Attached files
| file | filename |
|---|---|
| 10-K - FORM 10-K - XBiotech Inc. | f10k_031620p.htm |
| EX-32.2 - EXHIBIT 32.2 - XBiotech Inc. | exh_322.htm |
| EX-32.1 - EXHIBIT 32.1 - XBiotech Inc. | exh_321.htm |
| EX-31.2 - EXHIBIT 31.2 - XBiotech Inc. | exh_312.htm |
| EX-31.1 - EXHIBIT 31.1 - XBiotech Inc. | exh_311.htm |
| EX-23.1 - EXHIBIT 23.1 - XBiotech Inc. | exh_231.htm |
| EX-21.1 - EXHIBIT 21.1 - XBiotech Inc. | exh_211.htm |
| EX-10.15 - EXHIBIT 10.15 - XBiotech Inc. | exh_1015.htm |
| EX-4.1 - EXHIBIT 4.1 - XBiotech Inc. | exh_41.htm |
Exhibit 10.14

CLINICAL MANUFACTURING AGREEMENT BY AND BETWEEN [[5252615]] XBIOTECH USA, INC. AN D JANSSEN RESEARCH & DEVELOPMENT LLC

1 [[5252615]] TABLE OF CONTENTS P age Article 1 DEFINITIONS .........................................................................................................1 Article 2 CLINICAL PRODUCT MANUFACTURE AND SUPPLY......................................5 2.1 Manufacture and Supply. .....................................................................................5 2.2 Materials and Capacity.........................................................................................5 2.3 Forecasts..............................................................................................................6 2.4 Purchase Orders ...................................................................................................6 2.5 Packaging. ...........................................................................................................7 2.6 Delivery of Clinical Products. ..............................................................................7 Article 3 3 . 1 3 . 2 PRICING; PAYMENT .............................................................................................7 Supply Price.........................................................................................................7 Invoices and Payments. ........................................................................................8 Article 4 4 . 1 TRANSITION ..........................................................................................................8 Transition.............................................................................................................8 Article 5 SPECIFICATIONS AND QUALITY CONTROL MATTERS .................................9 5.1 Compliance with Law. .........................................................................................9 5.2 Clinical Product Requirements. ............................................................................9 5.3 Specifications.....................................................................................................10 5.4 Quality Agreement.............................................................................................10 5.5 Nonconforming Clinical Product........................................................................10 5.6 Clinical Product Actions. ...................................................................................12 5.7 Manufacturing Site Audits. ................................................................................12 5.8 Regulatory Matters.............................................................................................12 5.9 Person - In - Plant. .................................................................................................13 5.10 Records and Information Management. ..............................................................14 Article 6 6 . 1 6 . 2 6 . 3 6 . 4 REPRESENTATIONS, WARRANTIES AND COVENANTS ...............................14 Mutual Representations and Warranties. ............................................................14 Capacity.............................................................................................................15 Certain Compliance Matters...............................................................................15 No Other Representations or Warranties. ...........................................................15 Article 7 7 . 1 CONFIDENTIALITY AND PUBLICITY ..............................................................16 Confidentiality. ..................................................................................................16 Article 8 8 . 1 8 . 2 TERM AND TERMINATION................................................................................16 Term. .................................................................................................................16 Termination by Mutual Agreement. ...................................................................16

2 [[5252615]] 8.3 Termination for Material Breach. .......................................................................17 8.4 Termination for Convenience. ............................................................................17 8.5 Effects of Termination or Expiration. .................................................................17 8.6 Survival. ............................................................................................................17 Article 9 9 . 1 9 . 2 9 . 3 DISPUTE RESOLUTION.......................................................................................17 Dispute Resolution, Generally............................................................................17 Mediation...........................................................................................................18 Arbitration. ........................................................................................................18 Article 10 INDEMNIFICATION............................................................................................20 10.1 Incorporation of Asset Purchase Agreement Indemnification Provisions. ...........20 Article 11 11 . 1 11 . 2 11 . 3 11 . 4 11 . 5 11 . 6 11 . 7 11 . 8 11 . 9 MISCELLANEOUS ..............................................................................................20 N o t i c e s . ............................... . ............................... . ............................... . ............. . 20 Governing Law. .................................................................................................20 Assignment. .......................................................................................................20 Designation of Affiliates. ...................................................................................20 Relationship of the Parties..................................................................................21 Force Majeure....................................................................................................21 S e v e r a bil i t y . ............................... . ............................................................... . ...... . 21 English Language...............................................................................................21 Waiver and Non - Exclusion of Remedies. ...........................................................22 11.10 Further Assurance. .............................................................................................22 11.11 Headings............................................................................................................22 11.12 Construction.......................................................................................................22 11.13 Counterparts.......................................................................................................23 11.14 Entire Agreement; Amendments. .......................................................................23 11.15 Specific Performance. ........................................................................................23 E xhibit List Exhibit A Exhibit B Exhibit C Exhibit D Exhibit E Exhibit F Exhibit G Exhibit H Initial Forecast Schedule Specifications Compliance with Laws Policy on Employment of Young People Johnson & Johnson Policy for Wood Pallets Company Records and Information Requirements New Formulations Transition Matters

CLINICAL MANUFACTURING AGREEMENT This CLINICAL MANUFACTURING AGREEMENT (this “ Agreement ”) is entered into as of December 30 , 2019 (the “ CMA Effective Date ”), by and between JANSSEN RESEARCH & DEVELOPMENT LLC , a Pennsylvania corporation, having its principal place of business at 800 / 850 Ridgeview Drive, Horsham, PA 19044 (hereinafter “ Company ”), and XBIOTECH USA, INC . , a Delaware corporation, having its principal place of business at 5217 Winnebago Lane, Austin, TX 78744 (“ SUPPLIER ”) . Company and SUPPLIER are sometimes referred to herein individually as a “ Party ” and collectively as the “ Parties ” . WHEREAS, XBiotech Inc . , an Affiliate of SUPPLIER, and Janssen Biotech, Inc . , an Affiliate of Company, have entered into that certain Asset Purchase Agreement, dated as of December 7 , 2019 (the “ Asset Purchase Agreement ”) ; and WHEREAS, pursuant to Section 2 . 4 (b)(iv) and Section 2 . 4 (c)(v) of the Asset Purchase Agreement, the Parties desire to enter into this Agreement for the Manufacture and supply of Clinical Products by SUPPLIER to Company, subject to the terms and conditions set forth herein ; and WHEREAS, this Agreement constitutes the Clinical Manufacturing Agreement contemplated by the Asset Purchase Agreement . NOW, THEREFORE, in consideration of the foregoing and the premises and conditions set forth herein, the Parties agree as follows : Article 1 DEFINITIONS As used in this Agreement, the following terms shall have the meanings set forth in this Article 1 , or if not defined in this Article 1 , shall have the meanings set forth in the Asset Purchase Agreement : “ Additional Equipment ” shall have the meaning set forth in Section 2 . 2 . 2 . “ Additional Specification ” shall have the meaning set forth in Section 5 . 3 . 1 . “ Affiliate ” means, with respect to a particular Person and a particular time, another Person that controls, is controlled by or is under common control with such first Person at any such time during the Term . For the purposes of this definition, the word “control” (including, with correlative meaning, the terms “controlled by” or “under the common control with”) means the actual power, either directly or indirectly through one or more intermediaries, to direct or cause the direction of the management and policies of a Person, whether by the ownership of fifty percent ( 50 % ) or more of the voting stock of such Person, by contract, or otherwise . “ Agreement ” shall have the meaning set forth in the preamble . “ AKA ” shall have the meaning set forth in Exhibit C .

2 [[5252615]] “ Backup Equipment ” shall have the meaning set forth in Section 2.2.3 . “ Business Day ” means a day other than Saturday, Sunday or any other day on which banking institutions in New York, New York are closed for business. “ Clinical Product ” means Product and placebo for use in clinical trials, in the form of bulk drug substance, pre - filled syringes or vials, in each case as further described in the Specifications or the Additional Specifications . “ Clinical Product Action ” shall have the meaning set forth in Section 5.6.1 . “ Clinical Product Action Notice ” shall have the meaning set forth in Section 5.6.1 . “ Clinical Product Requirements ” shall have the meaning set forth in Section 5.2 . “ CMA Effective Date ” shall have the meaning set forth in the preamble. “ Company ” shall have the meaning set forth in the preamble. “ Compound ” means the monoclonal antibody known as bermekimab (MABp1), the sequence of which is set forth in Schedule 1.1(c) to the Asset Purchase Agreement. “ CPR Mediation Procedure ” shall have the meaning set forth in Section 9.2.1 . “ CPR Rules ” shall have the meaning set forth in Section 9.3.1 . “ Current Capacity ” means, in the case of Clinical Products in the form of pre - filled syringes or vials, up to 4 , 160 units per month (or up to 50 , 000 units per calendar year), in each case irrespective of the drug concentration in the formulation included in such syringes or vials . “ Dispute ” shall have the meaning set forth in Section 9.1 . “ EMA ” means the European Medicines Agency or any successor agency(ies) or authority having substantially the same function. “ FCA ” shall have the meaning set forth in Exhibit C . “ FCPA ” shall have the meaning set forth in Exhibit C . “ FDA ” means the U.S. Food and Drug Administration and any successor agency(ies) or authority having substantially the same function. “ FFDCA ” means the U.S. Federal Food, Drug, and Cosmetic Act (21 U.S.C. † 301 et seq.), as amended from time to time. “ Firm Order ” shall have the meaning set forth in Section 2.3.2 . “ Force Majeure ” means any event beyond the reasonable control of the affected Party, which may include embargoes; war or acts of war, including terrorism; insurrections, riots, or

3 [[5252615]] civil unrest ; strikes, lockouts or other labor disturbances ; epidemics, fire, floods, earthquakes or other acts of nature ; acts, omissions or delays in acting by any Governmental Authority (other than delays incident to the ordinary course of drug development) ; and failure of plant or machinery . “ Forecast Schedule ” shall have the meaning set forth in Section 2.3.1 . “ Good Manufacturing Practices ” or “ GMP ” means the then - current good manufacturing practices required by the FFDCA, as amended, and the regulations promulgated thereunder by the FDA at 21 C . F . R . Parts 210 and 211 , for the manufacture and testing of pharmaceutical materials, and comparable Law related to the manufacture and testing of pharmaceutical materials in jurisdictions outside the U . S . , including the quality guideline promulgated by the ICH designated ICH Q 7 A, titled “Q 7 A Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients” and the regulations promulgated thereunder, in each case as they may be updated from time to time . “ International Public Organization ” means any of the organizations listed in 8 C.F.R. † 316.20, as amended from time to time. “ Know - How ” shall have the meaning set forth in the IP License Agreement. “ Licensed Space ” shall have the meaning set forth in the License to Occupy. “ Licensed Rights ” shall have the meaning set forth in the IP License Agreement, in respect of licenses granted thereunder by SUPPLIER. “ Manufacture ” means all activities and processes related to the manufacturing of any pharmaceutical product, or any ingredient thereof, including purchasing raw materials and intermediates, producing active pharmaceutical ingredient, formulating, and all labeling, packaging, in - process and finished product testing, storage and release of pharmaceutical product or any component or ingredient thereof, performance of quality assurance activities related to manufacturing and release of pharmaceutical product, and the performance of ongoing stability tests and regulatory activities related to any of the foregoing . When used as a verb, to “Manufacture” means to engage in Manufacturing activities . “ Manufacturing Capacity ” means the Current Capacity unless the Parties agree upon a new Manufacturing Capacity, including in connection with the installation of Additional Equipment, as provided in Section 2 . 2 . 2 below . “ Manufacturing Process ” means the processes and activities (and each step in the processes and activities) planned to be used to Manufacture Clinical Products as described in the master batch record for such Clinical Products, which shall be mutually agreed by the Parties and documented in writing . “ Manufacturing Representative ” has the meaning set forth in Section 5 . 9 . “ Manufacturing Sites ” means facilities of SUPPLIER or its Affiliates where Clinical Products are Manufactured from time to time .

