Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - ASENSUS SURGICAL, INC. | trxc-ex322_6.htm |
| EX-32.1 - EX-32.1 - ASENSUS SURGICAL, INC. | trxc-ex321_11.htm |
| EX-31.2 - EX-31.2 - ASENSUS SURGICAL, INC. | trxc-ex312_10.htm |
| EX-31.1 - EX-31.1 - ASENSUS SURGICAL, INC. | trxc-ex311_7.htm |
| EX-23.1 - EX-23.1 - ASENSUS SURGICAL, INC. | trxc-ex231_8.htm |
| EX-21.1 - EX-21.1 - ASENSUS SURGICAL, INC. | trxc-ex211_9.htm |
| EX-10.17 - EX-10.17 - ASENSUS SURGICAL, INC. | trxc-ex1017_559.htm |
| EX-10.7 - EX-10.7 - ASENSUS SURGICAL, INC. | trxc-ex107_560.htm |
| EX-10.6 - EX-10.6 - ASENSUS SURGICAL, INC. | trxc-ex106_561.htm |
| EX-10.5 - EX-10.5 - ASENSUS SURGICAL, INC. | trxc-ex105_562.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 0-19437
TRANSENTERIX, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
11-2962080 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
635 Davis Drive, Suite 300, Morrisville, NC 27560
(Address of principal executive offices) (Zip Code)
Registrant’s telephone number, including area code: (919) 765-8400
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Name of each exchange on which registered |
|
Common Stock $0.001 par value per share |
|
NYSE American |
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒.
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐.
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐.
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K ☒.
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
|
☐ |
|
Accelerated filer |
|
☒ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☐ (Do not check if a smaller reporting company) |
|
Smaller reporting company |
|
☐ |
|
|
|
|
|
Emerging Growth Company |
|
☐ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act) Yes ☐ No ☒.
On June 30, 2017, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value (based on the average bid and asked price of its common stock on that date) of the voting stock held by non-affiliates of the registrant was $70.8 million.
The number of shares outstanding of the registrant’s common stock, as of March 1, 2018 was 200,049,326.
Documents Incorporated By Reference: Part III of this Annual Report on Form 10-K is incorporated by reference to our Definitive Proxy Statement on Schedule 14A to be filed in respect of our 2018 Annual Meeting of Stockholders.
ANNUAL REPORT ON FORM 10-K
DECEMBER 31, 2017
Table of Contents
|
|
|
|
|
Page |
|
|
|
|||
|
ITEM 1. |
|
|
1 |
|
|
ITEM 1.A. |
|
|
9 |
|
|
ITEM 1.B. |
|
|
22 |
|
|
ITEM 2. |
|
|
22 |
|
|
ITEM 3. |
|
|
22 |
|
|
ITEM 4. |
|
|
22 |
|
|
|
|
|
||
|
|
|
|||
|
ITEM 5. |
|
|
23 |
|
|
ITEM 6. |
|
|
25 |
|
|
ITEM 7. |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
|
26 |
|
ITEM 7.A. |
|
|
39 |
|
|
ITEM 8. |
|
|
40 |
|
|
ITEM 9. |
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
|
77 |
|
ITEM 9.A. |
|
|
77 |
|
|
ITEM 9.B. |
|
|
77 |
|
|
|
|
|
||
|
|
|
|||
|
ITEM 10. |
|
|
79 |
|
|
ITEM 11. |
|
|
79 |
|
|
ITEM 12. |
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
|
79 |
|
ITEM 13. |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE. |
|
79 |
|
ITEM 14. |
|
|
79 |
|
|
|
|
|
||
|
|
|
|||
|
ITEM 15. |
|
|
80 |
|
|
ITEM 16. |
|
|
83 |
|
i
This Annual Report on Form 10-K (“Annual Report”) contains certain “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995, Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Such forward-looking statements contain information about our expectations, beliefs or intentions regarding our product development and commercialization efforts, business, financial condition, results of operations, strategies or prospects. You can identify forward-looking statements by the fact that these statements do not relate strictly to historical or current matters. Rather, forward-looking statements relate to anticipated or expected events, activities, trends or results as of the date they are made. Because forward-looking statements relate to matters that have not yet occurred, these statements are inherently subject to risks and uncertainties that could cause our actual results to differ materially from any future results expressed or implied by the forward-looking statements.
Many factors could cause our actual operations or results to differ materially from the operations and results anticipated in forward-looking statements. These factors include, but are not limited to:
|
|
• |
our ability to successfully transition from a research and development company to a company focused on marketing, sales and distribution of our products; |
|
|
• |
our ability to successfully develop, clinically test and commercialize our products; |
|
|
• |
our ability to identify and pursue development of additional products; |
|
|
• |
the timing and outcome of the regulatory review process for our products; |
|
|
• |
competition from existing and new market entrants; |
|
|
• |
the impact of foreign currency fluctuations on our financial results; |
|
|
• |
our history of operating losses; |
|
|
• |
our need to obtain additional funding to continue our operations; |
|
|
• |
our ability to attract and retain key management, marketing and scientific personnel; |
|
|
• |
our ability to successfully prepare, file, prosecute, maintain, defend and enforce patent claims and other intellectual property rights; |
|
|
• |
changes in the health care and regulatory environments of the United States, Europe and other jurisdictions in which the Company operates; and |
|
|
• |
other factors contained in the section entitled “Risk Factors” contained in this Annual Report. |
We do not undertake any obligation to update our forward-looking statements, except as required by applicable law.
In this Annual Report we refer to TransEnterix, Inc. and its subsidiaries collectively as the “Company,” “it,” “we,” “our” or “us.” The Company’s subsidiaries are: TransEnterix International; TransEnterix Italia S.r.l.; TransEnterix Europe S.à.R.L; TransEnterix Europe S.à.R.L, Bertrange, Swiss Branch, Lugano; TransEnterix Asia Pte. Ltd.; and, TransEnterix Taiwan Ltd.
ii
Overview
TransEnterix is a medical device company that is digitizing the interface between the surgeon and the patient to improve minimally invasive surgery by addressing the clinical and economic challenges associated with current laparoscopic and robotic options in today's value-based healthcare environment. The Company is focused on the commercialization of the Senhance™ Surgical System, which digitizes laparoscopic minimally invasive surgery. The system allows for robotic precision, haptic feedback, surgeon camera control via eye sensing and improved ergonomics while offering responsible economics.
The Senhance System has been granted a CE Mark in Europe for laparoscopic abdominal and pelvic surgery, as well as limited thoracic operations excluding cardiac and vascular surgery. In April 2017, the Company submitted a 510(k) application to the FDA for the Senhance System. On October 13, 2017, the Company received 510(k) clearance from the FDA for use in laparoscopic colorectal and gynecologic surgery. These indications cover 23 procedures, including benign and oncologic procedures. We anticipate expanding the indications for use in the middle of 2018. The Senhance System is available for sale in the U.S., the EU and select other countries.
The Senhance System is a multi-port robotic surgery system which allows multiple robotic arms to control instruments and a camera. The system features advanced technology to enable surgeons with haptic feedback and the ability to move the camera via eye movement. The system replicates laparoscopic motion that is familiar to experienced surgeons, and integrates three-dimensional high definition, or 3DHD, vision technology. The Senhance System also offers responsible economics to hospitals by offering robotic technology with reusable instruments thereby reducing additional costs per surgery when compared to other robotic solutions.
The Company has also developed the SurgiBot™ System, a single-port, robotically enhanced laparoscopic surgical platform. On December 18, 2017, the Company announced that it had entered into an agreement with Great Belief International Limited, or GBIL, to advance the SurgiBot System towards global commercialization. The agreement transfers ownership of the SurgiBot System assets, while the Company retains the option to distribute or co-distribute the SurgiBot System outside of China. Upon completion of the transfer of all SurgiBot System assets, GBIL will have the SurgiBot System manufactured in China and obtain Chinese regulatory clearance from the China Food and Drug Administration, or CFDA, while entering into a nationwide distribution agreement with China National Scientific and Instruments and Materials Company, or CSIMC, for the Chinese market. The agreement provides the Company with proceeds of at least $29 million, of which $7.5 million was received in December 2017. An additional $7.5 million is expected to be received by March 31, 2018, which includes a $3.0 million equity investment at $2.33 per share. The remaining $14 million, representing minimum royalties, will be paid beginning at the earlier of receipt of Chinese regulatory approval or five years.
We believe that future outcomes of minimally invasive surgery will be enhanced through our combination of more advanced tools and robotic functionality, which are designed to: (i) empower surgeons with improved precision, dexterity and visualization; (ii) improve patient satisfaction and enable a desirable post-operative recovery; and (iii) provide a cost-effective robotic system, compared to existing alternatives today, for a wide range of clinical applications. Our strategy is to focus on the development and commercialization of the Senhance System.
The Company operates in one business segment. Please see the disclosure in Note 2 “Summary of Significant Accounting Policies – Segments” in the Notes to our Consolidated Financial Statements in Item 8 of this Annual Report regarding our business operations in the U.S. and elsewhere.
Market Overview
Over the past two decades, laparoscopic surgery has emerged as a minimally invasive alternative to open surgery. In laparoscopic surgery, multiple incisions are necessary to provide surgical access ports. Carbon dioxide gas insufflation is then used to create room in the body cavity, and long rigid instruments are introduced through ports placed in the incisions to perform surgical tasks. Millions of laparoscopic surgical procedures across a broad range of clinical applications are now performed each year worldwide, though many surgeries are still performed in an open fashion.
While laparoscopy has improved the invasive nature of many previously open procedures, it still has many limitations. Traditional, or rigid, laparoscopy still requires multiple incisions to achieve the visualization and instrument triangulation required to perform successful surgery. Rigid laparoscopy also creates physical challenges by forcing the surgeon’s hands and arms into awkward angles, requiring the surgeon to hold instruments in fixed positions for long periods of time, and requiring an assistant to stabilize and move a laparoscopic camera. Another challenge associated with rigid laparoscopic surgery is the creation of a cumbersome and potentially tissue-damaging fulcrum at the patient’s abdominal wall where instruments are manipulated. Nearly all laparoscopic instruments are rigid instruments that lack internal articulation to enhance dexterity in complex tasks. Most laparoscopic surgeries are performed with two-dimensional (2-D) visualization of the operative field, making depth perception difficult.
1
Despite such limitations, traditional laparoscopy remains the prevalent technique in minimally invasive surgery. We believe that robotic devices that replicate laparoscopic motion are more comfortable for surgeons to adopt, thereby increasing the opportunity to enhance traditional surgical methods with robotics. Our Senhance System mimics laparoscopic surgery.
Robotic and computer controlled assistance have developed as technologies that offer the potential to improve upon many aspects of the laparoscopic surgical experience. Hundreds of thousands of robotic-assisted surgical procedures are now performed each year worldwide, but they still represent a small fraction of the total laparoscopic procedures performed. While initial widespread adoption of robotic-assisted surgery was focused on urologic and gynecologic procedures that were primarily performed in an open fashion prior to robotics, recently developed robotic approaches have been applied to many other clinical applications, particularly in general surgery. Despite recent advances, we believe there remain many limitations associated with current robotic-assisted surgery systems used in connection with laparoscopic surgeries.
Product Overview
We are addressing the challenges in laparoscopy and robotic-assisted surgery with innovative products and product candidates that leverage the best features of both approaches to minimally invasive surgery.
Senhance System
On September 18, 2015, the Company entered into a Membership Interest Purchase Agreement, or the Purchase Agreement, with Sofar S.p.A., or Sofar, as seller, Vulcanos S.r.l., as the acquired company, and TransEnterix International, Inc., a direct, wholly owned subsidiary of the Company which was incorporated in September 2015, as buyer. The closing of the transactions occurred on September 21, 2015 pursuant to which the Company acquired all of the membership interests of Vulcanos from Sofar (now known as the Senhance Acquisition), and changed the name of Vulcanos to TransEnterix Italia S.r.l. For a description of the Senhance Acquisition and related transactions, see the disclosure titled “Senhance Acquisition and Related Transactions” under Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this Annual Report.
The Senhance System is a multi-port robotic surgery system which allows up to four arms to control robotic instruments and a camera. The system builds on the success of laparoscopy by enhancing the traditional features that surgeons have come to expect from existing products and by addressing some of the limitations associated with robotic surgery systems for laparoscopic procedures. The Senhance System also offers responsible economics to hospitals by offering robotic technology with reusable instruments with minimal additional costs per surgery when compared to laparoscopy. The Senhance System has been granted a CE Mark in Europe for laparoscopic abdominal and pelvic surgery, as well as limited thoracic operations excluding cardiac and vascular surgery. In April 2017, the Company submitted a 510(k) application to the FDA for the Senhance System. On October 13, 2017, the Company received 510(k) clearance from the FDA for use in laparoscopic colorectal and gynecologic surgery. The Senhance System is available for sale in the U.S., the EU and select other countries.
Key features of the Senhance System are:
|
|
• |
Haptic Feedback: The Senhance System’s haptic feedback feature provides the surgeon with the ability to feel the tissue response of the body during a procedure. |
|
|
• |
Enhanced Vision: The Senhance System is compatible with 3DHD vision technology and gives the surgeon the ability to move the camera via eye movement so that the camera is centered in the surgeon’s field of vision. |
|
|
• |
Laparoscopic Motion: The Senhance System utilizes laparoscopic motion that is similar to the motion used during traditional laparoscopic surgeries. |
|
|
• |
View of the Sterile Field: The Senhance System offers the user an open view of the operating room and sterile field from the console. |
|
|
• |
Reusable, Autoclavable Instrumentation: The Senhance System offers instrumentation that is cleaned and sterilized using current autoclave technology that does not require additional, less standard sterilization methods, and that has no pre-set limitation on number of uses that requires them to be disposed. |
The Senhance System is manufactured for us by third party contract manufacturers. We or our manufacturers acquire raw materials and components of the Senhance System from vendors, some of which are sole suppliers. We believe our relationships with our vendors and manufacturing contractors are good. We further believe that we have the manufacturing and inventory reserves to meet our anticipated Senhance System sales for the foreseeable future. We are currently taking steps to develop redundant manufacturing and supply alternatives.
2
The SurgiBot System is designed to utilize flexible instruments through articulating channels controlled directly by the surgeon, with robotic assistance, while the surgeon remains patient-side within the sterile field. In June 2015, the Company submitted a 510(k) application to the FDA for the SurgiBot System. On April 19, 2016, the FDA notified the Company that the SurgiBot System did not meet the criteria for substantial equivalence based on the data and information submitted by TransEnterix in the 510(k) submission. Following the FDA determination, in May 2016, the Company implemented a restructuring plan. The restructuring plan: (1) reduced the Company’s workforce; (2) abandoned certain equipment related to the SurgiBot System; (3) cancelled contracts related to the SurgiBot System; (4) wrote down inventory related to the SurgiBot System; and (5) wrote off certain patents. After interactions with the FDA, the Company determined that a new 510(k) application would need to be submitted in order to obtain clearance for the SurgiBot System.
Please see “Business - Overview” in this Item 1 of this Annual Report for a description of the 2017 agreement regarding the sale of the SurgiBot System assets.
Products in Development
We are working to expand our portfolio of surgical instruments, accessories and other products to compliment the Senhance System. We are currently focused on the development and regulatory clearance for advanced energy instruments, articulating instruments and 3 mm instruments for the Senhance System.
Revenues
In the year ended December 31, 2017, we had six customers in Europe, one customer in the U.S., and one customer in Asia, who accounted for 61%, 18%, and 21% of our revenue. In the year ended December 31, 2016, we had one European customer, Humanitas Hospital in Milan, Italy, who accounted for 100% of our revenue. In the year ended December 31, 2015, the Company had no revenue. The Company is not dependent on current customers. Please see the disclosure in Note 2 “Summary of Significant Accounting Policies – Segments” in the Notes to our Consolidated Financial Statements in Item 8 of this Annual Report regarding the geographic locations of our revenues and assets in the U.S. and elsewhere.
Business Strategy
Our current strategy is to focus our resources on the commercialization of the Senhance System.
We believe that:
|
|
• |
there are a number of hospitals and an increasing number of ambulatory surgery centers in the U.S. and internationally that could benefit from the addition of robotic-assisted minimally invasive surgery and, with the Senhance System, lower operational costs; |
|
|
• |
with the Senhance System, surgeons can benefit from the haptic feedback, enhanced 3DHD vision and open architecture consistent with current laparoscopic surgery procedures; |
|
|
• |
patients will continue to seek a minimally invasive option for many common general abdominal and gynecologic surgeries, which are addressed by the Senhance System; and |
|
|
• |
the addition of advanced energy instruments, articulating instruments and 3 mm instruments for the Senhance System will help to increase adoption of our products in the laparoscopic surgery market. |
Sales and Marketing
We have recruited a sales and marketing team and have initiated commercialization of the Senhance System in the United States, Europe, the Middle East, Africa and limited countries in Asia. We utilize distributors in a number of jurisdictions where we do not sell directly. Our distribution agreements typically provide exclusivity in a specific territory or jurisdiction.
We have initiated a Clinical Leadership Program with leading surgical centers in Europe and the United States to utilize the Senhance System. We believe the program helps improve our visibility and provides more widespread opportunity for observation of robotic surgery with the Senhance System. In addition, in December 2016 we opened a European training and research and development center in Milan, Italy, and in November 2017 we opened a training and research and development center at the Institute for Surgical Advancement at Florida Hospital Orlando.
3
During the fiscal years ended December 31, 2017, 2016 and 2015, we incurred research and development expenses of approximately $22.0 million, $29.3 million and $29.7 million, respectively. In 2017, such expenses primarily related to the Senhance System development, including the preparation and submission of regulatory filings with the FDA. In 2016, such expenses primarily related to the SurgiBot System development and the Senhance System development, including the preparation and submission of regulatory filings. In 2015, such expenses primarily related to the SurgiBot System development, including the preparation and submission of regulatory filings. We fund our research and development expenses primarily from proceeds raised from equity and debt financing transactions. We expect to continue to use equity and debt financing transactions to fund our research and development activities. No customers are obligated to pay any material portion of such research and development expenses.
Intellectual Property
We believe that our intellectual property and expertise is an important competitive resource. Our experienced research and development team has created a substantial portfolio of intellectual property, including patents, patent applications, trade secrets and proprietary know-how. We maintain an active program of intellectual property protection, both to assure that the proprietary technology developed by us is appropriately protected and, where necessary, to assure that there is no infringement of our proprietary technology by competitive technologies.
The following summarizes our current patent and patent application portfolio.
The Company’s patent portfolio includes 24 United States patents and 68 patents issued outside the United States, and more than 90 patent applications filed in the United States and abroad. We own all right, title and interest in approximately 140 of our patents and patent applications, and the rest are exclusively licensed to us. We have granted a security interest to our intellectual property, including patents and patent applications to the Lender under our existing loan agreement. See “Item 7 – Management’s Discussion and Analysis and Results of Operations – Debt Refinancing” for a description of our existing loan agreement.
Several of our issued patents resulted from filings related to the Senhance System. These include 5 United States patents, and 33 patents outside the United States. The earliest to expire US patents within this part of our portfolio will remain in force until 2030, and the earliest to expire non-US patents will expire in 2027. The patent applications include over 75 that relate to the Senhance System or other aspects of robotic-assisted surgery. We intend to continue to seek further patent and other intellectual property protection in the United States and internationally, where available and when appropriate, as we continue our product development efforts.
Some of our issued patents and pending applications for the Senhance System, as well as associated technology and know-how, are exclusively licensed to TransEnterix Italia from the European Union. The license agreement with the European Union has a term which runs until the final licensed patent expires, unless the agreement is terminated earlier by mutual consent of the parties or for breach. The Company is currently in compliance with the terms of this license agreement.
Competition
Our industry is highly competitive, subject to change and significantly affected by new product introductions and other activities of industry participants. Many of our competitors have significantly greater financial and human resources than we do and have established reputations with our target customers, as well as worldwide distribution channels that are more established and developed than ours.
There are many competitive offerings in the field of minimally invasive surgery. Several companies have launched devices that enable reduced incision or single incision laparoscopic surgery with or without robotic assistance. Our surgical competitors include, but are not limited to: Johnson & Johnson, Medtronic plc, Applied Medical and Intuitive Surgical.
In addition to surgical device manufacturer competitors, there are many products and therapies that are designed to reduce the need for or attractiveness of surgical intervention. These products and therapies may impact the overall volume of surgical procedures and negatively impact our business.
Our ability to compete may be affected by the failure to fully educate physicians in the use of our products and products in development, or by the level of physician expertise. This may have the effect of making our products less attractive. Among currently available surgical robotic systems, we expect the Senhance System to differentiate on the basis of overall attractiveness to laparoscopic surgeons due to its ability to provide robotic benefits while leveraging their laparoscopic training and experience lower per procedure costs when compared to other robotic systems on the market today; and we expect the Senhance System to differentiate, in most cases, its ability to provide the surgeon with tactile feedback. Several medical device companies are actively engaged in research and development of robotic systems or other medical devices and tools used in minimally invasive surgery procedures. We cannot predict the basis upon which we will compete with new products marketed by others.
4
Government Regulation of our Product Development Activities
The U.S. government and foreign governments regulate the medical device industry through various agencies, including but not limited to, the U.S. FDA, which administers the Federal Food, Drug and Cosmetic Act, or the FDCA. The design, testing, manufacturing, storage, labeling, distribution, advertising, and marketing of medical devices are subject to extensive regulation by federal, state and local governmental authorities in the United States, including the FDA, and by similar agencies in other countries, including the European Union. Any device product that we develop must receive all requisite regulatory approvals or clearances, as the case may be, before it may be marketed in a particular country.
Device Development, Marketing Clearance and Approval
Medical devices are subject to varying levels of pre-market regulatory requirements. The FDA classifies medical devices into one of three classes: (i) Class I devices are relatively simple and can be manufactured and distributed with general controls; (ii) Class II devices are somewhat more complex and receive greater scrutiny from the FDA and have heightened regulatory requirements; and (iii) Class III devices are new, high risk devices, and frequently are permanently implantable or help sustain life and generally require a Pre-Market Approval, or PMA, by the FDA.
In the United States, a company generally can obtain permission to distribute a new medical device in one of two ways. The first applies to any device that is substantially equivalent to a device first marketed prior to May 1976, or to another device marketed after that date, but which was substantially equivalent to a pre-May 1976 device. These devices are either Class I or Class II devices. To obtain FDA clearance to distribute the medical device, a company generally must submit a 510(k) notification, and receive an FDA order finding substantial equivalence to a predicate device (pre-May 1976 device or post-May 1976 device that was substantially equivalent to a pre-May 1976 device) and permitting commercial distribution of that medical device for its intended use. A 510(k) notification must provide information supporting a claim of substantial equivalence to a single medical device, the predicate device. If clinical data from human experience are required to support the 510(k) notification, these data must be gathered in compliance with investigational device exemption, or IDE, regulations for investigations performed in the United States. The 510(k) process is normally used for products of the type that we are developing and propose to market and sell. The FDA review process for premarket notifications submitted pursuant to Section 510(k) of the FDCA takes, pursuant to statutory requirements, 90 days, but it can take substantially longer if the FDA has questions regarding the regulatory submission. It is possible for 510(k) clearance procedures to take from six to eighteen months, depending on the concerns raised by the FDA and the complexity of the device. There is no guarantee that the FDA will “clear” a medical device for marketing, in which case the device cannot be distributed in the United States. There is also no guarantee that the FDA will deem the applicable device subject to the 510(k) process, as opposed to the more time-consuming, resource-intensive and problematic PMA process described below.
The second, more comprehensive, approval process applies to a new device that is not substantially equivalent to a pre-1976 product or that is to be used in supporting or sustaining life or preventing impairment. These devices are normally Class III devices. For example, most implantable devices are subject to the approval process as a Class III device. Two steps of FDA approval are generally required before a company can market a product in the United States that is subject to approval, as opposed to clearance, as a Class III device. First, a company must comply with IDE regulations in connection with any human clinical investigation of the device. These regulations permit a company to undertake a clinical study of a “non-significant risk” device without formal FDA approval. Prior express FDA approval is required if the device is a significant risk device. Second, the FDA must approve the company’s PMA application, which contains, among other things, clinical information acquired under the IDE. Additionally, devices subject to PMA approval may be subject to a panel review to obtain marketing approval and are required to pass a factory inspection in accordance with the current “good manufacturing practices” standards in order to obtain approval. The FDA will approve the PMA application if it finds there is reasonable assurance that the device is safe and effective for its intended use. The PMA process takes substantially longer than the 510(k) process, approximately one to two years or more.
However, in some instances the FDA may find that a device is new and not substantially equivalent to a predicate device but is also not a high risk device as is generally the case with Class III PMA devices. In these instances FDA may allow a device to be down classified from Class III to Class I or II. The de novo classification option is an alternate pathway to classify novel devices of low to moderate risk that had automatically been placed in Class III after receiving a “not substantially equivalent,” or NSE, determination in response to a 510(k) notification. The regulations have also been amended to allow a sponsor to submit a de novo classification request to the FDA for novel low to moderate risk devices without first being required to submit a 510(k) application. These types of applications are referred to as “Evaluation of Automatic Class III Designation” or “de novo.” In instances where a device is deemed not substantially equivalent to a Class II predicate device, the candidate device may be filed as a de novo application which may lead to delays in regulatory decisions by the FDA. FDA review of a de novo application may lead the FDA to identify the device as either a Class I or II device and worthy of either an exempt or 510(k) regulatory pathway.
The Company believes the Senhance System and many related products are Class II devices as evidenced by the Company’s recently cleared 510(k) premarket notification. The Company intends to further develop the product line by adding additional instrumentation
5
and accessories for use with the Senhance System. At this time, the Company believes that the items under development are Class II devices subject to 510(k) premarket notification. The FDA might find that the 510(k) submission does not provide the evidence required to prove that the additional instruments or accessories for use with the Senhance System are substantially equivalent to marketed Class II devices. If that were to occur, the Company would be required to undertake the more complex and costly PMA process or perhaps be considered for a de novo reclassification. For either the 510(k), de novo, or the PMA process, the FDA could require the Company to conduct clinical trials, which would take more time, cost more money and pose other risks and uncertainties.
Clinical studies conducted in the U.S. or used in any U.S. application on an unapproved medical device require approval from the FDA prior to initiation. Even when a clinical study has been approved by the FDA or deemed approved, the study is subject to factors beyond a manufacturer’s control, including, but not limited to, the fact that the institutional review board, or IRB, at a specified clinical site might not approve the study, might decline to renew approval, or might suspend or terminate the study before its completion. There is no assurance that a clinical study at any given site will progress as anticipated. In addition, there can be no assurance that the clinical study will provide sufficient evidence to assure the FDA that the product is safe and effective, a prerequisite for FDA approval of a PMA, or substantially equivalent in terms of safety and effectiveness to a predicate device, a prerequisite for clearance under Section 510(k). Even if the FDA approves or clears a device, it may limit its intended uses in such a way that manufacturing and distribution of the device may not be commercially feasible.
After clearance or approval to market is given, the FDA and foreign regulatory agencies, upon the occurrence of certain serious adverse events, are authorized under various circumstances to withdraw the clearance or approval of the device, or require changes to a device, its manufacturing process or its labeling or require additional proof that regulatory requirements have been met.
A manufacturer of a device approved through the PMA process is not permitted to make changes to the device which affect its safety or effectiveness without first submitting a supplement application to its PMA and obtaining FDA approval for that supplement, prior to marketing the modified device. In some instances, the FDA may require clinical trials to support a supplement application. A manufacturer of a device cleared through the 510(k) process must submit an additional premarket notification if it intends to make a change or modification in the device that could significantly affect the safety or effectiveness of the device, such as a significant change or modification in design, material, chemical composition, energy source, labeling or manufacturing process. Any change in the intended uses of a PMA device or a 510(k) device requires an approval supplement or new cleared premarket notification. Exported devices are subject to the regulatory requirements of each country to which the device is exported, as well as certain FDA export requirements.
Continuing FDA Regulation
After a device is placed on the market, numerous FDA and other regulatory requirements continue to apply. These include:
|
|
• |
establishment registration and device listing with the FDA; |
|
|
• |
quality system regulations, which require manufacturers to follow stringent design, testing, process control, documentation and other quality assurance procedures; |
|
|
• |
labeling regulations, which prohibit the promotion of products for unapproved, i.e. “off label,” uses and impose other restrictions on labeling; |
|
|
• |
Medical Device Reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
|
|
• |
corrections and removal reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; and |
|
|
• |
requirements to conduct postmarket surveillance studies to establish continued safety data. |
We are required to, and have, registered with the FDA as a medical device manufacturer. We must obtain all necessary permits and licenses to operate our business in all regions in which we do business. As manufacturers, we and our suppliers are subject to announced and unannounced inspections by the FDA to determine our compliance with the Quality System Regulation, or QSR, and other regulations.
In Europe, we need to comply with the requirements of the Medical Devices Directive, or MDD, and appropriately affix the CE Mark on our products to attest to such compliance. Our products marketed in the EU meet the “Essential Requirements” of the MDD relating to safety and performance. We have undergone verification of our regulatory compliance, or conformity assessment, by a notified body duly authorized by an EU country and must continue to do so as new products and changes to the products arise. The level of scrutiny of such
6
assessment depends on the regulatory class of the product. We are subject to continued surveillance by our notified body and will be required to report any serious adverse incidents to the appropriate authorities. We also must comply with additional requirements of individual countries in which our products are marketed. In the European Community, we are required to maintain certain quality system certifications in order to sell products. These regulations require us or our manufacturers to manufacture products and maintain documents in a prescribed manner with respect to design, manufacturing, testing, labeling and control activities. As manufacturers, we and our suppliers are subject to announced and unannounced inspections by the European Notified Bodies.
Impact of Regulation
Failure to comply with the applicable regulatory requirements can result in enforcement action by the FDA and other international regulatory bodies, which may include, among other things, any of the following sanctions:
|
|
• |
warning letters, fines, injunctions, consent decrees and civil penalties; |
|
|
• |
repair, replacement, refund or seizure of our products; |
|
|
• |
operating restrictions, partial suspension or total shutdown of production; |
|
|
• |
refusing our request for market access approvals of new products or modifications to existing products; |
|
|
• |
withdrawing or suspending clearances or approvals that are already granted; |
|
|
• |
criminal prosecution; and |
|
|
• |
disgorgement of profits. |
Further, the levels of revenues and profitability of medical device companies like us may be affected by the continuing efforts of government and third party payors to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing or profitability of products is subject to governmental control. In the United States, there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental controls.
Therefore, we cannot assure you that any of our products will be considered cost effective, or that, following any commercialization of our products, coverage and reimbursement will be available or sufficient to allow us to manufacture and sell them competitively and profitably.
Health Care Regulation
Our business activities are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician payment transparency laws. If our operations are found to be in violation of any of such laws that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
In the United States, there have been, and we expect there to continue to be, a number of legislative and regulatory initiatives, at both the federal and state government levels, to change the healthcare system in ways that, if approved, could affect our ability to sell our products profitably. At the current time, our products are not defined as durable medical equipment. Non-DME devices used in surgical procedures are normally paid directly by the hospital or health care provider and not reimbursed separately by third-party payors. Instead, the hospital or health care provider is reimbursed based on the procedure performed and the inpatient or outpatient stay. As a result, these types of devices are subject to intense price competition that can place a small manufacturer at a competitive disadvantage as hospitals, ambulatory surgery centers and health care providers attempt to negotiate lower prices for products such as the ones we develop and sell.
In March 2010, the Patient Protection and Affordable Care Act (the “Affordable Care Act”) and the reconciliation law known as Health Care and Education Reconciliation Act (the “Reconciliation Act,” and, with the Affordable Care Act, the “2010 Health Care Reform Legislation”) were enacted into law. The constitutionality of the 2010 Health Care Reform Legislation was confirmed twice by the Supreme Court of the United States. Both Congressional leaders and President Trump have announced plans to repeal or modify the 2010 Health Care Reform Legislation. At this time the Company is not certain as to the impact of federal health care legislation on its business.
7
The 2010 Health Care Reform Legislation subjects manufacturers of medical devices to an excise tax of 2.3% on certain U.S. sales of medical devices beginning in January 2013. This excise tax was suspended in December 2015 for two years, and again in January 2018 for an additional two years. If eventually implemented, this excise tax will likely increase our expenses in the future.
Further, the 2010 Health Care Reform Legislation includes the Open Payments Act (formerly referred to as the Physician Payments Sunshine Act), which, in conjunction with its implementing regulations, requires certain manufacturers of certain drugs, biologics, and devices that are reimbursed by Medicare, Medicaid and the Children’s Health Insurance Program to report annually certain payments or “transfers of value” provided to physicians and teaching hospitals and to report annually ownership and investment interests held by physicians and their immediate family members during the preceding calendar year. We have provided reports under the Open Payments Act to the Centers for Medicare & Medicaid Services since 2014. The failure to report appropriate data accurately, timely, and completely could subject us to significant financial penalties. Other countries and several states currently have similar laws and more may enact similar legislation.
International Regulation and Potential Impact
Through the Senhance Acquisition, the Company has expanded into international markets and intends to pursue continued expansion. Some of these markets maintain unique regulatory requirements outside of or in addition to those of the U.S. FDA and the European Union. The Senhance System is CE marked, which allows us to offer the product for sale in a number of jurisdictions, including select countries in Europe, the Middle East and Asia. Due to the variations in regulatory requirements within territories, the Company may be required to perform additional safety or clinical testing or fulfill additional agency requirements for specific territories. The Company may also be required to apply for registration using third parties within those territories and may be dependent upon the third parties’ successful regulatory processes to file, register and list the product applications and associated labeling, which could lead to significant investments and resource use. These additional requirements may result in delays in international registrations and commercialization of our products in certain countries.
In addition, we are utilizing distributors and sales agents in various territories throughout Europe, the Middle East and Africa, and need to ensure that our activities, and the activities of our distributors and sales agents, are compliant with local law and U.S. laws governing the sales of medical devices. The laws governing the registration, approval, clearance and sales of medical devices, such as the Senhance System, in multiple jurisdictions are complex, and the failure to comply with such laws in any given jurisdiction could subject us to financial penalties or suspension or termination of our ability to sell our products in the applicable jurisdiction.
Employees
As of December 31, 2017, we had 121 employees, including 119 full time employees. The Company considers its relationships with its employees to be good.
Corporate Information
The Company’s principal executive offices are located at 635 Davis Drive, Suite 300, Morrisville, NC 27560. TransEnterix Surgical was originally incorporated under the laws of the State of Delaware on July 12, 2006. On September 3, 2013, TransEnterix Surgical merged with and into a merger subsidiary of SafeStitch Medical, Inc. and became a wholly owned subsidiary of SafeStitch in a reverse merger transaction. SafeStitch was originally incorporated on August 19, 1988 as NCS Ventures Corp. under the laws of the State of Delaware. Its name was changed to Cellular Technical Services Company, Inc. on May 31, 1991. On September 4, 2007, SafeStitch acquired SafeStitch LLC, and, in January 2008, changed its name to SafeStitch Medical, Inc. On December 6, 2013, SafeStitch’s name was changed to TransEnterix, Inc. On September 21, 2015, TransEnterix International, a wholly owned subsidiary of the Company formed by the Company in conjunction with the Senhance Acquisition, acquired all of the membership interests of Vulcanos and changed the name of Vulcanos to TransEnterix Italia.
As of December 31, 2017, the active subsidiaries of the Company are TransEnterix International; TransEnterix Italia S.r.l.; TransEnterix Europe S.à.R.L; TransEnterix Europe S.à.R.L, Bertrange, Swiss Branch, Lugano; TransEnterix Asia Pte. Ltd.; and, TransEnterix Taiwan Ltd.
Available Information
The Company maintains a website at www.transenterix.com. Our Code of Business Conduct and Ethics, as reviewed and updated on November 8, 2017, is available on our website. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K, and amendments to those reports, filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, are available free of charge on our website as soon as practicable after electronic filing of such material with, or furnishing it to, the U.S. Securities and Exchange Commission (the “SEC”). This information may be read and copied at the Public Reference Room of the SEC at 100 F Street, N.E., Washington D.C. 20549. The SEC also maintains an internet website that contains reports, proxy statements, and other information about issuers, like TransEnterix, Inc., who file electronically with the SEC. The address of the site is http://www.sec.gov.
