Attached files
| file | filename |
|---|---|
| EX-99.7 - EX-99.7 - YUMANITY THERAPEUTICS, INC. | d141599dex997.htm |
| EX-99.6 - EX-99.6 - YUMANITY THERAPEUTICS, INC. | d141599dex996.htm |
| EX-99.4 - EX-99.4 - YUMANITY THERAPEUTICS, INC. | d141599dex994.htm |
| EX-99.2 - EX-99.2 - YUMANITY THERAPEUTICS, INC. | d141599dex992.htm |
| EX-99.1 - EX-99.1 - YUMANITY THERAPEUTICS, INC. | d141599dex991.htm |
| EX-23.1 - EX-23.1 - YUMANITY THERAPEUTICS, INC. | d141599dex231.htm |
| 8-K/A - 8-K/A - YUMANITY THERAPEUTICS, INC. | d141599d8ka.htm |
Exhibit 99.3
YUMANITY BUSINESS
Overview
Yumanity is a clinical stage biopharmaceutical company dedicated to accelerating the revolution in the treatment of neurodegenerative diseases. Neurodegenerative diseases cause a progressive loss of structure and function in the brain, leaving patients with devastating damage to their nervous system and widespread functional impairment. Although treatments may help relieve some of the physical or mental symptoms associated with neurodegenerative diseases, few of the currently available therapies slow or stop the continued loss of neurons, resulting in a critical unmet need. Yumanity is specifically focused on developing novel disease-modifying therapies to treat devastating conditions, either with large or orphan disease markets, including Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, or MSA, amyotrophic lateral sclerosis, or ALS (also known as Lou Gehrig’s disease), frontotemporal lobar degeneration, or FTLD, and Alzheimer’s disease.
Neurodegenerative diseases exert a heavy societal burden worldwide and represent one of the largest global healthcare challenges of our time. With an increasingly aging population, diseases affecting the brain and central nervous system are rising in prevalence, with overwhelming personal and economic consequences that exact a toll on patients, caregivers and treatment providers. The rising prevalence of neurodegenerative disease and a lack of disease-modifying treatments has resulted in a significant and growing unmet medical need. It is estimated that more than 60 million people worldwide suffer from neurodegenerative diseases, which is expected to almost double every 20 years. Global costs for treating these diseases are greater than $1 trillion annually.
Yumanity’s goal is to advance one new program into the clinic every year. Yumanity’s lead program, YTX-7739, is now in Phase 1 clinical trials for the potential treatment and disease modification of Parkinson’s disease. YTX-7739 targets an enzyme known as stearoyl-CoA desaturase, or SCD. Inhibition of SCD in multiple cellular systems, including patient-derived neurons, as well as in a novel mouse model of Parkinson’s disease, has been demonstrated to reverse the toxicity of misfolded alpha-synuclein, or α-synuclein, a protein strongly associated with Parkinson’s disease. Yumanity recently completed enrollment of a Phase 1 single ascending dose (SAD) study of YTX-7739 in healthy volunteers, which evaluated a broad range of doses of YTX-7739. Yumanity initiated a multiple ascending dose (MAD) study in healthy volunteers in the third quarter of 2020 with results anticipated in the first quarter of 2021. A Phase 1b clinical study of YTX-7739 in patients with Parkinson’s disease is planned as a continuation of the MAD study. The Phase 1b study will assess safety, tolerability and pharmacokinetics of YTX-7739 as well as proof of biology by exploring biomarkers of target engagement and potential correlative clinical parameters such as neuroimaging measurements to monitor for early effects of YTX-7739. Early results from the Phase 1b trial are anticipated in the second quarter of 2021. Yumanity’s second program, YTX-9184, also inhibits SCD but is chemically distinct from YTX-7739. Investigational new drug, or IND-enabling safety pharmacology and toxicological studies for YTX-9184 were initiated in the second quarter of 2020. Yumanity anticipates commencing the first in human studies of YTX-9184 in 2021, and intends to develop YTX-9184 for the potential treatment of dementia with Lewy bodies, which is another devasting neurodegenerative disease characterized by the abnormal accumulation of aggregates of α-synuclein.
At the center of Yumanity’s scientific foundation is its drug discovery engine, which is based on technology licensed from the Whitehead Institute, an affiliate of the Massachusetts Institute of Technology. This core technology, combined with investments and advancements by Yumanity, is designed to enable rapid screening to identify drugs with the potential to modify disease by overcoming toxicity in disease-causing gene networks. Toxicity in many neurodegenerative diseases results from an aberrant accumulation of misfolded proteins in the brain. Yumanity leverages its proprietary discovery engine to identify and screen novel drug targets and drug molecules for their ability to protect nerve cells from toxicity arising from misfolded proteins. To date, Yumanity has identified over one dozen targets, most of which have not previously been linked to neurodegenerative diseases. Yumanity believes this discovery platform will allow it to replenish its pipeline as programs graduate towards the clinic.
Yumanity is applying to neurodegeneration several research and development principles that have been proven to be successful in oncology and rare disease, including leveraging breakthroughs in genetics, use of advanced laboratory technology and techniques, and use of biomarkers to guide drug development, to inform the selection of specific and well-defined patient populations for clinical trials. To support this approach, Yumanity has assembled a management team with deep experience in neurology, neuroscience, rare disease, and oncology drug discovery and development.
1
Yumanity has built momentum across multiple dimensions of its business. In addition to advancing its clinical and preclinical pipeline, Yumanity has been successful in its business development efforts. In the second quarter of 2020, Yumanity secured a strategic research and development collaboration in ALS and FTLD with Merck Sharp & Dohme Corp., or Merck, with upfront and potential milestone payments of up to $530 million plus royalties. Yumanity anticipates scaling its business with a combination of internal programs and continued opportunities for biopharmaceutical company partnerships.
Neurodegenerative Disease Market and Challenges
Many factors, including too few disease-relevant biological hypotheses, the inherent complexity of the brain, and high patient heterogeneity, have led to a graveyard of failed approaches over the last several decades and have produced few approved disease-modifying therapies for neurodegenerative diseases to date.
Yumanity believes this is about to change. Recent scientific and technological advances include the improved understanding of the disease-relevant biology, innovative target discovery technologies, potentially better predictive animal models, new imaging approaches and identification of new biomarkers. These advances have ignited a renewed focus and commitment to neurodegenerative disease research and development, and Yumanity is one of the companies at the forefront of this emerging revolution.
Clinical study of neurodegenerative disease and evaluation of potential therapeutics has faced several hurdles. A primary consideration is the genetic heterogeneity of neurodegenerative diseases and subsequent variations in the disease biology in patients with similar clinical diagnoses. Yumanity believes this degree of heterogeneity is far greater than previously appreciated and is likely due to a unique combination of genetic and environmental factors which have important implications for development of therapies and their appropriate use by individual patients. As such, Yumanity plans to take a targeted approach to patient enrichment and stratification in its clinical trials. This approach further speaks to the need for a larger and more accurate set of biomarkers to aid in diagnosis as well as monitoring disease progression and treatment response in trials. Damage to brain cells early in the disease course and prior to the onset of symptoms presents further challenges for the design of clinical trials, as patients enrolling in clinical trials are typically selected based on expression of disease symptoms when significant damage to brain cells has already occurred. For example, increasingly sophisticated imaging studies have demonstrated that patients have lost at least 40% to 60% of dopaminergic neuronal integrity before qualifying for a diagnosis of Parkinson’s disease, indicating damage to brain cells begins long, often decades, before clinical symptoms manifest. As a result, many previous clinical trials in neurodegenerative disease included patients at a stage of the disease beyond which progression could no longer be modified. Thus, clinical trials would optimally be performed in patient populations that are at an early enough stage where potential disease-modifying therapies have an opportunity to preserve existing brain cells and function. Approaches to early diagnosis remain a focus in the neurodegenerative clinical research field.
In Parkinson’s disease, the cornerstone of pharmacological therapy for the past several decades has focused on either temporarily replenishing dopamine or mimicking the action of dopamine such as with the dopamine precursor levodopa. Levodopa, which is very helpful to patients in managing some of the motor symptoms of the disease, does not alter disease progression. Certain disease-modifying molecules are currently being investigated in early clinical trials for the potential of removing or reducing levels of α-synuclein. These programs, however, are predominantly focused on the development of antibodies. Therapeutic antibodies are large molecules that are administered systemically, and as such have significant challenges crossing the blood brain barrier and penetrating into the brain, which is the target tissue for neurodegenerative diseases. Even if some limited amount of antibody can penetrate into the brain, antibodies face a further challenge. α-Synuclein functions within cells to facilitate vesicle trafficking, however when α-synuclein misfolds it is thought to have an increased propensity to aggregate and disrupt multiple critical processes inside the cell. The ultimate expression of this pathology is the formation of Lewy bodies within neurons, which are a hallmark of dystrophic and degenerating cells. Therapeutics that target pathological processes within cells would be expected to prevent this toxic progression. α-Synuclein can also be secreted by neurons, and although the function is unclear, this results in α-synuclein outside of cells. It is this population that would be a target for antibody therapeutics which are generally believed to interact with protein extracellularly, or outside the
2
cell. Yumanity believes antibodies therefore have less access to α-synuclein, and recently, one antibody drug candidate that targets α-synuclein failed to meet primary clinical endpoints in a Phase 2 trial. By contrast, Yumanity is developing small molecules that the yeast platform pre-selects by design to cross the blood brain barrier and diffuse into the cell where α-synuclein causes cellular toxicity and damage. YTX-7739 and YTX-9184 both target the enzyme SCD, the inhibition of which has been shown to help overcome the toxicity of α-synuclein and promote protection of neurons.
Recent advances provide a rationale for optimism across the industry in the face of historic difficulties. A revolution in genetics over the past 15 years has identified specific genetic causes and genetic risk factors for several neurodegenerative diseases. Advanced research tools such as CRISPR and induced pluripotent stem cells, or iPSCs, allow scientists to probe core disease biology in the research laboratory in newfound ways, making inroads into the historic inaccessibility of the brain. Diagnostic methods are steadily improving, and the increased understanding of disease genetics helps to more precisely define disease subtypes and classify patients based on disease severity and likely rate of progression. Additionally, the use of imaging and digital technology, artificial intelligence and large analytical data repositories now allows for more precise monitoring of disease stage, progression, and response to treatment. These advancements will help clinicians enroll patients in clinical trials at a stage of disease when they are most likely to respond to neuroprotective therapy.
Yumanity’s Approach and Pipeline
Yumanity’s approach is to unlock the path to new therapies by addressing the fundamental and persistent barriers in neurodegeneration research: the poor understanding of disease mechanisms and lack of new biological targets. Yumanity believes that a dramatically expanded portfolio of programs focused on novel drug targets, which are grounded in an improved understanding of disease biology, will enable a higher likelihood of success in developing disease-modifying therapies.
Yumanity’s discovery engine is built upon core enabling technology that it exclusively licenses from the Whitehead Institute, an affiliate of the Massachusetts Institute of Technology. The core discovery technologies were created in the laboratory of Yumanity’s co-founder, Dr. Susan Lindquist. Dr. Lindquist and senior scientists from her team integrated multiple technology platforms to create a drug discovery engine designed to reliably generate new insights into fundamental mechanisms of neurodegenerative disease, and also reveal new potential drug targets and drug molecules that address neurodegeneration in a range of different ways, many of which were previously unknown.
The discovery engine is centered on the key insight that protein misfolding, a phenomenon at the root of virtually all neurodegenerative diseases, can be modeled in yeast cells. These yeast models are then screened against large chemical libraries using high throughput technology, selecting for chemical hits that protect cells from the toxicities created by the misfolded human disease-relevant proteins. The biological targets and pathways for these protective molecules are then uncovered using a series of chemical genetic techniques. Yumanity’s technology also allows for screening yeast collections that have individual genes deleted, such that, when rescue is observed, it can be inferred that the gene that was deleted in that yeast strain is involved in ameliorating the toxicity of the misfolded human disease-relevant protein. Since the only modification to the original yeast system was the introduction of the culprit misfolding proteins, any molecule or gene deletion that can protect cells from the resultant toxicity is of interest. The discovery of protective molecules and biological targets, especially when previously unknown, can reveal new or untapped areas for study. Yumanity believes that the complement and overlap between the small molecule and genetic rescue screens has the potential to create a powerful network of interlinked biological processes that can further identify previously unknown therapeutic targets. Yumanity explores these cell-protective discoveries from the yeast system for translation to human disease-relevant cells using informatics and cutting-edge stem cell and iPSC experimental techniques. The discovery engine is designed to ultimately output novel programs: molecules with novel biological targets that can then be progressed through the standard preclinical drug development processes.
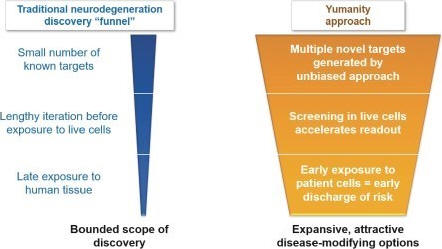
Yumanity believes its proprietary discovery engine has the potential to dramatically expand the knowledge around the complex biology of neurodegeneration, and further allows initiation of discovery programs outside of the traditional, limited set of hypotheses that exist today. Screening for hits in a living yeast system can save time by providing a biological-relevant readout sooner than some of the more typical practice of starting in non-live, test
3
tube systems. Additionally, shared features between yeast cell membranes and the blood brain barrier, such as comparable membrane permeability, polarity, and drug pumps for removal of non-native compounds, mean that molecules that can permeate a yeast cell to effect intracellular rescue may also be likely to penetrate the blood brain barrier. Furthermore, Yumanity’s molecules get tested in diseased human cells ex-vivo at the beginning rather than at the end of preclinical development. Yumanity believes that success in this setting confers increased confidence in programs compared to the more traditional paradigm of multiple rounds of animal studies before any actual testing in human tissues.
Key Differentiators of Yumanity Discovery Engine
Yumanity leverages the power of its discovery engine to generate a robust portfolio of promising novel drug targets and molecules. Yumanity then prioritizes the most promising targets to accelerate drug discovery programs and advance compounds into preclinical and ultimately clinical development. To date, this approach has already uncovered over one dozen novel targets, pathways, mechanisms, and molecules that Yumanity believes have the potential to ameliorate the fundamental cellular toxicities associated with neurodegenerative diseases. Yumanity expects that this list of new targets and subsequent programs will continue to grow as Yumanity iterates through its discovery engine. The list shown below illustrates Yumanity’s current 14 most advanced targets.
