Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - IMMUNOMEDICS INC | immu-20170630ex3222e676e.htm |
| EX-32.1 - EX-32.1 - IMMUNOMEDICS INC | immu-20170630ex32114ec3d.htm |
| EX-31.2 - EX-31.2 - IMMUNOMEDICS INC | immu-20170630ex312146a47.htm |
| EX-31.1 - EX-31.1 - IMMUNOMEDICS INC | immu-20170630ex31130c459.htm |
| EX-23.1 - EX-23.1 - IMMUNOMEDICS INC | immu-20170630ex2311dfb26.htm |
| EX-21.1 - EX-21.1 - IMMUNOMEDICS INC | immu-20170630ex2111d8d6d.htm |
| EX-10.37 - EX-10.37 - IMMUNOMEDICS INC | immu-20170630ex103752071.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark one)
[x] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended June 30, 2017.
or
[ ]TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d)
OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from ______ to ________.
Commission file number: 0-12104
IMMUNOMEDICS, INC.
(Exact name of registrant as specified in its charter)
|
Delaware |
61-1009366 |
|
(State of incorporation) |
(I.R.S. Employer Identification No.) |
|
|
|
|
300 The American Road, Morris Plains, New Jersey |
07950 |
|
(Address of principal executive offices) |
(Zip Code) |
Registrant's telephone number, including area code: (973) 605-8200
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Name of each exchange on which registered |
|
Common Stock, $0.01 par value |
|
NASDAQ Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ◻ No ☑
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ◻ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirement for the past 90 days. Yes ☑ No ◻
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☑ No ◻
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§299.405 of this chapter) is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See definitions of “large accelerated filer”, “accelerated filer”, “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large Accelerated Filer |
◻ |
Accelerated Filer |
☑ |
|
Non-Accelerated Filer |
◻ |
Smaller Reporting Company |
◻ |
|
Emerging Growth Company |
◻ |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ◻
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2 of the Act). Yes ◻ No ☑
The aggregate market value of the registrant’s common stock held by non-affiliates computed by reference to the price at which the common stock was last sold as of December 31, 2016 was $388,989,000. The number of shares of the registrant’s common stock outstanding as of August 15, 2017 was 111,279,090.
Documents Incorporated by Reference:
Certain information required in Part III of this Annual Report on Form 10-K will be set forth in, and incorporated from the registrant’s Proxy Statement for the 2017 Annual Meeting of Stockholders, which will be filed by the registrant with the Securities and Exchange Commission not later than 120 days after the end of the registrant’s fiscal year ended June 30, 2017.
In this Form 10-K, we use the words "Immunomedics, Inc." to refer to Immunomedics, Inc., a Delaware corporation, and we use the words "Company," "Immunomedics," "Immunomedics, Inc.," "we," "us" and "our" to refer to Immunomedics, Inc. and its subsidiaries.
PART I
Item 1. Business
Immunomedics is a clinical-stage biopharmaceutical company developing monoclonal antibody-based products for the targeted treatment of cancer, autoimmune disorders and other serious diseases. Our proprietary technologies allow us to create humanized antibodies that can be used either alone in unlabeled or “naked” form, or conjugated with radioactive isotopes, chemotherapeutics, cytokines or toxins. Using these technologies, we have built a pipeline of six clinical-stage product candidates. Our most advanced product candidate is IMMU-132 (sacituzumab govitecan), an antibody-drug conjugate (“ADC”) that has received Breakthrough Therapy Designation (“BTD”) from the United States Food and Drug Administration (“FDA”) for the treatment of patients with metastatic triple-negative breast cancer (“mTNBC”) who have failed at least two prior therapies for metastatic disease. BTD has provided us with ready access to the FDA to discuss its Expedited Programs for Serious Conditions for IMMU-132, including the Accelerated Approval Program.
At our Annual Meeting of Stockholders for 2016, a new Board of Directors was elected to embark on a new development plan for IMMU-132. After conducting a multifaceted assessment of the Company’s people, processes, and data to confirm the status of IMMU-132 development, the new Board, having recognized the significant potential of IMMU-132 and the Company’s capabilities, immediately adopted a new corporate strategy focused on bringing IMMU-132 to the market on its own for the benefit of patients with mTNBC and the creation of value for our shareholders.
To the best of our knowledge, IMMU-132 is the only antibody-based product candidate currently in clinical development that targets the cancer marker Trop-2. We chose this target because it internalizes rapidly into cancer cells following binding by the antibody, making it an ideal target for the delivery of toxic drugs with an ADC. Another reason is that Trop-2 is highly expressed by many and varied types of solid cancers. We believe that if IMMU-132 works in one of the Trop-2-expressing cancers, there is a good chance that it may work in other cancer types that contain the same marker. While this hypothesis is being evaluated in our Phase 2 basket trial, our current focus is to bring IMMU-132 to market for patients with mTNBC expeditiously via the FDA’s Accelerated Approval Program. To that end, our foremost goals for IMMU-132 during fiscal year 2018 are as follows:
|
1. |
Submit a Biologics License Application (“BLA”) to the FDA for accelerated approval of IMMU-132 in mTNBC between December 2017 and March 2018. Per FDA guidance, the following steps need to be addressed for the filing:; |
|
a. |
Complete the review of treatment responses by an independent, third-party blinded group of radiologists and finalize the full data set of approximately 100 assessable patients from the Phase 2 trial of IMMU-132 in mTNBC. Enrollment of the full complement of more than 100 assessable patients was completed in December 2016; |
|
b. |
Initiate the Phase 3 confirmatory trial of IMMU-132 in patients with mTNBC. We have a Special Protocol Assessment (“SPA”) in place for the study and clinical trial materials have been manufactured. In addition, we have engaged a Clinical Research Organization (“CRO”) and site selection and planning are currently underway; |
|
c. |
Validate all Chemistry, Manufacturing and Controls (“CMC”) processes on commercial manufacturing. We are required to validate all or part of CMC at the time of the BLA filing. The level of CMC validation required by the FDA at the time of submission will be a determining factor in the filing timeline. |
|
2. |
Demonstrate CMC preparedness for large-scale manufacturing in anticipation of a potential commercial launch in the U.S. for IMMU-132 in early 2019. Although we have enough capability with our in-house manufacturing facility to produce commercial materials to support the launch and initial commercial |
2
operations, our goal is to transition manufacturing of the antibody from in-house to a large-scale Contract Manufacturing Organizations (“CMO”) for longer term commercial production. |
We believe our current focus on commercializing IMMU-132 as a third-line therapy for patients with mTNBC is also the key to opening the door to further commercial opportunities in the future including developing IMMU-132 in earlier lines of therapy in mTNBC, as a monotherapy or in combination therapies, as well as expansion into other indications beyond TNBC, such as urothelial cancer (“UC”), small-cell lung cancer (“SCLC’), and non-small-cell lung cancer (“NSCLC”). It’s only by proving IMMU-132 in TNBC that we can explore, expand into, and potentially capitalize on these new opportunities. While our immediate focus on commercializing IMMU-132, on our own, in the U.S. and European markets; we are alert to opportunities to commercialize IMMU-132 in certain other regional markets; and we are also open to business development opportunities to develop other pipeline assets.
Our Clinical and Preclinical Programs
We believe that each of our antibodies has therapeutic potential either when administered as a naked antibody or when conjugated with chemotherapeutics, therapeutic radioisotopes (radiolabeled), cytokines or other toxins to create unique and potentially more effective treatment options. The attachment of various compounds to antibodies is intended to allow the delivery of these therapeutic agents to tumor sites with better specificity than conventional chemotherapy or radiation therapy approaches. This treatment method is designed to reduce the total exposure of the patient to the therapeutic agents, which ideally minimizes debilitating side effects.
Our portfolio of investigational products includes ADCs that are designed to deliver a specific payload of a chemotherapeutic directly to the tumor while reducing overall toxic effects that are usually found with conventional administration of these chemotherapy agents. In addition toIMMU-132, labetuzumab govitecan (“IMMU-130”), is in a Phase 2 trial for metastatic colorectal cancer (“mCRC”). These two ADCs facilitate targeted delivery of SN-38, the active metabolite of irinotecan, an effective, yet toxic chemotherapeutic, more directly to tumor cells. While sacituzumab govitecan and labetuzumab govitecan are circulating in the blood stream, our novel and proprietary ADC linking system keeps SN-38 conjugated to the antibody and in an inactive form, thereby reducing toxicity to normal tissues. The clinical safety and efficacy results obtained with sacituzumab govitecan and labetuzumab govitecan suggest that this half-life is long enough for the ADCs to reach their targets on the surface of tumor cells, without causing significant harm to the rest of the body. More importantly, the pH-sensitive nature of the linker allows the continuous release of SN-38 from the tumor-bound ADCs, regardless of whether the ADC is internalized or remains on the surface of the tumor cell leading to a locally enhanced concentration of SN-38 within or near the tumor. We believe this selective delivery enhances SN-38’s bioavailability at the tumor, which may improve efficacy while also reducing toxicity.
We have a research collaboration with Bayer to study epratuzumab as a thorium-227-labeled antibody. We have other ongoing collaborations in oncology with independent cancer study groups. The IntreALL Inter-European study group is conducting a large, randomized, Phase 3 trial combining epratuzumab with chemotherapy in children with relapsed acute lymphoblastic leukemia at clinical sites in Australia, Europe, and Israel.
We also have a number of other product candidates that target solid tumors and hematologic malignancies, as well as other diseases, in various stages of clinical and pre-clinical development. These include combination therapies involving antibody-drug conjugates, bispecific antibodies targeting cancers and infectious diseases as T-cell redirecting immunotherapies, as well as bispecific antibodies for next-generation cancer and autoimmune disease therapies, created using our patented DOCK-AND-LOCK® (“DNL®”) protein conjugation technology.
3
Below is our pipeline chart of late-stage antibody-based therapies including ADCs and other antibodies.

* The International clinical trial on childhood relapsed acute lymphoblastic leukemia (“IntReALL”) is funded by the European Commission.
Antibody-Drug Conjugates
The targeted delivery of drug by an antibody is an exciting approach in cancer treatment that has gained significant interest over the past few years. ADCs are designed to deliver a specific payload of a chemotherapeutic directly to the tumor while reducing overall toxic effects that are usually found with conventional administration of these chemotherapy agents. We have a unique approach to ADC design that has allowed us to develop a platform technology with solid tumor therapy in mind. ADCs have three critical components – the antibody, linker and payload. Beginning with antibody, we have a suite of proprietary humanized antibodies, which are the result of 30 years of research and development in the field, and have selected highly cancer-specific antibodies to deliver moderately potent drugs to the tumors. We believe this approach permits a greater delivery of drug over repeated cycles of therapy, thereby improving the therapeutic index. Secondly, our patented linker technology is designed specifically for SN-38, which keeps SN-38 conjugated to the antibody and in an inactive form, thereby reducing toxicity to normal tissues. In addition, the pH-sensitive nature of the linker allows for rapid and continuous release of the drug once the ADC reaches a tumor. A high drug-to-antibody ratio also enhances the drug’s bioavailability within or near the tumor, which may improve efficacy while also reducing toxicity. Our payload drug of choice is SN-38, which is the active form of irinotecan, but is about 1,000-fold more potent. SN-38 cannot be administered directly to patients because it is not water soluble, and is too toxic. However, our unique CL-2A linker allows antibody-SN-38 conjugates to be soluble in water. Furthermore, there is no loss of antibody binding or drug activity.
As a result, all of our ADC product candidates have relatively lower toxicity and are capable of delivering greater doses of drug to tumor. They offer patients and physicians an opportunity for longer-term, repeated treatments. Two of our ADCs have completed Phase 2 studies in solid cancers. IMMU-132 targets a variety of solid tumors that over-express Trop-2, while our second ADC, IMMU-130 (labetuzumab govitecan), binds the CEACAM5 antigen,
4
expressed on colorectal and other solid cancers. We are also positioned to expand our ADC Program to treat liquid cancers with the creation of IMMU-140 that targets HLA-DR.
Sacituzumab govitecan or IMMU-132
Sacituzumab govitecan is our lead investigational product for the treatment of patients with solid cancers. It is an ADC that contains SN-38, the active metabolite of irinotecan, approved by many health authorities, including the FDA, as a chemotherapeutic for patients with cancer. As noted earlier, SN-38 cannot be given directly to patients because of its toxicity and poor water solubility. Sacituzumab govitecan was created at Immunomedics by conjugating SN-38 site-specifically and at a high ratio of drug to hRS7, our anti-Trop-2 antibody. Trop-2 is a cell-surface receptor that while over-expressed by many human tumors, including cancers of the breast, urinary bladder, and lung, has limited expression in normal human tissues. It internalizes rapidly into cancer cells following binding by hRS7, making it a suitable target for the delivery of cytotoxic drugs.
Sacituzumab govitecan has received BTD from the FDA for the treatment of patients with mTNBC who have failed prior therapies for their disease. The regulatory agency has also granted this ADC Fast Track designation for patients with TNBC and for patients with SCLC, or NSCLC. Fast Track designation is designed to expedite the development and review of applications for products intended for the treatment of a serious or life-threatening disease or condition. This ADC has also been designated as an orphan drug by the FDA for the treatment of patients with SCLC or pancreatic cancer in the United States and by the European Medicines Agency (“EMA”) for the treatment of patients with pancreatic cancer in the European Union.
Our Phase 2 basket trial of sacituzumab govitecan has now treated over 500 patients in more than 15 types of solid cancers, with the dose of 10 mg/kg given on days 1 and 8 of repeated 21-day cycles being the established dose regimen. TNBC is the furthest along with the most patients enrolled. In December 2016, we achieved the goal of enrolling 100 TNBC patients as requested by the FDA for a BLA filing. Interim results in 85 assessable patients with mTNBC were presented by our clinical investigator at an Investor R&D Day conducted during January 2017. Overall, 81% of patients treated with IMMU-132 showed tumor shrinkage from baseline measurements. Two of these patients experienced complete responses (“CRs”) and 23 reported partial responses (“PRs”), while an additional three patients with initial PRs are awaiting confirmation. The clinical benefit rate (CR and PR, and patients with stable disease (“SD”)) at six months or later was 44% and the median duration of response (“DOR”) for those with objective responses was 10.8 months. Median progression-free survival (“PFS”) and median overall survival (“OS”) for all 85 patients were 6.0 months and 18.8 months, respectively. To put these results in perspective, based on what has been reported in the medical literature, the current standard of care offers mTNBC patients a PFS of 1.7 to 3.7 months and about 12 months of median survival.
The major toxicity (grade >3) has been neutropenia (39%) in this and most cancer patient cohorts, which has been managed by dose reduction, dose delays, or giving a hematopoietic cytokine. Diarrhea, which is the major side effect with irinotecan, has been much less, such as a grade >3 of 13%.
As per FDA requirements, radiological scans from patients with at least 20% tumor shrinkage are being reviewed by an independent third-party group in a blinded fashion. The centrally adjudicated results will be part of a BLA submission for the accelerated approval of sacituzumab govitecan in mTNBC. The FDA also requires a confirmatory Phase 3 trial to be underway at the time of BLA submission. We have a SPA in place for the study and clinical trial materials have been manufactured. In addition, we have engaged a CRO and site selection and planning are currently underway. Details of this confirmatory trial can be obtained from clinicaltrials.gov website using the identifier NCT02574455.
In addition to TNBC, we are making progress with IMMU-132 across three other advanced indications: UC, SCLC and NSCLC.
In patients with UC, especially metastatic urinary bladder cancer, updated results were reported at the 2017 Genitourinary Cancers Symposium by our clinical investigator. A total of 44 patients with metastatic UC had been enrolled into this open-label multicenter study. Sites of metastases included liver (N=9; 25%), lymph nodes (N=14; 39%), lungs (N=14; 39%, pelvis (N=9, 25%), and bone (N=4; 11%). Patients received a median of six doses (range, 1-50) of sacituzumab govitecan, which was administered at 8 or 10 mg/kg on days 1 and 8 of 3-week cycles. Despite
5
repeated dosing, grade 3 or higher adverse events were limited to neutropenia (30%), febrile neutropenia (11%), fatigue (11%), and diarrhea (3%). The objective response rate (“ORR”) in 36 response-assessable patients was 31%, including one confirmed CR and ten confirmed PRs. For the 41 intention-to-treat (“ITT”) patients, median PFS was 7.2 months and median OS was 15.5 months.
For SCLC, despite the aggressive nature of the disease, encouraging ORR in 50 ITT patients was published online in Clinical Cancer Research on July 5, 2017. The median number of prior chemotherapies for this group of patients was 2 (range, 1-7). All patients had metastatic (stage IV) disease and had been previously treated with platinum-based therapy and etoposide, with 11 having received topotecan. Notable findings from patients after receiving treatment with sacituzumab govitecan at the dose level of 8 mg/kg or 10 mg/kg include:
|
· |
Ninety-two percent of the patients evaluated for expression of the target for sacituzumab govitecan, Trop-2, had elevated levels in their archived tumor specimens; |
|
· |
Patients given repeated treatment cycles had manageable toxicity, mostly Grade >3 neutropenia (34%), and 60% of those patients experienced tumor shrinkage from baseline computed tomography (“CT”) measurements; |
|
· |
ORR was 17% at the optimal dose schedule; the median DOR and OS were 5.7 and 7.5 months, respectively; and |
|
· |
Activity was observed in patients who were chemosensitive or chemoresistant to frontline chemotherapy, in patients who failed second-line topotecan, and in a subset who relapsed after immune checkpoint inhibitor therapy. |
Phase 2 results with sacituzumab govitecan in patients with NSCLC were published online in the Journal of Clinical Oncology on May 26, 2017. In 54 heavily-pretreated patients with metastatic NSCLC who received either 8 or 10 mg/kg sacituzumab govitecan on days 1 and 8 of 21-day cycles. The primary endpoints were safety and ORR. PFS and OS were secondary endpoints. Notable findings from the study include:
|
· |
In the response-assessable study population (N = 47), which had a median of 3 prior therapies (range, 2-7), 67% of patients showed a shrinkage from baseline CT measurements; |
|
· |
The confirmed ORR was 19.1%, the median DOR 6.0 months, and the clinical benefit rate (CR+PR+SD>4 months) was 43%. Responses occurred with a median onset of 3.8 months, including patients who had relapsed or progressed after immune checkpoint inhibitor therapy; |
|
· |
Median ITT PFS was 5.2 months (95% CI, 3.2, 7.1), and median ITT OS was 9.5 months; |
|
· |
Grade 3 or higher adverse events included neutropenia (28%), diarrhea (7%), nausea (7%), fatigue (6%), and febrile neutropenia (4%); and |
|
· |
Over 90% of 26 assessable archival tumor specimens were highly positive for Trop-2 by immunohistochemistry. |
Sacituzumab govitecan has a tolerable safety profile in these patients with diverse, advanced, heavily-pretreated solid cancers. No prophylactic diarrhea or granulocyte colony-stimulating factor medication to stimulate the production of neutrophils was given. More importantly, repeated doses can be given over months without evoking interfering anti-sacituzumab govitecan antibodies from patients’ own immune system.
We have an extensive intellectual property portfolio protecting sacituzumab govitecan. Specifically, 35 patents were issued in the U.S. and 21 foreign patents were issued covering composition of matter, synthesis and uses. Certain patents relating to the protein sequence of the hRS7 antibody used in sacituzumab govitecan expire in 2017 in the United States and 2023 overseas. Patents to compositions and use of the CL2A linker incorporated in sacituzumab govitecan expire between 2023 and 2029 in the U.S. and overseas. Other patents relating to use of hRS7 for cancer therapy, including the SN-38 conjugated form of hRS7 used in sacituzumab govitecan, extend to 2033. Additionally, we are entitled to extend the term of our key patent for up to 5 more years. Outside the U.S., patents were issued in Australia, Canada, China, Europe, Israel, Japan, Mexico, South Korea and other key global markets.
IMMU-130 or Labetuzumab Govitecan
Our second investigational solid-tumor ADC involves our anti-CEACAM5 antibody, labetuzumab, conjugated to SN-38. The agent is currently being studied in patients with metastatic colorectal cancer (“mCRC”) who had received
6
at least one prior irinotecan-containing regimen and had an elevated blood titer of carcinoembryonic antigen (“CEA”). Several dosing schedules were evaluated in three Phase 1 studies. IMMU-130 showed therapeutic activity in all three trials, but a more frequent dosing schedule, with administrations of the ADC once or twice-weekly for two weeks followed by a week off, appeared to be more active in patients with mCRC than when administered every other week.
In the expanded Phase 2 study, patients were being treated in 3-week cycles, receiving IMMU-130 at 8 or 10 mg/kg once-weekly or twice a week at 4 or 6 mg/kg for the first two weeks followed by one week of rest. Updated results were presented at the 2016 American Association for Cancer Research Annual Meeting. A total of 82 patients were enrolled into the open-label study.
Since there was no significant difference in safety and efficacy between the two once-weekly dosing schedules, for patient’s convenience, the once-a-week dose of 10 mg/kg was chosen for future studies in mCRC patients.
Certain patents relating to labetuzumab used in IMMU-130 expired in 2016. Other patents relating to use of labetuzumab for cancer therapy, including the SN-38 conjugated form of labetuzumab used in labetuzumab govitecan, extend to 2033.
IMMU-140
Our third ADC, IMMU-140, is comprised of the humanized anti-HLA-DR antibody, IMMU-114, conjugated to SN-38, through the Company’s proprietary linker, CL2A. When given subcutaneously, the parental antibody, IMMU-114, has shown promising activity in patients with non-Hodgkin lymphoma (“NHL”) and chronic lymphocytic leukemia (“CLL”), and with a relatively safe profile. Thus, IMMU-140 is a dual-therapeutic, combining the signaling functions of the parental antibody, IMMU-114, with the cytotoxicity of SN-38. However, acute myelocytic leukemia (“AML”), despite having high expression levels of HLA-DR, has proven to be resistant to the antitumor effects of IMMU-114 in vitro. As a result, the potential treatment of AML, acute lymphocytic leukemia (“ALL”), and multiple myeloma (“MM”), continue to challenging for IMMU-114.
We conducted a preclinical study to determine if SN-38, a drug not commonly used in liquid malignancies, would prove to be an effective and safe therapeutic when targeted with the IMMU-114 antibody, which could then improve the antitumor activity of IMMU-114. A total of four human cancer cell lines, AML, ALL, MM, and CLL, were used to examine the in vitro and in vivo activity of IMMU-140 versus parental IMMU-114. The results were presented at the 2016 Annual Meeting of the American Society of Hematology.
In seven human disease models in mice ALL, CLL, MM, AML, diffuse large B-cell lymphoma (“DLBCL”), Hodgkin lymphoma (“HL”), and melanoma), IMMU-140 treatment at 25 mg/kg twice weekly for 4 weeks (human equivalent dose (“HED”) = 2 mg/kg) provided significant therapeutic efficacy compared to non-specific control ADCs. Of note, in intractable AML and ALL, IMMU-140 imparted a >140% and 80% increase in survival, respectively, compared to IMMU-114 therapy. Even a dose reduction to 12.5 mg/kg (HED = 1 mg/kg), produced significant antitumor effects compared to all controls in AML and malignant melanoma. Though not significant, in MM IMMU-140 improved survival approximately 60% compared to IMMU-114 while in CLL, this treatment was significantly better than IMMU-114. Therapy with IMMU-140 was well tolerated by the mice with no appreciable loss in body weight.
These preclinical results demonstrated IMMU-140’s higher potency than naked IMMU-114 in ALL and AML; and an added, if not significant, survival benefit in experimental MM and CLL. More importantly, the dual-therapeutic potential of IMMU-140 allows for the ability to treat a range of HLA-DR-positive hematopoietic and solid cancers, and therefore warrants further clinical development.
Other Product Candidates
We have additional potential products for the treatment of cancer and autoimmune diseases including epratuzumab, our anti-CD22 antibody; veltuzumab, our anti-CD20 antibody; milatuzumab, our anti-CD74 antibody; and IMMU-114, a humanized anti-HLA-DR antibody.
7
Epratuzumab
Epratuzumab is a humanized antibody that targets CD22, an antigen found on the surface of B lymphocytes, a type of white blood cell critical to proper immune system function. Elevated expression of CD22 and other B-cell receptor-associated (“BCR”) proteins on B lymphocytes has been associated with blood cancers and autoimmune diseases. Epratuzumab’s mechanism of action includes the transfer of BCR-proteins to helper cells called effector cells, thereby reducing B-cell destruction and the impact of epratuzumab on the immune system. We believe epratuzumab is the only antibody in development targeting the reduction of these proteins without severely depleting B-cells through a process known as trogocytosis.
We have a research collaboration with The Bayer Group (“Bayer”) to study epratuzumab as a thorium-227 labeled antibody. Bayer is currently enrolling patients with relapsed or refractory CD22-positive NHL into a Phase 1 study evaluating epratuzumab labeled with thorium-227. This study is focusing on patients with diffuse large B-cell lymphoma and potentially follicular lymphomas who have been previously treated with, or are not considered candidates for available therapies.
In addition, the IntreALL Inter-European study group is conducting a large, randomized, Phase 3 trial combining epratuzumab with chemotherapy in children with relapsed ALL at clinical sites in Australia, Europe, and Israel. This Phase 3 study, which is partially funded by the European Commission, assesses the efficacy and safety of this combination therapy using event-free survival as the surrogate for survival, the primary endpoint.
Although certain patents to the epratuzumab protein sequence expired in 2014 in the United States and in 2015 overseas, other patents issued to therapeutic use of epratuzumab extend to 2018-2023 for cancer and 2020 for autoimmune disease. The method of preparing concentrated epratuzumab for subcutaneous administration is covered by another patent family with expiration in the United States in 2032.
Veltuzumab
Veltuzumab is a humanized monoclonal antibody targeting CD20 receptors on B lymphocytes currently under development for the treatment of NHL and autoimmune diseases. The Office of Orphan Products Development of the FDA has granted orphan status for the use of veltuzumab for the treatment of patients with immune thrombocytopenia (“ITP”). We have studied the subcutaneous formulation of veltuzumab in patients with ITP in a Phase 1/2 trial, which was designed to evaluate different dosing schedules. This trial has completed patient accrual and patients are being followed for up to five years.
We are currently evaluating various options for further clinical development of veltuzumab in ITP and other autoimmune disease indications, as well as in oncology, including licensing arrangements and collaborations with outside study groups.
Milatuzumab
Milatuzumab is a humanized monoclonal antibody targeting tumors that express the CD74 antigen, which is present on a variety of hematological tumors and even on some solid cancers, with restricted expression by normal tissues. It has received orphan drug designation from the FDA for the treatment of patients with multiple myeloma or CLL. Milatuzumab is the first anti-CD74 antibody that has entered into human testing and we have completed initial Phase 1 studies in patients with relapsed multiple myeloma, NHL or CLL.
The anti-CD74 antibody is currently being studied subcutaneously in autoimmune disease in a Phase 1b study in patients with active SLE supported by a three-year research grant from the United States Department of Defense with a potential funding of $2 million.
Our interest in pursuing milatuzumab in immune diseases is driven by the observations that implicated CD74 in antigen presentation, particularly by dendritic and other immune cells—and as a survival factor for rapidly proliferating malignant cells. Recent findings have determined that CD74 is a receptor for the pro-inflammatory chemokine, macrophage migration-inhibitory factor, and that binding of the factor to CD74 initiates a signaling cascade resulting in proliferation and survival of normal and malignant B cells, such as in CLL. Migration-inhibitory factor is widely expressed by immune cells, particularly macrophages, and is known to play a role in autoimmune disease. Thus, we
8
believe that milatuzumab, by blocking the function of CD74, could be useful in the management of immune diseases either alone or in combination with other agents including other B-cell antibodies, such as epratuzumab and veltuzumab.
First results from the open-label Phase 1b study of subcutaneously administered milatuzumab in an initial cohort of ten adult patients with moderate lupus disease activity but not severe flares (at least 2 BILAG B scores, but no A's) were presented at a poster session during the 2016 annual European League Against Rheumatism (“EULAR”) Congress. Based on the early encouraging results, we have expanded the study into a double-blind, placebo-controlled 30-patient randomized trial to confirm the activity of milatuzumab in this population and have received approval from the Department of Defense for an increased budget to support the expansion.
IMMU-114
IMMU-114 is a novel humanized antibody directed against an immune response target, HLA-DR, under development for the treatment of patients with B-cell and other cancers. HLA-DR is a receptor located on the cell surface and its role is to present foreign objects to the immune system for the purpose of eliciting an immune response. Increased presence of HLA-DR in hematologic cancers has made it a prime target for antibody therapy.
Although other anti-HLA-DR antibodies have been developed, IMMU-114 is distinguished by having a different immunoglobulin class, IgG4, which does not function by the usual effector-cell activities of antibodies, such as complement-dependent cytotoxicity (“CDC”) and antibody-dependent cellular cytotoxicity (“ADCC”). As a result, IMMU-114 does not rely on an intact immune system in the patient to kill tumor cells. Furthermore, because ADCC and CDC are believed to play a major role in causing the side effects of antibody therapy, we expect IMMU-114 to be less toxic to patients.
By targeting HLA-DR, a receptor that is different from the antigen targeted by rituximab or other antibodies in development for NHL and other B-cell malignancies, IMMU-114 may represent a new tool in the arsenal to combat these cancers. The anti-HLA-DR antibody is being evaluated as a subcutaneously-administered monotherapy for patients with NHL or CLL in a Phase 1 study.
Subcutaneous injections of IMMU-114 produced encouraging efficacy in a Phase 1 first-in-man study in patients with advanced, relapsed NHL and CLL. The injections were well tolerated by patients, with only local skin reactions at the injection sites, which were all mild to moderate and transient. Furthermore, only one patient had evidence of immunogenicity of uncertain significance and no other cytopenias or changes in routine safety laboratory results occurred.
Our Platform Technologies
In our drive to improve targeted therapies of diseases, we have built significant expertise in antibody engineering, particularly proprietary CDR-grafting methods, antibody production and formulation, immunochemistry, molecular biology, antibody conjugation, peptide chemistry, synthetic organic chemistry, and protein engineering.
Beginning with our unique grafting technique to engineer humanized antibodies, our antibody humanization platform has produced a diverse portfolio of therapeutic agents that are in multiple stages of clinical trials for the therapy of cancer and autoimmune diseases, as detailed above. These humanized antibodies are well tolerated and also have a low incidence of immunogenicity.
With the successful humanized antibody platform as a foundation, we have built a robust ADC program using our own proprietary ADC linker technology. Finally, our protein engineering platform technology called DOCK-AND-LOCK® combines conjugation chemistry and genetic engineering to produce bioactive molecules of increasing complexity.
ADC Linker Technology
We developed a novel ADC platform using our proprietary linker, CL2A, which was designed with targeted delivery of SN-38 in mind. SN-38 is about 3 orders of magnitude (100 to 1,000 times) more potent than irinotecan, its
9
parent drug, but it cannot be administered systemically to patients because of its poor water solubility and toxicity. Our linker, CL2A, allows us to produce SN-38 conjugates that are soluble in water with excellent yields while preserving antibody binding and drug activity.
CL2A contains an antibody coupling group on one end and a chemical group on the other for binding with a drug. We have also added a short polyethylene glycol to improve the solubility of CL2A.
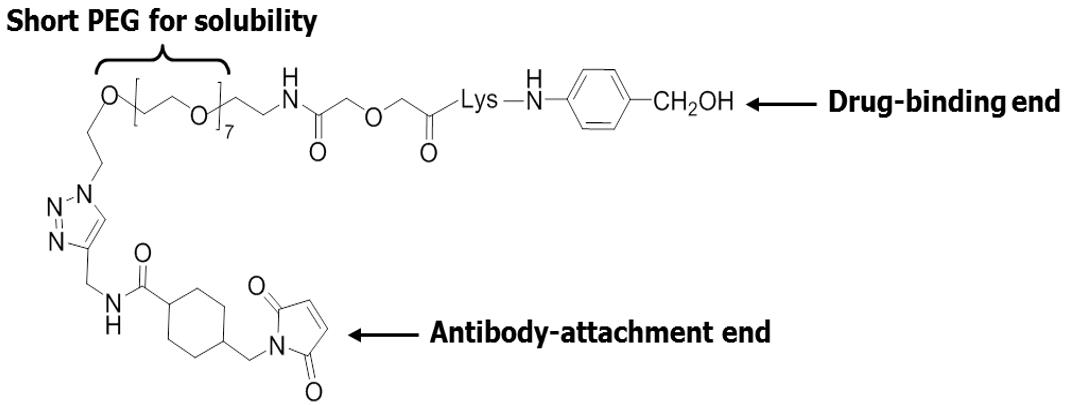
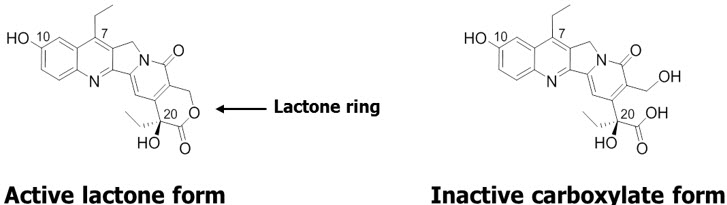
Furthermore, because SN-38 can be converted from its active lactone form to the inactive carboxylate form, CL2A was designed to attach close to the lactone ring to prevent it from opening up, thereby maintaining the activity of SN-38. Another key feature of our ADC platform is that the linkage between CL2A and SN-38 is sensitive to both acidic and alkaline conditions and will allow the detachment of SN-38 at a rate of about 50% per day in vivo.
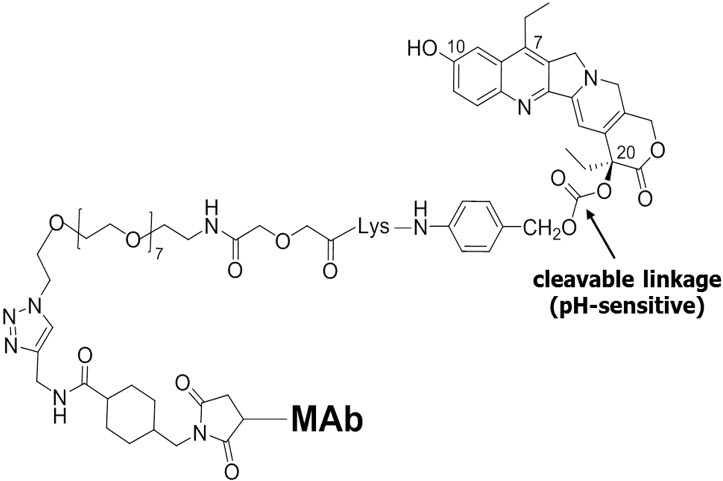
The final structure of our ADC is depicted below, with the pH-sensitive cleavable linkage highlighted. What differentiates our ADC platform from other companies is the high drug-to-antibody ratio of about seven to eight molecules of drug per antibody. That is to say, when our ADCs bind to their targets on cancer cells, they are delivering up to eight molecules of SN-38 per antibody molecule into the blood or at the vicinity of the tumor, which may explain why our ADCs can deliver more than 120-times the amount of SN-38 to the tumor when studied in an animal model, as compared to irinotecan, the parent compound. We can deliver this drug concentration because our drug is not supertoxic, thus permitting us to give higher antibody doses, in repeated therapy cycles, that we believe provide a better therapeutic index.
10
DOCK-AND-LOCK® Platform Technology
We developed a platform technology, called the DOCK-AND-LOCK® method, which has the potential for making a considerable number of bioactive molecules of increasing complexity. DNL® utilizes the natural interaction between two human proteins, cyclic AMP-dependent protein kinase A (“PKA”) and A-kinase anchoring proteins (“AKAPs”). The region that is involved in such interaction for PKA is called the dimerization and docking domain, (“DDD”), which always is produced in pairs. Its binding partner in AKAPs is the anchoring domain (“AD”). When mixed together, DDD and AD will bind with each other spontaneously to form a binary complex, a process termed docking. Once “docked,” certain amino acid residues incorporated into DDD and AD will react with each other to “lock” them into a stably-tethered structure. The outcome of the DNL® method is the exclusive generation of a stable complex, in a quantitative manner that retains the full biological activities of its individual components.
DNL® combines conjugation chemistry and genetic engineering to enable the creation of novel human therapeutics, and the potential construction of improved recombinant products over those currently on the market. Diverse drugs, chemical polymers, proteins, peptides, and nucleic acids are among suitable components that can be linked to either DDD or AD. Since the invention of DNL®, we have created multivalent, mono- or multi-specific antibodies, DNL-PEGylated cytokines; and cytokine-antibody conjugates.
We have employed DNL® to create bispecific antibodies targeting cancers as a T-cell redirecting immunotherapy. This is one of several new methods of cancer immunotherapy being studied both clinically and preclinically by many other commercial and academic groups. In contrast to hematological tumors, little progress has been made in this approach to treat the more challenging solid cancers, including pancreatic and gastric cancers, two malignancies with very high rates of mortality.
In this regard, we are developing a novel investigational T-cell redirecting bispecific antibody, (E1)-3s, created using DNL® for the potential treatment of pancreatic and gastric cancers. These and various other solid cancers express high-levels of Trop-2, a target recognized by the bispecific (E1)-3s, which also binds to the CD3 antigen on T cells. (E1)-3s effectively induced a potent and specific T-cell-mediated killing of human pancreatic and gastric cancer cell lines.
11
Furthermore, in animal models of human pancreatic or gastric cancer, treatment with (E1)-3s significantly inhibited tumor growth, which resulted in improved survival compared with the control groups. Adding IFNα enhanced the tumor-growth-inhibition activity of (E1)-3s.
As with all candidate therapeutic molecules developed by us, the safety and potential efficacy cannot be predicted until sufficient trials in humans have been conducted.
Immuno-Oncology
Harnessing the patient's own immune system to control metastatic disease has become an exciting approach in cancer therapy, particularly inhibitors of programmed cell death, such as PD-1. The approval of two such agents, as well as an antibody that inhibits T lymphocyte-associated antigen 4 (“CTLA4”), has spurred interest in their combination with other therapies in order to achieve synergy, whereby superior effects are achieved. We have begun to develop our own PD-1 antibody to evaluate its use in combination with our other anticancer agents in preclinical studies, such as the ADCs described above.
Another immunotherapy of current interest is utilizing chimeric antigen receptors (“CARs”) to direct T cells known as natural killer cells. The engineering of chimeric antigen receptors on the surface of such cells combines the potent functions of the effector cells with the tumor-targeting properties of the antibodies. To-date, clinical results using CAR-redirected immunotherapy have appeared to be more successful in liquid (hematological) tumors than in solid cancers. Using our own genetic engineering technology, our scientists have begun work on a more universal approach to direct effector cells to a variety of cancer types by a next-generation targeting model. Preclinical studies are in progress while patents to protect the intellectual property are being prosecuted.
Finally, as described above under DOCK-AND-LOCK® platform technology, we are developing an investigational T-cell redirected bispecific antibody that takes advantage of our Trop-2 antibody targeting, and has shown biological activity in our preclinical animal studies. We have now begun work to develop the constructs needed for translation into candidates for human clinical trials.
Diagnostic Imaging Products
We transitioned away from the development and commercialization of new diagnostic imaging products in order to accelerate the development of our therapeutic product candidates, although we continue to manufacture and sell, distribute and support LeukoScan® (sulesomab) in territories where regulatory approvals have previously been granted. LeukoScan® is indicated for diagnostic imaging to determine the location and extent of infection/inflammation in bone in patients with suspected osteomyelitis, including patients with diabetic foot ulcers.
