Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex322_47.htm |
| EX-32.1 - EX-32.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex321_48.htm |
| EX-31.2 - EX-31.2 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex312_49.htm |
| EX-31.1 - EX-31.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex311_50.htm |
| EX-23.1 - EX-23.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex231_52.htm |
| EX-21.1 - EX-21.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex211_51.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
|
☑ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2017
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-36407
ALNYLAM PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
|
Delaware |
|
77-0602661 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
|
(I.R.S. Employer Identification No.) |
300 Third Street, Cambridge, MA 02142
(Address of Principal Executive Offices) (Zip Code)
Registrant’s telephone number, including area code: (617) 551-8200
Securities registered pursuant to Section 12(b) of the Act:
|
Title of Each Class |
|
Name of Each Exchange on Which Registered |
|
Common Stock, $0.01 par value per share |
|
The Nasdaq Global Select Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☑ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ☐
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
☑ |
|
Accelerated filer |
☐ |
|
Non-accelerated filer |
☐ |
(Do not check if a smaller reporting company) |
Smaller reporting company |
☐ |
|
|
|
|
Emerging growth company |
☐ |
|
|
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☑
The aggregate market value of the registrant’s common stock, $0.01 par value per share (“Common Stock”), held by non-affiliates of the registrant, based on the last sale price of the Common Stock at the close of business on June 30, 2017, was $7,243,192,478. For the purpose of the foregoing calculation only, all directors and executive officers of the registrant are assumed to be affiliates of the registrant.
At January 31, 2018, the registrant had 99,867,820 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for its 2018 annual meeting of stockholders, which the registrant intends to file pursuant to Regulation 14A with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year end of December 31, 2017, are incorporated by reference into Part II, Item 5 and Part III of this Form 10-K.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2017
TABLE OF CONTENTS
|
ITEM 1. |
|
|
1 |
|
|
ITEM 1A. |
|
|
39 |
|
|
ITEM 1B. |
|
|
64 |
|
|
ITEM 2. |
|
|
65 |
|
|
ITEM 3. |
|
|
65 |
|
|
ITEM 4. |
|
|
65 |
|
|
|
|
|
|
|
|
ITEM 5. |
|
|
66 |
|
|
ITEM 6. |
|
|
68 |
|
|
ITEM 7. |
|
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
|
69 |
|
ITEM 7A. |
|
|
85 |
|
|
ITEM 8. |
|
|
86 |
|
|
ITEM 9. |
|
CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
|
126 |
|
ITEM 9A. |
|
|
126 |
|
|
ITEM 9B. |
|
|
126 |
|
|
|
|
|
|
|
|
ITEM 10. |
|
|
127 |
|
|
ITEM 11. |
|
|
127 |
|
|
ITEM 12. |
|
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
|
127 |
|
ITEM 13. |
|
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
|
127 |
|
ITEM 14. |
|
|
127 |
|
|
|
|
|
|
|
|
ITEM 15. |
|
|
128 |
|
|
ITEM 16. |
|
|
133 |
|
|
|
134 |
|||
This annual report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, that involve risks and uncertainties. All statements other than statements relating to historical matters should be considered forward-looking statements. When used in this report, the words “believe,” “expect,” “plan,” “anticipate,” “estimate,” “predict,” “may,” “could,” “should,” “intend,” “will,” “target,” “goal” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these words. Our actual results could differ materially from those discussed in the forward-looking statements as a result of a number of important factors, including the factors discussed in this annual report on Form 10-K, including those discussed in Item 1A of this report under the heading “Risk Factors,” and the risks discussed in our other filings with the Securities and Exchange Commission. Readers are cautioned not to place undue reliance on these forward-looking statements, which reflect management’s analysis, judgment, belief or expectation only as of the date hereof. We explicitly disclaim any obligation to update these forward-looking statements to reflect events or circumstances that arise after the date hereof.
Overview
We are a global biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. RNAi is a naturally occurring biological pathway within cells for sequence-specific silencing and regulation of gene expression. We are harnessing the RNAi pathway to develop a potential new class of innovative medicines, known as RNAi therapeutics. RNAi therapeutics are comprised of small interfering RNA, or siRNA, and function upstream of today’s medicines by potently silencing messenger RNA, or mRNA, that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
Our research and development strategy is to target genetically validated liver-expressed genes that have been implicated in the cause or pathway of human disease. We utilize a lipid nanoparticle, or LNP, or N-acetylgalactosamine, or GalNAc, conjugate approach to enable hepatic delivery of siRNAs. Our focus is on clinical indications where there is a high unmet need, early biomarkers for the assessment of clinical activity in Phase 1 clinical studies, and a definable path for drug development, regulatory approval, patient access and commercialization.
Specifically, our broad pipeline of investigational RNAi therapeutics is focused in three Strategic Therapeutic Areas, or “STArs:” Genetic Medicines; Cardio-Metabolic Diseases; and Hepatic Infectious Diseases. We are committed to the advancement of our Alnylam 2020 strategy, which is to achieve a company profile with three marketed products and ten RNAi therapeutic clinical programs, including four in late stages of development, across our three STArs by the end of 2020. In December 2017, we filed our first new drug application, or NDA, and marketing authorisation application, or MAA, for patisiran. In January 2018, we announced that the European Medicines Agency, or EMA, has accepted the MAA and initiated its review. Patisiran was previously granted an accelerated assessment by the EMA. In early February 2018, we announced that the United States Food and Drug Administration, or FDA, has accepted our NDA and granted our request for priority review, with an action date of August 11, 2018. If approved, we expect to launch patisiran and begin generating product revenues in 2018.
Finally, based on our expertise in RNAi therapeutics and broad intellectual property estate, we have formed alliances with leading pharmaceutical and life sciences companies to support our development and commercialization efforts, including Sanofi Genzyme, the specialty care global business unit of Sanofi, The Medicines Company, or MDCO, and Vir Biotechnology, Inc.
Key 2017 Highlights
Investigational Clinical Pipeline
|
|
• |
Patisiran - Hereditary Transthyretin-Mediated (hATTR) Amyloidosis, or hATTR Amyloidosis |
|
|
o |
Reported positive, final results from APOLLO Phase 3 study |
|
|
o |
EMA granted accelerated assessment status |
|
|
o |
FDA granted Breakthrough Therapy Designation |
|
|
o |
FDA expanded Orphan Drug Designation, or ODD, to treatment of ATTR amyloidosis |
|
|
o |
Completed submission of NDA with FDA |
|
|
o |
Completed submission of MAA with EMA |
1
|
|
o |
Aligned with FDA and other regulatory authorities on Phase 3 trial design, including, with the FDA, on an interim analysis based on reduction of urinary aminolevulinic acid, or ALA, as surrogate endpoint reasonably likely to predict clinical benefit |
|
|
o |
Initiated ENVISION Phase 3 clinical study with interim analysis data readout expected mid-2018 and potential for NDA filing at or around year-end 2018 |
|
|
• |
Fitusiran - Hemophilia A and B, With and Without Inhibitors |
|
|
o |
Initiated ATLAS Phase 3 study |
|
|
o |
Suspended dosing due to a fatal thrombotic event reported in September |
|
|
o |
Aligned with FDA on clinical risk mitigation measures; clinical hold lifted by FDA in December |
|
|
o |
Resumed dosing in Phase 2 open-label extension, or OLE; expect to dose in ATLAS Phase 3 study in early 2018 |
|
|
• |
Inclisiran - Hypercholesterolemia |
|
|
o |
Reported with MDCO positive data from the ORION-1 Phase 2 study |
|
|
o |
MDCO initiated comprehensive ORION Phase 3 program |
|
|
• |
Early to mid-stage programs |
|
|
o |
Reported positive preliminary results from Phase 1 study of ALN-TTRsc02 for the treatment of ATTR amyloidosis |
|
|
o |
Reported positive interim results from Phase 1/2 study for lumasiran (ALN-GO1) for treatment of primary hyperoxaluria type 1, or PH1 |
|
|
o |
Initiated Phase 2 trial for cemdisiran (ALN-CC5) for treatment of atypical hemolytic-uremic syndrome, or aHUS |
Corporate Highlights
|
|
• |
Finance |
|
|
o |
Completed two successful public offerings raising $1.14 billion in net proceeds |
|
|
o |
Ended 2017 with $1.73 billion in cash, cash equivalents, fixed income marketable securities and restricted investments |
|
|
• |
Business |
|
|
o |
Formed strategic alliance with Vir Biotechnology to advance RNAi therapeutics for infectious diseases |
|
|
• |
Commercial/Medical Affairs |
|
|
o |
Enabled our supply chain, medical, quality, compliance and commercial organizations in the U.S. and Western Europe |
|
|
o |
Established or enhanced existing diagnostic screening and patient/physician education initiatives to detect disease and raise awareness |
2
In preparation for a potential global commercial launch of patisiran in 2018 and potentially subsequent global product launches in 2019 and beyond, we are:
|
|
• |
Expanding the organization with approximately 250 new hires deployed in customer facing activities across the world, initially in the United States and major European countries, followed by Canada and Switzerland, with a phased approach to the Asia Pacific, Latin American and Middle Eastern regions, with broad experience in marketing, sales, patient access, patient services, distribution and product reimbursement, in particular for orphan diseases; |
|
|
• |
Expanding our commercial capabilities with incorporation of appropriate quality systems, compliance policies, systems and procedures, and implementation of internal systems and infrastructure in order to support global commercial sales and establish patient-focused programs; and |
|
|
• |
Planning to expand our global footprint in major European markets and beyond with the continued hiring of country general managers, medical experts, market access professionals and marketing and sales professionals. |
Recent Developments
On January 6, 2018, we and Sanofi Genzyme entered into an amendment to our 2014 collaboration, which is structured as an exclusive relationship for the worldwide development and commercialization of RNAi therapeutics in the field of Genetic Medicines. In connection and simultaneously with entering into the 2018 amendment to the 2014 Sanofi Genzyme collaboration, we and Sanofi Genzyme also entered into an Exclusive License Agreement with respect to all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, referred to as the Exclusive TTR License, and the ALN-AT3 Global License Terms with respect to fitusiran and any back-up products, referred to as the AT3 License Terms.
Under the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme has certain rights to our current and future Genetic Medicine programs that reach human proof-of-principal study completion, or Human POP, by the end of 2019, subject to extension to the end of 2021 in various circumstances. Under the 2014 Sanofi Genzyme collaboration, we were leading development and commercialization of patisiran in the United States, Canada and Western Europe (Alnylam Territory), while Sanofi Genzyme had rights to develop and commercialize the product in the rest of the world (Sanofi Genzyme Territory). Sanofi Genzyme also had a right to opt in to co-develop and co-promote ALN-TTRsc02 in the Alnylam Territory along with its regional opt-in rights. In addition, Sanofi Genzyme had opted in to co-develop and co-promote fitusiran in the Alnylam Territory, as well as develop and commercialize fitusiran in the Sanofi Genzyme Territory.
The 2018 amendment, together with the Exclusive TTR License and the AT3 License Terms, revise the terms and conditions of the 2014 collaboration to (i) provide us with the exclusive right to pursue the further global development and commercialization of all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, (ii) provide Sanofi Genzyme the exclusive right to pursue the further global development and commercialization of fitusiran and any back-up products and (iii) terminate the previous co-development and co-commercialization rights related to revusiran, ALN-TTRsc02 and fitusiran under the 2014 Sanofi Genzyme collaboration.
Sanofi Genzyme continues to have the right to opt into our other rare genetic disease programs for development and commercialization in territories outside of the Alnylam Territory as contemplated in the 2014 Sanofi Genzyme collaboration, as well as one right to a global license.
The transaction is subject to customary closing conditions and clearances, including clearance under the Hart-Scott-Rodino Antitrust Improvements Act. We expect the transaction to close during the first quarter of 2018.
The 2014 Sanofi Genzyme collaboration, as amended in January 2018, as well as the Exclusive TTR License and the AT3 License Terms, are described below under the heading “Strategic Alliances.”
3
In December 2017, we entered into a 'pre-competitive' consortium together with Regeneron Pharmaceuticals, Inc., or Regeneron, AbbVie Inc., AstraZeneca PLC, Biogen Inc. and Pfizer Inc., to fund the generation of exome sequence data from the 500,000 volunteer participants who make up the UK Biobank health resource by the end of 2019. We and each of the other collaborators agreed to commit $10.0 million to enable an acceleration of sequencing timelines. Regeneron will conduct the sequencing effort and the data will be paired with detailed, de-identified medical and health records within the UK Biobank resource, to create a comprehensive resource for linking human genetic variations to human biology and disease. All consortium members will have a limited period of exclusive access to the sequencing data, before the data will be made available to other health researchers by UK Biobank. Consortium members have committed to make all significant research findings public. We believe that the broad and ongoing access to detailed health and full exome sequencing data for the 500,000 UK Biobank participants will greatly enhance our target identification and validation efforts, contributing to the sustainability of our RNAi therapeutics product engine.
RNAi Therapeutics – A Potential New Class of Innovative Medicines
RNAi is a natural cellular process that was discovered in 1998 and was recognized with the award of the 2006 Nobel Prize for Physiology and Medicine to Dr. Andrew Fire and Dr. Craig Mello.
RNAi therapeutics harness the natural RNAi pathway to silence disease-associated genes and knock down production of disease-causing proteins, representing the opportunity to create a potential new class of innovative medicines. RNAi therapeutics exert their biological effects through a highly potent, catalytic mechanism. This unique mechanism of action confers a number of attributes that we believe have the potential to provide meaningful differentiation and distinct value for our investigational RNAi therapeutics relative to other drug classes.
|
|
|
|
Key Features of Alnylam Investigational RNAi Therapeutics Potential Attributes for Differentiation and Value |
|
|
|
• |
|
Potential to silence any disease-associated gene, including so-called “undruggable” targets, where conventional therapeutic modalities (e.g., small molecule drugs and biologics) have not been successful |
|
|
|
• |
|
Demonstrated potential in clinical trials to achieve robust clinical activity with up to 99 percent target gene knockdown in some cases |
|
|
|
• |
|
Clamped pharmacodynamic effect that has potential to provide improved and consistent efficacy compared with intermittent and transient effects often achieved with other drug classes |
|
|
|
• |
|
Demonstrated durability of effect in clinical trials that enables once-monthly, once-quarterly and, in some cases, possible bi-annual dose regimens |
|
|
|
• |
|
Ability to be administered via subcutaneous injection when using our proprietary GalNAc-conjugate delivery platform |
|
|
|
• |
|
Potential for room temperature stability, avoiding the inconveniences, costs and global challenges of a cold chain distribution |
|
We believe that the combination of these attributes represents a very promising profile for our investigational therapeutics, even in competitive markets, and in December 2017, we filed the first ever regulatory applications with the FDA and the EMA for approval of an RNAi therapeutic. We have reported on our advances in developing RNAi therapeutics as potential drugs in a large number of peer-reviewed publications and many scientific meetings, including publications by Alnylam scientists in the journals Nature, Nature Medicine, Nature Biotechnology, Cell, Proceedings of the National Academy of Sciences, The New England Journal of Medicine and The Lancet.
4
We believe that we have created a reproducible and modular platform for drug discovery, development and commercialization of innovative medicines.
|
Alnylam Reproducible and Modular Platform Strategic Framework for Innovative Medicines |
||
|
1 |
Genetically validated, liver-expressed target gene |
• High unmet need population • Opportunity for highly competitive profile • Delivery with GalNAc-conjugate platform |
|
2 |
Biomarker for human proof-of-concept in Phase 1 |
• Blood- or urine-based • Informative disease correlation • Establish dose/regimen for late stage development |
|
3 |
Definable path to potential approval and market |
• Clinical development plans with established endpoints • Demonstrable value for payors |
Delivery of RNAi Therapeutics
In recent years, a tremendous amount of progress has been made in effectively delivering RNAi therapeutics to targeted organs and cells, and we believe Alnylam has been the leader of this advancement. This delivery success is now enabling execution on our Alnylam 2020 strategy.
Early efforts focused on delivery of RNAi therapeutics utilizing LNPs, where siRNA molecules are encapsulated in specific lipid-based formulations. This technology enables systemic delivery with intravenous drug administration. Results with LNP-based investigational RNAi therapeutics demonstrated potent, rapid and durable target gene silencing in pre-clinical and clinical studies. Further, LNP-based investigational RNAi therapeutics have been found to be generally well tolerated in clinical studies conducted to date. Our lead product, patisiran, is formulated utilizing LNPs.
More recently, we began advancing proprietary technology that conjugates a sugar molecule called GalNAc to the siRNA molecule. This simpler delivery approach enables more convenient, subcutaneous administration of our drug candidates, a key aspect of our platform. Results from our Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform demonstrated a substantial increase in potency over our earlier standard template chemistry (STC)-GalNAc-conjugate approach in pre-clinical and clinical studies, and a durability of effect that we believe, based on our clinical results, supports once-monthly, once-quarterly, and in some cases, possibly even bi-annual subcutaneous dose regimens. Due to this increased potency and durability, as well as a wide therapeutic index, this conjugate platform has become our primary approach for development of investigational RNAi therapeutics. During 2017, we continued to invest in the enhancement of this platform and reported pre-clinical results from our ESC Plus (ESC+) GalNAc-conjugates. ESC+ GalNAc-conjugates utilize advanced design features to further improve specificity, including a glycol nucleic acid, or GNA, modification in the antisense seed region of the siRNA, while maintaining potency and durability, further improving our already wide therapeutic index by six-fold. The ESC+ design is now being applied to all of our pre-clinical programs and has shown successful translation of potency from rodents to non-human primates. We intend to employ our ESC+ GalNAc-conjugate platform in future development programs.
We have extensive human safety experience with our investigational RNAi therapeutics. Our data demonstrate that to date, RNAi therapeutics have been generally well tolerated with minimal platform-related safety findings. Based on data as of December 2016, in over 1,000 patients or volunteers dosed for over three years in more than ten clinical programs, the safety findings, set forth below, occur at a low incidence and are monitorable. They are also generally asymptomatic and reversible even with continued dosing. As noted, as of December 2016, these findings include:
|
|
• |
Low incidence (2.2 percent) of generally mild, asymptomatic, reversible liver function test increases greater than three times the upper limit of normal |
|
|
• |
Low incidence (15.2 percent) of generally mild, transient injection site reactions, or ISRs |
In our view, this is an acceptable tolerability profile in the high unmet need indications that we pursue. Continued review of data across our programs since December 2016 has not led to any meaningful change in human safety with respect to our RNAi platform.
5
In October 2016, we observed an apparent imbalance in mortality in the treatment arm of the ENDEAVOUR Phase 3 study for revusiran, our first generation, subcutaneously administered, investigational therapeutic for the treatment of cardiomyopathy due to hATTR amyloidosis, which utilized our STC-GalNAc-conjugate delivery platform. With patient safety at the forefront, the ENDEAVOUR study was discontinued and a comprehensive investigation into the causality of this imbalance was conducted.
The investigation revealed no overall baseline imbalance, although there was a greater number of older patients randomized to the revusiran arm. There was no clinical evidence for revusiran-mediated cardiotoxicity or findings that would suggest that the mortality imbalance was the result of revusiran pharmacokinetic or pharmacodynamic effects. While our investigation could not fully exclude a possible drug-related cause, there was some evidence for imbalance due to a lower-than-expected mortality rate in the placebo arm at the time of study discontinuation.
Due to lack of evidence of any broader platform issue, the decision to discontinue development of revusiran did not affect patisiran, which utilizes our LNP delivery platform, or any of our other investigational RNAi therapeutic programs in development, which utilize our ESC-GalNAc-conjugate or ESC+ GalNAc-conjugate delivery platform.
We believe RNAi therapeutics represent a simplified and efficient potential new class of innovative medicines. We have achieved human proof of concept in multiple clinical trials of our investigational candidates, providing strong support for our approach to drug development. Moreover, we believe that our reproducible and modular platform will support the achievement of our 2020 goals, such that by the end of 2020, we can grow into a multi-product commercial stage company with a deep and sustainable pipeline that can fuel continued growth for the future.
Our Product Pipeline
Our broad pipeline of investigational RNAi therapeutics is focused in three STArs: Genetic Medicines; Cardio-Metabolic Diseases; and Hepatic Infectious Diseases. The chart below is a summary of our product development programs as of January 31, 2018. It identifies those programs in which we have achieved human proof-of-concept, or POC, by demonstrating target gene knockdown and/or additional evidence of activity in clinical studies, those programs for which we have received Breakthrough Therapy Designation from the FDA, the development stage of our programs, and our commercial rights to such programs:
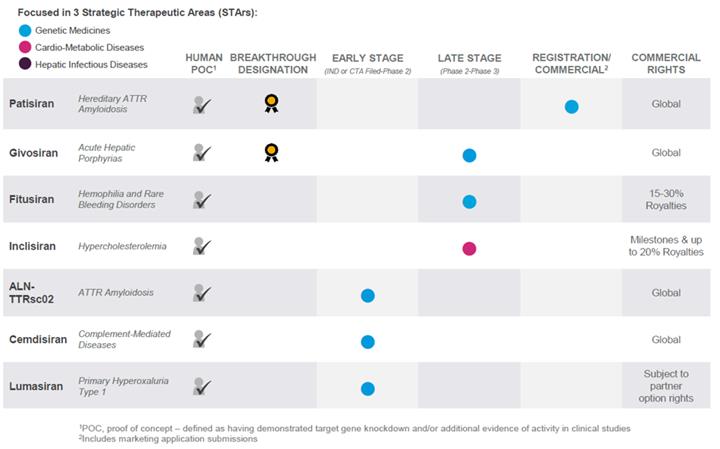
6
We have spent substantial funds over the past three years to develop our product pipeline and expect to continue to do so in the future. We incurred research and development expenses of $390.6 million in 2017, $382.4 million in 2016 and $276.5 million in 2015.
The investigational therapeutics described below are in various stages of clinical development and the scientific information included about these therapeutics is preliminary and investigative. None of our investigational therapeutics have been approved by the FDA, EMA or any other health authority and no conclusions can or should be drawn regarding the safety or efficacy of these investigational therapeutics.
Late Stage Clinical Development Programs
Patisiran — Hereditary Transthyretin-Mediated (hATTR) Amyloidosis
Patisiran, our most advanced investigational RNAi therapeutic, is currently under priority regulatory review in the United States and accelerated assessment in the European Union, or EU, for the treatment of hATTR amyloidosis. If approved, we expect to make patisiran commercially available to patients in the United States in mid-2018 and in the EU in late 2018. We also plan to file for regulatory approval in Japan in mid-2018 and in one or more additional countries by the end of the year.
hATTR amyloidosis is a rare, progressively debilitating and often fatal disease affecting approximately 50,000 patients worldwide. It is caused by deposition of wild-type and mutant transthyretin, or TTR, in peripheral tissues, such as the nerves, heart and gastrointestinal tract. Thus, polyneuropathy and cardiomyopathy are two cardinal manifestations of this disease, with most patients exhibiting a spectrum of both. TTR protein is produced primarily in the liver and is normally a carrier of vitamin A. Patisiran targets wild-type and all known mutant forms of TTR, including the V30M and V122I, the most common disease-causing mutations, and therefore it represents a potential therapeutic approach for the treatment of hATTR amyloidosis.
hATTR amyloidosis is a progressive disease, with an overall survival of two to 15 years from symptom onset, but only two to five years in patients presenting with cardiomyopathy. Orthotopic liver transplant, or OLT, and TTR tetramer stabilizers, such as tafamidis (approved in Europe, Japan and certain countries in Latin America, specific indication varies by region), are the only approved treatment options. However, availability is limited and most patients continue to experience significant morbidity and mortality associated with disease progression. Palliative therapies directed at specific symptoms such as pain, nausea, vomiting and diarrhea have been the mainstay of treatment despite their limited effectiveness. Other investigational drugs in development for the treatment of hATTR amyloidosis include the TTR-targeting antisense oligonucleotide, or ASO, inotersen, which showed statistically significant slowing of neuropathy progression and improvement in quality of life relative to placebo in a randomized Phase 3 trial and is currently under regulatory review in the United States and the EU, and a fibril-disrupting drug combination of doxycycline and tauroursodeoxycholic acid, which to date has only been tested in small single-arm Phase 2 studies. Thus, there remains a high unmet medical need for a safe and effective therapy that can benefit the broad hATTR amyloidosis population.
APOLLO Phase 3 Clinical Trial
Trial Design: Initiated in November 2013 and completed in September 2017, APOLLO is the largest pivotal study conducted to date in hATTR amyloidosis patients with polyneuropathy, with 225 patients enrolled, representing 39 TTR genotypes from across 19 countries. This randomized, double-blind, placebo-controlled, global study was designed to evaluate the efficacy and safety of patisiran in hATTR amyloidosis patients with polyneuropathy. The primary endpoint of the study was the change from baseline in the modified neuropathy impairment score, or mNIS+7, at 18 months. The mNIS+7 score is an evaluation of muscle weakness, sensory and autonomic function, and nerve conductance across a 304-point scale, where neuropathy progression leads to an increased score over time. A key secondary endpoint was the Norfolk Quality of Life Diabetic Neuropathy, or Norfolk QOL-DN, questionnaire, which is a validated instrument that measures clinical benefit. Additional secondary endpoints included: NIS-weakness, the subdomain of mNIS+7 assessing muscle strength; Rasch-built Overall Disability Scale, or R-ODS, a patient reported outcome measure of activities of daily living and disability; timed 10-meter walk test, assessing ambulatory ability and gait speed; modified body mass index, or mBMI, assessing nutritional status; and COMPASS-31, a patient questionnaire assessing autonomic disease symptoms. Exploratory endpoints measured the effects on cardiac structure and function. Patients were randomized 2:1, patisiran-to-placebo, with patisiran administered at 0.30 mg/kg once every three weeks for 18 months by intravenous infusion. 99 percent of patients who completed the APOLLO study rolled over into the Phase 3 OLE study, called the Global OLE.
7
Efficacy: Patisiran met the primary endpoint of mNIS+7 change from baseline at 18 months relative to placebo, and all secondary study endpoints. Specifically:
|
|
• |
Patisiran treatment (N=148) resulted in a negative 6.0-point mean change (improvement) in mNIS+7 score from baseline at 18 months as compared to a 28.0-point mean increase (worsening) reported for the placebo group (N=77), resulting in a 34.0-point mean difference relative to placebo (p=9.26 x 10-24). |
|
|
o |
Improvement in mNIS+7 from patisiran treatment was also consistently observed across all defined patient subgroups, including age, sex, race, geographic region, baseline neuropathy impairment, genotype, prior TTR stabilizer use, baseline Familial Amyloid Polyneuropathy, or FAP, stage, and inclusion in the pre-specified cardiac subpopulation. |
|
|
• |
Patisiran treatment resulted in a negative 6.7-point mean change (improvement) in Norfolk QoL-DN score from baseline at 18 months as compared to a 14.4-point mean increase (worsening) reported for the placebo group, resulting in a 21.1-point mean difference relative to placebo (p=1.10 x 10-10). |
|
|
• |
In a pre-specified binary analysis of neurological improvement, 56 percent (95 percent confidence interval, or CI: 48.1, 64.1) of patisiran patients had an improvement in mNIS+7 (less than 0-point change compared to baseline at 18 months), while 4 percent (95 percent CI: 0.0, 8.2) of placebo patients had an improvement (p=1.82 x 10-15). |
|
|
• |
Similarly, 51 percent (95 percent CI: 43.3, 59.4) of patisiran patients had an improvement in Norfolk QoL-DN (less than 0-point change compared to baseline at 18 months), versus 10 percent (95 percent CI: 3.6, 17.2) for placebo (p=1.95 x 10-10). |
|
|
• |
Patisiran also demonstrated statistically significant and clinically meaningful improvements over placebo in all other secondary endpoints at 18 months. |
Cardiac Subpopulation Results: Favorable and significant changes in several exploratory cardiac measures, including N-terminal pro b-type natriuretic peptide, or NT-proBNP, and certain echocardiographic parameters, were reported in patisiran-treated patients with pre-defined cardiac involvement (baseline left ventricular (LV) wall thickness ≥ 1.3 cm with no history of hypertension or aortic valve disease). Specifically:
|
|
• |
Patisiran treatment resulted in a median decrease (improvement) of 49.9 pg/ml in NT-proBNP levels as compared to a median increase (worsening) of 320 pg/ml reported for the placebo arm at 18 months (nominal p=7.74 x 10-8, based on analysis of log-transformed values). |
|
|
• |
Regarding echocardiographic measures, patisiran treatment resulted in a mean 0.93 mm reduction (improvement) in left ventricular (LV) wall thickness (nominal p=0.0173) and a mean absolute 1.37 percent improvement in longitudinal strain (nominal p=0.0154) relative to placebo. |
Safety and Tolerability: Patisiran showed an encouraging safety and tolerability profile relative to placebo with up to 18 months of dosing. Specifically:
|
|
• |
The most commonly reported adverse events, or AEs, that occurred more frequently in patisiran patients were peripheral edema (29.7 percent versus 22.1 percent in placebo) and infusion-related reactions, or IRRs (18.9 percent versus 9.1 percent in placebo). These were generally mild to moderate in severity and only one patient discontinued due to an IRR (0.7 percent). |
|
|
• |
Compared to placebo, patisiran treatment was associated with fewer treatment discontinuations (4.7 versus 14.3 percent) and fewer study withdrawals (4.7 versus 11.7 percent) due to AEs. |
|
|
• |
The incidence of serious adverse events, or SAEs, across the patisiran (36.5 percent) and placebo (40.3 percent) groups was similar. |
|
|
• |
SAEs reported in 2 or more patients in the patisiran group included: diarrhea (5.4 percent), cardiac failure, congestive cardiac failure, orthostatic hypotension, pneumonia, and atrioventricular block complete (2 percent each). These were all considered unrelated to patisiran, except for one SAE of diarrhea. SAEs occurred with similar frequency in the placebo group, except for diarrhea (1.3 percent in placebo group). |
|
|
• |
Deaths were recorded with a similar incidence across the patisiran (4.7 percent) and placebo (7.8 percent) treatment groups. |
|
|
o |
No deaths were considered related to study drug. |
|
|
• |
There were no safety signals with regard to hepatic or renal function, or evidence of thrombocytopenia, due to patisiran. |
8
Patisiran has been granted the following regulatory designations:
|
|
• |
Orphan Medicinal Product Designation (EMA) |
|
|
• |
Orphan Drug Designation (FDA) |
|
|
• |
Fast Track Designation (FDA) |
|
|
• |
Breakthrough Therapy Designation (FDA) |
|
|
• |
Orphan Drug Designation (Japanese Pharmaceuticals and Medical Devices Agency (PMDA)) |
|
|
• |
Promising Innovative Medicine (PIM) Designation (UK Medicines and Healthcare Products Regulatory Agency (MHRA)) |
As a result of the 2018 amendment to the Sanofi Genzyme collaboration and the Exclusive TTR License, following the closing of the transaction, we will have global rights for the development and commercialization of patisiran, together with ALN-TTRsc02, discussed below, and all back-up products. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, as well as the Exclusive TTR License, are described below under the heading “Strategic Alliances.”
Givosiran — Acute Hepatic Porphyrias
Givosiran is an investigational, subcutaneously administered RNAi therapeutic targeting aminolevulinate synthase-1, or ALAS1, for the treatment of acute hepatic porphyrias, or AHPs. It is designed to target and silence the ALAS1 mRNA, blocking the production of ALAS1 protein, the liver-expressed rate-limiting enzyme in the heme biosynthesis pathway. Lowering of ALAS1 may reduce the accumulation of neurotoxic heme intermediates, ALA and porphobilinogen, or PBG, that cause the clinical manifestations of AHPs, including acute debilitating neurovisceral attacks as well as chronic disabling disease symptoms. It is estimated that approximately 5,000 patients in the United States and Europe suffer sporadic AHP attacks annually, and approximately 1,000 patients are afflicted with recurrent, debilitating attacks. There are no approved therapies for prevention of acute attacks. Intravenous, or IV, hemin is currently approved for the treatment of acute AHP attacks, and is sometimes used prophylactically off-label by some porphyria specialists to prevent attacks, despite its unclear efficacy, short duration of action and association with significant side effects. There is a clear unmet need for novel therapeutics with an enhanced efficacy and safety profile and durable lowering of ALA and PBG, in order to prevent attacks, diminish chronic symptoms in between attacks, and improve patients’ quality of life.
Givosiran has the potential to be utilized for the chronic treatment of AHPs. In September 2017, we aligned with the FDA on a pivotal Phase 3 trial design that includes an interim analysis based on reduction of urinary ALA as a surrogate endpoint reasonably likely to predict clinical benefit. This will enable a potential NDA filing at or around year-end 2018, pending FDA review of the program at the time of interim analysis and assuming positive results. The ENVISION Phase 3 pivotal study for givosiran was initiated in November 2017 and we expect to report interim analysis data in mid-2018.
ENVISION Trial Design:
The ENVISION Phase 3 trial is a randomized, double-blind, placebo-controlled, global, multicenter study in more than 20 countries to evaluate the efficacy and safety of givosiran in approximately 75 patients with a documented diagnosis of AHPs. Patients will be randomized on a 1:1 basis to receive 2.5 mg/kg of givosiran or placebo subcutaneously administered monthly, over a six-month treatment period. The primary endpoint is the annualized rate of porphyria attacks requiring hospitalization, urgent healthcare visit or IV hemin administration at home over the six-month treatment period. The planned interim analysis will evaluate reduction of a urinary biomarker, ALA, in 30 patients after three months of dosing, as a surrogate endpoint reasonably likely to predict clinical benefit. The interim analysis is also designed to conduct a blinded assessment of the porphyria attack rate for the purpose of a study sample size adjustment from approximately 75 patients to up to approximately 94 patients. Key secondary and exploratory endpoints will evaluate reductions in the cardinal symptoms of AHPs, such as pain, nausea and fatigue, as well as impact on quality of life. All patients completing the six-month treatment period will be eligible to continue on an OLE study in which they will receive treatment with givosiran for up to 30 months.
Phase 1 Clinical Trial (Part C and OLE)
During 2017, we reported interim data from Part C of our Phase 1 clinical trial of givosiran, which was conducted as a randomized, double-blind, placebo-controlled study in up to 24 patients with acute intermittent porphyria, or AIP, the most common AHP sub-type, who experienced recurrent porphyria attacks, along with data from the Phase 1 OLE study of givosiran.
9
As of the data transfer date of April 21, 2017, givosiran achieved potent silencing of the ALAS1 mRNA, which resulted in robust and durable lowering of ALA and PBG. Data from the OLE study demonstrated that longer-term treatment with givosiran was associated with consistent reductions in the annualized porphyria attack rate, with preliminary evidence suggesting the potential for further reductions in the attack rate with extended dosing. As of the data cutoff date, givosiran administration was generally well tolerated in recurrent attack AIP patients in cohorts 1-3 in Part C of the Phase 1 study and in cohorts 1 and 2 of the ongoing OLE study, with a mean of 169 and 111 days on study, respectively, and up to 12 months on givosiran. As previously reported, one death occurred in a patient in cohort 3 in the givosiran arm due to hemorrhagic pancreatitis complicated by a pulmonary embolism and following a recent hospitalization for bacteremia; the death was considered to be unlikely related to study drug by the investigator and the study's Safety Review Committee.
Regulatory Designations
Givosiran has been granted the following regulatory designations for the treatment of AHPs:
|
|
• |
Orphan Drug Designation (FDA) |
|
|
• |
Orphan Medicinal Product Designation (EMA) |
|
|
• |
PRIME Designation (EMA) |
|
|
• |
Breakthrough Therapy Designation (FDA) |
During 2016, Sanofi Genzyme elected not to opt into the development and commercialization of givosiran in the Sanofi Genzyme Territory, providing us with full global control of the program for further development and commercialization, if approved. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, is described below under the heading “Strategic Alliances.”
Fitusiran — Hemophilia and Rare Bleeding Disorders
Fitusiran is a subcutaneously administered, investigational RNAi therapeutic targeting antithrombin, or AT, for the treatment of people with hemophilia A and B, with and without inhibitors. Fitusiran also has the potential to be used for rare bleeding disorders, or RBD. Fitusiran is designed to lower levels of AT with the goal of promoting sufficient thrombin generation to prevent bleeding. AT acts by inactivating thrombin and other coagulation factors, and plays a key role in normal hemostasis by helping to limit the process of fibrin clot formation. Hemophilia is a hereditary bleeding disorder characterized by an underlying defect in the ability to generate adequate levels of thrombin needed for effective fibrin clot formation, thereby resulting in recurrent bleeds into joints, muscles, and major internal organs. Lowering AT in the hemophilia setting may promote the generation of sufficient levels of thrombin needed to form an effective fibrin clot and prevent bleeding. This rationale is supported by human genetic data suggesting that co-inheritance of thrombophilic mutations, including AT deficiency, may ameliorate bleeding in hemophilia. We believe this approach is a unique and innovative strategy for preventing bleeding in people with hemophilia.
There are approximately 200,000 people living with hemophilia A and hemophilia B worldwide. Standard treatment for people with hemophilia currently involves replacement of the deficient clotting factor either as prophylaxis or on-demand therapy, which can lead to a temporary restoration of thrombin generation capacity. However, with current factor replacement treatments people with hemophilia are at risk of developing neutralizing antibodies, or inhibitors, to their replacement factor, a very serious complication affecting as many as one third of people with severe hemophilia A and a smaller fraction of people with hemophilia B. People who develop inhibitors become refractory to replacement factor therapy and are twice as likely to be hospitalized for a bleeding episode.
During 2017, we initiated the ATLAS Phase 3 program to evaluate the safety and efficacy of fitusiran in people with hemophilia A and B, with and without inhibitors. Fitusiran is also currently being evaluated in an ongoing Phase 2 OLE study. In September 2017, we reported a fatal thrombotic event in a patient with hemophilia A without inhibitors in the Phase 2 OLE study of fitusiran. As a result, we elected to suspend dosing in all ongoing fitusiran studies pending review of the safety event and development of a risk mitigation strategy. In early November 2017, we and the FDA reached alignment on clinical risk mitigation measures, including protocol-specified guidelines and additional investigator and patient education concerning reduced doses of replacement factor or bypassing agent to treat any breakthrough bleeds in fitusiran studies. Based on this, amended protocols were submitted to the regulatory authorities and in December 2017, the FDA lifted the clinical hold. Dosing has resumed in the Phase 2 OLE study and we expect to begin dosing patients in the ATLAS Phase 3 program in early 2018.
10
ATLAS Phase 3 Clinical Program
Trial Design: ATLAS is a global, multicenter program designed to evaluate the safety and efficacy of fitusiran in three separate trials, including patients with hemophilia A and B with or without inhibitors.
|
|
• |
ATLAS-INH, a nine-month, open-label, randomized, controlled trial designed to enroll approximately 50 patients with hemophilia A or B with inhibitors receiving prior on-demand therapy with bypassing agents. The study's primary endpoint is the annualized bleeding rate, or ABR, and secondary endpoints include the annualized spontaneous bleeding rate, annualized joint bleeding rate, and quality of life as measured by the Haem-A-QOL score. |
|
|
• |
ATLAS-A/B, a nine-month, open-label, randomized, controlled trial designed to enroll approximately 120 patients with hemophilia A or B without inhibitors receiving prior on-demand therapy with factor. The study's primary endpoint is the ABR, and secondary endpoints include the annualized spontaneous bleeding rate, annualized joint bleeding rate, and quality of life as measured by the Haem-A-QOL score. |
|
|
• |
ATLAS-PPX, an open-label, one-way crossover study designed to enroll approximately 30 patients with hemophilia A or B with inhibitors receiving prior prophylaxis therapy with bypassing agents. In this study, patients will receive standard of care bypassing agent prophylaxis therapy for six months and then transition to fitusiran treatment for seven months. The ABR will be prospectively measured in both periods. The study's primary endpoint is the ABR in the fitusiran period and in the bypassing agent prophylaxis period. Secondary endpoints include the annualized spontaneous bleeding rate, annualized joint bleeding rate, and quality of life as measured by the Haem-A-QOL score. |
Phase 2 OLE Clinical Study
In July 2017, we reported interim results from the ongoing fitusiran Phase 2 OLE study which includes patients with hemophilia A and B, and includes 14 patients with inhibitors, including one with hemophilia B. As of a June 15, 2017 data transfer date, once-monthly subcutaneous administration of fitusiran achieved lowering of AT, increases in thrombin generation, and, in a post-hoc exploratory analysis, reductions in the median estimated ABR in patients with and without inhibitors. With respect to safety, three SAEs were considered possibly related to study drug, including one fatal cerebral venous sinus thrombosis, as noted above, that occurred after the data cutoff date. The majority of AEs were mild or moderate in severity.
Regulatory Designations
Fitusiran has been granted the following regulatory designations for the treatment of hemophilia A and B:
|
|
• |
Orphan Drug Designation (FDA) |
|
|
• |
Orphan Medicinal Product Designation (EMA) |
As a result of the January 2018 amendment to the Sanofi Genzyme collaboration and the AT3 License Terms, following the closing of the transaction, Sanofi Genzyme will have global rights to develop and commercialize fitusiran. We expect to substantially complete the transition of the fitusiran program to Sanofi Genzyme by mid-2018. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, as well as the AT3 License Terms, are described below under the heading “Strategic Alliances.”
Inclisiran — Hypercholesterolemia
Inclisiran is a subcutaneously administered, investigational RNAi therapeutic targeting proprotein convertase subtilisin/kexin type 9, or PCSK9, for the treatment of hypercholesterolemia. PCSK9 is a protein involved in the regulation of low-density lipoprotein receptor, or LDL receptor, levels on hepatocytes and the metabolism of LDL cholesterol, or LDL-C, which is commonly referred to as “bad” cholesterol. PCSK9 is produced by the liver and circulates in the bloodstream. Both intracellular and extracellular PCSK9 reduce the liver’s capacity to absorb LDL-C by decreasing LDL receptor levels.
Despite advances in treatment, cardiovascular disease, or CVD, is the leading cause of death worldwide, resulting in over 17 million deaths annually. Eighty percent of all CVD deaths are due to coronary heart disease, or CHD, or strokes. Elevated LDL-C is a major risk factor for the development of CVD and has recently been described as causative. Lowering LDL-C has been shown to reduce the risk of cardiovascular death or heart attack, and within the range of effects achieved so far, the clinical risk reduction is linearly-proportional to absolute LDL-C reduction.
11
Approximately 100 million people worldwide are treated with lipid lowering therapies, predominantly statins, to reduce LDL-C and the associated risk of death, nonfatal myocardial infarction and nonfatal stroke or associated events. However, residual risk for cardiovascular events remains and statins are associated with well-known limitations. First, not all subjects reach LDL-C levels associated with optimal protection against clinical events. Second, not all subjects tolerate statins or are able to take statins at sufficiently-intensive doses. Third, observational studies have demonstrated that >50 percent of patients do not adhere to statin therapy for more than six months. Despite statins alone or in combination with other lipid lowering medications, current therapies for the management of elevated LDL-C remain insufficient in some subjects. This is particularly true in patients with pre-existing CHD and/or diabetes or a history of familial hypercholesterolemia, or FH, who are at the highest risk and require the most intensive management. There is an unmet need for additional treatment options beyond currently-available treatments for lowering of the LDL-C level to reduce cardiovascular risk.
In February 2013, we and MDCO entered into a license and collaboration agreement pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases. Under the terms of the agreement, MDCO assumed responsibility for the development and commercialization of inclisiran from Phase 2 forward. A description of our agreement with MDCO is included below under the heading “Strategic Alliances.”
In 2017, MDCO initiated the ORION Phase 3 program for inclisiran, a comprehensive set of clinical trials to assess LDL-C lowering and safety in a wide range of patients. The Phase 3 program includes the four pivotal Phase 3 clinical trials described below and represents the largest clinical experience for an investigational RNAi therapeutic to date:
|
|
• |
ORION-11 – a placebo-controlled, double-blind, randomized Phase 3 study of inclisiran versus placebo (1:1) in patients (N=1,500) with atherosclerotic cardiovascular disease, or ASCVD, or ASCVD-risk equivalents, and elevated LDL-C despite maximum tolerated doses of LDL-C lowering therapies, including statins. This pivotal trial was initiated in November 2017. In January 2018, MDCO announced that this trial had exceeded its target enrollment of 1,500 patients. |
|
|
• |
ORION-10 – a placebo-controlled, double-blind, randomized Phase 3 study of inclisiran versus placebo (1:1) in ASCVD patients (N=1,500). This pivotal trial was initiated in November 2017 and MDCO expects to complete enrollment during the first half of 2018. |
|
|
• |
ORION-9 – a placebo-controlled, double-blind, randomized Phase 3 study of inclisiran versus placebo (1:1) in patients (N=400) with heterozygous FH. This pivotal trial was initiated in November 2017 and MDCO expects to complete enrollment during the first half of 2018. |
|
|
• |
ORION-5 – a placebo-controlled, double-blind, randomized Phase 3 study of inclisiran versus placebo (1:1) in patients (N=60) with homozygous FH, or HoFH. MDCO expects to initiate this pivotal trial in 2018. |
ORION-1 Phase 2 Clinical Trial
During 2017, we and MDCO reported final results from ORION-1, a placebo-controlled, double-blind, randomized Phase 2 study of inclisiran in patients (N=501) with ASCVD or ASCVD-risk equivalents (e.g., diabetes and FH) and elevated LDL-C despite maximum tolerated doses of LDL-C lowering therapies. The Phase 2 study, conducted by MDCO, compared the effect of different doses of inclisiran and evaluated the potential for an infrequent dosing regimen. The primary endpoint of the study was the percentage change in LDL-C from baseline at Day-180. Inclisiran demonstrated significant and sustained reductions in LDL-C of over 50 percent. For all dose groups, at all time points, differences in the primary (LDL-C) and secondary (PCSK9) endpoints between inclisiran and placebo were statistically significant (p < 0.0001). Inclisiran represents the largest safety experience for one of our investigational RNAi therapeutics to date. Inclisiran was generally well tolerated and no material safety issue was observed, including no significant elevations of liver enzymes considered related to study medication and no neuropathy, change in renal function, thrombocytopenia or anti-drug antibodies.
The results from the Phase 2 study of inclisiran were published in April 2017 in The New England Journal of Medicine.
Regulatory Designations
Inclisiran has been granted the following regulatory designation for the treatment of HoFH:
|
|
• |
Orphan Drug Designation (FDA) |
12
Early Stage Clinical Development Programs
ALN-TTRsc02 — TTR-Mediated (ATTR) Amyloidosis
ALN-TTRsc02 is a subcutaneously administered, investigational RNAi therapeutic targeting TTR for the treatment of ATTR amyloidosis, representing an extension of our program for ATTR amyloidosis, discussed in more detail above under the heading “Patisiran.” We believe ALN-TTRsc02 has the potential to offer an attractive option for patients as a once-quarterly, low-volume, subcutaneously administered RNAi therapeutic for the treatment of ATTR amyloidosis.
We are evaluating ALN-TTRsc02 in a randomized, placebo-controlled, single ascending-dose Phase 1 study in healthy volunteers and we expect to advance ALN-TTRsc02 into a Phase 3 clinical trial in late 2018.
As a result of the 2018 amendment to the Sanofi Genzyme collaboration and the Exclusive TTR License, following the closing of the transaction, we will have global rights for the development and commercialization of patisiran, discussed above, together with ALN-TTRsc02 and all back-up products. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, as well as the Exclusive TTR License, are described below under the heading “Strategic Alliances.”
Lumasiran (ALN-GO1) — Primary Hyperoxaluria 1
Lumasiran is an investigational RNAi therapeutic targeting glycolate oxidase, or GO, in development for the treatment of primary hyperoxaluria type 1, or PH1. PH1 is an autosomal recessive disorder of glyoxylate metabolism, resulting in excessive oxalate production. Excess oxalate in PH1 patients results in the deposition of calcium oxalate crystals in the kidneys and urinary tract and can lead to the formation of recurrent kidney stones or nephrocalcinosis. Renal damage is caused by a combination of tubular toxicity from oxalate, calcium oxalate deposition in the kidneys, and urinary obstruction by calcium oxalate stones. Compromised kidney function exacerbates the disease as the excess oxalate can no longer be effectively excreted, resulting in subsequent accumulation and crystallization in bones, eyes, skin, and heart, leading to severe illness and death. Lumasiran is designed to reduce the hepatic levels of the GO enzyme, thereby depleting the substrate necessary for oxalate production, which directly contributes to the pathophysiology of PH1.
We are evaluating lumasiran for the treatment of patients with PH1 in a randomized, single-blind, placebo-controlled Phase 1/2 clinical trial.
Sanofi Genzyme has the right to opt in to develop and commercialize lumasiran in the Sanofi Genzyme Territory and could elect to exercise its one right to a global license for lumasiran. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, is described below under the heading “Strategic Alliances.”
Cemdisiran (ALN-CC5) — Complement-Mediated Diseases
Cemdisiran is an investigational RNAi therapeutic targeting the C5 component of the complement pathway in development for the treatment of complement-mediated diseases. The complement system plays a central role in immunity as a protective mechanism for host defense, but its dysregulation results in life-threatening complications in a broad range of human diseases including paroxysmal nocturnal hemoglobinuria, or PNH, and aHUS, amongst others. Complement component C5, which is predominantly expressed in liver cells, is a genetically and clinically validated target; loss-of-function human mutations are associated with an attenuated immune response against certain infections and intravenous anti-C5 monoclonal antibody, or mAb, therapy has demonstrated clinical activity and tolerability in a number of complement-mediated diseases. A subcutaneously administered RNAi therapeutic that silences C5 represents a novel approach for the potential treatment of complement-mediated diseases.
We are evaluating cemdisiran for the treatment of patients with aHUS in a Phase 2 clinical trial initiated in September 2017.
During 2016, Sanofi Genzyme elected not to opt into the development and commercialization of cemdisiran in the Sanofi Genzyme Territory, providing us with full global control of the program for further development and commercialization, if approved. The 2014 Sanofi Genzyme collaboration, as amended in January 2018, is described below under the heading “Strategic Alliances.”
Additional Early Stage and Pre-clinical Programs
In addition to the programs listed above, we are also advancing other earlier-stage clinical pipeline programs and expect multiple data read-outs throughout 2018. We also plan to file one or more new clinical trial applications, or CTAs, in 2018, and to advance our infectious disease collaboration with Vir Biotechnology.
13
Our Collaboration and Licensing Strategy
Our business strategy is to develop and commercialize a broad pipeline of RNAi therapeutic products directed towards our three STArs: Genetic Medicines; Cardio-Metabolic Diseases; and Hepatic Infectious Diseases. As part of this strategy, we have entered into, and expect to enter into additional, collaboration and licensing agreements as a means of obtaining resources, capabilities and funding to advance our investigational RNAi therapeutic programs.
Our collaboration strategy is to form alliances that create significant value for ourselves and our collaborators in the advancement of RNAi therapeutics as a potential new class of innovative medicines. Specifically, with respect to our Genetic Medicine pipeline, we formed a broad strategic alliance with Sanofi Genzyme in 2014 pursuant to which we retain development and commercial rights for our current and future Genetic Medicine products in the United States, Canada and Western Europe, and Sanofi Genzyme will develop and commercialize our current and future Genetic Medicine products for which it elects to opt-in, in the rest of the world, subject to certain broader rights. In January 2018, we and Sanofi Genzyme amended our 2014 collaboration to provide that we would develop and commercialize patisiran globally and Sanofi Genzyme would develop and commercialize fitusiran globally. With respect to our Cardio-Metabolic pipeline, we intend to seek future strategic alliances for these programs, under which we may retain certain product development and commercialization rights, or we may structure as global alliances, as we did in our collaboration with MDCO to advance inclisiran. With respect to our Hepatic Infectious Disease pipeline, in October 2017, we announced an exclusive licensing agreement with Vir Biotechnology for the development and commercialization of RNAi therapeutics for infectious diseases, including chronic hepatitis B virus infection.
We also have entered into license agreements to obtain rights to intellectual property in the field of RNAi. In addition, because delivery of RNAi therapeutics has historically been an important objective of our research activities, we have entered into various collaboration and licensing arrangements with other companies and academic institutions to gain access to delivery technologies, including various LNP delivery technologies.
Strategic Alliances
We have formed, and intend to continue to form, strategic alliances to gain access to the financial, technical, clinical and commercial resources necessary to develop and market RNAi therapeutics. We expect these alliances to provide us with financial support in the form of upfront cash payments, license fees, equity investments, research, development, and sales and marketing funding, milestone payments and/or royalties or profit sharing based on sales of RNAi therapeutics. Below is a brief description of our key strategic alliance and license agreements.
Product Alliances.
Sanofi Genzyme. In January 2014, we entered into a global, strategic collaboration with Sanofi Genzyme to discover, develop and commercialize RNAi therapeutics as Genetic Medicines to treat orphan diseases. On January 6, 2018, we and Sanofi Genzyme entered into an amendment to the 2014 Sanofi Genzyme collaboration, which is structured as an exclusive relationship for the worldwide development and commercialization of RNAi therapeutics in the field of Genetic Medicines. In connection and simultaneously with entering into the 2018 amendment to the 2014 Sanofi Genzyme collaboration, we and Sanofi Genzyme also entered into the Exclusive TTR License with respect to all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, and the AT3 License Terms with respect to fitusiran and any back-up products.
Under the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme has certain rights to our current and future Genetic Medicine programs that reach Human POP by the end of 2019, subject to extension to the end of 2021 in various circumstances. Under the 2014 Sanofi Genzyme collaboration, we were leading development and commercialization of patisiran in the Alnylam Territory, while Sanofi Genzyme had rights to develop and commercialize the product in the Sanofi Genzyme Territory. Sanofi Genzyme also had a right to opt in to co-develop and co-promote ALN-TTRsc02 in the Alnylam Territory along with its regional opt-in rights. In addition, Sanofi Genzyme had opted in to co-develop and co-promote fitusiran in the Alnylam Territory, as well as develop and commercialize fitusiran in the Sanofi Genzyme Territory.
The 2018 amendment, together with the Exclusive TTR License and the AT3 License Terms, revise the terms and conditions of the 2014 Sanofi Genzyme collaboration to (i) provide us with the exclusive right to pursue the further global development and commercialization of all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, (ii) provide Sanofi Genzyme the exclusive right to pursue the further global development and commercialization of fitusiran and any back-up products and (iii) terminate the previous co-development and co-commercialization rights related to revusiran, ALN-TTRsc02 and fitusiran under the 2014 Sanofi Genzyme collaboration.
14
Sanofi Genzyme continues to have the right to opt into our other rare genetic disease programs for development and commercialization in territories outside of the Alnylam Territory as contemplated in the 2014 Sanofi Genzyme collaboration, as well as one right to a global license.
The transaction is subject to customary closing conditions and clearances, including clearance under the Hart-Scott-Rodino Antitrust Improvements Act. We expect the transaction to close during the first quarter of 2018.
For more information regarding the 2014 Sanofi Genzyme collaboration, as amended in January 2018, as well as the Exclusive TTR License and the AT3 License Terms, including the ongoing or expected financial and accounting impact on our business, please read Note 3, Significant Agreements, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
The Medicines Company. In February 2013, we and MDCO entered into a license and collaboration agreement pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases. Under the MDCO agreement, we had responsibility for the development of inclisiran until Phase 1 Completion, as defined in the MDCO agreement, at our cost. In late 2015, MDCO assumed responsibility for all development and commercialization of inclisiran, at its sole cost, and is advancing inclisiran in a comprehensive Phase 3 development program. For more information regarding the MDCO agreement, including its ongoing financial and accounting impact on our business, please read Note 3, Significant Agreements, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
Platform Alliances.
