Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - Kindred Biosciences, Inc. | exhibit321-201710k.htm |
| EX-31.2 - EXHIBIT 31.2 - Kindred Biosciences, Inc. | exhibit312-201710k.htm |
| EX-31.1 - EXHIBIT 31.1 - Kindred Biosciences, Inc. | exhibit311-201710k.htm |
| EX-23.1 - EXHIBIT 23.1 - Kindred Biosciences, Inc. | exhibit231-201710k.htm |
| EX-21.1 - EXHIBIT 21.1 - Kindred Biosciences, Inc. | exhibit211listofsubsidaries.htm |
| EX-10.21 - EXHIBIT 10.21 - Kindred Biosciences, Inc. | exhibit1019-secondleaseadd.htm |
| EX-10.20 - EXHIBIT 10.20 - Kindred Biosciences, Inc. | exhbit1018-officeleaseadde.htm |
| EX-10.8 - EXHIBIT 10.8 - Kindred Biosciences, Inc. | exhibit108-2016equityincen.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
______________________
FORM 10-K
______________________
x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017 | |
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _____ to ______ | |
Commission file number: 001-36225
KINDRED BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
___________________
Delaware | 46-1160142 | |
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification Number) | |
1555 Bayshore Highway, Suite 200 Burlingame, California 94010 | ||
(Address of principal executive offices) | ||
(650) 701-7901 | ||
Registrant’s telephone number: | ||
Securities registered pursuant to Section 12(b) of the Act:
Title of each class | Name of each exchange on which registered | |
Common Stock, $0.0001 par value | The NASDAQ Stock Market LLC | |
Preferred Stock Purchase Rights | The NASDAQ Stock Market LLC | |
Securities registered pursuant to Section 12(g) of the Act: None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No þ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and, (2) has been subject to such filing requirements for the past 90 days.
Yes þ No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein and, will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | Accelerated filer þ | Non-accelerated filer o | Smaller reporting company o | ||
(Do not check if a smaller reporting company) | Emerging growth company þ | ||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. x
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes o No þ
As of June 30, 2017 (the last business day of the registrant’s most recently completed second fiscal quarter), the aggregate market value of the common stock of the registrant held by non-affiliates of the registrant was approximately $158.3 million.
The outstanding number of shares of the registrant’s common stock as of February 28, 2018 was 28,189,081.
Certain portions of the registrant’s Proxy Statement for the 2018 annual meeting of stockholders to be filed with the Securities and Exchange Commission pursuant to Regulation 14A, not later than 120 days after the close of the registrant’s fiscal year, are incorporated by reference into Part III of this Form 10-K.
TABLE OF CONTENTS | ||
Page | ||
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. All statements contained in this Annual Report that do not relate to matters of historical fact should be considered forward-looking statements, including, but not limited to, statements regarding our expectations about the trials, regulatory approval, manufacturing, distribution and commercialization of our current and future product candidates, and statements regarding our anticipated revenues, expenses, margins, profits and use of cash. These forward-looking statements are based on our current expectations. These statements are not promises or guarantees, but involve known and unknown risks, uncertainties and other important factors that may cause our actual results to be materially different from any future results expressed or implied by the forward-looking statements. These risks include, but are not limited to, the following: our limited operating history and expectations of losses for the foreseeable future; the absence of significant revenue from our product candidates for the foreseeable future; our potential inability to obtain any necessary additional financing; our substantial dependence on the success of our lead product candidates, which may not be successfully commercialized even if they are approved for marketing; the effect of competition; our potential inability to obtain regulatory approval for our existing or future product candidates; our dependence on third parties to conduct some of our development activities; our dependence upon third-party manufacturers for supplies of our product candidates; uncertainties regarding the outcomes of trials regarding our product candidates; our potential failure to attract and retain senior management and key scientific personnel; uncertainty about our ability to develop a satisfactory sales organization; our significant costs of operating as a public company; our potential inability to obtain patent protection and other intellectual property protection for our product candidates; potential claims by third parties alleging our infringement of their patents and other intellectual property rights; our potential failure to comply with regulatory requirements, which are subject to change on an ongoing basis; the potential volatility of our stock price; and the significant control over our business by our principal stockholders and management.
For a further description of these risks and other risks that we face, please see the risk factors described in Item 1A of this Annual Report under the caption “Risk Factors” and any subsequent updates that may be contained in our Quarterly Reports on Form 10-Q and other documents we file with the Securities and Exchange Commission, or SEC. As a result of these risks, actual results may differ materially from those indicated by the forward-looking statements made in this Annual Report. Forward-looking statements contained in this Annual Report speak only as of the date of this Annual Report, and we undertake no obligation to update or revise these statements except as may be required by law.
PART I.
ITEM 1. | BUSINESS. |
Overview
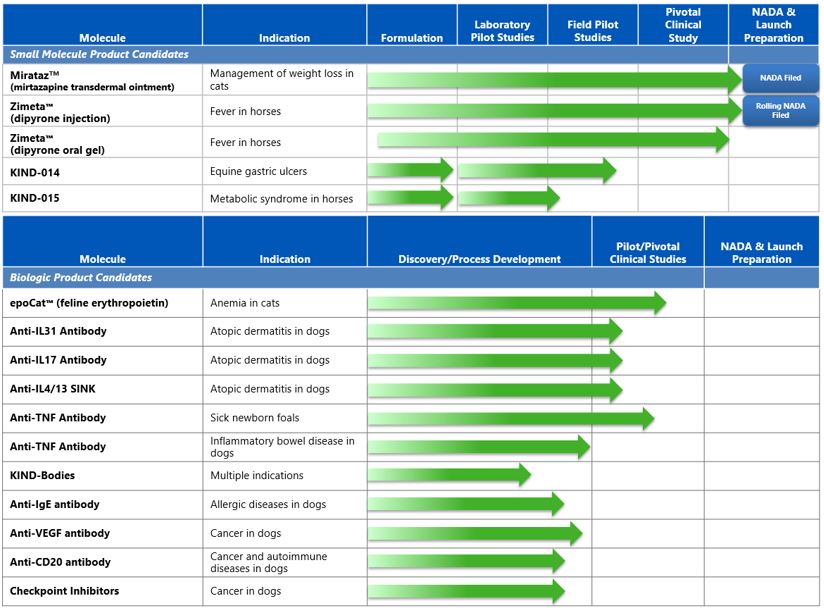
We are a pre-commercialization biopharmaceutical company focused on saving and improving the lives of pets. Our mission is to bring to our pets the same kinds of safe and effective medicines that our human family members enjoy. Our core strategy is to identify compounds and targets that have already demonstrated safety and efficacy in humans and to develop therapeutics based on these validated compounds and targets for pets, primarily dogs, cats and horses. We believe this approach will lead to shorter development times and higher approval rates than pursuing new, non-validated compounds and targets. Our current portfolio includes over 20 product candidates in development consisting of both small molecule pharmaceuticals and biologics.
We were incorporated in Delaware in September 2012. The address of our principal executive offices is 1555 Bayshore Highway, Suite 200, Burlingame, CA 94010. Unless the context requires otherwise, references to “KindredBio,” “the Company,” “we,” “us” or “our” in this Annual Report on Form 10-K for the fiscal year ended December 31, 2017 (2017 Annual Report) refer to Kindred Biosciences, Inc., a Delaware corporation, and its subsidiaries.
We have submitted all major technical sections of the New Animal Drug Application, or NADA, to the Food and Drug Administration, or FDA, for two product candidates, MiratazTM (mirtazapine transdermal ointment) for management of weight loss in cats and the intravenous, or IV, form of ZimetaTM (dipyrone injection) for the control of pyrexia (fever) in horses. In addition, we have multiple other product candidates, including several biologics, in various stages of development, with the potential to launch one or more products annually. We believe there are significant unmet medical needs for pets, and that the pet therapeutics segment of the animal health industry is likely to grow substantially as new therapeutics are identified, developed and marketed specifically for pets.
We plan to commercialize our feline, equine and canine products in the United States through a direct sales force complemented by selected distributor relationships, and in the European Union, or EU, through distributors and other third parties. Because we seek to identify product candidates that are not protected by third-party patents, we typically do not need to obtain licenses or make any upfront, milestone or royalty payments in connection with our product candidates.
Relative to human drug development, the development of pet therapeutics is generally faster and less expensive, since it requires fewer clinical studies involving fewer subjects and can be conducted directly in the target species. For example, studies that are typically required for approval of human drugs such as QTC studies, which detect cardiac irregularities, elderly patient studies, renal impairment studies, hepatic impairment studies or costly, long-term genotoxicity studies are not required for pet therapeutics. Based on our progress since inception in September 2012, we believe we can develop small molecule pet therapeutics from the Investigational New Animal Drug, or INAD, filing with the FDA to marketing approval in three to five years at an average cost of approximately $5 million per product candidate. The lower cost associated with the development of pet therapeutics permits us to pursue multiple product candidates simultaneously and avoid the binary outcome associated with the development of a single lead therapy by some human biotechnology companies. Because we typically develop drugs that have successfully been developed for humans, the active ingredients in many of our small molecule product candidates also have established Chemistry, Manufacturing and Controls, or CMC, which are important gating factors in the regulatory approval process. As a result, we usually do not need to invest in active pharmaceutical ingredient, or API, process development to comply with good manufacturing practices, or GMP, standards for our small molecule product candidates, and we can often advance our programs more rapidly than if we were pursuing new chemical entities.
Our biologics program, from INAD to marketing approval, is expected to take four to six years at an average cost of approximately $8 million per product candidate.
1
We estimate that the total U.S. market for veterinary care was approximately $69.4 billion in 2017, an increase of 68% from 2007. In 2017, 68% of households owned a pet, which translates to an estimated 89.7 million dogs and 94.2 million cats currently living in the United States. We believe there are many unmet or underserved medical needs and that the pet therapeutics portion of the market can grow significantly as new, safe and effective therapeutics are identified, developed and marketed. We expect continued market growth as new pet therapeutics are developed and owners grow more familiar with the treatment of pets with such therapeutics.
We are targeting the equine market in the near term because we have promising products for the equine sector and because we believe that it shares many similarities with the orphan human market. There are fewer horses than dogs or cats, but the willingness to pay is substantially higher. In addition, the cost of building a commercial infrastructure is much less for the equine market. We believe that a dozen or fewer sale representatives are sufficient to launch and support an equine product nationally.
Our management team’s extensive experience in both human and animal drug development has enabled us
to quickly establish our product pipeline, and should continue to enable us to promptly obtain Protocol
Concurrences from the FDA for our product candidates and to conduct the clinical trials. Our management team also has extensive experience in biologics, including in the development of antibodies such as Lucentis, Tysabri, Xolair, and Rituxan.
Richard Chin, M.D., our co-founder and Chief Executive Officer, was previously Head of Clinical Research for the Biotherapeutics Unit at Genentech, Inc., where he oversaw Phase I through Phase IV clinical programs for all products except oncology. Denise Bevers, our co-founder and Chief Operating Officer, has over 20 years of experience in clinical operations and medical affairs. Wendy Wee, our Chief Financial Officer, has over 30 years of experience and most recently was Vice President of Finance and Principal Accounting Officer at Telik, Inc. Hangjun Zhan, Ph.D., our Chief Scientific Officer, is a well-established protein biochemist and biophysicist with 21 years of drug discovery experience in the biotechnology industry.
Product Pipeline
Our current product pipeline consists of small molecules and biologics for a range of indications in dogs, cats and horses, with focus near-term on equine small molecule products and canine and feline biologics products.
The US Department of Agriculture, or USDA’s Center for Veterinary Biologics and the FDA’s Center for Veterinary Medicine have a memorandum of understanding under which animal products are to be regulated by the USDA as biologics, if they are intended for use to diagnose, cure, mitigate, treat, or prevent disease in animals and they work primarily through an immune process, or by the FDA as drugs, if they are intended for use in the diagnosis, cure, mitigation, treatment, or prevention of animal disease if the primary mechanism of action is not immunological or is undefined. Although we believe that most of our current animal biologics will be regulated by the USDA based on their mechanisms of action, it is possible that the agencies may determine that one or more of our animal biologics will be regulated by the FDA instead of the USDA.
We currently are pursuing over 20 indications with various small molecule and biologics product candidates. The following table illustrates some of the product candidates that we are developing.
2
Product Selection and Development
We utilize a rigorous screening and review process to identify compounds and targets that have demonstrated safety and efficacy in humans. Where possible, we try to identify compounds that have already demonstrated efficacy in the target companion animal species and address unmet medical needs in veterinary medicine. In some cases, we identify a chemical or functional equivalent of a validated human drug that addresses the same biological target or pathway. We review these compounds and targets with a view to differentiating them from existing treatments, including human products used extra-label in animals, based on ease of administration, method of delivery, dosing regimen, and other similar factors. We also try to identify product candidates that are free from any intellectual property rights of others, including drugs or dosage forms that are not marketed in the United States, or marketed only in a few countries, to minimize the potential for competition from human generics. For example, previously approved drugs that are found to have an idiosyncratic side effect in humans fit well with our target criteria, since such drugs are often no longer available for human use and could potentially be well suited for companion animals. We then develop these compounds for dogs, cats or horses for regulatory approval in the United States and the EU. As our product candidates are generally not protected by third-party patents, we typically do not need to obtain a license or make any upfront, milestone or royalty payments in connection with our product candidates.
For our small molecule product candidates, we customize the dosage, formulation, flavor and other characteristics of the product candidate before initiating pivotal clinical trials. In some cases, we reformulate the drug to have a longer half-life or into a form that is easier to administer for certain species. Pet therapeutics that are palatable to animals can command premium price and significant market share, as evidenced by the still-dominant position of Rimadyl compared to generic carprofen. Usually, the active ingredients in our small molecule product candidates are already available as a GMP-quality API. We target small molecule product candidates for which the active ingredient has not been previously approved for use in animals. If we are the first to gain approval for the use
3
of such active ingredient in animals, our small molecule product will enjoy five years of marketing exclusivity in the United States and ten years in the EU for the approved indication. Where appropriate, we also will seek patents and trademarks to provide added intellectual property protection in addition to the five-year or ten-year marketing exclusivity. In addition, we plan to introduce improved formulations, combination products and other product improvements in order to extend the lifecycle of our products.
Biologic therapies are typically derived from living organisms. A biologic can be defined as a large complex molecule (nucleic acid and protein platforms) produced from or extracted from a biological or living system. They are made by genetically engineering living cells, and a high level of precision is required in the manufacturing process to produce a consistent biologic product each time. A biologic product can be a monoclonal antibody, a vaccine, a tissue, or various proteins such as cytokines, enzymes, fusion proteins, whole cells, and viral and non-viral gene therapies. Our biologic product candidates are usually based on therapies and targets for which products have been successfully commercialized for humans. Human antibody therapies are expensive and are often ineffective in other species since they are usually immunogenic, or recognized as foreign bodies and rejected by the immune systems of dogs, cats, horses and other animals. We identify or create biologics, including antibodies that are fully or mostly canine, feline, or equine. We are developing a long-acting feline recombinant erythropoeitin. We are also developing antibodies that target canine IL-17a, IL13/IL4, IL-4Ra, IL-31, CD-20, IgE, TNF, and other validated targets.
In addition, our new scaffold technology, KIND-Bodies, which are non-antibodies with properties we believe will make them superior to antibodies, is proceeding on track. Specifically, we believe they can be bispecific, or bind to two different targets at the same time, and will not infringe antibodies patents against targets. This allows us to pursue molecules that may be superior to traditional antibodies, and to pursue high value targets before antibody patents expire.
We have an in-house laboratory capable of protein engineering, cell line development, analytics, and other activities necessary for advancing a world-class biologics pipeline. We believe that we have one of the best biologics teams in the pharmaceutical industry, drawn from some of the top biotechnology companies.
We have constructed a state-of-the-art manufacturing plant in Burlingame, California for our initial biologic product candidates, which we believe is one of the first GMP biologics plants for veterinary products. We started GMP manufacturing in January 2018 and believe that the plant will position the Company as a leader in the veterinary biologics field, and potentially afford us an advantage in cost of goods for our products. We acquired a second manufacturing plant in August 2017 in Elwood, Kansas and we have initiated the design and construction of our biologics manufacturing lines. The Elwood facility includes approximately 180,000 square feet with clean rooms, utility, equipment, and related quality documentation suitable for small molecule and biologics manufacturing.
We generally intend to seek composition-of-matter patents and other patents for these new chemical entities. Our biologic products, if approved, will not face generic competition or such generic competition may be significantly delayed, because there is presently no biosimilar pathway for veterinary biologics in the United States or in the EU.
Business Strategy
Key elements of our business strategy are as follows:
Obtain FDA approvals for Zimeta and Mirataz and launch products in the first half of 2018
We announced positive topline results in May 2016 upon the successful completion of a pharmacokinetic study and a randomized, placebo-controlled pivotal study of Mirataz (mirtazapine transdermal ointment), formerly known as KIND-010, for the management of weight loss in cats. All major technical sections of the NADA have been filed. We have received the technical section complete letter for effectiveness from the FDA and responded to the one remaining comment from the FDA regarding the CMC technical section. The FDA did not have any further comments on the safety technical section. Based on the feedback, we anticipate the approval of Mirataz and are
4
preparing for the commercial launch in the first half of 2018, assuming that the FDA finds our responses acceptable. The regulatory approval is expected, but is subject to the typical risks inherent in such a process.
The European Marketing Agency, or EMA, has accepted our European marketing authorization application for the review of Mirataz with the official acceptance date of December 21, 2017.
In November 2015, we announced a pivotal trial of Zimeta (dipyrone injection), previously known as KIND-012, for the control of pyrexia (fever) in horses with positive topline results. We submitted all major technical sections of the NADA for Zimeta to the FDA before the end of the first quarter of 2016. We have received the technical section complete letters for effectiveness and safety from the FDA and responded to additional comments from the FDA regarding the CMC technical section. We anticipate the approval of Zimeta in the first half of 2018, assuming that the FDA finds our responses acceptable. The regulatory approval is expected, but is subject to the typical risks inherent in such a process.
We have initiated pre-launch activities including build-out of a small commercial team, execution of distribution agreements and commercial scale-up and manufacturing.
We have also completed the pivotal field effectiveness study of ZimetaTM Oral (dipyrone oral gel) for the treatment of fever in horses and announced positive topline results in December 2017. The oral gel form of dipyrone is expected to be an additional valuable tool for equine veterinarians by providing an easy-to-administer fever reducing agent for the horse and horse owner.
We are currently developing KIND-014 for the treatment of equine gastric ulcers in horses, KIND-015 for the management of clinical signs associated with equine metabolic syndrome and epoCatTM (feline recombinant erythropoietin) for the control of non-regenerative anemia in cats. We expect to continue pilot field efficacy studies for the three product candidates in 2018. In addition to early stage pilot field studies of anti-cytokine antibodies for atopic dermatitis in dogs and anti-TNF in septic foals, we are also developing multiple other products, including interleukin antibodies and canine checkpoint inhibitors. In all, we have over 20 programs for various indications for dogs, cats, and horses.
Continue to focus on cost-effective research and development execution
In order to execute our studies rapidly and efficiently, we have built an experienced team drawn from both the veterinary and human pharmaceutical industries. We rely primarily on our own personnel or independent contractors, rather than on contract research organizations, or CROs, for many business-critical tasks, including protocol designs, regulatory interactions, statistics, data management and clinical operations. By doing so, we believe we can maintain higher quality, achieve lower costs and seek regulatory approval more quickly. Since our inception in September 2012, we have been able to quickly and efficiently build and advance our pipeline.
Leverage our antibody and biologics experience
Members of our team have extensive experience developing biologics such as antibodies. We are leveraging their expertise to identify and develop antibody-based therapies for pets based on approved human therapies, and to identify appropriate manufacturing technologies for these product candidates.
Leverage our current product pipeline in additional animal species
We intend to develop our product candidates primarily for approval in one or more indications in dogs, cats and horses. We believe the market for horse therapeutics may be particularly attractive, as it can be targeted by a limited sales force and has potentially less price sensitivity than therapeutic treatments for dogs and cats, because horse owners are willing to spend more on treatments for these more expensive companion animals. As an example, a one-month supply of omeprazole for a horse can cost over a thousand dollars. We may consider the development of our current or future product candidates for additional species in the future, but our pipeline currently is focused on dogs, cats and horses only.
5
Expand our pipeline with additional product candidates
We actively seek to identify small molecule and biologic therapeutics, or in some cases therapeutic targets, that have demonstrated safety and efficacy in humans, focusing on small molecules that are already marketed for humans or biologics for which there are no animal counterparts, and that are free from intellectual property rights of others in the United States. These therapeutics typically have been tested in animals such as dogs as part of standard toxicology studies in human clinical development. We have identified over 30 additional product candidates in the pre-INAD stage that we may potentially pursue. We will seek to protect our product candidates through a combination of regulatory exclusivity periods in the United States and in the EU, patents, know-how and other customary means.
Commercialize our equine products with our own direct sales force in the United States and with distributors in other regions
In conjunction with FDA approval of one or more of our lead product equine candidates, when approved, we intend to utilize a direct sales organization, complimented by select distributor relationships, to market our products directly to veterinarians in the United States. For our equine products, we believe we can accomplish this with a sales force of 4 to 12 sales representatives and reach most of the prescribing equine veterinarians in the United States. We also intend to establish collaborations with distributors to commercialize any of our products that may be approved by the EMA.
Commercialize our canine and feline products with our own direct sales force in the United States and with distributors in other regions
In conjunction with FDA approval of one or more of our lead product canine and feline candidates, when approved, we intend to utilize a direct sales organization, complimented by select distributor relationships to market our products directly to veterinarians in the United States. For our first lead product feline candidate, we believe we can start with 10 to 25 sales representatives supplemented by sales support from our distributors. We intend to grow our direct sales force incrementally if and as our product candidates are approved for marketing, and to utilize national and regional distributors to augment our sales force. We also intend to establish collaborations to commercialize any of our products that may be approved with the EMA.
Execute distribution agreements
In December 2017, we executed the first of our national distribution agreements with MWI Veterinary Supply Co.and Direct Vet Marketing, Inc. d/b/a Vets First Choice for distribution and sales support for our products to supplement the KindredBio sales force.
Pet Therapeutics Market
Overview
U.S. consumers spent an estimated $69.4 billion on their pets in 2017, according to the American Pet Products Association, or APPA, an increase of 68% from 2007. The veterinary care segment has been among the fastest growing segments of the overall U.S. pet market. This segment accounted for an estimated $16.6 billion spent on veterinary care in 2017, an increase of 65% from 2007.
We believe several factors will contribute to an increase in spending on pet therapeutics. Pets are generally
living longer, with the average lifespan for dogs increasing by nearly a year to 12 years for dogs and 13.1 years for cats between 2012 and 2016 according to a study by Banfield Pet Hospital. As a result, pets are increasingly exhibiting many of the same diseases associated with aging in humans such as cardiovascular disease, arthritis, and diabetes. For example, the incidence of diabetes in dogs has increased by 79.7% since 2006, while in felines, the prevalence of diabetes in cats has increased 18.1% over the same timeframe. The incidence of osteoarthritis in dogs has increased by 82% since 2006 according to the same study. As it is with human health, obesity is a growing concern for pets. Since 2006, overweight and obesity have increased by 74% in dogs and 179% in cats. The Association for Pet Obesity Prevention estimates that 54% of dogs and 59% of cats are overweight or obese, which
6
translates to 41.9 million dogs and 50.1 million cats. In addition, pet ownership numbers may increase as more people become aware of the myriad health benefits of pet ownership. According to the Human Animal Bond Research Institute, studies show that some of the benefits of having a dog include helping to lower your blood pressure, decrease your risk of heart disease, and preventing allergies in children.
Among pet owners, there is growing familiarity in treating these pet diseases with medications. According to the APPA, approximately 77% of U.S. dog owners treated their dogs with medications in 2015, an increase of over 50% from the level reported in 2004. In a 2010 poll by the Associated Press, 35% of pet owners are willing to spend $2,000 to treat their pet for a serious medical condition. More recently, a 2017 Harris Poll by the American Institute of Certified Public Accountants indicated that 76% of the U.S. adults (1,004, of which 526 identified as pet owners) surveyed would make financial sacrifices for their pets to pay for an emergency expense such as medical care. Additionally, 79% said they would stop eating at restaurants and 67% would give up a vacation to pay for pet-related expenses if they were in a difficult financial situation. Respondents also indicated that they would cancel cable and TV streaming services (61%), sacrifice contributions to their retirement account (37%), cancel a cell phone plan (35%), or forego paying a credit card bill (27%) to pay for their pet’s expenses. We expect pet owners to spend more on their pets’ health and welfare as new therapeutics are developed specifically for pets, particularly as 95% of pet owners considered their pet to be a member of their family, according to a 2015 survey by the Harris Poll of Harris Interactive.
Pet Therapeutics Market Dynamics
The respective businesses of developing and commercializing therapeutics for pets and for humans share a number of characteristics, including the need to demonstrate safety and efficacy in clinical trials, obtain FDA or other regulatory approval for marketing, manufacture the therapeutics in facilities compliant with GMP requirements and market the therapeutics only for their intended indication based on claims permitted in the product label, and not for other uses, which is referred to as extra-label use.
Despite their similarities, there are a number of important differences between the pet therapeutics and human therapeutics businesses, including:
• | Faster, less expensive and more predictable development. The development of pet therapeutics requires fewer clinical studies in fewer subject animals than the development of human therapeutics and, unlike human therapeutics, is conducted directly in the target animals. We believe our strategy of selecting compounds and targets with demonstrated efficacy and safety in humans enhances the predictability of results and probability of success of our pivotal trials relative to compounds and targets that have not been previously validated. |
• | Role and incentives for veterinary practices. In the United States, veterinarians generally serve the dual role of doctor and pharmacist, and pet owners typically purchase medicines directly from their veterinarians. Therapeutics specifically developed for pets enable veterinarians to provide potentially superior treatment options, while also increasing revenue from the sale of these therapeutics. |
• | Primarily private-pay nature of veterinary market. Pet owners in the United States generally pay for pet therapeutics out-of-pocket, and less than 5% of pet owners currently purchase pet insurance. As a result, pet owners must make decisions primarily on their veterinarians’ advice regarding available treatment options, rather than on the treatment options’ eligibility for reimbursement by insurance companies or government payers. We believe this results in less pricing pressure than in human healthcare, although the limited adoption of insurance may also reduce pet owners’ ability to pay for therapeutics recommended by their veterinarians. |
• | Less generic competition and strong brand loyalty. There is less generic competition in the pet therapeutics industry than in the human healthcare industry. Approximately 14% of veterinary drugs face generic competition, and the percentage of generic prescriptions in the veterinary space is only 7% as compared to approximately 81% for human drugs. For example, Rimadyl, the leading U.S. pet NSAID, lost regulatory exclusivity in 2001, but its sales continued to grow since generic competition was introduced in 2005. We believe that stronger brand loyalty and lack of mandatory generic drug substitution, as in human pharmaceuticals, partially explains the low penetration of generics in veterinary medicine. |
7
Unmet Medical Needs in the Pet Therapeutics Market
Despite the growing market for pet therapeutics, there are relatively few treatment options approved for use in pets as compared to human therapeutic treatments. As a result, veterinarians often must resort to prescribing products approved for use in humans but not approved, formulated or even formally studied in pets. Veterinarians must then rely upon trial and error or untested rules of thumb to assess the proper dosage needed to be effective in the particular species without undue risk of side effects. The veterinarian must also find a way to administer the human product in animals and determine the amount actually dosed, which are important and potentially overlooked practical considerations in the treatment of pets.
Even in disease categories with approved pet therapeutics, significant unmet medical needs remain. For example, the NSAID class of products, commonly prescribed for pain, have potentially serious side effects in dogs that limit their long-term use and may require ongoing monitoring by veterinarians. The treatment of pain in cats is further complicated as a result of their differing biology, which makes NSAIDs toxic.
Animal health companies have been relatively slow to develop new therapeutics for pets, and have tended to focus primarily on the larger market for the treatment of livestock and other farm animals. On average, for every 8 NDAs filed annually for human therapeutics, one NADA is filed for animal therapeutics. In 2017, human pharmaceutical companies received FDA approval for 46 novel drugs (non-generic), while pet therapeutic companies received only one novel FDA drug approval. In the EU, human pharmaceutical companies received EMA approval for 34 novel drugs in 2017, compared to only 3 novel drug approvals for pet therapeutic companies.
