Attached files
| file | filename |
|---|---|
| EX-99.4 - EX-99.4 - SYNLOGIC, INC. | d380947dex994.htm |
| EX-23.1 - EX-23.1 - SYNLOGIC, INC. | d380947dex231.htm |
| EX-99.5 - EX-99.5 - SYNLOGIC, INC. | d380947dex995.htm |
| EX-99.3 - EX-99.3 - SYNLOGIC, INC. | d380947dex993.htm |
| EX-99.2 - EX-99.2 - SYNLOGIC, INC. | d380947dex992.htm |
| 8-K/A - 8-K/A - SYNLOGIC, INC. | d380947d8ka.htm |
Exhibit 99.1
The following is an excerpt of portions of the prospectus contained in the Form S-4 registration statement (File No. 333-218885) as declared effective by the Securities and Exchange Commission on July 13, 2017. Such information is as of July 13, 2017 (unless an earlier date is indicated).
SYNLOGIC BUSINESS
Overview
Synlogic™ is pioneering the development of Synthetic Biotic™ medicines: a novel class of living medicines intended to treat a broad range of human diseases, ranging from genetic and acquired metabolic disorders to inflammation and cancer. Synthetic Biotic medicines are generated from Synlogic’s proprietary drug discovery and development platform. Synlogic applies the principles and tools of synthetic biology to engineer beneficial probiotic bacteria to perform or deliver critical therapeutic functions, compensating for missing or damaged pathways in patients with these serious diseases. As living medicines, Synthetic Biotic medicines are designed to sense a local disease context within a patient’s body and to respond by metabolizing toxic substances or delivering combinations of therapeutic factors.
Synlogic’s initial focus is on metabolic diseases with potential to be corrected following oral delivery of a living medicine to the gut. This includes a group of rare genetic diseases called inborn errors of metabolism (“IEMs”), as well as acquired metabolic diseases caused by organ dysfunction:
| • | Patients with certain IEMs are born with faulty genes that block the transformation of food into energy or prevent the elimination of toxic byproducts of metabolism. |
| • | Patients with acquired metabolic diseases have similar defects in the metabolism of food, but these defects arise due to the impaired function of organs responsible for food metabolism, such as the liver. |
In patients with these diseases, byproducts of failed metabolism can accumulate to toxic levels and cause serious health consequences throughout the body. Synthetic Biotic medicines are designed as oral therapies to act in the gut to convert toxic metabolites into non-toxic byproducts and, as a result, reduce toxic metabolite levels in the systemic circulation and tissues. Synthetic Biotic medicines are engineered to clear toxic metabolites specific to each metabolic disease and have the potential to provide meaningful benefits to patients suffering from these debilitating conditions.
Synlogic initiated a Phase 1 clinical trial for its lead Synthetic Biotic program, SYNB1020, in June 2017. SYNB1020 is in development as an oral treatment for patients with hyperammonemia. In patients with hyperammonemia, ammonia accumulates in the body and becomes toxic leading to neurocognitive crisis and risk of long-term cognitive or behavioral impairment, coma, or death. Hyperammonemic conditions include a group of IEMs known as Urea Cycle Disorders (“UCD”), and hepatic encephalopathy (“HE”) in liver disease patients. SYNB1020 is designed to remove excess ammonia from the gut by converting it into the beneficial amino acid arginine, with potential to result in lowered ammonia levels in the blood. Synlogic’s second program, SYNB1618, is an oral therapy intended for the treatment of phenylketonuria (“PKU”), an IEM in which the amino acid phenylalanine accumulates as a result of genetic defects, becoming toxic to the brain and leading to neurological dysfunction. SYNB1618 is designed to have activity in the gut of patients to reduce excess phenylalanine to result in normalization of levels in the blood and tissues. Synlogic is planning to initiate a Phase 1 clinical trial for SYNB1618 in the first half of 2018. Synlogic’s earlier metabolic disease pipeline includes discovery-stage product candidates for additional IEMs, such as maple syrup urine disease (“MSUD”), isovaleric acidemia (“IVA”) and organic acidemias.
Synlogic’s platform also has the potential to generate clinically meaningful therapies for patients affected by immune-mediated diseases and cancer. Synthetic Biotic medicines are designed to locally deliver combinations of complementary therapeutics to treat these complex disease states. Synlogic’s portfolio of immuno-oncology programs is designed to deliver a combination of activities to modify the tumor microenvironment, activate the immune system and result in tumor reduction. In addition, Synlogic has established a strategic collaboration with the integrated pharmaceutical company AbbVie to develop Synthetic Biotic-based treatments for inflammatory bowel disease (“IBD”) such as Crohn’s disease and ulcerative colitis. While Synlogic intends to develop and commercialize therapeutic candidates for the treatment of IEMs on its own, Synlogic may consider entering additional strategic partnerships in the future to maximize the value of Synlogic’s programs and its Synthetic Biotic platform.
To progress its pipeline, Synlogic collaborates with key disease experts who have developed robust models of relevant diseases to guide selection of Synlogic’s development candidates and to inform its translational medicine strategy. Synlogic focuses on indications with clear biomarkers associated with disease progression that enable straightforward, early and ongoing assessment of potential clinical benefit throughout the development process. Synlogic’s collaboration and intellectual property strategies additionally focus on building or leveraging existing third-party expertise in therapeutic research, pre-clinical and clinical development, regulatory affairs, manufacturing and commercialization, while also enhancing Synlogic’s industry-leading position in synthetic biology and metabolic engineering.
Synlogic has assembled a management team of seasoned biopharmaceutical executives with extensive, relevant experience at leading pharmaceutical companies such as Pfizer Inc. (“Pfizer”), GlaxoSmithKline, Biogen, Inc. (“Biogen”), AstraZeneca, Millennium Pharamceuticals, Inc. (“Millennium Pharmaceuticals”) (now Takeda Pharmaceutical Company Limited) and MedImmune, as well as the National Institute of Health. Synlogic is supported by the Synlogic Board of Directors and the Synlogic scientific advisory board, each of which offer complementary experience in drug discovery and development, as well as expertise in building public companies, management, and business development. Synlogic’s founding science came from the labs of Professors James Collins and Timothy Lu from the Massachusetts Institute of Technology (“MIT”), who remain highly engaged in guiding development and application of Synlogic’s platform.
Synlogic’s pipeline of programs is shown below.
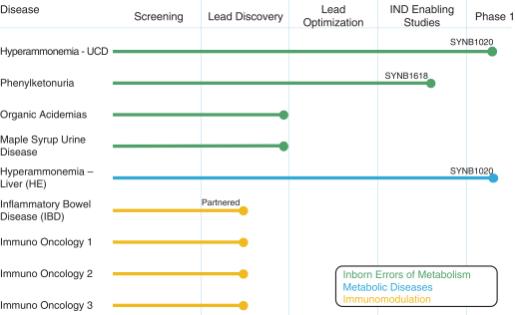
As Synlogic advances its lead programs, Synlogic continues to learn and improve the flexibility, manufacturability and translatability of its Synthetic Biotic platform, which will inform all future portfolio programs. Consequently, Synlogic believes it has a robust engine for building a sustainable pipeline of novel, living medicines across a range of diseases. Through the strength of Synlogic’s internal team and network of partners, Synlogic believes it can deliver on the promise of Synthetic Biotic medicines to improve the lives of patients with significant unmet medical needs.
Synlogic’s Strategy
Synlogic’s goal is to use its Synthetic Biotic platform to design, develop and commercialize living medicines to transform the lives of patients for whom conventional treatment approaches are either not available or have limited efficacy and safety. To achieve its goal, Synlogic is pursuing the following key strategies:
Rapidly Advance Clinical Development of the SYNB1020 Hyperammonemia Program. Synlogic’s lead Synthetic Biotic program is for the treatment of hyperammonemic conditions such as UCD and HE. SYNB1020 is an oral therapy designed to deliver a complementary metabolic pathway in the gut with the intended consequence of removing excess ammonia in the blood. SYNB1020 has received orphan drug designation and in June 2017 was granted Fast Track designation for UCD from the FDA. Synlogic initiated its first Phase 1 clinical trial to assess safety, tolerability and pharmacokinetics in healthy volunteers in June 2017. Assuming success in the Phase 1 clinical trial, Synlogic plans to initiate an HE study to better understand safety, tolerability and therapeutic potential of SYNB1020. Synlogic expects to start the study in the first half of 2018 and to have topline data by the end of 2018. Similarly, based on the results of the Phase 1 clinical trial, Synlogic expects to begin a clinical trial in UCD by mid-2018 with data expected in the first half of 2019.
Complete IND-Enabling Activities to Advance SYNB1618 into Clinical Development. Synlogic’s second IEM program is an oral therapy for PKU. SYNB1618 is designed to act from the gut to convert excess phenylalanine to non-toxic metabolites and thereby prevent phenylalanine from accumulating in the blood, becoming toxic and leading to neurological dysfunction. Synlogic expects to initiate a Phase 1 trial for this candidate in the first half of 2018. The Phase 1 design will include healthy volunteers, as well as an adult patient cohort, to assess safety, tolerability and pharmacodynamics. Synlogic expects to have final results from the healthy volunteer study, including insights from a mechanistic biomarker, by the end of 2018 and insights regarding therapeutic potential by the first half of 2019.
Expand Synlogic’s Pipeline by Targeting Additional Rare Genetic Metabolic Diseases. Synlogic plans to continue to leverage its expertise from its lead programs to accelerate development of Synlogic’s pipeline of clinical candidates for IEMs. For example, Synlogic’s portfolio includes two additional discovery-stage Synthetic Biotic programs in lead optimization, including one for MSUD/IVA and the other for propionic acidemia (“PA”)/methylmalonic acidemia (“MMA”), organic acidemias with high unmet need for which there are biomarkers that Synlogic believes can guide efficient product development programs.
Maximize the Value of the Synthetic Biotic Platform in Broader Metabolic and Inflammatory Diseases and in Immuno-Oncology Leveraging Strategic Partnerships. Synlogic’s Synthetic Biotic platform and product discovery and development capabilities offer the potential to generate multiple clinically meaningful treatments for a broad set of metabolic and inflammatory diseases as well as cancer. For these indications, there is opportunity to reset a metabolic or immune dysfunction with a lower risk of systemic toxicity than other modalities. To achieve this, oral Synthetic Biotic medicines may be designed to deliver a combination of mechanisms following oral administration for activity in the gut or intra-tumoral injection. For example, Synlogic is establishing a discovery-stage immuno-oncology portfolio.
Synlogic expects to continue to explore strategic partnerships that would leverage the complementary capabilities of its partners to develop Synthetic Biotic medicines for these broader groups of patients in need. Synlogic’s current partnership with AbbVie is focused on the discovery and development of Synthetic Biotic-based therapies for the treatment of IBD, and in June 2017 Synlogic announced its first milestone for this program. While Synlogic intends to develop and commercialize its programs for IEMs, Synlogic may consider entering into additional strategic partnerships to maximize the value of its Synthetic Biotic platform in these more common indications.
Expand the Synthetic Biotic Platform to Lead in the Discovery and Development of Additional Living Medicines and Enabling Technologies. Synlogic intends to advance in the field of living medicines by continuing to innovate and broaden the potential of its Synthetic Biotic platform to deliver clinically meaningful benefits for patients. Synlogic plans to build on its expertise in design, optimization and manufacturing to further develop the Synthetic Biotic platform as a reproducible and scalable engine for generating a pipeline of product candidates that address a broad range of diseases.
Protect and Leverage Synlogic’s Intellectual Property Portfolio and Patents. Synlogic believes that it has a broad intellectual property portfolio that includes patents and patent applications relevant to the engineering, development, manufacturing and formulation of human therapeutic products based on synthetic biology and the metabolic engineering of probiotics. Synlogic intends to continue to protect and leverage its intellectual property assets by maintenance and expansion of its worldwide portfolio of intellectual property, including through the pursuit of composition of matter and other intellectual property directed to its Synthetic Biotic programs and its technology platform.
Synlogic’s Focus: Living Medicines
Synlogic is developing and advancing a novel approach to creating living medicines—therapeutics designed to sense a local disease context within a patient’s body and to respond by metabolizing toxic substances or delivering combinations of therapeutic factors. Synlogic applies the tools and principles of synthetic biology to engineer beneficial probiotic bacteria to perform or deliver critical therapeutic functions, compensating for missing or damaged pathways in patients with metabolic diseases, inflammation and cancer.
Synlogic believes living medicines have unique advantages as potential therapeutics. Living biologic cells can carry out functions that cannot be performed by many conventional drug treatments, such as small molecules or antibodies. While many conventional treatments can address one molecular dysfunction, living medicines can compensate for the dysfunction of entire processes or pathways missing in disease and required for health. By contrast to conventional therapeutics that engage a single target, living medicines can be designed to dynamically sense diseased environments and respond with a programmed and combinatorial effect. Moreover, a living medicine can also function “catalytically,” as a single living cell can carry out multiple cycles of the intended therapeutic activity during its time in the patient.
There is opportunity to expand the impact that previous cell therapies have had by applying the well-established tools of synthetic biology to probiotic bacteria, converting them into efficient therapeutic engines. Probiotic bacteria are non-pathogenic bacteria isolated from the human microbiota widely used as supplements believed to provide health benefits. To confer a therapeutic effect, Synlogic leverages basic biological properties of bacteria to develop engineered probiotics. Bacteria have evolved over several billion years to adapt, survive, and carry out active metabolism in many different environments. They are also amenable to genetic manipulation. Synlogic’s intention is to lead in the discovery and development of Synthetic Biotic therapies as safe living medicines capable of robust and precise pathway complementation and therapeutic benefit.
Leveraging Synthetic Biology and Metabolic Engineering of Probiotic Bacteria to Produce Living Medicines
Synlogic’s proprietary Synthetic Biotic discovery and development platform combines synthetic biology and metabolic engineering to re-design the genetic circuitry of beneficial probiotic bacteria and generate living medicines.
