Attached files
| file | filename |
|---|---|
| EX-32.2 - EXHIBIT 32.2 - OMEROS CORP | omer-ex322_20171231.htm |
| EX-32.1 - EXHIBIT 32.1 - OMEROS CORP | omer-ex321_20171231.htm |
| EX-31.2 - EXHIBIT 31.2 - OMEROS CORP | omer-ex312_20171231.htm |
| EX-31.1 - EXHIBIT 31.1 - OMEROS CORP | omer-ex311_20171231.htm |
| EX-23.1 - EXHIBIT 23.1 - OMEROS CORP | omer-ex231_20171231.htm |
| EX-12.1 - EXHIBIT 12.1 - OMEROS CORP | omer-ex121_20171231.htm |
| EX-10.40 - EXHIBIT 10.40 - OMEROS CORP | omer-ex1040.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
________________________________________
FORM 10-K
________________________________________
(Mark One)
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-34475
________________________________________
OMEROS CORPORATION
(Exact name of registrant as specified in its charter)
________________________________________
Washington | 91-1663741 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) | |
201 Elliott Avenue West Seattle, Washington | 98119 | |
(Address of principal executive offices) | (Zip Code) | |
(206) 676-5000
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
Common Stock, $0.01 par value per share | The Nasdaq Stock Market LLC |
(Title of each class) | (Name of each exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act:
None
___________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes x No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer | x | Accelerated filer | ¨ | |||
Non-accelerated filer | ¨ (Do not check if a smaller reporting company) | Smaller reporting company | ¨ | |||
Emerging growth company | ¨ | |||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the voting and non-voting common stock held by non-affiliates of the registrant as of the last business day of the registrant’s most recently completed second fiscal quarter was $830,503,401.
As of February 26, 2018, the number of outstanding shares of the registrant’s common stock, par value $0.01 per share, was 48,285,978.
DOCUMENTS INCORPORATED BY REFERENCE
Specified portions of the registrant’s proxy statement with respect to the 2018 Annual Meeting of Shareholders to be held June 15, 2018, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2017, are incorporated by reference into Part III of this Form 10-K.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, or the Exchange Act, which are subject to the “safe harbor” created by those sections for such statements. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than statements of historical fact are “forward-looking statements.” Terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “goal,” “intend,” “likely,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would,” and similar expressions and variations thereof are intended to identify forward-looking statements, but these terms are not the exclusive means of identifying such statements. Examples of these statements include, but are not limited to, statements regarding:
• | our expectations regarding OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1%/0.3% product sales; |
• | our estimates regarding how long our existing cash, cash equivalents, short-term investments and revenues will be sufficient to fund our anticipated operating expenses and capital expenditures, as well as our interest and principal payments on our outstanding notes under our Term Loan Agreement, or the CRG Loan Agreement, with CRG Servicing LLC and the lenders identified therein, and the satisfaction of covenants thereunder; |
• | our expectations related to obtaining a reinstatement or extension of the pass-through period, or separate or similar reimbursement, for OMIDRIA from the Centers for Medicare and Medicaid Services, or CMS, and/or from Congress for periods beyond January 1, 2018 and our expectations regarding the per unit price and net product revenues we may receive for OMIDRIA; |
• | our plans for marketing and distribution of OMIDRIA; |
• | our expectations relating to OMIDRIA demand from wholesalers, ambulatory surgery centers, or ASCs, and hospitals, our expectations regarding the collection of accounts receivable and our estimates of chargebacks and rebates, distribution fees and product returns; |
• | our expectations regarding the clinical, therapeutic and competitive benefits of OMIDRIA and our product candidates; |
• | our ability to design, initiate and/or successfully complete clinical trials and other studies for our products and product candidates and our plans and expectations regarding our clinical trials, including our clinical trials for OMS721, for OMS527 and for OMS824; |
• | in our OMS721 program, our expectations regarding: whether enrollment in any or all ongoing and planned Phase 3 clinical trials will proceed as expected; whether accelerated approval, fast track designation, breakthrough therapy designation and/or orphan drug designation may be granted by the U.S. Food and Drug Administration, or FDA, or Priority Medicines status, conditional marketing authorization or orphan designation may be granted by the European Medicines Agency, or EMA, for indications for which we are pursuing such approval or designation; and potential label claims and assessments with respect to our Phase 3 clinical trial for patients with Immunoglobulin A nephropathy; |
• | our anticipation that we will rely on contract manufacturers to manufacture OMIDRIA for commercial sale and to manufacture our product candidates; |
• | our ability to enter into acceptable arrangements with potential corporate partners or contract service providers, including with respect to OMIDRIA, and our ability and plans to effect any such arrangement with respect to OMIDRIA in the European Union and place OMIDRIA on the market in at least one European Economic Area country prior to July 28, 2018 to preserve OMIDRIA marketing authorization in Europe; |
• | our ability to raise additional capital through the capital markets or through one or more corporate partnerships, equity offerings, debt financings, collaborations, licensing arrangements or asset sales; |
• | our expectations about the commercial competition that OMIDRIA and our product candidates, if commercialized, face or may face; |
• | the expected course and costs of existing claims, legal proceedings and administrative actions, our involvement in potential claims, legal proceedings and administrative actions, and the merits, potential outcomes and effects of both existing and potential claims, legal proceedings and administrative actions, as well as regulatory determinations, on our business, prospects, financial condition and results of operations, including but not limited to our patent infringement lawsuits against Sandoz, Inc., or Sandoz, and against Lupin Ltd. and Lupin Pharmaceuticals, Inc., which we refer to collectively as Lupin; |
• | the extent of protection that our patents provide and that our pending patent applications will provide, if patents issue from such applications, for our technologies, programs, products and product candidates; |
• | when or to what extent the dosing limitations in our OMS824 program may be removed, if at all; |
• | the factors on which we base our estimates for accounting purposes and our expectations regarding the effect of changes in accounting guidance or standards on our operating results; and |
i
• | our expected financial position, performance, revenues, growth, costs and expenses, magnitude of net losses and the availability of resources. |
Our actual results could differ materially from those anticipated in these forward-looking statements for many reasons, including the risks, uncertainties and other factors described in Item IA of Part I of this Annual Report on Form 10-K under the heading “Risk Factors” and in Item 7 of Part II under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and in our other filings with the Securities and Exchange Commission, or SEC. Given these risks, uncertainties and other factors, actual results or anticipated developments may not be realized or, even if substantially realized, they may not have the expected consequences to or effects on our company, business or operations. Accordingly, you should not place undue reliance on these forward-looking statements, which represent our estimates and assumptions only as of the date of the filing of this Annual Report on Form 10-K. You should read this Annual Report on Form 10-K completely and with the understanding that our actual results in subsequent periods may materially differ from current expectations. Except as required by applicable law, including the securities laws of the United States and the rules and regulations of the SEC, we assume no obligation to update or revise any forward-looking statements contained herein, whether as a result of any new information, future events or otherwise.
ii
OMEROS CORPORATION
ANNUAL REPORT ON FORM 10-K FOR THE YEAR ENDED DECEMBER 31, 2017
INDEX
Page | |
iii
PART I
This Annual Report on Form 10-K contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report. Please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K for further information.
ITEM 1. | BUSINESS |
Overview
We are a commercial-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting inflammation, complement-mediated diseases and disorders of the central nervous system.
Our drug product OMIDRIA® is marketed in the U.S. for use during cataract surgery or intraocular lens, or IOL, replacement. In our pipeline we have clinical-stage development programs focused on: complement-associated thrombotic microangiopathies; complement-mediated glomerulonephropathies; Huntington’s disease and cognitive impairment; and addictive and compulsive disorders. In addition, we have a diverse group of preclinical programs and two platforms: one capable of unlocking new G protein-coupled receptor, or GPCR, drug targets and the other used to generate antibodies. For OMIDRIA and each of our product candidates and our programs, other than OMS103, we have retained control of all commercial rights.
Commercial Product -- OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1%/0.3%
Overview. OMIDRIA is approved by the FDA for use during cataract surgery or IOL replacement to maintain pupil size by preventing intraoperative miosis (pupil constriction) and to reduce postoperative ocular pain, and is approved in all EU member states plus Iceland, Lichtenstein and Norway (together, the European Economic Area, or EEA) for use during cataract surgery and other IOL replacement procedures to maintain mydriasis (pupil dilation), to prevent miosis (pupil constriction), and to reduce postoperative eye pain. OMIDRIA is a proprietary drug product containing two active pharmaceutical ingredients, or APIs: ketorolac, an anti-inflammatory agent, and phenylephrine, a mydriatic, or pupil dilating, agent. Cataract and other lens replacement surgery involves replacement of the original lens of the eye with an artificial intraocular lens. These procedures are typically performed to replace a lens opacified by a cataract or to correct a refractive error. OMIDRIA is added to standard irrigation solution used during cataract and lens replacement surgery and is delivered intracamerally, or within the anterior chamber of the eye, to the site of the surgical trauma throughout the procedure. Preventing pupil constriction is essential for these procedures and, if miosis occurs, the risk of damaging structures within the eye and other complications increases as does the operating time required to perform the procedure.
United States. We launched OMIDRIA in the U.S. in the second quarter of 2015 and sell OMIDRIA primarily through wholesalers which, in turn, sell to ASCs and hospitals. In 2014, CMS, the federal agency responsible for administering the Medicare program, granted transitional pass-through reimbursement status for OMIDRIA, effective January 1, 2015. Pass-through status is designed to promote innovation and allows for separate payment (i.e., outside the packaged procedural payment) under Medicare Part B for certain new drugs and other medical technologies when used in hospital outpatient or ambulatory surgery centers and that meet well-established criteria specified by federal law and regulations governing Medicare spending. As of January 1, 2018, as scheduled, OMIDRIA is no longer subject to separate payment under Medicare Part B and, consequently, payment for the product is included as part of the packaged payment for the associated procedure for Medicare patients. Based on first quarter 2018 data to date, we believe that a substantial majority of facilities that were using OMIDRIA are awaiting resolution regarding reimbursement by CMS, or our decision to implement an alternative sales strategy, and, therefore, sales to our wholesalers during this period have been adversely affected. We expect this significant reduction in OMIDRIA sales to continue while the CMS reimbursement status of OMIDRIA remains uncertain. Both legislative and administrative means are being pursued to obtain permanent separate payment or similar reimbursement for OMIDRIA and/or to extend the pass-through reimbursement period from three to five years. For further discussion of OMIDRIA reimbursement and pricing, see Part II, Item 7, “Management’s Discussion and Analysis-Results of Operations.”
We have implemented a variety of programs and arrangements to facilitate the availability of OMIDRIA to cataract and IOL replacement patients in the U.S., including the following:
• | various purchase volume discount programs for OMIDRIA (for more information, see Part II, Item 7, “Management’s Discussion and Analysis--Results of Operations”); |
1
• | agreements to enable discounts on qualifying purchases of OMIDRIA by certain U.S. government purchasers and other eligible entities (e.g., 340B-eligible hospitals and clinics); and |
• | the OMIDRIAssure® Reimbursement Services Program, or OMIDRIAssure. |
The OMIDRIAssure coverage and reimbursement support services for surgeons and facilities help remove uncertainties about coding, billing and coverage of OMIDRIA. The “Equal Access” Patient Assistance and the “We Pay the Difference” Commercially Insured Patient Reimbursement Programs help address financial barriers restricting patient access to the drug. Under the “Equal Access” program, financially eligible uninsured and government-insured patients receive OMIDRIA free of charge for use during surgery. For commercially insured patients, through our “We Pay the Difference” program we pay the facility, on behalf of the patient, the difference between the facility’s acquisition cost for OMIDRIA, after accounting for any applicable volume discounts, and the amount covered by the patient’s insurance. We expect to evaluate these programs based on their ongoing effectiveness following any decision that is made regarding OMIDRIA reimbursement.
In December 2017, the FDA approved our supplemental new drug application, or sNDA, following review of efficacy and safety data from a pediatric clinical trial, expanding the indication for OMIDRIA to include use in pediatric patients (ages birth through 17 years old). In addition to the label expansion now including both pediatric and adult patients, the FDA also granted OMIDRIA an additional six months of U.S. market exclusivity, subject to any licenses that we may have (including pursuant to the Settlement Agreement with Par, described below) or into which we may enter. Under section 505A of the Federal Food, Drug, and Cosmetic Act, or FDCA, this six-month extension of market exclusivity is attached to the term of the drug’s patents listed in FDA’s Orange Book.
European Union and other International Territories. In 2015, we received approval from the European Commission, or EC, to market OMIDRIA in the EEA for use during cataract surgery and other IOL replacement procedures to maintain mydriasis (pupil dilation), to prevent miosis (pupil constriction), and to reduce postoperative eye pain. For the European OMIDRIA marketing authorization to remain valid, product must be placed on the market (i.e., released into the distribution chain) in at least one EEA country by July 28, 2018. Decisions about price and reimbursement for OMIDRIA are made on a country-by-country basis and may be required before marketing may occur in a particular country. We do not expect to see sales of OMIDRIA in any countries within the EEA and other international territories if we are unable to either enter into partnerships for the marketing and distribution of OMIDRIA or alternatively complete independent sales of the product in such countries within the EEA and other international territories. Timing of any such partnerships or independent sales depends on numerous factors, including completion of mutual diligence exercises or entry into suitable agreements with contract service vendors, respectively.
We have an exclusive supply and distribution agreement, or the ITROM Agreement, with ITROM Trading Drug Store, or ITROM, for the sale of OMIDRIA in the Kingdom of Saudi Arabia, the United Arab Emirates and certain other countries in the Middle East. Within the licensed territory, ITROM is responsible for obtaining marketing authorizations for OMIDRIA on our behalf and for promoting, marketing, selling and distributing product supplied by us. ITROM began selling OMIDRIA in December 2016 on a limited basis in the Kingdom of Saudi Arabia.
Par Settlement and Abbreviated New Drug Applications. In October 2017, we entered into a Settlement Agreement, or the Settlement Agreement, and consent judgment with Par Pharmaceutical, Inc. and its subsidiary, Par Sterile Products, LLC, or collectively, Par, resolving our patent litigation against Par that arose from Par’s filing of an Abbreviated New Drug Application, or ANDA, seeking approval from the FDA to market a generic version of OMIDRIA. Pursuant to the Settlement Agreement, Par, which had previously stipulated to infringement, acknowledged and confirmed the validity of each of the patents then listed in the Orange Book for OMIDRIA. In accordance with the terms of the Settlement Agreement, the U.S. District Court for the District of Delaware signed and entered a consent judgment on October 5, 2017, pursuant to which Par and its affiliates are prohibited from launching a generic version of OMIDRIA until the earlier of April 1, 2032 or a date on which the Company or a third party, through licensing or any future final legal judgment, should one ever exist, with respect to the Orange Book listed patents, is able to launch a generic version of OMIDRIA, as further detailed in the Settlement Agreement, or the Entry Date. Under the Settlement Agreement, we granted Par a non-exclusive, non-sublicensable license to make, sell and distribute a generic version of OMIDRIA between the Entry Date and the latest expiration of our U.S. patents related to OMIDRIA (i.e., October 23, 2033). During this period, Par will pay us a royalty equal to 15% of Par’s net sales of its generic version of OMIDRIA. For more information regarding this matter, see Part I, Item 3, “Legal Proceedings.”
In May 2017, we received a Notice Letter from Sandoz, Inc., or Sandoz, and a Notice Letter from Lupin Ltd. and Lupin Pharmaceuticals, Inc., which we refer to collectively as Lupin, stating that each of Sandoz and Lupin had filed with the FDA an ANDA containing a Paragraph IV Certification under the Hatch-Waxman Act seeking approval to market a generic version of OMIDRIA prior to the expiration of six Orange Book Patents. On June 21, 2017, Omeros filed a patent infringement lawsuit in the U.S. District Court for the District of Delaware against Sandoz and on June 22, 2017, Omeros filed a patent infringement lawsuit in the U.S. District Court for the District of Delaware against Lupin. The Delaware lawsuits against Sandoz and Lupin
2
were consolidated for all purposes by court order in October 2017. For more information regarding these matters, see Part I, Item 3, “Legal Proceedings.”
Our Product Candidates and Development Programs
Our product candidates currently in active clinical development include the following:
Product Candidate/Program | Targeted Disease | Development Status | Next Expected Milestone | Worldwide Rights |
Clinical | ||||
MASP-2 (OMS721) - Lectin Pathway Disorders | Immunoglobulin A (IgA) Nephropathy (IgAN) | Phase 3 | Complete Phase 3 Patient Enrollment | Omeros (In-licensed) |
MASP-2 (OMS721) - Lectin Pathway Disorders | Hematopoietic Stem-Cell Transplant-Associated Thrombotic Microangiopathy (HCT-TMA) | Phase 3 | Discuss Approval Pathway(s), Including Accelerated and Conditional Approvals, with FDA and EMA | Omeros (In-licensed) |
MASP-2 (OMS721) - Lectin Pathway Disorders | Atypical Hemolytic Uremic Syndrome (aHUS) | Phase 3 | Complete Phase 3 Patient Enrollment | Omeros (In-licensed) |
MASP-2 (OMS721) - Lectin Pathway Disorders | Lupus Nephritis and Other Renal Diseases | Phase 2 | Review Data; Determine Whether to Initiate Phase 3 Program | Omeros (In-licensed) |
PDE10 (OMS824) - CNS Disorders | Huntington’s Disease; Schizophrenia | Phase 2 (1) | Internal Review and Discussions with FDA | Omeros |
PPARγ (OMS405) - Addiction | Opioid and Nicotine Addiction | Phase 2 | Further Refine Development Path | Omeros |
(1) | Plans for continuation of the OMS824 program will be based on internal ongoing work and discussions with the FDA. Clinical trials in our Huntington’s program are approved by the FDA to progress subject to dosing limitations. Clinical trials evaluating OMS824 in schizophrenia remain suspended at the request of the FDA until we submit to the FDA a protocol for a schizophrenia trial and receive the Agency’s clearance to proceed. For additional information, see “Other Clinical Programs-PDE10 Programs-OMS824 for Huntington’s Disease and Schizophrenia.” |
3
Our programs currently in preclinical development, as well as our platforms, include the following:
Product Candidate/Program | Targeted Disease | Development Status | Next Expected Milestone | Worldwide Rights |
Preclinical / Platforms | ||||
PDE7 (OMS527) | Addictions and Compulsive Disorders; Movement Disorders | Preclinical | Submit CTA | Omeros (Compounds In-licensed) |
MASP-3 (OMS906) - Alternative Pathway Disorders | Paroxysmal Nocturnal Hemoglobinuria (PNH) and Other Alternative Pathway Disorders | Preclinical | Complete Manufacturing Scale-up of a Clinical Candidate for IND-Enabling Toxicology Studies | Omeros |
MASP-2 - Small-Molecule Inhibitors | aHUS, IgAN, HCT-TMA and Age-Related Macular Degeneration (AMD) | Preclinical | Continue Medicinal Chemistry and Co-Crystallization Efforts | Omeros |
MASP-3 - Small-Molecule Inhibitors | PNH and Other Alternative Pathway Disorders | Preclinical | Continue Medicinal Chemistry and Advance Co-Crystallization Efforts | Omeros |
GPCR Platform, including GPR174 and other Class A Orphan GPCRS | CNS, Metabolic, CV, Oncologic, Musculoskeletal & Other Disorders | Preclinical | Continue Drug Discovery and Selected Medicinal Chemistry for Class A and Class B Orphan and Non-Orphan GPCRs | Omeros |
Antibody Platform | Metabolic, CV, Oncologic, Musculoskeletal & Other Disorders | Preclinical | Continue Developing Antibodies Targeting Lectin and Alternative Pathway of Complement System and Expanding Antibody Library | Omeros (In-licensed) |
MASP Clinical and Preclinical Programs
MASP-2 Program - OMS721 - Lectin Pathway Disorders
Overview. Mannan-binding lectin-associated serine protease-2, or MASP-2, is a novel pro-inflammatory protein target involved in activation of the complement system, which is an important component of the immune system. The complement system plays a role in the inflammatory response and becomes activated as a result of tissue damage or trauma or microbial pathogen invasion. Inappropriate or uncontrolled activation of the complement system can cause diseases characterized by serious tissue injury. MASP-2 is recognized as the effector enzyme, and is required for the function, of the lectin pathway, one of the principal complement activation pathways. Importantly, inhibition of MASP-2 has been demonstrated not to interfere with the antibody-dependent classical complement activation pathway, which is a critical component of the acquired immune response to infection.
We are developing MASP-2 antibodies and small molecules and we expect that the intended therapeutic effect can be achieved through multiple routes of administration, including subcutaneous and intravenous, or IV, administration of our antibodies and oral and IV administration of our small molecules. OMS721 is our lead human monoclonal antibody targeting MASP-2. The current development focus for OMS721 is diseases in which the lectin pathway has been shown to contribute to significant tissue injury and pathology. These diseases are typically characterized by significant end organ injuries, such as kidney or central nervous system injury, when not treated. Phase 3 clinical programs are in process for OMS721 in Immunoglobulin A, or IgA, nephropathy, in hematopoietic stem cell transplant-associated thrombotic microangiopathy, or HCT-TMA, and in atypical hemolytic uremic syndrome, or aHUS.
Renal Disease
Phase 2 Clinical Trial - Renal Diseases. We have been conducting a Phase 2 clinical trial in patients with complement-associated renal diseases, specifically designed to cover: (1) IgA nephropathy; (2) membranous nephropathy; (3) lupus nephritis; and (4) complement component (C3) glomerulopathy. The open-label cohort completed in May 2017. No patients with C3 glomerulopathy were enrolled. The double-blind, placebo-controlled cohorts covering patients with IgA nephropathy are ongoing. All patients in the open-label cohort of the trial were required to have high levels of urinary protein (a marker used by nephrologists to assess disease activity) despite ongoing treatment with corticosteroids. These inclusion criteria were
4
intended to ensure that study patients are unlikely to improve spontaneously. Patients in the open-label cohort were treated with OMS721 for a total of 12 weeks: four weeks maintaining their entry corticosteroid dose; four weeks of corticosteroid tapering, if tolerated; and four weeks of resultant corticosteroid dose maintenance. Patients were then followed post-treatment for six weeks. During the trial, all patients with IgA nephropathy in the open-label cohort demonstrated clinically meaningful and statistically significant improvement in proteinuria.
In November 2017, we announced additional positive data that showed three of the four IgA nephropathy patients in the open label cohort maintained the proteinuria reduction demonstrated in the clinical trial during the follow-up period (at 12, 11 and three months, respectively, after cessation of dosing). Albumin/creatinine ratios, or uACRs, during the extended follow-up period remained decreased in these three patients. Numerical improvement in estimated glomerular filtration rate, or eGFR, a measure of renal function, was also observed in three of the four patients after the trial. OMS721 was well-tolerated in the clinical trial with fatigue and anemia as the most commonly reported adverse events. Based on the data in IgA nephropathy patients in the open-label cohort plus supporting data, the FDA granted breakthrough therapy designation to OMS721 in IgA nephropathy.
In the blinded, controlled cohort evaluating patients with IgA nephropathy in the U.S. and Hong Kong, patients must have elevated levels of urinary protein but are not treated with corticosteroids. Patients in the blinded, controlled cohort are treated with OMS721 or placebo for a total of 12 weeks, then followed for six weeks post-treatment. Data from the double-blind portion of the U.S. cohort are expected mid-year.
Also, in March 2017 we reported encouraging results in lupus nephritis patients in the Phase 2 renal disease trial. Four of five patients showed a substantial reduction in 24-hour urine protein excretion over the treatment period. The fifth patient experienced a systemic disease flare and demonstrated a substantial increase in 24-hour urine protein excretion. The majority of lupus responders were able to taper their steroid doses. We are reviewing the data from this particular clinical trial for possible further development.
Phase 3 Program - IgA Nephropathy. In our Phase 3 program for OMS721 in patients with IgA nephropathy, we have reached concurrence with the FDA on the study design for a Phase 3 clinical trial. The single Phase 3 trial design is a randomized, double-blind, placebo-controlled multicenter trial in patients at least 18 years of age with biopsy-confirmed IgA nephropathy and with 24-hour urine protein excretion greater than 1 g/day at baseline on optimized renin‑angiotensin system, or RAS, blockade. Initially, patients are expected to receive an IV dose of study drug each week for 12 weeks; additional weekly dosing can be administered for partial responders and relapsers. The primary endpoint, which potentially could suffice for full approval depending on the effect size, is reduction in proteinuria at 24 weeks after the start of dosing. The trial is structured to employ an adaptive design that will allow intra-trial adjustment in sample size. For purposes of safety and efficacy assessments, the initial sample size for the proteinuria endpoint is estimated at 140 patients in each of the treatment and placebo groups. This will include a subset of patients with high levels of proteinuria (i.e., equal to or greater than 2 g/day) at baseline, and a substantial improvement at 24 weeks in this subset of patients alone could potentially form the basis for full approval. We believe that the trial design will allow assessment for either full or accelerated approval at 24 weeks based on proteinuria results either (1) across the general population of study patients or (2) in the high-proteinuria subset of patients. In the event that the primary endpoint at 24 weeks results in accelerated approval from the FDA, change in eGFR is expected to be assessed at approximately three years after the start of dosing. These eGFR data, if satisfactory, would then likely form the basis for full approval. The initial sample size estimate for the eGFR endpoint is approximately 160 patients per group and also will be adjustable under the study’s adaptive design. Patient enrollment in this Phase 3 clinical trial is underway.
We also are planning to initiate a second OMS721 Phase 3 clinical trial focused solely on IgA nephropathy patients with high proteinuria. We expect that this trial would require significantly fewer patients than our trial in the IgA nephropathy general patient population, would include biopsy data to assess the disease modifying potential of OMS721, would likely enroll more quickly than our broader study and would provide a third approach to achieve full approval on 24-week proteinuria data alone.
The FDA has granted breakthrough therapy designation to OMS721 for the treatment of IgA nephropathy. In the EU, OMIDRIA has received orphan drug designation in patients with IgA nephropathy from the EMA, and we are requesting eligibility for the EMA’s Priority Medicines, or PRIME, program for OMS721 in this indication as well.
Thrombotic Microangiopathies
Phase 2 Clinical Trial - TMAs. We have an ongoing Phase 2 clinical trial in patients with TMAs, including initially aHUS, HCT-TMA and thrombotic thrombocytopenia, or TTP. Currently, patients with TTP or aHUS are no longer being enrolled in this study. To be eligible for enrollment, HCT-TMA patients are required to be adults with post-transplant TMA persisting at least two weeks following calcineurin inhibitor modification (conservative treatment). The primary efficacy
5
endpoint of the study is change in platelet count. Additional efficacy endpoints include changes in lactate dehydrogenase, or LDH, and haptoglobin levels.
In March 2017, we announced additional positive data in patients with HCT-TMA from the ongoing Phase 2 clinical trial. Statistically significant and clinically meaningful improvements in TMA disease activity were observed over the course of treatment, specifically in mean platelet count, mean LDH and mean haptoglobin. In October 2017, we announced the presentation by a trial investigator of a case report of a patient in this Phase 2 clinical trial whose post-transplant course was complicated by multiple episodes of steroid-refractory grade IV (life-threatening) graft-versus-host disease, or GvHD. The patient then presented with TMA and a recurrence of GVHD, which both resolved following OMS721 treatment.
In February 2018, we reported new results in patients with HCT-TMA from this ongoing Phase 2 study. The data demonstrate an increase in estimated median overall survival in HCT-TMA patients treated with OMS721 compared to a matched historical control (347 days vs. 21 days, respectively, by Kaplan-Meier analysis; p < 0.0001 by log-rank test). In addition to and consistent with the survival data reported, updated assessments of platelet count, lactate dehydrogenase, or LDH, and haptoglobin – all markers of TMA activity – continued to demonstrate clinically meaningful and statistically significant improvements in the HCT-TMA patients treated with OMS721. As of February 16, 2018, a total of 19 HCT-TMA patients had been treated with OMS721 (18 in the ongoing study and one patient under a compassionate use protocol), with OMS721 being well tolerated in the Phase 2 trial. No safety concerns were identified. The most commonly reported adverse events were diarrhea and neutropenia. Four deaths in HCT-TMA patients occurred during the study: one due to progression of acute myeloid leukemia, two due to neutropenic sepsis, and one due to acute renal and respiratory failure. Three of these deaths were deemed not to be related to OMS721 and only one of these deaths – the acute renal and respiratory failure, which is a common complication of HCT – was considered “possibly drug-related” because an association could not be definitively ruled out by the investigator.
In addition, we also observed significant improvement in transfusion requirements in HCT-TMA patients in this Phase 2 study. Eight of the 19 patients were receiving significant red blood cell and platelet transfusions at the time of study entry. The transfusions were either stopped completely or markedly reduced in seven of the eight patients. The eighth patient had ongoing acute myeloid leukemia, only received two doses of OMS721, discontinued the study and died shortly thereafter.
Phase 3 Program - HCT-TMA. We have initiated a Phase 3 program for OMS721 in HCT-TMA. We intend to amend the ongoing Phase 2 protocol following discussion with the FDA and/or the EMA regarding accelerated and/or conditional approval. Enrollment of HCT-TMA patients in the Phase 2 TMA trial has continued pending those discussions.
The FDA has granted to OMS721 orphan drug designation for the prevention (inhibition) of complement-mediated TMAs, which includes aHUS and HCT-TMA. We are also seeking breakthrough therapy designation from the FDA, as well as eligibility to the PRIME program from the EMA, for OMS721 in this indication. We are scheduled to meet with FDA and are requesting meetings with regulatory bodies in the EU to discuss the most expeditious approval path, including accelerated and conditional approvals, for OMS721 in HCT-TMA.
Phase 3 Program - aHUS. We have an ongoing Phase 3 clinical program in patients with aHUS with active sites in both the U.S. and Europe. The single-arm (i.e., no control arm), open-label Phase 3 clinical trial in patients with newly diagnosed or ongoing aHUS is enrolling. This trial is targeting approximately 40 patients for EMA approval and U.S. accelerated approval with 80 patients required for full approval in the U.S. Dosing consists of an initial IV loading followed by daily subcutaneous dosing. Based on discussions with the FDA and the EMA, we expect that the clinical package for the Biologics License Application, or BLA, would be similar to that which formed the basis of approval for Soliris® (eculizumab). We have also received feedback and reached concurrence from the FDA and from the EMA on our ongoing and planned manufacturing for both the Phase 3 program and commercialization of OMS721 as well as on our nonclinical safety and toxicology plan, most of which has already been successfully completed with no significant adverse findings.
The FDA has granted to OMS721 orphan drug designation for the prevention (inhibition) of complement-mediated TMAs, which includes aHUS and HCT-TMA, and fast track designation for the treatment of patients with aHUS.
Expanded Access / Compassionate Use. We have received requests from investigators and other physicians for expanded access to OMS721. Expanded access, sometimes called “compassionate use,” is the use of an investigational medical product outside of a clinical trial. Expanded access is permitted by the FDA and other regulatory agencies under specific circumstances. OMS721 has been provided to several patients through this program.
Other Preclinical Studies. For OMS721, we have generated positive preclinical data in in vivo models of AMD, myocardial infarction, diabetic neuropathy, stroke, ischemia-reperfusion injury, and other diseases and disorders.
6
Licensing Arrangements. We hold worldwide exclusive licenses to rights related to MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for those antibodies from the University of Leicester, from its collaborator, Medical Research Council at Oxford University, or MRC, and from Helion Biotech ApS, or Helion. For a more detailed description of these licenses, see “License and Development Agreements.”
MASP-3 Program - OMS906 - Alternative Pathway Disorders
Overview. As part of our MASP program, we have identified mannan-binding lectin-associated serine protease-3, or MASP-3, which has been shown to be the key activator of the complement system’s alternative pathway, or APC, and we believe that we are the first to make this and related discoveries associated with the APC. The complement system is part of the immune system’s innate response, and the APC is considered the amplification loop within the complement system. MASP-3 is responsible for the conversion of pro-factor D to factor D, and converted factor D is necessary for the activation of the APC. Based on our alternative pathway-related discoveries, we have expanded our intellectual property position to protect our inventions stemming from these discoveries beyond MASP-2-associated inhibition of the lectin pathway to include inhibition of the alternative pathway. In addition to our MASP-2 inhibitors of the lectin pathway, we are developing inhibitors of the alternative pathway as well as bispecific inhibitors of both the alternative and lectin pathways. For each of these targets, our efforts are directed to both antibody and small-molecule development.
Our current primary focus in this program is developing MASP-3 inhibitors for the treatment of disorders related to the APC. We believe that MASP-3 inhibitors may have the potential to treat patients suffering from a wide range of diseases and conditions, including: paroxysmal nocturnal hemoglobinuria, or PNH; C3 glomerulopathy; multiple sclerosis; arthritis; traumatic brain injury; neuromyelitis optica; pauci-immune necrotizing crescentic glomerulonephritis; disseminated intravascular coagulation; age-related macular degeneration; asthma; dense deposit disease; Bechet’s disease; aspiration pneumonia; TMA; ischemia-reperfusion injury; Guillain Barre syndrome; Alzheimer’s disease; amylotrophic lateral sclerosis; systemic lupus erythematosus; diabetic retinopathy; uveitis; chronic obstructive pulmonary disease; transplant rejection; acute respiratory distress syndrome; antineutrophil cytoplasmic antibody-associated vasculitis; anti-phospholipid syndrome; atherosclerosis; myasthenia gravis and others. In preparation for clinical trials, the manufacturing scale-up process is underway for a MASP-3 inhibitor antibody and we are currently targeting PNH as the first clinical indication for OMS906. We also are developing small-molecule inhibitors of MASP-3.
Our OMS906 program has generated positive data in a well-established animal model associated with PNH including in non-human primates. The program has also generated positive data in a well-established model of arthritis.
Licensing Arrangements. We jointly own and hold worldwide exclusive license rights related to therapeutic applications for inhibiting MASP-3 from the University of Leicester. For a more detailed description of these licenses, see “License and Development Agreements.”
