognition
During the fiscal year 2018, the Company adopted Accounting Standards Codification (“ASC”), Topic 606 (ASC 606), Revenue from Contracts with Customers, using the modified retrospective method to all contracts that were not completed as of January 1, 2018, and applying the new revenue standard as an adjustment to the opening balance of accumulated deficit at the beginning of 2018 for the cumulative effect. The results for the Company’s reporting periods beginning on and after January 1, 2018 are presented under ASC 606, while prior period amounts are not adjusted and continue to be reported under the accounting standards in effect for the prior period. Based on the Company’s review of existing collaborative agreements as of January 1, 2018, the Company concluded that the adoption of the new guidance did not have a significant change on the Company’s revenue during all periods presented.
Pursuant to ASC 606, the Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration that the Company expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that the Company determines is within the scope of ASC 606, the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the Company satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the Company will collect the consideration the Company is entitled to in exchange for the goods or services the Company transfers to the customers. At inception of the contract, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
Property and Equipment
Property and equipment is carried at cost net of accumulated depreciation. Repairs and maintenance are expensed as incurred. Expenditures that improve the functionality of the related asset or extend the useful life are capitalized. When property and equipment is retired or otherwise disposed of, the related gain or loss is included in operating income. Leasehold improvements are depreciated on the straight-line method over the shorter of the remaining lease term or estimated useful life of the asset. Depreciation is calculated on the straight-line method, including property and equipment under capital leases, generally based on the following useful lives:
| Estimated Life in Years | ||
| Buildings and leasehold improvements | 5 ~ 50 | |
| Machinery and equipment | 5 ~ 10 | |
| Office equipment | 3 ~ 6 |
58
Impairment of Long-Lived Assets
The Company has adopted Accounting Standards Codification subtopic 360-10, Property, Plant and Equipment (“ASC 360-10”). ASC 360-10 requires that long-lived assets and certain identifiable intangibles held and used by the Company be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company evaluates its long-lived assets for impairment annually or more often if events and circumstances warrant. Events relating to recoverability may include significant unfavorable changes in business conditions, recurring losses, or a forecasted inability to achieve break-even operating results over an extended period. Should impairment in value be indicated, the carrying value of intangible assets will be adjusted, based on estimates of future discounted cash flows resulting from the use and ultimate disposition of the asset. ASC 360-10 also requires assets to be disposed of be reported at the lower of the carrying amount or the fair value less costs to sell.
Long-term Equity Investment
The Company acquires the equity investments to promote business and strategic objectives. The Company accounts for non-marketable equity and other equity investments for which the Company does not have control over the investees as:
| ● | Equity method investments when the Company has the ability to exercise significant influence, but not control, over the investee. Its proportionate share of the income or loss is recognized monthly and is recorded in gains (losses) on equity investments. |
| ● | Non-marketable cost method investments when the equity method does not apply. |
Significant judgment is required to identify whether an impairment exists in the valuation of the Company’s non-marketable equity investments, and therefore the Company considers this a critical accounting estimate. Its yearly analysis considers both qualitative and quantitative factors that may have a significant impact on the investee’s fair value. Qualitative analysis of its investments involves understanding the financial performance and near-term prospects of the investee, changes in general market conditions in the investee’s industry or geographic area, and the management and governance structure of the investee. Quantitative assessments of the fair value of its investments are developed using the market and income approaches. The market approach includes the use of comparable financial metrics of private and public companies and recent financing rounds. The income approach includes the use of a discounted cash flow model, which requires significant estimates regarding the investees’ revenue, costs, and discount rates. The Company’s assessment of these factors in determining whether an impairment exists could change in the future due to new developments or changes in applied assumptions.
Other-Than-Temporary Impairment
The Company’s long-term equity investments are subject to a periodic impairment review. Impairments affect earnings as follows:
| ● | Marketable equity securities include the consideration of general market conditions, the duration and extent to which the fair value is below cost, and our ability and intent to hold the investment for a sufficient period of time to allow for recovery of value in the foreseeable future. The Company also considers specific adverse conditions related to the financial health of, and the business outlook for, the investee, which may include industry and sector performance, changes in technology, operational and financing cash flow factors, and changes in the investee’s credit rating. The Company records other-than-temporary impairments on marketable equity securities and marketable equity method investments in gains (losses) on equity investments. |
| ● | Non-marketable equity investments based on the Company’s assessment of the severity and duration of the impairment, and qualitative and quantitative analysis of the operating performance of the investee; adverse changes in market conditions and the regulatory or economic environment; changes in operating structure or management of the investee; additional funding requirements; and the investee’s ability to remain in business. A series of operating losses of an investee or other factors may indicate that a decrease in value of the investment has occurred that is other than temporary and that shall be recognized even though the decrease in value is in excess of what would otherwise be recognized by application of the equity method. A loss in value of an investment that is other than a temporary decline shall be recognized. Evidence of a loss in value might include, but would not necessarily be limited to, absence of an ability to recover the carrying amount of the investment or inability of the investee to sustain an earnings capacity that would justify the carrying amount of the investment. The Company records other-than-temporary impairments for non-marketable cost method investments and equity method investments in gains (losses) on equity investments. Other-than-temporary impairments of equity investments were $961,217 and $0 for the year ended December 31, 2020 and 2019, respectively. |
59
Goodwill
The Company evaluates goodwill for impairment annually or more frequently when an event occurs or circumstances change that indicate the carrying value may not be recoverable. In testing goodwill for impairment, the Company may elect to utilize a qualitative assessment to evaluate whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount. If the qualitative assessment indicates that goodwill impairment is more likely than not, the Company performs a two-step impairment test. The Company tests goodwill for impairment under the two-step impairment test by first comparing the book value of net assets to the fair value of the reporting units. If the fair value is determined to be less than the book value or qualitative factors indicate that it is more likely than not that goodwill is impaired, a second step is performed to compute the amount of impairment as the difference between the estimated fair value of goodwill and the carrying value. The Company estimates the fair value of the reporting units using discounted cash flows. Forecasts of future cash flows are based on our best estimate of future net sales and operating expenses, based primarily on expected category expansion, pricing, market segment share, and general economic conditions.
The Company completed the required testing of goodwill for impairment as of December 31, 2020, and determined that goodwill was impaired because of the current financial condition of the Company and the Company’s inability to generate future operating income without substantial sales volume increases, which are highly uncertain. Furthermore, the Company anticipates future cash flows indicate that the recoverability of goodwill is not reasonably assured.
Research and Development Expenses
The Company accounts for the cost of using licensing rights in research and development cost according to ASC Topic 730-10-25-1. This guidance provides that absent alternative future uses the acquisition of product rights to be used in research and development activities must be charged to research and development expenses when incurred.
For CDMO business unit, the Company accounts for R&D costs in accordance with Accounting Standards Codification (“ASC”) 730, Research and Development (“ASC 730”). Research and development expenses are charged to expense as incurred unless there is an alternative future use in other research and development projects or otherwise. Research and development expenses are comprised of costs incurred in performing research and development activities, including personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, research costs, and other consulting services. Non-refundable advance payment for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. In instances where the Company enters into agreements with third parties to provide research and development services, costs are expensed as services are performed.
Post-retirement and post-employment benefits
The Company’s subsidiaries in Taiwan adopted the government mandated defined contribution plan pursuant to the Labor Pension Act (the “Act”) in Taiwan. Such labor regulations require that the rate of contribution made by an employer to the Labor Pension Fund per month shall not be less than 6% of the worker’s monthly salaries. Pursuant to the Act, the Company makes monthly contribution equal to 6% of employees’ salaries to the employees’ pension fund. The Company has no legal obligation for the benefits beyond the contributions made. The total amounts for such employee benefits, which were expensed as incurred, were $13,961and $15,928 for the year ended December 31, 2020 and 2019, respectively. Other than the above, the Company does not provide any other post-retirement or post-employment benefits.
60
Stock-based Compensation
The Company measures expense associated with all employee stock-based compensation awards using a fair value method and recognizes such expense in the consolidated financial statements on a straight-line basis over the requisite service period in accordance with FASB ASC Topic 718 “Compensation-Stock Compensation”. Total employee stock-based compensation expenses were $1,623,102 and $0 for the years ended December 31, 2020 and 2019.
The Company accounted for stock-based compensation to non-employees in accordance with FASB ASC Topic 718 “Compensation-Stock Compensation” and FASB ASC Topic 505-50 “Equity-Based Payments to Non-Employees” which requires that the cost of services received from non-employees is measured at fair value at the earlier of the performance commitment date or the date service is completed and recognized over the period the service is provided. Total non-employee stock-based compensation expenses were $2,523,877 and $22,314 for the years ended December 31, 2020 and 2019, respectively.
Beneficial Conversion Feature
From time to time, the Company may issue convertible notes that may contain an imbedded beneficial conversion feature. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of the warrants, if related warrants have been granted. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid in capital. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
Income Taxes
The Company accounts for income taxes using the asset and liability approach which allows the recognition and measurement of deferred tax assets to be based upon the likelihood of realization of tax benefits in future years. Under the asset and liability approach, deferred taxes are provided for the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. A valuation allowance is provided for deferred tax assets if it is more likely than not these items will expire before the Company is able to realize their benefits, or future deductibility is uncertain.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The evaluation of a tax position is a two-step process. The first step is to determine whether it is more-likely-than-not that a tax position will be sustained upon examination, including the resolution of any related appeals or litigations based on the technical merits of that position. The second step is to measure a tax position that meets the more-likely-than-not threshold to determine the amount of benefits recognized in the financial statements. A tax position is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. Tax positions that previously failed to meet the more-likely-than-not recognition threshold should be recognized in the first subsequent period in which the threshold is met. Previously recognized tax positions that no longer meet the more-likely-than-not criteria should be de-recognized in the first subsequent financial reporting period in which the threshold is no longer satisfied. Penalties and interest incurred related to underpayment of income tax are classified as income tax expense in the year incurred. No significant penalty or interest relating to income taxes has been incurred for the years ended December 31, 2020 and 2019. GAAP also provides guidance on de-recognition, classification, interest and penalties, accounting in interim periods, disclosures and transition.
On December 22, 2017, the SEC issued Staff Accounting Bulletin (“SAB 118”), which provides guidance on accounting for tax effects of the Tax Act. SAB 118 provides a measurement period that should not extend beyond one year from the Tax Act enactment date for companies to complete the accounting under ASC 740. In accordance with SAB 118, a company must reflect the income tax effects of those aspects of the Act for which the accounting under ASC 740 is complete. To the extent that a company’s accounting for certain income tax effects of the Tax Act is incomplete but it is able to determine a reasonable estimate, it must record a provisional estimate to be included in the financial statements. If a company cannot determine a provisional estimate to be included in the financial statements, it should continue to apply ASC 740 on the basis of the provision of the tax laws that were in effect immediately before the enactment of the Tax Act. While the Company is able to make reasonable estimates of the impact of the reduction in corporate rate and the deemed repatriation transition tax, the final impact of the Tax Act may differ from these estimates, due to, among other things, changes in our interpretations and assumptions, additional guidance that may be issued by the I.R.S., and actions the Company may take. The Company is continuing to gather additional information to determine the final impact.
61
Valuation of Deferred Tax Assets
A valuation allowance is recorded to reduce the Company’s deferred tax assets to the amount that is more likely than not to be realized. In assessing the need for the valuation allowance, management considers, among other things, projections of future taxable income and ongoing prudent and feasible tax planning strategies. If the Company determines that sufficient negative evidence exists, then it will consider recording a valuation allowance against a portion or all of the deferred tax assets in that jurisdiction. If, after recording a valuation allowance, the Company’s projections of future taxable income and other positive evidence considered in evaluating the need for a valuation allowance prove, with the benefit of hindsight, to be inaccurate, it could prove to be more difficult to support the realization of its deferred tax assets. As a result, an additional valuation allowance could be required, which would have an adverse impact on its effective income tax rate and results. Conversely, if, after recording a valuation allowance, the Company determines that sufficient positive evidence exists in the jurisdiction in which the valuation allowance was recorded, it may reverse a portion or all of the valuation allowance in that jurisdiction. In such situations, the adjustment made to the deferred tax asset would have a favorable impact on its effective income tax rate and results in the period such determination was made.
Loss Per Share of Common Stock
The Company calculates net loss per share in accordance with ASC Topic 260, “Earnings per Share”. Basic loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common stock equivalents had been issued and if the additional common shares were dilutive. Diluted earnings per share excludes all dilutive potential shares if their effect is anti-dilutive.
Commitments and Contingencies
The Company has adopted ASC Topic 450 “Contingencies” subtopic 20, in determining its accruals and disclosures with respect to loss contingencies. Accordingly, estimated losses from loss contingencies are accrued by a charge to income when information available before financial statements are issued or are available to be issued indicates that it is probable that an assets had been impaired or a liability had been incurred at the date of the financial statements and the amount of the loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. If a loss contingency is not probable or reasonably estimable, disclosure of the loss contingency is made in the financial statements when it is at least reasonably possible that a material loss could be incurred.
Foreign-currency Transactions
For the Company’s subsidiaries in Taiwan, the foreign-currency transactions are recorded in New Taiwan dollars (“NTD”) at the rates of exchange in effect when the transactions occur. Gains or losses resulting from the application of different foreign exchange rates when cash in foreign currency is converted into New Taiwan dollars, or when foreign-currency receivables or payables are settled, are credited or charged to income in the year of conversion or settlement. On the balance sheet dates, the balances of foreign-currency assets and liabilities are restated at the prevailing exchange rates and the resulting differences are charged to current income except for those foreign currencies denominated investments in shares of stock where such differences are accounted for as translation adjustments under the Statements of Stockholders’ Equity (Deficit).
Translation Adjustment
The accounts of the Company’s subsidiaries in Taiwan were maintained, and their financial statements were expressed, in New Taiwan Dollar (“NT$”). Such financial statements were translated into U.S. Dollars (“$” or “USD”) in accordance ASC 830, “Foreign Currency Matters”, with the NT$ as the functional currency. According to the Statement, all assets and liabilities are translated at the current exchange rate, shareholder’s deficit are translated at the historical rates and income statement items are translated at an average exchange rate for the period. The resulting translation adjustments are reported under other comprehensive income (loss) as a component of shareholders’ equity (deficit).
Recent Accounting Pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, as part of its overall simplification initiative to reduce costs and complexity of applying accounting standards while maintaining or improving the usefulness of the information provided to users of financial statements. The FASB’s amendments primarily impact ASC 740, Income Taxes, and may impact both interim and annual reporting periods. ASU 2019-12 will be effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years and early adoption is permitted. The Company is currently evaluating the impact of adopting ASU 2019-12.
In August 2020, the FASB issued ASU 2020-06, Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging- Contracts in Entity’s Own Equity (Subtopic 815-40), to reduce the complexity associated with applying U.S. GAAP principles for certain financial instruments with characteristics of liabilities and equity. The amendments in this ASU reduce the number of accounting models for convertible instruments and expand the existing disclosure requirements over earnings per share as it relates to convertible instruments. This ASU will be effective for the fiscal year beginning January 1, 2022 and interim periods therein. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020. The amendments may be adopted through either a modified retrospective method, or a fully retrospective method. The Company is currently evaluating the impact of adopting ASU 2020-06.
62
Estimates and Assumptions
In preparing our consolidated financial statements, we use estimates and assumptions that affect the reported amounts and disclosures. Our estimates are often based on complex judgments, probabilities and assumptions that we believe to be reasonable, but that are inherently uncertain and unpredictable. We are also subject to other risks and uncertainties that may cause actual results to differ from estimated amounts.
Results of Operations — Year Ended December 31, 2020 Compared to Year Ended December 31, 2019.
Revenues. We generated $483,045 and $701,719 in revenues for the year ended December 31, 2020 and 2019, respectively. The decrease of $218,674, or approximately -31%, was primarily due to the impact of COVID-19 on our CDMO business sector. Even though we were not required to shut down our operations during the shelter-in-place period, several clients decided to postpone the progress of their current and new projects with us.
Operating Expenses. Our operating expenses were $8,970,105 in the year ended December 31, 2020 as compared to $4,140,360 in the year ended December 31, 2019. Our total operating expenses increased by $4,829,745 during the year ended December 31, 2020 from 2019. Such increase in operating expenses was mainly attributed to the increase in stock based compensation by $4,124,665 mainly due to the increase in professional service fees that relates to our up-list and general expenses that occurred for our CDMO business unit; and our recent action of converting unpaid employees’ salaries to common stock option.
Other Income (expense). The other expenses was ($2,308,160) in the year ended December 31, 2020 as compared to ($551,503) in the year ended December 31, 2019. The decrease of $1,756,657, or approximately 319%, was principally caused by the increase in impairment loss, investment loss, and loss on investment in equity securities, partially offset by the increase in interest income and other income.
Impairment loss was $961,217 for the year ended December 31, 2020 as compared to $0 for the year ended December 31, 2019. The increase of $961,217 was due to our re-assessment of valuation based on the latest market consensus and trading price.
Investment loss was $40,589 for the year ended December 31, 2020 as compared to $0 for the year ended December 31, 2019. The increase of $40,589 was mainly due to our liquidation of stocks at lower than historical book value based on the market condition.
Loss on investment in equity securities was $1,168,733 for the year ended December 31, 2020 as compared to $210,086 for the year ended December 31, 2019, representing an increase of $958,647, or 456%, which was mainly due to the investment in a related party, BioFirst, accounted for using the equity method accounting treatment.
Interest income was $71,045 for the year ended December 31, 2020 as compared to $23,344 for year ended December 31, 2019. The increase of $47,701, or approximately 204%, was primarily due to the interest income from various related-party loans.
Other income was $174,770 for the year ended December 31, 2020 as compared to $92,959 for the year ended December 31, 2019. The increase of $81,811, or approximately 88%, was primarily due to the adjustments of tax refund contributed by BioFirst based on the co-development agreement.
Net Loss. The net loss was $10,593,584 for the year ended December 31, 2020 compared to $3,933,240 for the year ended December 31, 2019. The Company’s net loss increased by $6,660,344 or approximately 169% during the year ended December 31, 2020 from 2019.
Liquidity and Capital Resources
Working Capital
| As
of December 31, 2020 | As
of December 31, 2019 | |||||||
| Current Assets | $ | 6,172,966 | $ | 878,238 | ||||
| Current Liabilities | $ | 4,844,391 | $ | 6,814,193 | ||||
| Working Capital (deficit) | $ | 1,328,575 | $ | (5,935,955 | ) | |||
63
Cash Flow from Operating Activities
During the year ended December 31, 2020 and 2019, the net cash used in operating activities were ($4,563,919) and ($3,134,526), respectively. The decrease in the amount of $1,429,393 was primarily due to the increased net loss and accrued expenses and other current liabilities, partially offset by the increase in stock based compensation for nonemployees, non-cash based investment loss, loss on investment in equity securities, and due to related parties during the year ended December 31, 2020.
Cash Flow from Investing Activities
During the year ended December 31, 2020 and 2019, the net cash used in investing activities was ($225,431) and the net cash generated was ($35,297), respectively. The decrease in the amount of $190,134 was primarily due to the loan to related parties and partially offset by the net proceeds from sale of investment during the year ended December 31, 2020.
Cash Flow from Financing Activities
During the year ended December 31, 2020 and 2019, the net cash provided by financing activities were $9,567,843 and $3,087,489, respectively. The net cash provided by financing activities increased by $6,480,354 during the compared periods because we obtained more funding through common stock, short-term bank loans and convertible notes during the year ended December 31, 2020 than in the year ended December 31, 2019.
Off-Balance Sheet Arrangements
As of December 31, 2020, we did not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures, or capital resources that is material to investors.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
64
Item 8. Financial Statements and Supplementary Data
Our Consolidated Financial Statements and Notes thereto and the report of KCCW Accountancy Corp, our independent registered public accounting firm, are set forth on pages F-1 through F-42 of this Report.
F-1

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of
American BriVision (Holding) Corporation and Subsidiaries
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of American BriVision (Holding) Corporation and subsidiaries (collectively “the Company”) as of December 31, 2020 and 2019, the related consolidated statements of operations, comprehensive income (loss), shareholders’ equity (deficit), and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2020 and 2019, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Change in Accounting Principle
As discussed in Note 16 to the consolidated financial statements, the Company has changed its method of accounting for leases in 2019 due to the adoption of Financial Accounting Standards Board Accounting Standards Codification Topic 842, Leases.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Recognition of Stock-Based Compensation Cost for Stock Options Issued
As described in Note 14 to the consolidated financial statements, the Company granted stock options to its employees and consultant for unpaid salaries and consulting fee and estimated total stock compensation expense related to the issuance of stock options of $1,706,419 for the year ended December 31, 2020. The stock compensation cost was valued at the grant date, and management evaluated the fair value of these stock options at the grant date and recognized based on the vesting schedule.
We identified the recognition of stock options as a critical audit matter due to the significant judgments made by management when developing underlying assumptions.
The following are the primary procedures we performed to address this critical audit matter. We obtained an understanding and evaluated the design and implementation of certain controls relating to significant judgments and assumptions developed by management. We evaluated and tested sources of data and assumptions used by management. In addition, we tested the completeness and accuracy of the underlying assumptions used by management.
/s/ KCCW Accountancy Corp.
We have served as the Company's auditor since 2019.
