Attached files
| file | filename |
|---|---|
| EX-32.1 - EX-32.1 - Bioverativ Inc. | bivv-20170331ex321c02007.htm |
| EX-31.2 - EX-31.2 - Bioverativ Inc. | bivv-20170331ex31226b303.htm |
| EX-31.1 - EX-31.1 - Bioverativ Inc. | bivv-20170331ex3117a9959.htm |
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-Q
|
(Mark one) |
|
|
☑ |
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
|
|
For the quarterly period ended March 31, 2017 |
|
|
|
|
|
|
or |
|
|
|
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(D) OF THE SECURITIES EXCHANGE ACT OF 1934 |
|
|
|
|
For the transition period from to . |
|
Commission file number 001-37859

Bioverativ Inc.
(Exact Name of Registrant as Specified in Its Charter)
|
Delaware |
81-3461310 |
|
225 Second Avenue, Waltham, |
02451 |
(781) 663‑4400
(Registrant’s Telephone Number, Including Area Code)
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
|
|
Yes ☑ No ☐ |
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files).
|
|
Yes ☑ No ☐ |
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
Non-accelerated filer ☑ (Do not check if a smaller reporting company) |
Smaller reporting company ☐ |
Emerging growth company ☑ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☑
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
|
|
Yes ☐ No ☑ |
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date:
The number of shares of the issuer’s common stock, $0.001 par value, outstanding as of May 1, 2017 was 108,159,852
BIOVERATIV INC.
FORM 10-Q – Quarterly Report
For the Quarterly Period Ended March 31, 2017
2
NOTE REGARDING FORWARD‑LOOKING STATEMENTS
This report contains forward‑looking statements. Use by Bioverativ of the words “may,” “will,” “would,” “could,” “should,” “believes,” “estimates,” “projects,” “potential,” “expects,” “plans,” “seeks,” “intends,” “evaluates,” “pursues,” “anticipates,” “continues,” “designs,” “impacts,” “affects,” “forecasts,” “target,” “outlook,” “initiative,” “objective,” “designed,” “priorities,” or the negative of those words or other similar expressions is intended to identify forward‑looking statements.
These forward‑looking statements may include statements with respect to:
|
· |
accounting estimates, assumptions and policies; |
|
· |
estimates of liabilities; |
|
· |
separation related adjustments and ongoing costs of certain services provided by Biogen post-separation; |
|
· |
expected timing of the completion of certain transition and other services provided by Biogen to us; |
|
· |
contingent payments, including milestone and royalty payment obligations; |
|
· |
our exposure to market volatility and foreign currency and interest rate risks; |
|
· |
revenue growth and associated costs, discounts or rebates in connection with our products; |
|
· |
tax rates; |
|
· |
future cash flows, working capital needs, capital expenditures, strategic investments and access to capital; |
|
· |
future transactions in our securities and debt issuances; |
|
· |
dividends; |
|
· |
litigation related matters, including outcomes; |
|
· |
the impact of healthcare reform; |
|
· |
business and strategic objectives; |
|
· |
business development activities; |
|
· |
our anticipated investments in infrastructure and research and development activities; |
|
· |
expected clinical trials; |
|
· |
our anticipated geographic expansion; |
|
· |
our growth, including patient share growth; |
|
· |
our manufacturing, supply and distribution arrangements; |
|
· |
the sufficiency of our facilities; |
|
· |
our relationships with third parties, collaborators and our employees; |
|
· |
our operation as a standalone company; |
|
· |
receipt of necessary regulatory authorization and approvals; |
|
· |
our ability to finance our operations and business initiatives and obtain funding for such activities; and |
|
· |
the adoption and impact of new laws and accounting standards. |
These forward-looking statements involve risks and uncertainties, including those that are described in Item 1A. Risk Factors and elsewhere in this report, that could cause actual results to differ materially from those reflected in such statements. You should not place undue reliance on these statements. Forward-looking statements speak only as of the date of this report. Except as required by law, we do not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise.
NOTE REGARDING PRESENTATION OF INFORMATION
Unless the context otherwise requires, references in this report to the following terms shall have the following respective meanings:
|
· |
“Biogen” refers to Biogen Inc., a Delaware corporation, and its consolidated subsidiaries; |
|
· |
“distribution” refers to the distribution by Biogen to Biogen stockholders of all of the outstanding shares of Bioverativ, as further described in this report; |
3
|
· |
“hemophilia business” includes Biogen’s hemophilia business and certain additional assets and liabilities associated with Biogen’s pipeline programs related to hemophilia and other blood disorders; |
|
· |
“separation” refers to the separation of Biogen’s hemophilia business from Biogen’s other businesses and the creation, as a result of the distribution, of an independent, publicly traded company, Bioverativ Inc., that holds the hemophilia business, as further described in this report; |
|
· |
“separation date” is February 1, 2017; and |
|
· |
“Bioverativ,” “we,” “us,” “our,” “our company” and “the company” refer to Bioverativ Inc., a Delaware corporation, or Bioverativ Inc., together with its subsidiaries, as the context requires. |
This report describes the business transferred to Bioverativ by Biogen in the separation as if the transferred business was Bioverativ’s business for all historical periods described. References in this report to Bioverativ’s historical assets, liabilities, products, businesses or activities of Bioverativ’s business are generally intended to refer to the historical assets, liabilities, products, businesses or activities of the transferred business as the business was conducted as part of Biogen prior to the separation. Since the separation date, Bioverativ has operated as standalone company.
NOTE REGARDING TRADEMARKS, TRADE NAMES AND SERVICE MARKS
Bioverativ owns or has rights to use the trademarks, service marks and trade names that it uses in conjunction with the operation of its business. Some of the trademarks that Bioverativ owns or has rights to use that appear in this report include: ALPROLIX® and ELOCTATE®, which may be registered or trademarked in the United States and other jurisdictions. Bioverativ’s rights to some of these trademarks may be limited to select markets. Each trademark, trade name or service mark of any other company appearing in this report is, to Bioverativ’s knowledge, owned by such other company. References to ELOCTATE in this report shall also refer to ELOCTA, the approved trade name for ELOCTATE in the European Union, as the context may require.