8
We are currently highly dependent on the commercial success of a single product, the Senhance System. We cannot give any assurance that the Senhance System can be successfully commercialized.
We are currently highly dependent on the commercial success of the Senhance System, which is FDA cleared and CE marked. We began our selling efforts for the Senhance System in the fourth quarter of 2015 in Europe and in the fourth quarter of 2017 in the United States. We have had limited commercial success to date. We are still in the process of establishing our commercial infrastructure in the U.S. We cannot assure you that we will be able to successfully commercialize the Senhance System, for a number of reasons, including, without limitation, failure in our sales and marketing efforts, the long sales cycle associated with the purchase of capital equipment, or the potential introduction by our competitors of more clinically effective or cost-effective alternatives. Failure to successfully commercialize the Senhance System would have a material and adverse effect on our business.
The sales cycle for the Senhance System is lengthy and unpredictable, which will make it difficult for us to forecast revenue and increase the magnitude of quarterly fluctuations in our operating results.
Purchase of a surgical robotic system such as the Senhance System represents a capital purchase by hospitals and other potential customers. The capital purchase nature of the transaction, the complexity of our product, the relative newness of surgical robotics and the competitive landscape requires us to spend substantial time and effort to assist potential customers in evaluating our robotic systems. We must communicate with multiple surgeons, administrative staff and executives within each potential customer in order to receive all approvals on behalf of such organizations. We may face difficulty identifying and establishing contact with such decision makers. Even after initial acceptance, the negotiation and documentation processes can be lengthy. Additionally, our customers may have strict limitations on spending depending on the current economic climate or trends in healthcare management.
We are also expanding the potential market for robotic surgical systems with our focus on laparoscopic surgery. Such expansion requires a different sales and marketing approach than a focus on open procedures. We expect our sales cycle to range between four to six quarters per sale. Each sale could take longer. Any delay in completing sales in a particular quarter could cause our operating results to fall below expectations. We also expect such a lengthy sales cycle makes it more difficult for us to accurately forecast revenue in future periods and may cause revenues and operating results to vary significantly in future periods.
Although we have expanded our commercial organization, we currently have limited marketing, sales and distribution capabilities. We are distributing our products through direct sales in the U.S. and select countries in Europe, and elsewhere through the use of independent contractor and distribution agreements with companies possessing established sales and marketing operations in the medical device industry. There can be no assurance that we will be successful in building our sales capabilities. To the extent that we enter into distribution, co-promotion or other arrangements, our product revenue is likely to be lower than if we directly market or sell our products. In addition, any revenue we receive will depend in whole or in part upon the efforts of such third parties, which may not be successful and are generally not within our control. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our products. If we are not successful in commercializing our existing and future products, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
We have procedures in place to require our distributors and sales agents to comply with applicable laws and regulations governing the sales of medical devices in the jurisdictions where they operate. Failure to meet such requirements could subject us to financial penalties or the suspension or termination of the ability to sell our products in such jurisdiction.
The surgical robotics industry is increasingly competitive, which can negatively impact our commercial opportunities.
The life sciences industry is highly competitive, and we face significant competition from many medical device companies that are researching and marketing products designed to address minimally invasive and robotic-assisted surgery, including new entrants in the competitive market. We are currently commercializing the Senhance System in the U.S. with FDA 510(k) clearance, in Europe which accepts a CE Mark, the Middle East and selected countries in Asia and face significant competition in such markets. Many of our competitors, including Intuitive Surgical, have significantly greater financial, manufacturing, marketing and product development resources than we do. Some of the medical device companies we compete with or expect to compete with include Johnson & Johnson, Medtronic plc, Applied Medical, Intuitive Surgical, Verb Surgical, Titan Medical and a number of minimally invasive surgical device and robotic surgical device manufacturers and providers of products and therapies that are designed to reduce the need for or attractiveness of surgical intervention. In addition, many other universities and private and public research institutions are or may become active in research involving surgical devices for minimally invasive and robotic-assisted surgery.
We are also expanding the potential market for robotic surgical systems with our focus on laparoscopic surgery. Such expansion may lead to additional competition with companies with sufficiently higher resources than ours.
9
We believe that our ability to successfully compete will depend on, among other things:
|
|
• |
the efficacy, safety and reliability of our products; |
|
|
• |
our ability to commercialize and market our cleared or approved products; |
|
|
• |
the completion of our development efforts and receipt of regulatory clearance or approval for instruments and accessories to support the use of the Senhance System; |
|
|
• |
the cost of ownership and use of our products in relation to alternative devices; |
|
|
• |
the timing and scope of regulatory clearances or approvals, including any expansion of the indications of use for our products; |
|
|
• |
whether our competitors substantially reduce the cost of ownership and use of an alternative device; |
|
|
• |
our ability to protect and defend intellectual property rights related to our products; |
|
|
• |
our ability to have our partners manufacture and sell commercial quantities of any cleared or approved products to the market; |
|
|
• |
the availability of adequate coverage and reimbursement by third-party payors for the procedures in which our products are used; |
|
|
• |
the effectiveness of our sales and marketing efforts; and |
|
|
• |
acceptance of future products by physicians and other health care providers. |
If our competitors market products that are more effective, safer, easier to use or less expensive than our products or future products, or that reach the market sooner than our products, we may not achieve commercial success. In addition, the medical device industry is characterized by rapid technological change. It may be difficult for us to stay abreast of the rapid changes in each technology. If we fail to stay at the forefront of technological change, we may be unable to compete effectively. Technological advances or products developed by our competitors may render our technologies or products obsolete or less competitive.
We anticipate that the highly competitive surgical robotics environment can lead our competitors to attempt to slow or derail our commercial progress. We are using our best efforts to enter the commercial markets effectively and efficiently while maintaining compliance with all regulatory and legal requirements. Responding to the actions of our competitors will require the attention of our management and may distract the management team from its focus on our commercial operations and lead to increased costs of commercialization, which could have a negative impact on our financial position.
Fluctuations in foreign currency exchange rates may adversely affect our financial results.
We conduct operations in several different countries, including the U.S. and throughout Europe, and portions of our revenues, expenses, assets and liabilities are denominated in U.S. dollars, Euros, and other currencies. Since our consolidated financial statements are presented in U.S. dollars, we must translate revenues, income and expenses, as well as assets and liabilities, into U.S. dollars at exchange rates in effect during or at the end of each reporting period. We have not historically hedged our exposure to foreign currency fluctuations. Accordingly, increases or decreases in the value of the U.S. dollar against the Euro and other currencies could materially affect our net operating revenues, operating income and the value of balance sheet items denominated in foreign currencies.
Our global operations expose us to additional risks and challenges associated with conducting business internationally.
The international expansion of our business may expose us to risks inherent in conducting foreign operations. These risks include:
|
|
• |
challenges associated with managing geographically diverse operations, which require an effective organizational structure and appropriate business processes, procedures and controls; |
|
|
• |
the increased cost of doing business in foreign jurisdictions, including compliance with international and U.S. laws and regulations that apply to our international operations; |
10
|
|
• |
changes in a specific country’s or region’s political or economic environment; |
|
|
• |
trade protection measures, import or export licensing requirements or other restrictive actions by U.S. or non-U.S. governments; |
|
|
• |
potentially adverse tax consequences; |
|
|
• |
complexities and difficulties in obtaining protection and enforcing our intellectual property; |
|
|
• |
compliance with additional regulations and government authorities in a highly regulated business; |
|
|
• |
difficulties associated with staffing and managing foreign operations, including differing labor relations; and |
|
|
• |
general economic and political conditions outside of the U.S. |
The risks that we face in our international operations may continue to intensify as we further develop and expand our international operations.
As we ramp up our manufacturing capabilities we face risks arising from sole suppliers of components and our ability to meet delivery schedules for sales of our products.
The Senhance System is manufactured for us under contract by a third party manufacturer. We or our manufacturer acquire raw materials and components of the Senhance System from vendors, some of which are sole suppliers. Although we believe that we have the manufacturing and inventory reserves to meet our anticipated Senhance System sales for the foreseeable future, we are currently taking steps to develop redundant manufacturing and supply alternatives. We cannot assure you that we will be successful in developing these redundant supply and manufacturing capabilities. If we are not successful, our business operations could suffer.
Because our design, development and manufacturing capabilities are limited, we may rely on third parties to design, develop, manufacture or supply some of our products. An inability to find additional or alternate sources for these services and products could materially and adversely affect our financial condition and results of operations.
We have used third-party design and development sources to assist in the design and development of our medical device products. In the future, we may choose to use additional third-party sources for the design and development of our products. If these design and development partners are unable to provide their services in the timeframe or to the performance level that we require, we may not be able to establish a contract and obtain a sufficient alternative supply from another supplier on a timely basis and in the manner that we require.
Our products require precise, high quality manufacturing. We and our contract manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and non-U.S. regulatory authorities to ensure strict compliance with the quality systems regulations, current “good manufacturing practices” and other applicable government regulations and corresponding standards. If we or our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with QSR, we may experience manufacturing errors resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our products, cost overruns or other problems that could seriously harm our business.
Any performance failure by us or on the part of our design and development partners or contract manufacturers could delay product development or regulatory clearance or approval of our products, or commercialization of our products and future products, depriving us of potential product revenue and resulting in additional losses. In addition, our dependence on any third party for design, development or manufacturing could adversely affect our future profit margins. Our ability to replace any then-existing manufacturer may be difficult because the number of potential manufacturers is limited and, in the case of Class III devices, the FDA must approve any replacement manufacturer before manufacturing can begin. It may be difficult or impossible for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or at all.
11
Our stock price has been volatile and may experience additional fluctuation in the future.
The market price of our common stock has been, and may continue to be, highly volatile, and such volatility could cause the market price of our common stock to decrease and could cause you to lose some or all of your investment in our common stock. During the two year period ended December 31, 2017, the market price of our common stock fluctuated from a high of $6.10 per share to a low of $0.45 per share. The market price of our common stock may continue to fluctuate significantly in response to numerous factors, some of which are beyond our control, such as:
|
|
• |
the announcement of favorable or unfavorable news regarding us, including our product development efforts and regulatory clearance activities; |
|
|
• |
the achievement of commercial sales of our products; |
|
|
• |
the announcement of new products or product enhancements by us or our competitors; |
|
|
• |
developments concerning intellectual property rights and regulatory approvals; |
|
|
• |
variations in our and our competitors’ results of operations; |
|
|
• |
changes in earnings estimates or recommendations by securities analysts, if our common stock is covered by analysts; |
|
|
• |
developments in surgical robotics; |
|
|
• |
the results of product liability or intellectual property lawsuits; |
|
|
• |
future issuances of common stock or other securities; |
|
|
• |
the addition or departure of key personnel; |
|
|
• |
announcements by us or our competitors of acquisitions, investments or strategic alliances; and |
|
|
• |
general market conditions and other factors, including factors unrelated to our operating performance. |
Further, the stock market in general, and the market for medical device companies in particular, can experience extreme price and volume fluctuations. Market fluctuations could result in extreme volatility in the price of our common stock, which could cause a decline in the value of our common stock and the loss of some or all of your investment.
We have a history of operating losses, and we may not be able to achieve or sustain profitability.
We have a limited operating history. We are not profitable and have incurred losses since our inception. Our net loss for the year ended December 31, 2017 was $144.8 million, and our accumulated deficit as of December 31, 2017 was $447.6 million. We believe that our existing cash and cash equivalents, together with cash received from sales of our products, will be sufficient to meet our anticipated cash needs for at least the next 12 months.
We expect to continue to incur losses for the foreseeable future. We will continue to incur research and development and general and administrative expenses related to our operations, and expect to increase our sales and marketing expenses as we increase our sales and marketing activities for the Senhance System in jurisdictions where FDA clearance and CE marking provides authorization for commercial activities. If our product candidates fail in development or do not gain regulatory clearance or approval, or if our products do not achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We will require substantial additional funding in the future, which may not be available to us on acceptable terms, or at all.
We do not anticipate that the net proceeds of equity financings in 2017 will be sufficient to support development of our products and product candidates and provide us with the necessary resources to commercialize the Senhance System and other products through the lengthy sales cycle. While we are currently focused on commercialization of our Senhance System, we intend to advance multiple additional products through clinical and pre-clinical development in the future. We believe we will need to raise substantial additional capital in order to continue our operations and achieve our business objectives.
We have an effective shelf registration statement. As of December 31, 2017, we had $100 million available for future financings under such shelf registration statement. Such capacity will expire in May 2020. We cannot assure you that we will be successful in obtaining such additional financing on terms acceptable to the Company or at all.
12
Our future funding requirements will depend on many factors, including, but not limited to:
|
|
• |
the costs of our Senhance System commercialization and development activities; |
|
|
• |
the costs and timing of seeking and obtaining FDA and other non-U.S. regulatory clearances and approvals for our products in development; |
|
|
• |
the costs associated with establishing a sales force and commercialization capabilities; |
|
|
• |
the costs associated with the expansion of our manufacturing capabilities; |
|
|
• |
our need to expand our research and development activities; |
|
|
• |
the costs of acquiring, licensing or investing in businesses, products and technologies; |
|
|
• |
the economic and other terms and timing of our existing licensing arrangement and any collaboration, licensing or other arrangements into which we may enter in the future; |
|
|
• |
our need and ability to hire additional management, scientific, medical and sales and marketing personnel; |
|
|
• |
the effect of competing technological and market developments; |
|
|
• |
our need to implement additional internal systems and infrastructure, including financial and reporting systems, quality systems and information technology systems; and |
|
|
• |
our ability to maintain, expand and defend the scope of our intellectual property portfolio. |
Until we generate a sufficient amount of revenue to finance our cash requirements, which may never occur, we expect to finance future cash needs primarily through public or private equity offerings, debt financings or strategic collaborations. We do not know whether additional funding will be available on acceptable terms, or at all. If we are not able to secure additional funding when needed, we may have to delay, reduce the scope of or eliminate one or more of our research and development programs. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience significant dilution; and debt financing, if available, may involve restrictive covenants that limit our operations. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our products or grant licenses on terms that may not be favorable to us.
Our stockholders have experienced dilution of their percentage ownership of our stock and may experience additional dilution in the future.
We have raised significant capital through the issuance of our common stock and warrants and anticipate that we will need to raise substantial additional capital in order to continue our operations and achieve our business objectives. We have an effective shelf registration statement under which we have the current ability to raise up to $100 million through the issuance of equity or debt securities. We cannot assure you that we will be able to sell shares or other securities in any offering at a price per share that is equal to or greater than the price per share paid by investors in previous offerings, and investors purchasing shares or other securities in the future could have rights superior to existing stockholders. The price per share at which we sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock in future transactions may be higher or lower than the price per share in previous offerings. The future issuance of the Company’s equity securities will further dilute the ownership of our outstanding common stock. The market price of our common stock has been, and may continue to be, highly volatile, and such volatility could cause the market price of our common stock to decrease and could cause you to lose some or all of your investment in our common stock.
Sales by stockholders of substantial amounts of our shares of common stock, the issuance of new shares of common stock by us or the perception that these sales may occur in the future could materially and adversely affect the market price of our common stock.
As of December 31, 2017, our directors, executive officers, principal stockholders and affiliated entities beneficially owned, in the aggregate, approximately 25% of our outstanding voting securities. As a result, if some or all of them acted together, they would have the ability to exert substantial influence over the election of our board of directors and the outcome of issues requiring approval by our stockholders. This concentration of ownership may also have the effect of delaying or preventing a change in control of the Company that may be favored by other stockholders. This could prevent transactions in which stockholders might otherwise recover a premium for their shares over current market prices.
13
The exercise of our outstanding options and warrants will dilute stockholders and could decrease our stock price.
The existence of our outstanding options and warrants, including the outstanding Series B Warrants, may adversely affect our stock price due to sales of a large number of shares or the perception that such sales could occur. These factors also could make it more difficult to raise funds through future offerings of common stock or warrants, and could adversely impact the terms under which we could obtain additional equity capital. Exercise of outstanding options and warrants, or any future issuance of additional shares of common stock or other equity securities, including but not limited to options, warrants or other derivative securities convertible into our common stock, may result in significant dilution to our stockholders and may decrease our stock price.
If we default on our existing indebtedness, such default would affect our financial condition.
We are party with Innovatus Life Sciences Lending Fund I, LP, or the Lender, and jointly and severally liable with certain of our U.S. subsidiaries for $14.0 million of outstanding debt under term loans issued under our Loan and Security Agreement, or the Innovatus Loan Agreement. The maturity date of the outstanding term loan is May 10, 2021. If we were to become unable to pay, when due, the principal of, interest on, or other amounts due in respect of, our indebtedness, our financial condition would be adversely affected. Further, under the Innovatus Loan Agreement, we are subject to certain restrictive covenants that, among other things, subject to exceptions, restrict the Company’s ability to do the following things: declare dividends or redeem or repurchase equity interests; incur additional liens; make loans and investments; incur additional indebtedness; engage in mergers, acquisitions, and asset sales; transact with affiliates; undergo a change in control; add or change business locations; and engage in businesses that are not related to its existing business. If we breach any of these restrictive covenants or are unable to pay our indebtedness under the Innovatus Loan Agreement when due, this could result in a default under the Innovatus Loan Agreement. In such event, the Lender may elect (after the expiration of any applicable notice or grace periods) to declare all outstanding borrowings, together with accrued and unpaid interest and other amounts payable under the Innovatus Loan Agreement, to be immediately due and payable. Any such occurrence would have an adverse impact on our financial condition. The Company’s obligations under the Innovatus Loan Agreement are secured by a security interest in all of the assets of the Company and its current and future domestic and material foreign subsidiaries, including a security interest in the intellectual property.
We issued 24,900,000 Series A Warrants and 24,900,000 Series B Warrants in May 2017; the outstanding warrants must be revalued each reporting period. In addition, we owe contingent consideration to Sofar under the Purchase Agreement that is also revalued each reporting period. Such assessments involve the use of estimates that could later be found to differ materially from actual results.
On April 28, 2017, we sold 24.9 million units, each consisting of one share of common stock, a Series A warrant to purchase one share of common stock, and a Series B warrant to purchase 0.75 shares of common stock, at a public offering price of $1.00 per unit for aggregate gross proceeds of $24.9 million in an underwritten firm commitment public offering. As of December 31, 2017, all Series A warrants were exercised. At December 31, 2017, Series B Warrants to acquire 9.8 million shares of common stock were outstanding. The outstanding Series B Warrants contain provisions, often referred to as “down-round protection” that may lead to adjustment of the exercise price and number of underlying warrant shares with respect to future issuances by the Company of its securities, including its common stock or convertible securities or debt securities. In addition, the third tranche of the contingent consideration to be paid to Sofar under the Purchase Agreement remains outstanding, to be paid if the designated milestone is met.
The Series B Warrants and the contingent consideration are each recorded as a liability on our financial statements, and we are required to revalue each of the outstanding Series B Warrants and the contingent consideration at each reporting period. Such revaluations necessarily involve the use of estimates, assumptions, probabilities and application of complex accounting principles. Actual value at the time the Series B Warrants are exercised or the contingent consideration paid could vary significantly from the value assigned to such liabilities on a quarterly basis. We cannot assure you that the revaluation of the Series B Warrants and contingent consideration will equal the value in the future, and know that the actual value could be significantly different, which could have a material adverse effect on us.
We sold our SurgiBot System assets in 2017, and we may not obtain the royalty income we anticipate from such sale.
In December 2017, we transferred ownership of the SurgiBot System assets to GBIL. The agreements provide rights to the purchaser to manufacture, or have manufactured, the SurgiBot System in China, and provides exclusive distribution rights to the Chinese market. The agreement provides us with minimum royalties of $14 million over a future five-year period. If the buyer is not successful in gaining Chinese regulatory approval or marketing the SurgiBot System, we will only receive such minimum royalties, decreasing the return on the funds expended in the development of the SurgiBot System.
14
Our commercial success depends significantly on our ability to operate without infringing the patents and other proprietary rights of third parties.
Other entities may have or obtain patents or proprietary rights that could limit our ability to manufacture, use, sell, offer for sale or import products or impair our competitive position. In addition, to the extent that a third party develops new technology that covers our products, we may be required to obtain licenses to that technology, which licenses may not be available or may not be available on commercially reasonable terms, if at all. If licenses are not available to us on acceptable terms, we will not be able to market the affected products or conduct the desired activities, unless we challenge the validity, enforceability or infringement of the third-party patent or circumvent the third-party patent, which would be costly and would require significant time and attention of our management. Third parties may have or obtain valid and enforceable patents or proprietary rights that could block us from developing products using our technology. Our failure to obtain a license to any technology that we require may materially harm our business, financial condition and results of operations.
If we become involved in patent litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, substantial liability for damages or be required to stop our product development and commercialization efforts, any of which could materially adversely affect our liquidity, business prospects and results of operations.
Third parties may sue us for infringing their patent rights. Likewise, we may need to resort to litigation to enforce a patent issued or licensed to us or to determine the scope and validity of proprietary rights of others. In addition, a third party may claim that we have improperly obtained or used its confidential or proprietary information. Furthermore, in connection with our third-party license agreements, we generally have agreed to indemnify the licensor for costs incurred in connection with litigation relating to intellectual property rights. The cost to us of any litigation or other proceeding relating to intellectual property rights, even if resolved in our favor, could be substantial, and the litigation would divert our management’s efforts. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of any litigation could limit our ability to continue our operations.
If any parties successfully claim that our creation or use of proprietary technologies infringes upon their intellectual property rights, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. In addition to any damages we might have to pay, a court could require us to stop the infringing activity or obtain a license. Any license required under any patent may not be made available on commercially acceptable terms, if at all. In addition, such licenses are likely to be non-exclusive and, therefore, our competitors may have access to the same technology licensed to us. If we fail to obtain a required license and are unable to design around a patent, we may be unable to effectively market some of our technology and products, which could limit our ability to generate revenues or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations.
For our Senhance System, we rely on our license from the European Union, and any loss of our rights under such license agreement, or failure to properly prosecute, maintain or enforce the patent applications underlying such license agreement, could materially adversely affect our business prospects for the Senhance System.
Some of the patents and patent applications in our patent portfolio related to the Senhance System are licensed to TransEnterix Italia under a license agreement with the European Union. Presently, we rely on such licensed technology for our Senhance System products and may license additional technology from the European Union or other third parties in the future. The EU license agreement gives us rights for the commercial exploitation of the licensed patents, patent applications and know-how, subject to certain provisions of the license agreement. Failure to comply with these provisions could result in the loss of our rights under the EU license agreement. Our inability to rely on these patents and patent applications which are the basis of certain aspects of our Senhance System technology would have an adverse effect on our business.
Further, our success will depend in part on the ability of us, the European Union and other third-party licensors to obtain, maintain and enforce patent protection for our licensed intellectual property and, in particular, those patents to which we have secured exclusive rights. We, the European Union or other third-party licensors may not successfully prosecute the patent applications which are licensed to us, may fail to maintain these patents, and may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than necessary to obtain an acceptable outcome from any such litigation. Without protection for the intellectual property we have licensed, other companies might be able to offer substantially identical products for sale, which could materially adversely affect our competitive business position, business prospects and results of operations.
If we or our licensors are unable to protect the confidentiality of our proprietary information and know-how, the value of our technology and products could be adversely affected.
In addition to patent protection, we also rely on other proprietary rights, including protection of trade secrets, know-how and confidential and proprietary information. To maintain the confidentiality of trade secrets and proprietary information, we will seek to
15
enter into confidentiality agreements with our employees, consultants and collaborators upon the commencement of their relationships with us. These agreements generally require that all confidential information developed by the individual or made known to the individual by us during the course of the individual’s relationship with us be kept confidential and not disclosed to third parties. Our agreements with employees also generally provide and will generally provide that any inventions conceived by the individual in the course of rendering services to us shall be our exclusive property. However, we may not obtain these agreements in all circumstances, and individuals with whom we have these agreements may not comply with their terms. In the event of unauthorized use or disclosure of our trade secrets or proprietary information, these agreements, even if obtained, may not provide meaningful protection, particularly for our trade secrets or other confidential information. To the extent that our employees, consultants or contractors use technology or know-how owned by third parties in their work for us, disputes may arise between us and those third parties as to the rights in related inventions. Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. The disclosure of our trade secrets would impair our competitive position and may materially harm our business, financial condition and results of operations.
If we are unable to obtain and enforce patent protection for our products, our business could be materially harmed.
Our success depends, in part, on our ability to protect proprietary methods and technologies that we develop or license under the patent and other intellectual property laws of the United States and other countries, so that we can prevent others from unlawfully using our inventions and proprietary information. However, we may not hold proprietary rights to some patents required for us to commercialize our proposed products. Because certain U.S. patent applications are confidential until patents issue, such as applications filed prior to November 29, 2000, or applications filed after such date which will not be filed in foreign countries, third parties may have filed patent applications for technology covered by our pending patent applications without our being aware of those applications, and our patent applications may not have priority over those applications. For this and other reasons, we or our third-party collaborators may be unable to secure desired patent rights, thereby losing desired exclusivity. If licenses are not available to us on acceptable terms, we will not be able to market the affected products or conduct the desired activities, unless we challenge the validity, enforceability or infringement of the third-party patent or otherwise circumvent the third-party patent.
Our strategy depends on our ability to promptly identify and seek patent protection for our discoveries. In addition, we may rely on third-party collaborators to file patent applications relating to proprietary technology that we develop jointly during certain collaborations. The process of obtaining patent protection is expensive and time-consuming. If our present or future collaborators fail to file and prosecute all necessary and desirable patent applications at a reasonable cost and in a timely manner, our business will be adversely affected. Despite our efforts and the efforts of our collaborators to protect our proprietary rights, unauthorized parties may be able to develop and use information that we regard as proprietary.
The issuance of a patent provides a presumption, but does not guarantee that it is valid. Any patents we have obtained, or obtain in the future, may be challenged or potentially circumvented. Moreover, the United States Patent and Trademark Office, or the USPTO, may commence interference proceedings involving our patents or patent applications. Any such challenge to our patents or patent applications would be costly, would require significant time and attention of our management and could have a material adverse effect on our business. In addition, future court decisions may introduce uncertainty in the enforceability or scope of any patent, including those owned by medical device companies.
Our pending patent applications may not result in issued patents. The patent position of medical device companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the USPTO and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in medical device patents. Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. The legal systems of certain countries do not favor the aggressive enforcement of patents, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. Therefore, the enforceability or scope of our owned or licensed patents in the United States or in foreign countries cannot be predicted with certainty, and, as a result, any patents that we own or license may not provide sufficient protection against competitors. We may not be able to obtain or maintain patent protection for our pending patent applications, those we may file in the future, or those we may license from third parties.
We cannot assure you that any patents that will issue, that may issue or that may be licensed to us will be enforceable or valid or will not expire prior to the commercialization of our products, thus allowing others to more effectively compete with us. Therefore, any patents that we own or license may not adequately protect our future products.
16
Even if we obtain regulatory clearances or approvals for our products, the terms thereof and ongoing regulation of our products may limit how we manufacture and market our products, which could materially impair our ability to generate anticipated revenues.
Once regulatory clearance or approval has been granted, the cleared or approved product and its manufacturer are subject to continual review. Any cleared or approved product may be promoted only for its indicated uses. In addition, if the FDA or other non-U.S. regulatory authorities clear or approve any of our products, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the product will be subject to extensive regulatory requirements. We and any outsourced manufacturers of our products are also required to comply with the FDA’s Quality System Regulation, or similar requirements of non-U.S. regulatory authorities which includes requirements relating to quality control and quality assurance, as well as the corresponding maintenance of records and documentation as well as other quality system requirements and regulations from non-U.S. regulatory authorities. Further, regulatory agencies must approve our manufacturing facilities for Class III devices before they can be used to manufacture our products, and all manufacturing facilities are subject to ongoing regulatory inspection. If we fail to comply with the regulatory requirements of the FDA, either before or after clearance or approval, or other non-U.S. regulatory authorities, or if previously unknown problems with our products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions, including:
|
|
• |
restrictions on the products, manufacturers or manufacturing process; |
|
|
• |
adverse inspectional observations (Form 483) warning letters, non-warning letters incorporating inspectional observations; |
|
|
• |
civil or criminal penalties or fines; |
|
|
• |
injunctions; |
|
|
• |
product seizures, detentions or import bans; |
|
|
• |
voluntary or mandatory product recalls and publicity requirements; |
|
|
• |
suspension or withdrawal of regulatory clearances or approvals; |
|
|
• |
total or partial suspension of production; |
|
|
• |
imposition of restrictions on operations, including costly new manufacturing requirements; |
|
|
• |
refusal to clear or approve pending applications or premarket notifications; and |
|
|
• |
import and export restrictions. |
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
In addition, the FDA and other non-U.S. regulatory authorities may change their policies and additional regulations may be enacted that could prevent or delay regulatory clearance or approval of our products. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to maintain regulatory compliance, we would likely not be permitted to market our future products and we may not achieve or sustain profitability.
17
The regulatory approval and clearance processes are expensive, time-consuming and uncertain and may prevent us from obtaining approvals or clearances, as the case may be, for the commercialization of some or all of our products.
Regulatory approval of a PMA, PMA supplement or clearance pursuant to a 510(k) premarket notification is not guaranteed, and the approval or clearance process, as the case may be, is expensive, uncertain and may, especially in the case of the PMA application, take several years. The FDA also has substantial discretion in the medical device clearance process or approval process. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to repeat or perform additional development, standardized testing, pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials that will be required for FDA clearance or approval varies depending on the medical device candidate, the disease or condition that the medical device candidate is designed to address, and the regulations applicable to any particular medical device candidate. The FDA or other non-U.S. regulatory authorities can delay, limit or deny clearance or approval of a medical device candidate for many reasons, including:
|
|
• |
a medical device candidate may not be deemed safe or effective, in the case of a PMA application; |
|
|
• |
a medical device candidate may not be deemed to be substantially equivalent to a device lawfully marketed either as a grandfathered device or one that was cleared through the 510(k) premarket notification process; |
|
|
• |
a medical device candidate may not be deemed to be in conformance with applicable standards and regulations; |
|
|
• |
FDA or other regulatory officials may not find the data from pre-clinical studies and clinical trials sufficient; |
|
|
• |
the FDA might not approve our processes or facilities or those of any of our third-party manufacturers for a Class III PMA device; |
|
|
• |
other non-U.S. regulatory authorities may not approve our processes or facilities or those of any of our third-party manufacturers, thereby restricting export; or |
|
|
• |
the FDA or other non-U.S. regulatory authorities may change clearance or approval policies or adopt new regulations. |
The laws governing the regulatory approval or clearance pathways in jurisdictions outside of the United States are complex. We need to ensure that our activities, and the activities of our distributors and agents, comply with such laws. If we do not comply with such laws, we may not be able to sell our products, including the Senhance System, in all jurisdictions we have targeted, which could have an adverse effect on our business operations and financial condition.
Once our products are cleared or approved, modifications to our products may require new 510(k) clearances, premarket approvals or new or amended CE Certificates of Conformity, and may require us to cease marketing or recall the modified products until clearances, approvals or the relevant CE Certificates of Conformity are obtained.
Any modification to a 510(k)-cleared device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, design, or manufacture, requires a new 510(k) clearance or, possibly, a PMA. The FDA requires every manufacturer to make this determination in the first instance, but the FDA may review such determinations. The FDA may not agree with our decisions regarding whether new clearances or approvals are necessary. If the FDA disagrees with our determinations for any future changes, or prior changes to previously marketed products, as the case may be, we may be required to cease marketing or to recall the modified products until we obtain clearance or approval, and we may be subject to significant regulatory fines or penalties.
Furthermore, the FDA’s ongoing review of the 510(k) program may make it more difficult for us to make modifications to our products, either by imposing more strict requirements on when a new 510(k) for a modification to a previously cleared product must be submitted, or applying more onerous review criteria to such submissions. In October 2017, the FDA issued guidance documents addressing when to submit a new 510(k) due to modifications to 510(k) cleared products and the criteria for evaluating substantial equivalence. The interpretation of the guidance document by the FDA staff could lead to instances where the FDA disagrees with the Company’s decision regarding a change, and could result in warning letters and other enforcement actions.
Even after clearance or approval for our products is obtained, we are subject to extensive post-market regulation by the FDA. Our failure to meet strict regulatory requirements could require us to pay fines, incur other costs or even close our facilities.
Even after we have obtained the proper regulatory clearance or approval to market a product, the FDA has the power to require us to conduct post-market studies. These studies can be very expensive and time-consuming to conduct. Failure to complete such studies in a timely manner could result in the revocation of clearance or approval and the recall or withdrawal of the product, which could prevent us from generating sales from that product in the United States. The FDA has broad enforcement powers, and any regulatory enforcement actions or inquiries, or other increased scrutiny on us, could dissuade some surgeons from using our products and adversely affect our reputation and the perceived safety and efficacy of our products.
18
We are also required to comply with the FDA’s QSR, which covers the methods used in, and the facilities and controls used for, the design, manufacture, quality assurance, labeling, packaging, sterilization, storage, shipping, installation and servicing of our marketed products. The FDA enforces the QSR through periodic announced and unannounced inspections of manufacturing facilities. In addition, in the future, regulatory authorities and/or customers may require specific packaging of sterile products, which could increase our costs and the price of our products. Later discovery of previously unknown problems with our products, including unanticipated adverse events or adverse events of unanticipated severity or frequency, manufacturing problems, or failure to comply with regulatory requirements such as QSR, may result in changes to labeling, restrictions on such products or manufacturing processes, withdrawal of the products from the market, voluntary or mandatory recalls, a requirement to repair, replace or refund the cost of any medical device we manufacture or distribute, fines, suspension of regulatory approvals, product seizures, injunctions or the imposition of civil or criminal penalties which would adversely affect our business, operating results and prospects.
If one of our products, or a malfunction of one of our products, causes or contributes to a death or a serious injury, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA’s medical device reporting, or MDR, regulations, we are required to report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Repeated product malfunctions may result in a voluntary or involuntary product recall, which could divert managerial and financial resources, impair our ability to manufacture our products in a cost-effective and timely manner, and have an adverse effect on our reputation, results of operations and financial condition. We are also required to follow detailed recordkeeping requirements for all firm-initiated medical device corrections and removals, and to report such corrective and removal actions to the FDA if they are carried out in response to a risk to health and have not otherwise been reported under the MDR regulations.
All manufacturers bringing medical devices to market in the European Economic Area, or EEA, are legally bound to report any incident that led or might have led to the death or serious deterioration in the state of health of a patient, user or other person, and which the manufacturer’s device is suspected to have caused, to the competent authority in whose jurisdiction the incident occurred. In such case, the manufacturer must file an initial report with the relevant competent authority, which would be followed by further evaluation or investigation of the incident and a final report indicating whether further action is required. Any adverse event involving our products could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Adverse events involving our products have been reported to us in the past, and we cannot guarantee that they will not occur in the future. Any corrective action, whether voluntary or involuntary, will require the dedication of our time and capital, distract management from operating our business and may harm our reputation and financial results.
A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities such as the competent authorities of the EEA countries have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture or in the event that a product poses an unacceptable risk to health. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our distributors could occur as a result of an unacceptable risk to health, component failures, manufacturing errors, design or labeling defects or other deficiencies and issues.
Any future recalls of any of our products would divert managerial and financial resources and could have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be required to bear other costs or take other actions that may have a negative impact on our future sales and our ability to generate profits.
U.S. legislative or FDA regulatory reforms may make it more difficult and costly for us to obtain regulatory approval of our product candidates and to manufacture, market and distribute our products after approval is obtained.
Legislative changes could significantly alter the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products. In addition, FDA regulations and guidance could be revised or reinterpreted by the FDA in ways that could significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of future products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be.
19
Even if we receive regulatory clearance or approval to market our products, the market may not be receptive to our products, which could undermine our financial viability.
Even if our products obtain regulatory clearance or approval, resulting products may not gain market acceptance among physicians, patients, health care payors and/or the medical community. We have experienced minimal sales of our Senhance System, to date. We believe that the degree of market acceptance will depend on a number of factors, including:
|
|
• |
timing of market introduction of competitive products; |
|
|
• |
safety and efficacy of our products; |
|
|
• |
physician training in the use of our products; |
|
|
• |
prevalence and severity of any side effects; |
|
|
• |
potential advantages or disadvantages over alternative treatments; |
|
|
• |
strength of marketing and distribution support; and |
|
|
• |
price of our future products, both in absolute terms and relative to alternative treatments. |
If applicable, availability of coverage and reimbursement from government and other third-party payors can also impact the acceptance of our product offerings.
