Attached files
| file | filename |
|---|---|
| 8-K - FORM 8-K - Regulus Therapeutics Inc. | d423592d8k.htm |
Exhibit 99.1
COMPANY OVERVIEW
We are a clinical-stage biopharmaceutical company focused on discovering and developing first-in-class drugs targeting microRNAs to treat diseases with significant unmet medical need. We were formed in 2007 when Alnylam Pharmaceuticals, Inc., or Alnylam, and Ionis Pharmaceuticals, Inc., or Ionis, contributed significant intellectual property, know-how and financial and human capital to pursue the development of drugs targeting microRNAs pursuant to a license and collaboration agreement. Our most advanced program, under our strategic alliance with Sanofi, is RG-012, an anti-miR targeting miR-21 for the treatment of Alport syndrome, a life-threatening kidney disease driven by genetic mutations, currently with no approved therapy available.
microRNAs are naturally occurring ribonucleic acid, or RNA, molecules that play a critical role in regulating key biological pathways. Scientific research has shown that an imbalance, or dysregulation, of microRNAs is directly linked to many diseases. Furthermore, many different infectious pathogens interact and bind to host microRNA to survive. To date, over 500 microRNAs have been identified in humans, each of which can bind to multiple messenger RNAs that control key aspects of cell biology. Since many diseases are multi-factorial, involving multiple targets and pathways, the ability to modulate multiple pathways by targeting a single microRNA provides a new therapeutic approach for treating complex diseases.
RNA plays an essential role in the process used by cells to encode and translate genetic information from DNA to proteins. RNA is comprised of subunits called nucleotides and is synthesized from a DNA template by a process known as transcription. Transcription generates different types of RNA, including messenger RNAs that carry the information for proteins in the sequence of their nucleotides. In contrast, microRNAs are RNAs that do not code for proteins but rather are responsible for regulating gene expression by modulating the translation and decay of target messenger RNAs. By interacting with many messenger RNAs, a single microRNA can regulate the expression of multiple genes involved in the normal function of a biological pathway. Many pathogens including viruses, bacteria and parasites also use host microRNAs to regulate the cellular environment for survival. In some instances, the host microRNAs are essential for the replication and/or survival of the pathogen. For example, miR-122 is a microRNA expressed in human hepatocytes and is a key factor for the replication of the hepatitis C virus, or HCV.
We believe that microRNA therapeutics have the potential to become a new and major class of drugs with broad therapeutic application for the following reasons:
| 1. | microRNAs play a critical role in regulating biological pathways by controlling the translation of many target genes; |
| 2. | microRNA therapeutics regulate disease pathways which may result in more effective treatment of complex multi-factorial diseases; |
| 3. | many human pathogens including viruses, bacteria and parasites use microRNAs (host and pathogen encoded) to enable their replication and suppression of host immune responses; and |
| 4. | microRNA therapeutics may be synergistic with other therapies because of their different mechanism of action. |
We believe we have assembled the leading position in the microRNA field, including expertise in microRNA biology and oligonucleotide chemistry, a broad intellectual property estate, relationships with key opinion leaders and a disciplined drug discovery and development process. We are using our microRNA expertise to develop chemically modified, single-stranded oligonucleotides that we call anti-miRs to modulate microRNAs and address underlying disease. We believe microRNAs may play a critical role in complex disease and that targeting them with anti-miRs may become a source of a new and major class of drugs with broad therapeutic application, much like small molecules, biologics and monoclonal antibodies.
We believe that microRNA biomarkers may be used to select optimal patient segments in clinical trials and to monitor disease progression or relapse. We believe these microRNA biomarkers can be applied toward drugs that we develop and drugs developed by other companies with which we partner or collaborate. We have completed a research collaboration with Biogen Inc. focused on the discovery of microRNAs as biomarkers for multiple sclerosis and have also completed research for another leading, commercial-stage pharmaceutical company to explore microRNAs as biomarkers for specific patient populations. We also maintain several academic research collaborations focused on the identification of microRNAs as biomarkers in multiple disease areas.
Development Stage Pipeline
We currently have multiple programs in various stages of clinical and preclinical development.
1
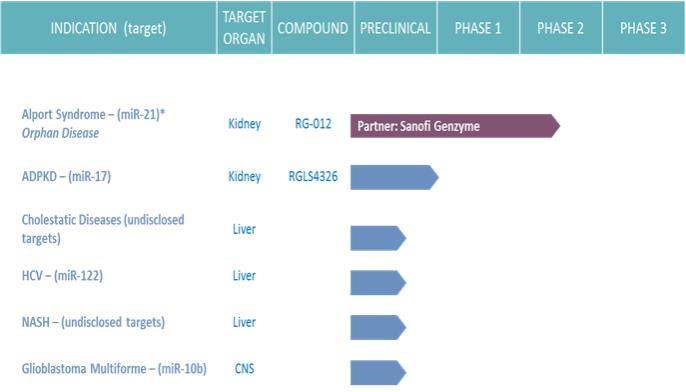
RG-012: In 2015, we completed a Phase I study to evaluate the safety, tolerability, and pharmacokinetics, or PK, of subcutaneous dosing of RG-012 in healthy volunteers. Forty healthy volunteer subjects were enrolled in this first-in-human, single ascending dose study. In May 2017, we completed a Phase I multiple-ascending dose, or MAD, study in 24 healthy volunteers (six-week repeat dosing) to determine safety, tolerability and PK of RG-012 prior to chronic dosing in patients. In both Phase I studies, RG-012 was well-tolerated, and there were no serious adverse events, or SAEs, reported. We also continue to enroll Alport syndrome patients in our global ATHENA natural history of disease study, which is designed to characterize the disease-related decline of renal function (as measured by established blood markers for renal function) in these patients over time. In mid-2017, we are planning to initiate HERA, the Phase II randomized (1:1), double-blinded, placebo-controlled study evaluating the safety and efficacy of RG-012 in 40 Alport syndrome patients. In parallel, a renal biopsy study is also planned to evaluate RG-012 renal tissue PK, target engagement and downstream effects on genomic disease biomarkers. Data from the renal biopsy study is anticipated by year-end and interim data from HERA is anticipated mid-2018.
RGLS4326: In December 2016, we nominated RGLS4326 as a clinical candidate targeting microRNA-17 (miR-17) for the treatment of autosomal dominant polycystic kidney disease, or ADPKD. IND-enabling toxicology, repeat pharmacology and manufacturing work have been completed as scheduled to support regulatory submissions as part of the investigational new drug application, or IND, package. We anticipate filing an IND or foreign equivalent regulatory filing by the end of 2017.
RG-101: In June 2016, we received verbal notice from the U.S. Food and Drug Administration, or FDA, that our IND for RG-101 for the treatment of chronic HCV infection was placed on clinical hold. The FDA initiated the clinical hold after a second RG-101 treated patient experienced an SAE of jaundice. In December 2016, we submitted a complete response to the FDA’s initial request for information, which included identification of a potential mechanism of hyperbilirubinemia. We also submitted a proposal to mitigate this risk. In January 2017, we received written communication from the FDA that the clinical development program for RG-101 remained on clinical hold. The FDA requested the complete safety and efficacy data from on-going RG-101 clinical and preclinical studies before reconsidering the clinical hold. The FDA also requested additional expert review of liver safety data considering the proposed mechanism of hyperbilirubinemia. In June 2017, we announced our plan to discontinue clinical development of RG-101 upon completion of the follow-up phase of the remaining RG-101 clinical study, which is expected to occur in July 2017. Comprehensive preclinical investigation and detailed analysis of clinical data from the RG-101 program have identified the direct inhibition of a hepatocyte conjugated bilirubin transporter as the likely mechanism for the cases of hyperbilirubinemia in the RG-101 program. We believe that a combination of factors, including inhibition of conjugated bilirubin transport by RG-101, impaired baseline bilirubin transport in HCV patients, and the preferential uptake of RG-101 by hepatocytes contributed to this mechanism. Additional patient-specific contributing factors cannot be excluded. Applying the learnings from the RG-101 program, alternative compounds targeting miR-122 have been identified that maintain potent HCV antiviral activity while lacking inhibition of the bilirubin transporter. We believe these compounds have the potential for rapid clinical proof-of-concept of a novel, markedly shortened treatment regimen for HCV and will be considered for further development pending an updated global commercial market assessment for HCV.
RG-125(AZD4076): In June 2017, AstraZeneca delivered written notice to us of its election to terminate the clinical development program for RG-125(AZD4076) for the treatment of non-alcoholic steatohepatitis, or NASH, in Type 2 Diabetes/Pre-diabetes. Pursuant to the terms of our August 2012 collaboration and license agreement with AstraZeneca, AstraZeneca’s rights with respect to RG-125(AZD4076) will revert to us when the termination becomes effective in June 2018.
RGLS5040: In November 2016, we nominated RGLS5040 as a clinical candidate targeting microRNA-27 (miR-27) for the treatment of cholestatic diseases. In June 2017, we discontinued development of RGLS5040 based on a positioning of the compound with respect to the competitive landscape coupled with the results from repeat pharmacology studies as part of IND-enabling work. We continue to work on developing therapeutics for genetic forms of cholestatic disease as part of our overall research activities targeting unmet diseases of the liver and kidney.
Preclinical Pipeline
A major focus of our preclinical research is targeting dysregulated microRNAs implicated in diseases of high unmet medical need where we know we can effectively deliver to the target tissue or organ, such as the liver and kidney. Multiple microRNAs have been identified as being dysregulated in NASH and these are in the process of target validation including the evaluation of tool compounds in animal models of NASH. Profiling of primary tumor cells from glioblastoma multiforme, or GBM, a rapidly fatal form of brain cancer, has identified miR-10b as a microRNA target with potential to inhibit tumor growth. We are investigating local and systemic delivery of anti-miR-10b oligonucleotides in preclinical models to evaluate potential for advancing this program to clinical testing in GBM. We also have early discovery programs investigating additional microRNA targets for infectious diseases.