.
4
PART I – FINANCIAL INFORMATION
BIOVERATIV INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF INCOME AND COMPREHENSIVE INCOME
(unaudited, in millions, except per share amounts)
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
|
|
2017 |
|
2016 |
||
|
Revenues: |
|
|
|
|
|
|
|
Product, net |
|
$ |
241.9 |
|
$ |
182.8 |
|
Collaboration |
|
|
17.2 |
|
|
8.9 |
|
Total revenues |
|
|
259.1 |
|
|
191.7 |
|
Cost and expenses: |
|
|
|
|
|
|
|
Cost of sales |
|
|
63.3 |
|
|
32.5 |
|
Research and development |
|
|
36.9 |
|
|
54.3 |
|
Selling, general and administrative |
|
|
47.0 |
|
|
39.0 |
|
Total cost and expenses |
|
|
147.2 |
|
|
125.8 |
|
Income from operations |
|
|
111.9 |
|
|
65.9 |
|
Other income (expense), net |
|
|
(0.4) |
|
|
(0.4) |
|
Income before income tax expense (benefit) |
|
|
111.5 |
|
|
65.5 |
|
Income tax expense (benefit) |
|
|
42.2 |
|
|
(1.2) |
|
Net income |
|
$ |
69.3 |
|
$ |
66.7 |
|
Net income per share: |
|
|
|
|
|
|
|
Basic earnings per share |
|
$ |
0.64 |
|
$ |
0.62 |
|
Diluted earnings per share |
|
$ |
0.64 |
|
$ |
0.62 |
|
Weighted average shares used in calculating: |
|
|
|
|
|
|
|
Basic earnings per share |
|
|
108.0 |
|
|
108.0 |
|
Diluted earnings per share |
|
|
108.2 |
|
|
108.0 |
|
Other comprehensive income: |
|
|
|
|
|
|
|
Net income |
|
$ |
69.3 |
|
$ |
66.7 |
|
Currency translation adjustment |
|
|
(2.5) |
|
|
1.4 |
|
Total other comprehensive income |
|
|
(2.5) |
|
|
1.4 |
|
Comprehensive income |
|
$ |
66.8 |
|
$ |
68.1 |
See accompanying notes to these unaudited condensed consolidated financial statements.
5
BIOVERATIV INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED BALANCE SHEETS
(unaudited, in millions, except share data)
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
|
|
2017 |
|
2016 |
||
|
ASSETS |
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
358.7 |
|
$ |
— |
|
Accounts receivable, net |
|
|
144.4 |
|
|
149.4 |
|
Inventory |
|
|
91.4 |
|
|
302.0 |
|
Due from Biogen |
|
|
45.9 |
|
|
— |
|
Other current assets |
|
|
38.3 |
|
|
24.2 |
|
Total current assets |
|
|
678.7 |
|
|
475.6 |
|
Property, plant and equipment, net |
|
|
23.1 |
|
|
28.4 |
|
Intangible assets, net |
|
|
50.2 |
|
|
51.7 |
|
Deferred tax assets |
|
|
22.6 |
|
|
154.2 |
|
Other long-term assets |
|
|
22.0 |
|
|
22.0 |
|
Total assets |
|
$ |
796.6 |
|
$ |
731.9 |
|
LIABILITIES AND EQUITY |
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
Accounts payable |
|
$ |
16.0 |
|
$ |
12.7 |
|
Accrued expenses and other current liabilities |
|
|
111.7 |
|
|
89.3 |
|
Due to Biogen |
|
|
10.7 |
|
|
— |
|
Total current liabilities |
|
|
138.4 |
|
|
102.0 |
|
Long-term liabilities |
|
|
73.1 |
|
|
63.7 |
|
Total liabilities |
|
$ |
211.5 |
|
$ |
165.7 |
|
Commitments and contingencies (Note 14) |
|
|
|
|
|
|
|
Equity: |
|
|
|
|
|
|
|
Preferred stock, $0.001 par value (shares authorized of 50,000,000 at March 31, 2017 and 0 at December 31, 2016; no shares issued and outstanding at March 31, 2017 or at December 31, 2016) |
|
|
— |
|
|
— |
|
Common stock, $0.001 par value (shares authorized of 800,000,000 at March 31, 2017 and 1,000 at December 31, 2016; shares issued and outstanding of 108,132,937 at March 31, 2017 and 1,000 at December 31, 2016) |
|
|
0.1 |
|
|
— |
|
Additional paid-in capital |
|
|
539.7 |
|
|
— |
|
Retained earnings |
|
|
46.0 |
|
|
— |
|
Net parent company investment |
|
|
— |
|
|
564.4 |
|
Accumulated other comprehensive income (loss) |
|
|
(0.7) |
|
|
1.8 |
|
Total equity |
|
|
585.1 |
|
|
566.2 |
|
Total liabilities and equity |
|
$ |
796.6 |
|
$ |
731.9 |
See accompanying notes to these unaudited condensed consolidated financial statements.
