Attached files
| file | filename |
|---|---|
| EX-32.1 - EXHIBIT 32.1 - PALISADE BIO, INC. | exh_321.htm |
| EX-31.1 - EXHIBIT 31.1 - PALISADE BIO, INC. | exh_311.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark one)
| ☒ | Quarterly Report Under Section 13 or 15(d) of the Securities Exchange Act of 1934 |
For the Quarterly Period Ended June 30, 2017
Or
| ☐ | Transition Report Under Section 13 or 15(d) of the Securities Exchange Act of 1934 |
Commission File Number 001-33672
NEURALSTEM, INC.
(Exact name of registrant as specified in its charter)
| Delaware | 52-2007292 | |
| State or other jurisdiction of | (I.R.S. Employer | |
| incorporation or organization | Identification No.) | |
| 20271 Goldenrod Lane | ||
| Germantown, Maryland | 20876 | |
| (Address of principal executive offices) | (Zip Code) |
Registrant’s telephone number, including area code (301)-366-4841
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). ☒Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Non-accelerated filer ☐ (Do not check if a small reporting company) | Smaller reporting company ☒ |
Emerging Growth Company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act) ☐ Yes ☒ No
As of July 31, 2017, there were 12,117,429 shares of common stock, $.01 par value, issued and outstanding.
1
Neuralstem, Inc.
Table of Contents
2
FINANCIAL INFORMATION
| ITEM 1. | UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS |
Unaudited Condensed Consolidated Balance Sheets
| June 30, 2017 | December 31, 2016 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash and cash equivalents | $ | 6,442,484 | $ | 15,194,949 | ||||
| Short-term investments | 5,000,000 | 5,000,000 | ||||||
| Trade and other receivables | 5,345 | 10,491 | ||||||
| Current portion of related party receivable, net of discount | - | 53,081 | ||||||
| Prepaid expenses | 239,309 | 646,195 | ||||||
| Total current assets | 11,687,138 | 20,904,716 | ||||||
| Property and equipment, net | 219,382 | 269,557 | ||||||
| Patents, net | 945,066 | 990,153 | ||||||
| Related party receivable, net of discount and current portion | 402,965 | 424,240 | ||||||
| Other assets | 13,696 | 15,662 | ||||||
| Total assets | $ | 13,268,247 | $ | 22,604,328 | ||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable and accrued expenses | $ | 2,235,908 | $ | 2,343,936 | ||||
| Accrued bonuses | - | 852,963 | ||||||
| Current portion of long-term debt, net of fees and discount | - | 3,705,787 | ||||||
| Other current liabilities | 67,992 | 430,738 | ||||||
| Total current liabilities | 2,303,900 | 7,333,424 | ||||||
| Derivative instruments | 3,267,408 | 3,921,917 | ||||||
| Other long-term liabilities | 4,136 | 18,209 | ||||||
| Total liabilities | 5,575,444 | 11,273,550 | ||||||
| Commitments and contingencies (Note 6) | ||||||||
| STOCKHOLDERS' EQUITY | ||||||||
| Convertible preferred stock, 7,000,000 shares authorized, $0.01 par value; 1,000,000 shares issued and outstanding at June 30, 2017 and December 31, 2016, respectively | 10,000 | 10,000 | ||||||
| Common stock, $0.01 par value; 300 million shares authorized, 12,012,877 and 11,032,858 shares issued and outstanding at June 30, 2017 and December 31, 2016, respectively | 120,129 | 110,329 | ||||||
| Additional paid-in capital | 212,809,063 | 204,239,837 | ||||||
| Accumulated other comprehensive income | 3,350 | 3,905 | ||||||
| Accumulated deficit | (205,249,739 | ) | (193,033,293 | ) | ||||
| Total stockholders' equity | 7,692,803 | 11,330,778 | ||||||
| Total liabilities and stockholders' equity | $ | 13,268,247 | $ | 22,604,328 | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
3
Unaudited Condensed Consolidated Statements of Operations and Comprehensive Loss
| Three Months Ended June 30, | Six Months Ended June 30, | |||||||||||||||
| 2017 | 2016 | 2017 | 2016 | |||||||||||||
| Revenues | $ | 2,500 | $ | 2,500 | $ | 5,000 | $ | 5,000 | ||||||||
| Operating expenses: | ||||||||||||||||
| Research and development expenses | 2,585,079 | 2,474,629 | 5,487,165 | 5,540,219 | ||||||||||||
| General and administrative expenses | 1,635,652 | 1,362,140 | 2,968,073 | 4,532,662 | ||||||||||||
| Total operating expenses | 4,220,731 | 3,836,769 | 8,455,238 | 10,072,881 | ||||||||||||
| Operating loss | (4,218,231 | ) | (3,834,269 | ) | (8,450,238 | ) | (10,067,881 | ) | ||||||||
| Other income (expense): | ||||||||||||||||
| Interest income | 14,013 | 13,433 | 34,896 | 24,569 | ||||||||||||
| Interest expense | (15,728 | ) | (322,407 | ) | (154,460 | ) | (708,913 | ) | ||||||||
| Change in fair value of derivative instruments | (341,611 | ) | 757,275 | (3,082,925 | ) | 757,275 | ||||||||||
| Fees related to issuance of derivative instruments, warrant inducement and other expenses | (87,635 | ) | (466,541 | ) | (563,719 | ) | (463,342 | ) | ||||||||
| Total other income (expense) | (430,961 | ) | (18,240 | ) | (3,766,208 | ) | (390,411 | ) | ||||||||
| Net loss | $ | (4,649,192 | ) | $ | (3,852,509 | ) | $ | (12,216,446 | ) | $ | (10,458,292 | ) | ||||
| Net loss per share - basic and diluted | $ | (0.39 | ) | $ | (0.47 | ) | $ | (1.06 | ) | $ | (1.37 | ) | ||||
| Weighted average common shares outstanding - basic and diluted | 11,906,334 | 8,141,198 | 11,525,730 | 7,606,725 | ||||||||||||
| Comprehensive loss: | ||||||||||||||||
| Net loss | $ | (4,649,192 | ) | $ | (3,852,509 | ) | $ | (12,216,446 | ) | $ | (10,458,292 | ) | ||||
| Foreign currency translation adjustment | (384 | ) | 3,268 | (555 | ) | 1,495 | ||||||||||
| Comprehensive loss | $ | (4,649,576 | ) | $ | (3,849,241 | ) | $ | (12,217,001 | ) | $ | (10,456,797 | ) | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
4
Unaudited Condensed Consolidated Statements of Cash Flows
| Six Months Ended June 30, | ||||||||
| 2017 | 2016 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (12,216,446 | ) | $ | (10,458,292 | ) | ||
| Adjustments to reconcile net loss to cash used in operating activities: | ||||||||
| Depreciation and amortization | 180,586 | 177,498 | ||||||
| Share-based compensation expense | 1,174,388 | 2,287,953 | ||||||
| Amortization of deferred financing fees and debt discount | 59,781 | 203,825 | ||||||
| Change in fair value of derivative instruments | 3,082,925 | (757,275 | ) | |||||
| Warrant inducement expense | 563,744 | - | ||||||
| Expenses related to issuance of derivative instrument | - | 466,541 | ||||||
| Loss on disposal of fixed assets | 8,128 | - | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Trade and other receivables | 5,146 | 32,231 | ||||||
| Related party receivable | 74,356 | - | ||||||
| Prepaid expenses | 459,398 | 391,077 | ||||||
| Other assets | 1,971 | 13,680 | ||||||
| Accounts payable and accrued expenses | (160,741 | ) | 955,159 | |||||
| Accrued bonuses | (852,963 | ) | (161,362 | ) | ||||
| Other current liabilities | (173,440 | ) | (58,640 | ) | ||||
| Other long term liabilities | (14,073 | ) | (152,319 | ) | ||||
| Net cash used in operating activities | (7,807,240 | ) | (7,059,924 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Maturity of short-term investments | 5,000,000 | 7,517,453 | ||||||
| Purchase of short-term investments | (5,000,000 | ) | - | |||||
| Patent costs | (83,774 | ) | (30,183 | ) | ||||
| Purchase of property and equipment | (9,674 | ) | (98,088 | ) | ||||
| Net cash (used in) provided by investing activities | (93,448 | ) | 7,389,182 | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from exercise of common stock purchase warrants, net | 3,006,410 | - | ||||||
| Proceeds from sale of common stock and warrants, net | 97,050 | 8,308,931 | ||||||
| Payments of long-term debt | (3,765,568 | ) | (2,225,987 | ) | ||||
| Payments of short-term notes payable | (189,306 | ) | - | |||||
| Net cash (used in ) provided by financing activities | (851,414 | ) | 6,082,944 | |||||
| Effects of exchange rates on cash | (363 | ) | 28 | |||||
| Net (decrease) increase in cash and cash equivalents | (8,752,465 | ) | 6,412,230 | |||||
| Cash and cash equivalents, beginning of period | 15,194,949 | 4,716,533 | ||||||
| Cash and cash equivalents, end of period | $ | 6,442,484 | $ | 11,128,763 | ||||
| Supplemental disclosure of cash flows information: | ||||||||
| Cash paid for interest | $ | 139,080 | $ | 718,989 | ||||
| Supplemental schedule of non cash investing and financing activities: | ||||||||
| Issuance of common stock for cashless exercise of options, warrants and RSUs | $ | - | $ | 8,936 | ||||
See accompanying notes to unaudited condensed consolidated financial statements.
5
NOTES TO UNAUDITED CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
JUNE 30, 2017AND 2016
Note 1. Business, Basis of Presentation and Liquidity
Neuralstem is a clinical stage biopharmaceutical company that is utilizing its proprietary human neural stem cell technology to create a comprehensive platform of therapies for the treatment of central nervous system diseases. The Company has utilized this technology as a tool for small-molecule drug discovery and to create cell therapy biotherapeutics to treat central nervous system diseases. The Company was founded in 1997 and currently has laboratory and office space in Germantown, Maryland and laboratory facilities in the People’s Republic of China. Our operations to date have been directed primarily toward developing business strategies, raising capital, research and development activities, and conducting pre-clinical testing and human clinical trials of our product candidates.
Neuralstem, Inc. and its subsidiary are referred to as “Neuralstem,” the “Company,” “us,” or “we” throughout this report. The operations of our wholly-owned and controlled subsidiary located in China are consolidated in our condensed consolidated financial statements and all intercompany activity has been eliminated. The Company operates in one business segment.
In management’s opinion, the accompanying interim condensed financial statements include all adjustments, consisting of normal recurring adjustments, which are necessary to present fairly our financial position, results of operations and cash flows. The condensed consolidated balance sheet at December 31, 2016, has been derived from audited financial statements as of that date. The interim results of operations are not necessarily indicative of the results that may occur for the full fiscal year. Certain information and footnote disclosure normally included in the financial statements prepared in accordance with generally accepted accounting principles in the United States of America (U.S. GAAP) have been condensed or omitted pursuant to instructions, rules and regulations prescribed by the U.S. Securities and Exchange Commission (SEC). We believe that the disclosures provided herein are adequate to make the information presented not misleading when these condensed financial statements are read in conjunction with the Financial Statements and Notes included in our Annual Report on Form 10-K for the year ended December 31, 2016, filed with the SEC, and as may be amended. Certain prior period amounts have been reclassified to conform to current year classifications.
The Board of Directors approved a 1-for-13 reverse stock split of the Company’s common stock effective January 6, 2017. Stockholders' equity and all references to share and per share amounts in the accompanying consolidated financial statements have been retroactively adjusted to reflect the 1-for-13 reverse stock split for all periods presented.
Liquidity
The Company has incurred losses since its inception and has not demonstrated an ability to generate significant revenues from the sales of its therapies or services and have not yet achieved profitable operations. There can be no assurance that profitable operations will ever be achieved, or if achieved, could be sustained on a continuing basis. In addition, development activities, clinical and pre-clinical testing, and commercialization of our products will require significant additional financing.
Our cash, cash equivalents and short-term investments balance at June 30, 2017 was approximately $11.4 million. On August 1, 2017, we closed a public offering of 3,000,000 shares of common stock and 2,250,000 common stock purchase warrants at a public purchase price of $2.00 per share and accompanying warrant. We received gross proceeds of $6.0 million and approximately $5.4 million of net proceeds from this offering.
We expect that our existing cash and cash equivalents will be sufficient to enable us to fund our anticipated level of operations based on our current operating plans into the fourth quarter of 2018. In the event we are not able to secure additional capital by such date we may not be able to continue as a going concern. Accordingly, we will require additional capital to continue to develop our pre-clinical and clinical development operations. To continue to fund our operations and the development of our product candidates, we anticipate raising additional cash through the private or public sales of equity or debt securities, collaborative arrangements, licensing agreements or a combination thereof. Although management believes that such funding sources will be available, there can be no assurance that any such collaborative or licensing arrangements will be entered into or that financing will be available to us when needed in order to allow us to continue our operations, or if available, on terms acceptable to us. If we do not raise sufficient funds in a timely manner, among other things, we may be forced to delay, scale back or eliminate some or all of our research and product development programs, planned clinical trials, and/or our capital expenditures or to license our potential products or technologies to third parties. We currently do not have commitments for future funding from any source.
We have spent and will continue to spend substantial funds in the research, development, pre-clinical and clinical testing of our small molecule and stem cell product candidates with the goal of ultimately obtaining approval from the United States Food and Drug Administration (the “FDA”) and its international equivalents, to market and sell our products. No assurance can be given that (i) FDA or other regulatory agency approval will ever be granted for us to market and sell our product candidates, or (ii) if regulatory approval is granted, that we will ever be able to sell our proposed products or be profitable.
6
Note 2. Significant Accounting Policies and Recent Accounting Pronouncements
Use of Estimates
The preparation of financial statements in accordance with United States of America generally accepted accounting principles (“U.S. GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The condensed consolidated financial statements include significant estimates for the expected economic life and value of our licensed technology and related patents, our net operating loss and related valuation allowance for tax purposes, the fair value of our derivative instruments and our stock-based compensation related to employees and directors, consultants and investment banks, among other things. Because of the use of estimates inherent in the financial reporting process, actual results could differ significantly from those estimates.
Fair Value Measurements
The carrying amounts of our short-term financial instruments, which primarily include cash and cash equivalents, short-term investments, accounts payable and accrued expenses, approximate their fair values due to their short maturities. The fair value of our long-term indebtedness is estimated based on the quoted prices for the same or similar issues or on the current rates offered to the Company for debt of the same remaining maturities and approximates the carrying value. The fair values of our derivative instruments were estimated using Level 3 unobservable inputs. See Note 3 for further details.
Foreign Currency Translation
The functional currency of our wholly owned foreign subsidiary is its local currency. Assets and liabilities of our foreign subsidiary are translated into United States dollars based on exchange rates at the end of the reporting period; income and expense items are translated at the weighted average exchange rates prevailing during the reporting period. Translation adjustments for subsidiaries that have not been sold, substantially liquidated or otherwise disposed of are accumulated in other comprehensive income or loss, a component of stockholders' equity. Transaction gains or losses are included in the determination of net loss.
Cash, Cash Equivalents, Short-Term Investments and Credit Risk
Cash equivalents consist of investments in low risk, highly liquid money market accounts and certificates of deposit with original maturities of 90 days or less. Cash deposited with banks and other financial institutions may exceed the amount of insurance provided on such deposits. If the amount of a deposit at any time exceeds the federally insured amount at a bank, the uninsured portion of the deposit could be lost, in whole or in part, if the bank were to fail.
Short-term investments consist entirely of fixed income certificates of deposit (“CDs”) with original maturities of greater than 90 days but not more than one year.
Financial instruments that potentially subject us to concentrations of credit risk consist primarily of cash equivalents and short-term investments. Our investment policy, approved by our Board of Directors, limits the amount we may invest in any one type of investment issuer, thereby reducing credit risk concentrations. In addition, our certificates of deposit are typically invested through the Certificate of Deposit Account Registry Service (“CDARS”) program which reduces or eliminates our risk related to concentrations of investments above FDIC insurance levels. We attempt to limit our credit and liquidity risks through our investment policy and through regular reviews of our portfolio against our policy. To date, we have not experienced any loss or lack of access to cash in our operating accounts or to our cash equivalents and short-term investments.
Research and Development
Research and development costs are expensed as they are incurred. Research and development expenses consist primarily of costs associated with the pre-clinical development and clinical trials of our product candidates.
Income (Loss) per Common Share
Basic income (loss) per common share is computed by dividing total net income (loss) available to common shareholders by the weighted average number of common shares outstanding during the period.