4
Novel Neurodegeneration Targets Discovered at Yumanity
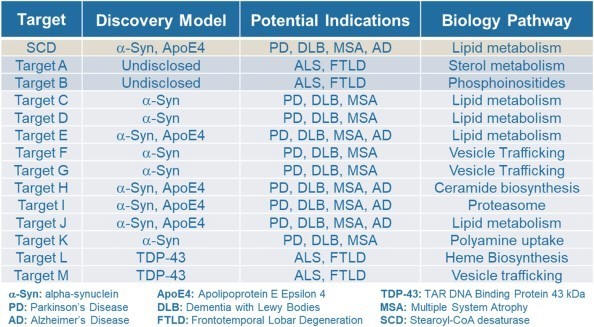
Discovered targets mature into programs as they advance through the discovery process. YTX-7739, Yumanity’s lead program, targets the enzyme SCD, one of the early targets identified and validated in Yumanity’s discovery engine and is being studied for the treatment of Parkinson’s disease. Yumanity’s second program, known as YTX-9184, is also a small molecule inhibitor of SCD and is being developed for the treatment of dementia with Lewy bodies as the likely indication. It can also serve as a back-up for YTX-7739. Yumanity is developing lead compounds and validating the targets for its potential third and fourth programs, which are represented as targets A and B in the chart above. These two targets are advancing through a research collaboration with Merck. Other targets for multiple potential indications are at varying stages of the discovery process, with several examples listed above for targets C through M.
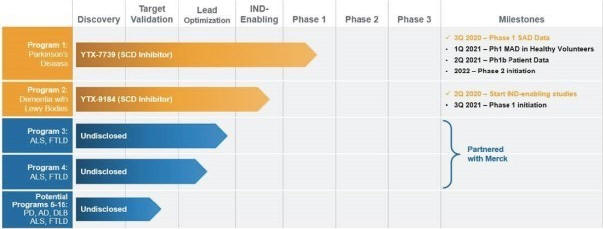
Yumanity’s Pipeline
Yumanity has set a goal of introducing one new program to the clinic every year. The following chart summarizes key information about Yumanity’s most advanced discovered targets. All of Yumanity’s therapeutic candidates are small molecules and are optimized and formulated for oral delivery. Yumanity owns both development and commercialization rights to its first and second programs as well as potential programs 5-15, the latter of which are in early preclinical studies and currently being evaluated in mammalian systems. Potential programs 3 and 4 are advancing as part of a research collaboration with Merck, who has licensed these potential programs and will conduct IND-enabling toxicology and safety pharmacology, clinical development, and commercialization.
5
Yumanity’s lead program, YTX-7739, is a novel small molecule for the potential treatment of Parkinson’s disease and related disorders of α-synuclein. The program that resulted in this lead compound was the first prioritized output program of Yumanity’s discovery engine. YTX-7739 is designed to ameliorate the consequences of α-synuclein toxicity in human cells that results in cellular dysfunction, specifically disruptions with the directed movement, or trafficking, of proteins or lipid-bound vesicles within cells. Yumanity’s second program, YTX-9184, is chemically distinct from YTX-7739 but is also designed to confer protection against α-synuclein toxicity. Both YTX-7739 and YTX-9184 target the enzyme SCD, that catalyzes a reaction in the lipid metabolism pathway.
α-Synuclein is a protein that is a prominent constituent of Lewy bodies, the abnormal protein aggregates that are the pathological hallmarks of Parkinson’s disease, dementia with Lewy bodies, MSA and other neurological disorders known collectively as “synucleinopathies”. Current treatments for Parkinson’s disease manage the early motor symptoms of the disease. The goal of Yumanity’s differentiated and potentially disease-modifying approach with YTX-7739 and YTX-9184 is to block the intracellular toxicity associated with α-synuclein misfolding and aggregation to allow the cell to continue to function normally, and to slow or possibly even halt the progressive degenerative consequences of the disease.
YTX-7739 is now in Phase 1 clinical development consisting of three initial studies: a SAD study in healthy volunteers, a MAD study in healthy volunteers, and a Phase 1b study in patients with Parkinson’s disease. The SAD study in healthy volunteers has completed enrollment and evaluates safety, tolerability, pharmacokinetics, pharmacodynamics parameters, and the effect of food on YTX-7739 pharmacokinetics over a broad range of doses of YTX-7739. The MAD study was initiated in the third quarter of 2020, with data anticipated for healthy volunteers in the first quarter of 2021. The Phase 1b study will be a continuation of the MAD study, conducted in patients with Parkinson’s disease, and will assess safety, tolerability and pharmacokinetics of YTX-7739 as well as proof of biology by exploring biomarkers of target engagement and potential correlative clinical parameters such as neuroimaging measurements to monitor for early effects of YTX-7739. Early results from the Phase 1b trial are expected to be available in the second quarter of 2021, assuming no further clinical trial delays due to the COVID-19 pandemic. Yumanity then plans to initiate a Phase 2 trial in patients with Parkinson’s disease in 2022. IND-enabling safety pharmacology and toxicological studies for YTX-9184, Yumanity’s second program, were initiated in the second quarter of 2020. YTX-9184 is being developed as a potential treatment for dementia with Lewy bodies and is anticipated to enter Phase 1 clinical development in 2021.
Yumanity’s potential third and fourth programs are novel targets for the treatment of ALS and FTLD. Activities for these potential programs are currently being conducted through a research collaboration with Merck, with up to $530 million in potential milestones for Yumanity plus royalties. If these potential programs are successful and achieve full target validation with small molecule agents, they will advance to IND-enabling safety pharmacology and toxicology studies to be conducted by Merck, who will also be responsible for any subsequent clinical development and commercialization.
6
Beyond Yumanity’s first two programs and two additional potential programs, Yumanity has additional targets that constitute a rich discovery pipeline. Targets C through M in the chart above are all progressing towards target validation in human neuron systems and development of small molecule inhibitors with drug-like properties.
Leadership team, Scientific Team, and Investors
Yumanity is led by a team of seasoned executives with prior experience in both public and private companies as well as small and large biopharmaceutical companies. The management team has deep expertise in neurology, neuroscience, rare diseases, and oncology.
Yumanity’s Chief Executive Officer is Richard Peters, M.D., Ph.D., the former President, Chief Executive Officer and director at Merrimack Pharmaceuticals, and former Senior Vice President and Head of Global Rare Diseases at Sanofi Genzyme. Paulash Mohsen is Yumanity’s Chief Business Officer, and he was the former Country Manager (Canada) at Cubist (acquired by Merck) and Vice President of Strategy and Business Operations at Optimer Pharmaceuticals (acquired by Cubist), and also a Vice President of Strategy and Multi-Channel Management at Pfizer. Yumanity’s Chief Medical Officer is Brigitte Robertson, M.D., who was previously the Vice President, Therapeutic Area Head of Neuroscience, Global Clinical Development at Shire/Takeda, the Chief Medical Officer at Neurovance and has held senior roles at Sunovion Pharmaceuticals in clinical development and experimental medicine, and at GlaxoSmithKline in the Center for Excellence in Drug Discovery. The scientific research team is led by Yumanity’s Head of Research, Robert Scannevin, Ph.D., who was formerly the Vice President of Discovery Biology at Yumanity and a Senior Director at Biogen, Johnson&Johnson and Wyeth.
Yumanity’s co-founder and Executive Chair of its board of directors is N. Anthony Coles, M.D., previously the Chief Executive Officer of Onyx Pharmaceuticals until its sale to Amgen in 2013, and current Chief Executive Officer of Cerevel Therapeutics. Yumanity’s other co-founder is the late Susan Lindquist, Ph.D., a renowned cell biologist, National Medal of Science recipient, Howard Hughes Medical Instituteinvestigator and former director of the Whitehead Institute. Yumanity’s investors include a leading group of healthcare-focused investors including Fidelity Investments, the Redmile Group, Alexandria Venture Investments, Merck, Pfizer, Biogen, and Sanofi.
Yumanity’s Priorities
Yumanity’s goal is to become a leading biopharmaceutical company to discover, develop, and commercialize disease-modifying therapies that employ novel approaches to treat neurodegenerative diseases. To achieve this goal, key elements of Yumanity’s priorities include:
| • | Advance lead program YTX-7739 through clinical development, regulatory approval, and commercialization. YTX-7739 is currently in Phase 1 trials. Yumanity aims to bring to patients a meaningful therapy that can modify the course of Parkinson’s disease and/or other disorders of misfolded α-synuclein. Yumanity’s near-term focus is to demonstrate safety, efficacy, and proof of biology (through target engagement) of YTX-7739 in patients with Parkinson’s disease through completion of a Phase 1b trial. |
| • | Progress YTX-9184 into clinical development. YTX-9184 is Yumanity’s second program. Like YTX-7739, YTX-9184 is also an inhibitor of the SCD enzyme. YTX-9184 is chemically distinct from YTX-7739 and may serve as a potential option for patients with other disorders of α-synuclein such as dementia with Lewy bodies as well as a potential back-up to YTX-7739. |
| • | Advance Yumanity’s potential third and fourth programs for ALS and FTLD in partnership with Merck. Yumanity expects to continue preclinical development of these two potential programs in partnership with Merck, from which Yumanity is eligible to receive potential milestone payments of up to $530 million plus royalties. |
7
| • | Continue to progress Yumanity’s early pipeline. Yumanity has discovered several other novel targets in neurodegeneration that are currently at early stages of validation. Yumanity’s goal is to advance its discovery pipeline through the preclinical stages and towards the clinic, either alone or in conjunction with potential partners. |
| • | Invest in and continue to innovate around Yumanity’s discovery engine. Yumanity’s discovery engine has been productive in identifying novel targets in neurodegeneration, often including chemical modifiers of these targets. Yumanity plans to continue to invest in its discovery engine to identify additional targets that will replenish its discovery pipeline as programs mature towards to clinic. |
Yumanity’s Discovery Engine Platform
Overview – Protein Misfolding and Toxicity Cascades
DNA is the foundational code for all proteins. The information held in DNA, in our genes, is transcribed first into RNA and then translated into linear strands of amino acids, the building blocks of all proteins found within cells. The linear strands of amino acids then fold in very precise, highly complex ways to form proteins, each having defined shapes and structures that enable them to carry out their normal biological function. When protein folding goes awry, critical functions of proteins may be lost, or new, abnormal functions may be gained.
Protein misfolding plays a key role in the initiation and progression of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, dementia with Lewy bodies, MSA, ALS and FTLD. In each of these diseases, as culprit proteins misfold, they form aggregates that may combine into plaques, which form sticky deposits in the brain cells or in brain tissue. These aggregates and plaques can interfere with normal cellular function in a number of ways. For example, they may interfere with the directed movement, or trafficking, of proteins or lipid-bound vesicles within cells, or they may trigger inflammatory reactions. They may also impede chemical and enzymatic processes. Ultimately, these aggregates and plaques result in nerve cell damage and cell death.
A revolution in genetics over the past 15 years has led to the identification of genetic risk factors for neurodegenerative diseases and a number of genes that can be causally tied to protein misfolding processes. Patients who inherit mutations in a single, specific gene generally present with early-onset and aggressive forms of disease. Genetic data have enabled the development of animal and cellular pathology models based on overexpression of disease-causing genes. While undoubtedly an important advance, these models often do not replicate the full features of disease pathology. As a result, there is no conclusive demonstration to date that simply reducing the levels of misfolded proteins reduces the neurodegenerative pathology or presents an efficacious therapeutic approach to the treatment of neurodegenerative disease in humans.
Protein aggregate formation occurs in different places. In Alzheimer’s disease, protein aggregates are found in extracellular plaques, but in diseases like Parkinson’s disease and ALS, misfolded proteins tend to aggregate and create toxic effects inside of brain cells. These intracellular protein aggregates are protected by a cell membrane and therefore lie beyond the typical reach of protein-based drugs, such as antibodies, that do not effectively cross cell membranes. Ongoing trials seeking to use antibody therapy to bind to the misfolded proteins can only do so while proteins are outside cell membranes, which is a relative minority of their life cycle.
The toxic consequences of protein misfolding and aggregation ultimately result in cell death, and the accumulation of dead cells within specific brain regions marks the progression of disease symptoms and severity. It is Yumanity’s goal to keep cells alive by protecting them from the consequences of these misfolded proteins, thereby slowing disease progression. Yumanity’s discovery engine is designed to better understand the processes through which protein misfolding and aggregation trigger cellular toxicity, and to do so in a manner that allows Yumanity to identify a network of new targets and biological processes which, when modulated using a therapeutic drug, Yumanity believes will ameliorate the toxicities initiated by protein misfolding, allow the cells to continue to function normally, and halt the progression of disease. Yumanity believes the identification of these new targets and close-in biology networks, and the molecules that modulate them, enables Yumanity to provide a new treatment approach to neurodegenerative drug discovery.
8
The Yeast Model
The core of Yumanity’s discovery engine are cellular models that allow for studying the consequences of protein misfolding in a way that improves the understanding of cellular pathways that trigger neurodegenerative disease and enables discovery of the molecules that inhibit specific drug targets within these pathways. Protein aggregation due to misfolding is an ancient cellular complication. All living cells have evolved mechanisms to deal with misfolded proteins. These mechanisms are highly conserved across evolution from simple cells such as yeast to complex cells like human neurons. It was the keen insight of Dr. Lindquist, Yumanity’s co-founder, at the Whitehead Institute that these parallels might be exploited to make yeast serve as a model for neurodegenerative drug discovery. When known genetic drivers of human disease, for example α-synuclein for Parkinson’s disease or beta-amyloid for Alzheimer’s disease, are introduced into a yeast cell, these proteins misfold and cause the yeast cells to stop growing or die. The cellular processes that cause the yeast cells to stop growing or die are largely indistinguishable from those that cause human neurons to die in neurodegenerative disease. For example, yeast cells with α-synuclein or beta-amyloid display disorders of vesicle trafficking, mitochondrial dysfunction and other toxicities observed in diseased human cells. Therefore, yeast cells, despite their simplicity, can function as effective models of disorders of protein misfolding, including neurodegenerative diseases.