Diamond Bar, California
March 15, 2021
KCCW Accountancy Corp. 3333 S. Brea Canyon Road, Suite 206, CA 91765 USA
Tel: +1 909 348 7228 • Fax: +1 909 895-4155s
F-2
AMERICAN BRIVISION (HOLDING) CORPORATION AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
| December
31, 2020 | December 31, 2019 | |||||||
| ASSETS | ||||||||
| Current Assets | ||||||||
| Cash and cash equivalents | $ | 4,273,208 | $ | 144,295 | ||||
| Restricted cash and cash equivalents | 728,163 | 16,148 | ||||||
| Accounts receivable, net | 159,712 | 163,566 | ||||||
| Accounts receivable - related parties, net | 143,435 | 143,278 | ||||||
| Due from related parties | 696,255 | 333,682 | ||||||
| Inventory, net | - | - | ||||||
| Prepaid expense and other current assets | 172,193 | 77,269 | ||||||
| Total Current Assets | 6,172,966 | 878,238 | ||||||
| Property and equipment, net | 514,834 | 520,930 | ||||||
| Operating lease right-of-use assets | 1,772,747 | 524,445 | ||||||
| Goodwill, net | - | - | ||||||
| Long-term investments | 1,190,727 | 3,364,619 | ||||||
| Deferred tax assets | 1,790,597 | 1,460,033 | ||||||
| Prepaid expenses – noncurrent | 119,315 | 135,443 | ||||||
| Security deposits | 45,519 | 44,103 | ||||||
| Total Assets | $ | 11,606,705 | $ | 6,927,811 | ||||
| LIABILITIES AND EQUITY | ||||||||
| Current Liabilities | ||||||||
| Accounts payable | $ | 23,044 | $ | 23,995 | ||||
| Short-term bank loans | 1,629,000 | 1,918,500 | ||||||
| Short-term loan | 100,000 | - | ||||||
| Long-term bank loans – current portion | - | 13,403 | ||||||
| Notes payable | 106,800 | 100,200 | ||||||
| Accrued expenses and other current liabilities | 2,118,854 | 2,007,573 | ||||||
| Advance from customers | 12,070 | 13,085 | ||||||
| Operating lease liabilities – current portion | 316,178 | 304,248 | ||||||
| Due to related parties | 288,445 | 425,689 | ||||||
| Convertible notes payable – current portion | - | 820,000 | ||||||
| Convertible notes payable – related parties, current portion | 250,000 | 1,187,500 | ||||||
| Total Current Liabilities | 4,844,391 | 6,814,193 | ||||||
| Paycheck Protection Program Loan Payable | 124,400 | - | ||||||
| Tenant security deposit | 19,280 | 2,880 | ||||||
| Operating lease liability – noncurrent portion | 1,456,567 | 235,555 | ||||||
| Convertible notes payable – noncurrent portion | 2,500,000 | - | ||||||
| Total Liabilities | 8,944,638 | 7,052,628 | ||||||
| Equity | ||||||||
| Preferred stock, $0.001 par value, 20,000,000 authorized, nil shares issued and outstanding | - | - | ||||||
| Common stock, $0.001 par value, 100,000,000 authorized, 24,420,526 and 19,478,168 shares issued and outstanding | 24,420 | 19,478 | ||||||
| Additional paid-in capital | 40,751,807 | 28,180,348 | ||||||
| Stock subscription receivable | (3,160,360 | ) | (4,063,320 | ) | ||||
| Accumulated deficit | (25,642,387 | ) | (15,851,223 | ) | ||||
| Accumulated other comprehensive income | 564,860 | 663,753 | ||||||
| Treasury stock | (9,100,000 | ) | (9,100,000 | ) | ||||
| Total Stockholders’ deficit | 3,438,340 | (150,964 | ) | |||||
| Noncontrolling interest | (776,273 | ) | 26,147 | |||||
| Total Equity (Deficit) | 2,662,067 | (124,817 | ) | |||||
| Total Liabilities and Equity (Deficit) | $ | 11,606,705 | $ | 6,927,811 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-3
AMERICAN BRIVISION (HOLDING) CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Revenues | $ | 483,045 | $ | 701,719 | ||||
| Cost of revenues | 18,716 | 20,137 | ||||||
| Gross profit | 464,329 | 681,582 | ||||||
| Operating expenses | ||||||||
| Selling, general and administrative expenses | 4,273,468 | 3,069,493 | ||||||
| Research and development expenses | 549,658 | 1,048,553 | ||||||
| Stock based compensation | 4,146,979 | 22,314 | ||||||
| Total operating expenses | 8,970,105 | 4,140,360 | ||||||
| Loss from operations | (8,505,776 | ) | (3,458,778 | ) | ||||
| Other income (expense) | ||||||||
| Interest income | 71,045 | 23,344 | ||||||
| Interest expense | (405,032 | ) | (482,014 | ) | ||||
| Rent income | 20,071 | 19,487 | ||||||
| Rent income – related parties | 4,800 | 4,400 | ||||||
| Impairment loss | (961,217 | ) | - | |||||
| Investment loss | (40,589 | ) | - | |||||
| Gain/Loss on foreign exchange changes | (3,275 | ) | 407 | |||||
| Gain/Loss on investment in equity securities | (1,168,733 | ) | (210,086 | ) | ||||
| Other income | 174,770 | 92,959 | ||||||
| Total other expenses | (2,308,160 | ) | (551,503 | ) | ||||
| Loss before provision income tax | (10,813,936 | ) | (4,010,281 | ) | ||||
| Provision for income tax | (220,352 | ) | (77,041 | ) | ||||
| Net loss | (10,593,584 | ) | (3,933,240 | ) | ||||
| Net loss attributable to noncontrolling interests | (802,420 | ) | (291,464 | ) | ||||
| Net loss attributed to ABVC and subsidiaries | (9,791,164 | ) | (3,641,776 | ) | ||||
| Foreign currency translation adjustment | (98,893 | ) | 7,902 | |||||
| Comprehensive loss | $ | (9,890,057 | ) | $ | (3,633,874 | ) | ||
| Net loss per share: | ||||||||
| Basic and diluted | $ | (0.50 | ) | $ | (0.21 | ) | ||
| Weighted average number of common shares outstanding: | ||||||||
| Basic and diluted | 19,715,559 | 17,498,543 | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
AMERICAN BRIVISION (HOLDING) CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEAR ENDED DECEMBER 31, 2020 AND 2019
| 2020 | 2019 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | (10,593,584 | ) | $ | (3,933,240 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation | 37,142 | 55,086 | ||||||
| Stock based compensation | 4,146,979 | 22,314 | ||||||
| Gain/Loss on investment in equity securities | 1,168,733 | 210,086 | ||||||
| Other non-cash income and expenses | (15,360 | ) | (5,747 | ) | ||||
| Investment loss | 1,001,806 | - | ||||||
| Deferred tax | (223,201 | ) | (80,692 | ) | ||||
| Changes in operating assets and liabilities: | ||||||||
| Decrease (increase) in accounts receivable | 3,696 | (120,739 | ) | |||||
| Decrease (increase) in prepaid expenses and deposits | (15,778 | ) | (27,617 | ) | ||||
| Decrease (increase) in due from related parties | 20,645 | (282,092 | ) | |||||
| Decrease (increase) in inventory | - | 1,306 | ||||||
| Increase (decrease) in accounts payable | (951 | ) | 50,244 | |||||
| Increase (decrease) in notes payable | - | (4,861 | ) | |||||
| Increase (decrease) in accrued expenses and other current liabilities | (359,822 | ) | 801,434 | |||||
| Increase (decrease) in advanced from others | (1,015 | ) | 1,909 | |||||
| Increase (decrease) in due to related parties | 266,791 | 178,083 | ||||||
| Net cash used in operating activities | (4,563,919 | ) | (3,134,526 | ) | ||||
| Cash flows from investing activities | ||||||||
| Net proceeds from sale of investment | 147,804 | - | ||||||
| Loan to related parties | (373,235 | ) | (17,496 | ) | ||||
| Long-term equity investment | - | (17,801 | ) | |||||
| Net cash used in investing activities | (225,431 | ) | (35,297 | ) | ||||
| Cash flows from financing activities | ||||||||
| Issuance of common stock for cash | 7,615,331 | - | ||||||
| Issuance of common shares for stock-based compensation | - | 552,962 | ||||||
| Proceeds from issuance of common stock for acquisition | - | 531,147 | ||||||
| Proceeds from convertible notes | 2,500,000 | 1,207,500 | ||||||
| Repayment of short-term bank loan | (350,000 | ) | - | |||||
| Proceeds from short-term loan | 100,000 | 1,000,000 | ||||||
| Proceeds from long-term loan | 124,400 | - | ||||||
| Net (repayments of) proceeds from short term borrowings from third parties | (480,989 | ) | 657,466 | |||||
| Proceeds from (repayment of) borrowings from related parties | 72,704 | (820,000 | ) | |||||
| Repayment of long-term bank loans | (13,603 | ) | (41,586 | ) | ||||
| Net cash provided by financing activities | 9,567,843 | 3,087,489 | ||||||
| Effect of exchange rate changes on cash and cash equivalents and restricted cash | 62,435 | (4 | ) | |||||
| Net increase (decrease) in cash and cash equivalents and restricted cash | 4,840,928 | (82,338 | ) | |||||
| Cash and cash equivalents and restricted cash | ||||||||
| Beginning | 160,443 | 242,781 | ||||||
| Ending | $ | 5,001,371 | $ | 160,443 | ||||
| Supplemental disclosure of cash flows | ||||||||
| Cash paid during the year for: | ||||||||
| Interest expense paid | $ | 208,556 | $ | 167,126 | ||||
| Income taxes paid | $ | - | $ | 2,050 | ||||
| Non-cash financing and investing activities | ||||||||
| Common shares issued for employees and consultants | $ | - | $ | 325,740 | ||||
| Capital contribution from related parties under common control | $ | - | $ | 7,872,340 | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
AMERICAN BRIVISION (HOLDING) CORPORATION AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
FOR THE YEAR ENDED DECEMBER 31, 2020 AND 2019
| Common Stock | Stock | Additional | Accumulated Other | Treasury Stock | Non | Stockholders’ | ||||||||||||||||||||||||||||||||||
| Number of shares | Amounts | Subscription Receivable | Paid-in Capital | Accumulated Deficit | Comprehensive Income | Number of Shares | Amount | controlling Interest | Equity (Deficit) | |||||||||||||||||||||||||||||||
| Balance at December 31, 2018 | 11,884,804 | $ | 11,885 | $ | - | $ | 14,983,714 | $ | (12,209,446 | ) | $ | 655,851 | (275,347 | ) | $ | (9,100,000 | ) | $ | 317,610 | $ | (5,340,386 | ) | ||||||||||||||||||
| Issuance of common shares | 7,593,364 | 7,593 | (4,063,360 | ) | 13,174,320 | - | - | - | - | 9,118,593 | ||||||||||||||||||||||||||||||
| Stock based compensation | - | - | - | 22,314 | - | - | - | - | 22,314 | |||||||||||||||||||||||||||||||
| Net loss for the year | - | - | - | - | (3,641,777 | ) | - | - | (291,463 | ) | (3,933,240 | ) | ||||||||||||||||||||||||||||
| Cumulative transaction adjustments | - | - | - | - | - | 7,902 | - | - | 7,902 | |||||||||||||||||||||||||||||||
| Balance at December 31, 2019 | 19,478,168 | 19,478 | (4,063,320 | ) | 28,180,348 | (15,851,223 | ) | 663,753 | (275,347 | ) | (9,100,000 | ) | 26,147 | (124,817 | ) | |||||||||||||||||||||||||
| Capital contribution | 3,384,615 | 3,385 | - | 7,611,946 | - | - | - | - | - | 7,615,331 | ||||||||||||||||||||||||||||||
| Stock based compensation for options granted | - | - | - | 1,706,419 | - | - | - | - | - | 1,706,419 | ||||||||||||||||||||||||||||||
| Stock based compensation for services | 641,887 | 642 | 902,960 | 1,536,958 | - | - | - | - | - | 2,440,560 | ||||||||||||||||||||||||||||||
| Debt conversion | 915,856 | 915 | - | 1,716,136 | - | - | - | - | - | 1,717,051 | ||||||||||||||||||||||||||||||
| Net loss for the year | - | - | - | - | (9,791,164 | ) | - | - | - | (802,420 | ) | (10,593,584 | ) | |||||||||||||||||||||||||||
| Cumulative transaction adjustments | - | - | - | - | - | (98,893 | ) | - | - | - | (98,893 | ) | ||||||||||||||||||||||||||||
| Balance at December 31, 2020 | 24,420,526 | $ | 24,420 | $ | (3,160,360 | ) | $ | 40,751,807 | $ | (25,642,387 | ) | $ | 564,860 | (275,347 | ) | $ | (9,100,000 | ) | $ | (776,273 | ) | $ | 2,662,067 | |||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
AMERICAN BRIVISION (HOLDING) CORPORATION AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. ORGANIZATION AND DESCRIPTION OF BUSINESS
American BriVision (Holding) Corporation (the “Company” or “Holding entity”), a Nevada corporation, through the Company’s operating entity, American BriVision Corporation (“BriVision”), which was incorporated in July 2015 in the State of Delaware, engages in biotechnology to fulfill unmet medical needs and focuses on the development of new drugs and medical devices derived from plants. BriVision develops its pipeline by carefully tracking new medical discoveries or medical device technologies in research institutions in the Asia-Pacific region. Pre-clinical, disease animal model and Phase I safety studies are examined closely by the Company to identify drugs that BriVision believes demonstrate efficacy and safety. Once a drug appears to be a good candidate for development and ultimately commercialization, BriVision licenses the drug or medical device from the original researchers and begins to introduce the drugs clinical plan to highly respected principal investigators in the United States, Australia and Taiwan to conduct a Phase II clinical trial. At present, clinical trials for the Company’s drugs and medical devices are being conducted at such world-famous institutions as Memorial Sloan Kettering Cancer Center (“MSKCC”) and MD Anderson Cancer Center. BriVision had no predecessor operations prior to its formation on July 21, 2015.
Reverse Merger
On February 8, 2016, a Share Exchange Agreement (the “Share Exchange Agreement”) was entered into by and among American BriVision (Holding) Corporation, American BriVision Corporation (“BriVision”), and Euro-Asia Investment & Finance Corp. Limited, a company incorporated under the laws of Hong Kong Special Administrative Region of the People’s Republic of China (“Euro-Asia”), being the owners of record of 164,387,376 (52,336,000 pre-stock split) shares of Common Stock of the Company, and the owners of record of all of the issued share capital of BriVision (the “BriVision Stock”).
Pursuant to the Share Exchange Agreement, upon surrender by the BriVision Shareholders and the cancellation by BriVision of the certificates evidencing the BriVision Stock as registered in the name of each BriVision Shareholder, and pursuant to the registration of the Company in the register of members maintained by BriVision as the new holder of the BriVision Stock and the issuance of the certificates evidencing the aforementioned registration of the BriVision Stock in the name of the Company, the Company issued 166,273,921(52,936,583 pre-stock split) shares (the “Acquisition Stock”) (subject to adjustment for fractionalized shares as set forth below) of the Company’s Common Stock to the BriVision Shareholders (or their designees), and 163,159,952 (51,945,225 pre-stock split) shares of the Company’s Common Stock owned by Euro-Asia were cancelled and retired to treasury. The Acquisition Stock collectively represented 79.70% of the issued and outstanding Common Stock of the Company immediately after the Closing, in exchange for the BriVision Stock, representing 100% of the issued share capital of BriVision in a reverse merger (the “Merger”).
Pursuant to the Merger, all of the issued and outstanding common shares of BriVision were converted, at an exchange ratio of 0.2536-for-1, into an aggregate of 166,273,921(52,936,583pre-stock split) common shares of the Company and BriVision had become a wholly owned subsidiary of the Company. The holders of Company’s Common Stock as of immediately prior to the Merger held an aggregate of 205,519,223(65,431,144 pre-stock split) shares of Company’s Common Stock. Because of the exchange of the BriVision Stock for the Acquisition Stock (the “Share Exchange”), BriVision had become a wholly owned subsidiary (the “Subsidiary”) of the Company and there was a change of control of the Company following the closing. There were no warrants, options or other equity instruments issued in connection with the share exchange agreement.
Upon the consummation of the Share Exchange, BriVision became a wholly owned subsidiary of the Company.
Following the Share Exchange, the Company has abandoned prior business plan and is now pursuing BriVision’s historically proposed businesses, which focus on the development of new drugs and innovative medical devices to fulfill unmet medical needs. The business model of the Company is to integrate research achievements from world-famous institutions, conduct clinical trials of translational medicine for Proof of Concept (“POC”), out-license to international pharmaceutical companies, and explore global markets.
F-7
Accounting Treatment of the Reverse Merger
For financial reporting purposes, the Share Exchange represents a “reverse merger” rather than a business combination and BriVision is deemed the accounting acquirer in the transaction. The Share Exchange is being accounted for as a reverse-merger and recapitalization. BriVision is the acquirer for financial reporting purposes and the Company is the acquired company. Consequently, the assets and liabilities and the operations reflected in the historical financial statements prior to the Share Exchange will be those of BriVision and recorded at the historical cost basis of BriVision. In addition, the consolidated financial statements after completion of the Share Exchange will include the assets and liabilities of the Company and BriVision, and the historical operations of BriVision and operations of the Combined Company from the closing date of the Share Exchange.
Merger
On February 8, 2019, the Company, BioLite Holding, Inc. (“BioLite”), BioKey, Inc. (“BioKey”), BioLite Acquisition Corp., a direct wholly-owned subsidiary of Parent (“Merger Sub 1”), and BioKey Acquisition Corp., a direct wholly-owned subsidiary of Parent (“Merger Sub 2”) (collectively referred to as the “Parties”) completed the business combination pursuant to the Agreement and Plan of Merger (the “Merger Agreement”) dated as of January 31, 2018 where ABVC acquired BioLite and BioKey via issuing additional Common Stock of ABVC to the shareholders of BioLite and BioKey.
Pursuant to the terms of the Merger Agreement, BioLite and BioKey became two wholly-owned subsidiaries of the Company on February 8, 2019. ABVC issued an aggregate of 104,558,777 shares (prior to the reverse stock split in 2019) to the shareholders of both BioLite and BioKey under a registration statement on Form S-4 (file number 333-226285), which became effective by operation of law on or about February 5, 2019.
BioLite Holding, Inc. (the “BioLite Holding”) was incorporated under the laws of the State of Nevada on July 27, 2016. BioLite BVI, Inc. (the “BioLite BVI”), a wholly owned subsidiary of BioLite Holding, was incorporated in the British Virgin Islands on September 13, 2016. BioLite Holding and BioLite BVI are holding companies and have not carried out substantive business operations of their own.
BioLite, Inc., (the “BioLite Taiwan”) was incorporated on February 13, 2006 under the laws of Taiwan. BioLite is in the business of developing and commercialization of new botanical drugs with application in central nervous system, autoimmunity, inflammation, hematology, and oncology. In addition, BioLite Taiwan distributes dietary supplements made from extracts of Chinese herbs and Maitake mushroom.
In January 2017, BioLite Holding, BioLite BVI, BioLite Taiwan, and certain shareholders of BioLite Taiwan entered into a share purchase / exchange agreement (the “BioLite Share Purchase / Exchange Agreement”). Pursuant to the BioLite Share Purchase / Exchange Agreement, the shareholder participants to the BioLite Share Purchase / Exchange Agreement have sold their equity in BioLite Taiwan and were using the proceeds from such sales to purchase shares of Common Stock of BioLite Holding at the same price per share, resulting in their owning the same number of shares of Common Stock as they owned in the BioLite Taiwan. Upon closing of the Share Purchase/ Exchange Agreement in August 2017, BioLite Holding ultimately owns via BioLite BVI approximately 73% of BioLite Taiwan. The other shareholders who did not enter this Share Purchase/ Exchange Agreement retain their equity ownership in BioLite Taiwan.
BioKey, Inc. was incorporated on August 9, 2000 in the State of California. BioKey provides a wide range of services, including, API characterization, pre-formulation studies, formulation development, analytical method development, stability studies, IND/NDA/ANDA/510K submissions, and manufacturing clinical trial materials (Phase I through phase III) and commercial manufacturing. It also licenses out its technologies and initiates joint research and development processes with other biotechnology, pharmaceutical, and nutraceutical companies.
F-8
Accounting Treatment of the Merger
The Company adopted ASC 805, “Business Combination” to record the merger transactions of BioKey. Since the Company and BioLite Holding are the entities under Dr. Tsung-Shann Jiang’s common control, the transaction is accounted for as a restructuring transaction. All the assets and liabilities of BioLite Holding, BioLite BVI, and BioLite Taiwan were transferred to the Company at their respective carrying amounts on the closing date of the Merger. The Company has recast prior period financial statements to reflect the conveyance of BioLite Holding’s common shares as if the restructuring transaction had occurred as of the earliest date of the financial statements. All material intercompany accounts, transactions, and profits have been eliminated in consolidation. The nature of and effects on earnings per share (EPS) of nonrecurring intra-entity transactions involving long-term assets and liabilities is not required to be eliminated and EPS amounts have been recast to include the earnings (or losses) of the transferred net assets.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with the generally accepted accounting principles in the United States of America (the “U.S. GAAP”). All significant intercompany transactions and account balances have been eliminated.
This basis of accounting involves the application of accrual accounting and consequently, revenues and gains are recognized when earned, and expenses and losses are recognized when incurred. The Company’s financial statements are expressed in U.S. dollars.
Fiscal Year
The Company changed its fiscal year from the period beginning on October 1st and ending on September 30th to the period beginning on January 1st and ending on December 31st, beginning January 1, 2018. All references herein to a fiscal year prior to December 31, 2017 refer to the twelve months ended September 30th of such year.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the amount of revenues and expenses during the reporting periods. Actual results could differ materially from those results.
Inventory
Inventory consists of raw materials, work-in-process, finished goods, and merchandise. Inventories are stated at the lower of cost or market and valued on a moving weighted average cost basis. Market is determined based on net realizable value. The Company periodically reviews the age and turnover of its inventory to determine whether any inventory has become obsolete or has declined in value, and incurs a charge to operations for known and anticipated inventory obsolescence.
Reclassifications
Certain classifications have been made to the prior year financial statements to conform to the current year presentation. The reclassification had no impact on previously reported net loss or accumulated deficit.
Forward Stock Split
On March 21, 2016, the Board of Directors of the Company approved an amendment to Articles of Incorporation to effect a forward split at a ratio of 1 to 3.141 and increase the number of our authorized shares of Common Stock, par value $0.001 per share, to 360,000,000, which was effective on April 8, 2016.
F-9
Stock Reverse Split
On March 12, 2019, the Board of Directors of the Company by unanimous written consent in lieu of a meeting approved to i) effect a stock reverse split at the ratio of 1-for-18 (the “Reverse Split”) of both the authorized common stock of the Company (the “Common Stock”) and the issued and outstanding Common Stock and ii) to amend the articles of incorporation of the Company to reflect the Reverse Split. The Board approved and authorized the Reverse Split without obtaining approval of the Company’s shareholders pursuant to Section 78.207 of Nevada Revised Statutes. On May 3, 2019, the Company filed a certificate of amendment to the Company’s articles of incorporation (the “Amendment”) to effect the Reverse Split with the Secretary of State of Nevada. The Financial Industry Regulatory Authority (“FINRA”) informed the Company that the Reverse Split was effective on May 8, 2019. All shares and related financial information in this Form 10-K reflect this 1-for-18 reverse stock split.
Fair Value Measurements
FASB ASC 820, “Fair Value Measurements” defines fair value for certain financial and nonfinancial assets and liabilities that are recorded at fair value, establishes a framework for measuring fair value and expands disclosures about fair value measurements. It requires that an entity measure its financial instruments to base fair value on exit price, maximize the use of observable units and minimize the use of unobservable inputs to determine the exit price. It establishes a hierarchy which prioritizes the inputs to valuation techniques used to measure fair value. This hierarchy increases the consistency and comparability of fair value measurements and related disclosures by maximizing the use of observable inputs and minimizing the use of unobservable inputs by requiring that observable inputs be used when available. Observable inputs are inputs that reflect the assumptions market participants would use in pricing the assets or liabilities based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances. The hierarchy prioritizes the inputs into three broad levels based on the reliability of the inputs as follows:
| ● | Level 1 - Inputs are quoted prices in active markets for identical assets or liabilities that the Company has the ability to access at the measurement date. Valuation of these instruments does not require a high degree of judgment as the valuations are based on quoted prices in active markets that are readily and regularly available. | |
| ● | Level 2 - Inputs other than quoted prices in active markets that are either directly or indirectly observable as of the measurement date, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. | |
| ● | Level 3 - Valuations based on inputs that are unobservable and not corroborated by market data. The fair value for such assets and liabilities is generally determined using pricing models, discounted cash flow methodologies, or similar techniques that incorporate the assumptions a market participant would use in pricing the asset or liability. |
The carrying values of certain assets and liabilities of the Company, such as cash and cash equivalents, restricted cash, accounts receivable, due from related parties, inventory, prepaid expenses and other current assets, accounts payable, accrued liabilities, and due to related parties approximate fair value due to their relatively short maturities. The carrying value of the Company’s short-term bank loan, convertible notes payable, and accrued interest approximates their fair value as the terms of the borrowing are consistent with current market rates and the duration to maturity is short. The carrying value of the Company’s long-term bank loan approximates fair value because the interest rates approximate market rates that the Company could obtain for debt with similar terms and maturities.
Cash and Cash Equivalents
The Company considers highly liquid investments with maturities of three months or less, when purchased, to be cash equivalents. As of December 31, 2020 and December 31, 2019, the Company’s cash and cash equivalents amounted 4,273,208 and $144,295, respectively. Some of the Company’s cash deposits are held in financial institutions located in Taiwan where there is currently regulation mandated on obligatory insurance of bank accounts. The Company believes this financial institution is of high credit quality.
F-10
Restricted Cash Equivalents
Restricted cash equivalents primarily consist of cash held in a reserve bank account in Taiwan. As of December 31, 2020 and December 31, 2019, the Company’s restricted cash equivalents amounted $728,163 and $16,148, respectively.
Concentration of Credit Risk
The Company’s financial instruments that are exposed to concentrations of credit risk consist primarily of cash and cash equivalents. The Company places its cash and temporary cash investments in high quality credit institutions, but these investments may be in excess of Taiwan Central Deposit Insurance Corporation and the U.S. Federal Deposit Insurance Corporation’s insurance limits. The Company does not enter into financial instruments for hedging, trading or speculative purposes.
Revenue Recognition
During the fiscal year 2018, the Company adopted Accounting Standards Codification (“ASC”), Topic 606 (ASC 606), Revenue from Contracts with Customers, using the modified retrospective method to all contracts that were not completed as of January 1, 2018, and applying the new revenue standard as an adjustment to the opening balance of accumulated deficit at the beginning of 2018 for the cumulative effect. The results for the Company’s reporting periods beginning on and after January 1, 2018 are presented under ASC 606, while prior period amounts are not adjusted and continue to be reported under the accounting standards in effect for the prior period. Based on the Company’s review of existing collaborative agreements as of January 1, 2018, the Company concluded that the adoption of the new guidance did not have a significant change on the Company’s revenue during all periods presented.
Pursuant to ASC 606, the Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration that the Company expects to receive in exchange for those goods or services. To determine revenue recognition for arrangements that the Company determines is within the scope of ASC 606, the Company performs the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the Company satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the Company will collect the consideration the Company is entitled to in exchange for the goods or services the Company transfers to the customers. At inception of the contract, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract, determines those that are performance obligations, and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
The following are examples of when the Company recognizes revenue based on the types of payments the Company receives.