Research and Development Expense
We have historically invested heavily in our research and development programs, spending approximately $51.8 million, $53.5 million and $41.7 million for these programs during the fiscal years ended June 30, 2017, 2016, and 2015, respectively. The expense decrease during the 2017 fiscal year resulted primarily from the closure of the Phase 3 pancreatic cancer “PANCRIT Trial” in the 2016 fiscal year, partially offset by higher spending in product development expense related to the manufacturing IMMU-132. The expense increases during the 2016 and 2015 fiscal years resulted primarily from higher spending for clinical trials, particularly for the PANCRIT Trial and the ADC clinical trials.
12
Patents and Proprietary Rights
Our Patents
We have accumulated a sizeable portfolio of patents and patent applications in the course of our research, which we believe constitutes a valuable business asset. Our key patents relate primarily to our therapeutic product candidates as well as our technologies and other discoveries for which no product candidate has yet been identified. As of August 4, 2017, our portfolio included approximately 316 active United States patents. In addition, as of such date, the portfolio included more than 400 foreign patents, with a number of United States and foreign patent applications pending.
The chart below highlights our material patents and product groups as of August 4, 2017, the major jurisdictions, and relevant expiration periods. Additional patents have been filed to extend the patent life on some of these products, but there can be no assurance that these will be issued as filed.
|
Program & Product Group |
|
Targeted |
|
Patent |
|
Major |
|
|
Antibody-Drug Conjugates |
|
Trop-2, CEA/CEACAM5 and HLA-DR |
|
2023-2033 |
|
U.S., Europe, Japan |
|
|
Subcutaneous Formulation |
|
All Antibodies |
|
2032 |
|
U.S., Europe, Japan |
|
|
Epratuzumab |
|
CD22 |
|
2017-2032 |
|
U.S., Europe, Japan |
|
|
Veltuzumab |
|
CD20 |
|
2023-2029 |
|
U.S., Europe, Japan |
|
|
Milatuzumab |
|
CD74 |
|
2018-2032 |
|
U.S., Europe, Japan |
|
|
IMMU-114 |
|
HLA-DR |
|
2026 |
|
U.S., Europe, Japan |
|
|
DNL® Program – (E1)-3s |
|
Trop-2 |
|
2033 |
|
U.S., Europe, Japan |
|
Our Licenses
We have obtained licenses from various parties for rights to use, develop and commercialize proprietary technologies and compounds. Currently, we have the following licenses:
Medical Research Council (“MRC”) – We entered into a license agreement with MRC in May 1994, whereby we have obtained a license for certain patent rights with respect to the genetic engineering on monoclonal antibodies. Our agreement does not require any milestone payments, nor have we made any payments to MRC to date. Our agreement with MRC, which expires at the expiration of the last of the licensed patents in 2020, provides for future royalty payments to be made based on a percentage of product sales.
Center for Molecular Medicine and Immunology (“CMMI”) – We entered into a license agreement with CMMI in December 2004, whereby we have licensed certain rights with respect to patents and patent applications owned by CMMI. Dr. Goldenberg, our Chief Scientific Officer and Chief Patent Officer and former Chairman of our Board of Directors, founded and was the President and member of the Board of Trustees of CMMI. No license or milestone payments are required under this agreement. Under the license agreement, which expires at the expiration of the last of the licensed patents in 2031, CMMI will receive future royalty payments in the low single digits based on a percentage of sales of products that are derived from the CMMI patents. Inventions made independently of us by CMMI are the property of CMMI. CMMI has ceased operations and is in the process of dissolution. Please see the section entitled “Other Collaborations” for a description of the current status of the relationship.
Our Trademarks
The mark “IMMUNOMEDICS” is registered in the United States and 19 foreign countries and a European Community Trademark has been granted. Our logo is also registered in the United States and in one foreign country. The mark “IMMUSTRIP” is registered in the United States and Canada. The mark “LEUKOSCAN” is registered in the
13
United States and eight foreign countries, and a European Community Trademark has been granted. In addition, we have applied for registration in the United States for several other trademarks for use on products now in development or testing, and for corresponding foreign and/or European Community Trademarks for certain of those marks. The marks “EPRATUCYN,” “VELTUCYN,” “CLIVATUCYN” and “MILATUCYN” have been registered in the U.S. International Trademark Registrations and Canadian applications which claim priority to the respective United States applications have been filed for “EPRATUCYN” and “VELTUCYN.” The International Registrations request registration in China, Japan and the European Union. The marks “DOCK-AND-LOCK,” “DNL,” and “PANCRIT” have been registered in the United States.
Our Trade Secrets
We also rely upon unpatented trade secrets, and there is no assurance that others will not independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets or disclose such technology, or that such rights can be meaningfully protected. We require our employees, consultants, outside scientific collaborators, sponsored researchers and other advisers to execute confidentiality agreements upon the commencement of employment or consulting relationships with us. These agreements provide that all confidential information developed or made known to the individual during the course of the individual's relationship with us is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of our employees, the agreement provides that all inventions conceived by such employees shall be our exclusive property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Third Party Rights
Our success also depends in part on our ability to gain access to third party patent and proprietary rights and to operate our business without infringing on third party patent rights. We may be required to obtain licenses to patents or other proprietary rights from third parties to develop, manufacture and commercialize our product candidates. Licenses required under third-party patents or proprietary rights may not be available on terms acceptable to us, if at all. If we do not obtain the required licenses, we could encounter delays in product development while we attempt to redesign products or methods or we could be unable to develop, manufacture or sell products requiring these licenses at all.
Corporate Collaboration
In January 2013, we entered into a collaboration agreement with Algeta ASA, subsequently acquired by Bayer, for the development of epratuzumab conjugated with Algeta’s proprietary thorium-227 alpha-pharmaceutical payload. Under the terms of this agreement, we have manufactured and supplied clinical-grade epratuzumab to Bayer, which has rights to evaluate the potential of a conjugated thorium-227 epratuzumab for the treatment of cancer. Bayer will fund all nonclinical and clinical development costs up to the end of Phase 1 clinical testing. Upon successful completion of Phase 1 clinical testing, the parties shall negotiate terms for a license agreement at Bayer’s request. We have agreed with Bayer to certain parameters to be included in the license agreement. This agreement has been extended to December 30, 2018.
Other Collaborations
In previous years, we conducted research on a number of our programs in collaboration with CMMI and its clinical unit, the Garden State Cancer Center. CMMI performed contracted pilot and pre-clinical trials in scientific areas of importance to us and also conducted basic research and pre-clinical evaluations in a number of areas of potential interest to us. Dr. David M. Goldenberg, our Chief Scientific Officer, Chief Patent Officer and former Chairman of our Board of Directors, was the President and a Member of the Board of Trustees of CMMI. CMMI has ceased operations and is in the process of dissolution.
We also collaborate with numerous other academic and research centers. Our academic collaborators have included such institutions as the Erasme University Hospital, Brussels, Belgium; University of Nijmegen, The Netherlands; Institut National de la Sante et de la Recherche Medicale, Nantes, France; University Medical Center Göttingen, Germany; Karolinska Institutet, Stockholm, Sweden; New York Presbyterian Hospital – Weill Cornell Medical College; University of Ohio Cancer Center; University of Texas M.D. Anderson Cancer Center. We believe such academic research collaboration may identify new and improved products and techniques for diagnosing and treating various cancers, autoimmune and infectious diseases.
14
Government Regulation
Regulatory Compliance
Our research and development activities, including testing in laboratory animals and in humans, our manufacture of antibodies, as well as the design, manufacturing, safety, efficacy, handling, labeling, storage, record-keeping, advertising, promotion and marketing of the product candidates that we are developing and our marketed products, are all subject to stringent regulation, primarily by the FDA in the United States under the Federal Food, Drug, and Cosmetic Act (“FFDCA”), and its implementing regulations, and the Public Health Service Act (“PHSA”), and its implementing regulations, and by comparable authorities under similar laws and regulations in other countries. If for any reason we do not comply with applicable requirements, such noncompliance can result in various adverse consequences, including one or more delays in approval of, or even the refusal to approve, product licenses or other applications, the suspension or termination of clinical investigations, the revocation of approvals previously granted, as well as fines, criminal prosecution, recall or seizure of products, injunctions against shipping products and total or partial suspension of production and/or refusal to allow us to enter into governmental supply contracts.
Product Approval
In the United States, our product candidates are regulated as biologic pharmaceuticals, or biologics. The process required by the FDA before biologic product candidates may be marketed in the United States generally involves the following:
|
· |
completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices (“GLP”) regulations; |
|
· |
submission to the FDA of an Investigational New Drug Application (“IND”), which must become effective before human clinical trials may begin and must be updated annually; |
|
· |
approval by an independent Institutional Review Board (“IRB”), the ethics committee at each clinical site before the trial is initiated. |
|
· |
performance of adequate and well-controlled clinical trials to establish the safety, purity and potency of the proposed biologic, and the safety and efficacy of the proposed drug for each indication; |
|
· |
preparation of and submission to the FDA of a BLA, for a new biologic, after completion of all pivotal clinical trials; |
|
· |
satisfactory completion of an FDA Advisory Committee review, if applicable; |
|
· |
a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
|
· |
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities to assess compliance with current Good Manufacturing Practice (“cGMP”), regulations; and |
|
· |
FDA review and approval of a BLA for a new biologic, prior to any commercial marketing or sale of the product in the United States. |
Preclinical tests assess the potential safety and efficacy of a product candidate in animal models. Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with current Good Clinical Practices (“cGCPs”), which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical trial site’s IRB before the trials may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
15
The clinical investigation of a pharmaceutical, including a biologic, is generally divided into three phases. Although the phases are usually conducted sequentially, they may overlap or be combined.
|
· |
Phase 1 studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational product in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. |
|
· |
Phase 2 includes controlled clinical trials conducted to preliminarily or further evaluate the effectiveness of the investigational product for a particular indication(s) in patients with the disease or condition under study, to determine dosage tolerance and optimal dosage, and to identify possible adverse side effects and safety risks associated with the product. |
|
· |
Phase 3 clinical trials are generally controlled clinical trials conducted in an expanded patient population generally at geographically dispersed clinical trial sites, and are intended to further evaluate dosage, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the investigational product, and to provide an adequate basis for product approval. |
The FDA may place clinical trials on hold at any point in this process if, among other reasons, it concludes that clinical subjects are being exposed to an unacceptable health risk. Trials may also be terminated by IRBs, which must review and approve all research involving human subjects. Side effects or adverse events that are reported during clinical trials can delay, impede or prevent marketing authorization.
The results of the preclinical and clinical testing, along with information regarding the manufacturing of the product and proposed product labeling, are evaluated and, if determined appropriate, submitted to the FDA through a BLA. The application includes all relevant data available from pertinent preclinical and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Once the BLA submission has been accepted for filing, the FDA’s standard goal is to review applications within ten months of the filing date or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months from the filing date. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA offers certain programs, such as Fast Track designation, designed to expedite the development and review of applications for products intended for the treatment of a serious or life-threatening disease or condition. If Fast Track designation is obtained, the FDA may initiate review of sections of a BLA before the application is complete, and the product may be eligible for accelerated approval. However, receipt of Fast Track designation for a product candidate does not ensure that a product will be developed or approved on an expedited basis, and such designation may be rescinded if the product candidate is found to no longer meet the qualifying criteria.
The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, pure and potent, which includes determining whether it is effective for its intended use, and whether the product is being manufactured in accordance with cGMP, to assure and preserve the product’s identity, strength, quality, potency and purity. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it typically follows such recommendations.
After the FDA evaluates the BLA and conducts inspections of manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the biologic with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria for approval. The FDA could approve the BLA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on,
16
among other things, changes to proposed labeling, development of adequate controls and specifications, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
The Biologics Price Competition and Innovation Act of 2009 (“BPCIA”) created an abbreviated pathway for the approval of biosimilar and interchangeable biologic products. The abbreviated pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. In March 2015, the FDA approved Novartis’s Zarxio as a biosimilar product to Amgen’s Neupogen. The approval, the first biosimilar product approved for distribution in the United States, could usher in lower prices for biologic products from increased competition.
Expedited Review and Approval
The FDA has four program designations — Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review — to facilitate and expedite development and review of new drugs to address unmet medical needs in the treatment of serious or life-threatening conditions. The Fast Track designation provides pharmaceutical manufacturers with opportunities for frequent interactions with FDA reviewers during the product’s development and the ability for the manufacturer to do a rolling submission of the BLA. A rolling submission allows completed portions of the application to be submitted and reviewed by the FDA on an ongoing basis. The Breakthrough Therapy designation provides manufacturers with all of the features of the Fast Track designation as well as intensive guidance on implementing an efficient development program for the product and a commitment by the FDA to involve senior managers and experienced review staff in the review. The Accelerated Approval designation allows the FDA to approve a product based on an effect on a surrogate or intermediate endpoint that is reasonably likely to predict a product’s clinical benefit and generally requires the manufacturer to conduct required post-approval confirmatory trials to verify the clinical benefit. The Priority Review designation means that the FDA’s goal is to take action on the BLA within six months, compared to ten months under standard review. In February 2016, sacituzumab govitecan was granted Breakthrough Therapy designation from the FDA for the treatment of patients with TNBC who have failed at least two prior therapies for metastatic disease.
Post-Approval Requirements
Any products manufactured or distributed by us or on our behalf pursuant to FDA approvals are subject to continuing regulation by the FDA and certain state agencies, including requirements for record-keeping, reporting of adverse experiences with the biologic, submitting biological product deviation reports to notify the FDA of unanticipated changes in distributed products, establishment registration, compliance with cGMP standards (including investigation and correction of any deviations from cGMP), and certain state chain of distribution pedigree requirements. Additionally, any significant change in the approved product or in how it is manufactured, including changes in formulation or the site of manufacture, generally require prior FDA approval. The packaging and labeling of all products developed by us are also subject to FDA approval and ongoing regulation. Noncompliance with any regulatory requirements can result in, among other things, issuance of warning letters, civil and criminal penalties, seizures, and injunctive action. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Orphan Drug Act
To date, we have successfully obtained Orphan Drug designation by the FDA under the Orphan Drug Act of 1983 for epratuzumab for NHL, yttrium-90-labeled clivatuzumab tetraxetan for pancreatic cancer, sacituzumab govitecan for SCLC and pancreatic cancer, labetuzumab for ovarian, pancreatic and SCLCs, milatuzumab for multiple myeloma and CLL, and veltuzumab for ITP and pemphigus. Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs intended to treat a rare disease or condition, which is generally defined as a disease or condition that affects fewer than 200,000 individuals in the United States. Orphan drug designation must be requested before submitting a BLA. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and user-fee waivers. Orphan drug
17
designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first BLA applicant to receive FDA approval for a particular active ingredient to treat a particular disease with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the United States for that product, for that indication. During the seven-year exclusivity period, the FDA may not approve any other applications to market the same drug for the same orphan indication, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or where the manufacturer of the approved product cannot assure sufficient quantities. As a result, there can be no assurance that our competitors will not receive approval of drugs or biologics that have a different active ingredient for treatment of the diseases for which our products and product candidates are targeted.
Foreign Regulation
In addition to regulations in the United States, we are subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates being developed, and products being marketed outside of the United States. We must obtain approval by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of our products in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required by the FDA for BLA licensure. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the United States, we are subject to post-approval regulatory requirements, such as those regarding product manufacturing, marketing, or distribution.
Other Regulatory Considerations
We are also subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, The Clean Air Act, New Jersey Department of Environmental Protection and other current and potential future federal, state, or local regulations. Our research and development activities involve the controlled use of hazardous materials, chemicals, biological materials and various radioactive compounds. We believe that our procedures comply with the standards prescribed by state and federal regulations; however, the risk of injury or accidental contamination cannot be completely eliminated.
We may also be subject to healthcare regulation and enforcement by the federal government and the states and foreign governments where we may market our products and product candidates, if approved. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy, and security and physician sunshine laws and regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs, such as the Medicare and Medicaid programs. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies, based on sham consulting and other financial arrangements with physicians. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. In addition, the government may assert that a claim, including items or services resulting from a violation of the federal Anti-Kickback Statute, constitutes a false or fraudulent claim for purposes of the federal False Claims Act. The majority of states also have anti-kickback laws, which establish similar prohibitions and, in some cases, may apply to items or services reimbursed by any third-party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the United States government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the United States, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the
18
government will continue to devote substantial resources to investigating compliance of healthcare providers and manufacturers with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) also created new federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act (“HITECH”), and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
We are subject to the United States Foreign Corrupt Practices Act, which prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. Under this act, it is illegal to pay, offer to pay, or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. Our present and future business has been and will continue to be subject to various other laws and regulations.
Pricing Controls
The levels of revenues and profitability of biopharmaceutical companies may be affected by the continuing efforts of government and third party payers to contain or reduce the costs of health care through various means. For example, in certain foreign markets, pricing reimbursement or profitability of therapeutic and other pharmaceutical products is subject to governmental control. In the U. S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether any such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Third Party Coverage and Reimbursement
In addition, in the United States and elsewhere, sales of therapeutic and other pharmaceutical products are dependent in part on the availability of reimbursement to the consumer from third party payers such as government and private insurance plans. Third party payers are increasingly challenging the prices charged for medical products and services. We cannot assure you that any of our products will be considered cost effective and that reimbursement to the consumer will be available or will be sufficient to allow us to sell our products on a competitive and profitable basis.
Competition
Competition in the biopharmaceutical industry is intense and based significantly on scientific and technological factors such as the availability of patent and other protection for technology and products, the ability to commercialize technological developments and the ability to obtain governmental approval for testing, manufacturing and marketing.
19
We compete with specialized biopharmaceutical firms in the United States, Europe and elsewhere, as well as a growing number of large pharmaceutical companies. A number of companies, including Amgen, AstraZeneca, Bayer Healthcare Pharmaceuticals, Biogen Idec, Bristol-Myers Squibb, Celgene, Eli Lilly, Genmab, GlaxoSmithKline, Immunogen, Johnson & Johnson, Merck, Merck Serono, Novartis, Pfizer, Roche, and Seattle Genetics Inc. (“Seattle Genetics”), are engaged in the development of therapeutic oncology and autoimmune products. Many of these companies have significantly greater financial, technical and marketing resources than we do. Many major pharmaceutical companies have developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies. These companies, as well as academic institutions, governmental agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific, technical and professional personnel and consultants. Our ability to compete successfully with other companies in the biopharmaceutical field will also depend to a considerable degree on the continuing availability of capital to us.
Marketing, Sales and Distribution
As noted above, we intend to bring IMMU-132 to the U.S. market on our own for patients with mTNBC. Should our efforts become successful, we will need to build a commercial operation with a sales force of approximately 50 to 100 agents anticipated. At present, we have limited marketing and sales capabilities as we focus on developing our therapeutic product candidates. However, we continue to manufacture and market LeukoScan® in certain European markets with our internal sales force and to provide technical support directly to customers. In the past, we had agreements with third parties to market and provide distribution and customer support services for LeukoScan®. However, these agreements will be terminated January 1, 2018 and we will assume responsibility to distribute and support the LeukoScan® product on our own.
Our European operations are headquartered in Rodermark, Germany. Our distribution agreement with Logosys Logistik GmbH to package and distribute LeukoScan® in the EU will be terminated effective December 31, 2017.
Manufacturing
We operate a recombinant monoclonal antibody manufacturing facility at our Morris Plains, New Jersey location. This facility is used for the production of all of our therapeutic product candidates for clinical trials, and potentially for commercial production as well.
For the commercial-scale manufacturing of sacituzumab govitecan (IMMU-132) we have contracted with two outside contract manufacturing organizations to provide drug for the planned Phase 3 clinical trial and to support the commercial launch of IMMU-132 in the United States. Accordingly, we have agreements with Johnson Matthey Pharma Services of Devens, Massachusetts for the manufacture of the linker-drug payload,); and, BSP Pharmaceuticals of Latina Scalo, Italy for the conjugation of the antibody with the linker-drug and fill/finish of the IMMU-132 drug product. Presently, we have the capacity at our Morris Plains facility to manufacture sufficient quantities of the Trop-2 antibody to support the commercial launch of IMMU-132 in the U.S.. Together with our CMO partners, we have already manufactured sufficient quantities of the drug product to supply our full needs to continue the Phase 2 basket trial and also to complete the confirmatory Phase 3 clinical trial of IMMU-132 as a third-line therapy for patients with mTNBC. Additionally, we are currently in discussions with other CMOs to support our longer term needs for commercial-scale antibody production.
We also manufacture LeukoScan® for commercial sale at our facility in Morris Plains, New Jersey. The Committee on Proprietary Medicinal Products of the European Commission approved the manufacturing facility and product manufacturing processes for LeukoScan® in May 1998. We perform antibody processing and purification of all our therapeutic product candidates at this facility. We scaled-up our antibody purification and fragmentation manufacturing processes for our diagnostic imaging agent to permit us to produce commercial levels of product. We have an agreement with BAG GmbH, Lich, Germany for the final formulation, fill and finish of LeukoScan®.
Manufacturing Regulatory Considerations
In addition to regulating and auditing human clinical trials, the FDA regulates and inspects equipment, facilities and processes used in the manufacturing of such products prior to providing approval to market a product. If, after receiving clearance from the FDA, a material change is made in manufacturing equipment, location, or process, additional regulatory review may be required. We must also adhere to cGMP and product-specific regulations enforced
20
by the FDA through its facilities inspection program. The FDA also conducts regular, periodic visits to re-inspect equipment, facilities, and processes following the initial approval. If, as a result of these inspections, the FDA determines that our equipment, facilities or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations.
LeukoScan® is derived from the fluids produced in mice. Regulatory authorities, particularly in Europe, have expressed concerns about the use of these fluids for the production of monoclonal antibodies. These regulatory authorities may determine that our quality control procedures for these products are inadequate. In the event we have to discontinue the use of mouse fluids, we may not have the resources at the time to acquire the necessary manufacturing equipment and expertise that we will need to make the changes in our development programs.
Employees
As of August 1, 2017, we employed 138 persons on a full-time basis, 12 of whom were in research and development departments, 22 of whom were engaged in clinical research and regulatory affairs, 79 of whom were engaged in operations and manufacturing and quality control, and 25 of whom were engaged in finance, administration, sales and marketing. Of these employees, 49 hold M.D., Ph.D. or other advanced degrees. We believe that while we have been successful to date in attracting skilled and experienced scientific personnel, competition for such personnel continues to be intense and there can be no assurance that we will continue to be able to attract and retain the professionals we will need to grow our business. Our employees are not covered by a collective bargaining agreement and we believe that our relationship with our employees is excellent.
Corporate Information
We were incorporated in Delaware in 1982. Our principal offices are located at 300 The American Road, Morris Plains, New Jersey 07950. Our telephone number is (973) 605-8200. We have two foreign subsidiaries, Immunomedics B.V. in The Netherlands and Immunomedics GmbH in Rodermark, Germany, to assist us in managing sales and marketing efforts and coordinating clinical trials in Europe. In addition, we have a majority-owned subsidiary, IBC Pharmaceuticals, Inc. (“IBC”). Immunomedics has incurred expenses on behalf of the IBC operations, including interest, over the past thirteen years. As of June 30, 2017, IBC has a liability to Immunomedics Inc. of approximately $16.8 million, which is eliminated in consolidation. Our web address is www.immunomedics.com. We have not incorporated by reference into this Annual Report on Form 10-K the information on our website and you should not consider it to be a part of this document.
Our reports that have been filed with the Securities and Exchange Commission (“SEC”), are available on our website free of charge, including our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, Forms 3, 4 and 5 filed on behalf of directors and executive officers and any amendments to such reports filed pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Copies of this Annual Report on Form 10-K may also be obtained without charge electronically or by paper by contacting Investor Relations, Immunomedics, Inc., 300 The American Road, Morris Plains, New Jersey 07950 or by calling (973) 605-8200.
In addition, we make available on our website (i) the charters for the committees of the Board of Directors, including the Audit Committee, Compensation Committee and Governance and Nominating Committee, and (ii) the Company’s Code of Business Conduct (the “Code of Conduct”) governing its directors, officers and employees. Within the time period required by the SEC, we will post on our website any modifications to the Code of Conduct, as required by the Sarbanes-Oxley Act of 2002, (“Sarbanes-Oxley Act”).
The public may also read and copy the materials we file with the SEC at its Public Reference Room at 100 F Street, N.E., Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains a web site at http://www.sec.gov that contains reports, proxy and information statements and other information regarding companies that file electronically with the SEC.
21
Item 1A.RISK FACTORS
Factors That May Affect Our Business and Results of Operations
Our business is subject to certain risks and uncertainties, each of which could materially adversely affect our business, financial condition, cash flows and results of operations.
Risks Relating to Our Business, Operations and Product Development
We have a long history of operating losses and it is likely that our operating expenses will continue to exceed our revenues for the foreseeable future.
We have incurred significant operating losses since our formation in 1982. As of June 30, 2017, we had an accumulated deficit of approximately $521.7 million. We continue to spend our cash resources to fund our research and development programs and, subject to adequate funding, we expect these expenses to increase for the foreseeable future. Our only significant sources of revenue in recent years have been derived from our collaboration agreement with Bayer and sales of our LeukoScan® product in certain European countries. There can be no assurance that we will be profitable in future quarters or other periods. Additionally, the only product sales we have earned to date have come from the limited sales of our diagnostic imaging product for which our patent protection has expired (which may leave us vulnerable to increased competition, for example, from biosimilar manufacturers). In addition, we have made the strategic decision to de-emphasize sales of our diagnostic product and focus on our therapeutic pipeline. We have never had product sales of any therapeutic product. Although we may have net income from time to time based on the timing and amount of proceeds received under collaborative or licensing agreements, we expect to experience significant operating losses as we invest further in our research and development activities while simultaneously attempting to develop and commercialize our other therapeutic product candidates. If we are unable to develop commercially viable therapeutic products or to license them to third parties, it is likely that we will never achieve significant revenues or become profitable, either of which would jeopardize our ability to continue as a going concern.
We have significant future capital needs and may be unable to raise capital when needed, which could force us to delay or reduce our clinical development efforts.
As of June 30, 2017 we have $154.9 million in cash, cash equivalents and marketable securities; which we believe is sufficient to support operations through September 2018, and which allows us to accomplish the goal of filing a BLA for Accelerated Approval of IMMU-132 in mTNBC from the FDA during the period between December 2017 and March 2018, subject to FDA input on the acceptance of the Company’s chemistry, manufacturing and controls filing plan.
The Company will require additional funding after September 2018 to secure regulatory approval from the FDA, complete commercial preparations to market IMMU-132 to mTNBC patients in the United States, complete its clinical trials currently planned or underway, continue research and new development programs, and continue operations. Potential sources of funding include the exercise of outstanding warrants, potential various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, and equity and potential debt financing.
Until the Company can generate significant cash through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, or commercial operations, it expects to continue to fund its operations with its current financial resources. These financial resources are adequate to sustain the Company’s operations at a level of activity sufficient to support the filing of the BLA with the FDA for accelerated approval of IMMU-132 for patients with mTNBC; to continue manufacturing IMMU-132 at large scale; to prepare for commercial operations in the U.S. marketplace; to initiate a Phase 3 clinical trial of IMMU-132 for mTNBC patients to support the filing of the BLA; to initiate preparations to market IMMU-132 to mTNBC patients in the U.S. and, subject to meeting all standards, completing review and final determination of the FDA, to secure accelerated regulatory approval to market IMMU-132 for the use of patients with mTNBC in the U.S.. After September 2018, if the Company cannot obtain sufficient funding through the exercise of outstanding warrants,
22
various potential strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, it could be required to finance future cash needs through the sale of additional equity and/or debt securities in capital markets. However, there can be no assurance that the Company will be able to raise the additional capital needed to complete its pipeline of research and development programs on commercially acceptable terms, if at all. The capital markets have experienced volatility in recent years, which has resulted in uncertainty with respect to availability of capital and hence the timing to meet an entity’s liquidity needs. The Company’s existing debt may also negatively impact the Company’s ability to raise additional capital. If the Company is unable to raise capital on acceptable terms, its ability to continue its business would be materially and adversely affected.
Our most advanced therapeutic product candidates are still only in the clinical development stage, and will require us to raise capital in the future in order to fund further expensive and time-consuming studies before they can even be submitted for final regulatory approval. A failure of a clinical trial could severely harm our business and results of operations.
Clinical trials involve the administration of a product candidate to patients who are already extremely ill, making patient enrollment often difficult and expensive. Moreover, even in ideal circumstances where the patients can be enrolled and then followed for the several months or more required to complete the study, the trials can be suspended, terminated, delayed or otherwise fail for any number of reasons, including:
|
· |
later-stage clinical trials may raise safety or efficacy concerns not readily apparent in earlier trials or fail to meet the primary endpoint; |
|
· |
unforeseen difficulties in manufacturing the product candidate in compliance with all regulatory requirements and in the quantities needed to complete the trial which may become cost-prohibitive; |
|
· |
we or our collaboration partner may experience delays in obtaining, or be unable to obtain, agreement for the conduct of our clinical trials from the FDA, IRBs, or other reviewing entities at clinical sites selected for participation in our clinical trials; |
|
· |
while underway, the continuation of clinical trials may be delayed, suspended or terminated due to modifications to the clinical trial’s protocols based on interim results obtained or changes required or conditions imposed by the FDA, an IRB, a data and safety monitoring board (“DSMB”), or any other regulatory authority; |
|
· |
our third-party contractors may fail to meet their contractual obligations to us in a timely manner; |
|
· |
the FDA or other regulatory authorities may impose a clinical hold, for example based an inspection of the clinical trial operations or trial sites; |
|
· |
we or our collaboration partner may suspend or cease trials in our or their sole discretion; |
|
· |
during the long trial process alternative therapies may become available which make further development of the product candidate impracticable; and |
|
· |
if we are unable to obtain the additional capital we need to fund all of the clinical trials we foresee, we may be forced to cancel or otherwise curtail such trials and other studies. |
Any substantial delay in successfully completing clinical trials for our product candidates, sacituzumab govitecan and labetuzumab govitecan, could severely harm our business and results of operations.
Moreover, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and receive compensation in connection with such services. Under certain circumstances, the Company may be required to report some of these relationships to the FDA. The FDA may conclude that a financial relationship between the company and a principal investigator has created a conflict of interest or otherwise affected interpretation of the study. The FDA may therefore question the integrity of the data generated at the applicable clinical trial site and the utility of the clinical trial itself may be jeopardized. This could result in a delay in approval, or rejection, of our marketing applications by the FDA and may ultimately lead to the denial of regulatory approval of one or more of our product candidates.
23
Our clinical trials may not adequately show that our drugs are safe or effective, and a failure to achieve the planned endpoints could result in termination of product development.
Progression of our drug products through the clinical development process is dependent upon our trials indicating our drugs have adequate safety and efficacy in the patients being treated by achieving pre-determined safety and efficacy endpoints according to the trial protocols. Failure to achieve either of these endpoints could result in delays in our trials; require the performance of additional unplanned trials or termination of any further development of the product for the intended indication.
These factors could result in delays in the development of our product candidates and could result in significant unexpected costs or the termination of programs.
Should the clinical development process be successfully completed, our ability to derive revenues from the sale of therapeutics will depend upon our first obtaining FDA as well as foreign regulatory approvals, all of which are subject to a number of unique risks and uncertainties.
Even if we are able to demonstrate the safety and efficacy of our product candidates in clinical trials, if we fail to gain timely approval to commercialize our product candidates from the FDA and other foreign regulatory authorities, we will be unable to generate the revenues we will need to build our business. These approvals may not be granted on a timely basis, if at all, and even if and when they are granted, they may not cover all the indications for which we seek approval. For example, while we may develop a product candidate with the intention of addressing a large, unmet medical need, the FDA may only approve the use of the drug for indications affecting a relatively small number of patients, thus greatly reducing the market size and our potential revenues. The approvals may also contain significant limitations in the form of warnings, precautions or contraindications with respect to conditions of use, which could further narrow the size of the market. In certain countries, even if the health regulatory authorities approve a drug, it cannot be marketed until pricing for the drug is also approved. Finally, even after approval can be obtained, we may be required to recall or withdraw a product as a result of newly discovered safety or efficacy concerns, either of which would have a materially adverse effect on our business and results of operations.
In order to fund future operations, we will need to raise significant amounts of additional capital. Because it can be difficult for a small-cap company like ours to raise equity capital on acceptable terms, we cannot assure you that we will be able to obtain the necessary capital when we need it, or on acceptable terms, if at all.
Even if our technologies and product candidates are superior, if we lack the capital needed to bring our future products to market, we will never be successful. We have obtained the capital necessary to fund our research and development programs to date primarily from the following sources:
|
· |
upfront payments, milestone payments, and payments for limited amounts of our antibodies received from licensing partners; |
|
· |
proceeds from the public and private sale of our equity or debt securities; and |
|
· |
limited product sales of LeukoScan®, licenses, grants and interest income from our investments |
Over the long term, we expect to commercialize IMMU-132 in mTNBC in the U.S. and globally, to expand IMMU-132 to treat patients with other solid tumors, including urinary bladder cancer, small cell lung cancer, non-small cell lung cancer, and others serious cancers, to expand research and development activities to continue to expand and we do not believe we will have adequate cash to continue commercial expansion and development of IMMU-132, or to complete development of product candidates in line with our pipeline included in our long term corporate strategy. Our capital requirements are dependent on numerous factors, including:
|
· |
the rate of progress of commercialization of IMMU-132 in mTNBC and develop it for other cancers |
|
· |
the rate at which we progress our research programs and the number of product candidates we have in pre-clinical and clinical development at any one time; |
|
· |
the cost of conducting clinical trials involving patients in the United States, Europe and possibly elsewhere; |
24
|
· |
our need to establish the manufacturing capabilities necessary to produce the quantities of our product candidates we project we will need; |
|
· |
the time and costs involved in obtaining FDA and foreign regulatory approvals; |
|
· |
the cost of first obtaining, and then defending, our patent claims and other intellectual property rights; |
|
· |
the ability and willingness of the holders of our 4.75% Convertible Senior Notes due 2020 (“Convertible Senior Notes”) to convert their Convertible Senior Notes to Immunomedics common stock; and |
|
· |
our ability to enter into licensing and other collaborative agreements to help offset some of these costs. |
There may be additional cash requirements for many reasons, including, but not limited to, changes in our commercial expansion plans, our research and development plans, the need for unexpected capital expenditures or costs associated with any acquisitions of other businesses, assets or technologies that we may choose to undertake and marketing and commercialization of our product candidates. If we deplete our existing capital resources, we will be required to either obtain additional capital quickly, or significantly reduce our operating expenses and capital expenditures, either of which could have a material adverse effect on us.
Until we can generate significant cash through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, we expect to continue to fund our operations with our current financial resources. These financial resources will not be adequate to sustain our operations beyond the third quarter 2018. Consequently, if we cannot obtain sufficient funding through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, we could be required to finance future cash needs through the sale of additional equity and/or debt securities in capital markets. However, there can be no assurance that we will be able to raise the additional capital needed to complete our pipeline of research and development programs on commercially acceptable terms, if at all. The capital markets have experienced volatility in recent years, which has resulted in uncertainty with respect to availability of capital and hence the timing to meet an entity’s liquidity needs. The Company’s existing debt will also negatively impact the Company’s ability to raise additional capital. If the Company is unable to raise capital on acceptable terms, its ability to continue its business would be materially and adversely affected. Having insufficient funds may require us to delay, scale-back, or eliminate some or all of our programs, or renegotiate less favorable terms than we would otherwise choose. Failure to obtain adequate financing also may adversely affect our ability to operate as a going concern.
Additionally, if we raise funds by issuing equity securities, dilution to existing stockholders would result; and if we raise funds by incurring additional debt financing, the terms of the debt may involve future cash payment obligations and/or conversion to equity as well as restrictions that may limit our ability to operate our business.
If we, or our collaboration partner, cannot successfully and efficiently manufacture the compounds that make up our products and product candidates, our ability, and the ability of our collaboration partner, to sell products and conduct clinical trials will be impaired.
Our ability to conduct our pre-clinical and clinical research and development programs depends, in large part, upon our ability to manufacture our proprietary compounds in accordance with the FDA and other regulatory requirements. We have limited historical experience in manufacturing these compounds in significant quantities, and we may not be able to do so in the quantities required to commercialize these products. Any interruption in manufacturing at this site, whether by natural acts or otherwise, could significantly and adversely affect our operations, and delay our research and development programs.
We and our collaboration partner also depend on third parties to provide certain raw materials, manufacturing and processing services. All manufacturers of pharmaceutical products must comply with current Good Manufacturing Practice regulations or cGMPs, required by the FDA and other regulatory agencies. Such regulations address, among other matters, controls in manufacturing processes, quality control and quality assurance requirements and the maintenance of proper records and documentation. The FDA and other regulatory agencies routinely inspect manufacturing facilities. The FDA generally will issue a notice on Form 483 if it finds issues with respect to its inspections. If our manufacturing facility or those facilities of our partner and our respective contract manufacturers or processors do not comply with applicable cGMPs and other regulatory requirements, we may be subject to product
25
liability claims, we may be unable to meet clinical demand for our products, and we could suffer delays in the progress of clinical trials for products under development.
Although historically we have been a research and development company, we plan to commercialize our lead product candidate internally rather than license such asset. There can be no assurance that we will be successful in developing and expanding commercial operations or balancing our research and development activities with our commercialization activities.
We have historically been engaged primarily in research and development activities, but plan to commercialize our lead product candidate, IMMU-132, ourselves. There can be no assurance that we will be able to successfully manage the balance of our research and development operations with our planned commercialization activities. Potential investors should be aware of the problems, delays, expenses and difficulties frequently encountered by companies balancing development of product candidates, which can include problems such as unanticipated issues relating to clinical trials and receipt of approvals from the FDA and foreign regulatory bodies, with commercialization efforts, which can include problems relating to managing manufacturing and supply, reimbursement, marketing problems and additional costs. Our product candidates will require significant additional research and clinical trials, and we will need to overcome significant regulatory burdens prior to commercialization in the U.S. and other countries. In addition, we may be required to spend significant funds on building out our commercial operations. There can be no assurance that after the expenditure of substantial funds and efforts, we will successfully develop and commercialize any of our product candidates, generate any significant revenues or ever achieve and maintain a substantial level of sales of our products.
We may not successfully establish and maintain collaborative and licensing arrangements, which could adversely affect our ability to develop and commercialize certain of our product candidates. Our future collaboration partners may not adequately perform their responsibilities under our agreement, which could adversely affect our development and commercialization program.
A key element of our business strategy has been to develop, market and commercialize our product candidates through collaborations with more established pharmaceutical companies. To the extent we continue to rely on this business strategy, we may not be able to maintain or expand these licenses and collaborations or establish additional licensing and collaboration arrangements necessary to develop and commercialize any of our product candidates. Even if we are able to maintain or establish licensing or collaboration arrangements, these arrangements may not be on favorable terms and may contain provisions that will restrict our ability to develop, test and market our product candidates. Any failure to maintain or establish licensing or collaboration arrangements on favorable terms could adversely affect our business prospects, financial condition or ability to develop and commercialize our product candidates.
We expect to rely at least in part on third party collaborators to perform a number of activities relating to the development and commercialization of certain of our product candidates, including the manufacturing of product materials, the design and conduct of clinical trials for certain of our product candidates, and potentially the obtaining of regulatory approvals and marketing and distribution of any successfully developed products. Our collaborative partners may also have or acquire rights to control aspects of our product development and clinical programs. As a result, we may not be able to conduct these programs in the manner or on the time schedule we currently contemplate. In addition, if any of these collaborative partners withdraw support for our programs or product candidates or otherwise impair their development, our business could be negatively affected. Our expenses may also increase as a result of our plan to undertake these activities internally to commercialize IMMU-132.
