Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - NovoCure Ltd | nvcr-ex322_14.htm |
| EX-32.1 - EX-32.1 - NovoCure Ltd | nvcr-ex321_11.htm |
| EX-31.2 - EX-31.2 - NovoCure Ltd | nvcr-ex312_13.htm |
| EX-31.1 - EX-31.1 - NovoCure Ltd | nvcr-ex311_10.htm |
| EX-23.1 - EX-23.1 - NovoCure Ltd | nvcr-ex231_773.htm |
| EX-21 - EX-21 - NovoCure Ltd | nvcr-ex21_8.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
|
☒ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-37565
NovoCure Limited
(Exact Name of Registrant as Specified in Its Charter)
|
Jersey (State or Other Jurisdiction of Incorporation or Organization) |
98-1057807 (I.R.S. Employer Identification No.) |
|
|
|
|
No. 4 The Forum Grenville Street St. Helier, Jersey JE2 4UF (Address of Principal Executive Offices)
|
|
Registrant’s telephone number, including area code: +44 (0) 15 3475 6700
Securities registered pursuant to Section 12(b) of the Act:
|
|
Title of each class |
|
|
Name of each exchange on which registered |
|
|
Ordinary shares, no par value per share |
NASDAQ Global Select Market |
||||
Securities registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
|
Large accelerated filer |
☒ |
|
Accelerated filer |
☐ |
|
Non-accelerated filer |
☐ |
(Do not check if a smaller reporting company) |
Smaller reporting company |
☐ |
|
|
|
|
Emerging Growth Company |
☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the outstanding common equity of the registrant held by non-affiliates as of the last business day of the registrant’s most recently completed second fiscal quarter was $909,514,284.
The number of shares of the registrant’s ordinary shares outstanding as of February 16, 2018 was 89,882,437.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for its 2018 annual meeting of shareholders are incorporated by reference into Items 10, 11, 12, 13, and 14 of Part III of this Form 10-K. Such definitive proxy statement will be filed with the Securities and Exchange Commission within 120 days after the end of the registrant’s fiscal year ended December 31, 2017.
|
|
|
Page |
|
ii |
||
|
Item 1. |
1 |
|
|
Item 1A. |
18 |
|
|
Item 1B. |
46 |
|
|
Item 2. |
46 |
|
|
Item 3. |
46 |
|
|
Item 4. |
46 |
|
|
Item 5. |
48 |
|
|
Item 6. |
51 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
53 |
|
Item 7A. |
65 |
|
|
Item 8. |
66 |
|
|
Item 9. |
Changes in and Disagreements With Accountants on Accounting and Financial Disclosure |
97 |
|
Item 9A. |
97 |
|
|
Item 9B. |
97 |
|
|
Item 10. |
98 |
|
|
Item 11. |
98 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
98 |
|
Item 13. |
Certain Relationships and Related Transactions, and Director Independence |
98 |
|
Item 14. |
98 |
|
|
Item 15. |
99 |
|
|
Item 16. |
102 |
|
|
103 |
||
i
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
In addition to historical facts or statements of current condition, this report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements contained in this report are based on our current plans, expectations, hopes, beliefs, intentions or strategies concerning future developments and their impact on us. Forward-looking statements contained in this report constitute our expectations or forecasts of future events as of the date this report was filed with the Securities and Exchange Commission and are not statements of historical fact. You can identify these statements by the fact that they do not relate strictly to historical or current facts. Such statements may include words such as “anticipate,” “will,” “estimate,” “expect,” “project,” “intend,” “should,” “plan,” “believe,” “hope,” and other words and terms of similar meaning in connection with any discussion of, among other things, future operating or financial performance, strategic initiatives and business strategies, regulatory or competitive environments, the commercialization of Optune and our other delivery systems, our intellectual property and delivery system research and development. In particular, these forward-looking statements include, among others, statements about:
|
|
• |
our research and development, clinical trial and commercialization activities and projected expenditures; |
|
|
• |
the further commercialization of Optune and our delivery system candidates; |
|
|
• |
our business strategies and the expansion of our sales and marketing efforts in the United States and in other countries; |
|
|
• |
the market acceptance of Optune and our other delivery systems by patients, physicians, third-party payers and others in the healthcare and scientific community; |
|
|
• |
our plans to pursue the use of Tumor Treating Fields delivery systems for the treatment of solid tumor cancers other than GBM; |
|
|
• |
our estimates regarding revenues, expenses, capital requirements and needs for additional financing; |
|
|
• |
our ability to obtain regulatory approvals for the use of Tumor Treating Fields in cancers other than GBM and any future delivery systems; |
|
|
• |
our ability to acquire the supplies needed to manufacture our delivery systems from third-party suppliers; |
|
|
• |
our ability to manufacture adequate supply; |
|
|
• |
our ability to secure adequate coverage from third-party payers to reimburse us for our delivery systems; |
|
|
• |
our ability to receive reimbursement from third-party payers for use of our delivery systems; |
|
|
• |
our ability to maintain and develop our intellectual property position; |
|
|
• |
our cash needs; and |
|
|
• |
our prospects, financial condition and results of operations. |
These forward-looking statements involve a number of risks and uncertainties (some of which are beyond our control) or other assumptions that may cause actual results or performance to be materially different from those expressed or implied by these forward-looking statements. Should one or more of these risks or uncertainties materialize, or should any of our assumptions prove incorrect, actual results may vary in material respects from those projected in these forward-looking statements. Some of these factors are described in Part I, Item IA, Risk Factors, of this Annual Report on Form 10-K. We do not intend to update publicly any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
ii
Overview
We are a global oncology company developing a proprietary platform technology called Tumor Treating Fields, the use of electric fields tuned to specific frequencies to disrupt solid tumor cancer cell division. Our key priorities are to drive commercial adoption of Optune, our first commercial Tumor Treating Fields delivery system, for the treatment of glioblastoma (“GBM”) and to advance programs testing the efficacy and safety of Tumor Treating Fields in multiple solid tumor indications through our clinical pipeline.
We were founded in 2000 and operated as a development stage company through December 31, 2011. We initially received U.S. Food and Drug Administration (“FDA”) approval for Optune in 2011 for use as a monotherapy treatment for adult patients with GBM following confirmed recurrence after chemotherapy. In October 2015, we received FDA approval to market Optune for the treatment of adult patients with newly diagnosed GBM in combination with temozolomide, a chemotherapy drug. We have also received approval to market Optune in the European Union (“EU”), Switzerland, Japan and certain other countries. We have built a commercial organization and launched Optune in the United States, Germany, Austria, Switzerland, Israel and Japan, which we refer to as our currently active markets. 2017 was marked by substantial growth in our business as compared to 2016, driven primarily by increasing awareness of Optune’s unprecedented five-year survival data in newly diagnosed GBM and increasing physician confidence and belief in Optune as a standard treatment for GBM.
We have researched the biological effects of Tumor Treating Fields extensively. Tumor Treating Fields uses electric fields tuned to specific frequencies to disrupt cancer cell division, inhibiting tumor growth and causing affected cancer cells to die. Because Tumor Treating Fields is delivered regionally, acts only on dividing cells (a biological process to as mitosis) and is frequency tuned to target cancer cells of a specific size, we believe there is minimal damage to healthy cells. We believe our preclinical and clinical research demonstrates that Tumor Treating Fields’ mechanism of action affects fundamental aspects of cell division and may have broad applicability across a variety of solid tumors. We have demonstrated in preclinical studies that Tumor Treating Fields can offer additive or synergistic benefits in combination with other anti-cancer agents, which may lead to greater efficacy without significantly increasing the side effects.
In addition to our clinical and commercial progress in GBM, we are currently planning or conducting clinical trials evaluating the use of Tumor Treating Fields in brain metastases, non-small-cell lung cancer (“NSCLC”), pancreatic cancer, ovarian cancer and mesothelioma. We anticipate expanding our clinical pipeline over time to study the safety and efficacy of Tumor Treating Fields for additional solid tumor indications.
We own all commercialization rights to Tumor Treating Fields in oncology. Our robust global patent and intellectual property portfolio consists of over 140 issued patents, with numerous additional patent applications pending worldwide. We believe we will maintain exclusive rights to market Tumor Treating Fields for all solid tumor indications in our key markets through the life of our patents.
We were incorporated in the Bailiwick of Jersey in 2000. Our U.S. operations are located in Portsmouth, New Hampshire, Malvern, Pennsylvania, and New York City. Additionally, we have offices in Germany, Switzerland and Japan, and a research center in Israel. We completed our initial public offering (“IPO”) of our ordinary shares in October 2015. Our ordinary shares are quoted on the NASDAQ Global Select Market under the symbol “NVCR.”
Our therapy
By using physics to influence biology, we discovered another way to treat solid tumor cancers.
Tumor Treating Fields is a cancer therapy that uses electric fields tuned to specific frequencies to disrupt cell division, inhibiting tumor growth and causing affected cancer cells to die. After more than a decade of preclinical research in 18 different cancer cell lines, Tumor Treating Fields has demonstrated a consistent anti-mitotic effect. In our clinical research and commercial experience to date, Tumor Treating Fields has exhibited no systemic toxicity, with mild to moderate skin irritation being the most common side effect.
Recognizing what electric fields are and how they can be utilized for medical applications is essential to understanding Tumor Treating Fields. An electric field is a field of force. Electric fields surround all charged objects. An electric field exerts forces on other charged objects within it. Tumor Treating Fields uses alternating electric fields specifically tuned to target cancer cells. Once the electric fields enter the cancer cell, they attract and repel charged proteins during cancer cell division.
1
Tumor Treating Fields utilizes the natural electrical properties of dividing cancer cells. While many intracellular molecules are slightly polarized or neutral, some are highly polarized and are strongly affected by Tumor Treating Fields. For example, tubulin is a highly polarized cellular protein that must orient spatially to form the mitotic spindle, which segregates chromosomes into two daughter cells during mitosis. In the presence of Tumor Treating Fields, tubulin aligns with the direction of the electric field, causing disruption of mitotic spindle formation and eventual cell death. Septin is another highly polarized molecule in cells that must orient spatially to form the contractile ring needed to split daughter cells during mitosis. In the presence of Tumor Treating Fields, septin aligns with the direction of the electric field, leading to improper localization of the contractile ring. This process causes membrane blebbing, a sign of cell damage, and eventual cell death.
Research is ongoing to further develop our understanding of the multi-pronged mechanism behind Tumor Treating Fields. In addition to its anti-mitotic effect, Tumor Treating Fields has been shown to interfere with DNA damage response, to induce autophagy and to reduce cell migration and invasion.
We believe Tumor Treating Fields causes minimal damage to healthy cells surrounding affected cancer cells. The biological effects of Tumor Treating Fields are dependent on the frequency of oscillation (kHz) and the field intensity (V/cm). The cell membrane serves as a filter for electric fields unless tuned to a specific frequency, with the frequency required to penetrate the membrane principally linked to cell size. Cancer cells tend to be a different size than surrounding normal healthy cells and, as a result, we believe treatment with Tumor Treating Fields selectively targets cancer cells while minimizing damage to normal cells. Additionally, since the molecules affected by Tumor Treating Fields are primarily those utilized during mitosis, proliferating cancer cells are affected more than resting, non-dividing normal cells. Tumor Treating Fields is regionally delivered to the tumor site rather than systemically delivered throughout the body and, as a result, the parts of the body not covered by Tumor Treating Fields are generally not affected.
Although it is currently only approved for the treatment of GBM, we believe the basic mechanism behind treatment with Tumor Treating Fields may be broadly applicable to solid tumors and is not limited to a specific tumor type or genetic marker. Tumor Treating Fields is intended principally for use in combination with other standard-of-care cancer treatments. Our preclinical experience to date has demonstrated that combining Tumor Treating Fields with radiation, chemotherapy or immunotherapy may lead to additive efficacy or stronger efficacy than the effect of either treatment alone, and in some cases synergistic efficacy, or stronger efficacy than the sum of the effects of both treatments. The synergistic effect is most pronounced in preclinical studies with certain taxane-based chemotherapies. Importantly, Tumor Treating Fields do not appear to increase the systemic toxicities of radiation, chemotherapy or immunotherapy. No dose-limiting cumulative toxicity has been reported with Tumor Treating Fields and we believe the basic mechanism of action is unlikely to result in a cumulative toxic effect. Treatment with Tumor Treating Fields is different than radiation, chemotherapy, and immunotherapy and we believe it can be combined with many of these therapies to enhance efficacy against multiple solid tumor types.
Our technology
Treatment with Tumor Treating Fields is delivered through a portable, medical device. The complete delivery system, called Optune for the treatment of GBM, includes a portable electric field generator, transducer arrays, rechargeable batteries and accessories. Sterile, single-use transducer arrays are placed directly on the skin in the region surrounding the tumor and connected to the electric field generator to deliver therapy. Transducer arrays are changed when hair growth or the hydrogel reduces array adhesion to the skin, which is typically two to three times per week for our GBM patients. The therapy is designed to be delivered continuously throughout the day and night and efficacy is strongly correlated to compliance. If the device is not on, the patient is not being treated. The electric field generator can be run from a standard power outlet or carried with a battery in a specially designed bag that we provide to patients. Including the battery, Optune weighs approximately 2.7 pounds.
Tumor Treating Fields penetrates the volume of tissue between the arrays. The distribution of the field within a certain part of the body depends on the exact layout of the transducer arrays and the passive electrical properties, mainly resistance, of the different tissues between them. Array placement is optimized for each patient using proprietary software called NovoTAL, based on morphometric measurements of the patient’s anatomy according to a recent MRI scan and the location of the tumor.
We plan to use the same field generator technology across all indications for which Tumor Treating Fields is approved. We plan to specifically target individual solid tumor types by tuning the field generator to the appropriate frequency based upon tumor cell size and adjusting the output power to treat the required tumor tissue volume. Our transducer arrays have been developed and are in use, either commercially or clinically, for application on the head, chest and abdomen.
2
We plan to continue to enhance our Tumor Treating Fields delivery systems to improve ease of use for patients. We recently developed a transducer array in a tan color (instead of white) for GBM patients which is intended to be less conspicuous for patients using Optune. The tan array has been available to certain patients in Europe, the Middle East and Africa (“EMEA”) since October 2017 and we expect all EMEA patients will transition to the tan array in the first half of 2018. Pending applicable regulatory approvals, we hope to launch the tan transducer array in the United States in the second half of 2018. We are also working to develop a next generation transducer array intended to minimize the impact of wires and improve overall aesthetics through the use of new materials.
We are developing a remote download capability for monthly treatment compliance reports to improve visibility of compliance data for patients and physicians. Over time, we may also have the opportunity to advance the engineering of our Tumor Treating Fields delivery systems to optimize the electric field distribution, frequency and intensity in individual patients, potentially improving efficacy. Any enhancements or development work will be subject to the applicable regulatory reviews and approvals.
Our commercial business
The first indication we pursued for Tumor Treating Fields was GBM, the most common form of primary brain cancer. GBM are tumors that arise from astrocytes – the star-shaped cells that make up the “glue-like,” or supportive tissue of the brain. These tumors are usually highly malignant because the cells reproduce quickly and they are supported by a large network of blood vessels. GBM is an aggressive disease for which there are few effective treatment options.
Since the approval of temozolomide as a chemotherapy treatment in 2005, standard treatment for GBM generally includes maximal debulking surgery, radiation therapy with concomitant low-dose temozolomide and post radiation, high dose temozolomide. Prior to the approval of Optune, the median overall survival for patients with newly diagnosed GBM was approximately 15 months with standard therapies, and two-year survival was approximately 30%. Five-year survival was under 10%.
Our markets
We have received approval to market Optune in the Unites States, EU, Switzerland, Japan and certain other countries. We have built a commercial organization and launched Optune for the treatment of GBM in the United States, Germany, Austria, Switzerland, Israel and Japan, which we refer to as our currently active markets. Refer to Part II, Item 8 “Financial Statements and Supplementary Data”, Note 18, “Supplemental information” for more information regarding our assets and net revenues.
We estimate that approximately:
|
• |
12,500 people are diagnosed with GBM or tumors that typically progress to GBM in the United States each year. Of this population, we estimate that approximately 9,300 patients are candidates for treatment with Optune based upon the rate of disease progression and medical eligibility. |
|
• |
3,600 people are diagnosed with GBM or tumors that typically progress to GBM in Germany each year. Of this population, we estimate that approximately 2,700 patients are candidates for treatment with Optune based upon the rate of disease progression and medical eligibility. |
|
• |
1,500 people are diagnosed with GBM or tumors that typically progress to GBM in Japan each year. Of this population, we estimate that approximately 1,100 patients are candidates for treatment with Optune based upon the rate of disease progression and medical eligibility. |
|
• |
1,000 people are diagnosed with GBM or tumors that typically progress to GBM in our other currently active markets: Austria, Switzerland and Israel. Of this population, we estimate that approximately 750 patients are candidates for treatment with Optune based upon the rate of disease progression and medical eligibility. |
EF-11 phase 3 pivotal clinical trial data for the treatment of recurrent GBM
We initially received FDA approval for Optune in 2011 for use as a monotherapy treatment for adult patients with GBM, following confirmed recurrence after chemotherapy. The FDA approved Optune based on the EF-11 trial (“EF-11”), a randomized, phase 3 pivotal clinical trial. While the trial did not achieve its primary endpoint of superiority, the trial results indicate that monotherapy treatment with Optune monotherapy provides patients with clinically comparable extension of survival compared to chemotherapy and that patients treated with Optune alone had significantly fewer side effects and an overall better quality of life than patients treated with chemotherapy alone.
3
EF-11 was a multicenter, active controlled clinical trial of 237 adults with recurrent GBM. Participants received either Optune as a monotherapy (n=120) or the physician’s choice of chemotherapy (n=117). Chemotherapies chosen for the active control arm included mainly bevacizumab, nitrosureas and temozolomide. The primary endpoint for the trial was overall survival. The secondary endpoints included progression free survival at six months, radiological response rate, one-year survival rate, adverse event severity and frequency and quality of life. Overall survival for patients treated with Optune alone and active chemotherapy were 6.6 months and 6.0 months, respectively (p=0.27: HR = 0.86). Progression free survival was not significantly different between the groups and progression free survival at six months was numerically higher in the Optune arm (21.4% vs. 15.2%).
Twice as many EF-11 patients responded to Optune than to active chemotherapy (12 patients versus 6 patients). Three patients in the Optune alone arm had a complete response versus no patients in the active chemotherapy arm.
EF-11 trial demonstrated that patient compliance is important for successful outcomes. Patients who used Optune more than 75% of the time had a significant survival advantage compared to those who used it less than 75% of the time (median survival was 7.8 months compared to 4.5 months, respectively; p<0.05).
EF-14 phase 3 pivotal clinical trial data for the treatment of newly diagnosed GBM
In October 2015, we received FDA approval to market Optune for the treatment of adult patients with newly diagnosed supratentorial GBM in combination with temozolomide. The FDA approved Optune for newly diagnosed GBM based on the EF-14 trial (“EF-14”), which was a randomized, phase 3 pivotal clinical trial which compared, post radiation, Optune plus temozolomide versus temozolomide alone for the treatment of newly diagnosed GBM. The primary endpoint of the trial was progression free survival and a powered secondary endpoint was overall survival.
In the EF-14 interim analysis of the per-protocol population of 315 patients, upon which FDA approval was based, Optune plus temozolomide significantly extended median overall survival by 4.9 months from 15.6 months for temozolomide alone to 20.5 months for Optune plus temozolomide (p=0.0042). Optune was the first treatment in more than ten years to increase overall survival in newly diagnosed GBM. Optune plus temozolomide also significantly improved progression free survival by 3.2 months (p=0.0013). The EF-14 interim analysis results were published in JAMA in 2015. In 2016, the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology® (NCCN Guidelines®) for Central Nervous Systems Cancers were updated and now include alternating electric fields therapy (Optune) in combination with temozolomide following standard brain radiation therapy with concurrent temozolomide as a Category 2A recommended postoperative adjuvant treatment option for patients with newly diagnosed supratentorial GBM.
Optune plus temozolomide has demonstrated consistent superiority versus temozolomide alone in all analyses since the interim results were reported. The EF-14 five-year analysis of the intent-to-treat population of 695 patients was presented at the American Association for Cancer Research Annual Meeting in April 2017. The EF-14 intent-to-treat five year analysis results were published in JAMA in December 2017.
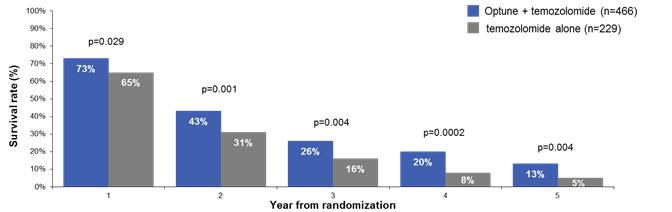
In that analysis, Optune plus temozolomide demonstrated unprecedented five-year survival results. Median overall survival was significantly extended by nearly five months (median overall survival of 20.9 months versus 16.0 months for temozolomide alone; p<0.001: HR 0.63). Optune plus temozolomide also significantly improved progression free survival. Median progression free survival was extended by 2.7 months from 4.0 months for temozolomide alone to 6.7 months for Optune plus temozolomide (p<0.001: HR 0.63). Optune plus temozolomide demonstrated consistently superior survival compared to temozolomide alone with 2.5 times greater overall survival at five years.
4
The following graph presents the overall survival data in the intent-to-treat population from our five-year analysis:
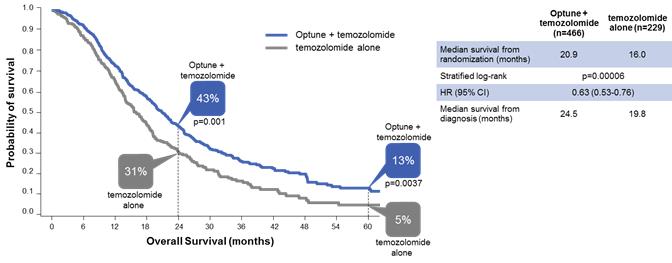
The following graph presents the survival rates by year in the intent-to-treat population from our five-year analysis:
In September 2017 at the American Society for Radiation Oncology Annual Meeting, we presented quality of life data from a prespecified analysis of our EF-14 trial demonstrating that the combination of Optune and temozolomide did not negatively influence health-related quality of life compared to temozolomide alone for newly diagnosed GBM patients except for itchy skin under Optune’s tranducer arrays. The analysis also demonstrated that a higher proportion of patients treated with Optune and temozolomide reported stable or improved quality life for global health status, pain, physical functioning and leg weakness.
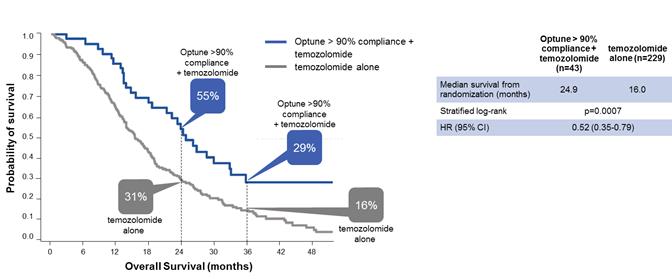
In November 2017, we presented a retrospective post-hoc analysis of our EF-14 trial at the Annual Meeting of the Society for Neuro-Oncology showing that increased compliance with Optune predicted increased survival in GBM patients. An Optune compliance threshold as low as 50 percent correlated with significantly improved outcomes in patients treated with Optune together with temozolomide compared to patients treated with temozolomide alone. The greater patients’ compliance with Optune, the better their outcomes. Patients who used Optune more than 90 percent of the time (n=43) had the greatest chance of survival: a median survival of 24.9 months from randomization and a five-year survival of 29.3 percent.
5
The following graph presents the effect of compliance on overall survival from our five-year analysis:

The following graph presents the overall survival data from our five-year analysis for patients who used Optune more than 90 percent of the time:
The significant extension of progression free and overall survival in patients receiving Optune in combination with temozolomide in the EF-14 trial was not specific to any prognostic subgroup or tumor genetic marker and was consistent regardless of MGMT methylation status, extent of resection, age, performance status or gender. Optune was safely combined with temozolomide with no significant increase in serious adverse events compared with temozolomide alone. The most common side effect related to Optune was mild to moderate skin irritation.
Commercial execution
Our first commercial priority in each market is to generate awareness of Optune and our unprecedented five-year survival data. We believe we have achieved high levels of awareness in our currently active markets amongst neuro-oncologists and neurosurgeons who treat GBM patients. Neuro-oncologists and neurosurgeons wrote approximately 59% of our prescriptions in 2017. We remain focused on developing awareness amongst radiation oncologists and medical oncologists who treat GBM patients. Radiation oncologists are our second largest and fastest growing customer segment and represented approximately 25% of our prescriptions in 2017. Healthcare providers must undergo a certification training in order to prescribe Optune. As of December 31, 2017, we had more than 60 sales force colleagues globally, responsible for promotion to certified prescribers at 1,122 clinical centers, including certified prescribers at 714 clinical centers in the United States, 245 clinical centers in Europe, and 163 clinical centers in Japan.
6
Once awareness has been established, our commercial priority shifts to increasing the percentage of physicians who routinely discuss Optune with their patients and increasing the percentage of physicians who confidently position Optune plus temozolomide as the treatment for newly diagnosed GBM that offers the best chance for long-term survival. We also work to ensure the prescriber has the necessary resources to effectively discuss Optune with their patients and to complete the prescription process. We believe that, unlike traditional cancer therapies, the patient perception of treatment plays a significant role in determining whether or not a prescription for Optune is written and subsequently filled. Therefore, we have also focused efforts on developing targeted tools to support the physician-patient dialogue and patient education. We currently operate as a direct-to-patient distributor of Optune in the United States and EMEA. Once an appropriate Optune patient is identified by a certified prescriber, the healthcare provider’s office submits a prescription order form and supporting documentation to us. We employ a team of device support specialists who provide technical training to the patient and caregiver. Once treatment is initiated, we provide 24/7 technical support for patients and caregivers as well as assistance with insurance reimbursement. We also provide the healthcare provider and the patient with a monthly compliance report for monitoring patient use of Optune. In Japan, we distribute our product through hospitals and provide patient support services under a contractual arrangement with the hospital. We believe we have the experience, expertise and infrastructure to scale our sales and marketing efforts in our key markets. In addition to our commercial organization, we believe we have established a scalable supply chain.
The number of active patients on therapy is our principal revenue driver. There were 1,834 active patients on Optune at December 31, 2017, an increase of 68% versus December 31, 2016. Of the global active patients, there were 1,320 active patients in the United States, 512 active patients in our EMEA markets and 2 active patients in Japan. An active patient is a patient who is receiving treatment with Optune under a commercial prescription order as of the measurement date, including patients who may be on a temporary break from treatment and who plan to resume treatment in less than 60 days. Growth in the number of active patients is a factor of both new patient starts and treatment duration. Median treatment duration differs based upon the clinical diagnosis of the patient. For the twelve months ended December 31, 2017, 62% of prescriptions received were for patients with newly diagnosed GBM. Median treatment duration for patients with recurrent GBM was 4.1 months in our published commercial registry data and 8.2 months in the intent-to-treat five-year analysis of our EF-14 trial in newly diagnosed GBM.
Prescriptions are a leading indicator of demand. In 2017, 4,119 prescriptions were received, an increase of 47% versus 2016. Of those prescriptions, 3,102 were received in the U.S. and 1,017 were received outside of the U.S., primarily in EMEA. A prescription is a commercial order for Optune that is received from a physician certified to treat patients with Optune for a patient not previously on Optune. Orders to renew or extend treatment are not included in this total.
The conversion of prescriptions to new patients is driven by the prescription fill rate and the time to fill. In 2017, our prescription fill rate was between 70-75% each quarter. Our estimated penetration rate considers our new patient starts over the estimated patients that are candidates for treatment with Optune in each of our currently active markets. For the twelve months ended December 31, 2017, we estimate a penetration rate of 20% in our currently active markets.
We believe there are many more patients who could benefit from treatment with Optune than are currently on therapy. In 2018, we plan to focus on increasing penetration in currently active markets and anticipate strategically expanding into additional geographic markets in the future.
Billing and reimbursement
We provide Optune directly to patients following receipt of a prescription order and a signed patient service agreement. We bill payers a single monthly fee for a month of therapy and we bear the financial risk of securing payment from patients and third-party payers in all markets except for Japan. We distribute our product through hospitals in Japan with the hospitals receiving reimbursement from the government mandated insurance program and in turn contracting with us for the equipment, supplies and services necessary to treat patients with Optune.
The monthly list price for Optune is $21,000 in the United States and we have set list prices in our other currently active markets that are approximately equivalent to this price subject to currency fluctuations. We typically negotiate discounts from our list price with healthcare payers in order to secure reimbursement for Optune.
As of December 31, 2017, more than 210 million Americans had coverage of Optune for newly diagnosed and/or recurrent GBM. Additionally, we had negotiated contracts to establish Optune as an in-network benefit for more than 178 million American lives. In 2017, 20- 25% of our active U.S. patients were beneficiaries of the Medicare fee-for-service program, which has denied coverage for our claims to date. We are actively appealing Medicare coverage denials through the Administrative Law Judge (“ALJ”) process with Centers for Medicare and Medicaid Services (“CMS”). Currently, there are significant delays in the assignment of ALJ cases and as of December 31, 2017, a de minimis number of Medicare fee-for-service cases have been fully adjudicated with an ALJ. Our ability to be appropriately reimbursed for Medicaid patients is impacted by the status of Medicare coverage.
7
We continue to engage in active discussions with the CMS administration regarding Medicare reimbursement for Optune. The discussion currently focuses on ensuring Medicare appropriately prices the billing code for Optune under its existing rules. Medicare does not have a defined timeline for establishing pricing for DME billing codes. We plan to request Medicare coverage for Optune after we understand how CMS and Medicare intend to calculate pricing for the Optune billing code. We continue to treat Medicare patients although we have not yet received any material payments from Medicare.
In Germany, we are able to bill healthcare payers for individual cases and each case is evaluated individually on its merits and under the payer’s specific rules for such cases. We have started the formal process for the German Federal Joint Committee (“G-BA”) to review Optune and determine whether to recommend Optune for national reimbursement. In September 2017, the G-BA published its decision to support a clinical trial studying Optune for the treatment of newly diagnosed GBM and has submitted the proposed trial protocol for public comment. We will continue to bill payers for individual cases as we advance through the review process in Germany.
In August 2017, we signed a contract with the Federation of Austrian Social Insurance Institutions that secures reimbursement for Optune for patients with newly diagnosed GBM in Austria. We are pursuing reimbursement for Optune in Switzerland and Israel. We continue to treat patients in both markets and do not expect material revenue in either market while our reimbursement applications are pending.
In December 2017, the Japanese Ministry of Health, Labour and Welfare (“MHLW”) approved the recommendation by Japan’s Central Social Insurance Medical Council to provide national reimbursement for Optune for the treatment of newly diagnosed glioblastoma.
Our clinical pipeline
Based on the results of our preclinical research, we have developed a pipeline strategy to advance Tumor Treating Fields through phase 2 pilot and phase 3 pivotal trials across multiple solid tumor types. We anticipate expanding our clinical pipeline over time for additional solid tumor indications.
Current Clinical Pipeline

The solid tumor types subject to our phase 2 pilot and phase 3 pivotal trials are described in greater detail below, as well as additional details regarding these trials.
Brain metastases
Metastatic cancer is cancer that has spread from the place where it first started to another place in the body. In metastasis, cancer cells break away from where they first formed (the primary cancer), travel through the blood or lymph system, and form new tumors (the metastatic tumors) in other parts of the body. The exact incidence of brain metastases is unknown because no national cancer registry documents brain metastases. However, it has been estimated that 170,000 new cases are diagnosed in the United States each year, 75,000 new cases are diagnosed in Europe each year, and 13,000 new cases are diagnosed in Japan each year. Brain metastases occur in roughly 15% of all cancer patients, and we believe that approximately 40% of brain metastases are a result of NSCLC.
As with GBM, brain metastases are commonly treated with a combination of surgery and radiation. Chemotherapy is often given for the primary tumor, but many chemotherapy agents do not cross the blood brain barrier and are thus ineffective in the treatment of brain metastases. When brain metastases appear, they are either surgically removed or treated with radiation using stereotactic radiosurgery (“SRS”) when possible. Whole brain radiation therapy, although effective in delaying progression or recurrence of brain metastases when given either before or after SRS, is associated with neurotoxicity with a significant decline in cognitive and emotional functioning. Thus, whole brain radiation therapy is often delayed until later in the disease course and is often used as a last resort. This practice results in a window of unmet need after localized surgery and SRS are used and before whole brain radiation therapy is administered to delay or prevent the additional spread of brain metastases.
8
In 2016, we enrolled the first patient in our METIS trial, a phase 3 pivotal trial testing the effectiveness of SRS plus Tumor Treating Fields compared to SRS alone in patients with brain metastases resulting from NSCLC. We have opened the trial to 270 patients and anticipate enrolling the last patient in 2019. We anticipate data will be available for presentation approximately 12 months following last patient enrollment.
Non-small cell lung cancer
Lung cancer is the most common cause of cancer-related death worldwide, and NSCLC accounts for approximately 85% of all lung cancers. The incidence of NSCLC is approximately 214,000 new cases annually in the United States, approximately 350,000 new cases annually in Europe, and approximately 95,000 new cases annually in Japan. Of the 214,000 Americans diagnosed with lung cancer annually, only 18% are alive five years later.
Physicians use different combinations of surgery, radiation and pharmacological therapies to treat NSCLC, depending on the stage of the disease. Surgery, which may be curative in a subset of patients, is usually used in early stages of the disease. Since 1991, radiation with a combination of platinum-based chemotherapy drugs has been the first line standard of care for locally advanced or metastatic NSCLC. Certain immune checkpoint inhibitors have recently been approved for the first line treatment of NSCLC and the standard of care in this setting appears to be evolving rapidly. The standard of care for second line treatment is also evolving and may include may include platinum based chemotherapy for patients who received immune checkpoint inhibitors as their first line regimen, pemetrexed, docetaxel or immune checkpoint inhibitors.
Phase 2 pilot trial
In 2013, we published the results of our phase 2 pilot trial evaluating the safety and efficacy of Tumor Treating Fields in the treatment of advanced NSCLC. The pilot study focused on the effects of treatment with Tumor Treating Fields in combination with standard of care pemetrexed chemotherapy. Results of the pemetrexed Phase 3 FDA registration trial were used as a historical control in this trial.
A total of 42 patients were recruited to the study with a minimum follow-up of six months. Efficacy results based on 41 evaluable patients showed both progression free survival and overall survival for patients receiving Tumor Treating Fields in combination with pemetrexed increased compared to historical control data for pemetrexed alone. Median progression free survival in the Tumor Treating Fields-treated group was 6.5 months (compared to 2.9 months in the historical control) and median overall survival was 13.8 months (compared to 8.3 months in the historical control). Adverse events reported in this combination study were comparable to those reported with pemetrexed alone, suggesting minimal added toxicities due to Tumor Treating Fields.
Phase 3 pivotal trial
In February 2017, we enrolled the first patient in our LUNAR trial, a phase 3 pivotal trial testing the effectiveness of Tumor Treating Fields in combination with immune checkpoint inhibitors or docetaxel versus immune checkpoint inhibitors or docetaxel alone. We believe our protocol incorporates the evolving standard of care for second-line treatment of NSCLC. We have opened the trial to 534 patients and anticipate enrolling the last patient in 2019. The protocol specifies an event-driven interim analysis. We anticipate final data will be available for presentation approximately 18 months following last patient enrollment.
Pancreatic cancer
Pancreatic cancer is one of the most lethal cancers and is the third most frequent cause of death from cancer in the United States. While overall cancer incidence and death rates are declining, the incidence and death rates for pancreatic cancer are increasing. The incidence of pancreatic cancer is 54,000 new cases annually in the United States, approximately 130,000 new cases annually in Europe, and approximately 39,000 new cases annually in Japan. Pancreatic cancer is the only major cancer with a five-year relative survival rate in the single digits, at just 8 percent.
Physicians use different combinations of surgery, radiation and pharmacological therapies to treat pancreatic cancer, depending on the stage of the disease. For patients with locally advanced pancreatic cancer involving encasement of arteries but no extra-pancreatic disease, the standard of care is surgery followed by chemotherapy with or without radiation. Unfortunately, the majority of locally advanced cases are diagnosed once the cancer is no longer operable, generally leaving chemotherapy with or without radiation as the only treatment option.
9
We have completed a phase 2 pilot trial in advanced pancreatic adenocarcinoma, the PANOVA trial, examining Tumor Treating Fields in combination with standard of care chemotherapy.
The first cohort was a single-arm, open-label, historically-controlled, multi-center trial designed to test the feasibility, safety and preliminary efficacy of Tumor Treating Fields in combination with the chemotherapy gemcitabine. This cohort included 20 patients with advanced pancreatic cancer whose tumors could not be removed surgically and who had not received chemotherapy or radiation therapy prior to the clinical trial with a minimum follow-up of six months. Results of the first cohort were presented at the American Society of Clinical Oncology Gastrointestinal Cancers Symposium in 2016. Results of the nab-paclitaxel phase 3 FDA registration trial were used as a historical control in this trial.
In the first cohort, efficacy results showed that progression free survival and overall survival of patients treated with Tumor Treating Fields combined with gemcitabine were more than double those of gemcitabine-treated historical controls. Median progression free survival in the Tumor Treating Fields-treated group was 8.3 months (compared to 3.7 months in the gemcitabine historical control), with locally advanced patients reaching a median progression free survival of 10.3 months and patients with metastatic disease reaching a median progression free survival of 5.7 months. The median overall survival for all patients was 14.9 months (compared to 6.7 months in the gemcitabine historical control). Median overall survival was longer than 15 months in locally advanced patients and 86% of patients were alive at end of follow up. Patients with metastatic disease experienced a median overall survival of 8.3 months. Median one-year survival was 55% (compared to 22% in the gemcitabine historical control). Thirty percent of the evaluable tumors, or 19 patients in total, had partial responses (compared to 7% with gemcitabine alone) and another 30% had stable disease, which means that the cancer is neither decreasing nor increasing in extent or severity.
Following the approval of nab-paclitaxel, a taxane-based chemotherapy, for the treatment of advanced pancreatic cancer, we expanded this study to include a second cohort of 20 patients that were treated with Tumor Treating Fields in combination with nab-paclitaxel and gemcitabine. Results of the second cohort were presented at the American Association for Cancer Research Annual in April 2017.
In the second cohort, efficacy results showed that progression free survival and overall survival of patients treated with Tumor Treating Fields combined with nab-paclitaxel plus gemcitabine were more than double those of nab-paclitaxel plus gemcitabine-treated historical controls. Median progression free survival in the Tumor Treating Fields-treated group was 12.7 months (compared to 5.5 months in the nab-paclitaxel plus gemcitabine historical control) and median overall survival was not yet reached. Median one-year survival was 72% (compared to 35% in nab-paclitaxel plus gemcitabine historical control). Forty percent of the evaluable tumors had partial responses (compared to 23% with the nab-paclitaxel plus gemcitabine alone) and another 47% had stable disease.
Safety results from both cohorts suggested that Tumor Treating Fields plus first-line chemotherapies nab-paclitaxel and/or gemcitabine may be tolerable and safe in patients with advanced pancreatic cancer. Patients reported no serious adverse events related to Tumor Treating Fields.
Phase 3 pivotal trial
In December 2017, we received FDA approval of our investigational device exemption (“IDE”) application to initiate our PANOVA 3 trial, a phase 3 pivotal trial testing the effectiveness of Tumor Treating Fields with nab-paclitaxel and gemcitabine as a front-line treatment for unresectable locally advanced pancreatic cancer. We have opened the trial to 556 patients and anticipate enrolling the last patient in 2020. The protocol specifies an event-driven interim analysis. We anticipate final data will be available for presentation approximately 18 months following last patient enrollment.
Ovarian cancer
In the United States, ovarian cancer ranks fifth in cancer deaths among women, accounting for more deaths than any other cancer of the female reproductive system. Ovarian cancer incidence increases with age, and the median age at time of diagnosis is 63 years old. The incidence of ovarian cancer is approximately 22,000 new cases annually in the United States, approximately 68,000 new cases annually in Europe, and approximately 10,000 new cases annually in Japan.