Collaborative Revenues — The Company recognizes collaborative revenues generated through collaborative research, development and/or commercialization agreements. The terms of these agreements typically include payment to the Company related to one or more of the following: non-refundable upfront license fees, development and commercial milestones, partial or complete reimbursement of research and development costs, and royalties on net sales of licensed products. Each type of payments results in collaborative revenues except for revenues from royalties on net sales of licensed products, which are classified as royalty revenues. To date, the Company has not received any royalty revenues. Revenue is recognized upon satisfaction of a performance obligation by transferring control of a good or service to the collaboration partners.
F-11
As part of the accounting for these arrangements, the Company applies judgment to determine whether the performance obligations are distinct, and develop assumptions in determining the stand-alone selling price for each distinct performance obligation identified in the collaboration agreements. To determine the stand-alone selling price, the Company relies on assumptions which may include forecasted revenues, development timelines, reimbursement rates for R&D personnel costs, discount rates and probabilities of technical and regulatory success.
The Company had multiple deliverables under the collaborative agreements, including deliverables relating to grants of technology licenses, regulatory and clinical development, and marketing activities. Estimation of the performance periods of the Company’s deliverables requires the use of management’s judgment. Significant factors considered in management’s evaluation of the estimated performance periods include, but are not limited to, the Company’s experience in conducting clinical development, regulatory and manufacturing activities. The Company reviews the estimated duration of its performance periods under its collaborative agreements on an annually basis, and makes any appropriate adjustments on a prospective basis. Future changes in estimates of the performance period under its collaborative agreements could impact the timing of future revenue recognition.
(i) Non-refundable upfront payments
If a license to the Company’s intellectual property is determined to be distinct from the other performance obligations identified in an arrangement, the Company recognizes revenue from the related non-refundable upfront payments based on the relative standalone selling price prescribed to the license compared to the total selling price of the arrangement. The revenue is recognized when the license is transferred to the collaboration partners and the collaboration partners are able to use and benefit from the license. To date, the receipt of non-refundable upfront fees was solely for the compensation of past research efforts and contributions made by the Company before the collaborative agreements entered into and it does not relate to any future obligations and commitments made between the Company and the collaboration partners in the collaborative agreements.
(ii) Milestone payments
The Company is eligible to receive milestone payments under the collaborative agreement with collaboration partners based on achievement of specified development, regulatory and commercial events. Management evaluated the nature of the events triggering these contingent payments, and concluded that these events fall into two categories: (a) events which involve the performance of the Company’s obligations under the collaborative agreement with collaboration partners, and (b) events which do not involve the performance of the Company’s obligations under the collaborative agreement with collaboration partners.
The former category of milestone payments consists of those triggered by development and regulatory activities in the territories specified in the collaborative agreements. Management concluded that each of these payments constitute substantive milestone payments. This conclusion was based primarily on the facts that (i) each triggering event represents a specific outcome that can be achieved only through successful performance by the Company of one or more of its deliverables, (ii) achievement of each triggering event was subject to inherent risk and uncertainty and would result in additional payments becoming due to the Company, (iii) each of the milestone payments is non-refundable, (iv) substantial effort is required to complete each milestone, (v) the amount of each milestone payment is reasonable in relation to the value created in achieving the milestone, (vi) a substantial amount of time is expected to pass between the upfront payment and the potential milestone payments, and (vii) the milestone payments relate solely to past performance. Based on the foregoing, the Company recognizes any revenue from these milestone payments in the period in which the underlying triggering event occurs.
(iii) Multiple Element Arrangements
The Company evaluates multiple element arrangements to determine (1) the deliverables included in the arrangement and (2) whether the individual deliverables represent separate units of accounting or whether they must be accounted for as a combined unit of accounting. This evaluation involves subjective determinations and requires management to make judgments about the individual deliverables and whether such deliverables are separate from other aspects of the contractual relationship. Deliverables are considered separate units of accounting provided that: (i) the delivered item(s) has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially within its control. In assessing whether an item under a collaboration has standalone value, the Company considers factors such as the research, manufacturing, and commercialization capabilities of the collaboration partner and the availability of the associated expertise in the general marketplace. The Company also considers whether its collaboration partners can use the other deliverable(s) for their intended purpose without the receipt of the remaining element(s), whether the value of the deliverable is dependent on the undelivered item(s), and whether there are other vendors that can provide the undelivered element(s).
F-12
The Company recognizes arrangement consideration allocated to each unit of accounting when all of the revenue recognition criteria in ASC 606 are satisfied for that particular unit of accounting. In the event that a deliverable does not represent a separate unit of accounting, the Company recognizes revenue from the combined unit of accounting over the Company’s contractual or estimated performance period for the undelivered elements, which is typically the term of the Company’s research and development obligations. If there is no discernible pattern of performance or objectively measurable performance measures do not exist, then the Company recognizes revenue under the arrangement on a straight-line basis over the period the Company is expected to complete its performance obligations. Conversely, if the pattern of performance in which the service is provided to the customer can be determined and objectively measurable performance measures exist, then the Company recognizes revenue under the arrangement using the proportional performance method. Revenue recognized is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line method or proportional performance method, as applicable, as of the period ending date.
At the inception of an arrangement that includes milestone payments, the Company evaluates whether each milestone is substantive and at risk to both parties on the basis of the contingent nature of the milestone. This evaluation includes an assessment of whether: (1) the consideration is commensurate with either the Company’s performance to achieve the milestone or the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from its performance to achieve the milestone, (2) the consideration relates solely to past performance and (3) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, clinical, regulatory, commercial, and other risks that must be overcome to achieve the particular milestone and the level of effort and investment required to achieve the particular milestone in making this assessment. There is considerable judgment involved in determining whether a milestone satisfies all of the criteria required to conclude that a milestone is substantive. Milestones that are not considered substantive are recognized as earned if there are no remaining performance obligations or over the remaining period of performance, assuming all other revenue recognition criteria are met.
(iv) Royalties and Profit Sharing Payments
Under the collaborative agreement with the collaboration partners, the Company is entitled to receive royalties on sales of products, which is at certain percentage of the net sales. The Company recognizes revenue from these events based on the revenue recognition criteria set forth in ASC 606. Based on those criteria, the Company considers these payments to be contingent revenues, and recognizes them as revenue in the period in which the applicable contingency is resolved.
Revenues Derived from Research and Development Activities Services — Revenues related to research and development and regulatory activities are recognized when the related services or activities are performed, in accordance with the contract terms. The Company typically has only one performance obligation at the inception of a contract, which is to perform research and development services. The Company may also provide its customers with an option to request that the Company provides additional goods or services in the future, such as active pharmaceutical ingredient, API, or IND/NDA/ANDA/510K submissions. The Company evaluates whether these options are material rights at the inception of the contract. If the Company determines an option is a material right, the Company will consider the option a separate performance obligation.
If the Company is entitled to reimbursement from its customers for specified research and development expenses, the Company accounts for the related services that it provides as separate performance obligations if it determines that these services represent a material right. The Company also determines whether the reimbursement of research and development expenses should be accounted for as revenues or an offset to research and development expenses in accordance with provisions of gross or net revenue presentation. The Company recognizes the corresponding revenues or records the corresponding offset to research and development expenses as it satisfies the related performance obligations.
F-13
The Company then determines the transaction price by reviewing the amount of consideration the Company is eligible to earn under the contracts, including any variable consideration. Under the outstanding contracts, consideration typically includes fixed consideration and variable consideration in the form of potential milestone payments. At the start of an agreement, the Company’s transaction price usually consists of the payments made to or by the Company based on the number of full-time equivalent researchers assigned to the project and the related research and development expenses incurred. The Company does not typically include any payments that the Company may receive in the future in its initial transaction price because the payments are not probable. The Company would reassess the total transaction price at each reporting period to determine if the Company should include additional payments in the transaction price.
The Company receives payments from its customers based on billing schedules established in each contract. Upfront payments and fees may be recorded as advance from customers upon receipt or when due, and may require deferral of revenue recognition to a future period until the Company performs its obligations under these arrangements. Amounts are recorded as accounts receivable when the right of the Company to consideration is unconditional. The Company does not assess whether a contract has a significant financing component if the expectation at contract inception is such that the period between payment by the customers and the transfer of the promised goods or services to the customers will be one year or less.
Property and Equipment
Property and equipment is carried at cost net of accumulated depreciation. Repairs and maintenance are expensed as incurred. Expenditures that improve the functionality of the related asset or extend the useful life are capitalized. When property and equipment is retired or otherwise disposed of, the related gain or loss is included in operating income. Leasehold improvements are depreciated on the straight-line method over the shorter of the remaining lease term or estimated useful life of the asset. Depreciation is calculated on the straight-line method, including property and equipment under capital leases, generally based on the following useful lives:
| Estimated Life in Years | ||
| Buildings and leasehold improvements | 5 ~ 50 | |
| Machinery and equipment | 5 ~ 10 | |
| Office equipment | 3 ~ 6 |
Impairment of Long-Lived Assets
The Company has adopted Accounting Standards Codification subtopic 360-10, Property, Plant and Equipment (“ASC 360-10”). ASC 360-10 requires that long-lived assets and certain identifiable intangibles held and used by the Company be reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The Company evaluates its long-lived assets for impairment annually or more often if events and circumstances warrant. Events relating to recoverability may include significant unfavorable changes in business conditions, recurring losses, or a forecasted inability to achieve break-even operating results over an extended period. Should impairment in value be indicated, the carrying value of intangible assets will be adjusted, based on estimates of future discounted cash flows resulting from the use and ultimate disposition of the asset. ASC 360-10 also requires assets to be disposed of be reported at the lower of the carrying amount or the fair value less costs to sell.
F-14
Long-term Equity Investment
The Company acquires the equity investments to promote business and strategic objectives. The Company accounts for non-marketable equity and other equity investments for which the Company does not have control over the investees as:
| ● | Equity method investments when the Company has the ability to exercise significant influence, but not control, over the investee. Its proportionate share of the income or loss is recognized monthly and is recorded in gains (losses) on equity investments. |
| ● | Non-marketable cost method investments when the equity method does not apply. |
Significant judgment is required to identify whether an impairment exists in the valuation of the Company’s non-marketable equity investments, and therefore the Company considers this a critical accounting estimate. Its yearly analysis considers both qualitative and quantitative factors that may have a significant impact on the investee’s fair value. Qualitative analysis of its investments involves understanding the financial performance and near-term prospects of the investee, changes in general market conditions in the investee’s industry or geographic area, and the management and governance structure of the investee. Quantitative assessments of the fair value of its investments are developed using the market and income approaches. The market approach includes the use of comparable financial metrics of private and public companies and recent financing rounds. The income approach includes the use of a discounted cash flow model, which requires significant estimates regarding the investees’ revenue, costs, and discount rates. The Company’s assessment of these factors in determining whether an impairment exists could change in the future due to new developments or changes in applied assumptions.
Other-Than-Temporary Impairment
The Company’s long-term equity investments are subject to a periodic impairment review. Impairments affect earnings as follows:
| ● | Marketable equity securities include the consideration of general market conditions, the duration and extent to which the fair value is below cost, and our ability and intent to hold the investment for a sufficient period of time to allow for recovery of value in the foreseeable future. The Company also considers specific adverse conditions related to the financial health of, and the business outlook for, the investee, which may include industry and sector performance, changes in technology, operational and financing cash flow factors, and changes in the investee’s credit rating. The Company records other-than-temporary impairments on marketable equity securities and marketable equity method investments in gains (losses) on equity investments. |
| ● | Non-marketable equity investments based on the Company’s assessment of the severity and duration of the impairment, and qualitative and quantitative analysis of the operating performance of the investee; adverse changes in market conditions and the regulatory or economic environment; changes in operating structure or management of the investee; additional funding requirements; and the investee’s ability to remain in business. A series of operating losses of an investee or other factors may indicate that a decrease in value of the investment has occurred that is other than temporary and that shall be recognized even though the decrease in value is in excess of what would otherwise be recognized by application of the equity method. A loss in value of an investment that is other than a temporary decline shall be recognized. Evidence of a loss in value might include, but would not necessarily be limited to, absence of an ability to recover the carrying amount of the investment or inability of the investee to sustain an earnings capacity that would justify the carrying amount of the investment. The Company records other-than-temporary impairments for non-marketable cost method investments and equity method investments in gains (losses) on equity investments. Other-than-temporary impairments of equity investments were $961,217 and $0 for the year ended December 31, 2020 and 2019, respectively. |
F-15
Goodwill
The Company evaluates goodwill for impairment annually or more frequently when an event occurs or circumstances change that indicate the carrying value may not be recoverable. In testing goodwill for impairment, the Company may elect to utilize a qualitative assessment to evaluate whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount. If the qualitative assessment indicates that goodwill impairment is more likely than not, the Company performs a two-step impairment test. The Company tests goodwill for impairment under the two-step impairment test by first comparing the book value of net assets to the fair value of the reporting units. If the fair value is determined to be less than the book value or qualitative factors indicate that it is more likely than not that goodwill is impaired, a second step is performed to compute the amount of impairment as the difference between the estimated fair value of goodwill and the carrying value. The Company estimates the fair value of the reporting units using discounted cash flows. Forecasts of future cash flows are based on our best estimate of future net sales and operating expenses, based primarily on expected category expansion, pricing, market segment share, and general economic conditions.
The Company completed the required testing of goodwill for impairment as of December 31, 2020, and determined that goodwill was impaired because of the current financial condition of the Company and the Company’s inability to generate future operating income without substantial sales volume increases, which are highly uncertain. Furthermore, the Company anticipates future cash flows indicate that the recoverability of goodwill is not reasonably assured.
Research and Development Expenses
The Company accounts for the cost of using licensing rights in research and development cost according to ASC Topic 730-10-25-1. This guidance provides that absent alternative future uses the acquisition of product rights to be used in research and development activities must be charged to research and development expenses when incurred.
For CDMO business unit, the Company accounts for R&D costs in accordance with Accounting Standards Codification (“ASC”) 730, Research and Development (“ASC 730”). Research and development expenses are charged to expense as incurred unless there is an alternative future use in other research and development projects or otherwise. Research and development expenses are comprised of costs incurred in performing research and development activities, including personnel-related costs, facilities-related overhead, and outside contracted services including clinical trial costs, manufacturing and process development costs for both clinical and preclinical materials, research costs, and other consulting services. Non-refundable advance payment for goods and services that will be used in future research and development activities are expensed when the activity has been performed or when the goods have been received rather than when the payment is made. In instances where the Company enters into agreements with third parties to provide research and development services, costs are expensed as services are performed.
Post-retirement and post-employment benefits
The Company’s subsidiaries in Taiwan adopted the government mandated defined contribution plan pursuant to the Labor Pension Act (the “Act”) in Taiwan. Such labor regulations require that the rate of contribution made by an employer to the Labor Pension Fund per month shall not be less than 6% of the worker’s monthly salaries. Pursuant to the Act, the Company makes monthly contribution equal to 6% of employees’ salaries to the employees’ pension fund. The Company has no legal obligation for the benefits beyond the contributions made. The total amounts for such employee benefits, which were expensed as incurred, were $13,961and $15,928 for the year ended December 31, 2020 and 2019, respectively. Other than the above, the Company does not provide any other post-retirement or post-employment benefits.
Stock-based Compensation
The Company measures expense associated with all employee stock-based compensation awards using a fair value method and recognizes such expense in the consolidated financial statements on a straight-line basis over the requisite service period in accordance with FASB ASC Topic 718 “Compensation-Stock Compensation”. Total employee stock-based compensation expenses were $1,623,102 and $0 for the year ended December 31, 2020 and 2019, respectively.
The Company accounted for stock-based compensation to non-employees in accordance with FASB ASC Topic 718 “Compensation-Stock Compensation” and FASB ASC Topic 505-50 “Equity-Based Payments to Non-Employees” which requires that the cost of services received from non-employees is measured at fair value at the earlier of the performance commitment date or the date service is completed and recognized over the period the service is provided. Total non-employee stock-based compensation expenses were $2,523,877 and $22,314 for the year ended December 31, 2020 and 2019, respectively.
F-16
Beneficial Conversion Feature
From time to time, the Company may issue convertible notes that may contain an imbedded beneficial conversion feature. A beneficial conversion feature exists on the date a convertible note is issued when the fair value of the underlying common stock to which the note is convertible into is in excess of the remaining unallocated proceeds of the note after first considering the allocation of a portion of the note proceeds to the fair value of the warrants, if related warrants have been granted. The intrinsic value of the beneficial conversion feature is recorded as a debt discount with a corresponding amount to additional paid in capital. The debt discount is amortized to interest expense over the life of the note using the effective interest method.
Income Taxes
The Company accounts for income taxes using the asset and liability approach which allows the recognition and measurement of deferred tax assets to be based upon the likelihood of realization of tax benefits in future years. Under the asset and liability approach, deferred taxes are provided for the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. A valuation allowance is provided for deferred tax assets if it is more likely than not these items will expire before the Company is able to realize their benefits, or future deductibility is uncertain.
Under ASC 740, a tax position is recognized as a benefit only if it is “more likely than not” that the tax position would be sustained in a tax examination, with a tax examination being presumed to occur. The evaluation of a tax position is a two-step process. The first step is to determine whether it is more-likely-than-not that a tax position will be sustained upon examination, including the resolution of any related appeals or litigations based on the technical merits of that position. The second step is to measure a tax position that meets the more-likely-than-not threshold to determine the amount of benefits recognized in the financial statements. A tax position is measured at the largest amount of benefit that is greater than 50 percent likely of being realized upon ultimate settlement. Tax positions that previously failed to meet the more-likely-than-not recognition threshold should be recognized in the first subsequent period in which the threshold is met. Previously recognized tax positions that no longer meet the more-likely-than-not criteria should be de-recognized in the first subsequent financial reporting period in which the threshold is no longer satisfied. Penalties and interest incurred related to underpayment of income tax are classified as income tax expense in the year incurred. No significant penalty or interest relating to income taxes has been incurred for the year ended December 31, 2020 and 2019. GAAP also provides guidance on de-recognition, classification, interest and penalties, accounting in interim periods, disclosures and transition.
On December 22, 2017, the SEC issued Staff Accounting Bulletin (“SAB 118”), which provides guidance on accounting for tax effects of the Tax Act. SAB 118 provides a measurement period that should not extend beyond one year from the Tax Act enactment date for companies to complete the accounting under ASC 740. In accordance with SAB 118, a company must reflect the income tax effects of those aspects of the Act for which the accounting under ASC 740 is complete. To the extent that a company’s accounting for certain income tax effects of the Tax Act is incomplete but it is able to determine a reasonable estimate, it must record a provisional estimate to be included in the financial statements. If a company cannot determine a provisional estimate to be included in the financial statements, it should continue to apply ASC 740 on the basis of the provision of the tax laws that were in effect immediately before the enactment of the Tax Act. While the Company is able to make reasonable estimates of the impact of the reduction in corporate rate and the deemed repatriation transition tax, the final impact of the Tax Act may differ from these estimates, due to, among other things, changes in our interpretations and assumptions, additional guidance that may be issued by the I.R.S., and actions the Company may take. The Company is continuing to gather additional information to determine the final impact.
Valuation of Deferred Tax Assets
A valuation allowance is recorded to reduce the Company’s deferred tax assets to the amount that is more likely than not to be realized. In assessing the need for the valuation allowance, management considers, among other things, projections of future taxable income and ongoing prudent and feasible tax planning strategies. If the Company determines that sufficient negative evidence exists, then it will consider recording a valuation allowance against a portion or all of the deferred tax assets in that jurisdiction. If, after recording a valuation allowance, the Company’s projections of future taxable income and other positive evidence considered in evaluating the need for a valuation allowance prove, with the benefit of hindsight, to be inaccurate, it could prove to be more difficult to support the realization of its deferred tax assets. As a result, an additional valuation allowance could be required, which would have an adverse impact on its effective income tax rate and results. Conversely, if, after recording a valuation allowance, the Company determines that sufficient positive evidence exists in the jurisdiction in which the valuation allowance was recorded, it may reverse a portion or all of the valuation allowance in that jurisdiction. In such situations, the adjustment made to the deferred tax asset would have a favorable impact on its effective income tax rate and results in the period such determination was made.
F-17
Loss Per Share of Common Stock
The Company calculates net loss per share in accordance with ASC Topic 260, “Earnings per Share”. Basic loss per share is computed by dividing the net loss by the weighted average number of common shares outstanding during the period. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common stock equivalents had been issued and if the additional common shares were dilutive. Diluted earnings per share excludes all dilutive potential shares if their effect is anti-dilutive.
Commitments and Contingencies
The Company has adopted ASC Topic 450 “Contingencies” subtopic 20, in determining its accruals and disclosures with respect to loss contingencies. Accordingly, estimated losses from loss contingencies are accrued by a charge to income when information available before financial statements are issued or are available to be issued indicates that it is probable that an assets had been impaired or a liability had been incurred at the date of the financial statements and the amount of the loss can be reasonably estimated. Legal expenses associated with the contingency are expensed as incurred. If a loss contingency is not probable or reasonably estimable, disclosure of the loss contingency is made in the financial statements when it is at least reasonably possible that a material loss could be incurred.
Foreign-currency Transactions
For the Company’s subsidiaries in Taiwan, the foreign-currency transactions are recorded in New Taiwan dollars (“NTD”) at the rates of exchange in effect when the transactions occur. Gains or losses resulting from the application of different foreign exchange rates when cash in foreign currency is converted into New Taiwan dollars, or when foreign-currency receivables or payables are settled, are credited or charged to income in the year of conversion or settlement. On the balance sheet dates, the balances of foreign-currency assets and liabilities are restated at the prevailing exchange rates and the resulting differences are charged to current income except for those foreign currencies denominated investments in shares of stock where such differences are accounted for as translation adjustments under the Statements of Stockholders’ Equity (Deficit).
Translation Adjustment
The accounts of the Company’s subsidiaries in Taiwan were maintained, and their financial statements were expressed, in New Taiwan Dollar (“NT$”). Such financial statements were translated into U.S. Dollars (“$” or “USD”) in accordance ASC 830, “Foreign Currency Matters”, with the NT$ as the functional currency. According to the Statement, all assets and liabilities are translated at the current exchange rate, stockholder’s deficit are translated at the historical rates and income statement items are translated at an average exchange rate for the period. The resulting translation adjustments are reported under other comprehensive income (loss) as a component of stockholders’ equity (deficit).
Recent Accounting Pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes, as part of its overall simplification initiative to reduce costs and complexity of applying accounting standards while maintaining or improving the usefulness of the information provided to users of financial statements. The FASB’s amendments primarily impact ASC 740, Income Taxes, and may impact both interim and annual reporting periods. ASU 2019-12 will be effective for fiscal years beginning after December 15, 2020, and interim periods within those fiscal years and early adoption is permitted. The Company is currently evaluating the impact of adopting ASU 2019-12.
In August 2020, the FASB issued ASU 2020-06, Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging- Contracts in Entity’s Own Equity (Subtopic 815-40), to reduce the complexity associated with applying U.S. GAAP principles for certain financial instruments with characteristics of liabilities and equity. The amendments in this ASU reduce the number of accounting models for convertible instruments and expand the existing disclosure requirements over earnings per share as it relates to convertible instruments. This ASU will be effective for the fiscal year beginning January 1, 2022 and interim periods therein. Early adoption is permitted, but no earlier than fiscal years beginning after December 15, 2020. The amendments may be adopted through either a modified retrospective method, or a fully retrospective method. The Company is currently evaluating the impact of adopting ASU 2020-06.
F-18
3. COLLABORATIVE AGREEMENTS
Collaborative agreements with BHK
(i) On February 24, 2015, BioLite Taiwan and BioHopeKing Corporation (the “BHK”) entered into a co-development agreement, (the “BHK Co-Development Agreement”), pursuant to which it is collaborative with BHK to develop and commercialize BLI-1401-2 (Botanical Drug) Triple Negative Breast Cancer (TNBC) Combination Therapy (BLI-1401-2 Products) in Asian countries excluding Japan for all related intellectual property rights, and has developed it for medicinal use in collaboration with outside researchers. The development costs shall be shared 50/50 between BHK and the Company. The BHK Co-Development Agreement will remain in effect for fifteen years from the date of first commercial sale of the Product in in Asia excluding Japan.