2
Our microRNA Product Platform
We believe we are the leading company in the field of microRNA therapeutics and are uniquely positioned to leverage oligonucleotide technologies developed by us and our founding companies.
We view the following as providing a competitive advantage for our microRNA product platform:
| • | a mature platform selectively producing multiple development candidates advancing to the clinic; |
| • | scientific advisors who are pioneers in the microRNA field; |
| • | exclusive access to proven RNA therapeutic technologies through our founding companies, such as GalNAc conjugation and the corresponding manufacturing rights licensed to us from Alnylam; |
| • | a leading microRNA intellectual property estate with over 300 patents and patent applications covering compositions and therapeutic uses related to microRNA and microRNA drug products, as well as access to numerous patents and patent applications relating to RNA technologies, including patent and patent applications relating to chemical modification of oligonucleotides that are useful for microRNA therapeutics; |
| • | development expertise and financial resources provided by our strategic alliances; and |
| • | numerous academic collaborations that help us identify new microRNA targets and support our early stage discovery efforts. |
Our Development Candidates
We are developing single-stranded oligonucleotides, which are chemically synthesized chains of nucleotides that are mirror images of specific target microRNAs. We incorporate proprietary chemical modifications to enhance drug properties such as potency, stability and tissue distribution. We refer to these chemically modified oligonucleotides as anti-miRs. Each anti-miR is designed to bind with and inhibit a specific microRNA target that is up-regulated in a cell and that is involved in the disease state. In binding to the microRNA, anti-miRs correct the dysregulation and return diseased cells to their healthy state. We have demonstrated the potential therapeutic benefit of inhibiting microRNA-122 in humans with RG-101 in HCV patients. In addition to these human proof-of-concept results, we have demonstrated therapeutic benefits of our anti-miRs in over 20 different preclinical models of human diseases.
We have identified and validated several microRNA targets across a number of disease categories and are working independently and with our strategic alliance partner to optimize anti-miR development candidates. We intend to pursue a balanced approach between product candidates that we develop ourselves and those that we develop with partners. We intend to focus our own resources on proprietary product opportunities in therapeutic areas where development and commercialization activities are appropriate for our size and financial resources. In therapeutic areas where costs are more significant, development timelines are longer or markets are too large for our capabilities, we may seek to secure partners with requisite expertise and resources.
3
| * | Sanofi will have the exclusive option, exercisable after proof-of-concept, to take over further development and commercialization of this program. At this stage, Regulus will have the option to co-promote this product in the United States. |
Our Strategy
We are focused on discovering and developing of first-in-class drugs based on our proprietary microRNA product platform. The key elements of our strategy are to (i) build a meaningful clinical portfolio by advancing our current clinical programs and rapidly advancing our preclinical programs into clinical development; (ii) focus our resources on developing drugs for indications that represent significant unmet medical need and where the development and commercialization activities are appropriate for our size and financial resources; (iii) selectively form strategic alliances to augment our expertise and accelerate development and commercialization; (iv) develop microRNA biomarkers to support our therapeutic product candidates; and (v) maintain our scientific and intellectual leadership in the microRNA field.
In June 2010, we formed a strategic alliance with Sanofi to discover and develop microRNA therapeutics for fibrotic diseases. In July 2012, we expanded the alliance to include potential microRNA therapeutics in oncology. The original research term for this strategic alliance expired in June 2013, upon which we and Sanofi entered into an option agreement pursuant to which we granted Sanofi an exclusive right to negotiate the co-development and commercialization of certain of our unencumbered microRNA programs, for which Sanofi paid us an upfront option fee of $2.5 million. In addition, Sanofi granted us an exclusive option to negotiate the co-development and commercialization of miR-21. In February 2014, we and Sanofi extended our strategic alliance and Sanofi concurrently made a $10.0 million investment in our common stock. Under the terms of our extended alliance, Sanofi will have opt-in rights to our RG-012 clinical fibrosis program targeting miR-21 for the potential treatment of Alport Syndrome, our preclinical program targeting miR-21 for oncology indications, and our preclinical programs targeting miR-221/222 for oncology indications, each of which is to be led by us. If Sanofi chooses to exercise its option on any of these programs, Sanofi will reimburse us for a significant portion of our preclinical and clinical development costs and will also pay us an option exercise fee for any such program, provided that $1.25 million of the $2.5 million upfront option fee paid to us by Sanofi in connection with the June 2013 option agreement will be creditable against such option exercise fee. In addition, we will be eligible to receive clinical and regulatory milestone payments under these programs and potentially commercial milestone payments. We also continue to be eligible to receive royalties on microRNA therapeutic products commercialized by Sanofi and have the right to co-promote these products.
Under this strategic alliance, we are eligible to receive milestone payments of up to $101.8 million for proof-of-concept option exercise fees (net of $1.25 million creditable, as noted above), $15.0 million for clinical milestones and up to $300.0 million for regulatory and commercial milestones. In addition, we are entitled to receive royalties based on a percentage of net sales of any products from the miR-21 and miR-221/222 programs which, in the case of sales in the United States, will be in the middle of the 10 to 20% range, and, in the case of sales outside of the United States, will range from the low end to the middle of the 10 to 20% range, depending upon the volume of sales. If we exercise our option to co-promote a product, we will continue to be eligible to receive royalties on net sales of each product in the United States at the same rate, unless we elect to share a portion of Sanofi’s profits from sales of such product in the United States in lieu of royalties.
4
In August 2012, we formed a strategic alliance with AstraZeneca to discover and develop microRNA therapeutics for cardiovascular diseases, metabolic diseases and oncology. In March 2015, we and AstraZeneca nominated RG-125(AZD4076), a GalNAc-conjugated anti-miR 103/107 oligonucleotide that has been observed to improve insulin sensitivity and glucose tolerance in animal models as a clinical development candidate in NAFLD in patients with type 2 diabetes/pre-diabetes and we earned a $2.5 million milestone. AstraZeneca became responsible for further development of RG-125(AZD4076) under our collaboration. In December 2015, AstraZeneca commenced the first-in-human dosing of RG-125(AZD4076) in healthy volunteers and we earned an additional $10.0 million milestone. AstraZeneca commenced dosing patients in a Phase IIa clinical trial in the third quarter of 2016. In June of 2017, AstraZeneca informed us of its election to terminate the agreement in its entirety. Under the agreement, the termination becomes effective 12 months after notice of termination. Effective upon the termination of the agreement, AstraZeneca’s rights with respect to RG-125(AZD4076) will revert to us.
5
RISK FACTORS
You should carefully consider the following risk factors, as well as the other information in this report, before deciding whether to purchase, hold or sell shares of our common stock. The occurrence of any of the following risks could harm our business, financial condition, results of operations and/or growth prospects or cause our actual results to differ materially from those contained in forward-looking statements we have made in this report and those we may make from time to time. You should consider all the factors described when evaluating our business. If any of the following risks actually occurs, our business, financial condition, results of operations and future growth prospects would likely be materially and adversely affected. In these circumstances, the market price of our common stock would likely decline.
RISKS RELATED TO OUR FINANCIAL CONDITION AND NEED FOR ADDITIONAL CAPITAL
We have a limited operating history, have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future.
We are a biopharmaceutical company, formed in 2007, with a limited operating history. Since inception, our operations have been primarily limited to acquiring and in-licensing intellectual property rights, developing our microRNA product platform, undertaking basic research around microRNA targets and conducting preclinical and clinical studies for our initial programs. We have not yet obtained regulatory approval for any product candidates. Consequently, any predictions about our future success or viability, or any evaluation of our business and prospects, may not be accurate. We have incurred losses in each year since our inception in September 2007.
We have devoted most of our financial resources to research and development, including our preclinical and clinical development activities. To date, we have financed our operations primarily through the sale of equity securities and convertible debt, through our secured term loan from Oxford and from revenue received from our strategic alliance partners. We have a strategic alliance with Sanofi relating to the development of our miR-21 programs for HCC and kidney fibrosis and our miR-221/222 program for oncology indications. In June 2017, AstraZeneca provided notice to us of its election to terminate the clinical development of RG-125(AZD4076) for NAFLD and terminate our collaboration and license agreement. Under the agreement with AstraZeneca, the termination will become effective on June 9, 2018, at which time AstraZeneca’s rights to RG-125(AZD4076) will revert to us. Under our agreement with Sanofi, Sanofi has an option to obtain exclusive worldwide licenses for the development, manufacture and commercialization of potential product candidates selected from our programs. If Sanofi exercises its option to obtain a license to develop, manufacture and commercialize any such product candidate, it will assume responsibility for funding and conducting further clinical development and commercialization activities for such product candidate. However, if Sanofi does not exercise its option within the timeframes that we expect, or at all, we will be responsible for funding further development of the applicable product candidate and may not have the resources to do so unless we are able to enter into another strategic alliance for such product candidate. The size of our future net losses will depend, in part, on the rate of future expenditures and our ability to obtain funding through equity or debt financings, strategic alliances or grants. We have initiated IND-enabling activities for RGLS4326. We have also initiated clinical development of RG-012, however, it will be several years, if ever, before we or our strategic alliance partners have a product candidate ready for commercialization. Even if we or our strategic alliance partners successfully obtain regulatory approval to market a product candidate, our revenues will also depend upon the size of any markets in which our product candidates have received market approval, and our ability to achieve sufficient market acceptance and adequate market share for our products.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if and as we: continue our research and preclinical and clinical development of our product candidates, both independently and under our strategic alliance agreements; seek to identify additional microRNA targets and product candidates; acquire or in-license other products and technologies; continue with clinical development of our product candidates; seek marketing approvals for our product candidates that successfully complete clinical trials; ultimately establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; maintain, expand and protect our intellectual property portfolio; hire additional clinical, regulatory, research and administrative personnel; and create additional infrastructure to support our operations and our product development and planned future commercialization efforts.