In addition, our success depends on the performance of our collaborators of their responsibilities under these arrangements. Some potential collaborators may not perform their obligations in a timely fashion or in a manner satisfactory to us. Because such agreements may be exclusive, we may not be able to enter into a collaboration agreement with any other company covering the same product field during the applicable collaborative period. In addition, our collaborators’ competitors may not wish to do business with us at all due to our relationship with our collaborators. If we are unable to enter into additional product discovery and development collaborations, our ability to sustain or expand our business will be significantly diminished.
26
Our future success will depend upon our ability to first obtain and then adequately protect our patent and other intellectual property rights, as well as avoiding the infringement of the rights of others.
Our future success will be highly dependent upon our ability to first obtain and then defend the patent and other intellectual property rights necessary for the commercialization of our product candidates. We have filed numerous patent applications on the technologies and processes that we use in the United States and certain foreign countries. Although we have obtained a number of issued U.S. patents to date, the patent applications owned or licensed by us may not result in additional patents being issued. Moreover, these patents may not afford us the protection we need against competitors with similar technologies or products. A number of jurisdictions where we have sought, or may in future choose to seek, intellectual property protection, have intellectual property laws and patent offices which are still developing. Accordingly, we may have difficulty obtaining intellectual property protection in these markets, and any intellectual property protections which we do obtain may be less protective than in the United States, which could have an adverse effect on our operations and financial prospects.
The successful development of therapeutic products frequently requires the application of multiple technologies that may be subject to the patent or other intellectual property rights of third parties. Although we believe it is likely we will need to license technologies and processes from third parties in the ordinary course of our business, we are not currently aware of any material conflict involving our technologies and processes with any valid patents or other intellectual property rights owned or licensed by others. In the event that a third party was to claim such a conflict existed, they could sue us for damages as well as seek to prevent us from commercializing our product candidates. It is possible that a third party could successfully claim that our products infringe on their intellectual property rights. Uncertainties resulting from the litigation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Any patent litigation or other proceeding, even if resolved in our favor, would require significant financial resources and management time.
Some of our competitors may be able to sustain these costs more effectively than we can because of their substantially greater financial and managerial resources. If a patent litigation or other proceeding is resolved unfavorably to us, we may be enjoined from manufacturing or selling our products without a license from the other party, in addition to being held liable for significant damages. We may not be able to obtain any such license on commercially acceptable terms, if at all.
In addition to our reliance on patents, we attempt to protect our proprietary technologies and processes by relying on trade secret laws, nondisclosure and confidentiality agreements and licensing arrangements with our employees and other persons who have access to our proprietary information. These agreements and arrangements may not provide meaningful protection for our proprietary technologies and processes in the event of unauthorized use or disclosure of such information. In addition, our competitors may independently develop substantially equivalent technologies and processes or otherwise gain access to our trade secrets or technology, either of which could materially and adversely affect our competitive position.
Expiry of our intellectual property rights could lead to increased competition
Even where we are able to obtain and then defend patent and other intellectual property rights necessary for research, development and commercialization of our product candidates, such intellectual property rights will be for a limited term. Where patents which we own or license expire, the technology the subject of the patent may be utilized by third parties in research and development or competing products (for example, biosimilars of a patented product may be manufactured by third parties once the patent expires). While we endeavor to maintain robust intellectual property protection, as our existing issued patents expire it may materially and adversely affect our competitive position.
We face substantial competition in the biotechnology industry and may not be able to compete successfully against one or more of our competitors.
The biotechnology industry is highly competitive, particularly in the area of diagnostic and therapeutic oncology and autoimmune disease products. In recent years, there have been extensive technological innovations achieved in short periods of time, and it is possible that future technological changes and discoveries by others could
27
result in our products and product candidates quickly becoming uncompetitive or obsolete. A number of companies, including Amgen, AstraZeneca, Bayer Healthcare Pharmaceuticals, Biogen Idec, Bristol-Myers Squibb, Celgene, Eli Lilly, Genmab, GlaxoSmithKline, Immunogen, Johnson & Johnson, Merck, Merck Serono, Novartis, Pfizer, Roche, and Seattle Genetics, are engaged in the development of therapeutic oncology products. Many of these companies have significantly greater financial, technical and marketing resources than we do. In addition, many of these companies have more established positions in the pharmaceutical industry and are therefore better equipped to develop, commercialize and market oncology and autoimmune disease products. Even some smaller competitors may obtain a significant competitive advantage over us if they are able to discover or otherwise acquire patentable inventions, form collaborative arrangements or merge with larger pharmaceutical companies. Further, even if we are able to successfully develop and commercialize products, other manufacturers operating in emerging markets may also have a competitive advantage over us with respect to competing products due to their ability to manufacture with a lower cost base.
We expect to face increasing competition from universities and other non-profit research organizations. These institutions carry out a significant amount of research and development in the field of antibody-based technologies and they are increasingly aware of the commercial value of their findings. As a result, they are demanding greater patent and other proprietary rights, as well as licensing and future royalty revenues. It is possible that such competition could come from universities with which we have, or have previously had, collaborative research and development relationships, notwithstanding our efforts to protect our intellectual property in the course of such relationships.
We may be liable for contamination or other harm caused by hazardous materials that we use in the operations of our business.
In addition to laws and regulations enforced by the FDA, we are also subject to regulation under various other foreign, federal, state and local laws and regulations. Our manufacturing and research and development programs involve the controlled use of viruses, hazardous materials, chemicals and various radioactive compounds. The risk of accidental contamination or injury from these materials can never be completely eliminated, and if an accident occurs we could be held liable for any damages that result, which could exceed our available resources.
The nature of our business exposes us to significant liability claims, and our insurance coverage may not be adequate to cover any future claims.
The use of our compounds in clinical trials and any future sale exposes us to liability claims that could be substantial. These claims might be made directly by healthcare providers, medical personnel, patients, consumers, pharmaceutical companies, and others selling or distributing our compounds. While we currently have product liability insurance that we consider adequate for our current needs, we may not be able to continue to obtain comparable insurance in the future at an acceptable cost, if at all. If for any reason we cannot maintain our existing or comparable liability insurance, our ability to clinically test and market products could be significantly impaired. Moreover, the amount and scope of our insurance coverage, as well as the indemnification arrangements with third parties upon which we rely, may be inadequate to protect us in the event of a successful product liability claim. Any successful claim in excess of our insurance coverage could materially and adversely affect our financial condition and operating results.
Certain potential for conflicts of interest, both real and perceived, exist which could result in expensive and time-consuming litigation.
Certain members of our senior management and Board of Directors have relationships and agreements, both with us as well as among themselves and their respective affiliates, which create the potential for both real, as well as perceived, conflicts of interest. These include Dr. David M. Goldenberg, a director and our Chief Scientific Officer and Chief Patent Officer, who was the former Chairman of our Board of Directors, Ms. Cynthia L. Sullivan, a director and our former President and Chief Executive Officer (who is also the wife of Dr. Goldenberg), and certain companies with which we do business, including the Center for Molecular Medicine and Immunology and the Garden State Cancer Center (which operated as the clinical arm of CMMI to facilitate the translation of CMMI’s research efforts in the treatment of patients), collectively defined as CMMI. For example, Dr. Goldenberg was the President and a Trustee of CMMI, a not-for-profit cancer research center that we used to conduct certain research activities. CMMI has ceased operations. Dr. Goldenberg is also a minority stockholder, director and officer of our majority-owned subsidiary, IBC.
28
Dr. Goldenberg is the primary inventor of new intellectual property for Immunomedics and IBC and is largely responsible for allocating ownership between the two companies. Immunomedics has incurred expenses on behalf of the IBC operations, including interest, over the past thirteen years. As of June 30, 2017, IBC has a liability to Immunomedics Inc. of approximately $16.8 million, which is eliminated in consolidation. Dr. Goldenberg also has primary responsibility for monitoring the market for incidences of potential infringement of the Company’s intellectual property by third parties.
As a result of these and other relationships, the potential for both real and perceived conflicts of interest exists and disputes could arise over the allocation of funds, research projects and ownership of intellectual property rights. In addition, in the event that we become involved in stockholder litigation regarding these potential conflicts, we might be required to devote significant resources and management time defending the company from these claims, which could adversely affect our results of operations.
Given that recent cancer therapeutics for solid cancers such as the ones we are developing can cost approximately in excess of $12,500 a month, even if our product candidates become available for sale it is likely that federal and state governments, insurance companies and other payers of health care costs will try to first limit the use of these drugs to certain patients, and may be reluctant to provide a level of reimbursement that permits us to earn a significant profit on our investment, if any.
Our ability to successfully commercialize therapeutic products will depend, in significant part, on the extent to which hospitals and physicians can obtain appropriate reimbursement levels for the cost of our products and related treatment. Third-party payers are increasingly challenging the prices charged for diagnostic and therapeutic products and related services. In addition, legislative proposals to reform health care or reduce government insurance programs may result in lower prices or the actual inability of prospective customers to purchase our products. Furthermore, even if reimbursement is available, it may not be available at price levels sufficient for us to realize a positive return on our investment.
A portion of our funding has come from federal government grants and research contracts. Due to reductions in funding, we may not be able to rely on these grants or contracts as a continuing source of funds.
During the last few years, we have generated revenues from awards made to us by the National Institutes of Health and the Department of Defense to partially fund some of our programs. We cannot rely on grants or additional contracts as a continuing source of funds. Funds available under these grants and contracts must be applied by us toward the research and development programs specified by the government rather than for all our programs generally. The government’s obligation to make payments under these grants and contracts is subject to appropriation by the United States Congress for funding in each year. It is possible that Congress or the government agencies that administer these government research programs will continue to scale back these programs or terminate them due to their own budgetary constraints, as they have recently been doing. Additionally, these grants and research contracts are subject to adjustment based upon the results of periodic audits performed on behalf of the granting authority. Consequently, the government may not award grants or research contracts to us in the future, and any amounts that we derive from existing awards may be less than those received to date. In those circumstances, we would need to provide funding on our own, obtain other funding, or scale back or terminate the affected program. In particular, we cannot assure you that any currently-contemplated or future efforts to obtain funding for our product candidate programs through government grants or contracts will be successful, or that any such arrangements which we do conclude will supply us with sufficient funds to complete our development programs without providing additional funding on our own or obtaining other funding. Where funding is obtained from government agencies or research bodies, our intellectual property rights in the research or technology funded by the grant are typically subject to certain licenses to such agencies or bodies, which could have an impact on our utilization of such intellectual property in future.
We face a number of risks relating to the maintenance of our information systems and our use of information relating to clinical trials.
In managing our operations, we rely on computer systems and electronic communications, including systems relating to record keeping, financial information, sourcing, and back-up and the internet (“Information Systems”). Our
29
Information Systems include the electronic storage of financial, operational, research, patient and other data. Our Information Systems may be subject to interruption or damage from a variety of causes, including power outages, computer and communications failures, system capacity constraints, catastrophic events (such as fires, tornadoes and other natural disasters), cyber risks, computer viruses and security breaches. If our Information Systems cease to function properly, are damaged or are subject to unauthorized access, we may suffer interruptions in our operations, be required to make significant investments to fix or replace systems and/or be subject to fines, penalties, lawsuits, or government action. The realization of any of these risks could have a material adverse effect on our business, financial condition and results of operations. Our clinical trials information and patient data (which may include personally identifiable information) is part of our Information Systems and is therefore subject to all of the risks set forth above, notwithstanding our efforts to code and protect such information.
Risks Related to Government Regulation of our Industry
Legislative or regulatory reform of the healthcare system may affect our ability to sell our products profitably.
In both the United States and certain foreign jurisdictions, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could impact our ability to sell our future products and profitability. On March 23, 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “PPACA”), which includes a number of health care reform provisions and requires most United States citizens to have health insurance. The new law, among other things, imposes a significant annual fee on companies that manufacture or import branded prescription drug products, addresses a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increases the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extends the rebate program to individuals enrolled in Medicaid managed care organizations, and establishes a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D. Substantial new provisions affecting compliance also have been added, which may require modification of business practices with health care practitioners.
In the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our future products, and we could be adversely affected by current and future health care reforms.
Our industry and we are subject to intense regulation from the United States Government and such other governments and quasi-official regulatory bodies where our products are and product candidates may be sold.
Both before and after regulatory approval to market a particular product candidate, including our biologic product candidates, the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, distribution and record keeping related to the product are subject to extensive, ongoing regulatory requirements, including, without limitation, submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP requirements and good clinical practice requirements for any clinical trials that we conduct post-approval. As a result, we are subject to a number of governmental and other regulatory risks, which include:
|
· |
clinical development is a long, expensive and uncertain process; delay and failure can occur at any stage of our clinical trials; |
|
· |
our clinical trials are dependent on patient enrollment and regulatory approvals; we do not know whether our planned trials will begin on time, or at all, or will be completed on schedule, or at all; |
|
· |
the FDA or other regulatory authorities may not approve a clinical trial protocol or may place a clinical trial on hold; |
|
· |
we rely on third parties, such as consultants, contract research organizations, medical institutions, and clinical investigators, to conduct clinical trials for our drug candidates and if we or any of our third-party contractors fail to |
30
comply with applicable regulatory requirements, such as cGCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials; |
|
· |
if the clinical development process is completed successfully, our ability to derive revenues from the sale of therapeutics will depend on our first obtaining FDA or other comparable foreign regulatory approvals, each of which are subject to unique risks and uncertainties; |
|
· |
there is no assurance that we will receive FDA or corollary foreign approval for any of our product candidates for any indication; we are subject to government regulation for the commercialization of our product candidates; |
|
· |
we have not received regulatory approval in the United States for the commercial sale of any of our biologic product candidates; |
|
· |
even if one or more of our product candidates does obtain approval, regulatory authorities may approve such product candidate for fewer or more limited indications than we request, may not approve the price we intend to charge for our products, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate; |
|
· |
undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities; |
|
· |
later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with the regulatory requirements of FDA and other applicable United States and foreign regulatory authorities could subject us to administrative or judicially imposed sanctions; |
|
· |
although several of our product candidates have received orphan drug designation in the United States and the EU for particular indications, we may not receive orphan drug exclusivity for any or all of those product candidates or indications upon approval, and even if we do obtain orphan drug exclusivity, that exclusivity may not effectively protect the product from competition; |
|
· |
even if one or more of our product candidates is approved in the United States, it may not obtain the 12 years of exclusivity from biosimilars for which innovator biologics are eligible, and even if it does obtain such exclusivity, that exclusivity may not effectively protect the product from competition; |
|
· |
the FDA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates, and if we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained; and |
|
· |
we may be liable for contamination or other harm caused by hazardous materials used in the operations of our business. |
In addition, our operations are also subject to various federal and state fraud and abuse, physician payment transparency and privacy and security laws, including, without limitation:
|
· |
The federal Anti-Kickback Statute, which prohibits, among other things, soliciting, receiving, offering or providing remuneration intended to induce the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare or Medicaid programs. This statute has been applied to pharmaceutical manufacturer marketing practices, educational programs, pricing policies and relationships with healthcare providers. A person or entity does not need to have actual knowledge of this statute or specific intent to violate it to have committed a violation; |
|
· |
Federal civil and criminal false claims laws and civil monetary penalty laws, including civil whistleblower or qui tam actions that prohibit, among other things, knowingly presenting, or causing to be presented, claims for payment or approval to the federal government that are false or fraudulent, knowingly making a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay or transmit money or property to the federal government. The government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the false claims statutes; |
31
|
· |
HIPAA and its implementing regulations, which created federal criminal laws that prohibit, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
|
· |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, also imposes certain regulatory and contractual requirements regarding the privacy, security and transmission of individually identifiable health information; |
|
· |
Federal “sunshine” requirements imposed by PPACA on drug manufacturers regarding any “transfer of value” made or distributed to physicians and teaching hospitals, and any ownership and investment interests held by such physicians and their immediate family members. Failure to submit the required information may result in civil monetary penalties of up an aggregate of $150,000 per year (and up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests not reported in an annual submission, and may result in liability under other federal laws or regulations; and |
|
· |
State and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require drug manufacturers to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of certain health information, many of which differ from each other in significant ways and often are not preempted by HIPAA. |
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under such laws, it is possible that some of our business activities, including certain sales and marketing practices and financial arrangements with physicians, could be subject to challenge under one or more of such laws. Any action against us, even if we successfully defend against it, could result in the commencement of civil and/or criminal proceedings, exclusion from governmental health care programs, substantial fines, penalties, and/or administrative remedies, any of which could have an adverse effect on our financial condition and results of operations.
Risks Related to Our Securities
Conversion of the Convertible Senior Notes will dilute the ownership interest of existing stockholders and could adversely affect the market price of our common stock.
The conversion of some or all of the Convertible Senior Notes will dilute the ownership interests of existing stockholders. Any sales in the public market of the common stock issuable upon such conversion and exercise could adversely affect prevailing market prices of our common stock. In addition, the existence of the Convertible Senior Notes may encourage short selling by market participants.
Our indebtedness and debt service obligations may adversely affect our cash flow.
As of June 30, 2017, our total consolidated indebtedness was $131.3 million, including our obligations under our Convertible Senior Notes and other liabilities. We intend to fulfill our current debt service obligations, including repayment of the principal from our existing cash and investments, as well as the proceeds from potential licensing agreements and any additional financing from equity or debt transactions. However, our ability to make scheduled payments of the principal of, to pay interest on or to refinance our indebtedness, including the Convertible Senior Notes, depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow to meet these obligations, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional equity capital on terms that may be onerous or highly dilutive, or delaying or curtailing research and development programs. Our ability to refinance our indebtedness will depend on the capital markets and our financial condition at such time. We
32
may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
Our common stock may be delisted from the NASDAQ Global Market, or NASDAQ.
If the bid price of our common stock falls below $1.00 for an extended period, or we are unable to continue to meet NASDAQ’s listing maintenance standards for any other reason, our common stock could be delisted from NASDAQ.
If our stock is delisted from NASDAQ, we will make every possible effort to have it listed on the Over the Counter Bulletin Board (the “OTC Bulletin Board”). If our common stock was to be traded on the OTC Bulletin Board, the Securities Exchange Act of 1934, as amended, and related SEC rules would impose additional sales practice requirements on broker-dealers that sell our securities. These rules may adversely affect the ability of stockholders to sell our common stock and otherwise negatively affect the liquidity, trading market and price of our common stock.
If our common stock would not be able to be traded on the OTC Bulletin Board, we would make every effort to have it available for trading on the National Quotation Bureau’s Pink Sheets (“the Pink Sheets”). The Pink Sheets market consists of security firms who act as market makers in the stocks, usually, of very small companies. The bid and asked prices are not quoted electronically, but are quoted daily in “hard copy” which is delivered to firms that subscribe. Stocks that trade in the Pink Sheets are usually not as liquid as those that trade in electronic markets and, often time, the difference between the bid and the asked prices are substantial. As a result, if our common stock were traded on the Pink Sheets, there would likely be a further negative affect on the liquidity, trading market and price of our common stock even compared to what we might suffer if we were traded on the OTC Bulletin Board.
As a result of the above, we cannot assure you that our common stock will be listed on a national securities exchange, a national quotation service, the OTC Bulletin Board or the Pink Sheets; or if it is to be listed, whether or not there would be an interruption in the trading of our common stock. We believe that the listing of our stock on a recognized national trading market, such as NASDAQ, is an important part of our business and strategy. Such a listing helps our stockholders by providing a readily available trading market with current quotations. Without that, stockholders may have a difficult time getting a quote for the sale or purchase of our stock, the sale or purchase of our stock would likely be made more difficult and the trading volume and liquidity of our stock would likely decline. The absence of such a listing may adversely affect the acceptance of our common stock as currency or the value accorded it by other parties. In that regard, listing on a recognized national trading market will also affect our ability to benefit from the use of its operations and expansion plans, including for use in licensing agreements, joint ventures, the development of strategic relationships and acquisitions, which are critical to our business and strategy and none of which is currently the subject of any agreement, arrangement or understanding, with respect to any future financing or strategic relationship it may undertake. The delisting from NASDAQ would result in negative publicity and would negatively impact our ability to raise capital in the future.
If we were delisted from NASDAQ, we may become subject to the trading complications experienced by “Penny Stocks” in the over-the-counter market.
Delisting from NASDAQ may depress the price of our common stock such that we may become a penny stock. The SEC generally defines a penny stock as an equity security that has a market price of less than $5.00 per share or an exercise price of less than $5.00 per share, subject to specific exemptions. We continue to be listed on NASDAQ. “Penny Stock” rules require, among other things, that any broker engaging in a purchase or sale of our securities provide its customers with: (i) a risk disclosure document; (ii) disclosure of market quotations, if any; (iii) disclosure of the compensation of the broker and its salespersons in the transaction; and (iv) monthly account statements showing the market values of our securities held in the customers’ accounts.
33
A broker would be required to provide the bid and offer quotations and compensation information before effecting the transaction. This information must be contained on the customers’ confirmation. Generally, brokers are less willing to effect transactions in penny stocks due to these additional delivery requirements. These requirements may make it more difficult for stockholders to purchase or sell our common stock. Because the broker, not us, prepares this information, we would not be able to assure that such information is accurate, complete or current.
We may add lease lines to finance capital expenditures and may obtain additional long‑term debt and lines of credit. If we issue other debt securities in the future, our debt service obligations will increase further.
Our indebtedness could have significant additional negative consequences, including, but not limited to:
|
· |
requiring the dedication of a substantial portion of our existing cash and marketable securities balances and, if available, future cash flow from operations to service our indebtedness, thereby reducing the amount of our expected cash flow available for other purposes, including capital expenditures; |
|
· |
increasing our vulnerability to general adverse economic and industry conditions; |
|
· |
limiting our ability to obtain additional financing; |
|
· |
limiting our ability to sell assets if deemed necessary; |
|
· |
limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
|
· |
placing us at a possible competitive disadvantage to less leveraged competitors and competitors that have better access to capital resources. |
We may not have the ability to raise funds necessary to purchase the Convertible Senior Notes upon a fundamental change and our future debt may contain limitations on our ability to repurchase the Convertible Senior Notes.
Following a fundamental change (which includes matters such as a change in control of the Company, approval by the Company’s stockholders of a plan of dissolution or liquidation of the Company, and the cessation of listing of the Company’s common stock on NASDAQ or The New York Stock Exchange, among others as further described in the indenture), holders of Convertible Senior Notes will have the right to require the Company to purchase their Convertible Senior Notes for cash. A fundamental change may also constitute an event of default or require prepayment under, and result in the acceleration of the maturity of, our other then-existing indebtedness. We cannot assure you that we will have sufficient financial resources, or will be able to arrange financing, to pay the fundamental change purchase price in cash with respect to any Convertible Senior Notes surrendered by holders for purchase upon a fundamental change. In addition, restrictions in the agreements governing our then-outstanding indebtedness, if any, may not allow us to purchase the Convertible Senior Notes upon a fundamental change. Our failure to purchase the Convertible Senior Notes upon a fundamental change when required would result in an event of default with respect to the Convertible Senior Notes which could, in turn, constitute a default under the terms of our other indebtedness, if any. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and purchase the Convertible Senior Notes, which could have a material and adverse impact on our financial condition and results of operations.
Shares eligible for future sale may adversely affect our ability to sell equity securities.
Sales of our common stock (including the issuance of shares upon conversion of convertible debt) in the public market could materially and adversely affect the market price of shares. We have outstanding $100 million principal amount of Convertible Senior Notes that convert to common stock at prices equivalent to $5.11 (subject to adjustment for certain dilutive events). Our obligation to convert the Convertible Senior Notes upon demand by the holders may depress the price of our common stock and also make it more difficult for us to sell equity securities or equity‑related securities in the future at a time and price that we deem appropriate.
As of June 30, 2017 we had 110,344,643 shares of common stock issued, plus (1) 1,000,000 shares of preferred stock issued, which is convertible into up to approximately 23,105,348 shares of common stock at the conversion price of $5.41, (2) $100 million of principal amount of Convertible Senior Notes convertible into up to approximately
34
19,583,360 shares of common stock at the conversion rate of $5.11 subject to adjustment as described in the indenture, (3) 2,893,240 options to purchase shares of common stock with a weighted‑average exercise price of $3.48 per share, (4) 1,831,329 restricted stock units, (5) 9,540,417 for potential future grants of options to purchase shares of common stock under the Plan, (6) warrants to purchase 10,000,000 shares of common stock with an exercise price of $3.75 and (7) warrants to purchase 8,655,804 shares of common stock with an exercise price of $4.90. Of the 250,000,000 shares of common stock authorized under our Certificate of Incorporation, there are 64,045,859 shares of common stock that remain available for future issuance.
Our outstanding Convertible Senior Notes, Convertible Preferred Shares, options and warrants may adversely affect our ability to consummate future equity‑based financings due to the dilution potential to future investors.
Due to the number of shares of common stock we are obligated to issue pursuant to outstanding Convertible Senior Notes, Convertible Preferred Shares, options and warrants, potential investors may not purchase our future equity offerings at market price because of the potential dilution such investors may suffer as a result of the exercise of the outstanding Convertible Senior Notes, options and warrants.
The market price of our common stock has fluctuated widely in the past, and is likely to continue to fluctuate widely based on a number of factors, many of which are beyond our control.
The market price of our common stock has been, and is likely to continue to be, highly volatile. Furthermore, the stock market and the market for stocks of relatively small biopharmaceutical companies like ours have from time to time experienced, and likely will again experience, significant price and volume fluctuations that are unrelated to actual operating performance.
From time to time, stock market analysts publish research reports or otherwise comment upon our business and future prospects. Due to a number of factors, we may fail to meet the expectations of securities analysts or investors and our stock price would likely decline as a result. These factors include:
|
· |
Announcements by us, our current collaboration partner, any future alliance partners or our competitors of pre-clinical studies and clinical trial results, regulatory developments, technological innovations or new therapeutic products, product sales, new products or product candidates and product development timelines; |
|
· |
The formation or termination of corporate alliances; |
|
· |
Developments in patent or other proprietary rights by us or our respective competitors, including litigation; |
|
· |
Developments or disputes concerning our patent or other proprietary rights, and the issuance of patents in our field of business to others; |
|
· |
Government regulatory action; |
|
· |
Period-to-period fluctuations in the results of our operations; and |
|
· |
Developments and market conditions for emerging growth companies and biopharmaceutical companies, in general. |
In addition, Internet “chat rooms” have provided forums where investors make predictions about our business and prospects, oftentimes without any real basis in fact, that readers may trade on.
In the past, following periods of volatility in the market prices of the securities of companies in our industry, securities class action litigation has often been instituted against those companies. Please see Item 3 (“Legal Proceedings”) for a description of such litigation. If we face such litigation in the future, it would result in substantial costs and a diversion of management’s attention and resources, which could negatively impact our business.
Our principal stockholders can significantly influence all matters requiring the approval by our stockholders.
As of June 30, 2017 venBio Select Advisor LLC, (‘venBio”) is the beneficial owner of approximately 11.1% of our outstanding common stock (assuming for these purposes only, the conversion of all outstanding shares of preferred
35
stock into common stock) and approximately 7.9% of our fully diluted common stock. venBio is our largest stockholder, and Dr. Behzad Aghazadeh, the Managing Partner and portfolio manager of the venBio Select Fund, serves as Chairman of our Board of Directors.
As of June 30, 2017, Dr. David M. Goldenberg, our Chief Scientific Officer, Chief Patent Officer and former Chairman of the Board, together with certain members of his family, including Ms. Cynthia L. Sullivan, our former President and Chief Executive Officer, who is Dr. Goldenberg’s wife, and other affiliates, controlled the right to vote approximately 6.9% of our outstanding common stock and approximately 4.1% of our fully diluted common stock.
As a result of this voting power, venBio and Dr. Goldenberg have the ability to significantly influence the outcome of substantially all matters that may be put to a vote of our stockholders, including the election of our directors.
There are limitations on the liability of our directors, and we may have to indemnify our officers and directors in certain instances.
Our certificate of incorporation limits, to the maximum extent permitted under Delaware law, the personal liability of our directors for monetary damages for breach of their fiduciary duties as directors. Our bylaws provide that we will indemnify our officers and directors and may indemnify our employees and other agents to the fullest extent permitted by law. These provisions may be in some respects broader than the specific indemnification provisions under Delaware law. The indemnification provisions may require us, among other things, to indemnify such officers and directors against certain liabilities that may arise by reason of their status or service as directors or officers (other than liabilities arising from willful misconduct of a culpable nature), to advance their expenses incurred as a result of certain proceedings against them as to which they could be indemnified and to obtain directors’ and officers’ insurance. Section 145 of the Delaware General Corporation Law provides that a corporation may indemnify a director, officer, employee or agent made or threatened to be made a party to an action by reason of the fact that he or she was a director, officer, employee or agent of the corporation or was serving at the request of the corporation, against expenses actually and reasonably incurred in connection with such action if he or she acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful. Delaware law does not permit a corporation to eliminate a director’s duty of care and the provisions of our certificate of incorporation have no effect on the availability of equitable remedies, such as injunction or rescission, for a director’s breach of the duty of care.
We believe that our limitation of officer and director liability assists us to attract and retain qualified employees and directors. However, in the event an officer, a director or the board of directors commits an act that may legally be indemnified under Delaware law, we will be responsible to pay for such officer(s) or director(s) legal defense and potentially any damages resulting there from. Furthermore, the limitation on director liability may reduce the likelihood of derivative litigation against directors and may discourage or deter stockholders from instituting litigation against directors for breach of their fiduciary duties, even though such an action, if successful, might benefit our stockholders and us. Given the difficult environment and potential for incurring liabilities currently facing directors of publicly-held corporations, we believe that director indemnification is in our and our stockholders’ best interests because it enhances our ability to attract and retain highly qualified directors and reduce a possible deterrent to entrepreneurial decision-making.
Nevertheless, limitations of director liability may be viewed as limiting the rights of stockholders, and the broad scope of the indemnification provisions contained in our certificate of incorporation and bylaws could result in increased expenses. Our board of directors believes, however, that these provisions will provide a better balancing of the legal obligations of, and protections for, directors and will contribute positively to the quality and stability of our corporate governance. Our board of directors has concluded that the benefit to stockholders of improved corporate governance outweighs any possible adverse effects on stockholders of reducing the exposure of directors to liability and broadened indemnification rights.
36
We are exposed to potential risks from legislation requiring companies to evaluate controls under Section 404 of the Sarbanes-Oxley Act.
The Sarbanes-Oxley Act requires that we maintain effective internal controls over financial reporting and disclosure controls and procedures. Among other things, we must perform system and process evaluation and testing of our internal controls over financial reporting to allow management to report on, and our independent registered public accounting firm to attest to, our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act (“Section 404”). Compliance with Section 404 requires substantial accounting expense and significant management efforts. Our testing, or the subsequent review by our independent registered public accounting firm, may reveal deficiencies in our internal controls that would require us to remediate in a timely manner so as to be able to comply with the requirements of Section 404 each year. If we are not able to comply with the requirements of Section 404 in a timely manner each year, we could be subject to sanctions or investigations by the SEC, the NASDAQ Stock Market or other regulatory authorities that would require additional financial and management resources and could adversely affect the market price of our common stock.
We do not intend to pay dividends on our common stock. Until such time as we pay cash dividends our stockholders, must rely on increases in our stock price for appreciation.
We have never declared or paid dividends on our common stock. We intend to retain future earnings to develop and commercialize our product candidates and therefore we do not intend to pay cash dividends in the foreseeable future. Until such time as we determine to pay cash dividends on our common stock, our stockholders must rely on increases in the market price of our common stock for appreciation of their investment.
37
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
Our headquarters is located at 300 The American Road, Morris Plains, New Jersey 07950, where we lease approximately 85,000 square feet of commercial office space, pursuant to a lease which is scheduled to expire in October 2031. The current base annual rate is $1.0 million, which is a fixed rate through October 2021 and increases thereafter every five years. Our manufacturing, regulatory, medical, research and development laboratories, and our finance, marketing and executive offices are currently located in this facility. We operate a 7,500 square-foot manufacturing facility within our Morris Plains headquarters, which consists of four independent antibody manufacturing suites, several support areas, and a quality control laboratory. See Item 1 Business, “Manufacturing.” In addition, our European subsidiary, Immunomedics GmbH, leases executive office space in Rodermark, Germany.
38
Item 3. Legal Proceedings
a. Patent litigation:
Immunomedics filed a first amended complaint on October 22, 2015, a second amended complaint on January 14, 2016, and a third amended complaint on October 12, 2016, in the United States District Court for the District of New Jersey, against Roger Williams Medical Center (“RWMC”), Richard P. Junghans, M.D., Ph.D., and Steven C. Katz, M.D. The third amended complaint alleges that RWMC and Dr. Junghans breached a Material Transfer Agreement (“MTA”) through which it provided to them a monoclonal antibody known as MN-14 and related materials. Defendants are alleged to have breached the MTA and to have been negligent by, among other things, using the materials beyond the agreed-upon Research Project, sharing confidential information, failing to provide Immunomedics with a right of first refusal, failing to notify Immunomedics of intended publications prior to publishing, and refusing to return the materials upon request. Immunomedics also asserts the following claims against some of these defendants: conversion, tortious interference, unjust enrichment, and infringement of three patents owned by Immunomedics. Defendants Junghans, Katz, and RWMC subsequently moved to dismiss for failure to state a claim on November 14, 2016, but this motion was denied on January 4, 2017. The third amended complaint also added parties named Sorrento, TNK, BDL, and CARgenix. On December 2, 2016, Sorrento, TNK, BDL, and CARgenix moved to dismiss for lack of personal jurisdiction over them in New Jersey. The court granted this motion on January 25, 2017. On January 20, 2017, the court held a Markman hearing to construe the claims in the patents in suit. On February 28, 2017, the court issued an opinion and order finding, inter alia, that the term “effective amount” in the patents in suit is not indefinite and should be given its plain and order meaning, as proposed by Immunomedics, of “an amount capable of producing the claim result.” All other terms in the patents were given their plain and ordinary meaning. On May 11, 2017, the Court ordered the parties to mediation with former New Jersey District Court Judge Garrett Brown, and stayed the case for 90 days. A mediation took place on June 28, 2017. The mediation was unsuccessful; and the stay of discovery was lifted on August 9.
b. Stockholder complaints:
Class Action Stockholder Federal Securities Cases
Two purported class action cases have been filed in the United States District Court for the District of New Jersey; namely, Fergus v. Immunomedics, Inc., et al., No. 2:16-cv-03335, filed June 9, 2016; and Becker v. Immunomedics, Inc., et al., No. 2:16-cv-03374, filed June 10, 2016. These cases arise from the same alleged facts and circumstances, and seek class certification on behalf of purchasers of our common stock between April 20, 2016 and June 2, 2016 (with respect to the Fergus matter) and between April 20, 2016 and June 3, 2016 (with respect to the Becker matter). These cases concern the Company's statements in press releases, investor conference calls, and SEC filings beginning in April 2016 that the Company would present updated information regarding its IMMU-132 breast cancer drug at the 2016 American Society of Clinical Oncology ("ASCO") conference in Chicago, Illinois. The complaints allege that these statements were false and misleading in light of June 2, 2016 reports that ASCO had cancelled the presentation because it contained previously reported information. The complaints further allege that these statements resulted in artificially inflated prices for our common stock, and that the Company and certain of its officers are thus liable under Sections 10(b) and 20(a) of the Securities Exchange Act of 1934. An order of voluntarily dismissal without prejudice was entered on November 10, 2016 in the Becker matter. An order granting motion to consolidate cases, appoint lead plaintiff, and approve lead and liaison counsel was entered on February 7, 2017 in the Fergus matter. As of the date hereof, service of the initiating papers in the Fergus matter has not been made on the Company.
Stockholder Derivative Action in the Superior Court of New Jersey
On October 3, 2016, plaintiff commenced an action captioned Rosenfeld v. Goldenberg, et al., No. L-2200-16, alleging the same underlying facts and circumstances as in the pending federal securities class action, the Fergus matter. Specifically, this action concerns the Company’s statements in press releases, investor conference calls, and SEC filings beginning in April 2016 that the Company would present updated information regarding its IMMU-132 breast cancer drug at the 2016 ASCO conference in Chicago, Illinois. The complaint alleges that these statements were false and misleading in light of the June 2, 2016 reports that ASCO had cancelled the presentation because it contained previously reported information. The complaint further alleges that these statements resulted in artificially inflated prices for our
39
common stock, and that certain directors and officers of the Company breached their fiduciary duties to the Company. In addition to monetary damages, the complaint seeks to require the Company to reform its corporate governance and internal procedures. Service was effectuated on all defendants on April 7, 2017. The defendants filed a motion to dismiss the complaint on June 19, 2017.
Class Action Stockholder Claim in the Court of Chancery of the State of Delaware
On December 13, 2016, plaintiff commenced an action captioned Desanctis v. Goldenberg, C.A. No. 12981-VCL (Del. Ch. Ct.), alleging that members of the Company's Board of Directors failed to comply with Delaware law and breached their fiduciary duties when it rescheduled the Immunomedics 2016 Annual Meeting of Stockholders from December 14, 2016 to February 16, 2017. On December 22, 2016, the Delaware Court of Chancery refused to schedule an expedited hearing in the action and concluded that plaintiff failed to carry his burden of demonstrating that he had pleaded a colorable claim and that there was a threat of irreparable harm. The Court further stated that the Complaint failed to demonstrate that the Board's actions were unreasonable when it rescheduled the Annual Meeting in response to venBio Select Advisor LLC's proxy contest.
Stockholder Claim in the Court of Chancery of the State of Delaware
On February 13, 2017, venBio commenced an action captioned venBio Select Advisor LLC v. Goldenberg, et al., C.A. No. 2017-0108-VCL (Del. Ch.) (the “venBio Action”), alleging that members of the Company’s Board breached their fiduciary duties when the Board (i) rescheduled the Company’s 2016 Annual Meeting of Stockholders (the “2016 Annual Meeting”) from December 14, 2016 to February 16, 2017, and then again to March 3, 2017, and (ii) agreed to the proposed licensing transaction with Seattle Genetics (the “Licensing Transaction”). venBio also named Seattle Genetics as a defendant and sought an injunction preventing the Company from closing the licensing transaction with Seattle Genetics. On March 6, 2017, venBio amended its complaint, adding further allegations, including that members of the Company’s Board breached their fiduciary duties when the Board amended the Company’s Amended and Restated By-laws (the “By-Laws”) to call for a plurality voting regime for the election of directors instead of majority voting, and providing for mandatory advancement of attorneys’ fees and costs for the Company’s directors and officers. The Court of Chancery entered a temporary restraining order on March 9, 2017, enjoining the closing of the Licensing Transaction. venBio amended its complaint a second time on April 19, 2017, this time adding as additional defendants the Company’s financial advisor on the Licensing Transaction, Greenhill & Co., LLC and Greenhill & Co. Inc. (together “Greenhill”).
On May 4, 2017, the Company entered into the Termination Agreement with Seattle Genetics, pursuant to which the Company and Seattle Genetics agreed to relinquish their respective rights under the Licensing Agreement and amend the term of the SGEN Warrant, and in connection therewith, the Company and venBio agreed to fully settle, resolve and release Seattle Genetics, and Seattle Genetics agreed to fully settle, resolve and release the Company and venBio, from all disputes, claims and liabilities arising from the Licensing Agreement and the transactions contemplated thereby, subject to the terms of the Termination Agreement and the related settlement agreement. The Termination Agreement will be effective thirty days following the entry on July 25, 2017 of a final judgment of the Court of Chancery approving the dismissal of Seattle Genetics from the venBio Action.