Other Clinical Programs
PDE10 Programs - OMS824 for Huntington’s Disease and Schizophrenia
Overview. Phosphodiesterase 10, or PDE10, is an enzyme that is expressed in areas of the brain strongly linked to diseases that affect cognition, including Huntington’s disease and schizophrenia. Cognitive dysfunction occurs early in these diseases and is responsible for substantial disability. PDE10 inhibitors have been shown to be effective in multiple animal models of behavior and cognition, and there remain substantial unmet clinical needs with current treatments. Our proprietary compound OMS824 inhibits PDE10 and is being developed in clinical programs for the treatment of cognitive disorders, including Huntington’s disease and schizophrenia. OMS824 has received orphan drug designation for the treatment of Huntington’s disease and fast track designation for the treatment of cognitive impairment in patients with Huntington’s disease.
Clinical Trials. OMS824 is in a Phase 2 clinical program for the treatment of Huntington’s disease and a Phase 2 clinical program evaluating OMS824 for the treatment of schizophrenia. We are also evaluating other neurological indications for OMS824. Plans for continuation of the OMS824 program will be based on internal ongoing work and subsequent discussions with the FDA. The FDA has approved the advancement of clinical trials in our Huntington’s program subject to dosing limitations pending further discussions potentially to remove those limitations. Clinical trials evaluating OMS824 in schizophrenia remain suspended at the request of the FDA until we submit to the FDA a protocol for a schizophrenia trial and receive the Agency’s clearance to proceed.
Funding Agreement with The Stanley Medical Research Institute. Our preclinical development of OMS824 was funded by The Stanley Medical Research Institute, or SMRI, a non-profit corporation that supports research on the causes and
7
treatment of schizophrenia and bipolar disorder. For a more detailed description of our agreement with SMRI, see “License and Development Agreements.”
PPARγ Program - OMS405
Overview. In our peroxisome proliferator-activated receptor gamma, or PPARγ, program, we are developing proprietary compositions that include PPARγ agonists for the treatment and prevention of addiction to substances of abuse, which may include opioids, nicotine and alcohol. We believe that Omeros is the first to demonstrate a link between PPARγ and addiction disorders. Data from clinical studies and from animal models of addiction suggest that PPARγ agonists could be efficacious in the treatment of a wide range of addictions.
Clinical trials. Our collaborators at The New York State Psychiatric Institute have completed two Phase 2 clinical trials related to our PPARγ program. These studies evaluated a PPARγ agonist, alone or in combination with other agents, for treatment of addiction to heroin and to nicotine. The National Institute on Drug Abuse provided substantially all of the funding for these clinical trials and solely oversaw the conduct of these trials. We have the right or expect to be able to reference the data obtained from these studies for subsequent submissions to the FDA and continue to retain all other rights in connection with the PPARγ program. We have also reported positive results (i.e., decreased craving and protection of brain white matter) from a Phase 2 clinical trial conducted by an independent investigator evaluating the effects of a PPARγ agonist in patients with cocaine use disorder.
Patent Assignment Agreement with Roberto Ciccocioppo, Ph.D. We acquired the patent applications and related intellectual property rights for our PPARγ program in February 2009 from Roberto Ciccocioppo, Ph.D., of the Università di Camerino, Italy, pursuant to a patent assignment agreement. For a more detailed description of our agreement with Dr. Ciccocioppo, see “License and Development Agreements.”
Preclinical Programs and Platforms
PDE7 Program - OMS527
Overview. Our phosphodiesterase 7, or PDE7, program is based on our discoveries of previously unknown links between PDE7 and any addiction or compulsive disorder and between PDE7 and any movement disorders, such as Parkinson’s disease. PDE7 appears to modulate the dopaminergic system, which plays a significant role in regulating both addiction and movement. We believe that PDE7 inhibitors could be effective therapeutics for the treatment of addiction and compulsive disorders as well as for movement disorders. Data generated in preclinical studies support the use of PDE7 inhibitors in both of these therapeutic areas. We have selected nicotine addiction as the initial indication and have initiated toxicology studies intended to support the submission of a Clinical Trial Application, or CTA, in the EU and subsequent clinical trials. We currently expect to submit a CTA for OMS527 by mid-2018.
Exclusive License Agreement with Daiichi Sankyo Co., Ltd. We hold an exclusive license to certain PDE7 inhibitors claimed in patents and pending patent applications owned by Daiichi Sankyo Co., Ltd. (successor-in-interest to Asubio Pharma Co., Ltd.), or Daiichi Sankyo, for use in the treatment of movement, addiction and compulsive disorders as well as other specified indications. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements.”
GPCR Platform
Overview. GPCRs, which are cell surface membrane proteins involved in mediating both sensory and nonsensory functions, comprise one of the largest families of proteins in the genomes of multicellular organisms. Sensory GPCRs are involved in the perception of light, odors, taste and sexual attractants. Non-sensory GPCRs are involved in metabolism, behavior, reproduction, development, hormonal homeostasis and regulation of the central nervous system. The vast majority of GPCR drug targets are non-sensory. Although GPCRs form a super-family of receptors, individual GPCRs display a high degree of specificity and affinity for the functionally active molecules, or ligands, that bind to a given receptor. Ligands can either activate the receptor (agonists) or inhibit it (antagonists and inverse agonists). When activated by its ligand, the GPCR interacts with intracellular G proteins, resulting in a cascade of signaling events inside the cell that ultimately leads to the particular function linked to the receptor. Without a known ligand, there is no template from which medicinal chemistry efforts can be readily initiated nor a means to identify the GPCR’s signaling pathway and, therefore, drugs are very difficult to develop against orphan GPCRs. “Unlocking” these orphan GPCRs by identifying one or more of their respective ligands could lead to the development of drugs that act at these new targets.
To our knowledge, despite efforts by others in the biopharmaceutical industry and academic community, Omeros’ technology is the first commercially viable technology capable of identifying ligands of orphan GPCRs in high throughput. We
8
have developed a proprietary cellular redistribution assay, or CRA, which we use in a high-throughput manner to identify synthetic ligands, including antagonists, agonists and inverse agonists, that bind to and affect the function of orphan GPCRs. We have screened Class A orphan GPCRs against our small-molecule chemical libraries using the CRA. As of February 16, 2018, we had identified and confirmed compounds that interact with 54 of the 81 Class A orphan GPCRs linked to a wide range of indications including cancer as well as metabolic, cardiovascular, immunologic, inflammatory and central nervous system disorders. We are conducting in vitro and in vivo preclinical efficacy studies and optimizing compounds for a number of targets including: GPR151, linked to schizophrenia and cognition; GPR161, which is associated with triple negative breast cancer and various sarcomas; GPR183, linked to osteoporosis and to Epstein-Barr virus infections and related diseases; GPR174, which appears to be involved in the modulation of the immune system and, in particular, increases cytokine production and inhibits production of regulatory T cells, or “T-regs,” and checkpoint molecules, all of which are known to be important in autoimmune disease, such as multiple sclerosis, in cancer and in organ transplantation; and OPN4, linked to seasonal affective disorder, mood disorders, sleep disorders and photophobia.
In addition to Class A orphan GPCRs, we have also begun screening orphan and non-orphan Class B receptors. Class B GPCRs have large extracellular domains and their natural ligands are generally large peptides, making the development of orally active, small-molecule drugs against these receptors, such as glucagon and parathyroid hormone, a persistent challenge. Our CRA technology finds functionally active small molecules for GPCRs, which we believe could lead to the development of oral medications for many of the Class B GPCRs. While our focus to date has remained on Class A orphan GPCRs, as of February 16, 2018, we had identified and confirmed sets of compounds that interact selectively with, and modulate signaling of, a small subset of Class B GPCRs, namely glucagon-like peptide-1 receptor, or GLP-1R, and parathyroid hormone 1 receptor, or PTH-1R.
GPCR Platform Funding Agreements with Vulcan Inc. and the Life Sciences Discovery Fund. In October 2010, we entered into funding agreements for our GPCR program with Vulcan Inc. and its affiliate, which we refer to collectively as Vulcan, and with the Life Sciences Discovery Fund Authority, a granting agency of the State of Washington, or LSDF. For a more detailed description of these agreements, see “License and Development Agreements.”
Antibody Platform
Overview. Our proprietary ex vivo platform for the discovery of novel, high-affinity monoclonal antibodies, which was in-licensed from the University of Washington and then further developed by our scientists, utilizes a chicken B-cell lymphoma cell line. It has successfully generated diverse antibodies that can be readily engineered. This platform offers several advantages over other antibody platforms. The ex vivo immunizations of our proprietary cell line are significantly more rapid than whole animal immunizations and conventional hybridoma technology. By avoiding immunization of mice or other animals, we believe the antibodies we generate from this platform are not limited by immunological tolerance. In addition, our platform is capable of producing novel antibodies against difficult targets, such as highly homologous proteins, enzymes, and receptors with short extracellular domains. Chicken antibodies also have unique features that enable binding capabilities distinct from mammalian antibodies.
Using our platform and other know-how and techniques, we have generated antibodies to several clinically significant targets, including highly potent antibodies against MASP-3 and MASP-1, and our platform continues to add to our pipeline antibodies against additional important targets.
Asset Purchase Agreement with Xori Corporation. In February 2012 we entered into an Asset Purchase Agreement, or the Xori APA, with Xori Corporation, or Xori, pursuant to which we acquired all of Xori’s rights and obligations in certain license and material transfer agreements, intellectual property, antibodies and other assets related to our antibody platform. We are obligated to make development and research-related milestone payments to Xori.
Exclusive License Agreement with the University of Washington. We hold a worldwide exclusive license to patent rights related to our antibody platform from the University of Washington. For a more detailed description of this agreement, see “License and Development Agreements.”
PharmacoSurgery®
We believe that current standards of care for the management and treatment of surgical trauma are limited in effectiveness. Surgical trauma causes a complex cascade of molecular signaling and biochemical changes, resulting in inflammation, pain, pupil constriction, muscle spasm, loss of function and other problems. As a consequence, multiple pharmacologic actions are required to manage the complexity and inherent redundancy of the cascade. Accordingly, we believe that single-agent treatments acting on single targets do not result in optimal therapeutic benefit. We have generated from our PharmacoSurgery platform proprietary products, such as OMIDRIA, and product candidates, such as OMS103 for use during
9
arthroscopic procedures, that are combinations of therapeutic agents designed to act simultaneously at multiple discrete targets to block preemptively the molecular-signaling and biochemical cascade caused by surgical trauma and to provide clinical benefits both during and after surgery. These products and product candidates are supplied in pre-dosed, pre-formulated, single-use containers and added to standard surgical irrigation solutions, delivered intraoperatively to the site of tissue trauma throughout the surgical procedure. This is expected to result in the delivery of low concentrations of agents with minimal systemic uptake and reduced risk of adverse side effects and does not require a surgeon to change his or her operating routine.
OMS103, part of our PharmacoSurgery platform, is a proprietary combination of anti-inflammatory/analgesic APIs, specifically amitriptyline, ketoprofen and oxymetazoline, each with well-known safety and pharmacologic profiles, and was designed to provide a multimodal approach to block preemptively the inflammatory cascade induced by arthroscopy. In June 2015, we entered into an exclusive licensing agreement, or the OMS103 Agreement, with Fagron Compounding Services, LLC, d/b/a Fagron Sterile Services, and JCB Laboratories, LLC, or collectively Fagron, an FDA-registered human drug outsourcing facility, under which Fagron is obligated to produce under Good Manufacturing Practice, or GMP, and to commercialize OMS103 in the U.S. Fagron has not met its performance diligence obligations under the OMS103 Agreement, including initiating sales, and we do not expect them to do so. We are currently evaluating our options regarding the OMS103 Agreement and our OMS103 program. For a more detailed description of this agreement, see “License and Development Agreements.”
Sales and Marketing
We have retained all worldwide marketing and distribution rights to OMIDRIA, our product candidates and our development programs, other than OMS103. This allows us the opportunity to market and sell independently OMIDRIA or, if approved, any of our product candidates, to make arrangements with third parties to perform these services for us, or both.
With respect to OMIDRIA in the U.S., we have developed our own internal marketing and sales capabilities and, as of February 16, 2018, employ 45 sales and reimbursement team members. For the EU OMIDRIA marketing authorization to remain valid, product must be placed on the market (i.e., released into the distribution chain) in at least one EEA country by July 28, 2018 and we expect that this will occur. Outside of the U.S. and EU, we have the ITROM Agreement for certain countries in the Middle East and we are exploring additional potential regional partnerships to make OMIDRIA available to ophthalmologists. Other than the ITROM Agreement, we have not yet entered into any agreements with third parties to market OMIDRIA outside of the U.S.
Manufacturing, Supply and Commercial Operations
OMIDRIA. We use third parties to produce, store and distribute OMIDRIA and currently do not own or operate manufacturing facilities. Our agreements with these third parties include confidentiality and intellectual property provisions to protect our proprietary rights related to OMIDRIA. We require manufacturers that produce APIs and finished drug products to operate in accordance with current Good Manufacturing Practices, or cGMPs, and all other applicable laws and regulations.
We have an agreement with Hospira Worldwide, Inc., a wholly owned subsidiary of Pfizer, Inc., or Hospira, to provide commercial supply of OMIDRIA. The Hospira manufacturing site for OMIDRIA cleared the FDA in December 2017. Under the agreement with Hospira, or the Hospira OMIDRIA Agreement, Hospira has agreed to manufacture and supply, and we have agreed to purchase, a minimum percentage of our requirements of OMIDRIA for commercial sales and clinical supplies for the development of additional therapeutic indications in the U.S. In addition, Hospira has agreed to manufacture and supply a portion of our requirements of OMIDRIA in the EU in an amount to be mutually agreed by the parties (not to exceed a maximum percentage of our EU requirements) by amendment, with there being no minimum purchase and supply requirement in the EU if the parties do not enter into such an amendment to the agreement. The Hospira OMIDRIA Agreement has an initial term of five years from the date of first commercial sale of OMIDRIA in the U.S. or in any country in the EU, and thereafter is renewed automatically for up to two additional one-year periods. The Hospira OMIDRIA Agreement may be terminated prior to the end of its term upon the occurrence of certain specified events, including without limitation an uncured breach of the agreement or bankruptcy or dissolution of a party. Upon termination of the Hospira OMIDRIA Agreement, except in the case of termination for an uncured breach by Hospira, we will be required to purchase all of Hospira’s inventory of OMIDRIA and, if applicable, all work-in-progress inventory and to reimburse Hospira for all supplies purchased or ordered based on firm purchase orders or our estimates of its requirements of OMIDRIA.
We have used multiple suppliers for the APIs for OMIDRIA in the past and we intend to leverage Hospira’s sourcing of APIs in the future under the Hospira OMIDRIA Agreement. Given the large amount of these APIs manufactured annually by these and other suppliers, and the quantities of these APIs we have on hand, we anticipate that we will be capable of addressing our commercial API supply needs for OMIDRIA in the near-term. We have not yet signed commercial agreements with suppliers for the supply of all of our anticipated commercial quantities of these APIs for OMIDRIA, although we may elect to do so in the future.
10
In the U.S., we sell OMIDRIA through a limited number of wholesalers that distribute the product to ASCs and hospitals. Title transfers upon delivery of OMIDRIA to the wholesaler. We use a single third-party logistics provider to handle warehousing and final packaging of our commercial supply of OMIDRIA in the U.S. and to ship OMIDRIA to our wholesalers. Our third-party logistics provider also performs certain support services on our behalf. Virtually all of our revenues for the last three fiscal years were generated from OMIDRIA product sales in the U.S. Our four major distributors -- AmerisourceBergen Corporation, Cardinal Health, Inc., McKesson Corporation and FFF Enterprises -- together with entities under their common control each accounted for 10% or more, and nearly 100% in aggregate, of our total revenue in 2017.
Product Candidates. We have laboratories in-house for analytical method development, bioanalytical testing, formulation, stability testing and small-scale compounding of laboratory supplies of product candidates. We utilize contract manufacturers to produce sufficient quantities of product candidates for use in preclinical and clinical studies and to store and distribute our product candidates, and we currently do not own or operate manufacturing facilities for our product candidates. We require manufacturers that produce APIs and finished drug products for clinical use to operate in accordance with cGMPs and all other applicable laws and regulations. We anticipate that we will rely on contract manufacturers to develop and manufacture our product candidates for commercial sale. We maintain agreements with potential and existing manufacturers that include confidentiality and intellectual property provisions to protect our proprietary rights related to our product candidates. We have not yet entered into a commercial supply agreement for any of our product candidates, although we intend to do so prior to the applicable product candidate’s commercial launch. Given the nature of the manufacturing processes of our product candidates, we anticipate that we will be capable of identifying contract manufacturers to produce these product candidates and of entering into agreements for the commercial supply of these drugs.
License and Development Agreements
MASP Program. Under our exclusive license agreements with the University of Leicester and MRC, we have agreed to pay royalties to each of the University of Leicester and MRC that are a percentage of any proceeds we receive from the licensed MASP-2 technology during the terms of the agreements. Our exclusive license agreement with the University of Leicester, but not our agreement with the MRC, also applies to other MASPs. The continued maintenance of these agreements requires us to undertake development activities. We must pay low single-digit percentage royalties with respect to proceeds that we receive from products incorporating certain intellectual property within the licensed technology that are used, manufactured, directly sold or directly distributed by us, and we must pay royalties, in the range of a low single-digit percentage to a low double-digit percentage, with respect to proceeds we receive from sublicense royalties or fees that we receive from third parties to which we grant sublicenses to certain intellectual property within the licensed technology. We did not make any upfront payments for these exclusive licenses nor are there any milestone payments or reversion rights associated with these license agreements. We also agreed to sponsor research of MASP-2 at these institutions at pre-determined rates for maximum terms of approximately three years. We have agreed to expand the scope of research at the University of Leicester to MASP-3 and continued the sponsorship of research at the University of Leicester on a year-by-year basis until the fall of 2017. We retain worldwide exclusive licenses from these institutions to develop and commercialize any intellectual property rights developed in the sponsored research. The term of each license agreement ends when there are no longer any pending patent applications, applications in preparation or unexpired issued patents related to any of the intellectual property rights we are licensing under the agreement. Both of these license agreements may be terminated prior to the end of their terms by us for convenience or by one party if the other party (1) breaches any material obligation under the agreement and does not cure such breach after notice and an opportunity to cure or (2) is declared or adjudged to be insolvent, bankrupt or in receivership and materially limited from performing its obligations under the agreement.
In April 2010, we entered into an exclusive license agreement with Helion Biotech ApS, or Helion, pursuant to which we received a royalty-bearing, worldwide exclusive license to all of Helion’s intellectual property rights related to MASP-2 antibodies, polypeptides and methods in the field of inhibition of mannan-binding lectin-mediated activation of the complement system for the prevention, treatment or diagnosis of any disease or condition. We are obligated to make remaining development and sales milestone payments to Helion of up to $6.1 million upon the achievement of certain events, such as the filing of an IND with the FDA, initiation of Phase 2 and 3 clinical trials, receipt of marketing approval, and reaching specified sales milestones. We are obligated to pay Helion a low single-digit percentage royalty on net sales of a MASP-2 inhibitor product covered by the patents licensed under the agreement. The term of the agreement continues so long as there is a valid, subsisting and enforceable claim in any patents or patent applications covered by the agreement. The agreement may be terminated sooner by either party following a material breach of the agreement by the other party that has not been cured within 90 days.
OMS824. We have agreed to pay royalties to SMRI based on any net income we receive from sales of a PDE10 product until we have paid a maximum aggregate amount that is a low single-digit multiple of the amount of grant funding that we have received from SMRI. This multiple increases as time elapses from the date we received the grant funding. There are no minimum payment obligations under our agreement with SMRI. Based on the amount of grant funding that we received from
11
SMRI, the maximum amount of royalties payable to SMRI is $12.8 million and payment is required only from any net income, after all related expenses, that we receive from sales of a PDE10 product. The funding agreement and our obligation to pay a royalty to SMRI terminate when we have repaid such amount in the form of royalties.
PPARγ. We acquired the patent applications and related intellectual property rights for our PPARγ program in February 2009 from Roberto Ciccocioppo, Ph.D. of the Università di Camerino, Italy, pursuant to a patent assignment agreement. In February 2011, we amended the agreement to include all intellectual property rights, including patent applications, related to nutraceuticals that increase PPARγ activity. Under the amended agreement, we have agreed to pay Dr. Ciccocioppo a low-single digit percentage royalty on net sales of any products that are covered by any patents that issue from the patent applications that we acquired from him. In addition, if we grant any third parties rights to manufacture, sell or distribute any such products, we must pay to Dr. Ciccocioppo a percentage of any associated fees we receive from such third parties in the range of low single-digits to low double-digits depending on the stage of development at which such rights are granted. We have also agreed to make total milestone payments of up to $3.8 million to Dr. Ciccocioppo upon the occurrence of certain development events, such as patient enrollment in a Phase 1 clinical trial and receipt of marketing approval of a product candidate covered by any patents that issue from the patent applications that we acquired from him. If we notify Dr. Ciccocioppo that we have abandoned all research and development and commercialization efforts related to the patent applications and intellectual property rights we acquired from him, Dr. Ciccocioppo has the right to repurchase those assets from us at a price equal to a double-digit percentage of our direct and indirect financial investments and expenditures in such assets. If he does not exercise his right to repurchase those assets within a limited period of time by paying the purchase price, we will have no further obligations to sell those assets to Dr. Ciccocioppo. The term of our agreement with Dr. Ciccocioppo ends when there are no longer any valid and enforceable patents related to the intellectual property rights we acquired from him, provided that either party may terminate the agreement earlier in case of an uncured breach by the other party. Under the terms of the agreement, we have agreed to pay a portion of the payments due to Dr. Ciccocioppo to the Università di Camerino without any increase to our payment obligations.
PDE7. Under an agreement with Daiichi Sankyo, we hold an exclusive worldwide license to PDE7 inhibitors claimed in certain patents and pending patent applications owned by Daiichi Sankyo for use in the treatment of (1) movement disorders and other specified indications, (2) addiction and compulsive disorders and (3) all other diseases except those related to dermatologic conditions. Under the agreement, we agreed to make milestone payments to Daiichi Sankyo of up to an aggregate total of $33.5 million upon the achievement of certain events in each of these three fields; however, if only one of the three indications is advanced through the milestones, the total milestone payments would be $23.5 million. The milestone payment events include successful completion of preclinical toxicology studies; dosing of human subjects in Phase 1, 2 and 3 clinical trials; receipt of marketing approval of a PDE7 inhibitor product candidate; and reaching specified sales milestones. In addition, Daiichi Sankyo is entitled to receive from us a low single-digit percentage royalty of any net sales of a PDE7 inhibitor licensed under the agreement by us and/or our sublicensee(s) provided that, if the sales are made by a sublicensee, then the amount payable by us to Daiichi Sankyo is capped at an amount equal to a low double-digit percentage of all royalty and specified milestone payments received by us from the sublicensee.
The term of the agreement with Daiichi Sankyo continues so long as there is a valid, subsisting and enforceable claim in any patents covered by the agreement. The agreement may be terminated sooner by us, with or without cause, upon 90 days advance written notice or by either party following a material breach of the agreement by the other party that has not been cured within 90 days or immediately if the other party is insolvent or bankrupt. Daiichi Sankyo also has the right to terminate the agreement if we and our sublicensee(s) cease to conduct all research, development and/or commercialization activities for a PDE7 inhibitor covered by the agreement for a period of six consecutive months, in which case all rights held by us under Daiichi Sankyo’s patents will revert to Daiichi Sankyo.
GPCR Platform Funding Agreements with Vulcan Inc. and the Life Sciences Discovery Fund. In October 2010, we entered into funding agreements for our GPCR program with Vulcan and LSDF. We received $20.0 million and $5.0 million, respectively, under the agreements with Vulcan and LSDF. Under these agreements, we have agreed to pay Vulcan and LSDF tiered percentages of the net proceeds, if any, that we derive from the GPCR program. The percentage rates of net proceeds payable to Vulcan and LSDF decrease as the cumulative net proceeds reach specified thresholds, and the blended percentage rate payable to Vulcan and LSDF in the aggregate is in the mid-teens with respect to the first approximately $1.5 billion of cumulative net proceeds that we receive from our GPCR program. If we receive cumulative net proceeds in excess of approximately $1.5 billion, the percentage rate payable to Vulcan and LSDF in the aggregate decreases to one percent. An acquirer of the assets in our GPCR program may be required, and an acquirer of our company would be required, to assume all of our payment and other obligations under our agreements with Vulcan and LSDF.
Under our agreement with Vulcan, we granted Vulcan a security interest in our personal property related to the GPCR program, other than intellectual property, which security interest is junior to any existing or future security interests granted in connection with a financing transaction and which will be released automatically after Vulcan receives $25.0 million under the
12
agreement. We also agreed not to grant any liens on intellectual property related to the GPCR program without Vulcan’s consent, subject to specified exceptions. These restrictions could limit our ability to pursue business opportunities involving the GPCR program or reduce the price for which a potential buyer would pay for the GPCR assets. If we default under our agreement with Vulcan, in certain circumstances Vulcan may, subject to the rights of any holders of senior security interests, take control of such pledged assets. If we are liquidated, Vulcan’s right to receive any payments then due under our agreement would be senior to the rights of the holders of our common stock to receive any proceeds from the liquidation of our GPCR program assets.
The term of our agreement with Vulcan is 35 years, provided that the term will automatically extend until the cumulative net proceeds that we receive from the GPCR program are approximately $1.5 billion. The term of our agreement with LSDF expires on the six-month anniversary following the last date that we deliver a report related to our incurrence of grant-funded expenses described in the agreement, provided that certain obligations will survive the expiration of the term. The term of our payment obligations to LSDF is the same as that under our agreement with Vulcan.
OMS103. In June 2015, we entered into the OMS103 Agreement under which Fagron is obligated to manufacture and commercialize OMS103. Pursuant to the OMS103 Agreement, we granted Fagron an exclusive, royalty-free license to the OMS103 intellectual property, manufacturing information and clinical data to produce, on a large-scale registered basis, and commercialize OMS103 in the United States. The OMS103 Agreement obligates Fagron to produce under GMP and to commercialize OMS103 in the U.S. Unless terminated earlier, the OMS103 Agreement will continue in effect until expiration of the last-to-expire of the patents in the licensed intellectual property or as otherwise provided under the terms of the OMS103 Agreement. Either party may terminate the OMS103 Agreement earlier if the other party materially breaches the OMS103 Agreement and does not cure the breach within a specified notice period or upon the other party’s insolvency. Additionally, we may terminate the OMS103 Agreement earlier if Fagron does not meet its performance diligence requirements, in response to a negative action by a regulatory authority, or if Fagron opposes or challenges any of the licensed patents for OMS103. Fagron has not met its performance diligence obligations under the OMS103 Agreement, including initiating sales, and we do not expect them to do so. We are evaluating our options regarding the OMS103 Agreement and our OMS103 program.
Antibody Platform. We hold a worldwide exclusive license to patent rights related to our antibody platform from the University of Washington, or UW. Pursuant to the Xori APA, we acquired all of Xori’s exclusive rights under a license agreement with the UW to certain patents and patent applications related to our antibody platform owned by the UW in exchange for our agreement to make royalty and development milestone payments to UW.
OMIDRIA. Our Settlement Agreement with Par includes a non-exclusive, nonsublicensable license to make, sell and distribute a generic version of OMIDRIA under certain circumstances. For more information, see Part I, Item 3, “Legal Proceedings.”
Competition
Overview. The pharmaceutical industry is highly competitive and characterized by a number of established, large pharmaceutical companies as well as smaller companies like ours. We expect to compete with other pharmaceutical and biotechnology companies, and our competitors may:
• | develop and market products that are less expensive, more effective or safer than our future products; |
• | commercialize competing products before we can launch our products; |
• | operate larger research and development programs, possess greater manufacturing capabilities or have substantially greater financial resources than we do; |
• | initiate or withstand substantial price competition more successfully than we can; |
• | have greater success in recruiting skilled technical and scientific workers from the limited pool of available talent; |
• | more effectively negotiate third-party licenses and strategic relationships; and |
• | take advantage of acquisition or other opportunities more readily than we can. |
We expect to compete for market share against large pharmaceutical and biotechnology companies, smaller companies that are collaborating with larger pharmaceutical companies, new companies, academic institutions, government agencies and other public and private research organizations. In addition, the pharmaceutical and biotechnology industry is characterized by rapid technological change. Because our research approach integrates many technologies, it may be difficult for us to remain current with the rapid changes in each technology. Further, our competitors may render our technologies obsolete by advancing their existing technological approaches or developing new or different approaches. If we fail to stay at the forefront of technological change, we may be unable to compete effectively.
13
OMIDRIA. We are not aware of any FDA-approved products comprised of two or more APIs that directly compete with OMIDRIA that are approved for intraoperative delivery in irrigation solutions during surgical procedures; however, OMIDRIA could compete with single API products that are delivered intraoperatively as well as preoperative and postoperative treatments for mydriasis, pain or inflammation. Our primary competition for OMIDRIA comes from surgeons’ current practices, which may include use of products obtained from distributors or compounding pharmacies at a relatively low cost. In addition, we anticipate that there are some surgeons who do not use intraoperative mydriatics and may not agree with the value proposition of maintaining pupil dilation and inhibiting miosis during the procedure or with the use of a nonsteroidal anti-inflammatory drug intraoperatively to reduce inflammation and postoperative pain. Although we are not aware of any companies developing similar combination approaches for maintenance of intraoperative pupil size and postoperative pain reduction as an FDA-approved product, such strategies may develop. In Europe, an inexpensive mydriatic and local anesthetic combination product is available but, unlike OMIDRIA, this product does not include an anti-inflammatory agent.
As described above, in October 2017, we entered into a Settlement Agreement with Par, resolving our patent litigation against Par that arose from Par’s filing of an Abbreviated New Drug Application, or ANDA, seeking approval from the FDA to market a generic version of OMIDRIA. In accordance with the terms of the Settlement Agreement, Par and its affiliates are prohibited from launching a generic version of OMIDRIA until the earlier of April 1, 2032 or a date on which we or a third party, through licensing or any future final legal judgment, should one ever exist, with respect to our Orange Book listed patents, is able to launch a generic version of OMIDRIA, as further detailed in the Settlement Agreement. As also described above, each of Sandoz and Lupin filed an ANDA containing a Paragraph IV Certification seeking approval to market a generic version of OMIDRIA prior to the expiration of the Orange Book Patents. An adverse outcome in our patent infringement lawsuit filed against Sandoz or Lupin following receipt of the applicable Notice Letter regarding the Paragraph IV Certification could, among other things, result in generic versions of OMIDRIA being launched, which could have a material negative impact on our financial condition and results of operations. In the future, other manufacturers may potentially file ANDAs seeking approval for the sale of generic versions of OMIDRIA before our relevant patents expire, or additional generic manufacturers may challenge one or more of the patents using U.S. Patent and Trademark Office, or USPTO, procedures. For more information regarding the ANDAs filed by Sandoz and by Lupin and our patent infringement lawsuits against Par, Sandoz and Lupin, see Part I, Item 3, “Legal Proceedings.”
Product Candidates, Development Programs and Platforms. Our product candidates that are eventually commercialized may face competing products being developed by other pharmaceutical or biotechnology companies that have significantly greater resources than we have. With respect to our complement system program, there are multiple companies developing potential therapies targeting the complement system, although none of which, to our knowledge, inhibit the lectin pathway. Soliris® is a monoclonal complement inhibitor administered intravenously and approved for commercial use that will compete with our lead MASP-2 inhibitor OMS721, and/or our MASP-3 inhibitor OMS906, if either is approved for any indication(s) for which Soliris® is also approved. We are also aware of two Soliris® biosimilar antibodies that are in development. Alexion, the manufacturer of Soliris®, has announced follow-on antibodies that are directed to the Soliris® target, but which will require less frequent dosing than Soliris® or that can be administered subcutaneously. With respect to our PDE10 inhibitor program, we are developing PDE10 inhibitors for use in the treatment of Huntington’s disease, schizophrenia and other diseases that affect cognition. Other pharmaceutical companies, many with significantly greater resources than we have, are also developing, or may develop, PDE10 inhibitors for the treatment of these indications, and these companies may be further along in development. In 2017, Pfizer announced negative results in a clinical trial of a PDE10 inhibitor for the treatment of Huntington’s disease. Also, Pfizer previously announced negative results from schizophrenia trials with a PDE10 inhibitor and Takeda Pharmaceuticals previously announced that the primary endpoint was not met in a clinical trial of a different PDE10 inhibitor in a schizophrenia trial. In 2018, Roche announced the termination of its small-molecule inhibitor of PDE10 program for the treatment of schizophrenia.
We are aware of other companies attempting to de-orphanize orphan GPCRs. If any of these companies is able to de-orphanize an orphan GPCR before we unlock this receptor, we may be unable to establish an exclusive or commercially valuable intellectual property position around that orphan GPCR.
Intellectual Property
As of February 16, 2018, we owned or held worldwide exclusive licenses to a total of 76 issued patents and 73 pending patent applications in the U.S. and 659 issued patents and 338 pending patent applications in foreign markets directed to therapeutic compositions and methods related to our development programs. For each program, our decision to seek patent protection in specific foreign markets, in addition to the U.S., is based on many factors, including one or more of the following: our available resources, the size of the commercial market, the presence of a potential competitor or a contract manufacturer in the market and whether the legal authorities in the market effectively enforce patent rights.