We have never generated any revenue from product sales and may never be profitable.
Our ability to generate revenue and achieve profitability depends on our ability, alone or with strategic alliance partners, to successfully complete the development of, obtain the necessary regulatory approvals for and commercialize product candidates. We do not anticipate generating revenues from sales of products for the foreseeable future, if ever. Our ability to generate future revenues from product sales depends heavily on our success in:
6
| • | identifying and validating new microRNAs as therapeutic targets; |
| • | completing our research and preclinical development of product candidates; |
| • | initiating and completing clinical trials for product candidates; |
| • | seeking and obtaining marketing approvals for product candidates that successfully complete clinical trials; |
| • | establishing and maintaining supply and manufacturing relationships with third parties; |
| • | launching and commercializing product candidates for which we obtain marketing approval, with an alliance partner or, if launched independently, successfully establishing a sales force, marketing and distribution infrastructure; |
| • | maintaining, protecting and expanding our intellectual property portfolio; and |
| • | attracting, hiring and retaining qualified personnel. |
Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to predict the timing or amount of increased expenses and when we will be able to achieve or maintain profitability, if ever. In addition, our expenses could increase beyond expectations if we are required by the FDA or foreign regulatory agencies to perform studies and trials in addition to those that we currently anticipate.
Even if one or more of the product candidates that we independently develop is approved for commercial sale, we anticipate incurring significant costs associated with commercializing any approved product. Even if we are able to generate revenues from the sale of any approved products, we may not become profitable and may need to obtain additional funding to continue operations.
We may need to raise additional capital, which may not be available on acceptable terms, or at all.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We expect our research and development expenses to substantially increase in connection with our ongoing activities, particularly as we advance our product candidates towards or through clinical trials. We will need to raise additional capital to support our operations and such funding may not be available to us on acceptable terms, or at all. As of June 30, 2017, we had cash, cash equivalents and short-term investments of approximately $40.1 million. We believe that our existing cash resources will be sufficient to fund our planned operations and expenditures into mid-2018. However, we cannot provide assurances that our plans will not change or that changed circumstances will not result in the depletion of our capital resources more rapidly than we currently anticipate.
As we move RGLS4326 and any other future lead compounds through toxicology and other preclinical studies, also referred to as nonclinical studies, required to file an IND, and as we conduct clinical development of RG-012 and any other future product candidates, we may have adverse results requiring mitigation strategies that may cause us to consume additional capital. Additionally, our strategic alliance partners may not elect to pursue the development and commercialization of any of our microRNA product candidates that are subject to their respective strategic alliance agreements with us. Any of these events may increase our development costs more than we expect. For example, AstraZeneca recently terminated its development of RG-125(AZD4076). Upon the effective date of termination, AstraZeneca’s rights to the program will revert to us and we may decide to continue with its development but will then be responsible for any continuing costs of development. We may need to raise additional capital or otherwise obtain funding through additional strategic alliances if we choose to initiate clinical trials for new product candidates other than programs currently partnered. In any event, we will require additional capital to obtain regulatory approval for, and to commercialize, future product candidates.
If we are required to secure additional financing, such additional fundraising efforts may divert our management from our day-to-day activities, which may adversely affect our ability to develop and commercialize future product candidates. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital when required or on acceptable terms, we may be required to:
| • | significantly delay, scale back or discontinue the development or commercialization of any future product candidates; |
| • | seek strategic alliances for research and development programs at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; or |
| • | relinquish or license on unfavorable terms, our rights to technologies or any future product candidates that we otherwise would seek to develop or commercialize ourselves. |
7
If we are required to conduct additional fundraising activities and we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing development and commercialization efforts, which will have a material adverse effect on our business, operating results and prospects.
Payments under the instruments governing our indebtedness may reduce our working capital. In addition, a default under our loan and security agreement could cause a material adverse effect on our financial position.
In June 2016, we entered into a loan and security agreement with Oxford. Under the terms of the loan agreement, Oxford provided us with a Term A Loan of $20.0 million, with an additional $10.0 million Term B Loan available to us upon the achievement of a milestone until the earlier of 60 days after the achievement of the milestone or March 31, 2017, subject to the non-occurrence of a prior event of default. The ability to borrow on the Term B Loan expired on March 31, 2017. Our obligations under the loan agreement are secured by a first priority security interest in substantially all of our current and future assets, other than our intellectual property. We have also agreed not to encumber our intellectual property assets, except as permitted by the loan agreement. Amounts outstanding under the loan agreement mature on June 1, 2020 and will be interest-only through June 1, 2018, followed by 24 equal monthly payments of principal and unpaid accrued interest. Payments under the loan agreement could result in a significant reduction of our working capital.
The loan agreement requires us, and any debt arrangements we may enter into in the future may require us, to comply with various covenants that limit our ability to, among other things:
| • | dispose of assets; |
| • | complete mergers or acquisitions; |
| • | incur indebtedness; |
| • | encumber assets; |
| • | pay dividends or make other distributions to holders of our capital stock; |
| • | make specified investments; and |
| • | engage in transactions with our affiliates. |
These restrictions could inhibit our ability to pursue our business strategies. If we default under our obligations under the loan agreement, the lender could proceed against the collateral granted to it to secure our indebtedness or declare all obligation under the loan agreement to be due and payable. In certain circumstances, procedures by the lenders could result in a loss by us of all of our equipment and inventory, which are included in the collateral granted to the lenders. If any indebtedness under the loan agreement were to be accelerated, there can be no assurance that our assets would be sufficient to repay in full that indebtedness. In addition, upon any distribution of assets pursuant to any liquidation, insolvency, dissolution, reorganization or similar proceeding, the holders of secured indebtedness will be entitled to receive payment in full from the proceeds of the collateral securing our secured indebtedness before the holders of other indebtedness or our common stock will be entitled to receive any distribution with respect thereto.
We may incur additional indebtedness in the future. The debt instruments governing such indebtedness may contain provisions that are as, or more, restrictive than the provisions governing our existing indebtedness under the loan agreement. If we are unable to repay, refinance or restructure our indebtedness when payment is due, the lenders could proceed against the collateral or force us into bankruptcy or liquidation.
RISKS RELATED TO THE DISCOVERY AND DEVELOPMENT OF PRODUCT CANDIDATES
Preclinical and clinical studies of our product candidates may not be successful. If we are unable to generate successful results from our preclinical and clinical studies of our product candidates, or experience significant delays in doing so, our business may be materially harmed.
We have invested a significant portion of our efforts and financial resources in the identification and development of product candidates that target microRNAs. Our ability to generate product revenues, which we do not expect will occur for many years, if ever, will depend heavily on the successful development and eventual commercialization of our product candidates.
The success of our product candidates will depend on several factors, including the following:
| • | successfully designing preclinical studies which may be predictive of clinical outcomes; |
| • | successful results from preclinical and clinical studies; |
8
| • | receipt of marketing approvals from applicable regulatory authorities; |
| • | obtaining and maintaining patent and trade secret protection for future product candidates; |
| • | establishing and maintaining manufacturing relationships with third parties or establishing our own manufacturing capability; and |
| • | successfully commercializing our products, if and when approved, whether alone or in collaboration with others. |
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully complete the development of, or commercialize, our product candidates, which would materially harm our business.
The approach we are taking to discover and develop drugs is novel and may never lead to marketable products.
We have concentrated our therapeutic product research and development efforts on microRNA technology, and our future success depends on the successful development of this technology and products based on our microRNA product platform. Neither we nor any other company has received regulatory approval to market therapeutics targeting microRNAs. The scientific discoveries that form the basis for our efforts to discover and develop product candidates are relatively new. The scientific evidence to support the feasibility of developing product candidates based on these discoveries is both preliminary and limited. If we do not successfully develop and commercialize product candidates based upon our technological approach, we may not become profitable and the value of our common stock may decline.
Further, our focus solely on microRNA technology for developing drugs as opposed to multiple, more proven technologies for drug development increases the risks associated with the ownership of our common stock. If we are not successful in developing any product candidates using microRNA technology, we may be required to change the scope and direction of our product development activities. In that case, we may not be able to identify and implement successfully an alternative product development strategy.
We may not be successful in our efforts to identify or discover potential product candidates.
The success of our business depends primarily upon our ability to identify, develop and commercialize microRNA therapeutics. Our research programs may initially show promise in identifying potential product candidates, yet fail to yield product candidates for clinical development for a number of reasons, including:
| • | our research methodology or that of our strategic alliance partners may be unsuccessful in identifying potential product candidates; |
| • | potential product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval; or |
| • | our strategic alliance partners may change their development profiles for potential product candidates or abandon a therapeutic area. |
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business and could potentially cause us to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. We may focus our efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
If clinical trials of our product candidates fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or do not otherwise produce positive results, we may incur additional costs or experience delays in completing, or ultimately be unable to complete, the development and commercialization of our product candidates.
Before obtaining marketing approval from regulatory authorities for the sale of product candidates, we or our strategic alliance partners must conduct extensive clinical trials to demonstrate the safety and efficacy of the product candidates in humans. Clinical trials are expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. The outcome of preclinical studies and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for their products.