Synthetic Biology
Synthetic biology is an emerging and rapidly-evolving discipline that applies engineering principles to biological systems to enable rational, design-based control of cellular function for a specific purpose. Biological systems are governed by DNA sequences, or genes, that code for the production and regulation of proteins, metabolites and other molecules. The regulation of the function of proteins occurs via complex biochemical and cellular reactions working through intricate signaling pathways. Synthetic biology allows manipulation of these pathways to direct a desired therapeutic outcome. While efforts have been made to apply these principles across industries, Synlogic believes it is a leader in deploying synthetic biology for the treatment of human disease.
Synlogic scientists genetically engineer a beneficial probiotic bacterium with “wiring” or biological circuits to direct cellular biological processes in a manner analogous to designing electrical circuits. The critical parts of an engineered Synthetic Biotic medicine include (1) the chassis, or probiotic bacterium, (2) the effector module, which is a gene or pathway encoding the core biological activity that provides the therapeutic function, and (3) tunable switches to precisely determine the circumstances under which the effector module will be activated, as well as the strength, performance and output of the effectors themselves. Synlogic aims to precisely control the amount, location and activity of its Synthetic Biotic medicines to address a broad range of disease.
Schematic of the Synthetic Biotic Platform Components: Chassis, Effector, Switch
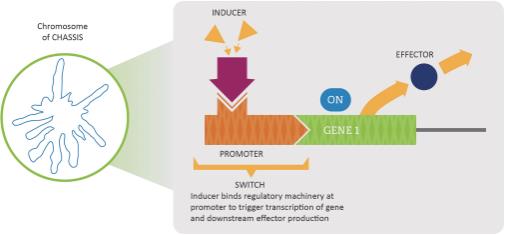
Metabolic Engineering of Probiotic Bacteria
(1) The Chassis: Synlogic’s Synthetic Biotic platform employs well-characterized bacteria used as probiotics to serve as the chassis upon which Synlogic builds its living medicines. Synlogic’s initial programs use E. coli Nissle, which is one of many non—pathogenic strains isolated from the human microbiota. E. coli Nissle has been used as a probiotic bacterial supplement for the last 20 years to promote gut health. E. coli Nissle is a non-colonizing probiotic in that it has recently been shown in the clinic to be rapidly cleared from most individuals with no significant safety issues. Synlogic believes E. coli Nissle’s widespread use as a probiotic is evidence of its utility as a safe background chassis to apply synthetic biology to confer a therapeutic effect.
(2) Building the Effector Module: E. coli Nissle’s metabolic systems are well-understood and extremely adaptable, making it an excellent organism for introducing new or enhanced activities to treat human disease. The highly flexible nature of its genetic and metabolic machinery provides a robust cellular context into which genetic information encoding proteins and pathways to correct for disease can be introduced with high efficiency and little or no damage to the fitness of the bacterium. This provides the potential for excellent reproducibility, stability, and activity during manufacturing. Moreover, the advanced nature of the synthetic biology toolkit available for E. coli Nissle enables the rapid iterative design, assembly, and testing of prototype product candidates and remains unique among other bacterial and cellular engineering approaches. Synlogic has leveraged proprietary tools, know how and intellectual property to build multiple Synthetic Biotic lead strains that produce a therapeutically relevant effect in laboratory experiments. Progression of these strains as product candidates in diseases with high unmet need is based on prioritizing those with feasible drug development paths in terms of availability of informative animal models and existence of biomarkers to guide efficient clinical development.
(3) Tunable Switches: Synlogic also designs and engineers proprietary switches to mediate the activity of the new pathways it introduces, with the aim of controlling the therapeutic output, or effector, of Synthetic Biotic medicines. To optimize the fitness of a Synthetic Biotic strain, it is critical that the effector is activated only at the proper time and place. The switches Synlogic has developed are based on engineering DNA elements call “inducible promoters” to sense and respond to disease states, specific environmental signals, or exogenously added inducing molecules. The goal is to discover and develop Synthetic Biotic medicines programmed with switches to produce its therapeutic effect at precisely the right time and location such as the anaerobic environment of the gut, in the context of local inflammation, and in response to other pathogenic factors.
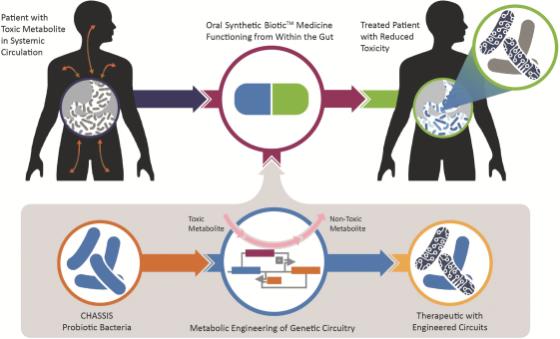
While applicable across metabolic, inflammatory and immuno-oncology indications, Synlogic’s initial Synthetic Biotic programs are designed for rare metabolic diseases in which a toxic metabolite accumulates in the body and causes systemic toxicity. Synlogic believes that the Synthetic Biotic platform can be leveraged to engineer a safe probiotic with enhanced genetic circuitry and the capability to transform a toxic metabolite into one that is non-toxic or even beneficial. The resulting Synthetic Biotic medicines are built to be taken orally and function from within the gut. Metabolites produced by both a person’s organs and by our endogenous flora circulate or flux between the human gastrointestinal (“GI”) tract and blood circulation and vice versa. As Synlogic’s Synthetic Biotic medicines transit through the GI tract, they are designed to have activity in the gut and to take advantage of this flux, ultimately reducing the systemic levels of toxic metabolites in the blood to treat rare metabolic diseases.
Schematic of the Synthetic Biotic Platform to Engineer Probiotic Bacteria
Advantages of Synlogic’s Synthetic Biotic Living Medicines
Synlogic believes its platform has the potential to provide safe and effective therapies for patients given several attributes of Synlogic’s Synthetic Biotic approach:
Unique Mechanisms to Treat Systemic Metabolic and Immune Dysfunction: Local Pathway Complementation or Therapeutic Delivery
Synlogic’s Synthetic Biotic platform allows it to engineer living medicines that act as engines capable of metabolic pathway compensation and essentially replace what a patient cannot do with his or her somatic organs, such as the liver. Unlike traditional small molecule and biologic therapeutics, Synthetic Biotic medicines can be designed with multiple pathway components optimized to consume or transform unwanted metabolites or produce those that are medically beneficial. This approach is well suited to regulate the amount of a metabolic byproduct in a patient’s body, particularly when there is unconstrained metabolite flux between the systemic circulation. Synlogic’s Synthetic Biotic programs for rare metabolic diseases are designed to be dosed orally, to act locally while transiting through the gut and, as a consequence, to decrease toxic metabolite levels in the blood, thereby providing a systemic therapeutic benefit to the patient.
In addition, Synlogic is developing Synthetic Biotic medicines with the potential to normalize function of a dysregulated immune system. In inflammatory and autoimmune indications, this may be achieved by producing anti-inflammatory metabolites and proteins particularly for diseases of the GI tract. Synthetic Biotic medicines can also be designed to consume or produce metabolites or secrete and display proteins that may shift the tumor microenvironment of the immune system towards anti-tumor activity.
Combination and Local Delivery of Multiple Mechanisms in One Therapy for Greater Efficacy and Enhanced Safety
Currently, many complex diseases, such as inflammatory and autoimmune indications and oncology, require that patients be treated with a combination of therapeutic agents, often resulting in poor tolerability, multiple adverse events and increased cost of therapy. Synlogic’s approach is to leverage the adaptability of E. coli Nissle to enable the combination of multiple activities into one therapy, which therefore could have greater efficacy while avoiding the impact of multiple separate systemic therapies.
Moreover, Synlogic’s Synthetic Biotic medicines are based on beneficial probiotic bacteria derived from the natural human gut. A chassis such as E. coli Nissle is suited for local delivery, either orally or through intra-tumoral injection. Synlogic believes that, when delivered locally, Synthetic Biotic medicines have the potential to avoid the risks of dose-limiting side effects often associated with systemic therapies, especially when combinations of systemic therapies are required.
Ability to Tune and Enhance Efficacy in Context of Disease
Synlogic’s Synthetic Biotic platform includes a suite of switches to permit precise control of the timing and amount of therapeutic effect produced. Synthetic Biotic therapies may be designed such that they are activated to produce the desired effect in a particular disease environment, such as sites of inflammation. This tuning has the potential to increase the therapeutic window by increasing the margin between the level of medicine needed for efficacy relative to the risk of systemic toxic side effects.
Advantages of Synlogic’s Synthetic Biotic Drug Development Platform
The Synthetic Biotic platform employs a well-characterized probiotic bacterium with a proven safety record that is readily modified using state-of-the-art synthetic biology tools. This unique combination of features allows Synlogic to rapidly develop prototypes for the treatment of human diseases with unmet medical need. Advantages to discovery, development, manufacturing and commercialization, include unique mechanisms of action enabling a broad range of therapeutic applications and rational design to achieve predictable drug-like properties:
Unique Mechanisms of Action Enabling a Broad Range of Therapeutic Applications
Synlogic’s approach allows it to engineer two types of mechanistic activities into Synlogic’s Synthetic Biotic medicines. These activities may be further improved for therapeutic effect when combined or when under the control of tunable switches that determine when the mechanisms should be activated.
| • | Metabolic Pathway Complementation : Synthetic Biotic medicines may be programmed with entire pathways to degrade unwanted molecules or produce those that are beneficial. Synlogic believes metabolic pathway complementation is advantageous as compared to gene, RNA or enzyme replacement therapies that are limited to targeting a single gene or protein defect and may require several unique drug products to address genetically heterogeneous patient populations. By compensating with an entire pathway, Synthetic Biotic medicines may provide a therapeutic solution to broader disease populations as a single engineered therapeutic. Moreover, in the IEMs space Synlogic believes its approach has advantages versus these other modalities that may be limited by delivery, transduction efficiency, duration of therapeutic expression and unclear potential for long-term dosing. Given the potential for chronic oral dosing, Synthetic Biotic medicines may have benefits in terms of prediction of dose, reversibility of activity and more traditional pricing strategies. |
| • | Production of One or More Protein Effectors at the Site of Disease : Combinations of cytokine, antibody and protein therapies have potential for great effect, but can be restricted by dose-limiting side effects when administered systemically. The potential to program the control of expression of one or more proteins at the local disease site represents a unique approach to targeted therapy. Synlogic has developed proprietary integration systems to direct stable insertion of multiple genetic circuits and pathways into optimal chromosomal locations, or “landing pads,” of E. coli Nissle. This enables efficient expression of multiple genes encoding desired enzymes and other proteins. Synlogic has also developed approaches to enhance the secretion of protein effectors to the extracellular environment. For example, in the case of inflammatory conditions, Synthetic Biotic medicines may be programmed to detect inflammation and respond with the production of one or more anti-inflammatory molecules. In oncology, Synlogic’s programs are being designed to secrete effectors to promote immune system activity against a tumor. These activities may further be combined with metabolic complementation pathways. By incorporating multiple actions, Synthetic Biotic medicines have the potential to address complex diseases while avoiding the risk of systemic toxicity and reducing development costs associated with combining systemic therapies. |
Rational Design to Achieve Predictable Drug-like Properties
Synlogic has demonstrated the ability to move a program from concept to clinical development in as little as three years for its lead program. Features of Synlogic’s Synthetic Biotic platform enable a highly efficient drug discovery and development process and have the potential to advance product candidates more rapidly and efficiently than is typically possible with other novel or emerging modalities. These include:
| • | Single Strain as Safe Chassis . There are several benefits of employing a single, safe and well-characterized probiotic bacterium such as E. coli Nissle as the background chassis. First, because Synlogic’s lead programs are based on E. coli Nissle, experience can be leveraged broadly across the portfolio, further optimizing the efficiency and reproducibility of discovery, development and manufacturing efforts. Next, the non-colonizing nature of E. coli Nissle can be combined with engineering approaches to optimize safety in terms of impact on the patient and the environment. E. coli Nissle can be engineered to require a specific exogenous nutrient supplement for growth, which limits the ability to replicate in the human body and environment. By controlling replication, Synlogic can control the number of cells being administered to a patient. Also, dependence on an essential nutritional supplement not available in the environment reduces biocontainment risk. Moreover, the risk of Synthetic Biotic medicines to the environment is further limited given that it is disadvantaged in terms of fitness due to its modifications. |
| • | Predictive Pharmacology and Biomarkers . Synthetic Biotic programs are designed to achieve a target activity, and the platform supports an iterative design-build-test cycle to improve performance for achieving this target. For example, Synthetic Biotic programs can be optimized by including multiple copies or regulated control of certain genes, by adding transporters for particular substrates or by optimizing enzymes for basic bacterial metabolism. These tools enable rational and iterative engineering cycles in the discovery phase. |
Biomarkers as indicators of mechanistic and clinical activity may also be engineered into Synthetic Biotic medicines from the beginning to drive optimization and decision-making. By assessing the activities of Synlogic’s Synthetic Biotic programs in in vitro and in vivo pre-clinical models, Synlogic can model activity in humans. As Synlogic progresses into clinical studies, Synlogic expects its predictive pharmacology models will be further refined to inform dosing and development decisions for its additional programs.
| • | Stability and Manufacturing . Synlogic’s lead Synthetic Biotic programs have advanced the platform by defining manufacturing processes that can be deployed against the entire portfolio. Manufacturing efforts have demonstrated reproducibility, yield and stability during small, medium and Phase 1 clinical-scale manufacturing efforts. Moreover, Synlogic’s use of synthetic biology switches permits the precise control of engineered metabolic pathway activation. This can be used to suppress effector activity during manufacturing, enabling generation of biomass and robust, cost-efficient scale up of product candidates. |
Synlogic’s Product Pipeline
Synlogic’s approach to selecting its initial programs is based on the potential of the Synthetic Biotic platform to uniquely address conditions in which there is (1) unmet medical need with (2) well understood biology that is (3) based on an imbalance of a metabolite and (4) where that metabolite is available within or originates from the gut lumen. Additional considerations include the availability of animal models, relevant biomarkers and feasible clinical development paths. Synlogic’s initial clinical and pre-clinical programs are focused on certain IEMs that share these characteristics. When delivered orally, Synthetic Biotic medicines are designed to act from the gut to compensate for the dysfunctional metabolic pathway with the intended consequence of reducing the levels of the toxic metabolites systemically. Synlogic believes success in IEMs will enable it to demonstrate the potential of its oral Synthetic Biotic medicines to address metabolic dysfunction, while bringing meaningful change to lives of patients suffering from these debilitating conditions.