6
BIOVERATIV INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(unaudited, in millions)
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
|
|
2017 |
|
2016 |
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
Net income |
|
$ |
69.3 |
|
$ |
66.7 |
|
Adjustments to reconcile net income to net cash flows from operating activities: |
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
5.7 |
|
|
4.1 |
|
Stock-based compensation |
|
|
6.0 |
|
|
2.5 |
|
Deferred taxes |
|
|
0.4 |
|
|
— |
|
Changes in operating assets and liabilities, net: |
|
|
|
|
|
|
|
Accounts receivable |
|
|
5.1 |
|
|
(17.0) |
|
Inventory |
|
|
31.6 |
|
|
(27.1) |
|
Due from (to) Biogen |
|
|
(35.2) |
|
|
— |
|
Other assets |
|
|
(25.5) |
|
|
(0.9) |
|
Accounts payable, accrued expenses and other current liabilities |
|
|
41.4 |
|
|
18.0 |
|
Other liabilities |
|
|
9.4 |
|
|
3.8 |
|
Net cash flows provided by operating activities |
|
|
108.2 |
|
|
50.1 |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
Purchases of property, plant and equipment |
|
|
(6.3) |
|
|
(0.9) |
|
Acquisition of intangible assets |
|
|
— |
|
|
— |
|
Net cash flows used in investing activities |
|
|
(6.3) |
|
|
(0.9) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
Transfers to Biogen |
|
|
(45.2) |
|
|
(49.2) |
|
Cash from Biogen |
|
|
325.0 |
|
|
— |
|
Working capital adjustments paid to Biogen, net |
|
|
(23.5) |
|
|
— |
|
Net cash flows provided by (used) in financing activities |
|
|
256.3 |
|
|
(49.2) |
|
Effect of foreign exchange rate changes on cash and equivalents |
|
|
0.5 |
|
|
|
|
Net increase in cash and cash equivalents |
|
|
358.7 |
|
|
— |
|
Cash and cash equivalents, beginning of the period |
|
$ |
— |
|
$ |
— |
|
Cash and cash equivalents, end of the period |
|
$ |
358.7 |
|
$ |
— |
See accompanying notes to these unaudited condensed consolidated financial statements.
7
BIOVERATIV INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENT OF EQUITY
(unaudited, in millions)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additional |
|
|
|
|
Net Parent |
|
Accumulated Other |
|
|
|
||
|
|
|
Common Stock |
|
|
Paid-In |
|
|
Retained |
|
Company |
|
Comprehensive |
|
|
|
|||||
|
|
|
Shares |
|
|
Amount |
|
|
Capital |
|
|
Earnings |
|
Investment |
|
Income (Loss) |
|
Total Equity |
|||
|
Balance, December 31, 2015 |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
384.4 |
|
|
0.3 |
|
$ |
384.7 |
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
66.7 |
|
|
— |
|
|
66.7 |
|
Transfers to Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(48.0) |
|
|
— |
|
|
(48.0) |
|
Foreign currency translation adjustments |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
1.4 |
|
|
1.4 |
|
Balance, March 31, 2016 |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
403.1 |
|
$ |
1.7 |
|
$ |
404.8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance, December 31, 2016 |
|
— |
|
$ |
— |
|
$ |
— |
|
$ |
— |
|
$ |
564.4 |
|
$ |
1.8 |
|
$ |
566.2 |
|
Net income |
|
— |
|
|
— |
|
|
— |
|
|
46.0 |
|
|
23.3 |
|
|
— |
|
|
69.3 |
|
Separation-related adjustments |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(312.4) |
|
|
— |
|
|
(312.4) |
|
Transfers to Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(43.0) |
|
|
— |
|
|
(43.0) |
|
Cash from Biogen |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
325.0 |
|
|
— |
|
|
325.0 |
|
Working capital adjustments paid to Biogen, net |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(23.5) |
|
|
— |
|
|
(23.5) |
|
Reclassification of net parent company investment to additional paid-in capital |
|
— |
|
|
— |
|
|
533.8 |
|
|
— |
|
|
(533.8) |
|
|
— |
|
|
— |
|
Issuance of common stock upon separation |
|
108.0 |
|
|
0.1 |
|
|
(0.1) |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Share-based compensation expense |
|
— |
|
|
— |
|
|
6.0 |
|
|
— |
|
|
— |
|
|
— |
|
|
6.0 |
|
Shares issued under employee benefit plans |
|
0.1 |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
Foreign currency translation adjustments |
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(2.5) |
|
|
(2.5) |
|
Balance, March 31, 2017 |
|
108.1 |
|
$ |
0.1 |
|
$ |
539.7 |
|
$ |
46.0 |
|
$ |
— |
|
$ |
(0.7) |
|
$ |
585.1 |
See accompanying notes to these unaudited condensed consolidated financial statements
8
BIOVERATIV INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited)
1. Nature of Business and Basis of Presentation
Nature of Business
Bioverativ Inc. (Bioverativ) separated from Biogen Inc. (Biogen) on February 1, 2017 as a result of a special dividend distribution of all the outstanding shares of common stock of Bioverativ to Biogen stockholders. The distribution was made to Biogen stockholders of record as of the close of business on January 17, 2017, who received one share of Bioverativ common stock for every two shares of Biogen common stock held as of such date. As a result of the distribution, Bioverativ became an independent public company.
Bioverativ holds the assets and liabilities of Biogen’s former hemophilia business. Bioverativ is focused on the discovery, research, development and commercialization of innovative therapies for the treatment of hemophilia and other blood disorders.
Bioverativ’s marketed products include ELOCTATE and ALPROLIX, extended half‑life factors for the treatment of hemophilia A and hemophilia B, respectively. Pursuant to a development and commercialization agreement, Bioverativ collaborates with Swedish Orphan Biovitrum AB (publ) (Sobi) to jointly develop and commercialize ELOCTATE and ALPROLIX globally. Sobi has assumed responsibility for commercialization of ELOCTATE and ALPROLIX in Europe, Russia and certain countries in Northern Africa and the Middle East, while Bioverativ retains rights to commercialize those therapies in the United States, Japan, Canada, Australia, Latin American countries and all other markets excluding Sobi’s commercialization territory. See Note 2, Collaborations, for further information on Bioverativ’s collaboration with Sobi.
Basis of Presentation
In the opinion of management, the accompanying unaudited condensed consolidated financial statements include all adjustments, consisting of normal recurring accruals, necessary for a fair presentation of our financial statements for interim periods in accordance with accounting principles generally accepted in the United States (U.S. GAAP).
We operate as one operating segment, which is discovering, researching, developing and commercializing innovative therapies for the treatment of hemophilia and other blood disorders.
The information included in this Quarterly Report on Form 10-Q should be read in conjunction with our audited consolidated financial statements and the accompanying notes included in our Annual Report on Form 10-K for the year ended December 31, 2016 (2016 Form 10-K). Our accounting policies are described in the “Notes to Consolidated Financial Statements” in our 2016 Form 10-K and updated, as necessary, in this Quarterly Report on Form 10-Q. The December 31, 2016 condensed consolidated balance sheet data presented for comparative purposes was derived from our audited financial statements, but does not include all disclosures required by U.S. GAAP. The results of operations for the three months ended March 31, 2017, are not necessarily indicative of the operating results for the full year or for any other subsequent interim period.
