Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - Novan, Inc. | a12312019-10kaexhibit322.htm |
| EX-32.1 - EXHIBIT 32.1 - Novan, Inc. | a12312019-10kaexhibit321.htm |
| EX-31.2 - EXHIBIT 31.2 - Novan, Inc. | a12312019-10kaexhibit312.htm |
| EX-31.1 - EXHIBIT 31.1 - Novan, Inc. | a12312019-10kaexhibit311.htm |
| EX-23.1 - EXHIBIT 23.1 - Novan, Inc. | a12312019-10kaexhibit231.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
___________________________________________________________________
FORM 10-K/A
(Amendment No. 1)
___________________________________________________________________
(Mark One)
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2019
OR
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 For the transition period from to |
Commission file number 001-37880
Novan, Inc.
(Exact name of registrant as specified in its charter)
___________________________________________________________________
Delaware | 20-4427682 |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
4105 Hopson Road Morrisville, North Carolina | 27560 |
(Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code: (919) 485-8080
Securities registered pursuant to Section 12(b) of the Act:
Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | |
Common Stock, $0.0001 par value | NOVN | Nasdaq Global Market | |
Securities registered pursuant to Section 12(g) of the Act: None
___________________________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ý No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ¨ | Accelerated filer | ¨ | |||
Non-accelerated filer | ý | Smaller reporting company | ý | |||
Emerging growth company | ý | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ý | ||||||
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No ý
As of June 28, 2019, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of common stock held by non-affiliates of the registrant was approximately $57.0 million (based on a closing price of $2.70 per share as reported by the Nasdaq Global Market on June 28, 2019). For purposes of this calculation, shares of common stock beneficially owned by the registrant’s officers, directors and certain stockholders as of June 30, 2019 have been excluded in that such persons may be deemed to be affiliates. This determination of affiliate status is not necessarily a conclusive determination for other purposes. The registrant has no non-voting common equity.
The number of shares of registrant’s common stock outstanding as of May 14, 2020 was 75,471,388.
Explanatory Note
Novan, Inc. (together with its subsidiaries, “we”, “our” or “us”) is filing this Annual Report on Form 10-K/A (Amendment No. 1), or this Annual Report, to amend our Annual Report on Form 10-K for the year ended December 31, 2019, originally filed with the Securities and Exchange Commission, or the SEC, on February 24, 2020, or the Original Filing, to restate our consolidated financial statements for the year ended December 31, 2018. We are also restating the condensed consolidated financial statements for the interim periods set forth below through expanded disclosure of our interim consolidated financial information in the accompanying consolidated financial statements included in this Annual Report, including describing the restatement and its impact on previously reported amounts.
We believe that presenting all of the restated information for the year ended December 31, 2018 and the interim periods ended March 31, 2019 and 2018, June 30, 2019 and 2018, and September 30, 2019 and 2018, or collectively the Affected Periods, in this Annual Report allows investors and others to review all pertinent data in a single presentation.
Background of Restatement
On May 14, 2020, the audit committee of our board of directors, or our audit committee, in consultation with management and BDO USA, LLP, concluded that, because of a misapplication of the accounting guidance applicable to the warrants we issued in January 2018, our previously issued consolidated financial statements for the Affected Periods should no longer be relied upon. As such, we determined that we would restate our consolidated financial statements for each of the Affected Periods. In addition, although we have determined the misapplication to be immaterial to our consolidated financial statements for the year ended December 31, 2019, we have also revised these financial statements in connection with the restatement of our consolidated financial statements for the Affected Periods.
As described more fully in “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements included in this Annual Report, the warrants we issued in January 2018 provide the warrant holders the option to receive a cash settlement, equal to the Black-Scholes value of the remaining unexercised portion of the warrant, in certain specified situations involving a “fundamental transaction” (as defined in the warrants). Due to the potential for cash settlement of the warrants, we have historically classified the warrants as liabilities in accordance with Financial Accounting Standards Board, or FASB, Accounting Standards Codification, or ASC, 480, Distinguishing Liabilities from Equity, or ASC 480. Upon further analysis of the terms of the warrants, including consideration of whether the specified fundamental transactions that permit net cash settlement are within or outside of the control of the Company, we have determined that the warrants meet the criteria for equity classification under ASC 480 and FASB ASC 815, Derivatives and Hedging, or ASC 815, and should be classified within stockholders’ equity (deficit).
Effect of Restatement and Revision
As described above, as a result of the misapplication of the accounting guidance applicable to the warrants, we are including in this Annual Report (i) restated consolidated financial statements for the year ended December 31, 2018, (ii) revised consolidated financial statements for the year ended December 31, 2019, and (iii) restated interim consolidated financial information for each interim period within the Affected Periods through expanded disclosure of interim consolidated financial information in “Note 2—Restatement of Consolidated Financial Statements” and “Note 17—Quarterly Financial Information (Unaudited)” to the accompanying consolidated financial statements included in this Annual Report. The cumulative effect of the change in the accounting treatment of the warrants and the resulting restatement and revision of our consolidated financial statements is an 8.0% increase in our accumulated deficit of approximately $16.3 million as of December 31, 2019.
The change in the accounting treatment for the warrants and the resulting restatement and revision of our consolidated financial statements include (i) the reclassification of the initial warrant fair value from warrant liability to additional paid-in capital within our consolidated balance sheets and (ii) the adjustment of previously reported non-cash changes in fair value of the warrant liability in our consolidated statements of operations and corresponding adjustments to accumulated deficit for each applicable period. There was no impact on revenues, operating expenses or operating loss for any period as the change in fair value of the warrant liability was presented within other income (expense) and not as a component of operating loss in our consolidated statements of operations for each applicable period. The restatement of the consolidated financial statements for the Affected Periods and the revision of the consolidated financial statements for the year ended December 31, 2019 had no impact on our liquidity or cash position. An explanation of the impact on our consolidated financial statements is contained in “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements included in this Annual Report.
2
As all material restatement information will be included in this Annual Report, we do not intend to amend our Annual Report on Form 10-K for the year ended December 31, 2018 or any of our previously filed Quarterly Reports on Form 10-Q. Accordingly, investors and others should rely only on the financial information and other disclosures regarding the periods described above in this Annual Report and in future filings with the SEC (as applicable) and should not rely on any previously issued or filed reports, press releases, corporate presentations or similar communications relating to the Affected Periods.
Internal Control Considerations
We have concluded that the errors in our accounting for the warrants resulted from a material weakness in our internal control over financial reporting that existed during and as of the end of each Affected Period and as of and for the year ended December 31, 2019, and which continues to exist. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim consolidated financial statements will not be prevented or detected on a timely basis. For a discussion of management’s consideration of the material weakness identified, see Part II, Item 9A: Controls and Procedures included in this Annual Report.
Items Amended in this Annual Report on Form 10-K/A
The following items of this Annual Report include restated or revised financial data: (i) Part II, Item 7: Management’s Discussion and Analysis of Financial Condition and Results of Operations and (ii) Part II, Item 8: Financial Statements. The following items of this Annual Report also include amendments due to the restatement: (i) Part I, Item 1A: Risk Factors and (ii) Part II, Item 9A: Controls and Procedures.
Our principal executive officer and principal financial officer are providing currently dated certifications in connection with this Annual Report. These certifications are filed as Exhibits 31.1, 31.2, 32.1 and 32.2.
Except for the foregoing amended and restated information required to reflect the effects of the restatement of the consolidated financial statements for the Affected Periods and the revised consolidated financial statements for the year ended December 31, 2019, and applicable cross-references within this Annual Report, no other changes have been made to the Original Filing. This Annual Report continues to describe conditions as of the date of the Original Filing, and the disclosures herein have not been updated to reflect events, results or developments that have occurred after the date of the Original Filing, or to modify or update those disclosures affected by subsequent events. Accordingly, forward looking statements included in this Annual Report represent management’s views as of the date of the Original Filing and should not be assumed to be accurate as of any date thereafter. This Annual Report should be read in conjunction with our filings made with the SEC subsequent to the Original Filing date.
3
Table of Contents
Page | ||
PART I | ||
Item 1. | ||
Item 1A. | ||
Item 1B. | ||
Item 2. | ||
Item 3. | ||
Item 4. | ||
PART II | ||
Item 5. | ||
Item 6. | ||
Item 7. | ||
Item 7A. | ||
Item 8. | ||
Item 9. | ||
Item 9A. | ||
Item 9B. | ||
PART III | ||
Item 10. | ||
Item 11. | ||
Item 12. | ||
Item 13. | ||
Item 14. | ||
PART IV | ||
Item 15. | ||
Item 16. | ||
4
SPECIAL NOTE ABOUT FORWARD-LOOKING STATEMENTS
This Annual Report (Amendment No. 1) on Form 10-K/A, or this Annual Report, contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Act of 1934, as amended, or the Exchange Act, that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this Annual Report are forward-looking statements. These statements are often identified by the use of words such as “may,” “will,” “expect,” “believe,” “anticipate,” “intend,” “could,” “should,” “potential,” “predict,” “project,” “estimate,” or “continue” and similar expressions or variations.
These statements are based on the beliefs and assumptions of our management based on information currently available to management. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Factors that could cause or contribute to these differences include those set forth in the “Risk Factors” section of this Annual Report.
You should not rely upon forward-looking statements as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Such forward-looking statements speak only as of the date of this Annual Report. Except as may be required by law, we undertake no obligation to update any forward-looking statements to reflect events or circumstances after the date of such statements.
5
PART I
Item 1. Business.
Overview
We are a clinical development-stage biotechnology company focused on leveraging nitric oxide’s naturally occurring anti-viral, anti-bacterial, anti-fungal and immunomodulatory mechanisms of action to treat a range of diseases with significant unmet needs. Nitric oxide plays a vital role in the natural immune system response against microbial pathogens and is a critical regulator of inflammation. Our ability to harness nitric oxide and its multiple mechanisms of action has enabled us to create a platform with the potential to generate differentiated product candidates.
The two key components of our nitric oxide platform are our proprietary Nitricil technology, which drives the creation of new chemical entities, or NCEs, and our formulation science, both of which we use to tune our product candidates for specific indications. Our ability to deploy nitric oxide in a solid form, on demand and in localized formulations allows us the potential to improve patient outcomes in a variety of diseases.
We have advanced strategic development programs in the field of dermatology, while also further expanding the platform into women’s health and gastroenterological, or GI, therapeutic areas. This decision was based on the connection between the multi-factorial pathologies of diseases in these areas and the demonstrable anti-microbial, anti-viral and anti-inflammatory properties of Novan’s nitric oxide technology. Our goal is to create the world’s leading macro-molecular nitric oxide-based science, technology, and clinical translation company that delivers safe and efficacious therapies for patients.
We have clinical-stage dermatology drug candidates with multi-factorial (SB204), anti-viral (SB206), anti-fungal (SB208) and anti-inflammatory (SB414) mechanisms of action. We have recently introduced SB207 as a possible product candidate for additional anti-viral programs. During 2019, our clinical-stage development efforts were focused on our molluscum contagiosum (SB206) and atopic dermatitis (SB414) programs. We also conducted preclinical work on NCEs and formulations for the treatment of human papilloma virus, or HPV, related illnesses in the women’s health field (WH504 and WH602) and inflammatory diseases in the GI field. During 2020, we intend to focus our clinical-stage development efforts on our molluscum contagiosum (SB206) program, subject to available capital and regulatory feedback. All other clinical-stage programs are currently on hold.
Further advancement of the molluscum contagiosum (SB206) program, or any other program across our platform, is dependent upon our ability to access additional capital. Additional capital may potentially include (i) non-dilutive sources, such as partnerships, collaborations, licensing, grants or other strategic relationships; or (ii) equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. We intend to pursue financing which may be dilutive, non-dilutive or both, in the near future. We intend to utilize one or more financial advisors to assist in the pursuit of optimal capital sourcing pathways, including those that are strategic in nature and center around our late-stage assets, and the broader dermatology platform and underlying Nitricil technology, as well as exploring other potential sources of financing and strategic alternatives.
Current Financial Status
As of December 31, 2019, we had a total cash and cash equivalents balance of $13.7 million and positive working capital of $2.8 million. We believe that our existing cash and cash equivalents balance will provide us with adequate liquidity to fund our planned operating needs into the early part of the second quarter of 2020. This projected cash runway excludes (i) potential costs associated with an additional confirmatory Phase 3 trial, which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (ii) any proceeds received subsequent to January 31, 2020 from potential future sales of common stock under the Aspire Common Stock Purchase Agreement, described below, if available.
We will need substantial additional funding to continue our operating activities and make further advancements in our drug development programs, as described in the section entitled “Our Product Candidates” below and in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Overview” of this Annual Report. Therefore, we will need to secure additional capital or financing and/or delay, defer, or reduce our cash expenditures by the early part of the second quarter of 2020, including those associated with our product development programs, or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. There can be no assurance that we will be able to obtain additional capital or financing on terms acceptable to us, on a timely basis or at all. If we are forced to terminate or eliminate our product development programs, wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders. Alternatively, we may seek to engage in one or more
6
potential transactions, such as the sale of the Company, or sale or divestiture of some of our assets, such as a sale of our dermatology platform assets, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us.
Please refer to “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” for further discussion of our current liquidity and our future funding needs.
Common Stock Purchase Agreement and Registration Rights Agreement with Aspire Capital
On August 30, 2019, we entered into a common stock purchase agreement, or the Aspire Common Stock Purchase Agreement with Aspire Capital Fund, LLC, or Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, partially described below and also explained in detail within “Note 10—Stockholders’ Equity (Deficit)” to the accompanying consolidated financial statements included in this Annual Report, Aspire Capital is committed to purchase up to a maximum of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement. The aggregate amount that we may raise through sales of common stock under the Aspire Common Stock Purchase Agreement is subject to certain limitations including, but not limited to: (i) the number of shares that may be sold will be limited to 5,211,339 shares, representing 19.99% of our outstanding shares of common stock on August 30, 2019, if the average price paid for all shares issued under the agreement is less than $2.17; and (ii) on any purchase date, the closing sale price of our common stock must be greater than or equal to $0.25. As of December 31, 2019, we had sold an aggregate of 300,000 shares of common stock at an average price of $2.49 per share under the Aspire Common Stock Purchase Agreement. As of January 31, 2020, we had sold an aggregate of 1,000,000 shares of common stock at an average price of $1.19 per share under the Aspire Common Stock Purchase Agreement. These amounts, combined with the 345,622 shares issued as part of the commitment fee related to the agreement’s execution, leads to a total of 1,345,622 shares to Aspire Capital under the agreement as of January 31, 2020.
Concurrently with entering into the Aspire Common Stock Purchase Agreement, we also entered into a registration rights agreement with Aspire Capital, or the Registration Rights Agreement, in which we agreed to file one or more registration statements, as permissible and necessary to register under the Securities Act, registering the sale of the shares of our common stock that have been and may be issued to Aspire Capital under the Aspire Common Stock Purchase Agreement. On September 16, 2019, we filed with the SEC a prospectus to our effective Registration Statement on Form S-1 (File No. 333-233632) registering 7,032,630 shares of common stock that have been and may be offered to Aspire Capital from time to time under the Aspire Common Stock Purchase Agreement.
There are no trading volume requirements or restrictions under the Aspire Common Stock Purchase Agreement, and we will control the timing and amount of sales of our common stock to Aspire Capital. Aspire Capital has no right to require any sales by us, but is obligated to make purchases from us as we may direct in accordance with the Aspire Common Stock Purchase Agreement. There are no limitations on use of proceeds, financial or business covenants, restrictions on future financing transactions, rights of first refusal, participation rights, penalties or liquidated damages in the Aspire Common Stock Purchase Agreement. In consideration for entering into the Aspire Common Stock Purchase Agreement, concurrently with the execution of the Aspire Common Stock Purchase Agreement, we issued to Aspire Capital 345,622 shares of our common stock as part of the commitment fee. The Aspire Common Stock Purchase Agreement may be terminated by us any time, at our discretion, without any penalty or additional cost to us. Any proceeds we receive under the Aspire Common Stock Purchase Agreement are expected to be used for working capital and general corporate purposes.
Please refer to “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” for further discussion of our current liquidity and our future funding needs, including the Aspire Common Stock Purchase Agreement and the Registration Rights Agreement.
The Novan Nitric Oxide Platform
Nitric oxide is one of the most researched molecules in human physiology and has been extensively studied in many areas of medicine including in microbial diseases and in the modulation of inflammation. The scarcity of nitric oxide-based therapeutic products is due to the challenges associated with controlling the release of a gas, the poor stability and low storage capacity of nitric oxide-loaded molecules, the inability to target specific tissues and the toxicity of several small molecules used as carriers to store nitric oxide.
The two key components of our nitric oxide platform are our proprietary Nitricil technology, which drives the creation of NCEs, and our formulation science, both of which we use to tune our product candidates for specific indications:
7
(1) | Novan’s Nitricil technology enables us to store large amounts of nitric oxide gas in a stable, solid form by chemically loading it on a macromolecule, or polymer. The advantages of our proprietary Nitricil technology include tunability, stability, high storage capacity, targeted delivery and what we believe is an attractive safety profile. Our ability to select from several nitric oxide-loaded materials has created our proprietary library of Nitricil compositions, each of which possesses a unique nitric oxide release profile. |
(2) | Our formulation science and expertise allow us to customize the drug delivery method for the relevant anatomical location of a variety of diseases. With our dermatological indications, the topical semi-solid formulations enable us to further tune the release of nitric oxide when applied by using proprietary combinations of inactive ingredients. This additional level of control enables us to use one NCE for multiple indications by altering the nitric oxide pharmacology with the composition of the topical formulation. This component of our nitric oxide platform creates an additional barrier to entry, which we believe positions us to prolong the period of market exclusivity for each of our product candidates. |
We believe that our ability to deploy nitric oxide in a solid form, on demand and in localized formulations allows us the potential to improve patient outcomes in a variety of dermatology, women’s health and GI diseases.
At present, our nitric oxide platform has produced a portfolio that includes the following clinical stage dermatology product candidates.
• | SB204 is a once-daily, topical monotherapy for the treatment of acne vulgaris, a multi-factorial disease with multiple aspects of the disease pathology (anti-inflammatory and anti-bacterial) potentially treatable with SB204. |
• | SB206 is a topical anti-viral gel for the treatment of viral skin infections, with a current focus on the treatment of molluscum contagiosum, a contagious skin infection caused by the molluscipoxvirus, and external genital and perianal warts caused by human papillomavirus, or HPV. |
• | SB208 is a topical broad-spectrum anti-fungal gel for the treatment of fungal infections of the skin and nails, including athlete’s foot (tinea pedis) and fungal nail infections (onychomycosis). |
• | SB414 is a topical cream-based product candidate for the treatment of inflammatory skin diseases, with a current focus on the treatment of atopic dermatitis and psoriasis. |
We presently maintain exclusive, worldwide commercial rights for all product candidates currently in our pipeline, with the exception of the rights we licensed to Sato Pharmaceutical Co., Ltd., or Sato, in January 2017 and October 2018 to develop, use and sell SB204 and SB206 in certain topical dosage forms in Japan for the treatment of acne vulgaris and viral skin infections, respectively, and to manufacture the finished form of such products for sale in Japan.
Nitric Oxide Background
Nitric oxide, or NO, is a two-atom molecule that is produced naturally by the human body. Since the Nobel Prize-winning discovery in 1998 that nitric oxide is responsible for regulating blood flow, or vasodilation, the effects of nitric oxide have been extensively studied in many areas of physiology.
As a fundamental component in host defense against invading organisms, cells of the immune system naturally generate nitric oxide using the enzyme nitric oxide synthase, or NOS, and the amino acid precursor L-arginine. Nitric oxide is released in a targeted manner to kill microbial pathogens, including bacteria, fungi and viruses. Nitric oxide and its metabolites drive cell death within bacteria and fungi by targeting metal centers or amino acids on proteins critical to sustaining microbial viability. In virally infected cells, nitric oxide inhibits viral replication by binding directly to free sulfurs or metals that are a part of key enzymes that can induce apoptosis, or programmed cell death, in cells where tumor suppressors have been degraded or disabled.
We believe that nitric oxide has potential to be a novel anti-microbial agent due to its multiple mechanisms of action and its ability as a gas to diffuse freely through cell membranes – unlike most other pharmaceutical agents. Importantly, the pharmacologic activity of nitric oxide is such that its production is localized at or near the site of infection. Because nitric oxide is a key component of the immune system’s natural response to invading organisms, it may provide a therapeutic solution for degrading and killing microorganisms without the development of anti-microbial resistance.
Nitric oxide and its multiple mechanisms of action have wide ranging possibilities to treat human disease. We believe that our expertise at developing nitric oxide NCEs and fine tuning the formulation technology to the targeted disease separates us from other drug development companies focused in this space. Nitric oxide is a naturally occurring chemical in the human body,
8
which enhances its safety profile. The proven anti-microbial and anti-inflammatory effects of nitric oxide, combined with its naturally strong safety profile and our ability to capture and deliver effective doses, positions Novan with the potential to bring multiple products to patients.
Limitations of Other Nitric Oxide-Based Approaches
Despite its therapeutic potential, there is currently only one use of nitric oxide approved by the U.S. Food and Drug Administration, or FDA, which is the use of nitric oxide gas for the treatment of pulmonary hypertension in neonatal infants. However, the delivery of nitric oxide from a gas tank is inconvenient and limits practical applications. The scarcity of nitric oxide-based products is due to the historical challenges associated with developing safe and effective approaches for the chemical storage and controlled release of a gas for therapeutic applications.
Advantages of Our Nitric Oxide Platform
We believe the Novan platform harnesses the potential of nitric oxide in a manner that leads to the creation of differentiated product candidates that address these limitations by (1) engineering tunable NCEs that store nitric oxide gas in solid form using our Nitricil technology and (2) using our formulation science to customize the drug delivery method for the anatomical location of a disease.
Our Product Candidates
We have advanced strategic development programs in the field of dermatology, with the intention of further expanding the platform into women’s health and GI therapeutic areas. We have clinical-stage dermatology drug candidates with multi-factorial (SB204), anti-viral (SB206), anti-fungal (SB208) and anti-inflammatory (SB414) mechanisms of action.
Advancement of our development programs is dependent upon our ability to access additional capital from non-dilutive sources, including partnerships, collaborations, licensing, grants or other strategic relationships, or through equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. Please refer to “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” for further discussion of our current liquidity and our future funding needs.
9
Our development pipeline is currently positioned as described in Figure 1 below.
Figure 1:
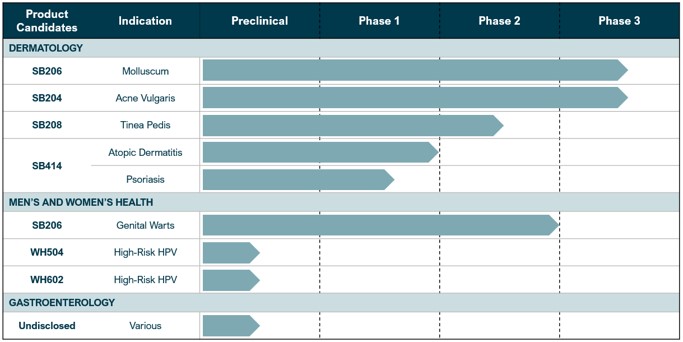
Dermatological clinical stage product candidate updates:
SB206, a Topical Anti-viral Treatment for Viral Skin Infections
We are developing SB206 as a topical anti-viral gel for the treatment of viral skin infections, with a current focus on molluscum contagiosum, and external genital and perianal warts caused by human papillomavirus.
Molluscum Contagiosum
At the end-of-Phase 2 meeting for SB206 in the external genital warts indication, we also had a constructive discussion with the FDA regarding expansion of the SB206 program into the treatment of molluscum contagiosum, or molluscum. Molluscum is a contagious skin infection caused by the molluscipoxvirus. Molluscum affects up to six million people in the U.S. annually. The greatest incidence is in children aged one to 14 years. The average time to resolution is 13 months, however, 13% of children experience lesions that may not resolve in 24 months. There is no FDA-approved treatment for molluscum. More than half of patients diagnosed with the infection are untreated. The majority of patients that receive treatment are treated with painful procedures and the remaining are often prescribed products indicated for the treatment of external genital warts.
We believed that observational learnings from an in-licensed topical nitric oxide technology study showing clinically meaningful complete clearance rates of baseline molluscum lesions, combined with our SB206 program knowledge, provided a logical pathway for SB206 development in the molluscum indication. We submitted an investigational new drug application, or IND, to the FDA in December 2017 and initiated a Phase 2 clinical trial utilizing SB206 for the treatment of molluscum in the first quarter of 2018. The Phase 2 multi-center, randomized, double-blind, vehicle-controlled, ascending dose clinical trial evaluated the efficacy, safety and tolerability of SB206 in 256 patients, ages 2 and above, with molluscum. Patients were treated with one of three concentrations of SB206 or vehicle for up to 12 weeks. The primary endpoint was the proportion of patients achieving complete clearance of all molluscum lesions at Week 12. We announced top-line results from this Phase 2 clinical trial in the fourth quarter of 2018. SB206 demonstrated statistically significant results in the clearance of all molluscum lesions at Week 12, with signs of efficacy evident as early as Week 2 with the 12% once-daily dose. The safety and tolerability profiles were favorable overall with no serious adverse events reported, including the most effective dose, SB206 12% once-daily.
10
With the full results from this Phase 2 trial made available, we held an end-of-Phase 2 (Type B) meeting with the FDA in early March 2019. Based on this meeting and the written minutes received, we commenced the Phase 3 development program for molluscum, primarily comprised of two pivotal clinical trials, in the second quarter of 2019 with SB206 12% once-daily as the active treatment arm. The “B-SIMPLE” (Berdazimer Sodium In Molluscum Patients with Lesions) Phase 3 pivotal trials consisted of two (B-SIMPLE1 and B-SIMPLE2) multi-center, randomized, double-blind, vehicle-controlled studies to evaluate the efficacy and safety of SB206 12% once-daily in approximately 680 patients (2:1 active:vehicle randomization), ages 6 months and above, with molluscum. Patients were treated once-daily with SB206 12% or Vehicle Gel once daily for a minimum of 4 weeks and up to 12 weeks to all treatable lesions (baseline and new). There were visits at Screening/Baseline, Week 2, Week 4, Week 8, Week 12 and a safety follow-up at Week 24. The primary endpoint was the proportion of patients achieving complete clearance of all molluscum lesions at Week 12. Both Phase 3 pivotal trials began dosing patients in June 2019 and we completed patient recruitment in August 2019. Top-line efficacy results from the Phase 3 trials were announced in January 2020.
SB206 did not achieve statistically significant results in the primary endpoint in both trials, which was the complete clearance of all molluscum lesions at Week 12. In B-SIMPLE2, SB206 was near statistical significance for the primary endpoint (p=0.062), and was statistically significant for the secondary endpoint, the complete clearance of all lesions at Week 8 (p=0.028), and all other pre-specified sensitivity analyses. We believe this confirms the robustness of the data in the B-SIMPLE2 trial. While the B-SIMPLE1 trial was not statistically significant for the primary endpoint (p=0.375) nor the secondary endpoint (p=0.202), all other pre-specified analyses trended in the same direction of improved treatment effect as the B-SIMPLE2 results.
In addition, the results of a statistical test of heterogeneity support that the two pivotal trials are not different from each other. Across both studies, the primary analysis odds ratio and standard error point estimates were similar and in a consistent direction with overlapping 95% confidence intervals. These statistical results are supported by an integrated analysis of the two pivotal trials, which demonstrated statistically significant complete clearance rates at Week 12 for SB206 (p=0.049). These additional analyses do not change the outcome of either B-SIMPLE trial, and the FDA may disagree with our conclusions from these analyses. P-value is a conventional statistical method for measuring the statistical significance of clinical results. A p-value of less than 0.050 is generally considered to represent statistical significance, meaning that there is a less than five percent likelihood that the observed results occurred by chance.
The last subject completed their final visit as part of the ongoing safety evaluation through Week 24 in February 2020 and full efficacy and safety data, including data from the safety evaluation through Week 24, are targeted to be available in March 2020.
Based on the results of the Phase 3 pivotal trials, discussed above, we target commencing an additional confirmatory Phase 3 trial in the second quarter of 2020, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020.
Execution of remaining Phase 3 pivotal development program activities for SB206 in molluscum continues into 2020 with receipt of pivotal trial safety data in the first quarter of 2020 and the completion of ancillary trials targeted prior to or during the second quarter of 2020.
We will need substantial additional funding to continue our operating activities and make further advancements in this program. Refer to the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” for further discussion of our current liquidity and our current and future funding needs.
11
External Genital Warts
Genital warts are among the world’s most common sexually transmitted diseases. Genital warts are usually flesh-colored growths that can be raised, flat or cauliflower-shaped and are typically found on the surface of the external genitalia or in and around the anus. In males, they can appear on the surface of the penis and scrotum, and in females inside the vagina or on the cervix. Genital warts carry a substantial psychosocial burden due to the shame and embarrassment related to having a sexually transmitted disease as well as the inconvenience and discomfort of current treatment modalities. Current treatment options for genital warts consist of ablative procedures that cut, burn or freeze the warts but do not address the underlying viral infection, and there are no currently approved oral or topical prescription products indicated for the treatment of genital warts with a direct anti-viral mechanism of action. Approximately 70% of patients treated for external genital warts receive locally destructive procedures, such as cryotherapy or curettage. Approximately 46% of patients are treated with prescription drugs alone or in combination with procedures. Both topical therapies and ablative procedures for genital warts remain largely ineffective in achieving long-term wart eradication and the average recurrence rates range from 30% to 70%. The approved drugs for the treatment of warts are pro-inflammatory in their mechanism of action and lead to ulcers, erosions and burning/stinging.
We evaluated SB206’s anti-viral activity in a Phase 2 randomized, double-blinded, vehicle-controlled clinical trial in 107 patients with genital warts caused by HPV. We announced top-line results from this Phase 2 clinical trial in the fourth quarter of 2016. SB206 demonstrated statistically significant results in the clearance of external genital and perianal warts. Once-daily treatment arms were generally well-tolerated, including the most effective dose, SB206 12% once-daily.
With the full results from this Phase 2 trial made available, a Type B meeting was held with the FDA in the second quarter of 2017 with minutes received shortly thereafter. SB206 is currently positioned for Phase 3 pivotal trials in patients with external genital warts, subject to obtaining additional financing or strategic partnering.
In October 2018, we entered into a second amendment to the Sato Agreement (as defined below), or the Sato Amendment, and collectively, with the Sato Agreement, the Amended Sato Agreement, whereby we licensed rights to Sato to develop, use, and sell SB206 in certain topical dosage forms in Japan for the treatment of viral skin infections, and to manufacture the finished form of SB206 for sale in Japan. The significant terms and the related accounting considerations of the Amended Sato Agreement, are further described in the “Collaboration and Licensing Agreements” section below and in “Note 4—Licensing Arrangements” to the accompanying consolidated financial statements included in this Annual Report.
SB204, for the Treatment of Acne Vulgaris
We are developing, SB204, as a once-daily, topical monotherapy for the treatment of acne vulgaris. Acne vulgaris is the most common skin condition in the U.S., affecting approximately 40 million to 50 million Americans annually. The disease ranges in severity from mild to severe cystic acne and causes both physical and psychological effects, including permanent scarring, anxiety, depression and poor self-esteem. Acne is a multi-factorial disease with several mechanistic contributors to the disease pathology, often requiring treatments that address more than one of the major causes of acne pathogenesis. Localized nitric oxide delivery may provide anti-inflammatory and anti-bacterial activity from a single active ingredient.
We believe that acne continues to be characterized as an unmet medical need due to the difficulty of balancing efficacy, systemic safety and cutaneous tolerability, as well as the growing concerns with anti-bacterial resistance with existing therapies. In our more than 3,200-patient SB204 clinical development program, topical application of SB204 has been well-tolerated with no significant safety concerns identified. In maximal-use pharmacokinetic trials that we have conducted in adult and pediatric patients with acne vulgaris, we observed no detectable systemic exposure from SB204 following its topical application.
In the first quarter of 2017, we reported top-line results from two identically designed Phase 3 pivotal clinical trials for SB204. SB204 demonstrated statistical significance compared to vehicle on all three co-primary endpoints in one of the trials but demonstrated statistical significance on only one of three co-primary endpoints in the other trial. We conducted an in-depth examination of the full data sets from these trials, including post hoc analyses in pooled and sub populations, with extensive assistance from third-party expert consultants in biostatistics and regulatory affairs.
In mid-2017 we completed our 40-week long term safety trial in eligible patients with acne who had previously completed 12 weeks of treatment in the related Phase 3 pivotal trials of SB204. No serious adverse events were observed with over 400 patients followed for six months and over 200 patients followed for one year.
We have had several interactions with the FDA since mid-2017 regarding SB204 and the acne indication. In September 2017, we conducted a guidance meeting with the FDA to obtain clinical and regulatory guidance by reviewing the previously completed parallel Phase 3 pivotal trials in patients with moderate-to-severe acne. The FDA’s specific feedback noted that there
12
were no additional safety requirements and that one additional pivotal trial, in moderate-to-severe acne, would be required for submission of a New Drug Application, or NDA.
In the second quarter of 2018, we conducted a Type C meeting to further discuss the Phase 3 program with the FDA and the potential for proceeding with a more narrowly defined patient segmentation. In that dialogue, our focus was centered specifically on the severe patient population. The FDA provided feedback in their minutes, received in the third quarter of 2018, on two paths forward for the acne indication, confirming the need for one additional pivotal trial for moderate-to-severe acne or, as an alternative, additional preliminary trials for a severe-only patient population.
Following receipt of FDA feedback via written minutes, we have determined that the most pragmatic development pathway for us will be to conduct one additional pivotal Phase 3 trial in moderate-to-severe acne patients. We have completed our clinical development plan for this additional trial, and further advancement of this program is subject to obtaining additional financing or strategic partnering.
In January 2017, we entered into a license agreement, and a related first amendment, with Sato, or, collectively, the Sato Agreement, whereby we licensed rights to develop, use, and sell SB204 in certain topical dosage forms in Japan for the treatment of acne vulgaris, and to manufacture the finished form of SB204 for sale in Japan. The significant terms and the related accounting considerations of the Sato Agreement are further described in the “Collaboration and Licensing Agreements” section below and in “Note 4—Licensing Arrangements” to the accompanying consolidated financial statements included in this Annual Report.
SB414, a Topical Cream for the Treatment of Inflammatory Skin Diseases
We are developing SB414 as a topical cream product candidate for the treatment of inflammatory skin diseases, such as atopic dermatitis and psoriasis. Inflammatory skin disorders are the results of immune system reactions that involve the skin. Biologic therapies are often used to treat patients with severe disease. A non-steroidal topical therapy that targets key inflammatory cytokines could address an unmet need for approximately 14 million atopic dermatitis patients and approximately 6 million psoriasis patients with less severe disease burden.
We submitted an IND with SB414 cream for the treatment of inflammatory skin diseases to the FDA during the third quarter of 2017. In 2018, we completed two complementary Phase 1b clinical trials with SB414 in patients with atopic dermatitis and psoriasis. The design of these complementary trials was to evaluate the safety, tolerability and pharmacokinetics of SB414. The trials were also designed to assess overall and specific target engagement through a reduction of key inflammatory biomarkers, also known as pharmacodynamic assessment.
Atopic Dermatitis
Atopic dermatitis, also known as atopic eczema, is the most common chronic relapsing inflammatory skin disease, affecting nearly 18 million people in the United States with no FDA-approved cure. Stabilizing the disease and reducing the number and severity of flares are the primary goals of current treatment options. The disease is characterized by recurrent red plaques, intense itching, dry skin with red papules and plaques, “weeping” clear fluid, crust and scaling. Immune cells in the deep layers of skin release inflammatory signals, causing an itchy rash. Scratching leads to defects in the skin barrier function, allowing environmental triggers, such as the bacteria Staphylococcus aureus, to penetrate the skin barrier and further exacerbate the immune cells. A recent study showed that the entry of S. aureus into the dermis triggers immune abnormalities seen in atopic dermatitis skin. Nearly 80% of the atopic dermatitis population suffers from mild-to-moderate disease and is treated with first-line monotherapies, such as corticosteroids and calcineurin inhibitors, however, corticosteroids and calcineruin inhibitors have side effects and are not well-suited for chronic use. Recently, the first biologic treatment for atopic dermatitis targeting interleukin-4, or IL-4, and IL-13 was approved, but it is reserved for patients with moderate to severe disease. Additionally, a topical PDE4 inhibitor was recently approved after more than a decade absent of any new mechanisms of action.
In two in vivo models that assess critical components of atopic dermatitis disease pathology, SB414 displayed potent anti-staphylococcal activity and dose-dependent inhibition of inflammation comparable to betamethasone, a mid-potency corticosteroid used to treat patients with atopic dermatitis. Based on preclinical data generated to date and documented literature on nitric oxide’s mechanisms of action, we believe that SB414 cream has the potential to offer non-steroidal, immunomodulatory activity and anti-staphylococcal activity for the treatment of atopic dermatitis. Additionally, SB414 cream is an occlusive formulation allowing for pH control in the skin and a possible reduction in trans-epidermal water loss, both important factors for treating the disease.
We initiated a Phase 1b trial with SB414 in adults with mild-to-moderate atopic dermatitis in December 2017. In the Phase 1b trial, 48 adults with mild-to-moderate atopic dermatitis with up to 30% body surface area at baseline, were randomized to
13
receive one of 2% SB414 cream, 6% SB414 cream, or vehicle, twice daily for two weeks. In the complementary Phase 1b trial for mild-to-moderate chronic plaque psoriasis, 36 adults received SB414 6% cream or vehicle twice daily for four weeks.
We received and analyzed the preliminary top line results from the Phase 1b clinical trials during the second and third quarters of 2018. In the atopic dermatitis trial, biomarkers from the Th2, Th17 and Th22 inflammatory pathways known to be highly relevant and indicative of atopic dermatitis, including Interleukin-13, or IL-13, IL-4R, IL-5, IL-17A and IL-22, were downregulated after two weeks of treatment with SB414 2%. The changes in Th2 and Th22 biomarkers and clinical efficacy assessed as the percent change in Eczema Area Severity Index scores were highly correlated in the SB414 2% group. Additionally, the proportion of patients achieving a greater than or equal to 3-point improvement on the pruritus (itch) numeric rating scale after two weeks of treatment was greater for patients treated with SB414 2% compared to patients treated with vehicle.
The 2% or 6% doses of SB414 in the trial did not result in any serious adverse events, and SB414 2% was more tolerable with no patients discontinuing treatment in the trial due to application site reactions. SB414 at the 6% dose was not consistently effective in reducing biomarkers across both the atopic dermatitis and psoriasis trials. This lack of consistent biomarker movement could potentially be explained by the increased irritation score experienced by patients treated with SB414 6%. Additionally, SB414 6% showed detectable systemic exposure in a subset of patients, which cleared in nearly all affected patients within 12 hours, in both the atopic dermatitis and psoriasis trials. Given the successful downregulation of key biomarkers, favorable tolerability and lack of systemic exposure with SB414 2%, we conducted non-clinical studies and completed our Phase 2 clinical development plan during 2019 to support a potential future Phase 2 clinical program launch. The SB414 program is currently on hold with further advancement subject to obtaining additional financing or strategic partnering.
Psoriasis
Psoriasis is a chronic inflammatory skin disease that affects approximately 7.5 million people in the United States. The disease is characterized by an errant immune-system response that drives inflammation and thickening of the skin caused by rapid turnover of skin cells. This typically results in patches of plaques, or thick, red raised skin with silvery-white scales. There is no cure for psoriasis. The healthcare market has seen an increase in the introduction of systemic therapies, including biologics, to treat patients with higher disease burden, but the current systemic therapies are indicated only for patients with moderate-to-severe disease. For the approximately 80% of patients with mild-to-moderate psoriasis, prescription treatment options include topical corticosteroids, retinoids and vitamin D3.
We initiated clinical development of SB414, our first use of our nitric oxide platform in the field of immunology by dosing the first patient in October 2017 in a Phase 1b clinical trial to evaluate SB414 in a cream for the treatment of psoriasis. Earlier in 2017, we presented mechanistic evidence for SB414, demonstrating a statistically significant reduction in composite psoriasis scores and an inhibition of IL-17A and IL-17F in an animal model.
In the Phase 1b trial for mild-to-moderate chronic plaque psoriasis, 36 adults received SB414 6% cream or vehicle twice daily for four weeks. We received and analyzed the preliminary top line results from this Phase 1b clinical trial during the second and third quarters of 2018. SB414 at the 6% dose did not result in any serious adverse events, but SB414 at the 6% dose was not consistently effective in reducing biomarkers across the trial. This lack of consistent biomarker movement could potentially be explained by the increased irritation score experienced by patients treated with SB414 6%. Additionally, SB414 6% showed detectable systemic exposure in a subset of patients, which cleared in nearly all affected patients within 12 hours. Based on the results of the Phase 1b trial in psoriasis, we will potentially explore the use of lower doses of SB414 in psoriasis, subject to obtaining additional financing or strategic partnering.
SB208, a Topical Anti-fungal for the Treatment of Athlete’s Foot (Tinea Pedis) and Fungal Nail Infections (Onychomycosis)
We are developing SB208 as a broad-spectrum anti-fungal gel for the treatment of superficial cutaneous fungal infections of the skin and nails, such as tinea pedis and onychomycosis. Recent studies suggest that both the nail plate, interdigital space and surrounding cutaneous tissue may serve as an overlooked reservoir of dermatophytes, perpetuating reinfection and coinfection of onychomycosis and tinea pedis. Additionally, studies have demonstrated enhanced efficacy when tinea pedis and onychomycosis are treated concurrently, suggesting that an effective topical treatment, suitable for simultaneous application to the nail plate and skin, may lead to lower rates of recurrence and enhanced efficacy.
Onychomycosis is a chronic fungal infection of the nails that affects approximately 40 million Americans and accounts for one-third of cutaneous fungal infections. The prevalence of disease increases with age, and more than 50% of patients are 70 years or older. The infection, caused by dermatophytes such as Trichophyton rubrum, often results in painful thickening and deformation of the nail and sometimes the separation of the nail plate from the nail bed, leading to an inability of the nail to
14
perform its natural protective function. Oral therapies used to treat the infection are associated with severe side effects, and topical therapies have modest efficacy profiles with complete cure rates of less than 20%.
Tinea pedis, often referred to as Athlete’s Foot, is a common fungal infection of the feet, affecting approximately 75 million Americans. Trichophyton rubrum is the most prominent dermatophyte in tinea pedis and also a causative pathogen in onychomycosis. Approximately one-third of onychomycosis patients also suffer from tinea pedis. Topical treatments are the first-line therapy for tinea pedis, while oral anti-fungals are prescribed when the infection is severe or the use of topical anti-fungals is not feasible. Currently, there is no approved single topical therapeutic agent that provides for the simultaneous treatment of the nail plate, bed, and surrounding cutaneous tissue.
In the ChubTur® infected human nail assay, a model utilized previously in the drug development of Kerydin® (tavaborole) Topical Solution, 5%, and Jublia® (efinaconazole) Topical Solution, 10%, nitric oxide-releasing formulations including SB208 demonstrated rapid penetration of the nail and effective fungal killing of Trichophyton rubrum in 24 hours following a single treatment application.
We conducted a Phase 2 proof-of-concept trial in patients with clinical signs and symptoms of tinea pedis and announced top-line results in the second quarter of 2017. SB208 demonstrated a statistically significant effect compared to vehicle in (i) the primary endpoint of achieving negative fungal culture at day 14 and (ii) the secondary endpoint of achieving mycological cure at the day 14 (mycological cure is defined by having a negative laboratory culture and negative fungal clinical diagnosis). At the end of a 4-week post treatment follow-up period, mycological cure was maintained at day 42 in both dose groups.
We conducted a Phase 1, single-center, double-blinded, randomized clinical trial in 32 adult females to evaluate the rate of fingernail growth associated with SB208 16% and the local tolerability of the gel when used over the course of 29 days. SB208 16% demonstrated a statistically significant greater mean daily nail growth rate for the treatment period when compared to the same patient’s own growth rate in the run-in period and was well tolerated by patients.
SB207, a Topical Anti-viral Product Candidate
In response to our identification of targeted viral opportunities of high unmet need where we believe our nitric oxide releasing technology could provide clinical benefit to patients, we developed SB207, a new anti-viral product candidate. The SB207 product candidate incorporates our existing drug substance, berdazimer sodium (NVN1000), with a new formulation specifically engineered for a number of anti-viral programs. In December 2019, we received written responses in response to a pre-IND meeting request with the FDA and, based on such FDA responses, have determined that further advancement of SB207 is subject to further evaluation of clinical plans and securing funding.
Women’s Health Business Unit
On October 25, 2018, we announced the formation of a dedicated women’s health business unit as well as a foundational collaboration with Health Decisions Inc., or Health Decisions. Health Decisions is a full-service contract research organization specializing in clinical studies of therapeutics for women’s health indications. Over the past twelve months, we have progressed our knowledge on the potential to utilize nitric oxide-based products in the field of women’s health, with an emphasis on oncovirus applications and our initial focus centering on persistent high-risk HPV. Central to our effort has been an ongoing, multi-year research collaboration with the University of Alabama-Birmingham studying the effects of nitric oxide-releasing compounds on HPV infections. Published clinical research on high-risk HPV infections has demonstrated a link to the development of malignant lesions and neoplasia, including female cancers in the cervix, vagina, vulva, anus and oral cavity. This foundational science advancement pairs with our previously announced Phase 2 data for the treatment of external genital warts, where SB206 12% demonstrated statistically significant clearance of baseline warts and was generally well-tolerated, provide a specific late stage clinical asset that targets HPV. We believe that our clinical collaboration with Health Decisions and our ongoing academic research collaboration with the University of Alabama-Birmingham provides us with a differentiated opportunity for advancement in the area of women’s health.
In August 2019, we received a Phase 1 Federal grant of approximately $0.2 million from the National Institutes of Health, or NIH. The funds are to be used to advance formulation development of a nitric oxide-containing intravaginal gel (WH602) designed to treat high-risk HPV infections that can lead to cervical intraepithelial neoplasia, or CIN. The specific focus is to ensure the nitric oxide delivery from the gel replicates doses of nitric oxide previously demonstrated to be effective against HPV in our clinical and in vitro studies.
In February 2020, following the successful progression of Phase 1, we were awarded a Phase 2 federal grant of approximately $1.0 million from the NIH that will enable the conduct of IND-enabling toxicology and pharmacology studies and other
15
preclinical activity with respect to WH602. These funds will be received by us in the form of periodic cost reimbursements as the underlying research and development activities are performed. We may be eligible to receive an additional $0.5 million in funding as part of this Phase 2 grant, subject to availability of NIH funds and satisfactory progress of the project during the initial 12-month term.
In September 2019, we received a Federal grant from the U.S. Department of Defense’s, or DoD, Congressionally Directed Medical Research Programs, or CDMRP, of approximately $1.1 million as part of its Peer Reviewed Cancer Research Program. The grant will support the development of a non-gel formulation product candidate (WH504) designed to treat high-risk HPV infections that can lead to CIN, with well-characterized physical chemical properties suitable for intravaginal administration. In addition, the grant will support the evaluation of the effect of varying concentrations and treatment durations of berdazimer sodium (NVN1000) against HPV-18 in human raft cell culture in vitro studies. This targeted research aims to create a disease-altering treatment that could be used upon detection and the early signs of high-risk HPV infection to intervene before progression to cervical cancer.
Under the terms of the aforementioned NIH and DoD grants, we are entitled to receive the grant funds in the form of periodic reimbursements of our allowable direct expenses, allocated overhead, general and administrative expenses and payment of other specified amounts.
These product candidates currently in development together represent the core of our Women’s Health business unit. This unit has continued to be supported through a collaboration with Health Decisions.
Our acquisition of exclusive worldwide rights for certain oncovirus applications of nitric oxide-based products from KNOW Bio, LLC, or KNOW Bio, in October 2017 enables the potential expansion into this therapeutic area. The terms of this intellectual property license transaction are further described in “Note 3—KNOW Bio, LLC” to the accompanying consolidated financial statements included in this Annual Report.
GI Therapeutic Focus
In January 2019, we announced the addition of GI diseases as a therapeutic focus area as part of our overall science and business strategy. This decision is based on the connection between the multi-factorial pathologies of GI diseases and the demonstrable anti-microbial and anti-inflammatory properties of Novan’s nitric oxide technology. Nitric oxide produced in the GI tract regulates many of its functions including the secretion of mucous for protection against physical, chemical, and microbial injury, perfusion of blood through the GI tissue, mitigation of white blood cell adherence to GI tissue to protect from injury and the healing and repair of ulcers. We believe that our initial expansion into GI will require minimal investment due to our ability to leverage current technology experience and assets.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We consider our primary potential competition to be a broad base of existing providers and drug developers of therapeutics to treat molluscum, acne vulgaris, genital warts, onychomycosis, psoriasis and atopic dermatitis. Additional providers and drug developers will become primary potential competition as we expand our platform to include the women’s health, GI and other potential therapeutic areas. Product competition includes pharmaceutical generics, branded generics, pharmaceutical brands, biologics as well as over-the-counter, or OTC, products. We expect continued future competition across research and drug development in various different fields of innovation; capital and resource allocation to many of these areas appears to be continuous and of a global nature. In addition, there are certain instances where competition extends into the medical procedure and the medical device spectrums of human health care. Any product candidates that we successfully develop and commercialize will compete with these existing therapies as well as new therapies that may become available in the future. Our success will be based in part on our ability to identify, develop and manage a portfolio of product candidates that are safer and more effective than competing products and therapies.
16
Intellectual Property
Our success depends in large part upon our ability to obtain and maintain proprietary protection for our products and technologies and to operate without infringing the proprietary rights of others. We seek to avoid the latter by monitoring patents and publications that may affect our business, and to the extent we identify such developments, evaluating and taking appropriate courses of action. With respect to the former, our policy is to protect our proprietary position by, among other methods, filing for patent applications on inventions that are important to the development and conduct of our business with the U.S. Patent and Trademark Office, or USPTO, and its foreign counterparts. We also use other forms of protection, such as trademark, copyright and trade secret protection, to protect our intellectual property, particularly where we do not believe patent protection is appropriate or obtainable.
We own or have an exclusive license to issued patents and pending patent applications in the United States and in foreign jurisdictions (including applications filed in foreign jurisdictions and international or Patent Cooperation Treaty, or PCT, applications that have not yet entered national phase). Patent coverage lasts for varying periods according to the date of filing of the patent application or the date of grant or issuance of the patent and the legal term of patents in various countries where patent protection is obtained. Generally, patents issued for regularly filed applications in the United States are granted a term of 20 years from the earliest filing date of a non-provisional patent application. In addition, in certain instances, the term of a patent can be extended to recapture a portion of the USPTO delay in issuing the patent or may be shortened if a patent is terminally disclaimed over another patent that expires earlier. The term of a patent may also be eligible for patent term extension to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. However, as to the FDA component, the extension term cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest filing date of a non-provisional patent application. However, the actual protection afforded by a patent varies on a product by product basis from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
Nitricil Technology
We exclusively license from the University of North Carolina at Chapel Hill, or UNC, issued patents and pending applications directed to our library of Nitricil compounds, including patents issued in the United States, Canada, Japan and Australia with claims intended to cover NVN1000, the NCE for our current clinical-stage product candidates. Additionally, one such issued patent in the United States has claims specifically directed to the composition of matter of NVN1000. These patents and pending applications, if issued, are projected to expire in 2026 without taking into account any patent term extensions that may be available to us. Additionally, NVN1000 has been classified as an NCE, and patent term extensions may be available to extend the life of a U.S. patent that covers NVN1000 beyond 2026. We also own patents issued in the United States, China, Germany, Spain, France, Great Britain, Ireland, Italy and Switzerland directed to methods of manufacturing Nitricil compounds. These patents are projected to expire in 2032.
Formulation Science and Therapeutic Uses
We own patents issued in the United States, Australia, Germany, Spain, France, Great Britain, Italy, China, Mexico, South Korea and Japan and pending applications filed in foreign jurisdictions, including Brazil and Canada directed to methods of reducing sebum production using nitric oxide-releasing macromolecules, including, in certain embodiments, through the use of Nitricil compounds. We also own issued patents in the United States, Australia and Japan and pending applications filed in the United States, Brazil, Canada, China, Europe and Japan directed to the alcohol gel component of SB204 and SB206 and/or the SB204 and SB206 two-component formulations. We are pursuing United States, Australia, Brazil, Canada, China, Europe, Japan and South Korea applications directed to the use of nitric oxide-releasing compounds, including, in certain embodiments, Nitricil compounds, for the treatment of viral skin infections.
Altogether, our issued U.S. and foreign patents and pending U.S. and foreign patent applications, if issued, relating to one or more of our clinical-stage product candidates are projected to expire between 2026 and 2037, without taking into account any patent term extensions that may be available to us and assuming that prosecution is pursued to issuance with no shortening of term.
Other Patented Technology
In addition to the patents and pending applications we own or have an exclusive license related to Nitricil and our product candidates, we also own or have exclusive licenses to issued patents and pending applications in the United States and in foreign jurisdictions covering other nitric oxide-based therapeutics and/or methods of use in indications for dermatological and oncovirus-mediated diseases.
17
Trade Secrets
We rely upon trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, by requiring our employees, consultants, contractors and other advisors to execute nondisclosure and assignment of invention agreements, or to include such provisions in their consulting agreement, upon commencement of their respective employment or engagement. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements and provisions, to grant us ownership of technologies that are developed through a relationship with a third party. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our commercial partners, collaborators, employees and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Trademarks
Novan® is a registered trademark of our company in the United States.
Research and Development Arrangements
Royalty and Milestone Payments Purchase Agreement with Reedy Creek Investments LLC
On April 29, 2019, we entered into a royalty and milestone payments purchase agreement, or the Purchase Agreement, with Reedy Creek Investments LLC, or Reedy Creek, pursuant to which Reedy Creek provided us funding in an initial amount of $25.0 million, which we will use primarily to pursue the development, regulatory approval and commercialization (including through out-license agreements and other third party arrangements) activities for SB206, for the treatment of molluscum, and advancing programmatically other activities with respect to SB414, for atopic dermatitis, and SB204, for acne. Reedy Creek was to provide $10.0 million of additional funding contingent upon our achievement of SB206 clinical trial success, defined as (i) the achievement, no later than March 31, 2020, of statistically significant rates of complete clearance of lesions for molluscum contagiosum in humans at week 12 in each of the two Phase 3 clinical trials or any other primary endpoint required or accepted by the FDA for the SB206 product; or (ii) equivalent achievement (as agreed upon by the parties). Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
Pursuant to the Purchase Agreement, we will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by us pursuant to any out-license agreement for SB204, SB206 and SB414 in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by us to third parties pursuant to any agreements under which we have in-licensed intellectual property with respect to such products.
The applicable percentage used for determining the ongoing quarterly payments, applied to amounts received directly by us pursuant to any out-license agreement for each product, ranges from 10% for SB206 to 20% for SB414 and SB204. However, the agreement provides that the applicable percentage for each product will be 25% for fees or milestone payments received by us (but not royalty payments received by us) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If we decide to commercialize SB204, SB206 or SB414 on our own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, we will be obligated to pay Reedy Creek a low single digits royalty on net sales of such products.
Development Funding and Royalties Agreement with Ligand Pharmaceuticals Incorporated
On May 4, 2019, we entered into a development funding and royalties agreement, or the Funding Agreement, with Ligand Pharmaceuticals Incorporated, or Ligand, pursuant to which Ligand provided us funding of $12.0 million, which we used to pursue the development and regulatory approval of SB206, for the treatment of molluscum.
Pursuant to the Funding Agreement, we will pay Ligand up to $20.0 million in milestone payments upon the achievement by us of certain regulatory and commercial milestones associated with SB206 or any product that incorporates or uses NVN1000, the active pharmaceutical ingredient for our clinical stage product candidates, for the treatment of molluscum. In addition to the milestone payments, we will pay Ligand tiered royalties ranging from 7% to 10% based on annual aggregate net sales of such products in the United States, Mexico or Canada.
18
For additional information about the Purchase Agreement and Funding Agreement, please see the sections entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Business Updates” and “Note 7—Research and Development Arrangements” to the accompanying consolidated financial statements included in this Annual Report.
Collaboration and Licensing Agreements
Amended Sato License Agreement
On January 12, 2017, we entered into the Sato Agreement relating to SB204. On October 5, 2018, we entered into the Sato Amendment, or collectively, with the Sato Agreement, the Amended Sato Agreement. The Sato Amendment expands the Amended Sato Agreement to include SB206, our drug candidate for the treatment of viral skin infections, including molluscum. Pursuant to the Amended Sato Agreement, we granted to Sato an exclusive, royalty-bearing, non-transferable license under certain of our intellectual property rights, with the right to sublicense with our prior written consent, to develop, use and sell products in Japan that incorporate SB206 or SB204 in certain topical dosage forms for the treatment of viral skin infections or acne vulgaris, respectively, and to make the finished form of such products. We, or our designated contract manufacturer will also supply finished product to Sato for use in development of SB204 or SB206 in the licensed territory. The rights granted to Sato do not include the right to manufacture the active pharmaceutical ingredient, or API, of SB206 or SB204; rather, the parties agreed to negotiate a commercial supply agreement pursuant to which we or a third-party contract manufacturer would be the exclusive supplier to Sato of the API for the commercial manufacture of licensed products in the licensed territory.
Pursuant to the terms of the Sato Agreement, Sato had an exclusive option to negotiate for the license rights in certain additional territories within Asia, subject to Sato’s payment of a specified option exercise fee. During the third quarter of 2017, Sato elected not to execute this option and, as a result, the option expired unexercised.
Under the terms of the Amended Sato Agreement, we also have exclusive rights to certain intellectual property that may be developed by Sato in the future, which we may choose to use for our own development and commercialization of SB204 or SB206 outside of Japan. The term of the Amended Sato Agreement (and the period during which Sato must pay royalties under the Amended Sato Agreement) expires on the twentieth anniversary of the first commercial sale of a licensed product in the licensed field in the licensed territory.
For additional information about the Amended Sato Agreement, please see the sections entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Business Updates” and “Note 4—Licensing Arrangements” to the accompanying consolidated financial statements included in this Annual Report.
UNC License Agreement
We acquired exclusive rights to our library of Nitricil compounds pursuant to license agreements with UNC entered into in July 2007 and October 2009, which were subsequently amended, restated and consolidated in June 2012. We amended the consolidated license agreement in November 2012 to expand the scope of licensed patents to cover additional nitric oxide technologies in consideration for an upfront cash payment. We may obtain similar amendments to the consolidated license agreement to expand the scope of licensed patents to cover future additional nitric oxide technologies or as improvements on licensed technology and, if such amendments were executed, we may be required to pay additional upfront cash payments. In April 2016, we amended the agreement to clarify the scope of the intellectual property of the consolidated license agreement. In November 2018, we amended the agreement to further clarify the scope of the intellectual property of the consolidated license agreement and to make modifications to certain milestones under the consolidated license agreement.
Under the consolidated license agreement with UNC, we are granted an exclusive, worldwide license, with the ability to sublicense, under the licensed UNC patents, including those directed to Nitricil compounds, to develop and commercialize products utilizing the licensed technology. As partial consideration for the consolidated license agreement, we issued 191,052 shares of our common stock to UNC and a nominal upfront cash payment. Additionally, under the consolidated license agreement, we are obligated to pay UNC a running royalty percentage in the low single digits on net sales of licensed products (by us or any of our sublicensees, such as Sato), and to pay up to $425,000 to UNC in regulatory and commercial milestones on a licensed product by licensed product basis.
Under the consolidated license agreement, UNC controls prosecution activities with respect to licensed patents owned solely by UNC, we control prosecution activities with respect to licensed patents jointly owned by us and UNC and we are obligated to reimburse UNC for reasonable prosecution and maintenance costs. Pursuant to the consolidated license agreement, we have the first right to defend against third-party claims of patent infringement with respect to the licensed products and to enforce the licensed patents against third-party infringers.
19
Unless earlier terminated by us at our election, or if we materially breach the agreement or become bankrupt, the consolidated license agreement remains in effect on a country by country and licensed product by licensed product basis until the expiration of the last to expire issued patent covering such licensed product in the applicable country, and upon such expiration, we receive a perpetual, unrestricted, fully-paid and royalty free right to develop and commercialize such licensed product in such country. As of December 31, 2019, the last to expire issued patent licensed to us under the consolidated license agreement is projected to expire in 2033. UNC may terminate the agreement or render the license granted thereunder non-exclusive for our material breach of the agreement that remains uncured after 90 days of receipt of written notice thereof from UNC and may also terminate the agreement or render the license granted thereunder non-exclusive upon providing written notice for our bankruptcy or insolvency-related events within 30 days of the occurrence of such events. We may terminate the agreement at any time for convenience upon providing written notice of not less than 30 days to UNC.
Separation Transaction and Licensing Arrangements with KNOW Bio, including Amendments
2015 Separation Transaction and Licensing Arrangements
In connection with the December 2015 separation of our non-dermatology assets to KNOW Bio, we granted to KNOW Bio, through two separate agreements, exclusive licenses, with the right to sublicense, to certain U.S. and foreign patents and patent applications controlled by us as of the execution date of the agreement, and, under one of the agreements, patents and patent applications which became controlled by us during the three years immediately following the execution date of such agreement, directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics.
Under the exclusive licenses, the following rights were retained by Novan or conveyed to KNOW Bio:
• | Novan retained exclusive development and commercialization rights in all fields for any products containing certain specified particles, referred to as the Novan Particles, including those in our NVN1000 API and in other NCEs we are developing for the GI therapeutic area. |
• | Novan retained exclusive rights to develop and commercialize products utilizing the licensed technology in the Retained Dermatology Field, which is defined as the diagnosis, treatment, prevention, and palliation of diseases, conditions, or disorders of the skin, nails, hair or scalp in humans or animals, and all cosmetic uses for the skin, nails, hair or scalp, other than (i) for wound care through formulations of therapeutic product specifically designed to treat chronic wounds, thermal burns, radiation injury, accidental injury, surgical sites or scars, and (ii) therapeutic uses for treating cancer, excluding basal cell carcinoma, squamous cell carcinoma, precancerous conditions of the skin, actinic keratosis, actinic cheilitis, cutaneous horn, Bowen disease, radiation dermatosis, and dysplastic nevi. The Retained Dermatology Field was amended in 2017 as described in the section entitled “2017 Amendments to KNOW Bio Licensing Arrangements.” |
• | KNOW Bio received exclusive rights to develop and commercialize products utilizing the licensed technology, excluding products containing the Novan Particles, in the KNOW Bio Field, which is defined as all fields of use except for the Retained Dermatology Field. The KNOW Bio Field was amended in 2017 as described in the section entitled “2017 Amendments to KNOW Bio Licensing Arrangements.” |
Under one of these exclusive license agreements, KNOW Bio granted to us an exclusive license, with the right to sublicense, under any patents and patent applications which became controlled by KNOW Bio during the three years immediately following the execution date of such agreement and directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, but not towards medical devices, for use in the diagnosis, treatment, prevention, and palliation of diseases, conditions, or disorders in the Retained Dermatology Field, including but not limited to SB204, SB206, SB208, SB414 and our other presently-contemplated dermatology pipeline candidates. KNOW Bio granted us a right of first negotiation to obtain a license under any patents and patent applications generated by KNOW Bio during the first three years following the execution date of the agreement and directed towards medical devices to develop and commercialize licensed products in the Retained Dermatology Field. Additionally, Novan and KNOW Bio also agreed that neither party would commercialize any products in the other’s field of use during the first three years following the execution date of the agreement. The three-year period in which new patents and patent applications controlled by us or by KNOW Bio are added to the exclusive licenses and the three-year term of the commercialization non-compete both expired on December 29, 2018. Neither we nor, to our knowledge, KNOW Bio commercialized a product in the other party’s field during this period.
Additionally, we granted to KNOW Bio exclusive sublicenses, with the ability to further sublicense, under certain of the U.S. and foreign patents and patent applications exclusively licensed to us from UNC and another third party directed towards nitric oxide-releasing compositions, including certain Nitricil compounds, to develop and commercialize products utilizing the
20
licensed technology in the KNOW Bio Field. Under the exclusive sublicense to the UNC patents and applications, KNOW Bio is subject to the terms and conditions under the consolidated license agreement with UNC, including diligence obligations and milestone payment obligations.
Under the exclusive license agreements and sublicense agreements, we retain all rights under our owned and exclusively licensed patents and patent applications with respect to development and commercialization of products for use in the Retained Dermatology Field. The exclusive license agreements and sublicense agreements will continue for so long as there is a valid patent claim under the respective agreement, unless earlier terminated, and upon expiration continues as a perpetual non-exclusive license. Under each agreement, Novan and KNOW Bio have the right to terminate the agreement by written notice for the other party’s material breach which remains uncured within 30 days of receipt of notice thereof. Novan also has the right to terminate each such agreement immediately upon written notice if KNOW Bio, its affiliates or sublicensees challenge the validity of any patent licensed in such agreement. KNOW Bio has the right to terminate each such agreement, with notice, for any reason upon ninety days advance written notice to the Company. The licenses granted by KNOW Bio to the Company in the agreements survive termination of the agreements.
For additional information about the Separation Transaction, please see “Note 3—KNOW Bio, LLC” to the accompanying consolidated financial statements included in this Annual Report.
2017 Amendments to KNOW Bio Licensing Arrangements
In October 2017, we entered into certain amendments, or the KNOW Bio Amendments, to the original license and sublicense agreements described above between us and KNOW Bio, or the Original KNOW Bio Agreements. Pursuant to the terms of the KNOW Bio Amendments, we re-acquired from KNOW Bio exclusive, worldwide rights under certain U.S. and foreign patents and patent applications controlled by us as of the execution date of the Original KNOW Bio Agreements, and patents and patent applications which became controlled by us during the three years immediately following the execution date of the Original KNOW Bio Agreements, directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, to develop and commercialize products for all diagnostic, therapeutic, prophylactic and palliative uses for any disease, condition or disorder caused by certain oncoviruses, or the Oncovirus Field. KNOW Bio also granted to us an exclusive license, with the right to sublicense, under any patents and patent applications which became controlled by KNOW Bio during the three years immediately following the execution date of the Original KNOW Bio Agreements and directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, but not towards medical devices, to develop and commercialize products for use in the Oncovirus Field. Additionally, KNOW Bio agreed that KNOW Bio would not commercialize any products in the Oncovirus Field during the first three years following the execution date of the Original KNOW Bio Agreements. The three-year period in which new patents and patent applications controlled by KNOW Bio are added to the exclusive license and the three-year term of the commercialization non-compete both expired on December 29, 2018.