4 [[5252615]] “ Materials ” means active pharmaceutical ingredients, raw ingredients, intermediaries, excipients, processing aids, packaging materials and any other components used in the Manufacture of Clinical Product . “ Nonconforming Clinical Product ” shall have the meaning set forth in Section 5.2 . “ Nonconformity ” shall have the meaning set forth in Section 5.2 . “ Officials ” shall have the meaning set forth in Section 6.2.2 . “ Party ” and “ Parties ” shall have the meaning set forth in the preamble. “ Payment ” shall have the meaning set forth in Section 6.2.2 . “ Person ” means an individual, sole proprietorship, partnership, limited partnership, limited liability partnership, corporation, limited liability company, business trust, joint stock company, trust, incorporated association, joint venture or similar entity or organization, including a government or political subdivision, department or agency of a government . “ Product ” means any pharmaceutical product containing the Compound, including all dosage forms, presentations, formulations and line extensions thereof, including a pharmaceutical product which is comprised of the Compound and other pharmaceutically active compound(s) and/or ingredients, any prototypes thereof and any variations thereof . “ Purchase Order ” shall have the meaning set forth in Section 2.4.1 . “ Quality Agreement ” shall have the meaning set forth in Section 5.4 . “ Quarterly Fee ” shall have the meaning set forth in Section 3.1 . “ Quarterly Manufacturing Capacity ” means, with respect to any calendar quarter, the Manufacturing Capacity for such calendar quarter, being calculated as three ( 3 ) times the Manufacturing Capacity per month . “ Regulatory Authority ” means any applicable Governmental Authority with authority over the Manufacture or Exploitation of a pharmaceutical product in a country or jurisdiction, including (a) in the U . S . , the FDA, and (b) in the European Union, the EMA . “ Specifications ” shall have the meaning set forth in Section 5.2 . “ SUPPLIER ” shall have the meaning set forth in the preamble. “ Term ” shall have the meaning set forth in Section 8.1 . “ Third Party ” means any Person other than (a) Company, (b) SUPPLIER, or (c) an Affiliate of either of Company or SUPPLIER. “ U.S. ” means the United States of America, including its territories and possessions.

5 [[5252615]] “ UKBA ” shall have the meaning set forth in Exhibit C . Article 2 CLINICAL PRODUCT MANUFACTURE AND SUPPLY 1. M anufacture and Supply . During the Term and pursuant to the terms of this Agreement, SUPPLIER shall supply to Company all of Company’s requirements of Clinical Products, except as otherwise permitted by this Agreement or mutually agreed by the Parties in accordance with this Agreement . 2. M aterials and Capacity . 1. Prioritization . During the Term, and subject to Company’s compliance with its obligations hereunder, SUPPLIER shall use reasonable best efforts to maintain capacity adequate to fulfill Company’s requirements of Clinical Products, as set forth in the applicable Forecast Schedule . If at any time during the Term, SUPPLIER is unable to Manufacture and supply all of the quantities of Clinical Products forecasted or ordered by Company hereunder, on the one hand, and all of the quantities of products desired by SUPPLIER, its Affiliates and their Third Party customers, on the other hand, due to (a) shortages of Materials that are used in both the Manufacture of Clinical Products and in the Manufacture of products for SUPPLIER, its Affiliates or their Third Party customers or (b) constraints on the capacity at the Manufacturing Sites, then SUPPLIER shall allocate Materials and capacity (including, for the avoidance of doubt, the use of any Additional Equipment) (i) first, to the Manufacture and supply of Clinical Products for Company and (ii) second, only to the extent of any remaining Materials and/or capacity, to the Manufacture and supply of products for SUPPLIER, its Affiliates or their Third Party customers . 2. Additional Equipment . If, at any time during the Term, notwithstanding SUPPLIER’S compliance with Section 2 . 2 . 1 , SUPPLIER is unable to Manufacture and supply all of the quantities of Clinical Products forecasted or ordered by Company hereunder due to constraints on the capacity at the Manufacturing Sites, SUPPLIER shall (i) promptly provide Company with written notice thereof and (ii) promptly provide such information as is reasonably requested by Company to enable Company to determine whether such constraints could be alleviated (in whole or in part) through the acquisition of additional equipment . If Company determines that such constraints could be alleviated (in whole or in part) through the acquisition of additional equipment, Company may, in its sole discretion, direct SUPPLIER to (and, upon such direction, SUPPLIER shall) acquire such additional equipment (“ Additional Equipment ”) at Company’s cost . In connection with the acquisition of any Additional Equipment, the Parties shall agree to an updated Manufacturing Capacity . SUPPLIER shall retain title to any Additional Equipment following any expiration or termination of this Agreement, other than the termination of this Agreement by Company pursuant to Section 8 . 3 (in which case Company shall be deemed to have been granted such title as of such termination) . 3. Backup Equipment . At any time during the Term, Company may, in its sole discretion, direct SUPPLIER to (and, upon such direction, SUPPLIER shall) acquire additional equipment to be used in the event that any of SUPPLIER’S equipment is temporarily or permanently rendered inoperative (“ Backup Equipment ”) at Company’s cost . SUPPLIER agrees

6 [[5252615]] that, during the Term, it shall use any such Backup Equipment only to the extent necessary to Manufacture and supply Clinical Products for Company, and for no other purpose . SUPPLIER shall retain title to any Backup Equipment following any expiration or termination of this Agreement, other than the termination of this Agreement by Company pursuant to Section 8 . 3 (in which case Company shall be deemed to have been granted such title as of such termination) . 2 . 2 . 4 Calculation of Costs ; Invoicing . Costs for Additional Equipment or Backup Equipment shall include, in addition to acquisition costs for such Additional Equipment or Backup Equipment, all reasonable documented out - of - pocket costs incurred by SUPPLIER with respect to the acquisition, installation, testing and validation of such Additional Equipment or Backup Equipment . SUPPLIER shall include any such costs actually incurred in the invoices delivered pursuant to Section 3 . 2 below, together with reasonably detailed supporting documentation therefor . 3. F orecasts . 1. Monthly Forecast Schedule . On the CMA Effective Date and within the first two weeks of each month commencing following the CMA Effective Date, Company shall submit to SUPPLIER a written, good faith rolling forecast of Company’s monthly requirements for Clinical Products for at least the following eighteen ( 18 ) months or such shorter period remaining under the Term (each such forecast, a “ Forecast Schedule ”) . The initial Forecast Schedule is attached hereto as Exhibit A . 2. Binding Commitment . The first four ( 4 ) months of each Forecast Schedule provided by Company shall be a binding commitment on Company to purchase from SUPPLIER, and, so long as such quantities are within the then - current Manufacturing Capacity, a binding commitment on SUPPLIER to sell to Company, the specified volume of Clinical Products set forth therein (each, a “ Firm Order ”) . For the avoidance of doubt, any months of a Forecast Schedule beyond the Firm Order period shall be non - binding . 4. P urchase Orders 1. All Clinical Product shall be supplied pursuant to purchase orders (each, a “ Purchase Order ”) submitted by Company to SUPPLIER . Each Purchase Order shall be consistent with the corresponding Firm Order and shall contain such Purchase Order number, quantities, order schedule, delivery locations, carrier information and other information reasonably necessary to permit correct delivery of Clinical Products for shipment, including such information and in a format as may be reasonably requested by SUPPLIER . 2. Exclusive Terms . This Agreement and the Quality Agreement set forth the exclusive contract terms between the Parties for, and shall apply to, all orders for Clinical Products . Any terms in any Firm Order, Purchase Order, invoice or other notice submitted by either Party to the other Party that are different from or additional to the provisions hereof shall be null and void notwithstanding SUPPLIER’s delivery of, and Company’s acceptance of, Clinical Products under any Firm Order, Purchase Order, invoice or other notice containing such terms .

7 [[5252615]] 5. P ackaging . SUPPLIER shall be responsible for packaging Clinical Products in accordance with the Specifications and the Quality Agreement . 6. D elivery of Clinical Products . SUPPLIER shall deliver Clinical Products to Company DAP (Incoterms 2010 ) at the location, and within five ( 5 ) days of (before or after) the delivery date, requested in the applicable Purchase Order . For the avoidance of doubt, SUPPLIER shall retain risk of loss to any Clinical Product unless and until such Clinical Product has been delivered to Company at such location as specified in the applicable Purchase Order . SUPPLIER shall provide Company notice of the anticipated delivery date at least three ( 3 ) days prior to delivery, and if such anticipated delivery date changes, SUPPLIER shall promptly provide Company notice of such change . Article 3 PRICING; PAYMENT 1. S upply Price . 1. Subject to Section 3 . 1 . 2 , for each calendar quarter during the Term, Company shall pay SUPPLIER in consideration for the Manufacture and supply of Clinical Products (which, for the avoidance of doubt, shall include Company’s right to occupy the Licensed Space pursuant to the License to Occupy) a fee of four million five hundred thousand dollars ( $ 4 , 500 , 000 ) for such quarter (the “ Quarterly Fee ”) . For purposes of this Agreement, references to “calendar quarters” in this Agreement shall include the calendar quarters (or partial calendar quarters) (i) beginning on the CMA Effective Date and (ii) ending on the last day of the Term . 2. If the Term includes any partial calendar quarter, the Quarterly Fee payable in respect of such partial calendar quarter shall be prorated based on the number of days in such partial calendar quarter (as compared to the number of days in a full calendar quarter) . 3. If, during any calendar quarter, SUPPLIER fails to deliver all of the Clinical Products specified in one or more Purchase Orders to be delivered to Company during such calendar quarter, the Quarterly Fee payable in respect of the next calendar quarter shall be reduced by the percentage of Clinical Products not so delivered as compared to the Clinical Products so specified for delivery ; provided that there shall be no such reduction to the extent such Clinical Products not so delivered were in excess of the Quarterly Manufacturing Capacity with respect to such calendar quarter . 4. If Company is entitled to a prorated or reduced Quarterly Fee pursuant to Section 3 . 1 . 2 , Section 3 . 1 . 3 and/or Section 5 . 5 . 2 (c), but has already paid such Quarterly Fee, SUPPLIER shall promptly reimburse Company for the difference between the Quarterly Fee paid and the prorated or reduced Quarterly Fee that was actually owed . If the application of Section 3 . 13 and/or Section 5 . 5 . 2 (c) would result in a reduction to a future Quarterly Fee, but no such future Quarterly Fee is payable hereunder, SUPPLIER shall promptly reimburse Company in an amount equal to the amount that such future Quarterly Fee would have been reduced .

8 [[5252615]] 2. I nvoices and Payments . 1. The first Quarterly Fee shall be payable within five ( 5 ) days of the CMA Effective Date . Thereafter, invoices with respect to each calendar quarter (or partial calendar quarter) shall be provided to Company no more than sixty ( 60 ) days prior to the first day of such calendar quarter . Payment terms will be net ninety ( 90 ) days after Company’s receipt of an uncontested invoice from SUPPLIER ; provided , however , the actual payment to SUPPLIER from Company or its designee will not be made until the next scheduled payment run as set forth at www . ap . jnj . com . Company may contest any invoice or portion thereof if (i) it reasonably believes that the charges reflected therein do not accurately reflect a proration or reduction in the Quarterly Fee required under Section 3 . 1 . 2 , 3 . 1 . 3 and/or 5 . 5 . 2 (c) or (ii) it disputes any of the costs included in such invoice pursuant to Section 2 . 2 . 4 , in each case by providing notice to SUPPLIER of such dispute within twenty ( 20 ) days of its receipt of such invoice Once the matter is resolved, Company shall pay the appropriate charges . SUPPLIER shall continue to perform its obligations under this Agreement during such dispute . If an invoice is disputed in part, SUPPLIER may issue a new invoice in compliance with this Section 3 . 2 . 1 reflecting solely the undisputed charges, and any such invoice shall be payable within ninety ( 90 ) days after receipt thereof ; provided , however , the actual payment to SUPPLIER from Company or its designee will not be made until the next scheduled payment run as set forth at www . ap . jnj . com . 2. SUPPLIER shall not invoice Company hereunder, and no claim for payments will be considered with respect to Clinical Products Manufactured hereunder prior to both Parties’ duly authorized representatives signing this Agreement and Company issuing a purchase order number to SUPPLIER with respect to the services provided hereunder, provided that Company shall use reasonable best efforts to issue such purchase orders at such times and in such manner as will facilitate payments in accordance with this Section 3 . 2 . Article 4 TRANSITION 1. T ransition. 1. During the Term and for the 12 months after the expiration or termination of this Agreement, the Parties shall cooperate and use reasonable best efforts to enable the prompt transition of the Manufacture and supply of Product from the existing Manufacturing processes of SUPPLIER at the Manufacturing Sites to new Manufacturing processes of Company at facilities designated by Company at no additional cost . SUPPLIER shall provide reasonable technical assistance, including (i) information and Know - How in its control and related to Product or the Compound (including any information described on Exhibit H ) and (ii) introductions and access to SUPPLIER’S suppliers of Materials, in each case as requested by Company to facilitate the foregoing . 2. In the event that SUPPLIER is unable to fulfill all of Company’s requirements for Clinical Products (as a result of constraints on capacity, Force Majeure, insolvency, bankruptcy or otherwise), upon Company’s request, SUPPLIER shall transfer existing