9
Events which may result in a delay or unsuccessful completion of clinical development include:
| • | delays in reaching an agreement with the FDA or other regulatory authorities on final trial design; |
| • | imposition of a clinical hold of our clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| • | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
| • | our inability to adhere to clinical trial requirements directly or with third parties such as CROs; |
| • | delays in obtaining required institutional review board approval at each clinical trial site; |
| • | delays in recruiting suitable patients to participate in a trial; |
| • | delays in the testing, validation, manufacturing and delivery of the product candidates to the clinical sites; |
| • | delays in having patients complete participation in a trial or return for post-treatment follow-up; |
| • | delays caused by patients dropping out of a trial due to protocol procedures or requirements, product side effects or disease progression; |
| • | clinical sites dropping out of a trial to the detriment of enrollment; |
| • | time required to add new clinical sites; or |
| • | delays by our contract manufacturers to produce and deliver sufficient supply of clinical trial materials. |
For example, in June 2016, the FDA placed a full clinical hold on our RG-101 clinical program after a second patient experienced an SAE of jaundice. In July 2016, we received a formal clinical hold letter from the FDA requesting additional studies, expert opinion and detailed safety data analysis. In December 2016, we submitted a complete response to the FDA’s initial request for information, however, the FDA later informed us that the full clinical hold placed on our RG-101 clinical development program in June 2016 would remain in effect pending the FDA’s review of all requested data. We thereafter decided to discontinue clinical development of RG-101 upon completion of the one remaining clinical study since our evaluation of the clinical data from RG-101 led to the identification of a bilirubin transport mechanism as the likely cause for the cases of SAEs of jaundice. As a result, our progress in the development of this program was significantly slowed and the associated costs increased, potentially adversely affecting our business and causing a drop in our stock price.
If we or our strategic alliance partners are required to conduct additional clinical trials or other testing of any product candidates beyond those that are currently contemplated, are unable to successfully complete clinical trials of any such product candidates or other testing, or if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we or our strategic alliance partners may:
| • | be delayed in obtaining marketing approval for our future product candidates; |
| • | not obtain marketing approval at all; |
| • | obtain approval for indications or patient populations that are not as broad as originally intended or desired; |
| • | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| • | be subject to additional post-marketing testing requirements; or |
| • | have the product removed from the market after obtaining marketing approval. |
Our product development costs will also increase if we experience delays in testing or marketing approvals. We do not know whether any clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which would impair our ability to successfully commercialize our product candidates and may harm our business and results of operations. Any inability to successfully complete preclinical and clinical development, whether independently or with our strategic alliance partners, could result in additional costs to us or impair our ability to generate revenues from product sales, regulatory and commercialization milestones and royalties.
Any of our product candidates may cause adverse effects or have other properties that could delay or prevent their regulatory approval or limit the scope of any approved label or market acceptance.
Adverse events, or AEs, caused by our product candidates could cause us, other reviewing entities, clinical trial sites or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval. Certain oligonucleotide therapeutics have shown injection site reactions and pro-inflammatory effects and may also lead to impairment of kidney or liver function. There is a risk that our future product candidates may induce similar AEs.
10
If AEs are observed in any clinical trials of our product candidates, including those that our strategic partners may develop under our alliance agreements, our or our partners’ ability to obtain regulatory approval for product candidates may be negatively impacted.
Further, if any of our future products, if and when approved for commercial sale, cause serious or unexpected side effects, a number of potentially significant negative consequences could result, including:
| • | regulatory authorities may withdraw their approval of the product or impose restrictions on its distribution in the form of a modified risk evaluation and mitigation strategy; |
| • | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| • | we may be required to change the way the product is administered or conduct additional clinical trials; |
| • | we could be sued and held liable for harm caused to patients; or |
| • | our reputation may suffer. |
Any of these events could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing our future products and impair our ability to generate revenues from the commercialization of these products either by us or by our strategic alliance partners.
Even if we complete the necessary preclinical studies and clinical trials, we cannot predict whether or when we will obtain regulatory approval to commercialize a product candidate and we cannot, therefore, predict the timing of any revenue from a future product.
Neither we nor our strategic alliance partners can commercialize a product until the appropriate regulatory authorities, such as the FDA, have reviewed and approved the product candidate. The regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA Advisory Committee recommends restrictions on approval or recommends non-approval. In addition, we or our strategic alliance partners may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical trials and the review process.
Even if we obtain regulatory approval for a product candidate, we will still face extensive regulatory requirements and our products may face future development and regulatory difficulties.
Even if we obtain regulatory approval in the United States, the FDA may still impose significant restrictions on the indicated uses or marketing of our product candidates, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. The holder of an approved NDA is obligated to monitor and report AEs and any failure of a product to meet the specifications in the NDA. The holder of an approved NDA must also submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling or manufacturing process. Advertising and promotional materials must comply with FDA rules and are subject to FDA review, in addition to other potentially applicable federal and state laws.
In addition, drug product manufacturers and their facilities are subject to payment of user fees and continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current good manufacturing practices, or cGMP, and adherence to commitments made in the NDA. If we or a regulatory agency discovers previously unknown problems with a product such as AEs of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we or our partners fail to comply with applicable regulatory requirements following approval of any of our product candidates, a regulatory agency may:
| • | issue a warning letter asserting that we are in violation of the law; |
| • | seek an injunction or impose civil or criminal penalties or monetary fines; |
| • | suspend or withdraw regulatory approval; |
11
| • | suspend any ongoing clinical trials; |
| • | refuse to approve a pending NDA or supplements to an NDA submitted by us; |
| • | seize product; or |
| • | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our future products and generate revenues.
We may not be successful in obtaining or maintaining necessary rights to microRNA targets, drug compounds and processes for our development pipeline through acquisitions and in-licenses.
Presently we have rights to the intellectual property, through licenses from third parties and under patents that we own, to modulate only a subset of the known microRNA targets. Because our programs may involve a range of microRNA targets, including targets that require the use of proprietary rights held by third parties, the growth of our business will likely depend in part on our ability to acquire, in-license or use these proprietary rights. In addition, our product candidates may require specific formulations to work effectively and efficiently and these rights may be held by others. We may be unable to acquire or in-license any compositions, methods of use, processes or other third-party intellectual property rights from third parties that we identify. The licensing and acquisition of third-party intellectual property rights is a competitive area, and a number of more established companies are also pursuing strategies to license or acquire third-party intellectual property rights that we may consider attractive. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities.
For example, we may collaborate with U.S. and foreign academic institutions to accelerate our preclinical research or development under written agreements with these institutions. Typically, these institutions provide us with an option to negotiate a license to any of the institution’s rights in technology resulting from the collaboration. Regardless of such right of first negotiation for intellectual property, we may be unable to negotiate a license within the specified time frame or under terms that are acceptable to us. If we are unable to do so, the institution may offer the intellectual property rights to other parties, potentially blocking our ability to pursue our program.
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain rights to required third-party intellectual property rights, our business, financial condition and prospects for growth could suffer.
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and human resources, we intend to leverage our existing strategic alliance agreements and may enter into new strategic alliance agreements for the development and commercialization of our programs and potential product candidates in indications with potentially large commercial markets such as HCC, fibrosis and HCV, while focusing our internal development resources and any internal sales and marketing organization that we may establish on research programs and product candidates for selected markets, such as orphan diseases. As a result, we may forego or delay pursuit of opportunities with other programs or product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on research and development programs and product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic alliance, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate, or we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce
12
hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties or other sanctions.
RISKS RELATED TO OUR RELIANCE ON THIRD PARTIES
We will depend upon our strategic alliances for the development and eventual commercialization of certain microRNA product candidates. If these strategic alliances are unsuccessful or are terminated, we may be unable to commercialize certain product candidates and we may be unable to generate revenues from our development programs.
We are likely to depend upon third party alliance partners for financial and scientific resources for the clinical development and commercialization of certain of our microRNA product candidates. These strategic alliances will likely provide us with limited control over the course of development of a microRNA product candidate, especially once a candidate has reached the stage of clinical development. For example, in our alliance with Sanofi, Sanofi has the option to obtain an exclusive worldwide license to develop, manufacture and commercialize product candidates upon the achievement of relevant endpoints in clinical trials. However, Sanofi is not under any obligation to exercise these options to progress any of our microRNA development candidates. While Sanofi has development obligations with respect to programs that it may elect to pursue under our agreement, our ability to ultimately recognize revenue from this and future relationships will depend upon the ability and willingness of our alliance partners to successfully meet their respective responsibilities under our agreements with them. Our ability to recognize revenues from successful strategic alliances may be impaired by several factors including:
| • | an alliance partner may shift its priorities and resources away from our programs due to a change in business strategies, or a merger, acquisition, sale or downsizing of its company or business unit; |
| • | an alliance partner may cease development in therapeutic areas which are the subject of our strategic alliances; |
| • | an alliance partner may change the success criteria for a particular program or potential product candidate thereby delaying or ceasing development of such program or candidate; |
| • | a significant delay in initiation of certain development activities by an alliance partner will also delay payment of milestones tied to such activities, thereby impacting our ability to fund our own activities; |
| • | an alliance partner could develop a product that competes, either directly or indirectly, with an alliance product; |
| • | an alliance partner with commercialization obligations may not commit sufficient financial or human resources to the marketing, distribution or sale of a product; |
| • | an alliance partner with manufacturing responsibilities may encounter regulatory, resource or quality issues and be unable to meet demand requirements; |
| • | an alliance partner may exercise its rights under the agreement to terminate a strategic alliance; |
| • | a dispute may arise between us and an alliance partner concerning the research, development or commercialization of a program or product candidate resulting in a delay in milestones, royalty payments or termination of a program and possibly resulting in costly litigation or arbitration which may divert management attention and resources; and |
| • | an alliance partner may use our proprietary information or intellectual property in such a way as to invite litigation from a third party or fail to maintain or prosecute intellectual property rights such that our rights in such property are jeopardized. |
Specifically, with respect to termination rights, Sanofi may terminate the entire alliance or its current alliance target program for any or no reason upon 30 days’ written notice to us. The agreement with Sanofi may also be terminated by either party for material breach by the other party, including a failure to comply with such party’s diligence obligations that remains uncured after 120 days. Depending on the timing of any such termination, we may not be entitled to receive the option exercise fees or milestone payments, as these payments terminate with termination of the respective program or agreement. In addition, AstraZeneca terminated the agreement in its entirety upon 12 months’ written notice to us. Upon expiration of the termination period, AstraZeneca’s rights to RG-125(AZD4076) will revert to us. We will be responsible for any further development costs at that time.