If we fail to attract and retain key management and professional personnel, we may be unable to successfully commercialize or develop our products.
We will need to effectively manage our operational, sales and marketing, development and other resources in order to successfully pursue our commercialization and research and development efforts for our existing and future products. Our success depends on our continued ability to attract, retain and motivate highly qualified personnel. If we are not successful in retaining and recruiting highly qualified personnel, our business may be harmed as a result.
We may be subject, directly or indirectly, to federal and state anti-kickback, fraud and abuse, false claims, privacy and security and physician payment transparency laws. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Our business activities are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which we conduct our business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician payment transparency laws. If our operations are found to be in violation of any of such laws that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Current legislation and future legislative or regulatory reform of the health care system may affect our ability to sell our products profitably.
In the United States, there have been, and we expect there to continue to be, a number of legislative and regulatory initiatives, at both the federal and state government levels, to change the healthcare system in ways that, if approved, could affect our ability to sell our products profitably. While many of the proposed policy changes require congressional approval to implement, we cannot assure you that reimbursement payments under governmental and private third-party payor programs to health care providers will remain at levels comparable to present levels or will be sufficient to cover the costs allocable to patients eligible for reimbursement under these programs. Any changes that lower reimbursement rates under Medicare, Medicaid or private payor programs could negatively affect our business.
To the extent that any of our products are deemed to be durable medical equipment, or DME, they may be subject to distribution under Medicare’s Competitive Acquisition regulations, which could adversely affect the amount that we can seek from payors. Non‑DME devices used in surgical procedures are normally paid directly by the hospital or health care provider and not reimbursed separately by third-party payors. As a result, these types of devices are subject to intense price competition that can place a small manufacturer at a competitive disadvantage as hospitals and health care providers attempt to negotiate lower prices for products such as the ones we develop and sell.
20
With the continued uncertainty regarding the status of the 2010 Health Care Reform Legislation, at this time, the Company is not certain as to the impact of federal health care legislation on its business.
The 2010 Health Care Reform Legislation subjects manufacturers of medical devices to an excise tax of 2.3% on certain U.S. sales of medical devices beginning in January 2013. This excise tax was suspended in December 2017 for two additional years, however, if eventually implemented, this excise tax will likely increase our expenses in the future.
Further, the 2010 Health Care Reform Legislation includes the Open Payments Act (formerly referred to as the Physician Payments Sunshine Act), which, in conjunction with its implementing regulations, requires certain manufacturers of certain drugs, biologics, and devices that are reimbursed by Medicare, Medicaid and the Children’s Health Insurance Program to report annually certain payments or “transfers of value” provided to physicians and teaching hospitals and to report annually ownership and investment interests held by physicians and their immediate family members during the preceding calendar year. We provided reports under the Open Payments Act to the Centers for Medicare & Medicaid Services, or CMS. The failure to report appropriate data accurately, timely, and completely could subject us to significant financial penalties. Other countries and several states currently have similar laws and more may enact similar legislation.
We are unable to predict what additional legislation or regulation relating to the health care industry or third-party coverage and reimbursement may be enacted in the future or what effect such legislation or regulation would have on our business. Any cost containment measures or other health care system reforms that are adopted could have a material and adverse effect on our ability to commercialize our existing and future products successfully.
Because our design, development and manufacturing capabilities are limited, we may rely on third parties to design, develop, manufacture or supply some of our products. An inability to find additional or alternate sources for these services and products could materially and adversely affect our financial condition and results of operations.
We have used third-party design and development sources to assist in the design and development of our medical device products. In the future, we may choose to use additional third-party sources for the design and development of our products. If these design and development partners are unable to provide their services in the timeframe or to the performance level that we require, we may not be able to establish a contract and obtain a sufficient alternative supply from another supplier on a timely basis and in the manner that we require.
Our products require precise, high quality manufacturing. We and our contract manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and non-U.S. regulatory authorities to ensure strict compliance with the quality systems regulations, current “good manufacturing practices” and other applicable government regulations and corresponding standards. If we or our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with QSR, we may experience manufacturing errors resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our products, cost overruns or other problems that could seriously harm our business.
Any performance failure by us or on the part of our design and development partners or contract manufacturers could delay product development or regulatory clearance or approval of our products, or commercialization of our products and future products, depriving us of potential product revenue and resulting in additional losses. In addition, our dependence on any third party for design, development or manufacturing could adversely affect our future profit margins. Our ability to replace any then-existing manufacturer may be difficult because the number of potential manufacturers is limited and, in the case of Class III devices, the FDA must approve any replacement manufacturer before manufacturing can begin. It may be difficult or impossible for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or at all.
We may become subject to potential product liability claims, and we may be required to pay damages that exceed our insurance coverage.
Our business exposes us to potential product liability claims that are inherent in the design, testing, manufacture, sale and distribution of our products and each of our product candidates that we are seeking to introduce to the market. Surgical medical devices involve significant risks of serious complications, including bleeding, nerve injury, paralysis, infection, and even death. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates or in our inability to secure coverage in the future on commercially reasonable terms, if at all. In addition, if our product liability insurance proves to be inadequate to pay a damage award, we may have to pay the excess of this award out of our cash reserves, which could significantly harm our financial condition. If longer-term patient results and experience indicate that our products or any component of a product causes tissue damage, motor impairment or other adverse effects, we could be subject to significant liability. A product liability claim, even one without merit, could harm our reputation in the industry, lead to significant legal fees, and result in the diversion of management’s attention from managing our business.
21
None.
Our principal corporate office is located at 635 Davis Drive, Suite 300, Morrisville, North Carolina. We lease this facility, which consists of 37,328 square feet, for a five-year term, under a lease that commenced on April 1, 2010. An amendment to this lease was signed on June 13, 2014, extending the lease term until June 30, 2018. Pursuant to a lease entered into on October 24, 2013, we also lease 24,000 square feet of warehouse and office space in Durham, North Carolina. That lease commenced in January 2014 and expired in January 2018.
Our Italian research and development and demonstration facilities are located at Viale dell'Innovazione 3, 20126 Milan, Italy. We lease these facilities, which consist of 11,273 square feet, for a six-year term ending on July 31, 2022, under a lease that commenced on May 12, 2016.
None.
Not applicable.
22
|
ITEM 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Since April 2, 2014, our common stock has been listed on the NYSE American under the symbol “TRXC.” The table below sets forth, for the respective periods indicated, the high and low bid prices for our common stock on the NYSE American. The bid prices represent inter-dealer transactions, without adjustments for retail mark-ups, mark-downs or commissions and may not necessarily represent actual transactions.
|
|
|
Bid Prices |
|
|||||
|
|
|
High |
|
|
Low |
|
||
|
2018 |
|
|
|
|
|
|
|
|
|
First Quarter (through February 27, 2018) |
|
$ |
2.19 |
|
|
$ |
1.27 |
|
|
2017 |
|
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
1.63 |
|
|
$ |
1.05 |
|
|
Second Quarter |
|
|
1.26 |
|
|
|
0.45 |
|
|
Third Quarter |
|
|
1.53 |
|
|
|
0.60 |
|
|
Fourth Quarter |
|
|
5.00 |
|
|
|
1.32 |
|
|
2016 |
|
|
|
|
|
|
|
|
|
First Quarter |
|
$ |
4.79 |
|
|
$ |
1.54 |
|
|
Second Quarter |
|
|
6.10 |
|
|
|
1.03 |
|
|
Third Quarter |
|
|
1.97 |
|
|
|
1.16 |
|
|
Fourth Quarter |
|
|
2.33 |
|
|
|
1.30 |
|
As of March 1, 2018, there were approximately 228 record holders of our common stock (counting all shares held in single nominee registration as one stockholder).
We paid no dividends or made any other distributions in respect of our common stock during our fiscal years ended December 31, 2017, 2016 and 2015, and we have no plans to pay any dividends or make any other distributions in the future. In addition, the terms of the Innovatus Loan Agreement prohibit the Company from paying any dividends without the consent of the Lender.
Securities Authorized for Issuance Under Equity Compensation Plans.
The Company currently has one equity compensation plan under which it makes awards, the TransEnterix, Inc. Amended and Restated Incentive Compensation Plan, (the “Plan”). The Plan was originally approved by the Board of Directors and adopted by the majority of our stockholders on November 13, 2007, and amended and restated and approved by the Board of Directors and approved by the majority of our stockholders on May 7, 2015 to increase the number of shares of common stock authorized under the Plan to 11,940,000 shares, and to make other changes. The Plan was amended on June 8, 2016 to increase the number of shares reserved for issuance under the Plan to 18,940,000 shares. The Plan was amended on May 25, 2017 to increase the number of shares reserved for issuance under the Plan to 25,940,000 shares. The Plan is used for plan-based awards for officers, other employees, consultants, advisors and non-employee directors. In connection with the 2013 merger transaction with SafeStitch Medical, Inc., or the Merger, we assumed all of the options that were issued and outstanding immediately prior to the Merger as issued by TransEnterix Surgical, and adjusted based on the Merger at the exchange ratio, which are now exercisable for approximately 1,203,825 shares of common stock. Such options were granted under the TransEnterix, Inc. 2006 Stock Plan (the “2006 Plan”) which was assumed by the Company in the Merger. The 2006 Plan is maintained solely for the purpose of the stock options granted under such 2006 Plan that remain outstanding; no future awards are authorized to be made under the 2006 Plan.
The following table gives information about the Company’s common stock that may be issued upon the exercise of options and other equity awards as of December 31, 2017:
|
Plan Category |
|
Number of securities to be issued upon exercise of outstanding options (1) |
|
|
Weighted average exercise price of outstanding options |
|
|
Number of securities remaining available for future issuance (2) |
|
|||
|
Equity compensation plans approved by security holders |
|
|
18,884,506 |
|
|
$ |
1.86 |
|
|
|
6,491,343 |
|
|
Equity compensation plans not approved by security holders (3) |
|
|
1,203,825 |
|
|
$ |
1.04 |
|
|
|
— |
|
|
Total |
|
|
20,088,331 |
|
|
|
|
|
|
|
6,491,343 |
|
23
|
(1) |
Includes 14,489,850 shares underlying outstanding stock options awarded under the Plan and 4,394,656 restricted stock units awarded under the Plan. |
|
(2) |
These shares are all available for future awards under the Plan. |
|
(3) |
Represents 1,203,825 shares underlying outstanding stock options awarded prior to the Merger under the 2006 Plan and assumed in the Merger. |
The graph below matches TransEnterix, Inc.'s cumulative 5-year total shareholder return on common stock with the cumulative total returns of the NYSE American index and the RDG SmallCap Medical Devices index. The graph tracks the performance of a $100 investment in our common stock and in each index (with the reinvestment of all dividends) from December 31, 2012 to December 31, 2017.
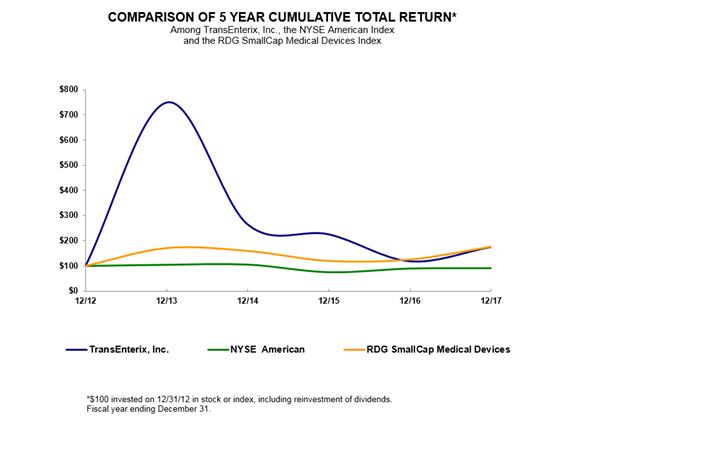
|
|
|
12/12 |
|
12/13 |
|
12/14 |
|
12/15 |
|
12/16 |
|
12/17 |
|
TransEnterix, Inc. |
|
100.00 |
|
750.00 |
|
264.55 |
|
225.45 |
|
118.18 |
|
175.45 |
|
NYSE American |
|
100.00 |
|
104.47 |
|
105.23 |
|
75.69 |
|
89.97 |
|
91.27 |
|
RDG SmallCap Medical Devices |
|
100.00 |
|
172.06 |
|
160.48 |
|
119.92 |
|
125.76 |
|
177.60 |
The stock price performance included in this graph is not necessarily indicative of future stock price performance.
Sales of Equity Securities and Use of Proceeds.
On September 7, 2017, the Board of Directors approved the issuance of common stock warrants to purchase 950,000 shares to a service provider to the Company. The issuance of the foregoing securities were exempt from the registration requirements of the Securities Act of 1933, as amended, or the Securities Act, afforded by Section 3(a)(9) or 4(a)(2) thereof and Regulation D promulgated thereunder, which exception we believe is available because the securities were not offered pursuant to a general solicitation and such issuances were otherwise made in compliance with the requirements of Regulation D and Rule 506. The securities issued in this transaction may not be resold except pursuant to an effective registration statement filed under the Securities Act or pursuant to a valid exemption from the registration requirements of the Securities Act.
24
On May 5, 2017, the Board of Directors approved the issuance of a common stock warrant to purchase 1,244,746 shares at $1.00 per share to Innovatus Life Sciences Lending Fund I, LP, a new Lender under a Loan Agreement entered into on May 10, 2017. The issuance of the foregoing securities were exempt from the registration requirements of the Securities Act afforded by Section 3(a)(9) or 4(a)(2) thereof, which exception we believe is available because the securities were not offered pursuant to a general solicitation. The securities issued in this transaction may not be resold except pursuant to an effective registration statement filed under the Securities Act or pursuant to a valid exemption from the registration requirements of the Securities Act.
On September 14, 2016, the Board of Directors approved the issuance of up to 150,000 shares of common stock to a vendor of the Company in lieu of a cash payment. To date, the Company has issued a total of 95,678 shares of the Company’s common stock to such vendor. The issuance of the foregoing securities were exempt from the registration requirements of the Securities Act afforded by Section 3(a)(9) or 4(a)(2) thereof and Regulation D promulgated thereunder, which exception we believe is available because the securities were not offered pursuant to a general solicitation and such issuances were otherwise made in compliance with the requirements of Regulation D and Rule 506. The securities issued in this transaction may not be resold except pursuant to an effective registration statement filed under the Securities Act or pursuant to a valid exemption from the registration requirements of the Securities Act.
The Company did not purchase any of its common stock during the quarter ended December 31, 2017.
The table below shows selected consolidated financial data. The statements of operations and comprehensive loss data for the years ended December 31, 2017, 2016 and 2015 and the balance sheet data at December 31, 2017 and 2016 are derived from our financial statements included elsewhere in this Annual Report. The statements of operations and comprehensive loss data for the years ended December 31, 2014 and 2013 and the balance sheet data at December 31, 2015, 2014 and 2013 are derived from our financial statements not included in this Annual Report. The historical results presented below are not necessarily indicative of financial results to be achieved in future periods.
|
Year ended December 31, |
|
2017(1) |
|
|
2016(1) |
|
|
2015 (1) |
|
|
2014 (2) |
|
|
2013 (3)(4) |
|
|||||
|
|
|
(in thousands) |
|
|||||||||||||||||
|
Statement of Operations and Comprehensive Loss Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sales |
|
$ |
7,111 |
|
|
$ |
1,519 |
|
|
$ |
— |
|
|
$ |
401 |
|
|
$ |
1,431 |
|
|
Income (loss) |
|
$ |
(144,796 |
) |
|
$ |
(119,980 |
) |
|
$ |
(46,948 |
) |
|
$ |
(37,652 |
) |
|
$ |
(28,358 |
) |
|
Income (loss) per common share |
|
$ |
(0.97 |
) |
|
$ |
(1.07 |
) |
|
$ |
(0.59 |
) |
|
$ |
(0.64 |
) |
|
$ |
(2.23 |
) |
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total assets |
|
$ |
250,251 |
|
|
$ |
176,249 |
|
|
$ |
248,602 |
|
|
$ |
135,111 |
|
|
$ |
116,714 |
|
|
Long-term obligations (5) |
|
$ |
28,473 |
|
|
$ |
27,690 |
|
|
$ |
40,253 |
|
|
$ |
9,175 |
|
|
$ |
4,602 |
|
|
(1) |
Includes the assets and liabilities of TransEnterix Italia acquired and assumed in the Senhance Acquisition, which occurred on September 21, 2015. See the description titled “Senhance Acquisition and Related Transactions” under Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in this Annual Report. |
|
(2) |
On March 31, 2014, we effectuated a reverse stock split of our issued and outstanding shares of common stock at a ratio of 1 for 5. As a result of the Reverse Stock Split, our issued and outstanding stock decreased from 244,276,923 to 48,855,255 shares of common stock, all with a par value of $0.001. All information related to common stock, preferred stock and earnings per share for prior periods has been retroactively adjusted to give effect to the Reverse Stock Split. |
|
(3) |
On September 3, 2013, TransEnterix Surgical, Inc. a Delaware corporation, and SafeStitch Medical, Inc., a Delaware corporation or SafeStitch consummated a merger transaction whereby TransEnterix Surgical merged with a merger subsidiary of SafeStitch, with TransEnterix Surgical as the surviving entity in the merger or Merger. As a result of the Merger, TransEnterix Surgical became a wholly owned subsidiary of SafeStitch. On December 6, 2013, SafeStitch changed its name to TransEnterix, Inc. The Merger was a reverse merger for accounting purposes with TransEnterix Surgical as the acquiring company. Therefore, from September 3, 2013 forward the financial statements of the Company are the historical financial statements of TransEnterix Surgical with the addition of SafeStitch as of the date of the Merger. |
|
(4) |
Represents the financial statements of TransEnterix Surgical for the period from January 1, 2013 to September 2, 2013. |
|
(5) |
Long-term obligations include: (1) cash consideration installments to be paid to Sofar in connection with the Senhance Acquisition; (2) outstanding amounts under a Loan Agreement, first entered into by TransEnterix Surgical in January 2012 and amended from time to time since such time and replaced in 2017; (3) in 2017, 2016 and 2015 net deferred tax liabilities; and (4) in 2013, promissory notes of SafeStitch, which were converted into equity securities of the Company in 2013. |
25
The following discussion of our financial condition and results of operations should be read in conjunction with our “Risk Factors” and our consolidated financial statements and the related notes to our consolidated financial statements included in this Annual Report. The following discussion contains forward-looking statements. See cautionary note regarding “Forward-Looking Statements” at the beginning of this Annual Report.
Overview
TransEnterix is a medical device company that is digitizing the interface between the surgeon and the patient to improve minimally invasive surgery by addressing the clinical and economic challenges associated with current laparoscopic and robotic options in today's value-based healthcare environment. The Company is focused on the commercialization of the Senhance™ Surgical System, which digitizes laparoscopic minimally invasive surgery. The system allows for robotic precision, haptic feedback, surgeon camera control via eye sensing and improved ergonomics while offering responsible economics.
The Senhance System has been granted a CE Mark in Europe for laparoscopic abdominal and pelvic surgery, as well as limited thoracic operations excluding cardiac and vascular surgery. In April 2017, the Company submitted a 510(k) application to the FDA for the Senhance System. On October 13, 2017, the Company received 510(k) clearance from the FDA for use in laparoscopic colorectal and gynecologic surgery. These indications cover 23 procedures, including benign and oncologic procedures. We anticipate expanding the indications for use in the middle of 2018. The Senhance System is available for sale in the U.S., the EU and select other countries.
The Senhance System is a multi-port robotic surgery system which allows multiple robotic arms to control instruments and a camera. The system features advanced technology to enable surgeons with haptic feedback and the ability to move the camera via eye movement. The system replicates laparoscopic motion that is familiar to experienced surgeons, and integrates three-dimensional high definition vision technology. The Senhance System also offers responsible economics to hospitals by offering robotic technology with reusable instruments thereby reducing additional costs per surgery when compared to other robotic solutions.
The Company has also developed the SurgiBot System, a single-port, robotically enhanced laparoscopic surgical platform. On December 18, 2017, the Company announced that it had entered into an agreement with Great Belief International Limited, or GBIL, to advance the SurgiBot System towards global commercialization. The agreement transfers ownership of the SurgiBot System assets, while the Company retains the option to distribute or co-distribute the SurgiBot System outside of China. Upon completion of the transfer of all SurgiBot System assets, GBIL will have the SurgiBot System manufactured in China and obtain Chinese regulatory clearance from the China Food and Drug Administration, or CFDA, while entering into a nationwide distribution agreement with China National Scientific and Instruments and Materials Company, or CSIMC, for the Chinese market. The agreement provides the Company with proceeds of at least $29 million, of which $7.5 million was received in December 2017. An additional $7.5 million is expected to be received by March 31, 2018, including a $3.0 million equity investment at $2.33 per share. The remaining $14 million, representing minimum royalties, will be paid beginning at the earlier of receipt of Chinese regulatory approval or five years.
We believe that future outcomes of minimally invasive surgery will be enhanced through our combination of more advanced tools and robotic functionality, which are designed to: (i) empower surgeons with improved precision, dexterity and visualization; (ii) improve patient satisfaction and enable a desirable post-operative recovery; and (iii) provide a cost-effective robotic system, compared to existing alternatives today, for a wide range of clinical applications. Our strategy is to focus on the commercialization and further development of the Senhance System.
From our inception, we devoted a substantial percentage of our resources to research and development and start-up activities, consisting primarily of product design and development, clinical studies, manufacturing, recruiting qualified personnel and raising capital.
Since inception, we have been unprofitable. As of December 31, 2017, we had an accumulated deficit of $447.6 million.
We expect to continue to invest in research and development and sales and marketing and increase selling, general and administrative expenses as we grow. As a result, we will need to generate significant revenue in order to achieve profitability.
We operate in one business segment.
26
On May 10, 2017, the Company and its domestic subsidiaries, as co-borrowers, entered into a Loan and Security Agreement, or the Innovatus Loan Agreement with Innovatus Life Sciences Lending Fund I, LP, as Lender and Collateral Agent, or the Lender. Under the Innovatus Loan Agreement, the Lender has agreed to make certain term loans in the aggregate principal amount of up to $17,000,000. Funding of the first $14,000,000 tranche occurred on May 10, 2017. The Company will be eligible to draw on the second tranche of $3,000,000 upon achievement of certain milestones. So long as the Company meets each Interest-Only Milestone (as defined below), the Company is entitled to make interest-only payments for up to twenty-four (24) months. At the end of the interest-only period, the Company will be required to repay the term loans over a two-year period, based on a twenty-four (24) month amortization schedule, with a final maturity date occurring on the fourth anniversary of the initial funding date. However, the interest-only period will end if the Company fails to meet any Interest-Only Milestone. Commencing on the first day of the month following such failure to achieve an Interest-Only Milestone, the Company will be required to repay the term loans over a two year period, based on a twenty-four (24) month amortization schedule. The Interest-Only Milestones require the Company to (i) achieve certain twelve month revenue targets, measured quarterly, commencing with quarter ending March 31, 2018, (ii) meet a minimum capital raising threshold through the sale and issuance of equity securities during the period from April 10, 2017 through May 31, 2018 and (iii) obtain clearance for commercialization of the Senhance System by the U.S. Food and Drug Administration, or the Senhance Clearance, by May 30, 2018. Each such milestone is referred to as an Interest-Only Milestone. In connection with its entrance into the Innovatus Loan Agreement, the Company repaid its existing credit facility with Silicon Valley Bank and Oxford Finance LLC under the SVB Loan Agreement.
The term loans bear interest at a fixed rate equal to 11% per annum, of which 2.5% can be paid in-kind and added to the outstanding principal amount of the term loans until the earlier of (i) the first anniversary following the funding date and (ii) the Company’s failure to achieve an Interest-Only Milestone. The Company will be required to repay the term loans if they are accelerated following an event of default. In addition, the Company is permitted to prepay the term loans in full at any time upon five (5) business days’ written notice to the Lender. Upon the earliest to occur of the maturity date, acceleration of the term loan, or prepayment of the term loan, the Company is required to make a final payment equal to the total term loan commitment multiplied by four percent (4%); provided, however, that in the event the Company refinances its obligations with the Lender after Senhance Clearance, no final fee or prepayment fee described below will be due; and provided, further, that if the Company elects to refinance its obligations prior to the funding of the second tranche, the final fee with respect to the second tranche shall be paid in full on the date of such refinancing. Any prepayment of the term loans in full, whether mandatory or voluntary, must include (i) the final fee, (ii) interest at the default rate (which is the rate otherwise applicable plus five percent (5%)) with respect to any amounts past due, (iii) the Lender’s expenses and all other obligations that are due and payable to the Lender and (iv) a prepayment fee of three percent (3%) if the term loan is paid in full on or before the first anniversary of the effective date, two percent (2%) if paid off after the first anniversary but on or before the second anniversary of the effective date and one percent (1%) if paid off after the second anniversary but on or before the third anniversary of the effective date.
In connection with the funding, the Company paid a facility fee of $170,000 on the date of funding of the first tranche. In addition, the Company issued warrants to the Lender to purchase shares of the Company’s common stock. Additional warrants will be issued on the funding date of each subsequent tranche and will expire five (5) years from such issue date. The warrants issued in connection with funding of the first tranche entitle the Lender to purchase up to 1,244,746 shares of the Company’s common stock at an exercise price of $1.00 per share.
The Company’s obligations under the Innovatus Loan Agreement are secured by a security interest in all of the assets of the Company and its current and future domestic and material foreign subsidiaries, including a security interest in the intellectual property. The Innovatus Loan Agreement contains customary representations and covenants that, subject to exceptions, restrict the Company’s ability to do the following things: declare dividends or redeem or repurchase equity interests; incur additional liens; make loans and investments; incur additional indebtedness; engage in mergers, acquisitions, and asset sales; transact with affiliates; undergo a change in control; add or change business locations; and engage in businesses that are not related to its existing business. Under the terms of the Innovatus Loan Agreement, the Company is required to maintain minimum unrestricted cash in an amount equal to (x) six million dollars ($6,000,000) at all times prior to Senhance Clearance; and (y) at all times thereafter, the lesser of (i) $6,000,000, (ii) the Company’s trailing three (3) months’ cash used to fund operating activities, as determined as of the most recent month end and (iii) the then outstanding principal amount of the term loans, together with accrued but unpaid interest.
27
On April 28, 2017, we entered into an underwriting agreement with Stifel, Nicolaus & Company, Incorporated, or the Underwriter, relating to an underwritten public offering of an aggregate of 24,900,000 Units, each consisting of one share of the Company’s Common Stock, a Series A Warrant to purchase one share of Common Stock and a Series B Warrant to purchase 0.75 shares of Common Stock at an offering price to the public of $1.00 per Unit. Certain of the Company’s officers, directors and existing stockholders purchased approximately $2.5 million of Units in the public offering. The closing of the public offering occurred on May 3, 2017.
Each Series A Warrant had an initial exercise price of $1.00 per share and was able to be exercised at any time beginning on the date of issuance, and from time to time thereafter, through and including the first anniversary of the issuance date, unless terminated earlier as provided in the Series A Warrant. Receipt of 510(k) clearance for the Senhance System on October 13, 2017, triggered the acceleration of the expiration date of the Series A Warrants to October 31, 2017. As of December 31, 2017, all of the Series A Warrants had been exercised.
Each Series B Warrant has an initial exercise price of $1.00 per share and may be exercised at any time beginning on the date of issuance and from time to time thereafter through and including the fifth anniversary of the issuance date, or by May 3, 2022. As of December 31, 2017, Series B Warrants representing approximately 8.9 million shares had been exercised.
The exercise prices and the number of shares issuable upon exercise of the outstanding Series B Warrants are subject to adjustment upon the occurrence of certain events, including, but not limited to, stock splits or dividends, business combinations, sale of assets, similar recapitalization transactions, or other similar transactions. The Series B Warrants are subject to adjustment in the event that the Company issues or is deemed to issue shares of common stock for less than the then applicable exercise price of the Series B Warrants. The exercisability of the Series B Warrants may be limited if, upon exercise, the holder or any of its affiliates would beneficially own more than 4.99% of our common stock. If, at any time Series B Warrants are outstanding, any fundamental transaction occurs, as described in the Series B Warrants and generally including any consolidation or merger into another corporation, the consummation of a transaction whereby another entity acquires more than 50% of the Company’s outstanding voting stock, or the sale of all or substantially all of its assets, the successor entity must assume in writing all of the obligations to the Series B Warrant holders. Additionally, in the event of a fundamental transaction, each Series B Warrant holder will have the right to require the Company, or its successor, to repurchase the Series B Warrants for an amount of cash equal to the Black-Scholes value of the remaining unexercised portion of such Series B Warrants.
The underwriting agreement contains customary representations, warranties and agreements by the Company, customary conditions to closing, indemnification obligations of the Company and the Underwriters, including for liabilities under the Securities Act of 1933, as amended, other obligations of the parties and termination provisions. The representations, warranties and covenants contained in the underwriting agreement were made only for purposes of such agreement and as of specific dates, were solely for the benefit of the parties to such agreement, and may be subject to limitations agreed upon by the contracting parties.
The net proceeds to the Company from the offering were approximately $23.2 million, prior to any exercise of the Series A Warrants or Series B Warrants, after deducting underwriting discounts and commissions and estimated offering expenses paid by the Company. The net proceeds to the Company from the exercise of all of the Series A Warrants and the Series B Warrants exercised prior to December 31, 2017 were approximately $33.6 million.
The Units were issued pursuant to a prospectus supplement dated April 28, 2017 and an accompanying base prospectus dated June 22, 2016 that form a part of the registration statement on Form S-3 that the Company filed with the SEC on November 7, 2014 and was declared effective on December 19, 2014 (File No. 333-199998), and post-effectively amended pursuant to Post-Effective Amendment No. 1 on Form S-3, as filed with the SEC on March 8, 2016 and declared effective on June 22, 2016 and a related registration statement filed pursuant to Rule 462(b) promulgated under the Securities Act of 1933.
On December 15, 2017, we filed a registration statement on Form S-3 (File No. 333-222103) to register shares of common stock underlying outstanding Series B Warrants previously issued as part of the Company’s May 3, 2017 public offering. The new registration statement replaced the registration statement on Form S-3 that expired on December 19, 2017 with respect to these securities. On January 26, 2018, we filed an Amendment No. 1 to such registration statement on Form S-3 to update the information, in the registration statement. The registration statement covers up to 9,579,884 shares of common stock underlying the outstanding Series B Warrants. This registration statement on Form S-3 was declared effective on January 29, 2018.
28
Lincoln Park Purchase Agreement
On December 16, 2016, we entered into a purchase agreement, or the LPC Purchase Agreement, with Lincoln Park Capital Fund, LLC, an Illinois limited liability company, or Lincoln Park, pursuant to which we had the right to sell to Lincoln Park up to an aggregate of $25,000,000 in shares of our common stock, subject to certain limitations and conditions set forth in the LPC Purchase Agreement. Effective April 27, 2017, we terminated the LPC Purchase Agreement. The LPC Purchase Agreement provided us with an election to terminate the Purchase Agreement for any reason or for no reason by delivering a notice to Lincoln Park, and we did not incur any early termination penalties in connection with the termination of the LPC Purchase Agreement. Prior to termination, we sold shares of our common stock to Lincoln Park under the LPC Purchase Agreement for gross proceeds of approximately $5.7 million.
At-the-Market Offerings
On February 9, 2016, we entered into a Controlled Equity Offering SM Sales Agreement, or the “2016 Sales Agreement, with Cantor Fitzgerald & Co., or Cantor, under which we could offer and sell, through Cantor, up to approximately $43.6 million in shares of common stock in an at-the market offering, or the 2016 ATM Offering. The 2016 Sales Agreement was terminated, effective September 10, 2017. On February 20, 2015, we had entered into a Controlled Equity Offering SM Sales Agreement, or the 2015 Sales Agreement, with Cantor, as sales agent, pursuant to which we offered and sold, through Cantor, $25.0 million in shares of common stock in an at-the-market offering from February 2015 through February 2016, or the 2015 ATM Offering. All sales of shares under these offerings were made pursuant to an effective shelf registration statement on Form S-3 filed with the SEC. We paid Cantor a commission of approximately 3% of the aggregate gross proceeds received from all sales of common stock under the 2015 Sales Agreement and the 2016 Sales Agreement.
On August 31, 2017, we entered into an At-the-Market Equity Offering Sales Agreement, or the 2017 Sales Agreement, with Stifel, Nicolaus & Company, Incorporated, or Stifel, under which we could offer and sell, through Stifel, up to approximately $50.0 million in shares of common stock in an at-the-market offering, or the 2017 ATM Offering. All sales of shares were made pursuant to an effective shelf registration statement on Form S-3 filed with the SEC. We paid Stifel a commission of approximately 3% of the aggregate gross proceeds received from all sales of common stock under the 2017 Sales Agreement. As of October 31, 2017, the 2017 ATM Offering was completed.
The following table summarizes the total sales under the 2015 Sales Agreement, 2016 Sales Agreement and 2017 Sales Agreement for the periods indicated (in thousands, except per share amounts):
|
|
|
2017 Sales Agreement |
|
|
2016 Sales Agreement |
|
|
2015 Sales Agreement |
|
|
|||||||
|
|
|
Year Ended December 31, 2017 |
|
|
Year Ended December 31, 2016 |
|
|
Year Ended December 31, 2016 |
|
|
Year Ended December 31, 2015 |
|
|
||||
|
Total shares of common stock sold |
|
|
15,998.5 |
|
|
|
8,763.4 |
|
|
|
5,710.2 |
|
|
|
2,014.3 |
|
|
|
Average price per share |
|
$ |
3.13 |
|
|
$ |
4.70 |
|
|
$ |
3.23 |
|
|
$ |
3.25 |
|
|
|
Gross proceeds |
|
$ |
50,000 |
|
|
$ |
41,156 |
|
|
$ |
18,454 |
|
|
$ |
6,546 |
|
|
|
Commissions earned by Stifel or Cantor |
|
$ |
1,500 |
|
|
$ |
1,235 |
|
|
$ |
553 |
|
|
$ |
197 |
|
|
|
Other issuance costs |
|
$ |
97 |
|
|
$ |
185 |
|
|
$ |
— |
|
|
$ |
259 |
|
|
Senhance Acquisition and Related Transactions
Membership Interest Purchase Agreement and Amendment
On September 21, 2015, the Company announced that it had entered into a Membership Interest Purchase Agreement, dated September 18, 2015 with Sofar S.p.A., as the Seller, Vulcanos S.r.l., as the acquired company, and TransEnterix International, Inc., a wholly owned subsidiary of the Company as the Buyer. The closing of the transactions contemplated by the Purchase Agreement occurred on September 21, 2015. The Buyer acquired all of the membership interests of the acquired company from the Seller, and changed the name of the acquired company to TransEnterix Italia S.r.l. On the closing date, pursuant to the Purchase Agreement, the Company completed the strategic acquisition from Sofar S.p.A. of all of the assets, employees and contracts related to the advanced robotic system for minimally invasive laparoscopic surgery now known as the Senhance System, or the Senhance Acquisition.
29
Under the terms of the Purchase Agreement, the consideration consisted of the issuance of 15,543,413 shares of the Company’s common stock, or the Securities Consideration, and approximately $25,000,000 U.S. Dollars and €27,500,000 Euro in cash consideration, or the Cash Consideration. The Securities Consideration was issued in full at closing of the acquisition; the Cash Consideration was or will be paid in four tranches, with US $25,000,000 paid at closing and the remaining Cash Consideration of €27,500,000 to be paid in three additional tranches based on achievement of negotiated milestones. On December 30, 2016, the Company and Sofar entered into an Amendment to the Purchase Agreement to restructure the terms of the second tranche of the Cash Consideration. Under the Amendment, the second tranche was restructured to reduce the contingent cash consideration by €5.0 million in exchange for the issuance of 3,722,685 shares of the Company’s common stock with an aggregate fair market value of €5.0 million, which were issued on January 4, 2017. The price per share was $1.404 and was calculated based on the average of the closing prices of the Company’s common stock on ten consecutive trading days ending one day before the execution of the Amendment.