Physicians use different combinations of surgery and pharmacological therapies to treat ovarian cancer, depending on the stage of the disease. Surgery is usually used in early stages of the disease and is usually combined with chemotherapy, including paclitaxel and platinum-based chemotherapy. Unfortunately, the majority of patients are diagnosed at an advanced stage when the cancer has spread outside of the ovaries to include regional tissue involvement and/or metastases. Platinum-based chemotherapy remains the standard of care in advanced ovarian cancer, but most patients with advanced ovarian cancer will have tumor progression or, more commonly, recurrence. Almost all patients with recurrent disease ultimately develop platinum resistance, and the prognosis for this population remains poor.
10
We have completed a 30 patient phase 2 pilot trial in recurrent ovarian cancer, the INNOVATE trial, examining Tumor Treating Fields in combination with standard of care chemotherapy. This trial was a single-arm, open-label, historically-controlled, multi-center study, designed to test the feasibility, safety and preliminary efficacy of Tumor Treating Fields in combination with weekly paclitaxel. The paclitaxel control arm from the bevacizumab phase 3 FDA registration trial was used as a historical control in this trial. Results were presented at the American Association for Cancer Research Annual in April 2017.
A total of 30 patients were recruited to the study with a minimum follow-up of six months. Safety results suggested that Tumor Treating Fields in combination with weekly paclitaxel may be tolerable and safe as first-line treatment for patients with recurrent ovarian cancer. Median progression free survival in the Tumor Treating Fields-treated group was 8.9 months (compared to 3.9 months in the paclitaxel-alone historical control) and median overall survival was not yet reached. Median one-year survival was 61%. Efficacy results based on the 30 evaluable patients suggested more than doubling of the progression free survival and an improvement in overall survival among patients who received Tumor Treating Fields therapy with paclitaxel compared to paclitaxel alone.
Phase 3 pivotal trial
Based on our phase 2 pilot trial results, we expect to commence a phase 3 pivotal trial in recurrent ovarian cancer in 2018.
Mesothelioma
Malignant mesothelioma is a rare thoracic solid tumor cancer that has been strongly linked to asbestos exposure. It has a long latency period of at least 20-30 years following exposure, and global incidence is still increasing in countries where asbestos is still in use. There are approximately 3,000 new cases of mesothelioma annually in the United States, an estimated incidence of 1,000 new cases annually in Japan and a predicted peak of approximately 9,000 male deaths from mesothelioma in Western Europe that may occur around the year 2018. The prognosis of mesothelioma patients is very poor, with a median overall survival of approximately 12 months in most reported studies. Of the 3,000 Americans diagnosed with mesothelioma annually, only 9% are alive five years later. Mesothelioma is often limited to the thoracic cavity and progresses regionally, making it an attractive target for Tumor Treating Fields.
Physicians use different combinations of surgery and pharmacological therapies to treat mesothelioma, depending on the stage of the disease. Surgery may be used for patients with early stage disease. However, most cases are diagnosed once the cancer is at a later stage, involving extensive tumor growth and regional lymph node spread, and surgical resection for the treatment of mesothelioma is feasible for only a minority of patients. First line standard of care treatment includes pemetrexed, a chemotherapy, in combination with platinum-based chemotherapy, including carboplatin or cisplatin. Second-line treatments may include the chemotherapies oxaliplatin, gemcitabine, vinorelbine or immunotherapies. Despite the many advances in chemotherapy made in recent decades, treatment effectiveness remains very limited.
Phase 2 pilot trial
We enrolled the last patient in our phase 2 pilot trial in mesothelioma, the STELLAR trial, in March 2017. There is a minimum 12 month follow up following last patient enrollment and we anticipate final data collection in the first half of 2018. The STELLAR trial is a single-arm, open-label, multi-center trial designed to test the efficacy and safety of Tumor Treating Fields in combination with pemetrexed combined with cisplatin or carboplatin in patients with unresectable, previously untreated malignant mesothelioma. The historical control for this trial is the results of the 2003 pemetrexed phase 3 FDA registration trial.
An interim analysis of the first 42 patients enrolled in the trial with an average follow-up time of 11.5 months was presented at the International Association for the Study of Lung Cancer in 2016. The one-year survival rate of patients treated with Tumor Treating Fields combined with pemetrexed and cisplatin or carboplatin was 80% (compared to 50% in the pemetrexed and cisplatin-alone historical controls). Median progression free survival in the Tumor Treating Fields-treated group was 7.3 months (compared to 5.7 months in the pemetrexed and cisplatin-alone historical control) and one-year survival rate was 79.7% (compared to 50.3% in the pemetrexed and cisplatin-alone historical control). Median overall survival had not yet been reached. No device-related serious adverse events had been reported to date.
In May 2017, we received humanitarian use device (“HUD”) designation for the use of Tumor Treating Fields for the treatment of pleural mesothelioma. The HUD designation is the first step in obtaining a Humanitarian Device Exemption (“HDE”) for the treatment of pleural mesothelioma with Tumor Treating Fields. Assuming the final data readout from our STELLAR trial is consistent with the interim analysis, we expect to submit an HDE application to the FDA for approval. An approved HDE would allow us to market Tumor Treating Fields in combination with standard of care chemotherapy as a treatment for pleural mesothelioma in the United States.
11
We outsource production of all of our system components to qualified partners. Disposable transducer array manufacturing, the dominant activity in our manufacturing supply chain, includes several specialized processes. Production of the durable system components follows standard electronic medical device methodologies.
We have supply agreements in place with our third-party manufacturing partners. We hold safety stocks of single source components in quantities we believe are sufficient to protect against possible supply chain disruptions.
We currently obtain the ceramic discs used in the transducer arrays for Optune from a single supplier. We have technically qualified and entered into a supply agreement with an additional supplier, and we have requested regulatory approval to use these ceramic disks in the EU. This source will be able to ship product for use within the United States and Japan at a future date pending regulatory approval.
We are developing second sources for all critical materials. We have qualified and entered into supply agreements with second source third party manufacturing partners for the transducer array subassemblies and transducer array final assemblies. They are currently approved to ship transducer array subassemblies for use in the United States and the EU and will be able to ship product for use in Japan pending regulatory approval. In addition, they are approved to ship transducer array final assemblies for use in the EU and will be able to ship product for use in the United States and Japan pending regulatory approval. We anticipate that the diversification of our supply chain will both ensure a continuity of supply and reduce costs.
Intellectual property
We own all commercialization rights to Tumor Treating Fields in oncology. Our robust global patent and intellectual property portfolio consists of over 140 issued patents. The patents have expected expiration dates between 2021 and 2035. We have also filed over 40 additional patent applications worldwide that, if issued, may protect aspects of our platform beyond 2035. We believe we will maintain exclusive rights to market Tumor Treating Fields for all solid tumor indications in our key markets through the life of our patents. However, our reliance on intellectual property involves certain risks, as described under the heading “Risk factors—Risks relating to intellectual property.”
In addition to our patent portfolio, we further protect our intellectual property by maintaining the confidentiality of our trade secrets, know-how and other confidential information. Given the length of time and expense associated with bringing delivery systems candidates through development and regulatory approval to the market place, the healthcare industry has traditionally placed considerable importance on obtaining patent protection and maintaining trade secrets, know-how and other confidential information for significant new technologies, products and processes.
Our policy is to require each of our employees, consultants and advisors to execute a confidentiality agreement before beginning their employment, consulting or advisory relationship with us. These agreements generally provide that the individual must keep confidential and not disclose to other parties any confidential information developed or learned by the individual during the course of their relationship with us except in limited circumstances. These agreements also generally provide that we own, or the individual is required to assign to us, all inventions conceived by the individual in the course of rendering services to us.
In 2015, we entered into a settlement agreement (the “Settlement Agreement”) with the Technion Research and Development Foundation (“Technion”) to resolve certain potential disputes regarding intellectual property developed by our founder and previously assigned to us. Pursuant to the Settlement Agreement, and in exchange for a release of potential disputes from the third party, the Company is obligated to pay a $5.5 million milestone payment (the “Milestone Payment”) to Technion in the quarter following the quarter in which the Company achieves $250.0 million of cumulative net sales (as defined in the Settlement Agreement) (the “Net Sales Milestone”). We achieved the Net Sales Milestone in the fourth quarter of 2017 and anticipate making the Milestone Payment in the first quarter of 2018.
In 2005, we granted an exclusive license to a third party, NovoBiotic LLC, to certain of our key intellectual property for use outside the field of oncology. We are not entitled to any future revenues from this license.
Competition
The market for cancer treatments is intensely competitive, subject to rapid change and significantly affected by new product and treatment introductions and other activities of industry participants. The general bases of competition are overall effectiveness, side effect profile, availability of reimbursement and general market acceptance of a product as a suitable cancer treatment.
12
We believe our intellectual property rights would provide an obstacle to the introduction of Tumor Treating Fields delivery systems by a competitor, and we intend to protect and enforce our intellectual property. In addition, even after the expiration of our U.S. patents, we believe that potential market entrants applying low-intensity, alternating electric fields to solid tumors in the United States will have to undertake their own clinical trials and regulatory submissions to prove equivalence to Tumor Treating Fields, a necessary step in receiving regulatory approvals for a competing product.
Presently, the traditional biotechnology, pharmaceutical and medical technology industries expend significant resources in developing novel and proprietary therapies for the treatment of solid tumors, including GBM and the other indications that we are currently investigating. As we work to increase market acceptance of Tumor Treating Fields, we compete with companies commercializing or investigating other anti-cancer therapies, some of which are in clinical trials for GBM that currently specifically exclude patients who have been or are being treated with Tumor Treating Fields.
Government regulation
Our delivery systems and operations are subject to extensive regulation by the FDA under the Federal Food, Drug, and Cosmetic Act (“FDCA”) and by agencies and notified bodies of the countries or regions in which we develop and market our delivery systems. In addition, our delivery systems must meet the requirements of a large and growing body of international standards that govern the preclinical and clinical testing, manufacturing, labeling, certification, storage, recordkeeping, advertising, promotion, export and marketing and distribution, among other things, of Tumor Treating Fields and our delivery systems.
In the United States, advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the Federal Trade Commission and by state regulatory and enforcement authorities. Promotional activities for FDA-regulated products of other companies have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. In addition, under the federal Lanham Act and similar state laws, competitors and others can initiate litigation relating to unfair competition based on advertising claims. In addition, we are required to meet regulatory requirements in countries outside the United States, which can change rapidly with relatively short notice.
Our research, development and clinical programs, as well as our manufacturing and marketing operations, are subject to extensive regulation.
Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in any number of regulatory enforcement actions, or civil or criminal liability.
Food and Drug Administration
The FDA regulates the development, testing, manufacturing, labeling, storage, recordkeeping, promotion, marketing, distribution and service of medical devices in the United States to ensure that medical products distributed domestically are safe and effective for their intended uses. In addition, the FDA regulates the export of medical devices manufactured in the United States to international markets and the importation of medical devices manufactured abroad. The FDA has broad post-market and regulatory enforcement powers to ensure compliance with the FDCA.
The FDA governs the following activities that we perform or that are performed on our behalf:
|
• |
product design, development and manufacture; |
|
• |
product safety, testing, labeling and storage; |
|
• |
record keeping procedures; |
|
• |
product marketing, sales and distribution; and |
|
• |
post-marketing surveillance, complaint handling, medical device reporting, reporting of deaths, serious injuries or device malfunctions and repair or recall of products. |
We have registered three of our facilities with the FDA. We are subject to announced and unannounced inspections by the FDA to determine our compliance with the Quality System Regulation (“QSR”) and other regulations and these inspections include the manufacturing facilities of our suppliers.
13
FDA’s premarket clearance and approval requirements
Unless an exemption applies, before we can commercially distribute medical devices in the United States, we must obtain, depending on the type of device, either prior 510(k) clearance or premarket approval (“PMA”) from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose lower risks are placed in either class I or II, which typically requires the manufacturer to submit to the FDA a premarket notification requesting permission to commercially distribute the device. This process is generally known as 510(k) clearance. Some low-risk devices are exempted from this requirement. Devices deemed by the FDA to pose the greatest risks, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in class III, requiring PMA approval.
Premarket approval (PMA) pathway
Optune, which is the only delivery system we have marketed in the United States, is classified as a Class III device as it is deemed a life-sustaining device. Accordingly, we were required to receive PMA approval for Optune, which the FDA granted in April 2011 and October 2015 for the treatment of recurrent and newly diagnosed supratentorial GBM, respectively, in adult patients. We expect that we will be required to receive PMA approval for future indications (and the applicable delivery systems for such indications) using Tumor Treating Fields.
A PMA must be supported by extensive data, including from technical tests, preclinical studies and clinical trials, manufacturing information and intended labeling to demonstrate, to the FDA’s satisfaction, the safety and effectiveness of a medical device for its intended use. During the PMA review period, the FDA will typically request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. The FDA may or may not accept the panel’s recommendation. In addition, the FDA will generally conduct a pre-approval inspection of the manufacturing facility or facilities to ensure compliance with QSRs. Prior to approval of the Optune PMA for the treatment of recurrent GBM, we and our critical component suppliers were each inspected by the FDA.
New PMAs or PMA supplements are required for modifications that affect the safety or effectiveness of our delivery systems, including, for example, certain types of modifications to a delivery system’s indication for use, manufacturing process, labeling and design. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA and may not require any or as extensive clinical data as the original PMA required, or the convening of an advisory panel. The FDA requires a company to make the determination as to whether a new PMA or PMA supplement application is to be filed. If a company determines that neither a new PMA nor a PMA supplement application is required for modifications, it must nevertheless notify the FDA of these modifications in its PMA Annual Report. The FDA may review a company’s decisions when reviewing the PMA Annual Report and require the filing of an application.
We have received approval for a number of PMA supplements since approval of the PMA for recurrent GBM, including for modifications to Optune’s electric field generator, transducer arrays, software, manufacturing processes and labeling. In 2015, we received FDA approval to expand our label for Optune to include the treatment of newly diagnosed GBM and in 2016, we received FDA approval for our second generation Optune system. Future modifications may be considered by us as the need arises, some of which we may deem to require a PMA supplement application and others to require reporting in our PMA Annual Report.
Clinical trials
Clinical trials are generally required to support a PMA. Such trials generally require an IDE approval from the FDA for a specified number of patients and study sites, unless the product is deemed a non-significant risk device eligible for more abbreviated IDE requirements. Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an institutional review board (“IRB”) for the relevant clinical trial sites and must comply with FDA regulations, including those relating to good clinical practices. To conduct a clinical trial, we also are required to obtain the patients’ informed consent in form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations. We, the FDA or the respective IRB could suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA approval to market the product in the United States.
Post-approval studies are also typically required as a condition of PMA approval to reinforce the reasonable assurance of safety and effectiveness. Such studies are conducted in the post-market setting with the approved device, often to address the long-term use of the device or other discrete questions that may have been raised based on the clinical data from the IDE clinical study. The FDA required a post-approval registry study as a condition of approval for Optune for recurrent GBM. We have obtained approval of the protocol for this study and the study is fully enrolled.
14
Sales and marketing of medical devices outside of the United States are subject to foreign regulatory requirements that vary widely from country to country. These include the requirement to affix a CE mark to our medical devices in the EU. Whether or not we have obtained FDA approval, our delivery systems must be subject to conformity assessment procedure in which a notified body can be involved. Apart from low risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue a declaration of conformity based on a self-assessment of the conformity of its products with the Essential Requirements laid down in the Medical Devices Directive, a conformity assessment procedure requires the intervention of a notified body. The notified body typically audits and examines products’ technical file and the quality system for the manufacture, design and final inspection of our devices before issuing a CE Certificate of Conformity demonstrating compliance with the relevant Essential Requirements or the quality system requirements laid down in the relevant Annexes to the Medical Devices Directive. Following the issuance of this CE Certificate of Conformity, we can draw up a declaration of conformity and affix the CE mark to the delivery systems covered by this CE Certificate of Conformity and the declaration of conformity. The time required to CE mark our delivery systems or to obtain approval from other foreign authorities may be longer or shorter than that required for FDA approval. Pursuant to a mutual recognition agreement, our products bearing a CE mark may be exported to Switzerland. In the EU, a clinical study must receive a positive opinion from a local ethics committee and approval from the competent authority in the applicable EU member states in which the clinical study is conducted. When a clinical study relates to a CE marked medical device that will be used as part of the study according to its CE mark intended purpose, the approval of the competent authorities is not required. In Japan, we must obtain approvals from the MHLW to market our delivery systems. Each regulatory approval process outside of the United States includes all the risks associated with FDA regulation, as well as country-specific regulations.
Pervasive and continuing regulation
After a device is placed on the market, numerous regulatory requirements apply depending upon the country in which the device is being marketed. These may include:
|
• |
product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
|
• |
QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process for products marketed in the United States; |
|
• |
labeling regulations and FDA and equivalent competent authority in other jurisdictions requiring promotion is truthful and non-misleading and prohibiting the promotion of products for uncleared, unapproved or off-label uses; |
|
• |
approval of product modifications that affect the safety or effectiveness of one of our delivery systems that has been approved or is the subject of a CE Certificate of Conformity; |
|
• |
Medical Device Reporting regulations of the FDCA and medical device vigilance, which require that manufacturers comply with FDA or equivalent competent authority requirements in other jurisdictions to report if their device may have caused or contributed to a death or serious injury, or has malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction of the device or a similar device were to recur; |
|
• |
post-approval restrictions or conditions, including post-approval study commitments; |
|
• |
post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device; |
|
• |
the FDA’s and equivalent competent authority’s recall authority, whereby they can ask, or under certain conditions order, device manufacturers to recall from the market a product that is in violation of governing laws and regulations; |
|
• |
regulations pertaining to voluntary recalls; and |
|
• |
notices of corrections or removals. |
Our delivery systems could be subject to voluntary recall if we, the FDA or another applicable regulatory authority determine, for any reason, that our delivery systems pose a risk of injury or are otherwise defective. Moreover, the FDA and other applicable regulatory authorities can order a mandatory recall if there is a reasonable probability that our delivery system would cause serious adverse health consequences or death.
15
The FDA has broad post-market and regulatory enforcement powers. We are subject to unannounced inspections by the FDA to determine our compliance with the QSR and other regulations, and these inspections include the manufacturing facilities of our subcontractors. We are also subject to FDA’s broad regulatory enforcement power around promotional activities. Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other applicable regulatory authorities, which may result in sanctions, including, but not limited to:
|
• |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
• |
unanticipated expenditures to address or defend such actions; |
|
• |
customer notifications for repair, replacement and/or refunds; |
|
• |
recall, detention or seizure of our delivery systems; |
|
• |
operating restrictions or partial suspension or total shutdown of production; |
|
• |
refusing or delaying our requests for approval of delivery system candidates or a modified version of Optune; |
|
• |
withdrawal of PMA approvals or suspension, variation or withdrawal of CE Certificates of Conformity that have already been granted; |
|
• |
refusal to grant export approval for our delivery systems; or |
|
• |
civil and/or criminal prosecution by the United States Department of Justice or other enforcement authorities outside of the Unites States. |
To date, our facility and those of our critical suppliers have been inspected by the FDA in order to obtain FDA approval of Optune. No inspectional observations were identified and no FDA Form 483s were issued following these inspections.
DME accreditation and licensing and other requirements
In the United States, we are subject to accreditation and licensing requirements as a DME supplier in most states and must meet the supplier standards of Medicare, Medicaid and other federal programs. Certain states require that DME providers maintain an in-state location. Although we believe we are in compliance with all applicable federal and state regulations regarding accreditation and licensure requirements and similar requirements in other jurisdictions, if were found to be noncompliant, we could lose our accreditation or licensure in that state or our supplier rights with that federal program, which could prohibit us from selling our current or future delivery systems to patients in that state or to that federal program.
Healthcare regulatory matters
In addition to FDA restrictions on the marketing of medical devices, several other types of U.S. federal and state laws have been applied to restrict certain business practices in the healthcare industry and penalize unlawful conduct. These laws include anti-kickback, self-referral and false claims statutes.
The U.S. federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between device manufacturers on one hand and prescribers and purchasers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly and practices that involve remuneration intended to induce ordering, purchasing or recommending of a medical device may be subject to scrutiny if they do not qualify for an exemption or safe harbor. In some cases, our practices may not meet all of the criteria for safe harbor protection from anti-kickback liability.
As a DME supplier, we also are subject to a U.S. federal self-referral law, commonly known as the Stark law, which prohibits Medicare payments for DME ordered by physicians who, personally or through an immediate family member, have ownership interests in or compensation arrangements with the furnishing supplier. The Stark law contains a number of specific exceptions that, if met, permit physicians who have certain financial relationships with a DME supplier to make referrals to that entity.
The False Claims Act prohibits any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. The government has pursued a number of cases under the False Claims Act in connection with the off-label promotion of medical products and various other health care law violations.
16
The majority of states also have statutes or regulations similar to the federal anti-kickback, self-referral and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, that apply regardless of the payer.
Numerous federal and state laws and regulations, including the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) as amended by the Health Information Technology for Economic and Clinical Health Act, govern the collection, dissemination, use, security and privacy of individually identifiable health information. We believe we are in substantial compliance with such applicable laws and regulations, including HIPAA.
HIPAA also included a number of federal criminal provisions, including for healthcare fraud and for false statements relating to healthcare matters. The healthcare fraud provision prohibits knowingly and willfully executing a scheme to defraud any healthcare benefit program, including private third-party payers. The false statements provision prohibits knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Many states have similar healthcare fraud laws or insurance fraud laws that apply to claims for healthcare reimbursement.
Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment.
Legislation similar to U.S. anti-kickback, self-referral and false claims statutes have been adopted in foreign countries, including a number of EU member states.
In the United States, the Sunshine Act requires manufacturers of drugs, medical devices, biologicals or medical supplies that participate in U.S. federal health care programs to track and then report certain payments and items of value given to U.S. physicians and U.S. teaching hospitals, which are defined as Covered Recipients. The Sunshine Act requires that manufacturers collect this information on a yearly basis and then report it to Centers for Medicare & Medicaid Services by the 90th day of each subsequent year. We have adopted policies and codes of conduct regarding our interactions with Covered Recipients and believe we are in material compliance with the Sunshine Act. However, our failure to adhere to these requirements could materially adversely impact our business and financial results. Additionally, regulations similar to the Sunshine Act have been adopted in foreign countries including a number of EU member states.
In addition, the U.S. Foreign Corrupt Practices Act (“FCPA”) prohibits corporations and individuals from engaging in certain activities to obtain or retain business outside the United States or to influence a person working in an official capacity in a foreign country. It is illegal to pay, offer to pay or authorize the payment of anything of value to any official of another country, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in that capacity. Legislation similar to the FCPA has been adopted in foreign countries, including a number of EU member states.
Employees
As of December 31, 2017, we had 495 employees. We believe relations with our employees are good.
Available information
Our corporate website address is www.novocure.com. Our website is an inactive textual reference and nothing on our website is incorporated by reference in this Annual Report. Our Annual Reports on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments to reports filed pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, are available free of charge on our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission.
We may use our website as a means of disclosing material information and for complying with our disclosure obligations under Regulation Fair Disclosure promulgated by the SEC. Accordingly, investors should monitor our website, in addition to following our press releases, SEC filings, public conference calls, webcasts and our social media accounts.
17
An investment in our ordinary shares involves a high degree of risk. Investors and prospective investors should carefully consider all of the information in this Annual Report on Form 10-K, including the risks and uncertainties described below. Any of the following risks could have a material adverse effect on our business, prospects, financial condition and results of operations. In any such case, the trading price of our ordinary shares could decline, and you could lose all or part of your investment. In assessing these risks, you should also refer to the other information contained in this Annual Report on Form 10-K, including our consolidated financial statements and the related notes thereto.
Risks relating to our business, Tumor Treating Fields and our delivery systems
Our business and prospects depend heavily on Optune, which is currently approved only for treatment of GBM. If we are unable to increase sales of Optune, obtain further regulatory approvals for and further commercialize Optune or our other delivery system candidates for the treatment of additional indications or are significantly delayed or limited in doing so, our business and prospects will be materially harmed.
To date we have received FDA regulatory approvals and certain approvals in other jurisdictions for the use of Optune for treatment of adult patients with newly diagnosed GBM in combination with temozolomide (a form of chemotherapy) and for treatment of adult patients with recurrent GBM, and have affixed a CE mark to our Tumor Treating Fields delivery systems for certain indications in the EU; however, such approvals and the CE mark affixed to Optune do not guarantee future revenues for these indications. Further, until we receive FDA and analogous approval in other jurisdictions for the use of Tumor Treating Fields for other indications through our delivery system candidates, almost all of our revenues will derive from sales of Optune for the treatment of newly diagnosed and recurrent GBM. The commercial success of Optune and any other delivery systems and our ability to generate and maintain revenues from the use of these delivery systems will depend on a number of factors, including:
|
• |
our ability to obtain additional regulatory approvals for and further commercialize Optune; |
|
• |
our ability to develop, obtain regulatory approval for and commercialize our other Tumor Treating Fields delivery system candidates for additional indications; |
|
• |
the acceptance of Tumor Treating Fields by patients and the healthcare community, including physicians and third-party payers (both private and public), as therapeutically effective and safe; |
|
• |
the relative cost, safety and efficacy of alternative therapies; |
|
• |
our ability to obtain and maintain sufficient coverage or reimbursement by private and public third-party payers; |
|
• |
the ability of our third-party manufacturers to manufacture Optune and other delivery system candidates in sufficient quantities with acceptable quality; |
|
• |
our ability to provide marketing and distribution support for Optune and our other delivery system candidates; |
|
• |
results of future clinical studies relating to Tumor Treating Fields or our competitors’ products; |
|
• |
compliance with applicable health care laws and regulations; |
|
• |
the maintenance of our existing regulatory approvals in the United States, the EU, Japan and other jurisdictions; and |
|
• |
the consequences of any reportable adverse events involving Optune or Tumor Treating Fields occurring in the United States, the EU, Switzerland, Israel, Japan or other jurisdictions. |
In addition, the promotion of Optune is limited to approved indications, which vary by geography, and the FDA label for Optune is limited in certain respects (for example, it is not approved for use in the brain stem, is not approved for use as monotherapy in newly diagnosed GBM and is limited for use by adults ages 22 and older), which may reduce the number of GBM patients to whom it may be prescribed.
In addition to Optune, our ability to generate future revenues will depend on achieving regulatory approval of, and eventual commercialization of, our delivery system candidates. However, obtaining regulatory approval of our delivery system candidates is not guaranteed. Our near-term prospects are substantially dependent on our ability to obtain regulatory approvals on the timetable we have anticipated, and thereafter to further successfully commercialize these delivery system candidates. Regulatory changes or actions in which we operate or propose to operate may further affect our ability to obtain regulatory approvals on the anticipated timetable. If we are not able to receive such approvals or to further commercialize our delivery system candidates, or are significantly delayed or limited in doing so, our business and prospects will be materially harmed and we may need to delay our initiatives or even significantly curtail operations.
18
To date, we have incurred substantial operating losses.
We were founded in 2000, operated as a development stage company through December 31, 2011 and have incurred substantial operating losses to date. In assessing our prospects, you must consider the risks and difficulties frequently encountered by companies in new and rapidly evolving markets, particularly companies engaged in the development and sales of oncology products. These risks include our ability to:
|
• |
continue to develop and enhance Optune and our delivery system candidates; |
|
• |
obtain regulatory approval to commercialize new delivery systems and enhance or modify our existing delivery systems; |
|
• |
increase our sales, marketing and distribution organization to commercialize our delivery systems; |
|
• |
perform clinical research and trials on Tumor Treating Fields; |
|
• |
establish and increase awareness and acceptance of our delivery systems; |
|
• |
implement and successfully execute our business and marketing strategy; |
|
• |
respond effectively to competitive pressures and developments; |
|
• |
maintain, protect and expand our intellectual property portfolio; |
|
• |
operate in compliance with applicable health care laws and regulations; |
|
• |
expand our presence in our key markets; |
|
• |
attract, retain and motivate qualified personnel; and |
|
• |
grow our organization to support our operations and our clinical pipeline and expand commercialization efforts. |
We anticipate continuing to incur significant costs associated with commercializing our delivery systems for approved indications including product sales, marketing, manufacturing and distribution expenses. We expect our research, development and clinical trials expenses to increase in connection with our ongoing activities and as additional indications enter late-stage clinical development. Our expenses could increase beyond expectations if, for example, we are required by the FDA, or other regulatory agencies to change manufacturing processes for our delivery systems, or to perform clinical, nonclinical or other types of studies in addition to those that we currently anticipate. Our revenues are dependent, in part, upon the size of the markets in the jurisdictions in which we receive regulatory approval, the accepted price for our delivery systems and the ability to obtain reimbursement at such price. If the number of addressable patients is not as significant as we estimate, the indications approved by regulatory authorities is narrower than we expect or the eligible population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenues. If we are not able to generate significant revenues, we may never become profitable.
If we do not achieve our projected research and development and commercialization goals in the timeframes we announce or expect, our business would be harmed and we may need to raise additional capital to fund our operations.
For planning purposes, we estimate the timing of the accomplishment of various scientific, clinical, regulatory and other goals, which we sometimes refer to as milestones. These milestones may include the commencement or completion of scientific studies and clinical trials, the submission of regulatory filings in the United States and other jurisdictions and the receipt of regulatory approvals in such jurisdictions. From time to time, we may publicly announce the expected timing of some of these milestones. All of these milestones are based on a variety of assumptions. The actual timing of the achievement of these milestones can vary dramatically from our estimates, in many cases for reasons beyond our control, depending on numerous factors, including:
|
• |
the rate of progress, costs and results of our research and development activities and clinical trials; |
|
• |
our ability to identify and engage appropriate health care professionals to conduct our clinical trials; |
|
• |
our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
|
• |
the extent of scheduling conflicts with participating clinicians and clinical institutions; |
|
• |
the occurrence of unanticipated adverse events during clinical trials; |
|
• |
the receipt of approvals by our competitors of competing products and by us of our delivery system candidates; |
|
• |
our ability to achieve coverage and reimbursement milestones with private and governmental third-party payers; |
|
• |
our ability to access sufficient, reliable and cost-effective supplies of components used in the manufacture of Optune and delivery system candidates, including the transducer arrays and other materials; |
19
|
• |
our ability to develop and maintain a sales and marketing organization and/or enter into sales and marketing collaborations for Optune and, if approved, our delivery system candidates; and |
|
• |
changes in regulations and other actions by regulators. |
For example, our key milestones include clinical development milestones for the use of Tumor Treating Fields to treat brain metastases, non-small cell lung cancer, pancreatic cancer, ovarian cancer and mesothelioma. We can provide no assurance that we will achieve these milestones on our expected timetable, or at all.
If we do not achieve these milestones in the timeframes we expect and generate substantial revenues, and/or if we are unable to obtain sufficient additional funds through financings, the proceeds from long-term loans, strategic collaborations or the license or sale of certain of our assets on a timely basis when necessary, we may be required to reduce expenses by delaying, reducing or curtailing the development of our delivery systems and we may need to raise additional capital to fund our operations, which we may not be able to obtain on favorable terms, if at all. If we fail to commence or complete, or experience delays in or are forced to curtail, our proposed clinical programs or otherwise fail to adhere to our projected development milestones in the timeframes we announce or expect (or within the timeframes expected by analysts or investors), or we fail to raise any required additional capital, any of such events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. We will need to generate significant revenues to achieve profitability, and we may never do so.
We may not be successful in our efforts to create a pipeline of delivery system candidates for future indications for Tumor Treating Fields and successfully commercialize them, or we may expend our resources on indications that do not yield a successful approval and fail to capitalize on other indications that may be more profitable or for which there is a greater likelihood of success.
We are pursuing clinical development of Tumor Treating Fields to treat a variety of solid tumors through our delivery system candidates. For these future indications, we are at varying stages of development and we generally do not have relevant regulatory approvals to market Tumor Treating Fields in these indications. Further, we do not currently intend to pursue indications involving solid tumors of the throat or extremities, and we do not believe Tumor Treating Fields would be efficacious for non-solid tumor cancers like lymphoma or other blood cancers.
Even if we are successful in continuing to build our pipeline, obtaining regulatory approvals and commercializing our delivery system candidates for additional indications are susceptible to risks of failure, including the significant risk that the development of our delivery system candidates for any potential indications will fail to demonstrate adequate efficacy or an acceptable safety profile, to gain regulatory approval and/or to become commercially viable. We cannot provide you any assurance that we will be able to advance any of these additional indications through the development and commercialization process. Our research programs may initially show promise in addressing additional indications, yet fail to yield approvals or commercialization for many reasons, including the following:
|
• |
we may not be able to assemble sufficient resources to pursue clinical trials for additional indications; |
|
• |
our delivery system candidates may not succeed in preclinical or clinical testing; |
|
• |
our delivery systems may, on further study be shown to have harmful side effects for other indications or other characteristics that indicate they are unlikely to be effective or otherwise do not meet applicable regulatory criteria for such indications; |
|
• |
competitors may develop alternative treatments that render our delivery systems obsolete or less attractive; |
|
• |
the market for Tumor Treating Fields may change so that the continued development of our pipeline as currently contemplated is no longer appropriate; |
|
• |
our delivery systems may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
|
• |
our delivery systems may not meet standards set by applicable regulatory authorities to obtain approval or clearance to market such delivery systems; |
|
• |
our delivery systems may not be accepted as safe, effective, convenient or otherwise desirable by patients, the medical community or third-party payers. |
If any of these events occur, we may be forced to delay or abandon our development efforts for our anticipated pipeline, which would have a material adverse effect on our business and prospects and could potentially cause our stock price to decline and cause us to cease operations. Moreover, any such events in respect of any particular indication and/or delivery system candidate may have a negative effect on the approval process for other indications and/or result in losing approval of approved delivery systems for other indications, which may exacerbate the harm to our business and prospects.
20
If we encounter difficulties enrolling patients in our clinical trials, our clinical trials could be delayed or otherwise adversely affected.
The timely completion of clinical trials depends, among other things, on our ability to enroll a sufficient number of patients who remain in the trial until its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons, including:
|
• |
the severity of the disease under investigation; |
|
• |
the limited size and nature of the patient population; |
|
• |
the patient eligibility criteria defined in our protocol and other clinical trial protocols; |
|
• |
the nature of the trial protocol, including the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; |
|
• |
clinicians’ and patients’ perceptions as to the potential advantages and side effects of Tumor Treating Fields in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are pursuing; |
|
• |
availability of other clinical trials; |
|
• |
the possibility or perception that enrolling in a Tumor Treating Fields clinical trial may limit the patient’s ability to enroll in future clinical trials for other therapies due to protocol restrictions; |
|
• |
patient referral practices of physicians; |
|
• |
the ability to monitor patients adequately during and after treatment; |
|
• |
the availability of appropriate clinical trial investigators, support staff and proximity of patients to clinical sites; |
|
• |
physicians’ or our ability to obtain and maintain patient consents; and |
|
• |
the risk that patients enrolled in clinical trials will choose to withdraw from or otherwise not be able to complete a clinical trial. |
Patients may be discouraged from enrolling in our clinical trials if the trial protocol requires them to undergo extensive follow-up to assess the safety and effectiveness of Tumor Treating Fields or if they determine that the treatments received under the trial protocols are not attractive or involve unacceptable risks or discomforts. Patients may also not participate in our clinical trials if they choose to participate in contemporaneous clinical trials of competing products. In addition, the inclusion of critically ill patients in our clinical trials may result in deaths or other adverse medical events for reasons that may not be related to Tumor Treating Fields, or, in those trials where Tumor Treating Fields is being tested in combination with one or more other therapies, for reasons that may be attributable to the other therapies, but which can nevertheless negatively affect clinical trial results. If we have difficulty enrolling a sufficient number or diversity of patients to conduct our clinical trials as planned, we may need to delay or terminate ongoing or planned clinical trials, either of which would have an adverse effect on our business.
If we are unable to continue the development of an adequate sales and marketing organization or contract with third parties to assist us, we may not be able to successfully commercialize our delivery systems that are and may be approved for commercial sale.
To achieve commercial success for Optune and our delivery system candidates, we must continue to develop and grow our sales and marketing organization and, as necessary, enter into sales and distribution relationships with third parties to market and sell Optune and our delivery system candidates. Developing and managing a sales and marketing organization is a difficult, expensive and time consuming process. To be successful we must:
|
• |
recruit and retain adequate numbers of effective and experienced sales personnel; |
|
• |
effectively train our sales personnel in the benefits and risks of Optune and our delivery system candidates; |
|
• |
establish and maintain successful sales, marketing and education programs that educate health care providers so they can appropriately inform their patients about Optune and our delivery system candidates; and |
|
• |
manage geographically disbursed sales and marketing operations. |
We may not be able to successfully develop adequate sales and marketing capabilities to achieve our growth objectives. We will have to compete with other medical device, pharmaceutical and life sciences companies to recruit, hire, train and retain the sales and marketing personnel that we anticipate we will need. In addition, because Optune requires, and we anticipate our delivery system candidates will require, physician training and education, our sales and marketing organization must grow substantially as we expand our approved indications and markets. As a consequence, our expenses associated with building up and maintaining our sales force and marketing capabilities may be disproportionate to the revenues we may be able to generate on sales of Optune and our delivery system candidates.
21
If we are unable to establish adequate sales and marketing capabilities or successful sales and distribution relationships, we may fail to realize the full revenue potential of Optune and some or all of our delivery system candidates, and we may not be able to achieve the necessary growth in a cost-effective manner or realize a positive return on our investment. If we establish sales and distribution agreements with other companies, we may not have control over the resources or degree of effort that any of these third parties may devote to our delivery systems, and if they fail to devote sufficient time and resources to the marketing of such delivery systems, or if their performance is substandard, it will adversely affect our revenues.
We may not be successful in achieving market acceptance of Tumor Treating Fields by healthcare professionals, patients and/or third-party payers in the timeframes we anticipate, or at all, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
Our business model is predicated on achieving market acceptance of Tumor Treating Fields as a monotherapy or in combination with well-established cancer treatment modalities like surgery, radiation and pharmacological therapies. We may not achieve market acceptance of Optune and other Tumor Treating Fields delivery systems we develop in the amount of time that we have anticipated, or at all, for a number of different reasons. As a general matter, we may not achieve market acceptance of Tumor Treating Fields because of the following factors, among others:
|
• |
it may be difficult to gain broad acceptance of Tumor Treating Fields because it is a new technology and involves a novel delivery system, and as such physicians may be reluctant to prescribe Tumor Treating Fields delivery systems without prior experience or additional data or training; |
|
• |
physicians may be reluctant to prescribe Tumor Treating Fields delivery systems due to their perception that a clinical trial is not appropriately designed; |
|
• |
it may be difficult to gain broad acceptance at community hospitals where the number of patients seeking cancer treatment may be more limited than at larger medical centers, and such community hospitals may not be willing to invest in the resources necessary for their physicians to become trained to use Tumor Treating Fields, which could lead to reluctance to prescribe our Tumor Treating Fields delivery systems; |
|
• |
patients may be reluctant to elect to use our Tumor Treating Fields delivery systems, including Optune, for various reasons, including a perception that the treatment is untested or difficult to use; |
|
• |
the delivery systems may have some side effects (for example, dermatitis where the transducer arrays are placed) and the delivery system cannot be worn in all circumstances (for example, it cannot get wet and is difficult to wear in high temperatures); and |
|
• |
the price of the Tumor Treating Fields delivery systems includes a monthly fee for use of the delivery system, so as the duration of the treatment course increases, the price will increase correspondingly, and, when used in combination with other treatments, the overall cost of treatment will be greater than using a single type of treatment. |
In particular, Optune may not achieve market acceptance because of the following additional factors (which also may apply to our future delivery systems, to varying degrees):
|
• |
achieving patient acceptance is difficult because GBM is a devastating disease with a poor prognosis, and not all patients with potentially short lifespans are willing to comply with requirements of treatment with Optune, such as extended use of Optune, carrying around a device and shaving their heads (which may be of particular concern to women), and other patients may forego Optune treatment for privacy, cosmetic visibility or mobility reasons; |
|
• |
achieving patient compliance is difficult because the recommended average daily use of Optune is at least 18 hours a day, requiring patients to wear the delivery system nearly continuously, which to some extent restricts physical mobility because the battery must be frequently recharged, and the patient or a caregiver must ensure that it remains continuously operable; |
|
• |
certain patients are not advised to use Optune, including patients who have an active electronic medical device, which include deep brain stimulators, spinal cord stimulators, vagus nerve stimulators, pacemakers, defibrillators and programmable shunts, because the use of Optune with these devices has not been tested and may lead to malfunctioning of these devices; patients who have a skull defect or bullet fragments are also not advised to use Optune because the use of Optune with these conditions has not been tested and may lead to tissue damage or render Optune ineffective; and patients who are sensitive to conductive hydrogels because skin contact with the gel used in Optune for patients that are sensitive to conductive hydrogels may commonly cause increased redness and itching, and in rare instances may lead to severe allergic reactions, such as shock or respiratory failure; |
22
|
• |
adverse events reported in clinical trials by GBM patients treated with Optune as monotherapy include medical device site reaction, headache, malaise, muscle twitching, fall and skin ulcer; additional adverse events reported in clinical trials by GBM patients treated with Optune in combination with temozolomide in addition to the above, were thrombocytopenia, nausea, constipation, vomiting, and fatigue. |
In addition, even if we are successful in achieving market acceptance of Optune for GBM, we may be unsuccessful in achieving market acceptance of Tumor Treating Fields as a treatment for other solid tumor cancers, such as brain metastases, NSCLC, pancreatic cancer, ovarian cancer, mesothelioma and other solid tumor cancers, because certain radiation, chemotherapies and/or systemic medical therapies may become or remain the preferred standard of care for these indications.