For periods of net income when the effects are dilutive, diluted earnings per share is computed by dividing net income available to common shareholders by the weighted average number of shares outstanding and the dilutive impact of all potential dilutive common shares. Potential dilutive common shares consist primarily of convertible preferred stock, stock options, restricted stock units and common stock purchase warrants. The dilutive impact of potential dilutive common shares resulting from common stock equivalents is determined by applying the treasury stock method. Our unvested restricted shares contain non-forfeitable rights to dividends, and therefore are considered to be participating securities; the calculation of basic and diluted income per share excludes net income attributable to the unvested restricted shares from the numerator and excludes the impact of the shares from the denominator.
7
For all periods of net loss, diluted loss per share is calculated similarly to basic loss per share because the impact of all potential dilutive common shares is anti-dilutive due to the net losses; accordingly, diluted loss per share is the same as basic loss per share for all periods presented. A total of approximately 8.1 million and 4.8 million potential dilutive shares have been excluded in the calculation of diluted net income per share for the three- and six-month periods ended June 30, 2017 and 2016, respectively, as their inclusion would be anti-dilutive.
Share-Based Compensation
We account for share-based compensation at fair value. Share-based compensation cost for stock options and stock purchase warrants granted to employees and board members is generally determined at the grant date while awards granted to non-employee consultants are generally valued at the vesting date using an option pricing model that uses Level 3 unobservable inputs; share-based compensation cost for restricted stock and restricted stock units is determined at the grant date based on the closing price of our common stock on that date. The value of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the requisite service period.
Intangible and Long-Lived Assets
We assess impairment of our long-lived assets using a "primary asset" approach to determine the cash flow estimation period for a group of assets and liabilities that represents the unit of accounting for a long-lived asset to be held and used. Long-lived assets to be held and used are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. The carrying amount of a long-lived asset is not recoverable if it exceeds the sum of the undiscounted cash flows expected to result from the use and eventual disposition of the asset. No significant impairment losses were recognized during the three or six months ended June 30, 2017 or 2016.
Income Taxes
We account for income taxes using the asset and liability approach, which requires the recognition of future tax benefits or liabilities on the temporary differences between the financial reporting and tax bases of our assets and liabilities. A valuation allowance is established when necessary to reduce deferred tax assets to the amounts expected to be realized. We also recognize a tax benefit from uncertain tax positions only if it is “more likely than not” that the position is sustainable based on its technical merits. Our policy is to recognize interest and penalties on uncertain tax positions as a component of income tax expense.
Significant New Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standard Update (“ASU”), No. 2014-09, Revenue from Contracts with Customers. This ASU consists of a comprehensive revenue recognition standard that will supersede nearly all existing revenue recognition guidance under U.S. GAAP. The issuance of ASU No. 2015-14 in August 2015 delays the effective date of the standard to interim and annual periods beginning after December 15, 2017. Either full retrospective adoption or modified retrospective adoption is permitted. In addition to expanded disclosures regarding revenue, this pronouncement may impact timing of recognition in some arrangements with variable consideration or contracts for the sale of goods or services. We do not expect the adoption of this guidance to have a significant impact on our current licensing arrangements.
In February 2016, the FASB issued ASU, No. 2016-02, Leases. This ASU consists of a comprehensive lease accounting standard. The guidance requires lessees to recognize assets and liabilities related to long-term leases on the balance sheet and expands disclosure requirements regarding leasing arrangements. The guidance is effective for reporting periods beginning after December 15, 2018 and early adoption is permitted. The guidance must be adopted on a modified retrospective basis and provides for certain practical expedients. We currently expect that the adoption of this guidance will likely change the way we account for our operating leases and will likely result in recording the future benefits of those leases and the related minimum lease payments on our consolidated balance sheets. We have not yet begun to evaluate the specific impacts of this guidance.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instrument’s – Credit Losses. This ASU relates to measuring credit losses on financial instruments, including trade receivables. The guidance eliminates the probable initial recognition threshold that was previously required prior to recognizing a credit loss on financial instruments. The credit loss estimate can now reflect an entity's current estimate of all future expected credit losses. Under the previous guidance, an entity only considered past events and current conditions. The guidance is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. Early adoption is permitted for fiscal years beginning after December 15, 2018, including interim periods within those fiscal years. The adoption of certain amendments of this guidance must be applied on a modified retrospective basis and the adoption of the remaining amendments must be applied on a prospective basis. We currently expect that the adoption of this guidance will likely change the way we assess the collectability of our receivables and recoverability of other financial instruments. We have not yet begun to evaluate the specific impacts of this guidance nor have we determined the manner in which we will adopt this guidance.
In November 2015, the FASB issued ASU No. 2015-17, Balance Sheet Classification of Deferred Taxes. This ASU eliminates the requirement for separate presentation of current and non-current portions of deferred tax. Subsequent to adoption, all deferred tax assets and deferred tax liabilities are presented as non-current on the balance sheet. The ASU became effective for us on January 1, 2017 and had no material effect on our consolidated financial statements.
8
In March 2016, the FASB issued ASU No. 2016-09, Improvements to Employee Share Based Payment Accounting. This guidance simplifies the accounting for and financial statement disclosure of stock-based compensation awards, consisting of changes in the accounting for excess tax benefits and tax deficiencies, and changes in the accounting for forfeitures associated with share-based awards, among other things. The ASU became effective for us on January 1, 2017. We no longer record estimate forfeitures on share-based awards. This adoption had no material effect on our consolidated financial statements.
In May 2017, the FASB issued ASU No. 2017-09, Compensation – Stock Compensation. This ASU provides clarification regarding when changes the terms or conditions of share-based payment awards should be accounted for as modifications. This guidance is effective for fiscal years beginning after December 15, 2017 and early adoption is permitted. This guidance must be applied prospectively to awards modified after the adoption date. We do not expect this guidance to have a material effect on our consolidated financial statements.
In July 2017, the FASB issued ASU No. 2017-11, I. Accounting for Certain Financial Instrument with Down Round Features II. Replacement of the Indefinite Deferral for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests with a Scope Exception. Part I of the ASU simplifies the accounting for certain equity-linked financial instruments and embedded features with down round features that reduce the exercise price when the pricing of a future round of financing is lower (“down round protection”). Current accounting guidance provides that instruments with down round protection be classified as derivative liabilities with changes in fair value recorded through earnings. The updated guidance provides that instruments with down round protection are no longer precluded from being classified as equity. This guidance is effective for fiscal years beginning after December 15, 2018 and early adoption is permitted. This guidance must be applied retrospectively. While we have not determined if we will adopt this guidance early, the adoption may have a material effect to our financial statements when it is adopted, as a result of changing the way we currently account for certain of our equity-linked securities that have down round features.
We have reviewed other recent accounting pronouncements released during the year and concluded that they are either not applicable to our business, or that no material effect is expected on the consolidated financial statements as a result of future adoption.
Note 3. Fair Value Measurements
Fair value is the price that would be received from the sale of an asset or paid to transfer a liability assuming an orderly transaction in the most advantageous market at the measurement date. U.S. GAAP establishes a hierarchical disclosure framework which prioritizes and ranks the level of observability of inputs used in measuring fair value. These levels are:
| · | Level 1 – inputs are based upon unadjusted quoted prices for identical instruments traded in active markets. |
| · | Level 2 – inputs are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, and model-based valuation techniques (e.g. the Black-Scholes model) for which all significant inputs are observable in the market or can be corroborated by observable market data for substantially the full term of the assets or liabilities. Where applicable, these models project future cash flows and discount the future amounts to a present value using market-based observable inputs including interest rate curves, foreign exchange rates, and forward and spot prices for currencies and commodities. |
| · | Level 3 – inputs are generally unobservable and typically reflect management's estimates of assumptions that market participants would use in pricing the asset or liability. The fair values are therefore determined using model-based techniques, including option pricing models and discounted cash flow models. |
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
We have segregated our financial assets and liabilities that are measured at fair value on a recurring into the most appropriate level within the fair value hierarchy based on the inputs used to determine the fair value at the measurement date.
The inputs used in measuring the fair value of cash and cash equivalents are considered to be Level 1 in accordance with the three-tier fair value hierarchy. The fair value of all other financial instruments (prepaid expenses, accounts payable and accrued expenses) approximate their carrying values because of their short-term nature. The fair value of our long-term indebtedness is estimated based on Level 2 quoted prices for the same or similar issues or on the current rates offered to the Company for debt of the same remaining maturities and approximates the carrying value.
At June 30, 2017 and December 31, 2016, we had certain common stock purchase warrants issued in connection with our May 2016 capital raises (See Note 5) that are accounted for as derivative instruments whose fair value was determined using Level 3 inputs. The following table identifies the carrying amounts of such liabilities:
9
| Level 1 | Level 2 | Level 3 | Total | |||||||||||||
| Liabilities | ||||||||||||||||
| Derivative instruments - stock purchase warrants | $ | - | $ | - | $ | 3,921,917 | $ | 3,921,917 | ||||||||
| Balance at December 31, 2016 | $ | - | $ | - | $ | 3,921,917 | $ | 3,921,917 | ||||||||
| Derivative instruments - stock purchase warrants | $ | - | $ | - | $ | 3,267,408 | $ | 3,267,408 | ||||||||
| Balance at June 30, 2017 | $ | - | $ | - | $ | 3,267,408 | $ | 3,267,408 | ||||||||
The following table presents the activity for those items measured at fair value on a recurring basis using Level 3 inputs for the six months ended June 30, 2017:
| Derivative instruments - stock purchase warrants | ||||
| Balance at December 31, 2016 | $ | 3,921,917 | ||
| Exercise of warrants | (3,737,434 | ) | ||
| Change in fair value - loss | 3,082,925 | |||
| Balance at June 30, 2017 | $ | 3,267,408 | ||
The following table presents the activity for those items measured at fair value on a recurring basis using Level 3 inputs for the six months ended June 30, 2016:
| Derivative instruments - stock purchase warrants | ||||
| Balance at December 31, 2015 | $ | - | ||
| Issuance of warrants | 4,582,170 | |||
| Change in fair value - gain | (757,275 | ) | ||
| Balance at June 30, 2016 | $ | 3,824,895 | ||
The (gains) losses resulting from the changes in the fair value of the derivative instruments are classified as the “change in the fair value of derivative instruments” in the accompanying condensed consolidated statements of operations. The fair value of the common stock purchase warrants is determined based on the Black-Scholes option pricing model for “plain vanilla” stock options and other pricing models as appropriate, and includes the use of unobservable inputs such as the expected term, anticipated volatility and expected dividends. Changes in any of the assumptions related to the unobservable inputs identified above may change the instrument’s fair value; increases in expected term, anticipated volatility and expected dividends generally result in increases in fair value, while decreases in these unobservable inputs generally result in decreases in fair value.
We do not have any financial assets and liabilities that are measured at fair value on a non-recurring basis.
Nonfinancial assets and liabilities measured at fair value
We do not have any non-financial assets and liabilities that are measured at fair value on a recurring basis.
We measure our long-lived assets, including property, plant, and equipment, and patents, at fair value on a non-recurring basis. These assets are recognized at fair value when they are deemed to be other-than-temporarily impaired. No such fair value impairment was recognized in the three or six months ended June 30, 2017 and 2016.
10
Note 4. Debt
In October 2014, we entered into an agreement to refinance and amend the terms of our March 2013 loan and security agreement. In conjunction with the loan amendment, we issued the lender a five-year common stock purchase warrant to purchase 5,784 shares of common stock at an exercise price of $34.58 per share. The warrant contains standard anti-dilution protection but does not contain any anti-dilution price protection for subsequent offerings and is classified in equity.
We also incurred expenses with various third parties in connection with the loan amendment, consisting of approximately $86,000 in cash, 2,163 shares of common stock valued at approximately $80,000, and a three-year common stock purchase warrant to purchase 4,475 shares at an exercise price of $34.58 per share. The warrant has terms substantially similar to the lender warrant and is classified as equity.
The amended loan was paid off in its entirety in April 2017, pursuant to its terms.
Note 5. Stockholders’ Equity
We have granted share-based compensation awards to employees, board members and service providers. Awards may consist of common stock, restricted common stock, restricted common stock units, common stock purchase warrants, or common stock options. Our stock options and stock purchase warrants have lives of up to ten years from the grant date. Awards vest either upon the grant date or over varying periods of time. The stock options provide for exercise prices equal to or greater than the fair value of the common stock at the date of the grant. Restricted stock units grant the holder the right to receive fully paid common shares with various restrictions on the holder’s ability to transfer the shares. As of June 30, 2017, we have approximately 5.1 million shares of common stock reserved for issuance upon the exercise of such awards.
We record share-based compensation expense on a straight-line basis over the requisite service period. Share-based compensation expense included in the statements of operations is as follows:
| Three Months Ended June 30, | ||||||||
| 2017 | 2016 | |||||||
| Research and development expenses | $ | 435,969 | $ | 264,137 | ||||
| General and administrative expenses | 215,980 | 552,930 | ||||||
| Total | $ | 651,949 | $ | 817,067 | ||||
| Six Months Ended June 30, | ||||||||
| 2017 | 2016 | |||||||
| Research and development expenses | $ | 808,435 | $ | 973,553 | ||||
| General and administrative expenses | 365,953 | 1,314,400 | ||||||
| Total | $ | 1,174,388 | $ | 2,287,953 | ||||
Included in the general and administrative expense for the six months ended June 30, 2016 is approximately $407,000 related to the acceleration of the vesting of options for the previous CEO whose employment was terminated during the first quarter of 2016. In addition, approximately $42,000 and $15,000 is included in research and development and general and administrative expenses, respectively for the three- and six-month periods ended June 30, 2016 related to the modification of certain awards in conjunction with our corporate reorganization.
Stock Options
A summary of stock option activity and related information for the six months ended June 30, 2017 follows:
| Number of Options | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life (in years) | Aggregate Intrinsic Value | |||||||||||||
| Outstanding at January 1, 2017 | 1,691,987 | $ | 22.60 | 5.1 | $ | - | ||||||||||
| Granted | 33,408 | $ | 4.96 | |||||||||||||
| Exercised | - | $ | - | |||||||||||||
| Forfeited | (15,386 | ) | $ | 36.40 | ||||||||||||
| Outstanding at June 30, 2017 | 1,710,009 | $ | 22.14 | 4.7 | $ | 287,704 | ||||||||||
| Exercisable at June 30, 2017 | 1,438,619 | $ | 24.68 | 4.0 | $ | 31,115 | ||||||||||
11
| Range of Exercise Prices | Number of Options Outstanding | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life (in years) | Aggregate Intrinsic Value | ||||||||||||||
| $3.50 | - | $13.00 | 751,480 | $ | 9.51 | 6.8 | $ | 287,704 | ||||||||||
| $13.01 | - | $26.00 | 379,774 | $ | 15.30 | 4.6 | - | |||||||||||
| $26.01 | - | $39.00 | 155,339 | $ | 32.52 | 2.2 | - | |||||||||||
| $39.01 | - | $65.00 | 423,416 | $ | 46.86 | 1.9 | - | |||||||||||
| 1,710,009 | $ | 22.14 | 4.7 | $ | 287,704 | |||||||||||||
The Company uses the Black-Scholes option pricing model for “plain vanilla” options and other pricing models as appropriate to calculate the fair value of options. Significant assumptions used in these models include:
| Six Months Ended June 30, | ||||||||||||
| 2017 | 2016 | |||||||||||
| Annual dividend | - | - | ||||||||||
| Expected life (in years) | 0.3 | - | 4.0 | 6.0 | - | 7.0 | ||||||
| Risk free interest rate | 0.80% | - | 1.72% | 1.35% | - | 1.75% | ||||||
| Expected volatility | 62.2% | - | 82.3% | 69.0% | - | 80.2% | ||||||
Options granted in the six months ended June 30, 2017 and 2016, had a weighted average grant date fair value of $2.99 and $7.28 per share, respectively. Unrecognized compensation cost for unvested stock option awards outstanding at June 30, 2017 was approximately $1,270,000 to be recognized over approximately 1.7 years.
RSUs
We have granted restricted stock units (RSUs) to certain employees and board members that entitle the holders to receive shares of our common stock upon vesting and subject to certain restrictions regarding the exercise of the RSUs. The grant date fair value of RSUs is based upon the market price of the underlying common stock on the date of grant.
At June 30, 2017, we had 1,924 vested and outstanding restricted stock units with a weighted average grant date fair value of $42.75 and a total intrinsic value of approximately $11,000. The total value of all restricted stock units that were converted in the six months ended June 30, 2017 was approximately $23,000.
Stock Purchase Warrants.
We have issued warrants to purchase common stock to certain officers, directors, stockholders and service providers as well as in conjunction with debt and equity offerings and at various times replacement warrants were issued as an inducement for warrant exercises.