We believe that therapeutics specifically developed for pets can extend and improve the quality of the lives of pets, help veterinarians achieve improved medical outcomes and make the process of administering therapeutics to pets much more convenient. Advances in human medicines have created new therapeutics for managing chronic diseases associated with aging, such as osteoarthritis, cancer, diabetes and cardiovascular diseases. Pets often suffer from the same disease as humans, including diabetes, arthritis, cancer, Alzheimer’s disease (canine cognitive dysfunction), lupus, Crohn’s disease, Lou Gehrig’s disease (degenerative myelopathy) and others. In most cases, the biologies of the diseases in pets are very similar to those in humans. Because of the similarity of the diseases, many human drugs, when formulated properly and administered in proper doses, are effective in pets. However, most human drugs are neither formulated nor approved for animals.
Products in Development
Mirataz (mirtazapine transdermal ointment)
Overview
Inappetence is a serious unmet medical need in cats, and may be caused by multiple factors, including chronic illness or infection. If untreated, it may lead to hepatic failure, which can be a life-threatening condition. In cats, mirtazapine, a noradrenergic and specific serotonergic antidepressant, increases the level of serotonin in the brain and balances the activity of serotonin in the gastro-intestinal tract, resulting in alleviation of nausea and appetite loss.
We are developing Mirataz, formerly known as KIND-010, for the management of weight loss in cats. Mirataz is a mirtazapine transdermal drug with innovative AccusorbTM transdermal technology which results in high and predictable absorption. As a transdermal product, Mirataz is an attractive alternative for veterinarians and owners to administering a drug orally to cats. Weight loss is the leading cause of veterinary visits among cat owners, and a veterinarian will see on average 7 or more cats per week with this condition. In many cases, significant weight loss can lead to liver failure and ultimately death. Currently, there are limited options for the management of weight loss in cats, none of which include an FDA-approved transdermal option.
We announced positive topline results in May 2016 upon the successful completion of a pharmacokinetic study and a randomized, placebo-controlled pivotal study of Mirataz. All major technical sections of the NADA have been filed. We have received the technical section complete letter for effectiveness from the FDA and responded to the one remaining comment from the FDA regarding the CMC technical section. The FDA did not
8
have any further comments on the safety technical section. Based on the feedback, we anticipate the approval of Mirataz and are preparing for the commercial launch in the first half of 2018, assuming that the FDA finds our responses acceptable.
In December 2017, we submitted the European Marketing Authorization application for Mirataz to the EMA. The EMA has accepted the submission for review, with the official acceptance date of December 21, 2017.
The active ingredient in Mirataz is available as GMP-grade material from one supplier and we believe our current manufacturer will be able to provide sufficient quantities for potential commercialization. In addition, we executed a commercial manufacturing agreement with DPT Laboratories, Ltd. for the manufacture of Mirataz. The agreement provides for production to supply KindredBio’s initial launch and future commercial campaigns upon regulatory approval, with capabilities to meet excess demand.
Clinical Data
The pivotal field study, KB105, was a multicenter, randomized, double-blind, placebo-controlled pivotal field study that enrolled 230 cats to assess the effectiveness of Mirataz in managing weight loss in cats. The primary endpoint was percentage change in body weight from Day 1 to Week 2. At Week 2, the mean percent increase in body weight from Day 1 was 3.94% in the KIND-010 group (n=83), versus 0.41% in the placebo group (n=94) (p<0.0001). In the target animal safety study, Mirataz was generally well-tolerated and no significant safety concerns were identified. At the proposed label dose, topical administration of mirtazapine ointment was associated with mild, reversible skin changes at the site of dose application (ear). Based on a preliminary review of the safety data, the drug appears to be well tolerated.
Zimeta (dipyrone injection)
Overview
Zimeta, formerly known as KIND-012, is a pyrazolone anti-inflammatory drug, with a mechanism of action unlike traditional NSAIDs. Zimeta is widely used both for horses and humans as an antipyretic outside of the United States. It is also considered to be an anti-spasmodic that in horses can be used without masking the surgical signs of colic.
In humans, the active ingredient in Zimeta can, in very rare cases, cause bone marrow suppression. In some countries, it is still available as prescription or over the counter medication, while in other countries, it has been withdrawn from market. However, the side effects are not seen in horses, and the product is widely used outside the U.S. by many equine veterinarians.
We are developing Zimeta for the control of pyrexia (fever) in horses. In March 2015, we announced a positive randomized, blinded, placebo-controlled pilot study. In November 2015, we announced positive results for a multicenter, randomized, blinded, placebo-controlled pivotal study of Zimeta. We submitted all major technical sections of the NADA for Zimeta to the FDA before the end of the first quarter of 2016. We have received the technical section complete letters for effectiveness and safety from the FDA and responded to comments from the FDA regarding the CMC technical section. We anticipate the approval of Zimeta including product launch in the first half of 2018, assuming that the FDA finds our responses acceptable. However, regulatory approval timelines can not be guaranteed.
The active ingredient in Zimeta is available as GMP-grade material from two suppliers, and we believe our current manufacturer or other suppliers will be able to provide sufficient quantities for commercialization. In addition, we executed a commercial manufacturing agreement with Corden Pharma SPA for the manufacture of Zimeta. The agreement provides for production to supply KindredBio’s initial launch and future commercial campaigns upon regulatory approval, with capabilities to meet excess demand.
Clinical Data
9
The pivotal field study was a multicenter, randomized, blinded, placebo-controlled pivotal study that enrolled 138 horses to assess the effectiveness of Zimeta. The primary endpoint was improvement (a 2°F or greater decrease in temperature from baseline) or resolution of fever (a return to normothermia (≤101.0°F)) at hour 6 following treatment. The success rate was approximately 75% in the KIND-012 group vs. approximately 20% in the placebo group (p < 0.0001). Based on preliminary review of the safety data, the drug appears to be well tolerated.
We have also completed the pivotal field effectiveness study of ZimetaTM Oral (dipyrone oral gel) for the treatment of fever in horses and announced positive topline results in December 2017. This study was a multicenter, randomized, blinded, placebo-controlled pivotal study that enrolled 139 horses to assess the effectiveness of Zimeta Oral. The primary endpoint was improvement or resolution of fever 6 hours after treatment. The success rate was approximately 78% in the Zimeta Oral group vs. approximately 18% in the placebo group (p = 0.0026). Based on preliminary review of the safety data, the drug appears to be well tolerated.
KindredBio has completed the in-life portion of the Target Animal Safety Study and is analyzing the data. We anticipate submitting the effectiveness and safety technical sections of the NADA in the first half of 2018, and the CMC technical section by the end of 2018, assuming the data are supportive of approval.
Other Product Candidates
KIND-014 is a small molecule product candidate we are developing for treatment of equine gastric ulcers in horses. We have completed dose range finding and palatability studies with KIND-014 and based on the study results, we have advanced two formulations into pilot field studies. The pilot studies have been initiated and we expect to complete the review of data by the end of the second quarter to determine which formulation will move into a pivotal study, assuming the data support further development.
Ulcers are a common medical condition in horses and foals. It is estimated that almost 50% of foals and 1/3 of adult horses confined in stalls may have mild ulcers. Up to 60% of show horses and 90% of racehorses may develop moderate to severe ulcers. Because they are so common, and can occur as a result of a number of factors, the condition is often called equine gastric ulcer syndrome or EGUS. Our development of KIND-014 provides another option for equine veterinarians to treat equine gastric ulcers.
KIND-015 is a small molecule product we are developing for the management of clinical signs associated with equine metabolic syndrome, or EMS. EMS is an endocrinopathy affecting horses and ponies. It is of primary concern due to its link to obesity, insulin resistance, and subsequent laminitis which is a painful and debilitating disease of the digital laminae (the tissue inter-connection between the coffin bone and hoof wall). Often by the time clinical signs are recognized, crippling body changes such as sinking and rotation of the coffin bone have occurred. KIND-015 delays absorption of glucose from the gastrointestinal tract, increases insulin sensitivity (enhancing peripheral tissue sensitivity to insulin) and glucose uptake into cells, and inhibits synthesis of glucose by the liver. We have optimized the formulation for KIND-015 and have initiated a pilot field effectiveness study.
In addition to small molecules, we have the following biologics programs:
epoCat, or KIND-510, is feline recombinant erythropoietin for the control of non-regenerative anemia in cats. Anemia is a common condition in older cats which is often associated with chronic kidney disease resulting in decreased levels of endogenous erythropoietin. epoCat is a recombinant protein that has been specially engineered by us with a prolonged half-life compared to endogenous feline erythropoietin. The PK data suggest that the molecule may have a sufficiently long half-life to allow for once-monthly dosing. The initial laboratory studies have been completed, with a positive efficacy signal, as evidenced by increased reticulocyte formation. Enrollment in a pilot field efficacy study is currently ongoing.
KIND-011 is an anti-Tumor Necrosis Factor, or anti-TNF, treatment for newborn foals. Sick newborn foals, defined as sepsis score ≥ 11 or positive blood culture, are challenging and difficult to treat and result in approximately 50% mortality. We have completed a pilot field study in sick or septic foals to assess safety and efficacy of anti-TNF monoclonal antibody, with positive results. By Kaplan-Meier analysis, the difference in survival between the control and placebo groups was statistically significant (p=0.0293). Sepsis in foals can cause
10
up to 50% mortality and is an important unmet medical need. There is currently no FDA-approved therapy. The Company plans to discuss the development plan with the FDA and will pursue additional development of the indication.
Canine atopic dermatitis, or CAD, is an immune-mediated inflammatory, chronic skin disease associated with allergies. Atopic dermatitis is a large market, with the leading two products on the market expected to sell over $500 million per year. It is the second most common allergic skin disease in dogs, sometimes so severe that dogs have to be put down. These allergic reactions can be brought on by normally harmless substances like grass, mold spores, house dust mites, and other environmental allergens. Based on positive results of previous pilot studies, KindredBio is in the process of initiating pilot field efficacy studies for several molecules for atopic dermatitis, including fully caninized anti-IL31 antibody, fully caninized anti-IL17 antibody and canine anti-IL4/IL13 SINK molecule. We are pursuing a multi-pronged approach toward atopic dermatitis, with a portfolio of promising biologics.
Our other promising biologic product candidates in our pipeline with key focus areas of atopic dermatitis and cancer include: KIND-502, a new biologic that targets the canine counterpart of the human target for Xolair, for allergic and immune-mediated diseases; KIND-0888, an antibody that targets CD20; several antibodies that target cytokines involved in atopic dermatitis; and KIND-509, an antibody that targets the canine TNF.
KIND-Bodies, a novel biologics scaffold with certain advantages over antibodies, including bispecific binding, is also proceeding on track.
Product Launches and Commercialization
Our executive management team has extensive experience with product launches. Richard Chin, M.D., and Denise Bevers, our co-founders and Chief Executive Officer and Chief Operating Officer, respectively, have each been involved in the launch of numerous products in humans, including such drugs as Tysabri and Xolair. Other members of the KindredBio team have experience with multiple product launches for companion animals.
If our equine product candidates are approved by the FDA, we intend to establish a direct sales organization market to our products directly to veterinarians in the United States. For our equine products, we believe we can do this with a sales force of 4 to 12 sales representatives and reach most of the prescribing equine veterinarians in the United States. We also intend to establish collaborations with distributors to commercialize any of our products that may be approved with the EMA.
In conjunction with FDA approval of one or more of our lead product canine and feline candidates, we intend to establish a direct sales force of 10 to 25 sales representatives, complimented by select distributor relationships, to market our products directly to veterinarians in the United States. We also intend to establish collaborations to commercialize any of our products that may be approved with the EMA.
Our direct sales force will sell products directly to veterinarians, who typically mark up the pet therapeutics they prescribe for pet owners. According to industry sources, approximately one-third of pet veterinary practice revenue comes from prescription drug sales, vaccinations and non-prescription medicines. We believe veterinarians are self-motivated to prescribe innovative therapeutics that are safe, effective and supported by reliable clinical data and regulatory approval in order to improve the health of pets, while also generating additional revenue.
Animal health companies commonly use wholesale veterinary distributors to inventory, sell, bill and ship products to independent veterinarians. We estimate that the top three national distributors are responsible for fulfillment of approximately 70% of U.S. pet sales by veterinarians. Each of these distributor organizations has a sales team of approximately 275 field sales representatives, 175 telesales representatives and a dozen distribution centers geographically placed throughout the United States. We intend to grow our direct sales force incrementally if and as our product candidates are approved for marketing, and to utilize national and regional distributors to augment our sales force.
11
Manufacturing
We have no internal manufacturing capabilities for small molecules. To ensure dependable and high quality supply of the API in our toxicology studies and pivotal trials, we rely on GMP-compliant contract manufacturers for small molecules rather than devote capital and resources toward developing or acquiring our own manufacturing facilities. Our selection process for small molecule products takes into account the availability of established and cost-effective GMP manufacturing before proceeding to INAD filing. We believe that contract manufacturers can manufacture these supplies more cheaply than we could on our own.
We have or expect to have a sufficient supply of formulated drugs for our potential launch of Zimeta and Mirataz. We also have identified multiple potential contract manufacturers to provide commercial supplies of the formulated drug product candidates if they are approved for marketing. We have or intend to secure contract manufacturers with established track records of quality product supply and significant experience with regulatory requirements of the FDA and the EMA.
For biologics, we have established our own GMP manufacturing capabilities in Burlingame, California and proceeded to GMP manufacturing in January 2018. In August 2017, we acquired a manufacturing facility in Elwood, Kansas and have completed the design and construction plans for biologics manufacturing lines. We expect to start construction shortly. The Elwood facility includes approximately 180,000 square feet with clean rooms, utility, equipment, and related quality documentation suitable for small molecule and biologics manufacturing. The USDA regulates the manufacture of pet biologics under standards that are less stringent than those for human biologics, which may reduce the cost of goods of our biologic product candidates relative to human biologics.
While we and our contract manufacturers have historically been able to obtain supplies of the API for development of our small molecule product candidates, neither we nor our contract manufacturers have long-term supply agreements with the API manufacturers. We also have no agreements for commercial-scale supply of the API or manufacture of any of our product candidates. As a result, we and our contract manufacturers may be unable to procure API in a timely manner on commercially reasonable terms, or at all.
Competition
While there are fewer competitors in the pet therapeutics industry than in the human pharmaceutical industry, the development and commercialization of new animal health medicines is highly competitive, and we expect considerable competition from major pharmaceutical, biotechnology and specialty animal health medicines companies.
Our potential competitors include large animal health companies, which currently derive the majority of their revenue from livestock medications. For example, in 2016 livestock accounted for 50%, and pets 50%, of sales for Zoetis, a large company focused on animal health. Within the pet therapeutics market, vaccines and parasiticides are currently the greatest sources of revenue.
Large animal health companies include Merck Animal Health, the animal health division of Merck & Co., Inc.; Elanco, the animal health division of Eli Lilly and Company; Bayer Animal Health, the animal health division of Bayer AG; Boehringer Ingelheim Animal Health, the animal health division of Boehringer Ingelheim GmbH; and Zoetis, Inc. We will also compete against several animal health companies in Europe, such as the Virbac Group, Ceva Animal Health and Dechra Pharmaceuticals PLC. We are also aware of smaller companies that are developing products for use in the pet therapeutics market, including Aratana Therapeutics, Inc.
At the product level, we will face competition for Zimeta from Banamine and Butazolidin even though they are not approved for control of fever in horses, and for KIND-014 from GastroGard and UlcerGard, as well as potentially from additional products in development including Entyce from Aratana, if approved for cats. In addition, we may face competition from various products including additional products in development. Our products may also face competition from generic medicines and products approved for use in humans that are used extra-label for pets. Some of our other products also may face competition from their human generic equivalents in countries where such equivalents are available.
12
Many of our competitors and potential competitors have substantially more financial, technical and human resources than we do. Many also have far more experience than we have in the development, manufacture, regulation and worldwide commercialization of animal health medicines, including pet therapeutics. In addition, these and other potential competing products may benefit from greater brand recognition and brand loyalty than any that our product candidates may achieve. Accordingly, there is no assurance that we and our products can compete effectively.
Intellectual Property
We intend to rely primarily upon a combination of regulatory exclusivity, proprietary know-how, and
confidentiality agreements to protect our product formulations, processes, methods and other technologies and to preserve any trade secrets and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. We currently have several provisional and nonprovisional patent applications, but no issued patents. Because most of our current product candidates, including all of our current small molecule product candidates, are based on generic human drugs, there is little, if any, composition-of-matter patent protection available for the API in such product candidates. Where feasible, however, we intend to pursue the broadest intellectual property protection possible for our current compounds and any future compounds and any proprietary technology through enhanced formulations of our products, both in the United States and abroad. For example, we are developing slow release oral formulations of some of our products, and we intend to develop combination therapies and slow release parenteral formulations of some of our products. In addition, we have filed composition of matter patents on many of our biologics. However, even intellectual property protection, if available to us, may not afford us with complete protection against competitors. See “Risk Factors-Risks Related to Intellectual Property.”
We depend upon the skills, knowledge and experience of our management personnel, as well as that of our other employees, advisors, consultants and contractors, none of which are patentable. To help protect our know-how, and any inventions for which patents may be difficult to obtain or enforce, we require all of our employees, consultants, advisors and other contractors to enter into customary confidentiality and inventions agreements that prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business.
Regulatory
The development, approval and sale of animal health products are governed by the laws and regulations of each country in which we intend to sell our products. To comply with these regulatory requirements, we have established processes and resources to provide oversight of the development and launch of our products and their maintenance in the market.
United States
Three federal regulatory agencies regulate the health aspects of animal health products in the United States: the FDA; the USDA; and the Environmental Protection Agency, or the EPA. In addition, the Drug Enforcement Administration, or DEA, regulates animal therapeutics that are classified as controlled substances.
The FDA Center for Veterinary Medicine, or CVM, regulates animal pharmaceuticals under the Federal Food, Drug and Cosmetic Act. The USDA Center for Veterinary Biologics, or CVB, regulates veterinary vaccines and certain biologics pursuant to the Virus, Serum, Toxin Act. The EPA Office of Pesticide Programs, or OPP, regulates veterinary pesticides under the Federal Insecticide, Fungicide and Rodenticide Act. Many topical products used for treatment of flea and tick infestations are regulated by the EPA.
All of our current product candidates are animal pharmaceuticals or biologics regulated by the CVM or the CVB, respectively. Manufacturers of animal health pharmaceuticals and biologics, including us, must show their products to be safe, effective and produced by a consistent method of manufacture. We will also be required to conduct post-approval monitoring of products and to submit reports of product quality defects, adverse events or unexpected results, and be subject to regulatory inspection from time to time. In addition, for our controlled substance product candidates, we will be required to comply with the Controlled Substances Act, or CSA, and
13
related state laws regarding manufacturing, labeling, packaging, testing, dispensing, production and procurement quotas, recordkeeping, reporting, handling, shipment and disposal.
Requirements for Approval of Veterinary Pharmaceuticals for Pets
As a condition to regulatory approval for sale of animal products, regulatory agencies worldwide generally require that a product to be used for pets be demonstrated to:
• be safe for the intended use in the intended species;
• | have substantial evidence of effectiveness for the intended use; |
• | have a defined manufacturing process that ensures the product can be made with high quality consistency; and |
• | be safe for humans handling the product and for the environment |
Safety. To determine that a new veterinary drug is safe for use, most regulatory authorities will require us to provide data from a safety study generated in laboratory cats, dogs, and horses tested at doses higher than the intended label dose, over a period of time determined by the intended length of dosing of the product. In the case of the FDA, the design and review of the safety study and the study protocol can be completed prior to initiation of the study to help assure that the data generated will meet FDA requirements. These studies are conducted under rigorous quality control, including GLP, to assure integrity of the data. They are designed to clearly define a safety margin, identify any potential safety concerns, and establish a safe dose for the product. In addition, safety data from pivotal field studies conducted under GCP standards are evaluated to assure that the product will be safe in the target population. Furthermore, because safety and effectiveness studies must conform to VICH guidelines, which are established under an international program aimed at harmonizing technical requirements for veterinary product registration, they can be utilized by regulatory bodies in the European Union, Japan, Canada, New Zealand and Australia.
Effectiveness. Early pilot studies may be conducted in laboratory cats, dogs, or horses to establish effectiveness and the dose range for each product. Data on how well the drug is absorbed when dosed by different routes of administration and the relationship of the dose to the effectiveness are studied. When an effective dose is established, a study protocol to test the product in real world conditions is developed prior to beginning the study. In the case of the FDA, the pivotal effectiveness field study protocol can be submitted for review and concurrence prior to study initiation, to help assure that the data generated will meet requirements.
The pivotal field effectiveness study must be conducted with the formulation of the product that is intended to be commercialized, and is a multi-site, randomized, controlled study, generally with a placebo control. To reduce bias in the study, individuals doing the assessment are not told whether the subject is in the group receiving the treatment being tested or the placebo group. In both the United States and the European Union, the number of subjects enrolled in pivotal field effectiveness studies is required to be approximately 100 to 150 animal subjects treated with the test product and a comparable number of subjects in the control group that receive the placebo. In many cases, a pivotal field study may be designed with clinical sites in both the European Union and the United States, and this single study may satisfy regulatory requirements in both jurisdictions.
Chemistry, Manufacturing and Controls, or CMC. To assure that the product can be manufactured consistently, regulatory agencies will require us to provide documentation of the process by which the API is made and the controls applicable to that process that assure the API and the formulation of the final commercial product meet certain criteria, including quality, purity and stability. After a product is approved, we will be required to communicate with the regulatory bodies any changes in the procedures or manufacturing site. Both API and commercial formulations are required to be manufactured at facilities that practice pharmaceutical GMP.
Environmental and Human Safety. We will not be required under United States law to provide an environmental impact statement for products currently in development if the products are given at the home of the pet’s owner or in a veterinary hospital. If products might result in some type of environmental exposure or release,
14
the environmental impact must be assessed. For approval in the EU, a risk assessment for potential human exposure will be required.
Labeling, All Other Information, and Freedom of Information Summary. We also will be required to submit the intended label for the product, and also any information regarding additional research that has been conducted with the drug, to the CVM and other regulatory bodies for review. We will draft, and submit for regulatory review, the Freedom of Information Summary for use in the United States. This summary outlines the studies and provides substantial information that the FDA uses to assess the drug’s safety and effectiveness and then publishes on its website.
Regulatory Process at the FDA
To begin the development process for products in the United States, we must file an Investigational New Animal Drug, or INAD, submission with the FDA. We will then usually hold a pre-development meeting with the FDA to reach a general agreement on the plans for providing the data necessary to fulfill requirements for an NADA. During development, we will usually submit pivotal protocols to the FDA for review and concurrence prior to conducting the required studies. We will gather and submit data on manufacturing, safety and effectiveness to the FDA for review, and this review will be conducted according to timelines specified in the Animal Drug User Fee Act. These are called technical sections, which collectively form the basis of the NADA. Once all data have been submitted and reviewed for each technical section - safety, effectiveness and CMC - the FDA will issue us a technical section complete letter as each section review is completed, and when the three letters have been issued, we will compile a draft of the Freedom of Information Summary, the proposed labeling, and all other relevant information, and submit these for FDA review. An administrative NADA is a NADA that is submitted after all of the technical sections that fulfill the requirements for the approval of the new animal drug have been reviewed by CVM and CVM has issued a technical section complete letter for each of those technical sections. Although this process is not required and submission of a non-administrative NADA is also acceptable, we plan to take advantage of the administrative NADA process to obtain a more timely, phased review. Because CVM has already reviewed the individual technical sections before the administrative NADA is filed, CVM is committed under its user fee agreements to reviewing and acting on 90% of administrative NADAs within 60 days after submission. The CVM user fee goal is to review and act on 90% of non-administrative NADAs within 180 days after submission. After approval, we will be required to collect reports of adverse events and submit them on a regular basis to the FDA.
Regulatory Process at the USDA
To begin the development process for veterinary biologics products in the United States, we typically file an Application for United States Veterinary Biological Product License with the USDA. For the biologics products that we develop, we may then meet with the USDA to reach a general agreement on the plans for providing the data necessary to fulfill requirements for an approval. During development, we gather and submit data on manufacturing, purity and potency to the USDA for review. Once all data have been submitted and reviewed, the USDA will issue its decision. Unlike the FDA, there are no timelines specified by law for the USDA’s review.
In some cases, it may be unclear whether our product candidates meet the definition of a biological product subject to regulation by the USDA or a drug subject to regulation by the FDA. The USDA’s Center for Veterinary Biologics and the FDA’s Center for Veterinary Medicine have a memorandum of understanding concerning their joint responsibilities for resolving jurisdictional issues over products of this nature. Under the memorandum of understanding, animal products are to be regulated by the USDA as biologics if they are intended for use to diagnose, cure, mitigate, treat, or prevent disease in animals and they work primarily through an immune process, or by the FDA as drugs, if they are intended for use in the diagnosis, cure, mitigation, treatment, or prevention of animal disease if the primary mechanism of action is not immunological or is undefined.
Regulatory Process at the EMA
The EMA is responsible for coordinating scientific evaluation of applications for marketing approval for pet therapeutics in the EU. Its veterinary review section is distinct from the review section for human drugs. To perform these evaluations the EMA established a specific scientific committee, the Committee for Medicinal Products for Veterinary Use, or CVMP. The CVMP considers applications submitted by companies for the
15
marketing approval of individual pet therapeutics and evaluates whether or not the medicines meet the necessary quality, safety and efficacy requirements. Assessments conducted by the CVMP are based on scientific criteria and are intended to ensure that pet therapeutics reaching the marketplace have a positive benefit-risk balance in the pet population for which they are intended. Based on the CVMP’s recommendation, a centralized marketing authorization is granted by the EMA, which allows the product to be marketed in any of the EU states, Norway, Lichtenstein and Iceland. The CVMP is also responsible for various post-authorization and maintenance activities, including the assessment of modifications or extensions to an existing marketing authorization.
To obtain authorization from the EMA, we must submit a marketing authorization application called a dossier. The dossier is the EMA’s equivalent of the FDA’s NADA and includes data from studies showing the quality, safety and efficacy of the product. The CVMP reviews and evaluates the dossier. For any dossier, a rapporteur and co-rapporteur are appointed from the members of the CVMP. Their role is to lead the scientific evaluation and prepare the assessment report. The rapporteur can utilize experts to assist it in performing its assessment. The report is critiqued by the co-rapporteur and other members of the CVMP before the CVMP makes its determination. The final opinion of the CVMP is generally given within 210 days of the submission of a dossier, but the EMA makes the final decision on the approval of products. In general, the requirements for regulatory approval of an animal health product in the EU are similar to those in the United States, requiring demonstrated evidence of purity, safety, efficacy and consistency of manufacturing processes.
Alternatively, product approval applications may be submitted directly to the regulatory authority in each country rather than by centralized approval by the EMA.
Regulatory Processes at the DEA
The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use, and may not be marketed or sold in the United States. An animal drug may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances. Certain of our product candidates are likely to be scheduled as controlled substances under the CSA. Consequently, their manufacture, shipment, storage, sale and use will be subject to a high degree of regulation.
Annual registration is required for any facility that manufactures, distributes, dispenses, imports or exports any controlled substance. The registration is specific to the particular location, activity and controlled substance schedule. For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
The DEA typically inspects a facility to review its security measures prior to issuing a registration. Security requirements vary by controlled substance schedule, with the most stringent requirements applying to Schedule I and Schedule II substances. Required security measures include background checks on employees and physical control of inventory through measures such as cages, surveillance cameras and inventory reconciliations. Records must be maintained for the handling of all controlled substances, and periodic reports must be made to the DEA, for example, distribution reports for Schedule II controlled substances, Schedule III substances that are narcotics, and other designated substances. Reports must also be made for thefts or losses of any controlled substance, and to obtain authorization to destroy any controlled substance. In addition, special authorization and notification requirements apply to imports and exports.
In addition, a DEA quota system controls and limits the availability and production of controlled substances in Schedule I or II. Distributions of any Schedule I or II controlled substance must also be accompanied by special order forms, with copies provided to the DEA. The DEA may adjust aggregate production quotas and individual production and procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments.
16
Other Regulatory Considerations
Regulatory rules relating to human food safety, food additives, or drug residues in food will not apply to the products we currently are developing because our products are not intended for use in food animals or food production animals, with the exception of horses, which qualify as food animals in Europe and Canada.
Advertising and promotion of animal health products is controlled by regulations in the United States and other countries. These rules generally restrict advertising and promotion to those claims and uses that have been reviewed and authorized by the applicable agency. We will conduct a review of advertising and promotional material for compliance with the local and regional requirements in the markets where we sell pet therapeutics.
