Attached files
| file | filename |
|---|---|
| EX-3.1 - COMPREHENSIVE ARTICLES AND CERTIFICATES OF INCORPORATION - Avid Bioservices, Inc. | peregrine_ex0301.htm |
| EX-32 - CERTIFICATION - Avid Bioservices, Inc. | peregrine_ex32.htm |
| EX-31.2 - CERTIFICATION - Avid Bioservices, Inc. | peregrine_ex3102.htm |
| EX-31.1 - CERTIFICATION - Avid Bioservices, Inc. | peregrine_ex3101.htm |
| EX-23.1 - CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM - Avid Bioservices, Inc. | peregrine_ex2301.htm |
| EX-21 - SUBSIDIARIES - Avid Bioservices, Inc. | peregrine_ex21.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended April 30, 2017
OR
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from _________to __________
Commission file number: 001-32839
PEREGRINE PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its charter)
| Delaware | 95-3698422 |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
| 14282 Franklin Avenue, Tustin, California | 92780 |
| (Address of principal executive offices) | (Zip Code) |
| (714) 508-6000 | |
| (Registrant’s telephone number, including area code) | |
| Securities registered pursuant to Section 12(b) of the Act: | |
| Title of Each Class | Name of Each Exchange on Which Registered |
|
Common Stock ($0.001 par value per share) Preferred Stock Purchase Rights 10.50% Series E Convertible Preferred Stock ($0.001 par value per share)
|
The NASDAQ Stock Market LLC
The NASDAQ Stock Market LLC |
| Securities registered pursuant to Section 12(g) of the Act: | |
| None | |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (check one):
| Large accelerated filer o | Accelerated filer x | Non-accelerated filer o (Do not check if a smaller reporting company) |
Smaller reporting company o |
| Emerging growth company o |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
The aggregate market value of the common stock held by non-affiliates as of October 31, 2016 was $84,450,000.
Number of shares of common stock outstanding as of July 10, 2017: 45,069,188
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this report incorporates certain information by reference from the registrant’s proxy statement for the annual meeting of stockholders, which proxy statement will be filed no later than 120 days after the close of the registrant’s fiscal year ended April 30, 2017.
PEREGRINE PHARMACEUTICALS, INC.
Fiscal Year 2017
Annual Report on Form 10-K
| i |
In this Annual Report on Form 10-K (the “Annual Report”), unless the context otherwise indicates, the terms “we,” “us,” “our,” “Company” and “Peregrine” refer to Peregrine Pharmaceuticals, Inc., and our wholly-owned subsidiary, Avid Bioservices, Inc. (“Avid”). This Annual Report contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that involve risks and uncertainties. The inclusion of forward-looking statements should not be regarded as a representation by us or any other person that the objectives or plans will be achieved because our actual results may differ materially from any forward-looking statement. The words “may,” “should,” “plans,” “believe,” “anticipate,” “estimate,” “expect,” their opposites and similar expressions are intended to identify forward-looking statements, but the absence of these words does not necessarily mean that a statement is not forward-looking. We caution readers that such statements are not guarantees of future performance or events and are subject to a number of factors that may tend to influence the accuracy of the statements, including but not limited to, those risk factors outlined in the section titled “Risk Factors” as well as those discussed elsewhere in this Annual Report. You should not duly rely on these forward-looking statements, which speak only as of the date of this Annual Report. We undertake no obligation to publicly revise any forward-looking statement to reflect circumstances or events after the date of this Annual Report or to reflect the occurrence of unanticipated events. You should, however, review the factors and risks we describe in the reports that we file from time to time with the Securities and Exchange Commission (“SEC”) after the date of this Annual Report.
Our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and all amendments to those reports filed with or furnished to the SEC are available, free of charge, through our website at www.peregrineinc.com as soon as reasonably practicable after such reports are electronically filed with or furnished to the SEC. The information on, or that can be accessed through, our website is not part of this Annual Report.
Peregrine® and Avid Bioservices® are registered trademarks of Peregrine Pharmaceuticals, Inc. All other brand names or trademarks appearing in this Annual Report are the property of their respective holders.
| Item 1. | Business |
Overview
We were originally incorporated in the State of California in June 1981 and reincorporated in the State of Delaware on September 25, 1996. Our principal executive offices are located at 14282 Franklin Avenue, Tustin, California, 92780 and our telephone number is (714) 508-6000. Our Internet website addresses are www.peregrineinc.com and www.avidbio.com. Information contained on, or accessed through, our websites does not constitute any part of this Annual Report.
We are a biopharmaceutical company committed to improving the lives of patients by manufacturing high quality pharmaceutical products through our contract manufacturing business, Avid Bioservices, Inc. (“Avid”), and by advancing and licensing our novel, development-stage immunotherapy product through our research and development business, Peregrine Pharmaceuticals, Inc. (“Peregrine”).
Peregrine is focused primarily on the research and development of immune stimulating therapies for the treatment of various cancers. Avid, a wholly-owned subsidiary of Peregrine, is our contract development and manufacturing organization (“CDMO”) business.
In June 2016, following the discontinuation of our bavituximab Phase III SUNRISE trial, we announced a shift in our corporate focus toward achieving profitability within two (2) years (during the quarter ending July 31, 2018). This was to be achieved by focusing on revenue growth from our contract manufacturing business, Avid, while also reducing our spending on research and development while continuing to collect and evaluate data from the Phase III SUNRISE trial. We believe we made significant progress towards this goal during the fiscal year ended April 30, 2017 by reducing research and development expenses by fifty-two percent (52%) while simultaneously growing Avid revenues by thirty percent (30%), as compared to the prior fiscal year. The result was a reduction of our net loss by 49% to $28,159,000 for the fiscal year ended April 30, 2017.
| 1 |
While reducing research and development expenses over the past fiscal year, we have also been able to allow the clinical data from the Phase III SUNRISE trial to mature, enabling us to conduct and present data analysis at key scientific meetings that support the clinical development of bavituximab with immune checkpoint inhibitors such as anti-PD-1/PD-L1 therapies. While we currently expect to reduce research and development spending by at least 40% in fiscal year 2018 compared to fiscal year 2017, this reduction in spending will not allow us to advance the bavituximab clinical program through a company sponsored trial. Instead, we are collaborating with leading immunotherapy experts and institutions to continue to drive forward our research and further guide our clinical development program.
With respect to our CDMO business, fiscal year 2017 was a record year for revenues, topping $57 million and representing 30% revenue growth over the prior fiscal year. While we are pleased at the continued year over year revenue growth, we have also recently seen unanticipated decreases in manufacturing demand from our largest customer and a recent regulatory filing delay from our second largest customer which will have some impact on our ability to grow the revenues from our CDMO business in fiscal year 2018 and could impact our ability to achieve overall profitability by the quarter ending July 31, 2018. However, we believe this to be temporary delay in revenue growth during fiscal year 2018 and have recently secured four new customers and are continuing to focus on securing additional customer business in order to better diversify our customer base. Our goal is to maintain profitability for Avid over the short term while positioning the business for long-term growth and attracting the resources necessary to continue to advance our promising research and development efforts.
Importantly, we continually evaluate various strategic options that we believe may enable us to enhance stockholder value, including the possible separation of these two distinct businesses.
Avid—Our CDMO Business
Avid provides fully-integrated current good manufacturing practices (“cGMP”) services from cell line development to commercial biomanufacturing of large molecules, such as monoclonal antibodies and recombinant proteins for third-party customers while also supporting Peregrine’s internal drug development business. We believe this integration offers considerable time and cost efficiencies for our internal drug development business.
We have been developing and manufacturing biologics since 1993 in our Franklin biomanufacturing facility (the “Franklin Facility”) located at our current headquarters in Tustin, California and formed Avid in 2002 to offer these services to third-party customers. These services include cGMP clinical and commercial product manufacturing utilizing stainless steel and single use bioreactor technology, purification, bulk packaging, stability testing and regulatory strategy and support. Avid also provides a variety of process development activities, including cell line development and optimization, cell culture and feed optimization, analytical methods development and product characterization.
In March 2016, Avid expanded its manufacturing capacity through the launch of its Myford biomanufacturing facility (the “Myford Facility”), which doubled our manufacturing capacity. The 42,000 square foot facility, which is our second manufacturing facility, can accommodate single-use bioreactors up to the 2,000-liter manufacturing scale. The Myford Facility was designed to accommodate a fully disposable manufacturing process for products in late stage clinical development to commercial. To date, the Myford Facility has been utilized to complete a number of process validation runs for our third-party customers, which may lead to future commercial production, and has supported the process validation of our lead immunotherapy candidate, bavituximab. The Myford Facility is located adjacent to our Franklin Facility.
As we look to expand the manufacturing capacity of our CDMO business, in February 2017, we leased an additional 42,000 square feet of vacant warehouse space within the same building as our existing Myford Facility. The proximity of this space will allow us to utilize existing manufacturing infrastructure that we believe should enhance our manufacturing efficiencies and reduce the overall cost and timeframe to construct a third biomanufacturing facility. Although we previously anticipated that the new manufacturing facility would be constructed and ready for manufacturing activities by mid-calendar year 2018, due to unanticipated changes in and/or timing of customer demand (as discussed above), we have decided to defer construction of this third facility until demand from existing or potential new customers is expected to exceed the current manufacturing capacity at our Franklin Facility and Myford Facility. Additionally, commencement of construction is also subject to our ability to raise sufficient additional capital to support this expansion effort. As a result, we presently do not expect to commence construction of this third facility prior to April 30, 2018.
| 2 |
To date, Avid has been audited and qualified by large and small, domestic and foreign, biotechnology companies interested in the production of biologic material for clinical and commercial use. Additionally, Avid has been audited by several regulatory agencies, including the U.S. FDA, the European Medicines Agency, the Brazilian Health Surveillance Agency (ANVISA), the Canadian Health Authority and the California Department of Health.
Fiscal Year 2018 Key Objectives—Our CDMO Business
Our key objectives for our contract manufacturing business in the coming fiscal year include:
| · | Expand our manufacturing capacity through the installation and validation of two 2,000 liter single use bioreactors in our Myford Facility to support the anticipated needs of a current customer; and |
| · | Continue to diversify our customer base by securing additional customers to support our future revenue growth beyond fiscal year 2018. |
Peregrine—Our Research and Development Business
Peregrine is developing therapeutics designed to fight cancer by reversing the immunosuppressive environment that tumors establish in order to proliferate. By doing so, these therapeutics allow the immune system to recognize and destroy tumor cells.
Bavituximab is our lead immunotherapy candidate and currently we are collaborating with the National Comprehensive Cancer Network® (“NCCN”) and Memorial Sloan Kettering Cancer Center (“MSKCC”), to evaluate the potential of bavituximab in combination with immune stimulating therapies.
Bavituximab is a monoclonal antibody that targets and binds to phosphatidylserine (“PS”), a highly immunosuppressive molecule that is usually located inside the membrane of healthy cells, but then “flips” and becomes exposed on the outside of cells in the tumor microenvironment, causing the tumor to evade immune detection. Bavituximab targets and binds to PS to block this immunosuppressive pathway and simultaneously activates adaptive immunity, thereby enabling the immune system to recognize and fight the tumor.
Clinical Development Strategy
In June 2016, we announced a clinical development strategy focused on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies. These trials may be conducted independently, in conjunction with our collaborators, or through investigator sponsored trials (“ISTs”). The goal of these trials is to generate compelling clinical and translational data demonstrating bavituximab’s immunotherapeutic mechanism of action in a combination treatment setting. Additionally, we are in the process of completing an extensive review and analysis of the available data from the discontinued Phase III SUNRISE trial discussed below and testing the numerous collected samples for potential biomarkers in order to determine if certain subgroups may have benefited more from the bavituximab treatment. We believe such information will assist us in guiding the bavituximab clinical program. In keeping with this strategy, we are evaluating options to advance clinical development of bavituximab in an efficient and cost-effective way.
We believe this strategy will allow us to (i) continue our research and development activities while avoiding costly, later stage clinical trials, thereby allowing us to achieve profitability sooner, and (ii) generate additional data that we believe, if positive, could create future potential value, including attracting potential licensing partners or re-engaging our collaboration with AstraZeneca.
| 3 |
NCCN Collaboration
In January 2016, we announced that we entered into a research collaboration with NCCN, a not-for-profit alliance of 27 of the world’s leading cancer centers, to expand the clinical research and development of bavituximab for the treatment of a range of tumors. Under this research collaboration, we are funding three ISTs and related correlative studies with bavituximab at NCCN member institutions and their affiliate community hospitals through a $2 million research grant to NCCN’s Oncology Research Program. NCCN is responsible for oversight and monitoring of the three clinical studies awarded under the research grant. In September 2016, NCCN announced that investigators at the following NCCN-affiliated institutions received the grant award:
| 1. | Moffitt Cancer Center—A Phase I Trial of Sorafenib and Bavituximab Plus Stereotactic Body Radiation Therapy for Unresectable Hepatitis C Associated Hepatocellular Carcinoma. This trial is open for enrollment. |
| 2. | Massachusetts General Hospital Cancer Center—Phase I/II Clinical Trial of Bavituximab with Radiation and Temozolomide for Patients with Newly Diagnosed Glioblastoma. This trial is open for enrollment. |
| 3. | The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins—Phase II Study of Pembrolizumab and Bavituximab for Progressive Recurrent/Metastatic Squamous Cell Carcinoma of the Head and Neck. We expect this trial to be initiated by the end of the calendar year 2017. |
MSKCC Collaboration
In May 2015, we entered into a sponsored research agreement with investigators at MSKCC to identify effective treatment combinations based on our PS-targeting agents, including bavituximab, with other checkpoint inhibitors or immune stimulating agents that will further guide our clinical development program.
In November 2016, we announced initial results from our collaboration with the investigators at MSKCC at the Society for Immunotherapy of Cancer (SITC) Annual Meeting. Study results highlighted that PS-targeting antibodies similar to bavituximab synergize with checkpoint inhibitors and radiation to improve anti-tumor activity in various animal tumor models.
A team of MSKCC researchers evaluated the effects of combining PS-targeting, anti-PD-1 and radiation therapies in the mouse B16 melanoma model. Study data showed that PS-targeting antibodies synergize with both anti-PD-1 and radiation therapy to improve anti-cancer activity. PS-targeting treatment in combination with radiation, as well as triple combination of PS-targeting treatment, anti-PD-1 and radiation, led to a reduction in tumor burden. Median survival for the triple combination treatment still had not been reached at the end of the 80-day observation period, while other arms in the study showed median survival that ranged from 24–70 days.
Researchers also evaluated the impact of the PS-targeting and radiation combination treatment on the level and type of immune activity. These results demonstrated that the combination led to a change in the tumor microenvironment, shifting it from immunosuppressive in which tumors are protected to immune-active in which tumors are more susceptible to treatment. Analysis of local immune responses in the tumors of the treated animals showed that the combination treatment increased the number of tumor associated macrophages and shifted the macrophage polarization from the immunosuppressive M2 type to the immune active M1 type. When systemic immune responses were analyzed following the triple combination of PS-targeting, anti-PD-1 and radiation therapies, researchers also saw evidence of increased immune activity. This was illustrated by key indicators of immune activity, including increases in CD8+ T–cell activation, effector cytokine production and differentiation into effector memory cells.
In April 2017, at the Annual Meeting of the American Association for Cancer Research (“AACR”), researchers from MSKCC presented preclinical study results highlighting the potential of our PS-targeting antibodies to enhance the anti-tumor activity of adoptive T cell transfer therapy without triggering off-target toxicities. The researchers evaluated and compared the anti-tumor activity and off-target toxicities of adoptive T cell transfer therapy in combination with either PS-targeting antibodies or anti-OX40 antibodies in mice with advanced melanomas. Whereas PS-targeting and anti-OX40 demonstrated comparable tumor regression when administered in combination with transferred adoptive T cells, only the PS-targeting combination achieved these results without any detectable off-target toxicities. By contrast, the anti-OX40 treatment combination triggered off-target inflammatory destruction of healthy tissues. Additional study results demonstrated that the PS-targeting antibodies decreased tumor-induced immunosuppression as evidenced by a decrease in immunosuppressive regulatory T cells and M2 macrophages.
| 4 |
The preclinical data from both of these studies has further supported our belief that bavituximab modulates the immunosuppressive tumor microenvironment and enhances the activity of other immunotherapy agents. MSKCC continues to evaluate bavituximab with other immunotherapy agents, which will further guide our clinical development program.
Phase III SUNRISE Trial
In December 2013, we initiated a randomized, double-blind, placebo-controlled Phase III trial evaluating bavituximab plus docetaxel, versus docetaxel plus placebo, for the treatment of previously-treated NSCLC (the “Phase III SUNRISE trial”).
In February 2016, we announced that we were discontinuing the Phase III SUNRISE trial based on the recommendation of the study’s Independent Data Monitoring Committee following a pre-specified interim analysis performed after 33% of targeted overall events (patient deaths). Results of the analysis demonstrated that the patients treated in the bavituximab plus docetaxel treatment arm did not show a sufficient improvement in overall survival as compared to the patients treated in the docetaxel plus placebo treatment arm to warrant continuation of the study. Patient enrollment was discontinued and existing patients in the trial were given the choice to continue chemotherapy and/or bavituximab, as appropriate. Patient treatment and follow-up data from the trial were collected through March 2017 and the trial is now closed.
Following the discontinuation of the Phase III SUNRISE trial, we promptly initiated and are nearing completion of an extensive review and analysis of the available data and testing the numerous collected samples for potential biomarkers in order to understand what subgroups may have benefited more from the bavituximab treatment. We believe such information will be important in supporting and refining our clinical strategy, as discussed above.
The most compelling data to date was presented in April 2017 at the Annual Meeting of the AACR. In a subgroup analysis, we looked at the outcome of 91 patients that were enrolled in the Phase III SUNRISE trial that were subsequently treated with immune checkpoint inhibitors (“ICI’s”) post study treatments. The results from this analysis demonstrated that the patients who received docetaxel plus bavituximab and subsequent ICI’s (anti-PD-1/PD-L1) had not yet reached median overall survival (“mOS”) compared to mOS of 13.0 months for patients who received docetaxel plus placebo (hazard ratio [HR], 0.43; p=0.005). The statistically significant difference between the two arms in the trial provides strong rationale for combining bavituximab with ICI’s and supports the hypothesis that bavituximab may modulate the tumor microenvironment to enhance the anti-tumor activity of ICI’s.
In June 2017, we announced additional supportive data at the Annual Meeting of the American Society of Clinical Oncology (“ASCO”) demonstrating that patients in the bavituximab containing arm who had low baseline PD-L1 expression on tumor cells (i.e., patients typically with poorer response to PD-1/PD-L1 checkpoint inhibitors) lived significantly longer than patients with high baseline PD-L1 expression. These data further support the hypothesis that bavituximab may modulate the tumor microenvironment to complement and enhance the anti-tumor activity of ICI’s.
Antibody Discovery Technology
Recently, our scientists have developed an antibody discovery and characterization platform through which we can generate antibodies against virtually any target. These capabilities are state-of-the-art and meant for rapid screening for high affinity antibodies as drug candidates. These capabilities are a natural extension of the services we already offer through Avid and it may represent a way to bring in customers at a much earlier stage of development with the potential to move them quickly into process development and cGMP manufacturing. Continuing to diversify our customer base is one of our key initiatives at Avid and we believe the antibody discovery capabilities will help attract new customers. The antibody discovery capabilities can also be used in our research and development business to rapidly identify antibodies against known targets as well as to identify novel targets. The antibody discovery platform is now fully functional and we are in the process of evaluating the technology through identifying several known targets and thus far, the results have looked promising. Once this process is completed, the technology platform will be marketed to potential customers as part of our CDMO service offerings.
| 5 |
Exosome Technology
We licensed from The University of Texas System, on behalf of The University of Texas Southwestern Medical Center (“UTSWMC”) in July 2016, proprietary exosome-based cancer diagnostic technology, with the goal of developing an optimized test for further clinical testing in collaboration with a potential partner. The platform is based on the diagnostic potential of tumor derived exosomes, which are small vesicles from tumor cells that are released into the blood as tumors grow. Tumor derived exosomes have PS on their surface as a potentially detectable marker. It is believed that even small tumors begin to release PS-positive exosomes and thus the ability to detect these exosomes in the blood may be an indicator of the presence of a tumor.
In the study published by Oncotarget, plasma samples from 34 patients with ovarian tumors and 10 healthy subjects were analyzed for the presence of PS-expressing exosomes in a blinded test. Results demonstrated that those patients with malignant ovarian cancer displayed significantly higher blood PS exosome levels than those with benign tumors (median 0.237 vs. -0.027, p=0.0001) and the malignant and benign groups displayed significantly higher blood PS exosome levels than the healthy subjects (median 0.237 vs -0.158, p < 0.0001 and -0.027 vs -0.158, p=0.0002, respectively).
Fiscal Year 2018 Objectives—Our Research and Development Business
Our objectives for our research and development business in the coming fiscal year include:
| · | Determine the optimal path forward for bavituximab based on the final evaluation of all data collected from our completed Phase III SUNRISE trial; |
| · | Support the advancement of our NCCN collaboration and the underlying clinical trials to evaluate bavituximab in combination with other therapies, including immune stimulating therapies; |
| · | Support any new potential ISTs, which could provide further confirmation of bavituximab’s immunotherapeutic mechanism of action in the clinic; |
| · | Generate additional preclinical, translational, and clinical data to further demonstrate the immunotherapeutic mechanism of action of bavituximab as we continue to identify new clinical indications, therapeutic combinations and potential partnerships; and, |
| · | Complete the evaluation process for the antibody discovery platform and begin offering as a CDMO service through Avid Bioservices. |
Reverse Stock Split
On July 7, 2017, we effected a reverse stock split of our outstanding shares of common stock at a ratio of one-for-seven, which took effect with the opening of trading on July 10, 2017. All common stock share and per share amounts in this Annual Report have been adjusted to give effect to the one-for-seven reverse stock split, retrospectively.
In-Licensing Agreements
The following is a summary of our key in-licensing agreements covering bavituximab, our lead immunotherapy product in clinical development.
In August 2001 and August 2005, we exclusively in-licensed the worldwide rights to the PS-targeting technology platform, including bavituximab, from The University of Texas System, on behalf of UTSWMC. In November 2003, we entered into a non-exclusive license agreement with Genentech, Inc. (“Genentech”), to license certain intellectual property rights covering methods and processes for producing antibodies used in connection with the development of our PS-targeting program. In December 2003, we entered into an exclusive commercial license agreement with Avanir Pharmaceuticals, Inc., (“Avanir”) covering the generation of a chimeric monoclonal antibody. In March 2005, we entered into a worldwide non-exclusive license agreement with Lonza Biologics (“Lonza”) for intellectual property and materials relating to the expression of recombinant monoclonal antibodies for use in the manufacture of bavituximab.
| 6 |
Under our in-licensing agreements relating to bavituximab, we are obligated to pay future milestone payments based on potential clinical development and regulatory milestones, plus a royalty on net sales and/or a percentage of sublicense income. The applicable royalty rate under each of the foregoing in-licensing agreements is in the low single digits. We did not incur any milestone related expenses during the three fiscal years ended April 30, 2017.
The following table provides certain information with respect to each of our in-licensing agreements relating to our bavituximab program.
| Licensor | Agreement Date | Total Milestone Obligations Expensed To Date |
Potential | |||||||
| UTSWMC | August 2001 | $ | 173,000 | $ | 300,000 | |||||
| UTSWMC | August 2005 | 85,000 | 375,000 | |||||||
| Lonza | March 2005 | 64,000 | – | (2) | ||||||
| Avanir | December 2003 | 100,000 | 1,000,000 | |||||||
| Genentech | November 2003 | 500,000 | 5,000,000 | |||||||
| Total | $ | 922,000 | $ | 6,675,000 | ||||||
______________
| (1) | Under our current agreements, potential future milestone obligations are due upon achieving certain clinical and regulatory milestones. Based on the current stage of clinical development for bavituximab, future milestone obligations would be due upon submission of a biologics license application in the U.S. and upon FDA approval, which events are currently uncertain and depend on positive clinical trial results. In addition, potential future milestone obligations vary by license agreement (as defined in each license agreement) and certain agreements depend on a valid patent claim, as defined in each of these underlying agreements, at the time the potential milestone is achieved. |
| (2) | In the event we utilize a third-party contract manufacturer other than Lonza to manufacture bavituximab for commercial purposes, we would owe Lonza 300,000 pounds sterling per year. |
We do not expect to incur any milestone related expenses regarding our bavituximab program during fiscal year 2018. In addition, of the total potential future milestone obligations of $6,675,000, up to $6,400,000 would be due upon the first commercial approval of bavituximab pursuant to these in-licensing agreements. However, given the uncertainty of the drug development and the regulatory approval process, we are unable to predict with any certainty when any of these future milestones will occur, if at all.
Government Regulation
Regulation by governmental authorities in the U.S. and other countries is a significant factor in our ongoing research and development activities and in the production of our products under development. Our products and our research and development activities are subject to extensive governmental regulation in the U.S., including the Federal Food, Drug, and Cosmetic Act, as amended, the Public Health Service Act, as amended, as well as to other federal, state and local statutes and regulations. These laws, and similar laws outside the U.S., govern the clinical and non-clinical testing, manufacture, safety, effectiveness, approval, labeling, distribution, sale, import, export, storage, record keeping, reporting, advertising and promotion of our products, if approved. Violations of regulatory requirements at any stage may result in various adverse consequences, including regulatory delay in approving or refusal to approve a product, enforcement actions, including withdrawal of approval, labeling restrictions, seizure of products, fines, injunctions and/or civil or criminal penalties. Any product that we develop must receive all relevant regulatory approvals or clearances before it may be marketed in a particular country.
The regulatory process, which includes extensive preclinical testing and clinical trials of each product candidate to study its safety and efficacy, is uncertain, takes many years and requires the expenditure of substantial resources.
| 7 |
The activities required before a product, such as bavituximab, may be marketed in the U.S. are generally performed in the following sequential steps:
| 1. | Preclinical testing. This generally includes evaluation of our products in the laboratory or in animals to characterize the product and determine safety and efficacy. Some preclinical studies must be conducted by laboratories that comply with FDA regulations regarding good laboratory practice. |
| 2. | Submission to the FDA of an Investigational New Drug (“IND”) application. The results of preclinical studies, together with manufacturing information, analytical data and proposed clinical trial protocols, are submitted to the FDA as part of an IND application, which must become effective before the clinical trials can begin. Once a new IND application is filed, the FDA has 30 days to review the IND application. The IND application will automatically become effective 30 days after the FDA receives the application, unless the FDA indicates prior to the end of the 30-day period that the application raises concerns that must be resolved to the FDA’s satisfaction before clinical trials may proceed. If the FDA raises concerns at any time, we may be unable to resolve the issues in a timely fashion, if at all. |
| 3. | Completion of clinical trials. Human clinical trials are necessary to seek approval for a new drug or biologic and typically involve a three-phase process. In Phase I, small clinical trials are generally conducted to determine the safety of the product. In Phase II, clinical trials are generally conducted to assess safety and acceptable dose and gain preliminary evidence of the efficacy of the product. In Phase III, clinical trials are generally conducted to provide sufficient data for the statistically valid proof of safety and efficacy. A clinical trial must be conducted according to good clinical practices under protocols that detail the trial’s objectives, inclusion and exclusion criteria, the parameters to be used to monitor safety and the efficacy criteria to be evaluated, and informed consent must be obtained from all study subjects. Each protocol involving U.S. trial sites must be submitted to the FDA as part of the IND application. The FDA may impose a clinical hold on an ongoing clinical trial if, for example, safety concerns arise, in which case the study cannot recommence without FDA authorization under terms sanctioned by the FDA. Similarly, trials conducted outside the U.S. require notification and/or approval by the governing health authority. In addition, before a clinical trial can be initiated, each clinical site or hospital administering the product must have the protocol reviewed and approved by an institutional review board (“IRB”) or independent ethics committee (“IEC”). The IRB/IEC will consider, among other things, ethical factors and the safety of human subjects. The IRB/IEC may require changes in a protocol, which may delay initiation or completion of a study. Phase I, Phase II or Phase III clinical trials may not be completed successfully within any specific period of time, if at all, with respect to any of our potential products. Furthermore, we, the governing health authority (including the FDA) or an IRB/IEC may suspend a clinical trial at any time for various reasons, including a finding that patients are being exposed to an unacceptable health risk. |
| 4. | Submission to the FDA of a Biologics License Application (“BLA”) or New Drug Application (“NDA”). After completion of clinical studies for an investigational product, a BLA or NDA is submitted to the FDA for product marketing approval. No action can be taken to market any new drug or biologic product in the U.S. until the FDA has approved an appropriate marketing application. |
| 5. | FDA review and approval of the BLA or NDA before the product is commercially sold or shipped. The results of preclinical studies, clinical trials and manufacturing information are submitted to the FDA in the form of a BLA or NDA for approval to manufacture, market and ship the product for commercial use. The FDA may take a number of actions after the BLA or NDA is filed, including but not limited to, denying the BLA or NDA if applicable regulatory criteria are not satisfied, requiring additional clinical testing or information, or requiring post-market testing and surveillance to monitor the safety or efficacy of the product. Adverse events that are reported after marketing approval can result in additional limitations being placed on the product’s use and, potentially, withdrawal of the product from the market. Any adverse event, either before or after marketing approval, can result in product liability claims against us. |
| 8 |
In addition, we are subject to regulation under state, federal, and international laws and regulations regarding occupational safety, laboratory practices, environmental protection and hazardous substance control, and other regulations. Our clinical trial and research and development activities involve the controlled use of hazardous materials, chemicals and radioactive compounds. Although we believe that our safety procedures for handling and disposing of such materials comply with the standards prescribed by state and federal regulations, the risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result and any such liability could exceed our financial resources. In addition, disposal of radioactive materials used in our clinical trials and research efforts may only be made at approved facilities. We believe that we are in material compliance with all applicable laws and regulations including those relating to the handling and disposal of hazardous and toxic waste.