9 [[5252615]] Manufacturing processes for the Compound and Products to Company (or its designee) and provide reasonable technical assistance to Company (or its designee), including introductions and access to SUPPLIER’S suppliers of Materials, to the extent reasonably necessary to enable Company to Manufacture the Compound and Products during the Term . 3. In the event that SUPPLIER is unable to fulfill all of Company’s requirements for Clinical Products (as a result of constraints on capacity, Force Majeure, insolvency, bankruptcy or otherwise), upon Company’s request, SUPPLIER will transfer any other information and Know - How in its control reasonably requested by Company in order to enable Company to Manufacture the Compound and Products during the Term, including (a) complete sets of any preclinical or clinical data generated by or on behalf of SUPPLIER with respect to the Compound or any Products, (b) raw data tables with respect to the data described in clause (a), (c) Chemistry, Manufacture and Control (CMC) data or information generated by or on behalf of SUPPLIER with respect to the Compound or any Product, (d) any information described on Exhibit H and (e) any other Know - How that is necessary or specifically useful for the Manufacture of the Compound or Products, in each case to the extent that such information or know - how was not previously provided by SUPPLIER to Company . 4. Any Know - How or Trade Secrets transferred or otherwise provided by SUPPLIER to Company or its designee pursuant to this Article 4 shall be deemed to be Licensed Rights, with respect to SUPPLIER as Licensor and Company as Licensee under the IP License Agreement, unless and solely to the extent such Know - How or Trade Secrets is Seller Intellectual Property . Article 5 SPECIFICATIONS AND QUALITY CONTROL MATTERS 1. C ompliance with Law . SUPPLIER will, and will cause its Affiliates to, comply with applicable Laws, including GMP, in performing Manufacturing activities with respect to Clinical Products . 2. C linical Product Requirements . SUPPLIER hereby represents, warrants and covenants to Company that Clinical Products supplied to Company under this Agreement shall be Manufactured in accordance with the Manufacturing Process, applicable Laws (including GMP), the Quality Agreement and the policies of Company set forth on Exhibit D and Exhibit E hereto (the “ Manufacturing Methods and Procedures ”) . SUPPLIER hereby further represents, warrants and covenants to Company that Clinical Products supplied to Company under this Agreement shall, at the time of delivery, (a) conform to the applicable Clinical Product specifications set forth on Exhibit B hereto or, to the extent applicable, the Quality Agreement (as such specifications may be amended from time to time in accordance with Section 5 . 3 . 2 or the Quality Agreement, the “ Specifications ”) or the applicable Additional Specifications, (b) have at least eighteen ( 18 ) months shelf life from the date of filling of the drug product into syringes or vials and (c) conform to the volume and form (i . e . , bulk drug substance, prefilled syringes or vials) ((a), (b) and (c) collectively, the “ Clinical

10 [[5252615]] Product Requirements ”) . Any supply of Clinical Products that does not satisfy the Clinical Product Requirements at the time that such supply is released by SUPPLIER or its Affiliate to Company is referred to in this Agreement as “ Nonconforming Clinical Product ” and shall be regarded as having a “ Nonconformity . ” 3. S pecifications . 1. During the Term, SUPPLIER shall use reasonable best efforts to develop additional formulations of Clinical Products as described on Exhibit G hereto . SUPPLIER and Company shall cooperate and use reasonable best efforts to agree on Clinical Product specifications for such additional formulations (such agreed specifications, as such specifications may be amended from time to time in accordance with Section 5 . 3 . 2 or the Quality Agreement, the “ Additional Specifications ”) . For the avoidance of doubt, once Additional Specifications have been agreed, Company shall be permitted to place Purchase Orders in respect of the Clinical Products represented by such Additional Specifications . If, during the Term, Company determines that additional formulations of Clinical Products not set forth on Exhibit G are necessary, SUPPLIER may from time to time cooperate with Company to develop such formulations and any Additional Specifications with respect thereto, subject to the mutual agreement of the Parties with respect to the terms and conditions applicable to the activities described in this sentence (it being understood that the additional formulations set forth on Exhibit G hereto are not subject to this sentence and are instead subject to the first sentence of this Section 5 . 3 . 1 and shall be at no additional cost to Company) . 2. During the Term, if Company proposes any change(s) to the Specifications or Additional Specifications based on any requirement, request or recommendation of a Governmental Authority, Company shall deliver a written request for such change(s) to SUPPLIER, and SUPPLIER shall reasonably consider such change(s) in good faith . SUPPLIER shall have final decision - making authority with respect to such proposed change(s) ; provided that SUPPLIER shall implement any such change(s) required, requested or recommended by a Governmental Authority if such change(s) would not reasonably be expected to adversely affect SUPPLIER or its Affiliates or any Regulatory Authorization held thereby . Company shall be responsible for the incremental costs of any additional resources required to implement any such change(s) requested by Company . 4. Q uality Agreement . SUPPLIER and Company have entered into that certain Quality Agreement, dated as of December 19 , 2019 , relating to the Clinical Products supplied hereunder (the “ Quality Agreement ”) . SUPPLIER, either by itself or through its Affiliates, shall perform such quality control or analytical tests on Clinical Products and provide to Company certifications or other documents, in each case, as may be provided for in the Quality Agreement, and shall maintain such records as are reasonably necessary to demonstrate compliance with GMP in the Manufacture of Clinical Products, as may be provided for in the Quality Agreement . 5. N onconforming Clinical Product . 1. Inspection of Clinical Products .

11 [[5252615]] (a) Company will inspect Clinical Products supplied under this Agreement promptly upon receipt thereof . Subject to the immediately following sentence, Company shall have ninety ( 90 ) days following the delivery of any order of Clinical Products to notify SUPPLIER that it has rejected all or any part of such order in its reasonable and good faith belief that such order contains Nonconforming Clinical Product, which notice shall be accompanied by a sample of the allegedly Nonconforming Clinical Product . With respect to latent Nonconformities and Nonconformities not discoverable by Company within ninety ( 90 ) days of delivery through the use of reasonable inspection methods and procedures (a “ Latent Nonconformity ”), Company shall give notice to SUPPLIER by the first to occur of eighteen ( 18 ) months after delivery thereof or within sixty ( 60 ) days following detection of any such Latent Nonconformity . (b) If Company gives timely notice of allegedly Nonconforming Clinical Products in accordance with Section 5 . 5 . 1 (a), such Clinical Products shall be conclusively deemed to be Nonconforming Clinical Products, unless SUPPLIER delivers a written notice of disagreement (a “ Nonconformity Disagreement Notice ”) to Company within fifteen ( 15 ) days of receiving notice of the allegedly Nonconforming Clinical Products from Company . If Company fails to give timely notice of allegedly Nonconforming Clinical Products in accordance with Section 5 . 5 . 1 (a), such Clinical Products shall be conclusively deemed to have been accepted by Company . 2. Nonconformity . The following terms shall apply for Nonconforming Clinical Product that have not been accepted, or deemed accepted, by Company: (a) Company shall destroy the Nonconforming Clinical Products or return them to SUPPLIER, in accordance with SUPPLIER’s written instructions and at SUPPLIER’s expense ; and (b) SUPPLIER shall, at Company’s request, replace the Nonconforming Clinical Products ; and (c) SUPPLIER shall reduce the Quarterly Fees owed by Company to SUPPLIER (or refund the Quarterly Fees paid by Company to SUPPLIER, as applicable), as if SUPPLIER failed to deliver the quantity of Nonconforming Clinical Products, in accordance with and to the extent required by Section 3 . 1 . 3 . (d) Disagreement Regarding Nonconformity . If SUPPLIER does not agree with Company’s determination that any Clinical Products are Nonconforming Clinical Products and timely delivers a Nonconformity Disagreement Notice, then the Parties will select an independent Third Party laboratory reasonably acceptable to each Party to evaluate if the allegedly Nonconforming Clinical Products meet such requirements . Absent manifest error or fraud, this evaluation will be binding on the Parties . If the evaluation certifies that the allegedly Nonconforming Clinical Products do not meet the Clinical Product Requirements, SUPPLIER will be responsible for the cost of the evaluation . If the evaluation certifies that the allegedly Nonconforming Clinical Products do meet the requirements of Sections 5 . 2 (a) and (b) , then ( 1 ) Company shall be responsible for the cost of the evaluation, ( 2 ) the Clinical Products shall be deemed accepted, and ( 3 ) if SUPPLIER replaced the Nonconforming Clinical Products and if, as

12 [[5252615]] a result of such replacement, the total Clinical Products supplied by SUPPLIER exceeded the Manufacturing Capacity (on an annual or monthly basis), Company shall pay SUPPLIER for such Clinical Products . 6. C linical Product Actions . 1. Notification . SUPPLIER shall notify Company in writing promptly following its determination that any event, incident or circumstance related to safety issues or regulatory concerns has occurred that is reasonably likely to result in the need for a recall or withdrawal of Clinical Products supplied under this Agreement (a “ Clinical Product Action ”), and shall include in such notice (a “ Clinical Product Action Notice ”) the reasoning behind such determination and any supporting facts . Such Clinical Product Action Notice shall be given no later than five ( 5 ) Business Days after such determination is made ; provided that if any Regulatory Authority (a) threatens or initiates any action to remove Clinical Products from use in clinical trials in any country, or (b) requires a Party, or any of its Affiliates or (sub)licensees, to distribute a “Dear Doctor” letter or its equivalent regarding the use of Clinical Products, then, in either case ((a) or (b)), the Clinical Product Action Notice shall be given within one ( 1 ) Business Day after SUPPLIER becomes aware of the action, threat or requirement (as applicable) . 2. Expenses . Unless and solely to the extent a Clinical Product Action is necessitated by a Latent Nonconformity (or by the bad faith, willful misconduct or gross negligence of, or the material breach of this Agreement by, SUPPLIER), Company shall be solely responsible for all costs arising out of such Clinical Product Action and shall reimburse SUPPLIER for any out - of - pocket expenses incurred by SUPPLIER in carrying out a Clinical Product Action . If and then solely to the extent a Clinical Product Action is necessitated by Latent Nonconformity (or by the bad faith, willful misconduct or gross negligence of, or the material breach of this Agreement by, SUPPLIER), SUPPLIER shall reimburse Company for any out - of - pocket expenses incurred by Company or its Affiliates in assisting SUPPLIER to carry out a Clinical Product Action . 7. M anufacturing Site Audits . SUPPLIER will permit Company to conduct quality assurance audits and inspections of SUPPLIER’s and its Affiliates’ records and facilities relating to Manufacture of Clinical Products, during normal business hours, in accordance with the terms of the Quality Agreement . Each Company representative participating in any such audit or inspection shall enter into a confidentiality agreement in a form reasonably acceptable to SUPPLIER and any applicable Affiliate . 8. R egulatory Matters . 1. SUPPLIER’s Obligations . SUPPLIER shall, and shall ensure that its Affiliates shall, at their own cost, obtain and maintain throughout the Term any certificates, permits, licenses and approvals issued by any relevant Governmental Authority required for the Manufacture of Clinical Products at the Manufacturing Sites in accordance with this Agreement . 2. Regulatory Approval Cooperation . SUPPLIER shall provide Company with all supporting data and information relating to the Chemistry, Manufacture and Control (CMC) of Clinical Products at the Manufacturing Sites that is in the possession and control of SUPPLIER and

13 [[5252615]] necessary for regulatory submissions by Company, including all records, raw data, reports, authorizations, certificates, methodologies, batch documentation, raw material specifications, standard operating procedures, standard test methods, certificates of analysis, certificates of compliance and other documentation in its possession or under its control relating to the Manufacturing of the Clinical Products . 5 . 8 . 3 Regulatory Inspections . SUPPLIER shall permit Company or its representatives to be present at any visit or inspection by any Regulatory Authority to the extent related to Clinical Products, or to any Manufacturing Site or other facility used to Manufacture, to test or to warehouse Clinical Products or specific to the systems or process used for the Manufacture of Clinical Products . SUPPLIER shall notify Company within three ( 3 ) Business Days of becoming aware of any planned inspection and within twenty - four ( 24 ) hours of any unplanned or ongoing inspection . SUPPLIER will provide Company copies of all regulatory reports of inspection, copies of all regulatory correspondence from Regulatory Authorities and copies of proposed written responses to be provided to Regulatory Authorities for Company’s review and comment before submission to any Regulatory Authority . SUPPLIER and Company will also provide daily inspection summary reports specific to Clinical Products or to any Manufacturing Site or other facility used to Manufacture, to test or to warehouse Clinical Products or specific to the systems or process used for the Manufacture of Clinical Products, in each case, in a format acceptable to both Parties each day of such an inspection . If SUPPLIER receives any observations or warning from any Regulatory Authority relating to any Clinical Products or Manufacturing Site or other facility (if it relates to the Manufacture of any Clinical Products) or to the systems or process used for the Manufacture of Clinical Products, SUPPLIER shall within ten ( 10 ) Business Days of the date such observations or warning is received by SUPPLIER, remedy or cause the remedy of the issues identified in such notice or warning or, if any such issues cannot reasonably be remedied within such ten ( 10 ) Business Day period the Parties will agree on a plan to resolve such issues within a mutually agreed time period . If the Parties cannot agree, the matter will be referred to the Head of Quality of each Party for resolution, by providing written notice to the appropriate contact person specified in the relevant Quality Agreement . 5 . 8 . 4 Additional Support . SUPPLIER may from time to time provide additional support in furtherance of the Manufacture and supply of Clinical Products, including support to assist Company in complying with applicable Law or the requirements of any Regulatory Authority (e . g . , the performance of analytical testing using certain assays in support of demonstrating Phase 2 and Phase 3 process comparability), subject to the mutual agreement of the Parties with respect to the terms and conditions applicable to the activities described in this sentence . 5.9 P erson - In - Plant . During the Term, SUPPLIER agrees to permit Company’s personnel or duly authorized representatives to observe and consult with respect to the Manufacturing of Clinical Products (each such employee or agent a “ Manufacturing Representative ”) . Each Manufacturing Representative will serve as the Company’s representative at the Manufacturing Sites during Manufacture of Clinical Products . SUPPLIER will allow each Manufacturing Representative reasonable access to (A) all data and information regarding Manufacture of Clinical Products and (B) to be present during the Manufacture of Clinical Products . Each Manufacturing Representative will have access only to those portions of the Manufacturing Sites reasonably