Monsanto Company. In August 2012, we and Monsanto Company, or Monsanto, entered into a license and collaboration agreement, pursuant to which we granted to Monsanto a worldwide, exclusive, royalty bearing right and license, including the right to grant sublicenses, to our RNAi platform technology and intellectual property controlled by us as of the date of the Monsanto agreement or during the 30 months thereafter, in the field of agriculture. The Monsanto agreement also included the transfer of technology from us to Monsanto and initially included a collaborative research project. Under the Monsanto agreement, Monsanto will be our exclusive collaborator in the agriculture field for a ten-year period. For more information regarding the Monsanto agreement, including its ongoing financial and accounting impact on our business, please read Note 3, Significant Agreements, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
Takeda Pharmaceutical Company Limited. In May 2008, we entered into a license and collaboration agreement with Takeda Pharmaceutical Company Limited, or Takeda, to pursue the development and commercialization of RNAi therapeutics. Under the Takeda agreement, we granted to Takeda a non-exclusive, worldwide, royalty-bearing license to our intellectual property, including delivery-related intellectual property, controlled by us as of the date of the Takeda agreement or during the five years thereafter, to develop, manufacture, use and commercialize RNAi therapeutics, subject to our existing contractual obligations to third parties. The license initially is limited to the fields of oncology and metabolic disease and may be expanded at Takeda’s option to include other therapeutic areas, subject to specified conditions. For more information regarding the Takeda agreement, including its ongoing financial and accounting impact on our business, please read Note 3, Significant Agreements, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
Other Strategic License Agreements.
Ionis Pharmaceuticals, Inc. (formerly Isis Pharmaceuticals, Inc.). In January 2015, we and Ionis Pharmaceuticals, Inc., or Ionis, entered into a second amended and restated strategic collaboration and license agreement, which we further amended in July 2015. The 2015 Ionis agreement provides for certain new exclusive target cross-licenses of intellectual property on eight disease targets, providing each company with exclusive RNA therapeutic license rights for four programs, and extends the parties’ existing non-exclusive technology cross-license, which was originally entered into in 2004 and was amended and restated in 2009, through April 2019. Under the original agreement, Ionis licensed to us its patent estate related to antisense motifs and mechanisms and oligonucleotide chemistry for double-stranded RNAi products. In turn, we non-exclusively licensed to Ionis our patent estate relating to antisense motifs and mechanisms and oligonucleotide chemistry to research, develop and commercialize single-stranded antisense therapeutics, single stranded RNAi therapeutics and to research double-stranded RNAi compounds. Ionis also received a license to develop and commercialize double-stranded RNAi drugs targeting a limited number of therapeutic targets on a non-exclusive basis. For more information regarding the 2015 Ionis agreement, including its ongoing financial and accounting impact on our business, please read Note 3, Significant Agreements, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
15
Intellectual Property Licenses
In December 2002, we entered into a co-exclusive license with Max Planck Innovation GmbH (formerly known as Garching Innovation GmbH), or Max Planck Innovation, for the worldwide rights to use and sublicense certain patented technology to develop and commercialize therapeutic products and related applications. We also obtained the rights to use, without the right to sublicense, the technology for all diagnostic uses other than for the purposes of therapeutic monitoring. We were also given the right to acquire the remaining 50 percent exclusive rights, which right we exercised upon our acquisition of Ribopharma AG in July 2003. In June 2005, we entered into an amendment to our agreement with Max Planck Innovation that secured our exclusivity to use and sublicense certain patented technology to develop and commercialize therapeutic products and related applications.
We are not obligated to pay any development or sales milestone payments to Max Planck Innovation, however, we will be required to pay Max Planck Innovation future single-digit royalties on net sales of all therapeutic and prophylactic products developed with the technology, if any.
Our agreements with Max Planck Innovation generally remain in effect until the expiration of the last-to-expire patent licensed thereunder. We estimate that the principal issued patents covered under the Max Planck Innovation agreements will expire both in and outside the United States during 2021, subject to any potential patent term extensions, restoration and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. We may terminate the agreements without cause with six months’ prior notice to Max Planck Innovation, and Max Planck Innovation may terminate the agreements in the event that we materially breach our obligations thereunder. Max Planck Innovation also has the right to terminate the agreements in the event that we, independently or through a third party, attack the validity of any of the licensed patents.
Delivery-Related License Agreements
Arbutus. In November 2012, we, Arbutus Biopharma Corporation, or ABC (formerly Tekmira Pharmaceuticals Corporation), and Protiva Biotherapeutics, Inc., or Protiva, a wholly owned subsidiary of ABC, and together with ABC, referred to as Arbutus, agreed to restructure our existing contractual relationship. In connection with this restructuring, the parties entered into a cross-license agreement that superseded the prior license and manufacturing agreements among us.
Under the 2012 cross-license agreement, the parties consolidated certain intellectual property related to LNP technology for the systemic delivery of RNAi therapeutics. Specifically, certain patents and patent applications, including the MC3 lipid family used with patisiran, were assigned by us to ABC. We retain rights to use this intellectual property for the research, development and commercialization of RNAi therapeutic products, including the rights to sublicense this intellectual property on a product-by-product basis. Arbutus has also granted us a worldwide license to its LNP technology for the research, development and commercialization of LNP-based RNAi therapeutics, which license shall be exclusive for up to eight targets designated by us, and otherwise shall be non-exclusive. We have the right to sublicense on a product-by-product basis.
In addition, we elected to buy out our manufacturing obligations to ABC with respect to our LNP-based pipeline programs. We made a one-time payment of $30.0 million to ABC for the termination of, and our release from, all of our obligations under the manufacturing agreement. We also have the right to manufacture and have manufactured our LNP-based RNAi therapeutics, which right may be sublicensed to our collaborators.
Further, pursuant to the 2012 cross-license agreement, we made a one-time payment of $35.0 million to ABC, which amount included a license termination payment, as well as the buy-down of certain milestone payments and the significant reduction of royalty rates for certain LNP-based products, including patisiran. In addition, we agreed to pay ABC an aggregate of $10.0 million in contingent milestone payments related to advancement of ALN-VSP and patisiran, representing the only remaining milestone obligations for these products. In December 2013, we paid to ABC $5.0 million in connection with the initiation of our APOLLO Phase 3 clinical trial for patisiran, fulfilling one of these milestone obligations. With respect to the second $5.0 million milestone, in August 2013, we initiated binding arbitration proceedings to resolve a disagreement with ABC regarding the achievement by ABC of this milestone under our cross-license agreement relating to the manufacture of ALN-VSP clinical trial material for use in China. We prevailed in the arbitration proceedings in March 2016 and were not required to pay the second milestone at that time.
Under the 2012 cross-license agreement, Arbutus has one exclusive and five non-exclusive licenses to research, develop and commercialize RNAi therapeutics directed to up to six gene targets. As of November 12, 2017, Arbutus’ right to select up to two additional exclusive targets and up to five additional non-exclusive targets expired. Arbutus may sublicense its rights on a product-by-product basis. We are eligible to receive from Arbutus up to an aggregate of $8.5 million in milestone payments for RNAi therapeutics directed to each of four of the targets for which we have granted licenses to Arbutus, together with single-digit royalties on annual product sales, if any, of RNAi therapeutic products directed to all six of the targets for which we have granted licenses to Arbutus. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Arbutus.
16
The term of the 2012 cross-license agreement generally ends upon the expiration of the last-to-expire royalty term. Royalties are payable on a product-by-product and country-by-country basis commencing on the first commercial sale of a product in a country and continuing during any period in which (a) in the case of us, a valid claim within the Arbutus Royalty-Bearing Patents (as defined in the 2012 cross-license agreement) covers our applicable product in such country of sale, or (b) in the case of Arbutus products, a valid claim within our patents covers the applicable Arbutus product in such country of sale. We estimate that our fundamental RNAi patents covered under the 2012 cross-license agreement will expire both in and outside the United States generally between 2019 and 2021, and that the Arbutus LNP patents covered under the 2012 cross-license agreement will expire both in and outside the United States generally between 2020 and 2030, subject in each case to any potential patent term extensions and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. Either party may terminate a license it granted to the other in the event that the other party fails to cure a material breach of its obligations relating to that license. Furthermore, either party may terminate the 2012 cross-license agreement in the event the other party fails to cure a material breach of an obligation under the agreement. In addition, either party may terminate the 2012 cross-license agreement upon patent-related challenges by the other party.
UBC and Acuitas. In July 2009, we entered into a research agreement with The University of British Columbia, or UBC, and Acuitas Therapeutics Inc., or Acuitas (formerly AlCana Technologies, Inc.), that was focused on the discovery of novel lipids, such as the MC3 lipid, which is employed in patisiran. Pursuant to the terms of the research agreement, we funded collaborative research through July 2012, which was conducted by our scientists, together with scientists at UBC and Acuitas. Under the research agreement, UBC and Acuitas are eligible to receive up to an aggregate of $1.3 million in milestone payments from us for each licensed product (as defined in the research agreement) directed to a particular target (as defined in the research agreement), together with single-digit royalty payments on annual product sales, if any.
Concurrent with the execution of the research agreement, we also entered into a supplemental agreement with ABC, Protiva, UBC and Acuitas, which contains additional terms regarding the intellectual property rights arising out of the research agreement. In connection with 2012 cross-license agreement with Arbutus described above, we and Arbutus agreed to supersede the rights and obligations under the supplemental agreement as between ourselves, with the rights and obligations set forth in the 2012 cross-license agreement.
Patents and Proprietary Rights
We have devoted considerable effort and resources to establish what we believe to be a strong intellectual property position relevant to RNAi therapeutic products and delivery technologies. In this regard, we have amassed a portfolio of patents, patent applications and other intellectual property covering:
|
|
• |
fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms; |
|
|
• |
chemical modifications to siRNAs that improve their suitability for therapeutic and other uses; |
|
|
• |
siRNAs directed to specific targets as treatments for particular diseases; |
|
|
• |
delivery technologies, such as in the fields of carbohydrate conjugates and cationic liposomes; and |
|
|
• |
all aspects of our specific development candidates. |
We believe that no other company possesses a portfolio of such broad and exclusive rights to the patents and patent applications required for the commercialization of RNAi therapeutics. Our intellectual property estate for RNAi therapeutics includes over 3,800 active cases and over 1,700 granted or issued patents, of which over 600 are issued or granted in the United States, the EU, including by the European Patent Office, or EPO, and Japan. Given the importance of our intellectual property portfolio to our business operations, we intend to vigorously enforce our rights and defend against challenges that have arisen or may arise in this area.
Intellectual Property Related to Fundamental Aspects and Uses of siRNA and RNAi-related Mechanisms
In this category, we include United States and certain foreign patents and patent applications that claim key aspects of siRNA architecture and RNAi-related mechanisms. Specifically included are patents and patent applications covering targeted cleavage of mRNA directed by RNA-like oligonucleotides and double-stranded RNAs of particular lengths and particular structural features, such as blunt and/or overhanging ends, as well as various types and patterns of chemical modifications. Our strategy has been to secure exclusive rights where possible and appropriate to key patents and patent applications that we believe cover fundamental aspects of RNAi.
17
The following table lists patents and/or patent applications to which we have secured rights that we regard as being fundamental for the use of siRNAs as therapeutics.
|
Patent Licensor/Owner |
|
Subject Matter |
|
First Priority Date |
|
Inventors |
|
Status |
|
Expiration Date* |
|
Alnylam Rights |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Carnegie Institution of Washington |
|
Double-stranded RNAs to induce RNAi |
|
12/23/1997 |
|
A. Fire, C. Mello |
|
U.S. 6,506,559, U.S. 7,560,438, U.S. 7,538,095, U.S. 7,622,633, U.S. 8,580,754, U.S. 8,283,329 & U.S. 9,102,939
Additional applications pending in the U.S. and several foreign jurisdictions |
|
12/18/2018 |
|
Non-exclusive |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Medical College of Georgia Research Institute, Inc. |
|
Methods for inhibiting gene expression using double-stranded RNA |
|
1/28/1999 |
|
Y. Li, M. Farrell, M. Kirby |
|
U.S. 7,888,325 & U.S. 8,148,345
AU 776150 (Australia)
Additional applications pending in the U.S., Europe and Canada |
|
1/28/2020 |
|
Exclusive rights |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Small double-stranded RNAs as therapeutic products |
|
1/30/1999 |
|
R. Kreutzer, S. Limmer |
|
U.S. 7,763,590, U.S. 7,829,697 & U.S. 7,994,309
EP 1798285, EP 2363479, EP 1144623, EP 1214945 (revoked/under appeal), EP 1550719 (revoked/under appeal), CA 2359180 (Canada), AU 778474 (Australia), ZA 2001/5909 (South Africa), DE 20023125 U1, DE 10066235 & DE 10080167 (Germany)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
1/29/2020 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Medicament for inhibiting the expression of a target gene and medicament for treating a tumor disease |
|
1/9/2001 |
|
R. Kreutzer, S. Limmer, H-P.Vornlocher, P. Hadwiger, A. Geick, M. Ocker, C. Herold, D. Schuppan |
|
U.S. 7,868,160 & U.S. 8,143,390
EP 1799270 & EP 1349927 (opposed and maintained in amended form) |
|
1/9/2022 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Method for inhibiting the expression of a wide variety of oncology target genes with double-stranded RNA between 15-49 nucleotides |
|
1/9/2001 |
|
R. Kreutzer, S. Limmer, P. Hadwiger |
|
U.S. 8,273,870, U.S. 8,546,143 & U.S. 9,074,213
EP 1352061 (opposed, maintained with no further right to appeal) |
|
1/9/2022 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Composition and methods for inhibiting a target nucleic acid with double-stranded RNA of between 20-49 base pairs wherein at least one end is blunt |
|
1/9/2001 |
|
R. Kreutzer, St. Limmer, Sy. Limmer, P. Hadwiger |
|
U.S. 9,587,240 |
|
1/9/2022 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Composition and methods for inhibiting a target nucleic acid with double-stranded RNA |
|
4/21/1999 |
|
C. Pachuk, C. Satishchandran |
|
EP 1171586, AU 781598 (Australia)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
4/19/2020 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
18
|
Patent Licensor/Owner |
|
Subject Matter |
|
First Priority Date |
|
Inventors |
|
Status |
|
Expiration Date* |
|
Alnylam Rights |
|
Technology Limited |
|
RNAi uses in mammalian oocytes, preimplantation embryos and somatic cells (EP only: wherein the RNAi compound is at least 25 base pairs) |
|
11/19/1999 |
|
M. Zernicka- Goetz, F. Wianny, M.J. Evans, D.M. Glover |
|
EP 1230375 (revoked/successfully appealed and granted in amended form), SG 89569 (Singapore), AU 774285 (Australia)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
11/17/2020 |
|
Exclusive rights |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Massachusetts Institute of Technology, Whitehead Institute for Biomedical Research, Max Planck Gesellschaft, University of Massachusetts ** |
|
Mediation of RNAi by small RNAs 21-23 base pairs long with claims directed to compositions, methods of use and manufacture |
|
3/30/2000 |
|
D.P. Bartel, P.A. Sharp, T. Tuschl, P.D. Zamore |
|
U.S. 8,790,922, U.S. 8,742,092, U.S. 8,632,997, U.S. 8,552,171, U.S. 8,420,391, U.S. 8,394,628, U.S. 8,957,157, U.S. 9,012,138, U.S. 9,012,621 & U.S. 9,193,753
EP 1309726 (opposed and maintained in amended form/under appeal), EP 2028278 (opposed), EP 2345742, EP 2360253 (opposed) & EP 2361981 (opposed), AU 2001249622 (Australia), NZ 522045 (New Zealand), KR 08724437 & KR 10-0909681 (Korea)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
3/30/2021 |
|
Exclusive rights |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Massachusetts Institute of Technology, Whitehead Institute, University of Massachusetts, Max Planck Gesellschaft (U.S.)***
Max Planck Gesellschaft (ex-U.S.), European Molecular Biology Laboratory (ex-U.S.)**** |
|
Synthetic and chemically modified siRNAs as therapeutic products including patents with claims including those directed to double-stranded RNA of between 19 to 23 or 19 to 25 nucleotides, with and without a 3’ overhang; claims directed to double-stranded RNA of between 19 to 52 nucleotides with a 3’ overhang; claims directed to double-stranded RNA of 14 to 24 base pairs or up to 25 base pairs with at least one nucleotide analogue, along with methods of using and making such double-stranded RNA |
|
12/1/2000 (EP), |
|
T. Tuschl, S. Elbashir, W. Lendeckel, M. Wilm#, R. Lührmann#
#EMBL inventors |
|
U.S. 7,056,704, U.S. 7,078,196, U.S. 8,329,463, U.S. 8,372,968, U.S. 8,362,231, U.S. 8,445,237, U.S. 8,765,930, U.S. 8,778,902, U.S. 8,796,016, U.S. 8,853,384, U.S. 8,895,721, U.S. 8,933,044, U.S. 8,895,718, U.S. 8,993,745 & U.S. 9,567,582
EP 1407044 (opposed and maintained in amended form/under appeal), EP 1873259, EP 2348133, EP 2348134, EP 2351852 (opposed) & EP 2813582 AU 2002235744 (Australia), ZA 2003/3929 (South Africa), SG 96891 (Singapore), NZ 52588 (New Zealand), JP 4 095 895 (Japan) (opposed and maintained), JP 4 494 392 (Japan), RU 2322500 (Russia), CN 1568373 (China)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
11/29/2021 |
|
Exclusive rights |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
19
|
Patent Licensor/Owner |
|
Subject Matter |
|
First Priority Date |
|
Inventors |
|
Status |
|
Expiration Date* |
|
Alnylam Rights |
|
|
Methods for inhibiting a target nucleic acid via the introduction of a vector encoding a double-stranded RNA |
|
1/31/2001 |
|
T. Giordano, C. Pachuk, C. Satishchandran |
|
U.S. 9,051,566
AU 785395 (Australia)
Additional applications pending in the U.S., Australia and Canada |
|
1/31/2021 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stanford University |
|
RNAi uses in vivo in mammalian liver |
|
7/23/2001 |
|
M.A. Kay, A.P. McCaffrey |
|
U.S. 9,018,179
EP 1409506, AU 2002326410 (Australia)
Additional applications pending in the U.S. and several foreign jurisdictions |
|
7/23/2021 |
|
Exclusive |
|
Alnylam |
|
Claims directed to carbohydrate conjugates linked to siRNA |
|
4/17/2003 |
|
M. Manoharan |
|
U.S. 7,723,509, U.S. 7,745,608, U.S. 7,851,615, U.S. 8,017,762, U.S. 8,507,661, U.S. 8,344,125, U.S. 8,796,436, U.S. 8,865,677 & U.S. 8,426,377
Additional applications pending in the U.S. and several foreign jurisdictions |
|
9/21/2024 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Alnylam |
|
Claims directed to GalNAc-conjugated siRNA |
|
12/4/2007 |
|
M. Manoharan |
|
U.S. 8,106,022, U.S. 8,450,467, U.S. 8,828,956 & U.S. 9,370,581
Additional applications pending in the U.S. and several foreign jurisdictions |
|
12/4/2028 |
|
Owned |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sirna***** |
|
Claims directed to highly chemically modified oligonucleotides with granted claims directed to double-stranded RNA of between 18 and 24 nucleotides with various combinations of chemical modifications |
|
2/20/2002 |
|
J. McSwiggen |
|
U.S. 7,923,547, U.S. 7,956,176, U.S. 7,989,612, U.S. 8,232,383, U.S. 8,268,986, U.S. 8,236,944, U.S. 8,272,979, U.S. 8,273,866, U.S. 8,242,257, U.S. 8,618,277, U.S. 8,846,894, U.S. 8,648,185, U.S. 9,181,551, U.S. 9,732,344 & U.S. 9,771,588
EP 1423406 (opposed and maintained), EP 2287306 (opposed and maintained in amended form), EP 2278004 (opposed, opposition withdrawn), EP 1627061, EP 1458741 (opposed, opposition withdrawn) & EP 1931781, AU 2003216324, AU 2006203725, CA 2526831 (Canada), JP 49481631 (Japan)
Additional cases pending in the US and Europe |
|
2/20/2023- |
|
Owned |
|
* |
For applications filed after June 7, 1995, the patent term generally is 20 years from the earliest application filing date. However, under the Drug Price Competition and Patent Term Extension Act of 1984, known as the Hatch-Waxman Act, we may be able to apply for patent term extensions for our U.S. patents. We cannot predict whether or not any patent term extensions will be granted or the length of any patent term extension that might be granted. |
20
|
*** |
We hold exclusive rights to the interest owned by all co-owners in the U.S. UMass had a right to sublicense the U.S. Tuschl II patent family to Merck but such right has been disclaimed by UMass. |
|
**** |
European Molecular Biology Laboratory, or EMBL, has a limited ownership interest in certain ex-US cases in this family with no rights to control or otherwise affect patent prosecution. |
|
***** |
Sirna is our wholly-owned subsidiary. |
We believe that we have a strong portfolio of broad rights to fundamental RNAi patents and patent applications. Many of these rights are exclusive, which we believe prevents potential competitors from commercializing products in the field of RNAi without taking a license from us. In securing these rights, we have focused on obtaining the strongest rights for those intellectual property assets we believe will be most important in providing competitive advantage with respect to RNAi therapeutic products.
We believe that the Crooke patent series, issued in several countries around the world, covers the use of modified oligonucleotides to achieve enzyme-mediated cleavage of a target mRNA. We have obtained rights to the Crooke patents for use with double-stranded RNA products, through a license agreement with Ionis. Under the terms of our agreement, Ionis agreed not to grant licenses under these patents to any other organization for double-stranded RNA products designed to work through an RNAi mechanism, except in the context of a collaboration in which Ionis plays an active role. Our agreement with Ionis was amended and restated in January 2015 to, among other things, extend the license for an additional five years, through April 2019.
Through our acquisition of Ribopharma AG, now known as Alnylam Europe AG, we own the entire Kreutzer-Limmer patent portfolio, which includes pending applications in the United States and many countries worldwide.
The Glover patent series has resulted in several patent grants, including in Europe (EP 1230375). We have an exclusive license to the Glover patent for therapeutic uses from Cancer Research Technology Limited, or CRT.
The Tuschl patent applications owned by Whitehead Institute for Biomedical Research, or Whitehead, the Massachusetts Institute of Technology, or MIT, UMass and Max Planck Gesellschaft zur Foerderung der Wissenschaften e.V. on the invention by Dr. Tuschl and his colleagues, which we call the Tuschl I patent series, cover compositions and methods important for RNAi discovery. We are the exclusive licensee of the Tuschl I patent series for RNAi therapeutics. The Tuschl patent applications owned by Max Planck Gesellschaft zur Foerderung der Wissenschaften e.V., Whitehead, MIT and UMass on the invention by Dr. Tuschl and his colleagues, which we call the Tuschl II patent series, cover what we believe are key structural features of siRNAs. Specifically, the Tuschl II patents and patent applications include claims directed to synthetic siRNAs and the use of chemical modifications to stabilize siRNAs. We have obtained an exclusive license to claims in the Tuschl II patent series uniquely covering the use of RNAi for therapeutic purposes. Collectively, the Tuschl I and II patent families cover a wide range of double-stranded RNA molecules between 19-52 nucleotides in length, including those unmodified and those comprising chemical modifications. Examples of those chemical modifications encompassed by the Tuschl claims include those modifications made in the ribose ring, e.g., at the 2’ position such as 2’-OMe, 2’-F or modifications such as those found in locked and unlocked (acyclic) nucleotides.
The Fire and Mello patent owned by the Carnegie Institution of Washington covers the use of double-stranded RNAs to induce RNAi. The Carnegie Institution has made this patent broadly available for licensing and we, like many companies, have taken a non-exclusive license to the patent for therapeutic purposes. We believe, however, that the claims of the Fire and Mello patent do not cover the structural features of double-stranded RNAs that are important for the biological activity of siRNAs in mammalian cells. We believe that these specific features are the subjects of the Crooke, Kreutzer-Limmer, Glover and Tuschl II patents and patent applications for which we have secured exclusive rights.
The other pending patent applications listed in the table above either provide further coverage for structural features of siRNAs or relate to the use of siRNAs in mammalian cells. For some of these, we have exclusive rights, and for others, we have non-exclusive rights. In addition, in December 2008, we acquired the intellectual property assets of Nucleonics, Inc., a privately held biotechnology company. This acquisition included over 100 active patent filings, including 15 patents that have been granted worldwide, of which five have been granted in the United States and Europe. With this acquisition, we obtained patents and patent applications with early priority dates, notably the “Li & Kirby,” “Pachuk I” and “Giordano” patent families, that cover broad structural features of RNAi therapeutics, thus extending the breadth of our fundamental intellectual property.
21
Intellectual Property Related to Chemical Modifications
Our amended and restated collaboration and license agreement with Ionis provided us with rights to practice the inventions covered by over 200 issued patents worldwide, as well as rights based on future chemistry patent applications through April 2014 for use with double-stranded RNA products. In January 2015, we entered into a second amended and restated agreement with Ionis to extend our rights to future chemistry applications through April 2019. These patents expire both in and outside the United States generally between 2015 and 2035, subject to any potential patent term extensions and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. These inventions cover chemical modifications we may wish to incorporate into double-stranded RNA therapeutic products designed to work through an RNAi mechanism. Under the terms of our agreement, Ionis agreed not to grant licenses under these patents to any other organization for double-stranded RNA products designed to work through an RNAi mechanism, except in the context of a collaboration in which Ionis plays an active role.
In addition to licensing these intellectual property rights from Ionis, we are also working to develop our own proprietary chemical modifications that may be incorporated into siRNAs to endow them with drug-like properties. We have filed a large number of patent applications relating to these novel and proprietary chemical modifications.
With the combination of the technology we have licensed from Ionis, various patents in the Tuschl II patent series and our own patent application filings, we possess issued claims that cover methods of making siRNAs that incorporate any of various chemical modifications, including the use of phosphorothioates, 2’-O-methyl and/or 2’-fluoro modifications and modifications such as those found in locked and unlocked (acyclic) nucleotides. These modifications are believed to be important for achieving “drug-like” properties for RNAi therapeutics. We hold exclusive worldwide rights to these claims for RNAi therapeutics.
In addition to the above, in March 2014, we acquired the RNAi assets from Merck, which included intellectual property developed at Sirna and Merck. The acquired patent portfolio includes the “McSwiggen” patent families with issued and pending claims covering highly chemically modified oligonucleotide compositions, both single- and double-stranded and independent of 5’ and 3’ architecture. Several patents have granted in the United States with claims directed to various combinations of chemical modifications to double-stranded RNA of between 18 and 24 nucleotides. Notably, U.S. 8,273,866 was granted in September 2012 with significant patent term adjustment extending the expiration of this patent to mid-2028. EP423406 was granted in September 2010 with claims directed to double-stranded RNA of between 18 and 24 nucleotides with ten or more chemical modifications on the pyrimidine residues of the sense and/or antisense strand. As indicated in the chart above, additional EP patents have granted with claims to various combinations of chemically modified compositions comprising double-stranded RNA of between 18 and 24 nucleotides and methods of making and using such combinations. In November 2015, U.S. 9,181,551 granted with claims directed to highly modified double-stranded RNA molecules comprising a ligand, with dependent claims wherein the ligand is chosen from a ligand for a cellular receptor, a protein localization sequence, an antibody, a nucleic acid aptamer, a vitamin, a co-factor, a phospholipid, a cholesterol, a polyamine, a galactose, a galactosamine, a folate, an N-acetyl-galactosamine (wherein the N-acetylgalactosamine is a mono-antennary, bi-antennary or a tri-antennary galactosamine). Additional dependent claims are directed to highly modified double-stranded RNA with modified nucleotides, including but not limited to unlocked (acyclic) and locked nucleotides. In addition, in August 2017 the United States Patent and Trademark Office, or USPTO, granted U.S. Patent No. 9,732,344 with claims directed to single-stranded antisense polynucleotide molecules of 18-20 nucleotides, comprising 10 or more phosphorothioates and 10 or more modified pyrimidine molecules.
Intellectual Property Related to the Delivery of siRNAs to Cells
We also pursue internal research and collaborative approaches regarding the delivery of siRNAs to mammalian cells. These approaches include exploring technology that may allow delivery of siRNAs to cells through the use of cholesterol and carbohydrate conjugation, cationic lipids, peptide and antibody-based targeting, and polymer conjugations. Our collaborative efforts have included working with academic and corporate third parties to examine specific embodiments of these various approaches to delivery of siRNAs to appropriate cell tissue, and in-licensing and/or acquiring the most promising technology.
In September 2014, the USPTO granted U.S. Patent No. 8,828,956 with claims directed to compositions including those comprising a modified RNA agent linked to a biantennary or triantennary ligand. Specifically, the granted patent includes claims that broadly cover single- or double-stranded, chemically modified RNA therapeutic molecules conjugated with a GalNAc ligand independent of length, sequence or disease target.
22
The acquisition of Sirna also accelerated our overall efforts to develop and commercialize siRNA delivery technologies, including GalNAc-siRNA and GalNAc-single stranded polynucleotide conjugate technology. As part of the Sirna acquisition, we obtained several patent families directed to various conjugate technologies including “tetra-GalNAc” compositions and methods. The tetra-GalNAc cases are pending worldwide and will expire May 1, 2033. Also included were patent families directed to novel lipid compositions and formulations that are pending worldwide and set to expire May 31, 2031.
In addition to the Sirna delivery technology, we have a license from UBC and Arbutus in the field of RNAi therapeutics to intellectual property covering cationic liposomes and their use to deliver nucleic acid to cells.
In addition, in April 2012, the USPTO granted U.S. Patent No. 8,158,601, covering composition of matter and formulations of the MC3 lipid, as well as methods of using these compositions and formulations. MC3 is being utilized in our patisiran development program. We assigned this patent, amongst other patents and patent applications relating to lipids and LNP technology, to Arbutus in connection with our November 2012 restructuring and cross-license agreement. We retain rights to use this intellectual property for the research, development and commercialization of RNAi therapeutic products, including the rights to sublicense this intellectual property on a product-by-product basis. A description of our 2012 restructuring and cross-license agreement with Arbutus is set forth above under “Strategic Alliances — Other Strategic License Agreements — Delivery-Related License Agreements — Arbutus.”
Intellectual Property Related to siRNAs Directed to Specific Targets
We have filed a number of patent applications claiming specific siRNAs directed to various gene targets that correlate to specific diseases. While there may be a significant number of competing applications filed by other organizations claiming siRNAs to treat the same gene target, we were among the first companies to focus and file on RNAi therapeutics, and thus, we believe that a number of our patent applications may predate competing applications that others may have filed. Reflecting this, in August 2005, the EPO granted a broad patent, which we call the Kreutzer-Limmer II patent, with 103 allowed claims on therapeutic compositions, methods and uses comprising siRNAs that are complementary to mRNA sequences in over 125 disease target genes. In July 2009, the EPO ruled in our favor in an opposition proceeding related to the Kreutzer-Limmer II patent. The decision had been appealed by Sirna and was subsequently withdrawn upon our acquisition of Sirna. No further appeal before the EPO is available. The Kreutzer-Limmer II patent will expire on January 9, 2022, subject to any potential patent term extensions and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. Some of these claimed gene targets are being pursued by our development and pre-clinical programs, such as those expressed by viral pathogens including respiratory syncytial virus and influenza virus. In addition, the claimed targets include oncogenes, cytokines, cell adhesion receptors, angiogenesis targets, apoptosis and cell cycle targets, and additional viral disease targets, such as hepatitis C virus and HIV. The Kreutzer-Limmer II patent series is pending in the United States and many foreign countries. Granted U.S. patent 8,618,277 obtained in the Sirna acquisition and set to expire on February 20, 2023, contains claims directed to a highly chemically modified double-stranded siRNA of between 18-24 nucleotides specifically targeting the hepatitis B virus in a sequence independent manner. Moreover, a patent in the Tuschl II patent series, U.S. Patent No. 7,078,196, claims methods of preparing siRNAs that mediate cleavage of an mRNA in mammalian cells and, therefore, covers methods of making siRNAs directed toward any and all target genes. We hold exclusive worldwide rights to these claims for RNAi therapeutics.
In 2016, we were granted U.S. Patent Nos. 9,370,581, 9,370,582 and 9,352,048 containing claims that broadly cover single- or double-stranded RNA therapeutic molecules conjugated with any biantennary or triantennary ligand (including but not limited to GalNAc) independent of length, specifically inhibiting TTR, PCSK9 or hepatitis b virus, respectively, wherein the HBV-specific RNA molecule is fully chemically modified.
In August 2017, we were granted U.S. Patent No. 9,738,899 with claims directed to single-stranded antisense polynucleotide molecules, capable of inhibiting expression of the human transthyretin gene, of 18-20 nucleotides, comprising 10 or more phosphorothioates and 10 or more modified pyrimidine molecules, 2’-deoxy,-O-Methyl, -Fluoro, -methoxyethoxy (MOE), pyrimidines, LNA-pyrimidines or a combination, with or without conjugation to a galactosamine or cholesterol.
Intellectual Property Related to Our Development Candidates
As our development pipeline matures, we have made and plan to continue to make patent filings that claim all aspects of our development candidates, including dose, method of administration and manufacture.
Intellectual Property Challenges
As the field of RNAi therapeutics is maturing, patent applications are being fully processed by national patent offices around the world. There is uncertainty about which patents will issue, and, if they do, as to when, to whom and with what claims. It is likely that there will be significant litigation and other proceedings, such as interference, reexamination, inter partes review, post-grant review
23
and opposition proceedings, in various patent offices relating to patent rights in the RNAi field. On September 16, 2012, the America Invents Act went into effect and provided for expanded patent challenge, i.e., inter partes review and post-grant review. These provide additional opportunities for third parties to challenge our patents. For example, as noted in the table above, various third parties have initiated oppositions to patents in our Kreutzer-Limmer and Tuschl II series in the EPO, as well as in other jurisdictions. We expect that additional oppositions will be filed in the EPO and elsewhere, and other challenges will be raised relating to other patents and patent applications in our portfolio. In many cases, the possibility of appeal exists for either us or our opponents, and it may be years before final, unappealable rulings are made with respect to these patents in certain jurisdictions. Given the importance of our intellectual property portfolio to our business operations, we intend to vigorously enforce our rights and defend against challenges that have arisen or may arise in this area. A description of ongoing legal matters relating to certain aspects of our intellectual property portfolio is set forth in Part I, Item 3, “Legal Proceedings,” of this annual report on Form 10-K.
Competition
The pharmaceutical marketplace is extremely competitive, with hundreds of companies competing to discover, develop and market new drugs. We face a broad spectrum of current and potential competitors, ranging from very large, global pharmaceutical companies with significant resources, to other biotechnology companies with resources and expertise comparable to our own, to smaller biotechnology companies with fewer resources and expertise than we have. We believe that for most or all of our drug development programs, there will be one or more competing programs under development at other companies. In many cases, the companies with competing programs will have access to greater resources and expertise than we do and may be more advanced in those programs.
Competition for Our Business in General
The competition we face can be grouped into three broad categories:
|
|
• |
other companies working to develop RNAi and microRNA therapeutic products; |
|
|
• |
companies developing technology known as antisense, which, like RNAi, attempts to silence the activity of specific genes by targeting the mRNAs copied from them; and |
|
|
• |
marketed products and development programs for therapeutics that treat the same diseases for which we may also be developing treatments. |
We are aware of several other companies that are working to develop RNAi therapeutic products. Some of these companies are seeking, as we are, to develop chemically synthesized siRNAs as drugs. Others are following a gene therapy approach, with the goal of treating patients not with synthetic siRNAs but with synthetic, exogenously-introduced genes designed to produce siRNA-like molecules within cells.
Companies working on chemically synthesized siRNAs include Takeda, Marina Biotech, Inc., or Marina, Arrowhead Pharmaceuticals, Inc., or Arrowhead, and its subsidiary, Calando Pharmaceuticals, Inc., or Calando, Quark Pharmaceuticals, Inc., or Quark, Silence Therapeutics plc, or Silence, Arbutus, Sylentis, S.A.U., Dicerna Pharmaceuticals, Inc., or Dicerna, WAVE Life Sciences Ltd., or WAVE, and Arcturus Therapeutics, Inc., or Arcturus. Many of these companies have licensed our intellectual property. Benitec Biopharma Ltd., or Benitec, is working on gene therapy approaches to RNAi therapeutics. Companies working on microRNA therapeutics include Regulus Therapeutics, Inc., or Regulus, Rosetta Genomics Ltd., F. Hoffmann-La Roche Ltd, or Roche, through its acquisition in 2014 of Santaris Pharma A/S, miRagen Therapeutics, Inc., Mirna Therapeutics, Inc. and Asuragen, Inc.
Antisense technology uses short, single-stranded, DNA-like molecules to block mRNAs encoding specific proteins. While we believe that RNAi drugs may potentially have significant advantages over ASOs, including greater potency and specificity, others are developing ASO drugs that are currently at a more advanced stage of development than RNAi drugs. For example, Ionis has developed several ASO drugs that have received regulatory approval. Ionis is also developing antisense drugs using ligand-conjugated GalNAc technology licensed from us, and these drugs have been shown to have increased potency at lower doses in clinical and pre-clinical studies, compared with antisense drugs that do not use such licensed GalNAc technology. In addition, a number of other companies have ASO-based product candidates in various stages of pre-clinical and clinical development, including Roche, Celgene Corporation, Akcea Therapeutics, Inc., or Akcea, Antisense Therapeutics, Ltd., WAVE and Sarepta Therapeutics, Inc.
The competitive landscape continues to expand and we expect that additional companies will initiate programs focused on the development of RNAi therapeutic products using the approaches described above as well as potentially new approaches that may result in the more rapid development of RNAi therapeutics or more effective technologies for RNAi drug development or delivery.
24
Competing Drugs for Our Investigational RNAi Therapeutics in Late Stage Clinical Development
Hereditary ATTR Amyloidosis. Until recently, organ transplantation was the only treatment option for patients with hATTR amyloidosis. Only a subset of patients with early stage disease qualify for this costly and invasive procedure, which carries significant morbidity and mortality. Even following liver transplantation, the disease continues to progress for many patients, presumably due to ongoing deposition of wild-type TTR protein. Currently there is only one approved drug for ATTR amyloidosis, as well as several investigational drugs in varying stages of clinical development. We believe that the following approved drugs and if approved, drug candidates, could compete with patisiran:
|
Drug |
Company |
Drug Description |
Phase |
Administration/Dosing |
|
Tafamidis |
Pfizer |
Small molecule drug to stabilize TTR protein |
Approved in the EU, Japan and certain countries in Latin America (indication varies by region) |
Daily oral capsule |
|
Inotersen |
Ionis |
ASO to reduce production of TTR Protein |
Registration |
Subcutaneous (SC) |
|
PRX004 |
Prothena Corporation plc |
Monoclonal antibody to clear amyloid deposits |
Preclinical |
Unknown |
|
Ionis-TTR-LRx |
Ionis |
ASO to reduce production of TTR Protein |
Preclinical |
SC |
|
GSK 2398852 + 2315698 |
GlaxoSmith Kline plc |
Antibody combination to clear amyloid deposits |
Phase 2 |
Intravenous (IV) |
|
Diflunisal |
N/A (generic) |
Non-steroid anti-inflammatory agent |
Approved |
Daily oral capsule/doses |
|
Tolcapone |
SOM Biotech |
Small molecule repurposed generic drug |
Phase 1/2 |
Daily oral dose |
|
AG10 |
Eidos Therapeutics |
Small molecule drug to stabilize TTR protein |
Phase 1 |
Daily oral dose |
Acute Hepatic Porphyrias. The only approved treatments for acute attacks are preparations of heme derived from human blood. The global market for AHP is made up of intravenous hemin in the United States and intravenous heme arginate in the EU. Both products are currently manufactured by Recordati S.p.A. Despite the lack of randomized studies demonstrating clinical efficacy, heme has been shown in case studies to hasten recovery from attacks and has been marketed since 1999 in the EU for the treatment of acute attacks of AIP, hereditary coproporphyria and variegate porphyria.
In addition to heme, the AIPGENE consortium, a European collaboration that included industry sponsors uniQure and Digna Biotech, was developing AMT-021, a gene therapy product for the treatment of AIP, but this development program is currently on hold. We are aware of other companies that have pre-clinical development programs for the treatment of AHPs.
Hemophilia. The global market for treatments of hemophilia and bleeding disorders is valued at more than $10.0 billion. Products on the market include: Factor VIII replacement products; Factor IX replacement products; and more recently, factor replacement products with extended half-lives. For the treatment of persons with inhibitors, there is an approved Factor VIIa replacement product and an activated prothrombin complex concentrate. In addition, new, innovative molecules are currently in development which may offer new treatments for people with hemophilia A and B, with and without inhibitors, including one such molecule that was recently approved for use in persons with hemophilia A with inhibitors in the United States. A number of companies are also actively developing gene therapy products that use virus-like particles to deliver a functional section of a particular gene into the liver cells of a person with hemophilia.
25
We believe that the following approved drugs and, if approved, drug candidates, could compete with fitusiran, along with additional approved drugs and drug candidates:
|
Drug (Company) |
Drug Description |
Phase |
Administration/ Dosing |
|
Hemophilia A |
|||
|
Advate (Shire), Adynovate (Shire), Kogenate (Bayer), Kovaltry (Bayer), Novoeight (Novo Nordisk), Xyntha (Pfizer), Nuwiq (Octapharma), Eloctate (Bioverativ) |
Recombinant FVIII factor products |
Approved |
IV |
|
Valoctocogene roxaparvovec (BioMarin) |
Gene therapy |
Phase 3 |
IV - Single Administration |
|
Emicizumab HemLibra, ACE-910 (Roche) |
Bispecific antibody mimetic of FVIII |
Phase 3 (Hemophilia A without inhibitors) |
SC |
|
Hemophilia B |
|||
|
Rixubis (Shire), Rebinyn (Novo Nordisk), BeneFIX (Pfizer), Alprolix (Bioverativ), Idelvion (CSL Behring) |
Recombinant FIX factor products |
Approved |
IV |
|
AMT-061, FIX (uniQure) |
rAAV5 FIX gene therapy |
Phase 2 |
IV - Single Administration |
|
SPK-9001 (Spark Therapeutics) |
Spark200 AAV FIX gene therapy |
Phase 2 |
IV - Single Administration |
|
Inhibitor Patients |
|||
|
Emicizumab HemLibra, ACE-910 (Roche) |
Bispecific antibody mimetic of FVIII |
Approved |
SC |
|
Feiba (Shire) |
Bypassing agent |
Approved |
IV |
|
NovoSeven (Novo Nordisk) |
Bypassing agent |
Approved |
IV |
|
Hemophilia A and B |
|||
|
Concizumab, anti-TFPI (Novo Nordisk) |
anti-TFPI antibody |
Phase 2 |
IV - Single Administration |
Hypercholesterolemia. The current standard of care for patients with hypercholesterolemia includes the use of dietary changes, lifestyle modification and the use of pharmacologic therapy. Front line therapy consists of HMG-CoA reductase inhibitors, commonly known as statins, which block production of cholesterol by the liver and increase clearance of LDL-C from the bloodstream. Several anti-PCSK9 antibodies have also been approved for the treatment of hypercholesterolemia in the United States and Europe. Other PCSK9-targeted approaches are in development at a number of companies.
We believe that the following approved drugs and if approved, drug candidates, could compete with inclisiran:
|
Drug |
Company |
Drug Description |
Phase |
Administration/Dosing |
|
Repatha |
Amgen |
Anti-PSCK9 mAb |
Marketed |
SC |
|
Praluent |
Sanofi |
Anti-PSCK9 mAb |
Marketed |
SC |
|
Bempedoic Acid (ETC-1002) |
Esperion |
Oral fatty acid and cholesterol synthesis dual inhibitor |
Phase 3 |
Oral |
|
REGN1500 (evinacumab) |
Regeneron |
Anti-ANGPTL3 mAb for antihypercholesterolemia |
Phase 2 |
SC |
|
Volanesorsen |
Akcea |
ASO therapy to reduce levels of APOC3 |
Registration |
SC |
|
Akcea-ANGPTL3-LRx |
Akcea |
ASO therapy to reduce levels of ANGPTL3 |
Phase 2 |
SC |
26
Finally, for many of the diseases that are the subject of our early stage clinical, pre-clinical development and discovery RNAi therapeutic programs, there are already drugs on the market or in development. However, notwithstanding the availability of existing drugs or drug candidates, we believe there currently exists sufficient unmet medical need to warrant the advancement of our investigational RNAi therapeutic programs.
Regulatory Matters
U.S. Regulatory Considerations
The research, testing, manufacture and marketing of drug products and their delivery systems are extensively regulated in the United States and the rest of the world. In the United States, drugs are subject to rigorous regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, or FDCA, and other federal and state statutes and regulations govern, among other things, the research, development, testing, approval, manufacture, storage, record keeping, reporting, labeling, marketing and distribution of drug products. Failure to comply with the applicable regulatory requirements may subject a company to a variety of administrative or judicially-imposed sanctions and the inability to obtain or maintain required approvals to test or market drug products. These sanctions could include, among other things, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, clinical holds, injunctions, fines, civil penalties or criminal prosecution.
The steps ordinarily required before a new drug product may be marketed in the United States include nonclinical laboratory tests, animal tests and formulation studies, the submission to the FDA of an investigational new drug, or IND, application, which must become effective prior to commencement of clinical testing, approval by an institutional review board, or IRB, at each clinical site before each trial may be initiated, completion of adequate and well-controlled clinical trials to establish that the drug product is safe and effective for the indication for which FDA approval is sought, submission to the FDA of an NDA and FDA review and approval of the NDA. Satisfaction of FDA pre-market approval requirements typically takes several years, but may vary substantially depending upon the complexity of the product and the nature of the disease. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on a company’s activities. Success in early stage clinical trials does not necessarily assure success in later stage clinical trials. Data obtained from clinical activities, including but not limited to the data derived from our clinical trials for patisiran, fitusiran, givosiran and inclisiran, are not always conclusive and may be subject to alternative interpretations that could delay, limit or even prevent regulatory approval. Even if a product receives regulatory approval, later discovery of previously unknown problems with a product, including new safety risks, may result in restrictions on the product or even complete withdrawal of the product from the market.
Nonclinical Tests and Clinical Trials.
Nonclinical tests include laboratory evaluation of product chemistry and formulation, as well as animal testing to assess the potential safety and efficacy of the product. The conduct of the nonclinical tests and formulation of compounds for testing must comply with federal regulations and requirements. The results of nonclinical testing are submitted to the FDA as part of an IND, together with chemistry, manufacturing and controls, or CMC, information, analytical and stability data, a proposed clinical trial protocol and other information. Clinical testing in humans may not commence until an IND is in effect.
An IND becomes effective 30 days after receipt by the FDA unless the FDA notifies the sponsor that the proposed investigation(s) are subject to a clinical hold. If the FDA imposes a clinical hold, the FDA’s concerns must be resolved prior to the commencement of clinical trials. The IND review process can result in substantial delay and expense. We, an IRB, or the FDA may, at any time, suspend, terminate or impose a clinical hold on ongoing clinical trials. For example, in October 2016, we decided to discontinue development of revusiran, an investigational RNAi therapeutic that was in development for the treatment of patients with cardiomyopathy due to hATTR amyloidosis, due to safety concerns. If the FDA imposes a clinical hold, clinical trials cannot commence or recommence without FDA authorization and then only under terms authorized by the FDA. For example, in September 2017, we temporarily suspended dosing in all ongoing fitusiran studies pending further review of a fatal thrombotic SAE and agreement with regulatory authorities on a risk mitigation strategy. We have reached alignment with study investigators and the FDA on safety measures and a risk mitigation strategy to enable resumption of dosing in clinical studies with fitusiran, including our Phase 2 OLE study and the ATLAS Phase 3 program.
27
Clinical trials involve the administration of an investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical studies are conducted under protocols detailing, among other things, the objectives of the trial and the safety and effectiveness criteria to be evaluated. Each protocol involving testing on human subjects in the United States must be submitted to the FDA as part of the IND. In addition, clinical trials must be conducted in compliance with federal regulations and requirements, commonly referred to as good clinical practice, or GCP, to assure data integrity and protect the rights, safety and well-being of trial participants. Among other things, GCP requires that all research subjects provide their informed consent prior to participating in any clinical study, and that an IRB at each institution participating in the clinical trial review and approve the plan for any clinical trial before it commences at that institution and conduct continuing review throughout the trial. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects.
Clinical trials to support NDAs are typically conducted in three sequential phases, which may overlap or be combined.
|
|
• |
In Phase 1, the initial introduction of the drug into healthy human subjects or patients, the drug is tested to primarily assess safety, tolerability, pharmacokinetics, pharmacological actions and metabolism associated with increasing doses. |
|
|
• |
Phase 2 usually involves trials in a limited patient population, to assess the optimum dosage and dose regimen, identify possible adverse effects and safety risks, and provide preliminary support for the efficacy of the drug in the indication being studied. |
|
|
• |
Phase 3 clinical trials further evaluate the drug’s clinical efficacy, side effects and safety in an expanded patient population, typically at geographically dispersed clinical trial sites, to establish the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the drug. |
Phase 1, Phase 2 or Phase 3 testing of any drug candidates may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted in the United States. The FDA may, at its discretion, reevaluate, alter, suspend or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the subject. An IRB or a clinical trial sponsor also may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request that additional clinical trials be conducted as a condition to product approval. Finally, sponsors are required to publicly disseminate information about certain ongoing and completed clinical trials on a government website administered by the National Institutes of Health, or NIH.
New Drug Applications.
We believe that any RNAi product candidate we develop, whether for the treatment of ATTR amyloidosis, AHPs, hemophilia, hypercholesterolemia or the various indications targeted in our development or nonclinical discovery programs, will be regulated as a non-biological new drug by the FDA. FDA approval of an NDA is required before commercial distribution of a non-biological new drug may begin in the United States. In December 2017, we submitted our first NDA to the FDA for patisiran. An NDA must include the results of extensive nonclinical, clinical and other testing, as described above, a compilation of data relating to the product’s pharmacology, chemistry, manufacture and controls, proposed labeling and other information. In addition, an NDA for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration typically must contain data assessing the safety and effectiveness for the claimed indication in all relevant pediatric subpopulations, although deferrals or full or partial waivers may be available in some circumstances.
The cost of preparing and submitting an NDA is substantial. Under the Prescription Drug User Fee Act, or PDUFA, as amended, each NDA must be accompanied by a user fee. For fiscal year 2018, the user fee for each NDA requiring clinical data is approximately $2.4 million. The PDUFA also imposes an annual program fee for each approved prescription drug, which is set at approximately $300,000 for fiscal year 2018. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication. The FDA conducts a preliminary review of all NDAs within the first 60 days after submission before accepting them for filing to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. If the submission is accepted for filing, the FDA begins an in-depth review of the NDA. The FDA has agreed to specified performance goals regarding the timing of its review of NDAs, although the FDA does not always meet these goals. The review process is often significantly extended by FDA requests for additional information or clarification regarding information already provided in the submission. The FDA may also refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes independent clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA normally conducts a pre-approval inspection to gain assurance that the manufacturing facility, methods and controls are
28
adequate to preserve the drug’s identity, strength, quality, purity and stability, and are in compliance with regulations governing current good manufacturing practice, or cGMP, requirements. In addition, the FDA often will conduct a bioresearch monitoring inspection of select clinical trial sites involved in conducting pivotal studies to assure data integrity and compliance with applicable GCP requirements.
If the FDA evaluation of the NDA and the various inspections are favorable, the FDA may issue an approval letter, which authorizes commercial marketing of the drug with specific prescribing information for a specific indication. As a condition of NDA approval, the FDA may require post-approval testing, sometimes referred to as Phase 4 trials and surveillance to monitor the drug’s safety or effectiveness and may impose other conditions, including labeling restrictions, such as a Boxed Warning, and/or distribution and use restrictions through a Risk Evaluation and Mitigation Strategy, or REMS, all of which can materially impact the potential market and profitability of the drug. Once granted, product approvals may be further limited or withdrawn if compliance with regulatory standards is not maintained or safety or other problems are identified following initial marketing.
Once an NDA is approved, a product will be subject to certain post-approval requirements, including requirements for registration and listing, AE reporting, submission of periodic reports, recordkeeping, product sampling and distribution. Additionally, the FDA also strictly regulates the promotional claims that may be made about prescription drug products and biologics. In particular, the FDA generally prohibits pharmaceutical companies from promoting their drugs or biologics for uses that are not approved by the FDA as reflected in the product’s approved labeling. In addition, the FDA requires substantiation of any safety or effectiveness claims, including claims that one product is superior in terms of safety or effectiveness to another. Superiority claims generally must be supported by two adequate and well-controlled head-to-head clinical trials. To the extent that market acceptance of our products depends on their superiority over existing therapies, any restriction on our ability to advertise or otherwise promote claims of superiority, or requirements to conduct additional expensive clinical trials to provide proof of such claims, could negatively affect the sales of our products or our costs. We must also notify the FDA of any change in an approved product beyond variations already allowed in the approval. Certain changes to the product, its labeling or its manufacturing require prior FDA approval and may require the conduct of further clinical investigations to support the change. Such approvals may be expensive and time-consuming and, if not approved, the FDA will not allow the product to be commercially distributed as modified.
If the FDA’s evaluation of the NDA submission or manufacturing facilities is not favorable, the FDA may refuse to approve the NDA or issue a complete response letter. The complete response letter describes the deficiencies that the FDA has identified in an application and, when possible, recommends actions that the applicant might take to allow FDA to approve the application. Such actions may include, among other things, conducting additional safety or efficacy studies. Even with the completion of this additional testing or the submission of additional requested information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. With limited exceptions, the FDA may withhold approval of an NDA regardless of prior advice it may have provided or commitments it may have made to the sponsor.
Some of our product candidates may need to be administered using specialized drug delivery systems that are considered to be medical devices. We may rely on drug delivery systems that are already approved to deliver drugs like ours to similar physiological sites or, in some instances, we may need to modify the design or labeling of the legally available device for delivery of our product candidate. The FDA may regulate our product candidate when used with a specialized drug delivery system as a combination product, which could permit the combination to be approved through a single application, such as an NDA. Alternatively, the FDA could require separate, additional approvals or clearances for the modified device. In addition, to the extent the delivery device is owned by another company, we would need that company’s cooperation to implement the necessary changes to the device and to obtain any additional approvals or clearances. Obtaining such additional approvals or clearances, and cooperation of other companies, when necessary, could significantly delay, and increase the cost of obtaining marketing approval, which could reduce the commercial viability of a product candidate. To the extent that we rely on previously unapproved drug delivery systems, we may be subject to additional testing and approval requirements from the FDA above and beyond those described above.
Abbreviated Applications.
Once an NDA is approved, the product covered thereby becomes a listed drug that can, in turn, be relied upon by potential competitors in support of approval of an abbreviated new drug application, or ANDA, or 505(b)(2) application. An ANDA generally provides an abbreviated approval pathway for a drug product that has the same active ingredients in the same strength, dosage form and route of administration as the listed drug and has been shown through appropriate testing (unless waived) to be bioequivalent to the listed drug. Drugs approved in this way are commonly referred to as generic equivalents to the listed drug and can often be substituted by pharmacists under prescriptions written for the original listed drug. A 505(b)(2) application is a type of NDA that relies, in part, upon data the applicant does not own and to which it does not have a right of reference. Such applications typically are submitted for changes to previously approved drug products.
29
The approval of ANDAs and 505(b)(2) applications can be delayed by patents and non-patent exclusivity covering the listed drug. Federal law provides for a period of three years of exclusivity following approval of a listed drug that contains a previously approved active ingredient if the FDA determines that new clinical investigations, other than bioavailability studies, were conducted or sponsored by the applicant and are essential to the approval of the application. This three-year exclusivity covers only the conditions of approval for which the new clinical investigations were essential, such as a new dosage form or indication. Accordingly, three-year exclusivity generally protects changes to a previously approved drug product that require clinical testing for approval and, as a general matter, does not prohibit the FDA from approving ANDAs or 505(b)(2) applications for generic versions of the original, unmodified drug product.