13
If any of our alliance partners do not elect to pursue the development and commercialization of our microRNA development candidates or if they terminate the strategic alliance, then, depending on the event:
| • | in the case of Sanofi, under certain circumstances, we may owe Sanofi royalties with respect to product candidates covered by our agreement with Sanofi that we elect to continue to commercialize, depending upon the stage of development at which such product commercialization rights reverted back to us, or additional payments if we license such product candidates to third parties; |
| • | the development of our product candidate subject to the AstraZeneca agreement or the product candidates subject to the Sanofi agreement, as applicable, may be terminated or significantly delayed; |
| • | our cash expenditures could increase significantly if it is necessary for us to hire additional employees and allocate scarce resources to the development and commercialization of product candidates that were previously funded, or expected to be funded, by AstraZeneca or Sanofi, as applicable; |
| • | we would bear all of the risks and costs related to the further development and commercialization of product candidates that were previously the subject of the AstraZeneca agreement or the Sanofi agreement, as applicable, including the reimbursement of third parties; in particular, upon expiration of the AstraZeneca 12-month termination period, we will be responsible for any further costs of development, and we may owe AstraZeneca certain consideration for use of any intellectual property generated by AstraZeneca; and |
| • | in order to fund further development and commercialization, we may need to seek out and establish alternative strategic alliances with third-party partners; this may not be possible, or we may not be able to do so on terms which are acceptable to us, in which case it may be necessary for us to limit the size or scope of one or more of our programs or increase our expenditures and seek additional funding by other means. |
Any of these events would have a material adverse effect on our results of operations and financial condition.
We rely on third parties to conduct some aspects of our compound formulation, research and preclinical studies, and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such formulation, research or testing.
We do not expect to independently conduct all aspects of our drug discovery activities, compound formulation research or preclinical studies of product candidates. We currently rely and expect to continue to rely on third parties to conduct some aspects of our preclinical studies and formulation development.
Any of these third parties may terminate their engagements with us at any time. If we need to enter into alternative arrangements, it would delay our product development activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibilities. For example, for product candidates that we develop and commercialize on our own, we will remain responsible for ensuring that each of our IND-enabling studies and clinical trials are conducted in accordance with the study plan and protocols for the trial.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines or conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, we will not be able to complete, or may be delayed in completing, the necessary preclinical studies to enable us or our strategic alliance partners to select viable product candidates for IND submissions and will not be able to, or may be delayed in our efforts to, successfully develop and commercialize such product candidates.
We rely on third-party manufacturers to produce our preclinical and clinical product candidates, and we intend to rely on third parties to produce future clinical supplies of product candidates that we advance into clinical trials and commercial supplies of any approved product candidates.
Reliance on third-party manufacturers entails risks, including risks that we would not be subject to if we manufactured the product candidates ourselves, including:
| • | the inability to meet any product specifications and quality requirements consistently; |
| • | a delay or inability to procure or expand sufficient manufacturing capacity; |
| • | manufacturing and product quality issues related to scale-up of manufacturing; |
14
| • | costs and validation of new equipment and facilities required for scale-up; |
| • | a failure to comply with cGMP and similar foreign standards; |
| • | the inability to negotiate manufacturing or supply agreements with third parties under commercially reasonable terms; |
| • | termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; |
| • | the reliance on a limited number of sources, and in some cases, single sources for raw materials, such that if we are unable to secure a sufficient supply of these product components, we will be unable to manufacture and sell future product candidates in a timely fashion, in sufficient quantities or under acceptable terms; |
| • | the lack of qualified backup suppliers for any raw materials that are currently purchased from a single source supplier; |
| • | operations of our third-party manufacturers or suppliers could be disrupted by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier; |
| • | carrier disruptions or increased costs that are beyond our control; and |
| • | the failure to deliver products under specified storage conditions and in a timely manner. |
Any of these events could lead to clinical study delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize future products. Some of these events could be the basis for FDA action, including injunction, recall, seizure or total or partial suspension of production.
We rely on limited sources of supply for the drug substance of product candidates and any disruption in the chain of supply may cause a delay in developing and commercializing these product candidates.
We have established manufacturing relationships with a limited number of suppliers to manufacture raw materials and the drug substance of any product candidate for which we are responsible for preclinical or clinical development. Each supplier may require licenses to manufacture such components if such processes are not owned by the supplier or in the public domain. As part of any marketing approval, a manufacturer and its processes are required to be qualified by the FDA prior to commercialization. If supply from the approved vendor is interrupted, there could be a significant disruption in commercial supply. An alternative vendor would need to be qualified through an NDA supplement which could result in further delay. The FDA or other regulatory agencies outside of the United States may also require additional studies if a new supplier is relied upon for commercial production. Switching vendors may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
In addition, if our alliance partners elect to pursue the development and commercialization of certain programs, we will lose control over the manufacturing of the product candidate subject to the agreement. For example, if Sanofi elects to develop and commercialize a product candidate targeting miR-21 or miR-221/222 for oncology indications or RG-012 for kidney fibrosis under its strategic alliance with us, Sanofi will be responsible for the manufacture of the product candidates for further clinical trials. Sanofi will be free to use a manufacturer of its own choosing or manufacture the product candidates in its own manufacturing facilities. In such a case, we will have no control over Sanofi’s processes or supply chains to ensure the timely manufacture and supply of the product candidates. In addition, we will not be able to ensure that the product candidates will be manufactured under the correct conditions to permit the product candidates to be used in such clinical trials.
These factors could cause the delay of clinical trials, regulatory submissions, required approvals or commercialization of our product candidates, cause us to incur higher costs and prevent us from commercializing our products successfully. Furthermore, if our suppliers fail to deliver the required commercial quantities of active pharmaceutical ingredients on a timely basis and at commercially reasonable prices, and we are unable to secure one or more replacement suppliers capable of production in a timely manner at a substantially equivalent cost, our clinical trials may be delayed or we could lose potential revenue.
Manufacturing issues may arise that could increase product and regulatory approval costs or delay commercialization.
As we scale-up manufacturing of product candidates and conduct required stability testing, product, packaging, equipment and process-related issues may require refinement or resolution in order to proceed with any clinical trials and obtain regulatory approval for commercial marketing. We may identify significant impurities, which could result in increased scrutiny by the regulatory agencies, delays in clinical programs and regulatory approval, increases in our operating expenses, or failure to obtain or maintain approval for product candidates or any approved products.
15
We rely on third parties to conduct, supervise and monitor our clinical trials, and if those third parties perform in an unsatisfactory manner, it may harm our business.
We or our strategic alliance partners rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials. While we will have agreements governing their activities, we and our strategic alliance partners have limited influence over their actual performance. We control only certain aspects of our CROs’ activities. Nevertheless, we or our strategic alliance partners are responsible for ensuring that each of our clinical trials are conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities.
We, our alliance partners and our CROs are required to comply with the FDA’s or other regulatory agency’s good clinical practices, or GCPs, for conducting, recording and reporting the results of IND-enabling studies and clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. The FDA and non-U.S. regulatory agencies enforce these GCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or applicable non-U.S. regulatory agency may require us to perform additional clinical trials before approving any marketing applications for the relevant jurisdiction. Upon inspection, the FDA or applicable non-U.S. regulatory agency may determine that our clinical trials did not comply with GCPs. In addition, our clinical trials will require a sufficiently large number of test subjects to evaluate the safety and effectiveness of a potential drug product. Accordingly, if our CROs fail to comply with these regulations or fail to recruit a sufficient number of patients, we may be required to repeat such clinical trials, which would delay the regulatory approval process.
Our CROs will not be our employees, and we will not be able to control whether or not they devote sufficient time and resources to our clinical and nonclinical programs. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials, or other drug development activities which could harm our competitive position. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for such products and any product candidates that we develop would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
We also rely on other third parties to store and distribute drug products for any clinical trials that we may conduct. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product revenue.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
If we are unable to obtain or protect intellectual property rights related to our future products and product candidates, we may not be able to compete effectively in our markets.
We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our future products and product candidates. The strength of patents in the biotechnology and pharmaceutical field involves complex legal and scientific questions and can be uncertain. The patent applications that we own or in-license may fail to result in patents with claims that cover the products in the United States or in other countries. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found; such prior art can invalidate a patent or prevent a patent from issuing based on a pending patent application. Even if patents do successfully issue, third parties may challenge their validity, enforceability or scope, which may result in such patents being narrowed or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims.
If the patent applications we hold or have in-licensed with respect to our programs or product candidates fail to issue or if their breadth or strength of protection is threatened, it could dissuade companies from collaborating with us to develop product candidates, and threaten our ability to commercialize, future products. We cannot offer any assurances about which, if any, patents will issue or whether any issued patents will be found invalid and unenforceable or will be threatened by third parties. A patent may be challenged through one or more of several administrative proceedings including post-grant challenges, re-examination or opposition before the U.S. PTO or foreign patent offices. For example, re-examination of, or oppositions to, patents owned by or licensed to us have previously been initiated, and while we believe these concluded proceedings did not result in a commercially relevant impact on the individual patents, any successful challenge of patents or any other patents owned by or licensed to us could deprive us of rights necessary for the successful commercialization of any product candidates that we or our strategic alliance partners may develop.