Our small molecule product candidates, if approved, may eventually face generic competition in the United States and in the EU. In the United States, a generic animal drug may be approved pursuant to an Abbreviated New Animal Drug Application, or ANADA. Instead of demonstrating the drug’s safety and effectiveness in the target species as required in a NADA, a generic applicant must only show that the proposed generic product is the same as, and bioequivalent to, the approved brand name product. However, if our product candidates are the first approved by the FDA or the EMA as applicable for use in animals, they will be eligible for five years of regulatory exclusivity in the United States and ten years in the EU. There is no comparable pathway for approval of a generic veterinary biologic regulated by the USDA.
Employees
As of December 31, 2017, we had 63 employees, including 21 employees with D.V.M., M.D. or Ph.D. degrees. Of our employees, 38 including Dr. Chin and Ms. Bevers are engaged in one or more aspects of our research and development activities. Dr. Chin and Ms. Bevers also are engaged in corporate and administrative activities. None of our employees are represented by labor unions or covered by collective bargaining agreements.
Corporate Information
We were incorporated on September 25, 2012 by our co-founder, Richard Chin, M.D., our President and Chief Executive Officer. Our principal executive offices are located at 1555 Bayshore Highway, Suite 200, Burlingame, California 94010, and our telephone number is (650) 701-7901. Our website address is www.kindredbio.com. The information contained in, or accessible through, our website should not be considered a part of this Annual Report on Form 10-K.
ITEM 1A. | RISK FACTORS |
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as the other information contained in or incorporated by reference into our other public filings with the Securities and Exchange Commission, before deciding whether to invest in our common stock. The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations and growth prospects. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations.
Risks Related to Our Business
We have a limited operating history, are not profitable and may never become profitable.
We are a development stage biopharmaceutical company. Since our formation in September 2012, our operations have been limited to the identification of product candidates and research and development of our product candidates, including our lead product candidates, Zimeta and Mirataz. As a result, we have limited historical operations upon which to evaluate our business and prospects and have not yet demonstrated an ability to obtain marketing approval for any of our product candidates or successfully overcome the risks and
17
uncertainties frequently encountered by companies in emerging fields such as the pet therapeutics industry. We also have not generated any revenue to date, and continue to incur significant research and development and other expenses. As of December 31, 2017, we had an accumulated deficit of $112.0 million. For the foreseeable future, we expect to continue to incur losses, which will increase significantly from historical levels as we expand our product development activities, seek regulatory approvals for our product candidates and begin to commercialize them if they are approved by the Center for Veterinary Medicine branch of the U.S. Food and Drug Administration, or FDA, the U.S. Department of Agriculture, or USDA, or the European Medicines Agency, or EMA. Even if we succeed in developing and commercializing one or more product candidates, we expect to continue to incur losses for the foreseeable future, and we may never become profitable. If we fail to achieve or maintain profitability, it would adversely affect the value of our common stock.
We will have no material product revenue for the foreseeable future, and we may need to raise additional capital to achieve our goals.
Until, and unless, we receive approval from the FDA, USDA or EMA, as applicable, for one or more of our product candidates, we cannot market or sell our products in the United States or in the European Union, or EU, and will have no material product revenue. We have submitted to the FDA all technical sections of the NADA for our drug product candidates, Zimeta and Mirataz. Although we received the technical sections complete letter for effectiveness and safety from the FDA for both Zimeta and Mirataz, we are still waiting for responses on our CMC technical section. Our other small molecule product candidates will require from three to five years of further development at a cost of an average of approximately $5 million per product candidate and our biologics candidates will require four to six years of further development at an average cost of approximately $8 million per product candidate before we expect to be able to apply for marketing approval in the United States. We also are actively involved in identifying additional human therapeutics for development and commercialization as pet therapeutics, and will continue to expend substantial resources for the foreseeable future to develop our current product candidates and any other product candidates we may develop or acquire. These expenditures will include: costs of identifying additional potential product candidates; costs associated with drug formulation; costs associated with conducting pilot, pivotal, and toxicology studies; costs associated with completing other research and development activities; costs associated with payments to technology licensors and maintaining other intellectual property; costs of obtaining regulatory approvals; costs associated with establishing commercial manufacturing and supply capabilities; and costs associated with marketing and selling any of our products approved for sale. We also may incur unanticipated costs. Because the outcome of our development activities and commercialization efforts is inherently uncertain, the actual amounts necessary to successfully complete the development and commercialization of our current or future product candidates may be greater or less than we anticipate.
We believe we have sufficient cash and cash equivalents to fund our operating plan through the anticipated approval and launch of our lead product candidates. However, we may seek additional funds through public or private equity or debt financings or other sources such as strategic collaborations. Additionally, we do not expect our existing cash and cash equivalents to be sufficient to complete the development of all of our current product candidates, or of any additional product candidates that we may identify, and we may need to raise additional capital to fund these activities. Even if we believe we have sufficient funds on hand for our current or planned future business and operations, we may seek from time to time to raise additional capital based upon favorable market conditions or strategic considerations such as potential acquisitions.
Our future capital requirements depend on many factors, including, but not limited to:
• | the scope, progress, results and costs of researching and developing our current or future product candidates; |
• | the timing of, and the costs involved in, obtaining regulatory approvals for any of our current or future product candidates; |
18
• | the number and characteristics of the product candidates we pursue; |
• | the cost of manufacturing our current and future product candidates and any products we successfully commercialize; |
• | the cost of commercialization activities if any of our current or future product candidates are approved for sale, including marketing, sales and distribution costs; |
• | the expenses needed to attract and retain skilled personnel; |
• | the costs associated with being a public company; |
• | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; and |
• | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation. |
Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, reduce or terminate one or more of our product development programs or any future commercialization efforts.
We are substantially dependent on the success of our lead product candidates, and cannot be certain that any of them will be approved for marketing or successfully commercialized even if approved.
We have no product approved for sale in any jurisdiction. Our current efforts are, and a substantial portion of our efforts over the foreseeable future will be, primarily focused on our lead product candidates, Zimeta and Mirataz, and on the progress of other product candidates that are in our pipeline. We discontinued our previous lead product candidates, SentiKind and CereKin, as a result of not meeting their primary endpoints. We also re-assessed our atopic dermatitis program and the atopic dermatitis market and decided to discontinue our AtoKin study in order to redirect our resources toward other programs in our portfolio. Accordingly, our near-term prospects, including our ability to generate material product revenue, obtain any new financing if needed to fund our business and operations, or enter into potential strategic transactions, will depend heavily on the successful development and commercialization of one or more of our lead candidates, which in turn will depend on a number of factors, including the following:
• | the successful completion of the pivotal trials and toxicology studies of one or more of our current product candidates, which may take significantly longer than we currently anticipate and will depend, in part, upon the satisfactory performance of third-party contractors; |
• | our ability to demonstrate to the satisfaction of the FDA, the USDA and the EMA the safety and efficacy of our product candidates and to obtain regulatory approvals; |
• | the ability of our third-party manufacturers to manufacture supplies of any of our product candidates and to develop, validate and maintain viable commercial manufacturing processes that are compliant with Good Manufacturing Practices, or GMP; |
• | our ability to successfully launch commercial sales of our current product candidates, assuming marketing approval is obtained, whether alone or in collaboration with others; |
• | the availability, perceived advantages, relative cost, relative safety and relative efficacy of our products compared to alternative and competing treatments; |
• | the acceptance of our product candidates as safe and effective by veterinarians, pet owners and the animal health community; |
19
• | our ability to achieve and maintain compliance with all regulatory requirements applicable to our business; and |
• | our ability to obtain and enforce our intellectual property rights and obtain marketing exclusivity for our product candidates, and avoid or prevail in any third-party patent interference, patent infringement claims or administrative patent proceedings initiated by third parties or the U.S. Patent and Trademark Office, or USPTO. |
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will be successful in developing or commercializing one or more of our lead product candidates. If we are unsuccessful or are significantly delayed in developing and commercializing Zimeta and Mirataz or any of our other current or future product candidates, our business and prospects will be materially adversely affected and you may lose all or a portion of the value of your investment in our common stock.
Most of our current and future small molecule product candidates are or will be based on generic human drugs, and other companies may develop substantially similar products that may compete with our products.
Most of the small molecule product candidates we are currently developing or expect to develop are based on generic human drugs. We do not engage in early-stage research or discovery with respect to our small molecule product candidates, but focus primarily on product candidates whose active pharmaceutical ingredient, or API, has been successfully commercialized or demonstrated to be safe or effective in human trials, which we sometimes refer to as validated. There is little, if any, third-party patent protection of the active ingredient in most of our current small molecule product candidates, and this means that our small molecule product candidates may face competition from their human generic equivalents in countries where such equivalents are available and used in unapproved animal indications, which is known as extra-label use.
While in most cases we select product candidates that are not available as a human generic in the United States, in cases where there is a human generic available there is no assurance that the eventual prices of our products will be lower than or competitive with the prices of human generic equivalents used extra-label, or that a palatable, easy-to-administer formulation such as the chewable, beef-flavored formulation that we utilize will be sufficient to differentiate them from their human equivalents. Human generics available outside the United States cannot be imported into the United States for use in animals, except on a case-by-case basis where the FDA determines it is medically necessary.
In most cases, we target small molecule product candidates for which the active ingredients have not been previously approved for use in animals. If we are the first to gain approval for the use of such active ingredients in animals, our small molecule products will enjoy five years of marketing exclusivity in the United States and ten years in the EU for the approved indication. We also plan to differentiate our products where possible with specific formulations, including flavors, methods of administration, new patents and other strategies, but we cannot assure you that we will be able to prevent competitors from developing substantially similar products and bringing those products to market earlier than we can. In addition, while we expect to have composition of matter patents on most of our biologic product candidates, we may not ultimately be able to obtain such patents. Although there are no generic regulatory approval pathways for animal biologics in the United States and European Economic Area, or EEA, our competitors may develop biologics that bind to the same target, but do not infringe any patents we may obtain. Thus, our competitors may be able to develop and market competing products if they are willing and able to conduct the full set of required studies, file a New Animal Drug Application, or NADA, with the FDA, or Application for United States Veterinary Biological Product License with the USDA, also called a Product License Application, or PLA, and obtain marketing approval. If such competing products achieve regulatory approval and commercialization prior to our product candidates, or if our intellectual property protection and efforts to obtain regulatory exclusivity fail to provide us with exclusive marketing rights for some of our products, then our business and prospects could be materially adversely affected.
20
If our product candidates are approved, they may face significant competition and may be unable to compete effectively.
The development and commercialization of pet therapeutics is highly competitive and our success depends on our ability to compete effectively with other products in the market. If our product candidates are approved, we expect to compete with animal health divisions of major pharmaceutical and biotechnology companies such as Merck Animal Health, Elanco, Bayer Animal Health, and Boehringer Ingelheim Animal Health, as well as specialty animal health medicines companies such as Zoetis, Virbac Group, Ceva Animal Health and Dechra Pharmaceuticals. Additionally, we are aware of several early-stage companies that are developing products for use in the pet therapeutics market, including Aratana Therapeutics Inc. and Jaguar Health, Inc. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health medicines.
If approved, Zimeta will face competition from existing products approved for treatment of horses such as Banamine. Mirtazapine, the active ingredient in Mirataz, is widely used today from human generic and compounded formulations. Many of our product candidates also will face competition from various products approved for use in humans that are used extra-label in animals, and all of our products will face potential competition from new products in development. These and other potential competing products may benefit from greater brand recognition and brand loyalty than our product candidates may achieve.
Many of our competitors and potential competitors have substantially more financial, technical and human resources than we do. Many also have far more experience than we have in the development, manufacture, regulation and worldwide commercialization of animal health medicines, including pet therapeutics. We also expect to compete with academic institutions, governmental agencies and private organizations that are conducting research in the field of animal health medicines.
For these reasons, there is no assurance that we and our products can compete effectively.
The development of our biologic product candidates is dependent upon relatively novel technologies and uncertain regulatory pathways.
We plan to develop biologics, including animal antibodies, for pets. Identification, optimization, and manufacture of therapeutic animal biologics is a relatively new field in which unanticipated difficulties or challenges could arise, and we expect the discovery, development, manufacturing and sale of biologic products to be a long, expensive and uncertain process. While many biologics have been approved for use in humans, apart from vaccines, relatively few recombinant proteins or antibodies have been approved for use in animals. There are unique risks and uncertainties with biologics, the development, manufacturing, and sale of which are subject to regulations that are often more complex and extensive than the regulations applicable to other small molecule products. We may be unable to identify biologics suitable for development or to achieve the potency and stability required for use in pets. In particular, canine, feline, and equine antibodies represent new types of product candidates that may be difficult to develop successfully.
In some cases, it may be unclear whether our product candidates meet the definition of a biological product subject to regulation by the USDA or a drug subject to regulation by the FDA. The USDA’s Center for Veterinary Biologics and the FDA’s Center for Veterinary Medicine have a memorandum of understanding concerning their joint responsibilities for resolving jurisdictional issues over products of this nature. Under the memorandum of understanding, animal products are to be regulated by the USDA as biologics, if they are intended for use to diagnose, cure, mitigate, treat, or prevent disease in animals and they work primarily through an immune process, or by the FDA as drugs, if they are intended for use in the diagnosis, cure, mitigation, treatment, or prevention of animal disease if the primary mechanism of action is not immunological or is undefined.
Although we believe that most of our current animal biologics will be regulated by the USDA based on their mechanisms of action, the USDA and the FDA may not agree with our assessment, or disputes may
21
arise between the USDA and the FDA over regulatory jurisdiction for one or more of such biologics. If so, the development of our biologics may be delayed while any such disputes are adjudicated by the agencies. Furthermore, if the agencies were to determine that one or more of our animal biologics will be regulated by the FDA instead of the USDA, the time and cost of developing such biologics may be longer and more expensive than we currently anticipate, and we may determine to discontinue development of such biologics. It is also possible that the USDA’s regulatory standards for novel biologics may be more difficult to satisfy than we anticipate. The current trend indicates that the FDA is attempting to exert jurisdiction over more biologics.
Because the regulatory standards for pet biologics are often less stringent than for small molecule animal drugs, we believe that some veterinarians prefer to see further efficacy data before making a new biologic product purchasing decision. Accordingly, we may also find it necessary to conduct additional studies of our biologic product candidates in order to achieve commercial success.
The results of earlier studies may not be predictive of the results of our pivotal trials, and we may be unable to obtain regulatory approval for our existing or future product candidates under applicable regulatory requirements. The denial or delay of any regulatory approval would prevent or delay our commercialization efforts and adversely affect our potential to generate material product revenue and our financial condition and results of operations.
The research, testing, manufacturing, labeling, approval, sale, marketing and distribution of pet therapeutics are subject to extensive regulation. We are usually not permitted to market our products in the United States until we receive approval of an NADA from the FDA or a PLA from the USDA, or in the EU or in other EEA countries until we receive marketing approval from the EMA. To gain approval to market a pet therapeutic for a particular species, we must provide the FDA, the USDA and the EMA, as applicable, with efficacy data from pivotal trials that adequately demonstrate that our product candidates are safe and effective in the target species (e.g., dogs, cats or horses) for the intended indications. In addition, we must provide manufacturing data. For the FDA and EMA, we must provide data from toxicology studies, also called target animal safety studies, and in some cases environmental impact data. We are conducting the pivotal trial of our compounds internally without significant outsourcing, but we rely on contract research organizations, or CROs, and other third parties to conduct our toxicology studies and for certain other development activities. The results of toxicology studies and other initial development activities, and of any previous studies in humans or animals conducted by us or third parties, may not be predictive of future results of pivotal trials or other future studies, and failure can occur at any time during the conduct of pivotal trials and other development activities by us or our CROs. Our pivotal trial may fail to show the desired safety or efficacy of our product candidates despite promising initial data or the results in previous human or animal studies conducted by others, and success of a product candidate in prior animal studies, or in the treatment of human beings, does not ensure success in subsequent studies. Clinical trials in humans and pivotal trials in animals sometimes fail to show a benefit even for drugs that are effective, because of statistical limitations in the design of the trials or other statistical anomalies. Therefore, even if our studies and other development activities are completed as planned, the results may not be sufficient to obtain regulatory approval for our product candidates.
The FDA, USDA or EMA can delay, limit or deny approval of any of our product candidates for many reasons, including:
• | if the FDA, USDA or EMA disagrees with our interpretation of data from our pivotal studies or other development efforts; |
• | if we are unable to demonstrate to the satisfaction of the FDA, USDA or EMA that the product candidate is safe and effective for the target indication; |
• | if the FDA, USDA or EMA requires additional studies or changes its approval policies or regulations; |
22
• | if the FDA, USDA or EMA does not approve of the formulation, labeling or the specifications of our current and future product candidates; and |
• | if the FDA, USDA or EMA fails to approve the manufacturing processes of our third-party contract manufacturers. |
Further, even if we receive approval of our product candidates, such approval may be for a more limited indication than we originally requested, and the FDA, USDA or EMA may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates.
Any delay or failure in obtaining applicable regulatory approval for the intended indications of our product candidates would delay or prevent commercialization of such product candidates and would materially adversely impact our business and prospects.
Our Protocol Concurrences with the FDA for our pivotal studies do not guarantee marketing approval in the United States.
We may conduct pivotal trials under Protocol Concurrences with the FDA. A Protocol Concurrence in animal drug development is analogous to a Special Protocol Assessment in human drug development, and means that the FDA agrees that the design and analyses proposed in a protocol are acceptable to support regulatory approval of the product candidate with respect to effectiveness of the indication studied and will not change its view of these matters, unless public or animal health concerns arise that were not recognized at the time of protocol concurrence or we change the protocol. Even under a Protocol Concurrence, approval of an NADA by the FDA is not guaranteed, because a final determination that the agreed-upon protocol satisfies a specific objective, such as the demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data submitted to the FDA.
Development of pet therapeutics is inherently expensive, time-consuming and uncertain, and any delay or discontinuance of our current or future pivotal trials would significantly harm our business and prospects.
Development of pet therapeutics remains an inherently lengthy, expensive and uncertain process, and there is no assurance that our development activities will be successful. We do not know whether the trials of our current or future product candidates will begin or conclude on time, and they may be delayed or discontinued for a variety of reasons, including if we are unable to:
• | address any safety concerns that arise during the course of the studies; |
• | complete the studies due to deviations from the study protocols or the occurrence of adverse events; |
• | add new study sites; |
• | address any conflicts with new or existing laws or regulations; or |
• | reach agreement on acceptable terms with study sites, which can be subject to extensive negotiation and may vary significantly among different sites. |
Any delays in completing our development efforts will increase our costs, delay our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenue. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, factors that may cause a delay in the commencement or completion of our development efforts may also ultimately lead to the denial of regulatory approval of our product candidates which, as described above, would materially, adversely impact our business and prospects.
We currently rely on third parties to conduct some of our development activities, and may rely more heavily on such third parties in the future. If these third parties do not successfully carry out their contractual
23
duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our current or future product candidates as planned.
We currently plan to conduct our own pivotal trials, but we rely upon CROs to conduct our toxicology studies and for other development activities. We also may rely on CROs in the future to conduct one or more pivotal trials. These CROs are not our employees, and except for contractual duties and obligations, we have limited ability to control the amount or timing of resources that they devote to our programs or manage the risks associated with their activities on our behalf. We are responsible to regulatory authorities for ensuring that each of our studies is conducted in accordance with the development plans and trial protocols, and any failure by our CROs to do so may adversely affect our ability to obtain regulatory approvals, subject us to penalties, or harm our credibility with regulators. The FDA and foreign regulatory authorities also require us and our CROs to comply with regulations and standards, commonly referred to as good clinical practices, or GCPs, or good laboratory practices, or GLPs, for conducting, monitoring, recording and reporting the results of our studies to ensure that the data and results are scientifically credible and accurate.
Our agreements with CROs may allow termination by the CROs in certain circumstances with little or no advance notice to us. These agreements generally will require our CROs to reasonably cooperate with us at our expense for an orderly winding down of the CROs’ services under the agreements. If the CROs conducting our studies do not comply with their contractual duties or obligations to us, or if they experience work stoppages, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our development protocols or GCPs or for any other reason, we may need to secure new arrangements with alternative CROs, which could be difficult and costly. In such event, our studies also may need to be extended, delayed or terminated as a result, or may need to be repeated. If any of the foregoing were to occur, regulatory approval and commercialization of our product candidates may be delayed and we may be required to expend substantial additional resources.
Even if we obtain regulatory approval of one or more of our current or future product candidates, they may never achieve market acceptance or commercial success.
If we obtain FDA, USDA or EMA approvals for one or more of our current or future product candidates, they may not achieve market acceptance among veterinarians and pet owners, and may not be commercially successful. Market acceptance of any of our current or future product candidates for which we may receive approval depends on a number of factors, including:
• | the indications for which our products are approved; |
• | the potential and perceived advantages of our product candidates over alternative treatments, including generic medicines and competing products currently prescribed by veterinarians, and products approved for use in humans that are used extra-label in animals; |
• | the cost of treatment in relation to alternative treatments and willingness on the part of veterinarians and pet owners to pay for our products, including other discretionary items, especially during economically challenging times; |
• | the prevalence and severity of any adverse side effects of our products; |
• | the relative convenience and ease of administration of our products; |
• | the effectiveness of our sales and marketing efforts; and |
• | the proper training and administration of our products by veterinarians and acceptance by veterinarians and pet owners of our products as safe and effective. |
24
Any failure by our product candidates that obtain regulatory approval to achieve market acceptance or commercial success would adversely affect our financial condition and results of operations.
Pet therapeutics, like human therapeutics, are subject to unanticipated post-approval safety or efficacy concerns, which may harm our business and reputation.
The success of our commercialization efforts will depend upon the perceived safety and effectiveness of pet therapeutics, in general, and of our products, in particular. Unanticipated safety or efficacy concerns can arise with respect to approved pet therapeutics after they enter into commerce, which may result in product recalls or withdrawals or suspension of sales, as well as product liability and other claims. Because reliable detection of rare events might require exposure of millions of animals, it is not possible to rule out the risk until well after the launch of the product.
It is also possible that the occurrence of significant adverse side effects in approved human generic compounds upon which our product candidates are based could impact our products. Any safety or efficacy concerns, or recalls, withdrawals or suspensions of sales of our products or other pet therapeutics, or of their human equivalents, could harm our reputation, in particular, or pet therapeutics, generally, and materially, adversely affect our business and prospects or the potential growth of the pet therapeutics industry, regardless of whether such concerns or actions are justified.
Future federal and state legislation may result in increased exposure to product liability claims, which could result in substantial losses to us.
Under current federal and state laws, pets are generally considered to be personal property of their pet owners and, as such, pet owners’ recovery for product liability claims involving their pets may be limited to the replacement value of the pets. Pet owners and their advocates, however, have filed lawsuits from time to time seeking non-economic damages such as pain and suffering and emotional distress for harm to their pets based on theories applicable to personal injuries to humans. If new legislation is passed to allow recovery for such non-economic damages, or if precedents are set allowing for such recovery, we could be exposed to increased product liability claims that could result in substantial losses to us if successful. In addition, some horses can be worth millions of dollars or more, and product liability for horses may be very high.
It is also possible that our product liability insurance will not be sufficient to cover any future product liability claims against us.
If we fail to retain current members of our senior management, or to attract and keep additional key personnel, our business and prospects could be materially adversely impacted.
Our success depends on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Richard Chin, M.D., our President and Chief Executive Officer, Denise Bevers, our Chief Operating Officer, Wendy Wee, our Chief Financial Officer and Hangjun Zhan, Ph.D., our Chief Scientific Officer. The loss of services of any of our key personnel could adversely affect our ability to successfully develop our current or future product pipeline and commercialize our product candidates. Although we have entered into employment agreements with these key members of senior management, such agreements generally do not prohibit them from leaving our employ at any time. We currently do not maintain “key man” life insurance on any of our senior management team. The loss of Dr. Chin or other members of our current senior management could adversely affect the timing or outcomes of our current and planned studies, as well as longer-term prospects for commercializing our product candidates.
In addition, competition for qualified personnel in the animal health fields is intense, because there is a limited number of individuals who are trained or experienced in the field. We will need to hire additional personnel as we expand our product development and commercialization activities, and we may not be able to attract and retain qualified personnel on acceptable terms, or at all.
25
We are dependent upon third-party manufacturers for supplies of our current product candidates, and intend to rely on third-party manufacturers for commercial quantities of any of our product candidates that may be approved.
We currently have no internal capability to manufacture the formulated small molecule product candidates for use in our studies or commercial supplies of any of our product candidates that may be approved, and will be dependent upon third-party manufacturers for such supplies. We and our contract manufacturers have historically been able to obtain supplies of the API for development of our small molecule product candidates, but neither we nor our contract manufacturers have long-term supply agreements with the API manufacturers. We also have no agreements for commercial-scale supply of the API or manufacture of any of our product candidates. As a result, we and our contract manufacturers may be unable to procure API in a timely manner on commercially reasonable terms, or at all. Any delay in identifying and contracting with third-party contract manufacturers on commercially reasonable terms would have an adverse impact upon our current product development activities and future commercialization efforts.
The facilities used by our contract manufacturers to manufacture the drugs are subject to inspections by the FDA, USDA, and the EMA, and we depend on our contract manufacturers to comply with GMP or other applicable regulatory standards. If our contract manufacturers cannot successfully manufacture material in compliance with these strict regulatory requirements, we and they will not be able to secure or maintain regulatory approval for their manufacturing facilities. In some cases, we also are dependent on our contract manufacturers to produce supplies in conformity to our specifications and maintain quality control and quality assurance practices and not to employ disqualified personnel. If the FDA or a comparable foreign regulatory authority does not approve the manufacturing facilities of our contract manufacturers, or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which could result in delays in, or adversely affect our ability to, develop or commercialize our product candidates. We and our contract manufacturers also may be subject to penalties and sanctions from the FDA and other regulatory authorities for any violations of applicable regulatory requirements. The USDA and EMA employ different regulatory standards than the FDA, so we may require multiple manufacturing processes and facilities for the same product candidate or any approved product.
The commercialization of any of our product candidates could be adversely affected if we are unable to secure sufficient quantities and quality of drug products in a timely manner.
The raw materials used to manufacture our current small molecule product candidates are generally readily available in commercial quantities from multiple suppliers, but we will be dependent upon our contract manufacturers to obtain these raw materials. If manufacturers are unable to do so as and when they are needed to supply our development and commercial needs, we will have no other means of producing our product candidates until they are able to do so or we or they procure alternative supplies of the API. If our third-party manufacturers suffer damage or destruction to their facilities or equipment, we may experience disruptions in supplies, or be unable to obtain supplies of product candidates on a timely basis. Any inability to secure sufficient quantities and quality of the API or other raw materials in our products candidates would adversely impact our development activities and commercialization efforts. In some cases, contract manufacturers may be reluctant to manufacture the API in pet therapeutics, because of regulatory or other concerns. This may make it more difficult for us to identify manufacturers needed to supply sufficient quantities of our product candidates for development.
Biologics manufacturing is difficult and costly, and may not be commercially viable.
There are no established sources of the active ingredients in our biologic product candidates, so we or our collaborators will be required to develop the manufacturing process, perform validation and in some cases establish new facilities to manufacture pet biologics. Manufacturing of pet biologics, apart from vaccines, is a relatively new field in which unanticipated difficulties or challenges could arise. Small changes in the manufacturing process can have significant impact on product quality, consistency and yield. Manufacturing biologics, especially in large quantities, is complex and may require the use of innovative technologies that we
26
may need to develop ourselves or in conjunction with third-party collaborators. Such manufacturing requires facilities specifically designed and validated for this purpose and sophisticated quality assurance and quality control procedures. Biologics are also usually costly to manufacture, because production usually requires the use of living organisms. Factors such as these may make it more technically challenging, time-consuming and expensive than we anticipate to manufacture biologics. Animal antibodies also must be manufactured at a sufficiently low cost that they are economically viable for us and for our customers.We are building a manufacturing plant for biologics manufacturing. There may be unanticipated issues in the construction and commissioning of the plant, including cost overruns, timeline delays, and other technical or regulatory issues. In addition, if we build a manufacturing plant and it is not utilized to full capacity, it may result in increased annual costs and increase in cost of goods. There is no assurance that we will be able to manufacture biologics at an economical cost, if at all.