14
• | OMIDRIA-Ophthalmology. OMIDRIA is encompassed by our PharmacoSurgery patent portfolio. The relevant patents and patent applications in this portfolio are directed to combinations of agents, generic and/or proprietary to us or to others, drawn from therapeutic classes such as pain and inflammation inhibitory agents, mydriatic agents and agents that reduce intraocular pressure, delivered locally and intraoperatively to the site of ophthalmological procedures, including cataract and lens replacement surgery. As of February 16, 2018, we owned seven issued U.S. patents and three pending U.S. patent applications and 57 issued patents and 60 pending patent applications in foreign markets that are directed to OMIDRIA. Our OMIDRIA patents have terms that will expire as late as October 23, 2033 and, if currently pending patent applications are issued, as late as November 30, 2035. |
• | MASP-2 Program - OMS721. We hold worldwide exclusive licenses to rights in connection with MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for those antibodies from the University of Leicester, MRC and Helion. As of February 16, 2018, we exclusively controlled 19 issued patents and 29 pending patent applications in the U.S., and 273 issued patents and 101 pending patent applications in foreign markets, related to our MASP-2 program. |
• | MASP-3 Program - OMS906. We own and exclusively control under a license from the University of Leicester all rights to methods of treating various disorders and diseases by inhibiting MASP-3. As of February 16, 2018, we exclusively controlled four pending patent applications in the U.S. and three issued and 43 pending patent applications in foreign markets that are directed to these therapeutic methods. |
• | PDE10 Program - OMS824. As of February 16, 2018, we owned 12 issued patents and six pending patent applications in the U.S., and 32 issued patents and 48 pending patent applications in foreign markets, that are directed to proprietary PDE10 inhibitors. |
• | PPARγ Program - OMS405. As of February 16, 2018, we owned one issued patent and two pending patent applications in the U.S., and 27 issued patents and 18 pending patent applications in foreign markets, directed to our discoveries linking PPARγ and addictive disorders. |
• | PDE7 Program - OMS527. As of February 16, 2018, we owned two issued patents and one pending patent application in the U.S., and 22 issued patents and 10 pending patent applications in foreign markets directed to our discoveries linking PDE7 to movement disorders, as well as one issued patent and two pending patent applications in the U.S., and eight issued patents and 23 pending patent applications in foreign markets directed to the link between PDE7 and addiction and compulsive disorders. Additionally, under a license from Daiichi Sankyo, we exclusively control rights to three issued U.S. patents and 58 issued and four pending patent applications in foreign markets that are directed to proprietary PDE7 inhibitors. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements.” |
• | GPCR Platform. As of February 16, 2018, we owned six issued patents and 14 pending patent applications in the U.S., and 54 issued patents and two pending patent applications in foreign markets, which are directed to previously unknown links between specific molecular targets in the brain and a series of CNS disorders, to our cellular redistribution assay and to other research tools that are used in our GPCR program, and to orphan GPCRs and other GPCRs for which we have identified functionally interacting compounds using our cellular redistribution assay. |
• | Antibody Platform. As of February 16, 2018, we owned and/or held worldwide exclusive license rights from the UW to eight issued patents and one pending patent application in the U.S., and 13 issued patents and nine pending patent applications in foreign markets, directed to our antibody platform and antibodies generated using our platform. |
• | OMS103-Arthroscopy. OMS103 is encompassed by our PharmacoSurgery patent portfolio. The relevant patents and patent applications in this portfolio are directed to combinations of agents, generic and/or proprietary to us or to others, drawn from therapeutic classes such as pain and inflammation inhibitory agents and vasoconstrictive agents, delivered locally and intraoperatively to the site of medical or surgical procedures, including arthroscopy. As of February 16, 2018, we owned three issued U.S. patents and three pending U.S. patent applications, together with 36 issued patents and eight pending patent applications in foreign markets, that are directed to OMS103. Our OMS103 patents have terms that will expire as late as September 24, 2022 and, if currently pending patent applications are issued, as late as August 3, 2032. |
All of our employees enter into our standard employee proprietary information and inventions agreement, which includes confidentiality provisions and provides us ownership of all inventions and other intellectual property made by our employees that pertain to our business or that relate to our employees’ work for us or that result from the use of our resources. Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of the use, formulation and structure of our products and product candidates and the methods used to manufacture them, as well as on our ability to defend successfully these patents against third-party challenges. Our ability to protect our products and product
15
candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities. In addition, we have granted a lien on substantially all of our assets, including intellectual property, to the administrative agent under the CRG Loan Agreement.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date in the U.S., and tests used for determining the patentability of patent claims in all technologies are in flux. The pharmaceutical, biotechnology and other life sciences patent situation outside the U.S. is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in the patents that we own or have licensed or in third-party patents.
We sell OMIDRIA under trademarks that we consider in the aggregate to be important to our operations. We have registered, and intend to maintain, the trademarks “OMEROS”, “OMIDRIA”, “OMIDRIASSURE” and “PHARMACOSURGERY” with the USPTO in connection with the products and services we offer. We are not aware of any material claims of infringement or other challenges to our right to use the “OMEROS”, “OMIDRIA”, “OMIDRIASSURE” or “PHARMACOSURGERY” trademarks in the U.S.
We have retained control of all worldwide manufacturing, marketing and distribution rights for OMIDRIA and each of our product candidates and programs (other than OMS103). Some of our products and product candidates and programs are based on inventions and other intellectual property rights that we acquired through assignments, exclusive licenses or acquisitions.
• | PharmacoSurgery Platform. Our scientific co-founders, Gregory A. Demopulos, M.D. and Pamela Pierce Palmer, M.D., Ph.D., conceived the initial invention underlying our PharmacoSurgery platform and transferred all of their related intellectual property rights to us in 1994. Other than their rights as shareholders, our scientific co-founders have not retained any rights to our PharmacoSurgery platform, except that if we file for liquidation under Chapter 7 of the U.S. Bankruptcy Act or voluntarily liquidate or dissolve, other than in connection with a merger, reorganization, consolidation or sale of assets, our scientific co-founders have the right to repurchase the initial PharmacoSurgery intellectual property at its then-current fair market value. Subsequent developments of the PharmacoSurgery intellectual property were assigned to us by Dr. Demopulos, Dr. Palmer and other of our employees and consultants, without restriction. |
• | MASP Program. We hold worldwide exclusive licenses to rights related to MASP-2, the antibodies targeting MASP-2 and the therapeutic applications for the antibodies from the University of Leicester, MRC and Helion. We jointly own and hold worldwide exclusive license rights related to therapeutic applications for inhibiting MASP-3 from the University of Leicester. For more detailed descriptions of these licenses, see “License and Development Agreements.” |
• | PDE10 and PDE7 Programs. We acquired our PDE10 and PDE7 programs and some of our related patents and other intellectual property rights as a result of our acquisition of nura, inc. We hold an exclusive license to certain PDE7 inhibitors claimed in patents and pending patent applications owned by Daiichi Sankyo for use in the treatment of movement, addiction and compulsive disorders as well as other specified indications. For a more detailed description of our agreement with Daiichi Sankyo, see “License and Development Agreements.” |
• | PPARγ Program. We acquired the patent applications and related intellectual property rights for our PPARγ program in 2009 from Roberto Ciccocioppo, Ph.D., of the Università di Camerino, Italy, pursuant to a patent assignment agreement. For a more detailed description of this agreement, see “License and Development Agreements.” |
• | GPCR Platform. We acquired our GPCR program and some of our related patents and other intellectual property rights as a result of our acquisition of nura, inc. In November of 2010 we acquired intellectual property rights related to an assay technology for our GPCR program from Patobios Limited for approximately $10.8 million. |
• | Antibody Platform. We hold a worldwide exclusive license to patent rights related to our antibody platform from the UW. For a more detailed description of this agreement, see “License and Development Agreements. |
Government Regulation
Government authorities in the U.S., the EU and other countries extensively regulate the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing, and export and import of drug and biologic products such as OMIDRIA and the product candidates that we are developing. Failure to comply with applicable requirements, both before and after receipt of regulatory approval, may subject us, our third-party manufacturers, and other partners to
16
administrative and judicial sanctions, such as warning letters, product recalls, product seizures, a delay in approving or refusal to approve pending applications, civil and other monetary penalties, total or partial suspension of production or distribution, injunctions, and/or criminal prosecutions.
In the U.S., our products and product candidates are regulated by the FDA as drugs or biologics under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations and, in the case of biologics, also under the Public Health Service Act, or PHSA. In Europe, our products and product candidates are regulated by the EMA and national medicines regulators under the rules governing medicinal products in the EU as well as national regulations in individual countries. OMIDRIA has received marketing approval from the FDA and from the applicable regulatory authorities in the EU. Our product candidates are in various stages of testing and none of our product candidates, nor OMS103, has received marketing approval from the FDA or the applicable regulatory authorities in the EU.
The steps required before a product may be approved for marketing by the FDA, or the applicable regulatory authorities outside of the U.S., typically include the following:
• | formulation development and manufacturing process development; |
• | preclinical laboratory and animal testing; |
• | submission to the FDA of an IND for human clinical testing, which must become effective before human clinical trials may begin; and in Europe, a CTA is filed according to the country’s local regulations; |
• | adequate and well-controlled human clinical trials to establish the efficacy and safety of the product for each indication for which approval is sought; |
• | adequate assessment of drug product stability to determine shelf life/expiry dating; |
• | in Europe, submission to the EMA or national regulatory authority of a marketing authorization application, or MAA, and in the U.S., submission to the FDA of a New Drug Application, or NDA, in the case of a drug product, or a BLA in the case of a biologic product; |
• | satisfactory completion of inspections of one or more clinical sites at which clinical trials with the product were carried out and of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good Clinical Practices, or cGCP, and cGMP; and |
• | FDA review and approval of an NDA or BLA, or review and approval of an MAA by the applicable regulatory authorities in the EU. |
Manufacturing. Manufacturing of drug products for use in clinical trials must be conducted according to relevant national and international guidelines, for example, cGMP. Process and formulation development are undertaken to design suitable routes to manufacture the drug substance and the drug product for administration to animals or humans. Analytical development is undertaken to obtain methods to quantify the potency, purity and stability of the drug substance and drug product as well as to measure the amount of the drug substance and its metabolites in biological fluids, such as the blood.
Preclinical Tests. Preclinical tests include laboratory evaluations and animal studies to assess efficacy, toxicity and pharmacokinetics. The results of the preclinical tests, together with manufacturing information, analytical data, clinical development plan, and other available information are submitted as part of an IND or CTA.
The IND/CTA Process. An IND or CTA must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA unless, before that time, the FDA raises concerns or questions and imposes a clinical hold. In that event, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before the clinical hold is lifted and clinical trials can proceed. Similarly, a CTA must be cleared by the local independent ethics committee and competent authority prior to conducting a clinical trial in the EU country in which it was submitted. This process can take from two weeks to several months. There can be no assurance that submission of an IND or CTA will result in authorization to commence clinical trials. Once an IND or CTA is in effect, there are certain reporting requirements.
Clinical Trials. Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified personnel and must be conducted in accordance with local regulations and cGCP. Clinical trials are conducted under protocols detailing, for example, the parameters to be used in monitoring patient safety and the efficacy criteria, or endpoints, to be evaluated. Each trial must be reviewed and approved by an independent institutional review board or ethics committee for each clinical site at which the trial will be conducted before it can begin. Clinical trials are typically conducted in three defined phases, but the phases may overlap or be combined:
17
• | Phase 1 usually involves the initial administration of the investigational product to human subjects, who may or may not have the disease or condition for which the product is being developed, to evaluate the safety, dosage tolerance, pharmacodynamics and, if possible, to gain an early indication of the effectiveness of the product. |
• | Phase 2 usually involves trials in a limited patient population with the disease or condition for which the product is being developed to evaluate appropriate dosage, to identify possible adverse side effects and safety risks, and to evaluate preliminarily the effectiveness of the product for specific indications. |
• | Phase 3 clinical trials usually further evaluate and confirm effectiveness and test further for safety by administering the product in its final form in an expanded patient population. |
We, our product development partners, institutional review boards or ethics committees, the FDA or other regulatory authorities may suspend clinical trials at any time on various grounds, including a belief that the subjects are being exposed to an unacceptable health risk.
Disclosure of Clinical Trial Information. Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed for up to two years if the sponsor certifies that it is seeking approval of an unapproved product or that it will file an application for approval of a new indication for an approved product within one year. Competitors may use this publicly available information to gain knowledge regarding the design and progress of our development programs.
The Application Process. If the necessary clinical trials are successfully completed, the results of the preclinical trials and the clinical trials, together with other detailed information, including information on the manufacture and composition of the product, are submitted to the FDA in the form of an NDA or a BLA, as applicable, and to the EMA or national regulators in the form of an MAA, requesting approval to market the product for a specified indication. In the EU, an MAA may be submitted to the EMA for review and, if the EMA gives a positive opinion, the EC may grant a marketing authorization that is valid across the EU (centralized procedure). Alternatively, an MAA may be submitted to one or more national regulators in the EU according to one of several national or decentralized procedures. The type of submission in Europe depends on various factors and must be cleared by the appropriate authority prior to submission. For most of our product candidates, the centralized procedure will be either mandatory or available as an option.
If the regulatory authority determines that the application is not acceptable, it may refuse to accept the application for filing and review, outlining the deficiencies in the application and specifying additional information needed to file the application. Notwithstanding the submission of any requested additional testing or information, the regulatory authority ultimately may decide that the application does not satisfy the criteria for approval. Before approving an NDA or BLA, or an MAA, the FDA or the EMA, respectively, may inspect one or more of the clinical sites at which the clinical study(ies) were conducted to assure that GCPs were followed and may inspect facility(ies) at which the product is manufactured to assure satisfactory compliance with cGMP. After approval, changes to the approved product such as adding new indications, manufacturing changes, or additional labeling claims will require submission of a supplemental application, referred to as a Variation in the EU, or, in some instances, a new application, for further review and approval. The testing and approval process requires substantial time, effort, and financial resources, and we cannot be sure that any future approval will be granted on a timely basis, if at all.
Some of our drug products may be eligible for submission to the FDA of NDAs for approval under the Section 505(b)(2) process. Section 505(b)(2) applications may be submitted for drug products that represent a modification, such as a new indication or new dosage form, of a previously approved drug. Section 505(b)(2) applications may rely on the FDA’s previous findings for the safety and effectiveness of the previously approved drug in addition to information obtained by the 505(b)(2) applicant to support the modification of the previously approved drug. Preparing Section 505(b)(2) applications may be less costly and time-consuming than preparing an NDA based entirely on new data and information.
The FDA regulates certain of our products and product candidates, such as OMIDRIA and OMS103, as fixed-dose combination drugs under its Combination Drug Policy (21 CFR Section 300.50) because they are comprised of two or more active ingredients. In addition to demonstrating that the drug product is safe and effective, the FDA’s Combination Drug Policy requires that we demonstrate that each active ingredient in a drug product contributes to the product’s effectiveness. The EMA has a similar Guideline for fixed-dose combination products. Satisfaction of the U.S. or EU requirements for fixed-dose combination products may involve substantial time, effort, and financial resources, and we cannot be sure that work conducted to satisfy these requirements will be deemed acceptable by the applicable regulatory authority.
18
Some of our product candidates, such as those from our MASP-2 and MASP-3 programs, are considered biologics because they are derived from natural sources as opposed to being chemically synthesized. The added complexity associated with manufacturing biologics may result in additional monitoring of the manufacturing process and product changes.
In addition, we, our suppliers, and our contract manufacturers are required to comply with extensive regulatory requirements both before and after approval. For example, we must establish a pharmacovigilance system and are required to report adverse reactions and production problems, if any, to the regulatory authorities. We must also comply with certain requirements concerning advertising and promotion for our products. The regulatory authorities may impose specific obligations as a condition of the marketing authorization, such as additional safety monitoring, or the conduct of additional clinical trials or post-marketing safety studies. Also, quality control and manufacturing procedures must continue to conform to cGMPs after approval. Accordingly, manufacturers must continue to expend time, money, and effort in all areas of regulatory compliance, including production and quality control to comply with cGMPs. In addition, discovery of problems such as safety issues may result in changes in labeling or restrictions on a product manufacturer or marketing authorization holder, including removal of the product from the market.
Fast Track and Priority Review Designations. Section 506(b) of the FDCA provides for the designation of a drug as a fast track product if it is intended, whether alone or in combination with one or more other drugs, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. A program with fast track status is afforded greater access to the FDA for the purpose of expediting the product’s development, review and potential approval. Many products that receive fast track designation are also considered appropriate to receive priority review, and their respective applications may be accepted by the FDA as a rolling submission in which portions of an NDA or BLA are reviewed before the complete application is submitted. Together, these may reduce time of development and FDA review time. In Europe, products that are considered to be of major public health interest are eligible for accelerated assessment, which shortens the review period substantially. The grant of fast track status, priority review or accelerated assessment does not alter the standard regulatory requirements for obtaining marketing approval, however.
Breakthrough Therapy Designation and PRIME. In 2012, Congress enacted the Food and Drug Administration Safety and Innovation Act. This law established a new regulatory scheme allowing for increased interactions with the FDA with the goal of expediting development and review of products designated as “breakthrough therapies.” A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an efficient manner. The EU has a somewhat similar program, referred to as Priority Medicines, or PRIME, to that of the U.S., and it is administered through the EMA.
Accelerated Approval. The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality and that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Studies that that are conducted to demonstrate a drug’s effect on a surrogate or intermediate clinical endpoint for accelerated approval must be adequate and well-controlled as required by the FDCA.
Following accelerated approval, the FDA may require that the company still conduct certain adequate and well-controlled post-marketing clinical studies to verify and describe clinical benefit of the product, and the FDA may impose restrictions on distribution to assure safe use. Post-marketing studies would usually be required to be studies already underway at the time of the accelerated approval. If the required post-marketing studies fail to verify the clinical benefit of the drug, or if the applicant fails to perform the required post-marketing studies with due diligence, the FDA may withdraw approval of the drug under streamlined procedures in accordance with the agency’s regulations. The agency may also withdraw approval of a drug if, among other things, other evidence demonstrates that the drug product is not shown to be safe or effective under its conditions of use.
The EU also has accelerated approval programs. In the EU, a marketing authorization may be granted on the basis of less complete data than are normally required in certain “exceptional circumstances,” such as when the product’s indication is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive data. Alternatively, a conditional marketing authorization may be granted prior to obtaining the comprehensive clinical data required for a full MAA
19
if a product fulfills an unmet medical need and the benefit to public health of the product’s immediate availability outweighs the risk inherent in the incomplete data.
Orphan Drug Designation. Under the Orphan Drug Act, or ODA, the FDA may grant orphan drug designation to drugs or biologics intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. for which the cost of developing and making the product available in the U.S. for this type of disease or condition is not likely to be recovered from U.S. sales for that product. The granting of orphan designation does not alter the standard regulatory requirements (other than payment of certain fees) and process for obtaining marketing approval. If a product that has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the sponsor of the product qualifies for various development incentives specified in the ODA, including tax credits for qualified clinical testing (although Congress recently reduced the tax credit from 50% to 25% in 2017 tax legislation). Furthermore, the product is entitled to an orphan drug exclusivity period, which means that the FDA may not grant approval to any other application to market the same drug for the same indication for a period of seven years except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. If the FDA designates an orphan drug based on a finding of clinical superiority, the FDA must provide a written notification to the sponsor that states the basis for orphan designation, including “any plausible hypothesis” relied on by the FDA. The FDA must also publish a summary of its clinical superiority findings upon granting orphan drug exclusivity based on clinical superiority. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. The EU has a similar Orphan Drug program to that of the U.S., and it is administered through the EMA’s Committee for Orphan Medicinal Products, or COMP.
Pediatric Testing and Exclusivity. In the U.S., NDAs and BLAs are subject to both mandatory pediatric testing requirements and voluntary pediatric testing incentives in the form of exclusivity. An additional six months of exclusivity in the U.S. may be granted to a sponsor of an NDA or BLA if the sponsor conducts certain pediatric studies. This process is initiated when the FDA issues a Written Request for pediatric studies to determine if the drug or biologic could have meaningful pediatric health benefits. If the FDA determines that the sponsor has conducted the requested pediatric studies in accordance with the written request, then an additional six months of exclusivity may attach in the case of a drug to any other regulatory exclusivity or patent protection applicable to the drug, and in the case of a biologic to any other regulatory exclusivity applicable to the biologic. The EU has a similar requirement and incentive for the conduct of pediatric studies according to the pediatric investigation plan, which must be adopted by the EMA before an MAA may be submitted.
Expanded Access. “Expanded access” refers to the use of an investigational drug where the primary purpose is to diagnose, monitor, or treat a patient’s disease or condition rather than to collect information about the safety or effectiveness of a drug. There are three FDA-recognized categories of expanded access trials: expanded access for individual patients, including for emergency use; expanded access for intermediate-size patient populations; and expanded access for large patient populations under a treatment IND or treatment protocol. For all types of expanded access, the FDA must determine prior to authorizing expanded access that: (1) the patient or patients to be treated have a serious or life threatening disease or condition and there is no comparable or satisfactory alternative therapy; (2) the potential patient benefit justifies the potential risks of use and that the potential risks are not unreasonable in the context of the disease or condition to be treated; and (3) granting the expanded access will not interfere with the initiation, conduct, or completion of clinical studies in support of the drug’s approval. Only a licensed physician or the drug’s manufacturer may apply for expanded access. Manufacturers are not required to supply the investigational product. The FDA has established streamlined processes for physicians to request individual patient expanded access whereby physicians can submit an abbreviated application. In cases of individual patient emergency expanded access, physicians can receive FDA approval for access by phone and follow up with the abbreviated form. In addition, the sponsor of an expanded access IND must submit IND safety reports and, in the cases of protocols continuing for one year or longer, annual reports to the FDA. Expanded access programs are not intended to yield information relevant to evaluating a drug’s effectiveness for regulatory purposes. INDs for expanded access trials may be sponsored by physicians or by manufacturers.
A manufacturer or distributor of an investigational drug for the diagnosis, monitoring or treatment of a serious disease or condition must make available its policy for evaluating and responding to requests for individual patient access to the investigational drug. A manufacturer or distributor must make its expanded access policy publicly available on: (1) the date of initiation of a Phase 2 or 3 study with respect to the investigational drug, or (2) if such date is applicable and earlier, 15 days after the drug receives a designation as a breakthrough therapy, fast track product or regenerative advanced therapy. The policy must be made public and readily available, such as by posting on the Internet, and may be generally applicable to all of the manufacturer’s or distributor’s investigational drugs. Posting a policy does not guarantee access to an investigational drug by any individual patient, and the manufacturer or distributor may revise the policy at any time.
20
U.S. Labeling, Marketing and Promotion. The FDA closely regulates the labeling, marketing and promotion of drugs. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties.
In addition to regulation by the FDA, in the U.S. the research, manufacturing, distribution, sale and promotion of drug products are potentially subject to regulation by various federal, state and local authorities, including CMS, other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice, state Attorneys General, and other state and local government agencies. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws. Violations of these laws are punishable by prison sentences, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information or impose other special requirements for the sale and marketing of drug products. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, federal and state “transparency laws” require manufacturers to track and report certain payments made to healthcare providers and, under some state laws, other information concerning our products. These laws may affect our sales, marketing and other promotional activities by imposing administrative and compliance burdens on us. In addition, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities.
Compounding Pharmacies and Registered Outsourcing Facilities. Title I (the Compounding Quality Act) of the Drug Quality Security Act, or DQSA, which was enacted in November 2013, amended the FDCA to establish a distinct category of drug compounders known as “outsourcing facilities.” A compounding pharmacy that elects to register with the FDA as an outsourcing facility is exempt from certain FDCA requirements, including the obligation to obtain FDA approval of an NDA, if the facility satisfies conditions set out in the statute. The DQSA also imposes restrictions on the materials that may be compounded at registered outsourcing facilities. Like “traditional” pharmacy compounders, outsourcing facilities may not compound drugs that are “essentially a copy of one or more approved drugs” or that present “demonstrable difficulties for compounding.” The statute also imposes conditions on the compounding of bulk substances. The FDA has identified compounding as a regulatory priority in 2018 and is currently taking steps to implement these provisions of the DQSA. To date, approximately 70 compounding pharmacies are registered as outsourcing facilities and most have been inspected by the FDA. None of those that have been inspected by the FDA for sterile compounding have passed the FDA’s inspection, with all inspected receiving either a Warning Letter or an FDA Form 483, which is a document that lists observed conditions or practices that, in the FDA inspector’s judgment, indicate violations of the FDA’s requirements. Across all compounding pharmacies -- traditional compounders and outsourcing facilities -- the FDA has sent over 150 Warning Letters and initiated over 140 recall events related to compounding since passage of the DQSA.
Drug Supply Chain Security Act. Title II (the Drug Supply Chain Security Act, or DSCSA), of the DQSA imposes on manufacturers of certain pharmaceutical products new obligations related to product tracking and tracing, among others, which began a several-year phase-in process in 2015. Among the requirements of this legislation, manufacturers subject to the DSCSA will be required to provide certain information regarding the drug product to individuals and entities to which product ownership is transferred, label drug product with a product identifier (i.e., serialize) and keep certain records regarding the drug product. The transfer of information to subsequent product owners by manufacturers must be done electronically. Covered manufacturers will also be required to verify that purchasers of the manufacturers’ products are appropriately licensed. Further, under the DSCSA, covered manufacturers have drug product investigation, quarantine, disposition, and notification responsibilities related to product suspected or reasonably believed to be counterfeit, diverted, stolen, intentionally adulterated the subject of fraudulent transactions or otherwise unfit for distribution such that they would be reasonably likely to result in serious health consequences or death.
Foreign Regulatory Requirements. Outside of the U.S., our ability to conduct clinical trials or market our products will also depend on receiving the requisite authorizations from the appropriate regulatory authorities. The foreign regulatory approval processes include similar requirements and many of the risks associated with the FDA and/or the EMA approval process described above, although the precise requirements may vary from country to country. In the EU, once an MAA is granted, the product must be “placed on the market” in at least one EEA country within three years of the date of authorization. “Placed on the market” is defined as when the medicinal product is “released into the distribution chain,” i.e., out the of the direct control of the marketing authorization holder, or MAH. With respect to OMIDRIA, this requires releasing the product into the distribution chain in at least one EEA country by July 28, 2018. In addition, a marketing authorization will cease to be valid if a product previously placed on the market is no longer actually present on the market for three consecutive years.
Hatch-Waxman Act. In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s drug or an approved method of use of the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the FDA’s Approved Drug Products with Therapeutic
21
Equivalence Evaluations, commonly known as the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or a 505(b)(2) application. An ANDA provides for marketing of a drug that has the same active ingredients and, in some cases (e.g., ophthalmology), also the same inactive ingredients, in the same strengths and dosage form as the listed drug and has been shown through testing to be bioequivalent to the listed drug or receives a waiver from bioequivalence testing. ANDA applicants are generally not required to conduct or submit results of pre-clinical or clinical tests to prove the safety or effectiveness of their drug, other than the requirement for bioequivalence testing. Drugs approved in this way based on a showing of sameness and bioequivalence to the listed drug are considered therapeutically equivalent, and are commonly referred to as “generic equivalents” to the listed drug. These drugs then generally can be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA or 505(b)(2) applicant is required to certify to the FDA concerning any patents listed for the referenced approved drug in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or unenforceable, or will not be infringed by the new drug. A certification that the new drug will not infringe the already approved drug’s listed patents or that such patents are invalid or unenforceable is called a Paragraph IV certification. If the ANDA or 505(b)(2) applicant does not challenge the listed patents, the ANDA or 505(b)(2) application will not be approved until all of the listed patents claiming the referenced drug have expired, except for any listed patents that only apply to uses of the drug not being sought by the ANDA or 505(b)(2) applicant.
If the ANDA or 505(b)(2) applicant has made a Paragraph IV certification, the applicant must also send notice of a Paragraph IV Notice Letter to the NDA and patent holders once the ANDA or 505(b)(2) application has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV Notice Letter. The filing of a patent infringement lawsuit within 45 days of the receipt of notice of a Paragraph IV Notice Letter automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, modification by a court or a decision in the infringement case that is favorable to the ANDA or 505(b)(2) applicant.
The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced drug has expired. The U.S. Drug Price Competition and Patent Term Restoration Act of 1984, more commonly known as the “Hatch-Waxman Act,” provides a period of five years following approval of a drug containing no previously approved active moiety, during which ANDAs for generic versions of those drugs and 505(b)(2) applications referencing those drugs cannot be submitted unless the submission contains a Paragraph IV challenge to a listed patent, in which case the submission may be made four years following the original drug approval. Federal law provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage form, route of administration or combination, or for a new use, the approval of which was required to be supported by new clinical trials, other than bioavailability studies, conducted by or for the sponsor, during which FDA cannot grant effective approval of an ANDA or 505(b)(2) application based on that listed drug.
For a discussion of our litigation under the Hatch-Waxman Act with respect to OMIDRIA, see Part I, Item 3, “Legal Proceedings.”
Biosimilars. In the U.S., the FDA regulates biologics under the FDCA, the PHSA, and implementing regulations. The enactment of federal health care reform legislation in March 2010 provided a new pathway for approval of follow-on biologics (i.e., biosimilars) under the PHSA. Licensure by the FDA is dependent upon many factors, including a showing that the proposed biosimilar is “highly similar” to the reference product, notwithstanding minor differences in clinically inactive components, and has no clinically meaningful differences from the reference product in terms of safety, purity, and potency. The types of data ordinarily required in a biosimilar application to show high similarity include analytical data, animal studies (including toxicity studies), and clinical studies (including immunogenicity and pharmacokinetic/pharmacodynamic studies). A biosimilar must seek licensure for a condition of use for which the reference product is licensed.
Furthermore, the PHSA provides that for a biosimilar to be considered “interchangeable” (i.e., the biological product may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product), the applicant must make a further safety showing. Specifically, the applicant must demonstrate that the biosimilar can be expected to produce the same clinical result as the reference product in any given patient, and if the product is administered more than once to a patient, that safety risks and risk for diminished efficacy of alternating or switching between the biological product and the reference product is no greater than the risk of using the reference product without switching. Although the FDA has provided guidance on what information and data an applicant should submit to enable an interchangeability determination, thus far the FDA has not licensed any biologic as being interchangeable with its reference product. The law is
22
beginning to be interpreted and implemented by the FDA, and its ultimate impact will likely be subject to substantial uncertainty for years to come.
In the EU, a pathway for the approval of biosimilars has existed since 2005.
Healthcare compliance laws. In the U.S., commercialization of OMIDRIA and our product candidates, if approved, is subject to regulation and enforcement under a number of federal and state healthcare compliance laws administered and enforced by various agencies. These include, but are not limited to, the following:
• | the federal Anti-Kickback Statute, which prohibits offering or paying anything of value to a person or entity to induce or reward referrals for goods or services reimbursed by a federal health care program such as Medicare or Medicaid; |
• | the federal False Claims Act, which prohibits presenting or causing to be presented a false claim for payment by a federal health care program, and which has been interpreted to also include claims caused by improper drug-manufacturer product promotion or the payment of kickbacks; |
• | a variety of governmental pricing, price reporting, and rebate requirements, including those under Medicaid and the Veterans Health Care Act; and |
• | the so-called Sunshine Act and certain provisions of the Affordable Care Act, which require that we report to the federal government information on financial payments that we make to physicians and certain healthcare institutions and also on drug samples that we distribute. |
In addition to these federal law requirements, there are related state law requirements. Also, if we receive protected patient health information, we may be subject to federal or state privacy laws.
Similar requirements apply to our operations outside of the U.S. Laws in the U.S. such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials for business, and this includes physicians or other medical professionals who are employees of public health care entities. In addition, many countries have their own laws similar to the healthcare compliance laws that exist in the U.S.
Pharmaceutical Pricing and Reimbursement
Overview. In both U.S. and foreign markets, our ability to commercialize our products and product candidates successfully, and to attract commercialization partners for our products and product candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payers including, in the U.S., managed care organizations and other private health insurers as well as governmental payers such as the Medicare and Medicaid programs. Reimbursement by a third-party payer may depend on a number of factors, including the payer’s determination that use of a product is
• | a covered benefit under its health plan; |
• | safe, effective and medically necessary; |
• | appropriate for the specific patient; |
• | cost-effective; and |
• | neither experimental nor investigational. |
Reimbursement by government payers is based on statutory authorizations and may depend on some of the same or similar factors as reimbursement by private third-party payers and also depends on complex regulations that may change with annual or more frequent rulemaking, as well as legislative reform measures.
Third-party private and governmental payers are increasingly challenging the prices charged for medicines and examining their cost effectiveness in addition to their safety and efficacy. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of our products or product candidates. Even with the availability of such studies, third-party private and/or governmental payers may not provide coverage and reimbursement for our products or product candidates, in whole or in part.
United States. Political, economic and regulatory influences are subjecting the healthcare industry in the U.S. to fundamental changes. There have been, and we expect there will continue to be, legislative and regulatory proposals to change the healthcare system in ways that could significantly affect our business. For example, in March 2010 President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively the ACA, which is intended to broaden access to health insurance, reduce or constrain the
23
growth of healthcare spending, enhance remedies against fraud and abuse, add transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Other legislative changes included across-the-board reductions to Medicare payments to providers of 2%, effective April 1, 2013, which, due to subsequent legislative amendments, will stay in effect through fiscal year 2025 unless additional congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, reduced Medicare payments to several providers, and increased the period for the government to recover overpayments to providers from three to five years. President Trump and various members of Congress have expressed a desire to repeal all or portions of the ACA and, in December 2017, portions of the ACA dealing with the individual mandate insurance requirement were effectively repealed by the Tax Cuts and Jobs Act of 2017. In addition, other legislative changes have been proposed and adopted since the ACA was enacted. We anticipate that the U.S. Congress, state legislatures, governmental agencies and the private sector will continue to consider and may adopt new measures and policies intended to curb rising healthcare costs. We are unable to predict what additional legislation, regulations or policies, if any, relating to the healthcare industry or coverage and reimbursement may be enacted or imposed in the future or what effect such legislation, regulations or policies would have on our business. Any cost-containment measures, including those listed above, or other healthcare system reforms that are adopted could have a material adverse effect on our business prospects and financial operations.
Please see “Business--Commercial Product -- OMIDRIA® (phenylephrine and ketorolac intraocular solution) 1%/0.3%” above, as well as Part II, Item 7, “Management’s Discussion and Analysis--Results of Operations,” regarding the reimbursement status for OMIDRIA.