Our product candidates, if approved, may also be subject to import laws in other countries, the food and drug laws in various states in which the products are or may be sold and subject to the export laws of agencies of the U.S. government.
In addition, we must also adhere to cGMP and product-specific regulations enforced by the FDA through its facilities inspection program. Failure to comply with manufacturing regulations can result in, among other things, warning letters, fines, injunctions, civil penalties, recall or seizure of products, total or partial suspension of production, refusal of the government to renew marketing applications and criminal prosecution.
Manufacturing and Raw Materials
We manufacture cGMP pharmaceutical-grade products for our customers and to supply our clinical trials through our wholly-owned subsidiary, Avid. The process for manufacturing generally uses commercially available raw materials from multiple suppliers, and in some instances, from a sole source supplier. We currently do not have long-term supply contracts with these suppliers, and accordingly, we may experience delays in receiving raw materials to support the manufacturing of these cGMP pharmaceutical-grade products. However, to date, we have not experienced any significant difficulty in obtaining these raw materials.
Patents and Trade Secrets
We continue to seek patents on inventions originating from our ongoing research and development activities and in collaboration with other companies and university researchers. In addition to seeking patent protection in the U.S., we typically file patent applications in Europe, Canada, Japan and additional countries on a selective basis. Patents, issued or applied for, cover inventions relating in general to cancer therapy and anti-viral therapy and in particular to different proteins, peptides, antibodies and conjugates, methods and devices for labeling antibodies, and therapeutic and diagnostic uses of the peptides, antibodies and conjugates. We intend to pursue opportunities to license these technologies and any advancements or enhancements, as well as to pursue the incorporation of our technologies in the development of our own products.
Our issued patents extend for varying periods according to the date of patent application filing and/or grant and the legal term of patents in the various countries where patent protection is obtained. In the U.S., patents issued on applications filed prior to June 8, 1995 have a term of 17 years from the issue date or 20 years from the earliest effective filing date, whichever is longer. U.S. patents issued on applications filed on or after June 8, 1995, have a term first calculated as 20 years from the earliest effective filing date, not counting any provisional application filing date. Certain U.S. patents issued on applications filed on or after June 8, 1995, and particularly on applications filed on or after May 29, 2000, are eligible for Patent Term Adjustment, which extends the term of the patent to compensate for delays in examination at the U.S. Patent and Trademark Office. The term of foreign patents varies in accordance with provisions of applicable local law, but is typically 20 years from the effective filing date, which is often the filing date of an application under the provisions of the Patent Cooperation Treaty.
In addition, in certain cases, the term of U.S. and foreign patents can be extended to recapture a portion of the term effectively lost as a result of health authority regulatory review. As such, certain U.S. patents may be eligible for Patent Term Extension under 35 U.S.C. § 156 (known as “the Hatch-Waxman Act”) to restore the portion of the patent term that has been lost as a result of review at the U.S. FDA. Such extensions, which may be up to a maximum of five years (but cannot extend the remaining term of a patent beyond a total of 14 years), are potentially available to one U.S. patent that claims an approved human drug product (including a human biological product), a method of using a drug product, a method of manufacturing a drug product, or a medical device.
| 9 |
We consider that in the aggregate our patents, patent applications and licenses under patents owned by third parties are of material importance to our operations. Of the patent portfolios that are owned, controlled by or exclusively licensed to us, those concerning our PS-targeting technology platform, including bavituximab are of particular importance to our operations and our clinical pipeline.
Our patent portfolios relating to the PS-targeting technology platform in oncology include U.S. and foreign patents and patent applications with claims directed to methods, compositions and combinations for targeting tumor vasculature and imaging and treating cancer using antibodies and conjugates that localize to the aminophospholipids, PS (Phosphatidylserine) and PE (Phosphatidylethanolamine), exposed on tumor vascular endothelial cells. These patents are currently set to expire between 2019 and 2021.
Our patent portfolios relating to the PS-targeting technology platform in the viral field include U.S. and foreign patents and patent applications with claims directed to methods, compositions and combinations for inhibiting viral replication or spread and for treating viral infections and diseases using antibodies, certain peptides and conjugates that localize to the aminophospholipids, PS and PE, exposed on viruses and virally-infected cells. Such anti-viral patents concerning antibodies and conjugates are currently set to expire in 2023.
Additionally, we have U.S. and foreign patents and patent applications relating more specifically to our product, bavituximab, including composition of matter, combinations and methods of use in treating angiogenesis and cancer and in treating viral infections and diseases, alone and in combination therapies. These patents that more specifically concern bavituximab compositions and their use in treating cancer, both alone and in combination therapies, are currently set to expire between 2023 and 2025.
The information given above is based on our current understanding of the patents and patent applications that we own, control, or have exclusively licensed. The information is subject to revision, for example, in the event of changes in the law or legal rulings affecting our patents, or if we become aware of new information. In particular, the expiry information given above does not account for possible extension of any U.S. or foreign patent to recapture patent term effectively lost as a result of FDA or other health authority regulatory review. We intend to seek such extensions, as appropriate to approved product(s), which may be up to a maximum of five years (but not extending the term of a patent beyond 14 years).
The actual protection afforded by a patent, which can vary from country to country, depends upon the type of patent, the scope of its coverage and the availability of legal remedies in the country. We have either been issued patents or have patent applications pending that relate to a number of current and potential products including products licensed to others. In general, we have obtained licenses from various parties that we deem to be necessary or desirable for the manufacture, use or sale of our products. These licenses (both exclusive and non-exclusive) generally require us to pay royalties to the parties.
We also own trademarks to protect the names of our products and services. Trademark protection continues in some countries so long as the trademark is used, and in other countries, so long as the trademark is registered. Trademark registration is for fixed terms and can be renewed indefinitely.
With respect to our contract manufacturing business, we have acquired and developed and continue to acquire and develop knowledge and expertise (“know-how”) and trade secrets in the provision of process development and manufacturing services. Our know-how and trade secrets may not be patentable, but they are valuable in that they enhance our ability to provide high-quality services to our customers. We also intend to continue to rely upon trade secrets and improvements, unpatented proprietary know-how, and continuing technological innovation to develop and maintain our competitive position in research and development of our therapeutic and diagnostic products. We typically place restrictions in our agreements with third-parties, which contractually restrict their right to use and disclose any of our proprietary technology with which they may be involved. In addition, we have internal non-disclosure safeguards, including confidentiality agreements, with our employees.
| 10 |
Segment Information
Our business is organized into two reportable operating segments. Peregrine is engaged in the research and development of monoclonal antibodies focused on the treatment of cancer and has not generated any product sales from any of its technologies under development. Our wholly-owned subsidiary, Avid, is engaged in providing fully-integrated cGMP biomanufacturing services for us and its third-party customers. In addition, we had no foreign based operations and no long-lived assets located in foreign countries as of and for the fiscal years ended April 30, 2017, 2016 and 2015. Refer to Note 10, “Segment Reporting” to the accompanying consolidated financial statements for additional financial information regarding our operating segments.
Customers
Contract manufacturing revenue has historically been derived from a small customer base. For the fiscal year ended April 30, 2017, approximately 98% of contract manufacturing revenue was generated from three customers. For the fiscal years ended April 30, 2016 and 2015, approximately 95% and 91%, respectively, of contract manufacturing revenue was generated from two customers. In addition, typically we have not entered into long-term commitments with these third-party customers because their need for product supply depends on a variety of factors, including the products stage of development, the timing of regulatory filings and approvals, the product needs of their collaborators, if applicable, their financial resources and the market demand with respect to commercial products. Our future results of operations could be adversely affected if revenue from any one of our primary customers is significantly reduced or eliminated. Refer to Note 10, “Segment Reporting” to the accompanying consolidated financial statements for additional financial information regarding Avid’s customer concentration and geographic areas of its customers.
Competition
The CDMO and pharmaceutical and biotechnology industries are intensely competitive. Our competition in the CDMO market includes full-service contract manufacturers and large pharmaceutical companies offering third-party manufacturing services to fill their excess capacity. Also, large pharmaceutical companies have been seeking to divest portions of their manufacturing capacity, and any such divested businesses may compete with us in the future. In addition, most of our competitors may have substantially greater financial, marketing, technical or other resources than we do. Moreover, additional competition may emerge and may, among other things, result in a decrease in the fees paid for our services, which would affect our results of operations and financial condition.
Our competition in the pharmaceutical and biotechnology industry includes several pharmaceutical and biotechnology companies actively engaged in research and development of immunotherapy-based products that have commenced clinical trials with, or have successfully commercialized, these products. Some or all of these companies may have greater financial resources, larger technical staffs and larger research budgets than we have, as well as greater experience in developing products and running clinical trials.
With respect to our lead immunotherapy candidate, bavituximab, we are not aware of any other company developing an investigational product similar to bavituximab, however, before we can more clearly define our competition, we will need to generate additional clinical data from our current or potential future planned trials.
Research and Development
The majority of our operating expenses to date have been related to research and development. Research and development expenses primarily include (i) payroll and related costs, including share-based compensation, associated with research and development personnel, (ii) costs related to clinical trials and preclinical testing of our technologies under development, (iii) costs to develop and manufacture the product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses. Research and development expenses are charged to expense as incurred when these expenditures relate to our research and development efforts and have no alternative future uses. Research and development expenses were $28,297,000 in fiscal year 2017, $59,529,000 in fiscal year 2016, and $42,996,000 in fiscal year 2015.
Human Resources
As of April 30, 2017, we employed 319 full-time employees and four part-time employees. None of our employees are covered by a collective bargaining agreement. We have never experienced employment-related work stoppages and consider our employee relations to be good.
| 11 |
| Item 1A. | Risk Factors |
You should carefully consider the risks and uncertainties described below, together with all of the other information contained in this report, including our financial statements and the related notes thereto, before making a decision to invest in our securities. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us, or that we currently believe are not material, also may become important factors that affect us and impair our business operations. The occurrence of any of the events or developments discussed in the risk factors below could have a material and adverse impact on our business, results of operations, financial condition and cash flows, and in such case, our future prospects would likely be materially and adversely affected. If any of such events or developments were to happen, the trading price of our common stock and the value of our 10.50% Series E Convertible Preferred Stock could decline, and you could lose part or all of your investment.
Risks Related to Our Business
If we cannot obtain additional funding, we may have to delay, scale back, or eliminate some or all our research and development efforts, and/or restructure our operations.
At April 30, 2017, we had $46,799,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced losses and negative cash flows from operations since our inception and we expect negative cash flows from operations to continue for the foreseeable future until we can generate sufficient revenue from Avid’s contract manufacturing services to achieve profitability. Therefore, unless and until we are able to generate sufficient revenue from Avid’s contract manufacturing services or from the sale or licensing of our product candidate under development, we expect such losses to continue through at least the fiscal year ending April 30, 2018, and as a result, we will require additional capital to fund our operations and to execute our business plans.
Our ability to continue to fund our operations is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, (i) raising additional capital in the equity markets, (ii) generating additional contract manufacturing revenue from Avid, or (iii) licensing or partnering our product candidate in development.
Historically, we have funded a significant portion of our operations through the issuance of equity. During fiscal year 2017, we raised $31,277,000 in aggregate gross proceeds from the sale of shares of our common stock and raised an additional $1,634,000 in aggregate gross proceeds from the sale of shares of our 10.50% Series E Convertible Preferred Stock (the “Series E Preferred Stock”) (as described in Note 5 to the accompanying consolidated financial statements). Subsequent to April 30, 2017 and through June 30, 2017, we raised an additional $4,304,000 in aggregate gross proceeds from the sale of shares of our common stock (as described in Note 12 to the accompanying consolidated financial statements). As of July 14, 2017, $67,674,000 remained available to us under our effective shelf registration statement, which allows us from time to time to offer and sell shares of our common stock, in one or more offerings, either individually or in combination.
Our ability to raise additional capital in the equity markets to fund our obligations in future periods is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse financial results, and negative research and development results. In addition, on July 7, 2017, we effected a reverse stock split of our issued and outstanding common stock at a ratio of one-for-seven, which took effect with the opening of trading on July 10, 2017 (as described in Note 1 to the accompanying consolidated financial statements). While the reverse stock split resulted in an increase in the market price of our common stock, it also reduced the number of shares outstanding, which could adversely affect the liquidity of our common stock. Because we have predominately raised capital through sales arrangements that are dependent on the trading volume of our common stock, any adverse effect on liquidity could negatively impact our ability to raise additional capital in the equity markets. In such a case, we may not be able to rely on the sale of shares of our common stock to fund our operations to the extent we have in prior years.
| 12 |
With respect to our ability to generate additional contract manufacturing revenue, Avid has a revenue backlog of $58 million under signed contracts from existing customers, which is consistent with the revenue backlog we reported as of April 30, 2016. As such, we may not be able to rely on generating additional contract manufacturing revenue from Avid in fiscal year 2018 to make up any capital raising shortfall experienced through the equity markets.
If we are unable to either (i) raise sufficient capital in the equity markets, (ii) generate additional contract manufacturing revenue from Avid, or (iii) license or partner our product in development, or any combination thereof, we may need to delay, scale back, or eliminate all our research and development efforts, or restructure our operations. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
As a result, we have concluded that there is substantial doubt about our ability to continue as a going concern within one year after the date that our financial statements are issued. Our independent registered public accounting firm included an explanatory paragraph highlighting this uncertainty in its “Report of Independent Registered Public Accounting Firm” dated July 14, 2017, which report is included in Item 15 of Part IV of this Annual Report.
We have had significant losses, anticipate future losses and may never achieve profitability.
We have incurred net losses in most fiscal years since we began operations in 1981. The following table represents net losses incurred for each of the past three fiscal years:
| Net Loss | |
| Fiscal Year 2017 | $ 28,159,000 |
| Fiscal Year 2016 | $ 55,652,000 |
| Fiscal Year 2015 | $ 50,358,000 |
As of April 30, 2017, we had an accumulated deficit of $537,435,000. In addition, we expect negative cash flows from operations to continue for the foreseeable future until we can generate sufficient revenue from Avid’s contract manufacturing services to achieve profitability. Further, if we fail to generate sufficient revenue from Avid’s contract manufacturing services or at all, we may never achieve profitability.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern, and absent additional financing, we may be unable to remain a going concern.
In its report accompanying our audited consolidated financial statements for the fiscal year ended April 30, 2017, our independent registered public accounting firm included an explanatory paragraph stating that our recurring losses from operations and negative cash flows from operating activities raise substantial doubt as to our ability to continue as a going concern. Our financial statements do not include any adjustments that may be necessary in the event we are unable to continue as a going concern. We may need to significantly modify our operation plans in an effort to continue as a going concern. Absent sufficient additional capital, which we may be unable to raise on commercially reasonable terms or at all, we may be unable to remain a going concern.
Failure to comply with existing and future regulatory requirements could adversely affect our business, results of operations and financial condition.
Our industry is highly regulated. We are required to comply with the regulatory requirements of various local, state, provincial, national and international regulatory bodies having jurisdiction in the countries or localities in which we manufacture products or in which our customers’ products are distributed. In particular, we are subject to laws and regulations concerning development, testing, manufacturing processes, equipment and facilities, including compliance with cGMPs, import and export, and product registration and listing, among other things. As a result, most of our facilities are subject to regulation by the FDA, as well as regulatory bodies of other jurisdictions such as the EMEA and/or Health Canada, depending on the countries in which our customers market and sell the products we manufacture on their behalf. As we expand our operations and geographic scope, we may be exposed to more complex and new regulatory and administrative requirements and legal risks, any of which may require expertise in which we have little or no experience. It is possible that compliance with new regulatory requirements could impose significant compliance costs on us. Such costs could have a material adverse effect on our business, financial condition and results of operations.
| 13 |
These regulatory requirements impact many aspects of our operations, including manufacturing, developing, storage, distribution, import and export and record keeping related to customers’ products. Noncompliance with any applicable regulatory requirements can result in government refusal to approve (i) facilities for testing or manufacturing products or (ii) products for commercialization. The FDA and other regulatory agencies can delay, limit or deny approval for many reasons, including:
| · | changes to the regulatory approval process, including new data requirements for product candidates in those jurisdictions, including the United States, in which our customers may be seeking approval; |
| · | that a customer’s product candidate may not be deemed to be safe or effective; |
| · | the ability of the regulatory agency to provide timely responses as a result of its resource constraints; and |
| · | that the manufacturing processes or facilities may not meet the applicable requirements. |
In addition, if new legislation or regulations are enacted or existing legislation or regulations are amended or are interpreted or enforced differently, we may be required to obtain additional approvals or operate according to different manufacturing or operating standards. This may require a change in our development and manufacturing techniques or additional capital investments in our facilities. Any related costs may be significant. If we fail to comply with applicable regulatory requirements in the future, then we may be subject to warning letters and/or civil or criminal penalties and fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, restrictions on the import and export of our products, debarment, exclusion, disgorgement of profits, operating restrictions and criminal prosecution and the loss of contracts and resulting revenue losses. Inspections by regulatory authorities that identify any deficiencies could result in remedial actions, production stoppages or facility closure, which would disrupt the manufacturing process and supply of product to our customers. In addition, such failure to comply could expose us to contractual and product liability claims, including claims by customers for reimbursement for lost or damaged active pharmaceutical ingredients (“APIs”) or recall or other corrective actions, the cost of which could be significant.
In addition, products we manufacture must undergo pre-clinical and clinical evaluations relating to product safety and efficacy before they are approved as commercial therapeutic products. The regulatory authorities having jurisdiction in the countries in which we or our customers intend to market their products may delay or put on hold clinical trials or delay approval of a product or determine that the product is not approvable. The FDA or other regulatory agencies can delay approval of a drug if our manufacturing facility, including any newly commissioned facility, is not able to demonstrate compliance with cGMPs, pass other aspects of pre-approval inspections or properly scale up to produce commercial supplies. The FDA and comparable government authorities having jurisdiction in the countries in which we or our customers intend to market their products have the authority to withdraw product approval or suspend manufacture if there are significant problems with raw materials or supplies, quality control and assurance or the product we manufacture is adulterated or misbranded. If our manufacturing facilities and services are not in compliance with FDA and comparable government authorities, we may be unable to obtain or maintain the necessary approvals to continue manufacturing products for our customers, which would materially adversely affect our results of operations and financial condition.
The failure to receive or maintain regulatory approval for our or our customers’ product candidates could negatively impact our revenue and profitability.
Our contract manufacturing business materially depends upon the regulatory approval of the products we manufacture. As such, any delay in, or failure to receive, approval for any of our customers’ product candidates or the failure to maintain regulatory approval for our or our customers’ products could negatively impact our revenue and profitability. If the FDA or a comparable foreign regulatory authority does not approve of our facilities for the manufacture of a customer product or if it withdraws such approval in the future, our customers may choose to identify alternative manufacturing facilities and/or relationships, which could significantly impact our ability to expand our CDMO capacity and capabilities and achieve profitability.
| 14 |
Our manufacturing services are highly complex, and if we are unable to provide quality and timely services to our customers, our business could suffer.
The manufacturing services we offer are highly complex, due in part to strict regulatory requirements. A failure of our quality control systems in our facilities could cause problems to arise in connection with facility operations for a variety of reasons, including equipment malfunction, viral contamination, failure to follow specific manufacturing instructions, protocols and standard operating procedures, problems with raw materials or environmental factors. Such problems could affect production of a single manufacturing run or a series of runs, requiring the destruction of products, or could halt manufacturing operations altogether. In addition, our failure to meet required quality standards may result in our failure to timely deliver products to our customers, which in turn could damage our reputation for quality and service. Any such incident could, among other things, lead to increased costs, lost revenue, reimbursement to customers for lost drug substance, damage to and possibly termination of existing customer relationships, time and expense spent investigating the cause and, depending on the cause, similar losses with respect to other manufacturing runs. With respect to our commercial manufacturing, if problems are not discovered before the product is released to the market, we may be subject to regulatory actions, including product recalls, product seizures, injunctions to halt manufacture and distribution, restrictions on our operations, civil sanctions, including monetary sanctions, and criminal actions. In addition, such issues could subject us to litigation, the cost of which could be significant.
Because a significant portion of our contract manufacturing revenue comes from a limited number of customers, any decrease in sales to these customers could harm our business, results of operations and financial condition.
Contract manufacturing revenue has historically been derived from a small customer base. For the fiscal year ended April 30, 2017, our top three customers accounted for approximately 98% of our revenues and for the fiscal years ended April 30, 2016 and 2015, approximately 95% and 91%, respectively, of contract manufacturing revenue was generated from two customers. In addition, typically we have not entered into long-term commitments with these third-party customers because their need for product supply depends on a variety of factors, including the product’s stage of development, the timing of regulatory filings and approvals, the product needs of their collaborators, if applicable, their financial resources and the market demand with respect to commercial products. The loss or a significant reduction of business from any of our major customers could have a material adverse effect on our business, results of operations and financial condition.
Our CDMO operates in a highly competitive market and competition may adversely affect our business.
We operate in a market that is highly competitive. Our competition in the CDMO market includes full-service contract manufacturers and large pharmaceutical companies offering third-party manufacturing services to fill their excess capacity. Also, large pharmaceutical companies have been seeking to divest portions of their manufacturing capacity, and any such divested businesses may compete with us in the future. In addition, most of our competitors may have substantially greater financial, marketing, technical or other resources than we do. Moreover, additional competition may emerge and may, among other things, result in a decrease in the fees paid for our services, which may adversely affect our results of operations and financial condition.
We rely on third parties to supply most of the necessary raw materials and supplies for the products we manufacture on behalf of our customers and our inability to obtain such raw materials or supplies may adversely impact our business, results of operations and financial condition.
Our CDMO operations require various raw materials, including proprietary media, resins, buffers, filters, in addition to numerous additional raw materials supplied primarily by third parties. We or our customers specify the raw materials and other items required to manufacture their product and, in some cases, specify the suppliers from whom we must purchase these raw materials. In certain instances, the raw materials and other items can only be supplied by a limited number of suppliers or in limited quantities. If third-party suppliers do not supply raw materials or other items on a timely basis, it may cause a manufacturing run to be delayed or canceled which would adversely impact our results of operations and financial condition.
| 15 |
Furthermore, third-party suppliers may fail to provide us with raw materials and other items that meet the qualifications and specifications required by us or our customers. If third-party suppliers are not able to provide us with raw materials that meet our or our customers’ specifications on a timely basis, we may be unable to manufacture their product or it could prevent us from delivering products to our customers within required timeframes. Any such delay in delivering our products may create liability for us to our customers for breach of contract or cause us to experience order cancellations and loss of customers. In the event that we manufacture products with inferior quality components and raw materials, we may become subject to product liability claims caused by defective raw materials or components from a third-party supplier or from a customer, or our customer may be required to recall its products from the market.
We have refocused our clinical development strategy in order to drive partnering interest, which may not be successful.
In June 2016, we announced our clinical development strategy focused on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies. These trials may be conducted independently, in conjunction with our collaborators, or through ISTs. The goal of these trials will be to generate compelling clinical and translational data demonstrating bavituximab’s immunotherapeutic mechanism of action in a combination treatment setting. We plan to leverage this data to drive partnering interest in our phosphatidylserine (PS)-targeting platform. Our clinical development strategy is subject to a number of risks and uncertainties, including but not limited to, the risk that:
| · | we may not secure sufficient capital to conduct any sponsored trial that may be necessary to support a potential partnership; |
| · | the early stage studies do not generate any compelling data; |
| · | the data, while compelling, is viewed by potential partners as being based on an insufficient sample size; and |
| · | the data, while compelling, is in tumor indications for which there is little or no partnering interest. |
Even if we are successful in generating compelling data that generates partnering interest, we may face additional challenges in consummating a licensing transaction, or on terms acceptable to us, considering the additional later stage clinical trials that such partner would need to conduct, and the time, costs and regulatory risks associated therewith, relative to the remaining life of our patents that cover bavituximab compositions and their use in treating cancer, both alone and in combination therapies, which are currently set to expire between 2023 and 2025, before any potential extensions. If we are unable to efficiently and quickly generate the type and quantity of data needed to drive future partnering interest, we may not be successful in our clinical development strategy, nor obtain any monetary value from our PS-targeting platform, including bavituximab.
We rely on third-parties to conduct our clinical trials and many of our preclinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our drug candidates may not advance in a timely manner or at all.
In the course of our discovery, preclinical testing and clinical trials, we rely on third parties, including universities, investigators and CROs, to perform critical services for us. For example, we rely on third parties to conduct our clinical trials and many of our preclinical studies. CROs and investigators are responsible for many aspects of the trials, including finding and enrolling patients for testing and administering the trials. We therefore must rely on third parties to conduct our clinical trials, but their failure to comply with all regulatory and contractual requirements, or to perform their services in a timely and acceptable manner, may compromise our clinical trials in particular or our business in general. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as good clinical practices (“GCPs”) for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the trial patients are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner. Any failings by these third parties may compromise our clinical trials in particular or our business in general. Similarly, we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. For example, if such third parties fail to perform their obligations in compliance with our clinical trial protocols, our clinical trials may not meet regulatory requirements or may need to be repeated. As a result of our dependence on third parties, we may face delays or failures outside of our direct control. These risks also apply to the development activities of our collaborators, and we do not control our collaborators’ research and development, clinical trials or regulatory activities.
| 16 |
Failure to recruit, enroll and retain patients for clinical trials may cause the development of our product candidates to be delayed or development costs to increase substantially.
While our clinical development strategy is focused on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies, we may none-the-less experience delays in patient enrollment in these clinical trials for a variety of reasons. The timely completion of any new potential clinical trials in accordance with their protocols depends on, among other things, our ability to enroll a sufficient number of patients who remain in the study until its conclusion. The enrollment of patients depends on many factors, including:
| · | the patient eligibility criteria defined in the protocol; |
| · | the size of the patient population required for analysis of the trial’s primary endpoints; |
| · | the proximity of patients to study sites; |
| · | the design of the trial; |
| · | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| · | our ability to obtain and maintain patient consents; |
| · | the risk that patients enrolled in clinical trials will drop out of the trials before completion; and |
| · | competition for patients by clinical trial programs for other competitive treatments. |
Our planned early stage clinical trials will compete with other clinical trials for product candidates that are in the same therapeutic areas as our product candidates, and this competition reduces the number and types of patients available to us, because some patients who might have opted to enroll in our trials opt to enroll in a trial being conducted by one of our competitors. In addition, some of these competing clinical trials will be for product candidates in later stage clinical trials with compelling data from earlier stage trials that could influence the patient’s decision to opt to enroll in the competitive trial. Since the number of qualified clinical investigators is limited, we conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which reduces the number of patients who are available for our clinical trials in such clinical trial site. Delays in patient enrollment in our planned early stage clinical trials may affect the timing or outcome of our clinical trials, which could prevent us from completing these trials and generating the compelling data necessary to drive partnering interest.
Success in early clinical trials may not be indicative of results obtained in later trials, which could hinder future partner interest.
Our clinical development strategy is dependent on our ability to generate compelling data from small, early stage studies of bavituximab in combination with immune stimulating therapies. A number of new drugs and biologics have shown promising results in initial clinical trials, but subsequently failed to establish sufficient safety and effectiveness data to obtain necessary regulatory approvals, as we evidenced with our Phase III SUNRISE trial. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit or prevent us from generating partnering interest.
If we do not establish additional collaborations, or if the trials conducted by our collaborators do not proceed efficiently, our clinical development strategy may not be successful.
Our clinical development strategy is focused on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies. We anticipate that a number of these trials will be conducted in conjunction with our collaborators, such as NCCN. In order to be successful, we may need to generate a significant amount of compelling data in a number of cancer indications and therefore, may need to enter into one or more of additional collaborations in the future. We face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms, or at all. Even if we successfully enter into a collaboration, our collaborator may not perform its contractual obligations or may terminate the agreement.
| 17 |
In addition, many of these early stage studies may be conducted by our existing or future collaborators as ISTs. While the use of ISTs will allow us to increase the number of early stage trials of bavituximab and conserve our financial and personnel resources, because we do not have control over the conduct of these trials, we do not have the ability to influence the speed at which these trials are conducted, including the rate at which patients are enrolled or data is analyzed.
Any one or a combination of these factors could adversely affect our ability to timely generate the compelling data that is necessary to attract potential partners to advance the bavituximab program.
If we use hazardous and biological materials in a manner that causes injury or violates applicable law, we may be liable for damages.
Our research and development activities and manufacturing operations involve the controlled use of hazardous materials and chemicals. We are subject to federal, state and local laws and regulations in the U.S. governing the use, manufacture, storage, handling and disposal of hazardous materials and chemicals. Although we believe that our procedures for using, handling, storing and disposing of these materials comply with legally prescribed standards, we may incur significant additional costs to comply with applicable laws in the future. Also, even if we are in compliance with applicable laws, we cannot completely eliminate the risk of contamination or injury resulting from hazardous materials or chemicals. As a result of any such contamination or injury, we may incur liability or local, city, state or federal authorities may curtail the use of these materials and interrupt our business operations. In the event of an accident, we could be held liable for damages or penalized with fines, and the liability could exceed our resources. Compliance with applicable environmental laws and regulations is expensive, and current or future environmental regulations may impair our research and development activities and manufacturing operations, which could materially harm our business, financial condition and results of operations.
We may have significant product liability exposure because we maintain only limited product liability insurance.
We face an inherent business risk of exposure to product liability claims in the event that the administration of one of our product candidates during a clinical trial adversely affects or causes the death of a patient. Although we maintain product liability insurance for clinical studies in the amount of $10,000,000 per occurrence or $10,000,000 in the aggregate on a claims-made basis, as well as country-specific coverage where required for clinical sites located in foreign countries, our coverage may not be adequate. Product liability insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms, if at all. Our inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims in excess of our insurance coverage, if any, or a product recall, could negatively impact our financial position and results of operations.