Federal law also provides a five-year period of NCE exclusivity following approval of a drug that contains a new chemical entity, or NCE. An NCE is a drug that contains an active moiety (the molecule or ion responsible for the action of the drug substance) that has never previously been approved by the FDA. If a listed drug has NCE exclusivity, ANDAs and 505(b)(2) applications referencing the listed drug cannot be submitted to the FDA for five years unless the application contains a certification challenging a listed patent, i.e., a paragraph IV certification (discussed further below), in which case the ANDA or 505(b)(2) application may be submitted four years following approval of the listed drug. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinical studies and clinical trials necessary to demonstrate safety and effectiveness.
Additionally, applicants submitting an ANDA or 505(b)(2) application referencing a listed drug generally are required to make a certification with respect to each patent listed in the FDA’s publication Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book, for the listed drug. The only exception is if the applicant is not seeking approval of a use claimed by a method-of-use patent, in which case the applicant can submit a statement to that effect. These certifications (and statements) determine when the FDA can approve the ANDA or 505(b)(2) application. If the ANDA or 505(b)(2) applicant certifies that it does not intend to market its generic product before a listed patent expires (i.e., a paragraph III certification), then the FDA cannot grant effective approval of the ANDA or 505(b)(2) application until the relevant patent expires. If the ANDA or 505(b)(2) applicant certifies that a listed patent is invalid, unenforceable, or will not be infringed by its proposed product, and thus that it is seeking approval prior to patent expiration (i.e., a paragraph IV certification), the statute provides a process for litigating the patent infringement issues during the FDA’s review of the ANDA or 505(b)(2) application. In particular, the applicant is required to provide notice of its patent challenge to the NDA sponsor and the patent holder within certain time limits. If the patent holder then initiates a suit for patent infringement within 45 days of receipt of the notice, the FDA cannot grant effective approval of the ANDA or 505(b)(2) application until either 30 months have passed (which may be extended or shortened in certain cases) or there has been a court decision or settlement order holding or stating that the patents in question are invalid, unenforceable or not infringed. If the patent holder does not initiate a suit for patent infringement within the 45 days, the ANDA or 505(b)(2) application may be approved immediately upon successful completion of FDA review, unless blocked by another listed patent or regulatory exclusivity period.
Orphan Drug Designation (ODD).
Under the Orphan Drug Act, as amended, the FDA may grant ODD to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States or for which there is no reasonable expectation of recovering drug development costs in the United States. ODD must be requested before submitting an NDA. After the FDA grants ODD, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. We intend to request ODD designation for our product candidates, if applicable. For example, the FDA has granted ODD for patisiran as a therapeutic approach for the treatment of ATTR amyloidosis, givosiran as a therapeutic approach for AHPs, fitusiran as a therapeutic approach for hemophilia A and B, and inclisiran as a therapeutic approach for HoFH.
If a product that has ODD subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve for seven years any other applications, including a full NDA, to market the same orphan drug for the same indication, except in very limited circumstances. For purposes of small molecule drugs, the FDA defines “same drug” as a drug that contains the same active moiety and is intended for the same use as the previously approved orphan drug. For purposes of large molecule drugs, the FDA defines “same drug” as a drug that contains the same principal molecular structural features, but not necessarily all of the same structural features, and is intended for the same use as the drug in question. Notwithstanding the above definitions, a drug that is clinically superior to an orphan drug will not be considered the “same drug” and thus will not be blocked by orphan drug exclusivity.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the drug to meet the needs of patients with the rare disease or condition.
30
The FDCA, as amended by the Food and Drug Administration Safety and Innovation Act, or FDASIA, requires that a sponsor who is planning to submit a marketing application for a drug or biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-phase 2 meeting or as may be agreed between the sponsor and the FDA. Drugs with ODD are exempt from these requirements. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from nonclinical studies, early phase clinical trials, and/or other clinical development programs.
Fast Track Program.
The FDA has a Fast Track program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. Fast Track designation applies to the product and the specific indication for which it is being studied. The sponsor of a new drug or biological product may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product, but ideally no later than the pre-NDA or –biologics license application, or BLA, meeting. Fast Track designation provides opportunities for frequent interactions with FDA to expedite drug development and review as well as the opportunity for priority and/or rolling review of the NDA. We intend to request Fast Track designation for our product candidates, if applicable. For example, the FDA granted Fast Track designation to patisiran for the treatment of hATTR amyloidosis.
Any product submitted to the FDA for marketing, including under a Fast Track program, may be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. Any product is eligible for priority review if it treats a serious condition and, if approved, would provide a significant improvement in the safety or effectiveness of treatment, diagnosis or prevention of a disease compared to marketed products. The FDA’s goal for taking action on an application with a Priority Review designation is six months instead of ten months. The FDA granted our request for priority review for patisiran and has set an action date of August 11, 2018 under the PDUFA. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefits. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical studies to verify the predicted clinical benefit. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Breakthrough Therapy Designation.
FDASIA also amended the FDCA to require the FDA to expedite the development and review of a “breakthrough therapy.” A drug or biological product can be designated as a breakthrough therapy if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. A sponsor may request that a drug or biological product be designated as a breakthrough therapy at any time during the clinical development of the product. If so designated, the FDA shall act to expedite the development and review of the product’s marketing application, including by meeting with the sponsor throughout the product’s development, providing timely advice to the sponsor to ensure that the development program to gather nonclinical, manufacturing/controls and clinical data is as efficient as practicable, involving senior managers and experienced review staff in a cross-disciplinary review, assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor, taking steps to ensure that the design of the clinical trials is as efficient as practicable, and allowing a rolling review. The FDA granted Breakthrough Therapy designation for patisiran and givosiran. We intend to request “breakthrough therapy” designation for our other product candidates, if applicable.
31
Pharmaceutical Coverage, Pricing and Reimbursement.
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we obtain regulatory approval. In the United States and markets in other countries, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government healthcare programs, managed care providers, private health insurers and other organizations. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Third-party payors may provide coverage, but place stringent limitations on such coverage, such as requiring alternative treatments to be tried first. These third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of our products, in addition to incurring the costs required to obtain FDA approvals. Our product candidates may not be considered medically reasonable or necessary or cost-effective. Even if a drug product is covered, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
Federal, state and local governments in the United States and foreign governments continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers. Future legislation could limit payments for pharmaceuticals such as the drug candidates that we are developing.
Different pricing and reimbursement schemes exist in other countries. In the EU, governments influence the price of drug products through their pricing and reimbursement rules and control of national health care systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate systems under which products may be marketed only after a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to set their own prices for medicines, but monitor and control company profits. The downward pressure on health care costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, in some countries, cross-border imports from low-priced markets exert competitive pressure that may reduce pricing within a country.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. In addition, the emphasis on managed care in the United States has increased and we expect will continue to exert downward pressure on pharmaceutical pricing. Coverage policies, third-party reimbursement rates and pharmaceutical pricing regulations may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
In March 2010, the Patient Protection and Affordable Care Act, also referred to as the Affordable Care Act or the PPACA, was enacted, which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers. Among the provisions of the PPACA of greatest importance to the pharmaceutical industry are the following:
|
|
• |
The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the Secretary of the Department of Health and Human Services as a condition for states to receive federal matching funds for the manufacturer’s outpatient drugs furnished to Medicaid patients. Effective in 2010, the PPACA made several changes to the Medicaid Drug Rebate Program, including increasing pharmaceutical manufacturers’ rebate liability by raising the minimum basic Medicaid rebate on most branded prescription drugs and biologic products from 15.1 percent of average manufacturer price, or AMP, to 23.1 percent of AMP and adding a new rebate calculation for “line extensions” (i.e., new formulations, such as extended release formulations) of solid oral dosage forms of branded products, as well as potentially impacting their rebate liability by modifying the statutory definition of AMP. In addition, the PPACA provides for the public availability of retail survey prices and certain weighted average AMPs under the Medicaid program. The implementation of this requirement by the Centers for Medicare and Medicaid Services, or CMS, may also provide for the public availability of pharmacy acquisition of cost data, which could negatively impact our sales. |
32
|
|
• |
Effective in 2011, the PPACA imposed a requirement on manufacturers of branded drugs and biologic products to provide a 50 percent discount off the negotiated price of branded drugs dispensed to Medicare Part D patients in the coverage gap (i.e., “donut hole”). |
|
|
• |
Effective in 2011, the PPACA imposed an annual, nondeductible fee on any entity that manufactures or imports certain branded prescription drugs and biologic products, apportioned among these entities according to their market share in certain government healthcare programs, although this fee would not apply to sales of certain products approved exclusively for orphan indications. |
|
|
• |
Effective in 2012, the PPACA required certain manufacturers to track certain financial arrangements with physicians and teaching hospitals, including any “transfer of value” made or distributed to such entities, as well as any investment interests held by physicians and their immediate family members. Manufacturers annually report this information to CMS, which posts this information on its website. |
|
|
• |
As of 2010, a new Patient-Centered Outcomes Research Institute was established pursuant to the PPACA to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. The research conducted by the Patient-Centered Outcomes Research Institute may affect the market for certain drug products. |
|
|
• |
The PPACA created the Independent Payment Advisory Board which, beginning in 2014, has authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription drugs. Under certain circumstances, these recommendations will become law unless Congress enacts legislation that will achieve the same or greater Medicare cost savings. |
|
|
• |
The PPACA established the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. Funding has been allocated to support the mission of the Center for Medicare and Medicaid Innovation from 2011 to 2019. |
|
|
• |
The law expands eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133 percent of the federal poverty level, thereby potentially increasing a manufacturer's Medicaid rebate liability. |
Possible Change in Laws or Policies.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency or reviewing courts in ways that may significantly affect our business and development of our product candidates and any products that we may commercialize. It is impossible to predict whether additional legislative changes will be enacted, or FDA regulations, guidance or interpretations will be changed, or what the impact of any such changes may be. Federal budget uncertainties or spending reductions may reduce the capabilities of the FDA, extend the duration of required regulatory reviews, and reduce the availability of clinical research grants.
EU Regulatory Considerations
In the EU medicinal products are subject to extensive pre- and post-market regulation by regulatory authorities at both the EU and national levels.
Clinical Trials.
Clinical trials of medicinal products in the EU must be conducted in accordance with EU and national regulations and the International Conference on Harmonization, or ICH, guidelines on GCP. If the sponsor of the clinical trial is not established within the EU, it must appoint an entity within the EU to act as its legal representative. The sponsor must take out a clinical trial insurance policy, and in most EU countries the sponsor is liable to provide ‘no fault’ compensation to any study subject injured in the clinical trial.
33
Prior to commencing a clinical trial, the sponsor must obtain a CTA from the competent authority, and a positive opinion from an independent ethics committee. The application for a CTA must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation. Any substantial changes to the trial protocol or other information submitted with the clinical trial applications must be notified to or approved by the relevant competent authorities and ethics committees.
Currently, CTAs must be submitted to the competent authority in each EU member state in which the trial will be conducted. Under the new Regulation on Clinical Trials, which is currently expected to come into application in the second half of 2019, there will be a centralized application procedure where one national authority leads the scientific review of the application leading to increased information-sharing and decision-making between member states. Each concerned member state will continue to complete an ethical review of any CTA.
Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is made public by the competent authority once the CTA is approved. The results of the clinical trial must be submitted by the sponsor to the competent authorities and, with the exception of non-pediatric Phase 1 trials, will be made public at the latest within six months of the end of a pediatric clinical trial, or otherwise within 12 months after the end of the trial.
During the development of a medicinal product, the EMA and national medicines regulators within the EU provide the opportunity for dialogue and guidance on the development program. At the EMA level, this is usually done in the form of scientific advice, which is given by the Scientific Advice Working Party of the Committee for Medicinal Products for Human Use, or CHMP. A fee is incurred with each scientific advice procedure. Advice from the EMA is typically provided based on questions concerning, for example, quality (chemistry, manufacturing and controls testing), nonclinical testing and clinical studies, and pharmacovigilance plans and risk-management programs. Advice is not legally binding with regard to any future marketing authorisation application of the product concerned.
Marketing Authorisations.
After completion of the required clinical testing, we must obtain a marketing authorisation before we may place a medicinal product on the market in the EU. There are various application procedures available, depending on the type of product involved. All application procedures require an application in the common technical document, or CTD, format, which includes the submission of detailed information about the manufacturing and quality of the product, and nonclinical study and clinical trial information. There is an increasing trend in the EU towards greater transparency and, while the manufacturing or quality information is currently generally protected as confidential information, the EMA and national regulatory authorities are now liable to disclose much of the nonclinical and clinical information in marketing authorisation dossiers, including the full clinical study reports, in response to freedom of information requests after the marketing authorisation has been granted. In October 2014, the EMA adopted a policy under which clinical study reports would be posted on the agency’s website following the grant, denial or withdrawal of a marketing authorisation application, subject to procedures for limited redactions and protection against unfair commercial use. A similar requirement is contained in the new Regulation on Clinical Trials that is currently expected to take effect in the second half of 2019.
The centralized procedure gives rise to marketing authorisations that are valid throughout the EU and, by extension (after national implementing decisions), in Norway, Iceland and Liechtenstein, which, together with the EU member states, comprise the European Economic Area, or EEA. Applicants file MAAs with the EMA, where they are reviewed by relevant scientific committees, including the CHMP. The EMA forwards CHMP opinions to the European Commission, or EC, which uses them as the basis for deciding whether to grant a marketing authorisation. The centralized procedure is compulsory for medicinal products that (1) are derived from biotechnology processes, (2) contain a new active substance (not yet approved on November 20, 2005) indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders, viral diseases or autoimmune diseases and other immune dysfunctions, (3) are orphan medicinal products or (4) are advanced therapy medicinal products, such as gene or cell therapy medicines. For medicines that do not fall within these categories, an applicant may voluntarily submit an application for a centralized marketing authorisation to the EMA, as long as the CHMP agrees that (i) the medicine concerned contains a new active substance (not yet approved on November 20, 2005), (ii) the medicine is a significant therapeutic, scientific, or technical innovation, or (iii) if its authorization under the centralized procedure would be in the interest of public health. In December 2017, we submitted our first MAA for patisiran under the centralized procedure, which has been accepted by the EMA.
For those medicinal products for which the centralized procedure is not available, the applicant must submit MAAs to the national medicines regulators through one of three procedures: (1) a national procedure, which results in a marketing authorisation in a single EU member state; (2) the decentralized procedure, in which applications are submitted simultaneously in two or more EU member states; and (3) the mutual recognition procedure, which must be used if the product has already been authorized in at least one other EU member state, and in which the EU member states are required to grant an authorization recognizing the existing authorization in the other EU member state, unless they identify a serious risk to public health. A national procedure is only possible
34
for one member state; as soon as an application is submitted in a second member state the mutual recognition or decentralized procedure will be triggered.
Under the centralized procedure in the EU, the maximum timeframe for the evaluation of an MAA is 210 days. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically takes a year or more. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major interest for public health and therapeutic intervention, defined by the absence or insufficiency of an appropriate alternative therapeutic approach for the disease to be treated; and anticipation of high therapeutic benefit of the new product. In this circumstance, EMA ensures that the opinion of the CHMP is given within 150 days. In November 2017, the EMA granted an accelerated assessment for patisiran.
Data Exclusivity.
MAAs for generic medicinal products do not need to include the results of pre-clinical studies and clinical trials, but instead can refer to the data included in the marketing authorisation of a reference product for which regulatory data exclusivity has expired. If a marketing authorisation is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic MAAs referring to the data of that product may not be accepted by the regulatory authorities, and a further two years of market exclusivity, during which such generic products may not be placed on the market. The two-year period may be extended to three years if during the first eight years a new therapeutic indication with significant clinical benefit over existing therapies is approved.
There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials or manufacturing processes. For such products, the results of appropriate pre-clinical studies or clinical trials must be provided, and guidelines from the EMA detail the type of quantity of supplementary data to be provided for different types of biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and regulatory experience gained at the time.
Orphan Medicinal Products.
The EMA’s COMP may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition and when, without incentives, it is unlikely that sales of the product in the EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication. Following a positive opinion by the COMP, the EC adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of an MAA and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria (for instance because in the meantime a new product was approved for the indication and no convincing data are available to demonstrate a significant benefit over that product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following marketing authorisation. During this period, the competent authorities may not accept or approve any similar medicinal product for the same therapeutic indication, unless the second medicinal product is safer, more effective or otherwise clinically superior. This period may be reduced to six years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of orphan designation. Patisiran, givosiran and fitusiran have been granted orphan medicinal product designation.
Post-Approval Controls.
The holder of a marketing authorisation must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance, or QPPV, who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorisation. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials
35
or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions.
All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another.
Manufacturing.
Medicinal products may only be manufactured in the EU, or imported into the EU from another country, by the holder of a manufacturing authorization from the competent national authority. The manufacturer or importer must have a qualified person, or QP, who is responsible for certifying that each batch of product has been manufactured in accordance with EU standards of cGMP before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with cGMP.
Pricing and Reimbursement.
Governments influence the price of medicinal products in the EU through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
Foreign Regulation of New Drug Compounds
In addition to regulations in the United States and the EU, we are subject to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products. In particular, we plan to file for regulatory approval in Japan in mid-2018 and in one or more additional countries by the end of the year, and will have to follow the specific regulations in Japan and such other countries, which are complex.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in all or most foreign countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the United States have a similar process that requires the submission of a CTA, much like the IND prior to the commencement of human clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed. Similarly, all clinical trials in Australia require, among other things, review and approval of clinical trial proposals by an ethics committee, which provides a combined ethical and scientific review process.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials must be conducted in accordance with GCP, which have their origin in the World Medical Association’s Declaration of Helsinki, the applicable regulatory requirements, and guidelines developed by the ICH for GCP in clinical trials.
The approval procedure also varies among countries and can involve requirements for additional testing. The time required may differ from that required for FDA approval and may be longer than that required to obtain FDA approval. Thus, there can be substantial delays in obtaining required approvals from foreign regulatory authorities after the relevant applications are filed.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Hazardous Materials
Our research, development and manufacturing processes involve the controlled use of hazardous materials, chemicals and radioactive materials and produce waste products. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We do not expect the cost of complying with these laws and regulations to be material.
36
To date, we have manufactured only limited supplies of drug substance for use in IND-enabling toxicology studies in animals at our own facility, as well as patisiran formulated bulk drug product for use in clinical trials, and if approved, for commercial sale. We have contracted with several third-party contract manufacturing organizations, or CMOs, for the supply of drug substance and finished product, other than patisiran, to meet our testing needs for pre-clinical toxicology and clinical testing. We expect to continue to rely on third-party CMOs for the supply of drug substance and certain drug product, including siRNAs and siRNA conjugates, for our product candidates for at least the next several years, including to support the launch of our first several products and to supply Sanofi Genzyme with fitusiran under the AT3 License Terms. During 2015, we amended our manufacturing agreement with Agilent Technologies, Inc., or Agilent, to provide for Agilent to supply, subject to any conflicting obligations under our third-party agreements, a specified percentage of the active pharmaceutical ingredients required for certain of our products in clinical development, as well as other products the parties may agree upon in the future, over an initial term of four years. We are required to provide rolling forecasts for products on a quarterly basis, a portion of which will be considered a binding, firm order. Agilent is required to reserve sufficient capacity to ensure that it can supply products in the amounts specified under such firm orders, as well as up to a certain percentage of the remaining, non-binding portions of each forecast. Subject to any conflicting obligations under our third-party agreements, we have also agreed to negotiate in good faith to enter into a separate commercial manufacturing supply agreement with Agilent for certain products, consistent with certain specified terms, including a specified minimum purchase commitment. Currently, Agilent is the sole manufacturer of the active pharmaceutical ingredients for patisiran for clinical and commercial use. In April 2016, we completed our purchase of a parcel of land in Norton, Massachusetts. We have commenced construction of a cGMP manufacturing facility at this site for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use, which we currently expect to be operational in 2020.
During 2012, we established a manufacturing facility and have developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late stage clinical trials and commercial use. During 2013, we manufactured our first cGMP batches of patisiran for use in our Phase 2 OLE and Phase 3 clinical trials. We will manufacture commercial supply for patisiran formulated bulk drug product in our facility for the foreseeable future. Commercial quantities of any drugs that we may seek to develop will have to be manufactured in facilities, and by processes, that comply with FDA regulations and other federal, state and local regulations, as well as comparable foreign regulations.
We believe we have sufficient manufacturing capacity through our third-party CMOs and our current internal cGMP manufacturing facility to meet our current research, clinical and commercial needs and the needs of Sanofi Genzyme for clinical supply. We believe that the supply capacity we have established externally, together with the internal capacity we developed to support pre-clinical trials, our existing facility for patisiran formulated bulk drug product and the new facility we are building, will be sufficient to meet our and Sanofi Genzyme’s anticipated needs for the next several years. We monitor the capacity availability for the manufacture of drug substance and drug product and believe that our supply agreements with our CMOs and the lead times for new supply agreements would allow us to access additional capacity to meet our and Sanofi Genzyme’s currently anticipated needs. We also believe that our products can be manufactured at a scale and with production and procurement efficiencies that will result in commercially competitive costs.
After successfully overcoming various challenges associated with developing a potential new class of innovative medicines - such as solving the issue of drug delivery, optimizing our RNAi therapeutics to exhibit potency and durability of effect, and designing and carrying out comprehensive clinical trials to demonstrate the safety and clinical efficacy of our investigational products - we now embark on the next part of the journey; introducing our RNAi therapeutics to as many eligible patients in need as possible. To meet that new challenge, we have started to build a global commercial operation which will be fully integrated and ready to sequentially manage the potential of multiple product launches across multiple geographies. As a commercial-stage biopharmaceutical company, we intend to leverage the internal knowledge accumulated at Alnylam as well as hire talented people from industry to commercialize our products ourselves in key countries globally. The conduct of these commercial activities will be dependent upon regulatory approvals and on agreements that we have made or may make in the future with strategic collaborators, currently as follows with respect to our late-stage clinical programs:
|
|
• |
For patisiran, we now have global rights to develop and commercialize patisiran, pending approval by regulatory authorities, and other TTR products, including ALN-TTRsc02 and any back-ups, as a result of the 2018 amendment to the Sanofi Genzyme collaboration and the related Exclusive TTR License; |
|
|
• |
For givosiran, we retain global rights to develop and commercialize; |
|
|
• |
For fitusiran, Sanofi Genzyme has global rights to develop and commercialize fitusiran and any back-ups as a result of the 2018 amendment to the Sanofi Genzyme collaboration and the related AT3 License Terms; and |
|
|
• |
For inclisiran, we have granted MDCO global rights to develop and commercialize. |
37
Throughout the development of our product candidates, we have remained focused on keeping patients at the center of everything we do. This patient focus will continue as we transition towards commercialization. Moreover, the late stage programs we are advancing to commercialization are focused on orphan diseases, and these patients and their families are often in need of more than just a product. It is our goal to identify information, education solutions and services that benefit these patients and their families, and to have a rich patient services approach in these orphan diseases. In addition, we are focused early in the product development cycle on establishing evidence that we can bring to payors about the overall burden of disease and pharmacoeconomic opportunities that our product candidates represent to ensure access for patients.
We are assembling the key components of a global commercial organization with a focus on preparation for the anticipated commercial launch of patisiran in 2018, assuming regulatory approval is obtained. We have already initiated a staged build of commercial capabilities with the planned hire of approximately 250 employees deployed in customer facing activities across the world, initially in the United States and major European countries, followed by Canada and Switzerland, with a phased approach to the Asia Pacific, Latin American and Middle Eastern regions. We plan to build a focused commercial team with broad experience in marketing, sales, patient access, patient services, distribution and product reimbursement, in particular for orphan diseases. In the coming months, we will continue to expand our footprint in major European markets and beyond. During 2017, as we prepared for a potential patisiran commercial launch in the United States and Europe in 2018, we continued to expand our commercial organization, incorporating the appropriate quality systems, compliance policies, systems and procedures, as well as implementing internal systems and infrastructure in order to support commercial sales, and the establishment of patient-focused programs. Given our accelerated global commercial opportunity, we intend to expand these capabilities more broadly during 2018 as we strategically assess the global opportunity for patisiran. Ultimately, we intend to leverage the commercial infrastructure that we build to support the potential launches of givosiran and ALN-TTRsc02. Our objective is to be ready to execute successful product launches. For many territories/countries, we may also elect to utilize strategic partners, distributors or contract sales forces to assist in the commercialization of our products.
Employees
At January 31, 2018, we had 749 employees. None of our employees are represented by a labor union or covered by a collective bargaining agreement, nor have we experienced work stoppages. We believe that relations with our employees are good.
Financial Information About Geographic Areas
See the section entitled “Segment Information” appearing in Note 2 to our consolidated financial statements for financial information about geographic areas. The Notes to our consolidated financial statements are contained in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K.
Corporate Information
Alnylam Pharmaceuticals, Inc. is a Delaware corporation that was formed in May 2003. Alnylam U.S., Inc., one of our wholly owned subsidiaries, is also a Delaware corporation that was formed in June 2002 as our initial corporate entity. Our principal executive office is located at 300 Third Street, Cambridge, Massachusetts 02142, and our telephone number is (617) 551-8200.
Investor Information
We maintain an internet website at http://www.alnylam.com. The information on our website is not incorporated by reference into this annual report on Form 10-K and should not be considered to be a part of this annual report on Form 10-K. Our website address is included in this annual report on Form 10-K as an inactive technical reference only. Our reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including our annual reports on Form 10-K, our quarterly reports on Form 10-Q and our current reports on Form 8-K, and amendments to those reports, are accessible through our website, free of charge, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the United States Securities and Exchange Commission, or SEC. We also make available on our website the charters of our audit committee, compensation committee, nominating and corporate governance committee, and science and technology committee, as well as our corporate governance guidelines and our code of business conduct and ethics. In addition, we intend to disclose on our web site any amendments to, or waivers from, our code of business conduct and ethics that are required to be disclosed pursuant to the SEC rules.
38
You may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet website that contains reports, proxy and information statements, and other information regarding Alnylam and other issuers that file electronically with the SEC. The SEC’s Internet website address is http://www.sec.gov.
Our business is subject to numerous risks. We caution you that the following important factors, among others, could cause our actual results to differ materially from those expressed in forward-looking statements made by us or on our behalf in filings with the SEC, press releases, communications with investors and oral statements. All statements other than statements relating to historical matters should be considered forward-looking statements. When used in this report, the words “believe,” “expect,” “plan,” “anticipate,” “estimate,” “predict,” “may,” “could,” “should,” “intend,” “will,” “target,” “goal” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these words. Any or all of our forward-looking statements in this annual report on Form 10-K and in any other public statements we make may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Many factors mentioned in the discussion below will be important in determining future results. Consequently, no forward-looking statement can be guaranteed. Actual future results may vary materially from those anticipated in forward-looking statements. We explicitly disclaim any obligation to update any forward-looking statements to reflect events or circumstances that arise after the date hereof. You are advised, however, to consult any further disclosure we make in our reports filed with the SEC.
Risks Related to Our Business
Risks Related to Being a Clinical Stage Company
Although we have several product candidates in late stage clinical development, including one in registration, there is limited information about our ability to successfully overcome many of the risks and uncertainties encountered by companies in the biopharmaceutical industry.
Although we have product candidates in late stage clinical development and one product under regulatory review for approval in the United States and the EU, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
|
|
• |
execute product development activities using unproven technologies related to both RNAi and to the delivery of siRNAs to the relevant tissues and cells; |
|
|
• |
build and maintain a strong intellectual property portfolio; |
|
|
• |
gain regulatory acceptance for the development and commercialization of our product candidates and market success for any products we commercialize; |
|
|
• |
develop and maintain successful strategic alliances; and |
|
|
• |
manage our spending as costs and expenses increase due to clinical trials, regulatory approvals and commercialization. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop product candidates, commercialize products, raise capital, expand our business or continue our operations.
The approach we are taking to discover and develop novel RNAi therapeutics is unproven and may never lead to marketable products.
We have concentrated our efforts and therapeutic product research and development on RNAi technology and our future success depends on the successful development of this technology and products based on it. Neither we nor any other company has received regulatory approval to market therapeutics utilizing siRNAs, the class of molecule we are trying to develop into drugs. The scientific discoveries that form the basis for our efforts to discover and develop new drugs are relatively new. The scientific evidence to support the feasibility of developing drugs based on these discoveries is still limited. Skepticism as to the feasibility of developing RNAi therapeutics has been expressed in scientific literature. For example, there are potential challenges to achieving safe RNAi therapeutics based on the so-called off-target effects and activation of the interferon response. In addition, decisions by other companies with respect to their RNAi development efforts or their adoption of different or related technologies and the potential success of any such different or related technologies may increase skepticism in the marketplace regarding the potential for RNAi therapeutics.
39
Relatively few product candidates based on these discoveries have ever been tested in humans. siRNAs may not naturally possess the inherent properties typically required of drugs, such as the ability to be stable in the body, or the ability to enter cells within relevant tissues in order to exert their effects. We have spent and expect to continue to spend large amounts of money developing siRNAs that possess the properties typically required of drugs, and to date, we have only taken one product candidate through Phase 3 development and filed for regulatory approval in the United States and the EU. In addition, these compounds may not demonstrate in patients the chemical and pharmacological properties ascribed to them in laboratory studies, and they may interact with human biological systems in unforeseen, ineffective or harmful ways. For example, in October 2016, we discontinued development of revusiran, an investigational RNAi therapeutic that was in development for the treatment of patients with cardiomyopathy due to hATTR amyloidosis, due to safety concerns. We conducted a comprehensive evaluation of the revusiran data and reported the results of this evaluation in August 2017, however, our investigation did not result in a conclusive explanation regarding the cause of the mortality imbalance observed in the ENDEAVOUR Phase 3 study. If we do not succeed in developing products that gain regulatory approval and succeed in the marketplace, we may not become profitable and the value of our common stock will decline.
Further, our focus solely on RNAi technology for developing drugs, as opposed to multiple, more proven technologies for drug development, increases the risks associated with the ownership of our common stock. If we are not successful in developing and commercializing one or more products using RNAi technology, we may be required to change the scope and direction of our product development activities. In that case, we may not be able to identify and implement successfully an alternative product development strategy.
Risks Related to Our Financial Results and Need for Financing
We have a history of losses and may never become and remain consistently profitable.
We have experienced significant operating losses since our inception. At December 31, 2017, we had an accumulated deficit of $2.15 billion. To date, we have not received regulatory approval to market or sell any products nor generated any revenues from the sale of products. Further, we do not expect to generate any product revenues until at the earliest mid- to late-2018, assuming we receive marketing approval for patisiran. We expect to continue to incur annual net operating losses over the next several years and will require substantial resources over the next several years as we expand our efforts to discover, develop and commercialize RNAi therapeutics. Until we are successful in obtaining regulatory approval for our product candidates and successful in commercializing such products, we anticipate that a significant portion of any revenues we generate over the next several years will be from alliances with pharmaceutical and biotechnology companies, but cannot be certain that we will be able to maintain our existing alliances or secure and maintain new alliances, or meet the obligations or achieve any milestones that we may be required to meet or achieve to receive payments. We anticipate that revenues derived from such sources will not be sufficient to make us consistently profitable.
We believe that to become and remain consistently profitable, we must succeed in discovering, developing and commercializing novel drugs with significant market potential. This will require us to be successful in a range of challenging activities, including pre-clinical testing and clinical trial stages of development, obtaining regulatory approval and reimbursement for these novel drugs and manufacturing, marketing and selling them. We may never succeed in these activities, and may never generate revenues that are significant enough to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. If we cannot become and remain consistently profitable, the market price of our common stock could decline. In addition, we may be unable to raise capital, expand our business, develop additional product candidates or continue our operations.
We will require substantial additional funds to complete our research, development and commercialization activities and if additional funds are not available, we may need to critically limit, significantly scale back or cease our operations.
We have used substantial funds to develop our RNAi technologies and will require substantial funds to conduct further research and development, including pre-clinical testing and clinical trials of our product candidates, and to manufacture, market and sell any products that are approved for commercial sale. Because we cannot be certain of the length of time or activities associated with successful development of our product candidates, we are unable to estimate the actual funds we will require to develop and commercialize them.
Our future capital requirements and the period for which we expect our existing resources to support our operations may vary from what we expect. We have based our expectations on a number of factors, many of which are difficult to predict or are outside of our control, including:
|
|
• |
our progress in demonstrating that siRNAs can be active as drugs and achieve desired clinical effects; |
|
|
• |
progress in our research and development programs, as well as what may be required by regulatory bodies to advance these programs; |
40
|
|
• |
the timing, receipt and amount of milestone and other payments, if any, from present and future collaborators, if any; |
|
|
• |
our ability to maintain and establish additional collaborative arrangements and/or new business initiatives; |
|
|
• |
the resources, time and costs required to successfully initiate and complete our pre-clinical and clinical studies, obtain regulatory approvals, prepare for commercialization of our product candidates and obtain and maintain licenses to third-party intellectual property; |
|
|
• |
our ability to establish, maintain and operate our own manufacturing facilities in a timely and cost effective manner; |
|
|
• |
our ability to manufacture, or contract with third parties for the manufacture of, our product candidates for clinical testing and commercial sale; |
|
|
• |
the resources, time and cost required for the preparation, filing, prosecution, maintenance and enforcement of patent claims; |
|
|
• |
the costs associated with legal activities, including litigation, arising in the course of our business activities and our ability to prevail in any such legal disputes; and |
|
|
• |
the timing, receipt and amount of sales and royalties, if any, from our potential products. |
If our estimates and predictions relating to these factors are incorrect, we may need to modify our operating plan.
Even if our estimates are correct, we will be required to seek additional funding in the future and intend to do so through either collaborative arrangements, public or private equity offerings or debt financings, or a combination of one or more of these funding sources. Additional funds may not be available to us on acceptable terms or at all.
In April 2016, our subsidiary, Alnylam U.S., Inc., entered into an aggregate of $150.0 million in term loan agreements related to the build out of our new drug substance manufacturing facility. In December 2017, we repaid in full $120.0 million outstanding under one such term loan agreement. We are the guarantor under the remaining term loan agreement, which matures in April 2021. Interest on the borrowings is calculated based on LIBOR plus 0.45 percent. During an event of default under the remaining agreement, the obligations under such agreement will bear interest at a rate per annum equal to the interest rate then in effect plus two percent. The obligations under the term loan agreement are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount outstanding under such agreement at such time. The remaining agreement includes restrictive covenants that could limit our flexibility in conducting future business activities and further limit our ability to change the nature of our business and, in the event of insolvency, the lenders would be paid before holders of equity securities received any distribution of corporate assets. If an event of default occurs, the interest rate would increase and the lender would be entitled to take various actions, including the acceleration of amounts due under the loan. Our ability to satisfy our obligations under this agreement and meet our debt service obligations will depend upon our future performance, which will be subject to financial, business and other factors affecting our operations, many of which are beyond our control.
In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our existing stockholders will result. In addition, as a condition to providing additional funding to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Moreover, our investor agreement with Sanofi Genzyme provides Sanofi Genzyme with the right, subject to certain exceptions, generally to maintain its ownership position in us until Sanofi Genzyme owns less than 7.5 percent of our outstanding common stock, subject to certain additional limited rights of Sanofi Genzyme to maintain its ownership percentage. In accordance with the investor agreement, to date, Sanofi Genzyme has exercised its right to purchase an additional 344,448 shares of our common stock in connection with our acquisition of Sirna in March 2014, an aggregate of 401,281 shares of our common stock based on its 2014 and 2015 compensation-related rights and an aggregate of 1,042,067 shares of our common stock in connection with our public offerings in January 2015 and May 2017. These purchases allowed Sanofi Genzyme to maintain its ownership level of our outstanding common stock. Sanofi Genzyme currently holds approximately 11 percent of our outstanding common stock. While the exercise of these rights by Sanofi Genzyme has provided us with an additional $147.7 million in cash to date, and while any exercise of these rights by Sanofi Genzyme in the future will provide us with further additional cash, these exercises have caused, and any future exercise of these rights by Sanofi Genzyme will also cause further, dilution to our stockholders. Sanofi Genzyme elected not to exercise its compensation-related rights for 2016 and 2017. Additionally, Sanofi Genzyme elected not to exercise its right to purchase additional shares in connection with our public offering in November 2017.
41
If we are unable to obtain additional funding on a timely basis, we may be required to significantly delay or curtail one or more of our research or development programs, delay the build-out of our global commercial infrastructure or undergo future reductions in our workforce or other corporate restructuring activities, and our ability to achieve our strategy for 2020 may be delayed or diminished. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own.
If the estimates we make, or the assumptions on which we rely, in preparing our consolidated financial statements prove inaccurate, our actual results may vary from those reflected in our projections and accruals.
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
The investment of our cash, cash equivalents and fixed income marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
At December 31, 2017, we had $1.70 billion in cash, cash equivalents and fixed income marketable securities, excluding the $30.0 million of restricted investments related to our term loan agreement. We historically have invested these amounts in high–grade corporate notes, commercial paper, securities issued or sponsored by the U.S. government, certificates of deposit and money market funds meeting the criteria of our investment policy, which is focused on the preservation of our capital. Corporate notes may also include foreign bonds denominated in U.S. dollars. These investments are subject to general credit, liquidity, market and interest rate risks. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our consolidated financial statements. In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The market risks associated with our investment portfolio may have an adverse effect on our results of operations, liquidity and financial condition.
The effect of comprehensive U.S. tax reform legislation on us, our subsidiaries and our affiliates, whether adverse or favorable, is uncertain.
Our business is subject to numerous international, federal, state, and other governmental laws, rules, and regulations that may adversely affect our operating results, including, taxation and tax policy changes, tax rate changes, new tax laws, or revised tax law interpretations, which individually or in combination may cause our effective tax rate to increase. For example, on December 22, 2017, the President of the United States signed into law the Tax Cuts and Jobs Act of 2017, or the TCJA. Among a number of significant changes to the current U.S. federal income tax rules, the TCJA reduces the marginal U.S. corporate income tax rate from 35 percent to 21 percent, introduces a capital investment deduction, limits the current deduction for net interest expense, limits the use of net operating losses to offset future taxable income, and makes extensive changes in the way in which income earned outside the United States is taxed in the United States. The TCJA is complex and far-reaching and we cannot predict with certainty the impact its enactment will have on us. Moreover, that effect, whether adverse or favorable, may not become evident for some period of time.
Risks Related to Our Dependence on Third Parties
We may not be able to execute our business strategy if we are unable to enter into alliances with other companies that can provide business and scientific capabilities and funds for the development and commercialization of our product candidates. If we are unsuccessful in forming or maintaining these alliances on terms favorable to us, our business may not succeed.
We currently are developing capabilities for sales or distribution and also have early capabilities for marketing, sales and market access, as well as limited capacity for drug development due to our growing pipeline of RNAi therapeutic opportunities. Accordingly, we have entered into alliances with other companies and collaborators that we believe can provide such capabilities in certain territories, and we intend to enter into additional such alliances in the future. Our collaboration strategy is to form alliances that create significant value for us and our collaborators in the advancement of RNAi therapeutics as a new class of innovative medicines. Specifically, with respect to our Genetic Medicine pipeline, we formed a broad strategic alliance with Sanofi Genzyme in 2014 pursuant to which we retain development and commercial rights for our current and future Genetic Medicine products in the United States, Canada and Western Europe, and Sanofi Genzyme has the right to develop and commercialize our current and future Genetic Medicine products principally in the rest of the world, subject to certain broader rights. In January 2018, we and Sanofi Genzyme amended our 2014 collaboration to provide that we would develop and commercialize patisiran globally and Sanofi Genzyme would develop and commercialize fitusiran globally. With respect to our Cardio-Metabolic Disease pipeline, we intend to seek future
42
strategic alliances for these programs, under which we may retain certain product development and commercialization rights, or we may structure as global alliances, as we did in our collaboration with MDCO to advance inclisiran. In October 2017, we announced an exclusive licensing agreement with Vir Biotechnology for the development and commercialization of RNAi therapeutics for infectious diseases, including chronic hepatitis B virus infection.
In such alliances, we expect our current, and may expect our future, collaborators to provide substantial capabilities in clinical development, regulatory affairs, and/or marketing, sales and distribution. Under certain of our alliances, we also may expect our collaborators to develop, market and/or sell certain of our product candidates. We may have limited or no control over the development, sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties. For example, we will rely entirely on (i) Sanofi Genzyme for the development and commercialization of fitusiran worldwide and potentially other of our Genetic Medicine programs in territories outside of the United States, Canada and Western Europe, and (ii) MDCO for all future development and commercialization of inclisiran worldwide. If Sanofi Genzyme and/or MDCO are not successful in their development and/or commercialization efforts, our future revenues from RNAi therapeutics for these indications may be adversely affected. Sanofi Genzyme also has the right to elect one global license for a future Genetic Medicine program that was not one of our defined Genetic Medicine programs as of the effective date of our 2014 collaboration. Sanofi Genzyme could elect its one global license for lumasiran, an investigational RNAi therapeutic targeting GO for the treatment of PH1. If Sanofi Genzyme elects to take a global license to lumasiran or another of our programs, we will no longer control the development and potential commercialization of such program and any revenues we receive will depend solely on the success of Sanofi Genzyme’s efforts. In addition, Sanofi Genzyme may elect not to opt into one or more of our Genetic Medicine programs. For example, during 2016, Sanofi Genzyme elected not to take a regional license for our givosiran and cemdisiran programs. While we intend to advance these programs independently, retaining global development and commercial rights, our ability to advance these programs and successfully develop and commercialize these product candidates may be adversely affected as a result of Sanofi Genzyme’s decision.
We may not be successful in entering into future alliances on terms favorable to us due to various factors, including our ability to successfully demonstrate proof-of-concept for our technology in humans, our ability to demonstrate the safety and efficacy of our specific drug candidates, our ability to manufacture or have third parties manufacture RNAi therapeutics, the strength of our intellectual property and/or concerns around challenges to our intellectual property. For example, our decision in October 2016 to discontinue development of revusiran could make it more difficult for us to attract collaborators due to concerns around the safety and/or efficacy of our technology platform or product candidates. In addition, our decision in September 2017 to temporarily suspend dosing in all ongoing fitusiran studies pending further review of a fatal thrombotic SAE and agreement with regulatory authorities on a risk mitigation strategy could, notwithstanding the alignment reached with the FDA on a risk mitigation strategy in November 2017, contribute to further concerns about the safety of our therapeutic candidates. Even if we do succeed in securing any such alliances, we may not be able to maintain them if, for example, development or approval of a product candidate is delayed, challenges are raised as to the validity or scope of our intellectual property, we are unable to secure adequate reimbursement from payors or sales of an approved drug are lower than we expected.
Furthermore, any delay in entering into collaboration agreements would likely either delay the development and commercialization of certain of our product candidates and reduce their competitiveness even if they reach the market, or prevent the development of certain product candidates. Any such delay related to our collaborations could adversely affect our business.
For certain product candidates that we may develop, we have formed collaborations to fund all or part of the costs of drug development and commercialization, such as our collaborations with Sanofi Genzyme, MDCO and Vir Biotechnology. We may not, however, be able to enter into additional collaborations for certain other programs, and the terms of any collaboration agreement we do secure may not be favorable to us. If we are not successful in our efforts to enter into future collaboration arrangements with respect to one or more of our product candidates, we may not have sufficient funds to develop that or other product candidates internally, or to bring our product candidates to market. If we do not have sufficient funds to develop and bring our product candidates to market, we will not be able to generate revenues from these product candidates, and this will substantially harm our business.
If any collaborator terminates or fails to perform its obligations under agreements with us, the development and commercialization of our product candidates could be delayed or terminated.
Our dependence on collaborators for capabilities and funding means that our business could be adversely affected if any collaborator terminates its collaboration agreement with us or fails to perform its obligations under that agreement. Our current or future collaborations, if any, may not be scientifically or commercially successful. Disputes may arise in the future with respect to the ownership of rights to technology or products developed with collaborators, which could have an adverse effect on our ability to develop and commercialize any affected product candidate.
43
Our current collaborations allow, and we expect that any future collaborations will allow, either party to terminate the collaboration for a material breach by the other party. In addition, our collaborators may have additional termination rights for convenience with respect to the collaboration or a particular program under the collaboration, under certain circumstances. For example, Sanofi Genzyme has the right to terminate our 2014 collaboration on a product-by-product basis in the event of certain safety concerns. Sanofi Genzyme also has the right to terminate its global license agreement for fitusiran at any time upon six months’ prior written notice. If Sanofi Genzyme were to terminate a particular program, we may have to expend significantly more on the development and commercialization of such product candidate. Moreover, our agreement with MDCO relating to the development and commercialization of inclisiran worldwide may be terminated by MDCO at any time upon four months’ prior written notice. If we were to lose a commercialization collaborator, we would have to attract a new collaborator or develop expanded sales, distribution and marketing capabilities internally, which would require us to invest significant amounts of financial and management resources.
In addition, if we have a dispute with a collaborator over the ownership of technology or other matters, or if a collaborator terminates its collaboration with us, for breach or otherwise, or determines not to pursue the research, development and/or commercialization of RNAi therapeutics, it could delay our development of product candidates, result in the need for additional company resources to develop product candidates, require us to expend time and resources to develop expanded sales and marketing capabilities on a more expedited timeline, make it more difficult for us to attract new collaborators and could adversely affect how we are perceived in the business and financial communities. For example, in March 2011, Arbutus filed a civil complaint against us claiming, among other things, misappropriation of its confidential and proprietary information and trade secrets. As a result of the litigation, which was settled in November 2012, we were required to expend resources and management attention that would otherwise have been engaged in other activities. In addition, in August 2013, we initiated binding arbitration proceedings to resolve a disagreement with Arbutus regarding the achievement by Arbutus of a $5.0 million milestone payment under our cross-license agreement relating to the manufacture of ALN-VSP clinical trial material for use in China. The Arbutus arbitration hearing was held in May 2015. In March 2016, the arbitration panel ruled in our favor and as a result, no milestone payment is due to Arbutus at this time. Arbutus did not appeal this ruling.
Moreover, a collaborator, or in the event of a change in control of a collaborator or the assignment of a collaboration agreement to a third party, the successor entity or assignee, could determine that it is in its interests to:
|
|
• |
pursue alternative technologies or develop alternative products, either on its own or jointly with others, that may be competitive with the products on which it is collaborating with us or which could affect its commitment to the collaboration with us; |
|
|
• |
pursue higher-priority programs or change the focus of its development programs, which could affect the collaborator’s commitment to us; or |
|
|
• |
if it has marketing rights, choose to devote fewer resources to the marketing of our product candidates, if any are approved for marketing, than it does for product candidates developed without us. |
If any of these occur, the development and commercialization of one or more product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, our development plans may be adversely affected.
We rely on independent clinical investigators, contract research organizations, or CROs, and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We have contracted, and we plan to continue to contract with, certain third parties to provide certain services, including site selection, enrollment, monitoring, auditing and data management services. Although we depend heavily on these parties, we control only certain aspects of their activity and therefore, we cannot be assured that these third parties will adequately perform all of their contractual obligations to us in compliance with regulatory and other legal requirements and our internal policies and procedures. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with GCP requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our CROs fail to comply with applicable GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA, the PMDA in Japan or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations.
44
If our third-party service providers cannot adequately and timely fulfill their obligations to us, or if the quality and accuracy of our clinical trial data is compromised due to failure by such third party to adhere to our protocols or regulatory requirements or if such third parties otherwise fail to meet deadlines, our development plans and/or regulatory reviews for marketing approvals may be delayed or terminated, including, for example, review of our NDA and MAA filings for patisiran. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
We have limited manufacturing experience and resources and we must incur significant costs to develop this expertise and/or rely on third parties to manufacture our products.
We have limited manufacturing experience. In order to develop our product candidates, apply for regulatory approvals and commercialize our products, if approved, we will need to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities. Historically, our internal manufacturing capabilities were limited to small-scale production of material for use in in vitro and in vivo experiments that is not required to be produced under cGMP standards. During 2012, we developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late stage clinical trial use and commercial supply. In addition, in April 2016, we completed our purchase of a parcel of land in Norton, Massachusetts, where we have commenced construction of a cGMP manufacturing facility for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use.
We may manufacture limited quantities of clinical trial materials ourselves, but otherwise we currently rely on third parties to manufacture the drug substance and, with the exception of patisiran, the finished product we will require for any clinical trials that we initiate and to support the commercial launch of our first several products. There are a limited number of manufacturers that supply synthetic siRNAs. We currently rely on a limited number of CMOs for our supply of synthetic siRNAs. For example, in July 2015, we amended our manufacturing agreement with Agilent, to provide for Agilent to supply, subject to any conflicting obligations under our third-party agreements, a specified percentage of the active pharmaceutical ingredients required for certain of our products in clinical development, as well as other products the parties may agree upon in the future. We will also rely on Agilent to supply the active pharmaceutical ingredients to support the commercial launch of patisiran. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our CMOs, including Agilent, to meet our delivery time requirements or provide adequate amounts of material to meet our needs. Included in these risks are potential synthesis and purification failures and/or contamination during the manufacturing process, as well as other issues with the CMO’s facility and ability to comply with the applicable manufacturing requirements, which could result in unusable product and cause delays in our manufacturing timelines and ultimately delay our clinical trials and potentially put at risk commercial supply, as well as result in additional expense to us. To fulfill our siRNA requirements, we will likely need to secure alternative suppliers of synthetic siRNAs and such alternative suppliers are limited and may not be readily available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner. As noted above, in order to ensure long-term supply capabilities for our RNAi therapeutics, we are developing our own capabilities to manufacture drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use.
In addition to the manufacture of the synthetic siRNAs, we may have additional manufacturing requirements related to the technology required to deliver the siRNA to the relevant cell or tissue type, such as LNPs or conjugates. In some cases, the delivery technology we utilize is highly specialized or proprietary, and for technical and/or legal reasons, we may have access to only one or a limited number of potential manufacturers for such delivery technology. In addition, the scale-up of our delivery technologies could be very difficult and/or take significant time. We also have very limited experience in such scale-up and manufacturing, requiring us to depend on a limited number of third parties, who might not be able to deliver in a timely manner, or at all. Failure by manufacturers to properly manufacture our delivery technology and/or formulate our siRNAs for delivery could result in unusable product. Furthermore, competition for supply from our manufacturers from other companies, a breach by such manufacturers of their contractual obligations or a dispute with such manufacturers would cause delays in our discovery and development efforts, as well as additional expense to us.
Given the limited number of suppliers for our delivery technology and drug substance, we have developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late stage clinical use and commercial supply. During 2015, we scaled our cGMP manufacturing capacity for patisiran and believe we should have adequate resources to supply our commercial needs. In addition, as noted above, we are developing our own capabilities to manufacture drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use. In developing these manufacturing capabilities by building our own manufacturing facilities, we have incurred substantial expenditures, and expect to incur significant additional expenditures in the future. In addition, the construction and qualification of our drug substance facility is expected to take several years to complete and there are many risks inherent in the construction of a new facility that could result in delays and additional costs, including the need to obtain access to necessary equipment and third-party technology, if any. Also, we have had to, and will likely need to continue to, hire and train qualified employees to staff our facilities. We do not currently have a second source of supply for patisiran formulated bulk
45
drug product. If we are unable to manufacture sufficient quantities of material or if we encounter problems with our facilities in the future, we may also need to secure alternative suppliers of patisiran formulated bulk drug product and drug substance, and such alternative suppliers may not be available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner. Any delay or setback in the manufacture of patisiran could, assuming approval, delay the launch or ongoing commercial supply, which could significantly impact our revenues and operating results.
The manufacturing process for any products that we may develop is subject to the FDA and foreign regulatory authority approval process and we will need to meet, and will need to contract with CMOs who can meet, all applicable FDA and foreign regulatory authority requirements on an ongoing basis. In addition, if we receive the necessary regulatory approval for any product candidate, we also expect to rely on third parties, including potentially our commercial collaborators, to produce materials required for commercial supply. We may experience difficulty in obtaining adequate manufacturing capacity for our needs and the needs of our collaborators, who we have, in some instances, the obligation to supply. If we are unable to obtain or maintain CMOs for these product candidates, or to do so on commercially reasonable terms, we may not be able to successfully develop and commercialize our products.
To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we depend, and will depend in the future, on these third parties, including Agilent, to perform their obligations in a timely manner and consistent with contractual and regulatory requirements, including those related to quality control and quality assurance. The failure of Agilent or any other CMO to perform its obligations as expected, or, to the extent we manufacture all or a portion of our product candidates ourselves, our failure to execute on our manufacturing requirements, could adversely affect our business in a number of ways, including:
|
|
• |
we or our current or future collaborators may not be able to initiate or continue clinical trials of product candidates that are under development; |
|
|
• |
we or our current or future collaborators may be delayed in submitting regulatory applications, or receiving regulatory approvals, for our product candidates, including patisiran; |
|
|
• |
we may lose the cooperation of our collaborators; |
|
|
• |
our facilities and those of our CMOs, and our products could be the subject of inspections by regulatory authorities that could have a negative outcome and result in delays in supply; |
|
|
• |
we may be required to cease distribution or recall some or all batches of our products or take action to recover clinical trial material from clinical trial sites; and |
|
|
• |
ultimately, we may not be able to meet commercial demands for our products. |
If any CMO with whom we contract, including Agilent, fails to perform its obligations, we may be forced to manufacture the materials ourselves, for which we may not have the capabilities or resources, or enter into an agreement with a different CMO, which we may not be able to do on reasonable terms, if at all. In either scenario, our clinical trials or commercial distribution could be delayed significantly as we establish alternative supply sources. In some cases, the technical skills required to manufacture our products or product candidates may be unique or proprietary to the original CMO and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills to a back-up or alternate supplier, or we may be unable to transfer such skills at all. In addition, if we are required to change CMOs for any reason, we will be required to verify that the new CMO maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product according to the specifications previously submitted to or approved by the FDA or another regulatory authority. The delays associated with the verification of a new CMO could negatively affect our ability to develop product candidates in a timely manner or within budget. Furthermore, a CMO may possess technology related to the manufacture of our product candidate that such CMO owns independently. This would increase our reliance on such CMO or require us to obtain a license from such CMO in order to have another CMO manufacture our products or product candidates.
We have no sales or distribution experience and only early capabilities for marketing, sales and market access, and expect to invest significant financial and management resources to establish these capabilities and to establish a global commercial infrastructure.
We have no sales or distribution experience and only early capabilities for marketing, sales and market access. We currently expect to rely heavily on third parties to launch and market certain of our product candidates in certain geographies, if approved. However, we intend to commercialize several of our late-stage products on our own globally, including patisiran, as a result of the January 2018 amendment to our Sanofi Genzyme collaboration, and givosiran. Accordingly, we will need to develop internal sales, distribution and marketing capabilities as part of our core product strategy initially in the United States and the EU, then Canada and Switzerland, Central and Eastern Europe, Japan and in other major markets in the rest of the world, which will require significant
46
financial and management resources. For those products for which we will perform sales, marketing and distribution functions ourselves, including patisiran and givosiran, if approved, and for future products we successfully develop where we may retain certain product development and commercialization rights, we could face a number of additional risks, including:
|
|
• |
we may not be able to attract and build a significant marketing or sales force; |
|
|
• |
we may not be able to establish our global capabilities and infrastructure in a timely manner; |
|
|
• |
the cost of establishing a marketing or sales force may not be justifiable in light of the revenues generated by any particular product and/or in any specific geographic region; and |
|
|
• |
our direct sales and marketing efforts may not be successful. |
If we are unable to develop our own global sales, marketing and distribution capabilities for patisiran and other products, we will not be able to successfully commercialize our products without reliance on third parties.