Europe. Governments in the various member states of the EU influence or control the price of medicinal products in their countries through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials or pharmacoeconomic studies that assess the cost-effectiveness of a product or product candidate relative to currently available therapies or relative to a specified standard. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Research and Development
We have built a research and development organization that includes expertise in discovery research, preclinical development, product formulation, analytical and medicinal chemistry, manufacturing, clinical development and regulatory and quality assurance. We operate cross-functionally and are led by an experienced research and development management team. We use rigorous project management techniques to assist us in making disciplined strategic research and development programmatic decisions and to limit the risk profile of our product pipeline. We also access relevant market information and key opinion leaders in creating target product profiles and, when appropriate, as we advance our programs to commercialization. We engage third parties on a limited basis to conduct portions of our preclinical research; however, we are not substantially dependent on any third parties for our preclinical research nor do any of these third parties conduct a major portion of our preclinical research. In addition, we engage multiple clinical sites to conduct our clinical trials; however, we are not substantially dependent upon any one of these sites for our clinical trials nor do any of them conduct a major portion of our clinical trials. Research and development expenses were $55.6 million, $50.7 million and $48.4 million in 2017, 2016 and 2015, respectively.
Employees
As of February 16, 2018, we had 173 full-time employees, 87 of whom are in research and development, 52 of whom are in sales and marketing and 34 of whom are in finance, legal, business development and administration. Our full-time employees include five with M.D.s and 22 with Ph.D.s., of whom four and 21, respectively, are in research and development. None of our employees is represented by a labor union, and we consider our employee relations to be good.
Executive Officers and Significant Employees
The following table provides information regarding our executive officers and significant employees as of March 1, 2018:
24
Name | Age | Position(s) | ||
Executive Officers: | ||||
Gregory A. Demopulos, M.D. | 59 | President, Chief Executive Officer and Chairman of the Board of Directors | ||
Michael A. Jacobsen | 59 | Vice President, Finance, Chief Accounting Officer and Treasurer | ||
Marcia S. Kelbon, J.D., M.S. | 58 | Vice President, Patent, General Counsel and Secretary | ||
Significant Employees: | ||||
Leonard Blum | 57 | Chief Business and Commercial Officer | ||
Christopher S. Bral, Ph.D. | 52 | Vice President, Nonclinical Development | ||
Daniel M. Canafax, Pharm.D., FCCP | 65 | Vice President, Medical Affairs and Clinical Research | ||
Timothy M. Duffy | 57 | Vice President, Business Development | ||
Timi Edeki, M.D., Ph.D. | 57 | Vice President, Clinical Development | ||
George A. Gaitanaris, M.D., Ph.D. | 61 | Vice President, Science and Chief Scientific Officer | ||
William J. Lambert, Ph.D. | 59 | Vice President, Chemistry, Manufacturing and Controls | ||
Catherine A. Melfi, Ph.D. | 58 | Vice President, Regulatory Affairs and Quality Systems and Chief Regulatory Officer | ||
J. Steven Whitaker, M.D., J.D. | 62 | Vice President, Clinical Development and Chief Medical Officer | ||
Gregory A. Demopulos, M.D. founded our company and has served as our president, chief executive officer and chairman of the board of directors since June 1994. He also served as our chief financial officer and treasurer from January 2009 to October 2013 in an interim capacity and as our chief medical officer from June 1994 to March 2010. Prior to founding Omeros, Dr. Demopulos completed his residency in orthopedic surgery at Stanford University and his fellowship training in hand and microvascular surgery at Duke University. Dr. Demopulos currently serves on the board of trustees of the Smead Funds Trust, an open-end mutual fund company registered under the Investment Company Act of 1940. His non-profit service includes the Seattle Community Development Round Table and the Northwest NeuroNeighborhood board of directors. Dr. Demopulos received his M.D. from the Stanford University School of Medicine and his B.S. from Stanford University. Dr. Demopulos is the brother of Peter A. Demopulos, M.D., a member of our board of directors.
Michael A. Jacobsen has served as our vice president, finance, chief accounting officer and treasurer since October 2013. Prior to joining Omeros, Mr. Jacobsen served as vice president of finance of Sarepta Therapeutics, Inc. from September 2011 to May 2013 and as its chief accounting officer from September 2011 to December 2012. From April 2007 to August 2011, Mr. Jacobsen was vice president and chief accounting officer at ZymoGenetics, Inc. Prior to his service with ZymoGenetics, Mr. Jacobsen held various roles at ICOS Corporation, including senior director of finance and corporate controller. From April 1995 to October 2001, Mr. Jacobsen held vice president of finance or chief financial officer roles at three companies in the software, computer hardware and internet retailing industries, two of which were publicly traded. Mr. Jacobsen is a certified public accountant and received his bachelor’s degree in accounting from Idaho State University.
Marcia S. Kelbon, J.D., M.S. has served as our vice president, patent and general counsel since October 2001 and as our secretary since September 2007. Prior to joining Omeros, Ms. Kelbon was a partner with the firm of Christensen O’Connor Johnson & Kindness, PLLC, where she specialized in U.S. and international intellectual property procurement, management, licensing and enforcement issues. Ms. Kelbon received her J.D. and her M.S. in chemical engineering from the University of Washington and her B.S. from The Pennsylvania State University.
Leonard M. Blum has served as our chief business and commercial officer since April 2016. Mr. Blum previously served as senior vice president, chief commercial officer from 2007 until March 2016 at Theravance, Inc., a publicly traded biopharmaceutical company, and its spin-off Theravance BioPharma. Prior to that, Mr. Blum founded and led the commercial functions at ICOS Corporation, a biotechnology company, ultimately as senior vice president, sales and marketing, from 2000 until the company’s acquisition by Eli Lilly and Company in 2007. Mr. Blum began his career in the pharmaceutical industry at Merck & Co. Inc. where he spent 13 years in positions of increasing responsibility in marketing and business unit leadership in the U.S. and Europe. Mr. Blum earned his A.B. in Economics at Princeton University and his M.B.A. at Stanford University’s
25
Graduate School of Business. Before beginning his career in the pharmaceutical industry, he served as an officer in the U.S. Army Special Forces.
Christopher S. Bral, Ph.D. has served as our vice president, nonclinical development in October 2015. From April 2014 to October 2015, Dr. Bral was the executive director, toxicology at Arrowhead Research Corporation, a biopharmaceutical company. From June 2008 to April 2014, Dr. Bral served as director, drug safety evaluation at Vertex Pharmaceuticals, a biotechnology company. Prior to Vertex, Dr. Bral held various pre-clinical drug safety positions of increasing responsibility at Schering-Plough Research Institute including associate director, drug safety evaluation. Dr. Bral received his Ph.D. in biochemistry and biophysics from Texas A&M University and his B.S. in chemistry from John Carroll University, and has been board-certified in toxicology through the American Board of Toxicology since 2000.
Daniel M. Canafax, Pharm.D., FCCP has served as our vice president, medical affairs and clinical research since June 2017. Dr. Canafax served as vice president of clinical development at Theravance, Inc./Theravance Biopharma, Inc., a biopharmaceutical company, from August 2011 through February 2017 and consulted for Theravance until joining Omeros. Prior to this period, Dr. Canafax was chief development officer and vice president of clinical development at ARYx Therapeutics, Inc. and also held medical affairs and clinical development positions with Xenoport, Inc., MedImmune, Inc., Elan Pharmaceuticals, Inc. and SangStat Medical Corporation. Before joining the pharmaceutical industry Dr. Canafax was a professor in the departments of pharmacy practice, surgery and otolaryngology at the University of Minnesota. Dr. Canafax is a fellow in the American College of Clinical Pharmacy and has a Bachelor of Pharmacy degree from Washington State University, a Doctor of Pharmacy degree from the University of Kentucky and completed a clinical residency at the Albert Chandler Medical Center, University of Kentucky.
Timothy M. Duffy has served as our vice president, business development since March 2010. From November 2008 to March 2010, Mr. Duffy served as the managing director of Pacific Crest Ventures, a life science consulting firm that he founded. From June 2004 through September 2008, Mr. Duffy served at MDRNA, Inc. (formerly Nastech Pharmaceutical Company, Inc.), a biotechnology company. At MDRNA, he held roles of increasing responsibility in marketing and business development, most recently as the chief business officer. Prior to MDRNA, Mr. Duffy served as vice president, business development at Prometheus Laboratories, Inc., a specialty pharmaceutical company, and as a customer marketing manager at The Procter & Gamble Company. Mr. Duffy received his B.S. from Loras College.
Timi Edeki, M.D., Ph.D. has served as our vice president, clinical development since May 2017. From 2006 through 2016, Dr. Edeki served as principal physician for AstraZeneca PLC, a biopharmaceutical company, and during most of that period also served as senior director, research and development. Prior to his service with AstraZeneca, Dr. Edeki was associate director at Abbott Laboratories from 2003 to 2006. Dr. Edeki is a fellow of the American College of Clinical Pharmacology and currently serves on the editorial board of Clinical Pharmacology and Therapeutics. Dr. Edeki currently holds academic appointments as adjunct Professor of Pharmacology, Physiology, and Internal Medicine at Drexel University and previously as Clinical Professor of Medicine at the Chicago Medical School. Dr. Edeki received his medical and Ph.D. degree from the University of Lagos and London respectively, and fellowship training at Vanderbilt University. He is a Diplomate of both the American Board of Internal Medicine and Clinical Pharmacology.
George A. Gaitanaris, M.D., Ph.D. has served as our vice president, science since August 2006 and as our chief scientific officer since January 2012. From August 2003 to our acquisition of nura, inc. in August 2006, Dr. Gaitanaris served as the chief scientific officer of nura, a company that he co-founded and that developed treatments for central nervous system disorders. From 2000 to 2003, Dr. Gaitanaris served as president and chief scientific officer of Primal, Inc., a biotechnology company that was acquired by nura in 2003. Prior to co-founding Primal, Dr. Gaitanaris served as staff scientist at the National Cancer Institute. Dr. Gaitanaris received his Ph.D. in cellular, molecular and biophysical studies and his M.Ph. and M.A. from Columbia University and his M.D. from the Aristotelian University of Greece.
William J. Lambert, Ph.D. has served as our vice president, chemistry, manufacturing and controls since January 2015. From October 2011 to January 2015, Dr. Lambert headed the Innovative Drug Delivery Group of MedImmune, the global biologics research and development arm of AstraZeneca PLC. From January 2006 to September 2011, Dr. Lambert served as senior vice president of pharmaceutical development at Pacira Pharmaceuticals. Prior to Pacira, Dr. Lambert directed drug delivery, product development and cGMP clinical supply groups at Eisai Inc. He has also held various pharmaceutical research positions at Pfizer Inc. and the Upjohn Company. Dr. Lambert received his Ph.D. in Pharmaceutics from the University of Utah and his B.S. in Pharmacy from the University of Rhode Island.
Catherine A. Melfi, Ph.D. has served as our vice president, regulatory affairs and quality systems since October 2012 and has served as our chief regulatory officer since April 2016. Dr. Melfi previously served from January 1996 to October 2012 at Eli Lilly and Company, where she held technical and leadership roles of increasing scope and responsibility, including as senior director and scientific director in Global Health Outcomes and Regulatory Affairs, respectively. Prior to joining Eli Lilly,
26
Dr. Melfi held various faculty and research positions at Indiana University, including appointments in its Economics Department, in the School of Public and Environmental Affairs, and in the Indiana University School of Medicine. Dr. Melfi received her Ph.D. in Economics from the University of North Carolina - Chapel Hill and B.S. in Economics from John Carroll University.
J. Steven Whitaker, M.D., J.D. has served as our vice president, clinical development and chief medical officer since March 2010. From May 2008 to March 2010, Dr. Whitaker served as the chief medical officer, vice president of clinical development at Allon Therapeutics, Inc., a biotechnology company focused on developing drugs for neurodegenerative diseases. From August 2007 to May 2008, he served as a medical consultant to Accelerator Corporation, a biotechnology-company investor and incubator. From May 1994 to May 2007, Dr. Whitaker served at ICOS Corporation, which was acquired by Eli Lilly and Company in 2007. At ICOS, he held roles of increasing responsibility in clinical research and medical affairs, most recently as divisional vice president, clinical research as well as medical director of the Cialis® global product team. Dr. Whitaker received his M.D. from the Indiana University School of Medicine, his J.D. from the University of Washington and his B.S. from Butler University.
Corporate Information
We were incorporated in 1994 as a Washington corporation. Our principal executive offices are located at 201 Elliott Avenue West, Seattle, Washington, 98119, and our telephone number is (206) 676-5000. Our website address is www.omeros.com. We make available, free of charge through our investor relations website at investor.omeros.com, our annual report on Form 10-K, our quarterly reports on Form 10-Q, our current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act, including exhibits to those reports, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. Our websites and the information contained therein or incorporated therein are not intended to be incorporated into this Annual Report on Form 10-K. In addition, the public may read and copy any materials we file or furnish with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549 or may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. Moreover, the SEC maintains a website that contains reports, proxy and information statements, and other information regarding reports that we file or furnish electronically with them at www.sec.gov.
ITEM 1A. RISK FACTORS
The risks and uncertainties described below may have a material adverse effect on our business, prospects, financial condition or operating results. In addition, we may be adversely affected by risks that we currently deem immaterial or by other risks that are not currently known to us. You should carefully consider these risks before making an investment decision. The trading price of our common stock could decline due to any of these risks and you may lose all or part of your investment. In assessing the risks described below, you should also refer to the other information contained in this Annual Report on Form 10-K.
Risks Related to Our Products, Programs and Operations
Our ability to achieve profitability is dependent on the commercial success of OMIDRIA. To the extent OMIDRIA is not successful, our business, financial condition and results of operations may be materially adversely affected and the price of our common stock may decline.
OMIDRIA is our only product that has been approved by the FDA for commercial sale in the U.S. For the three and twelve months ended December 31, 2017, we recorded net sales of OMIDRIA of $13.8 million and $64.8 million, respectively. We have not generated revenue from sales of OMIDRIA to date that are sufficient to fund fully our operations and cannot provide assurance that we will generate sufficient revenue from OMIDRIA in the future to fund fully our operations. We will need to generate substantially more product revenue from OMIDRIA to achieve and sustain profitability. Our ability to generate significant revenue from OMIDRIA product sales depends on our ability to achieve increased market acceptance of, and to otherwise market and sell effectively, OMIDRIA, which may not occur for a number of reasons, including:
• | the extent of coverage and reimbursement for OMIDRIA when used in Medicare patients following the expiration of pass-through reimbursement on January 1, 2018; |
• | pricing, coverage and reimbursement policies of government and private payers such as Medicare, Medicaid, the Department of Veterans Affairs, or VA, group purchasing organizations, insurance companies, health maintenance organizations and other plan administrators; |
• | a lack of acceptance by physicians, patients and other members of the healthcare community; |
27
• | the availability, relative price and efficacy of the product as compared to alternative treatment options or branded, compounded or generic competing products; |
• | an unknown safety risk; |
• | the failure to enter into and maintain acceptable partnering arrangements for marketing and distribution of OMIDRIA outside of the U.S.; |
• | changed or increased regulatory restrictions in the U.S., EU and other foreign territories; and |
• | a lack of adequate financial or other resources. |
Pass-through status for OMIDRIA under Medicare Part B terminated as scheduled effective January 1, 2018. If legislative and/or administrative means to extend separate payment for OMIDRIA continue to be delayed, or are not successful, we would need to pursue an alternative sales strategy, and our revenues, financial condition and prospects for profitability could be adversely and significantly affected.
Effective January 1, 2018, OMIDRIA no longer has separate payment under Medicare Part B when used in the hospital outpatient department or ambulatory surgery centers and, consequently, payment is included as part of the packaged items and services included in the payment for the procedure. Legislative and administrative means are being pursued to obtain a continuation of separate or similar reimbursement for OMIDRIA on and after January 1, 2018 and/or to extend the pass-through period from three to five years. An extension or separate reimbursement for OMIDRIA requires action from legislative and/or administrative authorities and, as a consequence, we can provide no assurance that any such action will be taken or, if taken, when such action will be effective, nor can we predict the actual reimbursement rate or the duration of the extension or separate reimbursement period.
Due to the scheduled expiration of pass-through reimbursement on January 1, 2018, we saw a significant reduction in ASC and hospital demand for OMIDRIA beginning in December 2017. We expect this reduction in OMIDRIA demand to continue while the CMS reimbursement status of OMIDRIA remains uncertain. Based on first quarter 2018 data to date, we believe that a substantial majority of facilities that were using OMIDRIA are awaiting resolution regarding reimbursement by CMS, or our decision to implement an alternative sales strategy, and, therefore, have largely suspended use of the product or are using the product on a selective basis only. As a result, sales to our wholesalers have decreased substantially from their peak levels in November 2017 as the wholesalers adjust on-hand inventory in light of the reduced ASC and hospital demand. Even if OMIDRIA receives separate or similar reimbursement in the future, we cannot predict how quickly and to what extent sales will resume, if at all. It is possible that some customers may defer OMIDRIA purchases until Medicare Part B reimbursement is obtained or until an alternative sales strategy is implemented. Further, some former customers could decide not to resume using OMIDRIA, which could inhibit or limit our potential sales growth.
In the event that neither the legislative or the administrative approaches are ultimately successful, we expect to pursue an alternative sales strategy. After implementing this strategy, we cannot predict how quickly, or if, our customers would increase their OMIDRIA utilization and the net revenues we receive for OMIDRIA could be reduced, potentially by a significant amount. A reduction in net OMIDRIA revenues for this or any other reason may also impair our ability to satisfy annual revenue covenants in the CRG Loan Agreement, which would constitute an event of default if the corresponding market capitalization threshold was not satisfied. Furthermore, we cannot guarantee that a substantial reduction in OMIDRIA revenues would not trigger “material adverse change” under the CRG Loan Agreement, which would also constitute an event of default.
Any of these risks, if realized, would adversely affect our ability to generate revenue and attain profitability and there would be a material adverse effect on our business, financial condition, results of operations and growth prospects and the trading price of our stock could decline.
If OMIDRIA or any other product that we develop and commercialize does not receive adequate coverage or reimbursement from governments or private payers, or if we do not establish and maintain market-acceptable pricing for OMIDRIA or those potential other commercialized products, our prospects for revenue and profitability would suffer.
Our revenues and profitability will depend heavily on the pricing, availability and duration of adequate coverage or reimbursement for the use of products that we or our third-party business partners commercialize, including OMIDRIA, from government, private and other third-party payers, both in the U.S. and in other countries. Any product that we bring to market may not be considered cost-effective and/or the amount reimbursed for any product may be insufficient to allow us to sell the product profitably. Obtaining coverage and reimbursement for any product from each government or third-party payer can be a time-consuming and costly process that may require expansion of staff and/or increased use of third parties and could require us to provide additional supporting scientific, clinical and cost-effectiveness data for the use of our approved products to each payer. We can provide no assurances at this time regarding the cost-effectiveness of OMIDRIA, OMS103 or any of our product
28
candidates. Further, we can provide no assurance that the amounts, if any, reimbursed to surgical facilities for utilization of any of our surgery-related products, including OMIDRIA, OMS103 or any of our product candidates, or to surgeons for the administration and delivery of these products or product candidates will be considered adequate to justify the use of these products or product candidates. In addition, obtaining acceptable coverage and reimbursement from one payer does not guarantee that we will obtain similar acceptable coverage or reimbursement from another payer.
There may be significant delays in obtaining coverage or reimbursement for newly approved products, and we may not be able to provide data sufficient to be granted coverage or reimbursement. Even when a payer determines that a product is eligible for reimbursement, coverage may be limited to the uses of a product that are either approved by the FDA (or, in other countries, the relevant country’s regulatory agency) and/or appear in a recognized drug compendium, and other conditions may apply. Moreover, eligibility for coverage does not mean that any product will be reimbursed at a rate that allows us to make a profit in all cases or at a rate that covers our costs, including research, development, manufacturing, sales and distribution. Increasingly, government and private third-party payers that reimburse for healthcare services and products are requiring that companies provide them with predetermined discounts from list prices and challenging the prices charged for medical products, which could adversely impact the pricing of our products. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. Pricing may also be adversely affected by changes in the terms, scope and/or complexity of government pricing requirements. Even if we achieve coverage or reimbursement for a product, the initial rate or method at which the product will be reimbursed could become unfavorable to us at the time reimbursement is initiated or in the future or may be of a limited duration.
In non-U.S. jurisdictions, we must obtain separate reimbursement approvals and comply with related foreign legal and regulatory requirements. In some countries, including those in the EU, our products may be subject to government price controls. Pricing negotiations with governmental authorities can take a considerable amount of time and expenditure of resources after the receipt of marketing approval for a product. We provide no assurances that the price of any product in one or more of these countries or regions will allow us to make a profit or cover our costs, including research, development, manufacturing, sales and distribution, and as a result we may decide to delay, potentially indefinitely, initiating sales in the particular country or region.
If the reimbursement or pricing that we are able to obtain and maintain for any product that we develop and commercialize, including OMIDRIA, is inadequate, is significantly delayed or is subject to overly restrictive conditions, our ability to generate revenue, attain profitability and/or commercialize our product candidates may be impaired and there could be a material adverse effect on our business, financial condition, results of operations and growth prospects and the trading price of our stock could decline.
Our operating results are unpredictable and may fluctuate.
Our operating results are difficult to predict and will likely fluctuate from quarter to quarter and year to year. We believe that our quarterly and annual results of operations may be affected by a variety of factors, including:
• | the level and timing of commercial sales of OMIDRIA, as well as our product candidates if and when approved or commercialized; |
• | the extent of coverage and reimbursement for OMIDRIA following the expiration of pass-through reimbursement on January 1, 2018; |
• | the amount of OMIDRIA chargebacks, rebates and product returns; |
• | the extent of any payments received from collaboration arrangements and development funding as well as the achievement of development and clinical milestones under collaboration and license agreements that we may enter into from time to time and that may vary significantly from quarter to quarter; and |
• | the timing, cost and level of investment in our research and development activities as well as expenditures we will or may incur to acquire or develop additional technologies, products and product candidates. |
In addition, the number of procedures in which OMIDRIA or any of our product candidates, if commercialized, would be used may be significantly less than the total number of such procedures performed or total possible market size. These and other factors, including our limited history of product sales, may make it difficult for us to forecast and provide accurate guidance (including updates to prior guidance) related to our expected financial performance. If our operating results are below the expectations of securities analysts or investors, the trading price of our stock could decline.
We have incurred cumulative operating losses since inception. If we are unable to raise additional capital when needed, our commercial operations may be limited and we may be unable to complete the development and commercialization of our product candidates or to continue our other preclinical development programs.
29
Our operations have consumed substantial amounts of cash since our incorporation and, as of December 31, 2017, we had an accumulated deficit of approximately $523.4 million. We expect to continue to spend substantial amounts to:
• | initiate and conduct clinical trials for our programs and product candidates; |
• | continue OMIDRIA sales and marketing; |
• | continue research and development in our programs; |
• | make principal, interest and fee payments under the CRG Loan Agreement; and |
• | commercialize and launch product candidates for which we may receive regulatory approval. |
We expect to continue to incur additional losses until such time as we generate significant revenue from the sale of OMIDRIA, other commercial products and/or significant partnering revenues. We are unable to predict the extent of any future losses and cannot provide assurance that we will generate sufficient revenue from OMIDRIA or other commercial products in the future to fund fully our operations. To date we have not generated revenue from sales of OMIDRIA that is sufficient to fund fully our operations. If we are unable to generate sufficient revenue from the sale of OMIDRIA, other commercialized products and/or partnering arrangements, we may never become and remain profitable and will be required to raise additional capital to continue to fund our operations. We cannot be certain that additional capital will be available to us on acceptable terms, if at all, when required. Adverse developments to our financial condition or business, as well as disruptions in the global equity and credit markets, may limit our ability to access capital. If we do not raise additional capital when needed through one or more funding avenues, such as corporate partnering or debt or equity financings, we may have to significantly delay, scale back or discontinue the development or commercialization of one or more of our product candidates or one or more of our preclinical programs or other research and development initiatives. In addition, we may be required to seek collaborators for one or more of our current or future products at an earlier stage than otherwise would be desirable or on terms that are less favorable than otherwise might be available or to relinquish or license on unfavorable terms our rights to technologies or products that we otherwise would seek to develop or commercialize ourselves. We also may have insufficient funds or otherwise be unable to advance our preclinical programs, such as potential new drug targets developed from our GPCR program, to a point where they can generate revenue through partnerships, collaborations or other arrangements. Any of these actions could limit the amount of revenue we are able to generate and harm our business and prospects.
Management, as well as our independent registered public accounting firm, have concluded that a substantial doubt is deemed to exist concerning our ability to continue as a going concern.
Accounting Standards Update, or ASU, 2014-15, requires management to assess our ability to continue as a going concern for one year after the date the financial statements are issued. As further discussed in Part II, Item 8, “Note 1--Organization and Basis of Presentation--Going Concern” to our Consolidated Financial Statements in this Annual Report on Form 10-K, substantial doubt is deemed to exist about the company’s ability to continue as a going concern through March 1, 2019. Our financial statements do not include any adjustment relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should we be unable to continue as a going concern. Our ability to continue as a going concern will require us to generate positive cash flow from operations, obtain additional financing, enter into strategic alliances and/or sell assets. The reaction of investors to the inclusion of a going concern statement in this Annual Report on Form 10-K, our current lack of cash resources and our potential inability to continue as a going concern may materially adversely affect our share price and our ability to raise new capital, enter into strategic alliances and/or make our scheduled debt payments on a timely basis or at all. If we become unable to continue as a going concern, we may have to liquidate our assets and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements.
We are subject to extensive government regulation and the failure to comply with these regulations may have a material adverse effect on our operations and business.
Both before and after approval of any product, we and our suppliers, contract manufacturers and clinical investigators are subject to extensive regulation by governmental authorities in the U.S. and other countries, covering, among other things, testing, manufacturing, quality control, clinical trials, post-marketing studies, labeling, advertising, promotion, distribution, import and export, governmental pricing, price reporting and rebate requirements. Failure to comply with applicable requirements could result in one or more of the following actions: warning letters; unanticipated expenditures; delays in approval or refusal to approve a product candidate; product recall or seizure; interruption of manufacturing or clinical trials; operating or marketing restrictions; injunctions; criminal prosecution and civil or criminal penalties including fines and other monetary penalties; adverse publicity; and disruptions to our business. Further, government investigations into potential violations of these laws would require us to expend considerable resources and face adverse publicity and the potential disruption of our business even if we are ultimately found not to have committed a violation.
30
Obtaining FDA approval of our product candidates requires substantial time, effort and financial resources and may be subject to both expected and unforeseen delays, and there can be no assurance that any approval will be granted on any of our product candidates on a timely basis, if at all. Even if we discuss with, and obtain feedback from, the FDA regarding our proposed clinical trials and nonclinical studies before initiating those trials or studies, the FDA may decide that the design of our clinical trials as actually run or our resulting data are insufficient for approval of our product candidates and require additional preclinical, clinical or other studies or additional work related to chemistry, manufacturing and controls. In addition, we, the FDA or an independent institutional review board or ethics committee may suspend or terminate human clinical trials at any time on various grounds, including a finding that the patients are or would be exposed to an unacceptable health risk or because of the way in which the investigators on which we rely carry out the trials. If we are required to conduct additional trials or to conduct other testing of our product candidates beyond that which we currently contemplate for regulatory approval, if we are unable to complete successfully our clinical trials or other testing, or if the results of these and other trials or tests fail to demonstrate efficacy or raise safety concerns, we may face substantial additional expenses, be delayed in obtaining marketing approval for our product candidates or may never obtain marketing approval.
We are also required to comply with extensive governmental regulatory requirements after a product has received marketing authorization. Governing regulatory authorities may require post-marketing studies that may negatively impact the commercial viability of a product. Once on the market, a product may become associated with previously undetected adverse effects and/or may develop manufacturing difficulties. We are required to comply with other post-marketing requirements including GMPs, advertising and promotion restrictions, reporting and recordkeeping obligations and other requirements. As a result of any of these or other problems, a product’s regulatory approval could be withdrawn, which could harm our business and operating results. In addition, we must establish and maintain an effective healthcare compliance program in order to comply with U.S. and other laws applicable to marketed drug products and, in particular, laws (such as the Anti-Kickback Statute, the False Claims Act and the Sunshine Act) applicable when drug products are purchased or reimbursed by a federal healthcare program. U.S. laws such as the Foreign Corrupt Practices Act prohibit the offering or payment of bribes or inducements to foreign public officials, including potentially physicians or other medical professionals who are employees of public health care entities. In addition, many countries have their own laws similar to the healthcare compliance laws that exist in the U.S. Implementing and maintaining an effective compliance program requires the expenditure of significant time and resources. If we are found to be in violation of any of these laws, we may be subject to significant penalties, including but not limited to civil or criminal penalties, damages and fines as well as exclusion from government healthcare programs.
We may face difficulties from changes to current regulations as well as future legislation.
Existing regulatory policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability.
There is uncertainty with respect to the impact that health care reform legislation may have on coverage and reimbursement for healthcare items and services covered by plans that are authorized by the ACA. President Trump and various members of Congress have expressed a desire to repeal all or portions of the ACA and in December 2017 portions of the ACA dealing with the individual mandate insurance requirement were effectively repealed by the Tax Cuts and Jobs Act of 2017. In addition, other legislative changes have been proposed and adopted since the ACA was enacted. President Trump and the Secretary of Health and Human Services have also made statements about controlling drug prices. We cannot predict the ultimate content, timing or effect of any healthcare reform legislation or executive order or the impact that the resulting changes may have on us.
We expect that the ACA, if it remains in effect, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate sufficient revenue, attain and/or maintain profitability or commercialize our product candidates. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on OMIDRIA or the marketing approvals of our product candidates, if any, may be.
31
U.S. federal income tax reform could adversely affect us.
On December 22, 2017, the Tax Cuts and Jobs Act, or 2017 Tax Act, was signed into law. The 2017 Tax Act, among other things, includes changes to U.S. federal tax rates, imposes additional limitations on the deductibility of interest, changes to the Orphan Drug Credit, allows for the expensing of capital expenditures, puts into effect the migration from a “worldwide” system of taxation to a territorial system and modifies or repeals many business deductions and credits. The estimated impact of the 2017 Tax Act is based on our management’s current knowledge and assumptions and recognized impacts could be materially different from current estimates based on further analysis of the new law. We have revalued our net deferred tax assets and liabilities at the newly enacted U.S. corporate rate, and the estimated impact was recognized in our financial statements for the year ended December 31, 2017.
Failure to obtain and maintain regulatory approval in foreign jurisdictions would prevent us from marketing our products internationally.
We intend to have OMIDRIA and our product candidates, if approved, marketed outside the U.S. In order to market our products in non-U.S. jurisdictions, we or our partners must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The regulatory approval procedure varies among countries and can involve additional testing and data review. The requirements governing marketing authorization, the conduct of clinical trials, pricing and reimbursement vary from country to country. Approval by the FDA or the EMA does not ensure approval by regulatory agencies in other jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory agencies in other foreign countries or by the FDA. The time required to obtain regulatory approval outside the U.S. and EU may differ from that required to obtain FDA or EMA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval discussed in these “Risk Factors” and we may not obtain foreign regulatory approvals on a timely basis, or at all. In addition, even if we were able to obtain regulatory approval for a product in one or more foreign jurisdictions, we may need to complete additional requirements to maintain that approval and our ability to market the product in the applicable jurisdiction. For example, OMIDRIA must be placed on the market (i.e., released into the distribution chain) in at least one EEA country by July 28, 2018 in order for our EU marketing authorization for OMIDRIA to remain valid.
OMIDRIA, as well as any of our product candidates, if approved, that are marketed outside of the United States, may face a variety of risks associated with international operations that, if realized, could materially adversely affect our business.
We may be subject to additional risks for OMIDRIA or any of our product candidates that are marketed outside the U.S., including:
• | reduced protection for intellectual property rights; |
• | unexpected changes in tariffs, trade barriers and regulatory requirements; |
• | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
• | foreign currency fluctuations and other obligations incident to doing business in another country; and |
• | business interruptions resulting from geopolitical actions, including war and terrorism or natural disasters including earthquakes, typhoons, floods and fires. |
Any of these risks, if realized, could increase our operating expenses and reduce our revenues.
We have no internal capacity to manufacture commercial or clinical supplies of OMIDRIA or our product candidates and intend to rely solely on third-party manufacturers. If the contract manufacturers that we rely on experience difficulties manufacturing and supplying OMIDRIA or our product candidates or fail FDA or other regulatory inspections, our clinical trials, regulatory submissions and ability to sell OMIDRIA or any other commercialized product and generate revenue may be significantly limited or delayed.
We rely and intend to continue to rely on third-party manufacturers to produce commercial quantities of OMIDRIA and clinical drug supplies of our product candidates that are needed for clinical trials. We cannot provide any assurance that we will be able to enter into or maintain these types of arrangements on commercially reasonable terms, or at all. If we or the manufacturer were to terminate one of these arrangements early, or the manufacturer was unable to supply product quantities sufficient to meet our requirements, we would be required to transfer the manufacturing to an approved alternative facility and/or establish additional manufacturing and supply arrangements. We may also need to establish additional or replacement manufacturers, potentially with little or no notice, in the event that one of our manufacturers fails to comply with FDA and/or other pharmaceutical manufacturing regulatory requirements. Even if we are able to establish additional or replacement manufacturers, identifying these sources and entering into definitive supply agreements and obtaining regulatory approvals may
32
require a substantial amount of time and cost and may create a shortage of the product. Such alternate supply arrangements may not be available on commercially reasonable terms, or at all. Additionally, if we are unable to engage multiple suppliers to manufacture our products, we may have inadequate supply to meet the demand of our product.
Our contract manufacturers may encounter difficulties with formulation, manufacturing, supply chain and/or release processes that could result in delays in clinical trials and/or regulatory submissions or that could impact adversely the commercialization of our products or product candidates, as well as in the initiation of enforcement actions by the FDA and other regulatory authorities. These difficulties also could result in the recall or withdrawal of a product from the market or a failure to have adequate supplies to meet market demand. If the safety of OMIDRIA or any product candidate supplied by contract manufacturers is compromised due to one or more of those contract manufacturers’ failure to adhere to applicable laws or for other reasons, we may not be able to maintain regulatory approval of OMIDRIA, to continue sales and marketing of OMIDRIA or to obtain and maintain regulatory approval for one or more of our product candidates, which would harm our business and prospects significantly.