In addition, the contract manufacturing services that we offer through Avid expose us to an inherent risk of liability as the antibodies or other substances manufactured by Avid, at the request and to the specifications of our customers, could possibly cause adverse effects or have product defects. We obtain agreements from our customers indemnifying and defending us from any potential liability arising from such risk. However, these indemnification agreements may not adequately protect us against potential claims relating to such contract manufacturing services or protect us from being named in a possible lawsuit. Although Avid has procured insurance coverage, we may not be able to maintain our existing coverage or obtain additional coverage on commercially reasonable terms, or at all, or such insurance may not provide adequate coverage against all potential claims to which we might be exposed. A partially successful or completely uninsured claim against Avid could materially harm our business, financial condition and results of operations.
| 18 |
Our research and development activities rely on technology licensed from third parties, and termination of any of those licenses would result in loss of significant rights to develop and market our products, which would impair our business, prospects, financial condition and results of operations.
We have been granted rights to a variety of technologies necessary for our research and development activities from third parties through license agreements. Each license generally may be terminated by the licensor if we fail to perform our obligations under the agreement, including obligations to develop the product candidates or technologies under license. If terminated, we would lose the right to develop the product candidates, which could adversely affect our business, prospects, financial condition and results of operations. The license agreements also generally require us to meet specified milestones or show reasonable diligence in development of the technology. If disputes arise over the definition of these requirements or whether we have satisfied the requirements in a timely manner, or if any other obligations in the license agreements are disputed by the other party, the other party could terminate the agreement, and we could lose our rights to develop the licensed technology.
In addition, if new technology is developed from these licenses, we may be required to negotiate certain key financial and other terms, such as milestone and royalty payments, for the licensing of this future technology with the third party licensors, and it might not be possible to obtain any such license on terms that are satisfactory to us, or at all.
If we are unable to obtain, protect and enforce our patent rights, we may be unable to effectively protect or exploit our proprietary technology, inventions and improvements.
Our success depends in part on our ability to obtain, protect and enforce commercially valuable patents. We try to protect our proprietary positions by filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to developing our business. However, if we fail to obtain and maintain patent protection for our proprietary technology, inventions and improvements, our competitors could develop and commercialize products that would otherwise infringe upon our patents.
Our patent position is generally uncertain and involves complex legal and factual questions. Legal standards relating to the validity and scope of claims in the biotechnology and biopharmaceutical fields are still evolving. Accordingly, the degree of future protection for our patent rights is uncertain. The risks and uncertainties that we face with respect to our patents include the following:
| · | the pending patent applications we have filed or to which we have exclusive rights may not result in issued patents or may take longer than we expect to result in issued patents; |
| · | the claims of any patents that issue may not provide meaningful protection; |
| · | we may be unable to develop additional proprietary technologies that are patentable; |
| · | the patents licensed or issued to us may not provide a competitive advantage; |
| · | other parties may challenge patents licensed or issued to us; |
| · | disputes may arise regarding the invention and corresponding inventorship and ownership rights in inventions and know-how resulting from the joint creation or use of intellectual property by us, our licensors, corporate partners and other scientific collaborators; and |
| · | other parties may design around our patented technologies. |
If we are unable to adequately protect our intellectual property rights, our business may be adversely impacted.
| 19 |
The patent protection for our product candidates may expire before we are able to commercialize those product candidates, which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue.
The patents for our product candidates have varying expiration dates and, if these patents expire, we may be subject to increased competition and we may not be able to recover our development costs or market any of our approved products profitably. For example, one of our licensed U.S. patents claims compounds encompassing bavituximab and is due to expire in 2024, and two of our other licensed U.S. patents claim treatment methods encompassing bavituximab and are due to expire in 2025. In some of the larger potential market territories, such as the United States and Europe, patent term extension or restoration may be available to compensate for time taken during aspects of the product’s development and regulatory review. However, such an extension may not be granted, or if granted, the applicable time period or the scope of patent protection afforded during any extension period may not be sufficient to maximize the commercial value of the patent(s). In addition, even though some regulatory authorities may provide some other exclusivity for a product under their own laws and regulations, we may not be able to qualify the product or obtain the exclusive time period. If we are unable to obtain patent term extension/restoration or some other exclusivity, we could be subject to increased competition and our opportunity to establish or maintain product revenue could be substantially reduced or eliminated. Furthermore, we may not have sufficient time to recover our development costs prior to the expiration of our U.S. and non-U.S. patents.
We may become involved in disputes or lawsuits pertaining to patent applications, or to protect or enforce our patents that would be expensive, time consuming and may lead to disclosure of our confidential information.
Disputes may arise in connection with the preparation, filing and examination of patent applications, including licensed patent applications, which may be difficult, time-consuming and expensive to resolve. In addition, we may become subject to interference, opposition or other post-grant review proceedings conducted in patent and trademark offices to determine the priority and/or patentability of inventions. In order to protect or enforce our patent rights, we may initiate patent litigation against third parties. The defense of intellectual property rights, including patent rights through lawsuits, interference, opposition or other post-grant review proceedings and other legal and administrative proceedings, would be costly and divert our technical and management personnel from their normal responsibilities. An adverse determination of any litigation or defense proceedings could put our pending patent applications at risk of not being issued.
Furthermore, there is a risk that some of our confidential information could be compromised by disclosure during patent disputes. Due to the substantial amount of discovery required in connection with intellectual property litigation, this type of litigation poses a particular risk. For example, during the course of this kind of litigation, confidential information may be inadvertently disclosed in the form of documents or testimony in connection with discovery requests, depositions or trial testimony. This disclosure could have a material adverse effect on our business and our financial results.
Business disruptions could seriously harm our future revenues and financial condition and increase our costs and expenses.
Our operations could be subject to earthquakes, power shortages and surges, telecommunications failures, water shortages, floods, fires, extreme weather conditions, medical epidemics and other natural or manmade disasters or business interruptions, for which we have limited insurance or are predominantly self-insured. The occurrence of any of these business disruptions could seriously harm our manufacturing operations and financial condition and increase our costs and expenses. Our ability to obtain raw materials, components and supplies for the manufacture, as well as the services of outside testing laboratories, of our third party customers’ products, for which we act as a contract manufacturer, could be disrupted, if the operations of these suppliers and/or labs is affected by a man-made or natural disaster or other business interruption. Our corporate headquarters and manufacturing facility is located in California near major earthquake faults. The ultimate impact on us, our significant suppliers and our general infrastructure of being located near major earthquake faults and being consolidated in certain geographical areas is unknown, but our operations and financial condition could suffer in the event of a major earthquake or other natural disaster.
| 20 |
We may not be able to compete with our competitors in the pharmaceutical and biotechnology industries because many of them have greater resources than we do and they are further along in their development efforts.
The pharmaceutical and biotechnology industry is intensely competitive and any product candidate developed by us would compete with existing drugs and therapies. There are many pharmaceutical companies, biotechnology companies, public and private universities, government agencies and research organizations that compete with us in developing various approaches to cancer therapy.
In addition, we are aware of several pharmaceutical and biotechnology companies actively engaged in research and development of immunotherapy-based products that have commenced clinical trials with, or have successfully commercialized, these products. Some or all of these companies may have greater financial resources, larger technical staffs and larger research budgets than we have, as well as greater experience in developing products and running clinical trials. We expect to continue to experience significant and increasing levels of competition in the future. In addition, there may be other companies which are currently developing competitive technologies and products or which may in the future develop technologies and products that are comparable or superior to our technologies and products.
If we lose qualified management, including manufacturing or scientific personnel or are unable to attract and retain such personnel, we may be unable to successfully develop or manufacture our products or our customers’ products.
Our success is dependent, in part, upon a limited number of key executive officers, each of whom is an at-will employee. For example, because of his extensive understanding of our contract manufacturing operations, technologies and product development programs, the loss of Mr. Steven W. King, our President and Chief Executive Officer, would adversely affect our contract manufacturing operations and product development efforts during the six- to twelve-month period that we estimate it would take to find a qualified replacement.
We also believe that our future success will depend largely upon our ability to attract and retain highly-skilled manufacturing, process development and research and development personnel. We face intense competition in our recruiting activities, including competition from larger companies with greater resources. We do not know if we will be successful in attracting or retaining skilled personnel. The loss of certain key employees or our inability to attract and retain other qualified employees could negatively affect our operations and financial performance.
We have federal and state net operating loss (“NOL”) carry forwards which, if we were to become profitable, could be used to offset/defer federal and state income taxes. Our ability to use such carry forwards to offset future taxable income may be subject to certain limitations related to changes in ownership of our stock.
As of April 30, 2017, we had federal and state NOL carry forwards of approximately $392 million and $264 million, respectively, expiring from 2018 to 2037. These NOL carry forwards could potentially be used to offset certain future federal and state income tax liabilities. However, utilization of NOL carry forwards may be subject to a substantial annual limitation pursuant to Section 382 of the Internal Revenue Code of 1986, as amended, as well as similar state provisions due to ownership changes that have occurred previously or that could occur in the future. In general, an ownership change, as defined by Section 382, results from transactions increasing the ownership of certain stockholders or public groups in the stock of a corporation by more than 50 percentage points over a three-year period. We performed a detailed analysis of our NOL carry forwards through April 30, 2017 and it was determined that no change in ownership had occurred. However, ownership changes occurring subsequent to April 30, 2017 may impact the utilization of our NOL carry forwards and other tax attributes. Any limitation may result in expiration of a portion of the carry forwards before utilization. If we were not able to utilize our carry forwards, we would be required to use our cash resources to pay taxes that would otherwise have been offset, thereby reducing our liquidity.
| 21 |
We have become increasingly dependent on information technology and any breakdown, interruption or breach of our information technology systems could subject us to liability or interrupt the operation of our business, which could have a material adverse effect on our business, financial condition, cash flows and results of operations.
We are increasingly dependent upon sophisticated information technology systems and infrastructure in connection with the conduct of our business. We must constantly update our information technology infrastructure and our various current information technology systems throughout the organization may not continue to meet our current and future business needs. Furthermore, modification, upgrade or replacement of such systems may be costly. In addition, due to the size and complexity of these systems, any breakdown, interruption, corruption or unauthorized access to or cyber-attack on these systems could create system disruptions, shutdowns or unauthorized disclosure of confidential information. While we attempt to take appropriate security and cyber-security measures to protect our data and information technology systems and to prevent such breakdowns and unauthorized breaches and cyber-attacks, these measures may not be successful and these breakdowns and breaches in, or attacks on, our systems and data may not be prevented. Such breakdowns, breaches or attacks may cause business interruption and could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares to decline, and we may suffer financial damage or other loss, including fines or criminal penalties because of lost or misappropriated information.
Our governance documents and state law provide certain anti-takeover measures which will discourage a third party from seeking to acquire us unless approved by the Board of Directors.
We have a rights plan that is designed to protect stockholders against unsolicited attempts to acquire control of us that do not offer a fair price to our stockholders as determined by our board of directors. Under the plan, the acquisition of 15% or more of our outstanding common stock by any person or group, unless approved by our board of directors, will trigger the right of our stockholders (other than the acquirer of 15% or more of our common stock) to acquire additional shares of our common stock, and, in certain cases, the stock of the potential acquirer, at a 50% discount to market price, thus significantly increasing the acquisition cost to a potential acquirer. In addition, our certificate of incorporation and by-laws contain certain additional anti-takeover protective devices. For example,
| · | no stockholder action may be taken without a meeting, without prior notice and without a vote; solicitations by consent are thus prohibited; |
| · | special meetings of stockholders may be called only by our board of directors; and |
| · | our board of directors has the authority, without further action by the stockholders, to fix the rights and preferences, and issue shares, of preferred stock. An issuance of preferred stock with dividend and liquidation rights senior to the common stock and convertible into a large number of shares of common stock could prevent a potential acquirer from gaining effective economic or voting control. |
Further, we are subject to Section 203 of the Delaware General Corporation Law, which, subject to certain exceptions, restricts certain transactions and business combinations between a corporation and a stockholder owning 15% or more of the corporation’s outstanding voting stock for a period of three years from the date the stockholder becomes a 15% stockholder.
Although we believe these provisions and our rights plan collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
Our bylaws, as amended, provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our bylaws, as amended, provide that, unless we consent in writing to an alternative forum, the Court of Chancery of the State of Delaware is the exclusive forum for any derivative action or proceeding brought on our behalf, any action asserting a breach of a fiduciary duty owed by any of our directors, officers, or other employees to us, any action asserting a claim against us arising pursuant to the Delaware General Corporation Law, our certificate of incorporation or our bylaws, any action to interpret, apply, enforce, or determine the validity of our certificate of incorporation or bylaws, or any action asserting a claim against us that is governed by the internal affairs doctrine. The choice of forum provision may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees.
| 22 |
Risks Related to the Ownership of Our Common Stock
The sale of substantial shares of our common stock may depress our stock price.
As of April 30, 2017, there were 44,014,040 shares of our common stock issued and outstanding (as adjusted to reflect the 1-for-7 reverse stock split of our issued and outstanding common stock that took effect on July 10, 2017). Substantially all of these shares are eligible for trading in the public market, subject in some cases to volume and other limitations. The market price of our common stock may decline if our common stockholders sell a large number of shares of our common stock in the public market, or the market perceives that such sales may occur.
In addition, our common stock issued and outstanding as of April 30, 2017 excludes the following common shares reserved for future issuance (as adjusted to reflect the 1-for-7 reverse stock split of our issued and outstanding common stock that took effect on July 10, 2017):
| · | 5,630,872 common shares reserved for issuance under outstanding option grants and available for issuance under our stock incentive plans; |
| · | 1,359,736 common shares reserved for and available for issuance under our 2010 Employee Stock Purchase Plan (the “Employee Stock Purchase Plan”); |
| · | 39,040 common shares issuable upon exercise of outstanding warrants; and |
| · | 6,826,435 common shares issuable upon conversion of our outstanding Series E Preferred Stock. |
In addition, we expect we will continue to need to raise substantial additional capital to fund our operations until we can generate sufficient revenue from Avid’s contract manufacturing services to achieve profitability. If we raise additional funds by issuing equity securities, the market price of our securities may decline and our existing stockholders may experience significant dilution.
Our highly volatile stock price may adversely affect the liquidity of our common stock.
The market price of our common stock and the market prices of securities of companies in the biotechnology sector have generally been highly volatile and are likely to continue to be highly volatile. For instance, the market price of our common stock has ranged from $1.97 to $14.00 per share over the last three fiscal years ended April 30, 2017 (as adjusted to reflect the 1-for-7 reverse stock split of our issued and outstanding common stock that took effect on July 10, 2017).
In addition, the market price of our common stock may be significantly impacted by many factors, including, but not limited to:
| · | the success or failure of our internal drug development efforts; |
| · | positive or negative data reported on programs in clinical trials we or our investigators are conducting; |
| · | announcements of technological innovations or new commercial products by us or our competitors; |
| · | Avid’s loss of a significant customer; |
| · | uncertainties about our ability to continue to fund our operations beyond the next twelve months; |
| · | significant changes in our financial results or that of our competitors, including our ability to continue as a going concern; |
| · | our ability to meet revenue projections; |
| · | the offering and sale of shares of our common stock, either sold at market prices or at a discount under an equity transaction; |
| · | significant changes in our capital structure; |
| · | published reports by securities analysts; |
| · | announcements of partnering transactions, licensing agreements, joint ventures, strategic alliances, and any other transaction that involves the development, sale or use of our technologies or competitive technologies; |
| · | developments and/or disputes concerning our patent or other proprietary rights; |
| · | regulatory developments, including possible delays, and product safety concerns; |
| · | outcomes of significant litigation, disputes and other legal or regulatory proceedings; |
| · | general stock trends in the biotechnology and pharmaceutical industry sectors; |
| · | public concerns as to the safety and effectiveness of our products; |
| · | economic trends and other external factors, including but not limited to, interest rate fluctuations, economic recession, inflation, foreign market trends, national crisis, and disasters; and |
| · | healthcare reimbursement reform and cost-containment measures implemented by government agencies. |
| 23 |
These and other external factors have caused and may continue to cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock, and may otherwise negatively affect the liquidity of our common stock.
If we fail to meet continued listing standards of NASDAQ, our common stock may be delisted, which could have a material adverse effect on the liquidity of our common stock.
Our common stock is currently traded on The NASDAQ Capital Market. As previously reported, on April 12, 2016 we received a letter from the staff of the Listing Qualifications Department (the “Staff”) of The NASDAQ Stock Market LLC (“NASDAQ”) notifying us that, for the previous 30 consecutive business days, the bid price for our common stock had closed below the minimum $1.00 per share requirement for continued listing on The NASDAQ Capital Market under NASDAQ’s Listing Rule 5550(a)(2), which requires a minimum bid price of $1.00 per share (the “Minimum Bid Price Requirement”). In accordance with NASDAQ Listing Rule 5810(c)(3)(A), we were automatically afforded an initial “compliance period” of 180 calendar days following the date of the notification, or until October 10, 2016, to regain compliance with the Minimum Bid Price Requirement. We did not regain compliance with the Minimum Bid Price Requirement by October 10, 2016; however, because (i) we met the continued listing requirement for market value of publicly held shares and all other applicable requirements for initial listing on The NASDAQ Capital Market with the exception of the Minimum Bid Price Requirement, as of the last day of the initial “compliance period”, and (ii) provided written notice to NASDAQ of our intention to regain compliance by effecting a reverse stock split, if necessary, the Staff afforded us an additional “compliance period” of 180 calendar days, or until April 10, 2017, to regain compliance with the Minimum Bid Price Requirement.
Although our stockholders approved, at our Annual Meeting of Stockholders on October 13, 2016, an amendment to our certificate of incorporation, as amended, to effect a reverse split of our issued and outstanding common stock at a ratio of up to 1-for-7 as a means to regain compliance with the Minimum Bid Price Requirement, we determined that it was in the best interests of the Company and our stockholders not to effect the reverse split prior to April 10, 2017. On April 11, 2017, the Staff notified us that our common stock would be delisted unless we requested a hearing before the NASDAQ Hearings Panel (the “Panel”). On April 13, 2017, we requested a hearing before the Panel, which was held on May 25, 2017. On June 1, 2017, we received a decision letter from the NASDAQ Office of General Counsel stating that the Panel had granted us until no later than July 21, 2017 to regain compliance with the Minimum Bid Price Requirement. Following authorization from our board of directors, we filed a certificate of amendment to our certificate of incorporation with the Secretary of State of Delaware to effect a reverse split of our issued and outstanding common stock at a ratio of one-for-seven, which took effect with the opening of trading on July 10, 2017. As of the close of the market on July 13, 2017, the bid closing price of our common stock has exceeded the $1.00 minimum closing bid price requirement for four (4) consecutive business days, and it must continue to exceed $1.00 for an additional six (6) consecutive business days, or through July 21, 2017, in order for us to regain compliance with the Minimum Bid Price Requirement. If we are unable to regain compliance with the Minimum Bid Price Requirement by the close of the market on July 21, 2017, our common stock will be delisted from the NASDAQ Capital Market.
If our common stock is delisted, the liquidity of our common stock would be adversely affected and the market price of our common stock could decrease.
We do not intend to pay dividends on our common stock so any returns will be limited to the value of our stock.
We have never declared or paid any cash dividend on our common stock. We currently anticipate that we will retain future earnings, if any, for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to stockholders will therefore be limited to the appreciation of their stock.
| 24 |
If securities or industry analysts do not publish research reports about us, or if they issue adverse opinions about our business, our stock price and trading volume could decline.
The research and reports that industry or securities analysts publish about us or our business will influence the market for our common stock. If one or more analysts who cover us issues an adverse opinion about us, our stock price would likely decline. If one or more of these analysts ceases research coverage of us or fails to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline. Further, if we fail to meet the market expectations of analysts who follow our stock, our stock price likely would decline.
additional Risks Related to the Ownership of our Series E Preferred Stock
We may not be able to pay dividends on the series e preferred stock.
We are incorporated in Delaware and governed by the Delaware General Corporation Law. Delaware law allows a corporation to pay dividends only out of surplus, as determined under Delaware law, or if there is no surplus, out of net profits for the fiscal year in which the dividend was declared and for the preceding fiscal year. Under Delaware law, however, we cannot pay dividends out of net profits if, after we pay the dividend, our capital would be less than the capital represented by the outstanding stock of all classes having a preference upon the distribution of assets. In addition, payment of our dividends depends upon our financial condition and other factors as our board of directors may deem relevant from time to time. Our business may not generate sufficient cash flow from operations or future borrowings may not be available to us in an amount sufficient to enable us to make distributions on our Series E Preferred Stock.
The market price of the Series E Preferred Stock could be substantially affected by various factors.
The market price of the Series E Preferred Stock will depend on many factors, which may change from time to time, including:
| · | prevailing interest rates, increases in which may have an adverse effect on the market price of the Series E Preferred Stock; |
| · | trading prices of common and preferred equity securities issued by other biopharmaceutical companies; |
| · | the annual yield from distributions on the Series E Preferred Stock as compared to yields on other financial instruments; |
| · | announcements of technological innovations or new commercial products by us or our competitors; |
| · | publicity regarding actual or potential company-sponsored clinical trial and investigator-sponsored clinical trial results relating to products under development by us or our competitors; |
| · | announcements of licensing agreements, joint ventures, strategic alliances, and any other transaction that involves the development, sale or use of our technologies; |
| · | regulatory developments and product safety concerns; |
| · | general economic and financial market conditions; |
| · | government action or regulation; |
| · | significant changes in the financial condition, performance and prospects of us and our competitors; |
| · | changes in financial estimates or recommendations by securities analysts with respect to us, our competitors in our industry; |
| · | our issuance of additional preferred equity or debt securities; and |
| · | actual or anticipated variations in quarterly operating results of us and our competitors. |
As a result of these and other factors, holders of our Series E Preferred Stock may experience a decrease, which could be substantial and rapid, in the market price of the Series E Preferred Stock, including decreases unrelated to our operating performance or prospects.
| 25 |
| Item 1B. | Unresolved Staff Comments |
Not applicable.
| Item 2. | Properties |
Our corporate offices, research and development, and manufacturing facilities are all located in close proximity in Tustin, California, which are shared by our contract manufacturing business and our drug development business. We currently lease an aggregate of approximately 196,000 square feet of office, warehouse, research and manufacturing space in six buildings under five separate lease agreements, as summarized in the following table:
|
Lease # |
Original Lease Execution Date |
Approximate Square Footage Leased |
# of Buildings Occupied |
Initial Lease Term Expiration Date |
# of Options to Extend Lease |
Extended Lease Term Expiration Date(1) |
| 1 | December 1998 | 48,000 | 2 | 12/31/27 | 2 | 12/31/37 |
| 2 | May 2010 | 13,000 | 1 | 12/31/17 | 1 | 12/31/22 |
| 3 | July 2014 | 84,000 | 1 | 1/31/27 | 2 | 1/31/37 |
| 4 | April 2016 | 26,000 | 1 | 8/31/23 | 2 | 8/31/35 |
| 5 | April 2016 | 25,000 | 1 | 8/31/23 | 2 | 8/31/35 |
____________________
| (1) | Extended lease term expiration date assumes we execute all available option(s) to extend lease in accordance with the terms of the lease agreement. |
We believe that the space we lease is adequate to meet our current needs and that, if necessary, additional space would be available to accommodate any future growth.
| Item 3. | Legal Proceedings |
In the ordinary course of business, we are at times subject to various legal proceedings and disputes. We make provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are reviewed at least quarterly and adjusted to reflect the impact of any settlement negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case.
On October 10, 2013, a derivative and class action complaint, captioned Michaeli v. Steven W. King, et al., C.A. No. 8994-VCL, was filed in the Court of Chancery of the State of Delaware (the “Court”), purportedly on behalf of the Company, which is named a nominal defendant, against certain of our executive officers and directors (collectively, the “Defendants”). On December 1, 2015, the plaintiffs filed an amended and supplemental derivative and class action complaint (the “Amended Complaint”). The Amended Complaint alleges that the Defendants breached their respective fiduciary duties in connection with certain purportedly improper compensation decisions made by our board of directors during the past four fiscal years ended April 30, 2015, including: (i) the grant of a stock option to Mr. King on May 4, 2012; (ii) the non-routine broad-based stock option grant to our directors, executives, all other employees and certain consultants on December 27, 2012; and (iii) the payment, during the past four fiscal years ended April 30, 2015, of compensation to our non-employee directors. In addition, the complaint alleges that our directors breached their fiduciary duty of candor by filing and seeking stockholder action on the basis of an allegedly materially false and misleading proxy statement for our 2013 annual meeting of stockholders. The plaintiffs are seeking, among other things, rescission of a portion of the stock option grant to Mr. King on May 4, 2012 and the stock options granted to the Defendants on December 27, 2012, as well as disgorgement of any excessive compensation paid to our non-employee directors during the four fiscal years ended April 30, 2015 and other monetary relief for our benefit. On May 15, 2017, the parties filed with the Court a Stipulation and Agreement of Compromise, Settlement and Release setting forth the terms of the proposed settlement of the claims in the Amended Complaint. A hearing on the proposed settlement will be held before the Court on July 27, 2017. We do not expect to incur a loss associated with this matter should the Court approve the terms of the proposed settlement.
| Item 4. | Mine Safety Disclosures |
Not applicable.
| 26 |
| Item 5. | Market For Registrant’s Common Equity, Related Stockholder Matters And Issuer Purchases Of Equity Securities |
Market Information
Our common stock is listed on The NASDAQ Capital Market under the trading symbol “PPHM.” The following table sets forth the high and low sales prices per share of our common stock for each quarter during the two years ended April 30, 2017, as adjusted to reflect the 1-for-7 reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017:
Common Stock Sales Price | ||||
| High | Low | |||
| Fiscal Year 2017 | ||||
| Quarter Ended April 30, 2017 | $5.42 | $2.07 | ||
| Quarter Ended January 31, 2017 | $2.85 | $1.97 | ||
| Quarter Ended October 31, 2016 | $3.64 | $2.19 | ||
| Quarter Ended July 31, 2016 | $4.69 | $2.05 | ||
| Fiscal Year 2016 | ||||
| Quarter Ended April 30, 2016 | $7.84 | $2.31 | ||
| Quarter Ended January 31, 2016 | $9.28 | $6.02 | ||
| Quarter Ended October 31, 2015 | $9.03 | $6.38 | ||
| Quarter Ended July 31, 2015 | $10.50 | $8.47 | ||
Holders of Common Stock
As of June 30, 2017, we had 4,018 stockholders of record of our common stock.
Dividends
No dividends on our common stock have been declared or paid by us. We intend to employ all available funds for the development of our business and, accordingly, do not intend to pay any cash dividends in the foreseeable future. In addition, the Certificate of Designations governing the Series E Preferred Stock restricts us from declaring and paying any dividends on our common stock unless full cumulative dividends on the Series E Preferred Stock have been or contemporaneously are declared and paid or declared and a sum sufficient for the payment thereof is set apart for payment for all past dividend periods. Any future determinations related to dividend policy will be made at the discretion of our board of directors.
Securities Authorized for Issuance under Equity Compensation
The information included under Item 12 of Part III of this Annual Report is hereby incorporated by reference into this Item 5 of Part II of this Annual Report.
Recent Sales of Unregistered Securities
None.
| 27 |
Performance Graph
Notwithstanding any statement to the contrary in any of our previous or future filings with the SEC, the following information relating to the price performance of our common stock shall not be deemed to be “filed” with the SEC or to be “soliciting material” under the Securities Exchange Act of 1934, as amended, or the Exchange Act, and it shall not be deemed to be incorporated by reference into any of our filings under the Securities Act of 1933, as amended, or the Exchange Act, except to the extent we specifically incorporate it by reference into such filing.
The following chart shows the performance from April 30, 2012 through April 30, 2017 of Peregrine Pharmaceuticals, Inc. common stock, compared with an investment in the stocks represented in the NASDAQ ICB: 4577 Pharmaceuticals Index and the NASDAQ U.S. Benchmark TR Index assuming the investment of $100 at the beginning of the period and the reinvestment of dividends, if any. The total return data for the comparative indexes were prepared by NASDAQ OMX Global Indexes.