There may be other factors that are presently unknown to us that also may negatively impact our ability to achieve market acceptance of Tumor Treating Fields delivery systems. If we do not achieve market acceptance of our delivery systems in the timeframes we anticipate, or are unable to achieve market acceptance at all, our business, prospects, financial condition and results of operations could be materially adversely affected, and our stock price could decline.
Failure to secure and maintain adequate coverage and reimbursement from third-party payers could adversely affect acceptance of our delivery systems and reduce our revenues.
We expect that the vast majority of our revenues will come from third-party payers either directly to us in markets where we provide Optune or plan to provide our delivery system candidates to patients or indirectly via payments made to hospitals or other entities providing Optune or which may in the future provide our delivery system candidates to patients.
In the United States, private payers cover the largest segment of the population, with the remainder either uninsured or covered by governmental payers. We anticipate that the majority of the third-party payers outside the United States will be government agencies, government sponsored entities or other payers operating under significant regulatory requirements from national or regional governments.
Third-party payers may decline to reimburse for procedures, supplies or services not under coverage policies. Additionally, some third-party payers may decline to reimburse us for a particular patient even with the existence of a coverage policy. Additionally, private commercial and government payers may be permitted to consider the cost of a treatment in approving coverage or in setting payment for the treatment.
Private and government payers around the world are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of governments around the world. Adoption of additional price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our revenues and operating results. If third-party payers do not consider our delivery system or the combination of our delivery system with additional treatments to be cost-justified under a required cost-testing model, they may not cover our delivery systems for their populations or, if they do, the level of payment may not be sufficient to allow us to sell our delivery systems on a profitable basis.
Reimbursement for the treatment of patients with medical devices around the world is governed by complex mechanisms established on a national level in each country. These mechanisms vary widely among countries and evolve constantly, reflecting the efforts of these countries to reduce public spending on healthcare. As a result, obtaining and maintaining reimbursement for the treatment of patients with medical devices has become more challenging globally. We cannot, therefore, guarantee that the treatment of patients with Optune or any of our future delivery systems would receive reimbursement approvals and cannot guarantee that our existing reimbursement approvals will be maintained in any country.
We provide financial assistance to patients in certain markets, primarily those where we have or are actively pursuing reimbursement coverage, to defray patients’ out-of-pocket costs for Optune, and therefore, absorb any unreimbursed costs of patients who begin treatment and are unable to pay for the costs of their treatment not covered by insurance. Our costs associated with this program could increase if payers increase the cost-sharing burden of patients or we do not obtain national reimbursement coverage and we elect to continue providing financial assistance in those markets.
23
Our failure to secure or maintain adequate coverage or reimbursement for Optune or any of our future delivery systems by third-party payers in the United States or in the other jurisdictions in which we market Optune or any of our future delivery systems, could have a material adverse effect on our business, financial condition and results of operations and cause our stock price to decline.
We may not be successful securing and maintaining reimbursement codes necessary to facilitate accurate and timely billing for Optune, future delivery systems and physician services attendant to Tumor Treating Fields therapy.
Third-party payers, healthcare systems, government agencies or other groups often issue reimbursement codes to facilitate billing for products and physician services used in the delivery of medicine. Within the United States, the billing codes most directly related to Optune and future delivery systems are contained in the Healthcare Common Procedure Coding System (“HCPCS code set”). The HCPCS code set contains Level I codes that describe physician services, also known as Common Procedural Terminology codes (“CPT codes”) and Level II codes that primarily describe products. The Centers for Medicare and Medicaid Services (“CMS”) is responsible for issuing the HCPCS Level II codes. The American Medical Association issues HCPCS Level I codes.
We have secured unique HCPCS Level II codes that describe Optune and we are able to use these codes in the United States to bill third-party payers. Loss of these codes or any alteration in the payment attached to these codes would materially impact our operating results.
Although we are attempting to secure CPT codes, no CPT codes currently exist to describe physician services related to the delivery of Tumor Treating Fields therapy. We may not be able to secure CPT codes for physician services related to Optune based on the relatively low incidence of GBM. Our future revenues and results may be affected by the absence of CPT codes, as physicians may be less likely to adopt the therapy when not adequately reimbursed for the time, effort, skill, practice expense and malpractice costs required to provide the therapy to patients.
We have not secured codes to describe our delivery systems or to document physician services related to the delivery of Tumor Treating Fields therapy in markets outside the United States. Absence of these codes may affect the future growth of our business.
There is no assurance that Medicare or the Medicare Administrative Contractors will provide coverage or adequate payment rates for Optune or our future delivery systems.
In 2017, approximately 20-25% of patients using Optune in the United States were beneficiaries under the Medicare fee-for-service program. Failure to secure coverage and adequate payment from Medicare would reduce our revenues and may also affect the coverage and payment decisions of other third-party payers in the United States.
Medicare has the authority to issue national coverage determinations or to defer coverage decisions to its regional Medicare Administrative Contractors (“MACs”). Medicare has not issued a national coverage determination for Optune. The two MACs that currently administer the durable medical equipment benefit for MACs have each issued local coverage determination policies stating that Optune is not reasonable and necessary for the treatment of recurrent GBM. The fact that only two MACs administer the entire DME program may negatively affect our ability to petition individual medical policy decision-makers at the MACs for coverage. The continuing absence of a positive coverage determination from Medicare or the DME MACs would materially affect our future revenues.
Additionally, Medicare has the authority to publish the price of durable medical equipment products. Medicare may publish prices for Optune or future delivery systems that do not reflect then current prices for Optune or future delivery systems. Medicare price schedules are frequently referenced by private payers in the United States and around the world. Medicare would materially reduce our revenues and operating results by publishing a price for Optune or future delivery systems that is not based on the actual price of Optune or future delivery systems within the private payer market.
CMS implemented a demonstration project in 2012 to require prior authorization for certain Durable Medical Equipment, Prosthetics, Orthotics and Supplies items. Claims for services that did not receive prior authorization before they were rendered will be automatically denied. In the event Medicare provides coverage for Optune in the future and Optune is added to the list of items requiring prior authorization that may reduce our ability to bill and secure payment for patients who would otherwise be covered to use Optune under the Medicare fee-for-service program.
The Medicare fee-for-service program has denied coverage for our claims to date. Although we are actively appealing these coverage denials, we are unable to bill our existing Medicare fee-for-service patients for amounts not paid by Medicare. Therefore, we are absorbing and may continue to absorb the costs of treatment for amounts not paid by Medicare.
24
We appeal Medicare coverage denials through the Medicare appeals process: redetermination by a MAC, reconsideration by a Qualified Independent Contractor, hearing before an Administrative Law Judge, or ALJ, at the Office of Medicare Hearings and Appeals, review by the Medicare Appeals Council, and judicial review in U.S. District Court. Currently, there is a considerable backlog of appeals at the ALJ level and there are significant delays in the assignment of ALJ cases. Thus, we anticipate that, even if we are successful in winning our appeals, we will experience a significant delay in securing payment for Medicare patients when Medicare’s DME MACs deny coverage for patients who start therapy.
We depend on single-source suppliers for some of our components. The loss of these suppliers could prevent or delay shipments of Optune, delay our clinical trials or otherwise adversely affect our business.
We source some of the key components of Optune from only a single vendor. If any one of these single-source suppliers were to fail to continue to provide components to us on a timely basis, or at all, our business and reputation could be harmed. For example, we currently obtain the ceramic discs used in the transducer arrays for Optune from a single supplier. We have technically qualified and entered into a supply agreement with an additional supplier, and we have requested regulatory approval to use these ceramic discs in the EU. This source will be able to ship product for use within the United States and Japan at a future date pending regulatory approval, which we may never receive.
Establishing additional or replacement suppliers for any components of our delivery systems, and obtaining any additional regulatory approvals required to add or replace suppliers, will take a substantial amount of time and could result in increased costs and impair our ability to produce Optune, which would have a material adverse effect on our business, prospects, financial condition and results of operations. We may have difficulty obtaining similar components from other suppliers that are acceptable to the FDA or other regulatory authorities, or to comply with the Essential Requirements laid down in Annex I to the Directive 93/42/EEC concerning medical devices, commonly known as the Medical Devices Directive, which are the minimum requirements governing design and manufacturing in the EU. The risks associated with the failure of our suppliers to comply with strictly enforced regulatory requirements as described below are exacerbated by our dependence on single-source suppliers. Furthermore, since some of these suppliers are located outside of the United States, we are subject to export laws in other jurisdictions and United States import and customs regulations, which complicate and could delay shipments of components to us. Changes in U.S. social, political, regulatory and economic conditions or in laws and policies governing foreign trade, manufacturing, development and investment in the territories and countries where we may develop and sell products, and any negative sentiments towards the United States as a result of such changes, could adversely affect our business.
Our policy is to seek second-source suppliers, but we can provide no assurance we will secure or maintain such suppliers. Various steps must be taken before securing these suppliers, including qualifying these suppliers in accordance with regulatory requirements.
If we experience any delay or deficiency in the quality of components supplied to us by third-party suppliers, or if we have to switch to replacement suppliers, we may face additional regulatory delays and the manufacture and delivery of Optune would be interrupted for an extended period of time, which could materially adversely affect our business, prospects, financial condition and results of operations. In addition, we may be required to obtain prior regulatory approval if we use different suppliers or components. Such changes could affect our FDA regulatory approvals and the compliance of our delivery systems with the Essential Requirements laid down in Annex I to the Medical Devices Directive and the validity of our current CE Certificates of Conformity. If we are required to obtain prior regulatory approval from the FDA or regulatory authorities in other jurisdictions or to conduct a new conformity assessment procedure and obtain new CE Certificates of Conformity in the EU to use different suppliers or components for our delivery systems, regulatory approval or the CE Certificates of Conformity for our delivery systems may not be received on a timely basis, or at all, which would have a material adverse effect on our business, prospects, financial condition and results of operations.
Quality control problems with respect to delivery systems and components supplied by third-party vendors could have a material adverse effect on our reputation, our clinical trials or the commercialization of Optune and our future delivery systems and, as a result, a material adverse effect on our business, prospects, financial condition and results of operations.
Our delivery systems, which are manufactured by third parties, are highly technical and are required to meet exacting specifications. Any quality control problems that we experience with respect to the delivery systems and components supplied by third-party vendors could have a material adverse effect on our reputation, our attempts to complete our clinical trials or the commercialization of Optune and our future delivery systems. The failure of our suppliers to comply with strictly enforced regulatory requirements could expose us to regulatory action, including warning letters, product recalls, suspension or termination of distribution, product seizures or civil penalties. If we experience any delay or deficiency in the quality of products supplied to us by third-party suppliers, or if we have to switch to replacement suppliers, we may face additional regulatory delays and the manufacture and delivery of our delivery systems would be interrupted for an extended period of time, which would materially adversely affect our business, prospects, financial condition and results of operations.
25
If the third parties on which we rely to conduct our clinical trials and to assist us with preclinical research and development do not perform as contractually required or expected, we may not be able to obtain regulatory approvals for our future delivery systems or commercialize our future delivery systems.
We do not have the ability to independently conduct some of our preclinical and all of clinical trials for our delivery systems and we must rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct such trials. We and these third parties are required to comply with current good clinical practices (“cGCPs“) which are regulations and guidelines enforced by the FDA and comparable regulatory authorities in other jurisdictions for clinical development. We and these third parties are also required to comply with current good laboratory practices (“cGLPs”) which are regulations and guidelines enforced by the FDA and comparable regulatory authorities in other jurisdictions for nonclinical laboratory studies. Regulatory authorities enforce these cGLPs and cGCPs through periodic inspections of trial sponsors, laboratories, principal investigators and trial sites. If we or any of these third parties fail to comply with applicable cGLP and cGCP regulations, the clinical data generated in our nonclinical studies and clinical trials may be deemed unreliable and the FDA or regulatory authorities in other jurisdictions may require us to perform additional nonclinical or clinical trials before approving our approved applications. We cannot be certain that, upon inspection or review of our files, such regulatory authorities will determine that any of our nonclinical studies or clinical trials comply with the applicable cGLP or cGCP regulations.
Any third parties conducting our nonclinical studies and clinical trials are not and will not be our employees and, except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our ongoing preclinical, clinical and nonclinical programs. These third parties may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting nonclinical studies, clinical studies or other cancer treatment development activities, which could affect their performance on our behalf. If these third parties do not successfully carry out their contractual duties or regulatory obligations or meet expected deadlines, if these third parties need to be replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements or for other reasons, our preclinical development activities or clinical trials may be extended, delayed, suspended or terminated, and we may not be able to obtain regulatory approval for our delivery systems or successfully commercialize our delivery systems on a timely basis, if at all, and our business, prospects and results of operations may be adversely affected.
Continued testing of Optune or our other delivery system candidates may not yield successful results and could reveal currently unknown safety hazards associated with Tumor Treating Fields.
Our research and development programs are designed to test the safety and efficacy of Tumor Treating Fields through extensive preclinical and clinical testing. Even if our ongoing and future clinical trials are completed as planned, we cannot be certain that their results will support our claims or that the FDA and other regulatory authorities will agree with our conclusions regarding them. Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and preclinical studies. The clinical trial process may fail to demonstrate that our delivery system candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a delivery system candidate and may delay development of others. It is also possible that patients enrolled in clinical trials will experience adverse side effects that have not been previously observed. In addition, our preclinical studies and clinical trials for our delivery system candidates involve a relatively small patient population, and as a result, these studies and trials may not be indicative of future results.
We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent further commercialization of Optune and any of our delivery system candidates, including the following:
|
• |
safety and efficacy results for Optune and any of our delivery system candidates obtained in our preclinical and clinical testing may be inconclusive or may not be predictive of results obtained in future clinical trials, following long-term use or in much larger populations; |
|
• |
unanticipated adverse events may occur during our clinical trials; |
|
• |
the data collected from clinical trials of our delivery system candidates may not reach statistical significance due to limited sample size or otherwise not be sufficient to support FDA or other regulatory approval; and |
|
• |
our delivery system candidates may not produce the desired effects or may result in adverse health effects or other characteristics that are not currently known that preclude additional regulatory approval or limit their commercial use if approved. |
To date, patients treated with Optune in our clinical trials have experienced treatment-related side effects, including dermatitis (including mild to moderate skin irritation) where the transducer arrays are placed, headaches, weakness, falls, fatigue, muscle twitching and skin ulcers. There may be additional side effects observed in future clinical trials and/or through real-world experience
26
with patients using Optune or our other Tumor Treating Fields delivery system candidates. Undesirable side effects caused by our delivery systems could cause us or regulatory authorities to interrupt, delay or terminate clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other regulatory authorities. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects.
If unacceptable side effects arise in the development of our delivery system candidates, we could suspend or terminate our clinical trials or the FDA or other regulatory authorities could order us to cease clinical trials or deny approval of our delivery system candidates for any or all targeted indications, narrow the approved indications for use or otherwise require restrictive product labeling or marketing, or require further clinical trials, which may be time-consuming and expensive and may not produce results supporting FDA or other regulatory approval of our delivery system candidates in a specific indication. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have to train medical personnel using our delivery system candidates to understand the side effect profiles for our clinical trials and upon any commercialization of any of our delivery system candidates. Inadequate training in recognizing or managing the potential side effects of our delivery system candidates could result in patient injury or death. Any of these occurrences may harm our business, prospects and financial condition significantly.
Any delay or termination of our clinical trials will delay the filing of our delivery systems submissions for regulatory approvals and ultimately our ability to commercialize our delivery systems and generate revenues. Furthermore, we may abandon delivery system candidates that we previously believed to be promising. Any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
We face competition from numerous competitors, most of whom have far greater resources than we have, which may make it more difficult for us to achieve significant market penetration and which may allow them to develop additional oncology treatments to compete with Tumor Treating Fields.
The oncology market is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. As a monotherapy, Tumor Treating Fields primarily competes with radiation and pharmacological therapies. We may face additional competition as advancements are made in the field of anti-cancer therapies. To date, we have conducted clinical trials where Tumor Treating Fields is used in combination with a certain subset of other anti-cancer therapies. Many of our competitors are large, well-capitalized companies with significantly more market share and resources than we have. As a consequence, they are able to spend more aggressively on product development, marketing, sales and other initiatives than we can. Many of these competitors have:
|
• |
significantly greater name recognition and experience; |
|
• |
established relations with healthcare professionals, patients and third-party payers; |
|
• |
established distribution networks; |
|
• |
additional product lines, and the ability to offer rebates or bundle products to offer higher discounts or more competitive pricing or other incentives to gain a competitive advantage; and/or |
|
• |
greater financial and human resources for research and development, sales and marketing, patent litigation and/or acquisitions. |
Although we believe Tumor Treating Fields represents a treatment modality that can be used in combination with other cancer treatment modalities, our current competitors or other companies may at any time develop additional drugs and devices for the treatment of GBM and other solid tumors that could be more effective than using our Tumor Treating Fields delivery systems. If an existing or future competitor develops a product that proves to be superior or comparable to Optune or any of our future delivery systems, our revenues may decline. In addition, some of our competitors may compete by changing the price of their cancer treatments. If these competitors’ products were to gain acceptance by healthcare professionals, patients or third-party payers, a downward pressure on prices could result. If prices were to fall, we may not be able to improve our gross margins or sales growth sufficiently to achieve profitability.
As we expand, we may experience difficulties managing our growth.
Our anticipated growth will place a significant strain on our management and on our operational and financial resources and systems. Failure to manage our growth effectively could materially adversely affect us. Additionally, our anticipated growth will increase the demands placed on our suppliers, resulting in an increased need for us to carefully monitor the available supply of components and quality assurance. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
27
Because of the specialized nature of our business, the termination of relationships with our key employees, consultants and advisors may prevent us from developing Tumor Treating Fields, conducting clinical trials and obtaining any necessary financing. Further, the inability to recruit and retain additional personnel may have an adverse effect on our ability to successfully operate our business.
We are highly dependent on members of our executive team, the loss of whose services may adversely impact the achievement of our objectives. While we have entered into employment agreements with each of our executive officers, any of them could leave our employment at any time. We do not have “key person” insurance on any of our employees. The loss of the services of one or more of our current employees might impede the achievement of our business objectives. The competition for qualified personnel in the oncology field is intense, and we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. Our future success depends upon our ability to attract, retain and motivate highly skilled employees. In order to commercialize Tumor Treating Fields successfully, we will be required to expand our workforce, particularly in the areas of research and development and clinical trials, sales and marketing and supply chain management. These activities will require the addition of new personnel and the development of additional expertise by existing management personnel. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. We may not be able to attract and retain these individuals on acceptable terms or at all. Failure to do so would materially harm our business.
Changes in tax or other laws, regulations or treaties, changes in our status under U.S. or non-U.S. laws or adverse determinations by taxing or other governmental authorities could increase our tax burden or otherwise affect our financial condition or results of operations, as well as subject our shareholders to additional taxes.
The amount of taxes we pay is subject to a variety of tax laws in the various jurisdictions in which we and our subsidiaries are organized and operate. Our domestic and international tax liabilities are dependent on the location of earnings among these various jurisdictions. Such tax liabilities could be affected by changes in tax or other laws, treaties and regulations as well as the interpretation or enforcement thereof by tax or other governmental entities in any relevant jurisdiction. The amount we pay in tax to any particular jurisdiction depends, in part, on the correct interpretation of the tax laws in such jurisdiction, and we have made a number of determinations as to the effect of such tax laws in our particular circumstances. In some cases, the determinations we have made as to the effect of the tax laws in a particular jurisdiction depend on the continuing effectiveness of administrative rulings we have received from the tax authorities in that jurisdiction, while in other cases, our determinations are based on the reasoned judgment of our tax advisors. Although we believe that we are in compliance with the administrative rulings we have received, that the assumptions made by our tax advisors in rendering their advice remain correct, and that as a result we are in compliance with applicable tax laws in the jurisdictions where we and our subsidiaries are organized and operate, a taxing authority in any such jurisdiction may challenge our interpretation of those laws and assess us or any of our subsidiaries with additional taxes.
Additionally, from time to time, proposals can be made and legislation can be introduced to change the tax laws, regulations or interpretations thereof (possibly with retroactive effect) of various jurisdictions or limit tax treaty benefits that, if enacted, could materially increase our tax burden, increase our effective tax rate or otherwise have a material adverse impact on our financial condition and results of operations. It is possible that these changes could adversely affect our business. While we monitor proposals and other developments that would materially impact our tax burden and effective tax rate and investigate our options accordingly, we could still be subject to increased taxation on a going forward and retroactive basis no matter what action we undertake if certain legislative proposals or regulatory changes are enacted, certain tax treaties are amended and/or our interpretation of applicable tax or other laws is challenged and determined to be incorrect. Any alternative interpretations of applicable tax laws asserted by a tax authority or changes in tax laws, regulations or accounting principles that limit our ability to take advantage of tax treaties between jurisdictions, modify or eliminate the deductibility of various currently deductible payments, increase the tax burden of operating or being resident in a particular country, result in transfer pricing adjustments or otherwise require the payment of additional taxes, may have a material adverse effect on our cash flows, financial condition and results of operations
The termination or revision of any of our tax rulings or indirect tax exemptions that we have or may have in the future may have a material adverse effect on our cash flows, financial condition and results of operations.
We believe our ordinary shares should not be treated as stock of a passive foreign investment company, or PFIC, for U.S. federal income tax purposes in the current taxable year or in a future taxable year, but this conclusion is a factual determination that is made annually and thus may be subject to change. If we were to be treated as a PFIC, this could result in adverse U.S. federal income tax consequences to U.S. persons that hold our ordinary shares.
Based on the composition of our assets and the nature of our income, we believe that our shares should not be treated as stock of a PFIC for U.S. federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change.
28
A non-U.S. corporation will be treated as a PFIC for U.S. federal income tax purposes in any taxable year in which a specified percentage of its gross income is “passive income” or a specified percentage of its assets produce or are held for the production of passive income (“passive assets”), including cash. If we are treated as a PFIC, and a U.S. person that holds our ordinary shares, either directly or indirectly, did not make one of the applicable available elections, such U.S. person would be subject to adverse U.S. federal income tax consequences on distributions with respect to the ordinary shares to the extent the distributions are “excess distributions,” which are generally distributions in excess of a normal rate of distribution as calculated for PFIC purposes. Gain realized on the sale or other disposition of the ordinary shares would generally not be treated as capital gain, but rather would be treated as if such U.S. person had realized such gain and certain “excess distributions” ratably over the holding period for the ordinary shares and would be taxed at the highest tax rate in effect for each such year to which the gain was allocated, together with an interest charge in respect of the tax attributable to each such year. Partial redemptions would also be treated as excess distributions. We will, upon request from any shareholder, prepare and provide information as necessary for “qualified electing fund” elections but we make no representation as to the availability of “mark to market” elections that may mitigate the consequences of our being a PFIC to any U.S. investor. Prospective U.S. investors should consult their own U.S. tax advisors regarding the potential application of the PFIC rules.
Product liability suits, whether or not meritorious, could be brought against us due to alleged defective delivery systems or for the misuse of our delivery systems. These suits could result in expensive and time-consuming litigation, payment of substantial damages and an increase in our insurance rates.
If our current or future delivery systems are defectively designed or manufactured, contain defective components or are misused, or if someone claims any of the foregoing, whether or not meritorious, we may become subject to substantial and costly litigation. For example, we may be sued if our delivery systems cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. This may occur if Optune is misused or damaged, has a sudden failure or malfunction (including with respect to safety features) or is otherwise impaired due to wear and tear. Even absent a product liability suit, malfunctions of the device or misuse by the physician or patient would need to be remedied swiftly in order to maintain continuous use and ensure efficacy of Tumor Treating Fields.
Any product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the delivery system, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of Optune and our delivery system candidates. Even successful defense may require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
|
• |
decreased demand for our Tumor Treating Fields delivery systems; |
|
• |
injury to our reputation; |
|
• |
withdrawal of clinical trial participants and inability to continue clinical trials; |
|
• |
initiation of investigations by regulators; |
|
• |
costs to defend the related litigation; |
|
• |
a diversion of management’s time and our resources; |
|
• |
substantial monetary awards to trial participants or patients; |
|
• |
product recalls, withdrawals or labeling, marketing or promotional restrictions; |
|
• |
loss of revenues; |
|
• |
exhaustion of any available insurance and our capital resources; |
|
• |
the inability to commercialize any delivery system candidate; and |
|
• |
a decline in our share price. |
Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizable damage awards against us. We may not have sufficient insurance coverage for all claims. Any product liability claims brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing continuing coverage, could harm our reputation in the industry and could reduce revenues. Product liability claims in excess of our insurance coverage would be paid out of cash reserves, if any, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Even if our agreements with our manufacturers and suppliers entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
29
Other future litigation and regulatory actions could have a material adverse impact on the Company.
From time to time, we may be subject to litigation and other legal and regulatory proceedings relating to our business or investigations or other actions by governmental agencies, including as described in Part I, Item 3 “Legal Proceedings” of this Annual Report on Form 10-K. No assurances can be given that the results of these or new matters will be favorable to us. An adverse resolution of lawsuits, arbitrations, investigations or other proceedings or actions could have a material adverse effect on our financial condition and results of operations, including as a result of non-monetary remedies. Defending ourselves in these matters may be time-consuming, expensive and disruptive to normal business operations and may result in significant expense and a diversion of management’s time and attention from the operation of our business, which could impede our ability to achieve our business objectives. Additionally, any amount that we may be required to pay to satisfy a judgment, settlement, fine or penalty may not be covered by insurance. Subject to the Jersey Companies Law, our articles of association permit us to indemnify any director against any liability, to purchase and maintain insurance against any liability for any director and to provide any director with funds (whether by loan or otherwise) to meet expenditures incurred or to be incurred by such director in defending any criminal, regulatory or civil proceedings or in connection with an application for relief (or to enable any such director to avoid incurring such expenditure). In addition, we have entered into indemnification agreements with each of our directors, and we anticipate entering into indemnification agreements with each of our officers, to indemnify them against certain liabilities and expenses arising from their being a director to the maximum extent permitted by Jersey law. In the event we are required to make such payments to our directors, there can be no assurance that any of these payments will not be material.
Global economic, political and industry conditions constantly change and unfavorable conditions, particularly in Israel, may have a material adverse effect on our business and results of operations.
We are a global oncology treatment company with worldwide operations. Volatile economic, political and market conditions, such as political or economic instability, majority hostilities or acts of terrorism, in the regions in which we operate may have a negative impact on our operating results and our ability to achieve our business objectives. We may not have insight into economic and political trends that could emerge and negatively affect our business. In addition, significant or volatile changes in exchange rates between the U.S. dollar and other currencies may have a material adverse impact upon our liquidity, revenues, costs and operating results.
In particular, we have research facilities located in Israel, and one of our key suppliers manufactures its goods in one physical location in Israel. Due to the high-conflict nature of this area, Israel could be subject to additional political, economic and military confines, which could result in a material adverse effect on our operations. Parties with whom we do business have sometimes declined to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in the agreements.
The vote by the United Kingdom electorate in favor of the United Kingdom’s exit from the European Union could adversely impact our business, results of operations and financial condition.
The passage of the referendum on the United Kingdom’s membership in the EU, referred to as “Brexit,” in favor of the exit of the United Kingdom from the EU, could cause disruption to and create uncertainty surrounding our business, which could have an adverse effect on our business, financial results and operations. A process of negotiation is now taking place to determine the future terms of the United Kingdom’s relationship with the EU, including trade terms between the United Kingdom and countries comprising the EU. The effects of Brexit will depend on any agreements the United Kingdom makes to retain access to markets in the EU, either during a transitional period or more permanently.
Depending on the outcome of these negotiations, we could face new challenges in our operations, such as instability in global financial and foreign exchange markets, including volatility in the value of the British pound and European euro, and increased trade barriers, all of which could result in restrictions on the movement of capital within our organization, the mobility of our personnel and the potential future commercialization of Optune or our delivery system candidates and could change our tax benefits or liabilities, any of which could have a material adverse effect on our business, results of operations or financial condition.
30
We are increasingly dependent on information technology systems and subject to privacy and security laws, and our systems and infrastructure face certain risks, including from cyber security breaches and data leakage.
We increasingly rely upon technology systems and infrastructure. Our technology systems are potentially vulnerable to breakdown or other interruption by fire, power loss, system malfunction, unauthorized access and other events. Likewise, data privacy breaches by employees and others with both permitted and unauthorized access to our systems may pose a risk that sensitive data (including protected health information (“PHI”)) may be exposed to unauthorized persons or to the public, or may be permanently lost. The increasing use and evolution of technology, including cloud-based computing, creates additional opportunities for the unintentional dissemination of information, intentional destruction of confidential information stored in our systems or in non-encrypted portable media or storage devices. We could also experience a business interruption, information theft of confidential information, or reputational damage from industrial espionage attacks, malware or other cyber incidents, which may compromise our system infrastructure or lead to data leakage, either internally or at our third-party service providers or other business partners. Additionally, we must comply with numerous laws and regulations governing the collection, dissemination, access, use, security and PHI, including, in the U.S., HIPAA and its implementing privacy and security regulations and applicable state laws, and currently in the EU, Directive 95/46/EC of the European Parliament and of the Council of October 24, 1995 and applicable national laws. The Data Protection Directive will be replaced in May 2018 by the EU's General Data Protection Regulation (“GDPR”) which is designed to harmonize data privacy laws for both patient and employee data across Europe. While we have invested heavily in the protection of data and information technology and in related training, there can be no assurance that our efforts will prevent significant breakdowns, breaches in our systems or other cyber incidents or ensure compliance with all applicable security and privacy laws, regulations and standards, including with respect to third-party service providers that utilize sensitive personal information, including PHI, on our behalf. Any such breakdown, breach, incident or failure to comply could have a material adverse effect upon our reputation, business, operations or financial condition. In addition, significant implementation issues may arise as we continue to consolidate and outsource certain computer operations and application support activities.
Changes in our technology could result in impairment charges in future periods.
United States generally accepted accounting principles (“GAAP”) require annual (or more frequently if events or changes in circumstances warrant) impairment tests of goodwill, intangible assets and other long-lived assets. Generally speaking, if the carrying value of the asset is in excess of the estimated fair value of the asset, the carrying value will be adjusted to fair value through an impairment charge. Circumstances such as changes in technology or in the way an asset is being used may trigger an impairment review. Any negative perception of such a deficit could have an adverse effect on the price of our ordinary shares and could impair our ability to obtain new financing or refinance existing indebtedness.
If any of our facilities are damaged or our clinical, research and development or other business processes interrupted, our business could be seriously harmed.
We conduct our business in a limited number of facilities in the United States, Germany, Switzerland, Israel and Japan. Damage or extended periods of interruption to our or our suppliers’ or manufacturers’ corporate, development or research facilities due to fire, natural disaster, power loss, communications failure, unauthorized entry, terrorist attacks or other events could cause us to cease or delay development and/or delivery of some or all of our delivery systems. Our internal computer systems may fail or suffer security breaches, which could result in a material disruption of our business. Our business may be seriously harmed by such delays and interruption.
Our research facilities are located in Israel, and one of our key suppliers manufactures its goods in one physical location in Israel. Due to the high-conflict nature of this area, one of our key suppliers manufactures its goods in one physical location in Israel. Although our facilities have not sustained any damage from such attacks, this is a high conflict area and any future attacks and resulting damage could adversely affect our operations. In addition, our business insurance only covers certain specified events associated with war or terrorism in the Middle East, and may not cover all such events. Although the Israeli government currently covers the reinstatement value of direct damages that are caused by terrorist attacks or acts of war, this government coverage may not be maintained, or may be insufficient to cover all losses we incur, even if available. Any losses or damages incurred by us could have a material adverse effect on our business.
We have significant debt service obligations and may incur additional indebtedness in the future, which could adversely affect our financial condition and results of operations and our ability to react to changes in our business.
We currently have $150.0 million of principal indebtedness outstanding under our Loan and Security Agreement dated as of February 7, 2018, between us, as borrower, and BioPharma Credit PLC, as lender (“2018 Credit Facility”). We may incur additional indebtedness in the future. Our existing indebtedness and any additional indebtedness we may incur could require us to divert funds identified for other purposes for debt service and impair our liquidity position.
31
The fact that a substantial portion of our cash flow from operations could be needed to make payments on our indebtedness could have important consequences, including the following:
|
• |
increasing our vulnerability to general adverse economic and industry conditions or increased interests rates; |
|
• |
reducing the availability of our cash flow for other purposes; |
|
• |
limiting our flexibility in planning for or reacting to changes in our business and the markets in which we operate, which would place us at a competitive disadvantage compared to our competitors that may have less debt; |
|
• |
limiting our ability to borrow additional funds for working capital, capital expenditures and other investments; and |
|
• |
failing to comply with the covenants in our debt agreements could result in all of our indebtedness becoming immediately due and payable. |
Our ability to obtain necessary funds through borrowing, as well as our ability to service our indebtedness, will depend on our ability to generate cash flow from operations. Our ability to generate cash is subject to general economic, financial, competitive, legislative, regulatory and other factors that are beyond our control. If our business does not generate sufficient cash flow from operations or if future borrowings are not available to us under our 2018 Credit Facility or otherwise in amounts sufficient to enable us to fund our liquidity needs, our financial condition and results of operations may be adversely affected. Our inability to make scheduled payments on our debt obligations in the future would require us to refinance all or a portion of our indebtedness on or before maturity, sell assets or seek additional equity investment. We may not be able to take any of such actions on a timely basis, on terms satisfactory to us or at all.
Covenants in our debt agreements restrict our operational flexibility.
Our 2018 Credit Facility contains usual and customary restrictive covenants relating to the operation of our business, including restrictions on our ability:
|
• |
to incur or guarantee additional indebtedness; |
|
• |
to incur or permit to exist certain liens; |
|
• |
to enter into certain sale and lease-back transactions; |
|
• |
to make certain investments, loans and advances; |
|
• |
to effect certain mergers, consolidations, asset sales and acquisitions; |
|
• |
to pay dividends on, or redeem or repurchase, capital stock, enter into transactions with affiliates or materially change our business; and |
|
• |
to repay or modify certain other agreements with respect to other material indebtedness or modify our organizational documents. |
Risks relating to regulation
Our delivery system candidates must undergo rigorous preclinical and clinical testing and we must obtain regulatory approvals, which could be costly and time-consuming and subject us to unanticipated delays or prevent us from marketing any delivery systems.
Our research and development activities, as well as the manufacturing and marketing of Optune and our delivery system candidates, are subject to regulation, including regulation for safety, efficacy and quality, by the FDA in the United States and comparable authorities in other countries. FDA regulations and the regulations of comparable regulatory authorities in other jurisdictions are wide-ranging and govern, among other things:
|
• |
the conduct of preclinical and clinical studies; |
|
• |
product design, development, manufacturing and testing; |
|
• |
product labeling; |
|
• |
product storage and shipping; |
|
• |
premarket clearance, approval and conformity assessment procedures; |
|
• |
premarket clearance, approval and conformity assessment procedures for modifications introduced in marketed products; |
32
|
• |
reporting of adverse events or incidents and implementation of corrective actions, including product recalls; |
|
• |
pricing and reimbursement; |
|
• |
interactions with healthcare professionals; |
|
• |
advertising and promotion; and |
|
• |
product sales and distribution. |
Clinical testing can be costly and take many years, and the outcome is uncertain and susceptible to varying interpretations. Moreover, success in preclinical and early clinical trials does not ensure that large-scale trials will be successful or predict final results. Acceptable results in early trials may not be replicable in later trials. A number of companies in the oncology industry have suffered significant setbacks in advanced clinical trials, even after promising results in earlier trials. Negative or inconclusive results or adverse medical events during a clinical trial could cause it to be redone or terminated. In addition, failure to construct appropriate clinical trial protocols could result in the test or control group experiencing a disproportionate number of adverse events and could cause a clinical trial to be suspended, redone or terminated. We cannot be certain if or when the FDA, a regulatory agency in another jurisdiction or our notified body (a private organization designated in an EU member state to conduct conformity assessment procedures under the Medical Devices Directive) might request additional studies, under what conditions such studies might be requested, or what the size or length of any such studies might be. The clinical trials of our delivery system candidates may not be completed on schedule, the FDA, regulatory agencies in other jurisdictions or our notified body may order us to stop or modify our research, or these agencies or our notified body may not ultimately approve or issue a CE Certificate of Conformity for any of our delivery system candidates for commercial sale. While we have received regulatory approval for Optune for treatment of adult patients with recurrent GBM in the United States, the FDA required us to initiate a post-approval study and we have met this requirement. The data collected from our clinical trials may not be sufficient to support regulatory approval in the United States, Japan and other countries or to obtain CE Certificate of Conformity in the European Union for our various future delivery system candidates. Even if we believe the data collected from our clinical trials are sufficient, the FDA, equivalent regulatory bodies in other jurisdictions and notified bodies have substantial discretion in the assessment and approval or conformity assessment processes and may disagree with our interpretation of the data. Our failure to adequately demonstrate the safety and efficacy of any of our delivery system candidates would delay or prevent regulatory approval in the United States, Japan and other countries or the CE marking in the European Union of our delivery system candidates, which could prevent us from achieving profitability.
We currently market Optune in the United States, as well as the EU, Switzerland, Israel and Japan. We intend to market our Tumor Treating Fields delivery systems in a number of additional international markets. Although certain of our delivery systems have been approved for commercialization in Australia, Switzerland and Israel and are CE marked in the EU, in order to market our delivery systems in other jurisdictions and for other indications, we must obtain separate regulatory approvals and CE Certificates of Conformity, as applicable. The requirements governing the conduct of clinical trials and manufacturing and marketing of our delivery system candidates outside the United States vary widely from country to country. CE Certificates of Conformity and regulatory approvals in other jurisdictions may take longer to obtain than FDA approvals and can require, among other things, additional testing and different clinical trial designs. CE marking processes and regulatory approvals in other jurisdictions include essentially all of the risks associated with the FDA approval processes. Some regulatory agencies in other jurisdictions must also approve prices of the delivery systems. Approval of a product by the FDA does not ensure approval of the same product by the health authorities of other countries or CE marking of Optune in the EU and vice versa. In addition, changes in regulatory policy in the United States or in other countries for the approval or CE marking of a medical device during the period of product development and regulatory agency review or notified body review of each submitted new application may cause delays or rejections.