In addition, any product candidate from certain of our programs, including but not limited to MASP-2 and MASP-3, could be a biologic drug product, and we do not have the internal capability to produce biologics for use in clinical trials or on a commercial scale. There are only a limited number of manufacturers of biologic drug products and we cannot be certain that we can enter into supply agreements with a sufficient number of them on commercially reasonable terms, if at all.
Any significant delays in the manufacture and/or supply of clinical or commercial supplies could materially harm our business, financial condition, results of operations and prospects.
Ingredients, excipients and other materials necessary to manufacture OMIDRIA or our product candidates may not be available on commercially reasonable terms, or at all, which may adversely affect the development and commercialization of OMIDRIA or those product candidates.
We and our third-party manufacturers must obtain from third-party suppliers the active pharmaceutical ingredients, excipients, and/or other raw materials plus primary and secondary packaging materials necessary for our contract manufacturers to produce OMIDRIA and our product candidates for our clinical trials and, to the extent approved or commercialized, for commercial distribution. Although we have entered or intend to enter into agreements with third-party suppliers that will guarantee the availability and timely delivery of active pharmaceutical ingredients, excipients and materials for OMIDRIA and our product candidates, we have not yet entered into agreements for the supply of all such ingredients, excipients or materials, and we may be unable to secure all such supply agreements or guarantees on commercially reasonable terms, if at all. Even if we were able to secure such agreements or guarantees, our suppliers may be unable or choose not to provide us the ingredients, excipients or materials in a timely manner or in the quantities required. If we or our third-party manufacturers are unable to obtain the quantities of these ingredients, excipients or materials that are necessary for the manufacture of commercial supplies of OMIDRIA, our ability to generate revenue from the sale of OMIDRIA would be materially and adversely affected. Further, if we or our third-party manufacturers are unable to obtain active pharmaceutical ingredients, excipients and materials as necessary for our clinical trials or for the manufacture of commercial supplies of our product candidates, if approved, potential regulatory approval or commercialization would be delayed, which would materially and adversely affect our ability to generate revenue from the sale of our product candidates.
If our clinical trials are delayed, suspended or terminated, we may be unable to develop our product candidates on a timely basis, which would adversely affect our ability to obtain regulatory approvals, increase our development costs and delay or prevent commercialization of approved products.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical trials that will cause regulatory agencies, institutional review boards or ethics committees, or us to delay our clinical trials or suspend or delay the analysis of the data from those trials. Clinical trials can be delayed for a variety of reasons, including:
• | discussions with the FDA, the EMA or other foreign authorities regarding the scope or design of our clinical trials; |
• | delays or the inability to obtain required approvals from institutional review boards, ethics committees or other responsible entities at clinical sites selected for participation in our clinical trials; |
• | delays in enrolling patients into clinical trials for any reason including disease severity, trial protocol design, study eligibility criteria, patient population size (e.g., for orphan diseases or for some pediatric indications), proximity and/or availability of clinical trial sites for prospective patients, availability of competing therapies and clinical trials, regional differences in diagnosis and treatment, perceived risks and benefits of the product or product candidate, physician patient referral practices or the ability to monitor patients adequately before and after treatment; |
• | lower than anticipated retention rates of patients in clinical trials; |
33
• | the need to repeat or conduct additional clinical trials as a result of inconclusive or negative results, failure to replicate positive early clinical data in subsequent clinical trials, failure to deliver an efficacious dose of a product candidate, poorly executed testing, a failure of a clinical site to adhere to the clinical protocol, an unacceptable study design or other problems; |
• | adverse findings in clinical or nonclinical studies related to the safety of our product candidates in humans; |
• | an insufficient supply of product candidate materials or other materials necessary to conduct our clinical trials; |
• | the need to qualify new suppliers of product candidate materials for FDA and foreign regulatory approval; |
• | an unfavorable inspection or review by the FDA or other regulatory authority of a clinical trial site or records of any clinical investigation; |
• | the occurrence of unacceptable drug-related side effects or adverse events experienced by participants in our clinical trials; |
• | the suspension by a regulatory agency of a trial put on a clinical hold; or |
• | the amendment of clinical trial protocols to reflect changes in regulatory requirements and guidance or other reasons as well as subsequent re-examination of amendments of clinical trial protocols by institutional review boards or ethics committees. |
In addition, a clinical trial or development program may be suspended or terminated by us, the FDA or other regulatory authorities, or institutional review boards or ethics committees due to a number of factors, including:
• | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
• | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
• | the failure to remove a clinical hold in a timely manner (which we cannot predict with certainty), if at all; |
• | unforeseen safety issues or any determination that a trial presents unacceptable health risks; |
• | inability to deliver an efficacious dose of a product candidate; or |
• | lack of adequate funding to continue the clinical trial or development program, including the incurrence of unforeseen costs due to enrollment delays, requirements to conduct additional trials and studies and increased expenses associated with the services of our contract research organizations, or CROs, and other third parties. |
If the results of our clinical trials are not available when we expect or if we encounter any delay in the analysis of data from our clinical trials, we may be unable to file for regulatory approval or conduct additional clinical trials on the schedule we currently anticipate. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate. Any delays in completing our clinical trials could increase our development costs, could slow down our product development and regulatory submission process, could delay our receipt of product revenue and could make it difficult to raise additional capital. In addition, significant clinical trial delays also could allow our competitors to bring products to market before we do and impair our ability to commercialize our future products, potentially harming our business.
Because we have a number of product candidates and development programs, we may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications for which there is a greater likelihood of obtaining regulatory approval and that may be more profitable, if approved.
We have limited resources and must focus on the product candidates and clinical and preclinical development programs that we believe are the most promising. As a result, we may forego or delay the pursuit of opportunities with other product candidates or other indications that later prove to have greater commercial potential and may not be able to progress development programs as rapidly as otherwise possible. Further, if we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product through collaboration, license or other royalty arrangements in cases in which it would have been advantageous for us to retain sole development and commercialization rights.
Our preclinical programs may not produce product candidates that are suitable for clinical trials, our product candidates may not successfully complete clinical development and/or our product candidates may not be suitable for successful commercialization or generation of revenue through partnerships.
We must complete successfully preclinical testing, which may include demonstrating efficacy and the lack of toxicity in established animal models, before commencing clinical trials for any product candidate. Many pharmaceutical and biological product candidates do not successfully complete preclinical testing. There can be no assurance that positive results from preclinical studies will be predictive of results obtained from subsequent preclinical studies or clinical trials. Even if preclinical
34
testing is successfully completed, we cannot be certain that any product candidates that do advance into clinical trials will successfully demonstrate safety and efficacy in clinical trials. Even if we achieve positive results in early clinical trials, they may not be predictive of the results in later trials.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and such costs or an adverse outcome in such a proceeding may have a material negative effect on our financial condition, results of operations and/or stock price.
If we choose to go to court or take other enforcement action to stop someone else from using our inventions, that individual or company has the right to ask the court to rule that our underlying patents are invalid or should not be enforced against that third-party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of our patents. In addition, a lawsuit could result in a finding that some or all of the claims of one or more of our relevant patents are invalid, unenforceable and/or not infringed, and could also result in a generic version of OMIDRIA being launched. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our patents. An adverse outcome in any such legal action could have a material negative effect on our financial condition, results of operations and/or stock price. See “Legal Proceedings” under Item 3 of Part I of this Annual Report on Form 10-K for further discussion of our patent infringement lawsuits against Sandoz and against Lupin.
It may not be feasible to detect and undertake patent enforcement action to stop infringing activity by a number of individual entities, each on a small scale, such as compounding pharmacies. Further, our industry has produced a large number of patents and it is not always clear which patents cover various types of products or methods of use. A third party may claim that we or our contract manufacturers are using inventions covered by the third party’s patent rights and may go to court to stop us from engaging in the alleged infringing activity, including making, using or selling our products and product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and technical personnel. There is a risk that a court would decide that we, or our contract manufacturers, are infringing the third party’s patents and would order us or our contractors to stop the activities covered by the patents. In addition, if we or our contract manufacturers are found to have violated a third party’s patent, we or our contract manufacturers could be ordered to pay damages to the other party. We have agreed to or may agree to indemnify our contract manufacturers against certain patent infringement claims and thus may be responsible for any of their costs associated with such claims and actions. If we were sued for patent infringement, we would need to demonstrate that our products and product candidates or methods of use either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we might be unable to do this. Proving invalidity, in particular, is difficult since it requires clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
Although we have conducted searches of third-party patents with respect to our programs, we have not obtained written freedom to operate opinions for our programs and may not have identified all relevant third-party patents. Consequently, we cannot be certain that third-party patents containing claims covering our products, product candidates, programs, technologies or methods do not exist, have not been filed, or could not be filed or issued.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the capital necessary to continue our operations.
It is difficult and costly to protect our intellectual property and our proprietary technologies, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection for the use, formulation and structure of our products and product candidates, the methods used to manufacture them, the related therapeutic targets and associated methods of treatment as well as on successfully defending these patents against potential third-party challenges. Our ability to protect our products and product candidates from unauthorized making, using, selling, offering to sell or importing by third parties is dependent on the extent to which we have rights under valid and enforceable patents that cover these activities.
The patent positions of pharmaceutical, biotechnology and other life sciences companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Further, the determination that a patent application or patent claim meets all of the requirements for patentability is a subjective determination based on the application of law and jurisprudence. The ultimate determination by the USPTO or by a court or other trier of fact in the U.S., or corresponding foreign national patent offices or courts, on whether a claim meets all requirements of patentability cannot be assured. Although we have conducted searches for third-party publications, patents and
35
other information that may affect the patentability of claims in our various patent applications and patents, we cannot be certain that all relevant information has been identified. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or patent applications, in our licensed patents or patent applications or in third-party patents.
We cannot provide assurances that any of our patent applications will be found to be patentable, including over our own prior art patents, or will issue as patents. Neither can we make assurances as to the scope of any claims that may issue from our pending and future patent applications nor to the outcome of any proceedings by any potential third parties that could challenge the patentability, validity or enforceability of our patents and patent applications in the U.S. or foreign jurisdictions. Any such challenge, if successful, could limit patent protection for our products and product candidates and/or materially harm our business.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
• | we may not be able to generate sufficient data to support full patent applications that protect the entire breadth of developments in one or more of our programs, including our GPCR program; |
• | it is possible that one or more of our pending patent applications will not become an issued patent or, if issued, that the patent(s) will be sufficient to protect our technology, provide us with a basis for commercially viable products or provide us with any competitive advantages; |
• | if our pending applications issue as patents, they may be challenged by third parties as not infringed, invalid or unenforceable under U.S. or foreign laws; or |
• | if issued, the patents under which we hold rights may not be valid or enforceable. |
In addition, to the extent that we are unable to obtain and maintain patent protection for one of our products or product candidates or in the event that such patent protection expires, it may no longer be cost-effective to extend our portfolio by pursuing additional development of a product or product candidate for follow-on indications.
We also may rely on trade secrets to protect our technologies or products, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisers may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third-party entity illegally obtained and is using any of our trade secrets is expensive and time-consuming, and the outcome is unpredictable. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
The terms of our debt facility place restrictions on our operating and financial flexibility and, if we raise additional capital through debt financing, the terms of any new debt could further restrict our ability to operate our business.
We have borrowed $80.0 million under the CRG Loan Agreement and pledged substantially all of our assets, including intellectual property, as collateral. The CRG Loan Agreement restricts our ability to, among other things, incur indebtedness, grant liens, dispose of assets, make investments, make acquisitions, enter into certain transactions with affiliates, pay cash dividends or make distributions, repurchase stock, license certain of our intellectual property on an exclusive basis and engage in significant business transactions such as a change of control. Any of these restrictions could significantly limit our operating and financial flexibility and ability to respond to changes in our business or competitive activities.
The CRG Loan Agreement requires us to achieve either (a) certain minimum net revenue amounts through the end of 2021, which are $65.0 million and $75.0 million for the 2018 and 2019 calendar years, respectively, or (b) a minimum market capitalization threshold equal to the product of 6.4 multiplied by the aggregate principal amount of loans outstanding under the CRG Loan Agreement determined as of the fifth business day following announcement of earnings results for the applicable year. In the event we do not achieve either of the minimum revenue amount or the minimum market capitalization threshold for a year, we can satisfy the requirement by raising additional funds through an equity or subordinated debt issuance and using the proceeds to pay down the loan balance by an amount equal to the difference between the minimum revenue amount for such year and the actual revenue amount for such year. We recorded net sales of OMIDRIA of $64.8 million for the year ended December 31, 2017, which satisfied the minimum net revenue amounts from OMIDRIA for 2017. However, we cannot guarantee that we will satisfy the 2018 annual revenue covenant in the CRG Loan Agreement or the alternative market capitalization covenant that will be calculated in February or March 2019, or that there would not otherwise be a “material adverse change” under the CRG Loan Agreement, each of which would constitute an event of default. We are also required to maintain $5.0 million in cash and cash equivalents during the term of the CRG Loan Agreement.
The failure to satisfy these or other obligations under the CRG Loan Agreement would constitute an event of default. An event of default under the CRG Loan Agreement also includes the occurrence of any material adverse effect upon our business,
36
condition (financial or otherwise), operations, performance or property taken as a whole. If there is an event of default under the CRG Loan Agreement, the lenders may have the right to accelerate all of our repayment obligations under the CRG Loan Agreement and to take control of our pledged assets, which include substantially all of our assets including our intellectual property. Upon the acceleration of the loan, we will be required to repay the loan immediately or to attempt to reverse the declaration through negotiation or litigation. Further, if we are liquidated, the lenders’ right to repayment would be senior to the rights of the holders of our common stock to receive any proceeds from the liquidation. Any declaration of an event of default could significantly harm our business and prospects and could cause our stock price to decline. If we raise any additional debt financing, the terms of such debt could further restrict our operating and financial flexibility.
Under the CRG Loan Agreement, we may borrow up to an additional $45.0 million on or before May 20, 2018 at our sole discretion, subject to customary closing conditions, including without limitation the lack of a default and the lack of an occurrence of a material adverse change in or on the business, condition (financial or otherwise), operations, performance or property of Omeros and its subsidiaries taken as a whole. If we are unable to satisfy any of these conditions, we will not be able to borrow any of the additional $45.0 million and it may be necessary for us to seek alternative sources of capital.
Our competitors may develop products that are less expensive, safer or more effective, or which may otherwise diminish or eliminate the success of any products that we may commercialize.
We may not achieve commercial success if our competitors, many of whom have significantly more resources and experience than we, market products that are safer, more effective, less expensive or faster to reach the market than OMIDRIA or any future products that we may develop and commercialize. Our competitors also may market a product that proves to be unsafe or ineffective, which may affect the market for our competing product, or future product, regardless of the safety or efficacy of our product. The failure of OMIDRIA or any other future product that we may market to compete effectively with products marketed by our competitors would impair our ability to generate revenue, which would have a material adverse effect on our future business, our financial condition and our results of operations.
The loss of members of our management team could substantially disrupt our business operations.
Our success depends to a significant degree on the continued individual and collective contributions of our management team. The members of our management team are at-will employees, and we do not maintain any key-person life insurance policies other than on the life of Gregory A. Demopulos, M.D., our president, chief executive officer and chairman of the board of directors. Losing the services of any key member of our management team, whether from death or disability, retirement, competing offers or other causes, without having a readily available and appropriate replacement could delay the execution of our business strategy, cause us to lose a strategic partner, or otherwise materially affect our operations.
We rely on highly skilled personnel and, if we are unable to retain or motivate key personnel or hire qualified personnel, we may not be able to maintain our operations or grow effectively.
Our performance is largely dependent on the talents and efforts of highly skilled individuals. Our future success depends on our continuing ability to identify, hire, develop, motivate and retain highly skilled personnel for all areas of our organization. If we are unable to hire and train a sufficient number of qualified employees for any reason, we may not be able to implement our current initiatives or grow effectively. We have in the past maintained a rigorous, highly selective and time-consuming hiring process. We believe that our approach to hiring has significantly contributed to our success to date. If we do not succeed in attracting qualified personnel and retaining and motivating existing personnel, our existing operations may suffer and we may be unable to grow effectively.
We may encounter difficulties managing our growth, which could delay our business plans or adversely affect our results of operations.
To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems and continue to recruit and train additional qualified personnel. Due to our limited financial resources, we may not be able to manage effectively the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations. Additionally, our inability to manage growth effectively could cause our operating costs to grow even faster than we currently are anticipating.
37
Product liability claims may damage our reputation and, if insurance proves inadequate, these claims may harm our business.
We may be exposed to the risk of product liability claims that is inherent in the biopharmaceutical industry. A product liability claim may damage our reputation by raising questions about our product’s safety and efficacy and could limit our ability to sell one or more products by preventing or interfering with commercialization of our products and product candidates. In addition, product liability insurance for the biopharmaceutical industry is generally expensive to the extent it is available at all. There can be no assurance that we will be able to obtain or maintain such insurance on acceptable terms or that we will be able to secure and maintain increased coverage for OMIDRIA or for our product candidates, if commercialization progresses, or that future claims against us will be covered by our product liability insurance. Further, our product liability insurance coverage may not reimburse us or may be insufficient to reimburse us for any or all expenses or losses we may suffer. A successful claim against us with respect to uninsured liabilities or in excess of insurance coverage could have a material adverse effect on our business, financial condition and results of operations.
We rely on third parties to conduct portions of our preclinical research and clinical trials. If these third parties do not perform as contractually required or otherwise expected, or if we fail to adequately supervise or monitor these parties, we may not be able to obtain regulatory approval for or commercialize our product candidates.
We rely on third parties, such as CROs, medical and research institutions and clinical investigators, to conduct a portion of our preclinical research and assist us in conducting our clinical trials. Nonetheless, we are responsible for confirming that our preclinical research and clinical trials are conducted in accordance with applicable regulations, the relevant trial protocol and within the context of approvals by an institutional review board or ethics committee, and we may not always be successful in ensuring such compliance. Our reliance on these third parties does not relieve us of responsibility for ensuring compliance with FDA and other regulations and standards for conducting, monitoring, recording and reporting the results of preclinical research and clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if the third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical and clinical development processes may be extended, delayed, suspended or terminated, and we may not be able to commercialize or obtain regulatory approval for our product candidates.
We may need to maintain licenses for active ingredients from third parties to develop and commercialize some of our product candidates, which could increase our development costs and delay our ability to commercialize those product candidates.
Should we decide to use active pharmaceutical ingredients in any of our product candidates that are proprietary to one or more third parties, such as our PDE7 program (OMS527), we would need to maintain licenses to those active ingredients from those third parties. If we are unable to continue to access rights to these active ingredients prior to conducting preclinical toxicology studies intended to support clinical trials, we may need to develop alternate product candidates from these programs by either accessing or developing alternate active ingredients, resulting in increased development costs and delays in commercialization of these product candidates. If we are unable to maintain continued access rights to the desired active ingredients on commercially reasonable terms or develop suitable alternate active ingredients, or if we do not meet diligence or other obligations under the corresponding licenses, we may not be able to commercialize product candidates from these programs.
We use hazardous materials in our business and must comply with environmental laws and regulations, which can be expensive.
Our research operations produce hazardous waste products, which include chemicals and radioactive and biological materials. We are subject to a variety of federal, state and local regulations relating to the use, handling, storage and disposal of these materials. Although we believe that our safety procedures for handling and disposing of these materials comply with applicable legal regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. We generally contract with third parties for the disposal of such substances and store our low-level radioactive waste at our facility until the materials are no longer considered radioactive. We may be required to incur further costs to comply with current or future environmental and safety regulations. In addition, although we carry insurance, in the event of accidental contamination or injury from these materials, we could be held liable for any damages that result and any such liability could exceed our insurance coverage and other resources.
Cyber-attacks or other failures in telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations.
38
We utilize information technology, or IT, systems and networks to process, transmit and store electronic information in connection with our business activities. As use of digital technologies has increased, cyber incidents, including deliberate attacks and attempts to gain unauthorized access to computer systems and networks, have increased in frequency and sophistication. These threats pose a risk to the security of our systems and networks, the confidentiality and the availability and integrity of our data. There can be no assurance that we will be successful in preventing cyber-attacks or successfully mitigating their effects. Similarly, there can be no assurance that our collaborators, CROs, third-party logistics providers, distributors and other contractors and consultants will be successful in protecting our clinical and other data that is stored on their systems. Any cyber-attack or destruction or loss of data could have a material adverse effect on our business and prospects. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyber-attacks or other data security breaches and may incur significant additional expense to implement further data protection measures.
We currently depend on a third-party for the commercialization of OMS103 and we cannot guarantee that such commercialization will occur or be successful.
In June 2015 we entered into the OMS103 Agreement pursuant to which we granted Fagron an exclusive, royalty-free license to the OMS103 intellectual property, manufacturing information and clinical data to manufacture and commercialize OMS103 in the U.S. in exchange for potential future payments based on product revenue and achievement of commercial milestones. As a result of entering into the OMS103 Agreement, we discontinued our OMS103 clinical development program and are dependent on Fagron to commercialize OMS103 in the U.S. We cannot control whether Fagron will fulfill its obligations under the OMS103 Agreement or whether the commercialization of OMS103 will be successful. Fagron has not performed its diligence milestones in the OMS103 Agreement, including initiating sales of OMS103, and we believe that it is unlikely they will do so. We continue to evaluate our options with respect to the OMS103 Agreement and the OMS103 program. If we elect to pursue arbitration with Fagron, and/or the OMS103 Agreement is terminated, we can provide no assurances that we will be able to enter into another licensing agreement or have sufficient resources to restart clinical development and conduct any clinical trials if desired. Any of the above risks, if realized, could adversely affect our results of operations.
Under Section 503B of the FDCA, registered outsourcing facilities are required to manufacture under GMP and are subject to FDA inspections and audits. They also are not allowed to manufacture a product that is essentially a copy of one or more FDA-approved drugs. If a licensed registered outsourcing facility such as Fagron is prohibited from commercializing or from continuing commercial sales of OMS103 following initial commercialization because of violations of any FDA regulations or any other reason, our ability to generate revenue from royalty payments from the licensed registered outsourcing facility and achieve profitability will be adversely affected and the market price of our common stock could decline.
Risks Related to Our Common Stock
Our stock price has been and may continue to be volatile, and the value of an investment in our common stock may decline.
During the 12-month period ended December 31, 2017, our stock traded as high as $27.09 per share and as low as $8.71 per share. The trading price of our common stock is likely to continue to be highly volatile and could be subject to wide fluctuations in response to numerous factors, many of which are beyond our control. In addition, the stock market has experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of publicly traded companies. Broad market and industry factors may seriously affect the market price of companies’ stock, including ours, regardless of actual operating performance. These fluctuations may be even more pronounced in the trading market for our stock. In addition, in the past, following periods of volatility in the overall market and the market price of a particular company’s securities, securities class action litigation has often been instituted against these companies. This litigation, if instituted against us, could result in substantial costs and a diversion of our management’s attention and resources.
If we issue additional shares of our common stock or other securities that may be convertible into, or exercisable or exchangeable for, our common stock, our existing shareholders would experience further dilution.
To the extent that we raise additional funds in the future by issuing equity securities, our shareholders would experience dilution, which may be significant and could cause the market price of our common stock to decline significantly. In addition, approximately 9,757,861 shares of common stock as of December 31, 2017 subject to outstanding options and warrants may become eligible for sale in the public market to the extent permitted by the provisions of various vesting agreements. Further, as of December 31, 2017 we also had approximately 3,513,540 shares of common stock reserved for future issuance under our employee benefit plans that are not subject to outstanding options. If the holders of these outstanding options or warrants elect
39
to exercise some or all of them, or if the shares subject to our employee benefit plans are issued and become eligible for sale in the public market, our shareholders would experience dilution and the market price of our common stock could decline.
Anti-takeover provisions in our charter documents and under Washington law could make an acquisition of us, which may be beneficial to our shareholders, difficult and prevent attempts by our shareholders to replace or remove our current management.
Provisions in our articles of incorporation and bylaws and under Washington law may delay or prevent an acquisition of us or a change in our management. These provisions include a classified board of directors, a prohibition on shareholder actions by less than unanimous written consent, restrictions on the ability of shareholders to fill board vacancies and the ability of our board of directors to issue preferred stock without shareholder approval. In addition, because we are incorporated in Washington, we are governed by the provisions of Chapter 23B.19 of the Washington Business Corporation Act, which, among other things, restricts the ability of shareholders owning 10% or more of our outstanding voting stock from merging or combining with us. Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some shareholders. In addition, these provisions may frustrate or prevent any attempts by our shareholders to replace or remove our current management by making it difficult for shareholders to replace members of our board of directors, which is responsible for appointing the members of our management.
We have never declared or paid dividends on our capital stock, and we do not anticipate paying dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings, if any, in the development and growth of our business. Additionally, under the CRG Loan Agreement, we have agreed not to pay any cash dividends. Therefore, we currently do not anticipate paying any cash dividends on our common stock in the foreseeable future. As a result, a rise in the market price of our common stock, which is uncertain and unpredictable, will be the sole source of potential gain for shareholders in the foreseeable future, and an investment in our common stock for dividend income should not be relied upon.
40
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
Not applicable.
ITEM 2. | PROPERTIES |
We lease approximately 82,000 square feet for our principal office and laboratory space in the building located at 201 Elliott Avenue West, Seattle, Washington, or The Omeros Building, which includes approximately 6,077 square feet of laboratory space that we are subleasing to third parties. The lease term for our space is through November 2027. We also have two options to extend the lease term, each by five years. The annual base rent due under the lease for our principal office and laboratory space is $4.6 million for 2018, $4.7 million for 2019 and $4.8 million for 2020 and will increase by approximately 2.3% each year thereafter. In addition, we are responsible for paying our proportionate share of the building’s utilities, taxes, insurance and maintenance as well as a property management fee.
We have a right of first refusal for the premises that we do not currently lease as well as a right of first offer for specified premises in The Omeros Building. If at any time during the remaining term of the lease the landlord is unable to accommodate our request for then-available additional space in The Omeros Building, the landlord is required to negotiate with us to relocate us to a new building under a build-to-suit lease with no termination penalty payable under our existing lease, subject to the negotiation of a mutually acceptable build-to-suit lease. In addition, beginning with the sixth year of the lease term, if we request from the landlord additional space in The Omeros Building that is otherwise available for leasing with a minimum square footage specified in the lease and the landlord is unable to provide such additional space to us, we may terminate the lease with 12 months’ prior notice without payment of any termination fees other than the unamortized portion of a $3.0 million lease incentive paid to us by the landlord when we entered the lease (plus accrued interest). We have the right to terminate the lease beginning with year nine of the lease term, subject to the payment of a lease termination fee. If we terminate the lease during years nine or 10, the termination fee is equal to 30% of the unamortized tenant improvements and 100% of the unamortized lease incentive. If we terminate the lease any time after year 10 of the term, the termination fee is equal to 20% of the unamortized tenant improvements and 100% of the unamortized lease incentive. We believe that these facilities we lease currently are sufficient for our anticipated near-term needs.
ITEM 3. LEGAL PROCEEDINGS
On October 4, 2017, we entered into a Settlement Agreement, or the Settlement Agreement, and consent judgment with Par, resolving our patent litigation against Par under the Hatch-Waxman Act in the U.S. District Court for the District of Delaware. The Settlement Agreement arose from Par’s filing of an Abbreviated New Drug Application, or ANDA, seeking approval from the FDA to market a generic version of OMIDRIA. Pursuant to the Settlement Agreement, Par, which had previously stipulated to infringement, acknowledged and confirmed the validity of each of the patents then listed in the Orange Book for OMIDRIA, which are U.S. Patent No. 8,173,707, U.S. Patent No. 8,586,633, U.S. Patent No. 9,066,856, U.S. Patent No. 9,278,101, U.S. Patent No. 9,399,040, and U.S. Patent No. 9,486,406. In accordance with the terms of the Settlement Agreement, the U.S. District Court for the District of Delaware entered a consent judgment on October 5, 2017, pursuant to which Par and its affiliates are prohibited from launching a generic version of OMIDRIA until the earlier of April 1, 2032 or a date on which we or a third party, through licensing or any future final legal judgment, should one ever exist, with respect to our Orange Book listed patents, is able to launch a generic version of OMIDRIA, as further detailed in the Settlement Agreement (such date referred to as the Entry Date). Under the Settlement Agreement, we granted Par a non-exclusive, nonsublicensable license to make, sell and distribute a generic version of OMIDRIA between the Entry Date and the latest expiration of our U.S. patents related to OMIDRIA (i.e., October 23, 2033). During this period, Par is required pay to us a royalty equal to 15% of Par’s net sales of its generic version of OMIDRIA.
In May 2017, we received Notice Letters from Sandoz and Lupin that each had filed an ANDA containing a Paragraph IV Certification under the Hatch-Waxman Act seeking approval from the FDA to market a generic version of OMIDRIA prior to the expiration of six Orange Book-listed patents covering OMIDRIA. On June 21, 2017, we filed a patent infringement lawsuit in the U.S. District Court for the District of Delaware and a patent infringement lawsuit in the U.S. District Court for the District of New Jersey against Sandoz and on June 22, 2017 we filed a patent infringement lawsuit in the U.S. District Court for the District of Delaware and a patent infringement lawsuit in the U.S. District Court for the District of New Jersey against Lupin. The Delaware lawsuits against Sandoz and Lupin were consolidated for all purposes by court order entered on October 16, 2017, and the New Jersey lawsuits were dismissed by agreement of the parties on October 13, 2017. Sandoz has filed an answer to our Delaware lawsuit asserting defenses of patent invalidity. Lupin has filed an answer to our Delaware lawsuit asserting defenses and counterclaims for declaratory judgment of patent invalidity and non-infringement. The lawsuits were filed under the Hatch-Waxman Act for Sandoz’s and Lupin’s respective infringement of six Omeros patents: U.S. Patent Nos. 8,173,707, 8,586,633, 9,066,856, 9,278,101, 9,399,040 and 9,486,406, which relate to OMIDRIA and are listed in the Orange
41
Book. On January 31, 2018, we filed an amended complaint against Lupin to assert the newly issued U.S. Patent No. 9,855,246, and Lupin answered the amended complaint on February 14, 2018, asserting counterclaims for noninfringement and invalidity. Omeros has also filed a motion for leave to amend the lawsuit against Sandoz to assert this seventh patent. The asserted patents were all granted following review by the U.S. Patent and Trademark Office, are presumed to be valid under governing law, and can only be invalidated in federal court with clear and convincing evidence. Under the Hatch-Waxman Act, we were permitted to file suit within 45 days from receipt of each Notice Letter and thereby trigger a 30-month stay of the FDA’s approval of the respective ANDAs. Each stay is expected to remain in effect until November 2019 while the lawsuits are pending. The assertions raised in Sandoz’s and Lupin’s Paragraph IV Notice Letters and their answers to our lawsuits are substantially similar to those raised by Par in the above-described patent litigation matter against Par. We believe the assertions in the Sandoz and Lupin Paragraph IV Notice Letters and their answers to our lawsuits do not have merit, and we intend to vigorously prosecute our infringement claims against each of Sandoz and Lupin.
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
42
PART II
ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED SHAREHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock is traded on The Nasdaq Global Market under the symbol “OMER.”
The following table sets forth the range of high and low sales prices of our common stock as quoted on The Nasdaq Global Market for the periods indicated.
Year Ended December 31, 2017 | High | Low | ||
4th Quarter | $24.45 | $12.45 | ||
3rd Quarter | $25.19 | $18.63 | ||
2nd Quarter | $27.09 | $13.56 | ||
1st Quarter | $16.40 | $8.71 | ||
Year Ended December 31, 2016 | High | Low | ||
4th Quarter | $14.15 | $7.20 | ||
3rd Quarter | $13.71 | $10.36 | ||
2nd Quarter | $16.38 | $9.46 | ||
1st Quarter | $16.80 | $8.90 | ||
Holders
As of February 16, 2018, there were approximately 107 holders of record of our common stock.
Dividends
We have never declared or paid any cash dividends on our capital stock, and under the CRG Loan Agreement we have agreed not to pay any cash dividends. We expect to retain all available funds and future earnings to fund the development and growth of our business and we do not anticipate paying any cash dividends in the foreseeable future.
Stock Performance Graph
The following graph compares the cumulative total shareholder return for our common stock (OMER), the Nasdaq Biotechnology Index (NBI) and the Nasdaq U.S. Benchmark TR Index (NQUSBT) for the period beginning December 31, 2012 and ending December 31, 2017. This graph assumes that $100 was invested on December 31, 2012 in our common stock, the Nasdaq Biotechnology Index and the Nasdaq U.S. Benchmark TR Index. It also assumes that any dividends were reinvested. The data shown in the following graph are not necessarily indicative of future stock price performance.
43
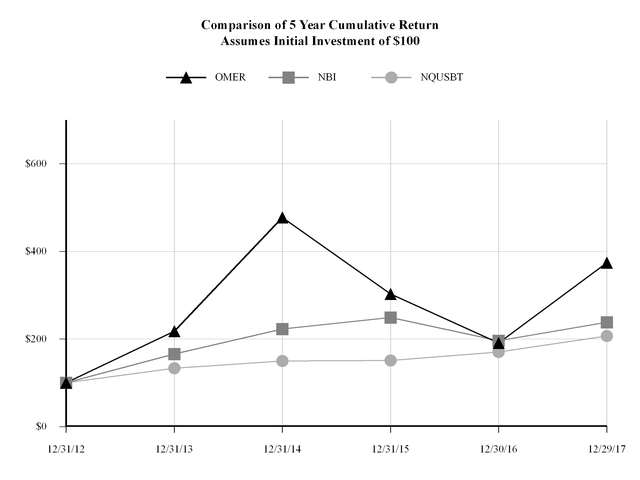
The foregoing information shall not be deemed to be “soliciting material” or to be “filed” for purposes of Section 18 of the Exchange Act or otherwise subject to liability under that Section. In addition, the foregoing information shall not be deemed to be incorporated by reference into any of our filings under the Exchange Act or the Securities Act of 1933, except to the extent that we specifically incorporate this information by reference.