COMPARISON OF FIVE-YEAR CUMULATIVE TOTAL
RETURN
VALUE OF INVESTMENT OF $100 ON APRIL 30, 2012
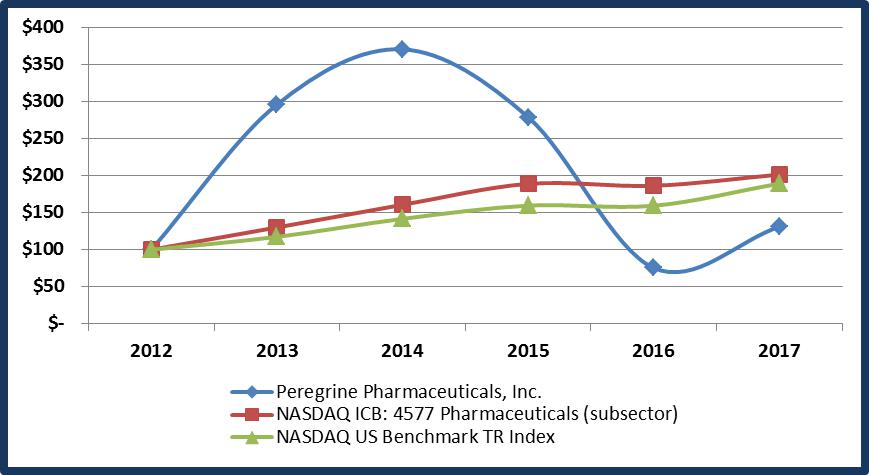
The underlying data for the foregoing graph is as follows:
| April 30, 2012 |
April 30, 2013 |
April 30, 2014 |
April 30 2015 |
April 30, 2016 |
April 30, 2017 | |
| Peregrine Pharmaceuticals, Inc. | $ 100.00 | $ 295.74 | $ 370.21 | $ 278.72 | $ 75.34 | $ 130.98 |
| NASDAQ ICB: 4577 Pharmaceuticals (subsector) | $ 100.00 | $ 130.03 | $ 160.55 | $ 188.82 | $ 186.26 | $ 201.35 |
| NASDAQ U.S. Benchmark TR Index | $ 100.00 | $ 117.32 | $ 141.62 | $ 159.47 | $ 159.32 | $ 189.23 |
| 28 |
| Item 6. | Selected Financial Data |
The selected consolidated financial data set forth below as of April 30, 2017 and 2016, and for the fiscal years ended April 30, 2017, 2016 and 2015, are derived from our audited consolidated financial statements included elsewhere in this Annual Report. This information should be read in conjunction with those consolidated financial statements, the notes thereto, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The selected consolidated financial data set forth below as of April 30, 2015, 2014 and 2013, and for the fiscal years ended April 30, 2014 and 2013, are derived from our audited consolidated financial statements that are contained in Annual Reports previously filed with the SEC, not included herein.
| CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS | ||||||||||||||||||||
| FISCAL YEAR ENDED APRIL 30, | ||||||||||||||||||||
| 2017 (a) | 2016 (b) | 2015 (c) | 2014 (d) | 2013 (e) | ||||||||||||||||
| Total revenues | $ | 57,630,000 | $ | 44,686,000 | $ | 26,781,000 | $ | 22,401,000 | $ | 21,683,000 | ||||||||||
| Net loss | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | $ | (35,362,000 | ) | $ | (29,780,000 | ) | |||||
| Series E preferred stock accumulated dividends | $ | (4,640,000 | ) | $ | (4,484,000 | ) | $ | (3,696,000 | ) | $ | (401,000 | ) | $ | – | ||||||
| Net loss attributable to common stockholders (f) | $ | (32,799,000 | ) | $ | (60,136,000 | ) | $ | (54,054,000 | ) | $ | (35,763,000 | ) | $ | (29,780,000 | ) | |||||
| Basic and diluted loss per common share (g) | $ | (0.88 | ) | $ | (1.95 | ) | $ | (2.07 | ) | $ | (1.55 | ) | $ | (1.73 | ) | |||||
| Weighted average common shares outstanding (g) | 37,109,493 | 30,895,089 | 26,079,762 | 23,082,807 | 17,195,762 | |||||||||||||||
| CONSOLIDATED BALANCE SHEET DATE | ||||||||||||||||||||
| AS OF APRIL 30, | ||||||||||||||||||||
| 2017 | 2016 | 2015 | 2014 | 2013 | ||||||||||||||||
| Cash and cash equivalents | $ | 46,799,000 | $ | 61,412,000 | $ | 68,001,000 | $ | 77,490,000 | $ | 35,204,000 | ||||||||||
| Working capital | $ | 26,169,000 | $ | 24,234,000 | $ | 43,192,000 | $ | 63,564,000 | $ | 21,353,000 | ||||||||||
| Total assets | $ | 118,112,000 | $ | 109,043,000 | $ | 97,464,000 | $ | 90,545,000 | $ | 45,058,000 | ||||||||||
| Long-term debt | $ | – | $ | – | $ | – | $ | – | $ | 13,000 | ||||||||||
| Accumulated deficit | $ | (537,435,000 | ) | $ | (509,276,000 | ) | $ | (453,624,000 | ) | $ | (403,266,000 | ) | $ | (367,904,000 | ) | |||||
| Stockholders' equity | $ | 53,582,000 | $ | 50,074,000 | $ | 59,035,000 | $ | 67,699,000 | $ | 23,760,000 | ||||||||||
__________
| (a) | Total revenues in fiscal year 2017 include contract manufacturing revenue of $57,630,000 and license revenue of nil. |
| (b) | Total revenues in fiscal year 2016 include contract manufacturing revenue of $44,357,000 and license revenue of $329,000. |
| (c) | Total revenues in fiscal year 2015 include contract manufacturing revenue of $26,744,000 and license revenue of $37,000. |
| (d) | Total revenues in fiscal year 2014 include contract manufacturing revenue of $22,294,000 and license revenue of $107,000. |
| (e) | Total revenues in fiscal year 2013 include contract manufacturing revenue of $21,333,000 and license revenue of $350,000. |
| (f) | Net loss attributable to common stockholders represents our net loss plus Series E preferred stock accumulated dividends. |
| (g) | All share and per share amounts of our common stock for all periods presented have been retroactively adjusted to reflect the one-for-seven reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017 (as described in Note 1 to the accompanying consolidated financial statements). |
| 29 |
| Item 7. | Management’s Discussion And Analysis Of Financial Condition And Results Of Operations |
The following discussion is included to describe our financial position and results of operations for each of the three years in the period ended April 30, 2017. The audited consolidated financial statements and notes thereto contain detailed information that should be referred to in conjunction with this discussion.
Overview
We are a biopharmaceutical company committed to improving the lives of patients by manufacturing high quality pharmaceutical products through our contract manufacturing business, Avid Bioservices, Inc. (“Avid”), and by advancing and licensing our novel, development-stage immunotherapy product through our research and development business, Peregrine Pharmaceuticals, Inc. (“Peregrine”).
Peregrine is focused primarily on the research and development of immune stimulating therapies for the treatment of various cancers. Avid, a wholly-owned subsidiary of Peregrine, is our contract development and manufacturing organization (“CDMO”) business.
In June 2016, following the discontinuation of our bavituximab Phase III SUNRISE trial, we announced a shift in our corporate focus toward achieving profitability within two (2) years (during the quarter ending July 31, 2018). This was to be achieved by focusing on revenue growth from our contract manufacturing business, Avid, while also reducing our spending on research and development while continuing to evaluate data from the Phase III SUNRISE trial. We believe we made significant progress towards this goal during the fiscal year ended April 30, 2017 by reducing research and development expenses by fifty-two percent (52%) while simultaneously growing Avid revenues by thirty percent (30%), as compared to the prior fiscal year. The result was a reduction of our net loss by 49% to $28,159,000 for the fiscal year ended April 30, 2017.
While reducing research and development expenses over the past fiscal year, we have also been able to allow the clinical data from the Phase III SUNRISE trial to mature, enabling us to conduct and present data analysis at key scientific meetings that support the clinical development of bavituximab with immune checkpoint inhibitors such as anti-PD-1/PD-L1 therapies. While we currently expect to reduce research and development spending by at least 40% in fiscal year 2018 compared to fiscal year 2017, this reduction in spending will not allow us to advance the bavituximab clinical program through a company sponsored trial. Instead, we are collaborating with leading immunotherapy experts and institutions to continue to drive forward our research and further guide our clinical development program.
With respect to our CDMO business, fiscal year 2017 was a record year for revenues, topping $57 million and representing 30% revenue growth over the prior fiscal year. While we are pleased at the continued year over year revenue growth, we have also recently seen unanticipated decreases in manufacturing demand from our largest customer and a recent regulatory filing delay from our second largest customer which will have some impact on our ability to grow the revenues from our CDMO business in fiscal year 2018 and could impact our ability to achieve overall profitability by the quarter ending July 31, 2018. However, we believe this to be temporary delay in revenue growth during fiscal year 2018 and have recently secured four new customers and are continuing to focus on securing additional customer business in order to better diversify our customer base. Our goal is to maintain profitability for Avid over the short term while positioning the business for long-term growth and attracting the resources necessary to continue to advance our promising research and development efforts.
Importantly, we continually evaluate various strategic options that we believe may enable us to enhance stockholder value, including the possible separation of these two distinct businesses.
Avid—Our CDMO Business
Avid provides fully-integrated cGMP services from cell line development to commercial biomanufacturing of large molecules, such as monoclonal antibodies and recombinant proteins for third-party customers while also supporting Peregrine’s internal drug development business. We believe this integration offers considerable time and cost efficiencies for our internal drug development business.
| 30 |
We have been developing and manufacturing biologics since 1993 in our Franklin biomanufacturing facility (the “Franklin Facility”) located at our current headquarters in Tustin, California and formed Avid in 2002 to offer these services to third-party customers using. In March 2016, we expanded our manufacturing capacity through the launch of our Myford biomanufacturing facility (the “Myford Facility”), which doubled our manufacturing capacity. The 42,000 square foot facility, which is our second biomanufacturing facility, can accommodate single-use bioreactors up to the 2,000-liter manufacturing scale. The Myford Facility was designed to accommodate a fully disposable biomanufacturing process for products in late stage clinical development to commercial. To date, Myford Facility has been utilized to complete a number of process validation runs for our third-party customers, which may lead to future commercial production, and has supported the process validation of our internal product, bavituximab. The Myford Facility is located adjacent to our Franklin Facility.
As we look to expand our CDMO business, in February 2017, we leased an additional 42,000 square feet of vacant warehouse space within the same building as our existing Myford Facility. The proximity of this space will allow us to utilize existing manufacturing infrastructure that we believe should enhance our manufacturing efficiencies and reduce the overall cost and timeframe to construct a third biomanufacturing facility. Although we previously anticipated that the new manufacturing facility would be constructed and ready for manufacturing activities by mid-calendar year 2018, due to unanticipated changes in and/or timing of customer demand (as discussed above), we have decided to defer construction of this third facility until demand from existing or potential new customers is expected to exceed the current capacity at our Franklin Facility and Myford Facility. Additionally, commencement of construction is also subject to our ability to raise sufficient additional capital to support this expansion effort. As a result, we presently do not expect to commence construction of this third facility prior to April 30, 2018.
Peregrine—Our Research and Development Business
Peregrine is developing therapeutics designed to fight cancer by reversing the immunosuppressive environment that tumors establish in order to proliferate. By doing so, these therapeutics allow the immune system to recognize and destroy tumor cells. Bavituximab is our lead immunotherapy candidate and currently we are collaborating with the National Comprehensive Cancer Network® (“NCCN”) and Memorial Sloan Kettering Cancer Center (“MSKCC”), to evaluate the potential of bavituximab in combination with immune stimulating therapies.
Clinical Development Strategy
In June 2016, we announced a clinical development strategy focused on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies. These trials may be conducted independently, in conjunction with our collaborators, or through investigator sponsored trials (“ISTs”). The goal of these trials is to generate compelling clinical and translational data demonstrating bavituximab’s immunotherapeutic mechanism of action in a combination treatment setting. Additionally, we are in the process of completing an extensive review and analysis of the available data from the discontinued Phase III SUNRISE trial discussed below and testing the numerous collected samples for potential biomarkers in order to determine if certain subgroups may have benefited more from the bavituximab treatment. We believe such information will assist us in guiding the bavituximab clinical program. In keeping with this strategy, we are evaluating options to advance clinical development bavituximab in an efficient and cost-effective way.
We believe this strategy will allow us to (i) continue our research and development activities while avoiding costly, later stage clinical trials, thereby allowing us to achieve profitability sooner, and (ii) generate additional data that we believe, if positive, could create future potential value, including attracting potential licensing partners or re-engaging our collaboration with AstraZeneca.
| 31 |
NCCN Collaboration
In January 2016, we announced that we entered into a research collaboration with NCCN, a not-for-profit alliance of 27 of the world’s leading cancer centers, to expand the clinical research and development of bavituximab for the treatment of a range of tumors. Under this research collaboration, we are funding three ISTs and related correlative studies with bavituximab at NCCN member institutions and their affiliate community hospitals through a $2 million research grant to NCCN’s Oncology Research Program. NCCN is responsible for oversight and monitoring of the three clinical studies awarded under the research grant. In September 2016, NCCN announced that investigators at the following NCCN-affiliated institutions received the grant award:
| 1. | Moffitt Cancer Center—A Phase I Trial of Sorafenib and Bavituximab Plus Stereotactic Body Radiation Therapy for Unresectable Hepatitis C Associated Hepatocellular Carcinoma. This trial is open for enrollment. |
| 2. | Massachusetts General Hospital Cancer Center—Phase I/II Clinical Trial of Bavituximab with Radiation and Temozolomide for Patients with Newly Diagnosed Glioblastoma. This trial is open for enrollment. |
| 3. | The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins—Phase II Study of Pembrolizumab and Bavituximab for Progressive Recurrent/Metastatic Squamous Cell Carcinoma of the Head and Neck. We expect this trial to be initiated by the end of the calendar year 2017. |
MSKCC Collaboration
In May 2015, we entered into a sponsored research agreement with investigators at MSKCC to identify effective treatment combinations based on our phosphatidylserine (“PS”)-targeting agents, including bavituximab, with other checkpoint inhibitors or immune stimulating agents that will further guide our clinical development program.
To date, preclinical data generated from the investigators at MSKCC have further supported our belief that bavituximab modulates the immunosuppressive tumor microenvironment and enhances the activity of other immunotherapy agents. MSKCC continues to evaluate bavituximab with other immunotherapy agents, which will further guide our clinical development program.
Phase III SUNRISE Trial
In December 2013, we initiated a randomized, double-blind, placebo-controlled Phase III trial evaluating bavituximab plus docetaxel, versus docetaxel plus placebo, for the treatment of previously-treated NSCLC (the “Phase III SUNRISE trial”).
In February 2016, we announced that we were discontinuing the Phase III SUNRISE trial based on the recommendation of the study’s Independent Data Monitoring Committee following a pre-specified interim analysis performed after 33% of targeted overall events (patient deaths). Results of the analysis demonstrated that the patients treated in the bavituximab plus docetaxel treatment arm did not show a sufficient improvement in overall survival as compared to the patients treated in the docetaxel plus placebo treatment arm to warrant continuation of the study. Patient enrollment was discontinued and existing patients in the trial were given the choice to continue chemotherapy and/or bavituximab, as appropriate. Patient treatment and follow-up data from the trial were collected through March 2017 and the trial is now closed.
Following the discontinuation of the Phase II SUNRISE trial, we promptly initiated and are nearing completion of an extensive review and analysis of the available data and testing the numerous collected samples for potential biomarkers in order to understand what subgroups may have benefited more from the bavituximab treatment. We believe such information will be important in supporting and refining our clinical strategy, as discussed above.
The most compelling data to date was presented in April 2017 at the Annual Meeting of the AACR. In a subgroup analysis, we looked at the outcome of 91 patients that were enrolled in the Phase III SUNRISE trial that were subsequently treated with immune checkpoint inhibitors (“ICI’s”) post study treatments. The results from this analysis demonstrated that the patients who received docetaxel plus bavituximab and subsequent ICI’s (anti-PD-1/PD-L1) had not yet reached median overall survival (“mOS”) compared to mOS of 13.0 months for patients who received docetaxel plus placebo (hazard ratio [HR], 0.43; p=0.005). The statistically significant difference between the two arms in the trial provides strong rationale for combining bavituximab with ICI’s and supports the hypothesis that bavituximab may modulate the tumor microenvironment to enhance the anti-tumor activity of ICI’s.
| 32 |
In June 2017, we announced additional supportive data at the Annual Meeting of the American Society of Clinical Oncology (“ASCO”) demonstrating that patients in the bavituximab containing arm who had low baseline PD-L1 expression on tumor cells (i.e., patients typically with poorer response to PD-1/PD-L1 checkpoint inhibitors) lived significantly longer than patients with high baseline PD-L1 expression. These data further support the hypothesis that bavituximab may modulate the tumor microenvironment to complement and enhance the anti-tumor activity of ICI’s.
Results of Operations
The following table compares the consolidated statements of operations and comprehensive loss for the fiscal years ended April 30, 2017, 2016 and 2015. This table provides you with an overview of the changes in the statements of operations and comprehensive loss for the comparative periods, which are further discussed below.
| Years Ended April 30, | Years Ended April 30, | |||||||||||||||||||||||
| 2017 | 2016 | $ Change | 2016 | 2015 | $ Change | |||||||||||||||||||
| REVENUES: | ||||||||||||||||||||||||
| Contract manufacturing | $ | 57,630,000 | $ | 44,357,000 | $ | 13,273,000 | $ | 44,357,000 | $ | 26,744,000 | $ | 17,613,000 | ||||||||||||
| License revenue | – | 329,000 | (329,000 | ) | 329,000 | 37,000 | 292,000 | |||||||||||||||||
| Total revenues | 57,630,000 | 44,686,000 | 12,944,000 | 44,686,000 | 26,781,000 | 17,905,000 | ||||||||||||||||||
| COST AND EXPENSES: | ||||||||||||||||||||||||
| Cost of contract manufacturing | 38,259,000 | 22,966,000 | 15,293,000 | 22,966,000 | 15,593,000 | 7,373,000 | ||||||||||||||||||
| Research and development | 28,297,000 | 59,529,000 | (31,232,000 | ) | 59,529,000 | 42,996,000 | 16,533,000 | |||||||||||||||||
| Selling, general and administrative | 19,334,000 | 18,551,000 | 783,000 | 18,551,000 | 18,691,000 | (140,000 | ) | |||||||||||||||||
| Total cost and expenses | 85,890,000 | 101,046,000 | (15,156,000 | ) | 101,046,000 | 77,280,000 | 23,766,000 | |||||||||||||||||
| LOSS FROM OPERATIONS | (28,260,000 | ) | (56,360,000 | ) | 28,100,000 | (56,360,000 | ) | (50,499,000 | ) | (5,861,000 | ) | |||||||||||||
| OTHER INCOME (EXPENSE): | ||||||||||||||||||||||||
| Interest and other income | 108,000 | 722,000 | (614,000 | ) | 722,000 | 142,000 | 580,000 | |||||||||||||||||
| Interest and other expense | (7,000 | ) | (14,000 | ) | 7,000 | (14,000 | ) | (1,000 | ) | (13,000 | ) | |||||||||||||
| NET LOSS | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | 27,493,000 | $ | (55,652,000 | ) | $ | (50,358,000 | ) | $ | (5,294,000 | ) | |||||||
| COMPREHENSIVE LOSS | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | 27,493,000 | $ | (55,652,000 | ) | $ | (50,358,000 | ) | $ | (5,294,000 | ) | |||||||
Contract Manufacturing Revenue
Fiscal Year 2017 Compared to Fiscal Year 2016:
The increase in contract manufacturing revenue of $13,273,000 (30%) during fiscal year 2017 was primarily due to manufacturing services provided to support the process validation of three separate customer products in the amount of $15,444,000, all of which were manufactured in our Myford Facility. No process validation services were provided in the prior year for third-party customers.
Excluding any future potential new business, we expect contract manufacturing revenue for fiscal year 2018 to slightly decline in comparison to fiscal year 2017. Part of this decline is due to lower anticipated commitments from Halozyme, Inc. (our largest customer) based on their most recent committed forecast (covering the three quarters ending March 2018), which amount is expected to be partially offset by revenue in the amount of $10 million that was expected to be recognized in fiscal year 2017, but has been shifted to fiscal year 2018 due to a delay in shipping product that was complete and ready for shipment as of the fiscal year ended April 30, 2017.
| 33 |
As we continue to seek to diversify our customer base, over the recent months, we have secured four new customers. These new customers are predominately in an earlier stage of development and, therefore, we expect that contract manufacturing revenue from these new customers during fiscal year 2018 will only partially offset the net anticipated decrease from our other existing customers.
Therefore, based on our current commitments for manufacturing services and the anticipated completion of in-process third-party customer manufacturing runs, we expect contract manufacturing revenue for fiscal year 2018 to range from $50 to $55 million.
Fiscal Year 2016 Compared to Fiscal Year 2015:
The increase in contract manufacturing revenue of $17,613,000 (66%) during fiscal year 2016 was primarily due to an increase in the number of manufacturing runs completed and shipped in fiscal year 2016 compared to fiscal year 2015, which was attributed to the increase in demand for contract manufacturing services from third-party customers.
License Revenue
Fiscal Years 2017 and 2016 Compared to Fiscal Years 2016 and 2015:
The changes in license revenue in fiscal years 2017 and 2016 compared to fiscal years 2016 and 2015, respectively, were directly related to revenue recognized in accordance with the terms of our existing license agreements. Based on our existing license agreements, we expect license revenue to be insignificant in fiscal year 2018.
Cost of Contract Manufacturing
Fiscal Year 2017 Compared to Fiscal Year 2016:
The increase in cost of contract manufacturing of $15,293,000 (67%) during fiscal year 2017 was primarily related to the fiscal year 2017 increase in contract manufacturing revenue. In addition, we saw a decline in our gross margin in fiscal year 2017, which was primarily attributed to higher operating costs associated with our Myford Facility to support the process validation of three separate customer products combined with the write-off of unusable work-in process inventory of $2,063,000.
Fiscal Year 2016 Compared to Fiscal Year 2015:
The increase in cost of contract manufacturing of $7,373,000 (47%) during fiscal year 2016 was directly related to the fiscal year 2016 increase in contract manufacturing revenue. In addition, we saw an improvement in our gross margin in fiscal year 2016, which was primarily attributed to greater utilization of our Franklin Facility, combined with a decrease in expenses associated with the write-off of unusable work-in process inventory.
Research and Development Expenses
Research and development expenses primarily include (i) payroll and related costs and share-based compensation expense (non-cash), associated with research and development personnel, (ii) costs related to clinical trials and preclinical testing, (iii) costs to develop and manufacture our product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses. Research and development expenses are charged to expense as incurred when these expenditures relate to our research and development efforts and have no alternative future uses.
For the years ended April 30, 2017, 2016 and 2015, approximately 91%, 100% and 98%, respectively, of our total research and development expenses related to our PS-targeting platform, which includes our lead immunotherapy candidate, bavituximab.
| 34 |
Fiscal Year 2017 Compared to Fiscal Year 2016:
In June 2016, we announced our clinical development strategy that will focus on conducting small, early stage studies of bavituximab in combination with immune stimulating therapies while we continued to wind down activities and related costs associated with the Phase III SUNRISE trial. As we executed on this clinical development strategy, research and development expenses decreased $31,232,000 (52%) during fiscal year 2017. The current fiscal year decrease in research and development expenses was primarily related to the following decreases associated with our PS-targeting platform:
| · | Decrease in third-party clinical trial costs of $15,290,000 primarily related to our discontinued Phase III SUNRISE trial (as discussed above) and previously planned Phase II trials in breast and lung cancers; |
| · | Decrease in manufacturing costs of $11,749,000 primarily related to internal and external costs and expenses incurred in the prior year associated with preparing bavituximab for commercial production; |
| · | Decrease in payroll and related expenses of $3,288,000 related to a reduction of research and development personnel combined with the reassignment of certain personnel to our contract manufacturing operations; and |
| · | Decreases in facility-related expenses and share-based compensation expense (non-cash) of $672,000 and $501,000, respectively. |
As we continue to execute our clinical development strategy, we currently expect research and development expenses for fiscal year 2018 to decrease at least 40% or more in comparison to fiscal year 2017.
Fiscal Year 2016 Compared to Fiscal Year 2015:
The increase in research and development expenses of $16,533,000 (38%) during fiscal year 2016 was directly related to the fiscal year 2016 increase in PS-targeting expenses of $16,970,000, offset by the fiscal year 2016 decrease in expenses related to our other technologies of $437,000. The fiscal year 2016 net increase in PS-targeting expenses was primarily attributed to:
| · | Increase in manufacturing costs of $10,053,000 primarily related to the internal and external costs and expenses associated with preparing bavituximab for commercial production; |
| · | Increase in third-party clinical trial costs of $6,014,000 primarily related to our discontinued Phase III SUNRISE trial (as discussed above) and previously planned Phase II trials in breast and lung cancers, which were also discontinued during fiscal year 2016; |
| · | Increase in payroll and related expenses of $1,314,000 primarily related to increased employee headcount to support the aforementioned discontinued Phase III SUNRISE trial and previously planned Phase II trials in breast and lung cancers; and |
| · | Decrease in share-based compensation expense (non-cash) of $769,000. |
Selling, General and Administrative Expenses
Selling, general and administrative (“SG&A”) expenses consist primarily of payroll and related expenses and share-based compensation expense (non-cash), for personnel in executive, finance, accounting, business development, legal, human resources, information technology, and other internal support functions. In addition, SG&A expenses include corporate and patent legal fees, audit and accounting fees, investor relation expenses, non-employee director fees, facility related expenses, and other expenses relating to our general management, administration, and business development activities.
| 35 |
Fiscal Year 2017 Compared to Fiscal Year 2016:
The increase in SG&A expenses of $783,000 (4%) during fiscal year 2017 compared to fiscal year 2016 was primarily due to current year increases in payroll and related expenses, facility related expenses and other general corporate expenses, offset by a current year decrease in share-based compensation expense (non-cash).
As discussed above, we are exploring various strategic options that we believe may enable us to enhance stockholder value, including the possible separation of our two distinct businesses. As a result, we currently expect SG&A expenses could increase up to 10% in fiscal year 2018 in comparison to fiscal year 2017.
Fiscal Year 2016 Compared to Fiscal Year 2015:
SG&A expenses for fiscal year 2016 remained consistent with fiscal year 2015, but decreased slightly by $140,000 (1%). The fiscal year 2016 decrease in SG&A expenses was primarily due to fiscal year 2016 decreases in share-based compensation expense (non-cash) and other general corporate expenses, offset by a fiscal year 2016 increase in payroll and related expenses.
Interest and Other Income
Fiscal Years 2017 and 2016 Compared to Fiscal Years 2016 and 2015:
The changes in interest and other income in fiscal years 2017 and 2016 compared to fiscal years 2016 and 2015, respectively, were directly related to the receipt in fiscal year 2016 of a $600,000 settlement payment from a former third-party vendor in accordance with the terms of the confidential settlement and release agreement we entered into during the quarter ended October 31, 2015.
Critical Accounting Policies
Our discussion and analysis of our consolidated financial position and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”). The preparation of our consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and related disclosures. We review our estimates and assumptions on an ongoing basis. We base our estimates on historical experience and on assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for our judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may vary from what we anticipate, and different assumptions or estimates about the future could change our reported results. We believe the following accounting policies to be critical to the assumptions and estimates used in the preparation of our consolidated financial statements.
Revenue Recognition
We currently derive revenue from our contract manufacturing services provided by Avid. We recognize revenue in accordance with the authoritative guidance for revenue recognition when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller’s price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple elements.
Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer or licensing partner. When deliverables are separable, consideration received is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units, which may require the use of significant judgement. Deliverables are considered separate units of accounting if (1) the delivered item(s) has value to the customer on a stand-alone basis and (2) the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
| 36 |
Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence (“VSOE”) of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, we use our best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement.
On occasion, we receive requests from customers to hold product manufactured by Avid on a “bill-and-hold” basis. Revenue is recognized for these “bill-and-hold” arrangements in accordance with the authoritative guidance, which requires, among other things, the existence of a valid business purpose for the arrangement; the “bill-and-hold” arrangement is at the request of the customer; title and risk of ownership must pass to the customer; the product is complete and ready for shipment; a fixed delivery date that is reasonable and consistent with the customer’s business practices; the product has been separated from our inventory; and no further performance obligations by us exist.
In addition, we also follow the authoritative guidance when reporting revenue as gross when we act as a principal versus reporting revenue as net when we act as an agent. For transactions in which we act as a principal, have discretion to choose suppliers, bear credit and inventory risk and perform a substantive part of the services, revenue is recorded at the gross amount billed to a customer and costs associated with these reimbursements are reflected as a component of cost of sales for contract manufacturing services.
Any amounts received prior to satisfying our revenue recognition criteria are recorded as deferred revenue or customer deposits in the accompanying consolidated financial statements. We also record a provision for estimated contract losses, if any, in the period in which they are determined.
Research and Development Expenses
Research and development expenses primarily include (i) payroll and related costs, including share-based compensation associated with research and development personnel, (ii) costs related to clinical trials and preclinical testing of our technologies under development, (iii) costs to develop and manufacture the product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses. Research and development expenses are charged to expense as incurred when these expenditures relate to our research and development efforts and have no alternative future uses.
Clinical trial costs are a significant component of our research and development expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the ongoing development of our product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow. Expenses related to clinical trials are accrued based on our estimates and/or representations from third parties (including clinical research organizations) regarding services performed. If the contracted amounts are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we modify our accruals accordingly on a prospective basis. Revisions in the scope of a contract are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. There were no material adjustments for a change in estimate to research and development expenses in the accompanying consolidated financial statements in any of the three years ended April 30, 2017.
Under certain research and development agreements, we are obligated to make certain advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities and are deferred and capitalized as prepaid research and development expenses. These advance payments are recognized as an expense in the period the related goods are delivered or the related services are performed. We assess our prepaid research and development expenses for impairment when events or changes in circumstances indicate that the carrying amount of the prepaid expense may not be recoverable or provide future economic benefit.
| 37 |
In addition, under certain in-licensing agreements associated with the research and development of our product candidates, we are obligated to pay certain milestone payments based on potential clinical development and regulatory milestones (as described in Note 4 to the accompanying consolidated financial statements). These milestone payments have no alternative future uses (in other research and development projects or otherwise) and therefore have no separate economic values and are expensed as research and development costs at the time the costs are incurred. We have no in-licensed product candidates that have alternative future uses in research and development projects or otherwise. In addition, we do not perform any research and development activities for any unrelated entities.
Share-based Compensation
We account for stock options and other share-based awards granted under our equity compensation plans in accordance with the authoritative guidance for share-based compensation. The estimated fair value of share-based payments to employees in exchange for services is measured at the grant date, using a fair value based method, such as a Black-Scholes option valuation model, and is recognized as expense on a straight-line basis over the requisite service periods. The fair value of modifications to share-based awards, if any, is generally estimated using a Black-Scholes option valuation model, unless a lattice model is required. Share-based compensation expense recognized during the period is based on the value of the portion of the share-based payment that is ultimately expected to vest during the period and is reduced for estimated forfeitures. The authoritative guidance requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. As of April 30, 2017, there were no outstanding share-based awards with market or performance conditions.
The estimated fair value of stock options are measured at the grant date, using a fair value based method, such as a Black-Scholes option valuation model, and is amortized as compensation expense on a straight-line basis over the requisite service period of the award, which is generally the vesting period. The use of a valuation model requires us to make certain estimates and assumptions with respect to selected model inputs. The expected volatility is based on the daily historical volatility of our common stock covering the estimated expected term. The expected term of options granted reflects actual historical exercise activity and assumptions regarding future exercise activity of unexercised, outstanding options. The risk-free interest rate is based on U.S. Treasury notes with terms within the contractual life of the option at the time of grant. The expected dividend yield assumption is based on our expectation of future dividend payouts. We have never declared or paid any cash dividends on our common stock and currently do not anticipate paying such cash dividends. In addition, guidance requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates.