Using yeast as a model system confers several noteworthy benefits. First, the yeast genome is highly tractable and well-characterized. At approximately 6,000 genes, it is less than a quarter of the size of the human genome. The yeast genome was sequenced much earlier than the human genome and has been studied extensively. Based on thousands of genome-wide studies, the yeast genome and proteome are well-annotated, including gene-gene interactions, biochemical processes, intracellular and cell-to-cell signaling cascades as well as gene to protein interaction networks. As a comparison, while sequenced, the functions of the majority of genes in the human genome are currently unknown. Second, core aspects of eukaryotic cell biology, such as organelle and cytoskeletal biology, protein homeostasis pathways, intracellular protein trafficking, lipid metabolism, RNA metabolism, and signal transduction, are well conserved from yeast to human cells. Critically, recent human genetic studies have strongly implicated distress of these conserved eukaryotic pathways in major neurodegenerative diseases. Third, there are existing research tools readily available in yeast, for example gene deletion libraries, that do not currently exist for human cells. Finally, experimentation in yeast cells tends to be easier to control and therefore more reproducible compared to human cells or cell lines.
As an additional benefit, yeast cells express many drug pumps and transporters that are responsible for removing potentially harmful molecules from the interior of the cell. These yeast pumps and transporters are believed to be very similar to the drug pumps and transporters found in the human blood brain barrier. This means that a compound, for example a potential drug, that can penetrate the yeast cell membrane is also likely to be able to cross the human blood brain barrier and not be pumped back out of the brain. Compounds that are able to permeate a yeast cell membrane need to have the right size and properties to diffuse into the cell, which are advantageous properties also needed to cross the blood brain barrier.
Because cellular pathologies of many neurodegenerative diseases are conserved across cell types, Yumanity’s co-founder hypothesized that proteins whose misfolding and aggregation drives neurodegeneration would disrupt similar cellular processes in yeast and human cells. Yumanity has studied the toxic consequences of misfolding of various neurodegenerative disease-related proteins, including α-synuclein, beta-amyloid, TDP-43, ApoE4, FUS, C9orf72, and huntingtin in yeast cells. The expression of these proteins in yeast causes cellular toxicities that are clear analogs of their human counterparts. The development of yeast as a model system allows Yumanity to recreate cellular toxicities caused by protein misfolding relevant to neurodegeneration in a simpler cell with unparalleled genetic tractability, which Yumanity then translates to human cell models.
Phenotypic Screening in Yeast
Since the dawn of the genomics revolution in biology and widespread availability of recombinant DNA methods, the conventional approach employed by the biopharmaceutical industry for drug discovery has been the target-based screen. In this approach, one begins with a known ‘target,’ or a specific protein involved in a defined cellular process considered to be relevant to disease pathology. Large chemical libraries are then screened for compounds that bind to or modulate the activity of this target. This screening is usually performed in a cell-free environment, meaning no membranes, with just the protein target in solution in a test tube or assay plate. The successful screening hit then undergoes iterative rounds of chemical optimization and refinement to improve affinity and activity at the
9
target, again in a cell free environment. Once a molecule with favorable properties is obtained, standard preclinical development processes occur, including further optimization, pharmacology and toxicity testing and formulation. In the field of neurodegeneration, the very small number of hypothesized targets has proven to be a significant limitation of the productivity of the target-based screening approach. Moreover, the iterative rounds of optimization and screening occasionally yield advanced compounds that do not readily cross the blood brain barrier or do not cross cell membranes efficiently.
In contrast to target-based screens, phenotypic screening seeks compounds or genetic factors that modify a specific observable trait, or phenotype, within the physiological context of an intact cell or organism. A large chemical or genetic deletion library can be screened for molecules or genes that modify the phenotype in question. The phenotype Yumanity seeks to correct is slow growth or cell death that results from pathologies driven by misfolded proteins. The primary benefit of phenotypic screening is the identification of compounds or genes that modify a specific trait without the bias of any pre-existing hypotheses about what may be the best molecule or target to modify the phenotype. Success in phenotypic screening can therefore point to previously unknown pharmacology and potential therapeutic compounds and their targets in an unbiased manner. Additionally, Yumanity can learn what pharmacology is relevant to the amelioration of disease phenotypes from the results of the screen.
Phenotypic screens are possible in some in vivo animal models, including certain worms, flies, zebrafish, mice and rats. Based on the complexities of working with larger multicellular species, phenotypic screening in unicellular yeast is expected to allow for higher throughput and exploration of more cellular phenotypes. Yeast cells also have an advantage of sophisticated genetic tools that are essential for downstream target identification. Using human neuronal cells in high throughput phenotypic screens is also believed to be challenging due to the inherent complexity in generating and maintaining robust and reproducible numbers of cells to support large-scale screening in a lab setting.
In contrast, phenotypic screening in yeast cells is inexpensive, extremely high throughput, and highly replicable. The robust cell growth and viability of phenotypes in yeast are well suited to phenotypic screening in high throughput formats. Large numbers of yeast cells with genetically introduced misfolding human proteins can be subjected to screening against large chemical libraries to isolate compounds which protect against cellular toxicity. While most of the yeast cells in the screening exercise will die as a result of the misfolding mediated toxicities, any cell that do not die as a result of exposure to specific compounds in the screening library results in ‘hits’, or compounds that warrant further investigation due to their ability to rescue cells from the effects of protein misfolding-induced toxicity.
Target Identification
Despite many advantages, phenotypic screens are beset with a fundamental limitation: while the screen identifies a compound that rescues the toxic phenotype, it does not identify the biological target implicated in the rescue. Phenotypic screening in yeast successfully identifies molecules that reduce protein misfolding toxicity in the yeast model, but it does not immediately define their biological targets.
The second technology platform in Yumanity’s discovery engine overcomes this limitation of phenotypic screening by uncovering the target implicated in the toxicity rescue. Yumanity’s target identification capability takes advantage of the fact that yeast is uniquely well-studied and characterized from a genetic perspective. Yumanity’s proprietary capability leverages pre-existing gene knockout libraries, gene-gene interaction networks, and other readily available tools in yeast to identify, in a high-throughput manner, the targets implicated in the phenotypic screen’s successful hits. This information, collected across hundreds of screening hits across multiple independent screening libraries, gives Yumanity a growing understanding of the genes and pathways involved in rescuing cells from the consequences of misfolding protein induced toxicity. Using the set of experimental tools available in yeast, Yumanity is able to narrow down and eventually identify the specific genetic targets associated with the hits that result from the phenotypic screens.
Once the targets have been identified within the yeast genome, Yumanity then uses informatics to discern the analogous targets within the human genome. Yumanity can compare the sequence, structure, and function of yeast genes with a database of human gene sequences to identify the closest yeast-to-human match. Yeast genes may have a range of relationships to their human counterparts, including one-to-one, one-to-many, or other configurations.
10
Importantly, like the phenotypic screening platform in yeast, the methods involved in target identification in yeast (and subsequent translation to human targets) are high throughput. Yumanity has used this process multiple times to identify a portfolio of potentially attractive novel targets.
Human Cell Translation
The third technology platform of Yumanity’s discovery engine translates its work from yeast into human cells. The outputs of the first two technology platforms are hit compounds that rescue toxicity in yeast and identification of the genetic targets implicated in that rescue in both yeast and human systems. The critical translational test is to see whether the observed rescue in yeast can be replicated in diseased human neurons.
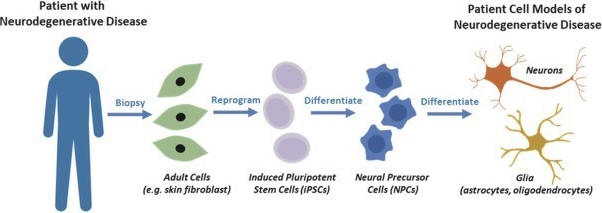
To accomplish this, Yumanity takes advantage of extraordinary advances in stem cell technology. Yumanity is able to reprogram adult somatic cells, specifically skin fibroblasts taken from a patient biopsy, to create induced pluripotent stem cells, or patient iPSCs. These patient iPSCs can be used to generate various types of neurons in the laboratory, with the genetic sequence of a patient with neurodegenerative disease. Yumanity can also induce toxicity in human neurons by forcing the elevated expression of disease-relevant misfolded proteins. In the case of both patient-derived neurons and inducted toxicity neuron models, Yumanity develops and maintains these cells to validate the rescue activity discovered in the yeast phenotypic screen. This translation from yeast to human cells, first published in Science, gives Yumanity confidence that the discovery in yeast is relevant in a human context.
Reprogramming patient cells to create human neurons in culture
Integration of Technology Platforms
Yumanity’s discovery engine integrates the three technology platforms described above: phenotypic screening in yeast, target identification, and human cell translation. Phenotypic screening provides unbiased hits that rescue toxicity via biological targets that may be previously unknown in neurodegeneration. The target identification platform allows Yumanity to understand the genes implicated in such rescue, both in yeast and their corresponding genes in humans. The human cell translation platform leverages iPSC technology to translate the rescue activity observed in yeast to human cells, specifically rescuing diseased patient neurons.
An important element of this design is that it enables Yumanity to move back and forth between yeast cells and human patient neurons in a highly iterative and parallel fashion. For example, if Yumanity identifies an interesting target through a library screening hit, but the compound used to discover the hit is not an optimal drug-like candidate, Yumanity can optimize the compound though chemistry or re-screen the target for better compounds. Also, if Yumanity has identified multiple closely related genes across different screening and target identification campaigns, Yumanity can profile them individually to determine which gene gives Yumanity the optimal rescue response. Thus, Yumanity can continually build on lessons learned from multiple protein pathologies in order to accelerate the discovery of novel therapies.
11
To date, Yumanity has successfully implemented its discovery engine to discover over one dozen new targets in neurodegeneration. Yumanity has also confirmed that some previously identified targets for other indications are in fact active within neurodegenerative toxicity cascades. Yumanity’s first target in Parkinson’s disease, SCD, is a novel target that was not previously known to be involved in Parkinson’s disease. Yumanity’s lead drug candidate, YTX-7739 and its second drug candidate, YTX-9184, address the toxicity caused by α-synuclein, the protein that causes some forms of Parkinson’s disease and dementia with Lewy bodies. Yumanity is also pursuing targets in ALS and FTLD that were not previously known to be involved in these indications, and Yumanity is moving forward to develop novel therapies for some of these targets in collaboration with Merck.
Drug Candidate Selection Process
Once Yumanity has identified a promising target and molecule that modulates that target using its discovery engine, Yumanity then uses chemistry to optimize the molecule to determine which ones have the most drug-like properties. Yumanity uses a wide array of tests to evaluate drug candidates and to identify those that generally have the desired molecular weight, solubility and other characteristics that Yumanity believes provides the potential to cross the blood brain barrier, access the targets and deliver the potent rescue to the cell. The optimized rescuing molecules are then advanced toward IND-enabling studies. Examples of such tests and general acceptability ranges are shown below.
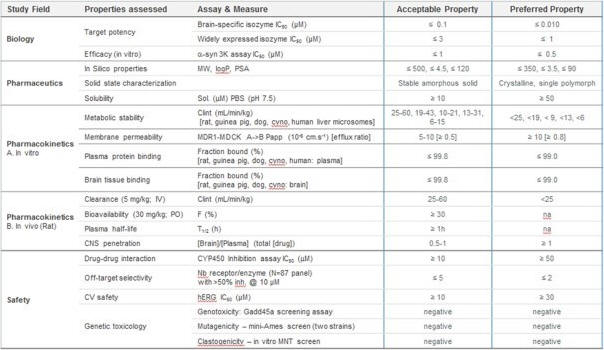
Yumanity’s Programs
Yumanity has leveraged its discovery engine to identify more than one dozen diverse biological targets not previously linked to neurodegenerative disease that Yumanity believes are suitable for a future disease-modifying drug discovery program. Yumanity’s lead program, YTX-7739 for the potential treatment and disease-modification of Parkinson’s disease, is currently in Phase 1 clinical trials. Yumanity’s second program, YTX-9184, has a distinct profile and is from a different chemical series than YTX-7739. YTX-9184 entered IND-enabling studies in the second quarter of 2020. In collaboration with Merck, Yumanity is also currently in the lead optimization process for Yumanity’s potential third program for the potential treatment of ALS and FTLD. Yumanity believes these potential programs and the others in its portfolio will enable Yumanity to reach its goal of one new program entering human clinical trials every year. The status of Yumanity’s pipeline is reflected below:
12
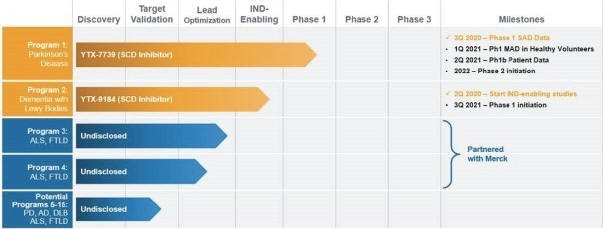
Yumanity’s Lead Program – YTX-7739
Yumanity is developing YTX-7739 for the treatment of Parkinson’s disease. YTX-7739 is the first prioritized drug candidate identified by Yumanity’s discovery engine and is designed to reduce α-synuclein toxicity by inhibiting SCD, an enzyme that metabolizes saturated fatty acids to their monounsaturated form.
Parkinson’s Disease Overview
Parkinson’s disease is a chronic, progressive neurological disorder of the central nervous system and the second most prevalent neurodegenerative disorder in the United States after Alzheimer’s disease, with an estimated 500,000 to one million prevalent cases. More than 10 million people worldwide are believed to have Parkinson’s disease. In patients with Parkinson’s disease, the premature death of neurons in the brain reduces levels of the neurotransmitter dopamine, causing motor dysfunction including tremor, slow movement, muscle rigidity, and difficulty with balance, falling, swallowing, speech, and writing. Other changes in the brain associated with the disease may result in cognitive and sensory symptoms. Additional features of the disease include disruptions in nerves connecting the brain to other systems such as cardiovascular, gastrointestinal, and urogenital systems, as well as sleep disturbances, constipation, and loss of sense of smell. Later-stage Parkinson’s disease is severely debilitating, and its symptoms make sufferers more likely to experience life-threatening medical issues. As a result, while Parkinson’s does not directly cause death, it is nevertheless the fourteenth leading cause of death in the United States.