On July 27, 2016, BioLite Taiwan and BHK agreed to amend the payment terms of the milestone payment in an aggregate amount of $10 million based on the following schedule:
| ● | Upon the signing of the BHK Co-Development Agreement: $1 million, or 10% of total payment |
| ● | Upon the first Investigational New Drug (IND) submission and BioLite Taiwan will deliver all data to BHK according to FDA Reviewing requirement: $1 million, or 10% of total payment |
| ● | At the completion of first phase II clinical trial: $1 million, or 10% of total payment |
| ● | At the initiation of phase III of clinical trial research: $3 million, or 30% of total payment |
| ● | Upon the New Drug Application (NDA) submission: $4 million, or 40% of total payment |
In December 2015, BHK has paid a non-refundable upfront cash payment of $1 million, or 10% of $10,000,000, upon the signing of BHK Co-Development Agreement. The Company concluded that the deliverables are considered separate units of accounting as the delivered items have value to the customer on a standalone basis and recognized this cash receipt as collaboration revenue when all research, technical, and development data was delivered to BHK in 2015. The receipt is for the compensation of past research efforts and contributions made by BioLite Taiwan before this collaborative agreement was signed and it does not relate to any future commitments made by BioLite Taiwan and BHK in this collaborative agreement. In August 2016, the Company has received the second milestone payment of NT$31,649,000, approximately equivalent to $1 million, and recognized collaboration revenue for the year ended December 31, 2016. As of the date of this report, the Company has not completed the first phase II clinical trial.
In addition to the milestone payments, BioLite Taiwan is entitled to receive royalty on 12% of BHK’s net sales related to BLI-1401-2 Products. As of December 31, 2020 and 2019, the Company has not earned the royalty under the BHK Co-Development Agreement.
(ii) On December 9, 2015, BioLite Taiwan entered into another two collaborative agreements (the “BHK Collaborative Agreements”), pursuant to which it is collaborative with BHK to co-develop and commercialize BLI-1005 for “Targeting Major Depressive Disorder” (BLI-1005 Products) and BLI-1006 for “Targeting Inflammatory Bowel Disease” (BLI-1006 Products) in Asia excluding Japan for all related intellectual property rights, and has developed it for medicinal use in collaboration with outside researchers. The development costs shall be shared 50/50 between BHK and the Company. The BHK Co-Development Agreement will remain in effect for fifteen years from the date of first commercial sale of the Product in in Asia excluding Japan.
In 2015, the Company recognized the cash receipt in a total of NT$50 million, approximately equivalent to $1.6 million, as collaboration revenue when all research, technical, and development data was delivered to BHK. The Company concluded that the deliverables are considered separate units of accounting as the delivered items have value to the customer on a standalone basis and recognized this payment as collaboration revenue when all research, technical, data and development data was delivered to BHK. The cash receipt is for the compensation of past research efforts and contributions made by BioLite Taiwan before this BHK Collaborative Agreements was signed and it does not relate to any future commitments made by BioLite Taiwan and BHK in this BHK Collaborative Agreements.
In addition to the total of NT$50 million, approximately equivalent to $1.60 million, BioLite Taiwan is entitled to receive 50% of the future net licensing income or net sales profit. As of December 31, 2020 and 2019, the Company has not earned the royalty under the BHK Collaborative Agreements.
F-19
Co-Development agreement with Rgene Corporation, a related party
On May 26, 2017, American BriVision Corporation entered into a co-development agreement (the “Co-Dev Agreement”) with Rgene Corporation (the “Rgene”), a related party under common control by controlling beneficiary shareholder of YuanGene Corporation and the Company (See Note 12). Pursuant to Co-Dev Agreement, BriVision and Rgene agreed to co-develop and commercialize ABV-1507 HER2/neu Positive Breast Cancer Combination Therapy, ABV-1511 Pancreatic Cancer Combination Therapy and ABV-1527 Ovary Cancer Combination Therapy. Under the terms of the Co-Dev Agreement, Rgene is required to pay the Company $3,000,000 in cash or stock of Rgene with equivalent value by August 15, 2017. The payment is for the compensation of BriVision’s past research efforts and contributions made by BriVision before the Co-Dev Agreement was signed and it does not relate to any future commitments made by BriVision and Rgene in this Co-Dev Agreement. In addition to $3,000,000, the Company is entitled to receive 50% of the future net licensing income or net sales profit earned by Rgene, if any, and any development costs shall be equally shared by both BriVision and Rgene.
On June 1, 2017, the Company has delivered all research, technical, data and development data to Rgene. Since both Rgene and the Company are related parties and under common control by a controlling beneficiary shareholder of YuanGene Corporation and the Company, the Company has recorded the full amount of $3,000,000 in connection with the Co-Dev Agreement as additional paid-in capital during the year ended December 31, 2017. During the year ended December 31, 2017, the Company has received $450,000 in cash. On December 24, 2018, the Company received the remaining balance of $2,550,000 in the form of newly issued shares of Rgene’s Common Stock, at the price of NT$50 (approximately equivalent to $1.60 per share), for an aggregate number of 1,530,000 shares, which accounted for equity method long-term investment as of December 31, 2018. During the year ended December 31, 2018, the Company has recognized investment loss of $549. On December 31, 2018, the Company determined to fully write off this investment based on the Company’s assessment of the severity and duration of the impairment, and qualitative and quantitative analysis of the operating performance of the investee, adverse changes in market conditions and the regulatory or economic environment, changes in operating structure of Rgene, additional funding requirements, and Rgene’s ability to remain in business. All projects that have been initiated will be managed and supported by the Company and Rgene.
The Company and Rgene signed an amendment to the Co-Dev Agreement on November 10, 2020, pursuant to which both parties agreed to delete AB-1507 HER2/neu Positive Breast Cancer Combination Therapy and AB 1527 Ovary Cancer Combination Therapy and add ABV-1519 EGFR Positive Non-Small Cell Lung Cancer Combination Therapy and ABV-1526 Large Intestine / Colon / Rectal Cancer Combination Therapy to the products to be co-developed and commercialized. Other provisions of the Co-Dev Agreement remain in full force and effect.
Collaborative agreement with BioFirst Corporation, a related party
On July 24, 2017, American BriVision Corporation entered into a collaborative agreement (the “BioFirst Collaborative Agreement”) with BioFirst Corporation (“BioFirst”), pursuant to which BioFirst granted the Company the global licensing right for medical use of the product (the “Product”): BFC-1401 Vitreous Substitute for Vitrectomy. BioFirst is a related party to the Company because a controlling beneficiary shareholder of YuanGene Corporation and the Company is one of the directors and Common Stock shareholders of BioFirst (See Note 12).
Pursuant to the BioFirst Collaborative Agreement, the Company will co-develop and commercialize the Product with BioFirst and pay BioFirst in a total amount of $3,000,000 in cash or stock of the Company before December 31, 2018. The amount of $3,000,000 is in connection with the compensation for BioFirst’s past research efforts and contributions made by BioFirst before the BioFirst Collaborative Agreement was signed and it does not relate to any future commitments made by BioFirst and BriVision in this BioFirst Collaborative Agreement. In addition, the Company is entitled to receive 50% of the future net licensing income or net sales profit, if any, and any development cost shall be equally shared by both BriVision and BioFirst.
F-20
On September 25, 2017, BioFirst has delivered all research, technical, data and development data to BriVision. The Company determined to fully expense the entire amount of $3,000,000 since currently the related licensing rights do not have alternative future uses. According to ASC 730-10-25-1, absent alternative future uses the acquisition of product rights to be used in research and development activities must be charged to research and development expenses immediately. Hence, the entire amount of $3,000,000 is fully expensed as research and development expense during the year ended December 31, 2017.
On June 30, 2019, BriVision entered into a Stock Purchase Agreement (the “Purchase Agreement”) with BioFirst Corporation (“BioFirst”). Pursuant to the Purchase Agreement, the Company issued 428,571 shares of the Company’s common stock (the “Shares”) to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst (the “Total Payment”) in connection with a certain collaborative agreement between the Company and BioFirst dated July 24, 2017 (the “Collaborative Agreement”). Pursuant to the Collaborative Agreement, BioFirst granted the Company the global licensing right to co-develop BFC-1401 or ABV-1701 Vitreous Substitute for Vitrectomy for medical purposes in consideration for the Total Payment.
On August 5, 2019, BriVision entered into a second Stock Purchase Agreement (“Purchase Agreement 2”) with BioFirst Corporation (“BioFirst”). Pursuant to Purchase Agreement 2, the Company issued 414,702 shares of the Company’s common stock (the “Shares”) to BioFirst in consideration for $2,902,911 owed by the Company to BioFirst (the “Total Payment”) in connection with a loan provided to BriVision from BioFirst.
4. INVENTORY
Inventory consists of the following:
| December
31, 2020 | December 31, 2019 | |||||||
| Finished goods | $ | 100,967 | $ | 94,727 | ||||
| Work-in-process | 22,038 | 20,676 | ||||||
| Raw materials | 61,718 | 57,904 | ||||||
| Allowance for inventory valuation and obsolescence loss | (184,723 | ) | (173,307 | ) | ||||
| Inventory, net | $ | - | $ | - | ||||
5. PROPERTY AND EQUIPMENT
Property and equipment as of December 31, 2020 and 2019 are summarized as follows:
| December
31, 2020 | December 31, 2019 | |||||||
| Land | $ | 395,645 | $ | 371,195 | ||||
| Buildings and leasehold improvements | 2,233,573 | 2,225,386 | ||||||
| Machinery and equipment | 994,544 | 987,234 | ||||||
| Office equipment | 189,760 | 178,409 | ||||||
| 3,813,522 | 3,762,224 | |||||||
| Less: accumulated depreciation | (3,298,688 | ) | (3,241,294 | ) | ||||
| Property and equipment, net | $ | 514,834 | $ | 520,930 | ||||
Depreciation expense were $37,142 and $55,086 for the year ended December 31, 2020 and 2019, respectively.
F-21
6. LONG-TERM INVESTMENTS
| (1) | The ownership percentages of each investee are listed as follows: |
| Ownership percentage | ||||||||||
| December 31, | December 31, | Accounting | ||||||||
| Name of related party | 2020 | 2019 | treatments | |||||||
| Braingenesis Biotechnology Co., Ltd. | 0.17 | % | 0.17 | % | Cost Method | |||||
| Genepharm Biotech Corporation | 0.70 | % | 0.72 | % | Cost Method | |||||
| BioHopeKing Corporation | 5.90 | % | 7.13 | % | Cost Method | |||||
| BioFirst Corporation | 15.99 |
% | 15.89 |
% | Equity Method | |||||
| Rgene Corporation | 31.62 | % | 31.61 | % | Equity Method | |||||
| (2) | The extent the investee relies on the company for its business are summarized as follows: |
| Name of related party | The extent the investee relies on the Company for its business | |
| Braingenesis Biotechnology Co., Ltd. | No specific business relationship | |
| Genepharm Biotech Corporation | No specific business relationship | |
| BioHopeKing Corporation | Collaborating with the Company to develop and commercialize drugs | |
| BioFirst Corporation | Loaned from the investee and provides research and development support service | |
| Rgene Corporation | Collaborating with the Company to develop and commercialize drugs |
| (3) | Long-term investment mainly consists of the following: |
| December 31, 2020 | December 31, 2019 | |||||||
| Non-marketable Cost Method Investments, net | ||||||||
| Braingenesis Biotechnology Co., Ltd. | $ | 7,853 | $ | 7,367 | ||||
| Genepharm Biotech Corporation | 23,974 | 22,493 | ||||||
| BioHopeKing Corporation | 890,564 | 1,998,310 | ||||||
| Sub total | 922,391 | 2,028,170 | ||||||
| Equity Method Investments, net | ||||||||
| BioFirst Corporation | 268,336 | 1,336,449 | ||||||
| Rgene Corporation | - | - | ||||||
| Total | $ | 1,190,727 | $ | 3,364,619 | ||||
| (a) | BioFirst Corporation (the “BioFirst”): |
The Company holds an equity interest in BioFirst Corporation, accounting for its equity interest using the equity method to accounts for its equity investment as prescribed in ASC 323, Investments—Equity Method and Joint Ventures (“ASC 323”). Equity method adjustments include the Company’s proportionate share of investee’s income or loss and other adjustments required by the equity method. As of December 31, 2020 and 2019, the Company owns 15.99% and 15.89% common stock shares of BioFirst, respectively.
F-22
Summarized financial information for the Company’s equity method investee, BioFirst, is as follows:
Balance Sheet
| December 31, 2020 | December 31, 2019 | |||||||
| Current Assets | $ | 1,299,822 | $ | 1,350,701 | ||||
| Noncurrent Assets | 2,540,041 | 7,450,032 | ||||||
| Current Liabilities | 1,986,340 | 2,060,460 | ||||||
| Noncurrent Liabilities | 73,197 | 78,888 | ||||||
| Stockholders’ Equity | 1,780,326 | 6,661,385 | ||||||
Statement of operation
| Year
Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Net sales | $ | 257,235 | $ | 43,975 | ||||
| Gross profit | 10,121 | (37,160 | ) | |||||
| Net loss | (5,401,074 | ) | (972,303 | ) | ||||
| Share of losses from investments accounted for using the equity method | (1,168,733 | ) | (210,086 | ) | ||||
| (b) | Rgene Corporation (the “Rgene”) |
Both Rgene and the Company are under common control by Dr. Tsung-Shann Jiang, the CEO and chairman of the BioLite Inc. Since Dr. Tsung-Shann Jiang is able to exercise significant influence, but not control, over the Rgene, the Company determined to use the equity method to accounts for its equity investment as prescribed in ASC 323, Investments—Equity Method and Joint Ventures (“ASC 323”). Equity method adjustments include the Company’s proportionate share of investee’s income or loss and other adjustments required by the equity method. As of December 31, 2020 and 2019, the Company owns 31.62% and 31.61% common stock shares of Rgene, respectively.
Summarized financial information for the Company’s equity method investee, Rgene, is as follows:
Balance Sheets
| December
31, 2020 | December 31, 2019 | |||||||
| Current Assets | $ | 123,958 | $ | 82,254 | ||||
| Noncurrent Assets | 412,342 | 62,768 | ||||||
| Current Liabilities | 1,392,756 | 312,950 | ||||||
| Noncurrent Liabilities | 38,953 | - | ||||||
| Shareholders’ Deficit | (895,409 | ) | (167,928 | ) | ||||
Statement of operations
| Year
Ended December 31, |
||||||||
| 2020 | 2019 | |||||||
| Net sales | $ | 16,595 | $ | - | ||||
| Gross Profit | (335,735 | ) | - | |||||
| Net loss | (641,636 | ) | (53,877 | ) | ||||
| Share of loss from investments accounted for using the equity method | - | - | ||||||
F-23
| (4) | Disposition of long-term investment |
During the year ended December 31, 2020, the Company sold 218,000 shares of common stock of BioHopeKing Corporation at price of NT$24, equivalent $0.85, to several individuals, and the percentage of ownership decreased to 5.90% as of December 31, 2020. As a result of the transactions, the Company recognized investment loss of $40,589 and impairment loss of $961,217 for the same period.
| (5) | Losses on Equity Investments |
The components of losses on equity investments for each period were as follows:
| Year
Ended December 31, |
||||||||
| 2020 | 2019 | |||||||
| Share of equity method investee losses | $ | (1,168,733 | ) | $ | (210,086 | ) | ||
7. CONVERTIBLE NOTES PAYABLE
On May 9, 2018, the Company issued an eighteen-month term unsecured convertible promissory note (the “Yu and Wei Note”) in an aggregate principal amount of $300,000 to Guoliang Yu and Yingfei Wei Family Trust (the “Yu and Wei”), pursuant to which the Company received $300,000. The Yu and Wei Note bears interest at 8% per annum. The Company shall pay to the Yu and Wei an amount in cash representing all outstanding principal and accrued and unpaid interest on the Eighteenth (18) month anniversary of the issuance date of the Yu and Wei Note, which is on November 8, 2019. In the event that the Company raises gross proceeds from the sale of its common stock of at least $5,000,000 (an “Equity Offering”) then within five days of the closing for such offering, the Company must repay the outstanding amount of this Yu and Wei Note. At any time from the date hereof until this Yu and Wei Note has been satisfied, the Yu and Wei may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $2.00 per share (the “Fixed Conversion Price”), subject to adjustment or (ii) 80% of the per share offering price (the “Alternative Conversion Price”) of any completed equity offering of the Company in an amount exceeding $500,000 that occurs when any part of the Yu and Wei Note is outstanding, subject to adjustments set forth in the Yu and Wei Note. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the Yu and Wei Note. On January 21, 2020, Yu and Wei entered into a new agreement that the new Note bears interest at 20% per annum. The Company shall pay to the Yu and Wei an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the new “Yu and Wei” Note, which is on January 20, 2021. On April 5, 2020, the Company entered into an exchange agreement with “Yu and Wei”. The aggregate principal amount plus accrued interest expenses were $354,722, and the Company agreed to issue to the Holders an aggregate of 192,784 shares of the Company’s common stock, and warrants to purchase 192,784 shares of the Company’s common stock. As of December 31, 2020, these common shares have been issued.
F-24
On June 27, 2018, the Company issued an eighteen-month term unsecured convertible promissory note (the “Keypoint Note”) in the aggregate principal amount of $250,000 to Keypoint Technology Ltd. (“Keypoint”), a related party, pursuant to which the Company received $250,000. The Keypoint Note bears interest at 8% per annum. The Company shall pay to the Keypoint an amount in cash representing all outstanding principal and accrued and unpaid interest on the Eighteenth (18) month anniversary of the issuance date of the Keypoint Note, which is on December 26, 2019. In the event that the Company raises gross proceeds from the sale of its common stock of at least $5,000,000 (an “Equity Offering”) then within five days of the closing for such offering, the Company must repay the outstanding amount of this Keypoint Note. At any time from the date hereof until this Keypoint Note has been satisfied, Keypoint may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $2.00 per share (the “Fixed Conversion Price”), subject to adjustment or (ii) 80% of the per share offering price (the “Alternative Conversion Price”) of any completed equity offering of the Company in an amount exceeding $500,000 that occurs when any part of the Keypoint Note is outstanding, subject to adjustments set forth in the Keypoint Note. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the Keypoint Note. On January 21, 2020, Keypoint entered into a new agreement that the new Note bears interest at 20% per annum. The Company shall pay to the Keypoint an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the new “Keypoint” Note, which is on January 20, 2021. On April 5, 2020, the Company entered into an exchange agreement with “Keypoint”. The aggregate principal amount plus accrued interest expenses were $292,826, and the Company agreed to issue to the Holders an aggregate of 159,145 shares of the Company’s common stock, and warrants to purchase 159,145 shares of the Company’s common stock. As of December 31, 2020, these common shares have been issued.
On August 25, 2018, the Company issued an eighteen-month term unsecured convertible promissory notes (the “Odaira Note”) in the aggregate principal amount of $250,000 to Yoshinobu Odaira. (“Odaira”), pursuant to which the Company received $250,000. The Odaira Note bears interest at 8% per annum. The Company shall pay to the Odaira an amount in cash representing all outstanding principal and accrued and unpaid interest on the Eighteenth (18) month anniversary of the issuance date of the Odaira Note, which is on February 24, 2020. In the event that the Company raises gross proceeds from the sale of its common stock of at least $5,000,000 (an “Equity Offering”) then within five days of the closing for such offering, the Company must repay the outstanding amount of this Odaira Note. At any time from the date hereof until this Odaira Note has been satisfied, Odaira may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $2.00 per share (the “Fixed Conversion Price”), subject to adjustment or (ii) 80% of the per share offering price (the “Alternative Conversion Price”) of any completed equity offering of the Company in an amount exceeding $500,000 that occurs when any part of the Odaira Note is outstanding, subject to adjustments set forth in the Odaira Note. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the Odaira Note. On January 21, 2020, Odaira entered into a new agreement that the new Note bears interest at 20% per annum. The Company shall pay to the Odaira an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the new “Odaira” Note, which is on January 20, 2021. On April 5, 2020, the Company entered into an exchange agreement with “Odaira”. The aggregate principal amount plus accrued interest expenses were $284,036, and the Company agreed to issue to the Holders an aggregate of 154,368 shares of the Company’s common stock, and warrants to purchase 154,368 shares of the Company’s common stock. As of December 31, 2020, these common shares have been issued.
On May 30 and July 10, 2019, the Company issued two (2) twelve-month term unsecured convertible promissory notes (the “KSL Note”) in an aggregate principal amount of $250,000 to Kuo Sheng Lung (the “KSL”), pursuant to which the Company received $160,000 and $90,000, respectively. The KSL Note bears interest at 20% per annum. The Company shall pay to KSL an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the KSL Note, which is on May 29, 2020 and July 9, 2020. At any time from the issuance date until the KSL Note has been satisfied, the KSL may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $0.50 per share (the “Fixed Conversion Price”), subject to adjustment, or (ii) 70% of the per share offering price (the “Alternative Conversion Price”) of the completed public equity offering of the Company in an amount exceeding $10,000,000 as stated on the registration statement on a Form S-1 filed with the Securities and Exchange Commission on November 14, 2018 (the “Public Offering”), as amended from time to time. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of the embedded beneficial conversion feature present in the KSL Note. On May 13, 2020, the Company received an acknowledgement letter from KSL that they will not claim the repayment of loan for 12 months. On November 9, 2020, the Company entered into an agreement with “KSL”. The aggregate principal amount plus accrued interest expenses are $270,272, and KSL agreed to use the full amount to purchase certain securities pursuant to a securities purchase agreement; KSL agreed to purchase and the Company agreed to issue 120,121 shares of the Company’s common stock and warrants for a purchase price of $270,272. As of December 31, 2020, the Company issued to the Holders an aggregate of 120,121 shares of the Company’s common stock.
F-25
On July 10, 2019, the Company issued a twelve-month term unsecured convertible promissory note (the “NEA Note”) in an aggregate principal amount of $250,000 to New Eastern Asia (the “NEA”), a related party, pursuant to which the Company received $250,000 on July 10, 2019. The NEA Note bears interest at 20% per annum. The Company shall pay to the NEA an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the NEA Note, which is on July 9, 2020. At any time from the date hereof until this NEA Note has been satisfied, the NEA may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $.50 per share (the “Fixed Conversion Price”), subject to adjustment, or (ii) 70% of the per share offering price (the “Alternative Conversion Price”) of the completed public equity offering of the Company in an amount exceeding $10,000,000 as stated on the registration statement on a Form S-1 filed with the Securities and Exchange Commission on November 14, 2018 (the “Public Offering”), as amended from time to time. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the NEA Note. As of December 31, 2020, the Company paid off the convertible promissory note of $306,667, including principal and accrued and unpaid interest expense.
On August 28, 2019, the Company issued a twelve-month term unsecured convertible promissory note (the “KLS Note”) in an aggregate principal amount of $200,000 to Kuo Li Shen (the “KLS”), pursuant to which the Company received $200,000 on August 28, 2019. The KLS Note bears interest at 20% per annum. The Company shall pay to the KLS an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the KLS Note, which is on August 27, 2020. At any time from the date hereof until this KLS Note has been satisfied, the KLS may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $.50 per share (the “Fixed Conversion Price”), subject to adjustment, or (ii) 70% of the per share offering price (the “Alternative Conversion Price”) of the completed public equity offering of the Company in an amount exceeding $10,000,000 as stated on the registration statement on a Form S-1 filed with the Securities and Exchange Commission on November 14, 2018 (the “Public Offering”), as amended from time to time. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the KLS Note. On April 20, 2020, the Company entered into an exchange agreement with KLS. The aggregate principal amount plus accrued interest expenses were $225,222, and the Company agreed to issue to the Holders an aggregate of 126,530 shares of the Company’s common stock, and warrants to purchase 126,530 shares of common stock. As of December 31, 2020, these common shares have been issued.
On September 4, 2019, the Company issued 3 twelve-month term unsecured convertible promissory note (the “C.L.L. Note”) in an aggregate principal amount of $257,500 to Chang Ping Shan, Lin Shan Tyan, and Liu Ching Hsuan (together the “C.L.L.”), pursuant to which the Company received $257,500 on September 4, 2019. Chang Ping Shan and Liu Ching Hsuan are related parties to the Company. The C.L.L. Note bears interest at 20% per annum. The Company shall pay to the C.L.L. an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the C.L.L. Note, which is on September 3, 2020. At any time from the date hereof until this C.L.L. Note has been satisfied, the C.L.L. may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $.50 per share (the “Fixed Conversion Price”), subject to adjustment, or (ii) 70% of the per share offering price (the “Alternative Conversion Price”) of the completed public equity offering of the Company in an amount exceeding $10,000,000 as stated on the registration statement on a Form S-1 filed with the Securities and Exchange Commission on November 14, 2018 (the “Public Offering”), as amended from time to time. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the C.L.L. Note. On April 20, 2020, the Company entered into an exchange agreement with C.L.L.. The aggregate principal amount plus accrued interest expenses were $289,974, and the Company agreed to issue to the Holders an aggregate of 162,908 shares of the Company’s common stock, and warrants to purchase 162,908 shares of common stock. As of December 31, 2020, these common shares have been issued.