The accompanying unaudited condensed consolidated interim financial statements reflect the consolidated financial position and consolidated results of operations of the company as an independent, publicly-traded company for the period after the February 1, 2017 separation. The unaudited condensed consolidated interim financial statements also reflect the consolidated financial position and consolidated results of operations of the company as a consolidated reporting entity of Biogen for periods prior to the separation.
9
During the three months ended March 31, 2017, the company recorded certain separation related adjustments in its statement of equity. The separation related adjustments primarily related to differences between assets and liabilities transferred to Bioverativ as a result of the separation and assets and liabilities reported in the company’s consolidated balance sheet as of January 31, 2017. Separation related adjustments for the three months ended March 31, 2017 totaled $312.4 million consisting primarily of inventory and deferred tax assets retained by Biogen upon separation. Additional separation related adjustments could be recorded in future periods.
Prior to the separation, the unaudited condensed consolidated interim financial statements were prepared on a standalone basis and were derived from Biogen’s consolidated financial statements and accounting records as if the former hemophilia business of Biogen had been standalone business. Accordingly, financial information for periods prior to the separation are shown on a carve-out basis for the hemophilia business as part of Biogen. The unaudited condensed consolidated interim financial statements reflected the company’s financial position, results of operations and cash flows as the business was operated as part of Biogen prior to the distribution.
Prior to the separation, the unaudited condensed consolidated interim financial statements included the attribution of certain assets and liabilities that were historically held at the Biogen corporate level but which were specifically identifiable or attributable to the company. All intercompany transactions and accounts within the company were eliminated. All transactions between the company and Biogen were considered to be effectively settled in the unaudited condensed consolidated interim financial statements at the time the transaction was recorded. The total net effect of the settlement of the transactions with Biogen are reflected in the unaudited condensed consolidated statements of cash flows in periods prior to the separation as a financing activity and in the unaudited condensed consolidated balance sheet as net parent company investment.
Prior to the separation, these unaudited condensed consolidated interim financial statements include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. The research and development costs include depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. The selling, general and administrative costs include certain services provided by Biogen, which include, but are not limited to, executive oversight, treasury, finance, legal, human resources, tax planning, internal audit, financial reporting, information technology, investor relations, shared services, insurance, employee benefits and incentives and share‑based compensation. Allocated amounts have been included in research and development, selling, general and administrative and other income and expense. These expenses have been allocated to the company based on direct usage or benefit where specifically identifiable, with the remainder allocated primarily based on hours or direct costs. The company considers the expense methodology and results to be reasonable for all periods presented. However, the allocations may not be indicative of the actual expense that would have been incurred had the company operated as an independent, publicly traded company for the periods presented.
In periods prior to the separation, Bioverativ’s employees participated in various benefit and share-based compensation plans maintained by Biogen. A portion of the cost of those plans was included in the company’s financial statements. However, the unaudited condensed consolidated balance sheets in periods prior to the separation did not include any equity related to share-based compensation plans.
Prior to the separation, the company’s equity balance represented the excess of total assets over total liabilities, including the due to/from balances between the company and Biogen (net parent company investment) and cumulative translation adjustment. In connection with the separation, the company’s net parent company investment balance was reclassified to additional paid-in capital.
Reclassifications
Certain prior year amounts have been reclassified for consistency with the current period presentation. These reclassifications had no effect on the reported results of operations. In first quarter of 2017, the company concluded that it was appropriate to classify costs associated with medical affairs as research and development expense to better align with Bioverativ’s organizational structure. Previously, such costs had been classified as selling, general and
10
administrative. As a result, the amounts that were previously presented in selling, general and administrative were reclassified in research and development for each of the following periods in 2016.
|
|
For the Three Months Ended |
|
For the Year Ended |
|||||||||||
|
(In millions) |
March 31, 2016 |
|
June 30, 2016 |
|
September 30, 2016 |
|
December 31, 2016 |
|
December 31, 2016 |
|||||
|
|
$ |
7.7 |
|
$ |
8.9 |
|
$ |
7.7 |
|
$ |
6.9 |
|
$ |
31.2 |
Use of Estimates
The preparation of our condensed consolidated financial statements requires us to make estimates, judgments, and assumptions that may affect the reported amounts of assets, liabilities, equity, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis we evaluate our estimates, judgments and methodologies. We base our estimates on historical experience and on various other assumptions that are believed to be reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenues and expenses. Actual results may differ from these estimates under different assumptions or conditions.
New Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board (FASB) or other standard setting bodies that we adopt as of the specified effective date.
The following new standards issued by FASB were adopted by the company on January 1, 2017:
|
· |
Accounting Standards Update (ASU) No. 2015‑11, Inventory (Topic 330): Simplifying the Measurement of Inventory. The new standard applies only to inventory for which cost is determined by methods other than last-in, first-out and the retail inventory method, which includes inventory that is measured using FIFO or average cost. Inventory within the scope of this standard is required to be measured at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less reasonably predictable costs of completion, disposal and transportation. The adoption of this standard did not have a material impact on our financial position, results of operations or statements of cash flows upon adoption. |
|
· |
ASU No. 2016‑09, Compensation‑Stock Compensation (Topic 718): Improvements to Employee Share‑Based Payment Accounting. The new standard requires recognition of the income tax effects of vested or settled awards in the income statement and involves several other aspects of the accounting for share-based payment transactions, including the income tax consequences, classification of awards as either equity or liabilities and classification on the statement of cash flows. The adoption of this standard did not have a material impact on our financial position, results of operations or statements of cash flows upon adoption. |
The following new standards have been issued by FASB but are not yet effective.