14 [[5252615]] related to the Manufacture of Clinical Products as well as reasonable access to office space, data and communication resources on an as - needed basis to enable such Manufacturing Representative to carry out the activities contemplated herein . In no event will any Manufacturing Representative interfere with, and SUPPLIER will remain fully responsible for, the Manufacture of Clinical Products . Each Manufacturing Representative will coordinate closely with SUPPLIER in order to minimize the impact of his/her presence on operations and will comply with all of SUPPLIER’S policies and procedures regarding their presence in the Facilities including any training requirements . 5.10 R ecords and Information Management . SUPPLIER shall comply with the records and information management provisions set forth on Exhibit F . Article 6 REPRESENTATIONS, WARRANTIES AND COVENANTS 1. M utual Representations and Warranties . Each of the Parties hereby represents and warrants to the other Party as of the CMA Effective Date that: 1. Organization . It is a corporation duly organized, validly existing, and in good standing under the Laws of the jurisdiction of its organization, and has all requisite corporate power and authority to execute, deliver, and perform this Agreement . 2. Binding Agreement . This Agreement is a legal and valid obligation binding upon such Party and enforceable in accordance with its terms, subject to the effects of bankruptcy, insolvency, or other Laws of general application affecting the enforcement of creditor rights, judicial principles affecting the availability of specific performance, and general principles of equity (whether enforceability is considered a proceeding at law or equity) . 3. Authorization . The execution, delivery, and performance of this Agreement by such Party have been duly authorized by all necessary corporate action and do not conflict with any agreement, obligation, instrument, or understanding, oral or written, to which it is a party or by which it is bound, nor violate any applicable Law or any order, writ, judgment, injunction, decree, determination, or award of any Governmental Authority presently in effect applicable to such Party . 4. No Further Approval . It is not aware of any government authorization, consent, approval, license, exemption of or filing or registration with any Governmental Authority under any applicable Law, currently in effect, necessary for the execution and delivery of this Agreement or any other agreement or instrument executed in connection herewith . 5. No Inconsistent Obligations . It is not under any obligation, contractual or otherwise, to any Person that conflicts with or is inconsistent in any material respect with the terms of this Agreement, or that would impede the diligent and complete fulfillment of its obligations hereunder .

15 [[5252615]] 2. C apacity . SUPPLIER hereby represents and warrants to Company that (a) as of the CMA Effective Date, the capacity at the Manufacturing Sites to Manufacture and supply Clinical Products in the form of pre - filled syringes or vials will be at least equal to the Current Capacity and (b) it will have sufficient capacity to Manufacture and supply all of the Clinical Products included on the initial Forecast Schedule attached hereto as Exhibit A . 3. C ertain Compliance Matters . 1. No Violation of Law . Notwithstanding any other provision of this Agreement, neither Party shall be required to undertake any activity or obligation under this Agreement which it has reason to believe may violate any applicable Laws ; provided , however , a Party which so believes shall promptly inform the other Party of such belief . 2. Anti - Bribery and Corruption . Neither SUPPLIER nor its Affiliates will make any payment, either directly or indirectly, of money or other assets, including the compensation SUPPLIER derives from this Agreement (collectively, a “ Payment ”), to government or political party officials, officials of International Public Organizations, candidates for public office, or representatives of other businesses or persons acting on behalf of any of the foregoing (collectively, “ Officials ”) or other individuals where such Payment would constitute violation of any applicable Law, including the Foreign Corrupt Practices Act of 1977 , 15 U . S . C . †† 78 dd - 1 , et seq . , and the United Kingdom Bribery Act . In addition regardless of legality, neither SUPPLIER nor its Affiliates will make any Payment either directly or indirectly to Officials or other individuals if such Payment is for the purpose of improperly influencing decisions or actions to secure a business advantage, including with respect to the subject matter of this Agreement . SUPPLIER shall have necessary procedures in place to prevent bribery and corrupt conduct by itself and each of its Affiliates and subcontractors . All activities will be conducted in compliance with the U . S . False Claims Act and the U . S . Anti - Kickback Statute . SUPPLIER and each of its Affiliates and subcontractors shall conduct its activities hereunder in accordance with the provisions of Exhibit C to this Agreement . 4. N o Other Representations or Warranties . EXCEPT AS EXPRESSLY SET FORTH IN THIS AGREEMENT OR IN ANY OTHER WRITTEN AGREEMENT EXECUTED BY EACH OF THE PARTIES, THE PARTIES MAKE NO REPRESENTATIONS OR WARRANTIES OF ANY KIND WHATSOEVER, EITHER EXPRESS OR IMPLIED, WRITTEN OR ORAL, IN FACT OR BY OPERATION OF LAW, BY STATUTE OR OTHERWISE, AND EACH PARTY SPECIFICALLY DISCLAIMS ANY OTHER WARRANTIES, INCLUDING ANY EXPRESS OR IMPLIED WARRANTY OF QUALITY, MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE, OR WARRANTY OF NON - INFRINGEMENT .

16 [[5252615]] Article 7 CONFIDENTIALITY AND PUBLICITY 7.1 C onfidentiality . SUPPLIER will, and will cause its Affiliates and its and their Representatives, to keep confidential and not disclose to any Person (i) the terms of this Agreement or (ii) any non - public, confidential or proprietary information of Company or its Affiliates (including information relating to the Business) obtained pursuant to or in connection with this Agreement and to not use any such information other than in furtherance of the performance of its obligations hereunder . The obligations of SUPPLIER under this Section 7 . 1 shall not apply to information to the extent such information (a) becomes generally available to the public without breach of SUPPLIER’S or its Affiliates’ obligations under this Section 7 . 1 or under the Asset Purchase Agreement or any Related Document or (B) is required to be disclosed by Law or any Order ; provided , however , that in the case of the foregoing clause (B), to the extent not prohibited by such Law or Order, SUPPLIER shall notify Company as early in advance of such disclosure as is practicable to allow Company to take appropriate measures (and SUPPLIER shall reasonably cooperate, at the expense of Company, in the taking of such measures) to preserve the confidentiality of such information . Article 8 TERM AND TERMINATION 1. T erm . This Agreement shall become effective as of the CMA Effective Date and, unless earlier terminated pursuant to this Article 8 , shall continue in full force and effect until December 31 , 2021 (the “ Term ”) ; provided that the foregoing shall not limit any ongoing obligations of SUPPLIER with respect to Clinical Products ordered prior to the end of the Term, such as the performance of quality assurance activities or ongoing stability tests, as set forth in this Agreement or the Quality Agreement, following the end of the Term . Without limiting the foregoing, if Company requires additional supply of Clinical Products following the Term to complete then - ongoing clinical trials, then upon Company’s request, the Parties shall discuss in good faith an extension to the Term in order for SUPPLIER to provide such additional supply, including with respect to any modifications to the terms of this Agreement that would apply to such Clinical Product supply during such extension as may be reasonably requested by either Party . Any such requested modifications shall be commercially reasonable, and any such extension shall be subject to the mutual written agreement of the Parties . 2. T ermination by Mutual Agreement . This Agreement may be terminated at any time upon the mutual written agreement of the Parties.

17 [[5252615]] 3. T ermination for Material Breach . This Agreement may be terminated by either Party if the other Party has committed a material breach and has failed to remedy such breach within thirty ( 30 ) Business Days following receipt of a written notice of such breach from the non - breaching Party . 4. T ermination for Convenience . This Agreement may be terminated at any time by Company upon providing at least sixty ( 60 ) days’ prior written notice to SUPPLIER . 5. E ffects of Termination or Expiration . Termination or expiration of this Agreement shall not relieve the Parties of any liability or obligation which accrued hereunder prior to the effective date of such termination or expiration . Each Party shall be free, pursuant to Article 9 , to seek, without restriction as to the number of times it may seek, damages, costs and remedies that may be available to it under applicable Law or in equity and shall be entitled, following final resolution of a Dispute in accordance with Article 9 , to offset the amount of any damages and costs awarded pursuant to a final determination under Article 9 against any amounts due to such other Party under this Agreement . Upon termination or expiration of this Agreement, SUPPLIER shall transfer to Company all right, title and interest in, to and under any Inventory (including Clinical Products) in the possession of SUPPLIER at no cost to Company, and Company will acquire from SUPPLIER good and marketable title to all such Inventory, free and clear of any Liens . 6. S urvival . In the event of termination or expiration of this Agreement, in addition to the provisions of this Agreement that continue in effect in accordance with their terms, the following provisions of this Agreement shall survive : Articles 1 (Definitions) (as applicable), 7 (Confidentiality), 9 (Dispute Resolution), 10 (Indemnification) (solely as to activities arising during the Term) and 11 (Miscellaneous) ; Sections 2 . 3 (Forecasts), 4 . 1 (Transition), 5 . 5 (Nonconforming Clinical Product), 5 . 6 (Clinical Product Actions), 6 . 4 (No Other Representations or Warranties), 8 . 5 (Effects of Termination or Expiration) and 8 . 6 (Survival) ; and any other provisions of this Agreement that are necessary to interpret or effectuate the intent of the foregoing provisions . Article 9 DISPUTE RESOLUTION 9.1 D ispute Resolution, Generally . The Parties recognize that a dispute may arise relating to this Agreement (a “ Dispute ”) . Any Dispute, including Disputes that may involve the parent company, subsidiaries or Affiliates under common control of any Party, shall be resolved in accordance with this Article 9 .

18 [[5252615]] 2. M ediation . 1. The Parties shall first attempt in good faith to resolve any Dispute by confidential mediation in accordance with the then current Mediation Procedure of the International Institute for Conflict Prevention and Resolution (“ CPR Mediation Procedure ”) (www . cpradr . org) before initiating arbitration . The CPR Mediation Procedure shall control, except where it conflicts with these provisions, in which case these provisions control . The mediator shall be chosen pursuant to CPR Mediation Procedure . The mediation shall be held in New York, New York . 2. Either Party may initiate mediation by written notice to the other Party of the existence of a Dispute . The Parties agree to select a mediator within 20 days of the notice and the mediation will begin promptly after the selection . The mediation will continue until the mediator, or either Party, declares in writing, no sooner than after the conclusion of one full day of a substantive mediation conference attended on behalf of each Party by a senior business person with authority to resolve the Dispute, that the Dispute cannot be resolved by mediation . In no event, however, shall mediation continue more than 60 days from the initial notice by a Party to initiate mediation unless the Parties agree in writing to extend that period . 3. Any period of limitations that would otherwise expire between the initiation of mediation and its conclusion shall be extended until 20 days after the conclusion of the mediation . 3. A rbitration . 1. If the Parties fail to resolve the Dispute in mediation, and a Party desires to pursue resolution of the Dispute, the Dispute shall be submitted by either Party for resolution in arbitration pursuant to the then current CPR Non - Administered Arbitration Rules (“ CPR Rules ”) (www . cpradr . org), except where they conflict with these provisions, in which case these provisions control . The arbitration will be held in New York, New York . All aspects of the arbitration shall be treated as confidential . 2. The arbitrators will be chosen form the CPR Panel of Distinguished Neutrals, unless a candidate not on such panel is approved by both Parties . Each arbitrator shall be a lawyer with at least 15 years experience with a law firm or corporate law department of over 25 lawyers or who was a judge of a court of general jurisdiction . To the extent that the Dispute requires special expertise, the Parties will so inform CPR prior to the beginning of the selection process . 3. The arbitration tribunal shall consist of three arbitrators, of whom each Party shall designate one in accordance with the “screened” appointment procedure provided in CPR Rule 5 . 4 . The chair will be chosen in accordance with CPR Rule 6 . 4 . 4. If, however, the aggregate award sought by the Parties is less than $ 5 million and equitable relief is not sought, a single arbitrator shall be chosen in accordance with the CPR Rules . 5. Candidates for the arbitrator position(s) may be interviewed by representatives of the Parties in advance of their selection, provided that all Parties are represented .