On May 3, 2017, venBio the Company and individual defendants Goldenberg, Sullivan and Markison (collectively, the “Individual Defendants”) entered into a binding Term Sheet, which is to be reduced to a definitive settlement agreement (“Settlement Agreement”), pursuant to which, among other things, venBio and the Company will release the Individual Defendants from certain claims described below and will submit the remaining claims against the non-settling defendants (including non-settling defendants and former directors Robert Forrester, Jason Aryeh, Geoff Cox and Bob Oliver, but excluding those claims with respect to Seattle Genetics, which the parties have agreed to settle pursuant to the Termination Agreement as described above) to non-binding mediation before a mediator mutually acceptable to the parties that remain in the venBio Action. Once venBio,the Company and the Individual Defendants execute the Settlement Agreement, it will be submitted to the Court of Chancery for approval.
On June 8, 2017, venBio, the Company and Greenhill entered into a binding term sheet (the “GHL Term Sheet”), which is to be reduced to a definitive settlement agreement (“GHL Settlement Agreement”), pursuant to which,
40
among other things, venBio and the Company will release Greenhill from “all direct and derivative claims that have been or could be asserted by or on behalf of venBio, the Company, or the directors, officers, employees, affiliates and related persons of venBio or the Company, whether known or unknown, against Greenhill and Greenhill’s affiliates and related persons in connection with the claims alleged in the venBio Action, the 225 Action, the Federal Action, the Licensing Transaction, the Financing, the Company’s 2017 annual stockholder meeting, Greenhill’s 9/23/2016 and 12/14/2016 engagement letters with the Company (the “Engagement Letters”) (including any claims related to or arising in any manner out of any activities performed or services furnished pursuant to the Engagement Letters, the transactions contemplated thereby or Greenhill’s role in connection therewith), and this settlement.” Greenhill similarly agreed to release “all direct and derivative claims that have been or could be asserted by or on behalf of Greenhill, or the directors, officers, employees, affiliates and related persons of Greenhill, whether known or unknown, against venBio or the Company and their current and former directors, officers, members, employees, affiliates and related persons in connection with the claims alleged in the venBio Action, the 225 Action, the Federal Action, the Licensing Transaction, the Financing, the Company’s 2017 annual stockholder meeting, the Engagement Letters, and this settlement.” Greenhill, the Company and venBio also agreed that the Engagement Letters would be terminated and that Greenhill would forgo and not seek any and all fees, expense, reimbursement or indemnification from the Company, except that, upon final Court approval of the GHL Settlement Agreement, the Company shall reimburse Greenhill up to $200,000 for reasonable and documented expenses that Greenhill incurred in connection with services provided under the Engagement Letters. Greenhill also consented to the settlement reflected in the Term Sheet and Greenhill, the Company and venBio agreed that Greenhill need not participate in the non-binding mediation contemplated by the Term Sheet.
Lawsuit Against venBio Select Advisor LLC in the U.S. District Court (Delaware) (the “District Court”)
On February 17, 2017, the Company commenced an action captioned Immunomedics, Inc. v. venBio Select Advisor LLC, No. 17-176-LPS (D. Del.) (the “Federal Action”), seeking for the District Court to invalidate the proxies solicited by venBio in furtherance of its contest for the election of directors of the Company. The Company named as defendants venBio and its then-nominees, Behzad Aghazadeh, Scott Canute, Peter Barton Hutt, and Khalid Islam. The Company alleged that venBio had conducted its proxy contest and solicited proxies in violation the federal securities laws and regulations, namely by failing timely file a Schedule 13D form indicating venBio’s intent to effectuate change at the Company, publishing early voting results of the Company’s annual election of directors, publishing improper statements about the then-incumbent Board, forming a “group” of like-minded stockholders without publicly disclosing the group, and soliciting proxies without disclosing the solicitations to the SEC. On February 21, 2017, the Company sought an injunction preventing, among other things, the venBio nominees from benefiting from an allegedly illegal shadow proxy contest, including, but not limited to, by asserting any claimed right to take office as a member of the Board until venBio made corrective disclosures and the stockholders were permitted time to consider them. On March 2, 2017, the District Court denied the Company the requested relief. On April 6, 2017, the District Court entered a stipulation and order pursuant to which the Company’s claims were voluntarily dismissed without prejudice. On April 17, 2017, Dr. Goldenberg, the Company’s Chief Scientific Officer and Chief Patent Officer and director, notified the District Court that he may maintain the claims initially brought by the Company. On May 3, 2017, Goldenberg and venBio entered into a binding Term Sheet which is to be reduced to a definitive Settlement Agreement, pursuant to which, among other things, the Parties have agreed to submit to the District Court a stipulation and proposed order dismissing all claims in the Federal Action with prejudice, including those against the individual defendants (the then-venBio nominees). The Settlement Agreement will also include a mutual release of claims that were or could have been asserted in the Federal Action.
Lawsuit Challenging the Results of the 2016 Election of Directors
On March 3, 2017, six of the seven then-incumbent members of the Company’s Board commenced an action captioned Goldenberg, et al. vs Aghazadeh, et al., C.A. No. 2017-0163-VCL (Del. Ch.) (the “225 Action”), challenging the results of the election of directors at the 2016 Annual Meeting that took place on March 3, 2017, in which all four of venBio’s nominees won seats on the Company’s Board. The director-plaintiffs named as defendants venBio and its then-nominees, Behzad Aghazadeh, Scott Canute, Peter Barton Hutt, and Khalid Islam. The incumbent directors alleged the same underlying facts as the Company alleged in its lawsuit against venBio in federal court. On March 13, 2017, the Court of Chancery entered an order (the “Status Quo Order”) seating all four venBio nominees (and the three incumbent
41
directors who secured a plurity of votes, the “Status Quo Board”) and limiting the Company’s Board to actions within the “ordinary course of business,” unless either waived by the parties on a case-by-case basis or by Order the Court of Chancery. On March 24, 2017, the defendants, venBio and its four nominees, moved to dismiss the action. The plaintiffs in the action opposed this motion to dismiss, which remains pending. On April 7, 2017, the three incumbent director plaintiffs not seated on the Status Quo Board voluntarily withdrew their claims, leaving Goldenberg, Sullivan and Markison as plaintiffs. On April 20, 2017, the parties agreed to permit the Status Quo Board to explore a potential financing plan for the Company and negotiate a termination of the Licensing Transaction. On May 3, 2017, the Parties entered into the Term Sheet, pursuant to which, among other things, the Parties agreed to submit to the Court of Chancery a stipulation and proposed order lifting the Status Quo Order. On May 4, 2017, the Parties submitted that stipulation, which confirmed that the Status Quo Board is the lawful Board of the Company, provided that if the 225 Action is not dismissed, the Parties shall be restored to their positions in the 225 Action as of immediately prior to execution of the Term Sheet. Once the Settlement Agreement is executed, the Parties will submit to the Court of Chancery another stipulation and proposed order dismissing the 225 Action with prejudice, including those against the individual defendants (the then-venBio nominees). The Settlement Agreement will also include a mutual release of all claims that were or could have been asserted in the 225 Action.
(c) Settlement Term Sheet and Settlement Agreement
On May 3, 2017, the Company entered the Term Sheet by and among the Parties in order to resolve certain legal actions among the Parties, as described below. The Parties also agreed to cooperate and use their best efforts to reduce the Term Sheet to a definitive Settlement Agreement and, to the extent necessary, obtain the approval of the Court of Chancery.
Resolution of Litigation
Pursuant to the Term Sheet, the Parties submitted a stipulation and proposed order to the Court of Chancery lifting the Status Quo Order seating all four venBio nominees (with the three incumbent directors who also won election (based on the plurality vote standard), the “Status Quo Board”) and confirming that the Status Quo Board is the lawful Board of the Company (provided however, if the 225 Action is not dismissed, the Parties will be restored to their positions in the 225 Action as of immediately prior to the execution of the Term Sheet). The Court of Chancery entered the proposed order on the afternoon of May 4, 2017. Pursuant to the Term Sheet, the Parties also agreed to submit a stipulation and proposed order to the Court of Chancery staying the venBio Action (as described below) and removing the trial dates from the calendar of the Court of Chancery.
The Company has further agreed to reimburse venBio for reasonable fees and expenses it incurred in connection with the proxy contest between venBio and the Company, the venBio Action, the 225 Action (as described below) and the Federal Action (as described below and, together with the venBio Action and the 225 Action, the “Actions”), and Goldenberg and Sullivan have agreed to not object to such reimbursement.
The Parties have agreed, immediately upon execution of the Settlement Agreement, to submit stipulations and proposed orders dismissing with prejudice both the 225 Action and the Federal Action. The Settlement Agreement will include (i) a mutual release of all claims that were or could have been asserted in the Federal Action or in the 225 Action and (ii) a release of all direct and derivative claims that have been or could be asserted by or on behalf of (a) venBio or the Company, whether known or unknown, against Goldenberg, Sullivan and Markison and their affiliates and related persons, and (b) Goldenberg, Sullivan or Markison, whether known or unknown, against venBio or the Company and their affiliates and related persons, in both cases in connection with the claims alleged in the venBio Action, the Financing, the settlement of the venBio Action, the Licensing Transaction and the Termination Agreement. The settlement of claims against Goldenberg, Sullivan and Markison in the venBio Action will be subject to approval of the Court of Chancery. venBio and the Company have agreed to stay the venBio Action and submit the claims asserted against the remaining individual defendants (former directors Robert Forrester, Jason Aryeh, Geoff Cox and Bob Oliver) to non-binding mediation. As part of the Termination Agreement, which is subject to the approval of the Court of Chancery, venBio will release SGEN from any claims in the venBio Action.
42
Financing and Termination of SGEN Transaction
On May 4, 2017 the Board of Directors, including, Goldenberg and Sullivan (i) voted to support the Financing, (ii) voted to terminate the Licensing Transaction with SGEN, as discussed above, pursuant to the terms of the Termination Agreement entered into with SGEN, (iii) voted to approve an amendment to the Company’s Amended and Restated Certificate of Incorporation, as amended, to increase the number of shares of authorized Common Stock by an aggregate number of shares of Common Stock to enable conversion of all of the Preferred Shares into shares of Common Stock (the “Charter Amendment”), (iv) approved the submission of a stipulation in the 225 Action to permit the Board to consummate and enter into both the Financing and the Termination Agreement Pursuant to the Term Sheet, Goldenberg and Sullivan agreed to not sell any shares of the Company (with certain exceptions) until the earlier of July 31, 2017 or the date on which the Charter Amendment is approved and the shares of Common Stock issuable upon conversion of the Preferred Shares were registered and issued.
Indemnification
The Term Sheet provides that the Company will, to the extent not covered by the Company’s insurance policies, (i) indemnify Dr. Goldenberg, Ms. Sullivan and Mr. Markison from attorneys’ fees and expenses or other losses in connection with the Actions, and (ii) reimburse and indemnify Dr. Goldenberg and Ms. Sullivan for legal fees for actions taken with respect to the Actions and negotiation of the Settlement Agreement. The Term Sheet provides that the indemnification agreements entered into between the Company and each of Dr. Goldenberg, Ms. Sullivan and Mr. Markison on or about February 9, 2017 shall be terminated and not apply to acts, transactions, legal fees or expenses incurred after approval of the Settlement Agreement by the Court of Chancery.
Intellectual Property Assignments
In accordance with the Term Sheet the Settlement Agreement shall provide that Dr. Goldenberg and Ms. Sullivan will assign all global intellectual property and any rights thereto, other than those subject to existing agreements with the Company and Dr. Goldenberg’s patent and related intellectual property relating to cyber space medicine, to the Company, and perform all acts reasonably requested by the Company to perfect title in and to all such assigned intellectual property.
Sullivan Resignation
Effective July 1, 2014, the Company entered into the Fifth Amended and Restated Employment Agreement with Cynthia L. Sullivan pertaining to Ms. Sullivan’s service to the Company as the Company’s President and Chief Executive Officer. This agreement was terminated effective July 1, 2017.
Upon execution of the contemplated Settlement Agreement, Ms. Sullivan has agreed to resign from director of the Company and any of its affiliates, effective as of the date of the Settlement Agreement. The Settlement Agreement will provide that Ms. Sullivan will abide by all post-termination covenants and obligations contemplated by the Amended Sullivan Agreement, (defined below). In exchange for a release of claims as required by the Amended Sullivan Agreement and subject to compliance with the terms of the Settlement Agreement, Sullivan will be entitled to (i) termination payments in accordance with the Amended Sullivan Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of the exercise period for equity awards already earned, pursuant to the Amended Sullivan Agreement, and (iii) COBRA payments. The Company and Sullivan disagree over the precise amount owed to Sullivan under the Amended Sullivan Agreement. The foregoing cash payments accumulate to approximately $3.1 million (with additional amounts in dispute).
Goldenberg Resignation
Upon execution of the Settlement Agreement, Dr. Goldenberg will remain a director of the Company, but has agreed to resign from all officer and other positions of the Company and all director, officer and other positions at any of the Company’s affiliates (other than Dr. Goldenberg’s position as a member of the board of directors of IBC Pharmaceuticals, the Company’s majority owned U.S. subsidiary), effective as of the date of the Settlement Agreement.
43
The Settlement Agreement will provide that Dr. Goldenberg will abide by all post-termination covenants and obligations contemplated by the Amended and Restated Goldenberg Agreement, (defined below). In exchange for a release of claims as required by the Amended and Restated Goldenberg Agreement and subject to compliance with the terms of the Settlement Agreement, Dr. Goldenberg will be entitled to (i) termination payments in accordance with the Amended and Restated Goldenberg Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of exercise period for equity awards already earned, pursuant to the Amended and Restated Goldenberg Agreement, (iii) COBRA payments, (iv) royalties and payments in accordance with existing agreements. The Company and Goldenberg disagree over the precise amounts owed to Goldenberg under the Amended and Restated Goldenberg Agreement. The foregoing cash payments accumulate to no more than $2.4 million (with additional amounts in dispute). The Company and Goldenberg also dispute the vesting of 1,500,000 Restricted Stock Units granted to Dr. Goldenberg under the terms of the Amended and Restated Goldenberg Agreement.
Arbitration of Disputed Matters
The Parties have agreed to arbitrate disputes relating to Dr. Goldenberg’s claimed entitlement to severance payments, and Dr. Goldenberg’s and Ms. Sullivan’s claimed rights to certain bonus and equity payments, to the extent the Parties cannot reach agreement on such issues before execution of the Settlement Agreement. The Company has agreed to pay in full the arbitrator in such arbitration as well as reasonable attorneys’ fees and expenses incurred by Dr. Goldenberg and/or Ms. Sullivan in connection with any such arbitration, up to a maximum amount of $650,000. As of June 30, 2017 no expenses have been incurred regarding such arbitration.
Directors and Officers Liability Insurance
The Company has filed claims with its insurance providers for various expenses incurred through June 30, 2017 for proxy defense-related expenses and reimbursement amounts payable to venBio for fees and expenses incurred by venBio in connection with the proxy contest between venBio and the Company.
Other matters:
Immunomedics is also a party to various claims and litigation arising in the normal course of business, which includes some or all of certain of its patents. While it is not possible to determine the outcome of these matters, the Company believes that the resolution of all such matters will not have a material adverse effect on its consolidated financial position or liquidity, but could possibly be material to its consolidated results of operations in any one accounting period.
Item 4. Mine Safety Disclosures
Not applicable.
44
PART II
Item 5. Market For Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Price and Dividend Information
Our common stock is quoted on the NASDAQ Global Market under the symbol “IMMU.” The following table sets forth the high and low sales prices for our common stock for each full quarterly period within the last two fiscal years, as reported by the NASDAQ Global Market:
|
Fiscal Quarter Ended |
|
High |
|
Low |
|
||
|
|
|
|
|
|
|
|
|
|
September 30, 2015 |
|
$ |
4.32 |
|
$ |
1.50 |
|
|
December 31, 2015 |
|
|
3.40 |
|
|
1.59 |
|
|
March 31, 2016 |
|
|
3.02 |
|
|
1.61 |
|
|
June 30, 2016 |
|
|
5.44 |
|
|
1.95 |
|
|
|
|
|
|
|
|
|
|
|
September 30, 2016 |
|
$ |
3.43 |
|
$ |
2.09 |
|
|
December 31, 2016 |
|
|
4.10 |
|
|
2.02 |
|
|
March 31, 2017 |
|
|
7.15 |
|
|
3.30 |
|
|
June 30, 2017 |
|
|
9.04 |
|
|
5.00 |
|
As of August 15, 2017, the closing sales price of our common stock on the NASDAQ Global Market was $8.04. As of August 15, 2017, there were approximately 364 stockholders of record of our common stock and, according to our estimates, approximately 20,535 beneficial owners of our common stock. We have not paid dividends on our common stock since inception and do not plan to pay cash dividends in the foreseeable future.
STOCK PERFORMANCE GRAPH
This graph is not “soliciting material,” and is not deemed filed with the SEC and not to be incorporated by reference in any filing by our Company under the Securities Act of 1933, as amended, or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing. The total return values data is prepared by the NASDAQ OMX Global Index Group. Total Return Indexes are posted on NASDAQ Online on a monthly basis.
45
The following graph compares the yearly change in cumulative total stockholder return on the Company’s common stock for the prior five fiscal years with the total cumulative return of the NASDAQ Composite Index and the NASDAQ Pharmaceutical Index. The returns are indexed to a value of $100 at June 30, 2012.
|
Company/Index |
|
Indexed Returns (years ending) |
|
||||||||||
|
|
|
6/30/12 |
|
6/30/13 |
|
6/30/14 |
|
6/30/15 |
|
6/30/16 |
|
6/30/17 |
|
|
Immunomedics |
|
100 |
|
153 |
|
103 |
|
114 |
|
65 |
|
248 |
|
|
NASDAQ Composite |
|
100 |
|
121 |
|
152 |
|
163 |
|
167 |
|
198 |
|
|
NASDAQ Pharmaceutical |
|
100 |
|
123 |
|
158 |
|
185 |
|
187 |
|
202 |
|
46
Item 6. Selected Financial Data
The following table sets forth our consolidated financial data as of and for each of the five fiscal years ended June 30, 2017. The following table sets forth our consolidated financial data as of and for each of the five fiscal years ended June 30, 2017 which has been derived from our audited consolidated financial statements. The audited consolidated financial statements as of June 30, 2017 and 2016 and for the three-year period ended June 30, 2017 are included elsewhere in this Annual Report on Form 10-K. The information below should be read in conjunction with the consolidated financial statements (and notes thereon) and Item 7, Management’s Discussion and Analysis of Financial Condition and Results of Operations.
|
|
|
Fiscal year ended June 30, |
|
|||||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|
|||||
|
|
|
(In thousands, except per share amounts) |
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
3,091 |
|
$ |
3,233 |
|
$ |
5,653 |
|
$ |
9,042 |
|
$ |
4,962 |
|
|
Costs and expenses |
|
|
82,241 |
|
|
62,241 |
|
|
51,873 |
|
|
44,622 |
|
|
35,754 |
|
|
Changes in fair market value of warrant liabilities |
|
|
(61,074) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Warrant related expenses |
|
|
(7,649) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Interest expense (1) |
|
|
(5,480) |
|
|
(5,480) |
|
|
(2,091) |
|
|
— |
|
|
— |
|
|
Interest and other income, net |
|
|
431 |
|
|
338 |
|
|
246 |
|
|
56 |
|
|
10 |
|
|
Other financing expenses |
|
|
(347) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Foreign currency transaction gain, (loss) net |
|
|
24 |
|
|
(40) |
|
|
(1) |
|
|
1 |
|
|
(37) |
|
|
Arbitration settlement, net |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
16,739 |
|
|
Insurance proceeds received |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
2,638 |
|
|
Loss before income tax |
|
|
(153,245) |
|
|
(64,190) |
|
|
(48,066) |
|
|
(35,523) |
|
|
(11,442) |
|
|
Income tax (expense) benefit |
|
|
(21) |
|
|
5,054 |
|
|
(58) |
|
|
(8) |
|
|
(44) |
|
|
Net loss |
|
|
(153,266) |
|
|
(59,136) |
|
|
(48,124) |
|
|
(35,531) |
|
|
(11,486) |
|
|
Less: Net loss attributable to noncontrolling interest |
|
|
(60) |
|
|
(99) |
|
|
(122) |
|
|
(105) |
|
|
(105) |
|
|
Net loss attributable to Immunomedics, Inc stockholders |
|
$ |
(153,206) |
|
$ |
(59,037) |
|
$ |
(48,002) |
|
$ |
(35,426) |
|
$ |
(11,381) |
|
|
Loss per common share attributable to Immunomedics, Inc stockholders (basic and diluted) |
|
$ |
(1.47) |
|
$ |
(0.62) |
|
$ |
(0.51) |
|
$ |
(0.42) |
|
$ |
(0.15) |
|
|
Weighted average shares outstanding used to calculate loss per common share (basic and diluted) |
|
|
104,536 |
|
|
94,770 |
|
|
93,315 |
|
|
84,632 |
|
|
78,040 |
|
|
|
|
As of June 30, |
|
|||||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
2014 |
|
2013 |
|
|||||
|
|
|
|
|
|
(In thousands) |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cash, cash equivalents and marketable securities |
|
$ |
154,902 |
|
$ |
50,628 |
|
$ |
99,618 |
|
$ |
41,833 |
|
$ |
41,326 |
|
|
Total assets |
|
|
162,573 |
|
|
56,950 |
|
|
105,780 |
|
|
47,486 |
|
|
47,927 |
|
|
Convertible senior notes, net of unamortized debt issuance costs |
|
|
98,084 |
|
|
97,354 |
|
|
96,625 |
|
|
— |
|
|
— |
|
|
Warrant liabilities |
|
|
90,706 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
Stockholders’ equity (deficit) (2) |
|
$ |
(59,463) |
|
$ |
(57,527) |
|
$ |
(4,525) |
|
$ |
38,859 |
|
$ |
39,795 |
|
|
(1) |
Interest expense represents the Convertible Senior Notes interest expense ($4.8 million, $4.8 million and $1.8 million for 2017, 2016 and 2015, respectively) and amortized debt issuance costs ($0.7 million, $0.7 million and $0.3 million for 2017, 2016 and 2015, respectively). |
|
(2) |
We have never paid cash dividends on our common stock. Stockholders’ equity (deficit) equity represents Immunomedics, Inc. stockholders equity and the non-controlling interest in subsidiary. |
47
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The SEC encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. This Annual Report on Form 10-K contains such “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. These statements may be made directly in this Annual Report, and they may also be made a part of this Annual Report on Form 10-K by reference to other documents filed with the SEC, which is known as “incorporation by reference”.
Words such as “may,” “anticipate,” “estimate,” “expects,” “projects,” “intends,” “plans,” “believes” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, are intended to identify forward-looking statements. All forward-looking statements are management’s present expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially from those described in the forward-looking statements. These risks and uncertainties include, among other things: our inability to further identify, develop and achieve commercial success for new products and technologies; the possibility of delays in the research and development necessary to select drug development candidates and delays in clinical trials; the risk that clinical trials may not result in marketable products; the risk that we may be unable to obtain additional capital through strategic collaborations, licensing, issuance of convertible debt securities or equity financing in order to continue our research and development programs as well as secure regulatory approval of and market our drug candidates; our dependence upon pharmaceutical and biotechnology collaborations; the levels and timing of payments under our collaborative agreements; uncertainties about our ability to obtain new corporate collaborations and acquire new technologies on satisfactory terms, if at all; the development of competing products; our ability to protect our proprietary technologies; patent-infringement claims; and risks of new, changing and competitive technologies and regulations in the United States and internationally. Please also see the discussion of risks and uncertainties under Item 1A. Risk Factors “Factors That May Affect Our Business and Results of Operations” in this Annual Report on Form 10-K.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this Annual Report on Form 10-K or in any document incorporated by reference might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this Annual Report on Form 10-K or the date of the document incorporated by reference in this Annual Report on Form 10-K, as applicable. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise except as may be required by applicable law. All subsequent forward-looking statements attributable to the Company or to any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
Overview
We are a clinical-stage biopharmaceutical company developing monoclonal antibody-based products for the targeted treatment of cancer, autoimmune and other serious diseases. We believe that each of our antibodies has therapeutic potential either when administered as a naked antibody or when conjugated with chemotherapeutics, therapeutic radioisotopes (radiolabeled), cytokines or other toxins to create unique and potentially more effective treatment options. The attachment of various compounds to antibodies is intended to allow the delivery of these therapeutic agents to tumor sites with better specificity than conventional chemotherapy or radiation therapy approaches. This treatment method is designed to reduce the total exposure of the patient to the therapeutic agents, which ideally minimizes debilitating side effects. We are currently prioritizing our efforts on antibodies conjugated with drugs.
Our most advanced product candidate is IMMU-132 (sacituzumab govitecan), an ADC that has received BTD from the FDA for the treatment of patients with mTNBC who have failed at least two prior therapies for metastatic disease. Following the election of a new Board of Directors at our Annual Meeting of Stockholders for 2016, we embarked on a new corporate strategy focused on bringing IMMU-132 to the market on our own for the benefit of patients with mTNBC and the creation of value for our stockholders. To that end, we plan to submit a BLA to the FDA for accelerated approval of IMMU-132 in mTNBC between December 2017 and March 2018. In addition to IMMU-132, our first-in-class ADC programs also include IMMU-130 (labetuzumab govitecan), which binds the CEACAM5 antigen
48
expressed on colorectal and other solid cancers, and IMMU-140 that targets HLA-DR for the potential treatment of liquid cancers. Additionally, our pipeline contains other potential antibody-based product candidates for the treatment of cancer and autoimmune diseases including epratuzumab, our anti-CD22 antibody; veltuzumab, our anti-CD20 antibody; milatuzumab, our anti-CD74 antibody; and IMMU-114, a humanized anti-HLA-DR antibody.
We transitioned away from the development and commercialization of new diagnostic imaging products in order to accelerate the development of our therapeutic product candidates, although we continue to manufacture and sell, distribute and support LeukoScan® (sulesomab) in territories where regulatory approvals have previously been granted. LeukoScan® is indicated for diagnostic imaging for determining the location and extent of infection/inflammation in bone in patients with suspected osteomyelitis, including patients with diabetic foot ulcers.
From our inception in 1982 through June 30, 2017, we had an accumulated deficit of approximately $521.7 million. In the absence of increased revenues from the sale of current or future products and licensing activities (the amount, timing, nature or source of which cannot be predicted), our losses will continue as we conduct our research, development, and commercialization activities. These activities are budgeted to expand over time and will require further resources if we are to be successful; especially costs in connection with the final development, and commercialization of IMMU-132. As a result, our operating losses are likely to be substantial over the next several years.
The development and commercialization of successful therapeutic products is subject to numerous risks and uncertainties including, without limitation, the following:
|
· |
the type of therapeutic compound under investigation and nature of the disease in connection with which the compound is being studied; |
|
· |
our ability, as well as the ability of our partners, to conduct and complete clinical trials on a timely basis; |
|
· |
the time required for us to comply with all applicable federal, state and foreign legal requirements, including, without limitation, our receipt of the necessary approvals of the FDA; |
|
· |
many other factors associated with the commercial development of therapeutic products outside of our control. (See Risk Factors under Item 1A in this Annual Report on Form 10-K for other factors.) |
Critical Accounting Policies and Accounting Estimates
A critical accounting policy is one that is both important to the portrayal of our financial condition and results of operation and requires management's most difficult, subjective or complex judgments, often as a result of the need to make estimates about the effect of matters that are inherently uncertain.
For a description of our significant accounting policies, see Notes to Consolidated Financial Statements – Note 2 Summary of Significant Accounting Policies. Of these policies, the following are considered critical to an understanding of the Company’s Consolidated Financial Statements as they require the application of the most difficult, subjective and complex judgments; (i) Revenue recognition, (ii) Research and development costs, (iii) Common stock warrants and (iv) Stock-based compensation.
The Company’s critical accounting estimates and assumptions impacting the consolidated financial statements relate to stock compensation expenses and determination of fair value of warrants. See Note 2 Summary of Significant Accounting Policies and Note 7 Estimated Fair Value of Financial Instruments, respectively, for the basis of related assumptions.
49
Results of Operations
Fiscal Year 2017 compared to Fiscal Year 2016
Revenues
Total revenue for the fiscal year ended June 30, 2017 was $3.1 million, compared to $3.2 million for the fiscal year ended June 30, 2016, a decrease of $0.1 million, or approximately 3.0%. The decrease was due primarily to a $0.2 million decrease in grant revenue offset partially a $0.1 million increase in LeukoScan® sales.
Costs and Expenses
Total costs and expenses were $82.2 million for the fiscal year ended June 30, 2017, an increase of $20.0 million, or approximately 32% compared to the same period last year. The increase was due primarily to a $22.5 million increase in general and administrative costs during fiscal 2017 including a $9.0 million increase in legal and advisory fees associated with the proxy contest and professional services in connection with the Licensing Agreement with Seattle Genetics (which was subsequently terminated), $6.9 million for executive severance, $4.5 million to reimburse venBio Select LLC for proxy related costs, $2.0 million for consulting services for strategic planning, and a $1.8 million increase in legal fees, offset partially by the elimination of $1.7 million deferred unearned executive bonuses from fiscal 2016 and 2015.
Research and development expenses were $51.5 million for the fiscal year ended June 30, 2017, a $1.7 million reduction, or approximately 3% compared to the same period in 2016; due primarily to a $11.4 million reduction in clinical trial costs resulting from the closure of the Phase 3 PANCRIT-1 clinical trial in 2016, offset partially by a $9.7 million increase in product development expense for IMMU-132 manufacturing.
Cost of goods sold for the LeukoScan® product was $0.5 million for the fiscal year ended June 30, 2017, a $0.7 million reduction, or approximately 58%, compared to the same period last year. The reduction was due primarily to a $0.6 million increase in fiscal 2016 as a result of the write-down of certain work-in-process inventory of LeukoScan® which was deemed to be unsaleable.
Sales and marketing expenses decreased from $1.0 million for the 2016 fiscal year to $0.9 million for the 2017 fiscal year.
Change in Fair Value of Warrant Liabilities
The Company recognized a $61.1 million non-cash expense during fiscal year ended June 30, 2017, arising from the $47.6 increase in warrant liability from the increase in the fair value of the public offering warrants issued in October 11, 2016, and a $13.5 million increase in warrant liability from the increase in the fair market value of the warrant issued to Seattle Genetics on February 10, 2017 (the “SGEN Warrant”), resulting from the increase in the share price of our common stock from the date of inception of each warrant through June 30, 2017. There was no warrant liability in fiscal 2016.
Warrant-Related Expense
The Company recognized a $7.6 million non-cash warrant-related expense during the year ended June 30, 2017 representing the excess of fair value of the SGEN Warrant issued on February 10, 2017 over proceeds received for the issuance of common stock and such Warrant. There was no warrant-related expense in fiscal 2016.
Interest Expense
Interest expense related to the 4.75% Convertible Senior Notes due 2020 was $5.5 million for both fiscal years ended June 30, 2017 and 2016, including the amortization of $0.7 million debt issuance costs.
Other Financing Expense
Other financing expense of $0.3 million for the fiscal year ended June 30, 2017 relates to expenses incurred in connection with the public offering we consummated on October 11, 2016 that were attributable to the warrant liability.
50
Income Tax (Expense) Benefit
The Company did not receive an income tax benefit for the year ended June 30, 2017, compared to a $5.1 million income tax benefit received in the year ended June 30, 2016. The income tax benefit received in fiscal 2016 was from the sale of a portion of our New Jersey State Tax NOLs and R&D tax credits. The Company did not receive an income tax benefit during the year ended June 30, 2017 because it had reached the maximum amount permissible under the New Jersey Business Tax Certificate Transfer Program for the sale of NOL’s. Income tax expense in 2017 related to net income in foreign operations. In fiscal 2017, foreign operations had net income, and associated income tax expense of $19 thousand. There was no federal income tax expense for domestic operations in either period due to losses in both fiscal years.
Net Loss Attributable to Immunomedics, Inc. Stockholders
Net loss attributable to Immunomedics, Inc., common stockholders was $153.2 million, or approximately $1.47 per share for fiscal year 2017, compared to net loss of $59.0 million, or approximately $0.62 per share, for fiscal year 2016, an increase of $94.2 million, or approximately 160%. The increase was due primarily to the $61.1 million increase in non-cash expense from the increase in fair value of warrant liabilities, the $22.5 million increase in general and administrative expenses, the $7.6 million increase in non-cash expense in excess of fair value of the SGEN Warrant, and the receipt of $5.1 million proceeds from the sale of non-recurring tax credits in 2016, offset partially by the $1.7 million decrease in research and development expenses.
Fiscal Year 2016 compared to Fiscal Year 2015
Revenues
Total revenues for the fiscal year ended June 30, 2016 were $3.2 million, compared to $5.7 million for the fiscal year ended June 30, 2015, a decrease of $2.5 million, or approximately 44%. The decrease was due primarily to $0.6 million research and development revenues of for the year ended June 30, 2016, a $1.2 million decrease, or approximately 67%, from the same period in 2015. The decrease was due primarily to a decrease in the number of grant programs and level of activity during the current year. License fee and other revenues were $0.4 million for the 2016 fiscal year, a $0.9 million decrease, or approximately 69%, from fiscal 2015. The decrease was due primarily to a $1.0 million clinical milestone payment related to collaboration agreement with Bayer (the Bayer Collaboration Agreement), which was received in fiscal year 2015. There was no similar milestone in fiscal year 2016. Product sales of LeukoScan® in Europe for the year ended June 30, 2016 were $2.3 million, a $0.3 million decrease, or approximately 12%, compared to $2.6 million for the year ended June 30, 2015. The decrease was due primarily to unfavorable fluctuations in the currency rates in Europe and sales volume decline of LeukoScan® in Europe.
Costs and Expenses
Total costs and expenses were $62.2 million for the fiscal year ended June 30, 2016, compared to $51.9 million in the fiscal year ended June 30, 2015, a $10.3 million increase, or approximately 20%. The increase was due primarily to a $12.5 million higher research and development expenses for fiscal 2016, or approximately 28%, to $53.5 million primarily from the increased clinical trial expenses and manufacturing costs for the antibody-drug conjugates’ clinical trials and the Phase 3 clinical trial of the clivatuzumab tetraxetan for the treatment of patients with pancreatic cancer.
Cost of goods sold for the LuekoScan® product was $1.2 million in fiscal year ended June 30, 2016, compared to $0.3 million in fiscal year ended June 30, 2015, a $0.9 million increase, or approximately 300%. During the 2016 fiscal year, cost of goods sold included a $0.3 million write down relating to LeukoScan® finished product inventories that were deemed to be unsaleable due to an excess of the finished product over anticipated sales forecasted through its effective shelf-life. In addition, during the year ended June 30, 2016, cost of goods sold increased $0.6 million as a result of the inventory reserve on certain of LeukoScan® work-in-process inventories which were deemed to be unsaleable due to a manufacturing process deviation that resulted in product that did not meet our quality control standards. Gross profit margins were 49% and 90% for fiscal years 2016 and 2015, respectively.
51
Sales and marketing expenses increased $0.2 million, or approximately 25%, $1.0 million for the 2016 fiscal year. The increase of was due primarily to employee related severance costs and the relocation of the Immunomedics GmbH offices. General and administrative expenses decreased $2.5 million for fiscal year 2016, or approximately 27%, to $6.6 million in fiscal year 2016. The decrease is due primarily to a $2.6 million lower legal and professional fees, principally from the conclusion of the arbitration proceedings during the 2015 fiscal year with Takeda-Nycomed, a former licensing partner.
Interest Expense
Interest expense related to the 4.75% Convertible Senior Notes issued in February 2015 was $5.5 million and $2.1 million, including the amortization of $0.7 million, and $0.3 million debt issuance costs, for the years ended June 30, 2016, and 2015, respectively.
Income Tax Benefit (Expense)
The Company realized a $5.1 million income tax benefit for the year ended June 30, 2016, compared to a $58 thousand income tax expense for in fiscal 2015. The income tax benefit in fiscal 2016 was due primarily to the sale of a portion of our New Jersey State Tax NOLs and R&D tax credits through the New Jersey Business Tax Certificate Transfer Program. There were no NOLs or R&D tax credits sold during fiscal 2015. Income tax expense in fiscal 2015 related to net income in foreign operations. In fiscal 2016 the foreign operations had a net loss, with no income tax expense. There was no federal income tax expense for both periods for domestic operations due to losses in both fiscal years.
Net Loss Attributable to Immunomedics, Inc. Stockholders
Net loss attributable to Immunomedics, Inc., common stockholders for fiscal year 2016 was $59.1 million, or $0.62 per share, compared to net loss of $48.0 million, or $0.51 per share, in fiscal year 2015, a $11.1 million increase, or approximately 23%. The increase was due primarily to increased research and development costs related to clinical trials and the associated manufacturing costs for the antibody-drug conjugates’ clinical trials, interest expense for the convertible senior notes for the full fiscal year, and decreased revenues related to the Bayer Collaboration Agreement, partially offset by the income tax benefit and reduced legal and professional fees.
Research and Development Expenses
Research and development expenses were $51.8 million for fiscal year ended June 30, 2017, $53.5 million for fiscal year ended June 30, 2016, and $41.7 million for fiscal year ended June 30, 2015. Research and development expenses decreased $1.7 million in fiscal year 2017, or 3%, compared to 2016. Research and development expenses increased by $11.8 million in fiscal year 2016, or 28%, compared to fiscal year 2015.
We do not track expenses on the basis of each individual compound under investigation and therefore we do not provide a breakdown of such historical information in that format. We evaluate projects under development from an operational perspective, including such factors as results of individual compounds from laboratory/animal testing, patient results and enrollment statistics in clinical trials. It is important to note that multiple product candidates are often tested simultaneously. It is not possible to calculate each antibody’s supply costs. There are many different development processes and test methods that examine multiple product candidates at the same time. We have, historically, tracked our costs in the categories discussed below, specifically “research costs” and “product development costs” and by the types of costs outlined below.
Our research costs consist of outside costs associated with animal studies and costs associated with research and testing of our product candidates prior to reaching the clinical stage. Such research costs primarily include personnel costs, facilities, including depreciation, lab supplies, funding of outside contracted research and license fees. Our product development costs consist of costs from preclinical development (including manufacturing), conducting and administering clinical trials and patent expenses.
52
The following table sets forth a breakdown of our research and development expenses by those associated with research and those associated with product development for the periods indicated.
|
|
|
Years Ended June 30, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
|
|
(in thousands) |
|
|||||||
|
Research Costs |
|
$ |
4,524 |
|
$ |
5,137 |
|
$ |
5,959 |
|
|
Product Development Costs |
|
|
47,252 |
|
|
48,355 |
|
|
35,777 |
|
|
Total |
|
$ |
51,776 |
|
$ |
53,492 |
|
$ |
41,736 |
|
Research Costs
Research costs decreased by $0.6 million, or 11.9%, for the year ended June 30, 2017 compared to June 30, 2016. Research costs decreased by $0.8 million, or 14%, for the year ended June 30, 2016 compared to June 30, 2015.
The $0.6 million reduction in research costs during 2017 compared to the same period in fiscal 2016 relate primarily to a $0.6 million reduction , or 23% in personnel costs, due primarily to the redeployment of employees from basic research to product development in fiscal 2017; and a $0.4 million, or 57% reduction in lab supplies and chemical reagents in fiscal 2017 compared to the same period in fiscal 2016, offset partially by a $0.4 million in pre-clinical toxicity studies costs for IMMU-132 development in fiscal 2017, which had not been undertaken in fiscal 2016.