Once completed, the facilities used to manufacture the biologics are subject to inspections by the FDA, USDA, and the EMA. If we cannot successfully manufacture material in compliance with these strict regulatory requirements, we will not be able to secure or maintain regulatory approval for the manufacturing facility. If the FDA or a comparable foreign regulatory authority does not approve the manufacturing facilities, or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which could result in delays in, or adversely affect our ability to, develop or commercialize our product candidates. We also may be subject to penalties and sanctions from the FDA and other regulatory authorities for any violations of applicable regulatory requirements. The USDA and EMA employ different regulatory standards than the FDA, so we may require multiple manufacturing processes and facilities for the same product candidate or any approved product.
If we are unable to establish sales capabilities on our own or through third parties, we may not be able to market and sell our current or future product candidates, if approved, and generate product revenue.
We currently have no proven sales, marketing or distribution capabilities. If our current or future product candidates receive regulatory approval, we expect to establish a direct sales organization in the United States and to utilize distributors to commercialize our products, which will be expensive and time-consuming. In jurisdictions outside of the United States we intend to utilize companies with an established commercial presence to market our products in those jurisdictions, but we may be unable to enter into such arrangements on acceptable terms, if at all. We have no prior experience in the marketing, sale and distribution of pet therapeutics or other products, and there are significant risks involved in building and managing a sales organization, including our potential inability to hire, retain and motivate qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively oversee a geographically-dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities and entry into adequate arrangements with distributors would adversely impact the commercialization of our product candidates. If we are not successful in commercializing any of our current or future product candidates, either on our own or through one or more distributors, we may never generate significant revenue and may continue to incur significant losses, which would adversely affect our financial condition and results of operations.
If we are not successful in identifying, developing, and commercializing additional product candidates, our ability to expand our business and achieve our strategic objectives would be impaired.
A key element of our strategy is to identify, develop and commercialize a portfolio of products to serve the emerging pet therapeutics market. We expect to identify additional potential pet therapeutic product candidates from targets, molecules, and compounds discovered or developed as part of human biopharmaceutical research. Ideally, we try to identify product candidates that are free from any intellectual property rights of others. If we are unable to identify human health-generated molecules and compounds to conduct research and development, our ability to develop new products could be limited. In addition, we may in the future enter into license agreements with third parties to provide us with rights to the compounds for purposes of our business. Even if we enter into these arrangements, we may not be able to maintain these relationships or establish new ones in the future on acceptable terms, or at all.
27
Even if we successfully identify or license potential product candidates, we may still fail to yield product candidates for development and commercialization for many reasons, including the following:
• | product candidates we develop may be covered by third parties’ patents or other exclusive rights unknown to us; |
• | a product candidate may on further study be shown to have harmful side effects in pets or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
• | a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; |
• | a product candidate may not be accepted as safe and effective by veterinarians, pet owners and the pet therapeutic community; and |
• | competitors may develop alternatives that render our product candidates obsolete. |
Failure to identify further product candidates ultimately suitable for development and commercialization would have an adverse impact on our growth strategy and future business prospects.
Changes in distribution channels for pet therapeutics may make it more difficult or expensive to distribute our products.
In the United States, pet owners typically purchase their pet therapeutics from their local veterinarians who also prescribe such therapeutics. There is a trend, however, toward increased purchases of pet therapeutics from Internet-based retailers, “big-box” retail stores and other over-the-counter distribution channels, which follows a significant shift in recent years away from the traditional veterinarian distribution channel in the sale of parasiticides and vaccines. It is also possible that pet owners may come to rely increasingly on internet-based animal health information rather than on their veterinarians. We currently expect to market our pet therapeutics directly to veterinarians, so any reduced reliance on veterinarians by pet owners could materially adversely affect our business and prospects. Pet owners also may substitute human health products for pet therapeutics if the human health products are less expensive or more readily available, which substitution also could adversely affect our business.
Legislation has been or may be proposed in the United States or abroad that would require veterinarians to provide pet owners with written prescriptions and disclosures that the pet owner has the right to fill the prescriptions through other means. If enacted, such legislation could lead to a reduction in the number of pet owners who purchase their pet therapeutics directly from veterinarians, which also could adversely affect our business.
While most of our biologic products will be delivered by injection and therefore may be insulated to a degree from competition from non-veterinary dispensing, for our small molecule products, over time, these and other competitive conditions may make us reliant upon Internet-based retailers, “big-box” retail stores or other over-the-counter distribution channels, for which we have no current or planned business relationships, to sell our pet products. Any of these events could materially adversely affect our business and prospects or require us to dramatically change our marketing and distribution strategies, which may not be feasible or successful.
Consolidation of our customers could negatively affect the pricing of our products.
Veterinarians will be our primary customers for any approved products. In recent years, there has been a trend towards the consolidation of veterinary clinics and animal hospitals. If this trend continues, these large clinics and hospitals could attempt to leverage their buying power to obtain favorable pricing from us and other pet therapeutics companies. Any resulting downward pressure on the prices of any of our approved products could have a material adverse effect on our results of operations and financial condition.
28
We will need to increase the size of our organization and may not successfully manage our growth.
As of February 28, 2018, we had 76 employees, and our management systems currently in place may not be adequate to support our future growth, if any. Our ability to manage our growth effectively will require us to hire, train, retain, manage and motivate additional employees and to implement and improve our operational, financial and management systems. These demands also may require the hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. If we fail to expand and enhance our operational, financial and management systems in conjunction with our potential future growth, it could have a material adverse effect on our business, financial condition and results of operations.
Our research and development relies on evaluations in animals, which is controversial and may become subject to bans or additional regulations.
The evaluation of our product candidates in target animals is required to develop and commercialize our product candidates. Although our animal testing will be subject to GLP and GCP requirements, as applicable, animal testing in the human pharmaceutical industry and in other industries has been the subject of controversy and adverse publicity. Some organizations and individuals have sought to ban animal testing or encourage the adoption of additional regulations applicable to animal testing. To the extent that such bans or regulations are imposed, our research and development activities, and by extension our operating results and financial condition, could be materially adversely affected. In addition, negative publicity about animal practices by us or in our industry could harm our reputation among potential customers for our products.
If approved, our product candidates may be marketed in the United States only in the target animals and for the indications for which they are approved, and if we want to expand the approved animals or indications, we will need to obtain additional FDA or USDA approvals, which may not be granted.
If our product candidates are approved by regulatory authorities, we may market or advertise them only in the specific species and for treatment of the specific indications for which they were approved, which could limit use of the products by veterinarians and pet owners. We intend to develop, promote and commercialize one or more of our current product candidates for other animals and new treatment indications in the future, but there is no assurance whether or at what additional time and expense we will be able to do so. If we do not obtain marketing approvals for other animals or for new indications, our ability to expand our business may be adversely affected.
Use of a drug outside its cleared or approved indications in the animal context is known as extra-label use. Under the Animal Medicinal Drug Use Clarification Act of 1994, or AMDUCA, veterinarians are permitted to prescribe extra-label uses of certain approved animal drugs and approved human drugs for animals under certain conditions. Thus, although veterinarians may in the future prescribe and use human-approved products or our products for extra-label uses, we may not promote our products for extra-label uses. If the FDA determines that any of our marketing activities constitute promotion of an extra-label use, it could subject us to regulatory enforcement, which could have an adverse impact on our reputation and potential liability to us.
The commercial potential of a product candidate in development is difficult to predict. The market for our product candidates, or for the pet therapeutics industry as a whole, is uncertain and may be smaller than we anticipate, which could significantly and negatively impact our revenue, results of operations and financial condition.
It is very difficult to estimate the commercial potential of any of our product candidates because of the emerging nature of our industry as a whole. The pet therapeutics market continues to evolve and it is difficult to predict the market potential for what we believe to be the unmet medical needs of pets. The market will depend on important factors such as safety and efficacy compared to other available treatments, including potential human generic therapeutic alternatives with similar efficacy profiles, changing standards of care,
29
preferences of veterinarians, the willingness of pet owners to pay for such products, and the availability of competitive alternatives that may emerge either during the product development process or after commercial introduction. If the market potential for our product candidates is less than we anticipate due to one or more of these factors, it could negatively impact our business, financial condition and results of operations. Further, the willingness of pet owners to pay for our product candidates, if approved, may be less than we anticipate, and may be negatively affected by overall economic conditions. The current penetration of pet insurance in the United States is low, pet owners are likely to have to pay for our products, if at all, out-of-pocket, and pet owners may not be willing or able to pay for any approved products of ours.
We may acquire other businesses or form joint ventures that may be unsuccessful and could adversely dilute your ownership of our company.
As part of our business strategy, we may pursue acquisitions of other complementary assets and businesses and may also pursue strategic alliances. Our company has no experience in acquiring other assets or businesses and has limited experience in forming such alliances. We may not be able to successfully integrate any acquisitions into our existing business, and we could assume unknown or contingent liabilities or become subject to possible stockholder claims in connection with any related-party or third-party acquisitions or other transactions. We also could experience adverse effects on our reported results of operations from acquisition-related charges, amortization of acquired technology and other intangibles and impairment charges relating to write-offs of goodwill and other intangible assets from time to time following an acquisition. Integration of an acquired company requires management resources that otherwise would be available for ongoing development of our existing business. We may not realize the anticipated benefits of any acquisition, technology license or strategic alliance.
To finance future acquisitions, we may choose to issue shares of our common stock as consideration, which would dilute your ownership interest in us. Alternatively, it may be necessary for us to raise additional funds through public or private financings. Additional funds may not be available on terms that are favorable to us and, in the case of equity financings, may result in dilution to our stockholders.
Risks Related to Intellectual Property
Our commercial success will depend, in part, on obtaining and maintaining patent protection for our products.
In so far as our business strategy is to develop successful human drugs and biologics for veterinary use, our ability to obtain a proprietary intellectual property position for our products is uncertain. We do not have any issued patents for our lead product candidates at this time. However, we have filed patent applications covering various aspects of our drug and biological candidates in animals. Our patent applications may never result in the issuance of patents, and/or patents issued to us may be dominated by the patents of third parties, including, for example, patents issued to analogous human drug or biological compositions and their usages. Furthermore, even if they are unchallenged by third parties, our patents, if issued, may not adequately protect our intellectual property or prevent others from designing around their claims. In order to commercialize our drug and biological candidates in one or more species, we could be required to enter into third party licenses or, if a license is not available on terms that we consider reasonable, we could be required to cease development or commercialization of one or more of our drug or biologic products or product candidates. Thus, if we cannot obtain ownership of issued patents covering our product candidates, our business and prospects would be adversely affected.
It is possible that no patents will issue to us to cover our approved products, and/ or that we will have little to no commercial protection against competing products. In such cases, we would then rely solely on other forms of exclusivity, such as regulatory exclusivity provided by the Federal Food, Drug and Cosmetic Act, if available, which may provide less protection to our competitive position.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that issue. On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act
30
includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art, may affect patent litigation, and switch the U.S. patent system from a “first-to-invent” system to a “first-to-file” system. Under a “first-to-file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor had made the invention earlier. The USPTO recently developed new regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first-to-file provisions, only became effective on March 16, 2013. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of any patents that issue, all of which could have a material adverse effect on our business and financial condition.
We may become subject to third parties’ claims alleging infringement of patents and proprietary rights or priority of invention, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of our current or future product candidates.
There has been substantial litigation regarding patents and other intellectual property rights in the field of therapeutics, as well as patent challenge proceedings, including interference and administrative law proceedings before the United States Patent and Trademark Office, or the USPTO, and oppositions and other comparable proceedings in foreign jurisdictions. Under U.S. patent reform laws, new procedures, including inter partes review and post-grant review, were implemented as of March 16, 2013, and the implementation of such reform laws presents uncertainty regarding the outcome of any challenges to our future patents, if any. We are aware of several issued patents and pending patent applications with claims directed to long-acting or extended-release pharmaceutical formulations and uses of the same small molecules as in some of our small molecule product candidates, and other patents and pending patent applications with claims directed to pharmaceutical formulations and use of human biologics conceptually similar to some of our biologics product candidates. There also may be other patents already issued of which we are unaware that might be infringed by one of our current or future product candidates. Because patent applications can take many years to issue and may be confidential for eighteen months or more after filing, there may be applications now pending of which we are unaware and which may later result in issued patents that may be infringed by our current or future product candidates. There is no assurance that our current or future product candidates will not infringe these or other existing or future third-party patents. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents.
To the extent we become subject to future third-party claims against us or our collaborators, we could incur substantial expenses and, if any such claims are successful, we could be liable to pay substantial damages, including treble damages and attorney’s fees if we or our collaborators are found to be willfully infringing a third-party’s patents. If a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product candidate that is the subject of the suit. Even if we are successful in defending such claims, infringement and other intellectual property claims can be expensive and time-consuming to litigate and divert management’s attention from our business and operations. As a result of or in order to avoid potential patent infringement claims, we or our collaborators may be compelled to seek a license from a third party for which we would be required to pay license fees or royalties, or both. Moreover, these licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain such a license, the rights may be nonexclusive, which could allow our competitors access to the same intellectual property. Any of these events could harm our business and prospects.
In addition to possible infringement claims against us, we may be subject to third-party preissuance submission of prior art to the USPTO, or become involved in opposition, derivation, reexamination, inter partes review, post-grant review, or other patent office proceedings or litigation in the United States or elsewhere, challenging our patent rights or the patent rights of others. If third parties have prepared and filed patent applications in the United States that also claim technology to which we have rights, we may have to
31
participate in interference proceedings in the USPTO to determine the priority of invention. We may also become involved in similar opposition proceedings in the European Patent Office or similar offices in other jurisdictions regarding our intellectual property rights with respect to our products and technology. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our future patent rights, if any, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights.
If our efforts to protect the proprietary nature of the intellectual property related to any of our current or future product candidates are not adequate, we may not be able to compete effectively in our market.
We intend to rely upon a combination of regulatory exclusivity periods, patents, trade secret protection, confidentiality agreements, and license agreements to protect the intellectual property related to our current product candidates and our development programs.
Composition-of-matter patents on the active ingredients in pharmaceutical products, including pet therapeutics, are generally considered to be the strongest form of intellectual property protection, since such patents provide protection without regard to any particular method of use or manufacture. Because all of our current small molecule product candidates are based on generic human drugs, we do not have composition-of-matter patents for the active pharmaceutical ingredient (API) in our small molecule product candidates, and there is little, if any, such composition-of-matter patent protection available for the API in such product candidates. Moreover, we cannot be certain that the claims in our patent applications covering composition-of-matter of our biologics product candidates will be considered patentable by the USPTO and courts in the United States, or by the patent offices and courts in foreign countries.
Method-of-use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from developing or marketing an identical product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted indications for which we may obtain patents, veterinarians may recommend that pet owners use these products extra-label, or pet owners may do so themselves. Although extra-label use may infringe or contribute to the infringement of method-of-use patents, the practice is common and such infringement is difficult to prevent or prosecute.
If the breadth or strength of protection provided by any patent applications or future patents we may own, in-license, or pursue with respect to any of our current or future product candidates is threatened, it could threaten our ability to commercialize any of our current or future product candidates. Further, if we encounter delays in our development efforts, the period of time during which we could market any of our current or future product candidates under any patent protection we obtain would be reduced.
Even where laws provide protection or we are able to obtain patents, costly and time-consuming litigation may be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have.
We also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable or for which we have not filed patent applications, processes for which patents are difficult to enforce and other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we require all of our employees to assign their inventions to us, and endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, or that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will
32
not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
We may be involved in lawsuits to protect or enforce any future patents issued to us, which could be expensive, time-consuming and unsuccessful.
Competitors may infringe any patents that may issue to us, or any patents that we may license. To counter infringement or unauthorized use of any patents we may obtain, we may be required to file infringement claims, which can be expensive and time-consuming to litigate. In addition, if we or one of our future collaborators were to initiate legal proceedings against a third party to enforce a patent covering our current product candidates, or one of our future products, the defendant could counterclaim that the patent is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a materially misleading statement, during prosecution. Third parties may also raise similar claims before the USPTO, even outside the context of litigation. The outcome following legal assertions of invalidity and unenforceability is unpredictable. We cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of any future patent protection on our current or future product candidates. Such a loss of patent protection could have a material adverse impact on our business.
Litigation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be unsuccessful, it could have an adverse effect on the price of our common stock. Finally, we may not be able to prevent, alone or with the support of our licensors, misappropriation of our trade secrets or confidential information, particularly in countries where the laws may not protect those rights as fully as in the United States.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable
33
ways that would weaken our ability to obtain new patents or to enforce patents that we might obtain in the future.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on product candidates throughout the world would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we may obtain patent protection, but where patent enforcement is not as strong as that in the United States. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and any future patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our future patents, if any, or marketing of competing products in violation of our proprietary rights generally. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. Proceedings to enforce our future patent rights, if any, in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
We have one registered US trademark and one allowed US trademark for our current product candidate KINDLET, as well as one registered trademark for KINDLET in Europe. Additionally, we have five other registered trademarks outside the US, nine other trademark applications pending in the US, two pending European trademark applications, one pending Japanese trademark application, and three pending Canadian trademark applications. We have no registered trademarks for our company name or for our other current product candidates in the United States or any other countries, and failure to obtain those registrations could adversely affect our business.
Six of our US pending trademark applications have received Notices of Allowance and are awaiting their first use in commerce prior to Registration. One US trademark application is currently being examined, one European trademark application is currently being examined, and the remaining two pending US trademark applications, the three Canadian trademark applications, the European trademark application, and the Japanese trademark application are awaiting examination. Accordingly, while various of our pending trademark applications have been allowed but not yet registered, various other of our pending US and foreign trademark applications have neither been allowed nor registered. During trademark registration proceedings, we may receive rejections. If so, we will have an opportunity to respond, but we may be unable to overcome such rejections. In addition, USPTO and comparable agencies in many foreign jurisdictions may permit third parties to oppose pending trademark applications and to seek to cancel registered trademarks. If opposition or cancellation proceedings are filed against any of our trademark applications or any registered trademarks, our trademarks may not survive such proceedings. Moreover, any name we propose to use with our product candidates in the United States must be approved by the FDA or the USDA, regardless of whether we have registered or applied to register as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA or the USDA objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA or USDA.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
34
We have received confidential and proprietary information from third parties. In addition, we employ individuals who were previously employed at other biotechnology, pharmaceutical or animal health companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise improperly used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against any such claims. Even if we are successful in defending against any such claims, such litigation could result in substantial cost and be a distraction to our management and employees.
Risks Related to Government Regulation
Even if we receive regulatory approval for any of our current or future product candidates, we will be subject to ongoing FDA, USDA, and EMA obligations and continued regulatory review, which may result in significant additional expense. Additionally, any product candidates, if approved, will be subject to labeling and manufacturing requirements and could be subject to other restrictions. Failure to comply with these regulatory requirements or the occurrence of unanticipated problems with our products could result in significant penalties.
If the FDA, USDA, or EMA approves any of our current or future product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, establishment registration, and product listing, as well as continued compliance with GMP, GLP and GCP for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
• | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary product recalls; |
• | fines, warning letters or holds on target animal studies; |
• | refusal by the FDA, USDA, or EMA to approve pending applications or supplements to approved applications filed by us or our strategic collaborators, or suspension or revocation of product license approvals; |
• | product seizure or detention, or refusal to permit the import or export of products; and |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA, USDA, or EMA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained and we may not achieve or sustain profitability, which would adversely affect our business.
If approved, any of our current or future products may cause or contribute to adverse medical events that we are required to report to regulatory authorities and, if we fail to do so, we could be subject to sanctions that would materially harm our business.
If we are successful in commercializing any of our current or future product candidates, at least certain regulatory authorities will require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail
35
to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the regulatory authorities could take action including criminal prosecution, seizure of our products or delay in approval or clearance of future products.
Legislative or regulatory reforms with respect to pet therapeutics may make it more difficult and costly for us to obtain regulatory clearance or approval of any of our current or future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress or EU that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, FDA and USDA regulations and guidance are often revised or reinterpreted by the FDA and USDA in ways that may significantly affect our business and our products. Similar changes in laws or regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of any of our current or future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
• | changes to manufacturing methods; |
• | recall, replacement, or discontinuance of certain products; and |
• | additional record keeping. |
Each of these would likely entail substantial time and cost and could materially harm our financial results. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products would harm our business, financial condition, and results of operations.
Certain of our product candidates currently in development may be classified as controlled substances, the manufacture, use, sale, importation, exportation, and distribution of which are subject to additional regulation by state, federal, and foreign law enforcement and other regulatory agencies.
Certain of our product candidates may be subject to regulation as controlled substances under the federal Controlled Substances Act of 1970, or CSA, and regulations of the U.S. Drug Enforcement Administration, or DEA. The DEA regulates controlled substances as Schedule I, II, III, IV or V substances. Schedule I substances by definition have no established medicinal use and may not be marketed or sold in the United States. An animal drug product may be listed as Schedule II, III, IV or V, with Schedule II substances considered to present the highest risk of abuse and Schedule V substances the lowest relative risk of abuse among such substances.
Various states also independently regulate controlled substances. Though state controlled substances laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule drugs as well. While some states automatically schedule a drug when the DEA does so, in other states there must be rulemaking or a legislative action. State scheduling may delay commercial sale of any controlled substance drug product for which we obtain federal regulatory approval and adverse scheduling could impair the commercial attractiveness of such product. We would also be required to obtain separate state registrations in order to be able to obtain, handle and distribute controlled substances for target animal studies, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions from the states in addition to those from the DEA or otherwise arising under federal law.
For any of our product candidates classified as controlled substances, we and our suppliers, manufacturers, contractors, customers and distributors will be required to obtain and maintain applicable registrations from state, federal and foreign law enforcement and regulatory agencies and comply with state,
36
federal and foreign laws and regulations regarding the manufacture, use, sale, importation, exportation and distribution of controlled substances. There is a risk that DEA regulations may limit the supply of the compounds used in pivotal trials of our product candidates, and, in the future, the ability to produce and distribute our products in the volume needed to meet commercial demand.
Regulations associated with controlled substances govern manufacturing, labeling, packaging, testing, dispensing, production and procurement quotas, recordkeeping, reporting, handling, shipment and disposal. These regulations increase the personnel needs and the expense associated with development and commercialization of product candidates containing controlled substances. The DEA and some states conduct periodic inspections of registered establishments that handle controlled substances. Failure to obtain and maintain required registrations or comply with any applicable regulations could delay or preclude us from developing and commercializing our product candidates containing controlled substances and subject us to enforcement action. The DEA may seek civil penalties, refuse to renew necessary registrations or initiate proceedings to revoke those registrations. In some circumstances, violations could lead to criminal proceedings. Because of their restrictive nature, these regulations could limit commercialization of any of our product candidates that are classified as controlled substances.
Risks Related to Our Common Stock
The price of our common stock could be subject to volatility related or unrelated to our operations.
Our stock prices and the market prices for securities of biotechnology companies in general have been highly volatile, with recent significant price and volume fluctuations, and may continue to be highly volatile in the future. Since our initial public offering in December 2013, the trading price of our common stock has ranged from a low of $2.90 to a high of $26.99. The trading price may continue to be subject to wide fluctuations in response to various factors, some of which are beyond our control. These factors include those discussed previously in this “Risk Factors” section of this Annual Report on Form 10-K and others, such as:
• | any delays in, or suspension or failure of, our current and future studies; |
• | announcements of regulatory approval or disapproval of any of our current or future product candidates or of regulatory actions affecting us or our industry; |
• | delays in the commercialization of our current or future product candidates; |
• | manufacturing and supply issues related to our development programs and commercialization of our current or future product candidates; |
• | quarterly variations in our results of operations or those of our competitors; |
• | changes in our earnings estimates or recommendations by securities analysts or adverse publicity regarding us or our product candidates; |
• | announcements by us or our competitors of new product candidates, significant contracts, commercial relationships, acquisitions or capital commitments; |
• | announcements relating to future development or license agreements including termination of such agreements; |
• | adverse developments with respect to our intellectual property rights or those of our principal collaborators; |
• | commencement of litigation involving us or our competitors; |
• | any major changes in our board of directors or management; |
37
• | new legislation in the United States relating to the prescription, sale, distribution or pricing of pet therapeutics; |
• | product liability claims, other litigation or public concern about the safety of our product candidates or future products; |
• | market conditions in the animal health industry, in general, or in the pet therapeutics sector, in particular, including performance of our competitors; and |
• | general economic conditions in the United States and abroad. |
In addition, the stock market, in general, or the market for stocks in our industry, in particular, may experience broad market fluctuations, which may adversely affect the market price or liquidity of our common stock. Any sudden decline in the market price of our common stock could trigger securities class-action lawsuits against us. If any of our stockholders were to bring such a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management would be diverted from our business and operations. We also could be subject to damages claims if we are found to be at fault in connection with a decline in our stock price.
If securities or industry analysts do not publish research or reports about our company, or if they issue adverse or misleading opinions regarding us or our stock, our stock price and trading volume could decline.
Although we have research coverage by securities and industry analysts, if coverage is not maintained, the market price for our stock may be adversely affected. Our stock price also may decline if any analyst who covers us issues an adverse or erroneous opinion regarding us, our business model, our intellectual property or our stock performance, or if our target animal studies and operating results fail to meet analysts’ expectations. If one or more analysts cease coverage of us or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline and possibly adversely affect our ability to engage in future financings.
Our principal stockholders and management own a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
As of December 31, 2017, our executive officers, directors, holders of 5% or more of our capital stock and their respective affiliates beneficially own in the aggregate approximately 42% of our outstanding shares of common stock, excluding shares they may acquire upon exercise of stock options they hold. As a result of their stock ownership, these stockholders may have the ability to influence our management and policies, and will be able to significantly affect the outcome of matters requiring stockholder approval such as elections of directors, amendments of our organizational documents or approvals of any merger, sale of assets or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may feel are in your best interest as one of our stockholders.
If we fail to maintain an effective system of disclosure controls and internal control over financial reporting, our ability to produce timely and accurate financial statements or comply with applicable regulations could be impaired.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, and the listing standards of the NASDAQ Stock Market. We expect that the requirements of these rules and regulations will continue to increase our legal, accounting and financial compliance costs, make some activities more difficult, time consuming and costly, and place significant strain on our personnel, systems and resources.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective disclosure controls and procedures and internal control over financial reporting. We are continuing to develop and refine our
38
disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we will file with the SEC is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms, and that information required to be disclosed in reports under the Exchange Act is accumulated and communicated to our principal executive and financial officers. We are also continuing to improve our internal control over financial reporting. In order to maintain and improve the effectiveness of our disclosure controls and procedures and internal control over financial reporting, we have expended, and anticipate that we will continue to expend, significant resources, including accounting-related costs and significant management oversight.
Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business. Further, weaknesses in our disclosure controls or our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls, or any difficulties encountered in their implementation or improvement, could harm our operating results or cause us to fail to meet our reporting obligations and may result in a restatement of our financial statements for prior periods. Any failure to implement and maintain effective internal control over financial reporting also could adversely affect the results of management evaluations of our internal control over financial reporting that we are required to include in our periodic reports that we file with the SEC. Ineffective disclosure controls and procedures and internal control over financial reporting could also cause investors to lose confidence in our reported financial and other information, which would likely have a negative effect on the trading price of our common stock. In addition, if we are unable to continue to meet these requirements, we may not be able to remain listed on the NASDAQ Stock Market.
Any failure to maintain effective disclosure controls and internal control over financial reporting could have a material and adverse effect on our business and operating results, and cause a decline in the price of our common stock. So long as we remain an emerging growth company, we will not be required to obtain an attestation report from our independent registered public accounting firm regarding our internal control over financial reporting, which might make it more difficult for us to identify weaknesses in our internal control over financial reporting.
Provisions in our charter documents and under Delaware law could discourage a takeover that stockholders may consider favorable and may lead to entrenchment of management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent changes in control or changes in our management without the consent of our board of directors. These provisions include the following:
• | a classified board of directors with three-year staggered terms, which may delay the ability of stockholders to change the membership of a majority of our board of directors; |
• | no cumulative voting in the election of directors, which limits the ability of minority stockholders to elect director candidates; |
• | the exclusive right of our board of directors to elect a director to fill a vacancy created by the expansion of the board of directors or the resignation, death or removal of a director, which prevents stockholders from being able to fill vacancies on our board of directors; |
• | the ability of our board of directors to authorize the issuance of shares of preferred stock and to determine the terms of those shares, including preferences and voting rights, without stockholder approval, which could adversely affect the rights of our common stockholders or be used to deter a possible acquisition of our company; |
• | the ability of our board of directors to alter our bylaws without obtaining stockholder approval; |
39
• | the required approval of the holders of at least two-thirds of the shares entitled to vote at an election of directors to adopt, amend or repeal our bylaws or repeal the provisions of our amended and restated certificate of incorporation regarding the election and removal of directors; |
• | a prohibition on stockholder action by written consent, which forces stockholder action to be taken at an annual or special meeting of our stockholders; |
• | the requirement that a special meeting of stockholders may be called only by the chairman of the board of directors, the chief executive officer, the president or the board of directors, which may delay the ability of our stockholders to force consideration of a proposal or to take action, including the removal of directors; and |
• | advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors or to propose matters to be acted upon at a stockholders’ meeting, which may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of us. |
These provisions could inhibit or prevent possible transactions that some stockholders may consider attractive.