F-26
On October 29, 2019, the Company issued a twelve-month term unsecured convertible promissory note (the “Lee Note”) in an aggregate principal amount of $250,000 to Hwalin Lee (the “Lee”), a related party, pursuant to which the Company received $250,000 on October 29, 2019. The Lee Note bears interest at 20% per annum. The Company shall pay to the Lee an amount in cash representing all outstanding principal and accrued and unpaid interest on the Twelve (12) month anniversary of the issuance date of the Lee Note, which is on October 28, 2020. At any time from the date hereof until this Lee Note has been satisfied, the Lee may convert the unpaid and outstanding principal plus any accrued and unpaid interest and or default interest, if any, into shares of the Company’s common stock at a conversion price (the “Conversion Price”) equal to the lower of (i) $.50 per share (the “Fixed Conversion Price”), subject to adjustment, or (ii) 70% of the per share offering price (the “Alternative Conversion Price”) of the completed public equity offering of the Company in an amount exceeding $10,000,000 as stated on the registration statement on a Form S-1 filed with the Securities and Exchange Commission on November 14, 2018 (the “Public Offering”), as amended from time to time. In accordance with FASB ASC 470-20, the Company recognized none of the intrinsic value of embedded beneficial conversion feature present in the Lee Note. On January 19, 2021, the Company paid off the convertible promissory note of $311,233, including principal and accrued and unpaid interest expense.
On October 23, 2020, the Company entered into a Securities Purchase Agreement (the “October SPA”) with one accredited investor. Pursuant to the October SPA, the Company sold and issued a convertible promissory note (the “October Note”) in the principal amount of $2,500,000 to the investor and received the payment from such investor on October 30, 2020. The October Note was issued on October 23, 2020 and the maturity date of the October Note is the twenty-four (24) month anniversary from the issuance date (the “Maturity Date”). Upon the Maturity Date, the Company shall pay to the holder, in cash, an amount representing all outstanding principal amount and accrued and unpaid interest under the October Note. The October Note bears an interest rate of ten percent (10%) per annum and may be convertible into shares of the Company’s common stock at a fixed conversion price of $2.25 per share. The holder of the October Note may elect to convert part or all of the outstanding balance of the October Note from the issuance date until the Maturity Date. The Company may prepay the outstanding amount at any time, in whole or in part, without any penalty.
As of December 31, 2020 and 2019, the aggregate carrying values of the convertible debentures were $2,750,000 and $2,007,500, respectively; and accrued convertible interest was $104,551 and $181,852, respectively.
Total interest expenses in connection with the above convertible note payable were $240,420 and $145,514 for the year ended December 31, 2020 and 2019, respectively.
8. BANK LOANS
| (1) | Short-term bank loan consists of the following: |
| December 31, | December 31, | |||||||
| 2020 | 2019 | |||||||
| Cathay United Bank | $ | 267,000 | $ | 250,500 | ||||
| CTBC Bank | 712,000 | 668,000 | ||||||
| Cathay Bank | 650,000 | 1,000,000 | ||||||
| Total | $ | 1,629,000 | $ | 1,918,500 | ||||
F-27
Cathay United Bank
On June 28, 2016, BioLite Taiwan and Cathay United Bank entered into a one-year bank loan agreement (the “Cathay United Loan Agreement”) in an amount of NT$7,500,000, equivalent to $267,000. The term started June 28, 2016 with maturity date at June 28, 2017. The loan balance bears interest at a floating rate of prime rate plus 1.15%. The prime rate is based on term deposit saving interest rate of Cathay United Bank. On September 6, 2017, BioLite Taiwan extended the Cathay United Loan Agreement for one year, which was due on September 6, 2018, with the principal amount of NT$7,500,000, equivalent to $267,000. On October 1, 2018, BioLite Taiwan extended the Cathay United Loan Agreement with the same principal amount of NT$7,500,000, equivalent to $267,000 for one year, which was due on September 6, 2019. On September 6, 2019, BioLite Taiwan extended the Cathay United Loan Agreement with the same principal amount of NT$7,500,000, equivalent to $267,000 for one year, which is due on September 6, 2020. On September 6, 2020, BioLite Taiwan extended the Cathay United Loan Agreement with the same principal amount of NT$7,500,000, equivalent to $267,000 for one year, which is due on September 6, 2021. As of December 31, 2020 and 2019, the effective interest rates per annum was 2.1% and 2.22%, respectively. The loan is collateralized by the building and improvement of BioLite Taiwan, and is also personal guaranteed by the Company’s chairman.
Interest expenses were $5,280 and $5,395 for the year ended December 31, 2020 and 2019, respectively.
CTBC Bank
On June 12, 2017 and July 19, 2017, BioLite Taiwan and CTBC Bank entered into short-term saving secured bank loan agreements (the “CTBC Loan Agreements”) in an amount of NT$10,000,000, equivalent to $356,000, and NT$10,000,000, equivalent to $356,000, respectively. Both two loans with the same maturity date at January 19, 2018. In February 2018, BioLite Taiwan combined two loans and extended the loan contract with CTBC for one year. On January 18, 2019, BioLite Taiwan and CTBC Bank agreed to extend the loan with a new maturity date, which was July 18, 2019. On July 18, 2019, BioLite Taiwan extended the CTBC Loan Agreement with the same principal amount of NT$20,000,000, equivalent to $712,000 for six months, which is due on January 17, 2020. On January 19, 2020, BioLite Taiwan extended the CTBC Loan Agreement with the same principal amount of NT$20,000,000, equivalent to $712,000 for six months, which is due on July 19, 2020. On July 17, 2020, BioLite Taiwan extended the CTBC Loan Agreement with the same principal amount of NT$20,000,000, equivalent to $712,000 for six months, which is due on January 15, 2021. On January 15, 2021, BioLite Taiwan extended the CTBC Loan Agreement with the same principal amount of NT$20,000,000, equivalent to $712,000 for six months, which is due on July 15, 2021. The loan balances bear interest at a fixed rate of 1.68% per annum. The loan is secured by the money deposited in a savings account with the CTBC Bank. This loan was also personal guaranteed by the Company’s chairman and BioFirst. During the year ended December 31, 2020, BioLite Taiwan has opened a TCD account with CTBC bank to guarantee the loan going forward.
Interest expenses were $11,395 and $10,563 for the year ended December 31, 2020 and 2019, respectively.
Cathay Bank
On January 21, 2019, the Company received a loan in the amount of $500,000 from Cathay Bank (the “Bank”) pursuant to a business loan agreement (the “Loan Agreement”) entered by and between the Company and Bank on January 8, 2019 and a promissory note (the “Note”) executed by the Company on the same day. The Loan Agreement provides for a revolving line of credit in the principal amount of $1,000,000 with a maturity date (the “Maturity Date”) of January 1, 2020. The Note executed in connection with the Loan Agreement bears an interest rate (the “Regular Interest Rate”) equal to the sum of one percent (1%) and the prime rate as published in the Wall Street Journal (the “Index”) and the accrued interest shall become payable each month from February 1, 2019. Pursuant to the Note, the Company shall pay the entire outstanding principal plus accrued unpaid interest on the Maturity Date and may prepay portion or all of the Note before the Maturity Date without penalty. If the Company defaults on the Note, the default interest rate shall become five percent (5%) plus the Regular Interest Rate.
In connection with the Note and Loan Agreement, on January 8, 2019, each of Dr. Tsung Shann Jiang and Dr. George Lee, executed a commercial guaranty (the “Guaranty”) to guaranty the loans for the Company pursuant to the Loan Agreement and Note, severally and individually, in the amount not exceeding $500,000 each until the entire Note plus interest are fully paid and satisfied. Dr. Tsung Shann Jiang is the Chairman and Chief Executive Officer of BioLite Holding, Inc. and Dr. George Lee serves as the Chairman of the board of directors of BioKey, Inc, which became a wholly-owned subsidiary of the Company effective by operation of law on or about February 5, 2019. On December 29, 2020, the Company entered into a new loan extension agreement and assignment of deposit account with the Bank, which allowed Dr. Tsung Shann Jiang and Dr. George Lee to be removed as guarantees from the list of Guaranty.
F-28
In addition, on January 8, 2019, each of the Company and BioKey, a wholly-owned subsidiary of the Company, signed a commercial security agreement (the “Security Agreement”) to secure the loans under the Loan Agreement and the Note. Pursuant to the Security Agreements, each of the Company and BioKey (each, a “Grantor”, and collectively, the “Grantors”) granted security interest in the collaterals as defined therein, comprised of almost all of the assets of each Grantor, to secure such loans for the benefit of the Bank. On March 31, 2020, the Company extended the Loan Agreement with the same term for seven months, which is due on October 31, 2020. On April 8, 2020 and October 3, 2020, the Company repaid an aggregated principal amount of $350,000. On December 3, 2020, The Company renewed the Loan Agreement with the principal amount of $650,000 for ten months, which is due on October 31, 2021. The outstanding loan balance was $650,000 as of December 31, 2020.
Interest expenses were $53,992 and $59,586 for the year ended December 31, 2020 and 2019, respectively.
| (2) | Long-term bank loan consists of the following: |
| December 31, | December 31, | |||||||
| 2020 | 2019 | |||||||
| Cathay United Bank | $ | - | $ | 13,403 | ||||
| Less: current portion of long-term bank loan | - | (13,403 | ) | |||||
| Total | $ | - | $ | - | ||||
Cathay United Bank
On April 30, 2010, BioLite Taiwan entered a seven-year bank loan of NT$8,900,000, equivalent to $288,360, with Cathay United Bank. The term started April 30, 2010 with maturity date at April 30, 2017. On April 30, 2017, BioLite Taiwan extended the original loan agreement for additional three years with the new maturity date at April 30, 2020. The loan balance bears interest at a floating rate of prime rate plus variable rates from 0.77% to 1.17%. The prime rate is based on term deposit saving interest rate of Cathay United Bank. The loan was collateralized by the building and improvement of BioLite Taiwan, and is also personal guaranteed by the Company’s chairman. On April 30, 2020, the loan was paid off in full.
Interest expenses were $63 and $859 for the year ended December 31, 2020 and 2019, respectively.
9. PAYCHECK PROTECTION PROGRAM LOAN PAYABLE
On April 14, 2020, the Company received a loan in the amount of $124,400 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from East West Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll.
The loan was granted pursuant to a promissory note dated April 14, 2020 issued by the Company, which matures on April 13, 2022 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is two years after the date of the promissory note. In addition, the Company will pay regular monthly payments in an amount equal to one month’s accrued interest commencing on the date that is seven months after the date of the promissory note, with all subsequent interest payments to be due on the same day of each month after that. No collateral or personal guarantees are required.
On February 27, 2021, the Company has submitted all required to East West Bank for the application of forgiveness.
F-29
10. NOTES PAYABLE
In January, 2019, BioLite Taiwan entered an unsecured loan agreement with one individual bearing interest at fixed rates at 12% per annum of NT$3,000,000, equivalent to $106,800, for working capital purpose. As of the date of this report, BioLite Taiwan is still in discussion with the individual with respect to the terms of the unsecured loans. As of December 31, 2020 and 2019, the balance due to this individual amounted to $106,800 and $100,200, respectively. Interest expense was $12,204 and $11,778 for the year ended December 31, 2020 and 2019, respectively.
11. SHORT-TERM LOAN
On February 18, 2020, the Company entered an unsecured loan agreement with a third-party in the amount of $100,000. This loan bears the interest rate of 1.5% per annum and will be matured on August 17, 2020. On August 18, 2020, the Company extended the contract for six months under the same term. Accrued interest expense was $1,302 as of and for the year ended December 31, 2020.
F-30
12. RELATED PARTIES TRANSACTIONS
The related parties of the company with whom transactions are reported in these financial statements are as follows:
| Name of entity or Individual | Relationship with the Company and its subsidiaries | |
| BioFirst Corporation (the “BioFirst”) | Entity controlled by controlling beneficiary shareholder of YuanGene | |
| BioFirst (Australia) Pty Ltd. (the “BioFirst (Australia)”) | 100% owned by BioFirst; Entity controlled by controlling beneficiary shareholder of YuanGene | |
| Rgene Corporation (the “Rgene”) | Shareholder of the Company; entity controlled by controlling beneficiary shareholder of YuanGene | |
| YuanGene Corporation (the “YuanGene”) | Controlling beneficiary shareholder of the Company | |
| AsiaGene Corporation (the “AsiaGene”) | Shareholder; entity controlled by controlling beneficiary shareholder of YuanGene | |
| Eugene Jiang | Former President and Chairman | |
| Keypoint Technology Ltd. (the “Keypoint’) | The Chairman of Keypoint is Eugene Jiang’s mother. | |
| Lion Arts Promotion Inc. (the “Lion Arts”) | Shareholder of the Company | |
| Yoshinobu Odaira (the “Odaira”) | Director of the Company | |
| GenePharm Inc. (the “GenePharm”) | Dr. George Lee, Board Director of Biokey, is the Chairman of GenePharm. | |
| Euro-Asia Investment & Finance Corp Ltd. (the “Euro-Asia”) | Shareholder of the Company | |
| LBG USA, Inc. (the “LBG USA”) | 100% owned by BioFirst; Entity controlled by controlling beneficiary shareholder of YuanGene | |
| LionGene Corporation (the “LionGene”) | Shareholder of the Company; Entity controlled by controlling beneficiary shareholder of YuanGene | |
| Kimho Consultants Co., Ltd. (the “Kimho”) | Shareholder of the Company | |
| Mr. Tsung-Shann Jiang, Ms. Shu-Ling Jiang, Mr. Chang-Jen Jiang, Ms. Mei-Ling Jiang, and Mr. Eugene Jiang (collectively the “Jiangs”) | Mr.
Tsung-Shann Jiang, the controlling beneficiary shareholder of the Company and Rgene, the Chairman and CEO of the BioLite Holding
Inc. and BioLite Inc. and the President and a member of board of directors of BioFirst Ms. Shu-Ling Jiang, Mr. Tsung-Shann Jiang’s wife, is the Chairman of Keypoint; and a member of board of directors of BioLite Inc. Mr. Eugene Jiang is Mr. and Ms. Jiang’s son. Mr. Eugene Jiang is the chairman, and majority shareholder of the Company and a member of board of directors of BioLite Inc. Mr. Chang-Jen Jiang is Mr. Tsung-Shann Jiang’s sibling and the director of the Company. Ms. Mei-Ling Jiang is Ms. Shu-Ling Jiang’s sibling. | |
| Amkey Ventures, LLC (“Amkey”) | An entity controlled by Dr. George Lee, who serves as one of the board directors of BioKey, Inc | |
| BioLite Japan | Entity controlled by controlling beneficiary shareholder of ABVC |
F-31
Accounts receivable - related parties
Accounts receivable due from related parties consisted of the following as of the periods indicated:
| December 31, | December 31, | |||||||
| 2020 | 2019 | |||||||
| GenePharm Inc. | $ | 142,225 | $ | 142,225 | ||||
| Rgene | - | 1,053 | ||||||
| Amkey | 1,210 | - | ||||||
| Total | $ | 143,435 | $ | 143,278 | ||||
Due from related parties
Amount due from related parties consisted of the following as of the periods indicated:
| December 31, | December 31, | |||||||
| 2020 | 2019 | |||||||
| Rgene | $ | 42,911 | $ | 36,332 | ||||
| AsiaGene | 4,241 | 3,578 | ||||||
| BioFirst | - | 137,151 | ||||||
| BioFirst (Australia) | 373,235 | 40,000 | ||||||
| BioHopeKing Corporation | 123,583 | 115,946 | ||||||
| LBG USA | 675 | 675 | ||||||
| BioLite Japan | 150,000 | - | ||||||
| Keypoint | 1,610 | - | ||||||
| Total | $ | 696,255 | $ | 333,682 | ||||
| (1) | As of December 31, 2020 and 2019, the Company has advanced an aggregate amount of $31,684 and $29,194 to Rgene for working capital purpose. Under the terms of the loan agreement, the loan bears interest at 1% per month (or equivalent to 12% per annum) and the loan will be matured on December 31, 2019. On January 1, 2020, the contract has been renewed for another year, when the new maturity date now is on December 31, 2020. As of December 31, 2020, and December 31, 2019, the outstanding loan balance was $31,684 and $29,194; and accrued interest was $11,227 and $7,138, respectively. |
| (2) | On May 27, 2019, the Company entered into loan agreements with AsiaGene for NT $100,000, equivalent to $3,560, to meet its working capital needs. Under the terms of the loan agreement, the loan bears interest at 1% per month (or equivalent to 12% per annum) and the loan will be matured on December 31, 2019. On January 1, 2020, the agreement has been renewed for another year, when the new maturity date now is on December 31, 2020. As of December 31, 2020 and 2019, the outstanding loan balance was $3,560 and $3,343, and accrued interest was $681 and $235, respectively. |
| (3) | On July 12, 2019, the Company had an aggregate amount of loan with BioFirst of $150,000 to meet its working capital needs, pursuant to which the interest bears at 12% per annum. The Company paid back $21,317 in 2019. The remaining loan balance was $128,683 as of December 31, 2019. This loan is matured on July 11, 2020 and bears interest at 1% per month (or equivalent to 12% per annum). As of December 31, 2020 and 2019, the outstanding loan balance was $0 and $128,683, and accrued interest was $0 and $8,468, respectively. |
F-32
| (4) | On May 11, 2018, the Company and BioFirst (Australia) entered into a loan agreement for a total amount of $40,000 to meet its working capital needs. The advances bear 0% interest rate and are due on demand prior to June 30, 2020. Afterwards, all outstanding load will bear interest rate at 12% per annum. On July 1, 2020, the Company entered into a loan agreement with BioFirst (Australia) for $321,487 to properly record R&D cost and tax refund allocation based on co-development contract executed on July 24, 2017. The loan will be matured on June 30, 2021 with an interest rate of 12% per annum. As of December 31, 2020 and 2019, the outstanding loan balances was $373,235 and $40,000, respectively. |
| (5) | On February 24, 2015, BioLite Taiwan and BioHopeKing Corporation (the “BHK”) entered into a co-development agreement, (the “BHK Co-Development Agreement”, see Note 3). The development costs shall be shared 50/50 between BHK and the Company. Under the term of the agreement, BioLite issued relevant development cost to BHK. As of December 31, 2020 and 2019, due from BHK was $123,583 and $115,946, respectively. |
| (6) | On February 27, 2019, the Company has advanced funds to LBG USA for working capital purpose. The advances bear 0% interest rate and are due on demand. As of December 31, 2020 and 2019, the outstanding advance balances were $675. |
| (7) | On May 8, 2020, the Company and Lucidaim entered into a Letter of Intent (LOI) in regard to a potential joint venture of BioLite Japan. Based on the LOI, each party will advance an aggregated amount of $150,000 to meet BioLite Japan’s working capital needs, which the Company advanced an amount of $150,000 and the advance bear 0% interest rate. As of December 31, 2020 and 2019, the outstanding advance balances was $150,000 and $0, respectively. |
| (8) | On October 31, 2020, the Company has advanced an aggregate amount of $1,610 to Keypoint for working capital purpose. Under the terms of the loan agreement, the loan bears interest at 6.5% per annum and the loan will be matured on October 30, 2021. As of December 31, 2020, the outstanding loan balance was $1,610. |
Due to related parties
Amount due to related parties consisted of the following as of the periods indicated:
| December 31, | December 31, | |||||||
| 2020 | 2019 | |||||||
| LionGene Corporation | $ | - | $ | 10,275 | ||||
| BioFirst Corporation | 23,647 | 24,182 | ||||||
| AsiaGene | - | 24,017 | ||||||
| YuanGene | 9,205 | 9,205 | ||||||
| The Jiangs | 16,627 | 40,031 | ||||||
| Kimho | - | 21,500 | ||||||
| Euro Asia | - | 12,000 | ||||||
| Due to shareholders | 166,261 | 284,479 | ||||||
| Due to employee | 72,704 | - | ||||||
| Total | $ | 288,445 | $ | 425,689 | ||||
| (1) | In November 2018, BioLite Taiwan has borrowed an aggregate amount of NT$13,295,000, equivalent to $430,817 from LionGene for working capital purpose. The advances bear 0% interest rate and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreements to convert the all of remaining balance of $428,099, to 61,157 shares of the Company’s common stock at a conversion price of $7.00 per share.
On October 15, 2019, LionGene has advanced funds to the Company for working capital purpose in an aggregate amount of NTD $300,000, equivalent to $10,020, The advances bear 1% interest rate per month. As of December 31, 2020 and 2019, the outstanding principal of loans was $0 and $10,020, and accrued interest was $0 and $255, respectively. Interest expenses in connection with these loans were $955 and $255 for the year ended December 31, 2020 and 2019, respectively. |
F-33
| (2) | On January 26, 2017, BriVision and BioFirst entered into a loan agreement for a total commitment (non-secured indebtedness) of $950,000 to meet its working capital needs. On February 2, 2019, BriVision and BioFirst agreed to extend the remaining loan balance of $693,000 for one year matured on February 1, 2020. Under the terms of the loan agreement, the loan bears interest at 12% per annum. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $693,000 to 99,000 shares of the Company’s common stock at a conversion price of $7.00 per share.
Since 2017, BioLite Taiwan and BioFirst entered into several loan agreements for an aggregate amount of NT$19,430,000, equivalent to $625,646, to meet its working capital needs. Under the terms of the loan agreements, the loans bear interest at 12% per annum. The term of the loans has various maturity dates through May 27, 2020. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $625,646 to 89,378 shares of the Company’s common stock at a conversion price of $7.00 per share.
Since 2017, BioFirst has also advanced funds to the Company for working capital purpose. The advances bear 0% interest rate and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $597,128 to 85,304 shares of the Company’s common stock at a conversion price of $7.00 per share.
On April 12, 2017, BioLite BVI and BioFirst entered into a loan agreement for NT$30,000,000, equivalent to $987,134 to meet its working capital needs. Under the terms of the loan agreement, the loan bears interest at 1% per month (or equivalent to 12% per annum). BioLite BVI and BioFirst extended the loan with the same interest rate and amount for one year. The loan will be matured on May 11, 2019. On May 12, 2019, the two parties extended the loan with the same interest rate and amount for one year. The loan will be matured on May 11, 2020. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $987,134 to 141,020 shares of the Company’s common stock at a conversion price of $7.00 per share.
On July 24, 2017, BriVision entered into a collaborative agreement (the “BioFirst Collaborative Agreement”) with BioFirst (See Note 3). On September 25, 2017, BioFirst has delivered all research, technical, data and development data to BriVision, and the Company has recorded the full amount of $3,000,000 due to BioFirst. On June 30, 2019, the Company entered into a Stock Purchase Agreement with BioFirst, pursuant to which the Company agreed to issue 428,571 shares of the Company’s common stock to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst.
Since 2019, BioFirst has also advanced funds to the Company for working capital purpose. The advances bear interest 1% per month (or equivalent to 12% per annum). As of December 31, 2020 and 2019, the aggregate amount of outstanding balance and accrued interest is $23,647 and $24,182, respectively. |
| (3) | In September 2017, AsiaGene entered an investment and equity transfer agreement (the “Investment and Equity Transfer Agreement”) with Everfront Biotech Inc. (the “Everfront”), a third party. Pursuant to the Investment and Equity Transfer Agreement, Everfront agreed to purchase 2,000,000 common shares of the Company owned by AsiaGene at $1.60 per share in a total amount of $3,200,000, of which $160,000 is due before September 15, 2017 and the remaining amount of $3,040,000 is due before December 15, 2017. AsiaGene also agreed to loan the proceeds to the Company for working capital purpose. The non-secured loan bears 0% interest rate and is due on demand. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $160,000 to 22,858 shares of the Company’s common stock at a conversion price of $7.00 per share.
As of December 31, 2020 and 2019, AsiaGene has advanced the Company an aggregate amount of $0 and $24,017, respectively for working capital purpose. This advance bears 0% interest rate and is due on demand. |
F-34
| (4) | On January 18, 2018, the Company and YuanGene entered into a loan agreement for a total of $50,000 to meet its working capital needs. Under the terms of the loan agreement, the loan bears interest at 1% per month (or equivalent to 12% per annum) and the Company is required to pay interest monthly to the lender. The maturity date of this loan is January 19, 2019. On January 20, 2019, the two parties extended the loan with the same interest rate and amount for one year. The loan will be matured on January 19, 2020. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $50,000 to 7,143 shares of the Company’s common stock at a conversion price of $7.00 per share.
In January 2018, YuanGene Corporation has advanced an aggregate amount of $42,690 to the Company for working capital purpose. The advances bear 0% interest rate and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $42,690 to 6,099 shares of the Company’s common stock at a conversion price of $7.00 per share.