19 [[5252615]] 6. The Parties agree to select the arbitrator(s) within 45 days of initiation of the arbitration . The hearing will be concluded within nine ( 9 ) months after selection of the arbitrator(s) and the award will be rendered within 60 days of the conclusion of the hearing, or of any post - hearing briefing, which briefing will be completed by both sides within 45 days after the conclusion of the hearing . In the event the Parties cannot agree upon a schedule, then the arbitrator(s) shall set the schedule following the time limits set forth above as closely as practical . 7. The hearing will be concluded in ten hearing days or less . Multiple hearing days will be scheduled consecutively to the greatest extent possible . A transcript of the testimony adduced at the hearing shall be made and shall be made available to each Party . 8. The arbitrator(s) shall be guided, but not bound, by the CPR Protocol on Disclosure of Documents and Presentation of Witnesses in Commercial Arbitration (www . cpradr . org) ("Protocol") . The Parties will attempt to agree on modes of document disclosure, electronic discovery, witness presentation, etc . within the parameters of the Protocol . If the Parties cannot agree on discovery and presentation issues, the arbitrator(s) shall decide on presentation modes and provide for discovery within the Protocol, understanding that the Parties contemplate reasonable discovery . 9. The arbitrator(s) shall decide the merits of any Dispute in accordance with the law governing this Agreement, without application of any principle of conflict of laws that would result in reference to a different law . The arbitrator(s) may not apply principles such as "amiable compositeur" or "natural justice and equity . " 10. The arbitrator(s) are expressly empowered to decide dispositive motions in advance of any hearing and shall endeavor to decide such motions as would a United States District Court Judge sitting in the jurisdiction whose substantive law governs . 11. The arbitrator(s) shall render a written opinion stating the reasons upon which the award is based . The Parties consent to the jurisdiction of the United States District Court for the district in which the arbitration is held for the enforcement of these provisions and the entry of judgment on any award rendered hereunder . Should such court for any reason lack jurisdiction, any court with jurisdiction may act in the same fashion . 12. Each Party has the right to seek from the appropriate court provisional remedies such as attachment, preliminary injunction, replevin, etc . to avoid irreparable harm, maintain the status quo , or preserve the subject matter of the Dispute . Rule 14 of the CPR Rules does not apply to this Agreement . 13. EACH PARTY HERETO WAIVES : ( 1 ) ITS RIGHT TO TRIAL OF ANY ISSUE BY JURY, ( 2 ) WITH THE EXCEPTION OF RELIEF MANDATED BY STATUTE OR RESULTING FROM THE WILLFUL MATERIAL BREACH OF THIS AGREEMENT, ANY CLAIM TO PUNITIVE, EXEMPLARY, MULTIPLIED, INDIRECT, CONSEQUENTIAL OR LOST PROFITS/REVENUES DAMAGES (EXCEPT, IN EACH CASE, TO THE EXTENT AWARDED TO A THIRD PARTY), AND ( 3 ) ANY CLAIM FOR ATTORNEY FEES, COSTS AND PREJUDGMENT INTEREST .

20 [[5252615]] Article 10 INDEMNIFICATION 10.1 I ncorporation of Asset Purchase Agreement Indemnification Provisions . This Agreement shall be deemed to be a “Related Document” for the purposes of Article VII of the Asset Purchase Agreement, and Article VII of the Asset Purchase Agreement will govern the indemnification obligations of the Parties with respect to any “Losses”, as such term is defined in the Asset Purchase Agreement, arising under this Agreement (including, for the avoidance of doubt, with respect to any “Losses” arising from, relating to or otherwise in connection with any breach of or failure to perform any covenant or agreement of SUPPLIER or Company, as applicable, contained in this Agreement) . Article 11 MISCELLANEOUS 1. N otices . All notices given by one Party to the other Party under this Agreement will follow the procedures and be delivered to the addresses set forth in Section 9 . 2 of the Asset Purchase Agreement . 2. G overning Law . THIS AGREEMENT SHALL BE GOVERNED BY, AND CONSTRUED IN ACCORDANCE WITH, THE LAWS OF THE STATE OF NEW YORK, REGARDLESS OF THE LAWS THAT MIGHT OTHERWISE GOVERN UNDER APPLICABLE PRINCIPLES OF CONFLICTS OF LAWS THEREOF . 3. A ssignment . Neither this Agreement nor any of the rights, interests or obligations hereunder shall be assigned, in whole or in part, by either of the Parties without the prior written consent of the other Party, and any assignment without such consent shall be null and void, except that either Party may, without the prior written consent of the other Party, assign (a) any or all of its rights and obligations under this Agreement to any of its Affiliates ( provided that the assigning Party shall remain responsible for the performance of such assignee Affiliate) or (b) this Agreement in its entirety to a Third Party acquirer of that portion of its business relating to the subject matter of this Agreement . Any successor or assignee of rights and/or obligations permitted hereunder shall, in writing, expressly assume performance of such rights and/or obligations . 4. D esignation of Affiliates . Each Party may discharge any obligation and exercise any right hereunder through delegation of its obligations or rights to any of its Affiliates . Each Party hereby guarantees the performance by its Affiliates of such Party’s obligations under this Agreement, and shall cause

21 [[5252615]] its Affiliates to comply with the provisions of this Agreement in connection with such performance . Any breach by a Party’s Affiliate of any of such Party’s obligations under this Agreement shall be deemed a breach by such Party, and the other Party may proceed directly against such Party without any obligation to first proceed against such Party’s Affiliate . 5. R elationship of the Parties . It is expressly agreed that SUPPLIER, on the one hand, and Company, on the other hand, are independent contractors, and it is further agreed that the Parties fully intend and expect that the relationship between the two Parties shall not constitute a partnership, joint venture or agency . Except as expressly provided herein, neither SUPPLIER nor Company shall have the authority to make any statements, representations or commitments of any kind, or to take any action which shall be binding on the other, without the prior written consent of the other Party to do so . All individuals employed by a Party shall be employees of that Party and not of the other Party and all costs and obligations incurred by reason of such employment shall be for the account and expense of such Party . 6. F orce Majeure . Both Parties shall be excused from the performance of their obligations under this Agreement to the extent that such performance is prevented by Force Majeure and the nonperforming Party promptly provides notice of such Force Majeure circumstances to the other Party . Such excuse shall be continued so long as the condition constituting Force Majeure continues and the nonperforming Party takes reasonable best efforts to remove the condition . Notwithstanding the foregoing, a Party shall not be excused from making payments owed hereunder because of a Force Majeure affecting such Party . If a Force Majeure persists for more than ninety ( 90 ) days, then the Parties shall discuss in good faith the modification of the Parties’ obligations under this Agreement in order to mitigate the delays caused by such Force Majeure . In the event a Party is prevented from performing its obligations under this Agreement due to Force Majeure for more than six ( 6 ) months according to this Section 11 . 6 , the other Party shall have the right to terminate this Agreement upon sixty ( 60 ) days’ notice after the expiration of such period . A termination under this Section 11 . 6 by either Party shall be treated as a termination under Section 8 . 2 . 7. S everability . If any one or more of the provisions of this Agreement is held to be invalid or unenforceable by any court of competent jurisdiction from which no appeal can be or is taken, the provision shall be considered severed from this Agreement and shall not serve to invalidate any remaining provisions hereof . The Parties shall make good faith efforts to replace any invalid or unenforceable provision with a valid and enforceable one such that the objectives contemplated by the Parties when entering this Agreement may be realized . 8. E nglish Language . This Agreement shall be written in and executed in, and all other communications under or in connection with this Agreement shall be in, the English language . Any translation into any other language shall not be an official version hereof or thereof, and in the event of any conflict

22 [[5252615]] in interpretation between the English version and such translation, the English version shall control. 9. W aiver and Non - Exclusion of Remedies . Any term or condition of this Agreement may be waived at any time by the Party that is entitled to the benefit thereof, but no such waiver shall be effective unless set forth in a written instrument duly executed by or on behalf of the Party waiving such term or condition . The waiver by either Party of any right hereunder or of the failure to perform or of a breach by the other Party shall not be deemed a waiver of any other right hereunder or of any other breach or failure by such other Party whether of a similar nature or otherwise . The rights and remedies provided herein are cumulative and do not exclude any other right or remedy provided by applicable Law or otherwise available except as expressly set forth herein . 10. F urther Assurance . Each Party shall duly execute and deliver, or cause to be duly executed and delivered, such further instruments and do and cause to be done such further acts and things, including the filing of such assignments, agreements, documents, and instruments, as may be necessary or as the other Party may reasonably request in connection with this Agreement to carry out more effectively the provisions and purposes hereof . 11. H eadings . The headings of each Article and Section in this Agreement have been inserted for convenience of reference only and are not intended to limit or expand on the meaning of the language contained in the particular Article or Section . 12. C onstruction . Whenever this Agreement refers to a number of days without using a term otherwise defined herein, such number refers to calendar days, whether or not “calendar days” is expressly stated . Except where the context otherwise requires, (a) wherever used, the singular shall include the plural, the plural shall include the singular ; (b) the use of any gender shall be applicable to all genders ; (c) the terms “including,” “include,” “includes” and “for example” shall not limit the generality of any description preceding such term and, as used herein, shall have the same meaning as “including, but not limited to,” and “including, without limitation” ; (d) the words “herein”, “hereof” and “hereunder”, and words of similar import, refer to this Agreement in its entirety and not to any particular provision hereof ; (e) the word “will” means “shall” ; (f) if a period of time is specified and dates from a given day or Business Day, or the day or Business Day of an act or event, it is to be calculated exclusive of that day or Business Day ; (g) “Dollar”, “USD” or “ $ ” means U . S . Dollars ; (h) references to a particular Person include such Person’s successors and assigns to the extent not prohibited by this Agreement ; (i) a capitalized term not defined herein but reflecting a different part of speech than a capitalized term which is defined herein shall be interpreted in a correlative manner ; (j) any definition of or reference to any agreement, instrument or other document herein shall be construed as referring to such agreement, instrument or other document as from time to time amended, supplemented or otherwise modified (subject to any restrictions on such amendments, supplements or

23 [[5252615]] modifications set forth herein) ; (k) any provision under this Agreement requiring the mutual agreement of the Parties or the consent or approval of a Party shall only be satisfied if made in writing signed by the relevant Party(ies) and (l) if this Agreement is terminated in accordance with its terms, the “Term” shall be deemed to end on the effective date of such termination . The language of this Agreement shall be deemed to be the language mutually chosen by the Parties and no rule of strict construction shall be applied against either Party . Each Party represents that it has been represented by legal counsel in connection with this Agreement and acknowledges that it has participated in the drafting hereof . 13. C ounterparts . This Agreement may be executed in two ( 2 ) or more counterparts, each of which shall be deemed an original, but all of which together shall constitute one and the same instrument . This Agreement may be executed by . pdf or other electronically transmitted signatures and such signatures shall be deemed to bind each Party as if they were the original signatures . 14. E ntire Agreement; Amendments . This Agreement, including the Exhibits hereto, sets forth the complete, final and exclusive agreement and all the covenants, promises, agreements, warranties, representations, conditions and understandings between the Parties with respect to the subject matter hereof and supersedes, as of the CMA Effective Date, all prior and contemporaneous agreements and understandings between the Parties with respect to the subject matter hereof . In the event of any inconsistency between the body of this Agreement or any Exhibits to this Agreement and the Asset Purchase Agreement or any other Related Document, this Agreement shall govern and control with respect to the supply of Clinical Product and the specific subject matter hereof, and the Asset Purchase Agreement and other Related Documents shall govern and control with respect to all other matters . There are no covenants, promises, agreements, warranties, representations, conditions or understandings, either oral or written, between the Parties with respect to the subject matter hereof other than as are set forth herein and therein . No subsequent alteration, amendment, change or addition to this Agreement shall be binding upon the Parties unless reduced to writing and signed by an authorized officer of each Party . In the event of any inconsistency between the body of this Agreement and any Exhibits to this Agreement, unless otherwise expressly stated to the contrary in such Exhibit, the terms contained in this Agreement shall govern and control . 15. S pecific Performance . The Parties agree that irreparable damage would occur and that the Parties would not have any adequate remedy at law in the event that any of the provisions of this Agreement were not performed in accordance with their specific terms or were otherwise breached . It is accordingly agreed that the Parties shall be entitled to an injunction or injunctions to prevent breaches of this Agreement and to enforce specifically the terms and provisions of this Agreement, this being in addition to any other remedy to which they are entitled at law or in equity and as further set forth in Article 9 . For the avoidance of doubt, this Section 11 . 15 shall not restrict any Party from asserting that the terms and provisions of this Agreement have not

24 [[5252615]] been breached (or would not be breached) by the actions or omissions (or intended actions or omissions) of such Party. [SIGNATURE PAGE FOLLOWS]

IN WITNESS WHEREOF, the Parties have signed this Clinical Manufacturing Agreement as of the date first set forth above . JANSSEN RESEARCH & DEVELOPMENT LLC By: Name: Title: XBIOTECH USA, INC. By: Name: Title; U5252615ll

IN WITNESS WHEREOF, the Parties have signed this Clinical Manufacturing Agreement as of the date first set forth above. JANSSEN RESEARCH & DEVELOPMENT LLC By: Name: Title: XBIOTECH USA, INC. \ By: Name: Title: Chief Executive Officer [[5252615]]

Exhibit A - 1 [[5252615]] Exhibit A Initial Forecast Schedule

[[5262776]] Exhibit A Initial Forecast Schedule 1 M o n t h s 18 - - - - 4100 - 17 - - - - 4100 - 16 - - - - 4100 - 15 - - - - 4100 - 14 - - - - 4100 - 13 - - - - 4100 - 12 - - - - 4100 - 11 - - - - 4100 - 10 - - - - 4100 - 9 - - - - 4100 - 8 - - - - 4100 - 7 - - - - 4100 - 6 - - - - 4100 - 5 - - - - 4100 - Binding Commitment 4 - - - - 4100 100 3 - - - - 4100 - 2 - - - - 4100 - 1 336 48 48 24 2100 500 Vials (units) 100 mg/ml 150 mg/ml 175 mg/ml 200 mg/ml Syringes (units) 2 Bulk Drug Substance (g) 1 Schedule does not include demand required for release testing, stability or similar studies. Seller will provide additional Clinical Products as necessary for such studies. 2 Syringe demand will be allocated between the formulations listed in Exhibit G and placebo.