The issuance of the initial Securities Consideration was effected as a private placement of securities under Section 4(a)(2) of the Securities Act, and Regulation D promulgated thereunder. The issuance of the additional shares in January 2017 was made under an existing shelf registration statement on Form S-3.
As of December 31, 2017, the Company has paid all Cash Consideration due under the second tranche and €1.8 million due under the fourth tranche.
The Purchase Agreement contains customary representations and warranties of the parties and the parties have customary indemnification obligations, which are subject to certain limitations described further in the Purchase Agreement.
Registration Rights and Lock-Up Agreements
In connection with the Senhance Acquisition, we also entered into a Registration Rights Agreement, dated as of September 21, 2015, with the Seller, pursuant to which we agreed to register the Securities Consideration shares for resale following the end of the lock-up periods described below. The resale Registration Statement has been filed and is effective.
In connection with the Senhance Acquisition, Sofar entered into a Lock-Up Agreement with us pursuant to which Sofar agreed, subject to certain exceptions, not to sell, transfer or otherwise convey any of the Securities Consideration over a two-year period following the Closing Date. As of September 21, 2017, all of the Securities Consideration was released from the lock-up restrictions and is eligible to be resold under the effective resale registration statement.
2015 Public Offering
On June 11, 2015, we sold 16,666,667 shares of common stock at a public offering price of $3.00 per share for aggregate gross proceeds of $50.0 million in an underwritten firm commitment public offering. We granted the underwriters an option, exercisable for 30 days, to purchase up to an additional 2,500,000 shares of common stock to cover over-allotments. The common stock was offered and sold pursuant to our shelf registration statement on Form S-3 (File No. 333-199998) registering an aggregate of $100.0 million of our designated securities. The closing of the public offering occurred on June 17, 2015. On July 10, 2015, the underwriters exercised a portion of their over-allotment option to acquire an additional 2,075,000 shares at the public offering price of $3.00 per share for aggregate additional gross proceeds of $6.2 million. The purchase of the over-allotment shares closed on July 15, 2015. Total proceeds were $52.2 million, net of issuance costs of $4.0 million.
Results of Operations
Our results of operations include the operations of TransEnterix Italia from the Senhance Acquisition date of September 21, 2015 forward.
Revenue
In 2017, our revenue consisted of product and service revenue resulting from a total of four Senhance Systems: Europe (two), Asia (one) and the United States (one), and related instruments, accessories and services. In 2016, our revenue consisted of product and service revenue resulting from the sale in Europe of a Senhance System, instruments and accessories, and related services. We recognize revenue when persuasive evidence that an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable, and collectability is reasonably assured. Amounts billed in excess of the associated revenue recognized are deferred.
30
We expect to experience some unevenness in the number and trend, and average selling price, of units sold on a quarterly basis given the early stage of commercialization of our products.
Product and service revenue for the year ended December 31, 2017 increased to $7.1 million compared to $1.5 million for the year ended December 31, 2016. The $5.6 million increase was the result of the revenue recognized on the sale of four Senhance Systems, net of deferred revenue.
Product and service revenue for the year ended December 31, 2016 increased to $1.5 million compared to $0 for the year ended December 31, 2015. The $1.5 million increase was the result of the revenue recognized on the sale of one Senhance System, net of deferred revenue, during the third quarter of 2016.
Cost of Revenue
Cost of revenues consists primarily of costs related to contract manufacturing, materials, and manufacturing overhead. We expense all inventory provisions as cost of revenues. The manufacturing overhead costs include the cost of quality assurance, material procurement, inventory control, facilities, equipment depreciation and operations supervision and management. We expect overhead costs as a percentage of revenues to become less significant as our production volume increases. We expect cost of revenues to increase in absolute dollars to the extent our revenues grow and as we continue to invest in our operational infrastructure to support anticipated growth.
Cost of revenue for the year ended December 31, 2017 increased to $6.7 million as compared to $1.1 million for the year ended December 31, 2016. This increase over the prior year period was the result of increased sales and costs for manufacturing overhead and field service.
Cost of revenue for the year ended December 31, 2016 increased to $1.1 million as compared to $0 for the year ended December 31, 2015. This increase over the prior year period was the result of the costs recognized in connection with the sale of one Senhance System during the third quarter of 2016. The $1.1 million cost of revenue primarily represents the fair value of the Senhance System determined using the acquisition method of accounting at the Senhance Acquisition date.
Research and Development
Research and development, or R&D expenses primarily consist of engineering, product development and regulatory expenses incurred in the design, development, testing and enhancement of our products and legal services associated with our efforts to obtain and maintain broad protection for the intellectual property related to our products. In future periods, we expect R&D expenses to remain consistent or be modestly lower as we continue to transition our investments into commercial activities. R&D expenses are expensed as incurred.
R&D expenses for the year ended December 31, 2017 decreased 25% to $22.0 million as compared to $29.3 million for the year ended December 31, 2016. The $7.3 million decrease resulted primarily from decreased personnel costs of $4.0 million, decreased supplies expense of $1.9 million, decreased contract engineering services, consulting and other outside services of $2.0 million and decreased other costs of $0.1 million, offset by increased stock compensation costs of $0.7 million
R&D expenses for the year ended December 31, 2016 decreased 1% to $29.3 million as compared to $29.7 million for the year ended December 31, 2015. The $0.4 million decrease resulted primarily from decreased supplies expense of $3.7 million and decreased contract engineering services, consulting and other outside services of $3.6 million, offset by increased preclinical lab expense of $3.8 million, increased facility costs of $0.9 million, increased stock compensation costs of $0.8 million, increased travel related expenses of $0.5 million, increased other costs of $0.6 million and increased personnel related costs of $0.3 million.
Sales and Marketing
Sales and marketing expenses include costs for sales and marketing personnel, travel, demonstration product, market development, physician training, tradeshows, marketing clinical studies and consulting expenses. We expect sales and marketing expenses to increase significantly in 2018 in support of our Senhance System commercialization.
Sales and marketing expenses for the year ended December 31, 2017 increased 90% to $17.5 million compared to $9.2 million for the year ended December 31, 2016. The $8.3 million increase was primarily related to increased personnel related costs of $3.0 million, increased consulting and outside service costs of $1.8 million, increased depreciation expense $1.0 million, increased travel related expenses of $0.8 million, increased stock compensation costs of $0.5 million, increased demonstration product costs of $0.5 million, increased tradeshow costs of $0.4 million and increased other costs of $0.3 million, as we increased our U.S. sales and marketing team following receipt of 510(k) clearance for the Senhance System.
31
Sales and marketing expenses for the year ended December 31, 2016 increased 217% to $9.2 million compared to $2.9 million for the year ended December 31, 2015. The $6.3 million increase was primarily related to increased personnel related costs of $2.6 million, increased travel related expenses of $1.0 million, increased consulting costs of $0.9 million, increased tradeshow costs of $0.5 million, increased stock compensation costs of $0.5 million, increased other costs of $0.6 million and increased depreciation expense of $0.2 million.
General and Administrative
General and administrative expenses consist of personnel costs related to the executive, finance and human resource functions, as well as professional service fees, legal fees, accounting fees, insurance costs, and general corporate expenses. In future periods, we expect general and administrative expenses to increase to support our sales, marketing, and research and development efforts.
General and administrative expenses for the year ended December 31, 2017 increased 14% to $12.3 million compared to $10.8 million for the year ended December 31, 2016. The $1.5 million increase was primarily due to increased stock compensation costs of $0.7 million, increased personnel costs of $0.5 million and increased other costs of $1.0 million offset by decreased legal, accounting, and investor relation fees and other public company costs of $0.7 million.
General and administrative expenses for the year ended December 31, 2016 increased 38% to $10.8 million compared to $7.8 million for the year ended December 31, 2015. The $3.0 million increase was primarily due to increased legal, accounting, and investor relation fees and other public company costs of $1.8 million, increased stock compensation costs of $0.5 million, increased personnel costs of $0.4 million, and increased other costs of $0.3 million.
Amortization of Intangible Assets
Amortization of intangible assets for the year ended December 31, 2017 increased to $7.9 million compared to $7.0 million for the year ended December 31, 2016. The $0.9 million increase was primarily the result of amortization of developed technology related to the acquisition of the Senhance System on September 21, 2015 and the amortization of in-process research and development transferred to intellectual property in October 2017.
Amortization of intangible assets for the year ended December 31, 2016 increased to $7.0 million compared to $2.2 million for the year ended December 31, 2015. The $4.8 million increase was primarily the result of amortization of developed technology related to the acquisition of the Senhance System on September 21, 2015.
Change in Fair Value of Contingent Consideration
The change in fair value of contingent consideration in connection with the Senhance Acquisition was $2.0 million for the year ended December 31, 2017 primarily related to the change in expected timelines for the achievement of milestones, the effect of the passage of time on the fair value measurement and the impact of foreign currency exchange rates.
The change in fair value of contingent consideration in connection with the Senhance Acquisition was $0.5 million for the year ended December 31, 2016 primarily related to the Amendment to the Purchase Agreement for the second tranche, the change in expected timelines for the achievement of milestones, the effect of the passage of time on the fair value measurement and the impact of foreign currency exchange rates.
Issuance costs for Warrants
Issuance costs of $0.6 million were allocated to the Series A Warrants and Series B Warrants issued in April 2017.
Inventory write-down related to restructuring
On April 19, 2016, the FDA notified the Company that the SurgiBot System did not meet the criteria for substantial equivalence based on the data submitted in the 510(k) submission. As a result, we reprioritized our near-term regulatory efforts to the 510(k) submission for the Senhance System. Consequently, in May 2016, the Company implemented a restructuring plan. Under this plan, we recorded a $2.6 million write-down of inventory related to the SurgiBot System.
32
Restructuring and other charges
Under the restructuring plan executed in May 2016, we recorded $3.1 million in restructuring and other charges. The restructuring charges included: (i) $0.5 million to be paid in cash, of which $0.4 million related to employee severance costs and $0.1 million related to cancellation of certain contracts; and (ii) $2.6 million for other non-cash charges, of which $1.0 million related to the write-off of long-lived assets for the abandonment of certain equipment and tooling and $1.6 million related to the write-off of intellectual property for certain patents.
Goodwill impairment
The Company performs an annual impairment test of goodwill at December 31, or more frequently if events or changes in circumstances indicate that the carrying value of our one reporting unit may not be recoverable. During the second quarter of 2016, we were notified by the FDA that the SurgiBot System did not meet the criteria for substantial equivalency, negatively impacting our market capitalization, and warranting an interim two-step quantitative impairment test. Based on the impairment test, we recorded goodwill impairment of $61.8 million during the second quarter of 2016. No charge for goodwill impairment was required as of December 31, 2017.
Acquisition Related Costs
Acquisition related costs consist primarily of legal, accounting and other professional fees related to the Senhance Acquisition. We incurred $4.2 million of acquisition-related expenses for the year ended December 31, 2015.
Change in Fair Value of Warrant Liabilities
The change in fair value of Series A Warrants and Series B Warrants issued in April 2017 was $83.7 million for the year ended December 31, 2017. The increase was primarily the result of our increased stock price at the time the warrants were exercised and increased stock price for the remaining outstanding warrants at December 31, 2017.
Interest Expense, Net
Interest expense for the year ended December 31, 2017 increased to $2.1 million compared to $1.9 million for the year ended December 31, 2016. The $0.2 million increase was primarily related to the increase in interest rate on refinanced notes payable.
Interest expense for the year ended December 31, 2016 increased to $1.9 million compared to $1.6 million for the year ended December 31, 2015. The $0.3 million increase was primarily related to the increase in notes payable of approximately $10.0 million in August 2015.
Income Tax Benefit
Income tax benefit consists primarily of taxes related to the amortization of purchase accounting intangibles in connection with the Italian taxing jurisdiction for TransEnterix Italia as a result of the acquisition of the Senhance System. We recognized $3.3 million, $5.5 million and $1.0 million of income tax benefit for the year ended December 31, 2017, 2016 and 2015, respectively.
Liquidity and Capital Resources
Sources of Liquidity
Since our inception we have incurred significant losses and, as of December 31, 2017, we had an accumulated deficit of $447.6 million. We have not yet achieved profitability and we cannot assure investors that we will achieve profitability with our existing capital resources. As of December 31, 2017, the Company's cash and restricted cash balance was approximately $97.6 million. We believe that our existing cash and cash equivalents, together with cash received from sales of our products, will be sufficient to fund operations through at least the next 12 months. We expect to continue to fund sales and marketing, research and development and general and administrative expenses at similar to current or higher levels and, as a result, we will need to generate significant revenues to achieve profitability. Our principal sources of cash to date have been proceeds from public offerings of common stock, private placements of common and preferred stock, incurrence of debt and the sale of equity securities held as investments.
33
We currently have one effective shelf registration statement on file with the SEC, which registers up to $150.0 million of debt securities, common stock, preferred stock, or warrants, or any combination thereof for future financing transactions. The shelf registration statement was declared effective by the SEC on May 19, 2017. We have raised $50.0 million in gross proceeds and approximately $48.5 in net proceeds under such shelf registration statement through the sale of all the shares available under the 2017 ATM Offering. As of December 31, 2017, we had $100.0 million available for future financings under such shelf registration statement.
At December 31, 2017, we had cash and cash equivalents, excluding restricted cash, of approximately $91.2 million.
Consolidated Cash Flow Data
|
|
|
Years Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
(in millions) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net cash (used in) provided by |
|
|
|
|
|
|
|
|
|
|
|
|
|
Operating activities |
|
$ |
(39.8 |
) |
|
$ |
(52.4 |
) |
|
$ |
(38.8 |
) |
|
Investing activities |
|
|
(2.0 |
) |
|
|
(1.4 |
) |
|
|
(26.2 |
) |
|
Financing activities |
|
|
104.4 |
|
|
|
49.9 |
|
|
|
68.4 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
0.4 |
|
|
|
— |
|
|
|
— |
|
|
Net increase (decrease) in cash, cash equivalents and restricted cash |
|
$ |
63.0 |
|
|
$ |
(3.9 |
) |
|
$ |
3.4 |
|
Operating Activities
For the year ended December 31, 2017, cash used in operating activities of $39.8 million consisted of net loss of $144.8 million offset by cash provided by working capital of $3.4 million and non-cash items of $101.6 million. The non-cash items primarily consisted of $83.7 million change in fair value of warrant liabilities, $7.1 million of stock-based compensation expense, $2.5 million of depreciation, $7.9 million of amortization, and $2.0 million change in fair value of contingent consideration, offset by $3.3 million deferred income tax benefit. The increase in cash from changes in working capital included $2.1 million increase in accrued expenses, $7.5 million increase for cash received for the sale of the SurgiBot assets and $1.1 million increase for deferred revenue, offset by $3.0 million increase in inventories, $0.5 million decrease in accounts payable, $3.3 million increase in other current and long term assets and $0.4 million increase in accounts receivable.
For the year ended December 31, 2016, cash used in operating activities of $52.4 million consisted of net loss of $120.0 million and cash used for working capital of $8.5 million, offset by non-cash items of $76.1 million. The non-cash items primarily consisted of $61.8 million goodwill impairment, $2.6 million inventory write-down related to restructuring, $2.6 million non-cash restructuring and other charges, $5.0 million of stock-based compensation expense, $1.9 million of depreciation, $7.1 million of amortization, and $0.5 million change in fair value of contingent consideration, offset by $5.6 million deferred income tax benefit. The decrease in cash from changes in working capital included $6.6 million increase in inventories, $0.4 million decrease in accounts payable, $1.5 million increase in other current and long term assets and $1.0 million increase in accounts receivable, offset by $1.1 million increase in accrued expenses.
For the year ended December 31, 2015, cash used in operating activities of $38.8 million consisted of net loss of $46.9 million, offset by non-cash items of $5.5 million and cash provided by working capital of $2.6 million. The non-cash items primarily consisted of $3.3 million of stock-based compensation expense, $1.3 million of depreciation, and $2.3 million of amortization, offset by $1.0 million deferred income tax benefit and $0.4 million change in fair value of contingent consideration. The increase in cash from changes in working capital included $1.9 million increase in inventories and $2.0 million increase in other current and long term assets. These amounts were partially offset by $1.1 million increase in accounts payable, $5.4 million increase in accrued expenses and $0.1 million decrease in accounts receivable.
Investing Activities
For the year ended December 31, 2017, net cash used in investing activities was $2.0 million. This amount reflected the purchases of property and equipment and intellectual property.
For the year ended December 31, 2016, net cash used in investing activities was $1.4 million. This amount reflected the purchases of property and equipment.
34
For the year ended December 31, 2015, net cash used in investing activities was $26.2 million. This amount reflected the $25.0 million payment for the Senhance Acquisition and $1.2 million paid for the purchases of property and equipment.
Financing Activities
For the year ended December 31, 2017, net cash provided by financing activities was $104.4 million. This amount was primarily related to $77.6 million in proceeds from the issuance of common stock and warrants, net of issuance costs, $13.0 million in proceeds from the issuance of debt and $34.5 million proceeds from the exercise of warrants, partially offset by $13.3 million in payments of debt and $7.2 million in payments of contingent consideration.
For the year ended December 31, 2016, net cash provided by financing activities was $49.9 million. This amount was primarily related to $58.0 million in proceeds from the issuance of common stock, net of issuance costs, partially offset by $6.9 million in payments of debt and $1.2 million in payments of contingent consideration.
For the year ended December 31, 2015, net cash provided by financing activities was $68.4 million. This amount was primarily related $58.3 million in proceeds from the issuance of common stock, net of issuance costs, and $9.9 million proceeds from the issuance of debt, and $0.2 million proceeds from the issuance of stock options and warrants.
Operating Capital and Capital Expenditure Requirements
We believe that our existing cash and cash equivalents, together with cash received from sales of our products, will be sufficient to meet our anticipated cash needs through at least the next 12 months. We intend to spend substantial amounts on commercial activities, on research and development activities, including product development, regulatory and compliance, clinical studies in support of our future product offerings, the enhancement and protection of our intellectual property, on notes payable payments as they come due, and on contingent consideration payments in connection with the acquisition of the Senhance System. We will need to obtain additional financing to pursue our business strategy, to respond to new competitive pressures or to take advantage of opportunities that may arise. To meet our capital needs, we are considering multiple alternatives, including, but not limited to, additional equity financings, debt financings, strategic collaborations and other funding transactions. There can be no assurance that we will be able to complete any such transaction on acceptable terms or otherwise. If we are unable to obtain the necessary capital, we will need to pursue a plan to license or sell our assets, seek to be acquired by another entity, cease operations and/or seek bankruptcy protection.
Cash and cash equivalents held by our foreign subsidiaries totaled $1.8 million at December 31, 2017, including restricted cash. We do not intend or currently foresee a need to repatriate cash and cash equivalents held by our foreign subsidiary. If these funds are needed in the U.S., we believe that the potential U.S. tax impact to repatriate these funds would be immaterial.
Innovatus Loan Agreement
On May 10, 2017, the Company and its domestic subsidiaries, as co-borrowers, entered into the Innovatus Loan Agreement with Innovatus Life Sciences Lending Fund I, LP, as Lender and Collateral Agent. Please see the description of the Innovatus Loan Agreement above in this “Management’s Discussion and Analysis of Financial Condition and Results of Operations‑Debt Refinancing.
SVB Loan Agreement
In connection with its entrance into the Innovatus Loan Agreement on May 10, 2017, the Company repaid its existing credit facility with Silicon Valley Bank and Oxford Finance LLC, or the Prior Lenders, under the SVB Loan Agreement, , initially entered into in January 2012, as subsequently amended or amended and restated, or, collectively, the SVB Loan Agreement. A number of the amendments related to the Senhance Acquisition or the growth of our business in non-U.S. jurisdictions. Under the SVB Loan Agreement, our current borrowing capacity was $20.0 million, all of which was borrowed under term loans. We had periods of interest-only payments during the SVB Loan Agreement, and had been making principal payments since January 2016. The maturity date of the term loans was July 1, 2018.
In connection with the entry into the Innovatus Loan Agreement, we were obligated to pay final payment and facility fees under the SVB Loan Agreement. The final payment fee obligation was $1.3 million.
35
Contractual Obligations and Commercial Commitments
The following table summarizes our contractual obligations as of December 31, 2017 (in millions):
|
|
|
Payments due by period |
|
|
|
|
|
|||||||||||||
|
|
|
Total |
|
|
Less than 1 year |
|
|
1 to 3 years |
|
|
3 to 5 years |
|
|
Thereafter |
|
|||||
|
Long-term debt obligations (1) |
|
$ |
16.5 |
|
|
$ |
5.5 |
|
|
$ |
11.0 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Operating leases |
|
$ |
2.2 |
|
|
$ |
0.9 |
|
|
$ |
1.0 |
|
|
$ |
0.3 |
|
|
$ |
— |
|
|
License and supply agreements |
|
$ |
9.6 |
|
|
$ |
3.6 |
|
|
$ |
1.8 |
|
|
$ |
1.2 |
|
|
$ |
3.0 |
|
|
Total contractual obligations (2) |
|
$ |
28.3 |
|
|
$ |
10.0 |
|
|
$ |
13.8 |
|
|
$ |
1.5 |
|
|
$ |
3.0 |
|
|
(1) |
Long-term debt obligations include principal and interest payments on our notes payable. |
|
(2) |
As of December 31, 2017, the contingent consideration that may be paid under the Purchase Agreement with Sofar upon the achievement of milestones is approximately €15.7 million. Due to uncertainty regarding the timing and amount of future payments related to these liabilities, these amounts are excluded from the contractual obligations table above. |
Long-term debt obligations include future payments under the Innovatus Loan Agreement.
Operating lease amounts include future minimum lease payments under all our non-cancelable operating leases with an initial term in excess of one year. We rent office space in North Carolina under an operating lease which expires in 2018, with options to extend the lease through 2021. We also rent space for a warehouse facility in North Carolina which expired in January 2018. In Italy, we rent space for research and development and demonstration facilities under an operating lease which expires in 2022. This table does not include obligations for any lease extensions.
License, supply and third party vendor agreements include agreements assumed as part of the Senhance Acquisition and other third party vendor agreements.
Off-Balance Sheet Arrangements
As of December 31, 2017, we did not have any off-balance sheet arrangements.
Critical Accounting Policies and Estimates
The discussion and analysis of our financial condition and results of operations set forth above under the headings “Results of Operations” and “Liquidity and Capital Resources” have been prepared in accordance with U.S. GAAP and should be read in conjunction with our financial statements and notes thereto appearing in Item 8 of this Annual Report. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, we evaluate our critical accounting policies and estimates, including identifiable intangible assets and goodwill, in-process research and development, contingent consideration, warrant liabilities, stock-based compensation, inventory, and revenue recognition. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. A more detailed discussion on the application of these and other accounting policies can be found in Note 2 in the Notes to the Financial Statements set forth in our financial statements for the years ended December 31, 2017, 2016, and 2015 which are included as Item 8 of this Annual Report. Actual results may differ from these estimates under different assumptions and conditions.
While all accounting policies impact the financial statements, certain policies may be viewed as critical. Critical accounting policies are those that are both most important to the portrayal of financial condition and results of operations and that require management’s most subjective or complex judgments and estimates. Our management believes the policies that fall within this category are the policies on accounting for identifiable intangible assets and goodwill, in-process research and development, contingent consideration, warrants liabilities, stock-based compensation, inventory and revenue recognition.
Identifiable Intangible Assets and Goodwill
Identifiable intangible assets consist of purchased patent rights recorded at cost and developed technology acquired as part of a business acquisition recorded at estimated fair value. Intangible assets are amortized over 5 to 10 years. We periodically evaluate identifiable intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
36
Indefinite-lived intangible assets, such as goodwill, are not amortized. We test the carrying amounts of goodwill for recoverability on an annual basis or when events or changes in circumstances indicate evidence of potential impairment exists by performing either a qualitative evaluation or a two-step quantitative test. The qualitative evaluation is an assessment of factors, including industry, market and general economic conditions, market value, and future projections to determine whether it is more likely than not that the fair value of a reporting unit is less than it’s carrying amount, including goodwill. Prior to the adoption of Accounting Standards Update (“ASU”) 2017-14, Simplifying the Test for Goodwill Impairment (“ASU 2017-04”), as of the beginning of fiscal year 2017, in certain instances, we elected to bypass this qualitative assessment and perform a two-step quantitative test. The quantitative goodwill impairment test was performed using a two-step approach. In the first step, the fair value of the reporting unit was determined and compared to the reporting unit's carrying value, including goodwill. If the fair value of the reporting unit was less than its carrying value, the second step of the goodwill impairment test was performed to measure the amount of impairment, if any. In the second step, the fair value of the reporting unit was allocated to the assets and liabilities of the reporting unit as if it had been acquired in a business combination and the purchase price was equivalent to the fair value of the reporting unit. The excess of the fair value of the reporting unit over the amounts assigned to its assets and liabilities was referred to as the implied fair value of goodwill. The implied fair value of the reporting unit's goodwill was then compared to the actual carrying value of goodwill. If the implied fair value of goodwill was less than the carrying value of goodwill, an impairment loss was recognized for the difference. ASU 2017-04 removes Step 2 of the goodwill impairment test.
During the second quarter of 2016, the FDA notified the Company that the SurgiBot System did not meet the criteria for substantial equivalency, negatively impacting the Company’s market capitalization, and warranting an interim two-step quantitative impairment test. We determined the fair value of our reporting unit using a discounted cash flow analysis derived from our long-term plans. The fair value of the reporting unit was corroborated using market prices for TransEnterix, Inc. The inputs used to determine the fair values were classified as Level 3 in the fair value hierarchy. Based on the impairment test, we recorded goodwill impairment of $61.8 million during the second quarter of 2016. We performed a qualitative assessment during the annual impairment review for fiscal 2016 as of December 31, 2016 and concluded that it is not more likely than not that the fair value of our single reporting unit was less than its carrying amount. Therefore, the two-step goodwill impairment test for the reporting unit was not necessary as of December 31, 2016.
During the second quarter of 2017, our stock price experienced a significant decline and as of June 30, 2017 we performed a Step 1 goodwill impairment test as of the second quarter. Our analysis included utilizing our market capitalization with a control premium. To determine the appropriate control premium, we considered recent merger and acquisition transaction activity of comparable public healthcare equipment companies. Based on this analysis, we determined a control premium range of approximately 19% to 46%, and selected the mid-range of approximately 32.5%. After applying a 32.5% control premium, our market value exceeded our carrying value by 13%. Based on this analysis, we determined that no charge to goodwill for impairment was required during the second quarter of 2017. As of December 31, 2017, we elected to bypass the qualitative assessment and calculated the fair value of our reporting unit, which exceeded the carrying amount. Accordingly, no charge for goodwill impairment was required as of December 31, 2017.
A significant amount of judgment is involved in determining if an indicator of goodwill impairment has occurred. Such indicators may include, among others: a significant decline in expected future cash flows; a sustained, significant decline in the Company's stock price and market capitalization; a significant adverse change in legal factors or in the business climate; adverse assessment or action by a regulator; and unanticipated competition. Key assumptions used in the annual goodwill impairment test are highly judgmental and include: selection of comparable companies and amount of control premium. Any change in these indicators or key assumptions could have a significant negative impact on the Company's financial condition, impact the goodwill impairment analysis or cause the Company to perform a goodwill impairment analysis more frequently than once per year.
In-Process Research and Development
In-process research and development, or IPR&D, assets are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. IPR&D assets represent the fair value assigned to technologies that we acquire, which at the time of acquisition have not reached technological feasibility and have no alternative future use. During the period that the assets are considered indefinite-lived, they are tested for impairment on an annual basis, or more frequently if we become aware of any events occurring or changes in circumstances that indicate that the fair value of the IPR&D assets are less than their carrying amounts. If and when development is complete, which generally occurs when we have regulatory approval and are able to commercialize products associated with the IPR&D assets, these assets are then deemed definite-lived and are amortized based on their estimated useful lives at that point in time. If development is terminated or abandoned, we may have a full or partial impairment charge related to the IPR&D assets, calculated as the excess of carrying value of the IPR&D assets over fair value.
37
Contingent consideration is recorded as a liability and measured at fair value using a discounted cash flow model utilizing significant unobservable inputs including the probability of achieving each of the potential milestones and an estimated discount rate associated with the risks of the expected cash flows attributable to the various milestones. Significant increases or decreases in any of the probabilities of success or changes in expected timelines for achievement of any of these milestones would result in a significantly higher or lower fair value of these milestones, respectively, and commensurate changes to the associated liability. The fair value of the contingent consideration at each reporting date will be updated by reflecting the changes in fair value in our statements of operations and comprehensive loss.
Warrant Liabilities
For the Series A Warrants and Series B Warrants, the warrants are recorded as liabilities and are revalued at each reporting period. The change in fair value is recognized in the consolidated statements of operations and comprehensive loss. The selection of the appropriate valuation model and the inputs and assumptions that are required to determine the valuation requires significant judgment and requires management to make estimates and assumptions that affect the reported amount of the related liability and reported amounts of the change in fair value. Actual results could differ from those estimates, and changes in these estimates are recorded when known. As the warrant liability is required to be measured at fair value at each reporting date, it is reasonably possible that these estimates and assumptions could change in the near term.
Stock-Based Compensation
We recognize as expense, the grant-date fair value of stock options and other stock based compensation issued to employees and non-employee directors over the requisite service periods, which are typically the vesting periods. We use the Black-Scholes-Merton model to estimate the fair value of our stock-based payments. The volatility assumption used in the Black-Scholes-Merton model is based on the calculated historical volatility based on an analysis of reported data for a peer group of companies as well as the Company’s historical volatility. The expected term of options granted by us has been determined based upon the simplified method, because we do not have sufficient historical information regarding our options to derive the expected term. Under this approach, the expected term is the mid-point between the weighted average of vesting period and the contractual term. The risk-free interest rate is based on U.S. Treasury rates whose term is consistent with the expected life of the stock options. We have not paid and do not anticipate paying cash dividends on our shares of common stock; therefore, the expected dividend yield is assumed to be zero. We estimate forfeitures based on our historical experience and adjust the estimated forfeiture rate based upon actual experience. We adopted ASU 2016-09, Compensation – Stock Compensation (Topic 718) – Improvements to Employee Share-Based Payment Accounting as of the beginning of fiscal year 2017 and did not elect to account for forfeitures when they occur, but will continue to estimate the number of awards that are expected to vest. The adoption of this ASU did not have a material impact on the consolidated financial statements.
Inventory
Inventory, which includes material, labor and overhead costs, is stated at the lower of cost, determined on a first-in, first-out basis, or net realizable value. We record reserves, when necessary, to reduce the carrying value of inventory to its net realizable value. At the point of loss recognition, a new, lower-cost basis for that inventory is established, and any subsequent improvements in facts and circumstances do not result in the restoration or increase in that newly established cost basis.
Revenue Recognition
Our revenue consists of product revenue resulting from the sales of systems, instruments and accessories, and service revenue. We recognize revenue when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable, and collectability is reasonably assured. Revenue is presented net of taxes collected from customers that are remitted to government authorities. We generally recognize revenue at the following points in time:
|
|
• |
System sales. For systems sold directly to end customers, revenue is recognized when acceptance occurs, which is deemed to have occurred upon customer acknowledgment of delivery or installation, depending on the terms of the arrangement. The Senhance Systems are delivered with a software component. However, because the software and non-software elements function together to deliver the system’s essential functionality, our arrangements are excluded from being accounted for under software revenue recognition guidance. |
|
|
• |
Instruments and accessories. Revenue from sales of instruments and accessories is generally recognized at the time of shipment. Revenue from services related to the supply and management of instruments and accessories is recognized as the services are rendered. |
|
|
• |
Service. Service revenue is recognized ratably over the term of the service period. Revenue related to services performed on a time-and-materials basis is recognized when it is earned and billable. |
38
Our system sale arrangements contain multiple elements including a system(s), instruments, accessories, and system service. We generally deliver all of the elements, other than service, within days of entering into the system sale arrangement. Each of these elements is a separate unit of accounting. System instruments, accessories and service are also sold on a stand-alone basis.
For multiple-element arrangements, revenue is allocated to each unit of accounting based on their relative selling prices. Relative selling prices are based first on vendor specific objective evidence of fair value (“VSOE”), then on third-party evidence of selling price (“TPE”) when VSOE does not exist, and then on management's best estimate of the selling price (“BESP”) when VSOE and TPE do not exist.
Our system sale arrangements generally include a one-year period of free service, and the right for the customer to purchase service annually thereafter. The revenue allocated to the free service period is deferred and recognized ratably over the free service period. Deferred revenue was primarily comprised of deferred revenue related to service contracts for the periods presented.
Because we have neither VSOE nor TPE for our systems, the allocation of revenue is based on BESP for the systems sold. The objective of BESP is to determine the price at which we would transact a sale, had the product been sold on a stand-alone basis. We determine BESP for our systems by considering multiple factors, including, but not limited to, features and functionality of the system, geographies, type of customer, and market conditions. We regularly review BESP and maintain internal controls over establishing and updating these estimates.
Recent Accounting Pronouncements
See “Note 2. Summary of Significant Accounting Policies” of the Notes to Consolidated Financial Statements in “Item 8. Financial Statements and Supplementary Data” of this Annual Report for a full description of recent accounting pronouncements including the respective expected dates of adoption and effects on our Consolidated Balance Sheets and Consolidated Statements of Operations and Comprehensive Loss.
General
We have limited exposure to market risks from instruments that may impact the Balance Sheets, Statements of Operations and Comprehensive Loss, and Statements of Cash Flows. Such exposure is due primarily to changing interest rates and foreign currency exchange rates.
Interest Rates
The primary objective for our investment activities is to preserve principal while maximizing yields without significantly increasing risk. This is accomplished by investing excess cash in money market funds and Treasury securities. As of December 31, 2017, approximately 100% of the investment portfolio was in cash equivalents with very short term maturities and therefore not subject to any significant interest rate fluctuations.
Foreign Currency Exchange Rate Risk
We conduct operations in several different countries, including the U.S. and throughout Europe, and portions of our revenues, expenses, assets and liabilities are denominated in U.S. dollars, Euros or other currencies. Since our consolidated financial statements are presented in U.S. dollars, we must translate revenues, income and expenses, as well as assets and liabilities, into U.S. dollars at exchange rates in effect during or at the end of each reporting period. We have not historically hedged our exposure to foreign currency fluctuations. Accordingly, increases or decreases in the value of the U.S. dollar against the Euro and other currencies could materially affect our net operating revenues, operating income and the value of balance sheet items denominated in foreign currencies.
During the year ended December 31, 2017, 82% of our revenue and approximately 39% of our expenses were denominated in currencies other than the U.S. dollar, most notably the Euro. Based on actual results over the past year, a hypothetical 10% increase or decrease in the U.S. dollar against the Euro would have increased or decreased revenue by approximately $0.6 million and operating expenses by approximately $2.4 million.
39
|
|
|
Page |
|
|
41 |
|
|
|
|
|
|
Consolidated Financial Statements : |
|
|
|
Consolidated Balance Sheets as of December 31, 2017 and 2016 |
|
43 |
|
|
44 |
|
|
|
45 |
|
|
|
46 |
|
|
|
47 |
40
Report of Independent Registered Public Accounting Firm
Stockholders and Board of Directors
TransEnterix, Inc.
Morrisville, North Carolina
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of TransEnterix, Inc. (the “Company”) and subsidiaries as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company and subsidiaries at December 31, 2017 and 2016, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and our report dated March 8, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ BDO USA, LLP
We have served as the Company's auditor since 2013.
Raleigh, North Carolina
March 8, 2018
41
Report of Independent Registered Public Accounting Firm
Stockholders and Board of Directors
TransEnterix, Inc.