If factors change and we employ different assumptions in the determination of fair value in future periods, the share-based compensation expense that we record may differ significantly from what we have recorded in the current period. There are a number of factors that affect the amount of share-based compensation expense, including the number of employee options granted during subsequent fiscal years, the price of our common stock on the date of grant, the volatility of our stock price, the estimate of the expected life of options granted and the risk-free interest rates.
In addition, we periodically grant stock options and other share-based awards to non-employee consultants, which we account for in accordance with the authoritative guidance for share-based compensation. The cost of non-employee services received in exchange for share-based awards are measured based on either the fair value of the consideration received or the fair value of the share-based award issued, whichever is more reliably measurable. In addition, guidance requires share-based compensation related to unvested options and awards issued to non-employees to be recalculated at the end of each reporting period based upon the fair market value on that date until the share-based award has vested, and any cumulative catch-up adjustment to share-based compensation resulting from the re-measurement is recognized in the current period.
| 38 |
Liquidity and Capital Resources
At April 30, 2017, we had $46,799,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced losses and negative cash flows from operations since our inception and we expect negative cash flows from operations to continue for the foreseeable future until we can generate sufficient revenue from Avid’s contract manufacturing services to achieve profitability. Therefore, unless and until we are able to generate sufficient revenue from Avid’s contract manufacturing services or from the sale or licensing of our product candidates under development, we expect such losses to continue through at least the fiscal year ending April 30, 2018, and as a result, we will require additional capital to fund our operations and to execute our business plans.
Our ability to continue to fund our operations is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, (i) raising additional capital in the equity markets, (ii) generating additional contract manufacturing revenue from Avid, or (iii) licensing or partnering our product in development.
Historically, we have funded a significant portion of our operations through the issuance of equity. During fiscal year 2017, we raised $31,277,000 in aggregate gross proceeds from the sale of shares of our common stock and raised an additional $1,634,000 in aggregate gross proceeds from the sale of shares of our 10.50% Series E Convertible Preferred Stock (the “Series E Preferred Stock”) (as described in Note 5 to the accompanying consolidated financial statements). Subsequent to April 30, 2017 and through June 30, 2017, we raised an additional $4,304,000 in aggregate gross proceeds from the sale of shares of our common stock (as described in Note 12 to the accompanying consolidated financial statements). As of July 14, 2017, $67,674,000 remained available to us under our effective shelf registration statement, which allows us from time to time to offer and sell shares of our common stock, in one or more offerings, either individually or in combination.
Our ability to raise additional capital in the equity markets to fund our obligations in future periods is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse financial results, and negative research and development results. In addition, on July 7, 2017, we effected a reverse stock split of our issued and outstanding common stock at a ratio of one-for-seven, which took effect with the opening of trading on July 10, 2017 (as described in Note 1 to the accompanying consolidated financial statements). While the reverse stock split resulted in an increase in the market price of our common stock, it also reduced the number of shares outstanding which could adversely affect the liquidity of our common stock. Because we have predominately raised capital through sales arrangements that are dependent on the trading volume of our common stock, any adverse effect on liquidity could negatively impact our ability to raise additional capital in the equity markets. In such a case, we may not be able to rely on the sale of shares of our common stock to fund our operations to the extent we have in prior years.
With respect to our ability to generate additional contract manufacturing revenue, Avid currently has a revenue backlog of $58 million under signed contracts from existing customers, which is consistent with the revenue backlog we reported as of April 30, 2016. As such, we may not be able to rely on generating additional contract manufacturing revenue from Avid in fiscal year 2018 to make up any capital raising shortfall experienced through the equity markets.
If we are unable to either (i) raise sufficient capital in the equity markets, (ii) generate additional contract manufacturing revenue from Avid, or (iii) license or partner our product in development, or any combination thereof, we may need to delay, scale back, or eliminate some or all our research and development efforts, or restructure our operations. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
As a result, we have concluded that there is substantial doubt about our ability to continue as a going concern within one year after the date that our financial statements are issued. Our independent registered public accounting firm included an explanatory paragraph highlighting this uncertainty in its “Report of Independent Registered Public Accounting Firm” dated July 14, 2017, which report is included in Item 15 of Part IV of this Annual Report.
| 39 |
Significant components of the changes in cash flows from operating, investing and financing activities for the fiscal years ended April 30, 2017, 2016 and 2015 are as follows:
Cash Used In Operating Activities. Net cash used in operating activities represents our (i) net loss, as reported, (ii) less non-cash operating expenses, and (iii) net changes in the timing of cash flows as reflected by the changes in operating assets and liabilities, as described in the below table:
| Fiscal Year Ended April 30, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Net loss, as reported | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | |||
| Less non-cash operating expenses: | ||||||||||||
| Share-based compensation | 3,363,000 | 4,898,000 | 6,702,000 | |||||||||
| Depreciation and amortization | 2,463,000 | 1,535,000 | 1,041,000 | |||||||||
| Loss on disposal of property and equipment | 1,000 | 14,000 | 2,000 | |||||||||
| Net cash used in operating activities before changes in operating assets and liabilities | $ | (22,332,000 | ) | $ | (49,205,000 | ) | $ | (42,613,000 | ) | |||
| Net change in operating assets and liabilities | $ | (17,454,000 | ) | $ | 9,614,000 | $ | 6,594,000 | |||||
| Net cash used in operating activities | $ | (39,786,000 | ) | $ | (39,591,000 | ) | $ | (36,019,000 | ) | |||
Net cash used in operating activities increased $195,000 to $39,786,000 for fiscal year 2017 compared to net cash used in operating activities of $39,591,000 for fiscal year 2016. This increase in net cash used in operating activities was due to a net change in operating assets and liabilities of $27,068,000 due to the timing of cash receipts and expenditures primarily associated with customer deposits, deferred revenue, accrued clinical trial and related fees, inventories, and trade and other receivables, offset by a decrease of $26,873,000 in net loss reported for fiscal year 2017 after deducting non-cash operating expenses as described in the above table.
Net cash used in operating activities increased $3,572,000 to $39,591,000 for fiscal year 2016 compared to net cash used in operating activities of $36,019,000 for fiscal year 2015. This increase in net cash used in operating activities was due to an increase of $6,592,000 in net loss reported for fiscal year 2016 after deducting non-cash operating expenses as described in the above table, offset by a net change in operating assets and liabilities of $3,020,000 due to the timing of cash receipts and expenditures.
Cash Used In Investing Activities. Net cash used in investing activities for the fiscal years ended April 30, 2017, 2016, and 2015, was $2,992,000, $8,791,000, and $8,449,000, respectively.
Cash used in investing activities during fiscal year 2017 consisted of property and equipment acquisitions of $1,501,000 related to our manufacturing operations combined with an increase in other assets of $1,491,000 primarily related to deposits and/or progress payments for certain equipment to support the growth of our manufacturing operations.
Cash used in investing activities during fiscal year 2016 consisted of property and equipment acquisitions of $9,324,000 offset by a decrease in other assets of $533,000. Property and equipment acquisitions during fiscal year 2016 primarily related to costs associated with the construction of our Myford Facility to support Avid’s projected revenue growth and to support the manufacturing of our product candidates. The construction of the Myford Facility was completed and placed into service during fiscal year 2016.
Cash used in investing activities during fiscal year 2015 consisted of property and equipment acquisitions of $9,047,000 offset by a decrease in other assets of $598,000. Property and equipment acquisitions during fiscal year 2015 primarily related to construction-in-progress associated with the construction of the aforementioned Myford Facility, the implementation of an enterprise resource planning system, and the acquisition of laboratory equipment.
| 40 |
Cash Provided By Financing Activities. Net cash provided by financing activities for the fiscal years ended April 30, 2017, 2016, and 2015, was $28,165,000, $41,793,000 and $34,979,000, respectively.
Net cash provided by financing activities during fiscal year 2017 consisted of (i) $17,759,000 in net proceeds from the sale of shares of our common stock under an At Market Issuance Sales Agreement, (ii) $12,691,000 in net proceeds from the sale of shares of our common stock under an Equity Distribution Agreement, (iii) $1,576,000 in net proceeds from the sale of shares of our Series E Preferred Stock under a separate At Market Issuance Sales Agreement, (iv) $526,000 in net proceeds from the purchase of shares of our common stock under our Employee Stock Purchase Plan (the “ESPP”), and (v) $31,000 in net proceeds from stock option exercises, which amounts were offset by dividends paid on our issued and outstanding Series E Preferred Stock of $4,279,000 and principal payments on a capital lease of $139,000.
Net cash provided by financing activities during fiscal year 2016 consisted of (i) $19,999,000 in net proceeds from the sale of shares of our common stock under a Common Stock Purchase Agreement, (ii) $18,402,000 in net proceeds from the sale of shares of our common stock under two separate At Market Issuance Sales Agreements, (iii) $6,794,000 in net proceeds from the sale of shares of our common stock under an Equity Distribution Agreement, (iv) $540,000 in net proceeds from the purchase of shares of our common stock under our ESPP, (v) $138,000 in net proceeds from stock option exercises, and (vi) $59,000 in net proceeds from the sale of shares of our Series E Preferred Stock under a separate At Market Issuance Sales Agreement, which amounts were offset by dividends paid on our issued and outstanding Series E Preferred Stock of $4,139,000.
Net cash provided by financing activities during fiscal year 2015 consisted of (i) $19,235,00 in net proceeds from the sale of shares of our common stock under two separate At Market Issuance Sales Agreements, (ii) $18,203,000 in net proceeds from the sale of shares of our Series E Preferred Stock under a separate At Market Issuance Sales Agreement (iii) $608,000 in net proceeds from the purchase of shares of our common stock under our ESPP, and (iv) $298,000 in net proceeds from stock option exercises, which amounts were offset by dividends paid on our issued and outstanding Series E Preferred Stock of $3,352,000 and principal payments on a capital lease of $13,000.
Contractual Obligations
Contractual obligations represent future cash commitments and liabilities under agreements with third parties, and exclude contractual liabilities already recorded on our consolidated balance sheet as current liabilities and contingent liabilities for which we cannot reasonably predict future payments. The following chart represents our contractual obligations as of April 30, 2017, aggregated by type:
| Payments Due by Period | ||||||||||||||||||||
| Total | < 1 year | 1-3 years | 4-5 years | After 5 years | ||||||||||||||||
| Operating leases (1) | $ | 26,099,000 | $ | 2,741,000 | $ | 6,091,000 | $ | 5,905,000 | $ | 11,362,000 | ||||||||||
| Capital lease obligation (2) | 180,000 | 180,000 | – | – | – | |||||||||||||||
| Purchase obligations (3) | 4,469,000 | 1,089,000 | 3,380,000 | – | – | |||||||||||||||
| Total contractual obligations | $ | 30,748,000 | $ | 4,010,000 | $ | 9,471,000 | $ | 5,905,000 | $ | 11,362,000 | ||||||||||
______________
| (1) | Represents future minimum lease payments under all non-cancelable operating leases including our facility operating leases as further described in Note 3 to the accompanying audited consolidated financial statements. |
| (2) | Represents a capital lease agreement to finance certain office equipment. Amounts include principal and interest. |
| (3) | Primarily represents non-cancellable purchase orders for certain consumables associated with our single use bioreactors in our Myford Facility. |
Off Balance Sheet Arrangements.
We do not have any off balance sheet arrangements, as defined in Item 303 of Regulation S-K.
Recently Issued Accounting Pronouncements
See Note 2, Summary of Significant Accounting Policies - Pending Adoption of Recent Accounting Pronouncements, in the accompanying Notes to Consolidated Financial Statements for a discussion of recent accounting pronouncements and their effect, if any, on our consolidated financial statements.
| 41 |
| Item 7A. | Quantitative And Qualitative Disclosures About Market Risk |
Our cash and cash equivalents are primarily invested in money market funds with one major commercial bank with the primary objective to preserve our principal balance. Our deposits held with this bank exceed the amount of government insurance limits provided on our deposits and, therefore, we are exposed to credit risk in the event of default by the major commercial bank holding our cash balances. However, these deposits may be redeemed upon demand and, therefore, bear minimal risk. In addition, while changes in U.S. interest rates would affect the interest earned on our cash balances at April 30, 2017, such changes would not have a material adverse effect on our financial position or results of operations based on historical movements in interest rates.
| Item 8. | Financial Statements And Supplementary Data |
The information required by this Item is incorporated by reference to the financial statements set forth in Item 15 of Part IV of this Annual Report, “Exhibits and Financial Statement Schedules.”
| Item 9. | Changes In And Disagreements With Accountants On Accounting And Financial Disclosures |
None.
| Item 9A. | Controls And Procedures |
(a) Evaluation of Disclosure Controls and Procedures. The term “disclosure controls and procedures” (defined in Rule 13a-15(e) under the Exchange Act refers to the controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files under the Exchange Act is recorded, processed, summarized and reported within the required time periods. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we have conducted an evaluation of the effectiveness of the design and operation of our disclosure controls and procedures as of April 30, 2017. Based on this evaluation, our president and chief executive officer and our chief financial officer concluded that our disclosure controls and procedures were effective as of April 30, 2017 to ensure the timely disclosure of required information in our SEC filings.
(b) Management’s Report on Internal Control Over Financial Reporting. Management’s Report on Internal Control Over Financial Reporting and the report of our independent registered public accounting firm on our internal control over financial reporting, which appear on the following pages, are incorporated herein by this reference.
(c) Changes in Internal Control over Financial Reporting. There have been no significant changes in our internal control over financial reporting during the fourth quarter of the fiscal year ended April 30, 2017 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| Item 9B. | Other Information |
None.
| 42 |
PEREGRINE PHARMACEUTICALS, INC.
MANAGEMENT’S REPORT ON
INTERNAL CONTROL OVER FINANCIAL REPORTING
The management of the Company is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. The Company’s internal control over financial reporting is a process designed, as defined in Rule 13a-15(f) and Rule 15d-15(f) under the Securities Exchange Act of 1934, as amended, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements for external purposes in accordance with generally accepted accounting principles.
The Company’s internal control over financial reporting is supported by written policies and procedures that:
| · | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the Company’s assets; |
| · | provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the Company are being made only in accordance with authorizations of the Company’s management and directors; and |
| · | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s assets that could have a material effect on the consolidated financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In connection with the preparation of the Company’s annual consolidated financial statements, management of the Company has undertaken an assessment of the effectiveness of the Company’s internal control over financial reporting based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework). Management’s assessment included an evaluation of the design of the Company’s internal control over financial reporting and testing of the operational effectiveness of the Company’s internal control over financial reporting.
Based on this assessment, management has concluded that the Company’s internal control over financial reporting was effective as of April 30, 2017.
Ernst & Young LLP, the independent registered public accounting firm that audited the company’s consolidated financial statements included in this Annual Report on Form 10-K, has issued an attestation report on the Company’s internal control over financial reporting which appears on the following page.
| By: | /s/ Steven W. King | By: | /s/ Paul J. Lytle | |||||
| Steven W. King | Paul J. Lytle | |||||||
| President and Chief Executive Officer | Chief Financial Officer |
July 14, 2017
| 43 |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Peregrine Pharmaceuticals, Inc.
We have audited Peregrine Pharmaceuticals, Inc.’s internal control over financial reporting as of April 30, 2017, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). Peregrine Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Peregrine Pharmaceuticals, Inc.’s Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Peregrine Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of April 30, 2017, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of Peregrine Pharmaceuticals, Inc. as of April 30, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended April 30, 2017 of Peregrine Pharmaceuticals Inc., and our report dated July 14, 2017 expressed an unqualified opinion thereon that included an explanatory paragraph regarding Peregrine Pharmaceuticals, Inc.’s ability to continue as a going concern.
/s/ Ernst & Young LLP
Irvine, California
July 14, 2017
| 44 |
| Item 10. | Directors, Executive Officers And Corporate Governance |
The information required by this Item regarding our directors, executive officers and committees of our board of directors is incorporated by reference to the information set forth under the captions “Election of Directors,” “Executive Compensation” and “Corporate Governance” in our 2017 Definitive Proxy Statement to be filed within 120 days after the end of our fiscal year ended April 30, 2017 (the “2017 Definitive Proxy Statement”).
Information required by this Item regarding Section 16(a) reporting compliance is incorporated by reference to the information set forth under the caption “Section 16(a) Beneficial Ownership Reporting Compliance” in our 2017 Definitive Proxy Statement.
Information required by this Item regarding our code of ethics is incorporated by reference to the information set forth under the caption “Corporate Governance” in our 2017 Definitive Proxy Statement.
| Item 11. | Executive Compensation |
The information required by this Item is incorporated by reference to the information set forth under the captions “Director Compensation,” “Compensation Discussion and Analysis” and “Executive Compensation” in our 2017 Definitive Proxy Statement to be filed within 120 days after the end of our fiscal year ended April 30, 2017.
| Item 12. | Security Ownership Of Certain Beneficial Owners And Management And Related Stockholder Matters |
Other than as set forth below, the information required by this Item is incorporated by reference to the information set forth under the caption “Security Ownership of Certain Beneficial Owners, Directors and Management” in our 2017 Definitive Proxy Statement to be filed within 120 days after the end of our fiscal year ended April 30, 2017.
Equity Compensation Plan Information
We currently maintain six equity compensation plans: the 2002 Stock Incentive Plan (the “2002 Plan”), the 2003 Stock Incentive Plan (the “2003 Plan”), the 2005 Stock Incentive Plan (the “2005 Plan”), the 2009 Stock Incentive Plan (the “2009 Plan”), the 2010 Stock Incentive Plan (the “2010 Plan”) and the 2011 Stock Incentive Plan, as amended on October 15, 2015 (the “2011 Plan”), in addition to which we maintain our Employee Stock Purchase Plan. The 2003 Plan, 2005 Plan, 2009 Plan, 2010 Plan and 2011 Plan, as well as the Employee Stock Purchase Plan, were approved by our stockholders, while we did not submit the 2002 Plan for stockholder approval.
The 2002 Plan, which expired in June 2012, was a broad-based non-qualified stock option plan for the issuance of up to 85,714 options. The 2002 Plan provided for the granting of options to purchase shares of our common stock at prices not less than the fair market value of our common stock at the date of grant and generally expired ten years after the date of grant. No additional options can be granted under the expired 2002 Plan, however, the terms of the 2002 Plan remain in effect with respect to outstanding options granted under the 2002 Plan until they are exercised, canceled or expired.
| 45 |
The following table sets forth certain information as of April 30, 2017 concerning our common stock that may be issued upon the exercise of options or pursuant to purchases of stock under all of our equity compensation plans approved by stockholders and equity compensation plans not approved by stockholders in effect as of April 30, 2017:
| Plan Category | (a) Number of Securities to be Issued Upon the Exercise of Outstanding Options, Warrants and Rights | (b) Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights ($/share) | (c) Number of Shares Remaining Available for Future Issuance Under Equity Compensation Plans (Excluding Securities Reflected in Column (a)) | |||||||||
| Equity compensation plans approved by stockholders (1) | 4,050,332 | 8.72 | 1,549,324 | |||||||||
| Equity compensation plans not approved by stockholders (1) | 31,216 | (2) | 14.79 | – | ||||||||
| Employee Stock Purchase Plan approved by stockholders (1) | – | – | 1,359,736 | |||||||||
| Total | 4,081,548 | (3) | 8.77 | (4) | 2,909,060 | |||||||
__________
| (1) | All share and per share amounts of our common stock for all periods presented have been retroactively adjusted to reflect the one-for-seven reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017 (as described in Note 1 to the accompanying consolidated financial statements). |
| (2) | Includes 5,130 options granted in a previous fiscal year to one of our named executive officers. |
| (3) | Represents shares to be issued upon the exercise of outstanding options. There were no shares of common stock subject to restricted stock awards as of April 30, 2017. |
| (4) | Represents the weighted-average exercise price of outstanding options. |
| Item 13. | Certain Relationships And Related Transactions, And Director Independence |
The information required by this Item is incorporated by reference to the information set forth under the captions “Certain Relationships and Related Transactions,” “Director Independence” and “Compensation Committee Interlocks and Insider Participation” in our 2017 Definitive Proxy Statement to be filed within 120 days after the end of our fiscal year ended April 30, 2017.
| Item 14. | Principal Accounting Fees and Services |
The information required by this Item is incorporated by reference to the information set forth under the caption “Independent Registered Public Accounting Firm Fees” in our 2017 Definitive Proxy Statement to be filed within 120 days after the end of our fiscal year ended April 30, 2017.
| 46 |
| Item 15. | Exhibits And Financial Statement Schedules |
| (a) | (1) | Consolidated Financial Statements |
| Index to consolidated financial statements filed as part of this Form 10-K: |
| (2) | Financial Statement Schedules |
All schedules are omitted as the required information is inapplicable, or the information is presented in the consolidated financial statements or related notes.
| 47 |
| (3) | Exhibits |
| Exhibit Number |
Description | |
| 3.1 | Certificate of Incorporation of Peregrine Pharmaceuticals, Inc., a Delaware corporation, as amended through July 7, 2017. *** | |
| 3.2 | Amended and Restated Bylaws of Peregrine Pharmaceuticals, Inc., a Delaware corporation (Incorporated by reference to Exhibit 3.2 to Registrant’s Current Report on Form 8-K as filed with the Commission on November 14, 2014). | |
| 4.1 | Form of Certificate for Common Stock (Incorporated by reference to Exhibit 4.1 to Registrant’s Annual Report on Form 10-K for the year end April 30, 1988). | |
| 4.2 | Form of Non-qualified Stock Option Agreement by and between Registrant, Director and certain consultants dated December 22, 1999 (Incorporated by reference to Exhibit 4.16 to Registrant’s Registration Statement on Form S-3 (File No. 333-40716)). * | |
| 4.3 | Peregrine Pharmaceuticals, Inc. 2002 Non-Qualified Stock Option Plan (Incorporated by reference to Exhibit 4.17 to Registrant’s Registration Statement on Form S-8 (File No. 333-106385)). * | |
| 4.4 | Form of 2002 Non-Qualified Stock Option Agreement (Incorporated by reference to Exhibit 4.18 to Registrant’s Registration Statement on Form S-8 (File No. 333-106385)). * | |
| 4.5 | Amended and Restated Rights Agreement, dated March 16, 2016, between Peregrine Pharmaceuticals, Inc. and Broadridge Corporate Issuer Solutions, Inc., as Rights Agent (Incorporated by reference to Exhibit 4.1 to Registrant’s Current Report on Form 8-K as filed with the Commission on March 17, 2016). | |
| 4.6 | 2003 Stock Incentive Plan Non-qualified Stock Option Agreement (Incorporated by reference to Exhibit 10.95 to Registrant’s Registration Statement on Form S-8 (File No. 333-121334)). * | |
| 4.7 | 2003 Stock Incentive Plan Incentive Stock Option Agreement (Incorporated by reference to Exhibit 10.96 to Registrant’s Registration Statement on Form S-8 (File No. 333-121334)). * | |
| 4.8 | Form of Incentive Stock Option Agreement for 2005 Stock Incentive Plan (Incorporated by reference to Exhibit 10.98 to Registrant’s Current Report on Form 8-K as filed with the Commission on October 28, 2005). * | |
| 4.9 | Form of Non-Qualified Stock Option Agreement for 2005 Stock Incentive Plan (Incorporated by reference to Exhibit 10.99 to Registrant’s Current Report on Form 8-K as filed with the Commission on October 28, 2005). * | |
| 4.10 | Peregrine Pharmaceuticals, Inc., 2005 Stock Incentive Plan (Incorporated by reference to Exhibit B to Registrant’s Definitive Proxy Statement filed with the Commission on August 29, 2005). * | |
| 4.11 | Form of Incentive Stock Option Agreement for 2009 Stock Incentive Plan (Incorporated by reference to Exhibit 4.14 to Registrant’s Current Report on Form 8-K as filed with the Commission on October 27, 2009). * | |
| 4.12 | Form of Non-Qualified Stock Option Agreement for 2009 Stock Incentive Plan (Incorporated by reference to Exhibit 4.15 to Registrant’s Current Report on Form 8-K as filed with the Commission on October 27, 2009). * | |
| 4.13 | Form of Restricted Stock Issuance Agreement dated February 1, 2010 (Incorporated by reference to Exhibit 4.15 to Registrant’s Annual Report on Form 10-K as filed with the Commission on July 14, 2011). * | |
| 4.14 | 2010 Stock Incentive Plan (Incorporated by reference to Exhibit A to Registrant’s Definitive Proxy Statement filed with the Commission on August 27, 2010). * | |
| 4.15 | Form of Stock Option Award Agreement under 2010 Stock Incentive Plan (Incorporated by reference to Exhibit 4.17 to Registrant’s Registration Statement on Form S-8 (File No. 333-171067)). * | |
| 4.16 | 2010 Employee Stock Purchase Plan (Incorporated by reference to Exhibit B to Registrant’s Definitive Proxy Statement filed with the Commission on August 27, 2010). * |
| 48 |
| 49 |
| Exhibit Number |
Description | |
| 10.6 | Amendment No. 1 to Exclusive Patent License agreement between The University of Texas System and Peregrine Pharmaceuticals, Inc., dated June 1, 2009 (Incorporated by reference to Exhibit 10.20 to Registrant’s Current Report on Form 8-K as filed with the Commission on April 14, 2010). ** | |
| 10.7 | Non-Exclusive Cabilly Patent License Agreement between Genentech, Inc. and Peregrine Pharmaceuticals, Inc., effective as of November 5, 2003 (Incorporated by reference to Exhibit 10.21 to Registrant’s Current Report on Form 8-K as filed with the Commission on April 14, 2010). ** | |
| 10.8 | Commercial License Agreement between Avanir Pharmaceuticals, Inc. and Peregrine Pharmaceuticals, Inc., dated December 1, 2003 (Incorporated by reference to Exhibit 10.22 to Registrant’s Current Report on Form 8-K as filed with the Commission on April 14, 2010). ** | |
| 10.9 | License Agreement between Lonza Biologics PLC and Peregrine Pharmaceuticals, Inc., dated March 1, 2005 (Incorporated by reference to Exhibit 10.24 to Registrant’s Current Report on Form 8-K as filed with the Commission on April 14, 2010). ** | |
| 10.10 | Annual Bonus Plan for Executive Officers adopted July 12, 2011(Incorporated by reference to Exhibit 10.29 to Registrant’s Annual Report on Form 10-K as filed with the Commission on July 14, 2011). * | |
| 10.11 | Warrant to Purchase Stock issued to Oxford Finance LLC, dated August 30, 2012 (Incorporated by reference to Exhibit 10.29 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on December 10, 2012). | |
| 10.12 | Warrant to Purchase Stock issued to Midcap Financial SBIC LP, dated August 30, 2012 (Incorporated by reference to Exhibit 10.30 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on December 10, 2012). | |
| 10.13 | Warrant to Purchase Stock issued to Silicon Valley Bank, dated August 30, 2012 (Incorporated by reference to Exhibit 10.31 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on December 10, 2012). | |
| 10.14 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Steven W. King, effective December 27, 2012 (Incorporated by reference to Exhibit 10.34 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on March 12, 2013). * | |
| 10.15 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Paul J. Lytle, effective December 27, 2012 (Incorporated by reference to Exhibit 10.35 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on March 12, 2013). * | |
| 10.16 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Shelley P.M. Fussey, Ph.D., effective December 27, 2012 (Incorporated by reference to Exhibit 10.36 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on March 12, 2013). * | |
| 10.17 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Joseph Shan, effective December 27, 2012 (Incorporated by reference to Exhibit 10.37 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on March 12, 2013). * | |
| 10.18 | Amended and Restated Employment Agreement by and between Peregrine Pharmaceuticals, Inc. and Mark R. Ziebell, effective December 27, 2012 (Incorporated by reference to Exhibit 10.38 to Registrant’s Quarterly Report on Form 10-Q as filed with the Commission on March 12, 2013). * | |
| 10.19 | At Market Issuance Sales Agreement, dated June 13, 2014, by and between Peregrine Pharmaceuticals, Inc. and MLV & Co. LLC (Incorporated by reference to Exhibit 10.28 to Registrant’s Current Report on Form 8-K as filed with the Commission on June 16, 2014). | |
| 10.20 | At Market Issuance Sales Agreement, dated August 7, 2015, by and between Peregrine Pharmaceuticals, Inc. and MLV & Co. LLC (Incorporated by reference to Exhibit 10.26 to Registrant’s Current Report on Form 8-K as filed with the Commission on August 7, 2015). |
| 50 |
| Exhibit Number |
Description | |
| 10.21 | Equity Distribution Agreement, dated August 7, 2015, by and between Peregrine Pharmaceuticals, Inc. and Noble International Investments, Inc., doing business as Noble Life Science Partners, a division of Noble Financial Capital Markets (Incorporated by reference to Exhibit 10.27 to Registrant’s Current Report on Form 8-K as filed with the Commission on August 7, 2015). | |
| 21 | Subsidiaries of Registrant. *** | |
| 23.1 | Consent of Independent Registered Public Accounting Firm. *** | |
| 24 | Power of Attorney (included on signature page of Annual Report). *** | |
| 31.1 | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended. *** | |
| 31.2 | Certification of Chief Financial Officer pursuant to Rule 13a-14(a)/15d-14(a) under the Securities Exchange Act of 1934, as amended. *** | |
| 32 | Certification of Chief Executive Officer and Chief Financial Officer pursuant to Rule 13a-14(b)/15d-14(b) under the Securities Exchange Act of 1934, as amended, and 18 U.S.C. Section 1350. *** | |
| 101.INS | XBRL Taxonomy Extension Instance Document. *** | |
| 101.SCH | XBRL Taxonomy Extension Schema Document. *** | |
| 101.CAL | XBRL Taxonomy Extension Calculation Linkbase Document. *** | |
| 101.DEF | XBRL Taxonomy Extension Definition Linkbase Document. *** | |
| 101.LAB | XBRL Taxonomy Extension Label Linkbase Document. *** | |
| 101.PRE | XBRL Presentation Extension Linkbase Document. *** | |
| _______________________________ | ||
|
* ** ***
|
This Exhibit is a management contract or a compensation plan or arrangement. Portions omitted pursuant to a request of confidentiality filed separately with the SEC. Filed herewith. | |
| 51 |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
| PEREGRINE PHARMACEUTICALS, INC. | |
| Dated: July 14, 2017 | By: /s/ Steven W. King |
| Steven W. King, | |
| President and Chief Executive Officer |
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Steven W. King, President and Chief Executive Officer, and Paul J. Lytle, Chief Financial Officer, and each of them, his true and lawful attorneys-in-fact and agents, with the full power of substitution and re-substitution, for him and in his name, place and stead, in any and all capacities, to sign any amendments to this report, and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto each said attorney-in-fact and agent full power and authority to do and perform each and every act in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or either of them, or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this Report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
| Signature | Capacity | Date |
| /s/ Steven W. King | President and Chief Executive | July 14, 2017 |
| Steven W. King | Officer (Principal Executive | |
| Officer), and Director | ||
| /s/ Paul J. Lytle | Chief Financial Officer | July 14, 2017 |
| Paul J. Lytle | (Principal Financial and | |
| Principal Accounting Officer) | ||
| /s/ Carlton M. Johnson | Director | July 14, 2017 |
| Carlton M. Johnson | ||
| /s/ David H. Pohl | Director | July 14, 2017 |
| David H. Pohl | ||
| /s/ Eric S. Swartz | Director | July 14, 2017 |
| Eric S. Swartz |
| 52 |
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders of Peregrine Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of Peregrine Pharmaceuticals, Inc. as of April 30, 2017 and 2016, and the related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended April 30, 2017. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Peregrine Pharmaceuticals, Inc. at April 30, 2017 and 2016, and the consolidated results of its operations and its cash flows for each of the three years in the period ended April 30, 2017, in conformity with U.S. generally accepted accounting principles.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has recurring losses from operations and negative cash flows from operating activities that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Peregrine Pharmaceuticals, Inc.’s internal control over financial reporting as of April 30, 2017, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated July 14, 2017, expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Irvine, California
July 14, 2017
| F-1 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
AS OF APRIL 30, 2017 AND 2016
| 2017 | 2016 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS: | ||||||||
| Cash and cash equivalents | $ | 46,799,000 | $ | 61,412,000 | ||||
| Trade and other receivables | 7,742,000 | 2,859,000 | ||||||
| Inventories | 33,099,000 | 16,186,000 | ||||||
| Prepaid expenses | 1,460,000 | 1,351,000 | ||||||
| Total current assets | 89,100,000 | 81,808,000 | ||||||
| PROPERTY AND EQUIPMENT: | ||||||||
| Leasehold improvements | 20,098,000 | 19,610,000 | ||||||
| Laboratory equipment | 10,777,000 | 10,257,000 | ||||||
| Furniture, fixtures, office equipment and software | 4,499,000 | 4,045,000 | ||||||
| 35,374,000 | 33,912,000 | |||||||
| Less accumulated depreciation and amortization | (11,700,000 | ) | (9,610,000 | ) | ||||
| Property and equipment, net | 23,674,000 | 24,302,000 | ||||||
| Restricted cash | 1,150,000 | 600,000 | ||||||
| Other assets | 4,188,000 | 2,333,000 | ||||||
| TOTAL ASSETS | $ | 118,112,000 | $ | 109,043,000 | ||||
| F-2 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
AS OF APRIL 30, 2017 AND 2016 (continued)
| 2017 | 2016 | |||||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| CURRENT LIABILITIES: | ||||||||
| Accounts payable | $ | 5,779,000 | $ | 8,429,000 | ||||
| Accrued clinical trial and related fees | 4,558,000 | 7,594,000 | ||||||
| Accrued payroll and related costs | 6,084,000 | 5,821,000 | ||||||
| Deferred revenue | 28,500,000 | 10,030,000 | ||||||
| Customer deposits | 17,017,000 | 24,212,000 | ||||||
| Other current liabilities | 993,000 | 1,488,000 | ||||||
| Total current liabilities | 62,931,000 | 57,574,000 | ||||||
| Deferred rent, less current portion | 1,599,000 | 1,395,000 | ||||||
| Commitments and contingencies | ||||||||
| STOCKHOLDERS' EQUITY (1): | ||||||||
| Preferred stock - $.001 par value; authorized 5,000,000 shares; issued and outstanding - 1,647,760 and 1,577,440, respectively | 2,000 | 2,000 | ||||||
| Common stock - $.001 par value; authorized 500,000,000 shares; issued and outstanding - 44,014,040 and 33,847,213, respectively | 44,000 | 34,000 | ||||||
| Additional paid-in-capital | 590,971,000 | 559,314,000 | ||||||
| Accumulated deficit | (537,435,000 | ) | (509,276,000 | ) | ||||
| Total stockholders' equity | 53,582,000 | 50,074,000 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS' EQUITY | $ | 118,112,000 | $ | 109,043,000 | ||||
| (1) | All share and per share amounts of our common stock for all periods presented have been retroactively adjusted to reflect the one-for-seven reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017 (Note 1). |
See accompanying notes to consolidated financial statements.