12
In May 2016, we issued 1,746,173 common stock purchase warrants in conjunction with our capital raising transactions. Such warrants were classified as derivative liabilities due to the existence of non-standard anti-dilution and certain other conditions contained in the warrants. At June 30, 2017, after giving effect of exercises, 800,017 remain outstanding and are recorded at fair value as derivative liabilities (see Note 3).
In March 2017, we entered into a letter agreement with an investor pursuant to which the investor agreed to exercise certain of their warrants to purchase 692,309 shares of the Company’s common stock; such warrants were originally issued on May 6, 2016 in the Company’s registered offering and contained a current exercise price of $3.25 per share. In exchange for and to induce the investor to exercise the warrants, we issued to the investors an inducement warrant to purchase 230,771 shares of the Company’s common stock.
The inducement warrants are exercisable through March 20, 2018 at an exercise price equal to $5.80 per share, and contain provisions providing for an adjustment in the underlying number of shares and exercise price in the event of stock splits or dividends, subsequent rights offerings, pro rata distributions, and fundamental transactions. In the event that the shares underlying the inducement warrants are not subject to an effective registration statement at the time of exercise, the inducement warrants may be exercised on a cashless basis at any time after six (6) months from the issuance date. The inducement warrants are classified in equity. The fair value of the inducement warrants of $476,084 was expensed as inducement expense in the accompanying condensed consolidated statement of operations for the six months ended June 30, 2017.
In April 2017, we executed a similar agreement with a different investor pursuant to which the investor agreed to exercise certain of their stock purchase warrants to purchase 153,847 shares of the Company’s common stock; such warrants were originally issued on May 6, 2016 in the Company’s registered offering and contained a current exercise price of $3.25 per share. In exchange for and to induce the investor to exercise the warrants, we issued to the investors an inducement warrant to purchase 51,283 shares of the Company common stock at $5.80 per share (the “Inducement Warrants”). The terms of the inducement warrants issued in April 2017 are substantially similar to the terms of the inducement warrants issued in March 2017 and are classified in equity. The fair value of the inducement warrants of $87,660 was expensed as inducement expense in the accompanying condensed consolidated statement of operations for the three and six months ended June 30, 2017.
A summary of outstanding warrants at June 30, 2017 follows:
| Range of Exercise Prices | Number of Warrants Outstanding | Range of Expiration Dates | ||||||
| $3.25 | - | $3.90 | 811,556 | May 2021 - July 2021 | ||||
| $5.80 | - | $6.50 | 282,054 | March 2018 | ||||
| $12.80 | - | $12.90 | 39,296 | January 2022 | ||||
| $13.20 | - | $13.30 | 314,246 | August 2017 | ||||
| $16.20 | - | $16.30 | 174,544 | March 2020 | ||||
| $18.60 | - | $19.80 | 12,309 | March 2018 - June 2018 | ||||
| $22.10 | - | $27.90 | 153,755 | March 2019 - January 2021 | ||||
| $34.50 | - | $39.00 | 164,114 | November 2017 - October 2019 | ||||
| $39.10 | - | $39.20 | 230,772 | October 2020 - October 2021 | ||||
| $47.30 | - | $52.20 | 275,897 | January 2019 - July 2019 | ||||
| 2,458,543 | ||||||||
Preferred and Common Stock
We have outstanding 1,000,000 shares of Series A 4.5% Convertible Preferred Stock issued in December 2016. Shares of the Series A 4.5% Convertible Preferred Stock are convertible into 3,887,387 shares of the Company’s common stock subject to certain ownership restrictions.
In March and April 2017, we issued 846,156 shares of our common stock upon the exercise of certain outstanding common stock purchase warrants. The warrants were exercised at $3.25 per share and we received approximately $2,700,000 in net proceeds. The exercises were pursuant to an inducement agreement entered into with the investors. In conjunction with the exercise we issued certain inducement warrants to the investors. (See “Stock Purchase Warrants” section of Note 5).
In June 2017, we issued 100,000 shares of our common stock upon the exercise of certain outstanding common stock purchase warrants. The warrants were exercised at $3.25 per share and we received approximately $32500,000 in net proceeds.
13
During the six months ended June 30, 2017, we issued 4,939 shares of our common stock upon the conversion of 4,939 outstanding restricted stock units.
Note 6. Commitments and Contingencies
We currently operate one facility located in the United States and one facility located in China. Our corporate offices and primary research facilities are located in Germantown, Maryland, where we license approximately 1,500 square feet. This license provides for monthly payments of approximately $5,500 per month with the term expiring on December 31, 2017.
In 2015, we entered into a lease consisting of approximately 3,100 square feet of research space in San Diego, California. This lease provides for current monthly payments of approximately $11,600 and expires on August 31, 2019. In May 2017, we ceased-use of this property and recognized a loss of approximately $92,000 representing the present value of the expected remaining net payments due under such lease and the costs to vacate the property. The loss is included in research and development expense on our statements of operations for the three- and six-month periods ended June 30, 2017. We are currently exploring opportunities to sub-lease the unused research space.
We also lease a research facility in People’s Republic of China. This lease expires on September 30, 2018 with lease payments of approximately $3,200 per month.
From time to time, we are parties to legal proceedings that we believe to be ordinary, routine litigation incidental to the business. We are currently not a party to any litigation or legal proceeding.
Note 7. Related Party Receivable
On August 10, 2016, we entered into a reimbursement agreement with a former executive officer. Pursuant to the reimbursement agreement, the former officer agreed to repay the Company, over a six-year period, approximately $658,000 in expenses that the Company determined to have been improperly paid under the Company's prior expense reimbursement policies. In addition to this reimbursement agreement, the Company has implemented and is continuing to implement enhanced policies and procedures for travel expense reimbursements and disbursements.
The $658,000 non-interest bearing receivable is recorded net of a discount to reflect the net present value of the future cash payments. The Company recorded a non-operating gain of $459,000 for the year ended December 31, 2016. The discount is being amortized through interest income using the effective interest method. The principal amount of $558,000, excluding discount remains outstanding at June 30, 2017 and is payable in $100,000 annual installments with a final balloon payment due six years from issuance.
Note 8. Subsequent Events
On August 1, 2017, we closed a public offering of 3,000,000 shares of common stock and 2,250,000 common stock purchase warrants at a public purchase price of $2.00 per share and accompanying warrant. We received gross proceeds of $6.0 million and approximately $5.4 million of net proceeds from this offering. The warrants have an exercise price of $2.00 per share of common stock and a term of seven (7) years. This offering was made pursuant to our shelf registration statement that was declared effective by the SEC on June 23, 2017 (Registration No. 333-218608).
| ITEM 2. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Statements in this Quarterly Report that are not strictly historical are forward-looking statements and include statements about products in development, results and analyses of pre-clinical studies, clinical trials and studies, research and development expenses, cash expenditures, and alliances and partnerships, among other matters. You can identify these forward-looking statements because they involve our expectations, intentions, beliefs, plans, projections, anticipations, or other characterizations of future events or circumstances. These forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties that may cause actual results to differ materially from those in the forward-looking statements as a result of any number of factors. These factors include, but are not limited to, risks relating to our ability to conduct and obtain successful results from ongoing clinical trials, commercialize our technology, obtain regulatory approval for our product candidates, contract with third parties to adequately test and manufacture our proposed therapeutic products, protect our intellectual property rights and obtain additional financing to continue our development efforts. Some of these factors are more fully discussed, as are other factors, in our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, filed with the SEC, as well as in the section of this Quarterly Report entitled “Risk Factors” and elsewhere herein. We do not undertake to update any of these forward-looking statements or to announce the results of any revisions to these forward-looking statements except as required by law.
14
We urge you to read this entire Quarterly Report on Form 10-Q, including the “Risk Factors” section, the financial statements, and related notes. As used in this Quarterly Report, unless the context otherwise requires, the words “we,” “us,” “our,” “the Company” and “Neuralstem” refers to Neuralstem, Inc. and its subsidiaries. Also, any reference to “common shares” or “common stock,” refers to our $.01 par value common stock. Any reference to “Series A Preferred Stock” refers to our Series A 4.5% Convertible Preferred Stock. The information contained herein is current as of the date of this Quarterly Report (June 30, 2017), unless another date is specified. We prepare our interim financial statements in accordance with U.S. GAAP. Our financials and results of operations for the three- and six-month periods ended June 30, 2017 are not necessarily indicative of our prospective financial condition and results of operations for the pending full fiscal year ending December 31, 2017. The interim financial statements presented in this Quarterly Report as well as other information relating to our Company contained in this Quarterly Report should be read in conjunction and together with the reports, statements and information filed by us with the United States Securities and Exchange Commission or SEC.
Our Management’s Discussion and Analysis of Financial Condition and Results of Operations or MD&A, is provided in addition to the accompanying financial statements and notes to assist readers in understanding our results of operations, financial condition and cash flows. Our MD&A is organized as follows:
| · | Executive Overview — Discussion of our business and overall analysis of financial and other highlights affecting the Company in order to provide context for the remainder of MD&A. |
| · | Trends & Outlook — Discussion of what we view as the overall trends affecting our business and overall strategy. |
| · | Critical Accounting Policies— Accounting policies that we believe are important to understanding the assumptions and judgments incorporated in our reported financial results and forecasts. |
| · | Results of Operations— Analysis of our financial results comparing the three- and six-month periods ended June 30, 2017 to the comparable periods of 2016. |
| · | Liquidity and Capital Resources— An analysis of cash flows and discussion of our financial condition and future liquidity needs. |
Executive Overview
We are focused on the research and development of nervous system therapies based on our proprietary human neural stem cells and our small molecule compounds with the ultimate goal of gaining approval from the United States Food and Drug Administration or FDA, and its international counterparts, to market and commercialize such therapies. We are headquartered in Germantown, Maryland.
Our technology has produced three primary assets: our NSI-189 small molecule program, our NSI-566 stem cell therapy program and our novel and proprietary chemical entity screening platform.
Our patented technologies enable the commercial-scale production of multiple types of central nervous system stem cells, which are under development for the potential treatment of nervous system diseases and conditions. In addition, this ability to generate human neural stem cell lines provides a platform for chemical screening and discovery of novel compounds that we believe may be used to stimulate the brain's capacity to regenerate neurons, thereby potentially treating or reversing pathologies associated with certain nervous system conditions.
We have developed and maintain what we believe is a strong portfolio of patents and patent applications that form the proprietary base for our research and development efforts. We own or exclusively license over 20 U.S. issued and pending patents and over 120 foreign issued and pending patents in the field of regenerative medicine, related to our stem cell technologies as well as our small molecule compounds.
We believe our technology, in combination with our expertise, and established collaborations with major research institutions, could facilitate the development and commercialization of products for use in the treatment of a wide array of nervous system disorders including neurodegenerative conditions and regenerative repair of acute and chronic disease.
Recent Clinical & Business Highlights
| · | On August 2, 2017, Neuralstem was awarded a Small Business Innovation Research (SBIR) grant by the National Institutes of Health (NIH) to evaluate in preclinical studies the potential of NSI-189, a novel small molecule compound, for the prevention and treatment of diabetic neuropathy. The award of approximately $1 million will be paid over a two-year period. |
15
| · | On July 25, 2017, we announced top-line results from our exploratory Phase 2 clinical trial examining the efficacy of NSI-189 at 40 mg once daily (QD) and 40 mg twice daily (BID) compared to placebo for the treatment of major depressive disorder (MDD). The study, which utilized the two-staged sequential parallel comparison design (SPCD), did not meet its primary efficacy endpoint of a statistically significant reduction in depression symptoms on the Montgomery-Asberg Depression Rating Scale (MADRS). However, the 40 mg QD dose was directionally positive on the MADRS. |
| · | Of two secondary efficacy endpoints in the Phase 2 MDD trial results analyzed so far, the patient-rated Symptoms of Depression Questionnaire (SDQ) achieved statistical significance (p=0.044) with NSI-189 40 mg QD compared to placebo in the overall SPCD analysis. Results were also directionally positive on the Hamilton Depression Rating Scale (HAM-D17) at both doses. Both the 40 mg QD and 40 mg BID doses were well-tolerated with no serious adverse events reported. The company will continue to evaluate the Phase 2 MDD data and provide a full update in the fourth quarter of 2017. |
| · | In June 2017, Neuralstem (NASDAQ: CUR) was added to the Russell Microcap® Index as part of the FTSE’s annual reconstitution of its family of U.S. indexes. The Russell Microcap® Index measures the performance of the microcap segment of the U.S. equity market. |
| · | On August 1, 2017, we closed a public offering of 3,000,000 shares of common stock and 2,250,000 common stock purchase warrants at a public purchase price of $2.00 per share and accompanying warrant. We received gross proceeds of $6.0 million and approximately $5.4 million of net proceeds from this offering. |
| · | From March through July 2017, we received approximately $3,238,000 upon the exercise of 996,156 common stock purchase warrants issued in our May 2016 registered offering at an exercise price of $3.25 per share. We expect that our existing cash and cash equivalents will be sufficient to enable us to fund our anticipated level of operations based on our current operating plans, into the fourth quarter of 2018. |
Clinical Development Program Review
We have devoted substantially all of our efforts and financial resources to the pre-clinical and clinical development of our small molecule compounds and our stem cell therapeutics. Below is a description of our most advanced clinical programs, their intended indication and current stage of development.
16
Clinical Pipeline:
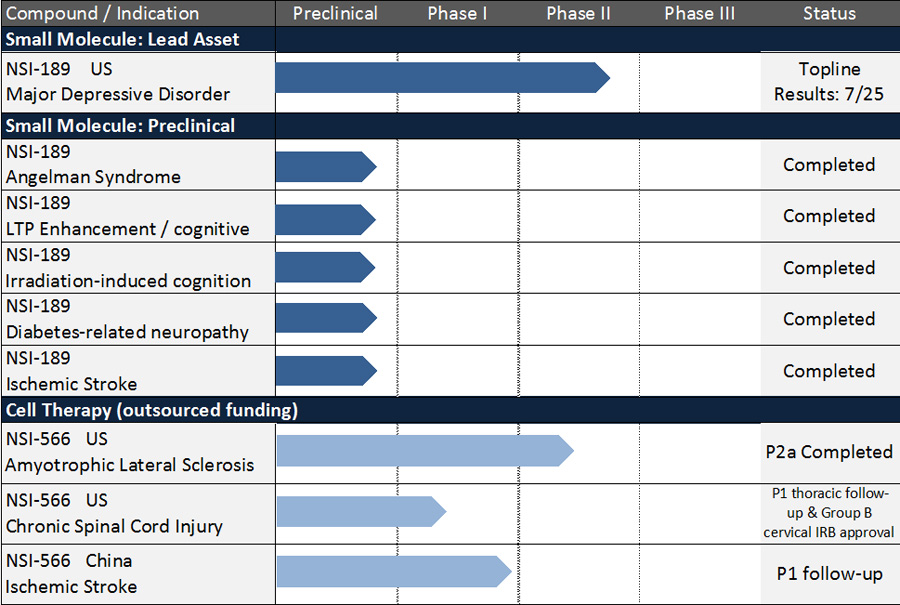
Pipeline Summary
NSI-189 Phase 2 randomized, placebo-controlled, double-blind clinical trial for the treatment of MDD
- In July 2017, the company announced, top-line results from its exploratory Phase 2 clinical trial examining the efficacy of NSI-189 at 40 mg once daily (QD) and 40 mg twice daily (BID) compared to placebo for the treatment of major depressive disorder (MDD). The study, which utilized the two-staged sequential parallel comparison design (SPCD), did not meet its primary efficacy endpoint of a statistically significant reduction in depression symptoms on the Montgomery-Asberg Depression Rating Scale (MADRS). However, the 40 mg QD dose was directionally positive on the MADRS. Two secondary efficacy endpoints analyzed so far, the patient-rated Symptoms of Depression Questionnaire (SDQ) achieved statistical significance (p=0.044) with NSI-189 40 mg QD compared to placebo in the overall SPCD analysis. Results were also directionally positive on the Hamilton Depression Rating Scale (HAM-D17) at both doses. Both the 40 mg QD and 40 mg BID doses were well-tolerated with no serious adverse events reported. The company will continue to evaluate the data and will provide an update in 4Q17. NSI-189 Phase 2 MDD clinical trial study results were announced 4 months ahead of schedule. The clinical trial was initiated in May 2016, The company announced 50% enrollment in September 2016 and last subject completed the study in May 2017. 220 subjects were randomized for a 12-week interventional study with NSI-189 or placebo. The study was conducted under the direction of Principal Investigator (PI) Maurizio Fava, MD, Executive Vice Chair, Department of Psychiatry and Executive Director, Clinical Trials Network and Institute, Massachusetts General Hospital.