Synlogic’s two lead therapeutic programs are being developed for the treatment of IEMs; UCD and PKU. There is unmet need for both indications, as well as an opportunity to reduce toxic metabolites that originate from the gut. Both also inform the potential of the Synthetic Biotic platform in unique ways. Synlogic’s lead product candidate, SYNB1020, is designed as an oral therapy to remove excess ammonia from the blood by accessing ammonia in the lower GI tract and converting it into arginine, a natural amino acid used in normal growth and metabolism. The conversion of ammonia into arginine is based on enhancing an enzyme pathway endogenous to E. coli Nissle. The program has clinical application in that multiple disease indications involve toxic ammonia levels. In addition to UCD, Synlogic is exploring SYNB1020 to treat patients with HE secondary to chronic liver disease to stave off episodes of cognitive dysfunction. SYNB1020 has also received orphan drug designation and in June 2017 was granted Fast Track designation for UCD from the FDA. Synlogic initiated a Phase 1 clinical trial of SYNB1020 in healthy volunteers in June 2017.
Synlogic’s second IEM program, SYNB1618 for PKU, is designed to act in the upper GI tract to reduce excess phenylalanine in the blood. Unlike SYNB1020, the engineering of SYNB1618 is based on leveraging enzymes from other bacterial species to optimize the conversion of phenylalanine to non-toxic metabolites. SYNB1618 has demonstrated activity in a rodent model of PKU. Synlogic expects to initiate a Phase 1 clinical trial for this program in the first half of 2018. Synlogic’s research-stage IEM portfolio includes Synthetic Biotic programs for (1) MSUD and IVA and (2) PA/MMA. These are rare metabolic deficiencies with no approved therapies in which the toxic accumulation of leucine and organic acids, respectively, can lead to neurological decline and death.
For more common metabolic, inflammatory and immuno-oncology indications with more complex biology, clinical and commercial paths, Synlogic will explore strategic partnerships to exploit the potential of the Synthetic Biotic platform. Synlogic’s collaboration with AbbVie for the discovery and development of Synthetic Biotic therapies for the treatment of IBD is one such example. Synlogic is also developing a portfolio of immuno-oncology programs using a rational approach to select combinations of relevant mechanisms to address specific tumor types. Synlogic’s strategy is to alter the state of the tumor microenvironment to one that is “anti-tumor” through Synthetic Biotic medicines that consume or combine effectors that promote immune system activation, reverse immunosuppression, expand tumor antigen-specific T cells and/or remodel the tumor protective stroma to tip the balance toward an anti-tumor effect. Synlogic is currently working on three discovery-stage programs, which are diversified in terms of indications, combinations of mechanisms and routes of administration.
Synlogic’s Initial Programs: Overview of IEMs
Patients with IEMs are born with faulty genes that result in the loss of a necessary enzyme function in an essential metabolic pathway and prevent the body from metabolizing commonly occurring byproducts of digestion. In patients with IEMs, these byproducts can accumulate to toxic levels in the gut and systemically throughout the body to cause serious health consequences, including irreversible neurological dysfunction. Although in some cases diet modification can be beneficial, unmet medical need remains as there are few current treatments for IEMs.
While there are hundreds of genetic conditions grouped as IEMs, individual IEMs are considered orphan diseases, with each disease affecting fewer than 200,000 patients in the United States and fewer than five per 10,000 people in the European Union. IEMs include diseases of the urea cycle, amino acid metabolism and organic acid accumulation, among others. Many IEMs are thought to be underdiagnosed given the rarity of the conditions, potential for infant death, lack of available diagnostics and limited therapies.
SYNB1020 for Hyperammonemia: Urea Cycle Disorder and Hepatic Encephalopathy
Hyperammonemia is a metabolic condition characterized by an excess of ammonia in the blood. In healthy individuals, ammonia is primarily produced in the intestine as a byproduct of protein metabolism and microbial degradation of nitrogenous-containing compounds. Ammonia itself is then converted to urea in the liver and is excreted in urine. However, if the liver’s ability to convert ammonia to urea is compromised, either due to a genetic defect or acquired liver disease, ammonia accumulates in the blood. Elevated blood ammonia levels are toxic to the brain and can have severe consequences including neurologic crises requiring hospitalization, irreversible cognitive damage and death.
SYNB1020, Synlogic’s lead Synthetic Biotic program, is a genetically engineered strain of E. coli Nissle designed to deliver a complementary metabolic pathway to the gut to reduce excess ammonia in the blood in individuals with disease. The SYNB1020 program offers potential in treating multiple indications associated with toxic ammonia levels, including UCD and HE, and has demonstrated reduction in blood ammonia levels in rodent models of hyperammonemia. SYNB1020 has received orphan drug designation and in June 2017 was granted Fast Track designation for UCD from the FDA. Synlogic initiated a Phase 1 clinical trial of SYNB1020 in healthy volunteers in June 2017. Assuming success in this study, Synlogic plans to initiate two studies in UCD and HE to better understand the safety and tolerability of SYNB1020 in patients. Synlogic intends to clinically explore ammonia lowering in these patients to drive design of confirmatory studies for SYNB1020.
Overview of UCD
UCDs are a group of rare but serious and potentially fatal, genetic diseases. The urea cycle is an enzymatic pathway in which waste nitrogen, produced as a byproduct of protein metabolism, is converted into urea by the liver and eliminated from the body through urine. Patients with a UCD carry a deficiency in one of the six enzymes necessary for completion of the urea cycle, resulting in accumulation of waste nitrogen throughout the body in the form of ammonia, a substance that is highly toxic even in small amounts.
Functional Urea Cycle
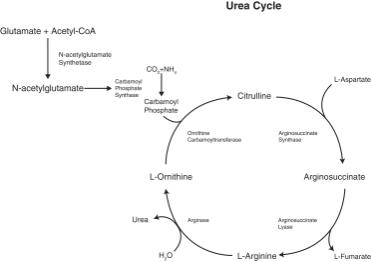
UCD patients have intermittent periods of hyperammonemia, the symptoms of which can range from mild (loss of appetite, vomiting, and lethargy) to a severe hyperammonemic crisis associated with long-term cognitive or behavioral impairment, toxic encephalopathy, and even death. Symptoms often depend on the severity of the enzyme deficiency, and patients with the most severe disease present shortly after birth. Hyperammonemia in newborn infants due to UCD could be catastrophic and is associated with 24% mortality. Patients with later onset disease could suffer from a period of hyperammonemia that is often triggered by stress or illness (surgery, trauma, or drugs) resulting in severe neurological symptoms and associated with a high risk of mortality.
While it is difficult to estimate the exact incidence and prevalence of UCD, as it is thought that many patients go undiagnosed, it is estimated that UCD occurs in approximately one in 35,000 births in the United States. Based on analysis of the newborn screening data and demographic data from the UCD Longitudinal Registry Study sponsored by the NIH, Synlogic believes the size of the diagnosed prevalent population in the United States to be approximately 2,000 patients and that approximately two-thirds of these patients are under 18 years of age.
The mainstay of management of UCD is dietary protein restriction. Patients must carefully balance their protein intake to ensure the body receives adequate nutrients for growth and development, while avoiding triggering hyperammonemia. However, varying protein requirements and variable growth and activity levels often elicit episodes of hyperammonemia that can result in irreversible neurological damage. To supplement for the lower protein intake, patients may incorporate amino acid dietary formulations, such as L-citrulline or L-arginine, into their diet. However, dietary management remains challenging, especially in infants and children.
The only available drugs, sodium phenylbutyrate (Buphenyl ® ) and glycerol phenylbutyrate (Ravicti ® ), are approved for the chronic management of patients with UCD and create an alternate pathway for nitrogen/ammonia elimination from the body, but patients maintain protein restricted diets. Use of sodium phenylbutyrate is limited by pill burden, taste, and tolerability issues that can make compliance challenging. These therapies are mechanistically similar treatment options with limitations on maximal effect due to dose-related neurological safety issues (e.g., vomiting, nausea, headache, somnolence, confusion, or sleepiness) and enzymatic saturation and, therefore, the unmet need remains high.
When these management approaches fail to control chronic UCD-induced hyperammonemia, patients may be candidates for liver transplantation, which is potentially curative as it may correct the enzyme deficiency that causes UCD. However, aside from being very costly, transplants are limited by availability of donor organs and are associated with potentially life-threatening risks and require life-long suppression of the immune system. Ultimately, morbidity and mortality remain high in UCD, and patients continue to suffer hyperammonemic crises. Synlogic believes that a truly transformative therapy for UCD would be an effective oral medicine without systemic toxicity that will maintain blood ammonia concentration at a safe level while allowing patients to eat a normal or only moderately restricted diet.
Overview of HE
The primary function of the liver is to filter out toxins, particularly ammonia, that are harmful if not sufficiently metabolized. In patients whose liver function is impaired, these toxins can accumulate in the blood stream and cause organ damage, particularly in the brain, which leads to a decline in brain function that is referred to as HE. Ammonia, a highly toxic substance produced in the body as a byproduct of protein metabolism, plays a key role in the development and prognosis of HE. While ammonia can be minimally metabolized by the brain in patients whose liver function is impaired, excessive ammonia levels can overwhelm the capacity of brain tissue and lead to a greater chance of developing brain swelling, coma and death for patients with HE. It is estimated that 30-45% of patients with chronic liver disease are affected by episodes of HE, and while many HE symptoms can be reversed with appropriate treatment, persistent impairment of memory and learning can occur.
HE severity is typically classified as covert or overt based largely on a patient’s mental state. Covert HE is difficult to diagnose and is often observed in patients with cirrhosis who appear to have no obvious disorientation, but who display mild to moderate symptoms, such as difficulty concentrating, forgetfulness, changes in personality or behavior, and poor sleep. Patients with covert disease are at a higher risk of developing the more severe overt HE and have increasingly been recognized as a cause of morbidity linked with increased risk of traffic accidents and unemployment. Overt HE is associated with obvious mental disorientation and physical symptoms such as lethargy, seizures, tremors, organ failure, or brain swelling, that arise suddenly and may induce a coma or even death, particularly if not adequately treated. Overt HE is associated with a poor prognosis, with one-year survival estimates of 20% to 55%.
The current standard of care for overt HE includes lactulose, a non-absorbable disaccharide that prevents the absorption of ammonia in the gut. Lactulose is associated with GI side effects including both painful abdominal cramping and diarrhea. Non-absorbable antibiotics are also used to treat HE, often concurrently with lactulose. Rifaximin (Xifaxan ® ), a broad-spectrum antibiotic used to reduce growth of bacteria that produce ammonia in the colon, was approved for HE based on improvements in the duration of remission, reduced hospitalizations over six months, and improved quality of life in patients with HE. Although rifaximin and lactulose are used therapeutically for overt HE, there are no approved treatments for covert HE.
Morbidity and mortality associated with overt HE remains high and hospitalizations for HE impose a high burden on community resources. When current therapies fail to control overt HE, patients may be candidates for a potentially curative liver transplantation. However, aside from being costly, transplants are limited by availability of donor organs and are associated with potentially life-threatening risks and require life-long suppression of the immune system. There is a need for an effective therapy for patients with HE to stave off episodes of cognitive dysfunction and hospitalizations.
Synlogic believes that because ammonia is produced in the GI tract, a Synthetic Biotic medicine could be an effective therapeutic to reduce the levels of excess ammonia in the blood of patients with UCD and HE without the need for severe protein restriction and risk of systemic toxicities.
SYNB1020 Design
SYNB1020, Synlogic’s lead Synthetic Biotic program, is an orally administered, engineered strain of E. coli Nissle. SYNB1020 was designed to complement the missing enzyme functions in patients with UCD with an enhanced pathway to consume ammonia, thus having the potential to treat the spectrum of enzyme deficiencies that underlie UCD. This mechanism also has applicability in liver disease where there is a need to reduce excess ammonia in the colon before it can be absorbed into the blood and cause HE episodes.
Synlogic’s approach was to create a Synthetic Biotic medicine that would continuously consume excess ammonia where it is naturally produced in the colon, before it can be absorbed into the blood, and produce arginine. Arginine production is deficient in UCD patients due to a defect in the urea cycle, and patients are often treated with arginine supplements. E. coli Nissle has an endogenous arginine production pathway that uses four molecules of ammonia for every new molecule of arginine produced. Synlogic modified this pathway to significantly enhance arginine production function through two key modifications: (1) deletion of a gene that represses the production of the arginine biosynthetic enzymes ( argR ) and (2) insertion of a gene that encodes a feedback-resistant enzyme in the arginine biosynthesis pathway (“ argA fbr ”). To enhance activity, argA fbr is placed under the control of an inducible promoter, FNR, to allow expression of the gene when the cell experiences micro-aerobic or anaerobic environments, such as the mammalian gut.
Schematic of SYNB1020
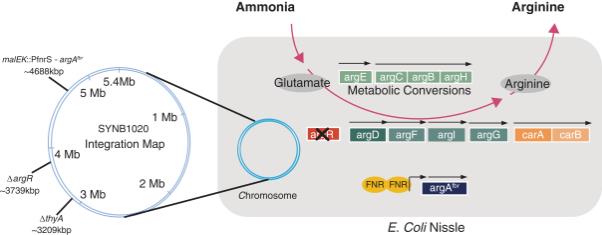
Abbreviations: argA = N-acetylglutamate synthase gene; argA fbr = feedback resistant N-acetylglutamate synthase; ArgB = acetylglutamate kinase; ArgC = N-acetyl glutamylphosphate reductase; ArgD = N-acetylornithine aminotransferase; ArgE = acetylornithine deacetylase; ArgFI = ornithine carbamoyltransferase; ArgG = arginosuccinate synthase; ArgH = arginosuccinate lyase; argR = arginine repressor gene; CarAB = carbamoylphosphate synthetase; FNR = fumarate and nitrate reductase; P fnrS = fumarate and nitrate reductase regulator sensor promoter; D thyA = thymidylate synthase such that the strain can only grow in thymidine-rich environments. Arrows denote operons.