Approximately 60,000 people are diagnosed with Parkinson’s disease in the United States each year, most often after the age of 50. The National Institutes of Health estimates the annual cost of treating Parkinson’s disease in the United States to be $14 billion, with indirect costs such as lost productivity adding at least another $6 billion. As with many other neurodegenerative diseases, the greatest risk factor for Parkinson’s disease is increasing age. The growing population of older adults and longer average lifespans are therefore likely to increase the number of Parkinson’s patients and the need for effective treatments. Currently, it is estimated that the number people diagnosed with Parkinson’s disease in the United States will double by the year 2040.
Although certain Parkinson’s disease cases have been associated with rare gene mutations, both hereditary and environmental factors are likely to contribute to the occurrence of Parkinson’s disease in the majority of cases. Additionally, while the biological cause of most cases of Parkinson’s disease is not clear, the core pathology of Parkinson’s disease is degeneration of the dopaminergic neurons in the midbrain. In certain cases, cell loss occurs in association with the formation of intraneuronal Lewy inclusion bodies. Abnormally aggregated α-synuclein is the principal component of Lewy bodies, which are the pathological hallmark of Parkinson’s disease. The presence of Lewy bodies and other associated fibrils is correlated with neuron loss and death, decline in motor function and cognitive dysfunction.
13
Limitations of Current Therapies
There is currently no known cure for Parkinson’s disease. Pharmacological therapies for Parkinson’s disease are aimed at either temporarily replenishing dopamine or mimicking the action of dopamine. They generally help reduce muscle rigidity, improve speed and coordination of movement and lessen tremor.
The dopamine precursor levodopa is the most commonly prescribed pharmacotherapy. While extremely helpful overall, some symptoms do not respond as well to levodopa, like difficulty with balance, falling, difficulty with speech and swallowing, and memory issues. It can also be challenging to titrate and find the optimal dose and patients may experience “on” and “off” periods when the drug concentration falls to below their individual needs. Unfortunately, the long-term use of levodopa is frequently associated with the development of additional motor complications, for example dyskinesias, or uncontrolled, involuntary movements. Additional therapies attempt to slow the degradation of dopamine using monoamine oxidase-B inhibitors. Catechol-o-methyltransferase inhibitors and carbidopa may be used to reduce levodopa degradation. Amantadine, an N-methyl-D-aspartate, or NMDA, receptor antagonist, is also used and may act through more than one mechanism. Drug treatment for Parkinson’s disease is commonly individualized by patient and disease characteristics, and patients may receive multiple drug therapies throughout their course of disease, including other medications for comorbid conditions and symptomatic management such as antidepressants, anxiolytics, and anti-psychotics. While these therapies address the symptoms of Parkinson’s disease, they are unable to halt disease progression over the longer-term. As a result, these symptomatic therapies lose their efficacy over time, leaving patients with few treatment options.
Outside of drug therapy, electrical deep brain stimulation is also be used to control motor symptoms of Parkinson’s disease, typically in the advanced stages of disease. In addition to pharmacotherapy, a holistic approach to treatment is encouraged and patients may gain benefit from regular exercise, psychological, physical, occupational and speech therapy, nutrition consultation, education, support groups, and the use of assistive devices and caregiver relief.
There are currently a small number of early clinical trials investigating the potential of directly reducing α-synuclein to change the course of the disease. These programs, however, are predominantly based on antibody therapy which, due to their large size, do not readily enter the brain or brain cells and are believed to interact only with extracellular α-synuclein that has been secreted or released from cells. Formation of pathological α-synuclein aggregates, known as Lewy bodies, occurs inside of cells, and the ability of therapeutic antibodies to impact consequent toxic α-synuclein cascades is unclear.
Yumanity’s Solution
Yumanity is developing YTX-7739 as a small molecule therapy to slow or halt disease progression in patients suffering with Parkinson’s disease. Using Yumanity’s discovery engine and Parkinson’s disease patient cell lines, Yumanity identified SCD as a biological target enzyme for diseases caused by α-synuclein-mediated toxicity. YTX-7739 is designed to inhibit SCD to reduce α-synuclein-mediated toxicity within cells.
The α-Synuclein Toxicity Cascade
Aggregated α-synuclein protein is the primary constituent of the pathological Lewy bodies formed in the brains of patients with Parkinson’s disease. Although its precise molecular function is poorly understood, α-synuclein is known to be a membrane-associated lipid binding protein and has been implicated in vesicle trafficking, a process by which cells transport materials between different cellular destinations, as well as in membrane curvature and fusion.
In yeast, the overexpression of α-synuclein causes severe cellular toxicity by disrupting multiple cellular mechanisms, including vesicle trafficking. These disruptions occur in both yeast models and mammalian systems, where mutations or overexpression of α-synuclein increases the amount of membrane-associated α-synuclein, which in turn impairs vesicle trafficking and increases cellular stress and toxicity. Importantly, human genetic studies indicate mutations and overexpression of α-synuclein lead to severe and rapidly progressing forms of Parkinson’s disease, and this relationship to disease severity almost certainly involves toxic effects of α-synuclein aggregates on the functions of cell membranes.
14
YTX-7739’s Target
Yumanity discovered SCD as a result of its unbiased phenotypic screening efforts, which identified a series of compounds that potently protected cells against α-synuclein-mediated toxicity. Using Yumanity’s discovery engine’s target identification capability, Yumanity was able to identify that the specific biological target implicated in this protection was inhibition of Ole1, the single yeast fatty-acid desaturase enzyme and direct counterpart of SCD in humans.
Ole1 in yeast and SCD in humans are enzymes that metabolize saturated lipids and break them down into their unsaturated lipid components. Unsaturated lipids are important components of cell membranes because of the processes they regulate, including membrane fluidity, curvature, and fusion. Paradoxically, the greater the level of unsaturated lipid in membranes, the greater the vesicle trafficking impairment and toxicity caused by α-synuclein. Inhibiting SCD enzymatic activity reduces the levels of unsaturated lipids, which ameliorates the detrimental vesicle trafficking defects associated with increased α-synuclein expression. Yumanity’s hypothesis is that reducing SCD activity will reduce abnormal vesicle trafficking within cells caused by α-synuclein, thereby reducing the accumulation of neurotoxicity and slowing the progression of neurologic impairment in patients with Parkinson’s disease and related disorders.
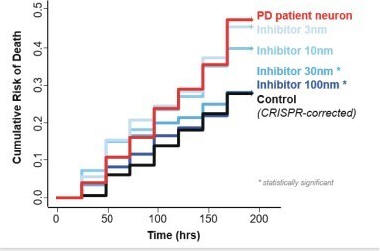
The chart below demonstrates the impact of inhibiting the SCD enzyme in a diseased human cell line prepared from a patient with Parkinson’s disease. This patient had a single amino acid mutation in the α-synuclein protein sequence. The red line shows the risk of cell death in cells containing the α-synuclein mutation. The black line, which shows lower risk of death, is a control cell line – genetically identical to the Parkinson’s disease patient cell line but with a correction of the α-synuclein mutation generated using CRISPR technology. The increasing shades of blue lines represent the survival of the mutation-containing cells upon exposure to increasing concentrations of a potent SCD inhibitor. As the chart shows, inhibition of SCD reduces cell death in the mutation-containing cell line down to the levels of the mutation-corrected control. The improved survival effect is clearly dependent upon the concentration of SCD inhibitor.
Cell death in Parkinson’s patient cell line corrected by inhibition of SCD
In addition to demonstrating that inhibition of SCD can protect human neurons grown in a dish, Yumanity has also explored the effects of SCD inhibition in a new mouse model of Parkinson’s disease. This mouse was engineered to express a mutant version of human α-synuclein, which leads to progressive motor deficits and pathological neuron cell loss in the brain that are similar to the progression seen in Parkinson’s disease. Dopamine replacement therapy, the standard of care in Parkinson’s therapy, can partially reverse the motor deficits in these engineered mice, demonstrating the disease-relevance of the model. When these mice were administered YTX-7739 for 4 months, the expected motor deficits never developed, whereas similar mice in the same study that received placebo evidenced the expected altered motor behaviors. Interestingly, when the SCD1 gene was removed in these engineered α-synuclein mice, known as a gene knockout, which would mimic the effects of an SCD inhibitor, these mice also had
15
significantly reduced motor deficits and pathological neuron loss. These studies demonstrate the consistent therapeutic effect of reducing SCD activity and validate SCD as a target for reducing the toxic effects of α-synuclein in disease-relevant models.
While Yumanity has established that SCD inhibition protects cells from α-synuclein toxicity, the precise mechanism of protection has not been defined. Yumanity believes there are at least three possible mechanisms of action: (1) SCD inhibition reverses a toxic increase in fatty acid desaturation triggered by α-synuclein aggregation, (2) SCD inhibition directly antagonizes toxic effects of α-synuclein on membrane properties and/or trafficking, or (3) reduced fatty acid desaturation ameliorates a direct toxic interaction of α-synuclein with cell membranes.
The knowledge of SCD enzyme biology also allows Yumanity to define a biomarker that can be used to measure target inhibition. Specifically, because the substrates for SCD are sixteen-carbon, or C16, or eighteen-carbon, or C18, saturated fatty acids, and the products are C16 and C18 monounsaturated fatty acids, Yumanity can therefore monitor drug effects on SCD by measuring the amount, or ratio, of the C16 and C18 precursors and their monounsaturated products. The result of this analysis gives Yumanity the fatty-acid desaturation index, or FA-DI, expressed as a ratio of the amount of monounsaturated C16 or C18 substrates divided by the amount of corresponding saturated fatty acid. The FA-DI gives Yumanity a biomarker that allows it to measure the effects of its compounds on SCD in vitro and in vivo.
Preclinical Studies
Preclinical in vivo pharmacokinetic and pharmacodynamics studies were conducted on YTX-7739 to demonstrate that YTX-7739 achieves sufficient exposure to inhibit the SCD enzyme in the brain, plasma, and other body tissues and biofluids.
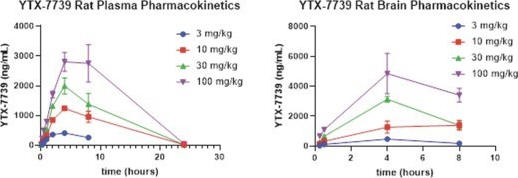
As shown below, single dose pharmacokinetic studies in rats demonstrate that YTX-7739 achieves sustained and dose-dependent exposures in both plasma and the brain. As the dose increases, the exposure levels increase. Yumanity has observed similar findings using twice-daily dose schedules, and in both single and repeat dose studies. At lower doses of YTX-7739, consistent dose-proportional exposure was achieved in both the plasma as well as the brain, and YTX-7739 also exhibited good brain penetration. In addition to rats, Yumanity has also performed these studies in other species, including mice, guinea pigs, dogs, and monkeys with similarly successful results.
Single dose pharmacokinetic data for YTX-7739, plasma (left) and brain (right)
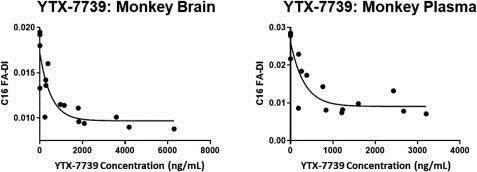
In addition, pharmacodynamics studies of YTX-7739 to date have shown that YTX-7739 inhibits SCD enzymatic activity as intended, which can be measured by the target engagement biomarker C16 FA-DI. As the chart below demonstrates, YTX-7739 has achieved clear, exposure-dependent target engagement in both the brain and plasma of monkeys treated for 14 days. As concentrations of YTX-7739 increase in either the brain or plasma, the levels of C16 FA-DI correspondingly decrease. These studies have been conducted across multiple species, with similar relationships between pharmacokinetics and pharmacodynamic responses. Moreover, at drug doses and exposures that produce substantial reductions in C16 FA-DI in the brain, treatment with YTX-7739 appears to be very well tolerated.
16
YTX-7739 Pharmacodynamics: exposure-dependent target inhibition in brain (left) and plasma (right)
Yumanity has a reliable and quality-controlled manufacturing process for the active pharmaceutical ingredient, or API, of YTX-7739 to support future clinical trials. IND-enabling safety pharmacology and toxicology studies have been completed and YTX-7739 is currently in Phase 1 clinical trials.
Clinical Trials
YTX-7739 is currently in Phase 1 clinical development consisting of a single ascending dose (SAD) study in healthy volunteers, a multiple ascending dose (MAD) study in healthy volunteers, and a Phase 1b study in patients with Parkinson’s disease. The SAD study has completed enrollment and was a randomized, double-blind, placebo-controlled single ascending dose study to investigate the safety, tolerability, and pharmacokinetics of YTX-7739 in healthy volunteers. The SAD study was conducted in three parts. The first part was to investigate the safety, tolerability and pharmacokinetics of increasing doses of YTX-7739 in healthy subjects. The second and third parts were to study the effect of food on the pharmacokinetics of YTX-7739 after administration in a fed state in healthy subjects.
Fifty-six healthy volunteers were dosed in the SAD study. Safety assessments included, but were not limited to, adverse events, serious adverse events, safety laboratory tests, vital signs and electrocardiograms (ECG). In addition, plasma drug levels were collected to assess pharmacokinetic variables and a pharmacodynamic biomarker was also included to explore the potential of YTX-7739 to change plasma levels of fatty acids.
While final results are pending, preliminary data indicate that all adverse events were mild or moderate, of short duration and self-limiting. There were no serious or unexpected adverse events reported.
The most frequently reported adverse event for subjects receiving YTX-7739 was headache, which was observed in 11 subjects. There were no clinically-relevant changes or dose related trends identified in blood chemistry, hematology, urinalysis, vital signs or ECGs. There were dose-linear increases in the maximum serum concentration achieved and the 24-hour area under the concentration-time curve observed at 5 mg, 10 mg, 30 mg and 250 mg dose levels in the fed state, suggesting dose-proportionality of YTX-7739 exposure in the 5 – 250 mg dose range, when administered in a fed state.
The safety and biopharmaceutical profile observed after single oral doses in healthy volunteers supported progression to a multiple ascending dose (MAD) study. Yumanity initiated a Phase 1 MAD study in healthy volunteers in the third quarter of 2020 with results anticipated in the first quarter of 2021. A subsequent Phase 1b trial in patients with Parkinson’s disease will assess the safety, tolerability and pharmacokinetics of YTX-7739 and will explore biomarkers of target engagement as well as potential correlative clinical markers such as neuroimaging measurements to monitor for early effects of YTX-7739. Early results from the Phase 1b trial are expected in the second quarter of 2021 assuming no delays due to the COVID-19 pandemic.