NSI-566 Phase 1 and 2 safety trials for the treatment of Amyotrophic Lateral Sclerosis (ALS)
- In September 2015, nine-month Phase 2 and combined Phase 1 and Phase 2 data from our ALS trials were presented at the American Neurological Association Meeting by Principal Investigator Eva Feldman, MD, PhD, Director of the A. Alfred Taubman Medical Research Institute and Director of Research of the ALS Clinic at the University of Michigan Health. The data showed that the intraspinal transplantation of the cells was safe and well tolerated. Subjects from both the Phase 1 and Phase 2 continue to be monitored for long-term follow-up evaluations. Long-term follow-up data on subjects from both the Phase 1 and Phase 2 safety trials showed an encouraging signal of continued therapeutic benefit versus historical control database, PRO-ACT. This data was presented by Dr. Feldman at the 2017 International Society For Stem Cell Research (ISSCR) Conference in June 2017.
17
NSI-566 Phase 1 safety trial for the treatment of motor deficits in stroke
- In March 2016, we completed dosing the final planned cohort, for a total of nine subjects. Subjects are currently being monitored through their 24-month observational follow-up period. The trial is being conducted by Suzhou Neuralstem, a wholly owned subsidiary of Neuralstem in China.
NSI-566 Phase 1 safety trial for the treatment of chronic Spinal Cord Injury (cSCI)
- In April 2017, the company announced that it had received FDA approval to recruit a new cohort (Group B) of four subjects with stable AISA-A complete, quadriplegic, cervical injuries to the ongoing Phase 1 human clinical trial evaluating the safety and feasibility of using NSI-566 spinal cord-derived neural stem cells to repair chronic cSCI. In January 2016, we reported on the interim status of the Phase 1 safety data on all four subjects with stable thoracic spinal cord injuries; the stem cell treatment demonstrated feasibility and safety. A self-reported ability to contract some muscles below the level of injury was confirmed via clinical and electrophysiological follow-up examinations in one of the four subjects treated. All subjects will be followed for five years. This study is being conducted with support from the University of California, San Diego (UCSD) School of Medicine.
Pre-Clinical Development Pipeline
Our preclinical research on NSI-189 is focused on identifying its mechanism of action and investigating its potential utility as a broad neuroregenerative drug that can prevent or reverse various types of central and peripheral nerve degeneration and that may have significant cognitive benefit in diseases that impact memory and cognition. Recent preclinical data support the potential benefits of NSI-189 in other indications beyond MDD.
Our preclinical studies with NSI-566 have served to provide a solid foundation for our ongoing clinical trials by demonstrating performance and efficacy of this cell line in animal models for ALS, spinal cord injury, and ischemic stroke, and demonstrated safety in large animals. Additional studies involving NSI-566 are directed at identifying new therapeutic indications.
In addition to NSI-566 we have developed an inventory of over 300 unique stem cell lines. These stem cell lines include cortex, hippocampus, midbrain, hindbrain, cerebellum, and spinal cord. We believe these lines possess unique properties and represent candidates for both transplantation-based strategies to treat disease and for screening of compound libraries to discover novel drug therapies.
Our Technologies
Our technology has produced three primary assets: our NSI-189 small molecule program, our NSI-566 stem cell therapy program and our novel and proprietary chemical entity screening platform.
Small Molecule Pharmaceutical Compounds.
Utilizing our proprietary stem cell-based screening capability, we have discovered and patented a series of small molecule compounds. We believe our low molecular weight organic compounds can efficiently cross the blood/brain barrier. In mice, research indicated that the small molecule compounds both stimulate neurogenesis of the hippocampus and increase its volume. We believe the small molecule compounds may promote synaptogenesis and neurogenesis in the human hippocampus in indications such as MDD and, also provide clinical benefit to patients in indications such as Angelman Syndrome, Diabetic Neuropathy, Cognition, Stroke and Radiation Induced Cognitive Deficit.
Our portfolio of small molecule compounds which includes NSI-189 are covered by 10 U.S. exclusively owned issued and pending patents and over 60 exclusively owned foreign issued and pending patents.
Stem Cells.
Our stem cell based technology has both therapeutic and screening characteristics.
From a therapeutic perspective, our stem cell based technology enables the isolation and large-scale expansion of regionally specific, human neural stem cells from all areas of the developing human brain and spinal cord thus enabling the generation of physiologically relevant human neurons of different types. We believe that our stem cell technology will enable the replacement of malfunctioning or dead cells or the protection of neurons as a way to treat disease and injury. Many significant and currently untreatable human diseases arise from the loss or malfunction of specific cell types in the body. Our focus is the development of effective methods to generate replacement cells from neural stem cells. We believe that replacing damaged, malfunctioning or dead neural cells with fully functional ones may be a useful therapeutic strategy in treating many diseases and conditions of the central nervous system.
Our Proprietary and Novel Screening Platform
Our human neural stem cell lines form the foundation for functional cell-based assays used to screen for small molecule compounds that can impact biologically relevant outcomes such as neurogenesis, synapse formation, and protection against toxic insults. We have developed over 300 unique stem cell lines representing multiple different regions of the developing brain and spinal cord at multiple different time points in development, enabling the generation of physiologically relevant human neural cells for screening, target validation, and mechanism-of-action studies. This platform provides us with a unique and powerful tool to identify new chemical entities to treat a broad range of nervous system conditions. NSI-189 was discovered using our stem cell-based screening platform.
18
Intellectual Property
We have developed and maintain what we believe is a strong portfolio of patents and patent applications that form the basis for our research and development efforts. We own or exclusively license over 10 U.S. issued and pending patents and over 60 foreign issued and pending patents related to our stem cell technologies for use in treating disease and injury. We own over 10 U.S. issued and pending patents and over 60 foreign issued and pending patents related to our small molecule compounds. Our issued patents have expiration dates ranging from 2017 through 2035. Two of our original patents covering methods and composition of matter associated with our stem cell technologies expired in 2016. In our opinion, the expiration of these patents is not material to our intellectual property.
Operating Strategy
We generally employ an outsourcing strategy where we outsource our preclinical and clinical development activities to contract research organizations and academic partners. Manufacturing of our small molecule portfolio is also outsourced to organizations with approved facilities and manufacturing practices. Manufacturing of our stem cells is proprietary and we operate a closed, in-house system to ensure the protection of all critical know-how associated with the technology. All non-critical corporate functions are outsourced as well. This model allows us to better manage cash on hand and minimize non-vital expenditures. It also allows for us to operate with relatively fewer employees and lower fixed costs than that required by other companies conducting similar business.
Employees
As of July 31, 2017, we had ten (10) full-time employees. Of these full-time employees, seven (7) work on research and development and clinical operations and three (3) work in administration. We also use the services of numerous outside consultants in business and scientific matters.
Our Corporate Information
We were incorporated in Delaware in 2001. Our principal executive offices are located at 20271 Goldenrod Lane, Germantown, Maryland 20876, and our telephone number is (301) 366-4841. Our website is located at www.neuralstem.com.
We have not incorporated by reference into this report the information in, or that can be accessed through, our website and you should not consider it to be a part of this report.
Trends & Outlook
Revenue
We generated no revenues from the sale of our proposed therapies for any of the periods presented. We are mainly focused on successfully managing our current clinical trials related to our small molecule compounds and seeking potential partnerships for our stem cell product candidates. We are also pursuing pre-clinical studies on other central nervous system indications in preparation for potential future clinical trials.
During the six months ended June 30, 2017 and 2016, we recognized approximately $5,000 of revenue in each period related to ongoing fees pursuant to certain licenses of our intellectual property to third parties.
On a long-term basis, we anticipate that our revenue will be derived primarily from licensing fees and sales of our small molecule compounds and licensing fees and royalties from our cell based therapies. Because we are at such an early stage in the clinical trials process, we are not yet able to accurately predict when we will have a product ready for commercialization, if ever.
Research and Development Expenses
Our research and development expenses consist primarily of clinical trial expenses, including; payments to clinical trial sites that perform our clinical trials and clinical research organizations (CROs) that help us manage our clinical trials, manufacturing of small molecule drugs and stem cells for both human clinical trials and for pre-clinical studies and research, personnel costs for research and clinical personnel, and other costs including research supplies and facilities.
We focus on the development of treatment candidates with potential uses in multiple indications, and use employee and infrastructure resources across several projects. Accordingly, many of our costs are not attributable to a specifically identified product and we do not account for internal research and development costs on a project-by-project basis.
19
We expect that research and development expenses, which include expenses related to our ongoing clinical trials, will increase in the future, as funding allows and we proceed later stage clinical trials.
We have formed a wholly owned subsidiary in the People’s Republic of China. We anticipate that this subsidiary will primarily: (i) conduct pre-clinical research with regard to proposed stem cells therapies, and (ii) oversee our approved future clinical trials in China, including the current trial to treat motor deficits due to ischemic stroke.
General and Administrative Expenses
General and administrative expenses are primarily comprised of salaries, benefits and other costs associated with our operations including, finance, human resources, information technology, public relations and costs associated with maintaining a public company listing, legal, audit and compliance fees, facilities and other external general and administrative services.
Critical Accounting Policies
Our condensed consolidated financial statements have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues and expenses. Note 2 of the Notes to Unaudited Condensed Consolidated Financial Statements included elsewhere herein describes the significant accounting policies used in the preparation of the financial statements. Certain of these significant accounting policies are considered to be critical accounting policies, as defined below.
A critical accounting policy is defined as one that is both material to the presentation of our financial statements and requires management to make difficult, subjective or complex judgments that could have a material effect on our financial condition and results of operations. Specifically, critical accounting estimates have the following attributes: (1) we are required to make assumptions about matters that are highly uncertain at the time of the estimate; and (2) different estimates we could reasonably have used, or changes in the estimate that are reasonably likely to occur, would have a material effect on our financial condition or results of operations.
Estimates and assumptions about future events and their effects cannot be determined with certainty. We base our estimates on historical experience and on various other assumptions believed to be applicable and reasonable under the circumstances. These estimates may change as new events occur, as additional information is obtained and as our operating environment changes. These changes have historically been minor and have been included in the financial statements as soon as they became known. Based on a critical assessment of our accounting policies and the underlying judgments and uncertainties affecting the application of those policies, management believes that our financial statements are fairly stated in accordance with U.S. GAAP, and present a meaningful presentation of our financial condition and results of operations. We believe the following critical accounting policies reflect our more significant estimates and assumptions used in the preparation of our consolidated financial statements:
Use of Estimates - Our financial statements prepared in accordance with U.S. GAAP require us to make estimates and assumptions that affect the reported amounts of assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Specifically, we have estimated the expected economic life and value of our patent technology, our net operating loss carryforward and related valuation allowance for tax purposes the fair value of our derivative instruments and our stock-based compensation expenses related to employees, directors, consultants and investment banks. Actual results could differ from those estimates.
Long Lived Intangible Assets - Our long lived intangible assets consist of our intellectual property patents including primarily legal fees associated with the filings and in defense of our patents. The assets are amortized on a straight-line basis over the expected useful life which we define as ending on the expiration of the patent group. These assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. We assess this recoverability by comparing the carrying amount of the asset to the estimated undiscounted future cash flows to be generated by the asset. If an asset is deemed to be impaired, we estimate the impairment loss by determining the excess of the asset’s carrying amount over the estimated fair value. These determinations use assumptions that are highly subjective and include a high degree of uncertainty. During the six months ended June 30, 2017 and 2016, no significant impairment losses were recognized.
Fair Value Measurements - The fair value of our short-term financial instruments, which primarily include cash and cash equivalents, other short-term investments, accounts payable and accrued expenses, approximate their carrying values due to their short maturities. The fair value of our long-term indebtedness is estimated based on the quoted prices for the same or similar issues or on the current rates offered to the Company for debt of the same remaining maturities which approximates the carrying value. The fair values of our derivative instruments are estimated using Level 3 unobservable inputs.
20
Share-Based Compensation - We account for share-based compensation at fair value; accordingly, we expense the estimated fair value of share-based awards over the requisite service period. Share-based compensation cost for stock options and warrants issued to employees and board members is determined at the grant date while awards granted to non-employee consultants are generally valued at the vesting date using an option pricing model. Option pricing models require us to make assumptions, including expected volatility and expected term of the options. If any of the assumptions we use in the model were to significantly change, stock based compensation expense may be materially different. Share-based compensation cost for restricted stock and restricted stock units issued to employees and board members is determined at the grant date based on the closing price of our common stock on that date. The value of the award that is ultimately expected to vest is recognized as expense on a straight-line basis over the requisite service period.
RESULTS OF OPERATIONS
Comparison of Three Months Ended June 30, 2017 and 2016
Revenue
During each of the three months ended June 30, 2017 and 2016 we recognized $2,500 of revenue related to ongoing fees pursuant to certain licenses of our intellectual property to third parties.
Operating Expenses
Operating expenses for the three months ended June 30, 2017 and 2016 were as follows:
| Three Months Ended June 30, | Increase (Decrease) | |||||||||||||||
| 2017 | 2016 | $ | % | |||||||||||||
| Operating Expenses | ||||||||||||||||
| Research and development expenses | $ | 2,585,079 | $ | 2,474,629 | $ | 110,450 | 4 | % | ||||||||
| General and administrative expenses | 1,635,652 | 1,362,140 | 273,512 | 20 | % | |||||||||||
| Total operating expenses | $ | 4,220,731 | $ | 3,836,769 | $ | 383,962 | 10 | % | ||||||||
Research and Development Expenses
The increase of approximately $110,000 or 4% in research and development expenses for the three months ended June 30, 2017 compared to the comparable period of 2016 was primarily attributable to $420,000 increase in clinical trial costs associated with our ongoing Phase 2 MMD study, $170,000 increase in non-cash stock based compensation expense partially offset by a $480,000 decrease in in personnel related costs, internal and external research expenditures and other expenses associated with our May 2016 restructuring.
General and Administrative Expenses
The increase of approximately $274,000 or 20% in general and administrative expenses for the three months ended June 30, 2017 compared to the comparable period of 2016 was primarily attributable to a $530,000 increase in personnel related expenses due to severances partially offset by $340,000 decrease in non-cash stock based compensation expense.
Other expense
Other expense, net totaled approximately $431,000 and $18,000 for the three months ended June 30, 2017 and 2016, respectively.
Other expense, net in 2017 consisted primarily of approximately $342,000 of losses related to the fair value adjustment of our derivative instruments and $88,000 of expense related to the issuance of inducement warrants.
Other expense, net in 2016 consisted of approximately $467,000 of fees related to the issuance of our derivative instruments and
$322,000 of interest expense related to our long-term debt, partially offset by a gain of approximately $757,000 related to the fair
value adjustment of our derivative instruments.
Comparison of Six Months Ended June 30, 2017 and 2016
Revenue
During each of the six months ended June 30, 2017 and 2016 we recognized $5,000 of revenue related to ongoing fees pursuant to certain licenses of our intellectual property to third parties.
21
Operating Expenses
Operating expenses for the six months ended June 30, 2017 and 2016 were as follows:
| Six Months Ended June 30, | Increase (Decrease) | |||||||||||||||
| 2017 | 2016 | $ | % | |||||||||||||
| Operating Expenses | ||||||||||||||||
| Research and development expenses | $ | 5,487,165 | $ | 5,540,219 | $ | (53,054 | ) | (1 | %) | |||||||
| General and administrative expenses | 2,968,073 | 4,532,662 | (1,564,589 | ) | (35 | %) | ||||||||||
| Total operating expenses | $ | 8,455,238 | $ | 10,072,881 | $ | (1,617,643 | ) | (16 | %) | |||||||
Research and Development Expenses
The decrease of approximately $53,000 or 1% in research and development expenses for the six months ended June 30, 2017 compared to the comparable period of 2016 was primarily attributable to a $2.0 million reduction in employment costs, internal and external research expenditures associated with our May 2016 restructuring almost entirely offset by an increase in clinical trial costs associated with our ongoing Phase 2 MDD study.
General and Administrative Expenses
The decrease of approximately $1,565,000 or 35% in general and administrative expenses for the six months ended June 30, 2017 compared to the comparable period of 2016 was primarily attributable to personnel related savings associated with our May, 2016 restructuring partially offset by current period severance expenses.
Other expense
Other expense, net totaled approximately $3,766,000 and $390,000 for the six months ended June 30, 2017 and 2016, respectively.
Other expense, net in 2017 consisted of approximately $3,083,000 of losses related to the fair value adjustment of our derivative instruments, $564,000 of expense related to the issuance of inducement warrants and $154,000 of interest expense primarily related to our long-term debt, partially offset by $35,000 of interest income.
Other expense, net in 2016 consisted of approximately $467,000 of fees related to the issuance of our derivative instruments and $709,000 of interest related to our long-term debt, partially offset by a gain of approximately $757,000 related to the fair value adjustment of our derivative instruments.