Credit and financial market conditions may exacerbate certain risks affecting our business from time to time.
Due to tightening of global credit, there may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators. We rely on third parties for several important aspects of our business, including significant portions of our manufacturing needs, development of product candidates and conduct of clinical trials. If such third parties are unable to satisfy their commitments to us, our business could be adversely affected.
Our ability to secure additional financing in addition to our term loan agreement and to satisfy our financial obligations under indebtedness outstanding from time to time will depend upon our future operating performance, which is subject to then prevailing general economic and credit market conditions, including interest rate levels and the availability of credit generally, and financial, business and other factors, many of which are beyond our control. In light of periodic uncertainty in the capital and credit markets, there can be no assurance that sufficient financing will be available on desirable or even any terms to fund investments, acquisitions, stock repurchases, dividends, debt refinancing or extraordinary actions.
Risks Related to Managing Our Operations
If we are unable to attract and retain qualified key management and scientists, development, medical and commercial staff, consultants and advisors, our ability to implement our business plan may be adversely affected.
We are highly dependent upon our senior management and our scientific, clinical and medical staff. The loss of the service of any of the members of our senior management, including Dr. John Maraganore, our Chief Executive Officer, may significantly delay or prevent the achievement of product development and commercialization, and other business objectives. Our employment arrangements with our key personnel are terminable without notice. We do not carry key person life insurance on any of our employees.
We have grown our workforce significantly over the past several years and anticipate continuing to add a significant number of additional employees as we focus on achieving our Alnylam 2020 strategy. We face intense competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities, governmental entities and other research institutions, many of which have substantially greater resources with which to reward qualified individuals than we do. In addition, due to the risks associated with developing a new class of medicine, we may experience disappointing results in a clinical program and our stock price may decline as a result, as was the case following our decision in October 2016 to discontinue our revusiran program, and, to less of an extent, following our temporary suspension of dosing in our fitusiran program in September 2017. As a result, we may face additional challenges in attracting and retaining employees. Accordingly, we may be unable to attract and retain suitably qualified individuals in order to support our growing research, development and commercialization efforts and initiatives, and our failure to do so could have an adverse effect on our ability to implement our future business plan.
We may have difficulty expanding our operations successfully as we evolve from a U.S.-based company primarily involved in discovery, pre-clinical testing and clinical development into a global company that develops and commercializes multiple drugs.
As we increase the number of product candidates we are developing we will also need to expand our operations in the United States and continue to build operations in the EU and other geographies, including Japan. Based on the positive data reported from our APOLLO Phase 3 study of patisiran, we filed an NDA and an MAA for patisiran in December 2017. Assuming regulatory approvals, we are preparing to commercialize patisiran in 2018 and now have global development and commercialization rights for patisiran as a result of the January 2018 amendment to our Sanofi Genzyme collaboration. We also plan to file for regulatory approval in Japan in mid-2018 and in one or more additional countries by the end of the year.
47
As noted above, we grew our workforce significantly during 2016 and 2017, and anticipate continuing to hire additional employees, including employees in the EU, Japan and other territories, as we focus on the potential commercial launch of patisiran and achieving our Alnylam 2020 strategy. This expected growth is placing a strain on our administrative and operational infrastructure, and we will need to develop additional and/or new infrastructure and capabilities to support our growth and obtain additional space to conduct our operations in the United States, the EU, Japan and other geographies. If we are unable to develop such additional infrastructure or obtain sufficient space to accommodate our growth in a timely manner and on commercially reasonable terms, our business could be negatively impacted. As product candidates we develop enter and advance through clinical trials, we will need to expand our global development, regulatory, manufacturing, quality, compliance, and marketing and sales capabilities, or contract with other organizations to provide these capabilities for us. In addition, as our operations expand due to our development progress, we expect that we will need to manage additional relationships with various collaborators, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls and systems, reporting systems and infrastructure, and policies and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
The increasing use of social media platforms presents new risks and challenges.
Social media is increasingly being used to communicate about our clinical development programs and the diseases our investigational RNAi therapeutics are being developed to treat, and we intend to utilize appropriate social media in connection with our commercialization efforts following approval of our drug candidates. Social media practices in the biopharmaceutical industry continue to evolve and regulations relating to such use are not always clear. This evolution creates uncertainty and risk of noncompliance with regulations applicable to our business. For example, patients may use social media channels to comment on their experience in an ongoing blinded clinical study or to report an alleged AE. When such disclosures occur, there is a risk that we fail to monitor and comply with applicable AE reporting obligations or we may not be able to defend our business or the public’s legitimate interests in the face of the political and market pressures generated by social media due to restrictions on what we may say about our investigational products. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social networking website. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions or incur other harm to our business.
Our business and operations could suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. Such events could cause interruption of our operations. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our product candidates could result in delays in our regulatory filings and development efforts, as well as delays in the commercialization of our products, and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development and potential commercialization of our product candidates could be delayed.
The results of the United Kingdom’s referendum on withdrawal from the EU may have a negative effect on global economic conditions, financial markets and our business.
In June 2016, the United Kingdom, or UK, held a referendum in which voters approved an exit from the EU, commonly referred to as “Brexit.” This referendum has created political and economic uncertainty, particularly in the UK and the EU, and this uncertainty may persist for years. A withdrawal could, among other outcomes, disrupt the free movement of goods, services and people between the UK and the EU, and result in increased legal and regulatory complexities, as well as potential higher costs of conducting business in Europe. The UK’s vote to exit the EU could also result in similar referendums or votes in other European countries in which we do business. Given the lack of comparable precedent, it is unclear what financial, trade and legal implications the withdrawal of the UK from the EU would have and how such withdrawal would affect us.
For example, Brexit could result in the UK or the EU significantly altering its regulations affecting the clearance or approval of our product candidates that are developed in the UK. Any new regulations could add time and expense to the conduct of our business, as well as the process by which our products receive regulatory approval in the UK, the EU and elsewhere. In addition, the announcement of Brexit and the withdrawal of the UK from the EU have had and may continue to have a material adverse effect on global economic conditions and the stability of global financial markets, and may significantly reduce global market liquidity and restrict the ability of key market participants to operate in certain financial markets. Any of these effects of Brexit, among others, could adversely affect our business, our results of operations, liquidity and financial condition.
48
Risks Related to Development, Clinical Testing and Regulatory Approval of Our Product Candidates
Any product candidates we develop may fail in development or be delayed to a point where they do not become commercially viable.
Before obtaining regulatory approval for the commercial distribution of our product candidates, we must conduct, at our own expense, extensive nonclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. Nonclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome, and the historical failure rate for product candidates is high. In October 2016, we discontinued development of one of our product candidates, which included a Phase 3 clinical trial. We currently have multiple other programs in clinical development, including several internal programs and two partnered programs currently in Phase 3 development, as well as several earlier stage clinical programs. In November 2017, we reported positive complete results from our APOLLO Phase 3 clinical trial for our lead product candidate, patisiran, and in December 2017, we filed an NDA and an MAA for patisiran. However, we may not be able to further advance these or any other product candidate through clinical trials and regulatory approval.
If we enter into clinical trials, the results from nonclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in subsequent subjects or in subsequent human clinical trials of that product candidate or any other product candidate. For example, in June 2017, we announced updated results from our Phase 1 clinical trial of givosiran. Although the clinical data from this trial are encouraging, the data are preliminary in nature, based on a limited number of patients with AIP. These data, or other positive data, may not continue for patients with AIP, and may not be repeated or observed in our ENVISION Phase 3 study. There can be no assurance that our studies with givosiran will ultimately be successful or support further clinical advancement or regulatory approval of this product candidate. There is a high failure rate for drugs proceeding through clinical studies. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies, and any such setbacks in our clinical development could have a material adverse effect on our business and operating results. Moreover, patisiran, givosiran, fitusiran, inclisiran and our other product candidates each employ novel delivery technologies that have yet to be extensively evaluated in human clinical trials and proven safe and effective.
In addition, we, the FDA or other applicable regulatory authorities, or an IRB or similar foreign review board or committee, may delay initiation of or suspend clinical trials of a product candidate at any time for various reasons, including if we or they believe the healthy volunteer subjects or patients participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a product candidate or related product on healthy volunteer subjects or patients in a clinical trial could result in our decision, or a decision by the FDA or foreign regulatory authorities, to suspend or terminate the trial, or, in the case of regulatory agencies, a refusal to approve a particular product candidate for any or all indications of use. For example, in October 2016, we announced our decision to discontinue development of revusiran, an investigational RNAi therapeutic that was being developed for the treatment of patients with cardiomyopathy due to hATTR amyloidosis. Our decision followed the recommendation of the revusiran ENDEAVOUR Phase 3 study Data Monitoring Committee, or DMC, to suspend dosing and the observation of an imbalance in mortality in revusiran-treated patients as compared to those on placebo. We conducted a comprehensive evaluation of the revusiran data and reported the results of our evaluation on August 9, 2017. Following our evaluation, we continue to believe that the decision to discontinue development of revusiran does not affect patisiran, which is under regulatory review for the treatment of hATTR amyloidosis, or any of our other investigational RNAi therapeutic programs in development. In September 2017, we announced that we had temporarily suspended dosing in all ongoing fitusiran studies pending further review of a fatal thrombotic SAE and agreement with regulatory authorities on a risk mitigation strategy. We have reached alignment with study investigators and the FDA on safety measures and a risk mitigation strategy to enable resumption of dosing in clinical studies with fitusiran, including our Phase 2 OLE study and the ATLAS Phase 3 program, including protocol-specified guidelines and additional investigator and patient education concerning reduced doses of replacement factor or bypassing agent to treat any breakthrough bleeds in fitusiran studies. Based on this, amended protocols were submitted to the regulatory authorities and in December 2017, the FDA lifted the clinical hold. Dosing has resumed in the Phase 2 OLE study and we expect to begin dosing patients in the ATLAS Phase 3 program in early 2018.
Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the age and condition of the patients, the stage and severity of disease, the availability of clinical trials for other investigational drugs for the same disease or condition, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, and the eligibility criteria for the clinical trial. For example, we or our partners may experience difficulty enrolling our clinical trials, including, but not limited to, our clinical trials for fitusiran, due to the availability of existing approved treatments, as well as other investigational treatments in development. Moreover, given the recent temporary suspension of dosing in our fitusiran studies due to a fatal thrombotic SAE, people with hemophilia may be more reluctant to enroll in the ATLAS Phase 3 program of fitusiran. Delays or difficulties in patient
49
enrollment or difficulties retaining trial participants, including as a result of the availability of existing or other investigational treatments or safety concerns, can result in increased costs, longer development times or termination of a clinical trial.
Although our investigational RNAi therapeutics have been generally well tolerated in our clinical trials to date, new safety findings may emerge. For example, as noted above, in September 2017, we announced that we had temporarily suspended dosing in all ongoing fitusiran studies pending further review of a fatal thrombotic SAE that occurred in a patient with hemophilia A without inhibitors who was receiving fitusiran in our Phase 2 OLE study. In addition, in October 2016, we made the decision to discontinue our revusiran program. Following reports in the revusiran Phase 2 OLE study of new onset or worsening peripheral neuropathy, the revusiran ENDEAVOUR Phase 3 study DMC assembled in early October 2016 at our request to review these reports and ENDEAVOUR safety data on an unblinded basis. The DMC did not find conclusive evidence for a drug-related neuropathy signal in the ENDEAVOUR trial, but informed us that the benefit-risk profile for revusiran no longer supported continued dosing. We subsequently reviewed unblinded ENDEAVOUR data which revealed an imbalance of mortality in the revusiran arm as compared to placebo. Further, a review by us in 2017 of the ENDEAVOUR results subsequent to the completion of follow-up of the patients post-dosing discontinuation revealed an imbalance in new onset or worsening peripheral neuropathy in the revusiran arm as compared to placebo. We had previously reported, in July 2016, preliminary data from our revusiran Phase 2 OLE study for 12 patients who had reached the 12-month endpoint as of the data transfer date of May 26, 2016. SAEs were observed in 14 patients, one of which, a case of lactic acidosis, was deemed possibly related to the study drug and the patient discontinued treatment. There were a total of seven deaths reported at that time in the revusiran OLE study, all of which were unrelated to the study drug. The majority of the AEs were mild or moderate in severity; ISRs were reported in 12 patients. In August 2015, we reported that three patients had discontinued from the revusiran Phase 2 OLE study due to recurrent localized reactions at the injection site or a diffuse rash; no further discontinuations due to ISRs had occurred as of May 26, 2016.
In our patisiran APOLLO Phase 3 study in patients with polyneuropathy due to hATTR amyloidosis, the most commonly reported AEs that occurred more frequently in patisiran patients were peripheral edema and IRRs. These were generally mild to moderate in severity and only one patient discontinued from the APOLLO study due to an IRR. Compared to placebo, patisiran treatment was associated with fewer treatment discontinuations and fewer study withdrawals due to AEs. The incidence of SAEs across the patisiran and placebo groups was similar and the SAEs reported in two or more patients in the patisiran group included: diarrhea, cardiac failure, congestive cardiac failure, orthostatic hypotension, pneumonia and atrioventricular block complete. These were all considered unrelated to patisiran, except for one SAE of diarrhea. SAEs occurred with similar frequency in the placebo group, except for diarrhea. Deaths were recorded with a similar incidence across the patisiran and placebo treatment groups and no deaths were considered related to the study drug.
In addition, in our ALN-VSP clinical trial, one patient with advanced pancreatic neuroendocrine cancer with extensive involvement of the liver developed hepatic failure five days following the second dose of ALN-VSP and subsequently died; this was deemed possibly related to the study drug. As demonstrated by the discontinuation of our revusiran program in October 2016 and the temporary suspension of dosing in September 2017 in our fitusiran studies, the occurrence of SAEs and/or AEs can result in the suspension or termination of clinical trials of a product candidate by us or the FDA or a foreign regulatory authority, or refusal to approve a particular product candidate for any or all indications of use.
Clinical trials also require the review, oversight and approval of IRBs or, outside of the United States, an independent ethics committee, which continually review clinical investigations and protect the rights and welfare of human subjects. Inability to obtain or delay in obtaining IRB or ethics committee approval can prevent or delay the initiation and completion of clinical trials, and the FDA or foreign regulatory authorities may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB or ethics committee review and approval, as the case may be, in support of a marketing application.
Our product candidates that we develop may encounter problems during clinical trials that will cause us, an IRB, ethics committee or regulatory authorities to delay, suspend or terminate these trials, or that will delay or confound the analysis of data from these trials. If we experience any such problems, we may not have the financial resources to continue development of the product candidate that is affected, or development of any of our other product candidates. We may also lose, or be unable to enter into, collaborative arrangements for the affected product candidate and for other product candidates we are developing.
A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, nonclinical testing and the clinical trial process that could delay or prevent regulatory approval or our ability to commercialize our product candidates, including:
|
|
• |
our nonclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional nonclinical testing or clinical trials, or we may abandon projects that we expect to be promising; |
50
|
|
• |
conditions imposed on us by an IRB or ethics committee, or the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
|
|
• |
problems in engaging IRBs or ethics committees to oversee clinical trials or problems in obtaining or maintaining IRB or ethics committee approval of trials; |
|
|
• |
delays in enrolling patients and volunteers into clinical trials, and variability in the number and types of patients and volunteers available for clinical trials; |
|
|
• |
high drop-out rates for patients and volunteers in clinical trials; |
|
|
• |
negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours; |
|
|
• |
inadequate supply or quality of product candidate materials or other materials necessary for the conduct of our clinical trials; |
|
|
• |
greater than anticipated clinical trial costs; |
|
|
• |
serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; |
|
|
• |
poor or disappointing effectiveness of our product candidates during clinical trials; |
|
|
• |
unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or records of any clinical or nonclinical investigation; |
|
|
• |
failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all; |
|
|
• |
governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or |
|
|
• |
varying interpretations of data by the FDA and similar foreign regulatory agencies. |
Even if we successfully complete clinical trials of our product candidates, as is the case with patisiran, any given product candidate may not prove to be a safe and effective treatment for the disease for which it was being tested.
We may be unable to obtain United States or foreign regulatory approval and, as a result, unable to commercialize our product candidates.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, pricing, marketing and distribution of drugs. Rigorous nonclinical testing and clinical trials and an extensive regulatory approval process are required to be successfully completed in the United States and in many foreign jurisdictions before a new drug can be marketed. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. It is possible that none of the product candidates we may develop will obtain the regulatory approvals necessary for us or our collaborators to begin selling them.
We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA. The time required to obtain FDA and other regulatory approvals is unpredictable but typically takes many years following the commencement of clinical trials, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when regulating us are not always applied predictably or uniformly and can change. Any analysis we perform of data from nonclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations, for example, from future legislation or administrative action, or from changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be.
51
Because the drugs we are developing may represent a new class of drug, the FDA and its foreign counterparts have not yet established any definitive policies, practices or guidelines in relation to these drugs. The lack of policies, practices or guidelines may hinder or slow review by the FDA of any regulatory filings that we may submit. Moreover, the FDA may respond to these submissions by defining requirements we may not have anticipated. Such responses could lead to significant delays in the clinical development of our product candidates. In addition, because there may be approved treatments for some of the diseases for which we may seek approval, in order to receive regulatory approval, we may need to demonstrate through clinical trials that the product candidates we develop to treat these diseases, if any, are not only safe and effective, but safer or more effective than existing products. Furthermore, in recent years, there has been increased public and political pressure on the FDA with respect to the approval process for new drugs, and the FDA’s standards, especially regarding drug safety, appear to have become more stringent.
In November 2017, we reported positive complete results from our APOLLO Phase 3 clinical trial and generally encouraging safety data, and in December 2017, we completed the submission of our first NDA and submitted our first MAA for patisiran. In January 2018, we announced that the EMA has accepted the MAA and initiated its review. Patisiran was previously granted an accelerated assessment by the EMA. In early February 2018, we announced that the FDA has accepted our NDA and granted our request for priority review, with an action date of August 11, 2018. We also plan to file for regulatory approval in Japan in mid-2018 and in one or more additional countries by the end of the year. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate revenues from patisiran or any product candidate for which we may seek approval in the future. Furthermore, any regulatory approval to market patisiran or any other product may be subject to limitations on the approved uses for which we may market the product or the labeling or other restrictions. In addition, the FDA has the authority to require a REMS plan as part of an NDA, or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. In the EU, we could be required to adopt a similar plan, known as a risk management plan, and our products could be subject to specific risk minimization measures, such as restrictions on prescription and supply, the conduct of post-marketing safety or efficacy studies, or the distribution of patient and/or prescriber educational materials. In either instance, these limitations and restrictions may limit the size of the market for the product and affect reimbursement by third-party payors.
We are also subject to numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing and marketing authorisation, pricing and third-party reimbursement. The foreign regulatory approval process varies among countries and includes all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not ensure approval by regulatory authorities outside the United States and vice versa.
Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory oversight. If we fail to comply with continuing U.S. and foreign requirements, our approvals could be limited or withdrawn, we could be subject to other penalties, and our business would be seriously harmed.
Following any initial regulatory approval of patisiran and any other drugs we may develop, we will also be subject to continuing regulatory oversight, including the review of adverse drug experiences and clinical results that are reported after our drug products are made commercially available. This would include results from any post-marketing tests or surveillance to monitor the safety and efficacy of patisiran or other drug products required as a condition of approval or agreed to by us. Any regulatory approvals that we receive for patisiran or our other product candidates may also be subject to limitations on the approved uses for which the product may be marketed. Other ongoing regulatory requirements include, among other things, submissions of safety and other post-marketing information and reports, registration and listing, as well as continued compliance with cGMP requirements and GCP requirements for any clinical trials that we conduct post-approval. In addition, we are conducting, and intend to continue to conduct, clinical trials for our product candidates, and we intend to seek approval to market our product candidates, in jurisdictions outside of the United States, and therefore will be subject to, and must comply with, regulatory requirements in those jurisdictions.
The FDA has significant post-market authority, including, for example, the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate serious safety risks related to the use of a drug and to require withdrawal of the product from the market. The FDA also has the authority to require a REMS plan after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug.
The CMO and manufacturing facilities we use to make our product candidates, including our Cambridge facility, our future Norton facility, and Agilent and other CMOs, will also be subject to periodic review and inspection by the FDA and other regulatory agencies. To date, our Cambridge manufacturing facility has not been subject to an inspection by any regulatory authority. We expect Agilent will undergo regulatory inspection by the FDA and potentially other regulatory authorities in connection with the review of our NDA and MAA, as well as any subsequent applications for regulatory approval in other territories. The discovery of any new or previously unknown problems with us or our CMOs, or our or their manufacturing processes or facilities, may result in restrictions on
52
the drug or CMO or facility, including delay in approval or, in the future, withdrawal of the drug from the market. We have developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for Phase 3 clinical and commercial use. In addition, in April 2016, we completed our purchase of a parcel of land in Norton, Massachusetts, where we have commenced construction of a cGMP manufacturing facility for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use. We may not have the ability or capacity to manufacture material at a broader commercial scale in the future. We may manufacture clinical trial materials or we may contract a third party to manufacture these materials for us. Reliance on CMOs entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the CMO for regulatory compliance. Our product promotion and advertising will also be subject to regulatory requirements and continuing regulatory review.
If we or our collaborators, CMOs or service providers fail to comply with applicable continuing regulatory requirements in the United States or foreign jurisdictions in which we may seek to market our products, we or they may be subject to, among other things, fines, warning letters, holds on clinical trials, refusal by the FDA or foreign regulatory authorities to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatory approval, product recalls and seizures, refusal to permit the import or export of products, operating restrictions, injunction, civil penalties and criminal prosecution.
Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our product candidates upon their commercial introduction, which will prevent us from becoming profitable.
The product candidates that we are developing are based upon new technologies or therapeutic approaches. Key participants in pharmaceutical marketplaces, such as physicians, third-party payors and consumers, may not accept a product intended to improve therapeutic results based on RNAi technology. As a result, it may be more difficult for us to convince the medical community and third-party payors to accept and use our product, or to provide favorable reimbursement.
Other factors that we believe will materially affect market acceptance of our product candidates include:
|
|
• |
the timing of our receipt of any marketing approvals, the terms of any approvals and the countries in which approvals are obtained; |
|
|
• |
the safety and efficacy of our product candidates, as demonstrated in clinical trials and as compared with alternative treatments, if any; |
|
|
• |
relative convenience and ease of administration of our product candidates; |
|
|
• |
the willingness of patients to accept potentially new routes of administration or new or different therapeutic approaches and mechanisms of action; |
|
|
• |
the success of our physician education programs; |
|
|
• |
the availability of adequate government and third-party payor reimbursement; |
|
|
• |
the pricing of our products, particularly as compared to alternative treatments, and the market perception of such prices and any price increase that we may implement in the future; and |
|
|
• |
availability of alternative effective treatments for the diseases that product candidates we develop are intended to treat and the relative risks, benefits and costs of those treatments. |
For example, patisiran utilizes an intravenous mode of administration that physicians and/or patients may not readily adopt or which may not compete with other potentially available options. In addition, fitusiran represents a new approach to treating hemophilia which may not be readily accepted by patients and their caregivers.
In addition, our estimates regarding the potential market size for patisiran or our other product candidates may be materially different from what we currently expect at the time we commence commercialization, which could result in significant changes in our business plan and may have a material adverse effect on our results of operations and financial condition.
If we or our collaborators, CMOs or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could affect our ability to develop, market and sell our products and may harm our reputation.
As a manufacturer of pharmaceuticals, we are subject to federal, state, and comparable foreign healthcare laws and regulations pertaining to fraud and abuse and patients’ rights. These laws and regulations include:
|
|
• |
The U.S. federal Anti-Kickback statute, which prohibits, among other things, persons from soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid. |
53
|
|
• |
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created federal criminal statutes that prohibit knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statements in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity can be found guilty of violating HIPAA without actual knowledge of the statute or specific intent to violate it. |
|
|
• |
HIPAA as amended by the Health Information Technology for Economic and Clinical Health Act, which impose requirements relating to the privacy, security, and transmission of individually identifiable health information; and require notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information. |
|
|
• |
The U.S. federal Open Payments requirements were implemented by the CMS, pursuant to the PPACA. Under the Open Payments Program, manufacturers of medical devices, medical supplies, biological products and drugs covered by Medicare, Medicaid and the Children’s Health Insurance Programs must report all transfers of value, including consulting fees, travel reimbursements, research grants, and other payments or gifts with values over $10 made to physicians and teaching hospitals. |
|
|
• |
Federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers. |
|
|
• |
State and foreign laws comparable to each of the above federal laws, including in the EU laws prohibiting giving healthcare professionals any gift or benefit in kind as an inducement to prescribe our products, national transparency laws requiring the public disclosure of payments made to healthcare professionals and institutions, and data privacy laws, in addition to anti-kickback and false claims laws applicable to commercial insurers and other non-federal payors, requirements for mandatory corporate regulatory compliance programs, and laws relating to government reimbursement programs, patient data privacy and security. |
If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, criminal prosecution, monetary damages, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, or the imposition of a corporate integrity agreement with the Office of Inspector General of the Department of Health and Human Services, any of which could adversely affect our financial results. We are establishing our global compliance infrastructure as we prepare for the potential launch of patisiran in 2018 in the United States and the EU. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
If we or our collaborators, CMOs or service providers fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market and sell patisiran or our other products successfully and could harm our reputation and lead to reduced acceptance of our products by the market. These enforcement actions include, among others:
|
|
• |
adverse regulatory inspection findings; |
|
|
• |
warning letters; |
|
|
• |
voluntary or mandatory product recalls or public notification or medical product safety alerts to healthcare professionals; |
54
|
|
• |
restrictions on, or prohibitions against, importation or exportation of our products; |
|
|
• |
suspension of review or refusal to approve pending applications or supplements to approved applications; |
|
|
• |
exclusion from participation in government-funded healthcare programs; |
|
|
• |
exclusion from eligibility for the award of government contracts for our products; |
|
|
• |
suspension or withdrawal of product approvals; |
|
|
• |
product seizures; |
|
|
• |
injunctions; and |
|
|
• |
civil and criminal penalties, up to and including criminal prosecution resulting in fines, exclusion from healthcare reimbursement programs and imprisonment. |
Moreover, federal, state or foreign laws or regulations are subject to change, and while we, our collaborators, CMOs and/or service providers currently may be compliant, that could change due to changes in interpretation, prevailing industry standards or the legal structure.
Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drugs vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. We are actively monitoring these regulations as we await potential regulatory approval for patisiran in the U.S. and the EU and several of our other programs move into late stages of development, however, a number of our programs are currently in the earlier stages of development and we will not be able to assess the impact of price regulations for such programs for a number of years. We might obtain regulatory approval for a product, including patisiran, in a particular country, but then be subject to price regulations that delay our commercial launch of the product and negatively impact the revenues we are able to generate from the sale of the product in that country and potentially in other countries due to reference pricing.
Our ability to commercialize patisiran or any other products successfully also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if we succeed in bringing patisiran or other products to the market, patisiran and such other products may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell patisiran or our other products on a competitive basis. Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts reimbursed for drug products. If the price we are able to charge for patisiran or any other products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, or if reimbursement is denied, our return on investment could be adversely affected. In addition, we have stated publicly that we intend to grow through continued scientific innovation rather than arbitrary price increases. Specifically, we have stated that we will not raise the price of any product for which we receive marketing approval over the rate of inflation, as determined by the consumer price index for urban consumers (approximately 2.2 percent currently). Our patient access philosophy could also negatively impact the revenues we are able to generate from the sale of one or more of our products in the future.
We currently expect that some of the drugs we develop may need to be administered under the supervision of a physician or other healthcare professional on an outpatient basis, including patisiran. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable drugs) may be eligible for coverage under the Medicare Part B program if:
|
|
• |
they are incident to a physician’s services; |
|
|
• |
they are reasonable and necessary for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standards of medical practice; and |
|
|
• |
they have been approved by the FDA and meet other requirements of the statute. |
55
There may be significant delays in obtaining coverage for newly-approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or foreign regulatory authorities. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution or that covers a particular provider’s cost of acquiring the drug. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on payments allowed for lower-cost drugs that are already reimbursed, may be incorporated into existing payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for patisiran or other new drugs that we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products, and our overall financial condition.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. Specifically, there have been several recent U.S. Congressional inquiries and proposed federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers.
A number of other legislative and regulatory changes in the healthcare system in the United States and other major healthcare markets have been proposed in recent years, and such efforts have expanded substantially in recent years. These developments have included prescription drug benefit legislation that was enacted in 2003 and took effect in January 2006, healthcare reform legislation enacted by certain states, and major healthcare reform legislation that was passed by Congress and enacted into law in the United States in 2010. These developments could, directly or indirectly, affect our ability to sell our products, if approved, at a favorable price.
In particular, in March 2010, the PPACA was signed into law. This legislation changed the system of healthcare insurance and benefits intended to broaden coverage and control costs. The law also contains provisions that affect companies in the pharmaceutical industry and other healthcare related industries by imposing additional costs and changes to business practices. Provisions affecting pharmaceutical companies include the following:
|
|
• |
Mandatory rebates for drugs sold into the Medicaid program were increased, and the rebate requirement was extended to drugs used in risk-based Medicaid managed care plans. |
|
|
• |
The 340B Drug Pricing Program under the Public Health Service Act was extended to require mandatory discounts for drug products sold to certain critical access hospitals, cancer hospitals and other covered entities. |
|
|
• |
Pharmaceutical companies are required to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “donut hole.” |
|
|
• |
Pharmaceutical companies are required to pay an annual non-tax deductible fee to the federal government based on each company’s market share of prior year total sales of branded products to certain federal healthcare programs, such as Medicare, Medicaid, Department of Veterans Affairs and Department of Defense. Since we expect our branded pharmaceutical sales to constitute a small portion of the total federal healthcare program pharmaceutical market, we do not expect this annual assessment to have a material impact on our financial condition. |
|
|
• |
The law provides that approval of an application for a follow-on biologic product may not become effective until 12 years after the date on which the reference innovator biologic product was first licensed by the FDA, with a possible six-month extension for pediatric products. After this exclusivity ends, it will be easier for generic manufacturers to enter the market, which is likely to reduce the pricing for such products and could affect our profitability. |
|
|
• |
The law creates a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected. |
|
|
• |
The law expands eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133 percent of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability. |
56
|
|
• |
The law expands the entities eligible for discounts under the Public Health Service Act pharmaceutical pricing program. |
|
|
• |
The law establishes new requirements to report financial arrangements with physicians and teaching hospitals and to annually report drug samples that manufacturers and distributors provide to physicians. |
|
|
• |
The law establishes a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
The full effects of the U.S. healthcare reform legislation cannot be known until the law is fully implemented through regulations or guidance issued by the CMS and other federal and state healthcare agencies. The financial impact of the U.S. healthcare reform legislation over the next few years will depend on a number of factors, including, but not limited, to the policies reflected in implementing regulations and guidance, and changes in sales volumes for products affected by the new system of rebates, discounts and fees. This legislation may also have a positive impact on our future net sales, if any, by increasing the aggregate number of persons with healthcare coverage in the United States.
As a result of the 2016 election in the United States, there is great political uncertainty concerning the fate of the PPACA and other healthcare laws. Members of the United States Congress and the Trump Administration have expressed an intent to pass legislation or adopt executive orders to fundamentally change or repeal parts of the PPACA. While Congress has not passed repeal legislation to date, the TCJA includes a provision repealing the individual insurance coverage mandate included in PPACA, effective January 1, 2019. Further, on January 20, 2017, the President signed an Executive Order directing federal agencies with authorities and responsibilities under the PPACA to waive, defer, grant exemptions from, or delay the implementation of any provision of the PPACA that would impose a fiscal burden on states or a cost, fee, tax, penalty or regulatory burden on individuals, healthcare providers, health insurers, or manufacturers of pharmaceuticals or medical devices. On October 13, 2017, the President signed an Executive Order terminating the cost-sharing subsidies that reimburse insurers under the PPACA. Several state Attorneys General filed suit to stop the administration from terminating the subsidies, but their request for a restraining order was denied by a federal judge in California on October 25, 2017. In addition, CMS has recently proposed regulations that would give states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the PPACA for plans sold through such marketplaces. Congress may consider other legislation to replace elements of the PPACA. The implications of the PPACA, its possible repeal, any legislation that may be proposed to replace the PPACA, or the political uncertainty surrounding any repeal or replacement legislation for our business and financial condition, if any, are not yet clear.
We cannot predict what healthcare reform initiatives may be adopted in the future. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the United States to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop drug candidates.
Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending.
Under the Budget Control Act of 2011, the failure of Congress to enact deficit reduction measures of at least $1.2 trillion for the years 2013 through 2021 triggered automatic cuts to most federal programs. These cuts included aggregate reductions to Medicare payments to providers of up to 2 percent per fiscal year, starting in 2013. Certain of these automatic cuts have been implemented resulting in reductions in Medicare payments to physicians, hospitals, and other healthcare providers, among other things. The full impact on our business of these automatic cuts is uncertain.
If other federal spending is reduced, any budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or NIH to continue to function. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
There is a substantial risk of product liability claims in our business. If we are unable to obtain sufficient insurance, a product liability claim against us could adversely affect our business.
Our business exposes us to significant potential product liability risks that are inherent in the development, testing, manufacturing and marketing of human therapeutic products. Product liability claims could delay or prevent completion of our clinical development programs. Following the decision to discontinue clinical development of revusiran, we conducted a comprehensive evaluation of available revusiran data. We reported the results of this evaluation on August 9, 2017, however, our investigation did not result in a conclusive explanation regarding the cause of the mortality imbalance observed in the ENDEAVOUR Phase 3 study. In addition, in September 2017, we announced that we had temporarily suspended dosing in all ongoing fitusiran studies pending further
57
review of a fatal thrombotic SAE and agreement with regulatory authorities on a risk mitigation strategy. Notwithstanding the risks undertaken by all persons who participate in clinical trials, and the information on risks provided to study investigators and patients participating in revusiran and fitusiran studies, it is possible that product liability claims will be asserted against us relating to the worsening of a patient’s condition, injury or death alleged to have been caused by revusiran or fitusiran. Such claims might not be fully covered by product liability insurance. If we succeed in marketing products, including patisiran, product liability claims could result in an FDA investigation of the safety and effectiveness of our products, our manufacturing processes and facilities or our marketing programs, and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used, or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients and a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of development and may need to obtain higher levels prior to marketing any of our product candidates. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and manufacturing involve the use of hazardous materials, chemicals and various radioactive compounds. We maintain quantities of various flammable and toxic chemicals in our facilities in Cambridge that are required for our research, development and manufacturing activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these materials in our Cambridge facilities comply with the relevant guidelines of the City of Cambridge, the Commonwealth of Massachusetts and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Risks Related to Patents, Licenses and Trade Secrets
If we are not able to obtain and enforce patent protection for our discoveries, our ability to develop and commercialize our product candidates will be harmed.
Our success depends, in part, on our ability to protect proprietary methods and technologies that we develop under the patent and other intellectual property laws of the United States and other countries, so that we can prevent others from unlawfully using our inventions and proprietary information. However, we may not hold proprietary rights to some patents required for us to manufacture and commercialize our proposed products. Because certain U.S. patent applications are confidential until the patents issue, such as applications filed prior to November 29, 2000, or applications filed after such date which will not be filed in foreign countries, third parties may have filed patent applications for technology covered by our pending patent applications without our being aware of those applications, and our patent applications may not have priority over those applications. For this and other reasons, we may be unable to secure desired patent rights, thereby losing desired exclusivity. Further, we may be required to obtain licenses under third-party patents to market our proposed products or conduct our research and development or other activities. If licenses are not available to us on acceptable terms, we may not be able to market the affected products or conduct the desired activities.
Our strategy depends on our ability to rapidly identify and seek patent protection for our discoveries. In addition, we may rely on third-party collaborators to file patent applications relating to proprietary technology that we develop jointly during certain collaborations. The process of obtaining patent protection is expensive and time-consuming. If our present or future collaborators fail to file and prosecute all necessary and desirable patent applications at a reasonable cost and in a timely manner, our business may be adversely affected. Despite our efforts and the efforts of our collaborators to protect our proprietary rights, unauthorized parties may
58
be able to obtain and use information that we regard as proprietary. While issued patents are presumed valid, this does not guarantee that the patent will survive a validity challenge or be held enforceable. Any patents we have obtained, or obtain in the future, may be challenged, invalidated, adjudged unenforceable or circumvented by parties attempting to design around our intellectual property. Moreover, third parties or the USPTO may commence interference proceedings involving our patents or patent applications. Any challenge to, finding of unenforceability or invalidation or circumvention of, our patents or patent applications, would be costly, would require significant time and attention of our management, could reduce or eliminate royalty payments to us from third party licensors and could have a material adverse effect on our business.
Our pending patent applications may not result in issued patents. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the USPTO and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. Similarly, the ultimate degree of protection that will be afforded to biotechnology inventions, including ours, in the United States and foreign countries, remains uncertain and is dependent upon the scope of the protection decided upon by patent offices, courts and lawmakers. Moreover, there are periodic discussions in the Congress of the United States and in international jurisdictions about modifying various aspects of patent law. For example, the America Invents Act included a number of changes to the patent laws of the United States. If any of the enacted changes do not provide adequate protection for discoveries, including our ability to pursue infringers of our patents for substantial damages, our business could be adversely affected. One major provision of the America Invents Act, which took effect in March 2013, changed United States patent practice from a first-to-invent to a first-to-file system. If we fail to file an invention before a competitor files on the same invention, we no longer have the ability to provide proof that we were in possession of the invention prior to the competitor’s filing date, and thus would not be able to obtain patent protection for our invention. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents.
Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. We also rely to a certain extent on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business and financial condition could be materially adversely affected.
We license patent rights from third-party owners. If such owners do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, our competitive position and business prospects may be harmed.
We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained licenses from, among others, CRT, Ionis, MIT, Whitehead, Max Planck Innovation and Arbutus. We also intend to enter into additional licenses to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects. In addition, we sublicense our rights under various third-party licenses to our collaborators. Any impairment of these sublicensed rights could result in reduced revenues under our collaboration agreements or result in termination of an agreement by one or more of our collaborators.
Other companies or organizations may challenge our patent rights or may assert patent rights that prevent us from developing and commercializing our products.
RNAi is a relatively new scientific field, the commercial exploitation of which has resulted in many different patents and patent applications from organizations and individuals seeking to obtain patent protection in the field. We have obtained grants and issuances of RNAi patents and have licensed many of these patents from third parties on an exclusive basis. The issued patents and pending patent applications in the United States and in key markets around the world that we own or license claim many different methods, compositions and processes relating to the discovery, development, manufacture and commercialization of RNAi therapeutics.
Specifically, we have a portfolio of patents, patent applications and other intellectual property covering: fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms; chemical modifications to siRNAs that improve their suitability for therapeutic and other uses; siRNAs directed to specific targets as treatments for particular diseases; delivery technologies, such as in the fields of carbohydrate conjugates and cationic liposomes; and all aspects of our specific development candidates.
59
As the field of RNAi therapeutics is maturing, patent applications are being fully processed by national patent offices around the world. There is uncertainty about which patents will issue, and, if they do, as to when, to whom, and with what claims. It is likely that there will be significant litigation and other proceedings, such as interference, reexamination and opposition proceedings, as well as inter partes and post-grant review proceedings introduced by provisions of the America Invents Act, which became available to third party challengers on September 16, 2012, in various patent offices relating to patent rights in the RNAi field. For example, various third parties have initiated oppositions to patents in our McSwiggen, Kreutzer-Limmer and Tuschl II series in the EPO and in other jurisdictions. We expect that additional oppositions will be filed in the EPO and elsewhere, and other challenges will be raised relating to other patents and patent applications in our portfolio. In many cases, the possibility of appeal exists for either us or our opponents, and it may be years before final, unappealable rulings are made with respect to these patents in certain jurisdictions. The timing and outcome of these and other proceedings is uncertain and may adversely affect our business if we are not successful in defending the patentability and scope of our pending and issued patent claims. In addition, third parties may attempt to invalidate our intellectual property rights. Even if our rights are not directly challenged, disputes could lead to the weakening of our intellectual property rights. Our defense against any attempt by third parties to circumvent or invalidate our intellectual property rights could be costly to us, could require significant time and attention of our management and could have a material adverse effect on our business and our ability to successfully compete in the field of RNAi.
There are many issued and pending patents that claim aspects of oligonucleotide chemistry and modifications that we may need for our siRNA therapeutic candidates. There are also many issued patents that claim targeting genes or portions of genes that may be relevant for siRNA drugs we wish to develop. In addition, there may be issued and pending patent applications that may be asserted against us in a court proceeding or otherwise based upon the asserting party’s belief that we may need such patents for our siRNA therapeutic candidates. Thus, it is possible that one or more organizations will hold patent rights to which we may need a license, or hold patent rights which could be asserted against us. If those organizations refuse to grant us a license to such patent rights on reasonable terms and/or a court rules that we need such patent rights that have been asserted against us and we are not able to obtain a license on reasonable terms, we may be unable to market products or perform research and development or other activities covered by such patents. For example, Silence issued and has now served, a claim in the High Court of England and Wales, naming us, our wholly owned subsidiary Alnylam UK Ltd., and The Medicines Company UK Ltd as co-defendants. The claim seeks a declaration that Silence is entitled to supplementary protection certificates, or SPCs, based on a European patent held by Silence, that Silence alleges covers certain of our product candidates. An SPC is an intellectual property right that could extend the life of the Silence patent in relation to a specified product for a period of up to five additional years bringing the potential expiration date to 2028. On October 27, 2017, we, through our affiliate Alnylam UK Ltd., and The Medicines Company UK Ltd filed and served a claim against Silence Therapeutics GmbH and Silence in the High Court of England and Wales seeking revocation of Silence’s patent, as well as a declaration of non-infringement by each of the products of such patent, and costs and interest among other potential remedies. On November 30, 2017, we and The Medicines Company UK Ltd filed our defense to Silence’s claim against us denying that the products that are the subject of Silence’s claim against us fall under the Silence patent or that they are entitled to an SPC based on that patent. Both cases are expected to be tried in the High Court of England and Wales in December 2018. Although we believe Silence’s patent is invalid and not infringed by our product candidates and that, therefore, Silence would not be entitled to obtain an SPC based on any of our product candidates, litigation is subject to inherent uncertainty, and a court could ultimately rule against us.
If we become involved in patent litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, substantial liability for damages or be required to stop our product development and commercialization efforts.
Third parties may sue us for infringing their patent rights. Likewise, we may need to resort to litigation to enforce a patent issued or licensed to us or to determine the scope and validity of proprietary rights of others or protect our proprietary information and trade secrets. For example, during the second quarter of 2015, we filed a trade secret misappropriation lawsuit against Dicerna , to protect our rights in the RNAi assets we purchased from Merck. A third party may also claim that we have improperly obtained or used its confidential or proprietary information. For example, in March 2011, Arbutus (formerly Tekmira) filed a civil complaint against us alleging, among other things, misappropriation of its confidential and proprietary information and trade secrets. In November 2012, we settled this litigation and restructured our contractual relationship with Arbutus. In connection with this restructuring, we incurred a $65.0 million charge to operating expenses during the quarter ended December 31, 2012.
In protecting our intellectual patent rights through litigation or other means, a third party may claim that we have improperly asserted our rights against them. For example, in August 2017, Dicerna successfully added counterclaims against us in the above-referenced trade secret lawsuit alleging that our lawsuit represented abuse of process and claiming tortious interference with its business. In addition, in August 2017, Dicerna filed a lawsuit against us in the United States District Court of Massachusetts alleging
attempted monopolization by us under the Sherman Antitrust Act. Although we believe we have meritorious claims against Dicerna and meritorious defenses and responses to the counterclaims and federal claim being asserted by Dicerna, litigation is subject to inherent uncertainty, we will incur significant costs in defending against such claims and a court could rule against us awarding unspecified money damages.
60
Furthermore, third parties may challenge the inventorship of our patents or licensed patents. For example, in March 2011, The University of Utah, or Utah, filed a complaint against us, Max Planck Gesellschaft Zur Foerderung Der Wissenschaften e.V. and Max Planck Innovation, together, Max Planck, Whitehead, MIT and UMass, claiming that a professor of Utah was the sole inventor, or in the alternative, a joint inventor of certain of our in-licensed patents. Utah was seeking correction of inventorship of the Tuschl patents, unspecified damages and other relief. After several years of court proceedings and discovery, the court granted our motions for summary judgment, and dismissed Utah’s state law damages claims as well. During the pendency of this litigation, as well as the Arbutus litigation described above, we incurred significant costs, and in each case, the litigation diverted the attention of our management and other resources that would otherwise have been engaged in other activities.
In addition, in connection with certain license and collaboration agreements, we have agreed to indemnify certain third parties for certain costs incurred in connection with litigation relating to intellectual property rights or the subject matter of the agreements. The cost to us of any litigation or other proceeding relating to intellectual property rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s efforts. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of any litigation could delay our research and development efforts and limit our ability to continue our operations.
If any parties successfully claim that our creation or use of proprietary technologies infringes upon or otherwise violates their intellectual property rights, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. In addition to any damages we might have to pay, a court could require us to stop the infringing activity or obtain a license. Any license required under any patent may not be made available on commercially acceptable terms, if at all. In addition, such licenses are likely to be non-exclusive and, therefore, our competitors may have access to the same technology licensed to us. If we fail to obtain a required license and are unable to design around a patent, we may be unable to effectively market some of our technology and products, which could limit our ability to generate revenues or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. Moreover, we expect that a number of our collaborations will provide that royalties payable to us for licenses to our intellectual property may be offset by amounts paid by our collaborators to third parties who have competing or superior intellectual property positions in the relevant fields, which could result in significant reductions in our revenues from products developed through collaborations.
If we fail to comply with our obligations under any licenses or related agreements, we may be required to pay damages and could lose license or other rights that are necessary for developing and protecting our RNAi technology and any related product candidates that we develop, or we could lose certain rights to grant sublicenses.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license or render the license non-exclusive, which could result in us being unable to develop, manufacture, market and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology. For example, in 2013, Arbutus (formerly Tekmira) notified us that it believed it had achieved a $5.0 million milestone payment under our cross-license agreement relating to the manufacture of ALN-VSP clinical trial material for use in China. We notified Arbutus that we did not believe that the milestone has been achieved under the terms of the cross-license agreement. In August 2013, we initiated binding arbitration proceedings seeking a declaratory judgment that Arbutus had not yet met the conditions of the milestone and was not entitled to payment at the time. The Arbutus arbitration hearing was held in May 2015. On March 9, 2016, the arbitration panel ruled in our favor and as a result, no milestone payment is due to Arbutus at this time. Arbutus did not appeal this ruling.
Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we will be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize products, we may be unable to achieve or maintain profitability.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, employees, consultants, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of
61
unauthorized disclosure of confidential information. In addition, others may independently discover trade secrets and proprietary information, and in such cases we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Risks Related to Competition
The pharmaceutical market is intensely competitive. If we are unable to compete effectively with existing drugs, new treatment methods and new technologies, we may be unable to commercialize successfully any drugs that we develop.
The pharmaceutical market is intensely competitive and rapidly changing. Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations are pursuing the development of novel drugs for the same diseases that we are targeting or expect to target. Many of our competitors have:
|
|
• |
much greater financial, technical and human resources than we have at every stage of the discovery, development, manufacture and commercialization of products; |
|
|
• |
more extensive experience in pre-clinical testing, conducting clinical trials, obtaining regulatory approvals, and in manufacturing, marketing and selling drug products; |
|
|
• |
product candidates that are based on previously tested or accepted technologies; |
|
|
• |
products that have been approved or are in late stages of development; and |
|
|
• |
collaborative arrangements in our target markets with leading companies and research institutions. |
We will face intense competition from drugs that have already been approved and accepted by the medical community for the treatment of the conditions for which we may develop drugs. We also expect to face competition from new drugs that enter the market. We believe a number of drugs are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop drugs. These drugs may be more effective, safer, less expensive, or marketed and sold more effectively, than any products we develop. For example, we are developing patisiran for the treatment of hATTR amyloidosis. In November 2017, we reported positive complete results from our APOLLO Phase 3 clinical trial and generally encouraging safety data, and in December 2017, we submitted our first NDA and MAA for patisiran. In January 2018, we announced that the EMA has accepted the MAA and initiated its review. Patisiran was previously granted an accelerated assessment by the EMA. In early February 2018, we announced that the FDA has accepted our NDA and granted our request for priority review, with an action date of August 11, 2018. We are aware of other approved products used to treat this disease, including tafamidis, marketed by Pfizer in Europe and certain countries outside the United States, as well as product candidates in various stages of clinical development, including an investigational drug being developed by Ionis. In October and November 2017, Ionis reported positive efficacy data as well as safety data from its Phase 3 clinical trial in hATTR amyloidosis, including thrombocytopenia and renal insufficiency SAEs. In November 2017, Ionis reported that it has filed its NDA and MAA for this investigational drug and in January 2018, reported that the FDA had accepted its NDA for priority review and set a PDUFA date of July 6, 2018. While we believe that patisiran will have a competitive product profile, it is possible it will not compete favorably with these products and product candidates, or others, and even if approved, it may not achieve commercial success.
If we successfully develop product candidates, and obtain approval for them, we will face competition based on many different factors, including:
|
|
• |
the safety and effectiveness of our products relative to alternative therapies, if any; |
|
|
• |
the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration; |
|
|
• |
the timing and scope of regulatory approvals for these products; |
|
|
• |
the availability and cost of manufacturing, marketing and sales capabilities; |
|
|
• |
price; |
|
|
• |
reimbursement coverage; and |
|
|
• |
patent position. |
Our competitors may develop or commercialize products with significant advantages over any products we develop based on any of the factors listed above or on other factors. Our competitors may therefore be more successful in commercializing their products than we are, which could adversely affect our competitive position and business. Competitive products may make any
62
products we develop obsolete or noncompetitive before we can recover the expenses of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and the ability to execute on our business plan. Furthermore, we also face competition from existing and new treatment methods that reduce or eliminate the need for drugs, such as the use of advanced medical devices. The development of new medical devices or other treatment methods for the diseases we are targeting could make our product candidates noncompetitive, obsolete or uneconomical.
We face competition from other companies that are working to develop novel drugs and technology platforms using technology similar to ours. If these companies develop drugs more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to successfully commercialize drugs may be adversely affected.
In addition to the competition we face from competing drugs in general, we also face competition from other companies working to develop novel drugs using technology that competes more directly with our own. We are aware of several other companies that are working to develop RNAi therapeutic products. Some of these companies are seeking, as we are, to develop chemically synthesized siRNAs as drugs. Others are following a gene therapy approach, with the goal of treating patients not with synthetic siRNAs but with synthetic, exogenously-introduced genes designed to produce siRNA-like molecules within cells. Companies working on chemically synthesized siRNAs include Takeda, Marina, Arrowhead, and its subsidiary, Calando, Quark, Silence, Arbutus, Sylentis, Dicerna, WAVE and Arcturus. In addition, we granted licenses or options for licenses to Ionis, Benitec, Arrowhead, and its subsidiary, Calando, Arbutus, Quark, Sylentis and others under which these companies may independently develop RNAi therapeutics against a limited number of targets. Any one of these companies may develop its RNAi technology more rapidly and more effectively than us.
In addition, as a result of agreements that we have entered into, Arrowhead, as the assignee of Roche, and Takeda have obtained non-exclusive licenses, and Arrowhead, as the assignee of Novartis Pharma AG, has obtained specific exclusive licenses for 30 gene targets, that include access to certain aspects of our technology that give them the right to compete with us in certain circumstances. We also compete with companies working to develop antisense-based drugs. Like RNAi therapeutics, antisense drugs target mRNAs in order to suppress the activity of specific genes. Ionis is currently marketing several antisense drugs and has multiple antisense product candidates in clinical trials, including one for the treatment of hATTR amyloidosis that is currently under regulatory review in the United States and the EU. Ionis is also developing antisense drugs using ligand-conjugated GalNAc technology licensed from us, and these drugs have been shown to have increased potency at lower doses in clinical and pre-clinical studies, compared with antisense drugs that do not use such licensed GalNAc technology. The development of antisense drugs is more advanced than that of RNAi therapeutics, and antisense technology may become the preferred technology for drugs that target mRNAs to silence specific genes.
In addition to competition with respect to RNAi and with respect to specific products, we face substantial competition to discover and develop safe and effective means to deliver siRNAs to the relevant cell and tissue types. Safe and effective means to deliver siRNAs to the relevant cell and tissue types may be developed by our competitors, and our ability to successfully commercialize a competitive product would be adversely affected. In addition, substantial resources are being expended by third parties in the effort to discover and develop a safe and effective means of delivering siRNAs into the relevant cell and tissue types, both in academic laboratories and in the corporate sector. Some of our competitors have substantially greater resources than we do, and if our competitors are able to negotiate exclusive access to those delivery solutions developed by third parties, we may be unable to successfully commercialize our product candidates.
Risks Related to Our Common Stock
If our stock price fluctuates, purchasers of our common stock could incur substantial losses.
The market price of our common stock has fluctuated significantly and may continue to fluctuate significantly in response to factors that are beyond our control. The stock market in general has from time to time experienced extreme price and volume fluctuations, and the biotechnology in particular has experienced extreme price and volume fluctuations. The market prices of securities of pharmaceutical and biotechnology companies have been extremely volatile, and have experienced fluctuations that often have been unrelated or disproportionate to the clinical development progress or operating performance of these companies, including as a result of adverse development events. These broad market and sector fluctuations have resulted and could in the future result in extreme fluctuations in the price of our common stock, which could cause purchasers of our common stock to incur substantial losses.
We may incur significant costs from class action litigation due to stock volatility.
Our stock price may fluctuate for many reasons, including as a result of public announcements regarding the progress of our development and commercialization efforts or the development and commercialization efforts of our collaborators and/or competitors, the addition or departure of our key personnel, variations in our quarterly operating results and changes in market valuations of
63
pharmaceutical and biotechnology companies. For example, in October 2016, we announced that we were discontinuing the development of revusiran and our stock price declined significantly as a result and in September 2017, following our temporary suspension of dosing in our fitusiran program, our stock also declined, although to a lesser extent. When the market price of a stock has been volatile as our stock price has been, holders of that stock have occasionally brought securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit is without merit, we could incur substantial costs defending the lawsuit. The lawsuit could also divert the time and attention of our management.
Sales of additional shares of our common stock, including by us or our directors and officers, could cause the price of our common stock to decline.
Sales of substantial amounts of our common stock in the public market, or the availability of such shares for sale, by us or others, including the issuance of common stock upon exercise of outstanding options, could adversely affect the price of our common stock.
Sanofi Genzyme’s ownership of our common stock could delay or prevent a change in corporate control.
Sanofi Genzyme currently holds approximately 11 percent of our outstanding common stock and has the right to increase its ownership up to 30 percent, as well as the right to maintain its then current ownership percentage through the term of our collaboration, subject to certain limitations. This concentration of ownership may harm the market price of our common stock by:
|
|
• |
delaying, deferring or preventing a change in control of our company; |
|
|
• |
impeding a merger, consolidation, takeover or other business combination involving our company; or |
|
|
• |
discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our company. |
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and our bylaws may delay or prevent an acquisition of us or a change in our management. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:
|
|
• |
a classified board of directors; |
|
|
• |
a prohibition on actions by our stockholders by written consent; |
|
|
• |
limitations on the removal of directors; and |
|
|
• |
advance notice requirements for election to our board of directors and for proposing matters that can be acted upon at stockholder meetings. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15 percent of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15 percent of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. These provisions would apply even if the proposed merger or acquisition could be considered beneficial by some stockholders.