The $0.8 million decrease in research costs in fiscal 2016 resulted from a $0.3 million reduction or 75%, in outside research and testing services from fiscal 2015, due to pre-clinical toxicity studies conducted in fiscal 2015 ($0.3 million) that were not undertaken in fiscal 2016. In addition, employee costs were $0.2 million lower in fiscal 2016 compared to fiscal 2015.
Indirect administrative and support services that are allocated to research based on research spending levels were approximately $0.5 million in 2017 and 2016 and $0.7 million in 2015.
Product Development Costs
Product development costs were $47.3 million for the year ended June 30, 2017, a decrease of $1.1 million, or 2.3%, compared to 2016, which increased $12.6 million, or 35%, to $48.4 million from $35.8 million in 2015.
Clinical trial expenses in fiscal year 2017 were $4.5 million compared to $14.7 million in fiscal year 2016, a reduction of $10.2 million or 69%, driven primarily from the closure of the Phase 3 PANCRIT-1 clinical trial in 2016. Clinical trial expenses in fiscal year 2016 were $14.7 million compared to $12.6 million in fiscal year 2015, a $2.1 million increase due primarily to increased activity in the clivatuzumab tetraxetan Phase 3 clinical trial and additional antibody-drug conjugates’ clinical trials in fiscal 2016.
Personnel costs in fiscal 2017 were $9.2 million compared to $8.6 million in 2016, an increase of $0.6 million, or 7%, due primarily to increased focus on product development efforts and salary and benefit increases. Personnel costs increased $1.0 million, or 13% in 2016, compared to 2015, due primarily to salary and benefit increases.
Lab supplies and chemical reagent costs were $5.4 million for fiscal 2017, compared to $6.6 million in fiscal 2016, a reduction of $1.2 million or 18%. The reduction was primarily a result of increased utilization of outside manufacturers for certain materials used for the antibody-drug conjugates’ clinical trial related expenses. Lab supplies and chemical reagent costs of $3.3 million or 100% over the previous fiscal year. The increase was primarily a result of increased manufacturing costs for material used for the antibody-drug conjugates’ clinical trial related expenses.
We utilized $13.4 million in services of outside manufacturers’ organizations services in fiscal 2017, an increase of $8.6 million or 179% over fiscal 2016 as we have ramped-up manufacturing of IMMU-132 for the Phase 3 clinical trial antibody-drug conjugates. There were no similar expenses in fiscal 2015 fiscal year.
For fiscal 2017 we utilized outside consulting services of $1.1 million to improve our manufacturing and regulatory functions associated with fulfilling the FDA requirements for the Phase 3 clinical trials of sacituzumab
53
govitecan in patients with metastatic triple negative breast cancer (“mTNBC”) and its eventual commercialization. Outside consultants were not utilized in fiscal 2016.
Depreciation expense for fiscal 2017 was $0.5 million, an increase of $0.2 million or 67% over fiscal 2016, a result of the increase in capital expenditures during fiscal 2017 and the latter half of fiscal 2016. Depreciation expense for fiscal 2016 and fiscal 2015 were $0.3 million.
Expenses for outside testing were $3.0 million in fiscal 2017, a reduction of $0.5 million or 14%, from fiscal 2016. The reduction was due to reduced material testing for process validation and offsite lyophilization relating to product development for manufacturing of material used for the antibody-drug conjugates. Expenses for outside testing were $3.5 million in fiscal 2016, an increase of $1.1 million or 46%, from fiscal 2015. The increase was due to increased material testing for process validation and offsite lyophilization relating to product development for manufacturing of material used for the antibody-drug conjugates.
Indirect administrative and support services that are allocated to development based on spending levels were approximately $4.8 million for fiscal 2017 and fiscal 2016. For fiscal 2016 indirect administrative and support services that are allocated to development based on spending levels increased by $0.6 million, or 14%, to $4.8 million due primarily to a greater emphasis on spending in the product development area as compared to the research area.
Completion of clinical trials may take several years or more. The length of time varies according to the type, complexity and the disease indication of the product candidate. We estimate that clinical trials of the type we generally conduct are typically completed over the following periods:
|
|
|
Estimated Completion Period |
|
|
Clinical Phase |
|
(Years) |
|
|
I |
|
0-1 |
|
|
II |
|
1-2 |
|
|
III |
|
1-4 |
|
The duration and cost of clinical trials through each of the clinical phases may vary significantly over the life of a particular project as a result of, among other things, the following factors:
|
· |
the length of time required to recruit qualified patients for clinical trials; |
|
· |
the duration of patient follow-up in light of trial results; |
|
· |
the number of clinical sites required for trials; and |
|
· |
the number of patients that ultimately participate. |
Liquidity and Capital Resources
Since its inception in 1982, Immunomedics’ principal sources of funds have been the private and public sale of equity and debt securities, and revenues from licensing agreements, including up-front and milestone payments, funding of development programs, and other forms of funding from collaborations.
As of June 30, 2017 we have $154.9 million in cash, cash equivalents and marketable securities; which we believe is sufficient to support operations through September 2018, and which allows us to accomplish the goal of filing a Biologics License Applications (“BLA”) for Accelerated Approval of IMMU-132 in mTNBC from the U.S. Food and Drug Administration (the “FDA”) during the period between December 2017 and March 2018, subject to FDA input on the acceptance of the Company’s chemistry, manufacturing and controls filing plan.
The Company will require additional funding after September 2018 to secure regulatory approval from the FDA, complete commercial preparations to market IMMU-132 to mTNBC patients in the United States, complete its clinical trials currently underway or planned, continue research and new development programs, and continue
54
operations. Potential sources of funding include the exercise of outstanding warrants, potential various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, and equity and potential debt financing.
Until the Company can generate significant cash through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, or commercial operations, it expects to continue to fund its operations with its current financial resources. These financial resources are adequate to sustain the Company’s operations at a level of activity sufficient to support the filing of the BLA with the FDA for accelerated approval of IMMU-132 for patients with mTNBC; to continue manufacturing IMMU-132 at large scale to prepare for commercial operations in the U.S. marketplace; to initiate a Phase 3 clinical trial of IMMU-132 for mTNBC patients to support the filing of the BLA, to initiate preparations to market IMMU-132 to mTNBC patients in the U.S. and, subject to meeting all standards, completing review and final determination of the FDA, to secure accelerated regulatory approval to market IMMU-132 for the use of patients with mTNBC in the U.S. After September 2018, if the Company cannot obtain sufficient funding through the exercise of outstanding Warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC and beyond, it could be required to finance future cash needs through the sale of additional equity and/or debt securities in capital markets. However, there can be no assurance that the Company will be able to raise the additional capital needed to complete its pipeline of research and development programs on commercially acceptable terms, if at all. The capital markets have experienced volatility in recent years, which has resulted in uncertainty with respect to availability of capital and hence the timing to meet an entity’s liquidity needs. The Company’s existing debt may also negatively impact the Company’s ability to raise additional capital. If the Company is unable to raise capital on acceptable terms, its ability to continue its business would be materially and adversely affected.
Actual results could differ materially from our expectations as a result of a number of risks and uncertainties, including the risks described in Item 1A Risk Factors, “Factors That May Affect Our Business and Results of Operations,” and elsewhere in this Annual Report on Form 10-K. Our working capital and working capital requirements are affected by numerous factors and such factors may have a negative impact on our liquidity. Principal among these are the success of product commercialization and marketing products, the technological advantages and pricing of our products, the impact of the regulatory requirements applicable to us, and access to capital markets that can provide us with the resources, when necessary, to fund our strategic priorities.
Discussion of Cash Flows
Cash flows from operating activities. Net cash used in operating activities for the year ended June 30, 2017 was $62.3 million, compared to cash used in operations of $48.5 million for the year ended June 30, 2016, an increase of $13.8 million, or approximately 28%. The increase in the current fiscal year’s cash flow used in operations principally is due to higher net loss of $94.1 million offset by an increase in non-cash expenses.
Net cash used in operating activities for the year ended June 30, 2016 was $48.5 million, compared to cash used in operations of $39.0 million for the year ended June 30, 2015, an increase of $9.5 million, due primarily to a higher net loss of $11.2 million offset by an increase in non-cash expenses in fiscal 2016.
Cash flows from investing activities. Net cash used in investing activities for the year ended June 30, 2017 was $76.3 million, compared to $45.9 million, net cash provided by investing activities for the year ended June 30, 2016. The increase in cash flow used in investing activities for fiscal 2017 of $122.2 million resulted from a $128.9 million increase in purchases of marketable securities from the previous fiscal year, offset partially by an increase in sales or maturities of marketable securities of $6.3 million and a decrease in capital expenditures of $0.4 million.
Net cash provided by investing activities for the year ended June 30, 2016 was $45.9 million, compared to $52.7 million net cash used in investing activities for the year ended June 30, 2015. The $98.6 million increase in cash flow provided by investing activities for fiscal 2016 resulted from an $83.6 million reduction in purchases of marketable securities from the previous fiscal year and a $13.6 million increase in sales or maturities of marketable securities, offset partially by an increase in capital expenditures of $1.3 million.
55
Cash flows from financing activities. Net cash provided by financing activities for the year ended June 30, 2017 was $168.8 million, compared to $2.4 million net cash provided by financing activities for the year ended June 30, 2016. The $165.1 million increase in cash provided by financing was due primarily to two public offerings conducted during fiscal 2017 and the sale of three million shares of common stock to Seattle Genetics. Net cash provided by financing activities for the year ended June 30, 2016 was $2.4 million, compared to $98.7 million net cash provided by financing activities for the year ended June 30, 2015. The $96.3 million decrease in cash provided by financing was due primarily to the issuance of $100.0 million Convertible Senior Notes in February 2015.
Working capital was $35.1 million as of June 30, 2017, compared to $37.5 million as of June 30, 2016, a $2.4 million decrease, or approximately 6%. The $165.1 million cash proceeds received from the sale of common and preferred shares were offset by $62.2 million cash used in operations and a $90.7 million increase in warrant liabilities in 2017. Working capital was $37.5 million as of June 30, 2016, compared to $91.4 million as of June 30, 2015, a decrease of $53.9 million, or 59%. The decrease was primarily the result of the loss on operations for the year, offset partially by proceeds received from the exercise of stock options.
Total cash, cash equivalents and marketable securities as of June 30, 2017 was $154.9 million, an increase of $104.3 million compared to $50.6 million as of June 30, 2016. The increase was primarily the result of a public offering and a private placement in October, 2016 and May, 2017, respectively, offset partially by the $5.1 million income tax benefit from the sale of a portion of our New Jersey State Tax NOLs and R&D tax credits in fiscal 2016.
Contractual Commitments
Our major contractual obligations relate to an operating lease for our facility and our convertible senior notes. We have quantified the significant commitments in the following table for the fiscal years ended June 30:
|
|
|
Payments Due by Period |
|
|||||||||||||||||||
|
|
|
(in thousands) |
|
|||||||||||||||||||
|
Contractual Obligation |
|
2018 |
|
2019 |
|
2020 |
|
2021 |
|
2022 |
|
Thereafter |
|
Total |
|
|||||||
|
Operating Lease(1) |
|
$ |
974 |
|
$ |
974 |
|
$ |
974 |
|
$ |
974 |
|
$ |
1,020 |
|
$ |
9,742 |
|
$ |
14,658 |
|
|
Convertible Senior Notes(2) |
|
|
4,750 |
|
|
4,750 |
|
|
102,941 |
|
|
— |
|
|
— |
|
|
— |
|
|
112,441 |
|
|
TOTAL |
|
$ |
5,724 |
|
$ |
5,724 |
|
$ |
103,915 |
|
$ |
974 |
|
$ |
1,020 |
|
$ |
9,742 |
|
$ |
127,099 |
|
|
(1) |
The operating lease for our Morris Plains, New Jersey facility expires in October 2031 and is at a base annual rental rate of $0.9 million, which has a fixed rate through October 2021 with increases thereafter every five years. |
|
(2) |
The $100,000,000 Convertible Senior Notes will mature on February 15, 2020, unless earlier purchased or converted, and bear interest at 4.75% semiannually on February 15 and August 15 each year. |
Recently Issued Accounting Pronouncements
Accounting Standard adopted during the year:
In August 2014, the FASB issued ASU 2014-15, “Presentation of Financial Statements – Going Concern: Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern”. This guidance clarifies that an entity’s management should evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. The amendments in this update are effective for annual reporting periods ending after December 15, 2016, and annual and interim periods thereafter, and early application is permitted. Note 1 of the notes to our consolidated financial statements appearing elsewhere in this Annual Report on Form 10-K incorporates the disclosure requirements from the adoption of ASU 2014-15.
Accounting Standards yet to be adopted:
In May 2017, the FASB issued ASU 2017-09, "Stock Compensation - Scope of Modification Accounting", guidance that clarifies that all changes to share-based payment awards are not necessarily accounted for as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions,
56
or the classification of the award changes as a result of the change in terms or conditions. The amendments in this guidance should be applied prospectively in annual periods beginning after December 15, 2017, including interim periods within those periods, with early adoption permitted. This guidance will apply to any future modifications. The Company is assessing ASU 2017-09’s impact and if applicable, will adopt it when effective.
In August 2016, the FASB issued ASU 2016-15, “Statement of Cash Flows: Clarification of Certain Cash Receipts and Cash Payments”, which eliminates the diversity in practice related to the classification of certain cash receipts and payments in the statement of cash flows, by adding or clarifying guidance on eight specific cash flow issues. ASU 2016-15 is effective for annual and interim reporting periods beginning after December 15, 2017 and early adoption is permitted. ASU 2016-15 provides for retrospective application for all periods presented. The Company is assessing the impact of ASU 2016-15 and will adopt it when effective.
In March 2016, the FASB issued ASU 2016-09, “Improvements to Employee Share-Based Payment Accounting” which simplified several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. Public companies will be required to adopt this standard in annual reporting periods beginning after December 15, 2016, and interim periods within those annual periods. Early adoption is permitted in any interim or annual period provided that the entire standard is adopted. The Company does not expect ASU 2016-09 to have a material impact on the consolidated financial statement presentation as (a) the Company classifies cash paid when directly withholding shares for tax withholding purposes as a financing activity and (b) the Company will make an accounting policy election to continue to use its current method of using historical data to estimate forfeitures.
In February 2016, the FASB issued ASU 2016-02, “Leases”. This standard requires a lessee to record on the balance sheet the assets and liabilities for the rights and obligations created by lease terms of more than 12 months. The amendments in this update are effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years, and early application is permitted. The Company is assessing the impact of ASU 2016-02 and will adopt it when effective.
On May 28, 2014, the FASB issued ASU 2014-09, “Revenue from Contracts with Customers,” which requires an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The ASU will replace most existing revenue recognition guidance in U.S. GAAP when it becomes effective. In August 2015, with the issuance of ASU 2015-14, the FASB amended the effective date of this ASU to fiscal years beginning after December 15, 2017, and early adoption is permitted only for fiscal years beginning after December 15, 2016. The standard permits the use of either the retrospective or cumulative effect transition method. The Company is assessing ASU 2014-09’s impact and will adopt it when effective.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
The following discussion about our exposure to market risk of financial instruments contains forward-looking statements under the Private Securities Litigation Reform Act of 1995. Actual results may differ materially from those described due to a number of factors, including uncertainties associated with general economic conditions and conditions impacting our industry.
We have not entered into and do not expect to enter into, financial instruments for trading or hedging purposes. We do not currently anticipate entering into interest rate swaps and/or similar instruments. One of our primary market risk exposure with regard to financial instruments is to changes in interest rates, which would impact interest income earned on such instruments. A one percent change (100 basis points) in interest rates on our investments would have impacted interest income by a nominal amount for the year ended June 30, 2016.
We also may be exposed to fluctuations in foreign currencies with regard to certain agreements with service providers relating to certain clinical trials that are in process. Depending on the strengthening or weakening of the U.S. dollar, realized and unrealized currency fluctuations could be significant.
57
Item 8. Financial Statements and Supplementary Data
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Immunomedics, Inc.:
We have audited the accompanying consolidated balance sheets of Immunomedics, Inc. and subsidiaries as of June 30, 2017 and 2016, and the related consolidated statements of comprehensive loss, changes in stockholders’ equity (deficit), and cash flows for each of the years in the three‑year period ended June 30, 2017. In connection with our audits of the consolidated financial statements, we also have audited financial statement schedule II. These consolidated financial statements and financial statement schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements and financial statement schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Immunomedics, Inc. and subsidiaries as of June 30, 2017 and 2016, and the results of their operations and their cash flows for each of the years in the three‑year period ended June 30, 2017, in conformity with U.S. generally accepted accounting principles. Also in our opinion, the related financial statement schedule, when considered in relation to the basic consolidated financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Immunomedics, Inc.’s internal control over financial reporting as of June 30, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO), and our report dated August 16, 2017 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
/s/ KPMG LLP
Short Hills, New Jersey
August 16, 2017
58
IMMUNOMEDICS, INC. AND SUBSIDIARIES
CONSOLIDATED BALANCE SHEETS
|
|
|
June 30 |
|
||||
|
|
|
2017 |
|
2016 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
Current Assets: |
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
43,393,570 |
|
$ |
13,203,625 |
|
|
Marketable securities |
|
|
111,508,225 |
|
|
37,424,221 |
|
|
Accounts receivable, net of allowance for doubtful accounts of $9,371 and $74,546 at June 30, 2017 and 2016, respectively |
|
|
488,723 |
|
|
513,992 |
|
|
Inventory |
|
|
580,016 |
|
|
350,524 |
|
|
Other receivables |
|
|
13,428 |
|
|
236,768 |
|
|
Prepaid expenses |
|
|
891,284 |
|
|
1,038,155 |
|
|
Other current assets |
|
|
422,916 |
|
|
183,820 |
|
|
Total current assets |
|
|
157,298,162 |
|
|
52,951,105 |
|
|
Property and equipment, net |
|
|
5,245,230 |
|
|
3,969,163 |
|
|
Other long-term assets |
|
|
30,000 |
|
|
30,000 |
|
|
Total Assets |
|
$ |
162,573,392 |
|
$ |
56,950,268 |
|
|
|
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) |
|
|
|
|
|
|
|
|
Current Liabilities: |
|
|
|
|
|
|
|
|
Accounts payable and accrued expenses |
|
$ |
31,366,976 |
|
$ |
15,188,189 |
|
|
Warrant liabilities |
|
|
90,706,206 |
|
|
— |
|
|
Deferred revenues |
|
|
170,967 |
|
|
235,372 |
|
|
Total current liabilities |
|
|
122,244,149 |
|
|
15,423,561 |
|
|
Convertible senior notes – net of unamortized debt issuance costs of $1,915,781 and $2,645,602 at June 30, 2017 and 2016 |
|
|
98,084,219 |
|
|
97,354,398 |
|
|
Other liabilities |
|
|
1,708,272 |
|
|
1,699,276 |
|
|
Commitments and Contingencies (Note 15) |
|
|
— |
|
|
— |
|
|
Stockholders’ Equity (Deficit): |
|
|
|
|
|
|
|
|
Convertible preferred stock, $.01 par value; authorized 10,000,000 shares; 1,000,000 shares issued and outstanding at June 30, 2017: no shares issued and outstanding at June 30, 2016 |
10,000 |
— |
|||||
|
Common stock, $.01 par value; authorized 250,000,000 shares issued 110,344,643 shares and outstanding 110,309,918 shares at June 30, 2017; authorized 155,000,000 shares, issued 95,867,298 shares and outstanding 95,832,573 shares at June 30, 2016 |
|
|
1,103,446 |
|
|
958,672 |
|
|
Capital contributed in excess of par |
|
|
462,666,366 |
|
|
311,320,651 |
|
|
Treasury stock, at cost: 34,725 shares at June 30, 2017 and 2016 |
|
|
(458,370) |
|
|
(458,370) |
|
|
Accumulated deficit |
|
|
(521,710,899) |
|
|
(368,504,954) |
|
|
Accumulated other comprehensive loss |
|
|
(302,710) |
|
|
(132,226) |
|
|
Total Immunomedics, Inc. stockholders’ deficit |
|
|
(58,692,167) |
|
|
(56,816,227) |
|
|
Noncontrolling interest in subsidiary |
|
|
(771,081) |
|
|
(710,740) |
|
|
Total stockholders’ deficit |
|
|
(59,463,248) |
|
|
(57,526,967) |
|
|
Total Liabilities and Stockholders' Deficit |
|
$ |
162,573,392 |
|
$ |
56,950,268 |
|
See accompanying notes to consolidated financial statements.
59
IMMUNOMEDICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF
COMPREHENSIVE LOSS
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years ended June 30, |
|
||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
||||
|
Revenues: |
|
|
|
|
|
|
|
|
|
|
|
|
Product sales |
|
$ |
2,443,388 |
|
$ |
2,260,994 |
|
$ |
2,648,657 |
|
|
|
License fee and other revenues |
|
|
284,290 |
|
|
386,941 |
|
|
1,250,000 |
|
|
|
Research and development |
|
|
363,572 |
|
|
585,312 |
|
|
1,754,434 |
|
|
|
Total revenues |
|
|
3,091,250 |
|
|
3,233,247 |
|
|
5,653,091 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Costs and Expenses: |
|
|
|
|
|
|
|
|
|
|
|
|
Costs of goods sold |
|
|
482,657 |
|
|
1,159,173 |
|
|
264,915 |
|
|
|
Research and development |
|
|
51,776,395 |
|
|
53,492,471 |
|
|
41,735,888 |
|
|
|
Sales and marketing |
|
|
873,154 |
|
|
1,027,139 |
|
|
768,871 |
|
|
|
General and administrative |
|
|
29,108,777 |
|
|
6,562,555 |
|
|
9,102,926 |
|
|
|
Total costs and expenses |
|
|
82,240,983 |
|
|
62,241,338 |
|
|
51,872,600 |
|
|
|
Operating loss |
|
|
(79,149,733) |
|
|
(59,008,091) |
|
|
(46,219,509) |
|
|
|
Changes in fair market value of warrant liabilities |
|
|
(61,073,808) |
|
|
— |
|
|
— |
|
|
|
Warrant related expenses |
|
|
(7,649,395) |
|
|
— |
|
|
— |
|
|
|
Interest expense |
|
|
(5,479,821) |
|
|
(5,479,821) |
|
|
(2,090,750) |
|
|
|
Interest and other income, net |
|
|
430,595 |
|
|
337,901 |
|
|
245,705 |
|
|
|
Other financing expenses |
|
|
(346,568) |
|
|
— |
|
|
— |
|
|
|
Foreign currency transaction gain (loss), net |
|
|
23,311 |
|
|
(39,538) |
|
|
(1,188) |
|
|
|
Loss before income tax |
|
|
(153,245,419) |
|
|
(64,189,549) |
|
|
(48,065,742) |
|
|
|
Income tax (expense) benefit |
|
|
(20,867) |
|
|
5,053,833 |
|
|
(58,229) |
|
|
|
Net loss |
|
|
(153,266,286) |
|
|
(59,135,716) |
|
|
(48,123,971) |
|
|
|
Less: Net loss attributable to noncontrolling interest |
|
|
(60,341) |
|
|
(98,766) |
|
|
(121,605) |
|
|
|
Net loss attributable to Immunomedics, Inc. stockholders |
|
$ |
(153,205,945) |
|
$ |
(59,036,950) |
|
$ |
(48,002,366) |
|
|
|
Loss per common share attributable to Immunomedics, Inc. stockholders (basic and diluted): |
|
$ |
(1.47) |
|
$ |
(0.62) |
|
$ |
(0.51) |
|
|
|
Weighted average shares used to calculate loss per common share (basic and diluted) |
|
|
104,535,577 |
|
|
94,770,172 |
|
|
93,314,872 |
|
|
|
Other comprehensive (loss) income, net of tax: |
|
|
|
|
|
|
|
|
|
|
|
|
Foreign currency translation adjustments |
|
|
(62,085) |
|
|
1,192 |
|
|
(434,617) |
|
|
|
Unrealized (loss) gain on securities available for sale |
|
|
(108,399) |
|
|
27,674 |
|
|
11,688 |
|
|
|
Other comprehensive (loss) income, net of tax: |
|
|
(170,484) |
|
|
28,866 |
|
|
(422,929) |
|
|
|
Comprehensive loss |
|
|
(153,436,770) |
|
|
(59,106,850) |
|
|
(48,546,900) |
|
|
|
Less comprehensive loss attributable to noncontrolling interest |
|
|
(60,341) |
|
|
(98,766) |
|
|
(121,605) |
|
|
|
Comprehensive loss attributable to Immunomedics, Inc. stockholders |
|
$ |
(153,376,429) |
|
$ |
(59,008,084) |
|
$ |
(48,425,295) |
|
|
See accompanying notes to consolidated financial statements.
60
IMMUNOMEDICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIT)
|
|
|
Immunomedics, Inc. Stockholders |
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
Convertible |
|
|
|
|
|
|
Capital |
|
|
|
|
|
|
|
Other |
|
|
|
|
|
|
|
|||||
|
|
|
Preferred Stock |
|
Common Stock |
|
Contributed in |
|
Treasury |
|
Accumulated |
|
Comprehensive |
|
Noncontrolling |
|
|
|
|
|||||||||||
|
|
|
Shares |
|
Amount |
|
Shares |
|
Amount |
|
Excess of Par |
|
Stock |
|
Deficit |
|
Income (Loss) |
|
Interest |
|
Total |
|
||||||||
|
Balance, at June 30, 2014 |
|
— |
|
$ |
— |
|
93,113,480 |
|
$ |
931,134 |
|
$ |
300,080,804 |
|
$ |
(458,370) |
|
$ |
(261,465,638) |
|
$ |
261,837 |
|
$ |
(490,369) |
|
$ |
38,859,398 |
|
|
Exercise of stock options, net |
|
|
|
|
|
|
1,202,575 |
|
|
12,026 |
|
|
2,947,904 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,959,930 |
|
|
Stock based compensation |
|
|
|
|
|
|
230,523 |
|
|
2,305 |
|
|
2,200,646 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,202,951 |
|
|
Other comprehensive income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(422,929) |
|
|
|
|
|
(422,929) |
|
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(48,002,366) |
|
|
|
|
|
(121,605) |
|
|
(48,123,971) |
|
|
Balance, at June 30, 2015 |
|
— |
|
$ |
— |
|
94,546,578 |
|
$ |
945,465 |
|
$ |
305,229,354 |
|
$ |
(458,370) |
|
$ |
(309,468,004) |
|
$ |
(161,092) |
|
$ |
(611,974) |
|
$ |
(4,524,621) |
|
|
Exercise of stock options, net |
|
|
|
|
|
|
1,097,500 |
|
|
10,975 |
|
|
2,721,987 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2,732,962 |
|
|
Stock based compensation |
|
|
|
|
|
|
223,220 |
|
|
2,232 |
|
|
3,369,310 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,371,542 |
|
|
Other comprehensive income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28,866 |
|
|
|
|
|
28,866 |
|
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(59,036,950) |
|
|
|
|
|
(98,766) |
|
|
(59,135,716) |
|
|
Balance, at June 30, 2016 |
|
— |
|
$ |
— |
|
95,867,298 |
|
$ |
958,672 |
|
$ |
311,320,651 |
|
$ |
(458,370) |
|
$ |
(368,504,954) |
|
$ |
(132,226) |
|
$ |
(710,740) |
|
$ |
(57,526,967) |
|
|
Issuance of preferred stock, net |
|
1,000,000 |
|
|
10,000 |
|
|
|
|
|
|
|
121,771,941 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
121,781,941 |
|
|
Issuance of common stock in public offering, net |
|
|
|
|
|
|
10,000,000 |
|
|
100,000 |
|
|
28,478,473 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
28,578,473 |
|
|
Proceeds of public offering allocated to warrant liability |
|
|
|
|
|
|
|
|
|
|
|
|
(6,966,435) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(6,966,435) |
|
|
Issuance of common stock to Seattle Genetics, Inc. |
|
|
|
|
|
|
3,000,000 |
|
|
30,000 |
|
|
14,670,000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
14,700,000 |
|
|
Proceeds of share issuance to Seattle Genetics, Inc. allocated to warrant liability |
|
|
|
|
|
|
|
|
|
|
|
|
(14,670,000) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(14,670,000) |
|
|
Exercise of stock options, net |
|
|
|
|
|
|
1,279,354 |
|
|
12,794 |
|
|
4,277,309 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4,290,103 |
|
|
Stock based compensation |
|
|
|
|
|
|
197,991 |
|
|
1,980 |
|
|
3,784,427 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3,786,407 |
|
|
Other comprehensive (loss) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(170,484) |
|
|
|
|
|
(170,484) |
|
|
Net loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(153,205,945) |
|
|
|
|
|
(60,341) |
|
|
(153,266,286) |
|
|
Balance, at June 30, 2017 |
|
1,000,000 |
|
$ |
10,000 |
|
110,344,643 |
|
$ |
1,103,446 |
|
$ |
462,666,366 |
|
$ |
(458,370) |
|
$ |
(521,710,899) |
|
$ |
(302,710) |
|
$ |
(771,081) |
|
$ |
(59,463,248) |
|
See accompanying notes to consolidated financial statements.
61
IMMUNOMEDICS, INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Years ended June 30, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(153,266,286) |
|
$ |
(59,135,716) |
|
$ |
(48,123,971) |
|
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
|
|
Change in fair value of warrant liabilities |
|
|
61,073,808 |
|
|
— |
|
|
— |
|
|
Warrant related expense |
|
|
7,649,395 |
|
|
— |
|
|
— |
|
|
Depreciation and amortization |
|
|
923,348 |
|
|
737,661 |
|
|
578,066 |
|
|
Amortization of deferred revenue |
|
|
(79,292) |
|
|
(202,088) |
|
|
(5,712) |
|
|
Amortization of bond premiums |
|
|
218,426 |
|
|
669,858 |
|
|
544,208 |
|
|
Amortization of debt issuance costs |
|
|
729,821 |
|
|
729,821 |
|
|
281,791 |
|
|
Amortization of deferred rent |
|
|
8,996 |
|
|
99,516 |
|
|
99,516 |
|
|
Loss (gain) on sale of marketable securities |
|
|
15,682 |
|
|
(1,844) |
|
|
(11,015) |
|
|
(Decrease) increase in allowance for doubtful accounts |
|
|
(61,932) |
|
|
20,369 |
|
|
(34,432) |
|
|
Non-cash expense related to stock compensation |
|
|
4,333,430 |
|
|
3,740,526 |
|
|
2,788,677 |
|
|
Non-cash decrease in value of life insurance policy |
|
|
— |
|
|
20,566 |
|
|
155,544 |
|
|
Non-cash financing expenses |
|
|
346,568 |
|
|
— |
|
|
— |
|
|
Changes in operating assets and liabilities |
|
|
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
102,640 |
|
|
(190,300) |
|
|
271,820 |
|
|
Inventory |
|
|
(195,958) |
|
|
256,381 |
|
|
328,126 |
|
|
Other receivables |
|
|
223,340 |
|
|
620,300 |
|
|
(553,966) |
|
|
Prepaid expenses |
|
|
146,871 |
|
|
97,948 |
|
|
478,794 |
|
|
Other current assets |
|
|
(241,775) |
|
|
761,803 |
|
|
(776,975) |
|
|
Accounts payable and accrued expenses |
|
|
15,807,887 |
|
|
3,147,606 |
|
|
4,946,099 |
|
|
Deferred revenues |
|
|
14,887 |
|
|
165,793 |
|
|
37,221 |
|
|
Net cash used in operating activities |
|
|
(62,250,144) |
|
|
(48,461,800) |
|
|
(38,996,209) |
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
|
|
Purchases of marketable securities |
|
|
(131,610,011) |
|
|
(2,749,117) |
|
|
(86,307,071) |
|
|
Proceeds from sales/maturities of marketable securities |
|
|
57,183,499 |
|
|
50,850,088 |
|
|
34,491,153 |
|
|
Purchases of property and equipment |
|
|
(1,837,167) |
|
|
(2,226,256) |
|
|
(924,429) |
|
|
Net cash (used in) provided by investing activities |
|
|
(76,263,679) |
|
|
45,874,715 |
|
|
(52,740,347) |
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
|
|
Sale of preferred stock, net of related expenses |
|
|
121,781,941 |
|
|
— |
|
|
— |
|
|
Proceeds from public offering of common stock, net of expenses |
|
|
28,578,473 |
|
|
— |
|
|
— |
|
|
Proceeds from sale of common stock to Seattle Genetics, Inc. |
|
|
14,700,000 |
|
|
— |
|
|
— |
|
|
Proceeds from issuance of convertible senior notes |
|
|
— |
|
|
— |
|
|
100,000,000 |
|
|
Payment of debt issuance costs |
|
|
— |
|
|
— |
|
|
(3,657,215) |
|
|
Exercise of stock options |
|
|
4,290,103 |
|
|
2,732,962 |
|
|
2,959,930 |
|
|
Tax withholding payments for stock compensation |
|
|
(547,021) |
|
|
(368,984) |
|
|
(585,725) |
|
|
Net cash provided by financing activities |
|
|
168,803,496 |
|
|
2,363,978 |
|
|
98,716,990 |
|
|
Effect of changes in exchange rates on cash and cash equivalents |
|
|
(99,728) |
|
|
(26,043) |
|
|
(489,153) |
|
|
Net increase (decrease) in cash and cash equivalents |
|
|
30,189,945 |
|
|
(249,150) |
|
|
6,491,281 |
|
|
Cash and cash equivalents beginning of the year |
|
|
13,203,625 |
|
|
13,452,775 |
|
|
6,961,494 |
|
|
Cash and cash equivalents end of the year |
|
$ |
43,393,570 |
|
$ |
13,203,625 |
|
$ |
13,452,775 |
|
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
|
|
Interest paid |
|
$ |
4,750,000 |
|
$ |
4,802,778 |
|
$ |
— |
|
|
Income taxes paid |
|
$ |
23,636 |
|
$ |
28,679 |
|
$ |
75,598 |
|
See accompanying notes to consolidated financial statements.
62
IMMUNOMEDICS, INC. AND SUBSIDIARIES
Notes to Consolidated Financial Statements
1. Business Overview
Immunomedics, Inc., a Delaware corporation (“Immunomedics” or the “Company”) is a clinical-stage biopharmaceutical company developing monoclonal antibody-based products for the targeted treatment of cancer, autoimmune and other serious diseases. The Company is focused on the acceleration of the development of its therapeutic product candidates and accordingly has transitioned away from the development and commercialization of new diagnostic imaging products to accelerate the development of its therapeutic product candidates, although the Company continues to manufacture and sell, distribute and support LeukoScan® (sulesomab) in territories where regulatory approvals have previously been granted. LeukoScan® is indicated for diagnostic imaging for determining the location and extent of infection/inflammation in bone in patients with suspected osteomyelitis, including patients with diabetic foot ulcers.
The Company has two foreign subsidiaries, Immunomedics B.V. in the Netherlands and Immunomedics GmbH in Rodermark, Germany, to assist the Company in managing sales efforts and coordinating clinical trials in Europe. In addition, included in the accompanying financial statements is the majority-owned U.S. subsidiary, IBC Pharmaceuticals, Inc. (“IBC”), which has been working since 1999 on the development of novel cancer radiotherapeutics using patented pretargeting technologies with proprietary, bispecific antibodies.
Immunomedics is subject to significant risks and uncertainties, including, without limitation, the risk that the Company may be unable to further identify, develop and achieve commercial success for new products and technologies; the possibility of delays in the research and development necessary to select drug development candidates and delays in clinical trials; the risk that clinical trials may not result in marketable products; the risk that the Company may be unable to successfully finance and secure regulatory approval of and market its drug candidates; its dependence upon pharmaceutical and biotechnology collaborations; the levels and timing of payments under its collaborative agreements; uncertainties about the Company’s ability to obtain new corporate collaborations and acquire new technologies on satisfactory terms, if at all; the development of competing products; its ability to protect its proprietary technologies; patent-infringement claims; and risks of new, changing and competitive technologies and regulations in the United States and internationally.
Since its inception in 1982, Immunomedics’ principal sources of funds have been the private and public sale of equity and debt securities and revenues from licensing agreements, including up-front and milestone payments, funding of development programs, and other forms of funding from collaborations.
As of June 30, 2017 the Company has $154.9 million in cash, cash equivalents and marketable securities; which the Company believes is sufficient to support operations through September 2018, and which allows the Company to accomplish the goal of filing a BLA for Accelerated Approval of IMMU-132 in mTNBC from the FDA during the period between December 2017 and March 2018, subject to FDA input on the acceptance of the Company’s chemistry, manufacturing and controls filing plan.
The Company will require additional funding after September 2018 to secure regulatory approval from the FDA, complete commercial preparations to market IMMU-132 to mTNBC patients in the United States, complete its clinical trials currently underway or planned, continue research and new development programs, and continue operations. Potential sources of funding include the exercise of outstanding warrants, potential various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC, and equity and potential debt financing.
Until the Company can generate significant cash through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC, or commercial operations, it expects to continue to fund its operations with its current financial resources. These financial resources are adequate to sustain the Company’s operations at a level of activity sufficient to support the filing of the BLA with the FDA for accelerated approval of IMMU-132 for patients with mTNBC; to continue manufacturing IMMU-132 at large scale to prepare for commercial operations in the U.S. marketplace; to initiate a Phase 3 clinical trial of IMMU-132 for mTNBC patients to support the filing of the BLA, to initiate preparations to market IMMU-132 to mTNBC patients in the U.S. and, subject to meeting all standards, completing review and final determination of the FDA, to secure accelerated regulatory approval to market IMMU-132 for the use of patients with mTNBC in the U.S.. After September
63
2018, if the Company cannot obtain sufficient funding through the exercise of outstanding warrants, various strategic partnership transactions towards advancing and maximizing the Company’s full pipeline for mTNBC, it could be required to finance future cash needs through the sale of additional equity and/or debt securities in capital markets. However, there can be no assurance that the Company will be able to raise the additional capital needed to complete its pipeline of research and development programs on commercially acceptable terms, if at all. The capital markets have experienced volatility in recent years, which has resulted in uncertainty with respect to availability of capital and hence the timing to meet an entity’s liquidity needs. The Company’s existing debt may also negatively impact the Company’s ability to raise additional capital. If the Company is unable to raise capital on acceptable terms, its ability to continue its business would be materially and adversely affected.
2. Summary of Significant Accounting Policies
Principles of Consolidation and Presentation
The consolidated financial statements include the accounts of Immunomedics and its majority-owned subsidiaries. Noncontrolling interests in consolidated subsidiaries in the Consolidated Balance Sheets represent minority stockholders' proportionate share of the equity (deficit) in such subsidiaries. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reported period. Actual results could differ from those estimates. The Company’s significant estimates and assumptions relate to revenue recognition, allowance for doubtful accounts, valuation of inventory and property and equipment, useful lives of property and equipment, accrued liabilities, stock compensation expenses, income tax uncertainties and other contingencies.
Foreign Currencies
For subsidiaries outside of the United States that operate in a local currency environment, income and expense items are translated to United States dollars at the monthly average rates of exchange prevailing during the year, assets and liabilities are translated at year-end exchange rates and equity accounts are translated at historical exchange rates. Translation adjustments are accumulated in a separate component of stockholders' equity in the Consolidated Balance Sheets and the Consolidated Statements of Changes in Stockholders’ Equity (Deficit) and are included in the determination of comprehensive (loss) income in the Consolidated Statements of Comprehensive Loss. Transaction gains and losses are included in the determination of net loss in the Consolidated Statements of Comprehensive Loss.
Cash and Cash Equivalents
The Company considers all liquid investments purchased with an original maturity of three months or less to be cash equivalents and all investments with maturities of greater than three months from date of purchase are classified as marketable securities available-for-sale.