As of December 31, 2020 and 2019, YuanGene has advanced the Company an aggregate amount of $9,205 for working capital purpose. This advance bears 0% interest rate and is due on demand. |
| (5) | Since 2018, Mr. Tsung-Shann Jiang, Mr. Chang-Jen Jiang, Ms. Shu-Ling Jiang, and Ms. Mei-Ling Jiang have entered into various loans with the Company for working capital purpose in an aggregate amount of $795,340. These loans bear interest at 12% per annum and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $837,726 to 119,675 shares of the Company’s common stock at a conversion price of $7.00 per share.
Since 2018, the Jiangs have advanced funds to the Company for working capital purpose in an aggregate amount of $353,050. The advances bear 0% interest rate and are due on demand. On August 4, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $353,050 to 50,436 shares of the Company’s common stock at a conversion price of $7.00 per share.
Since 2019, the Jiangs advanced funds to the Company for working capital purpose. As of December 31, 2020 and December 31, 2019, the outstanding balance due to the Jiangs amounted to $16,627 and $40,031, respectively. These loans bear interest rate of 0% to 1% per month, and are due on demand. |
| (6) | On July 2, 2019, the Company entered into an agreement with Kimho for consulting service, with such services to begin in September 2019. As of December 31, 2020 and 2019, the outstanding services charge was $0 and $21,500, respectively. |
| (7) | As of December 31, 2020 and 2019, Euro Asia has advanced of $0 and $12,000, respectively, to the Company for working capital purpose. The advances bear 0% interest rate. |
F-35
| (8) | Since 2018, the Company’s shareholders have advanced funds to the Company for working capital purpose. The advances bear interest rate from 12% to 13.6224% per annum. As of December 31, 2020 and 2019, the outstanding principal and accrued interest was $166,261 and $284,479, respectively. Interest expenses in connection with these loans were $21,520 and $14,910 for the year ended December 31, 2020 and 2019, respectively. |
| (9) | During the year Ended December 31, 2020, the Company had advances from one employee for working capital purpose. The outstanding balance due to this employee amounted to $72,704 as of December 31, 2020. This loan bears interest rate of 1.5% per annum, and is due on demand. |
13. EQUITY
During October 2015, $350,000 of subscription receivable was fully collected from the shareholders.
On February 8, 2016, a Share Exchange Agreement (“Share Exchange Agreement”) was entered into by and among American BriVision (Holding) Corporation (the “Company”), American BriVision Corporation (“BriVision”), Euro-Asia Investment & Finance Corp. Limited, a company incorporated under the laws of Hong Kong Special Administrative Region of People’s Republic of China (“Euro-Asia”), being the owners of record of 164,387,376 (52,336,000 pre-stock split) shares of Common Stock of the Company, and the owners of record of all of the issued share capital of BriVision (the “BriVision Stock”). Pursuant to the Share Exchange Agreement, upon surrender by the BriVision Shareholders and the cancellation by BriVision of the certificates evidencing the BriVision Stock as registered in the name of each BriVision Shareholder, and pursuant to the registration of the Company in the register of members maintained by BriVision as the new holder of the BriVision Stock and the issuance of the certificates evidencing the aforementioned registration of the BriVision Stock in the name of the Company, the Company should issue 166,273,921(52,936,583 pre-stock split) shares (the “Acquisition Stock”) (subject to adjustment for fractionalized shares as set forth below) of the Company’s Common Stock to the BriVision Shareholders (or their designees), and 163,159,952 (51,945,225 pre-stock split) shares of the Company’s Common Stock owned by Euro-Asia should be cancelled and retired to treasury. The Acquisition Stock collectively should represent 79.70% of the issued and outstanding Common Stock of the Company immediately after the Closing, in exchange for the BriVision Stock, representing 100% of the issued share capital of BriVision in a reverse merger, or the Merger. Pursuant to the Merger, all of the issued and outstanding shares of BriVision’s Common Stock were converted, at an exchange ratio of 0.2536-for-1, into an aggregate of 166,273,921(52,936,583 pre-stock split) shares of Company’s Common Stock and BriVision became a wholly owned subsidiary, of the Company. The holders of Company’s Common Stock as of immediately prior to the Merger held an aggregate of 205,519,223 (65,431,144 pre-stock split) shares of Company’s Common Stock, Because of the exchange of the BriVision Stock for the Acquisition Stock (the “Share Exchange”), BriVision became a wholly owned subsidiary (the “Subsidiary”) of the Company and there was a change of control of the Company following the closing. There were no warrants, options or other equity instruments issued in connection with the share exchange agreement.
On February 17, 2016, pursuant to the 2016 Equity Incentive Plan (the “2016 Plan”), 157,050 (50,000 pre-stock split) shares were granted to the employees.
On March 21, 2016, the Board of Directors of the Company approved an amendment to Articles of Incorporation to effect a forward split at a ratio of 1 to 3:141 (the “Forward Stock Split”) and increase the number of our authorized shares of Common Stock, par value $0.001 per share, to 360,000,000, which was effective on April 8, 2016.
F-36
On May 6, 2016, the Company and BioLite Taiwan agreed to amend the BioLite Collaborative Agreement, through entry into the Milestone Payment Agreement, whereby the Company has agreed to issue shares of Common Stock of the Company, at the price of $1.60 per share, for an aggregate number of 562,500 shares, as part of the Company’s first installation of payment pursuant to the Milestone Payment. The shares issuance was completed in June 2016. On August 26, 2016, the Company issued 1,468,750 shares (“Shares”) of the Company’s Common Stock, par value $0.001 (the “Offering”) to BioLite Taiwan pursuant to a certain Stock Purchase Agreement dated August 26, 2016 (the “SPA”). The Shares are exempt from the registration requirements of the Securities Act of 1933, as amended (the “Securities Act”), pursuant to Regulation S of the Securities Act promulgated thereunder. The purchase price per share of the Offering is $1.60. The net proceeds to the Company from the Offering are approximately $2,350,000. Pursuant to the BioLite Collaborative Agreement, BriVision should pay a total of $100,000,000 in cash or stock of the Company with equivalent value according to the milestone achieved. The agreement requires that 6.5% of total payment, $6,500,000 shall be made upon the first IND submission which was submitted in March 2016. In February 2017, the Company remitted this amount to BioLite with $650,000 in cash and $5,850,000 in the form of newly issued shares of the Company’s Common Stock, at the price of $2.0 per share, for an aggregate number of 2,925,000 shares. Upon the consummation of the restructuring transaction between the Company and BioLite on February 8, 2019, the Company’s Common Stock held by BioLite Taiwan was accounted for treasury stocks in the statement of equity (deficit). On February 8, 2019, after the Merger, the Company issued 74,997,546 shares to the shareholders of BioLite and 29,561,231 shares to the shareholders of BioKey.
On May 3, 2019, the Company filed a Certificate of Amendment with the Secretary of State of Nevada, which was effective May 8, 2019 upon its receipt of the written notice from Financial Industry Regulatory Authority (“FINRA”). Pursuant to the Certificate of Amendment, the Company effectuated a 1-for-18 reverse stock split of its issued and outstanding shares of common stock, $0.001 par value, whereby 318,485,252 outstanding shares of the Company’s common stock were exchanged for 17,693,625 shares of the Company’s Common Stock. All shares and related financial information in this Form 10-K reflect this 1-for-18 reverse stock split.
On October 1, 2016, the Company entered into a consulting agreement with Kazunori Kameyama (“Kameyama”) for the provision of services related to the clinical trials and other administrative work, public relation work, capital raising, trip coordination, In consideration for providing such services, the Company agreed to indemnify the consultant in an amount of $150 per hour in cash up to $3,000 per month, and issue to Kameyama the Company’s Common Stock at $1.00 per share for any amount exceeding $3,000. The Company’s stocks shall be calculated and issued in December every year. On November 21, 2020, the Company entered into an agreement with Kameyama, pursuant to which the Company granted and issued 24,694 stock options to Kameyama related to unpaid consulting fees of $49,388 (see Note 14).
During the year ended December 31, 2019, the Company entered into service agreements with Euro-Asia Investment & Finance Corp Ltd. (a related party), Ever Adventure inv. (Formosa) Consultant Co., Ltd., New Eastern Asia (a related party),and Kimho Consultants Co., Ltd. (a related party) for the maintenance of the listing in the U.S. stock exchange market, investor relations, and business development. Pursuant to the agreements, the Company issued 644,972 shares of the Company’s common stock for the consulting service from July 2019 to July 2024 for the service fee of $4,514,800 in aggregate, and recorded as stock subscription receivable. As of December 31, 2020 and 2019, stock subscription receivable was $3,160,360 and $4,063,320, respectively.
F-37
On July 24, 2017, BriVision entered into a collaborative agreement (the “BioFirst Collaborative Agreement”) with BioFirst (See Note 3). On September 25, 2017, BioFirst has delivered all research, technical, data and development data to BriVision, and the Company has recorded the full amount of $3,000,000 due to BioFirst. On June 30, 2019, the Company entered into a Stock Purchase Agreement with BioFirst, pursuant to which the Company agreed to issue 428,571 shares of the Company’s common stock to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst. These common shares have been issued during the year ended December 31, 2019.
As stated in Note 12, in August 2019, the Company entered into several Conversion Agreements to all creditors that are listed under below table of “due to related parties” in consideration for a total of $4,872,340 owed by the Company to various creditors based on outstanding loan agreements. Under the Conversion Agreements, creditor agrees to convert the amount of debt into the Company’s common stock at a price of $7.00 per share.
| Amount
of Debt Converted | Number
of Shares Issued | |||||||
| Lion Arts Promotion Inc | $ | 97,864 | 13,981 | |||||
| LionGene Corporation | 428,099 | 61,157 | ||||||
| BioFirst Corporation | 2,902,911 | 414,702 | ||||||
| AsiaGene Corporation | 160,000 | 22,858 | ||||||
| YuanGene Corporation | 92,690 | 13,242 | ||||||
| The Jiangs | 1,190,776 | 170,111 | ||||||
| Total | $ | 4,872,340 | 696,051 | |||||
On March 12, 2020, the board of directors of the Company approved and adopted an amendment to the Company’s Articles of Incorporation, to increase the authorized shares of its common stock, par value $0.001 per share, from 20,000,000 to 100,000,000 shares.
On July 8, 2020, the Company entered an agreement with View Trade Securities Inc. (“ViewTrade”) to engage ViewTrade as the placement agent and the Company’s advisor with respect to its ongoing capital events. Pursuant to the agreement, the Company agreed to pay View Trade 60,000 restricted common shares of the Company and 60,000 warrants to purchase common shares of the Company at an exercise price of $6 per share for a period of 5 years with cashless exercise provision. As of December 31, 2020, the Company has issued 60,000 shares of common stock to ViewTrade for the consulting fee with an estimated value of $135,000. The warrants were never issued and the parties mutually agreed to terminate the agreement on November 19, 2020. As a termination fee, the Company agreed to issue ViewTrade 50,000 restricted common shares of the Company.
Also on November 19, 2020, the Company and ViewTrade agreed to a new Advisory agreement under which ViewTrade was engaged to provide advisory services only. In addition to a retainer fee, the Company agreed to issue 200,000 warrants, with an exercise price of $2.25, an industry standard cashless exercise provision, and a term of 5 years from November 19, 2020.
During the year ended December 31, 2020, the Company entered into a consulting agreement with a service provider for consulting and advisory services, pursuant to which the Company agreed to pay the service fee by issuing 50,000 shares of unrestricted common shares, valued at the closing price of $2.9 per share on the grant date. As of December 31, 2020, these shares have been issued.
During the year ended December 31, 2020, the Company received aggregated capital contributions of $7,615,331 in cash from 45 investors through private placements of the sale of the Company’s common stock for the purchase price of $2.25 per share and a free warrant attached with each common stock purchased. As of December 31, 2020, 3,384,615 shares of the Company’s common stock have been issued.
During the year ended December 31, 2020, the Company entered into consulting agreements with four service providers for consulting and advisory services, pursuant to which the Company agreed to pay the service fee by issuing 521,887 shares of unrestricted common shares, valued at the closing price from $2 to $3.68 per share on the grant date. As of December 31, 2020, these shares have been issued.
F-38
During the year ended December 31, 2020, the Company issued aggregated common shares of 915,856 to six previous note holders, who had converted their outstanding principals and accrued and unpaid interests, including the debt conversion to the following:
| a. | Keypoint converted the aggregated amount of $292,826 at the conversion price of $1.84 on April 5, 2020, in exchange for 159,145 shares of the Company’s common stock, and warrants to purchase 159,145 shares of the Company’s common stock. |
| b. | Odaira converted the aggregated amount of $284,036 at the conversion price of $1.84 on April 5, 2020, in exchange for 154,368 shares of the Company’s common stock, and warrants to purchase 154,368 shares of the Company’s common stock. |
| c. | C.L.L. converted the aggregated amount of $289,974 at the conversion price of $1.78 on April 20, 2020, in exchange for 162,908 shares of the Company’s common stock, and warrants to purchase 162,908 shares of the Company’s common stock. |
| d. | KLS converted the aggregated amount of $225,222 at the conversion price of $1.78 on April 20, 2020, in exchange for 126,530 shares of the Company’s common stock, and warrants to purchase 126,530 shares of the Company’s common stock. |
| e. | Yu and Wei converted the aggregated amount of $354,722 at the conversion price of $1.84 on April 5, 2020, in exchange for 192,784 shares of the Company’s common stock, and warrants to purchase 192,784 shares of the Company’s common stock. |
| f. | KSL converted the aggregated amount of $270,272 at the conversion price of $2.25 on November 9, 2020, in exchange for 120,121 shares of the Company’s common stock, and warrants to purchase 120,121 shares of the Company’s common stock. |
See Note 7 for more details in connection with the above debt conversion.
14. STOCK OPTIONS
On October 30, 2020, the Company issued an aggregate of 545,182 shares of common stock in lieu of unpaid salaries of certain employees and unpaid consulting fees under the 2016 Equity Incentive Plan, as amended, at a conversion price of $2 per share; the total amount of converted salaries and consulting fees was $1,090,361. On November 21, 2020, the Company has entered into acknowledgement agreements and stock option purchase agreements with these employees and consultant; pursuant to which the Company granted stock options to purchase 545,182 shares of the Company’s common stock in lieu of common stock. The options were vested at the grant date and become exercisable for 10 years from the grant date.
F-39
Options issued and outstanding as of December 31, 2020, and their activities during the year then ended are as follows:
| Weighted- | ||||||||||||||||
| Weighted- | Average | |||||||||||||||
| Number of | Average | Contractual | ||||||||||||||
| Underlying | Exercise Price | Life Remaining | Aggregate | |||||||||||||
| Shares | Per Share | in Years | Intrinsic Value | |||||||||||||
| Outstanding as of January 1, 2020 | - | $ | - | $ | ||||||||||||
| Granted | 545,182 | 2.00 | 9.89 | 616,056 | ||||||||||||
| Forfeited | - | - | ||||||||||||||
| Outstanding as of December 31, 2020 | 545,182 | 2.00 | 9.89 | $ | 616,056 | |||||||||||
| Exercisable as of December 31, 2020 | 545,182 | 2.00 | 9.89 | $ | 616,056 | |||||||||||
| Vested and expected to vest | 545,182 | $ | 2.00 | 9.89 | $ | 616,056 | ||||||||||
The fair value of stock options granted for the year ended December 31, 2020 was calculated using the Black-Scholes option-pricing model applying the following assumptions:
| Year ended | ||||
| December 31, 2019 | ||||
| Risk free interest rate | 0.38 | % | ||
| Expected term (in years) | 5.00 | |||
| Dividend yield | 0 | % | ||
| Expected volatility | 89.01 | % | ||
The weighted average grant date fair value of options granted during 2020 was $3.13. There are 2,812,949 options available for grant under the 2016 Equity Incentive Plan as of December 31, 2020. Compensation costs associated with the Company’s stock options are recognized, based on the grant-date fair values of these options over vesting period. Accordingly, the Company recognized stock-based compensation expense of $1,706,419 and $0 for the years ended December 31, 2020 and 2019, respectively. As of December 31, 2020, there were no unvested options.
15. LOSS PER SHARE
Basic loss per share is computed by dividing net loss by the weighted-average number of common shares outstanding during the year. Diluted loss per share is computed by dividing net loss by the weighted-average number of common shares and dilutive potential common shares outstanding during the Year Ended December 31, 2020 and 2019.
| For the Year Ended | ||||||||
| December 31, 2020 | December 31, 2019 | |||||||
| Numerator: | ||||||||
| Net loss attributable to ABVC’s common stockholders | $ | (9,791,164 | ) | $ | (3,641,776 | ) | ||
| Denominator: | ||||||||
| Weighted-average shares outstanding: | ||||||||
| Weighted-average shares outstanding - Basic | 19,715,559 | 17,498,543 | ||||||
| Stock options | – | |||||||
| Weighted-average shares outstanding - Diluted | 19,715,559 | 17,498,543 | ||||||
| Loss per share | ||||||||
| -Basic | $ | (0.50 | ) | $ | (0.21 | ) | ||
| -Diluted | $ | (0.50 | ) | $ | (0.21 | ) | ||
F-40
Diluted loss per share takes into account the potential dilution that could occur if securities or other contracts to issue Common Stock were exercised and converted into Common Stock.
16. LEASE
The Company adopted FASB Accounting Standards Codification, Topic 842, Leases (“ASC 842”) using the modified retrospective approach, electing the practical expedient that allows the Company not to restate its comparative periods prior to the adoption of the standard on January 1, 2019. As such, the disclosures required under ASC 842 are not presented for periods before the date of adoption. For the comparative periods prior to adoption, the Company presented the disclosures which were required under ASC 840.
The Company applied the following practical expedients in the transition to the new standard and allowed under ASC 842:
| ● | Reassessment of expired or existing contracts: The Company elected not to reassess, at the application date, whether any expired or existing contracts contained leases, the lease classification for any expired or existing leases, and the accounting for initial direct costs for any existing leases. |
| ● | Use of hindsight: The Company elected to use hindsight in determining the lease term (that is, when considering options to extend or terminate the lease and to purchase the underlying asset) and in assessing impairment of right-to-use assets. |
| ● | Reassessment of existing or expired land easements: The Company elected not to evaluate existing or expired land easements that were not previously accounted for as leases under ASC 840, as allowed under the transition practical expedient. Going forward, new or modified land easements will be evaluated under ASU No. 2016-02. |
| ● | Separation of lease and non- lease components: Lease agreements that contain both lease and non-lease components are generally accounted for separately. |
| ● | Short-term lease recognition exemption: The Company also elected the short-term lease recognition exemption and will not recognize ROU assets or lease liabilities for leases with a term less than 12 months. |
The new leasing standard requires recognition of leases on the consolidated balance sheets as right-of-use (“ROU”) assets and lease liabilities. ROU assets represent the Company’s right to use underlying assets for the lease terms and lease liabilities represent the Company’s obligation to make lease payments arising from the leases. Operating lease ROU assets and operating lease liabilities are recognized based on the present value and future minimum lease payments over the lease term at commencement date. The Company’s future minimum based payments used to determine the Company’s lease liabilities mainly include minimum based rent payments. As most of Company’s leases do not provide an implicit rate, the Company uses its estimated incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments.
The Company recognized lease liabilities, with corresponding ROU assets, based on the present value of unpaid lease payments for existing operating leases longer than twelve months as of January 1, 2019. The ROU assets were adjusted per ASC 842 transition guidance for existing lease-related balances of accrued and prepaid rent, unamortized lease incentives provided by lessors, and restructuring liabilities.
The adoption of ASC 842 had a substantial impact on the Company’s consolidated balance sheets. The most significant impact was the recognition of the operating lease right-of-use assets and the liability for operating leases. Accordingly, adoption of this standard resulted in the recognition of operating lease right-of-use assets of $577,830 and operating lease liabilities of $598,937 comprised of $301,105 of current operating lease liabilities and $297,832 of non-current operating lease liabilities on the consolidated balance sheet as of January 1, 2019. The adoption of ASC 842 also resulted in a cumulative-effect adjustment of $(21,107) to the opening balance of accumulated deficit.
In addition, the adoption of the standard did not have a material impact on the Company’s results of operations or cash flows. Operating lease cost is recognized as a single lease cost on a straight-line basis over the lease term and is recorded in Selling, general and administrative expenses. Variable lease payments for common area maintenance, property taxes and other operating expenses are recognized as expense in the period when the changes in facts and circumstances on which the variable lease payments are based occur.
F-41
The Company has no finance leases. The Company’s leases primarily include various office and laboratory spaces, copy machine, and vehicles under various operating lease arrangements. The Company’s operating leases have remaining lease terms of up to approximately five years.
| December 31, 2020 | December 31, 2019 | |||||||
| ASSETS | ||||||||
| Operating lease right-of-use assets | $ | 1,772,747 | $ | 524,445 | ||||
| LIABILITIES | ||||||||
| Operating lease liabilities (current) | 316,178 | 304,248 | ||||||
| Operating lease liabilities (noncurrent) | 1,456,567 | 235,555 | ||||||
Supplemental Information
The following provides details of the Company’s lease expenses:
| Year
Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Operating lease expenses | $ | 337,282 | $ | 301,437 | ||||
Other information related to leases is presented below:
| Year
Ended December 31, |
||||||||
| 2020 | 2019 | |||||||
| Cash paid for amounts included in the measurement of operating lease liabilities | $ | 337,282 |
$ | 301,437 | ||||
| December
31, 2020 |
December 31, 2019 |
|||||||
| Weighted Average Remaining Lease Term: | ||||||||
| Operating leases | 3.08 years | 3.08 years | ||||||
| Weighted Average Discount Rate: | ||||||||
| Operating leases | 0.55 | % | 0.55 | % | ||||
The minimum future annual payments under non-cancellable leases during the next five years and thereafter, at rates now in force, are as follows:
| Operating leases | ||||
| 2021 | $ | 324,788 | ||
| 2022 | 341,527 | |||
| 2023 | 358,431 | |||
| 2024 | 375,337 | |||
| 2025 | 338,676 | |||
| Thereafter | 56,916 | |||
| Total future minimum lease payments, undiscounted | 1,795,675 | |||
| Less: Imputed interest | 22,930 | |||
| Present value of future minimum lease payments | $ | 1,772,745 | ||
F-42
17. BUSINESS COMBINATION
On February 8, 2019, the Company consummated the Merger transactions of BioLite and BioKey (See Note 1). Pursuant to the terms of the Merger Agreement, BioLite and BioKey became two wholly-owned subsidiaries of the Company on February 8, 2019. The Company adopted ASC 805, “Business Combination” to record the merger transactions of BioKey. The acquisition was accounted for as a business combination under the purchase method of accounting. BioKey’s results of operations were included in the Company’s results beginning February 8, 2019. The purchase price has been allocated to the assets acquired and the liabilities assumed based on their fair value at the acquisition date as summarized in the following:
| Purchase consideration: | ||||
| Common Stock (*) | $ | 44,341,847 | ||
| Allocation of the purchase price: | ||||
| Cash and cash equivalents | $ | 531,147 | ||
| Accounts receivable, net | 188,550 | |||
| Property and equipment, net | 56,075 | |||
| Operating lease right-of-use assets | 485,684 | |||
| Security deposits | 10,440 | |||
| Total assets acquired | 1,271,896 | |||
| Accounts payable | (56,204 | ) | ||
| Accrued expenses and other current liabilities | (251,335 | ) | ||
| Operating lease liability | (267,256 | ) | ||
| Tenant security deposit | (2,880 | ) | ||
| Total liabilities assumed | (577,675 | ) | ||
| Total net assets acquired | 694,221 | |||
| Goodwill as a result of the Merger | $ | 43,647,626 | ||
| * | 29,561,231 shares (1,642,291 after stock reverse split) of common stock of the Company was issued to BioKey in connection with the Merger. Those shares were valued at $1.50 per share, based on the bid-and-ask share price of common stock of the Company on the final day of trading, February 8, 2019. |
On February 8, 2019, the Company has recorded a 100% goodwill write-down of $43,647,626. Goodwill was determined to have been impaired because of the current financial condition of the Company and the Company’s inability to generate future operating income without substantial sales volume increases, which are highly uncertain. Furthermore, the Company’s anticipated future cash flows indicate that the recoverability of goodwill is not reasonably assured. The goodwill write-down was reflected as a decrease in additional paid-in capital in the statement of equity upon the consummation of the Merger.