We are also subject to the anti-takeover provisions contained in Section 203 of the Delaware General Corporation Law. Under Section 203, a corporation generally may not engage in a business combination with any holder of 15% or more of its capital stock unless the holder has held the stock for three years or, among other exceptions, the board of directors has approved the transaction.
Our stockholder rights agreement could discourage a takeover that stockholders may consider favorable and may lead to an entrenchment of management.
On May 19, 2017, our board of directors approved and adopted a rights agreement with American Stock Transfer & Trust Company, LLC, as rights agent, and, on July 24, 2017, our stockholders approved the adoption of the rights agreement. The rights agreement is intended to protect our stockholders from coercive or otherwise unfair proposals to acquire control of KindredBio by significantly diluting the ownership interest of any person who acquires at least 20% of our outstanding common stock by providing all other stockholders with the right to acquire additional shares of our preferred stock or common stock at a significant discount. Although the rights agreement is intended to encourage an acquiring person to negotiate a proposed merger or other business combination with our board of directors and management, it could discourage a takeover transaction that stockholders may consider favorable and may lead to an entrenchment of management.
Our amended and restated by-laws designate the Court of Chancery of the State of Delaware as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or other employees.
Our amended and restated by-laws provide that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware will be the sole and exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by any director, officer or other employee to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the Delaware General Corporation Law, or (iv) any action asserting a claim that is governed by the internal affairs doctrine. Any person purchasing or otherwise acquiring any interest in any shares of our capital stock shall be deemed to have notice of and to have consented to this provision of our amended and restated by-laws. This choice-of-forum provision may limit our stockholders’ ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits. Alternatively, if a court were to find this provision of our amended and restated by-laws inapplicable or unenforceable with respect to one or more of the specified types
40
of actions or proceedings, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business and financial condition.
We do not intend to pay dividends on our common stock, and the ability of investors in our common stock to achieve a return on their investment will depend on appreciation in the market price of our common stock.
We currently intend to invest any future earnings to fund our growth and not to pay any cash dividends on our common stock. Since we do not intend to pay dividends, the ability of investors in our common stock to receive a return on their investment will depend on any appreciation in the market price of our common stock. There is no assurance that our common stock will appreciate in price.
We are an “emerging growth company” and we cannot be certain if the reduced disclosure requirements applicable to “emerging growth companies” will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we may take advantage of certain exemptions and relief from various reporting requirements that are applicable to other public companies that are not “emerging growth companies.” In particular, while we are an “emerging growth company” (i) we will not be required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act, (ii) we will be exempt from any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotations or a supplement to the auditor’s report on financial statements, (iii) we will be subject to reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and (iv) we will not be required to hold nonbinding advisory votes on executive compensation or stockholder approval of any golden parachute payments not previously approved. In addition, the JOBS Act provides that an emerging growth company can delay its adoption of any new or revised accounting standards, but we have irrevocably elected not to avail ourselves of this exemption and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
We may remain an “emerging growth company” until as late as December 31, 2018 (the fiscal year-end following the fifth anniversary of the completion of our initial public offering), though we may cease to be an “emerging growth company” earlier under certain circumstances, including (i) if the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30, in which case we would cease to be an “emerging growth company” as of the following December 31, or (ii) if our gross revenue exceeds $1 billion in any fiscal year.
The exact implications of the JOBS Act are still subject to interpretations and guidance by the SEC and other regulatory agencies, and we cannot assure you that we will be able to take advantage of all of the benefits of the JOBS Act. In addition, investors may find our common stock less attractive if we rely on the exemptions and relief granted by the JOBS Act. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may decline and/or become more volatile.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes (such as research and development tax credits) to offset its post-change income and taxes may be limited. In general, an “ownership change” occurs if there is a cumulative change in our ownership by “5% shareholders” that exceeds 50 percentage points over a rolling three-year period. Similar rules may apply under state tax laws. If we experience one or more ownership changes as a result of future transactions in our stock, we may be limited in our ability to use our net operating loss carryforwards and other tax assets to reduce taxes owed on the net taxable income that we earn. Any such limitations on the ability to use our net operating loss carryforwards and other tax assets could potentially result in increased future tax liability to us.
41
ITEM 1B. | UNRESOLVED STAFF COMMENTS. |
Not Applicable.
ITEM 2. | PROPERTIES. |
Our corporate headquarters are located in Burlingame, California. In April 2014, we entered into new non-cancelable operating leases for 2,145 square feet of laboratory space through May 2017 and 6,900 square feet of office space through November 2017. In January, August and November 2015, we amended our original operating lease for laboratory space to expand the facility with an additional 2,431 square feet, 131 square feet and 123 square feet, respectively, of manufacturing space through May 2017. In August 2015, we entered into a new non-cancelable operating lease for 3,126 square feet of office space in San Diego, California through September 2019. In February and October 2016, we further amended our original operating lease for laboratory space to further expand the facility with an additional 3,599 square feet and 2,326 square feet, respectively, of quality control and laboratory space through May 2017. In February 2017, we further amended our existing lease for an expansion of an additional 721 square feet of laboratory space. Commencing on June 1, 2017, the non-cancelable operating lease for the entire existing laboratory space of a total 11,476 square feet is extended for another five years through May 2022. In April 2017, we renewed our headquarters office lease for 6,900 square feet of office space in Burlingame, California through November 30, 2020 and in June 2017, we amended the lease with an additional 1,190 square feet of office space through November 30, 2020.
On June 21, 2017, we entered into a purchase agreement with Strategic Veterinary Pharmaceuticals, Inc. ("SVP") for the purchase of an approximately 180,000 sq. ft. biologics plant ("the Plant") with clean rooms, utility, equipment, and related quality documentation suitable for small molecule and biologics manufacturing, that is located in Elwood, Kansas. The purchase was finalized on August 7, 2017 upon completion of the diligence period and satisfaction of the conditions of escrow. The Plant was purchased for $3,750,000, which includes approximately eight acres of land located at 1411 Oak Street, Elwood, Kansas, all improvements located at the Plant, and all personal property and intangible property owned by SVP and located at the Plant or used in connection with the operation of the Plant.
ITEM 3. | LEGAL PROCEEDINGS. |
We are not currently a party to any material legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES.
Not applicable.
42
PART II
ITEM 5. | MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES. |
Market Information
Since December 12, 2013, our common stock has been traded on The NASDAQ Capital Market under the symbol “KIN.” Prior to December 12, 2013, there was no public trading market for our common stock. The following table sets forth the high and low sale prices for our common stock for the periods indicated as reported on The NASDAQ Capital Market:
High | Low | ||||||
Fiscal Year 2017: | |||||||
Quarter ended March 31, 2017 | $7.45 | $4.35 | |||||
Quarter ended June 30, 2017 | $9.65 | $6.50 | |||||
Quarter ended September 30, 2017 | $8.95 | $6.55 | |||||
Quarter ended December 31, 2017 | $9.80 | $6.80 | |||||
Fiscal Year 2016: | |||||||
Quarter ended March 31, 2016 | $4.13 | $2.90 | |||||
Quarter ended June 30, 2016 | $4.29 | $3.12 | |||||
Quarter ended September 30, 2016 | $5.43 | $3.53 | |||||
Quarter ended December 31, 2016 | $5.65 | $3.90 | |||||
Common Stock Information
As of February 28, 2018, there were 28,189,081 outstanding shares of our common stock outstanding held of record by approximately 24 holders.
Dividends
We have never declared or paid any cash dividends on our capital stock. We intend to retain any future earnings and do not expect to pay any dividends in the foreseeable future. Any future determination to declare cash dividends will be made at the discretion of our board of directors, subject to applicable laws, and will depend on a number of factors, including our financial condition, results of operations, capital requirements, contractual restrictions, general business conditions and other factors that our board of directors may deem relevant.
Equity Compensation Plan Information
The following table sets forth certain information as of December 31, 2017 regarding securities authorized for issuance under our equity compensation plans:
43
Plan Category | Number of securities to be issued upon exercise of outstanding options, warrants and rights | Weighted-average exercise price of outstanding options, warrants and rights | Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) | ||||||
(a) | (b) | (c) | |||||||
Equity compensation plans approved by stockholders: | |||||||||
2012 Equity Incentive Plan (terminated) | 3,315,200 | $6.60 | — | ||||||
2016 Equity Incentive Plan | 1,258,425 | $6.47 | 1,488,450 | ||||||
Equity compensation plans not approved by stockholders | — | — | — | ||||||
Total | 4,573,625 | $6.57 | 1,488,450 | ||||||
Stock Performance Graph
The following line graph presentation compares cumulative total stockholder returns of Kindred Biosciences, Inc. with The NASDAQ Stock Market Index and The NASDAQ Biotechnology Index (the “Peer Index”) for the period from December 12, 2013 (the date that our common stock commenced trading on the NASDAQ Capital Market) to December 31, 2017. The graph and table assume that $100 was invested in each of our common stock, The NASDAQ Stock Market Index and the Peer Index on December 12, 2013, and that all dividends were reinvested. The returns shown are based on historical results and are not intended to suggest future performance. The graph shall not be deemed “soliciting material” or to be “filed” with the SEC for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liabilities under that Section, and shall not be deemed to be incorporated by reference into any of our filings of under the Securities Act of 1933, as amended, or the Exchange Act.
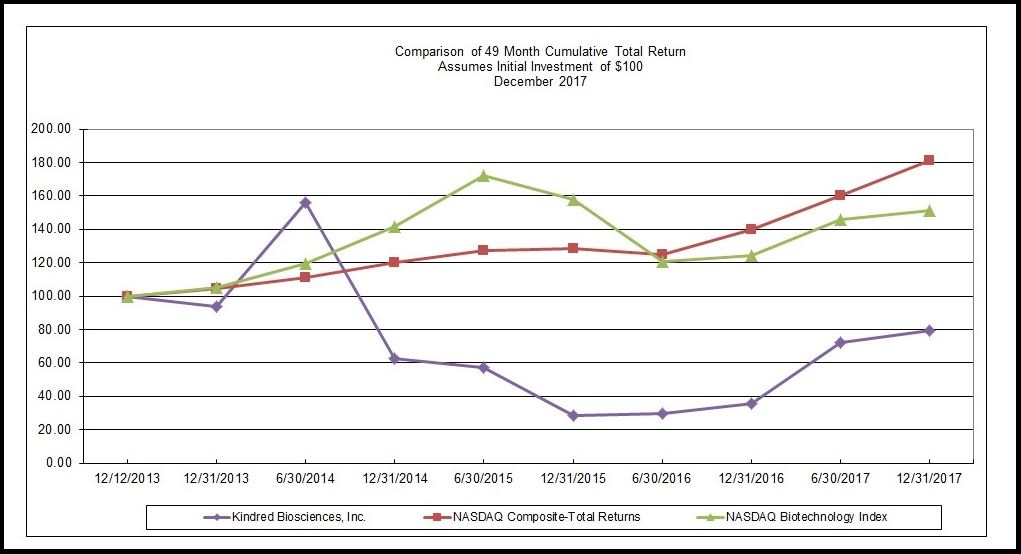
44
12/12/2013 | 12/31/2013 | 6/30/2014 | 12/31/2014 | 6/30/2015 | 12/31/2015 | 6/30/2016 | 12/31/2016 | 6/30/2017 | 12/31/2017 | |||||||||||||||||||
Kindred Biosciences, Inc. | 100.00 | 93.47 | 155.98 | 62.34 | 57.07 | 28.45 | 29.62 | 35.56 | 71.97 | 79.08 | ||||||||||||||||||
NASDAQ Composite-Total Returns | 100.00 | 104.50 | 110.96 | 119.91 | 126.98 | 128.26 | 124.85 | 139.63 | 160.17 | 181.01 | ||||||||||||||||||
NASDAQ Biotechnology Index | 100.00 | 105.15 | 119.35 | 141.32 | 172.12 | 157.95 | 120.44 | 124.23 | 145.81 | 151.14 | ||||||||||||||||||
Recent Sales of Securities
On December 12, 2013, our registration statement on Form S-1 (File No. 333-192242) was declared effective by the SEC for our initial public offering pursuant to which we sold an aggregate of 8,625,000 shares of our common stock at a price to the public of $7.00 per share. On April 8, 2014, we completed a follow-on public offering of 3,450,000 shares of our common stock at a price to the public of $18.00 per share.
We received net proceeds in the offering of approximately $54,871,000 after deduction of underwriting commissions and offering expenses of $5,504,000 from the initial public offering and approximately $58,065,000 after deduction of underwriting commission and offering expenses of $4,035,000 from the follow-on public offering.
In January 2015, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity securities in one or more offerings up to a total dollar amount of $150.0 million. During the six months ended June 30, 2017, we completed the sale of 4,501,985 shares of common stock under an At Market Issuance Sales Agreement with FBR Capital Markets & Co. Net proceeds, after deducting approximately $1,038,000 in underwriting commissions and offering costs, were approximately $28,962,000. In July 2017, we completed an underwritten public offering of common stock. After giving effect to the exercise of the over-allotment option by the underwriters, the total number of shares sold by us in the public offering totaled 3,314,000 shares with gross proceeds of $24,855,000. Net proceeds, after deducting underwriting commission and offering costs, were approximately $23,198,000.
During the fiscal years ended December 31, 2016 and 2015, we did not sell any securities that were not registered under the Securities Act.
Repurchase of Shares
We did not repurchase any of our shares of capital stock during the fiscal years ended December 31, 2017, 2016 and 2015.
ITEM 6. | SELECTED FINANCIAL DATA. |
The following selected historical information has been derived from the audited consolidated financial statements of Kindred Biosciences and should be read in conjunction with “Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Item 8. Financial Statements and Supplementary Data” included elsewhere in this Annual Report on Form 10-K. The Statement of Operations and Comprehensive Loss Data and the Balance Sheet Data are derived from the audited consolidated financial statements which are included in the Form 10-K. The historical results are not necessarily indicative of the results of operations to be expected in the future.
45
(In thousands, except per share amounts) | Years ended December 31, | ||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
Statement of Operations and Comprehensive Loss Data: | |||||||||||||||||||
Operating expenses: | |||||||||||||||||||
Research and development | $ | 17,665 | $ | 13,861 | $ | 19,412 | $ | 18,694 | $ | 3,140 | |||||||||
General and administrative | 13,988 | 8,308 | 7,850 | 8,539 | 1,079 | ||||||||||||||
Restructuring costs | — | 655 | — | — | — | ||||||||||||||
Total operating expenses | 31,653 | 22,824 | 27,262 | 27,233 | 4,219 | ||||||||||||||
Loss from operations | (31,653 | ) | (22,824 | ) | (27,262 | ) | (27,233 | ) | (4,219 | ) | |||||||||
Interest and other income, net | 774 | 325 | 132 | 94 | 6 | ||||||||||||||
Net loss | (30,879 | ) | (22,499 | ) | (27,130 | ) | (27,139 | ) | (4,213 | ) | |||||||||
Change in unrealized gains or losses on available-for-sale securities | — | 19 | (23 | ) | (27 | ) | — | ||||||||||||
Comprehensive loss | $ | (30,879 | ) | $ | (22,480 | ) | $ | (27,153 | ) | $ | (27,166 | ) | $ | (4,213 | ) | ||||
Net loss per share, basic and diluted (1) | $ | (1.23 | ) | $ | (1.13 | ) | $ | (1.37 | ) | $ | (1.44 | ) | $ | (1.13 | ) | ||||
Weighted-average number of common shares outstanding, basic and diluted | 25,084 | 19,873 | 19,773 | 18,782 | 3,732 | ||||||||||||||
(In thousands) | As of December 31, | ||||||||||||||||||
2017 | 2016 | 2015 | 2014 | 2013 | |||||||||||||||
Balance Sheet Data: | |||||||||||||||||||
Cash and cash equivalents | $ | 34,813 | $ | 6,687 | $ | 19,992 | $ | 12,969 | $ | 65,329 | |||||||||
Short-term investments | 46,207 | 50,068 | 53,051 | 88,058 | — | ||||||||||||||
Long-term investments | 1,499 | 1,052 | 4,590 | — | — | ||||||||||||||
Working capital | 75,790 | 54,170 | 70,547 | 98,652 | 63,267 | ||||||||||||||
Total assets | 90,822 | 61,576 | 79,619 | 101,920 | 65,489 | ||||||||||||||
Total liabilities | 6,142 | 3,896 | 3,248 | 2,896 | 2,210 | ||||||||||||||
Accumulated deficit | (111,980 | ) | (81,101 | ) | (58,602 | ) | (31,472 | ) | (4,333 | ) | |||||||||
Total stockholders' equity | 84,680 | 57,680 | 76,371 | 99,024 | 63,279 | ||||||||||||||
(1) See Note 12 of the notes to consolidated financial statements included elsewhere in this annual report for an explanation of the method used to calculate the basic and diluted net loss per share attributable to common stockholders and the number of shares used in the computation of the per share amounts.
46
ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis of our financial condition and results of operations together with our consolidated financial statements and the related notes and other financial information included elsewhere in this Annual Report on Form 10-K. Some of the information contained in this discussion and analysis, including information with respect to our plans and strategy for our business, includes forward-looking statements that involve risks and uncertainties. You should review the “Risk Factors” section of this Annual Report for a discussion of important factors that could cause our actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis or elsewhere in this Annual Report.
Overview
We are a pre-commercialization biopharmaceutical company focused on saving and improving the lives of pets. Our mission is to bring to our pets the same kinds of safe and effective medicines that our human family members enjoy. Our core strategy is to identify compounds and targets that have already demonstrated safety and efficacy in humans and to develop therapeutics based on these validated compounds and targets for pets, primarily dogs, cats and horses. We believe this approach will lead to shorter development times and higher approval rates than pursuing new, non-validated compounds and targets. Our current portfolio includes over 20 product candidates in development consisting of both small molecule pharmaceuticals and biologics.
We have submitted all major technical sections of the New Animal Drug Application, or NADA, to the Food and Drug Administration, or FDA, for two product candidates. In addition, we have multiple other product candidates, including several biologics, in various stages of development. We believe there are significant unmet medical needs for pets, and that the pet therapeutics segment of the animal health industry is likely to grow substantially as new therapeutics are identified, developed and marketed specifically for pets.
We announced positive topline results in May 2016 upon the successful completion of a pharmacokinetic study and a randomized, placebo-controlled pivotal study of Mirataz (mirtazapine transdermal ointment), formerly known as KIND-010, for the management of weight loss in cats. All major technical sections of the NADA have been filed. We have received the technical section complete letter for effectiveness from the FDA and responded to the one remaining comment from the FDA regarding the CMC technical section. The FDA did not have any further comments on the safety technical section. Based on the feedback, we anticipate the approval of Mirataz and are preparing for the commercial launch in the first half of 2018, assuming that the FDA finds our responses acceptable. The regulatory approval is expected, but is subject to the typical risks inherent in such a process.
The European Marketing Agency, or EMA, has accepted our European marketing authorization application for the review of Mirataz with the official acceptance date of December 21, 2017.
In November 2015, we completed a pivotal trial of Zimeta (dipyrone injection), previously known as
KIND-012, for the control of pyrexia (fever) in horses with positive topline results. We submitted all major technical sections of the NADA for Zimeta to the FDA before the end of the first quarter of 2016. We have received the technical section complete letters for effectiveness and safety from the FDA and responded to additional comments from the FDA regarding the CMC technical section. We anticipate the approval of Zimeta in the first half of 2018, assuming that the FDA finds our responses acceptable. The regulatory approval is expected, but is subject to the typical risks inherent in such a process.
47
We have initiated pre-launch activities for Mirataz and Zimeta, including build-out of a small commercial team, execution of distribution agreements and commercial scale-up and manufacturing. In addition, we executed a commercial manufacturing agreement with DPT Laboratories, Ltd. for the manufacture of Mirataz and a commercial manufacturing agreement with Corden Pharma SPA for the manufacture of Zimeta. The two agreements provide for production to supply KindredBio’s initial launch and future commercial campaigns upon regulatory approval, with capabilities to meet excess demand.
We have also completed the pivotal field effectiveness study of Zimeta Oral (dipyrone oral gel) for the treatment of fever in horses and announced positive topline results in December 2017. This study was a multicenter, randomized, blinded, placebo-controlled pivotal study that enrolled 139 horses to assess the effectiveness of Zimeta Oral. We have completed the in-life portion of the Target Animal Safety Study and are analyzing the data. We anticipate submitting the effectiveness and safety technical sections of the NADA in the first half of 2018, and the CMC technical section by the end of 2018, assuming the data are supportive of approval. The oral gel form of dipyrone is expected to be an additional valuable tool for equine veterinarians to provide horse owners with an easy-to-administer fever reducing agent for the horse.
We are currently developing KIND-014 for the treatment of equine gastric ulcers in horses, KIND-015 for the management of clinical signs associated with equine metabolic syndrome and epoCatTM (feline recombinant erythropoietin) for the control of non-regenerative anemia in cats. We expect to continue pilot field efficacy studies for the three product candidates in 2018. In addition to early stage pilot field studies of anti-cytokine antibodies for atopic dermatitis in dogs and anti-TNF in septic foals, we are also developing multiple other products, including interleukin antibodies and canine checkpoint inhibitors. In all, we have over 20 programs for various indications for dogs, cats, and horses.
In addition to the product candidates discussed above, we are in the early stages of development for multiple additional indications, including several biologics, with the potential to attain approval for one or more products annually for several years. We plan to commercialize our products in the United States through a direct sales force complemented by selected distributor relationships, and in the EU through distributors and other third parties. Because we seek to identify product candidates that are not protected by third-party patents, we typically do not need to obtain licenses or make any upfront, milestone or royalty payments in connection with our product candidates.
We have constructed a Good Manufacturing Practice, or GMP, biologics manufacturing plant in Burlingame, CA which is fully commissioned. We have proceeded to GMP manufacturing of our feline erythropoietin product candidate in January 2018. In addition, we acquired a second manufacturing plant in August 2017 in Elwood, Kansas and we have initiated the design and construction of our biologics manufacturing lines. The Elwood facility includes approximately 180,000 square feet with clean rooms, utility, equipment, and related quality documentation suitable for small molecule and biologics manufacturing.
We are a pre-commercialization company with no products approved for marketing and sale, and we have not generated any revenue. We have incurred significant net losses since our inception with cumulative net losses of $111,980,000 through December 31, 2017. These losses have resulted principally from costs incurred in connection with investigating and developing our product candidates, research and development activities and general and administrative costs associated with our operations.
Historically, our funding has been a combination of private and public offerings. Our initial public offering in December 2013 provided us with net proceeds of $54,871,000 and a follow-on public offering in April 2014 provided us with net proceeds of $58,065,000 after deducting underwriting discounts and commissions. During the six months ended June 30, 2017, we completed the sale of 4,501,985 shares of common stock under an At Market Issuance Sales Agreement, or ATM, with FBR Capital Markets & Co. Net proceeds, after deducting approximately $1,038,000 in underwriting commissions and offering costs, were approximately $28,962,000. In July 2017, we completed an underwritten public offering of common stock. After giving effect to the exercise of the over-allotment option by the underwriters, the total number of shares sold by us in the public offering totaled 3,314,000 shares with gross proceeds of $24,855,000. Net proceeds,
48
after deducting underwriting commission and offering costs, were approximately $23,198,000. As of December 31, 2017, we had cash, cash equivalents and investments in available-for-sale securities of $82,519,000.
For the foreseeable future, we expect to continue to incur losses, which will increase significantly from historical levels as we expand our product development activities, seek regulatory approvals for our product candidates and begin to commercialize them if they are approved by the Center for Veterinary Medicine branch of the U.S. Food and Drug Administration, or FDA, the U.S. Department of Agriculture, or USDA, or the European Medicines Agency, or EMA. If we are required to further fund our operations, we expect to do so through public or private equity offerings, debt financings, corporate collaborations and licensing arrangements. We cannot assure you that such funds will be available on terms favorable to us, if at all. Arrangements with collaborators or others may require us to relinquish rights to certain of our technologies or product candidates. In addition, we may never successfully complete development of, obtain adequate patent protection for, obtain necessary regulatory approval, or achieve commercial viability for any product candidate. If we are not able to raise additional capital on terms acceptable to us, or at all, as and when needed, we may be required to curtail our operations, and we may be unable to continue as a going concern.
Revenue
We do not have any products approved for sale, have not generated any revenue from product sales since our inception and do not expect to generate any material revenue from the sale of products in the near future. If our development efforts result in clinical success and regulatory approval or collaboration agreements with third parties for any of our product candidates, we may generate revenue from those product candidates.
Operating Expenses
The majority of our operating expenses to date have been for the research and development activities related to our lead product candidates.
Research and Development Expense
All costs of research and development are expensed in the period incurred. Research and development costs primarily consist of contracted development costs, manufacturing costs, salaries and related expenses for personnel, stock-based compensation expense, regulatory, outside service providers, professional and consulting services, travel costs and materials used in clinical trials and research and development.
We are currently pursuing over 20 indications. We typically use our employee and infrastructure resources across multiple development programs. We track outsourced development costs by development compound but do not allocate personnel or other internal costs related to development to specific programs or development compounds as these expenses are included in personnel costs and other internal costs.
General and Administrative Expense
General and administrative expense consists primarily of personnel costs, including salaries, related benefits and stock-based compensation for employees, consultants and directors. General and administrative expenses also include rent and other facilities costs, professional fees for legal, accounting, tax, information technology services, costs associated with being a public company and other general business services.
Interest and Other Income, Net
Consist of interest earned on our cash, cash equivalents and short-term investments and asset disposals.
49
Income Taxes
As of December 31, 2017, we had net operating loss carryforwards for federal and state income tax purposes of $93,253,000 and $93,172,000, respectively, which will begin to expire in fiscal year 2032. Our management has evaluated the factors bearing upon the realizability of our deferred tax assets, which are comprised principally of net operating loss carryforwards. Our management concluded that, due to the uncertainty of realizing any tax benefits as of December 31, 2017, a valuation allowance was necessary to fully offset our deferred tax assets.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or U.S. GAAP. The preparation of our consolidated financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, and revenue, costs and expenses and related disclosures during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those described below. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 of the notes to our consolidated financial statements appearing elsewhere in this document, we believe that the estimates and assumptions involved in the following accounting policies may have the greatest potential impact on our consolidated financial statements.
JOBS Act
On April 5, 2012, the Jumpstart Our Business Startups Act, or the JOBS Act, was signed into law. The JOBS Act contains provisions that, among other things, reduce certain reporting requirements for an “emerging growth company.” As an “emerging growth company” we are electing not to take advantage of the extended transition period afforded by the JOBS Act for the implementation of new or revised accounting standards, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision not to take advantage of the extended transition period is irrevocable.
We are currently relying on other exemptions and reduced reporting requirements provided by the JOBS Act which state, among other things, that we are not required to (i) provide an auditor’s attestation report on our system of internal control over financial reporting or (ii) comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the consolidated financial statements. These exemptions will apply for a period of five years following the completion of our initial public offering or until we no longer meet the requirements of being an “emerging growth company,” whichever is earlier.
Fair Value Measurements
We invest our cash in money market funds, cash deposits and debt instruments of the U.S. government agency securities. In the current market environment, the assessment of the fair value of the debt securities can be difficult and subjective. Accounting Standards Codification, or ASC, 820, “Fair Value Measurements and Disclosure" standard describes a fair value hierarchy based on three levels of inputs, of which the first two are considered observable and the last unobservable, that may be used to measure fair value which are the following:
50
Level 1 | Quoted prices in active markets for identical assets or liabilities; | |||
Level 2 | Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities; and | |||
Level 3 | Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. The determination of fair value for Level 3 instruments requires the most management judgment and subjectivity. | |||
Research and Development
As part of the process of preparing our consolidated financial statements, we are required to estimate accrued research and development expenses. Examples of estimated accrued expenses include fees paid to vendors and clinical sites in connection with our pivotal studies, to CROs in connection with our toxicology studies, and to contract manufacturers in connection with the production of API and formulated drug.