Marketable securities
Marketable securities, all of which are available-for-sale, consist of corporate debt securities, U.S. bonds, U.S. sponsored agencies and municipal bonds. Marketable securities are carried at fair value, with unrealized gains and losses, net of related income taxes, reported as accumulated other comprehensive (loss) income, except for losses from impairments which are determined to be other-than-temporary. Realized gains and losses, and declines in value judged to be other-than-temporary on available-for-sale securities are included in the determination of net loss and are included in interest and other income (net), at which time the average cost basis of these securities are adjusted to fair value. Fair
64
values are based on quoted market prices at the reporting date. Interest and dividends on available-for-sale securities are included interest and other income (net).
Accounts Receivable
Credit is extended to customers based upon an evaluation of the customer’s financial condition. Accounts receivable are recorded at net realizable value. The Company utilizes a specific identification accounts receivable reserve methodology based on a review of outstanding balances and previous activities to determine the allowance for doubtful accounts. The Company charges off uncollectible receivables at the time the Company determines the receivable is no longer collectible. The Company does not require collateral or other security to support financial instruments subject to credit risk.
Concentration of Credit Risk
Cash, cash equivalents and marketable securities are financial instruments that potentially subject the Company to concentration of credit risk. Immunomedics periodically invests its cash in corporate debt securities, U.S. bonds, U.S. sponsored agencies and municipal bonds with strong credit ratings. Immunomedics has established guidelines relative to diversification and maturities that are designed to help ensure safety and liquidity. These guidelines are periodically reviewed to take advantage of trends in yields and interest rates.
Inventory
Inventory, which consists of the raw materials, work-in-process and finished product of LeukoScan®, is stated at the lower of cost (on a first-in, first-out basis) or market, and includes materials, labor and manufacturing overhead.
Property and Equipment and Impairment of Assets
Property and equipment are stated at cost and are depreciated on a straight-line basis over the estimated useful lives (5 - 10 years) of the respective assets. Leasehold improvements are capitalized and amortized over the lesser of the remaining life of the lease or the estimated useful life of the asset. Immunomedics reviews long‑lived assets for impairment whenever events or changes in business circumstances occur that indicate that the carrying amount of the assets may not be recoverable. Immunomedics assesses the recoverability of long‑lived assets held and to be used based on undiscounted cash flows, and measures the impairment, if any, using discounted cash flows. To date the Company has not taken any impairment charges on property and equipment.
Revenue Recognition
The Company has accounted for revenue arrangements that include multiple deliverables as a separate unit of accounting if both of the following criteria are met: a) the delivered item has value to the customer on a standalone basis, and b) if the right of return exists, delivery of the undelivered items is considered probable and substantially in the control of the vendor. If these criteria are not met, the revenue elements must be considered a single unit of accounting for purposes of revenue recognition. The Company allocates revenue consideration, excluding contingent consideration, based on the relative selling prices of the separate units of accounting contained within an arrangement containing multiple deliverables. Relative selling prices are determined using vendor specific objective evidence, if it exists; otherwise third-party evidence or the Company’s best estimate of selling price is used for each deliverable.
Payments received under contracts to fund certain research activities are recognized as revenue in the period in which the research activities are performed. Payments received in advance that are related to future performance are deferred and recognized as revenue when the research projects are performed. Upfront nonrefundable fees associated with license and development agreements where the Company has continuing involvement in the agreement are recorded as deferred revenue and recognized over the estimated service period. The Company estimates the period of continuing involvement based on the best evidential matter available at each reporting period. If the estimated service period is subsequently modified, the period over which the upfront fee is recognized is modified accordingly on a prospective basis.
65
In order to determine the revenue recognition for contingent milestones, the Company evaluates the contingent milestones using the criteria as provided by the Financial Accounting Standards Boards (“FASB”) guidance on the milestone method of revenue recognition, as explained in ASU 2010-17, “Milestone Method of Revenue Recognition”, at the inception of a collaboration agreement. The criteria requires that (i) the Company determines if the milestone is commensurate with either its performance to achieve the milestone or the enhancement of value resulting from the Company’s activities to achieve the milestone, (ii) the milestone be related to past performance, and (iii) the milestone be reasonable relative to all deliverable and payment terms of the collaboration arrangement. If these criteria are met then the contingent milestones can be considered as substantive milestones and will be recognized as revenue in the period that the milestone is achieved. Royalties are recognized as earned in accordance with the terms of various research and collaboration agreements.
Revenue from the sale of diagnostic products is recorded when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed and determinable or collectability is reasonably assured. Allowances, if any, are established for uncollectible amounts, estimated product returns and discounts. Since allowances are recorded based on management’s estimates, actual amounts may be different in the future.
Research and Development Costs
Research and development costs are expensed as incurred. Costs incurred for clinical trials for patients and investigators are expensed as services are performed in accordance with the agreements in place with the institutions.
Reimbursement of Research and Development Costs
Reimbursement toward research and development costs under collaboration agreements are included as a reduction of research and development expenses. The Company records these reimbursements as a reduction of research and development expenses as the Company’s partner in the collaboration agreement has the financial risks and responsibility for conducting these research and development activities.
Common Stock Warrants
In connection with certain financing transactions in October 2016 and February 2017, the Company issued warrants and recorded them as liabilities due to certain net cash settlement provisions. The warrants were recorded at fair value using the Black-Scholes valuation model. The Black-Scholes valuation model takes into account, as of the valuation date, factors including the current exercise price, the term of the warrant, the current price of the underlying stock and its expected volatility, expected dividends on the stock, and the risk-free interest rate for the term of the warrant. These warrants are subject to re-measurement at each balance sheet date until the warrants are exercised or expired, and any change in fair value is recognized as “change in the fair value of warrant liability” in the consolidated statements of operations.
Income Taxes
The Company uses the asset and liability method to account for income taxes, including the recognition of deferred tax assets and deferred tax liabilities for the anticipated future tax consequences attributable to differences between financial statements amounts and their respective tax bases. The Company reviews its deferred tax assets for recovery. A valuation allowance is established when the Company believes that it is more likely than not that its deferred tax assets will not be realized. Changes in valuation allowances from period to period are included in the Company’s tax provision in the period of change.
The Company did not book an accrual for uncertain tax positions as of June 30, 2017 or 2016. The U.S. Federal statute of limitation remains open for the fiscal years 2013 onward. The Company’s tax returns filed in foreign jurisdictions remain open for the fiscal years 2013 onward. State income tax returns are generally subject to examination for a period of 3 - 5 years after filing of the respective return. The Company conducts business and files tax returns in New Jersey.
66
Net Loss Per Share Allocable to Common Stockholders
Basic net loss per share is calculated using the weighted average number of shares of common stock and vested restricted shares outstanding. Diluted net income per share is based upon the weighted average number of shares of common stock and dilutive potential shares of common stock outstanding. During fiscal years 2017, 2016, and 2015, no potential shares of common stock were included in the calculation since their affect would be anti-dilutive due to the operating losses. The common stock equivalents excluded from the earnings per share calculation are 66,069,081, 26,665,296 and 25,815,581 for the fiscal years ended June 2017, 2016, and 2015, respectively.
Net Comprehensive Loss
Net comprehensive loss consists of net loss, unrealized loss on available for sale securities and foreign exchange translation adjustments and is presented in the consolidated statements of comprehensive loss.
Stock-Based Compensation
The Company utilizes stock-based compensation in the form of stock options, stock appreciation rights, stock awards, stock unit awards, performance shares, cash-based performance units and other stock-based awards, each of which may be granted separately or in tandem with other awards.
The grant-date fair value of stock awards is based upon the underlying price of the stock on the date of grant. The grant-date fair value of stock option awards must be determined using an option pricing model. Option pricing models require the use of estimates and assumptions as to (a) the expected term of the option, (b) the expected volatility of the price of the underlying stock and (c) the risk-free interest rate for the expected term of the option. The Company uses the Black-Scholes-Merton option pricing formula for determining the grant-date fair value of such awards. The fair value of restricted stock units that vest based on achievement of certain market conditions are determined using a Monte Carlo simulation technique.
The expected term of the option is based upon the contractual term and expected employee exercise and expected post-vesting employment termination behavior. The expected volatility of the price of the underlying stock is based upon the historical volatility of the Company’s stock computed over a period of time equal to the expected term of the option. The risk free interest rate is based upon the implied yields currently available from the U.S. Treasury yield curve in effect at the time of the grant. Pre-vesting forfeiture rates are estimated based upon past voluntary termination behavior and past option forfeitures.
The fair value of each option granted during the years ended June 30, 2017, 2016, and 2015 is estimated on the date of grant using the Black‑Scholes option‑pricing model with the following weighted-average assumptions in the following table:
|
|
|
Years ended June 30, |
|
||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Expected dividend yield |
|
0% |
|
0% |
|
0% |
|
|
Expected option term (years) |
|
5.04 |
|
5.03 |
|
5.07 |
|
|
Expected stock price volatility |
|
63% |
|
58% |
|
57% |
|
|
Risk-free interest rate |
|
1.16% - 2.15% |
|
1.00% - 1.64% |
|
1.37% - 1.72% |
|
The Company uses historical data to estimate forfeitures. The expected term of options granted represents the period of time that options granted are expected to be outstanding. Expected stock price volatility was calculated based on the Company’s daily stock trading history. The risk-free rate for periods within the expected term of the option is based on the U.S. Treasury yield curve in effect at the time of grant.
Changes in any of these assumptions could impact, potentially materially, the amount of expense recorded in future periods related to stock-based awards.
67
Financial Instruments
The carrying amounts of cash and cash equivalents, other current assets and current liabilities approximate fair value due to the short‑term maturity of these instruments. The Company considers all highly liquid investments with an original maturity of three months or less when purchased to be cash equivalents.
Recently Issued Accounting Pronouncements
Accounting Standard adopted during the year:
In August 2014, the FASB issued ASU 2014-15, “Presentation of Financial Statements – Going Concern: Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern”. This guidance clarifies that an entity’s management should evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. The amendments in this update are effective for annual reporting periods ending after December 15, 2016, and annual and interim periods thereafter, and early application is permitted. Note 1 incorporates the disclosure requirements from the adoption of ASU 2014-15.
Accounting Standards yet to be adopted:
In May 2017, the FASB issued ASU 2017-09, "Stock Compensation - Scope of Modification Accounting", guidance that clarifies that all changes to share-based payment awards are not necessarily accounted for as a modification. Under the new guidance, modification accounting is required only if the fair value, the vesting conditions, or the classification of the award changes as a result of the change in terms or conditions. The amendments in this guidance should be applied prospectively in annual periods beginning after December 15, 2017, including interim periods within those periods, with early adoption permitted. This guidance will apply to any future modifications. The Company is assessing ASU 2017-09’s impact and if applicable, will adopt it when effective.
In August 2016, the FASB issued ASU 2016-15, “Statement of Cash Flows: Clarification of Certain Cash Receipts and Cash Payments”, which eliminates the diversity in practice related to the classification of certain cash receipts and payments in the statement of cash flows, by adding or clarifying guidance on eight specific cash flow issues. ASU 2016-15 is effective for annual and interim reporting periods beginning after December 15, 2017 and early adoption is permitted. ASU 2016-15 provides for retrospective application for all periods presented. The Company is assessing the impact of ASU 2016-15 and will adopt it when effective.
In March 2016, the FASB issued ASU 2016-09, “Improvements to Employee Share- Based Payment Accounting” which simplified several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. Public companies will be required to adopt this standard in annual reporting periods beginning after December 15, 2016, and interim periods within those annual periods. Early adoption is permitted in any interim or annual period provided that the entire standard is adopted. The Company does not expect ASU 2016-09 to have a material impact on the consolidated financial statement presentation as (a) the Company classifies cash paid when directly withholding shares for tax withholding purposes as a financing activity and (b) the Company will make an accounting policy election to continue to use its current method of using historical data to estimate forfeitures.
In February 2016, the FASB issued ASU 2016-02, “Leases”. This standard requires a lessee to record on the balance sheet the assets and liabilities for the rights and obligations created by lease terms of more than 12 months. The amendments in this update are effective for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years, and early application is permitted. The Company is assessing the impact of ASU 2016-02 and will adopt it when effective.
On May 28, 2014, the FASB issued ASU 2014-09, “Revenue from Contracts with Customers,” which requires an entity to recognize the amount of revenue to which it expects to be entitled for the transfer of promised goods or services to customers. The ASU will replace most existing revenue recognition guidance in U.S. GAAP when it becomes
68
effective. In August 2015, with the issuance of ASU 2015-14, the FASB amended the effective date of this ASU to fiscal years beginning after December 15, 2017, and early adoption is permitted only for fiscal years beginning after December 15, 2016. The standard permits the use of either the retrospective or cumulative effect transition method. The Company is assessing the impact of ASU 2014-09 and will adopt it when effective.
3. Marketable Securities
Immunomedics considers all of its current investments to be available-for-sale. Marketable securities at June 30, 2017 consist of the following (in thousands):
|
|
|
|
|
|
Gross |
|
Gross |
|
|
|
|
||
|
|
|
Amortized |
|
Unrealized |
|
Unrealized |
|
|
|
|
|||
|
|
|
Cost |
|
Gain |
|
(Loss) |
|
Fair Value |
|
||||
|
U.S. Treasury Bonds |
|
$ |
35,086 |
|
$ |
— |
|
$ |
(24) |
|
$ |
35,062 |
|
|
Certificate of Deposits |
|
|
15,298 |
|
|
— |
|
|
— |
|
|
15,298 |
|
|
U.S. Government Sponsored Agencies |
|
|
18,357 |
|
|
— |
|
|
(13) |
|
|
18,344 |
|
|
Corporate Debt Securities |
|
|
32,692 |
|
|
— |
|
|
(33) |
|
|
32,659 |
|
|
Commercial Paper |
|
|
10,144 |
|
|
1 |
|
|
— |
|
|
10,145 |
|
|
|
|
$ |
111,577 |
|
$ |
1 |
|
$ |
(70) |
|
$ |
111,508 |
|
Maturities of debt securities classified as available-for-sale were as follows at June 30, 2017 (in thousands):
|
|
|
|
|
|
Net Carrying |
|
|
|
|
|
Fair Value |
|
Amount |
|
||
|
Due within one year |
|
$ |
89,477 |
|
$ |
89,728 |
|
|
Due after one year through five years |
|
|
22,031 |
|
|
22,149 |
|
|
|
|
$ |
111,508 |
|
$ |
111,877 |
|
Marketable securities at June 30, 2016 consisted of the following (in thousands):
|
|
|
|
|
|
Gross |
|
Gross |
|
|
|
|
||
|
|
|
Amortized |
|
Unrealized |
|
Unrealized |
|
|
|
|
|||
|
|
|
Cost |
|
Gain |
|
(Loss) |
|
Fair Value |
|
||||
|
U.S. Treasury Bonds |
|
$ |
5,059 |
|
$ |
6 |
|
$ |
— |
|
$ |
5,065 |
|
|
Certificate of Deposits |
|
|
3,000 |
|
|
3 |
|
|
— |
|
|
3,003 |
|
|
U.S. Government Sponsored Agencies |
|
|
14,311 |
|
|
31 |
|
|
— |
|
|
14,342 |
|
|
Corporate Debt Securities |
|
|
15,014 |
|
|
2 |
|
|
(2) |
|
|
15,014 |
|
|
|
|
$ |
37,384 |
|
$ |
42 |
|
$ |
(2) |
|
$ |
37,424 |
|
4. Inventory
Inventory consisted of the following at June 30 (in thousands):
|
|
|
2017 |
|
2016 |
|
||
|
Raw Materials |
|
$ |
— |
|
$ |
68 |
|
|
Work-in-process |
|
|
— |
|
|
191 |
|
|
Finished goods |
|
|
580 |
|
|
92 |
|
|
|
|
$ |
580 |
|
$ |
351 |
|
69
5. Convertible Senior Notes
In February 2015, the Company issued $100.0 million of Convertible Senior Notes (net proceeds of $96.3 million after deducting the initial purchasers’ fees and offering expenses) in a private offering exempt from registration under the Securities Act of 1933, as amended (the “Securities Act”), in reliance upon Rule 144A under the Securities Act. The Convertible Senior Notes will mature on February 15, 2020, unless earlier purchased or converted. The debt issuance costs of approximately $3.7 million, primarily consisting of underwriting, legal and other professional fees, are amortized over the term of the Convertible Senior Notes. The Convertible Senior Notes are senior unsecured obligations of the Company. Interest at 4.75% is payable semiannually on February 15 and August 15 of each year. The effective interest rate on the Convertible Senior Notes was 5.48% for the period from the date of issuance through June 30, 2017.
The Convertible Senior Notes are convertible at the option of holders into approximately 19.6 million shares of Immunomedics common stock at any time prior to the close of business on the day immediately preceding the maturity date. The conversion rate will initially be 195.8336 shares of common stock per $1,000 principal amount of Convertible Senior Notes (equivalent to an initial conversion price of approximately $5.11 per share of Immunomedics common stock).
If the Company undergoes a fundamental change (as defined in the indenture governing the Convertible Senior Notes), holders may require Immunomedics to purchase for cash all or part of the Convertible Senior Notes at a purchase price equal to 100% of the principal amount of the Convertible Senior Notes to be purchased, plus accrued and unpaid interest, if any, to, but excluding, the fundamental change purchase date, subject to certain exceptions. In addition, if certain make-whole fundamental changes (as defined in the indenture governing the Convertible Senior Notes) occur, Immunomedics will, in certain circumstances, increase the conversion rate for any Convertible Note converted in connection with such make-whole fundamental change.
The indenture does not limit the amount of debt which may be issued by the Company under the indenture or otherwise, does not contain any financial covenants or restrict the Company from paying dividends or issuing or repurchasing its other securities. The indenture contains customary terms and covenants and events of default.
If an event of default with respect to the Convertible Senior Notes occurs, holders may, upon satisfaction of certain conditions, accelerate the principal amount of the Convertible Senior Notes plus premium, if any, and accrued and unpaid interest, if any. In addition, the principal amount of the Convertible Senior Notes plus premium, if any, and accrued and unpaid interest, if any, will automatically become due and payable in the case of certain types of bankruptcy or insolvency events of default involving the Company.
Total interest expense for the Convertible Senior Notes for the fiscal years ended June 30, 2017, 2016, and 2015 were $5.5 million, $5.5 million, and $2.1 million, respectively. Included in interest expense is the amortization of debt issuance costs of $0.7 million, $0.7 million, and $0.3 million for the fiscal years ended June 30, 2017, 2016, and 2015, respectively.
70
6. Warrant Liabilities
In connection with a public offering conducted during October 2016, the Company issued warrants that contain net cash settlement provisions. Additionally, in connection with a stock purchase agreement entered into with Seattle Genetics, Inc. during February 2017 (the “SGEN Warrant”), the Company issued warrants that also have similar net cash settlement provisions. In addition, the SGEN Warrant may only be exercised, upon receiving stockholder approval to increase its authorized capital and such shares being registered with the SEC. As stated in Note 10, at a special Stockholder Meeting on June 29, 2017 (“2017 Special Meeting”), the Company’s stockholders approved the amendment and restatement of the Company’s Certificate of Incorporation to increase the maximum number of shares of the Company’s stock authorized up to 260,000,000 shares of stock consisting of 250,000,000 shares of common stock and 10,000,000 shares of preferred stock. Such increase is considered to be sufficient for the Company to settle its obligation related to the warrants after considering all other commitments that may require the issuance of common stock through December 31, 2017, when these warrants will expire (See Note 10). The Company filed a registration statement with the SEC on Form S-3 on July 31, 2017, which is yet to be declared effective. Accordingly, as of June 30, 2017, both warrants do not meet the criteria for classification as equity and are recorded as liabilities on the Company’s balance sheet. The Company recorded these warrants as liabilities at their fair values as calculated at their respective dates of inception. The change in fair value of each warrant is measured, and booked as an income or expense to adjust the warrant liability on a periodic basis at the end of each fiscal quarter.
The Company uses Level 2 inputs for its valuation methodology for the warrant liabilities. The estimated fair value was determined using a Black-Scholes valuation model based on various assumptions. The warrant liabilities are adjusted to reflect estimated fair value at each period end, with any changes in the fair value being recorded in changes in fair value of warrant liabilities.
7. Estimated Fair Value of Financial Instruments
The Company’s financial instruments consist of cash and cash equivalents, marketable securities, accounts receivable, accounts payable and accrued expenses, and Convertible Notes. The carrying amount of accounts receivable, accounts payable and accrued expenses are generally considered to be representative of their respective fair values because of the short-term nature of those instruments as of June 30, 2017 and 2016.
The Company has categorized its other financial instruments, based on the priority of the inputs to the valuation technique, into a three-level fair value hierarchy as set forth below. If the inputs used to measure the financial instruments fall within different levels of the hierarchy, the categorization is based on the lowest level input that is significant to the fair value measurement of the instrument.
Financial instruments recorded on the consolidated balance sheets as of June 30, 2017 and 2016 are categorized based on the inputs to the valuation techniques as follows (in thousands):
|
· |
Level 1 – Values are based on unadjusted quoted prices for identical assets or liabilities in an active market which the Company has the ability to access at the measurement date (examples include active exchange-traded equity securities and most U.S. Government and agency securities). |
|
· |
Level 2 – Values are based on quoted market prices in markets where trading occurs infrequently or whose values are based on quoted prices of instruments with similar attributes in active markets. |
|
· |
Level 3 – Values are based on prices or valuation techniques that require inputs that are both unobservable and significant to the overall fair value measurement. These inputs reflect management’s own assumptions about the assumptions a market participant would use in pricing the asset. |
71
Cash equivalents and marketable securities:
|
|
|
($ in thousands) |
|
||||||||||
|
June 30, 2017 |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Money Market Funds Note (a) |
|
$ |
36,776 |
|
$ |
— |
|
$ |
— |
|
$ |
36,776 |
|
|
Marketable Securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury Bonds |
|
|
35,062 |
|
|
— |
|
|
— |
|
|
35,062 |
|
|
Certificate of Deposits |
|
|
15,298 |
|
|
— |
|
|
— |
|
|
15,298 |
|
|
U.S. Government Sponsored Agencies |
|
|
18,344 |
|
|
— |
|
|
— |
|
|
18,344 |
|
|
Corporate Debt Securities |
|
|
32,659 |
|
|
— |
|
|
— |
|
|
32,659 |
|
|
Commercial Paper |
|
|
10,145 |
|
|
— |
|
|
— |
|
|
10,145 |
|
|
Total |
|
$ |
148,284 |
|
$ |
— |
|
$ |
— |
|
$ |
148,284 |
|
|
|
|
($ in thousands) |
|
||||||||||
|
June 30, 2016 |
|
Level 1 |
|
Level 2 |
|
Level 3 |
|
Total |
|
||||
|
Money Market Funds Note (a) |
|
$ |
10,012 |
|
$ |
— |
|
$ |
— |
|
$ |
10,012 |
|
|
Marketable Securities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. Treasury Bonds |
|
|
5,065 |
|
|
— |
|
|
— |
|
|
5,065 |
|
|
Certificate of Deposits |
|
|
3,003 |
|
|
— |
|
|
— |
|
|
3,003 |
|
|
U.S. Government Sponsored Agencies |
|
|
14,342 |
|
|
— |
|
|
— |
|
|
14,342 |
|
|
Corporate Debt Securities |
|
|
15,014 |
|
|
— |
|
|
— |
|
|
15,014 |
|
|
Total |
|
$ |
47,436 |
|
$ |
— |
|
$ |
— |
|
$ |
47,436 |
|
|
(a) |
The money market funds noted above are included in cash and cash equivalents. |
Convertible Senior Notes
The carrying amounts and estimated fair values (Level 2) of debt instruments are as follows (in thousands):
|
|
|
As of June 30, 2017 |
|
As of June 30, 2016 |
|
||||||||
|
|
|
Carrying |
|
Estimated |
|
Carrying |
|
Estimated |
|
||||
|
|
|
Amount |
|
Fair Value |
|
Amount |
|
Fair Value |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Convertible Senior Notes |
|
$ |
98,084 |
|
$ |
180,950 |
|
$ |
97,354 |
|
$ |
71,359 |
|
The fair value of the Convertible Senior Notes, which differs from their carrying values, is influenced by interest rates, the Company’s stock price and stock price volatility and is determined by prices for the Convertible Senior Notes observed in market trading which are Level 2 inputs.
Warrant Liabilities
The Company has determined its warrant liabilities to be a Level 2 fair value measurement and used the Black Scholes valuation model to calculate the fair value as of June 30, 2017, February 10, 2017 (date of issuance of warrant liabilities in connection with stock purchase agreement) and October 11, 2016 (date of issuance of warrants in connection with public offering):
72
At the measurement dates, the Company estimated the fair value for the warrants based on Black-Scholes valuation model and using the following assumptions:
|
|
|
June 30, |
|
June 30, |
|
February 10, |
|
October 11, |
|
|
|
2017 (1) |
|
2017 (2) |
|
2017 |
|
2016 |
|
Risk-free interest rate |
|
1.14% |
|
1.38% |
|
1.47% |
|
0.87% |
|
Expected remaining term |
|
0.51 |
|
1.28 |
|
3.0 years |
|
2.0 years |
|
Expected volatility |
|
69.34% |
|
73.85% |
|
71.42% |
|
75.00% |
|
Dividend yield |
|
0% |
|
0% |
|
0% |
|
0% |
|
(1) |
Represents the fair value assumptions for the warrants issued in connection with February 10, 2017 stock purchase agreement. |
|
(2) |
Represents the fair value assumptions for the warrants issued in connection with October 11, 2016 public offering. |
The following table sets forth the warrant activity for the year ended June 30, 2017 ($ in thousands):
|
|
|
|
Estimated Fair |
|
|
|
Number of |
|
Value |
|
|
|
Warrants |
|
Level 2 |
|
|
Warrants outstanding as of July 1, 2016 |
— |
|
$ |
— |
|
Additions, pursuant to October 11, 2016 public offering |
10,000,000 |
|
|
7,313 |
|
Additions, pursuant to February 16, 2017 stock purchase agreement with Seattle Genetics, Inc. |
8,655,804 |
|
|
22,319 |
|
Change in fair value |
— |
|
|
61,074 |
|
Warrants outstanding as of June 30, 2017 |
18,655,804 |
|
$ |
90,706 |
8. Property and Equipment
Property and equipment consisted of the following at June 30 (in thousands):
|
|
|
2017 |
|
2016 |
|
||
|
Machinery and equipment |
|
$ |
9,353 |
|
$ |
9,071 |
|
|
Leasehold improvements |
|
|
21,602 |
|
|
19,863 |
|
|
Furniture and fixtures |
|
|
976 |
|
|
970 |
|
|
Computer equipment |
|
|
2,875 |
|
|
2,703 |
|
|
|
|
|
34,806 |
|
|
32,607 |
|
|
Accumulated depreciation and amortization |
|
|
(29,561) |
|
|
(28,638) |
|
|
|
|
$ |
5,245 |
|
$ |
3,969 |
|
Depreciation and amortization expense for the years ended June 30, 2017, 2016, and 2015 was $0.9 million, $0.7 million, and $0.6 million, respectively.
73
9. Accounts Payable and Accrued Expenses
Accounts payable and accrued expenses consisted of the following at June 30 (in thousands):
|
|
|
2017 |
|
2016 |
|
||
|
Trade accounts payable |
|
$ |
5,222 |
|
$ |
4,128 |
|
|
Contract manufacture organization expenses |
|
|
3,769 |
|
|
1,222 |
|
|
Clinical trial accruals |
|
|
2,865 |
|
|
6,087 |
|
|
Proxy defense-related expenses |
|
|
6,967 |
|
|
— |
|
|
Reimbursement for proxy expenses |
|
|
4,505 |
|
|
— |
|
|
Executive severance liabilities |
|
|
5,542 |
|
|
— |
|
|
Executive bonus |
|
|
— |
|
|
1,148 |
|
|
Accrued interest expense |
|
|
1,768 |
|
|
1,768 |
|
|
Miscellaneous other current liabilities |
|
|
729 |
|
|
835 |
|
|
|
|
$ |
31,367 |
|
$ |
15,188 |
|
10. Stockholders’ Deficit
At the June 29, 2017 Special Meeting, the Company’s stockholders approved the amendment and restatement of the Company’s Certificate of Incorporation to increase the maximum number of shares of the Company’s stock authorized up to 260,000,000 shares of stock consisting of 250,000,000 shares of common stock and 10,000,000 shares of preferred stock. Previously the Company’s Certificate of Incorporation authorized up to 165,000,000 shares of capital stock, consisting of 155,000,000 shares of common stock and 10,000,000 shares of preferred stock.
Preferred Stock
The Certificate of Incorporation of the Company authorizes 10,000,000 shares of preferred stock, $.01 par value per share. The preferred stock may be issued from time to time in one or more series, with such distinctive serial designations, rights and preferences as shall be determined by the Board of Directors.
On May 10, 2017, the Company issued in a private placement 1,000,000 shares (the “Preferred Shares”) of the Company’s Series A-1 Convertible Preferred Stock at a price of $125 per share for gross proceeds to the Company of $125 million, before deducting fees and expenses (the “Financing”). Each Preferred Share will be convertible into 23.10536 shares of common stock (or an aggregate of 23,105,348 shares of common stock). The conversion price per share of common stock is $5.41. For fiscal year ended June 30, 2016 the Company had no preferred stock outstanding.
Following the June 29 2017 Special Meeting and filing the Charter Amendment with the State of Delaware, the Company had authorized a sufficient number of unreserved shares of common stock to permit the conversion of the Preferred Shares. Preferred Shares will automatically convert to shares of common stock subsequent to the termination of the Licensing and Development Agreement entered into on February 10, 2017 by and between the Company and Seattle Genetics (the “License Agreement”); or, if such automatic conversion does not occur prior to January 1, 2018, the Purchasers may elect to convert at any time on or after January 1, 2018. On July 31, 2017, the Company filed a registration statement on Form S-3 to register the 23,105,348 shares of the Company’s common stock issuable upon the conversion of the Series A-1 Convertible Preferred Stock (in addition to 3,000,000 shares of Company’s common stock and 8,655,804 shares of common stock issuable upon the exercise of the warrants issued to Seattle Genetics, Inc as discussed below).
Common Stock
On February 10, 2017, in connection with the execution of a License Agreement, the Company entered into the Securities Purchase Agreement (“SPA”) with Seattle Genetics. Under the SPA, Seattle Genetics purchased 3,000,000 shares (the “Common Shares”) of the Company’s common stock at a price of $4.90 per share, for aggregate proceeds of $14.7 million. Concurrently with the sale of the Common Shares, pursuant to the SPA, the Company also agreed to issue the three-year warrant to purchase an aggregate of 8,655,804 shares of common stock. On July 31, 2017, the Company filed a registration statement on Form S-3 to register the 3,000,000 shares of Company’s common stock and 8,655,804 shares of common stock issuable upon the exercise of the warrants (in addition to the shares issuable upon the
74
conversion of our Series A-1 Convertible Preferred Stock, as discussed above). The warrant became exercisable for cash on February 16, 2017, and will expire on January 31, 2018. The warrant was issued on February 16, 2017 and was originally exercisable until February 10, 2020. On the date of issuance, the fair value of these warrants was determined to be $22.3 million. The difference between such fair value and the proceeds of $14.7 million has been recognized as an expense and presented in the consolidated statement operations as a “warrant related expense.” On May 4, 2017, the Company and Seattle Genetics entered into the Termination Agreement, pursuant to which the Company and Seattle Genetics relinquished their respective rights under the License Agreement and agreed to amend the terms of the warrant to amend the expiration date from February 10, 2020 to December 31, 2017.
On October 11, 2016, the Company completed an underwritten public offering of 10 million shares of its common stock and accompanying warrants to purchase 10 million shares of common stock at a purchase price of $3.00 per unit, comprising of one share of common stock and one warrant. The Company received gross and net proceeds of $30.0 million and approximately $28.6 million, respectively after deducting the underwriting discounts and commissions and estimated expenses related to the offering payable. The warrants became exercisable six months following the date of issuance, and will expire on the second anniversary of the date of issuance and have an exercise price of $3.75. On the date of issuance, the fair value of these warrants was determined to be $7.3 million and recognized as a liability. The shares of common stock were sold pursuant to an effective shelf registration statement filed with the SEC. The warrants under certain situations require cash settlement by the Company.
Stock Incentive Plans
At the Annual Stockholder Meeting on December 3, 2014, the Company’s stockholders approved the Immunomedics, Inc. 2014 Long-Term Incentive Plan (the “Plan”). The Plan replaced the Company’s 2006 Stock Incentive Plan (the “2006 Plan”), which terminated on December 3, 2014. The Plan was established to promote the interests of the Company, by providing eligible persons with the opportunity to acquire a proprietary interest in the Company as an incentive to remain with the organization and to align the employee’s interest with our stockholders. The approval authorized issuance of 9,000,000 shares plus the number of unallocated share available for issuance as of the effective date under the 2006 Plan that were not subject to outstanding awards.
As under the 2006 Plan, option awards under the Plan are generally granted with an exercise price equal to the market price of the Plan, the Company’s common stock at the date of grant; those option awards generally vest based on four years of continuous service and have seven year contractual terms. Option awards that are granted to non-employee Board members under the annual option grant program are granted with an exercise price equal to the market price of the Company’s common stock at the date of grant, are vested immediately and have seven year contractual terms. Certain options provide for accelerated vesting if there is a change in control (as defined in the Plan). At June 30, 2017, there were 14,264,986 shares of common stock reserved for possible future issuance under the Plan, both currently outstanding (4,724,569 shares) and which were available to be issued for future grants (9,540,417 shares).
The Plan is divided into three separate equity incentive programs. These incentive programs consist of:
|
· |
Discretionary Grant Program under which eligible persons may be granted options to purchase shares of common stock or stock appreciation rights tied to the value of the common stock; |
|
· |
Stock Issuance Program under which eligible persons may be issued shares of common stock pursuant to restricted stock awards, restricted stock shares, performance shares or other stock-based awards which vest upon completion of a designated service period or the attainment of pre-established performance milestones, or such shares of common stock may be a fully-vested bonus for services rendered; and |
|
· |
Automatic Grant Program under which eligible non-employee Board members will automatically receive grants at designated intervals over their period of continued Board service. |
For the 2016 and 2015 fiscal years each of the Company’s outside Directors who had been a Director prior to July 1st of each year were granted, at the annual stockholder meeting of each year, options to purchase shares of the Company’s common stock at fair market value on the grant date. For fiscal years 2016 and 2015, stock options were
75
granted to these outside directors to purchase an aggregate of 115,284 shares and 89,204 shares, respectively. The values of the granted options were $180 thousand for the fiscal years ended June 30, 2016 and 2015. Stock options granted to outside directors were vested when granted. No stock options were granted to outside directors in fiscal year 2017 as it was decided to defer the awarding of options until a revision to the program was completed in July, 2017. When an outside Director is elected to the Board of Directors, they are awarded options for 22,500 shares of the Company’s common stock. The Company recorded $184 thousand, $201 thousand and $180 thousand for stock-based compensation expense for these non-employee Board members stock options for the years ended June 30, 2017, 2016, and 2015, respectively. For new outside Directors elected on May 4, 2017, no options were awarded until a revision to the program was completed in July, 2017.
For the 2016 and 2015 fiscal years non-employee Board members who continue to serve would receive on the date of the annual stockholders meeting an annual grant of non-qualified stock options and restricted stock units, equal in value to $45 thousand. For fiscal years 2016 and 2015, restricted stock units were granted to these outside directors in an aggregate 57,876 units and 42,656 units, respectively. The value of the units granted were $180 thousand for the 2016 and 2015 fiscal years. Restricted stock units granted to outside directors become vested within one year of grant date. The Company recorded $31 thousand, $181 thousand, and $180 thousand for stock-based compensation expense for these non-employee Board members restricted stock units for the years ended June 30, 2017, 2016, and 2015, respectively. No restricted stock units or non-qualified stock options were granted to outside directors in fiscal year 2017, as it was decided to defer the awarding of options until a revision to the program was completed in July, 2017.
Information concerning options for the years ended June 30, 2017, 2016, and 2015 is summarized as follows:
|
|
|
Number of Shares |
|
Weighted Average Exercise Price |
|
|||||||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Options outstanding, beginning of year |
|
4,015,895 |
|
4,525,340 |
|
5,308,617 |
|
$ |
3.42 |
|
$ |
3.48 |
|
$ |
3.41 |
|
|
Options granted |
|
376,032 |
|
880,681 |
|
955,361 |
|
$ |
4.07 |
|
$ |
2.15 |
|
$ |
3.82 |
|
|
Options exercised |
|
(1,279,354) |
|
(1,097,500) |
|
(1,202,575) |
|
$ |
3.35 |
|
$ |
2.53 |
|
$ |
2.46 |
|
|
Options expired or forfeited |
|
(219,333) |
|
(292,626) |
|
(536,063) |
|
$ |
4.15 |
|
$ |
3.88 |
|
$ |
4.50 |
|
|
Options outstanding, end of year |
|
2,893,240 |
|
4,015,895 |
|
4,525,340 |
|
$ |
3.48 |
|
$ |
3.42 |
|
$ |
3.48 |
|
|
Options exercisable, end of year |
|
2,495,650 |
|
2,733,842 |
|
3,115,798 |
|
$ |
3.42 |
|
$ |
3.64 |
|
$ |
3.27 |
|
The weighted average fair value at the date of grant for options granted during the years ended June 30, 2017, 2016 and 2015 were $2.21, $1.08 and $1.91 per share, respectively.
The aggregate intrinsic value of the outstanding stock options as of June 30, 2017 and 2016 is $15.5 million and $0.3 million, respectively. The aggregate intrinsic value of the exercisable stock options as of June 30, 2017 and 2016 is $13.5 million and $25 thousand, respectively. The aggregate intrinsic value is the sum of the amounts by which the quoted market price of the Company’s common stock exceeded the exercise price of the options at June 30, 2017, for those options for which the quoted market price was in excess of the exercise price. The total intrinsic value of options exercised during the 2017, 2016, and 2015 fiscal years was $2.6 million, $1.2 million, and $1.8 million, respectively. Included in research and development and general and administrative expense categories the Company has recorded $1.8 million, $1.5 million, and $1.4 million for stock-based compensation expense related to these stock options during the 2017, 2016, and 2015 fiscal years, respectively.
76
The following table summarizes information concerning options outstanding under the Plan at June 30, 2017:
|
|
|
|
|
Weighted |
|
Weighted |
|
|
|
Weighted |
|
||
|
|
|
Number |
|
average |
|
average |
|
Number |
|
average |
|
||
|
Range of |
|
outstanding |
|
exercise |
|
remaining |
|
exercisable |
|
exercise |
|
||
|
exercise price |
|
at June 30, 2017 |
|
price |
|
term (yrs.) |
|
at June 30, 2017 |
|
price |
|
||
|
$1.59 - 3.00 |
|
655,163 |
|
$ |
1.98 |
|
5.30 |
|
491,533 |
|
$ |
1.88 |
|
|
3.01 - 5.00 |
|
1,950,233 |
|
|
3.65 |
|
3.54 |
|
1,763,729 |
|
|
3.59 |
|
|
5.01 - 7.00 |
|
242,844 |
|
|
5.15 |
|
3.16 |
|
240,388 |
|
|
5.15 |
|
|
7.01 - 9.50 |
|
45,000 |
|
|
8.70 |
|
7.00 |
|
— |
|
|
— |
|
|
|
|
2,893,240 |
|
$ |
3.48 |
|
3.96 |
|
2,495,650 |
|
|
3.42 |
|
At the Compensation Committee meeting held on August 14, 2014, the Company awarded an additional 226,657 restricted stock units to certain executive officers of the Company at the closing market price on that date ($3.32 per share). On August 20, 2015, the Company awarded an additional 214,205 restricted stock units to certain executive officers of the Company at the closing price on that date ($1.76 per share). On September 21, 2016, the Company awarded an additional 106,061 restricted stock units to an executive officer of the Company at the closing price on that date ($3.30 per share). These restricted stock units vest over a four year period. As of June 30, 2017 the 331,329 outstanding restricted stock units had vested in accordance with the terms of those agreements with the change in control which occurred on or before May 4, 2017. Consequently, as of June 30, 2017 there was no unrecognized compensation costs related to non-vested share-based compensation arrangements granted under the Plan for these restricted stock units. The Company recorded $1.2 million, $0.6 million, and $0.8 million for stock-based compensation expense for these executive officers for the fiscal years ended June 30, 2017, 2016, and 2015, respectively.