Liquidity and Capital Resources
Financial Condition
Since our inception, we have financed our operations through the sales of our securities, issuance of long-term debt, the exercise of investor warrants, and to a lesser degree from grants and research contracts as well as the licensing of our intellectual property to third parties.
We had cash, cash equivalents and short-term investments balances of approximately $11.4 million as at June 30, 2017. On August 1, 2017, we closed a public offering of 3,000,000 shares of common stock and 2,250,000 common stock purchase warrants at a public purchase price of $2.00 per share and accompanying warrant. We received gross proceeds of $6.0 million and approximately $5.4 million of net proceeds from this offering.
Based on our current expectations, we anticipate our average monthly cash burn rate will decrease to approximately (i) $0.8 million for the second half of the year due to completion of both our MDD study, and (ii) $0.6 million for the first half of 2018 due to completion of our observation-only, durability study of NSI-189 in subjects with MDD. Based upon our estimated cash burn rate, we expect to be able to fund our operations, into the fourth quarter of 2018.
We will require additional capital to continue to develop our pre-clinical and clinical development operations. To continue to fund our operations and the development of our product candidates we anticipate raising additional cash through the private or public sales of equity or debt securities, collaborative arrangements, licensing agreements or a combination thereof. Although management believes that such funding sources will be available, there can be no assurance that any such collaborative arrangement will be entered into or that financing will be available to us when needed in order to allow us to continue our operations, or if available, on terms acceptable to us. If we do not raise sufficient funds in a timely manner, we may be forced to curtail operations, delay or stop our ongoing clinical trials, cease operations altogether, or file for bankruptcy. We currently do not have commitments for future funding from any source. We cannot assure you that we will be able to secure additional capital or that the expected income will materialize. Several factors will affect our ability to raise additional funding, including, but not limited to market conditions, interest rates and, more specifically, our progress in our exploratory, preclinical and future clinical development programs.
22
| Six Months Ended June 30, | Increase (Decrease) | |||||||||||||||
| 2017 | 2016 | $ | % | |||||||||||||
| Net cash used in operating activities | $ | (7,807,240 | ) | $ | (7,059,924 | ) | $ | (747,316 | ) | (11 | %) | |||||
| Net cash (used in) provided by investing activities | $ | (93,448 | ) | $ | 7,389,182 | $ | (7,482,630 | ) | (101 | %) | ||||||
| Net cash (used in) provided by financing activities | $ | (851,414 | ) | $ | 6,082,944 | $ | (6,934,358 | ) | (114 | %) | ||||||
Net Cash Used in Operating Activities
We used approximately $7,807,000 and $7,060,000 of cash in our operating activities for the six months ended June 30, 2017 and 2016, respectively. The increase in our use of cash in operating activities of approximately $747,000 was primarily due to payment of accrued bonuses and accounts payable in 2017 partially offset by a decrease in our operating loss. The increase in our net loss was offset by adjustments including non-cash charges related to changes in fair value of derivative instruments, share-based compensation expense and warrant inducement expense.
Net Cash Provided by Investing Activities
For the six months ended June 30, 2017 we used cash of approximately $84,000 for costs related to our patent assets. For the six months ended June 30, 2016 we received approximately $7.5 million from the maturity of some of our short-term investments.
Net Cash Provided by Financing Activities
In the six months ended June 30, 2017, cash used in financing activities consisted of approximately $3,955,000 of payment on our short-term and long-term debt partially offset by $3,103,000 of net proceeds received from the sale of our common stock and exercise of common stock purchase warrants.
In the six months ended June 30, 2016, cash provided by financing activities was comprised of net proceeds from our two financing in May 2016 of approximately $8,309,000 partially offset by loan principal repayments of approximately $2,226,000.
Future Liquidity and Needs
We have incurred significant operating losses and negative cash flows since inception. We have not achieved profitability and may not be able to realize sufficient revenue to achieve or sustain profitability in the future. We do not expect to be profitable in the next several years, but rather expect to incur additional operating losses. We have limited liquidity and capital resources and must obtain significant additional capital resources in order to sustain our product development efforts, for acquisition of technologies and intellectual property rights, for preclinical and clinical testing of our anticipated products, pursuit of regulatory approvals, acquisition of capital equipment, laboratory and office facilities, establishment of production capabilities, for general and administrative expenses and other working capital requirements. We rely on cash balances and the proceeds from the offering of our securities, exercise of outstanding warrants and grants to fund our operations.
We intend to pursue opportunities to obtain additional financing in the future through the sale of our securities and additional research grants. On June 23, 2017, our shelf registration statement (Registration No. 333-218608), which replaced our prior expiring shelf registration statement, was declared effective by the SEC. Under such replacement shelf registration statement, we can offer and sell up to $100 million of our securities was declared effective by the SEC. To date, through August 1, 2017 we have sold and reserved for sale upon exercise of outstanding instruments approximately $10.5 million under this shelf registration statement.
The source, timing and availability of any future financing will depend principally upon market conditions, interest rates and, more specifically, current and future progress in our exploratory, preclinical and clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of necessary funds may require us, among other things, to delay, scale back or eliminate some or all of our research and product development programs, planned clinical trials, and/or our capital expenditures or to license our potential products or technologies to third parties.
| ITEM 3. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are not required to provide the information required by this item as we are considered a smaller reporting company, as defined by Rule 229.10(f)(1).
23
| ITEM 4. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
Disclosure controls and procedures are controls and other procedures that are designed to ensure that information required to be disclosed in our reports filed or submitted under the Securities Exchange Act of 1934, as amended (the "Exchange Act"), is recorded, processed, summarized, and reported within the time periods specified in the Securities and Exchange Commission's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed under the Exchange Act is accumulated and communicated to management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Based on an evaluation under the supervision and with the participation of the Company’s management, the Company’s principal executive officer, who is also our principal financial officer, has concluded that the Company’s disclosure controls and procedures as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act were effective as of June 30, 2017, to ensure that information required to be disclosed by the Company in reports that it files or submits under the Exchange Act is (i) recorded, processed, summarized and reported within the time periods specified in the SEC rules and forms and (ii) accumulated and communicated to the Company’s management, including its principal executive officer, who is also our principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
Changes in Internal Control Over Financial Reporting
Management has identified the following change in our internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) that occurred during the second quarter of 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
As reported in our Form 8-K filed with the Securities Exchange Commission on May 4, 2017, our former Chief Financial Officer and Principal Accounting Officer, is no longer employed by the Company effective April 30, 2017. Effective April 30, 2017, our current Chief Executive Officer will serve as the Company’s Principal Financial and Accounting Officer, on an interim basis, until a successor has been identified and retained.
We engaged third party financial reporting consultants to assist management in the preparation of the Company’s financial statements in accordance with U.S. GAAP and the rules and regulations promulgated by the Securities and Exchange Commission. We anticipate that these consultants will also assist in ensuring that our internal controls are effective to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in SEC rules and forms, and is accumulated and communicated to management, including our principal executive officer and principal financial officer, as appropriate, to allow timely decisions regarding required disclosure.
Inherent Limitations Over Internal Controls
The Company’s internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with U.S. generally accepted accounting principles (“GAAP”). The Company’s internal control over financial reporting includes those policies and procedures that:
(i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company’s assets;
(ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with GAAP, and that the Company’s receipts and expenditures are being made only in accordance with authorizations of the Company’s management and directors; and
(iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the Company’s assets that could have a material effect on the financial statements.
Management, including the Company’s principal executive officer and principal financial officer, does not expect that the Company’s internal controls will prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met. Further, the design of a control system must reflect the fact that there are resource constraints, and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of internal controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Also, any evaluation of the effectiveness of controls in future periods are subject to the risk that those internal controls may become inadequate because of changes in business conditions, or that the degree of compliance with the policies or procedures may deteriorate.
24
OTHER INFORMATION
| ITEM 1. | LEGAL PROCEEDINGS |
We are parties to legal proceedings that we believe to be ordinary, routine litigation incidental to the business of present or former operations. It is management’s opinion, based on the advice of counsel, that the ultimate resolution of such litigation will not have a material adverse effect on our financial condition, results of operations or cash flows.
| ITEM 1A. | RISK FACTORS |
Investing in our common stock involves a high degree of risk. We have described below a number of uncertainties and risks which, in addition to uncertainties and risks presented elsewhere in this Quarterly Report, may adversely affect our business, operating results and financial condition. The uncertainties and risks enumerated below as well as those presented elsewhere in this Quarterly Report, and those included in our Annual Report on Form 10-K for the year ended December 31, 2016, filed with the SEC should be considered carefully in evaluating our company and our business and the value of our securities.
Risks Relating to Our Stage of Development and Capital Structure
We have a history of losses.
Since inception in 1996 through June 30, 2017, we have accumulated losses totaling approximately $205,250,000. On June 30, 2017, we had a working capital surplus of approximately $9,383,000 and stockholders’ equity of approximately $7,693,000. Our net losses for the three most recent fiscal years have been approximately $21,075,000, $20,904,000 and $22,629,000 for 2016, 2015 and 2014, respectively. We have generated no significant revenue from the sales of our proposed products.
Our ability to generate revenues and achieve profitability will depend upon our ability to complete the development of our proposed products, obtain the required regulatory approvals, manufacture and market and sell our proposed products. To date, we have not generated any revenue from the commercial sale of our proposed products. No assurances can be given as to exactly when, if at all, we will be able to fully develop, commercialize, market, sell and/or derive any, let alone material, revenues from our proposed products.
We will need to raise additional capital to continue operations.
Since our inception, we have funded our operations through the sale of our securities, credit facilities, the exercise of options and warrants, and to a lesser degree, from grants and research contracts and other revenue generating activities such as licensing. As of June 30, 2017, we had cash, cash equivalents and short-term investments on hand of approximately $11.4 million. We cannot assure you that we will be able to secure additional capital through financing transactions, including issuance of debt, licensing agreements or grants. Our inability to license our intellectual property, obtain grants or secure additional financing will materially impact our ability to fund our current and planned operations.
We have spent and expect to continue spending substantial cash in the research, development, clinical and pre-clinical testing of our proposed products with the goal of ultimately obtaining FDA approval and equivalent international approvals to market such products. We will require additional capital to conduct research and development, establish and conduct clinical and pre-clinical trials, enter into commercial-scale manufacturing arrangements and to provide for marketing and distribution of our products. We cannot assure you that financing will be available if needed. If additional financing is not available, we may not be able to fund our operations, develop or enhance our technologies, take advantage of business opportunities or respond to competitive market pressures. If we exhaust our cash reserves and are unable to secure adequate additional financing, we may be unable to meet operating obligations which could result in us initiating bankruptcy proceedings or delaying, or eliminating some or all of our research and product development programs.
We may not be able to continue as a going concern if we do not obtain additional financing.
We have incurred losses since its inception and has not demonstrated an ability to generate revenues from sales or services. Our ability to continue as a going concern is dependent on generating cash from the sale of its common stock and/or obtaining debt financing. Our cash, cash equivalents and short-term investment balance at June 30, 2017 was approximately $11.4 million. Based on our current expected level of operating expenditures, we expect to be able to fund our operations into the fourth quarter of 2018. Our ability to remain a going concern is wholly dependent upon our ability to continue to obtain sufficient financing to fund our operations.
25
Accordingly, despite our ability to secure capital in the past, there is no assurance that additional equity or debt financing will be available to us when needed. In the event that we are not able to secure financing, we may be forced to curtail operations, delay or stop ongoing clinical trials, cease operations altogether or file for bankruptcy.
Risks Relating to Our Business
Following our announcements regarding the negative results from our Phase 2 study, we may not generate any future revenues from NSI-189 or its underlying intellectual property.
On July 25, 2017, we announced that our Phase 2 study of NSI-189 in subjects with MDD failed to achieve statistical significance on its primary endpoint. Following these clinical results, we may not generate any future revenues from NSI-189 or its underlying intellectual property.
Our business is dependent on the successful development of our product candidates and our ability to raise additional capital.
Our business is significantly dependent on our product candidates which are currently at different phases of pre-clinical and clinical development. The process to approve our product candidates is time-consuming, involves substantial expenditures of resources, and depends upon a number of factors, including the availability of alternative treatments, and the risks and benefits demonstrated in our clinical trials. Our success will depend on our ability to achieve scientific and technological advances and to translate such advances into FDA-approvable, commercially competitive products on a timely basis. Failure can occur at any stage of the process. On July 25, 2017, we announced that our Phase 2 clinical trial of NSI-189 in MDD failed to achieve statistical significance on its primary endpoint.. We are currently evaluating the Phase 2 trial data. If we are not successful in developing our product candidates, we will have invested substantial amounts of time and money without developing revenue-producing products. As we enter a more extensive clinical program for our product candidates, the data generated in these studies may not be as compelling as the earlier results. This, in turn, could adversely impact our ability to raise additional capital and pursue our business plan and planned research and development efforts.
Our proposed products are not likely to be commercially available for at least several years, if at all. Our development schedules for our proposed products may be affected by a variety of factors, including technological difficulties, clinical trial failures, regulatory hurdles, competitive products, intellectual property challenges and/or changes in governmental regulation, many of which will not be within our control. Any delay in the development, introduction or marketing of our product candidates could result either in such products being marketed at a time when their cost and performance characteristics would not be competitive in the marketplace or in the shortening of their commercial lives. In light of the long-term nature of our projects, the unproven technology involved and the other factors described elsewhere in this section, there can be no assurance that we will be able to successfully complete the development or marketing of any of our proposed product candidates.
Our business relies on technologies that we may not be able to commercially develop and we are unable to predict when or if we will be able to earn revenues.
We have allocated the majority of our resources to the development of our stem cell and small molecule technologies. Our ability to generate revenue and operate profitably will depend on being able to develop these technologies for human applications. These are emerging technologies that may have limited human application. On July 25, 2017, we announced that our Phase 2 clinical trial of NSI-189 in MDD failed to achieve statistical significance on its primary endpoint. We are currently evaluating the Phase 2 trial data. We cannot guarantee that we will be able to develop our technologies or that if developed, our technologies will result in commercially viable products or have any commercial utility or value. We anticipate that the commercial sale of our proposed products and/or royalty/licensing fees related to our technologies, will be our primary sources of revenue. We recognized revenue of approximately $16,000, $10,000 and $19,000 for the years ended December 31, 2016, 2015 and 2014, respectively, related to the licensing of certain intellectual property to third parties and certain subcontractor services that we provided. If we are unable to develop our technologies, we may never realize any significant revenue. Additionally, given the uncertainty of our technologies, product candidates and the need for government regulatory approval, we cannot predict when, or if ever, we will be able to realize revenues related to our products. As a result, we will be primarily dependent on our ability to raise capital through the sale of our securities for the foreseeable future.
Our product development programs are based on novel technologies in an emerging field and are inherently risky.
We are subject to the risks inherent in the development of products based on new technologies. The novel nature of therapies in the field of regenerative medicine creates significant challenges in regard to product development and optimization, manufacturing, government regulation, third party reimbursement, and market acceptance. For example, the pathway to regulatory approval for cell-based therapies, including our stem cell based product candidates, may be more complex and lengthy than the pathway for conventional drugs. These challenges may prevent us from developing and commercializing products on a timely or profitable basis or at all. Regenerative medicine is still an emerging field. There can be no assurances that we will ultimately produce any viable commercialized products and processes. Even if we are able to produce a commercially viable product, there may be strong competitors in this field and our products may not be able to successfully compete against them.
26
Our stem cell therapy programs rely on experimental surgical devices and experimental and highly invasive surgical procedures.
We are subject to the risks inherent in the use and development of experimental surgical devices and procedures. We have limited experience with medical devices and must rely on outside consultants and manufacturers to develop and seek any required approvals for the device we use in connection with our stem cell therapy program. Additionally, the surgical procedures required to administer our stem cell therapies are experimental, highly invasive and is required to be performed by highly experienced neurosurgeons who have received special training. We cannot guarantee consistent and safe performance of these devices or the surgical procedures. A surgery related adverse event may result in a clinical hold and may have long-term and damaging effects on our ability to complete development of the stem cell therapy programs, including the completion of any ongoing or planned clinical trials. Even if one or more of our programs is successful and receives marketing approval from a regulatory authority, due to the specialized nature of the device and surgical procedure, there may not be sufficient train surgeons to administer our therapy.
We are unable to predict when or if we will be able to earn revenues.
Given the uncertainty of our technologies and the need for government regulatory approval, we cannot predict when, or if ever, we will be able to realize revenues related to our products.