The rights granted to us in the Oncovirus Field in the KNOW Bio Amendments continue for so long as there is a valid patent claim under the Agreements, and upon expiration continue on a perpetual non-exclusive basis, and are subject to the termination rights of KNOW Bio and us that are set forth in the Original KNOW Bio Agreements. In addition, under the KNOW Bio Amendments, KNOW Bio may terminate the rights granted to us in the Oncovirus Field if: (i) we do not file a first IND application with the FDA for a product in the Oncovirus Field by October 2020; or (ii) we do not file a first NDA with the FDA by October 2025 for a product in the Oncovirus Field and do not otherwise have any active clinical programs related to the Oncovirus Field at such time.
Additional terms, including our financial obligations, under the KNOW Bio Amendments are described in further detail in “Note 3—KNOW Bio, LLC” to the accompanying consolidated financial statements included in this Annual Report.
Government Regulation
The FDA and comparable regulatory authorities in state and local jurisdictions and in other countries impose substantial and burdensome requirements upon companies involved in the clinical development, manufacture, marketing and distribution of drugs, such as those we are developing. These agencies and other federal, state and local entities regulate, among other things, the research and development, testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion, distribution, post-approval monitoring and reporting, sampling and export and import of our product candidates.
21
U.S. Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
• | completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s Good Laboratory Practice, or GLP, regulations; |
• | submission to the FDA of an IND which must become effective before human clinical trials may begin; |
• | approval by an independent Institutional Review Board, or IRB, at each clinical site before each trial may be initiated; |
• | performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, requirements to establish the safety and efficacy of the proposed drug product for each indication; |
• | submission to the FDA of an NDA after completion of all pivotal clinical trials; |
• | satisfactory completion of an FDA advisory committee review, if applicable; |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practice, or cGMP, requirements and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
• | FDA review and approval of the NDA to permit marketing of the product for particular indications for uses in the United States. |
Preclinical Studies
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. To support an IND to conduct clinical trials, a sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical Trials
Clinical trials involve the administration of the IND to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND submission. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their www.clinicaltrials.gov website.
Human clinical trials are typically conducted in three or four sequential phases, which may overlap or be combined:
• | Phase 1 clinical trial: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness. |
22
• | Phase 2 clinical trial: The drug is administered to a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
• | Phase 3 clinical trials: The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product and to provide adequate information for the labeling of the product. |
• | Phase 4 clinical trials: In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. In some cases, these Phase 4 studies are made a condition of approval of the NDA. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA, and more frequently if serious adverse events occur. Each of Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, or at all and favorable results in an earlier clinical or preclinical trial may not predict the outcomes of subsequent trials. Clinical trials may be delayed for a variety of reasons including unexpected safety or efficacy concerns, slow enrollment of subjects, unexpected shortages in the drug product, or other reasons. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Marketing Approval
Assuming successful completion of the required testing in accordance with all applicable regulatory requirements, the results of the preclinical studies and clinical trials, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications for use. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act, or PDUFA, guidelines that are currently in effect, the FDA has a goal of ten months from the date of “filing” of a standard NDA for a new molecular entity to review and act on the submission. This review typically takes twelve months from the date the NDA is submitted to the FDA because the FDA has approximately two months to make a “filing” decision as to whether it will accept the application for filing. The actual review time may be significantly longer, depending on the complexity of the review, FDA requests for additional information and the sponsor’s submission of additional information.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, as amended and reauthorized, certain NDAs or supplements to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, plan to ensure that the benefits of the drug outweigh its risks. The REMS plan could include medication guides, physician communication plans, assessment plans or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and whether the facility in which it is manufactured, processed, packaged or held meets standards designed to assure the product’s continued safety, quality and purity. During its review, the FDA may raise additional issues or request additional data or information, during which time, the review period is generally suspended until such requests are received. This can delay, sometimes substantially, the FDA’s review and potential approval of an application.
23
The FDA may refer an application for a novel drug to an advisory committee. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA may inspect one or more clinical trial sites to assure compliance with GCP requirements.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or preclinical testing in order for FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs or if unexpected safety or efficacy concerns arise. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could impact the timeline for regulatory approval or otherwise impact ongoing development programs. For example, in December 2016, the 21st Century Cures Act was signed into law. The Act is intended, among other things, to modernize the regulation of drugs and biologics and to encourage innovation.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including fast track designation, accelerated approval, priority review and breakthrough therapy designation, which are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life-threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
To be eligible for a fast track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if it will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. The FDA may review sections of the NDA for a fast track product on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA.
The FDA may give a priority review designation to drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. Under the new PDUFA agreement, these six and ten-month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. The review period may be suspended if the FDA requests additional information which may extend the timeline for review. Many products that are eligible for fast track designation are also likely to be considered appropriate to receive a priority review.
In addition, product candidates studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate
24
endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures in certain instances based on these studies.
Moreover, under the provisions of the Food and Drug Administration Safety and Innovation Act, or FDASIA, passed in July 2012, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. The FDA must take certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
Fast track designation, priority review and breakthrough therapy designation do not change the standards for approval and approval is not guaranteed. Such designation may, however, expedite the development or approval process. Even if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product candidate no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. We may explore some of these opportunities for our product candidates as appropriate.
Post-Approval Requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences associated with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual program fee requirements for any marketed products.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials, and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. The FDA may also limit the indications for use or may impose labeling or other requirements on the product.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP requirements and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
• | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
• | fines, warning letters or holds on post-approval clinical trials; |
• | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product approvals; |
• | product seizure or detention, or refusal to permit the import or export of products; or |
• | injunctions or the imposition of civil or criminal penalties. |
25
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties.
Other Health Care Laws
In addition to FDA restrictions on marketing of pharmaceutical products, other U.S. federal and state healthcare regulatory laws restrict business practices in the pharmaceutical industry, which include, but are not limited to, state and federal anti-kickback, false claims, physician payment and drug pricing transparency laws.
The federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value. The Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. In addition, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. The majority of states also have anti-kickback laws, which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
The federal False Claims Act prohibits any person or entity from, among other things, knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or from knowingly making a false statement to avoid, decrease or conceal an obligation. A claim includes “any request or demand” for money or property presented to the U.S. government. Violation of the federal Anti-Kickback Statute may also constitute a false or fraudulent claim for purposes of the federal civil False Claims Act. Actions under the civil False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the civil False Claims Act can result in very significant monetary penalties and treble damages. In addition, the civil monetary penalties statute imposes penalties against any person who is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Many states also have similar fraud and abuse statutes or regulations that apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Given the significant size of actual and potential settlements, it is expected that the government authorities will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, prohibits, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and certain other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA, imposed, among other things, new annual reporting requirements through the Physician Payments Sunshine Act for covered manufacturers for certain payments and “transfers of value” provided to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other health care professionals beginning in 2022, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. In addition, certain states require implementation of compliance programs and compliance with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, impose restrictions on marketing practices, and/or tracking and reporting of pricing information and marketing expenditure as well as gifts, compensation and other remuneration or items of value provided to physicians and other healthcare professionals and entities.
26
Violation of any of such laws or any other governmental regulations that may apply to us can result in penalties, including, without limitation, administrative, civil and criminal penalties, damages, fines, disgorgement, contractual damages, reputational harm, diminished profits and future earnings, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and individual imprisonment.
Coverage and Reimbursement
Sales of our product candidates, if approved, by us or any potential commercial partners will depend, in part, on the extent to which such products will be covered by third-party payors, such as government healthcare programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly limiting coverage or reducing reimbursements for medical products and services. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Sales of any products for which we receive regulatory approval for commercial sale will therefore depend, in part, on the availability of coverage and adequate reimbursement from third-party payors. Third-party payors include government authorities, managed care plans, private health insurers and other organizations.
The process for determining whether a third-party payor will provide coverage for a drug typically is separate from the process for setting the price of such product or for establishing the reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the FDA-approved products for a particular indication. A decision by a third-party payor not to cover our product candidates could reduce a physician’s willingness to prescribe our products once approved and have a material adverse effect on our sales, results of operations and financial condition. Moreover, a third-party payor’s decision to provide coverage for a drug does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Additionally, coverage and reimbursement for products can differ significantly from payor to payor. One third-party payor’s decision to cover a particular medical product or service does not ensure that other payors will also provide coverage for the medical product or service, or will provide coverage at an adequate reimbursement rate.
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost-effectiveness of drugs, in addition to questioning safety and efficacy. If these third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products after FDA approval or, if they do, the level of payment may not be sufficient to allow us to sell our products at a profit.
Healthcare reform
A primary trend in the U.S. healthcare industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products. For example, the ACA, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care plans; subjected drug manufacturers to new annual fees based on pharmaceutical companies’ share of sales to federal healthcare programs; and created a new Patient Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the ACA. By way of example, the Tax Cuts and Jobs Acts was enacted, which, among other things, removes penalties for not complying with the individual mandate to carry health insurance. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Cuts and Jobs Acts, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. It is unclear how these decisions, subsequent appeals, if any, and other efforts to challenge, repeal or replace the ACA will impact the law.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013
27
and, due to subsequent legislative amendments, will stay in effect through 2029 unless additional Congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals and imaging centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Recently there has also been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies.
We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products once approved or additional pricing pressures. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our drugs.
Data Privacy and Security
Pharmaceutical companies may be subject to U.S. federal and state and foreign health information privacy, security and data breach notification laws, which may govern the collection, use, disclosure and protection of health-related and other personal information. In the U.S., HIPAA imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon “covered entities” (health plans, health care clearinghouses and certain health care providers), and their respective business associates, individuals or entities that create, received, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HIPAA mandates the reporting of certain breaches of health information to HHS, affected individuals and if the breach is large enough, the media. Entities that are found to be in violation of HIPAA as the result of a breach of unsecured protected health information, a complaint about privacy practices or an audit by HHS, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Even when HIPAA does not apply, according to the Federal Trade Commission or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
In addition, certain state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same requirements, thus complicating compliance efforts. By way of example, the California Consumer Privacy Act, or CCPA, which went into effect January 1, 2020, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context.
In Europe, the European Union General Data Protection Regulation 2016/679, or GDPR, went into effect in May 2018 and introduces strict requirements for processing the personal data of European Union data subjects. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Moreover, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the EU will be regulated, especially following the United Kingdom's departure from the EU on January 31, 2020 without a deal. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom's departure from the EU.
Manufacturing and Supplies
We currently manufacture all drug substance, including NVN1000 (the API for all of our clinical stage product candidates), at our facility in Morrisville, North Carolina. In 2017, we also began manufacturing all drug product materials at our Morrisville, North Carolina facility, for use in our non-clinical studies and clinical trials.
28
We manufacture our investigational materials in accordance with cGMP required by the FDA, International Committee on Harmonization and other regulatory bodies. While our facilities have been audited by third-parties for cGMP and GLP compliance, we have not been audited by the FDA. In addition, our drug substance manufacturing processes and operating conditions have been evaluated and tested by qualified vendors to ensure a safe operating environment. These tests include raw materials and product handling, process chemistry, air quality and waste disposal and containment.
In June 2019, we executed a master contract manufacturing agreement with a full-scale active pharmaceutical ingredient (API) manufacturer. The agreement established an operating and business relationship for this manufacturer to become the primary external supplier of our proprietary berdazimer sodium (NVN1000) drug substance. Also incorporated in the agreement is the process and analytical method transfer necessary to advance the production of our berdazimer sodium (NVN1000) drug substance for future clinical trials and importantly, upon approval of any of our drug product candidates, for commercial purposes on a global basis. We are engaged in the transfer of the NVN1000 manufacturing technology and upon its completion intend for this API manufacturer to be able to manufacture NVN1000 in accordance with our established manufacturing processes, in compliance with applicable regulatory guidelines, as appropriate for clinical trials and alongside our current internal manufacturing capabilities.
In October 2018, we established a strategic alliance with Orion Corporation, or Orion, a Finnish full-scale pharmaceutical company with broad experience in drug manufacturing. The alliance enables Orion to manufacture our topical nitric oxide-releasing product candidates on our behalf and on the behalf of our global strategic partners. We have executed a master contract manufacturing agreement to enable technology transfer and manufacturing of clinical trial materials for future clinical trials with our topical product candidates. We are engaged in the transfer of technology for the manufacture of both SB204 and SB206, and upon its completion intend for Orion to be able to manufacture the drug product, or the finished dosage form of the gel, in accordance with our established manufacturing processes, in compliance with applicable regulatory guidelines, as appropriate for clinical trials and alongside our current internal manufacturing capabilities. A completed manufacturing technology transfer to Orion will enable the manufacture of multiple assets for clinical trial materials and, potentially, commercial quantities. Importantly, this alliance is being structured to support major global markets in which we and our partners pursue regulatory approvals for our product candidates and complements our present internal capability.
We intend for these, or potentially other, third parties to supply drug substance and drug product materials to support commercialization of any of our product candidates, subject to FDA approval. In such cases, they may be the primary suppliers for these product candidates. The progression of the relationships with the aforementioned third-party manufacturers is integral to the advancement of our dermatological platform, including our SB206 molluscum program, by us or through partnerships, collaborations, licensing or other strategic relationships. This strategy includes an increased utilization of and reliance upon third-party vendors and strategic partners for the performance of activities, processes and services that (i) do not result in the generation of significant new intellectual property; and (ii) can leverage existing robust infrastructure, systems, and facilities as well as associated subject matter expertise. A parallel and inter-related strategic objective is to reduce our own internal resources, facilities, and infrastructure capabilities that have historically performed such activities, processes and services. We believe this broad strategy can ultimately provide enhanced capabilities and operating efficiencies for us or any potential partnerships, collaborations, licensing or other strategic relationships we may enter. Although the third-party manufacturers are reducing their near-term activities and extending their time lines at our request in an effort to reduce our near-term cash utilization and extend our operating cash runway, we expect to incur certain incremental and discrete costs to effect this strategy upon resumption of the manufacturers’ transfer activities and as we seek to reduce our own internal resources, facilities, and infrastructure capabilities. We will need substantial additional funding to continue our operating activities, including these technical transfers, and to make further advancements in our drug development programs, as described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Liquidity and Capital Resources” of this Annual Report.
We currently rely on third-party suppliers to provide the raw materials that are used by us or our third-party manufacturers in the manufacture of our drugs. There are a limited number of suppliers for raw materials, including nitric oxide, that we use to manufacture our drugs.
Single Business Segment
We manage our operations and allocate resources as one reporting segment. For additional information, please refer to the notes to our consolidated financial statements included in this Annual Report.
Employees
As of December 31, 2019, we had 42 employees (including one part-time employee), including 20 dedicated to our Nitricil technology and formulation science research, development and manufacturing capability, 11 in clinical operations, non-clinical
29
development, and regulatory, and 11 in general and administrative functions. As a result of actions taken during February 2020 intended to reduce our internal resources in order to align with our business conditions and future operating strategy, we expect to have a total of 28 employees as of April 1, 2020 (including four part-time employees). We also utilize consultants and contractors from time to time to support our operating activities and our employees. None of our employees is subject to a collective bargaining agreement or represented by a labor or trade union. We believe that our relations with our employees are good.
Other Information
We were incorporated under the laws of the State of Delaware in 2006. Our principal executive offices are located at 4105 Hopson Road Morrisville, NC 27560, and our telephone number is 919-485-8080.
We maintain an internet website at www.novan.com and make available free of charge through our website our Annual Report, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Exchange Act. We make these reports available through our website as soon as reasonably practicable after we electronically file such reports with, or furnish such reports to, the SEC. Additionally, the SEC maintains an internet website at www.sec.gov that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The information contained on, or that can be accessible through, our website is not incorporated by reference into, and should not be considered to be a part of, this Annual Report.
Item 1A. Risk Factors.
Our operations and financial results are subject to a high degree of risk. These risks include, but are not limited to, those described below, each of which may have a material and adverse effect on our business, results of operations, cash flows, financial condition and the trading price of our common stock. You should carefully consider the risks described below, together with all of the other information included in this Annual Report. The realization of any of these risks could have a significant adverse effect on our reputation, business, including our financial condition, results of operations and growth, which we refer to collectively in this section as our business, and ability to accomplish our strategic objectives. In that event, the trading price of our common stock could decline, and you may lose part or all of your investment.
Risks Related to Our Current Financial Position and Need for Additional Capital
We have restated our consolidated financial statements for the Affected Periods, which may lead to additional risks and uncertainties, including loss of investor confidence and negative impacts on our stock price.
As discussed in the Explanatory Note and “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements included in this Annual Report, on May 14, 2020, we concluded that, because of a misapplication of the accounting guidance applicable to the warrants we issued in January 2018, our previously issued consolidated financial statements for the year ended December 31, 2018 and the interim periods ended March 31, 2019 and 2018, June 30, 2019 and 2018, and September 30, 2019 and 2018, or collectively the Affected Periods, should no longer be relied upon. As such, we determined that we would restate our consolidated financial statements for each of the Affected Periods and that we would revise our consolidated financial statements for the year ended December 31, 2019 in connection with the restatement of our consolidated financial statements for the Affected Periods. As a result of these events, we have become subject to a number of additional costs and risks, including unanticipated costs for accounting and legal fees in connection with the restatement and the remediation of our ineffective disclosure controls and procedures and material weakness in internal control over financial reporting. In addition, the attention of our management team has been diverted by these efforts. We could be subject to additional stockholder, regulatory or other actions in connection with the restatement or other matters. If any such actions occur, they will, regardless of the outcome, consume a significant amount of management’s time and attention and may result in additional legal, accounting, insurance and other costs. If we do not prevail in any such proceedings, we could be required to pay substantial damages or settlement costs. In addition, the restatement and related matters could impair our reputation or could cause our counterparties to lose confidence in us. Each of these occurrences could have a material adverse effect on our business, results of operations, financial condition and stock price.
We have identified a material weakness in our internal control over financial reporting. This material weakness could continue to adversely affect our ability to report our results of operations and financial condition accurately and in a timely manner.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with United States generally accepted accounting principles, or U.S. GAAP. Our management is likewise required, on a quarterly basis, to evaluate the effectiveness of our internal controls and to disclose any changes and material weaknesses identified through such evaluation in those internal controls. A material weakness is a
30
deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
As described elsewhere in this Annual Report, we identified a material weakness in our internal control over financial reporting related to the accounting for a significant and unusual transaction related to the warrants we issued in connection with the January 2018 Offering. As a result of this material weakness, our management concluded that our internal control over financial reporting was not effective as of December 31, 2019. This material weakness resulted in a material misstatement of our warrant liability, change in fair value of warrant liability, additional paid-in capital, accumulated deficit and related financial disclosures for the Affected Periods.
To respond to this material weakness, we have devoted, and plan to continue to devote, significant effort and resources to the remediation and improvement of our internal control over financial reporting. While we have processes to identify and appropriately apply applicable accounting requirements, we plan to enhance these processes to better evaluate our research and understanding of the nuances of the complex accounting standards that apply to our consolidated financial statements. Our plans at this time include providing enhanced access to accounting literature, research materials and documents and increased communication among our personnel and third party professionals with whom we consult regarding complex accounting applications. The elements of our remediation plan can only be accomplished over time, and we can offer no assurance that these initiatives will ultimately have the intended effects. For a discussion of management’s consideration of the material weakness identified related to our accounting for a significant and unusual transaction related to the warrants we issued in connection with the January 2018 Offering, see “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements, as well as Part II, Item 9A: Controls and Procedures included in this Annual Report.
Any failure to maintain such internal control could adversely impact our ability to report our financial position and results from operations on a timely and accurate basis. If our financial statements are not accurate, investors may not have a complete understanding of our operations. Likewise, if our financial statements are not filed on a timely basis, we could be subject to sanctions or investigations by the stock exchange on which our common stock is listed, the SEC or other regulatory authorities. In either case, there could result a material adverse effect on our business. Failure to timely file will cause us to be ineligible to utilize short form registration statements on Form S-3 or Form S-4, which may impair our ability to obtain capital in a timely fashion to execute our business strategies or issue shares to effect an acquisition. Ineffective internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our stock.
We can give no assurance that the measures we have taken and plan to take in the future will remediate the material weakness identified or that any additional material weaknesses or restatements of financial results will not arise in the future due to a failure to implement and maintain adequate internal control over financial reporting or circumvention of these controls. In addition, even if we are successful in strengthening our controls and procedures, in the future those controls and procedures may not be adequate to prevent or identify irregularities or errors or to facilitate the fair presentation of our consolidated financial statements.
If we are unable to secure additional capital and/or delay, defer, or reduce our cash expenditures, we estimate that our existing capital resources will only be sufficient to fund our operations into the early part of the second quarter of 2020.
As of December 31, 2019, we had a total cash and cash equivalents balance of $13.7 million and positive working capital of $2.8 million. As of the date of this filing, we believe that our existing cash and cash equivalents cash balance will provide us with adequate liquidity to fund our planned operating needs into the early part of the second quarter of 2020, as described in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Overview” of this Annual Report. We will need substantial additional funding to continue our operating activities and make further advancements in our drug development programs, as described in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Overview” of this Annual Report. Therefore, we will need to secure additional capital or financing and/or delay, defer, or reduce our cash expenditures by the early part of the second quarter of 2020, including those associated with our product development programs, or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. If we are forced to terminate or eliminate our product development programs or pursue other strategic alternatives or corporate transactions, there can be no assurance that such actions would result in any additional stockholder value. If we are forced to wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders, whereby, our stockholders may lose some or all of their investment.
31
We will need substantial additional funding to continue our business operations and for the advancement of our product development programs. If we are unable to raise capital, we will be forced to delay, reduce, terminate or eliminate our product development programs.
Our ability to continue to operate our business, including our ability to advance our development programs, is dependent upon our ability to access additional capital through non-dilutive sources, including partnerships, collaborations, licensing, grants or other strategic relationships, and/or through the issuance of debt or equity securities. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. There can be no assurance that we will be able to obtain additional capital on terms acceptable to us, on a timely basis or at all. A failure to obtain sufficient funds on acceptable terms when needed could impact our ability to execute on planned business and operational objectives, impact our ability to retain key personnel or cause us to alter or reduce our planned operating activities to conserve our cash and cash equivalents, including but not limited to delaying planned activities directly related to or in support of product candidate development, such as delaying the targeted launch of a confirmatory clinical trial for SB206 for the treatment of molluscum. Such actions could delay development timelines and have a material adverse effect on our results of operations, financial condition, and market valuation. As of December 31, 2019, we had an accumulated deficit of $220.0 million and there is substantial doubt about our ability to continue as a going concern.
Conducting preclinical studies and clinical trials is a time-consuming, expensive and uncertain process that takes years to complete, and we may never generate the necessary data or results required to obtain regulatory approval and achieve product sales. In addition, our product candidates, if approved, may not achieve commercial success by us or our potential partners. Our commercial-related cash flows, if any, will be derived from sales of products that we do not expect to be commercially available for several years, if at all. Accordingly, we will need to continue to rely on additional capital to achieve our business objectives. The magnitude and timing of our future capital requirements will depend on many factors, including:
• | the initiation, progress, timing, costs, results, and evaluation of results of trials for our clinical-stage product candidates, including trials conducted by us or potential future partners; |
• | the progress, timing, costs and results of development and preclinical study activities relating to other potential applications of our nitric oxide platform; |
• | the number and characteristics of product candidates that we pursue; |
• | our ability to enter into strategic relationships to support the continued development of certain product candidates and the success of those arrangements; |
• | our success in optimizing the size and capability of our current manufacturing facility and related processes to meet our strategic objectives; |
• | our success in the technical transfer of methods and processes related to our drug substance and drug product manufacturing with our current and/or potential future contract manufacturing partners; |
• | the outcome, timing and costs of seeking regulatory approvals; |
• | the occurrence and timing of potential development and regulatory milestones achieved by Sato, our licensee for SB204 and SB206 in Japan; |
• | the terms and timing of any future collaborations, licensing, consulting, financing or other arrangements that we may enter into; |
• | the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
• | the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights; |
• | defending against intellectual property related claims; |
• | the costs associated with any potential future securities litigation, and the outcome of that litigation; |
• | the extent to which we in-license or acquire other products and technologies; and |
• | subject to receipt of marketing approval, revenue received from commercial sales or out licensing of our product candidates. |
32
Our process of considering financial and strategic alternatives could adversely affect our business, financial condition, and results of operations.
We have announced that we are in the process of considering financial and strategic alternatives to deliver value to our stockholders. Such alternatives might include, among other things, out-licensing some or all of our product candidates, the sale of some or all of our assets, such as a sale of our dermatology platform assets, or a sale of our company, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us. We may pursue such alternatives at the same time as we seek to secure additional funding. This process could disrupt and create uncertainty concerning our business, regardless of whether we are able to obtain additional funding or complete any strategic alternatives, and poses other risks to our business, including:
• | potential uncertainty in the marketplace concerning our ongoing viability as a business; |
• | the possibility of disruption to our business and operations, including diversion of significant management time and resources towards the pursuit of funding and strategic alternatives; |
• | impairment of our ability to attract and retain key personnel who are necessary to the operation of the business and the development of its product candidates; |
• | restrictions on our business operations and ability to explore other strategic alternatives under any definitive agreement we may enter into as a result of this process; and |
• | potential future stockholder litigation relating to the strategic process that could prevent or delay the strategic process, and the related costs of such litigation. |
If any of the foregoing risks were realized, our business, financial condition, and results of operations could be adversely affected.
Raising additional capital may cause significant dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
We must secure, by the early part of the second quarter of 2020 and until such time, if ever, as we can generate substantial product revenues, additional capital through non-dilutive sources, including partnerships, collaborations, licensing, grants or other strategic relationships, and/or through the issuance of debt or equity securities (including any common stock issued to Aspire Capital pursuant to the Aspire Common Stock Purchase Agreement). Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders.
We are actively pursuing non-dilutive strategic funding transactions around certain of our late-stage product candidates, including SB206 for the treatment of molluscum contagiosum, and the broader dermatology platform as a whole. If we are able to enter into one or more such transactions, we may have to relinquish valuable rights to our technologies, future potential revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us, or we may be restricted operationally or in how we employ the resources provided through such an arrangement. For example, in late April 2019 and early May 2019, respectively, we entered into (i) the Purchase Agreement with Reedy Creek that provided $25.0 million of immediate funding; and (ii) the Funding Agreement with Ligand that provided $12.0 million of immediate funding to be used exclusively in the advancement of our Phase 3 molluscum program. We have also entered into an exclusive license agreement with Sato relating to SB204 and SB206 for the treatment of acne vulgaris and viral skin infections, respectively, in Japan.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, or if we agree to grant warrants or issue other equity to our strategic partners in connection with collaboration or other strategic arrangements, current ownership interest will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect current rights of stockholders. For example, in January 2018 we issued and sold 10.0 million shares of common stock and 10.0 million warrants to purchase shares of common stock in a public follow-on offering which resulted in dilution to our existing stockholders. There is a provision in each warrant that provides its holder the option to receive a cash settlement, equal to the Black-Scholes value of the remaining unexercised portion of the warrant, in certain specified situations involving a “fundamental transaction” (as defined in the warrants, which generally includes a merger with another entity, the sale, transfer or other disposition of all or substantially all of our assets to another entity, or the acquisition by a person of more than 50% of our common stock). Furthermore, it may be uncertain whether a particular transaction or series of transactions will trigger the cash-settlement provision or if the provision is triggered what the actual payment due to a warrant holder would be in such circumstance, but any such payment could be material to us and could materially and adversely affect our financial condition and may prevent or deter a third party from acquiring us. Moreover, we may find it more difficult to raise additional equity capital needed for our business or to pursue strategic alternatives or other corporate transactions while the warrants are
33
outstanding. Our common stock purchase agreement with Aspire Capital entered into in August 2019 provides that, upon the terms and subject to the conditions and limitations set forth therein, we may sell as much as an aggregate of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement.
Debt financing, if available, may involve agreements that include covenants requiring that we place liens on some or all of our assets or limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, effecting a change in control or declaring dividends.
The report of our independent registered public accounting firm on our consolidated financial statements for the years ended December 31, 2019 and 2018, contains an explanatory paragraph regarding going concern, and we will need additional financing to execute our business plan, to fund our operations and to continue as a going concern.
Since inception, we have experienced recurring operating losses and negative cash flows and we expect to continue to generate operating losses and consume significant cash resources in the foreseeable future. These conditions raise substantial doubt about our ability to continue as a going concern without additional financing. As a result, our independent registered public accounting firm included explanatory paragraphs in its report on our 2019 and 2018 consolidated financial statements, with respect to this uncertainty. Substantial doubt about our ability to continue as a going concern may materially and adversely affect the price per share of our common stock and we may have a more difficult time obtaining financing.
We have prepared our consolidated financial statements on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business. Our 2019 consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
The way in which we utilize our late-stage clinical dermatology product candidates to secure needed operating capital may significantly impact our business strategy, future operations and financial position.
We are currently considering and may engage in one or more potential transactions that could result in the licensing, sale or divestiture of some or all of our clinical-stage dermatology product candidates and related proprietary technologies. In certain potential scenarios, the counterparty(ies) to such a transaction may assume responsibility for the planning, execution, or oversight of the clinical development and regulatory requirements for the associated product candidates and/or the ultimate commercialization of the product candidates. If we decide to engage in such a transaction and, as a result, no longer have significant involvement or responsibility for late-stage clinical development activities or commercialization, we would adjust our business strategy, operating plans, resources and capabilities accordingly. Alternatively, we may pursue a transaction in which the counter-party agrees to finance the continued development of one or more product candidates in exchange for future milestone or royalty payments. Absent any such transaction and resulting change in strategic direction, we anticipate continuing to progress our late-stage dermatology product candidates through our existing business model to the extent funding is available. We cannot provide any commitment as to the timing of any such transaction or change in strategy we may adopt. If we determine to change our business strategy or to seek to engage in a strategic transaction, our future business, prospects, financial position and operating results could be significantly different from those in historical periods or projected by our management. Because of the significant uncertainty regarding our future plans, we are not able to accurately predict the impact of a potential change in our existing business strategy.
If we fail to meet the requirements for continued listing on the Nasdaq Global Market, our common stock could be delisted from trading, which would decrease the liquidity of our common stock and our ability to raise additional capital.
Although our common stock is currently listed on The Nasdaq Global Market, an active trading market for our shares may not be sustained. We are required to meet specified requirements to maintain our listing on The Nasdaq Global Market, including, among other things, a minimum $50.0 million market value of listed securities and a minimum bid price of $1.00 per share. On February 19, 2020, we received notice from the staff of the Nasdaq Stock Market LLC, or the Staff, notifying us that for the previous 30 consecutive business days, the minimum bid price of our common stock had not exceeded $1.00 per share and that the market value of our listed securities was below the minimum $50.0 million requirement, or the MVLS Requirement for continued inclusion on The Nasdaq Global Market. We have been provided a period of 180 calendar days, or until August 16, 2020, to regain compliance with the MVLS Requirement and the minimum bid price requirement. If we do not regain compliance with these various listing requirements, the Staff will provide written notification to us that our common stock is subject to delisting.
We are currently evaluating our options for regaining compliance, including the creation of shareholder value through the execution of business objectives described in “Management’s Discussion and Analysis of Financial Condition and Results of
34
Operations” of this Annual Report. However, we cannot guarantee that we will regain compliance with the minimum bid price requirement or the MVLS Requirement by August 16, 2020, or that we will be able to comply with the continued listing standards of The Nasdaq Global Market, and therefore our common stock may be subject to delisting.
If our common stock is delisted and there is no longer an active trading market for our shares, it may, among other things:
• | cause you difficulty in selling your shares without depressing the market price for the shares or sell your shares at all; |
• | substantially impair our ability to raise additional funds; |
• | result in a loss of institutional investor interest and fewer financing opportunities for us; and/or |
• | result in potential breaches of representations or covenants of agreements pursuant to which we made representations or covenants relating to our compliance with applicable listing requirements. Claims related to any such breaches, with or without merit, could result in costly litigation, significant liabilities and diversion of our management’s time and attention and could have a material adverse effect on our financial condition, business and results of operations. |
A delisting would also reduce the value of our equity compensation plans, which could negatively impact our ability to retain key employees.
Risks Related to the Development and Regulatory Approval of our Current and Future Product Candidates
Drug development involves a lengthy and expensive process with uncertain timelines and outcomes, and results from earlier studies and trials may not be predictive of future trial results. If development of our product candidates is unsuccessful or delayed, we may be unable to obtain required regulatory approvals and be unable to commercialize our product candidates on a timely basis, if at all.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure or delay can occur at any time during the clinical trial process. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical trials, even after obtaining promising results in earlier preclinical studies or clinical trials. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including previously unreported adverse events.
The results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. Product candidates in later stages of clinical trials may fail to show the required safety profile or meet the efficacy endpoints despite having progressed through preclinical studies and initial clinical trials. Notwithstanding any potential promising results in earlier testing, we cannot be certain that we will not face similar setbacks. Even if our clinical development is completed for any of our product candidates, the results may not be sufficient to obtain regulatory approval for our product candidates.
On January 2, 2020, we announced top-line results from the Phase 3 B-SIMPLE program with SB206 for the treatment of molluscum contagiosum. Statistical significance was not achieved for the primary endpoint in either B-SIMPLE1 or B-SIMPLE2, however multiple sensitivity analyses were consistent across both studies and we believe support a potential path forward for the asset. These additional analyses do not change the outcome of either B-SIMPLE trial, and the FDA may disagree with our conclusions from these analyses. At present, based on our conclusions from these analyses, we intend to support and confirm B-SIMPLE2 with an additional confirmatory Phase 3 trial targeted to commence in the second quarter of 2020, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020. If during the April 1, 2020 Type-C meeting the FDA disagrees with our conclusions that one additional confirmatory Phase 3 trial is needed for SB206, or if the FDA’s feedback lacks clarity and creates uncertainty around the required clinical development pathway for SB206, the cost to complete the clinical development of SB206 may increase significantly and we may not be able to obtain sufficient financing to advance the SB206 product candidate.
In the first quarter of 2017, we reported top-line results from two identically designed Phase 3 pivotal clinical trials for SB204 for the treatment of acne. SB204 demonstrated statistical significance compared to vehicle on all three co-primary endpoints in one of the trials but demonstrated statistical significance on only one of three co-primary endpoints in the other trial. We previously received feedback from the FDA with respect to potential paths forward for SB204 in 2018. Following the feedback on SB204 for the treatment of acne vulgaris, we have determined that the most pragmatic development pathway for us will be to conduct one additional pivotal Phase 3 trial in moderate-to-severe acne patients, subject to additional funding.
35
We cannot assure you that any additional clinical trials we may conduct for the SB206 and SB204 Phase 3 programs will achieve results that are sufficient to support an FDA submission for the applicable product candidates or regulatory approval of the product. We also cannot assure you that we will be able to obtain financing sufficient to advance development of one or more of our product candidates.
Delay or termination of planned clinical trials for our product candidates could result in unplanned expenses or significantly adversely impact our commercial prospects with respect to, and ability to generate revenues from, such product candidates.
We may experience delays in completing ongoing trials and initiating planned trials and we cannot be certain whether these trials or any other future clinical trials for our product candidates will begin on time, need to be redesigned, enroll an adequate number of patients on time or be completed on schedule, if at all. Clinical trials can be delayed or terminated for a variety of reasons, including delays or failures related to:
• | the FDA disagreeing as to the design or implementation of our clinical trials; |
• | reaching agreement on acceptable terms with prospective CROs, clinical trial sites and prospective strategic partners, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs, trial sites and partners; |
• | obtaining institutional review board, or IRB, approval at each site; |
• | the safety profiles of our product candidates; |
• | recruiting suitable patients to participate in a trial; |
• | having patients complete a trial or return for post-treatment follow-up; |
• | clinical sites deviating from trial protocol; |
• | addressing patient safety concerns that arise during the course of a trial; |
• | adding a sufficient number of clinical trial sites; |
• | manufacturing sufficient quantities of product candidate for use in clinical trials; |
• | utilizing an adequate container and delivery device for the product candidate; or |
• | changes to our financial priorities or insufficient capital available to fund clinical trials. |
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board, or DSMB, for such trial or by the FDA or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse events, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Even if we complete our trials on schedule, inconsistent trial results may result in a delay in our completion of an overall program for a product candidate.
If we experience delays in the completion, or termination, of any future clinical trials for our product candidates, we may experience increased costs, have difficulty raising capital through non-dilutive or dilutive sources, and have to slow down our product candidate development and regulatory approval process timelines. Further, the commercial prospects of our product candidates may be harmed and our ability to generate product revenues from any of these product candidates could be delayed or not realized at all. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
In addition, our ongoing or future preclinical studies may not prove successful in demonstrating proof-of concept, or may show adverse toxicological findings, and even, if successful, may not necessarily predict that subsequent clinical trials will show the requisite safety and efficacy of our product candidates.
We have entered into and may rely on strategic relationships for the further development and commercialization of our product candidates, and if we are unable to enter into such relationships, or if such relationships are unsuccessful, we may be unable to realize the potential economic benefit of those product candidates.
We have entered into various strategic relationships and are continuing to explore alternative pathways for continued development of our product candidates. For example, in 2019 we entered into the Purchase Agreement with Reedy Creek and
36
the Funding Agreement with Ligand Pharmaceuticals, and we are currently exploring and intend to advance certain clinical-stage dermatological product candidates through partnerships, collaborations or other strategic relationships, including those described in the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Overview” of this Annual Report. We cannot assure you that we will be able to complete any other strategic arrangements to finance and support the necessary development for our product candidates. If we are unable to enter into other strategic relationships on terms that are beneficial to us, or at all, we may not have sufficient capital to continue developing or commercialize our product candidates. When we enter into strategic relationships, we may have to relinquish a significant portion of the future economic value of the underlying product candidate(s) in connection with the applicable transactions and may be limited in our ability, or unable, to recover such value.
Our ability to be successful in our current strategic partnerships or enter into other strategic relationships for the continued development of one or more of our product candidates, or for the ultimate commercialization of a product candidate, may be impaired by several factors, including, among others, that:
• | we face significant competition in seeking appropriate strategic partners, and the negotiation process is likely to be time-consuming and complex; |
• | strategic partners who take over development of a product candidate may fail to secure sufficient capital resources to fund planned development activities; |
• | strategic partners may not devote the necessary resources to complete development activities because of limited financial or scientific resources or the belief that other product candidates may have a higher likelihood of obtaining approval or potentially generate a greater return on investment; |
• | strategic partners may fail to properly protect, maintain or defend our intellectual property rights, where applicable, or may use proprietary information in a way that may expose us to potential loss or liability; |
• | we are likely to have limited control over decisions of strategic partners that may result in significant delays or the termination of development and commercialization of our product candidates; |
• | strategic partners may develop a product that competes, directly or indirectly, with our product candidates, or may choose to pursue alternative technologies, including those of our competitors; |
• | disputes between us and our strategic partners concerning the research, development or commercialization of our product candidates or our arrangements with respect to our product candidates could lead to ligation or arbitration that would be costly and detract time from development; and |
• | we or our strategic partners may realize one or more of the risks described within this Item 1A related to the development, regulatory approval and commercialization of our current and future product candidates. |
Further, if a strategic relationship terminates or is otherwise unsuccessful, we may need to identify and establish an alternative arrangement. This may not be possible, or we may not be able to do so on terms which are acceptable to us, in which case, it may be necessary for us to cease the development of the applicable product candidate or candidates, or conduct the remaining clinical development on our own and with our own funds.
If we encounter difficulties or delays enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
The timely completion of clinical trials in accordance with their protocols depends on, among other things, the ability to enroll a sufficient number of patients who remain in the trial until its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons. The enrollment of patients depends on many factors, including:
• | the patient eligibility criteria defined in the protocol; |
• | the size of the patient population required for analysis of the trial’s primary endpoints; |
• | the proximity of patients to trial sites; |
• | the design of the trial; |
• | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
• | clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating; |
37
• | our ability to obtain patient consents; and |
• | the risk that patients enrolled in clinical trials will drop out of the trials before completion. |
In addition, our clinical trials will compete for the recruitment of patients with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition will reduce the number and types of patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors.
Delays in patient enrollment may result in increased costs, which would adversely impact our statement of operations and cash flows or may affect the timing or outcome of the planned clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our product candidates and hurt our competitive position.
Interim, top-line or preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose interim, top-line, or preliminary data from our clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a full analyses of all data related to the particular trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the interim, top-line, or preliminary results that we report may differ from future results of the same trials, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, top-line data should be viewed with caution until the final data are available. We may also disclose interim data from our clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Adverse differences between interim, top-line, or preliminary data and final data could significantly harm our business prospects.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our business in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is the material or otherwise appropriate information to include in our disclosure, and any information we determine not to disclose may ultimately be deemed significant with respect to future decisions, conclusions, views, activities or otherwise regarding a particular drug, product candidate or our business. If the interim, top-line, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for and commercialize our product candidates, our business, operating results, prospects or financial condition may be harmed.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we intend to focus on developing product candidates for specific indications that we identify as most likely to succeed, in terms of their potential both to gain regulatory approval and to achieve commercialization. As a result, we may forego or delay pursuit of opportunities with other product candidates or in other indications with greater commercial potential.
Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable product candidates. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to the product candidate.
38
Our product candidates may pose safety issues, cause adverse events or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
We, any partner with whom we may collaborate in the future, or the FDA may suspend, delay, require modifications to or terminate our clinical trials at any time, for various reasons, including the discovery of serious or unexpected toxicities or other safety issues experienced by trial participants.
In addition, adverse events caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or comparable foreign regulatory authorities. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of adverse events or unexpected characteristics. To date, patients treated with our product candidates have experienced instances of drug-related cutaneous intolerability observations, including dryness, scaling, burning, erythema, itching, pain or irritation, and adverse events, including irritation and contact dermatitis.
If safety issues or unacceptable adverse events arise in the development of our product candidates, we, the FDA, the IRBs at the institutions in which our trials are conducted, or the DSMB could suspend or terminate our clinical trials or the FDA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval of our product candidates for any or all targeted indications. Treatment-related adverse events could also affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. In addition, these adverse events may not be appropriately recognized or managed by the treating medical staff.
Any of the foregoing events could prevent us from achieving or maintaining market acceptance of the particular product candidate, if approved, and may result in the loss of significant revenues to us, which would materially and adversely affect our results of operations and business.
The regulatory approval processes of the FDA are lengthy, time-consuming and inherently unpredictable, and if we, or a potential future partner, are ultimately unable to obtain regulatory approval for our product candidates on a timely basis or at all, our business will be substantially harmed.
The time required to obtain approval by the FDA is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future ourselves or with a potential future strategic partner will ever obtain regulatory approval. Neither we nor any future collaborator is permitted to market any of our product candidates in the United States until we receive regulatory approval of an NDA from the FDA.
Prior to obtaining approval to commercialize a product candidate in the United States or abroad, we or our collaborators must demonstrate with substantial evidence from well-controlled clinical trials, and to the satisfaction of the FDA or foreign regulatory authorities, that such product candidates are safe and effective for their intended uses. Results from preclinical studies and clinical trials can be interpreted in different ways. Even if we believe the preclinical or clinical data for our product candidates are promising, such data may not be sufficient to support approval by the FDA and other regulatory authorities. For example, there are multiple methodologies for handling missing data and other statistical considerations to take into account that the FDA may utilize when analyzing the robustness of any data set during NDA review. The FDA may also require us to conduct additional preclinical studies or clinical trials for our product candidates either prior to or post-approval, or it may object to elements of our clinical development program.
The FDA can delay, limit or deny approval of our product candidates or require us to conduct additional preclinical or clinical testing or abandon a program for many reasons, including:
• | the FDA’s disagreement with the design or implementation of our clinical trials; |
• | unfavorable or ambiguous results from our clinical trials; |
• | results that may not meet the level of statistical significance required by the FDA for approval; |
• | serious and unexpected drug-related adverse events experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; |
39
• | our inability to demonstrate to the satisfaction of the FDA that our product candidates are safe and effective for the proposed indication; |
• | the FDA’s disagreement with the interpretation of data from preclinical studies or clinical trials; |
• | our inability to demonstrate that the clinical and other benefits of our product candidates outweigh any safety or other perceived risks; |
• | the FDA’s requirement for additional preclinical studies or clinical trials; |
• | the FDA’s disagreement regarding the formulation, container, dosing delivery device, labeling or the specifications of our product candidates; |
• | the FDA’s failure to approve the manufacturing processes or facilities of third-party manufacturers with which we contract; or |
• | the potential for approval policies or regulations of the FDA to significantly change in a manner rendering our clinical data insufficient for approval. |
Of the large number of drugs in development, only a small percentage successfully complete the FDA approval process and become commercialized. The lengthy approval process as well as the unpredictability of outcomes from future clinical trials may result in our failing to obtain regulatory approval to market our product candidates.
Even if we or a potential future partner, eventually complete clinical testing and receive approval of an NDA or foreign marketing application for our product candidates, the FDA may grant approval contingent on the performance of costly additional clinical trials, including Phase 4 clinical trials, or the implementation of a Risk Evaluation and Mitigation Strategy, or REMS, which may be required to ensure safe use of the drug after approval. The FDA also may approve a product candidate for a more limited indication or patient population than we originally requested, and the FDA may not approve the labeling that we believe is necessary or desirable for the successful commercialization of a product candidate. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of that product candidate.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise prevent new products and services from being developed or commercialized in a timely manner, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies, such as a federal government shutdown, may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, if a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
Regulatory approval of our product candidates by foreign regulatory authorities may be delayed or denied. We, or our current or potential future partners, may be subject to pricing controls imposed by foreign governments and regulatory authorities.
We, or any current or potential future partners, may seek regulatory approval of our product candidates from foreign regulatory authorities. Such regulatory authorities may impose additional regulations and guidelines that differ in form and substance from those imposed by their counterparts in the United States and with which we are more familiar. Accordingly, the regulatory approval of our product candidates in those foreign jurisdictions could be delayed, limited or denied altogether. This could limit the scope of or prevent the commercialization of our products in the future and adversely affect our financial performance.
40
Further, in some countries, the pricing of pharmaceutical prescriptions is subject to governmental control, including, for example, Japan. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after coverage and reimbursement have been obtained. Reference pricing used by various countries and parallel distribution or arbitrage between low-priced and high-priced countries can further reduce prices. To obtain reimbursement or pricing approval in some countries, we or our current or potential future partners may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies, which is time-consuming and costly. If coverage and reimbursement of our product candidates are unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be harmed.
We may face product liability exposure, and if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. This risk exists even if a product is approved for commercial sale by the FDA or an applicable foreign regulatory authority and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority. Our product candidates are designed to affect important bodily functions and processes. Any adverse events, manufacturing defects, misuse or abuse associated with our product candidates could result in injury to a patient or even death. We cannot offer any assurance that we will not face product liability suits in the future, nor can we assure you that our insurance coverage will be sufficient to cover our liability under any such cases.
In addition, a liability claim may be brought against us even if our product candidates merely appear to have caused an injury. Product liability claims may be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our product candidates, among others. If we cannot successfully defend ourselves against product liability claims we will incur substantial liabilities and reputational harm. In addition, regardless of merit or eventual outcome, product liability claims may result in:
• | withdrawal of clinical trial participants; |
• | decreased enrollment rates of clinical trial participants; |
• | termination of clinical trial sites or entire trial programs; |
• | the inability to commercialize our product candidates; |
• | decreased demand for our product candidates; |
• | impairment of our business reputation; |
• | product recall or withdrawal from the market or labeling, marketing or promotional restrictions; |
• | substantial costs of any related litigation or similar disputes; |
• | distraction of management’s attention and other resources from our primary business; |
• | substantial monetary awards to patients or other claimants against us that may not be covered by insurance; or |
• | loss of revenue. |
We have obtained product liability insurance coverage, with an aggregate limit of $5,000,000, for clinical trials. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated adverse events. Our insurance coverage may not be sufficient to cover all of our product liability related expenses or losses and may not cover us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost, in sufficient amounts or upon adequate terms to protect us against losses due to product liability. We will need to increase our product liability coverage if any of our product candidates receive regulatory approval, which will be costly, and we may be unable to obtain this increased product liability insurance on commercially reasonable terms, or at all. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash, negatively impact our statement of operations and harm our financial condition.
41
Risks Related to the Potential Future Commercialization of Our Product Candidates, if such Product Candidates Complete Development and Receive Regulatory Approval
If we, or a potential future partner, receive regulatory approval to market any of our product candidates, our relationships with healthcare providers, customers and third-party payors, as well as our general business operations, may be subject to applicable anti-kickback, fraud and abuse and other healthcare laws and regulations, and failure to comply with such regulations could expose us to penalties including criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm and diminished profits and future earnings.
Healthcare providers, customers and third-party payors will play a primary role in the recommendation and prescription of any product candidates for which we, or a potential future partner, may obtain marketing approval. Future arrangements with third-party payors, healthcare providers and customers and general operations may expose us, or a potential future partner, to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we, or a potential future partner, market, sell and distribute any product candidates for which we, or a potential future partner, obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations may include the following:
• | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it in order to have committed a violation. |
• | the federal false claims and civil monetary penalties laws, including the civil False Claims Act, which impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; in addition, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
• | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, executing or attempting to execute a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
• | the federal Physician Payments Sunshine Act, which requires manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report annually to the government information related to certain payments or other ‘‘transfers of value’’ made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other healthcare professionals beginning in 2022, and teaching hospitals, and requires applicable manufacturers to report annually to the government ownership and investment interests held by the physicians described above and their immediate family members and payments or other ‘‘transfers of value’’ to such physician owners; and |
• | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; and state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or report marketing expenditures and pricing information. |
42
Efforts to ensure that our internal operations and business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. The risk of our being found in violation of these laws is increased by the fact that many of them have not been fully interpreted by the regulatory authorities or the courts, and their provisions are open to a variety of interpretations. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under such laws, it is possible that some of our business activities, including our relationships with physicians and other healthcare providers, some of whom will recommend, purchase or prescribe our products, could be subject to challenge under one or more of such laws.
If our operations are found to be in violation of any of these laws or any other governmental laws and regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion of products from government funded healthcare programs, such as Medicare and Medicaid, disgorgement, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs, which would adversely impact our statement of operations and cash flows.
Even if our current product candidates or any future product candidates obtain regulatory approval, they may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success.
The commercial success of any of our current or future product candidates, if approved, will depend significantly on the broad adoption and use of the resulting product by physicians and patients for approved indications. Our product candidates may not be commercially successful. The degree and rate of physician and patient adoption of our current or future product candidates, if approved, will depend on a number of factors, including:
• | the clinical indications for which the product is approved and patient demand for approved products that treat those indications; |
• | the effectiveness of our product as compared to other available therapies; |
• | the availability of coverage and adequate reimbursement from managed care plans and other healthcare payors for any of our product candidates that may be approved; |
• | the cost of treatment with our product candidates in relation to alternative treatments and willingness to pay for the product, if approved, on the part of patients; |
• | acceptance by physicians, major operators of clinics and patients of the product as a safe and effective treatment; |
• | physician and patient willingness to adopt a new therapy over other available therapies to treat approved indications; |
• | overcoming any biases physicians or patients may have toward particular therapies for the treatment of approved indications; |
• | patient satisfaction with the results and administration of our product candidates and overall treatment experience; |
• | the willingness of patients to pay for certain of our product candidates relative to other discretionary items, especially during economically challenging times; |
• | the revenue and profitability that our product candidates may offer a physician as compared to alternative therapies; |
• | the prevalence and severity of adverse events; |
• | limitations or warnings contained in the FDA-approved labeling for our product candidates; |
• | any FDA requirement to undertake a REMS; |
• | the effectiveness of our sales, marketing and distribution efforts; |
• | adverse publicity about our product candidates or favorable publicity about competitive products; and |
• | potential product liability claims. |
43
If any of our current or future product candidates are approved for use but fail to achieve the broad degree of physician and patient adoption necessary for commercial success, our operating results and financial condition will be adversely affected, which may delay, prevent or limit our ability to generate revenue and continue our business.
Our product candidates may cause side effects which could delay or prevent their commercialization.
If any of our product candidates receives marketing approval, and we or other companies developing other nitric oxide-based therapies, later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
• | regulatory authorities may withdraw their approval of the product; |
• | we may be required to recall a product or change the way such product is administered to patients; |
• | additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof; |
• | regulatory authorities may require the addition of labeling statements, such as a ‘‘black box’’ warning or a contraindication; |
• | we may be required to implement a REMS or create a Medication Guide outlining the risks of such adverse events for distribution to patients; |
• | we could be sued and held liable for harm caused to patients; |
• | the product may become less competitive; and |
• | our reputation may suffer. |
We expect to educate and train medical personnel so they know how to use our product candidates to understand their potential side effect profiles. Inadequate training in recognizing or managing the potential side effects of our product candidates could result in patient injury.
If we are unable to establish sales, marketing and distribution capabilities for our product candidates or any future product candidate that receives regulatory approval, we may not be successful in commercializing those product candidates, if approved.
We do not currently have a sales, marketing or distribution infrastructure in place. To achieve commercial success for any product candidate for which we may obtain marketing approval, we will need to establish a sales, marketing and distribution framework internally or through a commercial partner or other form of strategic relationship for commercialization. In the future, we may build a focused sales, marketing and distribution infrastructure to market any of our product candidates in the United States. There are risks involved with establishing our own sales, marketing and distribution capabilities. For example, recruiting and training a sales force is expensive and time-consuming and could delay market uptake. If the commercial launch of a product candidate for which we recruit a sales force and establish marketing capabilities is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses. This may be costly, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
Factors that may inhibit our efforts to commercialize our products on our own include:
• | our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
• | the inability of sales personnel to obtain access to physicians or persuade adequate numbers of physicians to prescribe any future products; |
• | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
• | unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
We may enter into arrangements with third parties to perform sales, marketing and distribution services, which could decrease our revenue and our profitability. In addition, we may not be successful in entering into such arrangements with third parties or may be unable to do so on terms that are favorable to us. We may not have adequate control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. In addition, such third parties will be subject to the commercialization risks described above. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates.
44
Additionally, we have entered into an exclusive license agreement in Japan with Sato relating to SB204 and SB206 for the treatment of acne vulgaris and viral skin infections, respectively, and we expect to continue to evaluate strategic partnerships to commercialize our dermatology products in select international markets. We may not be sufficiently familiar or have the requisite resources to penetrate international markets where some of our competitors have already achieved broad recognition and have established commercialization strategies in place. Moreover, we may not succeed in targeting healthcare providers, including physicians, who may not be familiar with our product candidates.
Our product candidates, if approved, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration.
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on developing proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing and marketing of healthcare products competitive with those that we are developing. We face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies and academic and research institutions, many of which have greater financial resources, marketing capabilities, sales forces, manufacturing capabilities, research and development capabilities, clinical trial expertise, intellectual property portfolios, experience in obtaining patents and regulatory approvals for product candidates and other resources than we do. Some of the companies that offer competing products also have a broad range of other product offerings, large direct sales forces and long-term customer relationships with our target physicians, which could inhibit our market penetration efforts. In addition, certain of our product candidates, if approved, may compete with other products, including OTC treatments, for a share of some patients’ discretionary budgets and for physicians’ attention within their clinical practices.
Many pharmaceutical companies currently offer products and continue to develop additional alternative product candidates and technologies for indications similar to those targeted by our product candidates, as described in the section entitled “Business—Competition” in this Annual Report. The markets in which we compete, particularly the market for dermatological therapies, are competitive and are characterized by significant technological development and new product introduction. We anticipate that, if our product candidates obtain regulatory approval, the products will face significant competition from other approved therapies. If approved, our product candidates may also compete with unregulated, unapproved and off-label treatments. To compete successfully in the marketplace, our approved products, if any, will have to demonstrate that the relative cost, safety and efficacy of our approved products, if any, provide an attractive alternative to existing and other new therapies. Such competition could lead to reduced market share for our product candidates and contribute to downward pressure on the pricing of our product candidates.
Due to less stringent regulatory requirements in certain foreign countries, there are many more products and procedures available for use in those international markets than are approved for use in the United States. In certain international markets, there are also fewer limitations on the claims that our competitors can make about the effectiveness of their products and the manner in which they can market them. As a result, we expect our product candidates will face more competition in these markets than in the United States.
Even if any of our product candidates obtain marketing approval, the products may become subject to unfavorable third-party coverage or reimbursement policies, which would harm our business.
The success of our product candidates, if approved, depends on the availability of adequate coverage and reimbursement from government authorities and third-party payors, such as private health insurers and health maintenance organizations. Patients who are provided medical treatment for their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors is critical to product acceptance.
Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs and treatments they will cover and the amount of reimbursement that will be provided. Coverage decisions may depend on clinical and economic standards that disfavor new products when more established or lower cost therapeutic alternatives are already available or subsequently become available. Third-party payors may refuse to include a particular branded product in their formularies or lists of medications for which third-party payors provide coverage and reimbursement, or otherwise restrict patient access through formulary controls or otherwise to a branded product when a less costly generic equivalent or alternative is available. Coverage may be more limited than the purposes for which a product is approved by the FDA or similar regulatory authorities outside the United States.
Assuming that we obtain coverage for a given product, the resulting reimbursement rates might not be adequate to cover our costs, including research, development, manufacture, sale and distribution, or achieve or sustain profitability, or may require co-payments that patients find unacceptably high. Patients are unlikely to use our products unless coverage is provided and
45
reimbursement is adequate to cover a significant portion of the cost of our product candidates. Increasingly, third-party payors are requiring that pharmaceutical companies provide them with predetermined discounts from list prices and are challenging the prices charged for products. There is significant uncertainty related to insurance coverage and reimbursement of newly approved products. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates.
In the United States, no uniform policy of coverage and reimbursement for products exists among third-party payors. Therefore, coverage and reimbursement for a product can differ significantly from payor to payor. As a result, obtaining and maintaining coverage and reimbursement for a product from a government or other third-party payor is a time-consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor separately, with no assurance that adequate coverage and reimbursement will be applied consistently or obtained in the first instance.
Governmental and third-party payors in the United States and abroad are developing increasingly sophisticated methods of controlling healthcare costs. Further, we believe that future coverage and reimbursement will likely be subject to increased restrictions both in the United States and in international markets. Third-party coverage and reimbursement for our product candidates for which we may receive regulatory approval may not be available, limited, or adequate in either the United States or international markets.
Risks Related to Our Reliance on Third-Party Service Providers, Manufacturers, Collaborators and Partners
We may not be successful in continuing to establish or maintain development and commercialization collaborations, which could adversely affect, and potentially prohibit, our ability to develop our product candidates.
We intend to continue to enter into strategic partnerships with third parties to develop and commercialize our product candidates. There can be no assurance that we will be able to establish such collaborations on favorable terms, if at all, or that our current or future collaborative arrangements will be successful. If we are unable to reach successful agreements with suitable collaborators for our product candidates, we would face significant incremental costs, we may be required to limit the scope and number of our product candidates we can commercially develop or the territories in which we commercialize them or we might fail to commercialize products or programs for which a suitable collaborator cannot be found. Our current and future collaboration partners may not dedicate sufficient resources to the development and commercialization of our product candidates or may otherwise fail in their development and commercialization due to factors beyond our control. If we fail to achieve successful collaborations, we may incur additional product development and commercialization expenses and our operating results and financial condition will be materially and adversely affected. If we breach or fail to comply with any provision of a collaboration agreement, a collaborator may have the right to terminate, in whole or in part, such agreement or to seek damages. Some of our collaboration agreements are complex and involve sharing of certain data, know-how and intellectual property rights amongst the parties. Additionally, these potential collaborators may not accept the transfer of critical methods and processes in order for development and commercialization work for our drug product candidates to take place. Our collaborators could interpret certain provisions differently than we do, which could lead to unexpected or inadvertent disputes with our collaborators. Any one of our collaborators could breach obligations, covenants or restrictions in our agreements, leading us into disputes and potential breaches of our agreements with other collaborators, which could have direct or indirect financial implications.
We rely on third parties to conduct preclinical studies and clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize any of our product candidates.
We currently do not have the ability to independently conduct preclinical studies that comply with the regulatory requirements known as good laboratory practice, or GLP, requirements. We also do not currently have the ability to independently conduct any clinical trials. The FDA and regulatory authorities in other jurisdictions require us to comply with regulations and standards, commonly referred to as good clinical practice, or GCP, requirements for conducting, monitoring, recording and reporting the results of clinical trials, in order to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. We rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conduct GLP-compliant preclinical studies and GCP-compliant clinical trials on our product candidates properly and on time. While we will have agreements governing their activities, we control only certain aspects of their activities and have limited influence over their actual performance. The third parties with whom we contract for execution of our GLP preclinical studies and our GCP clinical trials play a significant role in the conduct of these studies and trials and the subsequent collection and analysis of data. These third parties are not our employees and, except for restrictions imposed by our contracts with such third parties, we have limited ability to control the amount or timing of resources that they devote to our programs. Although we rely on these third parties to conduct our GLP-
46
compliant preclinical studies and GCP-compliant clinical trials, we remain responsible for ensuring that each of our GLP preclinical studies and GCP clinical trials is conducted in accordance with its investigational plan and protocol and applicable laws and regulations, and our reliance on the third parties does not relieve us of our regulatory responsibilities. In addition, if any of our third parties terminate their involvement with us for any reason, we may not be able to enter into similar arrangements with alternative third parties within a short period of time or do so on commercially reasonable terms.
Many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. In addition, since the number of qualified clinical investigators is limited, we expect to conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials in such clinical trial site. If the third parties conducting our GLP preclinical studies or our GCP clinical trials do not perform their contractual duties or obligations, experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical trial protocols, GLPs or GCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties. This could be difficult, costly or impossible, and our preclinical studies or clinical trials may need to be extended, delayed, terminated or repeated. As a result, we may not be able to obtain regulatory approval in a timely fashion, or at all, for the applicable product candidate, our financial results and the commercial prospects for our product candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or the FDA concludes that the financial relationship may have affected the interpretation of the trial, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection by the FDA of any NDA we submit. Any such delay or rejection could prevent us from commercializing our future product candidates.
Unexpected results in the analysis of raw materials, the API or drug product or problems with the quality systems supporting analytical work, whether conducted internally or by third-party service providers, could adversely affect our development and commercialization timelines and result in increased costs of our development programs.
We currently rely on third parties to test most of the raw materials necessary to produce our API and drug products. In the future, third parties engaged directly by us or by our API and drug product contract manufacturing organizations, or CMOs, may test all such raw materials. It is a regulatory requirement that raw materials are tested and there are a limited number of suppliers for testing these raw materials. There may be a need to assess alternate suppliers to prevent a possible disruption of the supply of these raw materials for the manufacture of API or drug product. Additionally, the analytical equipment used by these third-parties must be maintained and operational. Except for the terms established within our, or our CMOs, contracts with the third parties responsible for testing raw materials, we have limited ability to control the process or timing of their testing work. Additionally, if the results do not meet specifications, then obtaining additional raw materials may jeopardize the CMOs’ ability to manufacture API and/or drug product and the start or overall conduct of preclinical studies and clinical trials which could result in the delay of developing or commercializing our product candidates.
We currently perform internal tests, and in the future our CMOs will perform tests, to ensure the API and drug product meets quality specifications. The analytical equipment used by us or our CMOs to perform these tests must be maintained, qualified, calibrated and operational. If there are equipment problems or if the results of the analytical testing do not meet our quality specifications, then manufacturing additional API or drug product may increase costs and may jeopardize the CMOs’ ability to manufacture API and/or drug product and the start or overall conduct of preclinical studies and clinical trials which could result in the delay of developing or commercializing our product candidates.
Unexpected delays in the manufacture of our (i) APIs, including NVN1000 API or any other Nitricil NCEs, or (ii) clinical trial materials or drug products, if any, whether by us or any third-party manufacturer, could adversely affect our development and commercialization timelines and result in increased costs of our development programs or in our breaching our obligations to others.
We currently manufacture the NVN1000 API, one of our Nitricil NCEs, for all of our current clinical stage product candidates at our facility in Morrisville, North Carolina. We have a limited number of personnel that have experience in drug substance manufacturing and who possess the expertise necessary to manufacture NVN1000. If our facility were to sustain significant damage, or if we had significant attrition in our manufacturing personnel, or if we have substantial problems with our equipment, our manufacturing operations could be delayed for an extended period of time. If our existing inventories of API are depleted or damaged, we may be unable to supply necessary materials for preclinical studies and clinical trials, causing longer
47
timelines, increased costs and delays in the development and commercialization of drug products, if approved by the FDA or other regulatory authorities.
We intend to outsource to third parties the manufacture of API for our own use and intend to rely on third parties for API that we may provide to others for development and commercial purposes, including Sato, our Japanese market commercial partner. In June 2019, we executed a master contract manufacturing agreement with a full-scale API CMO for the technology transfer and production of our proprietary drug substance. If this CMO or other potential future third-party manufacturers are unable to perform and complete the required technology transfer of the manufacturing processes and analytical methods for API development and commercial manufacturing under cGMP guidelines and regulations, we could experience delays in the development and commercialization timelines of our product candidates, as well as increased costs. Further, if we do not appropriately coordinate with, project manage, or provide adequate internal expertise, resources and documentation to the API CMO, we may not be successful, or may be significantly delayed, in transferring the activities, processes, capabilities and services.
While we have manufactured the finished drug product for clinical trials in our own facilities to date, we have established a strategic alliance with Orion to enable Orion to manufacture our topical nitric oxide-releasing product candidates on our behalf and on behalf of our global strategic partners. If Orion, or any other third-party manufacturer, is unable to perform and complete the required technology transfer of the manufacturing processes and analytical methods for API and drug product development, as applicable, and commercial manufacturing under cGMP guidelines and regulations, we could experience delays in the development and commercialization timelines of our product candidates, as well as increased costs. Further, if we do not appropriately coordinate with, project manage, or provide adequate internal expertise, resources and documentation to our third-party manufacturers, we may not be successful, or may be significantly delayed, in transferring the activities, processes, capabilities and services.