44
ITEM 6. | SELECTED CONSOLIDATED FINANCIAL DATA |
The following selected consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the accompanying notes included elsewhere in this Annual Report on Form 10-K. Our historical results are not necessarily indicative of the results to be expected in any future period due in part to the expiration of pass-through reimbursement for OMIDRIA effective January 1, 2018.
.
Year Ended December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
(In thousands, except per share and share data) | |||||||||||||||||||
Consolidated Statements of Operations and Comprehensive Loss Data: | |||||||||||||||||||
Revenues | |||||||||||||||||||
Product sales, net | $ | 64,826 | $ | 41,444 | $ | 13,264 | $ | — | $ | — | |||||||||
Grant revenue | — | 173 | 245 | 539 | 1,600 | ||||||||||||||
Total revenue | 64,826 | 41,617 | 13,509 | 539 | 1,600 | ||||||||||||||
Costs and expenses | |||||||||||||||||||
Cost of product sales | 1,078 | 1,412 | 1,041 | — | — | ||||||||||||||
Research and development | 55,599 | 50,699 | 48,379 | 47,946 | 36,297 | ||||||||||||||
Selling, general and administrative | 52,044 | 43,782 | 35,327 | 22,601 | 15,819 | ||||||||||||||
Total costs and expenses | 108,721 | 95,893 | 84,747 | 70,547 | 52,116 | ||||||||||||||
Loss from operations | (43,895 | ) | (54,276 | ) | (71,238 | ) | (70,008 | ) | (50,516 | ) | |||||||||
Litigation settlement | — | — | — | — | 12,500 | ||||||||||||||
Interest expense | (11,030 | ) | (7,819 | ) | (3,573 | ) | (3,470 | ) | (2,366 | ) | |||||||||
Other income (expense) | 1,444 | 945 | 1,030 | (195 | ) | 586 | |||||||||||||
Loss on early extinguishment of debt | — | (5,595 | ) | (1,315 | ) | — | — | ||||||||||||
Net loss | $ | (53,481 | ) | $ | (66,745 | ) | $ | (75,096 | ) | $ | (73,673 | ) | $ | (39,796 | ) | ||||
Comprehensive loss | $ | (53,481 | ) | $ | (66,745 | ) | $ | (75,096 | ) | $ | (73,673 | ) | $ | (39,796 | ) | ||||
Basic and diluted net loss per share | $ | (1.17 | ) | $ | (1.65 | ) | $ | (2.00 | ) | $ | (2.22 | ) | $ | (1.39 | ) | ||||
Weighted-average shares used to compute basic and diluted net loss per share | 45,539,362 | 40,446,410 | 37,560,257 | 33,234,294 | 28,560,360 | ||||||||||||||
As of December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
(In thousands) | |||||||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||||
Cash, cash equivalents and short-term investments | $ | 83,749 | $ | 45,331 | $ | 28,263 | $ | 6,886 | $ | 14,101 | |||||||||
Working capital (deficit) | 82,065 | 44,191 | 20,893 | (9,274 | ) | 2,944 | |||||||||||||
Restricted cash and investments | 5,835 | 5,835 | 10,679 | 679 | 679 | ||||||||||||||
Total assets | 116,328 | 67,278 | 48,995 | 10,834 | 16,535 | ||||||||||||||
Notes payable and lease financing obligations, net | 84,117 | 79,512 | 49,842 | 32,453 | 20,498 | ||||||||||||||
Accumulated deficit | (523,368 | ) | (469,887 | ) | (403,142 | ) | (328,046 | ) | (254,373 | ) | |||||||||
Total shareholders’ deficit | (2,814 | ) | (37,447 | ) | (26,234 | ) | (42,654 | ) | (18,384 | ) | |||||||||
45
ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis should be read in conjunction with the audited annual consolidated financial statements and the related notes thereto included elsewhere in this Annual Report on Form 10-K. This discussion contains forward-looking statements reflecting our current expectations that involve risks and uncertainties. Actual results may differ materially from those discussed in these forward-looking statements due to a number of factors, including those set forth in the section entitled “Risk Factors” and elsewhere in this Annual Report on Form 10-K. For further information regarding forward-looking statements, please refer to the special note regarding forward-looking statements at the beginning of this Annual Report on Form 10-K. Throughout this discussion, unless the context specifies or implies otherwise, the terms “Company,” “we,” “us” and “our” refer to Omeros Corporation and our wholly owned subsidiaries.
Overview
We are a commercial-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting inflammation, complement-mediated diseases and disorders of the central nervous system.
Our drug product OMIDRIA® is marketed in the U.S. for use during cataract surgery or intraocular lens, or IOL, replacement. In our pipeline we have clinical-stage development programs focused on: complement-associated thrombotic microangiopathies; complement-mediated glomerulonephropathies; Huntington’s disease and cognitive impairment; and addictive and compulsive disorders. In addition, we have a diverse group of preclinical programs and two platforms: one capable of unlocking new G protein-coupled receptor, or GPCR, drug targets and the other used to generate antibodies. For OMIDRIA and each of our product candidates and our programs, other than OMS103, we have retained control of all commercial rights.
Financial Summary
We recognized net losses of $53.5 million, $66.7 million, and $75.1 million for the years ended December 31, 2017, 2016 and 2015, respectively.
During the years ended December 31, 2017, 2016 and 2015, OMIDRIA revenues of $64.8 million, $41.4 million, and $13.3 million respectively, helped offset a portion of our 2017 and 2016 operating expenses. We expect our net losses will continue until such time as we derive sufficient revenues from sales of OMIDRIA and/or other sources, such as licensing, product sales and other revenues from our product candidates, that are sufficient to cover our expenses.
As of December 31, 2017, we had $83.7 million in cash and cash equivalents and short-term investments available for general corporate use and $17.1 million in accounts receivable that we anticipate collecting in the first quarter of 2018. In addition, we had restricted cash and investments of $5.8 million that we were required to maintain pursuant to (a) the CRG Loan Agreement with CRG, and the lenders identified therein, which requires us to maintain a balance of cash and cash equivalents of $5.0 million, and (b) our lease related to the Omeros Building.
Results of Operations
Revenue
Our revenue consists almost entirely of OMIDRIA product sales to ambulatory surgery centers, or ASCs, and hospitals in the U.S. Our product sales, net are as follows:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Product sales, net | $ | 64,826 | $ | 41,444 | $ | 13,264 | |||||
In 2017, OMIDRIA revenue increased $23.4 million, or 56.4%, as compared to the year ended December 31, 2016. The increase in OMIDRIA revenue in 2017 as compared to 2016 was primarily attributable to continued acceptance of OMIDRIA in the ophthalmic surgery community, as evidenced by an increase in new customers and increased OMIDRIA purchases from our existing customers, due in part to increased participation in the OMIDRIA purchase volume discount program for which
46
ASCs may qualify by meeting or exceeding quarterly purchase volumes of OMIDRIA. Separate payment for OMIDRIA under Medicare Part B terminated as scheduled on January 1, 2018 and, as further discussed below, sales are reduced in the first quarter of 2018 due to uncertainty around OMIDRIA reimbursement. Under our accounting policies, we are not able to recognize revenue in the fourth quarter of 2017 for wholesaler on-hand OMIDRIA inventory at December 31, 2017 in excess of eight weeks of projected ASC and hospital demand.
In 2016, OMIDRIA revenue increased $28.2 million, or 212.5%, as compared to the year ended December 31, 2015. The increase in OMIDRIA revenue in 2016 as compared to 2015 was primarily due to the continued acceptance of OMIDRIA in the ophthalmic surgery community, as evidenced by an increase in the number of ASCs and hospitals purchasing OMIDRIA and increased penetration into existing customer accounts.
CMS granted transitional pass-through reimbursement status for OMIDRIA through January 1, 2018, which allowed for separate payment (i.e., outside the packaged procedural payment) under Medicare Part B for new drugs and other medical technologies. As scheduled, on January 1, 2018, OMIDRIA no longer had separate payment under Medicare Part B and consequently is included as part of the packaged procedural payment for Medicare patients. Legislative and administrative means are being pursued to obtain a continuation of separate or similar reimbursement for OMIDRIA and/or to extend the pass-through period; however, that has not yet occurred.
We also accept returns from our ASCs and hospitals that have purchased OMIDRIA from our wholesalers. Due to the expiration of pass-through, we expect the ASCs and hospitals will return a portion of their OMIDRIA on hand at December 31, 2017 to us for a full refund of the purchase price. We have recorded a $2.4 million reserve as of December 31, 2017 in anticipation of these returns.
We expect the significantly reduced OMIDRIA demand to continue while the reimbursement status of OMIDRIA remains uncertain. Based on first quarter 2018 data to date, we believe that a substantial majority of facilities that were using OMIDRIA are awaiting resolution regarding reimbursement by CMS, or our decision to implement an alternative sales strategy and, therefore, have largely suspended use of the product or are using it on a selective basis only.
Once the uncertainties relating to separate reimbursement are resolved, or we implement an alternative sales strategy, and we are assured that our wholesalers will sell the inventory they have on hand to ASCs and hospitals, we will recognize wholesaler on-hand inventory at December 31, 2017 as revenue in 2018. Similarly, as the ASCs and hospitals increase their usage of OMIDRIA, they will re-establish an on-hand supply of OMIDRIA. This replenishment will also increase our future revenues.
We are currently unable to predict whether separate or similar reimbursement will be granted or, if granted, the effective date of such separate or similar reimbursement or the actual reimbursement rate. We currently expect that any extension of pass-through or administrative action, if either occurs, would be effective for two additional years and would become effective between now and mid-year, but could apply retroactively. In the event that neither the legislative nor the administrative approaches are ultimately successful, we expect to pursue an alternative sales strategy. After implementing this strategy, we cannot predict how quickly, or if, our customers would increase their OMIDRIA utilization.
Until we have additional clarity on reimbursement for OMIDRIA, we cannot determine the impact on OMIDRIA product sales trends through the remainder of 2018.
47
Gross-to-Net Deductions
We record OMIDRIA product sales net of estimated chargebacks, rebates, distribution fees and product returns. These deductions are generally referred to as gross-to-net deductions. Our total gross-to-net provisions for the years ended December 31, 2017, 2016 and 2015 were 27.8%, 12.0% and 6.2%, respectively of gross OMIDRIA product sales. The primary reason for the increases in gross-to-net deductions from 2015 through 2017 is due to increased sales subject to chargeback deductions, rebates under our OMIDRIAssure Reimbursement Program and rebates under our volume-purchase discount program.
A summary of our gross-to-net provision and payments for the years ended December 31, 2017, 2016 and 2015 are as follows:
Chargebacks and Rebates | Distribution Fees and Product Return Allowances | Total | ||||||||||
(In thousands) | ||||||||||||
Balance as of December 31, 2014 | $ | — | $ | — | $ | — | ||||||
Provisions | 320 | 555 | 875 | |||||||||
Payments | (140 | ) | (278 | ) | (418 | ) | ||||||
Balance as of December 31, 2015 | 180 | 277 | 457 | |||||||||
Provisions | 4,203 | 1,434 | 5,637 | |||||||||
Payments | (2,754 | ) | (1,230 | ) | (3,984 | ) | ||||||
Balance as of December 31, 2016 | 1,629 | 481 | 2,110 | |||||||||
Provisions | 19,188 | 5,741 | 24,929 | |||||||||
Payments | (15,264 | ) | (2,687 | ) | (17,951 | ) | ||||||
Balance as of December 31, 2017 | $ | 5,553 | $ | 3,535 | $ | 9,088 | ||||||
Chargebacks and Rebates
We record a provision for estimated chargebacks and rebates at the time we recognize OMIDRIA product sales revenue and reduce the accrual when payments are made or credits are granted. Our chargebacks are related to a Pharmaceutical Pricing Agreement, a Federal Supply Schedule agreement, a 340B prime vendor agreement and a Medicaid Drug Rebate Agreement. We also record a provision for estimated rebates for our OMIDRIAssure Reimbursement Services Program and our purchase volume discount programs.
Distribution Fees and Product Return Allowances
We pay our wholesalers a distribution fee for services that they perform for us based on the dollar value of their purchases of OMIDRIA. We record a provision for these charges as a reduction to revenue at the time of sale to the wholesaler and make payments to our wholesalers based on contractual terms.
We allow for the return of product up to 12 months past its expiration date or for product that is damaged or not used by our customers. We record a provision for returns upon sale of OMIDRIA to our wholesaler. When a return or claim is received, we issue a credit memo to the wholesaler against its outstanding receivable to us or we reimburse the ASC or hospital.
At December 31, 2017, we have recorded a product return allowance of $2.4 million reflecting payments we anticipate making to ASCs and hospitals for OMIDRIA that they have on hand and the portion of that inventory that could likely be returned to us.
48
Research and Development Expenses
Our research and development expenses can be divided into three categories: direct external expenses, which include clinical research and development and preclinical research and development activities; internal, overhead and other expenses; and stock-based compensation expense. Direct external expenses consist primarily of expenses incurred pursuant to agreements with third-party manufacturing organizations, contract research organizations, or CROs, clinical trial sites, collaborators, and licensors and consultants. Costs are reported in preclinical research and development until the program enters the clinic. Internal, overhead and other expenses consist of personnel costs, overhead costs such as rent, utilities and depreciation and other miscellaneous costs. We do not generally allocate our internal resources, employees and infrastructure to any individual research project because we deploy them across multiple clinical and preclinical projects that we are advancing in parallel.
The following table illustrates our expenses associated with these activities:
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Direct external expenses: | |||||||||||
Clinical research and development: | |||||||||||
MASP-2 Program - OMS721 | $ | 19,557 | $ | 17,241 | $ | 15,852 | |||||
OMIDRIA - Ophthalmology | 3,458 | 3,864 | 4,396 | ||||||||
Other clinical programs | 1,714 | 500 | 1,545 | ||||||||
Total clinical research and development | 24,729 | 21,605 | 21,793 | ||||||||
Preclinical research and development | 4,269 | 1,731 | 1,383 | ||||||||
Total direct external expenses | 28,998 | 23,336 | 23,176 | ||||||||
Internal, overhead and other expenses | 21,361 | 21,059 | 20,226 | ||||||||
Stock-based compensation expense | 5,240 | 6,304 | 4,977 | ||||||||
Total research and development expenses | $ | 55,599 | $ | 50,699 | $ | 48,379 | |||||
The $5.7 million, or 24.3%, increase in direct external expenses for the year ended December 31, 2017 as compared to the year ended December 31, 2016 was due primarily to higher third-party manufacturing scale up costs for our OMS721 program as we continue to increase our production capacity to meet anticipated clinical and commercial requirements and to incremental clinical costs as our OMS721 clinical trials advance. In addition, higher third-party development expenses were incurred as we continue to advance toward the clinic our preclinical product candidates including OMS527, OMS906 and small-molecule inhibitors in our MASP-2 program. These increases were partially offset by the completion of a post-marketing OMIDRIA pediatric trial in 2016 and decreased manufacturing costs associated with the product validation of OMIDRIA in connection with the transfer of production to a new commercial manufacturing facility.
Direct external clinical research and development expense stayed consistent during the year ended December 31, 2016 compared to 2015. During the year ended December 31, 2016, we incurred increased clinical trial costs for our MASP-2 program as we advanced our Phase 2 trials and initiated technical transfer to a new manufacturing facility capable of providing commercial product. This increase was partially offset by reduced costs for our PDE10 program and lower OMIDRIA clinical trial costs due to completing in 2016 our FDA-required pediatric post-marketing trial and to reduced manufacturing costs.
The increase in stock-based compensation during the year ended December 31, 2016 compared to 2015 was primarily due to stock option grants in February 2016 and in December 2016 relating to annual reviews of 2014 and 2015 employee performance, respectively. Two cycles of awards were granted during 2016 because no 2014 annual option grants were made to employees in 2015.
During 2018, the majority of our research and development expenses will be related to OMS721. We expect OMS721 costs to continue to increase in 2018 given our ongoing Phase 3 clinical programs and our commercial manufacturing efforts.
At this time, we are unable to estimate with any certainty the longer-term costs we will incur in the continued development of our product candidates due to the inherently unpredictable nature of our preclinical and clinical development activities. Clinical development timelines, the probability of success and development costs can differ materially as new data become available and as expectations change. While we are focused currently on advancing our product development programs, our future research and development expenses will depend, in part, on the preclinical or clinical success of each product candidate as well as ongoing assessments of each program’s commercial potential. In addition, we cannot forecast with
49
any degree of certainty which product candidates, if any, may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
We are required to expend substantial resources in the development of our product candidates due to the lengthy process of completing clinical trials and seeking regulatory approval. Any failure or delay in completing clinical trials, or in obtaining regulatory approvals, could delay our generation of product revenue and increase our research and development expenses and, in turn, could have a material adverse effect on our operations, financial condition and liquidity. Because of the factors above, we are not able to estimate with any certainty when or if we would recognize any net cash inflows from our research and development projects.
Selling, General and Administrative Expenses
Our selling, general and administrative, or SG&A, expenses are comprised primarily of salaries, benefits, stock-compensation costs for sales, marketing, and other personnel not directly engaged in research and development, marketing and selling expenses, professional and legal services, general corporate costs and an allocation of our occupancy costs.
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Selling, general and administrative expenses, excluding stock-based compensation expense | $ | 44,596 | $ | 36,504 | $ | 30,723 | |||||
Stock-based compensation expense | 7,448 | 7,278 | 4,604 | ||||||||
Total selling, general and administrative expenses | $ | 52,044 | $ | 43,782 | $ | 35,327 | |||||
The increase in selling, general and administrative expenses during the year ended December 31, 2017 compared to 2016 was primarily due to increased legal costs associated with pursuing our patent infringement claims related to Par’s effort to obtain FDA approval for a generic version of OMIDRIA, which was resolved in October 2017, and to a lesser degree, increased employee costs. For more information regarding the conclusion of the Par lawsuit, see Part I, Item 3, “Legal Proceedings.”
The increase in selling, general and administrative expenses during the year ended December 31, 2016 compared to 2015 was primarily due to increased legal costs associated with the Par lawsuit and increased stock compensation expense due to stock option grants in February 2016 and in December 2016 relating to annual reviews of employee performance in the 2014 and 2015 calendar years, respectively.
We expect that our selling, general and administrative expenses for 2018 may increase slightly from 2017, primarily due to pre-marketing activities associated with OMS721. The actual expense is also dependent on the timing of costs associated with the Lupin and Sandoz lawsuits versus the amount we spent in 2017 litigating the Par lawsuit.
Interest Expense
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Interest Expense | $ | 11,030 | $ | 7,819 | $ | 3,573 | |||||
The increase in interest expense for all periods was primarily due to the increase in our outstanding notes payable balance under the CRG Loan Agreement as compared to our notes payable balance with former lenders during the comparative periods. The effective interest rate on our notes payable remained relatively constant throughout the period.
50
Loss on Early Extinguishment of Debt
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Loss on Early Extinguishment of Debt | $ | — | $ | 5,595 | $ | 1,315 | |||||
In November 2016, we entered into the CRG Loan Agreement and repaid our then-outstanding loan under our loan and security agreement with Oxford and East West Bank, or the Oxford/EWB Loan Agreement. We incurred a loss of $5.6 million associated with the unamortized loan maturity fee and the prepayment fee related to the then-outstanding loans under the Oxford/EWB Loan Agreement.
In December 2015, we entered into the Oxford/EWB Loan Agreement. We incurred a loss of $1.3 million associated with the unamortized loan maturity fee and the prepayment related to then-outstanding loans from Oxford and another lender.
For more information regarding our notes payable, see Part II, Item 8, Note 7 to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Other Income
Years Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Other Income | $ | 1,444 | $ | 945 | $ | 1,030 | |||||
Other income principally includes sublease rental income and interest earned. The increase during the year ended December 31, 2017 is due to incremental subleased space as compared to 2016. Other income remained consistent during the year ended December 31, 2016 compared to 2015.
Financial Condition - Liquidity and Capital Resources
As of December 31, 2017, we had $83.7 million in cash, cash equivalents and short-term investments available for general corporate use held primarily in money-market accounts as compared to $45.3 million at December 31, 2016. In addition, as of December 31, 2017 we had $17.1 million in accounts receivable that we anticipate collecting in the first quarter of 2018. In 2017, we raised $63.6 million in an equity offering in which we sold 3.0 million shares of our common stock at a public offering price of $22.75 per share.
Our notes payable and lease financing obligation increased $4.9 million to $84.6 million as of December 31, 2017, compared to $79.7 million as of December 31, 2016. The increase was due to our election to defer a portion of interest payments under our CRG Loan Agreement and to entering into additional capital leases for equipment. For more information regarding the CRG Loan Agreement, see Part II, Item 8, Note 7 “Notes Payable and Lease Financing Obligations” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
We have historically generated net losses and incurred negative cash flows. For the years ended December 31, 2017, 2016, and 2015 we incurred net losses of $53.5 million, $66.7 million and $75.1 million, respectively, and also incurred negative cash flows from operations of $36.2 million, $51.5 million and $65.2 million in 2017, 2016 and 2015, respectively.
As described earlier in this section under “Results of Operations - Revenue” pass-through reimbursement status for OMIDRIA expired as scheduled on January 1, 2018. Due to the expiration of pass-through reimbursement, we saw a significant reduction in ASC and hospital demand for OMIDRIA beginning in December 2017 as those facilities utilized their existing inventories. Based on first quarter 2018 data to date, we believe that a substantial majority of facilities who were using OMIDRIA are awaiting resolution regarding reimbursement by CMS, or our decision to implement an alternative sales strategy and, therefore, have largely suspended use of the product or are using it on a selective basis only. Until we have additional clarity on reimbursement for OMIDRIA and/or additional history on OMIDRIA usage, we cannot determine the impact on OMIDRIA product sales trends through the remainder of 2018.
We expect to continue to incur negative cash flows until OMIDRIA product sales or other sources of revenue (e.g., corporate partnering, licensing or product sales) generate sufficient cash inflows to finance our operations and debt service requirements. We are pursuing continued separate reimbursement (or equivalent reimbursement treatment) for OMIDRIA and may borrow up to an additional $45.0 million under our CRG Loan Agreement (see Part II, Item 8, Note 7
51
“Notes Payable and Lease Financing Obligations” for further detail) but unless and until we are cash-flow positive, we will need to continue to raise operating funds through the issuance of public or private equity securities, incurring additional debt and/or pursuing partnering and licensing opportunities.
On an interim and annual basis we are required to assess our ability to continue as a going concern for one year after the date the financial statements are issued using rules defined by ASC No. 205-40 - Going Concern, or the Standard. In performing the assessment, we are required to evaluate whether our plans to mitigate the conditions above alleviate the substantial doubt about our ability to meet our obligations as they become due within one year after the date of the financial statements are issued. In performing this assessment, we are limited to the restrictions and definitions in the Standard. As such, we did not consider any future sources of working capital that we may otherwise be able to access. Consequently, based on this assessment performed using the associated limitations required by the Standard, we have concluded there is substantial doubt about our ability to continue as a going concern through March 1, 2019.
Cash Flow Data
Years ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Cash provided by (used in): | |||||||||||
Operating activities | $ | (36,227 | ) | $ | (51,504 | ) | $ | (65,209 | ) | ||
Investing activities | (37,598 | ) | (16,335 | ) | (20,606 | ) | |||||
Financing activities | 74,995 | 68,698 | 86,826 | ||||||||
Operating Activities. Net cash used in operating activities decreased for the year ended December 31, 2017 by $15.3 million as compared to the same period in 2016. The reduction in cash used in operating activities in 2017 largely resulted from the $13.3 million decline in our net loss from 2016 due primarily to an increase in OMIDRIA product sales, net of $23.4 million, which was partially offset by a $12.8 million increase in total costs and expenses. In addition, in 2017, non-cash charges which are primarily stock-based compensation, loss on early extinguishment of debt and non-cash interest expense, decreased by $4.0 million when compared to 2016, which negatively impacted the change in our cash used in operating activities. A $6.0 million net change in operating assets and liabilities between 2017 and 2016 also positively affected our cash used in operations.
Net cash used in operating activities decreased for the year ended December 31, 2016 by $13.7 million as compared to the same period in 2015. Our 2016 net loss decreased by $8.4 million from 2015 due primarily to increased OMIDRIA product sales, net of $28.2 million being offset by a $11.1 million increase in total costs and expenses. In addition, in 2016 non-cash charges increased by $9.3 million when compared to 2016 due primarily to the $5.6 million loss we incurred on the early extinguishment of loans under the Oxford East/West Loan Agreement, which further decreased our cash used in operating activities. A $4.0 million net change in operating assets and liabilities between 2016 and 2015 also negatively affected our cash used in operations
Investing Activities. Cash flows from investing activities primarily reflect cash used to purchase short-term investments and proceeds from the sale and maturity of short-term investments, thus causing a shift between our cash and cash equivalents and short-term investment balances. Because we manage our cash usage with respect to our total cash, cash equivalents and short-term investments, we do not consider the fluctuations between cash, cash equivalents and our short-term investment balances to be important to the understanding of our liquidity and capital resources. The remaining component of cash flows from investing activities is the purchase of property and equipment.
Net cash used in investing activities in the year ended December 31, 2017 was $37.6 million, an increase of $21.3 million from 2016, primarily due to the purchase of short-term investments for $65.3 million with the $63.7 million of net proceeds received from the sale of common stock in our August 2017 public offering. These purchases were partially offset by the sale and maturity of $28.1 million of short-term investments to provide cash for operating activities.
Net cash used in investing activities in the year ended December 31, 2016 was $16.3 million, a decrease of $4.3 million from 2015, primarily due to the purchase of short-term investments for $74.0 million with the $38.0 million of net proceeds received from the sale of common stock from our public offering in August 2016 and the sale of common stock under an At Market Issuance Sales Agreement, $3.0 million in net proceeds from the CRG Loan Agreement and the purchase of short-term investments upon the maturity of other short-term investments. These purchases were partially offset by the sale of $57.8 million of short-term investments.
52
Financing Activities. Net cash provided by financing activities in the year ended December 31, 2017 was $75.0 million, an increase of $6.3 million over the same period in 2016 primarily due to a higher amount of equity financing offset by reduction in debt financing. In 2017, we received $63.7 million of net proceeds from the sale of common stock in our public offering in August compared to $38.0 million of net proceeds from the sale of common stock and pre-funded warrants in our public offering in August 2016. In 2016, we also received $22.8 million in net additional borrowings under the Oxford/EWB Loan Agreement and the CRG Loan Agreement and did not have any similar borrowings in 2017. During 2017, we also received $11.8 million from the exercise of employee stock options and warrants as compared to $3.2 million in 2016.
Net cash provided by financing activities in the year ended December 31, 2016 was $68.7 million, a decrease of $18.1 million from 2015 primarily due to $38.0 million of net proceeds received from the sale of common stock and pre-funded warrants in our public offering in August 2016 as compared to $79.1 million in February 2015. In addition, we also received net additional borrowings of $22.8 million under the Oxford/EWB Loan Agreement and the CRG Loan Agreement compared to $14.9 million in 2015.
2016 CRG Loan Agreement
In October 2016, we entered into the CRG Loan Agreement, pursuant to which we pledged substantially all of our assets, including intellectual property, as collateral. As of December 31, 2017, we had $83.3 million outstanding under the CRG Loan Agreement. In February 2018, we and CRG amended the CRG Loan Agreement so that we are permitted to borrow, at our discretion and subject to customary closing conditions, including the absence of a “material adverse effect,” up to an additional $45.0 million available through May 20, 2018.
The CRG Loan Agreement accrues interest at an annual rate of 12.25% (4.0% of which can be deferred at our option through December 31, 2020 by adding such amount to the aggregate principal amount). As of December 31, 2017, as allowed under the CRG Loan Agreement, we have deferred $3.8 million of interest due by increasing the principal amount outstanding. The CRG Loan Agreement requires us to achieve either certain annual minimum net revenue thresholds from any sources, including product sales, licensing and partnering, ($65.0 million for 2018) or minimum market capitalization thresholds ($512.0 million based on the amount borrowed as of December 31, 2017). We satisfied the annual minimum revenue threshold for calendar year 2017, and the minimum market capitalization requirement applicable to calendar year 2018 will be determined in February or March 2019. For more information regarding the CRG Loan Agreement, see Part II, Item 8, Note 7, “Notes Payable and Lease Financing Obligations” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
As of December 31, 2017, we were not, and to date we are not, in default under the CRG Loan Agreement.
Contractual Obligations and Commitments
The following table presents a summary of our contractual obligations and commitments as of December 31, 2017.
Payments Due Within | |||||||||||||||||||
1 Year | 2-3 Years | 4-5 Years | More than 5 Years | Total | |||||||||||||||
(In thousands) | |||||||||||||||||||
Operating leases | $ | 4,564 | $ | 9,429 | $ | 9,875 | $ | 25,840 | $ | 49,708 | |||||||||
Capital leases (principal and interest) | 593 | 712 | 191 | — | 1,496 | ||||||||||||||
Notes payable (principal and interest) * | 7,012 | 25,379 | 82,438 | — | 114,829 | ||||||||||||||
Goods & Services | 4,974 | 439 | 119 | — | 5,532 | ||||||||||||||
Total | $ | 17,143 | $ | 35,959 | $ | 92,623 | $ | 25,840 | $ | 171,565 | |||||||||
*Assumes full deferral of interest at our option (Refer to “Financial Condition - Liquidity and Capital Resources-2016 CRG Loan Agreement” above) | |||||||||||||||||||
Operating Leases
We lease our office and laboratory space in The Omeros Building under a lease agreement with BMR - 201 Elliott Avenue LLC. The initial term of the lease ends in November 2027 and we have two options to extend the lease term, each by five years. We lease office and laboratory equipment under various operating lease agreements with initial terms of five years or less.
53
As of December 31, 2017, the remaining aggregate non-cancelable rent payable under the initial term of the lease, excluding common area maintenance and related operating expenses, is $49.7 million.
Notes Payable
Refer to “Financial Condition - Liquidity and Capital Resources - 2016 CRG Loan Agreement” above.
Goods & Services
We have certain non-cancelable obligations under other agreements for the acquisitions of goods and services associated with the manufacturing of our product candidates which contain firm commitments. As of December 31, 2017, our aggregate firm commitments are $5.5 million.
We may also be required, in connection with in-licensing or asset acquisition agreements, to make certain royalty and milestone payments and we cannot, at this time, determine when or if the related milestones will be achieved or whether the events triggering the commencement of payment obligations will occur. Therefore, such payments are not included in the table above. For information regarding agreements that include these royalty and milestone payment obligations, see Part II, Item 8, Note 8, “Commitments and Contingencies” to our Consolidated Financial Statements in this Annual Report on Form 10-K.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of our consolidated financial statements, in conformity with U.S. generally accepted accounting principles, or GAAP, requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; however, actual results could differ from those estimates. An accounting policy is considered critical if it is important to a company’s financial condition and results of operations and if it requires the exercise of significant judgment and the use of estimates on the part of management in its application. Although we believe that our judgments and estimates are appropriate, actual results may differ materially from our estimates.
We believe the following to be our critical accounting policies because they are both important to the portrayal of our financial condition and results of operations and they require critical judgment by management and estimates about matters that are uncertain:
• | revenue recognition; |
• | research and development expenses, primarily clinical trial expenses and manufacturing of drug product and clinical drug supply; and |
• | stock-based compensation. |
If actual results or events differ materially from those contemplated by us in making these estimates, our reported financial condition and results of operations for future periods could be materially affected.
Revenue Recognition
Our revenues are comprised primarily of product sales of OMIDRIA. Revenue is recognized when there is persuasive evidence that an arrangement exists, product title and risk of loss is passed to the customer or the service has been provided, the price is fixed or determinable and collection is reasonably assured. We record OMIDRIA product revenue upon delivery to our wholesalers. Product sales to a wholesaler are not recorded if we determine that the wholesaler’s on-hand OMIDRIA inventory, based on sell-through and inventory information we regularly receive from our wholesalers, exceeds approximately eight weeks of projected demand.
Product sales are recorded net of wholesaler distribution fees and estimated chargebacks, product returns, rebates and purchase volume discounts. Accruals or allowances are established for these deductions in the same period when revenue is recognized, and actual amounts incurred are offset against the applicable accruals or allowances. We reflect each of these accruals or allowances as either a reduction in the related account receivable or as an accrued liability, depending on how the amount is expected to be settled.
Chargebacks and Rebates: Provisions for chargebacks are determined utilizing historical and projected payer mix and sale-through and inventory on-hand information received directly from wholesalers. Chargebacks are generally settled within four weeks of recording product sales revenue.
We provide reimbursement support services and financial assistance in the form of a rebate to patients whose commercial insurance is inadequate to cover the full cost of OMIDRIA. We apply an experience ratio to product sales to determine the rebate accrual. This experience ratio is reviewed and updated periodically to reflect actual results.
54
We provide rebate payments for which ASCs qualify by meeting or exceeding purchase volumes of OMIDRIA under our purchase volume discount program. We calculate rebate payment amounts due under this program based on actual qualifying purchase volumes and apply a contractual discount rate. For purchases of OMIDRIA not yet reported as sold-through to the ASC by our wholesalers, we apply an experience ratio to product sales to determine the rebate accrual. This experience ratio is being reviewed and updated periodically to reflect actual results.
Distribution Fees and Product Return Allowances: We pay our wholesalers a distribution fee for services that they perform for us based on the WAC value of their purchases of OMIDRIA. We record a provision against product sales for these charges at the time of sale to the wholesaler.
We allow for the return of product up to 12 months past its expiration date or for product that is damaged. In estimating product returns, we take into consideration our return experience to date, the remaining shelf life of product we have previously sold, inventory in the wholesale channel and our expectation that product is typically not held by the healthcare providers based on the frequency of their reorders.
Research and Development Expenses
Research and development costs are comprised primarily of costs for personnel, including salaries, benefits and stock compensation; an allocation of our occupancy costs; clinical study costs; contracted research; manufacturing; consulting arrangements; depreciation; materials and supplies; milestones; and other expenses incurred to sustain our overall research and development programs. Clinical trial expenses for investigational sites require certain estimates. We estimate these costs based on a cost per patient that varies depending on the clinical trial site. As actual costs become known to us, we adjust our estimates; these changes in estimates may result in understated or overstated expenses at a given point in time. Research and development costs are expensed as incurred.