We review new and open contracts and communicate with applicable internal and vendor personnel to identify services that have been performed on our behalf and estimate the level of service performed and the associated costs incurred for the service when we have not yet been invoiced or otherwise notified of the actual cost for accrued expenses. The majority of our service providers invoice us monthly in arrears for services performed or as milestones are achieved in relation to our contract manufacturers. We make estimates of our accrued expenses as of each balance sheet date.
We base our accrued expenses related to pivotal studies on our estimates of the services received and efforts expended pursuant to contracts with vendors, our internal resources, and payments to clinical sites based on enrollment projections. The financial terms of the vendor agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of animals and the completion of development milestones. We estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we adjust the related expense accrual accordingly on a prospective basis. If we do not identify costs that have been incurred or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. To date, we have not made any material adjustments to our estimates of accrued research and development expenses or the level of services performed in any reporting period presented.
Stock-Based Compensation
We measure stock-based awards granted to employees and directors at fair value on the date of grant and recognize the corresponding compensation expense of the awards, net of estimated forfeitures, over the requisite service periods, which correspond to the vesting periods of the awards. Generally, we issue stock-based awards with only service-based vesting conditions, and record compensation expense for these awards using the straight-line method. Our intention is to grant stock-based awards with exercise prices equivalent to the fair value of our common stock as of the date of grant.
We account for all stock-based awards issued to non-employees based on the fair value of the award on each measurement date. Stock-based awards granted to non-employees are subject to revaluation at each reporting date over their vesting terms or until approved by our board of directors and settled. As a result, the charge to operations for non-employee awards with vesting conditions or awards which have not been approved and settled is affected each reporting period by changes in the fair value of our common stock.
The fair value of each stock-based award is estimated using the Black-Scholes option-pricing model. Due to insufficient company-specific historical and implied stock price volatility information, we estimate our expected stock price volatility based on the historical volatility of publicly-traded peer companies and expect
51
to continue to do so until such time as we have adequate historical data regarding the volatility of our common stock price. The expected terms of our awards have been determined utilizing the “simplified” method, since our historical experience for option grants is not relevant to our expectations for recent grants. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is zero, based on the fact that we have never paid cash dividends and do not expect to pay any cash dividends in the foreseeable future. See Note 8 in Notes to Consolidated Financial Statements for further information.
Results of Operations
The following table summarizes the results of our operations for the periods indicated:
(In thousands, except per share amounts) | Years Ended December 31, | ||||||||||
2017 | 2016 | 2015 | |||||||||
Operating expenses: | |||||||||||
Research and development | $ | 17,665 | $ | 13,861 | 19,412 | ||||||
General and administrative | 13,988 | 8,308 | 7,850 | ||||||||
Restructuring costs | — | 655 | — | ||||||||
Total operating expenses | 31,653 | 22,824 | 27,262 | ||||||||
Loss from operations | (31,653 | ) | (22,824 | ) | (27,262 | ) | |||||
Interest and other income, net | 774 | 325 | 132 | ||||||||
Net loss | (30,879 | ) | (22,499 | ) | (27,130 | ) | |||||
Change in unrealized gains or losses on available-for-sale securities | — | 19 | (23 | ) | |||||||
Comprehensive loss | $ | (30,879 | ) | $ | (22,480 | ) | $ | (27,153 | ) | ||
Net loss per share attributable to common stockholders, basic and diluted | $ | (1.23 | ) | $ | (1.13 | ) | $ | (1.37 | ) | ||
Weighted-average number of common shares outstanding, basic and diluted | 25,084 | 19,873 | 19,773 | ||||||||
Revenue
We do not have any products approved for sale, have not generated any revenue since our inception and do not expect to generate any material revenue in the near future. If our development efforts result in clinical success and regulatory approval or collaboration agreements with third parties for any of our product candidates, we may generate revenue from those product candidates.
Research and Development Expense
All costs of research and development are expensed in the period incurred. Research and development costs consist primarily of salaries and related expenses for personnel, stock-based compensation expense, fees paid to consultants, outside service providers, professional services, travel costs and materials used in clinical trials and research and development. We are currently pursuing multiple product candidates for over 20 indications. We typically use our employee and infrastructure resources across multiple development programs.
Research and development expense was as follows for the periods indicated:
52
(In thousands except percentages) | Years ended December 31, | Annual percent change | |||||||||||||||
2017 | 2016 | 2015 | 2017/2016 | 2016/2015 | |||||||||||||
Payroll and related | $ | 6,517 | $ | 5,794 | $ | 8,473 | 12 | % | (32 | )% | |||||||
Consulting | 1,669 | 1,204 | 1,144 | 39 | % | 5 | % | ||||||||||
Field trial costs, including materials | 3,675 | 3,015 | 5,428 | 22 | % | (44 | )% | ||||||||||
Stock-based compensation | 1,650 | 1,464 | 1,857 | 13 | % | (21 | )% | ||||||||||
Other | 4,154 | 2,384 | 2,510 | 74 | % | (5 | )% | ||||||||||
$ | 17,665 | $ | 13,861 | $ | 19,412 | 27 | % | (29 | )% | ||||||||
During the year ended December 31, 2017, research and development expense related primarily to advancing the development of Zimeta Oral and our leading small molecule and biologics product candidates. During this period we successfully completed and announced positive topline results on the pivotal trial of Zimeta Oral. We have responded to all comments from the FDA related to the NADA for Zimeta as well as Mirataz. We continue to advance additional product candidates in our small molecule programs as well as continue to advance our biologics program by building an in-house team to finalize the set-up of a GMP manufacturing process for our potential biologic candidates.
Research and development expenses for the year ended December 31, 2017 increased by 27% to $17.7 million compared with $13.9 million for the same period in 2016. Payroll and related expenses increased by $0.7 million due to increase in headcount as we advance our biologics programs. Our consulting and field trial costs increased by $0.5 million and $0.7 million, respectively, due to the pivotal field effectiveness study of Zimeta Oral for the treatment of fever in horses. In addition, stock-based compensation expense increased by approximately $0.2 million. Other costs have increased by $1.8 million as we have increased our rented lab spaces and incurring higher maintenance and depreciation expenses, as well as higher biologics manufacturing and lab supplies expenses. Outsourced research and development expense related to our Zimeta (IV and Oral), KIND-014, Mirataz and other product development programs for the year ended December 31, 2017 were $1.7 million, $0.6 million, $0.3 million and $1.9 million, respectively.
Research and development expenses for the year ended December 31, 2016 decreased by 29% to $13.9 million compared with $19.4 million for the same period in 2015. Payroll and related expenses decreased by $2.7 million due to reduction in headcount. Our field trial costs decreased by $2.4 million as the pivotal study for Mirataz was completed earlier than expected, offset in part by costs associated with the pilot field study costs for KIND-014 and epoCat. The 2015 expenses included pivotal field trial costs for SentiKind and Zimeta, which we did not have in 2016. In addition, stock-based compensation expense decreased by approximately $0.4 million. Outsourced research and development expense related to our Mirataz, Zimeta (IV and Oral), KIND-014 and other product development programs for the year ended December 31, 2016 were $1.0 million, $1.0 million, $0.7 million and $1.2 million, respectively.
We expect research and development expense to increase for the foreseeable future as we continue to further develop our small molecule compounds and biologics development programs. Due to the inherently unpredictable nature of our development, we cannot reasonably estimate or predict the nature, specific timing or estimated costs of the efforts that will be necessary to complete the development of our product candidates.
General and Administrative Expense
The composition of general and administrative expense was as follows for the periods indicated:
53
(In thousands except percentages) | Years ended December 31, | Annual percent change | |||||||||||||||
2017 | 2016 | 2015 | 2017/2016 | 2016/2015 | |||||||||||||
Payroll and related | $ | 4,391 | $ | 2,583 | $ | 1,944 | 70 | % | 33 | % | |||||||
Consulting, professional and legal fees | 2,011 | 996 | 1,461 | 102 | % | (32 | )% | ||||||||||
Stock-based compensation | 3,557 | 2,164 | 2,300 | 64 | % | (6 | )% | ||||||||||
Corporate and marketing expenses | 2,084 | 1,468 | 992 | 42 | % | 48 | % | ||||||||||
Other | 1,945 | 1,097 | 1,153 | 77 | % | (5 | )% | ||||||||||
$ | 13,988 | $ | 8,308 | $ | 7,850 | 68 | % | 6 | % | ||||||||
General and administrative expenses for the year ended December 31, 2017 increased by 68% to $14.0 million compared with $8.3 million for the same period in 2016. The increase in general and administrative expense was related to increased headcount including higher marketing and corporate expenses as we have initiated pre-launch activities and build-out of a small commercial team. In addition, higher stock-based compensation expense along with higher consulting and professional fees as we build out our corporate infrastructure, also contributed to the increase in expenses .
General and administrative expenses for the year ended December 31, 2016 increased by 6% to $8.3 million compared with $7.9 million for the same period in 2015. The increase in general and administrative expense was related to increased headcount including higher marketing and corporate expenses as we have initiated pre-launch activities and build-out of a small commercial team. The increase was offset in part by lower consulting and professional fees as well as lower stock-based compensation expense.
We expect general and administrative expense to increase going forward as we prepare for the commercial launches of Zimeta and Mirataz and the expansion of our sales force and medical affairs personnel including corporate infrastructure in the expected commercialization of the two products.
Restructuring Costs
We recorded a restructuring charge of $655,000 in the first quarter of 2016 in order to streamline our focus on our development programs and to ensure remaining funds are sufficient to fund our planned operations through 2018. Eighteen employees were impacted by the restructuring and all restructuring charges were paid as of March 2016. There was no further restructuring charge for the remainder of 2016.
Interest and Other Income, Net
(In thousands) | Years ended December 31, | ||||||||||
2017 | 2016 | 2015 | |||||||||
Interest and other income, net | $ | 774 | $ | 325 | $ | 132 | |||||
The increase of approximately $449,000 in 2017 compared to 2016 and $193,000 in 2016 compared to 2015 was due to higher interest income from higher cash balances in 2017 as a result of the completion of our ATM financing and follow-on public offering of common stock, and higher interest income from better market performance in 2016.
Liquidity and Capital Resources
We have incurred losses and negative cash flows from operations and have not generated any revenue since our inception in September 2012 through December 31, 2017. As of December 31, 2017, we had an accumulated deficit of $112.0 million. During the year ended December 31, 2013, we raised approximately $66.0 million, net of offering costs, primarily in connection with our initial public offering and through the sale
54
of preferred stock (subsequently converted to common stock at the time of our initial public offering). On April 8, 2014, we completed a follow-on public offering of common stock, resulting in net proceeds of approximately $58.1 million. In 2017, we completed the sale of common stock under an ATM financing as well as an underwritten public offering, resulting in net proceeds of approximately $28,962,000 and $23,198,000, respectively. As of December 31, 2017, we had cash, cash equivalents and investments in available-for-sale securities of approximately $82.5 million, which we believe are sufficient to fund our planned operations for at least the next 18 months.
Cash Flows
The following table shows a summary of our cash flows for the periods set forth below:
(In thousands) | Years ended December 31, | ||||||||
2017 | 2016 | 2015 | |||||||
Cash flows used in operating activities | $ | (21,878 | ) | (18,781 | ) | (22,757 | ) | ||
Cash flows provided by (used in) investing activities | $ | (2,668 | ) | 5,314 | 29,437 | ||||
Cash flows provided by financing activities | $ | 52,672 | 162 | 343 | |||||
Net cash used in operating activities
During the year ended December 31, 2017, net cash used in operating activities was $21.9 million. Our net loss of $30.9 million included non-cash charges primarily in the form of share-based compensation of $5.2 million, depreciation expense of $475,000 and accretion of discounts and amortization of premiums on investments of $162,000. The non-cash charges were partly impacted by changes in operating assets and liabilities that resulted in approximately $3.1 million of cash provided by operating activities.
During the year ended December 31, 2016, net cash used in operating activities was $18.8 million. Our net loss of $22.5 million included non-cash charges primarily in the form of share-based compensation of $3.6 million, depreciation expense of $155,000 and accretion of discounts and amortization of premiums on investments of $274,000. The non-cash charges were partly offset by changes in operating assets and liabilities that resulted in $341,000 of cash used in operating activities.
During the year ended December 31, 2015, net cash used in operating activities was $22.8 million. Our net loss of $27.1 million included non-cash charges primarily in the form of share-based compensation of $4.2 million, depreciation expense of $146,000 and accretion of discounts and amortization of premiums on investments of $228,000. The non-cash charges were partly offset by changes in operating assets and liabilities that resulted in $158,000 of cash used in operating activities.
Net cash provided by (used in) investing activities
During the year ended December 31, 2017, net cash used in investing activities was $2.7 million, which resulted from $3.3 million related to the proceeds of sales and maturities of investments, net of purchases of investments, offset by $6.2 million in purchases of property and equipment, of which $0.3 million is included in accounts payable and accrued liabilities at December 31, 2017.
During the year ended December 31, 2016, net cash provided by investing activities was $5.3 million, which resulted from $6.3 million related to the maturities of investments, net of purchases of investments, partially offset by $1.6 million in purchases of property and equipment, of which $0.7 million is included in accounts payable and accrued liabilities at December 31, 2016.
During the year ended December 31, 2015, net cash provided by investing activities was $29.4 million, which resulted from $30.2 million related to the proceeds of sales and maturities of investments, net of purchases of investments, partially offset by $1.0 million in purchases of property and equipment, of which $0.3 million is included in accounts payable and accrued liabilities at December 31, 2015.
55
Net cash provided by financing activities
During the year ended December 31, 2017, net cash provided by financing activities consisted of approximately $52.2 million in net proceeds from the sale of common stock through an ATM and follow-on public offering, and approximately $0.5 million from the exercise of stock options and purchase of ESPP shares.
During the year ended December 31, 2016, net cash provided by financing activities consisted of proceeds from stock option exercises and purchase of ESPP shares.
During the year ended December 31, 2015, net cash provided by financing activities consisted of proceeds from stock option exercises and purchase of ESPP shares.
Future Funding Requirements
We anticipate that we will continue to incur losses for the next several years due to expenses relating to:
• | pivotal trials of our product candidates; |
• | toxicology studies for our product candidates; |
• | biologics manufacturing; and |
• | commercialization of two or more of our product candidates, if approved. |
We believe our existing cash, cash equivalents and investments in available-for-sale securities will be sufficient to fund our operating plan for at least the next 18 months and through the anticipated approvals and launches of our lead product candidates Zimeta and Mirataz. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. Such financing may result in dilution to stockholders, imposition of debt covenants and repayment obligations or other restrictions that may affect our business. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including, but not limited to:
• | the scope, progress, results and costs of researching and developing our current or future product candidates; |
• | the timing of, and the costs involved in, obtaining regulatory approvals for any of our current or future product candidates; |
• | the number and characteristics of the product candidates we pursue; |
• | the cost of manufacturing our current and future product candidates and any products we successfully commercialize, including cost of building internal biologics manufacturing capacity; |
• | the cost of commercialization activities if any of our current or future product candidates are approved for sale, including marketing, sales and distribution costs; |
• | the expenses needed to attract and retain skilled personnel; |
• | the costs associated with being a public company; |
• | our ability to establish and maintain strategic collaborations, licensing or other arrangements and the financial terms of such agreements; and |
• | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing possible patent claims, including litigation costs and the outcome of any such litigation. |
Contractual Obligations
56
We have non-cancelable operating leases for two office spaces and expanded laboratory space under which we are obligated to make minimum lease payments totaling $3.0 million through May 2022, the timing of which is described in more detail in the notes to the consolidated financial statements. In addition, we have three operating leases for equipment under which we are obligated to make minimum lease payments totaling $46,000 through 2020.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in the use of any off-balance sheet arrangements, such as structured finance entities, special purpose entities or variable interest entities.
Recently Issued Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update ("ASU") No. 2014-09, "Revenue from Contracts with Customers". This new standard will replace most of the existing revenue recognition guidance in U.S. GAAP when it becomes effective and permits the use of either the retrospective or cumulative effect transition method. The new standard, as amended, becomes effective in the first quarter of fiscal year 2018, but allows the adoption of the standard one year earlier if we so choose. The analysis identifying areas that will be impacted by the new guidance is complete. Additionally, we will continue to monitor modifications, clarifications, and interpretations issued by the FASB that may impact its assessment. We do not currently have and have never had any contracts that are within the scope of ASC 606, "Revenue from Contracts with Customers" or its predecessor guidance, ASC 605, "Revenue Recognition". Accordingly, based on our current assessment as of December 31, 2017, we do not believe adopting this guidance in the first quarter of 2018 will have a material impact on our consolidated financial statements as we are not currently generating revenues.
In January 2016, the FASB issued ASU No. 2016-01, "Financial Instruments—Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities", which amends the guidance in U.S. GAAP on the classification and measurement of financial instruments and also amends certain disclosure requirements associated with the fair value of financial instruments. The new guidance is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. We have adopted the new guidance and it did not have a material impact on our consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02, "Leases (Topic 842)", requiring organizations that lease assets—referred to as “lessees”—to recognize on the consolidated balance sheet the assets and liabilities for the rights and obligations created by those leases. Under the new guidance, a lessee will be required to recognize assets and liabilities for leases with lease terms of more than 12 months. The ASU on leases will take effect for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. We are currently evaluating the new guidance and have not determined the impact this standard may have on our consolidated financial statements.
In May 2017, the FASB issued ASU No. 2017-09, “Scope of Modification Accounting”, which amends ASC Topic 718, “Compensation - Stock Compensation”. The ASU includes provisions intended to (1) provide clarity and (2) reduce diversity in practice and reduce cost and complexity when calculate stock compensation, on a change to the terms or conditions of a share-based payment award. ASU 2017-09 is effective for public business entities for annual reporting periods, and interim periods within those annual periods, beginning after December 15, 2017. Early adoption will be permitted in any interim or annual period, with any adjustments reflected as of the beginning of the fiscal year of adoption. We have adopted the new guidance and it did not have a material impact on our consolidated financial statements.
We do not believe there are any other recently issued standards not yet effective that will have a material impact on our consolidated financial statements when the standards become effective.
57
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Interest Rate Fluctuation Risk
The primary objective of our investment activities is to preserve capital. We do not utilize hedging contracts or similar instruments.
We are exposed to certain market risks relating primarily to (1) interest rate risk on our cash and cash equivalents, (2) market price risk on our short-term investments, and (3) risks relating to the financial viability of the institutions which hold our capital and through which we have invested our funds. We manage such risks by investing in short-term, liquid, highly-rated instruments. As of December 31, 2017, our cash equivalents, short-term and long-term investments are invested in money market funds, U.S. treasury bills, U.S. treasury bonds, U.S. government agencies, commercial paper and high grade corporate notes. We do not believe we have any material exposure to interest rate risk due to the extremely low interest rate environment, the short duration of the securities we hold and our ability to hold our investments to maturity if necessary. Declines in interest rates would reduce investment income, but would not have a material effect on our financial condition or results of operations.
We do not currently have exposure to foreign currency risk.
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA. |
Our consolidated financial statements appear commencing on page F-1 of this Annual Report on Form 10-K, which information is incorporated herein by reference.
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
None.
ITEM 9A. | CONTROLS AND PROCEDURES. |
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Exchange Act Rule 13a-15(e)) that are designed to assure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to management, including our Chief Executive Officer and our Chief Financial Officer (the “Certifying Officers"), as appropriate, to allow timely decisions regarding required disclosures.
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide reasonable assurance only of achieving the desired control objectives, and management necessarily is required to apply its judgment in weighing the costs and benefits of possible new or different controls and procedures. Limitations are inherent in all control systems, so no evaluation of controls can provide absolute assurance that all control issues and any fraud within the company have been detected.
As required by Exchange Act Rule 13a-15(b), as of the end of the period covered by this Annual Report on Form 10-K, management, under the supervision and with the participation of our Certifying Officers, evaluated the effectiveness of our disclosure controls and procedures. Based on this evaluation, the Certifying Officers have concluded that, as of the end of the period covered by this Annual Report on Form 10-K, our disclosure controls and procedures were effective at a reasonable assurance level.
58
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Internal control over financial reporting is a process designed by, or under the supervision of, our Chief Executive Officer and Chief Financial Officer and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations (COSO). Based on such evaluation, our management concluded that our internal control over financial reporting was effective at a reasonable assurance level as of December 31, 2017.
This Annual Report on Form 10-K does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting due to a transition period established by the SEC for newly public companies under the JOBS Act.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2017 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. | OTHER INFORMATION |
None.
PART III
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required to be disclosed by this item will be contained in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC no later than April 30, 2018 and is incorporated herein by reference.
ITEM 11. | EXECUTIVE COMPENSATION |
The information required to be disclosed by this item will be contained in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC no later than April 30, 2018 and is incorporated herein by reference.
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
59
The information required to be disclosed by this item will be contained in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC no later than April 30, 2018 and is incorporated herein by reference.
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required to be disclosed by this item will be contained in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC no later than April 30, 2018 and is incorporated herein by reference.
ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required to be disclosed by this item will be contained in our Definitive Proxy Statement for the Annual Meeting of Stockholders to be filed with the SEC no later than April 30, 2018 and is incorporated herein by reference.
PART IV
ITEM 15. | EXHIBITS AND FINANCIAL STATEMENT SCHEDULES. |
Our financial statements and related notes thereto are listed and included in this Annual Report on Form 10-K beginning on page F-1. The following exhibits are filed with, or are incorporated by reference into, this Annual Report:
60
EXHIBIT INDEX
Exhibit No. | Description | |
1.1 | ||
3.1 | ||
3.2 | ||
3.3 | ||
4.1 | ||
4.2 | ||
10.1 | ||
10.2 | ||
10.3 | ||
10.4 | ||
10.5 | ||
10.6 | ||
10.7 | ||
10.8 | ||
10.9 | ||
10.10 | ||
10.11 | ||
10.12 | ||
10.13 | ||
10.14 | ||
10.15 | ||
10.16 | ||
10.17 | ||
61
Exhibit No. | Description | |
10.18 | ||
10.19 | ||
10.20 | ||
10.21 | ||
10.22 | ||
10.23 | ||
10.24 | ||
21.1 | ||
23.1 | ||
31.1 | ||
31.2 | ||
32.1 | ||
101.INS | XBRL Instance Document | |
101.SCH | XBRL Taxonomy Extension Schema Document | |
101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF | XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB | XBRL Taxonomy Extension Labels Linkbase Document | |
101.PRE | XBRL Taxonomy Extension Presentation Linkbase Document | |
† | Indicates a management contract or compensatory plan or arrangement. |
(1) | Previously filed on December 17, 2013 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference. |
(2) | Previously filed on December 2, 2013 as an exhibit to Registrant’s Amendment No. 4 to Registration Statement on Form S‑1 (File No. 333‑192242) and incorporated herein by reference. |
(3) | Previously filed on November 8, 2013 as an exhibit to Registrant’s Registration Statement on Form S-1 (File No. 333-192242) and incorporated herein by reference. |
(4) | Previously filed on November 13, 2013 as an exhibit to Registrant’s Amendment No. 1 to Registration Statement on Form S-1 (File No. 333-192242) and incorporated herein by reference. |
(5) Previously filed on May 14, 2014 as an exhibit to Registrant’s Quarterly Report on Form 10-Q and incorporated herein by reference.
62
(6) Previously filed on August 13, 2014 as an exhibit to Registrant’s Quarterly Report on Form 10-Q and incorporated herein by reference.
(7) Previously filed on October 14, 2014 as an appendix to Registrant’s Definitive Proxy Statement on Schedule 14A and incorporated herein by reference.
(8) | Previously filed on March 13, 2015 as an exhibit to Registrant’s Annual Report on Form 10-K and incorporated herein by reference. |
(9) | Previously filed on June 4, 2015 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference. |
(10) | Previously filed on April 8, 2016 as Appendix A to the Registrant’s Definitive Proxy Statement on Schedule 14A, and incorporated herein by reference. |
(11) Previously filed on June 3, 2016 as an exhibit to Registrant’s Report to Registration Statement on Form S‑8 (File No. 333‑211839) and incorporated herein by reference.
(12) Previously filed on May 24, 2017 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference.
(13) Previously filed on June 26, 2017 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference.
(14) Previously filed on July 12, 2017 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference.
(15) Previously filed on July 24, 2017 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference.
(16) Previously filed on November 7, 2017 as an exhibit to Registrant’s Report on Form 8-K and incorporated herein by reference.
(17) Previously filed on March 1, 2017 as an exhibit to Registrant’s Annual Report on Form 10-K and incorporated herein by reference.
ITEM 16. | FORM 10-K SUMMARY |
None
63
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
KINDRED BIOSCIENCES, INC. | ||
Date: March 1, 2018 | By: | /s/ Wendy Wee |
Wendy Wee | ||
Chief Financial Officer and Principal Financial and Accounting Officer | ||
Pursuant to the requirements of Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date |
/s/ Richard Chin, M.D. | President, Chief Executive Officer and Director | March 1, 2018 |
Richard Chin, M.D. | (Principal Executive Officer) | |
/s/ Wendy Wee | Chief Financial Officer | March 1, 2018 |
Wendy Wee | (Principal Financial and Accounting Officer) | |
/s/ Herbert Montgomery | Director | March 1, 2018 |
Herbert Montgomery | ||
/s/ Raymond Townsend | Director | March 1, 2018 |
Raymond Townsend | ||
/s/ Ervin Veszprémi | Director | March 1, 2018 |
Ervin Veszprémi | ||
64
Index to Consolidated Financial Statements
Page | |
Financial Statements: | |
F- 1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of Kindred Biosciences, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Kindred Biosciences, Inc. and Subsidiary (the “Company”) as of December 31, 2017 and 2016, the related consolidated statements of operations and comprehensive loss, changes in stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2017, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ KMJ Corbin & Company LLP
We have served as the Company's auditor since 2013.
Costa Mesa, California
March 1, 2018
F- 2
Kindred Biosciences, Inc.
Consolidated Balance Sheets
(In thousands, except share and per share amounts)
December 31, | |||||||
2017 | 2016 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | 34,813 | $ | 6,687 | |||
Short-term investments | 46,207 | 50,068 | |||||
Prepaid expenses and other | 797 | 1,282 | |||||
Total current assets | 81,817 | 58,037 | |||||
Property and equipment, net | 7,457 | 2,441 | |||||
Long-term investments | 1,499 | 1,052 | |||||
Other assets | 49 | 46 | |||||
Total assets | $ | 90,822 | $ | 61,576 | |||
LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | 1,439 | $ | 410 | |||
Accrued compensation | 2,688 | 1,807 | |||||
Accrued liabilities | 1,900 | 1,650 | |||||
Total current liabilities | 6,027 | 3,867 | |||||
Long-term liability | 115 | 29 | |||||
Total liabilities | 6,142 | 3,896 | |||||
Commitments and contingencies (Note 9) | |||||||
Stockholders’ equity: | |||||||
Common stock; $0.0001 par value; 100,000,000 shares authorized; 28,182,563 shares and 19,916,290 shares issued and outstanding at December 31, 2017 and 2016, respectively | 3 | 2 | |||||
Additional paid-in capital | 196,688 | 138,810 | |||||
Accumulated other comprehensive loss | (31 | ) | (31 | ) | |||
Accumulated deficit | (111,980 | ) | (81,101 | ) | |||
Total stockholders’ equity | 84,680 | 57,680 | |||||
Total liabilities and stockholders’ equity | $ | 90,822 | $ | 61,576 | |||
The accompanying notes are integral part of these consolidated financial statements.
F- 3
Kindred Biosciences, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except per share amounts)
Years Ended December 31, | ||||||||||||
2017 | 2016 | 2015 | ||||||||||
Operating expenses: | ||||||||||||
Research and development | $ | 17,665 | $ | 13,861 | $ | 19,412 | ||||||
General and administrative | 13,988 | 8,308 | 7,850 | |||||||||
Restructuring costs | — | 655 | — | |||||||||
Total operating expenses | 31,653 | 22,824 | 27,262 | |||||||||
Loss from operations | (31,653 | ) | (22,824 | ) | (27,262 | ) | ||||||
Interest and other income, net | 774 | 325 | 132 | |||||||||
Net loss | (30,879 | ) | (22,499 | ) | (27,130 | ) | ||||||
Change in unrealized gains or losses on available-for-sale securities | — | 19 | (23 | ) | ||||||||
Comprehensive loss | $ | (30,879 | ) | $ | (22,480 | ) | $ | (27,153 | ) | |||
Net loss per share, basic and diluted | $ | (1.23 | ) | $ | (1.13 | ) | $ | (1.37 | ) | |||
Weighted-average number of common shares outstanding, basic and diluted | 25,084 | 19,873 | 19,773 | |||||||||
The accompanying notes are integral part of these consolidated financial statements.