18. SUBSEQUENT EVENTS
On February 7, 2021, the entity of ABVC received a loan in the amount of $104,167 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from Cathay Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll. The loan was granted pursuant to a promissory note dated February 7, 2021 issued by the Company, which matures on February 6, 2026 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is five years after the date of the promissory note. In addition, the Company will start the application with the US Government regarding the loan forgiveness program as soon as the US Government allows. No collateral or personal guarantees are required.
On January 29, 2021, the entity of BioKey received a loan in the amount of $132,331 under the Paycheck Protection Program (“PPP”) administered by the United States Small Business Administration (the “SBA”) from East West Bank. According to the Coronavirus Aid, Relief, and Economic Security Act (the “Cares Act”), PPP loan provides for forgiveness of up to the full principal amount and accrued interest if the funds are used for payroll costs, interest on mortgages, rent, and utilities. However, at least 60% of the forgiven amount must have been used for payroll. The loan was granted pursuant to a promissory note dated January 27, 2021 issued by the Company, which matures on January 28, 2026 and bears interest at a rate of 1.00% per annum. The Company will pay the principal in one payment of all outstanding principal plus all accrued unpaid interest on that date that is five years after the date of the promissory note. In addition, the Company will start the application with the US Government regarding the loan forgiveness program as soon as the US Government allows. No collateral or personal guarantees are required.
The Company has evaluated subsequent events through the date which the financial statements were available to be issued. All subsequent events requiring recognition as of December 31, 2020 have been incorporated into these financial statements and there are no subsequent events that require disclosure in accordance with FASB ASC Topic 855, “Subsequent Events.”
F-43
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we have evaluated the effectiveness of the design and operation of our “disclosure controls and procedures,” as such term is defined in Rule 13a-15(e) or Rule 15d-15(e) promulgated under the Exchange Act as of the end of the period covered by this report. Based upon that evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures were not effective as of the end of the period covered by this report to provide reasonable assurance that material information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f) or Rule 15d-15(f). Our management conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 Framework). Based on this evaluation, our management concluded that our internal controls over financial reporting were not effective as of December 31, 2020 because of the lack in segregation of duties in financial reporting due to the small size of our financial staff.
This annual report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our independent registered public accounting firm pursuant to rules of the Securities and Exchange Commission that permit us to provide only management’s report in this annual report.
Changes in Internal Control over Financial Reporting
There has been no change in our internal control over financial reporting during year ended December 31, 2020.
None.
65
ITEM 10. DIRECTORS AND EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE.
The following table sets forth as of the date of this annual report, the name, age, and position of each executive officer and director and the term of office of each such person.
Set forth below is certain biographical information regarding each of our directors and officers as of the date of this annual report.
| Name | Age | Title | ||
| Eugene Jiang | 34 | Chairman of the Board | ||
| Chihliang An | 40 | Chief Financial Officer (“CFO”) | ||
| Dr. Tsang Ming Jiang | 60 | Director | ||
| Dr. Ming-Fong Wu | 45 | Independent Director | ||
| Norimi Sakamoto | 50 | Independent Director | ||
| Yen-Hsin Chou | 32 | Independent Director | ||
| Dr. Tsung-Shann (T.S.) Jiang | 67 | Chief Strategy Officer (“CSTRO”) and Director | ||
| Dr. Chang-Jen Jiang | 65 | Director | ||
| Dr. Shin-Yu Miao | 57 | Independent Director | ||
| Yoshinobu Odaira | 72 | Independent Director | ||
| Shih-Chen Tzeng | 63 | Independent Director | ||
| Dr. Hwalin Lee | 84 | Director | ||
| Dr. Howard Doong | 63 | Chief Executive Officer (“CEO”) | ||
| Dr. Chi-Hsin (Richard) King | 71 | Chief Scientific Officer (“CSO”) |
Eugene Jiang has served as our CEO and President since the Company’s inception in July 2015 until he resigned on September 15, 2017. He remains the Chairman of the Board. From June 2015 until present, Mr. Jiang also serves as Director for BioLite Incorporation. He also serves as CEO for Genepro Investment Company since March 2010. Mr. Jiang obtained an EMBA degree from the University of Texas in Arrington in 2009. And in 2008, Mr. Jiang received a bachelor’s degree in Physical Education from Fu-Jen Catholic University.
Dr. Howard Doong, was appointed as the Company’s new CEO on September 15, 2017. In addition to the position at the Company, Dr. Doong also serves as the CEO and Chief Scientific Officer (“CSO”) of LifeCode Biotechnology Company (“LifeCode”), a Taiwan company in the biotechnology business, since 2017. At the same time, he also serves as the CSO of Wuhan Frasergen Genomic Medicine Company (“Wuhan Frasergen Genomic”), a Chinese company in the biotechnology business, since 2016. He served as the CSO of Cold Spring Biotech Corporation, a Taiwan corporation in the biotechnology business from 2014 to 2016. He served as the CEO of iKnowledge-Care Bioscience Corp, a Taiwan company in the biotechnology business from 2014 to 2015. He served as the director of Taipei Veteran General Hospital-LihPao Laboratory of Cancer Genomic Medicine from 2012 to 2013. He served as the Vice President and director of Quality Assurance, TrimGen Corporation, a Maryland corporation in the biotechnology business from 2006 to 2011. Before 2006, Dr. Doong was a professor at the University of Maryland School of Medicine and Biotechnology Institute, and a researcher at National Cancer Institute (NCI) of the National Institutes of Health (NIH). Dr. Doong received his Ph.D. degree from University of Chicago, the Department of Organismal Biology and Anatomy and the Department of Surgery. He received his M.D and Ph.D. degree from Harvard-MIT Division of Health Sciences and Technology. He received his M.S. degree from the University of New Hampshire, Genetics Program and B.S. degree from Fu-Jen Catholic University, Taiwan, Department of Biology.
66
Mr. Chihliang An, was the Managing Director of Investment Team at Yinyai Investment (Hong Kong) since September 2018 to June 2019. Prior to that, Mr. An served as a Senior Director of Sales and Operations at Goertek (USA) from March 2017 to August 2018 and a Director of Finance at BioKey, Inc., which is a wholly-owned subsidiary of the Company now, from March 2015 to February 2017. Mr. Chihliang An received a Bachelor of Art degree in Statistics from Tamkang University in Taiwan in 2003 and an MBA in Finance from University of Kentucky in 2007.
Dr. T.S. Jiang, has been the chairman of BioLite, Inc., a subsidiary of BioLite, Inc., since January 2010. Prior to BioLite, Dr. Jiang served as the president and/or chairman of multiple biotech companies in Taiwan, including PhytoHealth Corporation from 1998 to 2009 and AmCad BioMed Corporation from 2008 to 2009. In addition, Dr. Jiang is a director on various biotech associations, such as the Taiwan Bio Industry Organization (Taiwan) from 2006 to 2008 and the Chinese Herbs and Biotech Development Association in Taiwan from 2003 to 2006. Dr. Jiang was an assistant professor at University of Illinois from 1981 to 1987 and an associate professor at Rutgers, the State University of New Jersey from 1987 to 1990 and served as a professor at a few Taiwanese universities during a period from 1990 to 1993, such as National Taiwan University, National Cheng Kung University and Tunghai University. Dr. Jiang obtained his bachelor degree in Engineering and Chemical Engineering from National Taiwan University in Taiwan in 1976, masters and Ph.D. from Northwestern University in the U.S. in 1981 and Executive Master of Business Administration (“EMBA”) from National Taiwan University in Taiwan in 2007. As a successful entrepreneur, Dr. Jiang has developed and commercialized PG2 Lyo Injection, a new drug to treat cancer related fatigue. From 1998 to 2009, Dr. T. S. Jiang served as President of Phyto Health Corporation where he led a project team to develop PG2 Injectable. This product was extracted, isolated and purified from a type of Traditional Chinese Medicine. PG2 Injection was intended for cancer patients who had trouble recovering from severe fatigue. Dr. Jiang oversaw and managed the R&D department, daily corporate operations and business of Phyto Health Corporation when he was the President. PG2 Lyo Injection received approval on its NDA from Taiwan Food and Drug Administration in 2010 and later was launched into the Taiwan market in 2012. We believe that Dr. Jiang provides leadership and technological guidance on our strategic development and operations.
Dr. Tsang Ming Jiang, has served as a technical director at the Industrial Technology Research Institute in Taiwan since January 2017. Prior to joining the Industrial Technology Research Institute as a technical director, Dr. Jiang worked at the Company as chief information officer from November 2016 to January 2017, Ericsson as engineering manager from 2013 to 2016 and the Industrial Technology Research Institute as deputy director from October 2011 to February 2013. In addition, Dr. Jiang worked at several other research institutes, including University of Alaska Fairbanks, National Taiwan University and Chung Cheng University, with his research interest in cloud computing and Internet security, especially in the areas of virtualization, software-defined data centers, SDN enabled networks and big data analytics. Dr. Jiang received his Bachelor of Science in electrical engineering in 1982 and Master of Science in electrical engineering in 1984, both from National Taiwan University, and his Ph.D. in electrical engineering and computer science from University of Illinois at Chicago in 1988. Dr. Tsang Ming Jiang is a brother of Dr. Tsung-Shann Jiang, who together with his wife collectively owns 80% of Lion Arts Promotion, Inc. which has approximately 69.3% of ownership interest in the Company through YuanGene Corporation, a wholly-owned subsidiary of Lion Arts Promotion, Inc.
Dr. Ming-Fong Wu, is a senior physician at Taoyuan Hanqun Orthopedic Clinic from 2012. Prior to Taoyuan Hanqun Orthopedic Clinic, Dr. Wu worked as a physician at various private and public hospitals and clinics, such as National Taiwan University Hospital. Dr. Wu graduated from National Taiwan University College of Medicine in 2000 and has obtained his license to practice medicine and orthopedist’s license in Republic of China.
Norimi Sakamoto, currently serves at four enterprises, Shogun Maitake Canada Co., Ltd. as an executive officer and business development manager from 2015, Shogun Maitake Odaira Enterprise Ltd as an executive officer from 2017, Odaira Corporation Co., Ltd. as chief executive officer since 2014 and MyLife Corporation as president and chief executive officer since 2012. Ms. Sakamoto started her career in 1997 from Sumitomo Corporation Hokkaido Co., Ltd. in Japan. Ms. Sakamoto received her Bachelor Degree of Arts in travel and tourism from Davis and Elkins College in 1993 and Master of Science in urban studies from the University of New Orleans in 1995.
67
Yen-Hsin Chou, has served as a clerk at Mega Securities Co., Ltd. since 2011. Ms. Chou’s responsibilities primarily include selling various types of securities, including futures, funds and insurance, managing clients’ accounts and business development. Ms. Chou received a Bachelor Degree from Yuan Chi University School of Economics in 2011.
Dr. Chang-Jen Jiang, has been an attending doctor at the department of pediatrics of Eugene Women and Children Clinic since 2009. Previously, Dr. Chang-Jen worked as an attending doctor at the department of pediatrics of Keelung Hospital, the Ministry of Health and Welfare in Taiwan from 1994 to 2009. Before his position at Keelung Hospital, he was a chief doctor at the department of pediatrics, hematology and oncology of Mackay Memorial Hospital in Taiwan for three years until 1994. Dr. Chang-Jen Jiang obtained his doctor of medicine degree (the Taiwanese equivalent degree of MD) from Taipei Medical University in Taiwan in 1982 and started his career in Mackay Memorial Hospital. We believe that the Company will benefit from Dr. Jiang’s knowledge in biology and experiences in medical practice.
Dr. Shin-Yu Miao, has served as an associate professor at Ling Tung University Department of Applied Foreign Languages since 2004. She served as a lecturer from 1996 to 2004. Ms. Miao received her M.S. in Adult Education from the University of Manchester in 1995 and Ph.D. in Adult Education from the University of South Australia in 2004. We believe that Ms. Miao’s familiarity with biotech research centers will be a valuable resource for our drug development.
Yoshinobu Odaira, is an entrepreneur and has founded a number of Japanese agricultural companies, including Yukiguni Maitake, our licensing partner. In 1983, Mr. Odaira established Yukiguni Maitake, which became a public company in Japan in 1994. In 2015, Bain Capital Private Equity purchased Yukiguni Maitake through a tender offer. In addition to his success with Yukiguni Maitake, Mr. Odaira served as the CEO of Yukiguni Shoji Co., Ltd. since 1988 and the CEO of Odaira Shoji Co., Ltd. from 1989. In 2015, Mr. Odaira founded two new companies, Shogun Maitake Canada Co., Ltd. in Canada and Odaira Kinoko Research Co., Ltd. in Japan. Yoshinobu Odaira graduated from the Ikazawa Junior High School in 1963. We believe that we will benefit from Mr. Odaira’s successful business experience.
Shih-Chen Tzeng, has served as a sales manager at SinoPac Securities Corp. (“SinoPac Securities”), a well-established brokerage firm in Taiwan, since 2000. SinoPac Securities has fifty-eight (58) branch offices in Taiwan and subsidiaries in Hong Kong, Shanghai and London. Shih-Chen Tszeng graduated from Dam Kang University in 1978 with a bachelor degree in Accounting. We believe the Company will benefit from Ms. Tszeng’s knowledge and experience with the securities industries.
Dr. Hwalin Lee, serves as the chairman of Phoeng Foundation since 2011 and will become the director and chairman of the board of directors of BioKey Surviving Corporation after the closing of the BioKey Merger. From 1986, Dr. Lee has been the chairman of the Chuan Lyu Foundation. From 1973 to 1989, Dr. Lee was the president of Deltan Corporation and prior to that he was senior research chemist at a couple of chemical companies. Dr. Hwalin Lee obtained a B.S. in pharmacy from National Taiwan University in 1957 and a Ph.D. in Pharmaceutical Chemistry from University of California, San Francisco in 1966. Dr. Lee qualifies as a director of the Company because he has extensive work experience in chemical companies and educational background in pharmaceutical chemistry.
Significant Employees
The following are employees who are not executive officers, but who are expected to make significant contributions to our business:
Dr. Chi-Hsin Richard King—Chief Scientific Officer
Effective September 15, 2017, the Board appointed Dr. Chi-Hsin Richard King as the CSO of the Company. Dr. Chi-Hsin Richard King, 71, retired since July 2017. He served as the consultant at TaiGen Biotechnology Co. Ltd (“TaiGen”), a Taiwan company in the biotechnology business, from August 2016 to July 2017, the Senior Vice President at TaiGen from July 2008 to August 2016 and as the Vice President at Research and Development of TaiGen from June 2005 to July 2008. Dr. King served as the Director at Albany Molecular Research Inc. (“AMRI”), a New York corporation, from January 2003 to June 2005, the Assistant Director at Medicinal Chemistry Department of AMRI from January 2000 to December 2002 and the Assistant Director at Chemical Development Department of AMRI from August 1997 to January 2000. Dr. King received the Ph. D. degree of organic chemistry from University of Utah in March 1980, and B.S. degree of chemistry from National Taiwan Normal University in July 1972.
68
Compliance with Section 16(a) of the Exchange Act
Our common stock is not registered pursuant to Section 12 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Accordingly, officers, directors and principal shareholders are not subject to the beneficial ownership reporting requirements of Section 16(a) of the Exchange Act.
ITEM 11. EXECUTIVE COMPENSATION
The following tables set forth, for each of the last two completed fiscal years of us, the total compensation awarded to, earned by or paid to any person who was a principal executive officer during the preceding fiscal year and every other highest compensated executive officers earning more than $100,000 during the last fiscal year (together, the “Named Executive Officers”). The tables set forth below reflect the compensation of the Named Executive Officers.
Summary Compensation Table
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Stock
Awards ($) | Option
Awards ($) (7) | Non-Equity
Incentive Plan Compensation ($) | Change
in Pension Value and Nonqualified Deferred Compensation Earnings ($) | All
Other Compensation ($) | Total ($) | |||||||||||||||||||||||||
| Howard Doong (1) | 2020 | 200,000 | 234,750 | 434,750 | ||||||||||||||||||||||||||||||
| 2019 | 133,332 | 150,003 | 283,335 | |||||||||||||||||||||||||||||||
| Chun Mu Hung (2) | 2020 | - | - | - | ||||||||||||||||||||||||||||||
| 2019 | 14,434 | - | - | - | - | - | 14,434 | |||||||||||||||||||||||||||
| Chihliang An (3) | 2020 | 200,000 | 156,500 | 356,500 | ||||||||||||||||||||||||||||||
| 2019 | 133,332 | 100,002 | 233,334 | |||||||||||||||||||||||||||||||
| Tsung-Shann Jiang (4) | 2020 | 200,000 | 106,749 | 306,749 | ||||||||||||||||||||||||||||||
| 2019 | 133,332 | - | 133,332 | |||||||||||||||||||||||||||||||
| Richard Chi-Hsin King (5) | 2020 | 200,000 | 234,750 | 434,750 | ||||||||||||||||||||||||||||||
| 2019 | 133,332 | 150,010 | 283,342 | |||||||||||||||||||||||||||||||
| Eugene Jiang (6) | 2020 | 200,000 | 207,585 | 407,585 | ||||||||||||||||||||||||||||||
| 2019 | 133,332 | 128,030 | 261,362 | |||||||||||||||||||||||||||||||
| (1) | Dr. Doong was appointed as the CEO on September 15, 2017. |
| (2) | Mr. Hung was appointed as the CFO, Secretary and Treasury on September 15, 2017 and resigned as the CFO, Secretary and Treasurer on May 4, 2018. |
| (3) | Mr. An was appointed as the CFO on September 1, 2019. |
| (4) | Dr. Jiang was appointed as the CSTRO on September 1, 2019 |
| (5) | Dr. King was appointed as the CSO on September 15, 2017 |
| (6) | Eugene Jiang was appointed as CBO on September 1, 2019 |
| (7) | All options disclosed in this table were issued in exchange for previously unpaid and accrued salary owed to each such individual, following the individuals agreement to convert such salary into options. The total amount of salary that was converted into options was $600,850. |
Narrative Disclosure to Summary Compensation Table
Other than set out below, there are no arrangements or plans in which we provide pension, retirement or similar benefits for directors or executive officers. Our directors and executive officers may receive share options at the discretion of our board of directors in the future. We do not have any material bonus or profit sharing plans pursuant to which cash or non-cash compensation is or may be paid to our directors or executive officers, except that share options may be granted at the discretion of our board of directors.
69
Stock Option Plan
Our board approved and adopted the Amended and Restated 2016 Equity Incentive Plan on September 12, 2020 (the “Plan”), a copy of which is attached hereto as exhibit 10.17.
Grants of Plan-Based Awards
On May 29, 2020, the Company’s Board of Directors approved the following future option awards pursuant to the Plan, but such options may not be issued until the Compensation Committee or the Board further approves such issuance:
| ● | 20,000 options to each director; such options are exercisable at $2.00 per share. |
| ● | Options for 266,667 shares, 155,556 shares, and 211,111 shares to the CEO, CFO and CSO, respectively; the options are exercisable at $2.00 per share. |
On November 21, 2020, the Company issued an aggregate of 545,182 options to purchase shares of Common Stock in lieu of unpaid salaries of certain employees (other than Officers and Directors) and unpaid consulting fees under the Plan, as amended; the total converted salaries was $1,090,361. The options are exercisable at $2.00 per share.
As of the date of this report, we have granted options under the Plan that can be exercised for an aggregate of 545,182 shares of Common Stock.
Outstanding Equity Awards at Fiscal Year End
The following table summarizes outstanding unexercised options, unvested stocks and equity incentive plan awards held by each of our named executive officers, as of December 31, 2020:
OUTSTANDING EQUITY AWARDS AT FISCAL YEAR-END
| OPTION AWARDS | STOCK AWARDS | |||||||||||||||||||||||||||||||||
| Name | Number
of Securities Underlying Unexercised Options (#) Exercisable | Number
of Securities Underlying Unexercised Options (#) Unexercisable | Equity
Incentive Plan Awards: Number of Securities Underlying Unexercised Unearned Options (#) | Options Exercise Prices ($) | Option Expiration Date | Number
of Shares or Units of Stock That Have Not Vested (#) | Market Value of Shares or Units of Stock That Have Not Vested ($) | Equity Incentive Plan Awards: Number of Unearned Shares, Units or Other Rights That Have Not Been Issued (#) | Equity
Incentive Plan Awards: Market or Payout Value of Unearned Shares, Units or Other Rights That Have Not Been Issued ($) | |||||||||||||||||||||||||
| Howard Doong | 85,715 | 10,715 | - | 2.00 | Nov 20, 2031 | - | - | - | - | |||||||||||||||||||||||||
| - | 266,667 | (1) | 1,360,002 | (2) | ||||||||||||||||||||||||||||||
| Chihliang An | 54,762 | 9,524 | 2.00 | Nov 20, 2031 | 155,556 | (1) | 793,336 | (2) | ||||||||||||||||||||||||||
| Tsung-Shann Jiang | 34,105 | - | 2.00 | Nov 20, 2031 | ||||||||||||||||||||||||||||||
| Richard Chi-Hsin King | 82,144 | 14,286 | 2.00 | Nov 20, 2031 | 211,111 | (1) | 1,076,666 | (2) | ||||||||||||||||||||||||||
| Eugene Jiang | 72,418 | 12,193 | 2.00 | Nov 20, 2031 | ||||||||||||||||||||||||||||||
| (1) | The Board approved granting these future options, but they have not been issued as of the date of this Report and may not be issued until the Compensation Committee or the Board further approves such issuance. |
| (2) | Based on $5.10 per share, which is the closing market price of our Common Stock as of December 31, 2020. |
70
Compensation of Directors
We did not pay any compensation to our non-executive directors in fiscal year 2020.
Pension, Retirement or Similar Benefit Plans
There are no arrangements or plans in which we provide pension, retirement or similar benefits for directors or executive officers. We have no material bonus or profit sharing plans pursuant to which cash or non-cash compensation is or may be paid to our directors or executive officers, except that stock options may be granted at the discretion of the board of directors or a committee thereof.
Employment Contracts
Dr. Howard Doong has entered into an employment agreements (“Doong Employment Agreement”) with the Company, pursuant to which he shall receive an annual base salary of $100,000. As of December 31, 2017, we paid Mr. Doong 20,833 shares of the Company’s common stock at a per share price of $1.60 as opposed to cash compensation. Under Doong Employment Agreement, Dr. Doong is employed as our CEO and President of the Company. We may terminate the employment for cause, at any time, without notice or remuneration, for certain acts of the executive officer, such as conviction or plea of guilty to a felony or grossly negligent or dishonest acts to our detriment, or misconduct or a failure to perform agreed duties. In such case, the executive officer will not be entitled to receive payment of any severance benefits or other amounts by reason of the termination, and the executive officer’s right to all other benefits will terminate, except as required by any applicable law. We may also terminate an executive officer’s employment without cause upon one-month advance written notice. In such case of termination by us, we are required to provide compensation to the executive officer, including severance pay equal to 12 months of base salary. The executive officer may terminate the employment at any time with a one-month advance written notice if there is any significant change in the executive officer’s duties and responsibilities or a material reduction in the executive officer’s annual salary. In such case, the executive officer will be entitled to receive compensation equivalent to 12 months of the executive officer’s base salary. On August 21, 2019, all of the Board members present at the Meeting, unanimously reelected Dr. Howard Doong as the Chief Executive Officer (“CEO”), which shall become effective on September 1, 2019 for a term of three years.
On August 21, 2019, all of the Board members present at the Meeting unanimously appointed Mr. Chihliang An as the Company’s Chief Financial Officer (“CFO”) effective from September 1, 2019 for a term of three years.
Mr. Chun Mu Hung has entered into an employment agreement (“Hung Employment Agreement”) with the Company, pursuant to which he shall receive an annual base salary of $40,000. On May 4, 2018, Mr. Hung resigned as the CFO, Secretary and Treasurer of the Company, effective immediately.
Dr. Chi-Hsin Richard King has entered into an employment agreements (“King Employment Agreement”) with the Company, pursuant to which he shall receive an annual base salary of $50,000. As of December 31, 2017, we paid Mr. King 10,416 shares of the Company’s common stock at a per share price of $1.60 as opposed to cash compensation. Under King Employment Agreement, Dr. King is employed as the CSO of the Company. We may terminate the employment for cause, at any time, without notice or remuneration, for certain acts of the executive officer, such as conviction or plea of guilty to a felony or grossly negligent or dishonest acts to our detriment, or misconduct or a failure to perform agreed duties. In such case, the executive officer will not be entitled to receive payment of any severance benefits or other amounts by reason of the termination, and the executive officer’s right to all other benefits will terminate, except as required by any applicable law. We may also terminate an executive officer’s employment without cause upon one-month advance written notice. In such case of termination by us, we are required to provide compensation to the executive officer, including severance pay equal to 12 months of base salary. The executive officer may terminate the employment at any time with a one-month advance written notice if there is any significant change in the executive officer’s duties and responsibilities or a material reduction in the executive officer’s annual salary. In such case, the executive officer will be entitled to receive compensation equivalent to 12 months of the executive officer’s base salary. On August 21, 2019, all of the Board members present at the Meeting, unanimously reelected Dr. Richard King as the Chief Scientific Officer (“CSO”), which shall become effective on September 1, 2019 for a term of three years
On August 21, 2019, all of the Board members present at the Meeting, except Eugene Jiang, appointed Mr. Eugene Jiang, the current Chairman of the Board, as the Chief Business Officer, effective since September 1, 2019 for a term of three years. Mr. Eugene Jiang excused himself from the discussion regarding his appointment as the Chief Business Officer of the Company during the Board meeting.