Advanced payments for goods or services that will be used or rendered for future research and development activities are deferred and then recognized as an expense as the related goods are delivered or as the services are performed, or when the goods or services are no longer expected to be provided.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments made to employees, directors and non-employees based on estimated fair values. The fair value of our stock options is calculated using the Black-Scholes option valuation model, which requires assumptions, including volatility, forfeiture rates and expected option life. If any of the assumptions used in the Black-Scholes model change significantly, stock-based compensation expense for new awards may differ materially from that recorded for existing awards and stock-based compensation for non-employees will vary as the awards are re-measured over the vesting term.
As stock-based compensation expense is based on options ultimately expected to vest, the expense has been reduced for estimated forfeitures. We estimate forfeitures for expense recognition based on our historical experience. Groups of employees that have similar historical forfeiture behavior are considered separately. We use the straight-line method to allocate compensation cost to reporting periods over each optionee’s respective requisite service period for employees and directors, which is generally the vesting period.
Stock options granted to non-employees are accounted for using the fair-value approach using the Black-Scholes option-pricing model and are re-measured over the vesting term as earned. The estimated fair value is charged to expense over the applicable service period.
Recent Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In 2017, we adopted Accounting Standards Update (ASU) 2016-09 related to stock compensation, which simplifies several aspects of the accounting for share-based payment transactions. Excess tax benefits or deficiencies are now reflected in the Statement of Operations whereas they previously were recognized in equity. We have elected to continue to account for forfeitures based on estimated expected forfeitures. As of December 31, 2017, we recognized the previously unrecognized excess tax benefits of $4.5 million through a cumulative-effect adjustment to accumulated deficit. The previously unrecognized excess tax effects were recorded as a deferred tax asset, which was fully offset by a valuation allowance.
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU No. 2014-09 (Topic 606) "Revenue from Contracts with Customers." Topic 606 supersedes the revenue recognition requirements in Topic 605 “Revenue Recognition” (Topic 605), and requires entities to
55
recognize revenue when control of the promised goods or services is transferred to customers at an amount that reflects the consideration to which the entity expects to be entitled to in exchange for those goods or services. We adopted Topic 606 as of January 1, 2018 using the modified retrospective transition method. Upon adoption, we evaluated our contracts with customers broadly, including our rebate program with qualifying surgery centers, and returns. The adoption of the standard will not change the timing of the recognition of our product sales revenue and will have no material impact on our ongoing results from operations.
In February 2016, the FASB issued ASU 2016-02 related to lease accounting. This standard requires lessees to recognize a right-of-use asset and a lease liability for most leases. This standard must be applied using a modified retrospective transition method and is effective for all annual and interim periods beginning after December 15, 2018. Earlier adoption is permitted. While we are still in the process of evaluating the effect of adoption on our consolidated financial statements and are currently assessing our leases, we expect to adopt the standard January 1, 2019. The adoption will lead to an increase in the assets and liabilities recorded on our Condensed Consolidated Balance Sheets primarily due to the lease agreements for our office building and vehicle leases. We continue to monitor business activity to ensure we capture all new leasing arrangements upon adoption.
In May 2016, the FASB issued ASU 2017-09 related to stock-based compensation, which effectively amends previously issued guidance and provides clarity and consistency in practice on the accounting for changes to the terms and conditions of stock-based payment arrangement, or modifications. This standard is effective for all annual and interim periods beginning after December 15, 2017 and is applied prospectively to modifications occurring after the adoption date. We have adopted the guidance January 1, 2018 and the adoption will not have a material impact on our stock-based compensation expense.
Off-Balance Sheet Arrangements
We have not engaged in any off-balance sheet arrangements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Our exposure to market risk is primarily confined to our investment securities and notes payable. The primary objective of our investment activities is to preserve our capital to fund operations and we do not enter into financial instruments for trading or speculative purposes. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of investments in high-credit-quality securities. As of December 31, 2017, we had cash, cash equivalents and short-term investments of $83.7 million. In accordance with our investment policy, we invest funds in highly liquid, investment-grade securities. The securities in our investment portfolio are not leveraged and are classified as available-for-sale. We currently do not hedge interest rate exposure. Because of the short-term maturities of our investments, we do not believe that an increase in market rates would have a material negative impact on the realized value of our investment portfolio. We actively monitor changes in interest rates and, with our current portfolio of short term investments, we are not exposed to potential loss due to changes in interest rates.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Index to Consolidated Financial Statements
56
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Omeros Corporation
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Omeros Corporation (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, shareholders' deficit and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company at December 31, 2017 and 2016, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated March 1, 2018 expressed an unqualified opinion thereon.
The Company's Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has stated that substantial doubt exists about the Company’s ability to continue as a going concern. Management's evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 1998.
Seattle, Washington
March 1, 2018
57
OMEROS CORPORATION
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
December 31, | |||||||
2017 | 2016 | ||||||
Assets | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 3,394 | $ | 2,224 | |||
Short-term investments | 80,355 | 43,107 | |||||
Receivables, net | 17,144 | 12,037 | |||||
Inventory | 443 | 1,128 | |||||
Prepaid expense | 7,036 | 1,766 | |||||
Total current assets | 108,372 | 60,262 | |||||
Property and equipment, net | 2,121 | 1,181 | |||||
Restricted investments | 5,835 | 5,835 | |||||
Total assets | $ | 116,328 | $ | 67,278 | |||
Liabilities and shareholders’ deficit | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 6,691 | $ | 2,519 | |||
Accrued expenses | 19,126 | 13,354 | |||||
Current portion of lease financing obligations | 490 | 198 | |||||
Total current liabilities | 26,307 | 16,071 | |||||
Notes payable and lease financing obligations, net | 84,117 | 79,512 | |||||
Deferred rent | 8,718 | 9,142 | |||||
Commitments and contingencies (Note 8) | |||||||
Shareholders’ deficit: | |||||||
Preferred stock, par value $0.01 per share, 20,000,000 authorized and none issued at December 31, 2017 and 2016. | — | — | |||||
Common Stock, par value $0.01 per share, 150,000,000 shares authorized at December 31, 2017 and 2016; 48,211,226 and 43,819,133 issued and outstanding at December 31, 2017 and December 31, 2016, respectively. | 482 | 438 | |||||
Additional paid-in capital | 520,072 | 432,002 | |||||
Accumulated deficit | (523,368 | ) | (469,887 | ) | |||
Total shareholders’ deficit | (2,814 | ) | (37,447 | ) | |||
Total liabilities and shareholders’ deficit | $ | 116,328 | $ | 67,278 | |||
See notes to consolidated financial statements
58
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except share and per share data)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Revenues: | |||||||||||
Product sales, net | $ | 64,826 | $ | 41,444 | $ | 13,264 | |||||
Grant revenue | — | 173 | 245 | ||||||||
Total revenue | 64,826 | 41,617 | 13,509 | ||||||||
Costs and expenses: | |||||||||||
Cost of product sales | 1,078 | 1,412 | 1,041 | ||||||||
Research and development | 55,599 | 50,699 | 48,379 | ||||||||
Selling, general and administrative | 52,044 | 43,782 | 35,327 | ||||||||
Total costs and expenses | 108,721 | 95,893 | 84,747 | ||||||||
Loss from operations | (43,895 | ) | (54,276 | ) | (71,238 | ) | |||||
Interest expense | (11,030 | ) | (7,819 | ) | (3,573 | ) | |||||
Other income | 1,444 | 945 | 1,030 | ||||||||
Loss on early extinguishment of debt | — | (5,595 | ) | (1,315 | ) | ||||||
Net loss | $ | (53,481 | ) | $ | (66,745 | ) | $ | (75,096 | ) | ||
Comprehensive loss | $ | (53,481 | ) | $ | (66,745 | ) | $ | (75,096 | ) | ||
Basic and diluted net loss per share | $ | (1.17 | ) | $ | (1.65 | ) | $ | (2.00 | ) | ||
Weighted-average shares used to compute basic and diluted net loss per share | 45,539,362 | 40,446,410 | 37,560,257 | ||||||||
See notes to consolidated financial statements
59
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ DEFICIT
(In thousands, except share data)
Common Stock | Additional Paid-in Capital | Accumulated Deficit | Total Shareholders’ Deficit | |||||||||||||||
Shares | Amount | |||||||||||||||||
Balance at December 31, 2014 | 34,185,464 | $ | 342 | $ | 285,050 | $ | (328,046 | ) | $ | (42,654 | ) | |||||||
Issuance of common stock and pre-funded warrants, net of offering costs | 3,444,831 | 34 | 79,042 | — | 79,076 | |||||||||||||
Issuance of common stock upon exercise of warrants | 133,240 | 1 | 1,435 | — | 1,436 | |||||||||||||
Issuance of common stock upon exercise of stock options | 277,356 | 3 | 1,420 | — | 1,423 | |||||||||||||
Stock-based compensation | — | — | 9,581 | — | 9,581 | |||||||||||||
Net loss | — | — | — | (75,096 | ) | (75,096 | ) | |||||||||||
Balance at December 31, 2015 | 38,040,891 | 380 | 376,528 | (403,142 | ) | (26,234 | ) | |||||||||||
Issuance of common stock in direct offering, net of offering costs | 3,478,260 | 35 | 37,279 | — | 37,314 | |||||||||||||
Issuance of common stock upon exercise of stock options | 1,486,167 | 15 | 3,131 | — | 3,146 | |||||||||||||
Issuance of common stock upon exercise of warrants | 749,250 | 7 | — | — | 7 | |||||||||||||
Issuance of common stock under the ATM Agreement, net of offering costs | 64,565 | 1 | 724 | — | 725 | |||||||||||||
Stock-based compensation | — | — | 13,582 | — | 13,582 | |||||||||||||
Warrants issued in connection with amendment to notes payable | — | — | 758 | — | 758 | |||||||||||||
Net loss | — | — | — | (66,745 | ) | (66,745 | ) | |||||||||||
Balance at December 31, 2016 | 43,819,133 | 438 | 432,002 | (469,887 | ) | (37,447 | ) | |||||||||||
Issuance of common stock in direct offering, net of offering costs | 3,000,000 | 30 | 63,627 | — | 63,657 | |||||||||||||
Issuance of common stock upon exercise of stock options | 1,392,093 | 14 | 11,755 | — | 11,769 | |||||||||||||
Stock-based compensation | — | — | 12,688 | — | 12,688 | |||||||||||||
Net loss | — | — | — | (53,481 | ) | (53,481 | ) | |||||||||||
Balance at December 31, 2017 | 48,211,226 | $ | 482 | $ | 520,072 | $ | (523,368 | ) | $ | (2,814 | ) | |||||||
See notes to consolidated financial statements
60
OMEROS CORPORATION
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Operating activities: | |||||||||||
Net loss | $ | (53,481 | ) | $ | (66,745 | ) | $ | (75,096 | ) | ||
Adjustments to reconcile net loss to net cash used in operating activities: | |||||||||||
Stock-based compensation expense | 12,688 | 13,582 | 9,581 | ||||||||
Non-cash interest expense | 4,187 | 1,977 | 1,045 | ||||||||
Depreciation and amortization | 551 | 300 | 209 | ||||||||
Loss on early extinguishment of debt | — | 5,595 | 1,315 | ||||||||
Changes in operating assets and liabilities: | |||||||||||
Receivables | (5,107 | ) | (5,520 | ) | (6,125 | ) | |||||
Inventory | 685 | (656 | ) | 96 | |||||||
Prepaid expenses | (5,270 | ) | 347 | (586 | ) | ||||||
Accounts payable, accrued expenses and other | 9,520 | (384 | ) | 4,352 | |||||||
Net cash used in operating activities | (36,227 | ) | (51,504 | ) | (65,209 | ) | |||||
Investing activities: | |||||||||||
Purchases of property and equipment | (350 | ) | (126 | ) | (240 | ) | |||||
Purchases of investments | (65,326 | ) | (73,966 | ) | (91,766 | ) | |||||
Proceeds from the sale and maturities of investments | 28,078 | 57,757 | 71,400 | ||||||||
Net cash used in investing activities | (37,598 | ) | (16,335 | ) | (20,606 | ) | |||||
Financing activities: | |||||||||||
Proceeds from issuance of common stock and pre-funded warrants, net | 63,657 | 38,039 | 79,076 | ||||||||
Proceeds from borrowings under notes payable | — | 100,000 | 50,000 | ||||||||
Payments on notes payable and lease financing obligations | (431 | ) | (70,137 | ) | (32,000 | ) | |||||
Payments on debt prepayment and extinguishment | — | (5,700 | ) | (2,673 | ) | ||||||
Payments for debt issuance costs | — | (1,501 | ) | (436 | ) | ||||||
Decrease (increase) in restricted investments | — | 4,844 | (10,000 | ) | |||||||
Proceeds upon exercise of stock options and warrants | 11,769 | 3,153 | 2,859 | ||||||||
Net cash provided by financing activities | 74,995 | 68,698 | 86,826 | ||||||||
Net increase in cash and cash equivalents | 1,170 | 859 | 1,011 | ||||||||
Cash and cash equivalents at beginning of period | 2,224 | 1,365 | 354 | ||||||||
Cash and cash equivalents at end of period | $ | 3,394 | $ | 2,224 | $ | 1,365 | |||||
Supplemental cash flow information | |||||||||||
Cash paid for interest | $ | 6,895 | $ | 5,293 | $ | 4,236 | |||||
Conversion of accrued interest to notes payable | $ | 3,315 | $ | 516 | $ | — | |||||
Property acquired under capital lease | $ | 1,141 | $ | 404 | $ | 137 | |||||
Issuance of warrants in connection with amendment to notes payable | $ | — | $ | 758 | $ | — | |||||
See notes to consolidated financial statements
61
OMEROS CORPORATION
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1—Organization and Basis of Presentation
Organization
We are a commercial-stage biopharmaceutical company committed to discovering, developing and commercializing small-molecule and protein therapeutics for large-market as well as orphan indications targeting inflammation, complement-mediated diseases and disorders of the central nervous system. Our first drug product, OMIDRIA, is marketed in the United States (U.S.) for use during cataract surgery or intraocular lens replacement.
Basis of Presentation
Our consolidated financial statements include the financial position and results of operations of Omeros Corporation (Omeros) and our wholly owned subsidiaries. All inter-company transactions have been eliminated. The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (GAAP). Certain prior year amounts in the statement of cash flows and the income tax footnote have been reclassified in the consolidated financial statements to conform to the current year presentation.
Segments
We operate in one segment. Management uses cash flow as the primary measure to manage our business and does not segment our business for internal reporting or decision-making.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Significant items subject to such estimates include revenue recognition, stock-based compensation expense and accruals for clinical trials, manufacturing of drug product and clinical drug supply and contingencies. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances; however, actual results could differ from these estimates.
Going Concern
On an interim and annual basis we are required to assess our ability to continue as a going concern for one year after the date the financial statements are issued using rules defined by ASC No. 205-40 - Going Concern (the Standard). As required by the Standard, management’s evaluation shall initially not take into consideration the potential mitigating effects of management’s plans that have not been fully implemented as of the date the financial statements are issued. In the second step of this evaluation, management’s assumptions and plans are derived according to restrictions and definitions in the Standard. As such, for purposes of this exercise, the following assumptions (which are discussed in further detail following this summary) were made:
• | No revenues from sales of OMIDRIA. We are unable at this time to predict accurately revenue from sales of OMIDRIA given the loss of CMS reimbursement and, therefore, no OMIDRIA revenues are included for this exercise. We are pursuing continued separate payment for OMIDRIA and, in the event that we are unsuccessful, we would implement in the near-term an alternative sales strategy for OMIDRIA; |
• | No additional draws on our CRG debt facility. As disclosed in Note 7, we are in compliance with all covenants under our CRG Loan Agreement. On February 26, 2018, we amended our CRG Loan Agreement to extend our ability to borrow up to $45.0 million through May 20, 2018 subject only to customary closing conditions. However, given the existence of customary closing conditions, including a typical material adverse event clause in the CRG Loan Agreement, the draw on this facility was not considered for purposes of this exercise; and |
• | No public or private equity transactions can be considered for purposes of this exercise in the absence of any existing or committed arrangements to raise additional capital. |
In performing the first step of the assessment, we concluded that the following conditions raise substantial doubt about our ability to meet our financial obligations as they become due. As of December 31, 2017, we had $83.7 million in cash, cash equivalents and short-term investments and $17.1 million of accounts receivable. We have a history of net losses ($53.5 million in 2017) and use of cash for operations ($36.2 million in 2017). In addition, on January 1, 2018, transitional pass-through reimbursement for our only commercial product, OMIDRIA, which allowed for separate payment (i.e., outside the packaged procedural payment) under Medicare Part B expired as scheduled and we cannot predict future revenues.
62
In performing the second step of this assessment, we are required to evaluate whether our plans to mitigate the conditions above alleviate the substantial doubt about our ability to meet our obligations as they become due within one year after the date the financial statements are issued. In performing this second step of the assessment, we are limited to those assumptions listed above and the restrictions and definitions in the Standard. As such, we did not consider any future sources of working capital that we may otherwise be able to access. Consequently, based on this assessment performed using the associated limitations required by the Standard, we have concluded there is substantial doubt about our ability to continue as a going concern through March 1, 2019.
If we are unable to raise additional equity, debt or partnering capital when needed through one or more of the avenues previously listed, or upon acceptable terms, such failure would have a significant negative impact on our financial condition. Should it be necessary to manage our operating expenses, we would reduce our projected cash requirements through reduction of our expenses by delaying clinical trials, reducing selected research and development efforts, or implementing other restructuring activities.
The accompanying consolidated financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The accompanying consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from uncertainty related to our ability to continue as a going concern.
Note 2—Significant Accounting Policies
Cash and Cash Equivalents, Short-Term Investments and Restricted Cash and Investments
Cash and cash equivalents include highly liquid investments with a maturity of three months or less on the date of purchase. Short-term investment securities are classified as available-for-sale and are carried at fair value. Unrealized gains and losses, if any, are reported as a separate component of shareholders’ deficit. Amortization, accretion, interest and dividends, realized gains and losses and declines in value judged to be other-than-temporary are included in other income. The cost of securities sold is based on the specific-identification method. Investments in securities with maturities of less than one year, or those for which management intends to use the investments to fund current operations, are included in current assets. We evaluate whether an investment is other-than-temporarily impaired based on the specific facts and circumstances. Factors that are considered in determining whether an other-than-temporary decline in value has occurred include: the market value of the security in relation to its cost basis; the financial condition of the investee; and the intent and ability to retain the investment for a sufficient period of time to allow for recovery in the market value of the investment. Restricted cash and investments are held in certificates of deposit and money-market funds.
As of December 31, 2017 and 2016, all investments are classified as short-term and available-for-sale on the accompanying Consolidated Balance Sheets. Investment income, which is included as a component of other income, consists primarily of interest earned.
Inventory
Inventory is stated at the lower of cost or market determined on a specific identification basis in a manner which approximates the first-in, first-out (FIFO) method. Costs include amounts related to third party manufacturing, transportation and internal labor and overhead. Capitalization of costs as inventory begins when the product candidate receives regulatory approval in the U.S. or the European Union (EU). We expense inventory costs related to product candidates as research and development expenses prior to receiving regulatory approval in the respective territory. Inventory is reduced to net realizable value for excess and obsolete inventories based on forecasted demand.
Receivables, Net
Receivables relate primarily to sales of OMIDRIA to wholesalers and include reductions for estimated chargebacks and product returns from wholesalers which are expected to be settled through reductions in receivables. Remaining receivables consist of amounts from subleases for space in The Omeros Building. Considering the nature and historic collectability of our receivables, we concluded an allowance for doubtful accounts is not necessary as of December 31, 2017 and 2016.
Property and Equipment, Net
Property and equipment are stated at cost and depreciation is calculated using the straight-line method over the estimated useful life of the assets, which is generally three to 10 years. Equipment financed under capital leases is recorded as property and equipment and is amortized over the shorter of the useful lives of the related assets or the lease term. Expenditures for repairs and maintenance are expensed as incurred.
63
Impairment of Long-Lived Assets
The carrying amount of long-lived assets is reviewed whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. Recoverability of these assets is measured by comparing the carrying value to future undiscounted cash flows the asset is expected to generate. If the asset is considered to be impaired, the amount of any impairment will be reflected in the results of operations in the period of impairment. We have not recognized any impairment losses for the years ending December 31, 2017, 2016 and 2015.
Deferred Rent
We recognize rent expense on a straight-line basis over the noncancelable term of The Omeros Building operating lease and, accordingly, record the difference between cash rent payments and the recognition of rent expense as an increase or decrease in deferred rent liability. We also record landlord-funded lease incentives, such as reimbursable leasehold improvements, as an increase in deferred rent liability which is amortized as a reduction of rent expense over the noncancelable terms of The Omeros Building operating lease.
Revenue Recognition
Our revenues are primarily comprised of product sales of OMIDRIA. Revenue is recognized when there is persuasive evidence that an arrangement exists, product title and risk of loss is passed to the customer or the service has been provided, the price is fixed or determinable and collection is reasonably assured.
Product Sales, Net
We generally record revenue from product sales when the product is delivered to our wholesalers. Product sales to a wholesaler are not recorded as revenue if we determine that the wholesaler’s on-hand OMIDRIA inventory, based on sell-through and inventory information we regularly receive from our wholesalers, exceeds approximately eight weeks of projected demand.
The Centers for Medicare and Medicaid Services (CMS) granted transitional pass-through reimbursement status for OMIDRIA through January 1, 2018. Pass-through status for OMIDRIA allowed for reimbursement payment to Ambulatory Surgery Centers (ASCs) and hospitals using OMIDRIA in procedures involving patients covered by Medicare Part B. As a result of this expiration, we saw a significant reduction in ASC and hospital demand for OMIDRIA beginning in December 2017 as surgical facilities utilized inventories and this reduction has continued in 2018 due to uncertainty around OMIDRIA reimbursement. Consequently, we did not recognize revenue for wholesaler on-hand OMIDRIA inventory at December 31, 2017 in excess of eight weeks of projected ASC and hospital demand.
Product sales are recorded net of wholesaler distribution fees and estimated chargebacks, product returns, rebates and purchase volume discounts. Accruals or allowances are established for these deductions in the same period when revenue is recognized, and actual amounts incurred are offset against the applicable accruals or allowances. We reflect each of these accruals or allowances as either a reduction in the related account receivable or as an accrued liability, depending on how the amount is expected to be settled.
We also accept returns from our ASCs and hospitals who have purchased OMIDRIA from our wholesalers. Due to the expiration of pass-through, we expect the ASCs and hospitals will return a portion of their OMIDRIA on hand at December 31, 2017 to us for a full refund of the purchase price.
Research and Development
Research and development costs are comprised primarily of costs for personnel, including salaries, benefits and stock compensation; an allocation of our occupancy costs; clinical study costs; contracted research; manufacturing; consulting arrangements; depreciation; materials and supplies; milestones; and other expenses incurred to sustain our overall research and development programs. Research and development costs are expensed as incurred.
Advance payments for goods or services that will be used or rendered for future research and development activities are deferred and then recognized as an expense as the related goods are delivered or the services are performed, or when the goods or services are no longer expected to be provided.
Patents
We generally apply for patent protection on processes and product candidates we or our licensors conceive or develop. Patent costs are comprised primarily of external legal fees, filing fees incurred to file patent applications, and periodic renewal fees to keep the patent in force and are expensed as incurred as a component of general and administrative expense.
64
Selling, General and Administrative
Selling, general and administrative (SG&A) expenses are comprised primarily of salaries, benefits, and stock-compensation costs for sales, marketing, and other personnel not directly engaged in research and development. Additionally, SG&A includes marketing and selling expenses, professional and legal services; patent costs; depreciation, an allocation of our occupancy costs; and other general corporate expenses. Advertising costs, which we consider to be media and marketing materials, are expensed as incurred and were $328,000, $672,000 and $885,000 during the years ended December 31, 2017, 2016 and 2015, respectively.
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their tax bases. Deferred tax assets and liabilities are measured using enacted tax rates applied to taxable income in the years in which those temporary differences are expected to be recovered or settled. We recognize the effect of income tax positions only if those positions are more likely than not of being sustained upon an examination. A valuation allowance is established when it is more likely than not that the deferred tax assets will not be realized.
In December 2017, the Tax Cuts and Jobs Act (the Tax Act) was enacted into law and the new legislation contains certain tax provisions that affected us, including a reduction of the federal corporate income tax rate to 21.0% effective January 1, 2018, among others. We are required to recognize the effect of the tax law changes in the period of enactment, such as re-measuring its deferred tax assets and liabilities as well as our valuation allowance against our net deferred tax assets. In December 2017, the SEC staff issued Staff Accounting Bulletin No. 118 (SAB 118), “Income Tax Accounting Implications of the 2017 Tax Cuts and Jobs Act”, which require us to record provisional amounts during a measurement period not to extend beyond one year of the enactment date. Since the Tax Act was passed in December 2017, and ongoing guidance and accounting interpretation are expected over the next 12 months, we consider the accounting of the deferred tax re-measurements to be incomplete. We expect to complete our analysis within the one year measurement period.
Stock-Based Compensation
Stock-based compensation expense is recognized for all share-based payments made to employees, directors and non-employees based on estimated fair values. The fair value of our stock options is calculated using the Black-Scholes option-pricing model which requires judgmental assumptions including volatility, forfeiture rates and expected option life. For employees and directors, we use the straight-line method to allocate compensation cost to reporting periods over each optionee’s requisite service period, which is generally the vesting period. Stock options granted to non-employees are accounted for using the fair-value approach and are subject to periodic revaluation over their vesting terms as earned. The stock compensation cost related to non-employee stock options is based on changes in estimated fair value and is charged to expense over the applicable service period.
Accumulated Other Comprehensive Loss
Accumulated other comprehensive loss is comprised of net loss and certain changes in equity that are excluded from net loss. There was no difference between comprehensive loss and net loss for the years ended December 31, 2017, 2016 or 2015.
Financial Instruments and Concentrations of Credit Risk
Cash and cash equivalents, receivables, accounts payable and accrued liabilities, which are recorded at invoiced amount or cost, approximate fair value based on the short-term nature of these financial instruments. The fair value of short-term investments is based on quoted market prices. Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash and cash equivalents, short-term investments and receivables. Cash and cash equivalents are held by financial institutions and are federally insured up to certain limits. At times, our cash and cash equivalents balance exceeds the federally insured limits. To limit the credit risk, we invest our excess cash in high quality securities such as money market mutual funds, certificates of deposit and commercial paper.
Our notes payable includes features that meet the requirements under existing accounting guidance to be bifurcated from the respective notes payable agreement and to be recorded at fair value as a derivative asset or liability on the Consolidated Balance Sheet with the corresponding change in fair value recognized in other income on the Consolidated Statement of Operations. As of December 31, 2017 and 2016, the fair value of the embedded derivatives was not material.
65
Major Customers
We sell OMIDRIA through a limited number of wholesalers. Each of these wholesalers, together with entities under their common control, accounted for greater than 10% of total revenues for the years ended December 31, 2017, 2016 and 2015 and greater than 10% of accounts receivable as of December 31, 2017 and 2016 as noted below.
2017 | 2016 | 2015 | |||||||||||||
Percentage of Total Revenue | Percentage of Accounts Receivable | Percentage of Total Revenue | Percentage of Accounts Receivable | Percentage of Total Revenue | |||||||||||
Distributor A | 29 | % | 31 | % | 32 | % | 29 | % | 31 | % | |||||
Distributor B | 26 | % | 23 | % | 31 | % | 27 | % | 37 | % | |||||
Distributor C | 22 | % | 26 | % | 28 | % | 24 | % | 28 | % | |||||
Distributor D | 23 | % | 20 | % | * | 19 | % | * | |||||||
* Distributor did not account for greater than 10% of total revenues for the year ended December 31, 2016 or 2015. | |||||||||||||||
Major Suppliers
We use a single contract manufacturer to supply OMIDRIA, and generally one to two contract manufacturers to produce clinical trial material for each of our clinical trials which creates a concentration of risk for us.
We endeavor to maintain reasonable levels of drug supply for our commercial and clinical trial use and other manufacturers are available should we need to change suppliers. A change in suppliers, however, could cause a delay in delivery of OMIDRIA or our clinical trial material that would adversely affect our business.
Recently Adopted Accounting Pronouncements
In 2017, we adopted Accounting Standards Update (ASU) 2016-09 related to stock compensation, which simplifies several aspects of the accounting for share-based payment transactions. Excess tax benefits or deficiencies are now reflected in the Statement of Operations whereas they previously were recognized in equity. We have elected to continue to account for forfeitures based on estimated expected forfeitures.
Recent Accounting Pronouncements
In May 2014, the FASB issued ASU No. 2014-09 (Topic 606) "Revenue from Contracts with Customers." Topic 606 supersedes the revenue recognition requirements in Topic 605 “Revenue Recognition” (Topic 605), and requires entities to recognize revenue when control of the promised goods or services is transferred to customers at an amount that reflects the consideration to which the entity expects to be entitled to in exchange for those goods or services. We adopted Topic 606 as of January 1, 2018 using the modified retrospective transition method. Upon adoption, we evaluated our contracts with customers broadly, including our rebate program with qualifying surgery centers, and returns. The adoption of the standard will not change the timing of the recognition of our product sales revenue and will have no material impact on our ongoing results from operations.
In February 2016, the FASB issued ASU 2016-02 related to lease accounting. This standard requires lessees to recognize a right-of-use asset and a lease liability for most leases. This standard must be applied using a modified retrospective transition method and is effective for all annual and interim periods beginning after December 15, 2018. Earlier adoption is permitted. While we are still in the process of evaluating the effect of adoption on our consolidated financial statements and are currently assessing our leases, we expect to adopt the standard January 1, 2019. The adoption will lead to an increase in the assets and liabilities recorded on our Condensed Consolidated Balance Sheets primarily due to the lease agreements for our office building lease. We continue to monitor business activity to ensure we capture all new leasing arrangements upon adoption.
In May 2016, the FASB issued ASU 2017-09 related to stock-based compensation which effectively amends previous issued guidance and provides clarity and consistency in practice on the accounting for changes to the terms and conditions of stock-based payment arrangement, or modifications. This standard is effective for all annual and interim periods beginning after December 15, 2017 and is applied prospectively to modifications occurring after the adoption date. We have adopted the guidance January 1, 2018 and the adoption will not have a material impact on our stock-based compensation expense.
66
Note 3—Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted-average number of common shares outstanding for the period. Diluted net loss per share is computed by dividing the net loss by the weighted-average number of common shares and dilutive common share equivalents outstanding for the period, determined using the treasury-stock method. Common share equivalents are excluded from the diluted net loss per share computation if their effect is anti-dilutive.
The basic and diluted net loss per share amounts for the years ended December 31, 2017, 2016 and 2015 were computed based on the shares of common stock outstanding during the respective periods. Potentially dilutive securities excluded from the diluted loss per share calculation are as follows:
Year Ended December 31, | ||||||||
2017 | 2016 | 2015 | ||||||
Outstanding options to purchase common stock | 9,657,259 | 9,809,374 | 8,310,235 | |||||
Warrants and pre-funded warrants to purchase common stock | 100,602 | 100,602 | 749,250 | |||||
Total potentially dilutive securities | 9,757,861 | 9,909,976 | 9,059,485 | |||||
Note 4—Accounts Receivable, Net
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Trade receivables, net | $ | 17,079 | $ | 11,937 | |||
Sublease and other receivables | 65 | 100 | |||||
Total accounts receivables net | $ | 17,144 | $ | 12,037 | |||
Trade receivables are shown net of $198,000 and $297,000 of chargeback and product return allowances as of December 31, 2017 and 2016, respectively.
67
Note 5—Fair-Value Measurements
On a recurring basis, we measure certain financial assets at fair value. Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. The accounting standard establishes a fair value hierarchy that requires an entity to maximize the use of observable inputs, where available. The following summarizes the three levels of inputs required:
Level 1—Observable inputs for identical assets or liabilities, such as quoted prices in active markets;
Level 2—Inputs other than quoted prices in active markets that are either directly or indirectly observable; and
Level 3—Unobservable inputs in which little or no market data exists, therefore they are developed using estimates and assumptions developed by us, which reflect those that a market participant would use.
Our fair-value hierarchy for our financial assets measured at fair value on a recurring basis are as follows:
December 31, 2017 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
(In thousands) | |||||||||||||||
Assets: | |||||||||||||||
Money-market funds classified as non-current restricted cash and investments | $ | 5,835 | $ | — | $ | — | $ | 5,835 | |||||||
Money-market funds classified as short-term investments | 80,355 | — | — | 80,355 | |||||||||||
Total | $ | 86,190 | $ | — | $ | — | $ | 86,190 | |||||||
December 31, 2016 | |||||||||||||||
Level 1 | Level 2 | Level 3 | Total | ||||||||||||
(In thousands) | |||||||||||||||
Assets: | |||||||||||||||
Money-market funds classified as non-current restricted cash and investments | $ | 5,835 | $ | — | $ | — | $ | 5,835 | |||||||
Money-market funds classified as short-term investments | 43,107 | — | — | 43,107 | |||||||||||
Total | $ | 48,942 | $ | — | $ | — | $ | 48,942 | |||||||
Cash held in demand deposit accounts of $3.4 million and $2.2 million is excluded from our fair-value hierarchy disclosure as of December 31, 2017 and 2016, respectively. There were no unrealized gains or losses associated with our short-term investments as of December 31, 2017 or 2016. The carrying amounts reported in the accompanying Consolidated Balance Sheets for receivables, accounts payable, other current monetary assets and liabilities and notes payable and lease financing obligations approximate fair value.
Note 6—Certain Balance Sheet Accounts
Inventory
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Raw materials | $ | 83 | $ | 101 | |||
Work-in-process | — | 854 | |||||
Finished goods | 360 | 173 | |||||
Total inventory cost | $ | 443 | $ | 1,128 | |||
Work-in-process consists of manufactured vials of OMIDRIA that have not been packaged into finished goods.