Not applicable.
64
Our operations are based primarily in Cambridge, Massachusetts; Zug, Switzerland; and Maidenhead, United Kingdom. A description of certain of the facilities we lease as of January 31, 2018 is included in the table below.
|
Location |
|
Primary Use |
|
Approximate |
|
|
Lease |
|
Renewal Option |
|
300 Third Street Cambridge, Massachusetts |
|
Corporate headquarters and primary research facility |
|
129,000 |
|
|
September 2021 |
|
One five-year term |
|
101 Main Street Cambridge, Massachusetts |
|
Additional office space |
|
72,000 |
|
|
March 2019 and June 2021 |
|
One five-year term on each lease |
|
675 West Kendall Street Cambridge, Massachusetts |
|
Future corporate headquarters and research facility* |
|
295,000 |
|
|
On or around February 2034 |
|
Two five-year terms |
|
665 Concord Avenue Cambridge, Massachusetts |
|
cGMP manufacturing |
|
15,000 |
|
|
August 2022 |
|
One five-year term |
|
Grafenauweg 4 6300 Zug |
|
International headquarters |
|
14,500 |
|
|
March 2023 |
|
One five-year term |
|
Braywick Gate Braywick Road, Maidenhead Berkshire, United Kingdom |
|
Office space |
|
21,500 |
|
|
May 2026 |
|
None |
|
* |
We intend to move our corporate headquarters and research facility to this location in early 2019. The term will commence on May 1, 2018 and rent payments will become due commencing upon substantial completion of the building improvements, which is currently expected to be on or around February 2019, and will continue for 15 years from the rent commencement date. |
In addition to the locations above, we also maintain small offices in multiple locations in and outside of the United States to support our operations and growth.
In April 2016, we completed the purchase of 12 acres of undeveloped land in Norton, Massachusetts. We have commenced construction of a manufacturing facility at this site for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use.
In the future, we may lease, operate, purchase or construct additional facilities in which to conduct expanded research, development and manufacturing activities and support future commercial operations. We believe that the total space available to us under our current leases will meet our needs for the foreseeable future and that additional space would be available to us on commercially reasonable terms if required.
For a discussion of material pending legal proceedings, please read Note 7, Commitments and Contingencies – Litigation, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K, which is incorporated into this item by reference.
Not applicable.
65
|
ITEM 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock trades on The NASDAQ Global Select Market under the symbol “ALNY.” The following table sets forth the high and low sale prices per share for our common stock on The NASDAQ Global Select Market for the periods indicated:
|
Year Ended December 31, 2016: |
|
High |
|
|
Low |
|
||
|
First Quarter |
|
$ |
98.00 |
|
|
$ |
51.51 |
|
|
Second Quarter |
|
$ |
75.08 |
|
|
$ |
49.96 |
|
|
Third Quarter |
|
$ |
80.11 |
|
|
$ |
53.56 |
|
|
Fourth Quarter |
|
$ |
71.67 |
|
|
$ |
31.38 |
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended December 31, 2017: |
|
High |
|
|
Low |
|
||
|
First Quarter |
|
$ |
60.41 |
|
|
$ |
35.98 |
|
|
Second Quarter |
|
$ |
86.92 |
|
|
$ |
46.90 |
|
|
Third Quarter |
|
$ |
119.02 |
|
|
$ |
70.76 |
|
|
Fourth Quarter |
|
$ |
147.63 |
|
|
$ |
111.25 |
|
Holders of record
At January 31, 2018, there were 33 holders of record of our common stock. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of beneficial holders represented by these record holders.
Dividends
We have never paid or declared any cash dividends on our common stock. We currently intend to retain any earnings for future growth and, therefore, do not expect to pay cash dividends in the foreseeable future.
Securities Authorized for Issuance Under Equity Compensation Plans
We intend to file with the SEC a definitive Proxy Statement, which we refer to herein as the Proxy Statement, not later than 120 days after the close of the fiscal year ended December 31, 2017. The information required by this item relating to our equity compensation plans is incorporated herein by reference to the information contained under the section captioned “Equity Compensation Plan Information” of the Proxy Statement.
66
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
The comparative stock performance graph below compares the five-year cumulative total stockholder return (assuming reinvestment of dividends, if any) from investing $100 on December 31, 2012, to the close of the last trading day of 2017, in each of our common stock and selected indices. We changed the comparison, for the years presented, from the NASDAQ US Benchmark TR Index and the NQ US Benchmark Pharma TR Index, to the NASDAQ Composite Total Return Index and NASDAQ Biotechnology Total Return, respectively, because we believe these indices reflect a better comparison of our performance relative to the broader market and our peers. In this transition year, we have retained the previous indices for comparison but will not include them in our stock performance graph in subsequent annual filings. The stock price performance reflected in the graph below is not necessarily indicative of future price performance.
Comparison of Five-Year Cumulative Total Return
Among Alnylam Pharmaceuticals, Inc.,
NASDAQ US Benchmark TR Index, NQ US Benchmark Pharma TR Index,
NASDAQ Composite Total Return and NASDAQ Biotechnology Total Return
|
|
12/31/2012 |
|
|
12/31/2013 |
|
|
12/31/2014 |
|
|
12/31/2015 |
|
|
12/30/2016 |
|
|
12/29/2017 |
|
||||||
|
Alnylam Pharmaceuticals, Inc. |
$ |
100.00 |
|
|
$ |
352.33 |
|
|
$ |
531.51 |
|
|
$ |
515.84 |
|
|
$ |
205.15 |
|
|
$ |
696.16 |
|
|
NASDAQ US Benchmark TR Index |
$ |
100.00 |
|
|
$ |
133.48 |
|
|
$ |
150.12 |
|
|
$ |
150.84 |
|
|
$ |
170.46 |
|
|
$ |
206.91 |
|
|
NQ US Benchmark Pharma TR Index |
$ |
100.00 |
|
|
$ |
135.68 |
|
|
$ |
165.28 |
|
|
$ |
174.27 |
|
|
$ |
172.37 |
|
|
$ |
205.33 |
|
|
NASDAQ Composite Total Return |
$ |
100.00 |
|
|
$ |
140.12 |
|
|
$ |
160.78 |
|
|
$ |
171.97 |
|
|
$ |
187.22 |
|
|
$ |
242.71 |
|
|
NASDAQ Biotechnology Total Return |
$ |
100.00 |
|
|
$ |
165.97 |
|
|
$ |
223.07 |
|
|
$ |
249.32 |
|
|
$ |
196.09 |
|
|
$ |
238.51 |
|
67
The following selected consolidated financial data for each of the five years in the period ended December 31, 2017 are derived from our audited consolidated financial statements. The selected consolidated financial data set forth below should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the financial statements, and the related Notes, included elsewhere in this annual report on Form 10-K. Historical results are not necessarily indicative of future results.
Selected Consolidated Financial Data
(In thousands, except per share data)
|
|
|
Year Ended December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Statements of Comprehensive Loss Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net revenues from collaborators |
|
$ |
89,912 |
|
|
$ |
47,159 |
|
|
$ |
41,097 |
|
|
$ |
50,561 |
|
|
$ |
47,167 |
|
|
Operating expenses(1) (2) |
|
|
590,000 |
|
|
|
471,746 |
|
|
|
337,105 |
|
|
|
455,541 |
|
|
|
140,109 |
|
|
Loss from operations |
|
|
(500,088 |
) |
|
|
(424,587 |
) |
|
|
(296,008 |
) |
|
|
(404,980 |
) |
|
|
(92,942 |
) |
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
$ |
(360,395 |
) |
|
$ |
(89,225 |
) |
|
Net loss per common share — basic and diluted |
|
$ |
(5.42 |
) |
|
$ |
(4.79 |
) |
|
$ |
(3.45 |
) |
|
$ |
(4.85 |
) |
|
$ |
(1.45 |
) |
|
Weighted-average common shares outstanding — basic and diluted |
|
|
90,554 |
|
|
|
85,596 |
|
|
|
83,992 |
|
|
|
74,278 |
|
|
|
61,551 |
|
|
__________ (1) Stock-based compensation expenses included in operating expenses |
|
$ |
92,819 |
|
|
$ |
75,528 |
|
|
$ |
45,783 |
|
|
$ |
33,061 |
|
|
$ |
20,703 |
|
(2) Operating expenses for the year ended December 31, 2014 included a $220.8 million charge to in process research and development expenses in connection with our acquisition of the Sirna RNAi assets from Merck.
|
|
|
December 31, |
|
|||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2014 |
|
|
2013 |
|
|||||
|
Balance Sheet Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and fixed income marketable securities |
|
$ |
1,704,537 |
|
|
$ |
942,601 |
|
|
$ |
1,280,951 |
|
|
$ |
881,929 |
|
|
$ |
350,472 |
|
|
Restricted investments |
|
|
30,000 |
|
|
|
150,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Working capital |
|
|
1,620,489 |
|
|
|
540,178 |
|
|
|
1,043,289 |
|
|
|
651,033 |
|
|
|
200,164 |
|
|
Total assets |
|
|
1,994,730 |
|
|
|
1,262,810 |
|
|
|
1,386,510 |
|
|
|
1,079,595 |
|
|
|
420,530 |
|
|
Long-term debt |
|
|
30,000 |
|
|
|
150,000 |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Total stockholders’ equity |
|
|
1,766,431 |
|
|
|
920,221 |
|
|
|
1,264,714 |
|
|
|
936,267 |
|
|
|
270,347 |
|
68
Overview
We are a global biopharmaceutical company developing novel therapeutics based on RNAi. RNAi is a naturally occurring biological pathway within cells for sequence-specific silencing and regulation of gene expression. We are harnessing the RNAi pathway to develop a potential new class of innovative medicines, known as RNAi therapeutics. RNAi therapeutics are comprised of siRNA, and function upstream of today’s medicines by potently silencing mRNA that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
Our research and development strategy is to target genetically validated liver-expressed genes that have been implicated in the cause or pathway of human disease. We utilize a LNP or GalNAc conjugate approach to enable hepatic delivery of siRNAs. Our focus is on clinical indications where there is a high unmet need, early biomarkers for the assessment of clinical activity in Phase 1 clinical studies, and a definable path for drug development, regulatory approval, patient access and commercialization.
Specifically, our broad pipeline of investigational RNAi therapeutics is focused in three STArs: Genetic Medicines; Cardio-Metabolic Diseases; and Hepatic Infectious Diseases. We are committed to the advancement of our Alnylam 2020 strategy, which is to achieve a company profile with three marketed products and ten RNAi therapeutic clinical programs, including four in late stages of development, across our three STArs by the end of 2020. In December 2017, we filed our first NDA and MAA for patisiran. In January 2018, we announced that the EMA has accepted the MAA and initiated its review. Patisiran was previously granted an accelerated assessment by the EMA. In early February 2018, we announced that the FDA has accepted our NDA and granted our request for priority review, with an action date of August 11, 2018. If approved, we expect to launch patisiran and begin generating product revenues in 2018.
In May 2017, we sold an aggregate of 5,000,000 shares of our common stock through an underwritten public offering at a price to the public of $71.87 per share. As a result of the offering, we received aggregate net proceeds of $355.2 million, after deducting underwriting discounts and commissions and other offering expenses of $4.2 million. In November 2017, we sold an aggregate of 6,440,000 shares of our common stock through an underwritten public offering at a price to the public of $125.00 per share. As a result of the offering, which included the full exercise of the underwriters’ option to purchase additional shares, we received aggregate net proceeds of $784.5 million, after deducting underwriting discounts and commissions and other offering expenses of $20.5 million. We have used and intend to continue to use these proceeds for general corporate purposes, focused on achieving our Alnylam 2020 strategy.
In addition, in April 2016, our subsidiary, Alnylam U.S., Inc., entered into an aggregate of $150.0 million of term loan agreements with Bank of America N.A., or BOA, and Wells Fargo Bank, National Association, or Wells, related to the build out of our new drug substance manufacturing facility. In December 2017, we repaid in full $120.0 million outstanding under the BOA term loan agreement. Please read Note 7 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for a description of these term loan agreements.
We have incurred significant losses since we commenced operations in 2002 and expect such losses to continue for the foreseeable future. At December 31, 2017, we had an accumulated deficit of $2.15 billion. Historically, we have generated losses principally from costs associated with research and development activities, acquiring, filing and expanding intellectual property rights and general administrative costs. As a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the establishment of late stage clinical and commercial capabilities, including global operations, continued management and growth of our patent portfolio, collaborations and general corporate activities, we expect to incur additional operating losses for the foreseeable future. We also anticipate that our operating results will fluctuate for the foreseeable future. Therefore, period-to-period comparisons should not be relied upon as predictive of the results in future periods.
We currently have programs focused on a number of therapeutic areas and, in December 2017, submitted our first NDA and MAA for marketing approval for patisiran. However, our development efforts may not be successful and we may not be able to commence sales of patisiran or any other product. If we gain approval for and successfully launch patisiran in 2018, we may begin to generate net revenues from product sales. A substantial portion of our total revenues in recent years has been derived from collaboration revenues from strategic alliances with Sanofi Genzyme and MDCO. In addition to potential revenues from the commercial sale of patisiran and future product candidates, we expect our sources of potential funding for the next several years to be derived primarily from existing and new strategic alliances, which may include license and other fees, funded research and development, milestone payments and royalties on product sales by our licensors, and proceeds from the sale of equity or debt.
69
Since our inception, we have focused on drug discovery and development programs. Research and development expenses represent a substantial percentage of our total operating expenses, as reflected by our broad pipeline of clinical development programs, which includes several programs in late-stage development and one product in registration in the United States and the EU.
There is a risk that any drug discovery or development program may not produce revenue for a variety of reasons, including the possibility that we will not be able to adequately demonstrate the safety and effectiveness of the product candidate. For example, in October 2016, we announced the discontinuation of our revusiran clinical development program due to safety concerns and in September 2017, we announced that we had temporarily suspended dosing in all ongoing fitusiran studies. Moreover, there are uncertainties specific to any new field of drug discovery, including RNAi. The success of any product candidate we develop is highly uncertain. Due to the numerous risks associated with developing drugs, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts necessary to complete the development of, or the period, if any, in which material net cash inflows will commence from, any potential product candidate.
Any failure to complete any stage of the development of any potential products in a timely manner could have a material adverse effect on our operations, financial position and liquidity. A discussion of some of the risks and uncertainties associated with completing our projects on schedule, or at all, and the potential consequences of failing to do so, are set forth in Part I, Item 1A of this annual report on Form 10-K under the heading “Risk Factors.”
Strategic Alliances
A significant component of our business plan is to enter into strategic alliances and collaborations with leading pharmaceutical and life sciences companies, academic institutions, research foundations and others, as appropriate, to gain access to funding, capabilities, technical resources and intellectual property to further our research, development and commercialization efforts and to generate revenues. We may also seek to form or advance new ventures and opportunities in areas outside our primary focus on RNAi therapeutics.
Our collaboration strategy is to form alliances that create significant value for ourselves and our collaborators in the advancement of RNAi therapeutics as a potential new class of innovative medicines. Specifically, with respect to our Genetic Medicine pipeline, we formed a broad strategic alliance with Sanofi Genzyme in 2014 pursuant to which we retain development and commercial rights for our current and future Genetic Medicine products in the United States, Canada and Western Europe, and Sanofi Genzyme will develop and commercialize our current and future Genetic Medicine products for which it elects to opt-in, in the rest of the world, subject to certain broader rights. In January 2018, we and Sanofi Genzyme amended our 2014 Sanofi Genzyme collaboration to provide that we would develop and commercialize patisiran globally and Sanofi Genzyme would develop and commercialize fitusiran globally. With respect to our Cardio-Metabolic pipeline, we intend to seek future strategic alliances for these programs, under which we may retain certain product development and commercialization rights, or we may structure as global alliances, as we did in our collaboration with MDCO to advance inclisiran. With respect to our Hepatic Infectious Disease pipeline, in October 2017, we announced an exclusive licensing agreement with Vir Biotechnology for the development and commercialization of RNAi therapeutics for infectious diseases, including chronic hepatitis B virus infection.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses and disclosure of contingent assets and liabilities in our consolidated financial statements. Actual results may differ from these estimates under different assumptions or conditions and could have a material impact on our reported results. While our significant accounting policies are more fully described in the Notes to our consolidated financial statements included elsewhere in this annual report on Form 10-K, we believe the following accounting policies to be the most critical in understanding the judgments and estimates we use in preparing our consolidated financial statements:
Our business strategy includes entering into collaborative license and development agreements with leading pharmaceutical and life sciences companies for the development and commercialization of our product candidates. For example, we have entered into collaboration agreements with Takeda, Kyowa Hakko Kirin Co., Ltd., or Kyowa Hakko Kirin, Monsanto, Sanofi Genzyme and MDCO. The terms of the agreements typically include deliverables such as non-refundable license fees, funding of research and development, payments based upon achievement of clinical and pre-clinical development milestones, regulatory milestones, manufacturing services, sales milestones and royalties on product sales. These agreements are generally referred to as multiple element arrangements.
70
We apply the accounting standard on revenue recognition for multiple element arrangements. The fair value of deliverables under the arrangement may be derived using a “best estimate of selling price” if vendor specific objective evidence and third-party evidence is not available. Deliverables under the arrangement will be separate units of accounting provided that (i) a delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the control of the vendor.
We recognize upfront license payments as revenue upon delivery of the license only if the license has standalone value of the undelivered performance obligations, typically including research and/or steering committee services, that can be determined. If the fair value of the undelivered performance obligations can be determined, such obligations would then be accounted for separately as performed. If the license is considered to not have standalone value, the arrangement would then be accounted for as a single unit of accounting and the license payments and payments for performance obligations are recognized as revenue over the estimated period of when the performance obligations are performed or deferred until the undelivered performance obligation can be determined. As a biotechnology entity with unique and specialized delivered and undelivered performance obligations, we have been unable to demonstrate standalone value in our multiple element arrangements.
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we must determine the period over which the performance obligations will be performed and revenue will be recognized. We recognize revenue using either proportional performance or a straight-line method. We recognize revenue using the proportional performance method when we can reasonably estimate the level of effort required to complete our performance obligations under an arrangement and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents are typically used as the measure of performance. The amount of revenue recognized under the proportional performance method is determined by multiplying the total payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of level of effort incurred to date to estimated total level of effort required to complete our performance obligations under the arrangement. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the proportional performance method, as of the period ending date.
If we cannot reasonably estimate the level of effort required to complete our performance obligations under an arrangement, we recognize revenue under the arrangement on a straight-line basis over the period we expect to complete our performance obligations. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line method, as of the period ending date.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement. Steering committee services that are not inconsequential or perfunctory and that are determined to be performance obligations are combined with other research services or performance obligations required under an arrangement, if any, in determining the level of effort required in an arrangement and the period over which we expect to complete our aggregate performance obligations.
Many of our collaboration agreements entitle us to additional payments upon the achievement of performance-based milestones. These milestones are generally categorized into three types; development milestones which are generally based on the advancement of our pipeline and initiation of clinical trials, regulatory milestones which are generally based on the submission, filing or approval of regulatory applications such as an NDA in the United States, and commercialization milestones which are generally based on meeting specific thresholds of sales in certain geographic areas. If the achievement of a milestone is considered probable at the inception of the collaboration, the related milestone payment is included with other collaboration consideration, such as upfront fees and research funding, in our revenue model. Milestones that are tied to regulatory approval are not considered probable of being achieved until such approval is received. Milestones tied to counter-party performance are not included in our revenue model until the performance conditions are met. Upfront and ongoing development milestones per our collaboration agreements are not subject to refund if the development activities are not successful.
We perform an assessment to determine whether a substantive milestone exists at the inception of our collaborative arrangements. In evaluating if a milestone is substantive, we consider whether uncertainty exists as to the achievement of the milestone event at the inception of the arrangement, the achievement of the milestone involves substantive effort and can only be achieved based in whole or part on the performance or the occurrence of a specific outcome resulting from our performance, the amount of the milestone payment appears reasonable either in relation to the effort expected to be expended or to the projected enhancement of the value of the delivered items, there is any future performance required to earn the milestone, and the consideration is reasonable relative to all deliverables and payment terms in the arrangement. When a substantive milestone is achieved, the accounting rules permit us to recognize revenue related to the milestone payment in its entirety.
71
To date, we have not recorded any substantive milestones under our collaborations because we have not identified any milestones that meet the required criteria listed above. We have deferred recognition of payments for achievement of non-substantive milestones and recognized revenue over the estimated period of performance applicable with each collaborative arrangement. As these milestones are achieved, we will recognize as revenue a portion of the milestone payment, which is equal to the percentage of the performance period completed, when the milestone is achieved, multiplied by the amount of the milestone payment, upon achievement of such milestone. We will recognize the remaining portion of the milestone payment over the remaining performance period under the proportional performance method or on a straight-line basis.
For revenue generating arrangements where we, as a vendor, provide consideration to a licensor or collaborator, as a customer, we apply the accounting standard that governs such transactions. This standard addresses the accounting for revenue arrangements where both the vendor and the customer make cash payments to each other for services and/or products. A payment to a customer is presumed to be a reduction of the selling price unless we receive an identifiable benefit for the payment and we can reasonably estimate the fair value of the benefit received. Payments to a customer that are deemed a reduction of selling price are recorded first as a reduction of revenue, to the extent of both cumulative revenue recorded to date and probable future revenues, which include any unamortized deferred revenue balances, under all arrangements with such customer, and then as an expense. Payments that are not deemed to be a reduction of selling price are recorded as an expense.
We evaluate our collaborative agreements for proper classification in our consolidated statements of comprehensive loss based on the nature of the underlying activity. Transactions between collaborators recorded in our consolidated statements of comprehensive loss are recorded on either a gross or net basis, depending on the characteristics of the collaborative relationship. We generally reflect amounts due under our collaborative agreements related to cost-sharing of development activities as revenue.
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets. Although we follow detailed guidelines in measuring revenue, certain judgments affect the application of our revenue policy. For example, in connection with our existing collaboration agreements, we have recorded on our consolidated balance sheet short-term and long-term deferred revenue based on our best estimate of when such revenue will be recognized. Short-term deferred revenue consists of amounts that are expected to be recognized as revenue in the next 12 months. Amounts that we expect will not be recognized prior to the next 12 months are classified as long-term deferred revenue. However, this estimate is based on our current operating plan and, if our operating plan should change in the future, we may recognize a different amount of deferred revenue over the next 12-month period.
The estimate of deferred revenue also reflects management’s estimate of the periods of our involvement in certain of our collaborations. Our performance obligations under these collaborations consist of participation on steering committees and the performance of other research and development services. In certain instances, the timing of satisfying these obligations can be difficult to estimate. Accordingly, our estimates may change in the future. Such changes to estimates would result in a change in revenue recognition amounts. If these estimates and judgments change over the course of these agreements, it may affect the timing and amount of revenue that we recognize and record in future periods. For example, in connection with our collaboration with Sanofi Genzyme, our estimate of the period to satisfy our performance obligations changed from approximately six years to approximately five years, effective October 2016. This change in our estimate was due to the discontinuation of our revusiran clinical development program. Beginning in the fourth quarter of 2016, due to the adjustment to the performance period made on a prospective basis in October 2016, we began to recognize an additional $1.8 million of revenue per quarter related to the consideration earned in connection with these programs related to the license to our patisiran and revusiran clinical programs. At December 31, 2017, we had short-term and long-term deferred revenue of $41.7 million and $43.1 million, respectively, related to our collaborations.
The new accounting standard related to revenue recognition effective as of January 1, 2018 will have a material impact on our consolidated financial statements. Please read Note 2 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for our discussion of recent accounting pronouncements for the expected impact of this new accounting standard.
Sanofi Genzyme. In January 2014, we entered into a global, strategic collaboration with Sanofi Genzyme to discover, develop and commercialize RNAi therapeutics as Genetic Medicines to treat orphan diseases, referred to as the 2014 Sanofi Genzyme collaboration. It superseded and replaced the previous collaboration between us and Sanofi Genzyme entered into in October 2012, referred to as the 2012 Sanofi Genzyme agreement, to develop and commercialize RNAi therapeutics targeting TTR for the treatment of hATTR amyloidosis, including patisiran and revusiran, in Japan and the Asia-Pacific region. In January 2018, we and Sanofi Genzyme amended the 2014 Sanofi Genzyme collaboration and entered into the Exclusive TTR License and the AT3 License Terms. The January 2018 transaction is subject to customary closing conditions and clearances, including clearance under the Hart-Scott-Rodino Antitrust Improvements Act. We expect the transaction to close during the first quarter of 2018.
72
Sanofi Genzyme paid us an upfront cash payment of $22.5 million under the 2012 Sanofi Genzyme agreement. We were also entitled to receive certain milestone payments under the 2012 Sanofi Genzyme agreement. In the fourth quarter of 2013, we earned two development milestones totaling $11.0 million. We determined that the deliverables under the 2012 Sanofi Genzyme agreement included the license, a joint steering committee and any additional TTR-specific RNAi therapeutic compounds that comprised the ALN-TTR program. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered joint steering committee and any additional TTR-specific RNAi therapeutic compounds did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme had the ability to grant sublicenses, it could not sublicense all or substantially all of its rights under the 2012 Sanofi Genzyme agreement. The uniqueness of our services and the limited sublicense right were indicators that standalone value was not present in the arrangement. Therefore the deliverables were not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. We were unable to reasonably estimate the period of performance under the 2012 Sanofi Genzyme agreement, as we were unable to estimate the timeline of our deliverables related to the deliverable for any additional TTR-specific RNAi therapeutic compounds. Through December 31, 2013, we had deferred all revenue, or $33.5 million, under the 2012 Sanofi Genzyme agreement.
In January 2014, we entered into the 2014 Sanofi Genzyme collaboration. As noted above, the 2014 Sanofi Genzyme collaboration superseded and replaced the 2012 Sanofi Genzyme agreement and was amended in January 2018, at which time we also entered into the Exclusive TTR License and the AT3 License Terms. Under the 2014 Sanofi Genzyme collaboration, we retain full product rights in the Alnylam Territory, while Sanofi Genzyme will obtain exclusive rights to develop and commercialize collaboration products in the Sanofi Genzyme Territory, together with worldwide rights for one product. Upon the effective date of the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme expanded the scope of its regional license and collaboration for patisiran for the Sanofi Genzyme Territory. We and Sanofi Genzyme also expanded our existing collaboration on revusiran, to include a co-development/co-commercialize license and collaboration in the Alnylam Territory. In October 2016, we discontinued our revusiran clinical development program. In September 2015, Sanofi Genzyme elected to opt into our fitusiran clinical development program under the regional license terms and began funding the program under the regional license terms in January 2016. In November 2016, Sanofi Genzyme exercised its right to co-develop and co-commercialize fitusiran in the Alnylam Territory, while retaining its rights to exclusively develop and commercialize the product in the Sanofi Genzyme Territory. Sanofi Genzyme shared in fifty percent of the global development costs for fitusiran in accordance with the co-development/co-commercialize license terms. In connection with the exercise of this right, Sanofi Genzyme paid us approximately $6.0 million in January 2017 for its incremental share of co-development costs incurred from January 2016 through September 2016.
Sanofi Genzyme’s rights with respect to patisiran and fitusiran will be modified in connection with the 2018 amendment, the Exclusive TTR License and the AT3 License Terms. Sanofi Genzyme continues to have the right to opt into our future rare genetic disease programs for development and commercialization in the Sanofi Genzyme Territory as contemplated in the 2014 Sanofi Genzyme collaboration, as well as one right to a global license. In connection with the 2018 amendment, the Exclusive TTR License and the AT3 License Terms, we and Sanofi Genzyme agreed to terminate the co-development and co-commercialization rights related to revusiran, ALN-TTRsc02 and fitusiran under the original 2014 Sanofi Genzyme collaboration. No future rights will be granted to Sanofi Genzyme for co-development and co-commercialization under the 2014 Sanofi Genzyme collaboration, as amended. Please read Note 3 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for our discussion of the terms of the Exclusive TTR License and the AT3 License Terms.
In connection with the 2014 Sanofi Genzyme collaboration, we sold to Sanofi Genzyme 8,766,338 shares of our common stock and Sanofi Genzyme paid us $700.0 million in aggregate cash consideration. Based on the common stock price of $85.72, the fair value of the shares issued was $751.5 million, which was $51.5 million in excess of the proceeds received from Sanofi Genzyme for the issuance of our common stock. This $51.5 million is being amortized on a straight-line basis over the performance period for the ALN-TTR programs.
Sanofi Genzyme will be required to make payments totaling up to $75.0 million per regional product, consisting of up to $55.0 million in development milestones and $20.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for each regional product based on annual net sales, if any, of such regional product by Sanofi Genzyme, its affiliates and sublicensees. In consideration for the rights granted to Sanofi Genzyme under the co-development/co-commercialize license terms, Sanofi Genzyme was required to make certain milestone payments for fitusiran, and, prior to the discontinuation of our revusiran clinical development program, was required to make certain milestone payments for revusiran. In December 2014, we earned a development milestone payment of $25.0 million based upon the initiation of the first global Phase 3 clinical trial for revusiran. Finally, with respect to its one global product right, Sanofi Genzyme will be required to make payments totaling up to $200.0 million for such global product, including up to $100.0 million in development milestones and $100.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for such global product based on annual net sales, if any, of such global product by Sanofi Genzyme, its affiliates and sublicensees.
73
Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Sanofi Genzyme under the 2014 Sanofi Genzyme collaboration, as amended, or the AT3 License Terms.
We determined that the deliverables for the programs on which Sanofi Genzyme was collaborating with us upon initiation of the 2014 collaboration included the licenses to our patisiran and revusiran clinical programs, which licenses were delivered to Sanofi Genzyme upon the closing date of the transaction, and the associated development activities, joint steering committee participation and information exchange for these clinical programs. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and associated undelivered development activities, joint steering committee participation and information exchange activities did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme has the ability to grant sublicenses, it cannot sublicense all or substantially all of its rights under the 2014 Sanofi Genzyme collaboration. The uniqueness of our services and the limited sublicense rights are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition model on the final deliverable. Under the 2014 Sanofi Genzyme collaboration, the last deliverables for patisiran and revusiran were expected to be completed within approximately six years from the closing date of the transaction and the last deliverables for fitusiran were expected to be completed within approximately five years from the date Sanofi Genzyme elected to opt into our fitusiran program under the regional license terms. Our estimate regarding the performance period under the 2014 Sanofi Genzyme collaboration related to the license to our patisiran and revusiran clinical programs was adjusted in October 2016 due to our decision to discontinue development of revusiran. As a result, with respect to these programs, we currently expect the last deliverables to be completed within approximately five years from the closing date of the transaction as compared to an initial expectation of approximately six years. Our estimate regarding the performance period under the 2014 Sanofi Genzyme collaboration related to the license to our fitusiran program was adjusted in September 2017 due to our temporary suspension of dosing in all ongoing fitusiran studies. As a result, with respect to the fitusiran program, we currently expect the last deliverables to be completed within approximately six years from the date Sanofi Genzyme elected to opt into the program under the regional license terms as compared to an initial expectation of approximately five years. Beginning in September 2017, we are prospectively recognizing the remaining deferred revenue as of August 31, 2017 related to the license to our fitusiran program over this adjusted performance period.
We determined that the total cash received from Sanofi Genzyme under the now superseded 2012 Sanofi Genzyme agreement reflects consideration for certain of the performance obligations for ALN-TTR programs included in the 2014 Sanofi Genzyme collaboration. Therefore, we are recognizing the $33.5 million of deferred revenue under the 2012 Sanofi Genzyme agreement on a straight-line basis over the period of performance of the ALN-TTR programs which, as noted above, is currently approximately five years. Through December 31, 2017, we have earned a milestone payment of $25.0 million and an aggregate of $140.1 million of expense reimbursement due to us from Sanofi Genzyme. We recognized these amounts as revenue equal to the percentage of the performance period completed when the milestone or development cost reimbursement was earned. As future consideration, including any milestones or reimbursement for development activities, are achieved, we recognize as revenue a portion of these payments equal to the percentage of the performance period completed when the milestone or activities have been satisfied, multiplied by the amount of the payment. We recognize the remaining portion of consideration received over the remaining performance period on a straight-line basis. At December 31, 2017, deferred revenue under the 2014 Sanofi Genzyme collaboration was $49.2 million.
We determined that the opt-in rights that Sanofi Genzyme has for future Genetic Medicine programs represent separate and additional deliverables that Sanofi Genzyme may receive from us in future periods. Upon each initial opt-in by Sanofi Genzyme, we have determined that each program and the related activities will represent a single unit of accounting and, consistent with our accounting policies, we base our revenue recognition period on the final deliverable associated with each future opt-in.
The Medicines Company. In February 2013, we and MDCO entered into a license and collaboration agreement pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases, including inclisiran. MDCO paid us an upfront cash payment of $25.0 million. In addition, MDCO is required to make payments to us upon achievement of certain milestones, up to an aggregate of $180.0 million, including up to $30.0 million in specified development milestones, $50.0 million in specified regulatory milestones and $100.0 million in specified commercialization milestones. In December 2014, we earned a development milestone payment of $10.0 million under the MDCO agreement based upon the initiation of our Phase 1 clinical trial for inclisiran. In November 2017, we earned a development milestone payment of $20.0 million under the MDCO agreement based upon the initiation by MDCO of a pivotal study for inclisiran. In addition, in 2017, 2016 and 2015, we were reimbursed an aggregate of $12.2 million of development costs from MDCO. We could potentially earn the next development milestone payment of $25.0 million based upon regulatory approval of an NDA for inclisiran in the United States. In addition, we will be entitled to royalties ranging from the low- to high- teens based on annual worldwide net sales, if any, of inclisiran by MDCO, its affiliates and sublicensees, subject to reduction under specified circumstances. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from MDCO.
74
Under the MDCO agreement, we were responsible for the development of inclisiran until Phase 1 Completion (as defined in the MDCO agreement) at our cost, up to an agreed upon initial development cost cap. MDCO is responsible for leading and funding development from Phase 2 forward, as well as potential commercialization, at its sole cost. Under the terms of the MDCO agreement, during 2015 we transferred the development leadership of inclisiran to MDCO. The collaboration between us and MDCO is governed by a joint steering committee that is comprised of an equal number of representatives from each party. We were solely responsible for obtaining supply of finished product reasonably required for the conduct of our obligations through Phase 1 Completion, and were responsible for supplying MDCO with finished product reasonably required for the first Phase 2 clinical trial of inclisiran conducted by MDCO, at our expense, subject to certain caps. In April 2016, we and MDCO entered into a supply and technical transfer agreement to provide for our supply of inclisiran to MDCO, in accordance with the terms of the MDCO agreement. MDCO now has the sole right and responsibility to manufacture and supply inclisiran for development and commercialization under the MDCO development plan, subject to the terms of the MDCO agreement and the supply and technical transfer agreement.
We have determined that the significant deliverables under the MDCO agreement include the license, the joint steering committee, technology transfer obligations, development activities through Phase 1 Completion and supply of product for a Phase 2 clinical trial. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and collective undelivered activities and services do not have standalone value due to the specialized nature of the activities and services to be provided by us. In addition, while MDCO has the ability to grant sublicenses, it must receive our prior written consent to sublicense all or substantially all of its rights. The uniqueness of our services and the limited sublicense right are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered services are being treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition pattern on the final deliverable. Under the MDCO agreement, all deliverables are expected to be completed within approximately five years. We are recognizing revenue under the MDCO agreement on a straight-line basis over approximately five years. We are not utilizing a proportional performance model since we are unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the effort required under the development activities is largely unknown.
The initial upfront payment of $25.0 million from MDCO was initially recorded as deferred revenue. During the fourth quarter of 2014, we recognized as revenue a portion of the $10.0 million milestone payment earned in December 2014 equal to the percentage of the performance period completed when the milestone was earned. During the fourth quarter of 2017, we recognized as revenue a portion of the $20.0 million milestone payment earned in November 2017 equal to the percentage of the performance period completed when the milestone was earned. During 2017, 2016 and 2015, we also recognized as revenue a portion of the $5.4 million, $3.0 million and $3.8 million, respectively, of expense reimbursement due to us under the terms of the MDCO agreement equal to the percentage of the performance period completed upon the invoice date. As future consideration, including any milestones or reimbursement for development activities, are earned, we will recognize as revenue a portion of these payments equal to the percentage of the performance period completed when the milestone is achieved or service has been provided, multiplied by the amount of the payment. We recognize the remaining portion of consideration received over the remaining performance period on a straight-line basis. At December 31, 2017, deferred revenue under the MDCO agreement was $5.7 million.
Monsanto. In August 2012, we and Monsanto entered into a license and collaboration agreement, pursuant to which we granted to Monsanto a worldwide, exclusive, royalty bearing right and license, including the right to grant sublicenses, to our RNAi platform technology and intellectual property controlled by us as of the date of the Monsanto agreement or during the 30 months thereafter, in the field of agriculture. The Monsanto agreement also included the transfer of technology from us to Monsanto and initially included a collaborative research project. Under the Monsanto agreement, Monsanto will be our exclusive collaborator in the agriculture field for a ten-year period.
Monsanto paid us $29.2 million in upfront cash payments, and was also required to make near-term milestone payments to us upon the achievement of specified technology transfer and patent-related milestones. We were also entitled to receive additional funding for collaborative research efforts. In the aggregate, we had the ability to earn up to $5.0 million in milestone payments and research funding under the Monsanto alliance. We received a total of $4.0 million in milestone payments from Monsanto based upon the achievement of a specified patent-related event and the completion of technology transfer activities. In September 2014, we and Monsanto mutually determined not to pursue the discovery collaboration originally contemplated under the terms of the Monsanto agreement. Accordingly, Monsanto will not be required to pay us the final milestone of $1.0 million. There are no remaining milestones under the Monsanto agreement. Monsanto is required to pay to us a percentage of specified fees from certain sublicense agreements Monsanto may enter into that include access to our intellectual property, as well as low single-digit royalty payments on worldwide, net sales by Monsanto, its affiliates and sublicensees of certain licensed products, as defined in the Monsanto agreement, if any. Due to the uncertainty of the application of RNAi technology in the field of agriculture, we may not receive any license fees or royalty payments from Monsanto.
75
Under the terms of the Monsanto agreement, in the event that during the exclusivity period we cease to own or otherwise exclusively control certain licensed patent rights in the agriculture field, for any reason other than Monsanto’s breach of the Monsanto agreement or its negligence or willful misconduct, resulting in the loss of exclusivity with respect to Monsanto’s rights to such patent rights, and such loss of exclusivity has a material adverse effect on the licensed products, then we would be required to pay Monsanto liquidated damages of up to $2.5 million, which amount was reduced from $5.0 million during the fourth quarter of 2017, and Monsanto’s royalty obligations to us under the Monsanto agreement would be reduced or, under certain circumstances, terminated. We have the right to cure any such loss of patent rights under the Monsanto agreement.
We initially determined that the significant deliverables under the Monsanto agreement included the license, the technology transfer activities and the services that we would be obligated to perform under the Monsanto discovery collaboration. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered technical transfer activities and Monsanto discovery collaboration services did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Monsanto has the ability to grant sublicenses, it cannot grant access to certain of our proprietary technology. The uniqueness of our services and the limited sublicense right are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered technical transfer activities and Monsanto discovery collaboration services were being treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition model on the final deliverable. Under the Monsanto agreement, the last deliverable expected to be completed was the discovery collaboration, which was originally to be completed within five years. Therefore, prior to the September 2014 amendment, we were recognizing revenue under the Monsanto agreement on a straight-line basis over five years. However, as a result of the September 2014 amendment, we determined that the final deliverable in the collaboration was the technology transfer activities, know-how exchange and access to intellectual property controlled by us as of the date of the Monsanto agreement or during the 30 months thereafter, in the field of agriculture. Consequently, we recognized the remaining deferred revenue of $16.8 million at the date of the amendment on a prospective basis from September 2014 through February 2015, the date which was the end of the 30-month obligation, which excluded $5.0 million related to a potential refund due to Monsanto under certain circumstances pursuant to the original terms of the Monsanto agreement. In 2017, we recognized an additional $2.5 million in revenue following the reduction of such potential refund by $2.5 million. We could not use a proportional performance model since we were unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the potential effort required was unknown. At December 31, 2017, deferred revenue under the Monsanto agreement was $2.5 million that will be recognized when the potential refund obligation ceases and can be considered fixed or determinable.
Takeda. In May 2008, we entered into a license and collaboration agreement with Takeda to pursue the development and commercialization of RNAi therapeutics. Under the Takeda agreement, we granted to Takeda a non-exclusive, worldwide, royalty-bearing license to our intellectual property, including delivery-related intellectual property, controlled by us as of the date of the Takeda agreement or during the five years thereafter, to develop, manufacture, use and commercialize RNAi therapeutics, subject to our existing contractual obligations to third parties. The license initially is limited to the fields of oncology and metabolic disease and may be expanded at Takeda’s option to include other therapeutic areas, subject to specified conditions.
Takeda paid us an upfront payment of $100.0 million and an additional $50.0 million upon achievement of specified technology transfer milestones. In addition, for each RNAi therapeutic product developed by Takeda, its affiliates and sublicensees, we are entitled to receive specified development, regulatory and commercialization milestone payments, totaling up to $171.0 million per product, together with a double-digit percentage royalty payment based on worldwide annual net sales, if any. The potential future milestone payments per product include up to $26.0 million for the achievement of specified development milestones, up to $40.0 million for the achievement of specified regulatory milestones and up to $105.0 million for the achievement of specified commercialization milestones. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Takeda.
Pursuant to the Takeda agreement, we and Takeda also agreed to collaborate on the research of RNAi therapeutics directed to one or two disease targets agreed to by the parties, subject to our existing contractual obligations with third parties. The collaboration is governed by a joint technology transfer committee, a joint research collaboration committee and a joint delivery collaboration committee, each of which is comprised of an equal number of representatives from each party.
We determined that the deliverables under the Takeda agreement included the license, the joint committees, the technology transfer activities and the services that we were obligated to perform under the research collaboration with Takeda. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered services (i.e., the joint committees and the research collaboration) were not separable and, accordingly, the license and services were being treated as a single unit of accounting. Under the Takeda agreement, the last elements to be delivered were the joint technology transfer committee and joint delivery collaboration committee services, each of which had a life of no more than seven years. We have fully recognized the upfront payment of $100.0 million and the technology transfer milestones of $50.0 million,
76
the receipt of which we believed was probable at the commencement of the collaboration, on a straight-line basis over seven years because we were unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the effort required under the research collaboration was largely unknown, and therefore, we could not utilize a proportional performance model. As future milestones are achieved, we recognize as revenue the milestone amount in its entirety because all performance obligations for the Takeda agreement have been delivered. As of December 31, 2017, there was no remaining deferred revenue balance under the Takeda agreement as all of our contractual performance obligations were met in May 2015.
Accounting for Income Taxes
We recognize the tax benefit from an uncertain tax position only if it is more likely than not that the tax position will be sustained upon examination by the taxing authorities, based on the technical merits of the tax position. The tax benefits recognized in our financial statements from such a position are measured based on the largest benefit that has a greater than 50 percent likelihood of being realized upon ultimate resolution. Our policy is to accrue interest and penalties related to unrecognized tax positions in income tax expense. As of December 31, 2017, we have not recorded significant interest and penalty expense related to uncertain tax positions.
On December 22, 2017, the President of the United States signed into law the TCJA tax reform legislation. The TCJA makes significant changes in U.S. tax law including a reduction in the corporate tax rates, changes to net operating loss carryforwards and carrybacks, and a repeal of the corporate alternative minimum tax. The TCJA reduced the U.S. corporate tax rate from the current rate of 35 percent down to 21 percent starting on January 1, 2018. As a result of the enacted law, we were required to revalue deferred tax assets and liabilities at 21 percent. This revaluation resulted in a provision of $227.9 million to income tax expense in continuing operations and a corresponding reduction in the valuation allowance. As a result, there was no impact to our consolidated statements of comprehensive loss due to the reduction in tax rates. The other provisions of the TCJA did not have a material impact on our consolidated financial statements.
Our preliminary estimate of the TCJA and the remeasurement of our deferred tax assets and liabilities is subject to the finalization of management’s analysis related to certain matters, such as developing interpretations of the provisions of the TCJA, changes to certain estimates and the filing of our tax returns. U.S. Treasury regulations, administrative interpretations or court decisions interpreting the TJCA may require further adjustments and changes in our estimates. The final determination of the TCJA and the remeasurement of our deferred assets and liabilities will be completed as additional information becomes available, but no later than one year from the enactment of the TCJA.
We operate in the United States, as well as in several countries outside of the United States, where our income tax returns are subject to audit and adjustment by local tax authorities. The nature of the uncertain tax positions is often very complex and subject to change, and the amounts at issue can be substantial. We develop our cumulative probability assessment of the measurement of uncertain tax positions using internal experience, judgment and assistance from professional advisors. We refine estimates as we become aware of additional information. Any outcome upon settlement that differs from our current estimate may result in additional tax expense in future periods. At December 31, 2017, we had no unrecognized tax benefits.
We recognize income taxes when transactions are recorded in our consolidated statements of comprehensive loss, with deferred taxes provided for items that are recognized in different periods for financial statement and tax reporting purposes. We record a valuation allowance to reduce the deferred tax assets to the amount that is more likely than not to be realized.
There was no benefit to income taxes recorded during the years ended December 31, 2017, 2016 or 2015.
At December 31, 2017, we had a valuation allowance against our net deferred tax assets to the extent it is more likely than not that the assets will not be realized. At December 31, 2017, we had federal and state net operating loss carryforwards of $1.47 billion and $1.55 billion, respectively, to reduce future taxable income that will expire at various dates through 2037. At December 31, 2017, we had federal and state research and development and investment tax credit carryforwards of $180.0 million and $15.3 million, respectively, available to reduce future tax liabilities that expire at various dates through 2037. At December 31, 2017, we had alternative minimum tax credits of $0.8 million that will either be available to reduce future regular tax liabilities or be fully refundable in 2021. We have a valuation allowance against the net operating loss and credit deferred tax assets as it is unlikely that we will realize these assets. Ownership changes, as defined in the Internal Revenue Code, including those resulting from the issuance of common stock in connection with our public offerings, may limit the amount of net operating loss and tax credit carryforwards that can be utilized to offset future taxable income or tax liability. The amount of the limitation is determined in accordance with Section 382 of the Internal Revenue Code. We have performed an analysis of ownership changes through December 31, 2017. Based on this analysis, we do not believe that any of our tax attributes will expire unutilized due to Section 382 limitations.
77
Accounting for Stock-Based Compensation
We have stock incentive plans and an employee stock purchase plan under which we grant equity instruments. We may also grant inducement stock grants outside of our stock incentive plans. We account for all stock-based awards granted to employees at their fair value and generally recognize compensation expense over the vesting period of the award. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of fair values of stock options as of the grant date. We calculate the grant date fair values of stock options using the Black-Scholes valuation model. Our expected stock price volatility assumption is based on the historical volatility of our publicly traded stock.
For stock option awards granted during the year ended December 31, 2017, we used a weighted-average expected stock-price volatility assumption of 66 percent. Our expected life assumption is based on our historical data. Our weighted-average expected term was 5.8 years for the year ended December 31, 2017. We utilize a dividend yield of zero based on the fact that we have never paid cash dividends and currently have no intention to pay cash dividends. The risk-free interest rate used for each grant is based on the U.S. Treasury yield curve in effect at the time of grant for instruments with a similar expected life.
The fair value of restricted stock awards granted to employees is based upon the quoted closing market price per share on the date of grant. Expense for time-based restricted stock awards is recognized over the vesting period.
We have performance conditions included in certain of our stock option and restricted stock awards that are based upon the achievement of pre-specified clinical development, regulatory and/or commercial events. As the outcome of each event has inherent risk and uncertainties and a positive outcome may not be known until the event is achieved, we begin to recognize the value of the performance-based stock option and restricted stock awards when we determine the achievement of each performance condition is deemed probable, which often is not until the condition is achieved. This determination requires significant judgment by management. At the probable date, we record a cumulative expense catch-up, with remaining expense amortized over the remaining service period.
At December 31, 2017, the estimated fair value of time-based unvested employee stock options was $124.3 million, net of estimated forfeitures. We will recognize this amount over the weighted-average remaining vesting period of approximately three years for these awards. At December 31, 2017, the estimated fair value of performance-based unvested employee stock options and restricted stock units was $74.1 million, net of estimated forfeitures. Stock-based employee compensation expense was $92.8 million for the year ended December 31, 2017. However, we cannot currently predict the total amount of stock-based compensation expense to be recognized in any future period because such amounts will depend on levels of stock-based payments granted in the future as well as the portion of the awards that actually vest, including our performance-based awards. We estimate forfeitures at the time of grant and revise, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The term “forfeitures” is distinct from “cancellations” or “expirations” and represents only the unvested portion of the surrendered stock option. We have applied an annual forfeiture rate to all unvested employee stock options and restricted stock awards at December 31, 2017 based on an analysis of our historical forfeitures. Ultimately, the actual expense recognized over the vesting period will only be for those shares that vest.
Estimated Liability for Development Costs
We record accrued liabilities related to expenses for which service providers have not yet billed us with respect to products we have received or services that we have incurred, specifically related to ongoing pre-clinical studies and clinical trials. These costs primarily relate to third-party clinical management costs, laboratory and analysis costs, toxicology studies and investigator fees. We have multiple product candidates in concurrent pre-clinical studies and clinical trials at multiple clinical sites throughout the world. In order to ensure that we have adequately provided for ongoing pre-clinical and clinical development costs during the period in which we incur such costs, we maintain an accrual to cover these expenses. We update our estimate for this accrual on at least a quarterly basis. The assessment of these costs is a subjective process that requires judgment. Upon settlement, these costs may differ materially from the amounts accrued in our consolidated financial statements. Our historical accrual estimates have not been materially different from our actual costs.
78
The following data summarizes the results of our operations for the periods indicated, in thousands:
|
|
|
Year Ended December 31, |
|||||||||||
|
Description |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
|||
|
Net revenues from collaborators |
|
$ |
89,912 |
|
|
$ |
47,159 |
|
|
$ |
41,097 |
|
|
|
Operating expenses |
|
|
590,000 |
|
|
|
471,746 |
|
|
|
337,105 |
|
|
|
Loss from operations |
|
|
(500,088 |
) |
|
|
(424,587 |
) |
|
|
(296,008 |
) |
|
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
Discussion of Results of Operations
Net revenues from collaborators
We generate revenues through research and development collaborations. The following table summarizes our total consolidated net revenues from collaborators, for the periods indicated, in thousands, together with the changes, in thousands:
|
|
|
Year Ended December 31, |
|
|
Dollar Change |
|
||||||||||||||
|
Description |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
2017 compared to 2016 |
|
|
2016 compared to 2015 |
|
|||||
|
Sanofi Genzyme |
|
$ |
54,625 |
|
|
$ |
32,015 |
|
|
$ |
11,005 |
|
|
$ |
22,610 |
|
|
$ |
21,010 |
|
|
MDCO |
|
|
30,217 |
|
|
|
11,220 |
|
|
|
10,301 |
|
|
|
18,997 |
|
|
|
919 |
|
|
Takeda |
|
|
— |
|
|
|
— |
|
|
|
8,867 |
|
|
|
— |
|
|
|
(8,867 |
) |
|
Monsanto |
|
|
2,500 |
|
|
|
— |
|
|
|
5,621 |
|
|
|
2,500 |
|
|
|
(5,621 |
) |
|
Other |
|
|
2,570 |
|
|
|
3,924 |
|
|
|
5,303 |
|
|
|
(1,354 |
) |
|
|
(1,379 |
) |
|
Total net revenues from collaborators |
|
$ |
89,912 |
|
|
$ |
47,159 |
|
|
$ |
41,097 |
|
|
$ |
42,753 |
|
|
$ |
6,062 |
|
Net revenues from collaborators increased during the year ended December 31, 2017 as compared to the year ended December 31, 2016 due primarily to increased services performed by us in connection with our clinical development programs for which Sanofi Genzyme had opted in and the achievement of a $20.0 million milestone under our agreement with MDCO upon initiation of its Phase 3 study for inclisiran in early November 2017.
Net revenues from collaborators increased for the year ended December 31, 2016 as compared to the year ended December 31, 2015 due primarily to services performed by us in connection with our clinical development programs for which Sanofi Genzyme had opted in, partially offset by the completion of our performance obligations under the Monsanto agreement in February 2015 and the completion of our revenue amortization under the Takeda agreement in May 2015.
We expect net revenues from collaborators to decrease during 2018 as compared to 2017 due primarily to decreased revenues under the MDCO agreement. We had $84.8 million and $82.9 million of deferred revenue at December 31, 2017 and 2016, respectively, which consists primarily of payments we have received from collaborators, primarily Sanofi Genzyme, MDCO and Kyowa Hakko Kirin, but have not yet recognized pursuant to our revenue recognition policies. As a result of our adoption of the new standard related to revenue recognition on January 1, 2018, we expect to record a cumulative reduction of $68.3 million of deferred revenue with a corresponding adjustment to accumulated deficit in the first quarter of 2018. Please read Note 2 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for our discussion of recent accounting pronouncements for the expected impact of this new accounting standard.
During 2018, if we are successful in obtaining regulatory approval and commercializing patisiran, we expect to begin to recognize net product revenues. Until we are successful in obtaining regulatory approval for patisiran and our other product candidates and in commercializing such products, we expect our revenues to continue to be derived primarily from our alliances with Sanofi Genzyme and MDCO, as well as other strategic alliances and potential new collaborations and licensing activities, which may include license and other fees, funded research and development, milestone payments and royalties on product sales by our licensors.