The FDA requires API and finished drug product to be manufactured in accordance with cGMP and be approved by the FDA pursuant to inspections that will be conducted after we, or a potential future partner, submit an NDA to the FDA. Our North Carolina facility has been audited for cGMP compliance by third parties but has not been inspected by the FDA. Orion and the API CMO have been inspected by the FDA and other foreign regulatory authorities, but future inspections could identify findings that could require remediation actions and cause delays to our regulatory approval process. In addition, our manufacturing processes and operating conditions have been evaluated and tested by qualified vendors to ensure a safe operating environment. These tests include raw materials and product handling, process chemistry, air quality and waste disposal and containment. However, if our facilities, or the facilities of a third-party manufacturer are found to be noncompliant with our specifications and the strict regulatory requirements of the FDA or others, we or our third-party manufacturers may be required to take remedial actions, causing further delays and increased costs.
In addition, except for the terms and conditions specified in our contractual arrangements with our contract manufacturers, we have no control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of our API or drug products or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We currently contract with multiple labeling and packaging materials suppliers for our drug products. If we or our labeling and packaging materials suppliers were unable to manufacture and provide the necessary drug product supplies to conduct our clinical trials, we may not be able to contract with another third party in a timely manner to meet our product candidate specifications and supply needs. As a result, we could experience delays in the development and commercialization timelines of our product candidates, as well as increased costs.
Delays and increased costs resulting from any of the above risks would negatively impact our ability to realize operating efficiencies and use of our capital resources.
We rely on third parties to supply raw materials necessary to manufacture our API and drug products. If these third parties do not successfully carry out their contractual duties or meet expected deadlines for raw materials, we may be unable to manufacture API or drug product which could jeopardize the start of preclinical studies or clinical trials and potentially delay or cause failure to obtain regulatory approval for or commercialize any of our product candidates.
We rely on third-party suppliers for the raw materials necessary to produce the API and drug products we require. There are a limited number of suppliers for raw materials, including nitric oxide, that are used in the manufacture of our product candidates, drugs (once approved by the FDA or comparable regulatory authority) or the drug products we supply to others, and
48
there may be a need to assess alternate suppliers to prevent a possible disruption of the manufacture of the materials, importantly nitric oxide, necessary to produce our product candidates for our clinical trials, and if approved, ultimately for commercial sale, or to satisfy our obligations to others. We have not entered into long-term agreements with our current suppliers or with any alternate suppliers. We currently obtain our raw material supplies for finished drug products through individual purchase orders. With future third-party manufacturers of our product candidates, we will not have any control over the process or timing of the acquisition of these raw materials. Moreover, we currently do not have any agreements for the commercial production of these raw materials, including nitric oxide. Although we generally do not begin a clinical trial unless we believe we have a sufficient supply of a product candidate to complete the clinical trial, any significant delay in the supply of the raw material components to manufacture drug products for an ongoing clinical trial due to the need to replace a raw material supplier could considerably delay completion of our clinical trials, product testing and potential regulatory approval of our product candidates. If we or our future third-party manufacturers are unable to purchase these raw materials, including nitric oxide, after regulatory approval has been obtained for our product candidates, the commercial launch of our product candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our product candidates.
Our employees, independent contractors, principal investigators, CMOs, CROs, consultants, commercial partners and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could expose us to liability and hurt our reputation.
We are exposed to the risk that our employees, independent contractors, principal investigators, CMOs, CROs, consultants, commercial partners and vendors may engage in fraudulent conduct or other illegal activity. Misconduct by these parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates: (i) FDA laws and regulations, including those laws that require the reporting of true, complete and accurate information to the FDA, (ii) manufacturing standards, (iii) federal, state and foreign data privacy, security, fraud and abuse and other healthcare laws, or (iv) laws that require the true, complete and accurate reporting of financial information or data. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials, creating fraudulent data in our preclinical studies or clinical trials or illegal misappropriation of drug product, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. Additionally, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and financial results, including, without limitation, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, reputational harm, diminished profits and future earnings, and curtailment of our operations.
Risks Related to Our Operations
Our future success depends on our ability to retain our current executive officers and to attract, retain and motivate qualified personnel.
We are highly dependent on our current executive leadership team, including Paula Brown Stafford, formerly our Chief Operating Officer, and as of February 2, 2020, our President and Chief Executive Officer, and other executive officers and principal members of our management and scientific teams. Although we have a formal employment agreement with Ms. Stafford, this agreement does not prevent her from terminating her employment with us at any time. The loss of the services of any of these persons could impede the achievement of our research, development and commercialization objectives.
Recruiting and retaining qualified personnel is critical to our success. We may not be able to attract and retain these personnel on acceptable terms given our current financial position, recent actions taken to align our resources with our operating strategy, and the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
Changes to our leadership team could prove disruptive to our operations and have adverse consequences for our business and operating results.
During 2018 and 2019, we announced several changes to our executive leadership team. Managing transitions in our executive leadership team may divert our existing management team’s attention from our core operations, and the recent transitions we
49
have experienced may make it more difficult for us to retain existing employees. In addition, the recent transitions we have experienced have increased our dependency on the remaining members of the senior executive team and other key employees within the organization. We have incurred costs related to transitions in our management team, including severance payments, and have required departing executives to agree to certain obligations in their separation agreements. We also expect to incur recruitment costs related to the hiring of new executives from time to time.
Our operating strategy includes the increased use of third-party vendors and strategic partners for the conduct of certain activities, processes, and services that are not part of our primary business strategy, including the large-scale commercial manufacture of our APIs and drug products. If, as a result of the activities, processes, and services being transferred to and performed by third parties and strategic partners, we experience (i) delays or failures (ii) reduced quality, (iii) delayed receipt of goods or services, or (iv) increased and unexpected costs, our clinical development and regulatory timelines and/or our financial position may be adversely affected.
Our operating strategy includes an increased utilization of and reliance upon third-party vendors and strategic partners for the performance of activities, processes and services that (i) do not result in the generation of significant new intellectual property; and (ii) can leverage existing robust infrastructure, systems, and facilities as well as associated subject matter expertise. For example, we have engaged a CMO to perform the large-scale manufacture of our active pharmaceutical ingredients (APIs), including NVN1000, and Orion to perform the large-scale manufacture of our formulated drug products containing NVN1000 and other APIs, respectively, for use in late-stage clinical trials and potential commercialization. When coupled with a parallel strategy to reduce our own internal resources, facilities, and infrastructure capabilities, we believe this broad strategy can ultimately provide enhanced capabilities and operating efficiencies for us or any potential partnerships, collaborations, licensing or other strategic relationships we may enter. However, we may not be successful in realizing the intended operating efficiencies from these arrangements based on a number of factors, including (i) delays or failures, including delays in our ability to transition applicable technology and processes to our vendors or partners, (ii) reduced quality, (iii) delayed receipt of goods or services, (iv) increased and unexpected costs on the part of the third-party vendors or strategic partners, (v) our requested near-term reduction in transfer activities, in an effort to reduce our near-term cash utilization, and (vi) certain incremental and discrete costs to effect this strategy upon resumption of the manufacturers’ transfer activities.
We have recently taken actions to reduce our internal resources, and we may encounter difficulties in managing our business as a result of these actions, or the attrition that may occur following these actions, which could disrupt our operations. In addition, we may not achieve anticipated benefits from these actions.
From time to time, including in November 2018 and February 2020, we have taken actions intended to reduce our internal resources in order to align with our business and operating strategy. We have experienced additional employee attrition following these actions. As of December 31, 2019, we had 41 full-time employees and one part-time employee. As of February 18, 2020, we had 39 full-time employees and one part-time employee. As a result of actions taken during February 2020, we expect to have a total of 28 employees as of April 1, 2020. These actions and any further actions and/or attrition that may occur in the future, result in the loss of institutional knowledge and expertise and the reallocation and combination of certain roles and responsibilities across the organization, all of which could adversely affect our operations. In addition, the actions we have taken and may take in the future may not achieve anticipated benefits or may not enable achievement of our operating strategy. Our management may need to divert a disproportionate amount of its attention away from our day-to-day strategic and operational activities, and devote a substantial amount of time to managing these organizational changes. Due to our limited resources, we may not be able to effectively manage our operations or recruit and retain qualified personnel, which may result in weaknesses in our infrastructure and operations, risks that we may not be able to comply with legal and regulatory requirements, loss of business opportunities, loss of employees and reduced productivity among remaining employees. If our management is unable to effectively manage this transition, our expenses may be more than expected, and we may not be able to implement our business strategy.
Our business involves the use of hazardous materials and we and our third-party suppliers and manufacturers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our manufacturing activities, and the manufacturing activities of our third-party suppliers and manufacturers, involve the controlled storage, use and disposal of hazardous materials, including the components of our product candidates such as nitric oxide and other hazardous compounds. Further, our manufactured drug substance and drug products may be considered hazardous materials under applicable laws and regulations. Our manufacturing activities, whether conducted by us or our third-party suppliers and manufacturers, like all manufacturing processes that utilize hazardous materials, including those under high pressures, must be properly controlled to avoid unintended reactions or other accidents that could cause injury or damage to personnel, equipment or property. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, transportation, handling and disposal of these hazardous materials. In some cases, these hazardous materials and various wastes resulting from their use are transported and stored at our suppliers’ or manufacturers’ facilities
50
pending use and disposal. We and our suppliers and manufacturers cannot completely eliminate the risk of contamination, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, injury to our service providers and others and environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the manufacturing controls and safety procedures utilized by us and our third-party suppliers and manufacturers for handling, transporting and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk (i) that the laws and regulations will not restrict our or our third-party suppliers’ or manufacturers’ ability to use, manufacture, store, transport, handle or dispose of such materials or (ii) of accidental contamination or injury from these hazardous materials and processes. If these risks were to materialize, we could experience an interruption of our business operations and we may be held liable for any resulting damages and such liability could exceed our financial resources.
We currently specialize solely in developing nitric oxide-based topical therapeutics to treat a range of diseases with significant unmet needs, and if we do not successfully achieve regulatory approval for any of our product candidates or successfully commercialize them, we may not be able to continue as a business.
All of our clinical development efforts to date have focused on the development of nitric oxide-based topical therapies. There can be no assurance that the intended or anticipated results from the use of nitric oxide-based therapies will be reaped, and that we, or our existing or potential future commercial partners, will successfully bring our product candidates to market. Because all of our current product candidates are based on nitric oxide and our Nitricil technology, the failure of our Nitricil technology to be safe or efficacious generally will have adverse implications for our entire product candidate pipeline. If, for any reason, our intended use of nitric oxide does not materialize, we may not be able to redeploy our resources to alternative components or raw materials, efficiently or at all.
We have a limited operating history and no history of commercializing drugs, which may make it difficult for you to evaluate the success of our business to date and to assess our future viability.
We commenced operations in 2006, and our operations to date have been largely focused on developing our Nitricil technology and platform of product candidates. We have not yet demonstrated our ability to obtain regulatory approvals, manufacture a drug on a commercial scale, or arrange for a third-party to do so on our behalf, or conduct sales and marketing activities necessary for successful commercialization. Consequently, any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing drugs.
We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives. We will need to transition at some point from a company with a development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
Our business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security.
We maintain sensitive company data on our computer networks, including our intellectual property and proprietary business information. Despite the implementation of security measures, our internal computer systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, natural disasters, terrorism, war, telecommunication and electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs, significant data losses or theft of our intellectual property or proprietary business information. For example, the loss of clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur material legal claims and liability or other negative consequences, including increased cybersecurity protection costs, damage to our reputation, disruption of our internal operations and delays in the further development of our product candidates.
Our disclosure controls and procedures address cybersecurity and include elements intended to ensure that there is an analysis of potential disclosure obligations arising from security breaches. We also maintain compliance programs to address the potential applicability of restrictions against trading while in possession of material, nonpublic information generally and in
51
connection with a cyber-security breach. However, a breakdown in existing controls and procedures around our cyber-security environment may prevent us from detecting, reporting or responding to cyber incidents in a timely manner and could have a material adverse effect on our financial position and value of our stock.
We may be adversely affected by natural disasters and other catastrophic events, and by man-made problems such as terrorism, that could disrupt our business operations and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our corporate headquarters are located in Morrisville, North Carolina, near major hurricane and tornado zones. If a disaster, power outage or other event occurred that prevented us from using all or a significant portion of our headquarters, that damaged critical infrastructure, such as enterprise financial systems, manufacturing resource planning or enterprise quality systems, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Our manufacturers’ and suppliers’ facilities are located in multiple locations, where other natural disasters or similar events, such as blizzards, tornadoes, fires, explosions or large-scale accidents or power outages, could severely disrupt their operations. In addition, acts of terrorism, pandemic illness and other geo-political unrest could cause disruptions in our business or the businesses of our collaborators, manufacturers or the economy as a whole. All of the aforementioned risks may be further increased if we do not implement a disaster recovery plan or our collaborators’ or manufacturers’ disaster recovery plans prove to be inadequate. Any of the above could result in delays in the regulatory approval, manufacture, distribution or commercialization of our product candidates.
Risks Related to Government Regulation
Even if we receive regulatory approval of our product candidates, we will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense, and we may be subject to penalties, if we fail to comply with regulatory requirements or experience unanticipated problems with our product candidates.
Any regulatory approvals that we receive for our product candidates may be subject to limitations on the approved indicated uses for which the product may be marketed or the conditions of approval or contain requirements for potentially costly post-market testing and surveillance to monitor the safety and efficacy of the product candidate. The FDA may also require a REMS as a condition of approval of our product candidates, which could include requirements for a medication guide, physician communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. In addition, if the FDA or a comparable foreign regulatory authority approves our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, and continued compliance with cGMPs and GCP requirements for any clinical trials that we conduct post-approval. Later discovery of previously unknown problems with our product candidates, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• | restrictions on the marketing or manufacturing of our product candidates, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
• | fines, warning letters or holds on clinical trials; |
• | refusal by the FDA to approve pending applications or supplements to approved applications filed by us or suspension or revocation of approvals; |
• | product seizure or detention, or refusal to permit the import or export of our product candidates; and |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may fail to obtain any marketing approvals, lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. For example, certain policies of the Trump administration may impact our business and industry. Namely, the Trump administration has taken several executive actions, including the issuance of a number of executive orders, that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and oversight activities such as implementing statutes through rulemaking, issuance of guidance,
52
and review and approval of marketing applications. It is difficult to predict how these or future executive actions will be implemented, and the extent to which they will impact the FDA’s ability to exercise its regulatory authority. In addition, if we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
If we obtain regulatory approval for our product candidates in the United States, any such approval will be limited to the specific indication authorized by the FDA. If we are found to be in violation of FDA and other regulations restricting the promotion of any approved products for unapproved uses, we could be subject to criminal penalties, substantial fines or other sanctions and damage awards.
If our clinical trials are successful, we intend to seek approval for our product candidates for various indications for use. If we obtain regulatory approval to market any of our product candidates with an indication statement for the treatment of one or more of these indications, we will likely be prohibited from marketing any approved products for uses outside of those for which we have received approval.
The regulations relating to the promotion of products for unapproved uses are complex and subject to substantial interpretation by the FDA and other government agencies. Products may not be promoted for uses that are not approved in the labeling by the FDA or EMA. Physicians may, following FDA approval, nevertheless prescribe our products off-label to their patients in a manner that is inconsistent with the approved label. We intend to implement compliance and training programs designed to ensure that our sales and marketing practices comply with applicable regulations. Notwithstanding these programs, the FDA or other government agencies may allege or find that our practices constitute prohibited promotion of our products for unapproved uses. We also cannot be sure that our employees will comply with company policies and applicable regulations regarding the promotion of products for unapproved uses, but we may nevertheless be deemed responsible for their marketing activities.
In recent years, a significant number of pharmaceutical and biotechnology companies have been the target of inquiries and investigations by various federal and state regulatory, investigative, prosecutorial and administrative entities in connection with the promotion of products for unapproved uses and other sales practices, including the Department of Justice and various U.S. Attorneys’ Offices, the Office of Inspector General of the Department of Health and Human Services, the FDA, the Federal Trade Commission and various state Attorneys General offices. These investigations have alleged violations of various federal and state laws and regulations, including claims asserting antitrust violations, violations of the Federal Food, Drug and Cosmetic Act, the False Claims Act, the Prescription Drug Marketing Act, anti-kickback laws and other alleged violations in connection with the promotion of products for unapproved uses, pricing and Medicare and/or Medicaid reimbursement. Many of these investigations originate as “qui tam” actions under the False Claims Act. Under the False Claims Act, any individual can bring a claim on behalf of the government alleging that a person or entity has presented a false claim, or caused a false claim to be submitted, to the government for payment. The person bringing a qui tam suit is entitled to a share of any recovery or settlement. Qui tam suits, also commonly referred to as “whistleblower suits,” are often brought by current or former employees. In a qui tam suit, the government must decide whether to intervene and prosecute the case. If it declines, the individual may pursue the case alone.
If the FDA or any other governmental agency initiates an enforcement action against us or if we are the subject of a qui tam suit and it is determined that we violated prohibitions relating to the promotion of products for unapproved uses, we could be subject to warning letters, untitled letters, substantial civil or criminal fines or damage awards and other sanctions such as consent decrees and corporate integrity agreements pursuant to which our activities would be subject to ongoing scrutiny and monitoring to ensure compliance with applicable laws and regulations. Any such fines, awards or other sanctions would have an adverse effect on our revenue, business, financial prospects and reputation.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize our product candidates and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval. In 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively the ACA. However, there have been significant ongoing efforts to modify or eliminate the ACA. For example, the Tax Cuts and Jobs Act, enacted on December 22, 2017, repealed the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code, commonly referred to as the individual mandate, beginning in 2019. Further legislative changes to and regulatory changes under the ACA remain possible. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of
53
the ACA, and therefore, because it was repealed as part of the Tax Cuts and Jobs Act, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit ruled that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. It is unclear how these decisions, subsequent appeals, if any, and other efforts to challenge, repeal or replace the ACA will impact the law or our business in the future.
We expect that the ACA, as well as other healthcare reform measures that have been adopted and may be adopted in the future, may, among other things, result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability, or commercialize our product candidates, if approved. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of our product candidates, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
We are subject to governmental economic sanctions and export and import controls that could impair our ability to compete in international markets or subject us to liability if we are not in compliance with applicable laws.
As a U.S. company, we are subject to U.S. import and export controls and economic sanctions laws and regulations, and we are required to import and export our product candidates, technology and services in compliance with those laws and regulations, including the U.S. Export Administration Regulations, the International Traffic in Arms Regulations, and economic embargo and trade sanction programs administered by the Treasury Department’s Office of Foreign Assets Control.
U.S. economic sanctions and export control laws and regulations prohibit the shipment of certain products and services to countries, governments and persons targeted by U.S. sanctions. While we are currently taking precautions to prevent doing any business, directly or indirectly, with countries, governments and persons targeted by U.S. sanctions and to ensure that our product candidates, if approved, are not exported or used by countries, governments and persons targeted by U.S. sanctions, such measures may be circumvented.
Furthermore, if we export our product candidates, if approved, the exports may require authorizations, including a license, a license exception or other appropriate government authorization. Complying with export control and sanctions regulations for a particular sale may be time-consuming and may result in the delay or loss of sales opportunities. Failure to comply with export control and sanctions regulations for a particular sale may expose us to government investigations and penalties.
If we are found to be in violation of U.S. sanctions or import or export control laws, it could result in civil and criminal, monetary and non-monetary penalties, including possible incarceration for those individuals responsible for the violations, the loss of export or import privileges and reputational harm.
We are subject to anti-corruption and anti-money laundering laws with respect to our operations and non-compliance with such laws can subject us to criminal or civil liability and harm our business.
We are subject to the U.S. Foreign Corrupt Practices Act of 1977, as amended, or the FCPA, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act and possibly other anti-bribery and anti-money laundering laws in countries in which we may conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees and third-party intermediaries from authorizing, offering or providing, directly or indirectly, improper payments or benefits to recipients in the public or private sector. As we commercialize our product candidates and eventually commence international sales and business, we may engage with collaborators and third-party intermediaries to sell our products abroad and to obtain necessary permits, licenses and other regulatory approvals. We or our third-party intermediaries may have direct or indirect interactions with officials and employees of government agencies or state-owned or affiliated entities. We may be held liable for the corrupt or other illegal activities of these third-party intermediaries, our employees, representatives, contractors, partners and agents, even if we do not explicitly authorize such activities.
54
Noncompliance with anti-corruption and anti-money laundering laws could subject us to whistleblower complaints, investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension or debarment from contracting with certain persons, the loss of export privileges, reputational harm, adverse media coverage and other collateral consequences. Responding to any action will likely result in a materially significant diversion of management’s attention and resources and significant defense costs and other professional fees.
Changes in and failures to comply with U.S. and foreign privacy and data protection laws, regulations and standards may adversely affect our business, operations and financial performance.
We are subject to or affected by numerous federal, state and foreign laws and regulations, as well as regulatory guidance, governing the collection, use, disclosure, retention, and security of personal data, such as information that we collect about patients and healthcare providers in connection with clinical trials in the United States and abroad. The global data protection landscape is rapidly evolving, and implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future. This evolution may create uncertainty in our business, affect our or our collaborators’, service providers’ and contractors’ ability to operate in certain jurisdictions or to collect, store, transfer use and share personal information, necessitate the acceptance of more onerous obligations in our contracts, result in liability or impose additional costs on us. The cost of compliance with these laws, regulations and standards is high and is likely to increase in the future. Any failure or perceived failure by us or our collaborators, service providers and contractors to comply with federal, state or foreign laws or regulation, our internal policies and procedures or our contracts governing processing of personal information could result in negative publicity, diversion of management time and effort and proceedings against us by governmental entities or others. In many jurisdictions, enforcement actions and consequences for noncompliance are rising.
In the United States, HIPAA imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon “covered entities” (health plans, health care clearinghouses and certain health care providers), and their respective business associates, individuals or entities that create, received, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HIPAA mandates the reporting of certain breaches of health information to HHS, affected individuals and if the breach is large enough, the media. Entities that are found to be in violation of HIPAA as the result of a breach of unsecured protected health information, or PHI, a complaint about privacy practices or an audit by the Department of Health and Human Services, or HHS, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Even when HIPAA does not apply, according to the Federal Trade Commission or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent, broader in scope or offer greater individual rights with respect to PHI than HIPAA and many of which may differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For example, California enacted the California Consumer Privacy Act, or the CCPA, on June 28, 2018, which took effect on January 1, 2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability. Some observers have noted that the CCPA could mark the beginning of a trend toward more stringent privacy legislation in the United States, which could increase our potential liability and adversely affect our business.
Our operations abroad may also be subject to increased scrutiny or attention from data protection authorities. Many countries in these regions have established or are in the process of establishing privacy and data security legal frameworks with which we, our collaborators, service providers, including our CRO, and contractors must comply. For example, the EU has adopted the EU General Data Protection Regulation (EU) 2016/679, or GDPR, which went into effect in May 2018 and introduces strict requirements for processing the personal information of EU subjects, including clinical trial data. The GDPR has and will continue to increase compliance burdens on us, including by mandating potentially burdensome documentation requirements and granting certain rights to individuals to control how we collect, use, disclose, retain and process information about them.
55
The processing of sensitive personal data, such as physical health condition, may impose heightened compliance burdens under the GDPR and is a topic of active interest among foreign regulators. In addition, the GDPR provides for more robust regulatory enforcement and fines of up to €20 million or 4% of the annual global revenue of the noncompliant company, whichever is greater. Moreover, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the EU will be regulated, especially following the United Kingdom's departure from the EU on January 31, 2020 without a deal. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom's departure from the EU. As we expand into other foreign countries and jurisdictions, we may be subject to additional laws and regulations that may affect how we conduct business.
Risks Related to Our Intellectual Property
If we fail to comply with our obligations under any license, collaboration or other agreements, it could have a material adverse effect on our, or potential future commercial partners’, commercialization efforts for our product candidates.
Our current licenses impose, and any future licenses we enter into may impose, various development, commercialization, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for our product candidates, we also rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position.
We seek to protect our trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees (including through specific provisions in employment contracts), corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be materially impaired.
Our reliance on third parties requires us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we expect to rely on third parties to manufacture any of our current or future product candidates, we must, at times, share trade secrets with them. These agreements typically limit the rights of the third parties to use or disclose our confidential information, including our trade secrets. Despite the contractual provisions employed when working with third parties, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, a competitor’s discovery of our trade secrets or other unauthorized use or disclosure would impair our competitive position and may adversely impact our business.
If we are unable to obtain and maintain patent protection for our product candidates, or if the scope of the patent protection obtained is not sufficiently broad, our competitors could develop and commercialize technology and products similar or identical to ours, and our ability to successfully commercialize our technology and product candidates may be impaired.
We rely upon a combination of patents, trade secret protection, and confidentiality agreements to protect the intellectual property related to our product candidates. Our success depends in large part on our ability to obtain and maintain patent protection in the United States and other countries with respect to our product candidates. We seek to protect our proprietary position by filing patent applications in the United States and abroad related to our product candidates.
56
The patent prosecution process is expensive and time-consuming, however, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our technology platform or product candidates before it is too late to obtain patent protection. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents licensed to or from third parties. In particular, certain patents and patent applications covering our core technology platform are exclusively licensed from the University of North Carolina, or UNC, and under our license agreement with UNC, we rely on UNC to prosecute and maintain such patents and applications. Therefore, these patents and applications, and any other patents and applications that we may license from or to third parties, may not be prosecuted and enforced in a manner consistent with the best interests of our business.
If the patent applications we hold or have in-licensed with respect to our product candidates fail to issue, if their breadth or strength of protection is threatened, or if they fail to provide meaningful exclusivity for our current or any future product candidates, it could have a materially adverse effect on our business. Even if our owned and licensed patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our owned and licensed patents by developing similar or alternative technologies or products in a non-infringing manner.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions and has in recent years been the subject of much litigation. In addition, the laws of foreign countries may not protect our rights to the same extent as the laws of the United States or vice versa. For example, European patent law restricts the patentability of methods of treatment of the human body more than United States law does. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot know with certainty whether we were the first to make the inventions claimed in our owned and licensed patents or pending patent applications, or that we were the first to file for patent protection of such inventions. As a result, the issuance, scope, validity, enforceability and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued that protect our technology or products, in whole or in part, or which effectively prevent others from commercializing competitive technologies and products. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our owned and licensed patents or narrow the scope of our patent protection while patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
Changes to patent laws in the United States or other countries could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. For example, changes to the United States patent system have come into force under the Leahy-Smith America Invents Act, or the Leahy-Smith Act, which was signed into law in September 2011. The Leahy-Smith Act included a number of significant changes to United States patent law. Under the Leahy-Smith Act, the United States transitioned in March 2013 to a “first to file” system in which the first inventor to file a patent application will be entitled to the patent. Third parties are allowed to submit prior art before the issuance of a patent by the USPTO, and may become involved in opposition, derivation, reexamination, inter partes review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, which could adversely affect our competitive position. While we cannot predict with certainty the impact the Leahy-Smith Act or any potential future changes to the United States or foreign patent systems will have on the operation of our business, the Leahy-Smith Act and such future changes could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, results of operations, financial condition and prospects. Additionally, the first to file system under the Leahy-Smith Act may incentivize companies like us in the biopharmaceutical industry to file patent applications as soon as possible, and filing applications as soon as possible runs the risk that the application will not have the supporting data to claim the broadest protection possible in the United States.
Moreover, we may be subject to a third-party preissuance submission of prior art to the USPTO or become involved in opposition, derivation, reexamination, inter partes review, post-grant review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our owned and licensed patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
57
In addition, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our owned and licensed patents may be challenged in the courts or patent offices in the United States and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our technology and products. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
Finally, certain of our activities and our licensors’ activities have been funded, and may in the future be funded, by the U.S. federal government. When new technologies are developed with U.S. federal government funding, the government obtains certain rights in any resulting patents, including a nonexclusive license authorizing the government to use the invention for non-commercial purposes. These rights may permit the government to disclose our confidential information to third parties and to exercise “march-in” rights to use or allow third parties to use our patented technology. The government can exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the U.S. government-funded technology, because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations or to give preference to U.S. industry. In addition, U.S. government-funded inventions must be reported to the government, U.S. government funding must be disclosed in any resulting patent applications, and our rights in such inventions may be subject to certain requirements to manufacture products in the United States.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and other foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign national or international patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of patent rights include, but are not limited to, failure to timely file national and regional stage patent applications based on our international patent application, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we or our licensors fail to maintain the patents and patent applications covering our technology platform or product candidates, our competitors might be able to enter the market, which would have an adverse effect on our business.
Changes in U.S. patent laws could diminish the value of patents in general, thereby impairing our ability to protect our products.
The United States has recently enacted and implemented wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances, modifying some legal standards applied by the USPTO in examination of patent applications or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on actions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents, increase the likelihood of challenges to patents we obtain or license or weaken our ability to enforce patents that we have licensed or that we might obtain in the future.
58
We may be involved in lawsuits to protect or enforce our owned and licensed patents, which could be expensive, time-consuming and unsuccessful. Further, our issued patents could be found invalid or unenforceable if challenged in court.
If we were to initiate legal proceedings against a third-party to enforce a patent directed to our product candidates, or one of our future product candidates, the defendant could counterclaim that our patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, non-enablement or insufficient written description. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO or made a misleading statement during prosecution. Third parties may also raise similar claims before the USPTO, even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such a loss of patent protection would harm our business.
Interference proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our owned and licensed patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms, or at all.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Most of our competitors are larger than we are and have substantially greater resources than we do. They are, therefore, likely to be able to sustain the costs of complex patent or other intellectual property rights litigation longer than we could. Accordingly, despite our efforts, we may not be able to prevent third parties from infringing upon or misappropriating our intellectual property. Litigation could result in substantial costs and diversion of management resources, which could harm our business. In addition, the uncertainties associated with litigation could compromise our ability to raise the funds necessary to continue our clinical trials, continue our internal research programs, or in-license needed technology or other product candidates. There could also be public announcements of the results of the hearing, motions, or other interim proceedings or developments. If securities analysts or investors perceive those results to be negative, it could cause the price of shares of our common stock to decline.
We may not be able to protect our intellectual property rights throughout the world, which could impair our business.
Filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our invention in such countries. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may export otherwise infringing products to territories where we have patent protection, but enforcement rights are not as strong as those in the United States. These products may compete with our product candidates and our owned and licensed patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of some countries do not favor the enforcement of patents and other intellectual property protection, which could make it difficult for us to stop the infringement of our owned and licensed patents generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our owned and licensed patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful.
Many countries, including European Union countries, India, Japan and China, have compulsory licensing laws under which a patent owner may be compelled under specified circumstances to grant licenses to third parties. In those countries, we may have limited remedies if patents are infringed or if we are compelled to grant a license to a third party, which could materially diminish the value of those patents. This could limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
59
We may not be able to obtain licenses to third-party intellectual property. Third parties may initiate legal proceedings alleging infringement of their intellectual property rights.
A third party may hold intellectual property, including patent rights that are important or necessary to the development or commercialization of our product candidates. However, we may not be able to obtain such licenses on commercially reasonable terms, or at all. In addition, our existing licenses may be terminated or may not be renewed, which could hurt our business.
In addition, our commercial success depends upon our ability to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. There is considerable intellectual property litigation in the biotechnology and pharmaceutical industries. We may become party to, or threatened with, future adversarial proceedings or litigation regarding intellectual property rights with respect to our products and technology, including interference or derivation proceedings before the USPTO. Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, and as we gain greater visibility and market exposure as a public company, the risk increases that our product candidates or other business activities may be subject to claims of infringement of the patent and other proprietary rights of third parties. Third parties may assert infringement claims against us based on existing patents or patents that may be granted in the future. We have conducted searches for information in support of patent protection and otherwise evaluating the patent landscape for nitric oxide releasing materials and products, and, based on these searches and evaluations to date, we do not believe that there are valid patents which contain granted claims that could be asserted with respect to our nitric oxide-based product candidates.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates or force us to cease some of our business operations. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. If we are found to infringe a third party’s intellectual property rights, we could be required to redesign our infringing products or obtain a license from such third party to continue developing and marketing our products and technology. However, we may not be able to obtain any required license on commercially reasonable terms or at all. Even if we were able to obtain a license, it could be non-exclusive, thereby giving our competitors access to the same technologies licensed to us. Moreover, we could be forced, including by court order, to cease commercializing the infringing technology or product. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business.
We may be subject to claims by third parties asserting that we or our employees have misappropriated their intellectual property or claiming ownership of what we regard as our own intellectual property.
Many of our employees were previously employed at other biotechnology or pharmaceutical companies or universities. Although we try to ensure that our employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that these employees or we have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such employee’s former employer. Litigation may be necessary to defend against these claims.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own. Our and their assignment agreements may not be self-executing or may be breached, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property.
If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to management.
60
Any trademarks we have obtained or may obtain may be infringed or successfully challenged, materially harming to our business.
We expect to rely on trademarks as one means to distinguish any of our product candidates that are approved for marketing from the products of our competitors. Once we select new trademarks and apply to register them, our trademark applications may not be approved. Third parties may oppose or attempt to cancel our trademark applications or trademarks, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. Further, our competitors may infringe our trademarks, including with respect to our Nitricil technology and we may not have adequate resources to enforce our trademarks.
Outside of the United States we cannot be certain that any country’s patent or trademark office will not implement new rules that could seriously affect how we draft, file, prosecute and maintain patents, trademarks and patent and trademark applications.
We cannot be certain that the patent or trademark offices of countries outside the United States will not implement new rules that increase costs for drafting, filing, prosecuting and maintaining patents, trademarks and patent and trademark applications or that any such new rules will not restrict our ability to file for patent protection. For example, we may elect not to seek patent protection in some jurisdictions or for some product candidates in order to save costs. We may be forced to abandon or return the rights to specific patents due to a lack of financial resources.
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and may not adequately protect our business, or permit us to maintain our competitive advantage, for reasons including but not limited to the following:
• | others may be able to make formulations or compositions that are the same as or similar to certain of our product candidates but that are not covered by the claims of the patents that we own or license; |
• | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our trade secret or similar rights; |
• | issued patents that we own or license may not provide us with any competitive advantages, or may be held invalid or unenforceable as a result of legal challenges; |
• | our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for certain research and development activities, as well as in countries where we do not have patent rights, and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; and |
• | we may not develop additional proprietary technologies that are patentable. |
Risks Related to Our Financial Results and Obligations and to Our Common Stock
We have incurred significant losses since our inception. We expect to incur losses for at least the next several years and may never achieve or maintain profitability.
Since inception, we have incurred significant operating losses. Our net loss was $30.4 million for the year ended December 31, 2019, and $29.2 million for the year ended December 31, 2018. As of December 31, 2019, we had an accumulated deficit of $220.0 million. As a result of our historical operating losses, current lack of liquidity and expected future negative cash flows from operations, we have concluded that there is substantial doubt about our ability to continue as a going concern. Similarly, the report of our independent registered public accounting firm on our December 31, 2019 financial statements includes an explanatory paragraph indicating that there is substantial doubt about our ability to continue as a going concern. To date, we have financed our operations primarily through the sale of our securities in public offerings, upfront and milestone payments from licensing and other strategic partnering agreements, private placements of our preferred stock, convertible notes and proceeds from government research contracts and grants. We have devoted substantially all of our efforts to research and development, including clinical trials. We have not completed development of any product candidates. We expect to continue to incur significant expenses and operating losses for at least the next several years. We anticipate that we will continue to incur substantial expenses if and as we:
• | continue to conduct clinical trials for our existing clinical stage product candidates; |
• | initiate clinical trials for other future product candidates and new chemical entities; |
61
• | seek regulatory approvals for our product candidates that complete clinical trials; |
• | qualify contract manufacturing organizations for the manufacture of drug product for the commercial launch of our product candidates; |
• | establish a sales, marketing and distribution infrastructure or partnership to commercialize products for which we may obtain regulatory approval; |
• | maintain, expand and protect our intellectual property portfolio; |
• | continue our research and development efforts; |
• | exercise any development or commercialization rights we may have under any arrangements with collaborators or partners; |
• | hire additional scientific, clinical and management personnel; |
• | add, modify or enhance executive, operational, financial and management information systems and personnel; |
• | incur costs associated with any potential future securities litigation, and the outcome of that litigation; and |
• | incur additional legal, accounting and other expenses in operating as a public company. |
To become and remain profitable, we must independently, or in collaboration with our current and potential future partners, develop and eventually commercialize a product or products with significant market potential. This development and commercialization will require us or our current and potential future partners to be successful in a range of challenging activities, including successfully completing clinical trials of our product candidates, obtaining regulatory approval for these product candidates, and marketing and selling those products that may obtain regulatory approval. We are only in the preliminary stages of some of these activities. We or our current and potential future partners may never succeed in these activities and may never generate revenues that are significant or large enough to enable us to achieve profitability.
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would decrease the value of the company and could impair our ability to raise capital, expand our business, maintain our research and development efforts or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We have broad discretion in the use of our financial resources, including our cash and cash equivalents, and may not use them effectively.
Except for the limitations imposed by the Purchase Agreement with Reedy Creek and the Funding Agreement with Ligand, our management has broad discretion in the application of our financial resources, including our cash and cash equivalents, and could spend our cash in ways that do not improve our results of operations or enhance the value of our common stock. Our future use of our financial resources may differ substantially from our current plans. The failure by our management to apply our financial resources effectively could result in financial losses that could have a material adverse effect on our business and cause the price of our common stock to decline. Pending their use, we may invest our cash and cash equivalents in a manner that does not produce income or that loses value.
In August 2018, our board approved and established the Tangible Stockholder Return Plan, a performance-based long-term incentive plan with two distinct share price targets. We may not be able to achieve the applicable targets, and even if they are achieved, we may not have the financial resources available to make the bonus payments contemplated by the plan.
On August 2, 2018, our board approved and established the Tangible Stockholder Return Plan, or the Performance Plan, which is a performance-based long-term incentive plan.
The Performance Plan is tiered, with two separate tranches, each of which has a distinct share price target (measured as the average publicly traded share price of our stock on the Nasdaq stock exchange for a thirty consecutive trading day period) that will trigger a distinct fixed bonus pool. The share price targets for the first and second tranches are $11.17 per share and $25.45 per share, respectively. The bonus pools for the first and second tranche are $25.0 million and $50.0 million, respectively. The compensation committee has discretion to distribute the bonus pool related to each tranche among eligible participants by establishing individual minimum bonus amounts before, as well as by distributing the remainder of the applicable pool after, the achievement of each tranche specific share price target. Otherwise, if we do not achieve one or both related share price
62
targets, as defined, no portion of the bonus pools will be paid. See “Note 12—Tangible Stockholder Return Plan” to the accompanying consolidated financial statements included in this Annual Report for details regarding the Performance Plan.
Management intends to continue to assess the facts and circumstances, in addition to its capital structure and liquidity, with regards to our potential obligations related to the Performance Plan and the likelihood of future payment. There can be no assurance that we will achieve either or both share price targets during the term of the Performance Plan, that we will have sufficient cash on hand to pay cash bonuses under the Performance Plan at the time any share price target is achieved or within the time frames described above for payment of the bonuses, or that we will receive stockholder approval to pay bonuses in shares of our common stock in lieu of some or all of such cash payment, if sought. These factors may impact our business, financial condition, ability to retain key employees and ability to obtain additional capital. In addition, in the event of a change in control, the plan provides that a bonus pool will become due and payable to participants on a pro rata basis, as calculated and determined by the compensation committee based upon our progress toward the share price target as of the date of the change in control and subject to adjustment by the compensation committee as permitted under the plan, which could increase the cost to acquire our company and prevent or delay a change in control.
Our ability to utilize our net operating loss, or NOL, carryforwards may be limited.
As of December 31, 2019, we had NOL carryforwards available to reduce future taxable income, if any, for federal and state income tax purposes of $165.6 million and $165.1 million, respectively. If not utilized, the federal and state NOL carryforwards will begin expiring in 2028 and 2023 for federal and state tax purposes, respectively. Our ability to utilize NOL carryforward amounts to reduce taxable income in future years may be limited for various reasons, including if future taxable income is insufficient to recognize the full benefit of such NOL carryforward amounts prior to their expiration. Additionally, our ability to fully utilize these U.S. tax assets can also be adversely affected by “ownership changes” within the meaning of Section 382 of the Internal Revenue Code of 1986, as amended, or the Code, in a three-year period. Any ownership change is generally defined as a greater than 50% increase in equity ownership by “5% stockholders,” as that term is defined for purposes of Section 382 of the Code in any three-year period. Although we have not completed a full analysis under Section 382, our initial public offering, or IPO, combined with our public offering in January 2018 may have resulted in an ownership change as defined in Section 382. Further, we may experience an ownership change in the future as a result of further shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change NOL carryforwards to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the state level, there may be periods during which the use of NOL carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
The price of our common stock may be volatile and fluctuate significantly, which could result in substantial losses for our existing stockholders.
Our stock price has in the past been, and is likely to be in the future, volatile. The stock market in general has experienced extreme volatility that has often been unrelated to the operating performance of particular companies. During the period from January 1, 2017 to February 18, 2020, the closing sales price of our common stock ranged from a high of $26.86 per share to a low of $0.44 per share. As a result of this volatility, our existing stockholders may not be able to sell their stock at a favorable price. The market price for our common stock may be influenced by many factors, including:
• | reports of clinical trial results or steps in the regulatory approval process; |
• | actual or anticipated fluctuations in our financial condition and operating results; |
• | actual or anticipated changes in our growth rate relative to our competitors; |
• | potential competition from existing products or new products that may emerge; |
• | development of new technologies that may address our markets and may make our technology less attractive; |
• | changes in physician, hospital or healthcare provider practices that may make our product candidates less attractive; |
• | announcements by us, our partners or our competitors regarding significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
• | developments or disputes concerning proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
• | the recruitment or departure of key personnel; |
63
• | failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public; |
• | actual or anticipated changes in estimates as to financial results, development timelines or recommendations by securities analysts; |
• | variations in our financial results or those of companies that are perceived to be similar to us; |
• | changes to reimbursement levels by commercial third-party payors and government payors, including Medicare, and negative announcements relating to reimbursement levels; |
• | public market’s assessment of our ability to raise additional capital; |
• | general economic, industry and market conditions; and |
• | the other factors described in this “Risk Factors” section. |
A certain degree of stock price volatility can be attributed to limited trading volume; our average daily trading volume during 2019 was approximately 165,000 shares, or less than one percent of the weighted average number of common shares outstanding during that period. This lack of liquidity in the marketplace has and may continue to cause significant volatility in the price of our common stock.
In addition, the stock market in general and emerging growth companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. These broad market and industry fluctuations may negatively impact the price or liquidity of our common stock, regardless of our operating performance. Any actual or perceived negative operational developments or market or industry fluctuations may compound each other’s negative impacts on the price of liquidity of our common stock.
We could again be subject to securities class action litigation, which could result in substantial damages and may divert management’s time and attention from our business.
Putative stockholder class action lawsuits were filed against us and certain of our current and former directors and officers in 2017. Although the court has dismissed those putative stockholder class actions with prejudice, we have concluded that these matters are closed, and we currently have no other pending or threated litigation against us, we may face similar securities class action litigation in the future. If we face similar litigation again in the future, it could result in substantial costs and a diversion of management’s attention and resources.
Our executive officers, directors and principal stockholders, if they choose to act together, will have the ability to control or significantly influence matters submitted to stockholders for approval.
Our executive officers, directors and stockholders who owned more than 5% of our outstanding common stock and their respective affiliates, in the aggregate, hold shares representing approximately 27% of our outstanding voting common stock as of February 3, 2020. As a result, if these stockholders were to choose to act together, they would be able to significantly influence matters submitted to our stockholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, would control or significantly influence the election of directors and approval of any merger, consolidation or sale of all or substantially all of our assets. This concentration of ownership control may:
• | delay, defer or prevent a change in control; |
• | entrench our management and the board of directors; or |
• | impede a merger, consolidation, takeover or other business combination involving us that other stockholders may desire. |
The significant concentration of stock ownership may negatively impact the price of our common stock due to investors’ perception that conflicts of interest may exist or arise.
64
Provisions in our amended and restated certificate of incorporation and amended and restated bylaws under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may discourage, delay or prevent a merger, acquisition or other change in control of our company that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. In addition, because our board of directors is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Among other things, these provisions include those establishing:
• | a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; |
• | no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
• | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from filling vacancies on our board of directors; |
• | the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could be used to significantly dilute the ownership of a hostile acquirer; |
• | the ability of our board of directors to alter our amended and restated bylaws without obtaining stockholder approval; |
• | the required approval of the holders of at least two-thirds of the shares entitled to vote at an election of directors to adopt, amend or repeal our amended and restated bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors; |
• | a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
• | the requirement that a special meeting of stockholders may be called only by the chief executive officer, the chairman, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and |
• | advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the General Corporation Law of the State of Delaware, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner.
Our restated certificate of incorporation provides that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, or employees or agents.
Our restated certificate of incorporation provides that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on behalf of us; (ii) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers or other employees or agents to us or our stockholders; (iii) any action asserting a claim against us arising pursuant to any provision of the Delaware General Corporation Law or our amended and restated certificate of incorporation or amended and restated bylaws; or (iv) any action asserting a claim against us governed by the internal affairs doctrine. These choice of forum provisions do not preclude or contract the scope of exclusive federal or concurrent jurisdiction for any actions brought under
65
the Securities Act or the Exchange Act. Accordingly, our choice of forum provisions will not relieve us of our duties to comply with the federal securities laws and the rules and regulations thereunder, and our stockholders will not be deemed to have waived our compliance with these laws, rules and regulations.
Any person or entity purchasing or otherwise acquiring any interest in any of our securities shall be deemed to have notice of and consented to these provisions. These choice of forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum of its choosing for disputes with us or our directors, officers or other employees or agents, which may discourage lawsuits against us and our directors, officers and other employees or agents.
If a court were to find the choice of forum provision contained in our restated certificate of incorporation to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our business, results of operations, and financial condition. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management and other employees.
Future sales of our common stock, or the perception that future sales may occur could cause our stock price to decline.
If our existing stockholders sell, or indicate an intent to sell, substantial amounts of our common stock that are eligible for sale in the public market, in some cases subject to compliance with the requirements of Rule 144, the trading price of our common stock could decline significantly. As of February 3, 2020, we had approximately 27.4 million shares of common stock outstanding and exercisable warrants to purchase approximately 10 million shares of common stock outstanding. Certain other of our stockholders hold substantial amounts of our common stock. If substantial amounts of shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
We may sell up to $25.0 million of our shares of common stock to Aspire Capital pursuant to our common stock purchase agreement with Aspire Capital. The sale of a substantial number of shares of our common stock by Aspire Capital, or anticipation of such sales, could cause the trading price of our common stock to decline or make it more difficult for us to sell equity or equity-related securities in the future at a time and at a price that we might otherwise desire. However, we have the right to control the timing and amount of sales of our shares to Aspire Capital, and we may terminate the financing arrangement at any time at our discretion without any penalty or cost to us.
The issuance of shares upon exercise of our outstanding warrants and options may cause substantial dilution to our existing stockholders and reduce the trading price of our common stock.
We have outstanding and exercisable warrants and options that if exercised may result in dilution to the interests of other stockholders and may reduce the trading price of our common stock. We presently have warrants to purchase 10 million shares of common stock outstanding and exercisable with an exercise price of $4.66 per share. In addition, we had outstanding and exercisable options to purchase approximately 1.0 million shares of common stock as of December 31, 2019 with a weighted average exercise price of $4.81 per share.
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and may remain an emerging growth company until the earliest of (i) the end of the fiscal year in which the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the end of the second fiscal quarter, (ii) the end of the fiscal year in which we have total annual gross revenue of $1.07 billion or more during such fiscal year, (iii) the date on which we issue more than $1.0 billion in non-convertible debt in a three-year period or (iv) December 31, 2021, the end of the fiscal year following the fifth anniversary of the completion of our IPO. For so long as we remain an emerging growth company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include:
• | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; |
• | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements; |
• | reduced disclosure obligations regarding executive compensation; and |
• | exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
66
We have taken advantage of reduced reporting requirements in this Annual Report. In particular, we do not intend to provide all of the executive compensation related information that would be required if we were not an emerging growth company. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be reduced or more volatile. In addition, the JOBS Act provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an emerging growth company to delay the adoption of these accounting standards until they would otherwise apply to private companies. We have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards and the accompanying demands on time and resources as other public companies that are not emerging growth companies face.
We have and expect to continue to incur substantial costs as a result of operating as a public company, and our management has and will continue to be required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, and particularly after we are no longer an emerging growth company, we have and will continue to incur significant legal, accounting and other expenses. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of The Nasdaq Global Market and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. Our management and other personnel devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations have and will continue to require substantial legal and financial compliance costs and make some activities more time-consuming and costly. For example, these rules and regulations make it more difficult and more expensive for us to obtain director and officer liability insurance, which in turn could make it more difficult for us to attract and retain qualified members of our board of directors.
We continue to evaluate these rules and regulations and cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
If securities or industry analysts do not publish research or reports about our business, or if they issue an adverse or misleading opinion regarding our stock, our stock price and trading volume could decline.
The trading market for our common stock is influenced by the research and reports that industry or securities analysts publish about us or our business. If any of the analysts who cover us issue an adverse or misleading opinion regarding us, our business model, our intellectual property or our stock performance, or if our regulatory clearance timelines, clinical trial results or operating results fail to meet the expectations of analysts, our stock price would likely decline. If one or more of these analysts ceases coverage of us or fail to publish reports on us regularly, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
Because we do not anticipate paying any cash dividends on our capital stock in the foreseeable future, capital appreciation, if any, will be our stockholders’ sole source of gain.
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all of our future earnings, if any, to finance the growth and development of our business. Any future debt agreements may preclude us from paying dividends. As a result, capital appreciation, if any, of our common stock will be our stockholders’ sole source of gain for the foreseeable future.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 2. Properties.
We currently operate out of our corporate headquarters is in Morrisville, North Carolina, where we lease an existing 51,350 square foot facility under a lease with an initial term expiring in 2026. This facility was designed and upfit specifically for our nitric oxide research and development activities. We have an option to extend the lease agreement by five years upon completion of the initial lease term. We use our facility for primary research, development, and drug compound and product manufacturing activities, as well as general and administrative purposes, to support our nitric oxide technology and drug development programs.
67
In May 2018, we entered into an agreement whereby we, as sublessor, subleased 6,400 square feet of office space to a third party from our existing facility square footage. In October 2019, we executed a termination agreement with the subtenant for this sublease, with an effective date of December 31, 2019.
As part of our current operating strategy described in the section entitled “Business—Manufacturing and Supplies” and the section entitled “Management’s Discussion and Analysis and Results of Operations—Overview—Business Updates—Drug Substance and Drug Product Agreements” in this annual report, we continue to explore further opportunities to potentially sublease additional space within our facility. We have selected contract manufacturing organizations (CMOs) and have begun transferring the manufacture and production technology for our drug product candidates and our NVN1000 API to these CMOs for clinical development and potential future commercial purposes. The progression of the relationships with the aforementioned third-party manufacturers is integral to the advancement of our dermatological platform, including our SB206 molluscum program, by us or through partnerships, collaborations, licensing or other strategic relationships. We believe this strategy can ultimately provide enhanced capabilities and operating efficiencies for us or any potential partnerships, collaborations, licensing or other strategic relationships we may enter.
Item 3. Legal Proceedings.
We are not currently a party to any material legal proceedings and are not aware of any claims or actions pending or threatened against us that we believe could have a material adverse effect on our business, operating results, cash flows or financial statements. In the future, we may from time to time become involved in litigation relating to claims arising from our ordinary course of business.
Item 4. Mine Safety Disclosures.
Not applicable.
68
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
Market Information
Our common stock has traded on the Nasdaq Global Market under the symbol “NOVN” since September 21, 2016. Prior to that time, there was no public market for our common stock.
Holders
As of February 18, 2020, there were approximately 131 stockholders of record of our common stock. Holders of record are defined as those stockholders whose shares are registered in their names in our stock records and do not include beneficial owners of common stock whose shares are held in the names of brokers, dealers or clearing agencies.
Dividends
We have never paid cash dividends and do not anticipate paying cash dividends in the foreseeable future.
Recent Sales of Unregistered Securities
During the quarter ended December 31, 2019, we sold an aggregate of 200,000 shares of common stock to Aspire Capital under the Aspire Common Stock Purchase Agreement, generating aggregate proceeds of $0.5 million. This is in addition to the 100,000 shares of common stock sold to Aspire Capital in the third quarter of 2019 as reported in our Form 10-Q for such period, for an aggregate of 300,000 shares sold under the Aspire Common Stock Purchase Agreement for the year ended December 31, 2019. Each issuance of these unregistered shares qualifies as an exempt transaction pursuant to Section 4(a)(2) of the Securities Act of 1933 because none involved a public offering. Each offering was not a public offering due to the number of persons involved, the manner of the issuance and the number of securities issued. Such shares are registered for re-sale by Aspire Capital on our Registration Statement on Form S-1 (File No. 333-233632) registering 7,032,630 shares of common stock that have been or may be offered to Aspire Capital from time to time under the Aspire Common Stock Purchase Agreement.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
We did not purchase any of our equity securities during the fourth quarter of 2019.
69
Item 6. Selected Financial Data.
Not applicable.
70
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
This Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read with our consolidated financial statements and notes thereto included elsewhere in this Annual Report. In addition to historical information, the following discussion contains forward-looking statements that involve risks, uncertainties and assumptions. Where possible, we have tried to identify these forward-looking statements by using words such as “believe,” “contemplate,” “continue,” “due,” “goal,” “objective,” “plan,” “seek,” “target,” “expect,” “believe,” “anticipate,” “intend,” “may,” “will,” “would,” “could,” “should,” “potential,” “predict,” “project,” or “estimate,” and similar expressions or variations. These statements are based on the beliefs and assumptions of our management based on information currently available to management. Forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Except as may be required by law, we undertake no obligation to update any forward-looking statements to reflect events or circumstances after the date of such statements. These forward-looking statements are subject to numerous risks including, but not limited to, those set forth in the “Risk Factors” in Part I, Item 1A of this Annual Report.
Restatement of Previously Issued Consolidated Financial Statements
This Management’s Discussion and Analysis of Financial Condition and Results of Operations has been amended and restated to give effect to the restatement and revision of our consolidated financial statements as more fully described in the Explanatory Note and in “Note 2—Restatement of Consolidated Financial Statements” to our accompanying consolidated financial statements. For further detail regarding the restatement adjustments, see Explanatory Note and Item 9A: Controls and Procedures, both contained herein.
Overview
We are a clinical development-stage biotechnology company focused on leveraging nitric oxide’s naturally occurring anti-viral, anti-bacterial, anti-fungal and immunomodulatory mechanisms of action to treat a range of diseases with significant unmet needs. Nitric oxide plays a vital role in the natural immune system response against microbial pathogens and is a critical regulator of inflammation. Our ability to harness nitric oxide and its multiple mechanisms of action has enabled us to create a platform with the potential to generate differentiated product candidates.
The two key components of our nitric oxide platform are our proprietary Nitricil technology, which drives the creation of new chemical entities, or NCEs, and our formulation science, both of which we use to tune our product candidates for specific indications. Our ability to deploy nitric oxide in a solid form, on demand and in localized formulations allows us the potential to improve patient outcomes in a variety of diseases.
We have advanced strategic development programs in the field of dermatology, while also further expanding the platform into women’s health and gastroenterological, or GI, therapeutic areas. We have clinical-stage dermatology drug candidates with multi-factorial (SB204), anti-viral (SB206), anti-fungal (SB208) and anti-inflammatory (SB414) mechanisms of action. We have recently introduced SB207 as a possible product candidate for additional anti-viral programs. During 2019, our clinical-stage development efforts were focused on our molluscum contagiosum (SB206) and atopic dermatitis (SB414) programs. We also conducted preclinical work on NCEs and formulations for the treatment of human papilloma virus, or HPV, related illnesses in the women’s health field (WH504 and WH602) and inflammatory diseases in the GI field. During 2020, we intend to focus our clinical-stage development efforts on our molluscum contagiosum (SB206) program, subject to available capital and regulatory feedback. All other clinical-stage programs are currently on hold. Further advancement of the molluscum contagiosum (SB206) program, or any other program across our platform, is dependent upon our ability to access additional capital. Additional capital may potentially include (i) non-dilutive sources, such as partnerships, collaborations, licensing, grants or other strategic relationships; or (ii) equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. We intend to pursue financing which may be dilutive, non-dilutive or both, in the near future. We intend to utilize one or more financial advisors to assist in the pursuit of optimal capital sourcing pathways, including those that are strategic in nature and center around our late-stage assets, and the broader dermatology platform and underlying Nitricil technology, as well as exploring other potential sources of financing and strategic alternatives.
As of December 31, 2019, we had a total cash and cash equivalents balance of $13.7 million and positive working capital of $2.8 million. We believe that our existing cash and cash equivalents balance will provide us with adequate liquidity to fund our planned operating needs into the early part of the second quarter of 2020. This projected cash runway excludes (i) potential costs associated with an additional confirmatory Phase 3 trial, which is subject to additional funding and feedback from a Type
71
C meeting with the FDA scheduled for April 1, 2020, and (ii) any proceeds received subsequent to January 31, 2020 from potential future sales of common stock under the Aspire Common Stock Purchase Agreement, described below, if available.
We will need substantial additional funding to continue our operating activities and make further advancements in our drug development programs, as described below in “Liquidity and Capital Resources”. Therefore, we will need to secure additional capital or financing and/or delay, defer, or reduce our cash expenditures by the early part of the second quarter of 2020, including those associated with our product development programs, or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. There can be no assurance that we will be able to obtain additional capital or financing on terms acceptable to us, on a timely basis or at all. If we are forced to terminate or eliminate our product development programs, wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders. Alternatively, we may seek to engage in one or more potential transactions, such as the sale of the Company, or sale or divestiture of some of our assets, such as a sale of our dermatology platform assets, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us.
As described below in “Business Updates,” in August 2019 we entered into the Aspire Common Stock Purchase Agreement with an institutional investor, Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase shares of our common stock at our request from time to time during the 30-month term of the agreement.
As also described below in “Business Updates,” in late April 2019 and early May 2019 we entered into (i) a royalty and milestone payments purchase agreement with a stockholder that provided $25.0 million of immediate funding; and (ii) a development funding and royalties agreement with a corporate partner that provided $12.0 million of immediate funding.
We will need additional funding to continue our operating activities and make further advancements in our drug development programs. Please refer to “Liquidity and Capital Resources” for further discussion of our current liquidity and our future funding needs.
During 2019, our primary programmatic focus was on our molluscum contagiosum (SB206) and atopic dermatitis (SB414) programs.
We conducted and completed our SB206 Phase 2 trial for the treatment of molluscum contagiosum in December 2018 and in early March 2019, completed an end-of Phase 2 meeting with the FDA and received written minutes. During the second quarter of 2019, we commenced the Phase 3 pivotal development program for the treatment of molluscum contagiosum. Patient recruitment for the Phase 3 trials was completed in August 2019. Top-line efficacy results from the Phase 3 trials were announced in January 2020. SB206 did not achieve statistically significant results in the primary endpoint in both trials, which was the complete clearance of all molluscum lesions at Week 12. Based on the results of the Phase 3 pivotal trials described below in “Key Clinical Stage Product Candidate Development Updates,” we target commencing an additional confirmatory Phase 3 trial in the second quarter of 2020, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020. Execution of remaining Phase 3 pivotal development program activities continues into 2020 with receipt of pivotal trial safety data in the first quarter of 2020 and completion of ancillary trials targeted prior to or during the second quarter of 2020.
Following the completion of two complementary Phase 1b clinical trials with SB414 in patients with psoriasis and atopic dermatitis in 2018, we conducted non-clinical studies with SB414 for atopic dermatitis and completed our Phase 2 clinical development plan during 2019 to support a potential future Phase 2 clinical program launch. The SB414 program is currently on hold with further advancement subject to obtaining additional financing or strategic partnering.
Key Clinical Stage Product Candidate Development Updates
SB206, a Topical Anti-viral Treatment for Viral Skin Infections
We are developing SB206 as a topical anti-viral gel for the treatment of viral skin infections, with a current focus on molluscum contagiosum. Molluscum is a contagious skin infection caused by the molluscipoxvirus. Molluscum affects up to six million people in the U.S. annually. The greatest incidence is in children aged one to 14 years. The average time to resolution is 13 months, however, 13% of children experience lesions that may not resolve in 24 months. There is no FDA-approved treatment for molluscum. More than half of patients diagnosed with the infection are untreated. The majority of patients that receive treatment are treated with painful procedures and the remaining are often prescribed products indicated for the treatment of external genital warts.
72
We believed that observational learnings from an in-licensed topical nitric oxide technology study showing clinically meaningful complete clearance rates of baseline molluscum lesions, combined with our SB206 program knowledge, provided a logical pathway for SB206 development in the molluscum indication. We submitted an investigational new drug application, or IND, to the FDA in December 2017 and initiated a Phase 2 clinical trial utilizing SB206 for the treatment of molluscum in the first quarter of 2018. The Phase 2 multi-center, randomized, double-blind, vehicle-controlled, ascending dose clinical trial evaluated the efficacy, safety and tolerability of SB206 in 256 patients, ages 2 and above, with molluscum. Patients were treated with one of three concentrations of SB206 or vehicle for up to 12 weeks. The primary endpoint was the proportion of patients achieving complete clearance of all molluscum lesions at Week 12. We announced top-line results from this Phase 2 clinical trial in the fourth quarter of 2018. SB206 demonstrated statistically significant results in the clearance of all molluscum lesions at Week 12, with signs of efficacy evident as early as Week 2 with the 12% once-daily dose. The safety and tolerability profiles were favorable overall with no serious adverse events reported, including the most effective dose, SB206 12% once-daily.
With the full results from this Phase 2 trial made available, we held an end-of-Phase 2 (Type B) meeting with the FDA in early March 2019. Based on this meeting and the written minutes received, we commenced the Phase 3 development program for molluscum, primarily comprised of two pivotal clinical trials, in the second quarter of 2019 with SB206 12% once-daily as the active treatment arm. The “B-SIMPLE” (Berdazimer Sodium In Molluscum Patients with Lesions) Phase 3 pivotal trials consisted of two (B-SIMPLE1 and B-SIMPLE2) multi-center, randomized, double-blind, vehicle-controlled studies to evaluate the efficacy and safety of SB206 12% once-daily in approximately 680 patients (2:1 active:vehicle randomization), ages 6 months and above, with molluscum. Patients were treated once-daily with SB206 12% or Vehicle Gel once daily for a minimum of 4 weeks and up to 12 weeks to all treatable lesions (baseline and new). There were visits at Screening/Baseline, Week 2, Week 4, Week 8, Week 12 and a safety follow-up at Week 24. The primary endpoint was the proportion of patients achieving complete clearance of all molluscum lesions at Week 12. Both Phase 3 pivotal trials began dosing patients in June 2019 and we completed patient recruitment in August 2019. Top-line efficacy results from the Phase 3 trials were announced in January 2020.
SB206 did not achieve statistically significant results in the primary endpoint in both trials, which was the complete clearance of all molluscum lesions at Week 12. In B-SIMPLE2, SB206 was near statistical significance for the primary endpoint (p=0.062), and was statistically significant for the secondary endpoint, the complete clearance of all lesions at Week 8 (p=0.028), and all other pre-specified sensitivity analyses. We believe this confirms the robustness of the data in the B-SIMPLE2 trial. While the B-SIMPLE1 trial was not statistically significant for the primary endpoint (p=0.375) nor the secondary endpoint (p=0.202), all other pre-specified analyses trended in the same direction of improved treatment effect as the B-SIMPLE2 results.
In addition, the results of a statistical test of heterogeneity support that the two pivotal trials are not different from each other. Across both studies, the primary analysis odds ratio and standard error point estimates were similar and in a consistent direction with overlapping 95% confidence intervals. These statistical results are supported by an integrated analysis of the two pivotal trials, which demonstrated statistically significant complete clearance rates at Week 12 for SB206 (p=0.049). These additional analyses do not change the outcome of either B-SIMPLE trial, and the FDA may disagree with our conclusions from these analyses. P-value is a conventional statistical method for measuring the statistical significance of clinical results. A p-value of less than 0.050 is generally considered to represent statistical significance, meaning that there is a less than five percent likelihood that the observed results occurred by chance.
The last subject completed their final visit as part of the ongoing safety evaluation through Week 24 in February 2020 and full efficacy and safety data, including data from the safety evaluation through Week 24, are targeted to be available in March 2020.
Based on the results of the Phase 3 pivotal trials, discussed above, we target commencing an additional confirmatory Phase 3 trial in the second quarter of 2020, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020.
Execution of remaining Phase 3 pivotal development program activities for SB206 in molluscum continues into 2020 with receipt of pivotal trial safety data in the first quarter of 2020 and the completion of ancillary trials targeted prior to or during the second quarter of 2020.
SB414, a Topical Cream for the Treatment of Inflammatory Skin Diseases
In 2018, we completed two complementary Phase 1b clinical trials with SB414 in patients with atopic dermatitis and psoriasis. The design of these complementary trials was to evaluate the safety, tolerability and pharmacokinetics of SB414. The trials
73
were also designed to assess overall and specific target engagement through a reduction of key inflammatory biomarkers, also known as pharmacodynamic assessment.
Atopic Dermatitis
We initiated a Phase 1b trial with SB414 in adults with mild-to-moderate atopic dermatitis in December 2017. In the Phase 1b trial, 48 adults with mild-to-moderate atopic dermatitis with up to 30% body surface area at baseline, were randomized to receive one of 2% SB414 cream, 6% SB414 cream, or vehicle, twice daily for two weeks. In the complementary Phase 1b trial for mild-to-moderate chronic plaque psoriasis, 36 adults received SB414 6% cream or vehicle twice daily for four weeks.