68
Property and Equipment, Net
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Laboratory equipment | $ | 2,180 | $ | 1,830 | |||
Capital lease equipment | 1,915 | 774 | |||||
Computer equipment | 684 | 684 | |||||
Office equipment and furniture | 625 | 625 | |||||
Total cost | 5,404 | 3,913 | |||||
Less accumulated depreciation and amortization | (3,283 | ) | (2,732 | ) | |||
Total property and equipment, net | $ | 2,121 | $ | 1,181 | |||
For the years ended December 31, 2017, 2016 and 2015, depreciation and amortization expense was $551,000, $300,000 and $209,000, respectively.
Accrued Expenses
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Sales rebates, fees and discounts | $ | 6,561 | $ | 1,773 | |||
Contract research and development | 4,251 | 3,030 | |||||
Employee compensation | 2,178 | 4,551 | |||||
ASC/hospital product return liability | 2,350 | — | |||||
Consulting and professional fees | 1,758 | 2,223 | |||||
Clinical trials | 1,026 | 1,167 | |||||
Other accruals | 1,002 | 610 | |||||
Total accrued liabilities | $ | 19,126 | $ | 13,354 | |||
Note 7—Notes Payable and Lease Financing Obligations
Notes payable and lease financing obligations consist of the following:
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Notes payable | $ | 83,831 | $ | 80,516 | |||
Lender facility fee payable upon maturity | 4,192 | 4,025 | |||||
Lease financing obligations | 1,300 | 522 | |||||
Notes payable, facility fee and lease financing obligations | 89,323 | 85,063 | |||||
Unamortized debt discount | (3,527 | ) | (3,958 | ) | |||
Unamortized debt issuance costs | (1,189 | ) | (1,395 | ) | |||
Current portion of lease financing obligations | (490 | ) | (198 | ) | |||
Non-current portion of notes payable and lease financing obligations, net | $ | 84,117 | $ | 79,512 | |||
2015 Oxford/EWB Loan Agreement
In December 2015, we entered into a Loan and Security Agreement (the Oxford/EWB Loan Agreement) with Oxford Finance, LLC (Oxford) and East West Bank (EWB) pursuant to which we borrowed $50.0 million. We used $27.3 million of the loan proceeds to repay all of the amounts owed by us under a previously outstanding loan.
69
After deducting all loan initiation costs and outstanding interest on the Oxford/MidCap Loan Agreement, we received $22.3 million in net proceeds. We accounted for the termination of the Oxford/Midcap Loan Agreement as a debt extinguishment and, accordingly, incurred a loss of $1.3 million associated with the unamortized loan maturity fee and the prepayment fee.
The Oxford/EWB Loan Agreement required a $3.8 million loan maturity fee upon full repayment of the initial $50.0 million borrowed and $525,000 for each additional $10.0 million borrowed. We had the option to prepay the outstanding principal balance in its entirety at any time if we pay a prepayment equal to 1.0% of the then-outstanding principal balance.
In May 2016, we entered into the First Amendment to the Oxford/EWB Loan Agreement (the Amendment) and borrowed an additional $20.0 million. After deducting all loan initiation costs, we received $19.9 million in net proceeds. In connection with the Amendment, we issued warrants to purchase an aggregate of 100,602 shares of Omeros common stock (the Warrants) to Oxford and EWB at the then current market price of $9.94 per share. We accounted for the Warrants as a discount to our notes payable (see Note 9 for further discussion of the Warrants).
We accounted for the Amendment as a debt modification and, accordingly, the unamortized discount and debt issuance costs associated with the Oxford/EWB Loan Agreement were being amortized to interest expense using the effective interest method over the remaining term of the Oxford/EWB Loan Agreement.
2016 CRG Loan Agreement
In October 2016, we entered into the CRG Loan Agreement with CRG Servicing LLC (CRG), as administrative and collateral agent, and the lenders identified therein and, in November 2016, borrowed $80.0 million. We used $75.7 million of the loan proceeds to repay all amounts owed by us under our then-outstanding Oxford/EWB Loan Agreement. After deducting the loan initiation costs and related fees on the CRG Loan Agreement, we received $3.0 million in net proceeds. We accounted for the termination of the Oxford/EWB Loan Agreement as a debt extinguishment and, accordingly, incurred a loss of $5.6 million associated with the unamortized loan maturity fee, loan initiation costs and the prepayment fee.
In October 2017, we and CRG amended the CRG Loan Agreement so that we will be permitted to borrow, at our sole discretion and subject to customary closing conditions, up to an additional $45.0 million. In February 2018, we and CRG amended the CRG Loan Agreement to extend the borrowing date for the additional $45.0 million through May 20, 2018; none of the other terms or conditions of the CRG Loan Agreement were modified.
The CRG Loan Agreement accrues interest at an annual rate of 12.25% (4.00% of which can be deferred at our option through December 31, 2020 by adding such amount to the aggregate principal amount). Subject to the achievement of certain milestones, this interest-only period could be extended through the maturity date of September 30, 2022. As of December 31, 2017, we have deferred $3.8 million of interest due by increasing the principal amount outstanding. The CRG Loan Agreement requires us to maintain cash and cash equivalents of $5.0 million during the term of the agreement which is recorded as restricted cash and investments in our Consolidated Balance Sheet.
If an OMIDRIA net revenue milestone is satisfied during the 12-month period ending on December 31, 2019, the interest rate may be reduced to 11.50%, 3.50% of which may be deferred at our option and added to the principal amount outstanding. In addition, if either the OMIDRIA net revenue milestone is satisfied during such period or a market capitalization milestone is achieved during the fourth quarter of 2020, the loan would convert to interest-only until the September 30, 2022 maturity.
We are required to pay a facility fee equal to 5.00% of the aggregate principal amount borrowed (including principal additions related to deferred interest) on repayment of the CRG Loan Agreement. The $4.2 million related to the facility fee is being accreted to notes payable using the effective interest method over the term of the CRG Loan Agreement.
We may prepay all or a portion of the outstanding principal under the CRG Loan Agreement at any time subject to a prepayment fee through September 30, 2019, with no prepayment fee thereafter. In certain circumstances, including a change of control and certain asset sales or licensing transactions, we are required to prepay all or a portion of the loan, including the applicable prepayment premium of on the amount of the outstanding principal to be prepaid.
The CRG Loan Agreement also requires us to achieve either (a) certain minimum net revenue amounts from any sources, including product sales, licensing and partnering, through the end of 2021, which are $65.0 million and $75.0 million for the 2018 and 2019 calendar years, respectively, or (b) a minimum market capitalization threshold equal to the product of 6.4 multiplied by the aggregate principal amount of loans outstanding under the CRG Loan Agreement ($512.0 million required market capitalization based on the amount borrowed at December 31, 2017) determined as of the fifth business day following announcement of earnings results for the applicable year. If we are unable to satisfy each of the minimum annual revenue requirement and the market capitalization threshold for any given year, we may avoid a related default by repaying the
70
shortfall between actual revenues and the minimum revenue requirement for such year using proceeds generated by an equity or subordinated debt issuance.
The CRG Loan Agreement includes customary events of default that include, among other things, non-payment, inaccuracy of representations and warranties, covenant breaches, cross default to material indebtedness or material agreements, bankruptcy and insolvency, material judgments and a change of control. An event of default under the CRG Loan Agreement also includes the occurrence of any material adverse effect upon our business, condition (financial or otherwise), operations, performance or property taken as a whole. If there is an event of default under the CRG Loan Agreement, the lenders may have the right to accelerate all of our repayment obligations under the CRG Loan Agreement and to take control of our pledged assets, which include substantially all of our assets including our intellectual property. Under certain circumstances, a default interest rate of an additional 4.00% per annum will apply on all outstanding obligations during the existence of an event of default under the Loan Agreement.
As of December 31, 2017, we were not in default under the CRG Loan Agreement.
Capital Lease Financing Obligations
We have capital leases for certain laboratory and office equipment that have lease terms expiring through December 2021. Equipment costs related to these capital leases of $1.9 million and $774,000 is included in our property and equipment as of December 31, 2017 and December 31, 2016, respectively and the accumulated depreciation on this equipment was $530,000 and $230,000, respectively. The remaining principal payments under these capital leases totaled $1.3 million as of December 31, 2017.
Future Principal Payments
Future principal payments as of December 31, 2017 under the CRG Loan Agreement and our capital equipment financing leases, based on stated contractual maturities, are as follows:
Year Ending December 31, | Notes Payable | Capital Lease Financing Obligations | Total | ||||||||
(In thousands) | |||||||||||
2018 | $ | — | $ | 492 | $ | 492 | |||||
2019 | — | 391 | 391 | ||||||||
2020 | 10,479 | 232 | 10,711 | ||||||||
2021 | 41,915 | 99 | 42,014 | ||||||||
2022 | 31,437 | 86 | 31,523 | ||||||||
Total future principal payments | $ | 83,831 | $ | 1,300 | $ | 85,131 | |||||
The principal payments reflected in the table above exclude the $4.2 million lender’s facility fee due on repayment of the CRG Loan Agreement.
Note 8—Commitments and Contingencies
Operating Lease Obligations
We lease office and laboratory spaces in The Omeros Building. The initial term of the real estate lease ends in November 2027 and we have two options to extend the lease term, each by five years. As of December 31, 2017, the remaining aggregate non-cancelable rent under the initial terms of the real estate lease, excluding common area maintenance and related operating expenses, was $49.7 million. The deferred rent balance of $9.1 million relates to rent deferrals and landlord funded lease incentives and is being amortized to research and development and selling, general and administrative expense on a straight-line basis through the initial term of the lease.
Rent expense, including the amortization of lease incentives and rent deferrals, totaled $4.4 million, $4.4 million and $4.5 million for the years ended December 31, 2017, 2016 and 2015, respectively.
We sublease unused space in The Omeros Building to third-party tenants. Rental income received under these subleases was $886,000, $737,000 and $889,000 for the years ended December 31, 2017, 2016 and 2015, respectively. Rental income is recorded as other income in the accompanying Consolidated Statements of Operations and Comprehensive Loss.
71
We had no material non-cancelable rental payments relating to equipment at December 31, 2017.
Future minimum payments related to our leases at December 31, 2017, are as follows:
Year Ending December 31, | The Omeros Building Lease | Building Sublease Income | Net Operating Lease Payments | |||||||||
(In thousands) | ||||||||||||
2018 | $ | 4,564 | $ | 558 | $ | 4,006 | ||||||
2019 | 4,660 | — | 4,660 | |||||||||
2020 | 4,769 | — | 4,769 | |||||||||
2021 | 4,880 | — | 4,880 | |||||||||
2022 | 4,995 | — | 4,995 | |||||||||
Thereafter | 25,840 | — | 25,840 | |||||||||
Total | $ | 49,708 | $ | 558 | $ | 49,150 | ||||||
Contracts
We have various agreements with third parties that collectively require payment of termination fees totaling $5.5 million as of December 31, 2017 if we cancel the work within specific time frames, either prior to commencing or during performance of the contracted services. This is in addition to fees associated with the CRG Loan Agreement (see Note 7).
Development Milestones and Product Royalties
We have licensed a variety of intellectual property from third parties that we are currently developing or may develop in the future. These licenses may require milestone payments during the clinical development processes as well as low single to low double-digit royalties on the net income or net sales of the product. For the years ended December 31, 2017, 2016 and 2015, we did not owe any development milestones or royalties.
Litigation
In 2015, Par Pharmaceutical, Inc. and its subsidiary, Par Sterile Products, LLC, (collectively, Par) filed an Abbreviated New Drug Application (ANDA) seeking approval from the FDA to market a generic version of OMIDRIA prior to the expiration of our issued patents and we filed a patent infringement lawsuit under the Hatch-Waxman Act against Par.
In July 2017, a bench trial was held on this matter. In October 2017, we entered into a settlement agreement with Par pursuant to which Par acknowledged and confirmed the validity of our issued U.S. OMIDRIA patents and Par and its affiliates are prohibited from launching a generic version of OMIDRIA until the earlier of April 1, 2032 or a date on which we or a third party is able to launch a generic version of OMIDRIA. Under the settlement agreement, Par is granted a non-exclusive, non-sublicensable license to make, sell and distribute a generic version of OMIDRIA between the permitted launch date and the latest expiration of our U.S. patents related to OMIDRIA (i.e., October 23, 2033). During this period, Par is required to pay us a royalty equal to 15% of Par’s net sales of its generic version of OMIDRIA.
In May 2017, we received Notice Letters from Sandoz Inc. (Sandoz) and Lupin Ltd. and Lupin Pharmaceuticals, Inc. (collectively, Lupin), respectively, that Sandoz and Lupin had each filed an ANDA seeking approval from the FDA to market a generic version of OMIDRIA prior to the expiration our patents covering OMIDRIA. In June 2017, we filed patent infringement lawsuits against Sandoz and Lupin. We believe the assertions in the Sandoz and Lupin are substantially similar to those filed by Par and do not have merit. We intend to prosecute vigorously our infringement claims against each of Sandoz and Lupin.
72
Note 9—Shareholders’ Equity
Common Stock
As of December 31, 2017, we had reserved shares of common stock for the following purposes:
Options granted and outstanding | 9,657,259 | |
Options available for future grant | 3,513,540 | |
Common stock warrants | 100,602 | |
Total shares reserved | 13,271,401 | |
At Market Issuance Sales Agreement - In 2016, under an At Market Issuance Sales Agreement, we sold 64,565 shares of our common stock at an average price of $11.41 and received net proceeds of $724,000.
Securities Offerings - In August 2017, we sold 3.0 million shares of our common stock at a public offering price of $22.75 per share. After deducting underwriter discounts and offering expenses of $4.6 million, we received net proceeds from the transaction of $63.6 million.
In August 2016, we sold 3.5 million shares of our common stock at a public offering price of $11.50 per share. After deducting underwriter discounts and offering expenses, we received net proceeds from the offering of $37.3 million.
In February 2015, we sold 3.4 million shares of our common stock at a public offering price of $20.03 and sold pre-funded warrants to purchase up to 749,250 shares of our common stock at a public offering price of $20.02 per pre-funded warrant share. The public offering price for the pre-funded warrants was equal to the public offering price of the common stock, less the $0.01 per share exercise price of each pre-funded warrant. After deducting underwriter discounts and offering expenses of $4.9 million, we received net proceeds from the transaction of $79.1 million.
Warrants
The following table summarizes our outstanding warrants at December 31, 2017:
Outstanding At December 31, 2017 | Expiration Date | Exercise Price | ||
100,602 | May 18, 2023 | $9.94 | ||
In connection with the Amendment to the Oxford/EWB Loan Agreement in May 2016, we issued warrants to purchase an aggregate of 100,602 shares of our common stock. As of December 31, 2017, these warrants remain outstanding and are exercisable through May 18, 2023 at an exercise price per share of $9.94 per share.
In March 2016, we received cash proceeds of approximately $7,500 upon the cash exercise of our then-outstanding pre-funded warrants related to our securities offering in February 2015. The warrants had an exercise price of $0.01 per share, and the exercise resulted in the issuance of 749,250 shares of our common stock.
In March 2009, we issued warrants with an exercise price of $12.25 per share to brokers who assisted us in our Series E financing (the Series E Warrants). For the year ended December 31, 2015, we received cash proceeds of $1.4 million upon the cash and cashless exercise of Series E Warrants which resulted in the issuance of 133,240 shares of our common stock. All of the unexercised warrants expired during the year ended December 31, 2015.
Note 10—Stock-Based Compensation
On June 16, 2017, our shareholders approved the Omeros Corporation 2017 Omnibus Incentive Compensation Plan (the 2017 Plan), which provides for the grant of incentive and non-statutory stock options, stock appreciation rights, restricted stock, restricted stock units, performance units, performance shares and other stock and cash awards to employees, directors and consultants and subsidiary corporations’ employees and consultants. The 2017 Plan replaces the Omeros Corporation 2008 Equity Incentive Plan (the 2008 Plan) and as a result we will not grant any new awards under the 2008 Plan. Any stock option awards granted under the 2008 Plan that were outstanding as of the effective date of the 2017 Plan remain in effect pursuant to their terms and, if the award is canceled or is repurchased, the shares underlying such award become available for grant under the 2017 Plan. Under the 2017 Plan, stock options must be granted with exercise prices not less than the fair market value of the common stock subject to the stock option on the date of the grant and the options may not exceed 10 years.
73
Under the 2008 Plan, we granted incentive and non-statutory stock options to employees, directors and non-employees. Options were granted with exercise prices equal to the closing fair market value of the common stock on the date of the grant. The options granted were generally for 10-year terms and vested over a four-year period.
As of December 31, 2017, a total of 13,271,887 shares were reserved for issuance under our stock plans, of which 3,513,540 were available for future grants.
Stock-based compensation expense includes amortization of stock options granted to employees, directors and non-employees and has been reported in our Consolidated Statements of Operations and Comprehensive Loss as follows:
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
(In thousands) | |||||||||||
Research and development | $ | 5,240 | $ | 6,304 | $ | 4,977 | |||||
Selling, general and administrative | 7,448 | 7,278 | 4,604 | ||||||||
Total stock-based compensation expense | $ | 12,688 | $ | 13,582 | $ | 9,581 | |||||
The fair value of each option grant to employees and directors is estimated on the date of grant using the Black-Scholes option-pricing model. The following assumptions were applied to employee and director stock option grants during the periods ended:
Year Ended December 31, | |||||||||||
2017 | 2016 | 2015 | |||||||||
Estimated weighted-average fair value | $ | 8.66 | $ | 6.89 | $ | 11.31 | |||||
Weighted-average assumptions: | |||||||||||
Expected volatility | 74 | % | 74 | % | 71 | % | |||||
Expected term, in years | 6.0 | 5.7 | 6.0 | ||||||||
Risk-free interest rate | 2.05 | % | 1.63 | % | 1.68 | % | |||||
Expected dividend yield | — | % | — | % | — | % | |||||
During the years ended December 31, 2017, 2016 and 2015, we granted to non-employees options to purchase 25,000 shares, 38,000 shares and 4,200 shares of common stock, respectively.
In connection with the non-employee options, we recognized expense of $549,000, $313,000 and $492,000 during the years ended December 31, 2017, 2016 and 2015, respectively.
Stock option activity for all stock plans is as follows:
Options Outstanding | Weighted- Average Exercise Price per Share | Remaining Contractual Life (in years) | Aggregate Intrinsic Value (In thousands) | |||||||||
Balance at December 31, 2016 | 9,809,374 | $ | 9.66 | |||||||||
Granted | 1,825,140 | 13.14 | ||||||||||
Exercised | (1,392,093 | ) | 8.45 | |||||||||
Forfeited/expired | (585,162 | ) | 11.13 | |||||||||
Balance at December 31, 2017 | 9,657,259 | $ | 10.39 | 6.77 | $ | 88,096 | ||||||
Vested and expected to vest at December 31, 2017 | 9,362,096 | $ | 10.32 | 6.71 | $ | 85,998 | ||||||
Exercisable at December 31, 2017 | 6,811,348 | $ | 9.57 | 5.93 | $ | 67,373 | ||||||
The total intrinsic value of options exercised during the years ended December 31, 2017, 2016 and 2015 was $16.4 million, $13.6 million and $3.9 million, respectively.
At December 31, 2017, there were 2,845,911 unvested options outstanding that will vest over a weighted-average period of 2.4 years. Excluding non-employee stock options, the remaining estimated compensation expense to be recognized in connection with these unvested options is $18.6 million.
74
Note 11—Income Taxes
We have a history of losses and therefore have made no provision for income taxes. Deferred income taxes reflect the tax effect of net operating loss and tax credit carryforwards and the net temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes.
Significant components of deferred income taxes are as follows:
December 31, | |||||||
2017 | 2016 | ||||||
(In thousands) | |||||||
Deferred tax assets: | |||||||
Net operating loss carryforwards | $ | 90,498 | $ | 126,410 | |||
Tax credit carryforwards | 26,748 | 18,741 | |||||
Stock-based compensation | 7,829 | 11,102 | |||||
Deferred rent | 2,123 | 3,318 | |||||
Other | 4,749 | 4,401 | |||||
Total deferred tax assets | 131,947 | 163,972 | |||||
Less valuation allowance | (131,947 | ) | (163,972 | ) | |||
Net deferred tax assets | $ | — | $ | — | |||
As of December 31, 2017 and 2016, we had federal net operating loss carryforwards of approximately $414.5 million and $378.9 million, respectively, state net operating losses of approximately $68.9 million and $50.0 million, respectively.
As of December 31, 2017, we remeasured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21.0%. However, we are still analyzing certain aspects of the Tax Act, which could potentially affect the measurement of these assets and liabilities or potentially give rise to new deferred tax assets and liabilities. The provisional amount recorded related to the remeasurement of our deferred tax balance was $61.3 million.
In certain circumstances, due to ownership changes, our net operating loss and tax credit carryforwards may be subject to limitations under Section 382 of the Internal Revenue Code. To date, we have not completed a Section 382 study. Unless previously utilized, our net operating loss and research and development tax credit carryforwards expire between 2018 and 2036.
We have established a 100% valuation allowance due to the uncertainty of our ability to generate sufficient taxable income to realize the deferred tax assets. Our valuation allowance decreased $32.0 million, increased $24.9 million and increased $30.4 million in 2017, 2016 and 2015, respectively, primarily due to net operating losses incurred during these periods.
Reconciliation of income tax computed at federal statutory rates to the reported provisions for income taxes is as follows:
Year ended December 31, | ||||||||
2017 | 2016 | 2015 | ||||||
U.S. Federal statutory rate on net loss | (34 | )% | (34 | )% | (34 | )% | ||
State tax, net of federal tax benefit | (2 | )% | (2 | )% | (2 | )% | ||
Effects of statutory rate change | 115 | % | — | % | — | % | ||
Change in valuation allowance | (60 | )% | 37 | % | 41 | % | ||
Tax credits | (11 | )% | (4 | )% | (5 | )% | ||
Other | (8 | )% | 3 | % | — | % | ||
Effective tax rate | — | % | — | % | — | % | ||
We file federal and certain state income tax returns, which provides varying statutes of limitations on assessments. However, because of net operating loss carryforwards, substantially all of our tax years remain open to federal and state tax examination.
75
We recognize interest and penalties related to the underpayment of income taxes as a component of income tax expense. To date, there have been no interest or penalties charged to us in relation to the underpayment of income taxes.
Note 12—401(k) Retirement Plan
We have adopted a 401(k) plan. Beginning in 2017, our 401(k) retirement plan provides for an annual company match on employee contributions, initially set at a maximum of 4.0% of each participating employee’s eligible earnings, with a maximum company match of $4,000 per employee per year. All employees are eligible to participate, provided they meet the requirements of the plan.
Note 13—Quarterly Information (Unaudited)
The following table summarizes the unaudited statements of operations and comprehensive loss for each quarter of 2017 and 2016 (in thousands, except per share amounts):
2017 | For the Quarter Ended | |||||||||||||||
March 31, | June 30, | September 30, | December 31, | |||||||||||||
Revenue | $ | 12,257 | $ | 17,151 | $ | 21,658 | $ | 13,760 | ||||||||
Total costs and expenses | 24,982 | 29,090 | 26,768 | 27,881 | ||||||||||||
Loss from operations | (12,725 | ) | (11,939 | ) | (5,110 | ) | (14,121 | ) | ||||||||
Net loss | (15,089 | ) | (14,359 | ) | (7,482 | ) | (16,551 | ) | ||||||||
Basic and diluted net loss per share | $ | (0.34 | ) | $ | (0.33 | ) | $ | (0.16 | ) | $ | (0.34 | ) | ||||
2016 | For the Quarter Ended | |||||||||||||||
March 31, | June 30, | September 30, | December 31, | |||||||||||||
Revenue | $ | 7,419 | $ | 10,004 | $ | 11,289 | $ | 12,905 | ||||||||
Total costs and expenses | 26,871 | 20,933 | 23,327 | 24,762 | ||||||||||||
Loss from operations | (19,452 | ) | (10,929 | ) | (12,038 | ) | (11,857 | ) | ||||||||
Net loss | (20,539 | ) | (12,612 | ) | (13,962 | ) | (19,632 | ) | ||||||||
Basic and diluted net loss per share | $ | (0.54 | ) | $ | (0.32 | ) | $ | (0.34 | ) | $ | (0.45 | ) | ||||
76
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, as of December 31, 2017. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our principal executive and principal financial officers concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Internal control over financial reporting cannot provide absolute assurance of achieving financial reporting objectives because of its inherent limitations. Internal control over financial reporting is a process that involves human diligence and compliance and is subject to lapses in judgment and breakdowns resulting from human failures. Internal control over financial reporting also can be circumvented by collusion or improper management override. Because of such limitations, there is a risk that material misstatements may not be prevented or detected on a timely basis by internal control over financial reporting. However, these inherent limitations are known features of the financial reporting process. Therefore, it is possible to design into the process safeguards to reduce, though not eliminate, this risk.
Our management, with the participation of our principal executive and principal financial officers, conducted an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 framework). Based on the results of this assessment and on those criteria, our management concluded that our internal control over financial reporting was effective as of December 31, 2017.
Ernst & Young LLP has independently assessed the effectiveness of our internal control over financial reporting as of December 31, 2017 and its report is included below.
There was no change in our internal control over financial reporting identified in connection with the evaluation required by Rules 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during our fourth fiscal quarter of 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
77
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Board of Directors of Omeros Corporation
Opinion on Internal Control over Financial Reporting
We have audited Omeros Corporation’s internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), (the COSO criteria). In our opinion, Omeros Corporation (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Omeros Corporation (the Company) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, shareholders' deficit and cash flows for each of the three years in the period ended December 31, 2017, and the related notes of the Company and our report dated March 1, 2018 expressed an unqualified opinion thereon that included an explanatory paragraph regarding the Company’s ability to continue as a going concern.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Seattle, Washington
March 1, 2018
78
ITEM 9B. | OTHER INFORMATION |
On February 26, 2018, we and CRG entered into Amendment No. 2 to the CRG Loan Agreement, or Amendment No. 2. For a discussion of Amendment No. 2, see Part II, Item 7, “Management’s Discussion and Analysis--Financial Condition - Liquidity and Capital Resources.”
79
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2018 Annual Meeting of Shareholders and is incorporated herein by reference. Certain information required by this item concerning executive officers is set forth in Part I of this Annual Report on Form 10-K in “Business-Executive Officers and Key Employees.”
ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED SHAREHOLDER MATTERS |
Except for the information set forth below, the information required by this item will be contained in our definitive proxy statement issued in connection with the 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides certain information regarding our equity compensation plans in effect as of December 31, 2017:
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights | Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights | Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans | |||||||
Equity compensation plans approved by security holders: | |||||||||
2017 Omnibus Incentive Compensation Plan (1) | 159,707 | $ | 21.22 | 3,513,540 | |||||
2008 Equity Incentive Plan (2) | 9,497,552 | $ | 10.21 | — | |||||
Total | 9,657,259 | $ | 10.30 | 3,513,540 | |||||
(1) Our 2017 Omnibus Incentive Compensation Plan, or the 2017 Plan, provides for the grant of incentive and nonstatutory stock options, restricted stock, restricted stock units, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants. The 2017 Plan replaced the Omeros Corporation 2008 Equity Incentive Plan, or the 2008 Plan, and as a result we will not grant any new awards under the 2008 Plan. Any stock option awards granted under the 2008 Plan that were outstanding as of the effective date of the 2017 Plan remain in effect pursuant to their terms and, if the award is canceled or is repurchased, the shares underlying such award become available for grant under the 2017 Plan.
(2) The 2008 Plan provided for the grant of incentive and nonstatutory stock options, restricted stock, stock appreciation rights, performance units and performance shares to employees, directors and consultants and subsidiary corporations’ employees and consultants.
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
80
ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required by this item will be contained in our definitive proxy statement issued in connection with the 2018 Annual Meeting of Shareholders and is incorporated herein by reference.
81
PART IV
ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
1. Financial Statements
See the Index to Consolidated Financial Statements in Part II, Item 8 of this Form 10-K.
2. Financial Statement Schedules
All schedules have been omitted as the required information is either not required, not applicable or otherwise included in the Financial Statements and notes thereto.
3. Exhibits
The following list of exhibits includes exhibits submitted with this Form 10-K as filed with the SEC and those incorporated by reference to other filings.
Exhibit Description | Incorporated by Reference | |||||
Exhibit No. | Form | File No. | Exhibit No. | Filing Date | Filed Herewith | |
3.1 | 10-K | 001-34475 | 3.1 | 03/31/2010 | ||
3.2 | 10-K | 001-34475 | 3.2 | 03/31/2010 | ||
4.1 | S-1/A | 333-148572 | 4.1 | 10/02/2009 | ||
4.2 | 8-K | 001-34475 | 10.3 | 05/19/2016 | ||
10.1* | S-1 | 333-148572 | 10.1 | 01/09/2008 | ||
10.2* | 10-K | 001-34475 | 10.6 | 03/16/2017 | ||
10.3* | 10-Q | 001-34475 | 10.2 | 11/07/2013 | ||
10.4* | S-8 | 333-218882 | 4.3 | 06/21/2017 | ||
10.5* | S-8 | 333-218882 | 4.4 | 06/21/2017 | ||
10.6* | 8-K | 001-34475 | 10.1 | 04/12/2010 | ||
10.7* | S-1 | 333-148572 | 10.12 | 01/09/2008 | ||
10.8* | S-1 | 333-148572 | 10.14 | 01/09/2008 | ||
10.9 | S-1 | 333-148572 | 10.15 | 01/09/2008 | ||
10.10* | S-1 | 333-148572 | 10.16 | 01/09/2008 | ||
82
10.11 | S-1 | 333-148572 | 10.17 | 01/09/2008 | ||
10.12* | 10-Q | 001-34475 | 10.3 | 08/08/2017 | ||
10.13 | 8-K | 001-34475 | 10.1 | 02/01/2012 | ||
10.14 | 10-Q | 001-34475 | 10.2 | 11/09/2012 | ||
10.15 | 10-K | 001-34475 | 10.18 | 03/18/2013 | ||
10.16 | 10-K | 001-34475 | 10.18 | 03/13/2014 | ||
10.17 | 10-Q | 001-34475 | 10.3 | 11/09/2015 | ||
10.18 | 10-Q | 001-34475 | 10.1 | 05/10/2017 | ||
10.19† | S-1/A | 333-148572 | 10.29 | 09/16/2009 | ||
10.20† | S-1 | 333-148572 | 10.30 | 01/09/2008 | ||
10.21† | 10-K | 001-34475 | 10.24 | 03/16/2015 | ||
10.22† | S-1/A | 333-148572 | 10.31 | 09/16/2009 | ||
10.23† | S-1 | 333-148572 | 10.32 | 01/09/2008 | ||
10.24† | S-1/A | 333-148572 | 10.33 | 05/15/2009 | ||
10.25† | S-1/A | 333-148572 | 10.47 | 09/16/2009 | ||
10.26† | 10-K | 001-34475 | 10.28 | 03/18/2013 | ||
10.27† | 10-Q | 001-34475 | 10.1 | 05/12/2010 | ||
83
10.28† | 10-Q | 001-34475 | 10.1 | 05/10/2011 | ||
10.29† | 10-Q | 001-34475 | 10.1 | 05/09/2013 | ||
10.30† | 10-Q | 001-34475 | 10.2 | 08/10/2010 | ||
10.31† | 10-K | 001-34475 | 10.44 | 03/15/2011 | ||
10.32† | 10-K | 001-34475 | 10.45 | 03/15/2011 | ||
10.33† | 10-K | 001-34475 | 10.46 | 03/16/2015 | ||
10.34† | 10-Q | 001-34475 | 10.1 | 11/09/2015 | ||
10.35† | 10-Q | 001-34475 | 10.1 | 08/10/2015 | ||
10.36 | 8-K | 001-34475 | 1.1 | 01/06/2016 | ||
10.37 | 10-Q | 001-34475 | 10.2 | 11/09/2016 | ||
10.38 | 8-K | 001-34475 | 10.2 | 10/27/2016 | ||
10.39 | 8-K | 001-34475 | 10.1 | 10/17/2017 | ||
10.40 | X | |||||
10.41 | 8-K | 001-34475 | 10.1 | 10/05/2017 | ||
12.1 | X | |||||
23.1 | X | |||||
31.1 | X | |||||
84
31.2 | X | |||||
32.1 | X | |||||
32.2 | X | |||||
101.INS | XBRL Instance Document | X | ||||
101.SCH | XBRL Taxonomy Extension Schema Document | X | ||||
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | X | ||||
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | X | ||||
101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | X | ||||
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | X | ||||
* | Indicates management contract or compensatory plan or arrangement. |
† | Portions of this exhibit are redacted in accordance with a grant of confidential treatment. |
85
ITEM 16. | FORM 10-K SUMMARY |
Not included.
86
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
OMEROS CORPORATION |
/s/ GREGORY A. DEMOPULOS, M.D. |
Gregory A. Demopulos, M.D. |
President, Chief Executive Officer and Chairman of the Board of Directors |
Dated: March 1, 2018
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date |
/s/ GREGORY A. DEMOPULOS, M.D. | President, Chief Executive Officer and Chairman of the Board of Directors (Principal Executive Officer) | March 1, 2018 |
Gregory A. Demopulos, M.D. | ||
/s/ MICHAEL A. JACOBSEN | Vice President, Finance, Chief Accounting Officer and Treasurer (Principal Financial Officer and Principal Accounting Officer) | March 1, 2018 |
Michael A. Jacobsen | ||
/s/ RAY ASPIRI | Director | March 1, 2018 |
Ray Aspiri | ||
/s/ THOMAS J. CABLE | Director | March 1, 2018 |
Thomas J. Cable | ||
/s/ PETER A. DEMOPULOS, M.D. | Director | March 1, 2018 |
Peter A. Demopulos, M.D. | ||
/s/ ARNOLD C. HANISH | Director | March 1, 2018 |
Arnold C. Hanish | ||
/s/ LEROY E. HOOD, M.D., PH.D. | Director | March 1, 2018 |
Leroy E. Hood, M.D., Ph.D. | ||
/s/ RAJIV SHAH, M.D. | Director | March 1, 2018 |
Rajiv Shah, M.D. | ||
87