Our proposed products are not likely to be commercially available for at least several or more years, if ever. Accordingly, we do not foresee generating any significant revenue during such time. As a result, we will be primarily dependent on our ability to raise capital through the sale of our securities to fund our operations for the foreseeable future.
Our reliance on third parties to manufacture and store our stem cells and small molecule compounds could adversely impact our business.
We currently outsource most of the manufacturing of our stem cells and small molecule pharmaceutical compounds to third party contractors and as such have limited ability to adequately control the manufacturing process and the safe storage thereof. Any manufacturing or storage irregularity, error, or failure to comply with applicable regulatory procedure would require us to find new third parties to outsource our manufacturing and storage responsibilities or our business would be impacted.
The manufacture of our therapeutic products is a complicated and difficult process, dependent upon substantial know-how and subject to the need for continual process improvements. In addition, our suppliers’ ability to scale-up manufacturing to satisfy the various requirements of our planned clinical trials is uncertain. Additionally, many of the materials that we use to prepare our cell-based products are highly specialized, complex and available from only a limited number of suppliers. The loss of one or more of these sources would likely delay our ability to conduct planned clinical trials and otherwise adversely affect our business.
If we are unable to complete pre-clinical and clinical testing and trials or if clinical trials of our product candidates are prolonged, delayed, suspended or terminated, our business and results of operations could be materially harmed.
Although we have commenced a number of trials, the ultimate outcome of the trials is uncertain. On July 25, 2017, we announced that our Phase 2 clinical trial of NSI-189 in MDD failed to achieve statistical significance on its primary endpoint. We are currently evaluating the Phase 2 trial data. If we are unable to satisfactorily complete our other trials, or if such trials also yield unsatisfactory results, we may be unable to obtain regulatory approval for and commercialize our proposed products. No assurances can be given that our clinical trials will be completed or result in successful outcomes. A number of events, including any of the following, could delay the completion of our planned clinical trials and negatively impact our ability to obtain regulatory approval for, and to market and sell, a particular product candidate:
| · | conditions imposed on us by the FDA or any foreign regulatory authority regarding the scope or design of our clinical trials; |
| · | delays in obtaining, or our inability to obtain, required approvals from institutional review boards, or IRBs, or other reviewing entities at clinical sites selected for participation in our clinical trials; |
| · | insufficient supply or deficient quality of our product candidates or other materials necessary to conduct our clinical trials; |
| · | delays in obtaining regulatory agency agreement for the conduct of our clinical trials; |
| · | lower than anticipated enrollment and retention rate of subjects in clinical trials; |
| · | serious and unexpected side effects experienced by patients in our clinical trials which are related to the use of our product candidates; or |
| · | failure of our third-party contractors to meet their contractual obligations to us in a timely manner. |
27
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, clinical trial site IRB’s, or a data safety monitoring board, or DSMB, overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors. Additionally, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for reexamination, which may impact the cost, timing or successful completion of a clinical trial. We do not know whether our clinical trials will be conducted as planned, will need to be restructured or will be completed on schedule, if at all. Delays in our clinical trials will result in increased development costs for our drug candidates. In addition, if we experience delays in the completion of, or if we terminate, any of our clinical trials, the commercial prospects for our drug candidates may be harmed and our ability to generate product revenues will be jeopardized. Furthermore, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a drug candidate. If regulatory authorities do not approve our products or if we fail to maintain regulatory compliance, we would be unable to commercialize our proposed products, and our business and results of operations could be materially harmed.
The results of pre-clinical studies and clinical trials may not be predictive of the results of our later-stage clinical trials and our proposed products may not have favorable results in later-stage clinical trials or receive regulatory approval.
Seemingly positive results from pre-clinical studies or clinical studies should not be relied upon as evidence that our clinical trials will succeed. Even if our product candidates achieve positive results in pre-clinical studies or during our Phase 1 and Phase 2 studies, we will be required to demonstrate through further clinical trials that our product candidates are safe and effective for use in a diverse population before we can seek regulatory approvals for their commercial sale. There is typically an extremely high rate of attrition from the failure of product candidates as they proceed through clinical trials. If any product candidate fails to demonstrate sufficient safety and efficacy in any clinical trial, then we may experience potentially significant delays in, or be required to abandon development of that product candidate. Additionally, failure to demonstrate safety and efficacy results acceptable to the FDA in later stage trials could impair our development prospects and even prevent regulatory approval of our current and future product candidates. Any such delays or abandonment in our development efforts of any of our product candidates would materially impair our ability to generate revenues.
We are subject to numerous risks inherent in conducting clinical trials.
We outsource the management of our clinical trials to third parties. Agreements with clinical investigators and medical institutions for clinical testing and with other third parties for data management services, place substantial responsibilities on these parties that, if unmet, could result in delays in, or termination of, our clinical trials. For example, if any of our clinical trial sites fail to comply with FDA-approved good clinical practices, we may be unable to use the data gathered at those sites. If these clinical investigators, medical institutions or other third parties do not carry out their contractual duties or obligations or fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to their failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated, and we may be unable to obtain regulatory approval for, or successfully commercialize, our proposed products. Delays in recruitment, lack of clinical benefit or unacceptable side effects would delay or prevent the completion of our clinical trials.
We or our regulators may suspend or terminate our clinical trials for a number of reasons. We may voluntarily suspend or terminate our clinical trials if at any time we believe they present an unacceptable risk to the patients enrolled in our clinical trials or do not demonstrate clinical benefit. In addition, regulatory agencies may order the temporary or permanent discontinuation of our clinical trials at any time if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements or that they present an unacceptable safety risk to the patients enrolled in our clinical trials.
Our clinical trial operations are subject to regulatory inspections at any time. If regulatory inspectors conclude that we or our clinical trial sites are not in compliance with applicable regulatory requirements for conducting clinical trials, we may receive reports of observations or warning letters detailing deficiencies, and we will be required to implement corrective actions. If regulatory agencies deem our responses to be inadequate, or are dissatisfied with the corrective actions we or our clinical trial sites have implemented, our clinical trials may be temporarily or permanently discontinued, we may be fined, we or our investigators may be precluded from conducting any ongoing or any future clinical trials, the government may refuse to approve our marketing applications or allow us to manufacture or market our products, and we may be criminally prosecuted.
The lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval for our proposed products, which would materially harm our business, results of operations and prospects.
We may be subject to litigation that will be costly to defend or pursue and uncertain in its outcome.
Our business may bring us into conflict with licensees, licensors, or others with whom we have contractual or other business relationships or with our competitors or others whose interests differs from ours. If we are unable to resolve these conflicts on terms that are satisfactory to all parties, we may become involved in litigation brought by or against such parties. Any litigation is likely to be expensive and may require a significant amount of management's time and attention, at the expense of other aspects of our business. The outcome of litigation is always uncertain, and in some cases, could include judgments against us which could have a materially adverse effect on our business.
28
We may not be able to obtain government or third-party payor coverage and reimbursement.
Our ability to successfully commercialize our product candidates, if approved, depends to a significant degree on the ability of patients to be reimbursed for the costs of such products and related treatments. We cannot assure you that reimbursement in the U.S. or in foreign countries will be available for any products developed, or, if available, will not decrease in the future, or that reimbursement amounts will not reduce the demand for, or the price of, our products. There is considerable pressure to reduce the cost of therapeutic products. Government and other third-party payors are increasingly attempting to contain health care costs by limiting both coverage and the level of reimbursement for new therapeutic products and by refusing, in some cases, to provide any coverage for uses of approved products for disease indications for which the FDA or other relevant authority has not granted marketing approval. Moreover, in some cases, government and other third-party payors have refused to provide reimbursement for uses of approved products for disease indications for which the FDA or other relevant authority has granted marketing approval. Significant uncertainty exists as to the reimbursement status of newly approved health-care products or novel therapies such as ours. We cannot predict what additional regulation or legislation relating to the health care industry or third-party coverage and reimbursement may be enacted in the future or what effect such regulation or legislation may have on our business. If additional regulations are overly onerous or expensive or if healthcare related legislation makes our business more expensive or burdensome than originally anticipated, we may be forced to significantly downsize our business plans or completely abandon the current business model.
Our products may not be profitable due to manufacturing costs and our inability to receive favorable pricing.
Our products may be significantly more expensive to manufacture than other drugs or therapies currently on the market today due to a fewer number of potential manufacturers, greater level of needed expertise and other general market conditions affecting manufacturers of our proposed products. Even if we are able to receive approval for the reimbursement of our proposed products the amount of reimbursement may be significantly less than the manufacturing costs of our products. Additionally, other market factors may limit the price which we can charge for our proposed products while still being competitive. Accordingly, even if we are successful in developing our proposed products, we may not be able to charge a high enough price for us to earn a profit.
We are dependent on the acceptance of our products by the healthcare community.
Our product candidates, if approved for marketing, may not achieve market acceptance since hospitals, physicians, patients or the medical community, in general, may decide not to accept and utilize these products. The products that we are attempting to develop represent substantial departures from established treatment methods and will compete with a number of more conventional therapies marketed by major pharmaceutical companies. If the healthcare community does not accept our products for any reason, our business will be materially harmed.
We depend on a limited number of employees and consultants for our continued operations and future success.
We are highly dependent on a limited number of employees and outside consultants. Although we have entered into employment and consulting agreements with these parties, these agreements can be terminated at any time. The loss of any of our employees or consultants could adversely affect our opportunities and materially harm our future prospects. In addition, we anticipate growth and expansion into areas and activities requiring additional expertise, such as clinical testing, regulatory compliance, manufacturing and marketing. We anticipate the need for additional management personnel as well as the development of additional expertise by existing management personnel. There is intense competition for qualified personnel in the areas of our present and planned activities, and there can be no assurance that we will be able to attract and retain the qualified personnel necessary for the development our business.
The employment contracts of certain key employees contain significant anti-termination provisions which could make changes in management difficult or expensive.
We have entered into employment agreements with Mr.. Daly and Dr. Johe. Each of these employment agreements require the payment of severance, in the event certain conditions are met, if these individuals are terminated. These provisions will make the replacement of these employees very costly and could cause difficulty in effecting a change in control.
Our competition has significantly greater experience and financial resources.
The biotechnology industry is characterized by rapid technological developments and a high degree of competition. We compete against numerous companies, many of which have substantially greater resources. Several such enterprises have initiated cell therapy research programs and/or efforts to treat the same diseases which we target. Given our current stage of development and resources, it may be extremely difficult for us to compete against more developed companies.
29
As a result, our proposed products could become obsolete before we recoup any portion of our related research and development and commercialization expenses. Competition in the biopharmaceutical industry is based significantly on scientific and technological factors. These factors include the availability of patent and other protection for technology and products, the ability to commercialize technological developments and the ability to obtain governmental approval for testing, manufacturing and marketing. We compete with specialized biopharmaceutical firms in the United States, Europe and elsewhere, as well as a growing number of large pharmaceutical companies that are applying biotechnology to their operations. Many major pharmaceutical companies have developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies. These companies, as well as academic institutions and governmental agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific personnel and consultants. Our ability to compete successfully with other companies in the pharmaceutical field will also depend to a considerable degree on the continuing availability of capital to us.
We believe that our proposed products under development and in pre-clinical testing and clinical trials will address unmet medical needs for those indications for which we are focusing our development efforts. Our competition will be determined in part by the potential indications for which our proposed products are developed and ultimately approved by regulatory authorities. Additionally, the timing of market introduction of some of our proposed products or of competitors’ products may be an important competitive factor. Accordingly, the relative speed with which we can develop our proposed products, complete preclinical testing, clinical trials and approval processes and supply commercial quantities to market is expected to be important competitive factors. We expect that competition among products approved for sale will be based on various factors, including product efficacy, safety, reliability, availability, price and patent position.
Our outsource model depends on third parties to assist in developing and testing our proposed products.
Our strategy for the development, clinical and pre-clinical testing and commercialization of our proposed products is based in large part on an outsource model. This model requires us to engage third parties in order to further develop our technology and products as well as for the day to day operations of our business. In the event we are not able to enter into such relationships in the future, our ability to operate and develop products may be seriously hindered or we may be required to spend considerable time and resources to bring such functions in-house. Either outcome could result in our inability to develop a commercially feasible product or in the need for substantially more working capital to complete the research in-house.
The commercialization of therapeutic products exposes us to product liability claims.
Product liability claims could result in substantial litigation costs and damage awards against us. We attempt to mitigate this risk by obtaining and maintaining appropriate insurance coverage. Historically, we have obtained liability insurance that covers our clinical trials. If we begin commercializing products, we will need to increase our insurance coverage. We may not be able to obtain insurance on acceptable terms, if at all, and the policy limits on our insurance policies may be insufficient to cover our potential liabilities.
We currently rely heavily upon third party FDA-regulated manufacturers and suppliers for our products
We currently manufacture our cells both in-house and on an outsource basis. We outsource the manufacturing of our pharmaceutical compound to third party manufacturers. We manufacture cells in-house which are not required to meet stringent FDA requirements. We use these cells in our research and collaborative programs. At present, we outsource all the manufacturing and storage of our stem cells and pharmaceuticals compound to be used in clinical testing, and which are subject to higher FDA requirements, to Charles River Laboratories, Inc., of Wilmington, Massachusetts (stem cells) and Albany Molecular Resources, Inc. (small molecule). Failure by our contract manufacturer to achieve and maintain high manufacturing standards could result in patient injury or death, product recalls or withdrawals, delays or failures in testing or delivery, cost overruns, or other problems that could seriously hurt our business. Contract manufacturers may encounter difficulties involving production yields, quality control, and quality assurance. These manufacturers are subject to ongoing periodic and unannounced inspections by the FDA and corresponding state and foreign agencies to ensure strict compliance with cGMPs, GTPs and other applicable government regulations and corresponding foreign standards; however, we do not have control over third-party manufacturers’ compliance with these regulations and standards.
Because manufacturing facilities are subject to regulatory oversight and inspection, failure to comply with regulatory requirements could result in material manufacturing delays and product shortages, which could delay or otherwise negatively impact our clinical trials and product development. Moreover, we do not have quantity or volume commitment orders from these manufacturers and we cannot assure you that the manufacturers will be able to manufacture in the quantity we require on a timely basis or at all. In the event we are required to seek alternative third-party suppliers or manufacturers, they may require us to purchase a minimum amount of materials or could require other unfavorable terms. Any such event would materially impact our business prospects and could delay the development of our products. Moreover, there can be no assurance that any manufacturer or supplier that we select will be able to supply our products in a timely or cost-effective manner or in accordance with applicable regulatory requirements or our specifications. In addition, due to the novelty of our products and product development, there can be no assurances that we would be able to find other suitable third-party FDA-regulated manufacturers on a timely basis and at terms reasonable to us. Even if we were to locate alternative manufacturers there may be delays before they are able to begin manufacturing. Failure to secure such third-party manufacturers or suppliers would materially impact our business.
30
We rely on third parties to conduct our clinical trials and perform data collection and analysis, which may result in costs and delays that prevent us from successfully commercializing our product candidates.
We do not have the in-house capability to conduct clinical trials for our product candidates. We rely, and will rely in the future, on medical institutions, clinical investigators, contract research organizations, contract laboratories, and collaborators to perform data collection and analysis and other aspects of our clinical trials. Our reliance on these third parties for clinical development activities results in reduced control over these activities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. Our preclinical activities or clinical trials conducted in reliance on third parties may be delayed, suspended, or terminated if:
| · | the third parties do not successfully carry out their contractual duties; |
| · | the third parties fail to meet FDA and other regulatory obligations or expected deadlines; |
| · | we replace a third party for any reason; or |
| · | the quality or accuracy of the data obtained by third parties is compromised due to their failure to adhere to clinical protocols, regulatory requirements, or for other reasons. |
Third party performance failures may increase our development costs, delay our ability to obtain regulatory approval, and delay or prevent the commercialization of our product candidates. While we believe that there are numerous alternative sources to provide these services, in the event that we seek such alternative sources, we may not be able to enter into replacement arrangements without incurring delays or additional costs.
Risks Relating to Intellectual Property
We may not be able to withstand challenges to our intellectual property rights.
We rely on our intellectual property, including issued and applied-for patents, as the foundation of our business. Our intellectual property rights may come under challenge. No assurances can be given that our current and potential future patents will survive such challenges. For example, in 2005 one of our patents was challenged in the USPTO. Although we prevailed in this particular matter, these cases are complex, lengthy, expensive, and could potentially be adjudicated adversely to our interests, removing the protection afforded by an issued patent. The viability of our business would suffer if such patent protection were limited or eliminated. Moreover, the costs associated with defending or settling intellectual property claims would likely have a material adverse effect on our business and future prospects.
We may not be able to adequately protect against the piracy of the intellectual property in foreign jurisdictions.