In May 2014 the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606), which supersedes all existing revenue recognition requirements, including most industry-specific guidance. The new standard requires a company to recognize revenue when it transfers goods or services to customers in an amount that reflects the consideration that the company expects to receive for those goods or services. The FASB has subsequently issued amendments to ASU No. 2014-09 that have the same effective date and transition date of January 1, 2018. We expect to adopt these standards using the modified retrospective method and continue to evaluate the potential impact that this standard may have on our financial position, results of operations and disclosures.
In January 2016 the FASB issued ASU No. 2016-01, Financial Instruments - Overall (Subtopic 825-10): Recognition and Measurement of Financial Assets and Financial Liabilities. The new standard amends certain aspects of accounting and disclosure requirements of financial instruments, including the requirement that equity investments with readily determinable fair values be measured at fair value with changes in fair value recognized in a company's results of
11
operations. The new standard does not apply to investments accounted for under the equity method of accounting or those that result in consolidation of the investee. Equity investments that do not have readily determinable fair values may be measured at fair value or at cost minus impairment adjusted for changes in observable prices. A financial liability that is measured at fair value in accordance with the fair value option is required to be presented separately in other comprehensive income for the portion of the total change in the fair value resulting from change in the instrument-specific credit risk. In addition, a valuation allowance should be evaluated on deferred tax assets related to available-for-sale debt securities in combination with other deferred tax assets. The new standard will be effective for us on January 1, 2018. As of March 31, 2017, the adoption of this standard is not expected to have a material impact on our financial position or results of operations.
In February 2016 the FASB issued ASU No. 2016-02, Leases (Topic 842). The new standard requires that all lessees recognize the assets and liabilities that arise from leases on their balance sheet and disclose qualitative and quantitative information about their leasing arrangements. The new standard will be effective for us on January 1, 2019. We are currently evaluating the impact that this standard may have on our results of operations, financial position and disclosures. As of March 31, 2017, the adoption of this standard is not expected to have a material impact on our net financial position, but may materially impact the reported amount of total assets and total liabilities.
In October 2016 the FASB issued ASU No. 2016-16, Income Taxes (Topic 740): Intra-Entity Transfer of Assets Other Than Inventory. This new standard eliminates the deferral of the tax effects of intra-entity transfers of an asset other than inventory. Under the new standard, entities should recognize the income tax consequences on an intra-entity transfer of an asset other than inventory when the transfer occurs. This new standard will be effective for us on January 1, 2018 and will be applied on a modified retrospective basis through a cumulative-effect adjustment directly to retained earnings as of the beginning of the period of adoption. As of March 31, 2017, the adoption of this standard is not expected to have a material impact on our financial position.
In January 2017 the FASB issued ASU No. 2017-01, Business Combinations (Topic 805): Clarifying the Definition of a Business. This new standard clarifies the definition of a business and provides a screen to determine when an integrated set of assets and activities is not a business. The screen requires that when substantially all of the fair value of the gross assets acquired (or disposed of) is concentrated in a single identifiable asset or a group of similar identifiable assets, the set is not a business. This new standard will be effective for us on January 1, 2018, however early adoption is permitted. As of March 31, 2017, the adoption of this standard is not expected to have a material impact on our financial position.
2. Collaborations
In connection with the company’s business strategy, the company has entered into various collaboration agreements which provide the company with rights to develop, produce and market products using certain know‑how, technology and patent rights maintained by the company’s collaborative partners. Terms of the various collaboration agreements may require the company to make milestone payments upon the achievement of certain product research and development objectives and pay royalties on future sales, if any, of commercial products resulting from the collaboration.
Swedish Orphan Biovitrum AB (publ)
In January 2007, Biogen acquired 100% of the stock of Syntonix Pharmaceuticals (Syntonix). Syntonix, now known as Bioverativ Therapeutics Inc. (formerly Biogen Hemophilia Inc.), had previously entered into a development and commercialization agreement with Sobi to jointly develop and commercialize Factor VIII and Factor IX hemophilia products, including ELOCTATE and ALPROLIX. Under the development and commercialization agreement, as has been amended and restated, Bioverativ has commercial rights for North America (the Bioverativ North America Territory) and for all other markets outside of the Sobi territory (the Bioverativ Direct Territory), which consists of Europe, Russia and certain countries in Northern Africa and the Middle East (the Sobi Territory).
Under the development and commercialization agreement, prior to May 5, 2024, either Bioverativ or Sobi may present a compound construct as a potential product candidate that the parties may consider developing and
12
commercializing under the collaboration. Upon Sobi's election to treat a compound construct as a product, and in the case of a novel compound construct Sobi's payment of an upfront fee to us, Sobi is granted the right to opt-in to such compound construct and become responsible for final development and commercialization of that compound construct in Sobi's commercialization territory. Generally, upon opt-in, Sobi becomes obligated to make an advance payment and reimburse Bioverativ for certain development expenses incurred with respect to the compound construct. Until Sobi's portion of the development expenses are fully paid, Sobi's royalty rate payable to Bioverativ is increased, and the royalty payment payable by Bioverativ to Sobi for the sale of products in Bioverativ's territory is decreased.
In November 2014, Sobi exercised its option under the agreement to assume final development and commercialization activities in the Sobi Territory for ELOCTA (the trade name for ELOCTATE in the European Union). In July 2015, Sobi exercised its option under the agreement to assume final development and commercialization of ALPROLIX within the Sobi Territory. Upon each exercise of opt‑in right under the terms of the development and commercialization agreement, Sobi made a $10.0 million payment.
Upon European Medicines Agency (EMA) regulatory approval of each of ELOCTA and ALPROLIX, Sobi was obligated to reimburse Bioverativ 50% of all shared manufacturing and development expenses incurred by Bioverativ from October 1, 2009 through the earlier of the date on which Sobi is registered as the marketing authorization holder for the applicable product or 90 days post‑regulatory approval, as well as 100% of certain development expenses incurred exclusively for the benefit of the Sobi Territory (the Opt‑In Consideration).
ELOCTA was approved by the European Commission (EC) in November 2015. The Opt‑In Consideration and aggregate amount reimbursable by Sobi to us for ELOCTA was $211.0 million. As of March 31, 2017, approximately $126.6 million remained reimbursable to us by Sobi.