We received and analyzed the preliminary top line results from the Phase 1b clinical trials during the second and third quarters of 2018. In the atopic dermatitis trial, biomarkers from the Th2, Th17 and Th22 inflammatory pathways known to be highly relevant and indicative of atopic dermatitis, including Interleukin-13, or IL-13, IL-4R, IL-5, IL-17A and IL-22, were downregulated after two weeks of treatment with SB414 2%. The changes in Th2 and Th22 biomarkers and clinical efficacy assessed as the percent change in Eczema Area Severity Index scores were highly correlated in the SB414 2% group. Additionally, the proportion of patients achieving a greater than or equal to 3-point improvement on the pruritus (itch) numeric rating scale after two weeks of treatment was greater for patients treated with SB414 2% compared to patients treated with vehicle.
The 2% or 6% doses of SB414 in the trial did not result in any serious adverse events, and SB414 2% was more tolerable with no patients discontinuing treatment in the trial due to application site reactions. SB414 at the 6% dose was not consistently effective in reducing biomarkers across both the atopic dermatitis and psoriasis trials. This lack of consistent biomarker movement could potentially be explained by the increased irritation score experienced by patients treated with SB414 6%. Additionally, SB414 6% showed detectable systemic exposure in a subset of patients, which cleared in nearly all affected patients within 12 hours, in both the atopic dermatitis and psoriasis trials. Given the successful downregulation of key biomarkers, favorable tolerability and lack of systemic exposure with SB414 2%, we conducted non-clinical studies and completed our Phase 2 clinical development plan during 2019 to support a potential future Phase 2 clinical program launch. The SB414 program is currently on hold with further advancement subject to obtaining additional financing or strategic partnering.
Psoriasis
We initiated clinical development of SB414, our first use of our nitric oxide platform in the field of immunology by dosing the first patient in October 2017 in a Phase 1b clinical trial to evaluate SB414 in a cream for the treatment of psoriasis. Earlier in 2017, we presented mechanistic evidence for SB414, demonstrating a statistically significant reduction in composite psoriasis scores and an inhibition of IL-17A and IL-17F in an animal model.
In the Phase 1b trial for mild-to-moderate chronic plaque psoriasis, 36 adults received SB414 6% cream or vehicle twice daily for four weeks. We received and analyzed the preliminary top line results from this Phase 1b clinical trial during the second and third quarters of 2018. SB414 at the 6% dose did not result in any serious adverse events, but SB414 at the 6% dose was not consistently effective in reducing biomarkers across the trial. This lack of consistent biomarker movement could potentially be explained by the increased irritation score experienced by patients treated with SB414 6%. Additionally, SB414 6% showed detectable systemic exposure in a subset of patients, which cleared in nearly all affected patients within 12 hours. Based on the results of the Phase 1b trial in psoriasis, we will potentially explore the use of lower doses of SB414 in psoriasis, subject to obtaining additional financing or strategic partnering.
SB204, for the Treatment of Acne Vulgaris
In the second quarter of 2018, we conducted a Type C meeting to further discuss the path forward for our SB204 candidate and possible Phase 3 programs for the treatment of acne vulgaris with the FDA, and the potential for proceeding with a more narrowly defined patient segmentation. In that meeting, our focus was centered specifically on the severe patient population. In the third quarter of 2018, the FDA provided feedback in their minutes on two paths forward for the acne indication, confirming the need for one additional pivotal trial for moderate-to-severe acne patients prior to a NDA submission or, as an alternative, additional preliminary trials for a severe-only patient population.
Following receipt of FDA feedback via written minutes, we have determined that the most pragmatic development pathway for us will be to conduct one additional pivotal Phase 3 trial in moderate-to-severe acne patients. We have completed our clinical development plan for this additional trial, and further advancement of this program is subject to obtaining additional financing or strategic partnering.
74
Key Preclinical Stage Product Candidate Development Updates
Expansion of Nitric Oxide Platform
In December 2019, we received written responses in response to a pre-IND meeting request with the FDA for a new anti-viral product candidate, SB207. We have identified targeted viral opportunities of high unmet need where we believe our nitric oxide releasing technology could provide clinical benefit to patients. The SB207 product incorporates our existing drug substance, berdazimer sodium (NVN1000), with a new formulation specifically engineered for a number of anti-viral programs.
Advancement in Women’s Health
In August 2019, we received a Phase 1 Federal grant of approximately $0.2 million from the National Institutes of Health. The funds are to be used to advance formulation development of a nitric oxide-containing intravaginal gel (WH602) designed to treat high-risk HPV infections that can lead to cervical intraepithelial neoplasia, or CIN. The specific focus is to ensure the nitric oxide delivery from the gel replicates doses of nitric oxide previously demonstrated to be effective against HPV in our clinical and in vitro studies.
In February 2020, following the successful progression of Phase 1, we were awarded a Phase 2 federal grant of approximately $1.0 million from the NIH that will enable the conduct of IND-enabling toxicology and pharmacology studies and other preclinical activity with respect to WH602. We may be eligible to receive an additional $0.5 million in funding as part of this Phase 2 grant, subject to availability of NIH funds and satisfactory progress of the project during the initial 12-month term.
In September 2019, we received a Federal grant from the U.S. Department of Defense’s, or DoD, Congressionally Directed Medical Research Programs, or CDMRP, of approximately $1.1 million as part of its Peer Reviewed Cancer Research Program. The grant will support the development of a non-gel formulation product candidate (WH504) designed to treat high-risk HPV infections that can lead to CIN, with well-characterized physical chemical properties suitable for intravaginal administration. In addition, the grant will support the evaluation of the effect of varying concentrations and treatment durations of berdazimer sodium (NVN1000) against HPV-18 in human raft cell culture in vitro studies. This targeted research aims to create a disease-altering treatment that could be used upon detection and the early signs of high-risk HPV infection to intervene before progression to cervical cancer.
Under the terms of the aforementioned NIH and DoD grants, we are entitled to receive the grant funds in the form of periodic reimbursements of our allowable direct expenses, allocated overhead, general and administrative expenses and payment of other specified amounts.
These product candidates currently in development together represent the core of our Women’s Health business unit. This unit has continued to be supported through a collaboration with Health Decisions, Inc., or Health Decisions.
Addition of Gastrointestinal Disease as a Therapeutic Focus
In January 2019, we announced the addition of GI diseases as a therapeutic focus area as part of our overall science and business strategy. This decision is based on the connection between the multi-factorial pathologies of GI diseases and the demonstrable anti-microbial and anti-inflammatory properties of Novan’s nitric oxide technology. Nitric oxide produced in the GI tract regulates many of its functions including the secretion of mucous for protection against physical, chemical, and microbial injury, perfusion of blood through the GI tissue, mitigation of white blood cell adherence to GI tissue to protect from injury and the healing and repair of ulcers. We believe that our initial expansion into GI will require minimal investment due to our ability to leverage current technology, experience and assets.
Business Updates
Common Stock Purchase Agreement and Registration Rights Agreement with Aspire Capital
On August 30, 2019, we entered into the Aspire Common Stock Purchase Agreement with Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement. The aggregate amount that we may raise through sales of common stock under the Aspire Common Stock Purchase Agreement is subject to certain limitations including, but not limited to: (i) the number of shares that may be sold will be limited to 5,211,339 shares, representing 19.99% of our outstanding shares of common stock on August 30, 2019, if the average price paid for all shares issued under the agreement is less than $2.17; and (ii) on any purchase date, the closing sale price of our common stock must be greater than or equal to $0.25. As of December 31, 2019, we had sold an aggregate of 300,000 shares of common stock at an average price of $2.49 per share under the Aspire Common Stock Purchase Agreement. As of January 31, 2020, we had sold an aggregate of 1,000,000 shares of common stock at an average
75
price of $1.19 per share under the Aspire Common Stock Purchase Agreement. These amounts, combined with the 345,622 shares issued as part of the commitment fee related to the agreement’s execution, leads to a total of 1,345,622 shares issued to Aspire Capital under the agreement as of January 31, 2020.
Concurrently with entering into the Aspire Common Stock Purchase Agreement, we also entered into a registration rights agreement with Aspire Capital, or the Registration Rights Agreement, in which we agreed to file one or more registration statements, as permissible and necessary to register under the Securities Act of 1933, as amended, or the Securities Act, registering the sale of the shares of our common stock that have been and may be issued to Aspire Capital under the Aspire Common Stock Purchase Agreement. On September 16, 2019, we filed with the Securities and Exchange Commission, or the SEC, a prospectus to our effective Registration Statement on Form S-1 (File No. 333-233632) registering 7,032,630 shares of common stock that have been and may be offered to Aspire Capital from time to time under the Aspire Common Stock Purchase Agreement.
There are no trading volume requirements or restrictions under the Aspire Common Stock Purchase Agreement, and we will control the timing and amount of sales of our common stock to Aspire Capital. Aspire Capital has no right to require any sales by us, but is obligated to make purchases from us as we may direct in accordance with the Aspire Common Stock Purchase Agreement. There are no limitations on use of proceeds, financial or business covenants, restrictions on future financing transactions, rights of first refusal, participation rights, penalties or liquidated damages in the Aspire Common Stock Purchase Agreement. In consideration for entering into the Aspire Common Stock Purchase Agreement, concurrently with the execution of the Aspire Common Stock Purchase Agreement, we issued to Aspire Capital 345,622 shares of our common stock as part of the commitment fee. The Aspire Common Stock Purchase Agreement may be terminated by us any time, at our discretion, without any penalty or additional cost to us. Any proceeds we receive under the Aspire Common Stock Purchase Agreement are expected to be used for working capital and general corporate purposes.
Please refer to “Liquidity and Capital Resources” for further discussion of the Aspire Common Stock Purchase Agreement and the Registration Rights Agreement.
Royalty and Milestone Payments Purchase Agreement with Reedy Creek Investments LLC
On April 29, 2019, we entered into the Purchase Agreement with Reedy Creek, pursuant to which Reedy Creek provided us funding in an initial amount of $25.0 million, which we will use primarily to pursue the development, regulatory approval and commercialization (including through out-license agreements and other third-party arrangements) activities for SB206, for the treatment of molluscum, and advancing programmatically other activities with respect to SB414, for atopic dermatitis, and SB204, for acne. Reedy Creek was to provide $10.0 million of additional funding contingent upon our achievement of SB206 clinical trial success, defined as (i) the achievement, no later than March 31, 2020, of statistically significant rates of complete clearance of lesions for molluscum contagiosum in humans at week 12 in each of the two Phase 3 clinical trials or any other primary endpoint required or accepted by the FDA for the SB206 product; or (ii) equivalent achievement (as agreed upon by the parties). Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
Pursuant to the Purchase Agreement, we will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by us pursuant to any out-license agreement for SB204, SB206 and SB414 in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by us to third parties pursuant to any agreements under which we have in-licensed intellectual property with respect to such products.
The applicable percentage used for determining the ongoing quarterly payments, applied to amounts received directly by us pursuant to any out-license agreement for each product, ranges from 10% for SB206 to 20% for SB414 and SB204. However, the agreement provides that the applicable percentage for each product will be 25% for fees or milestone payments received by us (but not royalty payments received by us) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If we decide to commercialize SB204, SB206 or SB414 on our own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, we will be obligated to pay Reedy Creek a low single digits royalty on net sales of such products.
Development Funding and Royalties Agreement with Ligand Pharmaceuticals Incorporated
On May 4, 2019, we entered into the Funding Agreement, with Ligand, pursuant to which Ligand provided us funding of $12.0 million, which we used to pursue the development and regulatory approval of SB206, for the treatment of molluscum.
76
Pursuant to the Funding Agreement, we will pay Ligand up to $20.0 million in milestone payments upon the achievement by us of certain regulatory and commercial milestones associated with SB206 or any product that incorporates or uses NVN1000, the active pharmaceutical ingredient for our clinical stage product candidates, for the treatment of molluscum. In addition to the milestone payments, we will pay Ligand tiered royalties ranging from 7% to 10% based on annual aggregate net sales of such products in the United States, Mexico or Canada.
Drug Substance and Drug Product Agreements
On October 15, 2018, we established a strategic alliance with Orion, a Finnish full-scale pharmaceutical company with broad experience in drug manufacturing. The alliance enables Orion to manufacture our topical nitric oxide-releasing product candidates on our behalf and on the behalf of our global strategic partners. We have executed a master contract manufacturing agreement to enable technology transfer and manufacturing of clinical trial materials for future clinical trials with our topical product candidates. We are engaged in the transfer of technology for the manufacture of both SB204 and SB206, and upon its completion intend for Orion to be able to manufacture the drug product, or the finished dosage form of the gel, in accordance with our established manufacturing processes, in compliance with applicable regulatory guidelines, as appropriate for clinical trials and alongside our current internal manufacturing capabilities. A completed manufacturing technology transfer to Orion will enable the manufacture of multiple assets for clinical trial materials and, potentially, commercial quantities. Importantly, this alliance is being structured to support major global markets in which we and our partners pursue regulatory approvals for our product candidates and complements our present internal capability.
In June 2019, we executed a master contract manufacturing agreement with a full-scale active pharmaceutical ingredient (API) manufacturer. The agreement established an operating and business relationship for this manufacturer to become the primary external supplier of our proprietary berdazimer sodium (NVN1000) drug substance. Also incorporated in the agreement is the process and analytical method transfer necessary to advance the production of our berdazimer sodium (NVN1000) drug substance for future clinical trials and importantly, upon approval of any of our drug product candidates, for commercial purposes on a global basis. We are engaged in the transfer of the NVN1000 manufacturing technology and upon its completion intend for this API manufacturer to be able to manufacture NVN1000 in accordance with our established manufacturing processes, in compliance with applicable regulatory guidelines, as appropriate for clinical trials and alongside our current internal manufacturing capabilities.
The progression of the relationships with the aforementioned third-party manufacturers is integral to the advancement of our dermatological platform, including our SB206 molluscum program, by us or through partnerships, collaborations, licensing or other strategic relationships. This strategy includes an increased utilization of and reliance upon third-party vendors and strategic partners for the performance of activities, processes and services that (i) do not result in the generation of significant new intellectual property; and (ii) can leverage existing robust infrastructure, systems, and facilities as well as associated subject matter expertise. A parallel and inter-related strategic objective is to reduce our own internal resources, facilities, and infrastructure capabilities that have historically performed such activities, processes and services. As part of our strategic objective to reduce our own internal resources, facilities, and infrastructure capabilities, we took actions in February 2020 that are intended to reduce our internal resources from a total of 42 employees as of December 31, 2019 to a total of 28 employees as of April 1, 2020.
We believe this broad strategy can ultimately provide enhanced capabilities and operating efficiencies for us or any potential partnerships, collaborations, licensing or other strategic relationships we may enter. Although the third-party manufacturers are reducing their near-term activities and extending their time lines at our request in an effort to reduce our near-term cash utilization and extend our operating cash runway, we expect to incur certain incremental and discrete costs to effect this strategy upon resumption of the manufacturers’ transfer activities. Similarly, we expect to incur certain incremental and discrete costs as we seek to reduce our own internal resources, facilities, and infrastructure capabilities, including those actions we took in February 2020 to reduce our internal resources. We will need substantial additional funding to continue our operating activities, including these technical transfer projects and internal cost structure changes, and to make further advancements in our drug development programs, as described below in “Liquidity and Capital Resources”.
77
Corporate Updates
Chief Executive Officer Transition
In December 2019, we announced that Paula Brown Stafford, our President and Chief Operating Officer, would succeed G. Kelly Martin as our Chief Executive Officer, effective February 2, 2020. Mr. Martin had a fixed term employment contract that expired on February 1, 2020. Mr. Martin also resigned from the Board of Directors, effective February 3, 2020. Ms. Stafford remains a member of the Board of Directors.
Stock Appreciation Rights
As described in “Note 11—Share-Based Compensation” to the accompanying consolidated financial statements included in this Annual Report, on August 8, 2018, we entered into an employment agreement with G. Kelly Martin, or the Martin Employment Agreement. The Martin Employment Agreement provided for 1,000,000 stock appreciation rights, or SARs, granted on a contingent basis that would have been considered irrevocably forfeited and voided in full if we failed to obtain stockholder approval for an amendment to the 2016 Incentive Award Plan, or the 2016 Plan. If such approval had not been obtained, we would have been required to pay Mr. Martin the cash equivalent of the value of the SARs.
On July 31, 2019, at our 2019 Annual Meeting of Stockholders, our stockholders approved a proposal to amend the 2016 Plan, to (i) increase the number of shares of our common stock reserved for issuance under the 2016 Plan by 1,000,000 shares; and (ii) increase the limit on the number of awards that may be granted to any one person in any year. As such, with stockholder approval of the amendment to the 2016 Plan, the SARs detailed within the Martin Employment Agreement were no longer considered granted on a contingent basis. The SARs entitled Mr. Martin to a payment (in cash, shares of common stock or a combination of both) equal to the fair market value of one share of our common stock on the date of exercise less the exercise price of $3.80 per share. The SARs were to be deemed automatically exercised and settled as of February 1, 2020, provided Mr. Martin remained continuously employed with the Company through such date unless vesting was otherwise expressly accelerated pursuant to the Martin SAR Award. The SARs vested in full on February 1, 2020. On February 1, 2020, the fair market value of our common stock was $0.52 per share, and as such, the SARs expired unexercised and 1,000,000 shares became available to be granted under the 2016 Plan.
Effective December 17, 2019, we entered into an amended and restated employment agreement with Paula Brown Stafford, or the Amended and Restated Stafford Employment Agreement. On January 6, 2020, following our release of top-line results of the Phase 3 molluscum clinical program as provided by the Amended and Restated Stafford Employment Agreement, 600,000 SARs were granted to Ms. Stafford with an exercise price of $0.82 per share, or the Stafford SAR Award. The Stafford SAR Award was granted on a contingent basis and would have been considered irrevocably forfeited and voided in full if sufficient shares of our common stock were not available under the 2016 Plan or if we failed to obtain stockholder approval for amendments to the 2016 Plan at the next annual stockholders’ meeting to provide sufficient shares for the Stafford SAR Award. If such approval had not been obtained, we would have been required to pay Ms. Stafford the cash equivalent of the value of the SARs. Such shares became available under the 2016 Plan on February 1, 2020 and the Stafford SAR Award was no longer considered granted on a contingent basis.
Board of Directors
On January 29, 2020, Dr. Eugene Sun, one of the members of our board of directors, notified us of his resignation from the board and any committees thereof, effective January 29, 2020.
Financial Overview
Since our inception in 2006, we have devoted substantially all of our efforts to developing our nitric oxide platform technology and resulting product candidates, including conducting preclinical and clinical trials and providing general and administrative support for these operations. We conduct these activities in a single operating segment. We have not generated any revenue from product sales and, to date, have funded our operations through a variety of sources described in further detail within the “Liquidity and Capital Resources” section below. From inception through December 31, 2019, we have raised total equity and debt proceeds of $184.8 million to fund our operations.
In addition, through December 31, 2019, we have also generated additional liquidity and capital through other sources including (i) governmental research contracts and grants totaling $11.8 million; (ii) our licensing and supply arrangements with Sato, totaling $24.2 million, described below; and (iii) $37.0 million in proceeds from two funding transactions during the second quarter of 2019, also described below.
78
The approximately $24.2 million we have received from Sato from January 2017 through December 31, 2019 under our amended license agreement includes a $10.8 million upfront payment received following the execution of the agreement in January 2017, a $2.2 million payment related to the initiation of a Phase 1 trial in Japan in the third quarter of 2018, and $11.2 million of installment payments received following the October 2018 amendment to our amended license agreement with Sato.
As noted above, in April 2019 and May 2019, respectively, we entered into the Purchase Agreement with Reedy Creek, which provided $25.0 million of immediate funding, and the Funding Agreement with Ligand, which provided $12.0 million of immediate funding. Reedy Creek was to provide an additional $10.0 million contingent upon our achievement of the primary end points in each of the two SB206 Phase 3 clinical trials no later than March 31, 2020. Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
As also noted above, in August 2019, we entered into the Aspire Common Stock Purchase Agreement with Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement. The aggregate amount that we may raise through sales of common stock under the Aspire Common Stock Purchase Agreement is subject to certain limitations including, but not limited to: (i) the number of shares that may be sold will be limited to 5,211,339 shares, representing 19.99% of our outstanding shares of common stock on August 30, 2019, if the average price paid for all shares issued under the agreement is less than $2.17; and (ii) on any purchase date, the closing sale price of our common stock must be greater than or equal to $0.25. As of December 31, 2019, we had sold an aggregate of 300,000 shares of common stock at an average price of $2.49 per share under the Aspire Common Stock Purchase Agreement. As of January 31, 2020, we had sold an aggregate of 1,000,000 shares of common stock at an average price of $1.19 per share under the Aspire Common Stock Purchase Agreement. These amounts, combined with the 345,622 shares issued as part of the commitment fee related to the agreement’s execution, leads to a total of 1,345,622 shares issued to Aspire Capital under the agreement as of January 31, 2020.
To date, we have focused our funding activities on equity, debt and strategic relationships. However, other historical forms of funding have included payments received from licensing and supply arrangements, as well as government research contracts.
We have never generated revenue from product sales and have incurred net losses in each year since inception. As of December 31, 2019, we had an accumulated deficit of $220.0 million. We incurred net losses of $30.4 million and $29.2 million in the years ended December 31, 2019 and 2018, respectively. We expect to continue to incur substantial losses in the future as we conduct our planned operating activities. We do not expect to generate revenue from product sales unless and until we obtain regulatory approval from the FDA for our clinical-stage product candidates. If we obtain regulatory approval for any of our product candidates, we and/or our commercial partners would expect to incur significant expenses related to product sales, marketing, manufacturing and distribution.
We expect that we will continue to incur substantial expenses as we continue, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, to commence an additional confirmatory Phase 3 trial in the second quarter of 2020 and fund our operations. At present, we have temporarily suspended development activities and taken measures to reduce our expenses. Presently, we only have sufficient cash to fund our operations until the early part of the second quarter of 2020. As a result, we need substantial additional funding to support our planned and future operating activities and make further advancements in our drug development programs. Adequate future funding may not be available to us on acceptable terms, or at all. The current market value of our common stock may negatively impact funding options and the acceptability of funding terms. Additionally, we expect future advancement of our product candidates to occur after the formation of partnering, collaborations, licensing, grants or other strategic relationships or through equity or debt financings. Our failure to enter into such relationships, or our failure to obtain sufficient additional funds on acceptable terms as and when needed could cause us to alter or reduce our planned operating activities, including but not limited to delaying, reducing, terminating or eliminating planned product candidate development activities, to conserve our cash and cash equivalents or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. Such actions could delay development timelines and have a material adverse effect on our business, results of operations, financial condition and market valuation. If we are forced to terminate or eliminate our product development programs, wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders, whereby, our stockholders may lose some or all of their investment. If we are forced to terminate or eliminate our product development programs or pursue other strategic alternatives or corporate transactions, there can be no assurance that such actions would result in any additional stockholder value. Alternatively, we may seek to engage in one or more potential transactions, such as the sale of our company, or sale or divestiture of some of our assets, such as a sale of our dermatology platform assets, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us. As further
79
discussed in our audited consolidated financial statements and related footnotes included in this Annual Report, these matters raise substantial doubt about our ability to continue as a going concern.
Please refer to “Liquidity and Capital Resources” for further discussion of our current liquidity and our future funding needs.
Components of our Results of Operations
Revenue
License and collaboration revenue consists of the amortization of certain fixed and variable consideration under the Amended Sato Agreement, that (i) has been received to date in the form of upfront and milestone payments; or (ii) are future, non-contingent milestone payments that become payable upon the earlier occurrence of specified fixed dates in the future or the achievement of specified milestone events.
This consideration is being recognized on a straight-line basis over the estimated performance period of approximately 7.5 years, from February 2017 through the third quarter of 2024. We monitor and reassess the estimated performance period for purposes of revenue recognition during each reporting period. We expect to reassess the estimated performance period during the first quarter of 2020, as we consider how the combined SB204 and SB206 development program timeline in Japan may potentially be affected by various factors, including (i) the recent results from our SB206 Phase 3 trials in the U.S., including but not limited to top-line efficacy results announced in January 2020, (ii) our plans and timelines for potential further clinical development of SB206 in the U.S., which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (iii) our in-house drug manufacturing capabilities and the progression of our manufacturing technology transfer projects with third-party contract manufacturing organizations. Therefore, if the duration of the combined SB204 and SB206 development program timeline is affected by the establishment or subsequent adjustments to a mutually agreed upon SB204 and SB206 development plan in the Japan territory, we will adjust its estimated performance period for revenue recognition purposes accordingly, as needed.
The material terms of the Amended Sato Agreement and related revenue recognition are described above and within “Note 4—Licensing Arrangements” and “Note 6—Revenue Recognition” to the accompanying consolidated financial statements included in this Annual Report.
Government research contracts and grant revenue relates to the research and development of our nitric oxide platform for preclinical advancement of NCEs and formulations related to potential treatments for illnesses in the women’s health field. Revenue related to conditional government contracts and grants is recognized when qualifying expenses are incurred.
Research and Development Expenses
Since our inception, we have focused our resources on our research and development activities, including conducting preclinical studies and clinical trials, manufacturing development efforts and activities related to regulatory filings for our product candidates. Research and development expenses, including those paid to third parties for which there is no alternative use, are expensed as they are incurred. Research and development expenses include:
• | external research and development expenses incurred under agreements with contract research organizations, investigative sites and consultants to conduct our clinical trials and preclinical studies; |
• | costs to acquire, develop and manufacture supplies for clinical trials and preclinical studies at our facilities; |
• | costs to establish drug substance and drug product manufacturing capabilities with external contract manufacturing organizations and to enhance drug delivery device technologies through partnerships with technology manufacturing vendors; |
• | legal and other professional fees related to compliance with FDA requirements; |
• | licensing fees and milestone payments incurred under license agreements; |
• | salaries and related costs, including share-based compensation, for personnel in our research and development functions; and |
• | facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent, maintenance of facilities, utilities, equipment and other supplies. |
From inception through December 31, 2019, we have incurred approximately $162.1 million in research and development expenses to develop, expand or otherwise improve our nitric oxide platform and resulting product candidates, as well as costs
80
incurred to generate research and development services revenue. This amount is net of $8.2 million of contra-research and development expense recorded for the year ended December 31, 2019 representing amortization of the liability related to the $12.0 million of funding received from Ligand to pursue the development and regulatory approval of SB206. The calculation used to recognize the ratable amortization of this liability includes an estimate of the total cost to progress the SB206 program to a regulatory approval in the U.S. During the first quarter of 2020, we expect to reassess this estimate and will take into consideration how such estimated costs may potentially be affected by recent development program events, as well as developmental and operational evaluations currently taking place.
For additional information about the Funding Agreement with Ligand, please see “Note 7—Research and Development Arrangements” to the accompanying consolidated financial statements included in this Annual Report.
The table below sets forth our research and development expenses incurred for external clinical programs and the related product candidates, and other research and development expenses for the years ended December 31, 2019 and 2018. Other research and development expenses include: (i) all preclinical program and development costs, including WH504 and WH602, (ii) manufacturing capability and campaign costs, (iii) external costs to establish drug substance and drug product manufacturing capabilities at third-party CMOs, (iv) facility and infrastructure costs, and (v) costs related to all research and development salaries and related personnel costs.
Year Ended December 31, | ||||||||
2019 | 2018 | |||||||
(in thousands) | ||||||||
External clinical programs: | ||||||||
SB204 | $ | 212 | $ | 1,116 | ||||
SB206 | 7,860 | (1) | 5,107 | |||||
SB208 | 8 | — | ||||||
SB414 | 1,836 | 1,772 | ||||||
Other research and development | 15,256 | 15,050 | ||||||
Total research and development expenses | $ | 25,172 | $ | 23,045 | ||||
(1) | Amount shown net of $8.2 million of contra-research and development expense recorded for the year ended December 31, 2019, respectively, related to the Funding Agreement with Ligand described in “Note 7—Research and Development Arrangements” to the accompanying consolidated financial statements included in this Annual Report. |
During the year ended December 31, 2019, our major clinical development activities were primarily associated with our SB206 molluscum program, where we completed our Phase 2 clinical program activities, held an end-of-Phase 2 meeting with the FDA, and commenced the Phase 3 development program for molluscum, including the initiation and progression of two pivotal clinical trials. We expect that for the foreseeable future, the substantial majority of our research and development efforts will be focused on the completion our current Phase 3 clinical program activities related to SB206 and, subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, additional costs related to one potential additional confirmatory Phase 3 trial with SB206 for molluscum.
We expect to incur substantial research and development expenses in the future as we develop our clinical product candidates. In particular, subject to obtaining additional capital resources or strategic partnership arrangements, we expect to continue to incur substantial external development service provider fees and other research and development costs in 2020 to: (i) complete the SB206 molluscum Phase 3 program, including ancillary supporting trials and studies; (ii) wind down our SB414 atopic dermatitis Phase 2 trial preparatory activities; (iii) conduct certain API manufacturing capability transfer activities to our external third-party CMO; and (iv) progress certain drug product manufacturing capability transfer activities to Orion. We expect to incur substantial costs in 2020 associated with our research and development personnel and our in-house facility and manufacturing capabilities that support the aforementioned external development activities. We may decide to revise our development and operating plans or the related timing, depending on information we learn through our research and development activities, our ability to access additional capital, our ability to enter into strategic arrangements and our financial priorities.
The successful development of our product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing or costs required to complete the remaining development of our current product candidates or any future product candidates. This is due to the numerous risks and uncertainties associated with the development of product candidates. See the
81
section entitled “Risk Factors” in this Annual Report for a discussion of the risks and uncertainties associated with our research and development projects.
General and Administrative Expenses
Our general and administrative expenses consist primarily of salaries and related costs, including share-based compensation expenses for personnel in our executive, finance, corporate development and other administrative functions. Other general and administrative expenses include allocated depreciation and facility-related costs, legal costs of pursuing patent protection of our intellectual property, insurance coverage and professional services fees for auditing, tax, general legal, business development, litigation defense and other corporate and administrative services.
We expect to continue to incur substantial general and administrative expenses in 2020 in support of our operating activities and as necessary to operate in a public company environment. Significant general and administrative expenses associated with operations in a public company environment include legal, accounting, regulatory and tax-related services associated with maintaining compliance with exchange listing and SEC requirements, directors’ and officers’ liability insurance premiums and investor relations activities. We experienced a decrease in litigation defense fees during 2019 as we concluded that the putative stockholder class action lawsuits that were filed in November 2017, which have since been dismissed by the court with prejudice, are substantially complete.
Other Income (Expense), net
Other income (expense), net consists primarily of (i) interest income earned on cash and cash equivalents; (ii) lease interest expense on our primary facility lease financing obligation during 2018 prior to the January 1, 2019 adoption of FASB Accounting Standards Update, or ASU, No. 2016-02, Leases (Topic 842), or Topic 842; and (iii) other miscellaneous income and expenses.
Results of Operations
Comparison of the Years Ended December 31, 2019 and 2018
The following table sets forth our results of operations for the periods indicated:
Year Ended December 31, | ||||||||||||||
2019 | 2018 | $ Change | % Change | |||||||||||
(Restated) | ||||||||||||||
(in thousands, except percentages) | ||||||||||||||
License and collaboration revenue | $ | 4,477 | $ | 5,982 | $ | (1,505 | ) | (25 | )% | |||||
Government research contracts and grants revenue | 419 | — | 419 | 100 | % | |||||||||
Research and development services revenue | — | 9 | (9 | ) | (100 | )% | ||||||||
Total revenue | 4,896 | 5,991 | (1,095 | ) | (18 | )% | ||||||||
Operating expenses: | ||||||||||||||
Research and development | 25,172 | 23,045 | 2,127 | 9 | % | |||||||||
General and administrative | 10,412 | 11,507 | (1,095 | ) | (10 | )% | ||||||||
Total operating expenses | 35,584 | 34,552 | 1,032 | 3 | % | |||||||||
Operating loss | (30,688 | ) | (28,561 | ) | (2,127 | ) | 7 | % | ||||||
Other income (expense), net: | ||||||||||||||
Interest income | 177 | 297 | (120 | ) | (40 | )% | ||||||||
Interest expense | (2 | ) | (1,047 | ) | 1,045 | (100 | )% | |||||||
Other income, net | 136 | 72 | 64 | 89 | % | |||||||||
Total other income (expense), net | 311 | (678 | ) | 989 | (146 | )% | ||||||||
Net loss and comprehensive loss | $ | (30,377 | ) | $ | (29,239 | ) | $ | (1,138 | ) | 4 | % | |||
Revenue
License and collaboration revenue of $4.5 million and $6.0 million for the years ended December 31, 2019 and 2018, respectively, was associated with our performance during the period and the related amortization of the non-refundable upfront and expected milestone payments under the Amended Sato Agreement.
82
Government research contracts and grants revenue of $0.4 million for the year ended December 31, 2019 includes (i) $0.3 million of revenue recognized related to the $1.1 million grant we received in September 2019 from the DoD’s CDMRP as part of its Peer Reviewed Cancer Research Program and (ii) $0.1 million of revenue recognized related to the $0.2 million grant received in September from the National Institutes of Health. For additional information regarding our government research contracts and grants revenue, see “Note 6—Revenue Recognition” to the accompanying consolidated financial statements included in this Annual Report.
Research and development expenses
Research and development expenses were $25.2 million for the year ended December 31, 2019, compared to $23.0 million for the year ended December 31, 2018. The net increase of $2.1 million, or 9% was primarily related to (i) a net $2.8 million increase in the SB206 program, (ii) a $0.2 million increase in other research and development expenses, (iii) a $0.1 million increase in our SB414 program due to the continued conduct of non-clinical studies during the third and fourth quarters of 2019, partially offset by (iv) a $0.9 million decrease in our SB204 program due to the completion of certain chemistry, manufacturing, and control activities that took place during the comparative period.
In the SB206 program, we experienced a $10.9 million increase in gross costs incurred due to the continued conduct of two Phase 3 pivotal trials and the initiation of other Phase 3 development activities, including ancillary supporting trials and studies, for the molluscum indication in the third quarter of 2019. This increase was partially offset by $8.2 million of contra-research and development expense representing the amortization of the liability related to the Funding Agreement with Ligand, which contributes to the clinical development and regulatory approval of SB206 for the treatment of molluscum.
The $0.2 million increase in other research and development expenses was primarily driven by costs associated with our increased utilization of third-party manufacturers and other strategic partners during 2019. Specifically, we experienced (i) a $2.1 million increase in service and support costs associated with our manufacturing technology and process engineering transfer projects ongoing with our third party API and drug product CMOs as well as a drug delivery device technology enhancement project ongoing with a technology manufacturing vendor, (ii) a $0.4 million increase in development service fees associated with preclinical development of new drug formulations and NCEs for use in the fields of women’s health and GI, (iii) a $0.4 million increase in allocated rental expense following the January 1, 2019 adoption of Topic 842, which was reported as interest expense in previous reporting periods prior to adoption of Topic 842, and (iv) a $0.3 million increase in allocated non-cash depreciation expense primarily due to a Topic 842 adoption requirement that increased the depreciation rate on our leasehold improvement assets.
These other research and development expense increases were partially offset by (i) a $0.9 million decrease in internal facility and manufacturing costs associated with manufacturing campaigns and safety and quality activities occurring in the comparative 2018 period that did not recur or were significantly reduced in 2019 and (ii) a $2.0 million decrease in research and development personnel-related costs. The decrease in facility and manufacturing costs is comprised of (i) a $0.6 million decrease in equipment repair, maintenance and support costs and (ii) a $0.3 million decrease in manufacturing safety, quality and efficiency initiatives at our Morrisville, NC manufacturing facility. The decrease in personnel-related costs is comprised of (i) a $1.4 million decrease in recurring salary and benefit expenses due to reductions in research and development personnel between the comparative periods, (ii) a $0.6 million decrease in annual discretionary bonus compensation expense in 2019 as the Compensation Committee of the Board of Directors determined that the associated corporate performance objectives were not achieved during 2019, (iii) a $0.4 million net decrease in non-cash performance-based compensation expense, partially offset by (iv) a $0.4 million discrete severance expense charge in the first quarter of 2019 associated with the departure of our former chief scientific officer. Non-cash performance based compensation included a $0.5 million decrease in stock option related compensation expense associated with (i) the forfeiture of stock options previously held by former officers and employees who departed the Company prior to or during 2019; and (ii) expense associated with the annual grant of stock option awards to all employees during 2018, which did not recur in 2019 after the establishment of the Tangible Stockholder Return Plan in the third quarter of 2018. This decrease was partially offset by a $0.1 million increase associated with the change in the fair value of the liability related to our Tangible Stockholder Return Plan, or the Performance Plan, which was established in the third quarter of 2018. The Performance Plan liability valuation increased during 2019 due to appreciation in our common stock’s market price, which is a key input to the Performance Plan’s valuation model.
General and administrative expenses
General and administrative expenses were $10.4 million for the year ended December 31, 2019, compared to $11.5 million during the year ended December 31, 2018. The decrease of approximately $1.1 million, or 10% was primarily due to (i) a $0.9 million decrease in general and administrative personnel and related costs, (ii) a $0.7 million reduction in legal fees primarily related to litigation defense fees for the putative stockholder class action lawsuits incurred in 2018 that did not recur in 2019, partially offset by (iii) a $0.3 million increase in facility costs and (iv) a $0.2 million increase in insurance expense.
83
The $0.9 million decrease in general and administrative personnel and related costs is primarily due to (i) the expense recognition of a one-time signing bonus in the third quarter of 2018 of $0.6 million in accordance with the Martin Employment Agreement, (ii) a $0.2 million decrease in annual discretionary bonus compensation expense in 2019 as the Compensation Committee of the Board of Directors determined that the associated corporate performance objectives were not achieved during 2019, and (iii) a decrease in salary and benefits costs of $0.2 million. These decreases were partially offset by an increase in non-cash stock compensation expense of $0.1 million.
Other income (expense), net
We reported other income, net of $0.3 million for the year ended December 31, 2019, compared to other expense, net of $0.7 million for the year ended December 31, 2018. The net increase in other income of approximately $1.0 million is primarily related to a $1.0 million decrease in interest expense associated with our Morrisville, North Carolina facility lease. The decrease in interest expense is due to the adoption of Topic 842 on January 1, 2019, whereby we no longer report a portion of our lease costs as interest expense as of the adoption date.
Comparison of Other Income (Expense), Net, as Restated, during the Affected Periods
As described in “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements included in this Annual Report, our consolidated financial statements for the Affected Periods are restated in this Annual Report to correct the misapplication of accounting guidance related to warrants in our previously issued consolidated financial statements for such periods. The restated consolidated financial statements are indicated as “Restated” in the accompanying consolidated financial statements. Although the impact of the restatement on the consolidated financial statements and the related disclosures for the year ended December 31, 2019 was immaterial, we have revised those financial statements as well in connection with the restatement of our consolidated financial statements noted above. See “Note 2—Restatement of Consolidated Financial Statements” for further description of the restatement background and explanation, as well as detailed tables describing the adjustments made to all affected previously reported balances. Further, see “Note 17—Quarterly Financial Information (Unaudited)” for tables containing unaudited consolidated quarterly financial information for 2019 and 2018, updated to reflect the aforementioned restatement.
Within our reported consolidated results of operations, the restatement had no impact on reported 2019 and 2018 revenue or operating expenses, including no impact on research and development expenses and general and administrative expenses. The restatement did impact other income (expense), net because this line item had previously included the change in fair value of warrant liability. As a result, we have presented the restated other income (expense), net for each Affected Period in the following table, along with corresponding restated narrative descriptions of the change in other income (expense), net for each comparative period.
84
Current Period | Comparative Period | $ Change | % Change | Note | ||||||||||||
(in thousands, except percentages) | ||||||||||||||||
Three months ended March 31, 2018 (current period) compared to three months ended March 31, 2017 (comparative period) | $ | (218 | ) | $ | (230 | ) | $ | 12 | (5 | )% | (1) | |||||
Three months ended June 30, 2018 (current period) compared to three months ended June 30, 2017 (comparative period) | (142 | ) | (233 | ) | 91 | (39 | )% | (2) | ||||||||
Three months ended September 30, 2018 (current period) compared to three months ended September 30, 2017 (comparative period) | (151 | ) | (239 | ) | 88 | (37 | )% | (3) | ||||||||
Six months ended June 30, 2018 (current period) compared to six months ended June 30, 2017 (comparative period) | (360 | ) | (463 | ) | 103 | (22 | )% | (4) | ||||||||
Nine months ended September 30, 2018 (current period) compared to nine months ended September 30, 2017 (comparative period) | (511 | ) | (702 | ) | 191 | (27 | )% | (5) | ||||||||
Year ended December 31, 2018 (current period) compared to year ended December 31, 2017 (comparative period) | (678 | ) | (942 | ) | 264 | (28 | )% | (6) | ||||||||
Three months ended March 31, 2019 (current period) compared to three months ended March 31, 2018 (comparative period) | 84 | (218 | ) | 302 | (139 | )% | (7) | |||||||||
Three months ended June 30, 2019 (current period) compared to three months ended June 30, 2018 (comparative period) | 103 | (142 | ) | 245 | (173 | )% | (8) | |||||||||
Three months ended September 30, 2019 (current period) compared to three months ended September 30, 2018 (comparative period) | 76 | (151 | ) | 227 | (150 | )% | (9) | |||||||||
Six months ended June 30, 2019 (current period) compared to six months ended June 30, 2018 (comparative period) | 187 | (360 | ) | 547 | (152 | )% | (10) | |||||||||
Nine months ended September 30, 2019 (current period) compared to nine months ended September 30, 2018 (comparative period) | 263 | (511 | ) | 774 | (151 | )% | (11) | |||||||||
(1) | Other income (expense), net was $0.2 million in net other expense for both the three months ended March 31, 2018 and the comparative period of the three months ended March 31, 2017. There were no substantive changes in reported interest income or interest expense between the two periods. |
(2) | Other income (expense), net was $0.1 million in net other expense for the three months ended June 30, 2018, compared to $0.2 million in net other expense for the three months ended June 30, 2017. The net expense decrease of approximately $0.1 million was primarily due to an increase in interest income of $0.1 million. |
(3) | Other income (expense), net was approximately $0.2 million in net other expense for the three months ended September 30, 2018, compared to approximately $0.2 million expense for the three months ended September 30, 2017. The net expense decrease of approximately $0.1 million was primarily due to an increase in interest income of approximately $0.1 million. |
(4) | Other income (expense), net was $0.4 million in net other expense for the six months ended June 30, 2018, compared to $0.5 million in net other expense for the six months ended June 30, 2017. The net expense decrease of approximately $0.1 million was primarily due to an increase in interest income of approximately $0.1 million. |
(5) | Other income (expense), net was $0.5 million in net other expense for the nine months ended September 30, 2018, compared to $0.7 million in net other expense for the nine months ended September 30, 2017. The net expense decrease of approximately $0.2 million was primarily due to an increase in interest income of $0.2 million. |
(6) | Other income (expense), net was $0.7 million in net other expense for the year ended December 31, 2018, compared to $0.9 million in net other expense for the year ended December 31, 2017. The net expense decrease of approximately $0.3 million was primarily due to an increase in interest income of $0.2 million. |
(7) | Other income (expense), net was $0.1 million in net other income for the three months ended March 31, 2019, compared to $0.2 million in net other expense for the three months ended March 31, 2018. The net income increase of approximately $0.3 million was primarily due to a $0.3 million decrease in interest expense associated with our Morrisville, North Carolina facility lease. Following the adoption of Topic 842 on January 1, 2019, we no longer report a portion of our lease costs as interest expense as of the adoption date. |
(8) | Other (expense) income, net was $0.1 million in net other income for the three months ended June 30, 2019, compared to $0.1 million in net other expense for the three months ended June 30, 2018. The net income increase of approximately $0.2 million was primarily due to a $0.3 million decrease in interest expense associated with our Morrisville, North Carolina facility lease due to the adoption of Topic 842 on January 1, 2019, whereby we no longer report a portion of our lease costs as interest expense as of the adoption date. |
(9) | Other (expense) income, net was approximately $0.1 million in net other income for the three months ended September 30, 2019, compared to approximately $0.2 million in net other expense for the three months ended September 30, 2018. The net income increase of approximately $0.2 million was primarily due to a less than $0.3 million decrease in interest expense associated with our |
85
Morrisville, North Carolina facility lease due to the adoption of Topic 842 on January 1, 2019, whereby we no longer report a portion of our lease costs as interest expense as of the adoption date.
(10) | Other (expense) income, net was approximately $0.2 million in net other income for the six months ended June 30, 2019, compared to approximately $0.4 million in net other expense for the six months ended June 30, 2018. The other income increase of approximately $0.5 million was primarily due to (i) a decrease in interest expense of approximately $0.5 million associated with our Morrisville, North Carolina facility lease due to the adoption of Topic 842 on January 1, 2019, whereby we no longer report a portion of our lease costs as interest expense as of the adoption date, and (ii) an increase in other income, net of approximately $0.1 million. These changes were partially offset by a decrease in interest income of approximately $0.1 million. |
(11) | Other (expense) income, net was $0.3 million in net other income for the nine months ended September 30, 2019, compared to $0.5 million in net other expense for the nine months ended September 30, 2018. The other income increase of approximately $0.8 million was primarily due to (i) a decrease in interest expense of approximately $0.8 million associated with our Morrisville, North Carolina facility lease due to the adoption of Topic 842 on January 1, 2019, whereby we no longer report a portion of our lease costs as interest expense as of the adoption date, and (ii) an increase in other income, net of approximately $0.1 million. These changes were partially offset by a decrease in interest income of approximately $0.1 million. |
Liquidity and Capital Resources
As of December 31, 2019, we had a total cash and cash equivalents balance of $13.7 million and positive working capital of $2.8 million.
Since our inception through December 31, 2019, we have financed our operations primarily with $184.8 million in net proceeds from the issuance and sale of equity securities and convertible debt securities, including $35.2 million in net proceeds from a public offering of common stock and accompanying warrants pursuant to our effective shelf registration statement and completed on January 9, 2018, or the January 2018 Offering, and $44.6 million in net proceeds from the sale of common stock in our 2016 initial public offering and $0.7 million in proceeds from the sale of common stock under the Aspire Common Stock Purchase Agreement. In addition, through December 31, 2019, we have also generated additional liquidity and capital through other sources including (i) governmental research contracts and grants totaling $11.8 million; (ii) our licensing and supply arrangements with Sato Pharmaceutical Co., Ltd., or Sato, totaling $24.2 million; and (iii) $37.0 million in proceeds from two funding transactions during the second quarter of 2019 described below.
In the first quarter of 2017, we received an upfront payment of approximately $10.8 million following the execution of the Sato Agreement for the exclusive right to develop, use and sell SB204 in certain topical dosage forms in Japan for the treatment of acne vulgaris. In addition, we received a milestone payment of approximately $2.2 million in the fourth quarter of 2018, related to the initiation of a Phase 1 trial in Japan in the third quarter of 2018. Under the terms of the Sato Amendment which expanded the Sato Agreement to include SB206, we also received a payment of $2.2 million (or 0.25 billion JPY) in October 2018, a payment of $4.5 million (or 0.5 billion JPY) in March 2019, and a payment of $4.6 million (or 0.5 billion JPY) in November 2019, representing the three installments of an upfront payment of 1.25 billion JPY.
As also described below, in August 2019, we entered into the Aspire Common Stock Purchase Agreement, which provides that, upon the terms and subject to the conditions and limitations set forth therein, and described below, Aspire Capital is committed to purchase up to an aggregate of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement.
As described below, in late April 2019 and early May 2019, respectively, we entered into (i) the Purchase Agreement with Reedy Creek, which provided $25.0 million of immediate funding; and (ii) the Funding Agreement with Ligand, which provided $12.0 million of immediate funding.
We believe that our existing cash and cash equivalents balance will provide us with adequate liquidity to fund our planned operating needs into the early part of the second quarter of 2020. We will need substantial additional funding to continue our operating activities and make further advancements in our drug development programs. This projected cash runway excludes (i) potential costs associated with an additional confirmatory Phase 3 trial, which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (ii) any proceeds received subsequent to January 31, 2020 from potential future sales of common stock under the Aspire Common Stock Purchase Agreement, described below, if available.
86
Our ability to continue to operate our business, including our ability to advance our development programs, is dependent upon our ability to access additional sources of capital, including, but not limited to (i) non-dilutive sources, such as partnerships, collaborations, licensing, grants or other strategic relationships; or (ii) equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. We may revise our development and operating activities or their timing depending on the availability of additional funding, partnership opportunities and our financial priorities. Our assumptions and plans may change and could impact the magnitude and/or timing of development and operating expenses and therefore our cash runway. In addition, we have continued to explore both financial as well as strategic options in order to continue operations and to progress SB206 for the molluscum indication, subject to FDA feedback. Alternatively, we may seek to engage in one or more potential transactions, such as the sale of our company, or sale or divestiture of some of our assets, such as a sale of our dermatology platform assets, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us.
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate revenue from product sales unless, and until, we obtain regulatory approval of one of our current or future product candidates and achieve successful commercialization by a strategic partner or by ourselves. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates and begin any commercialization activities. We are subject to all of the risks inherent in the development of new pharmaceutical products, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business.
As we continue to endeavor to raise additional capital, there can be no assurance that we will be able to obtain new funding on terms acceptable to us, on a timely basis, or at all. Our failure to obtain sufficient additional funds on acceptable terms as and when needed could cause us to alter or reduce our planned operating activities, including but not limited to delaying, reducing, terminating or eliminating planned product candidate development activities, to conserve our cash and cash equivalents or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. Such actions could delay development timelines and have a material adverse effect on our business, results of operations, financial condition and market valuation. If we are forced to terminate or eliminate our product development programs, wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders. A failure to obtain sufficient funds on acceptable terms when needed, including the inability to utilize the amount available under the Aspire Common Stock Purchase Agreement, could cause us to alter or reduce our planned operating activities to conserve our cash and cash equivalents, including but not limited to delaying planned activities directly related to or in support of product candidate development. Our anticipated expenditure levels may change if we adjust our current operating plan. Such actions could delay development timelines and have a material adverse effect on our results of operations, financial condition and market valuation. As of December 31, 2019, we had an accumulated deficit of $220.0 million and there is substantial doubt about our ability to continue as a going concern.
Our cash and cash equivalents are held in a variety of interest-bearing instruments, including money market accounts. Cash in excess of immediate requirements is invested with a view toward liquidity and capital preservation, and we seek to minimize the potential effects of concentration and degrees of risk.
Aspire Common Stock Purchase Agreement
On August 30, 2019, we entered into the Aspire Common Stock Purchase Agreement, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $25.0 million of shares of our common stock at our request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement. The aggregate amount that we may raise through sales of common stock under the Aspire Common Stock Purchase Agreement is subject to certain limitations including, but not limited to: (i) the number of shares that may be sold will be limited to 5,211,339 shares, representing 19.99% of our outstanding shares of common stock on August 30, 2019, if the average price paid for all shares issued under the agreement is less than $2.17, which was the closing sale price of our common stock immediately preceding the execution of the Aspire Common Stock Purchase Agreement; and (ii) on any purchase date, the closing sale price of our common stock must be greater than or equal to $0.25.
Under the Aspire Common Stock Purchase Agreement, on any trading day we select, we have the right, in our sole discretion, to present Aspire Capital with a purchase notice, or the Purchase Notice, directing Aspire Capital (as principal) to purchase up to 100,000 shares of our common stock per business day, up to an aggregate of $25.0 million of our common stock, at a per share price, or the Purchase Price, equal to the lesser of (i) the lowest sale price of our common stock on the purchase date, or (ii) the arithmetic average of the three (3) lowest closing sale prices for our common stock during the ten (10) consecutive trading days
87
ending on the trading day immediately preceding the purchase date. The aggregate purchase price payable by Aspire Capital on any one purchase date may not exceed $0.5 million. In addition, on any date on which we submit a Purchase Notice to Aspire Capital in an amount equal to 100,000 shares and our stock price is not less than $0.25 per share, we can also, in our sole discretion, present Aspire Capital with a volume-weighted average price purchase notice, or a VWAP Purchase Notice, directing Aspire Capital to purchase an amount of our common stock equal to up to 30% of the aggregate shares of our common stock traded on its principal market on the next trading day, or the VWAP Purchase Date, as determined by us.
There are no trading volume requirements or restrictions under the Aspire Common Stock Purchase Agreement, and we will control the timing and amount of sales of our common stock to Aspire Capital. Aspire Capital has no right to require any sales by us, but is obligated to make purchases from us as directed by the Company in accordance with the Aspire Common Stock Purchase Agreement. There are no limitations on use of proceeds, financial or business covenants, restrictions on future financing transactions, rights of first refusal, participation rights, penalties or liquidated damages in the Aspire Common Stock Purchase Agreement. In consideration for entering into the Aspire Common Stock Purchase Agreement, concurrently with the execution of the Aspire Common Stock Purchase Agreement, we issued to Aspire Capital 345,622 shares of our common stock as part of the commitment fee. The Aspire Common Stock Purchase Agreement may be terminated by us any time, at our discretion, without any penalty or additional cost to us. Any proceeds we receive under the Aspire Common Stock Purchase Agreement are expected to be used for working capital and general corporate purposes.
Royalty and Milestone Payments Purchase Agreement with Reedy Creek Investments LLC
On April 29, 2019, we entered into the Purchase Agreement with Reedy Creek, pursuant to which Reedy Creek provided us funding in an initial amount of $25.0 million, which we will use primarily to pursue the development, regulatory approval and commercialization (including through out-license agreements and other third-party arrangements) activities for SB206, for the treatment of molluscum, and advancing programmatically other activities with respect to SB414, for atopic dermatitis, and SB204, for acne. Reedy Creek was to provide $10.0 million of additional funding contingent upon our achievement of the primary endpoints in each of the two SB206 Phase 3 clinical trials no later than March 31, 2020. Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
Pursuant to the Purchase Agreement, we will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by us pursuant to any out-license agreement for the products in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by us to third parties pursuant to any agreements under which we have in-licensed intellectual property with respect to the products.
The applicable percentage used for determining the ongoing quarterly payments, applied to amounts received directly by us pursuant to any out-license agreement for each product, ranges from 10% for SB206 to 20% for SB414 and SB204. However, the agreement provides that the applicable percentage for each product will be 25% for fees or milestone payments received by us (but not royalty payments received by us) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If we decide to commercialize any product on its own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, we will be obligated to pay Reedy Creek a low single digits royalty on net sales of the products.
Development Funding and Royalties Agreement with Ligand Pharmaceuticals Incorporated
On May 4, 2019, we entered into the Funding Agreement with Ligand, pursuant to which Ligand provided us funding of $12.0 million, which we used to pursue the development and regulatory approval of SB206, for the treatment of molluscum.
Pursuant to the Funding Agreement, we will pay Ligand up to $20.0 million in milestone payments upon the achievement by us of certain regulatory and commercial milestones associated with SB206 or any product that incorporates or uses NVN1000, the active pharmaceutical ingredient for our clinical stage product candidates, for the treatment of molluscum. In addition to the milestone payments, we will pay Ligand tiered royalties ranging from 7% to 10% based on annual aggregate net sales of the products in the United States, Mexico or Canada.
Expansion of Partnership with Sato in Japanese Territory
On October 5, 2018, we and Sato entered into the Sato Amendment. The initial Sato Agreement had focused on the development and commercialization of SB204 for the treatment of acne vulgaris in Japan. The Sato Amendment also provides Sato with the exclusive rights to develop and commercialize SB206 and related dosage forms for the treatment of viral skin infections, including but not limited to molluscum contagiosum and external genital warts, in Japan. Under the terms of the
88
Sato Amendment, we received an upfront payment from Sato totaling 1.25 billion JPY (approximately $11.3 million USD) to be paid in three installments over a 12 months period. We received the first installment of 0.25 billion JPY (approximately $2.2 million USD) in October 2018, the second installment of 0.5 billion JPY (approximately $4.5 million USD) in March 2019 and the third installment of 0.5 billion JPY (approximately $4.6 million USD) in November 2019. The Sato Amendment also provides for an aggregate of 1.0 billion JPY in additional non-contingent milestone payments that become payable upon the earlier occurrence of specified fixed dates in the future or the achievement of specified milestone events.
January 2018 Offering
On January 9, 2018, we completed a public offering of our common stock and warrants under our effective shelf registration statement. We sold an aggregate of 10,000,000 shares of common stock and warrants to purchase up to 10,000,000 shares of our common stock at a public offering price of $3.80 per share of common stock and accompanying warrant. The warrant exercise price is $4.66 per share and the warrants will expire four years from the date of issuance. Net proceeds from the offering were approximately $35.2 million after deducting underwriting discounts and commissions and offering expenses of approximately $2.8 million. The warrants sold in the January 2018 Offering are classified as equity in the accompanying consolidated balance sheets.
Primary Facility Lease Financing
Our approximately 51,000 square foot leased facility in Morrisville, North Carolina serves as our corporate headquarters and sole research, development and manufacturing facility. We entered into the ten-year, non-cancellable lease agreement in 2016, currently have approximately six and a half years remaining under the lease term and currently have approximately $8.5 million in remaining minimum lease payments.
As part of our broader strategic plan to shift our operating cost structure characteristics from fixed to variable, we are actively pursuing efforts to reduce or offset our remaining fixed lease obligation. We have engaged a commercial real estate broker and are currently marketing our Morrisville, North Carolina headquarters facility for sublease or assignment.
In July 2018, the Company and a third-party tenant entered into a sublease of approximately 6,400 square feet of office space, or approximately 12% of total facility square footage, at our Morrisville, North Carolina headquarters. In October 2019, we executed a termination agreement with the subtenant for this sublease, with an effective date of December 31, 2019.
We assess the carrying value of long-lived assets whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. As of December 31, 2019, we concluded there were no such events or changes in circumstances requiring review of the carrying amount of the Company’s long-lived assets.
Cash Flows
The following table sets forth our cash flows for the periods indicated:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(in thousands) | |||||||
Net cash (used in) provided by: | |||||||
Operating activities | $ | (19,876 | ) | $ | (28,625 | ) | |
Investing activities | (422 | ) | (1,058 | ) | |||
Financing activities | 25,816 | 35,353 | |||||
Net increase in cash, cash equivalents and restricted cash | $ | 5,518 | $ | 5,670 | |||
89
Net Cash Used in Operating Activities
During the year ended December 31, 2019, net cash used in operating activities was $19.9 million and consisted primarily of a net loss of $30.4 million, with adjustments for non-cash amounts related primarily to depreciation expense of $2.0 million, share-based compensation expense of $1.8 million and a favorable change related to changes in other operating assets and liabilities of $6.6 million. The favorable net change in assets and liabilities was primarily due to a $12.0 million increase related to the advanced payment for the research and development service obligation associated with the Funding Agreement with Ligand, offset by a $8.2 million decrease in research and development service obligation liabilities related to the amortization of the liability associated with Ligand, a $4.5 million increase in deferred revenue following the receipt of an additional upfront installment payment under the Amended Sato Agreement during the first quarter of 2019, a $0.4 million increase in accounts payable and a $0.5 million increase in accrued outside research and development services. The increase in accounts payable and accruals was primarily related to the Phase 3 development program for molluscum and timing of payments related to our operating activities. These favorable changes were partially offset by a $1.0 million decrease in accrued compensation following the Compensation Committee of the Board of Directors’ determination that the associated corporate performance objectives were not achieved during 2019, and a decrease in other operating assets and liabilities totaling $1.5 million. The decrease in other operating assets and liabilities include an increase in prepaid expenses and contracts and grants receivable and a decrease in other long-term liabilities, primarily related to amortization of lease liabilities.
During the year ended December 31, 2018, net cash used in operating activities was $28.6 million and consisted primarily of a net loss of $29.2 million, with adjustments for non-cash amounts related primarily to depreciation expense of $1.7 million, share-based compensation expense of $2.2 million and a net decrease in other operating assets and liabilities of $3.4 million. The net decrease in assets and liabilities was primarily due to a $0.7 million decrease in accrued compensation following the payment of annual employee bonuses in the first quarter of 2018, a $1.3 million decrease in other accrued expenses following the payment of various accrued expenses during the period, including $0.2 million in travel costs paid to Malin (as reimbursement of out-of-pocket expenses for our Chief Executive Officer and a number of Malin employees who supported us with certain strategic and tactical initiatives and activities in 2017), and a $1.6 million decrease in deferred revenue associated with the continued recognition of licensing revenues from the Amended Sato Agreement during 2018. These decreases were partially offset by a favorable change in prepaid expenses and other current assets, other assets and accounts payable of $0.2 million.
Net Cash Used in Investing Activities
During the year ended December 31, 2019, net cash used in investing activities was $0.4 million, which primarily related to purchases of laboratory equipment and leasehold improvements at our facility in Morrisville, North Carolina.
During the year ended December 31, 2018, net cash used in investing activities was $1.1 million, which primarily related to purchases of laboratory equipment and leasehold improvements at our facility in Morrisville, North Carolina.
Net Cash Provided by Financing Activities
During the year ended December 31, 2019, net cash provided by financing activities was $25.8 million and consisted of $25.0 million of funding received pursuant to the Purchase Agreement with Reedy Creek, $0.7 million of proceeds from the sale of our common stock pursuant to the Aspire Common Stock Purchase Agreement and $0.1 million of proceeds from the exercise of stock options.
During the year ended December 31, 2018, net cash provided by financing activities was $35.4 million, consisting primarily of net proceeds from the January 2018 Offering after deducting underwriting discounts and offering expenses.
Capital Requirements
As of December 31, 2019, we had a total cash and cash equivalents balance of $13.7 million and positive working capital of $2.8 million. As of the date of this filing, we believe that our existing cash and cash equivalents balance will provide us with adequate liquidity to fund our planned operating needs into the early part of the second quarter of 2020. This projected cash runway excludes (i) potential costs associated with an additional confirmatory Phase 3 trial, which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (ii) any proceeds received subsequent to January 31, 2020 from potential future sales of common stock under the Aspire Common Stock Purchase Agreement, described above, if available.
We are utilizing our existing capital resources to fund the ongoing and near-term operating and development activities, as described in the “Overview” section above. We will need substantial additional funding to continue our operating activities and make further advancements in our drug development programs. Therefore, we will need to secure additional capital and/or
90
delay, defer, or reduce our cash expenditures by the early part of the second quarter of 2020, including those associated with our product development programs, or we may need to dissolve and liquidate our assets or seek protection under bankruptcy laws. If we are forced to terminate or eliminate our product development programs, wind down our operations, liquidate or seek bankruptcy protection, it is unclear to what extent we will be able to pay our obligations, and, accordingly, it is further unclear whether and to what extent any resources would be available for distributions to our stockholders. Alternatively, we may seek to engage in one or more potential transactions, such as the sale of our company, or sale or divestiture of some of our assets, such as a sale of our dermatology platform assets, but there can be no assurance that we will be able to enter into such a transaction or transactions on a timely basis or at all or on terms that are favorable to us.
Our ability to continue to operate our business, including our ability to advance our development programs, is dependent upon our ability to access additional sources of capital, including, but not limited to (i) non-dilutive sources, such as partnerships, collaborations, licensing, grants or other strategic relationships; or (ii) equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. We may revise our development and operating activities or their timing depending on the availability of additional funding, partnership opportunities and our financial priorities. Our assumptions and plans may change and could impact the magnitude and/or timing of development and operating expenses and therefore our cash runway. We continue to explore other potential non-dilutive business development activities around the developmental and commercial rights to the clinical-stage assets in our platform, including various geographic and indication-specific opportunities.
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate revenue from product sales unless, and until, we obtain regulatory approval of one of our current or future product candidates and achieve successful commercialization by a strategic partner or by ourselves. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our product candidates and begin any commercialization activities. We are subject to all of the risks inherent in the development of new pharmaceutical products, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may adversely affect our business.
As we continue to attempt to raise additional capital, there can be no assurance that we will be able to obtain it on terms acceptable to us, on a timely basis, or at all. A failure to obtain sufficient funds on acceptable terms when needed, including the inability to utilize the amount available under the Aspire Common Stock Purchase Agreement, could cause us to alter or reduce our planned operating activities to conserve our cash and cash equivalents, including but not limited to delaying planned activities directly related to or in support of product candidate development. Our anticipated expenditure levels may change if we adjust our current operating plan. Such actions could delay development timelines and have a material adverse effect on our results of operations, financial condition and market valuation. As of December 31, 2019, we had an accumulated deficit of $220.0 million and there is substantial doubt about our ability to continue as a going concern.
We have based our projections of operating capital requirements on assumptions that may prove to be incorrect and we may use all of our available capital resources sooner than we expect. Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amount or timing of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
• | the initiation, progress, timing, costs, results, and evaluation of results of trials for our clinical-stage product candidates, including trials conducted by us or potential future partners; |
• | the progress, timing, costs and results of development and preclinical study activities relating to other potential applications of our nitric oxide platform; |
• | the number and characteristics of product candidates that we pursue; |
• | our ability to enter into strategic relationships to support the continued development of certain product candidates and the success of those arrangements; |
• | our success in optimizing the size and capability of our current manufacturing facility and related processes to meet our strategic objectives; |
• | our success in the technical transfer of methods and processes related to our drug substance and drug product manufacturing with our current and/or potential future contract manufacturing partners; |
• | the outcome, timing and costs of seeking regulatory approvals; |
• | the occurrence and timing of potential development and regulatory milestones achieved by Sato, our licensee for SB204 and SB206 in Japan; |
91
• | the terms and timing of any future collaborations, licensing, consulting, financing or other arrangements that we may enter into; |
• | the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
• | the costs of preparing, filing and prosecuting patent applications, maintaining and protecting our intellectual property rights; |
• | defending against intellectual property related claims; |
• | the costs associated with any potential future securities litigation, and the outcome of that litigation; |
• | the extent to which we in-license or acquire other products and technologies; and |
• | subject to receipt of marketing approval, revenue received from commercial sales or out licensing of our product candidates. |
Contractual Obligations and Contingent Liabilities
Primary Facility Lease
We entered into a lease agreement in August 2015 for a facility totaling approximately 51,000 square feet in Morrisville, North Carolina and began to occupy and utilize the facility in October 2016. The term of the lease commenced April 1, 2016, and terminates June 2026. The remaining estimated lease payments for this facility over the term of the lease are approximately $8.5 million. We have an option to extend this facility lease by five years upon completion of the initial lease term.