On August 16, 2013, the Company awarded certain executive officers Performance Units of up to 389,864 of restricted stock units which are subject to attainment of certain performance milestones as well as certain continued service requirements. All or a portion of the Performance Units would vest based upon the level of achievement of the milestones set forth in each agreement, which was expected to be achieved within five years of the grant date. The Performance Units had vested were based upon attainment of the Performance milestone and were exercised based on a percentage basis on the attainment of anniversary dates. As of June 30, 2017, all of these Performance Units have vested. The Company recorded $0.1 million, $0.3 million, and $0.4 million for the stock-based compensation for the fiscal years ended June 30, 2017, 2016, and 2015, respectively.
As part of the Amended and Restated Employment Agreement with Dr. Goldenberg which became effective July 1, 2015 (see Note 13), Dr. Goldenberg received a grant of 1,500,000 Restricted Stock Units, which would vest, if at all, after the three (3) year period commencing on the grant date of July 14, 2015, provided the applicable milestones based on achievement of certain market conditions (stock prices) were met and conditioned upon Dr. Goldenberg's continued employment through the vesting period, subject to the terms and conditions of the Restricted Stock Units Notice and the Restricted Stock Units Agreement and such other terms and conditions as set forth in the grant agreement. The Company recorded $1.1 million for the stock-based compensation for the fiscal years ended June 30, 2017 and 2016 for this agreement. The Company believes that a change in control occurred on or before May 4, 2017, as defined in Dr. Goldenberg's employment agreement as a result of the new Board of Directors being seated. According to the terms of his employment agreement and notice of award, the Company believes that these 1.5 million restricted stock units did not vest since at the time of the change in control the actual price per share of the common stock had not achieved the specified target price required to trigger the vesting of the Restricted Stock Units. The Company understands that Dr. Goldenberg contests the Company’s interpretation of both the timing of the change in control and the vesting requirements of the Restricted Stock Units upon a change in control. The 1.5 million Restricted Stock Units are the subject of arbitration, discussed above.
77
The Restricted Stock Units granted to Dr. Goldenberg were valued using Monte Carlo simulation technique using the following assumptions:
|
Expected dividend |
|
0 |
% |
|
Expected option term (years) |
|
5 |
|
|
Expected stock price volatility |
|
49.6 |
% |
|
Risk-free interest rate |
|
1.32 |
% |
A summary of the Company’s non-vested restricted stock units at June 30, 2017, and changes during the year ended June 30, 2017 is presented below:
|
|
|
|
|
Weighted-Average |
|
|
|
|
|
|
|
per Share of |
|
|
|
|
|
|
|
Market Value |
|
|
|
Non-Vested Restricted Stock |
|
Number of Awards |
|
on Grant Date |
|
|
|
Non-vested at June 30, 2016 |
|
2,066,041 |
|
$ |
2.57 |
|
|
Restricted Units Granted(a) |
|
106,061 |
|
|
3.30 |
|
|
Cancelled |
|
(14,469) |
|
|
3.11 |
|
|
Vested/Exercised(b) |
|
(657,633) |
|
|
3.33 |
|
|
Non-vested at June 30, 2017 |
|
1,500,000 |
|
$ |
2.28 |
|
|
(a) |
For the year ended June 30, 2017, 106,061 restricted stock units were awarded to the Company’s former President and Chief Executive Officer. |
|
(b) |
Represents restricted stock units to the Company’s former President and Chief Executive Officer which vested upon her termination and pursuant to the Company’s change of control, and the Chairman which vested upon the Company’s change of control. |
As of June 30, 2017, the Company has 1,897,590 non-vested options and restricted stock units outstanding. As of June 30, 2017, 2016, and 2015 there was $2.0 million, $5.2 million, and $4.5 million, respectively, of total unrecognized compensation cost related to non-vested share-based compensation arrangements granted under the Plan. That cost is being recognized over a weighted-average period of 2.0 years. The weighted average remaining contractual terms of the exercisable shares is 1.92 years and 3.15 years as of June 30, 2017 and 2016, respectively.
The following table summarizes the stock-based compensation expense by the consolidated statements of comprehensive loss line items for the fiscal years ended June 30, 2017, 2016 and 2015 (in thousands):
|
|
|
Fiscal Year Ended June 30, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Research and development |
|
$ |
2,600 |
|
$ |
2,245 |
|
$ |
1,673 |
|
|
General and administrative |
|
|
1,733 |
|
|
1,496 |
|
|
1,116 |
|
|
Total expense |
|
$ |
4,333 |
|
$ |
3,741 |
|
$ |
2,789 |
|
78
11. Accumulated Other Comprehensive (Loss) Income
The components of accumulated other comprehensive (loss) income were as follows (in thousands):
|
|
|
Currency |
|
Net Unrealized Gains |
|
Accumulated Other |
|
|||
|
|
|
Translation |
|
(Losses) on Available- |
|
Comprehensive |
|
|||
|
|
|
Adjustments |
|
for-Sale Securities |
|
(Loss) Income |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, June 30, 2014 |
|
|
262 |
|
|
— |
|
|
262 |
|
|
Other comprehensive (loss) income before reclassifications |
|
|
(435) |
|
|
23 |
|
|
(412) |
|
|
Amounts reclassified from accumulated other comprehensive (loss)(a) |
|
|
— |
|
|
(11) |
|
|
(11) |
|
|
Net other comprehensive (loss) income for the year |
|
|
(435) |
|
|
12 |
|
|
(423) |
|
|
Balance, June 30, 2015 |
|
|
(173) |
|
|
12 |
|
|
(161) |
|
|
Other comprehensive income before reclassifications |
|
|
1 |
|
|
30 |
|
|
31 |
|
|
Amounts reclassified from accumulated other comprehensive income(a) |
|
|
— |
|
|
(2) |
|
|
(2) |
|
|
Net other comprehensive income for the year |
|
|
1 |
|
|
28 |
|
|
29 |
|
|
Balance, June 30, 2016 |
|
$ |
(172) |
|
$ |
40 |
|
$ |
(132) |
|
|
Other comprehensive loss before reclassifications |
|
|
(62) |
|
|
(125) |
|
|
(187) |
|
|
Amounts reclassified from accumulated other comprehensive loss(a) |
|
|
— |
|
|
16 |
|
|
16 |
|
|
Net other comprehensive loss for the year |
|
|
(62) |
|
|
(109) |
|
|
(171) |
|
|
Balance, June 30, 2017 |
|
$ |
(234) |
|
$ |
(69) |
|
$ |
(303) |
|
|
(a) |
For the fiscal years ended June 30, 2017, 2016 and 2015, $16 thousand, $2 thousand, and $11 thousand, respectively, were reclassified from accumulated other comprehensive (loss) income to interest and other income, respectively. |
All components of accumulated other comprehensive (loss) income are net of tax, except currency translation adjustments, which exclude income taxes related to indefinite investments in foreign subsidiaries.
79
12. Income Taxes
The expense (benefit) for income taxes is as follows (in thousands):
|
|
|
Year Ended June 30, |
|
|||||||
|
|
|
2017 |
|
2016 |
|
2015 |
|
|||
|
Federal |
|
|
|
|
|
|
|
|
|
|
|
Current |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
|
Deferred |
|
|
— |
|
|
— |
|
|
— |
|
|
Total Federal |
|
|
— |
|
|
— |
|
|
— |
|
|
State |
|
|
|
|
|
|
|
|
|
|
|
Current |
|
|
2 |
|
|
(5,054) |
|
|
2 |
|
|
Deferred |
|
|
— |
|
|
— |
|
|
— |
|
|
Total State |
|
|
2 |
|
|
(5,054) |
|
|
2 |
|
|
Foreign |
|
|
|
|
|
|
|
|
|
|
|
Current |
|
|
19 |
|
|
— |
|
|
56 |
|
|
Deferred |
|
|
— |
|
|
— |
|
|
— |
|
|
Total Foreign |
|
|
19 |
|
|
— |
|
|
56 |
|
|
Total Expense (Benefit) |
|
$ |
21 |
|
$ |
(5,054) |
|
$ |
58 |
|
A reconciliation of the statutory tax rates and the effective tax rates for each of the years ended June 30 is as follows:
|
|
|
2017 |
|
2016 |
|
2015 |
|
|
Statutory rate |
|
(34.0) |
% |
(34.0) |
% |
(34.0) |
% |
|
Foreign income tax |
|
— |
% |
— |
% |
0.1 |
% |
|
Change in valuation allowance |
|
21.9 |
% |
30.4 |
% |
34.7 |
% |
|
State income taxes, (net of federal tax benefit) |
|
(4.8) |
% |
(2.8) |
% |
— |
% |
|
Permanent differences, (primarily warrant-related expenses) |
|
15.3 |
% |
— |
% |
— |
% |
|
Other |
|
1.6 |
% |
(1.6) |
% |
(0.7) |
% |
|
Effective rate |
|
— |
% |
(8.0) |
% |
0.1 |
% |
For fiscal year 2016, the Company sold certain State of New Jersey State Net Operating Losses (“NOL”) and Research and Development (“R&D”) tax credits through the New Jersey Economic Development Authority Technology Business Tax Certificate Transfer Program. Pursuant to such sale, for the year ended June 30, 2016, the Company recorded a tax benefit of $5.1 million, as a result of its sale of approximately $66.2 million, of New Jersey State NOL and $1.5 million of New Jersey R&D tax credits. There were no sales of NOL or R&D for the 2017 or 2015 fiscal years.
The tax effects of temporary differences that give rise to significant portions of the Company’s deferred tax assets as of June 30, 2017 and 2016 are presented below (in thousands):
|
|
|
2017 |
|
2016 |
|
||
|
Deferred tax assets: |
|
|
|
|
|
|
|
|
NOL carry forwards |
|
$ |
134,476 |
|
$ |
103,171 |
|
|
Research and development credits |
|
|
14,357 |
|
|
15,322 |
|
|
Property and equipment |
|
|
3,406 |
|
|
3,693 |
|
|
Other |
|
|
7,335 |
|
|
3,734 |
|
|
Total |
|
|
159,574 |
|
|
125,920 |
|
|
Valuation allowance |
|
|
(159,574) |
|
|
(125,920) |
|
|
Net deferred taxes |
|
$ |
— |
|
$ |
— |
|
A valuation allowance is provided when it is more likely than not that some portion or all of the deferred tax assets will not be realized. The valuation allowances for fiscal years 2017 and 2016 have been applied to offset the deferred tax assets in recognition of the uncertainty that such tax benefits will be realized as the Company continues to
80
incur losses. The differences between book income and tax income primarily relate to the temporary differences from depreciation and stock compensation expenses.
At June 30, 2017, the Company has available net operating loss carry forwards for federal income tax reporting purposes of approximately $371.1 million and for state income tax reporting purposes of approximately $186.0 million, which expire at various dates between fiscal 2019 and 2037. Pursuant to Section 382 of the Internal Revenue Code of 1986, as amended, the annual utilization of a company’s net operating loss and research credit carry forwards may be limited if the Company experiences a change in ownership as defined in Section 382 of the Internal Revenue Code. The Company’s net operating loss carry forwards available to offset future federal taxable income arising before such ownership changes may be limited. Similarly, the Company may be restricted in using its research credit carry forwards arising before such ownership changes to offset future federal income tax expense.
At June 30, 2017, the Company did not have any material unrecognized tax benefits and the Company does not anticipate that its unrecognized tax benefits will significantly change in the next twelve months. The Company will recognize potential interest and penalties related to income tax positions as a component of the provision for income taxes on the Consolidated Statements of Comprehensive Loss in any future periods in which the Company must record a liability. The Company is subject to examination for U.S. Federal and Foreign tax purposes for 2012 and forward and for New Jersey 2013 and forward. The Company conducts business and files tax returns in New Jersey.
13. Related Party Transactions
Certain of the Company’s affiliates, including members of its senior management and Board of Directors, as well as their respective family members and other affiliates, have relationships and agreements among themselves as well as with the Company and its affiliates, that create the potential for both real, as well as perceived, conflicts of interest. These include Dr. David M. Goldenberg, the Company’s Chief Scientific Officer, Chief Patent Officer and former Chairman, Ms. Cynthia L. Sullivan, a director and the former President and Chief Executive Officer, who is the wife of Dr. David M. Goldenberg, and certain companies with which the Company does or has done business with, including the Center for Molecular Medicine and Immunology (“CMMI”), which has ceased operations, and IBC, the Company’s majority-owned subsidiary.
Dr. David M. Goldenberg founded Immunomedics in 1982 and was the Company’s Chairman of the Board of Directors through April 4, 2017. He continues to play a critical role in the Company’s business and currently serves as the Chief Scientific Officer and Chief Patent Officer. Dr. Goldenberg is a party to a number of agreements with the Company involving not only his services, but intellectual property owned by him.
Relationships with The Center for Molecular Medicine and Immunology
The Company’s product development has involved, to varying degrees, CMMI, for the performance of certain basic research and patient evaluations, the results of which are made available to the Company pursuant to a collaborative research and license agreement. Dr. Goldenberg was the founder, President and a member of the Board of Trustees of CMMI.
In fiscal years ended June 30, 2017, 2016 and 2015, the Company incurred $6 thousand, $27 thousand, and $33 thousand, respectively, of legal expenses for patent related matters for patents licensed to Immunomedics from CMMI. However, any inventions made independently of the Company at CMMI are the property of CMMI. CMMI has ceased operations and is in the process of dissolution.
IBC is a majority-owned subsidiary of Immunomedics, Inc.
81
As of June 30, 2017, the shares of IBC were held as follows:
|
Stockholder |
|
Holdings |
|
Percentage of Total |
|
|
Immunomedics, Inc. |
|
5,615,124 shares of Series A Preferred Stock |
|
73.46 |
% |
|
Third Party Investors |
|
628,282 shares of Series B Preferred Stock |
|
8.22 |
% |
|
David M. Goldenberg Millennium Trust |
|
1,399,926 shares of Series C Preferred Stock |
|
18.32 |
% |
|
|
|
|
|
100.00 |
% |
In the event of a liquidation, dissolution or winding up of IBC, the Series A, B and C Preferred Stockholders would be entitled to $0.6902, $5.17 and $0.325 per share (subject to adjustment), respectively. The Series A and B stockholders would be paid ratably until fully satisfied. The Series C stockholders would be paid only after the Series A and B stockholders have been fully repaid. These liquidation payments would be made only to the extent the assets of IBC are sufficient to make such payments.
In each of the fiscal years 2017, 2016, and 2015, Dr. Goldenberg received $41 thousand, $87 thousand, and $84 thousand, respectively, in compensation for his services to IBC. At June 30, 2017, Dr. Goldenberg was a director of IBC, and Cynthia L. Sullivan served as the President of IBC.
14. License and Collaboration Agreement
The Bayer Group (formerly Algeta ASA)
In January 2013 the Company entered into a collaboration agreement, (the “Collaboration Agreement”), with Algeta ASA (subsequently acquired by The Bayer Group (“Bayer”), for the development of epratuzumab to be conjugated with Algeta’s proprietary thorium-227 alpha-pharmaceutical payload. Under the terms of the Collaboration Agreement, the Company manufactured and supplied clinical-grade epratuzumab to Bayer, which has rights to evaluate the potential of a Targeted Thorium Conjugate (“TTC”), linking thorium-227 to epratuzumab, for the treatment of patients with cancer. Bayer has the right to terminate the Collaboration Agreement with three months prior written notice, subject to certain provisions. Bayer will fund all non-clinical and clinical development costs up to the end of Phase 1 clinical testing. Upon successful completion of Phase 1 testing, the parties shall negotiate terms for a license agreement at Bayer’s request. The Company and Bayer have agreed to certain parameters in the Collaboration Agreement. Under the terms of the Collaboration Agreement, as amended, Immunomedics received an upfront cash payment and other payments aggregating $6.0 million, which have been recognized in prior periods upon the Company fulfilling its obligations under the Collaboration Agreement.
For the year ended June 30, 2015, the Company recognized $1.0 million in license and other revenue for the completion of the clinical development milestone as described in the Collaboration Agreement, which required the shipment of sufficient quantities of clinical grade material to Bayer to complete their Phase 1 clinical trial. In addition, in January 2017, 2016, and 2015, the Company recorded revenue of $0.3 million representing an anniversary payment under the agreement. This agreement has been extended to December 30, 2018.
15. Commitments and Contingencies
a. Employment Agreements
Effective July 1, 2015, the Company entered into the Amended and Restated Employment Agreement with Dr. Goldenberg pertaining to Dr. Goldenberg’s service to the Company as the Company’s Chairman of the Board, Chief Scientific Officer and Chief Patent Officer (the “Amended and Restated Goldenberg Agreement”). The Amended and Restated Goldenberg Agreement was to continue until July 1, 2020.
On May 3, 2017 Dr. Goldenberg and other parties entered into a binding Term Sheet, to resolve certain legal actions among the parties. Upon execution of the contemplated Settlement Agreement, Dr. Goldenberg will remain a
82
director of the Company, but has agreed to resign from all officer and other positions of the Company and all director, officer and other positions at any of the Company’s affiliates (other than Dr. Goldenberg’s position as a member of the board of directors of IBC Pharmaceuticals, the Company’s majority owned U.S. subsidiary), effective as of the date of the Settlement Agreement. The Settlement Agreement will provide that Dr. Goldenberg will abide by all post-termination covenants and obligations contemplated by the Amended and Restated Goldenberg Agreement. In exchange for a release of claims as required by the Amended and Restated Goldenberg Agreement and subject to compliance with the terms of the Settlement Agreement, Dr. Goldenberg will be entitled to (i) termination payments in accordance with the Amended and Restated Goldenberg Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of exercise period for equity awards already earned, pursuant to the Amended and Restated Goldenberg Agreement, (iii) COBRA payments, and (iv) royalties or payment in accordance with existing agreements. As of June 30, 2017 the Company recorded a $2.7 million expense for this settlement cost. In addition to this amount an additional cash payment of approximately $1.8 million is in dispute. The 1.5 million Restricted Stock Units to Dr. Goldenberg under the terms of the Amended and Restated Goldenberg Agreement are the subject of arbitration, discussed above.
To the extent the Parties cannot reach agreement on such issues before execution of the Settlement Agreement, the Parties to the Term Sheet have agreed to arbitrate disputes relating to Dr. Goldenberg’s claims to certain equity awards and other severance payments. The Company has agreed to pay the arbitrator in full for such arbitration, as well as reasonable attorneys’ fees and expenses incurred by Dr. Goldenberg and Ms. Sullivan in connection with any such arbitration, up to a maximum amount of $650,000 combined. As of June 30, 2017 no expenses have been incurred regarding such arbitration.
Under the existing agreements, Dr. Goldenberg is eligible to receive royalty payments on royalties received by the Company. For each fiscal year the Company shall pay Dr. Goldenberg a sum equal to a percentage of the annual royalties the Company receives on each of the products for which Dr. Goldenberg is an Inventor, and all products using, related to or derived from products for which Dr. Goldenberg is an Inventor. The percentage of royalties that the Company will pay to Dr. Goldenberg on each patented product will be determined based on the percentage of royalties that the Company must pay to external third parties and payments are to continue for the life of the patent, as defined in his employment agreement.
Dr. Goldenberg is also eligible to receive minimum payments of $150 thousand during each of the fiscal years, payable in equal quarterly payments, as an advance against the amounts due as additional incentive compensation, royalty payments and dispositions of undeveloped assets. In the event the Company completes a disposition of the Company’s undeveloped assets for which Dr. Goldenberg was an Inventor, the Company will pay Dr. Goldenberg a sum equal to at least twenty percent or more of the consideration the Company receives from each disposition. The Company’s obligation to compensate Dr. Goldenberg upon dispositions of undeveloped assets applies to all dispositions completed within the contract term or within three years thereafter even if the Company actually receives the consideration at some time after the three (3) year period elapses.
For the 2017, 2016 and 2015 fiscal years, Dr. Goldenberg received the minimum payment under the employment agreement. Dr. Goldenberg also is compensated by IBC Pharmaceuticals as discussed in greater detail below.
Cynthia L. Sullivan
Effective July 1, 2014, the Company entered into the Fifth Amended and Restated Employment Agreement with Cynthia L. Sullivan pertaining to Ms. Sullivan’s service to the Company as the Company’s President and Chief Executive Officer (the “Amended Sullivan Agreement”). This agreement was terminated effective July 1, 2017.
On May 3, 2017 Ms. Sullivan and other parties entered into the Term Sheet to resolve certain legal actions among the parties as described in Note 15 below. Upon execution of the contemplated Settlement Agreement, Ms. Sullivan has agreed to resign from director of the Company and any of its affiliates, effective as of the date of the Settlement Agreement. The Settlement Agreement will provide that Ms. Sullivan will abide by all post-termination covenants and obligations contemplated by the Amended Sullivan Agreement. In exchange for a release of claims as required by the Sullivan Agreement and subject to compliance with the terms of the Settlement Agreement, Ms. Sullivan
83
will be entitled to (i) termination payments in accordance with the Amended Sullivan Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of the exercise period for equity awards already earned, pursuant to the Amended Sullivan Agreement, and (iii) COBRA payments. As of June 30, 2017 the Company recorded a $4.2 million expense for all costs that would be due to Ms. Sullivan under the terms of the Settlement Agreement. In addition to this amount, a cash payment of $0.9 million is in dispute.
The Parties to the Term Sheet have agreed to arbitrate disputes relating to Ms. Sullivan’s claimed entitlement to certain equity awards and severance payments, and Ms. Sullivan’s claimed rights to certain bonus payments, to the extent the Parties cannot reach agreement on such issues before execution of the Settlement Agreement. The Company has agreed to pay in full the arbitrator in such arbitration as well as reasonable attorneys’ fees and expenses incurred by Ms. Sullivan and Dr. Goldenberg in connection with any such arbitration, up to a maximum amount of $650,000 combined. As of June 30, 2017 no expenses have been incurred regarding such arbitration.
b. Operating Lease
Immunomedics is obligated under an operating lease for facilities used for research and development, manufacturing and office space, expiring in October 2031 at a base annual rate of $1.0 million, which is fixed through October 2021 and increases thereafter every five years. Rental expense related to this lease was approximately $0.9 million, $0.8 million and $0.8 million for fiscal years 2017, 2016, and 2015, respectively.
The minimum lease commitments for the non-cancelable term of the facility lease described above are as follows for fiscal years (in thousands):
|
2018 |
$ |
974 |
||
|
2019 |
|
$ |
974 |
|
|
2020 |
|
$ |
974 |
|
|
2021 |
|
$ |
974 |
|
|
2022 |
|
$ |
1,020 |
|
|
Thereafter |
|
$ |
9,742 |
|
c. Change of Control Agreements
Certain employees have Change of Control Agreements, whereby if a majority of a new board of directors is constituted by newly elected board members not endorsed by the Company’s current Board of Directors, and if, subsequent to such a change, there is a significant change in the responsibilities or employment status of these executives, then severance provisions included in their Change of Control Agreements could be triggered. These severance provisions could result in accelerated vesting of equity compensation and significant, unbudgeted, cash severance payments.
d. Legal Matters
Patent litigation:
Immunomedics filed a first amended complaint on October 22, 2015, a second amended complaint on January 14, 2016, and a third amended complaint on October 12, 2016, in the United States District Court for the District of New Jersey, against Roger Williams Medical Center (“RWMC”), Richard P. Junghans, M.D., Ph.D., and Steven C. Katz, M.D. The third amended complaint alleges that RWMC and Dr. Junghans breached a Material Transfer Agreement (“MTA”) through which it provided to them a monoclonal antibody known as MN-14 and related materials. Defendants are alleged to have breached the MTA and to have been negligent by, among other things, using the materials beyond the agreed-upon Research Project, sharing confidential information, failing to provide Immunomedics with a right of first refusal, failing to notify Immunomedics of intended publications prior to publishing, and refusing to return the materials upon request. Immunomedics also asserts the following claims against some of these defendants: conversion, tortious interference, unjust enrichment, and infringement of three patents owned by Immunomedics. Defendants Junghans, Katz, and RWMC subsequently moved to dismiss for failure to state a claim on November 14, 2016, but this motion was denied on January 4, 2017. The third amended complaint also added parties named Sorrento, TNK, BDL, and CARgenix. On December 2, 2016, Sorrento, TNK, BDL, and CARgenix moved to dismiss for lack of personal
84
jurisdiction over them in New Jersey. The court granted this motion on January 25, 2017. On January 20, 2017, the court held a Markman hearing to construe the claims in the patents in suit. On February 28, 2017, the court issued an opinion and order finding, inter alia, that the term “effective amount” in the patents in suit is not indefinite and should be given its plain and order meaning, as proposed by Immunomedics, of “an amount capable of producing the claim result.” All other terms in the patents were given their plain and ordinary meaning. On May 11, 2017, the Court ordered the parties to mediation with former New Jersey District Court Judge Garrett Brown, and stayed the case for 90 days. A mediation took place on June 28, 2017. The mediation was unsuccessful; and the stay of discovery will be lifted on August 9.
Stockholder complaints:
Class Action Stockholder Federal Securities Cases
Two purported class action cases have been filed in the United States District Court for the District of New Jersey; namely, Fergus v. Immunomedics, Inc., et al., No. 2:16-cv-03335, filed June 9, 2016; and Becker v. Immunomedics, Inc., et al., No. 2:16-cv-03374, filed June 10, 2016. These cases arise from the same alleged facts and circumstances, and seek class certification on behalf of purchasers of our common stock between April 20, 2016 and June 2, 2016 (with respect to the Fergus matter) and between April 20, 2016 and June 3, 2016 (with respect to the Becker matter). These cases concern the Company's statements in press releases, investor conference calls, and SEC filings beginning in April 2016 that the Company would present updated information regarding its IMMU-132 breast cancer drug at the 2016 American Society of Clinical Oncology ("ASCO") conference in Chicago, Illinois. The complaints allege that these statements were false and misleading in light of June 2, 2016 reports that ASCO had cancelled the presentation because it contained previously reported information. The complaints further allege that these statements resulted in artificially inflated prices for our common stock, and that the Company and certain of its officers are thus liable under Sections 10(b) and 20(a) of the Securities Exchange Act of 1934. An order of voluntarily dismissal without prejudice was entered on November 10, 2016 in the Becker matter. An order granting motion to consolidate cases, appoint lead plaintiff, and approve lead and liaison counsel was entered on February 7, 2017 in the Fergus matter. As of the date hereof, service of the initiating papers in the Fergus matter has not been made on the Company.
Stockholder Derivative Action in the Superior Court of New Jersey
On October 3, 2016, plaintiff commenced an action captioned Rosenfeld v. Goldenberg, et al., No. L-2200-16, alleging the same underlying facts and circumstances as in the pending federal securities class action, the Fergus matter. Specifically, this action concerns the Company’s statements in press releases, investor conference calls, and SEC filings beginning in April 2016 that the Company would present updated information regarding its IMMU-132 breast cancer drug at the 2016 ASCO conference in Chicago, Illinois. The complaint alleges that these statements were false and misleading in light of the June 2, 2016 reports that ASCO had cancelled the presentation because it contained previously reported information. The complaint further alleges that these statements resulted in artificially inflated prices for our common stock, and that certain directors and officers of the Company breached their fiduciary duties to the Company. In addition to monetary damages, the complaint seeks to require the Company to reform its corporate governance and internal procedures. Service was effectuated on all defendants on April 7, 2017. The defendants filed a motion to dismiss the complaint on June 19, 2017.
Class Action Stockholder Claim in the Court of Chancery of the State of Delaware
On December 13, 2016, plaintiff commenced an action captioned Desanctis v. Goldenberg, C.A. No. 12981-VCL (Del. Ch. Ct.), alleging that the Company's Board of Directors failed to comply with Delaware law and breached their fiduciary duties when it rescheduled the Immunomedics 2016 Annual Meeting of Stockholders from December 14, 2016 to February 16, 2017. On December 22, 2016, the Delaware Court of Chancery refused to schedule an expedited hearing in the action and concluded that plaintiff failed to carry his burden of demonstrating that he had pleaded a colorable claim and that there was a threat of irreparable harm. The Court further stated that the Complaint failed to demonstrate that the Board's actions were unreasonable when it rescheduled the Annual Meeting in response to venBio Select Advisor LLC's proxy contest.
85
Stockholder Claim in the Court of Chancery of the State of Delaware
On February 13, 2017, venBio commenced an action captioned venBio Select Advisor LLC v. Goldenberg, et al., C.A. No. 2017-0108-VCL (Del. Ch.) (the “venBio Action”), alleging that members of the Company’s Board breached their fiduciary duties when the Board (i) rescheduled the Company’s 2016 Annual Meeting of Stockholders (the “2016 Annual Meeting”) from December 14, 2016 to February 16, 2017, and then again to March 3, 2017, and (ii) agreed to the proposed Licensing Transaction with Seattle Genetics. venBio also named Seattle Genetics as a defendant and sought an injunction preventing the Company from closing the licensing transaction with Seattle Genetics. On March 6, 2017, venBio amended its complaint, adding further allegations, including that members of the Company’s Board breached their fiduciary duties when the Board amended the Company’s Amended and Restated By-laws (the “By-Laws”) to call for a plurality voting regime for the election of directors instead of majority voting, and providing for mandatory advancement of attorneys’ fees and costs for the Company’s directors and officers. The Court of Chancery entered a temporary restraining order on March 9, 2017, enjoining the closing of the Licensing Transaction. venBio amended its complaint a second time on April 19, 2017, this time adding as an additional defendant the Company’s financial advisor on the Licensing Transaction, Greenhill.
On May 4, 2017, the Company entered into the Termination Agreement with Seattle Genetics, pursuant to which the Company and Seattle Genetics agreed to relinquish their respective rights under the Licensing Agreement and amend the term of the SGEN Warrant, and in connection therewith, the Company and venBio agreed to fully settle, resolve and release Seattle Genetics, and Seattle Genetics agreed to fully settle, resolve and release the Company and venBio, from all disputes, claims and liabilities arising from the Licensing Agreement and the transactions contemplated thereby, subject to the terms of the Termination Agreement and the related settlement agreement. The Termination Agreement will be effective thirty days following the entry on July 25, 2017 of a final judgment of the Court of Chancery approving the dismissal of Seattle Genetics from the venBio Action.
On May 3, 2017, venBio and the Company and individual defendants Goldenberg, Sullivan and Markison (collectively, the “Individual Defendants”) entered into a binding Term Sheet, which is to be reduced to a definitive settlement agreement (“Settlement Agreement”), pursuant to which, among other things, venBio and the Company will release the Individual Defendants from all certain claims described below and will submit the remaining claims against the non-settling defendants (including non-settling defendants and former directors Robert Forrester, Jason Aryeh, Geoff Cox and Bob Oliver, but excluding those claims with respect to Seattle Genetics, which the parties have agreed to settle pursuant to the Termination Agreement as described above) to non-binding mediation. Once the Parties execute the Settlement Agreement, it will be submitted to the Court of Chancery for approval.
On June 8, 2017, venBio, the Company and Greenhill entered into a binding term sheet (the “GHL Term Sheet”), which is to be reduced to a definitive settlement agreement (“GHL Settlement Agreement”), pursuant to which, among other things, venBio and the Company will release Greenhill from “all direct and derivative claims that have been or could be asserted by or on behalf of venBio, the Company, or the directors, officers, employees, affiliates and related persons of venBio or the Company, whether known or unknown, against Greenhill and Greenhill’s affiliates and related persons in connection with the claims alleged in the venBio Action, the 225 Action, the Federal Action, the Licensing Transaction, the Financing, the Company’s 2017 annual stockholder meeting, Greenhill’s 9/23/2016 and 12/14/2016 engagement letters with the Company (the “Engagement Letters”) (including any claims related to or arising in any manner out of any activities performed or services furnished pursuant to the Engagement Letters, the transactions contemplated thereby or Greenhill’s role in connection therewith), and this settlement.” Greenhill similarly agreed to release “all direct and derivative claims that have been or could be asserted by or on behalf of Greenhill, or the directors, officers, employees, affiliates and related persons of Greenhill, whether known or unknown, against venBio or the Company and their current and former directors, officers, members, employees, affiliates and related persons in connection with the claims alleged in the venBio Action, the 225 Action, the Federal Action, the Licensing Transaction, the Financing, the Company’s 2017 annual stockholder meeting, the Engagement Letters, and this settlement.” Greenhill, the Company and venBio also agreed that the Engagement Letters would be terminated and that Greenhill would forgo and not seek any and all fees, expense, reimbursement or indemnification from the Company, except that, upon final Court approval of the GHL Settlement Agreement, the Company shall reimburse Greenhill up to $200,000 for reasonable and documented expenses that Greenhill incurred in connection with services provided under the Engagement Letters. Greenhill also consented to the settlement reflected in the Term Sheet and Greenhill, the
86
Company and venBio agreed that Greenhill need not participate in the non-binding mediation contemplated by the Term Sheet.
Lawsuit Against venBio Select Advisor LLC in the U.S. District Court (Delaware) (the “District Court”)
On February 17, 2017, the Company commenced an action captioned Immunomedics, Inc. v. venBio Select Advisor LLC, No. 17-176-LPS (D. Del.) (the “Federal Action”), seeking for the District Court to invalidate the proxies solicited by venBio in furtherance of its contest for the election of directors of the Company. The Company named as defendants venBio and its then-nominees, Behzad Aghazadeh, Scott Canute, Peter Barton Hutt, and Khalid Islam. The Company alleged that venBio had conducted its proxy contest and solicited proxies in violation the federal securities laws and regulations, namely by failing timely file a Schedule 13D form indicating venBio’s intent to effectuate change at the Company, publishing early voting results of the Company’s annual election of directors, publishing improper statements about the then-incumbent Board, forming a “group” of like-minded stockholders without publicly disclosing the group, and soliciting proxies without disclosing the solicitations to the SEC. On February 21, 2017, the Company sought an injunction preventing, among other things, the venBio nominees from benefiting from allegedly illegal shadow proxy contest, including, but not limited to, by asserting any claimed right to take office as a member of the Board until venBio made corrective disclosures and the stockholders were permitted time to consider them. On March 2, 2017, the District Court denied the Company the requested relief. On April 6, 2017, the District Court entered a stipulation and order pursuant to which the Company’s claims were voluntarily dismissed without prejudice. On April 17, 2017, Dr. Goldenberg, the Company’s Chief Scientific Officer and Chief Patent Officer and director, notified the District Court that he may maintain the claims initially brought by the Company. On May 3, 2017, Goldenberg and venBio entered into a binding Term Sheet which is to be reduced to a difinitive Settlement Agreement, pursuant to which, among other things, the Parties have agreed to submit to the District Court a stipulation and proposed order dismissing all claims in the Federal Action with prejudice, including those against the individual defendants (the then-venBio nominees). The Settlement Agreement will also include a mutual release of claims that were or could have been asserted in the Federal Action.
Lawsuit Challenging the Results of the 2016 Election of Directors
On March 3, 2017, six of the seven then-incumbent members of the Company’s Board commenced an action captioned Goldenberg, et al. vs Aghazadeh, et al., C.A. No. 2017-0163-VCL (Del. Ch.) (the “225 Action”), challenging the results of the election of directors at the 2016 Annual Meeting that took place on March 3, 2017, in which all four of venBio’s nominees won seats on the Company’s Board. The director-plaintiffs named as defendants venBio and its then-nominees, Behzad Aghazadeh, Scott Canute, Peter Barton Hutt, and Khalid Islam. The incumbent directors alleged the same underlying facts as the Company alleged in its lawsuit against venBio in federal court. On March 13, 2017, the Court of Chancery entered an order (the “Status Quo Order”) seating all four venBio nominees (with the three incumbent directors who secured a plurity of votes, (the “Status Quo Board”) and limiting the Company’s Board to actions within the “ordinary course of business,” unless either waived by the parties on a case-by-case basis or ordered by the Court of Chancery. On March 24, 2017, the defendants, venBio and its four nominees, moved to dismiss the action. The plaintiffs in the action have opposed this motion to dismiss, which remains pending. On April 7, 2017, the three incumbent director plaintiffs not seated on the Status Quo Board voluntarily withdrew their claims, leaving Goldenberg, Sullivan and Markison as plaintiffs. On April 20, 2017, the parties agreed to permit the Status Quo Board to explore a potential financing plan for the Company and negotiate a termination of the Licensing Transaction. On May 3, 2017, the Parties entered into the Term Sheet, pursuant to which, among other things, the Parties agreed to submit to the Court of Chancery a stipulation and proposed order lifting the Status Quo Order. On May 4, 2017, the Parties submitted that stipulation, which confirmed that the Status Quo Board is the lawful Board of the Company, provided that if the 225 Action is not dismissed, the Parties shall be restored to their positions in the 225 Action as of immediately prior to the execution of the Term Sheet. Once the Settlement Agreement is executed, the Parties will submit to the Court of Chancery another stipulation and proposed order dismissing the 225 Action with prejudice, including those against the individual defendants (the then-venBio nominees). The Settlement Agreement will also include a mutual release of all claims that were or could have been asserted in the 225 Action.
87
Settlement Term Sheet and Settlement Agreement
On May 3, 2017, the Company entered the Term Sheet by and among the Parties in order to resolve certain legal actions among the Parties, as described below. The Parties also agreed to cooperate and use their best efforts to reduce the Term Sheet to a definitive Settlement Agreement and, to the extent necessary, obtain the approval of the Court of Chancery.
Resolution of Litigation
Pursuant to the Term Sheet, the Parties submitted a stipulation and proposed order to the Court of Chancery lifting the Status Quo Order seating all four venBio nominees (with the three incumbent directors who also won election (based on the plurality vote standard), the “Status Quo Board”) and confirming that the Status Quo Board is the lawful Board of the Company (provided however, if the 225 Action is not dismissed, the Parties will be restored to their positions in the 225 Action as of immediately prior to the execution of the Term Sheet). The Court of Chancery entered the proposed order on the afternoon of May 4, 2017. Pursuant to the Term Sheet, the Parties also agreed to submit a stipulation and proposed order to the Court of Chancery staying the venBio Action (as described below) and removing the trial dates from the calendar of the Court of Chancery.
The Company has further agreed to reimburse venBio for reasonable fees and expenses it incurred in connection with the proxy contest between venBio and the Company, the venBio Action, the 225 Action (as described below) and the Federal Action (as described below and, together with the venBio Action and the 225 Action, the “Actions”), and Goldenberg and Sullivan have agreed to not object to such reimbursement.