ALPROLIX was approved by the EC in February 2016. The Opt‑In Consideration and aggregate amount reimbursable by Sobi to us for ALPROLIX was $184.7 million. As of March 31, 2017, approximately $113.2 million remained reimbursable to us by Sobi.
The Opt‑In Consideration for each product is being paid by Sobi using a cross‑royalty cash payment structure for sales in each company’s respective territories. If the reimbursement of the Opt‑in Consideration for a product has not been achieved within six years of the first commercial sale of such product (the Reimbursement Period), the company maintains the right to require Sobi to pay any remaining balances due to us within 90 days of the six-year anniversary date of the first commercial sales. After Sobi’s Opt‑In Consideration has been repaid, the royalty paid and received by the company resets to the contractual royalty rate of 12%.
Upon Sobi’s first commercial sale in 2016, and during the Reimbursement Period, the royalty rate the company will pay Sobi on sales of ELOCTATE and ALPROLIX in the Bioverativ North American Territory is 7% and Bioverativ Direct Territory is 12%. After the Reimbursement Period concludes, the royalty rate we pay to Sobi increases by 5%. For the three months ended March 31, 2017 and for the expected term of the agreement, we are recording cost of sales at the effective royalty rate of approximately 11%. For the three months ended March 31, 2016, the effective royalty rate was approximately 7%.
The company is accounting for the development and commercialization agreement under a right to use model and is recognizing revenue over the term of the commercialization period.
The royalty rate received by the company, during the Reimbursement Period on sales of ELOCTATE and ALPROLIX in Sobi’s territory is 17%. After the Reimbursement period concludes, the royalty we receive decreases to 12%. We are recording revenue at the effective royalty rate expected over the term of the agreement of approximately 14%.
In September 2014, Sobi elected to treat BIVV 001 (rFVIIIFc‑VWF‑XTEN), a preclinical compound construct developed using the XTEN technology licensed by Bioverativ from Amunix, as subject to the collaboration. In February 2017, Sobi elected to treat BIVV 002 (rFIXFc-XTEN), a preclinical compound construct developed using the XTEN technology licensed by Bioverativ from Amunix, as subject to the collaboration. In consideration for its election,
13
Sobi will pay us $6.2 million, which has been included as a component of research and development expense. If Sobi exercises its opt-in right for BIVV 001 or BIVV 002, as the case may be, it will become responsible for final development and commercialization of the applicable product in the Sobi Territory. Further, in general upon opt-in, Sobi will be obligated to reimburse Bioverativ 50% of all shared manufacturing and development expenses incurred by Bioverativ from the period of opt-in through the earlier of the date on which Sobi is registered as the marketing authorization holder for the applicable product or 90 days post‑regulatory approval, as well as 100% of certain development expenses incurred exclusively for the benefit of the Sobi Territory.
3. Financial instruments
Cash and Cash Equivalents
We consider only those investments which are highly liquid, readily convertible to cash and that mature within three months from date of purchase to be cash equivalents. As of March 31, 2017, cash equivalents were comprised of money market funds with maturities less than 90 days from the date of purchase and totaled approximately $328.8 million. The carrying values of money market funds approximate fair value due to their short-term maturities.
In accordance with the accounting standard for fair value measurements, we have classified our financial assets as Level 1, 2 or 3 within the fair value hierarchy. Fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets that we have the ability to access. Fair values determined by Level 2 inputs utilize data points that are observable such as quoted prices, interest rates, yield curves and foreign currency spot rates. Fair values determined by Level 3 inputs utilize unobservable data points for the asset. All of our financial assets have been classified as Level 2. These assets have been initially valued at the transaction price and subsequently valued, at the end of each reporting period, utilizing market observable data. These observable market inputs include reportable trades, benchmark yields, credit spreads, broker/dealer quotes, bids, offers, current spot rates and other industry and economic events.
The table below presents information about our assets that are regularly measured and carried at fair value and indicate the level within the fair value hierarchy of the valuation techniques we utilized to determine such fair value:
|
(In millions) |
|
Total |
|
Quoted Prices |
|
Significant Other |
|
Significant |
||||
|
Cash equivalents |
|
$ |
328.8 |
|
$ |
— |
|
$ |
328.8 |
|
$ |
— |
|
Plan assets for deferred compensation |
|
|
16.6 |
|
|
— |
|
|
16.6 |
|
|
|
|
Total |
|
$ |
345.4 |
|
$ |
— |
|
$ |
345.4 |
|
$ |
— |
There were no liabilities that are measured at fair value as of March 31, 2017.
Undesignated Derivative Instruments
In April 2017, the company entered into undesignated forward contracts to hedge effects of foreign exchange relating to the company’s intercompany loan denominated in Japanese Yen. These derivative instruments are not formally designated as hedges and the terms of these instruments generally do not exceed one month. The notional amount of undesignated derivative instruments is approximately $71 million.
4. Reserves for Discounts and Allowances
Following the company’s product launches, the company began recognizing reserves for discounts and allowances related to products revenue.
14
An analysis of the change in reserves is summarized as follows:
|
|
|
|
|
|
Contractual |
|
|
|
|
|
|
|
|
(In millions) |
|
Discounts |
|
Adjustments |
|
Returns |
|
Total |
||||
|
Balance at December 31, 2016 |
|
$ |
6.0 |
|
$ |
26.5 |
|
$ |
0.3 |
|
$ |
32.8 |
|
Provision related to current period sales |
|
|
40.8 |
|
|
62.7 |
|
|
— |
|
|
103.5 |
|
Adjustment related to prior period sales |
|
|
(1.3) |
|
|
2.3 |
|
|
— |
|
|
1.0 |
|
Credits/payments made |
|
|
(41.6) |
|
|
(62.5) |
|
|
— |
|
|
(104.1) |
|
Balance at March 31, 2017 |
|
$ |
3.9 |
|
$ |
29.0 |
|
$ |
0.3 |
|
$ |
33.2 |
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Reduction of accounts receivable |
|
$ |
8.0 |
|
$ |
11.1 |
|
Current liability |
|
|
25.2 |
|
|
21.7 |
|
Total reserves |
|
$ |
33.2 |
|
$ |
32.8 |
5. Inventory
The components of inventory are summarized as follows:
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Raw materials |
|
$ |
— |
|
$ |
42.9 |
|
Work in process |
|
|
69.0 |
|
|
227.2 |
|
Finished goods |
|
|
22.4 |
|
|
31.9 |
|
Total inventory |
|
$ |
91.4 |
|
$ |
302.0 |
Inventory amounts written down as a result of excess, obsolescence, unmarketability or other reasons are charged to cost of sales. There were no write downs of inventory in either of the three months ended March 31, 2017 and 2016.