79
The following table summarizes our operating expenses for the periods indicated, in thousands and as a percentage of total operating expenses, together with the changes, in thousands:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dollar Change |
|
|||||
|
Description |
|
2017 |
|
|
% of Total Operating Expenses |
|
|
2016 |
|
|
% of Total Operating Expenses |
|
|
2015 |
|
|
% of Total Operating Expenses |
|
|
2017 compared to 2016 |
|
|
2016 compared to 2015 |
|
||||||||
|
Research and development |
|
$ |
390,635 |
|
|
|
66 |
% |
|
$ |
382,392 |
|
|
|
81 |
% |
|
$ |
276,495 |
|
|
|
82 |
% |
|
$ |
8,243 |
|
|
$ |
105,897 |
|
|
General and administrative |
|
|
199,365 |
|
|
|
34 |
% |
|
|
89,354 |
|
|
|
19 |
% |
|
|
60,610 |
|
|
|
18 |
% |
|
|
110,011 |
|
|
|
28,744 |
|
|
Total operating expenses |
|
$ |
590,000 |
|
|
|
100 |
% |
|
$ |
471,746 |
|
|
|
100 |
% |
|
$ |
337,105 |
|
|
|
100 |
% |
|
$ |
118,254 |
|
|
$ |
134,641 |
|
Research and development. The following table summarizes the components of our research and development expenses for the periods indicated, in thousands and as a percentage of total research and development expenses, together with the changes, in thousands:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dollar Change |
|
|||||
|
Description |
|
2017 |
|
|
% of Expense Category |
|
|
2016 |
|
|
% of Expense Category |
|
|
2015 |
|
|
% of Expense Category |
|
|
2017 compared to 2016 |
|
|
2016 compared to 2015 |
|
||||||||
|
Research and development |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Compensation and related |
|
$ |
100,728 |
|
|
|
26 |
% |
|
$ |
87,124 |
|
|
|
23 |
% |
|
$ |
60,803 |
|
|
|
22 |
% |
|
$ |
13,604 |
|
|
$ |
26,321 |
|
|
Clinical trial |
|
|
87,730 |
|
|
|
22 |
% |
|
|
92,383 |
|
|
|
24 |
% |
|
|
68,240 |
|
|
|
25 |
% |
|
|
(4,653 |
) |
|
|
24,143 |
|
|
Manufacturing |
|
|
54,681 |
|
|
|
14 |
% |
|
|
56,348 |
|
|
|
15 |
% |
|
|
45,388 |
|
|
|
16 |
% |
|
|
(1,667 |
) |
|
|
10,960 |
|
|
Stock-based compensation |
|
|
51,872 |
|
|
|
13 |
% |
|
|
42,946 |
|
|
|
11 |
% |
|
|
27,086 |
|
|
|
10 |
% |
|
|
8,926 |
|
|
|
15,860 |
|
|
External services |
|
|
38,675 |
|
|
|
10 |
% |
|
|
48,624 |
|
|
|
13 |
% |
|
|
35,259 |
|
|
|
13 |
% |
|
|
(9,949 |
) |
|
|
13,365 |
|
|
Facilities-related |
|
|
31,022 |
|
|
|
8 |
% |
|
|
30,032 |
|
|
|
8 |
% |
|
|
21,525 |
|
|
|
8 |
% |
|
|
990 |
|
|
|
8,507 |
|
|
Lab supplies and materials |
|
|
10,513 |
|
|
|
3 |
% |
|
|
8,917 |
|
|
|
2 |
% |
|
|
7,494 |
|
|
|
2 |
% |
|
|
1,596 |
|
|
|
1,423 |
|
|
Other |
|
|
15,414 |
|
|
|
4 |
% |
|
|
16,018 |
|
|
|
4 |
% |
|
|
10,700 |
|
|
|
4 |
% |
|
|
(604 |
) |
|
|
5,318 |
|
|
Total research and development expenses |
|
$ |
390,635 |
|
|
|
100 |
% |
|
$ |
382,392 |
|
|
|
100 |
% |
|
$ |
276,495 |
|
|
|
100 |
% |
|
$ |
8,243 |
|
|
$ |
105,897 |
|
Research and development expenses increased slightly during the year ended December 31, 2017 as compared to the year ended December 31, 2016 due primarily to increased compensation and related expenses as a result of an increase in headcount during the period as we expand and advance our development pipeline, partially offset by decreases in external services expenses related to pre-clinical activities and clinical trial expenses as a result of our decision in October 2016 to discontinue development of revusiran. In addition, stock-based compensation expenses increased during the year ended December 31, 2017 as a result of increased expense related to the accounting for performance-based stock option awards.
Research and development expenses increased during the year ended December 31, 2016 as compared to the year ended December 31, 2015 due primarily to additional clinical trial and manufacturing and external services expenses as a result of the advancement of our Genetic Medicine pipeline. In addition, compensation and related expenses and stock-based compensation expenses increased during the year ended December 31, 2016 as compared to the prior year as a result of an increase in headcount during the period as we advanced our pipeline into later-stage development, as well as the vesting of certain performance-based stock option awards during 2016.
80
During the years ended December 31, 2017, 2016 and 2015, in connection with advancing activities under our significant agreements, we incurred significant research and development expenses, primarily related to external development and manufacturing services. The 2018 amendment, together with the Exclusive TTR License and the AT3 License Terms, revise the terms and conditions of our 2014 Sanofi Genzyme collaboration to provide us with the exclusive right to pursue the further global development and commercialization of all TTR products and any back-up products and provide Sanofi Genzyme with the exclusive right to pursue the further global development and commercialization of all fitusiran and any back-up products. As a result, we expect costs incurred under our significant agreements to decrease. The following table summarizes the expenses incurred under our significant agreements by collaboration partner for the periods indicated, in thousands:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Sanofi Genzyme |
|
$ |
184,703 |
|
|
$ |
160,580 |
|
|
$ |
115,768 |
|
|
MDCO |
|
|
5,527 |
|
|
|
1,275 |
|
|
|
4,575 |
|
|
Ionis |
|
|
3,250 |
|
|
|
525 |
|
|
|
3,300 |
|
|
Total |
|
$ |
193,480 |
|
|
$ |
162,380 |
|
|
$ |
123,643 |
|
We expect to continue to devote a substantial portion of our resources to research and development expenses to support our goals for 2020. We expect that research and development expenses will increase significantly in 2018 as compared to 2017 as we continue to develop our pipeline and advance our product candidates into later-stage development, hire additional employees and prepare regulatory submissions. However, we expect that certain expenses will be variable depending on the timing of manufacturing batches, clinical trial enrollment and results, regulatory review of our product candidates, and stock-based compensation expenses due to our determination regarding the probability of vesting for performance-based awards.
A significant portion of our research and development costs are not tracked by project as they benefit multiple projects or our technology platform. However, certain of our collaboration agreements contain cost-sharing arrangements pursuant to which certain costs incurred under the project are reimbursed. Costs reimbursed under the agreements typically include certain direct external costs and a negotiated full-time equivalent labor rate for the actual time worked on the project. As a result, although a significant portion of our research and development expenses are not tracked on a project-by-project basis, we do track direct external costs attributable to, and the actual time our employees worked on, our collaborations.
General and administrative. The following table summarizes the components of our general and administrative expenses for the periods indicated, in thousands and as a percentage of total general and administrative expenses, together with the changes, in thousands:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dollar Change |
|
|||||
|
Description |
|
2017 |
|
|
% of Expense Category |
|
|
2016 |
|
|
% of Expense Category |
|
|
2015 |
|
|
% of Expense Category |
|
|
2017 compared to 2016 |
|
|
2016 compared to 2015 |
|
||||||||
|
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Consulting and professional services |
|
$ |
68,847 |
|
|
|
35 |
% |
|
$ |
25,310 |
|
|
|
28 |
% |
|
$ |
21,451 |
|
|
|
35 |
% |
|
$ |
43,537 |
|
|
$ |
3,859 |
|
|
Compensation and related |
|
|
60,289 |
|
|
|
30 |
% |
|
|
20,967 |
|
|
|
24 |
% |
|
|
12,721 |
|
|
|
21 |
% |
|
|
39,322 |
|
|
|
8,246 |
|
|
Stock-based compensation |
|
|
40,947 |
|
|
|
21 |
% |
|
|
32,582 |
|
|
|
36 |
% |
|
|
18,697 |
|
|
|
31 |
% |
|
|
8,365 |
|
|
|
13,885 |
|
|
Facilities-related |
|
|
11,130 |
|
|
|
6 |
% |
|
|
5,547 |
|
|
|
6 |
% |
|
|
3,705 |
|
|
|
6 |
% |
|
|
5,583 |
|
|
|
1,842 |
|
|
Other |
|
|
18,152 |
|
|
|
8 |
% |
|
|
4,948 |
|
|
|
6 |
% |
|
|
4,036 |
|
|
|
7 |
% |
|
|
13,204 |
|
|
|
912 |
|
|
Total general and administrative expenses |
|
$ |
199,365 |
|
|
|
100 |
% |
|
$ |
89,354 |
|
|
|
100 |
% |
|
$ |
60,610 |
|
|
|
100 |
% |
|
$ |
110,011 |
|
|
$ |
28,744 |
|
General and administrative expenses increased significantly during the year ended December 31, 2017 as compared to the year ended December 31, 2016 due primarily to an increase in commercial and medical affairs headcount and commercial-related services to support corporate growth and prepare for potential commercial product launches. In addition, stock-based compensation expenses increased during the year ended December 31, 2017 as a result of increased expense related to the accounting for performance-based stock option awards.
General and administrative expenses increased during the year ended December 31, 2016 as compared to the year ended December 31, 2015 due primarily to an increase in compensation and related expenses and stock-based compensation expenses resulting from an increase in headcount, as well as the vesting of certain performance-based stock option awards during 2016 and a one-time expense recorded in connection with the acceleration of a stock option award under the terms of our stock plan as the result of the unexpected death of an employee.
81
We expect that general and administrative expenses will increase significantly in 2018 as compared to 2017 as we continue to grow our operations, including the continued build-out of our global commercial infrastructure and field team, in preparation for our anticipated first product launch in 2018, but expect that stock-based compensation expenses will be variable due to our determination regarding the probability of vesting for performance-based awards.
Liquidity and Capital Resources
The following table summarizes our cash flow activities for the periods indicated, in thousands:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities |
|
|
110,990 |
|
|
|
85,188 |
|
|
|
65,817 |
|
|
Changes in operating assets and liabilities |
|
|
(2,902 |
) |
|
|
17,219 |
|
|
|
35,116 |
|
|
Net cash used in operating activities |
|
|
(382,786 |
) |
|
|
(307,701 |
) |
|
|
(189,140 |
) |
|
Net cash (used in) provided by investing activities |
|
|
(290,361 |
) |
|
|
142,591 |
|
|
|
(321,321 |
) |
|
Net cash provided by financing activities |
|
|
1,124,891 |
|
|
|
177,832 |
|
|
|
616,177 |
|
|
Net increase in cash and cash equivalents |
|
|
451,744 |
|
|
|
12,722 |
|
|
|
105,716 |
|
|
Cash and cash equivalents, beginning of period |
|
|
193,617 |
|
|
|
180,895 |
|
|
|
75,179 |
|
|
Cash and cash equivalents, end of period |
|
$ |
645,361 |
|
|
$ |
193,617 |
|
|
$ |
180,895 |
|
Since we commenced operations in 2002, we have generated significant losses. At December 31, 2017, we had an accumulated deficit of $2.15 billion. At December 31, 2017, we had cash, cash equivalents and fixed income marketable securities of $1.70 billion, compared to $942.6 million at December 31, 2016, in each case excluding the restricted investments related to our term loan agreements.
In January 2015, we sold an aggregate of 5,447,368 shares of our common stock through an underwritten public offering at a price to the public of $95.00 per share. As a result of the offering, which included the full exercise of the underwriters’ option to purchase additional shares, we received aggregate net proceeds of $496.4 million, after deducting underwriting discounts and commissions and other offering expenses of $21.1 million. In May 2017, we sold an aggregate of 5,000,000 shares of our common stock through an underwritten public offering at a price to the public of $71.87 per share. As a result of the offering, we received aggregate net proceeds of $355.2 million, after deducting underwriting discounts and commissions and other offering expenses of $4.2 million. In November 2017, we sold an aggregate of 6,440,000 shares of our common stock through an underwritten public offering at a price to the public of $125.00 per share. As a result of the offering, which included the full exercise of the underwriters’ option to purchase additional shares, we received aggregate net proceeds of $784.5 million, after deducting underwriting discounts and commissions and other offering expenses of $20.5 million.
We have used and intend to continue to use these proceeds for general corporate purposes, including clinical trial costs and other research and development expenses, continued growth of our manufacturing, quality, commercial and medical affairs capabilities to support our transition toward a commercial-stage biopharmaceutical company, the anticipated global commercial launches of patisiran and givosiran and other potential future products, the potential expansion of patisiran commercialization efforts in mixed phenotype populations, assuming consistent product labelling, potential repayment of outstanding indebtedness, potential acquisitions, investments or licenses in businesses, products or technologies that are complementary to our own, working capital, capital expenditures and general and administrative expenses.
Sanofi Genzyme has certain rights to purchase additional shares from us under our investor agreement. In connection with our public offerings described above, Sanofi Genzyme exercised its right to purchase directly from us, in concurrent private placements, 744,566 shares of common stock in January 2015 at the public offering price resulting in $70.7 million in proceeds to us and 297,501 shares of common stock in May 2017 at the public offering price resulting in $21.4 million in proceeds to us. In addition, Sanofi Genzyme also has the right at the beginning of each year to purchase a number of shares of our common stock based on the number of shares we issued during the previous year for compensation-related purposes. Sanofi Genzyme exercised this right to purchase directly from us 196,251 shares of our common stock in January 2015 for $18.3 million and 205,030 shares of our common stock in February 2016 for $14.3 million. Sanofi Genzyme currently holds approximately 11 percent of our outstanding common stock.
We invest primarily in money market funds, U.S. government-sponsored enterprise securities, U.S. treasury securities, high-grade corporate notes, certificates of deposit and commercial paper. Corporate notes may also include foreign bonds denominated in U.S. dollars. Our investment objectives are, primarily, to assure liquidity and preservation of capital and, secondarily, to obtain
82
investment income. All of our investments in debt securities are recorded at fair value and are available-for-sale. Fair value is determined based on quoted market prices and models using observable data inputs. We have not recorded any impairment charges to our fixed income marketable securities during the three years ended December 31, 2017.
Operating activities
We have required significant amounts of cash to fund our operating activities as a result of net losses since our inception. Cash used in operating activities is adjusted for non-cash items to reconcile net loss to net cash used in operating activities. These non-cash adjustments have historically included stock-based compensation and depreciation and amortization.
We expect that we will require significant amounts of cash to fund our operating activities for the foreseeable future as we continue to execute on our Alnylam 2020 strategy through the advancement of our research, development, pre-commercial and potentially commercial initiatives. The actual amount of overall expenditures will depend on numerous factors, including the timing of expenses, the timing and terms of collaboration agreements or other strategic transactions, if any, and the timing and progress of our research, development and commercialization efforts.
The increase in net cash used in operating activities for the year ended December 31, 2017 compared to the year ended December 31, 2016 and for the year ended December 31, 2016 compared to the year ended December 31, 2015 was due primarily to our net loss.
Investing activities
For the years ended December 31, 2017, 2016 and 2015, net cash provided by or used in investing activities was due primarily to net purchases of fixed income marketable securities in accordance with management of our liquidity needs. For the year ended December 31, 2017 and 2016, there were purchases of property, plant and equipment of $104.2 million and $64.6 million, respectively, primarily in connection with the construction of our drug substance manufacturing facility. For the year ended December 31, 2016, there were $150.0 million of purchases of restricted investments related to our term loan agreements with BOA and Wells. In December 2017, we repaid in full $120.0 million outstanding under the BOA term loan agreement.
Financing activities
For the year ended December 31, 2017, net cash of $1.12 billion provided by financing activities was due primarily to proceeds of $1.14 billion received from our May and November 2017 underwritten public offerings. For the year ended December 31, 2016, net cash of $177.8 million provided by financing activities was due primarily to our term loan agreements with BOA and Wells. For the year ended December 31, 2015, net cash of $616.2 million provided by financing activities was due primarily to proceeds of $496.4 million received from our January 2015 underwritten public offering, as well as proceeds of $89.0 million received from our issuances of common stock to Sanofi Genzyme in January 2015.
Operating Capital Requirements
We currently have programs focused on a number of therapeutic areas and, in December 2017, submitted our first NDA and MAA for marketing approval for patisiran. However, our development efforts may not be successful and we may not be able to commence sales of patisiran or any other product. If we are able to gain approval and successfully launch patisiran in 2018, we may begin to generate net revenues from product sales. Therefore, we anticipate that we will continue to generate significant losses for the foreseeable future as a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the establishment of late stage clinical and commercial capabilities, including global operations, continued management and growth of our intellectual property including our patent portfolio, collaborations and general corporate activities. In addition, we are expanding our manufacturing capabilities, including through construction of a drug substance manufacturing facility in Norton, Massachusetts. In April 2016, our subsidiary, Alnylam U.S., Inc., entered into an aggregate of $150.0 million in term loan agreements with BOA and Wells, related to the build out of our new drug substance manufacturing facility. In December 2017, we repaid in full $120.0 million outstanding under the BOA term loan agreement. Interest on the borrowings under the BOA term loan agreement was, and interest under the Wells term loan agreement is, calculated based on LIBOR plus 0.45 percent. The obligations under the term loan agreements were, with respect to BOA, and are, with respect to Wells, secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of all term loans outstanding under the agreements at such time. We are the guarantor under the Wells term loan agreement, which matures in April 2021.
Based on our current operating plan, we believe that our existing cash, cash equivalents and fixed income marketable securities, together with the cash we expect to generate under our current alliances, will be sufficient to enable us to advance our Alnylam 2020
83
strategy for at least the next few years. For reasons discussed below, we may require significant additional funds earlier than we currently expect in order to develop, conduct clinical trials for, manufacture and commercialize any product candidates.
In the future, we may seek additional funding through additional collaborative arrangements and public or private financings. Additional funding may not be available to us on acceptable terms or at all. Moreover, the terms of any additional financing may further adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our existing stockholders will result. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. If we are unable to obtain funding on a timely basis, we may be required to significantly delay or curtail one or more of our research or development programs and our ability to achieve our goals for 2020 may be delayed or diminished. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own.
Even if we are able to raise additional funds in a timely manner, our future capital requirements may vary from what we expect and will depend on many factors, including:
|
|
• |
our progress in demonstrating that siRNAs can be active as drugs and achieve desired clinical effects; |
|
|
• |
progress in our research and development programs, as well as what may be required by regulatory bodies to advance these programs; |
|
|
• |
the timing, receipt and amount of milestone and other payments, if any, from present and future collaborators, if any; |
|
|
• |
our ability to maintain and establish additional collaborative arrangements and/or new business initiatives; |
|
|
• |
the resources, time and costs required to successfully initiate and complete our pre-clinical and clinical trials, obtain regulatory approvals, prepare for commercialization of our product candidates and obtain and maintain licenses to third-party intellectual property; |
|
|
• |
our ability to establish, maintain and operate our own manufacturing facilities in a timely and cost effective manner; |
|
|
• |
our ability to manufacture, or contract with third-parties for the manufacture of, our product candidates for clinical testing and commercial sale; |
|
|
• |
the resources, time and cost required for the preparation, filing, prosecution, maintenance and enforcement of patent claims; |
|
|
• |
the costs associated with legal activities, including litigation, arising in the course of our business activities and our ability to prevail in any such legal disputes; and |
|
|
• |
the timing, receipt and amount of sales and royalties, if any, from our potential products. |
Off-Balance Sheet Arrangements
In connection with our license agreements with Max Planck relating to the Tuschl I and II patent applications, we are required to indemnify Max Planck for certain damages arising in connection with the intellectual property rights licensed under the agreements. Under this indemnification agreement with Max Planck, we are responsible for paying the costs of any litigation relating to the license agreements or the underlying intellectual property rights. In connection with the settlement of the litigation regarding the Tuschl patents, we also agreed to indemnify Whitehead, MIT and UMass for certain costs associated with defending the University of Utah litigation. In connection with our research agreement with Acuitas, we agreed to indemnify Acuitas for certain legal costs, subject to certain exceptions and limitations. These indemnification costs are charged to general and administrative expense. In addition, we are a party to a number of agreements entered into in the ordinary course of business, which contain typical provisions that obligate us to indemnify the other parties to such agreements upon the occurrence of certain events. These indemnification obligations are considered off-balance sheet arrangements in accordance with GAAP. To date, other than certain costs associated with certain previously settled litigation related to the Tuschl patents and our disputes with Arbutus, and certain defense costs related to the University of Utah litigation, we have not encountered material costs as a result of such obligations and have not accrued any liabilities related to such obligations in our consolidated financial statements. Please read Note 7 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for further discussion of these indemnification agreements.
84
In the table below, we set forth our enforceable and legally binding obligations and future commitments at December 31, 2017. Some of the figures that we include in this table are based on management’s estimates and assumptions about these obligations, including their duration, the possibility of renewal, anticipated actions by third parties and other factors. Because these estimates and assumptions are necessarily subjective, the obligations we will actually pay in future periods may vary from those reflected in the table.
|
|
|
Payments Due by Period |
|
|||||||||||||||||
|
Contractual Obligations |
|
2018 |
|
|
2019 and 2020 |
|
|
2021 and 2022 |
|
|
After 2022 |
|
|
Total |
|
|||||
|
Facility lease obligations(1) |
|
$ |
14,464 |
|
|
$ |
67,016 |
|
|
$ |
53,978 |
|
|
$ |
297,726 |
|
|
$ |
433,184 |
|
|
Long-term debt(2) |
|
|
614 |
|
|
|
1,229 |
|
|
|
30,252 |
|
|
|
— |
|
|
|
32,095 |
|
|
Technology license and other commitments(3) |
|
|
26,012 |
|
|
|
18,072 |
|
|
|
1,390 |
|
|
|
1,500 |
|
|
|
46,974 |
|
|
Total contractual cash obligations |
|
$ |
41,090 |
|
|
$ |
86,317 |
|
|
$ |
85,620 |
|
|
$ |
299,226 |
|
|
$ |
512,253 |
|
|
(1) |
Relates primarily to our Cambridge, Massachusetts non-cancelable facility lease agreements. |
|
(2) |
In April 2016, our subsidiary, Alnylam U.S., Inc., entered into a $30.0 million term loan agreement with Wells, for which we are the guarantor, related to the build out of our new drug substance manufacturing facility, that matures in April 2021. Interest payments are included in the table above and are calculated based on LIBOR plus 0.45 percent. The obligations under the term loan agreement are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of the term loan outstanding at such time. We include estimates for interest in “Long-term debt,” which are equivalent to our expectations for the probable outcome of variable interest rates that are dependent on various future events and market interest rates. |
|
(3) |
Relates to our fixed payment obligations under license agreements, as well as other payments related to technology research and development. |
The table above excludes approximately $333.3 million of cancellable commitments related to clinical and manufacturing-related agreements, including certain costs related to investment in the construction of our drug substance manufacturing facility and commitments that may be transitioned to Sanofi Genzyme under the AT3 License Terms. We in-license technology from a number of sources, including Ionis and Merck. Pursuant to these in-license agreements, we will be required to make additional payments if and when we achieve specified development, regulatory and commercialization milestones. To the extent we are unable to reasonably predict the likelihood, timing or amount of such payments, we have excluded them from the table above.
Recent Accounting Pronouncements
Please read Note 2 to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data,” of this annual report on Form 10-K for a description of recent accounting pronouncements applicable to our business.
As part of our investment portfolio, we own financial instruments that are sensitive to market risks. The investment portfolio is used to preserve our capital until it is required to fund operations, including our research, development and early commercial activities. Our fixed income marketable securities consist of primarily U.S. government-sponsored enterprise securities, U.S. treasury securities, high-grade corporate notes, and commercial paper. Corporate notes may also include foreign bonds denominated in U.S. dollars. All of our investments in debt securities are classified as available-for-sale and are recorded at fair value. Our available-for-sale investments in debt securities are sensitive to changes in interest rates and changes in the credit ratings of the issuers. Interest rate changes would result in a change in the net fair value of these financial instruments due to the difference between the market interest rate and the market interest rate at the date of purchase of the financial instrument. If market interest rates were to increase immediately and uniformly by 50 basis points, or one-half of a percentage point, from levels at December 31, 2017, the net fair value of our interest-sensitive financial instruments would have resulted in a hypothetical decline of $2.7 million. We currently do not seek to hedge this exposure to fluctuations in interest rates. A downgrade in the credit rating of an issuer of a debt security or further deterioration of the credit markets could result in a decline in the fair value of the debt instruments. Our investment guidelines prohibit investment in auction rate securities and we do not believe we have any direct exposure to losses relating from mortgage-based securities or derivatives related thereto such as credit-default swaps. Historically, foreign currency fluctuations have not been material. We did not record any impairment charges to our fixed income marketable securities during the year ended December 31, 2017.
85
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
86
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
|
|
• |
Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets; |
|
|
• |
Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of management and directors; and |
|
|
• |
Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2017. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013).
Based on our assessment, our management concluded that, as of December 31, 2017, our internal control over financial reporting is effective based on those criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2017 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report. This report appears on page 88.
87
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Alnylam Pharmaceuticals, Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of Alnylam Pharmaceuticals, Inc. and its subsidiaries as of December 31, 2017 and 2016, and the related consolidated statements of comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2017, including the related notes (collectively referred to as the “consolidated financial statements”). We also have audited the Company's internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2017 and 2016, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2017 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2017, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company's internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
88
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/PricewaterhouseCoopers LLP
Boston, Massachusetts
February 15, 2018
We have served as the Company’s auditor since 2003.
89
(In thousands, except share and per share amounts)
|
|
|
December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
645,361 |
|
|
$ |
193,617 |
|
|
Marketable securities |
|
|
1,045,257 |
|
|
|
424,185 |
|
|
Investment in equity securities of Regulus Therapeutics Inc. |
|
|
— |
|
|
|
8,997 |
|
|
Billed and unbilled collaboration receivables |
|
|
34,002 |
|
|
|
23,334 |
|
|
Prepaid expenses and other current assets |
|
|
40,120 |
|
|
|
21,744 |
|
|
Total current assets |
|
|
1,764,740 |
|
|
|
671,877 |
|
|
Marketable securities |
|
|
13,919 |
|
|
|
324,799 |
|
|
Property, plant and equipment, net |
|
|
181,900 |
|
|
|
114,572 |
|
|
Restricted investments |
|
|
30,000 |
|
|
|
150,000 |
|
|
Other assets |
|
|
4,171 |
|
|
|
1,562 |
|
|
Total assets |
|
$ |
1,994,730 |
|
|
$ |
1,262,810 |
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
28,355 |
|
|
$ |
54,465 |
|
|
Accrued expenses |
|
|
72,203 |
|
|
|
42,118 |
|
|
Deferred rent |
|
|
1,988 |
|
|
|
1,576 |
|
|
Deferred revenue |
|
|
41,705 |
|
|
|
33,540 |
|
|
Total current liabilities |
|
|
144,251 |
|
|
|
131,699 |
|
|
Deferred rent, net of current portion |
|
|
6,626 |
|
|
|
8,431 |
|
|
Deferred revenue, net of current portion |
|
|
43,075 |
|
|
|
49,392 |
|
|
Long-term debt |
|
|
30,000 |
|
|
|
150,000 |
|
|
Other liabilities |
|
|
4,347 |
|
|
|
3,067 |
|
|
Total liabilities |
|
|
228,299 |
|
|
|
342,589 |
|
|
Commitments and contingencies (Note 7) |
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Preferred stock, $0.01 par value per share, 5,000,000 shares authorized and no shares issued and outstanding at December 31, 2017 and 2016 |
|
|
— |
|
|
|
— |
|
|
Common stock, $0.01 par value per share, 125,000,000 shares authorized; 99,666,549 shares issued and outstanding at December 31, 2017; 85,941,344 shares issued and outstanding at December 31, 2016 |
|
|
997 |
|
|
|
859 |
|
|
Additional paid-in capital |
|
|
3,947,552 |
|
|
|
2,609,614 |
|
|
Accumulated other comprehensive loss |
|
|
(34,433 |
) |
|
|
(33,441 |
) |
|
Accumulated deficit |
|
|
(2,147,685 |
) |
|
|
(1,656,811 |
) |
|
Total stockholders’ equity |
|
|
1,766,431 |
|
|
|
920,221 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
1,994,730 |
|
|
$ |
1,262,810 |
|
The accompanying notes are an integral part of these consolidated financial statements.
90
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands, except per share amounts)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Net revenues from collaborators |
|
$ |
89,912 |
|
|
$ |
47,159 |
|
|
$ |
41,097 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Research and development(1) |
|
|
390,635 |
|
|
|
382,392 |
|
|
|
276,495 |
|
|
General and administrative(1) |
|
|
199,365 |
|
|
|
89,354 |
|
|
|
60,610 |
|
|
Total operating expenses |
|
|
590,000 |
|
|
|
471,746 |
|
|
|
337,105 |
|
|
Loss from operations |
|
|
(500,088 |
) |
|
|
(424,587 |
) |
|
|
(296,008 |
) |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
|
12,236 |
|
|
|
8,308 |
|
|
|
5,859 |
|
|
Other (expense) income |
|
|
(3,022 |
) |
|
|
6,171 |
|
|
|
76 |
|
|
Total other income |
|
|
9,214 |
|
|
|
14,479 |
|
|
|
5,935 |
|
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
Net loss per common share — basic and diluted |
|
$ |
(5.42 |
) |
|
$ |
(4.79 |
) |
|
$ |
(3.45 |
) |
|
Weighted-average common shares used to compute basic and diluted net loss per common share |
|
|
90,554 |
|
|
|
85,596 |
|
|
|
83,992 |
|
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
Unrealized loss on marketable securities, net of tax |
|
|
(2,886 |
) |
|
|
(30,833 |
) |
|
|
(44,394 |
) |
|
Reclassification adjustment for realized loss (gain) on marketable securities included in net loss |
|
|
1,894 |
|
|
|
(6,977 |
) |
|
|
— |
|
|
Comprehensive loss |
|
$ |
(491,866 |
) |
|
$ |
(447,918 |
) |
|
$ |
(334,467 |
) |
|
(1) |
Stock-based compensation expenses included in operating expenses are as follows: |
|
Research and development |
|
$ |
51,872 |
|
|
$ |
42,946 |
|
|
$ |
27,086 |
|
|
General and administrative |
|
|
40,947 |
|
|
|
32,582 |
|
|
|
18,697 |
|
The accompanying notes are an integral part of these consolidated financial statements.
91
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
(In thousands, except share amounts)
|
|
|
Common Stock |
|
|
Additional Paid-in |
|
|
Accumulated Other Comprehensive |
|
|
Accumulated |
|
|
Total Stockholders’ |
|
|||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Income (Loss) |
|
|
Deficit |
|
|
Equity |
|
||||||
|
Balance at December 31, 2014 |
|
|
77,202,753 |
|
|
$ |
772 |
|
|
$ |
1,843,362 |
|
|
$ |
48,763 |
|
|
$ |
(956,630 |
) |
|
$ |
936,267 |
|
|
Exercise of common stock options |
|
|
1,461,237 |
|
|
|
15 |
|
|
|
29,410 |
|
|
|
— |
|
|
|
— |
|
|
|
29,425 |
|
|
Issuance of common stock under other types of equity plans |
|
|
32,427 |
|
|
|
1 |
|
|
|
2,665 |
|
|
|
— |
|
|
|
— |
|
|
|
2,666 |
|
|
Issuance of common stock under equity plans, net of tax withholdings |
|
|
6,366 |
|
|
|
— |
|
|
|
(378 |
) |
|
|
— |
|
|
|
— |
|
|
|
(378 |
) |
|
Issuance of common stock to Sanofi Genzyme |
|
|
940,817 |
|
|
|
9 |
|
|
|
89,009 |
|
|
|
— |
|
|
|
— |
|
|
|
89,018 |
|
|
Issuance of common stock, net of offering costs |
|
|
5,447,368 |
|
|
|
54 |
|
|
|
496,346 |
|
|
|
— |
|
|
|
— |
|
|
|
496,400 |
|
|
Stock-based compensation expense related to equity-classified awards |
|
|
— |
|
|
|
— |
|
|
|
45,783 |
|
|
|
— |
|
|
|
— |
|
|
|
45,783 |
|
|
Other comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(44,394 |
) |
|
|
— |
|
|
|
(44,394 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(290,073 |
) |
|
|
(290,073 |
) |
|
Balance at December 31, 2015 |
|
|
85,090,968 |
|
|
|
851 |
|
|
|
2,506,197 |
|
|
|
4,369 |
|
|
|
(1,246,703 |
) |
|
|
1,264,714 |
|
|
Exercise of common stock options, net of tax withholdings |
|
|
559,344 |
|
|
|
5 |
|
|
|
11,603 |
|
|
|
— |
|
|
|
— |
|
|
|
11,608 |
|
|
Issuance of common stock under other types of equity plans |
|
|
75,453 |
|
|
|
1 |
|
|
|
3,647 |
|
|
|
— |
|
|
|
— |
|
|
|
3,648 |
|
|
Issuance of common stock under equity plans, net of tax withholdings |
|
|
10,549 |
|
|
|
— |
|
|
|
(314 |
) |
|
|
— |
|
|
|
— |
|
|
|
(314 |
) |
|
Issuance of common stock to Sanofi Genzyme |
|
|
205,030 |
|
|
|
2 |
|
|
|
14,299 |
|
|
|
— |
|
|
|
— |
|
|
|
14,301 |
|
|
Stock-based compensation expense related to equity-classified awards |
|
|
— |
|
|
|
— |
|
|
|
74,182 |
|
|
|
— |
|
|
|
— |
|
|
|
74,182 |
|
|
Other comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(37,810 |
) |
|
|
— |
|
|
|
(37,810 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
(410,108 |
) |
|
|
(410,108 |
) |
|
Balance at December 31, 2016 |
|
|
85,941,344 |
|
|
|
859 |
|
|
|
2,609,614 |
|
|
|
(33,441 |
) |
|
|
(1,656,811 |
) |
|
|
920,221 |
|
|
Exercise of common stock options |
|
|
1,841,566 |
|
|
|
19 |
|
|
|
80,527 |
|
|
|
— |
|
|
|
— |
|
|
|
80,546 |
|
|
Issuance of common stock under other types of equity plans |
|
|
134,615 |
|
|
|
2 |
|
|
|
5,905 |
|
|
|
— |
|
|
|
— |
|
|
|
5,907 |
|
|
Issuance of common stock under equity plans, net of tax withholdings |
|
|
11,523 |
|
|
|
— |
|
|
|
(707 |
) |
|
|
— |
|
|
|
— |
|
|
|
(707 |
) |
|
Issuance of common stock to Sanofi Genzyme |
|
|
297,501 |
|
|
|
3 |
|
|
|
21,378 |
|
|
|
— |
|
|
|
— |
|
|
|
21,381 |
|
|
Issuance of common stock, net of offering costs |
|
|
11,440,000 |
|
|
|
114 |
|
|
|
1,139,511 |
|
|
|
— |
|
|
|
— |
|
|
|
1,139,625 |
|
|
Stock-based compensation expense related to equity-classified awards |
|
|
— |
|
|
|
— |
|
|
|
91,324 |
|
|
|
— |
|
|
|
— |
|
|
|
91,324 |
|
|
Other comprehensive loss, net of tax |
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
(992 |
) |
|
|
— |
|
|
|
(992 |
) |
|
Net loss |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
(490,874 |
) |
|
|
(490,874 |
) |
|
Balance at December 31, 2017 |
|
|
99,666,549 |
|
|
$ |
997 |
|
|
$ |
3,947,552 |
|
|
$ |
(34,433 |
) |
|
$ |
(2,147,685 |
) |
|
$ |
1,766,431 |
|
The accompanying notes are an integral part of these consolidated financial statements.
92
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
13,367 |
|
|
|
15,130 |
|
|
|
19,050 |
|
|
Stock-based compensation |
|
|
92,819 |
|
|
|
75,528 |
|
|
|
45,783 |
|
|
Charge for 401(k) company stock match |
|
|
2,302 |
|
|
|
1,507 |
|
|
|
984 |
|
|
Realized loss (gain) on sale of marketable equity securities |
|
|
1,894 |
|
|
|
(6,977 |
) |
|
|
— |
|
|
Other |
|
|
608 |
|
|
|
— |
|
|
|
— |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from landlord tenant improvements |
|
|
— |
|
|
|
2,145 |
|
|
|
374 |
|
|
Billed and unbilled collaboration receivables |
|
|
(10,668 |
) |
|
|
(15,036 |
) |
|
|
31,639 |
|
|
Prepaid expenses and other assets |
|
|
(20,711 |
) |
|
|
(5,276 |
) |
|
|
(6,820 |
) |
|
Accounts payable |
|
|
(4,939 |
) |
|
|
10,098 |
|
|
|
1,676 |
|
|
Accrued expenses and other |
|
|
31,568 |
|
|
|
10,673 |
|
|
|
6,343 |
|
|
Deferred revenue |
|
|
1,848 |
|
|
|
14,615 |
|
|
|
1,904 |
|
|
Net cash used in operating activities |
|
|
(382,786 |
) |
|
|
(307,701 |
) |
|
|
(189,140 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Purchases of property, plant and equipment |
|
|
(104,209 |
) |
|
|
(64,557 |
) |
|
|
(12,950 |
) |
|
Purchases of restricted investments and increase in restricted cash |
|
|
— |
|
|
|
(150,000 |
) |
|
|
(1,471 |
) |
|
Purchases of marketable securities |
|
|
(903,457 |
) |
|
|
(759,310 |
) |
|
|
(1,033,843 |
) |
|
Sales and maturities of marketable securities |
|
|
597,305 |
|
|
|
1,116,458 |
|
|
|
726,943 |
|
|
Sale of restricted investments |
|
|
120,000 |
|
|
|
— |
|
|
|
— |
|
|
Net cash (used in) provided by investing activities |
|
|
(290,361 |
) |
|
|
142,591 |
|
|
|
(321,321 |
) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Proceeds from exercise of stock options and other types of equity |
|
|
84,142 |
|
|
|
14,127 |
|
|
|
31,137 |
|
|
Proceeds from issuance of common stock, net of offering costs |
|
|
1,139,625 |
|
|
|
— |
|
|
|
496,400 |
|
|
Proceeds from issuance of common stock to Sanofi Genzyme |
|
|
21,381 |
|
|
|
14,301 |
|
|
|
89,018 |
|
|
Proceeds from issuance of long-term debt |
|
|
— |
|
|
|
150,000 |
|
|
|
— |
|
|
Repayment of long-term debt |
|
|
(120,000 |
) |
|
|
— |
|
|
|
— |
|
|
Payments for repurchase of common stock for employee tax withholding |
|
|
(257 |
) |
|
|
(596 |
) |
|
|
(378 |
) |
|
Net cash provided by financing activities |
|
|
1,124,891 |
|
|
|
177,832 |
|
|
|
616,177 |
|
|
Net increase in cash and cash equivalents |
|
|
451,744 |
|
|
|
12,722 |
|
|
|
105,716 |
|
|
Cash and cash equivalents, beginning of period |
|
|
193,617 |
|
|
|
180,895 |
|
|
|
75,179 |
|
|
Cash and cash equivalents, end of period |
|
$ |
645,361 |
|
|
$ |
193,617 |
|
|
$ |
180,895 |
|
|
Supplemental disclosure of cash flows: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash paid for interest |
|
$ |
2,430 |
|
|
$ |
1,096 |
|
|
$ |
— |
|
|
Cash paid for income taxes |
|
$ |
114 |
|
|
$ |
111 |
|
|
$ |
66 |
|
|
Supplemental disclosure of noncash investing and financing activities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Fixed asset expenditures included in accounts payable and accrued expenses |
|
$ |
8,176 |
|
|
$ |
33,153 |
|
|
$ |
1,333 |
|
|
Fair value of common stock received for collaboration agreement in other assets |
|
$ |
2,700 |
|
|
$ |
— |
|
|
$ |
— |
|
|
Receipt of common stock for exercises of stock options |
|
$ |
653 |
|
|
$ |
1,260 |
|
|
$ |
686 |
|
|
Repurchase of common stock for employee tax withholding in accrued expenses |
|
$ |
450 |
|
|
$ |
— |
|
|
$ |
— |
|
The accompanying notes are an integral part of these consolidated financial statements.
93
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
|
1. |
NATURE OF BUSINESS |
We commenced operations on June 14, 2002 as a biopharmaceutical company seeking to develop and commercialize novel therapeutics based on RNA interference, or RNAi. We are focused on discovering, developing and commercializing RNAi therapeutics by establishing strategic alliances with leading pharmaceutical and life sciences companies, establishing and maintaining a strong intellectual property position in the RNAi field, generating revenues through licensing agreements, and ultimately developing and commercializing RNAi therapeutics globally. We have devoted substantially all of our efforts to business planning, research, development and early commercial efforts, acquiring, filing and expanding intellectual property rights, recruiting management and technical staff, and raising capital. In late 2017, we filed a new drug application, or NDA, and a marketing authorisation application, or MAA, seeking regulatory approval of our first product in the United States and Europe, respectively. If one or both of such applications is approved, we expect to begin commercializing and generating product revenues in 2018.
We are subject to risks common to companies in our industry including, but not limited to, uncertainties relating to conducting clinical research and development, the manufacture and supply of products for clinical and commercial use, obtaining and maintaining regulatory approvals and pricing and reimbursement for our products, market acceptance, managing global growth and operating expenses, availability of additional capital, competition, obtaining and enforcing patents, stock price volatility, dependence on collaborative relationships and third-party service providers, dependence on key personnel, potential litigation, product liability claims and government investigations.
|
2. |
SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements reflect the operations of Alnylam and our wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Concentrations of Credit Risk and Significant Customers
Financial instruments that potentially expose us to concentrations of credit risk consist primarily of cash, cash equivalents and fixed income marketable securities. At December 31, 2017 and 2016, substantially all of our cash, cash equivalents and fixed income marketable securities were invested in money market funds, certificates of deposit, commercial paper, corporate notes, U.S. government-sponsored enterprise securities and U.S. treasury securities through highly rated financial institutions. Corporate notes may also include foreign bonds denominated in U.S. dollars. Investments are restricted, in accordance with our investment policy, to a concentration limit per issuer.
In recent periods, our revenues from collaborations have been generated primarily from Sanofi Genzyme, the specialty care global business unit of Sanofi, The Medicines Company, or MDCO, Takeda Pharmaceutical Company Limited, or Takeda, and Monsanto Company, or Monsanto. For the year ended December 31, 2017, our billed and unbilled collaboration receivables were composed primarily of a milestone payment due from MDCO and expense reimbursement due from Sanofi Genzyme. For the year ended December 31, 2016, our billed and unbilled collaboration receivables were composed primarily of expense reimbursement due from Sanofi Genzyme.
94
The following table summarizes customers that represent greater than 10 percent of our net revenues from collaborators, for the periods indicated:
|
|
|
Year Ended December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Sanofi Genzyme |
|
|
61 |
% |
|
|
68 |
% |
|
|
27 |
% |
|
MDCO |
|
|
34 |
% |
|
|
24 |
% |
|
|
25 |
% |
|
Takeda |
|
* |
|
|
* |
|
|
|
22 |
% |
||
|
Monsanto |
|
* |
|
|
* |
|
|
|
14 |
% |
||
The following table summarizes customers with amounts due that represent greater than 10 percent of our billed and unbilled collaboration receivables balance, at the periods indicated:
|
|
|
At December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
MDCO |
|
|
59 |
% |
|
* |
|
|
|
Sanofi Genzyme |
|
|
39 |
% |
|
|
97 |
% |
|
* |
Represents 10 percent or less |
Fair Value Measurements
The fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In general, fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities. Fair values determined by Level 2 inputs utilize data points that are observable, such as quoted prices (adjusted), interest rates and yield curves. Fair values determined by Level 3 inputs utilize unobservable data points for the asset or liability, and include situations where there is little, if any, market activity for the asset or liability. The fair value hierarchy level is determined by the lowest level of significant input.
Investments in Marketable Securities and Cash Equivalents
We invest our excess cash balances in short-term and long-term marketable debt and equity securities. We classify our investments in marketable debt securities as either held-to-maturity or available-for-sale based on facts and circumstances present at the time we purchased the securities. At each balance sheet date presented, we classified all of our investments in debt and equity securities as available-for-sale. We report available-for-sale investments at fair value at each balance sheet date and include any unrealized holding gains and losses (the adjustment to fair value) in accumulated other comprehensive income (loss), a component of stockholders’ equity. At December 31, 2017, the balance in our accumulated other comprehensive loss was composed solely of activity related to our available-for-sale marketable securities, including our investment in equity securities of Regulus Therapeutics Inc., or Regulus. Realized gains and losses are determined using the specific identification method and are included in other income (expense). We recognized $1.9 million of realized losses and $7.0 million of realized gains from sales of our Regulus available-for-sale securities as other income (expense) in our consolidated statements of comprehensive loss during the years ended December 31, 2017 and 2016, respectively. If any adjustment to fair value reflects a decline in the value of the investment, we consider all available evidence to evaluate the extent to which the decline is “other than temporary,” including our intention to sell and, if so, mark the investment to market through a charge to our consolidated statements of comprehensive loss. We did not record any impairment charges related to our fixed income marketable securities during the years ended December 31, 2017, 2016 or 2015. Our marketable securities are classified as cash equivalents if the original maturity, from the date of purchase, is 90 days or less, and as marketable securities if the original maturity, from the date of purchase, is in excess of 90 days. Our cash equivalents are composed of commercial paper, corporate notes, U.S. government-sponsored enterprise securities, U.S. treasury securities and money market funds.
During the second quarter of 2017, we sold all our remaining holdings in Regulus. We accounted for our investment in Regulus as an available-for-sale marketable security. Intraperiod tax allocation rules require us to allocate our provision for income taxes between continuing operations and other categories of earnings, such as other comprehensive income. In periods in which we have a year-to-date pre-tax loss from continuing operations and pre-tax income in other categories of earnings, such as other comprehensive income, we must allocate the tax provision to the other categories of earnings. We then record a related tax benefit in continuing operations. Upon sales of our available-for-sale marketable securities, we apply the aggregate portfolio approach to recognize the related tax provision or benefit into income (loss) from continuing operations. As a result, the disproportionate tax effect remains in accumulated other comprehensive income (loss) as long as we maintain an investment portfolio.
95
Property, plant and equipment are stated at cost, net of accumulated depreciation. Depreciation expense is recorded on a straight-line basis over the estimated useful life of the asset. Leasehold improvements are amortized over the shorter of the asset’s estimated useful life or the lease term. Construction in progress reflects amounts incurred for construction or improvements of property, plant or equipment that have not been placed in service. The cost and accumulated depreciation of assets retired or sold are removed from the respective asset category, and any gain or loss is recognized in our consolidated statements of comprehensive loss.
The estimated useful lives of property, plant and equipment are as follows:
|
Asset Category |
|
Useful Life |
|
|
Laboratory equipment |
|
5 years |
|
|
Computer equipment and software |
|
3-10 years |
|
|
Furniture and fixtures |
|
5 years |
|
|
Leasehold improvements |
|
Shorter of asset life or lease term |
|
|
Land |
|
|
— |
|
Construction in progress |
|
|
— |
Estimated Liability for Development Costs
We record accrued liabilities related to expenses for which service providers have not yet billed us with respect to products we have received or services that we have incurred, specifically related to ongoing pre-clinical studies and clinical trials. These costs primarily relate to third-party clinical management costs, laboratory and analysis costs, toxicology studies and investigator fees. We have multiple product candidates in concurrent pre-clinical studies and clinical trials at multiple clinical sites throughout the world. In order to ensure that we have adequately provided for ongoing pre-clinical and clinical development costs during the period in which we incur such costs, we maintain an accrual to cover these expenses. We update the estimate for this accrual on at least a quarterly basis. The assessment of these costs is a subjective process that requires judgment. Upon settlement, these costs may differ materially from the amounts accrued in our consolidated financial statements. Our historical accrual estimates have not been materially different from our actual costs.
Revenue Recognition
We have entered into collaboration agreements with leading pharmaceutical and life sciences companies, including Takeda, Kyowa Hakko Kirin Co., Ltd., or Kyowa Hakko Kirin, Monsanto, Sanofi Genzyme and MDCO. The terms of our collaboration agreements typically include deliverables such as non-refundable license fees, funding of research and development, payments based upon achievement of clinical and pre-clinical development milestones, regulatory milestones, manufacturing services, sales milestones and royalties on product sales. These agreements are generally referred to as multiple element arrangements.
We apply the accounting standard on revenue recognition for multiple element arrangements. The fair value of deliverables under the arrangement may be derived using a “best estimate of selling price” if vendor specific objective evidence and third-party evidence is not available. Deliverables under the arrangement will be separate units of accounting provided that (i) a delivered item has value to the customer on a standalone basis and (ii) if the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered item is considered probable and substantially in the control of the vendor.
We recognize upfront license payments as revenue upon delivery of the license only if the license has standalone value of the undelivered performance obligations, typically including research and/or steering committee services, that can be determined. If the fair value of the undelivered performance obligations can be determined, such obligations would then be accounted for separately as performed. If the license is considered to not have standalone value, the arrangement would then be accounted for as a single unit of accounting and the license payments and payments for performance obligations are recognized as revenue over the estimated period of when the performance obligations are performed or deferred until the undelivered performance obligation can be determined. As a biotechnology entity with unique and specialized delivered and undelivered performance obligations, we have been unable to demonstrate standalone value in our multiple element arrangements.
Whenever we determine that an arrangement should be accounted for as a single unit of accounting, we determine the period over which the performance obligations will be performed and revenue will be recognized. Revenue is recognized using either proportional performance or a straight-line method. We recognize revenue using the proportional performance method when the level of effort required to complete our performance obligations under an arrangement can be reasonably estimated and such performance obligations are provided on a best-efforts basis. Direct labor hours or full-time equivalents are typically used as the measure of performance. The amount of revenue recognized under the proportional performance method is determined by multiplying the total
96
payments under the contract, excluding royalties and payments contingent upon achievement of milestones, by the ratio of level of effort incurred to date to estimated total level of effort required to complete our performance obligations under the arrangement. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the proportional performance method, as of the period ending date.
If we cannot reasonably estimate the level of effort to complete our performance obligations under an arrangement, we recognize revenue under the arrangement on a straight-line basis over the period we are expected to complete our performance obligations. Revenue is limited to the lesser of the cumulative amount of payments received or the cumulative amount of revenue earned, as determined using the straight-line method, as of the period ending date.
Significant management judgment is required in determining the level of effort required under an arrangement and the period over which we are expected to complete our performance obligations under an arrangement. Steering committee services that are not inconsequential or perfunctory and that are determined to be performance obligations are combined with other research services or performance obligations required under an arrangement, if any, in determining the level of effort required in an arrangement and the period over which we expect to complete our aggregate performance obligations.
Many of our collaboration agreements entitle us to additional payments upon the achievement of performance-based milestones. These milestones are generally categorized into three types; development milestones which are generally based on the advancement of our pipeline and initiation of clinical trials, regulatory milestones which are generally based on the submission, filing or approval of regulatory applications such as an NDA in the United States, and commercialization milestones which are generally based on meeting specific thresholds of sales in certain geographic areas. If the achievement of a milestone is considered probable at the inception of the collaboration, the related milestone payment is included with other collaboration consideration, such as upfront fees and research funding, in our revenue model. Milestones that are tied to regulatory approval are not considered probable of being achieved until such approval is received. Milestones tied to counter-party performance are not included in our revenue model until the performance conditions are met. Upfront and ongoing development milestones per our collaboration agreements are not subject to refund if the development activities are not successful.
We perform an assessment to determine whether a substantive milestone exists at the inception of our collaborative arrangements. In evaluating if a milestone is substantive, we consider whether uncertainty exists as to the achievement of the milestone event at the inception of the arrangement, the achievement of the milestone involves substantive effort and can only be achieved based in whole or part on the performance or the occurrence of a specific outcome resulting from our performance, the amount of the milestone payment appears reasonable either in relation to the effort expected to be expended or to the projected enhancement of the value of the delivered items, there is any future performance required to earn the milestone, and the consideration is reasonable relative to all deliverables and payment terms in the arrangement. When a substantive milestone is achieved, the accounting rules permit us to recognize revenue related to the milestone payment in its entirety.
To date, we have not recorded any substantive milestones under our collaborations because we have not identified any milestones that meet the required criteria listed above. We have deferred recognition of payments for achievement of non-substantive milestones and recognized revenue over the estimated period of performance applicable with each collaborative arrangement. As these milestones are achieved, we will recognize as revenue a portion of the milestone payment, which is equal to the percentage of the performance period completed when the milestone is achieved, multiplied by the amount of the milestone payment, upon achievement of such milestone. We will recognize the remaining portion of the milestone payment over the remaining performance period under the proportional performance method or on a straight-line basis.
For revenue generating arrangements where we, as a vendor, provide consideration to a licensor or collaborator, as a customer, we apply the accounting standard that governs such transactions. This standard addresses the accounting for revenue arrangements where both the vendor and the customer make cash payments to each other for services and/or products. A payment to a customer is presumed to be a reduction of the selling price unless we receive an identifiable benefit for the payment and it can reasonably estimate the fair value of the benefit received. Payments to a customer that are deemed a reduction of selling price are recorded first as a reduction of revenue, to the extent of both cumulative revenue recorded to date and probable future revenues, which include any unamortized deferred revenue balances, under all arrangements with such customer, and then as an expense. Payments that are not deemed to be a reduction of selling price are recorded as an expense.
We evaluate our collaborative agreements for proper classification in our consolidated statements of comprehensive loss based on the nature of the underlying activity. Transactions between collaborators recorded in our consolidated statements of comprehensive loss are recorded on either a gross or net basis, depending on the characteristics of the collaborative relationship. We generally reflect amounts due under our collaborative agreements related to cost-sharing of development activities as revenue.
97
Amounts received prior to satisfying the above revenue recognition criteria are recorded as deferred revenue in the accompanying consolidated balance sheets. Although we follow detailed guidelines in measuring revenue, certain judgments affect the application of our revenue policy. For example, in connection with our existing collaboration agreements, we have recorded on our consolidated balance sheet short-term and long-term deferred revenue based on our best estimate of when such revenue will be recognized. Short-term deferred revenue consists of amounts that are expected to be recognized as revenue in the next 12 months. Amounts that we expect will not be recognized within the next 12 months are classified as long-term deferred revenue. However, this estimate is based on our current operating plan and, if our operating plan should change in the future, we may recognize a different amount of deferred revenue over the next 12-month period.
The estimate of deferred revenue also reflects management’s estimate of the periods of our involvement in certain of our collaborations. Our performance obligations under these collaborations consist of participation on steering committees and the performance of other research and development services. In certain instances, the timing of satisfying these obligations can be difficult to estimate. Accordingly, our estimates may change in the future. Such changes to estimates would result in a change in revenue recognition amounts. If these estimates and judgments change over the course of these agreements, it may affect the timing and amount of revenue that we recognize and record in future periods. At December 31, 2017, we had short-term and long-term deferred revenue of $41.7 million and $43.1 million, respectively, related to our collaborations. The new accounting standard related to revenue recognition effective as of January 1, 2018, discussed below under the heading “Recent Accounting Pronouncements,” will have a material impact on our consolidated financial statements.
Income Taxes
We use the asset and liability method of accounting for income taxes. Under the asset and liability method, deferred tax assets and liabilities reflect the impact of temporary differences between amounts of assets and liabilities for financial reporting purposes and such amounts as measured under enacted tax laws. A valuation allowance is required to offset any net deferred tax assets if, based upon the available evidence, it is more likely than not that some or all of the deferred tax asset will not be realized.
We account for uncertain tax positions using a “more-likely-than-not” threshold for recognizing and resolving uncertain tax positions. The evaluation of uncertain tax positions is based on factors that include, but are not limited to, changes in tax law, the measurement of tax positions taken or expected to be taken in tax returns, the effective settlement of matters subject to audit, new audit activity and changes in facts or circumstances related to a tax position. Our policy is to accrue interest and penalties related to unrecognized tax positions in income tax expense. As of December 31, 2017, we have not recorded significant interest and penalty expense related to uncertain tax positions.
Research and Development Expenses
We record research and development expenses as incurred. Included in research and development expenses are wages, stock-based compensation expenses, benefits and other operating costs, facilities, supplies, external services, clinical trial and manufacturing costs, and overhead directly related to our research and development operations, as well as costs to acquire technology licenses.
We have entered into several license agreements for rights to utilize certain technologies. The terms of the licenses may provide for upfront payments, annual maintenance payments, milestone payments based upon certain specified events being achieved and royalties on product sales. We charge costs to acquire and maintain licensed technology that has not reached technological feasibility and does not have alternative future use to research and development expense as incurred. During the years ended December 31, 2017, 2016 and 2015, we charged to research and development expense costs associated with license fees of $7.7 million, $2.4 million and $3.5 million, respectively.
Stock-Based Compensation
We have stock incentive plans and an employee stock purchase plan under which we grant equity instruments. We may also grant inducement stock grants outside of our stock incentive plans. We account for all stock-based awards granted to employees at their fair value and generally recognize compensation expense over the vesting period of the award. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of fair values of stock options as of the grant date. We calculate the grant date fair values of stock options using the Black-Scholes valuation model. Our expected stock price volatility assumption is based on the historical volatility of our publicly traded stock.