Yumanity’s Second Program
Yumanity’s second program in development, known as YTX-9184, also inhibits SCD and is being investigated as a potential treatment for dementia with Lewy Bodies and may also provide a potential back-up to YTX-7739. YTX-
17
9184 started IND-enabling toxicology and safety pharmacology in the second quarter of 2020. Yumanity plans to continue development for YTX-9184 with the goal of advancing it as its second drug candidate into the clinic in 2021. YTX-9184 is chemically distinct from YTX-7739 and has different properties, but functions by inhibiting the same enzyme, SCD. Pharmacokinetic and pharmacodynamic studies have been completed with YTX-9184, and YTX-9184 exhibits similar dose and exposure dependent effects on the brain and plasma C16 FA-DI across several small and large animal species.
Yumanity’s Potential Third and Fourth Programs
Yumanity is currently performing lead optimization to identify compounds that inhibit the activity of the enzyme that is the target of Yumanity’s potential third program as a potential treatment for ALS and FTLD. The undisclosed target in Yumanity’s potential third program is implicated in the rescue of toxicity created by a gene linked to ALS and FTLD. Yumanity’s potential fourth program is currently in the hit-to-lead stage of identifying small molecule inhibitors for its target enzyme. Inhibition of this target has been correlated with prevention of neurodegeneration associated with alterations in a gene, which has been correlated with increased risk of developing ALS. Both of these potential programs are being progressed through a research collaboration with Merck, and Merck will be responsible for IND-enabling toxicology and safety pharmacology studies, as well as subsequent clinical development and commercialization.
ALS and FTLD Disease Overview
ALS, also referred to as Lou Gehrig’s disease, is a neurodegenerative disease that affects nerve cells in the brain and the spinal cord. In healthy individuals, upper motor neurons in the brain send signals to lower motor neurons in the spinal cord and brainstem, which send signals to muscles, thereby generating body movement. In ALS, both the upper motor neurons and the lower motor neurons degenerate or die, resulting in loss of muscle function with progression to severe impairment of mobility, speech, and communication.
Early symptoms of ALS usually include muscle weakness or stiffness, and over time, all muscles under voluntary control are affected. As ALS progresses, individuals lose their strength and the ability to speak, eat and move. Many sufferers lose the muscular ability to maintain breathing, requiring permanent ventilatory support. Individuals with ALS usually retain their ability to perform higher mental processes such as reasoning, remembering, understanding, and problem solving, so they are entirely aware of their progressive loss of muscle function. Continued deterioration of muscle control leads to respiratory failure and death, with an average survival time of three years. About 20 percent of people with ALS live five years, 10 percent will survive 10 years and 5 percent will live 20 years or longer.
According to the ALS Association, in 2016, between 14,000 and 15,000 Americans had ALS. ALS is most commonly diagnosed between the ages of 55 and 75, and only ten percent of people with ALS will survive for ten years or more. Medical and non-medical costs of ALS, including lost income, range between $256 million and $433 million each year in the United States, with annual costs from ALS exceeding $60,000 per patient.
FTLD is an umbrella term for a group of related syndromes and processes, often also called frontotemporal lobar dementia, frontotemporal degeneration or dementia or Pick’s disease, that impacts the frontal and temporal lobes of the brain. Formally, the process of frontotemporal degeneration results in the condition of frontotemporal dementia. Disease processes cause the degeneration of neurons and shrinking of the frontal and temporal brain regions, which causes progressive alterations in personality, behavior, and language. There are different types of FTLD, which manifest as a frontal or behavioral variant affecting behavior and personality, or as a primary progressive aphasia variant, which results in difficulty communicating due to loss of speech and inability to use and understand language. FTLD patients often exhibit aggressive and compulsive behaviors and have various changes in sexual behaviors.
It is believed that the prevalence of FTLD in the United States is around 60,000 cases. There is a wide range of onset, from 21 to 80 years of age, with most cases occurring between 45 and 64 years of age. Given the younger age of onset as compared to Alzheimer’s disease, FTLD has been cited as the most common form of dementia in people under 60. Annual medical and nonmedical costs of FTLD are estimated at approximately $120,000 per patient, indicating a societal disease impact in the United States alone of over $7 billion annually.
18
These conditions are generally believed to exist as a spectrum disorder, with “pure” ALS as a neuromuscular disorder at one end of the spectrum, and “pure” FTLD as a dementia-related disorder at the other. Many patients exhibit varying degrees of both types of symptoms on this spectrum.
Limitations of Current Therapies
There is no known cure for ALS. The cause of ALS in 90% or more of cases is unknown. Known as sporadic ALS, genetic and environmental factors may play a role in these cases. In contrast, up to 10% of all ALS cases are of an inherited familial form. A number of genetic mutations have been implicated in familial ALS, most frequently C9orf72 and SOD1. In all cases of ALS, upper and lower motor neurons are lost, causing muscle dysfunction and atrophy. Two drugs have been approved in the United States for the treatment of ALS, riluzole and edaravone. Riluzole has demonstrated a survival benefit and edaravone delayed decline in an assessment of daily functioning.
There are also no known cures for FTLD, nor are there any approved medications for this disease. Patients are sometimes proscribed Riluzole, although its effectiveness in this indication is uncertain. To manage quality of life, antidepressants are prescribed to help with anxiety and obsessive-compulsive behaviors, and anti-psychotics can sometimes help control irrational and risky behavior. Sleep aids are also prescribed to help with insomnia and sleep disturbances.
Yumanity’s Solution
Yumanity is currently optimizing small molecule therapies to potentially slow or halt disease progression in patients suffering with ALS and FTLD. Yumanity has identified two undisclosed targets involved in the neurotoxic cascades which are hallmark toxicities observed in ALS and FTLD. Yumanity is advancing these two undisclosed targets through its collaboration with Merck.
Competition
The biotechnology and pharmaceutical industries, including in the neurodegenerative disease field, are highly competitive and subject to rapid and significant technological change. While Yumanity believes that its discovery engine platform and its employees and consultants, scientific knowledge and development experience provide Yumanity with competitive advantages, Yumanity faces potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical, and biotechnology companies, academic institutions, governmental agencies, and public and private research institutions. Several of these entities have commercial products, robust drug pipelines, readily available capital, and established research and development organizations. Many of Yumanity’s competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved products than Yumanity does. Mergers and acquisitions in the pharmaceutical, biotechnology, and diagnostic industries may result in even more resources being concentrated among a smaller number of Yumanity’s competitors. These competitors also compete with Yumanity in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, Yumanity’s programs. Small or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. The key competitive factors affecting the success of Yumanity’s product candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of branded and generic competition, and the availability of reimbursement from government and other third-party payors.
Any product candidates that Yumanity successfully develops and commercializes will compete with existing therapies and new therapies that may become available in the future, including but not limited to:
19
| • | Parkinson’s disease: Currently available therapies for Parkinson’s disease include Levodopa, D2/D3-preferring agonists, monoamine oxidase B inhibitors as monotherapy or in combination, anticholinergics as well as deep brain stimulation devices by Medtronic Inc. and St. Jude Medical Inc., among others. Yumanity is also aware of several large and specialty pharmaceutical and biotechnology companies developing potentially disease modifying therapeutics for Parkinson’s disease, including Denali, Prothena, F. Hoffman-la Roche, Novartis, AbbVie (in partnership with BioArctic AB), Voyager Therapeutics, Prevail Therapeutics, Sage Therapeutics, Sanofi, Neurocrine Biosciences, Eli Lilly, Biogen (in partnership with Ionis and Neurimmune), AstraZeneca, Takeda, IRLAB Therapeutics, Avanir Pharmaceuticals, Lysosomal Therapeutics and Lundbeck, that are in various stages of clinical development. |
| • | Dementia with Lewy bodies: Currently available therapies to alleviate symptoms in dementia with Lewy bodies include cholinesterase inhibitors, carbidopa/levodopa, memantine, “atypical” antipsychotics, melatonin, and clonazepam. Yumanity is also aware of several large and specialty pharmaceutical and biotechnology companies and academic institutes developing potentially disease modifying therapeutics for dementia with Lewy bodies, including Lawson Health Research Institute, Sun Pharma Advanced Research Company, Georgetown University, Pfizer, Eisai, Allergan and Novartis, that are in various stages of clinical development. |
| • | ALS: Currently available therapies for ALS include riluzole (Rilutek®) and edaravone (Radicava®). Yumanity is also aware of several large and specialty pharmaceutical and biotechnology companies and academic institutions developing potentially disease modifying therapeutics for ALS, including Denali, Avanir Pharmaceuticals, Amylyx Pharmaceuticals, Biogen (in partnership with Ionis), Neuropore Therapies, Cytokinetics and Mallinckrodt, that are in various stages of clinical development. |
| • | FTLD: There are no currently available therapies indicated for FTLD, however some patients are prescribed riluzole (Rilutek®) and other medications to manage symptoms such as antidepressants, antipsychotics and sleep aids. Yumanity is also aware of several large and specialty pharmaceutical and biotechnology companies developing potentially disease modifying therapeutics for FTLD, including Alector, Bristol-Myers Squibb/Biogen, TauRx Therapeutics and Bayer, that are in various stages of clinical development. |
| • | Alzheimer’s disease: Currently available therapies for Alzheimer’s disease include donepezil (AriceptTM), galantamine (RazadyneTM), memantine (EbixaTM), rivastigmine (Exelon™), suvorexant (Belsomra) and tacrine (CognexTM).Yumanity is also aware of several large and specialty pharmaceutical and biotechnology companies developing potentially disease modifying therapeutics for Alzheimer’s disease, including Axon Neuroscience, AbbVie, Aracion Biotech, AC Immune, Janssen, Alector, AstraZeneca, Allergan, Affiris, Pfizer, Biogen (in partnership with Neurimmune), Eli Lilly, GlaxoSmithKline, Novartis, and F. Hoffman-la Roche (including Genentech, its wholly owned subsidiary), that are in various stages of clinical trials. |
Collaboration Agreement with Merck
In June 2020, Yumanity entered into an exclusive license and research collaboration agreement, or the Merck Collaboration Agreement, with Merck to support the research, development and commercialization of products for the treatment of ALS and FTLD.
Pursuant to the Merck Collaboration Agreement, Yumanity granted Merck an exclusive, worldwide license with the right to grant and authorize sublicenses, under certain intellectual property rights related to two certain undisclosed targets in connection with Yumanity’s ALS and FTLD programs to make, have made, use, import, offer to sell and sell compounds and products covered by such intellectual property rights. In the event that the exploitation of such compound or product would infringe during the term of the Merck Collaboration Agreement a claim of an issued patent controlled by Yumanity, Yumanity also granted Merck a non-exclusive, sublicensable, royalty-free license under such issued patent to exploit such compound and product.
Under the terms of the Merck Collaboration Agreement, Yumanity and Merck are each responsible to perform certain research activities in accordance with a mutually agreed upon research plan. Upon the completion of certain
20
stages of the research plan, Merck will elect to either advance or terminate the applicable research program. Following completion of the research program, Merck is responsible for the development and commercialization of the compounds developed pursuant to the research program and any product containing such compounds.
As consideration for the licenses granted to Merck under the Merck Collaboration Agreement, Merck paid Yumanity a one-time upfront payment and also purchased Class C preferred units of Holdings. Under the terms of the Merck Collaboration Agreement, Yumanity is eligible to receive up to $280 million upon achievement of specified research and development milestones, and up to $250 million upon achievement of specified sales-based milestones as well as a tiered, mid-single digit royalty on net sales of licensed products, subject to customary reductions. Merck’s royalty obligations for each licensed product continue on a country-by-country basis until the later of (i) the last-to-expire valid claim of the patent rights in such country or (ii) the tenth anniversary of the first commercial sale of such product in such country.
Unless terminated earlier, the Merck Collaboration Agreement will continue in full force and effect until one or more products has received marketing authorization and, thereafter, until expiration of all royalty obligations under the Merck Collaboration Agreement. Yumanity or Merck may terminate the Merck Collaboration Agreement upon an uncured material breach by the other party or insolvency of the other party. Merck may also terminate the Merck Collaboration Agreement for any reason upon certain notice to Yumanity.
License Agreement with Whitehead Institute
In February 2016, Yumanity entered into a tangible property and exclusive patent license agreement or the Whitehead License Agreement, with the Whitehead Institute which was subsequently amended in April 2016, August 2016 and July 2018. Pursuant to the Whitehead License Agreement, the Whitehead Institute granted Yumanity a worldwide license under certain patent rights to develop, commercialize and sell products and to develop and perform processes covered by such patents for the treatment of any disease in humans other than certain specified treatments for infectious diseases and cancer. The Whitehead Institute also granted Yumanity a non-exclusive, worldwide license to use certain know-how and to use and make certain biological materials.
The patent rights licensed to Yumanity under the Whitehead License Agreement relate to Yumanity’s discovery engine and are directed to compositions and methods for identifying novel disease-modifying therapies for neurodegenerative diseases. Such patent rights include patent rights developed or co-developed by Dr. Lindquist as an employee of Howard Hughes Medical Institute, as well as patents owned or jointly owned by the University of Chicago, the University of Washington, the Massachusetts Institute of Technology, the Curators of the University of Missouri and Pfizer, Inc. Yumanity’s license under such patent rights is exclusive, subject to certain retained rights and certain patent rights previously licensed by the Whitehead Institute. Additionally, Yumanity’s exclusivity with respect to patent rights jointly owned by Pfizer, Inc. only applies to the Whitehead Institute’s right as a joint owner of such patents.
Under the terms of the Whitehead License Agreement, Yumanity must use commercially reasonable efforts to develop licensed products or processes and to introduce such licensed products or processes into the commercial market. Thereafter, Yumanity is required to use commercially reasonable efforts to make such licensed products or processes reasonably available to the public. In addition, in any given year until the first regulatory approval of a licensed product, Yumanity has diligence requirements to achieve a certain development milestone with respect to a licensed product or expend a minimum amount of money for platform development and/or development of licensed products or processes.