| F-3 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017
| 2017 | 2016 | 2015 | ||||||||||
| REVENUES: | ||||||||||||
| Contract manufacturing revenue | $ | 57,630,000 | $ | 44,357,000 | $ | 26,744,000 | ||||||
| License revenue | – | 329,000 | 37,000 | |||||||||
| Total revenues | 57,630,000 | 44,686,000 | 26,781,000 | |||||||||
| COSTS AND EXPENSES: | ||||||||||||
| Cost of contract manufacturing | 38,259,000 | 22,966,000 | 15,593,000 | |||||||||
| Research and development | 28,297,000 | 59,529,000 | 42,996,000 | |||||||||
| Selling, general and administrative | 19,334,000 | 18,551,000 | 18,691,000 | |||||||||
| Total costs and expenses | 85,890,000 | 101,046,000 | 77,280,000 | |||||||||
| LOSS FROM OPERATIONS | (28,260,000 | ) | (56,360,000 | ) | (50,499,000 | ) | ||||||
| OTHER INCOME (EXPENSE): | ||||||||||||
| Interest and other income | 108,000 | 722,000 | 142,000 | |||||||||
| Interest and other expense | (7,000 | ) | (14,000 | ) | (1,000 | ) | ||||||
| NET LOSS | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | |||
| COMPREHENSIVE LOSS | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | |||
| Series E preferred stock accumulated dividends | (4,640,000 | ) | (4,484,000 | ) | (3,696,000 | ) | ||||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | $ | (32,799,000 | ) | $ | (60,136,000 | ) | $ | (54,054,000 | ) | |||
| WEIGHTED AVERAGE COMMON SHARES OUTSTANDING: | ||||||||||||
| Basic and Diluted (1) | 37,109,493 | 30,895,089 | 26,079,762 | |||||||||
| BASIC AND DILUTED LOSS PER COMMON SHARE (1) | $ | (0.88 | ) | $ | (1.95 | ) | $ | (2.07 | ) | |||
| (1) | All share and per share amounts of our common stock for all periods presented have been retroactively adjusted to reflect the one-for-seven reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017 (Note 1). |
See accompanying notes to consolidated financial statements.
| F-4 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017
| Preferred Stock | Common Stock (1) | Additional Paid-In | Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||||||||
| BALANCES, April 30, 2014 | 775,000 | $ | 1,000 | 25,553,024 | $ | 26,000 | $ | 470,938,000 | $ | (403,266,000 | ) | $ | 67,699,000 | |||||||||||||||
| Series E preferred stock issued for cash under June 13, 2014 Financing, net of issuance costs of $1,002,000 | 799,764 | 1,000 | – | – | 18,202,000 | – | 18,203,000 | |||||||||||||||||||||
| Series E preferred stock dividends | – | – | – | – | (3,352,000 | ) | – | (3,352,000 | ) | |||||||||||||||||||
| Common stock issued for cash under December 27, 2012 Financing, net of issuance costs of $161,000 | – | – | 569,052 | 1,000 | 6,042,000 | – | 6,043,000 | |||||||||||||||||||||
| Common stock issued for cash under June 13, 2014 Financing, net of issuance costs of $352,000 | – | – | 1,383,109 | 1,000 | 13,191,000 | – | 13,192,000 | |||||||||||||||||||||
| Common stock issued under Employee Stock Purchase Plan | – | – | 71,063 | – | 608,000 | – | 608,000 | |||||||||||||||||||||
| Common stock issued upon exercise of options | – | – | 44,699 | – | 298,000 | – | 298,000 | |||||||||||||||||||||
| Share-based compensation | – | – | – | – | 6,702,000 | – | 6,702,000 | |||||||||||||||||||||
| Net loss | – | – | – | – | – | (50,358,000 | ) | (50,358,000 | ) | |||||||||||||||||||
| BALANCES, April 30, 2015 | 1,574,764 | 2,000 | 27,620,947 | 28,000 | 512,629,000 | (453,624,000 | ) | 59,035,000 | ||||||||||||||||||||
| Series E preferred stock issued for cash under June 13, 2014 Financing, net of issuance costs of $1,000 | 2,676 | – | – | – | 59,000 | – | 59,000 | |||||||||||||||||||||
| Series E preferred stock dividends | – | – | – | – | (4,139,000 | ) | – | (4,139,000 | ) | |||||||||||||||||||
| Common stock issued for cash under June 13, 2014 Financing, net of issuance costs of $311,000 | – | – | 1,232,821 | 1,000 | 11,144,000 | – | 11,145,000 | |||||||||||||||||||||
| Common stock issued for cash under August 7, 2015 Financing, net of issuance costs of $190,000 | – | – | 964,523 | 1,000 | 7,256,000 | – | 7,257,000 | |||||||||||||||||||||
| Common stock issued for cash under August 7, 2015 Financing, net of issuance costs of $175,000 | – | – | 1,210,328 | 1,000 | 6,793,000 | – | 6,794,000 | |||||||||||||||||||||
| Common stock issued for cash under October 30, 2015 Financing, net of issuance costs of $1,000 | – | – | 2,645,503 | 3,000 | 19,996,000 | – | 19,999,000 | |||||||||||||||||||||
| Common stock issued under Employee Stock Purchase Plan | – | – | 147,769 | – | 540,000 | – | 540,000 | |||||||||||||||||||||
| Common stock issued upon exercise of options | – | – | 25,322 | – | 138,000 | – | 138,000 | |||||||||||||||||||||
| Share-based compensation | – | – | – | – | 4,898,000 | – | 4,898,000 | |||||||||||||||||||||
| Net loss | – | – | – | – | – | (55,652,000 | ) | (55,652,000 | ) | |||||||||||||||||||
| BALANCES, April 30, 2016 | 1,577,440 | 2,000 | 33,847,213 | 34,000 | 559,314,000 | (509,276,000 | ) | 50,074,000 | ||||||||||||||||||||
| Series E preferred stock issued for cash under June 13, 2014 Financing, net of issuance costs of $58,000 | 70,320 | – | – | – | 1,576,000 | – | 1,576,000 | |||||||||||||||||||||
| Series E preferred stock dividends | – | – | – | – | (4,279,000 | ) | – | (4,279,000 | ) | |||||||||||||||||||
| Common stock issued for cash under August 7, 2015 Financing, net of issuance costs of $487,000 | – | – | 6,137,403 | 6,000 | 17,753,000 | – | 17,759,000 | |||||||||||||||||||||
| Common stock issued for cash under August 7, 2015 Financing, net of issuance costs of $340,000 | – | – | 3,750,323 | 4,000 | 12,687,000 | – | 12,691,000 | |||||||||||||||||||||
| Common stock issued under Employee Stock Purchase Plan | – | – | 270,075 | – | 526,000 | – | 526,000 | |||||||||||||||||||||
| Common stock issued upon exercise of options | – | – | 9,026 | – | 31,000 | – | 31,000 | |||||||||||||||||||||
| Share-based compensation | – | – | – | – | 3,363,000 | – | 3,363,000 | |||||||||||||||||||||
| Net loss | – | – | – | – | – | (28,159,000 | ) | (28,159,000 | ) | |||||||||||||||||||
| BALANCES, April 30, 2017 | 1,647,760 | $ | 2,000 | 44,014,040 | $ | 44,000 | $ | 590,971,000 | $ | (537,435,000 | ) | $ | 53,582,000 | |||||||||||||||
| (1) | All share and per share amounts of our common stock for all periods presented have been retroactively adjusted to reflect the one-for-seven reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017 (Note 1). |
See accompanying notes to consolidated financial statements.
| F-5 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017
| 2017 | 2016 | 2015 | ||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||
| Net loss | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||
| Share-based compensation | 3,363,000 | 4,898,000 | 6,702,000 | |||||||||
| Depreciation and amortization | 2,463,000 | 1,535,000 | 1,041,000 | |||||||||
| Loss on disposal of property and equipment | 1,000 | 14,000 | 2,000 | |||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Trade and other receivables | (4,883,000 | ) | 954,000 | (2,481,000 | ) | |||||||
| Inventories | (16,913,000 | ) | (8,832,000 | ) | (1,824,000 | ) | ||||||
| Prepaid expenses | (109,000 | ) | 4,000 | 64,000 | ||||||||
| Restricted cash | (550,000 | ) | (600,000 | ) | – | |||||||
| Other non-current assets | 278,000 | (325,000 | ) | 12,000 | ||||||||
| Accounts payable | (3,308,000 | ) | (3,521,000 | ) | 3,278,000 | |||||||
| Accrued clinical trial and related fees | (3,036,000 | ) | 3,684,000 | (523,000 | ) | |||||||
| Accrued payroll and related costs | 263,000 | 1,215,000 | 769,000 | |||||||||
| Deferred revenue | 18,470,000 | 3,400,000 | 1,097,000 | |||||||||
| Customer deposits | (7,195,000 | ) | 12,849,000 | 5,603,000 | ||||||||
| Other accrued expenses and current liabilities | (675,000 | ) | 1,051,000 | (52,000 | ) | |||||||
| Deferred rent, less current portion | 204,000 | (265,000 | ) | 651,000 | ||||||||
| Net cash used in operating activities | (39,786,000 | ) | (39,591,000 | ) | (36,019,000 | ) | ||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||
| Property and equipment acquisitions | (1,501,000 | ) | (9,324,000 | ) | (9,047,000 | ) | ||||||
| (Increase) decrease in other assets | (1,491,000 | ) | 533,000 | 598,000 | ||||||||
| Net cash used in investing activities | (2,992,000 | ) | (8,791,000 | ) | (8,449,000 | ) | ||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||
| Proceeds from issuance of common stock, net of issuance costs of $827,000, $677,000, and $513,000, respectively | 30,450,000 | 45,195,000 | 19,235,000 | |||||||||
| Proceeds from issuance of Series E preferred stock, net of issuance costs of $58,000, $1,000, and $1,002,000, respectively | 1,576,000 | 59,000 | 18,203,000 | |||||||||
| Proceeds from issuance of common stock under Employee Stock Purchase Plan | 526,000 | 540,000 | 608,000 | |||||||||
| Proceeds from exercise of stock options | 31,000 | 138,000 | 298,000 | |||||||||
| Dividends paid on preferred stock | (4,279,000 | ) | (4,139,000 | ) | (3,352,000 | ) | ||||||
| Principal payments on capital lease | (139,000 | ) | – | (13,000 | ) | |||||||
| Net cash provided by financing activities | 28,165,000 | 41,793,000 | 34,979,000 | |||||||||
| F-6 |
PEREGRINE PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 2017 | 2016 | 2015 | ||||||||||
| NET DECREASE IN CASH AND CASH EQUIVALENTS | $ | (14,613,000 | ) | $ | (6,589,000 | ) | $ | (9,489,000 | ) | |||
| CASH AND CASH EQUIVALENTS, beginning of period | 61,412,000 | 68,001,000 | 77,490,000 | |||||||||
| CASH AND CASH EQUIVALENTS, end of period | $ | 46,799,000 | $ | 61,412,000 | $ | 68,001,000 | ||||||
| SUPPLEMENTAL INFORMATION: | ||||||||||||
| Cash paid for interest | $ | 6,000 | $ | – | $ | – | ||||||
| SCHEDULE OF NON-CASH INVESTING AND FINANCING ACTIVITIES: | ||||||||||||
| Accounts payable and other liabilities for purchase of property and equipment and other assets | $ | 658,000 | $ | 1,565,000 | $ | 4,673,000 | ||||||
| Lease incentives | $ | – | $ | 562,000 | $ | 100,000 | ||||||
| Property and equipment acquired under capital lease | $ | 319,000 | $ | – | $ | – | ||||||
See accompanying notes to consolidated financial statements.
| F-7 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017
| 1. | ORGANIZATION AND BUSINESS DESCRIPTION |
Organization – In this Annual Report, “Peregrine,” “Company,” “we,” “us,” and “our,” refer to Peregrine Pharmaceuticals, Inc., and our wholly-owned subsidiary, Avid Bioservices, Inc. (“Avid”). Peregrine was incorporated under the laws of the state of California in June 1981, reincorporated in Delaware in September 1996 and commenced operations of Avid in January 2002.
Business Description – We are a biopharmaceutical company committed to improving the lives of patients by manufacturing pharmaceutical products through, Avid, our contract development and manufacturing organization (“CDMO”) and through advancing and licensing our novel, development-stage immunotherapy product.
Reverse Stock Split – On July 7, 2017, we effected a reverse stock split of our outstanding shares of common stock at a ratio of one-for-seven pursuant to our filed Certificate of Amendment to our Certificate of Incorporation with the Secretary of State of the State of Delaware. The reverse stock split took effect with the opening of trading on July 10, 2017. The primary purpose of the reverse stock split, which was approved by our stockholders at our 2016 Annual Meeting on October 13, 2016, was to enable us to regain compliance with the $1.00 minimum bid price requirement for continued listing on The NASDAQ Capital Market. Pursuant to the reverse stock split, every seven shares of our issued and outstanding shares of common stock were automatically combined into one issued and outstanding share of common stock, without any change in the par value per share of our common stock. All share and per share amounts of our common stock included in the accompanying consolidated financial statements have been retrospectively adjusted to give effect to the reverse stock split for all periods presented, including reclassifying an amount equal to the reduction in par value to additional paid-in capital. No fractional shares were issued in connection with the reverse stock split. Any fractional share of common stock created by the reverse stock split was rounded up to the nearest whole share. The number of authorized shares of our common stock remained unchanged.
The reverse stock split affected all issued and outstanding shares of our common stock, as well as the shares of common stock underlying our stock options, employee stock purchase plan, warrants and the general conversion right with respect to our 10.50% Series E Convertible Preferred Stock (the “Series E Preferred Stock”).
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
Basis of Presentation – The accompanying consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles and include the accounts of Peregrine and its wholly-owned subsidiary, Avid. All intercompany balances and transactions have been eliminated.
Use of Estimates – The preparation of our financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ materially from these estimates.
Going Concern – For the year ended April 30, 2017, we adopted Financial Accounting Standards Board (“FASB”) Accounting Standards Update (“ASU”) 2014-15, Presentation of Financial Statements – Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which requires us to evaluate whether there are conditions or events that, in the aggregate, raise substantial doubt about our ability to continue as a going concern and to meet our obligations as they become due within one year after the date that the financial statements are issued.
| F-8 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Our consolidated financial statements have been prepared on a going concern basis, which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The financial statements do not include any adjustments relating to the recoverability of the recorded assets or the classification of liabilities that may be necessary should it be determined that we are unable to continue as a going concern.
At April 30, 2017, we had $46,799,000 in cash and cash equivalents. We have expended substantial funds on the research and development of our product candidates, and funding the operations of Avid. As a result, we have historically experienced losses and negative cash flows from operations since our inception and we expect negative cash flows from operations to continue for the foreseeable future until we can generate sufficient revenue from Avid’s contract manufacturing services to achieve profitability. Therefore, unless and until we are able to generate sufficient revenue from Avid’s contract manufacturing services or from the sale or licensing of our product candidate under development, we expect such losses to continue through at least the fiscal year ending April 30, 2018, and as a result, we will require additional capital to fund our operations and to execute our business plans.
Our ability to continue to fund our operations is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, (i) raising additional capital in the equity markets, (ii) generating additional revenue from Avid, or (iii) licensing or partnering our product candidate in development.
Historically, we have funded a significant portion of our operations through the issuance of equity. During fiscal year 2017, we raised $31,277,000 in aggregate gross proceeds from the sale of shares of our common stock and raised an additional $1,634,000 in aggregate gross proceeds from the sale of shares of our Series E Preferred Stock (Note 5). Subsequent to April 30, 2017 and through June 30, 2017, we raised an additional $4,304,000 in aggregate gross proceeds from the sale of shares of our common stock (Note 12). As of July 14, 2017, $67,674,000 remained available to us under our effective shelf registration statement, which allows us from time to time to offer and sell shares of our common stock, in one or more offerings, either individually or in combination.
Our ability to raise additional capital in the equity markets to fund our obligations in future periods is dependent on a number of factors, including, but not limited to, the market demand for our common stock. The market demand or liquidity of our common stock is subject to a number of risks and uncertainties, including but not limited to, negative economic conditions, adverse market conditions, adverse financial results, and negative research and development results. If we are unable to either (i) raise sufficient capital in the equity markets, (ii) generate additional revenue from Avid, or (iii) license or partner our product candidate in development, or any combination thereof, we may need to delay, scale back, or eliminate all our research and development efforts, or restructure our operations. In addition, even if we are able to raise additional capital, it may not be at a price or on terms that are favorable to us.
As a result, we have concluded that there is substantial doubt about our ability to continue as a going concern within one year after the date that our financial statements are issued.
Cash and Cash Equivalents – We consider all short-term investments readily convertible to cash with an initial maturity of three months or less to be cash equivalents.
Restricted Cash – Under the terms of three separate operating leases related to our facilities, we are required to maintain, as collateral, letters of credit during the terms of such leases (Note 3). At April 30, 2017 and 2016, restricted cash of $1,150,000 and $600,000, respectively, was pledged as collateral under these letters of credit.
| F-9 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Trade and Other Receivables – Trade receivables are recorded at the invoiced amount net of an allowance for doubtful accounts, if necessary. Other receivables are reported at amounts expected to be collected net of an allowance for doubtful accounts, if necessary. Trade and other receivables, net, at April 30, consist of the following:
| 2017 | 2016 | |||||||
| Trade receivables(1) | $ | 7,274,000 | $ | 2,494,000 | ||||
| Other receivables | 468,000 | 365,000 | ||||||
| Trade and other receivables | $ | 7,742,000 | $ | 2,859,000 | ||||
______________
(1) Represents amounts billed for contract manufacturing services provided by Avid.
Allowance for Doubtful Accounts – We continually monitor our allowance for doubtful accounts for all receivables. We apply judgment in assessing the ultimate realization of our receivables and we estimate an allowance for doubtful accounts based on various factors, such as, the aging of accounts receivable balances, historical experience, and the financial condition of our customers. Based on our analysis of our receivables as of April 30, 2017 and 2016, we determined no allowance for doubtful accounts was necessary.
Inventories – Inventories are recorded at the lower of cost or market (net realizable value) and primarily include raw materials, work-in-process (comprised of raw materials, direct labor and overhead costs associated with in-process manufacturing services), and finished goods (representing manufacturing services completed and ready for shipment) associated with our wholly-owned subsidiary, Avid. Cost is determined by the first-in, first-out method. Inventories consist of the following at April 30,:
| 2017 | 2016 | |||||||
| Raw materials | $ | 11,304,000 | $ | 10,911,000 | ||||
| Work-in-process | 13,755,000 | 5,275,000 | ||||||
| Finished goods | 8,040,000 | – | ||||||
| Total inventories | $ | 33,099,000 | $ | 16,186,000 | ||||
Property and Equipment, net – Property and equipment is recorded at cost, less accumulated depreciation and amortization. Depreciation and amortization are computed using the straight-line method over the estimated useful lives of the related asset, generally ranging from three to ten years. Amortization of leasehold improvements is calculated using the straight-line method over the shorter of the estimated useful life of the asset or the remaining lease term.
Concentrations of Credit Risk and Customer Base – Financial instruments that potentially subject us to a significant concentration of credit risk consist of cash and cash equivalents, restricted cash and trade receivables. We maintain our cash and restricted cash balances primarily with one major commercial bank and our deposits held with the bank exceed the amount of government insurance limits provided on our deposits. We are exposed to credit risk in the event of default by the major commercial bank holding our cash and restricted cash balances to the extent of the cash and restricted cash amounts recorded on the accompanying consolidated balance sheet.
Our trade receivables from amounts billed for contract manufacturing services provided by Avid have historically been derived from a small customer base. Most contracts require up-front payments and installment payments during the service period. We perform periodic evaluations of the financial condition of our customers and generally do not require collateral, but we can terminate any contract if a material default occurs. At April 30, 2017 and 2016, approximately 93% and 98%, respectively, of our trade receivables were due from four customers.
In addition, contract manufacturing revenue generated by Avid has historically been derived from a small customer base (Note 10). These customers typically do not enter into long-term contracts because their need for drug supply depends on a variety of factors, including the drug’s stage of development, their financial resources, and, with respect to commercial drugs, demand for the drug in the market.
| F-10 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Our future results of operations could be adversely affected if revenue from any one of our primary customers is significantly reduced or eliminated.
Comprehensive Loss – Comprehensive loss is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Comprehensive loss is equal to our net loss for all periods presented.
Impairment – Long-lived assets are reviewed for impairment in accordance with authoritative guidance for impairment or disposal of long-lived assets. Long-lived assets are reviewed for events or changes in circumstances, which indicate that their carrying value may not be recoverable. Long-lived assets are reported at the lower of carrying amount or fair value less cost to sell. For the fiscal years ended April 30, 2017 and 2016, there was no impairment of the value of our long-lived assets.
Fair Value of Financial Instruments – The carrying amounts in the accompanying consolidated balance sheet for cash and cash equivalents, restricted cash, trade and other receivables, accounts payable and accrued liabilities approximate their fair values due to their short-term maturities.
Fair Value Measurements – Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The guidance prioritizes the inputs used in measuring fair value into the following hierarchy:
| · | Level 1 – Observable inputs, such as unadjusted quoted prices in active markets for identical assets or liabilities. |
| · | Level 2 – Observable inputs other than quoted prices included in Level 1, such as assets or liabilities whose values are based on quoted market prices in markets where trading occurs infrequently or whose values are based on quoted prices of instruments with similar attributes in active markets. |
| · | Level 3 – Unobservable inputs that are supported by little or no market activity and significant to the overall fair value measurement of the assets or liabilities; therefore, requiring the company to develop its own valuation techniques and assumptions. |
As of April 30, 2017 and 2016, we do not have any Level 2 or Level 3 financial assets or liabilities and our cash equivalents, which are primarily invested in money market funds with one major commercial bank, are carried at fair value based on quoted market prices for identical securities (Level 1 input).
Customer Deposits – Customer deposits primarily represents advance billings and/or payments received from Avid’s third-party customers prior to the initiation of contract manufacturing services.
Deferred Rent – Rent expense is recorded on a straight-line basis over the initial term of our operating lease agreements and the difference between rent expense and the amounts paid is recorded as a deferred rent liability. Incentives granted under our operating leases, including tenant improvements and landlord-funded lease incentives, are recorded as a deferred rent liability, which is amortized as a reduction to rent expense over the term of the operating lease (Note 3).
Revenue Recognition – We currently derive revenue from our contract manufacturing services provided by Avid. We recognize revenue in accordance with the authoritative guidance for revenue recognition when all of the following criteria are met: (i) persuasive evidence of an arrangement exists, (ii) delivery (or passage of title) has occurred or services have been rendered, (iii) the seller’s price to the buyer is fixed or determinable, and (iv) collectability is reasonably assured. We also comply with the authoritative guidance for revenue recognition regarding arrangements with multiple elements.
Revenue arrangements with multiple elements are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer or licensing partner. When deliverables are separable, consideration received is allocated among the separate units based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units, which may require the use of significant judgement. Deliverables are considered separate units of accounting if (1) the delivered item(s) has value to the customer on a stand-alone basis and (2) the arrangement includes a general right of return relative to the delivered item(s), delivery or performance of the undelivered item(s) is considered probable and substantially in our control.
| F-11 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Arrangement consideration is allocated at the inception of the agreement to all identified units of accounting based on their relative selling price. The relative selling price for each deliverable is determined using vendor specific objective evidence (“VSOE”) of selling price or third-party evidence of selling price if VSOE does not exist. If neither VSOE nor third-party evidence of selling price exists, we use our best estimate of the selling price for the deliverable. The amount of allocable arrangement consideration is limited to amounts that are fixed or determinable. The consideration received is allocated among the separate units of accounting, and the applicable revenue recognition criteria are applied to each of the separate units. Changes in the allocation of the sales price between delivered and undelivered elements can impact revenue recognition but do not change the total revenue recognized under any agreement.
On occasion, we receive requests from customers to hold product manufactured by Avid on a “bill-and-hold” basis. Revenue is recognized for these “bill-and-hold” arrangements in accordance with the authoritative guidance, which requires, among other things, the existence of a valid business purpose for the arrangement; the “bill-and-hold” arrangement is at the request of the customer; title and risk of ownership must pass to the customer; the product is complete and ready for shipment; a fixed delivery date that is reasonable and consistent with the customer’s business practices; the product has been separated from our inventory; and no further performance obligations by us exist.
In addition, we also follow the authoritative guidance when reporting revenue as gross when we act as a principal versus reporting revenue as net when we act as an agent. For transactions in which we act as a principal, have discretion to choose suppliers, bear credit and inventory risk and perform a substantive part of the services, revenue is recorded at the gross amount billed to a customer and costs associated with these reimbursements are reflected as a component of cost of sales for contract manufacturing services.