Morrisville, North Carolina
Opinion on Internal Control over Financial Reporting
We have audited TransEnterix, Inc.’s (the “Company’s”) internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated balance sheets of the Company and subsidiaries as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, stockholders’ equity, and cash flows for each of the three years in the period ended December 31, 2017, and the related notes and our report dated March 8, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Item 9A, “Management’s Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ BDO USA, LLP
Raleigh, North Carolina
March 8, 2018
42
(in thousands, except share amounts)
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current Assets |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
91,217 |
|
|
$ |
24,165 |
|
|
Accounts receivable, net |
|
|
1,536 |
|
|
|
621 |
|
|
Inventories |
|
|
10,817 |
|
|
|
7,883 |
|
|
Interest receivable |
|
|
80 |
|
|
|
12 |
|
|
Other current assets |
|
|
9,344 |
|
|
|
5,335 |
|
|
Total Current Assets |
|
|
112,994 |
|
|
|
38,016 |
|
|
Restricted cash |
|
|
6,389 |
|
|
|
10,425 |
|
|
Accounts receivable, net of current portion |
|
|
— |
|
|
|
266 |
|
|
Property and equipment, net |
|
|
6,670 |
|
|
|
5,772 |
|
|
Intellectual property, net |
|
|
52,638 |
|
|
|
37,090 |
|
|
In-process research and development |
|
|
— |
|
|
|
15,920 |
|
|
Goodwill |
|
|
71,368 |
|
|
|
68,697 |
|
|
Other long term assets |
|
|
192 |
|
|
|
63 |
|
|
Total Assets |
|
$ |
250,251 |
|
|
$ |
176,249 |
|
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
|
|
|
|
Current Liabilities |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
3,771 |
|
|
$ |
3,984 |
|
|
Accrued expenses |
|
|
10,974 |
|
|
|
8,206 |
|
|
Deferred revenue |
|
|
1,088 |
|
|
|
— |
|
|
Deferred gain on sale of SurgiBot assets |
|
|
7,500 |
|
|
|
— |
|
|
Contingent consideration – current portion |
|
|
719 |
|
|
|
10,502 |
|
|
Notes payable - current portion, net of debt discount |
|
|
4,788 |
|
|
|
7,997 |
|
|
Total Current Liabilities |
|
|
28,840 |
|
|
|
30,689 |
|
|
Long Term Liabilities |
|
|
|
|
|
|
|
|
|
Contingent consideration – less current portion |
|
|
11,699 |
|
|
|
12,298 |
|
|
Notes payable - less current portion, net of debt discount |
|
|
8,385 |
|
|
|
4,995 |
|
|
Warrant liabilities |
|
|
14,090 |
|
|
|
— |
|
|
Net deferred tax liabilities |
|
|
8,389 |
|
|
|
10,397 |
|
|
Total Liabilities |
|
|
71,403 |
|
|
|
58,379 |
|
|
Commitments and Contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ Equity |
|
|
|
|
|
|
|
|
|
Common stock $0.001 par value, 750,000,000 shares authorized at December 31, 2017 and 2016, respectively; 199,282,003 and 115,781,030 shares issued at December 31, 2017 and 2016, respectively; and 199,282,003 and 115,687,351 shares outstanding at December 31, 2017 and 2016, respectively |
|
|
199 |
|
|
|
115 |
|
|
Additional paid-in capital |
|
|
621,261 |
|
|
|
426,609 |
|
|
Accumulated deficit |
|
|
(447,640 |
) |
|
|
(302,844 |
) |
|
Treasury stock at cost, 0 and 93,679 shares at December 31, 2017 and 2016, respectively |
|
|
— |
|
|
|
(241 |
) |
|
Accumulated other comprehensive income (loss) |
|
|
5,028 |
|
|
|
(5,769 |
) |
|
Total Stockholders’ Equity |
|
|
178,848 |
|
|
|
117,870 |
|
|
Total Liabilities and Stockholders’ Equity |
|
$ |
250,251 |
|
|
$ |
176,249 |
|
See accompanying notes to consolidated financial statements.
43
Consolidated Statements of Operations and Comprehensive Loss
(in thousands except per share amounts)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Revenue |
|
$ |
7,111 |
|
|
$ |
1,519 |
|
|
$ |
— |
|
|
Cost of revenue |
|
|
6,727 |
|
|
|
1,069 |
|
|
|
— |
|
|
Gross profit |
|
|
384 |
|
|
|
450 |
|
|
|
— |
|
|
Operating Expenses |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development |
|
|
21,989 |
|
|
|
29,273 |
|
|
|
29,669 |
|
|
Sales and marketing |
|
|
17,536 |
|
|
|
9,151 |
|
|
|
2,855 |
|
|
General and administrative |
|
|
12,275 |
|
|
|
10,813 |
|
|
|
7,831 |
|
|
Amortization of intangible assets |
|
|
7,858 |
|
|
|
6,967 |
|
|
|
2,185 |
|
|
Change in fair value of contingent consideration |
|
|
2,026 |
|
|
|
482 |
|
|
|
(400 |
) |
|
Issuance costs for warrants |
|
|
627 |
|
|
|
— |
|
|
|
— |
|
|
Inventory write-down related to restructuring |
|
|
— |
|
|
|
2,565 |
|
|
|
— |
|
|
Restructuring and other charges |
|
|
— |
|
|
|
3,064 |
|
|
|
— |
|
|
Goodwill impairment |
|
|
— |
|
|
|
61,784 |
|
|
|
— |
|
|
Acquisition related costs |
|
|
— |
|
|
|
— |
|
|
|
4,231 |
|
|
Total Operating Expenses |
|
|
62,311 |
|
|
|
124,099 |
|
|
|
46,371 |
|
|
Operating Loss |
|
|
(61,927 |
) |
|
|
(123,649 |
) |
|
|
(46,371 |
) |
|
Other Expense |
|
|
|
|
|
|
|
|
|
|
|
|
|
Change in fair value of warrant liabilities |
|
|
(83,734 |
) |
|
|
— |
|
|
|
— |
|
|
Interest expense, net |
|
|
(2,135 |
) |
|
|
(1,889 |
) |
|
|
(1,601 |
) |
|
Other (expense) income |
|
|
(300 |
) |
|
|
35 |
|
|
|
— |
|
|
Total Other Expense, net |
|
|
(86,169 |
) |
|
|
(1,854 |
) |
|
|
(1,601 |
) |
|
Loss before income taxes |
|
$ |
(148,096 |
) |
|
$ |
(125,503 |
) |
|
$ |
(47,972 |
) |
|
Income tax benefit |
|
|
3,300 |
|
|
|
5,523 |
|
|
|
1,024 |
|
|
Net loss |
|
$ |
(144,796 |
) |
|
$ |
(119,980 |
) |
|
$ |
(46,948 |
) |
|
Other comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation gain (loss) |
|
|
10,797 |
|
|
|
(2,603 |
) |
|
|
(3,166 |
) |
|
Comprehensive loss |
|
$ |
(133,999 |
) |
|
$ |
(122,583 |
) |
|
$ |
(50,114 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(0.97 |
) |
|
$ |
(1.07 |
) |
|
$ |
(0.59 |
) |
|
Weighted average common shares outstanding - basic and diluted |
|
|
148,744 |
|
|
|
112,185 |
|
|
|
79,628 |
|
See accompanying notes to consolidated financial statements.
44
Consolidated Statements of Stockholders’ Equity
(in thousands)
|
|
|
Common Stock |
|
|
Treasury Stock |
|
|
Additional Paid-in |
|
|
Accumulated |
|
|
Accumulated Other Comprehensive |
|
|
Total Stockholders’ |
|
||||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Deficit |
|
|
Loss |
|
|
Equity |
|
||||||||
|
Balance, December 31, 2014 |
|
|
63,183 |
|
|
$ |
63 |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
257,642 |
|
|
$ |
(135,916 |
) |
|
$ |
— |
|
|
$ |
121,789 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
3,311 |
|
|
|
— |
|
|
|
— |
|
|
|
3,311 |
|
|
Issuance of common stock, net of issuance costs |
|
|
20,756 |
|
|
|
21 |
|
|
|
— |
|
|
|
— |
|
|
|
58,310 |
|
|
|
— |
|
|
|
— |
|
|
|
58,331 |
|
|
Issuance of common stock, acquisition |
|
|
15,543 |
|
|
|
15 |
|
|
|
— |
|
|
|
— |
|
|
|
43,662 |
|
|
|
— |
|
|
|
— |
|
|
|
43,677 |
|
|
Exercise of stock options and restricted stock units |
|
|
698 |
|
|
|
1 |
|
|
|
— |
|
|
|
— |
|
|
|
258 |
|
|
|
— |
|
|
|
— |
|
|
|
259 |
|
|
Return of common stock to pay withholding taxes on restricted stock |
|
|
— |
|
|
|
— |
|
|
|
(31 |
) |
|
|
(73 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(73 |
) |
|
Issuance of warrants |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
97 |
|
|
|
— |
|
|
|
— |
|
|
|
97 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(3,166 |
) |
|
|
(3,166 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(46,948 |
) |
|
|
— |
|
|
|
(46,948 |
) |
|
Balance, December 31, 2015 |
|
|
100,180 |
|
|
$ |
100 |
|
|
|
(31 |
) |
|
$ |
(73 |
) |
|
$ |
363,280 |
|
|
$ |
(182,864 |
) |
|
$ |
(3,166 |
) |
|
$ |
177,277 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
5,033 |
|
|
|
— |
|
|
|
— |
|
|
|
5,033 |
|
|
Issuance of common stock, net of issuance costs |
|
|
15,086 |
|
|
|
15 |
|
|
|
— |
|
|
|
— |
|
|
|
58,014 |
|
|
|
— |
|
|
|
— |
|
|
|
58,029 |
|
|
Exercise of stock options and restricted stock units |
|
|
419 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
166 |
|
|
|
— |
|
|
|
— |
|
|
|
166 |
|
|
Return of common stock to pay withholding taxes on restricted stock |
|
|
— |
|
|
|
— |
|
|
|
(63 |
) |
|
|
(168 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(168 |
) |
|
Issuance of common stock for services |
|
|
96 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
116 |
|
|
|
— |
|
|
|
— |
|
|
|
116 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(2,603 |
) |
|
|
(2,603 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(119,980 |
) |
|
|
— |
|
|
|
(119,980 |
) |
|
Balance, December 31, 2016 |
|
|
115,781 |
|
|
$ |
115 |
|
|
|
(94 |
) |
|
$ |
(241 |
) |
|
$ |
426,609 |
|
|
$ |
(302,844 |
) |
|
$ |
(5,769 |
) |
|
$ |
117,870 |
|
|
Stock-based compensation |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
7,078 |
|
|
|
— |
|
|
|
— |
|
|
|
7,078 |
|
|
Non-employee warrant awards |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
838 |
|
|
|
— |
|
|
|
— |
|
|
|
838 |
|
|
Issuance of common stock and treasury stock, net of issuance costs |
|
|
44,689 |
|
|
|
45 |
|
|
|
213 |
|
|
|
409 |
|
|
|
68,410 |
|
|
|
— |
|
|
|
— |
|
|
|
68,864 |
|
|
Exercise of stock options and warrants |
|
|
34,749 |
|
|
|
35 |
|
|
|
— |
|
|
|
— |
|
|
|
112,803 |
|
|
|
— |
|
|
|
— |
|
|
|
112,838 |
|
|
Award of restricted stock units |
|
|
340 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Return of common stock to pay withholding taxes on restricted stock |
|
|
— |
|
|
|
— |
|
|
|
(119 |
) |
|
|
(168 |
) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(168 |
) |
|
Issuance of common stock in exchange for contingent consideration |
|
|
3,723 |
|
|
|
4 |
|
|
|
— |
|
|
|
— |
|
|
|
5,223 |
|
|
|
— |
|
|
|
— |
|
|
|
5,227 |
|
|
Relative fair value of warrants issued with debt |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
300 |
|
|
|
— |
|
|
|
— |
|
|
|
300 |
|
|
Other comprehensive loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
10,797 |
|
|
|
10,797 |
|
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(144,796 |
) |
|
|
— |
|
|
|
(144,796 |
) |
|
Balance, December 31, 2017 |
|
|
199,282 |
|
|
$ |
199 |
|
|
|
— |
|
|
$ |
— |
|
|
$ |
621,261 |
|
|
$ |
(447,640 |
) |
|
$ |
5,028 |
|
|
$ |
178,848 |
|
See accompanying notes to consolidated financial statements.
45
Consolidated Statements of Cash Flows
(in thousands)
|
|
|
Twelve Months Ended |
|
|||||||||
|
|
|
December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Operating Activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(144,796 |
) |
|
$ |
(119,980 |
) |
|
$ |
(46,948 |
) |
|
Adjustments to reconcile net loss to net cash and cash equivalents used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation |
|
|
2,486 |
|
|
|
1,942 |
|
|
|
1,248 |
|
|
Amortization of intangible assets |
|
|
7,858 |
|
|
|
6,967 |
|
|
|
2,185 |
|
|
Amortization of debt discount and debt issuance costs |
|
|
510 |
|
|
|
177 |
|
|
|
142 |
|
|
Stock-based compensation |
|
|
7,078 |
|
|
|
5,033 |
|
|
|
3,311 |
|
|
Non-employee warrant awards |
|
|
838 |
|
|
|
— |
|
|
|
— |
|
|
Common stock issued for services |
|
|
— |
|
|
|
116 |
|
|
|
— |
|
|
Inventory write-down related to restructuring |
|
|
— |
|
|
|
2,565 |
|
|
|
— |
|
|
Loss on disposal of property |
|
|
— |
|
|
|
— |
|
|
|
34 |
|
|
Non-cash restructuring and other charges |
|
|
— |
|
|
|
2,556 |
|
|
|
— |
|
|
Goodwill impairment |
|
|
— |
|
|
|
61,784 |
|
|
|
— |
|
|
Deferred tax benefit |
|
|
(3,300 |
) |
|
|
(5,562 |
) |
|
|
(1,024 |
) |
|
Loss on extinguishment of debt |
|
|
308 |
|
|
|
— |
|
|
|
— |
|
|
Change in fair value of warrant liabilities |
|
|
83,734 |
|
|
|
— |
|
|
|
— |
|
|
Change in fair value of contingent consideration |
|
|
2,026 |
|
|
|
482 |
|
|
|
(400 |
) |
|
Changes in operating assets and liabilities, net of effect of acquisition: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
(381 |
) |
|
|
(1,041 |
) |
|
|
133 |
|
|
Interest receivable |
|
|
23 |
|
|
|
(6 |
) |
|
|
(5 |
) |
|
Inventories |
|
|
(2,981 |
) |
|
|
(6,647 |
) |
|
|
(4,630 |
) |
|
Other current and long term assets |
|
|
(3,348 |
) |
|
|
(1,528 |
) |
|
|
728 |
|
|
Accounts payable |
|
|
(531 |
) |
|
|
(356 |
) |
|
|
1,096 |
|
|
Accrued expenses |
|
|
2,093 |
|
|
|
1,112 |
|
|
|
5,371 |
|
|
Deferred revenue |
|
|
1,088 |
|
|
|
— |
|
|
|
— |
|
|
Deferred gain on sale of SurgiBot assets |
|
|
7,500 |
|
|
|
— |
|
|
|
— |
|
|
Net cash and cash equivalents used in operating activities |
|
|
(39,795 |
) |
|
|
(52,386 |
) |
|
|
(38,759 |
) |
|
Investing Activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Payment for acquisition of a business |
|
|
— |
|
|
|
— |
|
|
|
(25,000 |
) |
|
Purchase of property and equipment |
|
|
(1,566 |
) |
|
|
(1,361 |
) |
|
|
(1,234 |
) |
|
Purchase of intellectual property |
|
|
(425 |
) |
|
|
— |
|
|
|
— |
|
|
Net cash and cash equivalents used in investing activities |
|
|
(1,991 |
) |
|
|
(1,361 |
) |
|
|
(26,234 |
) |
|
Financing Activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Payment of debt |
|
|
(13,343 |
) |
|
|
(6,902 |
) |
|
|
— |
|
|
Proceeds from issuance of debt and warrants, net of issuance costs |
|
|
13,005 |
|
|
|
— |
|
|
|
9,887 |
|
|
Payment of contingent consideration |
|
|
(7,181 |
) |
|
|
(1,182 |
) |
|
|
— |
|
|
Proceeds from issuance of common stock and warrants, net of issuance costs |
|
|
77,579 |
|
|
|
58,029 |
|
|
|
58,331 |
|
|
Taxes paid related to net share settlement of vesting of restricted stock units |
|
|
(168 |
) |
|
|
(168 |
) |
|
|
(73 |
) |
|
Proceeds from exercise of stock options and warrants |
|
|
34,479 |
|
|
|
166 |
|
|
|
259 |
|
|
Net cash and cash equivalents provided by financing activities |
|
|
104,371 |
|
|
|
49,943 |
|
|
|
68,404 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
431 |
|
|
|
(55 |
) |
|
|
22 |
|
|
Net increase (decrease) in cash, cash equivalents and restricted cash |
|
|
63,016 |
|
|
|
(3,859 |
) |
|
|
3,433 |
|
|
Cash, cash equivalents and restricted cash, beginning of period |
|
|
34,590 |
|
|
|
38,449 |
|
|
|
35,016 |
|
|
Cash, cash equivalents and restricted cash, end of period |
|
$ |
97,606 |
|
|
$ |
34,590 |
|
|
$ |
38,449 |
|
|
Supplemental Disclosure for Cash Flow Information |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest paid |
|
$ |
899 |
|
|
$ |
1,289 |
|
|
$ |
973 |
|
|
Supplemental Schedule of Noncash Investing and Financing Activities |
|
|
|
|
|
|
|
|
|
|
|
|
|
Transfer of inventory to property and equipment |
|
$ |
1,258 |
|
|
$ |
3,198 |
|
|
$ |
— |
|
|
Issuance of common stock as contingent consideration |
|
$ |
5,227 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Relative fair value of warrants issued with debt |
|
$ |
300 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Reclass of warrant liability to common stock and additional paid in capital |
|
$ |
78,359 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Transfer of in-process research and development to intellectual property |
|
$ |
17,913 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Cashless exercise of warrants |
|
$ |
149 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Issuance of common stock warrants |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
97 |
|
|
Contingent consideration related to acquisition |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
23,900 |
|
|
Issuance of common stock related to acquisition |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
43,677 |
|
See accompanying notes to consolidated financial statements.
46
Notes to Consolidated Financial Statements
|
1. |
Organization and Capitalization |
TransEnterix is a medical device company that is digitizing the interface between the surgeon and the patient to improve minimally invasive surgery by addressing the clinical and economic challenges associated with current laparoscopic and robotic options in today's value-based healthcare environment. The Company is focused on the commercialization of the Senhance™ Surgical System, which digitizes laparoscopic minimally invasive surgery. The system allows for robotic precision, haptic feedback, surgeon camera control via eye sensing and improved ergonomics while offering responsible economics.
The Senhance System has been granted a CE Mark in Europe for laparoscopic abdominal and pelvic surgery, as well as limited thoracic operations excluding cardiac and vascular surgery. In April 2017, the Company submitted a 510(k) application to the FDA for the Senhance System. On October 13, 2017, the Company received 510(k) clearance from the FDA for use in laparoscopic colorectal and gynecologic surgery. The Senhance System is available for sale in the U.S., the EU and select other countries.
The Senhance System is a multi-port robotic surgery system which allows multiple robotic arms to control instruments and a camera. The system features advanced technology to enable surgeons with haptic feedback and the ability to move the camera via eye movement. The system replicates laparoscopic motion that is familiar to experienced surgeons, and integrates three-dimensional high definition (“3DHD”) vision technology. The Senhance System also offers responsible economics to hospitals by offering robotic technology with reusable instruments thereby reducing additional costs per surgery when compared to other robotic solutions.
The Company also developed the SurgiBot™ System, a single-port, robotically enhanced laparoscopic surgical platform. On December 18, 2017, the Company announced that it had entered into an agreement with Great Belief International Limited (“GBIL”) to advance the SurgiBot System towards global commercialization. The agreement transfers ownership of the SurgiBot System assets, while the Company retains the option to distribute or co-distribute the SurgiBot System outside of China. Upon completion of the transfer of all SurgiBot System assets, GBIL will have the SurgiBot System manufactured in China and obtain Chinese regulatory clearance from the China Food and Drug Administration (“CFDA”), while entering into a nationwide distribution agreement with China National Scientific and Instruments and Materials Company (“CSIMC”) for the Chinese market. The agreement provides the Company with proceeds of at least $29 million, of which $7.5 million was received in December 2017. An additional $7.5 million is expected to be received by March 31, 2018, which includes a $3.0 million equity investment at $2.33 per share. The remaining $14 million, representing minimum royalties, will be paid beginning at the earlier of receipt of Chinese regulatory approval or five years.
On September 18, 2015, the Company entered into a Membership Interest Purchase Agreement, (the “Purchase Agreement”) with Sofar S.p.A., (“Sofar”) as seller, Vulcanos S.r.l. (“Vulcanos”), as the acquired company, and TransEnterix International, Inc. (“TransEnterix International”), a direct, wholly owned subsidiary of the Company which was incorporated in September 2015, as buyer. The closing of the transactions occurred on September 21, 2015 (the “Closing Date”) pursuant to which the Company acquired all of the membership interests of Vulcanos from Sofar (now known as the “Senhance Acquisition”), and changed the name of Vulcanos to TransEnterix Italia S.r.l (“TransEnterix Italia”). The Senhance Acquisition included all of the assets, employees and contracts related to the Senhance System. See Note 3 for a description of the related transactions.
On September 3, 2013, TransEnterix Surgical, Inc. a Delaware corporation (“TransEnterix Surgical”), and SafeStitch Medical, Inc., a Delaware corporation (“SafeStitch”) consummated a merger transaction whereby TransEnterix Surgical merged with a merger subsidiary of SafeStitch, with TransEnterix Surgical as the surviving entity in the merger (the “Merger”). As a result of the Merger, TransEnterix Surgical became a wholly owned subsidiary of SafeStitch. On December 6, 2013, SafeStitch changed its name to TransEnterix, Inc. and increased the authorized shares of common stock from 225,000,000 to 750,000,000, and authorized 25,000,000 shares of preferred stock, par value $0.01 per share.
As used herein, the term “Company” refers to the combination of SafeStitch and TransEnterix Surgical after giving effect to the Merger, and includes TransEnterix International, Inc.; TransEnterix Italia S.r.l.; TransEnterix Europe S.à.R.L; Bertrange, Swiss Branch, Lugano; TransEnterix Asia Pte. Ltd.; and TransEnterix Taiwan Ltd. after giving effect to the Senhance Acquisition, the term “SafeStitch” refers to the historic business of SafeStitch Medical, Inc. prior to the Merger, and the term “TransEnterix Surgical” refers to the historic business of TransEnterix Surgical, Inc. prior to the Merger.
47
Basis of Presentation
The accompanying Consolidated Financial Statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”) and include the accounts of the Company and its direct and indirect wholly owned subsidiaries, SafeStitch LLC, TransEnterix Surgical, Inc., TransEnterix International, Inc., TransEnterix Italia S.r.l., TransEnterix Europe S.à.R.L, Bertrange, Swiss Branch, Lugano, TransEnterix Asia Pte. Ltd. and TransEnterix Taiwan Ltd. All inter-company accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant items subject to such estimates and assumptions include identifiable intangible assets and goodwill, contingent consideration, warrant liabilities, stock compensation expense, restructuring and other charges, excess and obsolete inventory reserves, and deferred tax asset valuation allowances.
Cash and Cash Equivalents and Restricted Cash
The Company considers all highly liquid investments with original maturities of 90 days or less at the time of purchase to be cash equivalents.
Restricted cash at December 31, 2017 includes $6.0 million in a money market account, held in connection with the Company’s notes payable (see Note 13) and $389,000 in cash accounts held as collateral primarily under the terms of an office operating lease, credit cards and automobile leases.
Concentrations and Credit Risk
The Company’s principal financial instruments subject to potential concentration of credit risk are cash and cash equivalents, including amounts held in money market accounts. The Company places cash deposits with a federally insured financial institution. The Company maintains its cash at banks and financial institutions it considers to be of high credit quality; however, the Company’s cash deposits may at times exceed the FDIC insured limit. Balances in excess of federally insured limitations may not be insured. The Company has not experienced losses on these accounts, and management believes that the Company is not exposed to significant risks on such accounts.
The Company’s accounts receivable are derived from net revenue to customers located throughout the world. The Company evaluates its customers’ financial condition and, generally, requires no collateral from its customers. The Company provides reserves for potential credit losses but has not experienced significant losses to date. The Company had eight customers who constituted 100% of the Company’s net accounts receivable at December 31, 2017. The Company had one customer who constituted 100% of the Company’s net accounts receivable at December 31, 2016 and one customer who constituted 100% of the Company’s net accounts receivable at December 31, 2015. The Company had eight customers who accounted for 100% of sales in 2017, one customer who accounted for 100% of sales in 2016. There were no sales in 2015.
Accounts Receivable
Accounts receivable are recorded at net realizable value, which includes an allowance for estimated uncollectable accounts. The allowance for uncollectible accounts was determined based on historical collection experience.
Inventories
Inventories are stated at the lower of cost (determined on a first-in, first-out basis) or net realizable value. Inventory costs include direct materials, direct labor, and normal manufacturing overhead. The Company records reserves, when necessary, to reduce the carrying value of inventory to its net realizable value. Management considers forecast demand in relation to the inventory on hand, competitiveness of product offerings, market conditions and product life cycles when determining excess and obsolescence and net realizable value adjustments. At the point of loss recognition, a new, lower-cost basis for that inventory is established, and any subsequent improvements in facts and circumstances do not result in the restoration or increase in that newly established cost basis. Any inventory on hand at the measurement date in excess of the Company’s current requirements based on anticipated levels of sales is classified as long-term on the Company’s consolidated balance sheets.
48
Identifiable Intangible Assets and Goodwill
Identifiable intangible assets are recorded at cost, or when acquired as part of a business acquisition, at estimated fair value. Certain intangible assets are amortized over 5 to 10 years. Similar to tangible personal property and equipment, the Company periodically evaluates identifiable intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
Intellectual property consists of purchased patent rights and developed research and development acquired as part of a business acquisition. Amortization of the patent rights is recorded using the straight-line method over the estimated useful life of the patents of 10 years. Amortization of the developed research and development is recorded using the straight-line method over the estimated useful life of 5 to 7 years. This method approximates the period over which the Company expects to receive the benefit from these assets. See Note 17 for additional information related to the write-off of purchased patents in connection with the restructuring plan executed in May 2016. No impairment existed at December 31, 2017.
Indefinite-lived intangible assets, such as goodwill, are not amortized. The Company tests the carrying amounts of goodwill for recoverability on an annual basis at December 31 or when events or changes in circumstances indicate evidence a potential impairment exists, using a fair value based test. The Company continues to operate in one segment, which is considered to be the sole reporting unit and therefore, goodwill is tested for impairment at the enterprise level. See Note 10 for additional information related to goodwill impairment recorded during the second quarter of 2016. No impairment existed at December 31, 2017 or 2015.
In-Process Research and Development
In-process research and development (“IPR&D”) assets represent the fair value assigned to technologies that were acquired, which at the time of acquisition have not reached technological feasibility and have no alternative future use. IPR&D assets are considered to be indefinite-lived until the completion or abandonment of the associated research and development projects. During the period that the IPR&D assets are considered indefinite-lived, they are tested for impairment on an annual basis, or more frequently if the Company becomes aware of any events occurring or changes in circumstances that indicate that the fair value of the IPR&D assets are less than their carrying amounts. If and when development is complete, which generally occurs upon regulatory approval, and the Company is able to commercialize products associated with the IPR&D assets, these assets are then deemed definite-lived and are amortized based on their estimated useful lives at that point in time. If development is terminated or abandoned, the Company may have a full or partial impairment charge related to the IPR&D assets, calculated as the excess of carrying value of the IPR&D assets over fair value. The IPR&D was acquired on September 21, 2015. No impairment existed at December 31, 2016 and 2015.
On October 13, 2017, upon regulatory approval and the ability to commercialize the products associated with the IPR&D assets, the assets were deemed definite-lived, reclassified to intellectual property and are now amortized based on their estimated useful lives.
Property and Equipment
Property and equipment consists primarily of machinery, manufacturing equipment, demonstration equipment, computer equipment, furniture, and leasehold improvements, which are recorded at cost.
Depreciation is recorded using the straight-line method over the estimated useful lives of the assets as follows:
|
Machinery, manufacturing and demonstration equipment |
|
3-5 years |
|
Computer equipment |
|
3 years |
|
Furniture |
|
5 years |
|
Leasehold improvements |
|
Lesser of lease term or 3 to 10 years |
Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repairs and maintenance costs are expensed as incurred.
Impairment of Long-Lived Assets
The Company reviews its long-lived assets for possible impairment whenever events or changes in circumstances indicate that the carrying amount of the assets may not be fully recoverable. To determine the recoverability of its long-lived assets, the Company evaluates the probability that future estimated undiscounted net cash flows will be less than the carrying amount of the assets. If such estimated cash flows are less than the carrying amount of the long-lived assets, then such assets are written down to their fair value. The Company’s estimates of anticipated cash flows and the remaining estimated useful lives of long-lived assets could be reduced in the future, resulting in a reduction to the carrying amount of long-lived assets.
49
Contingent consideration is recorded as a liability and is the estimate of the fair value of potential milestone payments related to business acquisitions. Contingent consideration is measured at fair value using a discounted cash flow model utilizing significant unobservable inputs including the probability of achieving each of the potential milestones and an estimated discount rate associated with the risks of the expected cash flows attributable to the various milestones. Significant increases or decreases in any of the probabilities of success or changes in expected timelines for achievement of any of these milestones would result in a significantly higher or lower fair value of these milestones, respectively, and commensurate changes to the associated liability. The contingent consideration is revalued at each reporting period and changes in fair value are recognized in the consolidated statements of operations and comprehensive loss.
Deferred Gain on Sale of SurgiBot Assets
In conjunction with the agreement with GBIL in relation to the transfer of the SurgiBot System assets, the Company received $7.5 million in December 2017. This amount was included in deferred gain in sale of SurgiBot assets on the consolidated balance sheet pending transfer of the assets.
Warrant Liabilities
The Company’s Series A Warrants and Series B Warrants (see Note 16) are measured at fair value using a simulation model which takes into account, as of the valuation date, factors including the current exercise price, the expected life of the warrant, the current price of the underlying stock, its expected volatility, holding cost and the risk-free interest rate for the term of the warrant (see Note 5). The warrant liability is revalued at each reporting period and changes in fair value are recognized in the consolidated statements of operations and comprehensive loss. The selection of the appropriate valuation model and the inputs and assumptions that are required to determine the valuation requires significant judgment and requires management to make estimates and assumptions that affect the reported amount of the related liability and reported amounts of the change in fair value. Actual results could differ from those estimates, and changes in these estimates are recorded when known. As the warrant liability is required to be measured at fair value at each reporting date, it is reasonably possible that these estimates and assumptions could change in the near term.
Translation of Foreign Currencies
The functional currency of the Company’s operational foreign subsidiaries is Euros. The assets and liabilities of the Company’s foreign subsidiaries are translated into U.S. dollars at exchange rates in effect at the balance sheet date. Income and expense items are translated at the average exchange rates prevailing during the period. The cumulative translation effect for a subsidiary using a functional currency other than the U.S. dollar is included in accumulated other comprehensive income or loss as a separate component of stockholders’ equity.
The Company’s intercompany accounts are denominated in the functional currency of the foreign subsidiary. Gains and losses resulting from the remeasurement of intercompany receivables that the Company considers to be of a long-term investment nature are recorded as a cumulative translation adjustment in accumulated other comprehensive income or loss as a separate component of stockholders’ equity, while gains and losses resulting from the remeasurement of intercompany receivables from a foreign subsidiary for which the Company anticipates settlement in the foreseeable future are recorded in the consolidated statement of operations and comprehensive loss. The net gains and losses included in net loss in the consolidated statements of operations and comprehensive loss for the years December 31, 2017, 2016, and 2015 were not significant.
Business Acquisitions
Business acquisitions are accounted for using the acquisition method of accounting in accordance with Accounting Standards Codification (“ASC”) 805, “Business Combinations.” ASC 805 requires, among other things, that assets acquired and liabilities assumed be recognized at their fair values, as determined in accordance with ASC 820, “Fair Value Measurements,” as of the acquisition date. For certain assets and liabilities, book value approximates fair value. In addition, ASC 805 establishes that consideration transferred be measured at the closing date of the acquisition at the then-current market price. Under ASC 805, acquisition related costs (i.e., advisory, legal, valuation and other professional fees) and certain acquisition-related restructuring charges impacting the target company are expensed in the period in which the costs are incurred. The application of the acquisition method of accounting requires the Company to make estimates and assumptions related to the estimated fair values of net assets acquired.
Significant judgments are used during this process, particularly with respect to intangible assets. Generally, intangible assets are amortized over their estimated useful lives. Goodwill and other indefinite-lived intangibles are not amortized, but are annually assessed for impairment. Therefore, the purchase price allocation to intangible assets and goodwill has a significant impact on future operating results.
50
The Company is subject to a number of risks similar to other similarly-sized companies in the medical device industry. These risks include, without limitation, the historical lack of profitability; the Company’s ability to raise additional capital; its ability to successfully develop, clinically test and commercialize its products; the timing and outcome of the regulatory review process for its products; changes in the health care and regulatory environments of the United States, Italy, other countries in the European Union, and other countries in which the Company intends to operate; its ability to attract and retain key management, marketing and scientific personnel; competition from new entrants; its ability to successfully prepare, file, prosecute, maintain, defend and enforce patent claims and other intellectual property rights; its ability to successfully transition from a research and development company to a marketing, sales and distribution concern; competition in the market for robotic surgical devices; and its ability to identify and pursue development of additional products.
Revenue Recognition
The Company’s revenue consists of product revenue resulting from the sales of systems, instruments and accessories, and service revenue. The Company recognizes revenue when persuasive evidence of an arrangement exists, delivery has occurred or service has been rendered, the price is fixed or determinable, and collectability is reasonably assured. Revenue is presented net of taxes collected from customers that are remitted to government authorities. The Company generally recognizes revenue at the following points in time:
|
|
• |
System sales. For systems sold directly to end customers, revenue is recognized when acceptance occurs, which is deemed to have occurred upon customer acknowledgment of delivery or installation, depending on the terms of the arrangement. The Senhance Systems are delivered with a software component. However, because the software and non-software elements function together to deliver the system’s essential functionality, the Company's arrangements are excluded from being accounted for under software revenue recognition guidance. |
|
|
• |
Instruments and accessories. Revenue from sales of instruments and accessories is generally recognized at the time of shipment. Revenue from services related to the supply and management of instruments and accessories is recognized as the services are rendered. |
|
|
• |
Service. Service revenue is recognized ratably over the term of the service period. Revenue related to services performed on a time-and-materials basis is recognized when it is earned and billable. |
The Company's system sale arrangements contain multiple elements including a system(s), instruments, accessories, and system service. The Company generally delivers all of the elements, other than service, within days of entering into the system sale arrangement. Each of these elements is a separate unit of accounting. System accessories, instruments, and service are also sold on a stand-alone basis.
For multiple-element arrangements, revenue is allocated to each unit of accounting based on their relative selling prices. Relative selling prices are based first on vendor specific objective evidence of fair value (“VSOE”), then on third-party evidence of selling price (“TPE”) when VSOE does not exist, and then on management's best estimate of the selling price (“BESP”) when VSOE and TPE do not exist.
The Company’s system sale arrangements generally include a one-year period of free service, and the right for the customer to purchase service annually thereafter. The revenue allocated to the free service period is deferred and recognized ratably over the free service period.
Because the Company has neither VSOE nor TPE for its systems, the allocation of revenue is based on BESP for the systems sold. The objective of BESP is to determine the price at which the Company would transact a sale, had the product been sold on a stand-alone basis. The Company determines BESP for its systems by considering multiple factors, including, but not limited to, features and functionality of the system, geographies, type of customer, and market conditions. The Company regularly reviews BESP and maintains internal controls over establishing and updating these estimates.
Cost of Revenue
Cost of revenue sold consists of contract manufacturing, materials, labor and manufacturing overhead incurred internally to produce the products. Shipping and handling costs incurred by the Company are included in cost of revenue.
51
Research and Development Costs
Research and development expenses primarily consist of engineering, product development and regulatory expenses, incurred in the design, development, testing and enhancement of our products. Research and development costs are expensed as incurred.
Stock-Based Compensation
The Company follows ASC 718 (“Stock Compensation”) and ASC 505-50 (“Equity-Based Payments to Non-employees”), which provide guidance in accounting for share-based awards exchanged for services rendered and requires companies to expense the estimated fair value of these awards over the requisite service period. For awards granted to non-employees, the Company determines the fair value of the stock-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. If the fair value of the equity instruments issued is used, it is measured using the stock price and other measurement assumptions as of the earlier of either (1) the date at which a commitment for performance by the counterparty to earn the equity instruments is reached, or (2) the date at which the counterparty’s performance is complete.