In connection with the separation, certain raw material and work in process inventory totaling $179.0 million was retained by Biogen. This inventory is subject to the terms and conditions of our manufacturing and supply agreement with Biogen.
6. Acquired Intangible Assets
Acquired intangibles primarily relate to approval milestones for ALPROLIX paid to the former Syntonix shareholders. In 2014, upon the U.S. Food and Drug Administration’s approval of ALPROLIX for the treatment of hemophilia B, a $20.0 million milestone was paid to the former Syntonix shareholders. This $20.0 million milestone and the corresponding deferred tax liability of $7.3 million were capitalized as an acquired intangible asset. In 2016, upon EMA approval of ALPROLIX in the European Union, an additional $20.0 million milestone was paid to the former Syntonix shareholders. This $20.0 million milestone and the corresponding deferred tax liability of $6.5 million were capitalized as an acquired intangible asset.
Acquired intangible assets as of March 31, 2017 and December 31, 2016, net of accumulated amortization, are $50.2 million and $51.7 million, respectively. During the three-month periods ending March 31, 2017 and 2016, amortization expense associated with acquired intangible assets was $1.4 million and $0.7 million, respectively.
15
7. Property, Plant and Equipment
Property, plant and equipment are recorded at historical cost, net of accumulated depreciation. Components of property, plant and equipment, net are summarized as follows:
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Buildings |
|
$ |
— |
|
$ |
8.0 |
|
Leasehold improvements |
|
|
— |
|
|
— |
|
Machinery and equipment |
|
|
27.1 |
|
|
21.0 |
|
Computer software and hardware |
|
|
5.5 |
|
|
5.9 |
|
Furniture and fixtures |
|
|
0.3 |
|
|
0.5 |
|
Construction in progress |
|
|
9.6 |
|
|
6.9 |
|
Total cost |
|
|
42.5 |
|
|
42.3 |
|
Less: accumulated depreciation |
|
|
19.4 |
|
|
13.9 |
|
Total property, plant and equipment, net |
|
$ |
23.1 |
|
$ |
28.4 |
In connection with the separation, certain property, plant and equipment, mainly buildings and associated accumulated depreciation totaling $9.8 million was retained by Biogen.
8. Income Taxes
Prior to the separation, the company’s income tax expense and deferred tax balances were calculated on a separate tax return basis although the company’s operations had historically been included in the tax returns filed by the respective Biogen entities, of which the company’s business was a part. For periods subsequent to the separation, Bioverativ files tax returns on its own behalf and its income tax expense and deferred income tax balances are recorded in accordance with the company’s standalone income tax positions. Biogen and Bioverativ entered into a tax matters agreement effective as of the date of separation.
Income Tax Expense Reconciliation
|
|
|
For the Three Months Ended |
||
|
|
|
March 31, |
||
|
|
|
2017 |
|
2016 |
|
Statutory rate |
|
35.0% |
|
35.0% |
|
State taxes |
|
2.1% |
|
0.9% |
|
Taxes on foreign earnings |
|
(0.1%) |
|
(0.1%) |
|
Credits and net operating losses |
|
(0.3%) |
|
(0.1%) |
|
Changes in valuation allowance |
|
— |
|
(37.5%) |
|
Other permanent items |
|
(0.9%) |
|
(0.1%) |
|
Others |
|
2.0% |
|
0.1% |
|
Effective tax rate |
|
37.8% |
|
(1.8%) |
The company’s effective income tax rate was 37.8% and (1.8)% during the three months ended March 31, 2017 and 2016. The company’s effective income tax rate for the three months ended March 31, 2017 differs from the U.S. federal statutory rate mainly due to state and local taxes. The company’s effective income tax rate for the three months ended March 31, 2016 differs from the U.S. federal statutory rate mainly due to the release of a valuation allowance associated with deferred tax assets resulting from our net losses and business credit carryforwards.
The effective income tax rate increased during the three months ended March 31, 2017 as compared to the prior year period primarily due to the release of a valuation allowance associated with deferred tax assets resulting from our net losses and business credit carryforwards in the three months ended March 31, 2016. Substantially all of these assets will not be available in the future since those attributes have already been utilized in the tax returns of Biogen.
16
9. Earnings per Share
The denominator for basic earnings per common share (EPS) is the weighted-average number of common shares outstanding during the period. The dilutive effect of outstanding stock options and time vested restricted stock units is reflected in the denominator for diluted EPS using the treasury stock method. The numerator for both basic and diluted EPS is net income.
On February 1, 2017, Biogen distributed approximately 108 million shares of Bioverativ common stock to its shareholders. The computation of basic and dilutive EPS for periods prior to the separation was calculated using the shares distributed.
The following is a reconciliation of basic shares to diluted shares.
Basic and diluted earnings per share are calculated as follows:
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Numerator: |
|
|
|
|
|
|
|
Net income |
|
$ |
69.3 |
|
$ |
66.7 |
|
Denominator: |
|
|
|
|
|
|
|
Weighted average number of common shares outstanding |
|
|
108.0 |
|
|
108.0 |
|
Effect of dilutive securities: |
|
|
|
|
|
|
|
Stock options |
|
|
— |
|
|
— |
|
Time-vested restricted stock units |
|
|
0.2 |
|
|
— |
|
Dilutive potential common shares |
|
|
0.2 |
|
|
— |
|
Shares used in calculating diluted earnings per share |
|
|
108.2 |
|
|
108.0 |
The computation of diluted earnings per share excludes 2.0 million and 2.8 million of outstanding common share equivalents for stock options and time vested restricted stock units for the three months ended March 31, 2017 and 2016, respectively, as the effect is anti-dilutive.