16
Since patent applications in the United States and most other countries are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we were the first to file any patent application related to a product candidate. Furthermore, in certain situations, if we and one or more third parties have filed patent applications in the United States and claiming the same subject matter, an administrative proceeding, known as an interference, can be initiated to determine which applicant is entitled to the patent on that subject matter. Such an interference proceeding provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications, or those of our alliance partners or licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of a patent or patent application in such a proceeding may not be successful and, even if successful, may result in substantial costs and distract our management and other employees.
In addition, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed. Various extensions may be available however the life of a patent, and the protection it affords, is limited. Once the patent life has expired for a product, we may be open to competition from generic medications. Further, if we encounter delays in regulatory approvals, the period of time during which we could market a product candidate under patent protection could be reduced.
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our drug discovery and development processes that involve proprietary know-how, information or technology that is not covered by patents. Although each of our employees agrees to assign their inventions to us through an employee inventions agreement, and all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed or that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all.
Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and abroad. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market, which could materially adversely affect our business, results of operations and financial condition.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our strategic alliance partners are pursuing development candidates. For example, we are aware that Roche Innovation Center Copenhagen has patents and patent applications in the microRNA therapeutics space, including patents and patent applications related to targeting microRNAs, such as miR-122, for the treatment of disease. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in patents that our product candidates may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our product
17
candidates, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all.
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our product candidates. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
If we fail to comply with our obligations in the agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We are a party to a number of intellectual property license agreements that are important to our business and expect to enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty and other obligations on us. For example, under our exclusive license agreement for Max-Planck-Innovation GmbH’s proprietary technology and know-how covering microRNA sequences, we are required to use commercially reasonable diligence to develop and commercialize a product and to satisfy specified payment obligations. If we fail to comply with our obligations under our agreement with Max-Planck-Innovation GmbH or our other license agreements, or we are subject to a bankruptcy, the licensor may have the right to terminate the license, in which event we, or our strategic alliance partners, would not be able to market products covered by the license. In addition, our exclusive license agreements with our founding companies, Alnylam and Ionis, provide us with rights to nucleotide technologies in the field of microRNA therapeutics based on oligonucleotides that modulate microRNAs. Some of these technologies, such as intellectual property relating to the chemical modification of oligonucleotides, are relevant to our product candidate development programs. If our license agreements with Alnylam or Ionis are terminated, or our business relationships with either of these companies or our other licensors are disrupted by events that may include the acquisition of either company, our access to critical intellectual property rights will be materially and adversely affected.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our product candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we would be unable to further develop and commercialize one or more of our product candidates, which could harm our business significantly. We cannot provide any assurances that third-party patents do not exist which might be enforced against our future products, resulting in either an injunction prohibiting our sales, or, with respect to our sales, an obligation on our part to pay royalties and/or other forms of compensation to third parties.
We may be involved in lawsuits to protect or enforce our patents or the patents of our licensors, which could be expensive, time consuming and unsuccessful.
Competitors may infringe our patents or the patents of our licensors. To counter infringement or unauthorized use, we may be required to file infringement claims, which can be expensive and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours or our licensors is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Our defense in a litigation may fail and, even if successful, may result in substantial costs and distract our management and other employees. We may not be able to prevent, alone or with our licensors, misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
18
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties.
We employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed confidential information of our employees’ former employers or other third parties. We may also be subject to claims that former employers or other third parties have an ownership interest in our patents. Litigation may be necessary to defend against these claims. There is no guarantee of success in defending these claims, and if we are successful, litigation could result in substantial cost and be a distraction to our management and other employees.
RISKS RELATED TO COMMERCIALIZATION OF PRODUCT CANDIDATES
The commercial success of our programs that are part of our strategic alliance agreements with Sanofi or others will depend in large part on the development and marketing efforts of our alliance partners. If our alliance partners are unable or unwilling to perform in accordance with the terms of our agreements, our potential to generate future revenue from these programs would be significantly reduced and our business would be materially and adversely harmed.
If or when Sanofi elects to further pursue the development and commercialization of any of the microRNA product candidates that are subject to its strategic alliance agreement with us, we will have limited influence and/or control over their approaches to development and commercialization. If Sanofi or any potential future strategic alliance partners do not perform in the manner that we expect or fail to fulfill their responsibilities in a timely manner, or at all, the clinical development, regulatory approval and commercialization efforts related to product candidates we have licensed to such strategic alliance partners could be delayed or terminated. If we terminate any of our strategic alliances or any program thereunder due to a material breach by Sanofi, we have the right to assume the responsibility at our own expense for the development of the applicable microRNA product candidates. Assuming sole responsibility for further development will increase our expenditures, and may mean we will need to limit the size and scope of one or more of our programs, seek additional funding and/or choose to stop work altogether on one or more of the affected product candidates. This could result in a limited potential to generate future revenue from such microRNA product candidates and our business could be materially and adversely affected. Further, under certain circumstances, we may owe Sanofi royalties on any product candidate that we may successfully commercialize.
We face significant competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive. We have competitors both in the United States and internationally, including major multinational pharmaceutical companies, biotechnology companies and universities and other research institutions. Our competitors may have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Additional mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated in our competitors. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our competitors may succeed in developing, acquiring or licensing on an exclusive basis, drug products that are more effective or less costly than any product candidate that we may develop.
Most of our programs are targeted toward indications for which there are approved products on the market or product candidates in clinical development. We will face competition from other drugs currently approved or that will be approved in the future for the same therapeutic indications. Our ability to compete successfully will depend largely on our ability to leverage our experience in drug discovery and development to:
| • | discover and develop therapeutics that are superior to other products in the market; |
| • | attract qualified scientific, product development and commercial personnel; |
| • | obtain patent and/or other proprietary protection for our microRNA product platform and future product candidates; |
| • | obtain required regulatory approvals; and |
| • | successfully collaborate with pharmaceutical companies in the discovery, development and commercialization of new therapeutics. |
The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize. We will not achieve our business plan if the acceptance of any of these products is inhibited by price competition or the reluctance of physicians to switch from existing drug products to our products, or if physicians switch to other new drug products or choose to reserve our future products for use in limited circumstances. The inability to compete with existing or subsequently introduced drug products would have a material adverse impact on our business, financial condition and prospects.
19
Established pharmaceutical companies may invest heavily to accelerate discovery and development of novel compounds or to in-license novel compounds that could make our product candidates less competitive. In addition, any new product that competes with an approved product must demonstrate compelling advantages in efficacy, convenience, tolerability and safety in order to overcome price competition and to be commercially successful. Accordingly, our competitors may succeed in obtaining patent protection, receiving FDA approval or discovering, developing and commercializing product candidates before we do, which would have a material adverse impact on our business.
The commercial success of our product candidates will depend upon the acceptance of these product candidates by the medical community, including physicians, patients and healthcare payors.
The degree of market acceptance of any product candidates will depend on a number of factors, including:
| • | demonstration of clinical safety and efficacy compared to other products; |
| • | the relative convenience, ease of administration and acceptance by physicians, patients and healthcare payors; |
| • | the prevalence and severity of any AEs; |
| • | limitations or warnings contained in the FDA-approved label for such products; |
| • | availability of alternative treatments; |
| • | pricing and cost-effectiveness; |
| • | the effectiveness of our or any collaborators’ sales and marketing strategies; |
| • | our ability to obtain hospital formulary approval; |
| • | our ability to obtain and maintain sufficient third party coverage or adequate reimbursement; and |
| • | the willingness of patients to pay out-of-pocket in the absence of third party coverage. |
Unless other formulations are developed in the future, we expect our compounds to be formulated in an injectable form. Injectable medications may be disfavored by patients or their physicians in the event drugs which are easy to administer, such as oral medications, are available. If a product is approved, but does not achieve an adequate level of acceptance by physicians, patients and healthcare payors, we may not generate sufficient revenues from such product and we may not become or remain profitable. For example, several new antivirals and antiviral combinations have been approved for the treatment of the Hepatitis C infection since we commenced our HCV program. Such increased competition may decrease any future potential revenue for future product candidates due to increasing pressure for lower pricing and higher discounts in the commercialization of our product.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate any revenues.
We currently do not have an organization for the sales, marketing and distribution of pharmaceutical products and the cost of establishing and maintaining such an organization may exceed the cost-effectiveness of doing so. In order to market any products that may be approved, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. For example, in order to exercise our co-promotion rights with Sanofi with respect to our miR-21 and miR-221/222 programs, we would need to build our sales, marketing, managerial and other non-technical capabilities in order to effectively carry out sales or co-promotion activities with respect to any approved products that are developed through these programs. With respect to certain of our current programs as well as future programs, we may rely completely on an alliance partner for sales and marketing. In addition, we intend to enter into strategic alliances with third parties to commercialize other product candidates, including in markets outside of the United States or for other large markets that are beyond our resources. Although we intend to establish a sales organization if we are able to obtain approval to market any product candidates for niche markets in the United States, we will also consider the option to enter into strategic alliances for future product candidates in the United States if commercialization requirements exceed our available resources. This will reduce the revenue generated from the sales of these products.
Our current and any future strategic alliance partners may not dedicate sufficient resources to the commercialization of our product candidates or may otherwise fail in their commercialization due to factors beyond our control. If we are unable to establish effective alliances to enable the sale of our product candidates to healthcare professionals and in geographical regions, including the United States, that will not be covered by our own marketing and sales force, or if our potential future strategic alliance partners do not successfully commercialize the product candidates, our ability to generate revenues from product sales will be adversely affected.