The fair value of restricted stock awards granted to employees is based upon the quoted closing market price per share on the date of grant. Expense for time-based restricted stock awards is recognized over the vesting period.
98
For performance-based stock option and restricted stock awards, we begin to recognize expense when we determine that the achievement of such performance conditions is deemed probable. This determination requires significant judgment by management. At the probable date, we record a cumulative expense catch-up, with remaining expense amortized over the remaining service period.
Comprehensive Loss
Comprehensive loss is comprised of net loss and certain changes in stockholders’ equity that are excluded from net loss. We include foreign currency translation adjustments in other comprehensive loss if the functional currency is not the United States dollar. We include unrealized gains and losses on certain marketable securities in other comprehensive loss.
Net Loss Per Common Share
We compute basic net loss per common share by dividing net loss by the weighted-average number of common shares outstanding. We compute diluted net loss per common share by dividing net loss by the weighted-average number of common shares and dilutive potential common share equivalents then outstanding. Potential common shares consist of shares issuable upon the exercise of stock options (the proceeds of which are then assumed to have been used to repurchase outstanding shares using the treasury stock method). Because the inclusion of potential common shares would be anti-dilutive for all periods presented, diluted net loss per common share is the same as basic net loss per common share.
The following table sets forth for the periods presented the potential common shares (prior to consideration of the treasury stock method) excluded from the calculation of net loss per common share because their inclusion would be anti-dilutive, in thousands:
|
|
|
At December 31, |
|
|||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Options to purchase common stock |
|
|
11,239 |
|
|
|
12,270 |
|
|
|
9,960 |
|
|
Unvested restricted common stock |
|
149 |
|
|
171 |
|
|
|
19 |
|
||
|
|
|
|
11,388 |
|
|
|
12,441 |
|
|
|
9,979 |
|
Segment Information
We operate in a single reporting segment, the discovery, development and commercialization of RNAi therapeutics.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued a new revenue recognition standard which amends revenue recognition principles and provides a single, comprehensive set of criteria for revenue recognition within and across all industries. The new standard provides a five step framework whereby revenue is recognized when control of promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. In August 2015, the FASB deferred the effective date of the new revenue standard from January 1, 2017 to January 1, 2018. In March 2016, the FASB issued amendments to clarify the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued amendments to clarify the guidance on accounting for licenses of intellectual property and identifying performance obligations. In May 2016, the FASB issued amendments related to collectibility, non-cash consideration, the presentation of sales and other similar taxes collected from customers and transition. The standard allows for adoption using a full retrospective method or a modified retrospective method. On January 1, 2018, we adopted this standard using the modified retrospective method. Our implementation approach included performing a detailed review of our collaboration agreements. In addition, we designed internal controls to enable the preparation of financial information and have reached conclusions on key accounting assessments related to the new standard, including our assessment that the impact of accounting for costs incurred to obtain a contract is immaterial. We completed our assessment to quantify the expected impact that the new standard will have on our consolidated financial statements and related disclosures. We expect the adoption of the new standard to result in a cumulative reduction of $68.3 million of deferred revenue with a corresponding adjustment to the opening balance of accumulated deficit to be recorded in the first quarter of 2018. This adjustment is due primarily to the application of the new standard to our collaboration agreements with Sanofi Genzyme, MDCO and Kyowa Hakko Kirin. A substantial portion of the incremental $68.3 million adjustment is the result of the application of the new guidance regarding how entities should measure progress in satisfying performance obligations and the contract’s transaction price. In addition, as a result of the cumulative reduction in deferred revenue, our corresponding deferred tax asset will be reduced by approximately $13.6 million, which will be offset by a corresponding decrease to our valuation allowance. These offsetting adjustments will be recorded to accumulated deficit in the first quarter of 2018. We do not expect an impact to cash from or used in operating, financing or investing on our consolidated statement of cash flows as a result of the adoption of the new standard.
99
In January 2016, the FASB issued new guidance on recognition and measurement of financial assets and financial liabilities. The new guidance will impact the accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. All equity investments in unconsolidated entities (other than those accounted for under the equity method of accounting) will generally be measured at fair value with changes in fair value recognized through earnings. There will no longer be an available-for-sale classification (changes in fair value reported in other comprehensive income (loss)) for equity securities with readily determinable fair values. In addition, the FASB clarified the need for a valuation allowance on deferred tax assets resulting from unrealized losses on available-for-sale debt securities. In general, the new guidance will require modified retrospective application to all outstanding instruments, with a cumulative effect adjustment recorded to opening retained earnings. This guidance became effective for us on January 1, 2018. We currently do not expect this guidance to have a significant impact on our consolidated financial statements and related disclosures.
In February 2016, the FASB issued a new leasing standard that requires that all lessees recognize the assets and liabilities that arise from leases on the consolidated balance sheet and disclose qualitative and quantitative information about its leasing arrangements. The new standard will be effective for us on January 1, 2019. Early adoption is permitted. We are currently evaluating the timing of our adoption and the expected impact that this standard could have on our consolidated financial statements and related disclosures.
In October 2016, the FASB issued guidance that an entity should recognize the income tax consequences of an intra-entity transfer of an asset other than inventory when the transfer occurs instead of deferring the income tax effects. The new guidance became effective for us on a modified retrospective basis on January 1, 2018. The adoption of this guidance did not have an impact on our consolidated financial statements and related disclosures.
In November 2016, the FASB issued guidance that requires restricted cash and restricted cash equivalents be included with cash and cash equivalents when reconciling the total beginning and ending amounts for the periods shown on the consolidated statement of cash flows. The new standard became effective for us on January 1, 2018 using a retrospective transition method for each period presented. For the years ended December 31, 2017 and 2016, our restricted cash and restricted cash equivalents were not significant. We currently do not expect this guidance to have a significant impact on our consolidated financial statements and related disclosures.
In March 2017, the FASB issued guidance that amends the amortization period for certain purchased callable debt securities held at a premium by shortening the amortization period for the premium to the earliest call date. The new standard will be effective for us on January 1, 2019. Early adoption is permitted. We are currently evaluating the timing of our adoption and the expected impact that this standard could have on our consolidated financial statements and related disclosures.
Subsequent Events
We did not have any material recognized subsequent events. However, we did have a nonrecognized subsequent event with respect to our collaboration with Sanofi Genzyme, which is more fully described in Note 3.
|
3. |
SIGNIFICANT AGREEMENTS |
The following table summarizes our total consolidated net revenues from collaborators, for the periods indicated, in thousands:
|
|
|
Year Ended December 31, |
|||||||||||
|
Description |
|
2017 |
|
|
2016 |
|
|
2015 |
|
|
|||
|
Sanofi Genzyme |
|
$ |
54,625 |
|
|
$ |
32,015 |
|
|
$ |
11,005 |
|
|
|
MDCO |
|
|
30,217 |
|
|
|
11,220 |
|
|
|
10,301 |
|
|
|
Takeda |
|
|
— |
|
|
|
— |
|
|
|
8,867 |
|
|
|
Monsanto |
|
|
2,500 |
|
|
|
— |
|
|
|
5,621 |
|
|
|
Other |
|
|
2,570 |
|
|
|
3,924 |
|
|
|
5,303 |
|
|
|
Total net revenues from collaborators |
|
$ |
89,912 |
|
|
$ |
47,159 |
|
|
$ |
41,097 |
|
|
100
The following table provides the research and development expenses incurred by type that are directly attributable to each significant agreement for the periods indicated, in thousands:
|
|
|
Year Ended December 31, |
|
|||||||||||||||||||||||||||||||||
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||||||||||||||||||||||||||
|
|
|
Sanofi Genzyme |
|
|
MDCO |
|
|
Ionis |
|
|
Sanofi Genzyme |
|
|
MDCO |
|
|
Ionis |
|
|
Sanofi Genzyme |
|
|
MDCO |
|
|
Ionis |
|
|||||||||
|
Research and development |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Clinical trial and manufacturing |
|
$ |
174,901 |
|
|
$ |
5,421 |
|
|
$ |
— |
|
|
$ |
150,179 |
|
|
$ |
1,211 |
|
|
$ |
— |
|
|
$ |
108,903 |
|
|
$ |
3,308 |
|
|
$ |
— |
|
|
External services |
|
|
4,475 |
|
|
|
— |
|
|
|
3,250 |
|
|
|
7,366 |
|
|
|
— |
|
|
|
— |
|
|
|
3,151 |
|
|
|
770 |
|
|
|
— |
|
|
Other |
|
|
5,327 |
|
|
|
106 |
|
|
|
— |
|
|
|
3,035 |
|
|
|
64 |
|
|
|
525 |
|
|
|
3,714 |
|
|
|
497 |
|
|
|
3,300 |
|
|
Total research and development expenses |
|
$ |
184,703 |
|
|
$ |
5,527 |
|
|
$ |
3,250 |
|
|
$ |
160,580 |
|
|
$ |
1,275 |
|
|
$ |
525 |
|
|
$ |
115,768 |
|
|
$ |
4,575 |
|
|
$ |
3,300 |
|
The research and development expenses incurred for each significant agreement consist of costs incurred for external development and manufacturing services for which we are reimbursed and licensing payments made to the counterparty to such agreement. In addition, these expenses include a reasonable estimate of compensation and related costs as billed to our counterparties. There were no material research and development expenses incurred for Monsanto or Takeda during the years ended December 31, 2017, 2016 and 2015. For the years ended December 31, 2017, 2016 and 2015, we did not incur material general and administrative expenses related to our significant agreements.
Product Alliances
Sanofi Genzyme Collaboration
In January 2014, we entered into a global, strategic collaboration with Sanofi Genzyme to discover, develop and commercialize RNAi therapeutics as Genetic Medicines to treat orphan diseases, referred to as the 2014 Sanofi Genzyme collaboration. The 2014 Sanofi Genzyme collaboration superseded and replaced the previous collaboration between us and Sanofi Genzyme entered into in October 2012 to develop and commercialize RNAi therapeutics targeting transthyretin, or TTR, for the treatment of hereditary ATTR amyloidosis, including patisiran and revusiran, in Japan and the Asia-Pacific region.
On January 6, 2018, we and Sanofi Genzyme entered into an amendment to our 2014 Sanofi Genzyme collaboration. In connection and simultaneously with entering into the amendment to the 2014 Sanofi Genzyme collaboration, we and Sanofi Genzyme also entered into an Exclusive License Agreement with respect to all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, referred to as the Exclusive TTR License, and the ALN-AT3 Global License Terms with respect to fitusiran and any back-up products, referred to as the AT3 License Terms. As a result, we will have the exclusive right to pursue the further global development and commercialization of all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, and Sanofi Genzyme will have the exclusive right to pursue the further global development and commercialization of fitusiran and any back-up products.
The January 2018 transaction is subject to customary closing conditions and clearances, including clearance under the Hart-Scott-Rodino Antitrust Improvements Act. We expect the transaction to close during the first quarter of 2018.
2012 Sanofi Genzyme Agreement
Under the 2012 Sanofi Genzyme agreement, Sanofi Genzyme paid us an upfront cash payment of $22.5 million. We were also entitled to receive certain milestone payments under the 2012 Sanofi Genzyme agreement. In the fourth quarter of 2013, we earned $11.0 million in patisiran development milestones under the 2012 Sanofi Genzyme agreement.
We determined that the deliverables under the 2012 Sanofi Genzyme agreement included the license, a joint steering committee and any additional TTR-specific RNAi therapeutic compounds that comprised the ALN-TTR program. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered joint steering committee and any additional TTR-specific RNAi therapeutic compounds did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme had the ability to grant sublicenses, it could not sublicense all or substantially all of its rights under the 2012 Sanofi Genzyme agreement. The uniqueness of our services and the limited sublicense right were indicators that standalone value was not present in the arrangement. Therefore the deliverables were not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. We were unable to reasonably estimate the period of performance under the 2012 Sanofi Genzyme agreement, as we were unable to estimate the timeline
101
of our deliverables related to the deliverable for any additional TTR-specific RNAi therapeutic compounds. Through December 31, 2013, we had deferred all revenue, or $33.5 million, under the 2012 Sanofi Genzyme agreement.
2014 Sanofi Genzyme Collaboration, as amended in January 2018
In January 2014, we entered into the 2014 Sanofi Genzyme collaboration. As noted above, the 2014 Sanofi Genzyme collaboration superseded and replaced the 2012 Sanofi Genzyme agreement and was amended in January 2018, at which time we also entered into the Exclusive TTR License and the AT3 License Terms.
The 2014 Sanofi Genzyme collaboration is structured as an exclusive relationship for the worldwide development and commercialization of RNAi therapeutics in the field of Genetic Medicines, which includes our current and future Genetic Medicine programs that reach Human Proof-of-Principle Study Completion (as defined in the Sanofi Genzyme master agreement), or Human POP, by the end of 2019, subject to extension to the end of 2021 in various circumstances. We will retain product rights in the United States, Canada and Western Europe, referred to as the Alnylam Territory, while Sanofi Genzyme will obtain exclusive rights to develop and commercialize collaboration products in the rest of the world, referred to as the Sanofi Genzyme Territory, together with worldwide rights for one product. Sanofi Genzyme’s rights under the 2014 Sanofi Genzyme collaboration, described in detail below, are structured as an opt-in that is triggered upon achievement of Human POP. We maintain development control for all programs prior to Sanofi Genzyme’s opt-in and maintain development and commercialization control after Sanofi Genzyme’s opt-in for all programs in the Alnylam Territory. We will retain global rights to any RNAi therapeutic Genetic Medicine program that does not reach Human POP by the end of 2019, subject to certain limited exceptions. We retain full rights to all current and future RNAi therapeutic programs outside of the field of Genetic Medicines, including the right to form new collaborations.
Under the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme’s specific license rights and the programs which Sanofi Genzyme opted into prior to the 2018 amendment include the following:
|
|
• |
Regional license terms and programs — Upon opt-in, we will retain product rights in the Alnylam Territory, while Sanofi Genzyme will obtain exclusive rights to develop and commercialize the product in the Sanofi Genzyme Territory. Sanofi Genzyme can elect this license for any of our current and future Genetic Medicine programs that complete Human POP by the end of 2019, subject to limited extension. Development costs for products once Sanofi Genzyme exercises an option will be shared between Sanofi Genzyme and us, with Sanofi Genzyme responsible for twenty percent of the global development costs. Sanofi Genzyme will be required to make payments totaling up to $75.0 million per regional product, consisting of up to $55.0 million in development milestones and $20.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for each regional product based on annual net sales, if any, of such regional product by Sanofi Genzyme, its affiliates and sublicensees. Upon the effective date of the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme expanded the scope of its regional license and collaboration for patisiran, which was originally established under the 2012 Sanofi Genzyme agreement. In September 2015, Sanofi Genzyme elected to opt into our fitusiran clinical development program for the treatment of hemophilia and other rare bleeding disorders under the regional license terms. Cost-sharing for the fitusiran program began in January 2016 under the regional license terms. Sanofi Genzyme also had the right to elect to co-develop and co-commercialize fitusiran in the Alnylam Territory pursuant to the co-development/co-commercialize license terms described below. In November 2016, Sanofi Genzyme exercised this right and elected to co-develop and co-commercialize fitusiran in the Alnylam Territory. In addition, during 2016, Sanofi Genzyme elected not to opt into the development and commercialization of givosiran or cemdisiran in the Sanofi Genzyme Territory. |
Sanofi Genzyme’s rights with respect to patisiran and fitusiran will be modified in connection with the 2018 amendment, the Exclusive TTR License and the AT3 License Terms, as described below. Sanofi Genzyme continues to have the right to opt into our future rare genetic disease programs for development and commercialization in the Sanofi Genzyme Territory under the regional license terms.
102
|
|
Genzyme paid us approximately $6.0 million in January 2017 for its incremental share of co-development costs incurred from January 2016 through September 2016. Sanofi Genzyme was required to make certain milestone payments for fitusiran, and, prior to the discontinuation of the revusiran program, was required to make certain milestone payments for revusiran. In December 2014, we earned a development milestone payment of $25.0 million based upon the initiation of the first global Phase 3 clinical trial for revusiran. Sanofi Genzyme was also obligated to pay us a milestone of $25.0 million upon the dosing of the first patient in our ATLAS Phase 3 program for fitusiran. In addition, Sanofi Genzyme was required to pay tiered double-digit royalties up to twenty percent for each co-development/co-commercialize product based on annual net sales, if any, in the Sanofi Genzyme Territory for such co-development/co-commercialize product by Sanofi Genzyme, its affiliates and sublicensees. The parties were to share profits equally and we expected to book product sales in the Alnylam Territory. |
In connection with the 2018 amendment, the Exclusive TTR License and the AT3 License Terms, as described below, we and Sanofi Genzyme agreed to terminate the co-development and co-commercialization rights related to revusiran, ALN-TTRsc02 and fitusiran under the 2014 Sanofi Genzyme collaboration. No future rights will be granted to Sanofi Genzyme for co-development and co-commercialization under the 2014 Sanofi Genzyme collaboration, as amended by the 2018 amendment.
|
|
• |
Global license terms and programs — Sanofi Genzyme continues to have one right to a global license through 2019, subject to limited extension, for a future Genetic Medicine program that was not one of our defined Genetic Medicine programs as of the effective date of the 2014 Sanofi Genzyme collaboration. Sanofi Genzyme could elect its global license for lumasiran. Upon opt-in, Sanofi Genzyme will obtain a worldwide license to develop and commercialize the product. Sanofi Genzyme shall be responsible for one hundred percent of global development costs for a global license product. Sanofi Genzyme will be required to make payments totaling up to $200.0 million for such global product, including up to $100.0 million in development milestones and $100.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for such global product based on annual net sales, if any, of such global product by Sanofi Genzyme, its affiliates and sublicensees. |
Exclusive TTR License and AT3 License Terms
As noted above, the 2018 amendment, together with the Exclusive TTR License and the AT3 License Terms, revise the terms and conditions of the 2014 Sanofi Genzyme collaboration to (i) provide us the exclusive right to pursue the further global development and commercialization of all TTR products, including patisiran, ALN-TTRsc02 and any back-up products, (ii) provide Sanofi Genzyme the exclusive right to pursue the further global development and commercialization of fitusiran and any back-up products and (iii) terminate the previous co-development and co-commercialization rights related to revusiran, ALN-TTRsc02 and fitusiran under the 2014 Sanofi Genzyme collaboration. Going forward, we will fund all development and commercialization costs for patisiran and ALN-TTRsc02. We also will fund development and commercialization costs for fitusiran through the transition period, up to a cap of $50.0 million, after which Sanofi Genzyme will fund all development and commercialization costs for fitusiran. We expect to substantially complete the transition of the fitusiran program to Sanofi Genzyme by mid-2018. Each party will be responsible for its costs associated with the transfer of the respective program to the other party.
Under the 2018 amendment and the Exclusive TTR License, Sanofi Genzyme will be eligible to receive (i) royalties up to twenty-five percent, increasing over time, based on annual net sales of patisiran in territories excluding the United States, Canada and Western Europe, provided royalties on annual net sales of patisiran in Japan will be twenty-five percent beginning as of the effective date of the Exclusive TTR License, (ii) tiered royalties of fifteen to thirty percent based on global annual net sales of ALN-TTRsc02 (consistent with the royalties due to us from Sanofi Genzyme on fitusiran), and (iii) tiered royalties of up to fifteen percent based on global annual net sales of any back-up products, in each case by us, our affiliates and our sublicensees. Except as described below, there will be no additional milestones due to either party with respect to patisiran, ALN-TTRsc02 or fitusiran.
In consideration for the rights granted to Sanofi Genzyme under the 2018 amendment and the AT3 License Terms, Sanofi Genzyme is required to make one milestone payment of $50.0 million following the dosing of the first patient in the ATLAS Phase 3 program for fitusiran. In addition, we will be eligible to receive tiered royalties of fifteen to thirty percent based on global annual net sales of fitusiran and up to fifteen percent based on global annual net sales of any back-up products, in each case by Sanofi Genzyme, its affiliates and its sublicensees. We and Sanofi Genzyme intend to enter into a supply agreement to provide for the supply of fitusiran to Sanofi Genzyme for ongoing clinical studies, and, at Sanofi Genzyme’s request, commercial sales. Sanofi Genzyme also has the right to manufacture fitusiran.
Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Sanofi Genzyme under the 2014 Sanofi Genzyme collaboration, as amended, or the AT3 License Terms.
103
The 2014 Sanofi Genzyme collaboration, as amended, will continue to be governed by an alliance joint steering committee that is comprised of an equal number of representatives from each party. There are additional committees to manage various aspects of each regional and global program and to oversee certain matters, including transition planning, that may arise under the Exclusive TTR License and the AT3 License Terms.
The original master agreement (including the license terms appended thereto), as well as the Exclusive TTR License and the AT3 License Terms, contain certain termination provisions, including for material breach by the other party. In addition, we have the right to terminate Exclusive TTR License without cause with respect to any or all licensed products at any time upon six months’ prior written notice and Sanofi Genzyme has the right to terminate the AT3 License Terms without cause with respect to any particular licensed product at any time upon six months’ prior written notice.
Unless terminated earlier pursuant to its terms, the master agreement will terminate upon the last to expire of any of the option periods under the master agreement or the license terms appended thereto. The term of the Exclusive TTR License expires on a licensed product-by-licensed product and country-by-country basis upon expiration of the last royalty term to expire under the agreement, where a royalty term is defined as the latest to occur of (a) expiration of the last valid claim of patent rights covering a licensed product; (b) the expiration of Regulatory Exclusivity for a licensed product, as defined in the Exclusive TTR License; or (c) the twelfth anniversary of the first commercial sale of the licensed product in such country. The term of the AT3 License Terms expires on a licensed product-by-licensed product and country-by-country basis upon expiration of the last royalty term to expire under the agreement, where a royalty term is defined as the latest to occur of (x) the expiration of the last valid claim of patent rights covering a licensed product; (y) the expiration of Regulatory Exclusivity for a licensed product, as defined in the AT3 License Terms; or (z) the twelfth anniversary of the first commercial sale of the licensed product in such country.
Upon the closing of the equity transaction in February 2014, we sold to Sanofi Genzyme 8,766,338 shares of our common stock and Sanofi Genzyme paid $700.0 million in aggregate cash consideration to us. As a condition to the closing of the equity transaction, Sanofi Genzyme entered into an investor agreement with us. Under the investor agreement, until the earlier of the fifth anniversary of the expiration or earlier termination of the 2014 Sanofi Genzyme collaboration and the date on which Sanofi Genzyme and its affiliates cease to beneficially own at least 5 percent of our outstanding common stock, Sanofi Genzyme and its affiliates are bound by certain “standstill” provisions. The standstill provisions include agreements not to acquire more than 30 percent of our outstanding common stock, call stockholder meetings, nominate directors other than those approved by our board of directors, subject to certain limited exceptions, or propose or support a proposal to acquire us. Further, Sanofi Genzyme has agreed to vote, and cause its affiliates to vote, all shares of our voting securities they are entitled to vote, up to a maximum of 20 percent of our outstanding common stock, in a manner either as recommended by our board of directors or proportionally with the votes cast by our other stockholders, except with respect to certain change of control transactions or our liquidation or dissolution. Until Sanofi Genzyme owns less than 7.5 percent of our outstanding common stock, subject to Sanofi Genzyme’s limited right to maintain its ownership percentage as described below, if we issue common stock or securities convertible into or exercisable for common stock to a third party that holds at least 30 percent of our outstanding common stock or, in connection with a collaboration or license transaction, to a third party that will initially hold at least the percentage of our outstanding common stock represented by the shares purchased by Sanofi Genzyme at the closing of the equity transaction, we will offer Sanofi Genzyme an opportunity to amend the standstill and voting provisions in the investor agreement to be consistent with the terms provided to such third party.
Under the investor agreement, Sanofi Genzyme has also agreed not to dispose of any shares of common stock beneficially owned by it immediately after the closing of the stock purchase until the earlier of (i) December 31, 2019 (subject to extension by up to two years if Sanofi Genzyme’s option to select additional compounds under the master agreement is extended beyond December 31, 2019) and (ii) six months after the expiration or earlier valid termination of the collaboration, in each case subject to earlier termination in the event certain clinical activities under the collaboration fail to occur. Following the expiration of this lock-up period, Sanofi Genzyme will be permitted to sell such shares of common stock subject to certain limitations, including volume and manner of sale restrictions. Notwithstanding the foregoing, in the event that the market price per share of our common stock is at least 100 percent higher than the market price per share of our common stock at closing of the stock purchase (in each case based upon a ten-day trailing average), Sanofi Genzyme may sell up to 25 percent of its initial shares, subject to certain restrictions on post-lock-up period dispositions as described above.
Under the investor agreement, following the lock-up period, Sanofi Genzyme will have three demand rights to require us to conduct a registered underwritten public offering with respect to the shares of common stock beneficially owned by Sanofi Genzyme immediately after the closing of the stock purchase, subject to certain conditions. In addition, following the lock-up period, subject to certain conditions, Sanofi Genzyme will be entitled to participate in registered underwritten public offerings by us if other selling stockholders are included in the registration.
104
The investor agreement provides that, until Sanofi Genzyme owns less than 7.5 percent of our outstanding common stock, subject to Sanofi Genzyme’s limited right to maintain its ownership percentage as described herein, in connection with new issuances of common stock, subject to certain exceptions, Sanofi Genzyme will be entitled to a right of first offer to participate proportionally to maintain its then-current ownership percentage of our common stock. If Sanofi Genzyme is not entitled to a right of first offer with respect to a new issuance, Sanofi Genzyme will have the opportunity, on a post-transaction basis, to purchase additional shares sufficient to maintain its pre-transaction ownership percentage of our common stock (subject to the same 7.5 percent ownership threshold).
Finally, in the event Sanofi Genzyme and its affiliates acquire at least 20 percent or more of our outstanding common stock, Sanofi Genzyme will be entitled to appoint one individual to our board of directors. Sanofi Genzyme will also be entitled to certain information rights, including with respect to financial information in the event Sanofi Genzyme or its affiliates require such information for its own financial reporting purposes. The rights and restrictions under the investor agreement are subject to termination upon the occurrence of certain events.
We recorded the issuance of 8,766,338 shares of our common stock under the stock purchase agreement using the price of our common stock on the date the shares were issued to Sanofi Genzyme. Based on the common stock price of $85.72, the fair value of the shares issued was $751.5 million, which was $51.5 million in excess of the proceeds received from Sanofi Genzyme for the issuance of our common stock. This $51.5 million is being amortized on a straight-line basis over the performance period for the ALN-TTR programs. In addition, due to intraperiod tax allocation rules, upon closing of the equity transaction we recorded a benefit from income taxes of $15.2 million due to the Sanofi Genzyme equity purchase being recorded in additional paid-in capital, net of tax.
In accordance with the investor agreement, as a result of our issuance of shares in connection with our acquisition of Sirna Therapeutics, Inc., or Sirna, in March 2014, Sanofi Genzyme exercised its right to purchase an additional 344,448 shares of our common stock for $23.0 million. In addition, in connection with our public offerings, Sanofi Genzyme exercised its right to purchase directly from us, in concurrent private placements, 744,566 shares of common stock in January 2015 at the public offering price resulting in $70.7 million in proceeds to us and 297,501 shares of common stock in May 2017 at the public offering price resulting in $21.4 million in proceeds to us. Sanofi Genzyme elected not to purchase shares in connection with our November 2017 offering. The sales of common stock to Sanofi Genzyme were not registered as part of these public offerings, though they were consummated simultaneously with such public offerings.
Sanofi Genzyme also has the right at the beginning of each year to purchase a number of shares of our common stock based on the number of shares we issued during the previous year for compensation-related purposes. Sanofi Genzyme exercised this right to purchase directly from us 196,251 shares of our common stock on January 22, 2015 for $18.3 million and 205,030 shares of our common stock on February 1, 2016 for $14.3 million. Sanofi Genzyme elected not to exercise its compensation-related right for 2016 or 2017. The sales of these shares to Sanofi Genzyme were consummated as private placements.
Sanofi Genzyme currently holds approximately 11 percent of our outstanding common stock.
We determined that the deliverables for the programs on which Sanofi Genzyme was collaborating with us upon initiation of the 2014 Sanofi Genzyme collaboration included the licenses to our patisiran and revusiran clinical programs, which licenses were delivered to Sanofi Genzyme upon the closing date of the transaction, and the associated development activities, joint steering committee participation and information exchange for these clinical programs. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and associated undelivered development activities, joint steering committee participation and information exchange activities did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme has the ability to grant sublicenses, it cannot sublicense all or substantially all of its rights under the 2014 Sanofi Genzyme collaboration. The uniqueness of our services and the limited sublicense rights are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition model on the final deliverable. Under the 2014 Sanofi Genzyme collaboration, the last deliverables for patisiran and revusiran were expected to be completed within approximately six years from the closing date of the transaction and the last deliverables for fitusiran were expected to be completed within approximately five years from the date Sanofi Genzyme elected to opt into our fitusiran program under the regional license terms. Our estimate regarding the performance period under the 2014 Sanofi Genzyme collaboration related to the license to our patisiran and revusiran clinical programs was adjusted in October 2016 due to our decision to discontinue development of revusiran. As a result, with respect to these programs, we currently expect the last deliverables to be completed within approximately five years from the closing date of the transaction as compared to an initial expectation of approximately six years. Our estimate regarding the performance period under the 2014 Sanofi Genzyme collaboration related to the license to our fitusiran program was adjusted in September 2017 due to our temporary suspension of dosing in all ongoing fitusiran studies. As a result, with respect to the fitusiran program, we currently expect the last deliverables to be completed within approximately six years from the date Sanofi Genzyme
105
elected to opt into the program under the regional license terms as compared to an initial expectation of approximately five years. Beginning in September 2017, we are prospectively recognizing the remaining deferred revenue as of August 31, 2017 related to the license to our fitusiran program over this adjusted performance period.
We determined that the total cash received from Sanofi Genzyme under the now superseded 2012 Sanofi Genzyme agreement reflects consideration for certain of the performance obligations for ALN-TTR programs included in the 2014 Sanofi Genzyme collaboration. Therefore we are recognizing the $33.5 million of deferred revenue under the 2012 Sanofi Genzyme agreement on a straight-line basis over the period of performance of the ALN-TTR programs. As consideration is achieved, including any milestones or reimbursement for development activities, we recognize as revenue a portion of these payments equal to the percentage of the performance period completed when the milestone or activities have been satisfied, multiplied by the amount of the payment. We recognize the remaining portion of consideration received over the remaining performance period on a straight-line basis.
The following table presents information related to the 2014 Sanofi Genzyme collaboration, in thousands:
|
Excess of fair value of our common stock issued to Sanofi Genzyme in February 2014 |
|
$ |
(51,450 |
) |
|
Deferred revenue remaining under the 2012 Sanofi Genzyme agreement upon execution of the 2014 Sanofi Genzyme collaboration |
|
|
33,500 |
|
|
Milestone payment received: |
|
|
|
|
|
Year-ended December 31, 2014 |
|
|
25,000 |
|
|
Development expense reimbursement from Sanofi Genzyme: |
|
|
|
|
|
Year-ended December 31, 2015 |
|
|
33,949 |
|
|
Year-ended December 31, 2016 |
|
|
54,337 |
|
|
Year-ended December 31, 2017 |
|
|
51,846 |
|
|
Total consideration at December 31, 2017 |
|
$ |
147,182 |
|
|
Cumulative revenue recognized at December 31, 2017 |
|
$ |
98,014 |
|
|
Deferred revenue at December 31, 2017 |
|
$ |
49,168 |
|
We determined that the opt-in rights that Sanofi Genzyme has for future Genetic Medicine programs represent separate and additional deliverables that Sanofi Genzyme may receive from us in future periods. Upon each initial opt-in by Sanofi Genzyme, we have determined that each program and the related activities will represent a single unit of accounting and, consistent with our accounting policies, we will base our revenue recognition period on the final deliverable associated with each future opt-in.
The Medicines Company Alliance
In February 2013, we and MDCO entered into a license and collaboration agreement pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases, including inclisiran. MDCO paid us an upfront cash payment of $25.0 million. Upon achievement of certain events, we will be entitled to receive milestone payments, up to an aggregate of $180.0 million, including up to $30.0 million in specified development milestones, $50.0 million in specified regulatory milestones and $100.0 million in specified commercialization milestones. In addition, we will be entitled to royalties ranging from the low- to high- teens based on annual worldwide net sales, if any, of licensed products by MDCO, its affiliates and sublicensees, subject to reduction under specified circumstances. In December 2014, we earned a development milestone payment of $10.0 million under the MDCO agreement based upon the initiation of our Phase 1 clinical trial for inclisiran. In November 2017, we earned a development milestone payment of $20.0 million under the MDCO agreement based upon the initiation by MDCO of a pivotal study for inclisiran. In addition, in 2017, 2016 and 2015, we were reimbursed an aggregate of $12.2 million for costs incurred for certain development activities. We could potentially earn the next development milestone payment of $25.0 million based upon regulatory approval of an NDA for inclisiran in the United States. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from MDCO.
Under the MDCO agreement, we and MDCO will collaborate in the further development of inclisiran. We had responsibility for the development of inclisiran until Phase 1 Completion, as defined in the MDCO agreement, at our cost, up to an agreed upon initial development cost cap. In late 2015, MDCO assumed responsibility for all development and commercialization of inclisiran, at its sole cost. The collaboration between us and MDCO is governed by a joint steering committee comprised of an equal number of representatives from each party.
106
We were solely responsible for obtaining supply of finished product reasonably required for the conduct of our obligations under the initial development plan through Phase 1 Completion, and for supplying MDCO with finished product reasonably required for the first Phase 2 clinical trial of inclisiran conducted by MDCO, at our expense, subject to certain caps. In April 2016, we and MDCO entered into a supply and technical transfer agreement to provide for our supply of inclisiran to MDCO, in accordance with the terms of the MDCO agreement. MDCO now has the sole right and responsibility to manufacture and supply inclisiran for development and commercialization under the MDCO development plan, subject to the terms of the MDCO agreement and the supply and technical transfer agreement.
Unless terminated earlier in accordance with the terms of the agreement, the MDCO agreement expires on a licensed product-by-licensed product and country-by-country basis upon expiration of the last royalty term for any licensed product in any country, where a royalty term is defined as the latest to occur of (1) the expiration of the last valid claim of patent rights covering a licensed product, (2) the expiration of the Regulatory Exclusivity, as defined in the MDCO agreement, and (3) the twelfth anniversary of the first commercial sale of the licensed product in such country. We estimate that our fundamental RNAi patents covering licensed products under the MDCO agreement will expire both in and outside of the United States generally between 2016 and 2028. We also estimate that our ALN-PCS product-specific patents covering licensed products under the MDCO agreement in the United States and elsewhere will expire at the end of 2033. These patent rights are subject to potential patent term extensions and/or supplemental protection certificates extending such terms in countries where such extensions may become available. In addition, more patent filings relating to the collaboration may be made in the future.
Either party may terminate the MDCO agreement in the event the other party fails to cure a material breach or upon patent-related challenges by the other party. In addition, MDCO has the right to terminate the agreement without cause at any time upon four months’ prior written notice.
During the term of the MDCO agreement, neither party will, alone or with an affiliate or third party, research, develop or commercialize, or grant a license to any third party to research, develop or commercialize, in any country, any product directed to the PCSK9 gene, other than a licensed product, without the prior written agreement of the other party, subject to the terms of the MDCO agreement.
We have determined that the significant deliverables under the MDCO agreement include the license, the joint steering committee, technology transfer obligations, development activities through Phase 1 Completion and supply of product for a Phase 2 clinical trial. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and collective undelivered activities and services do not have standalone value due to the specialized nature of the activities and services to be provided by us. In addition, while MDCO has the ability to grant sublicenses, it must receive our prior written consent to sublicense all or substantially all of its rights. The uniqueness of our services and the limited sublicense right are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered services are being treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition pattern on the final deliverable. Under the MDCO agreement, all deliverables are expected to be completed within approximately five years. We are recognizing revenue under the MDCO agreement on a straight-line basis over approximately five years. We are not utilizing a proportional performance model since we are unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the effort required under the development activities is largely unknown.
We received the upfront payment of $25.0 million from MDCO in February 2013, which was initially recorded as deferred revenue. During the fourth quarter of 2014, we recognized as revenue a portion of the $10.0 million milestone payment earned in December 2014 equal to the percentage of the performance period completed when the milestone was earned. During the fourth quarter of 2017, we recognized as revenue a portion of the $20.0 million milestone payment earned in November 2017 equal to the percentage of the performance period completed when the milestone was earned. During the years ended December 31, 2017, 2016 and 2015, we also recognized as revenue a portion of the $5.4 million, $3.0 million and $3.8 million, respectively, of expense reimbursement due to us under the terms of the MDCO agreement equal to the percentage of the performance period completed upon the invoice date. As future consideration, including any milestones or reimbursement for development activities, are earned, we will recognize as revenue a portion of these payments equal to the percentage of the performance period completed when the milestone is achieved or service has been provided, multiplied by the amount of the payment. We recognize the remaining portion of consideration received over the remaining performance period on a straight-line basis. At December 31, 2017, deferred revenue under the MDCO agreement was $5.7 million.
107
Vir Biotechnology, Inc. Alliance
In October 2017, we formed an exclusive licensing agreement with Vir Biotechnology, Inc. for the development and commercialization of RNAi therapeutics for infectious diseases, including chronic hepatitis B virus infection. We received an upfront payment, comprised of cash and shares of Vir Biotechnology common stock, which currently is not material to our consolidated financial statements.
Platform Alliances
Monsanto Alliance
In August 2012, we and Monsanto entered into a license and collaboration agreement, pursuant to which we granted to Monsanto a worldwide, exclusive, royalty bearing right and license, including the right to grant sublicenses, to our RNAi platform technology and intellectual property controlled by us as of the date of the Monsanto agreement or during the 30 months thereafter, in the field of agriculture. The Monsanto agreement also included the transfer of technology from us to Monsanto and initially included a collaborative research project. Under the Monsanto agreement, Monsanto will be our exclusive collaborator in the agriculture field for a ten-year period.
Monsanto paid us $29.2 million in upfront cash payments, and was also required to make near-term milestone payments to us upon the achievement of specified technology transfer and patent-related milestones. We were also entitled to receive additional funding for collaborative research efforts. In the aggregate, we had the ability to earn up to $5.0 million in milestone payments and research funding under the Monsanto alliance. We received a total of $4.0 million in milestone payments from Monsanto based upon the achievement of a specified patent-related event and the completion of technology transfer activities. In September 2014, we and Monsanto mutually determined not to pursue the discovery collaboration originally contemplated under the terms of the Monsanto agreement. Accordingly, Monsanto was not required to pay us the final milestone of $1.0 million. There are no remaining milestones under the Monsanto agreement. Monsanto is required to pay to us a percentage of specified fees from certain sublicense agreements Monsanto may enter into that include access to our intellectual property, as well as low single-digit royalty payments on worldwide, net sales by Monsanto, its affiliates and sublicensees of certain licensed products, as defined in the Monsanto agreement, if any. Due to the uncertainty of the application of RNAi technology in the field of agriculture, we may not receive any license fees or royalty payments from Monsanto.
The term of the Monsanto agreement generally ends upon the expiration of the last-to-expire patent licensed under the agreement. We estimate that our fundamental RNAi patents licensed under the Monsanto agreement will expire both in and outside the United States generally between 2016 and 2025, subject to any potential patent term extensions and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. Monsanto may terminate the Monsanto agreement in its entirety upon 30-days’ prior written notice to us, provided, however, that Monsanto is required to continue to make royalty payments to us if any royalties were payable on net sales of a licensed product during the previous 24 months. The Monsanto agreement may also be terminated by either party in the event the other party fails to cure a material breach under the Monsanto agreement.
Under the terms of the Monsanto agreement, in the event that during the exclusivity period we cease to own or otherwise exclusively control certain licensed patent rights in the agriculture field, for any reason other than Monsanto’s breach of the Monsanto agreement or its negligence or willful misconduct, resulting in the loss of exclusivity with respect to Monsanto’s rights to such patent rights, and such loss of exclusivity has a material adverse effect on the licensed products, then we would be required to pay Monsanto up to $2.5 million as liquidated damages, which amount was reduced from $5.0 million during the fourth quarter of 2017, and Monsanto’s royalty obligations to us under the Monsanto agreement would be reduced or, under certain circumstances, terminated. We have the right to cure any such loss of patent rights under the Monsanto agreement.
Under the Monsanto agreement, the last deliverable expected to be completed was the discovery collaboration, which was originally expected to be completed within five years. Therefore, prior to the September 2014 amendment, we were recognizing revenue under the Monsanto agreement on a straight-line basis over five years. However, as a result of the September 2014 amendment, we determined that the final deliverable in the collaboration is the technology transfer activities, know-how exchange and access to intellectual property controlled by us as of the date of the Monsanto agreement or during the 30 months thereafter, in the field of agriculture. Consequently, we recognized the remaining deferred revenue of $16.8 million at the date of the amendment on a prospective basis from September 2014 through February 2015, the date which was the end of the 30 month obligation, which excluded $5.0 million related to a potential refund due to Monsanto under certain circumstances pursuant to the original terms of the Monsanto agreement. In 2017, we recognized an additional $2.5 million in revenue following the reduction of such potential refund by $2.5 million. We could not use a proportional performance model since we were unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the potential effort required was unknown. At December 31, 2017, deferred revenue under the Monsanto agreement was $2.5 million and will be recognized when the potential refund obligation ceases and can be considered fixed or determinable.
108
In May 2008, we entered into a license and collaboration agreement with Takeda to pursue the development and commercialization of RNAi therapeutics. Under the Takeda agreement, we granted to Takeda a non-exclusive, worldwide, royalty-bearing license to our intellectual property, including delivery-related intellectual property, controlled by us as of the date of the Takeda agreement or during the five years thereafter, to develop, manufacture, use and commercialize RNAi therapeutics, subject to our existing contractual obligations to third parties. The license initially is limited to the fields of oncology and metabolic disease and may be expanded at Takeda’s option to include other therapeutic areas, subject to specified conditions.
Takeda paid us an upfront payment of $100.0 million and an additional $50.0 million upon achievement of specified technology transfer milestones. In addition, for each RNAi therapeutic product developed by Takeda, its affiliates and sublicensees, we are entitled to receive specified development, regulatory and commercialization milestone payments, totaling up to $171.0 million per product, together with a double-digit percentage royalty payment based on worldwide annual net sales, if any. The potential future milestone payments per product include up to $26.0 million for the achievement of specified development milestones, up to $40.0 million for the achievement of specified regulatory milestones and up to $105.0 million for the achievement of specified commercialization milestones. We could potentially earn the next milestone payment of $2.0 million based upon the achievement of a specified pre-clinical event by Takeda for an RNAi therapeutic product. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Takeda.
Pursuant to the Takeda agreement, we and Takeda also agreed to collaborate on the research of RNAi therapeutics directed to one or two disease targets agreed to by the parties, subject to our existing contractual obligations with third parties. The collaboration is governed by a joint technology transfer committee, a joint research collaboration committee and a joint delivery collaboration committee, each of which is comprised of an equal number of representatives from each party. The term of the Takeda agreement generally ends upon the later of (i) the expiration of our last-to-expire patent covering a licensed product and (ii) the last-to-expire term of a profit sharing agreement in the event we elect to enter into such an agreement. We estimate that our fundamental RNAi patents covered under the Takeda agreement will expire both in and outside the United States generally between 2016 and 2025, subject to any potential patent term extensions and/or supplemental protection certificates extending such term extensions in countries where such extensions may become available. The Takeda agreement may be terminated by either party in the event the other party fails to cure a material breach under the agreement. In addition, Takeda may terminate the agreement on a licensed product-by-licensed product or country-by-country basis upon 180-days’ prior written notice to us, provided, however, that Takeda is required to continue to make royalty payments to us for the duration of the royalty term with respect to a licensed product.
We determined that the deliverables under the Takeda agreement included the license, the joint committees, the technology transfer activities and the services that we were obligated to perform under the research collaboration. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered services (i.e., the joint committees and the research collaboration) were not separable and, accordingly, the license and services were being treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition pattern on the final deliverable. Under the Takeda agreement, the last elements to be delivered were the joint technology transfer committee and joint delivery collaboration committee services, each of which had a life of no more than seven years.
We have fully recognized the upfront payment of $100.0 million and the technology transfer milestones of $50.0 million, the receipt of which we believed was probable at the commencement of the collaboration, on a straight-line basis over seven years because we were unable to reasonably estimate the level of effort to fulfill these obligations, primarily because the effort required under the research collaboration was largely unknown, and therefore, we could not utilize a proportional performance model. As future milestones are achieved, if any, we will recognize as revenue the milestone amount in its entirety because all performance obligations for the Takeda agreement have been delivered. At December 31, 2017, there was no deferred revenue under the Takeda agreement as all of our contractual performance obligations were met in May 2015.
Other Strategic License Agreements
Ionis Collaboration and License Agreement
In January 2015, we and Ionis Pharmaceuticals, Inc., or Ionis (formerly Isis Pharmaceuticals, Inc.) entered into a second amended and restated strategic collaboration and license agreement, which we further amended in July 2015. The 2015 Ionis agreement provides for certain new exclusive target cross-licenses of intellectual property on eight disease targets, providing each company with exclusive RNA therapeutic license rights for four programs, and extends the parties’ existing non-exclusive technology cross-license, which was originally entered into in 2004 and was amended and restated in 2009, through April 2019.
109
Pursuant to the 2015 Ionis agreement, Ionis granted to us an exclusive, low single-digit royalty-bearing license to its chemistry, motif, mechanism and target-specific intellectual property for oligonucleotide therapeutics against four targets. In exchange, we granted to Ionis an exclusive, low single-digit royalty-bearing license to our chemistry, motif, mechanism and target-specific intellectual property for oligonucleotide therapeutics against four targets.
In addition, under the 2015 Ionis agreement, the parties agreed to extend the existing non-exclusive technology cross-license through April 2019. Specifically, Ionis granted us a low single-digit royalty-bearing, non-exclusive license to new platform technology arising from May 2014 through April 2019 for double-stranded RNAi therapeutics. In turn, we granted Ionis a low single-digit royalty-bearing, non-exclusive license to new platform technology arising from May 2014 through April 2019 for single-stranded antisense therapeutics. This broad, non-exclusive cross-license includes chemistry, motif and mechanism patents, but excludes patent claims on formulations, manufacturing and specific targets.
Under the original 2004 agreement, Ionis licensed to us its patent estate related to antisense motifs and mechanisms and oligonucleotide chemistry for double-stranded RNAi products in exchange for a previously disclosed technology access fee, participation in fees for our partnering programs and future milestone and royalty payments from us for programs that incorporate Ionis’ intellectual property. We have the right to use Ionis’ intellectual property in our development programs or in collaborations and Ionis agreed not to grant licenses under these patents to any other organization for the discovery, development and commercialization of double-stranded RNA products designed to work through an RNAi mechanism, except in the context of a collaboration in which Ionis plays an active role.
In turn, under the original 2004 agreement, we non-exclusively licensed to Ionis our patent estate relating to antisense motifs and mechanisms and oligonucleotide chemistry to research, develop and commercialize single-stranded antisense therapeutics, single stranded RNAi therapeutics and to research double-stranded RNAi compounds. Ionis also received a license to develop and commercialize double-stranded RNAi drugs targeting a limited number of therapeutic targets on a non-exclusive basis. We granted these licenses for RNAi therapeutics in exchange for option fees, and future milestone and royalty payments from Ionis for RNAi programs that incorporate certain of our intellectual property.
In August 2012, we and Ionis amended the agreement to provide certain terms for the discovery, development and commercialization of double-stranded RNA products by us or our sublicensees in the field of agriculture.
As set forth in the 2015 Ionis agreement, under the original 2004 agreement, we paid Ionis an upfront license fee of $5.0 million and we agreed to pay Ionis milestone payments, totaling up to approximately $3.4 million, upon the occurrence of specified development and regulatory events, and low single-digit royalties on sales, if any, for each product that we or a collaborator develop using Ionis intellectual property. In addition, we agreed to pay to Ionis a percentage of specified fees from strategic collaborations we may enter into that include access to Ionis’ intellectual property.
Ionis has the right to elect up to ten non-exclusive target licenses under the agreement and has the right to purchase one additional non-exclusive target per year during the term of the collaboration. Ionis agreed to pay us, per therapeutic target, a license fee of $0.5 million, milestone payments for double-stranded RNAi products totaling approximately $3.4 million, payable upon the occurrence of specified development and regulatory events, and low single-digit royalties on sales, if any, for each double-stranded RNAi or single-stranded RNAi product developed by Ionis or a collaborator that utilizes our intellectual property. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Ionis.
The term of the 2015 Ionis agreement generally ends upon the expiration of the last-to-expire patent licensed thereunder, whether such patent is a patent licensed by us to Ionis, or vice versa. Either party may terminate the 2015 Ionis agreement on 90 days’ prior written notice if the other party materially breaches the agreement and fails to cure the breach within the 90-day notice period and no substantial steps have otherwise been taken to cure the breach, provided, however, that neither party may terminate licenses granted to the other party to the extent necessary to develop or sell products that have at least reached investigational new drug-enabling studies (except for a party’s uncured failure of its payment obligations). Either party may also terminate the agreement in the event the other party undergoes specified bankruptcy events.
During the years ended December 31, 2017, 2016 and 2015, as result of certain payments received by us in connection with the Sanofi Genzyme and MDCO alliances, we paid $0.2 million, $0.2 million and $1.9 million to Ionis, respectively. In addition, as of December 31, 2017 and 2016, we owed Ionis $1.0 million and $0.2 million, respectively, for amounts incurred but not yet paid. These license fees were charged to research and development expense.
110
The following tables present information about our assets that are measured at fair value on a recurring basis at December 31, 2017 and 2016, and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value, in thousands:
|
Description |
|
At December 31, 2017 |
|
|
Quoted Prices in Active Markets (Level 1) |
|
|
Significant Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
||||
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Commercial paper |
|
$ |
82,262 |
|
|
$ |
— |
|
|
$ |
82,262 |
|
|
$ |
— |
|
|
Corporate notes |
|
|
18,116 |
|
|
|
— |
|
|
|
18,116 |
|
|
|
— |
|
|
U.S. government-sponsored enterprise securities |
|
|
231,122 |
|
|
|
— |
|
|
|
231,122 |
|
|
|
— |
|
|
U.S. treasury securities |
|
|
62,855 |
|
|
|
— |
|
|
|
62,855 |
|
|
|
— |
|
|
Money market funds |
|
|
122,986 |
|
|
|
122,986 |
|
|
|
— |
|
|
|
— |
|
|
Marketable securities (fixed income): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
Certificates of deposit |
|
|
30,200 |
|
|
|
— |
|
|
|
30,200 |
|
|
|
— |
|
|
Commercial paper |
|
|
56,951 |
|
|
|
— |
|
|
|
56,951 |
|
|
|
— |
|
|
Corporate notes |
|
|
373,252 |
|
|
|
— |
|
|
|
373,252 |
|
|
|
— |
|
|
U.S. government-sponsored enterprise securities |
|
|
398,298 |
|
|
|
— |
|
|
|
398,298 |
|
|
|
— |
|
|
U.S. treasury securities |
|
|
200,475 |
|
|
|
— |
|
|
|
200,475 |
|
|
|
— |
|
|
Restricted cash (Money market funds) |
|
|
1,471 |
|
|
|
1,471 |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
1,577,988 |
|
|
$ |
124,457 |
|
|
$ |
1,453,531 |
|
|
$ |
— |
|
|
Description |
|
At December 31, 2016 |
|
|
Quoted Prices in Active Markets (Level 1) |
|
|
Significant Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
||||
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Commercial paper |
|
$ |
17,199 |
|
|
$ |
— |
|
|
$ |
17,199 |
|
|
$ |
— |
|
|
Money market funds |
|
|
151,479 |
|
|
|
151,479 |
|
|
|
— |
|
|
|
— |
|
|
Marketable securities (fixed income): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
— |
|
|
Certificates of deposit |
|
|
17,999 |
|
|
|
— |
|
|
|
17,999 |
|
|
|
— |
|
|
Commercial paper |
|
|
59,340 |
|
|
|
— |
|
|
|
59,340 |
|
|
|
— |
|
|
Corporate notes |
|
|
333,872 |
|
|
|
— |
|
|
|
333,872 |
|
|
|
— |
|
|
U.S. government-sponsored enterprise securities |
|
|
297,773 |
|
|
|
— |
|
|
|
297,773 |
|
|
|
— |
|
|
U.S. treasury securities |
|
|
40,000 |
|
|
|
— |
|
|
|
40,000 |
|
|
|
— |
|
|
Marketable securities (Regulus equity holdings) |
|
|
8,997 |
|
|
|
8,997 |
|
|
|
— |
|
|
|
— |
|
|
Restricted cash (Money market funds) |
|
|
1,471 |
|
|
|
1,471 |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
928,130 |
|
|
$ |
161,947 |
|
|
$ |
766,183 |
|
|
$ |
— |
|
For the years ended December 31, 2017 and 2016, there were no transfers between Level 1 and Level 2 financial assets. The carrying amounts reflected in our consolidated balance sheets for cash, billed and unbilled collaboration receivables, other current assets, accounts payable and accrued expenses approximate fair value due to their short-term maturities. The fair value of our long-term debt at December 31, 2017, computed pursuant to a discounted cash flow technique using a market interest rate, was $30.1 million and is considered a Level 3 fair value measurement. The effective interest rate reflects the current market rate.
111
The following tables summarize the fair value, accumulated other comprehensive income (loss) and intraperiod tax allocation regarding our investment in Regulus available-for-sale marketable securities at December 31, 2017 and 2016, and for the activity recorded in each year, in thousands:
|
Description |
|
At December 31, 2016 |
|
|
Sales of Regulus Shares During Year Ended December 31, 2017 |
|
|
All Other Activity During Year Ended December 31, 2017 |
|
|
Balance at December 31, 2017 |
|
||||
|
Carrying value |
|
$ |
8,093 |
|
|
$ |
(7,485 |
) |
|
$ |
(608 |
) |
|
$ |
— |
|
|
Accumulated other comprehensive income (loss), before tax |
|
|
904 |
|
|
|
1,894 |
|
|
|
(2,798 |
) |
|
|
— |
|
|
Investment in equity securities of Regulus Therapeutics Inc., as reported |
|
$ |
8,997 |
|
|
$ |
(5,591 |
) |
|
$ |
(3,406 |
) |
|
$ |
— |
|
|
Accumulated other comprehensive income (loss), before tax |
|
$ |
904 |
|
|
$ |
1,894 |
|
|
$ |
(2,798 |
) |
|
$ |
— |
|
|
Intraperiod tax allocation recorded as a benefit from income taxes |
|
|
(32,792 |
) |
|
|
— |
|
|
|
— |
|
|
|
(32,792 |
) |
|
Accumulated other comprehensive income (loss), net of tax |
|
$ |
(31,888 |
) |
|
$ |
1,894 |
|
|
$ |
(2,798 |
) |
|
$ |
(32,792 |
) |
|
Description |
|
At December 31, 2015 |
|
|
Sales of Regulus Shares During Year Ended December 31, 2016 |
|
|
All Other Activity During Year Ended December 31, 2016 |
|
|
Balance at December 31, 2016 |
|
||||
|
Carrying value |
|
$ |
11,935 |
|
|
$ |
(3,842 |
) |
|
$ |
— |
|
|
$ |
8,093 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
|
39,484 |
|
|
|
(6,977 |
) |
|
|
(31,603 |
) |
|
|
904 |
|
|
Investment in equity securities of Regulus Therapeutics Inc., as reported |
|
$ |
51,419 |
|
|
$ |
(10,819 |
) |
|
$ |
(31,603 |
) |
|
$ |
8,997 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
$ |
39,484 |
|
|
$ |
(6,977 |
) |
|
$ |
(31,603 |
) |
|
$ |
904 |
|
|
Intraperiod tax allocation recorded as a benefit from income taxes |
|
|
(32,792 |
) |
|
|
— |
|
|
|
— |
|
|
|
(32,792 |
) |
|
Accumulated other comprehensive income (loss), net of tax |
|
$ |
6,692 |
|
|
$ |
(6,977 |
) |
|
$ |
(31,603 |
) |
|
$ |
(31,888 |
) |
We obtain fair value measurement data for our marketable securities from independent pricing services. We perform validation procedures to ensure the reasonableness of this data. This includes meeting with the independent pricing services to understand the methods and data sources used. Additionally, we perform our own review of prices received from the independent pricing services by comparing these prices to other sources and confirming those securities are trading in active markets.