The Company recognizes compensation expense for stock-based awards based on estimated fair values on the date of grant for awards granted to employees. The Company uses the Black-Scholes-Merton option pricing model to determine the fair value of stock options. The fair value of restricted stock units is determined by the market price of the Company’s common stock on the date of grant. The expense associated with stock-based compensation is recognized on a straight-line basis over the requisite service period of each award.
The Company records as expense the fair value of stock-based compensation awards, including stock options and restricted stock units. Compensation expense for stock-based compensation was approximately $7,078,000, $5,033,000 and $3,311,000 for the years ended December 31, 2017, 2016 and 2015, respectively.
Income Taxes
The Company accounts for income taxes using the asset and liability method, which requires the recognition of deferred tax assets or liabilities for the temporary differences between financial reporting and tax basis of the Company’s assets and liabilities, and for tax carryforwards at enacted statutory rates in effect for the years in which the asset or liability is expected to be realized. The effect on deferred taxes of a change in tax rates is recognized in income during the period that includes the enactment date. In addition, valuation allowances are established when necessary to reduce deferred tax assets and liabilities to the amounts expected to be realized.
On December 22, 2017, the Tax Cuts and Jobs Act (“Tax Legislation”) was enacted into law, which reduced the US federal corporate income tax rate to 21% for tax years beginning after December 31, 2017. As a result of the newly enacted tax rate, the Company adjusted its U.S. deferred tax assets as of December 31, 2017, by applying the new 21% rate, which resulted in a decrease to the deferred tax assets and a corresponding decrease to the valuation allowance of approximately $36.1 million.
The Tax Legislation also implements a territorial tax system. Under the territorial tax system, in general, the Company's foreign earnings will no longer be subject to tax in the U.S. As part of transition to the territorial tax system the Tax Legislation includes a mandatory deemed repatriation of all undistributed foreign earnings that are subject to a U.S. income tax. The Company estimates that the deemed repatriation will not result in any additional U.S. income tax liability as it estimates it currently has no undistributed foreign earnings.
The SEC staff issued Staff Accounting Bulletin (“SAB”) No. 118 which will allow the Company to record provisional amounts related to accounting for the Tax Legislation during a measurement period which is similar to the measurement period used when accounting for business combinations. The Company is following the guidance set forth by SAB 118 and any amounts calculated are provisional estimates and will be reevaluated as more information or guidance becomes available. The Company will continue to assess the impact of the Tax Legislation on its business and consolidated financial statements.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources.
52
The Company operates in one business segment—the research, development and sale of medical device robotics to improve minimally invasive surgery. The Company’s chief operating decision maker (determined to be the Chief Executive Officer) does not manage any part of the Company separately, and the allocation of resources and assessment of performance are based on the Company’s consolidated operating results. Approximately 60% and 49% of the Company’s total consolidated assets are located within the U.S. as of December 31, 2017 and 2016, respectively. The remaining assets are mostly located in Europe and are primarily related to the Company’s facility in Italy, and include goodwill, intellectual property, other current assets, property and equipment, cash, accounts receivable and inventory of $99.9 million and $90.4 million at December 31, 2017 and 2016, respectively associated with the Senhance Acquisition in September 2015. Total assets outside of the U.S. excluding goodwill amounted to 31% and 40% of total consolidated assets at December 31, 2017 and 2016, respectively. The Company recognizes sales by geographic area based on the country in which the customer is based. For the years ended December 31, 2017, 2016, and 2015, 18%, 0%, and 0%, respectively, of net revenue were generated in the United States; and 61%, 100%, and 0% were generated in Europe; and 21%, 0% and 0% were generated in Asia.
Impact of Recently Issued Accounting Standards
In July 2017, the Financial Accounting Standards Board (“FASB”) issued ASU 2017-11, Earnings Per Share (Topic 260); Distinguishing Liabilities from Equity (Topic 480); Derivatives and Hedging (Topic 815): (Part I) Accounting for Certain Financial Instruments with Down Round Features, (Part II) Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception. The amendments in this update are intended to simplify the accounting for certain equity-linked financial instruments and embedded features with down round features that result in the strike price being reduced on the basis of the pricing of future equity offerings. Under the new guidance, a down round feature will no longer need to be considered when determining whether certain financial instruments or embedded features should be classified as liabilities or equity instruments. That is, a down round feature will no longer preclude equity classification when assessing whether an instrument or embedded feature is indexed to an entity's own stock. In addition, the amendments clarify existing disclosure requirements for equity-classified instruments. These amendments are effective for fiscal years, and interim periods within those years, beginning after December 15, 2018, with early adoption permitted. The adoption of this ASU should not have a material impact on the consolidated financial statements.
In January 2017, the FASB issued Accounting Standards Update (“ASU”) 2017-04, Simplifying the Test for Goodwill Impairment. Under the new standard, goodwill impairment would be measured as the amount by which a reporting unit’s carrying value exceeds its fair value, not to exceed the carrying value of goodwill. This ASU eliminates existing guidance that requires an entity to determine goodwill impairment by calculating the implied fair value of goodwill by hypothetically assigning the fair value of a reporting unit to all of its assets and liabilities as if that reporting unit had been acquired in a business combination. This ASU is effective prospectively to annual and interim impairment tests beginning after December 15, 2019, with early adoption permitted. The Company early adopted this ASU as of the beginning of fiscal year 2017. The adoption of this ASU did not have a material impact on the consolidated financial statements.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230) which addresses changes to reduce the presentation diversity of certain cash receipts and cash payments in the statement of cash flows, including debt prepayment or extinguishment costs, settlement of certain debt instruments, contingent consideration payments made after a business combination, proceeds from the settlement of insurance claims, and distributions received from equity method investees. The guidance becomes effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years, with early adoption permitted. An entity that elects early adoption must adopt all of the amendments in the same period. The new standard will be applied retrospectively, but may be applied prospectively if retrospective application would be impracticable. The Company expects certain reclassifications within the consolidated statements of cash flows related to payments of contingent consideration.
In March 2016, the FASB issued ASU 2016-09, Compensation – Stock Compensation (Topic 718) – Improvements to Employee Share-Based Payment Accounting. Under ASU 2016-09, the tax effects of stock compensation will be recognized as income tax expense or benefit in the income statement and the tax effects of exercised or vested awards will be treated as discrete items in the reporting period in which they occur. Along with other income tax cash flows, excess tax benefits will be classified as operating activities, and cash paid by an employer when directly withholding shares for tax withholding purposes will be classified as financing activities. Entities may make an entity-wide accounting policy election to either estimate the number of awards that are expected to vest (current GAAP) or account for forfeitures when they occur. The threshold to qualify for equity classification permits withholding up to the maximum statutory tax rates in the applicable jurisdictions. For public companies, ASU 2016-09 is effective for annual periods beginning after December 15, 2016, and interim periods within those annual periods. Early adoption is permitted, however, an entity that elects early adoption must adopt all amendments under the new standard in the same period. The Company adopted this ASU as of the beginning of fiscal year 2017 and did not elect to account for forfeitures when they occur, but will continue to estimate the number of awards that are expected to vest. The adoption of this ASU did not have a material impact on the consolidated financial statements.
53
In February 2016, the FASB issued ASU 2016-02, Leases. The new standard establishes a right-of-use (ROU) model that requires a lessee to record a ROU asset and a lease liability on the balance sheet for all leases with terms longer than 12 months. Leases will be classified as either finance or operating, with classification affecting the pattern of expense recognition in the income statement. The new standard is effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. A modified retrospective transition approach is required for lessees for capital and operating leases existing at, or entered into after, the beginning of the earliest comparative period presented in the financial statements, with certain practical expedients available. The Company currently expects that upon adoption, ROU assets and lease liabilities will be recognized in the balance sheet in amounts that the Company does not expect will have a material impact on the consolidated financial statements based on the Company’s current leases.
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers, which sets forth a single, comprehensive revenue recognition model for all contracts with customers to improve comparability. Subsequently, the FASB has issued several standards related to ASU 2014-09 (collectively, the “New Revenue Standard”). The New Revenue Standard requires revenue recognition to depict the transfer of goods or services to customers in an amount that reflects the consideration that the entity expects to receive in exchange for those goods or services. In addition, the New Revenue Standard requires expanded disclosures. This New Revenue Standard permits the use of either the retrospective or cumulative effect transition method when adopted. The New Revenue Standards becomes effective for the Company in the first quarter of fiscal year 2018.
The Company will adopt the New Revenue Standard in the first quarter of fiscal year 2018 using the modified retrospective method resulting in a cumulative catch-up adjustment to opening retained earnings in the period of adoption. The Company has substantially completed its evaluation of the impact of the New Revenue Standard on its historical financial statements. The Company’s performance obligations under its existing contracts primarily include the sale of systems, instruments and accessories, as well as services. The product revenues will be recognized at a point in time upon delivery or installation, depending on the terms of the agreement. The service revenue will be recognized ratably over time. The Company has concluded that the timing and measurement of revenue recognition will be materially consistent under the New Revenue Standard, except for the future billings related to future service included in its multi-year contracts that should be part of the consideration allocated to all performance obligations under the New Revenue Standard. Under the current standard, future service billings are considered to be contingent revenue, and therefore, are not included in the consideration allocated. Accordingly, the amount of consideration allocated to the performance obligations identified in the Company’s system arrangements will be different under the New Revenue Standard than the amount allocated under the current standard. In general, this will result in an acceleration of the amount of revenue recognized for system sales with multi-year service contracts. Due to limited sales to date, the Company has evaluated its contracts and has quantified an immaterial cumulative catch-up adjustment upon adoption. The Company continues to evaluate the required disclosures.
The New Revenue Standard is principles based and interpretation of those principles may vary from company to company based on their unique circumstances. It is possible that interpretation, industry practice, and guidance may evolve as companies and the accounting profession work to implement this new standard. While substantially complete, the Company is still in the process of finalizing its evaluation of the effect of the New Revenue Standard on the Company’s historical financial statements and disclosures. The Company will finalize its accounting assessment and quantitative impact of the adoption of the New Revenue Standard during the first quarter of fiscal year 2018. As the Company completes its evaluation of this new standard, new information may arise that could change the Company’s current understanding of the impact to revenue and expense recognized. Additionally, the Company will continue to monitor industry activities and any additional guidance provided by regulators, standards setters, or the accounting profession and adjust the Company’s assessment and implementation plans accordingly.
In July 2015, the FASB issued ASU 2015-11, Simplifying the Measurement of Inventory (Topic 330). This update requires inventory within the scope of the standard to be measured at the lower of cost or net realizable value. Previous guidance required inventory to be measured at the lower of cost or market (where market was defined as replacement cost, with a ceiling of net realizable value and floor of net realizable value less a normal profit margin). This update is effective for annual and interim periods beginning after December 15, 2016. The Company adopted these provisions in the first quarter of fiscal year 2017 with no material impact on its consolidated financial statements.
|
3. |
Acquisition of Senhance Surgical Robotic System |
On September 21, 2015, the Company completed the strategic acquisition, through its wholly owned subsidiary TransEnterix International, from Sofar, of all of the assets, employees and contracts related to the advanced robotic system for minimally invasive laparoscopic surgery now known as the Senhance System and changed the name of the acquired company from Vulcanos S.r.l. to TransEnterix Italia S.r.l.
54
Under the terms of the Purchase Agreement, the consideration consisted of the issuance of 15,543,413 shares of the Company’s common stock (the “Securities Consideration”) and approximately $25.0 million U.S. Dollars and €27.5 million Euro in cash consideration (the “Cash Consideration”). The Securities Consideration was issued in full at the closing of the Senhance Acquisition; the Cash Consideration was or will be paid in four tranches, as follows:
(1) $25.0 million of the Cash Consideration was paid at closing.
(2) On December 30, 2016, the Company and Sofar entered into an Amendment to the Purchase Agreement (the “Amendment”) to restructure the terms of the second tranche of the Cash Consideration (the “Second Tranche”). Under the Amendment, the Second Tranche was restructured to be paid through the (A) the issuance of 3,722,685 shares of the Company’s common stock with an aggregate fair market value of €5.0 million and (B) the payment of €5.0 million in cash upon the occurrence of either (i) receipt of clearance from the FDA for the Senhance System; or (ii) the Company having cash on hand of at least $50.0 million, or (iii) successfully completing a financing, raising at least $50.0 million in gross proceeds after September 2015, exclusive of any financing proceeds related to the December 2016 purchase agreement between the Company and Lincoln Park Capital Fund, LLC.; with payment of simple interest at a rate of 9.0% per annum beginning on December 31, 2016. The Five Million Euro (€ 5,000,000) cash payment began to accrue simple interest at a rate of 9% per annum beginning on December 31, 2016 and continued to accrue interest until November 15, 2017 when it was paid in full. Prior to December 30, 2016, the Second Tranche of the Cash Consideration of €10.0 million was payable after the achievement of both of the following milestones (i) the earlier of approval from the FDA for the Senhance System or December 31, 2016, and (ii) the Company having cash on hand of at least $50.0 million, or successfully completing a financing, raising at least $50.0 million in gross proceeds; with payment of simple interest at a rate of 9.0% per annum between the achievement of the first milestone event and the payment date.
(3) The third tranche of the Cash Consideration (the “Third Tranche”) of €15.0 million shall be payable upon achievement of trailing revenues from sales or services contracts of the Senhance System of at least €25.0 million over a calendar quarter.
(4) The fourth tranche of the Cash Consideration of €2.5 million shall be payable by December 31 of each year as reimbursement for certain debt payments made by Sofar under an existing Sofar loan agreement in such year, with payments beginning as of December 31, 2016. As of December 31, 2017, the Company had paid €1.8 million of the fourth tranche.
The Third Tranche would have been payable even if the Second Tranche was not then payable. In addition, the Third Tranche payments will be accelerated in the event that (i) the Company or TransEnterix International is acquired, (ii) the Company significantly reduces or suspends selling efforts of the Senhance System, or (iii) the Company acquires a business that offers alternative products that are directly competitive with the Senhance System.
Under the Purchase Agreement, 10% of the Securities Consideration was being held in escrow to support Sofar’s representations and warranties under the Purchase Agreement. In accordance with a related escrow agreement, the escrowed shares were released in September 2016. The Company, a subsidiary and Sofar also entered into a Security Agreement, which provides that 10% of the membership interests of TransEnterix Italia have a lien placed thereon by and in favor of Sofar to support the Company’s representations and warranties under the Purchase Agreement. The security interest period was twenty-four months after the closing of the Senhance Acquisition and expired on September 21, 2017.
The Purchase Agreement contains customary representations and warranties of the parties and the parties have customary indemnification obligations, which are subject to certain limitations described further in the Purchase Agreement.
In connection with the Senhance Acquisition, the Company also entered into a Registration Rights Agreement, dated as of September 21, 2015, with Sofar, pursuant to which the Company agreed to register the Securities Consideration shares for resale following the end of the lock-up periods described below. The resale registration statement has been filed and is effective.
In connection with the Senhance Acquisition, Sofar entered into a Lock-Up Agreement with the Company pursuant to which Sofar agreed, subject to certain exceptions, not to sell, transfer or otherwise convey any of the Securities Consideration over a two-year period following the Closing Date. As of September 21, 2017, all of the Securities Consideration was released from the lock-up restrictions and is eligible to be resold under the effective resale registration statement.
The Senhance Acquisition was accounted for as a business combination utilizing the methodology prescribed in ASC 805. The purchase price for the Senhance Acquisition has been allocated to the assets acquired and liabilities assumed based on their estimated fair values.
55
The Senhance Acquisition-date fair value of the consideration is as follows (in thousands, except for per share amounts):
|
Common shares issued |
|
|
15,543 |
|
|
Closing price per share |
|
$ |
2.81 |
|
|
|
|
$ |
43,677 |
|
|
Cash consideration |
|
|
25,000 |
|
|
Contingent consideration |
|
|
23,900 |
|
|
Total consideration |
|
$ |
92,577 |
|
The following table summarizes the estimated fair values of the assets acquired and liabilities assumed on September 21, 2015, the date of acquisition (in thousands):
|
Accounts receivable |
|
$ |
78 |
|
|
Inventories |
|
|
2,800 |
|
|
Current deferred tax asset |
|
|
526 |
|
|
Other current assets |
|
|
4,180 |
|
|
Property and equipment |
|
|
1,384 |
|
|
Intellectual property |
|
|
48,500 |
|
|
In-process research and development |
|
|
17,100 |
|
|
Goodwill |
|
|
38,348 |
|
|
Total assets acquired |
|
$ |
112,916 |
|
|
Accounts payable and other liabilities |
|
|
1,915 |
|
|
Long-term deferred tax liabilities |
|
|
18,424 |
|
|
Net assets acquired |
|
$ |
92,577 |
|
The Company allocated $48.5 million of the purchase price to identifiable intangible assets of intellectual property that met the separability and contractual legal criterion of ASC 805. The intellectual property is being amortized using the straight-line method over 7 years.
IPR&D is principally the estimated fair value of the Senhance System technology which had not reached commercial technological feasibility nor had alternative future use at the time of the acquisition and therefore the Company considered IPR&D, with assigned values to be allocated among the various IPR&D assets acquired.
Goodwill is calculated as the difference between the acquisition-date fair value of the consideration transferred and the fair values of the assets acquired and liabilities assumed. The goodwill resulting from this acquisition arises largely from synergies expected from combining the operations of TransEnterix Italia with the Company’s existing operations. The goodwill is not deductible for income tax purposes.
All legal, consulting and other costs related to the acquisition, aggregating approximately $4.2 million, have been expensed as incurred and are included in operating expenses in the Company’s consolidated statement of operations and comprehensive loss for the year ended December 31, 2015. The results of operations for TransEnterix Italia are included in the Company’s consolidated statements of operations and comprehensive loss for the period from the September 21, 2015 acquisition date.
56
The following unaudited pro forma information presents the combined results of operations for the years ended December 31, 2015 and 2014, as if the Company had completed the Senhance Acquisition at the beginning of fiscal 2014. The pro forma financial information is provided for comparative purposes only and is not necessarily indicative of what actual results would have been had the acquisition occurred on the date indicated, nor does it give effect to synergies, cost savings, fair market value adjustments, immaterial amortization expense and other changes expected to result from the acquisition. Accordingly, the pro forma financial results do not purport to be indicative of consolidated results of operations as of the date hereof, for any period ended on the date hereof, or for any other future date or period. The pro forma consolidated financial information has been calculated after applying the Company’s accounting policies and includes adjustments for transaction-related costs and amortization of intellectual property.
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
|
|
(In thousands except per share amounts) |
|
|||||
|
Revenue |
|
$ |
77 |
|
|
$ |
401 |
|
|
Net loss |
|
|
53,994 |
|
|
|
46,874 |
|
|
Net loss per share |
|
$ |
0.57 |
|
|
$ |
0.63 |
|
|
4. |
Cash, Cash Equivalents, and Restricted Cash |
Cash, cash equivalents and restricted cash consist of the following:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Cash |
|
$ |
4,039 |
|
|
$ |
1,975 |
|
|
Money market |
|
|
87,178 |
|
|
|
22,190 |
|
|
Total cash and cash equivalents |
|
$ |
91,217 |
|
|
$ |
24,165 |
|
|
Restricted cash |
|
$ |
6,389 |
|
|
$ |
10,425 |
|
|
Total |
|
$ |
97,606 |
|
|
$ |
34,590 |
|
Restricted cash at December 31, 2017 includes $6.0 million in a money market account, held in connection with the Company’s notes payable and $389,000 in cash accounts held as collateral primarily under the terms of an office operating lease, credit card agreement and automobile leases. Restricted cash at December 31, 2016 includes $10.0 million in a money market account, held in connection with the Company’s notes payable and $425,000 in cash accounts held as collateral primarily under the terms of an office operating lease, credit card agreement and automobile leases.
|
5. |
Fair Value |
The Company held certain assets and liabilities that are required to be measured at fair value on a recurring basis. These assets and liabilities include cash and cash equivalents, restricted cash, contingent consideration and warrant liabilities. ASC 820-10 (“Fair Value Measurement Disclosure”) requires the valuation using a three-tiered approach, which requires that fair value measurements be classified and disclosed in one of three tiers. These tiers are: Level 1, defined as quoted prices in active markets for identical assets or liabilities; Level 2, defined as valuations based on observable inputs other than those included in Level 1, such as quoted prices for similar assets and liabilities in active markets, or other inputs that are observable or can be corroborated by observable input data; and Level 3, defined as valuations based on unobservable inputs reflecting the Company’s own assumptions, consistent with reasonably available assumptions made by other market participants. The Company did not have any transfers of assets and liabilities between Level 1, Level 2, and Level 3 of the fair value hierarchy during the years ended December 31, 2017 and 2016.
For assets and liabilities recorded at fair value, it is the Company’s policy to maximize the use of observable inputs and minimize the use of unobservable inputs when developing fair value measurements, in accordance with the fair value hierarchy. Fair value measurements for assets and liabilities where there exists limited or no observable market data and therefore, are based primarily upon estimates, are often calculated based on the economic and competitive environment, the characteristics of the asset or liability and other factors. Therefore, the results cannot be determined with precision and may not be realized in an actual sale or immediate settlement of the asset or liability. Additionally, there may be inherent weaknesses in any calculation technique, and changes in the underlying assumptions used, including discount rates and estimates of future cash flows, could significantly affect the results of current or future values. The Company utilizes fair value measurements to record fair value adjustments to certain assets and liabilities and to determine fair value disclosures.
57
As prescribed by U.S. GAAP, the Company groups assets and liabilities at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value. An adjustment to the pricing method used within either Level 1 or Level 2 inputs could generate a fair value measurement that effectively falls in a lower level in the hierarchy.
The determination of where an asset or liability falls in the hierarchy requires significant judgment. The Company evaluates its hierarchy disclosures and based on various factors, it is possible that an asset or liability may be classified differently from period to period. However, the Company expects changes in classifications between levels will be rare.
The carrying values of accounts receivable, interest receivable, accounts payable, and certain accrued expenses at December 31, 2017 and 2016, approximate their fair values due to the short-term nature of these items. The Company’s notes payable balance also approximates fair value as of December 31, 2017 and 2016, as the interest rates on the notes payable approximate the rates available to the Company as of these dates.
The following are the major categories of assets measured at fair value on a recurring basis as of December 31, 2017 and 2016, using quoted prices in active markets for identical assets (Level 1); significant other observable inputs (Level 2); and significant unobservable inputs (Level 3):
|
|
|
December 31, 2017 |
|
|||||||||||||
|
|
|
(In thousands) |
|
|||||||||||||
|
|
|
(unaudited) |
|
|||||||||||||
|
Description |
|
Quoted Prices in Active Markets for Identical Assets (Level 1) |
|
|
Significant Other Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
|
Total |
|
||||
|
Assets measured at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
91,217 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
91,217 |
|
|
Restricted cash |
|
|
6,389 |
|
|
|
— |
|
|
|
— |
|
|
|
6,389 |
|
|
Total Assets measured at fair value |
|
$ |
97,606 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
97,606 |
|
|
Liabilities measured at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent consideration |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
12,418 |
|
|
$ |
12,418 |
|
|
Warrant liabilities |
|
|
— |
|
|
|
— |
|
|
$ |
14,090 |
|
|
$ |
14,090 |
|
|
Total liabilities measured at fair value |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
26,508 |
|
|
$ |
26,508 |
|
|
|
|
December 31, 2016 |
|
|||||||||||||
|
|
|
(In thousands) |
|
|||||||||||||
|
Description |
|
Quoted Prices in Active Markets for Identical Assets (Level 1) |
|
|
Significant Other Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
|
Total |
|
||||
|
Assets measured at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
24,165 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
24,165 |
|
|
Restricted cash |
|
|
10,425 |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
10,425 |
|
|
Total Assets measured at fair value |
|
$ |
34,590 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
34,590 |
|
|
Liabilities measured at fair value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Contingent consideration |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
22,800 |
|
|
$ |
22,800 |
|
|
Total liabilities measured at fair value |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
22,800 |
|
|
$ |
22,800 |
|
The Company’s financial liabilities consisted of contingent consideration potentially payable to Sofar related to the Senhance Acquisition in September 2015 (Note 3). This liability is reported as Level 3 as estimated fair value of the contingent consideration related to the acquisition requires significant management judgment or estimation and is calculated using the income approach, using various revenue and cost assumptions and applying a probability to each outcome. The change in fair value of the contingent consideration of $2.0 million for the year ended December 31, 2017 was primarily due to the change in expected timelines for the achievement of milestones, the effect of the passage of time on the fair value measurement and the impact of foreign currency exchange rates. The change in fair value of the contingent consideration of $0.5 million for the year ended December 31, 2016 was primarily due to the Amendment to the Purchase Agreement for the Second Tranche (Note 3), the change in expected timelines for the achievement of milestones, the effect of the passage of time on the fair value measurement and the impact of foreign currency exchange rates. Adjustments associated with the change in fair value of contingent consideration are included in the Company’s consolidated statements of operations and comprehensive loss.
58
On April 28, 2017, the Company sold 24.9 million units (the “Units”), each consisting of one share of the Company’s common stock, par value $0.001 per share (the “Common Stock”), a Series A warrant to purchase one share of Common Stock with an exercise price of $1.00 per share (the “Series A Warrants”), and a Series B warrant to purchase 0.75 shares of Common Stock with an exercise price of $1.00 per share (the “Series B Warrants,” together with the Series A Warrants, the “Warrants”), at an offering price of $1.00 per Unit. Each Series A Warrant was exercisable at any time beginning on the date of issuance, and from time to time thereafter, through and including the first anniversary of the issuance date, unless terminated earlier as provided in the Series A Warrant. Receipt of 510(k) clearance for the Senhance System on October 13, 2017, triggered the acceleration of the expiration date of the Series A Warrants to October 31, 2017 (see Note 19). Each Series B Warrant may be exercised at any time beginning on the date of issuance and from time to time thereafter through and including the fifth anniversary of the issuance date.
The fair value of the Series A Warrants of $2.5 million at the date of issuance was estimated using the Black-Scholes Merton model which used the following inputs: term of 1 year, risk free rate of 1.07%, no dividends, volatility of 73.14%, and share price of $0.65 per share based on the trading price of the Company’s common stock. The fair value of the Series B Warrants of $6.2 million at the date of issuance was estimated using the Black-Scholes Merton model which used the following inputs: term of 5 years, risk free rate of 1.81%, no dividends, volatility of 73.14%, and share price of $0.65 per share based on the trading price of the Company’s common stock. All Series A Warrants were exercised as of December 31, 2017. The fair value of the Series B Warrants of $14.1 million at December 31, 2017 was estimated using the Monte Carlo valuation model which used the following inputs: term of 4.33 years, risk free rate of 2.13%, volatility of 80.6%, share price of $1.93 per share based on the trading price of the Company’s common stock, and probability of additional financing in 2018 of 25% and 2019 of 75%. The change in fair value of warrants for the year ended December 31, 2017 of $83.7 million was included in the Company’s consolidated statements of operations and comprehensive loss.
The following table presents quantitative information about the inputs and valuation methodologies used for the Company’s fair value measurements classified in Level 3 as of December 31, 2016 and 2017:
|
|
|
Valuation Methodology |
|
Significant Unobservable Input |
|
Weighted Average (range, if applicable) |
|
Contingent consideration |
|
Probability weighted income approach |
|
Milestone dates |
|
2018 to 2020 |
|
|
|
|
|
Discount rate Probability of occurrence |
|
7.5% to 12% 100% |
The following table summarizes the change in fair value, as determined by Level 3 inputs, for all assets and liabilities using unobservable Level 3 inputs for the years ended December 31, 2017, 2016 and 2015:
|
|
|
Fair Value Measurement at Reporting Date (Level 3) |
|
|||||
|
|
|
(In thousands) |
|
|||||
|
|
|
Common stock |
|
|
Contingent |
|
||
|
|
|
warrants |
|
|
consideration |
|
||
|
Balance at December 31, 2014 |
|
$ |
— |
|
|
$ |
— |
|
|
Additions for contingent consideration |
|
|
— |
|
|
|
23,900 |
|
|
Change in fair value |
|
|
— |
|
|
|
(400 |
) |
|
Balance at December 31, 2015 |
|
|
— |
|
|
|
23,500 |
|
|
Payment for contingent consideration |
|
|
— |
|
|
|
(1,182 |
) |
|
Change in fair value |
|
|
— |
|
|
|
482 |
|
|
Balance at December 31, 2016 |
|
|
— |
|
|
|
22,800 |
|
|
Issuance of common stock in exchange for contingent consideration |
|
|
— |
|
|
|
(5,227 |
) |
|
Issuance of warrants |
|
|
8,715 |
|
|
|
— |
|
|
Payment for contingent consideration |
|
|
— |
|
|
|
(7,181 |
) |
|
Exercise of warrants |
|
|
(78,359 |
) |
|
|
— |
|
|
Change in fair value |
|
|
83,734 |
|
|
|
2,026 |
|
|
Balance at December 31, 2017 |
|
|
14,090 |
|
|
$ |
12,418 |
|
|
Current portion |
|
|
— |
|
|
|
719 |
|
|
Long-term portion |
|
|
14,090 |
|
|
|
11,699 |
|
|
Balance at December 31, 2017 |
|
$ |
14,090 |
|
|
$ |
12,418 |
|
59
|
6. |
Accounts Receivable, Net |
The following table presents the components of accounts receivable:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Gross accounts receivable |
|
$ |
1,609 |
|
|
$ |
960 |
|
|
Allowance for uncollectible accounts |
|
|
(73 |
) |
|
|
(73 |
) |
|
Total accounts receivable, net |
|
$ |
1,536 |
|
|
$ |
887 |
|
|
Short-term portion |
|
$ |
1,536 |
|
|
$ |
621 |
|
|
Long-term portion |
|
|
— |
|
|
|
266 |
|
|
Total accounts receivable |
|
$ |
1,536 |
|
|
$ |
887 |
|
|
7. |
Inventories |
The components of inventories are as follows:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Finished goods |
|
$ |
4,432 |
|
|
$ |
4,698 |
|
|
Raw materials |
|
|
6,385 |
|
|
|
3,185 |
|
|
Total inventories |
|
$ |
10,817 |
|
|
$ |
7,883 |
|
As disclosed in Note 17, the Company executed a restructuring plan in May 2016 and wrote down inventory related to the SurgiBot System. The write down of inventory of $2.6 million is included in the accompanying consolidated statement of operations and comprehensive loss for the year ended December 31, 2016. There were no such write-downs for the year ended December 31, 2017 or 2015.
|
8. |
Other Current Assets |
The following table presents the components of other current assets:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Prepaid expenses |
|
$ |
1,519 |
|
|
$ |
2,186 |
|
|
Advances to vendors |
|
|
6,403 |
|
|
|
1,806 |
|
|
Other receivables |
|
|
1,422 |
|
|
|
1,343 |
|
|
Total |
|
$ |
9,344 |
|
|
$ |
5,335 |
|
|
9. |
Property and Equipment |
Property and equipment consisted of the following:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Machinery, manufacturing and demonstration equipment |
|
$ |
10,866 |
|
|
$ |
7,579 |
|
|
Computer equipment |
|
|
2,187 |
|
|
|
2,124 |
|
|
Furniture |
|
|
598 |
|
|
|
614 |
|
|
Leasehold improvements |
|
|
2,237 |
|
|
|
2,028 |
|
|
Total property and equipment |
|
|
15,888 |
|
|
|
12,345 |
|
|
Accumulated depreciation and amortization |
|
|
(9,218 |
) |
|
|
(6,573 |
) |
|
Property and equipment, net |
|
$ |
6,670 |
|
|
$ |
5,772 |
|
60
As disclosed in Note 17, the Company executed a restructuring plan in May 2016 and disposed of certain long-lived assets, primarily equipment and fixtures related to the SurgiBot System. The disposal of long-lived assets of $1.0 million is included as a component of restructuring and other charges in the accompanying consolidated statement of operations and comprehensive loss for the year ended December 31, 2016. There were no such disposals for the year ended December 31, 2017 or 2015.
Depreciation expense was $2,486,000, $1,942,000 and $1,248,000, for the years ended December 31, 2017, 2016 and 2015, respectively.
|
10. |
Goodwill, In-Process Research and Development and Intellectual Property |
Goodwill
Goodwill of $93.8 million was recorded in connection with the Merger, as described in Note 1, and goodwill of $38.3 million was recorded in connection with the Senhance Acquisition, as described in Note 3. The carrying value of goodwill and the change in the balance for the years ended December 31, 2017 and 2016 is as follows:
|
|
|
Goodwill |
|
|
|
|
|
(In thousands) |
|
|
|
Balance at December 31, 2015 |
|
$ |
130,869 |
|
|
Foreign currency translation impact |
|
|
(388 |
) |
|
Impairment loss |
|
|
(61,784 |
) |
|
Balance at December 31, 2016 |
|
|
68,697 |
|
|
Foreign currency translation impact |
|
|
2,671 |
|
|
Balance at December 31, 2017 |
|
$ |
71,368 |
|
Accumulated impairment of goodwill as of December 31, 2017 and 2016 was $61.8 million.
The Company performs an annual impairment test of goodwill at December 31, or more frequently if events or changes in circumstances indicates that the carrying value of the Company’s one reporting unit may not be recoverable. During the second quarter of 2016, the FDA notified the Company that the SurgiBot System did not meet the criteria for substantial equivalency, negatively impacting the Company’s market capitalization, and warranting an interim two-step quantitative impairment test. Prior to adopting ASU 2017-04 as of the beginning of fiscal year 2017, goodwill was tested for impairment using a two-step approach. In the first step, the fair value of the reporting unit was determined and compared to the reporting unit's carrying value, including goodwill. If the fair value of the reporting unit was less than its carrying value, the second step of the goodwill impairment test was performed to measure the amount of impairment, if any. In the second step, the fair value of the reporting unit was allocated to the assets and liabilities of the reporting unit as if it had been acquired in a business combination and the purchase price was equivalent to the fair value of the reporting unit. The excess of the fair value of the reporting unit over the amounts assigned to its assets and liabilities was referred to as the implied fair value of goodwill. The implied fair value of the reporting unit's goodwill was then compared to the actual carrying value of goodwill. If the implied fair value of goodwill was less than the carrying value of goodwill, an impairment loss was recognized for the difference. ASU 2017-04 removes Step 2 of the goodwill impairment test.
The Company determined the fair value of the reporting unit using a discounted cash flow analysis derived from the Company’s long-term plans. The fair value of the reporting unit was corroborated using market prices for TransEnterix, Inc. The inputs used to determine the fair values were classified as Level 3 in the fair value hierarchy. Based on the impairment test, the Company recorded goodwill impairment of $61.8 million during the second quarter of 2016. No impairment was recorded as of December 31, 2017 or 2015.
The Company performed a qualitative assessment during the annual impairment review for fiscal 2016 as of December 31, 2016 and concluded that it is not more likely than not that the fair value of the Company’s single reporting unit is less than its carrying amount. Therefore, the two-step goodwill impairment test for the reporting unit was not necessary at December 31, 2016. During the second quarter of 2017, the Company’s stock price experienced a significant decline. The Company performed a Step 1 goodwill impairment test as of the second quarter and determined that no charge to goodwill for impairment was required during the second quarter of 2017. As of December 31, 2017, the Company elected to bypass the qualitative assessment and calculated the fair value of the Company’s reporting unit, which exceeded the carrying amount. Accordingly, no charge for goodwill impairment was required as of December 31, 2017.
61
In-Process Research and Development
As described in Note 3, on September 21, 2015, the Company acquired all of the assets related to the Senhance System and recorded $17.1 million of IPR&D. The estimated fair value of the IPR&D was determined using a probability-weighted income approach, which discounts expected future cash flows to present value. The projected cash flows were based on certain key assumptions, including estimates of future revenue and expenses, taking into account the stage of development of the technology at the acquisition date and the time and resources needed to complete development. The Company used a discount rate of 45% and cash flows that have been probability adjusted to reflect the risks of product commercialization, which the Company believes are appropriate and representative of market participant assumptions.
On October 13, 2017, upon receipt of regulatory clearance to commercialize the products associated with the IPR&D assets in the United States, the assets were deemed definite-lived, transferred to developed technology and are amortized based on their estimated useful lives.