SYNB1020 Nonclinical Program
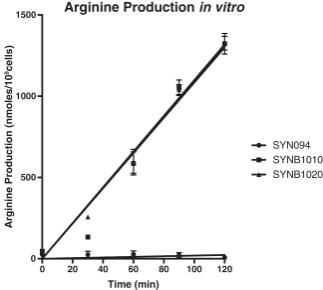
In an in vitro study, SYNB1020 and a related research strain SYNB1010 (identical to SYNB1020 except designed to grow in the presence of kanamycin for selection and use in pre-clinical studies) consumed ammonia and produced arginine at substantially higher rates compared with a control strain of E. coli Nissle that had not been engineered (“SYN94”). Arginine production was 650.1 and 658.7 nmol/10 9 cells/hour for SYNB1010 and SYNB1020, respectively, and only 11.8 nmol/10 9 cells/hour for the control strain. Similarly, conversion of ammonia to arginine was 2545 and 2570 nmol/10 9 cells/hour for SYNB1010 and SYNB1020, respectively, and 46 nmol/10 9 cells/hour for the control strain.
Pre-Clinical Efficacy Study
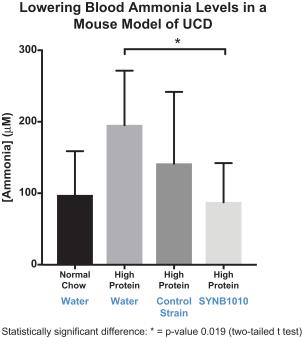
To test the in vivo activity in a setting of hyperammonemia, the spf-ash/F1 mouse was adapted from a published model based on a mutation in the gene for ornithine transcarbamylase (“OTC”), a common deficiency in human UCDs. The activity of the research strain SYNB1010 was compared to a non-arginine producing control strain of E. coli Nissle, and to water as an additional control. All mice were dosed orally, twice daily beginning on Day 1. Hyperammonemia was induced on Day 3 by switching animals to a high-protein diet. SYNB1010 reduced Day 5 blood ammonia levels in comparison with water and the non-arginine producing control strain of E. coli Nissle. This reduction in blood ammonia resulted in improved survival of animals dosed with SYNB1010 compared to animals given water or the non-arginine producing control strain.
Pre-Clinical Safety Study
In a GLP 28-day mouse toxicology study, SYNB1020 was safe and well tolerated. No toxicity was detected at the highest feasible dose, and there was no evidence of distribution of SYNB1020 outside the GI tract. Consequently, the no observed effect level was equal to the maximum feasible dose that could be administered, a threshold defined by volume limitations permitted in animals. This represents a greater than 1,000-fold safety margin over the starting dose planned in the Phase 1 study.
SYNB1020 Clinical Development Plan
Synlogic initiated a Phase 1, randomized, double-blinded, placebo-controlled study in June 2017 to evaluate the safety, tolerability, and gastrointestinal clearance of single and multiple doses of SYNB1020 in healthy volunteers. Synlogic expects approximately 50 subjects will be enrolled. The starting oral dose and subsequent range for dose escalation were selected based on the nonclinical toxicology and efficacy experiments and are expected to span the projected efficacious dose range in humans. The primary outcome measures will evaluate the safety and tolerability of SYNB1020 by assessing the nature and frequency of adverse events, laboratory assessments and electrocardiogram. Secondary measures will investigate the gastrointestinal tolerability and the kinetics of SYNB1020. In addition, blood, urine, and fecal samples will be collected and evaluated for exploratory biomarkers to gain mechanistic insights regarding ammonia consumption. If SYNB1020 appears well tolerated and safe in healthy subjects, Synlogic plans to evaluate SYNB1020 for the management of hyperammonemic patients, such as UCD and HE.
SYNB1618 for PKU
PKU is a rare IEM caused by a genetic defect in the gene phenylalanine hydroxylase (“PAH”) leading to phenylalanine (“Phe”) accumulation in the blood and brain, where it is neurotoxic and can lead to neurological deficits and even death. Current disease management of PKU involves dietary protein restriction with the consumption of phenylalanine-free protein supplements. The only approved medication, Kuvan ® (sapropterin dihydrochloride) is indicated for a subgroup of patients and does not eliminate the need for ongoing dietary management. Despite recommendations supporting life-long control of phenylalanine levels, compliance is challenging due to the highly restrictive nature of the diet, putting patients at risk for cognitive and psychiatric disease and supporting the need for novel treatment approaches.
Synlogic’s Synthetic Biology platform is well-suited to complement the missing enzyme function in PKU patients by providing alternative metabolic pathways to consume Phe. Synlogic’s second IEM program, SYNB1618 for PKU, is designed to remove excess phenylalanine from the blood by transforming it into non-toxic metabolites. SYNB1618 has demonstrated activity in a rodent model of PKU. Synlogic expects to initiate a Phase 1 clinical trial for SYNB1618 in the first half of 2018.
Overview of PKU
Phenylalanine is an essential amino acid that enters the body primarily through dietary protein, and can be toxic if not sufficiently broken down and eliminated. The metabolism of phenylalanine by the liver is dependent on adequate function of the liver enzyme PAH and the cofactor tetrahydrobiopterin (“BH4”) necessary for its activity. When the PAH gene is mutated and/or the production of BH4 is blocked, phenylalanine cannot be sufficiently broken down and accumulates to toxic levels (i.e., hyperphenylalaninemia), which can cause irreversible brain damage. PKU is an inherited metabolic disease that presents as a severe form of hyperphenylalaninemia.
The disease course of PKU typically involves worsening neurological function that begins in infancy or early childhood. The clinical manifestations vary depending on severity of the enzyme mutation, the time of diagnosis and treatment initiation, and compliance. Symptoms may be extensive, such as severe mental retardation, or they may reflect more moderate neurocognitive or physical issues, such as below average intelligence, behavioral or mood disorders, memory loss, difficulty concentrating, decreased motor function, eczema, body odor, and tremors or seizures. A woman with PKU who becomes pregnant could develop maternal PKU if her diet is not strictly controlled, and there is a risk that the baby will be born with one or more birth defects such as mental retardation, microcephaly or congenital heart disease.
Based on the success of newborn screening efforts that began in developed countries in the 1960s, it is believed that nearly all PKU patients under the age of 40 have been diagnosed at birth. The National PKU Alliance estimates that in the United States there are currently 16,500 people living with PKU.
Currently, management of PKU requires a heavily modified diet that restricts protein intake, in order to minimize consumption, combined with essential amino acid and vitamin supplementation. Special medical foods, including phenylalanine-free protein formula, provide patients with dietary protein and fulfill other nutrient needs. However, it is challenging for most PKU patients, even with the efforts of supportive family and social networks, to adhere to the restricted diet to the level that provides the necessary control of phenylalanine levels. Patients often have trouble adhering to the diet from a young age, with particular challenges arising during times of increasing independence during adolescence. Furthermore, access to low protein foods can be challenging, as they are costlier and less nutritious than their higher protein, non-modified counterparts.
Kuvan ® (sapropterin dihydrochloride) was the first drug approved for the treatment of PKU in 2007. It is indicated for the reduction of blood phenylalanine in patients with hyperphenylalaninemia with residual PAH activity as it is a synthetic form of the BH4 cofactor. Oral administration of Kuvan, along with protein restriction, has lowered phenylalanine levels in patients who have residual PAH activity and/or mild forms of the disease, which accounts for approximately 20-50% of the PKU population. However, Kuvan does not eliminate the need for ongoing dietary management in all patients . Large neutral amino acids have also demonstrated activity in blocking absorption of excess phenylalanine by the intestines and brain, but are currently only administered in adolescents and adults.
A pegylated form of recombinant phenylalanine ammonia lyase (“PAL”), called Pegvaliase, an enzyme that metabolizes phenylalanine but does not require cofactor activity, is in clinical development for PKU and is not yet approved. While Pegvaliase injections one to two times daily have been proven to lower phenylalanine levels regardless of whether patients are following a low protein diet or not, patients may experience injection site reactions and/or develop antibodies to the enzyme, which limits its effectiveness.
Despite recent improvements in PKU therapy, patients continue to suffer from poor outcomes. Even patients who are diagnosed and treated early have increased risk of neurocognitive abnormalities and psychiatric complications and are burdened by the life-long struggle to comply with strict dietary modifications. Available drug therapies demonstrate limited effectiveness. are accompanied by immunologic and other toxicities, and may still require patients to maintain a heavily restricted diet. Synlogic believes a truly transformative therapy would be orally-dosed and provide sustained, safe concentrations of phenylalanine while allowing for a normal or only moderately restricted diet. Synlogic believes that a Synthetic Biotic medicine could be an effective oral therapeutic that acts from the gut to transform excess phenylalanine with the consequent effect of reducing levels in the blood without the need for severe phenylalanine restriction or risk of systemic toxicities.
SYNB1618 Design
SYNB1618 is a genetically-modified strain of E. coli Nissle engineered to express a synthetic pathway for transporting and metabolizing phenylalanine in patients with PKU following oral administration. SYNB1618 was designed to overcome the missing enzyme function in patients with PKU with an alternative pathway to reduce phenylalanine levels.
In designing SNYB1618, Synlogic integrated genes encoding the phenylalanine transporter (“PheP”), PAL derived from Photorhabdus luminescens and L-amino acid deaminase (“LAAD”) derived from the organism Proteus mirabilis into the E. coli Nissle genome. PheP transports phenylalanine into the Synthetic Biotic bacterial cell with high efficiency, while within the cell PAL converts phenylalanine to the non-toxic byproduct trans- cinnamate (“TCA”). The inclusion of multiple copies of these genes further enhanced activity. Similar to PAL, LAAD converts phenylalanine to a non-toxic byproduct, phenylpyruvate.
SYNB1618 Nonclinical Program
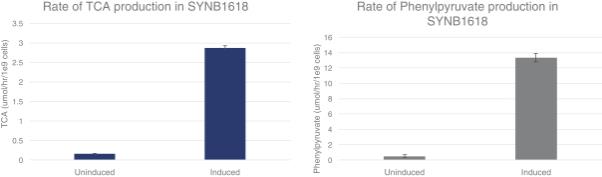
Synlogic has demonstrated that SYNB1618 can metabolize phenylalanine in vitro using both the PAL and LAAD enzymes by measuring their respective non-toxic byproducts. Synlogic compared the activity of SYNB1618 under conditions in which the Synthetic Biotic strain is induced (in the “ON” state) versus when uninduced when metabolic activity is suppressed. As shown in the graphs below, i n vitro activation of PAL led to an 18.5-fold increase in production of the TCA metabolite over uninduced levels, and in vitro activation of LAAD led to production of phenylpyruvate levels at 26.7-fold over uninduced levels.
Pre-Clinical Efficacy Studies
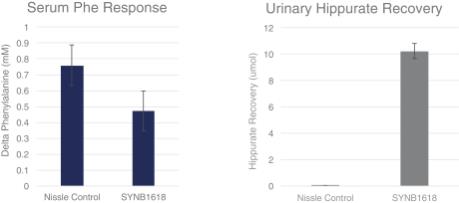
In vivo studies have focused on the enu2-/- mouse model that contains a mutation in the gene coding for phenylalanine hydroxylase, the same enzyme that is deficient in PKU patients. Mice with this genetic defect maintained on normal chow accumulate phenylalanine in their blood at concentrations greater than 2000 µM, which is similar to blood concentrations found in humans with PKU. On a phenylalanine-restricted diet, blood phenylalanine levels can be maintained at the healthier range of 100-200 µM. Subcutaneous injection of phenylalanine-restricted mice with phenylalanine (0.1 mg/g body weight) results in a rapid increase in blood phenylalanine concentrations. As shown in the graph below, this increase associated with this phenylalanine challenge was significantly blunted (as reported as the delta, or reduction, from peak phenylalanine levels) upon oral administration of SYNB1618, compared to administration of the non-engineered control strain that did not have the phenylalanine degradation pathway (39% blunting of serum phenylalanine, p = 0.0002).
To further support the development of SYNB1618, hippuric acid was followed as a urinary biomarker of phenylalanine degradation. One product of phenylalanine degradation by SYNB1618, TCA, is converted to hippurate by liver enzymes and excreted in the urine. Following treatment of enu2-/- mice with SYN1618, urinary hippurate concentration increased 270-fold compared to mice treated with unengineered E. coli Nissle controls. Taken together, these data show that SYNB1618 has activity in the GI tract, and that degradation of recirculating phenylalanine is effective in decreasing the levels found in blood, independent of dietary intake.
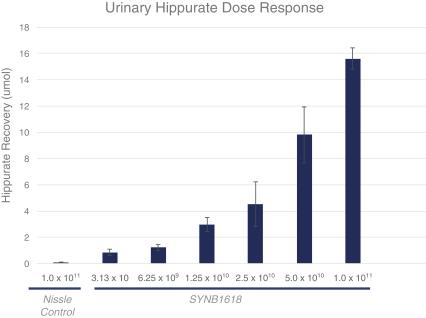
Synlogic has also demonstrated a dose response in the same animal model with its clinical candidate strain SYNB1618. With increasing oral doses of this single strain, Synlogic observes increasing levels of urinary hippurate in mice.
Moreover, in preliminary primate studies, administration of SYN1618 to cynomolgus monkeys following an oral high protein challenge resulted in elevated levels of urinary hippurate recovery compared to the protein challenge alone. These data indicate that SYN1618 is functional in the environment of the primate gut.
SYNB1618 Clinical Development Plan
Synlogic is currently conducting IND-enabling studies and scaling up manufacturing to support initiation of clinical studies of SYNB1618. Synlogic is planning a Phase 1, randomized, double-blinded, placebo-controlled study to evaluate the safety, tolerability, and gastrointestinal clearance of SYNB1618. In such study, healthy adult volunteers would be treated with single- or multiple-ascending doses of SYNB1618. Synlogic expects approximately 50 subjects will be enrolled.