The following tables summarize our marketable securities, other than our holdings in Regulus noted above, at December 31, 2017 and 2016, in thousands:
|
|
|
At December 31, 2017 |
|
|||||||||||||
|
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
|
Certificates of deposit |
|
$ |
30,200 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
30,200 |
|
|
Commercial paper |
|
|
56,951 |
|
|
|
— |
|
|
|
— |
|
|
|
56,951 |
|
|
Corporate notes |
|
|
373,736 |
|
|
|
11 |
|
|
|
(495 |
) |
|
|
373,252 |
|
|
U.S. government-sponsored enterprise securities |
|
|
399,281 |
|
|
|
— |
|
|
|
(983 |
) |
|
|
398,298 |
|
|
U.S. treasury securities |
|
|
200,649 |
|
|
|
1 |
|
|
|
(175 |
) |
|
|
200,475 |
|
|
Total |
|
$ |
1,060,817 |
|
|
$ |
12 |
|
|
$ |
(1,653 |
) |
|
$ |
1,059,176 |
|
112
|
|
|
At December 31, 2016 |
|
|||||||||||||
|
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
|
Certificates of deposit |
|
$ |
17,999 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
17,999 |
|
|
Commercial paper |
|
|
59,340 |
|
|
|
— |
|
|
|
— |
|
|
|
59,340 |
|
|
Corporate notes |
|
|
334,266 |
|
|
|
47 |
|
|
|
(441 |
) |
|
|
333,872 |
|
|
U.S. government-sponsored enterprise securities |
|
|
298,910 |
|
|
|
9 |
|
|
|
(1,146 |
) |
|
|
297,773 |
|
|
U.S. treasury securities |
|
|
40,022 |
|
|
|
1 |
|
|
|
(23 |
) |
|
|
40,000 |
|
|
Total |
|
$ |
750,537 |
|
|
$ |
57 |
|
|
$ |
(1,610 |
) |
|
$ |
748,984 |
|
We classify our debt security investments based on their contractual maturity dates. The following table summarizes our available-for-sale debt securities by contractual maturity, at December 31, 2017, in thousands:
|
|
|
At December 31, 2017 |
|
|||||
|
|
|
Amortized Cost |
|
|
Fair Value |
|
||
|
Less than one year |
|
$ |
1,046,834 |
|
|
$ |
1,045,257 |
|
|
Greater than one year but less than two years |
|
|
13,983 |
|
|
|
13,919 |
|
|
Total |
|
$ |
1,060,817 |
|
|
$ |
1,059,176 |
|
|
6. |
PROPERTY, PLANT AND EQUIPMENT, NET |
Property, plant and equipment consist of the following at December 31, 2017 and 2016, in thousands:
|
|
|
At December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Laboratory equipment |
|
$ |
40,462 |
|
|
$ |
37,303 |
|
|
Computer equipment and software |
|
|
9,608 |
|
|
|
9,650 |
|
|
Furniture and fixtures |
|
|
5,844 |
|
|
|
6,002 |
|
|
Leasehold improvements |
|
|
50,612 |
|
|
|
45,362 |
|
|
Land |
|
|
9,080 |
|
|
|
9,080 |
|
|
Construction in progress |
|
|
133,645 |
|
|
|
68,182 |
|
|
|
|
|
249,251 |
|
|
|
175,579 |
|
|
Less: accumulated depreciation |
|
|
(67,351 |
) |
|
|
(61,007 |
) |
|
|
|
$ |
181,900 |
|
|
$ |
114,572 |
|
During the years ended December 31, 2017, 2016 and 2015, we recorded $11.9 million, $9.6 million and $6.7 million, respectively, of depreciation expense related to our property, plant and equipment.
Manufacturing Facility
In April 2016, we purchased 12 acres of undeveloped land in Norton, Massachusetts. We are constructing a manufacturing facility at this site for drug substance, including small interfering RNAs, or siRNAs, and siRNA conjugates, for clinical and commercial use. At December 31, 2017 and 2016, property, plant and equipment, net, on our consolidated balance sheets reflects $140.5 million and $73.2 million, respectively, of land and associated costs related to the construction of our drug substance manufacturing facility.
113
Technology License and Other Commitments
We have licensed from third parties the rights to use certain technologies and information in our research processes as well as in any products that we may develop. In accordance with the related license or technology agreements, we are required to make certain fixed payments to the counterparty over various agreement terms. Many of these agreement terms are consistent with the remaining lives of the underlying intellectual property that we have licensed. At December 31, 2017, we were committed to make the following fixed, estimated and cancelable payments under existing license agreements, in thousands:
|
Year Ending December 31, |
|
|
|
|
|
2018 |
|
$ |
21,012 |
|
|
2019 |
|
|
12,277 |
|
|
2020 |
|
|
795 |
|
|
2021 |
|
|
795 |
|
|
2022 |
|
|
595 |
|
|
Thereafter |
|
|
1,500 |
|
|
Total |
|
$ |
36,974 |
|
At December 31, 2017, we were committed to make fixed, non-cancelable payments of $5.0 million in each of 2018 and 2019 under an agreement that will provide us access to information during this time that we can use in our future research and any products that we may develop.
We in-license technology from a number of sources, including Ionis and Merck. Pursuant to these in-license agreements, we will be required to make additional payments if and when we achieve specified development, regulatory and commercialization milestones. To the extent we are unable to reasonably predict the likelihood, timing or amount of such payments, we have excluded them from the table above.
Facility Leases
300 Third Street
We lease office and laboratory space located at 300 Third Street, Cambridge, Massachusetts, for our corporate headquarters and primary research facility under a non-cancelable real property lease agreement, or the Third Street Lease, with ARE-MA Region No. 28 LLC, or the Landlord. Under the Third Street Lease, we lease a total of approximately 129,000 square feet of office and laboratory space. The term of the Third Street Lease was set to expire in September 2016. In March 2014, we and the Landlord amended the Third Street Lease to extend the term for an additional five years, through September 30, 2021. Under the amended Third Street Lease, we have the option to extend the term for one additional five-year period.
665 Concord Avenue
On February 10, 2012, we entered into a non-cancelable real property lease agreement, or the BMR-665 Concord Avenue Lease, with BMR-Fresh Pond Research Park LLC for our manufacturing facility for patisiran formulated bulk drug product. Under the BMR-665 Concord Avenue Lease, we lease approximately 15,000 square feet of office and laboratory space located at 665 Concord Avenue, Cambridge, Massachusetts. The term of the BMR-665 Concord Avenue Lease was set to expire in August 2017. In August 2016, we and BMR-Fresh Pond Research Park LLC amended the BMR-665 Concord Avenue Lease to extend the term for an additional five years, through August 31, 2022. Under the amended BMR-665 Concord Avenue Lease, we have the option to extend the term for one additional five-year period.
675 West Kendall Street
In April 2015, we entered into a non-cancelable real property lease, or the BMR-675 West Kendall Lease, with BMR-675 West Kendall Street, LLC, or BMR, for laboratory and office space located at 675 West Kendall Street, Cambridge, Massachusetts. We intend to move our corporate headquarters and research facility to this location in early 2019.
Under the terms of the BMR-675 West Kendall Lease, we will lease approximately 295,000 square feet of laboratory and office space. The term of the BMR-675 West Kendall Lease will commence on May 1, 2018 and rent payments will become due commencing upon substantial completion of the building improvements, which is currently expected to be on or around February 1,
114
2019, and will continue for 15 years from the rent commencement date, with options to renew for two terms of five years each, subject to the terms of the BMR-675 West Kendall Lease.
Annual rent under the BMR-675 West Kendall Lease, exclusive of operating expenses and real property taxes, will be $19.8 million for the first year, with annual increases of 3 percent thereafter. Under the terms of the BMR-675 West Kendall Lease, BMR will contribute a total of $56.1 million toward the cost of base building and tenant improvements.
101 Main Street
In May 2015, we entered into a non-cancelable real property lease agreement with RREEF America REIT II CORP. PPP, or RREEF, for office space located on several floors at 101 Main Street, Cambridge, Massachusetts. This lease supplements the initial lease entered into in March 2015 between us and RREEF for office space on a separate floor at the 101 Main Street location.
Under the terms of the 101 Main Street leases, we lease approximately 72,000 square feet of office space at the 101 Main Street location. The terms of the initial 101 Main Street lease and the additional 101 Main Street lease commenced in March 2015 and January 2016, and continue for four years, with an option to renew for one five-year term, and five and a half years, with an option to renew for one five-year term, respectively.
Initial annual rent for the initial lease and the additional lease, exclusive of operating expenses and real property taxes, was $1.7 million and $3.5 million, respectively, with annual increases of $1/square foot under each lease thereafter. Rent payments commenced in May 2015 under the initial lease and rent payments commenced in May 2016 under the additional lease.
We have $1.5 million in restricted cash that is recorded in long-term other assets as of December 31, 2017 and 2016 in connection with an irrevocable standby letter of credit with RREEF.
Our facility leases described above generally contain customary provisions allowing the landlords to terminate the leases if we fail to remedy a breach of any of our obligations under any such lease within specified time periods, or upon our bankruptcy or insolvency.
Total rent expense, including operating expenses, under our real property leases was $18.7 million, $15.9 million and $10.5 million for the years ended December 31, 2017, 2016 and 2015, respectively.
In addition to the lease agreements described above, we also lease additional office space in several locations in and outside of the United States to support our operations and growth.
Future minimum payments under our non-cancelable facility leases, including rent payments for the BMR-675 West Kendall Lease which are expected to commence in early 2019, are approximately as follows, in thousands:
|
Year Ending December 31, |
|
|
|
|
|
2018 |
|
$ |
14,464 |
|
|
2019 |
|
|
32,446 |
|
|
2020 |
|
|
34,570 |
|
|
2021 |
|
|
30,928 |
|
|
2022 |
|
|
23,050 |
|
|
Thereafter |
|
|
297,726 |
|
|
Total |
|
$ |
433,184 |
|
Credit Agreements
On April 29, 2016, we entered into (i) a Credit Agreement, or the BOA Credit Agreement, with Alnylam U.S., Inc., our wholly-owned subsidiary, as the borrower, us, as a guarantor, and Bank of America N.A., or BOA, as the lender and (ii) a Credit Agreement, or the Wells Credit Agreement, together with the BOA Credit Agreement, the Credit Agreements, by and among Alnylam U.S., Inc., as the borrower, us, as a guarantor, and Wells Fargo Bank, National Association, or Wells, as the lender. The Credit Agreements were entered into in connection with the planned build out of our new drug substance manufacturing facility.
The BOA Credit Agreement provided for a $120.0 million term loan facility and was scheduled to mature on April 29, 2021. On December 27, 2017, we repaid in full the $120.0 million outstanding principal amount under the BOA Credit Agreement and the BOA Credit Agreement terminated in accordance with its terms upon repayment of the outstanding indebtedness. The Wells Credit
115
Agreement provides for a $30.0 million term loan facility and matures on April 29, 2021. The proceeds of the borrowing under the BOA Credit Agreement were, and the Wells Credit Agreement are, to be used for working capital and general corporate purposes. Interest on borrowings under the BOA Credit Agreement was, and under the Wells Credit Agreement is calculated based on LIBOR plus 0.45 percent, except in the event of default. The borrower may prepay loans under the Wells Credit Agreement at any time, without premium or penalty, subject to certain notice requirements and LIBOR breakage costs.
The obligations of the borrower and us under the BOA Credit Agreement were, and under the Wells Credit Agreement are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of all term loans outstanding under such Credit Agreement at such time. At December 31, 2017 and 2016, we have recorded $30.0 million and $150.0 million, respectively, of cash collateral in connection with the Credit Agreements as restricted investments on our consolidated balance sheets. Wells and the borrower have agreed to consider the appropriateness of a change in the type of approved collateral on a periodic basis throughout the term of the Wells Credit Agreement; provided that any such change to the type of such approved collateral shall be made only upon each of the lender’s and the borrower’s consent.
The Wells Credit Agreement contains limited representations and warranties and limited affirmative and negative covenants, including quarterly reporting obligations. The Wells Credit Agreement also contains certain customary events of default, including nonpayment of principal or interest, material inaccuracy of representations, failure to comply with covenants, cross-defaults to certain other indebtedness, invalidity of any loan document relating to such Credit Agreement, judgments having a material adverse effect, insolvency events and change of control. If an event of default occurs and is continuing under the Wells Credit Agreement, the entire outstanding balance may become immediately due and payable.
Several of the lenders under each of the Credit Agreements, as well as their affiliates, have various relationships with us and our subsidiaries involving the provision of financial services, such as investment banking, commercial banking, advisory, cash management, custody and corporate credit card services for which they receive customary fees and may do so in the future.
During the years ended December 31, 2017 and 2016, we recorded $0.8 million and $1.2 million, respectively, of interest expense related to the Credit Agreements that is reflected in other income (expense) on our consolidated statements of comprehensive loss.
Litigation
From time to time, we are a party to legal proceedings in the course of our business, including the matters described below. The claims and legal proceedings in which we could be involved include challenges to the scope, validity or enforceability of patents relating to our product candidates, and challenges by us to the scope, validity or enforceability of the patents held by others. These include claims by third parties that we infringe their patents. The outcome of any such legal proceedings, regardless of the merits, is inherently uncertain. In addition, litigation and related matters are costly and may divert the attention of our management and other resources that would otherwise be engaged in other activities. If we were unable to prevail in any such legal proceedings, our business, results of operations, liquidity and financial condition could be adversely affected. Our accounting policy for accrual of legal costs is to recognize such expenses as incurred.
Silence Litigation
On October 17, 2017, Silence Therapeutics plc, or Silence, served its previously announced claim in the High Court of England and Wales, or the High Court, issued in the name of Silence Therapeutics GmbH against Alnylam UK Ltd., Alnylam Pharmaceuticals, Inc., and The Medicines Company UK Ltd, referred to collectively as the Defendants. The claim seeks a declaration that patisiran, fitusiran, givosiran and inclisiran, together, the Products, are protected by Silence’s European Patent No. 2 258 847, or the ‘847 patent, within the meaning of the Supplementary Protection Certificate, or SPC, Regulation of the European Union. The claim alleges that any marketing authorization for any of these Products granted to any of the Defendants is a valid authorization within the meaning of the SPC Regulation, to support an application for an SPC by Silence for each of the Products, allegedly allowing Silence to extend the expiration date of their ‘847 patent on a Product by Product basis, based on the amount of time in regulatory review for each of the Products, again on a Product by Product basis, up to a statutory maximum. In addition, Silence is seeking costs, interest and other unspecified relief.
On October 31, 2017, the Defendants acknowledged service of the claim served by Silence contesting jurisdiction of the High Court. On November 30, 2017, the Defendants submitted substantive defenses to the claim.
On October 27, 2017, we, through our affiliate Alnylam UK Ltd., and The Medicines Company UK Ltd filed and served a claim against Silence Therapeutics GmbH and Silence in the High Court seeking revocation of the ‘847 patent, as well as a declaration of
116
non-infringement by each of the Products of the ‘847 patent, and costs and interest among other potential remedies. On November 14, 2017, Silence filed a defense to our claim along with counterclaims alleging infringement of the ‘847 patent by our Products. On December 11, 2017, we filed an answer and defense to the counter claims. The High Court has set a trial date of December 3, 2018 for all claims between Silence and the Defendants.
Although we believe the ‘847 patent is invalid and not infringed by our Products and that, therefore, Silence would not be entitled to obtain an SPC based on any of our Products, litigation is subject to inherent uncertainty, as noted above, and a court could ultimately rule against us.
Dicerna Litigation
On June 10, 2015, we filed a trade secret misappropriation lawsuit against Dicerna Pharmaceuticals, Inc., or Dicerna, in the Superior Court of Middlesex County, Massachusetts, or the Court, seeking to stop misappropriation by Dicerna of our confidential, proprietary and trade secret information related to the RNAi assets we purchased from Merck, including certain N-acetylgalactosamine, or GalNAc, conjugate technology. In addition to permanent injunctive relief, we are also seeking monetary damages from Dicerna. On July 10, 2015, Dicerna filed its answer to our complaint, in which it denied our claims, following which discovery proceeded. Fact discovery on our claims against Dicerna ended on August 16, 2017, and expert discovery on those claims is underway. In August 2017, Dicerna successfully added counterclaims against us in the trade secret lawsuit alleging that our lawsuit represented abuse of process and claiming tortious interference with its business. On September 27, 2017, we filed a motion to dismiss Dicerna’s counterclaims. The motion was denied on October 24, 2017, and we intend to vigorously defend against those claims. The trial for this lawsuit now is scheduled for April 23, 2018.
In addition, on August 8, 2017, Dicerna filed a lawsuit against us in the United States District Court of Massachusetts alleging attempted monopolization by us under the Sherman Antitrust Act. Dicerna’s allegations related to its new claim largely overlap with its counterclaims in the state court action. We do not believe the claim is meritorious and on October 23, 2017, we filed a motion to dismiss the antitrust lawsuit. On November 20, 2017, Dicerna filed an amended complaint adding the fact that the motion to dismiss Dicerna’s counterclaims had been denied. On December 4, 2017, we filed a renewed motion to dismiss Dicerna’s complaint, which Dicerna has opposed.
Although we believe we have meritorious claims against Dicerna and meritorious defenses and responses to the counterclaims and federal claim now being asserted by Dicerna, as noted above, litigation is subject to inherent uncertainty, we will incur significant costs in defending against such claims, and a court could ultimately rule against us.
University of Utah Litigation
On March 22, 2011, The University of Utah, or Utah, filed a civil complaint in the United States District Court for the District of Massachusetts, or the MA District Court, against us, Max Planck Gesellschaft Zur Foerderung Der Wissenschaften e.V. and Max Planck Innovation GmbH, together, Max Planck, the Whitehead Institute for Biomedical Research, or Whitehead, the Massachusetts Institute of Technology, or MIT, and the University of Massachusetts, or UMass, claiming a professor at Utah is the sole inventor or, in the alternative, a joint inventor, of the Tuschl patents. Utah was seeking changes to the inventorship of the Tuschl patents, unspecified damages and other relief. After several years of court proceedings and discovery, on September 28, 2015, the MA District Court granted both of our motions for summary judgment, finding that there was no collaboration between Dr. Bass and Dr. Tuschl, which is a pre-requisite for co-inventorship, and dismissing Utah’s state law damages claims as well.
On October 28, 2015, Utah filed a notice of appeal to the United States Court of Appeals for the Federal Circuit, or the CAFC. On December 18, 2015, the CAFC entered an order dismissing Utah’s appeal following a joint motion filed by us and Utah seeking dismissal of the appeal with prejudice. This disposed of Utah’s inventorship claims and its state law claims for damages. On October 14, 2015, we filed a motion with the MA District Court seeking reimbursement of costs and fees associated with defending this action in the amount of approximately $8.0 million. On November 30, 2015, the MA District Court denied our motion and on December 15, 2015, we filed a notice of appeal of this ruling with the CAFC. Oral arguments on our appeal were heard at the CAFC on January 12, 2017. On March 23, 2017, the CAFC denied our appeal and we decided not to appeal this ruling any further. Final judgment in our favor on the merits has been entered by the MA District Court.
Indemnifications
In connection with our license agreements with Max Planck relating to the Tuschl I and Tuschl II patent applications, we are required to indemnify Max Planck for certain damages arising in connection with the intellectual property rights licensed under the agreements. Under the Max Planck indemnification agreement, we are responsible for paying the costs of any litigation relating to the
117
license agreements or the underlying intellectual property rights, including the costs associated with certain litigation regarding the Tuschl patents, which was settled during 2011, as well as certain costs associated with defending the University of Utah litigation described above. In connection with the settlement of the litigation regarding the Tuschl patents, we also agreed to indemnify Whitehead, MIT and UMass for certain costs associated with defending the University of Utah litigation. In connection with our research agreement with Acuitas Therapeutics Inc., or Acuitas (formerly AlCana Technologies, Inc.), we agreed to indemnify Acuitas for certain legal costs, subject to certain exceptions and limitations, associated with certain litigation with Arbutus Biopharma Corporation, or ABC (formerly Tekmira Pharmaceuticals Corporation), and Protiva Biotherapeutics, Inc., a wholly owned subsidiary of ABC, and together with ABC, Arbutus, which has been settled. These indemnification costs were charged to general and administrative expense. We are also a party to a number of agreements entered into in the ordinary course of business, which contain typical provisions that obligate us to indemnify the other parties to such agreements upon the occurrence of certain events. Such indemnification obligations are usually in effect from the date of execution of the applicable agreement for a period equal to the applicable statute of limitations.
Our maximum potential future liability under any such indemnification provisions is uncertain. However, to date, other than certain costs associated with certain previously settled litigation related to the Tuschl patents and the litigation with Arbutus referenced above, and certain defense costs related to the University of Utah litigation described above, we have not incurred material costs to defend lawsuits or settle claims related to these indemnification provisions. We have determined that the estimated aggregate fair value of our potential liabilities under all such indemnification provisions is minimal and have not recorded any liability related to such indemnification provisions at December 31, 2017 or 2016.
|
8. |
STOCKHOLDERS’ EQUITY |
Preferred Stock
We have authorized up to 5,000,000 shares of preferred stock, $0.01 par value per share, for issuance. The preferred stock will have such rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, as shall be determined by our board of directors upon its issuance. At December 31, 2017 and 2016, there were no shares of preferred stock outstanding.
Public Offerings
In November 2017, we sold an aggregate of 6,440,000 shares of our common stock through an underwritten public offering at a price to the public of $125.00 per share. As a result of the offering, which included the full exercise of the underwriters’ option to purchase additional shares, we received aggregate net proceeds of $784.5 million, after deducting underwriting discounts and commissions and other offering expenses of $20.5 million.
In May 2017, we sold an aggregate of 5,000,000 shares of our common stock through an underwritten public offering at a price to the public of $71.87 per share. As a result of the offering, we received aggregate net proceeds of $355.2 million, after deducting underwriting discounts and commissions and other offering expenses of $4.2 million.
In January 2015, we sold an aggregate of 5,447,368 shares of our common stock through an underwritten public offering at a price to the public of $95.00 per share. As a result of the offering, which included the full exercise of the underwriters’ option to purchase additional shares, we received aggregate net proceeds of $496.4 million, after deducting underwriting discounts and commissions and other offering expenses of $21.1 million.
118
Stock Plans
In June 2009, our stockholders approved an amendment and restatement of our 2004 Stock Incentive Plan, or the Amended and Restated 2004 Plan, which replaced our 2004 Stock Incentive Plan, as amended. At December 31, 2017, the Amended and Restated 2004 Plan provided for the granting of stock options to purchase up to 12,366,485 shares of common stock.
In June 2009, our stockholders also approved our 2009 Stock Incentive Plan. In May 2015, our stockholders approved an amendment and restatement of the 2009 Stock Incentive Plan, which increased the number of shares of common stock authorized for issuance from 5,900,000 to 11,700,000. In May 2017, our stockholders approved a second amendment and restatement of the 2009 Stock Incentive Plan, or the Second Amended and Restated 2009 Plan, which increased the number of shares of common stock authorized for issuance from 11,700,000 to 15,480,000. The Second Amended and Restated 2009 Plan provides for the granting of stock options, restricted stock and restricted stock units (together, restricted stock awards), stock appreciation rights and other stock-based awards. The Second Amended and Restated 2009 Plan has a fungible share pool. Any award that is not a full value award is counted against the authorized share limits specified in the Second Amended and Restated 2009 Plan as one share for each share of common stock subject to the award, and all full value awards, defined in the Second Amended and Restated 2009 Plan as restricted stock awards or other stock-based awards, are counted as one and a half shares for each one share of common stock subject to such full value award.
At December 31, 2017, an aggregate of 15,556,526 shares of common stock were reserved for issuance under our stock plans, including outstanding stock options to purchase 11,239,128 shares of common stock, 148,670 outstanding restricted stock units, 3,641,501 of common stock available for additional equity awards and 527,227 shares available for future grant under our Amended and Restated 2004 Employee Stock Purchase Plan, or the Amended and Restated ESPP. Each stock option shall expire within ten years of issuance. Time-based stock options granted by us to employees generally vest as to 25 percent of the shares on the first anniversary of the grant date and 6.25 percent of the shares at the end of each successive three-month period thereafter until fully vested. Performance-based stock options granted to employees in 2015 and 2016 vest, with respect to each year, as to one-fourth of the shares upon the later of the one-year anniversary of the grant date and the achievement of each of four specific clinical development, regulatory or commercial events, as approved by our compensation committee. Performance-based stock options granted to employees in 2013 and 2014 vest, with respect to each year, as to one-third of the shares upon the achievement of each of three specific clinical development or regulatory events, as approved by our compensation committee. Historically, stock option awards to employees for annual performance, including performance-based stock option awards beginning in 2013, were approved for grant by the compensation committee of our board of directors at year-end. However, with respect to 2017 annual awards as well as future annual awards, our compensation committee intends to approve the grant of any such awards in the first quarter of the following year.
Change in Control Agreements
On November 7, 2017, we entered into a Change in Control, or CIC, Agreement with each member of our management board. If a member of our management board is terminated by us without Cause (as defined in the CIC Agreement) or if a management board member terminates his or her employment for Good Reason (as defined in the CIC Agreement), in either case, within 12 months following a CIC, such management board member will be entitled to receive certain benefits, including the immediate acceleration of all outstanding unvested stock options and other stock-based awards. In accordance with accounting guidance for stock-based compensation expense, we expect to record the modification date fair value for any equity grants that were not considered probable of vesting as of November 7, 2017 that ultimately vest.
Inducement Equity Grants
Effective as of each of May 8, 2017 and September 19, 2016, respectively, our compensation committee approved a grant to a newly hired executive of non-qualified stock options to purchase an aggregate of 150,000 shares of common stock. For each executive, time-vested options to purchase 125,000 shares of common stock will vest as to 25 percent of the shares on the first anniversary of the respective grant date and 6.25 percent of the shares at the end of each successive three-month period thereafter until fully vested. For each executive, performance-based stock options to purchase 25,000 shares of common stock will vest upon the later of the one-year anniversary of the respective grant date and the launch of our first internally developed product. In addition, effective as of February 6, 2017, our compensation committee approved a grant to a newly hired vice president level employee, of non-qualified stock options to purchase an aggregate of 50,000 shares of common stock. This time-vested option grant will vest as to 25 percent of the shares on the first anniversary of the grant date and 6.25 percent of the shares at the end of each successive three-month period thereafter until fully vested. Each of the May 8, 2017, February 6, 2017 and September 19, 2016 option grants was granted as an inducement grant outside of our stockholder approved stock plans in accordance with NASDAQ Listing Rule 5635(c)(4). These stock options have a ten-year term and an exercise price equal to the closing price of our common stock on the respective grant date.
119
The following table summarizes stock-based compensation expense by type of award during the three years ended December 31, 2017, in thousands:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Stock-based compensation expense by type of award: |
|
|
|
|
|
|
|
|
|
|
|
|
|
Time-based stock options |
|
$ |
61,802 |
|
|
$ |
62,800 |
|
|
$ |
43,078 |
|
|
Performance-based stock options |
|
|
23,260 |
|
|
|
8,337 |
|
|
|
— |
|
|
Restricted stock awards |
|
|
541 |
|
|
|
497 |
|
|
|
555 |
|
|
ESPP share issuances |
|
|
2,155 |
|
|
|
1,360 |
|
|
|
694 |
|
|
Other equity programs |
|
|
1,946 |
|
|
|
1,846 |
|
|
|
— |
|
|
Non-employee stock options |
|
|
3,115 |
|
|
|
688 |
|
|
|
1,456 |
|
|
|
|
$ |
92,819 |
|
|
$ |
75,528 |
|
|
$ |
45,783 |
|
The following table summarizes our unrecognized stock-based compensation expense, net of estimated forfeitures, at December 31, 2017 by type of awards, and the weighted-average period over which that expense is expected to be recognized:
|
|
|
At December 31, 2017 |
|
|||||
|
|
|
Unrecognized Expense, Net of Estimated Forfeitures |
|
|
Weighted- average Recognition Period |
|
||
|
|
|
(in thousands) |
|
|
(in years) |
|
||
|
Type of award: |
|
|
|
|
|
|
|
|
|
Time-based stock options |
|
$ |
124,295 |
|
|
|
2.60 |
|
|
Performance-based stock options |
|
|
59,416 |
|
|
* |
|
|
|
Performance-based restricted stock units |
|
|
14,649 |
|
|
* |
|
|
|
ESPP share issuances |
|
|
970 |
|
|
0.33 |
|
|
|
* |
Performance-based stock options and performance-based restricted stock units are recorded as expense beginning when vesting events are determined to be probable. |
Valuation Assumptions for Stock Options
The fair value of stock options, at date of grant, based on the following assumptions, was estimated using the Black-Scholes option-pricing model. Our expected stock-price volatility assumption is based on the historical volatility of our publicly traded stock. The expected life assumption is based on our historical data. The dividend yield assumption is based on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. The risk-free interest rate used for each grant is equal to the zero coupon rate for instruments with a similar expected life.
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Risk-free interest rate |
|
1.9-2.3% |
|
|
0.9-2.2% |
|
|
1.0-2.0% |
|
|||
|
Expected dividend yield |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
Expected option life |
|
5.7-7.2 years |
|
|
3.5-7.5 years |
|
|
3.5-7.5 years |
|
|||
|
Expected volatility |
|
61-67% |
|
|
55-65% |
|
|
53-60% |
|
|||
120
The following table summarizes the activity of our stock option plans and the inducement grants described above, excluding performance-based stock options:
|
|
|
Number of Options (in thousands) |
|
|
Weighted- average Exercise Price |
|
|
Weighted- average Remaining Contractual Term (in years) |
|
Aggregate Intrinsic Value (in thousands) |
|
|||
|
Outstanding, December 31, 2016 |
|
|
9,913 |
|
|
$ |
56.05 |
|
|
|
|
|
|
|
|
Granted |
|
|
1,664 |
|
|
|
75.66 |
|
|
|
|
|
|
|
|
Exercised |
|
|
(1,796 |
) |
|
|
42.81 |
|
|
|
|
|
|
|
|
Cancelled |
|
|
(680 |
) |
|
|
83.70 |
|
|
|
|
|
|
|
|
Outstanding, December 31, 2017 |
|
|
9,101 |
|
|
$ |
60.18 |
|
|
6.92 |
|
$ |
609,620 |
|
|
Exercisable at December 31, 2017 |
|
|
5,294 |
|
|
$ |
51.54 |
|
|
5.59 |
|
$ |
400,390 |
|
|
Vested or expected to vest at December 31, 2017 |
|
|
8,670 |
|
|
$ |
59.64 |
|
|
6.82 |
|
$ |
587,480 |
|
The weighted-average fair value of stock options granted was $44.76, $31.66 and $51.28 per share for the years ended December 31, 2017, 2016 and 2015, respectively. The intrinsic value of stock options exercised was $111.3 million, $22.0 million and $130.0 million for the years ended December 31, 2017, 2016 and 2015, respectively. We satisfy stock option exercises with newly issued shares of our common stock.
Performance-Based Stock Options
We granted performance-based stock options to employees that vest as to one-fourth of the shares upon the later of the one-year anniversary of the grant date and the achievement of each of four specific clinical development, regulatory or commercial events, as approved by our compensation committee, with respect to 2015 and 2016 grants, and that vest as to one-third of the shares upon the later of the one-year anniversary of the grant date and the achievement of each of three specific clinical development or regulatory events, as approved by our compensation committee, with respect to 2013 and 2014 grants. During each of the years ended December 31, 2017 and 2016, we also granted an option to purchase 25,000 shares of common stock to a newly hired executive that vests upon the later of the one-year anniversary of the grant date and the achievement of a commercial event.
The following table summarizes the activity of our performance-based stock options granted under our equity plans and the performance-based portion of the inducement grants described above:
|
|
|
Number of Options (in thousands) |
|
|
Weighted- average Exercise Price |
|
|
Weighted- average Remaining Contractual Term (in years) |
|
Aggregate Intrinsic Value (in thousands) |
|
|||
|
Outstanding, December 31, 2016 |
|
|
2,357 |
|
|
$ |
69.61 |
|
|
|
|
|
|
|
|
Granted |
|
|
25 |
|
|
|
52.61 |
|
|
|
|
|
|
|
|
Exercised |
|
|
(54 |
) |
|
|
80.46 |
|
|
|
|
|
|
|
|
Cancelled |
|
|
(190 |
) |
|
|
69.59 |
|
|
|
|
|
|
|
|
Outstanding, December 31, 2017 |
|
|
2,138 |
|
|
$ |
69.14 |
|
|
7.79 |
|
$ |
123,795 |
|
|
Exercisable at December 31, 2017 |
|
|
813 |
|
|
$ |
73.66 |
|
|
7.23 |
|
$ |
43,416 |
|
During the years ended December 31, 2017 and 2016, there were 614,796 and 159,122 performance-based stock options that vested, respectively. There were no performance-based stock options that vested during the year ended December 31, 2015. The weighted-average grant-date fair value for the performance-based stock options that vested during the years ended December 31, 2017 and 2016 was $37.86 and $52.29 per share, respectively. The intrinsic value of performance-based stock options exercised was $1.8 million, $35,000 and $1.4 million for the years ended December 31, 2017, 2016 and 2015, respectively. We satisfy performance-based stock option exercises with newly issued shares of our common stock. During the three years ended December 31, 2017, each performance-based stock option that vested was determined to be probable of vesting, and we began to record expense, during the year the performance-based stock option vested based on achievement of the respective performance-criteria. As of the grant date and December 31, 2017, we had determined that the remaining performance criteria for all outstanding performance-based stock options reflected in the table above as of December 31, 2017 were not probable of being achieved. As a result, we have not recorded stock-based compensation expense for these performance-based stock options as of December 31, 2017.
121
Contingent Stock Option Awards
On December 17, 2014, the compensation committee of our board of directors approved the grant of stock options to purchase 612,085 shares of our common stock, at an exercise price of $96.45 per share, to members of our management team. These stock option grants were approved subject to and contingent upon approval by our stockholders at our 2015 annual meeting of the Amended and Restated 2009 Plan, to, among other things, increase the shares authorized for issuance thereunder, which approval was obtained in May 2015. One-half of the contingent stock options granted are time-based stock options and one-half are performance-based stock options. The grant date fair value of the contingent stock options is based on a Black-Scholes valuation model based on the fair market value of the stock on May 1, 2015, the date of such stockholder approval. We began recording stock-based compensation expense relating to this contingent stock option grant on May 1, 2015.
Performance-Based Restricted Stock Units
In January 2016, we granted 172,718 shares of performance-based restricted stock units to certain employees, excluding our chief executive officer and president. These performance-based restricted stock units were valued at $16.1 million on the grant date. The vesting of these performance-based restricted stock units is predicated on the launch of our first internally developed product. As of the grant date and December 31, 2017, for accounting purposes we had determined that the performance criteria for these performance-based restricted stock units was not probable of being achieved. As a result, we have not recorded stock-based compensation expense for these performance-based restricted stock units as of December 31, 2017.
Employee Stock Purchase Plan
In 2004, we adopted the 2004 Employee Stock Purchase Plan with 315,789 shares authorized for issuance. In June 2010, our stockholders approved an amendment to the 2004 Employee Stock Purchase Plan, which increased the shares authorized for issuance from 315,789 shares to 715,789 shares. In May 2017, our stockholders approved the Amended and Restated ESPP, which further increased the shares authorized for issuance from 715,789 shares to 1,215,789 shares. Under the Amended and Restated ESPP, each offering period is six months, at the end of which employees may purchase shares of common stock through payroll deductions made over the term of the offering. The per-share purchase price at the end of each offering period is equal to the lesser of 85 percent of the closing price of our common stock at the beginning or end of the offering period. We issued 103,666, 53,499 and 22,639 shares during the years ended December 31, 2017, 2016 and 2015, respectively, and at December 31, 2017, 527,227 shares were available for issuance under the Amended and Restated ESPP.
The weighted-average fair value of stock purchase rights granted as part of the Amended and Restated ESPP was $17.10, $24.90 and $29.96 per share for the years ended December 31, 2017, 2016 and 2015, respectively. The fair value was estimated using the Black-Scholes option-pricing model. During the year ended December 31, 2017, we used a weighted-average stock-price volatility of 91 percent, expected option life assumption of six months and a risk-free interest rate of 0.7 percent. During the years ended December 31, 2016 and 2015, we used a weighted-average stock-price volatility of approximately 55 percent, expected option life assumption of six months and a risk-free interest rate of approximately 0.1 percent.
|
10. |
INCOME TAXES |
The domestic and foreign components of loss before income taxes are as follows, in thousands:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
Domestic |
|
$ |
(378,293 |
) |
|
$ |
(350,704 |
) |
|
$ |
(245,681 |
) |
|
Foreign |
|
|
(112,581 |
) |
|
|
(59,404 |
) |
|
|
(44,392 |
) |
|
Loss before income taxes |
|
$ |
(490,874 |
) |
|
$ |
(410,108 |
) |
|
$ |
(290,073 |
) |
122
Deferred income taxes reflect the tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and income tax purposes. We establish a valuation allowance when uncertainty exists as to whether all or a portion of the net deferred tax assets will be realized. Components of the net deferred tax (liability) asset at December 31, 2017 and 2016 are as follows, in thousands:
|
|
|
2017 |
|
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
$ |
405,784 |
|
|
$ |
346,965 |
|
|
Research and development credits |
|
|
192,094 |
|
|
|
111,394 |
|
|
AMT credits |
|
|
788 |
|
|
|
788 |
|
|
Foreign tax credits |
|
|
— |
|
|
|
3,196 |
|
|
Capitalized research and development and start-up costs |
|
|
13,163 |
|
|
|
18,138 |
|
|
Deferred revenue |
|
|
13,576 |
|
|
|
20,853 |
|
|
Deferred compensation |
|
|
42,990 |
|
|
|
51,355 |
|
|
Intangible assets |
|
|
9,018 |
|
|
|
14,725 |
|
|
Partnership interest |
|
|
— |
|
|
|
1,623 |
|
|
Other |
|
|
11,608 |
|
|
|
7,845 |
|
|
Total deferred tax assets |
|
|
689,021 |
|
|
|
576,882 |
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Unrealized gain on available-for-sale securities |
|
|
(765 |
) |
|
|
(1,524 |
) |
|
Deferred tax asset valuation allowance |
|
|
(688,256 |
) |
|
|
(575,358 |
) |
|
Net deferred tax liability |
|
$ |
— |
|
|
$ |
— |
|
Our effective income tax rate differs from the statutory federal income tax rate as follows for the years ended December 31, 2017, 2016 and 2015:
|
|
|
2017 |
|
|
2016 |
|
|
2015 |
|
|||
|
At U.S. federal statutory rate |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
|
35.0 |
% |
|
State taxes, net of federal effect |
|
|
3.8 |
|
|
|
3.7 |
|
|
|
4.0 |
|
|
Stock-based compensation |
|
|
3.3 |
|
|
|
(1.4 |
) |
|
|
(1.1 |
) |
|
Tax credits |
|
|
9.9 |
|
|
|
11.8 |
|
|
|
8.0 |
|
|
Orphan drug credit |
|
|
(3.4 |
) |
|
|
(3.5 |
) |
|
|
(2.0 |
) |
|
Other permanent items |
|
|
(1.0 |
) |
|
|
(0.4 |
) |
|
|
(0.1 |
) |
|
Foreign rate differential |
|
|
(8.1 |
) |
|
|
(5.1 |
) |
|
|
(5.4 |
) |
|
Tax reform change |
|
|
(46.5 |
) |
|
|
— |
|
|
|
— |
|
|
Other |
|
|
(0.9 |
) |
|
|
— |
|
|
|
— |
|
|
Valuation allowance |
|
|
7.9 |
|
|
|
(40.1 |
) |
|
|
(38.4 |
) |
|
Effective income tax rate |
|
|
— |
% |
|
|
— |
% |
|
|
— |
% |
We have evaluated the positive and negative evidence bearing upon the realizability of our deferred tax assets. We have concluded, in accordance with the applicable accounting standards, that it is more likely than not that we may not realize the benefit of all of our deferred tax assets. Accordingly, we have recorded a valuation allowance against the deferred tax assets that management believes will not be realized. We reevaluate the positive and negative evidence on a quarterly basis. The valuation allowance increased by $112.9 million, $177.9 million and $129.1 million for the years ended December 31, 2017, 2016 and 2015, respectively, due primarily to additional operating losses.
On December 22, 2017, the President of the United States signed into law the Tax Cuts and Jobs Act, or TCJA, tax reform legislation. The TCJA makes significant changes in U.S. tax law including a reduction in the corporate tax rates, changes to net operating loss carryforwards and carrybacks, and a repeal of the corporate alternative minimum tax. The TCJA reduced the U.S. corporate tax rate from the current rate of 35 percent down to 21 percent starting on January 1, 2018. As a result of the TCJA, we were required to revalue deferred tax assets and liabilities at 21 percent. This revaluation resulted in a provision of $227.9 million to income tax expense in continuing operations and a corresponding reduction in the valuation allowance. As a result, there was no impact to our consolidated statements of comprehensive loss as a result of the reduction in tax rates. The other provisions of the TCJA did not have a material impact on our consolidated financial statements.
123
Our preliminary estimate of the TCJA and the remeasurement of our deferred tax assets and liabilities is subject to the finalization of management’s analysis related to certain matters, such as developing interpretations of the provisions of the TCJA, changes to certain estimates and the filing of our tax returns. U.S. Treasury regulations, administrative interpretations or court decisions interpreting the TJCA may require further adjustments and changes in our estimates. The final determination of the TCJA and the remeasurement of our deferred assets and liabilities will be completed as additional information becomes available, but no later than one year from the enactment of the TCJA.
There was no benefit from income taxes recorded during the years ended December 31, 2017, 2016, and 2015.
On January 1, 2017, we adopted new accounting guidance released in March 2016 that updates the accounting for certain aspects of share-based payments to employees, including the income tax consequences, classification of awards as either equity or liabilities and classification on the consolidated statement of cash flows. On January 1, 2017, the deferred tax assets associated with net operating losses increased by $122.2 million and the deferred tax asset associated with federal and state research credit increased by $30.8 million. These amounts were offset by a corresponding increase in the valuation allowance. The adoption of this standard did not impact our consolidated financial statements.
At December 31, 2017, we had federal and state net operating loss carryforwards of $1.47 billion and $1.55 billion, respectively, to reduce future taxable income that will expire at various dates through 2037. At December 31, 2017, we had federal and state research and development and investment tax credit carryforwards of $180.0 million and $15.3 million, respectively, available to reduce future tax liabilities that expire at various dates through 2037. At December 31, 2017, we had alternative minimum tax credits of $0.8 million that will either be available to reduce future regular tax liabilities or be fully refundable in 2021. We have a valuation allowance against the net operating loss and credit deferred tax assets as it is unlikely that we will realize these assets. Ownership changes, as defined in the Internal Revenue Code, including those resulting from the issuance of common stock in connection with our public offerings, may limit the amount of net operating loss and tax credit carryforwards that can be utilized to offset future taxable income or tax liability. The amount of the limitation is determined in accordance with Section 382 of the Internal Revenue Code. We have performed an analysis of ownership changes through December 31, 2017. Based on this analysis, we do not believe that any of our tax attributes will expire unutilized due to Section 382 limitations.
At December 31, 2017, 2016 and 2015, we had no unrecognized tax benefits.
The tax years 2014 through 2017 remain open to examination by major taxing jurisdictions to which we are subject, which are primarily in the United States, as carryforward attributes generated in years past may still be adjusted upon examination by the Internal Revenue Service or state tax authorities if they have or will be used in a future period. However, the statute of limitations remains open to the extent we utilize net operating losses or credits from earlier years. We have not recorded any interest and penalties on any unrecognized tax benefits since its inception.
|
11. |
ACCRUED EXPENSES |
Accrued expenses consist of the following at December 31, 2017 and 2016, in thousands:
|
|
|
At December 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Compensation and related |
|
$ |
33,826 |
|
|
$ |
13,689 |
|
|
Clinical trial and manufacturing |
|
|
20,004 |
|
|
|
11,756 |
|
|
Consulting and professional services |
|
|
7,276 |
|
|
|
2,788 |
|
|
Pre-clinical |
|
|
239 |
|
|
|
1,257 |
|
|
Other |
|
|
10,858 |
|
|
|
12,628 |
|
|
|
|
$ |
72,203 |
|
|
$ |
42,118 |
|
124
The following information has been derived from unaudited consolidated financial statements that, in the opinion of management, include all recurring adjustments necessary for a fair statement of such information.
|
|
|
Three Months Ended |
|
|||||||||||||
|
|
|
March 31, 2017 |
|
|
June 30, 2017 |
|
|
September 30, 2017 |
|
|
December 31, 2017 |
|
||||
|
|
|
(In thousands, except per share data) |
|
|||||||||||||
|
Revenues |
|
$ |
18,960 |
|
|
$ |
15,932 |
|
|
$ |
17,096 |
|
|
$ |
37,924 |
|
|
Operating expenses |
|
|
125,471 |
|
|
|
136,406 |
|
|
|
142,896 |
|
|
$ |
185,227 |
|
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(118,420 |
) |
|
$ |
(122,937 |
) |
|
$ |
(142,227 |
) |
|
Net loss per common share — basic and diluted |
|
$ |
(1.25 |
) |
|
$ |
(1.34 |
) |
|
$ |
(1.34 |
) |
|
$ |
(1.48 |
) |
|
Weighted-average common shares — basic and diluted |
|
|
86,027 |
|
|
|
88,098 |
|
|
|
91,828 |
|
|
|
96,139 |
|
|
|
|
Three Months Ended |
|
|||||||||||||
|
|
|
March 31, 2016 |
|
|
June 30, 2016 |
|
|
September 30, 2016 |
|
|
December 31, 2016 |
|
||||
|
|
|
(In thousands, except per share data) |
|
|||||||||||||
|
Revenues |
|
$ |
7,345 |
|
|
$ |
8,709 |
|
|
$ |
13,651 |
|
|
$ |
17,454 |
|
|
Operating expenses |
|
|
117,373 |
|
|
|
101,159 |
|
|
|
120,327 |
|
|
|
132,887 |
|
|
Net loss |
|
$ |
(102,974 |
) |
|
$ |
(90,129 |
) |
|
$ |
(104,071 |
) |
|
$ |
(112,934 |
) |
|
Net loss per common share — basic and diluted |
|
$ |
(1.21 |
) |
|
$ |
(1.05 |
) |
|
$ |
(1.21 |
) |
|
$ |
(1.32 |
) |
|
Weighted-average common shares — basic and diluted |
|
|
85,277 |
|
|
|
85,545 |
|
|
|
85,716 |
|
|
|
85,843 |
|
125
None.
Our management, with the participation of our chief executive officer (principal executive officer) and senior vice president, chief financial officer (principal financial officer), evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2017, our chief executive officer and senior vice president, chief financial officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s report on our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) and the independent registered public accounting firm’s report on the effectiveness of our internal control over financial reporting are included in Item 8, “Financial Statements and Supplementary Data” of this annual report on Form 10-K and are incorporated herein by reference.
No change in our internal control over financial reporting occurred during the fiscal quarter ended December 31, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
None.
126
Incorporated by reference from the information in our Proxy Statement for our 2018 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Incorporated by reference from the information in our Proxy Statement for our 2018 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
|
ITEM 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Incorporated by reference from the information in our Proxy Statement for our 2018 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Incorporated by reference from the information in our Proxy Statement for our 2018 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
Incorporated by reference from the information in our Proxy Statement for our 2018 Annual Meeting of Stockholders, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
127
(a) (1) Financial Statements
The following consolidated financial statements are filed as part of this report under “Item 8 — Financial Statements and Supplementary Data”:
(a) (2) List of Schedules
All schedules to the consolidated financial statements are omitted as the required information is either inapplicable or presented in the consolidated financial statements.
(a) (3) List of Exhibits
|
Exhibit No. |
|
Exhibit |
|
|
|
|
|
|
|
2.1†* |
|
||
|
|
|
|
|
|
3.1 |
|
||
|
|
|
|
|
|
3.2 |
|
||
|
|
|
|
|
|
4.1 |
|
||
|
|
|
|
|
|
10.1** |
|
||
|
|
|
|
|
|
10.2** |
|
||
|
|
|
|
|
|
10.3** |
|
||
|
|
|
|
|
|
10.4** |
|
||
|
|
|
|
|
|
10.5** |
|
||
|
|
|
|
|
128
|
Exhibit No. |
|
Exhibit |
|
|
|
|||
|
|
|
|
|
|
10.7** |
|
||
|
|
|
|
|
|
10.8** |
|
||
|
|
|
|
|
|
10.9** |
|
||
|
|
|
|
|
|
10.10** |
|
||
|
|
|
|
|
|
10.11** |
|
||
|
|
|
|
|
|
10.12** |
|
||
|
|
|
|
|
|
10.13** |
|
||
|
|
|
|
|
|
10.14** |
|
||
|
|
|
|
|
|
10.15** |
|
||
|
|
|
|
|
|
10.16** |
|
||
|
|
|
|
|
|
10.17 |
|
||
|
|
|
|
|
|
10.18 |
|
||
|
|
|
|
|
|
10.19 |
|
||
|
|
|
|
|
|
10.20 |
|
||
|
|
|
|
|
|
10.21 |
|
||
|
|
|
|
|
129
|
Exhibit No. |
|
Exhibit |
|
|
|
|||
|
|
|
|
|
|
10.23† |
|
||
|
|
|
|
|
|
10.24 |
|
||
|
|
|
|
|
|
10.25 |
|
||
|
|
|
|
|
|
10.26 |
|
||
|
|
|
|
|
|
10.27 |
|
||
|
|
|
|
|
|
10.28 |
|
||
|
|
|
|
|
|
10.29† |
|
||
|
|
|
|
|
|
10.30† |
|
||
|
|
|
|
|
|
10.31† |
|
||
|
|
|
|
|
|
10.32 |
|
||
|
|
|
|
|
|
10.33 |
|
||
|
|
|
|
|
|
10.34 |
|
||
|
|
|
|
|
|
10.35† |
|
||
|
|
|
|
|
130
|
Exhibit No. |
|
Exhibit |
|
|
|
|||
|
|
|
|
|
|
10.37† |
|
||
|
|
|
|
|
|
10.38† |
|
||
|
|
|
|
|
|
10.39† |
|
||
|
|
|
|
|
|
10.40† |
|
||
|
|
|
|
|
|
10.41† |
|
||
|
|
|
|
|
|
10.42† |
|
||
|
|
|
|
|
|
10.43† |
|
||
|
|
|
|
|
|
10.44† |
|
||
|
|
|
|
|
|
10.45† |
|
||
|
|
|
|
|
|
10.46† |
|
||
|
|
|
|
|
|
10.47 |
|
||
|
|
|
|
|
|
10.48† |
|
||
|
|
|
|
|
131
|
Exhibit No. |
|
Exhibit |
|
|
|
|||
|
|
|
|
|
|
10.50† |
|
||
|
|
|
|
|
|
10.51† |
|
||
|
|
|
|
|
|
10.52† |
|
||
|
|
|
|
|
|
10.53† |
|
||
|
|
|
|
|
|
10.54 |
|
||
|
|
|
|
|
|
10.55 |
|
||
|
|
|
|
|
|
21.1# |
|
||
|
|
|
|
|
|
23.1# |
|
Consent of PricewaterhouseCoopers LLP, an Independent Registered Public Accounting Firm |
|
|
|
|
|
|
|
31.1# |
|
||
|
|
|
|
|
|
31.2# |
|
||
|
|
|
|
|
|
32.1# |
|
||
|
|
|
|
|
|
32.2# |
|
||
|
|
|
|
|
132
|
Exhibit No. |
|
Exhibit |
|
|
|
The following materials from Registrant’s Annual Report on Form 10-K for the year ended December 31, 2017, formatted in XBRL (Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Comprehensive Loss, (iii) the Consolidated Statements of Stockholders’ Equity, (iv) the Consolidated Statements of Cash Flows and (v) Notes to Consolidated Financial Statements.
|
||
|
|
|
||
|
* |
|
Schedules, exhibits and similar supporting attachments or agreements to the Stock Purchase Agreement are omitted pursuant to Item 601(b)(2) of Regulation S-K. The Registrant agrees to furnish a supplemental copy of any omitted schedule or similar attachment to the Securities and Exchange Commission upon request. |
|
|
** |
|
Management contracts or compensatory plans or arrangements required to be filed as an exhibit hereto pursuant to Item 15(a) of Form 10-K. |
|
|
† |
|
Indicates confidential treatment requested as to certain portions, which portions were omitted and filed separately with the Securities and Exchange Commission pursuant to a Confidential Treatment Request. |
|
|
# |
|
Filed herewith. |
|
Not applicable.
133
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized, on February 15, 2018.
|
ALNYLAM PHARMACEUTICALS, INC. |
||
|
|
|
|
|
By: |
|
/s/ John M. Maraganore, Ph.D. |
|
|
|
John M. Maraganore, Ph.D. |
|
|
|
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, the Report has been signed below by the following persons on behalf of the Registrant and in the capacities indicated as of February 15, 2018.
|
Name |
|
Title |
|
|
|
|
|
/s/ John M. Maraganore, Ph.D. |
|
Director and Chief Executive Officer (Principal Executive Officer) |
|
John M. Maraganore, Ph.D. |
|
|
|
|
|
|
|
/s/ Manmeet S. Soni |
|
Senior Vice President, Chief Financial Officer |
|
Manmeet S. Soni |
|
(Principal Financial Officer) |
|
|
|
|
|
/s/ Michael P. Mason |
|
Vice President of Finance and Treasurer (Principal Accounting Officer) |
|
Michael P. Mason |
|
|
|
|
|
|
|
/s/ Dennis A. Ausiello, M.D. |
|
Director |
|
Dennis A. Ausiello, M.D. |
|
|
|
|
|
|
|
/s/ Michael W. Bonney |
|
Director |
|
Michael W. Bonney |
|
|
|
|
|
|
|
/s/ John K. Clarke |
|
Director |
|
John K. Clarke |
|
|
|
|
|
|
|
/s/ Marsha H. Fanucci |
|
Director |
|
Marsha H. Fanucci |
|
|
|
|
|
|
|
/s/ Steven M. Paul, M.D. |
|
Director |
|
Steven M. Paul, M.D. |
|
|
|
|
|
|
|
/s/ David E. I. Pyott |
|
Director |
|
David E. I. Pyott |
|
|
|
|
|
|
|
/s/ Paul R. Schimmel, Ph.D. |
|
Director |
|
Paul R. Schimmel, Ph.D. |
|
|
|
|
|
|
|
/s/ Amy W. Schulman |
|
Director |
|
Amy W. Schulman |
|
|
|
|
|
|
|
/s/ Phillip A. Sharp, Ph.D. |
|
Director |
|
Phillip A. Sharp, Ph.D. |
|
|
|
|
|
|
|
/s/ Kevin P. Starr |
|
Director |
|
Kevin P. Starr |
|
134