Upcoming changes in the EU rules governing the placing on the market of medical devices will have impact on the CE marking of Optune and our delivery system candidates in the EU. In April 2017, the EU adopted the new Medical Devices Regulation, replacing the two existing directives, the Medical Devices Directive and the Active Implantable Medical Devices Directive. The new regulation will enter into force after a three-year transition period ending in spring 2020. When applicable, the regulations will change the regulatory system for medical devices in the EU, which may prevent or delay the CE marking of our delivery system candidates or impact our ability to modify Optune for CE marking purposes on a timely basis.
We may choose to, or may be required to, suspend, repeat or terminate our clinical trials if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive or the trials are not well designed.
Clinical trials must be conducted in accordance with the FDA’s cGCPs and the equivalent laws and regulations applicable in other jurisdictions in which the clinical trials are conducted. The clinical trials are subject to oversight by the FDA, regulatory agencies in other jurisdictions, ethics committees and institutional review boards at the medical institutions where the clinical trials are conducted.
33
In addition, clinical trials must be conducted with delivery system candidates produced under the FDA’s Good Manufacturing Practices (“GMP”) and in accordance with the applicable regulatory requirements in the other jurisdictions in which the clinical trials are conducted. The conduct of clinical trials may require large numbers of test patients. Patient enrollment is a function of many factors, including the size of the patient population for the target indication, the proximity of patients to clinical sites, the eligibility criteria for the trial, the existence of competing clinical trials and the availability of alternative or new treatments. Clinical trials may be suspended by the FDA or by a regulatory agency in another jurisdiction at any time if the FDA or the regulatory agency finds deficiencies in the conduct of these trials or it is believed that these trials expose patients to unacceptable health risks.
We, the FDA or regulatory agencies in other jurisdictions might delay or terminate our clinical trials of a delivery system candidate for various reasons, including:
|
• |
the delivery system candidate may have unforeseen adverse side effects; |
|
• |
the time required to determine whether the delivery system candidate is effective may be longer than expected; |
|
• |
we may not agree with the FDA, a regulatory authority in another jurisdiction or an ethics committee regarding the protocol for the conduct of a clinical trial; |
|
• |
new therapies may become the standard of care while we are conducting our clinical trials, which may require us to revise or amend our clinical trial protocols or terminate a clinical trial; |
|
• |
fatalities may occur during a clinical trial due to medical problems that may or may not be related to clinical trial treatments; |
|
• |
the delivery system candidate may not appear to be more effective than current therapies; |
|
• |
there may be insufficient patient enrollment in the clinical trials; or |
|
• |
we may not be able to produce sufficient quantities of the delivery system candidate to complete the trials. |
Furthermore, the process of obtaining and maintaining regulatory approvals in the United States and other jurisdictions and CE Certificates of Conformity in the EU for new therapeutic products is lengthy, expensive and uncertain. It can vary substantially, based on the type, complexity and novelty of the product involved. Accordingly, any of our delivery system candidates could take a significantly longer time than we expect to, or may never, gain regulatory approval or obtain CE Certificates of Conformity in the EU, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Healthcare reform and other legislative and regulatory changes in the United States and in other countries may adversely affect our business and financial results.
In response to perceived increases in healthcare costs in recent years, there have been and continue to be proposals by the U.S. federal government, state governments, regulators and third-party payers to control these costs and, more generally, to reform the United States healthcare system. In the United States, the Patient Protection and Affordable Care Act (the “PPACA”), was enacted in 2010 with a goal of reducing the cost of healthcare and substantially changing the way healthcare is financed by both government and private insurers.
In addition, other legislative changes have been proposed and adopted in the United States since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011 created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This included aggregate reductions of Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013. On January 2, 2013, the American Taxpayer Relief Act (the “ATRA”), was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals.
More recently, the new United States Administration and members of the U.S. Congress have stated that they will seek to modify, repeal, or otherwise invalidate all, or certain provisions of, the PPACA. On January 20, 2017, President Donald Trump signed an executive order, which stated that it is the policy of his Administration to seek the prompt repeal of the PPACA and directed executive departments and federal agencies to waive, defer, grant exemptions from, or delay the implementation of the provisions of the PPACA to the maximum extent permitted by law. Additionally, the House and Senate attempted, but failed, to pass legislation to repeal all or portions of the PPACA and these efforts may be resumed.
34
There is uncertainty with respect to the impact the United States Administration, the executive order and the attempted legislation may have, if any, and any changes will likely take time to unfold and could have an impact on coverage and reimbursement for healthcare items and services, including Optune and our delivery system candidates. For example, PPACA requires that health insurance plans sold to individuals and small businesses provide coverage for “essential health benefits” (EHBs), which are defined according to state-specific benchmark plans. The department of Health and Human Services has proposed a rule that has the potential to modify the implementation of the PPACA by changing the definitions of EHBs as defined in the PPACA. Changes to the definitions of the EHB, or providing states with increased flexibility to define EHBs, may have the effect of decreasing coverage for anti-cancer devices such as Optune.
In the future, the U.S. Congress could also pass additional healthcare laws and CMS could implement regulatory changes. Further, various healthcare reform proposals have emerged at the state level. These laws and regulations could potentially affect coverage and reimbursement for Optune and our delivery system candidates. However, we cannot predict the ultimate content, timing or effect of any future federal or state healthcare initiatives or the impact any future legislation or regulation will have on us.
In 2017 CMS published the Medicare Program: Changes to the Medicare Claims and Entitlement, Medicare Advantage Organization Determination, and Medicare Prescription Drug Coverage Determination Appeals Procedures final rule. This final rule aims to streamline the Medicare appeals process and includes changes such as permitting the designation of Medicare Appeals Council decisions as precedential, expanding the Office of Medicare Hearings and Appeals’ available adjudicator pool, and simplifying proceedings when CMS or CMS contractors are involved, among others. The final rule became effective on March 20, 2017. We are monitoring the implementation of this final rule and cannot predict to what extent CMS may or may not use this final rule in denying coverage for Optune.
Additionally, the process governing Medicare appeals and the significant backlog of appeals at the Administrative Law Judge, or ALJ level is the subject of active litigation in the D.C. federal courts. We are monitoring the impact of this litigation on our ability to secure payment from the Medicare program and can provide no assurance that this case will result in payments for amounts that we have and will bill to Medicare.
We believe that substantial uncertainty remains regarding the net effect of the PPACA, or its repeal and potential replacement, on our business, including uncertainty over how benefit plans purchased on exchanges will cover our products, how the expansion or contraction of the Medicaid program will affect access to our products, the effect of risk-sharing payment models such as Accountable Care Organizations and other value-based purchasing programs on coverage for our product, and the effect of the general increase or decrease in Federal oversight of healthcare payers. The taxes imposed and the expansion in government’s role in the U.S. healthcare industry under the PPACA, if unchanged, may result in decreased revenues, lower reimbursements by payers for our delivery systems and reduced medical procedure volumes, all of which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Specifically, beginning in January 2013, PPACA imposed a 2.3% excise tax on the constructive sale price in the United States of certain medical devices by a manufacturer, producer or importer of such devices. This tax was initially suspended for two years beginning January 1, 2016 and ending December 31, 2017. New legislation was passed such that this tax will continue to be suspended until January 1, 2020.
The competent authorities in the EU member states, Switzerland, Israel, Japan, and other jurisdictions are increasingly active in their goal of reducing public spending on healthcare. We cannot, therefore, guarantee that the treatment of patients with Optune would be reimbursed in any particular country or, if successfully included on reimbursement lists will remain thereon. In May 2017, new rules governing medical devices in the EU were adopted which will take effect in May 2020 and will impose additional requirements on manufacturers of medical devices placed on the market in the EU. Failure to comply with these new requirements may affect our ability to market our delivery systems in the EU.
We are subject to extensive regulation by the FDA and equivalent authorities in other jurisdictions, which could restrict the sales and marketing of Optune and could cause us to incur significant costs. In addition, we may become subject to additional regulation in other jurisdictions as we increase our efforts to market and sell Optune outside of the United States.
We market and sell Optune, and expect to market and sell our delivery system candidates, subject to extensive regulation by the FDA and numerous other federal, state and governmental authorities in other jurisdictions. These regulations are broad and relate to, among other things, the conduct of preclinical and clinical studies, product design, development, manufacturing, labeling, testing, product storage and shipping, premarket clearance and approval, conformity assessment procedures, premarket clearance and approval of modifications introduced in marketed products, post-market surveillance and monitoring, reporting of adverse events and incidents, pricing and reimbursement, interactions with healthcare professionals, advertising and promotion and product sales and distribution. Although we have received FDA approval to market Optune in the United States for the treatment of adult patients with newly diagnosed GBM (in combination with temozolomide) and recurrent GBM, we will require additional FDA approval to market Optune for other indications. We may be required to obtain approval of a new PMA or PMA supplement application for modifications made
35
to Optune. This approval process is costly and uncertain, and it could take one to three years, or longer, from the time the application is filed with the FDA. We may make modifications in the future that we believe do not or will not require additional approvals. If the FDA disagrees, and requires new PMAs or PMA supplements for the modifications, we may be required to recall and to stop marketing the modified versions of Optune.
In addition, before our delivery systems can be marketed in the EU, they must obtain a CE Certificate of Conformity from a notified body. New therapeutic uses of CE marked medical devices falling outside the scope of the current CE Certificate of Conformity require a completely new conformity assessment before the device can be CE marked and marketed in the EU for the new intended purpose.
These processes can be expensive and lengthy and entail significant fees. The process preceding CE marking of a medical device in the EU or approval in Japan could also be expensive and lengthy and its outcome would be uncertain. We may make modifications in the future that we believe do not or will not require additional approvals or the notification of our notified body and potentially additional conformity assessment to permit the maintenance of our current CE Certificate of Conformity. If the competent authorities of the EU member states or our notified body disagree and require the conduct of a new conformity assessment procedure and the modification of the existing CE Certificate of Conformity or the issuance of a new certificate, we may be required to recall or suspend the marketing of the modified versions of Optune.
In Japan, new medical devices or new therapeutic uses of medical devices falling outside the scope of the existing approval by the MHLW require a new assessment and approval for each such new device or use. Accordingly, we may be required to obtain a new approval from MHLW before we launch of a modified version of Optune or the use of Tumor Treating Fields for additional indications. Approval time frames from the MHLW vary from simple notifications to review periods of one or more years, depending on the complexity and risk level of the device. In addition, importation into Japan of medical devices is subject to “Quality Management System (QMS) Ordinance,” which includes the equivalent of “Good Import Practices” regulations in the United States. As with any highly regulated market, significant changes in the regulatory environment could adversely affect our ability to commercialize Optune and our other delivery systems in Japan.
In the United States and other jurisdictions, we also are subject to numerous post-marketing regulatory requirements, which include quality system regulations related to the manufacturing of our delivery systems, labeling regulations and medical device reporting regulations, which require us to report to the FDA or other regulatory authorities in other jurisdictions and notified bodies if our delivery systems cause or contribute to a death or serious injury, or malfunction in a way that would likely cause or contribute to a death or serious injury. In addition, these regulatory requirements may change in the future in a way that adversely affects us. If we fail to comply with present or future regulatory requirements that are applicable to us, we may be subject to enforcement action by the FDA or other regulatory authorities in other jurisdictions and notified bodies, which may include any of the following sanctions:
|
• |
untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
|
• |
unanticipated expenditures to address or defend such actions; |
|
• |
patient notification, or orders for repair, replacement or refunds; |
|
• |
voluntary or mandatory recall or seizure of our current or future delivery systems; |
|
• |
administrative detention by the FDA or other regulatory authority in another jurisdiction of medical devices believed to be adulterated or misbranded; |
|
• |
operating restrictions, suspension or shutdown of production; |
|
• |
refusal or delay of our requests for PMA or analogous approval for new intended uses or modifications to Optune; |
|
• |
refusal or delay of our requests for PMA or analogous approval of new delivery systems; |
|
• |
refusal or delay in obtaining CE Certificates of Conformity for new intended uses or modifications to Optune; |
|
• |
suspension, variation or withdrawal of the CE Certificates of Conformity granted by our notified body in the EU member states; |
|
• |
operating restrictions; |
|
• |
suspension or withdrawal of PMA or analogous approvals that have already been granted; |
|
• |
refusal to grant export approval for Optune or any delivery system candidates; or |
|
• |
criminal prosecution. |
36
The occurrence of any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Modifications to Optune or any of our delivery system candidates approved in the future may require approvals of new PMA or PMA supplement applications, modified or new CE Certificates of Conformity and analogous regulatory approvals in other jurisdictions or even require us to cease promoting or to recall the modified versions of Optune until such clearances, approvals or modified or new CE Certificates of Conformity are obtained, and the FDA, regulatory authorities in other jurisdictions or our notified body may not agree with our conclusions regarding whether new approvals are required.
Any modification to a device approved through the PMA pathway that impacts the safety or effectiveness of the device requires submission to the FDA and FDA approval of a PMA supplement application or even a new PMA application, as the case may be. The FDA requires a company to make the determination as to whether a new PMA or PMA supplement application is to be filed, but the FDA may review the company’s decision. For example, in the past, we have made initial determinations that certain modifications did not require the filing of a new PMA or PMA supplement application and have notified the FDA of these changes in our PMA Annual Report, but after its review of our PMA Annual Report, the FDA requested that we submit these modifications to the FDA as a PMA supplement application. From time to time, we may make other changes to the delivery systems, software, packaging, manufacturing facilities and manufacturing processes and may submit additional PMA supplement applications for these changes. FDA may conduct a facility inspection as part of its review and approval process. In addition, it is possible that the FDA will require a human factors (user interface) study. It is also possible that the FDA may require additional clinical data. We can provide no assurance that we will receive FDA approval for these changes on a timely basis, or at all. We also may make additional changes in the future that we may determine do not require the filing of a new PMA or PMA supplement application. The FDA may not agree with our decisions regarding whether the filing of new PMAs or PMA supplement applications are required.
In addition, any substantial change introduced to a medical device CE marked in the European Union or to the quality system review by our notified body requires a new conformity assessment of the device and can lead to changes to the CE Certificates of Conformity or the preparation of a new CE Certificate of Conformity. Substantial changes include, among others, the introduction of a new intended purpose of the device, a change in its design or a change in the company’s suppliers. Responsibility for determination that a modification constitutes a substantial change lies with the manufacturer of the medical device. We must inform the notified body that conducted the conformity assessment of the delivery systems we market or sell in the European Union of any planned substantial changes to our quality system or changes to our devices which could affect compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the devices’ intended purpose. The notified body will then assess the changes and verify whether they affect the delivery system’s conformity with the Essential Requirements laid down in Annex I to the Medical Devices Directive or the conditions for the use of the device. If the assessment is favorable, the notified body will issue a new CE Certificate of Conformity or an addendum to the existing CE Certificate of Conformity attesting compliance with the Essential Requirements laid down in Annex I to the Medical Devices Directive. There is a risk that the competent authorities of the EU member states or our notified body may disagree with our assessment of the changes introduced to our delivery systems. The competent authorities of the EU member states or our notified body also may come to a different conclusion than the FDA on any given product modification.
If the FDA disagrees with us and requires us to submit a new PMA or PMA supplement application for then-existing modifications and/or the competent authorities of the EU member states or our notified body disagree with our assessment of the change introduced in a product, we may be required to cease promoting or to recall the modified product until we obtain approval and/or until a new conformity assessment has been conducted in relation to the product, as applicable. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our delivery systems could be subject to recall if the FDA, analogous regulatory authorities in other jurisdictions, or the competent authorities of the EU member states or our notified body determine, for any reason, that our delivery systems are not safe or effective or that appropriate regulatory submissions were not made. Any recall or requirement that we seek additional approvals or clearances could result in significant delays, fines, increased costs associated with modification of a product, loss of revenues and potential operating restrictions imposed by the FDA, analogous foreign regulatory authorities, or the competent authorities of the EU member states or our notified body. Delays in receipt or failure to receive approvals, the loss of previously received approvals, the failure to conduct appropriate conformity assessments prior to CE marking of our delivery systems, or the failure to comply with any other existing or future regulatory requirements, could reduce our sales, profitability and future growth prospects.
37
We will spend considerable time and money complying with federal, state and regulations in other jurisdictions in addition to FDA regulations, and, if we are unable to fully comply with such regulations, we could face substantial penalties.
We are subject to extensive regulation by the U.S. federal government and the states and other countries in which we conduct our business. U.S. federal government healthcare laws apply when we submit a claim on behalf of a U.S. federal healthcare program beneficiary, or when a customer submits a claim for an item or service that is reimbursed under a U.S. federal government-funded healthcare program, such as Medicare or Medicaid. The laws that affect our ability to operate our business in addition to the Federal Food, Drug, and Cosmetic Act and FDA regulations include, but are not limited to, the following:
|
• |
the federal anti-kickback statute, which prohibits offering or providing remuneration of any kind for the purpose of inducing or rewarding referrals for items or services reimbursable by a federal healthcare program; |
|
• |
the U.S. federal False Claims Act (the “False Claims Act”) which prohibits submitting false claims or causing the submission of false claims to the federal government; |
|
• |
Medicare laws and regulations that prescribe requirements for coverage and payment, including the conditions of participation for DME suppliers, and laws prohibiting false claims for reimbursement under Medicare and Medicaid; |
|
• |
healthcare fraud statutes that prohibit false statements and improper claims to any third-party payer; |
|
• |
the federal physician self-referral prohibition, commonly known as the Stark law, which prohibits physicians from referring Medicare patients to an entity for the provision of certain designated health services (including DME) if the physician (or a member of the physician’s immediate family) has an impermissible financial relationship with that entity; |
|
• |
similar state anti-kickback, false claims, insurance fraud and self-referral laws, which may not be limited to government-reimbursed items, as well as state laws that require us to maintain permits or licenses to distribute durable medical equipment; |
|
• |
federal and state accreditation and licensing requirements applicable to DME providers and equivalent requirements in other jurisdictions; |
|
• |
the U.S. Foreign Corrupt Practices Act, which can be used to prosecute companies in the United States for arrangements with physicians or other parties outside the United States if the physician or party is a government official of another country and the arrangement violates the law of that country; |
|
• |
the Federal Trade Commission Act, the Lanham Act and similar federal and state laws regulating advertising and consumer protection; |
|
• |
the Physician Payments Sunshine Act (the “Sunshine Act”) and similar state and foreign laws, which require reporting of payments and other transfers of value to health care practitioners periodically; and |
|
• |
the laws and codes of practices applicable in the EU member states, Switzerland, Japan and other jurisdictions concerning the marketing and promotion of medical devices, interactions with healthcare professionals, consumer protection, comparative advertising and unfair commercial practices, data protection, anti-corruption, bribery and reimbursement of medical devices. |
The laws and codes of practices applicable to us are subject to evolving interpretations. Moreover, certain federal and state laws regarding healthcare fraud and abuse and certain laws in other jurisdictions regarding interactions with healthcare professionals are broad and we may be required to restrict certain of our practices to be in compliance with these laws. Similar law exists in the EU, individual EU member states and other countries. These laws are complemented by EU or national profession codes of practices. Healthcare fraud and abuse laws also are complex and even minor, inadvertent irregularities can potentially give rise to claims that a statute has been violated. If a governmental authority were to conclude that we are not in compliance with applicable laws and regulations, we and our officers and employees could be subject to severe criminal and civil penalties, including, for example, exclusion from participation as a supplier of delivery systems to beneficiaries covered by federal healthcare programs. For example, most states require us to maintain a license as a DME provider. The Medicare program requires that we maintain accreditation with an independent quality body. Loss of this accreditation would result in loss of our billing privileges to Medicare.
Any violation of these laws or equivalent laws and codes of practices in other jurisdictions regarding interactions with healthcare professionals and bribery could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Similarly, if there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which likewise could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. In addition, although we believe that we have the required licenses, permits and accreditation to dispense Optune and to dispense our delivery system candidates in the future, a regulator could find that we need to obtain additional licenses or permits. We also may be subject to audits, mandatory reaccreditation and other requirements in order to maintain our billing privileges. Failure to satisfy those requirements or successfully address any issues identified in an audit could cause us to lose our privileges to bill public
38
and private payers. If we are required to obtain permits or licenses that we do not already possess, we also may become subject to substantial additional regulation or incur significant expense. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business, our prospects and our financial results. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
If we, our contract manufacturers or our component suppliers fail to comply with the FDA’s quality system regulations or equivalent regulations established in other countries, the manufacturing and distribution of our delivery systems could be interrupted, and our delivery system sales and results of operations could suffer.
We, our contract manufacturers and our component suppliers are required to comply with the FDA’s quality system regulations and the equivalent quality system requirements imposed by the laws and regulations in other jurisdictions, which are a complex regulatory framework that covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our delivery systems. All aspects of our supply chain are subject to periodic inspections and audits by the FDA, notified bodies and other regulatory authorities to ensure continuing compliance. We cannot assure you that our facilities or our contract manufacturers’ or component suppliers’ facilities would pass any future quality system inspection. If our or any of our contract manufacturers’ or component suppliers’ facilities fails a quality system inspection, the manufacturing or distribution of our delivery systems could be interrupted and our operations disrupted. Failure to take adequate and timely corrective action in response to an adverse quality system inspection could force a suspension or shutdown of our packaging and labeling operations or the manufacturing operations of our contract manufacturers, and lead to suspension, variation or withdrawal of our regulatory approvals or a recall of our delivery systems. If any of these events occurs, we may not be able to provide our customers with Tumor Treating Fields delivery systems that they require on a timely basis, our reputation could be harmed and we could lose customers, any or all of which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Our delivery systems may in the future be subject to recalls that could harm our reputation, business and financial results.
The FDA and similar governmental authorities in other jurisdictions have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. In addition, governmental bodies in other jurisdictions have the authority to require the recall of our delivery systems in the event of material deficiencies or defects in design or manufacture. Distributors and manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our manufacturers could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated. Requirements for the reporting of product recalls to the competent authorities are imposed in other jurisdictions in which our delivery systems are or would be marketed in the future. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or to the competent authorities of other countries. In the future, we may initiate voluntary recalls involving our delivery systems that we determine do not require notification of the FDA or to other equivalent non-U.S. authorities. If the FDA or the equivalent non-U.S. authorities disagree with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA and the equivalent non-U.S. authorities could take enforcement action if we fail to report the recalls when they were conducted. Recalls of any of our delivery systems would divert managerial and financial resources and could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
If our delivery systems cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations and the equivalent regulations applicable in other jurisdictions in which our delivery systems are or may be marketed in the future, medical device manufacturers are required to report to the FDA and to the equivalent non-U.S. authorities information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA or to the equivalent authorities in other jurisdictions within the required timeframes, or at all, the FDA or the equivalent authorities in other jurisdictions could take enforcement action against us. Any such adverse event involving our delivery systems also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
39
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our delivery systems for unapproved or off-label uses.
Medical devices may be marketed only for the indications for which they are approved. Our promotional materials and training materials must comply with FDA regulations and other applicable laws and regulations governing the promotion of our delivery systems in the United States and other jurisdictions. Currently, Optune is approved for treatment of adult patients with newly diagnosed GBM (in combination with temozolomide) and recurrent GBM in the United States and Japan. In the European Union and Switzerland, we have CE marked the Optune delivery system for the treatment of newly diagnosed GBM (in combination with temozolomide), recurrent GBM, and advanced NSCLC (in combination with standard-of-care chemotherapy). Optune is also approved in Israel and in Australia for the treatment of recurrent GBM and newly diagnosed GBM (in combination with temozolomide).
If the FDA or the competent authorities in other jurisdictions, including the European Union member states, determine that our promotional materials or training constitutes promotion of an unapproved use, they could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled letter, a warning letter, an injunction, seizure, civil fines and criminal penalties. It is also possible that other federal, state or national enforcement in other jurisdictions authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and commercialization of Optune and future delivery systems would be impaired.
U.S. legislative or FDA regulatory reforms may make it more difficult and costly for us to obtain regulatory approval of our delivery system candidates and to manufacture, market and distribute our delivery systems after approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our delivery systems. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of future delivery system candidates. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our delivery systems. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be. Any change in the laws or regulations that govern the clearance and approval processes relating to our current and future delivery systems could make it more difficult and costly to obtain clearance or approval for new delivery systems, or to produce, market and distribute Optune. Significant delays in receiving clearance or approval, or the failure to receive clearance or approval for our new delivery systems would have an adverse effect on our ability to expand our business in the United States.
As a DME supplier, if we are found to have violated laws protecting the confidentiality of patient health information, we could be subject to civil or criminal penalties, which could increase our liabilities and harm our reputation or our business.
There are a number of federal and state laws protecting the confidentiality of certain patient health information, including patient records, and restricting the use and disclosure of that protected information, as well as data protection laws applicable in other jurisdictions, such as the European Union member states. In particular, the U.S. Department of Health and Human Services promulgated patient privacy rules under HIPAA. These privacy rules protect medical records and other personal health information by limiting their use and disclosure, giving individuals the right to access, amend and seek accounting of their own health information and limiting most use and disclosures of health information to the minimum amount reasonably necessary to accomplish the intended purpose. If we are found to be in violation of the privacy rules under HIPAA, we could be subject to civil or criminal penalties, which could increase our liabilities, harm our reputation and have a material adverse effect on our business, financial condition and results of operations.
The collection and use of personal health data in the European Union is currently governed by the provisions of Directive 95/46/EC of the European Parliament and of the Council of October 24, 1995, on the protection of individuals with regard to the processing of personal data and on the free movement of such data, commonly known as the Data Protection Directive. The European Union has adopted national laws and regulations transposing the EU Data Protection Directive into their national laws. The Data Protection Directive will be replaced in May 2018 by the GDPR which is designed to harmonize data privacy laws for both patient and employee data across Europe. The aim of the GDPR is to protect all European Union citizens from privacy and data breaches in an increasingly data-driven world that is vastly different from the time in which the 1995 directive was established. Although the key principles of data privacy still hold true to the previous directive, the new regulation will require us to make operational changes relating to our receipt and processing of personal data of European Union citizens. Important components of the new regulation include, but are not limited to, increased territorial scope; increased penalties up to 4% of annual gross revenue; stricter requirements for written privacy consent and notification language and specific and documented processes related to breach notification; right to be forgotten and right to access data; assignment of a Data Protection Officer.
40
We are affected by and subject to environmental laws and regulations that could be costly to comply with or that may result in costly liabilities.
We are subject to environmental laws and regulations, including those that impose various environmental controls on the manufacturing, transportation, storage, use and disposal of batteries and hazardous chemicals and other materials used in, and hazardous waste produced by, the manufacturing of our delivery systems. We incur and expect to continue to incur costs to comply with these environmental laws and regulations. Additional or modified environmental laws and regulations, including those relating to the manufacture, transportation, storage, use and disposal of materials used to manufacture our delivery systems or restricting disposal or transportation of batteries, may be imposed that may result in higher costs.
In addition, we cannot predict the effect that additional or modified environmental laws and regulations may have on us, our third-party suppliers of equipment, batteries and our delivery systems or our customers. For example, we and our suppliers rely on an exemption from the European Directive 2011/65/EU relating to the restriction of the use of certain hazardous substances in electrical and electronic equipment relating to lead content in our transducer arrays. To the extent this exemption is revoked, it may have a material impact on our business and results of operations.
Regulations on the transportation of lithium ion batteries may affect our business.
Our delivery systems use lithium ion batteries. The transportation of lithium and lithium-ion batteries is regulated worldwide. Laws regulating the transportation of batteries have been and may be enacted which could impose additional costs that could harm our ability to be profitable.
Under recommendations adopted by the International Air Transport Association ("IATA"), our batteries currently require a Class 9 designation for transportation. Our larger first generation delivery system batteries must be properly packaged and labeled in order to be shipped by air transport as cargo. Our smaller second generation delivery system batteries can be shipped without the class 9 sticker if shipped with the device but require the class 9 sticker if shipped by air separately. The larger batteries are not allowed on passenger aircraft according to the IATA standards. The smaller batteries are allowed as carry on only and cannot be checked as luggage. Consequently, we offer to ship batteries for patients who are traveling by air.
If additional restrictions are put in place that limit our ability to ship our delivery systems by air freight or on water borne cargo, it could have an adverse effect on our supply chain, our inventory management procedures and processes and our ability to fill prescriptions and service patients in a timely manner, which could have a material adverse effect on our business, prospects, financial condition and results of operations. In addition, compliance with future worldwide or IATA approval process and regulations could require significant time and resources from our technical staff and, if redesign were necessary, could delay the introduction of new products.
Risks relating to intellectual property
If we are unable to protect our proprietary technology, trade secrets or know-how, we may not be able to operate our business profitably. Similarly, if we fail to sustain, further build and enforce our intellectual property rights, competitors may be able to develop competing therapies.
Our success depends, in part, on our ability to obtain and maintain protection for our delivery systems and technologies under the patent laws or other intellectual property laws of the United States and other countries. The standards that the U.S. Patent and Trademark Office (“USPTO“) and its counterparts in other jurisdictions use to grant patents are not always applied predictably or uniformly and can change. Consequently, we cannot be certain as to whether pending patent applications will result in issued patents, and we cannot be certain as to the type and extent of patent claims that may be issued to us in the future. Any issued patents may not contain claims that will permit us to stop competitors from using similar technology.
Our current intellectual property portfolio consists of over 140 issued patents. The patents have expected expiration dates between 2021 and 2035. As such patents expire we will be subject to additional risks. Patent expiration could adversely affect our ability to protect future product development and our competitors may develop and market competing products. We have also filed additional patent applications worldwide that may never be issued. Consequently, our operating results and financial position could be materially adversely affected. In addition, due to the extensive time needed to develop, test and obtain regulatory approval for our treatment therapies, any patents that protect our product candidates may expire early during commercialization. This may reduce or eliminate any market advantages that such patents may give us and harm our financial position. If we fail to develop and successfully launch new products prior to the expiration of patents for our existing products, our sales and profits with respect to those products could decline significantly. We may not be able to develop and successfully launch more advanced replacement products before these and other patents expire.
41
Our existing and future patent portfolio also may be vulnerable to legal challenges. The standards that courts use to interpret patents are not always applied predictably or uniformly and can change, particularly as new technologies develop. On September 16, 2011, President Obama signed into law the Leahy-Smith America Invents Act (“AIA”) a significant patent law reform. The AIA implements a first-inventor-to-file standard for patent approval, changes the legal standards for patentability and creates a post-grant review system. As a result of the uncertainties of patent law in general, and surrounding the interpretation of the AIA in particular, we cannot predict with certainty how much protection, if any, will be given to our patents if we attempt to enforce them and they are challenged in court. Any attempt to enforce our intellectual property rights may also be time-consuming and costly, may divert the attention of management from our business, may ultimately be unsuccessful or may result in a remedy that is not commercially valuable. Such attempts may also provoke third parties to assert claims against us or result in our intellectual property being narrowed in scope or declared to be invalid or unenforceable.
In addition, we rely on certain proprietary trade secrets, know-how and other confidential information. We have taken measures to protect our unpatented trade secrets, know-how and other confidential information, including the use of confidentiality and assignment of inventions agreements with our employees, consultants and certain contractors. It is possible, however, that these persons may breach or challenge the agreements, that our trade secrets may otherwise be misappropriated or that competitors may independently develop or otherwise discover our trade secrets. There is therefore no guarantee that we will be able to obtain, maintain and enforce the intellectual property rights that may be necessary to protect and grow our business and to provide us with a meaningful competitive advantage, and our failure to do so could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
The oncology industry is characterized by patent and other intellectual property litigation and disputes, and any litigation, dispute or claim against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our business, harm our reputation and require us to remove certain delivery systems from the market.
Whether a product infringes a patent or violates other intellectual property rights involves complex legal and factual issues, the determination of which is often uncertain. Any intellectual property dispute, even a meritless or unsuccessful one, would be time consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, the disruption of research and development and marketing efforts, injury to our reputation and loss of revenues. Any of these events could negatively affect our business, prospects, financial condition and results of operations.
Third parties may assert that Tumor Treating Fields, Optune, our other delivery system candidates, the methods employed in the use of our delivery systems or other activities infringe on their patents. Such claims may be made by competitors seeking to obtain a competitive advantage or by other parties, many of whom have significantly larger intellectual property portfolios than we have. Additionally, in recent years, individuals and groups have begun purchasing intellectual property assets for the purpose of making claims of infringement and attempting to extract settlements from companies like ours. The risk of infringement claims is exacerbated by the fact that there are numerous issued and pending patents relating to the treatment of cancer. Because patent applications can take many years to issue, and in many cases remain unpublished for many months after filing, there may be applications now pending of which we are unaware that may later result in issued patents that our delivery systems may infringe. There could also be existing patents that one or more components of our delivery systems may inadvertently infringe. As the number of competitors in the market for the treatment of cancer grows, the possibility of inadvertent patent infringement by us or a patent infringement claim against us increases. To the extent we gain greater market visibility, our risk of being subject to such claims is also likely to increase.
If a third party’s patent was upheld as valid and enforceable and we were found to be infringing, we could be prevented from making, using, selling, offering to sell or importing Optune or other delivery system candidates, unless we were able to obtain a license under that patent or to redesign our systems to avoid infringement. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our delivery systems to avoid any infringement. Modification of our delivery systems or development of delivery system candidates to avoid infringement could require us to conduct additional clinical trials and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. If we are not successful in obtaining a license or redesigning our delivery systems, we may be unable to make, use, sell, offer to sell or import our delivery systems and our business could suffer. We may also be required to pay substantial damages and undertake remedial activities, which could cause our business to suffer.
We may also be subject to claims alleging that we infringe or violate other intellectual property rights, such as copyrights or trademarks, may have to defend against allegations that we misappropriated trade secrets, and may face claims based on competing claims of ownership of our intellectual property. The confidentiality and assignment of inventions agreements that our employees, consultants and other third parties sign may not in all cases be enforceable or sufficient to protect our intellectual property rights. In addition, we may face claims from third parties based on competing claims to ownership of our intellectual property.
42
We also employ individuals who were previously employed at other medical device companies, and as such we may be subject to claims that such employees have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of their former employers. Any such litigation, dispute or claim could be costly to defend and could subject us to substantial damages, injunctions or other remedies, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
The patent rights on which we rely to protect the intellectual property underlying Tumor Treating Fields delivery systems may not be adequate, which could enable third parties to use our technology or market competing products, which would harm our continued ability to compete in the market.
Our success will depend in part on our continued ability to develop or acquire commercially valuable patent rights and to protect these rights adequately. The scope of some of our patents is limited to certain ranges. For example, some of our patents protect low-intensity (1-3 V/cm), intermediate frequency (100-300 kHz) alternating electric fields, but do not cover intensities and frequencies for electric fields that are outside of these ranges. While intensities and frequencies of electric fields outside of these ranges have not yet proven to be effective treatment modalities, that may not be the case in the future. Our patent position is generally uncertain and involves complex legal and factual questions. The risks and uncertainties that we face with respect to our patents and other related rights include the following:
|
• |
the pending patent applications we have filed may not result in issued patents or may take longer than we expect to result in issued patents; |
|
• |
the pending patent applications and patents we own may be subject to interference proceedings or similar disputes over the priority of the inventions claimed; |
|
• |
the claims of any patents that are issued may not provide meaningful protection; |
|
• |
we may not be able to develop additional proprietary technologies that are patentable; |
|
• |
changes in patent laws or their interpretation in the United States and other countries (including the recently enacted AIA) could diminish the value of our patents, narrow the scope of our patent protection or adversely affect our ability to obtain new patents; |
|
• |
obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements; |
|
• |
other parties may challenge patents, patent claims or patent applications licensed or issued to us, and such patents, patent claims or patent applications may be narrowed or found to be invalid or unenforceable; and |
|
• |
other companies may design around or expand upon technologies we have patented or developed. |
We also may fail to apply for or be unable to obtain patent rights in some other countries. In addition, the legal systems of certain countries may not protect our rights to the same extent as the laws of the United States, which could affect our ability to enforce patent rights effectively in such other countries. For a variety of reasons, we may decide not to file for patent protection for certain of our intellectual property. Our patent rights underlying Tumor Treating Fields, Optune and our other delivery systems may not be adequate, and our competitors or customers may design around our proprietary technologies or independently develop similar or alternative technologies or products that are equal or superior to ours without infringing on any of our patent rights. In addition, the patents licensed or issued to us may not provide a competitive advantage, and may be insufficient to prevent others from commercializing products similar or identical to ours. The occurrence of any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
We have limited intellectual property rights in other jurisdictions outside of our key markets and may not be able to protect our intellectual property rights throughout the world.
We have limited intellectual property rights outside of our key markets. In some countries outside the United States, we do not have any intellectual property rights, and our intellectual property rights in other countries outside the United States have a different scope and strength compared to our intellectual property rights in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but where enforcement rights are not as strong as those in the United States. These products may compete with our delivery systems, and our patents or other intellectual property rights may not be effective or adequate to prevent such competition.
43
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our delivery systems.
As is the case with other medical device companies, our success is heavily dependent on our intellectual property rights, and particularly on our patent rights. Obtaining and enforcing patents in the medical device industry involves both technological and legal complexity, and is therefore costly, time consuming and inherently uncertain. In addition, the United States has recently enacted and is currently implementing wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could further negatively impact the value of our patents, narrow the scope of available patent protection or weaken the rights of patent owners.
Risks relating to our ordinary shares
The market price for our ordinary shares may be volatile, which could result in substantial losses to you.
The market price for our ordinary shares may be volatile and subject to wide fluctuations in response to factors such as publication of clinical studies relating to Optune, our other delivery system candidates or a competitor’s product, actual or anticipated fluctuations in our quarterly results of operations, changes in financial estimates by securities research analysts, negative publicity, studies or reports, changes in the economic performance or market valuations of other companies that operate in our industry, changes in the availability of third-party reimbursement in the United States or other countries, changes in governmental regulations or in the status of our regulatory approvals or applications, announcements by us or our competitors of material acquisitions, strategic partnerships, joint ventures or capital commitments, intellectual property litigation, release of lock-up or other transfer restrictions on our outstanding ordinary shares, and economic or political conditions in the United States, Israel or elsewhere. In addition, the performance, and fluctuation in market prices, of companies in other jurisdictions that have listed their securities in the United States may affect the volatility in the price of and trading volumes of our ordinary shares. Volatility in global capital markets, as was experienced during the global financial crisis beginning in 2008 and during the recent European sovereign debt crisis, as well as volatility resulting from the recent economic slowdown in Asia, could also have an adverse effect on the market price of our ordinary shares. Furthermore, the securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price and liquidity of our ordinary shares.
Our ordinary shares are issued under the laws of Jersey, which may not provide the level of legal certainty and transparency afforded by incorporation in a U.S. state.
We are incorporated under the laws of the Bailiwick of Jersey, Channel Islands. Jersey legislation regarding companies is largely based on English corporate law principles. However, there can be no assurance that Jersey law will not change in the future or that it will serve to protect investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of investors.
U.S. shareholders may not be able to enforce civil liabilities against us.
We are a Jersey entity with most of our assets located outside of the United States. Although we have appointed an agent for service of process in the United States for purposes of U.S. federal securities laws, a number of our directors and executive officers and a number of directors of each of our subsidiaries are not residents of the United States, and all or a substantial portion of the assets of such persons are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or to enforce against them judgments obtained in U.S. courts predicated upon the civil liability provisions of the federal securities laws of the United States.
We have been advised by our Jersey lawyers that the courts of Jersey would recognize any final and conclusive judgment under which a sum of money is payable (not being a sum payable in respect of taxes or other charges of a like nature or in respect of a fine or other penalty) obtained against us in the courts of any other territory in respect of certain enforceable obligations in accordance with the principles of private international law as applied by Jersey law (which are broadly similar to the principles accepted under English common law) and such judgment would be sufficient to form the basis of proceedings in the Jersey courts for a claim for liquidated damages in the amount of such judgment. In such proceedings, the Jersey courts would not re-hear the case on its merits save in accordance with such principles of private international law. Obligations may not necessarily be enforceable in Jersey in all circumstances or in accordance with their terms; and in particular, but without limitation: (a) any agreement purporting to provide for a payment to be made in the event of a breach of such agreement would not be enforceable to the extent that the Jersey courts were to construe such payment to be a penalty that was excessive, in that it unreasonably exceeds the maximum damages that an obligee could
44
have suffered as a result of the breach of an obligation; (b) the Jersey courts may refuse to give effect to any provision in an agreement that would involve the enforcement of any revenue or penal laws in other jurisdictions; and (c) the Jersey courts may refuse to allow unjust enrichment or to give effect to any provisions of an agreement (including provisions relating to contractual interest on a judgment debt) that it considers usurious.