As part of our broader strategic plan to shift our operating cost structure characteristics from fixed to variable, we are actively pursuing efforts to reduce or offset our remaining fixed lease obligation. We have engaged a commercial real estate broker and are currently marketing our Morrisville, North Carolina headquarters facility for sublease or assignment.
In July 2018, the Company and a third-party tenant entered into a sublease of approximately 6,400 square feet of office space, or approximately 12% of total facility square footage, at our Morrisville, North Carolina headquarters. In October 2019, we executed a termination agreement with the subtenant for this sublease, with an effective date of December 31, 2019.
Amended Sato Agreement
Pursuant to the Amended Sato Agreement, we are obligated to supply Sato with all quantities of licensed products required by Sato for their development activities in Japan. As part of the Amended Sato Agreement, we and Sato also agreed to negotiate a commercial supply agreement pursuant to which we or a third-party contract manufacturer would be the exclusive supplier to Sato of the API of licensed products for the commercial manufacture of licensed products in the licensed territory. Additionally, we have agreed to perform certain oversight, review and supporting activities for Sato, including: (i) using commercially reasonable efforts to obtain marketing approval of SB204 and SB206 in the U.S, (ii) sharing all future scientific information we may obtain during the term of the Amended Sato Agreement pertaining to SB204 and SB206, (iii) performing certain additional preclinical studies if such studies are deemed necessary by the Japanese regulatory authority, up to and not to exceed a total cost of $1.0 million, and (iv) participating in a joint committee that oversees, reviews, and approves Sato’s development and commercialization activities under the Amended Sato Agreement. Additionally, we have granted Sato the option to use our trademarks in connection with the commercialization of licensed products in the licensed territory for no additional consideration, subject to our approval of such use. We cannot estimate if, when or in what amounts such payments will become due under the Amended Sato Agreement.
The intellectual property rights granted to Sato under the Amended Sato Agreement include certain intellectual property rights which we have licensed from UNC. Under our license agreement with UNC described in “Note 4—Research and Development Licenses” to the accompanying consolidated financial statements included in this Annual Report, we are obligated to pay UNC a running royalty percentage in the low single digits on net sales of licensed products, including net sales that may be generated by Sato. Additionally, we are obligated to make payments to UNC that represent the portion of the Sato upfront and milestone payments that were estimated to be directly attributable to the UNC intellectual property rights included in the license to Sato.
We had also previously entered into an agreement with a third party to assist us in exploring the licensing opportunity which led to the execution of the Sato Agreement. We are obligated to pay the third party a low-single-digit percentage of all upfront and milestone payments the Company receives from Sato under the Amended Sato Agreement.
92
We have accrued certain fees that we will pay to UNC and a third party in the future upon receipt of non-contingent installment and milestone payments from Sato. As of December 31, 2019, we had recorded capitalized contract acquisition costs of $0.5 million in other assets and had accrued $0.2 million in the accompanying consolidated balance sheets. For the years ended December 31, 2019 and 2018 we paid fees totaling $0.2 million and $0.1 million, respectively.
See “Note 6—Revenue Recognition” to the accompanying consolidated financial statements included in this Annual Report for additional information on the Amended Sato Agreement.
Amendments to Sublicense Agreements with KNOW Bio
Pursuant to the terms of the amendments to the KNOW Bio Agreements that we entered in October 2017, we re-acquired from KNOW Bio exclusive, worldwide rights under certain U.S. and foreign patents and patent applications controlled by us as of the execution date of the KNOW Bio Agreements, and patents and patent applications which became controlled by us during the three years immediately following the execution date of the KNOW Bio Agreements, directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, to develop and commercialize products for all diagnostic, therapeutic, prophylactic and palliative uses for any disease, condition or disorder caused by certain oncoviruses, or the Oncovirus Field. KNOW Bio also granted to us an exclusive license, with the right to sublicense, under any patents and patent applications which became controlled by KNOW Bio during the three years immediately following the execution date of the KNOW Bio Agreements and directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, but not towards medical devices, to develop and commercialize products for use in the Oncovirus Field. Additionally, KNOW Bio agreed that KNOW Bio would not commercialize any products in the Oncovirus Field during the first three years following the execution date of the KNOW Bio Agreements. The three-year period in which new patents and patent applications are added to the exclusive license and the three-year term of the commercialization non-compete both expired on December 29, 2018.
In addition to the $0.3 million non-refundable upfront payment we made upon execution of the KNOW Bio Amendments, we are obligated to make the following contingent payments in exchange for the rights granted to us in the Oncovirus Field:
For products that incorporate a certain nitric oxide-releasing composition specified in the KNOW Bio Amendments and (i) are covered by KNOW Bio patents or (ii) materially use or incorporate know-how of KNOW Bio or us related to such composition that is created during the three years immediately following the execution date of the KNOW Bio Agreements, or the Covered Products, we must make the following payments to KNOW Bio:
o | A milestone payment upon the first time each Covered Product is approved by the FDA for marketing in the Oncovirus Field; |
o | A royalty in the low single digits on net sales of Covered Products in the Oncovirus Field until the later of the expiration of the KNOW Bio patents covering the applicable Covered Product or the expiration of regulatory exclusivity on the applicable Covered Product; and |
o | In the event we sublicense the rights to a Covered Product to a third party in the Oncovirus Field, the Company must pay KNOW Bio a low double-digit percentage of any clinical development or NDA approval milestones we receive from the sublicensee for the Covered Product in the Oncovirus Field. |
Nitricil is not the nitric oxide-releasing composition specified in the KNOW Bio Amendments as the subject of the foregoing payments. As such, products based on Nitricil are not subject to the foregoing milestone, royalty and sublicensing payment obligations.
The rights granted to us in the Oncovirus Field in the KNOW Bio Amendments continue for so long as there is a valid patent claim under the KNOW Bio Agreements, and upon expiration continue on a perpetual non-exclusive basis, and are subject to the termination rights of KNOW Bio and us that are set forth in the KNOW Bio Agreements. In addition, under the KNOW Bio Amendments, KNOW Bio may terminate the rights granted to us in the Oncovirus Field if: (i) we do not file a first IND with the FDA for a product in the Oncovirus Field by October 2020; or (ii) we do not file a first NDA with the FDA by October 2025 for a product in the Oncovirus Field and does not otherwise have any active clinical programs related to the Oncovirus Field at such time. We also obtained a three-year exclusive option, subject to payment of separate option exercise fees, to include up to four additional specified oncoviruses in the Oncovirus Field.
See “Note 3—KNOW Bio, LLC” to the accompanying consolidated financial statements included in this Annual Report for additional information on the sublicense agreement and our assessment of KNOW Bio under the variable interest consolidation model pursuant to FASB ASC 810, Consolidation.
93
Royalty and Milestone Payments Purchase Agreement with Reedy Creek Investments LLC
On April 29, 2019, we entered into the Purchase Agreement with Reedy Creek, pursuant to which Reedy Creek provided us funding in an initial amount of $25.0 million, which we will use primarily to pursue the development, regulatory approval and commercialization (including through out-license agreements and other third party arrangements) activities for SB206, for the treatment of molluscum, and advancing programmatically other activities with respect to SB414, for atopic dermatitis, and SB204, for acne. Reedy Creek was to provide $10.0 million of additional funding contingent upon our achievement of the primary endpoints in each of the two SB206 Phase 3 clinical trials no later than March 31, 2020. Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
Pursuant to the Purchase Agreement, we will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by us pursuant to any out-license agreement for the products in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by us to third parties pursuant to any agreements under which we have in-licensed intellectual property with respect to the products.
The applicable percentage used for determining the ongoing quarterly payments, applied to amounts received directly by us pursuant to any out-license agreement for each product, ranges from 10% for SB206 to 20% for SB414 and SB204. However, the agreement provides that the applicable percentage for each product will be 25% for fees or milestone payments received by us (but not royalty payments received by us) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If we decide to commercialize any product on its own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, we will be obligated to pay Reedy Creek a low single digits royalty on net sales of the products.
Development Funding and Royalties Agreement with Ligand Pharmaceuticals Incorporated
On May 4, 2019, we entered into the Funding Agreement with Ligand, pursuant to which Ligand provided us funding of $12.0 million, which we used to pursue the development and regulatory approval of SB206, for the treatment of molluscum.
Pursuant to the Funding Agreement, we will pay Ligand up to $20.0 million in milestone payments upon the achievement by us of certain regulatory and commercial milestones associated with SB206 or any product that incorporates or uses NVN1000, the active pharmaceutical ingredient for our clinical stage product candidates, for the treatment of molluscum. In addition to the milestone payments, we will pay Ligand tiered royalties ranging from 7% to 10% based on annual aggregate net sales of the products in the United States, Mexico or Canada.
Tangible Stockholder Return Plan, or Performance Plan
In August 2018, our board of directors approved and established the Performance Plan. We believe that the Performance Plan will help us attract, retain and incentivize the highly qualified resources that are and will be necessary to execute on our operating strategy. Executive management and the board of directors believe this plan clearly and directly ties long-term employee incentive compensation to specific, significant increases in our underlying common stock price and thus directly aligns employee and stockholder objectives. Unlike our historical practice of providing long-term incentives to our employees through annual stock option grants under the 2016 Plan at the then current market price of our common stock, the Performance Plan only provides for employees to receive long-term incentive compensation payments if the established stock price targets ($11.17 per share and $25.45 per share, subject to adjustment) are achieved.
The Performance Plan is tiered, with two separate tranches, each of which has a distinct share price target (measured as the average publicly traded share price of our common stock on the Nasdaq stock exchange for a thirty consecutive trading day period) that will trigger a distinct fixed bonus pool. The share price target for the first tranche is $11.17 per share. The share price target for the second tranche is $25.45 per share. The related contingent bonus pools for the first and second tranches are $25.0 million and $50.0 million, respectively. The compensation committee has discretion to distribute the bonus pool related to each tranche among eligible participants by establishing individual minimum bonus amounts before, as well as by distributing the remainder of the applicable pool after, the achievement of each tranche specific share price target. Otherwise, if we do not achieve one or both related share price targets, as defined, no portion of the bonus pools will be paid.
The Performance Plan provides for the bonus pool to generally be paid in the form of cash. However, the compensation committee has discretion to pay any bonus award under the Performance Plan in the form of cash, shares of our common stock or a combination thereof, provided that our board and stockholders have approved the reservation of such shares of our common stock for such payment. The share price targets will be adjusted in the event of any stock splits, cash dividends, stock
94
dividends, combinations, reorganizations, reclassifications, or similar events. In addition, in the event of a change in control, the plan provides that a bonus pool will become due and payable to participants on a pro rata basis, as calculated and determined by the compensation committee based upon our progress toward the share price target as of the date of the change in control and subject to adjustment by the compensation committee as permitted under the plan.
The Performance Plan was effective immediately upon approval, expires on March 1, 2022, and covers all employees, including our executive officers, consultants and other persons deemed eligible by our compensation committee. The Performance Plan was subsequently amended and restated to reflect minor changes in the timing for establishing minimum bonus amounts.
See “Note 12—Tangible Stockholder Return Plan” to the accompanying consolidated financial statements included in this Annual Report for additional information on the Performance Plan.
Stock Appreciation Rights
On August 8, 2018, we entered into the Martin Employment Agreement with G. Kelly Martin. The Martin Employment Agreement provided for 1,000,000 SARs granted on a contingent basis that would have been irrevocably forfeited and voided in full if we failed to obtain stockholder approval for an amendment to the 2016 Plan to allow such stock award. If such approval had not been obtained, we would have been required to pay Mr. Martin the cash equivalent of the value of the SARs.
On July 31, 2019, at our 2019 Annual Meeting of Stockholders, our stockholders approved a proposal to amend the 2016 Plan, to (i) increase the number of shares of our common stock reserved for issuance under the 2016 Plan by 1,000,000 shares; and (ii) increase the limit on the number of awards that may be granted to any one person in any year. As such, with stockholder approval of the amendment to the 2016 Plan, the SARs detailed within the Martin Employment Agreement were no longer considered granted on a contingent basis.
The SARs entitled Mr. Martin to a payment (in cash, shares of common stock or a combination of both) equal to the fair market value of one share of our common stock on the date of exercise less the exercise price of $3.80 per share. The SARs were to be deemed automatically exercised and settled as of February 1, 2020, provided Mr. Martin remained continuously employed with us through such date unless vesting was otherwise expressly accelerated pursuant to the Martin SAR Award. The SARs vested in full on February 1, 2020. On February 1, 2020, the fair market value of our common stock was $0.52 per share, and as such, the SARS expired unexercised and 1,000,000 shares became available to be granted under the 2016 Plan.
Effective December 17, 2019, we entered into the Amended and Restated Stafford Employment Agreement with Paula Brown Stafford. On January 6, 2020, as provided by the Amended and Restated Stafford Employment Agreement, 600,000 SARs were granted to Ms. Stafford with an exercise price of $0.82 per share, following our release of top-line results of our Phase 3 molluscum clinical program. The Stafford SAR Award was granted on a contingent basis and would have been considered irrevocably forfeited and voided in full if sufficient shares of our common stock were not available under the 2016 Plan or if we failed to obtain stockholder approval for amendments to the 2016 Plan at the next annual stockholders’ meeting to provide sufficient shares for the Stafford SAR Award. If such shares were not available, we would have been required to pay Ms. Stafford the cash equivalent of the value of the SARs. Such shares became available under the 2016 Plan on February 1, 2020 and the SARs were no longer considered granted on a contingent basis.
See “Note 11—Share-Based Compensation” to the accompanying consolidated financial statements included in this Annual Report for additional information on the SARs.
Other
We enter into contracts in the normal course of business with clinical research organizations for clinical trials and clinical supply manufacturing and with vendors for preclinical research studies, research supplies and other services and products for operating purposes. These contracts generally provide for termination on notice, and therefore we believe that our non-cancelable obligations under these agreements are not material.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules.
95
Net Operating Loss and Research and Development Tax Credit Carryforwards
As of December 31, 2019, we had federal and state net operating loss carryforwards of approximately of $165.6 million and $165.1 million, respectively. The net operating loss carryforwards begin to expire in 2028 and 2023 for federal and state tax purposes, respectively. We have research and development tax credits of approximately $8.1 million to offset future federal taxes. These credits begin to expire in 2028.
We record a valuation allowance to offset any net deferred tax assets if, based upon the available evidence, it is more likely than not that we will not recognize some or all of the deferred tax assets. We have had a history of net losses since inception, and, as a result, we have established a 100% valuation allowance of $54.4 million for our net deferred tax assets as of December 31, 2019. If circumstances change and we determine that we will be able to realize some or all of these net deferred tax assets in the future, we will record an adjustment to the valuation allowance.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events including significant changes in ownership interests. In accordance with Section 382 of the Code, a change in equity ownership of greater than 50% within a three-year period results in an annual limitation on our ability to utilize our NOL carryforwards created during the tax periods prior to the change in ownership. We have not determined whether ownership changes exceeding this threshold, including our IPO and the January 2018 offering, have occurred. If a change in equity ownership has occurred which exceeds the Section 382 threshold, a portion of our NOL carryforwards may be limited. If our net operating loss carryforwards are limited, and we have taxable income which exceeds the permissible yearly net operating loss carryforwards, we would incur a federal income tax liability even though net operating loss carryforwards would be available in future years.
Jumpstart Our Business Startups Act of 2012 (JOBS Act)
In April 2012, the JOBS Act was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” As an “emerging growth company,” we are electing not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and, as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth public companies. Section 107 of the JOBS Act provides that our decision not to take advantage of the extended transition period is irrevocable. We have chosen to rely on the other exemptions and reduced reporting requirements provided by the JOBS Act. Subject to certain conditions set forth in the JOBS Act, as an “emerging growth company” we are not required to, among other things, (i) provide an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act, (ii) provide all of the compensation disclosure that may be required of non-emerging growth public companies under the Dodd-Frank Act, (iii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements (auditor discussion and analysis) and (iv) disclose certain executive compensation-related items, such as the correlation between executive compensation and performance and comparisons of the chief executive officer’s compensation to median employee compensation. We may remain an emerging growth company until the last day of 2021. However, if certain events occur prior to such date, including if we become a “large accelerated filer,” our annual gross revenue equals or exceeds $1.07 billion or we issue more than $1.0 billion of non-convertible debt in any three-year period, we will cease to be an emerging growth company prior to such date.
Recent Accounting Pronouncements
Recently issued accounting pronouncements that we have adopted or are currently evaluating are described in detail within “Note 1—Organization and Significant Accounting Policies” to the accompanying consolidated financial statements included in this Annual Report.
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
There were no changes in or disagreements with accountants on accounting and financial disclosures.
96
Critical Accounting Policies and Use of Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue and expenses during the reporting periods. These items are monitored and analyzed by us for changes in facts and circumstances, and material changes in these estimates could occur in the future. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Changes in estimates are reflected in reported results for the period in which they become known. Actual results may differ materially from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in the notes to our financial statements included elsewhere in this annual report, we believe that the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements and understanding and evaluating our reported financial results.
Leases
We lease office space and certain equipment under non-cancelable lease agreements. Prior to January 1, 2019, we applied the accounting guidance in ASC 840, Leases, to our lease agreements. The leases were reviewed for classification as operating or capital leases. For operating leases, rent was recognized on a straight-line basis over the lease period. For capital leases, we recorded the leased asset with a corresponding liability and amortized the asset over the lease term. Payments were recorded as reductions to the liability with an appropriate interest charge recorded based on the then-outstanding remaining liability.
Effective January 1, 2019, we adopted ASU No. 2016-02, Leases (Topic 842) using the modified retrospective transition method and established our lease accounting policy pursuant to this new standard. We initially applied the transition provisions at January 1, 2019, which allowed us to continue to apply the legacy guidance in ASC 840 for periods prior to 2019. Based on the new guidance, we assess all arrangements, that convey the right to control the use of property, plant and equipment, at inception, to determine if it is, or contains, a lease based on the unique facts and circumstances present in that arrangement. For those leases identified, we determine the lease classification, recognition, and measurement at the lease commencement date. For arrangements that contain a lease we: (i) identify lease and non-lease components; (ii) determine the consideration in the contract; (iii) determine whether the lease is an operating or financing lease; and (iv) recognize lease Right of Use (“ROU”) assets and corresponding lease liabilities. Lease liabilities are recorded based on the present value of lease payments over the expected lease term. The corresponding ROU asset is measured from the initial lease liability, adjusted by (i) accrued or prepaid rents; (ii) remaining unamortized initial direct costs and lease incentives; and (iii) any impairments of the ROU asset. The interest rate implicit in our lease contracts is typically not readily determinable and as such, we use our incremental borrowing rate based on the information available at the lease commencement date, which represents an internally developed rate that would be incurred to borrow, on a collateralized basis, over a similar term, an amount equal to the lease payments in a similar economic environment.
Revenue Recognition
Beginning in 2017, we began to generate revenue from (i) non-refundable upfront fees, milestone payments and royalties earned under license agreements and (ii) providing research and development services.
Effective January 1, 2018, we adopted ASC Topic 606, Revenue from Contracts with Customers, using the full retrospective adoption method and established our revenue recognition accounting policy pursuant to this new standard. See “Note 1—Organization and Significant Accounting Policies” and “Note 6—Revenue Recognition” to the accompanying consolidated financial statements included in this Annual Report for further information and accounting considerations related to revenue recognition, including revenue recognition pertaining to licensing arrangements.
Licensing Arrangements
We entered into the Sato Agreement in the first quarter of 2017, and the Sato Amendment in October 2018, and may enter into additional licensing arrangements in the future, in exchange for non-refundable upfront payments and potential future milestone and royalty payments.
If the license of our Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable, upfront fees allocated to the license when the
97
license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are bundled with other promises, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the estimated performance period and the appropriate method of measuring progress during the performance period for purposes of recognizing revenue. We re-evaluate the estimated performance period and measure of progress each reporting period and, if necessary, adjust related revenue recognition accordingly.
At the inception of each arrangement that includes development milestone payments, we evaluate whether the milestones are considered probable of being reached and estimate the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which we recognize revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such development milestones and any related constraint, and if necessary, adjust our estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect license and collaboration revenue and earnings in the period of adjustment.
Amounts received prior to satisfying all revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets.
Specifically related to the Sato Agreement, as amended, we recognize revenue using a time-based input method that results in straight-line recognition over the Company’s performance period. We monitor and reassess the estimated performance period for purposes of revenue recognition during each reporting period. We expect to reassess the estimated performance period during the first quarter of 2020, as we consider how the combined SB204 and SB206 development program timeline in Japan may potentially be affected by various factors, including (i) the recent results from our SB206 Phase 3 trials in the U.S., including but not limited to top-line efficacy results announced in January 2020, (ii) our plans and timelines for potential further clinical development of SB206 in the U.S., which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (iii) our in-house drug manufacturing capabilities and the progression of our manufacturing technology transfer projects with third-party contract manufacturing organizations. Therefore, if the duration of the combined SB204 and SB206 development program timeline is affected by the establishment or subsequent adjustments to a mutually agreed upon SB204 and SB206 development plan in the Japan territory, we will adjust its estimated performance period for revenue recognition purposes accordingly, as needed.
Government research contracts and grants revenue.
Under the terms of the contracts and grants awarded, we are entitled to receive reimbursement of our allowable direct expenses, allocated overhead, general and administrative expenses and payment of other specified amounts. Revenues from development and support activities under government research contracts and grants are recorded in the period in which the related costs are incurred. Associated expenses are recognized when incurred as research and development expense. Revenue recognized in excess of amounts collected are recorded as contracts and grants receivable. Any of the funding sources may, at their discretion, request reimbursement for expenses or return of funds, or both, as a result of noncompliance by us with the terms of the grants. No reimbursement of expenses or return of funds has been requested or made since inception of the contracts and grants. See “Note 6—Revenue Recognition” to the accompanying consolidated financial statements included in this Annual Report for information regarding government grants.
Research and Development Expenses
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses. This process involves reviewing open contracts and purchase orders, communicating with applicable vendor personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued expenses include fees incurred by CROs in connection with clinical trials, fees paid to investigative sites in connection with clinical trials, professional service fees and unpaid salaries, wages and benefits.
98
We accrue our expenses related to clinical trials based on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we will adjust the accrual accordingly. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. We do not anticipate the future settlement of existing accruals to differ materially from our estimates.
Reedy Creek Purchase Agreement
We have determined that the Reedy Creek Purchase Agreement is within the scope of ASC 730-20, Research and Development Arrangements. We concluded that there has not been a substantive and genuine transfer of risk related to the Purchase Agreement as (i) Reedy Creek has the opportunity to recover its investment regardless of the outcome of the research and development programs within the scope of the agreement (prior to commercialization of any in scope assets through potential out-licensing agreements and related potential future milestone payments); and (ii) there is a presumption that we are obligated to pay Reedy Creek amounts equal to its investment based on the related party relationship at the time the parties entered into the Purchase Agreement. The Purchase Agreement is a broad funding arrangement, due to (i) the multi-asset, or portfolio approach including three developmental assets that are within the scope of the arrangement; and (ii) Reedy Creek’s approximate 15% ownership of our outstanding shares of common stock.
As such, we have determined that the appropriate accounting treatment under ASC 730-20 is to record the initial proceeds of $25,000 as cash and cash equivalents, as we have the ability to direct the usage of funds, and a long-term liability within our classified balance sheet. The long-term liability will remain until we receive future milestones from other potential third parties, as defined within the Purchase Agreement, of which 25% will be contractually owed to Reedy Creek. If potential future milestones are received by us, and become partly due to Reedy Creek, the corresponding partial repayment to Reedy Creek will result in a ratable reduction of the total long-term obligation to repay the initial purchase price.
Ligand Funding Agreement
We have determined that the Ligand transaction is within the scope of ASC 730-20 as it represents an obligation to perform contractual services for the development of SB206 using commercially reasonable efforts. In addition, the Funding Agreement also states that if all development of SB206 is ceased prior to the first regulatory approval, we must pay to Ligand an amount equal to the purchase price less the amount spent in accordance with the development budget on development activities conducted prior to such cessation. As such, we concluded that the appropriate accounting treatment under ASC 730-20 was to record the initial proceeds of $12,000, as a liability and as restricted cash on our consolidated balance sheet, as the funds could only be used for the progression of SB206.
We amortize the liability ratably during each reporting period, based on the Ligand funding as a percentage of the total direct costs we incur during the reporting period related to the estimated total cost to progress the SB206 program to a regulatory approval in the U.S. The ratable Ligand funding is presented within the consolidated statement of operations as an offset to research and development expenses associated with the SB206 program. During the first quarter of 2020, we expect to reassess the estimated total cost to progress the SB206 program to a U.S. regulatory approval, as we consider how such estimated costs may potentially be affected by various factors, including (i) the recent results from our SB206 Phase 3 trials in the U.S., including but not limited to top-line efficacy results announced in January 2020, (ii) our plans and timelines for potential further clinical development of SB206 in the U.S., which is subject to additional funding and FDA feedback from the Type C meeting with the FDA on April 1, 2020, and (iii) our in-house drug manufacturing capabilities and the progression of our manufacturing technology transfer projects with third-party contract manufacturing organizations.
The initial restricted cash balance was also reduced ratably during interim reporting periods in 2019 in a manner consistent with the amortization method for the Ligand funding liability balance. As of December 31, 2019, the aggregate amount spent in accordance with the SB206 development budget on SB206 development activities had exceeded the $12,000 purchase price, causing the aforementioned repayment provision provided for in the Funding Agreement to no longer be enforceable. Therefore, we reported no restricted cash balance related to the Funding Agreement, as of December 31, 2019 in our consolidated balance sheet.
99
Classification of Warrants Issued in Connection with Offerings of Common Stock
On January 9, 2018, we issued warrants to purchase 10,000,000 shares of common stock at an exercise price of $4.66, which expire four years from the date of issuance. The warrants provide each warrant holder with the right to require net cash settlement of the warrants upon the occurrence of certain fundamental transactions, provided that such transactions are within our control. For any fundamental transaction that is not within our control, including a fundamental transaction not approved by our board of directors, the warrant holder will only be entitled to receive from us or any successor entity the same type or form of consideration (and in the same proportion) that is being offered and paid to our common stockholders in connection with the fundamental transaction, whether that consideration be in the form of cash, stock or any combination thereof. In the event of any fundamental transaction, and regardless of whether it is within our control, the settlement amount of the warrants (whether in cash, stock or a combination thereof) is determined based upon a Black-Scholes value that is calculated using inputs as specified in the warrants, including a defined volatility input equal to the greater of our 100-day historical volatility or 100%.
We assessed the warrants for appropriate equity or liability classification pursuant to our accounting policy described in “Note 1—Organization and Significant Accounting Policies” to our consolidated financial statements. During this assessment, we determined that (i) the warrants do not constitute a liability under ASC 480; (ii) the warrants meet the definition of a derivative under ASC 815; (iii) the warrant holder’s option to receive a net cash settlement payment only becomes exercisable upon the occurrence of certain specified fundamental transactions that are within our control; (iv) upon the occurrence of a fundamental transaction that is not within our control, the warrant holder would receive the same type or form of consideration offered and paid to common stockholders; (v) the warrants are indexed to our common stock; and (vi) the warrants meet all other conditions for equity classification under ASC 480 and ASC 815. Based on the results of this assessment, we concluded that the warrants are freestanding equity-linked derivative instruments that meet the criteria for the own-equity scope exception to derivative accounting under ASC 815. Accordingly, the warrants are classified as equity and are accounted for as a component of additional paid-in capital at the time of issuance.
See “Note 10—Stockholders’ Equity (Deficit)” to the accompanying consolidated financial statements included in this Annual Report for additional discussion regarding the terms of the warrants and the applicable accounting treatment.
Share-Based Compensation
Determination of the Fair Value of Stock-based Compensation Grants
We record the fair value of stock options, and other stock-based compensation issued to employees and non-employees as of the grant date as stock-based compensation expense. We typically recognize compensation expense over the requisite service period, which is typically the vesting period. We recorded non-cash stock-based compensation expense for employee and nonemployee stock option grants of $1.8 million and $2.2 million for the years ended December 31, 2019 and 2018, respectively.
We estimate the fair value of our stock-based awards to employees and non-employees using the Black-Scholes option-pricing model, which requires the input of assumptions, some of which are highly subjective, including (i) the fair value of our common stock on the date of grant, (ii) the expected volatility of our stock, (iii) the expected term of the award, (iv) the risk-free interest rate and (v) expected dividends. In applying these assumptions, we considered the following factors:
• | Due to the lack of company-specific historical and implied volatility data, we have based our estimate of expected volatility on the historical volatility of a group of similar companies that are publicly traded. We also considered characteristics such as industry, stage of life cycle, financial leverage, enterprise value, risk profiles and position within the industry, along with historical share price information sufficient to meet the expected life of the stock-based awards. We compute the historical volatility data using the daily closing prices for the selected companies’ shares during the equivalent period of the calculated expected term of our stock-based awards. We will continue to apply this process until a sufficient amount of historical information regarding the volatility of our own stock price becomes available. |
• | We have estimated the expected term of our employee stock options using the “simplified” method, whereby, the expected life equals the average of the vesting term and the original contractual term of the option. |
• | The risk-free interest rate is based on the yields of U.S. Treasury securities with maturities similar to the expected term of granted stock-based awards. |
• | We have never declared or paid any cash dividends to common stockholders and do not presently plan to pay cash dividends in the foreseeable future. Consequently, we use an expected dividend yield of zero. |
100
See “Note 11—Share-Based Compensation” to the accompanying consolidated financial statements included in this Annual Report for the weighted average assumptions used in the Black-Scholes option-pricing model for awards granted in the years ended December 31, 2019 and 2018.
We are also required to estimate forfeitures at the time of grant, and to revise those estimates in subsequent periods if actual forfeitures differ from estimates. We use historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those awards that are expected to vest. To the extent that actual forfeitures differ from our estimates, the difference is recorded as a cumulative adjustment in the period the estimates were revised. Stock-based compensation expense recognized in the financial statements is based on awards that are ultimately expected to vest.
Tangible Stockholder Return Plan, or Performance Plan
On August 2, 2018, our board of directors approved and established the Tangible Stockholder Return Plan, which is a performance-based long-term incentive plan. The Performance Plan is tiered, with two separate tranches, each of which has a distinct share price target (measured as the average publicly traded share price of our common stock on the Nasdaq stock exchange for a 30 consecutive trading day period) that will, if achieved, trigger a distinct fixed bonus pool. The share price target for the first tranche and related bonus pool are $11.17 per share and $25.0 million, respectively. The share price target for the second tranche and related bonus pool are $25.45 per share and $50.0 million, respectively.
We have concluded that the Performance Plan is within the scope of ASC Topic 718, Compensation—Stock Compensation as the underlying plan obligations are based on the potential attainment of certain market share price targets of our common stock. Any awards under the Performance Plan would be payable, at the discretion of our compensation committee following the achievement of the applicable share price target, in cash, shares of our common stock, or a combination thereof, provided that, prior to any payment in common stock, our stockholders have approved the reservation of shares of our common stock for such payment.
ASC 718 requires that a liability-based award should be classified as a liability on our consolidated balance sheets and the amount of compensation cost recognized should be based on the fair value of the liability. When a liability-based award includes both a service and market condition, the market condition is taken into account when determining the appropriate method to estimate fair value and the compensation cost is amortized over the estimated service period. Therefore, the liability associated with the Performance Plan obligation is recorded within other long-term liabilities on our consolidated balance sheets at the estimated fair value on the date of issuance and is re-valued each subsequent reporting period end with adjustments to the fair value recognized as share-based compensation expense within operating expenses in the consolidated statements of operations.
The fair value of obligations under the Performance Plan are estimated using a Monte Carlo simulation approach. Our common stock price is simulated under the Geometric Brownian Motion framework under each simulation path. The other assumptions for the Monte Carlo simulation include the risk-free interest rate, estimated volatility and the expected term. Expected stock price volatility is based on the actual historical volatility of a group of comparable publicly traded companies observed over a historical period equal to the expected remaining life of the plan. The fair value of the underlying common stock is the published closing market price on the Nasdaq Global Market as of each reporting date, as adjusted for significant events, as necessary. The risk-free interest rate is based on the U.S. Treasury yield curve in effect on the date of valuation equal to the remaining expected life of the plan. The dividend yield percentage is zero because we do not currently pay dividends, nor do we intend to do so during the expected term of the plan. The expected life of bonus awards under the Performance Plan is assumed to be equivalent to the remaining contractual term based on the estimated service period including the service inception date of the plan participants and the contractual end of the Performance Plan.
Our estimates underlying the assumptions used in the Monte Carlo simulation valuation model are subject to risks and uncertainties and may change over time. Such changes could have a significant effect on our reported net losses in future periods. See “Note 12—Tangible Stockholder Return Plan” to the accompanying consolidated financial statements included in this Annual Report for the significant assumptions used in estimating the fair value of the Performance Plan and see “Note 1—Organization and Significant Accounting Policies” to the accompanying consolidated financial statements included in this Annual Report for our accounting policy pertaining to the fair value of financial instruments.
Stock Appreciation Rights
SARs that include cash settlement features are accounted for as liability-based awards pursuant to ASC 718 Compensation—Stock Compensation. The fair value of such SARs is estimated using a Black-Scholes option-pricing model on each financial reporting date using expected volatility, risk-free interest rate, expected life and fair value per share assumptions.
101
The fair value of each liability award is estimated with a valuation model that uses certain assumptions, such as the award date, expected volatility, risk-free interest rate, expected life of the award and fair value per share assumptions. Due to limited historical data, we estimate stock price volatility based on the actual volatility of comparable publicly traded companies over the expected term. In evaluating similarity, we considered factors such as industry, stage of life cycle, financial leverage, size and risk profile. The expected term for liability-based awards is the estimated contractual life. The risk-free rate is based on the U.S. Treasury yield curve during the expected life of the award. See “Note 11—Share-Based Compensation” to the accompanying consolidated financial statements included in this Annual Report for the significant assumptions used in estimating the fair value of SARs.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
Not applicable.
102
Item 8. Financial Statements and Supplementary Data.
NOVAN, INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
103
Report of Independent Registered Public Accounting Firm
Stockholders and Board of Directors
Novan, Inc.
Morrisville, North Carolina
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Novan, Inc. (the “Company”) as of December 31, 2019 and 2018, the related consolidated statements of operations and comprehensive loss, stockholders’ equity (deficit), and cash flows for each of the two years in the period ended December 31, 2019, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2019, in conformity with accounting principles generally accepted in the United States of America.
Restatement
As discussed in Note 2 to the consolidated financial statements, the 2018 consolidated financial statements have been restated to correct misstatements.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has suffered recurring losses from operations and has not generated significant revenue or positive cash flows from operations. These factors raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Change in Accounting Principle
As discussed in Notes 1 and 9 to the consolidated financial statements, the Company changed its method of accounting for leases in the year ended December 31, 2019 upon adoption of Accounting Standards Codification (ASC) Topic 842, Leases, using the modified retrospective method.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ BDO USA, LLP
104
We have served as the Company's auditor since 2018.
Raleigh, North Carolina
February 24, 2020, except for Notes 1, 2, 10 and 13, as to which the date is May 20, 2020
105
NOVAN, INC.
Consolidated Balance Sheets
(in thousands, except share and per share amounts)
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
ASSETS | (Restated) | ||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 13,711 | $ | 8,194 | |||
Contracts and grants receivable | 419 | — | |||||
Deferred offering costs | 49 | 49 | |||||
Prepaid expenses and other current assets | 1,545 | 1,107 | |||||
Total current assets | 15,724 | 9,350 | |||||
Restricted cash | 540 | 539 | |||||
Intangible assets | 75 | 75 | |||||
Other assets | 419 | 530 | |||||
Property and equipment, net | 10,506 | 15,868 | |||||
Right-of-use lease assets | 1,833 | — | |||||
Total assets | $ | 29,097 | $ | 26,362 | |||
LIABILITIES AND STOCKHOLDERS’ (DEFICIT) EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 1,602 | $ | 1,250 | |||
Accrued compensation | 437 | 1,467 | |||||
Accrued outside research and development services | 1,013 | 563 | |||||
Accrued legal and professional fees | 616 | 498 | |||||
Other accrued expenses | 553 | 871 | |||||
Deferred revenue, current portion | 4,428 | 4,401 | |||||
Research and development service obligation liability, current portion | 3,088 | — | |||||
Lease liabilities, current portion | 1,162 | 11 | |||||
Total current liabilities | 12,899 | 9,061 | |||||
Deferred revenue, net of current portion | 7,076 | 2,566 | |||||
Lease liabilities, net of current portion | 5,100 | 10 | |||||
Research and development service obligation liability, net of current portion | 727 | — | |||||
Research and development funding arrangement liability, related party | 25,000 | — | |||||
Other long-term liabilities | 578 | 289 | |||||
Facility financing obligation | — | 7,998 | |||||
Total liabilities | 51,380 | 19,924 | |||||
Commitments and contingencies (Note 9) | |||||||
Stockholders’ equity (deficit) | |||||||
Common stock $0.0001 par value; 200,000,000 shares authorized as of December 31, 2019 and 2018; 26,744,300 and 26,066,235 shares issued as of December 31, 2019 and 2018, respectively; 26,734,800 and 26,056,735 shares outstanding as of December 31, 2019 and 2018, respectively | 3 | 3 | |||||
Additional paid-in-capital | 197,853 | 195,483 | |||||
Treasury stock at cost, 9,500 shares as of December 31, 2019 and 2018 | (155 | ) | (155 | ) | |||
Accumulated deficit | (219,984 | ) | (188,893 | ) | |||
Total stockholders' (deficit) equity | (22,283 | ) | 6,438 | ||||
Total liabilities and stockholders’ (deficit) equity | $ | 29,097 | $ | 26,362 | |||
The accompanying notes are an integral part of these consolidated financial statements
106
NOVAN, INC.
Consolidated Statements of Operations and Comprehensive Loss
(in thousands, except share and per share amounts)
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(Restated) | |||||||
License and collaboration revenue | $ | 4,477 | $ | 5,982 | |||
Government research contracts and grants revenue | 419 | — | |||||
Research and development services revenue | — | 9 | |||||
Total revenue | 4,896 | 5,991 | |||||
Operating expenses: | |||||||
Research and development | 25,172 | 23,045 | |||||
General and administrative | 10,412 | 11,507 | |||||
Total operating expenses | 35,584 | 34,552 | |||||
Operating loss | (30,688 | ) | (28,561 | ) | |||
Other income (expense), net: | |||||||
Interest income | 177 | 297 | |||||
Interest expense | (2 | ) | (1,047 | ) | |||
Other income, net | 136 | 72 | |||||
Total other income (expense), net | 311 | (678 | ) | ||||
Net loss and comprehensive loss | $ | (30,377 | ) | $ | (29,239 | ) | |
Net loss per share, basic and diluted | $ | (1.16 | ) | $ | (1.13 | ) | |
Weighted-average common shares outstanding, basic and diluted | 26,254,119 | 25,795,721 | |||||
The accompanying notes are an integral part of these consolidated financial statements
107
NOVAN, INC.
Consolidated Statements of Stockholders’ Equity (Deficit)
(in thousands, except share amounts)
Additional Paid-In Capital | Treasury Stock | |||||||||||||||||||||
Common Stock | Accumulated | |||||||||||||||||||||
Shares | Amount | Deficit | Total | |||||||||||||||||||
Balance as of December 31, 2017 | 16,005,408 | $ | 2 | $ | 158,091 | $ | (155 | ) | $ | (159,654 | ) | $ | (1,716 | ) | ||||||||
Share-based compensation | — | — | 2,139 | — | — | 2,139 | ||||||||||||||||
Exercise of stock options | 51,327 | — | 60 | — | — | 60 | ||||||||||||||||
Common stock and warrants issued through public offering, net of underwriting discounts, commissions and offering costs | 10,000,000 | 1 | 35,193 | — | — | 35,194 | ||||||||||||||||
Net loss | — | — | — | — | (29,239 | ) | (29,239 | ) | ||||||||||||||
Balance as of December 31, 2018 (Restated) | 26,056,735 | $ | 3 | $ | 195,483 | $ | (155 | ) | $ | (188,893 | ) | $ | 6,438 | |||||||||
Share-based compensation | — | — | 1,188 | — | — | 1,188 | ||||||||||||||||
Exercise of stock options | 32,443 | — | 69 | — | — | 69 | ||||||||||||||||
Adoption of new accounting standards (Note 1) | — | — | — | — | (714 | ) | (714 | ) | ||||||||||||||
Liability-based awards reclassified to additional paid-in capital | — | — | 366 | — | — | 366 | ||||||||||||||||
Common stock issued pursuant to common stock purchase agreement | 645,622 | — | 747 | — | — | 747 | ||||||||||||||||
Net loss | — | — | — | — | (30,377 | ) | (30,377 | ) | ||||||||||||||
Balance as of December 31, 2019 | 26,734,800 | $ | 3 | $ | 197,853 | $ | (155 | ) | $ | (219,984 | ) | $ | (22,283 | ) | ||||||||
The accompanying notes are an integral part of these consolidated financial statements
108
NOVAN, INC.
Consolidated Statements of Cash Flows
(in thousands)
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(Restated) | |||||||
Cash flow from operating activities: | |||||||
Net loss | $ | (30,377 | ) | $ | (29,239 | ) | |
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||
Depreciation and amortization | 2,033 | 1,664 | |||||
Share-based compensation | 1,838 | 2,204 | |||||
Loss on disposal and write-offs of property and equipment | 36 | 154 | |||||
Changes in operating assets and liabilities: | |||||||
Contracts and grants receivable | (419 | ) | — | ||||
Prepaid expenses and other current assets | (438 | ) | (224 | ) | |||
Accounts payable | 352 | 777 | |||||
Accrued compensation | (1,030 | ) | (701 | ) | |||
Accrued outside research and development services | 450 | (829 | ) | ||||
Accrued legal and professional fees | 159 | 132 | |||||
Other accrued expenses | (397 | ) | (615 | ) | |||
Deferred revenue | 4,537 | (1,610 | ) | ||||
Advanced payment for research and development service obligation | 12,000 | — | |||||
Research and development service obligation liabilities | (8,185 | ) | — | ||||
Other long-term assets and liabilities | (435 | ) | (338 | ) | |||
Net cash used in operating activities | (19,876 | ) | (28,625 | ) | |||
Cash flow from investing activities: | |||||||
Purchases of property and equipment | (422 | ) | (1,107 | ) | |||
Proceeds from the sale of property and equipment | — | 49 | |||||
Net cash used in investing activities | (422 | ) | (1,058 | ) | |||
Cash flow from financing activities: | |||||||
Proceeds from research and development funding arrangement | 25,000 | — | |||||
Proceeds from issuance of common stock, net of underwriting fees and commissions | 747 | 35,625 | |||||
Payments related to public offering costs | — | (321 | ) | ||||
Proceeds from exercise of stock options | 69 | 60 | |||||
Payments on capital lease obligation | — | (11 | ) | ||||
Net cash provided by financing activities | 25,816 | 35,353 | |||||
Net increase in cash, cash equivalents and restricted cash | 5,518 | 5,670 | |||||
Cash, cash equivalents and restricted cash as of beginning of period | 8,733 | 3,063 | |||||
Cash, cash equivalents and restricted cash as of end of period | $ | 14,251 | $ | 8,733 | |||
Supplemental disclosure of cash flow information: | |||||||
Cash paid for interest | $ | 2 | $ | 1,043 | |||
Supplemental disclosure of non-cash investing and financing activities: | |||||||
Purchases of property and equipment with accounts payable and accrued expenses | $ | 79 | $ | — | |||
Right of use assets obtained in exchange for lease liabilities | $ | 1,827 | $ | — | |||
Liability-based awards reclassified to additional paid-in capital | $ | 366 | $ | — | |||
Common stock issued for payment of commitment fee | $ | 750 | $ | — | |||
Deferred offering costs reclassified to additional paid-in capital | $ | — | $ | 431 | |||
Reconciliation to consolidated balance sheets: | |||||||
Cash and cash equivalents | $ | 13,711 | $ | 8,194 | |||
Restricted cash included in noncurrent assets | 540 | 539 | |||||
Total cash, cash equivalents and restricted cash shown in the statement of cash flows | $ | 14,251 | $ | 8,733 | |||
The accompanying notes are an integral part of these consolidated financial statements
109
NOVAN, INC
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
(dollar values in thousands, except per share data)
Note 1: Organization and Significant Accounting Policies
Business Description and Basis of Presentation
Novan, Inc. (“Novan” and together with its subsidiaries, the “Company”), is a North Carolina-based clinical development-stage biotechnology company focused on leveraging nitric oxide’s naturally occurring anti-viral, anti-bacterial, anti-fungal and immunomodulatory mechanisms of action to treat a range of diseases with significant unmet needs. Novan was incorporated in January 2006 under the state laws of Delaware. Its wholly-owned subsidiary, Novan Therapeutics, LLC was organized in 2015 under the state laws of North Carolina. On March 14, 2019, the Company completed registration of a wholly-owned Ireland-based subsidiary, Novan Therapeutics, Limited.
The accompanying consolidated financial statements of the Company have been prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”). Additionally, each of the two reports of the Company’s independent registered public accounting firm on the Company’s consolidated financial statements as of and for the years ended December 31, 2019 and December 31, 2018, respectively, included an explanatory paragraph indicating that there is substantial doubt about the Company’s ability to continue as a going concern.
Basis of Consolidation
The accompanying consolidated financial statements reflect the operations of the Company and its wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated in consolidation.
As described in Note 2—Restatement of Consolidated Financial Statements, the Company’s consolidated financial statements for the year ended December 31, 2018, and all quarterly periods of 2019 and 2018 (collectively, the “Affected Periods”), are restated in this Annual Report on Form 10-K/A (Amendment No. 1) (this “Annual Report”) to correct the misapplication of accounting guidance related to the Company’s warrants in the Company’s previously issued consolidated financial statements for such periods. The restated consolidated financial statements are indicated as “Restated” in the consolidated financial statements and accompanying notes, as applicable. Although the impact of the restatement on the consolidated financial statements and the related disclosures for the year ended December 31, 2019 was immaterial, the Company has revised those financial statements as well in connection with the restatement of our consolidated financial statements noted above. See Note 2—Restatement of Consolidated Financial Statements for further discussion.
Liquidity and Ability to Continue as a Going Concern
The Company’s consolidated financial statements have been prepared assuming that the Company will continue as a going concern, which contemplates the realization of assets and the settlement of liabilities and commitments in the normal course of business. The accompanying consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from uncertainty related to the Company’s ability to continue as a going concern.
The Company has evaluated principal conditions and events that may raise substantial doubt about its ability to continue as a going concern within one year from the date that these financial statements are issued. The Company identified the following conditions:
• | The Company has reported a net loss in all fiscal periods since inception and, as of December 31, 2019, the Company had an accumulated deficit of $219,984. |
• | As described in Note 10—Stockholders’ Equity (Deficit), in August 2019 the Company entered into a common stock purchase agreement (the “Aspire Common Stock Purchase Agreement”) with Aspire Capital Fund, LLC, (“Aspire Capital”) which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $25,000 of shares of the Company’s common stock at the Company’s request from time to time during the 30-month term of the Aspire Common Stock Purchase Agreement. The aggregate amount available to the Company through sales of common stock under the Aspire Common Stock Purchase Agreement is subject to certain limitations including, but not limited to: (i) the number of shares that may be sold will be limited to 5,211,339 shares, representing 19.99% of the Company’s outstanding shares of common stock on August 30, 2019, if the average price paid for all shares issued under the agreement is less than $2.17; and (ii) on any purchase date, the closing sale price of the |
110
Company’s common stock must be greater than or equal to $0.25. As of December 31, 2019, the Company had sold 300,000 shares of common stock at an average price of $2.49 per share under the Aspire Common Stock Purchase Agreement. As of January 31, 2020, the Company had sold an aggregate of 1,000,000 shares of common stock at an average price of $1.19 per share under the Aspire Common Stock Purchase Agreement. These amounts, combined with the 345,622 shares issued as part of the commitment fee related to the agreement’s execution, leads to a total of 1,345,622 shares issued to Aspire Capital under the Aspire Common Stock Purchase Agreement as of January 31, 2020.
• | As of December 31, 2019, the Company had a total cash and cash equivalents balance of $13,711. |
The Company has concluded that the prevailing conditions and ongoing liquidity risks faced by the Company raise substantial doubt about its ability to continue as a going concern.
Based on its current cash flow forecast, the Company does not currently have sufficient cash and cash equivalents to continue its business operations beyond the early part of the second quarter of 2020. Therefore, the Company will need to raise substantial additional funding by the early part of the second quarter of 2020 in order to continue its operating activities and make further advancements in its drug development programs. There can be no assurance that the Company will be able to obtain additional capital on terms acceptable to the Company, on a timely basis or at all.
The failure of the Company to obtain sufficient funds on acceptable terms could have a material adverse effect on the Company’s business and cause the Company to alter or reduce its planned operating activities, including but not limited to delaying, reducing, terminating or eliminating planned product candidate development activities, to conserve its cash and cash equivalents. The Company needs and intends to secure additional capital from non-dilutive sources, including partnerships, collaborations, licensing, grants or other strategic relationships, or through equity or debt financings. Any issuance of equity or debt that could be convertible into equity would result in significant dilution to our existing stockholders. Alternatively, the Company may seek to engage in one or more potential transactions, such as the sale of the Company, or sale or divestiture of some of its assets, such as a sale of its dermatology platform assets, but there can be no assurance that the Company will be able to enter into such a transaction or transactions on a timely basis or at all on terms that are favorable to the Company. Under these circumstances, the Company may instead determine to dissolve and liquidate its assets or seek protection under the bankruptcy laws. If the Company decides to dissolve and liquidate its assets or to seek protection under the bankruptcy laws, it is unclear to what extent the Company will be able to pay its obligations, and, accordingly, it is further unclear whether and to what extent any resources will be available for distributions to stockholders.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. Actual results could differ from these estimates.
Cash and Cash Equivalents
The Company considers all highly liquid instruments purchased with a maturity of three months or less to be cash equivalents. Cash and cash equivalents include deposits and money market accounts.
Restricted Cash
Restricted cash as of December 31, 2019 and 2018 includes funds maintained in a separate deposit account to secure a letter of credit for the benefit of the lessor of facility space leased by the Company. See Note 7—Research and Development Arrangements for a discussion of the restricted cash presentation associated with the Ligand Funding Agreement during interim reporting periods in 2019.
Concentration of Credit Risk
Financial instruments that potentially subject the Company to a concentration of credit risk consist principally of cash and cash equivalents. The Company places its cash and cash equivalents with financial institutions and these deposits may at times be in excess of insured limits.
Contracts and Grants Receivable
The Company carries its contracts and grants receivable net of an allowance for doubtful accounts. All receivables or portions thereof that are deemed to be uncollectible or that require excessive collection costs are written off to the allowance for
111
doubtful accounts when it is probable that the receivable is unrecoverable. The Company actively reviews and evaluates its contracts and grants receivable, but no allowance for doubtful accounts has been considered necessary as of December 31, 2019. Actual results could differ from the estimates that were used. The Company did not have a contracts and grants receivable balance as of December 31, 2018.
Intangible Assets
Intangible assets represent the cost to obtain and register the Company’s internet domain. Indefinite-lived intangible assets are not amortized and are assessed for impairment at least annually.
Property and Equipment
Property and equipment are recorded at cost and depreciated using the straight-line method over their estimated useful lives as follows:
Computer and office equipment | 3 years |
Furniture and fixtures | 5-7 years |
Laboratory equipment | 7 years |
Building asset under facility lease | 25 years |
Leasehold improvements are amortized over the shorter of the life of the lease or the useful life of the improvements. Expenditures for maintenance and repairs are expensed as incurred. Improvements and betterments that add new functionality or extend the useful life of an asset are capitalized.
Intellectual Property
The Company’s policy is to file patent applications to protect technology, inventions and improvements that are considered important to its business. Patent positions, including those of the Company, are uncertain and involve complex legal and factual questions for which important legal principles are largely unresolved. Due to the uncertainty of future value to be realized from the expenses incurred in developing the Company’s intellectual property, the cost of filing, prosecuting and maintaining internally developed patents are expensed as general and administrative costs as incurred.
Leases
The Company leases office space and certain equipment under non-cancelable lease agreements. Prior to January 1, 2019, the Company applied the accounting guidance in ASC 840, Leases, to its lease agreements. The leases were reviewed for classification as operating or capital leases. For operating leases, rent was recognized on a straight-line basis over the lease period. For capital leases, the Company recorded the leased asset with a corresponding liability and amortized the asset over the lease term. Payments were recorded as reductions to the liability with an appropriate interest charge recorded based on the then-outstanding remaining liability.
Beginning January 1, 2019, the Company applies the accounting guidance in ASC 842, Leases. As such, the Company assesses all arrangements, that convey the right to control the use of property, plant and equipment, at inception, to determine if it is, or contains, a lease based on the unique facts and circumstances present in that arrangement. For those leases identified, the Company determines the lease classification, recognition, and measurement at the lease commencement date. For arrangements that contain a lease the Company: (i) identifies lease and non-lease components; (ii) determines the consideration in the contract; (iii) determines whether the lease is an operating or financing lease; and (iv) recognizes lease Right of Use (“ROU”) assets and corresponding lease liabilities. Lease liabilities are recorded based on the present value of lease payments over the expected lease term. The corresponding ROU asset is measured from the initial lease liability, adjusted by (i) accrued or prepaid rents; (ii) remaining unamortized initial direct costs and lease incentives; and (iii) any impairments of the ROU asset. The interest rate implicit in the Company’s lease contracts is typically not readily determinable and as such, the Company uses its incremental borrowing rate based on the information available at the lease commencement date, which represents an internally developed rate that would be incurred to borrow, on a collateralized basis, over a similar term, an amount equal to the lease payments in a similar economic environment.
112
Impairment of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized for an amount by which the carrying amount of the asset exceeds the fair value of the asset. There were no impairments of long-lived assets during the years ended December 31, 2019 and 2018.
Deferred Offering Costs
Deferred offering costs consist of legal, accounting, filing and other fees directly related to offerings or the Company’s shelf registration. These costs are offset against proceeds from each offering as applicable. Offering costs incurred prior to the completion of an offering are initially capitalized as assets, evaluated each period for likelihood of completion and subsequently reclassified to additional paid-in capital upon completion of the offering. Deferred costs associated with the shelf registration will be reclassified to additional paid in capital on a pro-rata basis in the event the Company completes an offering under the shelf registration, with any remaining deferred offering costs charged to general and administrative expense at the end of the three-year life of the shelf registration.
Revenue Recognition
Effective January 1, 2018, the Company adopted ASC Topic 606, Revenue from Contracts with Customers, using the full retrospective transition method. To determine revenue recognition for arrangements that the Company determines are within the scope of Topic 606, the Company performs the following five steps: (i) identify the contracts with a customer; (ii) identify the performance obligations in the contract; (iii) determine the transaction price; (iv) allocate the transaction price to the performance obligations in the contract; and (v) recognize revenue when (or as) the entity satisfies a performance obligation. The Company only applies the five-step model to contracts when it is probable that the entity will collect the consideration it is entitled to in exchange for the goods or services it transfers to the customer.
At contract inception, once the contract is determined to be within the scope of Topic 606, the Company assesses the goods or services promised within each contract and determines those that are performance obligations and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price that is allocated to the respective performance obligation when (or as) the performance obligation is satisfied.
Upon occurrence of a contract modification, the Company conducts an evaluation pursuant to the modification framework in Topic 606 to determine the appropriate revenue recognition. The framework centers around key questions, including (i) whether the modification adds additional goods and services, (ii) whether those goods and services are distinct, and (iii) whether the contract price increases by an amount that reflects the standalone selling price for the new goods or services. The resulting conclusions will determine whether the modification is treated as a separate, standalone contract or if it is combined with the original contract and accounted for in that manner. In addition, some modifications are accounted for on a prospective basis and others on a cumulative catch-up basis.
The Company’s agreements may contain some or all the following types of provisions or payments:
Licenses of Intellectual Property: If the license of the Company’s intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, the Company recognizes revenues from non-refundable, upfront fees allocated to the license when the license is transferred to the customer and the customer is able to use and benefit from the license. For licenses that are bundled with other promises, the Company utilizes judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time and, if over time, the estimated performance period and the appropriate method of measuring progress during the performance period for purposes of recognizing revenue. The Company re-evaluates the estimated performance period and measure of progress each reporting period and, if necessary, adjusts related revenue recognition accordingly.
113
Milestone Payments: At the inception of each arrangement that includes development milestone payments, the Company evaluates whether the milestones are considered probable of being reached and estimates the amount to be included in the transaction price using the most likely amount method. If it is probable that a significant revenue reversal would not occur, the associated milestone value is included in the transaction price. The transaction price is then allocated to each performance obligation on a relative stand-alone selling price basis, for which the Company recognizes revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, the Company re-evaluates the probability of achievement of such development milestones and any related constraint, and if necessary, adjusts its estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect license and collaboration revenue and earnings in the period of adjustment.
Manufacturing Supply Services: Arrangements that include a promise for future supply of drug substance or drug product for either clinical development or commercial supply at the customer’s discretion are generally considered as options. The Company assesses if these options provide a material right to the licensee and if so, they are accounted for as separate performance obligations. If the Company is entitled to additional payments when the customer exercises these options, any additional payments are recorded in license and collaboration revenue when the customer obtains control of the goods, which is upon delivery.
Royalties: For arrangements that include sales-based royalties, including milestone payments based on the level of sales, and the license is deemed to be the predominant item to which the royalties relate, the Company recognizes revenue at the later of (i) when the related sales occur, or (ii) when the performance obligation to which some or all of the royalty has been allocated has been satisfied (or partially satisfied). To date, the Company has not recognized any royalty revenue resulting from any of its licensing arrangements.
The Company’s revenue also includes research revenue earned under contracts and grants with Federal government agencies, which relates to the research and development of its nitric oxide platform.
Government research contracts and grants revenue. Under the terms of the contracts and grants awarded, the Company is entitled to receive reimbursement of its allowable direct expenses, allocated overhead, general and administrative expenses and payment of other specified amounts. Revenues from development and support activities under government research contracts and grants are recorded in the period in which the related costs are incurred. Associated expenses are recognized when incurred as research and development expense. Revenue recognized in excess of amounts collected from funding sources are recorded as contracts and grants receivable. Any of the funding sources may, at their discretion, request reimbursement for expenses or return of funds, or both, as a result of noncompliance by the Company with the terms of the grants. No reimbursement of expenses or return of funds has been requested or made since inception of the contracts and grants. See Note 6—Revenue Recognition for information regarding two government grants the Company received in the third quarter of 2019.
Research and Development Expenses
Research and development expenses include all direct and indirect development costs incurred for the development of the Company’s drug candidates. These expenses include salaries and related costs, including share-based compensation and travel costs for research and development personnel, allocated facility costs, laboratory and manufacturing materials and supplies, consulting fees, product development, preclinical studies, clinical trial costs, licensing fees and milestone payments under license agreements and other fees and costs related to the development of drug candidates. The cost of tangible and intangible assets that are acquired for use on a particular research and development project, have no alternative future uses, and are not required to be capitalized in accordance with the Company’s capitalization policy, are expensed as research and development costs as incurred.
Accrued Outside Research and Development Accruals
The Company is required to estimate its expenses resulting from its obligations under contracts with clinical research organizations, clinical site agreements, vendors, and consultants in connection with conducting clinical trials and preclinical development. The financial terms of these contracts are subject to negotiations which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided to the Company under such contracts. The Company’s objective is to reflect the appropriate development and clinical trial expenses in its financial statements by matching those expenses with the period in which the services and efforts are expended.
For clinical trials, the Company accounts for these expenses according to the progress of the trial as measured by actual hours expended by contract research organization personnel, investigator performance or completion of specific tasks, patient progression, or timing of various aspects of the trial. During the course of a clinical trial, the Company adjusts its rate of clinical trial expense recognition if actual results differ from its estimates. The Company utilizes judgment and experience to
114
estimate its accrued expenses as of each balance sheet date in its financial statements based on facts and circumstances known at that time. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of status and timing of services performed relative to the actual status and timing of services performed may vary and may result in increases or decreases in research and development expenses in future periods when the actual results become known.
For preclinical development services performed by outside service providers, the Company determines accrual estimates through financial models, considering development progress data received from outside service providers and discussions with applicable Company and service provider personnel.
Classification of Warrants Issued in Connection with Offerings of Common Stock
The Company accounts for common stock warrants as either equity-classified or liability-classified instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance in Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) 480, Distinguishing Liabilities from Equity (“ASC 480”) and ASC 815, Derivatives and Hedging (“ASC 815”). The assessment considers whether the warrants are freestanding financial instruments pursuant to ASC 480, meet the definition of a liability pursuant to ASC 480, and whether the warrants meet all of the requirements for equity classification under ASC 815, including whether the warrants are indexed to the Company’s own common stock and whether the warrant holders could potentially require “net cash settlement” in a circumstance outside of the Company’s control, among other conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of warrant issuance and as of each subsequent quarterly period end date while the warrants are outstanding.
For issued or modified warrants that meet all of the criteria for equity classification, the warrants are required to be recorded as a component of additional paid-in capital at the time of issuance. For issued or modified warrants that do not meet all the criteria for equity classification, the warrants are required to be recorded at their initial fair value on the date of issuance, and each balance sheet date thereafter. Changes in the estimated fair value of the warrants are recognized as a non-cash gain or loss on the consolidated statements of operations and comprehensive loss.
Fair Value of Financial Instruments
The carrying values of cash equivalents, accounts payable and accrued liabilities as of December 31, 2019 and 2018 approximated their fair values due to the short-term nature of these items.
The Company has categorized its financial instruments, based on the priority of the inputs used to value the investments, into a three-level fair value hierarchy. The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets or liabilities (Level 1) and lowest priority to unobservable inputs (Level 3). If the inputs used to measure the investments fall within different levels of the hierarchy, the categorization is based on the lowest level input that is significant to the fair value measurement of the investment. Financial instruments recorded in the accompanying consolidated balance sheets are categorized based on the inputs to valuation techniques as follows:
Level 1 - Observable inputs that reflect unadjusted quoted market prices for identical assets or liabilities in active markets.
Level 2 - Observable inputs other than Level 1 that are observable, either directly or indirectly, in the marketplace for identical or similar assets and liabilities.
Level 3 - Unobservable inputs that are supported by little or no market data, where values are derived from techniques in which one or more significant inputs are unobservable.
115
Share-Based Compensation
Equity-Based Awards
The Company applies the fair value method of accounting for share-based compensation, which requires all such compensation to employees, including the grant of employee stock options, to be recognized in the consolidated statements of operations and comprehensive loss based on its fair value at the measurement date (generally the grant date). The expense associated with share-based compensation is recognized over the requisite service period of each award. For awards with only service conditions and graded-vesting features, the Company recognizes compensation cost on a straight-line basis over the requisite service period. For awards with performance conditions, once achievement of the performance condition becomes probable, compensation cost is recognized over the expected period from the date the performance condition becomes probable to the date the performance condition is expected to be achieved. The Company will reassess the probability of vesting at each reporting period for performance awards and adjust compensation cost based on its probability assessment. Share-based awards granted to non-employee directors as compensation for serving on the Company’s board of directors are accounted for in the same manner as employee share-based compensation awards.
The fair value of each option grant is estimated using a Black-Scholes option-pricing model on the grant date using expected volatility, risk-free interest rate, expected life of options and fair value per share assumptions. Due to limited historical data, the Company estimates stock price volatility based on the actual volatility of comparable publicly traded companies over the expected life of the option. In evaluating similarity, the Company considered factors such as industry, stage of life cycle, financial leverage, size and risk profile.
The Company does not have sufficient stock option exercise history to estimate the expected term of employee stock options and thus continues to calculate expected life based on the mid-point between the vesting date and the contractual term, which is in accordance with the simplified method. The expected term for share-based compensation granted to non-employees is the contractual life. The risk-free rate is based on the U.S. Treasury yield curve during the expected life of the option. The Company estimates forfeitures based on the historical experience of the Company and adjusts the estimated forfeiture rate based upon actual experience.
Liability-Based Awards
Stock appreciation rights (“SARs”) that include cash settlement features are accounted for as liability-based awards pursuant to ASC 718 Share Based Payments. The fair value of such SARs is estimated using a Black-Scholes option-pricing model on each financial reporting date using expected volatility, risk-free interest rate, expected life and fair value per share assumptions.
The fair value of obligations under the Tangible Stockholder Return Plan are estimated using a Monte Carlo simulation approach. The Company’s common stock price is simulated under the Geometric Brownian Motion framework under each simulation path. The other assumptions for the Monte Carlo simulation include the risk-free interest rate, estimated volatility and the expected term.
The fair value of each liability award is estimated with a valuation model that uses certain assumptions, such as the award date, expected volatility, risk-free interest rate, expected life of the award and fair value per share assumptions. Due to limited historical data, the Company estimates stock price volatility based on the actual volatility of comparable publicly traded companies over the expected term. In evaluating similarity, the Company considered factors such as industry, stage of life cycle, financial leverage, size and risk profile. The expected term for liability-based awards is the estimated contractual life. The risk-free rate is based on the U.S. Treasury yield curve during the expected life of the award.
Income Taxes
Deferred tax assets and liabilities are determined based on the temporary differences between the financial statement carrying amounts and the tax bases of assets and liabilities using the enacted tax rates in effect in the years in which the differences are expected to reverse. In estimating future tax consequences, all expected future events are considered other than enactment of changes in the tax law or rates.
The Company did not record a federal or state income tax benefit for the years ended December 31, 2019 and 2018 due to its conclusion that a full valuation allowance is required against the Company’s deferred tax assets.
The determination of recording or releasing a tax valuation allowance is made, in part, pursuant to an assessment performed by management regarding the likelihood that the Company will generate future taxable income against which benefits of its deferred tax assets may or may not be realized. This assessment requires management to exercise judgment and make estimates with respect to its ability to generate taxable income in future periods.
116
The Company recognizes the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained on examination by the taxing authorities based on the technical merits of the position.
The Company’s policy for recording interest and penalties is to record them as a component of general and administrative expenses. As of December 31, 2019 and 2018, the Company accrued no interest and penalties related to uncertain tax positions.
Tax years 2016-2018 remain open to examination by the major taxing jurisdictions to which the Company is subject. Additionally, years prior to 2016 are also open to examination to the extent of loss and credit carryforwards from those years.