The carrying value of the Company’s IPR&D assets and the change in the balance for the years ended December 31, 2016 and 2017 is as follows:
|
|
|
In-Process Research and Development |
|
|
|
|
|
(In thousands) |
|
|
|
Balance at December 31, 2015 |
|
|
16,511 |
|
|
Foreign currency translation impact |
|
|
(591 |
) |
|
Balance at December 31, 2016 |
|
|
15,920 |
|
|
Foreign currency translation impact |
|
|
1,993 |
|
|
Transfer to developed technology |
|
|
(17,913 |
) |
|
Balance at December 31, 2017 |
|
$ |
— |
|
Intellectual Property
In 2009, the Company purchased certain patents from an affiliated company for $5.0 million in cash and concurrently terminated a license agreement related to the patents. The patent expiration dates begin in 2027. In addition, as described in Note 3, on September 21, 2015, the Company acquired all of the developed technology related to the Senhance System and recorded $48.5 million of intellectual property. The estimated fair value of the intellectual property was determined using a probability-weighted income approach, which discounts expected future cash flows to present value. The projected cash flows were based on certain key assumptions, including estimates of future revenue and expenses, taking into account the stage of development of the technology at the acquisition date and the time and resources needed to complete development. The Company used a discount rate of 45% and cash flows that have been probability adjusted to reflect the risks of product commercialization, which the Company believes are appropriate and representative of market participant assumptions.
In November 2016, the Company agreed to enter into a technology and patents purchase agreement with Sofar to acquire from Sofar certain technology and intellectual property rights related to the Senhance Acquisition, and formerly licensed by the Company. The technology and patents were acquired in 2017 at an acquisition price of $400,000.
As disclosed in Note 17, the Company executed a restructuring plan in May 2016 and wrote-off certain intellectual property consisting of patents related to the SurgiBot System. The write-off of intellectual property of $1.6 million is included as a component of restructuring and other charges in the accompanying consolidated statement of operations and comprehensive losses for the year ended December 31, 2016. There were no such write offs for the year ended December 31, 2015 or 2017.
62
The components of gross intellectual property, accumulated amortization, and net intellectual property as of December 31, 2017 and 2016 are as follows:
|
|
|
December 31, 2017 |
|
|
|
December 31, 2016 |
|
||||||||||||||||||||||||||||||
|
|
|
(In thousands) |
|
|
|
(In thousands) |
|
||||||||||||||||||||||||||||||
|
|
|
Gross Carrying Amount |
|
|
Accumulated Amortization |
|
|
Foreign currency translation impact |
|
|
Net Carrying Amount |
|
|
|
Gross Carrying Amount |
|
|
Accumulated Amortization |
|
|
Foreign currency translation impact |
|
|
Write-off |
|
|
Net Carrying Amount |
|
|||||||||
|
Patents |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
$ |
5,000 |
|
|
$ |
(3,438 |
) |
|
$ |
— |
|
|
$ |
(1,562 |
) |
|
$ |
— |
|
|
Developed technology |
|
|
66,413 |
|
|
|
(19,724 |
) |
|
|
5,529 |
|
|
|
52,218 |
|
|
|
|
48,500 |
|
|
|
(8,458 |
) |
|
|
(2,952 |
) |
|
|
— |
|
|
|
37,090 |
|
|
Technology and patents purchased |
|
|
400 |
|
|
|
(30 |
) |
|
|
50 |
|
|
|
420 |
|
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total intellectual property |
|
$ |
66,813 |
|
|
$ |
(19,754 |
) |
|
$ |
5,579 |
|
|
$ |
52,638 |
|
|
|
$ |
53,500 |
|
|
$ |
(11,896 |
) |
|
$ |
(2,952 |
) |
|
$ |
(1,562 |
) |
|
$ |
37,090 |
|
The weighted average remaining useful life of the developed technology and technology and patents purchased was 4.8 years and 9.3 years, respectively as of December 31, 2017.
The estimated future amortization expense of intangible assets as of December 31, 2017 is as follows:
|
|
|
Years ending December 31, |
|
|
|
|
|
(In thousands) |
|
|
|
2018 |
|
$ |
10,552 |
|
|
2019 |
|
|
10,552 |
|
|
2020 |
|
|
10,552 |
|
|
2021 |
|
|
10,552 |
|
|
2022 |
|
|
10,210 |
|
|
Thereafter |
|
|
220 |
|
|
Total |
|
$ |
52,638 |
|
|
11. |
Income Taxes |
The components for the income tax expense (benefit) are as follows for the years ended December 31 (in thousands):
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Current income taxes |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
State |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Deferred income taxes |
|
|
|
|
|
|
|
|
|
|
|
|
|
Federal |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
State |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Foreign |
|
|
(3,300 |
) |
|
|
(5,523 |
) |
|
|
(1,024 |
) |
|
Total income tax expense (benefit) |
|
$ |
(3,300 |
) |
|
$ |
(5,523 |
) |
|
$ |
(1,024 |
) |
The United States and foreign components of loss from operations before taxes are as follows for the years ended December 31 (in thousands):
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
(124,418 |
) |
|
$ |
(88,624 |
) |
|
$ |
(44,438 |
) |
|
Foreign |
|
|
(23,678 |
) |
|
|
(36,879 |
) |
|
|
(3,534 |
) |
|
Total loss from operations before taxes |
|
$ |
(148,096 |
) |
|
$ |
(125,503 |
) |
|
$ |
(47,972 |
) |
63
Significant components of the Company’s deferred tax assets consist of the following at December 31 (in thousands):
|
|
|
2017 |
|
|
2016 |
|
||
|
Noncurrent deferred tax assets: |
|
|
|
|
|
|
|
|
|
Stock-based compensation |
|
|
2,216 |
|
|
|
2,300 |
|
|
Inventory |
|
|
375 |
|
|
|
204 |
|
|
Accrued expenses and other |
|
|
637 |
|
|
|
564 |
|
|
Research credit carryforward |
|
|
5,540 |
|
|
|
4,970 |
|
|
Fixed assets |
|
|
450 |
|
|
|
557 |
|
|
Capitalized start-up costs and other intangibles |
|
|
2,130 |
|
|
|
3,586 |
|
|
Net operating loss carryforwards |
|
|
64,300 |
|
|
|
82,298 |
|
|
|
|
|
75,648 |
|
|
|
94,479 |
|
|
Valuation allowance |
|
|
(71,520 |
) |
|
|
(91,885 |
) |
|
Net noncurrent deferred tax asset |
|
|
4,128 |
|
|
|
2,594 |
|
|
Noncurrent deferred tax liabilities |
|
|
|
|
|
|
|
|
|
Fixed assets |
|
|
(334 |
) |
|
|
(292 |
) |
|
Purchase accounting intangibles |
|
|
(12,183 |
) |
|
|
(12,699 |
) |
|
Net noncurrent deferred tax liability |
|
|
(12,517 |
) |
|
|
(12,991 |
) |
|
Net deferred tax asset (liability) |
|
$ |
(8,389 |
) |
|
$ |
(10,397 |
) |
At December 31, 2017 and 2016, the Company has provided a full valuation allowance against its net deferred assets in the U.S. Luxembourg, and Swiss tax jurisdiction, since realization of these benefits is not more likely than not. The valuation allowance decreased approximately $20.4 million from the prior year. At December 31, 2017, the Company had federal and state net operating loss tax carryforwards of approximately $254.6 million and $204.8 million, respectively. These net operating loss carryforwards expire in various amounts starting in 2027 and 2022, respectively. At December 31, 2017, the Company had federal research credit carryforwards in the amount of $5.5 million. These carryforwards begin to expire in 2027. The utilization of the federal net operating loss carryforwards and credit carryforwards will depend on the Company’s ability to generate sufficient taxable income prior to the expiration of the carryforwards. In addition, the maximum annual use of net operating loss and research credit carryforwards is limited in certain situations where changes occur in stock ownership.
At December 31, 2017, the Company had foreign operating loss carryforwards in Italy of approximately $15.6 million, which can be carried forward indefinitely; foreign operating loss carryforwards in Luxembourg of approximately $0.2 million, which can be carried forward indefinitely; and foreign operating loss carryforwards in Switzerland of approximately $16.7 million, which begin to expire in 2023.
As of December 31, 2017 the Company has adopted ASU 2016-09 which is effective for public companies for annual periods beginning after December 15, 2016. The ASU requires all excess tax benefits and tax deficiencies to be recognized as income tax expense or benefit in the income statement in the year in which they occur. As such, the Company has grossed up its net operating loss deferred tax asset to include all excess tax benefits as of December 31, 2017.
The Company has evaluated its tax positions to consider whether it has any unrecognized tax benefits. As of December 31, 2017, the Company had gross unrecognized tax benefits of approximately $1.2 million. Of the total, none would reduce the Company’s effective tax rate if recognized. The Company does not anticipate a significant change in total unrecognized tax benefits or the Company’s effective tax rate due to the settlement of audits or the expiration of statutes of limitations within the next twelve months. Furthermore, the Company does not expect any cash settlement with the taxing authorities as a result of these unrecognized tax benefits as the Company has sufficient unutilized carryforward attributes to offset the tax impact of these adjustments.
The following is a tabular reconciliation of the Company’s change in gross unrecognized tax positions at December 31 (in thousands):
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Beginning balance |
|
$ |
1,048 |
|
|
$ |
862 |
|
|
$ |
606 |
|
|
Gross increases for tax positions related to current periods |
|
|
143 |
|
|
|
186 |
|
|
|
256 |
|
|
Gross increases for tax positions related to prior periods |
|
|
11 |
|
|
|
— |
|
|
|
— |
|
|
Ending balance |
|
$ |
1,202 |
|
|
$ |
1,048 |
|
|
$ |
862 |
|
The Company recognizes interest and penalties related to uncertain tax positions in the provision for income taxes. As of December 31, 2017 and 2016, the Company had no accrued interest or penalties related to uncertain tax positions.
64
The Company has analyzed its filing positions in all significant federal, state, and foreign jurisdictions where it is required to file income tax returns, as well as open tax years in these jurisdictions. With few exceptions, the Company is no longer subject to United States Federal, state, and local tax examinations by tax authorities for years before 2014, although carryforward attributes that were generated prior to 2014 may still be adjusted upon examination by the taxing authorities if they either have been or will be used in a future period. No income tax returns are currently under examination by taxing authorities. The North Carolina Department of Revenue recently completed an examination of the North Carolina state income tax returns for the 2013, 2014, and 2015 tax years for the Company’s subsidiary, TransEnterix Surgical, Inc. No material changes were made as a result of the audit, and those tax years are now effectively settled.
Taxes computed at the then-current statutory federal income tax rate of 34% are reconciled to the provision for income taxes as follows for the years ended December 31:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||||||||||||||
|
|
|
|
|
|
|
% of Pretax |
|
|
|
|
|
|
% of Pretax |
|
|
|
|
|
|
% of Pretax |
|
|||
|
|
|
Amount |
|
|
Earnings |
|
|
Amount |
|
|
Earnings |
|
|
Amount |
|
|
Earnings |
|
||||||
|
United States federal tax at statutory rate |
|
$ |
(50,352 |
) |
|
|
34.0 |
% |
|
$ |
(42,671 |
) |
|
|
34.0 |
% |
|
$ |
(16,311 |
) |
|
|
34.0 |
% |
|
State taxes (net of deferred benefit) |
|
|
(4,663 |
) |
|
|
3.1 |
% |
|
|
(2,487 |
) |
|
|
2.0 |
% |
|
|
(1,121 |
) |
|
|
2.3 |
% |
|
Nondeductible expenses |
|
|
466 |
|
|
|
(0.3 |
%) |
|
|
667 |
|
|
|
(0.5 |
%) |
|
|
1,797 |
|
|
|
(3.7 |
%) |
|
Change in fair market value of contingent consideration |
|
|
777 |
|
|
|
(0.5 |
%) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Warrant remeasurement and financing costs |
|
|
32,348 |
|
|
|
(21.8 |
%) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Research & Development credits |
|
|
(712 |
) |
|
|
0.5 |
% |
|
|
(922 |
) |
|
|
0.7 |
% |
|
|
(1,281 |
) |
|
|
2.7 |
% |
|
Change in unrecognized tax benefits |
|
|
142 |
|
|
|
(0.1 |
%) |
|
|
186 |
|
|
|
(0.1 |
%) |
|
|
256 |
|
|
|
(0.5 |
%) |
|
Foreign tax rate differential |
|
|
3,619 |
|
|
|
(2.4 |
%) |
|
|
3,969 |
|
|
|
(3.2 |
%) |
|
|
175 |
|
|
|
(0.4 |
%) |
|
Goodwill impairment |
|
|
— |
|
|
|
0.0 |
% |
|
|
20,816 |
|
|
|
(16.6 |
%) |
|
|
— |
|
|
|
— |
|
|
Change in enacted tax rates and other, net |
|
|
35,440 |
|
|
|
(24.1 |
%) |
|
|
(1,069 |
) |
|
|
0.8 |
% |
|
|
532 |
|
|
|
(1.2 |
%) |
|
Change in valuation allowance |
|
|
(20,365 |
) |
|
|
13.8 |
% |
|
|
15,988 |
|
|
|
(12.7 |
%) |
|
|
14,929 |
|
|
|
(31.1 |
%) |
|
Income tax benefit |
|
$ |
(3,300 |
) |
|
|
2.2 |
% |
|
$ |
(5,523 |
) |
|
|
4.4 |
% |
|
$ |
(1,024 |
) |
|
|
2.1 |
% |
On December 22, 2017, the Tax Cuts and Jobs Act (“Tax Legislation”) was enacted into law, which reduced the US federal corporate income tax rate to 21% for tax years beginning after December 31, 2017. As a result of the newly enacted tax rate, the Company adjusted its U.S. deferred tax assets as of December 31, 2017, by applying the new 21% rate, which resulted in a decrease to the deferred tax assets and a corresponding decrease to the valuation allowance of approximately $36.1 million. The newly enacted tax rate had no impact on deferred tax liabilities as they do not relate to U.S. amounts.
The Tax Legislation also implements a territorial tax system. Under the territorial tax system, in general, the Company's foreign earnings will no longer be subject to tax in the U.S. As part of transition to the territorial tax system the Tax Legislation includes a mandatory deemed repatriation of all undistributed foreign earnings that are subject to a U.S. income tax. The Company estimates that the deemed repatriation will not result in any additional U.S. income tax liability as it estimates it currently has no undistributed foreign earnings.
The SEC staff issued SAB 118 which will allow the Company to record provisional amounts related to accounting for the Tax Legislation during a measurement period which is similar to the measurement period used when accounting for business combinations. The Company is following the guidance set forth by SAB 118 and any amounts calculated are provisional estimates and will be reevaluated as more information or guidance becomes available. The Company will continue to assess the impact of the Tax Legislation on its business and consolidated financial statements.
65
The following table presents the components of accrued expenses:
|
|
|
December 31, |
|
|
December 31, |
|
||
|
|
|
2017 |
|
|
2016 |
|
||
|
|
|
(In thousands) |
|
|||||
|
Compensation and benefits |
|
$ |
4,533 |
|
|
$ |
2,328 |
|
|
Taxes and other assessments |
|
|
3,192 |
|
|
|
2,676 |
|
|
Consulting and other vendors |
|
|
1,414 |
|
|
|
1,428 |
|
|
Deferred rent |
|
|
595 |
|
|
|
323 |
|
|
Other |
|
|
504 |
|
|
|
49 |
|
|
Legal and professional fees |
|
|
386 |
|
|
|
243 |
|
|
Interest and final payment fee |
|
|
309 |
|
|
|
1,000 |
|
|
Royalties |
|
|
41 |
|
|
|
159 |
|
|
Total |
|
$ |
10,974 |
|
|
$ |
8,206 |
|
|
13. |
Notes Payable |
On May 10, 2017, the Company and its domestic subsidiaries, as co-borrowers, entered into a Loan and Security Agreement (the “Innovatus Loan Agreement”) with Innovatus Life Sciences Lending Fund I, LP, as Lender and Collateral Agent (the “Lender”). Under the Innovatus Loan Agreement, the Lender agreed to make certain term loans in the aggregate principal amount of up to $17,000,000. Funding of the first $14,000,000 tranche occurred on May 10, 2017. The Company will be eligible to draw on the Second Tranche of $3,000,000 upon achievement of certain milestones, including Senhance Clearance (as defined below). So long as the Company meets each Interest-Only Milestone (as defined below), the Company is entitled to make interest-only payments for up to twenty-four (24) months. At the end of the interest-only period, the Company will be required to repay the term loans over a two-year period, based on a twenty-four (24) month amortization schedule, with a final maturity date occurring on the fourth anniversary of the initial funding date. However, the interest-only period will end if the Company fails to meet any Interest-Only Milestone. Commencing on the first day of the month following such failure to achieve an Interest-Only Milestone, the Company will be required to repay the term loans over a two year period, based on a twenty-four (24) month amortization schedule. The Interest-Only Milestones require the Company to (i) achieve certain twelve month revenue targets, measured quarterly, commencing with the quarter ending March 31, 2018, (ii) meet a minimum capital raising threshold through the sale and issuance of equity securities during the period from April 10, 2017 through May 31, 2018 and (iii) obtain clearance for commercialization of the Senhance System by the FDA (“Senhance Clearance”) by May 30, 2018 (each such milestone, an “Interest-Only Milestone”).
The term loans bear interest at a fixed rate equal to 11% per annum, of which 2.5% can be paid in-kind and added to the outstanding principal amount of the term loans until the earlier of (i) the first anniversary following the funding date and (ii) the Company’s failure to achieve an Interest-Only Milestone. The Company will be required to repay the term loans if they are accelerated following an event of default. In addition, the Company is permitted to prepay the term loans in full at any time upon five (5) business days’ written notice to the Lender. Upon the earliest to occur of the maturity date, acceleration of the term loan, or prepayment of the term loan, the Company is required to make a final payment equal to the total term loan commitment multiplied by four percent (4%) (the “Final Fee”); provided, however, that in the event the Company refinances its obligations with the Lender after Senhance Clearance, no Final Fee or Prepayment Fee (as defined below) will be due thereunder; and provided, further, that if the Company elects to refinance its obligations prior to the funding of the Second Tranche, the Final Fee with respect to the Second Tranche shall be paid in full on the date of such refinancing. Any prepayment of the term loans in full, whether mandatory or voluntary, must include (i) the Final Fee, (ii) interest at the default rate (which is the rate otherwise applicable plus five percent (5%)) with respect to any amounts past due, (iii) the Lender’s expenses and all other obligations that are due and payable to the Lender and (iv) a prepayment fee of three percent (3%) if the term loan is paid in full on or before the first anniversary of the effective date, two percent (2%) if paid off after the first anniversary but on or before the second anniversary of the effective date and one percent (1%) if paid off after the second anniversary but on or before the third anniversary of the effective date (the “Prepayment Fee”).
In connection with the funding, the Company paid a facility fee of $170,000 on the date of funding of the first tranche and incurred additional debt issuance costs of approximately $1.2 million, recorded as debt discount. In addition, the Company issued warrants to the Lender to purchase shares of the Company’s common stock. Additional warrants will be issued on the funding date of each subsequent tranche and will expire five (5) years from such issue date. The warrants issued in connection with funding of the first tranche entitle the Lender to purchase up to 1,244,746 shares of the Company’s common stock at an exercise price of $1.00 per share. The Company estimated the fair value of the warrants to be $300,000. The value of the warrants was classified as equity and recorded as a discount to the loan. The debt discount is amortized as interest expense using the effective interest method over the life of the loan. As of December 31, 2017, the unamortized debt discount was $209,000.
66
The Company’s obligations under the Innovatus Loan Agreement are secured by a security interest in all of the assets of the Company and its current and future domestic and material foreign subsidiaries, including a security interest in the intellectual property. The Innovatus Loan Agreement contains customary representations and covenants that, subject to exceptions, restrict the Company’s ability to do the following things: declare dividends or redeem or repurchase equity interests; incur additional liens; make loans and investments; incur additional indebtedness; engage in mergers, acquisitions, and asset sales; transact with affiliates; undergo a change in control; add or change business locations; and engage in businesses that are not related to its existing business. Under the terms of the Innovatus Loan Agreement, the Company is required to maintain minimum unrestricted cash in an amount equal to (x) six million dollars ($6,000,000), at all times prior to Senhance Clearance; and (y) at all times thereafter, the least of (i) $6,000,000, (ii) the Company’s trailing three (3) months’ cash used to fund operating activities, as determined as of the most recent month end and (iii) the then outstanding principal amount of the term loans, together with accrued but unpaid interest.
As of December 31, 2017 future principal payments, including paid in-kind interest, under the Innovatus Loan Agreement are as follows:
|
Years ending December 31, |
|
|
|
|
|
(In thousands) |
|
|
|
|
|
2018 |
|
$ |
4,788 |
|
|
2019 |
|
|
7,181 |
|
|
2020 |
|
|
2,394 |
|
|
Total |
|
$ |
14,363 |
|
In connection with its entrance into the Innovatus Loan Agreement, the Company repaid its existing credit facility with Silicon Valley Bank and Oxford Finance LLC (the “Prior Lenders”), which loan and security agreement, as subsequently amended and restated is referred to as the “SVB Loan Agreement.” The Company recognized a loss of $308,000 on the extinguishment of notes payable for the year ended December 31, 2017, which is included in interest expense on the consolidated statements of operations and comprehensive loss. The Company paid $1.3 million in final payment obligations and $255,000 in facility fees under the SVB Loan Agreement upon repayment.
The SVB Loan Agreement was initially entered into on January 17, 2012. In connection with the Merger, the Company assumed and became the borrower under the SVB Loan Agreement.
On August 14, 2015, the Company entered into the First Amendment to the SVB Loan Agreement (the “First Amendment”) with the Prior Lenders. The first tranche of the First Amendment increased the Company’s borrowings at August 14, 2015 from $10,000,000 to $20,000,000. The First Amendment allowed for interest-only payments at 7.5% per annum through April 30, 2016 and had a maturity date of October 1, 2018.
On September 18, 2015, in connection with entry into the Purchase Agreement with Sofar S.p.A. (see Note 3 for a description of the related transactions), the Company and the Prior Lenders entered into the Consent and Second Amendment (the “Second Amendment”) to the SVB Loan Agreement. The Second Amendment modified the period in which the Company could make interest-only payments at 7.5% per annum on the term loans until January 31, 2016. The Second Amendment had a maturity date of July 1, 2018.
In addition, in connection with the borrowings under the SVB Loan Agreement, the Company issued warrants to the Prior Lenders to purchase shares of the Company’s common stock amounting to an aggregate of 430,815 warrants under the SVB Loan Agreement. The warrants expire seven years from their respective issue date.
In accordance with ASC 470-50 Debt – Modifications and Extinguishments, it was determined that a debt refinancing of the SVB Loan Agreement on September 26, 2014, was considered to be a debt modification. Accordingly, the Company recorded approximately $129,000 of debt discount, consisting of the $75,000 facility fee and the relative fair value of warrants on the issue date of $54,000. Additionally, approximately $30,000 of legal fees was recorded as a result of the transaction. The debt discount and deferred financing costs were amortized over the life of the new debt agreement using the effective interest method into interest expense, net, until the debt was extinguished in May 2017.
In accordance with ASC 470-50 Debt – Modifications and Extinguishments, it was determined that debt refinancings of the SVB Loan Agreement on August 14, 2015, September 18, 2015, April 19, 2016 and September 7, 2016 were considered to be debt modifications. The Company recorded a debt discount of approximately $210,000 for these amendments. Accordingly, the unamortized debt discount was presented as a reduction of the related debt liability in the Company’s consolidated balance sheet. The debt discount was amortized over the life of the new debt agreement using the effective interest method into interest expense, net, until the debt was extinguished in May 2017.
67
In connection with the issuance of the notes payable and amendments under the SVB Loan Agreement, TransEnterix Surgical incurred approximately $371,000 in debt issuance costs paid to the Prior Lenders and third parties and $280,000 in debt issuance costs related to issuance of warrants to the Prior Lenders. The unamortized balance of $107,000 as of December 31, 2016, was amortized using the effective interest method, until the debt was extinguished in May 2017. At the time of extinguishment in May 2017, $63,000 of unamortized debt issuance costs were included in the loss on extinguishment of notes payable.
|
14. |
Stock-Based Compensation |
The Company’s stock-based compensation plans include the TransEnterix, Inc. Amended and Restated Incentive Compensation Plan, previously named the TransEnterix, Inc. 2007 Incentive Compensation Plan (the “Plan”), as well as options outstanding under the TransEnterix, Inc. Stock Option Plan (the “2006 Plan”). As part of the Merger, options outstanding, whether vested or unvested, under the 2006 Plan were adjusted by the Exchange Ratio of 1.1533, and assumed by the Company concurrent with the closing of the Merger.
The Plan was initially approved by the majority of the stockholders on November 13, 2007. The Plan was amended on June 19, 2012 to increase the number of shares of common stock available for issuance to 1,000,000 and was amended on October 29, 2013 to (a) increase the number of shares of common stock authorized for issuance under the Plan from 1,000,000 shares of common stock to 4,940,000 shares of common stock, (b) increase the per-person award limitations for options or stock appreciation rights from 200,000 to 1,000,000 shares and for restricted stock, deferred stock, performance shares and/or other stock-based awards from 100,000 to 500,000 shares, and (c) change the name of the Plan to reflect the Merger-related change. The Plan was again amended on May 7, 2015 to (i) increase the number of shares reserved for issuance under the Plan to 11,940,000 shares; (ii) extend the term of the Plan until May 7, 2025; and (iii) make other changes and updates to the Plan and was further amended in October 2015 to add French Sub-Plan amendments applicable to awards made to France-based employees. The Plan was further amended on June 8, 2016 to (a) approve an increase in the number of shares reserved for issuance under the Plan to 18,940,000 shares and (b) establish maximum equity award limits for initial awards and annual awards to non-employee directors. The Plan was subsequently amended as of May 25, 2017, increasing the number of shares of Common Stock authorized under the Plan to 25,940,000.
The October 2013, May 2015, June 2016 and May 2017 amendments were approved by the Board of Directors and stockholders; the French Sub-Plan was approved by the Board of Directors. Under the Plan, which is administered by the Compensation Committee, the Company may grant stock options, stock appreciation rights, restricted stock and/or deferred stock to employees, officers, directors, consultants and vendors. The exercise price of stock options or stock appreciation rights may not be less than the fair market value of the Company’s shares at the date of grant. Additionally, no stock options or stock appreciation rights granted under the Plan may have a term exceeding ten years.
The 2006 Plan was adopted and approved by stockholders in September 2006 and provided for the granting of up to 80,000 stock options to employees, directors, and consultants. Under the 2006 Plan, both employees and non-employees were eligible for such stock options. In 2009, the 2006 Plan was amended to increase the total options pool to 1,110,053. In 2011, the 2006 Plan was amended to increase the total options pool to 3,378,189. The amendments were approved by the Board of Directors and stockholders. The Board of Directors had the authority to administer the plan and determine, among other things, the exercise price, term and dates of the exercise of all options at their grant date. Under the 2006 Plan, options become vested generally over four years, and expire not more than 10 years after the date of grant. As part of the Merger, options outstanding under the 2006 Plan were adjusted by the Conversion Ratio, and remain in existence as options of TransEnterix.
During the years ended December 31, 2017, 2016 and 2015, the Company recognized $7,078,000, $5,033,000 and $3,311,000, respectively, of stock-based compensation expense, including stock options and restricted stock units.
The Company recognizes as expense, the grant-date fair value of stock options and other stock based compensation issued to employees and non-employee directors over the requisite service periods, which are typically the vesting periods. The Company uses the Black-Scholes-Merton model to estimate the fair value of its stock-based payments. The volatility assumption used in the Black-Scholes-Merton model is based on the calculated historical volatility based on an analysis of reported data for a peer group of companies as well as the Company’s historical volatility. The expected term of options granted by the Company has been determined based upon the simplified method, because the Company does not have sufficient historical information regarding its options to derive the expected term. Under this approach, the expected term is the mid-point between the weighted average of vesting period and the contractual term. The risk-free interest rate is based on U.S. Treasury rates whose term is consistent with the expected life of the stock options. The Company has not paid and does not anticipate paying cash dividends on its shares of common stock; therefore, the expected dividend yield is assumed to be zero. The Company estimates forfeitures based on the historical experience of the Company and adjusts the estimated forfeiture rate based upon actual experience.
68
The fair value of options granted were estimated using the Black-Scholes-Merton option pricing model based on the assumptions in the table below:
|
|
|
Years ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Expected dividend yield |
|
|
0% |
|
|
|
0% |
|
|
|
0% |
|
|
Expected volatility |
|
70% - 72% |
|
|
47% |
|
|
45% - 56% |
|
|||
|
Risk-free interest rate |
|
1.84% - 2.29% |
|
|
1.13% - 2.09% |
|
|
1.44% - 1.95% |
|
|||
|
Expected life (in years) |
|
5.5 - 6.3 |
|
|
5.5 - 6.3 |
|
|
5.5 - 6.3 |
|
|||
The following table summarizes the Company’s stock option activity, including grants to non-employees, for the year ended December 31, 2017:
|
|
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
|
|
Weighted- |
|
|
Remaining |
|
||
|
|
|
Number of |
|
|
Average |
|
|
Contractual |
|
|||
|
|
|
Shares |
|
|
Exercise Price |
|
|
Term (Years) |
|
|||
|
Options outstanding at December 31, 2016 |
|
|
12,488,551 |
|
|
$ |
2.66 |
|
|
|
8.08 |
|
|
Granted |
|
|
4,746,250 |
|
|
|
1.25 |
|
|
|
|
|
|
Forfeited |
|
|
(574,773 |
) |
|
|
2.35 |
|
|
|
|
|
|
Cancelled |
|
|
(227,419 |
) |
|
|
4.16 |
|
|
|
|
|
|
Exercised |
|
|
(738,934 |
) |
|
|
0.59 |
|
|
|
|
|
|
Options outstanding at December 31, 2017 |
|
|
15,693,675 |
|
|
$ |
2.32 |
|
|
|
7.78 |
|
The following table summarizes information about stock options outstanding at December 31, 2017:
|
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
|
|
|
|
|
|
Average |
|
|
|
|
|
|
|
|
|
Weighted |
|
|
Remaining |
|
||
|
|
|
Number of |
|
|
Average |
|
|
Contractual |
|
|||
|
|
|
Shares |
|
|
Exercise Price |
|
|
Term (Years) |
|
|||
|
Exercisable at December 31, 2017 |
|
|
7,113,007 |
|
|
$ |
2.78 |
|
|
|
6.78 |
|
|
Vested or expected to vest at December 31, 2017 |
|
|
15,195,280 |
|
|
$ |
2.34 |
|
|
|
7.75 |
|
The aggregate intrinsic value of stock options outstanding, exercisable, and vested or expected to vest at December 31, 2017 was approximately $5.2 million, $1.6 million, and $4.9 million, respectively. This amount is before applicable income taxes and represents the closing market price of the Company’s common stock at December 31, 2017 less the exercise price, multiplied by the number of stock options that had an exercise price that is less than the closing market price. This amount represents the amount that would have been received by the optionees had these stock options been exercised on that date.
The total intrinsic value of options exercised during 2017, 2016 and 2015 was approximately $2,179,000, $519,000 and $1,603,000, respectively.
The Company granted 4,746,250, 5,368,755 and 4,407,758 options to employees and non-employees during the years ended December 31, 2017, 2016 and 2015, respectively, with a weighted-average grant date fair value of $0.82, $1.30 and $1.37, respectively.
As of December 31, 2017, the Company had future employee stock-based compensation expense of approximately $8,457,000 related to unvested share awards, which is expected to be recognized over an estimated weighted-average period of 2.5 years.
|
15. |
Restricted Stock Units |
In 2015, 2016 and 2017, the Company issued Restricted Stock Units (“RSUs”) to certain employees which vest over three years. The RSUs vest on defined vesting dates, subject to the continuous service with the Company at the applicable vesting event. Vesting can be accelerated by upon a change in control under the Plan if the RSUs are not assumed by the successor company. When vested, the RSUs represent the right to be issued the number of shares of the Company’s common stock that is equal to the number of RSUs granted. The fair value of each RSU is estimated based upon the closing price of the Company’s common stock on the grant date. Share-based compensation expense related to RSUs is recognized over the requisite service period as adjusted for estimated forfeitures.
69
The following is a summary of the RSU activity for the years ended December 31, 2017, 2016 and 2015:
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
Number of |
|
|
Average |
|
||
|
|
|
Restricted |
|
|
Grant |
|
||
|
|
|
Stock Units |
|
|
Date Fair |
|
||
|
|
|
Outstanding |
|
|
Value |
|
||
|
Unvested, December 31, 2014 |
|
|
140,000 |
|
|
$ |
7.19 |
|
|
Granted |
|
|
380,000 |
|
|
|
2.94 |
|
|
Vested |
|
|
(70,000 |
) |
|
|
7.19 |
|
|
Forfeited |
|
|
(27,500 |
) |
|
|
2.94 |
|
|
Unvested, December 31, 2015 |
|
|
422,500 |
|
|
$ |
3.64 |
|
|
Granted |
|
|
660,331 |
|
|
|
3.74 |
|
|
Vested |
|
|
(187,503 |
) |
|
|
4.53 |
|
|
Unvested, December 31, 2016 |
|
|
895,328 |
|
|
$ |
3.53 |
|
|
Granted |
|
|
3,873,000 |
|
|
|
0.82 |
|
|
Vested |
|
|
(337,618 |
) |
|
|
3.46 |
|
|
Forfeited |
|
|
(36,054 |
) |
|
|
3.60 |
|
|
Unvested, December 31, 2017 |
|
|
4,394,656 |
|
|
$ |
1.15 |
|
As of December 31, 2017, 2016 and 2015, the Company recorded approximately $1,751,000, $1,463,000 and $816,000, respectively, in compensation expense for the RSUs. As of December 31, 2017, the unrecognized stock-based compensation expense related to unvested RSUs was approximately $3.2 million, which is expected to be recognized over a weighted average period of approximately 2.3 years. The weighted average grant date fair value of the RSUs granted in 2015 was $2.94. The weighted average grant date fair value of the RSUs granted in 2016 was $3.74. The weighted average grant date fair value of the RSUs granted in 2017 was $0.82.
|
16. |
Warrants |
On March 22, 2013, SafeStitch entered into a stock purchase agreement with approximately 17 investors (the “2013 PIPE Investors”) pursuant to which the 2013 PIPE Investors purchased an aggregate of approximately 2,420,000 shares of common stock at a price of $1.25 per share for aggregate consideration of approximately $3.0 million. Included in this private placement was the issuance of warrants to purchase approximately 1,209,600 common shares, representing one warrant for every two common shares purchased, with an exercise price of $1.65 per share and five year expiration. Among the 2013 PIPE Investors purchasing shares were related parties who purchased 1.28 million shares and received 640,000 warrants. There were approximately 1.2 million warrants outstanding that were assumed as of the Merger. During the year ended December 31, 2017, 240,000 of these warrants were exercised. During the years ended December 31, 2016 and 2015, none of these warrants were exercised.
On January 17, 2012, TransEnterix Surgical entered into the original Loan Agreement with the Prior Lenders. Pursuant to such agreement, TransEnterix Surgical issued preferred stock warrants to the Prior Lenders on January 17, 2012 and December 21, 2012, respectively, to purchase shares of TransEnterix Surgical preferred stock. The preferred stock warrants expire 10 years from the issue date. The preferred stock warrants were remeasured immediately prior to the Merger. As of the Merger, the preferred stock warrants converted to common stock warrants, adjusted based on a Merger exchange ratio of 1.1533, and the preferred stock warrant liability was reclassified to additional paid-in capital. These warrants are exercisable for an aggregate of approximately 279,588 shares of common stock, with an exercise price of $1.45 per share. During the year ended December 31, 2013, 139,794 of these warrants were exercised in a cashless transaction for 112,766 shares of common stock. None of these warrants were exercised during the years ended December 31, 2017, 2016 or 2015.