20
If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate sufficient product revenue and may not become profitable. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies.
If we obtain approval to commercialize any approved products outside of the United States, a variety of risks associated with international operations could materially adversely affect our business.
Under our existing strategic alliance agreement, Sanofi will be responsible for the commercialization of current and any future product candidates developed under our programs. If any other product candidates that we develop are approved for commercialization, we may also enter into agreements with third parties to market them on a worldwide basis or in more limited geographical regions. We expect that we will be subject to additional risks related to entering into international business relationships, including:
| • | different regulatory requirements for drug approvals in foreign countries; |
| • | reduced protection for intellectual property rights; |
| • | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| • | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| • | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| • | foreign taxes, including withholding of payroll taxes; |
| • | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; |
| • | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| • | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| • | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods and fires. |
Coverage and adequate reimbursement may not be available for our product candidates, which could make it difficult for us to sell products profitably.
Market acceptance and sales of any product candidates that we develop will depend on coverage and reimbursement policies and may be affected by future healthcare reform measures. Government authorities and third party payors, such as private health insurers, government payors and health maintenance organizations, decide which drugs they will pay for and establish reimbursement levels. We cannot be sure that coverage and adequate reimbursement will be available for any future product candidates. Also, inadequate reimbursement amounts may reduce the demand for, or the price of, our future products. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage for the product. If reimbursement is not available, or is available only at limited levels, we may not be able to successfully commercialize product candidates that we develop.
In addition, we cannot be certain if and when we will obtain formulary approval to allow us to sell any products that we may develop and commercialize into our target markets. Obtaining formulary approval from hospitals and from payors can be an expensive and time consuming process. Failure to obtain timely formulary approval will limit our commercial success.
There have been a number of legislative and regulatory proposals to change the healthcare system in the United States and in some foreign jurisdictions that could affect our ability to sell products profitably. These legislative and/or regulatory changes may negatively impact the reimbursement for drug products, following approval. The availability of numerous generic treatments may also substantially reduce the likelihood of reimbursement for our future products. The potential application of user fees to generic drug products may expedite the approval of additional generic drug treatments. We expect to experience pricing pressures in connection with the sale of any products that we develop, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. If we fail to successfully secure and maintain reimbursement coverage for our future products or are significantly delayed in doing so, we will have difficulty achieving market acceptance of our future products and our business will be harmed.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the EU provides options
21
for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the EU do not follow price structures of the U.S. and generally tend to be priced significantly lower.
RISKS RELATED TO OUR BUSINESS OPERATIONS AND INDUSTRY
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on principal members of our executive team, the loss of whose services may adversely impact the achievement of our objectives. While we have entered into employment agreements with each of our executive officers, any of them could leave our employment at any time, as all of our employees are “at will” employees. Recruiting and retaining other qualified employees for our business, including scientific and technical personnel, will also be critical to our success. There is currently a shortage of skilled executives in our industry, which is likely to continue. As a result, competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical companies for individuals with similar skill sets. In addition, failure to succeed in preclinical studies and clinical trials may make it more challenging to recruit and retain qualified personnel. The inability to recruit or loss of the services of any executive or key employee might impede the progress of our research, development and commercialization objectives.
We may need to expand our organization and may experience difficulties in managing this growth, which could disrupt our operations.
As of June 30, 2017 we had 63 employees. In connection with our May 2017 corporate restructuring plan, we committed to a reduction in our total workforce by approximately 30% percent to 65 employees. In the future we may expand our employee base to increase our managerial, scientific, operational, commercial, financial and other resources and to hire more consultants and contractors. Future growth would impose significant additional responsibilities on our management, including the need to identify, recruit, maintain, motivate and integrate additional employees, consultants and contractors. Also, our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. Moreover, if our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate and/or grow revenues could be reduced, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize product candidates and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
22
We may undertake internal restructuring activities in the future that could result in disruptions to our business or otherwise materially harm our results of operations or financial condition.
From time to time we may undertake internal restructuring activities as we continue to evaluate and attempt to optimize our cost and operating structure in light of developments in our business strategy and long-term operating plans. For example, we initiated a corporate restructuring in May 2017 that we expect to result in a reduction in our workforce. Any such restructuring activities may result in write-offs or other restructuring charges. There can be no assurance that any restructuring activities that we have undertaken or undertake in the future will achieve the cost savings, operating efficiencies or other benefits that we may initially expect. Restructuring activities may also result in a loss of continuity, accumulated knowledge and inefficiency during transitional periods and thereafter. In addition, internal restructurings can require a significant amount of time and focus from management and other employees, which may divert attention from commercial operations. If any internal restructuring activities we have undertaken or undertake in the future fail to achieve some or all of the expected benefits therefrom, our business, results of operations and financial condition could be materially and adversely affected.
Certain current and future relationships with customers and third party payors as well as certain of our business operations may be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws and health information privacy and security laws. If we are unable to comply, or have not fully complied, with such laws, we could face criminal sanctions, civil penalties, contractual damages, reputational harm and diminished profits and future earnings.
If we obtain FDA approval for any of our product candidates and begin commercializing those products in the United States, our operations may be directly, or indirectly through our customers, further subject to various federal and state fraud and abuse laws, including, without limitation, the federal Anti-Kickback Statute and the federal False Claims Act. These laws may impact, among other things, our proposed sales, marketing and education programs. In addition, we may be subject to patient privacy regulation by the federal government and by the U.S. states and foreign jurisdictions in which we conduct our business. The healthcare laws and regulations that may affect our ability to operate include:
| • | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs; |
| • | federal civil and criminal false claims laws and civil monetary penalty laws, including the civil False Claims Act, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to the federal government, including Medicare or Medicaid, that are false or fraudulent; |
| • | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes that prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their implementing regulations, which imposes certain requirements on certain types of individuals and entities relating to the privacy, security and transmission of individually identifiable health information; |
| • | the federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or CMS, information related to payments or other transfers of value made to physicians, and further requires applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members; and |
| • | state and foreign law equivalents of each of the above federal laws, such as: anti-kickback and false claims laws which may apply to items or services reimbursed by any third party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. |
23
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, possible exclusion from Medicare, Medicaid and other government healthcare programs, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits and future earnings, and curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Recent and future healthcare legislation may further impact our business operations.
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
We expect that healthcare reform measures that may be adopted in the future may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors.
We cannot predict what healthcare reform initiatives may be adopted in the future. Further federal, state and foreign legislative and regulatory developments are likely, and we expect ongoing initiatives to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop product candidates.
We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our products. Certain oligonucleotide therapeutics have shown injection site reactions and pro-inflammatory effects and may also lead to impairment of kidney or liver function. There is a risk that our current and future product candidates may induce similar adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| • | impairment of our business reputation; |
| • | withdrawal of clinical trial participants; |
| • | costs due to related litigation; |
| • | distraction of management’s attention from our primary business; |
| • | substantial monetary awards to patients or other claimants; |
| • | the inability to commercialize our product candidates; and |
| • | decreased demand for our product candidates, if approved for commercial sale. |
We maintain product liability insurance relating to the use of our therapeutics in clinical trials. However, such insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and in the future we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If and when we obtain marketing approval for product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain product liability insurance on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated adverse effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could adversely affect our results of operations and business.
Cyber security risks and the failure to maintain the confidentiality, integrity, and availability of our computer hardware, software, and Internet applications and related tools and functions could result in damage to our reputation and/or subject us to costs, fines or lawsuits.
24
Our business requires manipulating, analyzing and storing large amounts of data. In addition, we rely on a global enterprise software system to operate and manage our business. We also maintain personally identifiable information about our employees. Our business therefore depends on the continuous, effective, reliable, and secure operation of our computer hardware, software, networks, Internet servers, and related infrastructure. To the extent that our hardware or software malfunctions or access to our data by internal research personnel is interrupted, our business could suffer. The integrity and protection of our employee and company data is critical to our business and employees have a high expectation that we will adequately protect their personal information. The regulatory environment governing information, security and privacy laws is increasingly demanding and continues to evolve. Maintaining compliance with applicable security and privacy regulations may increase our operating costs. Although our computer and communications hardware is protected through physical and software safeguards, it is still vulnerable to fire, storm, flood, power loss, earthquakes, telecommunications failures, physical or software break-ins, software viruses, and similar events. These events could lead to the unauthorized access, disclosure and use of non-public information. The techniques used by criminal elements to attack computer systems are sophisticated, change frequently and may originate from less regulated and remote areas of the world. As a result, we may not be able to address these techniques proactively or implement adequate preventative measures. If our computer systems are compromised, we could be subject to fines, damages, litigation and enforcement actions, and we could lose trade secrets, the occurrence of which could harm our business. In addition, any sustained disruption in internet access provided by other companies could harm our business.
Business interruptions could delay us in the process of developing our future products.
Our headquarters are located in San Diego County. We are vulnerable to natural disasters such as earthquakes and wild fires, as well as other events that could disrupt our operations. We do not carry insurance for earthquakes or other natural disasters and we may not carry sufficient business interruption insurance to compensate us for losses that may occur. Any losses or damages we incur could have a material adverse effect on our business operations.
RISKS RELATED TO OUR COMMON STOCK
The market price of our common stock may be highly volatile.
Since January 1, 2014, our closing stock price as reported on The NASDAQ Global Market has ranged from $0.8604 to $22.08, through July 18, 2017. The trading price of our common stock is likely to continue to be volatile.