In accordance with Section 382 of the Internal Revenue Code of 1986, as amended, a change in equity ownership of greater than 50% within a three-year period results in an annual limitation on the Company’s ability to utilize its net operating loss carryforwards created during the tax periods prior to the change in ownership. The Company has not determined whether ownership changes exceeding this threshold, including the Company’s IPO and the January 2018 Offering, have occurred. If a change in equity ownership has occurred which exceeds the Section 382 threshold, a portion of the Company’s net operating loss carryforwards may be limited.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. For the years ended December 31, 2019 and 2018, comprehensive loss was equal to net loss.
Net Loss Per Share
Basic net loss per share is calculated by dividing net loss by the weighted average shares outstanding during the period, without consideration of common stock equivalents. Diluted net loss per share is calculated by adjusting weighted average shares outstanding for the dilutive effect of common stock equivalents outstanding for the period. Diluted net loss per share is the same as basic net loss per share, since the effects of potentially dilutive securities are anti-dilutive for all periods presented.
The following securities, presented on a common stock equivalent basis, have been excluded from the calculation of weighted average common shares outstanding for the years ended December 31, 2019 and 2018 because the effect is anti-dilutive due to the net loss reported in each of those periods. All share amounts presented in the table below represent the total number outstanding as of the end of each period.
December 31, | |||||
2019 | 2018 | ||||
Warrants to purchase common stock associated with January 2018 public offering (Note 10) | 10,000,000 | 10,000,000 | |||
Stock options outstanding under the 2008 and 2016 Plans (Note 11) | 1,663,803 | 1,671,666 | |||
Stock appreciation rights outstanding under the 2016 Plan (Note 11) | 1,000,000 | — | |||
Inducement options outstanding (Note 11) | 125,500 | 100,500 | |||
Segment and Geographic Information
The Company has determined that it operates in one segment. The Company uses its nitric oxide-based technology to develop product candidates. The Chief Executive Officer (“CEO”), who is the Company’s chief operating decision maker, reviews financial information on an aggregate basis for purposes of allocating resources and evaluating financial performance. The Company has only had limited revenue since its inception, but substantially all revenue was derived from licensing agreements originating in the United States. All of the Company’s long-lived assets are maintained in the United States.
Although all operations are based in the United States, the Company generated revenue from its licensing partner in Japan of $4,477, or approximately 91% of total revenue during the year ended December 31, 2019, and $5,982, or 100% of total revenue during the year ended December 31, 2018.
117
Recently Issued Accounting Standards
Accounting Pronouncements Adopted
In February 2016, the FASB issued Accounting Standards Update (“ASU”) No. 2016-02, Leases (Topic 842). This guidance revises the accounting related to leases by requiring lessees to recognize a lease liability and a right-of-use asset for all leases. The new lease guidance also simplifies the accounting for sale and leaseback transactions. In July 2018, the FASB issued ASU No. 2018-10, Codification Improvements to Topic 842, Leases and ASU 2018-11, Leases (Topic 842): Targeted Improvements, and in March 2019, the FASB issued ASU 2019-01, Leases (Topic 842): Codification Improvements. These additional ASUs were issued to provide expanded or clarifying guidance associated with the application of certain principles. Under the guidance, lessees are required to recognize assets and lease liabilities on the balance sheet for most leases including operating leases and provide enhanced disclosures. There are optional practical expedients that a company may elect to apply. The guidance was effective for the Company beginning in its first quarter of 2019.
The Company adopted Topic 842 as of January 1, 2019 using the modified alternative retrospective transition method and initially applied the transition provisions as of January 1, 2019. This transition method allowed the Company to continue to apply the legacy guidance in ASC 840 for periods prior to 2019 and recognize a cumulative-effect adjustment to the opening balance of accumulated deficit as of the date of adoption.
The Company elected the package of transition practical expedients, which, among other things, allowed the Company to keep the historical lease classifications and not have to reassess the lease classification and initial direct costs for any existing or expired leases as of the date of adoption. The Company also made an accounting policy election to apply the short-term lease exception, which allows the Company to exclude leases with an initial term of twelve months or less from the consolidated balance sheets. Lease expense for leases with an initial term of twelve months or less will be recognized over the lease term, similar to the accounting treatment under ASC 840.
As a result of the adoption of Topic 842, the Company derecognized $10,557 of building assets (property, plant and equipment), and the $7,998 facility financing obligation associated with the previously existing build-to-suit arrangement related to its sole corporate and manufacturing facility. The Company also capitalized leasehold improvements and ROU assets of $5,885 and $1,827, respectively, and recorded lease liabilities for operating leases totaling $6,786, as of January 1, 2019. The capitalized leasehold improvement assets recorded as part of the adoption of Topic 842 were previously included within the derecognized building asset as part of the previous build-to-suit arrangement. The Company also recognized an increase of $714 to accumulated deficit related to its de-recognition of its previously recorded build-to-suit arrangement. The impact of the adoption of this guidance is non-cash in nature and did not affect the Company’s cash flows. See Note 9—Commitments and Contingencies for additional information related to the adoption of Topic 842.
In June 2018 the FASB issued ASU 2018-08 Not-for-Profit Entities (Topic 958): Clarifying the Scope and the Accounting Guidance for Contributions Received and Contributions Made. This guidance clarifies and improves the scope and the accounting guidance for contributions received and contributions made, in order to reduce diversity in practice for grants and other similar contracts. For contributions (nonreciprocal transactions), an entity should follow the guidance in ASC 958-605 Not-for-Profit Entities - Revenue Recognition, and for exchange (reciprocal) transactions an entity should follow other guidance. This standard is effective for annual reporting periods beginning after June 15, 2018, including interim reporting periods within those annual reporting periods, with early adoption permitted. This ASU was effective for the Company as of January 1, 2019. The adoption of this new accounting guidance did not have a material impact on the Company’s consolidated financial statements. See Note 6—Revenue Recognition for information related to the adoption of Topic 958.
In June 2018, the FASB issued ASU No. 2018-07 Compensation-Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting. This guidance simplifies the accounting for non-employee share-based payment transactions by expanding the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from non-employees. Under the new standard, most of the guidance on stock compensation payments to non-employees would be aligned with the requirements for share-based payments granted to employees. This standard is effective for annual reporting periods beginning after December 15, 2018, including interim reporting periods within those annual reporting periods, with early adoption permitted. This ASU was effective for the Company as of January 1, 2019. The adoption of this new accounting guidance did not have a material impact on the Company’s consolidated financial statements.
Accounting Pronouncements Being Evaluated
In August 2018, the FASB issued ASU No. 2018-13 Fair Value Measurement (Topic 820): Disclosure Framework—Changes to the Disclosure Requirements for Fair Value Measurement. This guidance is intended to improve the effectiveness of disclosure requirements on fair value measurements in Topic 820. The new standard modifies certain disclosure requirements and will be
118
effective for annual reporting periods beginning after December 15, 2019. The new guidance is not expected to have a material impact on the Company’s consolidated financial statements.
In October 2018, the FASB issued ASU No. 2018-17 Consolidation (Topic 810): Targeted Improvements to Related Party Guidance for Variable Interest Entities. This guidance is intended to improve the accounting for variable interest entities and whether the entity should be consolidated. This guidance is effective for annual reporting periods beginning after December 15, 2019, including interim reporting periods within those annual reporting periods, with early adoption permitted. The new guidance is not expected to have a material impact on the Company’s consolidated financial statements.
In November 2018, the FASB issued ASU No. 2018-18 Collaborative Arrangements (Topic 808): Clarifying the Interaction between Topic 808 and Topic 606. This guidance is intended to reduce diversity in practice and clarify the interaction between Topic 808, Collaborative Arrangements, and Topic 606, Revenue from Contracts with Customers. This ASU provided guidance on whether certain transactions between collaborative arrangement participants should be accounted for with revenue under Topic 606. This guidance is effective for annual reporting periods beginning after December 15, 2019, including interim reporting periods within those annual reporting periods, with early adoption permitted. The new guidance is not expected to have a material impact on the Company’s consolidated financial statements.
In December 2019, the FASB issued ASU No. 2019-12 Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes. This guidance is intended to improve consistent application and simplify the accounting for income taxes. This ASU removes certain exceptions to the general principles in Topic 740 and clarifies and amends existing guidance. This standard is effective for annual reporting periods beginning after December 15, 2020, including interim reporting periods within those annual reporting periods, with early adoption permitted. The Company is currently evaluating the impact of adoption of this ASU and does not expect the adoption of this new standard to have a material impact on its consolidated financial statements.
119
Note 2: Restatement of Consolidated Financial Statements
Restatement Background and Explanation
On May 14, 2020, the Company concluded that, because of a misapplication of the accounting guidance related to the warrants the Company issued in January 2018, the Company’s previously issued consolidated financial statements for the Affected Periods should no longer be relied upon. As such, the Company is restating its consolidated financial statements for the Affected Periods included in this Annual Report. In addition, although immaterial to the Company’s consolidated financial statements for the year ended December 31, 2019, the Company has also revised these financial statements in connection with the restatement of its consolidated financial statements for the Affected Periods.
The warrants subject to the misapplication of the applicable accounting guidance were issued in connection with a public offering of the Company’s common stock and warrants completed on January 9, 2018 pursuant to the Company’s effective shelf registration statement (the “January 2018 Offering”). In this offering, the Company sold an aggregate of 10,000,000 shares of common stock and issued warrants to purchase up to 10,000,000 shares of common stock at a public offering price of $3.80 per share of common stock and accompanying warrant. Pursuant to the terms of the warrants, the warrant exercise price is $4.66 per share and the warrants will expire four years from the date of issuance. The material terms of the warrants are more fully described in Note 10—Stockholders’ Equity (Deficit).
Upon issuance of the warrants in the first quarter of 2018, and during each quarterly and annual period thereafter through December 31, 2019, the Company conducted an evaluation pursuant to applicable guidance in ASC 480 and ASC 815 to determine the appropriate balance sheet classification for the warrants. The Company concluded during such historical assessments that the warrants should be classified as liabilities in the consolidated balance sheets with subsequent changes in the fair value of the warrants reported in the consolidated statements of operations for the relevant periods. This historical liability classification conclusion was based primarily on a determination that the warrant holder may have the option to receive a cash settlement, equal to the Black-Scholes value of the remaining unexercised portion of the warrant as determined in accordance with the warrant, in the event of a fundamental transaction (as that term is defined in the warrant).
In connection with the initial classification and accounting assessment of the warrants issued during the quarterly period ended March 31, 2020, the Company re-assessed the accounting treatment for the warrants issued in January 2018 as part of its periodic warrant classification and accounting assessment, including consideration of all classification criteria under ASC 480 and ASC 815 pursuant to the accounting policy described in Note 1—Organization and Significant Accounting Policies.
During this re-assessment of the warrants issued in January 2018, the Company further examined the fundamental transaction settlement provisions of the warrants. Pursuant to the terms of the warrants, a warrant holder may only require cash settlement of a warrant in connection with certain fundamental transactions that are within the control of the Company. Upon the occurrence of a fundamental transaction that is not within the control of the Company, the warrant holder would receive the same type or form of consideration offered and paid to common stockholders. The Company recognizes that ASC 480 and ASC 815 do not preclude equity classification when the only events that can trigger a net cash settlement payment to a warrant holder are within the control of the Company. Additionally, ASC 815 does not preclude equity classification when the warrant holder’s settlement, upon the occurrence of an event outside the control of the Company, is in the same type or form of consideration that is provided to common stockholders.
The Company then applied such examinations and observations during its assessment and determined that (i) the warrants do not constitute a liability under ASC 480; (ii) the warrants meet the definition of a derivative under ASC 815; (iii) the warrant holder’s option to receive a net cash settlement payment only becomes exercisable upon the occurrence of certain specified fundamental transactions that are within the control of the Company; (iv) upon the occurrence of a fundamental transaction that is not within the control of the Company, the warrant holder would receive the same type or form of consideration offered and paid to common stockholders; (v) the warrants are indexed to the Company’s common stock; and (vi) the warrants meet all other conditions for equity classification under ASC 480 and ASC 815.
Therefore, based on the results of the Company’s assessment, the Company has now concluded that the warrants issued in January 2018 are freestanding equity-linked derivative instruments that meet the criteria for the own-equity scope exception to derivative accounting under ASC 815. Accordingly, the warrants should have been classified as equity and should have been reported at the time of issuance as a component of additional paid-in capital rather than as a warrant liability on the consolidated balance sheets. Furthermore, the Company should not have reported any subsequent changes in the warrants’ estimated fair value within its consolidated statements of operations.
120
Impact of the Restatement
The impact of the restatement on the consolidated balance sheets, statements of operations and statements of cash flows for the Affected Periods is presented below. The restatement had no impact on net cash flows from operating, investing or financing activities.
Impact of the Restatement and Revision - 2019
($ in thousands except share and per share amounts)
Consolidated Statements of Operations and Comprehensive Loss
Year Ended December 31, 2019 | |||||||||||
As Reported | Adjustments | As Revised | |||||||||
Change in fair value of warrant liability | $ | (264 | ) | $ | 264 | $ | — | ||||
Total other income (expense), net | 47 | 264 | 311 | ||||||||
Net loss and comprehensive loss | (30,641 | ) | 264 | (30,377 | ) | ||||||
Net loss per share, basic and diluted | $ | (1.17 | ) | $ | 0.01 | $ | (1.16 | ) | |||
Weighted-average common shares outstanding, basic and diluted | 26,254,119 | — | 26,254,119 | ||||||||
Consolidated Balance Sheets
December 31, 2019 | |||||||||||
As Reported | Adjustments | As Revised | |||||||||
Warrant liability | $ | 1,504 | $ | (1,504 | ) | $ | — | ||||
Total liabilities | 52,884 | (1,504 | ) | 51,380 | |||||||
Additional paid-in-capital | 180,047 | 17,806 | 197,853 | ||||||||
Accumulated deficit | (203,682 | ) | (16,302 | ) | (219,984 | ) | |||||
Total stockholders' (deficit) equity | (23,787 | ) | 1,504 | (22,283 | ) | ||||||
Consolidated Statements of Cash Flows
Year Ended December 31, 2019 | |||||||||||
As Reported | Adjustments | As Revised | |||||||||
Net loss | $ | (30,641 | ) | $ | 264 | $ | (30,377 | ) | |||
Change in fair value of warrant liability | 264 | (264 | ) | — | |||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended September 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 1,160 | $ | (1,160 | ) | $ | — | ||||
Total other income (expense), net | 1,236 | (1,160 | ) | 76 | |||||||
Net loss and comprehensive loss | (8,336 | ) | (1,160 | ) | (9,496 | ) | |||||
Net loss per share, basic and diluted | $ | (0.32 | ) | $ | (0.04 | ) | $ | (0.36 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 26,189,454 | — | 26,189,454 | ||||||||
121
Condensed Consolidated Statements of Operations and Comprehensive Loss
Nine Months Ended September 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | (9,030 | ) | $ | 9,030 | $ | — | ||||
Total other income (expense), net | (8,767 | ) | 9,030 | 263 | |||||||
Net loss and comprehensive loss | (33,459 | ) | 9,030 | (24,429 | ) | ||||||
Net loss per share, basic and diluted | $ | (1.28 | ) | $ | 0.34 | $ | (0.94 | ) | |||
Weighted-average common shares outstanding, basic and diluted | 26,108,870 | — | 26,108,870 | ||||||||
Condensed Consolidated Balance Sheets
September 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 10,270 | $ | (10,270 | ) | $ | — | ||||
Total liabilities | 64,322 | (10,270 | ) | 54,052 | |||||||
Additional paid-in-capital | 179,047 | 17,806 | 196,853 | ||||||||
Accumulated deficit | (206,500 | ) | (7,536 | ) | (214,036 | ) | |||||
Total stockholders' (deficit) equity | (27,605 | ) | 10,270 | (17,335 | ) | ||||||
Condensed Consolidated Statements of Cash Flows
Nine Months Ended September 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (33,459 | ) | $ | 9,030 | $ | (24,429 | ) | |||
Change in fair value of warrant liability | 9,030 | (9,030 | ) | — | |||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended June 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | (9,802 | ) | $ | 9,802 | $ | — | ||||
Total other income (expense), net | (9,699 | ) | 9,802 | 103 | |||||||
Net loss and comprehensive loss | (18,098 | ) | 9,802 | (8,296 | ) | ||||||
Net loss per share, basic and diluted | $ | (0.69 | ) | $ | 0.37 | $ | (0.32 | ) | |||
Weighted-average common shares outstanding, basic and diluted | 26,069,734 | — | 26,069,734 | ||||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Six Months Ended June 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | (10,190 | ) | $ | 10,190 | $ | — | ||||
Total other income (expense), net | (10,003 | ) | 10,190 | 187 | |||||||
Net loss and comprehensive loss | (25,123 | ) | 10,190 | (14,933 | ) | ||||||
Net loss per share, basic and diluted | $ | (0.96 | ) | $ | 0.39 | $ | (0.57 | ) | |||
Weighted-average common shares outstanding, basic and diluted | 26,067,909 | — | 26,067,909 | ||||||||
122
Condensed Consolidated Balance Sheets
June 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 11,430 | $ | (11,430 | ) | $ | — | ||||
Total liabilities | 67,725 | (11,430 | ) | 56,295 | |||||||
Additional paid-in-capital | 178,100 | 17,806 | 195,906 | ||||||||
Accumulated deficit | (198,164 | ) | (6,376 | ) | (204,540 | ) | |||||
Total stockholders' (deficit) equity | (20,216 | ) | 11,430 | (8,786 | ) | ||||||
Condensed Consolidated Statements of Cash Flows
Six Months Ended June 30, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (25,123 | ) | $ | 10,190 | $ | (14,933 | ) | |||
Change in fair value of warrant liability | 10,190 | (10,190 | ) | — | |||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended March 31, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | (388 | ) | $ | 388 | $ | — | ||||
Total other income (expense), net | (304 | ) | 388 | 84 | |||||||
Net loss and comprehensive loss | (7,025 | ) | 388 | (6,637 | ) | ||||||
Net loss per share, basic and diluted | $ | (0.27 | ) | $ | 0.02 | $ | (0.25 | ) | |||
Weighted-average common shares outstanding, basic and diluted | 26,066,064 | — | 26,066,064 | ||||||||
Condensed Consolidated Balance Sheets
March 31, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 1,628 | $ | (1,628 | ) | $ | — | ||||
Total liabilities | 24,156 | (1,628 | ) | 22,528 | |||||||
Additional paid-in-capital | 177,855 | 17,806 | 195,661 | ||||||||
Accumulated deficit | (180,066 | ) | (16,178 | ) | (196,244 | ) | |||||
Total stockholders' (deficit) equity | (2,363 | ) | 1,628 | (735 | ) | ||||||
Condensed Consolidated Statements of Cash Flows
Three Months Ended March 31, 2019 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (7,025 | ) | $ | 388 | $ | (6,637 | ) | |||
Change in fair value of warrant liability | 388 | (388 | ) | — | |||||||
123
Impact of the Restatement - 2018
($ in thousands except share and per share amounts)
Consolidated Statements of Operations and Comprehensive Loss
Year Ended December 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 16,566 | $ | (16,566 | ) | $ | — | ||||
Total other income (expense), net | 15,888 | (16,566 | ) | (678 | ) | ||||||
Net loss and comprehensive loss | (12,673 | ) | (16,566 | ) | (29,239 | ) | |||||
Net loss per share, basic and diluted | $ | (0.49 | ) | $ | (0.64 | ) | $ | (1.13 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 25,795,721 | — | 25,795,721 | ||||||||
Consolidated Balance Sheets
December 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 1,240 | $ | (1,240 | ) | $ | — | ||||
Total liabilities | 21,164 | (1,240 | ) | 19,924 | |||||||
Additional paid-in-capital | 177,677 | 17,806 | 195,483 | ||||||||
Accumulated deficit | (172,327 | ) | (16,566 | ) | (188,893 | ) | |||||
Total stockholders' equity | 5,198 | 1,240 | 6,438 | ||||||||
Consolidated Statements of Cash Flows
Year Ended December 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (12,673 | ) | $ | (16,566 | ) | $ | (29,239 | ) | ||
Change in fair value of warrant liability | (16,566 | ) | 16,566 | — | |||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended September 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 1,464 | $ | (1,464 | ) | $ | — | ||||
Total other income (expense), net | 1,313 | (1,464 | ) | (151 | ) | ||||||
Net loss and comprehensive loss | (7,031 | ) | (1,464 | ) | (8,495 | ) | |||||
Net loss per share, basic and diluted | $ | (0.27 | ) | $ | (0.06 | ) | $ | (0.33 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 26,046,666 | — | 26,046,666 | ||||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Nine Months Ended September 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 5,733 | $ | (5,733 | ) | $ | — | ||||
Total other income (expense), net | 5,222 | (5,733 | ) | (511 | ) | ||||||
Net loss and comprehensive loss | (19,826 | ) | (5,733 | ) | (25,559 | ) | |||||
Net loss per share, basic and diluted | $ | (0.77 | ) | $ | (0.22 | ) | $ | (0.99 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 25,707,978 | — | 25,707,978 | ||||||||
124
Condensed Consolidated Balance Sheets
September 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 12,073 | $ | (12,073 | ) | $ | — | ||||
Total liabilities | 31,991 | (12,073 | ) | 19,918 | |||||||
Additional paid-in-capital | 177,336 | 17,806 | 195,142 | ||||||||
Accumulated deficit | (179,480 | ) | (5,733 | ) | (185,213 | ) | |||||
Total stockholders' (deficit) equity | (2,296 | ) | 12,073 | 9,777 | |||||||
Condensed Consolidated Statements of Cash Flows
Nine Months Ended September 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (19,826 | ) | $ | (5,733 | ) | $ | (25,559 | ) | ||
Change in fair value of warrant liability | (5,733 | ) | 5,733 | — | |||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended June 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 711 | $ | (711 | ) | $ | — | ||||
Total other income (expense), net | 569 | (711 | ) | (142 | ) | ||||||
Net loss and comprehensive loss | (7,578 | ) | (711 | ) | (8,289 | ) | |||||
Net loss per share, basic and diluted | $ | (0.29 | ) | $ | (0.03 | ) | $ | (0.32 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 26,039,169 | — | 26,039,169 | ||||||||
Condensed Consolidated Statements of Operations and Comprehensive Loss
Six Months Ended June 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 4,269 | $ | (4,269 | ) | $ | — | ||||
Total other income (expense), net | 3,909 | (4,269 | ) | (360 | ) | ||||||
Net loss and comprehensive loss | (12,795 | ) | (4,269 | ) | (17,064 | ) | |||||
Net loss per share, basic and diluted | $ | (0.50 | ) | $ | (0.17 | ) | $ | (0.67 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 25,535,827 | — | 25,535,827 | ||||||||
Condensed Consolidated Balance Sheets
June 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 13,537 | $ | (13,537 | ) | $ | — | ||||
Total liabilities | 34,359 | (13,537 | ) | 20,822 | |||||||
Additional paid-in-capital | 176,953 | 17,806 | 194,759 | ||||||||
Accumulated deficit | (172,449 | ) | (4,269 | ) | (176,718 | ) | |||||
Total stockholders' equity | 4,352 | 13,537 | 17,889 | ||||||||
Condensed Consolidated Statements of Cash Flows
Six Months Ended June 30, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (12,795 | ) | $ | (4,269 | ) | $ | (17,064 | ) | ||
Change in fair value of warrant liability | (4,269 | ) | 4,269 | — | |||||||
125
Condensed Consolidated Statements of Operations and Comprehensive Loss
Three Months Ended March 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Change in fair value of warrant liability | $ | 3,558 | $ | (3,558 | ) | $ | — | ||||
Total other income (expense), net | 3,340 | (3,558 | ) | (218 | ) | ||||||
Net loss and comprehensive loss | (5,217 | ) | (3,558 | ) | (8,775 | ) | |||||
Net loss per share, basic and diluted | $ | (0.21 | ) | $ | (0.14 | ) | $ | (0.35 | ) | ||
Weighted-average common shares outstanding, basic and diluted | 25,026,890 | — | 25,026,890 | ||||||||
Condensed Consolidated Balance Sheets
March 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Warrant liability | $ | 14,248 | $ | (14,248 | ) | $ | — | ||||
Total liabilities | 34,923 | (14,248 | ) | 20,675 | |||||||
Additional paid-in-capital | 176,402 | 17,806 | 194,208 | ||||||||
Accumulated deficit | (164,871 | ) | (3,558 | ) | (168,429 | ) | |||||
Total stockholders' equity | 11,379 | 14,248 | 25,627 | ||||||||
Condensed Consolidated Statements of Cash Flows
Three Months Ended March 31, 2018 | |||||||||||
As Reported | Adjustments | As Restated | |||||||||
Net loss | $ | (5,217 | ) | $ | (3,558 | ) | $ | (8,775 | ) | ||
Change in fair value of warrant liability | (3,558 | ) | 3,558 | — | |||||||
126
Note 3: KNOW Bio, LLC
On December 30, 2015, the Company completed the distribution of 100% of the outstanding member interests of KNOW Bio, LLC (“KNOW Bio”), a former wholly owned subsidiary of the Company, to Novan’s stockholders (the “Distribution”), pursuant to which KNOW Bio became an independent privately held company.
KNOW Bio is an independent, privately held company with a portfolio of operating subsidiaries that are advancing nitric oxide-based therapies using technology that is proprietary and/or in fields where they have exclusive intellectual property rights. The Company does not own any equity interest in KNOW Bio, has no common management or board representation at KNOW Bio, and the contractual arrangements between the two entities do not provide the Company with decision-making authority or power to influence KNOW Bio’s drug and medical device development activities.
The Company conducted an initial assessment of KNOW Bio under the variable interest consolidation model pursuant to FASB ASC 810, Consolidation (“ASC 810”), at the time of the Distribution in 2015 and has monitored KNOW Bio during each subsequent reporting period, including two required ASC 810 reassessments performed during 2017. The Company has consistently determined that KNOW Bio should not be consolidated in its consolidated financial statements. In the fourth quarter of 2018, KNOW Bio and its operating subsidiaries received significant additional equity investments that enable progression of their technology. These events required the Company to conduct another reassessment of variable interest entity characteristics, pursuant to FASB ASC 810-10, Consolidation, in which it determined that KNOW Bio should not be consolidated in its consolidated financial statements.
KNOW Bio Technology Agreements
In connection with the Distribution, the Company entered into exclusive license agreements and sublicense agreements with KNOW Bio, as described below. The agreements will continue for so long as there is a valid patent claim under the respective agreement, unless earlier terminated, and upon expiration, will continue as perpetual non-exclusive licenses. KNOW Bio has the right to terminate each such agreement, for any reason upon 90 days advance written notice to the Company.
License of existing and potential future intellectual property to KNOW Bio. The Company and KNOW Bio entered into an exclusive license agreement dated December 29, 2015 (the “KNOW Bio License Agreement”). Pursuant to the terms of the KNOW Bio License Agreement, the Company granted to KNOW Bio exclusive licenses, with the right to sublicense, under certain U.S. and foreign patents and patent applications that were controlled by the Company as of December 29, 2015 or that became controlled by the Company between that date and December 29, 2018, directed towards nitric-oxide releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds and other nitric oxide-based therapeutics.
Sublicense of UNC and other third party intellectual property to KNOW Bio. The Company and KNOW Bio also entered into sublicense agreements dated December 29, 2015 (the “KNOW Bio Sublicense Agreements” and together with the KNOW Bio License Agreement, the “Original KNOW Bio Agreements”). Pursuant to the terms of the KNOW Bio Sublicense Agreements, the Company granted to KNOW Bio exclusive sublicenses, with the ability to further sublicense, under certain of the U.S. and foreign patents and patent applications exclusively licensed to the Company from UNC under the Amended, Restated and Consolidated License Agreement dated June 27, 2012, as amended (the “UNC License Agreement”), and another third party directed towards nitric oxide-releasing compositions, to develop and commercialize products utilizing the licensed technology. Under the exclusive sublicense to the UNC patents and applications (the “UNC Sublicense Agreement”), KNOW Bio is subject to the terms and conditions under the UNC License Agreement, including milestone and diligence payment obligations. However, pursuant to the terms of the UNC License Agreement, the Company is directly obligated to pay UNC any future milestones or royalties, including those resulting from actions conducted by the Company’s sublicensees, including KNOW Bio. Therefore, in the event of KNOW Bio non-performance with respect to its obligations under the UNC Sublicense Agreement, the Company would be obligated to make such payments to UNC. KNOW Bio would then become obligated to repay the Company pursuant to the UNC Sublicense Agreement, otherwise KNOW Bio would be in breach of its agreements with the Company and intellectual property rights would revert back to the Company. There were no milestone or royalty payments required during the years ended December 31, 2019 and 2018.
127
Amendments to License and Sublicense Agreements with KNOW Bio
On October 13, 2017, the Company and KNOW Bio entered into certain amendments to the Original KNOW Bio Agreements (the “KNOW Bio Amendments”). Pursuant to the terms of the KNOW Bio Amendments, the Company re-acquired from KNOW Bio exclusive, worldwide rights under certain U.S. and foreign patents and patent applications controlled by the Company as of December 29, 2015, and that became controlled by the Company between December 29, 2015 and December 29, 2018, directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, to develop and commercialize products for all diagnostic, therapeutic, prophylactic and palliative uses for any disease, condition or disorder caused by certain oncoviruses (the “Oncovirus Field”). The Company also obtained a three-year exclusive option, subject to payment of separate option exercise fees, to include up to four additional specified oncoviruses in the Oncovirus Field.
KNOW Bio also granted to the Company an exclusive license, with the right to sublicense, under any patents and patent applications which became controlled by KNOW Bio during the three-year period between December 29, 2015 and December 29, 2018 and directed towards nitric oxide-releasing compositions and methods of manufacturing thereof, including methods of manufacturing Nitricil compounds, and other nitric oxide-based therapeutics, but not towards medical devices, to develop and commercialize products for use in the Oncovirus Field.
Upon execution of the KNOW Bio Amendments, in exchange for the Oncovirus Field rights, the Company paid a non-refundable upfront payment of $250. Products the Company develops in the Oncovirus Field based on Nitricil will not be subject to any further milestones, royalties or sublicensing payment obligations to KNOW Bio under the KNOW Bio Amendments. However, if the Company develops products in the Oncovirus Field that incorporate a certain nitric oxide-releasing composition specified in the KNOW Bio Amendments and (i) are covered by KNOW Bio patents or (ii) materially use or incorporate know-how of KNOW Bio or the Company related to such composition that was created between December 29, 2015 and December 29, 2018, the Company would be obligated to make the certain contingent milestone and royalty payments to KNOW Bio under the KNOW Bio Amendments.
The rights granted to the Company in the Oncovirus Field in the KNOW Bio Amendments continue for so long as there is a valid patent claim under the Original KNOW Bio Agreements, and upon expiration continue on a perpetual non-exclusive basis, and are subject to the termination rights of KNOW Bio and the Company that are set forth in the Original KNOW Bio Agreements. In addition, under the KNOW Bio Amendments, KNOW Bio may terminate the rights granted to the Company in the Oncovirus Field if: (i) the Company does not file a first investigational new drug (“IND”) application with the FDA for a product in the Oncovirus Field by October 2020; or (ii) the Company does not file a first new drug application (“NDA”) with the FDA by October 2025 for a product in the Oncovirus Field and does not otherwise have any active clinical programs related to the Oncovirus Field at such time.
The KNOW Bio Amendments also provide a mechanism whereby either party can cause a new chemical entity (“NCE”) covered by the Original KNOW Bio Agreements to become exclusive to such party by filing an IND on the NCE. An NCE that becomes exclusive to a party under this provision may not be commercialized by the other party until the later of expiration of patents covering the NCE or regulatory exclusivity covering the NCE. A party who obtains exclusivity for an NCE must advance development of the NCE pursuant to terms of the KNOW Bio Amendments in order to maintain such exclusivity; otherwise, such exclusivity will expire.
The terms of the KNOW Bio Amendments were negotiated at arms-length and do not provide the Company with an ability to significantly influence KNOW Bio or its operations.
Note 4: Research and Development Licenses
The Company has entered into various licensing agreements with universities and other research institutions under which the Company receives the rights, and in some cases substantially all of the rights, of the inventors, assignees or co-assignees to produce and market technology protected by certain patents and patent applications. The Company’s primary license agreement is with UNC and has been described in further detail within the subsection below. The counterparties to the Company’s various other licensing agreements are the University of Akron Research Foundation, Hospital for Special Surgery, Strakan International S.a.r.l., which is a licensee of the University of Aberdeen, KIPAX AB and KNOW Bio. The Company is generally required to make milestone payments based on development milestones and will be required to make royalty payments based on a percentage of future sales of covered products or a percentage of sublicensing revenue. Costs to acquire rights under license agreements and pre-commercialization milestone payments are classified as research and development expenses in the consolidated statements of operations. Research and development expense recognized in connection with the incurrence of such costs totaled zero and $20 during the years ended December 31, 2019 and 2018, respectively.
128
The Company is generally required by the various licensing agreements to reimburse the licensor for certain legal and other patent related costs. These costs are expensed as incurred and are classified as general and administrative expenses in the consolidated statements of operations. General and administrative expense recognized in connection with the incurrence of such costs totaled $93 and $74 during the years ended December 31, 2019 and 2018, respectively.
These license arrangements could require the Company to make payments upon achievement of certain milestones by the Company. As future royalty payments are directly related to future revenues (either sales or sublicensing), future commitments cannot be determined. No accrual for future payments under these agreements has been recorded, as the Company cannot estimate if, when or in what amount payments may become due.
UNC License Agreement
The UNC License Agreement provides the Company with an exclusive license to issued patents and pending applications directed to the Company’s library of Nitricil compounds, including patents issued in the U.S., Japan and Australia, with claims intended to cover NVN1000, the NCE for the Company’s current product candidates. The UNC License Agreement requires the Company to pay UNC up to $425 in regulatory and commercial milestones on a licensed product by licensed product basis and a running royalty percentage in the low single digits on net sales of licensed products. Licensed products include any products being developed by the Company or by its sublicensees.
Unless earlier terminated by the Company at its election, or if the Company materially breaches the agreement or becomes bankrupt, the UNC License Agreement remains in effect on a country by country and licensed product by licensed product basis until the expiration of the last to expire issued patent covering such licensed product in the applicable country. The projected date of expiration of the last to expire of the patents issued under the UNC Agreement is 2033.
Note 5: Licensing Arrangements
Sato License Agreement
Significant Terms
On January 12, 2017, the Company entered into a license agreement, and related first amendment, with Sato Pharmaceutical Co., Ltd. (“Sato”), relating to SB204, its drug candidate for the treatment of acne vulgaris in Japan (the “Sato Agreement”). Pursuant to the Sato Agreement, the Company granted to Sato an exclusive, royalty-bearing, non-transferable right and license under certain of the Company’s intellectual property rights, with the right to sublicense with the Company’s prior written consent, to develop, use and sell products in Japan that incorporate SB204 in certain topical dosage forms for the treatment of acne vulgaris, and to make the finished form of such products.
On October 5, 2018, the Company and Sato entered into the second amendment (the “Sato Amendment”) to the Sato Agreement (collectively, the “Amended Sato Agreement”). The Sato Amendment expanded the Sato Agreement to include SB206, the Company’s drug candidate for the treatment of viral skin infections. Pursuant to the Amended Sato Agreement, the Company granted to Sato an exclusive, royalty-bearing, non-transferable license under certain of its intellectual property rights, with the right to sublicense with the Company’s prior written consent, to develop, use and sell products in Japan that incorporate SB204 or SB206 in certain topical dosage forms for the treatment of acne vulgaris or viral skin infections, respectively, and to make the finished form of such products. The Company or its designated contract manufacturer will supply finished product to Sato for use in the development of SB204 and SB206 in the licensed territory. The rights granted to Sato do not include the right to manufacture the active pharmaceutical ingredient (“API”) of SB204 or SB206; rather, the parties agreed to negotiate a commercial supply agreement pursuant to which the Company or its designated contract manufacturer would be the exclusive supplier to Sato of the API for the commercial manufacture of licensed products in the licensed territory. Under the terms of the Amended Sato Agreement, the Company also has exclusive rights to certain intellectual property that may be developed by Sato in the future, which the Company could choose to use for its own development and commercialization of SB204 or SB206 outside of Japan.
Under the Amended Sato Agreement, in exchange for the SB204 and SB206 license rights granted to Sato, Sato agreed to pay the Company the following:
• | An upfront payment of 1.25 billion Japanese Yen, or “JPY”, payable in installments of 0.25 billion JPY, 0.5 billion JPY and 0.5 billion JPY on October 5, 2018, February 14, 2019 and September 13, 2019, respectively. This is in addition to the 1.25 billion JPY (approximately $10,813 USD) paid on January 19, 2017 following the execution of the Sato Agreement on January 12, 2017. On October 23, 2018, the Company received the first installment from the Amended Sato Agreement of 0.25 billion JPY (approximately $2,224 USD). On March 14, 2019, the Company received the second installment payment related to the Amended Sato Agreement of 0.5 billion JPY (approximately |
129
$4,460 USD). On November 7, 2019, the Company received the third installment payment related to the Amended Sato Agreement of 0.5 billion JPY (approximately $4,554 USD).
• | Up to an aggregate of 1.75 billion JPY (adjusted from 2.75 billion JPY in the Sato Agreement) upon the achievement of various development and regulatory milestones, including (i) a 0.25 billion JPY (approximately $2,162 USD) milestone payment received during the fourth quarter of 2018 following Sato’s initiation of a Phase 1 trial in Japan and (ii) an aggregate of 1.0 billion JPY that becomes payable upon the earlier occurrence of specified fixed future dates or the achievement of milestone events. |
• | Up to an aggregate of 3.9 billion JPY (adjusted from 0.9 billion JPY in the Sato Agreement) upon the achievement of various commercial milestones. |
• | A tiered royalty ranging from a mid-single digit to a low-double digit percentage (adjusted from a mid-single digit percentage in the Sato Agreement) of net sales of licensed products in the licensed territory, subject to a reduction in the royalty payments in certain circumstances. |
The term of the Amended Sato Agreement (and the period during which Sato must pay royalties under the amended license agreement) expires on the twentieth anniversary of the first commercial sale of a licensed product in the licensed field in the licensed territory (adjusted from the tenth anniversary of the first commercial sale in the license agreement). The term of the Amended Sato Agreement may be renewed with respect to a licensed product by mutual written agreement of the parties for additional two-year periods following expiration of the initial term. All other material terms of the license agreement remain unchanged by the Sato Amendment.
Sato is responsible for funding the development and commercial costs for the program that are specific to Japan. The Company is obligated to perform certain oversight, review and supporting activities for Sato, including: (i) using commercially reasonable efforts to obtain marketing approval of SB204 and SB206 in the U.S; (ii) sharing all future scientific information the Company may obtain during the term of the Amended Sato Agreement pertaining to SB204 and SB206; (iii) performing certain additional preclinical studies if such studies are deemed necessary by the Japanese regulatory authority, up to and not to exceed a total cost of $1,000; and (iv) participating in a joint committee that oversees, reviews and approves Sato’s development and commercialization activities under the Amended Sato Agreement. Additionally, the Company has granted Sato the option to use the Company’s trademarks in connection with the commercialization of licensed products in the licensed territory for no additional consideration, subject to the Company’s approval of such use.
The Amended Sato Agreement may be terminated by (i) Sato without cause upon 120 days’ advance written notice to the Company; (ii) either party in the event of the other party’s uncured material breach upon 60 days’ advance written notice; (iii) force majeure; (iv) either party in the event of the other party’s dissolution, liquidation, bankruptcy or insolvency; and (v) the Company immediately upon written notice if Sato challenges the validity, patentability, or enforceability of any of the Company’s patents or patent applications licensed to Sato under the Amended Sato Agreement. In the event of a termination, no portion of the upfront fees received from Sato are refundable.
Note 6: Revenue Recognition
Sato Agreement
The Company assessed the Sato Agreement in accordance with Topic 606 and concluded that the contract counterparty, Sato, is a customer within the scope of Topic 606. The Company identified the following promises under the Sato Agreement: (i) the grant of the intellectual property license to Sato; (ii) the obligation to participate in a joint committee that oversees, reviews, and approves Sato’s research and development activities and provides advisory support during Sato’s development process; (iii) the obligation to manufacture and supply Sato with all quantities of licensed product required for development activities in Japan; and (iv) the stand-ready obligation to perform any necessary repeat preclinical studies, up to $1,000 in cost. The Company determined that these promises were not individually distinct because Sato can only benefit from these licensed intellectual property rights and services when bundled together; they do not have individual benefit or utility to Sato. As a result, all promises have been combined into a single performance obligation.
The Sato Agreement also provides that the two parties agree to negotiate in good faith the terms of a commercial supply agreement pursuant to which the Company or a third party manufacturer would be the exclusive supplier to Sato of the API for the commercial manufacture of licensed products in the licensed territory. The Company concluded this obligation to negotiate the terms of a commercial supply agreement does not create (i) a legally enforceable obligation under which the Company may have to perform and supply Sato with API for commercial manufacturing; or (ii) a material right because the incremental commercial supply fee consideration agreed upon between the parties in the Sato Agreement is representative of a stand-alone selling price for the supply of API and does not represent a discount. Therefore, this contract provision is not considered to be a
130
promise to deliver goods or services and is not a performance obligation or part of the combined single performance obligation described above.
Amended Sato Agreement
On October 5, 2018, the Company and Sato entered into the Amended Sato Agreement. The Sato Amendment expanded the Sato Agreement to include SB206, the Company’s drug candidate for the treatment of viral skin infections. The Company assessed the Amended Sato Agreement in accordance with Topic 606 and concluded the contract modification should incorporate the additional goods and services provided for in the Amendment into the existing, partially satisfied single bundled performance obligation that will continue to be delivered to Sato over the remaining development period. This contract modification accounting is concluded to be appropriate as the additional goods and services conveyed under the Sato Amendment were determined to not be distinct from the single performance obligation, and the additional consideration provided did not reflect the standalone selling price of those additional goods and services. As such, the Company recorded a cumulative adjustment as of the amendment execution date to reflect revenue that would have been recognized cumulatively for the partially completed bundled performance obligation.
The Company concluded that the following consideration would be included in the transaction price as they were (i) received prior to December 31, 2019, or (ii) payable upon specified fixed dates in the future and are not contingent upon clinical or regulatory success in Japan:
• | The 1.25 billion JPY (approximately $10,813 USD) original upfront payment received on January 19, 2017 following the execution of the Sato Agreement on January 12, 2017. |
• | A milestone payment of 0.25 billion JPY (approximately $2,162 USD) received during the fourth quarter of 2018 following Sato’s initiation of a Phase 1 trial in Japan. |
• | The Sato Amendment upfront payment of 1.25 billion JPY, payable in installments of 0.25 billion JPY, 0.5 billion JPY and 0.5 billion JPY on October 5, 2018, February 14, 2019 and September 13, 2019, respectively. On October 23, 2018, the Company received the first installment from the Amended Sato Agreement of 0.25 billion JPY (approximately $2,224 USD). On March 14, 2019, the Company received the second installment payment related to the Amended Sato Agreement of 0.5 billion JPY (approximately $4,460 USD). On November 7, 2019, the Company received the third installment payment related to the Amended Sato Agreement of 0.5 billion JPY (approximately $4,554 USD). |
• | An aggregate of 1.0 billion JPY in non-contingent milestone payments that become payable upon the earlier occurrence of specified fixed dates in the future or the achievement of specified milestone events. |
The following table presents the Company’s contract assets and contract liabilities balances for the periods indicated.
Contract Asset | Contract Liability | Net Deferred Revenue | |||||||||
December 31, 2018 | $ | 17,790 | $ | 24,757 | $ | 6,967 | |||||
December 31, 2019 | $ | 8,974 | $ | 20,478 | $ | 11,504 | |||||
Short-term Deferred Revenue | Long-term Deferred Revenue | Net Deferred Revenue | |||||||||
December 31, 2018 | $ | 4,401 | $ | 2,566 | $ | 6,967 | |||||
December 31, 2019 | $ | 4,428 | $ | 7,076 | $ | 11,504 | |||||
The Company has recorded the Sato Agreement and Amended Sato Agreement transaction price, including the upfront payments received and the unconstrained variable consideration, as deferred revenue (comprised of (i) a contract liability; net of (ii) a contract asset).
The change in the net deferred revenue balances during the year ended December 31, 2019 was associated with the receipts of the second and third installment payments of 0.5 billion JPY (approximately $4,460 USD), and 0.5 billion JPY (approximately $4,554 USD), respectively, and recognition of license and collaboration revenue associated with the Company’s performance during the period (continued amortization of deferred revenue). During the years ended December 31, 2019 and 2018, the Company recognized $4,477 and $5,982, respectively, in license and collaboration revenue under this agreement.
131
The Company has concluded that the above consideration is probable of not resulting in a significant revenue reversal and therefore included in the transaction price and is allocated to the single performance obligation. No other variable consideration under the Amended Sato Agreement is probable of not resulting in a significant revenue reversal as of December 31, 2019 and therefore, is currently fully constrained and excluded from the transaction price.
The Company evaluated the timing of delivery for each of the obligations and concluded that a time-based input method is most appropriate because Sato is accessing and benefiting from the intellectual property and technology (the predominant items of the combined performance obligation) ratably over the duration of Sato’s estimated development period in Japan. Although the Company concluded that the intellectual property is functional rather than symbolic, the services provided under the performance obligation are provided over time. Therefore, the allocated transaction price will be recognized using a time-based input method that results in straight-line recognition over the Company’s performance period.
Prior to the Sato Amendment, the Company estimated the Sato Agreement development timeline for the SB204 product candidate to be approximately 5 years, starting in February 2017 and completing in the first quarter of 2022. With the Amended Sato Agreement, the Company and Sato are now developing both the SB204 and SB206 product candidates for the Japan territory. The parties continue to work collaboratively to reach agreement, but have not yet reached agreement, with respect to the combined SB204 and SB206 development plan for the Japan territory, including a corresponding timeline and estimated duration for the combined development program. The Company’s current estimated timeline is 7.5 years, starting in February 2017 and completing in the third quarter of 2024. The Company monitors and reassesses the estimated performance period for purposes of revenue recognition during each reporting period. The Company expects to reassess the estimated performance period during the first quarter of 2020, as the Company considers how the combined SB204 and SB206 development program timeline in Japan may potentially be affected by various factors, including (i) the recent results from the Company’s SB206 Phase 3 trials in the U.S., including but not limited to top-line efficacy results announced in January 2020, (ii) the Company’s plans and timelines for potential further clinical development of SB206 in the U.S., which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (iii) the Company’s in-house drug manufacturing capabilities and the progression of the Company’s manufacturing technology transfer projects with third-party contract manufacturing organizations. Therefore, if the duration of the combined SB204 and SB206 development program timeline is affected by the establishment or subsequent adjustments to a mutually agreed upon SB204 and SB206 development plan in the Japan territory, the Company will adjust its estimated performance period for revenue recognition purposes accordingly, as needed.
In future periods, the Company will lift the variable consideration constraint from each contingent payment when there is no longer a probable likelihood of significant revenue reversal. When the constraint is lifted from a milestone payment, the Company will recognize the incremental transaction price using the same time-based input method that is being used to recognize the revenue, which results in straight-line recognition over the performance period. If the Company’s performance is not yet completed at the time that the constraint is lifted, a cumulative catch-up adjustment will be recognized in the period. If no other performance is required by the Company at the time the constraint is lifted, the Company expects to recognize all revenue associated with such milestone payments at the time that the constraint is lifted.
Contract costs—Sato Agreement
The Company has incurred certain fees and costs in the process of obtaining the Amended Sato Agreement that were payable upon contract execution and, therefore, have been recognized as other assets and amortized as general and administrative expense on a straight-line basis over the same estimated performance period being used to recognize the associated revenue. These fees are associated with the following two arrangements and are described as follows:
• | The Company entered into an agreement with a third party to assist the Company in exploring the licensing opportunity which led to the execution of the Sato Agreement. The Company is obligated to pay the third party a low-single-digit percentage of all upfront and milestone payments the Company receives from Sato under the Amended Sato Agreement. |
• | The intellectual property rights granted to Sato under the Sato Agreement include certain intellectual property rights which the Company has licensed from UNC. Under the UNC License Agreement described in Note 4—Research and Development Licenses, the Company is obligated to pay UNC a running royalty percentage in the low single digits on net sales of licensed products, including net sales that may be generated by Sato. Additionally, the Company is obligated to make payments to UNC that represent the portion of the Sato upfront and milestone payments that were estimated to be directly attributable to the UNC intellectual property rights included in the license to Sato. |
132
The Company has also accrued certain fees that it will pay to the third party and to UNC in the future upon receipt of non-contingent installment and milestone payments from Sato. As of December 31, 2019, the Company had recorded capitalized contract acquisition costs of $533 in other assets and had accrued $230 in the accompanying balance sheet. For the years ended December 31, 2019 and 2018 the Company paid fees totaling $228 and $111, respectively.
Performance Obligations under the Sato Agreement
The net amount of existing performance obligations under long-term contracts unsatisfied as of December 31, 2019 was $11,504. The Company expects to recognize approximately 22% of the remaining performance obligations as revenue over the next 12 months, and the balance thereafter. The Company applied the practical expedient and does not disclose information about variable consideration related to sales-based or usage-based royalties promised in exchange for a license of intellectual property. This expedient specifically applied to the sales-based milestone payments that are present in the Amended Sato Agreement (3.9 billion JPY), as well as percentage-based royalty payments in the Sato Agreement that are contingent upon future sales.
Government Contracts and Grant Revenue
The Company assessed the following Federal grants in accordance with Topic 958 and concluded that both represent conditional non-exchange transactions.
In August 2019, the Company received a Phase 1 Federal grant of approximately $223 from the National Institutes of Health. The funds are to be used to advance formulation development of a nitric oxide-containing intravaginal gel (WH602) designed to treat high-risk human papilloma virus (“HPV”) infections that can lead to cervical intraepithelial neoplasia (“CIN”). The specific focus is to ensure the nitric oxide delivery from the gel replicates doses of nitric oxide previously demonstrated to be effective against HPV in the Company’s clinical and in vitro studies. Upon completion of grant Phase 1 preclinical work, the Company may be eligible to receive further awards for Phase 2 and 3 extensions under this grant. Revenue recognized under this grant was $83 during the year ended December 31, 2019.
In September 2019, the Company received a grant from the U.S. Department of Defense’s Congressionally Directed Medical Research Programs of approximately $1,113 as part of its Peer Reviewed Cancer Research Program. The grant will support the development of a non-gel formulation product candidate (WH504) designed to treat high-risk HPV infections that can lead to CIN, with well-characterized physical chemical properties suitable for intravaginal administration. In addition, the grant will support the evaluation of the effect of varying concentrations and treatment durations of berdazimer sodium (NVN1000) against HPV-18 in human raft cell culture in vitro studies. Revenue recognized under this grant was $336 during the year ended December 31, 2019.
Research and Development Services to KNOW Bio
The Company entered into a services agreement with KNOW Bio (the “KNOW Bio Services Agreement”) during 2017 and provided research and development services on a fee-for-service basis. After assessing revenue according to the five-step model of ASC 606, the Company determined that contract research and development services revenue should be recognized in the period in which the services are performed. During the year ended December 31, 2018, the Company recognized $9 in research and development services revenue for services performed under the KNOW Bio Services Agreement. There was no research and development services revenue recognized during the year ended December 31, 2019.
133
Note 7: Research and Development Arrangements
Royalty and Milestone Payments Purchase Agreement with Reedy Creek Investments LLC
On April 29, 2019, the Company entered into a royalty and milestone payments purchase agreement (the “Purchase Agreement”) with Reedy Creek Investments LLC (“Reedy Creek”), pursuant to which Reedy Creek provided funding to the Company in an initial amount of $25,000, which the Company will use primarily to pursue the development, regulatory approval and commercialization (including through out-license agreements and other third party arrangements) activities for SB206, a topical anti-viral gel being developed for the treatment of molluscum contagiosum, and advancing programmatically such activities with respect to SB204, a once-daily, topical monotherapy being developed for the treatment of acne vulgaris, and SB414, a topical cream-based product candidate being developed for the treatment of atopic dermatitis. Reedy Creek was to provide additional funding to the Company of $10,000 contingent upon the achievement by the Company of SB206 clinical trial success, defined as (i) the achievement, no later than March 31, 2020, of statistically significant rates of complete clearance of lesions for molluscum contagiosum in humans at week 12 in each of the two Phase 3 clinical trials or any other primary endpoint required or accepted by the FDA for the SB206 product; or (ii) equivalent achievement (as agreed upon by the parties). See Note 16—Subsequent Events regarding the January 2020 announcement of top-line results from two pivotal Phase 3 clinical trials of SB206 for the treatment of molluscum contagiosum. SB206 did not achieve statistically significant results in the primary endpoint in both trials, which was the complete clearance of all molluscum lesions at Week 12. Based on the top line efficacy results from the Phase 3 SB206 program released in January 2020, the Company understands that Reedy Creek will not be paying the Company the contingent $10,000 of additional funding.
Pursuant to the Purchase Agreement, the Company will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by the Company pursuant to any out-license agreement for SB204, SB206 or SB414 in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by the Company to third parties pursuant to any agreements under which the Company has in-licensed intellectual property with respect to such products in the United States, Mexico or Canada. The applicable percentage used for determining the ongoing quarterly payments, applied to amounts received directly by the Company pursuant to any out-license agreement for each product, ranges from 10% for SB206 to 20% for SB204 and SB414. However, the agreement provides that the applicable percentage for each product will be 25% for fees or milestone payments received by the Company (but not royalty payments received by the Company) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If the Company decides to commercialize any product on its own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, the Company will be obligated to pay Reedy Creek a low single digits royalty on net sales of such products.
The Company has determined that the Reedy Creek Purchase Agreement is within the scope of ASC 730-20, Research and Development Arrangements. The Company concluded that there has not been a substantive and genuine transfer of risk related to the Purchase Agreement as (i) Reedy Creek has the opportunity to recover its investment regardless of the outcome of the research and development programs within the scope of the agreement (prior to commercialization of any in scope assets through potential out-licensing agreements and related potential future milestone payments); and (ii) there is a presumption that the Company is obligated to pay Reedy Creek amounts equal to its investment based on the related party relationship at the time the parties entered into the Purchase Agreement. The Purchase Agreement is a broad funding arrangement, due to (i) the multi-asset, or portfolio approach including three developmental assets that are within the scope of the arrangement; and (ii) Reedy Creek’s approximate 15% ownership of the outstanding shares of common stock of the Company.
As such, the Company has determined that the appropriate accounting treatment under ASC 730-20 is to record the initial proceeds of $25,000 as cash and cash equivalents, as the Company has the ability to direct the usage of funds, and a long-term liability within its classified balance sheet. The long-term liability will remain until the Company receives future milestones from other potential third parties, as defined within the Purchase Agreement, of which 25% will be contractually owed to Reedy Creek. If potential future milestones are received by the Company, and become partly due to Reedy Creek, the corresponding partial repayment to Reedy Creek will result in a ratable reduction of the total long-term obligation to repay the initial purchase price.
Development Funding and Royalties Agreement with Ligand Pharmaceuticals Incorporated
On May 4, 2019, the Company entered into a development funding and royalties agreement (the “Funding Agreement”) with Ligand, pursuant to which Ligand provided funding to the Company of $12,000, which the Company will use to pursue the development and regulatory approval of SB206, a topical anti-viral gel being developed for the treatment of molluscum contagiosum.
134
Pursuant to the Funding Agreement, the Company will pay Ligand up to $20,000 in milestone payments upon the achievement by the Company of certain regulatory and commercial milestones associated with SB206 or any product that incorporates or uses NVN1000, the active pharmaceutical ingredient for the Company’s clinical stage product candidates, for the treatment of molluscum contagiosum. In addition to the milestone payments, the Company will pay Ligand tiered royalties ranging from 7% to 10% based on annual aggregate net sales of such products in the United States, Mexico or Canada.
The Company has determined that the Ligand transaction is within the scope of ASC 730-20 as it represents an obligation to perform contractual services for the development of SB206 using commercially reasonable efforts. In addition, the Funding Agreement also states that if all development of SB206 is ceased prior to the first regulatory approval, the Company must pay to Ligand an amount equal to the purchase price less the amount spent in accordance with the development budget on development activities conducted prior to such cessation. As such, the Company has concluded that the appropriate accounting treatment under ASC 730-20 is to record the initial proceeds of $12,000, as a liability and as restricted cash on its consolidated balance sheet, as the funds can only be used for the progression of SB206.
The Company amortizes the liability ratably during each reporting period, based on the Ligand funding as a percentage of the total direct costs incurred by the Company during the reporting period related to the estimated total cost to progress the SB206 program to a regulatory approval in the U.S. The ratable Ligand funding is presented within the consolidated statement of operations as an offset to research and development expenses associated with the SB206 program. During the first quarter of 2020, the Company expects to reassess the estimated total cost to progress the SB206 program to a U.S. regulatory approval, as the Company considers how such estimated costs may potentially be affected by various factors, including (i) the recent results from the Company’s SB206 Phase 3 trials in the U.S., including but not limited to top-line efficacy results announced in January 2020, (ii) the Company’s plans and timelines for potential further clinical development of SB206 in the U.S., which is subject to additional funding and feedback from a Type C meeting with the FDA scheduled for April 1, 2020, and (iii) the Company’s in-house drug manufacturing capabilities and the progression of the Company’s manufacturing technology transfer projects with third-party contract manufacturing organizations.
The initial restricted cash balance was also reduced ratably during interim reporting periods in 2019 in a manner consistent with the amortization method for the Ligand funding liability balance. As of December 31, 2019, the aggregate amount spent in accordance with the SB206 development budget on SB206 development activities had exceeded the $12,000 purchase price, causing the aforementioned repayment provision provided for in the Funding Agreement to no longer be enforceable. Therefore, the Company reported no restricted cash balance related to the Funding Agreement, as of December 31, 2019 in its consolidated balance sheet.
For the year ended December 31, 2019, the Company recorded $8,185 as contra-research and development expense related to the SB206 developmental program, funded by Ligand.
Note 8: Property and Equipment, Net
Property and equipment consisted of the following:
December 31, | |||||||
2019 | 2018 | ||||||
Computer equipment | $ | 575 | $ | 577 | |||
Furniture and fixtures | 305 | 312 | |||||
Laboratory equipment | 7,898 | 7,442 | |||||
Office equipment | 339 | 400 | |||||
Building related to facility lease obligation | — | 10,557 | |||||
Leasehold improvements | 7,068 | 1,168 | |||||
Property and equipment, gross | 16,185 | 20,456 | |||||
Less: Accumulated depreciation and amortization | (5,679 | ) | (4,588 | ) | |||
Total property and equipment, net | $ | 10,506 | $ | 15,868 | |||
Depreciation and amortization expense was $2,033 and $1,664 for the years ended December 31, 2019 and 2018, respectively.
See Note 1—Organization and Significant Accounting Policies and Note 9—Commitments and Contingencies regarding the adoption of Topic 842, Leases, and its impact to property and equipment, net for the year ended December 31, 2019.
135
Note 9: Commitments and Contingencies
Lease Obligations
The Company leases office space and certain equipment under non-cancelable lease agreements.
Prior to January 1, 2019, the Company applied the accounting guidance in ASC 840, Leases, to its lease agreements. The leases were reviewed for classification as operating or capital leases. For operating leases, rent was recognized on a straight-line basis over the lease period. For capital leases, the Company recorded the leased asset with a corresponding liability and amortized the asset over the lease term. Payments were recorded as reductions to the liability with an appropriate interest charge recorded based on the then-outstanding remaining liability.
The Company considered the nature of the renovations and the Company’s involvement during the construction period of previously leased office space to determine if it was considered to be the owner of the construction project during the construction period. If the Company determined that it was the owner of the construction project, it was required to capitalize the fair value of the building as well as the construction costs incurred, including capitalized interest, on its consolidated balance sheet along with a corresponding financing liability (“build-to-suit accounting”). Upon completion of the construction of the facility under a build-to-suit lease, the Company assessed whether the circumstances qualified for sales recognition under the sale-leaseback accounting guidance. If the lease met the sale-leaseback criteria, the Company would remove the asset and related financial obligation from the balance sheet and evaluate the lease for treatment as a capital or operating lease. If upon completion of construction, the project did not meet the sale-leaseback criteria, the leased property was treated as an asset financing for financial reporting purposes. The portion of the facility financing obligation representing the principal that was to be repaid in the following 12 months was classified as a current liability in the consolidated balance sheets, with the remaining portion of the obligation classified as a noncurrent liability.
Beginning January 1, 2019, the Company applies the accounting guidance in ASC 842, Leases. As such, the Company assesses all arrangements, that convey the right to control the use of property, plant and equipment, at inception, to determine if it is, or contains, a lease based on the unique facts and circumstances present in that arrangement. For those leases identified, the Company determines the lease classification, recognition, and measurement at the lease commencement date. For arrangements that contain a lease the Company: (i) identifies lease and non-lease components; (ii) determines the consideration in the contract; (iii) determines whether the lease is an operating or financing lease; and (iv) recognizes lease ROU assets and corresponding lease liabilities. Lease liabilities are recorded based on the present value of lease payments over the expected lease term. The corresponding ROU asset is measured from the initial lease liability, adjusted by (i) accrued or prepaid rents; (ii) remaining unamortized initial direct costs and lease incentives; and (iii) any impairments of the ROU asset. The interest rate implicit in the Company’s lease contracts is typically not readily determinable and as such, the Company uses its incremental borrowing rate based on the information available at the lease commencement date, which represents an internally developed rate that would be incurred to borrow, on a collateralized basis, over a similar term, an amount equal to the lease payments in a similar economic environment. The weighted average discount rate utilized on the Company’s operating lease liabilities as of December 31, 2019 was 9.85%. The weighted average remaining lease term for the Company’s operating leases as of December 31, 2019 was 6.50 years.
Primary Facility Lease
In August 2015, the Company entered into a lease agreement for approximately 51,000 rentable square feet of facility space in Morrisville, North Carolina, commencing in April 2016 (the “Primary Facility Lease”). The initial term of the Primary Facility Lease extends through June 30, 2026. The Company has an option to extend the Primary Facility Lease by five years upon completion of the initial lease term, however, the renewal period was not included in the calculation of the lease obligation. Current contractual base rent payments are $95 per month, subject to a three percent increase annually over the term of the Primary Facility Lease.
Prior to January 1, 2019, the Company applied the accounting guidance in ASC 840. Based on that guidance, the facility was accounted for as an asset financing, with the building asset and related facility financing obligation remaining on the Company’s balance sheet. The building asset was being depreciated over a 25 year period and the facility financing obligation was amortized so that the net carrying value of the building asset and the facility financing obligation were to be equivalent at the end of the initial term of the lease agreement. Monthly rental payments were allocated between principal and interest expense associated with the facility financing obligation, as well as grounds rent expense of $8 per month.
136
The Company had recorded an asset related to the building and construction costs within property and equipment of $10,557 as of December 31, 2018. The non-current facility lease obligation on the Company’s consolidated balance sheet was $7,998 as of December 31, 2018. During the year ended December 31, 2018, the Company recognized interest expense related to the primary facility lease of $1,044 and there was $41 of accrued interest included in other accrued expenses as of December 31, 2018
The Company adopted Topic 842 as of January 1, 2019 using the modified retrospective transition method and initially applied the transition provisions as of January 1, 2019. This transition method allowed the Company to continue to apply the legacy guidance in ASC 840 for periods prior to 2019 and recognize a cumulative-effect adjustment to the opening balance of accumulated deficit as of the date of adoption.
The Company elected the package of transition practical expedients, which, among other things, allowed the Company to keep the historical lease classifications and not have to reassess the lease classification for any existing leases as of the date of adoption. The Company also made an accounting policy election to apply the short-term lease exception, which allows the Company to exclude leases with an initial term of twelve months or less from the consolidated balance sheets.
As a result of the adoption of Topic 842, the Company derecognized $10,557 of building asset (property, plant and equipment), and $7,998 of facility financing obligation associated with previously existing build-to-suit arrangement related to its sole corporate and manufacturing facility. The Company also capitalized leasehold improvements and ROU assets of $5,885 and $1,827, respectively, and recorded lease liabilities for operating leases totaling $6,786, as of January 1, 2019. The capitalized leasehold improvement assets recorded as part of the adoption of Topic 842 were previously included within the derecognized building asset as part of the previous build-to-suit arrangement. The Company also recognized an increase of $714 to accumulated deficit related to its de-recognition of its previously recorded build-to-suit arrangement.
The Company has elected to separate lease components (fixed rent payments) with non-lease components (common-area maintenance costs) on its real estate assets. Fixed lease payments on operating leases are recognized over the expected term of the lease on a straight-line basis. Variable lease expenses that are not considered fixed are expensed as incurred. Fixed and variable lease expense on operating leases is recognized within operating expenses within our consolidated statements of operations. The Company has elected the short-term lease exemption and, therefore, does not recognize a ROU asset or corresponding liability for lease arrangements with an original term of 12 months or less.
Rent expense, including both short-term and variable lease components associated with the primary facility lease, was $880 and $308 for the years ended December 31, 2019 and 2018, respectively.
The Company’s supplemental non-cash disclosure for its ROU assets obtained in exchange for lease liabilities was $1,827 for the year ended December 31, 2019.
At December 31, 2019, maturities of operating lease liabilities over each of the next five years and thereafter are as follows:
Operating Leases | |||
2020 | $ | 1,215 | |
2021 | 1,241 | ||
2022 | 1,278 | ||
2023 | 1,317 | ||
2024 | 1,356 | ||
Thereafter | 2,111 | ||
Total minimum lease payments | $ | 8,518 | |
Less imputed interest | $ | (2,256 | ) |
Total lease liability | $ | 6,262 | |
Primary Facility Sublease
In July 2018, the Company and a third-party tenant commenced a sublease of approximately 6,400 square feet of office space at the Company’s headquarters. The sublease has a three-year, non-cancellable term and provides for monthly rental income to the Company of approximately $12 per month through July 2021. The Company has classified the sublease as an operating lease pursuant to classification criteria in ASC 842 and is recognizing the rental income on a straight-line basis over the lease term as a component of other income and expense in the Company’s consolidated statements of operations and comprehensive loss. In
137
October 2019, the Company executed a termination agreement with the subtenant for this sublease, with an effective date of December 31, 2019.