On September 26, 2014, the Company entered into an amendment to the SVB Loan Agreement with the Prior Lenders. In connection with the first tranche borrowings under such amendment, the Company issued 38,324 common stock warrants to the Prior Lenders to purchase shares of the Company’s common stock, with an exercise price of $4.015 per share. The warrants expire seven years from their respective issue date. The Company concluded that the warrants are considered equity instruments. The warrants were recognized at the relative fair value on the issuance date as a debt discount and will be amortized using the effective interest method from issuance to the maturity of the term loans. None of these warrants were exercised during the year ended December 31, 2017, 2016 or 2015.
On August 14, 2015, in connection with an amendment to the SVB Loan Agreement and first tranche borrowings thereunder, the Company issued 112,903 common stock warrants to the Prior Lenders to purchase shares of the Company’s common stock, with an exercise price of $3.10 per share. The warrants expire seven years from their respective issue date. The Company concluded that the warrants are considered equity instruments. The warrants were recognized at the relative fair value on the issuance date as a debt discount and will be amortized using the effective interest method from issuance to the maturity of the note. None of these warrants were exercised during the year ended December 31, 2017, 2016 or 2015.
70
On April 28, 2017, the Company sold 24.9 million Units, each consisting of one share of Common Stock, a Series A Warrant to purchase one share of Common Stock with an exercise price of $1.00 per share, and a Series B Warrant to purchase 0.75 shares of Common Stock with an exercise price of $1.00 per share at an offering price of $1.00 per Unit. Each Series A Warrant may be exercised at any time beginning on the date of issuance, and from time to time thereafter, through and including the first anniversary of the issuance date, unless terminated earlier as provided in the Series A Warrant. Receipt of 510(k) clearance for the Senhance System on October 13, 2017, triggered the acceleration of the expiration date of the Series A Warrants to October 31, 2017. All of the Series A Warrants were exercised prior to the expiration date.
Each Series B Warrant has an initial exercise price of $1.00 per share and may be exercised at any time beginning on the date of issuance and from time to time thereafter through and including the fifth anniversary of the issuance date.
The exercise prices and the number of shares issuable upon exercise of each of the Series B Warrants are subject to adjustment upon the occurrence of certain events, including, but not limited to, stock splits or dividends, business combinations, sale of assets, similar recapitalization transactions, or other similar transactions. The Series B Warrants are subject to adjustment in the event that the Company issues or is deemed to issue shares of Common Stock for less than the then applicable exercise prices of each of the Series B Warrants. The exercisability of the Series B Warrants may be limited if, upon exercise, the holder or any of its affiliates would beneficially own more than 4.99% of the Common Stock. If, at any time Series B Warrants are outstanding, any fundamental transaction occurs, as described in the Series B Warrants and generally including any consolidation or merger into another corporation, the consummation of a transaction whereby another entity acquires more than 50% of the Company’s outstanding voting stock, or the sale of all or substantially all of its assets, the successor entity must assume in writing all of the obligations to the Series B Warrant holders. Additionally, in the event of a fundamental transaction, each Series B Warrant holder will have the right to require the Company, or its successor, to repurchase the Series B Warrants for an amount of cash equal to the Black-Scholes value of the remaining unexercised portion of such Series B Warrants. During the year ended December 31, 2017, 8,893,700 Series B Warrants were exercised.
On May 10, 2017, in connection with the entry into the Innovatus Loan Agreement, the Company issued warrants to the Lender to purchase shares of the Company’s common stock. The warrants are issued on the funding date of each tranche and will expire five (5) years from such issue date. The warrants issued in connection with funding of the first tranche will entitle the Lender to purchase up to 1,244,746 shares of the Company’s common stock at an exercise price of $1.00 per share. None of these warrants were exercised as of December 31, 2017.
On September 12, 2017, the Company entered into a service agreement with a third party vendor. In connection with the service agreement, the Company issued 950,000 common stock warrants (“Service Warrants”) to purchase shares of the Company’s common stock, with an exercise price of $1.00 per share. The Service Warrants vest as follow: (a) twenty-five percent (25%) on the date of execution of the services agreement; (b) fifty percent (50%) upon completion of hiring the sales team; and (c) the remaining twenty-five percent (25%) upon achieving cumulative product revenue of $15.0 million. The Service Warrants expire ten years from their issue date. The Company concluded that the Service Warrants are considered equity instruments. The fair value of the Service Warrants on the issuance date was determined using a Black-Scholes Merton model. The initial expense of $0.6 million was recognized during the year ended December 31, 2017. The fair value of the remaining Service Warrants will be updated each reporting period and the expense will be recorded over the service period. None of these warrants were exercised as of December 31, 2017.
|
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
Average |
|
|
|
|
|
||
|
|
|
|
|
|
|
Average |
|
|
Remaining |
|
|
Weighted |
|
|||
|
|
|
Number of |
|
|
Exercise |
|
|
Contractual |
|
|
Average |
|
||||
|
|
|
Warrants |
|
|
Price |
|
|
Life (in years) |
|
|
Fair Value |
|
||||
|
Outstanding at December 31, 2014 |
|
|
1,313,719 |
|
|
$ |
1.70 |
|
|
|
3.9 |
|
|
$ |
1.75 |
|
|
Granted |
|
|
112,903 |
|
|
|
3.10 |
|
|
|
6.6 |
|
|
|
0.86 |
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Outstanding at December 31, 2015 |
|
|
1,426,622 |
|
|
$ |
1.81 |
|
|
|
3.2 |
|
|
$ |
1.54 |
|
|
Granted |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Exercised |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Outstanding at December 31, 2016 |
|
|
1,426,622 |
|
|
$ |
1.81 |
|
|
|
2.2 |
|
|
$ |
1.54 |
|
|
Granted |
|
|
45,769,746 |
|
|
|
1.00 |
|
|
|
4.8 |
|
|
|
0.22 |
|
|
Exercised |
|
|
(34,033,700 |
) |
|
|
1.00 |
|
|
|
— |
|
|
|
— |
|
|
Outstanding at December 31, 2017 |
|
|
13,162,668 |
|
|
$ |
1.08 |
|
|
|
4.5 |
|
|
$ |
0.39 |
|
71
The aggregate intrinsic value of the common stock warrants in the above table was $11.2 million, $0 and $1.0 million at December 31, 2017, 2016 and 2015, respectively. The aggregate intrinsic value is before applicable income taxes and is calculated based on the difference between the exercise price of the warrants and the estimated fair market value of the applicable stock as of the respective dates.
|
17. |
Restructuring |
On April 19, 2016, the FDA notified the Company that the SurgiBot System did not meet the criteria for substantial equivalence based on the data submitted in the 510(k) submission. In May 2016, the Company implemented a restructuring plan. Under the restructuring plan, the Company reduced headcount, discontinued efforts on the SurgiBot System, and cancelled certain contracts. The restructuring charges amounted to $5.7 million, of which $2.6 million was included as inventory write down related to restructuring and $3.1 million was included as restructuring and other charges in the consolidated statements of operations and comprehensive loss, during the second quarter of 2016.
The restructuring and other charges of $3.1 million included: (i) $0.5 million to be paid in cash, of which $0.4 million related to employee severance costs and $0.1 million related to cancellation of certain contracts; and (ii) $2.6 million for other non-cash charges, of which $1.0 million related to the disposal of long-lived assets for the abandonment of certain equipment and tooling directly relating to the SurgiBot System and $1.6 million related to the write-off of intellectual property for certain patents also relating to the SurgiBot System. There were no future payments under the restructuring plan as of December 31, 2017 or 2016.
|
18. |
Purchase Agreement, Controlled Equity Offering and Public Offering of Common Stock |
On April 28, 2017, the Company sold 24.9 million units, each consisting of one share of the Company’s common stock, a Series A warrant to purchase one share of common stock, and a Series B warrant to purchase 0.75 shares of common stock, at a public offering price of $1.00 per unit for aggregate gross proceeds of $24.9 million in an underwritten firm commitment public offering. Net proceeds after issuance costs were $23.2 million, assuming no exercise of the warrants. The closing of the public offering occurred on May 3, 2017.
On December 16, 2016, the Company entered into a purchase agreement (the “LPC Purchase Agreement”) with Lincoln Park Capital Fund, LLC, (“Lincoln Park”), pursuant to which the Company had the right to sell to Lincoln Park up to an aggregate of $25.0 million in shares of the Company’s common stock, (the “Common Stock”), subject to certain limitations and conditions set forth in the LPC Purchase Agreement. The Company issued to Lincoln Park 345,421 shares of Common Stock as commitment shares in consideration for the LPC Purchase Agreement through April 27, 2017. Sales under the LPC Purchase Agreement for the year ended December 31, 2016 were 300,000 shares, with gross proceeds of $412,500 and net proceeds of $392,500. Sales under the LPC Purchase Agreement for the year ended December 31, 2017 were 3,972,741 shares, with gross and net proceeds of $5,304,000. Effective April 27, 2017, the Company terminated the LPC Purchase Agreement. The LPC Purchase Agreement provided the Company with an election to terminate the Purchase Agreement for any reason or for no reason by delivering a notice to Lincoln Park, and the Company did not incur any early termination penalties in connection with the termination of the LPC Purchase Agreement.
On June 11, 2015, the Company sold 16,666,667 shares of common stock at a public offering price of $3.00 per share for aggregate gross proceeds of $50.0 million in an underwritten firm commitment public offering. Net proceeds after issuance costs were $46.4 million. The closing of the public offering occurred on June 17, 2015. The Company granted the underwriters an option, exercisable for 30 days, to purchase up to an additional 2,500,000 shares of Common Stock.
On July 10, 2015, the underwriters exercised a portion of their option to acquire an additional 2,075,000 shares at the public offering price of $3.00 per share for aggregate additional gross proceeds of $6.2 million. Net proceeds after issuance costs were $5.8 million. The purchase of the option shares closed on July 15, 2015. Total proceeds (including the option) were $52.2 million, net of issuance costs of $4.0 million. The common stock was offered and sold pursuant to the Shelf Registration Statement filed in November 2014 (the “November 2014 Shelf Registration Statement”), which was declared effective on December 19, 2014. The November 2014 Shelf Registration Statement allowed the Company to raise up to $100.0 million through the sale of debt securities, common stock, preferred stock, warrants, or any combination thereof. On March 3, 2016, the Company filed an amendment to the November 2014 Shelf Registration Statement increasing the amount available from $100.0 million to $150.0 million.
On February 20, 2015, the Company entered into a Controlled Equity Offering SM Sales Agreement (the “2015 Sales Agreement”) with Cantor Fitzgerald & Co. (“Cantor”), as sales agent, pursuant to which the Company sold through Cantor, from time to time, up to $25.0 million in shares of common stock in an at-the-market offering. The Company pays Cantor a commission of approximately 3% of the aggregate gross proceeds received from all sales of common stock under the Sales Agreement. Sales under the 2015 Sales Agreement have been fully sold as of February 9, 2016, with cumulative shares of 7,724,488, gross proceeds of $25.0 million and net proceeds of $24.0 million.
72
On February 9, 2016, the Company entered into a Controlled Equity Offering SM Sales Agreement (the “2016 Sales Agreement”) with Cantor, as sales agent, pursuant to which the Company can sell through Cantor, from time to time, up to $43.6 million in shares of common stock in an at-the-market offering. The Company pays Cantor a commission of approximately 3% of the aggregate gross proceeds received from all sales of common stock under the 2016 Sales Agreement.
On August 31, 2017, the Company entered into an At-the-Market Equity Offering Sales Agreement (the “2017 Sales Agreement”) with Stifel, Nicolaus & Company, Incorporated (“Stifel”), as sales agent, pursuant to which the Company can sell through Stifel, from time to time, up to $50.0 million in shares of common stock in an at-the-market offering. The Company pays Stifel a commission of approximately 3% of the aggregate gross proceeds received from all sales of common stock under the 2017 Sales Agreement. Unless otherwise terminated earlier, the 2017 Sales Agreement continues until all shares available under the Sales Agreement have been sold.
The following table summarizes the total sales under the 2015 Sales Agreement, 2016 Sales Agreement and the 2017 Sales Agreement for the periods indicated (in thousands, except per share amounts):
|
|
2017 Sales Agreement |
|
|
2016 Sales Agreement |
|
|
2015 Sales Agreement |
|
|||||||
|
|
Year Ended December 31, 2017 |
|
|
Year Ended December 31, 2016 |
|
|
Year Ended December 31, 2016 |
|
|
Year Ended December 31, 2015 |
|
||||
|
Total shares of common stock sold |
|
15,998.5 |
|
|
|
8,763.4 |
|
|
|
5,710.2 |
|
|
|
2,014.3 |
|
|
Average price per share |
$ |
3.13 |
|
|
$ |
4.70 |
|
|
$ |
3.23 |
|
|
$ |
3.25 |
|
|
Gross proceeds |
$ |
50,000 |
|
|
$ |
41,156 |
|
|
$ |
18,454 |
|
|
$ |
6,546 |
|
|
Commissions earned by Cantor |
$ |
1,500 |
|
|
$ |
1,235 |
|
|
$ |
553 |
|
|
$ |
197 |
|
|
Other issuance costs |
$ |
97 |
|
|
$ |
185 |
|
|
$ |
— |
|
|
$ |
259 |
|
|
19. |
Basic and Diluted Net Loss per Share |
Basic net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted average number of common shares outstanding during the period. Diluted net loss per common share is computed giving effect to all dilutive potential common shares that were outstanding during the period. Diluted potential common shares consist of incremental shares issuable upon exercise of stock options, warrants and restricted stock units. In computing diluted net loss per share for the years ended December 31, 2017, 2016, and 2015, no adjustment has been made to the weighted average outstanding common shares as the assumed exercise of outstanding options, warrants and restricted stock units would be anti-dilutive.
Potential common shares not included in calculating diluted net loss per share are as follows:
|
|
|
December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Stock options |
|
|
15,693,675 |
|
|
|
12,488,551 |
|
|
|
8,300,819 |
|
|
Stock warrants |
|
|
13,162,668 |
|
|
|
1,426,622 |
|
|
|
1,426,622 |
|
|
Nonvested restricted stock units |
|
|
4,394,656 |
|
|
|
895,328 |
|
|
|
422,500 |
|
|
Total |
|
|
33,250,999 |
|
|
|
14,810,501 |
|
|
|
10,149,941 |
|
|
20. |
Related Person Transactions |
Synergy Life Science Partners, L.P. and Synecor, LLC collectively owned approximately 2.9% and 5% of the Company’s common stock at December 31, 2017 and 2016, respectively. A member of the Company’s Board of Directors is managing partner of Synergy Life Science Partners, L.P. and an executive officer of Synecor, LLC. Various research and development services were purchased by the Company from Synecor, LLC and its wholly owned subsidiary Synchrony Labs LLC pursuant to arms’ length terms approved by the Audit Committee and totaled approximately $0, $5,000 and $435,000 for the years ended December 31, 2017, 2016 and 2015, respectively.
On September 18, 2015, TransEnterix Italia entered into a services agreement for receipt of administrative services from Sofar and payment of rent to Sofar, a stockholder that owned approximately 9.7% and 13% of the Company’s common stock at December 31, 2017 and 2016, respectively. Expenses under this agreement were approximately $55,000, $232,000 and $89,000 for the years ended December 31, 2017, 2016 and 2015, respectively. The services agreement terminated in 2017.
73
In November 2016, the Company agreed to enter into a technology and patents purchase agreement with Sofar to acquire from Sofar certain technology and intellectual property rights related to the Senhance Acquisition, and formerly licensed by the Company. The acquisition price was $400,000.
As discussed in Note 3, in September 2015, the Company completed the Senhance Acquisition using a combination of cash, stock and potential post-acquisition milestone payments. On December 30, 2016, the Company entered into an Amendment to the Senhance Acquisition purchase agreement with Sofar to restructure the terms of the Second Tranche of the Cash Consideration. Under the Amendment, the Second Tranche was restructured to reduce the contingent cash consideration by €5.0 million in exchange for the issuance of 3,722,685 shares of the Company’s common stock with an aggregate fair market value of €5.0 million. On January 4, 2017, the Company issued to Sofar 3,722,685 shares of the common stock with a fair value of €5.0 million. The price per share was $1.404 and was calculated based on the average of the closing prices of the Company’s common stock on ten consecutive trading days ending one day before the execution of the Amendment.
In March 2018, TransEnterix Europe entered into a Service Supply Agreement with 1Med S.A. for certain regulatory consulting services. Andrea Biffi, a current member of the Company’s Board of Directors, owns a non-controlling interest in 1Med S.A.
|
21. |
Commitments and Contingencies |
Contingent Consideration
As discussed in Note 3, in September 2015, the Company completed the Senhance Acquisition using a combination of cash, stock and potential post-acquisition milestone payments. These milestone payments may be payable in the future, depending on the achievement of certain regulatory and commercial milestones. On December 30, 2016, the Company entered into an Amendment to restructure the terms of the Second Tranche of the Cash Consideration. Under the Amendment, the Second Tranche was restructured to reduce the contingent cash consideration by €5.0 million in exchange for the issuance of 3,722,685 shares of the Company’s common stock with an aggregate fair market value of €5.0 million. As of December 31, 2017, the fair value of the contingent consideration was $12.4 million.
Legal Proceedings
When determining the estimated probable loss or range of losses, significant judgment is required to be exercised in order to estimate the amount and timing of the loss to be recorded. Estimating an amount or range of possible losses resulting from litigation proceedings is inherently difficult and requires an extensive degree of judgment, particularly where the matters involve indeterminate claims for monetary damages, are in the early stages of the proceedings, and are subject to appeal. In addition, because most legal proceedings are resolved over extended periods of time, potential losses are subject to change due to, among other things, new developments, changes in legal strategy, the outcome of intermediate procedural and substantive rulings and other parties’ settlement posture and their evaluation of the strength or weakness of their case against the Company. For these reasons, the Company is currently unable to predict the ultimate timing or outcome of, or reasonably estimate the possible losses or a range of possible losses resulting from, the matters described above. Based on information currently available, the Company does not believe that any reasonably possible losses arising from currently pending legal matters will be material to the Company’s results of operations or financial condition. However, in light of the inherent uncertainties involved in such matters, an adverse outcome in one or more of these matters could materially and adversely affect the Company’s financial condition, results of operations or cash flows in any particular reporting period.
No liability or related charge was recorded to earnings in the Company’s consolidated financial statements for legal contingencies for the year ended December 31, 2017, as all pending litigation, including two putative derivative claims were dismissed in 2017 with prejudice in the Company's favor.
.
74
On November 2, 2009, TransEnterix Surgical entered into an operating lease for its corporate offices for a period of five years commencing in April 2010. On June 12, 2014, the Company entered into a lease amendment extending the term of the lease for a period of 3 years and 2 months commencing on May 1, 2015 and expiring on June 30, 2018, with an option to renew for an additional three years. On January 8, 2018, the Company entered into a lease amendment extending the term of the lease for a period of eighteen months commencing on July 1, 2018 and expiring on December 31, 2019, with an option to renew for an additional five years. On October 25, 2013, the Company entered into an operating lease for its warehouse for a period of four years and four months commencing in January 2014, with an option to renew for an additional six years. On December 27, 2017 the Company entered into an agreement to terminate this lease effective January 31, 2018. On May 12, 2016 TransEnterix Italia entered into an operating lease for research and development and demonstration facilities for a period of 6 years commencing in July 2016. Rent expense was approximately $1,135,000, $907,000 and $513,000 for the years ended December 31, 2017, 2016 and 2015, respectively. The Company’s approximate future minimum payments for its operating lease obligations that have initial or remaining noncancelable terms in excess of one year are as follow:
|
|
|
Years ending December 31, |
|
|
|
|
|
(In thousands) |
|
|
|
2018 |
|
|
800 |
|
|
2019 |
|
|
789 |
|
|
2020 |
|
|
260 |
|
|
2021 |
|
|
260 |
|
|
2022 |
|
|
87 |
|
|
Total |
|
$ |
2,196 |
|
License and Supply Agreements
As discussed in Note 3, in September 2015, the Company completed the Senhance Acquisition. As part of this transaction, the Company assumed certain license and supply agreements. Commitments under these agreements amount to approximately $2,966,000 in 2018, $1,152,000 in 2019, $600,000 in 2020, $600,000 in 2021, $600,000 in 2021 and $3.0 million thereafter until termination in 2027.
On February 13, 2014, TransEnterix Surgical, Inc., a wholly owned subsidiary of the Company, entered into a Robotic Development and Supply Agreement (the “Robotic Agreement”) with Microline Surgical, Inc. (“Microline”). Under the Robotic Agreement, Microline was developing a flexible sealer product for exclusive use by the Company with the SurgiBot System in open, minimally invasive and laparoscopic surgery. Payments under the Robotic Agreement were $0 and $400,000 for the year ended December 31, 2016 and 2015, respectively. As part of the restructuring related to the SurgiBot System, the Robotic Agreement was terminated in 2016.
The Company has placed orders with various suppliers for the purchase of certain tooling, supplies and contract engineering and research services. Each of these orders has a duration or expected completion within the next twelve months.
75
The following is a summary of the Company’s unaudited quarterly results of operations for the fiscal years ended December 31, 2017 and 2016 (in thousands, except per share amounts):
|
|
|
Fiscal Year Ended December 31, 2017 |
|
|||||||||||||||||
|
|
|
1st |
|
|
2nd |
|
|
3rd |
|
|
4th |
|
|
Total |
|
|||||
|
|
|
Quarter |
|
|
Quarter |
|
|
Quarter |
|
|
Quarter |
|
|
Year |
|
|||||
|
Total revenues |
|
$ |
1,946 |
|
|
$ |
1,584 |
|
|
$ |
183 |
|
|
|
3,398 |
|
|
$ |
7,111 |
|
|
Cost of revenue |
|
|
1,334 |
|
|
|
972 |
|
|
|
921 |
|
|
|
3,500 |
|
|
|
6,727 |
|
|
Amortization of intangible assets |
|
|
1,636 |
|
|
|
1,687 |
|
|
|
1,821 |
|
|
|
2,714 |
|
|
|
7,858 |
|
|
Change in fair value of contingent consideration |
|
|
1,227 |
|
|
|
(774 |
) |
|
|
773 |
|
|
|
800 |
|
|
|
2,026 |
|
|
Issuance costs for warrants |
|
|
— |
|
|
|
627 |
|
|
|
— |
|
|
|
— |
|
|
|
627 |
|
|
Other operating expenses |
|
|
13,627 |
|
|
|
11,538 |
|
|
|
12,337 |
|
|
|
14,298 |
|
|
|
51,800 |
|
|
Change in fair value of warrant liabilities |
|
|
— |
|
|
|
2,326 |
|
|
|
22,887 |
|
|
|
58,521 |
|
|
|
83,734 |
|
|
Interest expense, net |
|
|
394 |
|
|
|
662 |
|
|
|
695 |
|
|
|
684 |
|
|
|
2,435 |
|
|
Loss before income taxes |
|
|
(16,272 |
) |
|
|
(15,454 |
) |
|
|
(39,251 |
) |
|
|
(77,119 |
) |
|
|
(148,096 |
) |
|
Income tax benefit |
|
|
858 |
|
|
|
741 |
|
|
|
738 |
|
|
|
963 |
|
|
|
3,300 |
|
|
Net loss |
|
$ |
(15,414 |
) |
|
$ |
(14,713 |
) |
|
$ |
(38,513 |
) |
|
$ |
(76,156 |
) |
|
$ |
(144,796 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(0.13 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.26 |
) |
|
$ |
(0.40 |
) |
|
$ |
(0.97 |
) |
|
|
|
Fiscal Year Ended December 31, 2016 |
|
|||||||||||||||||
|
|
|
1st |
|
|
2nd |
|
|
3rd |
|
|
4th |
|
|
Total |
|
|||||
|
|
|
Quarter |
|
|
Quarter |
|
|
Quarter |
|
|
Quarter |
|
|
Year |
|
|||||
|
Total revenues |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
1,466 |
|
|
$ |
53 |
|
|
$ |
1,519 |
|
|
Cost of revenue |
|
|
— |
|
|
|
— |
|
|
|
1,031 |
|
|
|
38 |
|
|
|
1,069 |
|
|
Amortization of intangible assets |
|
|
1,817 |
|
|
|
1,786 |
|
|
|
1,709 |
|
|
|
1,655 |
|
|
|
6,967 |
|
|
Goodwill impairment |
|
|
— |
|
|
|
61,784 |
|
|
|
— |
|
|
|
— |
|
|
|
61,784 |
|
|
Restructuring and other charges |
|
|
— |
|
|
|
3,085 |
|
|
|
— |
|
|
|
(21 |
) |
|
|
3,064 |
|
|
Inventory write-down related to restructuring |
|
|
— |
|
|
|
2,565 |
|
|
|
— |
|
|
|
— |
|
|
|
2,565 |
|
|
Other operating expenses |
|
|
13,163 |
|
|
|
11,509 |
|
|
|
12,278 |
|
|
|
12,769 |
|
|
|
49,719 |
|
|
Interest expense, net |
|
|
578 |
|
|
|
394 |
|
|
|
462 |
|
|
|
420 |
|
|
|
1,854 |
|
|
Loss before income taxes |
|
$ |
(15,558 |
) |
|
$ |
(81,123 |
) |
|
$ |
(14,014 |
) |
|
$ |
(14,808 |
) |
|
$ |
(125,503 |
) |
|
Income tax benefit |
|
$ |
2,645 |
|
|
$ |
992 |
|
|
$ |
1,070 |
|
|
$ |
816 |
|
|
$ |
5,523 |
|
|
Net loss |
|
$ |
(12,913 |
) |
|
$ |
(80,131 |
) |
|
$ |
(12,944 |
) |
|
$ |
(13,992 |
) |
|
$ |
(119,980 |
) |
|
Net loss per share - basic and diluted |
|
$ |
(0.12 |
) |
|
$ |
(0.70 |
) |
|
$ |
(0.11 |
) |
|
$ |
(0.12 |
) |
|
$ |
(1.07 |
) |
76
None.
Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2017. We maintain disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in our reports filed or submitted under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate, to allow for timely decisions regarding required disclosure. Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on such evaluation, our Chief Executive Officer and Chief Financial Officer concluded that, as of December 31, 2017, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the Company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of management and directors of the Company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
For the year ended December 31, 2017, pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, management (with the participation of our principal executive officer and principal financial officer) conducted an evaluation of the effectiveness of our internal control over financial reporting based on the original framework established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this evaluation, management concluded that, as of December 31, 2017, our internal control over financial reporting was effective.
The Company’s independent registered public accounting firm, BDO USA, LLP, audited the effectiveness of the Company’s internal control over financial reporting as of December 31, 2017. BDO USA, LLP’s report on the effectiveness of the Company’s internal control over financial reporting as of December 31, 2017 is set forth herein.
Changes in Internal Controls Over Financial Reporting
There were no changes in the Company’s internal control over financial reporting during the last quarter that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
On March 6, 2018, the Company entered into new employment agreements with each of Todd Pope, the Company’s Chief Executive Officer, Joseph Slattery, the Company’s Executive Vice President and Chief Financial Officer and Anthony Fernando, the Company’s Chief Operating Officer. The new agreements provide for the following changes: (1) standardization among Messrs. Slattery and Fernando of the post-termination compensation to be paid, both in the event of a termination without Cause or for Good Reason (as each term in defined in the agreements) prior to a Change in Control (as defined in the agreements) or in the event of a termination without Cause or for Good Reason in the one year period following, or six months prior to and in connection with a Change in
77
Control; and (2) the addition of target bonus to the severance benefits to be paid to each of the executive officers in a covered termination of employment. With these changes, the severance paid on a termination without Cause or for Good Reason prior to a Change in Control is the sum of base salary and target bonus for nine (9) months for Messrs. Slattery and Fernando and twelve (12) months for Mr. Pope, plus reimbursement of health care benefit costs for such nine- and twelve-month periods. The period for payment of severance benefits increases to eighteen (18) months for Messrs. Slattery and Fernando and twenty-four (24) months for Mr. Pope following a termination without Cause or for Good Reason in the covered period if a Change in Control occurs, plus the acceleration of all outstanding unvested equity awards. No compensation is paid single-trigger upon the occurrence of a Change in Control. The employment agreements each have an effective date of March 1, 2018 and a two-year term, subject to automatic renewal unless terminated with at least six months’ prior written notice prior to the expiration of the term or any extension. The entry into the new employment agreements caused a termination and replacement of the prior employment agreements between each such executive officer and the Company.
This summary of the employment agreements is not complete, and reference is made to the agreements as filed as exhibits to this Form 10-K and incorporated by reference herein.
78
The information required by this item is incorporated by reference from the information contained in our Proxy Statement for the Annual Meeting of Shareholders expected to be filed with the SEC on or prior to April 30, 2018.
The information required by this item is incorporated by reference from the information contained in our Proxy Statement for the Annual Meeting of Shareholders expected to be filed with the SEC on or prior to April 30, 2018.
|
ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
The information required by this item is incorporated by reference from the information contained in our Proxy Statement for the Annual Meeting of Shareholders expected to be filed with the SEC on or prior to April 30, 2018.
The information required by this item is incorporated by reference from the information contained in our Proxy Statement for the Annual Meeting of Shareholders expected to be filed with the SEC on or prior to April 30, 2018.
The information required by this item is incorporated by reference from the information contained in our Proxy Statement for the Annual Meeting of Shareholders expected to be filed with the SEC on or prior to April 30, 2018.
79
|
|
(a) |
(1) The following consolidated financial statements are filed as a part of this Annual Report: |
|
|
|
Page |
|
Consolidated Financial Statements : |
|
|
|
|
|
|
|
|
41 |
|
|
Consolidated Balance Sheets as of December 31, 2017 and 2016 |
|
43 |
|
|
44 |
|
|
|
45 |
|
|
|
46 |
|
(2) |
Consolidated Financial Statement Schedules: The information required by this item is included in the consolidated financial statements contained in Item 8 of this Annual Report. |
|
(3) |
Exhibits: The following exhibits are filed as part of, or incorporated by reference into, this Annual Report. |
|
Exhibit No. |
|
Description |
|
|
|
|
|
|
|
1.1 |
|
||
|
|
|
||
|
1.2 |
|
||
|
|
|
|
|
|
1.3 |
|
||
|
|
|
|
|
|
1.4 |
|
||
|
|
|
|
|
|
1.5 |
|
||
|
|
|
||
|
2.1 |
|
||
|
|
|
|
|
|
2.1(a) |
|
||
|
|
|
||
|
3.1 |
|
||
|
|
|
||
|
3.1.1 |
|
||
|
|
|
||
|
3.2 |
|
||
|
|
|
||
|
4.1 |
|
||
80
|
Exhibit No. |
|
Description |
|
|
|
|
||
|
4.2 |
|
||
|
|
|
|
|
|
4.3 |
|
||
|
|
|
||
|
4.4 |
|
||
|
|
|
|
|
|
4.5 |
|
||
|
|
|
|
|
|
4.6 |
|
||
|
|
|
|
|
|
4.7 |
|
||
|
|
|
|
|
|
4.8 |
|
||
|
|
|
|
|
|
10.1 |
|
||
|
|
|
||
|
10. 2++ |
|
||
|
|
|
||
|
10.3 |
|
||
|
|
|
||
|
10.4 |
|
||
|
|
|
||
|
10.5 + * |
|
||
|
|
|
|
|
|
|
|
||
|
10.6 + * |
|
||
|
|
|
||
|
|
|
||
|
10.7 + * |
|
||
|
|
|
||
|
|
|
||
|
10.8+ |
|
||
|
|
|
|
|
|
10.9 + |
|
||
|
|
|
||
|
10.10 + |
|
||
|
|
|
||
|
10.11 + |
|
||
|
|
|
||
81
|
Exhibit No. |
|
Description |
|
|
|
|||
|
|
|
||
|
10.13 + |
|
||
|
|
|
||
|
10.14 |
|
||
|
|
|
||
|
10.14.1 |
|
||
|
|
|
||
|
10.14.2 |
|
||
|
|
|
||
|
10.14.3 |
|
||
|
|
|
|
|
|
10.14.4 |
|
||
|
|
|
|
|
|
10.14.5 |
|
||
|
|
|
||
|
10.15 |
|
||
|
|
|
||
|
10.15.1 |
|
||
|
|
|
|
|
|
10.16 |
|
||
|
|
|
|
|
|
10.17*+++ |
|
||
|
|
|
||
|
21.1 * |
|
||
|
|
|
||
|
23.1 * |
|
||
|
|
|
||
|
31.1 * |
|
Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a). |
|
|
|
|
||
|
31.2 * |
|
Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a). |
|
|
|
|
||
|
32.1 * |
|
Certification of Chief Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
||
|
32.2 * |
|
Certification of Chief Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
|
||
|
101.INS * |
|
XBRL Instance Document. |
|
82
|
Exhibit No. |
|
Description |
|
|
|
|
||
|
101.SCH * |
|
XBRL Taxonomy Extension Schema Document. |
|
|
|
|
||
|
101.CAL * |
|
XBRL Taxonomy Extension Calculation Linkbase Document. |
|
|
|
|
||
|
101.DEF * |
|
XBRL Taxonomy Extension Definition Linkbase Document. |
|
|
|
|
|
|
|
101.LAB * |
|
XBRL Taxonomy Extension Label Linkbase Document. |
|
|
|
|
||
|
101.PRE * |
|
XBRL Taxonomy Extension Presentation Linkbase Document. |
|
|
+ |
A management contract, compensatory plan or arrangement required to be separately identified. |
|
++ |
Confidential treatment has been granted for certain portions of the agreement pursuant to a confidential treatment request filed with the Commission on November 9, 2015. Such provisions have been filed separately with the Commission. |
|
+++ |
Confidential treatment has been requested for certain portions of this agreement pursuant to an application for confidential treatment filed with the Securities and Exchange Commission on March 8, 2018. Such provisions have been filed separately with the Commission. |
|
* |
Filed herewith. |
Registrants may voluntarily include a summary of information required by Form 10-K under this Item 16. The Company has elected not to include such summary information.
83
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
|
Date: March 8, 2018 |
|
TransEnterix, Inc. |
||
|
|
|
|
|
|
|
|
|
By: |
|
/s/ Todd M. Pope |
|
|
|
|
|
Todd M. Pope |
|
|
|
|
|
President, Chief Executive Officer |
|
|
|
|
|
and a Director |
|
|
|
|
|
(principal executive officer) |
POWER OF ATTORNEY
We, the undersigned officers and directors of TransEnterix, Inc., hereby severally constitute and appoint Todd M. Pope and Joseph P. Slattery, our true and lawful attorney-in-fact and agent, with full power of substitution and resubstitution in him for him and in his name, place and stead, and in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as full to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the Registrant in the capacities and on the dates indicated.
|
Signature |
|
Title(s) |
|
Date |
|
|
|
|
||
|
/s/ Todd M. Pope |
|
President, Chief Executive Officer and a Director (principal executive officer) |
|
March 8, 2018 |
|
Todd M. Pope |
|
|
|
|
|
|
|
|
||
|
/s/ Joseph P. Slattery |
|
Executive Vice President and Chief Financial Officer (principal financial officer and principal accounting officer) |
|
March 8, 2018 |
|
Joseph P. Slattery |
|
|
|
|
|
|
|
|
|
|
|
/s/ Paul A. LaViolette |
|
Chairman of the Board and a Director |
|
March 8, 2018 |
|
Paul A. LaViolette |
|
|
|
|
|
|
|
|
||
|
/s/ Andrea Biffi |
|
Director |
|
March 8, 2018 |
|
Andrea Biffi |
|
|
|
|
|
|
|
|
||
|
/s/ Jane H. Hsaio |
|
Director |
|
March 8, 2018 |
|
Jane H. Hsaio, Ph.D. |
|
|
|
|
|
|
|
|
||
|
/s/ William N. Kelley |
|
Director |
|
March 8, 2018 |
|
William N. Kelley, M.D. |
|
|
|
|
|
|
|
|
||
|
/s/ Aftab R. Kherani |
|
Director |
|
March 8, 2018 |
|
Aftab R. Kherani |
|
|
|
|
|
|
|
|
||
|
/s/ David B. Milne |
|
Director |
|
March 8, 2018 |
|
David B. Milne |
|
|
|
|
|
|
|
|
||
|
/s/ Richard C. Pfenniger, Jr. |
|
Director |
|
March 8, 2018 |
|
Richard C. Pfenniger, Jr. |
|
|
|
|
|
|
|
|
||
|
/s/ William N. Starling, Jr. |
|
Director |
|
March 8, 2018 |
|
William N. Starling, Jr. |
|
|
|
|
84