Our annual and quarterly results may fluctuate due to a number of factors and, as a result, could fall below investor expectations or estimates by securities research analysts, which may cause the trading price of our ordinary shares to decline.
Our revenues and results of operations are difficult to predict, and potentially may vary significantly from period to period. As a result of a number of factors, many of which are beyond our control, it is possible that results of operations for future periods may be below the expectations of public market analysts and investors, which could cause our stock price to decline. Factors that may affect our quarterly results include, but are not limited to:
|
• |
failure to obtain regulatory approval for our delivery systems; |
|
• |
failure to effectively commercialize our delivery systems; |
|
• |
competition; and |
|
• |
changes in the laws and regulations that affect our operations. |
As a result, investors should not rely on year-to-year or quarter-to-quarter comparisons of results of operations as an indication of future performance.
Substantial future sales of our ordinary shares in the public market, or the perception that such sales may occur, could cause the price of our ordinary shares to decline.
Sales of our ordinary shares in the public market, or the perception that these sales may occur, could cause the market price of our ordinary shares to decline. All ordinary shares sold in our IPO (other than any shares acquired by our affiliates) are freely transferable without restriction or additional registration under the Securities Act of 1933, as amended, or the Securities Act.
Our memorandum and articles of association contain anti-takeover provisions that could adversely affect the rights of holders of our ordinary shares.
Our amended and restated memorandum and articles of association, referred to as the memorandum and articles of association, contain certain provisions that could limit the ability of third parties to acquire control of our company, including a provision for a classified board of directors and a provision that grants authority to our board of directors to issue from time to time one or more classes of preferred shares without action by our shareholders and to determine, with respect to any class of preferred shares, the terms and rights of that class. The provisions could have the effect of depriving our shareholders of the opportunity to sell their ordinary shares at a premium over the prevailing market price by discouraging third parties from seeking to obtain control of our company in a tender offer or similar transactions.
If securities or industry analysts do not publish research or publish unfavorable or inaccurate research about our business, our share price and trading volume could decline.
The trading market for our ordinary shares will continue to depend, in part, on the research and reports that securities or industry analysts publish about us or our business. We may be unable to sustain coverage by well-regarded securities and industry analysts. If either none or only a limited number of securities or industry analysts maintain coverage of our company, or if these securities or industry analysts are not widely respected within the general investment community, the trading price for our ordinary shares would be negatively impacted. In the event we obtain securities or industry analyst coverage, if one or more of the analysts who cover us downgrade our ordinary shares or publish inaccurate or unfavorable research about our business, the price of our ordinary shares would likely decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our ordinary shares could decrease, which might cause the price of our ordinary shares and trading volume to decline.
45
If we fail to maintain proper and effective internal controls, our ability to produce accurate and timely financial statements could be impaired and investors’ views of us could be harmed.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. We have spent considerable resources since our IPO designing, implementing and testing our internal controls over financial reporting and developing our internal audit function. If we or our independent registered public accounting firm identify deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of our ordinary shares could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
We currently lease space in several countries, including the following properties with square footage in the approximate amounts noted below:
|
• |
35,000 square feet of office and warehouse space in Portsmouth, New Hampshire which houses our U.S. operations center; |
|
• |
18,900 square feet of office and warehouse space in Root, Switzerland which houses our global supply chain and European commercial operations; |
|
• |
15,700 square feet of office and laboratory space in Haifa, Israel which houses our research and development operations; |
|
• |
12,850 square feet of office space in New York, New York which houses our U.S. commercial operations; |
|
• |
11,600 square feet of office space in Malvern, Pennsylvania which houses our finance, legal and information technology operations; |
|
• |
4,500 square feet of office space in Tokyo, Japan which houses our Japanese operations; and |
|
• |
4,400 square feet of office space in Munich, Germany which houses our commercial operations in Germany. |
We believe that our current facilities are adequate for our present purposes, but we may need additional space as we continue to grow and expand our operations. We believe that suitable additional or alternative office, laboratory, and manufacturing space would be available as required in the future on commercially reasonable terms.
As previously disclosed, in January 2017, two putative class action lawsuits were filed against us, our directors and certain of our officers, as well as the underwriters in our October 2015 initial public offering. The complaints, which purported to be brought on behalf of a class of persons and/or entities who purchased or otherwise acquired our ordinary shares pursuant and/or traceable to the registration statement and prospectus issued in connection with our initial public offering, alleged material misstatements and/or omissions in our initial public offering materials in alleged violation of the federal securities laws and sought compensatory damages, among other remedies. The two actions were subsequently consolidated, and the defendants moved to dismiss on multiple grounds. On December 4, 2017, the defendants’ motion to dismiss these actions was granted on the ground that the court lacked personal jurisdiction over any of the defendants. The plaintiffs did not appeal the dismissal and the deadline for filing an appeal has expired.
In addition, from time to time, we are involved in claims, proceedings, and litigation arising in the ordinary course of business. At this time, we do not expect litigation matters arising in the ordinary course of business to have a material adverse impact on our business, financial position or results of operations.
ITEM 4. MINE SAFETY DISCLOSURES
None.
46
Executive Officers of the Registrant
Our executive officers are elected by and serve at the discretion of our board of directors. The following table lists information about our executive officers:
|
Name |
|
Age |
|
Position |
|
Asaf Danziger |
|
51 |
|
Chief Executive Officer and Director |
|
|
|
|
|
|
|
William Doyle |
|
55 |
|
Executive Chairman |
|
|
|
|
|
|
|
Michael Ambrogi |
|
54 |
|
Chief Operating Officer |
|
|
|
|
|
|
|
Wilhelmus Groenhuysen |
|
60 |
|
Chief Financial Officer |
|
|
|
|
|
|
|
Eilon Kirson, M.D., Ph.D. |
|
49 |
|
Chief Science Officer and Head of Research and Development |
|
|
|
|
|
|
|
Todd Longsworth |
|
43 |
|
General Counsel |
|
|
|
|
|
|
Asaf Danziger has served as our Chief Executive Officer since 2002 and has been a director of NovoCure since 2012. From 1998 to 2002, Mr. Danziger was CEO of Cybro Medical, a subsidiary of Imagyn Medical Technologies, Inc. Mr. Danziger holds a B.Sc. in material engineering from Ben Gurion University of the Negev, Israel.
William Doyle has served as our Executive Chairman since 2016, as Chairman of the Board since 2009 and has been a director of NovoCure since 2004. Mr. Doyle has served as a director of Minerva Neurosciences, Inc., a clinical-stage biopharmaceutical company, since 2017. Mr. Doyle has also been the managing director of WFD Ventures LLC, a private venture capital firm he co-founded, since 2002 and was formerly a member of the investment team at Pershing Square Capital Management L.P., a private investment firm. Prior to 2002, Mr. Doyle was a member of Johnson & Johnson’s Medical Devices and Diagnostics Group Operating Committee and was vice president, Licensing and Acquisitions. While at Johnson & Johnson, Mr. Doyle was also chairman of the Medical Devices Research and Development Council, Worldwide president of Biosense-Webster, Inc. and a member of the board of directors of Cordis Corporation and Johnson & Johnson Development Corporation, Johnson & Johnson’s venture capital subsidiary. From 1992 to 1995, Mr. Doyle was a management consultant in the healthcare group of McKinsey & Company. Mr. Doyle holds an S.B. in materials science and engineering from the Massachusetts Institute of Technology and an M.B.A. from Harvard Business School.
Michael Ambrogi has been our Chief Operating Officer since 2010 and previously served as our U.S. General Manager from 2006 to 2010. Mr. Ambrogi has overall responsibility for our ongoing operations, engineering, manufacturing, service and human resources activities worldwide. From 1991 to 2006, Mr. Ambrogi worked for Deka Research and Development Corporation, inventor Dean Kamen’s research and development firm, last serving as general manager. Mr. Ambrogi led Deka’s teams on many products including the Baxter HomeChoice peritoneal dialysis machine, the Davol Hydroflex surgical irrigation device, the Cordis Crowne Stent and the J&J IBOT 3000 mobility system. Earlier in his career, from 1988 to 1990, Mr. Ambrogi was a consultant with McKinsey & Company, a global management consultant firm. Mr. Ambrogi holds a S.B. in mechanical engineering from MIT.
Wilhelmus Groenhuysen has been our Chief Financial Officer since 2012. He has served on the Board of Optinose Inc., a specialty pharmaceutical company, since October 2017. From 2007 to 2011, Mr. Groenhuysen worked for Cephalon, Inc., a U.S. biopharmaceutical company, last serving as executive vice president and chief financial officer, where he had responsibility for worldwide finance, commercial operations and risk management. From 1987 to 2007, Mr. Groenhuysen worked for Philips Group in various assignments in Europe, Asia and the United States, the latest of which started in 2002 when he was promoted to chief financial officer and senior vice president of Philips Electronics North America Corporation. Mr. Groenhuysen holds a Master’s Degree in Business Economics from VU University Amsterdam and graduated as a Registered Public Controller at VU University Amsterdam.
Eilon Kirson, M.D., Ph.D. has been our Chief Science Officer and Head of Research and Development since 2012 and previously served as our Chief Medical Officer from 2006 to 2012. Dr. Kirson has led the development of Tumor Treating Fields from preclinical testing to large, multi-center phase 3 studies and through multiple regulatory approvals. Dr. Kirson previously served as head of electrophysiology at Carmel Biosensors Ltd., a company which developed medical monitoring sensors. Dr. Kirson received his B.Med.Sc., M.D. and Ph.D. in Physiology-Biophysics from the Hebrew University of Jerusalem and served his residency in neurology at Sheba Medical Center, Tel Ha-Shomer Hospital, Israel.
Todd Longsworth joined Novocure in 2012 and serves as General Counsel and Chief Compliance Officer. Mr. Longsworth worked for Cephalon, Inc., a U.S. biopharmaceutical company, from 2005 to 2012, last serving as Mergers and Acquisitions, Securities and Corporate Governance Counsel. Prior to joining Cephalon, he was an associate at WilmerHale LLP, a global law firm from 2001 to 2005. Mr. Longsworth earned his B.A. from Duke University and his J.D. from the University of Pennsylvania.
47
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our ordinary shares are quoted on the NASDAQ Global Select Market under the symbol “NVCR.” Our ordinary shares began to be quoted on the NASDAQ Global Select Market on October 2, 2015. The following table sets forth the range of high and low sale prices for our ordinary shares as reported on the NASDAQ Global Select Market for the period indicated below.
|
|
|
High |
|
|
Low |
|
||
|
2017 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
22.11 |
|
|
$ |
16.10 |
|
|
Third Quarter |
|
$ |
22.30 |
|
|
$ |
17.15 |
|
|
Second Quarter |
|
$ |
17.90 |
|
|
$ |
9.85 |
|
|
First Quarter |
|
$ |
8.35 |
|
|
$ |
6.00 |
|
|
2016 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
10.29 |
|
|
$ |
5.95 |
|
|
Third Quarter |
|
$ |
12.49 |
|
|
$ |
7.30 |
|
|
Second Quarter |
|
$ |
17.00 |
|
|
$ |
9.87 |
|
|
First Quarter |
|
$ |
23.31 |
|
|
$ |
10.15 |
|
|
2015 |
|
|
|
|
|
|
|
|
|
Fourth Quarter |
|
$ |
30.89 |
|
|
$ |
15.01 |
|
Holders of Ordinary Shares
As of February 15, 2018, there were 57 holders of record of our ordinary shares. On February 15, 2018, the last reported sale price of our ordinary shares as reported on the NASDAQ Global Select Market was $21.15 per share.
Dividend Policy
We have not paid any dividends on our ordinary shares since our inception and do not anticipate paying any dividends on our ordinary shares in the foreseeable future.
48
The following performance graph is being furnished as part of this annual report and shall not be deemed “filed” with the SEC or incorporated by reference into any of our filings under the Securities Act of 1933, as amended (the “Securities Act”), or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing.
The following graph shows the total shareholder return of an investment of $100 in cash at market close on October 2, 2015 (the first day of trading of our ordinary shares) through December 31, 2017 for (1) our ordinary shares, (2) the Russell 2000 Index, and (3) the Nasdaq Biotechnology Index. Pursuant to applicable SEC rules, all values assume reinvestment of the full amount of all dividends; however, no dividends have been declared on our ordinary shares to date. The shareholder return shown on the graph below is not necessarily indicative of future performance, and we do not make or endorse any predictions as to future stockholder returns.

|
|
|
|
|
10/2/2015 |
|
|
12/31/2015 |
|
|
03/31/2016 |
|
|
03/60/2016 |
|
|
9/30/2016 |
|
|
12/31/2016 |
|
|
3/31/2017 |
|
|
03/60/2017 |
|
|
9/30/2017 |
|
|
12/31/2017 |
|
|||||||||||
|
NovoCure Limited |
|
Return % |
|
|
|
|
|
|
22.32 |
|
|
|
-35.24 |
|
|
|
-19.41 |
|
|
|
-26.82 |
|
|
|
-8.08 |
|
|
|
3.18 |
|
|
|
113.58 |
|
|
|
14.74 |
|
|
|
1.76 |
|
|
|
|
|
Cum $ |
|
|
100.00 |
|
|
|
122.32 |
|
|
|
79.21 |
|
|
|
63.84 |
|
|
|
46.72 |
|
|
|
42.94 |
|
|
|
44.31 |
|
|
|
94.64 |
|
|
|
108.59 |
|
|
|
110.50 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NASDAQ Biotechnology Index |
|
Return % |
|
|
|
|
|
|
7.29 |
|
|
|
-22.88 |
|
|
|
-1.12 |
|
|
|
12.50 |
|
|
|
-8.31 |
|
|
|
10.84 |
|
|
|
5.89 |
|
|
|
7.74 |
|
|
|
-3.79 |
|
|
|
|
|
Cum $ |
|
|
100.00 |
|
|
|
107.29 |
|
|
|
82.74 |
|
|
|
81.81 |
|
|
|
92.03 |
|
|
|
84.38 |
|
|
|
93.53 |
|
|
|
99.04 |
|
|
|
106.71 |
|
|
|
102.66 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Russell 2000 Index |
|
Return % |
|
|
|
|
|
|
2.33 |
|
|
|
-1.52 |
|
|
|
3.79 |
|
|
|
9.05 |
|
|
|
8.83 |
|
|
|
2.47 |
|
|
|
2.46 |
|
|
|
5.67 |
|
|
|
3.34 |
|
|
|
|
|
Cum $ |
|
|
100.00 |
|
|
|
102.33 |
|
|
|
100.78 |
|
|
|
104.60 |
|
|
|
114.06 |
|
|
|
124.14 |
|
|
|
127.20 |
|
|
|
130.33 |
|
|
|
137.72 |
|
|
|
142.32 |
|
|
49
Recent Sales of Unregistered Securities
From January 1, 2015 to December 31, 2017, we have issued the following securities in unregistered transactions, which include warrants and options to acquire our ordinary shares. We believe that each of the following instances was exempt from registration under the Securities Act in reliance on Regulation S under the Securities Act, under Section 4(a)(2) of the Securities Act regarding transactions not involving a public offering and under Rule 701 promulgated under the Securities Act:
|
Issuance |
|
Date of sale or issuance |
|
Number of securities |
|
|
Consideration |
|
||
|
Ordinary Shares (1) |
|
March 2015 |
|
1,005,210 |
|
|
|
- |
|
|
|
Series J Preferred Shares (2) |
|
June 2015 |
|
4,068,500 |
|
|
$94.60 million |
|
||
|
Options to Purchase Ordinary Shares (3) |
|
January 1, 2015 to December 31, 2015 |
|
|
4,113,603 |
|
|
|
- |
|
|
Exercise of Ordinary Share Options and Warrants |
|
January 1, 2015 to December 31, 2015 |
|
|
731,665 |
|
|
$2.04 million |
|
|
|
Options to Purchase Ordinary Shares |
|
January 1, 2016 to December 31, 2016 |
|
|
2,596,600 |
|
|
|
- |
|
|
Exercise of Ordinary Share Options and Warrants |
|
January 1, 2016 to December 31, 2016 |
|
|
2,517,690 |
|
|
$0.85 million |
|
|
|
Exercise of Warrants |
|
January 1, 2017 to December 31, 2017 |
|
|
809,636 |
|
|
$0.02 million |
|
|
|
(1) |
Ordinary shares issued to Technion. |
|
(2) |
The Series J Preferred Shares converted into ordinary shares in connection with the IPO in October 2015. |
|
(3) |
Number of securities includes an option to purchase 1,005,210 ordinary shares issued to Technion in March 2015. On October 7, 2015, we closed our IPO, in which we sold an aggregate of 7,500,000 ordinary shares at a price to the public of $22.00 per share. On October 19, 2015, the underwriters to the IPO partially exercised their over-allotment option, in which we sold an aggregate of 376,195 ordinary shares at a price to the public of $22.00 per share. We received net proceeds from the IPO and the over-allotment option of $157.5 million, after deducting the underwriting discounts, commissions and offering expenses payable by us. The offer and sale of all of the ordinary shares in the IPO and the over-allotment option were registered under the Securities Act pursuant to a registration statement on Form S-1 (File No. 333-206681), which was declared effective by the SEC on October 1, 2015 (the “Registration Statement”). |
There has been no material change in the planned use of proceeds from our IPO as described in the Registration Statement. As of December 31, 2017, we have used all of the net proceeds from the IPO primarily to fund the costs of our clinical studies, advance our second generation Optune system, expand commercial operations in the US, Germany and Japan, payment of interest on our Term Loan Credit Facility (as defined below), to further develop additional product candidates, to expand our internal research and development capabilities, and other general corporate purposes. We primarily invested the proceeds received in short-term, interest-bearing investment-grade securities and government securities. None of the offering proceeds were paid directly or indirectly to any of our directors or officers (or their associates) or persons owning 10.0% or more of any class of our equity securities or to any other affiliates.
Issuer Purchases of Equity Securities
Not applicable.
50
Securities Authorized for Issuance Under Equity Compensation Plans
The following table gives information about our ordinary shares that may be issued upon the exercise of stock options and vesting of restricted stock units, as applicable, under all of our existing equity compensation plans as of December 31, 2017, including the 2003 Share Option Plan (the “2003 Plan”), the 2013 Share Option Plan (the “2013 Plan”) and the 2015 Omnibus Incentive Plan (the “2015 Plan”). Each of the 2003 Plan, the 2013 Plan and the 2015 Plan has been approved by the Company’s shareholders.
Equity Compensation Plan Information
|
Plan Category |
|
(a) Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
|
|
(b) Weighted Average Exercise Price of Outstanding Options, Warrants and Rights |
|
|
(c) Number of Securities Remaining Available for Future Issuance (Excludes Securities Reflected in Column (a)) |
|
|||
|
Equity compensation plans approved by shareholders |
|
|
16,457,246 |
|
|
$ |
9.76 |
|
|
|
12,971,921 |
|
|
Equity compensation plans not approved by shareholders |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Total |
|
|
16,457,246 |
|
|
$ |
9.76 |
|
|
|
12,971,921 |
|
ITEM 6. SELECTED FINANCIAL DATA
The following tables set forth our consolidated statements of operations data:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net revenues |
|
$ |
177,026 |
|
|
$ |
82,888 |
|
|
$ |
33,087 |
|
|
Cost of revenues |
|
|
55,609 |
|
|
|
39,870 |
|
|
|
20,610 |
|
|
Impairment of field equipment |
|
|
- |
|
|
|
6,412 |
|
|
|
- |
|
|
Gross profit |
|
|
121,417 |
|
|
|
36,606 |
|
|
|
12,477 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
38,103 |
|
|
|
41,467 |
|
|
|
43,748 |
|
|
Sales and marketing |
|
|
63,528 |
|
|
|
59,449 |
|
|
|
38,861 |
|
|
General and administrative |
|
|
59,114 |
|
|
|
51,007 |
|
|
|
33,864 |
|
|
Total operating costs and expenses |
|
|
160,745 |
|
|
|
151,923 |
|
|
|
116,473 |
|
|
Operating loss |
|
|
(39,328 |
) |
|
|
(115,317 |
) |
|
|
(103,996 |
) |
|
Financial expenses, net |
|
|
(9,169 |
) |
|
|
(6,147 |
) |
|
|
(3,151 |
) |
|
Loss before income taxes |
|
|
(48,497 |
) |
|
|
(121,464 |
) |
|
|
(107,147 |
) |
|
Income taxes |
|
|
13,165 |
|
|
|
10,381 |
|
|
|
4,434 |
|
|
Net loss |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(0.70 |
) |
|
$ |
(1.54 |
) |
|
$ |
(3.67 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
88,546,719 |
|
|
|
85,558,448 |
|
|
|
30,401,603 |
|
Non-cash share-based compensation expense included in costs and expenses:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cost of revenues |
|
$ |
467 |
|
|
$ |
623 |
|
|
$ |
174 |
|
|
Research, development and clinical trials |
|
|
3,587 |
|
|
|
3,155 |
|
|
|
2,529 |
|
|
Sales and marketing |
|
|
3,784 |
|
|
|
5,111 |
|
|
|
2,496 |
|
|
General and administrative |
|
|
19,278 |
|
|
|
12,552 |
|
|
|
6,661 |
|
|
Total share-based compensation expense |
|
$ |
27,116 |
|
|
$ |
21,441 |
|
|
$ |
11,860 |
|
51
Consolidated balance sheet data:
|
|
|
December 31, |
|
|||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
||
|
Cash and cash equivalents |
|
$ |
78,592 |
|
|
$ |
99,780 |
|
|
Short-term investments |
|
|
104,719 |
|
|
|
119,854 |
|
|
Total assets |
|
|
265,298 |
|
|
|
282,081 |
|
|
Working capital |
|
|
194,932 |
|
|
|
224,991 |
|
|
Current liabilities |
|
|
50,202 |
|
|
|
36,882 |
|
|
Long term liabilities |
|
|
101,532 |
|
|
|
102,854 |
|
|
Total shareholders' equity |
|
$ |
113,564 |
|
|
$ |
142,345 |
|
Condensed cash flow data:
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net cash used in operating activities |
|
$ |
(33,134 |
) |
|
$ |
(107,592 |
) |
|
$ |
(99,884 |
) |
|
Net cash provided by (used in) investing activities |
|
|
6,770 |
|
|
|
12,815 |
|
|
|
(115,295 |
) |
|
Net cash provided by financing activities |
|
|
5,168 |
|
|
|
75,124 |
|
|
|
276,989 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
8 |
|
|
|
10 |
|
|
|
- |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
(21,188 |
) |
|
$ |
(19,643 |
) |
|
$ |
61,810 |
|
52
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) is intended to provide information to assist you in better understanding and evaluating our financial condition and results of operations. Certain statements in this MD&A are forward-looking within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, as further explained under the heading “Cautionary Note Regarding Forward-Looking Statements.” Investors and prospective investors should understand that many factors govern whether any forward-looking statement contained herein will be or can be realized. Any one of those factors could cause actual results to differ materially from those projected. We encourage you to read this MD&A in conjunction with our consolidated financial statements included in Part II, Item 8 of this Annual Report on Form 10-K and the “Risk Factors” contained in Part I, Item 1A of this Annual Report on Form 10-K.
Overview
We are a global oncology company developing a proprietary platform technology called Tumor Treating Fields, the use of electric fields tuned to specific frequencies to disrupt solid tumor cancer cell division. Our key priorities are to drive commercial adoption of Optune, our first commercial Tumor Treating Fields delivery system, for the treatment of glioblastoma (“GBM”), and to advance programs testing the efficacy and safety of Tumor Treating Fields in multiple solid tumor indications through our clinical pipeline.
We were founded in 2000 and operated as a development stage company through December 31, 2011. We initially received U.S. Food and Drug Administration (“FDA”) approval for Optune in 2011 for use as a monotherapy treatment for adult patients with GBM following confirmed recurrence after chemotherapy. In October 2015, we received FDA approval to market Optune for the treatment of adult patients with newly diagnosed GBM in combination with temozolomide, a chemotherapy drug. We have also received approval to market Optune in the European Union (“EU”), Switzerland, Japan and certain other countries. We have built a commercial organization and launched Optune in the United States, Germany, Austria, Switzerland, Israel and Japan, which we refer to as our currently active markets.
We have researched the biological effects of Tumor Treating Fields extensively. Tumor Treating Fields uses electric fields tuned to specific frequencies to disrupt cancer cell division, inhibiting tumor growth and causing affected cancer cells to die. Because Tumor Treating Fields is delivered regionally, acts only on dividing cells (a biological process known as mitosis) and is frequency tuned to target cancer cells of a specific size, we believe there is minimal damage to healthy cells. We believe our preclinical and clinical research demonstrates that Tumor Treating Fields’ mechanism of action affects fundamental aspects of cell division and may have broad applicability across a variety of solid tumors. We have demonstrated in preclinical studies that Tumor Treating Fields can offer additive or synergistic benefits in combination with other anti-cancer agents, which may lead to greater efficacy without significantly increasing the side effects.
In addition to our clinical and commercial progress in GBM, we are currently planning or conducting clinical trials evaluating the use of Tumor Treating Fields in brain metastases, non-small-cell lung cancer (NSCLC), pancreatic cancer, ovarian cancer and mesothelioma. We anticipate expanding our clinical pipeline over time to study the safety and efficacy of Tumor Treating Fields for additional solid tumor indications.
The following table presents an overview of our ongoing clinical trials:
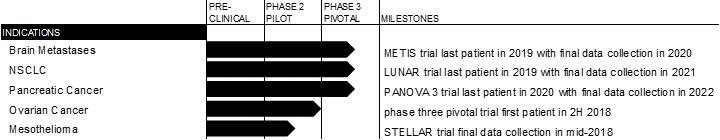
We own all commercialization rights to Tumor Treating Fields in oncology. Our robust global patent and intellectual property portfolio consists of over 140 issued patents, with numerous additional patent applications pending worldwide. We believe we will maintain exclusive rights to market Tumor Treating Fields for all solid tumor indications in our key markets through the life of our patents.
53
We view our operations and manage our business in one operating segment. Our net revenues were $177.0 million for the year ended December 31, 2017, $82.9 million for the year ended December 31, 2016 and $33.1 million for the year ended December 31, 2015. Our net loss was $61.7 million for the year ended December 31, 2017, $131.8 million for the year ended December 31, 2016 and $111.6 million for the year ended December 31, 2015. As of December 31, 2017, we had an accumulated deficit of $582.3 million. Our net loss primarily resulted from costs incurred in connection with our preclinical and clinical trial programs, costs incurred to commercialize Optune and general and administrative costs necessary to operate as a global oncology business.
Commentary on Results of Operations
Net revenues
Substantially all of our revenues are derived from patients using Optune in our currently active markets. We charge patients or their third-party healthcare payers on a monthly basis. Our potential net revenues per patient are determined by our ability to secure payment from payers, the monthly fee we collect and the number of months that the patient remains on therapy.
Cost of revenues
We contract with third-party manufacturers that manufacture Optune. Our cost of revenues is primarily comprised of the following:
|
|
• |
disposable transducer arrays; |
|
|
• |
depreciation expense for the field equipment, including the electric field generator used by patients; and |
|
|
• |
personnel, warranty and overhead costs such as facilities, freight and depreciation of property, plant and equipment associated with managing our inventory, warehousing and order fulfillment functions. |
Operating expenses
Our operating expenses consist of research, development and clinical trials, sales and marketing and general and administrative expenses. Personnel costs are a significant component for each category of operating expenses and consist of wages, benefits and bonuses. Personnel costs also include share-based compensation.
Research, development and clinical trials
Our research, development and clinical trials activity is focused on advancing Tumor Treating Fields through clinical trials across multiple solid tumor types and improving our delivery systems. Research, development and clinical trials costs, including direct and allocated expenses, are expensed as incurred and consist primarily of the following:
|
|
• |
personnel costs (including share-based compensation) for those employees involved in our research, development, clinical trial, regulatory and medical affairs activities; |
|
|
• |
costs to conduct research, development and clinical trial activity through agreements with contract research organizations and other third parties; |
|
|
• |
facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation of leasehold improvements and equipment and laboratory and other supplies; |
|
|
• |
manufacturing expenses associated with Tumor Treating Fields delivery systems, including durable components and disposable arrays, utilized in clinical trials and other research; and |
|
|
• |
professional fees related to regulatory approvals and conformity assessment procedures. |
54
We have incurred significant expenditures related to conducting clinical studies to develop Tumor Treating Fields in multiple solid tumor indications. The following table summarizes our principal clinical programs for Tumor Treating Fields for the years ended December 31, 2017, 2016 and 2015.
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Personnel costs |
|
$ |
17,725 |
|
|
$ |
18,620 |
|
|
$ |
15,194 |
|
|
General research and development |
|
|
11,450 |
|
|
|
12,156 |
|
|
|
13,198 |
|
|
Materials |
|
|
2,203 |
|
|
|
2,270 |
|
|
|
5,267 |
|
|
Phase II clinical trials |
|
|
983 |
|
|
|
1,915 |
|
|
|
1,362 |
|
|
Phase III clinical trials |
|
|
|
|
|
|
|
|
|
|
|
|
|
EF-14 |
|
|
704 |
|
|
|
4,508 |
|
|
|
8,726 |
|
|
METIS |
|
|
2,614 |
|
|
|
1,484 |
|
|
|
- |
|
|
LUNAR |
|
|
1,929 |
|
|
|
514 |
|
|
|
- |
|
|
PANOVA 3 |
|
|
495 |
|
|
|
- |
|
|
|
- |
|
|
Research, development and clinical trials |
|
$ |
38,103 |
|
|
$ |
41,467 |
|
|
$ |
43,748 |
|
Personnel costs include all preclinical, clinical, medical affairs and other research and development company personnel. General research and development costs include costs related to preclinical, engineering, regulatory, post-authorization studies, investigational sponsored trials, intellectual property, advisors and subcontractors, travel and other. Materials include the costs of all equipment, arrays and other disposables for use in the clinical trials. Clinical trial costs in these periods include contract research organization services, data managing services, clinic and lab costs, as well as clinical sites costs.
We expect our research and development expenses to increase in absolute dollars as we continue to advance Tumor Treating Fields and develop new delivery systems to address current and possible future indications.
Sales and marketing
Sales and marketing expenses consist primarily of personnel costs (including share-based compensation), travel expenses, marketing and promotional activities, commercial shipping and facilities costs. Over the next few years, we expect to continue to make significant expenditures associated with selling and marketing our delivery systems, primarily in connection with the continued commercialization of Optune in the United States, EU and Japan for the treatment of our approved indications.
General and administrative
General and administrative expenses consist primarily of personnel costs (including share-based compensation), professional fees and facilities costs. General and administrative personnel costs include our executive, finance, human resources, information technology and legal functions. These costs also include our contributions to support industry and patient groups. Our professional fees consist primarily of accounting, legal and other consulting costs. We expect that general and administrative expenses will increase in absolute dollars to support our growth. In addition, we incur significant legal and accounting costs related to compliance with SEC rules and regulations, including the costs of achieving and maintaining compliance with Section 404 of the Sarbanes-Oxley Act of 2002 (“Sarbanes-Oxley Act”) and compliance with rules of the NASDAQ Stock Market, as well as insurance, investor relations and other costs associated with being a public company.
Financial expenses, net
Financial expenses, net primarily consists of credit facility interest expense and related debt issuance costs, interest income from cash balances and short-term investments and gains (losses) from foreign currency transactions. Our reporting currency is the U.S. dollar. We have historically held substantially all of our cash balances in U.S. dollar denominated accounts to minimize the risk of translational currency exposure.
Critical accounting policies and estimates
In accordance with U.S. GAAP, in preparing our financial statements, we must make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of net revenues and expenses during the reporting period. We develop and periodically change these estimates and assumptions based on historical experience and on various other factors that we believe are reasonable under the circumstances. Actual results may differ from these estimates.
55
The critical accounting policies requiring estimates, assumptions and judgments that we believe have the most significant impact on our consolidated financial statements are described below.
Revenue recognition
Optune is comprised of two main components: (1) an electric field generator and (2) transducer arrays and related accessories that are disposable supplies to the device, or the transducer arrays. We retain title to the electric field generator, and the patient is provided replacement transducer arrays and technical support for the device during the term of treatment. The electric field generator and transducer arrays are always supplied and function together and are not sold on a standalone basis.
Revenues are recognized when persuasive evidence of an arrangement exists, delivery of Optune has occurred, the fee is fixed or determinable and collectability is reasonably assured. The evidence of an arrangement generally consists of a prescription, a patient service agreement and the verification of eligibility and insurance with the patient’s third-party insurance company. We assess whether the fee is fixed or determinable based on whether there is sufficient history with payers to reliably estimate their individual payment patterns or contractual arrangements exist and whether we can reliably estimate the amount that would be ultimately collected. Once we can reliably estimate the amounts that would be ultimately collected per payer and the above criteria are met, we recognize revenues net of allowances from the use of Optune on an accrual basis ratably over the lease term. The allowances are determined based on defined payment terms and historical collection data by payer. Allowance adjustments related to final settlements for the reported periods are insignificant. When the revenue criteria above are not met, such as when the price is not fixed or determinable or the collectability cannot be reasonably assured, revenues are recognized when cash is collected. Patients often have out-of-pocket costs for the amount not covered by their third-party payer and we bill the patient directly for the amounts of their co-pays and deductibles, subject to our patient assistance programs. To date, we recognize revenue from these patient payments at the time cash is collected.
Revenues are presented net of indirect taxes incurred in the reported periods, including the U.S. medical device excise tax, regardless of whether the revenues associated with those taxes are reported on a cash basis.
In May 2014, FASB issued ASU 2014-09, Revenue from Contracts with Customers (Topic 606) (ASU 2014-09), an updated standard on revenue recognition and issued subsequent amendments to the initial guidance in March 2016, April 2016, May 2016 and December 2016 within ASU 2016-08, 2016-10, 2016-12 and 2016-20, respectively. The Company has adopted the standard effective January 1, 2018. The Company does not anticipate that the adoption of this standard will have a material impact on its financial position, results of operations or cash flows. For additional information, see Note 2(x) to the Consolidated Financial Statements.
Share-based compensation
Under the Financial Accounting Standards Board’s (“FASB”) Accounting Standards Codification (“ASC”) 718, Compensation—Stock Compensation, we measure and recognize compensation expense for share options granted to our employees and directors based on the fair value of the awards on the date of grant. The fair value of share options is estimated at the date of grant using the Black-Scholes option pricing model that requires management to apply judgment and make estimates, including:
|
|
• |
the fair value of our ordinary shares on the date of grant until our IPO determined as discussed below; |
|
|
• |
the expected term of the stock option award, which we calculate using the simplified method, in accordance with ASC No.718-10-S99-1 (SAB No. 110) as we have insufficient historical information regarding our stock options to provide a basis for an estimate; |
|
|
• |
the expected share price volatility of our underlying ordinary shares, which we estimate based on the historical volatility of a representative group of publicly traded biopharmaceutical and medical technology companies with similar characteristics to us for a period matching the expected term assumption; |
|
|
• |
the risk-free interest rate, which we base on the yield curve of U.S. Treasury securities with periods commensurate with the expected term of the options being valued; and |
|
|
• |
the expected dividend yield, which we estimate to be zero based on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. |
For information about our ESPP, see Note 14(e) to the Consolidated Financial Statements.
56
We recognize share-based compensation costs only for those shares expected to vest over the requisite vesting period of the award, which is generally the option vesting term of four years, using the accelerated method. ASC 718 requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company changed its accounting policy in January 2017 (Accounting Standards Update (“ASU”) 2016-09, Compensation-Stock Compensation -Topic 718), recognizing forfeitures of awards as they occur. The Company changed its accounting policy to account for forfeitures as they occur for the year ended December 31, 2017 as further detailed in Note 2(w) to the Consolidated Financial Statements.
The table below summarizes the assumptions that were used to estimate the fair value of the options granted to employees during the periods presented:
|
|
|
Year ended December 31, |
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
Expected term (years) |
|
5.50-6.25 |
|
6.25 |
|
6.25 |
|
Expected volatility |
|
56.74%-59.45% |
|
58.4%-61.7% |
|
59%-65.8% |
|
Risk-free interest rate |
|
1.97%-2.23% |
|
1.23%-1.88% |
|
1.7%-2.1% |
|
Dividend yield |
|
0.00% |
|
0% |
|
0% |
If any of the assumptions used in the Black-Scholes option pricing model change significantly, share-based compensation for future awards may differ materially from the awards granted previously.
Our management and board of directors determined the fair value of our ordinary shares prior to our IPO based on a number of objective and subjective factors consistent with the methodologies, approaches and assumptions consistent with the American Institute of Certified Public Accountants’ Accounting and Valuation Guide, Valuation of Privately-Held-Company Equity Securities Issued as Compensation (the “AICPA Guide”). These factors included the hiring of key personnel, contemporaneous third-party valuations of our ordinary shares, our financial condition and prospects as of such date, the status of our research and development efforts, the public trading price of comparable companies for the March 5, 2015 grant, the lack of marketability of our ordinary shares as a private company, risk factors relevant to our business, capital markets conditions generally and the prices of our preferred shares sold to investors in arm’s-length transactions, and the rights, preferences and privileges of our preferred shares relative to our ordinary shares.
So long as our ordinary shares are publicly traded in a liquid market, we will rely on the daily trading price of our ordinary shares when we estimate the fair value of options granted.
We incurred share-based compensation expense of $27.1 million, $21.4 million and $11.9 million during the years ended December 31, 2017, 2016 and 2015, respectively. As of December 31, 2017, we have unrecognized compensation expense of $39.3 million, which is expected to be recognized over a weighted average period of approximately 2.86 years. We expect to continue to grant equity awards in the future, and to the extent that we do, our recognized share-based compensation expense will likely increase.
Long-lived assets
Property and equipment and field equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful life of the relevant asset. We make estimates of the useful life of our property and equipment and field equipment, based on similar assets purchased in the past and our historical experience with such similar assets, in order to determine the depreciation expense to be recorded for each reporting period.
Our field equipment consists of equipment being utilized under rental agreements accounted for on a monthly basis as an operating lease, as well as service pool equipment. Service pool equipment is equipment owned and maintained by us that is swapped for equipment that needs repair or maintenance by us while being used by a patient. We record a provision for any excess, lost or damaged equipment when warranted based on an assessment of the equipment.
We assess impairment whenever events or changes in circumstances indicate that the carrying amount of the asset is impaired or the estimated useful life is no longer appropriate. Circumstances such as changes in technology or in the way an asset is being used may trigger an impairment review. For additional information, see Notes 2(i) and 2(j) to the Consolidated Financial Statements.
57
Inventories are stated at the lower of cost or net realizable value. We regularly evaluate the ability to realize the value of inventory. If the inventories are deemed damaged, if actual demand for our delivery systems declines, or if market conditions are less favorable than those projected, inventory write-offs may be required. For additional information, see Note 2(g) to the Consolidated Financial Statements.
Income taxes
As part of the process of preparing our consolidated financial statements, we are required to calculate our income taxes based on taxable income by jurisdiction. We make certain estimates and judgments in determining our income taxes, including assessment of our uncertain tax positions, for financial statement purposes. Significant changes to these estimates may result in an increase or decrease to our tax provision in the subsequent period when such a change in estimate occurs.
Uncertain tax positions are based on estimates and assumptions that have been deemed reasonable by management. Our estimates of unrecognized tax benefits and potential tax benefits may not be representative of actual outcomes.
Recently issued accounting pronouncements
For a description of our recently issued accounting pronouncements, see Note 2(x) to the Consolidated Financial Statements.