Contingencies
From time to time, the Company may have certain contingent liabilities that arise in the ordinary course of business activities. The Company accrues a liability for such matters when it is probable that future expenditures will be made and such expenditures can be reasonably estimated. See Legal Proceedings below for further discussion of pending legal claims.
The Company has entered into, and expects to continue to enter into, contracts in the normal course of business with various third parties who support its clinical trials, preclinical research studies and other services related to its development activities. The scope of the services under these agreements can generally be modified at any time, and these agreements can generally be terminated by either party after a period of notice and receipt of written notice. There have been no material contract terminations as of December 31, 2019.
See Note 4—Research and Development Licenses regarding the Company’s research and development license agreements.
See Note 7—Research and Development Arrangements regarding the Purchase Agreement with Reedy Creek and the Funding Agreement with Ligand.
Legal Proceedings
In prior filings, the Company reported that it was subject to putative stockholder class action lawsuits that were filed in November 2017 in the United States District Court for the Middle District of North Carolina against the Company and certain of its current and former directors and officers, which were consolidated under the case name In re Novan, Inc. Securities Litigation. The consolidated amended complaint filed by the designated lead plaintiff asserted claims for violation of Sections 11 and 15 of the Securities Act of 1933 and Sections 10(b) and 20(a) of the Exchange Act and SEC Rule 10b-5 promulgated thereunder, in connection with statements related to the Company’s Phase 3 clinical trials of SB204. On June 14, 2018, the Company filed a motion to dismiss the consolidated amended complaint. On November 30, 2018, a federal magistrate judge entered an order recommending that the district court grant the Company’s motion. The plaintiff filed objections to this recommendation and the Company filed a response. On January 28, 2019, the district court adopted the magistrate judge’s recommendation, dismissed the action with prejudice and entered judgment in favor of the Company and against the plaintiff. The plaintiff did not appeal this dismissal and judgment. As such, the Company has concluded that this matter is closed.
Other than as described above, the Company is not currently a party to any material legal proceedings and is not aware of any claims or actions pending or threatened against the Company that the Company believes could have a material adverse effect on the Company’s business, operating results, cash flows or financial statements. In the future, the Company might from time to time become involved in litigation relating to claims arising from its ordinary course of business.
Compensatory Obligations
In conjunction with the departures of three former Company officers in 2019 and 2018, the Company entered into separation and general release agreements that included separation benefits consistent with the Company’s obligations under their previously existing employment agreements for “separation from service” for “good reason.” The Company recognized related severance expense of $878 and $332 during the years ended December 31, 2019 and 2018, respectively. The remaining accrued severance obligation in respect of the three former officers was $33 as of December 31, 2019, which is included in accrued compensation in the accompanying consolidated balance sheet. The Company also recognized non-cash stock compensation expense of $212 during the year ended December 31, 2018, respectively, related to the accelerated vesting of the former officers’ stock options. There was no non-cash stock compensation expense in relation to these departures during the year ended December 31, 2019.
In November 2018, the Company realigned its overall employee headcount to reduce certain fixed costs. The Company recognized related severance expense of $67 and $196 during the years ended December 31, 2019 and 2018, respectively. As of December 31, 2019, severance costs of $52 were accrued in the accompanying consolidated balance sheet.
See Note 11—Share-Based Compensation regarding the SARs granted in August 2018.
See Note 12—Tangible Stockholder Return Plan regarding the Tangible Stockholder Return Plan adopted in August 2018.
138
Note 10: Stockholders’ Equity (Deficit)
Capital Structure
In conjunction with the completion of the Company’s initial public offering in September 2016, the Company further amended its amended and restated certificate of incorporation and amended and restated its bylaws. The amendment provides for 210,000,000 authorized shares of capital stock, of which 200,000,000 shares have been designated as $0.0001 par value common stock, and 10,000,000 shares have been designated as $0.0001 par value preferred stock.
Aspire Common Stock Purchase Agreement
On August 30, 2019, the Company entered into the Aspire Common Stock Purchase Agreement with Aspire Capital, which provides that, upon the terms and subject to the conditions and limitations set forth therein, Aspire Capital is committed to purchase up to an aggregate of $25,000 of shares of the Company’s common stock at the Company’s request from time to time during the 30-month term of the Purchase Agreement. Concurrently with entering into the Purchase Agreement, the Company also entered into a registration rights agreement with Aspire Capital (the “Registration Rights Agreement”), in which the Company agreed to file one or more registration statements, as permissible and necessary to register under the Securities Act of 1933, as amended (the “Securities Act”), registering the sale of the shares of the Company’s common stock that have been and may be issued to Aspire Capital under the Aspire Common Stock Purchase Agreement. On September 16, 2019, the Company filed with the SEC, a prospectus to the effective Registration Statement on Form S-1 (File No. 333-233632) registering 7,032,630 shares of common stock that have been and may be offered to Aspire Capital from time to time under the Aspire Common Stock Purchase Agreement.
Under the Aspire Common Stock Purchase Agreement, on any trading day selected by the Company, the Company has the right, in its sole discretion, to present Aspire Capital with a purchase notice (each, a “Purchase Notice”), directing Aspire Capital (as principal) to purchase up to 100,000 shares of the Company’s common stock per business day, up to $25,000 of the Company’s common stock in the aggregate at a per share price (the “Purchase Price”) equal to the lesser of (i) the lowest sale price of the Company’s common stock on the purchase date, or (ii) the arithmetic average of the three (3) lowest closing sale prices for the Company’s common stock during the ten (10) consecutive trading days ending on the trading day immediately preceding the purchase date. The aggregate purchase price payable by Aspire Capital on any one purchase date may not exceed $500.
In addition, on any date on which the Company submits a Purchase Notice to Aspire Capital in an amount equal to 100,000 shares, the Company also has the right, in its sole discretion, to present Aspire Capital with a volume-weighted average price purchase notice (each, a “VWAP Purchase Notice”) directing Aspire Capital to purchase an amount of stock equal to up to 30% of the aggregate shares of the Company’s common stock traded on its principal market on the next trading day (the “VWAP Purchase Date”), subject to a maximum number of shares the Company may determine. The purchase price per share pursuant to such VWAP Purchase Notice is generally 97% of the volume-weighted average price for the Company’s common stock traded on its principal market on the VWAP Purchase Date.
The Purchase Price will be adjusted for any reorganization, recapitalization, non-cash dividend, stock split, or other similar transaction occurring during the period(s) used to compute the Purchase Price. The Company may deliver multiple Purchase Notices and VWAP Purchase Notices to Aspire Capital from time to time during the term of the Aspire Common Stock Purchase Agreement, so long as the most recent purchase has been completed.
The Aspire Common Stock Purchase Agreement provides that the Company and Aspire Capital shall not effect any sales under the Aspire Common Stock Purchase Agreement on any purchase date where the closing sale price of the Company’s common stock is less than $0.25. There are no trading volume requirements or restrictions under the Aspire Common Stock Purchase Agreement, and the Company will control the timing and amount of sales of the Company’s common stock to Aspire Capital. Aspire Capital has no right to require any sales by the Company, but is obligated to make purchases from the Company as directed by the Company in accordance with the Aspire Common Stock Purchase Agreement. There are no limitations on use of proceeds, financial or business covenants, restrictions on future financing transactions, rights of first refusal, participation rights, penalties or liquidated damages in the Aspire Common Stock Purchase Agreement. The Aspire Common Stock Purchase Agreement may be terminated by the Company at any time, at its discretion, without any penalty or additional cost to the Company. Aspire Capital has agreed that neither it nor any of its agents, representatives and affiliates shall engage in any direct or indirect short-selling or hedging of the Company’s common stock during any time prior to the termination of the Aspire Common Stock Purchase Agreement. Any proceeds the Company receives under the Aspire Common Stock Purchase Agreement are expected to be used for working capital and general corporate purposes.
The Aspire Common Stock Purchase Agreement provides that the number of shares that may be sold pursuant to the Aspire Common Stock Purchase Agreement will be limited to 5,211,339 shares (the “Exchange Cap”), which represents 19.99% of the Company’s outstanding shares of common stock on August 30, 2019, unless stockholder approval or an exception pursuant to the
139
rules of the Nasdaq Global Market is obtained to issue more than 19.99%. This limitation will not apply if, at any time the Exchange Cap is reached and at all times thereafter, the average price paid for all shares issued under the Aspire Common Stock Purchase Agreement is equal to or greater than $2.17, which was the closing sale price of the Company’s common stock immediately preceding the execution of the Aspire Common Stock Purchase Agreement. The Company is not required or permitted to issue any shares of common stock under the Aspire Common Stock Purchase Agreement if such issuance would breach its obligations under the rules or regulations of the Nasdaq Global Market. The Company may, in its sole discretion, determine whether to obtain stockholder approval to issue more than 19.99% of its outstanding shares of Common Stock hereunder if such issuance would require stockholder approval under the rules or regulations of the Nasdaq Global Market.
In consideration for entering into the Aspire Common Stock Purchase Agreement, concurrently with the execution of the Aspire Common Stock Purchase Agreement, the Company issued to Aspire Capital 345,622 shares of the Company’s common stock (the “Commitment Shares”). These Commitment Shares valued at $750 were recorded in August 2019 as non-cash costs of equity financing and charged against additional paid-in capital.
As of December 31, 2019, the Company has sold 300,000 shares of common stock at an average price of $2.49 per share under the Aspire Common Stock Purchase Agreement. The Company has remaining availability for sales of its common stock under the Aspire Common Stock Purchase Agreement, subject to the limitations described above, of up to $24,253 as of December 31, 2019.
January 2018 Offering
On January 9, 2018, the Company completed the January 2018 Offering, pursuant to which it sold an aggregate of 10,000,000 shares of common stock and warrants to purchase up to 10,000,000 shares of the Company’s common stock at a public offering price of $3.80 per share of common stock and accompanying warrant. The warrant exercise price is $4.66 per share and will expire four years from the date of issuance. Net proceeds from the offering were approximately $35,194 after deducting underwriting discounts and commissions and offering expenses of approximately $2,806.
The Company incurred costs directly related to (i) the shelf registration statement filing totaling $110 and (ii) the January 2018 Offering completed in January 2018 totaling $370, all of which were initially capitalized and included in deferred offering costs. A pro-rata portion of the shelf registration offering costs and all of the January 2018 Offering costs were reclassified to additional paid-in capital upon completion of the January 2018 Offering.
The warrants issued in the January 2018 Offering include certain provisions that establish certain warrant holder settlement rights that take effect upon the occurrence of certain fundamental transactions. The warrants define a fundamental transaction to generally include any consolidation or merger whereby another entity acquires more than 50% of the Company’s outstanding common stock or the sale of all or substantially all of the Company’s assets. The fundamental transaction provision provides the warrant holders with the option to settle any unexercised warrants for cash in the event of certain fundamental transactions that are within the control of the Company. For any fundamental transaction that is not within the control of the Company, including a fundamental transaction not approved by the Company’s board of directors, the warrant holder will only be entitled to receive from the Company or any successor entity the same type or form of consideration (and in the same proportion) that is being offered and paid to the stockholders of the Company in connection with the fundamental transaction, whether that consideration be in the form of cash, stock or any combination thereof. In the event of any fundamental transaction, and regardless of whether it is within the control of the Company, the settlement amount of the warrants (whether in cash, stock or a combination thereof) is determined based upon a Black-Scholes value that is calculated using inputs as specified in the warrants, including a defined volatility input equal to the greater of the Company’s 100-day historical volatility or 100%.
The warrants also include a provision whereby the exercisability of the warrants may be limited if, upon exercise, the warrant holder or any of its affiliates would beneficially own more than 4.99% (or an amount up to 9.99% if the holder so elects) of the Company’s common stock. The warrants also provide that this exercise limitation provision is not applicable to any warrant holder that beneficially owns 10.0% or more of the Company’s outstanding common stock immediately following the closing of the January 2018 Offering and the issuance of the accompanying warrants.
There were no exercises of warrants during the years ended December 31, 2019 or 2018.
The Company has assessed the warrants for appropriate equity or liability classification pursuant to the Company’s accounting policy described in Note 1—Organization and Significant Accounting Policies. During this assessment, the Company determined that (i) the warrants do not constitute a liability under ASC 480; (ii) the warrants meet the definition of a derivative under ASC 815; (iii) the warrant holder’s option to receive a net cash settlement payment only becomes exercisable upon the occurrence of certain specified fundamental transactions that are within the control of the Company; (iv) upon the occurrence
140
of a fundamental transaction that is not within the control of the Company, the warrant holder would receive the same type or form of consideration offered and paid to common stockholders; (v) the warrants are indexed to the Company’s common stock; and (vi) the warrants meet all other conditions for equity classification under ASC 480 and ASC 815.
Based on the results of this assessment, the Company concluded that the warrants issued in January 2018 are freestanding equity-linked derivative instruments that meet the criteria for the own-equity scope exception to derivative accounting under ASC 815. Accordingly, the warrants are classified as equity and are accounted for as a component of additional paid-in capital at the time of issuance.
Common Stock
The Company’s common stock has a par value of $0.0001 per share and consists of 200,000,000 authorized shares as of December 31, 2019 and 2018. There were 26,734,800 and 26,056,735 shares of common stock outstanding as of December 31, 2019 and 2018, respectively.
The Company had reserved shares of common stock for future issuance as follows:
December 31, | |||||
2019 | 2018 | ||||
Outstanding stock options (Note 11) | 1,789,303 | 1,671,666 | |||
Warrants to purchase common stock issued in January 2018 Offering | 10,000,000 | 10,000,000 | |||
Outstanding stock appreciation rights (Note 11) | 1,000,000 | — | |||
For possible future issuance under 2016 Stock Plan (Note 11) | 388,463 | 699,376 | |||
13,177,766 | 12,371,042 | ||||
Related Party Stock Repurchase
In April 2016, the Company repurchased 9,500 shares of common stock for an aggregate price of $155 from an executive of the Company who was also a member of the Company’s board of directors at that time. The repurchase of these shares is recorded as treasury stock on the Company’s consolidated balance sheet as of December 31, 2019 and 2018.
Preferred Stock
The Company’s amended and restated certificate of incorporation provides the Company’s board of directors with the authority to issue $0.0001 par value preferred stock from time to time in one or more series by adopting a resolution and filing a certificate of designations. Voting powers, designations, preferences, dividend rights, conversion rights and liquidation preferences shall be stated and expressed in such resolutions. There were 10,000,000 shares designated as preferred stock and no shares outstanding as of December 31, 2019 and 2018.
Note 11: Share-Based Compensation
2008 Stock Plan
During 2008, the Company adopted the 2008 Stock Plan (the “2008 Plan”). As amended, a total of 1,416,666 shares of common stock were reserved for issuance under the 2008 Plan. Eligible plan participants included employees, directors, and consultants. The 2008 Plan permitted the granting of incentive stock options, nonqualified stock options, and other stock-based awards. As further described below, as of September 20, 2016, no additional awards will be granted under the 2008 Plan.
2016 Stock Plan
Effective September 20, 2016 (the “Effective Date”), the Company adopted the 2016 Incentive Award Plan (the “2016 Plan”). The 2016 Plan is the successor to the 2008 Plan. As of the Effective Date, no additional awards will be granted under the 2008 Plan, but all stock awards granted under the 2008 Plan prior to the Effective Date will remain subject to the terms of the 2008 Plan. Any shares associated with stock awards previously granted under the 2008 Plan that are forfeited subsequent to the Effective Date of the 2016 Plan are not eligible for future issuance under the 2016 Plan. All awards granted on and after the Effective Date will be subject to the terms of the 2016 Plan. The 2016 Plan provides for the grant of the following awards: (i) incentive stock options, (ii) nonstatutory stock options, (iii) SARs, (iv) restricted stock awards, (v) restricted stock unit awards and (vi) other stock awards. Eligible plan participants include employees, directors, and consultants. An aggregate of 833,333 shares of the Company’s common stock were initially available for issuance under awards granted pursuant to the 2016 Plan, which shares may be authorized but unissued shares, treasury shares, or shares purchased in the open market.
141
On June 5, 2017, the Company’s stockholders approved an amendment to the 2016 Plan to increase the aggregate number of shares of common stock that may be issued pursuant to awards under the 2016 Plan by an additional 1,200,000 shares. All other material terms of the 2016 Plan otherwise remained unchanged.
On July 31, 2019, the Company’s stockholders approved an amendment to the 2016 Plan (“the 2016 Plan Amendment”), to increase the number of shares reserved under the 2016 Plan by 1,000,000 and to increase the award limit on the maximum aggregate number of shares of the Company’s common stock that may be granted to any one person during any calendar year from 250,000 to 1,000,000 shares of the Company’s common stock. All other material terms of the 2016 Plan otherwise remain unchanged.
As of December 31, 2019, there were 388,463 shares available for future issuance under the 2016 Plan.
Under both the 2008 Plan and the 2016 Plan, options to purchase the Company’s common stock may be granted at a price no less than the fair value of a common stock share on the date of grant. The fair value shall be the closing sales price for a share as quoted on any established securities exchange for such grant date or the last preceding date for which such quotation exists. Vesting terms of options issued are determined by the board of directors or compensation committee of the board. The Company’s stock options vest based on terms in the stock option agreements and have a maximum term of ten years.
Stock Appreciation Rights
On August 8, 2018, the Company entered into an employment agreement with G. Kelly Martin (the “Martin Employment Agreement”). The Martin Employment Agreement provided for 1,000,000 SARs (the “Martin SAR Award”) granted on a contingent basis that would have been irrevocably forfeited and voided in full if the Company had failed to obtain stockholder approval for the 2016 Plan Amendment. If such approval had not been obtained, the Company would have been required to pay Mr. Martin the cash equivalent of the value of the SARs. Following stockholder approval of the 2016 Plan Amendment, the SARs detailed within the Martin Employment Agreement were no longer considered to be granted on a contingent basis.
The SARs entitle Mr. Martin to a payment (in cash, shares of common stock or a combination of both) equal to the fair market value of one share of the Company’s common stock on the date of exercise less the exercise price of $3.80 per share. The SARs will be deemed automatically exercised and settled as of February 1, 2020, provided Mr. Martin remains continuously employed with the Company through such date unless vesting is otherwise expressly accelerated pursuant to the Martin SAR Award.
Due to the contingent nature of the SAR grant, prior to stockholder approval on July 31, 2019, these share-based payment awards were classified as liabilities and the amount of compensation cost recognized was based on the fair value of those liabilities. The corresponding obligation was recorded within other long-term liabilities on the Company’s consolidated balance sheet at the estimated fair value on the date of issuance and was re-valued each subsequent reporting period with adjustments to the fair value recognized as share-based compensation expense in the consolidated statements of operations.
As the Company has the sole discretion to settle any awards with cash, common stock or a combination of both, subsequent to stockholder approval as of July 31, 2019, the SARs were reclassified from liability to equity-based awards at fair value and reclassified to additional paid-in capital. The fair value of the SARs was estimated using the Black-Scholes option pricing model on the July 31, 2019 remeasurement date. The reclassification from other long-term liabilities to additional paid-in capital in the consolidated balance sheet was $366 as of July 31, 2019.
The fair value of the SARs was estimated using the Black-Scholes option-pricing model for the respective periods, using the following assumptions:
July 31, 2019 | December 31, 2018 | ||||||
Estimated dividend yield | — | — | |||||
Expected volatility | 115.82 | % | 86.71 | % | |||
Risk-free interest rate | 2.07 | % | 2.63 | % | |||
Expected term (years) | 0.51 | 1.09 | |||||
Fair value per share of common stock underlying the SAR | $ | 2.66 | $ | 0.83 | |||
SAR exercise price | $ | 3.80 | $ | 3.80 | |||
142
During the years ended December 31, 2019 and 2018, the Company recorded employee share-based compensation expense related to the SARs of $507 and $8, respectively. As of December 31, 2019, total unrecognized compensation expense related to non-vested SARs was $33, which is expected to be fully recognized by the February 1, 2020 vesting date.
As described in Note 16—Subsequent Events, on February 1, 2020, the fair market value of our common stock was $0.52 per share. As a result, the SARs expired unexercised and 1,000,000 shares became available for grant under the 2016 Plan.
Inducement Grants
During the years ended December 31, 2019 and 2018, the Company awarded nonstatutory stock options to purchase shares of common stock to newly-hired employees, not previously employees or directors of the Company, as inducements material to the individuals’ entering into employment with the Company within the meaning of Nasdaq Listing Rule 5635(c)(4) (the “Inducement Grants”). On May 31, 2018, the Company awarded 100,500 Inducement Grants with an exercise price of $3.15 per share, and on September 6, 2019, the Company awarded 25,000 Inducement Grants with an exercise price of $2.62 per share. The Inducement Grants were awarded outside of the Company’s 2016 Plan, pursuant to Nasdaq Listing Rule 5635(c)(4), but have terms and conditions generally consistent with the Company’s 2016 Plan and vest over three years, subject to the employee’s continued service as an employee or consultant through the vesting period.
Stock Compensation Expense
During the years ended December 31, 2019 and 2018, the Company recorded employee share-based compensation expense of $1,838 and $2,204, respectively. Total share-based compensation expense included in the consolidated statements of operations is as follows:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
Research and development | $ | 710 | $ | 1,144 | |||
General and administrative | 1,128 | 1,060 | |||||
$ | 1,838 | $ | 2,204 | ||||
The fair value of each option grant is estimated on the grant date using the Black-Scholes option-pricing model, and the following weighted average assumptions:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
Estimated dividend yield | 0.00 | % | 0.00 | % | |||
Expected volatility | 102.14 | % | 81.73 | % | |||
Risk-free interest rate | 1.87 | % | 2.75 | % | |||
Expected life of options (in years) | 5.20 | 5.68 | |||||
Weighted-average fair value per share | $ | 1.77 | $ | 2.06 | |||
143
Stock option activity for the periods indicated is as follows:
Shares Available for Grant | Shares Subject to Outstanding Options | Weighted- Average Exercise Price Per Share | Weighted- Average Remaining Contractual Term (in years) | Aggregate Intrinsic Value | |||||||||||
Options outstanding as of December 31, 2017 | 1,023,378 | 1,399,484 | $ | 7.17 | |||||||||||
Additional shares reserved under plan | — | — | |||||||||||||
Options granted | (626,757 | ) | 727,257 | 2.97 | |||||||||||
Options forfeited | 302,755 | (403,748 | ) | 7.61 | |||||||||||
Options exercised | — | (51,327 | ) | 1.16 | |||||||||||
Options outstanding as of December 31, 2018 | 699,376 | 1,671,666 | $ | 5.42 | |||||||||||
Options granted | (633,030 | ) | 658,030 | 2.36 | |||||||||||
Options forfeited | 322,117 | (507,950 | ) | 7.07 | |||||||||||
Options exercised | — | (32,443 | ) | 2.12 | |||||||||||
Options outstanding as of December 31, 2019 | 388,463 | 1,789,303 | $ | 3.89 | 8.03 | $ | 638 | ||||||||
Vested and expected to vest as of December 31, 2018 | 1,585,689 | $ | 5.53 | 8.11 | $ | 2 | |||||||||
Exercisable as of December 31, 2018 | 1,007,870 | $ | 6.58 | 7.54 | $ | 2 | |||||||||
Vested and expected to vest as of December 31, 2019 | 1,731,437 | $ | 3.94 | 7.99 | $ | 597 | |||||||||
Exercisable as of December 31, 2019 | 1,032,801 | $ | 4.81 | 7.20 | $ | 190 | |||||||||
The total intrinsic value of options exercised during the years ended December 31, 2019 and 2018 was $15 and $95, respectively.
As of December 31, 2019 and 2018, total unrecognized compensation expense related to non-vested stock options was $965 and $1,036, respectively, which is expected to be recognized over a weighted average period of 0.99 and 1.79 years, respectively.
Note 12: Tangible Stockholder Return Plan
Performance Plan
On August 2, 2018, the Company’s board of directors approved and established the Tangible Stockholder Return Plan, which is a performance-based long-term incentive plan (the “Performance Plan”). The Performance Plan was effective immediately upon approval and expires on March 1, 2022. The Performance Plan covers all employees, including the Company’s executive officers, consultants and other persons deemed eligible by the Company’s compensation committee. The core underlying metric of the Performance Plan is the achievement of two share price goals for the Company’s common stock, which if achieved, would represent measurable increases in stockholder value.
The Performance Plan is tiered, with two separate tranches, each of which has a distinct share price target (measured as the average publicly traded share price of the Company’s common stock on the Nasdaq stock exchange for a 30 consecutive trading day period) that will, if achieved, trigger a distinct fixed bonus pool. The share price target for the first tranche and related bonus pool are $11.17 per share and $25,000, respectively. The share price target for the second tranche and related bonus pool are $25.45 per share and $50,000, respectively. The compensation committee has discretion to distribute the bonus pool related to each tranche among eligible participants by establishing individual minimum bonus amounts before, as well as by distributing the remainder of the applicable pool after, the achievement of each tranche specific share price target. Otherwise, if the Company does not achieve one or both related share price targets, as defined, no portion of the bonus pools will be paid.
The Performance Plan provides for the distinct fixed bonus pools to be paid in the form of cash. However, the compensation committee has discretion to pay any bonus due under the Performance Plan in the form of cash, shares of the Company’s common stock or a combination thereof, provided that the Company’s stockholders have approved the reservation of shares of the Company’s common stock for such payment.
144
The Performance Plan permits the compensation committee to make bonus awards subject to varying payment terms, including awards that vest and are payable immediately upon achieving an applicable share price target as well as awards that pay over an extended period (either with or without ongoing employment requirements). The Performance Plan contemplates that no bonus award payments will be delayed beyond 24 months for named executive officers or more than 12 months for all other participants.
For purposes of determining whether a share price target has been met, the share price targets will be adjusted in the event of any stock splits, cash dividends, stock dividends, combinations, reorganizations, reclassifications or similar events. In the event of a change in control, as defined in the Performance Plan, during the term of the Performance Plan, a performance bonus pool will become due and payable to participants on a pro rata basis, as calculated and determined by the compensation committee based upon the Company’s progress toward the share price target as of the date of the change in control and subject to adjustment by the compensation committee as permitted under the Performance Plan.
The Company has concluded that the Performance Plan is within the scope of ASC 718, Compensation—Stock Compensation as the underlying plan obligations are based on the potential attainment of certain market share price targets of the Company’s common stock. Any awards under the Performance Plan would be payable, at the discretion of the Company’s compensation committee following the achievement of the applicable share price target, in cash, shares of the Company’s common stock, or a combination thereof, provided that, prior to any payment in common stock, the Company’s stockholders have approved the reservation of shares of the Company’s common stock for such payment.
ASC 718 requires that a liability-based award should be classified as a liability on the Company’s consolidated balance sheets and the amount of compensation cost recognized should be based on the fair value of the liability. When a liability-based award includes both a service and market condition, the market condition is taken into account when determining the appropriate method to estimate fair value and the compensation cost is amortized over the estimated service period. Therefore, the liability associated with the Performance Plan obligation is recorded within other long-term liabilities on the Company’s consolidated balance sheets at the estimated fair value on the date of issuance and is re-valued each subsequent reporting period end. The Company recognizes share-based compensation expense within operating expenses in the consolidated statements of operations, including adjustments to the fair value of the liability-based award, on a straight-line basis over the requisite service period.
The fair value of obligations under the Performance Plan are estimated using a Monte Carlo simulation approach. The Company’s common stock price is simulated under the Geometric Brownian Motion framework under each simulation path. The other assumptions for the Monte Carlo simulation include the risk-free interest rate, estimated volatility and the expected term. Expected stock price volatility is based on the actual historical volatility of a group of comparable publicly traded companies observed over a historical period equal to the expected remaining life of the plan. The fair value of the underlying common stock is the published closing market price on the Nasdaq Global Market as of each reporting date, as adjusted for significant results, as necessary. The risk-free interest rate is based on the U.S. Treasury yield curve in effect on the date of valuation equal to the remaining expected life of the plan. The dividend yield percentage is zero because the Company does not currently pay dividends, nor does it intend to do so during the expected term of the plan. The expected life of bonus awards under the Performance Plan is assumed to be equivalent to the remaining contractual term based on the estimated service period including the service inception date of the plan participants and the contractual end of the Performance Plan.
The fair value of the Performance Plan is estimated at each financial reporting date using the Monte Carlo simulation model and the following assumptions:
December 31, | |||||||
2019 | 2018 | ||||||
Estimated dividend yield | — | — | |||||
Expected volatility | 128.30 | % | 87.19 | % | |||
Risk-free interest rate | 1.53 | % | 2.47 | % | |||
Expected term (years) | 2.17 | 3.17 | |||||
Fair value per share of common stock underlying the Performance Plan | $ | 0.86 | $ | 0.83 | |||
During the years ended December 31, 2019 and 2018, the Company recorded employee share-based compensation expense related to the Performance Plan of $291 and $57, respectively.
145
Note 13: Income Taxes
There was no income tax benefit recognized for the years ended December 31, 2019 and 2018 due to the Company’s history of net losses combined with an inability to confirm recovery of the tax benefits from the Company’s losses and other net deferred tax assets. The Company has established a valuation allowance against its deferred tax assets due to the uncertainty surrounding the realization of such assets.
The reasons for the difference between actual income tax benefit for the years ended December 31, 2019 and 2018, and the amount computed by applying the statutory federal income tax rate to losses before income tax benefit are as follows:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(Restated) | |||||||
Income tax benefit at federal statutory rate | $ | (6,379 | ) | $ | (6,140 | ) | |
State income taxes, net of federal benefit | (582 | ) | (570 | ) | |||
Non-deductible expenses | 193 | 154 | |||||
Federal rate impact | — | — | |||||
Research and development tax credits | (1,225 | ) | (1,254 | ) | |||
Other | 330 | 380 | |||||
Change in valuation allowance | 7,663 | 7,430 | |||||
Total income tax provision | $ | — | $ | — | |||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and deferred tax liabilities are as follows:
As of December 31, | |||||||
2019 | 2018 | ||||||
Deferred tax assets: | |||||||
Accrued compensation | $ | 13 | $ | 184 | |||
Accrued liabilities | 235 | 149 | |||||
Tax loss carryforwards | 38,042 | 37,986 | |||||
Intangible assets | 268 | 286 | |||||
Share-based compensation | 666 | 814 | |||||
Tax credits | 8,141 | 6,917 | |||||
Facility financing lease obligation | — | 1,847 | |||||
Research and development service obligation | 6,620 | — | |||||
Right-of-use lease liabilities | 1,436 | — | |||||
Deferred revenue | 572 | 588 | |||||
Other | 54 | 10 | |||||
Total deferred tax assets | 56,047 | 48,781 | |||||
Less valuation allowance | (54,430 | ) | (46,604 | ) | |||
Net deferred tax asset | 1,617 | 2,177 | |||||
Deferred tax liabilities: | |||||||
Fixed assets | (1,014 | ) | (2,032 | ) | |||
Right-of-use lease assets | (421 | ) | — | ||||
Other | (182 | ) | (145 | ) | |||
Net noncurrent deferred tax asset (liability) | $ | — | $ | — | |||
146
In December 2017, the Tax Cuts and Jobs Act, or TCJA, was signed into law. Among other things, the TCJA permanently lowers the corporate federal income tax rate to 21% from the existing maximum rate of 35%, effective for tax years including or commencing January 1, 2018. Based on provisions of the TCJA, the Company remeasured its deferred tax assets and liabilities to reflect the lower statutory tax rate, which resulted in a provision of $18,894 to income tax expense. However, there is no impact to the Company’s effective tax rate because a corresponding and offsetting reduction was made in the valuation allowance. The other provisions of the TCJA did not have a material impact on the consolidated financial statements. The Company’s deferred tax remeasurement was complete and all tax effects of the TCJA were reflected in the Company’s income tax provision for the year ended December 31, 2019.
As of December 31, 2019, the Company had federal and state net operating loss carryforwards of $165,623 and $165,115, respectively. The net operating loss carryforwards begin to expire in 2028 and 2023 for federal and state tax purposes, respectively. As of December 31, 2019, the Company had government research and development tax credits of approximately $8,141 to offset future federal taxes which begin to expire in 2028.
The Company had no unrecognized tax benefits as of December 31, 2019 and 2018. The Company does not anticipate a significant change in total unrecognized tax benefits within the next 12 months. Tax years 2016-2018 remain open to examination by the major taxing jurisdictions to which the Company is subject. Additionally, years prior to 2016 are also open to examination to the extent of loss and credit carryforwards from those years.
The Tax Reform Act of 1986 contains provisions which limit the ability to utilize the net operating loss carryforwards in the case of certain events including significant changes in ownership interests. If the Company’s net operating loss carryforwards are limited, and the Company has taxable income which exceeds the permissible yearly net operating loss carryforwards, the Company would incur a federal income tax liability even though net operating loss carryforwards would be available in future years.
Note 14: Retirement Plan
The Company maintains a defined contribution savings plan under Section 401(k) of the Internal Revenue Code. This plan covers all employees who meet minimum age requirements and allows participants to defer a portion of their annual compensation on a pre-tax basis. The Company has made discretionary matching contributions, up to 3% of gross wages, during 2019 and 2018. The Company contributed $160 and $208, for the years ended December 31, 2019 and 2018, respectively.
Note 15: Related Party Transactions
Members of the Company’s board of directors held 1,002,776 and 782,083 shares of the Company’s common stock as of December 31, 2019 and 2018, respectively.
Malin Corporation
In June 2017, G. Kelly Martin was appointed as the Company’s Interim CEO before being named as the Company’s CEO in April 2018. Mr. Martin continued to serve as a member of the Company’s board of directors following his appointment as Interim CEO and continued to serve as chief executive officer of Malin Corporation plc (“Malin”) until October 1, 2017. Malin is the parent company of Malin Life Sciences Holdings Limited, which beneficially owns approximately 10% of the Company’s outstanding common stock.
See Note 16—Subsequent Events regarding the Company’s CEO transition in February 2020.
Two of the Company’s directors during 2018 were also affiliated with Malin. Sean Murphy, who resigned from the Company’s board in September 2018, was an executive officer and a director of Malin, and an executive vice president of Malin Corporation plc. In addition, Robert A. Ingram, the Company’s executive chairman of the board, was also a director of Malin Corporation plc until July 2018.
Cilatus BioPharma
In August 2019, Malin completed the sale of its former subsidiary, Cilatus BioPharma AG (“Cilatus”). Prior to this disposition, Cilatus was majority-owned by Malin. During the nine months ended September 30, 2019 and the year ended and December 31, 2018, respectively, the Company incurred costs of $250 and $601 in relation to a development and manufacturing consulting agreement with Cilatus. These costs were expensed as incurred and are classified as research and development expenses in the accompanying consolidated statements of operations and comprehensive loss.
147
Health Decisions
On October 25, 2018, the Company announced the formation of a dedicated women’s health business unit as well as a foundational collaboration with Health Decisions, Inc. (“Health Decisions”). Health Decisions is a full-service contract research organization specializing in clinical studies of therapeutics for women’s health indications. The Company’s women’s health business unit is led by Paula Brown Stafford, who also is a shareholder and serves on the board of directors of Health Decisions.
Reedy Creek
Reedy Creek beneficially owns approximately 15% of the Company’s outstanding common stock and approximately 3.9 million warrants, all of which was acquired during the January 2018 Offering. Accordingly, Reedy Creek is a related party of the Company. The purchase agreement with Reedy Creek, described in Note 7—Research and Development Arrangements, was evaluated and approved pursuant to the Company’s existing related party transactions policy.
Note 16: Subsequent Events
Reedy Creek
In accordance with the Purchase Agreement with Reedy Creek, Reedy Creek was to provide $10,000 of additional funding contingent upon the Company’s achievement of the primary endpoints in each of the two SB206 clinical trials no later than March 31, 2020. On January 2, 2020, the Company announced top-line results from two pivotal Phase 3 clinical trials of SB206 for the treatment of molluscum contagiosum. SB206 did not achieve statistically significant results in the primary endpoint in both trials, which was the complete clearance of all molluscum lesions at Week 12. Based on such results, the Company understands that Reedy Creek will not be providing the Company with the contingent $10,000 of additional funding.
Chief Executive Officer Transition
In December 2019 the Company announced that Paula Brown Stafford, the Company’s President and Chief Operating Officer, would succeed G. Kelly Martin as CEO, effective February 2, 2020. Mr. Martin had a fixed term employment contract that expired on February 1, 2020, and completed his service as CEO after fulfilling his term. Mr. Martin also stepped off the Board of Directors, effective February 3, 2020. Ms. Stafford remains a member of the Board of Directors.
Board of Directors
On January 29, 2020, Dr. Eugene Sun, one of the members of the Company’s board of directors, notified the Company of his resignation from the board and any committees thereof, effective January 29, 2020.
Stock Appreciation Rights
As described in Note 11—Share-Based Compensation, on August 8, 2018, the Company entered into the Martin Employment Agreement with G. Kelly Martin. The Martin Employment Agreement provided for 1,000,000 SARs and entitled Mr. Martin to a payment (in cash, shares of common stock or a combination of both) equal to the fair market value of one share of the Company’s common stock on the date of exercise less the exercise price of $3.80 per share. The SARs were to be deemed automatically exercised and settled as of February 1, 2020, provided Mr. Martin remained continuously employed with the Company through such date unless vesting was otherwise expressly accelerated pursuant to the Martin SAR Award. The SARs vested in full on February 1, 2020. On February 1, 2020, the fair market value of our common stock was $0.52 per share, and as such, the SARs expired unexercised and 1,000,000 shares became available to be granted under the 2016 Plan.
Effective December 17, 2019, the Company entered into an amended and restated employment agreement with Paula Brown Stafford, or the Amended and Restated Stafford Employment Agreement. On January 6, 2020, following the release of top-line results of the Company’s Phase 3 molluscum clinical program as provided in the Amended and Restated Stafford Employment Agreement, 600,000 SARs were granted to Ms. Stafford with an exercise price of $0.82 per share (the fair market value of the Company’s common stock on the grant date) and with a ten year term (the “Stafford SAR Award”). The Stafford SAR Award was granted on a contingent basis and would have been considered irrevocably forfeited and voided in full if sufficient shares of the Company’s common stock were not available under the 2016 Plan or if the Company failed to obtain stockholder approval for amendments to the 2016 Plan at the next annual stockholders’ meeting to provide sufficient shares for the Stafford SAR Award. Such shares became available under the 2016 Plan on February 1, 2020, and the SARs were no longer considered granted on a contingent basis.
Workforce Reduction
148
As part of a strategic objective to reduce the Company’s costs related to internal resources, facilities, and infrastructure capabilities, the Company took actions in February 2020 that are intended to reduce the Company’s internal resources from a total of 42 employees as of December 31, 2019 to an expected total of 28 employees as of April 1, 2020.
Equity Compensation Grant
On February 13, 2020, the Company issued 383,000 stock options from the 2016 Plan, (the “Retention Grants”). These Retention Grants were issued to certain employees, vest quarterly and will be fully vested on December 31, 2020, provided that the grantee remains an employee or consultant to the Company as of each vesting date.
Women’s Health Business Unit
In February 2020, following the successful progression of the Company’s Phase 1 WH602 program, the Company was awarded a Phase 2 federal grant of approximately $1.0 million from the NIH that will enable the conduct of IND-enabling toxicology and pharmacology studies and other preclinical activity with respect to WH602. These funds will be received by the Company in the form of periodic cost reimbursements as the underlying research and development activities are performed. The Company may be eligible to receive an additional $0.5 million in funding as part of this Phase 2 grant, subject to availability of NIH funds and satisfactory progress of the project during the initial 12-month term.
Continued Listing Standard
On February 19, 2020, the Company received notice from the staff of the Nasdaq Stock Market LLC (“Nasdaq”) notifying the Company that, for the last 30 consecutive business days, the market value of the Company’s listed securities has been below the minimum $50.0 million requirement for continued inclusion on The Nasdaq Global Market pursuant to Nasdaq Listing Rule 5450(b)(2)(A). The staff also noted that the Company did not meet alternative requirements for satisfying continued listing criteria found in Nasdaq Listing Rule 5450(b)(3)(A). The Company has 180 calendar days, or until August 16, 2020, to regain compliance with the market value of listed securities requirement. If, at any time before August 16, 2020, the market value of the Company’s listed securities closes at $50.0 million or more for a minimum of 10 consecutive business days, Nasdaq will provide written notification to the Company that it complies with the market value of listed securities requirement.
On February 19, 2020, the Company also received notice from the staff of Nasdaq notifying the Company that, for the last 30 consecutive business days, the minimum bid price of the Company’s common stock has not exceeded $1.00 per share and that the Company was therefore not in compliance with the minimum bid price requirement under Nasdaq Listing Rule 5450(a)(1). The Company has 180 calendar days, or until August 16, 2020, to regain compliance with the minimum bid price requirement. To regain compliance, the closing bid price of the common stock must meet or exceed $1.00 per share for a minimum of 10 consecutive business days prior to August 16, 2020. The Company intends to continue to monitor the bid price levels for the common stock, and will consider appropriate alternatives to achieve compliance within the 180-day compliance period (or such subsequent date if the compliance period is extended).
149
Note 17: Quarterly Financial Information (Unaudited)
The following tables contain unaudited consolidated quarterly financial information for 2019 and 2018 that has been updated to reflect the restatement and revision of the Company’s consolidated financial statements as described in Note 2—Restatement of Consolidated Financial Statements. The restatement and revision had no impact on net cash flows from operating, investing or financing activities. The Company has not amended its previously filed Annual Report on Form 10-K for the year ended December 31, 2018 or its Quarterly Reports on Form 10-Q for the Affected Periods. The financial information that has been previously filed or otherwise reported for the Affected Periods is superseded by the information in this Annual Report, and the financial statements and related financial information for the Affected Periods contained in such previously filed reports should no longer be relied upon.
Condensed Consolidated Statements of Operations
Three Months Ended | Three Months Ended | Six Months Ended | Three Months Ended | Nine Months Ended | Three Months Ended | ||||||||||||||||||
March 31, 2019 | June 30, 2019 | June 30, 2019 | September 30, 2019 | September 30, 2019 | December 31, 2019 | ||||||||||||||||||
(Restated) | (Restated) | (Restated) | (Restated) | (Restated) | |||||||||||||||||||
License and collaboration revenue | $ | 1,100 | $ | 1,101 | $ | 2,201 | $ | 1,100 | $ | 3,301 | $ | 1,176 | |||||||||||
Government research contracts and grants revenue | — | — | — | 216 | 216 | 203 | |||||||||||||||||
Total revenue | 1,100 | 1,101 | 2,201 | 1,316 | 3,517 | 1,379 | |||||||||||||||||
Operating expenses: | |||||||||||||||||||||||
Research and development | 4,827 | 6,189 | 11,016 | 8,598 | 19,614 | 5,558 | |||||||||||||||||
General and administrative | 2,994 | 3,311 | 6,305 | 2,290 | 8,595 | 1,817 | |||||||||||||||||
Total operating expenses | 7,821 | 9,500 | 17,321 | 10,888 | 28,209 | 7,375 | |||||||||||||||||
Operating loss | (6,721 | ) | (8,399 | ) | (15,120 | ) | (9,572 | ) | (24,692 | ) | (5,996 | ) | |||||||||||
Other income (expense), net: | |||||||||||||||||||||||
Interest income | 28 | 68 | 96 | 53 | 149 | 28 | |||||||||||||||||
Interest expense | — | (1 | ) | (1 | ) | — | (1 | ) | (1 | ) | |||||||||||||
Other income, net | 56 | 36 | 92 | 23 | 115 | 21 | |||||||||||||||||
Total other income (expense), net | 84 | 103 | 187 | 76 | 263 | 48 | |||||||||||||||||
Net loss and comprehensive loss | $ | (6,637 | ) | $ | (8,296 | ) | $ | (14,933 | ) | $ | (9,496 | ) | $ | (24,429 | ) | $ | (5,948 | ) | |||||
Net loss per share, basic and diluted | $ | (0.25 | ) | $ | (0.32 | ) | $ | (0.57 | ) | $ | (0.36 | ) | $ | (0.94 | ) | $ | (0.22 | ) | |||||
Weighted-average common shares outstanding, basic and diluted | 26,066,064 | 26,069,734 | 26,067,909 | 26,189,454 | 26,108,870 | 26,685,133 | |||||||||||||||||
150
Three Months Ended | Three Months Ended | Six Months Ended | Three Months Ended | Nine Months Ended | Three Months Ended | ||||||||||||||||||
March 31, 2018 | June 30, 2018 | June 30, 2018 | September 30, 2018 | September 30, 2018 | December 31, 2018 | ||||||||||||||||||
(Restated) | (Restated) | (Restated) | (Restated) | (Restated) | |||||||||||||||||||
License and collaboration revenue | $ | 649 | $ | 649 | $ | 1,298 | $ | 648 | $ | 1,946 | $ | 4,036 | |||||||||||
Research and development services revenue | 9 | — | 9 | — | 9 | — | |||||||||||||||||
Total revenue | 658 | 649 | 1,307 | 648 | 1,955 | 4,036 | |||||||||||||||||
Operating expenses: | |||||||||||||||||||||||
Research and development | 6,335 | 6,176 | 12,511 | 5,697 | 18,208 | 4,837 | |||||||||||||||||
General and administrative | 2,880 | 2,620 | 5,500 | 3,295 | 8,795 | 2,712 | |||||||||||||||||
Total operating expenses | 9,215 | 8,796 | 18,011 | 8,992 | 27,003 | 7,549 | |||||||||||||||||
Operating loss | (8,557 | ) | (8,147 | ) | (16,704 | ) | (8,344 | ) | (25,048 | ) | (3,513 | ) | |||||||||||
Other income (expense), net: | |||||||||||||||||||||||
Interest income | 44 | 115 | 159 | 83 | 242 | 55 | |||||||||||||||||
Interest expense | (262 | ) | (261 | ) | (523 | ) | (262 | ) | (785 | ) | (262 | ) | |||||||||||
Other income, net | — | 4 | 4 | 28 | 32 | 40 | |||||||||||||||||
Total other income (expense), net | (218 | ) | (142 | ) | (360 | ) | (151 | ) | (511 | ) | (167 | ) | |||||||||||
Net loss and comprehensive loss | $ | (8,775 | ) | $ | (8,289 | ) | $ | (17,064 | ) | $ | (8,495 | ) | $ | (25,559 | ) | $ | (3,680 | ) | |||||
Net loss per share, basic and diluted | $ | (0.35 | ) | $ | (0.32 | ) | $ | (0.67 | ) | $ | (0.33 | ) | $ | (0.99 | ) | $ | (0.14 | ) | |||||
Weighted-average common shares outstanding, basic and diluted | 25,026,890 | 26,039,169 | 25,535,827 | 26,046,666 | 25,707,978 | 26,056,504 | |||||||||||||||||
151
Condensed Consolidated Balance Sheets
March 31, 2019 | June 30, 2019 | September 30, 2019 | December 31, 2019 | ||||||||||||
(Restated) | (Restated) | (Restated) | |||||||||||||
ASSETS | |||||||||||||||
Current assets: | |||||||||||||||
Cash and cash equivalents | $ | 6,077 | $ | 22,438 | $ | 15,549 | $ | 13,711 | |||||||
Restricted cash | — | 8,286 | 5,651 | — | |||||||||||
Contracts and grants receivable | — | — | 216 | 419 | |||||||||||
Deferred offering costs | 49 | 49 | 49 | 49 | |||||||||||
Prepaid expenses and other current assets | 1,062 | 1,237 | 982 | 1,545 | |||||||||||
Total current assets | 7,188 | 32,010 | 22,447 | 15,724 | |||||||||||
Restricted cash | 539 | 2,060 | 1,345 | 540 | |||||||||||
Intangible assets | 75 | 75 | 75 | 75 | |||||||||||
Other assets | 501 | 473 | 444 | 419 | |||||||||||
Property and equipment, net | 11,657 | 11,054 | 10,569 | 10,506 | |||||||||||
Right-of-use lease assets | 1,833 | 1,837 | 1,837 | 1,833 | |||||||||||
Total assets | $ | 21,793 | $ | 47,509 | $ | 36,717 | $ | 29,097 | |||||||
LIABILITIES AND STOCKHOLDERS’ (DEFICIT) EQUITY | |||||||||||||||
Current liabilities: | |||||||||||||||
Accounts payable | $ | 1,510 | $ | 957 | $ | 2,495 | $ | 1,602 | |||||||
Accrued compensation | 2,082 | 1,412 | 1,507 | 437 | |||||||||||
Accrued outside research and development services | 817 | 789 | 1,382 | 1,013 | |||||||||||
Accrued legal and professional fees | 258 | 295 | 168 | 616 | |||||||||||
Other accrued expenses | 516 | 633 | 878 | 553 | |||||||||||
Deferred revenue, current portion | 4,401 | 4,401 | 4,401 | 4,428 | |||||||||||
Research and development service obligation liability, current portion | — | 8,286 | 5,651 | 3,088 | |||||||||||
Lease liabilities, current portion | 1,139 | 1,151 | 1,156 | 1,162 | |||||||||||
Total current liabilities | 10,723 | 17,924 | 17,638 | 12,899 | |||||||||||
Deferred revenue, net of current portion | 5,926 | 4,825 | 3,725 | 7,076 | |||||||||||
Lease liabilities, net of current portion | 5,544 | 5,395 | 5,249 | 5,100 | |||||||||||
Research and development service obligation liability, net of current portion | — | 1,521 | 806 | 727 | |||||||||||
Research and development funding arrangement liability, related party | — | 25,000 | 25,000 | 25,000 | |||||||||||
Other long-term liabilities | 335 | 1,630 | 1,634 | 578 | |||||||||||
Total liabilities | 22,528 | 56,295 | 54,052 | 51,380 | |||||||||||
Commitments and contingencies | |||||||||||||||
Stockholders’ equity (deficit) | |||||||||||||||
Common stock $0.0001 par value | 3 | 3 | 3 | 3 | |||||||||||
Additional paid-in-capital | 195,661 | 195,906 | 196,853 | 197,853 | |||||||||||
Treasury stock at cost | (155 | ) | (155 | ) | (155 | ) | (155 | ) | |||||||
Accumulated deficit | (196,244 | ) | (204,540 | ) | (214,036 | ) | (219,984 | ) | |||||||
Total stockholders' (deficit) equity | (735 | ) | (8,786 | ) | (17,335 | ) | (22,283 | ) | |||||||
Total liabilities and stockholders’ (deficit) equity | $ | 21,793 | $ | 47,509 | $ | 36,717 | $ | 29,097 | |||||||
152
March 31, 2018 | June 30, 2018 | September 30, 2018 | December 31, 2018 | ||||||||||||
(Restated) | (Restated) | (Restated) | (Restated) | ||||||||||||
ASSETS | |||||||||||||||
Current assets: | |||||||||||||||
Cash and cash equivalents | $ | 28,100 | $ | 20,984 | $ | 12,173 | $ | 8,194 | |||||||
Deferred offering costs | 49 | 49 | 49 | 49 | |||||||||||
Prepaid expenses and other current assets | 889 | 576 | 584 | 1,107 | |||||||||||
Total current assets | 29,038 | 21,609 | 12,806 | 9,350 | |||||||||||
Restricted cash | 539 | 539 | 539 | 539 | |||||||||||
Intangible assets | 75 | 75 | 75 | 75 | |||||||||||
Other assets | 176 | 161 | 146 | 530 | |||||||||||
Property and equipment, net | 16,474 | 16,327 | 16,129 | 15,868 | |||||||||||
Total assets | $ | 46,302 | $ | 38,711 | $ | 29,695 | $ | 26,362 | |||||||
LIABILITIES AND STOCKHOLDERS’ (DEFICIT) EQUITY | |||||||||||||||
Current liabilities: | |||||||||||||||
Accounts payable | $ | 828 | $ | 1,056 | $ | 386 | $ | 1,250 | |||||||
Accrued compensation | 977 | 1,380 | 1,689 | 1,467 | |||||||||||
Accrued outside research and development services | 1,258 | 1,408 | 1,571 | 563 | |||||||||||
Accrued legal and professional fees | 458 | 267 | 305 | 498 | |||||||||||
Other accrued expenses | 1,194 | 1,443 | 1,247 | 871 | |||||||||||
Deferred revenue, current portion | 2,638 | 2,595 | 2,595 | 4,401 | |||||||||||
Capital lease obligation, net of current portion | 11 | 11 | 11 | 11 | |||||||||||
Total current liabilities | 7,364 | 8,160 | 7,804 | 9,061 | |||||||||||
Deferred revenue, net of current portion | 5,294 | 4,648 | 4,000 | 2,566 | |||||||||||
Capital lease obligation, net of current portion | 19 | 16 | 13 | 10 | |||||||||||
Other long-term liabilities | — | — | 103 | 289 | |||||||||||
Facility financing obligation | 7,998 | 7,998 | 7,998 | 7,998 | |||||||||||
Total liabilities | 20,675 | 20,822 | 19,918 | 19,924 | |||||||||||
Commitments and contingencies | |||||||||||||||
Stockholders’ equity (deficit) | |||||||||||||||
Common stock $0.0001 par value | 3 | 3 | 3 | 3 | |||||||||||
Additional paid-in-capital | 194,208 | 194,759 | 195,142 | 195,483 | |||||||||||
Treasury stock at cost | (155 | ) | (155 | ) | (155 | ) | (155 | ) | |||||||
Accumulated deficit | (168,429 | ) | (176,718 | ) | (185,213 | ) | (188,893 | ) | |||||||
Total stockholders' (deficit) equity | 25,627 | 17,889 | 9,777 | 6,438 | |||||||||||
Total liabilities and stockholders’ (deficit) equity | $ | 46,302 | $ | 38,711 | $ | 29,695 | $ | 26,362 | |||||||
153
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
Evaluation of Disclosure Controls and Procedures
The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act refers to controls and procedures that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that such information is accumulated and communicated to a company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure.
In designing and evaluating our disclosure controls and procedures, management recognizes that disclosure controls and procedures, no matter how well conceived and operated, cannot provide absolute assurance that the objectives of the disclosure controls and procedures are met. Additionally, in designing disclosure controls and procedures, our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible disclosure controls and procedures. Our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives. The design of any system of controls also is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a control system, misstatements due to error or fraud may occur and not be detected.
As of December 31, 2019, our management, with the participation of our principal executive and financial officers, has evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934). Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Based upon such evaluation, our principal executive and financial officers concluded in the Original Filing that, as of December 31, 2019, our disclosure controls and procedures were effective at the reasonable assurance level. Subsequent to the evaluation made in connection with the Original Filing and in connection with the restatement and revision of our consolidated financial statements included in this Annual Report, our management has re-evaluated, under the supervision and with the participation of our principal executive and financial officers, the effectiveness of our disclosure controls and procedures. Based upon that re-evaluation, our principal executive and financial officers concluded that because of the material weakness in our internal control over financial reporting discussed below, our disclosure controls and procedures were not effective as of December 31, 2019.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in the Exchange Act Rule 13a-15(f). Our internal control over financial reporting is designed to provide reasonable assurance to our management and board of directors regarding the preparation and fair presentation of published financial statements. A control system, no matter how well designed and operated, can only provide reasonable, not absolute, assurance that the objectives of the control system are met. Because of these inherent limitations, management does not expect that our internal control over financial reporting will prevent all error and all fraud. Management conducted an evaluation of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework issued in 2013 by the Committee of Sponsoring Organizations of the Treadway Commission (the “2013 Framework”). Based on our evaluation under the 2013 Framework, management concluded that our internal control over financial reporting was not effective as of December 31, 2019.
At the time of the Original Filing on February 24, 2020, our principal executive and financial officers concluded that our internal control over financial reporting was effective as of December 31, 2019. Subsequent to the evaluation made in connection with the Original Filing and in connection with the restatement of our consolidated financial statements included in this Annual Report, our management, including our principal executive and financial officers, has re-evaluated the effectiveness of our internal control over financial reporting and concluded that we did not maintain effective internal control over financial reporting as of December 31, 2019 because of a material weakness in our internal control over financial reporting described below related to the accounting for a significant and unusual transaction related to the warrants we issued in connection with
154
the January 2018 Offering. Notwithstanding the material weakness described below, our management has concluded that our restated and revised audited consolidated financial statements included in this Annual Report are fairly stated in all material respects in accordance with U.S. GAAP for each of the periods presented herein.
In connection with the restatement described in “Note 2—Restatement of Consolidated Financial Statements” to the accompanying consolidated financial statements included in this Annual Report, management identified a material weakness in our internal control over financial reporting related to the accounting for a significant and unusual transaction related to the warrants we issued in connection with the January 2018 Offering. This material weakness resulted in a material misstatement of our warrant liability, change in fair value of warrant liability, additional paid-in capital and accumulated deficit for the Affected Periods.
To respond to this material weakness, we have devoted, and plan to continue to devote, significant effort and resources to the remediation and improvement of our internal control over financial reporting. While we have processes to identify and appropriately apply applicable accounting requirements, we plan to enhance these processes to better evaluate our research and understanding of the nuances of the complex accounting standards that apply to our consolidated financial statements. Our plans at this time include providing enhanced access to accounting literature, research materials and documents and increased communication among our personnel and third party professionals with whom we consult regarding complex accounting applications. The elements of our remediation plan can only be accomplished over time, and we can offer no assurance that these initiatives will ultimately have the intended effects.
This Annual Report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting due to a transition period established by the rules of the SEC for newly public companies. We are an “emerging growth company” as defined in the JOBS Act. For as long as we remain an “emerging growth company,” we are exempt from the auditor attestation requirement in the assessment of the effectiveness of our internal control over financial reporting.
Restatement of Previously Issued Financial Statements
On May 14, 2020, we revised our prior position on accounting for warrants and concluded that our previously issued consolidated financial statements for the Affected Periods should not be relied on because of a misapplication in the guidance on warrant accounting. However, the non-cash adjustments to the financial statements, in all of the Affected Periods, do not impact the amounts previously reported for our consolidated cash and cash equivalents, total assets, revenue or cash flows.
Changes in Internal Control over Financial Reporting
There were no changes in the Company’s internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) during the fourth quarter ended December 31, 2019 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
Compliance with the Nasdaq Market Value of Listed Shares Requirement
On February 19, 2020, we received notice from the staff of the Nasdaq Stock Market LLC, or Nasdaq, notifying us that, for the last 30 consecutive business days, the market value of our listed securities has been below the minimum $50.0 million requirement for continued inclusion on the Nasdaq Global Market pursuant to Nasdaq Listing Rule 5450(b)(2)(A), or the MVLS Requirement. The staff also noted that the Company did not meet alternative requirements for satisfying continued listing criteria found in Nasdaq Listing Rule 5450(b)(3)(A).
We have 180 calendar days, or until August 16, 2020, to regain compliance with the MVLS Requirement. If, at any time before August 16, 2020, the market value of our listed securities closes at $50.0 million or more for a minimum of 10 consecutive business days, Nasdaq will provide written notification to us that we comply with the MVLS Requirement.
If we do not regain compliance with the MVLS Requirement by August 16, 2020, Nasdaq will provide written notification to us that our common stock is subject to delisting. At that time, we may either apply for listing on the Nasdaq Capital Market, provided that we meet the continued listing requirements of that market, or appeal the decision to a Nasdaq Listing Qualifications Panel, or the Panel. In the event of an appeal, our common stock would remain listed on the Nasdaq Global Market pending a decision by the Panel following the hearing. However, there can be no assurance Nasdaq would grant our request for continued listing.
155
Compliance with the Nasdaq Minimum Bid Price Requirement
On February 19, 2020, we also received notice from Nasdaq that as of February 18, 2020, the closing bid price for our common stock on the Nasdaq Global Market was below $1.00 for the last 30 consecutive business days and that we were therefore not in compliance with the minimum bid price requirement under Nasdaq Listing Rule 5450(a)(1), or the Minimum Bid Price Requirement.
In accordance with Nasdaq Listing Rule 5810(c)(3)(A), we have 180 calendar days, or until August 16, 2020, to regain compliance with the Minimum Bid Price Requirement. To regain compliance, the closing bid price of the common stock must meet or exceed $1.00 per share for a minimum of ten consecutive business days prior to August 16, 2020.
If we are unable to regain compliance by August 16, 2020, we may be eligible for an additional 180 calendar day compliance period to demonstrate compliance with the Minimum Bid Price Requirement. To qualify, we will be required to meet the continued listing requirement for market value of listed shares and all other initial listing standards for The Nasdaq Global Market, with the exception of the Minimum Bid Price Requirement, and will need to provide written notice to Nasdaq of our intention to cure the deficiency during the second compliance period. If we do not qualify for the second compliance period or fail to regain compliance during the second 180 calendar day period, Nasdaq will notify us of its determination to delist the Common Stock, at which point we would have an opportunity to appeal the delisting determination to a Panel.
Our common stock will continue to trade on Nasdaq under the symbol NOVN during the 180-day compliance period for both the MVLS Requirement and the Minimum Bid Price Requirement. We are presently evaluating potential actions to regain compliance with all applicable requirements for continued listing on the Nasdaq Global Market or, alternatively, transferring our listing to the Nasdaq Capital Market, if possible. We cannot provide any assurances, however, that we will be able to regain compliance, including, if necessary, transferring to the Nasdaq Capital Market.
156
PART III
Item 10. Directors, Executive Officers and Corporate Governance.
Directors
Our board of directors consists of six directors and is divided into three classes with staggered, three-year terms. The terms of office of directors in Class I will expire at our annual meeting of stockholders to be held in 2020, or the 2020 Annual Meeting, or when such director’s successor is elected and qualified, or upon such director’s death, resignation or removal, and our Class I directors are expected to stand for re-election at the 2020 Annual Meeting. The terms of office of directors in Class II and Class III do not expire until the annual meetings of stockholders to be held in 2021 and 2022, respectively, and until his or her successor is elected and qualified, or until his or her death, resignation or removal.
Information about our directors, their ages as of February 3, 2020, occupations and length of board service are provided in the table below. Additional biographical descriptions are set forth in the text below the tables and include the primary individual experience, qualifications, qualities and skills of each director that led to the conclusion that such director should serve as a member of our board of directors at this time.
Name of Director | Age | Principal Occupation | Director Since | |||
Class I Directors: | ||||||
John Palmour, Ph.D. (1)(3) | 59 | Vice President and Chief Technology Officer, Wolfspeed, a Cree, Inc. company | 2010 | |||
Paula Brown Stafford | 55 | President and Chief Executive Officer, Novan, Inc. | 2017 | |||
Class II Directors: | ||||||
Robert A. Ingram (3) | 77 | General Partner, Hatteras Venture Advisors III, LLC | 2011 | |||
Machelle Sanders (2) | 56 | Secretary of the N.C. Department of Administration | 2017 | |||
Class III Directors: | ||||||
W. Kent Geer (1)(2) | 65 | Managing Director—Finance and Investor Relations, Med1 Ventures, LLC | 2015 | |||
Robert J. Keegan (1)(2) | 72 | Retired Chief Executive Officer, Goodyear Tire and Rubber Co. | 2016 | |||
(1) | Member of our audit committee |
(2) | Member of our compensation committee |
(3) | Member of our nominating and corporate governance committee |
John Palmour has served as a member of our board of directors since 2010. Since 1987, Dr. Palmour has worked at Cree, Inc., a company he co-founded and for which he currently serves as Vice President and the Chief Technology Officer of the Wolfspeed Division. Dr. Palmour served on Cree’s board of directors from 1995 to 2010. He is currently on the board of directors of Goodzer, Inc., a privately held company focused on local services Internet advertising. We believe that Dr. Palmour’s significant experience and leadership in the technology field and the advancement of innovation to broad-scale product commercialization qualifies him to serve on our board of directors.
Paula Brown Stafford is our President and was recently appointed as our Chief Executive Officer effective February 2, 2019, after previously serving as our Chief Operating Officer from January 2019 to December 2019, and after serving as our Chief Development Officer from March 2017 to January 2019. Ms. Stafford has served as a member of our board of directors since August 2017. Prior to joining Novan, Ms. Stafford held various roles of increasing importance at Quintiles Transnational Holdings Inc. (now IQVIA Holdings Inc.), a leading multinational provider of biopharmaceutical development services and commercial outsourcing services, since 1985, including serving as President of Clinical Development from 2010 to 2015, where she was responsible for all Phase I-IV clinical development operations globally and served on the Quintiles Executive Committee. Ms. Stafford also serves as a director of Health Decisions, Inc., a contract research organization for women’s health and diagnostics, serves as an adjunct professor in Public Health Leadership at the Gillings School of Global Public Health at the University of North Carolina, Chapel Hill, and operates her own third-party consulting business. We believe that Ms. Stafford’s extensive experience and leadership in clinical research and pharmaceutical product development, along with her extensive executive experience, qualifies her to serve on our board of directors.
157
Robert A. Ingram is the Executive Chairman of our board of directors and has served as a member of our board of directors since 2011. Since 2007, he has been a general partner at Hatteras Venture Advisors III, LLC, a venture capital firm. Prior to Hatteras Venture Advisors, Mr. Ingram held the roles of Chief Executive Officer and Chairman of the Board of GlaxoWellcome until his retirement in 2009 when he became the Strategic Advisor to the Chief Executive Officer, GlaxoSmithKline Plc. Mr. Ingram retired as Chairman of the board of directors of Cree, Inc., in August 2018, having served on its board of directors for 10 years. Mr. Ingram currently serves as Chairman of the board of directors of BioCryst Pharmaceuticals, Inc., HBM Healthcare Investments and Selenity Pharmaceuticals, Inc. (formerly Viamet Pharmaceuticals Inc.), a private company focused on endocrine and neurologic disorders. We believe that Mr. Ingram’s significant experience and leadership in the pharmaceutical industry qualifies him to serve as chairman of our board of directors.
Machelle Sanders joined our board of directors in September 2017 and is a seasoned executive with over 29 years of progressive pharmaceutical and biotechnology experience. Ms. Sanders is currently serving as the Secretary of the N.C. Department of Administration, appointed by Governor Roy Cooper. In the private sector, Ms. Sanders was most recently responsible for the pharmaceutical operations and technology operational strategy at Biogen, Inc., a multinational biotechnology company, as vice president of quality assurance and vice president of manufacturing and general manager from 2009 to 2015. Ms. Sanders has also held leadership positions in manufacturing, global quality assurance and quality control at Biogen, Inc., Purdue Pharmaceuticals, a pharmaceutical company, and Diosynth-Akzu Nobel, a company that develops and offers manufacturing processes for active ingredients for pharmaceutical companies. We believe that Ms. Sanders’s broad and extensive knowledge of pharmaceutical manufacturing and quality systems and leadership experience qualifies her to serve on our board of directors.