Our stock price could be subject to wide fluctuations in response to a variety of factors, including the following:
| • | adverse results or delays in preclinical studies or clinical trials; |
| • | inability to obtain additional funding; |
| • | any delay in filing an IND or NDA for any of our product candidates and any adverse development or perceived adverse development with respect to the FDA’s review of that IND or NDA; |
| • | failure to maintain our existing strategic alliances or enter into new alliances; |
| • | failure of our strategic alliance partners to elect to develop and commercialize product candidates under our alliance agreements or the termination of any programs under our alliance agreements; |
| • | failure by us or our licensors and strategic alliance partners to prosecute, maintain or enforce our intellectual property rights; |
| • | failure to successfully develop and commercialize our product candidates; |
| • | changes in laws or regulations applicable to our preclinical and clinical development activities, product candidates or future products; |
| • | inability to obtain adequate product supply for our product candidates or the inability to do so at acceptable prices; |
| • | adverse regulatory decisions; |
| • | introduction of new products, services or technologies by our competitors; |
| • | failure to meet or exceed financial projections we may provide to the public; |
| • | failure to meet or exceed the estimates and projections of the investment community; |
| • | the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our strategic alliance partners or our competitors; |
25
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| • | additions or departures of key scientific or management personnel; |
| • | significant lawsuits, including patent or stockholder litigation; |
| • | changes in the market valuations of similar companies; |
| • | sales of our common stock by us or our stockholders in the future; and |
| • | trading volume of our common stock. |
In addition, companies trading in the stock market in general, and The NASDAQ Global Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
We are an “emerging growth company,” and the reduced reporting requirements applicable to emerging growth companies could make our common stock less attractive to investors.
We are currently an “emerging growth company,” as defined in the JOBS Act. As an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We will likely be an “emerging growth company” through December 31, 2017, at which time we will lose that status. We cannot predict if investors will find our common stock less attractive because we may rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
Under the JOBS Act, emerging growth companies can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
The requirements of being a publicly traded company may strain our resources and divert management’s attention.
As a publicly traded company, we have incurred, and will continue to incur, significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and The NASDAQ Global Market have imposed various requirements on public companies. In July 2010, the Dodd-Frank Wall Street Reform and Consumer Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation related provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access. As an “emerging growth company” we are permitted to implement many of these requirements over a longer period and up to five years from the pricing of our initial public offering. We have taken advantage of this new legislation but cannot guarantee that we will not be required to implement these requirements sooner than budgeted or planned and thereby incur unexpected expenses. Stockholder activism, the current political environment and the current high level of government intervention and regulatory reform may lead to substantial new regulations and disclosure obligations, which may lead to additional compliance costs and impact the manner in which we operate our business in ways we cannot currently anticipate. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect these rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance and we may be required to incur substantial costs to maintain our current levels of such coverage.
26
Sales of a substantial number of shares of our common stock in the public market by our existing stockholders could cause our stock price to fall.
If our existing stockholders sell, or indicate an intention to sell, substantial amounts of our common stock in the public market, the trading price of our common stock could decline. In addition, shares of common stock that are either subject to outstanding options or reserved for future issuance under our employee benefit plans are or may become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules and Rule 144 under the Securities Act. If these additional shares of common stock are sold, or if it is perceived that they will be sold, in the public market, the trading price of our common stock could decline.
Certain holders of our securities are entitled to rights with respect to the registration of their shares under the Securities Act. Registration of these shares under the Securities Act would result in the shares becoming freely tradable without restriction under the Securities Act, except for shares held by our affiliates as defined in Rule 144 under the Securities Act. Pursuant to our registration statements on Form S-3 which became effective in April 2014 and in April 2015, up to 1,272,000 shares held by certain of our stockholders remain available for resale thereunder. We may file additional registration statements in the future to provide for the further sale of shares of common stock by our stockholders. Any further sales of securities by these stockholders could have a material adverse effect on the trading price of our common stock.
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time, any of which may result in material dilution to investors and/or our existing stockholders. New investors could also be issued securities with rights superior to those of our existing stockholders.
Pursuant to our 2012 Equity Incentive Plan, or the 2012 Plan, our management is authorized to grant stock options and other equity-based awards to our employees, directors and consultants. The number of shares available for future grant under the 2012 Plan will automatically increase each year by up to 4% of all shares of our capital stock outstanding as of December 31st of the preceding calendar year, subject to the ability of our board of directors to take action to reduce the size of the increase in any given year. In addition, we may grant or provide for the grant of rights to purchase shares of our common stock pursuant to our 2012 Employee Stock Purchase Plan, or the ESPP. The number of shares of our common stock reserved for issuance under the ESPP will automatically increase on January 1 of each calendar year by the lessor of 1% of the total number of shares of our common stock outstanding on December 31st of the preceding calendar year and 500,000 shares, subject to the ability of our board of directors to take action to reduce the size of the increase in any given year. Any such increase, of the maximum amount or a lesser amount, may cause our stockholders to experience additional dilution, which could cause our stock price to fall. Currently, we plan to register the increased number of shares available for issuance under the 2012 Plan and the ESPP each year.
In addition, we previously adopted an Inducement Plan pursuant to which our management had the ability to grant stock options exercisable for up to an aggregate of 1,000,000 shares of our common stock to new employees as inducements material to such new employees entering into employment with us. The number of shares which may be granted under the Inducement Plan may be increased in the future by our board of directors. In the event we increase the number of shares which may be granted under the Inducement Plan, or adopt another inducement plan for which no stockholder approval is required under applicable rules and regulations, and grant options pursuant to such plan, our stockholders may experience additional dilution, which could cause our stock price to fall.
We may be unable to comply with the applicable continued listing requirements of The NASDAQ Global Market.
Our common stock is currently listed on The NASDAQ Global Market, or NASDAQ. In order to maintain this listing, we must satisfy minimum financial and other continued listing requirements and standards, including a minimum closing bid price requirement for our common stock of $1.00 per share. There can be no assurance that we will be able to comply with the applicable listing standards. For example, if we were to fail to meet the minimum bid price requirement for 30 consecutive business days, we could become subject to delisting. Although NASDAQ may provide us with a compliance period in which to regain compliance with the minimum bid price requirement, we cannot assure you that we would be able to regain compliance within the period provided by NASDAQ. In order to regain compliance with such requirement, the closing bid price of our common stock would need to meet or exceed $1.00 per share for at least 10 consecutive business days during the compliance
27
period. If we were not able to regain compliance within the allotted compliance period for this requirement or any other applicable listing standard, including any extensions that may be granted by NASDAQ, our shares of common stock would be subject to delisting. In the event that our common stock is delisted from NASDAQ and is not eligible for quotation or listing on another market or exchange, trading of our common stock could be conducted only in the over-the-counter market or on an electronic bulletin board established for unlisted securities such as the Pink Sheets or the OTC Bulletin Board. In such event, it could become more difficult to dispose of, or obtain accurate price quotations for our common stock and there would likely also be a reduction in our coverage by securities analysts and the news media, which could cause the price of our common stock to decline further.
We are the subject of a putative securities class action lawsuit, and additional securities litigation may be brought against us in the future.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because pharmaceutical companies have experienced significant stock price volatility in recent years. On January 31, 2017, a putative class action complaint was filed in the United States District Court for the Southern District of California against us, Paul C. Grint (our former Chief Executive Officer), and Joseph P. Hagan (then our Chief Operating Officer and currently our Chief Executive Officer). The complaint includes claims asserted, on behalf of certain purchasers of our securities, under Sections 10(b) and 20(a) of the Securities Exchange Act of 1934, as amended. In general, the complaint alleges that between January 21, 2016, and June 27, 2016, the defendants violated the federal securities laws by making materially false and misleading statements regarding our business and the prospects for RG-101, thereby artificially inflating the price of our securities. A second action has subsequently been filed making the same allegations but extending the period of alleged violations to January 27, 2017 and also naming our Chief Research & Development Officer, Timothy Wright as a defendant. The plaintiffs seeks unspecified monetary damages and other relief. It is possible that additional lawsuits will be filed, or allegations made by stockholders, with respect to these same or other matters and also naming us and/or our officers and directors as defendants. Due to the early stage of these proceedings, we are not able to predict or reasonably estimate the ultimate outcome or possible losses relating to these claims. While we carry liability insurance, there is no assurance that any losses we incur in connection with the current lawsuits or any future lawsuits will be covered or that coverage, if any, will be sufficient. In addition, the current lawsuits and similar future litigation could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change,” generally defined as a greater than 50% change (by value) in its equity ownership over a three year period, the corporation’s ability to use its pre-change net operating loss carryforwards, or NOLs, and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We triggered an “ownership change” limitation at the completion of our initial public offering in October 2012 and again in July 2015. We may also experience ownership changes in the future as a result of subsequent shifts in our stock ownership. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to offset U.S. federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividends on our common stock. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. In addition, our ability to pay cash dividends is currently prohibited by the terms of our secured debt, and any future debt financing arrangement may contain terms prohibiting or limiting the amount of dividends that may be declared or paid on our common stock. Any return to stockholders will therefore be limited to the appreciation of their stock.
Provisions in our amended and restated certificate of incorporation and bylaws, as well as provisions of Delaware law, could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our stockholders or remove our current management.
Some provisions of our charter documents and Delaware law may have anti-takeover effects that could discourage an acquisition of us by others, even if an acquisition would be beneficial to our stockholders and may prevent attempts by our stockholders to replace or remove our current management. These provisions include:
28
| • | authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
| • | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
| • | eliminating the ability of stockholders to call a special meeting of stockholders; |
| • | establishing the state of Delaware as the sole forum for certain legal actions against the Company, its officers and directors; and |
| • | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. |
In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by our board of directors. This provision could have the effect of delaying or preventing a change in control, whether or not it is desired by or beneficial to our stockholders. Further, other provisions of Delaware law may also discourage, delay or prevent someone from acquiring us or merging with us.
29