Results of operations
The following tables set forth our consolidated statements of operations data:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net revenues |
|
$ |
177,026 |
|
|
$ |
82,888 |
|
|
$ |
33,087 |
|
|
Cost of revenues |
|
|
55,609 |
|
|
|
39,870 |
|
|
|
20,610 |
|
|
Impairment of field equipment |
|
|
- |
|
|
|
6,412 |
|
|
|
- |
|
|
Gross profit |
|
|
121,417 |
|
|
|
36,606 |
|
|
|
12,477 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
38,103 |
|
|
|
41,467 |
|
|
|
43,748 |
|
|
Sales and marketing |
|
|
63,528 |
|
|
|
59,449 |
|
|
|
38,861 |
|
|
General and administrative |
|
|
59,114 |
|
|
|
51,007 |
|
|
|
33,864 |
|
|
Total operating costs and expenses |
|
|
160,745 |
|
|
|
151,923 |
|
|
|
116,473 |
|
|
Operating loss |
|
|
(39,328 |
) |
|
|
(115,317 |
) |
|
|
(103,996 |
) |
|
Financial expenses, net |
|
|
(9,169 |
) |
|
|
(6,147 |
) |
|
|
(3,151 |
) |
|
Loss before income taxes |
|
|
(48,497 |
) |
|
|
(121,464 |
) |
|
|
(107,147 |
) |
|
Income taxes |
|
|
13,165 |
|
|
|
10,381 |
|
|
|
4,434 |
|
|
Net loss |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(0.70 |
) |
|
$ |
(1.54 |
) |
|
$ |
(3.67 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
88,546,719 |
|
|
|
85,558,448 |
|
|
|
30,401,603 |
|
Share-based compensation expense included in costs and expenses:
|
|
|
Year ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cost of revenues |
|
$ |
467 |
|
|
$ |
623 |
|
|
$ |
174 |
|
|
Research, development and clinical trials |
|
|
3,587 |
|
|
|
3,155 |
|
|
|
2,529 |
|
|
Sales and marketing |
|
|
3,784 |
|
|
|
5,111 |
|
|
|
2,496 |
|
|
General and administrative |
|
|
19,278 |
|
|
|
12,552 |
|
|
|
6,661 |
|
|
Total share-based compensation expense |
|
$ |
27,116 |
|
|
$ |
21,441 |
|
|
$ |
11,860 |
|
58
The following table includes certain commercial patient operating statistics for and as of the end of the periods presented.
|
|
|
December 31, |
|
|||||||||
|
Operating statistics |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Active patients at period end (1) |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
|
1,320 |
|
|
|
835 |
|
|
|
529 |
|
|
EMEA (*) |
|
|
512 |
|
|
|
256 |
|
|
|
74 |
|
|
Japan |
|
|
2 |
|
|
|
- |
|
|
|
2 |
|
|
|
|
|
1,834 |
|
|
|
1,091 |
|
|
|
605 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(*) including Germany |
|
|
356 |
|
|
|
186 |
|
|
|
51 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Prescriptions received in period (2) |
|
|
|
|
|
|
|
|
|
|
|
|
|
United States |
|
|
3,102 |
|
|
|
2,344 |
|
|
|
1,607 |
|
|
EMEA (*) |
|
|
1,011 |
|
|
|
463 |
|
|
|
167 |
|
|
Japan |
|
|
6 |
|
|
|
1 |
|
|
|
3 |
|
|
|
|
|
4,119 |
|
|
|
2,808 |
|
|
|
1,777 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(*) including Germany |
|
|
731 |
|
|
|
354 |
|
|
|
127 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
_______________________
|
(1) |
An “active patient” is a patient who is on Optune under a commercial prescription order as of the measurement date, including patients who may be on a temporary break from treatment and who plan to resume treatment in less than 60 days. |
|
(2) |
A “prescription received” is a commercial order for Optune that is received from a physician certified to treat patients with Optune for a patient not previously on Optune. Orders to renew or extend treatment are not included in this total. |
Year ended December 31, 2017 compared to year ended December 31, 2016
(All dollar figures in tables are in thousands unless otherwise indicated)
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
Change |
|
|
% Change |
|
||||
|
Net revenues |
|
$ |
177,026 |
|
|
$ |
82,888 |
|
|
$ |
94,138 |
|
|
|
114 |
% |
Net revenues. Net revenues increased by $94.1 million, or 114%, to $177.0 million for the year ended December 31, 2017 from $82.9 million for the year ended December 31, 2016. This was primarily due to an increase of 743 active patients, representing 68% growth, in our currently active markets and an increase in the net revenues per active patient. The increase in net revenues per active patient was primarily driven by an increase in positive coverage policies in the United States, improving reimbursement approval rates in Germany, and a one-time benefit from the transition to accrual-based revenue for a portion of our payers.
The following is a summary of gross billings and revenues recorded on an accrual basis and a cash basis by quarters (unaudited):
|
|
|
2017 |
|
|
2016 |
|
||||||||||||||||||||||||||
|
U.S. dollars in millions |
|
Q4 |
|
|
Q3 |
|
|
Q2 |
|
|
Q1 |
|
|
Q4 |
|
|
Q3 |
|
|
Q2 |
|
|
Q1 |
|
||||||||
|
Gross billings |
|
$ |
113.8 |
|
|
$ |
101.9 |
|
|
$ |
87.2 |
|
|
$ |
73.2 |
|
|
$ |
63.8 |
|
|
$ |
57.5 |
|
|
$ |
54.0 |
|
|
$ |
45.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenue recorded on accrual basis |
|
$ |
39.5 |
|
|
$ |
35.7 |
|
|
$ |
19.1 |
|
|
$ |
14.7 |
|
|
$ |
8.5 |
|
|
$ |
0.0 |
|
|
$ |
0.0 |
|
|
$ |
0.0 |
|
|
Cash basis revenue for therapy provided in the period |
|
|
4.8 |
|
|
|
3.1 |
|
|
|
5.7 |
|
|
|
5.9 |
|
|
|
6.3 |
|
|
|
8.9 |
|
|
|
7.6 |
|
|
|
5.6 |
|
|
Cash basis revenue for therapy provided in previous period |
|
$ |
9.3 |
|
|
$ |
11.3 |
|
|
$ |
13.6 |
|
|
$ |
14.3 |
|
|
$ |
15.5 |
|
|
$ |
12.7 |
|
|
$ |
10.3 |
|
|
$ |
7.4 |
|
|
Net revenues |
|
$ |
53.7 |
|
|
$ |
50.1 |
|
|
$ |
38.4 |
|
|
$ |
34.9 |
|
|
$ |
30.2 |
|
|
$ |
21.7 |
|
|
$ |
17.9 |
|
|
$ |
13.1 |
|
59
We began recognizing a portion of our net revenues on an accrual basis in the fourth quarter 2016. In the table above, gross billings reflect the total charges for active patients on therapy without any deductions or adjustments for payer discounts and allowances, patient financial assistance, charitable care or other similar items. The subsequent table line items detail the three sources of net revenue in the applicable reporting period. Revenue recorded on an accrual basis during the year ended December 31, 2017 was $109.0 million, an increase of $100.5 million, or 1182%, compared to the same period in 2016. The increase in revenue recorded on an accrual basis was primarily due to an increase in positive coverage polices and negotiated contracts with health plans in the United States and an increase in the approval rate for German claims, which enabled us to reliably estimate a fixed and determinable amount that would ultimately be collected from these payers.
Revenue recorded on a cash basis during the year ended December 31, 2017 was $68.0 million, a decrease of $6.4 million, or 9%, compared to the same period in 2015. The decrease in revenue recorded on a cash basis reflects the transition of payers from whom we reliably collect a fixed and determinable amount to accrual-based revenue recognition.
Cost of revenues. Our cost of revenues increased by $15.7 million, or 39%, to $55.6 million for the year ended December 31, 2017 from $39.9 million for the year ended December 31, 2016. The increase in cost of revenues was primarily due to the cost of shipping transducer arrays to a higher volume of commercial patients, as well as an increase in field equipment depreciation. Gross margin was 69% for the year ended December 31, 2017 and 44% for the year ended December 31, 2016.
Impairment of field equipment. In 2016, we recorded an impairment loss with respect to the write-off of our first generation Optune System field equipment (finished goods and production stage) in the amount of $6.4 million that is not recoverable.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
Change |
|
|
% Change |
|
||||
|
Research, development and clinical trials |
|
$ |
38,103 |
|
|
$ |
41,467 |
|
|
$ |
(3,364 |
) |
|
|
-8 |
% |
|
Sales and marketing |
|
|
63,528 |
|
|
|
59,449 |
|
|
$ |
4,079 |
|
|
|
7 |
% |
|
General and administrative |
|
|
59,114 |
|
|
|
51,007 |
|
|
$ |
8,107 |
|
|
|
16 |
% |
|
Total operating expenses |
|
$ |
160,745 |
|
|
$ |
151,923 |
|
|
$ |
8,822 |
|
|
|
6 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share-based compensation expense |
|
$ |
26,649 |
|
|
$ |
20,818 |
|
|
$ |
5,831 |
|
|
|
28 |
% |
|
Other non-cash expenses |
|
|
2,390 |
|
|
|
1,949 |
|
|
|
441 |
|
|
|
23 |
% |
|
Total non-cash expenses |
|
$ |
29,039 |
|
|
$ |
22,767 |
|
|
$ |
6,272 |
|
|
|
28 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses, net of non-cash expenses * |
|
$ |
131,706 |
|
|
$ |
129,156 |
|
|
$ |
2,550 |
|
|
|
2 |
% |
* This non-GAAP metric has been included because management believes that it provides for a more accurate year to year comparison of our operating expenses without the impact of non-cash items. This measure allows investors to better understand and evaluate our operating results in the same manner as management, to compare financial results across accounting periods and to better understand the long-term performance of our core business in future periods. In addition, management finds it useful to exclude certain non-cash expenses to assist in budgeting, planning and forecasting future periods. Management discusses this measure with the Audit Committee of our Board of Directors, when appropriate, for the purposes of reviewing our performance and the use of its cash resources.
Research, development and clinical trials expenses. Research, development and clinical trials expenses decreased by $3.4 million, or 8%, to $38.1 million for the year ended December 31, 2017 from $41.5 million for the year ended December 31, 2016. The change is primarily due to a decrease of $1.7 million in clinical trial expenses, resulting from the conclusion of our EF-14 phase 3 pivotal trial partially offset by start-up expenses for our LUNAR and METIS trials, and a $1.8 million decrease in other R&D expenses.
Sales and marketing expenses. Sales and marketing expenses increased by $4.1 million, or 7%, to $63.5 million for the year ended December 31, 2017 from $59.4 million for the year ended December 31, 2016. The increase was driven by an increase in field-based sales costs of $4.2 million to support Optune growth, an increase of $2.0 million in commercial shipping charges and an increase of $1.4 million in other expenses, partially offset by a $3.4 million decrease in marketing expenses related to launch activities.
General and administrative expenses. General and administrative expenses increased by $8.1 million, or 16%, to $59.1 million for the year ended December 31, 2017 from $51.0 million for the year ended December 31, 2016. The increase was primarily due to an increase of $11.8 million in personnel costs (including an increase of $6.7 million in share-based compensation) partially offset by a one-time net charge of $3.6 million in 2016 related to the 2015 Technion settlement.
60
Financial expenses, net. Financial expenses, net increased by $3.0 million, or 49%, to $9.2 million for the year ended December 31, 2017 from $6.1 million for the year ended December 31, 2016. The change is primarily due to additional interest expense, including amortization of the discount and deferred issuance costs, related to our July 2016 withdrawal of the remaining $75 million in available funds under our Term Loan Credit Facility, partially offset by higher interest earned on cash and investment balances.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
Change |
|
|
% Change |
|
||||
|
Income taxes |
|
$ |
13,165 |
|
|
$ |
10,381 |
|
|
$ |
2,784 |
|
|
|
27 |
% |
Income taxes. Income taxes increased by $2.8 million, or 27%, to $13.2 million for the year ended December 31, 2017 from $10.4 million for the year ended December 31, 2016. The change was primarily attributable to an increase in the statutory tax provisions related to an increase in activities in certain jurisdictions such as Switzerland, the United States and Israel.
Year ended December 31, 2016 compared to year ended December 31, 2015
(All dollar figures in tables are in thousands unless otherwise indicated)
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
Change |
|
|
% Change |
|
||||
|
Net revenues |
|
$ |
82,888 |
|
|
$ |
33,087 |
|
|
$ |
49,801 |
|
|
|
151 |
% |
Net revenues. Net revenues increased by $49.8 million, or 151%, to $82.9 million for the year ended December 31, 2016 from $33.1 million for the year ended December 31, 2015. This was primarily due to an increase of 486 active patients, representing 80% growth, in our currently active markets and an increase in the net revenues per active patient driven by an increase in positive coverage policies in the United States.
Cost of revenues. Our cost of revenues increased by $19.3 million, or 93%, to $39.9 million for the year ended December 31, 2016 from $20.6 million for the year ended December 31, 2015. The increase was primarily due to the cost of shipping transducer arrays to a higher volume of commercial patients, as well as an increase in field equipment depreciation.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
Change |
|
|
% Change |
|
||||
|
Research, development and clinical trials |
|
$ |
41,467 |
|
|
$ |
43,748 |
|
|
$ |
(2,281 |
) |
|
|
-5 |
% |
|
Sales and marketing |
|
|
59,449 |
|
|
|
38,861 |
|
|
$ |
20,588 |
|
|
|
53 |
% |
|
General and administrative |
|
|
51,007 |
|
|
|
33,864 |
|
|
$ |
17,143 |
|
|
|
51 |
% |
|
Total operating expenses |
|
$ |
151,923 |
|
|
$ |
116,473 |
|
|
$ |
35,450 |
|
|
|
30 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-cash expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Share-based compensation expense |
|
$ |
20,818 |
|
|
$ |
11,686 |
|
|
$ |
9,132 |
|
|
|
78 |
% |
|
Other non-cash expenses |
|
|
1,949 |
|
|
|
1,398 |
|
|
|
551 |
|
|
|
39 |
% |
|
Total non-cash expenses |
|
$ |
22,767 |
|
|
$ |
13,084 |
|
|
$ |
9,683 |
|
|
|
74 |
% |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total operating expenses, net of non-cash expenses * |
|
$ |
129,156 |
|
|
$ |
103,389 |
|
|
$ |
25,767 |
|
|
|
25 |
% |
(*) This non-GAAP metric has been included because management believes that it provides for a more accurate year to year comparison of our operating expenses without the impact of non-cash items. This measure allows investors to better understand and evaluate our operating results in the same manner as management, to compare financial results across accounting periods and to better understand the long-term performance of our core business in future periods. In addition, management finds it useful to exclude certain non-cash expenses to assist in budgeting, planning and forecasting future periods. Management discusses this measure with the Audit Committee of our Board of Directors, when appropriate, for the purposes of reviewing our performance and the use of its cash resources
Research, development and clinical trials expenses. Research, development and clinical trials expenses decreased by $2.3 million, or 5%, to $41.5 million for the year ended December 31, 2016 from $43.7 million for the year ended December 31, 2015. The change is primarily due to a decrease of $5.7 million in clinical trial expenses resulting from the conclusion of our EF-14 phase 3 pivotal trial in newly diagnosed GBM, as well as a reduction in expenses related to the development of our second generation Optune system, partially offset by an increase of $3.4 million in personnel costs.
61
Sales and marketing expenses. Sales and marketing expenses increased by $20.6 million, or 53%, to $59.4 million for the year ended December 31, 2016 from $38.9 million for the year ended December 31, 2015. The increase was driven by an increase of $13.1 million of personnel costs and an increase of $7.4 million in marketing expenses, reflecting our expanding commercial operations in the United States and Germany and our ongoing efforts to establish commercial operations in Switzerland and Japan.
General and administrative expenses. General and administrative expenses increased by $17.1 million, or 51%, to $51.0 million for the year ended December 31, 2016 from $33.9 million for the year ended December 31, 2015. The increase was primarily due to an increase of $9.3 million in personnel costs, an increase of $3.7 million in professional services to support our enterprise resource planning system implementation and public company-related activities and a one-time net charge of $3.6 million related to the 2015 Technion settlement.
Financial expenses, net. Financial expenses, net increased by $3.0 million, or 95%, to $6.1 million for the year ended December 31, 2016 from $3.1 million for the year ended December 31, 2015. The change is primarily due to interest expense, including amortization expense of the discount and deferred issuance costs, related to our July 2016 withdrawal of the remaining $75 million in available funds under our Term Loan Credit Facility.
|
|
|
Year Ended December 31, |
|
|||||||||||||
|
|
|
2016 |
|
|
2015 |
|
|
Change |
|
|
% Change |
|
||||
|
Income taxes |
|
$ |
10,381 |
|
|
$ |
4,434 |
|
|
$ |
5,947 |
|
|
|
134 |
% |
Income taxes. Income taxes increase by $5.9 million, or 13%, to $10.4 million for the year ended December 31, 2016 from $4.4 million for the year ended December 31, 2015. The change was primarily attributable to an increase in the statutory tax provision related to Switzerland, the United States and Japan.
Liquidity and capital resources
We have incurred significant losses and cumulative negative cash flows from operations since our founding in 2000. As of December 31, 2017, we had an accumulated deficit of $582.3 million. To date, we have primarily financed our operations through the issuance and sale of our convertible preferred shares, our ordinary shares and the proceeds from long-term loans. As of December 31, 2017, we had received a total of $717.2 million from the sale of our convertible preferred shares, including the sale of our Series J convertible preferred stock in 2015 for net proceeds of $94.6 million, all of which were converted into our ordinary shares after the consummation of our IPO, and including $157.5 million proceeds from our IPO including the exercise of the underwriters' overallotment option and the exercise of ordinary share warrants and options.
As of December 31, 2017 and December 31, 2016, we had $183.3 million and $219.6 million, respectively, of cash, cash equivalents, and short-term investments. In 2015, we entered into a Loan and Security Agreement between us, as borrower, and Biopharma Secured Investments III Holdings Cayman LP, as lender (the “Term Loan Credit Facility”) for up to $100.0 million, of which we drew $25.0 million on entering into the facility and the remaining $75.0 million in 2016. In 2015, we raised net proceeds of $94.6 million through the issuance of our Series J convertible preferred shares. Our IPO closed in 2015 and we issued and sold 7,876,195 ordinary shares. We received cash proceeds of $157.5 million from the IPO and the partial exercise of the underwriters’ overallotment option, net of underwriting discounts and commissions and offering expenses.
On February 7, 2018, we prepaid our Term Loan Credit Facility in full, terminated the Term Loan Credit Facility and entered into the 2018 Credit Facility. The Company used a portion of the proceeds from the 2018 Credit Facility to repay in full the Company’s obligations under the Term Loan Credit Facility and will continue to use proceeds to fund general corporate purposes.
We believe our cash, cash equivalents and short-term investments as of December 31, 2017 are sufficient for our operations for at least the next 12 months based on our existing business plan and our ability to control the timing of significant expense commitments. We expect that our research, development and clinical trials expenses, sales and marketing expenses and general and administrative expenses will continue to increase over the next several years and may outpace our gross profits. As a result, we may need to raise additional capital to fund our operations.
62
The following summary of our cash flows for the periods indicated has been derived from our consolidated financial statements, which are included elsewhere in this Annual Report:
|
|
|
Year Ended December 31, |
|
|||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net cash used in operating activities |
|
$ |
(33,134 |
) |
|
$ |
(107,592 |
) |
|
$ |
(99,884 |
) |
|
Net cash provided by (used in) investing activities |
|
|
6,770 |
|
|
|
12,815 |
|
|
|
(115,295 |
) |
|
Net cash provided by financing activities |
|
|
5,168 |
|
|
|
75,124 |
|
|
|
276,989 |
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
|
8 |
|
|
|
10 |
|
|
|
- |
|
|
Net increase (decrease) in cash and cash equivalents |
|
$ |
(21,188 |
) |
|
$ |
(19,643 |
) |
|
$ |
61,810 |
|
Operating activities
Net cash used in operating activities primarily represents our net loss for the periods presented. Adjustments to net loss for non-cash items include share-based compensation, depreciation, asset write-downs and accrued interest. Operating cash flows are also impacted by changes in operating assets and liabilities, principally trade receivables, inventories, prepaid expenses, trade payables, other payables and accrued expenses.
Net cash used in operating activities was $33.1 million for the year ended December 31, 2017, as compared to $107.6 million for the year ended December 31, 2016, reflecting a net loss of $61.7 million, and an increase of $6.7 million in our net operating assets and liabilities offset by non-cash charges of $35.3 million. The change in our net operating assets and liabilities was primarily the result of an increase in our trade receivables of $23.3 million and a decrease in trade payables and other long-term liabilities of $3.8 million, partially offset by an increase in other payables of $14.4 million, a decrease in inventories of $3.5 million and a decrease in receivables and prepaid expenses of $1.8 million. Non-cash charges included $27.1 million of share-based compensation and $7.7 million of depreciation.
Net cash used in operating activities was $107.6 million for the year ended December 31, 2016, as compared to $99.9 million for the year ended December 31, 2015, reflecting a net loss of $131.8 million, and an increase of $9.4 million in our net operating assets and liabilities offset by non-cash charges of $33.7 million. The change in our net operating assets and liabilities was primarily the result of an increase in our inventories of $12.0 million necessary to meet anticipated demand, an increase in trade receivables of $6.3 million, offset by an increase in trade payables of $1.6 million and other payables of $6.6 million. Non-cash charges included $21.4 million (net of excess tax benefit from share-based award activity) of share-based compensation, $6.4 million in impairment of field equipment and $5.7 million of depreciation and amortization.
Investing activities
Our investing activities consist primarily of capital expenditures to purchase property and equipment and field equipment, as well as investments in and redemptions of our short-term investments.
Net cash provided by investing activities was $6.8 million in the year ended December 31, 2017 attributable to our receipt of $120.0 million from the maturity of short-term investments, offset by the purchase of new short-term investments of $104.0 million, purchases of $5.0 million of field equipment, purchases of $2.5 million of property and equipment and an increase in restricted cash of $1.9 million.
Net cash provided by investing activities was $12.8 million in the year ended December 31, 2016 attributable to our receipt of $270.0 million from the maturity of short-term investments, offset by the purchase of new short-term investments of $239.3 million, purchases of $5.7 million of property and equipment and purchases of $12.0 million of field equipment, and an increase in restricted cash of $0.2 million.
Financing activities
To date, our primary financing activities have been the sale of our convertible preferred shares, our IPO and the proceeds from long-term loans.
Net cash provided by financing activities was $5.2 million for the year ended December 31, 2017, attributable to proceeds of $3.7 million from the exercise of options and warrants and $1.5 million net proceeds from the issuance of shares under our ESPP.
63
Net cash provided by financing activities was $75.1 million for the year ended December 31, 2016, attributable to borrowings under our Term Loan Credit Facility of $72.9 million, proceeds of $1.0 million from the exercise of options and warrants and $0.6 million net proceeds from the issuance of shares under our ESPP.
Term Loan Credit Facility
As of December 31, 2017, our material outstanding indebtedness consisted of our Term Loan Credit Facility, which provided for up to $100.0 million of borrowings in up to four draws, the first of which was made in January 2015 in the amount of $25.0 million and the second of which was made in July 2016 in the amount of $75.0 million. Interest on the outstanding loan was 10% annually, payable quarterly in arrears. As of December 31, 2017, the aggregate principal balance of amounts outstanding under the Term Loan Credit Facility was $100.0 million. We had the option to prepay the balance under the Term Loan Credit Facility, in whole, at any time, and we were required to prepay in the event of a change of control, in each case, subject to a pay-down fee, prepayment premium and/or make-whole payment. The funding fee payable on the amount drawn on the funding date was 1.5%, the pay-down fee on all principal payments to be paid on the date such payments are made was 0.75% and the pre-payment fee if we prepaid outstanding loan amounts prior to the first, second or third year from the initial funding date was 3.0%, 2.0% or 1.0%, respectively.
All obligations under the Term Loan Credit Facility were guaranteed by certain of our domestic direct and indirect subsidiaries. In addition, the obligations under the Term Loan Credit Facility were secured by a first-priority security interest in substantially all of the property and assets of, as well as the equity interests owned by, us and the other guarantors.
The Term Loan Credit Facility had a minimum liquidity covenant, which was tested quarterly. In addition, we were required to meet certain pro forma net sales requirements. The Term Loan Credit Facility contained other customary covenants. As of December 31, 2017, we were in compliance with such covenants.
On February 7, 2018, we prepaid our Term Loan Credit Facility in full and entered into a new term loan credit facility with BioPharma Credit PLC. For additional information, see Note 20 to the Consolidated Financial Statements.
Contractual obligations and commitments
The following summarizes our significant contractual obligations and commitments as of December 31, 2017:
|
|
|
December 31, |
|
|||||||||||||||||||||||||
|
Contractual Obligations: |
|
2018 |
|
|
2019 |
|
|
2020 |
|
|
2021 |
|
|
2022 |
|
|
After |
|
|
Total |
|
|||||||
|
|
|
(in thousands) |
|
|||||||||||||||||||||||||
|
Operating leases and other loans |
|
$ |
3,544 |
|
|
$ |
2,559 |
|
|
$ |
2,078 |
|
|
$ |
1,636 |
|
|
$ |
1,404 |
|
|
$ |
1,368 |
|
|
$ |
12,589 |
|
|
Term loan credit facility (1)(2) |
|
|
- |
|
|
|
- |
|
|
|
100,750 |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
100,750 |
|
|
Term loan credit facility interest (1)(2) |
|
|
10,000 |
|
|
|
10,000 |
|
|
|
833 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
20,833 |
|
|
Technion settlement (3) |
|
$ |
5,500 |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
5,500 |
|
__________________________
|
|
(1) |
The Term Loan Credit Facility has a fixed per annum interest rate of 10.0%. On February 7, 2018, the Term Loan Credit Facility was terminated upon the Company’s repayment in full of the term loan issued thereunder. For additional information, see Note 20 to the Consolidated Financial Statements. |
|
|
(2) |
On February 7, 2018, the Company and certain of its subsidiaries entered into a Loan and Security Agreement (“2018 Loan Agreement”) with a new lender pursuant to which such lender made a term loan to the Company in the principal amount of $150 million (the “2018 Credit Facility”). The term loan has a fixed per annum interest rate of 9.0%. For additional information, see Note 20 to the Consolidated Financial Statements. The following summarizes our significant contractual obligations with regards to this term loan as of the date hereof: |
|
|
|
December 31, |
|
|||||||||||||||||||||||||
|
Contractual Obligations: |
|
2018 |
|
|
2019 |
|
|
2020 |
|
|
2021 |
|
|
2022 |
|
|
After |
|
|
Total |
|
|||||||
|
|
|
(in thousands) |
|
|||||||||||||||||||||||||
|
2018 Credit Facility |
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
- |
|
|
$ |
150,000 |
|
|
$ |
150,000 |
|
|
2018 Credit Facility interest |
|
$ |
12,263 |
|
|
$ |
13,500 |
|
|
$ |
13,500 |
|
|
$ |
13,500 |
|
|
$ |
13,500 |
|
|
$ |
1,388 |
|
|
$ |
67,651 |
|
64
|
|
the Settlement Agreement, and in exchange for a release of potential disputes from the third party, the Company is obligated to pay the Milestone Payment to Technion in the quarter following the quarter in which the Company achieves the Net Sales Milestone. The Company achieved the Net Sales Milestone in the fourth quarter of 2017. Accordingly, in the first quarter of 2018, the Company anticipates making the Milestone Payment to Technion, which is included in other payables and accrued expenses as of December 31, 2017 and 2016. |
Except as described above, there were no material changes in our commitments under contractual obligations in the year ended December 31, 2017.
The total amount of unrecognized tax expenses for uncertain tax positions was $2.8 million and $2.4 million at December 31, 2017 and December 31, 2016, respectively. Payment of these obligations would result from settlements with taxing authorities. Due to the difficulty in determining the timing of settlements, these obligations are not included in the above table. We do not expect a significant tax payment related to these obligations within the next year.
We also have employment agreements with certain employees that require the funding of a specific level of payments if certain events, such as a change in control or termination without cause, occur. In the course of normal business operations, we also have agreements with contract service providers to assist in the performance of our research and development (including clinical trials) and manufacturing activities. We could also enter into additional collaborative research, contract research, manufacturing and supplier agreements in the future, which may require up-front payments and even long-term commitments of cash.
Off-balance sheet arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements as defined under SEC rules.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are exposed to market risks in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is primarily a result of fluctuations in interest rates and foreign currency exchange rates. We do not hold or issue financial instruments for trading purposes. There were no material quantitative changes in our market risk exposures between the year ended December 31, 2017 and the year ended December 31, 2016.
Interest rate sensitivity
Our exposure to market risk for changes in interest rates relates primarily to our investment portfolio. Our cash, cash equivalents and short-term investment accounts as of December 31, 2017 totaled $183.3 million and consist of cash, cash equivalents and short-term investments with maturities of less than one year from the date of purchase. Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of the interest rates in the United States. However, because of the short-term nature of the instruments in our portfolio and our intent to hold instruments to maturity, a 10% change in market interest rates would not be expected to have a material impact on our financial condition or our results of operations.
Foreign currency exchange risk
Our consolidated results of operations and cash flow are subject to fluctuations due to changes in foreign currency exchange rates. All of our revenues are generated in the local currency for commercial markets. Our expenses are generally denominated in the currencies in which our operations are located, which is primarily in the United States, Switzerland, Germany, Israel and Japan. Our consolidated results of operations and cash flows are, therefore, subject to fluctuations due to changes in foreign currency exchange rates and may be adversely affected in the future due to changes in foreign exchange rates. The effect of a hypothetical 10% change in foreign currency exchange rates applicable to our business would not have a material impact on our historical consolidated financial statements. We do not hedge our foreign currency exchange risk.
65
NovoCure Limited
Index to consolidated financial statements
66
Report of independent registered public accounting firm
To the board of directors and shareholders of
NovoCure Limited
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of NovoCure Limited and its subsidiaries (the “Company”) as of December 31, 2017 and 2016 and the related consolidated statements of operations, statements of comprehensive loss, changes in shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2017 and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the consolidated financial position of the Company at December 31, 2017 and 2016, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2017, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 22, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
KOST FORER GABBAY & KASIERER
A Member of Ernst & Young Global
We have served as the Company‘s auditor since 2003.
Tel-Aviv, Israel
February 22, 2018
67
Report of independent registered public accounting firm
To the board of directors and shareholders of
NovoCure Limited
Opinion on Internal Control over Financial Reporting
We have audited NovoCure Limited and its subsidiaries (the "Company") internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the "COSO Criteria"). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2017 and 2016, the related consolidated statements of operations, statements of comprehensive loss, changes in shareholders’ equity and cash flows for each of the three years in the period ended December 31, 2017 and the related notes, and our report dated February 22, 2018 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management's Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
KOST FORER GABBAY & KASIERER
A Member of Ernst & Young Global
Tel-Aviv, Israel
February 22, 2018
68
NovoCure Limited and subsidiaries
Consolidated balance sheets
|
|
December 31, |
|
||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
||
|
Assets |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
78,592 |
|
|
$ |
99,780 |
|
|
Short-term investments |
|
|
104,719 |
|
|
|
119,854 |
|
|
Restricted cash |
|
|
2,126 |
|
|
|
267 |
|
|
Trade receivables, net |
|
|
29,567 |
|
|
|
6,339 |
|
|
Receivables and prepaid expenses |
|
|
8,105 |
|
|
|
10,084 |
|
|
Inventories |
|
|
22,025 |
|
|
|
25,549 |
|
|
Total current assets |
|
|
245,134 |
|
|
|
261,873 |
|
|
Long-term assets: |
|
|
|
|
|
|
|
|
|
Property and equipment, net |
|
|
9,031 |
|
|
|
9,812 |
|
|
Field equipment, net |
|
|
9,036 |
|
|
|
8,808 |
|
|
Severance pay fund |
|
|
111 |
|
|
|
88 |
|
|
Other long-term assets |
|
|
1,986 |
|
|
|
1,500 |
|
|
Total long-term assets |
|
|
20,164 |
|
|
|
20,208 |
|
|
Total assets |
|
$ |
265,298 |
|
|
$ |
282,081 |
|
The accompanying notes are an integral part of the consolidated financial statements.
69
NovoCure Limited and subsidiaries
|
|
December 31, |
|
||||||
|
U.S. dollars in thousands, except share and per share data |
|
2017 |
|
|
2016 |
|
||
|
Liabilities and shareholders’ equity |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Trade payables |
|
$ |
17,206 |
|
|
$ |
18,356 |
|
|
Other payables and accrued expenses |
|
|
32,996 |
|
|
|
18,526 |
|
|
Total current liabilities |
|
|
50,202 |
|
|
|
36,882 |
|
|
Long-term liabilities: |
|
|
|
|
|
|
|
|
|
Long-term loan, net of discount and issuance costs |
|
|
97,342 |
|
|
|
96,231 |
|
|
Employee benefit liabilities |
|
|
2,453 |
|
|
|
2,590 |
|
|
Other long-term liabilities |
|
|
1,737 |
|
|
|
4,033 |
|
|
Total long-term liabilities |
|
|
101,532 |
|
|
|
102,854 |
|
|
Total liabilities |
|
|
151,734 |
|
|
|
139,736 |
|
|
Commitments and contingencies |
|
|
|
|
|
|
|
|
|
Shareholders’ equity: |
|
|
|
|
|
|
|
|
|
Share capital - |
|
|
|
|
|
|
|
|
|
Ordinary shares - No par value, Unlimited shares authorized; Issued and outstanding: 89,478,032 shares and 87,066,446 shares at December 31, 2017 and December 31, 2016 respectively; |
|
|
- |
|
|
|
- |
|
|
Additional paid-in capital |
|
|
697,165 |
|
|
|
664,154 |
|
|
Accumulated other comprehensive loss |
|
|
(1,343 |
) |
|
|
(1,883 |
) |
|
Accumulated deficit |
|
|
(582,258 |
) |
|
|
(519,926 |
) |
|
Total shareholders’ equity |
|
|
113,564 |
|
|
|
142,345 |
|
|
Total liabilities and shareholders’ equity |
|
$ |
265,298 |
|
|
$ |
282,081 |
|
The accompanying notes are an integral part of the consolidated financial statements.
70
NovoCure Limited and subsidiaries
Consolidated statements of operations
|
|
Year ended December 31, |
|
||||||||||
|
U.S. dollars in thousands, except share and per share data |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net revenues |
|
$ |
177,026 |
|
|
$ |
82,888 |
|
|
$ |
33,087 |
|
|
Cost of revenues |
|
|
55,609 |
|
|
|
39,870 |
|
|
|
20,610 |
|
|
Impairment of field equipment |
|
|
- |
|
|
|
6,412 |
|
|
|
- |
|
|
Gross profit |
|
|
121,417 |
|
|
|
36,606 |
|
|
|
12,477 |
|
|
Operating costs and expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research, development and clinical trials |
|
|
38,103 |
|
|
|
41,467 |
|
|
|
43,748 |
|
|
Sales and marketing |
|
|
63,528 |
|
|
|
59,449 |
|
|
|
38,861 |
|
|
General and administrative |
|
|
59,114 |
|
|
|
51,007 |
|
|
|
33,864 |
|
|
Total operating costs and expenses |
|
|
160,745 |
|
|
|
151,923 |
|
|
|
116,473 |
|
|
Operating loss |
|
|
(39,328 |
) |
|
|
(115,317 |
) |
|
|
(103,996 |
) |
|
Financial expenses, net |
|
|
(9,169 |
) |
|
|
(6,147 |
) |
|
|
(3,151 |
) |
|
Loss before income taxes |
|
|
(48,497 |
) |
|
|
(121,464 |
) |
|
|
(107,147 |
) |
|
Income taxes |
|
|
13,165 |
|
|
|
10,381 |
|
|
|
4,434 |
|
|
Net loss |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted net loss per ordinary share |
|
$ |
(0.70 |
) |
|
$ |
(1.54 |
) |
|
$ |
(3.67 |
) |
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
88,546,719 |
|
|
|
85,558,448 |
|
|
|
30,401,603 |
|
The accompanying notes are an integral part of the consolidated financial statements.
71
NovoCure Limited and subsidiaries
Consolidated statements of comprehensive loss
|
|
Year ended December 31, |
|
||||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net loss |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
Other comprehensive loss, net of tax : |
|
|
|
|
|
|
|
|
|
|
|
|
|
Change in foreign currency translation adjustments |
|
8 |
|
|
|
10 |
|
|
|
- |
|
|
|
Pension benefit plan |
|
532 |
|
|
|
(388 |
) |
|
|
(1,505 |
) |
|
|
Total comprehensive loss |
|
$ |
(61,122 |
) |
|
$ |
(132,223 |
) |
|
$ |
(113,086 |
) |
The accompanying notes are an integral part of the consolidated financial statements.
72
NovoCure Limited and subsidiaries
Statements of changes in shareholders’ equity
|
|
Ordinary shares |
|
|
Preferred shares |
|
|
Additional capital paid-in |
|
|
Accumulated other comprehensive loss |
|
|
Accumulated deficit |
|
|
Total shareholders’ equity |
|
|||||||
|
U.S. dollars in thousands, except share data |
|
(Shares) |
|
|
(Shares) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
Balance as of December 31, 2014 |
|
|
13,431,414 |
|
|
|
58,676,017 |
|
|
|
374,375 |
|
|
|
- |
|
|
|
(276,500 |
) |
|
|
97,875 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
- |
|
|
|
11,860 |
|
|
|
- |
|
|
|
- |
|
|
|
11,860 |
|
|
Exercise of options and warrants |
|
|
731,665 |
|
|
|
- |
|
|
|
2,038 |
|
|
|
- |
|
|
|
- |
|
|
|
2,038 |
|
|
Issuance of Series J preferred shares, net (a) |
|
|
- |
|
|
|
4,068,500 |
|
|
|
94,599 |
|
|
|
- |
|
|
|
- |
|
|
|
94,599 |
|
|
Issuance of shares and options in respect of settlement, net of fair value of shares provided as indemnification (Note 14c) |
|
|
(1,005,210 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Issuance of ordinary shares upon IPO and exercise of over-allotment, net (b) |
|
|
7,876,195 |
|
|
|
- |
|
|
|
157,534 |
|
|
|
- |
|
|
|
- |
|
|
|
157,534 |
|
|
Conversion of preferred shares to ordinary shares |
|
|
62,744,517 |
|
|
|
(62,744,517 |
) |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
Other comprehensive loss, net of tax benefit of $165 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(1,505 |
) |
|
|
- |
|
|
|
(1,505 |
) |
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(111,581 |
) |
|
|
(111,581 |
) |
|
Balance as of December 31, 2015 |
|
|
83,778,581 |
|
|
|
- |
|
|
|
640,406 |
|
|
|
(1,505 |
) |
|
|
(388,081 |
) |
|
|
250,820 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
- |
|
|
|
21,441 |
|
|
|
- |
|
|
|
- |
|
|
|
21,441 |
|
|
Exercise of options and warrants |
|
|
3,195,477 |
|
|
|
- |
|
|
|
993 |
|
|
|
- |
|
|
|
- |
|
|
|
993 |
|
|
Issuance of shares in connection with employee stock purchase plan |
|
|
92,388 |
|
|
|
- |
|
|
|
616 |
|
|
|
- |
|
|
|
- |
|
|
|
616 |
|
|
Tax benefit from share-based award activity |
|
|
- |
|
|
|
- |
|
|
|
698 |
|
|
|
- |
|
|
|
- |
|
|
|
698 |
|
|
Other comprehensive loss, net of tax benefit of $38 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(378 |
) |
|
|
- |
|
|
|
(378 |
) |
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(131,845 |
) |
|
|
(131,845 |
) |
|
Balance as of December 31, 2016 |
|
|
87,066,446 |
|
|
|
- |
|
|
|
664,154 |
|
|
|
(1,883 |
) |
|
|
(519,926 |
) |
|
|
142,345 |
|
|
Share-based compensation to employees |
|
|
- |
|
|
|
- |
|
|
|
27,116 |
|
|
|
- |
|
|
|
- |
|
|
|
27,116 |
|
|
Exercise of options and warrants |
|
|
2,244,153 |
|
|
|
- |
|
|
|
3,685 |
|
|
|
- |
|
|
|
- |
|
|
|
3,685 |
|
|
Issuance of shares in connection with employee stock purchase plan |
|
|
167,433 |
|
|
|
- |
|
|
|
1,540 |
|
|
|
- |
|
|
|
- |
|
|
|
1,540 |
|
|
Cumulative effect adjustment resulting from ASU 2016-09 adoption (See Note 2) |
|
|
- |
|
|
|
- |
|
|
|
670 |
|
|
|
- |
|
|
|
(670 |
) |
|
|
- |
|
|
Other comprehensive loss, net of tax expense of $68 |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
540 |
|
|
|
- |
|
|
|
540 |
|
|
Net loss |
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
- |
|
|
|
(61,662 |
) |
|
|
(61,662 |
) |
|
Balance as of December 31, 2017 |
|
|
89,478,032 |
|
|
|
- |
|
|
$ |
697,165 |
|
|
$ |
(1,343 |
) |
|
$ |
(582,258 |
) |
|
$ |
113,564 |
|
|
(a) |
Net of issuance expenses of $319 |
|
(b) |
Net of issuance expenses (including underwriter fees) of $15,742 |
The accompanying notes are an integral part of the consolidated financial statements.