The Parties have agreed, immediately upon execution of the Settlement Agreement, to submit stipulations and proposed orders dismissing with prejudice both the 225 Action and the Federal Action. The Settlement Agreement will include (i) a mutual release of all claims that were or could have been asserted in the Federal Action or in the 225 Action and (ii) a release of all direct and derivative claims that have been or could be asserted by or on behalf of (a) venBio or the Company, whether known or unknown, against Goldenberg, Sullivan and Markison and their affiliates and related persons, and (b) Goldenberg, Sullivan or Markison, whether known or unknown, against venBio or the Company and their affiliates and related persons, in both cases in connection with the claims alleged in the venBio Action, the Financing, the settlement of the venBio Action, the Licensing Transaction and the Termination Agreement. The settlement of claims against Goldenberg, Sullivan and Markison in the venBio Action will be subject to approval of the Court of Chancery. venBio and the Company have agreed to stay the venBio Action and submit the claims asserted against the remaining individual defendants (former directors Robert Forrester, Jason Aryeh, Geoff Cox and Bob Oliver) to non-binding mediation. As part of the Termination Agreement, which is subject to the approval of the Court of Chancery, venBio will release SGEN from any claims in the venBio Action.
Financing and Termination of SGEN Transaction
Pursuant to the Term Sheet, Goldenberg and Sullivan have or will (i) vote to support the Financing, (ii) vote to terminate the Licensing Transaction with SGEN, as further discussed below, pursuant to the terms of the Termination Agreement entered into with SGEN, (iii) vote to approve an amendment to the Company’s Amended and Restated Certificate of Incorporation, as amended, to increase the number of shares of authorized Common Stock by an aggregate number of shares of Common Stock to enable conversion of all of the Preferred Shares into shares of Common Stock (the “Charter Amendment”), (iv) approve the submission of a stipulation in the 225 Action to permit the Board to consummate and enter into both the Financing and the Termination Agreement, and (v) agree to not sell any shares of the Company (with certain exceptions) until the date which is the earlier of July 31, 2017 or the date on which the Charter Amendment is approved and the shares of Common Stock issuable upon conversion of the Preferred Shares are registered and issued.
Indemnification
The Term Sheet provides that the Company will, to the extent not covered by the Company’s insurance policies, (i) indemnify Dr. Goldenberg, Ms. Sullivan and Mr. Markison from attorneys’ fees and expenses or other losses
88
in connection with the Actions, and (ii) reimburse and indemnify Dr. Goldenberg and Ms. Sullivan for legal fees for actions taken with respect to the Actions and negotiation of the Settlement Agreement. The Term Sheet provides that the indemnification agreements entered into between the Company and each of Dr. Goldenberg, Ms. Sullivan and Mr. Markison on or about February 9, 2017 shall be terminated and not apply to acts, transactions, legal fees or expenses incurred after approval of the Settlement Agreement by the Court of Chancery.
Intellectual Property Assignments
The Settlement Agreement shall provide that Dr. Goldenberg and Ms. Sullivan will assign global intellectual property rights, other than those subject to existing agreements with the Company and Dr. Goldenberg’s patent and related intellectual property relating to cyber space medicine, to the Company, and perform all acts reasonably requested by the Company to perfect title in and to all such assigned intellectual property.
Sullivan Resignation
The Amended Sullivan Agreement was terminated effective July 1, 2017.
Upon execution of the contemplated Settlement Agreement, Ms. Sullivan has agreed to resign from director of the Company and any of its affiliates, effective as of the date of the Settlement Agreement. The Settlement Agreement will provide that Ms. Sullivan will abide by all post-termination covenants and obligations contemplated by the Amended Sullivan Agreement. In exchange for a release of claims as required by the Amended Sullivan Agreement and subject to compliance with the terms of the Settlement Agreement, Sullivan will be entitled to (i) termination payments in accordance with the Amended Sullivan Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of the exercise period for equity awards already earned, pursuant to the Amended Sullivan Agreement, and (iii) COBRA payments. The Company and Sullivan disagree over the precise amounts owed to Sullivan under the Sullivan Agreement. The foregoing cash payments accumulate to approximately $3.1 million with additional amounts in dispute).
Goldenberg Resignation
Upon execution of the Settlement Agreement, Dr. Goldenberg will remain a director of the Company, but has agreed to resign from all officer and other positions of the Company and all director, officer and other positions at any of the Company’s affiliates (other than Dr. Goldenberg’s position as a member of the board of directors of IBC Pharmaceuticals, the Company’s majority owned U.S. subsidiary), effective as of the date of the Settlement Agreement. The Settlement Agreement will provide that Dr. Goldenberg will abide by all post-termination covenants and obligations contemplated by the Amended and Restated Goldenberg Agreement. In exchange for a release of claims as required by the Amended and Restated Goldenberg Agreement and subject to compliance with the terms of the Settlement Agreement, Dr. Goldenberg will be entitled to (i) termination payments in accordance with the Amended and Restated Goldenberg Agreement for a termination without Good Cause after a Change in Control, (ii) accelerated vesting or extension of exercise period for equity awards already earned, pursuant to the Amended and Restated Goldenberg Agreement, (iii) COBRA payments, (iv) royalties and payments in accordance with existing agreements. The Company and Goldenberg disagree over the precise amount owed to Goldenberg under the Amended and Restated Goldenberg Agreement. The foregoing cash payments accumulate to approximately $2.4 million (with additional amounts in dispute). The Company and Goldenberg also dispute the vesting of 1,500,000 Restricted Stock Units granted to Dr. Goldenberg under the terms of the Amended and Restated Goldenberg Agreement.
Arbitration of Disputed Matters
The Parties have agreed to arbitrate disputes relating to Dr. Goldenberg’s claimed entitlement to certain severance payments, and Dr. Goldenberg’s and Ms. Sullivan’s claimed rights to certain bonus and equity payments, to the extent the Parties cannot reach agreement on such issues before execution of the Settlement Agreement. The Company has agreed to pay in full the arbitrator in such arbitration as well as reasonable attorneys’ fees and expenses incurred by Dr. Goldenberg and/or Ms. Sullivan in connection with any such arbitration, up to a maximum amount of $650,000. As of June 30, 2017 no expenses have been incurred regarding such arbitration.
89
Termination of the SGEN Licensing Agreement
The Company entered into the Licensing Agreement with SGEN, granting SGEN a worldwide, exclusive license, including the right to sublicense subject to the terms and conditions of the License Agreement, to develop, manufacture and commercialize IMMU-132.
On May 4, 2017, the Company and SGEN entered into the Termination Agreement, pursuant to which the Company and SGEN relinquished their respective rights under the Licensing Agreement.
The Termination Agreement constitutes an agreement to terminate the License Agreement and is not in any way an admission of liability or breach by either the Company or SGEN. The Termination Agreement between the Company and SGEN and the settlement of the venBio lawsuit against SGEN remain subject to court approval of the dismissal of the venBio Action. The termination of the Licensing Transaction will be effective as of the date of the approval by the Court of Chancery. In the event the Court of Chancery declines to dismiss the venBio Action against SGEN, or if the effective date of the Termination Agreement does not occur on or before October 1, 2017, any party to the Termination Agreement may terminate the Termination Agreement upon written notice to such other party.
Directors and Officers Liability Insurance
The Company has filed claims with its insurance providers for various expenses incurred through June 30, 2017 for proxy defense-related expenses and reimbursement amounts payable to venBio for fees and expenses incurred by venBio in connection with the proxy contest between venBio and the Company.
Other matters:
Immunomedics is also a party to various claims and litigation arising in the normal course of business, which includes some or all of certain of its patents. While it is not possible to determine the outcome of these matters, the Company believes that the resolution of all such matters will not have a material adverse effect on its consolidated financial position or liquidity, but could possibly be material to its consolidated results of operations in any one accounting period.
16. Geographic Segments
Immunomedics manages its operations as one line of business of researching, developing, manufacturing and marketing biopharmaceutical products, particularly antibody-based products for cancer, autoimmune and other serious diseases, and it currently reports as a single industry segment. Immunomedics conducts its research and development activities primarily in the United States. Immunomedics markets and sells LeukoScan® throughout Europe and in certain other countries outside the United States.
The following table presents financial information based on the geographic location of the facilities of Immunomedics as of and for the years ended (in thousands):
|
|
|
As of and for the year ended |
|
|||||||
|
|
|
June 30, 2017 |
|
|||||||
|
|
|
United |
|
|
|
|
|
|||
|
|
|
States |
|
Europe |
|
Total |
|
|||
|
Total assets |
|
$ |
161,484 |
|
$ |
1,089 |
|
$ |
162,573 |
|
|
Property and equipment, net |
|
|
5,166 |
|
|
79 |
|
|
5,245 |
|
|
Revenues |
|
|
648 |
|
|
2,443 |
|
|
3,091 |
|
|
(Loss) income before taxes |
|
|
(153,348) |
|
|
103 |
|
|
(153,245) |
|
90
|
|
|
As of and for the year ended |
|
|||||||
|
|
|
June 30, 2016 |
|
|||||||
|
|
|
United |
|
|
|
|
|
|||
|
|
|
States |
|
Europe |
|
Total |
|
|||
|
Total assets |
|
$ |
55,451 |
|
$ |
1,499 |
|
$ |
56,950 |
|
|
Property and equipment, net |
|
|
3,895 |
|
|
74 |
|
|
3,969 |
|
|
Revenues |
|
|
972 |
|
|
2,261 |
|
|
3,233 |
|
|
Loss before taxes |
|
|
(63,688) |
|
|
(502) |
|
|
(64,190) |
|
|
|
|
|
As of and for the year ended |
|||||||
|
|
|
|
June 30, 2015 |
|||||||
|
|
|
|
United |
|
|
|
|
|
|
|
|
|
|
|
States |
|
Europe |
|
Total |
|
||
|
Total assets |
|
$ |
104,168 |
|
$ |
1,612 |
|
$ |
105,780 |
|
|
Property and equipment, net |
|
|
2,234 |
|
|
8 |
|
|
2,242 |
|
|
Revenues |
|
|
3,054 |
|
|
2,599 |
|
|
5,653 |
|
|
(Loss) income before taxes |
|
|
(48,192) |
|
|
126 |
|
|
(48,066) |
|
17. Defined Contribution Plans
U.S. employees are eligible to participate in the Company’s 401(k) plan, while employees in international locations are eligible to participate in other defined contribution plans. Aggregate Company contributions to its benefit plans totaled approximately $104 thousand, $99 thousand and $99 thousand for the years ended June 30, 2017, 2016 and 2015, respectively.
91
18. Quarterly Results of Operations (Unaudited)
The following table present summarized unaudited quarterly financial data:
|
|
|
Three Months Ended |
|
||||||||||
|
|
|
June 30, |
|
March 31, |
|
December 31, |
|
September 30, |
|
||||
|
|
|
2017 |
|
2017 |
|
2016 |
|
2016 |
|
||||
|
|
|
(In thousands, except for per share amounts) |
|
||||||||||
|
Consolidated Statements of Comprehensive Loss Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
642 |
|
$ |
1,323 |
|
$ |
384 |
|
$ |
742 |
|
|
Net loss attributable to Immunomedics, Inc. stockholders |
|
|
(53,255) |
|
|
(59,306) |
|
|
(24,447) |
|
|
(16,198) |
|
|
Loss per common share attributable to Immunomedics Inc. stockholders – (basic and diluted) |
|
$ |
(0.48) |
|
$ |
(0.56) |
|
$ |
(0.25) |
|
$ |
(0.18) |
|
|
Weighted average shares used to calculate loss per common share – (basic and diluted) |
|
|
109,891 |
|
|
107,840 |
|
|
104,657 |
|
|
95,884 |
|
|
|
|
Three Months Ended |
|
||||||||||
|
|
|
June 30, |
|
March 31, |
|
December 31, |
|
September 30, |
|
||||
|
|
|
2016 |
|
2016 |
|
2015 |
|
2015 |
|
||||
|
|
|
(In thousands, except for per share amounts) |
|
||||||||||
|
Consolidated Statements of Comprehensive Loss Data: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Revenues |
|
$ |
932 |
|
$ |
899 |
|
$ |
671 |
|
$ |
731 |
|
|
Net loss attributable to Immunomedics, Inc. stockholders |
|
|
(15,901) |
|
|
(13,996) |
|
|
(13,746) |
|
|
(15,394) |
|
|
Loss per common share attributable to Immunomedics Inc. stockholders – (basic and diluted) |
|
$ |
(0.16) |
|
$ |
(0.15) |
|
$ |
(0.15) |
|
$ |
(0.16) |
|
|
Weighted average shares used to calculate loss per common share – (basic and diluted) |
|
|
94,770 |
|
|
94,748 |
|
|
94,665 |
|
|
94,596 |
|
92
Immunomedics, Inc. and Subsidiaries
Schedule II – Valuation and Qualifying Reserves
For the Fiscal Years Ended June 30, 2017, 2016 and 2015
(in thousands)
Allowance for Doubtful Accounts
|
|
|
Balance at |
|
|
|
|
|
|
|
|
|
|
Balance at |
|
||
|
|
|
Beginning of |
|
Changes to |
|
Credits to |
|
Other |
|
End of |
|
|||||
|
Year ended: |
|
Year |
|
Reserve |
|
Expense |
|
Charges |
|
Year |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
June 30, 2015 |
|
$ |
(89) |
|
$ |
35 |
|
$ |
— |
|
$ |
— |
|
$ |
(54) |
|
|
June 30, 2016 |
|
$ |
(54) |
|
$ |
(21) |
|
$ |
— |
|
$ |
— |
|
$ |
(75) |
|
|
June 30, 2017 |
|
$ |
(75) |
|
$ |
66 |
|
$ |
— |
|
$ |
— |
|
$ |
(9) |
|
93
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures:
Disclosure Controls and Procedures: We maintain controls and procedures designed to ensure that we are able to collect the information we are required to disclose in the reports we file with the SEC, and to record, process, summarize and disclose this information within the time periods specified in the rules promulgated by the SEC. Our Principal Executive Officer and Chief Financial Officer is responsible for establishing and maintaining these disclosure controls and procedures and as required by the rules of the SEC, to evaluate their effectiveness. Based on his evaluation of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K, our Principal Executive Officer and Chief Financial Officer believes that these procedures are functioning effectively to provide reasonable assurance that the information required to be disclosed by us in reports filed under the Securities Exchange Act of 1934 is (i) recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and (ii) accumulated and communicated to our management, including our Principal Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding disclosures.
Management’s Report on Internal Control Over Financial Reporting: Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of Immunomedics; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and our directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of June 30, 2017. In making this assessment, management used the criteria in the Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”). Based on its assessment and those criteria, our management has concluded we maintained effective internal control over financial reporting as of June 30, 2017.
Our independent registered public accounting firm has issued an attestation report on the effectiveness of Immunomedics’ internal control over financial reporting.
Changes in internal controls over financial reporting: There were no significant changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act), identified in connection with the evaluation of such internal control that occurred during our last fiscal quarter, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
94
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
Immunomedics, Inc.:
We have audited Immunomedics, Inc.’s internal control over financial reporting as of June 30, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Immunomedics Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Immunomedics, Inc. maintained, in all material respects, effective internal control over financial reporting as of June 30, 2017, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) .
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Immunomedics, Inc. and subsidiaries as of June 30, 2017 and 2016, and the related consolidated statements of comprehensive loss, changes in stockholders’ equity (deficit) and cash flows for each of the years in the three-year period ended June 30, 2017, and our report dated August 16, 2017 expressed an unqualified opinion on those consolidated financial statements.
/s/ KPMG LLP
Short Hills, New Jersey
August 16, 2017
95
None.
PART III
Item 10.Directors, Executive Officers, and Corporate Governance
Information required by this item is incorporated in this Annual Report on Form 10-K by reference from the sections entitled “Nominees for Directors,” “Executive Officers,” “Director Experience, Qualifications, Attributes and Skills,” “Section 16(a) Beneficial Ownership Reporting Compliance,” “Business Ethics and Compliance,” and “Committees of the Board,” contained in our definitive proxy statement for our 2017 annual meeting of stockholders scheduled to be held on December 13, 2017, which we intend to file within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
The text of our Code of Business Conduct, which applies to our directors and employees (including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions) is posted in the “Corporate Governance” section of our website, www.immunomedics.com. A copy of the Code of Business Conduct can be obtained free of charge on our website. We intend to disclose on our website any amendments to, or waivers from, our Code of Business Conduct that are required to be disclosed pursuant to the rules of the SEC and NASDAQ.
Item 11.Executive Compensation
Information required to be disclosed by this Item is incorporated in this Annual Report on Form 10-K by reference from the sections entitled “Compensation Discussion and Analysis,” “Compensation Committee Report,” “Summary Compensation Table,” “Grants of Plan Based Awards in Fiscal Year 2017,” “Outstanding Equity Awards at Fiscal Year-End 2017 Table,” “Fiscal Year 2017 Option Exercises and Stock Vested Table,” “Employment Contracts, Termination of Employment and Change in Control Agreements” contained in our definitive proxy statement for our 2017 annual meeting of stockholders scheduled to be held on December 13, 2017, which we intend to file within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
Item 12.Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Securities Authorized for Issuance Under Equity Compensation Plans
The following table provides information with respect to our compensation plans under which equity compensation is authorized as of June 30, 2017.
|
|
|
Number of securities to be |
|
|
|
|
|
|
|
|
|
issued upon vesting of |
|
Weighted-average |
|
Number of securities |
|
|
|
|
|
restricted shares and |
|
exercise price of |
|
remaining available for |
|
|
|
|
|
exercise of outstanding |
|
outstanding options |
|
future issuance under |
|
|
|
Plan Category |
|
options and rights |
|
and rights |
|
equity compensation plans |
|
|
|
Equity compensation plans approved by security holders(1) |
|
4,724,569 |
|
$ |
3.45 |
|
9,540,417 |
|
|
Equity compensation plans not approved by security holders |
|
— |
|
|
— |
|
— |
|
|
Total |
|
4,724,569 |
|
$ |
3.45 |
|
9,540,417 |
|
|
(1) |
Refers to Immunomedics, Inc. 2014 Long-Term Incentive Plan. |
Other information required to be disclosed by this Item is incorporated in this Annual Report on Form 10-K by reference from the sections entitled “Equity Compensation Plans,” “Ownership of Our Common Stock,” “Compensation for Executive Officers” and “Director Compensation,” contained in our definitive proxy statement for our 2017 annual meeting of stockholders scheduled to be held on December 13, 2017, which we intend to file within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
96
Item 13.Certain Relationships and Related Transactions and Director Independence
The information required to be disclosed by this Item is incorporated in this Annual Report on Form 10-K by reference from the section(s) entitled “Certain Relationships and Related Transactions,” “Our Corporate Governance,” “Compensation for Executive Officers,” “Director Compensation,” “Compensation Committee Interlocks and Insider Participation,” and “Compensation Committee Report” contained in our definitive proxy statement for our 2017 annual meeting of stockholders scheduled to be held on December 13, 2017, which we intend to file within 120 days of the end of the fiscal year covered by this Annual Report on From 10-K.
Item 14.Principal Accounting Fees and Services.
This information required to be disclosed by this Item is incorporated in this Annual Report on Form 10-K by reference from the section entitled “Independent Registered Public Accounting Firm” contained in our definitive proxy statement for our 2017 annual meeting of stockholders scheduled to be held on December 13, 2017, which we intend to file within 120 days of the end of the fiscal year covered by this Annual Report on Form 10-K.
PART IV
Item 15. Exhibits, Financial Statement Schedules
|
(a) Documents filed as part of this Report: |
||
|
1. |
Consolidated Financial Statements: |
|
|
Consolidated Balance Sheets – June 30, 2017 and 2016 |
||
|
Consolidated Statements of Comprehensive Loss for the years ended June 30, 2017, 2016 and 2015 |
||
|
Consolidated Statements of Changes in Stockholders’ Equity (Deficit) for the years ended June 30, 2017, 2016 and 2015 |
||
|
Consolidated Statements of Cash Flows for the years ended June 30, 2017, 2016 and 2015 |
||
|
Notes to Consolidated Financial Statements |
||
|
Reports of Independent Registered Public Accounting Firm – KPMG LLP |
||
|
2. |
Financial Statement Schedule: |
|
|
Schedule II – Valuation and Qualifying Reserves |
||
|
3. |
List of Exhibits |
|
|
Exhibit No. |
|
Description |
|
3.(i).1 |
|
Amended and Restated Certificate of Incorporation, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K as filed with the Commission on June 29, 2017. |
|
3.(i).2 |
|
Form of Certificate of Designation of Series A-1 Convertible Preferred Stock, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on May 5, 2017. |
|
3.(iii).1 |
|
Second Amended and Restated By-Laws of the Company, incorporated by reference from the Exhibits to the Company’s Current Report on Form 8-K as filed with the Commission on August 27, 2007. |
|
3.(iii).2 |
|
Amendment to Second Amended and Restated By-Laws of Immunomedics, Inc., incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on November 28, 2016. |
97
|
3.(iii).3 |
|
Second Amendment to Second Amended and Restated By-Laws of Immunomedics, Inc., incorporated by reference from Exhibit 3.3 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
4.1 |
|
Indenture, dated as of February 11, 2015, by and between the Company and Wells Fargo Bank, National Association, incorporated by reference from Exhibit 4.1 to the Company’s Current Report on Form 8-K as filed with the Commission on February 12, 2015. |
|
4.2 |
|
Form of 4.75% Convertible Senior Note due 2020 incorporated by reference from Exhibit 4.1 to the Company’s Current Report on Form 8-K as filed with the Commission on February 12, 2015. |
|
4.3 |
|
Warrant Agreement, dated as of October 11, 2016, between the Company and Broadridge Financial Solutions, Inc., as warrant agent , incorporated by reference to exhibit 4.1 to the Company’s current report on Form 8-K, as filed with the Commission on October 12, 2016. |
|
4.4 |
|
Warrant Agreement, dated as of February 16, 2017, between the Company and Broadridge Financial Solutions, Inc., as warrant agent, incorporated by reference to exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
4.5 |
|
Registration Rights Agreement, dated as of February 10, 2017, between the Company and Seattle Genetics, Inc., incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-3, as filed with the Commission on July 31, 2017 (Commission File No. 333-219594). |
|
10.1 |
|
Amended and Restated License Agreement among the Company, David M. Goldenberg and the Center for Molecular Medicine and Immunology, Inc., dated December 11, 1990, incorporated by reference from the Exhibits to the Company’s Registration Statement on Form S-2 effective July 24, 1991 (Commission File No. 33-41053). |
|
10.2 |
|
Amendment, dated March 13, 1995, to the Amended and Restated License Agreement among the Company, David M. Goldenberg and the Center for Molecular Medicine and Immunology, Inc., dated December 11, 1990, incorporated by reference from the Exhibits to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 1995. |
|
10.3 |
|
License Agreement, dated as of January 21, 1997, between the Company and the Center for Molecular Medicine and Immunology, Inc., incorporated by reference from Exhibit 10.25 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended December 31, 1996. |
|
10.4 |
|
License Agreement, dated March 5, 1999, between the Company and IBC Pharmaceuticals, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K as filed with the Commission on March 24, 1999. |
|
10.5 |
|
Contract for Services effective as of January 1, 2002 between the Company and Logosys Logistik GmbH, incorporated by reference from Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2001. |
|
10.6 |
|
Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from the Exhibits to the Company’s Registration Statement on Form S-2 (Commission File No. 33-44750), effective January 30, 1992. |
|
10.7 |
|
First Addendum, dated May 5, 1993, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.31 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.8 |
|
Second Addendum, dated March 29, 1995, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.32 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.9 |
|
Letter Amendment, dated October 5, 1998, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.33 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.10 |
|
Fourth Amendment Expansion/Extension Agreement dated August 15, 2001, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.34 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.11 |
|
Fifth Amendment Expansion Agreement dated June 18, 2009 of the Lease with WU/LH 300 American L.L.C. a successor-in-interest to Baker Properties Limited Partnership, incorporated by reference from Exhibit 10.36 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2009. |
98
|
10.12 |
|
Sixth Amendment Extension Agreement dated February 11, 2011 of the Lease with WU/LH 300 American L.L.C. a successor-in-interest to Baker Properties Limited Partnership, incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2011. |
|
10.13# |
|
Immunomedics, Inc. 2006 Stock Incentive Plan, incorporated by reference from Exhibit 99.1 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-143420), as filed with the Commission on May 31, 2007. |
|
10.14# |
|
Amendment 2007-1 to the Immunomedics, Inc. 2006 Stock Incentive Plan, incorporated by reference from Exhibit 99.2 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-143420), as filed with the Commission on May 31, 2007. |
|
10.15# |
|
Form of Stock Option Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.24 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.16# |
|
Form of Change of Control Addendum to the Stock Option Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.25 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.17# |
|
Form of Notice of Grant of Stock Option under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.26 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.18# |
|
Form of RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.27 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.19# |
|
Form of Change of Control Addendum to RSU Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.28 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.20# |
|
Form of Initial Director RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.29 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.21# |
|
Form of Annual Director RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.30 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.22# |
|
Form of Restricted Stock Unit Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.1 to the Company’s current report on Form 8-K, as filed with the Commission on August 22, 2013. |
|
10.23# |
|
Form of Performance-Based Restricted Stock Unit Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.2 to the Company’s current report on Form 8-K, as filed with the Commission on August 22, 2013. |
|
10.24# |
|
Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.1 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-201470), as filed with the Commission on January 13, 2015. |
|
10.25# |
|
Forms of Incentive Stock Option Notice and Incentive Stock Option Agreement under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.2 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-201470), as filed with the Commission on January 13, 2015. |
|
10.26# |
|
Forms of Nonqualified Stock Option Notice and Nonqualified Stock Option Agreement under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.3 to the Company’s Registration Statement on Form S-8 as filed with the Commission on January 13, 2015. |
|
10.27# |
|
Forms of Restricted Stock Units Notice and Restricted Stock Units Agreement (for Officers/Employees) under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.4 to the Company’s Registration Statement on Form S-8 as filed with the Commission on January 13, 2015. |
99
|
10.28# |
|
Forms of Restricted Stock Units Notice and Restricted Stock Units Agreement (for Directors) under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.5 to the Company’s Registration Statement on Form S-8 as filed with the Commission on January 13, 2015. |
|
10.29# |
|
Amended and Restated Employment Agreement, entered into on July 14, 2015 and effective as of July 1, 2015, between the Company and Dr. David M. Goldenberg, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on July 16, 2015. |
|
10.30# |
|
Restricted Stock Units Notice, entered into on July 14, 2015, between the Company and Dr. David M. Goldenberg., incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K, as filed with the Commission on July 16, 2015. |
|
10.31# |
|
Amendment No. 1 to Amended and Restated Employment Agreement, effective as of November 30, 2015, between the Company and Dr. David M. Goldenberg, incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended December 31, 2015. |
|
10.32# |
|
Fifth Amended and Restated Employment Agreement, dated July 1, 2011, between the Company and Cynthia L. Sullivan, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on June 25, 2014. |
|
10.33† |
|
Development and License Agreement, dated as of February 10, 2017, by and between the Company and Seattle Genetics, Inc., incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017. |
|
10.34 |
|
Stock Purchase Agreement, dated as of February 10, 2017, by and between the Company and Seattle Genetics, Inc., incorporated by reference from Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017. |
|
10.35 |
|
Form of Indemnification Agreement by and between the Company and each of its directors, executive officers, and certain of its former directors and executive officers, incorporated by reference to exhibit 10.1 to the Company’s current report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
10.36 |
|
Securities Purchase Agreement between the Company and the Purchasers, dated as of May 4, 2017, incorporated by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-3, as filed with the Commission on July 31, 2017 (Commission File No. 333-219594). |
|
10.37* † |
|
Termination Agreement, dated May 4, 2017, between the Company and Seattle Genetics, Inc. |
|
21.1* |
|
Subsidiaries of the Company. |
|
23.1* |
|
Consent of Independent Registered Public Accounting Firm – KPMG LLP. |
|
31.1* |
|
Certification of the Principal Executive Officer pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
31.2* |
|
Certification of the Principal Financial Officer pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
32.1* |
|
Certification of the Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
32.2* |
|
Certification of the Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
101* |
|
The following financial information from the Annual report on Form 10-K for the fiscal year ended June 30, 2017, formatted in XBRL (eXtensible Business Reporting Language) and furnished electronically herewith: (i) the Consolidated Balance Sheets; (ii) the Consolidated Statements of Comprehensive Loss; (iii) the Consolidated Statements of Changes in Stockholders’ Equity (Deficit); (iv) the Consolidated Statements of Cash Flows; and (v) the Notes to Consolidated Financial Statements. |
* Filed herewith.
# Management contract or compensatory plan or arrangement required to be filed as an exhibit to this Form 10-K pursuant to Item 15(a)(3) of Form 10-K.
† Confidential treatment has been granted for certain portions of this exhibit.
(Exhibits available upon request)
100
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
IMMUNOMEDICS, INC. |
|||
|
|
|
|
|
|
|
Date: August 16, 2017 |
By: |
/s/ MICHAEL R. GARONE |
||
|
|
|
Michael R. Garone |
||
|
Principal Executive Officer, Vice President Finance, and Chief Financial Officer |
||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
|
|
|
|
|
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/Dr. BEHZAD AGHAZADEH |
|
Chairman of the Board, Director |
|
August 16, 2017 |
|
Dr. Behzad Aghazadeh |
|
|
|
|
|
|
|
|
|
|
|
/s/Dr. KHALID ISLAM |
|
Director |
|
August 16, 2017 |
|
Dr. Khalid Islam |
|
|
|
|
|
|
|
|
|
|
|
/s/SCOTT CANUTE |
|
Director |
|
August 16, 2017 |
|
Scott Canute |
|
|
|
|
|
/s/PETER BARTON HUTT |
|
Director |
|
August 16, 2017 |
|
Peter Barton Hutt |
|
|
|
|
|
|
|
|
|
|
|
/s/BRIAN A. MARKISON |
|
Director |
|
August 16, 2017 |
|
Brian A. Markison |
|
|
|
|
|
|
|
|
|
|
|
|
|
Director |
|
|
|
David M. Goldenberg |
|
|
|
|
|
|
|
|
|
|
|
|
|
Director |
|
|
|
Cynthia L. Sullivan |
|
|
|
|
|
|
|
|
|
|
|
/s/MICHAEL R. GARONE |
|
Principal Executive Officer, Vice President, Finance and Chief Financial |
|
August 16, 2017 |
|
Michael R. Garone |
|
Officer (Principal Financial and |
|
|
|
|
|
Accounting Officer) |
|
|
101
EXHIBIT LIST
|
Exhibit No. |
|
Description |
|
3(i).1 |
|
Amended and Restated Certificate of Incorporation, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K as filed with the Commission on June 29, 2017. |
|
3(i).2 |
|
Form of Certificate of Designation of Series A-1 Convertible Preferred Stock, incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on May 5, 2017. |
|
3(ii).1 |
|
Second Amended and Restated By-Laws of the Company, incorporated by reference from the Exhibits to the Company’s Current Report on Form 8-K as filed with the Commission on August 27, 2007. |
|
3(ii).2 |
|
Amendment to Second Amended and Restated By-Laws of Immunomedics, Inc., incorporated by reference from Exhibit 3.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on November 28, 2016. |
|
3(ii).3 |
|
Second Amendment to Second Amended and Restated By-Laws of Immunomedics, Inc., incorporated by reference from Exhibit 3.3 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
4.1 |
|
Indenture, dated as of February 11, 2015, by and between the Company and Wells Fargo Bank, National Association, incorporated by reference from Exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 12, 2015. |
|
4.2 |
|
Form of 4.75% Convertible Senior Note due 2020 incorporated by reference from Exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 12, 2015. |
|
4.3 |
|
Warrant Agreement, dated as of October 11, 2016, between the Company and Broadridge Financial Solutions, Inc., as warrant agent , incorporated by reference to exhibit 4.1 to the Company’s current report on Form 8-K, as filed with the Commission on October 12, 2016. |
|
4.4 |
|
Warrant Agreement, dated as of February 16, 2017, between the Company and Broadridge Financial Solutions, Inc., as warrant agent, incorporated by reference to exhibit 4.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
4.5 |
|
Registration Rights Agreement, dated as of February 10, 2017, between the Company and Seattle Genetics, Inc., incorporated by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-3, as filed with the Commission on July 31, 2017 (Commission File No. 333-219594). |
|
10.1 |
|
Amended and Restated License Agreement among the Company, David M. Goldenberg and the Center for Molecular Medicine and Immunology, Inc., dated December 11, 1990, incorporated by reference from the Exhibits to the Company’s Registration Statement on Form S-2 effective July 24, 1991 (Commission File No. 33-41053). |
|
10.2 |
|
Amendment, dated March 13, 1995, to the Amended and Restated License Agreement among the Company, David M. Goldenberg and the Center for Molecular Medicine and Immunology, Inc., dated December 11, 1990, incorporated by reference from the Exhibits to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 1995. |
|
10.3 |
|
License Agreement, dated as of January 21, 1997, between the Company and the Center for Molecular Medicine and Immunology, Inc., incorporated by reference from Exhibit 10.25 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended December 31, 1996. |
|
10.4 |
|
License Agreement, dated March 5, 1999, between the Company and IBC Pharmaceuticals, incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K as filed with the Commission on March 24, 1999. |
|
|
|
|
|
10.5 |
|
Contract for Services effective as of January 1, 2002 between the Company and Logosys Logistik GmbH, incorporated by reference from Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended September 30, 2001. |
|
10.6 |
|
Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from the Exhibits to the Company’s Registration Statement on Form S-2 (Commission File No. 33-44750), effective January 30, 1992. |
|
10.7 |
|
First Addendum, dated May 5, 1993, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.31 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.8 |
|
Second Addendum, dated March 29, 1995, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.32 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.9 |
|
Letter Amendment, dated October 5, 1998, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.33 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.10 |
|
Fourth Amendment Expansion/Extension Agreement dated August 15, 2001, of the Lease Agreement with Baker Properties Limited Partnership, dated January 16, 1992, incorporated by reference from Exhibit 10.34 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.11 |
|
Fifth Amendment Expansion Agreement dated June 18, 2009 of the Lease with WU/LH 300 American L.L.C. a successor-in-interest to Baker Properties Limited Partnership, incorporated by reference from Exhibit 10.36 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2009. |
|
10.12 |
|
Sixth Amendment Extension Agreement dated February 11, 2011 of the Lease with WU/LH 300 American L.L.C. a successor-in-interest to Baker Properties Limited Partnership, incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2011. |
|
|
|
|
|
10.13# |
|
Immunomedics, Inc. 2006 Stock Incentive Plan, incorporated by reference from Exhibit 99.1 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-143420), as filed with the Commission on May 31, 2007. |
|
10.14# |
|
Amendment 2007-1 to the Immunomedics, Inc. 2006 Stock Incentive Plan, incorporated by reference from Exhibit 99.2 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-143420), as filed with the Commission on May 31, 2007. |
|
10.15# |
|
Form of Stock Option Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.24 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.16# |
|
Form of Change of Control Addendum to the Stock Option Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.25 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.17# |
|
Form of Notice of Grant of Stock Option under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.26 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.18# |
|
Form of RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.27 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.19# |
|
Form of Change of Control Addendum to RSU Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.28 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.20# |
|
Form of Initial Director RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.29 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.21# |
|
Form of Annual Director RSU Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.30 to the Company’s Annual Report on Form 10-K for the fiscal year ended June 30, 2007. |
|
10.22# |
|
Form of Restricted Stock Unit Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.1 to the Company’s current report on Form 8-K, as filed with the Commission on August 22, 2013. |
|
10.23# |
|
Form of Performance-Based Restricted Stock Unit Issuance Agreement under the Immunomedics, Inc. 2006 Stock Incentive Plan, as amended, incorporated by reference from Exhibit 10.2 to the Company’s current report on Form 8-K, as filed with the Commission on August 22, 2013. |
|
10.24# |
|
Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.1 to the Company’s Registration Statement on Form S-8 (Commission File Number 333-201470), as filed with the Commission on January 13, 2015. |
|
10.25# |
|
Forms of Incentive Stock Option Notice and Incentive Stock Option Agreement under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.2 to the Company’s Registration Statement on Form S-8, (Commission File Number 333-201470), as filed with the Commission on January 13, 2015. |
|
10.26# |
|
Forms of Nonqualified Stock Option Notice and Nonqualified Stock Option Agreement under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.3 to the Company’s Registration Statement on Form S-8, as filed with the Commission on January 13, 2015. |
|
10.27# |
|
Forms of Restricted Stock Units Notice and Restricted Stock Units Agreement (for Officers/Employees) under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.4 to the Company’s Registration Statement on Form S-8, as filed with the Commission on January 13, 2015. |
|
10.28# |
|
Forms of Restricted Stock Units Notice and Restricted Stock Units Agreement (for Directors) under the Immunomedics, Inc. 2014 Long-Term Incentive Plan, incorporated by reference from Exhibit 99.5 to the Company’s Registration Statement on Form S-8, as filed with the Commission on January 13, 2015. |
|
10.29# |
|
Amended and Restated Employment Agreement, entered into on July 14, 2015 and effective as of July 1, 2015, between the Company and Dr. David M. Goldenberg, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on July 16, 2015. |
|
10.30# |
|
Restricted Stock Units Notice, entered into on July 14, 2015, between the Company and Dr. David M. Goldenberg., incorporated by reference from Exhibit 10.2 to the Company’s Current Report on Form 8-K, as filed with the Commission on July 16, 2015. |
|
10.31# |
|
Amendment No. 1 to Amended and Restated Employment Agreement, effective as of November 30, 2015, between the Company and Dr. David M. Goldenberg, incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended December 31, 2015. |
|
10.32# |
|
Fifth Amended and Restated Employment Agreement, effective as of July 1, 2014, between the Company and Cynthia L. Sullivan, incorporated by reference from Exhibit 10.1 to the Company’s Current Report on Form 8-K, as filed with the Commission on June 25, 2014. |
|
|
|
|
|
10.33† |
|
Development and License Agreement, dated as of February 10, 2017, by and between the Company and Seattle Genetics, Inc., incorporated by reference from Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017. |
|
10.34 |
|
Stock Purchase Agreement, dated as of February 10, 2017, by and between the Company and Seattle Genetics, Inc., incorporated by reference from Exhibit 10.2 to the Company’s Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2017. |
|
10.35 |
|
Form of Indemnification Agreement by and between the Company and each of its directors, executive officers, and certain of its former directors and executive officers, incorporated by reference to exhibit 10.1 to the Company’s current report on Form 8-K, as filed with the Commission on February 16, 2017. |
|
10.36 |
|
Securities Purchase Agreement between the Company and the Purchasers, dated as of May 4, 2017, incorporated by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-3, as filed with the Commission on July 31, 2017 (Commission File No. 333-219594). |
|
10.37* † |
|
Termination Agreement, dated May 4, 2017, between the Company and Seattle Genetics, Inc. |
|
21.1* |
|
Subsidiaries of the Company. |
|
23.1* |
|
Consent of Independent Registered Public Accounting Firm – KPMG LLP. |
|
31.1* |
|
Certification of the Principal Executive Officer pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
31.2* |
|
Certification of the Principal Financial Officer pursuant to Section 302(a) of the Sarbanes-Oxley Act of 2002. |
|
32.1* |
|
Certification of the Principal Executive Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
32.2* |
|
Certification of the Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
101* |
|
The following financial information from the Annual report on Form 10-K for the fiscal year ended June 30, 2017, formatted in XBRL (eXtensible Business Reporting Language) and furnished electronically herewith: (i) the Consolidated Balance Sheets; (ii) the Consolidated Statements of Comprehensive Loss; (iii) the Consolidated Statements of Changes in Stockholders’ Equity (Deficit); (iv) the Consolidated Statements of Cash Flows; and (v) the Notes to Consolidated Financial Statements. |
* Filed herewith.
# Management contract or compensatory plan or arrangement required to be filed as an exhibit to this Form 10-K pursuant to Item 15(a)(3) of Form 10-K.
† Confidential treatment has been granted for certain portions of this exhibit.
(Exhibits available upon request)