As consideration for the licenses granted to Yumanity under the Whitehead License Agreement, Yumanity paid an initial license fee and reimbursed the Whitehead Institute for certain expenses incurred in connection with the patent rights. Holdings also issued 3,000 common units (which, following a 100:1 unit split, currently represent 300,000 units) to the Whitehead Institute and certain persons and entities as directed by the Whitehead Institute in satisfaction of the Whitehead Institute’s policy on equity sharing. Under the terms of the Whitehead License Agreement, Yumanity is also required to pay an annual license maintenance fee which is creditable against royalties on net sales earned during the same calendar year. In addition, Yumanity is obligated to make payments to the Whitehead Institute upon achievement of certain milestones, the amount of which depends on the licensed product and indication, with up to an aggregate of $1.9 million for each of the first two licensed products for the first
21
indication and less for subsequent licensed products and additional indications. Yumanity is also required to pay the Whitehead Institute a low single digit royalty percentage of net sales of licensed products and a low single digit royalty on net sales of products determined to have biological activity or utility by the use of a licensed product or process, or Identified Products. Additionally, Yumanity is required to pay a mid single to low double digit percentage of certain income received from sublicensees and certain partners. Yumanity’s royalty obligation continues on a licensed product-by-licensed product and country-by-country basis for so long as the manufacture, use or sale of such licensed product in such country infringes a valid claim of the patent rights or, with respect to each Identified Product, for ten years after the first sale for consumption by an end user patient of such Identified Product.
Unless terminated earlier, the Whitehead License Agreement will expire upon the expiration or abandonment of all issued patents and filed patent applications within the licensed patent rights, which is currently projected to occur in 2035. The Whitehead Institute may terminate the Whitehead License Agreement upon notice to Yumanity if Yumanity ceases to carry on its business related to the Whitehead License Agreement, if Yumanity fails to make required payments within a certain period time or if Yumanity commits a material breach and fails to cure such breach within a certain period of time. The Whitehead Institute may also terminate the Whitehead License Agreement and/or the licenses granted to Yumanity if Yumanity brings or assists others in bringing a patent challenge against the Whitehead Institute. Yumanity may terminate the Whitehead License Agreement for any reason upon certain notice to the Whitehead Institute.
Intellectual Property
The proprietary nature of, or protection for, Yumanity’s product candidates and methods of manufacture and clinical use are an important part of Yumanity’s strategy to develop and commercialize novel therapies. Yumanity has filed numerous patent applications pertaining to its product candidates and clinical use. Yumanity strives to protect and enhance the proprietary technology, inventions, and improvements that are commercially important to the development of its business by seeking, maintaining and defending Yumanity’s intellectual property, whether developed internally or licensed from third parties. Yumanity also relies on trade secrets, know-how, continuing technological innovation, and potential in-licensing opportunities to develop, strengthen, and maintain its proprietary position in the field of neurodegenerative diseases and protein misfolding. Additionally, Yumanity intends to rely on regulatory protection afforded through data exclusivity and market exclusivity, as well as patent term extensions, where available.
As of September 4, 2020, Yumanity’s patent portfolio as it pertains to certain of its product candidates included:
| • | Yumanity’s Lead Program – YTX-7739:one pending international patent (PCT) application, one pending U.S. non-provisional patent application, and fourteen pending patent applications outside the U.S., which, if pursued and granted, would be expected to expire in 2038-2039, without taking a potential patent term adjustment or extension into account,with composition of matter claims directed to the YTX-7739 compounds and method claims directed to the treatment of neurological disorders, for example SCD-associated disorders, or inhibiting toxicity in a cell relating to a protein; |
| • | Yumanity’s Second Program – YTX-9184:three pending U.S. non-provisional patent applications, one pending international patent (PCT) application, one pending U.S. provisional patent application, and nineteen pending patent applications outside the U.S., which, if pursued and granted in the U.S., would be expected to expire in2037-2040, without taking a potential patent term adjustment or extension into account, with composition of matter claims directed to YTX-9184 and method claims directed to the treatment of neurological disorders, for example SCD-associated disorders, or inhibiting toxicity in a cell relating to a protein; |
| • | Yumanity’s Potential Third Program: five pending U.S. provisional patent applications, which, if pursued and granted in the U.S., would be expected to expire in 2041, without taking a potential patent term adjustment or extension into account, with composition of matter and claims directed to compounds and method claims directed to the treatment of neurological disorders using inhibitors of Yumanity’s undisclosed target. |
22
| • | Yumanity’s Potential Fourth Program: four pending U.S. provisional patent applications, which, if pursued and granted in the U.S., would be expected to expire in 2041, without taking a potential patent term adjustment or extension into account, with composition of matter claims directed to compounds and methods claims directed to the treatment of neurological disorders using inhibitors of Yumanity’s undisclosed target. |
In addition, Yumanity has in-licensed an estate of patents and patent applications relating to Yumanity’s discovery engine from the Whitehead Institute which is directed to compositions and methods for identifying novel disease-modifying therapies for neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and ALS. For example, this estate includes granted and pending claims to a number of yeast models of protein misfolding and methods of use thereof. As of September 4, 2020, this estate includes twenty-two granted U.S. patents, projected to expire between 2022-2035; three pending U.S. patent applications, which if granted in the U.S., would be expected to expire between 2034-2038, without taking a potential patent term adjustment or extension into account; thirty-three granted foreign patents, projected to expire between 2022-2035; and fourteen pending foreign patent applications, which if granted, would be expected to expire between 2034-2038. Yumanity does not control the prosecution and maintenance of all of its in-licensed patents and patent applications, and Yumanity’s rights to enforce the patents are limited in certain ways. For additional detail regarding the risks associated with Yumanity’s license agreements, see “Risk Factors—Risks Related to Yumanity’s Intellectual Property.”
The term of individual patents may vary based on the countries in which they are obtained. Generally, patents issued for applications filed in the United States are effective for 20 years from the earliest effective non-provisional filing date in the absence, for example, of a terminal disclaimer shortening the term of the patent or patent term adjustment increasing the term of the patent. In addition, in certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of FDA regulatory review periods. The restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed 14 years following FDA approval. The duration of patents outside of the United States varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective non-provisional filing date.
In addition to patents and patent applications that Yumanity owns and license, Yumanity relies on trade secrets and know-how to develop and maintain its competitive position. However, trade secrets can be difficult to protect. Yumanity seeks to protect its proprietary technology and processes, and obtain and maintain ownership of certain technologies, in part, through confidentiality agreements and invention assignment agreements with its employees, consultants, scientific advisors, contractors, and commercial partners.
Yumanity’s future commercial success depends, in part, on Yumanity’s ability to obtain and maintain patent and other proprietary protection for commercially important technology, inventions and know-how related to Yumanity’s business; defend and enforce Yumanity’s patents; preserve the confidentiality of Yumanity’s trade secrets; and operate without infringing valid enforceable patents and proprietary rights of third parties. Yumanity’s ability to stop third parties from making, using, selling, offering to sell, or importing its products may depend on the extent to which Yumanity has rights under valid and enforceable patents or trade secrets that cover these activities. With respect to Yumanity’s intellectual property, Yumanity cannot be sure that patents will issue from any of the pending patent applications to which Yumanity owns or that Yumanity may file in the future, nor can Yumanity be sure that any patents that may be issued in the future to Yumanity will be commercially useful in protecting Yumanity’s product candidates and methods of using or manufacturing the same. Moreover, Yumanity may be unable to obtain patent protection for certain aspects of its product candidates generally, as well as with respect to certain indications. See the section titled “Risk Factors—Risks Related to Yumanity’s Intellectual Property” for a more comprehensive description of risks related to Yumanity’s intellectual property.
Manufacturing
Yumanity does not have any manufacturing facilities or personnel. Yumanity currently relies, and expects to continue to rely, on third parties for the manufacturing of its product candidates for preclinical and clinical testing, as well as for commercial manufacturing if Yumanity’s product candidates receive marketing approval.
Currently,all of Yumanity’s product candidates are small molecules and are manufactured in reliable and reproducible synthetic processes from readily available starting materials. The chemistry does not require unusual equipment in the manufacturing process. Yumanity expects to continue to develop product candidates that can be produced at contract manufacturing facilities.
23
Commercialization
Yumanity intends to develop and, if approved by the FDA, to commercialize its product candidates alone or in collaboration with others. Yumanity may work in combination with one or more large pharmaceutical partners for certain indications, where specialist capabilities are needed. For specialized indications, Yumanity intends to commercialize its product candidates independently. For example, Yumanity believes the patient and prescriber populations for Parkinson’s disease are relatively concentrated and can be addressed with a focused sales team. Yumanity also does not believe any existing pharmaceutical companies have significant expertise in the commercialization of disease-modifying therapies for neurodegenerative diseases. Yumanity will, however, continuously review its partnering strategy in the light of new clinical data and market understanding. Yumanity may enter into distribution or licensing arrangements for commercialization rights for other regions outside the United States.
Government Regulation
Government authorities in the United States at the federal, state and local level and in other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug products. Generally, before a new drug can be marketed, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Drug Development
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. Drugs are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Additionally, a manufacturer may need to recall a product from the market. Any agency or judicial enforcement action could have a material adverse effect on Yumanity.
Yumanity’s product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| • | completion of extensive nonclinical laboratory tests, animal studies and formulation studies in accordance with applicable regulations, including the FDA’s Good Laboratory Practice, or GLP, regulations; |
| • | submission to the FDA of an IND application, which must become effective before human clinical trials may begin; |
| • | approval by an independent institutional review board, or IRB, or ethics committee at each clinical trial site before each study may be initiated; |
| • | performance of adequate and well-controlled human clinical trials in accordance with applicable IND and other clinical trial-related regulations, referred to as Good Clinical Practices, or GCPs, to establish the safety and efficacy of the proposed drug for each proposed indication; |
| • | submission to the FDA of an NDA for a new drug; |
24
| • | a determination by the FDA within 60 days of its receipt of an NDA to file the NDA for review; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug is produced to assess compliance with current Good Manufacturing Practices, or cGMP, requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
| • | potential FDA audit of the nonclinical study and/or clinical trial sites that generated the data in support of the NDA; and |
| • | FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the United States. |
The nonclinical and clinical testing and approval process requires substantial time, effort and financial resources, and Yumanity cannot be certain that any approvals for Yumanity’s product candidates will be granted on a timely basis, if at all.
The data required to support an NDA are generated in two distinct development stages: nonclinical and clinical. For new chemical entities, the nonclinical development stage generally involves synthesizing the active component, developing the formulation and determining the manufacturing process, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies in the laboratory, which support subsequent clinical testing. These nonclinical tests include laboratory evaluation of product chemistry, formulation, stability and toxicity, as well as animal studies to assess the characteristics and potential safety and efficacy of the product. The conduct of the nonclinical tests must comply with federal regulations, including GLPs. The sponsor must submit the results of the nonclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. Some nonclinical testing may continue even after the IND is submitted, but an IND must become effective before human clinical trials may begin. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials, including concerns that human research subjects will be exposed to unreasonable health risks, and places the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a drug candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, Yumanity cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that could cause the study to be suspended or terminated.
The clinical stage of development involves the administration of the drug candidate to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by an IRB at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completion. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries.
As part of the 21st Century Cures Act, or the Cures Act, which was signed into law on December 13, 2016, upon request, the FDA is to establish a process for the qualification of drug development tools. A drug development tool includes a biomarker including a surrogate endpoint, a clinical outcome assessment including a patient-reported outcome, and any other method, material or measure that the FDA determines aids drug development and regulatory review. A drug development tool is qualified if the FDA has determined that the tool and its proposed context of use
25
can be relied upon to have a specific interpretation and application in drug development and regulatory review. A qualified drug development tool may be used to support the investigational use of a drug or support or obtain NDA approval.
A sponsor who wishes to conduct a clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of an NDA so long as the clinical trial is conducted in compliance with GCP and the FDA is able to validate the data through an onsite inspection if the agency deems it necessary.
Clinical Trials
Clinical trials are generally conducted in three sequential phases that may overlap, known as Phase 1, Phase 2 and Phase 3 clinical trials.
| • | Phase 1 clinical trials generally involve a small number of healthy volunteers who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. |
| • | Phase 2 clinical trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits and provide a preliminary evaluation of efficacy. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks. |
| • | Phase 3 clinical trials generally involve large numbers of patients at multiple sites (from several hundred to several thousand subjects) and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product and provide an adequate basis for physician labeling. Phase 3 clinical trials may include comparisons with placebo and/or comparator treatments. |
Post-approval studies, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA. Written IND safety reports must be submitted to the FDA and the investigators within 15 calendar days for serious and unexpected suspected adverse events, finding from other studies or animal or in vitro testing that suggests a significant risk for human subjects, and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. Additionally, a sponsor must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study.
Pursuant to the Cures Act, the manufacturer of an investigational drug for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the earlier of the first initiation of a Phase 2 or Phase 3 study of the investigational drug, or as applicable, 15 days after the drug receives a designation as a breakthrough therapy, fast track product, or regenerative advanced therapy.
26
Concurrently with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality and purity of the final drug product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
NDA and FDA Review Process
The results of the nonclinical studies and clinical trials, together with other detailed information, including extensive manufacturing information and information on the composition of the drug and proposed labeling, are submitted to the FDA in the form of an NDA requesting approval to market the drug for one or more specified indications. The FDA reviews an NDA to determine, among other things, whether a drug is safe and effective for its intended use and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality, and purity. FDA approval of an NDA must be obtained before a drug may be offered for sale in the United States.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA must contain data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of pediatric data or full or partial waivers.
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each NDA must be accompanied by a user fee. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fee schedule, effective from October 1, 2020 through September 30, 2021, the user fee for an application requiring clinical data, such as an NDA, is $2,875,842. PDUFA also imposes an annual prescription drug product program fee for human drugs ($336,432). Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business.
The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. The FDA must make a decision on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA, for drugs that contain a new chemical entity, or NCE, the FDA has 10 months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months from the filing date for a priority NDA. For drugs that do not contain an NCE, these 10 and six month review timeframes are from the receipt date of an NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMPs. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. Additionally, the FDA may refer applications for novel drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. The review and evaluation of an NDA by the FDA is extensive and time consuming and may take longer than originally planned to complete, and Yumanity may not receive a timely approval, if at all.
27
After the FDA evaluates an NDA, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may resubmit the NDA addressing all of the deficiencies identified in the letter, withdraw the application, or request an opportunity for a hearing. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than Yumanity interprets the same data.