73
NovoCure Limited and subsidiaries
Consolidated statements of cash flows
|
|
Year ended December 31, |
|
||||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
7,677 |
|
|
|
5,652 |
|
|
|
3,153 |
|
|
Asset write-downs and impairment of field equipment |
|
|
241 |
|
|
|
6,446 |
|
|
|
46 |
|
|
Increase in accrued interest expense |
|
|
- |
|
|
|
- |
|
|
|
672 |
|
|
Share-based compensation to employees |
|
|
27,116 |
|
|
|
22,139 |
|
|
|
11,860 |
|
|
Excess tax benefits from share-based award activity |
|
|
- |
|
|
|
(698 |
) |
|
|
- |
|
|
Increase in trade receivables, net |
|
|
(23,228 |
) |
|
|
(6,339 |
) |
|
|
- |
|
|
Amortization of discount, net |
|
|
252 |
|
|
|
155 |
|
|
|
329 |
|
|
Decrease (increase) in receivables and prepaid expenses |
|
|
1,979 |
|
|
|
243 |
|
|
|
(5,088 |
) |
|
Decrease (increase) in inventories |
|
|
3,524 |
|
|
|
(11,955 |
) |
|
|
(10,148 |
) |
|
Increase in other long-term assets |
|
|
(554 |
) |
|
|
(692 |
) |
|
|
(381 |
) |
|
Increase (decrease) in trade payables |
|
|
(1,150 |
) |
|
|
1,601 |
|
|
|
6,961 |
|
|
Increase in other payables and accrued expenses |
|
|
14,460 |
|
|
|
6,647 |
|
|
|
3,579 |
|
|
Increase in employee benefit liabilities, net |
|
|
440 |
|
|
|
97 |
|
|
|
133 |
|
|
Increase (decrease) in other long-term liabilities |
|
|
(2,229 |
) |
|
|
957 |
|
|
|
581 |
|
|
Net cash used in operating activities |
|
$ |
(33,134 |
) |
|
$ |
(107,592 |
) |
|
$ |
(99,884 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchase of property and equipment |
|
|
(2,459 |
) |
|
|
(5,674 |
) |
|
|
(4,667 |
) |
|
Purchase of field equipment |
|
|
(4,907 |
) |
|
|
(11,990 |
) |
|
|
(5,604 |
) |
|
Increase in restricted cash |
|
|
(1,858 |
) |
|
|
(180 |
) |
|
|
(26 |
) |
|
Proceeds from maturity of short-term investments |
|
|
120,000 |
|
|
|
270,000 |
|
|
|
104,000 |
|
|
Purchase of short-term investments |
|
|
(104,006 |
) |
|
|
(239,341 |
) |
|
|
(208,998 |
) |
|
Net cash provided by (used in) investing activities |
|
$ |
6,770 |
|
|
$ |
12,815 |
|
|
$ |
(115,295 |
) |
The accompanying notes are an integral part of the consolidated financial statements.
74
NovoCure Limited and subsidiaries
Consolidated statements of cash flows
|
|
Year ended December 31, |
|
||||||||||
|
U.S. dollars in thousands |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from issuance of shares, net |
|
$ |
1,540 |
|
|
$ |
616 |
|
|
$ |
252,133 |
|
|
Proceeds from long-term loan, net |
|
|
- |
|
|
|
72,887 |
|
|
|
22,886 |
|
|
Excess tax benefits from share-based award activity |
|
|
- |
|
|
|
698 |
|
|
|
- |
|
|
Proceeds from issuance of other long-term loans |
|
|
19 |
|
|
|
- |
|
|
|
- |
|
|
Repayment of other long-term loans |
|
|
(76 |
) |
|
|
(70 |
) |
|
|
(63 |
) |
|
Exercise of options and warrants |
|
|
3,685 |
|
|
|
993 |
|
|
|
2,038 |
|
|
Purchase of shares in respect of settlement |
|
|
- |
|
|
|
- |
|
|
|
(5 |
) |
|
Net cash provided by financing activities |
|
$ |
5,168 |
|
|
$ |
75,124 |
|
|
$ |
276,989 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Effect of exchange rate changes on cash and cash equivalents |
|
$ |
8 |
|
|
$ |
10 |
|
|
$ |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Increase (decrease) in cash and cash equivalents |
|
$ |
(21,188 |
) |
|
|
(19,643 |
) |
|
|
61,810 |
|
|
Cash and cash equivalents at the beginning of the year |
|
$ |
99,780 |
|
|
|
119,423 |
|
|
|
57,613 |
|
|
Cash and cash equivalents at the end of the year |
|
$ |
78,592 |
|
|
$ |
99,780 |
|
|
$ |
119,423 |
|
|
Supplemental cash flow activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid during the year for: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Income taxes |
|
$ |
10,286 |
|
|
$ |
9,447 |
|
|
$ |
1,489 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest |
|
$ |
10,162 |
|
|
$ |
6,595 |
|
|
$ |
1,688 |
|
The accompanying notes are an integral part of the consolidated financial statements.
75
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 1: Organization and basis of presentation
NovoCure Limited (including its consolidated subsidiaries, the “Company”) was incorporated in the Bailiwick of Jersey and is principally engaged in the development, manufacture and commercialization of tumor treating fields (“Tumor Treating Fields”) for the treatment of solid tumors. The Company has regulatory approvals and clearances in certain countries for Optune, its first Tumor Treating Fields delivery system, to treat adult patients with glioblastoma (“GBM”).
In September 2015, the Company’s shareholders approved the restructuring of the Company’s share capital by converting the Company’s ordinary and preferred shares to no par value shares and by effecting a sub division of the issued and outstanding share capital of the Company based on a proportion of 1: 5.913 (“Share Split Ratio”), such that each ordinary and preferred share nominal value of £0.01 of the Company, was divided into 5.913 shares of such applicable class of shares of the Company each with no par value. It was also resolved to apply the Split Ratio to the Company’s outstanding options and warrants, in accordance with their terms. All share and per share information included in these consolidated financial statements has been retroactively adjusted to reflect the conversion to no par value shares and the Share Split Ratio.
Note 2: Significant accounting policies
The consolidated financial statements are prepared according to United States generally accepted accounting principles (“U.S. GAAP”).
a. Use of estimates:
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. The Company evaluates on an ongoing basis its assumptions, including those related to contingencies, deferred taxes, tax liabilities, useful-life of field equipment, revenue recognition and the estimations required in accrual base accounting, and share-based compensation costs. The Company’s management believes that the estimates, judgment and assumptions used are reasonable based upon information available at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities at the dates of the consolidated financial statements, and the reported amounts of net revenue and expenses during the reporting period. Actual results could differ from those estimates.
b. Financial statements in U.S. dollars:
The accompanying financial statements have been prepared in U.S. dollars in thousands, except for share and per-share data.
The Company finances its operations in U.S. dollars and a substantial portion of its costs and revenues from its primary markets is incurred in U.S. dollars. As such, the Company’s management believes that the U.S. dollar is the currency of the primary economic environment in which NovoCure Limited and certain subsidiaries operate. The Company’s reporting currency is U.S. dollars.
Transactions and balances denominated in U.S. dollars are presented at their original amounts. Monetary accounts maintained in currencies other than the U.S. dollar are re-measured into dollars in accordance with Accounting Standards Codification (ASC) No. 830-10, “Foreign Currency Matters.” All transaction gains and losses of the re-measurement of monetary balance sheet items are reflected in the consolidated statements of operations as financial income or expenses, as appropriate.
For a subsidiary whose functional currency has been determined to be its local currency, assets and liabilities are translated at year-end exchange rates and statement of operations items are translated at average exchange rates prevailing during the year. Such translation adjustments are recorded as a separate component of accumulated other comprehensive income (loss) in shareholders' equity.
c. Principles of consolidation:
The consolidated financial statements include the accounts of the Company and its wholly owned subsidiaries. Intercompany transactions and balances, including profits from intercompany sales not yet realized outside the Company, have been eliminated upon consolidation.
76
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Cash equivalents are short-term, highly liquid investments that are readily convertible into cash with an original maturity of three months or less at the date acquired.
e. Short-term investments and restricted cash:
1. Short-term investments:
The Company accounts for investments in debt securities in accordance with ASC 320, “Investments-Debt and Equity Securities.” Management determines the appropriate classification of its investments in marketable debt securities at the time of purchase and reevaluates such determinations at each balance sheet date. For the years ended December 31, 2017 and 2016, all securities are classified as held-to-maturity since the Company has the intent and ability to hold the securities to maturity and, accordingly, debt securities are stated at amortized cost.
The amortized cost of held-to-maturity securities is adjusted for amortization of premiums and accretion of discounts to maturity and any other than temporary impairment losses. Such amortization and interest are included in the consolidated statement of operations as financial income or expenses, as appropriate.
For the three years ended December 31, 2017, no impairment losses have been identified.
2. Restricted cash:
The Company has restricted cash used as security for the use of Company credit cards, presented in short-term assets. Additionally, the Company has pledged bank deposits to cover bank guarantees related to facility rental agreements, fleet lease agreements and customs payments presented in other long-term assets (see Note 12).
f. Trade receivables:
Revenues from the use of Optune are recorded on an accrual basis for payers that meet the revenue recognition criteria for accrual basis where an agreement exists and collectability is reasonably assured. Trade receivables are presented net of allowances and allowance for doubtful accounts of $3,453 and $0, as of December 31, 2017 and 2016, respectively. In order to provide for trade receivables that could become uncollectible in the future, the Company establishes an allowance for doubtful accounts to reduce the carrying value of such receivables to their estimated net realizable value. The Company considers receivables past due based on payment terms and historical cash collection experience. The Company evaluates such reserves on a regular basis and adjusts its reserves as needed. Once a receivable is deemed uncollectible, such balance is charged against the reserve. As of December 31, 2017 and 2016, the allowance for doubtful accounts was de minimis.
Trade receivables include unbilled receivables for therapy provided and not invoiced in the reported period.
g. Inventories:
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the weighted average method. The Company regularly evaluates its ability to realize the value of inventory. If the inventories are deemed damaged, if actual demand for the Company’s delivery systems deteriorates, or if market conditions are less favorable than those projected, inventory write-offs may be required.
Inventory write-offs of $489, $ 774 and $0, respectively, were identified for the years ended December 31, 2017, 2016 and 2015.
77
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful lives of the assets at the following rates:
|
|
|
% |
|
Computers and laboratory equipment |
|
15 - 33 |
|
Office furniture |
|
6 - 33 |
|
Production equipment |
|
20 |
|
Leasehold improvements |
|
Over the shorter of the term of the lease or its useful life |
i. Field equipment:
Field equipment is stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the estimated useful life of the field equipment which was determined to be 18 to 36 months. Field equipment is equipment being utilized under service agreements, and accounted for in accordance with ASC 840 on a monthly basis as an operating lease. The Company records a write-off provision for any excess, lost or damaged equipment when warranted based on an assessment of the equipment. Write-offs for equipment are included in cost of revenues. During the years ended December 31, 2017, 2016 and 2015, write-offs for $195, $ 6,436 and $36, respectively, were recorded (see Note 7).
j. Impairment of long-lived assets:
The Company’s long-lived assets are reviewed for impairment in accordance with ASC 360-10, “Property, Plant and Equipment,” whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. Recoverability of an asset to be held and used is measured by a comparison of the carrying amount of an asset to the future undiscounted cash flows expected to be generated by the asset. If such asset is considered to be impaired, the impairment to be recognized is measured by the amount by which the carrying amount of the asset exceeds its fair value. During the three years ended December 31, 2017, no impairment losses have been identified other than the impairment of field equipment described below in Note 7.
k. Other long-term assets:
Long term lease deposits in respect of office rent and vehicles under operating leases and restricted deposits are presented in other long-term assets.
l. Revenue recognition:
The Tumor Treating Fields delivery system for GBM, Optune, is comprised of two main components: (1) an electric field generator (the “device”) and (2) transducer arrays and related accessories that are disposable supplies to the device (“disposables”). Title is retained by the Company for the device and the patient is provided replacement disposables and technical support for the device during the rental period. The device and disposables are always supplied and functioning together and are not sold on a standalone basis.
Revenues are recognized when persuasive evidence of an arrangement exists, delivery of Optune has occurred, the fee is fixed or determinable and collectability is reasonably assured. The evidence of an arrangement generally consists of a prescription, a patient service agreement and the verification of eligibility and insurance with the patient’s third-party insurance company (“Payer”). The Company assesses whether the fee is fixed or determinable based on whether there is sufficient history with Payers to reliably estimate their individual payment patterns or contractual arrangements exist and whether it can reliably estimate the amount that would be ultimately collected. Once the Company can reliably estimate the amounts that would be ultimately collected per Payer and the above criteria are met, the Company recognizes revenues net of allowances from the use of Optune on an accrual basis ratably over the lease term. The allowances are determined based on defined payment terms and historical collection data by Payer. Allowance adjustments related to final settlements for the reported periods are insignificant. Revenues are recognized when cash is collected when the revenue criteria above are not met, such as when the price is not fixed or determinable or the collectability cannot be reasonably assured. Patients have out-of-pocket costs for the amount not covered by their Payer and the Company bills the patient directly for the amounts of their co-pays and deductible, subject to the Company’s patient assistance programs. The Company currently recognizes revenue from patients at the time cash is collected.
78
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Deferred revenues include amounts invoiced for days of therapy to be provided in future periods.
Unbilled revenues include revenues recognized for therapy provided and not invoiced in the reported period, and are presented as part of accounts receivable.
Revenues are presented net of indirect taxes of $1,293, $ 972 and $ 2,275 for the years ended December 31, 2017, 2016 and 2015, respectively.
m. Charitable care:
The Company provides Optune treatment at no charge to patients who meet certain criteria under its charitable care policy. Because the Company does not pursue collection of amounts determined to qualify as charity, they are not reported as revenue. The Company's costs of care provided under charitable care were $1,483, $1,675 and $ 1,376 for the years ended December 31, 2017, 2016 and 2015, respectively. These amounts were determined by applying charitable care as a percentage of total billings to total cost of goods sold.
n. Shipping and handling costs:
The Company does not bill its customers for shipping and handling costs associated with shipping Optune to its customers. These direct shipping and handling costs of $5,322, $3,389 and $ 1,385 for the years ended December 31, 2017, 2016 and 2015, respectively are included in selling and marketing costs.
o. Accounting for share-based payments:
The Company accounts for share-based compensation in accordance with ASC 718, “Compensation—Stock Compensation.” ASC 718 requires companies to estimate the fair value of equity-based payment awards on the date of grant using an option-pricing model. The value of the portion of the award that is ultimately expected to vest is recognized as an expense over the requisite service periods in the Company’s consolidated statements of operations.
The Company recognizes compensation costs for the value of awards granted using the accelerated method over the requisite service period of the award, which is generally the option vesting term of four years.
The Company selected the Black-Scholes model as the most appropriate fair value method for all equity awards and the Employee Share Purchase Plan (the “ESPP”). For market condition awards, the Company also applied the Monte-Carlo simulation model. The Black-Scholes model requires a number of assumptions, of which the most significant are the share price, expected volatility and the expected equity award term.
Prior to NovoCure Limited’s initial public offering (“IPO”), the fair value of ordinary shares underlying the options was historically determined by management and the board of directors. Because there was no public market for the Company’s ordinary shares, the board of directors determined fair value of an ordinary share at the time of grant of the option by considering a number of objective and subjective factors including operating and financial performance, the lack of liquidity of share capital, general and industry specific economic outlook and valuations performed amongst other factors. For the period from January 1, 2015 through the IPO, the Company’s board of directors determined the fair value of ordinary shares for the reported periods, among other factors, based on valuations performed using the hybrid method, which is the hybrid between the probability weighted expected return method (PWERM) and the option pricing method.
The computation of expected volatility is based on actual historical share price volatility of comparable companies. Expected term of options granted is calculated using the average between the vesting period and the contractual term to the expected term of the options in effect at the time of grant. The Company has historically not paid dividends and has no foreseeable plans to pay dividends and, therefore, uses an expected dividend yield of zero in the option pricing model. The risk-free interest rate is based on the yield of U.S. treasury bonds with equivalent terms.
79
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
p. Fair value of financial instruments:
The carrying amounts of cash and cash equivalents, short-term investments, restricted cash, receivables and prepaid expenses, trade receivables, trade payables and other accounts payable and accrued expenses approximate their fair value due to the short-term maturity of such instruments. Based upon the borrowing terms and conditions currently available to the Company, the carrying values of the long-term loans approximate fair value.
The Company accounts for certain assets and liabilities at fair value under ASC 820, “Fair Value Measurements and Disclosures.” Fair value is an exit price, representing the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants. As such, fair value is a market-based measurement that should be determined based on assumptions that market participants would use in pricing an asset or a liability.
The hierarchy below lists three levels of fair value based on the extent to which inputs used in measuring fair value are observable in the market. The Company categorizes each of its fair value measurements in one of these three levels based on the lowest level input that is significant to the fair value measurement in its entirety.
The three levels of inputs that may be used to measure fair value are as follows:
Level 1 - Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets;
Level 2 - Includes other inputs that are directly or indirectly observable in the marketplace, other than quoted prices included in Level 1, such as quoted prices for similar assets or liabilities in active markets, quoted prices for identical or similar assets or liabilities in markets with insufficient volume or infrequent transactions, or other inputs that are observable (model-derived valuations in which significant inputs are observable), or can be derived principally from or corroborated by observable market data; and
Level 3 - Unobservable inputs which are supported by little or no market activity.
The availability of observable inputs can vary from instrument to instrument and is affected by a wide variety of factors, including, for example, the type of instrument, the liquidity of markets and other characteristics particular to the transaction. To the extent that valuation is based on models or inputs that are less observable or unobservable in the market, the determination of fair value requires more judgment and the instrument are categorized as Level 3.
q. Basic and diluted net loss per share:
The Company applies the two class method as required by ASC 260-10, “Earnings per Share.” ASC 260-10 requires the income or loss per share for each class of shares (ordinary and preferred shares) to be calculated assuming 100% of the Company’s earnings are distributed as dividends to each class of shares based on their contractual rights. No dividends were declared or paid during the reported periods.
According to the provisions of ASC 260-10, the Company’s pre-IPO preferred shares were not participating securities in losses and, therefore, are not included in the computation of net loss per share. Post-IPO, there are no preferred shares outstanding.
Basic and diluted net loss per share is computed based on the weighted average number of ordinary shares outstanding during each year. Diluted loss per share is computed based on the weighted average number of ordinary shares outstanding during the period, plus dilutive potential shares considered outstanding during the period, in accordance with ASC 260-10. Basic and diluted net loss per ordinary share was the same for each period presented as the inclusion of all potential ordinary shares (all options and warrants) outstanding was anti-dilutive.
r. Income taxes:
The Company accounts for income taxes in accordance with ASC 740-10, “Income Taxes.” ASC 740-10 prescribes the use of the liability method whereby deferred tax asset and liability account balances are determined based on differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected to reverse. The Company provides a valuation allowance, to reduce deferred tax assets to their estimated realizable value, if needed.
80
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The Company established reserves for uncertain tax positions based on the evaluation of whether or not the Company’s uncertain tax position is “more likely than not” to be sustained upon examination. The Company records interest and penalties pertaining to its uncertain tax positions in the financial statements as income tax expense.
s. Concentration of risks:
Financial instruments that potentially subject the Company to concentration of credit risk consist principally of cash and cash equivalents, restricted cash, short-term investments and trade receivables.
Cash and cash equivalents and restricted cash are invested at top tier banks or financial institutions in Jersey, the United States, Israel, Luxemburg, Switzerland, Japan and Germany. Such investments may be in excess of insured limits and are not insured in other jurisdictions. Generally, these investments may be redeemed upon demand and, therefore, bear minimal risk.
The Company has no off-balance sheet concentrations of credit risk such as foreign exchange contracts, option contracts or other foreign hedging arrangements.
In 2017, one payer represented $15,479, or 9 %, of net revenues. In 2016, the same payer represented $10,393, or 13%, of net revenues. In 2015, the same payer represented $5,595, or 17%, of net revenues. Credit risk with respect to trade receivables is limited.
t. Retirement, pension and severance plans:
The Company has a 401(k) retirement savings plan for its U.S. employees. Each eligible employee may elect to contribute a portion of the employee’s compensation to the plan. The Company historically has not and currently does not make any matching contributions to this plan.
The Company has a defined benefit plan with a pension fund for its Swiss employees, whereby the employee and the Company contribute to the pension fund. The Company accounts for its obligation, in accordance with ASC 715, "Compensation – Retirement Benefits" (see Note 9).
The pension expense for the years ended December 31, 2017, 2016 and, 2015 was $1,036, $ 529 and $404, respectively.
Israeli law generally requires payment of severance pay upon dismissal of an employee or upon termination of employment in certain other circumstances. The Company makes ongoing deposits into employee pension plans to fund its severance liabilities. According to Section 14 of Israel Severance Pay Law, the Company makes deposits on behalf of its employees with respect to the Company’s severance liability and therefore no obligation is provided for in the financial statements. Severance pay liabilities with respect to employees who are not subject to Section 14, are provided for in the financial statements based upon the number of years of service and the latest monthly salary and the related deposits are recorded as an asset based on the cash surrender value. Severance expense for the years ended December 31, 2017, 2016 and 2015 amounted to $506, $430 and $356, respectively.
u. Contingent liabilities:
The Company accounts for its contingent liabilities in accordance with ASC 450, “Contingencies.” A provision is recorded when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated (see Note 14(c)).
With respect to legal matters, provisions are reviewed and adjusted to reflect the impact of negotiations, estimated settlements, legal rulings, advice of legal counsel and other information and events pertaining to a particular matter. As of December 31, 2017 and 2016, the Company was not a party to any ligation that could have a material adverse effect on the Company’s business, financial position, results of operations or cash flows.
v. Other comprehensive income (loss):
The Company accounts for comprehensive income (loss) in accordance with ASC 220, "Comprehensive Income". ASC 220 establishes standards for the reporting and display of comprehensive income (loss) and its components. Comprehensive income (loss) generally represents all changes in shareholders' equity during the period except those resulting from investments by, or distributions to, shareholders. The accumulated other comprehensive income (loss), net of taxes, relates to a pension liability and foreign currency translation adjustments.
81
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
w. Recently adopted accounting pronouncements:
In March 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2016-09, Compensation-Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. The amendments in ASU 2016-09 affect all entities that issue share-based payment awards to their employees and involve multiple aspects of the accounting for share-based payment transactions, including income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. ASU 2016-09 is effective for annual periods beginning after December 15, 2016, and interim periods within those annual periods. The Company adopted ASU 2016-09 during the quarter ended March 31, 2017, at which time it changed its accounting policy to account for forfeitures as they occur. The change was applied on a modified retrospective basis with a cumulative effect adjustment to accumulated deficit of $670 as of January 1, 2017. In addition, excess tax benefits for share-based payments are now presented as an operating activity in the statements of cash flows rather than financing activity. The changes have been applied prospectively in accordance with the ASU and prior periods have not been adjusted.
x. Recently issued accounting pronouncements:
In May 2014, FASB issued ASU 2014-09, Revenue from Contracts with Customers (Topic 606) (ASU 2014-09), an updated standard on revenue recognition and issued subsequent amendments to the initial guidance in March 2016, April 2016, May 2016 and December 2016 within ASU 2016-08, 2016-10, 2016-12 and 2016-20, respectively. The core principle of the new standard is for companies to recognize revenue to depict the transfer of goods and services to customers in amounts that reflect the consideration to which the company expects to be entitled in exchange for those goods and services. In addition, the new standard requires expanded disclosures. The Company has adopted the standard effective January 1, 2018 using the modified retrospective method applied to all contracts. In preparation for adoption of the standard, the Company has implemented internal controls and key system functionality to enable the preparation of financial information including the assessment of the impact of the standard. The Company uses the portfolio approach to apply the standard to portfolios of contracts with similar characteristics. Adoption of the standard will result in an increase to trade receivables of $3,215, deferred revenues of $645 and a cumulative impact to the Company's accumulated deficit as of January 1, 2018 of $2,570.
In February 2016, FASB issued ASU 2016-02-Leases (ASC 842), which sets out the principles for the recognition, measurement, presentation and disclosure of leases for both parties to a contract (i.e. lessees and lessors). The new standard requires lessees to apply
a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight line basis over the term of the lease, respectively. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than twelve months regardless of their classification. Leases with a term of twelve months or less will be accounted for similar to existing guidance for operating leases. The new standard requires lessors to account for leases using an approach that is substantially equivalent to existing guidance for sales-type leases, direct financing leases and operating leases. ASC 842 supersedes the previous leases standard, ASC 840. The standard is effective on January 1, 2019, with early adoption permitted. The Company currently anticipates adopting the new standard effective January 1, 2019 and is evaluating the impact of the adoption of this standard on its consolidated financial statements.
In June 2016, FASB issued ASU 2016-13, Financial Instruments - Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. ASU 2016-13 amends the impairment model to utilize an expected loss methodology in place of the currently used incurred loss methodology, which will result in the more timely recognition of losses. ASU 2016-13 also applies to employee benefit plan accounting, with an effective date of the first quarter of fiscal 2020. The amendments in this update are effective for fiscal years beginning after December 31, 2019, including interim periods within those fiscal years. The Company is currently assessing the impact of the adoption of this standard on its consolidated financial statements, footnote disclosures and employee benefit plans’ accounting.
In August 2016, FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230) Classification of Certain Cash Receipts and Cash Payments. ASU 2016-15 eliminates the diversity in practice related to the classification of certain cash receipts and payments for debt prepayment or extinguishment costs, the maturing of a zero coupon bond, the settlement of contingent liabilities arising from a business combination, proceeds from insurance settlements, distributions from certain equity method investees and beneficial interests obtained in a financial asset securitization. ASU 2016-15 designates the appropriate cash flow classification, including requirements to allocate certain components of these cash receipts and payments among operating, investing and financing activities. The retrospective transition method, requiring adjustment to all comparative periods presented, is required unless it is impracticable for some of the amendments, in which case those amendments would be prospectively as of the earliest date practicable. The standard is effective on January 1, 2018. The Company is currently assessing the impact of the adoption of this standard on its consolidated financial statements and footnote disclosures.
82
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
In November 2016, FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash. This standard requires the presentation of the statement of cash flows to show the changes in the total of cash, cash equivalents, restricted cash and restricted cash equivalents. The standard is effective for fiscal years and the interim periods within those fiscal years beginning after December 15, 2017. Early adoption is permitted. The Company is currently evaluating the effects of the adoption of this ASU on the consolidated financial statements.
Note 3: Cash and Cash equivalents and Short-term investments
|
|
a. |
Cash and cash equivalents: |
Cash equivalents include items almost as liquid as cash, such as certificates of deposit and time deposits with maturity periods of three months or less when purchased.
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Cash |
|
$ |
5,522 |
|
|
$ |
29,915 |
|
|
Money market funds |
|
|
73,070 |
|
|
|
69,865 |
|
|
Total cash and cash equivalents |
|
$ |
78,592 |
|
|
$ |
99,780 |
|
|
|
b. |
Short-term investments |
The Company invests in marketable U.S. Treasury Bills (“T-bills”) that are classified as held-to-maturity securities. The amortized cost and recorded basis of the T-bills are presented as short-term investments in the amount of $104,719 and $119,854, as of December 31, 2017 and 2016, respectively and their estimated fair value as of December 31, 2017 and 2016 was $104,655 and $119,825, respectively.
Note 4: Receivables and prepaid expenses
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Advances to and receivables from suppliers |
|
$ |
2,924 |
|
|
$ |
5,829 |
|
|
Government authorities |
|
|
2,006 |
|
|
|
1,867 |
|
|
Prepaid expenses |
|
|
2,890 |
|
|
|
2,238 |
|
|
Others |
|
|
285 |
|
|
|
150 |
|
|
|
|
$ |
8,105 |
|
|
$ |
10,084 |
|
Note 5: Inventories
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Raw materials |
|
$ |
4,276 |
|
|
$ |
5,243 |
|
|
Work in process |
|
|
8,435 |
|
|
|
8,292 |
|
|
Finished goods |
|
|
9,314 |
|
|
|
12,014 |
|
|
|
|
$ |
22,025 |
|
|
$ |
25,549 |
|
83
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 6: Property and equipment, net
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Cost: |
|
|
|
|
|
|
|
|
|
Computers and laboratory equipment |
|
$ |
10,833 |
|
|
$ |
10,121 |
|
|
Office furniture |
|
|
2,303 |
|
|
|
1,931 |
|
|
Production equipment |
|
|
1,222 |
|
|
|
1,179 |
|
|
Leasehold improvements |
|
|
3,614 |
|
|
|
2,885 |
|
|
Total cost |
|
$ |
17,972 |
|
|
$ |
16,116 |
|
|
Accumulated depreciation and amortization |
|
|
(8,941 |
) |
|
|
(6,304 |
) |
|
Depreciated cost |
|
$ |
9,031 |
|
|
$ |
9,812 |
|
Depreciation expense was $1,968, $1,673 and $ 1,348 for the years ended December 31, 2017, 2016 and 2015, respectively.
The Company capitalized software costs according to FASB ASC 350-40, "Accounting for the costs of Computer Software Developed or Obtained for Internal Use". As of December 31, 2017 and 2016, the Company capitalized an accumulated amount of $5,576 and $4,742, respectively. Amortization for the year ended December 31, 2017 and 2016 was $1,226 and $731, respectively.
Note 7: Field equipment, net
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Field equipment |
|
$ |
15,020 |
|
|
$ |
11,167 |
|
|
Accumulated depreciation |
|
|
(5,984 |
) |
|
|
(2,359 |
) |
|
Field equipment, net |
|
$ |
9,036 |
|
|
$ |
8,808 |
|
Depreciation expense was $4,483, $3,248 and $1,555 for the years ended December 31, 2017, 2016 and 2015, respectively. Write downs of $195, $6,436, and $36 were identified for the years ended December 31, 2017, 2016 and 2015, respectively.
The Company made the second generation Optune system available to all patients in the United States in 2016 and manufacturing of the first generation Optune system has been terminated. In 2016, the Company recorded an impairment loss with respect to the write-down of first generation Optune system field equipment in the amount of $6,412 (finished goods and production stage goods in the amount of $4,830 and $1,582, respectively) presented in cost of revenues.
Note 8: Other payables and accrued expenses
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Employees and payroll accruals |
|
$ |
13,283 |
|
|
$ |
7,541 |
|
|
Taxes payable and others |
|
|
9,110 |
|
|
|
3,142 |
|
|
Provision for settlement (Note 12) |
|
|
5,500 |
|
|
|
5,500 |
|
|
Deferred revenues |
|
|
4,959 |
|
|
|
2,267 |
|
|
Other |
|
|
144 |
|
|
|
76 |
|
|
|
|
$ |
32,996 |
|
|
$ |
18,526 |
|
84
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 9: Employee benefit obligations
The Company sponsors a defined benefit plan (the “Swiss Plan”) for all its employees in Switzerland for retirement benefits, as well as benefits on death or long-term disability. The liability in respect of the Swiss Plan is the projected benefit obligation calculated using the projected unit credit method. The projected benefit obligation as of December 31, 2017 represents the actuarial present value of the estimated future payments required to settle the obligation that is attributable to employee service rendered before that date. Swiss Plan assets are recorded at fair value. Pension expense is presented in the payroll expenses in the various functions in which the employees are engaged. Actuarial gains and losses arising from differences between the actual and the expected return on the Swiss Plan assets are recognized in accumulated other comprehensive income (loss) and amortized over the requisite service period. The plan is part of a collective pension foundation run by an insurance company. The Company and the employees pay retirement contributions, which are defined as a percentage of the employees’ covered salaries. The foundation, in turn, has all its risks (disability, death, longevity) and future benefits managed and guaranteed by the insurance company. Interest is credited to the employees’ account at the minimum rate provided in the Swiss Plan, payment which is guaranteed by the insurance contract, which represents the Swiss Plan’s primary asset. The targeted allocation for these funds is as follows:
|
Asset Allocation by Category as of December 31, 2017: |
|
|
|
|
|
Asset Category: |
|
Asset allocation (%) |
|
|
|
Debt Securities |
|
|
27 |
|
|
Real Estate |
|
|
22 |
|
|
Equity Securities |
|
|
30 |
|
|
Others |
|
|
21 |
|
|
Total |
|
|
100 |
|
85
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The following table sets forth the Swiss Plan’s funded status and amounts recognized in the consolidated financial statements for the year ended December 31, 2017 and 2016:
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Change in Benefit Obligation |
|
|
|
|
|
|
|
|
|
Projected benefit obligation at beginning of year |
|
$ |
8,241 |
|
|
$ |
6,223 |
|
|
Interest cost |
|
|
54 |
|
|
|
64 |
|
|
Company service cost |
|
|
878 |
|
|
|
498 |
|
|
Employee contributions |
|
|
417 |
|
|
|
321 |
|
|
Prior service cost |
|
|
(314 |
) |
|
|
- |
|
|
Benefits paid |
|
|
341 |
|
|
|
422 |
|
|
Actuarial loss |
|
|
700 |
|
|
|
713 |
|
|
Projected benefit obligation at end of year |
|
$ |
10,317 |
|
|
$ |
8,241 |
|
|
Change in Plan Assets |
|
|
|
|
|
|
|
|
|
Fair value of plan assets at beginning of year |
|
$ |
5,978 |
|
|
$ |
4,433 |
|
|
Actual return on plan assets |
|
|
882 |
|
|
|
320 |
|
|
Employer contributions |
|
|
625 |
|
|
|
482 |
|
|
Employee contributions |
|
|
417 |
|
|
|
321 |
|
|
Benefits paid |
|
|
341 |
|
|
|
422 |
|
|
Fair value of plan assets at end of year |
|
$ |
8,243 |
|
|
$ |
5,978 |
|
|
|
|
|
|
|
|
|
|
|
|
Funded Status at End of year |
|
|
|
|
|
|
|
|
|
Excess of obligation over assets |
|
$ |
(2,074 |
) |
|
$ |
(2,263 |
) |
|
|
|
|
|
|
|
|
|
|
|
Change in Accrued Benefit Liability |
|
|
|
|
|
|
|
|
|
Accrued benefit liability at beginning of year |
|
$ |
(2,263 |
) |
|
$ |
(1,790 |
) |
|
Company contributions made during year |
|
|
625 |
|
|
|
482 |
|
|
Net periodic benefit cost for year |
|
|
(1,036 |
) |
|
|
(529 |
) |
|
Net decrease (increase) in accumulated other comprehensive loss |
|
|
600 |
|
|
|
(426 |
) |
|
Accrued benefit liability at end of year |
|
$ |
(2,074 |
) |
|
$ |
(2,263 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Non - current plan assets |
|
$ |
8,243 |
|
|
$ |
5,979 |
|
|
Non - current liability |
|
|
10,317 |
|
|
|
8,242 |
|
|
Accrued benefit liability at end of year |
|
$ |
(2,074 |
) |
|
$ |
(2,263 |
) |
|
Projected Benefit Payments |
|
|
|
|
|
|
|
|
|
Projected year 1 |
|
$ |
166 |
|
|
$ |
148 |
|
|
Projected year 2 |
|
|
168 |
|
|
|
150 |
|
|
Projected year 3 |
|
|
172 |
|
|
|
152 |
|
|
Projected year 4 |
|
|
1,124 |
|
|
|
155 |
|
|
Projected year 5 |
|
|
163 |
|
|
|
1,069 |
|
|
Projected year 6-10 |
|
$ |
1,053 |
|
|
$ |
928 |
|
86
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The fair value of the plan assets is the estimated cash surrender value of the insurance contract at December 31, 2017. The level of inputs used to measure fair value was Level 2.
|
|
Year ended December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net Periodic Benefit Cost |
|
|
|
|
|
|
|
|
|
Service cost |
|
$ |
878 |
|
|
$ |
498 |
|
|
Interest cost (income) |
|
|
62 |
|
|
|
(21 |
) |
|
Expected return on plan assets |
|
|
(42 |
) |
|
|
(49 |
) |
|
Amortization of prior service costs |
|
|
124 |
|
|
|
87 |
|
|
Amortization of transition obligation |
|
|
14 |
|
|
|
14 |
|
|
Total net periodic benefit cost |
|
$ |
1,036 |
|
|
$ |
529 |
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average assumptions: |
|
|
|
|
|
|
|
|
|
Discount rate as of December 31 |
|
0.60% |
|
|
0.60% |
|
||
|
Expected long-term rate of return on assets |
|
0.60% |
|
|
0.60% |
|
||
|
Rate of compensation increase |
|
1.00% |
|
|
1.00% |
|
||
|
Mortality and disability assumptions (*) |
|
BVG 2015 GT |
|
|
BVG 2015 GT |
|
||
|
(*) |
Mortality data used for actuarial calculation. |
Note 10: Long-term loan, net of discount and issuance costs
In January 2015, the Company entered into a five-year term loan agreement (the “Term Loan Credit Facility”) with a lender to draw up to $100,000. In January 2015, the Company drew $25,000 from the lender and the remaining $75,000 was drawn in July 2016. As of December 31, 2017 and 2016, there was $100,000 principal outstanding under the Term Loan Credit Facility.
Interest on the Term Loan Credit Facility is 10% annually, payable quarterly in arrears. In addition, there is a 1.5% funding fee payable on the amount drawn on the funding date, a 0.75% pay-down fee on all principal amount repayments to be paid on the date such payments of principal are made and a pre-payment fee of 3.0%, 2.0% or 1.0% if the Company prepays outstanding loan amounts prior to the first, second or third year anniversaries, respectively, from the initial funding date. The entire outstanding principal loan is due in January 2020. The loan is secured by a first priority security interest in substantially all assets of the Company. The Term Loan Credit Facility sets forth certain affirmative and negative covenants with which the Company must comply on a quarterly basis commencing March 31, 2015 through the term of loan. As of December 31, 2017, the Company was in compliance with such covenants.
As of December 31, 2017 and 2016, the total discount of $1,204 and $1,699, respectively, and additional issuance costs of $1,454 and $2,070, respectively, are presented net of the loan and are amortized to interest expense over the five year term of the loan using the effective interest method. On February 7, 2018, the Company prepaid our Term Loan Facility in full from the proceeds of a new term loan credit facility with a new lender. For additional information, see Note 20.
Note 11: Other long-term liabilities
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred rent liability |
|
$ |
746 |
|
|
$ |
906 |
|
|
Leasehold improvements financing and other (see a and b below) |
|
|
128 |
|
|
|
193 |
|
|
Unrecognized tax benefits (Note 13e) |
|
|
244 |
|
|
|
2,400 |
|
|
Term Loan Credit Facility repayment fee (Note 10) |
|
|
619 |
|
|
|
534 |
|
|
|
|
$ |
1,737 |
|
|
$ |
4,033 |
|
87
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
a. In July 2013, the Company entered into a loan agreement with the landlord of its facility in Switzerland whereby the landlord will offer a loan of up to CHF 400 for the purpose of financing leasehold improvements in the facility. As of December 31, 2016 and 2015, the Company received CHF 220 ($232) of this financing. The principal and interest is due in monthly payments from January 1, 2014 through December 31, 2018 and bears an annual interest of 5%.
b. In May 2013, the Company entered into an agreement with the landlord of one of its facilities in the United States and in January 2014, the Company entered into an agreement with a leasing company for an aggregate of $226 for the purpose of financing leasehold improvements in the facility and a lease of machinery, respectively. The loan and interest is due in monthly payments from June 1, 2013 through May 1, 2023 and bears an annual interest of 7%.