W. Kent Geer has served as a member of our board of directors since 2015 and as our Lead Independent Director since June 2017. Since 2016, Mr. Geer has served as managing director, finance and investor relations for Med1 Ventures, LLC, an early stage medical device development company. Mr. Geer was an audit partner with Ernst & Young LLP from 1989 to 2011. Beginning in 2012, Mr. Geer served as the chairman of the board of directors of PowerSecure International, Inc. until the successful sale of the company in May 2016. Mr. Geer also serves on the board of governors of North Raleigh Christian Academy. We believe that Mr. Geer’s significant experience and leadership in public accounting and the biotechnology, pharmaceutical and technology industries qualifies him to serve on our board of directors.
Robert J. Keegan has served as a member of our board of directors since 2016. Mr. Keegan held the roles of chief executive officer and chairman of the board of directors of Goodyear Tire and Rubber Co. from 2000 to 2010. Most recently, he served as the non-executive chairman of the board of directors of Xerox Corporation and was an operating partner of the San Francisco-based private equity firm Friedman, Fleischer & Lowe. From 1972 to 2000, Mr. Keegan held various marketing, financial and managerial posts at Eastman Kodak, except for a two-year period from 1995 to 1997 when he worked as an executive vice president of the Avery Dennison Corporation. Mr. Keegan serves on the board of directors of the Heart Center of Duke University and the Duke Health Board of Visitors. Mr. Keegan is a partner of L&K Properties of North Carolina, LLC. We believe that Mr. Keegan’s broad business experience, executive leadership expertise and extensive knowledge of financial and operational matters qualifies him to serve on our board of directors.
Executive Officers
Certain information regarding our executive officers is set forth below as of February 3, 2020. Executive officers are appointed by our board of directors to hold office until their successors are duly appointed and qualified, or until their resignation or removal.
Name | Age | Position(s) | ||
Paula Brown Stafford | 55 | President, Chief Executive Officer and Director | ||
John M. Gay | 43 | Vice President, Finance and Corporate Controller | ||
For information regarding Ms. Stafford, please refer to “Directors,” above.
John M. Gay is currently our Vice President, Finance and Corporate Controller and serves as our Principal Financial Officer and Corporate Secretary. He joined Novan in May of 2018 and previously held the position of Senior Director of Finance and Corporate Controller until his promotion to his current role in January 2019. Prior to Novan, Mr. Gay held previous director positions, including Director of SEC Reporting, with Valassis Digital and MaxPoint Inc., from May 2014 to April 2018. Mr. Gay also served as Corporate Controller of Furiex Pharmaceuticals, Inc. from June 2010 to May 2014, including from its initial listing on the Nasdaq stock market through the execution of an acquisition agreement of the company by Actavis plc (Forest Laboratories, Inc.) in an all-cash transaction valued at approximately $1.1 billion. Prior to joining Furiex Pharmaceuticals, Inc., Mr. Gay served as Audit Senior Manager and in other roles of increasing responsibilities at Deloitte and Arthur Andersen from September 2000 to
158
May 2010. Mr. Gay is a certified public accountant and holds Bachelor’s degrees in Economics and History, and a Master of Accounting degree from the University of North Carolina at Chapel Hill.
Audit Committee and Audit Committee Financial Experts
Our board of directors has a standing audit committee, which consists of W. Kent Geer, Robert J. Keegan and John Palmour. The chair of our audit committee is W. Kent Geer, who our board of directors has determined is an “audit committee financial expert,” as that term is defined by the rules of the SEC implementing Section 407 of the Sarbanes-Oxley Act, and possesses financial sophistication, as defined under the listing standards of The Nasdaq Global Market. Our board of directors has also determined that each member of our audit committee can read and understand fundamental financial statements in accordance with applicable SEC and Nasdaq requirements. To arrive at these determinations, our board of directors has examined each audit committee member’s scope of experience and the nature of his experience in the corporate finance sector.
Codes of Conduct
We have adopted a Code of Business Conduct and Ethics that applies to our directors, officers (including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions) and other employees. Our Code of Business Conduct and Ethics is available on the “Corporate Governance” page of the “Investor Relations” section of our website, which may be accessed by navigating to http://investors.novan.com/, by clicking the link under “Corporate Governance” and then by clicking on “Code of Business Conduct and Ethics” under “Governance Documents.” We intend to post on our website and (if required) file on Form 8-K all disclosures that are required by applicable law, the rules of the SEC or the Nasdaq listing standards, concerning any amendment to, or waiver from, our Code of Business Conduct and Ethics. However, the reference to our website does not constitute incorporation by reference of the information contained on or available through our website, and you should not consider it to be a part of this report.
159
Item 11. Executive Compensation.
This section discusses the material components of the executive compensation program with respect to the 2019 fiscal year for the individual who served as our principal executive officer during the year and our two other most highly compensated executive officers who were serving as executive officers as of December 31, 2019. We refer to these persons as our “named executive officers” elsewhere in this report.
Our named executive officers for the 2019 fiscal year were:
• | Paula Brown Stafford, President and Chief Operating Officer (currently our Chief Executive Officer); |
• | John M. Gay, Vice President, Finance and Corporate Controller; and |
• | G. Kelly Martin, Chief Executive Officer (through February 1, 2020) |
Summary Compensation Table
The following table sets forth information concerning the compensation of our named executive officers for the years ended December 31, 2019 and December 31, 2018.
Name and Principal Position | Fiscal Year | Salary ($) | Bonus ($) | Stock Awards ($)(1) | Option Awards ($)(2) | Non-Equity Incentive Plan Compensation ($)(3) | All Other Compensation ($)(4) | Total ($) | ||||||||||||||||||||||
Paula Brown Stafford (5) | 2019 | $ | 443,326 | $ | — | $ | 17,364 | $ | 322,880 | — | $ | 7,155 | $ | 790,725 | ||||||||||||||||
President and Chief Executive Officer; Former Chief Operating Officer | 2018 | 288,000 | 134,400 | (6) | 7,021 | 25,513 | — | — | 454,934 | |||||||||||||||||||||
John M. Gay (7) | 2019 | 245,753 | — | 8,103 | 45,457 | — | 10,059 | 309,372 | ||||||||||||||||||||||
Vice President, Finance and Corporate Controller | ||||||||||||||||||||||||||||||
G. Kelly Martin | 2019 | 480,000 | — | — | — | — | 59,571 | 539,571 | ||||||||||||||||||||||
Former Chief Executive Officer | 2018 | 170,909 | 560,000 | (8) | 105,534 | 593,010 | — | 30,522 | 1,459,975 | |||||||||||||||||||||
(1) | Amounts reflect the grant-date fair value of minimum bonus amounts established by our compensation committee for our named executive officers under our Tangible Stockholder Return Plan, which is a performance-based long-term incentive plan (the “Performance Plan”) that directly ties compensation to the performance of our common stock. Minimum bonus amounts under the Performance Plan are contingent and only become payable if the Company achieves the Performance Plan’s established share price targets of $11.17 and $25.45. See the section entitled “Narrative to Summary Compensation Table—Performance Plan” for a further description of the Performance Plan. Performance Plan minimum bonus award fair values are estimated using a Monte Carlo simulation approach in accordance with Financial Accounting Standards Board Accounting Standards Codification (“ASC”) Topic 718, rather than the amounts payable to or realized by the named individual. For a discussion of the assumptions used to estimate the value of the Performance Plan awards made to our named executive officers, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Use of Estimates—Share-Based Compensation” and Notes 1 and 12 to the accompanying consolidated financial statements included in this Annual Report. |
(2) | Amounts reflect the grant-date fair value of equity-based awards granted to our named executive officers, as applicable, including: (i) stock options in 2019 and 2018; and (ii) SARs in 2018. Both stock option and SARs fair values are estimated using the Black Scholes Option Pricing Model in accordance with ASC Topic 718, rather than the amounts paid to or realized by the named individual. For a discussion of the assumptions used to estimate the value of the options and SARs made to our named executive officers, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Use of Estimates—Share-Based Compensation” and Notes 1 and 11 to the accompanying consolidated financial statements included in this Annual Report. |
(3) | The Company did not award performance-based cash bonuses under the Company’s Senior Executive Annual Incentive Plan in 2019 or 2018. For a description of the named executive officers’ annual bonus opportunities, please review the section entitled “Executive Compensation—Narrative to Summary Compensation Table—Annual Bonuses.” |
160
(4) | All other compensation includes matching contributions made under our 401(k) plan for Ms. Stafford and Mr. Gay, payout of accrued vacation in lieu of time-off for Mr. Martin and Mr. Gay and premiums for executive life insurance and a housing allowance for Mr. Martin. In addition, in 2018 Mr. Martin’s amount includes compensation paid pursuant to our non-employee director compensation policy through the second quarter of 2018. |
(5) | Ms. Stafford served as our Chief Development Officer on a part-time basis in 2018 and received base compensation at a rate of $288,000, equivalent to 75% of $384,000 on a full-time basis. Ms. Stafford became our President and Chief Operating Officer on a full-time basis effective January 2, 2019 and entered into a new employment agreement effective January 29, 2019, or the Stafford COO Employment Agreement, as described in further detail within the section entitled “Executive Compensation—Arrangements with our Named Executive Officers—Arrangements with Paula Brown Stafford.” Ms. Stafford entered into the Amended and Restated Stafford Employment Agreement in connection with becoming our Chief Executive Officer effective February 2, 2020. |
(6) | The amount disclosed as bonus represents bonus compensation paid to Ms. Stafford in accordance with the terms of her executed 2017 offer letter, as amended, for her service as our Chief Development Officer in 2018. |
(7) | Mr. Gay was appointed as our Principal Financial Officer effective January 31, 2019, and assumed the role of Vice, President Finance while continuing in the role of Corporate Controller. |
(8) | The amount disclosed as bonus represents a one-time signing bonus in August 2018 in conjunction with the execution of Mr. Martin’s employment agreement following his April 2018 appointment as our Chief Executive Officer. In determining the amount of the one-time signing bonus, our compensation committee considered the fact that Mr. Martin received no executive compensation while serving as our interim Chief Executive Officer from June 2017 through April 2018, nor did he receive executive compensation from April 2018 until his employment agreement became effective in August 2018. |
Narrative to Summary Compensation Table
Elements of Compensation
During 2019, we compensated our named executive officers through a combination of base salary, cash bonuses, long-term performance-based awards under our Performance Plan and 2016 Incentive Award Plan and other perquisites and benefits as described below.
Annual Base Salaries
The named executive officers receive a base salary to compensate them for services rendered to us. The base salary payable to each named executive officer is intended to provide a fixed component of compensation reflecting the executive’s skill set, experience, role and responsibilities. In 2019, our named executive officers were entitled to the following total base salaries:
• | Mr. Martin was entitled to $480,000 pursuant to his employment agreement. |
• | Ms. Stafford was entitled to $443,326, which reflects the prorated amounts of (i) Ms. Stafford’s $590,000 annual salary for services rendered from the date of December 17, 2019 (the effective date of the Amended and Restated Stafford Employment Agreement) through December 31, 2019, (ii) Ms. Stafford’s $450,000 annual salary for services rendered from January 29, 2019 (the effective date of her COO Employment Agreement) through December 16, 2019, and (iii) prior to January 29, 2019, the prorated amount of the $288,000 part-time equivalent to a full-time annual salary of $384,000; and |
• | Mr. Gay was entitled to $245,753 which reflects the prorated amount of Mr. Gay’s $250,000 annual salary for services rendered from January 31, 2019 (the effective date of his appointment as our principal financial officer by our Board of Directors) through December 31, 2019, and prior to January 31, 2019, the prorated amount of Mr. Gay’s $200,000 annual salary. |
Please see the section entitled “Executive Compensation—Arrangements with our Named Executive Officers” for further description of each named executive officer’s employment arrangement, including those entered into with Ms. Stafford in January 2019 and December 2019.
Bonuses
Each named executive officer’s employment arrangement (other than Mr. Martin) provided for certain cash bonuses in 2019, as described below:
• | In 2019, Ms. Stafford’s employment agreement provides for an annual target cash bonus opportunity of up to 50% of her base salary, payable based on performance criteria. Our compensation committee has determined that Ms. Stafford will |
161
not receive any bonus for 2019 after determining that the associated corporate performance objectives were not achieved during 2019.
• | Mr. Gay was eligible for a 2019 target bonus opportunity equal to 25% of his base salary, payable based on performance criteria. Our compensation committee has determined that Mr. Gay will not receive any bonus for 2019 after determining that the associated corporate performance objectives were not achieved during 2019. |
Please see the section entitled “Executive Compensation—Arrangements with our Named Executive Officers” for further description of each named executive officer’s employment agreement, including the Amended and Restated Stafford Employment Agreement entered into with Ms. Stafford in December 2019.
Long-term Performance-based Compensation—Performance Plan
In August 2018, our board of directors approved and established the Performance Plan, which is a performance-based long-term incentive plan. The Performance Plan is intended to tie long-term employee incentive compensation to specific, significant increases in our underlying common stock price and thus directly aligns employee and stockholder objectives. Unlike our historical practice of providing long-term incentives to our employees through annual stock option grants under the 2016 Plan at the then-current market price of our common stock, the Performance Plan only provides for employees to receive long-term incentive compensation payments if the established stock price targets ($11.17 per share and $25.45 per share, subject to adjustment as described below) are achieved.
The core underlying metric of the Performance Plan is the achievement of two share price goals for our common stock, which if achieved, would represent measurable increases in stockholder value. The Performance Plan is intended to align the interests of plan participants with those of our stockholders in a manner that is intended to be constructive, direct and transparent, in that if we do not achieve one or both related distinct share price targets, no portion of the potential bonus pools will be distributed.
The Performance Plan is tiered, with two separate tranches, each of which has a distinct share price target (measured as the average publicly traded share price of our common stock on the Nasdaq stock exchange for a thirty consecutive trading day period) that will trigger a distinct fixed bonus pool. The share price target for the first tranche is $11.17 per share. The share price target for the second tranche is $25.45 per share. In August 2018, when the Performance Plan was approved and the share price targets were set by our board of directors, the closing price for our common stock ranged from $2.37 to $2.90. The related contingent bonus pools for the first and second tranches are $25.0 million and $50.0 million, respectively. Our compensation committee has discretion to distribute the bonus pool related to each tranche among eligible participants by establishing individual minimum bonus amounts before, as well as by distributing the remainder of the applicable pool after the achievement of each tranche specific share price target. Otherwise, if we do not achieve one or both related share price targets, as defined, prior to the Performance Plan’s expiration date of March 1, 2022, no portion of the bonus pools will be paid, including the established minimum bonus amounts. The share price targets will be adjusted in the event of any stock splits, cash dividends, stock dividends, combinations, reorganizations, reclassifications, or similar events. In addition, in the event of a change in control, the plan provides that a bonus pool will become due and payable to participants on a pro rata basis, as calculated and determined by the compensation committee based upon our progress toward the share price target as of the date of the change in control and subject to adjustment by the compensation committee as permitted under the plan.
The Performance Plan provides for the bonus pool to generally be paid in the form of cash. However, our compensation committee has discretion to pay any bonus award under the Performance Plan in the form of cash, shares of our common stock or a combination thereof, but only if our board of directors and stockholders approve the reservation of shares of our common stock for such payment. To date, our board of directors has not approved the reservation of any shares for issuance under the Performance Plan or requested that our stockholders approve any such reservation, and accordingly, no shares of our common stock may be issued thereunder unless and until such approvals occur.
The Performance Plan was effective immediately upon approval, expires on March 1, 2022, and covers all employees, including our executive officers, consultants and other persons deemed eligible by our compensation committee. If the Performance Plan’s share price targets are not achieved by the expiration date of March 1, 2022, no established bonus awards will be disbursed under the plan. The Performance Plan was subsequently amended and restated to reflect minor changes in the timing for establishing minimum bonus amounts.
Our compensation committee has established that our named executive officers will receive the following minimum bonus amounts under the Performance Plan if the share price targets are achieved:
• | In August 2018, our compensation committee established that Mr. Martin would receive the following minimum bonus amounts: |
162
o | If the Performance Plan’s first share price target of $11.17 per share is achieved, Mr. Martin would receive a minimum bonus amount under the Performance Plan of $5,250,000. If the Performance Plan’s first share price target is not achieved, no bonus award would be disbursed. |
o | If the Performance Plan’s second share price target of $25.45 per share is achieved and Mr. Martin is serving as our Chief Executive Officer, he would receive a minimum bonus amount of $10,500,000 or, if the Performance Plan’s second share price target of $25.45 per share is achieved and he is serving as a director but is no longer serving as our Chief Executive Officer, he would instead receive a minimum bonus amount of $8,000,000. |
o | If the Performance Plan’s second share price target is not achieved or if Mr. Martin is not serving as either Chief Executive Officer or a director at the time the target is achieved, no bonus award would be disbursed. Following his resignation in February 2020, Mr. Martin is no longer entitled to any bonus amount under the Performance Plan. |
o | Mr. Martin’s minimum bonus amount under the Performance Plan was a contingent, performance-based award that, together with Mr. Martin’s SAR Award (as defined below), was implemented by our compensation committee in lieu of a stock option or other form of equity grant and targeted to be commensurate with an equity position typically granted to the chief executive officer of comparable life sciences companies. |
• | In November 2018, our compensation committee established that, if the Performance Plan’s first share price target of $11.17 per share is achieved, Ms. Stafford would receive a minimum bonus amount under the Performance Plan of $500,000. If the Performance Plan’s first share price target is not achieved, no bonus award will be disbursed. In January 2019, our compensation committee established that Ms. Stafford would be entitled to an additional minimum bonus amount of $250,000, bringing her total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $750,000. In June 2019, our compensation committee established that Ms. Stafford would be entitled to an additional minimum bonus amount of $500,000, bringing her total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $1,250,000. |
• | In January 2019, our compensation committee established that Mr. Gay would be entitled to an additional minimum bonus amount of $100,000, bringing his total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $250,000. In June 2019, our compensation committee established that Mr. Gay would be entitled to an additional minimum bonus amount of $250,000, bringing his total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $500,000. |
Additionally, Ms. Stafford and Mr. Gay will also be eligible for consideration for a discretionary bonus under the Performance Plan to be determined by our compensation committee in connection with each share price target being earned.
Long-term Performance-based Compensation—2016 Incentive Award Plan
We currently sponsor the 2016 Incentive Award Plan, or the 2016 Plan, for purposes of granting stock options, SARs, and other equity-based instruments to our executive officers, directors and employees.
Initial and promotion option grants to our executive officers are generally set forth in their employment agreements. These initial and promotion grants are the product of negotiation with the executive officer, but we generally seek to establish equity ownership levels that we believe are commensurate with the equity positions held by executive officers serving in similar roles at comparable biopharmaceutical companies. Stock option grants made to our executive officers include (i) time-based vesting awards with vesting provisions ranging from six months to three years and (ii) awards that have also included performance-based vesting conditions.
Mr. Martin was not granted any stock options during 2019. In August 2018, in connection with entering into his employment agreement, Mr. Martin was awarded 1,000,000 SARs with an exercise price of $3.80 and a vesting date of February 1, 2020, or the Martin SAR Award. The Martin SAR Award was a performance-based award that, together with Mr. Martin’s contingent, performance-based minimum bonus amount under the Performance Plan, was implemented by our compensation committee in lieu of a stock option or other form of equity grant and targeted to be commensurate with an equity position typically granted to the chief executive officer of comparable life sciences companies. The SARs were granted by our board of directors on a contingent basis that would have been irrevocably forfeited and voided in full if we had failed to obtain stockholder approval of an amendment to the 2016 Plan that authorizes underlying common shares for the SARs. On July 31, 2019, at the Company’s 2019 Annual Meeting of Stockholders, stockholders approved an amendment to the 2016 Plan authorizing underlying common shares for the SARs, and the SARs were no longer considered to be granted on a contingent basis. The SARs vested in full and expired unexercised on February 1, 2020.
163
In January 2019, in connection with Ms. Stafford’s promotion to President and Chief Operating Officer and the execution of the Stafford COO Employment Agreement, Ms. Stafford received an option to purchase 55,000 shares of common stock. In September 2019, after a compensation review, Ms. Stafford received an option to purchase 130,000 shares of common stock. In connection with entering into the Amended and Restated Stafford Employment Agreement, as discussed below, Ms. Stafford was entitled to receive 600,000 SARs, however, the Stafford SAR Award was granted in the first quarter of 2020 following our release of top-line results of our Phase 3 molluscum clinical program.
In January 2019, in connection with Mr. Gay’s appointment as our Principal Financial Officer, Mr. Gay received an option to purchase 35,000 shares of common stock. In September 2019, after a compensation review, Mr. Gay received an option to purchase 5,000 shares of common stock.
Other Elements of Compensation
Retirement Plans
We currently maintain the Novan, Inc. 401(k) Plan, a defined contribution retirement savings plan, or the 401(k) Plan, for the benefit of our employees, including our named executive officers, who satisfy certain eligibility requirements. Our named executive officers were eligible to participate in the 401(k) Plan on the same terms as our other full-time employees. The Internal Revenue Code allows eligible employees to defer a portion of their compensation, within prescribed limits, on a pre-tax basis through contributions to the 401(k) Plan. In 2018, each participant in the 401(k) Plan was eligible to receive matching contributions of up to 3% of such participant’s gross wages. These matching contributions are fully vested after one full year of employment. We believe that providing a vehicle for retirement savings though our 401(k) Plan and making matching contributions adds to the overall desirability of our executive compensation package and further incentivizes our employees, including our named executive officers.
Employee Benefits and Perquisites
All of our full-time employees, including our named executive officers, are eligible to participate in our health and welfare plans, including:
• | medical, dental and vision benefits; |
• | medical and dependent care flexible spending accounts; |
• | short-term and long-term disability insurance; and |
• | life insurance. |
In addition to the health and welfare benefits described above, certain named executive officers participate in a company-paid executive life insurance plan. We generally do not provide any other perquisites to our named executive officers, except for certain travel and living expenses under our employment agreement with Mr. Martin, as described below.
We believe the benefits and perquisites described above are necessary and appropriate to provide a competitive compensation package to our named executive officers.
No Tax Gross-Ups
We do not make gross-up payments to cover our named executive officers’ personal income taxes that may pertain to any of the compensation or perquisites paid or provided by us.
164
Outstanding Equity Awards at Fiscal Year End
The following table provides information regarding outstanding equity awards held by our named executive officers as of December 31, 2019.
Option Awards | Stock Awards | |||||||||||||||||||||
Name | Grant Date | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($/Share) | Option Expiration Date | Equity Incentive Plan Awards: Number of unearned shares, units or other right that have not vested (#) | Equity Incentive Plan Awards: Payout value of unearned shares, units or other right that have not vested ($) | |||||||||||||||
Paula Brown Stafford | 03/20/17 | (1) | 54,000 | — | $ | 6.53 | 03/20/27 | |||||||||||||||
President and Chief Executive Officer; Former Chief Operating Officer | 08/25/17 | (2) | 30,500 | — | 4.27 | 08/14/27 | ||||||||||||||||
10/12/17 | (3) | 68,401 | — | 5.03 | 09/14/27 | |||||||||||||||||
02/12/18 | (4) | 7,760 | 4,386 | 3.03 | 02/11/28 | |||||||||||||||||
01/28/19 | (5) | 50,417 | 4,583 | 1.35 | 01/01/29 | |||||||||||||||||
09/06/19 | (6) | — | 130,000 | 2.68 | 09/05/29 | |||||||||||||||||
11/13/18 | (7) | (8) | $ | 1,250,000 | (7) | |||||||||||||||||
John M. Gay | 05/31/18 | (9) | 4,168 | 8,332 | 3.15 | 05/20/28 | ||||||||||||||||
Vice President, Finance and Corporate Controller | 11/16/18 | (10) | 834 | 1,666 | 2.43 | 11/12/28 | ||||||||||||||||
01/28/19 | (11) | — | 35,000 | 1.35 | 01/27/29 | |||||||||||||||||
09/06/19 | (6) | — | 5,000 | 2.68 | 09/05/29 | |||||||||||||||||
01/30/19 | (12) | (8) | 500,000 | (12) | ||||||||||||||||||
G. Kelly Martin | 09/20/16 | (13) | 14,484 | — | 11.00 | 09/19/26 | ||||||||||||||||
Former Chief Executive Officer | 06/05/17 | (14) | 34,014 | — | 4.64 | 06/04/27 | ||||||||||||||||
08/16/18 | (15) | — | 1,000,000 | 3.80 | 02/01/20 | |||||||||||||||||
08/08/18 | (8) | 15,750,000 | (16) | |||||||||||||||||||
(1) | The option was granted under the 2016 Plan and vested six months from March 20, 2017. |
(2) | The option was granted under the 2016 Plan and vested in four equal quarterly installments, with the first installment vesting on September 5, 2017. |
(3) | The option was granted under the 2016 Plan and vested six months from vesting commencement date of September 15, 2017. |
(4) | The option was granted under the 2016 Plan and vests in thirty-six equal monthly installments on the first day of each month following February 12, 2018. |
(5) | This option was granted under the 2016 Plan, one-half vested six months from the January 2, 2019 vesting commencement date, and subsequent to the six-month anniversary of the vesting commencement date, one-twelfth vests each successive monthly anniversary following July 2, 2019. |
(6) | The option was granted under the 2016 Plan and vests in its entirety on June 25, 2020. |
(7) | The amount reflects the minimum bonus amount payable to Ms. Stafford as of December 31, 2019 under the Performance Plan if the first share price target of $11.17 per share is achieved. If the Performance Plan’s first share price target is not achieved, no bonus award will be disbursed. See the section entitled “Executive Compensation—Narrative to Summary Compensation Table—Long-term Performance-based Compensation—Performance Plan” for further information regarding the Performance Plan. In November 2018, our compensation committee established a minimum bonus amount under the Performance Plan of $500,000. In January 2019, our compensation committee established that Ms. Stafford would be entitled to an additional minimum bonus amount of $250,000 and in June 2019, our compensation committee established that Ms. |
165
Stafford would be entitled to an additional minimum bonus amount of $500,000, bringing her total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $1,250,000.
(8) | Minimum bonus amounts established by our compensation committee under the Performance Plan—the Performance Plan provides for the bonus pool to generally be paid in the form of cash, and awards are denominated in cash. Our compensation committee has discretion to pay any bonus award under the Performance Plan in the form of cash, shares of our common stock or a combination thereof, provided that our board and stockholders have approved the reservation of shares of our common stock for such payment. |
(9) | The option was granted as an inducement grant in accordance with Nasdaq Listing Rule 5635(c)(4), and vests in three equal annual installments with the first installment vesting on May 21, 2019. |
(10) | The option was granted under the 2016 Plan and vests in three equal annual installments with the first installment vesting on November 13, 2019. |
(11) | The option was granted under the 2016 Plan and vests in three equal annual installments with the first installment vesting on January 28, 2020. |
(12) | The amount reflects the minimum bonus amount payable to Mr. Gay as of December 31, 2019 under the Performance Plan if the first share price target of $11.17 per share is achieved. See the section entitled “Executive Compensation—Narrative to Summary Compensation Table—Long-term Performance-based Compensation—Performance Plan” for further information regarding the Performance Plan. In November 2018, our compensation committee established a minimum bonus amount under the Performance Plan of $150,000. In January 2019, our compensation committee established that Mr. Gay would be entitled to an additional minimum bonus amount of $100,000 and in June 2019, our compensation committee established that Mr. Gay would be entitled to an additional minimum bonus amount of $250,000, bringing his total potential minimum bonus amount upon achievement of the first share price of $11.17 per share of common stock to $500,000. |
(13) | The option was granted under the 2016 Plan and vested in four equal quarterly installments, with the first installment vesting on December 20, 2016. |
(14) | The option was granted under the 2016 Plan and vested in four equal quarterly installments, with the first installment vesting on September 5, 2017. |
(15) | The SARs were granted on a contingent basis by our board under the 2016 Plan and were subject to stockholder approval of an amendment to the 2016 Plan. On July 31, 2019, at the Company’s 2019 Annual Meeting of Stockholders, stockholders approved an amendment to the 2016 Plan authorizing underlying common shares for the SARs, and the SARs were no longer considered to be granted on a contingent basis. The SARs vested in full and expired unexercised on February 1, 2020. See the section entitled “ Executive Compensation—Narrative to Summary Compensation Table—Long-term Performance-based Compensation—2016 Incentive Award Plan” for further details regarding the SARs’ terms. |
(16) | The amount reflects the minimum bonus amount payable to Mr. Martin as of December 31, 2019 under the Performance Plan if the first share price target of $11.17 and second share price target of $25.45 per share is achieved. If the Performance Plan’s first share price target is not achieved, no bonus award will be disbursed under the Performance Plan. See the section entitled “Executive Compensation—Narrative to Summary Compensation Table—Long-term Performance-based Compensation—Performance Plan” for further information regarding the Performance Plan. Following his resignation in February 2020, Mr. Martin is no longer entitled to any bonus amount under the Performance Plan. |
Arrangements with our Named Executive Officers
We have entered into employment arrangements with our named executive officers that set forth certain terms and conditions of their employment, including base salary and employee benefits.
166
Arrangements with Paula Brown Stafford
As of February 2, 2020, Ms. Stafford serves as our President and Chief Executive Officer and is compensated pursuant to the Amended and Restated Stafford Employment Agreement. Pursuant to the Amended and Restated Stafford Employment Agreement, Ms. Stafford receives an annual base salary of $590,000 and is eligible to receive an annual performance-based bonus with a target bonus of 55% to 75% of her base salary. For 2019, Ms. Stafford is eligible for an annual performance-based bonus with a target bonus of 50% or more of her annual base salary. Ms. Stafford is also eligible to participate in our incentive award plans. Ms. Stafford continues to be eligible to participate in standard benefit plans as well as an executive life insurance plan and reimbursement of reasonable business expenses. In addition, our board of directors approved a stock appreciation right, or the Stafford SAR Award, for Ms. Stafford under the 2016 Plan covering 600,000 shares of our common stock. The Stafford SAR Award was granted on a contingent basis and would have been considered irrevocably forfeited and voided in full if sufficient shares of our common stock were not available under the 2016 Plan or if we failed to obtain stockholder approval for amendments to the 2016 Plan at the next annual stockholders’ meeting to provide sufficient shares for the Stafford SAR Award. In such event, we would have been required to pay Ms. Stafford the cash-equivalent value of the amount that would have been due and payable per the Stafford SAR Award upon any properly noticed exercise of any vested portion of the Stafford SAR Award. Such condition was satisfied, and the SARs were no longer considered to be granted on a contingent basis, as of February 1, 2020.
In the event of Ms. Stafford’s termination of employment either upon nonrenewal by the Company of the term of the Amended and Restated Stafford Employment Agreement, by the Company without “cause” or by Ms. Stafford for “good reason” (except as set forth below), then in addition to any accrued amounts and subject to Ms. Stafford timely delivering an effective release of claims in the Company’s favor and her continued compliance with the previously signed Restrictive Covenants Agreement between the Company and Ms. Stafford, Ms. Stafford will be entitled to receive payment of her then-current base salary, plus a prorated annual bonus calculated at the minimum target level of the calendar year in which the “separation date,” as defined in the Amended and Restated Stafford Employment Agreement, occurs based on the percentage of the calendar year actually worked by Ms. Stafford as of the separation date, each multiplied by 1.5, plus the amount of any unpaid Annual Bonus for the prior calendar year. Such amounts will be paid in equal monthly installments over 12 months in accordance with standard payroll practices and provided, that to the extent that any such cash award constitutes nonqualified deferred compensation under Section 409A, the cash payment will be paid subject to any delay required by Section 409A. Ms. Stafford will also be entitled to vesting of any then unvested portion of the Stafford SAR Award that would have otherwise vested through the calendar quarter following the calendar quarter in which the separation date occurs. In the event of certain changes to the Company’s board of directors that constitute “good reason” under the Amended and Restated Stafford Employment Agreement, Ms. Stafford will be entitled to receive payment of her then-current base salary for a one-year period and the amount of any unpaid Annual Bonus for the prior calendar year, if any, and vesting of any then unvested portion of the Stafford SAR Award that would have otherwise vested through the calendar quarter in which the separation date occurs. Upon termination of employment by Ms. Stafford other than for good reason or due to her death or disability, or by the Company for cause, Ms. Stafford will not be entitled to any additional compensation beyond any accrued amounts.
Notwithstanding the foregoing, the Amended and Restated Stafford Employment Agreement further provides that, in the event of a “double trigger” event, Ms. Stafford will be entitled to receive payment of her then-current base salary, plus a prorated annual bonus calculated at the minimum target level of the calendar year in which the separation date occurs based on the percentage of the calendar year actually worked by Ms. Stafford as of the separation date, each multiplied by 2.5, plus the amount of any unpaid Annual Bonus for the prior calendar year. Such amounts will be paid in equal monthly installments over 24 months in accordance with standard payroll practices and provided, that to the extent that any such cash award constitutes nonqualified deferred compensation under Section 409A, the cash payment will be paid subject to any delay required by Section 409A. Ms. Stafford will also be entitled to vesting of any then unvested portion of the Stafford SAR Award and any other equity grant as of the separation date.
The following circumstances are considered a “double trigger” event:
(i) a “change in control,” as defined in the Amended and Restated Stafford Employment Agreement (which incorporates the definition from the 2016 Plan), and
(ii) Ms. Stafford is terminated from employment by the Company without cause or upon the nonrenewal by the Company of the term of the Amended and Restated Stafford Employment Agreement or by Ms. Stafford for good reason (other than due to certain changes on the Company’s board of directors) within 12 months after a change in control, subject to Ms. Stafford timely delivering an effective release of claims in the Company’s favor and her continued compliance with the Restrictive Covenants Agreement between the Company and Ms. Stafford
Prior to the Amended and Restated Stafford Employment Agreement, Ms. Stafford served as President and Chief Operating Officer pursuant to the Stafford COO Employment Agreement. Under that agreement, Ms. Stafford received an
167
annual base salary of $450,000 and was eligible to receive an annual performance-based bonus with a target bonus equal of 50% of her base salary. Ms. Stafford was also eligible to participate in our incentive award plans, our standard benefit plans, an executive life insurance plan and reimbursement of reasonable business expenses.
Arrangements with John M. Gay
Mr. Gay currently serves as our Vice President, Finance and Corporate Controller and is an at-will employee of the Company. On January 26, 2019, the Board of Directors approved certain compensatory matters for Mr. Gay and pursuant to these terms, Mr. Gay receives an annual salary of $250,000 and is eligible to receive annual performance-based bonus with a target bonus equal to 25% of his annual base salary. Mr. Gay is also eligible to participate in our incentive award plans, our standard benefit plans and reimbursement of reasonable business expenses.
Arrangements with G. Kelly Martin
Mr. Martin began serving as our Chief Executive Officer in April 2018, and prior to the employment agreement entered into on August 8, 2018, or the Martin Employment Agreement, he was only compensated pursuant to our Non-Employee Director Compensation Policy, as described in the section entitled “Director Compensation.”
Pursuant to the Martin Employment Agreement, Mr. Martin received an annual base salary of $480,000 and received a signing bonus in the amount of $560,000. Mr. Martin was also eligible to participate in the standard benefit plans as well as an executive life insurance plan and reimbursement of reasonable business expenses. In addition, we agreed to pay for or reimburse Mr. Martin for his extra living and travel expenses beginning in June 2017 associated with the fact that Mr. Martin’s primary residence is in Connecticut, and Mr. Martin was also eligible to earn awards equal to certain minimum bonus amounts, along with any discretionary awards, under and in accordance with the terms of the Performance Plan. In addition, our board of directors approved a stock appreciation right, or the Martin SAR Award, for Mr. Martin under the 2016 Plan covering 1,000,000 shares of our common stock. This award was considered a contingent award and would have been be forfeited had we failed to obtain stockholder approval for amendments to the 2016 Plan required to permit the grant of the Martin SAR Award. In such event, we would have paid Mr. Martin the cash-equivalent value of the amount that would have been due and payable per the Martin SAR Award as of February 1, 2020. On July 31, 2019, at the Company’s 2019 Annual Meeting of Stockholders, stockholders approved an amendment to the 2016 Plan authorizing additional common shares under the 2016 Plan, and the SARs were no longer considered to be granted on a contingent basis. The SARs vested in full and expired unexercised on February 1, 2020.
The Martin Employment Agreement had a fixed term that expired on February 1, 2020, and Mr. Martin completed his service as our Chief Executive Officer after fulfilling his term. Concurrent with the end of his term as Chief Executive Officer, Mr. Martin also resigned from our board of directors, effective February 3, 2020.
Director Compensation
The following table sets forth information concerning the compensation of our directors, other than Mr. Martin and Ms. Stafford, for the year ended December 31, 2019.
Name | Fees Earned or Paid in Cash | Option Awards (1) | Total | ||||||
W. Kent Geer | $ | 76,250 | $ | 41,799 | $ | 118,049 | |||
Robert A. Ingram | 70,000 | 41,799 | 111,799 | ||||||
Robert J. Keegan | 57,500 | 41,799 | 99,299 | ||||||
John Palmour | 47,500 | 41,799 | 89,299 | ||||||
Machelle Sanders | 47,250 | 41,799 | 89,049 | ||||||
Eugene Sun | 60,000 | 41,799 | 101,799 | ||||||
168
(1) | Amounts reflect the grant-date Black-Scholes value of stock awards and stock options granted during 2019, computed in accordance with ASC Topic 718, rather than the amounts paid to or realized by the named individual. For a discussion of the assumptions used to calculate the value of all stock awards and option awards made to our directors, see the section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Critical Accounting Policies and Use of Estimates—Share-Based Compensation” and Notes 1 and 11 to the accompanying consolidated financial statements included in this Annual Report. These amounts do not necessarily correspond to the actual value that may be recognized from the option awards by the applicable directors. |
The table below shows the aggregate numbers of option awards (exercisable and unexercisable) held as of December 31, 2019, by each director who was serving as of December 31, 2019, other than Mr. Martin and Ms. Stafford. No such director held any other equity awards.
Name | Options Outstanding at Fiscal Year End December 31, 2019 | |
W. Kent Geer | 103,748 | |
Robert A. Ingram | 88,498 | |
Robert J. Keegan | 88,498 | |
John Palmour | 88,498 | |
Machelle Sanders | 59,240 | |
Eugene Sun | 53,078 | |
Non-Employee Director Compensation Policy
On May 19, 2018, we amended the Novan, Inc. Non-Employee Director Compensation Policy, or the Director Compensation Policy, for our non-employee directors that consists of annual retainer fees and equity awards that will be paid or made automatically and without further action by our board of directors. Pursuant to the Director Compensation Policy, subject to continued service on our board, (i) each non-employee director receives an annual cash retainer of $35,000; (ii) each non-employee director serving as a committee chair receives an additional annual retainer between $10,000 and $20,000; (iii) each non-employee director serving as a committee member receives an additional annual retainer between $5,000 and $7,500; (iv) the non-employee chairman of our board of directors receives an additional annual retainer of $25,000; and (v) the lead independent director receives an additional annual retainer of $20,000. The Director Compensation Policy also provides each non-employee director with an annual equity award, subject to continued service on the board, equal to the lesser of 20,000 shares or the number of shares that have an aggregate grant-date fair value of $100,000 (and each non-employee director who is initially elected or appointed on any date other than the date of an annual meeting of stockholders will receive a prorated portion of such annual equity award for the year of such election or appointment). Notwithstanding the foregoing, our board of directors in its sole discretion may determine that the annual equity award for any year be granted in the form of restricted stock units with equivalent value on the date of grant (with the number of shares of common stock underlying each such award not to exceed 20,000 shares and subject to adjustment as provided in the 2016 Plan). Each director equity award will vest and become exercisable in four equal quarterly installments, such that each such award shall be fully vested and exercisable on the first anniversary of the date of grant, subject to the director’s continued service on our board of directors through each applicable vesting date.
Directors have been and will continue to be reimbursed for expenses directly related to their activities as directors, including attendance at board and committee meetings. Directors are also entitled to the protection provided by their indemnification agreements and the indemnification provisions in our certificate of incorporation and bylaws.
169
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
EQUITY COMPENSATION PLAN INFORMATION
The following table presents information as of December 31, 2019, with respect to compensation plans under which shares of our common stock may be issued. The category “Equity Compensation Plans approved by security holders” in the table below consists of the 2016 Plan and the 2008 Plan. The table does not include the Performance Plan as no shares of our common stock have been authorized for issuance under that plan.
Plan Category | Number of Securities to be Issued upon Exercise of Outstanding Options and SARs | Weighted Average Exercise Price of Outstanding Options and SARs | Number of Securities Remaining Available for Future Issuances under Equity Compensation Plans (excluding securities reflected in column (a)) | ||||||||
(a) | ($)(b) | (c) | |||||||||
Equity Compensation Plans approved by security holders (4) | 2,663,803 | (1) | $ | 3.90 | (2) | 388,463 | (3) | ||||
Equity Compensation Plans not approved by security holders (5) | 125,500 | 3.04 | - | ||||||||
Total | 2,789,303 | 3.86 | 388,463 | ||||||||
(1) | Includes shares of common stock issuable upon exercise of outstanding options under the 2008 Plan – 45,077 shares; and outstanding options and SARs under the 2016 Plan – 2,618,726 shares. |
(2) | The weighted-average remaining contractual term (in years) was 8.58. |
(3) | Includes shares remaining for future issuance under the 2016 Plan. |
(4) | During the first quarter of 2020, Mr. Martin’s SARs expired unexercised, we issued 600,000 SARs to Ms. Stafford, we issued equity compensation awards to certain employees, and certain equity compensation awards were forfeited or expired. As a result, as of February 14, 2020, there were 2,646,803 securities to be issued upon exercise of outstanding options and SARs, including 45,077 shares under the 2008 Plan and 2,601,726 shares under the 2016 Plan. The weighted average exercise price of outstanding options and SARs as of February 14, 2020, was $2.74, and the weighted average remaining contractual term (in years) was 8.50. As of February 14, 2020, there were 405,463 shares remaining for future issuance under the 2016 Plan. |
(5) | During the years ended December 31, 2019 and 2018, we awarded nonstatutory stock options to purchase shares of common stock to newly-hired employees, not previously employees or directors of Novan, as inducements material to the individuals’ entering into employment with us within the meaning of Nasdaq Listing Rule 5635(c)(4) (the “Inducement Grants”). On May 31, 2018, the Company awarded 100,500 Inducement Grants with an exercise price of $3.15 per share, and on September 6, 2019, the Company awarded 25,000 Inducement Grants with an exercise price of $2.62 per share. The Inducement Grants were awarded outside of the 2016 Plan, pursuant to Nasdaq Listing Rule 5635(c)(4), but have terms and conditions generally consistent with our 2016 Plan and vest over three years, subject to the employee’s continued service as an employee or consultant through the vesting period. All 125,500 Inducement Grants were outstanding as of December 31, 2019. As of February 14, 2020, there were 100,500 Inducement Grants outstanding. |
170
SECURITY OWNERSHIP OF MANAGEMENT AND CERTAIN BENEFICIAL OWNERS
The following table sets forth information regarding the beneficial ownership of our common stock as of February 3, 2020, by the following:
• | each stockholder known by us to be the beneficial owner of more than 5% of our common stock; |
• | each of our directors; |
• | each of our named executive officers; and |
• | all of our directors and executive officers as a group. |
Applicable percentages are based on 27,434,800 shares outstanding on February 3, 2020, adjusted as required by rules promulgated by the SEC.
The number of shares beneficially owned by each stockholder is determined under rules issued by the SEC. Under these rules, beneficial ownership includes any shares as to which the individual or entity has sole or shared voting power or investment power. The following table is based upon information supplied by officers, directors and principal stockholders and Schedules 13D and 13G filed with the SEC. In computing the number of shares beneficially owned by an individual or entity and the percentage ownership of that person, shares of common stock issuable upon the exercise of stock options or SARs or warrants exercisable within 60 days of February 3, 2020, are considered outstanding, although these shares are not considered outstanding for purposes of computing the percentage ownership of any other person. Unless otherwise indicated, the address of each of the individuals and entities named below is c/o Novan, Inc., 4105 Hopson Road, Morrisville, NC 27560. Each of the stockholders listed has sole voting and investment power with respect to the shares beneficially owned by the stockholder unless noted otherwise, subject to community property laws where applicable.
Name of Beneficial Owner | Number of Shares Beneficially Owned | Percentage of Outstanding Shares | ||
5% Stockholders: | ||||
Reedy Creek Investments LLC (1) | 7,894,736 | 25.16% | ||
Malin Life Sciences Holdings Limited (2) | 2,598,485 | 9.47% | ||
Directors and Named Executive Officers: | ||||
G. Kelly Martin (3) | 153,498 | * | ||
Paula Brown Stafford (4) | 372,366 | 1.34% | ||
John Gay (5) | 56,671 | * | ||
Robert A. Ingram (6) | 248,275 | * | ||
W. Kent Geer (7) | 109,576 | * | ||
Robert J. Keegan (8) | 141,533 | * | ||
John Palmour (9) | 646,941 | 2.35% | ||
Machelle Sanders (10) | 49,240 | * | ||
All current directors and executive officers, as a group (7 persons) (11) | 1,624,602 | 5.06% | ||
* | Represents beneficial ownership of less than one percent. |
(1) | Reedy Creek Investments LLC (“Reedy Creek”) is the direct owner of 3,947,368 shares of common stock and 3,947,368 shares of common stock issuable upon exercise of outstanding warrants. Mr. Donald R. Parker is the sole member of the board of managers and the president and chief executive officer, treasurer and chief financial officer of Reedy Creek. The James H. Goodnight Management Trust (the “Trust”) owns a majority of the equity interests in Reedy Creek and has the right to appoint a majority of the members of the board of managers of Reedy Creek. Dr. James H. Goodnight is the sole trustee of the Trust and directs the voting and investment activities of the Trust. Each of Mr. Parker, the Trust and Dr. Goodnight may be deemed to share voting and dispositive power with respect to the securities owned by Reedy Creek. As such, Mr. Parker, the Trust and Dr. Goodnight may be deemed to be the indirect beneficial owners of the securities owned by Reedy Creek. Each of Mr. Parker, the Trust and Dr. Goodnight disclaims beneficial ownership of the securities owned by Reedy Creek, except to the extent of his and, with respect to the Trust, its, pecuniary interest therein, if any. The mailing address of Reedy Creek, the Trust and each of the foregoing individuals is 100 SAS Campus Drive, Cary, NC 27513. |
171
(2) | Malin Life Sciences Holdings Limited is a wholly owned subsidiary of Malin Corporation plc. Malin Corporation plc may be deemed to beneficially own the shares and may be deemed to share voting and dispositive power over these shares. The mailing address of Malin Life Sciences Holdings Limited is 2 Harbour Square, Crofton Road, Dun Laoghaire, Co., Dublin, Ireland. |
(3) | Consists of (i) 105,000 shares of common stock, of which 45,000 are held by the George Kelly Martin IRRA FBO George Kelly Martin and (ii) options to purchase 48,498 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(4) | Consists of (i) 80,693 shares of common stock held by Ms. Stafford (ii) options to purchase 216,673 shares of common stock that are exercisable within 60 days of February 3, 2020 and (iii) 75,000 SARs exercisable within 60 days of February 3, 2020. |
(5) | Consists of (i) 15,000 shares of common stock held by Mr. Gay, (ii) warrants to purchase 25,000 shares of common stock that are exercisable within 60 days of February 3, 2020 and (iii) options to purchase 16,671 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(6) | Consists of (i) 169,777 shares of common stock held by Mr. Ingram and (ii) options to purchase 78,498 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(7) | Consists of (i) 15,828 shares of common stock held by Mr. Geer and (ii) options to purchase 93,748 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(8) | Consists of (i) 63,035 shares of common stock held by the Robert J. Keegan Trust, with Mr. Keegan as trustee, and (ii) options to purchase 78,498 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(9) | Consists of (i) 568,443 shares of common stock, of which 274,875 are held by the Palmour 2012 Irrevocable Children’s Trust, with Dr. Palmour as trustee, and (ii) options to purchase 78,498 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(10) | Consists of options to purchase 49,240 shares of common stock that are exercisable within 60 days of February 3, 2020. |
(11) | Consists of (i) 912,776 common shares held by our current executive officers and current directors, (ii) warrants to purchase 25,000 shares of common stock that are exercisable within 60 days of February 3, 2020, and (iii) options and SARs to purchase 686,826 shares of common stock exercisable within 60 days of February 3, 2020. |
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Policies and Procedures for Related Party Transactions
Our board of directors has adopted a written related person transaction policy setting forth the policies and procedures for the review and approval or ratification of related person transactions. This policy covers, with certain exceptions set forth in Item 404 of Regulation S-K under the Securities Act, any transaction, arrangement or relationship, or any series of similar transactions, arrangements or relationships, in which we were or are to be a participant, the amount involved exceeds the lesser of (i) $120,000 or (ii) one percent of the average of our total assets at year-end for the last two completed fiscal years, and in which a related person had, has or will have a direct or indirect material interest, including without limitation, purchases of goods or services by or from the related person or entities in which the related person has a material interest, indebtedness, guarantees of indebtedness and employment by us of a related person. In reviewing and approving any such transactions, our audit committee is tasked to consider all relevant facts and circumstances, including, but not limited to, whether the transaction is on terms comparable to those that could be obtained in an arm’s length transaction and the extent of the related person’s interest in the transaction. All of the transactions described in this section either were approved or ratified pursuant to this policy or occurred prior to the adoption of this policy.
Certain Relationships and Related Transactions
The following includes a summary of transactions since January 1, 2017, to which we were or are to be a participant, in which the amount involved exceeded or will exceed the lesser of (i) $120,000 or (ii) one percent of the average of our total assets at year-end for the last two completed fiscal years, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described in “Executive Compensation.” We also describe below certain other transactions with our directors, executive officers and stockholders.
172
Reedy Creek Investments
On April 29, 2019, we entered into a royalty and milestone payments purchase agreement, or the Purchase Agreement, with Reedy Creek Investments LLC, or Reedy Creek, which is a greater than 5% stockholder. Pursuant to the Purchase Agreement, Reedy Creek provided funding in an initial amount of $25.0 million, which we will use primarily to pursue the development, regulatory approval and commercialization activities (including through out-license agreements and other third-party arrangements) for certain of our product candidates for certain indications, namely SB206, SB414 and SB204, which we refer to as the Products. Reedy Creek would have also been required to provide additional funding of $10.0 million contingent upon our achievement of SB206 clinical trial success, as defined in the Purchase Agreement; however, based on the top line efficacy results from the Phase 3 SB206 program available released in January 2020, we understand that Reedy Creek will not be paying us the contingent $10.0 million of additional funding.
Pursuant to the Purchase Agreement, we will pay Reedy Creek ongoing quarterly payments, calculated based on an applicable percentage per product, of any upfront fees, milestone payments, royalty payments or equivalent payments received by us pursuant to any out-license agreement for the Products in the United States, Mexico or Canada, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by us to third parties pursuant to any agreements under which we have in-licensed intellectual property with respect to the Products in the United States, Mexico or Canada. The applicable percentage used for determining the ongoing quarterly payments for each Product ranges from 10% for SB206 to 20% for SB204 and SB414, provided that the applicable percentage for each Product will be 25% for fees or milestone payments received by us (but not royalty payments received by us) until Reedy Creek has received payments under the Purchase Agreement equal to the total funding amount provided by Reedy Creek under the Purchase Agreement. If we decide to commercialize any of the relevant products on our own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, we will be obligated to pay Reedy Creek a low single digits royalty on net sales of the relevant products.
Unless earlier terminated, the Purchase Agreement will continue for so long as payments are due or payable under the Purchase Agreement. Reedy Creek may terminate the Purchase Agreement in the event of an uncured material breach by us, which, in certain circumstances, could cause us to be required to repay the amount paid by Reedy Creek under the Purchase Agreement, less any payments made to Reedy Creek by us under the Purchase Agreement as of the effective date of the termination.
Malin Life Sciences Holdings and Majority-owned Subsidiaries
On September 26, 2016, we completed our initial public offering and issued a total of 4,715,000 shares of common stock at a public offering price of $11.00 per share, including 800,000 shares sold to Malin Life Sciences Holding Limited, a greater than 5% stockholder at the time of our initial public offering.
In June 2017, G. Kelly Martin assumed the role of our Chief Executive Officer on an interim basis before being appointed as our Chief Executive Officer in April 2018, while also serving as a member of our board of directors. Until October 1, 2017, Mr. Martin served as chief executive officer of Malin Corporation plc, the parent company of Malin. Mr. Martin is no longer our Chief Executive Officer as of February 1, 2020 and is no longer a member of our board of directors as of February 3, 2020.
Upon stepping into our Chief Executive Officer role on an interim basis, Mr. Martin engaged a number of Malin employees to assist him in certain strategic and tactical initiatives and activities. We agreed to reimburse Malin for its out-of-pocket expenses for Mr. Martin and other Malin employees related to this effort. During the year ended December 31, 2017, we recognized $230,000 in out-of-pocket travel expenses owed to Malin and reimbursed in the first quarter of 2018. There were no such expenses for the year ended December 31, 2018.
Two of our directors during 2018 were also affiliated with Malin. Sean Murphy, who resigned from our board in September 2018, was an executive officer and a director of Malin, and an executive vice president of Malin Corporation plc. In addition, Robert A. Ingram, executive chairman of our board of directors, was also a director of Malin Corporation plc until July 2018.
In August 2019, Malin Corporation plc completed the sale of its former subsidiary, Cilatus BioPharma AG, or Cilatus. Prior to this disposition, Cilatus was majority-owned by Malin Corporation plc. During the nine months ended September 30, 2019 and the year ended and December 31, 2018, respectively, we incurred costs of $250,000 and $601,000 in relation to a development and manufacturing consulting agreement with Cilatus.
173
KNOW Bio
On December 30, 2015, we completed the distribution, or the Distribution, of all of the outstanding member interests of KNOW Bio, LLC, or KNOW Bio, our former wholly owned subsidiary, pro rata to our stockholders. We do not own an equity interest in KNOW Bio. However, certain of our current and former directors and executive officers, including Mr. Murphy and Dr. Stasko, as well as Malin, received equity interests in KNOW Bio as a result of the Distribution, and Mr. Murphy and Dr. Stasko both served as directors of KNOW Bio while they served as directors of the Company.
In April 2017, we entered into a master development services and clinical supply agreement with KNOW Bio and entered into related statements of work in the second quarter and second half of 2017, or collectively the KNOW Bio Services Agreement. Under the KNOW Bio Services Agreement, we provided certain development and manufacturing services to KNOW Bio’s respiratory drug development subsidiary. During the year ended December 31, 2017, we recognized $375,000 in research and development services revenue for services performed under the KNOW Bio Services Agreement. In January 2018, upon request by KNOW Bio, we stopped performing remaining development or manufacturing services contemplated under the KNOW Bio Services Agreement after reporting revenues of $9,000 in 2018.
In October 2017, we entered into amendments to certain licensing arrangements with KNOW Bio that were originally entered at the time of the Distribution. In connection with those amendments, we made an upfront payment to KNOW Bio of $250,000, and we will be obligated to make certain contingent payments in exchange for the rights granted under the licensing arrangements, as amended.
Health Decisions
On October 25, 2018, we announced the formation of a dedicated women’s health business unit as well as a foundational collaboration with Health Decisions, Inc., or Health Decisions. Health Decisions is a full-service contract research organization specializing in clinical studies of therapeutics for women’s health indications. Our women’s health business unit is led by Paula Brown Stafford, who also is a shareholder and serves on the board of directors of Health Decisions.
Arrangements with Executive Officers and Directors
We have entered into employment arrangements with our named executive officers. For more information regarding our arrangements with our named executive officers, see the section entitled “Executive Compensation—Arrangements with our Named Executive Officers.”
We have entered, or will enter, into an indemnification agreement with each of our directors and executive officers. The indemnification agreements and our bylaws require us to indemnify our directors and officers to the fullest extent permitted by Delaware law.
Independence of Directors
Our common stock is listed on The Nasdaq Global Market. Under the listing requirements and rules of The Nasdaq Global Market, independent directors must comprise a majority of our board of directors, and each member of our audit committee, compensation committee and nominating and governance committee must be independent. Under the rules of The Nasdaq Global Market, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Audit committee members must also satisfy independence criteria set forth in Rule 10A-3 under the Exchange Act. To be considered independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of a company’s audit committee, the company’s board of directors or any other board committee, (i) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (ii) be an affiliated person of the listed company or any of its subsidiaries.
Our board of directors has undertaken a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships, our board of directors has determined that Robert A. Ingram, W. Kent Geer, Robert J. Keegan, John Palmour and Machelle Sanders do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the applicable rules and regulations of the listing requirements and rules of The Nasdaq Global Market. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with us and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director.
174
Our board of directors determined that W. Kent Geer, Robert J. Keegan and John Palmour, each of the three members of our audit committee, satisfy the independence standards for our audit committee established by applicable SEC rules and the listing standards of The Nasdaq Global Market and Rule 10A-3.
Our board of directors has determined that Robert J. Keegan, W. Kent Geer and Machelle Sanders, each of the three members of our compensation committee, satisfy the independence standards for our compensation committee established by applicable SEC Rules and the listing standards of The Nasdaq Global Market, taking into consideration all factors specified in the applicable standards.
Our board of directors has determined that Robert A. Ingram and John Palmour, the two members of our nominating and corporate governance committee, are independent within the meaning of the applicable listing standards of The Nasdaq Global Market.
175
Item 14. Principal Accounting Fees and Services.
Principal Accountant Fees and Services
The following table represents the aggregate fees and expenses for services provided by BDO USA, LLC, or BDO, our independent registered public accounting firm for the fiscal years ended December 31, 2019 and 2018.
Fiscal Year Ended | ||||||||
2019 | 2018 | |||||||
(in thousands) | ||||||||
Audit Fees (1) | $ | 253 | $ | 257 | ||||
Audit-Related Fees | — | — | ||||||
Tax Fees | — | — | ||||||
All Other Fees | — | — | ||||||
Total Fees | $ | 253 | $ | 257 | ||||
(1) | Audit fees consist of fees billed, or expected to be billed, for professional services rendered for the audit of our consolidated annual financial statements, review of the interim consolidated financial statements, the issuance of consent and comfort letters in connection with registration statement filings with the SEC and all services that are normally provided by the accounting firm in connection with statutory and regulatory filings or engagements. |
All fees described above were approved by our audit committee.
Pre-Approval Policies and Procedures
Our audit committee has adopted a policy and procedures for the pre-approval of audit and non-audit services rendered by our independent registered public accounting firm. The policy generally pre-approves specified services in the defined categories of audit services, audit-related services and tax services up to specified amounts. Pre-approval may also be given as part of our audit committee’s approval of the scope of the engagement of the independent auditor or on an individual, explicit, case-by-case basis before the independent auditor is engaged to provide each service. The pre-approval of services may be delegated to one or more of our audit committee’s members, but the decision must be reported to the full audit committee at its next scheduled meeting.
Our audit committee has determined that the rendering of services other than audit services by BDO are compatible with maintaining the principal accountant’s independence.
176
PART IV
Item 15. Exhibits, Financial Statement Schedules.
(a) | The following financial statements are included in this Annual Report on Form 10-K/A: |
(1) | List of Financial Statements: |
The financial statements required by this item are listed in Item 8, “Financial Statements and Supplementary Data” herein.
(2) | List of Financial Statement Schedules: |
All financial statement schedules have been omitted because they are not applicable, not required or the information required is shown in the financial statements or notes thereto.
(3) | List of Exhibits. |
177
INCORPORATED BY REFERENCE | ||||||||||||
EXHIBIT NO. | DESCRIPTION | Filed Herewith | FORM | File No. | Exhibit | Filing Date | ||||||
3.1 | 8-K | 001-37880 | 3.1 | September 27, 2016 | ||||||||
3.2 | 8-K | 001-37880 | 3.2 | September 27, 2016 | ||||||||
4.1 | S-1 | 333-213276 | 4.1 | September 8, 2016 | ||||||||
4.2 | 10-K | 001-37880 | 4.2 | February 24, 2020 | ||||||||
4.3 | 8-K | 001-37880 | 4.1 | January 9, 2018 | ||||||||
4.4 | 8-K | 001-378880 | 4.1 | September 5, 2019 | ||||||||
10.1 | # | S-1 | 333-213276 | 10.1 | August 24, 2016 | |||||||
10.2 | # | S-1 | 333-213276 | 10.2 | August 24, 2016 | |||||||
10.3 | # | 8-K | 001-37880 | 10.1 | September 5, 2019 | |||||||
10.4 | # | 10-K | 001-37880 | 10.4 | March 20, 2017 | |||||||
10.5 | # | 10-Q | 001-37880 | 10.4 | November 8, 2018 | |||||||
10.6 | # | 10-Q | 001-37880 | 10.1 | November 14, 2016 | |||||||
10.7 | # | 10-Q | 001-37880 | 10.2 | November 14, 2016 | |||||||
10.8 | # | 10-Q | 001-37880 | 10.3 | November 14, 2016 | |||||||
10.9 | # | 10-Q | 001-37880 | 10.3 | August 8, 2018 | |||||||
178
INCORPORATED BY REFERENCE | ||||||||||||
EXHIBIT NO. | DESCRIPTION | Filed Herewith | FORM | File No. | Exhibit | Filing Date | ||||||
10.10 | # | 10-K | 001-37880 | 10.16 | March 27, 2018 | |||||||
10.11 | # | 10-K | 001-37880 | 10.14 | March 27, 2019 | |||||||
10.12 | # | 10-K | 001-37880 | 10.12 | February 24, 2020 | |||||||
10.13 | # | 10-K | 001-37880 | 10.13 | February 24, 2020 | |||||||
10.14 | # | 10-Q | 001-37880 | 10.2 | November 8, 2018 | |||||||
10.15 | # | 10-Q | 001-37880 | 10.3 | November 8, 2018 | |||||||
10.16 | # | 10-Q | 001-37880 | 10.1 | August 8, 2018 | |||||||
10.17 | † | S-1/A | 333-213276 | 10.7 | September 8, 2016 | |||||||
10.18 | † | 10-Q | 001-37880 | 10.4 | November 14, 2016 | |||||||
10.19 | † | 10-K | 001-37880 | 10.23 | March 27, 2019 | |||||||
10.20 | † | S-1 | 333-213276 | 10.8 | August 24, 2016 | |||||||
10.21 | † | 10-K | 001-37880 | 10.21 | March 27, 2018 | |||||||
179
INCORPORATED BY REFERENCE | ||||||||||||
EXHIBIT NO. | DESCRIPTION | Filed Herewith | FORM | File No. | Exhibit | Filing Date | ||||||
10.22 | † | 10-K | 001-37880 | 10.26 | March 27, 2019 | |||||||
10.23 | † | S-1 | 333-213276 | 10.9 | August 24, 2016 | |||||||
10.24 | † | 10-K | 001-37880 | 10.23 | March 27, 2018 | |||||||
10.25 | † | 10-K | 001-37880 | 10.29 | March 27, 2019 | |||||||
10.26 | † | 10-K | 001-37880 | 10.17 | March 20, 2017 | |||||||
10.27 | † | 10-K | 001-37880 | 10.18 | March 20, 2017 | |||||||
10.28 | † | 10-Q | 001-37880 | 10.1 | November 5, 2018 | |||||||
10.29 | S-1 | 333-213276 | 10.11 | August 24, 2016 | ||||||||
10.30 | 10-Q | 001-37880 | 10.7 | November 14, 2016 | ||||||||
10.31 | † | 10-Q | 001-37880 | 10.1 | August 13, 2019 | |||||||
180
INCORPORATED BY REFERENCE | ||||||||||||
EXHIBIT NO. | DESCRIPTION | Filed Herewith | FORM | File No. | Exhibit | Filing Date | ||||||
10.32 | † | 10-Q | 001-37880 | 10.2 | August 13, 2019 | |||||||
10.33 | 8-K | 001-37880 | 10.1 | September 5, 2019 | ||||||||
23.1 | X | |||||||||||
31.1 | X | |||||||||||
31.2 | X | |||||||||||
32.1 | X | |||||||||||
32.2 | X | |||||||||||
101.INS | XBRL Instance Document. | X | ||||||||||
101.SCH | XBRL Taxonomy Extension Schema Document. | X | ||||||||||
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. | X | ||||||||||
101.DEF | XBRL Taxonomy Extension Definition Document. | X | ||||||||||
101.LAB | XBRL Taxonomy Extension Label Linkbase Document. | X | ||||||||||
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document. | X | ||||||||||
† | Portions of this exhibit (indicated by asterisks) have been omitted pursuant to a request for confidential treatment pursuant to Rule 24b-2 under the Securities Exchange Act of 1934. |
# | Indicates management contract or compensatory plan. |
181
Item 16. Form 10-K Summary.
None.
182
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
Novan, Inc. | |||
Date: May 20, 2020 | By: | /s/ Paula Brown Stafford | |
Paula Brown Stafford | |||
President and Chief Executive Officer (Principal Executive Officer) | |||
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
Name | Title | Date | ||
/s/ Paula Brown Stafford | President, Chief Executive Officer and Director | May 20, 2020 | ||
Paula Brown Stafford | ||||
/s/ John M. Gay | Vice President, Finance and Corporate Controller (Principal Financial Officer) | May 20, 2020 | ||
John M. Gay | ||||
/s/ Andrew J. Novak | Vice President, Accounting and Business Operations (Principal Accounting Officer) | May 20, 2020 | ||
Andrew J. Novak | ||||
/s/ Robert A. Ingram | Chairman of the Board | May 20, 2020 | ||
Robert A. Ingram | ||||
/s/ W. Kent Geer | Director | May 20, 2020 | ||
W. Kent Geer | ||||
/s/ Robert J. Keegan | Director | May 20, 2020 | ||
Robert J. Keegan | ||||
/s/ John Palmour | Director | May 20, 2020 | ||
John Palmour | ||||
/s/ Machelle Sanders | Director | May 20, 2020 | ||
Machelle Sanders | ||||
183