We conduct research in countries outside of the U.S., including through our subsidiary in the People’s Republic of China. A number of our competitors are located in these countries and may be able to access our technology or test results. The laws protecting intellectual property in some of these countries may not adequately protect our trade secrets and intellectual property. The misappropriation of our intellectual property may materially impact our position in the market and any competitive advantages, if any, that we may have.
We may infringe the intellectual property rights of others and may not be able to obtain necessary licenses to third-party patents and other rights.
A number of companies, universities and research institutions have filed patent applications or have received patents relating to technologies in our field. We cannot predict which, if any, of these applications will issue as patents or how many of these issued patents will be found valid and enforceable. There may also be existing issued patents on which we would infringe by the commercialization of our product candidates. If so, we may be prevented from commercializing these products unless the third party is willing to grant a license to us. We may be unable to obtain licenses to the relevant patents at a reasonable cost, if at all, and may also be unable to develop or obtain alternative non-infringing technology. If we are unable to obtain such licenses or develop non-infringing technology at a reasonable cost, our business could be significantly harmed. Also, any infringement lawsuits commenced against us may result in significant costs, divert our management’s attention and result in an award against us for substantial damages, or potentially prevent us from continuing certain operations.
31
Risks Relating to Our Common Stock
The market price for our common shares is particularly volatile.
The market for our common shares is characterized by significant price volatility when compared to seasoned issuers, and we expect that our share price will continue to be more volatile than those of a seasoned issuer. The volatility in our share price is attributable to a number of factors. Mainly however, we are a speculative or “risky” investment due to our limited operating history, lack of significant revenues to date and the uncertainty of FDA approval. As a consequence of this enhanced risk, more risk-adverse investors may, under the fear of losing all or most of their investment in the event of negative news or lack of progress, be more inclined to sell their shares on the market more quickly and at greater discounts than would be the case with the stock of a seasoned issuer. Additionally, in the past, plaintiffs have often initiated securities class action litigation against a company following periods of volatility in the market price of its securities. We may in the future be the target of similar litigation. Securities litigation could result in substantial costs and liabilities and could divert management’s attention and resources.
The following factors may add to the volatility in the price of our common shares: actual or anticipated variations in our quarterly or annual operating results; the results of clinical trials for our product candidates; FDA’s determination with respect to filings for new clinical studies, new drug applications and new indications; government regulations; announcements of significant acquisitions, strategic partnerships or joint ventures; our capital commitments; offerings of our securities and additions or departures of our key personnel. Many of these factors are beyond our control and may decrease the market price of our common shares, regardless of our operating performance. We cannot make any predictions or projections as to what the prevailing market price for our common shares will be at any time, including as to whether our common shares will sustain their current market prices, or as to what effect the sale of shares or the availability of common shares for sale at any time will have on the prevailing market price.
If we are unable to satisfy NASDAQ maintenance requirements, our common stock may be delisted from NASDAQ, which could impair the liquidity and the value of our common stock.
Continued listing on NASDAQ generally requires that meet certain listing maintenance requirements. If we are unable to satisfy NASDAQ’S maintenance requirements, our common stock may be delisted from NASDAQ. In such event, trading in our common stock would likely take place in the over-the-counter market on the “OTC Markets” or the “OTC Bulletin Board.” Consequently, the liquidity of our common stock could be impaired, not only in the number of shares of common stock which could be bought and sold, but also through delays in the timing of transactions, a reduction in security analysts and new media coverage and lower prices for our common stock than might otherwise be obtained. While the shares of our common stock meet current NASDAQ listing requirements and are currently listed on The Nasdaq Capital Market, there can be no assurance that we will meet the criteria for continued listing.
While we continue to monitor our compliance with the requirements for continued listing on The Nasdaq Capital Market, we cannot assure you that we will not fail to satisfy one of the criteria in the future. If that were to occur, NASDAQ may take steps to delist our common stock. A delisting would likely have a negative effect on the price of our common stock and would likely impair your ability to sell or purchase our common stock if and when you wish to do so. In the event of a delisting, we cannot assure you that any action we take to restore listing would be successful. Even if successful, we cannot assure you that any such action would stabilize the market price of our common stock, improve the liquidity of our common stock, or prevent our future non-compliance with NASDAQ listing requirements. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would no longer be recognized as a “covered security” and we would be subject to regulation in each state in which we offer our securities. Thus, delisting from NASDAQ could adversely affect our ability to raise additional financing through the public or private sale of equity securities, would significantly impact the ability of investors to trade our securities and would negatively impact the value and liquidity of our common stock. Delisting could also have other negative results, including the potential loss of confidence by employees, the loss of institutional investor interest and fewer business development opportunities.
If our common stock were delisted from NASDAQ, the Company would be subject to the risks relating to penny stocks .
If our common stock were to be delisted from trading on NASDAQ and the trading price of the common stock were below $5.00 per share on the date the common stock were delisted, trading in our common stock would also be subject to the requirements of certain rules promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These rules require additional disclosure by broker-dealers in connection with any trades involving a stock defined as a "penny stock" and impose various sales practice requirements on broker-dealers who sell penny stocks to persons other than established customers and accredited investors, generally institutions. These additional requirements may discourage broker-dealers from effecting transactions in securities that are classified as penny stocks, which could severely limit the market price and liquidity of such securities and the ability of purchasers to sell such securities in the secondary market. A penny stock is defined generally as any non-exchange listed equity security that has a market price of less than $5.00 per share, subject to certain exceptions.
32
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain qualified board members.
As a public company, we incur significant legal, accounting and other expenses that we would not incur as a private company, including costs associated with public company reporting requirements. We also incur costs associated with the Sarbanes-Oxley Act of 2002, as amended, the Dodd-Frank Wall Street Reform and Consumer Protection Act and related rules implemented or to be implemented by the SEC and the Nasdaq. The expenses incurred by public companies generally for reporting, insurance and corporate governance purposes have been increasing. We expect these rules and regulations to increase our legal and financial compliance costs and to make some activities more time-consuming and costly. These laws and regulations could also make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. These laws and regulations could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as our executive officers and may divert management’s attention. Furthermore, if we are unable to satisfy our obligations as a public company, we could be subject to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
We have never paid a cash dividend and do not intend to pay cash dividends on our common stock in the foreseeable future.
We have never paid cash dividends nor do we anticipate paying cash dividends in the foreseeable future. Accordingly, any return on your investment will be as a result of stock appreciation if any.
Our anti-takeover provisions may delay or prevent a change of control, which could adversely affect the price of our common stock.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that may make it difficult to remove our board of directors and management and may discourage or delay “change of control” transactions, which could adversely affect the price of our common stock. These provisions include, among others:
| · | our board of directors is divided into three classes, with each class serving for a staggered three-year term, which prevents stockholders from electing an entirely new board of directors at an annual meeting; |
| · | advance notice procedures that stockholders must comply with in order to nominate candidates to our board of directors and propose matters to be brought before an annual meeting of our stockholders may discourage or deter a potential acquirer from conducting a solicitation of proxies to elect the acquirer’s own slate of directors or otherwise attempting to obtain control of our company; and |
| · | our board of directors may, without stockholder approval, issue series of preferred stock, or rights to acquire preferred stock, that could dilute the interest of, or impair the voting power of, holders of our common stock or could also be used as a method of discouraging, delaying or preventing a change of control. |
If securities or industry analysts do not publish research reports, or publish unfavorable research about our business, the price and trading volume of our common stock could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us and our business. We currently have limited research coverage by securities and industry analysts. In the event an analyst downgrades our securities the price of our securities would likely decline. If analysts cease to cover us or fails to publish regular reports on us, interest in our securities could decrease, which could cause the price of our common stock and other securities and their trading volume to decline.
Our board of directors has broad discretion to issue additional securities which might dilute the net tangible book value per share of our common stock for existing stockholders.
We are entitled under our certificate of incorporation to issue up to 300,000,000 shares of common stock and 7,000,000 “blank check” shares of preferred stock. Shares of our blank check preferred stock provide our board of directors with broad authority to determine voting, dividend, conversion, and other rights. As of June 30, 2017, we have issued and outstanding 12,012,877 shares of common stock and we have 9,004,887 shares of common stock reserved for future grants under our equity compensation plans and for issuances upon the exercise or conversion of currently outstanding options, warrants and convertible securities. As of June 30, 2017, we had 1,000,000 shares of preferred stock issued and outstanding. Accordingly, we are entitled to issue up to 278,982,236 additional shares of common stock and 6,000,000 additional shares of “blank check” preferred stock. Our board may generally issue those common and preferred shares, or convertible securities to purchase those shares, without further approval by our shareholders. Any preferred shares we may issue will have such rights, preferences, privileges and restrictions as may be designated from time-to-time by our board, including preferential dividend rights, voting rights, conversion rights, redemption rights and liquidation provisions. It is likely that we will be required to issue a large amount of additional securities to raise capital in order to further our development and marketing plans. It is also likely that we will be required to issue a large amount of additional securities to directors, officers, employees and consultants as compensatory grants in connection with their services, both in the form of stand-alone grants or under our various stock plans. The issuance of additional securities may cause substantial dilution to our shareholders.
33
Risks Related to Government Regulation and Approval of our Product Candidates.
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and our products may not receive regulatory approval.
The time required to obtain approval by the FDA and comparable foreign authorities is inherently unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a drug candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Our drug candidates could fail to receive regulatory approval for many reasons, including the following:
| · | the FDA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| · | we may be unable to demonstrate to the satisfaction of the FDA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication; |
| · | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| · | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| · | the FDA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| · | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a BLA, NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| · | the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; or |
| · | the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
We are currently undertaking clinical trials for our lead products candidates NSI-189 and NSI-566. We cannot assure you that we will successfully complete any clinical trials in connection with such INDs. Further, we cannot predict when we might first submit any product license application (NDA or BLA) for FDA approval or whether any such product license application will be granted on a timely basis, if at all. Any delay in obtaining, or failure to obtain, such approvals could have a material adverse effect on the marketing of our products and our ability to generate product revenue.
Development of our product candidates is subject to extensive government regulation.
Our research and development efforts, as well as any future clinical trials, and the manufacturing and marketing of any products we may develop, will be subject to, and restricted by, extensive regulation by governmental authorities in the U.S. and other countries. The process of obtaining FDA and other necessary regulatory approvals is lengthy, expensive and uncertain. FDA and other legal and regulatory requirements applicable to our proposed products could substantially delay or prevent us from initiating additional clinical trials. We may fail to obtain the necessary approvals to commence clinical testing or to manufacture or market our potential products in reasonable time frames, if at all. In addition, the U.S. Congress and other legislative bodies may enact regulatory reforms or restrictions on the development of new therapies that could adversely affect the regulatory environment in which we operate or the development of any products we may develop.
A substantial portion of our research and development entails the use of stem cells obtained from human tissue. The U.S. federal and state governments and other jurisdictions impose restrictions on the acquisition and use of human tissue, including those incorporated in federal Good Tissue Practice, or “GTP,” regulations. These regulatory and other constraints could prevent us from obtaining cells and other components of our products in the quantity or of the quality needed for their development or commercialization. These restrictions change from time to time and may become more onerous. Additionally, we may not be able to identify or develop reliable sources for the cells necessary for our potential products — that is, sources that follow all state and federal laws and guidelines for cell procurement. Certain components used to manufacture our stem and progenitor cell product candidates will need to be manufactured in compliance with the FDA’s GMP. Accordingly, we will need to enter into supply agreements with companies that manufacture these components to GMP standards. There is no assurance that we will be able to enter into any such agreements.
Noncompliance with applicable regulatory requirements can subject us, our third party suppliers and manufacturers and our other collaborators to administrative and judicial sanctions, such as, among other things, warning letters, fines and other monetary payments, recall or seizure of products, criminal proceedings, suspension or withdrawal of regulatory approvals, interruption or cessation of clinical trials, total or partial suspension of production or distribution, injunctions, limitations on or the elimination of claims we can make for our products, refusal of the government to enter into supply contracts or fund research, or government delay in approving or refusal to approve new drug applications.
34
We cannot predict if or when we will be able to commercialize our products due to regulatory constraints.
Federal, state and local governments and agencies in the U.S. (including the FDA) and governments in other countries have significant regulations in place that govern many of our activities. We are, or may become, subject to various federal, state and local laws, regulations and recommendations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances used in connection with its research and development work. The preclinical testing and clinical trials of our proposed products are subject to extensive government regulation that may prevent us from creating commercially viable products. In addition, our sale of any commercially viable product will be subject to government regulation from several standpoints, including manufacturing, advertising, marketing, promoting, selling, labeling and distributing. If, and to the extent that, we are unable to comply with these regulations, our ability to earn revenues, if any, will be materially and negatively impacted.
If our clinical trials fail to demonstrate that any of our product candidates are safe and effective for the treatment of particular diseases, the FDA may require us to conduct additional clinical trials or may not grant us marketing approval for such product candidates for those diseases.
We are not permitted to market our product candidates in the United States until we receive approval of a BLA or NDA from the FDA. Before obtaining regulatory approvals for the commercial sale of any product candidate for a target indication, we must demonstrate with evidence gathered in preclinical and well-controlled clinical trials, and, with respect to approval in the United States, to the satisfaction of the FDA and, with respect to approval in other countries, similar regulatory authorities in those countries, that the product candidate is safe and effective for use for that target indication and that the manufacturing facilities, processes and controls used to produce the product are compliant with applicable statutory and regulatory requirements. Our failure to adequately demonstrate the safety and effectiveness of any of our product candidates for the treatment of particular diseases may delay or prevent our receipt of the FDA’s approval and, ultimately, may prevent commercialization of our product candidates for those diseases. The FDA has substantial discretion in deciding whether, based on the benefits and risks in a particular disease, any of our product candidates should be granted approval for the treatment of that particular disease. Even if we believe that a clinical trial or trials has demonstrated the safety and statistically significant efficacy of any of our product candidates for the treatment of a disease, the results may not be satisfactory to the FDA. Preclinical and clinical data can be interpreted by the FDA and other regulatory authorities in different ways, which could delay, limit or prevent regulatory approval. If regulatory delays are significant or regulatory approval is limited or denied altogether, our financial results and the commercial prospects for those of our product candidates involved will be harmed, and our prospects for profitability will be significantly impaired.
Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain, and subject to unanticipated delays. Despite our efforts, our drug candidates may not:
| · | offer improvement over existing comparable products; |
| · | be proven safe and effective in clinical trials; or |
| · | meet applicable regulatory standards. |
In addition, in the course of its review of a BLA or NDA or other regulatory application, the FDA or other regulatory authorities may conduct audits of the practices and procedures of a company and its suppliers and contractors concerning manufacturing, clinical study conduct, non-clinical studies and several other areas. If the FDA and/or other regulatory authorities conducts an audit relating to a BLA, NDA or other regulatory application and finds a significant deficiency in any of these or other areas, the FDA or other regulatory authorities could delay or not approve such BLA, NDA or other regulatory application. If regulatory delays are significant or regulatory approval is limited or denied altogether, our financial results and the commercial prospects for those of our products or product candidates involved will be harmed, and our prospects for profitability will be significantly impaired.
Both before and after marketing approval, our product candidates are subject to extensive and rigorous ongoing regulatory requirements and continued regulatory review, and if we fail to comply with these continuing requirements, we could be subject to a variety of sanctions.
Both before and after the approval of our product candidates, we, our product candidates, our operations, our facilities, our suppliers, and our contract manufacturers, contract research organizations, and contract testing laboratories are subject to extensive regulation by governmental authorities in the United States and other countries, with regulations differing from country to country. In the United States, the FDA regulates, among other things, the pre-clinical testing, clinical trials, manufacturing, safety, efficacy, potency, labeling, packaging, adverse event reporting, storage, record keeping, quality systems, advertising, promotion, sale and distribution of therapeutic products. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP, requirements and current good clinical practice, or cGCP, requirements for any clinical trials that we conduct post-approval. Failure to comply with applicable requirements could result in, among other things, one or more of the following actions: restrictions on the marketing of our products or their manufacturing processes, notices of violation, untitled letters, warning letters, civil penalties, fines and other monetary penalties, unanticipated expenditures, delays in approval or refusal to approve a product candidate, suspension or withdrawal of regulatory approvals, product, seizure or detention, voluntary or mandatory product recalls and related publicity requirements, interruption of manufacturing or clinical trials, operating restrictions, injunctions, import or export bans, and criminal prosecution. We or the FDA, or an institutional review board, may suspend or terminate human clinical trials at any time on various grounds, including a finding that subjects are being exposed to an unacceptable health risk.
35
The FDA’s policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. If we are slow or unable to adapt to changes in existing or new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
If side effects are identified during the time our drug candidates are in development or after they are approved and on the market, we may choose to or be required to perform lengthy additional clinical trials, discontinue development of the affected drug candidate, change the labeling of any such products, or withdraw any such products from the market, any of which would hinder or preclude our ability to generate revenues.