Any amounts received prior to satisfying our revenue recognition criteria are recorded as deferred revenue or customer deposits in the accompanying consolidated financial statements. We also record a provision for estimated contract losses, if any, in the period in which they are determined.
Research and Development Expenses – Research and development expenses primarily include (i) payroll and related costs, including share-based compensation associated with research and development personnel, (ii) costs related to clinical trials and preclinical testing of our technologies under development, (iii) costs to develop and manufacture the product candidates, including raw materials and supplies, product testing, depreciation, and facility related expenses, (iv) expenses for research services provided by universities and contract laboratories, including sponsored research funding, and (v) other research and development expenses. Research and development expenses are charged to expense as incurred when these expenditures relate to our research and development efforts and have no alternative future uses.
Clinical trial costs are a significant component of our research and development expenses. We have a history of contracting with third parties that perform various clinical trial activities on our behalf in the ongoing development of our product candidates. The financial terms of these contracts are subject to negotiations and may vary from contract to contract and may result in uneven payment flow. Expenses related to clinical trials are accrued based on our estimates and/or representations from third parties (including clinical research organizations) regarding services performed. If the contracted amounts are modified (for instance, as a result of changes in the clinical trial protocol or scope of work to be performed), we modify our accruals accordingly on a prospective basis. Revisions in the scope of a contract are charged to expense in the period in which the facts that give rise to the revision become reasonably certain. There were no material adjustments for a change in estimate to research and development expenses in the accompanying consolidated financial statements in any of the three years ended April 30, 2017.
Under certain research and development agreements, we are obligated to make certain advance payments, including nonrefundable amounts, for goods or services that will be used or rendered for future research and development activities and are deferred and capitalized as prepaid research and development expenses. These advance payments are recognized as an expense in the period the related goods are delivered or the related services are performed. We assess our prepaid research and development expenses for impairment when events or changes in circumstances indicate that the carrying amount of the prepaid expense may not be recoverable or provide future economic benefit.
| F-12 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
In addition, under certain in-licensing agreements associated with the research and development of our product candidates, we are obligated to pay certain milestone payments based on potential clinical development and regulatory milestones (Note 4). These milestone payments have no alternative future uses (in other research and development projects or otherwise) and therefore have no separate economic values and are expensed as research and development costs at the time the costs are incurred. We have no in-licensed product candidates that have alternative future uses in research and development projects or otherwise. In addition, we do not perform any research and development activities for any unrelated entities.
Share-based Compensation – We account for stock options and other share-based awards granted under our equity compensation plans in accordance with the authoritative guidance for share-based compensation. The estimated fair value of share-based payments to employees in exchange for services is measured at the grant date, using a fair value based method, such as a Black-Scholes option valuation model, and is recognized as expense on a straight-line basis over the requisite service periods. The fair value of modifications to share-based awards, if any, is generally estimated using a Black-Scholes option valuation model, unless a lattice model is required. Share-based compensation expense recognized during the period is based on the value of the portion of the share-based payment that is ultimately expected to vest during the period and is reduced for estimated forfeitures. The authoritative guidance requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. As of April 30, 2017, there were no outstanding share-based awards with market or performance conditions.
Periodically, we grant stock options and other share-based awards to non-employee consultants, which we account for in accordance with the authoritative guidance for share-based compensation. The cost of non-employee services received in exchange for share-based awards are measured based on either the fair value of the consideration received or the fair value of the share-based award issued, whichever is more reliably measurable. In addition, authoritative guidance requires share-based compensation related to unvested options and awards issued to non-employees to be recalculated at the end of each reporting period based upon the fair market value on that date until the share-based award has vested, and any cumulative catch-up adjustment to share-based compensation resulting from the re-measurement is recognized in the current period (Note 6).
Income Taxes – We utilize the liability method of accounting for income taxes in accordance with authoritative guidance for accounting for income taxes. Under the liability method, deferred taxes are determined based on the differences between the consolidated financial statements and tax basis of assets and liabilities using enacted tax rates. A valuation allowance is provided when it is more likely than not that some portion or the entire deferred tax asset will not be realized (Note 8). In addition, we recognize the impact of an uncertain tax position only when it is more likely than not the tax position will be sustained upon examination by the tax authorities. We are also required to file federal, state and foreign income tax returns in various jurisdictions. The preparation of these returns requires us to interpret the applicable tax laws in effect in such jurisdictions, which could affect the amount paid by us.
Basic and Dilutive Net Loss Per Common Share – Basic net loss per common share is computed by dividing our net loss attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period excluding the dilutive effects of stock options, shares of common stock expected to be issued under our Employee Stock Purchase Plan (the “ESPP”), warrants, and Series E Preferred Stock outstanding during the period. Diluted net loss per common share is computed by dividing our net loss attributable to common stockholders by the sum of the weighted average number of shares of common stock outstanding during the period plus the potential dilutive effects of stock options, shares of common stock expected to be issued under our ESPP, warrants, and Series E Preferred Stock outstanding during the period. Net loss attributable to common stockholders represents our net loss plus Series E Preferred Stock accumulated dividends. Series E Preferred Stock accumulated dividends include dividends declared for the period (regardless of whether or not the dividends have been paid) and dividends accumulated for the period (regardless of whether or not the dividends have been declared).
The potential dilutive effect of stock options, shares of common stock expected to be issued under our ESPP, and warrants outstanding during the period are calculated in accordance with the treasury stock method, but are excluded if their effect is anti-dilutive. The potential dilutive effect of our Series E Preferred Stock outstanding during the period is calculated using the if-converted method assuming the conversion of our Series E Preferred Stock as of the earliest period reported or at the date of issuance, if later, but are excluded if their effect is anti-dilutive. However, because the impact of stock options, shares of common stock expected to be issued under our ESPP, warrants, and Series E Preferred Stock are anti-dilutive during periods of net loss, there was no difference between basic and diluted loss per common share amounts for the three years ended April 30, 2017.
| F-13 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
The calculation of weighted average diluted shares outstanding excludes the dilutive effect of the following weighted average outstanding stock options and shares of common stock expected to be issued under our ESPP since their impact are anti-dilutive during periods of net loss:
| 2017 | 2016 | 2015 | ||||||||||
| Stock options | – | 252,098 | 547,599 | |||||||||
| ESPP | 45,767 | 37,862 | 6,714 | |||||||||
| Total | 45,767 | 289,960 | 554,313 | |||||||||
The calculation of weighted average diluted shares outstanding also excludes the following weighted average outstanding stock options, warrants, and Series E Preferred Stock (assuming the if-converted method), as their exercise prices or conversion price were greater than the average market price of our common stock during the respective periods, resulting in an anti-dilutive effect:
| 2017 | 2016 | 2015 | ||||||||||
| Stock options | 4,156,421 | 2,740,922 | 1,210,144 | |||||||||
| Warrants | 39,040 | 39,040 | 39,040 | |||||||||
| Series E Preferred Stock | 1,955,588 | 1,893,122 | 1,411,362 | |||||||||
| Total | 6,151,049 | 4,673,084 | 2,660,546 | |||||||||
Subsequent to April 30, 2017 and through June 30, 2017, we sold an aggregate of 1,051,258 shares of our common stock (Note 12), which are not included in the calculation of basic and dilutive net loss per common share for the fiscal year ended April 30, 2017.
Recently Adopted Accounting Pronouncements
For the year ended April 30, 2017, we adopted FASB ASU 2014-15, Presentation of Financial Statements – Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, which requires us to evaluate whether there are conditions or events that, in the aggregate, raise substantial doubt about our ability to continue as a going concern and to meet our obligations as they become due within one year after the date that the financial statements are issued. We have included the disclosures required by ASU 2014-15 in the “Going Concern” section of Note 2 to our consolidated financial statements.
Pending Adoption of Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) 2014-09, Revenue from Contracts with Customers (Topic 606): Revenue from Contracts with Customers, which, along with subsequent amendments issued in 2015 and 2016, will replace substantially all current US GAAP revenue recognition guidance. ASU 2014-09, as amended, is based on the principle that revenue is recognized to depict the transfer of goods or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The new guidance also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from customer contracts, including significant judgments and changes in judgments and assets recognized from costs incurred to obtain or fulfill a contract. ASU 2014-09, as amended, is effective for annual reporting periods beginning after December 15, 2017, including interim periods within that reporting period, which will be our fiscal year 2019 beginning May 1, 2018. The new guidance permits adoption either by using (i) a full retrospective approach for all periods presented in the period of adoption or (ii) a modified retrospective approach where the new standard is applied in the financial statements starting with the year of adoption. Under both approaches, cumulative impact of the adoption is reflected as an adjustment to retained earnings (accumulated deficit) as of the earliest date presented in accordance with the new standard. We are continuing to assess the impact of the new guidance on our accounting policies and procedures and are evaluating the new requirements as applied to existing manufacturing contracts under our contract manufacturing business. Although we are continuing to assess the impact of the new guidance, we have identified our revenue streams and based on our preliminary assessment we believe the most significant impact may relate to the recognition of contract manufacturing revenue over a period of time rather than at a point in time. We plan to adopt ASU 2014-09, as amended, on May 1, 2018 and have not yet determined which transition method will be elected.
| F-14 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
In July 2015, the FASB issued ASU 2015-11, Inventory (Topic 330): Simplifying the Measurement of Inventory. ASU 2015-11 requires that inventory should be measured at the lower of cost and net realizable value for entities that measure inventory using the first-in, first-out method. ASU 2015-11 defines net realizable value as the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal and transportation. ASU 2015-11 is effective for fiscal years beginning after December 15, 2016, which will be our fiscal year 2018 beginning May 1, 2017, and interim periods within those fiscal years. We do not expect the adoption of ASU 2015-11 to have a material impact on our consolidated financial statements and related disclosures.
In November 2015, the FASB issued ASU 2015-17, Income Taxes (Topic 740): Balance Sheet Classification of Deferred Taxes. Under existing standards, deferred taxes for each tax-paying jurisdiction are presented as a net current asset or liability and net long-term asset or liability. To simplify presentation, the new guidance will require that all deferred tax assets and liabilities, along with related valuation allowances, be classified as long-term on the balance sheet. As a result, each tax-paying jurisdiction will now only have one net long-term deferred tax asset or liability. The new guidance does not change the existing requirement that prohibits offsetting deferred tax liabilities from one jurisdiction against deferred tax assets of another jurisdiction. ASU 2015-17 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2016, which will be our fiscal year 2018 beginning May 1, 2017. Due to the full valuation allowance on our U.S. deferred tax assets, we do not expect the adoption of ASU 2015-17 to have a material impact on our consolidated financial statements and related disclosures.
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842). ASU 2016-2 requires an entity to recognize right-of-use assets and lease liabilities on its balance sheet and disclose key information about leasing arrangements. ASU 2016-02 offers specific accounting guidance for a lessee, a lessor and sale and leaseback transactions. Lessees and lessors are required to disclose qualitative and quantitative information about leasing arrangements to enable a user of the financial statements to assess the amount, timing and uncertainty of cash flows arising from leases. ASU 2016-02 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2018, which will be our fiscal year 2020 beginning May 1, 2019. Early adoption is permitted. We are currently in the process of evaluating the impact of adoption of ASU 2016-02 on our consolidated financial statements and related disclosures.
In March 2016, FASB issued ASU 2016-09, Compensation - Stock Compensation (Topic 718): Improvements to Employee Share-Based Payment Accounting. ASU 2016-09 changes certain aspects of accounting for share-based payments to employees and involves several aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities, and classification on the statement of cash flows. Specifically, ASU 2016-09 requires that all income tax effects of share-based awards be recognized as income tax expense or benefit in the reporting period in which they occur. Additionally, ASU 2016-09 amends existing guidance to allow forfeitures of share-based awards to be recognized as they occur. Previous guidance required that share-based compensation expense include an estimate of forfeitures. ASU 2016-09 is effective for annual and interim periods beginning after December 15, 2016, which will be our fiscal year beginning May 1, 2017. We do not expect the adoption of ASU 2016-09 to have a material impact on our consolidated financial statements and related disclosures.
In November 2016, FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230): Restricted Cash, which addresses diversity in practice related to the classification and presentation of changes in restricted cash on the statement of cash flows. ASU 2016-18 will require that a statement of cash flows explain the change during the period in the total of cash, cash equivalents, and amounts generally described as restricted cash or restricted cash equivalents. Therefore, amounts generally described as restricted cash and restricted cash equivalents should be included with cash and cash equivalents when reconciling the beginning-of-period and end-of-period total amounts shown on the statement of cash flows. ASU 2016-18 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2017, which will be our fiscal year 2019 beginning May 1, 2018. Early adoption is permitted. We are currently evaluating the impact the adoption of ASU 2016-18 will have on our consolidated financial statements and related disclosures.
In May 2017, the FASB issued ASU 2017-09, Compensation - Stock Compensation (Topic 718): Scope of Modification Accounting, which provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. This pronouncement is effective for annual reporting periods beginning after December 15, 2017, which will be our fiscal year 2019 beginning May 1, 2018. Early adoption is permitted. We do not expect the adoption of ASU 2016-09 to have a material impact on our consolidated financial statements and related disclosures.
| F-15 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 3. | COMMITMENTS AND CONTINGENCIES |
Operating Leases – Our corporate offices, research and development, and manufacturing facilities are all located in close proximity in Tustin, California. We currently lease an aggregate of approximately 196,000 square feet of office, warehouse, research and manufacturing space in six buildings under five separate lease agreements, as summarized in the following table:
|
Lease # |
Original Lease Execution Date |
Approximate Square Footage Leased |
# of Buildings Occupied |
Initial Lease Term Expiration Date |
# of Options to Extend Lease |
Extended Lease Term Expiration Date(1) | |
| 1 | December 1998 | 48,000 | 2 | 12/31/27 | 2 | 12/31/37 | |
| 2 | May 2010 | 13,000 | 1 | 12/31/17 | 1 | 12/31/22 | |
| 3 | July 2014 | 84,000 | 1 | 1/31/27 | 2 | 1/31/37 | |
| 4 | April 2016 | 26,000 | 1 | 8/31/23 | 2 | 8/31/35 | |
| 5 | April 2016 | 25,000 | 1 | 8/31/23 | 2 | 8/31/35 |
______________
| (1) | Extended lease term expiration date assumes we execute all available option(s) to extend lease in accordance with the terms of the lease agreement. |
The following represents additional information for each of the lease agreements included in the above table:
In December 1998, we entered into our first lease agreement (the “First Lease”) with an original lease term of 12 years with two 5-year renewal options and includes scheduled rental increases of approximately 3% every two years. In December 2005, we entered into an amendment to the First Lease that extended the original lease term for seven additional years to expire on December 31, 2017. In November 2016, we entered into a second amendment to the First Lease that extends the lease term through December 31, 2027, while also maintaining our two 5-year renewal options that could extend the lease term to December 31, 2037.
In May 2010, we entered into a second lease agreement (the “Second Lease”) to lease additional office and research space. The Second Lease includes a 5-year option to extend the lease to December 31, 2022 and includes annual scheduled rental increases of $0.05 per square foot per year. In addition, a tenant improvement allowance of $125,000 associated with the Second Lease were recorded as leasehold improvements and are being amortized over the shorter of the estimated useful life of the improvements or the remaining life of the Second Lease.
In July 2014, we entered into a third lease agreement (the “Third Lease”) to lease approximately 42,000 square feet of vacant warehouse space to expand our manufacturing capacity to support the manufacturing of products in late-stage clinical development to commercial. The Third Lease includes an option to extend the lease term in two 5-year periods to extend the lease to July 31, 2031 and includes scheduled annual rent increases of approximately 3%. In addition, the Third Lease provided for 12.5 months of free rent, lessor improvements of $250,000 and a tenant improvement allowance of $365,000. Upon completion of the manufacturing facility build-out during fiscal year 2016 (the “Myford Facility”), certain of these improvements were classified as leasehold improvements and are being amortized over the shorter of the estimated useful life of the improvements or the remaining life of the Third Lease, as amended.
In February 2017, we entered into a lease amendment to the Third Lease (the “Third Lease Amendment”), pursuant to which we secured approximately 42,000 square feet of additional vacant warehouse space (the “Expansion Space”) within the same building as our existing Myford Facility. The purpose of the Expansion Space is to expand our biomanufacturing capacity, which we believe will further support the growth of our contract manufacturing business. Under the Third Lease Amendment, our Myford Facility now encompasses approximately 84,000 square feet. The Third Lease Amendment also extends the initial lease term to January 31, 2027 and maintains our two 5-year renewal options that could extend the lease term to January 31, 2037. Our scheduled annual rent increases of approximately 3% are also maintained under the Third Lease Amendment. In addition, with respect to the Expansion Space, the Third Lease Amendment provides for eight (8) months of free rent and a tenant improvement allowance of $1,269,000, which is subject to certain performance contingencies, as defined in the Third Lease Amendment. As a result, the tenant improvement allowance, is accounted for as contingent rent and will be recorded when the tenant improvement allowance is received. Additionally, under the terms of the Third Lease Amendment, we are required to maintain, as collateral for the lease, a letter of credit in the amount of $550,000 during the entire term of the Third Lease, as amended, which amount is included in restricted cash in the accompanying consolidated balance sheet as of April 30, 2017.
| F-16 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
In April 2016, we entered into a fourth lease agreement (the “Fourth Lease”) to lease additional office space. The Fourth Lease includes two separate option periods to extend the lease term to August 31, 2035 and includes annual scheduled rent increases of approximately 3%. In addition, the Fourth Lease provided for four months of free rent and a tenant improvement allowance of $562,000. The tenant improvements classified as leasehold improvements are being amortized over the shorter of the estimated useful life of the improvements or the remaining life of the Fourth Lease Additionally, under the terms of the Fourth Lease, we are required to maintain, as collateral for the lease, a letter of credit in the amount of $350,000 during the entire term of the Fourth Lease, which amount is included in restricted cash in the accompanying consolidated balance sheets as of April 30, 2017 and 2016.
In April 2016, we entered into a fifth lease agreement (the “Fifth Lease”) to support our manufacturing operations. The Fifth Lease includes two separate option periods to extend the lease term to August 31, 2035 and includes annual scheduled rent increases of approximately 3%. In addition, under the terms of the Fifth Lease, we are required to maintain, as collateral for the lease, a letter of credit in the amount of $250,000 during the entire term of the Fifth Lease, which amount is included in restricted cash in the accompanying consolidated balance sheets as of April 30, 2017 and 2016.
Under each of the aforementioned facility operating leases, we record rent expense on a straight-line basis over the initial term of the lease. The difference between rent expense and the amounts paid under the operating leases is recorded as a deferred rent liability in the accompanying consolidated financial statements. Annual rent expense under the aforementioned facility operating lease agreements totaled $2,180,000, $1,265,000, and $1,197,000 for the fiscal years ended April 30, 2017, 2016, and 2015, respectively.
At April 30, 2017, future minimum lease payments under all non-cancelable operating leases are as follows:
| Year ending April 30,: | Minimum Lease Payments | |||||
| 2018 | $ | 2,741,000 | ||||
| 2019 | 3,054,000 | |||||
| 2020 | 3,037,000 | |||||
| 2021 | 3,116,000 | |||||
| 2022 | 2,789,000 | |||||
| Thereafter | 11,362,000 | |||||
| $ | 26,099,000 | |||||
Legal Proceedings - In the ordinary course of business, we are at times subject to various legal proceedings and disputes. We make provisions for liabilities when it is both probable that a liability has been incurred and the amount of the loss can be reasonably estimated. Such provisions are reviewed at least quarterly and adjusted to reflect the impact of any settlement negotiations, judicial and administrative rulings, advice of legal counsel, and other information and events pertaining to a particular case.
On October 10, 2013, a derivative and class action complaint, captioned Michaeli v. Steven W. King, et al., C.A. No. 8994-VCL, was filed in the Court of Chancery of the State of Delaware (the “Court”), purportedly on behalf of the Company, which is named a nominal defendant, against certain of our executive officers and directors (collectively, the “Defendants”). On December 1, 2015, the plaintiffs filed an amended and supplemental derivative and class action complaint (the “Amended Complaint”). The Amended Complaint alleges that the Defendants breached their respective fiduciary duties in connection with certain purportedly improper compensation decisions made by our Board of Directors during the past four fiscal years ended April 30, 2015, including: (i) the grant of a stock option to Mr. King on May 4, 2012; (ii) the non-routine broad-based stock option grant to our directors, executives, all other employees and certain consultants on December 27, 2012; and (iii) the payment, during the past four fiscal years ended April 30, 2015, of compensation to our non-employee directors. In addition, the complaint alleges that our directors breached their fiduciary duty of candor by filing and seeking stockholder action on the basis of an allegedly materially false and misleading proxy statement for our 2013 annual meeting of stockholders. The plaintiffs are seeking, among other things, rescission of a portion of the stock option grant to Mr. King on May 4, 2012 and the stock options granted to the Defendants on December 27, 2012, as well as disgorgement of any excessive compensation paid to our non-employee directors during the four fiscal years ended April 30, 2015 and other monetary relief for our benefit. On May 15, 2017, the parties filed with the Court a Stipulation and Agreement of Compromise, Settlement and Release setting forth the terms of the proposed settlement of the claims in the Amended Complaint. A hearing on the proposed settlement will be held before the Court on July 27, 2017. We do not expect to incur a loss associated with this matter should the Court approve the terms of the proposed settlement.
| F-17 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 4. | LICENSing AGREEMENTS |
The following represents a summary of our key in-licensing agreements covering bavituximab, our lead investigational immunotherapy product in clinical development.
In August 2001 and August 2005, we exclusively in-licensed the worldwide rights to the phosphatidylserine (“PS”)-targeting technology platform, including bavituximab, from The University of Texas System, on behalf of The University of Texas Southwestern Medical Center at Dallas (“UTSWMC”). In November 2003, we entered into a non-exclusive license agreement with Genentech, Inc. (“Genentech”), to license certain intellectual property rights covering methods and processes for producing antibodies used in connection with the development of our PS-targeting program. In December 2003, we entered into an exclusive commercial license agreement with Avanir Pharmaceuticals, Inc. (“Avanir”) covering the generation of a chimeric monoclonal antibody. In March 2005, we entered into a worldwide non-exclusive license agreement with Lonza Biologics (“Lonza”) for intellectual property and materials relating to the expression of recombinant monoclonal antibodies for use in the manufacture of bavituximab.
Under our in-licensing agreements relating to bavituximab, we are obligated to pay future milestone payments based on potential clinical development and regulatory milestones, plus a royalty on net sales and/or a percentage of sublicense income. The applicable royalty rate under each of the foregoing in-licensing agreements is in the low single digits. We did not incur any milestone related expenses during the three fiscal years ended April 30, 2017.
The following table provides certain information with respect to each of our in-licensing agreements relating to our bavituximab program.
| Licensor | Agreement Date | Total Milestone Obligations Expensed To Date | Potential Future Milestone Obligations (1) | |||||||||
| UTSWMC | August 2001 | $ | 173,000 | $ | 300,000 | |||||||
| UTSWMC | August 2005 | 85,000 | 375,000 | |||||||||
| Lonza | March 2005 | 64,000 | – (2) | |||||||||
| Avanir | December 2003 | 100,000 | 1,000,000 | |||||||||
| Genentech | November 2003 | 500,000 | 5,000,000 | |||||||||
| Total | $ | 922,000 | $ | 6,675,000 | ||||||||
______________
| (1) | Under our current agreements, potential future milestone obligations are due upon achieving certain clinical and regulatory milestones. Based on the current stage of clinical development for bavituximab, future milestone obligations would be due upon submission of a biologics license application in the U.S. and upon FDA approval, which events are currently uncertain and depend on positive clinical trial results. In addition, potential future milestone obligations vary by license agreement (as defined in each license agreement) and certain agreements depend on a valid patent claim, as defined in each of these underlying agreements, at the time the potential milestone is achieved. |
| (2) | In the event we utilize a third-party contract manufacturer other than Lonza to manufacture bavituximab for commercial purposes, we would owe Lonza 300,000 pounds sterling per year. |
We do not expect to incur any milestone related expenses regarding our bavituximab program during fiscal year 2018. In addition, of the total potential future milestone obligations of $6,675,000, up to $6,400,000 would be due upon the first commercial approval of bavituximab pursuant to these in-licensing agreements. However, given the uncertainty of the drug development and the regulatory approval process, we are unable to predict with any certainty when any of these future milestones will occur, if at all.
| F-18 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 5. | STOCKHOLDERS’ EQUITY |
Stockholder Rights Agreement
On March 16, 2006, our Board of Directors adopted a Stockholder Rights Agreement, which was amended and restated on March 16, 2016 (the “Rights Agreement”), that is designed to strengthen the ability of the Board of Directors to protect the interests of our stockholders against potential abusive or coercive takeover tactics and to enable all stockholders the full and fair value of their investment in the event that an unsolicited attempt is made to acquire Peregrine. The Rights Agreement is not intended to prevent an offer the Board of Directors concludes is in the best interest of Peregrine and its stockholders.
Under the Rights Agreement, the Board of Directors declared a dividend of one preferred share purchase right (a “Right”) for each share of our common stock held by stockholders of record as of the close of business on March 27, 2006. Each Right entitles holders of each share of our common stock to buy seven one thousandths (7/1,000th) of a share of Peregrine’s Series D Participating Preferred Stock, par value $0.001 per share, at an exercise price of $77.00 per share, subject to adjustment. The Rights are neither exercisable nor traded separately from our common stock. The Rights will become exercisable and will detach from the common shares if a person or group acquires 15% or more of our outstanding common stock, without prior approval from our Board of Directors, or announces a tender or exchange offer that would result in that person or group owning 15% or more of our common stock. Each Right, when exercised, entitles the holder (other than the acquiring person or group) to receive our common stock (or in certain circumstances, voting securities of the acquiring person or group) with a value of twice the Rights’ exercise price upon payment of the exercise price of the Rights.
Peregrine will be entitled to redeem the Rights at $0.007 per Right at any time prior to a person or group achieving the 15% threshold. The Rights will expire on March 16, 2021.
Sales of Common Stock and Series E Preferred Stock
Our ability to continue fund our operations is highly dependent on the amount of cash and cash equivalents on hand combined with our ability to raise additional capital to support our future operations through one or more methods, including but not limited to, issuing additional equity.
Sale of Common Stock
During the three fiscal years ended April 30, 2017, we issued shares of our common stock under various financing transactions, as summarized in the following table:
| Description of Financing Transaction | Number of Common Stock Shares Issued | Gross Proceeds Raised | ||||||
| Fiscal Year 2015 | ||||||||
| At Market Issuance Sales Agreement dated December 27, 2012 | 569,052 | $ | 6,204,000 | |||||
| At Market Issuance Sales Agreement dated June 13, 2014 | 1,383,109 | $ | 13,544,000 | |||||
| 1,952,161 | $ | 19,748,000 | ||||||
| Fiscal Year 2016 | ||||||||
| At Market Issuance Sales Agreement dated June 13, 2014 | 1,232,821 | $ | 11,456,000 | |||||
| At Market Issuance Sales Agreement dated August 7, 2015 | 964,523 | $ | 7,447,000 | |||||
| Equity Distribution Agreement dated August 7, 2015 | 1,210,328 | $ | 6,969,000 | |||||
| Common Stock Purchase Agreement dated October 30, 2015 | 2,645,503 | $ | 20,000,000 | |||||
| 6,053,175 | $ | 45,872,000 | ||||||
| Fiscal Year 2017 | ||||||||
| At Market Issuance Sales Agreement dated August 7, 2015 | 6,137,403 | $ | 18,246,000 | |||||
| Equity Distribution Agreement dated August 7, 2015 | 3,750,323 | $ | 13,031,000 | |||||
| 9,887,726 | $ | 31,277,000 | ||||||
| F-19 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
The following represents additional information for each of the financing transactions included in the above table:
December 2012 AMI Sales Agreement – On December 27, 2012, we entered into an At Market Issuance Sales Agreement (“December 2012 AMI Sales Agreement”) with MLV & Co. LLC (“MLV”), pursuant to which we were able to sell shares of our common stock through MLV, as agent, for aggregate gross proceeds of up to $75,000,000, in registered transactions from our shelf registration statement on Form S-3 (File No. 333-180028), which was declared effective by the Securities and Exchange Commission (“SEC”) on April 12, 2012. Sales of our common stock through MLV were made by any method that was deemed an “at the market offering” as defined in Rule 415 of the Securities Act of 1933, as amended (the “Securities Act”). We paid MLV a commission equal to 2.5% of the gross proceeds from the sale of our common stock pursuant to the December 2012 AMI Sales Agreement. As of April 30, 2015, we had raised the full amount of gross proceeds available to us under the December 2012 AMI Sales Agreement.
June 2014 AMI Sales Agreement – On June 13, 2014, we entered into an At Market Issuance Sales Agreement with MLV, as amended on April 13, 2015 (“June 2014 AMI Sales Agreement”), pursuant to which we were able to sell shares of our common stock through MLV, as agent, for aggregate gross proceeds of up to $25,000,000 in registered transactions from our shelf registration statement on Form S-3 (File No. 333-201245), which was declared effective by the SEC on January 15, 2015 (“January 2015 Shelf”). Sales of our common stock through MLV were made by any method that was deemed an “at the market offering” as defined in Rule 415 of the Securities Act. We paid MLV a commission equal to 2.5% of the gross proceeds from the sale of our common stock pursuant to the June 2014 AMI Sales Agreement. As of April 30, 2016, we had raised the full amount of gross proceeds available to us under the June 2014 AMI Sales Agreement.