F- 4
Kindred Biosciences, Inc.
Consolidated Statements of Changes in Stockholders' Equity
(In thousands)
Common Stock | Additional Paid-In Capital | Accumulated Other Comprehensive Loss | Accumulated Deficit | Total Stockholders' Equity | ||||||||||||||||||
Shares | Amount | |||||||||||||||||||||
Balance at December 31, 2014 | 19,724 | $ | 2 | $ | 130,521 | $ | (27 | ) | $ | (31,472 | ) | $ | 99,024 | |||||||||
Comprehensive loss | ||||||||||||||||||||||
Net loss | — | — | — | — | (27,130 | ) | (27,130 | ) | ||||||||||||||
Change in unrealized losses on available-for-sale securities | — | — | — | (23 | ) | — | (23 | ) | ||||||||||||||
Total comprehensive loss | (27,153 | ) | ||||||||||||||||||||
Stock-based compensation | — | — | 4,157 | — | — | 4,157 | ||||||||||||||||
Exercise of common stock options | 58 | — | 105 | — | — | 105 | ||||||||||||||||
Common stock issued under ESPP | 54 | — | 238 | — | — | 238 | ||||||||||||||||
Balance at December 31, 2015 | 19,836 | 2 | 135,021 | (50 | ) | (58,602 | ) | 76,371 | ||||||||||||||
Comprehensive loss | ||||||||||||||||||||||
Net loss | — | — | — | — | (22,499 | ) | (22,499 | ) | ||||||||||||||
Change in unrealized losses on available-for-sale securities | — | — | — | 19 | — | 19 | ||||||||||||||||
Total comprehensive loss | (22,480 | ) | ||||||||||||||||||||
Stock-based compensation | — | — | 3,627 | — | — | 3,627 | ||||||||||||||||
Exercise of common stock options | 40 | — | 23 | — | — | 23 | ||||||||||||||||
Common stock issued under ESPP | 40 | — | 139 | — | — | 139 | ||||||||||||||||
Balance at December 31, 2016 | 19,916 | 2 | 138,810 | (31 | ) | (81,101 | ) | 57,680 | ||||||||||||||
Comprehensive loss | ||||||||||||||||||||||
Net loss | — | — | — | — | (30,879 | ) | (30,879 | ) | ||||||||||||||
Change in unrealized losses on available-for-sale securities | — | — | — | — | — | — | ||||||||||||||||
Total comprehensive loss | (30,879 | ) | ||||||||||||||||||||
Restricted stock awards, unvested | 250 | — | — | — | — | — | ||||||||||||||||
Stock-based compensation | — | — | 5,207 | — | — | 5,207 | ||||||||||||||||
Exercise of common stock options | 157 | — | 311 | — | — | 311 | ||||||||||||||||
At-the-Market issuance of common stock, net of $1,038 of issuance costs | 4,502 | 1 | 28,961 | — | — | 28,962 | ||||||||||||||||
Public offering of common stock, net of $1,657 of offering costs | 3,314 | — | 23,198 | — | — | 23,198 | ||||||||||||||||
Common stock issued under ESPP | 44 | — | 201 | — | — | 201 | ||||||||||||||||
Balance at December 31, 2017 | 28,183 | $ | 3 | $ | 196,688 | $ | (31 | ) | $ | (111,980 | ) | $ | 84,680 | |||||||||
The accompanying notes are integral part of these consolidated financial statements.
F- 5
Kindred Biosciences, Inc.
Consolidated Statements of Cash Flows
(In thousands)
Years Ended December 31, | ||||||||||||
2017 | 2016 | 2015 | ||||||||||
Cash Flows from Operating Activities | ||||||||||||
Net loss | $ | (30,879 | ) | $ | (22,499 | ) | $ | (27,130 | ) | |||
Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
Stock-based compensation expense | 5,207 | 3,627 | 4,157 | |||||||||
Depreciation and amortization expense | 475 | 155 | 146 | |||||||||
Loss on disposal of property and equipment | 27 | 3 | — | |||||||||
Amortization of premium on marketable securities | 162 | 274 | 228 | |||||||||
Changes in operating assets and liabilities: | ||||||||||||
Prepaid expenses and other | 485 | (570 | ) | (235 | ) | |||||||
Other assets | (3 | ) | (16 | ) | (8 | ) | ||||||
Accounts payable | 1,280 | (710 | ) | 30 | ||||||||
Accrued liabilities and accrued compensation | 1,368 | 955 | 55 | |||||||||
Net cash used in operating activities | (21,878 | ) | (18,781 | ) | (22,757 | ) | ||||||
Cash Flows from Investing Activities | ||||||||||||
Purchase of investments | (70,110 | ) | (72,047 | ) | (102,195 | ) | ||||||
Sale of investments | 4,897 | — | 3,000 | |||||||||
Maturities of investments | 68,465 | 78,313 | 129,361 | |||||||||
Purchase of property and equipment | (5,920 | ) | (952 | ) | (729 | ) | ||||||
Net cash provided by (used in) investing activities | (2,668 | ) | 5,314 | 29,437 | ||||||||
Cash Flows from Financing Activities | ||||||||||||
Exercise of stock options and purchase of ESPP shares | 512 | 162 | 343 | |||||||||
Net proceeds from sale of common stock | 52,160 | — | — | |||||||||
Net cash provided by financing activities | 52,672 | 162 | 343 | |||||||||
Net change in cash and cash equivalents | 28,126 | (13,305 | ) | 7,023 | ||||||||
Cash and cash equivalents at beginning of year | 6,687 | 19,992 | 12,969 | |||||||||
Cash and cash equivalents at end of year | $ | 34,813 | $ | 6,687 | $ | 19,992 | ||||||
Supplemental disclosure of non-cash financing activities: | ||||||||||||
Purchase of property and equipment included in accounts payable and accrued liabilities | $ | 266 | $ | 670 | $ | 267 | ||||||
The accompanying notes are integral part of these consolidated financial statements.
F- 6
Notes to Consolidated Financial Statements
1. Organization and Description of Business
Kindred Biosciences, Inc. (“we”, "us" or "our") was incorporated on September 25, 2012 (inception) in the State of Delaware. On April 25, 2016, we filed a Certificate of Incorporation with the State of Delaware for a wholly owned subsidiary, KindredBio Equine, Inc. ("Subsidiary"). The Subsidiary has one class of capital stock which is designated common stock, $0.0001 par value per share. The authorized number of shares of common stock for the Subsidiary is 1,000.
We are a pre-commercialization biopharmaceutical company focused on saving and improving the lives of pets. Our activities since inception have consisted principally of raising capital, establishing facilities, recruiting management and technical staff and performing research and development and advancing our product candidates seeking regulatory approval. Our headquarters are in Burlingame, California.
We are subject to risks common to companies in the biotechnology and pharmaceutical industries. There can be no assurance that our research and development will be successfully completed, that adequate protection for our technology will be obtained, that any products developed will obtain necessary government regulatory approval or that any approved products will be commercially viable. We operate in an environment of substantial competition from other animal health companies. In addition, we are dependent upon the services of our employees and consultants, as well as third-party contract research organizations and manufacturers.
Liquidity
We have incurred losses and negative cash flows from operations and have not generated any revenue since our inception and had an accumulated deficit of $111,980,000 as of December 31, 2017. We expect to continue to incur losses and negative cash flows, which will increase significantly from historical levels as we expand our product development activities, seek regulatory approvals for our product candidates, establish a biologics manufacturing capability, and begin to commercialize any approved products. To date, we have been funded primarily through sales of convertible preferred stock, the sale of our common stock in our initial public offering in December 2013, the sale of our common stock in our April 2014 follow-on public offering, periodic sales of our common stock under the ATM in the first half year of 2017 and the sale of our common stock in a follow-on public offering in the third quarter of 2017. We might require additional capital until such time as we can generate operating revenues in excess of operating expenses. In December 2016, we entered into an At Market Issuance Sales Agreement with FBR Capital Markets & Co., or FBR, whereby we were able to issue and sell shares of our common stock having an aggregate offering price up to $30.0 million. During the six months ended June 30, 2017, we sold 4,501,985 shares through FBR under the sales agreement and received approximately $28,962,000 in net proceeds after deducting commissions and other related expenses. On July 12, 2017, we completed an underwritten public offering of 3,000,000 shares of common stock at an offering price of $7.50 per share for total gross proceeds of $22,500,000. On August 11, 2017, we completed the closing of the exercise of the underwriter's option to purchase an additional 314,000 shares of common stock at the public offering price of $7.50 per share, resulting in additional gross proceeds of $2,355,000. After giving effect to the exercise of the over-allotment option, the total number of shares sold by us in the public offering increased to 3,314,000 shares and gross proceeds increased to $24,855,000. Net proceeds, after deducting underwriting commission and offering costs, were approximately $23,198,000. We believe our cash, cash equivalents, short-term and long-term investments of $82,519,000 at December 31, 2017 are sufficient to fund our operations for at least the next 18 months.
If we require additional funding for operations, we may seek such funding through public or private equity or debt financings or other sources, such as corporate collaborations and licensing arrangements. We may not be able to obtain financing on acceptable terms, or at all, and we may not be able to enter into corporate collaborations or licensing arrangements. The terms of any financing may result in dilution or otherwise adversely affect the holdings or the rights of our stockholders. If we are unable to obtain funding, we could be
F- 7
forced to delay, reduce or eliminate our research and development programs or commercialization efforts, which could adversely affect our business prospects.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and include the accounts of the Company and its wholly owned Subsidiary. All intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of consolidated financial statements and related disclosures in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements, and the reported amounts of revenues and expenses during the reporting periods. Significant estimates and assumptions reflected in these consolidated financial statements include, but are not limited to, the valuation of stock-based awards, the realization of deferred tax assets, the recoverability of long-lived assets and the accrual of research and development expenses. Estimates are periodically reviewed in light of changes in circumstances, facts and experience. Actual results could differ from those estimates.
Cash, Cash Equivalents and Investments
We consider all highly liquid investments purchased with an original maturity of three months or less at the date of acquisition to be cash equivalents. Debt securities with original maturities greater than three months and remaining maturities less than one year are classified as short-term investments. We classify all investments as available-for-sale. Available-for-sale securities are carried at estimated fair value, with accumulated unrealized gains and losses reported as a component of accumulated other comprehensive income (loss) in the accompanying consolidated balance sheets.
Realized gains or losses on the sale of investments are determined on a specific identification method, and such gains and losses are reflected as a component of interest and other income, net in the accompanying consolidated statements of operations and comprehensive loss.
Marketable securities investments are evaluated periodically for impairment. We take into account general market conditions, changes in the economic environment as well as specific investment attributes, such as credit downgrade or illiquidity for each investment, the expected cash flows from the securities, our intent to sell the securities and whether or not we will be required to sell the securities before the recovery of their amortized cost, to estimate the fair value of our investments and to determine whether impairment is other than temporary. If it is determined that a decline in fair value of any investment is other than temporary, then the unrealized loss related to credit risk would be included in interest and other income, net.
Concentration of Credit Risk and of Significant Suppliers
Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash, cash equivalents and investments. From time to time, we maintain cash and cash equivalent balances in excess of amounts insured by the Federal Deposit Insurance Corporation (“FDIC”) and the Securities Investor Protection Corporation ("SIPC"). Primarily all of our cash, cash equivalents and investments at December 31, 2017 were in excess of amounts insured by the FDIC and SIPC. We do not believe that we are subject to unusual credit risk beyond the normal credit risk associated with commercial banking relationships.
F- 8
We are dependent on third-party manufacturers to supply products for research and development activities in our programs. In particular, we rely on a small number of manufacturers to supply us with our requirements for the active pharmaceutical ingredients, or API, and formulated drugs related to some of these programs. These programs would be adversely affected by a significant interruption in the supply of API.
Fair Value Measurements
We use the provisions of Accounting Standards Codification ("ASC") 820, “Fair Value Measurements and Disclosure", to determine the fair values of our financial and nonfinancial assets and liabilities where applicable. ASC 820 defines fair value, establishes a framework for measuring fair value in U.S. GAAP and expands disclosure about fair value measurements. The objective of fair value measurement is to determine the price that would be received to sell the asset or paid to transfer the liability (an exit price) in an orderly transaction between market participants at the measurement date. ASC 820 emphasizes that fair value is a market-based measurement, not an entity-specific measurement, and that market participant assumptions include assumptions about risk and effect of a restriction on the sale or use of an asset. To increase consistency and comparability in fair value measurement and related disclosures, ASC 820 establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value into three broad levels: (1) Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities that the reporting entity has the ability to access at the measurement date; (2) Level 2 inputs are inputs other than quoted prices included in Level 1 that are observable for the asset or liability, either directly or indirectly through corroboration with observable market data; and (3) Level 3 inputs are unobservable inputs for the asset or liability that reflect the reporting entity’s own assumptions about risk and the assumptions market participants would use in pricing the asset or liability developed based on the best information available in the circumstances.
Government agency notes, corporate notes and commercial papers are recorded at their estimated fair value. Since these available-for-sale securities generally have market prices from multiple sources and it can be difficult to select the best individual price directly from the quoted prices in the active markets, we use Level 2 inputs for the valuation of these securities. Using the Level 2 inputs, a “consensus price” or a weighted average price for each of these securities can be derived from a distribution-curve-based algorithm which includes market prices obtained from a variety of industrial standard data providers (e.g. Bloomberg), security master files from large financial institutions, and other third-party sources.
The carrying amount of financial instruments, including cash, accounts payable and accrued liabilities approximate fair value due to the short maturities of these financial instruments. Financial assets, which consist of money market funds and available-for-sale securities, are measured at fair value on a recurring basis (see Note 3).
Property and Equipment
On June 21, 2017, we entered into a purchase agreement with Strategic Veterinary Pharmaceuticals, Inc. ("SVP") for the purchase of an approximately 180,000 sq. ft. biologics plant ("the Plant") with clean rooms, utility, equipment, and related quality documentation suitable for small molecule and biologics manufacturing, that is located in Elwood, Kansas. The purchase was finalized on August 7, 2017 upon completion of the diligence period and satisfaction of the conditions of escrow. The Plant was purchased for $3,750,000, which includes approximately eight acres of land located at 1411 Oak Street, Elwood, Kansas, all improvements located at the Plant, and all personal property and intangible property owned by SVP and located at the Plant or used in connection with the operation of the Plant.
Property and equipment are stated at cost less accumulated depreciation and amortization. We calculate depreciation using the straight-line method over the estimated useful lives of the assets, which range
F- 9
from two to five years for furniture, fixtures, lab and computer equipment and software, and fifteen to thirty-nine years for land improvements and real property. Land and assets held within construction in progress are not depreciated. Construction in progress is related to the construction or development of property and equipment that have not yet been placed in service for their intended use. Expenditures for repairs and maintenance of assets are charged to expense as incurred. We amortize leasehold improvements using the straight-line method over the estimated useful lives of the respective assets or the lease term, whichever is shorter. Upon retirement or sale, the cost and related accumulated depreciation and amortization of assets disposed of are removed from the accounts and any resulting gain or loss is included in other income/expense.
Licenses
The costs incurred for the rights to use licensed technologies in the research and development process, including licensing fees and milestone payments, are charged to research and development expense as incurred in situations where we have not identified an alternative future use for the acquired rights, and are capitalized in situations where we have identified an alternative future use. No costs associated with the use of licensed technologies have been capitalized to date.
Impairment of Long-Lived Assets
We review long-lived assets, including property and equipment, for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. Factors that we consider in deciding when to perform an impairment review include significant underperformance of the business in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in the use of the assets. If an impairment review is performed to evaluate a long-lived asset for recoverability, we compare forecasts of undiscounted cash flows expected to result from the use and eventual disposition of the long-lived asset to its carrying value. An impairment loss would be recognized when estimated undiscounted future cash flows expected to result from the use of an asset are less than its carrying amount. The impairment loss would be based on the excess of the carrying value of the impaired asset over its fair value, determined based on discounted cash flows. To date, we have not recorded any impairment losses on long-lived assets.
Revenue Recognition
We are in the research and development stage and have not generated any revenues since inception.
Research and Development Costs
All costs of research and development are expensed in the period incurred. Research and development costs primarily consist of salaries and related expenses for personnel, stock-based compensation expense, fees paid to consultants, outside service providers, professional services, travel costs and materials used in clinical trials and research and development.
Patent Costs
All patent-related costs incurred in connection with filing patent applications are recorded in research and development expenses when incurred, as recoverability of such expenditures is uncertain.
Stock-Based Compensation
Our stock-based compensation plan (see Note 8) provides for the grant of stock options, restricted common stock and stock appreciation rights. The estimated fair values of employee stock option grants are determined
F- 10
as of the date of grant using the Black-Scholes option pricing model. This method incorporates the fair value of our common stock at the date of each grant and various assumptions such as the risk-free interest rate, expected volatility based on the historic volatility of publicly-traded peer companies, expected dividend yield, and expected term of the options. The estimated fair values of restricted stock awards are determined based on the fair value of our common stock on the date of grant. The estimated fair values of stock-based awards, including the effect of estimated forfeitures, are expensed over the requisite service period, which is generally the awards’ vesting period. We classify stock-based compensation expense in the consolidated statements of operations and comprehensive loss in the same manner in which the award recipient’s payroll costs are classified.
Our accounting policy for equity instruments issued to consultants and vendors in exchange for goods and services follows Financial Accounting Standards Board ("FASB") guidance. All transactions in which goods or services are the consideration received for the issuance of equity instruments are accounted for based on the fair value of the consideration received or the fair value of the equity instrument issued, whichever is more reliably measurable. The measurement date of the fair value of the equity instrument issued is the earlier of the date on which the counterparty’s performance is complete or the date at which a commitment for performance is reached. For transactions in which the fair value of the equity instrument issued to non-employees is the more reliable measurement and a measurement date has not been reached, the fair value is re-measured at each vesting and reporting date using the Black-Scholes option pricing model. Compensation expense for these share-based awards is recognized over the term of the consulting agreement or until the award is approved and settled.
Income Taxes
We account for income taxes using the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the consolidated financial statements or in our tax returns. Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. Changes in deferred tax assets and liabilities are recorded in the provision for income taxes. We assess the likelihood that our deferred tax assets will be recovered from future taxable income and, to the extent we believe, based upon the weight of available evidence, that it is more likely than not that all or a portion of deferred tax assets will not be realized, a valuation allowance is established through a charge to income tax expense. Potential for recovery of deferred tax assets is evaluated by estimating the future taxable profits expected and considering prudent and feasible tax planning strategies.
We account for uncertainty in income taxes recognized in the consolidated financial statements by applying a two-step process to determine the amount of tax benefit to be recognized. First, the tax position must be evaluated to determine the likelihood that it will be sustained upon external examination by the taxing authorities. If the tax position is deemed more-likely-than-not to be sustained, the tax position is then assessed to determine the amount of benefit to recognize in the consolidated financial statements. The amount of the benefit that may be recognized is the largest amount that has a greater than 50% likelihood of being realized upon ultimate settlement. The provision for income taxes includes the effects of any resulting tax reserves, or unrecognized tax benefits, that are considered appropriate as well as the related net interest and penalties.
Comprehensive Loss
Our comprehensive loss includes the change in unrealized gains or losses on available-for-sale securities. The cumulative amount of gains or losses is reflected as a separate component of stockholders' equity in the accompanying consolidated balance sheets as accumulated other comprehensive loss.
F- 11
Segment Data
We manage our operations as a single segment for the purposes of assessing performance and making operating decisions. We are a veterinary biotechnology company focusing on developing therapies for pets. Our chief operating decision maker is our Chief Executive Officer. All assets are held in the United States.
Basic and Diluted Net Loss Per Common Share
Basic net loss per common share is computed by dividing net loss attributable to common stockholders for the period by the weighted average number of common shares outstanding during the period. Diluted net loss per share is computed by dividing the net loss attributable to common stockholders for the period by the weighted average number of common shares, including potential dilutive shares of common stock assuming the dilutive effect of potentially dilutive securities. For periods in which we have reported a net loss, diluted net loss per common share is the same as basic net loss per common share, since the impact of the potentially dilutive securities would be anti-dilutive to the calculation of net loss per common share (see Note 12).
Recently Issued Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board ("FASB") issued Accounting Standards Update ("ASU") No. 2014-09, "Revenue from Contracts with Customers". This new standard will replace most of the existing revenue recognition guidance in U.S. GAAP when it becomes effective and permits the use of either the retrospective or cumulative effect transition method. The new standard, as amended, becomes effective in the first quarter of fiscal year 2018, but allows the adoption of the standard one year earlier if we so choose. The analysis identifying areas that will be impacted by the new guidance is complete. Additionally, we will continue to monitor modifications, clarifications, and interpretations issued by the FASB that may impact its assessment. We do not currently have and have never had any contracts that are within the scope of ASC 606, "Revenue from Contracts with Customers" or its predecessor guidance, ASC 605, "Revenue Recognition". Accordingly, based on our current assessment as of December 31, 2017, we do not believe adopting this guidance in the first quarter of 2018 will have a material impact on our consolidated financial statements as we are not currently generating revenues.
In January 2016, the FASB issued ASU No. 2016-01, "Financial Instruments—Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities", which amends the guidance in U.S. GAAP on the classification and measurement of financial instruments and also amends certain disclosure requirements associated with the fair value of financial instruments. The new guidance is effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. We have adopted the new guidance and it did not have a material impact on our consolidated financial statements.
In February 2016, the FASB issued ASU No. 2016-02, "Leases (Topic 842)", requiring organizations that lease assets—referred to as “lessees”—to recognize on the consolidated balance sheet the assets and liabilities for the rights and obligations created by those leases. Under the new guidance, a lessee will be required to recognize assets and liabilities for leases with lease terms of more than 12 months. The ASU on leases will take effect for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018. We are currently evaluating the new guidance and have not determined the impact this standard may have on our consolidated financial statements.
In May 2017, the FASB issued ASU No. 2017-09, “Scope of Modification Accounting”, which amends ASC Topic 718, “Compensation - Stock Compensation”. The ASU includes provisions intended to (1) provide clarity and (2) reduce diversity in practice and reduce cost and complexity when calculate stock compensation, on a change to the terms or conditions of a share-based payment award. ASU 2017-09 is effective for public business entities for annual reporting periods, and interim periods within those annual periods, beginning after
F- 12
December 15, 2017. Early adoption will be permitted in any interim or annual period, with any adjustments reflected as of the beginning of the fiscal year of adoption. We have adopted the new guidance and it did not have a material impact on our consolidated financial statements.
We do not believe there are any other recently issued standards not yet effective that will have a material impact on our consolidated financial statements when the standards become effective.
3. Fair Value Measurements
We measure certain financial assets at fair value on a recurring basis, including cash equivalents and available-for-sale securities. The fair value of these financial assets was determined based on a three-tier fair value hierarchy as described in Note 2, which prioritizes the inputs used in measuring fair value.
The following table presents information about our financial assets that are measured at fair value on a recurring basis as of December 31, 2017 and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value:
(In thousands) | Fair Value Measurements as of December 31, 2017 | |||||||||||||||
Description | Total | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Unobservable Inputs (Level 3) | ||||||||||||
Cash equivalents: | ||||||||||||||||
Money market funds | $ | 801 | $ | 801 | $ | — | $ | — | ||||||||
Commercial paper | 31,977 | — | 31,977 | — | ||||||||||||
Corporate notes | 1,500 | — | 1,500 | — | ||||||||||||
Short-term investments: | ||||||||||||||||
Commercial paper | 22,052 | — | 22,052 | — | ||||||||||||
U.S. government agency notes | 6,746 | — | 6,746 | — | ||||||||||||
U.S. treasury bonds and notes | 3,482 | 3,482 | — | — | ||||||||||||
Corporate notes | 13,927 | — | 13,927 | — | ||||||||||||
Long-term investments: | ||||||||||||||||
Corporate notes | 1,499 | — | 1,499 | — | ||||||||||||
$ | 81,984 | $ | 4,283 | $ | 77,701 | $ | — | |||||||||
The following table presents information about our financial assets that are measured at fair value on a recurring basis as of December 31, 2016 and indicates the fair value hierarchy of the valuation techniques utilized to determine such fair value:
F- 13
(In thousands) | Fair Value Measurements as of December 31, 2016 | |||||||||||||||
Description | Total | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Unobservable Inputs (Level 3) | ||||||||||||
Cash equivalents: | ||||||||||||||||
Money market funds | $ | 3,157 | $ | 3,157 | $ | — | $ | — | ||||||||
Commercial paper | 850 | — | 850 | — | ||||||||||||
Short-term investments: | ||||||||||||||||
U.S. treasury bills | 5,997 | 5,997 | — | — | ||||||||||||
Commercial paper | 4,228 | — | 4,228 | — | ||||||||||||
U.S. government agency notes | 13,550 | — | 13,550 | — | ||||||||||||
U.S. treasury bonds and notes | 11,015 | 11,015 | — | — | ||||||||||||
Corporate notes | 15,278 | — | 15,278 | — | ||||||||||||
Long-term investments: | ||||||||||||||||
Corporate notes | 1,052 | — | 1,052 | — | ||||||||||||
$ | 55,127 | $ | 20,169 | $ | 34,958 | $ | — | |||||||||
There were no other transfers of assets between Level 1, Level 2 or Level 3 of the fair value hierarchy during the years ended December 31, 2017.
During the year ended December 31, 2016, U.S. treasuries were transferred from Level 2 to Level 1. With the exception of U.S. treasuries, there were no other transfers of assets between Level 1, Level 2 or Level 3 of the fair value hierarchy during the years ended December 31, 2016.
At December 31, 2017 and 2016, we did not have any financial liabilities which were measured at fair value on a recurring basis.
4. Investments
The following tables summarize our investments in available-for-sale securities by significant investment category reported as short-term or long-term investments as of December 31, 2017 and 2016 (in thousands):
F- 14
December 31, 2017 | ||||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | |||||||||||||
Short-term investments: | ||||||||||||||||
Commercial paper | $ | 22,052 | $ | — | $ | — | $ | 22,052 | ||||||||
U.S. government agency notes | 6,750 | — | (4 | ) | 6,746 | |||||||||||
U.S. treasury bonds and notes | 3,483 | — | (1 | ) | 3,482 | |||||||||||
Corporate notes | 13,946 | — | (19 | ) | 13,927 | |||||||||||
46,231 | — | (24 | ) | 46,207 | ||||||||||||
Long-term investments: | ||||||||||||||||
Corporate notes | 1,506 | — | (7 | ) | 1,499 | |||||||||||
Total available-for-sale investments | $ | 47,737 | $ | — | $ | (31 | ) | $ | 47,706 | |||||||
December 31, 2016 | ||||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | |||||||||||||
Short-term investments: | ||||||||||||||||
U.S. treasury bills | $ | 5,996 | $ | 1 | $ | — | $ | 5,997 | ||||||||
Commercial paper | 4,228 | — | — | 4,228 | ||||||||||||
U.S. government agency notes | 13,552 | 1 | (3 | ) | 13,550 | |||||||||||
U.S. treasury bonds and notes | 11,015 | 1 | (1 | ) | 11,015 | |||||||||||
Corporate notes | 15,305 | — | (27 | ) | 15,278 | |||||||||||
50,096 | 3 | (31 | ) | 50,068 | ||||||||||||
Long-term investments: | ||||||||||||||||
Corporate notes | 1,055 | — | (3 | ) | 1,052 | |||||||||||
Total available-for-sale investments | $ | 51,151 | $ | 3 | $ | (34 | ) | $ | 51,120 | |||||||
The following table summarizes the contractual maturities of our available-for-sale securities at December 31, 2017 (in thousands):
Amortized Cost | Fair Value | ||||||
Mature in less than one year | $ | 46,231 | $ | 46,207 | |||
Mature in one year or more | $ | 1,506 | $ | 1,499 | |||
5. Property and Equipment, Net
F- 15
Property and equipment consisted of the following:
As of December 31, | |||||||
(in thousands) | 2017 | 2016 | |||||
Computer and lab equipment | $ | 2,303 | $ | 565 | |||
Furniture & fixtures | 46 | 41 | |||||
Leasehold improvements | 930 | 82 | |||||
Construction-in-process | 4,969 | 2,073 | |||||
Total | 8,248 | 2,761 | |||||
Less accumulated depreciation and amortization | (791 | ) | (320) | ||||
Property and equipment, net | $ | 7,457 | $ | 2,441 | |||
Construction-in-process is comprised of a building, land, land improvement and equipment that have not been put into services for its intended use as of December 31, 2017. As disclosed in Note 2, the Plant was purchased for $3,750,000, which includes approximately eight acres of land, all improvements located at the Plant, and all personal property and intangible property located at the Plant or used in connection with the operation of the Plant. We also purchased approximately $1,219,000 additional manufacturing and lab equipment that have not been put into service, resulting in a construction-in-process balance of $4,969,000 at December 31, 2017.