On August 21, 2019, all of the Board members present at the Meeting, except Dr. Tsung-Shann Jiang, reelected Dr. Tsung-Shann Jiang as the Chief Strategy Officer, effective since September 1, 2019 for a term of three years. Dr. Tsung-Shann Jiang excused himself from the discussion regarding his appointment as the Chief Strategy Officer of the Company during the Board meeting.
71
ITEM 12. SECURITY OWENERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Beneficial Owners
The following table sets forth certain information regarding beneficial ownership of our common stock as of March 15, 2021 (i) each person (or group of affiliated persons) who is known by us to own more than five percent (5%) of the outstanding shares of our common stock, (ii) each director, executive officer and director nominee, and (iii) all of our directors, executive officers and director nominees as a group.
Beneficial ownership is determined in accordance with SEC rules and generally includes voting or investment power with respect to securities. For purposes of this table, a person or group of persons is deemed to have “beneficial ownership” of any shares of common stock that such person has the right to acquire within 60 days of the date of the respective table. For purposes of computing the percentage of outstanding shares of our common stock held by each person or group of persons named above, any shares that such person or persons has the right to acquire within 60 days of the date of the respective table is deemed to be outstanding for such person, but is not deemed to be outstanding for the purpose of computing the percentage ownership of any other person. The inclusion herein of any shares listed as beneficially owned does not constitute an admission of beneficial ownership.
Unless otherwise noted, the business address of each beneficial owner listed is 44370 Old Warm Springs Blvd., Fremont, CA 94538. Except as otherwise indicated, the persons listed below have sole voting and investment power with respect to all shares of our common stock owned by them, except to the extent that power may be shared with a spouse.
As of March 15, 2021, we had 24,420,526 shares of common stock issued and outstanding.
| Name of Beneficial Owner | Amount
and Nature of Beneficial Ownership | Percent Class |
| |||||
| Dr. Howard Doong | 18,404 | * | % | |||||
| Eugene Jiang (1) | 732,716 | * | ||||||
| Chihliang An | 13,334 | * | ||||||
| Chi-Hsin (Richard) King | 869 | * | ||||||
| Yen-Hsin Chou | 5,679 | * | ||||||
| Dr. Shin-Yu Miao (2) | 67,378 | * | ||||||
| Dr. Tsang-Ming Jiang | 6,067 | * | ||||||
| Dr. Ming-Fong Wu | - | * | ||||||
| Norimi Sakamoto | - | * | ||||||
| Dr. Tsung-Shann Jiang (3)(5) | 12,103,971 | 49.6 | % | |||||
| Dr. Chang-Jen Jiang (4) | 5,949 | * | ||||||
| Yoshinobu Odaira | 163,702 | * | ||||||
| Shih-Chen Tzeng | - | * | ||||||
| Dr. Hwalin Lee | 133,314 | * | ||||||
| All officers and directors as a group (Fourteen (14) persons) | 13,251,383 | 54.3 | % | |||||
| YuanGene Corporation (5) | 8,296,968 | 34 | % | |||||
| * | less than 1%. |
| (1) | Eugene Jiang held 701,989 shares of the Company’s common stock through his ownership in AsianGene, 5,352 shares of the Company’s common stock through his ownership in BioFirst, 202 shares of the Company’s common stock through his ownership in Rgene, and the rest of 25,173 shares through direct ownership. |
| (2) | Dr. Shin-Yu Miao held 1,705 shares of the Company’s common stock through her ownership in BioFirst, 7 shares of the Company’s common stock through his ownership in Rgene, and the rest of 65,666 through direct ownership. |
| (3) | Dr. Tsung-Shann Jiang held 8,296,968 shares of common stock through his ownership in YuanGene Corporation, 11,603 shares of the Company’s common stock through BioLite, 19,609 shares through Rgene Corporation, 143,264 shares through BioFirst, 674,723 shares through Lion Arts, 509,877 shares through LionGene, 8,850 shares through Genepro Investment, 213,120 shares through Keypoint, 64,215 shares through Shu-Ling Chiang, and the rest of 2,161,742 shares through direct ownership. |
| (4) | Dr. Chang-Jen Jiang held 1,343 shares of common stock in the Company through his ownership in BioFirst, 6 shares of the Company’s common stock through Rgene, and the rest of 4,600 shares through direct ownership. |
| (5) | YuanGene Corporation is a company wholly-owned by Lion Arts, which is owned by Shu-Ling Chiang (80%) and Dr. Tsung-Shann Jiang (20%); however, YuanGene appointed Eugene Jiang to have sole voting control over the shares held by YuanGene, the principal office address of which is 2nd floor, Building B, SNPF Plaza, Savalalo, Apia, Samoa. |
72
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, DIRECTOR INDEPENDENCE
Except as disclosed herein, no director, executive officer, shareholder holding at least 5% of shares of our common stock, or any family member thereof, had any material interest, direct or indirect, in any transaction, or proposed transaction since January 1, 2019, in which the amount involved in the transaction exceeds the lesser of $120,000 or one percent of the average of our total assets at the year-end for the last two completed fiscal years.
Co-Development agreement with Rgene Corporation
On November 10, 2020, the Company and Rgene signed an amendment to the Co-Dev Agreement dated May 26, 2017, pursuant to which both parties agreed to delete AB-1507 HER2/neu Positive Breast Cancer Combination Therapy and AB 1527 Ovary Cancer Combination Therapy and add ABV-1519 EGFR Positive Non-Small Cell Lung Cancer Combination Therapy and ABV-1526 Large Intestine / Colon / Rectal Cancer Combination Therapy to the products to be co-developed and commercialized. Other provisions of the Co-Dev Agreement remain in full force and effect.
Collaborative agreement with BioFirst Corporation
On November 4, 2020, we executed an amendment to our collaboration agreement with BioFirst dated July 24, 2017, to add BFC-1403 Intraocular Irrigation Solution and BFC-1404 Corneal Storage Solution to our agreement. BFC-1404 is utilized during a corneal transplant procedure to replace a damaged or diseased cornea while BFC-1403 has broader utilization during a variety of ocular procedures.
Initially ABVC will focus on BFC-1404, a solution utilized to store a donor cornea prior to either penetrating keratoplasty (full thickness cornea transplant) or endothelial keratoplasty (back layer cornea transplant). Designated ABV-2002 under ABVC’s product identification system, the solution is comprised of a specific poly amino acid that protects ocular tissue from damage caused by external osmolarity exposure during pre-surgery storage. The specific polymer in ABV 2002 can adjust osmolarity to maintain a range of 330 to 390 mOsM thereby permitting hydration within the corneal stroma during the storage period. Stromal hydration results in (a) maintaining acceptable corneal transparency and (b) prevents donor cornea swelling. ABV-2002 also contains an abundant phenolic phytochemical found in plant cell walls that provides antioxidant antibacterial properties and neuroprotection.
Early testing by BioFirst indicates that ABV-2002 may be more effective for protecting the cornea and retina during long-term storage than other storage media available today and can be manufactured at lower cost. Categorized as a Class I Medical Device that has the lowest risk to patients, ABVC intends to submit a Premarket Notification 510(K) submission to the FDA before the end of 2021 to demonstrate the device is at least as safe and effective as current products on the market.
On July 12, 2019, the Company had an aggregate amount of loan with BioFirst of $150,000 to meet its working capital needs, pursuant to which the interest bears at 12% per annum. The Company paid back $21,317 in 2019. The remaining loan balance was $128,683 as of December 31, 2019. The loan matured on July 11, 2020.
On May 11, 2018, the Company and BioFirst (Australia) Pty Ltd. (the “BioFirst (Australia)”) (an entity 100% owned by BioFirst and controlled by controlling beneficiary shareholder of YuanGene), entered into a loan agreement for a total amount of $40,000 to meet its working capital needs. The advances bear 0% interest rate and are due on demand prior to June 30, 2020. Afterwards, all outstanding load will bear interest rate at 12% per annum. On July 1, 2020, the Company entered into another loan agreement with BioFirst (Australia) for $321,487 to properly record R&D cost and tax refund allocation based on a co-development contract executed on July 24, 2017. The loan will mature on June 30, 2021 with an interest rate of 12% per annum. As of December 31, 2020, the outstanding loan balances was $373,235.
73
Other related party transactions
| ● | On February 24, 2015, BioLite Taiwan and BioHopeKing Corporation (the “BHK”) entered into a co-development agreement, (the “BHK Co-Development Agreement”, see Note 3). The development costs shall be shared 50/50 between BHK and the Company. Under the terms of the agreement, BioLite issued relevant development cost to BHK. As of December 31, 2020, the amount due was $123,583. |
| ● | On May 8, 2020, the Company and Lucidaim entered into a Letter of Intent (LOI) in regard to a potential joint venture of BioLite Japan. Based on the LOI, each party will advance an aggregated amount of $150,000 to meet BioLite Japan’s working capital needs, which the Company advanced an amount of $100,000 and the advance bear 0% interest rate. As of December 31, 2020 and December 31, 2019, the outstanding advance balances was $150,000 and $0, respectively |
| ● | On January 26, 2017, BriVision and BioFirst entered into a loan agreement for a total commitment (non-secured indebtedness) of $950,000 to meet its working capital needs. On February 2, 2019, BriVision and BioFirst agreed to extend the remaining loan balance of $693,000 for one year matured on February 1, 2020. Under the terms of the loan agreement, the loan bears interest at 12% per annum. On August 1, 2019, the Company entered into a Conversion Agreements to convert the remaining balance of $693,000 to 99,000 shares of the Company’s common stock at a conversion price of $7.00 per share. |
Since 2017, BioLite Taiwan and BioFirst entered into several loan agreements for an aggregate amount of NT$19,430,000, equivalent to $625,646, to meet its working capital needs. Under the terms of the loan agreements, the loans bear interest at 12% per annum. The term of the loans has various maturity dates through May 27, 2020. On August 1, 2019, the Company entered into a Conversion Agreements to convert the remaining balance of $625,646 to 89,378 shares of the Company’s common stock at a conversion price of $7.00 per share.
Since 2017, BioFirst has also advanced funds to the Company for working capital purpose. The advances bear 0% interest rate and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreements to convert the remaining balance of $597,128 to 85,304 shares of the Company’s common stock at a conversion price of $7.00 per share.
On April 12, 2017, BioLite BVI and BioFirst entered into a loan agreement for NT$30,000,000, equivalent to $987,134 to meet its working capital needs. Under the terms of the loan agreement, the loan bears interest at 1% per month (or equivalent to 12% per annum). BioLite BVI and BioFirst extended the loan with the same interest rate and amount for one year. The loan will be matured on May 11, 2019. On May 12, 2019, the two parties extended the loan with the same interest rate and amount for one year. The loan will be matured on May 11, 2020. On August 1, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $987,134 to 141,020 shares of the Company’s common stock at a conversion price of $7.00 per share.
On July 24, 2017, BriVision entered into a collaborative agreement (the “BioFirst Collaborative Agreement”) with BioFirst (See Note 3). On September 25, 2017, BioFirst has delivered all research, technical, data and development data to BriVision, and the Company has recorded the full amount of $3,000,000 due to BioFirst. On June 30, 2019, the Company entered into a Stock Purchase Agreement with BioFirst, pursuant to which the Company agreed to issue 428,571 shares of the Company’s common stock to BioFirst in consideration for $3,000,000 owed by the Company to BioFirst.
Since 2019, BioFirst has also advanced funds to the Company for working capital purpose. The advances bear interest 1% per month (or equivalent to 12% per annum). As of December 31, 2020, the aggregate amount of outstanding balance and accrued interest is $23,647.
| ● | Since 2018, Mr. Tsung-Shann Jiang (Director), Mr. Chang-Jen Jiang (Director), Ms. Shu-Ling Jiang, and Ms. Mei-Ling Jiang (collectively, the “Jiangs”) have entered into various loans with the Company for working capital purpose in an aggregate amount of $795,340. These loans bear interest at 12% per annum and are due on demand. On August 1, 2019, the Company entered into a Conversion Agreements to convert the remaining balance of $837,726 to 119,675 shares of the Company’s common stock at a conversion price of $7.00 per share. |
Since 2018, the Jiangs have advanced funds to the Company for working capital purpose in an aggregate amount of $353,050. The advances bear 0% interest rate and are due on demand. On August 4, 2019, the Company entered into a Conversion Agreement to convert the remaining balance of $353,050 to 50,436 shares of the Company’s common stock at a conversion price of $7.00 per share.
74
On November 15, 2019, the Jiangs advanced funds to the Company for working capital purpose in an aggregate amount of NTD $300,000, equivalent to $10,680, The advances bear 1% interest rate per month. As of December 31, 2020, the outstanding principal of loans was $10,680 and accrued interest was $5,947.
As of December 31, 2020, the outstanding balance due to the Jiangs amounted to $16,627. These loans bear interest rate of 0% to 1% per month, and are due on demand.
| ● | On December 31, 2018, one of company’s shareholders had advanced funds to the Company for working capital purpose in an aggregate amount of NTD $4,660,000, equivalent to $165,896. The advances bear 13.6224% interest rate per annum. As of December 31, 2020, the outstanding principal of loans was $165,896 and accrued interest was $366. |
| ● | On January 21, 2019, the Company received a loan in the amount of $500,000 from Cathay Bank (the “Bank”) pursuant to a business loan agreement (the “Loan Agreement”) entered by and between the Company and Bank on January 8, 2019 and a promissory note (the “Note”) executed by the Company on the same day. The Loan Agreement provides for a revolving line of credit in the principal amount of $1,000,000 with a maturity date (the “Maturity Date”) of January 1, 2020. The Note executed in connection with the Loan Agreement bears an interest rate (the “Regular Interest Rate”) equal to the sum of one percent (1%) and the prime rate as published in the Wall Street Journal (the “Index”) and the accrued interest shall become payable each month from February 1, 2019. If the Company defaults on the Note, the default interest rate shall become five percent (5%) plus the Regular Interest Rate. |
In connection with the Note and Loan Agreement, on January 8, 2019, each of Dr. Tsung Shann Jiang (the Company’s majority shareholder, Chief Strategy Officer and a Director, who is also the Chairman and Chief Executive Officer of BioLite Holding, Inc the Chairman and Chief Executive Officer of BioLite Holding, Inc.) and Dr. George Lee (Chairman of the board of directors of BioKey, Inc.), executed a commercial guaranty (the “Guaranty”) to guaranty the loans for the Company pursuant to the Loan Agreement and Note, severally and individually, in the amount not exceeding $500,000 each until the entire Note plus interest are fully paid and satisfied. On December 29, 2020, the Company entered into a new loan extension agreement and assignment of deposit account with the Bank, which allowed Dr. Tsung Shann Jiang and Dr. George Lee to be removed as guarantees from the list of Guaranty.
In addition, on January 8, 2019, each of the Company and BioKey, signed a commercial security agreement (the “Security Agreement”) to secure the loans under the Loan Agreement and the Note. Pursuant to the Security Agreements, each of the Company and BioKey (each, a “Grantor”, and collectively, the “Grantors”) granted security interest in the collaterals as defined therein, comprised of almost all of the assets of each Grantor, to secure such loans for the benefit of the Bank. On March 31, 2020, the Company extended the Loan Agreement with the same term for seven months, which is due on October 31, 2020. On April 8, 2020 and October 3, 2020, the Company repaid an aggregated principal amount of $350,000. The outstanding loan balance was $650,000 as of December 31, 2020.
| ● | On May 8, 2020, the Company and Lucidaim entered into a Letter of Intent (LOI) in regard to a potential joint venture of BioLite Japan, an entity controlled by the Company’s controlling beneficiary shareholder. Based on the LOI, each party will advance an aggregated amount of $150,000 to meet BioLite Japan’s working capital needs, which the Company advanced an amount of $150,000 and the advance bears 0% interest rate. |
Promoters and Certain Control Persons
None of our management or other control persons were “promoters” (within the meaning of Rule 405 under the Securities Act), and none of such persons took the initiative in the formation of our business or received any of our debt or equity securities or any of the proceeds from the sale of such securities in exchange for the contribution of property or services, during the last five years.
Doctor Independence
The Company currently has six independent directors, Dr. Ming-Fong Wu, Norimi Sakamoto, Yen-Hsin Chou, Dr. Shin-Yu Miao, Yoshinobu Odaira, and Shih-Chen Tzeng, as that term is defined under the Nasdaq listing standard.
75
Audit Committee & Financial Expert
The Audit Committee of the Board of Directors currently consists of Ms. Chou (Chair), Mr. Wu and Ms. Miao. The functions of the Audit Committee include the retention of our independent registered public accounting firm, reviewing and approving the planned scope, proposed fee arrangements and results of the Company’s annual audit, reviewing the adequacy of the Company’s accounting and financial controls and reviewing the independence of the Company’s independent registered public accounting firm. The Board has determined that Ms. Chou, Mr. Wu and Ms. Maio are each an “independent director” under the listing standards of The NASDAQ Stock Market. The Board of Directors has also determined Ms. Chou is an “audit committee financial expert” within the applicable definition of the SEC. The Audit Committee is governed by a written charter approved by the Board of Directors, a copy of which is available on our website at www.abvcpharma.com. Information contained on our website are not incorporated by reference into and do not form any part of this prospectus. We have included the website address as a factual reference and do not intend it to be an active link to the website.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Various audit, audit related and non-audit services to us is as follows:
| For
the Year Ended December 31, | ||||||||
| 2020 | 2019 | |||||||
| Audit Fees | $ | 280,000 | $ | 83,000 | ||||
| Audit Related Fees | - | - | ||||||
| Tax Fees | - | - | ||||||
| All Other Fees | - | - | ||||||
| Total Fees | $ | 280,000 | $ | 83,000 | ||||
Audit Fees. Audit Fees consists of fees for professional services rendered by our principal accountants for the contemporaneous audit of our annual financial statements and the review of quarterly financial statements or services that are normally provided by our principal accountants in connection with statutory and regulatory filings or engagements.
Audit Related Fees. Audit Related Fees consists of fees for assurance and related services by our principal accountants that are reasonably related to the performance of the audit or review of our financial statements and are not reported under “Audit Fees.”
Tax Fees and All Other Fees. Tax Fees and All Other Fees Consists of fees for products and services provided by our principal accountants, other than the services reported under “Audit Fees,” “Audit-Related Fees” and “Tax Fees” above.
76
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
(a)(1) List of Financial statements included in Part II hereof
| Report of Independent Registered Public Accounting Firm | F-2 |
| Balance Sheets as of December 31, 2020 | F-3 |
| Statements of Operations for the years ended December 31, 2020 and 2019 | F-4 |
| Statements of Stockholders’ Equity (Deficit) for the years ended December 31, 2020 and 2019 | F-6 |
| Statements of Cash Flows for the years ended December 31, 2020 and 2019 | F-5 |
| Notes to the Financial Statements | F-7 |
(a)(2) List of Financial Statement schedules included in Part IV hereof: None.
(a)(3) Exhibits
The following exhibits are included herewith:
| + | Filed herewith |
77
| (1) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on February 16, 2016. |
| (2) | Incorporated by reference to Exhibit 3.01 to the Company’s Form SB-2 filed on June 28, 2002 |
| (3) | Incorporated by reference to Exhibit 3.02 to the Company’s Form SB-2, filed on June 28, 2002 |
| (4) | Incorporated by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K, filed on March 28, 2016. |
| (5) | Incorporated by reference to Exhibit 3.4 to the Company’s Form S-1, filed on September 13, 2016. (3.4) |
| (6) | Incorporated by reference to Exhibit 3.1 to the Company’s Form 8-K, filed on April 7, 2020 |
| (7) | Incorporated by reference to Exhibit 4.1 the Company’s Current Report on Form 8-K, filed on April 24, 2020 |
| (8) | Incorporated by reference to Exhibit 10.2 the Company’s Current Report on Form 8-K, filed on February 16, 2016. |
| (9) | Incorporated by reference to Exhibit 99.1 to the Company’s Current Report on Form 8-K, filed on June 9, 2016. |
| (10) | Incorporated by reference to Exhibit 10.3 to the Company’s Annual Report on Form 10-K, filed on January 12, 2017. |
| (11) | Incorporated by reference to Exhibit 99.1 to the Company’s Current Report on Form 8-K, filed on February 22, 2017. |
| (12) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on July 24, 2017. |
| (13) | Incorporated by reference to Exhibit 99.1 to the Company’s Current Report on Form 8-K, filed on May 30, 2017. |
| (14) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on September 20, 2017. |
| (15) | Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K, filed on September 20, 2017. |
| (16) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on February 1, 2019. |
| (17) | Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K, filed on February 1, 2019. |
| (18) | Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K, filed on February 1, 2019. |
| (19) | Incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on April 24, 2020. |
| (20) | Incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K, filed April 14, 2020. |
| (21) | Incorporated by reference to Exhibit 10.15 to the Company’s Annual Report on Form 10-K, filed May 15, 2020. |
| (22) | Incorporated by reference to Exhibit 10.16 to the Company’s Annual Report on Form 10-K, filed May 15, 2020 |
| (23) | Incorporated by reference to Exhibit 14.1 to the Company’s Amendment No.1 to Form S-1, filed on November 14, 2016. |
| (24) | Incorporated by reference to 21.1 to the Company’s Form S-1, filed on September 13, 2016. |
| (25) | Incorporated by reference to Exhibit 10.9 to the Company’s Annual Report on Form 10-K, filed May 15, 2020. |
Not applicable.
78
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized on March 16, 2021.
| American BriVision (Holding) Corporation |
| By: | /s/ Howard Doong | |
| Howard Doong | ||
| Chief Executive Officer and President |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| Signature | Title | Date | ||
| /s/ Howard Doong | President and Chief Executive Officer | March 16, 2021 | ||
| Howard Doong | ||||
| /s/ Eugene Jiang | Chairman of the Board of Directors | March 16, 2021 | ||
| Eugene Jiang | ||||
| /s/ Chihliang An | Chief Financial Officer | March 16, 2021 | ||
| Chihliang An | ||||
| /s/ Yen-Hsin Chou | Director | March 16, 2021 | ||
| Yen-Hsin Chou | ||||
| /s/ Shin-Yu Miao | Director | March 16, 2021 | ||
| Shin-Yu Miao | ||||
| /s/ Tsang-Ming Jiang | Director | March 16, 2021 | ||
| Tsang-Ming Jiang | ||||
| /s/ Ming-Fong Wu | Director | March 16, 2021 | ||
| Ming-Fong Wu | ||||
| /s/ Norimi Sakamoto | Director | March 16, 2021 | ||
| Norimi Sakamoto | ||||
| /s/ Tsung-Shann Jiang | Director | March 16, 2021 | ||
| Tsung-Shann Jiang | ||||
| /s/ Chang-Jen Jiang | Director | March 16, 2021 | ||
| Chang-Jen Jiang | ||||
| /s/ Yoshinobu Odaira | Director | March 16, 2021 | ||
| Yoshinobu Odaira | ||||
| /s/ Shih-Chen Tzeng | Director | March 16, 2021 | ||
| Shih-Chen Tzeng | ||||
| /s/ Hwalin Lee | Director | March 16, 2021 | ||
| Hwalin Lee |
79
SUPPLEMENTAL INFORMATIONTO BE FURNISHED WITH REPORTS FILED PURSUANT TO
SECTION 15(d) OF THE ACT BY REGISTRANTS WHICH HAVE NOT REGISTERED
SECURITIES PURSUANT TO SECTION 12 OF THE ACT.
On February 11, 2021, the Company filed a Current Report on Form 8-K to disclose the shareholder meeting it held on February 8, 2021; the related shareholder notice and proxy card were attached as an exhibit to the Current Report on Form 8-K filed on January 8, 2021, which Report was furnished to the SEC. The Company did not send any other annual report or proxy material to its shareholders during the fiscal year ended December 31, 2019.
80