Exhibit B - 1 [[5252615]] Exhibit B Specifications Specifications of existing strengths must meet all specifications as listed in the Product IND/IMPD. Specifications of the new formulations will be adjusted accordingly. [See attached.]
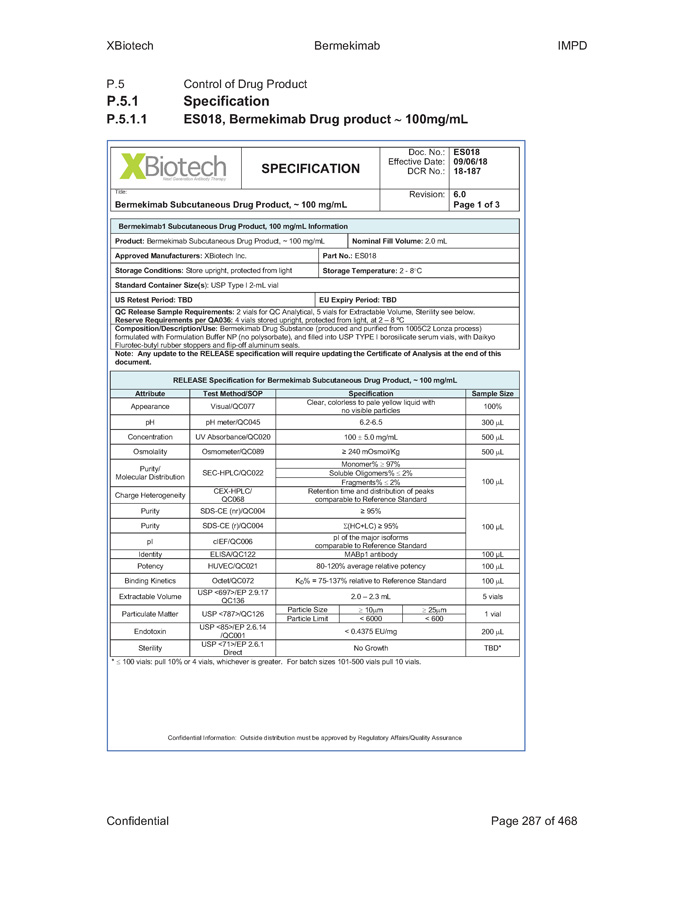
XBiotech Bermekimab I M PD P.5 P.5.1 P .5.1.1 Control of Drug Product Specification ES018, Bermekimab Drug product 100mg/ml Confidential Information: Outside distribution must be approved by Regulatory Affairs/Quality Assurance B i N o ext G e t nera t e ion Ant i c body T h herapy SPECIFICATION Doc. No.: Effective Date: DCR No.: ES018 09 / 0 6 / 18 18 - 187 Title: Bermekimab Subcutaneous Drug Product, ~ 100 mg/ml Revision: 6.0 Page 1 of 3 Bermekimab1 Subcutaneous Drug Product, 100 mg/ml Information Product: Bermekimab Subcutaneous Drug Product, ~ 100 mg/mL Nominal Fill Volume: 2.0 mL Approved Manufacturers: XBiotech Inc. Part No.: ES018 Storage Conditions: Store upright, protected from light Storage Temperature: 2 - 8 C Standard Container Size(s ): USP Type I 2 - mL vial US Retest Period: TBD EU Expiry Period: TBD QC Release Sample Requirements: 2 vials for QC Analytical, 5 vials for Extractable Volume, Sterility see below. Reserve Requirements per QA036: 4 vials stored upright, protected from light, at 2 - 8 ƒ C Composition/Description/Use: Bermekimab Drug Substance (produced and purified from 1005C2 Lonza process) formulated with Formulation Buffer NP (no polysorbate), and filled into USP TYPE I borosilicate serum vials, with Daikyo Flurotec - butyl rubber stoppers and flip - off aluminum seals. Note: Any update to the RElEASE specification will require updating the Certificate of Analysis at the end of this document. RElEASE Specification for Bermekimab Subcutaneous Drug Product, ~ 100 mg/ml Attribute Test Method/SOP Specification Sample Size Appearance Visual/QC077 Clear, colorless to pale yellow liquid with no visible particles 100% pH pH meter/QC045 6.2 - 6.5 300 L Concentration UV Absorbance/QC020 100 5.0 mg/mL 500 L Osmolality Osmometer/QC089 ೦ 240 mOsmol/Kg 500 L Purity/ Molecular Distribution SEC - HPLC/QC022 Monomer% 97% 100 L Soluble Oligomers% 2% Fragments% 2% Charge Heterogeneity CEX - HP L C / QC068 Retention time and distribution of peaks comparable to Reference Standard Purity SDS - CE (nr)/QC004 ೦ 95% 100 µL Purity SDS - CE (r)/QC004 (HC+LC) ೦ 95% pI cIEF/QC006 pI of the major isoforms comparable to Reference Standard Identity ELISA/QC122 MABp1 antibody 100 µL Potency HUVEC/QC021 80 - 120% average relative potency 100 L Binding Kinetics Octet/QC072 K D % = 75 - 137% relative to Reference Standard 100 L Extractable Volume USP <697>/EP 2.9.17 QC136 2.0 - 2.3 mL 5 vials Particulate Matter USP <787>/QC126 Particle Size 10 m 25 m 1 vial Particle Limit < 6000 < 600 Endotoxin USP <85>/EP 2.6.14 /QC001 < 0.4375 EU/mg 200 L Sterility USP <71>/EP 2.6.1 Direct No Growth TBD* * 100 vials: pull 10% or 4 vials, whichever is greater. For batch sizes 101 - 500 vials pull 10 vials. Confidential Page 287 of 468
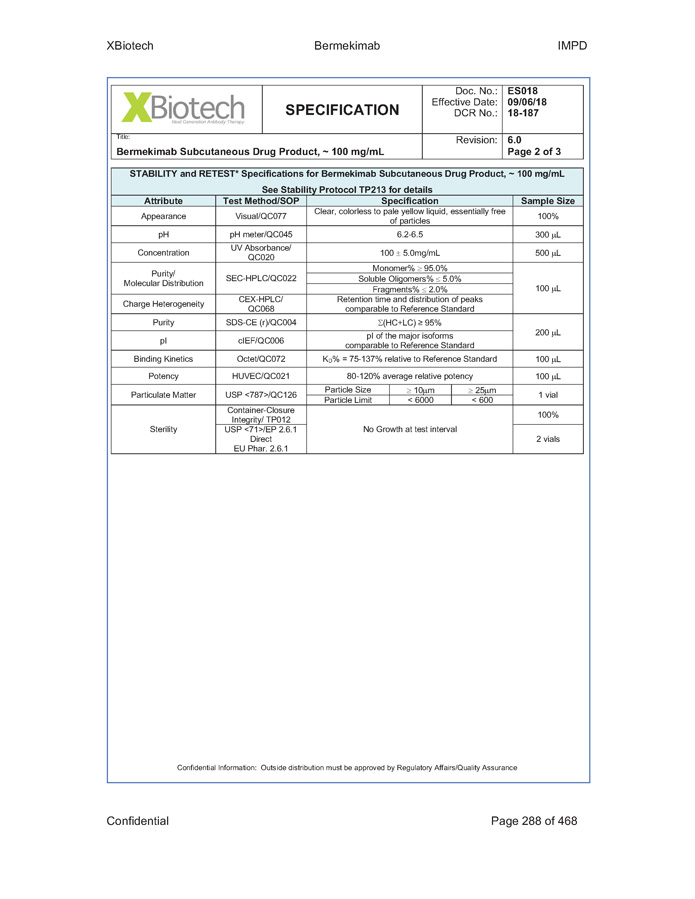
XBiotech Bermekimab I M PD Confidential lnformation: Outside distribution must be approved by Regulatory Affairs/Quality Assurance B i N o ext G e t nera t e ion Ant i c body T h herapy SPECIFICATION Doc. No.: Effective Date: DCR No.: ES018 09 / 0 6 / 1 8 18 - 187 Title: Bermekimab Subcutaneous Drug Product, ~ 100 mg/ml Revision: 6.0 Page 2 of 3 STABIlITY and RETEST* Specifications for Bermekimab Subcutaneous Drug Product, ~ 100 mg/ml See Stability Protocol TP213 for details Attribute Test Method/SOP Specification Sample Size Appearance Visual/QC077 Clear, colorless to pale yellow liquid, essentially free of particles 100% pH pH meter/QC045 6.2 - 6.5 300 L Concentration UV Absorbance/ QC020 100 5.0mg/mL 500 L Purity/ Molecular Distribution SEC - HPLC/QC022 Monomer% 95.0% 100 L Soluble Oligomers% 5.0% Fragments% 2.0% Charge Heterogeneity CEX - HP L C / QC068 Retention time and distribution of peaks comparable to Reference Standard Purity SDS - CE (r)/QC004 (HC+LC) ೦ 95% 200 L pl clEF/QC006 pl of the major isoforms comparable to Reference Standard Binding Kinetics Octet/QC072 K D % = 75 - 137% relative to Reference Standard 100 L Potency HUVEC/QC021 80 - 120% average relative potency 100 L Particulate Matter USP <787>/QC126 Particle Size 10 m 25 m 1 vial Particle Limit < 6000 < 600 Sterility Container - Closure lntegrity/ TP012 No Growth at test interval 100% USP <71>/EP 2.6.1 Direct EU Phar. 2.6.1 2 vials Confidential Page 288 of 468
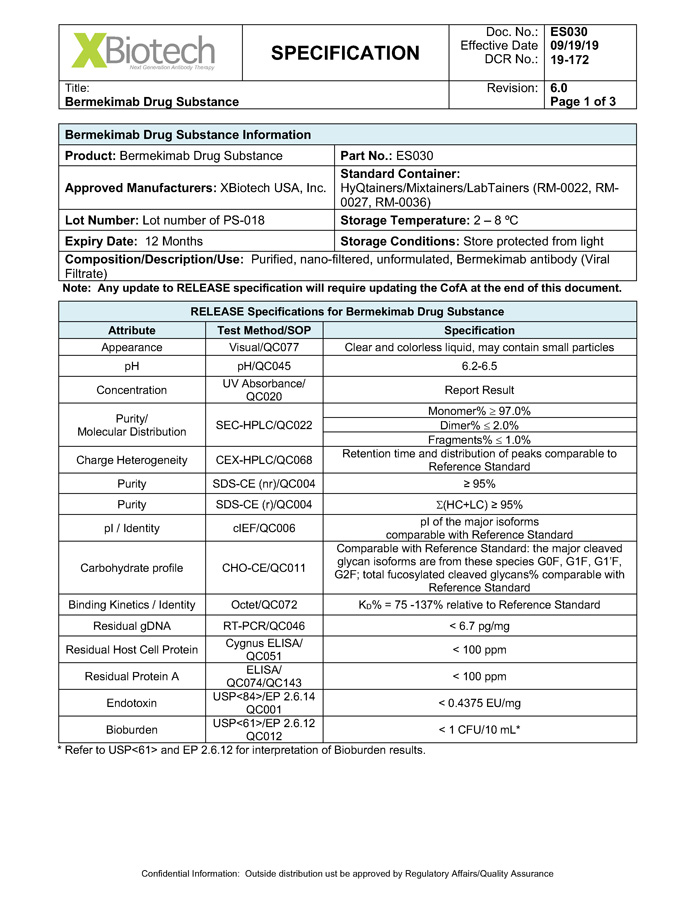
Confidential Information: Outside distribution ust be approved by Regulatory Affairs/Quality Assurance B i N o ext G e t nera t e ion Ant i c body T h herapy SPECIFICATION Doc. No.: Effective Date DCR No.: ES030 09/19/19 19 - 172 Title: Bermekimab Drug Substance Revision: 6.0 Page 1 of 3 Bermekimab Drug Substance Information Product: Bermekimab Drug Substance Part No.: ES030 Approved Manufacturers: XBiotech USA, Inc. Standard Container: HyQtainers/Mixtainers/LabTainers (RM - 0022, RM - 0027, RM - 0036) Lot Number: Lot number of PS - 018 Storage Temperature: 2 – 8 ºC Expiry Date: 12 Months Storage Conditions: Store protected from light Composition/Description/Use: Purified, nano - filtered, unformulated, Bermekimab antibody (Viral Filtrate) Note: Any update to RELEASE specification will require updating the CofA at the end of this document. RELEASE Specifications for Bermekimab Drug Substance Attribute Test Method/SOP Specification Appearance Visual/QC077 Clear and colorless liquid, may contain small particles pH pH/QC045 6.2 - 6.5 Concentration UV Absorbance/ QC020 Report Result Purity/ Molecular Distribution SEC - HPLC/QC022 Monomer% 97.0% Dimer% 2.0% Fragments% 1.0% Charge Heterogeneity CEX - HPLC/QC068 Retention time and distribution of peaks comparable to Reference Standard Purity SDS - CE (nr)/QC004 ≥ 95% Purity SDS - CE (r)/QC004 (HC+LC) ≥ 95% pI / Identity cIEF/QC006 pI of the major isoforms comparable with Reference Standard Carbohydrate profile CHO - CE/QC011 Comparable with Reference Standard: the major cleaved glycan isoforms are from these species G0F, G1F, G1’F, G2F; total fucosylated cleaved glycans% comparable with Reference Standard Binding Kinetics / Identity Octet/QC072 K D % = 75 - 137% relative to Reference Standard Residual gDNA RT - PCR/QC046 < 6.7 pg/mg Residual Host Cell Protein Cygnus ELISA/ QC051 < 100 ppm Residual Protein A ELISA/ QC 07 4 / QC 14 3 < 100 ppm Endotoxin USP<84>/EP 2.6.14 QC001 < 0.4375 EU/mg Bioburden USP<61>/EP 2.6.12 QC012 < 1 CFU/10 mL* * Refer to USP<61> and EP 2.6.12 for interpretation of Bioburden results.
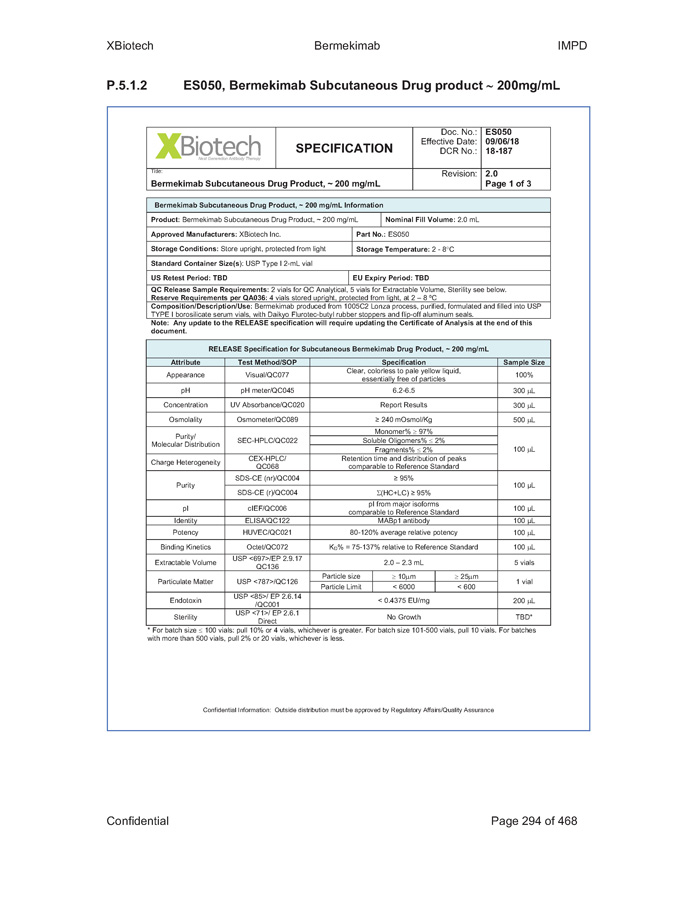
XBiotech Bermekimab I M PD P .5.1.2 ES050, Bermekimab Subcutaneous Drug product 200mg/ml Confidential Information: Outside distribution must be approved by Regulatory Affairs/Quality Assurance Bi N o ext G e t nera t e ion Ant i c body T h herapy SPECIFICATION Doc. No.: Effective Date: DCR No.: ES050 09 / 0 6 / 1 8 18 - 187 Title: Bermekimab Subcutaneous Drug Product, ~ 200 mg/ml Revision: 2.0 Page 1 of 3 Bermekimab Subcutaneous Drug Product, ~ 200 mg/ml Information Product: Bermekimab Subcutaneous Drug Product, ~ 200 mg/mL Nominal Fill Volume: 2.0 mL Approved Manufacturers: XBiotech Inc. Part No.: ES050 Storage Conditions: Store upright, protected from light Storage Temperature: 2 - 8 C Standard Container Size(s ): USP Type I 2 - mL vial US Retest Period: TBD EU Expiry Period: TBD QC Release Sample Requirements: 2 vials for QC Analytical, 5 vials for Extractable Volume, Sterility see below. Reserve Requirements per QA036: 4 vials stored upright, protected from light, at 2 - 8 ƒ C Composition/Description/Use: Bermekimab produced from 1005C2 Lonza process, purified, formulated and filled into USP TYPE I borosilicate serum vials, with Daikyo Flurotec - butyl rubber stoppers and flip - off aluminum seals. Note: Any update to the RElEASE specification will require updating the Certificate of Analysis at the end of this document. RElEASE Specification for Subcutaneous Bermekimab Drug Product, ~ 200 mg/ml Attribute Test Method/SOP Specification Sample Size Appearance Visual/QC077 Clear, colorless to pale yellow liquid, essentially free of particles 100% pH pH meter/QC045 6.2 - 6.5 300 L Concentration UV Absorbance/QC020 Report Results 300 L Osmolality Osmometer/QC089 ೦ 240 mOsmol/Kg 500 L Purity/ Molecular Distribution SEC - HPLC/QC022 Monomer% 97% 100 L Soluble Oligomers% 2% Fragments% 2% Charge Heterogeneity CEX - HP L C / QC068 Retention time and distribution of peaks comparable to Reference Standard Purity SDS - CE (nr)/QC004 ೦ 95% 100 µL SDS - CE (r)/QC004 (HC+LC) ೦ 95% pI cIEF/QC006 pI from major isoforms comparable to Reference Standard 100 µL Identity ELISA/QC122 MABp1 antibody 100 µL Potency HUVEC/QC021 80 - 120% average relative potency 100 L Binding Kinetics Octet/QC072 K D % = 75 - 137% relative to Reference Standard 100 L Extractable Volume USP <697>/EP 2.9.17 QC136 2.0 - 2.3 mL 5 vials Particulate Matter USP <787>/QC126 Particle size 10 m 25 m 1 vial Particle Limit < 6000 < 600 Endotoxin USP <85>/ EP 2.6.14 /QC001 < 0.4375 EU/mg 200 L Sterility USP <71>/ EP 2.6.1 Direct No Growth TBD* * For batch size 100 vials: pull 10% or 4 vials, whichever is greater. For batch size 101 - 500 vials, pull 10 vials. For batches with more than 500 vials, pull 2% or 20 vials, whichever is less. Confidential Page 294 of 468
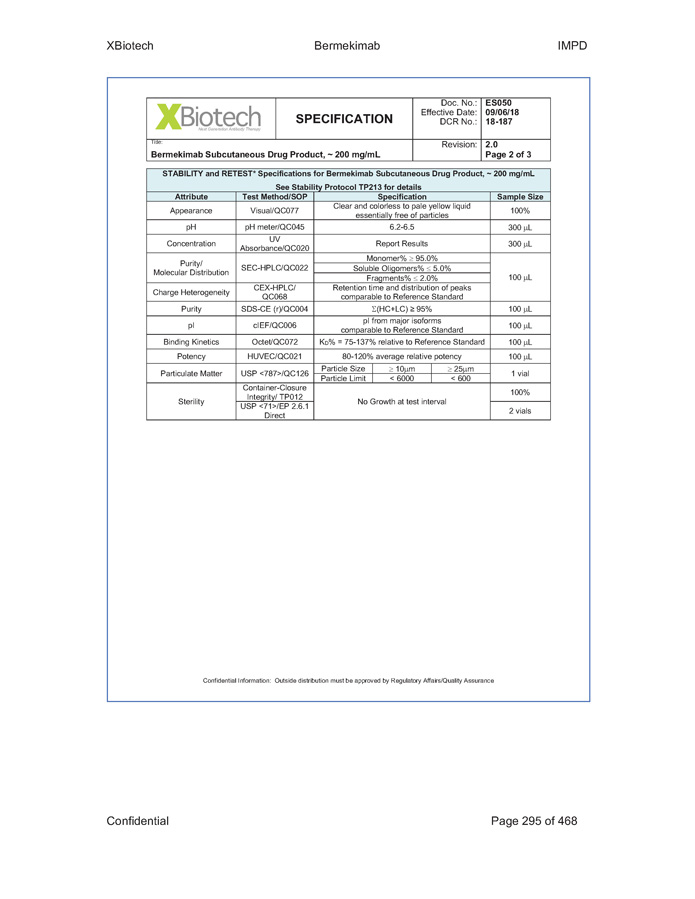
XBiotech Bermekimab I M PD Confidential lnformation: Outside distribution must be approved by Regulatory Affairs/Quality Assurance Bi N o ext G e t nera t e ion Ant i c body T h herapy SPECIFICATION Doc. No.: Effective Date: DCR No.: ES050 09 / 0 6 / 1 8 18 - 187 Title: Bermekimab Subcutaneous Drug Product, ~ 200 mg/ml Revision: 2.0 Page 2 of 3 STABIlITY and RETEST* Specifications for Bermekimab Subcutaneous Drug Product, ~ 200 mg/ml See Stability Protocol TP213 for details Attribute Test Method/SOP Specification Sample Size Appearance Visual/QC077 Clear and colorless to pale yellow liquid essentially free of particles 100% pH pH meter/QC045 6.2 - 6.5 300 L Concentration UV Absorbance/QC020 Report Results 300 L Purity/ Molecular Distribution SEC - HPLC/QC022 Monomer% 95.0% 100 L Soluble Oligomers% 5.0% Fragments% 2.0% Charge Heterogeneity C E X - H P LC/ QC068 Retention time and distribution of peaks comparable to Reference Standard Purity SDS - CE (r)/QC004 (HC+LC) ೦ 95% 100 L pl clEF/QC006 pl from major isoforms comparable to Reference Standard 100 L Binding Kinetics Octet/QC072 K D % = 75 - 137% relative to Reference Standard 100 L Potency HU VE C /Q C021 80 - 120% average relative potency 100 L Particulate Matter USP <787>/QC126 Particle Size 10 m 25 m 1 vial Particle Limit < 6000 < 600 Sterility Con t aine r - Clo s u re lntegrity/ TP012 No Growth at test interval 100% USP <71>/EP 2.6.1 Direct 2 vials Confidential Page 295 of 468