There is no assurance that the FDA will ultimately approve a drug product for marketing in the United States and Yumanity may encounter significant difficulties or costs during the review process. If a product receives marketing approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings, or precautions be included in the product labeling or may condition the approval of the NDA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-marketing testing or clinical trials and surveillance to monitor the effects of the approved product. For example, the FDA may require Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and efficacy and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. The FDA also may place other conditions on approvals including the requirement for a risk evaluation and mitigation strategy, or REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United
States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product.
Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications to market the same drug for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety, or providing a major contribution to patient care, or in instances of drug supply issues. Competitors, however, may receive approval of either a different product for the same indication or the same product for a different indication but that could be used off-label in the orphan indication. Orphan drug exclusivity also could block the approval of one of Yumanity’s products for seven years if a competitor obtains approval before Yumanity does for the same product, as defined by the FDA, for the same indication Yumanity is seeking approval, or if Yumanity’s product is determined to be contained within the scope of the competitor’s product for the same indication or disease. If one of Yumanity’s products designated as an orphan drug receives marketing approval for an indication broader than that which is designated, it may not be entitled to orphan drug exclusivity. Orphan drug status in the European Union has similar, but not identical, requirements and benefits.
28
Expedited Development and Review Programs
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs that meet certain criteria. Specifically, new drugs are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new drug may request the FDA to designate the drug as a Fast Track product at any time during the clinical development of the product. Unique to a Fast Track product, the FDA may review sections of the marketing application on a rolling basis before the complete NDA is submitted, if the sponsor provides a schedule for the submission of the sections of the application, the FDA agrees to accept sections of the application and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the application.
Any product submitted to the FDA for marketing, including under the Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review. A product is eligible for priority review if it has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or offers a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug designated for priority review in an effort to facilitate the review.
Additionally, a drug may be eligible for designation as a breakthrough therapy if the drug is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The benefits of breakthrough therapy designation include the same benefits as fast track designation, plus intensive guidance from FDA to ensure an efficient drug development program. Fast Track designation, priority review, and breakthrough designation do not change the standards for approval but may expedite the development or approval process.
Pediatric Trials
The FDCA requires that a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase 2 meeting or as may be agreed between the sponsor and the FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials and/or other clinical development programs.
Post-Marketing Requirements
Following approval of a new product, a pharmaceutical company and the approved product are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the FDA of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses. Prescription drug promotional materials must be submitted to the FDA in
29
conjunction with their first use. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new NDA or NDA supplement, which may require the applicant to develop additional data or conduct additional nonclinical studies and clinical trials. As with new NDAs, the review process is often significantly extended by FDA’s requests for additional information or clarification. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, or the PDMA, a part of the FDCA.
In the United States, once a product is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that products be manufactured in specific facilities and in accordance with cGMP. Yumanity relies, and expects to continue to rely, on third parties for the production of clinical and commercial quantities of its products in accordance with cGMP regulations. NDA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These manufacturers must comply with cGMP regulations that require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance. The discovery of violative conditions, including failure to conform to cGMP, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them. Discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved NDA, including, among other things, recall or withdrawal of the product from the market.
Discovery of previously unknown problems with a product or the failure to comply with applicable FDA requirements can have negative consequences, including judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of Yumanity’s products under development. Changes in statutes, regulations, or the interpretation of existing regulations could impact Yumanity’s business in the future by requiring, for example: (i) changes to Yumanity’s manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of Yumanity’s products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of Yumanity’s business.
Orange Book Listing
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A Section 505(b)(2) NDA is an application in which the applicant, in part, relies on investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. Section 505(j) establishes an abbreviated approval process for a generic version of approved drug products through the submission of an Abbreviated New Drug Application, or ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to a previously approved product. Limited changes must be preapproved by the FDA via a suitability petition. ANDAs are termed “abbreviated” because they are generally not required to include nonclinical and clinical data to establish safety and efficacy. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug through in vitro, in vivo, or other testing. The generic version must deliver the same amount of active ingredients into a subject’s bloodstream in the same amount of time as the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug.
30
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to list with the FDA certain patents having claims that cover the applicant’s product and method of use. Upon approval of an NDA, each of the patents listed in the application for the drug is then published in Approved Drug Products with Therapeutic Equivalence Evaluations, also known as the Orange Book. These products may be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
Any applicant who files an ANDA seeking approval of a generic equivalent version of a drug listed in the Orange Book or a 505(b)(2) NDA referencing a drug listed in the Orange Book must make patent certifications to the FDA that (1) no patent information on the drug or method of use that is the subject of the application has been submitted to the FDA; (2) the patent has expired; (3) the date on which the patent has expired and approval will not be sought until after the patent expiration; or (4) the patent is invalid or will not be infringed upon by the manufacture, use, or sale of the drug product for which the application is submitted. The last certification is known as a paragraph IV certification. Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except where the ANDA or 505(b)(2) NDA applicant challenges a listed patent through a paragraph IV certification or if the applicant is not seeking approval of a patented method of use. If the applicant does not challenge the listed patents or does not indicate that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all of the listed patents claiming the referenced product have expired.
If the competitor has provided a paragraph IV certification to the FDA, the competitor must also send notice of the paragraph IV certification to the holder of the NDA for the reference listed drug and the patent owner within 20 days after the application has been accepted for filing by the FDA. The NDA holder or patent owner may then initiate a patent infringement lawsuit in response to the notice of the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a paragraph IV certification notice prevents the FDA from approving the ANDA or 505(b)(2) application until the earlier of 30 months from the date of the lawsuit, expiration of the patent, settlement of the lawsuit, a decision in the infringement case that is favorable to the applicant or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the 30-month stay.
In instances where an ANDA or 505(b)(2) NDA applicant files a paragraph IV certification, the NDA holder or patent owners regularly take action to trigger the 30-month stay, recognizing that the related patent litigation may take many months or years to resolve. Thus, approval of an ANDA or 505(b)(2) NDA could be delayed for a significant period of time depending on the patent certification the applicant makes and the reference drug sponsor’s decision to initiate patent litigation.
U.S. Marketing Exclusivity
Marketing exclusivity provisions under the FDCA can also delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for an NCE. A drug is an NCE if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovator drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder. The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA, if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the active agent for the original indication or condition of use. Three-year and five-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
31
U.S. Patent-Term Extension
Depending upon the timing, duration and specifics of FDA approval of Yumanity’s current product candidates or any future product candidate, some of Yumanity’s U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch Waxman Act. The Hatch Waxman Act permits extension of the patent term of up to five years as compensation for patent term lost during FDA regulatory review process. Patent term extension, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term extension period is generally one half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved drug is eligible for the extension (and only those patent claims covering the approved drug, a method for using it or a method for manufacturing it may be extended), and the application for the extension must be submitted prior to the expiration of the patent. A patent that covers multiple products for which approval is sought can only be extended in connection with one of the approvals. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension. In the future, Yumanity may apply for extension of a patent term for its currently owned patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA. However, there can be no assurance that the USPTO will grant Yumanity any requested patent term extension, either for the length Yumanity requests or at all.
Healthcare Reform
In the United States and some foreign jurisdictions, there have been, and continue to be, several legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of product candidates, restrict or regulate post-approval activities, and affect the ability to profitably sell product candidates for which marketing approval is obtained. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, ACA, enacted in the United States in March 2010, has already had, and is expected to continue to have, a significant impact on the healthcare industry. The ACA expanded coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, the ACA, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% (increased to 70% in 2019 pursuant to subsequent legislation) point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D.
Since its enactment there have been judicial, administrative, executive, and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the current administration to repeal or replace certain aspects of the ACA. For example, various portions of the ACA are currently undergoing legal and constitutional challenges in the United States Supreme Court, and since January 2017, President Trump has signed various Executive Orders and other directives which eliminated cost sharing subsidies and various provisions that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. Additionally, Congress has introduced several pieces of
32
legislation aimed at significantly repealing or repealing and replacing all or part of the ACA. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the ACA such as removing penalties, starting January 1, 2019, for not complying with the ACA’s individual mandate to carry health insurance, delaying the implementation of certain ACA-mandated fees, and increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. It unclear whether the ACA will be overturned, repealed, replaced, or further amended.
More recently, in December 2018, the Centers for Medicare and Medicaid Services, or CMS, published a final rule permitting further collections and payments to and from certain ACA-qualified health plans and health insurance issuers under the ACA risk adjustment program in response to the outcome of federal district court litigation regarding the method CMS uses to determine this risk adjustment. Since then, the ACA risk adjustment program payment parameters have been updated annually. In addition, CMS published a final rule that would give states greater flexibility, starting in 2020, in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. For example, on August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. Specifically, the Joint Select Committee on Deficit Reduction was created to recommend to Congress proposals in spending reductions. The Joint Select Committee on Deficit Reduction did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2012 through 2021, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, started in April 2013 and which, due to subsequent legislative amendments will stay in effect through 2030 unless additional Congressional action is taken. However, pursuant to the Coronavirus Aid, Relief and Economic Security Act, or CARES Act, the 2% Medicare sequester reductions have been suspended from May 1, 2020 through December 31, 2020 due to the COVID-19 pandemic. Additionally, on January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA. The ATRA, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Recently, there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products. Such scrutiny has resulted in several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, reduce the cost of drugs under Medicare and reform government program reimbursement methodologies for drug products. At the federal level, the Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Additionally, the current administration previously released a ‘‘Blueprint’’ to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products and reduce the out of pocket costs of drug products paid by consumers. On July 24, 2020, President Trump signed four Executive Orders aimed at lowering drug prices. The Executive Orders direct the Secretary of HHS to: (1) eliminate protection under an Anti-Kickback Statute safe harbor for certain retrospective price reductions provided by drug manufacturers to sponsors of Medicare Part D plans or pharmacy benefit managers that are not applied at the point-of-sale; (2) allow the importation of certain drugs from other countries through individual waivers, permit the re-importation of insulin products, and prioritize finalization of FDA’s December 2019 proposed rule to permit the importation of drugs from Canada; (3) ensure that payment by the Medicare program for certain Medicare Part B drugs is not higher than the payment by other comparable countries (depending on whether pharmaceutical manufacturers agree to other measures); and (4) allow certain low-income individuals receiving insulin and epinephrine purchased by a Federally Qualified Health Center, or FQHC, as part of the 340B drug program to purchase those drugs at the discounted price paid by the FQHC. On August 6, 2020, President Trump signed an additional Executive Order directing U.S. government agencies to encourage the domestic procurement of Essential Medicines, Medical Countermeasures, and
33
Critical Inputs, which include among other things, active pharmaceutical ingredients and drugs intended for use in the diagnosis, cure, mitigation, treatment, or prevention of COVID-19. FDA must release a full list of Essential Medicines, Medical Countermeasures, and Critical Inputs affected by this Order by November 5, 2020. Although a number of these, and other proposed measures will require authorization through additional legislation to become effective, Congress and the current administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Additionally, on May 30, 2018, the Right to Try Act was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase I clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a drug manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act.
Other U.S. Healthcare Laws and Compliance Requirements
Manufacturing, sales, promotion, and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, CMS, other divisions of HHS including the Office of the Inspector General, the U.S. Department of Justice, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local regulatory authorities. For example, sales, marketing and scientific/educational grant programs may have to comply with state and federal fraud and abuse laws, the privacy and security provisions of the Health Insurance Portability and Accountability Act of 1996, or HIPAA, and similar state laws, each as amended.
The federal Anti-Kickback Statute, makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration (including any kickback, bribe, or rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce, or in return for, the referral of an individual, or the purchase, lease, order, or recommendation of any good, facility, item, or service (including the purchase, order, or prescription of a particular drug), for which payment may be made, in whole or in part, under a federal healthcare program, such as Medicare or Medicaid. “Remuneration” has been interpreted broadly to include anything of value. Violations of this law are punishable by up to five years in prison, criminal fines, administrative civil money penalties and exclusion from participation in federal healthcare programs. In addition, the ACA, among other things, amended the intent requirement of the federal Anti-Kickback Statute. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. Further, courts have found that if “one purpose” of renumeration is to induce referrals, the federal Anti-Kickback statute is violated. Moreover, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. There are a number of statutory exceptions and regulatory safe harbors protecting some common activities.
Although Yumanity would not submit claims directly to payors, drug manufacturers can be held liable under the federal civil and criminal false claims laws and civil monetary penalty laws, including the False Claims Act, which prohibit, among other things, anyone from knowingly presenting, or causing to be presented, for payment to or approval by federal programs (including Medicare and Medicaid) claims for items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services, knowingly concealing or knowingly and improperly avoiding or decreasing or concealing an obligation to pay money to the federal government. The government may deem manufacturers to have “caused” the submission of false or fraudulent claims by, for example, providing inaccurate billing or coding information to customers or promoting a product off-label. In addition, Yumanity’s activities relating to the reporting of wholesaler or estimated retail prices for its products, the reporting of prices used to calculate Medicaid rebate information and other information affecting federal, state and third-party reimbursement for Yumanity’s products, and the sale and
34
marketing of its products, are subject to scrutiny under this law. Penalties for a False Claims Act violation include three times the actual damages sustained by the government, plus mandatory civil penalties for each separate false claim, the potential for exclusion from participation in federal healthcare programs, and, although the federal False Claims Act is a civil statute, conduct that results in a False Claims Act violation may also implicate various federal criminal statutes. If the government were to allege that Yumanity were, or convict Yumanity of, violating these false claims laws, Yumanity could be subject to a substantial fine and may suffer a decline in its stock price. In addition, private individuals have the ability to bring actions under the federal False Claims Act and certain states have enacted laws modeled after the federal False Claims Act.
HIPAA created new federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private), willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the federal Anti-Kickback Statute a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under Medicaid and other state programs. Additionally, to the extent that any of Yumanity’s product candidates, if approved, are sold in a foreign country, Yumanity may be subject to similar foreign laws.
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, impose requirements relating to the privacy, security and transmission of individually identifiable health information on certain healthcare providers, healthcare clearinghouses, and health plans, known as covered entities, as well as independent contractors, or agents of covered entities that create, receive or obtain individually identifiable health information in connection with providing a service on behalf of a covered entity, known as a business associates. Among other things, HITECH makes HIPAA’s security standards directly applicable to business associates, defined as independent contractors or agents of covered entities that create, receive or obtain protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities and business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and criminal penalties.