Upon determination of the maximum tolerated dose, Synlogic expects an expansion cohort of up to 16 adult subjects with PKU will be treated. In addition to the primary endpoint of safety and tolerability, this study will evaluate the change from baseline in several pharmacodynamic parameters compared to placebo in order to characterize the kinetics of SYNB1618 in humans, and provide mechanistic and clinical insights regarding urinary hippurate production and phenylalanine reduction. Synlogic expects to initiate this Phase 1 trial in the first half of 2018.
Synthetic Biotic Medicines for Additional IEMs
Learnings from the design, pre-clinical research, clinical planning and scalable manufacturing of the lead programs have already informed development of future clinical candidates. Synlogic’s initial programs were selected based on applicability of the Synthetic Biotic platform to provide pathway complementation in IEMs in which the toxic metabolite was known to be associated with the relevant clinical endpoint and to be accessible in the GI tract. Additional examples in which there is opportunity to expand the potential of Synthetic Biotic medicines include discovery-stage programs for (1) MSUD and IVA and (2) PA and MMA. These are
rare metabolic deficiencies in which a toxic metabolite can accumulate and lead to neurological decline and death. There is no approved therapy for either disease and these patients are managed with dietary modifications, supportive care, and liver transplant when available.
A Synthetic Biotic Program for Maple Syrup Urine Disease and Isovaleric Acidemia
MSUD is an IEM that was first described in the 1950s as an inherited progressive neurological degenerative disorder. Patients with this disease have mutations in one of the protein subunits of the mitochondrial multi-enzyme complex called branched-chain alpha-ketoacid dehydrogenase. These mutations cause the patients to accumulate high levels of the branched chain amino acids (“BCAA”) leucine, isoleucine or valine that are neurotoxic and cause severe neurological pathologies characterized by brain edema, seizure, spasticity and respiratory irregularities that can lead to death. The MSUD name derives from the strong maple syrup odor in the urine of these patients. Similarly, IVA can result from a genetic defect leading to leucine accumulation. It is difficult to estimate the prevalence of these rare indications given few longitudinal studies. Based on estimates of the live birth rate of MSUD of 1:185,000 and IVA of 1:250,000, respectively, and applying assumptions to account for mortality and survival rates, it is estimated that there may be approximately 2,500 MSUD or IVA patients in the United States.
Currently-available treatments for disorders involving the catabolism of BCAA are inadequate for the long-term management of the disorders and have severe limitations. A low protein/BCAA-restricted diet, with micronutrient and vitamin supplementation as necessary, is the widely-accepted long-term disease management strategy. However, BCAA-intake restrictions can be problematic since these amino acids are also essential nutrients that can only be acquired through diet and are necessary for metabolic activities such as protein synthesis. Even with proper monitoring and patient compliance, branched chain amino acid dietary restrictions result in a high incidence of mental retardation and mortality. MSUD is cured by liver transplantation; however, limited availability of donor organs, costs, and the need to rely on life-long immunosuppressant therapy are limiting. Therefore, there is significant unmet need for an effective, reliable, and/or long-term treatment for disorders involving the catabolism of branched chain amino acids.
Synlogic has built a Synthetic Biotic discovery program to modulate the expression of two BCAA transporters and three BCAA-degrading enzymes. Results in vitro demonstrate the efficient degradation of BCAAs into non-toxic branched-chain alcohols that can then be further metabolized and eliminated from the body. In preliminary studies in a mouse model of MSUD, the oral delivery of the Synthetic Biotic strain suppresses the increase in blood BCAA levels induced by a high-protein diet and prevents the associated waning, or moribund, phenotype as measured by improved locomotor activity. Based on the in vivo therapeutic effects observed, Synlogic continues to improve this approach as a potential promising therapy for MSUD and IVA patients.
Synlogic’s Synthetic Biotic Program for Propionic Acidemia and Methylmalonic Acidemia
Organic acidemias are a group of rare IEMs in which amino acid metabolism is disrupted, causing an accumulation of toxins. Normally, the human body converts certain amino acids, such as isoleucine, valine, threonine, and methionine, into a derivative of propionic acid to create energy. Patients with PA and MMA have enzyme deficiencies caused by mutations in the pathway for propionate catabolism that lead to the toxic accumulation of propionic acid or methylmalonic acid-related metabolites in the blood stream, leading to damage of the brain, heart, and liver. Clinical manifestations of the disease vary depending on the degree of enzyme deficiency and include seizures, vomiting, lethargy, hypotonia, encephalopathy, developmental delay, failure to thrive, and secondary hyperammonemia. It is difficult to estimate the prevalence of these indications given few longitudinal studies. The live birth rates are estimated as 1:105,000-1:130,000 for PA and 1:50,000-100,000 for MMA. Applying assumptions to account for mortality and survival rates, it is estimated that there may be 2,000-3,000 PA or MMA patients in the United States.
Currently available treatments for disorders involving propionate catabolism are inadequate and have severe limitations. Patients may present acutely at birth with metabolic acidosis and hyperammonemia, or later in life with more heterogeneous clinical symptoms, and run the risk of early death or severe neurologic damage. Mental outcomes tend to be worse in PA, and patients who can also experience late complications like cardiomyopathy. Late complications for MMA patients include chronic kidney disease. Except for MMA patients who are responsive to vitamin B12, there is significant unmet need for effective, reliable and/or long-term treatment for disorders involving the catabolism of propionate.
Propionate is produced naturally in the gut by bacterial metabolism, and therefore a Synthetic Biotic medicine that consumes propionate in that environment could be an attractive approach to treating these disorders. Synlogic has constructed two discovery-stage Synthetic Biotic strains that have each demonstrated degradation of propionate into non-toxic metabolites in vitro . In a preliminary experiment in a mouse model of propionic acidemia, the oral delivery of both Synthetic Biotic strains independently suppressed the plasma concentration of disease-related toxic metabolites. Synlogic is planning to continue assessing these strains in animal models and improving them as potential promising therapies for PA and MMA patients.
Synthetic Biotic Medicines for Broader Metabolic Disease
Synlogic’s Synthetic Biotic platform combined with its product discovery and development capabilities drive the potential for multiple clinically meaningful opportunities for patients affected by a broad set of metabolic diseases such as Nonalcoholic Steatohepatitis (“NASH”). For these indications, there is need for a safe, oral therapy with local activity in the gut to reset a metabolic dysfunction. Synlogic’s approach is amenable to enabling combination therapy, which is increasingly recognized as a necessary component of effective treatment. Synlogic continues to explore strategic partnerships that would leverage the complementary capabilities of partners in order to develop Synthetic Biotic medicines for these broader groups of patients in need.
Synthetic Biotic Medicines for Immunomodulation
Synlogic’s Synthetic Biotic platform has the potential to generate clinically meaningful therapies for patients affected by immune-mediated diseases. Among these conditions, IBD is particularly attractive, as it allows Synlogic to leverage knowledge and expertise gleaned from Synlogic’s metabolic programs to develop living medicines that can act locally at the site of disease in the gut. Because Synlogic’s approach is based on local delivery to the site of inflammation and not on systemic administration, Synlogic anticipates that its Synthetic Biotic medicines may offer an attractive safety profile in this setting. In 2015, Synlogic entered into a multi-year global collaboration with AbbVie focused on the discovery and development of a Synthetic Biotic medicines for the treatment of IBD.
Synlogic’s Synthetic Biotic Medicines for Inflammatory Bowel Disease
IBD is a group of diseases characterized by significant local inflammation in the GI tract typically driven by T cells, activated macrophages and compromised function of the epithelial barrier. IBD pathogenesis is linked to both genetic and environmental factors and may be caused by altered interactions between gut microbes and the intestinal immune system. Current approaches to treat IBD are focused on therapeutics that modulate the immune system and suppress inflammation. These therapies include steroids, such as prednisone, and tumor necrosis factor inhibitors, such as Humira ® . Drawbacks from these approaches are associated with systemic immunosuppression, which includes greater susceptibility to infectious diseases and cancer. It is estimated that between 1.0-1.3M patients have IBD in the United States.
Compromised gut barrier function also plays a central role in autoimmune diseases pathogenesis. A single layer of epithelial cells separates the luminal contents of the gut from the host circulatory system and the immune cells in the body. Disrupting the epithelial layer can lead to pathological exposure of foreign antigens from the lumen resulting in increased susceptibility to autoimmune disorders. The interplay between the gut microbiota and the host is thought to play key roles in both the maintenance of the epithelial barrier as well as homeostatic immunity. Thus, enhancing barrier function and reducing inflammation in the gastrointestinal tract are potential therapeutic mechanisms for the treatment or prevention of autoimmune disorders. Synlogic’s Synthetic Biotic platform allows for the effective programming of E. coli Nissle to execute these functions, including the metabolic production of factors such short chain fatty acids to enhance barrier function and secreting proteins, such as immunomodulatory cytokines.
Synlogic’s Synthetic Biotic Medicines for Immuno-Oncology
Synlogic believes boosting the body’s immune response against tumor cells is one of the most promising advances in the treatment of cancer. The so-called “hot tumors”, those with robust immune cell infiltration, specifically by T cells, respond well to immunotherapies such as the PD-1 and CTLA-4 checkpoint inhibitors. Checkpoint inhibitors work by blocking pathways that inhibit T cells thus enabling them to recognize and destroy the tumor. Checkpoint inhibitors have significantly extended the lives of patients with several cancer types and, in some cases, have resulted in complete clinical responses. However, a large proportion of tumors are “cold” (i.e., they lack T cells), and respond poorly to immunotherapy.
Synlogic’s goal is to leverage its Synthetic Biotic platform to design living medicines that can modify the tumor microenvironment to convert “cold” tumors into “hot.” Synlogic believes that this transition will dramatically expand the patient population amenable to clinical benefit by immunotherapy. Synlogic’s approach is designed to deliver robust therapeutic combinations to the tumors, without significant systemic exposure. Synthetic Biotic medicines are being developed to be administered by an intra-tumor injection or, in the case of GI cancers, by oral administration and can be engineered to perform three types of functions: metabolic conversions, secretions of proteins or bacterial surface display of specific single chain antibody domains, known as scFvs.
Synlogic’s Synthetic Biotic platform allows it to approach “cold” tumors in a rational, mechanistic way, and can deliver multiple validated mechanisms to elicit specific immune responses in the tumor microenvironment. Synlogic’s main mechanistic areas of focus in the context of tumor immunology include:
| • | Immune activation and priming: Synlogic’s bacterial Synthetic Biotic chassis is predicted to engage innate immune cells in the tumor microenvironment, thereby initiating an immune cascade to activate and direct T cells to the tumor. Lack of effective presentation of tumor-specific antigens to T cells is recognized as a significant limitation to the initiation of immune responses in tumors. Synlogic is building and optimizing Synthetic Biotics medicines with the potential of addressing this issue. |
| • | Immune augmentation/Reversal of immunosuppression: Synlogic has developed strains that actively consume and transform immunosuppressive metabolites in the tumor microenvironment, with the goal of setting up a milieu conducive to immune activation and tumor destruction. |
| • | T cell expansion: Tumor antigen-specific T cell expansion and prevention of exhaustion are recognized as key objectives for successful cancer immunotherapy. Synlogic is developing Synthetic Biotic medicines programs to secrete specific cytokines to promote T cell survival and expansion. |
| • | Stromal modulation: The physical structure of tumors is receiving increasing attention as emerging data demonstrate its importance in orchestrating tumor growth, immune evasion and resistance to chemotherapy, such as in pancreatic ductal adenocarcinoma. Tumor-derived extracellular matrix proteins can limit the perfusion of drugs or antibodies, contributing to the remarkable resistance of this tumor type to therapy. Synlogic has developed strains that secrete active enzymes with the capacity to remodel extracellular matrix proteins to make the tumor more permeable. |
Synlogic’s product vision for immuno-oncology is to use a rational approach to selecting and combining relevant mechanisms of action for the microenvironment of specific tumor types. Synlogic will focus on tumor types with high unmet medical need, including colorectal and hepatocellular carcinomas, pancreatic cancer and melanomas refractory to current immunotherapies. Currently three programs are in the early pipeline and are diversified in terms of indication, combinations of mechanisms, and route of administration.
Collaboration Agreements
To accelerate the development and commercialization of Synthetic Biotic medicines to patients in therapeutic areas outside of IEMs, Synlogic has formed, and intends to seek other opportunities to form, strategic alliances with collaborators that can expand Synlogic’s pipeline of therapeutic development and product candidates. Synlogic also works, and intends to seek additional opportunities to work, with multiple academic, research and translational medicine organizations and entities to deepen its understanding and development of living medicines with the potential to treat disease and disorders.
AbbVie
In July 2015, Synlogic entered into a license agreement with its subsidiary Synlogic IBDCo, Inc. (“IBDCo”) and an Agreement and Plan of Merger with AbbVie (together, the “AbbVie Agreements”) to collaborate on the discovery and development of Synthetic Biotic medicines for the treatment of IBD. The AbbVie Agreements provide AbbVie with an exclusive option to acquire IBDCo, which would then have an exclusive worldwide license to develop and commercialize up to three specified Synthetic Biotic medicines for the treatment of IBD.
Under the terms of the collaboration with AbbVie, Synlogic has the responsibility to discover, characterize and optimize one lead Synthetic Biotic product candidate to the point of a IND-enabling package, together with two backup product candidates, through a research and development program covering a limited number of effectors that modulate the IBD pathophysiology. The multi-year collaboration combines AbbVie’s expertise in inflammatory diseases with Synlogic’s expertise in synthetic biology and metabolic engineering. AbbVie agreed to pay IBDCo an upfront payment of $2.0 million, received in December 2015, and up to $16.5 million upon the achievement of certain research and development milestones. In May 2017, IBDCo achieved one of these research and development milestones under the AbbVie Agreement for which it will receive $2.0 million.