August 2015 AMI Sales Agreement – On August 7, 2015, we entered into an At Market Issuance Sales Agreement (“August 2015 AMI Sales Agreement”) with MLV, pursuant to which we may sell shares of our common stock through MLV, as agent, for aggregate gross proceeds of up to $30,000,000, in registered transactions from our January 2015 Shelf. Sales of our common stock through MLV may be made by any method that is deemed an “at the market offering” as defined in Rule 415 of the Securities Act. We pay MLV a commission equal to 2.5% of the gross proceeds from the sale of our common stock pursuant to the August 2015 AMI Sales Agreement. As of April 30, 2017, aggregate gross proceeds of up to $4,307,000 remained available to us under the August 2015 AMI Sales Agreement.
Equity Distribution Agreement – On August 7, 2015, we entered into an Equity Distribution Agreement, with Noble International Investments, Inc., doing business as Noble Life Science Partners, a division of Noble Financial Capital Markets (“Noble”), pursuant to which we were able to sell shares of our common stock through Noble, as agent, for aggregate gross proceeds of up to $20,000,000, in registered transactions from our January 2015 Shelf. Sales of our common stock through Noble were made by any method that was deemed an “at the market offering” as defined in Rule 415 of the Securities Act. We paid Noble a commission equal to 2.5% of the gross proceeds from the sale of our common stock pursuant to the Equity Distribution Agreement. As of April 30, 2017, we had raised the full amount of gross proceeds available to us under the Equity Distribution Agreement.
Common Stock Purchase Agreement – On October 30, 2015, we entered into a Common Stock Purchase Agreement with Eastern Capital Limited, pursuant to which we issued and sold 2,645,503 shares of our common stock, at a purchase price of $7.56 per share for aggregate gross proceeds of $20,000,000 before deducting issuance costs of $1,000. These shares of common stock were sold under our January 2015 Shelf pursuant to a prospectus supplement filed with the SEC on October 30, 2015.
| F-20 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Sale of Series E Preferred Stock
During the three fiscal years ended April 30, 2017, we issued shares of our Series E Preferred Stock under an At Market Issuance Sales Agreement, as summarized in the following table:
| Description of Financing Transaction | Number of Series E Preferred Stock Shares Issued | Gross Proceeds Raised | ||||||
| Fiscal Year 2015 | ||||||||
| At Market Issuance Sales Agreement dated June 13, 2014 | 799,764 | $ | 19,205,000 | |||||
| Fiscal Year 2016 | ||||||||
| At Market Issuance Sales Agreement dated June 13, 2014 | 2,676 | $ | 60,000 | |||||
| Fiscal Year 2017 | ||||||||
| At Market Issuance Sales Agreement dated June 13, 2014 | 70,320 | $ | 1,634,000 | |||||
The following represents additional information for the At Market Issuance Sales Agreement included in the above table:
On June 13, 2014, we entered into an At Market Issuance Sales Agreement (“Series E AMI Sales Agreement”) with MLV, pursuant to which we were able to sell shares of our Series E Preferred Stock through MLV, as agent, for aggregate gross proceeds of up to $30,000,000, in registered transactions from our shelf registration statement on Form S-3 (File No. 333-193113), which was declared effective by the SEC on January 16, 2014 (“January 2014 Shelf”). Sales of our Series E Preferred Stock through MLV were be made by any method that was deemed an “at the market offering” as defined in Rule 415 of the Securities Act. We paid MLV a commission of up to 5% of the gross proceeds from the sale of our Series E Preferred Stock pursuant to the Series E AMI Sales Agreement. During January 2017, the underlying January 2014 Shelf expired, and therefore, we do not plan to issue and sell any additional shares of our Series E Preferred Stock under the Series E AMI Sales Agreement.
Series E Preferred Stock Rights and Preferences
On February 12, 2014, we filed with the Secretary of State of the State of Delaware a Certificate of Designations of Rights and Preferences (the “Certificate of Designations”) to designate the Series E Preferred Stock. The Certificate of Designations designated 2,000,000 shares of Series E Preferred Stock out of our 5,000,000 shares of authorized but unissued shares of preferred stock. In addition, the Series E Preferred Stock is classified as permanent equity in accordance with FASB Accounting Standards Codification Topic 480, Distinguishing Liabilities from Equity. Certain terms of the Series E Preferred Stock include:
(i) The holders are entitled to receive a 10.50% per annum cumulative quarterly dividend, payable in cash, on or about the 1st day of each of January, April, July, and October;
(ii) The dividend may increase to a penalty rate of 12.50% if: (a) we fail to pay dividends for any four consecutive or nonconsecutive quarterly dividend periods, or (b) once the Series E Preferred Stock becomes initially eligible for listing on a national securities exchange, we fail, for 180 or more consecutive days, to maintain such listing;
(iii) Following a change of control of the Company (as defined in the Certificate of Designations) by a person or entity, we (or the acquiring entity) may, at our option, redeem the Series E Preferred Stock, in whole but not in part, within 120 days after the date on which the change of control has occurred for cash, at the redemption price;
(iv) On and after February 11, 2017, we may redeem the Series E Preferred Stock for cash at our option, from time to time, in whole or in part, at the redemption price;
(v) The redemption price is $25.00 per share, plus any accrued and unpaid dividends (whether or not earned or declared) to, but excluding, the redemption date;
(vi) The liquidation preference is $25.00 per share, plus any accrued and unpaid dividends (whether or not earned or declared);
| F-21 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
(vii) The Series E Preferred Stock has no stated maturity date or mandatory redemption and is senior to all of the Company’s other securities;
(viii) There is a general conversion right with respect to the Series E Preferred Stock with a current conversion price of $21.00 (as adjusted to reflect the 1-for-7 reverse stock split of our issued and outstanding common stock, which took effect on July 10, 2017), a special conversion right upon a change of control, and a market trigger conversion at our option in the event of Market Trigger (as defined in the Certificate of Designations); and
(ix) The holders of the Series E Preferred Stock have no voting rights, except as defined in the Certificate of Designations.
Series E Preferred Stock Dividends
The following table summarizes the Series E Preferred Stock quarterly dividend payments during the three fiscal years ended April 30, 2017:
| Declaration Date | Record Date | Payment Date | Dividends Paid | Dividend Per Share | ||||||||||||
| Fiscal Year 2015 | ||||||||||||||||
| 6/10/2014 | 6/20/2014 | 7/1/2014 | $ | 771,000 | $ | 0.65625 | ||||||||||
| 9/8/2014 | 9/19/2014 | 10/1/2014 | $ | 773,000 | $ | 0.65625 | ||||||||||
| 12/9/2014 | 12/19/2014 | 1/2/2015 | $ | 774,000 | $ | 0.65625 | ||||||||||
| 3/10/2015 | 3/20/2015 | 4/1/2015 | $ | 1,034,000 | $ | 0.65625 | ||||||||||
| $ | 3,352,000 | $ | 2.62500 | |||||||||||||
| Fiscal Year 2016 | ||||||||||||||||
| 6/5/2015 | 6/19/2015 | 7/1/2015 | $ | 1,034,000 | $ | 0.65625 | ||||||||||
| 9/8/2015 | 9/18/2015 | 10/1/2015 | $ | 1,035,000 | $ | 0.65625 | ||||||||||
| 12/7/2015 | 12/18/2015 | 1/4/2016 | $ | 1,035,000 | $ | 0.65625 | ||||||||||
| 3/7/2016 | 3/18/2016 | 4/1/2016 | $ | 1,035,000 | $ | 0.65625 | ||||||||||
| $ | 4,139,000 | $ | 2.62500 | |||||||||||||
| Fiscal Year 2017 | ||||||||||||||||
| 6/2/2016 | 6/17/2016 | 7/1/2016 | $ | 1,036,000 | $ | 0.65625 | ||||||||||
| 9/6/2016 | 9/16/2016 | 10/3/2016 | $ | 1,081,000 | $ | 0.65625 | ||||||||||
| 12/6/2016 | 12/16/2016 | 1/3/2017 | $ | 1,081,000 | $ | 0.65625 | ||||||||||
| 3/9/2017 | 3/20/2017 | 4/3/2017 | $ | 1,081,000 | $ | 0.65625 | ||||||||||
| $ | 4,279,000 | $ | 2.62500 | |||||||||||||
Shares of Common Stock Authorized and Reserved For Future Issuance
We are authorized to issue up to 500,000,000 shares of our common stock. As of April 30, 2017, 44,014,040 shares of our common stock were issued and outstanding. In addition, our common stock outstanding as of April 30, 2017 excluded the following shares of common stock reserved for future issuance:
| · | 5,630,872 shares of common stock reserved for issuance under outstanding option grants and available for issuance under our stock incentive plans; |
| · | 1,359,736 shares of common stock reserved for and available for issuance under our ESPP; |
| · | 39,040 shares of common stock issuable upon exercise of outstanding warrants; and |
| · | 6,826,435 shares of common stock issuable upon conversion of our outstanding Series E Preferred Stock (1). |
_____________
| (1) | The Series E Preferred Stock is convertible into a number of shares of our common stock determined by dividing the liquidation preference of $25.00 per share by the conversion price, currently $21.00 per share. If all of our outstanding Series E Preferred Stock were converted at the $21.00 per share conversion price, the holders of our Series E Preferred Stock would receive an aggregate of 1,961,619 shares of our common stock. However, we have reserved the maximum number of shares of our common stock that could be issued upon a change of control event assuming our shares of common stock are acquired for consideration of $5.985 per share or less. In this scenario, each outstanding share of our Series E Preferred Stock could be converted into 4.18 shares of our common stock, representing the Share Cap. |
| F-22 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 6. | EQUITY COMPENSATION PLANS |
Stock Incentive Plans
We currently maintain six stock incentive plans referred to as the 2011 Plan, the 2010 Plan, the 2009 Plan, the 2005 Plan, the 2003 Plan, and the 2002 Plan (collectively referred to as the “Stock Plans”). The 2011, 2010, 2009, 2005 and 2003 Plans were approved by our stockholders while the 2002 Plan was not submitted for stockholder approval. The Stock Plans provide for the granting of stock options, restricted stock awards and other forms of share-based awards to purchase shares of our common stock at exercise prices not less than the fair market value of our common stock at the date of grant.
As of April 30, 2017, we had an aggregate of 5,630,872 shares of our common stock reserved for issuance under the Stock Plans, of which, 4,081,548 shares were subject to outstanding options and 1,549,324 shares were available for future grants of share-based awards.
Stock Options – Stock options granted under our Stock Plans are granted at an exercise price not less than the fair market value of our common stock on the date of grant. The options generally vest over a two to four year period and expire ten years from the date of grant, if unexercised. However, certain option awards provide for accelerated vesting if there is a change in control (as defined in the Stock Plans).
The estimated fair value of stock options are measured at the grant date, using a fair value based method, such as a Black-Scholes option valuation model, and is amortized as compensation expense on a straight-line basis over the requisite service period of the award, which is generally the vesting period. The use of a valuation model requires us to make certain estimates and assumptions with respect to selected model inputs. The expected volatility is based on the daily historical volatility of our common stock covering the estimated expected term. The expected term of options granted reflects actual historical exercise activity and assumptions regarding future exercise activity of unexercised, outstanding options. The risk-free interest rate is based on U.S. Treasury notes with terms within the contractual life of the option at the time of grant. The expected dividend yield assumption is based on our expectation of future dividend payouts. We have never declared or paid any cash dividends on our common stock and currently do not anticipate paying such cash dividends. The fair value of stock options on the date of grant and the weighted-average assumptions used to estimate the fair value of the stock options using the Black-Scholes option valuation model for fiscal years ended April 30, 2017, 2016 and 2015, were as follows:
| Fiscal Year Ended April 30, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Risk-free interest rate | 1.32% | 1.66% | 1.95% | |||||||||
| Expected life (in years) | 6.12 | 5.96 | 5.74 | |||||||||
| Expected volatility | 111.30% | 104.74% | 111.78% | |||||||||
| Expected dividend yield | – | – | – | |||||||||
The following summarizes our stock option transaction activity for fiscal year ended April 30, 2017:
| Stock Options | Shares | Weighted Average Exercisable Price | Weighted Average Remaining Contractual Term (years) | Aggregate Intrinsic Value (1) | ||||||||||||
| Outstanding, May 1, 2016 | 3,393,037 | $ | 10.37 | |||||||||||||
| Granted | 1,009,085 | $ | 3.42 | |||||||||||||
| Exercised | (9,026 | ) | $ | 3.44 | ||||||||||||
| Canceled or expired | (311,548 | ) | $ | 9.06 | ||||||||||||
| Outstanding, April 30, 2017 | 4,081,548 | $ | 8.77 | 6.36 | $ | 1,267,665 | ||||||||||
| Exercisable and expected to vest | 4,067,608 | $ | 8.78 | 6.35 | $ | 1,252,814 | ||||||||||
| Exercisable, April 30, 2017 | 3,343,699 | $ | 9.74 | 5.82 | $ | 687,283 | ||||||||||
______________
| (1) | Aggregate intrinsic value represents the difference between the exercise price of an option and the closing market price of our common stock on April 28, 2017 (the last trading day of fiscal year 2017), which was $4.31 per share. |
| F-23 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
The weighted-average grant date fair value of options granted to employees during the fiscal years ended April 30, 2017, 2016 and 2015 was $2.86, $7.09 and $9.99 per share, respectively.
The aggregate intrinsic value of stock options exercised during the fiscal years ended April 30, 2017, 2016 and 2015 was $11,000, $93,000 and $192,000, respectively. Cash received from stock options exercised during fiscal years ended April 30, 2017, 2016 and 2015, totaled $31,000, $138,000 and $298,000, respectively, net of issuance costs of nil, $1,000 and $3,000, respectively.
We issue shares of common stock that are reserved for issuance under the Stock Plans upon the exercise of stock options, and we do not expect to repurchase shares of common stock from any source to satisfy our obligations under our compensation plans.
As of April 30, 2017, the total estimated unrecognized compensation cost related to non-vested employee stock options was $2,215,000. This cost is expected to be recognized over a weighted average vesting period of 1.43 years based on current assumptions.
Employee Stock Purchase Plan
We have reserved a total of 2,142,857 shares of our common stock to be purchased under our Employee Stock Purchase Plan (the “ESPP”), of which 1,359,736 shares remained available to purchase at April 30, 2017, and are subject to adjustment as provided in the ESPP for stock splits, stock dividends, recapitalizations and other similar events. Under the ESPP, we sell shares to participants at a price equal to the lesser of 85% of the fair market value of our common stock at the (i) beginning of a six-month offering period, or (ii) end of the six-month offering period. The ESPP provides for two six-month offering periods each fiscal year; the first offering period begins on the first trading day on or after each May 1; the second offering period begins on the first trading day on or after each November 1. During the fiscal years ended April 30, 2017, 2016 and 2015, 270,075, 147,769 and 71,063 shares of our common stock were purchased, respectively, under the ESPP at a weighted average purchase price per share of $1.95, $3.65 and $8.56, respectively.
The fair value of the shares purchased under the ESPP were determined using a Black-Scholes option pricing model (see explanation of valuation model inputs above under “Stock Options”), and is recognized as expense on a straight-line basis over the requisite service period (or six-month offering period). The weighted average grant date fair value of purchase rights under the ESPP during fiscal years ended April 30, 2017, 2016 and 2015 was $1.07, $2.40 and $3.91, respectively, based on the following Black-Scholes option valuation model inputs:
| Fiscal Year Ended April 30, | ||||||||||||
| 2017 | 2016 | 2015 | ||||||||||
| Risk-free interest rate | 0.46% | 0.18% | 0.06% | |||||||||
| Expected life (in years) | 0.50 | 0.50 | 0.50 | |||||||||
| Expected volatility | 105.27% | 46.14% | 63.54% | |||||||||
| Expected dividend yield | – | – | – | |||||||||
| F-24 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Share-based Compensation Expense
Total share-based compensation expense related to share-based awards issued under our equity compensation plans for the fiscal years ended April 30, 2017, 2016 and 2015 was comprised of the following:
| 2017 | 2016 | 2015 | ||||||||||
| Cost of contract manufacturing | $ | 108,000 | $ | 41,000 | $ | 59,000 | ||||||
| Research and development | 1,615,000 | 2,124,000 | 2,904,000 | |||||||||
| Selling, general and administrative | 1,640,000 | 2,733,000 | 3,739,000 | |||||||||
| Total | $ | 3,363,000 | $ | 4,898,000 | $ | 6,702,000 | ||||||
| Share-based compensation from: | ||||||||||||
| Stock options | $ | 3,094,000 | $ | 4,720,000 | $ | 6,465,000 | ||||||
| ESPP | 269,000 | 178,000 | 237,000 | |||||||||
| $ | 3,363,000 | $ | 4,898,000 | $ | 6,702,000 | |||||||
Share-based compensation expense recorded during fiscal years ended April 30, 2017, 2016 and 2015 associated with share-based awards granted to non-employees amounted to $20,000, $109,000 and $289,000, respectively. As of April 30, 2017, the total estimated unrecognized compensation cost related to non-vested stock options granted to non-employees was $13,000 based on an April 30, 2017 measurement date. This cost is expected to be recognized over a weighted average vesting period of 1.10 years.
Due to our net loss position, no tax benefits have been recognized in the consolidated statements of cash flows.
| 7. | WARRANTS |
No warrants were issued or exercised during fiscal years ended April 30, 2017, 2016 and 2015. As of April 30, 2017, warrants to purchase 39,040 shares of our common stock at an exercise price of $17.29 were outstanding and are exercisable through August 30, 2018.
| 8. | INCOME TAXES |
We are primarily subject to U.S. federal and California state jurisdictions. To our knowledge, all tax years remain open to examination by U.S. federal and state authorities.
In addition, in accordance with authoritative guidance, we are required to recognize the impact of an uncertain tax position in the consolidated financial statements when it is more likely than not the position will be sustained upon examination by the tax authorities. An uncertain tax position will not be recognized if it has less than a 50% likelihood of being sustained upon examination by the tax authorities. We had no unrecognized tax benefits from uncertain tax positions as of April 30, 2017 and 2016. It is also our policy, in accordance with authoritative guidance, to recognize interest and penalties related to income tax matters in interest and other expense in our consolidated statements of operations and comprehensive loss. We did not recognize interest or penalties related to income taxes for fiscal years ended April 30, 2017, 2016, and 2015, and we did not accrue for interest or penalties as of April 30, 2017 and 2016.
At April 30, 2017, we had net deferred tax assets of $178,400,000. Due to uncertainties surrounding our ability to generate future taxable income to realize these tax assets, a full valuation has been established to offset our net deferred tax assets. Additionally, the future utilization of our net operating loss carry forwards to offset future taxable income may be subject to an annual limitation, pursuant to Internal Revenue Code Section 382, as a result of ownership changes that may have occurred previously or that could occur in the future. A Section 382 analysis was completed as of the fiscal year ended April 30, 2017 and it was determined that no change in ownership had occurred. Ownership changes occurring subsequent to April 30, 2017 may impact the utilization of net operating loss carry forwards and other tax attributes.
| F-25 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
At April 30, 2017, we had federal net operating loss carry forwards of approximately $391,878,000. The net operating loss carry forwards expire in fiscal years 2019 through 2037. We also have California state net operating loss carry forwards of approximately $263,812,000 at April 30, 2017, which begin to expire in fiscal year 2018. In addition, we have approximately $5,956,000 of net operating loss attributable to excess tax deductions on share-based compensation that when utilized, if any, the tax benefit will be booked to additional paid-in-capital.
The provision for income taxes consists of the following for the three years ended April 30,:
| 2017 | 2016 | 2015 | ||||||||||
| Federal income taxes at statutory rate | $ | (9,573,000 | ) | $ | (18,921,000 | ) | $ | (17,122,000 | ) | |||
| State income taxes | (2,264,000 | ) | (4,824,000 | ) | (4,450,000 | ) | ||||||
| Expiration and adjustments of deferred tax assets | 1,693,000 | 1,580,000 | 1,790,000 | |||||||||
| Change in valuation allowance | 10,005,000 | 21,871,000 | 19,532,000 | |||||||||
| Other, net | 139,000 | 294,000 | 250,000 | |||||||||
| Income tax (expense) benefit | $ | – | $ | – | $ | – | ||||||
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts for income tax purposes. Significant components of our deferred tax assets and deferred tax liabilities at April 30, 2017 and 2016 are as follows:
| 2017 | 2016 | |||||||
| Share-based compensation | $ | 9,583,000 | $ | 8,806,000 | ||||
| Deferred revenue | 12,157,000 | 4,296,000 | ||||||
| Deferred rent | 738,000 | 470,000 | ||||||
| Other | 2,984,000 | 2,486,000 | ||||||
| Net operating losses | 154,030,000 | 152,746,000 | ||||||
| Total deferred tax assets | 179,492,000 | 168,804,000 | ||||||
| Less valuation allowance | (178,400,000 | ) | (168,395,000 | ) | ||||
| Total deferred tax assets, net of valuation allowance | $ | 1,092,000 | $ | 409,000 | ||||
| Deferred tax liabilities: | ||||||||
| Fixed assets | (1,092,000 | ) | (409,000 | ) | ||||
| Total deferred tax liabilities | (1,092,000 | ) | (409,000 | ) | ||||
| Net deferred tax assets | $ | – | $ | – | ||||
| 9. | BENEFIT PLAN |
During fiscal year 1997, we adopted a 401(k) benefit plan (the “Plan”) for all full-time employees who are at least the age of 21 and have three or more months of continuous service. The Plan provides for employee contributions of up to 100% of their compensation on a tax deferred basis up to the maximum amount permitted by the Internal Revenue Code. We are not required to make matching contributions under the Plan, and prior to January 2010, we did not make any matching contributions from the Plan’s inception. Presently, we have voluntarily agreed to match, depending on an employee’s years of continuous service, between 50% and 100% of an employee’s contributions up to 6% of their annual eligible compensation, subject to certain IRS limitations.
Under the Plan, each participating employee is fully vested in his or her contributions to the Plan and our contributions to the Plan will fully vest after six years of service. The expense related to our matching contributions to the Plan was $845,000, $543,000, and $454,000 for the fiscal years ended April 30, 2017, 2016, and 2015, respectively.
| F-26 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
| 10. | SEGMENT REPORTING |
Our business is organized into two reportable operating segments and both operate in the U.S. Peregrine is engaged in the research and development of monoclonal antibodies for the treatment of cancer. Avid is engaged in providing contract manufacturing services for third-party customers on a fee-for-service basis while also supporting our internal drug development efforts.
The accounting policies of the operating segments are the same as those described in Note 2. We evaluate the performance of our contract manufacturing services segment based on gross profit or loss from third-party customers. However, our products in the research and development segment are not evaluated based on gross profit or loss, but rather based on scientific progress of the technologies. As such, gross profit or loss is only provided for our contract manufacturing services segment in the below table. All revenues shown below are derived from transactions with third-party customers.
Segment information for the fiscal years ended April 30, 2017, 2016 and 2015 is summarized as follows:
| 2017 | 2016 | 2015 | ||||||||||
| Contract manufacturing services revenue | $ | 57,630,000 | $ | 44,357,000 | $ | 26,744,000 | ||||||
| Cost of contract manufacturing services | 38,259,000 | 22,966,000 | 15,593,000 | |||||||||
| Gross profit | $ | 19,371,000 | $ | 21,391,000 | $ | 11,151,000 | ||||||
| Revenue from products in research and development | $ | – | $ | 329,000 | $ | 37,000 | ||||||
| Research and development expense | (28,297,000 | ) | (59,529,000 | ) | (42,996,000 | ) | ||||||
| Selling, general and administrative expense | (19,334,000 | ) | (18,551,000 | ) | (18,691,000 | ) | ||||||
| Other income (expense), net | 101,000 | 708,000 | 141,000 | |||||||||
| Net loss | $ | (28,159,000 | ) | $ | (55,652,000 | ) | $ | (50,358,000 | ) | |||
Revenue generated from our contract manufacturing services segment during fiscal years ended April 30, 2017, 2016 and 2015 was derived from a limited number of customers. The percentages below represent revenue derived from each customer (and geographical location) as a percentage of total contract manufacturing services revenue:
| Customer | Geographic Location | 2017 | 2016 | 2015 | ||||||||||||
| Halozyme Therapeutics, Inc. | U.S. | 58% | 69% | 79% | ||||||||||||
| Customer A | U.S. | 26 | 26 | 12 | ||||||||||||
| Customer B | U.S. | 14 | – | – | ||||||||||||
| Other customers | U.S./non-U.S. | 2 | 5 | 9 | ||||||||||||
| Total | 100% | 100% | 100% | |||||||||||||
In addition, we attribute contract manufacturing services revenue to the individual countries where the customer is headquartered. Contract manufacturing services revenue from customers are summarized by geographic location in the following table:
| 2017 | 2016 | 2015 | ||||||||||||
| U.S. | $ | 57,630,000 | $ | 44,357,000 | $ | 26,715,000 | ||||||||
| Non-U.S. | – | – | 29,000 | |||||||||||
| Total | $ | 57,630,000 | $ | 44,357,000 | $ | 26,744,000 | ||||||||
Revenue generated from our products in our research and development segment during fiscal years ended April 30, 2016 and 2015 were directly related to license revenue recognized under licensing agreements with unrelated entities.
| F-27 |
PEREGRINE PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
FOR EACH OF THE THREE YEARS IN THE PERIOD ENDED APRIL 30, 2017 (continued)
Our long-lived assets are located in the U.S. and consist of leasehold improvements, laboratory equipment, furniture and fixtures, office equipment and software and are net of accumulated depreciation. Long-lived assets by segment as of April 30, 2017 and 2016 consist of the following:
| 2017 | 2016 | |||||||
| Long-lived Assets, net: | ||||||||
| Contract manufacturing services | $ | 22,599,000 | $ | 22,783,000 | ||||
| Products in research and development | 1,074,000 | 1,519,000 | ||||||
| Total | $ | 23,673,000 | $ | 24,302,000 | ||||
| 11. | SELECTED QUARTERLY FINANCIAL DATA (UNAUDITED) |
Selected quarterly financial information for each of the two most recent fiscal years is as follows:
| Quarter Ended | ||||||||||||||||||||||||||||||||
| April 30, | January 31, | October 31, | July 31, | April 30, | January 31, | October 31, | July 31, | |||||||||||||||||||||||||
| 2017 | 2017 | 2016 | 2016 | 2016 | 2016 | 2015 | 2015 | |||||||||||||||||||||||||
| Net revenues | $ | 17,904,000 | $ | 10,747,000 | $ | 23,370,000 | $ | 5,609,000 | $ | 18,783,000 | $ | 6,709,000 | $ | 9,523,000 | $ | 9,671,000 | ||||||||||||||||
| Gross profit (a) | $ | 6,122,000 | $ | 2,773,000 | $ | 7,929,000 | $ | 2,547,000 | $ | 9,062,000 | $ | 2,776,000 | $ | 4,782,000 | $ | 4,771,000 | ||||||||||||||||
| Loss from operations | $ | (5,304,000 | ) | $ | (7,797,000 | ) | $ | (4,077,000 | ) | $ | (11,082,000 | ) | $ | (11,915,000 | ) | $ | (16,867,000 | ) | $ | (13,824,000 | ) | $ | (13,754,000 | ) | ||||||||
| Net loss | $ | (5,272,000 | ) | $ | (7,774,000 | ) | $ | (4,056,000 | ) | $ | (11,057,000 | ) | $ | (11,884,000 | ) | $ | (16,847,000 | ) | $ | (13,198,000 | ) | $ | (13,723,000 | ) | ||||||||
| Series E preferred stock accumulated dividends (b) | $ | (1,442,000 | ) | $ | (1,442,000 | ) | $ | (1,442,000 | ) | $ | (1,380,000 | ) | $ | (1,380,000 | ) | $ | (1,380,000 | ) | $ | (1,380,000 | ) | $ | (1,378,000 | ) | ||||||||
| Net loss attributable to common stockholders | $ | (6,714,000 | ) | $ | (9,216,000 | ) | $ | (5,498,000 | ) | $ | (12,437,000 | ) | $ | (13,264,000 | ) | $ | (18,227,000 | ) | $ | (14,578,000 | ) | $ | (15,101,000 | ) | ||||||||
| Basic and diluted loss per common share | $ | (0.16 | ) | $ | (0.25 | ) | $ | (0.16 | ) | $ | (0.36 | ) | $ | (0.40 | ) | $ | (0.56 | ) | $ | (0.50 | ) | $ | (0.54 | ) | ||||||||
_____ _________
| (a) | Gross profit represents contract manufacturing revenue less cost of contract manufacturing. |
| (b) | Series E preferred stock accumulated dividends include dividends declared for the period (regardless of whether or not the dividends have been paid) and dividends accumulated for the period (regardless of whether or not the dividends have been declared). |
| 12. | SUBSEQUENT EVENTS |
Sale of Common Stock
August 2015 AMI Sales Agreement - Subsequent to April 30, 2017 and through June 30, 2017, we sold 1,051,258 shares of common stock at market prices under the August 2015 AMI Sales Agreement (Note 5) for aggregate gross proceeds of $4,304,000. As of June 30, 2017, we had raised the full amount of the gross proceeds available to us under the August 2015 AMI Sales Agreement.
Series E Preferred Stock Dividend
On June 6, 2017, our Board of Directors declared a quarterly cash dividend of $0.65625 per share on our Series E Preferred Stock. The dividend payment is equivalent to an annualized 10.50% per share, based on the $25.00 per share stated liquidation preference, accruing from April 1, 2017 through June 30, 2017. The cash dividend of $1,081,000 was paid on July 3, 2017 to holders of the Series E Preferred Stock of record on June 19, 2017.
Reverse Stock Split
On July 7, 2017, we effected a reverse stock split of our outstanding shares of common stock at a ratio of one-for-seven, which took effect with the opening of trading on July 10, 2017. The primary purpose of the reverse stock split, which was approved by our stockholders at our 2016 Annual Meeting on October 13, 2016, was to enable us to regain compliance with the $1.00 minimum bid price requirement for continued listing on The NASDAQ Capital Market. All fractional shares created by the reverse stock split were rounded up to the nearest whole share. In addition, all share and per share amounts in the accompanying consolidated financial statements have been adjusted to give effect to the one-for-seven reverse stock split, retrospectively (Note 1).
| F-28 |