Undesirable side effects caused by our drug candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA or other comparable foreign authorities. Drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete a trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly. Even if any of our drug candidates receives marketing approval, as greater numbers of patients use a drug following its approval, an increase in the incidence of side effects or the incidence of other post-approval problems that were not seen or anticipated during pre-approval clinical trials could result in a number of potentially significant negative consequences, including:
| · | regulatory authorities may withdraw their approval of the product; |
| · | regulatory authorities may require the addition of labeling statements, such as warnings or contradictions; |
| · | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| · | we could be sued and held liable for harm caused to patients; and |
| · | our reputation may suffer. |
Any of these events could substantially increase the costs and expenses of developing, commercializing and marketing any such drug candidates or could harm or prevent sales of any approved products.
Even if our product candidates receive regulatory approval in the United States, we may never receive approval or commercialize our products outside of the United States.
In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval would impair our ability to develop foreign markets for our drug candidates.
Our product candidates for which we intend to seek approval as biologic products may face competition sooner than anticipated.
We expect our stem cell product candidates to be regulated by the FDA as biologic products and we intend to seek approval for these products pursuant to the BLA pathway. The Biologics Price Competition and Innovation Act of 2009, or BPCIA, created an abbreviated pathway for the approval of biosimilar and interchangeable biologic products. The abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. Under the BPCIA, an application for a biosimilar product cannot be approved by the FDA until 12 years after the original branded product was approved under a BLA. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty. While it is uncertain when such processes intended to implement BPCIA may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for our biologic products.
36
We believe that any of our product candidates approved as a biologic product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our drug candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biologic products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
| ITEM 2. | UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS |
The following information is given with regard to unregistered securities sold from January 1, 2017 to July 31, 2017. The following securities were issued in private offerings pursuant to the exemption from registration contained in the Securities Act and the rules promulgated thereunder in reliance on Section 4(2) thereof, relating to offers of securities by an issuer not involving any public offering:
In March 2017, we issued warrants to purchase 230,770 shares of common stock. The warrants have an exercise price of $5.80 per share and a term of one year. The warrants were issued as an inducement for the exercise of 692,309 outstanding warrants resulting in gross proceeds of approximately $2,250,000.
In April 2017, we issued warrants to purchase 51,283 shares of common stock. The warrants have an exercise price of $5.80 per share and a term of one year. The warrants were issued as an inducement for the exercise of 153,847 outstanding warrants resulting in gross proceeds of approximately $500,000.
In the first and second quarters of 2017, we sold an aggregate of 10,887 shares of commons stock to certain members of our management. The average price for the shares was $4.59 based on the closing price of our common stock on each respective purchase date. The sales resulted in gross proceeds of $50,000.
In July 2017, we issued one of our legal advisors a common stock purchase warrant to purchase 11,539 shares of our common stock at an exercise price of $5.79 per share as partial compensation for legal work. The warrant vests monthly over one year from the grant date, has a term of 5 years and will expire on June 30, 2022.
In July of 2017, we issued an aggregate of: (i) 37,244 shares of restricted stock, (ii) 9,311 restricted stock units and (iii) 13,574 common stock purchase options. The securities were issued to our non-executive directors pursuant to our non-executive director compensation policy. The respective grants vest quarterly over the grant year. With respect to the options, they have an exercise price of $5.37 and a term of 10 years. All the securities were issued pursuant to our 2010 Equity Compensation Plan, as amended.
| ITEM 3. | DEFAULT UPON SENIOR SECURITIES |
None
| ITEM 4. | MINE SAFETY DISCLOSURE |
Not Applicable
| ITEM 5. | OTHER INFORMATION |
Not Applicable
| ITEM 6. | EXHIBITS |
The exhibits listed in the accompanying index to exhibits are filed or incorporated by reference as part of this Form 10-Q.
37
In accordance with the requirements of the Securities Exchange Act of 1934, the Registrant has caused this report to be signed by the undersigned hereunto duly authorized.
| NEURALSTEM, INC. | ||
| Date: August 8, 2017 | /s/ Richard Daly | |
| Chief Executive Officer | ||
| /s/ Richard Daly | ||
| Chief Financial Officer | ||
| (Principal Accounting Officer) | ||
38
INDEX TO EXHIBITS
| Incorporated by Reference | ||||||||||||
| Filed/ | ||||||||||||
| Exhibit | Furnished | Exhibit | ||||||||||
| No. | Description | Herewith | Form | No. | File No. | Filing Date | ||||||
| 3.01(i) | Amended and Restated Certificate of Incorporation of Neuralstem, Inc. filed on 1/5/2017 | 8-K | 3.01(i) | 001-33672 | 1/6/17 | |||||||
| 3.02(i) | Certificate of Designation of Series A 4.5% Convertible Preferred Stock | 8-K | 3.01 | 001-33672 | 12/12/16 | |||||||
| 3.03(ii) | Amended and Restated Bylaws of Neuralstem, Inc. adopted on 11/10/2015 | 8-K | 3.01 | 001-33672 | 11/16/15 | |||||||
| 4.01** | Amended and Restated 2005 Stock Plan adopted on 6/28/07 | 10-QSB | 4.2(i) | 333-132923 | 8/14/07 | |||||||
| 4.02** | Non-qualified Stock Option Agreement between Neuralstem, Inc. and Richard Garr dated 7/28/05 | SB-2 | 4.4 | 333-132923 | 6/21/06 | |||||||
| 4.03** | Non-qualified Stock Option Agreement between Neuralstem, Inc. and Karl Johe dated 7/28/05 | SB-2 | 4.5 | 333-132923 | 6/21/06 | |||||||
| 4.04** | Neuralstem, Inc. 2007 Stock Plan | 10-QSB | 4.21 | 333-132923 | 8/14/07 | |||||||
| 4.05 | Form of Common Stock Purchase Warrant Issued to Karl Johe on 6/5/07 | 10-KSB | 4.22 | 333-132923 | 3/27/08 | |||||||
| 4.06 | Form of Placement Agent Warrant Issued to Midtown Partners & Company on 12/18/08 | 8-K | 4.1 | 001-33672 | 12/18/08 | |||||||
| 4.07 | Form of Consultant Common Stock Purchase Warrant issued on 1/5/09 | S-3/A | 10.1 | 333-157079 | 02/3/09 | |||||||
| 4.08 | Form of Series D, E and F Warrants | 8-K | 4.01 | 001-33672 | 7/1/09 | |||||||
| 4.09 | Form of Placement Agent Warrant | 8-K | 4.02 | 001-33672 | 7/1/09 | |||||||
| 4.10 | Form of Consultant Warrant Issued 1/8/10 | 10-K | 4.20 | 001-33672 | 3/31/10 | |||||||
| 4.11 | Form of Replacement Warrant Issued 1/29/10 | 10-K | 4.21 | 001-33672 | 3/31/10 | |||||||
| 4.12 | Form of Series C Replacement Warrant Issued March of 2010 and May, June and July of 2013 (Original Ex. Price $2.13 and $1.25) | 10-K | 4.22 | 001-33672 | 3/31/10 | |||||||
| 4.13 | Form of employee and consultant option grant pursuant to our 2007 Stock Plan and 2010 Equity Compensation Plan | 10-K | 4.23 | 001-33672 | 3/31/10 | |||||||
| 4.14 | Form of Warrants dated 6/29/10 | 8-K | 4.01 | 001-33672 | 6/29/10 | |||||||
39
| 4.15** | Amended Neuralstem 2010 Equity Compensation Plan adopted on June 22, 2017 | DEF 14A | Appendix I | 001-33672 | 5/1/17 | |||||||
| 4.16 | Form of Consultant Warrant issued 10/1/09 and 10/1/10 | S-3 | 4.07 | 333-169847 | 10/8/10 | |||||||
| 4.17** | Form of Restricted Stock Award Agreement pursuant to our 2007 Stock Plan and 2010 Equity Compensation Plan | S-8 | 4.06 | 333-172563 | 3/1/11 | |||||||
| 4.18** | Form of Restricted Stock Unit Agreement | S-8 | 4.08 | 333-172563 | 3/1/11 | |||||||
| 4.19 | Form of Common Stock Purchase Warrant issued pursuant to February 2012 registered offering | 8-K | 4.01 | 001-33672 | 2/8/12 | |||||||
| 4.20 | Form of Common Stock Purchase Warrant issued to Consultants in June of 2012 and March 19, 2013 | 10-Q | 4.20 | 001-33672 | 8/9/12 | |||||||
| 4.21 | Form of Underwriter Warrant issued to Aegis Capital Corp. on 8/20/12 | 8-K | 4.1 | 001-33672 | 8/17/12 | |||||||
| 4.22 | Form of Placement Agent Warrant issued to Aegis Capital Corp. on 9/13/12 | 8-K | 4.1 | 001-33672 | 9/19/12 | |||||||
| 4.23 | Form of Consulting Warrant issued January 2011 and March 2012 | S-3 | 4.01 | 333-188859 | 5/24/13 | |||||||
| Form of Replacement Warrant issued January, February and May of 2013 (Original Ex. Prices $3.17 and $2.14) | ||||||||||||
| 4.24 | Form of Lender Warrant issued March 22, 2013 | 8-K | 4.01 | 001-33672 | 3/27/13 | |||||||
| 4.25 | Form of Advisor Warrant issued March 22, 2013 | 8-K | 4.02 | 001-33672 | 3/27/13 | |||||||
| 4.26 | Form of Warrant issued June of 2013 and July of 2014 to Legal Counsel | 10-Q | 4.26 | 001-33672 | 8/8/13 | |||||||
| 4.27 | Form of Warrant issued in September 2013 in connection with Issuer’s registered direct offering | 8-K | 4.01 | 011-33672 | 9/10/13 | |||||||
| 4.28 | Form of Warrant issued to strategic advisor in August 2013 | 10-Q | 4.28 | 001-33672 | 11/12/13 | |||||||
| 4.29 | Form of Investor Warrant issued January 2014 | 8-K | 4.01 | 001-33672 | 1/6/14 |
40
| 4.30 | Form of Lender Warrant Issued October 28, 2014 | 8-K | 4.01 | 001-33672 | 10/29/14 | |||||||
| 4.31** | Inducement Stock Option Plan adopted 2/15/2016 | 8-K | 4.01 | 001-33672 | 2/19/16 | |||||||
| 4.32** | Form of Inducement Award Non-Qualified Stock Option Grant pursuant to Inducement Stock Option Plan | 8-K | 4.02 | 001-33672 | 2/19/16 | |||||||
| 4.33 | Form of Common Stock Purchase Warrant From May 2016 Public Offering dated May 6, 2016 | 8-K | 4.01 | 001-33672 | 5/3/16 | |||||||
| 4.34 | Form of Common Stock Purchase Warrant from May 2016 Private Offering Dated May 12, 2016 | 8-K | 4.01 | 001-33672 | 5/13/16 | |||||||
| 4.35 | Form of Series A Preferred Stock Certificate | 8-K | 4.01 | 001-33672 | 9/12/16 | |||||||
| 4.36 | Form of Inducement Warrant issued March 20, 2017 and March 31, 2017 | 8-K | 4.01 | 001-33672 | 3/20/17 | |||||||
| 4.37 | Form of Common Stock Purchase Warrant from August 2017 Public Offering Dated August 1, 2017 | 8-K | 4.01 | 001-33672 | 7/28/17 | |||||||
| 10.01** | Employment Agreement with I. Richard Garr dated January 1, 2007 and amended as of November 1, 2005 | SB-2 | 10.1 | 333-132923 | 6/21/06 | |||||||
| 10.02** | Amended terms to the Employment Agreement of I Richard Garr dated January 1, 2008 | 10-K | 10.02 | 001-33672 | 3/31/09 | |||||||
| 10.03** | Amended terms to the employment Agreement of I. Richard Garr dated March 1, 2015 | 8-K | 10.01 | 001-33672 | 3/2/15 | |||||||
| 10.04** | Employment Agreement with Karl Johe dated January 1, 2007 and amended as of November 1, 2005 | SB-2 | 10.2 | 333-132923 | 6/21/06 | |||||||
| 10.05** | Amended terms to the Employment Agreement of Karl Johe dated January 1, 2009 | 10-K | 10.04 | 001-33672 | 3/31/09 | |||||||
| 10.06** | Employment Agreement with Thomas Hazel, Ph.D dated August 11, 2008 | 10-K/A | 10.05 | 001-33672 | 10/5/10 | |||||||
| 10.07** | Employment Agreement with Richard Daly dated February 15, 2016 | 8-K | 10.01 | 001-33672 | 2/19/16 | |||||||
| 10.08 | Consulting Agreement dated January 2010 between Market Development Consulting Group and the Company and amendments No. 1 and 2. | 10-K | 10.07 | 001-33672 | 3/16/11 | |||||||
41
| 10.09** | Renewal of I. Richard Garr Employment Agreement dated 7/25/12 | 8-K | 10.01 | 001-33672 | 7/27/12 | |||||||
| 10.10** | Renewal of Dr. Karl Johe Employment Agreement dated 7/25/12 | 8-K | 10.02 | 001-33672 | 7/27/12 | |||||||
| 10.11** | Renewal of Dr. Tom Hazel Employment Agreement dated 7/25/12 | 8-K | 10.03 | 001-33672 | 7/27/12 | |||||||
| 10.12** | Amendment of terms of Karl Johe Employment Agreement dated 9/17/14 | 8-K | 10.01 | 001-33672 | 9/18/14 | |||||||
| 10.13 | Loan and Security Agreement dated March 2013 | 8-K | 10.01 | 001-33672 | 3/27/13 | |||||||
| 10.14 | Intellectual Property and Security Agreement dated March 2013 | 8-K | 10.02 | 001-33672 | 3/27/13 | |||||||
| 10.15 | At the Market Offering Agreement entered into on October 25, 2013 | 8-K | 10.01 | 001-33672 | 10/25/13 | |||||||
| 10.16** | Form of Amendment to Karl Johe Employment Agreement | 8-K | 10.01 | 001-33672 | 9/18/14 | |||||||
| 10.17 | Form of Second Amendment to Loan and Security Agreement dated March of 2013 that was entered into on October 28, 2014 | 8-K | 10.01 | 001-33672 | 10/29/14 | |||||||
| 10.18** | Offer Letter Between Neuralstem, Inc. and Jonathan Lloyd Jones | 8-K | 10.01 | 001-33672 | 5/11/15 | |||||||
| 10.19** | General Release and Waiver of Claims with I. Richard Garr dated 3/2/2016 | 8-K | 10.01 | 001-33672 | 3/4/16 | |||||||
| 10.20 | Form of Securities Purchase Agreement from May 2016 Private Offering | 8-K | 10.01 | 001-33672 | 5/13/16 | |||||||
| 10.21** | Amendment to General Release and Waiver of claims with I. Richard Garr dated 6/6/16 | 8-K | 10.01 | 001-33672 | 6/16/16 | |||||||
| 10.22 | Form of Securities Purchase Agreement between Issuer and Tianjin Pharmaceuticals Holdings, Ltd. | 8-K | 10.01 | 001-33672 | 9/12/16 | |||||||
| 10.23** | Form of Securities Purchase Agreement between Issuer and Jonathan Lloyd Jones | 10-Q | 10.22 | 001-33672 | 11/8/16 | |||||||
| 10.24 | Form of Securities Purchase Agreement between Issuer and Richard Daly | 10-Q | 10.23 | 001-33672 | 11/8/16 | |||||||
| 10.25 | Form of Letter Agreement for Warrant Exercises on March 20, 2017 and March 30, 2017 | 8-K | 10.01 | 001-33672 | 3/20/17 | |||||||
| 10.26** | Form of Separation Agreement and Release with Jonathan Lloyd Jones dated April 30, 2017 | 8-K | 10.01 | 001-33672 | 5/4/17 | |||||||
| 31.1 | Certification of the Principal Executive Officer and Principal Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | * | ||||||||||
42
| 32.1 | Certification of Principal Executive Officer and Principal Financial Officer Pursuant to 18 U.S.C. § 1350 |
*
|
||||||||||
| 101.INS | XBRL Instance Document | * | ||||||||||
| 101.SCH | XBRL Taxonomy Extension Schema | * | ||||||||||
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase | * | ||||||||||
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase | * | ||||||||||
| 101.LAB | XBRL Taxonomy Extension Label Linkbase | * | ||||||||||
| 101.PRE | XBRL Taxonomy Extension Presentation Linkbase | * |
* Filed herein
** Management contracts or compensation plans or arrangements in which directors or executive officers are eligible to participate.
43