If AbbVie accepts Synlogic’s IND-enabling package covering the lead Synthetic Biotic product candidate, AbbVie may exercise its exclusive option to acquire IBDCo, which would house the lead and two backup product candidates. If this option is exercised, AbbVie would pay Synlogic an option exercise fee upon the closing of the IBDCo merger and Synlogic would be eligible to receive future development, regulatory and commercial milestone payments, and low single digit royalties on sales of the Synthetic Biotic medicines. In addition, AbbVie would then assume full control of all further clinical development and commercial activity, including responsibility for all expenses and decisions.
Potential Future Collaborations
Synlogic views strategic partnerships as important drivers for helping accelerate its goal of effectively treating patients, and Synlogic will continue to seek strategic alliances with collaborators who can help fund, develop and commercialize its novel therapeutic development and product candidates, particularly in large metabolic indications and immune-oncology. As the potential application of its Synthetic Biotics platform is extremely broad, Synlogic also plans to continue to identify academic, research and translational medicine organizations and entities that can contribute expertise and resources to its programs, to allow it to more rapidly expand Synlogic’s impact to broader patient populations.
Intellectual Property
Synlogic strives to protect and enhance the proprietary technology, inventions, and improvements that are commercially important to its business, including seeking, maintaining, and defending patent rights, whether developed internally or licensed from Synlogic’s collaborators or other third parties. Synlogic’s policy is to seek to protect Synlogic’s proprietary position by, among other methods, filing patent applications in the United States and in jurisdictions outside of the United States related to Synlogic’s proprietary technology, inventions, improvements, and product candidates that are important to the development and implementation of Synlogic’s business. Synlogic also relies on trade secrets and know-how relating to its proprietary technology and product candidates, continuing innovation, and in-licensing opportunities to develop, strengthen, and maintain its proprietary position in the field of synthetic biology. Synlogic additionally relies on data exclusivity, market exclusivity, and patent term extensions when available, and plans to seek and rely on regulatory protection afforded through orphan drug designations. Synlogic’s commercial success may depend in part on its ability to obtain and maintain patent and other proprietary protection for its technology, inventions, and improvements; to preserve the confidentiality of its trade secrets; to maintain Synlogic’s licenses to use intellectual property owned by third parties; to defend and enforce Synlogic’s proprietary rights, including its patents; and to operate without infringing on the valid and enforceable patents and other proprietary rights of third parties.
Synlogic believes it is well positioned in terms of intellectual property because Synlogic:
| • | has built and expanded, and intends to continue expansion in, a broad worldwide portfolio of intellectual property, including patents and patent applications, in areas relevant to the development, manufacturing and formulation of human therapeutic products using live biotherapeutics based on synthetic biology; |
| • | intends to take additional steps, where appropriate, to further protect Synlogic’s intellectual property rights, including, for example, through the use of copyright and trademark protection, as well as regulatory protection available via orphan drug designations, data exclusivity, market exclusivity and patent term extensions. |
Synlogic believes its intellectual property portfolio provides broad coverage of its Synthetic Biotic platform and applicable disease-related technologies, which are directed to diseases and conditions associated with hyperammonemia, hyperphenylalanemia, other IEMs and acquired metabolic disorders, autoimmune and other inflammatory disorders and oncology. As of June 2017, Synlogic’s intellectual property portfolio consists of 193 Synlogic-owned and in-licensed patents and patent applications in U.S. and foreign jurisdictions, including 11 issued patents.
Disease-related applications.
The disease-related applications in Synlogic’s intellectual property portfolio relate to certain pathological conditions and provide coverage for engineered bacteria having genetic circuitry designed to specifically address those conditions and the associated disease states. Disease related applications relate to pathological conditions and include:
Hyperammonemia
| • | Synlogic’s lead program addresses conditions associated with hyperammonemia, for which it has developed engineered bacterial strains containing genetic circuitry specifically designed to metabolize ammonia. |
| • | The intellectual property portfolio provides robust coverage for compositions directed to engineered bacterial strains, methods of making the bacterial strains and methods for treating diseases that involve accumulation of ammonia (e.g., UCD, HE). Synlogic’s intellectual property provides coverage for the lead product candidate SYNB1020 and methods of its manufacture and use. In addition to UCD, SYNB1020 could be useful for the treatment of hyperammonemia in HE patients with cirrhosis of the liver, which indication is also covered by Synlogic’s intellectual property. |
| • | Currently, intellectual property relating to this technology includes ten pending applications in U.S. and foreign jurisdictions, as well as one issued and one allowed U.S. patent directed to composition of matter and pharmaceutical composition claims covering Synlogic’s clinical candidate. The patent term for this IP will expire in December 2035, excluding any patent term adjustments or extensions. |
Hyperphenylalanemia
| • | Synlogic’s program addresses conditions associated with hyperphenylalanemia, for which it has developed engineered bacterial strains containing genetic circuitry specifically designed to metabolize phenylalanine. |
| • | Synlogic’s intellectual property portfolio provides coverage for compositions directed to engineered bacterial strains, methods of making the bacterial strains and methods for treating diseases that involve accumulation of phenylalanine. Synlogic’s intellectual property provides coverage for the lead product candidate SYNB1618 and methods of its manufacture and use. |
| • | Currently, intellectual property relating to this technology includes three pending U.S. patent applications and two international patent applications directed to composition of matter and pharmaceutical compositions covering Synlogic’s lead product candidate. The patent term for this intellectual property will expire in May 2036, excluding any patent term adjustments or extensions. |
Other Inborn Errors of Metabolism
| • | Additional disease-related intellectual property includes patent applications directed to Synlogic’s Synthetic Biotic technology for use in treating diseases and conditions arising from IEMs. |
| • | Synlogic’s intellectual property provides coverage of compositions of engineered bacteria, methods of making the bacterial strains and methods of treating diseases associated with accumulation of BCAA (e.g., leucine, isoleucine and valine), including MSUD. Synlogic currently has one U.S. application and one PCT application directed to diseases involving accumulation of BCAA. The patent term for this intellectual property will expire in June 2036, excluding any patent term adjustments or extensions. |
| • | Additional Synlogic intellectual property covers compositions of engineered bacteria, methods of making the bacterial strains and methods of treating organic acidemias, including those associated with accumulation of propionic acid and related toxic metabolites, such as PA and MMA. Synlogic currently has one U.S. application and one PCT application directed to diseases involving accumulation of organic acid metabolites. The patent term for this intellectual property will expire in July 2036, excluding any patent term adjustments or extensions. |
Metabolic Disorders
| • | In addition to IEMs, other disease-related intellectual property includes patent applications directed to Synlogic’s Synthetic Biotic technology for use in treating diseases and conditions associated with acquired metabolic disorders, including, but not limited to NASH. |
| • | Synlogic’s intellectual property provides broad coverage of compositions of engineered bacteria, methods of making the bacterial strains and methods of treating various metabolic diseases. Synlogic’s current intellectual property consists of two PCT applications relating to this technology. The patent term for this intellectual property has expiration dates ranging from June 2036 to December 2036, excluding any patent term adjustments or extensions. |
Inflammatory and Autoimmune Diseases
| • | Additional disease-related intellectual property includes numerous patent applications directed to Synlogic’s Synthetic Biotic technology for use in treating diseases and conditions associated with an inflammatory state, including, but not limited to, diseases associated with gut inflammation, compromised gut mucosal barrier (leaky gut), and various autoimmune disorders. |
| • | Synlogic’s intellectual property provides broad coverage of compositions of engineered bacteria, methods of making the bacterial strains and methods of treating diseases associated with gut inflammation, leaky gut, and autoimmune disorders, such as Inflammatory Bowel Disease, including Crohn’s Disease, ulcerative colitis, and other diseases. Synlogic’s current intellectual property consists of three U.S. applications and three PCT applications relating to this technology, which is being developed in collaboration with AbbVie and which intellectual property is Synlogic-owned. The patent term for this intellectual property has expiration dates ranging from December 2035 to March 2036, excluding any patent term adjustments or extensions. In addition, Synlogic has one PCT application relating to this technology which is jointly owned by Synlogic and MIT, which expires in December 2035, excluding any patent term adjustments or extensions. |
Immuno-Oncology
| • | In addition, Synlogic has disease-related intellectual property directed to its Synthetic Biotic technology for use in immuno-oncology, which intellectual property covers bacterial strains engineered to metabolize and/or produce biomolecules that modify the tumor microenvironment and immune response, resulting in an array of mechanistic functions, including immune activation and priming, immune augmentation and/or reversal of immunosuppression, T-cell expansion, and tumor stromal modulation. |
| • | Synlogic’s intellectual property provides broad coverage of compositions of engineered bacteria, methods of making the bacterial strains and methods of treating various cancers. Synlogic’s current intellectual property consists of two PCT applications with expiration dates ranging from January 2037 to February 2037, excluding any patent term adjustments or extensions. |
Platform Technology Applications.
In addition to the disease-related technology, Synlogic’s intellectual property portfolio also includes applications directed to platform technologies developed internally by Synlogic. Exemplary platform technologies include bacterial chassis-related and genetic circuitry-related technological developments, including, for example, improvements in inducible gene regulation, control of bacterial cell growth, including auto-regulation thereof, and systems for importing metabolites, as well as secreting therapeutic effectors. These platform technologies, and Synlogic’s intellectual property coverage thereof, are broadly applicable to Synlogic’s therapeutic Synthetic Biotic medicines.
In addition to Synlogic’s own patent applications, Synlogic has licensed patents and patent applications from MIT and Trustees of Boston University (“BU”) to access intellectual property covering synthetic biology circuitry that Synlogic is exploring and developing. The intellectual property licensed from MIT and BU relates to genetic circuitry (designed to be modular components for integration into biological systems), cells containing the genetic circuitry, and methods and systems for gene regulation using the genetic circuitry.
The intellectual property licensed from MIT includes applications related to genome editing systems used to target specific genes for recombination and methods for delivering a gene editing system to endogenous bacteria. It also includes applications directed to genetic circuits and biological systems for regulating gene expression using various recombinase-based and other promoter systems, including promoter systems that respond to different levels of an input signal. The MIT intellectual property also covers methods for identifying mutant promoters that have an altered level of response to an input signal and methods of controlling gene expression in certain bacteria. In addition, the MIT intellectual property includes a PCT application jointly owned by Synlogic and MIT, directed to engineered bacteria and methods for treating inflammatory bowel disease. The licensed patents and applications from the MIT have expiration dates ranging from 2033 to 2037, excluding any patent term adjustments or extensions.
The intellectual property licensed from BU includes patents and applications relates to genetic circuitry and biological systems for controlling gene expression employing the genetic circuits, detecting the production of a target gene product, and delivering genetic circuits to endogenous bacteria. The various genetic circuits are designed to respond to external cues and also designed to tighten control of gene expression regulated by inducible and constitutive promoter systems using a variety of genetic components, for example, sensors, inducers, repressors, antisense, stem-loop sequences, recombinases, RNAi, inverted sequences, and ribosome-binding site sequences, to generate various promoter toggle switches, adjustable threshold switches, and oscillator switches, among others. In addition, the BU intellectual property covers biocontainment systems that couple environmental sensing with circuit-based control of cell viability. The licensed patents and applications from BU have expiration dates ranging from 2019 to 2036, excluding any patent term adjustments or extensions.
Massachusetts Institute of Technology (“MIT”) License
Synlogic entered into a license agreement with MIT, effective November 2015 and amended as of July 2016. Under this license agreement, MIT granted Synlogic a worldwide license under certain patents and patent applications that is exclusive in the therapeutics and theranostics fields and non-exclusive in the internal research field. The license grants Synlogic rights to develop, make, have made, use, import, sell, and offer to sell licensed products and processes. Synlogic does not have the right to control prosecution of these licensed patents and patent applications and its rights to enforce the in-licensed patent rights are subject to certain limitations.
Under the terms of the MIT license agreement, as consideration for the license, Synlogic paid to MIT an upfront license fee and is eligible to receive an annual maintenance fee, milestone fees , sublicense fees if Synlogic should ever grant a sublicense to the licensed patents or patent applications and low single-digit royalty percentages on net sales of licensed products. MIT also receives reimbursement from Synlogic for patent prosecution expenses. Synlogic is subject to diligence requirements to develop licensed products in accordance with certain development milestones.
BU and MIT License
Synlogic entered into a license agreement with BU and MIT effective October 2015 and signed April 2017. Howard Hughes Medical Institute (“HHMI”) has an ownership interest in certain patent rights licensed to Synlogic under this license, which interest HHMI assigned to BU. HHMI is not a party to the license agreement, but receives the benefit of certain terms. Under this license agreement, BU and MIT granted Synlogic a worldwide license under certain patents and patent applications that is exclusive in the therapeutics and theranostics fields and non-exclusive in the diagnostic and internal research field. The license grants Synlogic rights to make, have made, use, lease, import, sell, and offer to sell licensed products and processes. Synlogic does not have the right to control prosecution of the licensed patents and patent applications, and Synlogic’s rights to enforce the licensed patent rights are subject to certain limitations. Under the terms of this license agreement, as partial consideration for the license, BU, MIT and MIT’s agent Omega Cambridge SPV, L.P. were issued an aggregate of 325,377 shares of Synlogic Common Stock. In addition, Synlogic paid an upfront fee, and reimbursed past patent prosecution costs, and the licensors are eligible to receive from Synlogic an annual maintenance fee, milestone fees, sublicense fees if Synlogic should ever grant a sublicense to the licensed patents and patent applications and low single-digit royalty percentages on net sales of licensed products. BU also receives reimbursement from Synlogic for patent prosecution expenses. Synlogic is subject to diligence requirements to develop licensed products in accordance with certain development milestones.
Individual patents extend for varying periods of time, depending upon the date of filing of the patent application, the date of patent issuance, and the legal term of patents in the countries in which they are obtained. Generally, patents issued for applications filed in the United States are effective for 20 years from the earliest effective non-provisional filing date. In addition, in certain instances, a patent term can be extended to account for delays in prosecution at the U.S. Patent and Trademark Office and/or to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. For regulatory delays, the restoration period cannot be longer than five years and the total patent term, including the restoration period, must not exceed 14 years following FDA approval. The duration of patents outside of the United States varies in accordance with provisions of applicable local law, but typically is also 20 years from the earliest effective non-provisional filing date. However, the actual protection afforded by a patent varies on a product-by-product basis, from country-to-country, and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country, and the validity and enforceability of the patent.