Depreciation and amortization expense was $475,000, $155,000 and $146,000 for the years ended December 31, 2017, 2016 and 2015, respectively.
6. Accrued Liabilities
Accrued liabilities consisted of the following as of December 31, 2017 and 2016:
(In thousands) | December 31, 2017 | December 31, 2016 | |||||
Accrued consulting | $ | 335 | $ | 106 | |||
Accrued research and development costs | 919 | 1,390 | |||||
Accrued other | 646 | 124 | |||||
Deferred rent | 115 | 59 | |||||
2,015 | 1,679 | ||||||
Less current portion | (1,900 | ) | (1,650 | ) | |||
Long-term liability (deferred rent) | $ | 115 | $ | 29 | |||
7. Stockholders' Equity
Preferred Stock
Our Certificate of Incorporation, as amended, authorizes us to issue 10,000,000 shares of $0.0001 par value preferred stock. At December 31, 2017, 100,000 unissued shares of our preferred stock are designated as Series A Preferred Stock, and the remaining 9,900,000 unissued shares of our preferred stock are undesignated.
Common Stock
Our Certificate of Incorporation, as amended and restated, authorizes us to issue 100,000,000 shares of
F- 16
$0.0001 par value common stock.
Each share of common stock entitles the holder to one vote on all matters submitted to a vote of our stockholders, provided, however, that, except as otherwise required by law, holders of common stock shall not be entitled to vote on any amendment to the Certificate of Incorporation that relates solely to the terms of one or more outstanding shares of preferred stock if the holders of such affected series are entitled, either separately or together with the holders of one or more other series, to vote thereon pursuant to the Certificate of Incorporation or pursuant to the Delaware General Corporation Law.
In 2015, we issued 58,126 shares of common stock upon exercise of stock options for total proceeds of $105,000. In addition, we issued 53,752 shares of common stock to employees in connection with our employee stock purchase program for total proceeds of $238,000.
In 2016, we issued 39,501 shares of common stock upon exercise of stock options for total proceeds of $23,000. In addition, we issued 40,429 shares of common stock to employees in connection with our employee stock purchase program for total proceeds of $139,000.
In 2017, we issued 156,927 shares of common stock upon exercise of stock options for total proceeds of $311,000. In addition, we issued 43,561 shares of common stock to employees in connection with our employee stock purchase program for total proceeds of $201,000.
As of December 31, 2017, we had 28,182,563 shares of common stock outstanding.
Stock Offerings
In January 2015, we filed a shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $150.0 million. On December 19, 2016, we entered into an At Market Issuance Sales Agreement with FBR Capital Markets & Co., or FBR, pursuant to which we were able to issue and sell shares of our common stock having an aggregate offering price up to $30.0 million, through FBR as our sales agent. In conjunction with the sales agreement, FBR received compensation based on an aggregate of 3% of the gross proceeds on the sale price per share of our common stock. Any sales made pursuant to the sales agreement were deemed an “at-the-market” offering and were made pursuant to the shelf registration statement on Form S-3. For the year ended December 31, 2016, we did not sell any shares through FBR under the sales agreement. During the six months ended June 30, 2017, we completed the sale of 4,501,985 shares of common stock under the Sales Agreement. Net proceeds, after deducting approximately $906,000 in commissions and fees and approximately $132,000 in offering costs, were approximately $28,962,000. On July 12, 2017, we completed an underwritten public offering of 3,000,000 shares of common stock at an offering price of $7.50 per share for total gross proceeds of $22,500,000. On August 11, 2017, we completed the closing of the exercise of the underwriter's option to purchase an additional 314,000 shares of common stock at the public offering price of $7.50 per share, resulting in additional gross proceeds of $2,355,000. After giving effect to the exercise of the over-allotment option, the total number of shares sold by us in the public offering increased to 3,314,000 shares and gross proceeds increased to $24,855,000. Net proceeds, after deducting underwriting commission and offering costs, were approximately $23,198,000.
In January 2018, we filed a new shelf registration statement on Form S-3 to offer and sell, from time to time, equity and debt securities in one or more offerings up to a total dollar amount of $150.0 million.
8. Stock-Based Awards and Benefit Plan
On November 4, 2012, our board of directors adopted the Kindred Biosciences, Inc. 2012 Equity Incentive
F- 17
Plan or the “2012 Plan". The 2012 Plan provided for our board of directors to grant incentive stock options or non-qualified stock options for the purchase of common stock, to issue or sell shares of restricted common stock and to grant stock appreciation rights (“SARs”) to our employees, directors, consultants and advisers of the Company. Pursuant to the terms of the 2012 Plan, no options or SARs shall be granted under the 2012 Plan after 10 years from the date of adoption of the 2012 Plan. We have reserved 4,000,000 shares of our common stock for issuance under the 2012 Plan. The 2012 Plan terminated in May 2016 and 3,315,200 stock option shares which had been granted prior to the plan’s expiration remaining outstanding as of December 31, 2017.
In May 2016, we adopted the 2016 Equity Incentive Plan or the “2016 Plan”, and reserved 3,000,000 shares of our common stock for issuance under the 2016 Plan. The 2016 Plan is the successor to our 2012 Plan and all awards made under the 2012 Plan shall remain subject to the terms of that plan. Options granted under the 2016 Plan may be either incentive stock options or nonstatutory stock options. The 2016 Plan also provides for the grant of stock appreciation rights, restricted stock awards, restricted stock unit awards, performance stock awards, performance cash awards and other stock awards. The exercise price of a stock option may not be less than 100% of the closing price of our common stock on the date of the grant. If, at any time we grant an option, and the optionee directly or by attribution owns stock possessing more than 10% of the total combined voting power of all classes of our stock, the option price shall be at least 110% of the fair value and shall not be exercisable more than five years after the date of grant. Options generally vest over a period of one or four years from the date of grant. Options granted under the 2016 Plan expire no later than 10 years from the date of grant. As of December 31, 2017, there were 1,258,425 option shares outstanding, 250,000 restricted stock awards issued but unvested, and 1,488,450 shares available for future grants under the 2016 Plan.
2014 Employee Stock Purchase Plan
In December 2014, our board of directors adopted the Kindred Biosciences, Inc. 2014 Employee Stock Purchase Plan (the “Purchase Plan”). A total of 200,000 shares of our common stock are authorized for issuance under the Purchase Plan. The Purchase Plan permits eligible employees to purchase common stock at a discount through payroll deductions during defined six months consecutive offering periods beginning on December 1 with the exception of our first offering period which commenced on January 1, 2015 for a five months duration. The price at which the stock is purchased is equal to the lower of 85% of the fair market value of the common stock on the first day of the offering or 85% of the fair market value of our common stock on the purchase date. A participant may purchase a maximum of 2,000 shares of common stock during each offering period, not to exceed $25,000 worth of common stock on the offering date during each calendar year. We use the Black-Scholes option pricing model, in combination with discounted employee price, in determining the value of the Purchase Plan expense to be recognized during each offering period. The weighted-average grant date fair value per share using the Black-Scholes option pricing model was $1.73 during the year ended December 31, 2017.
As of December 31, 2017, there were 137,542 shares of common stock issued under the Purchase Plan and 62,458 shares available for future issuance under the Purchase Plan. At December 31, 2017 and 2016, we had an outstanding liability of $27,000 and $17,000, respectively, which is included in accrued compensation on the consolidated balance sheets, for employee contributions to the Purchase Plan for shares pending issuance at the end of the next offering period.
Reserved Shares
At December 31, 2017, shares of common stock reserved for future issuance inclusive of outstanding option shares are as follows:
F- 18
2016 Equity Incentive Plan | 2,746,875 | |
2014 Employee Stock Purchase Plan | 62,458 | |
2,809,333 | ||
Stock Option Plan Activity Summary
A summary of activity under our stock option plans is as follows:
Shares Available For Grant | Shares Issuable Under Options | Weighted Average Exercise Price | Weighted Average Remaining ContractualTerm (In Years) | Aggregate Intrinsic Value | |||||||
Balance, December 31, 2014 | 1,571,298 | 2,329,415 | $7.60 | ||||||||
Granted | (922,083 | ) | 922,083 | $6.54 | |||||||
Exercised | (58,126 | ) | $1.81 | ||||||||
Expired | 5,104 | (5,104 | ) | $19.96 | |||||||
Forfeited - stock options | 72,083 | (72,083 | ) | $12.48 | |||||||
Balance, December 31, 2015 | 726,402 | 3,116,185 | $7.26 | 8.2 | $2,908,000 | ||||||
2012 Plan terminated (a) | (307,107 | ) | |||||||||
2016 Plan authorized (b) | 3,000,000 | ||||||||||
Granted | (847,683 | ) | 847,683 | $3.48 | |||||||
Exercised | (39,501 | ) | $0.57 | ||||||||
Expired | 43,818 | (43,818 | ) | $14.52 | |||||||
Forfeited - stock options | 312,220 | (312,220 | ) | $7.06 | |||||||
Balance, December 31, 2016 | 2,927,650 | 3,568,329 | $6.36 | 7.5 | $4,353,000 | ||||||
2012 Plan true up retired shares (c) | (26,977 | ) | |||||||||
2016 Plan issued RSA shares (d) | (250,000 | ) | |||||||||
Granted | (1,208,200 | ) | 1,208,200 | $6.62 | |||||||
Exercised | (156,927 | ) | $1.98 | ||||||||
Expired | 7,267 | (7,267 | ) | $14.54 | |||||||
Forfeited - stock options | 38,710 | (38,710 | ) | $6.60 | |||||||
Balance, December 31, 2017 | 1,488,450 | 4,573,625 | $6.57 | 7.2 | $18,745,000 | ||||||
Options vested and expected to vest, December 31, 2017 | 4,573,625 | $6.57 | 7.2 | $18,745,000 | |||||||
Options exercisable, December 31, 2017 | 2,962,057 | $6.80 | 6.4 | $13,208,000 | |||||||
(a) The 2012 Equity Incentive Plan terminated in May 2016. All shares available for grant under this Plan expired.
(b) The 2016 Equity Incentive Plan was adopted and approved by stockholders in May 2016.
(c) True up of all expired shares available for grant under the 2012 Equity Incentive Plan, which was terminated in May 2016.
(d) Issued 250,000 RSA shares on 01/23/2017 under the 2016 Equity Incentive Plan.
The aggregate intrinsic value of options is calculated as the difference between the exercise price of options and the fair value of our common stock for those options that had exercise prices lower than the fair value of our common stock on December 31, 2017, 2016 and 2015.
The aggregate intrinsic value of stock options exercised during the years ended December 31, 2017, 2016 and
F- 19
2015 was $911,000, $117,000 and $266,000, respectively.
We received proceeds of $311,000, $23,000 and $105,000 from the exercise of common stock options during the years ended December 31, 2017, 2016 and 2015, respectively.
The weighted-average grant date fair value of options granted during the years ended December 31, 2017, 2016 and 2015 was $4.21, $2.45 and $5.03 per share, respectively.
Restricted Stock
On January 23, 2017, we granted 250,000 shares of restricted stock awards to four employees. Shares will vest 25% on each one year anniversary of the grant date provided that the employee is in the employment of the Company on such vesting date. The total stock-based compensation expense related to these awards is $1,600,000. As of December 31, 2017, we have an aggregate of approximately $1,225,000 unrecognized stock-based compensation expense for restricted stock awards outstanding which is expected to be recognized over a weighted-average period of 3.1 years. We did not grant any restricted common stock in 2016 and 2015.
Restricted stock activity for the year ended December 31, 2017, was as follows:
Restricted Stock Award | Shares | Weighted Average Grant Date Fair Value | |||
Unvested balance at December 31, 2016 | — | — | |||
Granted | 250,000 | $6.40 | |||
Vested | — | — | |||
Forfeited | — | — | |||
Unvested balance at December 31, 2017 | 250,000 | $6.40 | |||
Stock-Based Compensation
We recognize stock-based compensation expense for only the portion of awards that are expected to vest. In developing a forfeiture rate estimate, we have considered our historical experience to estimate pre-vesting forfeitures for service-based awards. The impact of a forfeiture rate adjustment will be recognized in full in the period of adjustment, and if the actual forfeiture rate is materially different from our estimate, we may be required to record adjustments to stock-based compensation expense in future periods.
The fair value of each stock option grant is estimated on the date of grant using the Black-Scholes option-pricing model. Due to insufficient company-specific historical and implied volatility information, we estimate the expected stock price volatility based on the historical volatility of publicly traded peer companies and expect to continue to do so until such time as we have adequate historical data regarding the volatility of our common stock price. The expected term of our common stock options has been determined utilizing the “simplified” method as we have insufficient historical experience for options grants overall, rendering existing historical experience irrelevant to expectations for current grants. The risk-free interest rate is determined by reference to the U.S. Treasury yield curve in effect at the time of grant of the award for time periods approximately equal to the expected term of the award. Expected dividend yield is based on the fact that we have never paid cash dividends and does not expect to pay any cash dividends in the foreseeable future.
F- 20
Total stock-based compensation expense, related to all of our share-based payment awards, is comprised of the following:
(In thousands) | Years Ended December 31, | |||||||
2017 | 2016 | 2015 | ||||||
Research and development | $1,650 | $1,463 | $1,857 | |||||
General and administrative | 3,557 | 2,164 | 2,300 | |||||
$5,207 | $3,627 | $4,157 | ||||||
Total stock-based compensation expense includes stock options, restricted stock award and expense from the Purchase Plan for the years ended December 31, 2017, 2016, and 2015.
We had an aggregate of approximately $5,524,000 of unrecognized stock-based compensation expense for options outstanding and the Purchase Plan as of December 31, 2017, which is expected to be recognized over a weighted average period of 2.6 years.
Valuation assumptions
The relevant data used to determine the fair value of stock options earned or granted and the Purchase Plan is as follows:
Years Ended December 31, | |||||
2017 | 2016 | 2015 | |||
Stock options: | |||||
Weighted average risk-free interest rate | 1.98% | 1.61% | 1.51% | ||
Weighted average expected term (in years) | 6.0 | 6.3 | 6.1 | ||
Weighted average expected volatility | 70.4% | 83.4% | 94.8% | ||
Weighted average expected dividend yield | — | — | — | ||
Fair value at grant date | $4.21 | $2.45 | $5.03 | ||
Employee stock purchase plan: | |||||
Weighted average risk-free interest rate | 1.04% | 0.50% | 0.08% | ||
Weighted average expected term (in years) | 0.5 | 0.5 | 0.5 | ||
Weighted average expected volatility | 56.7% | 84.4% | 80.7% | ||
Weighted average expected dividend yield | — | — | — | ||
Fair value at grant date | $1.73 | $1.42 | $2.08 | ||
Restricted Stock Award: | |||||
Fair value at grant date | $6.40 | — | — | ||
9. Commitments and Contingencies
F- 21
Operating Leases
In April 2014, we entered into non-cancelable operating leases for 2,145 square feet of laboratory space through May 2017 and 6,900 square feet of office space through November 2017. In January, August and November 2015, we amended our original operating lease for laboratory space to expand the facility with an additional 2,431 square feet, 131 square feet and 123 square feet, respectively, of manufacturing space through May 2017. In August 2015, we entered into a new non-cancelable operating lease for 3,126 square feet of office space in San Diego, California through September 2019. In February and October 2016, we further amended our original operating lease for laboratory space to further expand the facility with an additional 3,599 square feet and 2,326 square feet, respectively, of laboratory space through May 2017. Commencing on June 1, 2017, the non-cancellable operating lease for the entire existing laboratory space of a total 10,755 square feet is extended for another 5 years through May 2022. In February 2017, we further amended the operating lease for laboratory space with an additional 721 square feet through May 2022. In April 2017, we renewed our headquarters office lease for 6,900 square feet of office space in Burlingame, California through November 30, 2020 and in June 2017, we amended the lease with an additional 1,190 square feet of office space through November 30, 2020. In addition, we have three equipment leases expiring through 2020.
Rent expense for the years ended December 31, 2017, 2016 and 2015 was $679,000, $522,000 and $339,000, respectively. In addition, we have three operating leases for equipment through 2020. As of December 31, 2017, we are obligated to make minimum lease payments under all of our operating leases as follows (in thousands):
Year ending December 31, | Lease Payments | |||
2018 | $ | 812 | ||
2019 | 810 | |||
2020 | 726 | |||
2021 | 459 | |||
2022 | 194 | |||
Total | $ | 3,001 | ||
Indemnities and Guarantees
We have made certain indemnities and guarantees, under which we may be required to make payments to a guaranteed or indemnified party, in relation to certain transactions. We indemnify our officers and directors to the maximum extent permitted under the laws of the State of Delaware. The duration of these indemnities and guarantees varies and, in certain cases, is indefinite. These indemnities and guarantees do not provide for any limitation of the maximum potential future payments we could be obligated to make. Historically, we have not been obligated to make any payments for these obligations and no liabilities have been recorded for these indemnities and guarantees in the accompanying consolidated balance sheets.
Legal Matters
In the ordinary course of business, we may face various claims brought by third parties and may, from time to time, make claims or take legal actions to assert our rights, including intellectual property disputes, contractual disputes and other commercial disputes. Any of these claims could subject us to litigation. Management believes there are currently no claims that are likely to have a material effect on our consolidated financial position and results of operations.
F- 22
10. Restructuring Plan
On January 8, 2016, our Board of Directors committed to a restructuring plan intended to better align our workforce to our revised operating needs and program development plans. On January 11, 2016, we implemented the restructuring plan that focused on streamlining our development programs and to ensure our remaining funds are sufficient to fund our planned operations through 2018. To match these priorities, we reduced our workforce by 18 positions, or approximately 31% of our workforce, resulting in a total workforce of 39 positions. As a result of the restructuring plan which has been completed, we recorded a restructuring charge of approximately $655,000 related to severance payments which was entirely paid in the quarter ended March 31, 2016. There were no further restructuring charges since then.
11. Income Taxes
There is no provision for income taxes because we have historically incurred operating losses and we maintain a full valuation allowance against our net deferred tax assets.
Differences between the provision (benefit) for income taxes and income taxes at the statutory federal income tax rate are as follows:
(In thousands, except percentages) | For the years ended December 31, | |||||||||||||||||||
2017 | 2016 | 2015 | ||||||||||||||||||
Income tax expense (benefit) at statutory federal rate | $ | (10,499 | ) | 34.0 | % | $ | (7,651 | ) | 34.0 | % | $ | (9,226 | ) | 34.0 | % | |||||
State income tax, net of federal benefit | (1,880 | ) | 6.1 | (1,389 | ) | 6.2 | (1,678 | ) | 6.2 | |||||||||||
Permanent items | 63 | (0.2 | ) | 38 | (0.2 | ) | 58 | (0.2 | ) | |||||||||||
Research credits | (1,172 | ) | 3.8 | (970 | ) | 4.3 | (1,379 | ) | 5.1 | |||||||||||
Stock-based compensation | (122 | ) | 0.4 | (29 | ) | 0.1 | — | — | ||||||||||||
ASC 740-10 | 469 | (1.5 | ) | 388 | (1.7 | ) | 552 | (2 | ) | |||||||||||
Change in valuation allowance | (128 | ) | 0.4 | 9,602 | (42.6 | ) | 11,924 | (43.9 | ) | |||||||||||
Tax Cuts and Jobs Act | 13,092 | (42.4 | ) | — | — | — | — | |||||||||||||
Other | 177 | (0.6 | ) | 11 | (0.1 | ) | (251 | ) | 0.8 | |||||||||||
Provision (benefit) for income taxes | $ | — | — | % | $ | — | — | % | $ | — | — | % | ||||||||
Deferred tax assets are recognized for temporary differences that will result in deductible amounts in future periods. The components of the deferred tax assets are as follows at December 31, 2017 and 2016:
F- 23
December 31, | |||||||
(In thousands) | 2017 | 2016 | |||||
Deferred tax assets: | |||||||
Net operating loss carryforwards | $ | 26,369 | $ | 27,093 | |||
Research & development credits | 3,044 | 2,173 | |||||
Accrued expenses | 753 | 720 | |||||
Amortization and depreciation | (132 | ) | 2,738 | ||||
Stock-based compensation | 4,860 | 2,296 | |||||
Other | 12 | 14 | |||||
34,906 | 35,034 | ||||||
Valuation Allowance | (34,906 | ) | (35,034 | ) | |||
Net current deferred tax assets | $ | — | $ | — | |||
At December 31, 2017, we had net deferred tax assets of $34,906,000. Due to uncertainties surrounding our ability to generate future taxable income to realize these assets, a full valuation allowance has been established to offset the net deferred tax asset.
Additionally, the future utilization of our net operating loss and research and development tax credits carryforwards is subject to annual limitation under Sections 382 and 383 of the Internal Revenue Code of 1986, and similar state tax provisions due to ownership change limitations that have occurred previously or that could occur in the future. These ownership changes limit the amount of the net operating loss and research and development tax credit carryforward and other deferred tax assets that can be utilized to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Sections 382 and 383, results from transactions increasing ownership of certain stockholders or public groups in the stock of the corporation by more than 50 percent points over a three-year period. We believe we incurred ownership changes since April 2014, however, we have not completed an analysis yet to determine the impact of our ability to use net operating losses and research and development credits as of December 31, 2017.
At December 31, 2017, we had federal and California net operating loss carryovers of $93,253,000 and $93,172,000, respectively. The federal and California net loss carryforwards will begin to expire in 2032.
At December 31, 2017, we had federal and state research tax credit carryovers of approximately $2,972,000 and $2,660,000, respectively. The federal research and development tax credit carryforwards will begin to expire in 2033. The California research and development credit carryforwards are available indefinitely.
The impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more likely than not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. There were no unrecognized tax benefits recorded by us as of the date of adoption. As a result of the implementation, we did not recognize an increase in the liability for unrecognized tax benefits.
A rollforward of changes in our unrecognized tax benefits is shown below.
F- 24
(In thousands) | December 31, | ||||||
2017 | 2016 | ||||||
Balance at beginning of year | $ | 1,698 | $ | 1,234 | |||
Additions based on tax positions related to the current year | 554 | 464 | |||||
Additions for tax positions of prior years | — | — | |||||
Balance at end of year | $ | 2,252 | $ | 1,698 | |||
The amount of unrecognized tax benefits that would impact the effective tax rate if recognized and realized is $2,029,000.
Our practice is to recognize interest and/or penalties related to income tax matters as income tax expense. We had no accrual for interest or penalties on our accompanying consolidated balance sheets at December 31, 2017 and 2016, and have not recognized interest and/or penalties in our consolidated statements of operations and comprehensive loss for the years ended December 31, 2017, 2016 and 2015.
We do not anticipate a significant change to our unrecognized tax benefits during the next twelve months.
We file tax returns as prescribed by tax laws of the jurisdictions in which we operate. In the normal course of business, we are subject to examination by federal and state jurisdictions, where applicable. There are currently no pending tax examinations. Our federal and state tax returns are still open under statute from 2012 to present.
On December 22, 2017, the President of the United States signed into law the Tax Cuts and Jobs Act (the "Act"). The Act amends the Internal Revenue Code to reduce tax rates and modify policies, credits, and deductions for individuals and businesses. For businesses, the Act reduces the corporate tax rate from a maximum of 35% to a flat 21% rate. The rate reduction is effective on January 1, 2018.
As a result of the rate reduction, the Company has reduced the deferred tax asset balance as of December 31, 2017 by $13.1 million.
Due to the Company's full valuation allowance position, the Company has also reduced the valuation allowance by the same amount.
Due to uncertainties which currently exist in the interpretation of the provisions of the Tax Cuts and Jobs Act of 2017 regarding Internal Revenue Code Section 162(m), the Company has not evaluated the potential impacts of IRC Section 162(m) as amended by the Tax Cuts and Jobs Act of 2017 on its consolidated financial statements.
12. Net Loss Per Share
Basic and diluted net loss per share was calculated as follows for the years ended December 31, 2017, 2016 and 2015:
F- 25
(In thousands, except per share amounts) | Years Ended December 31, | ||||||||||
2017 | 2016 | 2015 | |||||||||
Basic and diluted net loss per share attributable to common stockholders: | |||||||||||
Numerator: | |||||||||||
Net loss attributable to common stockholders | $ | (30,879 | ) | $ | (22,499 | ) | $ | (27,130 | ) | ||
Denominator: | |||||||||||
Weighted-average number of common shares outstanding, basic and diluted | 25,084 | 19,873 | 19,773 | ||||||||
Net loss per common share attributable to common stockholders, basic and diluted | $ | (1.23 | ) | $ | (1.13 | ) | $ | (1.37 | ) | ||
There was no difference between our net loss and the net loss attributable to common stockholders for all periods presented.
Stock options to purchase 4,573,625 shares, 3,568,329 shares and 3,116,185 shares of common stock as of December 31, 2017, 2016 and 2015, respectively, were excluded from the computation of diluted net loss per share attributable to common stockholders because their effect was anti-dilutive. 250,000 shares unvested restricted stock award as of December 31, 2017 were also excluded from the computation of diluted net loss per share calculations because their effect was anti-dilutive.
13. Employee Savings Plan
We have established an employee savings plan pursuant to Section 401(k) of the Internal Revenue Code, effective May 1, 2014. The plan allows participating employees to deposit into tax deferred investment accounts up to 90% of their salary, subject to annual limits. We make contributions to the plan in an amount equal to 50% on the first 6% for a maximum of 3% of the participant’s compensation which is deferred. We contributed approximately $149,000, $101,000 and $164,000 to the plan during the years ended December 31, 2017, 2016 and 2015, respectively.
14. Selected Quarterly Financial Information (unaudited)
The following table presents selected unaudited quarterly financial data for each of the quarters in the years ended December 31, 2017 and 2016.
F- 26
(In thousands, except per share amounts) | 2017 | 2016 | |||||||||||||||||||||||||||||
Quarter ended | Dec. 31 | Sep. 30 | Jun. 30 | Mar. 31 | Dec. 31 | Sep. 30 | Jun. 30 | Mar. 31 | |||||||||||||||||||||||
Operating costs and expenses | |||||||||||||||||||||||||||||||
Research and development | $ | 5,142 | $ | 4,877 | $ | 3,866 | $ | 3,780 | $ | 3,509 | $ | 3,754 | $ | 3,161 | $ | 3,437 | |||||||||||||||
General and administrative | 4,820 | 3,269 | 3,056 | 2,843 | 2,401 | 2,022 | 1,865 | 2,020 | |||||||||||||||||||||||
Restructuring costs | — | — | — | — | — | — | — | 655 | |||||||||||||||||||||||
Total operating cost and expenses | 9,962 | 8,146 | 6,922 | 6,623 | 5,910 | 5,776 | 5,026 | 6,112 | |||||||||||||||||||||||
Loss from operations | (9,962 | ) | (8,146 | ) | (6,922 | ) | (6,623 | ) | (5,910 | ) | (5,776 | ) | (5,026 | ) | (6,112 | ) | |||||||||||||||
Interest and other income, net | 232 | 256 | 155 | 131 | 101 | 93 | 79 | 52 | |||||||||||||||||||||||
Net loss | $ | (9,730 | ) | $ | (7,890 | ) | $ | (6,767 | ) | $ | (6,492 | ) | $ | (5,809 | ) | $ | (5,683 | ) | $ | (4,947 | ) | $ | (6,060 | ) | |||||||
Net loss per share, basic and diluted (1) | $ | (0.35 | ) | $ | (0.29 | ) | $ | (0.29 | ) | $ | (0.30 | ) | $ | (0.29 | ) | $ | (0.29 | ) | $ | (0.25 | ) | $ | (0.31 | ) | |||||||
Weighted average shares used in computing net loss per share, basic and diluted | 27,915 | 27,400 | 23,409 | 21,516 | 19,900 | 19,891 | 19,865 | 19,836 | |||||||||||||||||||||||
(1) Net loss per share for each quarter is calculated as a discrete period; the sum of four quarters may not equal the calculated full year amount. | |||||||||||||||||||||||||||||||
15. Subsequent Events
We have evaluated subsequent events through the filing date of this annual report on Form 10-K, and determined that no subsequent events have occurred that would require recognition in the financial statements or disclosure in the notes thereto.
F- 27