Exhibit C - 1 [[5252615]] E xhibit C Compliance with Laws 1. SUPPLIER acknowledges that Company aims to perform its activities, and to have other parties (such as SUPPLIER) with which it enters into business arrangements to perform their activities under such arrangements, in accordance with the highest ethical standards and best industry practices, including any voluntary codes of practice applicable in the industry . SUPPLIER agree to use reasonable efforts to help ensure that it does not fail to meet such aim with respect to activities hereunder through any violation of the U . S . Foreign Corrupt Practices Act (the “ FCPA ”), the U . S . False Claims Act (the “ FCA ”), the U . S . Anti - Kickback Law (the “ AKA ”), the United Kingdom Bribery Act (the “ UKBA ”), or any laws or regulations regarding governmental research grants (e . g . , rules governing grants from the National Institutes of Health, or other governmental agencies) . 2. SUPPLIER shall comply with all laws and regulations concerning its efforts in any country or jurisdiction where it is providing work hereunder or otherwise applying to any of its activities under this Agreement . SUPPLIER shall use reasonable efforts to ensure that its personnel performing hereunder become reasonably familiar with the FCPA, the FCA, the UKBA and the AKA, and their prohibitions and purposes, and that they will not undertake any actions that would violate the FCPA, the FCA, the UKBA and the AKA . Accordingly, SUPPLIER hereby warrants that : ( i) ( ii) ( iii) ( iv) neither it nor its agents or employees whose duties pertain to this Agreement are excluded from a federal health care program as outlined in Sections 1128 and 1156 of the Social Security Act (see the Office of Inspector General of the Department of Health and Human Services List of Excluded Individuals / Entities at http : //oig . hhs . gov/exclusions/exclusions_list . asp) ; neither it nor its agents or employees whose duties pertain to this Agreement are debarred by the FDA under 21 U.S.C. 335a (see the FDA Office of Regulatory Affairs Debarment List at http://www.fda.gov/ICECI/EnforcementActions/FDADebarmentList/default.htm); neither it, nor its agents or employees are otherwise excluded from contracting with the federal government (see the System for Award Management at https : //www . sam . gov/portal/SAM/# 1 ) ; it, or its agents or employees, if required, are duly licensed and in good standing in accordance with applicable U . S . , state or other applicable governmental licensing requirements ; (v) no payment or offer to pay, or the giving or offering to give, anything of value to an official or employee of any government or any department, agency or instrumentality thereof (including any health or medical providers owned or controlled by the government), or to any political party or any candidate for political office, shall be made with the purpose of influencing any decisions favorable to SUPPLIER or its Affiliates and the business resulting therefrom in