The patent positions of companies like Synlogic are generally uncertain and involve complex legal and factual questions. No consistent policy regarding the scope of claims allowable in patents in the field of synthetic biology has emerged in the United States. The patent situation outside of the United States is even more uncertain. With respect to both licensed and company-owned intellectual property, Synlogic cannot be sure that patents will be granted with respect to any of Synlogic’s pending patent applications or with respect to any patent applications filed by it in the future, nor can Synlogic be sure that any of its existing patents or any patents that may be granted to in the future will be commercially useful in protecting Synlogic’s products and the methods used to manufacture those products. For additional risks, please see the section entitled “ Risk Factors—Risks Related to Intellectual Property ” in this proxy statement/prospectus/information statement.
Trademarks
Synlogic’s registered trademark portfolio currently contains 31 registered trademark applications, consisting of seven (7) pending trademark applications in the United States and 24 pending trademark applications in Australia, Canada, China, Europe, India, Japan, Mexico and New Zealand and under the Madrid Protocol. Synlogic may also rely, in some circumstances, on trade secrets to protect its technology.
Other
Generally, Synlogic seeks to protect its technology and product candidates, in part, by entering into confidentiality agreements with those who have access to Synlogic’s confidential information, including employees, contractors, consultants, collaborators, and advisors. In some circumstances, Synlogic may rely on trade secrets to protect its technology. Synlogic seeks to preserve the integrity and confidentiality of its proprietary technology, trade secrets and processes by maintaining physical security of Synlogic’s premises and physical and electronic security of its information technology systems. Although Synlogic has confidence in these individuals, organizations, and systems, agreements or security measures may be breached and Synlogic may not have adequate remedies for any breach. In addition, Synlogic’s trade secrets may otherwise become known or may be independently discovered by competitors. To the extent that company employees, contractors, consultants, collaborators, and advisors use intellectual property owned by others in their work for Synlogic, disputes may arise as to the rights in related or resulting know-how and inventions. For this and more
comprehensive risks related to Synlogic’s proprietary technology, inventions, improvements and products, please see the section entitled “ Risk Factors—Risks Related to Intellectual Property, ” in this proxy statement/prospectus/information statement.
Regulatory Matters
Government Regulation and Product Approval
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record keeping, promotion, advertising, distribution, marketing and export and import of products such as those Synlogic is developing. A new drug must be approved by the FDA through the NDA process and a new biologic must be approved by the FDA through the biologics license application (“BLA”), process before it may be legally marketed in the United States
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the federal Food, Drug and Cosmetic Act (“FDCA”) and in the case of biologics, also under the Public Health Service Act (“PHSA”), and implementing regulations. Synlogic’s product candidates will be regulated by the FDA as biologics. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on Synlogic. The process required by the FDA before a drug or biologic may be marketed in the United States generally involves the following:
| • | completion of pre-clinical laboratory tests, animal studies and formulation studies according to cGLP other applicable regulations; |
| • | submission to the FDA of an IND which must become effective before human clinical trials may begin; |
| • | performance of adequate and well controlled human clinical trials according to cGCP to establish the safety and efficacy of the proposed drug for its intended use; |
| • | development and approval of a companion diagnostic device if the FDA or the sponsor believes that its use is essential for the safe and effective use of a corresponding product; |
| • | submission to the FDA of a BLA; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| • | FDA review and approval of the BLA. |
Once a pharmaceutical candidate is identified for development, it enters the pre-clinical testing stage. Pre-clinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the pre-clinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. In June 2016, the FDA issued an updated guidance for the industry entitled “Early Clinical Trials with Live Biotherapeutic Products: Chemistry, Manufacturing and Control Information,” which included recommendations from the FDA regarding the chemistry, manufacturing and control information that should be included in an IND for early clinical trials with live biotheraeutic products. Guidances such as this one reflect the FDA’s thinking on a topic at the time that they are issued and although this guidance is not binding on the FDA or a sponsor, it provided Synlogic with additional information about what should be included in Synlogic’s IND. The sponsor will also include a protocol detailing, among other things, the objectives of the first phase of the clinical trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated, if the first phase lends itself to an efficacy evaluation. Some pre-clinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns about on-going or proposed clinical trials or non-compliance with specific FDA requirements, and the trials may not begin or continue until the FDA notifies the sponsor that the hold has been lifted.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with cGCP regulations. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and timely safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. An institutional review board (“IRB”) at each institution participating in the clinical trial must review and approve each protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each trial subject or his or her legal representative, monitor the study until completed and otherwise comply with IRB regulations.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1: The product candidate is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase 2: This phase involves clinical trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| • | Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These clinical trials are intended to establish the overall risk benefit ratio of the product candidate and provide, if appropriate, an adequate basis for product labeling. |
Post-approval trials, sometimes referred to as Phase 4, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of a BLA.
The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the sponsor, known as a data safety monitoring board or committee. Depending on its charter, this group may determine whether a trial may move forward at designated check points based on access to certain data from the trial. Phase 1, Phase 2, and Phase 3 testing may not be completed successfully within any specified period, if at all.
During the development of a new drug, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to submission of an IND, at the end of Phase 2, and before a BLA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide advice, and for the sponsor and FDA to reach agreement on the next phase of development. Sponsors typically use the end of Phase 2 meeting to discuss their Phase II clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support approval of the new drug. If this type of discussion occurs, a sponsor may be able to request a Special Protocol Assessment (“SPA”), the purpose of which is to reach agreement with the FDA on the design of the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
According to FDA guidance for industry on the SPA process, a sponsor that meets the prerequisites may make a specific request for a special protocol assessment and provide information regarding the design and size of the proposed clinical trial. The FDA is required to evaluate the protocol within 45 days of the request to assess whether the proposed trial is adequate, and that evaluation may result in discussions and a request for additional information. An SPA request must be made before the proposed trial begins, and all open issues must be resolved before the trial begins. If a written agreement is reached, it will be documented and made part of the record. The agreement will be binding on the FDA and may not be changed by the sponsor or the FDA after the trial begins except with the written agreement of the sponsor and the FDA or if the FDA determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began. If the sponsor makes any unilateral changes to the approved protocol, the agreement will be invalidated.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things the manufacturer must develop methods for testing the identity, strength, quality and purity of the final drug. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
While the IND is active and before approval, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
There are also requirements governing the reporting of ongoing clinical trials and completed trial results to public registries. Sponsors of certain clinical trials of FDA-regulated products are required to register and disclose specified clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved. However, there are evolving rules and increasing requirements for publication of all trial related information, and it is possible that data and other information from trials involving drugs that never garner approval could require disclosure in the future.
U.S. Review and Approval Processes
The results of product development, pre-clinical and other non-clinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling, and other relevant information are submitted to the FDA as part of a BLA requesting approval to market the product. The submission of a BLA is subject to the payment of user fees; a waiver of such fees may be obtained under certain limited circumstances. The user fee for FY 2017 is $2,038,100 for an application containing clinical data. The FDA reviews all BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept a BLA for filing. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in depth substantive review. FDA may refer the BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The approval process is lengthy and often difficult, and the FDA may refuse to approve a BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than Synlogic interprets the same data. The FDA may issue a complete response letter, which may require additional clinical or other data or impose other conditions that must be met in order to secure final approval of the BLA, or an approved letter following satisfactory completion of all aspects of the review process. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP compliant to assure and preserve the product’s identity, strength, quality and purity. The FDA reviews a BLA to determine, among other things whether the product is safe, pure and potent and the facility in which it is manufactured, processed, packed or held meets standards designed to assure the product’s continued safety, purity and potency. Before approving a BLA, the FDA will inspect the facility or facilities where the product is manufactured.
BLAs receive either standard or priority review. A drug representing a significant improvement in treatment, prevention or diagnosis of disease may receive priority review. Priority review for an original BLA will be six months from the date that the BLA is filed. In addition, products studied for their safety and effectiveness in treating serious or life threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval and may be approved on the basis of adequate and well controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform adequate and well controlled Phase 4 clinical trials. Priority review and accelerated approval do not change the standards for approval, but may expedite the approval process.
After the FDA evaluates a BLA, it will issue an approval letter or a Complete Response Letter (“CRL”). An approval letter authorizes commercial marketing of the drug with prescribing information for specific indications. A CRL indicates that the review cycle of the application is complete and the application will not be approved in its present form. A CRL usually describes the specific deficiencies in the BLA identified by the FDA and may require additional clinical data, such as an additional pivotal Phase 3 trial or other significant and time-consuming requirements related to clinical trials, nonclinical studies or manufacturing. If a CRL is issued, the sponsor must resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the BLA does not satisfy the criteria for approval.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may require a sponsor to conduct Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after BLA approval, and may require testing and surveillance programs to monitor the safety of approved products which have been commercialized. The FDA may also place other conditions on approval including the requirement for a Risk Evaluation and Mitigation Strategy (“REMS”), to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve the BLA without an approved REMS, if required. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Marketing approval may be withdrawn for non-compliance with regulatory requirements or if problems occur following initial marketing.
The Pediatric Research Equity Act (“PREA”), requires a sponsor to conduct pediatric clinical trials for most drugs and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original BLAs and supplements thereto, must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must evaluate the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The sponsor or FDA may request a deferral of pediatric clinical trials for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the drug or biologic is ready for approval for use in adults before pediatric clinical trials are complete or that additional safety or effectiveness data needs to be collected before the pediatric clinical trials begin. Orphan indications are exempt from PREA. The FDA must send a non-compliance letter to any sponsor that fails to submit the required assessment, keep a deferral current or fails to submit a request for approval of a pediatric formulation.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA approval of Synlogic’s drugs, some of Synlogic’s U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (referred to as the “Hatch Waxman Amendments”). The Hatch Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term
restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one half the time between the effective date of an IND, and the submission date of a BLA, plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension, and the extension must be applied for prior to expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, Synlogic intends to apply for restorations of patent term for some of its currently-owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the filing of the relevant NDA.
Pediatric exclusivity is a type of marketing exclusivity available in the United States. Under the Best Pharmaceuticals for Children Act (the “BPCA”), an additional six months of marketing exclusivity may be available if a sponsor conducts clinical trials in children in response to a written request from the FDA. If a written request does not include clinical trials in neonates, the FDA is required to include its rationale for not requesting those clinical trials. The FDA may request studies on approved or unapproved indications in separate written requests. The issuance of a written request does not require the sponsor to undertake the described clinical trials. To date, Synlogic has not received any written requests.
Biologics Price Competition and Innovation Act of 2009
The ACA, which included the BPCIA, amended the PHSA to create an abbreviated approval pathway for two types of “generic” biologics, biosimilars and interchangeable biologic products, and provides for a 12-year data exclusivity period for the first approved biological product, or reference product, against which a biosimilar or interchangeable application is evaluated; however if pediatric clinical trials are performed and accepted by the FDA, the 12-year data exclusivity period will be extended for an additional six months. Because Synlogic’s product candidates will be regulated as biologics, if they are approved they may be subject to competition from biosimilars. A biosimilar product is defined as one that is highly similar to a reference product notwithstanding minor differences in clinically-inactive components and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product. An interchangeable product is a biosimilar product that may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The biosimilar applicant must demonstrate that the product is biosimilar based on data from (1) analytical studies showing that the biosimilar product is highly similar to the reference product; (2) animal studies (including toxicity); and (3) one or more clinical trials to demonstrate safety, purity and potency in one or more appropriate conditions of use for which the reference product is approved. In addition, the applicant must show that the biosimilar and reference products have the same mechanism of action for the conditions of use on the label, route of administration, dosage and strength, and the production facility must meet standards designed to assure product safety, purity and potency.
An application for a biosimilar product may not be submitted until four years after the date on which the reference product was first approved. The first approved interchangeable biologic product will be granted an exclusivity period of up to one year after it is first commercially marketed, but the exclusivity period may be shortened under certain circumstances.
The FDA has issued several final and draft guidance documents that provide FDA’s current thinking on approaches to demonstrating that a proposed biological product is biosimilar to a reference product. The FDA intends to issue additional guidance documents in the future. Nonetheless, the absence of final guidance documents covering all biosimilars issues does not prevent a sponsor from seeking licensure of a biosimilar under the BPCIA, and the FDA has already approved a few biosimilar applications in the United States.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for this type of disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use will be disclosed publicly by the FDA; the posting will also indicate whether a drug is no longer designated as an orphan drug. More than one product candidate may receive an orphan drug designation for the same indication. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to seven years of orphan product exclusivity, except in very limited circumstances. The FDA issued a final rule, effective August 12, 2013, intended to clarify several regulatory provisions, among which was a clarification of some of those limited circumstances. One of the provisions makes clear that the FDA will not recognize orphan drug exclusive approval if a sponsor fails to demonstrate upon approval that the drug is clinically superior to a previously approved drug, regardless of whether or not the approved drug was designated an orphan drug or had orphan drug exclusivity. Thus orphan drug exclusivity could also block the approval of one of Synlogic’s products for seven years if a competitor obtains approval of the same drug as defined by the FDA and Synlogic is not able to show the clinical superiority of its drug or if Synlogic’s product candidate is determined to be contained within the competitor’s product for the same indication or disease.
In August 2016, the FDA granted Synlogic orphan drug designation for its lead product candidate E. coli Nissle bacterium modified to metabolize ammonia for the treatment of urea cycle disorders. Orphan drug designation will provide Synlogic with seven years of market exclusivity that begins when the BLA for the drug receives FDA marketing approval for the use for which the orphan drug status was granted.