Other regulations may affect other aspects of Yumanity’s business. For example, pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in ACA. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws. There has also been a recent trend of increased federal and state regulation of payments made to physicians. Certain states mandate implementation of compliance programs, impose restrictions on drug manufacturers’ marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians. For example, the federal Physician Payment Sunshine Act, or Sunshine Act, created under the ACA, requires manufacturers of drugs, devices, biologicals and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to HHS information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and transfers of value provided, as well as ownership and
35
investment interests held, during the previous year to certain other healthcare professionals, including physician assistants and nurse practitioners. As noted, many states also govern the reporting of payments or other transfers of value, many of which differ from each other in significant ways, are often not pre-empted, and may have a more prohibitive effect than the Sunshine Act, thus further complicating compliance efforts.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. If Yumanity’s operations are found to be in violation of any of such laws or any other governmental regulations that apply to Yumanity, Yumanity may be subject to penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of Yumanity’s operations, exclusion from participation in federal and state healthcare programs and individual imprisonment, any of which could adversely affect Yumanity’s ability to operate its business and its financial results. Any action against Yumanity for violation of these laws, even if Yumanity successfully defend against it, could cause Yumanity to incur significant legal expenses and divert its management’s attention from the operation of its business.
Coverage and Reimbursement
Sales of Yumanity’s drugs will depend, in part, on the extent to which Yumanity’s drugs will be covered by third-party payors, such as government health programs, commercial insurance, and managed healthcare organizations. Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we may obtain regulatory approval. Coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid in the United States, and commercial payors are critical to new product acceptance. Third-party payors decide which drugs they will pay for and establish reimbursement levels. In the United States, the principal decisions about reimbursement for new medicines are typically made by CMS. CMS decides whether and to what extent our products will be covered and reimbursed under Medicare and private payors tend to follow CMS to a substantial degree. Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a therapeutic is:
| • | a covered benefit under its health plan; |
| • | safe, effective and medically necessary; |
| • | appropriate for the specific patient; |
| • | cost-effective; and |
| • | neither experimental nor investigational. |
We cannot be sure that reimbursement will be available for any product that we commercialize and, if coverage and reimbursement are available, we cannot be sure that the level of reimbursement will be adequate. Coverage may also be more limited than the purposes for which the product is approved by the FDA or comparable foreign regulatory authorities. Limited coverage and less than adequate reimbursement may reduce the demand for, or the price of, any product for which we obtain regulatory approval.
These third-party payors are increasingly reducing reimbursements for medical drugs and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures, and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic drugs. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit Yumanity’s net revenue and results. Decreases in third-party reimbursement for Yumanity’s drug candidates, if approved, or a decision by a third-party payor to not cover Yumanity’s drug candidates could reduce physician usage of such drugs and have a material adverse effect on Yumanity’s sales, results of operations and financial condition.
36
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for drugs for which Yumanity obtains marketing approval. However, any negotiated prices for Yumanity’s drugs covered by a Part D prescription drug plan will likely be lower than the prices Yumanity might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal drugs for which their national health insurance systems provide reimbursement and to control the prices of medicinal drugs for human use. A member state may approve a specific price for the medicinal drug or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal drug on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical drugs will allow favorable reimbursement and pricing arrangements for any of Yumanity’s drugs. Historically, drugs launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
European Union Drug Development
In the European Union, Yumanity’s product candidates may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of nonclinical and clinical research in the European Union are subject to significant regulatory controls. Although the EU Clinical Trials Directive 2001/20/EC has sought to harmonize the European Union clinical trials regulatory framework, setting out common rules for the control and authorization of clinical trials in the European Union, the European Union Member States have transposed and applied the provisions of the Directive differently. This has led to significant variations in the Member State regimes. Under the current regime, before a clinical trial can be initiated it must be approved in each of the European Union Member States where the study is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one or more Ethics Committees, or ECs. Under the current regime all suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred.
In April 2014, the European Union adopted a new Clinical Trials Regulation (EU) No 536/2014, which is set to replace the current EU Clinical Trials Directive 2001/20/EC. It is expected that the new Clinical Trials Regulation (EU) No 536/2014 will apply following confirmation of full functionality of the Clinical Trials Information System, or CTIS, the centralized European Union portal and database for clinical trials foreseen by the regulation, through an independent audit. The regulation becomes applicable six months after the European Commission publishes notice of this confirmation with a three-year transition period. It will overhaul the current system of approvals for clinical trials in the European Union. Specifically, the new regulation, which will be directly applicable in all member states, aims at simplifying and streamlining the approval of clinical trials in the European Union. For instance, the new Clinical Trials Regulation provides for a streamlined application procedure via a single point and strictly defined deadlines for the assessment of clinical trial applications.
37
European Union Drug Review and Approval
In the European Economic Area, or EEA, comprising the 27 Member States of the European Union plus Norway, Iceland and Liechtenstein, medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. There are two types of marketing authorizations: The Community MA is issued by the European Commission through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the European Medicines Agency, or EMA, and is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as human medicines derived from biotechnology processes or advanced therapy medicinal products (such as gene therapy, somatic cell therapy and tissue engineered products), products that contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, neurodegenerative disorders, diabetes, autoimmune diseases and other immune dysfunctions, viral diseases, and officially designated orphan medicines. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European Union.
National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in another Member State through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. Under the Decentralized Procedure an identical dossier is submitted to the competent authorities of each of the Member States in which the MA is sought, one of which is selected by the applicant as the Reference Member State, or RMS. The competent authority of the RMS prepares a draft assessment report, a draft summary of the product characteristics, or SPC, and a draft of the labeling and package leaflet, which are sent to the other Member States (referred to as the Member States Concerned) for their approval. If the Member States Concerned raise no objections, based on a potential serious risk to public health, to the assessment, SPC, labeling, or packaging proposed by the RMS, the product is subsequently granted a national MA in all the Member States (i.e., in the RMS and the Member States Concerned).
Under the above described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
European Union New Chemical Entity Exclusivity
In the European Union, new chemical entities, sometimes referred to as new active substances, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. The data exclusivity, if granted, prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic application for eight years, after which generic marketing authorization can be submitted, and the innovator’s data may be referenced, but not approved for two years. The overall ten year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are determined to bring a significant clinical benefit in comparison with currently approved therapies.
European Union Orphan Designation and Exclusivity
In the European Union, the European Commission, based on the recommendation of the EMA’s Committee for Orphan Medicinal Products, grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the European Union (or where it is unlikely that the development of the medicine would generate sufficient return to justify the investment) and for which no satisfactory method of diagnosis, prevention or treatment has been authorized (or, if a method exists, the product would be a significant benefit to those affected).
38
In the European Union, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following medicinal product approval. This period is extended by two years for compliance with an agreed upon pediatric investigation plan granted at the time of review of the orphan drug designation. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time, if (i) the holder of the marketing authorization for the original orphan medicinal product consents to a second orphan medicinal product application, (ii) the holder of the marketing authorization for the original orphan medicinal product cannot supply sufficient quantities of the orphan medicinal product, or (iii) the second applicant can establish that the second medicinal product, although similar, is safer, more effective or otherwise clinically superior to the authorized orphan medicinal product. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Brexit and the Regulatory Framework in the United Kingdom
In June 2016, the electorate in the United Kingdom voted in favor of leaving the European Union (commonly referred to as “Brexit”). Thereafter, in March 2017, the country formally notified the European Union of its intention to withdraw pursuant to Article 50 of the Lisbon Treaty. The United Kingdom formally left the European Union on January 31, 2020. A transition period began on February 1, 2020, during which European Union pharmaceutical law remains applicable to the United Kingdom. This transition period is due to end on December 31, 2020. Since the regulatory framework for pharmaceutical products in the United Kingdom covering quality, safety and efficacy of pharmaceutical products, clinical trials, marketing authorization, commercial sales and distribution of pharmaceutical products is derived from European Union directives and regulations, Brexit could materially impact the future regulatory regime which applies to products and the approval of product candidates in the United Kingdom. It remains to be seen how, if at all, Brexit will impact regulatory requirements for product candidates and products in the United Kingdom.
European Union Data Collection
The collection and use of personal health data in the European Economic Area (EEA) is governed by the EU General Data Protection Regulation 2016/679, or GDPR, which went into effect on May 25, 2018 and superseded the Data Protection Directive. The GDPR applies to any company established in the EEA and to companies established outside the EEA that provides goods or services to residents in the EEA. This would include companies that process personal data in connection with the offering of goods or services to data subjects in the EU or the monitoring of the behavior of data subjects in the EU. The GDPR enhances data protection obligations for data controllers of personal data (including stringent requirements relating to the consent of data subjects, expanded disclosures about how personal data is used, requirements to conduct privacy impact assessments for “high risk” processing, limitations on retention of personal data, mandatory data breach notification and “privacy by design” requirements), creates direct obligations on service providers acting as data processors, and imposes special protections for “sensitive information,” which includes health and genetic information of data subjects residing in the EU. The GDPR grants individuals the opportunity to object to the processing of their personal information, and allows them to request deletion of personal information in certain circumstances. Additionally, the GDPR also imposes strict rules on the transfer of personal data outside of the EEA to countries that do not ensure an adequate level of protection, like the U.S. Failure to comply with the requirements of the GDPR and the related national data protection laws of the EEA Member States may result in fines of up to 20 million Euros or 4% of a company’s global annual revenues for the preceding financial year, whichever is higher. Moreover, the GDPR grants data subjects the right to claim material and non-material damages resulting from infringement of the GDPR. Given the breadth and depth of changes in data protection obligations, maintaining compliance with the GDPR will require significant time, resources and expense, and Yumanity may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. This may be onerous and adversely affect Yumanity’s business, financial condition, results of operations and prospects. Further, the United Kingdom’s decision to leave the European Union has created uncertainty with regard to data protection regulation in the United Kingdom. In particular, it is unclear how data transfers to and from the United Kingdom will be regulated now that the United Kingdom has left the European Union.
39
Rest of the World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki. If Yumanity fails to comply with applicable foreign regulatory requirements, Yumanity may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Employees and Human Capital Resources
As of September 1, 2020, Yumanity employed 41 full-time employees, including 30 in research, 2 in clinical development and 9 in general and administrative, and 3 part-time employees. Twenty of Yumanity’s full-time employees hold M.D. or Ph.D. degrees. Yumanity is highly dependent on its management and scientific and medical personnel, and it is crucial that it continues to attract and retain valuable employees. To facilitate attraction and retention, Yumanity strives to make Yumanity a diverse, inclusive and safe workplace, with opportunities for its employees to grow and develop in their careers, supported by strong compensation and benefits programs. Yumanity has never had a work stoppage, and none of its employees is represented by a labor organization or under any collective-bargaining arrangements. Yumanity considers its relationship with its employees to be good.
Facilities
Yumanity currently has one location in Boston, Massachusetts. Yumanity licenses approximately 27,000 square feet of office and laboratory space from Smart Labs located at 40 Guest Street, Suite 4410, Boston, Massachusetts. Yumanity’s lease on its corporate headquarters expires in April 2023. Yumanity believes its facilities are adequate for its current needs and that it will be able to find suitable additional substitute space when needed.
Legal Proceedings
Between October 14 and December 7, 2020, following the announcement of the proposed merger among Proteostasis Therapeutics, Inc. (“PTI”), Yumanity, Inc. (f/k/a Yumanity Therapeutics, Inc) and Pangolin Merger Sub, Inc. (“Merger Sub”), a wholly owned subsidiary of PTI (the “Merger”), nine lawsuits were filed by purported stockholders of Proteostasis Therapeutics, Inc., challenging the Merger. The first lawsuit, brought as a putative class action, is captioned Aniello v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-08578 (S.D.N.Y. filed Oct. 14, 2020). The remaining eight lawsuits, brought by the plaintiffs individually, are captioned Culver v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-08595 (S.D.N.Y. filed Oct. 15, 2020); Donolo v. Proteostasis Therapeutics, Inc. et al., 1:20-cv-01400 (D. Del. filed Oct. 16, 2020); Straube v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-08653 (S.D.N.Y. filed Oct. 16, 2020); Beck v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-08783 (S.D.N.Y. filed Oct. 21, 2020); Dreyer v. Proteostasis Therapeutics, Inc., et al., 1:20-cv-05193 (E.D.N.Y. filed Oct. 28, 2020); Kopkin v. Proteostasis Therapeutics, Inc. et al., No. 1:20-cv-12103 (D. Mass. filed Nov. 23, 2020); Merritt v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-10275 (S.D.N.Y. filed Dec. 6, 2020); and Koh v. Proteostasis Therapeutics, Inc., et al., No. 1:20-cv-10296 (S.D.N.Y. filed Dec. 7, 2020). All of the complaints named PTI and the individual members of PTI’s board of directors as defendants. The Aniello complaint also named Yumanity, Inc. as an additional defendant, and the Donolo complaint named Yumanity. Inc. and Merger Sub as additional defendants. The complaints asserted violations of Section 14(a) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and Rule 14a-9 promulgated thereunder against PTI and its directors, and violations of Section 20(a) of the Exchange Act against PTI’s directors. The Donolo complaint asserted an additional violation of Section 20(a) of the Exchange Act against Yumanity, Inc. The Aniello complaint asserted additional claims for breach of fiduciary duty against PTI’s directors and aiding and abetting against PTI and Yumanity, Inc. The plaintiffs contended that the registration statement on Form S-4 filed by PTI with the Securities and Exchange Commission on September 23, 2020 (the “Registration Statement”) or the proxy statement/prospectus on Form 424B3 filed by PTI with the SEC on November 12, 2020 (the “Definitive Proxy”) omitted or misrepresented certain material information regarding the Merger. The complaints sought injunctive relief, rescission, or rescissory damages, dissemination certain information requested by the plaintiffs, and an award of plaintiffs’ costs, including attorneys’ fees and expenses. While PTI and Yumanity, Inc. believed that the disclosures set forth in the Registration Statement and Definitive Proxy complied fully with all applicable law and denied the allegations in the pending actions described
40
above, in order to moot plaintiffs’ disclosure claims, avoid nuisance and possible expense and business delays, and provide additional information to its stockholders, on December 9, 2020, PTI filed a Form 8-K voluntarily to supplement certain disclosures in the Definitive Proxy related to plaintiffs’ claims with the supplemental disclosures (the “Supplemental Disclosures”). Following the filing of the Supplemental Disclosures, all of the actions discussed above were voluntarily dismissed by the respective plaintiffs, with the exception of the Merritt action, in which the defendants have not yet been served with the complaint.
41