Exhibit C - 2 [[5252615]] ( vi) ( vii) contravention of the FCPA or the laws of the country in which it is providing work ; it has not paid, nor offered or agreed to pay, nor caused to be paid, directly or indirectly, any political contributions, fees or commissions to any governmental employee or representative (including any employee of any health or medical provider owned or controlled by the government) that would appear to cause a violation of the FCPA ; it will not directly or indirectly offer, pay, promise to pay, or authorize the giving of money or anything of value to any governmental official or representative, to any political party or official thereof, or to any candidate for political office, or to any other person, for the purpose of : a. inappropriately influencing any act or decisions of such person, official, political party, party official, or candidate in, if applicable, its official capacity, including a decision to fail to perform official functions ; or b. inducing such person, official, political party, party official, or candidate to use influence with the government, any instrumentality thereof, or any other entity to affect or influence any act or decision of such government or instrumentality, or entity, in order to assist SUPPLIER in obtaining or retaining business for or with, or directing business to, any Affiliate or Third Party . SUPPLIER further agrees that if subsequent developments cause the certifications and information reported herein to be no longer accurate or complete, it will immediately so advise Company in writing . 1 . 3 In the event of a claim or investigation, or an official request for Company to cooperate with respect to any such claim or investigation, by a Regulatory Authority or other legal authority having jurisdiction over either Party, of an alleged violation of the FCPA arising from any activities conducted by the SUPPLIER relating specifically to this Agreement or to the Clinical Products, SUPPLIER shall reasonably provide such Regulatory Authority or other legal authority having jurisdiction with access to SUPPLIER’s facilities, records (financial and otherwise), and supporting documentation, as reasonably requested by Company or its agents in order to cooperate in connection with such claim or investigation . 1 . 4 During the Term, SUPPLIER shall maintain true, accurate and complete books and records, including those : (i) documenting its interactions with any government or its officials or employees relating to its activities in connection with any Clinical Product ; (ii) payments made to any officials or employees of any government or any department, agency or instrumentality thereof ; and (iii) political contributions . SUPPLIER shall also maintain a reasonable system of internal accounting controls sufficient to satisfy the FCPA . 1 . 5 SUPPLIER acknowledges and agrees that any breach of its obligations under this Exhibit C shall be a basis for notification of a material breach of this Agreement.

Exhibit C - 3 [[5252615]] 1 . 6 Notwithstanding anything to the contrary in this Agreement, a Party may disclose its terms and conditions (including any financial terms) to any governmental authorities in the U . S . or those in a country where it performs activities in connection with any Clinical Product that such Party determines in good faith has a legitimate need for access to such information for purposes of investigating or determining either Party’s compliance with applicable Law . 1 . 7 The Parties acknowledge that certain laws, now or in the future, may require pharmaceutical, medical device and other companies to disclose information on compensation, gifts or other remuneration provided to physicians, institutions, and other health care professionals . Either Party may report information about remuneration provided under this Agreement . Once reported, such information may be publicly accessible .

Exhibit D [[5252615]] E xhibit D Policy on Employment of Young People J&J Policy on the Employment of Young Persons This policy applies to the employment of persons under the age of 18 ("young persons") in the manufacture of any product, or any component of a product, by or for Company or any of its Affiliates worldwide. Age, Health & Safety – No person under the age of 16 shall be employed. No person between the ages of 16 and 18 shall be employed unless such employment is in compliance with the health, safety and morals provisions of the International Labour Organization Convention 138 Concerning Minimum Age ("ILO Convention 138"), a summary of which is attached hereto. Hours – No young person shall be required to work more than 48 hours of regularly scheduled time and 12 hours of overtime per week nor more than six days per week. Laws & Regulations – No young person shall be employed unless such employment is in compliance with all applicable laws and regulations concerning age, hours, compensation, health and safety. External Manufacturers – No manufacturer shall be engaged to manufacture any product, or any component of a product, for Company or any of its Affiliates worldwide unless such manufacturer has entered into an enforceable written agreement to comply with this policy, submit to periodic compliance inspections, and maintain the records necessary to demonstrate compliance. If any such manufacturer shall be found to be in breach of such agreement, the manufacturer's engagement shall be terminated.

Exhibit E [[5252615]] E xhibit E Johnson & Johnson Policy for Wood Pallets This clause applies to all products and/or materials shipped to Company or its affiliates or authorized locations on wood pallets . Wood pallets must be constructed from lumber sourced from countries that prohibit the treatment of wood with any form of halophenol based chemicals (including but not limited to 2 , 4 , 6 trichlorophenol, 2 , 4 , 6 tribromophenol, any of the tetrachlorophenols, any of the tetrabromophenols and pentachlorophenol) . Wood pallets used must have been heat treated only in accordance with the Heat Treatment standards set forth in International Standards for Phytosanitary Measures Publication No . 15 , 2009 Revision (ISPM 15 ) . Additionally, the sourced lumber or finished pallets shall not be shipped or stored with pallets or materials that may contain the chemicals mentioned above . While ISPM 15 currently provides for the use of Methyl Bromide (MB), the use of pallets fumigated with Methyl Bromide is also prohibited . All wood pallets must be labeled with the HT stamp in accordance with ISPM 15 Annex II . This requirement is effective immediately . Failure to meet the above requirements of this paragraph may lead to rejection of shipments at SUPPLIER’s expense .

Exhibit F [[5252615]] E xhibit F Company Records and Information Requirements 1 . 1 Company’s Records and Information . All records and information, in any format, that SUPPLIER receives from, creates, or edits on behalf of Company or Company’s Affiliates will be referred to herein as “ Company Records and Information ” . For the avoidance of doubt, Company Records and Information does not include records and information created by SUPPLIER as part of SUPPLIER’S business processes (e . g . , invoices, internal reports, etc . ) . (a) SUPPLIER shall maintain, manage and protect Company Records and Information pursuant to this Agreement and any applicable Purchase Orders (i) in accordance with Company’s records retention policies and (ii) in accordance with all applicable statutes and regulations . (b) SUPPLIER shall not transfer Company Records and Information to any other entity unless directed by Company . For avoidance of doubt, this means no SUPPLIER employee or contractor may take Company Records and Information with them upon leaving SUPPLIER . (c) SUPPLIER shall maintain and manage Company Records and Information such that Company Records and Information is not commingled with records and information generated, managed or maintained by SUPPLIER under agreements with other customers . (d) SUPPLIER shall retain electronic backups of Company Records and Information (i) for disaster recovery purposes only and (ii) for no longer than 120 days from the creation date . 2. Preservation and Production . SUPPLIER shall comply promptly and fully with any request from Company to preserve Company Records and Information (or parts thereof) . SUPPLIER shall deliver promptly Company Records and Information requested to be searched, retrieved and produced, all as part of the services SUPPLIER provides under this Agreement in accordance with Section 1 . 8 of this Exhibit F . 3. Third Party Requests . Within 48 hours of SUPPLIER receiving from anyone other than Company a request, demand, notice, subpoena, order or other legal request (a “ Third - Party Request ”) for Company Records and Information, SUPPLIER shall notify Company and provide Company with a copy of the Third - Party Request (unless legally prohibited) . SUPPLIER shall confer with Company to identify, document and implement procedures to comply with the request . SUPPLIER shall take all reasonable steps to protect Company’s legal rights when responding to a Third - Party Request . 4. Training . All employees and contractors of SUPPLIER with access to the Johnson & Johnson Enterprise Network (JJNET) shall annually complete Records and Information Management training as specified by Company .

Exhibit F [[5252615]] 5. Destruction . SUPPLIER shall not destroy any Company Records and Information without first having received Company’s written confirmation that the Company Records and Information is not subject to any pending preservation obligation or retention requirement . 6. Transfer . When a transfer of Company Records and Information from SUPPLIER is required, SUPPLIER shall (i) transfer Company Records and Information to Company or an entity specified by Company in accordance with Section 1 . 8 of this Exhibit F , (ii) take no action on Company Records and Information until written notification from Company confirming accurate and complete transfer is received, and (iii) if directed by Company to permanently delete transferred Company Records and Information, SUPPLIER shall certify in writing that Company Records and Information has been permanently deleted as specified by Company . 7. Termination . Upon termination of this Agreement and at Company’s direction, SUPPLIER shall (i) transfer Company Records and Information to Company or an entity specified by Company in accordance with Section 1 . 6 of this Exhibit F or (ii) obtain Company’s written approval to destroy Company Records and Information in accordance with Section 1 . 5 of this Exhibit F and (iii) upon completion of the steps directed by Company, certify in writing that SUPPLIER has permanently deleted all Company Records and Information as specified by Company . 8. Format of Company Records and Information . As part of the services SUPPLIER provides under this Agreement, SUPPLIER will meet with Company as reasonably requested to identify, document and implement procedures to deliver to Company or an entity specified by Company, Company Records and Information in the format directed by Company . To meet Company’s records retention and other legal preservation obligations, Company may require SUPPLIER to provide Company Records and Information in a structured format maintaining the relationships that exist in the database underlying SUPPLIER’S application .

Exhibit G [[5252615]] E xhibit G New Formulations New formulations are requested to change the bermekimab concentrations to 150 mg/mL and 175 mg/mL. The new formulations may be filled in either vials or pre - filled syringes. There are no changes requested to the excipients. Specifications for the new formulations should meet all specifications as listed in Exhibit B, and adjusted for the concentration as appropriate.

Exhibit H [[5252615]] E xhibit H Transition Matters R egulatory All Health Authority interactions and correspondence, IND and IMPD. Health Authorities GMP inspection documentation, if available C ell Line Reports detailing the history of the host CHO cell line, creation of the plasmid DNA construct, generation of the manufacturing cell line, steps taken to assure monoclonality, MCB/WCB testing and results, and any genetic characterization studies performed An aliquot of the plasmid DNA used at transfection and the full nucleotide sequence of the plasmid DNA (gb file) D rug Substance For each unit operation: Summary of purpose for each unit operation Evidence of performance o Impact on quality attributes o Yield (and volumes and concentration used to calculate yields) o Titer o Chromatograms (for purification unit operations) including: ▪ OD ▪ pH ▪ conductivity ▪ pressure drop o UF/DF profile ▪ Pressure profile (inlet, outlet, TMP) ▪ Flow rate profile (feed, cross - flow) ▪ Conductivity, pH Risk assessment of known and plausible failure modes: Potential for failure to achieve purpose of the unit operation Potential for failure resulting in negative impact on drug substance/ drug product quality or stability Intermediate hold times and supporting data Drug substance stability data Virus clearance and inactivation data Limit of in vitro cell age information Details of how LIVCA was performed Genetic stability Virus load (virus - like particle) Samples of process intermediates, drug substance and drug product from across clinical manufacturing history D rug Product

Exhibit H [[5252615]] Retains of samples of filled syringes and vials at all concentrations Formulation development reports Process development reports Manufacturing flow diagrams and equipment used for each unit operation. Unit operation process parameters and targets Risk assessments Stability data from GMP and non - GMP fills. Table of GMP lots with genealogy table identifying lot no and corresponding clinical trials that the DP was used for, date of manufacturing, scale of manufacturing. Drug Delivery Syringe selection rational and syringe evaluation and performance data Syringe F/F process Viscosity and glide force data for formulations used in the development and in clinical trials during the PFS stability; Viscosity vs temperature if such data exist Any feasibility report on drug delivery and injection device A nalytical All methods SOPs, validation reports and any development reports for release in process and characterization methods All critical reagents for methods o Inventory of these critical reagents o Qualification reports/protocols for critical reagents Reference standards (current and any past ref std.) with clear inventory of material and clear history of the reference standard program including qualification protocols and reports o The complete history of the reference standard to link potency from the beginning of the program to the current ref std. Inventory and access to sample retains DS and DP both T=0 and stability samples o Samples as requested to cover method bridging and comparability (amounts of each sample TBD). Inventory and access to in process sample retains Method bridging support o Perform testing of appropriate DS and DP batches using Phase 2 methods Regulatory filings Characterization reports for Elucidation of structure Reports on CQAs Reports on extinction coefficient (theoretical and experimental) Stability protocols, reports and results Per Janssen guidance, continuation of ongoing stability plans or closing out or bridge studies execution Specification documents Agency feedback on analytical methods to understand any outstanding commitments Batch history tables to understand what batches were used from GLP Tox to current clinical trials

Exhibit H [[5252615]] o Results for all batches and clear understanding of what methods were used to generate these results History of comparability studies with data/reports O ther List of raw materials o CoA of raw materials o Testing strategy of raw materials