Expedited Review and Approval
The FDA has various programs, including Fast Track, priority review, and accelerated approval, which are intended to expedite or simplify the process for reviewing drugs, and/or provide for approval on the basis of surrogate endpoints. Even if a drug qualifies for one or more of these programs, the FDA may later decide that the drug no longer meets the conditions for qualification or that the time period for FDA review or approval will not be shortened. Generally, drugs that may be eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs, and those that offer meaningful benefits over existing treatments. For example, Fast Track is a process designed to facilitate the development, and expedite the review, of drugs to treat serious diseases and fill an unmet medical need. The request may be made at the time of IND submission and generally no later than the pre-BLA meeting. The FDA will respond within 60 calendar days of receipt of the request. Priority review, which is requested at the time of BLA submission, is designed to give drugs that offer major advances in treatment or provide a treatment where no adequate therapy exists an initial review within six months as compared to a standard review time of 10 months. Although Fast Track and priority review do not affect the standards for approval, the FDA will attempt to facilitate early and frequent meetings with a sponsor of a Fast Track designated drug and expedite review of the application for a drug designated for priority review. Accelerated approval provides an earlier approval of drugs that treat serious diseases, and that fill an unmet medical need based on a surrogate endpoint, which is a laboratory measurement or physical sign used as an indirect or substitute measurement representing a clinically meaningful outcome. Discussions with the FDA about the feasibility of an accelerated approval typically begin early in the development of the drug in order to identify, among other things, an appropriate endpoint. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing clinical trials to confirm the appropriateness of the surrogate marker trial.
In the Food and Drug Administration Safety and Improvement Act (“FDASIA”), Congress encouraged the FDA to utilize innovative and flexible approaches to the assessment of products under accelerated approval. The law required the FDA to issue related draft guidance within a year after the law’s enactment and also promulgate confirming regulatory changes. The FDA published a final guidance on May 30, 2014, entitled “Expedited Programs for Serious Conditions—Drugs and Biologics.” One of the expedited programs added by FDASIA is that for Breakthrough Therapy. A Breakthrough Therapy designation is designed to expedite the development and review of drugs that are intended to treat a serious condition where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint. A sponsor may request Breakthrough Therapy designation at the time that the IND is submitted, or no later than at the end of Phase 2 meeting. The FDA will respond to a Breakthrough Therapy designation request within 60 days of receipt of the request. A drug that receives Breakthrough Therapy designation is eligible for all Fast Track designation features, intensive guidance on an efficient drug development program, beginning as early as Phase 1 and commitment from the FDA involving senior managers. FDA has already granted this designation to several new biologics and two have received approval as of the end of March 2017.
In June 2017, the FDA granted Synlogic Fast Track designation for the use of a genetically modified strain of E. coli Nissle, SYNB1020, for the treatment of urea cycle disorders.
Post-Approval Requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, certain manufacturing changes and additional labeling claims, are subject to further FDA review and approval. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws and regulations. Synlogic relies, and expects to continue to rely, on third parties for the production of clinical and commercial quantities of its products. Future inspections by the FDA and other regulatory agencies may identify compliance issues at the facilities of Synlogic’s contract manufacturers that may disrupt production or distribution, or require substantial resources to correct.
Any drug products manufactured or distributed by Synlogic or Synlogic’s partners pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, record keeping requirements, reporting of adverse experiences with the drug, providing the FDA with updated safety and efficacy information, drug sampling and distribution requirements, complying with certain electronic records and signature requirements, and complying with FDA promotion and advertising requirements. FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label.
From time-to-time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. It is impossible to predict whether further legislative changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Foreign Regulation
In addition to regulations in the United States, Synlogic will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of its products. Whether or not Synlogic obtains FDA approval for a product, Synlogic must obtain approval by the comparable regulatory authorities of foreign countries or economic areas, such as the European Union, before it may commence clinical trials or market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicinal products produced by biotechnology or those medicinal products containing new active substances for specific indications such as the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, viral diseases and designated orphan medicines, and optional for other medicines which are highly innovative. Under the centralized procedure, a marketing application is submitted to the European Medicines Agency where it will be evaluated by the Committee for Medicinal Products for Human Use and a favorable opinion typically results in the grant by the European Commission of a single marketing authorization that is valid for all European Union member states within 67 days of receipt of the opinion. The initial marketing authorization is valid for five years, but once renewed is usually valid for an unlimited period. The decentralized procedure provides for approval by one or more “concerned” member states based on an assessment of an application performed by one member state, known as the “reference” member state. Under the decentralized approval procedure, an applicant submits an application, or dossier, and related materials to the reference member state and concerned member states. The reference member state prepares a draft assessment and drafts of the related materials within 120 days after receipt of a valid application. Within 90 days of receiving the reference member state’s assessment report, each concerned member state must decide whether to approve the assessment report and related materials. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
As in the United States, Synlogic may apply for designation of a product as an orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to 10 years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
Reimbursement
Sales of pharmaceutical products depend in significant part on the availability of third party reimbursement. Third party payors include government healthcare programs such as Medicare, managed care providers, private health insurers and other organizations. Synlogic anticipates third party payors will provide reimbursement for its products. However, these third party payors are increasingly challenging the price and examining the cost effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly-approved healthcare products. Synlogic may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost effectiveness of its products. Synlogic’s product candidates may not be considered cost effective. It is time consuming and expensive for Synlogic to seek reimbursement from third party payors. Reimbursement may not be available or sufficient to allow Synlogic to sell its products on a competitive and profitable basis.
Medicare is a federal healthcare program administered by the federal government that covers individuals age 65 and over as well as individuals with certain disabilities. Drugs may be covered under one or more sections of Medicare depending on the nature of the drug and the conditions associated with and site of administration. For example, under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which provide coverage for outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Parts A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level.
Medicare Part B covers most injectable drugs given in an in-patient setting and some drugs administered by a licensed medical provider in hospital outpatient departments and doctors’ offices. Medicare Part B is administered by Medicare Administrative Contractors, which generally have the responsibility of making coverage decisions. Subject to certain payment adjustments and limits, Medicare generally pays for a Part B-covered drug based on a percentage of manufacturer-reported average sales price, which is regularly updated. Synlogic believes that its product candidates that are intended to be administered intratumorally will be subject to the Medicare Part B rules.
Synlogic expects that there will continue to be a number of federal and state proposals to implement governmental pricing controls and limit the growth of healthcare costs, including the cost of prescription drugs. For example, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, “ACA”) enacted in March 2010, was expected to have a significant impact on the health care industry. ACA resulted in expanded coverage for the previously uninsured, however, President Trump ran for office on a platform that supported the repeal of the ACA and one of his first actions after his inauguration was to sign an Executive Order commanding federal agencies to try to waive or delay requirements of the ACA that impose economic or regulatory burdens on states, families, the health care industry and others. In March 2017, following the passage of the budget resolution for fiscal year 2017, the U.S. House of Representatives passed legislation known as the American Health Care Act, which, if enacted, would amend or repeal significant portions of the ACA. The U.S. Senate is currently considering its own legislation, and while Synlogic believes that the Senate is unlikely to adopt the American Health Care Act as passed by the House of Representatives, Synlogic cannot predict whether the Senate will ever introduce its own bill, what a Senate bill will contain if it does, or whether the House of Representatives and the Senate will ultimately agree on a joint bill.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of Synlogic placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of Synlogic’s products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
Other Regulatory Matters
Synlogic is subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. These operations may involve the use of hazardous and flammable materials, including chemicals and biological materials. Synlogic’s operations may also produce hazardous waste products. Synlogic contracts with third parties for the disposal of these materials and wastes.
Manufacturing
Synlogic has made and continues to make significant investments to develop manufacturing processes designed to allow it to reproducibly manufacture high quality living medicines at clinical scale and, later, at commercial scale to enable approval of its product candidates. Synlogic has a small-scale internal development group to support discovery and pre-clinical research and is building the organization to support scale-up and development towards commercialization. Synlogic currently works with contract manufacturing organizations (“CMOs”) for clinical material and formulation development work.
Synlogic has successfully transferred its manufacturing process for its lead hyperammonemia program to a CMO where it was used to manufacture Phase 1 clinical material pursuant to FDA’s cGMP requirements. Synlogic is similarly undertaking manufacturing technology transfer for its PKU program to supply material for its IND-enabling studies and subsequently for Phase 1 clinical trials.
These first clinical materials use a liquid formulation. Synlogic is investing in formulation development in parallel with Phase 1 clinical trial progress with the goal of providing a solid dose oral formulation (tablets or capsules) for later stage clinical development and commercial presentation, likely with a sachet formulation for pediatric use.
To enable the production of high levels of cells, or biomass, that can be administered as activated living medicines to perform metabolic functions, Synlogic can engineer its Synthetic Biotic medicines with switches. These switches are comprised of transcription factor and promoter pairs that allow for controlled expression of the therapeutic effectors produced by its Synthetic Biotic medicines. To ensure the metabolic capacity of the cells is allotted to the production of a high level of biomass during manufacturing, the effector circuits in the Synthetic Biotic programs are not expressed during this growth phase. At the end of the manufacturing process, the circuits are then induced, or activated. This two-step approach was designed to enable a high level of biomass production as well as to deliver the required activity necessary at the time of administration.
As Synlogic progresses in clinical development, it will need to scale up from Phase 1 clinical-scale to commercial-scale manufacturing. Synlogic is in the process of assessing CMOs who meet its criteria to supply its later-stage clinical development and commercial supply. Synlogic plans to compare the merits of working with one or more CMOs who meet its criteria with the possibility of building cGMP manufacturing capacity and capabilities internally.
Competition
The biotechnology industry is extremely competitive in the race to develop new products. While Synlogic believes it has significant competitive advantages with its industry-leading expertise in synthetic biology and metabolic engineering of probiotic bacteria, its clinical development expertise, and dominant intellectual property position, Synlogic currently faces and will continue to face competition for its development programs from companies that use synthetic biology or cell therapy development platforms and from companies focused on more conventional therapeutic modalities such as small molecules and antibodies. The competition is likely to come from multiple sources, including larger pharmaceutical companies, biotechnology companies and academia. Many of these competitors may have access to greater capital and resources than Synlogic. These competitors also compete with Synlogic in recruiting and retaining qualified scientific and management personnel, in establishing clinical trial sites and patient registration for clinical trials, and in accessing technologies to enable Synlogic’s programs. For any products that Synlogic may ultimately commercialize, not only will Synlogic compete with any existing therapies and those therapies currently in development, but it will also have to compete with new therapies that may become available in the future.
Competitors to Synlogic’s efforts to provide living medicines to patients with a wide range of indications include other synthetic biology companies developing other synthetic biology methods, cellular and microbiome-based companies, DNA and RNA-based companies, as well as companies developing small molecules or other biologics. In the case of indications that Synlogic is targeting with Synlogic’s own Synthetic Biotic medicines, competitors include, but are not limited to:
| • | UCD |
| • | Horizon Pharma plc has a licensed product; and |
| • | Dimension Therapeutics, Inc., Aeglea Biotherapeutics, Inc., Arcturus Therapeutics Inc., Castle Creek Pharma LLC, PhaseRx, Inc., RaNA Therapeutics and Selecta Biosciences, Inc. are each involved with discovery or pre-clinical stage product candidates. |
| • | HE |
| • | Valeant Pharmaceuticals International, Inc. has a licensed product; and |
| • | Ocera Therapeutics, Inc., Umecrine Cognition AB and Salix Pharmaceuticals, Ltd, as well as other pre-clinical and discovery stage companies are each developing product candidates. |
| • | PKU |
| • | BioMarin, Inc. has a licensed and development stage product; and |
| • | MipSalus ApS, Codexis, Inc., Dimension Therapeutics, Inc. and Synthetic Biologics, Inc. are each developing product candidates. |
The Synlogic Team: Executives, Founders and Scientific Advisors
Synlogic’s team of executives has proven track records of successfully translating scientific visions into successful commercial therapeutic products, solving complex issues in developing novel therapeutics and progressing new and novel products through regulatory approval. Synlogic’s scientific founders, Timothy Lu, M.D., Ph.D., and James Collins, Ph.D., are experts in the emerging field of synthetic biology. In addition to Synlogic’s management team and founders, it has established advisory relationships with researchers and clinicians dedicated to the development of Synthetic Biotic therapeutic products for patients with significant unmet medical needs and whose expertise spans synthetic biology, metabolic engineering, metabolism, immuno-modulation and immune-oncology arenas. Synlogic’s scientific advisors include Dr. Lu and Dr. Collins; Christopher Voigt, Ph.D., Cammie Lesser, M.D., Ph.D. and Kristala Prather, Ph.D., experts in synthetic biology and bacterial metabolism; and Charles Mackay, Ph.D., Ulrich von Andrian, M.D., Ph.D. and Sangeeta Bhatia, M.D., Ph.D., experts in immunomodulation and oncology. Synlogic intends to expand its advisory boards as Synlogic grows. All of Synlogic’s founders and advisors are equity holders in Synlogic and receive compensation as scientific advisors. Although they are regularly available for scientific consultation, Synlogic’s arrangements with these individuals do not entitle Synlogic to any of their existing or future intellectual property derived from their independent research or research with other third parties.
Employees
As of July 1, 2017, Synlogic had 38 full-time employees, 25 of whom have an M.D. or Ph.D. Of Synlogic’s full-time employees, 28 were primarily engaged in research and development activities. None of Synlogic’s employees are subject to a collective bargaining agreement. Synlogic believes that it has good relations with its employees.
Facilities
Synlogic currently leases facilities at 200 Sidney Street, Suite 320, Cambridge, Massachusetts 02139 containing its research and development, laboratory and office spaces. This facility consists of approximately 14,390 square feet. Synlogic’s lease expires in April 2021. However, this lease is likely to be terminated prematurely by agreement as Synlogic negotiates to enter into a new lease to replace the current Sidney Street facilities with increased occupancy in the first quarter of 2018.
Legal Proceedings
Synlogic is not currently a party to any material legal proceedings.
Corporate Information
Synlogic was incorporated in Delaware on March 14, 2014 under the name TMC Therapeutics, Inc. Its principal executive offices are located at 200 Sidney Street, Suite 320, Cambridge, Massachusetts 02139 and its telephone number is (617)-401-9947. Synlogic’s website is www.synlogictx.com. References to Synlogic’s website are inactive textual references only and the content of Synlogic’s website should not be deemed incorporated by reference into this proxy statement/prospectus/information statement.