The above principal leasehold improvement financing repayments as of December 31, 2017 are as follows:
|
2018 |
|
$ |
32 |
|
|
2019 |
|
|
34 |
|
|
2020 |
|
|
29 |
|
|
2021 |
|
|
26 |
|
|
2022 |
|
|
27 |
|
|
Thereafter |
|
|
12 |
|
|
|
|
|
160 |
|
|
Less: current portion of long-term loans |
|
|
(32 |
) |
|
Long-term loans, net of current portion |
|
$ |
128 |
|
Note 12: Commitments and contingent liabilities
The facilities of the Company are leased under various operating lease agreements for periods ending no later than 2024. The Company also leases motor vehicles under various operating leases, which expire on various dates, the latest of which is in 2020.
Future minimum lease payments under non-cancelable operating leases as of December 31, 2017, are as follows:
|
2018 |
$ |
3,512 |
|
|
2019 |
|
2,525 |
|
|
2020 |
|
2,049 |
|
|
2021 |
|
1,610 |
|
|
2022 |
|
1,377 |
|
|
Thereafter |
|
1,356 |
|
|
|
$ |
12,429 |
|
Lease and rental expense for the years ended December 31, 2017, 2016 and 2015 was $3,474, $2,748, and $2,194, respectively.
As of December 31, 2017 and 2016 the Company pledged bank deposits of $1,038 and $807, respectively, to cover bank guarantees in respect of its leases of operating facilities and obtained guarantees by the bank for the fulfillment of the Company’s lease commitments of $1,202 and $955, respectively.
In February 2015, the Company entered into a settlement agreement (the “Settlement Agreement”) with the Technion Research and Development Foundation (“Technion”) to resolve certain potential disputes regarding intellectual property developed by the Company’s founder and previously assigned to the Company. Pursuant to the Settlement Agreement, and in exchange for a release of potential disputes from Technion, the Company is obligated to pay a $5.5 million milestone payment (the “Milestone Payment”) to Technion in the quarter following the quarter in which the Company achieves $250.0 million of cumulative net sales (as defined in the Settlement Agreement) (the “Net Sales Milestone”). The Company achieved the Net Sales Milestone in the fourth quarter of 2017. Accordingly, in the first quarter of 2018, the Company anticipates making the Milestone Payment to Technion.
88
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
a. The provision for income taxes from continuing operations is comprised of:
Income (loss) before income taxes:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States (U.S.) |
|
$ |
(77,654 |
) |
|
$ |
(80,972 |
) |
|
$ |
(55,087 |
) |
|
Non-U.S. |
|
|
29,157 |
|
|
|
(40,492 |
) |
|
|
(52,060 |
) |
|
|
|
$ |
(48,497 |
) |
|
$ |
(121,464 |
) |
|
$ |
(107,147 |
) |
Income taxes expense:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Current: |
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. |
|
$ |
8,491 |
|
|
$ |
6,501 |
|
|
$ |
891 |
|
|
Non-U.S. |
|
|
5,028 |
|
|
|
3,863 |
|
|
|
3,678 |
|
|
Total current |
|
|
13,519 |
|
|
|
10,364 |
|
|
|
4,569 |
|
|
Deferred: |
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. |
|
$ |
(3 |
) |
|
$ |
1 |
|
|
$ |
- |
|
|
Non-U.S. |
|
|
(351 |
) |
|
|
16 |
|
|
|
(135 |
) |
|
Total deferred |
|
|
(354 |
) |
|
|
17 |
|
|
|
(135 |
) |
|
Total income taxes provision |
|
$ |
13,165 |
|
|
$ |
10,381 |
|
|
$ |
4,434 |
|
b. For purposes of comparability, the Company uses the notional U.S. federal income tax rate of 35% when presenting the Company's reconciliation of the income tax provision. The Company is a resident taxpayer in Jersey and as such is not generally subject to Jersey tax on remitted foreign earnings. A reconciliation of the provision for income taxes compared with the amounts at the notional federal statutory rate was:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
U.S statutory income taxes rate |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
Non-deductible expenses |
|
|
(6.8 |
) |
|
|
(2.5 |
) |
|
|
(2.4 |
) |
|
Foreign taxes rate differential |
|
|
15.1 |
|
|
|
(14.2 |
) |
|
|
(19.2 |
) |
|
Change in valuation allowance (1) |
|
|
(11.9 |
) |
|
|
(30.0 |
) |
|
|
(18.2 |
) |
|
State income taxes (1) |
|
|
18.7 |
|
|
|
2.3 |
|
|
|
1.8 |
|
|
Share based compensation |
|
|
(4.5 |
) |
|
|
1.2 |
|
|
|
- |
|
|
Change in unrecognized taxes expense |
|
|
(0.8 |
) |
|
|
(0.7 |
) |
|
|
(1.2 |
) |
|
Other (1) |
|
|
(71.9 |
) |
|
|
0.4 |
|
|
|
0.1 |
|
|
Effective taxes rate |
|
|
(27.1 |
)% |
|
|
(8.5 |
)% |
|
|
(4.1 |
)% |
_____________________________
|
|
(1) |
For additional information, see the table below reflecting the net impact of the TCJA. |
The Company's tax rate is affected by the tax rates in the jurisdictions outside the U.S. in which the Company operates. The jurisdictional location of earnings is a significant component of our effective tax rate as the tax rates outside of the U.S. are generally lower than the U.S. tax rate of 35% and the relative amount of losses or income for which no tax benefit or expense was recognized due to a valuation allowance.
89
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
On December 22, 2017, the Tax Cuts and Jobs Act of 2017 (the “TCJA”) was signed into law making significant changes to the Internal Revenue Code. Changes include, but are not limited to, a corporate tax rate decrease from 35% to 21% effective for tax years beginning after December 31, 2017. On the same date, Staff Accounting Bulletin No. 118 ("SAB 118") was issued to address the application of US GAAP in situations when a registrant does not have the necessary information available, prepared, or analyzed (including computations) in reasonable detail to complete the accounting for certain income tax effects of the Act. In accordance with SAB 118, we have determined that the $34.8 million of the deferred tax expense recorded in connection with the remeasurement of certain deferred tax assets and liabilities earnings was a provisional amount and a reasonable estimate at December 31, 2017. This remeasurement was fully offset by a valuation allowance resulting in no impact to the Company’s income tax expense for the year ended December 31, 2017. The Company’s subsidiary in the United States does not have any foreign subsidiaries and, therefore, the remaining provisions of the TCJA have no material impact on the Company's results of operations. Additional work is necessary for a more detailed analysis of our deferred tax assets and liabilities as well as potential correlative adjustments. Any subsequent adjustment to these amounts will be recorded in the quarter of 2018 when the analysis is complete.
The table below reflects the net impact of the TCJA:
|
|
|
Year ended December 31, 2017 |
|
|||||||||
|
|
|
ETR before TCJA |
|
|
US Tax Cuts & Jobs Act Impact |
|
|
Reported ETR |
|
|||
|
U.S statutory income taxes rate |
|
|
35.0 |
% |
|
|
0.0 |
% |
|
|
35.0 |
% |
|
Non-deductible expenses |
|
|
(6.8 |
) |
|
|
0.0 |
|
|
|
(6.8 |
) |
|
Foreign taxes rate differential |
|
|
15.1 |
|
|
|
0.0 |
|
|
|
15.1 |
|
|
Change in valuation allowance |
|
|
(83.4 |
) |
|
|
71.5 |
|
|
|
(11.9 |
) |
|
State income taxes |
|
|
12.8 |
|
|
|
5.9 |
|
|
|
18.7 |
|
|
Share based compensation |
|
|
2.0 |
|
|
|
(6.5 |
) |
|
|
(4.5 |
) |
|
Change in unrecognized taxes expense |
|
|
(0.9 |
) |
|
|
0.1 |
|
|
|
(0.8 |
) |
|
Other |
|
|
(0.9 |
) |
|
|
(71.0 |
) |
|
|
(71.9 |
) |
|
Effective taxes rate |
|
|
(27.1 |
)% |
|
|
0.0 |
% |
|
|
(27.1 |
)% |
c. Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets and liabilities are as follows:
|
|
December 31, |
|
||||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts |
|
$ |
6,797 |
|
|
$ |
18,770 |
|
|
Revenue recognition |
|
|
60,099 |
|
|
|
46,953 |
|
|
Net operating loss carryforwards |
|
|
972 |
|
|
|
577 |
|
|
Share based compensation |
|
|
7,544 |
|
|
|
3,510 |
|
|
Deferred revenue |
|
|
1,340 |
|
|
|
879 |
|
|
Other temporary differences |
|
|
1,147 |
|
|
|
1,481 |
|
|
Total gross deferred taxes assets |
|
$ |
77,899 |
|
|
$ |
72,170 |
|
|
Less: valuation allowance |
|
|
(75,804 |
) |
|
|
(70,061 |
) |
|
Total deferred taxes assets |
|
$ |
2,095 |
|
|
$ |
2,109 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Fixed assets |
|
|
1,486 |
|
|
|
1,789 |
|
|
Total gross deferred taxes liabilities |
|
$ |
1,486 |
|
|
$ |
1,789 |
|
|
|
|
|
|
|
|
|
|
|
|
Net deferred taxes assets |
|
$ |
609 |
|
|
$ |
320 |
|
d. Carryforward loss:
As of December 31, 2017, one of the Company's Luxembourg subsidiaries has $3.6 million of net operating loss carry forwards (NOLs) available for utilization in future years.
90
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
e. A reconciliation of the beginning and ending balances of uncertain tax benefits is as follows:
|
|
December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Balance at beginning of the year |
|
$ |
2,400 |
|
|
$ |
1,565 |
|
|
$ |
308 |
|
|
Additions for taxes positions related current year |
|
|
55 |
|
|
|
1,088 |
|
|
|
848 |
|
|
Additions for taxes positions related to prior years |
|
|
372 |
|
|
|
58 |
|
|
|
409 |
|
|
Reduction related to lapse of applicable statute of limitations |
|
|
- |
|
|
|
(311 |
) |
|
|
- |
|
|
Balance at the end of the year |
|
$ |
2,827 |
|
|
$ |
2,400 |
|
|
$ |
1,565 |
|
The Company recognizes interest and penalties related to unrecognized tax benefits in tax expense. During the years ended December 31, 2017, 2016 and 2015, the Company accrued $125, $ 31 and $26, respectively, for interest and penalties expenses related to uncertain tax positions.
The Company's Israeli subsidiary is currently under an income tax audit for the tax years 2013 through 2016. There are no other ongoing income tax audits.
Note 14: Share capital
Share capital is composed as follows:
|
|
Issued and outstanding |
|
||||||
|
|
|
Number of shares |
|
|||||
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Ordinary shares no par value |
|
|
89,478,032 |
|
|
|
87,066,446 |
|
a. Investment rounds:
In June 2015, the Company sold to investors 4,068,500 Series J Convertible Preferred shares at a price per share of $23.33, for a total consideration of $94,599 (net of issuance expenses of $319). Prior to conversion of the Series J Convertible Preferred shares into ordinary shares as a result of the IPO, such shares were senior to the other series of preferred shares on payment of the liquidation preference (equal to $23.33 per share), but otherwise had similar participating preferred rights, dividend rights and voting rights of the other series of preferred shares.
b. Rights, preferences and restrictions:
On October 7, 2015, the Company completed the IPO of its ordinary shares by issuing 7,876,195 ordinary shares (including exercise of overallotments) and raising net proceeds of $157,534, at which time the Series A through J Convertible Preferred shares converted into ordinary shares and ceased to exist. Each holder of ordinary shares is entitled to one vote per ordinary share.
c. Warrants:
As part of the Series D and E Convertible Preferred share investment agreements, the investors received warrants to purchase ordinary shares. The Company accounted for these warrants as equity instruments based on the guidance of ASC 815, “Derivatives and Hedging”, ASC 480-10, “Distinguishing Liabilities from Equity”, its related FASB staff positions, ASC 815-40 “Contracts in Entity’s Own Stock” and the AICPA Technical Practice Aid for accounting for preferred shares and warrants, including the roadmap for accounting for freestanding financial instruments indexed to, and potentially settled in, a company’s own stock.
91
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Significant terms of the warrants to purchase ordinary shares that were issued to purchasers of the Series D and E Convertible Preferred shares are as follows as of December 31, 2017 and 2016:
|
|
Warrants for ordinary shares |
|
|
|
|
|
||||||
|
|
|
December 31, |
|
|
Exercise price |
|
||||||
|
Expiration date |
|
2017 |
|
|
2016 |
|
|
per share |
|
|||
|
July 31, 2017 |
|
|
- |
|
|
|
547,478 |
|
|
$ |
3.59 |
|
|
January 22, 2018 |
|
|
203,241 |
|
|
|
554,331 |
|
|
|
3.59 |
|
|
July 21, 2018 |
|
|
304,863 |
|
|
|
831,504 |
|
|
$ |
3.59 |
|
|
|
|
|
508,104 |
|
|
|
1,933,313 |
|
|
|
|
|
In the years ended December 31, 2017 and 2016, warrants to purchase 1,418,711 and 902,132 ordinary shares, respectively, were cashlessly exercised, resulting in the issuance of 803,138 and 864,341 ordinary shares, respectively. Also, in the year ended December 31, 2017 and 2016 warrants to purchase 6,498 and 220,316 ordinary shares, respectively, with an exercise price of $3.59 per share were exercised for cash
Pursuant to a credit facility that the Company entered into in January 2013 (the “Credit Agreement”) which was fully paid in December 2013, the Company issued to the lenders under the Credit Agreement 975,644 warrants to purchase Series H Convertible Preferred shares at an exercise price of $18.77 per share. The warrants were exercised on a cashless basis in January 2016, resulting in the issuance of 315,155 ordinary shares.
d. Share option plans and ESPP:
Until the IPO in October 2015, the Company maintained and granted option awards under the 2003 Share Option Plan (the “2003 Plan”) and the 2013 Equity Incentive Share Option Plan (the “2013 Plan”) for the Company’s officers, directors, employees and advisors. The 2003 Plan and the 2013 Plan terminated as of the IPO as to future awards, but they continue to govern option awards previously granted thereunder.
In August 2015, the Company’s board of directors adopted and established the 2015 Omnibus Incentive Plan (the “2015 Plan”). The Company’s shareholders approved the 2015 Plan in September 2015. Under the 2015 Plan, the Company can issue various types of equity compensation awards such as restricted shares, performance shares, restricted stock units (“RSUs”), performance units, long-term cash award and other share-based awards. The options granted generally have a four-year vesting period and expire ten years after the date of grant. Options granted under the 2015 Plan generally have a four-year vesting period and expire ten years after the date of grant. Options granted under the 2015 Plan that are cancelled or forfeited before expiration become available for future grants. RSUs granted under the 2015 Plan vest in equal installments over a three-year period.
On December 31, 2017, in accordance with the terms of the 2015 Plan, the number of shares available for issuance under the 2015 Plan automatically increased by 4% of the Company’s outstanding ordinary shares as of December 30, 2017. As a result, the number of shares available for issuance under the 2015 Plan increased from 19,730,105 shares to 23,302,529 shares. As of December 31, 2017, 12,971,921 ordinary shares are available for grant under the 2015 Plan.
In August, 2015, the Company’s board of directors adopted the ESPP, which was approved by the Company’s shareholders in September, 2015. The Company adopted the ESPP to encourage and enable eligible employees to acquire ownership of the Company’s ordinary shares purchased through accumulated payroll deductions on an after-tax basis. The ESPP is intended to be an “employee stock purchase plan” within the meaning of Section 423 of the Code and the provisions of the ESPP will be construed in a manner consistent with the requirements of such section. The Company began its offerings under the ESPP on August 1, 2016. The Company issued 259,821 ordinary shares for the plan periods ended till December 31, 2017.
Under the ESPP, initially an aggregate of 830,000 ordinary shares could be purchased by eligible employees who become participants in the ESPP; which amount shall be automatically increased on December 31 of each year during the term of the ESPP to an amount equal to 1% of the total number of ordinary shares outstanding on December 30 of such year unless otherwise determined by the board of directors. As of December 31, 2017, 2,277,705 ordinary shares are available for offering under the ESPP.
92
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
The fair value of share-based awards was estimated using the Black-Scholes model for all equity grants. For market condition awards, the Company also applied the Monte-Carlo simulation model, with the following underlying assumptions:
|
|
Year ended December 31, |
|
||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
|
Stock Option Plans |
|
|
|
|
|
|
|
|
|
Expected term (years) |
|
5.50-6.25 |
|
6.25 |
|
6.25 |
|
|
|
Expected volatility |
|
56.74%-59.45% |
|
58.4%-61.70% |
|
59.0%-65.80% |
|
|
|
Risk-free interest rate |
|
1.97%-2.23% |
|
1.23%-1.88% |
|
1.74%-2.05% |
|
|
|
Dividend yield |
|
0.00% |
|
0.00% |
|
0.00% |
|
|
|
ESPP |
|
|
|
|
|
|
|
|
|
Expected term (years) |
|
0.50 |
|
0.42 |
|
|
- |
|
|
Expected volatility |
|
76.37%-82.00% |
|
70.45% |
|
|
- |
|
|
Risk-free interest rate |
|
0.62%-1.13% |
|
0.4% |
|
|
- |
|
|
Dividend yield |
|
0.00% |
|
0.00% |
|
|
- |
|
A summary of the status of the Company’s options to purchase ordinary shares as of December 31, 2017 and changes during the year ended on that date is presented below:
|
|
Year ended December 31, 2017 |
|
||||||||||
|
|
|
Number of options |
|
|
Weighted average exercise price |
|
|
Aggregate intrinsic value |
|
|||
|
Outstanding at beginning of year |
|
|
11,377,354 |
|
|
$ |
9.76 |
|
|
|
|
|
|
Granted |
|
|
5,381,613 |
|
|
$ |
10.53 |
|
|
|
|
|
|
Exercised |
|
|
(1,442,522 |
) |
|
$ |
2.64 |
|
|
|
|
|
|
Forfeited and cancelled |
|
|
(510,418 |
) |
|
$ |
12.54 |
|
|
|
|
|
|
Outstanding at end of year |
|
|
14,806,027 |
|
|
$ |
10.64 |
|
|
$ |
145,755 |
|
|
Exercisable options |
|
|
6,389,813 |
|
|
$ |
8.64 |
|
|
|
75,547 |
|
A summary of the status of the Company’s RSUs as of December 31, 2017 and changes during the year ended on that date is presented below:
|
|
|
Year ended December 31, 2017 |
|
|||||||||
|
|
|
Number of RSUs |
|
|
Weighted average grant date fair value price |
|
|
Aggregate intrinsic value |
|
|||
|
Unvested at beginning of year |
|
- |
|
|
$ |
- |
|
|
|
|
|
|
|
Granted |
|
|
1,661,619 |
|
|
|
9.64 |
|
|
|
|
|
|
Vested |
|
- |
|
|
|
- |
|
|
|
|
|
|
|
Forfeited and cancelled |
|
|
(10,400 |
) |
|
|
7.15 |
|
|
|
|
|
|
Unvested as of December 31, 2017 |
|
|
1,651,219 |
|
|
$ |
9.66 |
|
|
$ |
33,354 |
|
The total equity-based compensation expense related to all of the Company’s equity-based awards recognized for the years ended December 31, 2017, 2016 and 2015, was comprised as follows:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cost of revenues |
|
$ |
467 |
|
|
$ |
623 |
|
|
$ |
174 |
|
|
Research, development and clinical trials |
|
|
3,587 |
|
|
|
3,155 |
|
|
|
2,529 |
|
|
Sales and marketing |
|
|
3,784 |
|
|
|
5,111 |
|
|
|
2,496 |
|
|
General and administrative |
|
|
19,278 |
|
|
|
12,552 |
|
|
|
6,661 |
|
|
Total share-based compensation expense |
|
$ |
27,116 |
|
|
$ |
21,441 |
|
|
$ |
11,860 |
|
93
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
As of December 31, 2017, there were unrecognized compensation costs of $39,253, which are expected to be recognized over a weighted average period of approximately 2.86 years.
The weighted average grant date fair values of the Company’s options granted during the years ended December 31, 2017, 2016 and 2015 were $10.53, $7.37 and $10.64 per share, respectively.
The weighted average grant date fair values of the Company’s options forfeited and cancelled during the years ended December 31, 2017, 2016 and 2015 were $ 12.54, $9.72 and $5.73, respectively.
The aggregate intrinsic values for the options exercised during the years ended December 31, 2017, 2016 and 2015 were $17,945, $7,673 and $3,546, respectively. The aggregate intrinsic value is calculated as the difference between the per share exercise price and the deemed fair value of the Company’s ordinary shares for each share subject to an option multiplied by the number of shares subject to options at the date of exercise. The Company deemed the fair value of the Company’s ordinary shares to be $20.20, $7.85 and $22.36 per share as of December 31, 2017, 2016, and 2015, respectively.
The options outstanding as of December 31, 2017 are as follows:
|
Exercise price |
|
Number of options outstanding |
|
|
Weighted average remaining contractual term |
|
|
Number of options exercisable |
|
|
Weighted average remaining contractual term |
|
||||
|
$ |
|
|
|
|
|
(years) |
|
|
|
|
|
|
(years) |
|
||
|
0.23 - 1.00 |
|
|
338,906 |
|
|
|
1.95 |
|
|
|
338,906 |
|
|
|
1.95 |
|
|
1.01 - 7.00 |
|
|
2,627,476 |
|
|
|
4.29 |
|
|
|
2,491,388 |
|
|
|
4.05 |
|
|
7.01 - 11.00 |
|
|
4,395,057 |
|
|
|
7.56 |
|
|
|
1,589,345 |
|
|
|
5.20 |
|
|
11.01 - 16.00 |
|
|
5,260,210 |
|
|
|
8.21 |
|
|
|
1,305,796 |
|
|
|
6.94 |
|
|
16.01 - 20.00 |
|
|
351,750 |
|
|
|
9.57 |
|
|
|
- |
|
|
|
- |
|
|
20.01 - 27.50 |
|
|
1,832,628 |
|
|
|
8.15 |
|
|
|
664,378 |
|
|
|
7.83 |
|
|
|
|
|
14,806,027 |
|
|
|
7.20 |
|
|
|
6,389,813 |
|
|
|
5.21 |
|
Note 15: Financial expenses, net
The following table sets forth the Company’s total financial expenses, net:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Financial expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest expense |
|
$ |
(10,261 |
) |
|
$ |
(5,937 |
) |
|
$ |
(2,373 |
) |
|
Amortization of credit facility costs |
|
|
(1,111 |
) |
|
|
(667 |
) |
|
|
(329 |
) |
|
Foreign currency transaction losses |
|
|
- |
|
|
|
(396 |
) |
|
|
(356 |
) |
|
Others |
|
|
(321 |
) |
|
|
(318 |
) |
|
|
(177 |
) |
|
|
|
$ |
(11,693 |
) |
|
$ |
(7,318 |
) |
|
$ |
(3,235 |
) |
|
Financial income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Amortization of treasury bills premium |
|
$ |
859 |
|
|
$ |
512 |
|
|
$ |
- |
|
|
Foreign currency transaction gains |
|
|
549 |
|
|
|
- |
|
|
|
- |
|
|
Interest income |
|
|
1,116 |
|
|
|
659 |
|
|
|
84 |
|
|
|
|
$ |
2,524 |
|
|
$ |
1,171 |
|
|
$ |
84 |
|
|
Total financial expenses, net |
|
$ |
(9,169 |
) |
|
$ |
(6,147 |
) |
|
$ |
(3,151 |
) |
94
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 16: Basic and diluted net loss per share
The following table sets forth the computation of the Company’s basic and diluted net loss per ordinary share:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net loss attributable to ordinary shares as reported |
|
$ |
(61,662 |
) |
|
$ |
(131,845 |
) |
|
$ |
(111,581 |
) |
|
Shares used in computing net loss per ordinary share, basic and diluted |
|
|
88,546,719 |
|
|
|
85,558,448 |
|
|
|
30,401,603 |
|
|
Net loss per ordinary share, basic and diluted |
|
$ |
(0.70 |
) |
|
$ |
(1.54 |
) |
|
$ |
(3.67 |
) |
For the years ended December 31, 2017, 2016 and 2015, all outstanding preferred shares, options and warrants have been excluded from the calculation of the diluted net loss per share since their effect was anti-dilutive.
In certain markets and for certain key components, the Company is currently dependent upon sole source suppliers used in its delivery systems. The Company’s management believes that in most cases other suppliers could provide similar components at comparable terms. A change of suppliers which requires FDA or other regulatory approval, however, could cause a material delay in manufacturing and a possible loss of sales, which could adversely affect the Company’s operating results and financial position.
Note 18: Supplemental information
The following table presents long-lived assets by location:
|
|
December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
10,372 |
|
|
$ |
11,981 |
|
|
$ |
6,600 |
|
|
Switzerland |
|
|
5,114 |
|
|
|
4,346 |
|
|
|
4,204 |
|
|
Israel |
|
|
2,081 |
|
|
|
1,915 |
|
|
|
1,376 |
|
|
Others |
|
|
500 |
|
|
|
378 |
|
|
|
401 |
|
|
|
|
$ |
18,067 |
|
|
$ |
18,620 |
|
|
$ |
12,581 |
|
The Company’s net revenues by geographic region, based on the patient’s location are summarized as follows:
|
|
Year ended December 31, |
|
||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
United States |
|
$ |
134,688 |
|
|
$ |
72,771 |
|
|
$ |
30,961 |
|
|
EMEA (*) |
|
|
42,035 |
|
|
|
10,028 |
|
|
|
2,070 |
|
|
Japan |
|
|
303 |
|
|
|
89 |
|
|
|
56 |
|
|
|
|
$ |
177,026 |
|
|
$ |
82,888 |
|
|
$ |
33,087 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(*) including Germany |
|
$ |
40,215 |
|
|
$ |
9,799 |
|
|
$ |
1,803 |
|
95
NovoCure Limited and subsidiaries
Notes to consolidated financial statements
U.S. dollars in thousands (except share and per share data)
Note 19: Selected quarterly financial information (Unaudited)
The following table sets forth selected financial information for the Company:
|
|
|
2017 |
|
|||||||||||||
|
|
|
Three months ended |
|
|||||||||||||
|
|
|
December 31 |
|
|
September 30 |
|
|
June 30 |
|
|
March 31 |
|
||||
|
Net revenues |
|
$ |
53,661 |
|
|
$ |
50,109 |
|
|
$ |
38,376 |
|
|
$ |
34,880 |
|
|
Gross profit |
|
$ |
38,021 |
|
|
$ |
34,956 |
|
|
$ |
25,224 |
|
|
$ |
23,216 |
|
|
Operating loss |
|
$ |
(4,506 |
) |
|
$ |
(5,919 |
) |
|
$ |
(15,530 |
) |
|
$ |
(13,373 |
) |
|
Net loss |
|
$ |
(10,945 |
) |
|
$ |
(11,498 |
) |
|
$ |
(21,174 |
) |
|
$ |
(18,045 |
) |
|
Basic and diluted net loss per ordinary share |
|
$ |
(0.12 |
) |
|
$ |
(0.13 |
) |
|
$ |
(0.24 |
) |
|
$ |
(0.21 |
) |
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
89,389,364 |
|
|
|
89,125,646 |
|
|
|
88,218,868 |
|
|
|
87,452,983 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2016 |
|
|||||||||||||
|
|
|
Three months ended |
|
|||||||||||||
|
|
|
December 31 |
|
|
September 30 |
|
|
June 30 |
|
|
March 31 |
|
||||
|
Net revenues |
|
$ |
30,242 |
|
|
$ |
21,674 |
|
|
$ |
17,919 |
|
|
$ |
13,053 |
|
|
Gross profit |
|
$ |
19,268 |
|
|
$ |
10,556 |
|
|
$ |
1,710 |
|
|
$ |
5,071 |
|
|
Operating loss |
|
$ |
(17,877 |
) |
|
$ |
(28,265 |
) |
|
$ |
(37,237 |
) |
|
$ |
(31,938 |
) |
|
Net loss |
|
$ |
(22,168 |
) |
|
$ |
(33,628 |
) |
|
$ |
(40,612 |
) |
|
$ |
(35,437 |
) |
|
Basic and diluted net loss per ordinary share |
|
$ |
(0.26 |
) |
|
$ |
(0.39 |
) |
|
$ |
(0.48 |
) |
|
$ |
(0.42 |
) |
|
Weighted average number of ordinary shares used in computing basic and diluted net loss per share |
|
|
86,760,316 |
|
|
|
85,774,874 |
|
|
|
85,274,683 |
|
|
|
84,397,164 |
|
Note 20: Subsequent event
On February 7, 2018, the Company and certain of its subsidiaries entered into a Loan and Security Agreement (“2018 Loan Agreement”) with BioPharma Credit PLC pursuant to which such lender made a term loan to the Company in the principal amount of $150 million (the “2018 Credit Facility”). The term loan, which was drawn in full upon execution of the 2018 Loan Agreement, bears interest at 9.0% per annum, payable quarterly in arrears. The Company used a portion of the proceeds of the 2018 Credit Facility to repay in full the Company’s obligations under the Term Loan Credit Facility and will continue to use the proceeds to fund general corporate purposes.
The 2018 Credit Facility will mature on February 7, 2023, at which time any unpaid principal and accrued unpaid interest in respect of the term loan will be due and payable. The Company may prepay the term loan, in full, at any time. The Company must prepay the term loan (i) in full or in part upon the entry into certain licensing arrangements and (ii) in full in the event of a change of control. In each case, any prepayment (whether permitted or mandatory) is subject to a prepayment premium and/or make-whole payment. The pre-payment fee if the Company prepays outstanding loan amounts prior to February 7, 2021 is 2.0% and is 1.0% if made after the February 7, 2021 but prior to February 7, 2022.
All obligations under the 2018 Credit Facility are guaranteed by the Company’s current and future direct and indirect subsidiaries. In addition, the obligations under the 2018 Credit Facility are secured by a first-priority security interest in substantially all of the property and assets of, as well as the equity interests owned by, the Company and certain of the other guarantors.
On February 7, 2018, the Term Loan Credit Facility was terminated upon the Company’s repayment in full of the term loan issued thereunder.
96
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on that evaluation, the Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K have been designed and are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934 is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. We believe that a controls system, no matter how well designed and operated, cannot provide absolute assurance that the objectives of the controls system are met, and no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, within a company have been detected.
(b) Management’s Annual Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d- 15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with general accepted accounting principles. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, it used the criteria established in Internal Control- Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on such assessment, management has concluded that, as of December 31, 2017, our internal control over financial reporting is effective based on those criteria.
(c) Attestation Report of the Registered Public Accounting Firm
The effectiveness of our internal control over financial reporting as of December 31, 2017, has been audited by Kost Forer Gabbay & Kasierer, a member of Ernst & Young Global, an independent registered public accounting firm, as stated in their attestation report, which is included herein.
(d) Changes in Internal Control over Financial Reporting
There have been no changes in our internal control over financial reporting during our most recent fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
None.
97
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Directors
The information required by Item 10 is incorporated herein by reference to the information contained under the caption “Proposal 1—Election of Directors” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
Executive Officers
The information concerning our executive officers required by this Item 10 is provided under the caption “Executive Officers of the Registrant” in Part I hereof.
Section 16(a) Beneficial Ownership Reporting Compliance
The information concerning Section 16(a) Beneficial Ownership Reporting Compliance by our directors and executive officers is incorporated by reference to the information contained under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
Audit Committee
The information required by this Item 10 is incorporated by reference to the information contained under the caption “Corporate Governance” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
Code of Ethics
The information concerning our Code of Ethics is incorporated by reference to the information contained under the caption “Corporate Governance” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item 11 is incorporated by reference to the information contained under the caption “2017 Director Compensation” and “Executive Compensation” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 regarding the ownership of our ordinary shares is incorporated by reference to the information contained under the caption “Information About Stock Ownership” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
The information required by Item 12 with respect to securities authorized for issuance under our equity compensation plans is provided under the caption “Equity Compensation Plan Information” in Part II, Item 5 hereof.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Item 13 is incorporated by reference to the information contained under the captions “Proposal 1 – Election of Directors,” “Corporate Governance,” and “Certain Relationships and Related Party Transactions” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by Item 14 is incorporated by reference to the information contained under the caption “Proposal 2 – Approval and Ratification of Appointment of Independent Registered Public Accounting Firm” in our definitive proxy statement related to the 2018 annual meeting of shareholders.
98
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) DOCUMENTS FILED AS PART OF THIS REPORT
The following is a list of our consolidated financial statements and our subsidiaries and supplementary data included in this Annual Report on Form 10-K under Item 8 of Part II hereof:
|
1. FINANCIAL STATEMENTS AND SUPPLEMENTAL DATA |
|
|
|
|
|
|
Report of Independent Registered Public Accounting Firm. |
|
|
|
|
|
Consolidated Balance Sheets as of December 31, 2017, 2016 and 2015. |
|
|
|
|
|
Consolidated Statements of Operations for the years ended December 31, 2017, 2016 and 2015. |
|
|
|
|
|
Consolidated Statement of Comprehensive Loss for the years ended December 31, 2017, 2016 and 2015. |
|
|
|
|
|
Consolidated Statements of Shareholders’ Equity for the years ended December 31, 2017, 2016 and 2015. |
|
|
|
|
|
Consolidated Statements of Cash Flows for the years ended December 31, 2017, 2016 and 2015. |
|
|
|
|
|
Notes to Consolidated Financial Statements. |
|
|
|
|
2. FINANCIAL STATEMENT SCHEDULES |
|
|
|
|
Schedules are omitted because they are not applicable or are not required, or because the required information is included in the consolidated financial statements or notes thereto.
99
The following is a list of exhibits filed as part of this Annual Report on Form 10-K. Where so indicated by footnote, exhibits that were previously filed are incorporated by reference. For exhibits incorporated by reference, the location of the exhibit in the previous filing is indicated.
EXHIBIT INDEX
|
Exhibit Number |
|
|
|
Incorporated by Reference |
|
Filed Herewith |
||||
|
|
Exhibit Description |
|
Form |
|
Date |
|
Number |
|
||
|
|
|
|
|
|
|
|
|
|
||
|
3.1 |
|
|
S-1/A |
|
9/21/15 |
|
3.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.1 |
|
|
S-1/A |
|
9/21/15 |
|
4.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.2 |
|
Eleventh Amended and Restated Investors Rights Agreement, dated June 1, 2015 |
|
DRS |
|
6/24/15 |
|
4.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4.3 |
|
Tenth Amended and Restated Registration Rights Agreement, dated June 1, 2015 |
|
DRS |
|
6/24/15 |
|
4.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.1 |
|
|
DRS |
|
6/24/15 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.2 |
|
|
DRS |
|
6/24/15 |
|
10.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.3 |
|
|
DRS |
|
6/24/15 |
|
10.4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.4 |
|
|
S-1/A |
|
9/21/15 |
|
10.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.5 |
|
Consulting Agreement with Palti Consultants Ltd. and the Company, dated May 1, 2002†# |
|
DRS |
|
6/24/15 |
|
10.10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.6 |
|
Settlement Agreement with the Technion, dated February 10, 2015 |
|
DRS/A |
|
8/11/2015 |
|
10.13 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.7 |
|
|
S-1/A |
|
9/21/15 |
|
10.14 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.8 |
|
|
S-1/A |
|
9/21/15 |
|
10.15 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.9 |
|
|
S-1/A |
|
9/21/15 |
|
10.17 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.10 |
|
|
S-1/A |
|
9/21/15 |
|
10.18 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.11 |
|
|
8-K |
|
12/22/15 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.12 |
|
|
8-K |
|
12/22/15 |
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.13 |
|
|
8-K |
|
12/22/15 |
|
10.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.14 |
|
|
8-K |
|
12/22/15 |
|
10.4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.15 |
|
|
8-K |
|
12/22/15 |
|
10.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.16 |
|
Form of Stock Option Award Agreement based on the 2015 Omnibus Incentive Plan for use in Germany# |
|
10-K |
|
03/01/16 |
|
10.25 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.17 |
|
Form of Incentive Stock Option Agreement pursuant to the 2015 Omnibus Incentive Plan |
|
8-K |
|
5/12/17 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.18 |
|
|
8-K |
|
3/22/16 |
|
10.1 |
|
|
|
100
|
Exhibit Number |
|
|
|
Incorporated by Reference |
|
Filed Herewith |
||||
|
|
Exhibit Description |
|
Form |
|
Date |
|
Number |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
10.19 |
|
Employment Agreement, dated as of May 11, 2016, by and between Novocure USA LLC and William F. Doyle |
|
8-K |
|
5/13/16 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.20 |
|
Israeli SubPlan to the NovoCure Limited Employee Share Purchase Plan |
|
8-K |
|
6/30/16 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.21 |
|
|
10-Q |
|
7/28/16 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.22 |
|
|
8-K |
|
10/14/16 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.23 |
|
|
8-K |
|
10/14/16 |
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.24 |
|
|
8-K |
|
10/14/16 |
|
10.3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.25 |
|
|
8-K |
|
12/30/16 |
|
10.2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.26 |
|
|
8-K |
|
3/31/17 |
|
10.1 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.27 |
|
|
10-K |
|
2/23/17 |
|
10.27 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.28 |
|
|
10-K |
|
2/23/17 |
|
10.28 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.29 |
|
|
10-K |
|
2/23/17 |
|
10.29 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.30 |
|
|
10-K |
|
2/23/17 |
|
10.30 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10.31 |
|
|
10-K |
|
2/23/17 |
|
10.31 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
21 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.1 |
|
Consent of Independent Registered Public Accounting Firm |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
31.1 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
31.2 |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.1* |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
32.2* |
|
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
101.INS |
|
XBRL Instance Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Extension Definition Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
|
|
|
|
X |
|
|
|
|
|
|
|
|
|
|
|
|
|
101.PRE |
|
XBRL Extension Presentation Linkbase Document |
|
|
|
|
|
|
|
X |
101
|
under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date of this Form 10-K, irrespective of any general incorporation language contained in such filing. |
|
† |
Confidential treatment has been granted for certain information set forth in this exhibit. Such information has been omitted and filed separately with the Securities and Exchange Commission. |
|
# |
Compensation plans and arrangements for executive officers and others. |
This Annual Report on Form 10-K includes trademarks of NovoCure Limited and other persons. All trademarks or trade names referred to herein are the property of their respective owners.
None.
102
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
Date: February 22, 2018
|
|
|
Novocure limited |
|
|
|
|
|
|
|
|
|
By: |
/s/ Asaf Danziger |
|
|
|
|
Asaf Danziger |
|
|
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
|
Date: |
|
Signature |
|
Title |
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Asaf Danziger |
|
Chief Executive Officer and Director (Principal Executive Officer) |
|
|
|
Asaf Danziger |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Wilhelmus Groenhuysen |
|
Chief Financial Officer (Principal Financial and Accounting Officer) |
|
|
|
Wilhelmus Groenhuysen |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ William F. Doyle |
|
Executive Chairman and Director |
|
|
|
William F. Doyle |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Kinyip Gabriel Leung |
|
Vice Chairman and Director |
|
|
|
Kinyip Gabriel Leung |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Yoram Palti, M.D., Ph.D. |
|
Director |
|
|
|
Yoram Palti, M.D., Ph.D. |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ William Burkoth |
|
Director |
|
|
|
William Burkoth |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Louis J. Lavigne, Jr. |
|
Director |
|
|
|
Louis J. Lavigne, Jr. |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Martin J. Madden |
|
Director |
|
|
|
Martin J. Madden |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Gert Lennart Perlhagen |
|
Director |
|
|
|
Gert Lennart Perlhagen |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ Charles G. Phillips III |
|
Director |
|
|
|
Charles G. Phillips III |
|
|
|
|
|
|
|
|
|
February 22, 2018 |
|
/s/ William A. Vernon |
|
Director |
|
|
|
William A. Vernon |
|
103