10. Share‑Based Compensation
Bioverativ Share‑Based Compensation Plans
In connection with the separation, we adopted our own share‑based compensation plans. Specifically, we adopted the Bioverativ Inc. 2017 Omnibus Equity Plan (the Omnibus Plan); the Bioverativ Inc. 2017 Non‑Employee Directors Equity Plan (the Directors Plan); and the Bioverativ Inc. 2017 Employee Stock Purchase Plan (ESPP). Cash-settled performance units, market stock units, performance‑vested restricted stock units and restricted stock units (RSUs) granted under Biogen 2008 Omnibus Equity Plan to Biogen employees who became employees of Bioverativ were converted into Bioverativ RSUs, and stock options held by Biogen employees who became employees of Bioverativ were converted into Bioverativ stock options, each according to the terms of an employee matters agreement entered into between Biogen and Bioverativ in connection with the separation (converted awards). Converted awards, as well as future awards issued to Bioverativ officers, directors and employees, will be awarded under, and subject to the terms of, the Omnibus Plan, the Directors Plan and the ESPP, as the case may be.
17
The following table provides share-based compensation expense by statement of income line item for the three months ended March 31, 2017 and 2016:
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Cost of sales |
|
$ |
— |
|
$ |
0.9 |
|
Research and development |
|
|
0.8 |
|
|
0.8 |
|
Selling, general and administrative |
|
|
5.2 |
|
|
0.8 |
|
Total share-based compensation expense |
|
$ |
6.0 |
|
$ |
2.5 |
During the three months ended March 31, 2017 and subsequent to the separation, Bioverativ awarded share-based compensation grants consisting of 2.0 million stock options and 0.3 million RSUs, with respect to the company’s employees. In addition, Biogen market stock units, performance‑vested restricted stock units and RSUs converted into 0.5 million Bioverativ RSUs.
Stock Options
Post-Separation
The weighted-average Black-Scholes assumptions used in estimating the fair value of stock options granted by Bioverativ following the separation during the three months ended March 31, 2017, along with the weighted-average grant-date fair values, were as follows:
|
|
|
For the Three Months Ended |
|
|
|
|
March 31, 2017 |
|
|
Expected volatility |
|
|
40.0% |
|
Expected life (in years) |
|
|
5 |
|
Range of risk-free interest rates |
|
|
1.80% ̶ 1.99% |
|
Dividend yield |
|
|
̶ |
|
Range of fair value per stock option |
|
|
$16.78 ̶ $20.15 |
Expected volatility was determined using an average of peer companies’ expected volatility. As of March 31, 2017, the unrecognized compensation cost related to all unvested stock options held by Bioverativ’s employees of $30.5 million is expected to be recognized as expense over a weighted-average period of 2.9 years.
RSUs
As of March 31, 2017, the unrecognized compensation cost related to all unvested RSUs held by Bioverativ employees of $26.6 million is expected to be recognized as expense over a weighted-average period of 2.0 years.
11. Related Parties
Under the terms of the transition services agreement entered into with Biogen in connection with the separation, Biogen provides various services on an interim, transitional basis. The services provided by Biogen to Bioverativ consist of certain information technology, quality, regulatory, research and supply chain. These services are generally provided on a cost-plus basis. The services generally extend for approximately 9 months to 2 years following the separation. For
18
the period from the separation date to March 31, 2017, the company incurred the following costs under the transition services agreement:
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Cost of sales |
|
$ |
0.6 |
|
|
** |
|
Research and development |
|
|
4.7 |
|
|
** |
|
Selling, general and administrative |
|
|
3.2 |
|
|
** |
** Amount not applicable.
For the company’s operations in the United States and Canada transfer of marketing authorizations and other regulatory requirements did not occur by the separation date of February 1, 2017. As a result, the company entered into a commercial operations agreement with Biogen for the United States and Canada. Under the terms of the commercial operations agreements, until the Company has obtained the various required authorizations, Biogen will perform certain services related to the distribution of finished goods to the company’s customers. The company is responsible for the business activities conducted by Biogen on its behalf, and is subject to the risks and entitled to the benefits generated by these operations and assets. As a result, the related assets and liabilities and results of operations have been reported in the company’s condensed consolidated interim financial statements as of and for the three months ended March 31, 2017. Net sales related to these operations totaled approximately $147.4 million during the three months ended March 31, 2017. At March 31, 2017, the assets and liabilities consisted of certain work in process and finished goods inventories which are reported in inventories on the condensed consolidated balance sheet, and accounts receivable, which is reported as due from Biogen, on the condensed consolidated balance sheet. The majority of these operations are expected to be transferred to the company in mid to late 2017.
The company and Biogen also entered into a manufacturing and supply agreement whereby Biogen will continue to produce ELOCTATE and ALPROLIX for us on a cost‑plus basis. As products were historically transferred at cost between Biogen and Bioverativ, these manufacturing and supply arrangements will result in changes to cost of goods sold in future periods.
The company and Biogen also entered into a separation and distribution agreement, tax matters agreement, an employee matters agreement.
The following is a summary of the amounts in the unaudited condensed consolidated balance sheets due to or from Biogen, including the assets and liabilities of certain of the company’s operations that have not yet transferred to Bioverativ and are held by Biogen as of the balance sheet dates:
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Receivables to be transferred to Bioverativ, held by Biogen |
|
$ |
45.9 |
|
|
** |
|
Amounts due to Biogen associated with transition service agreements |
|
|
(10.7) |
|
|
** |
|
Net |
|
$ |
35.2 |
|
|
** |
** Amount not applicable.
19
Corporate Overhead and Other Allocations from Biogen
Prior to the separation, the company did not historically operate as a standalone business and had various relationships with Biogen whereby Biogen provided services to the company. These unaudited condensed consolidated financial statements include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. For more information, see Note 1, Nature of Business and Basis of Presentation. For the three months ended March 31, 2017, the allocations are for the period from January 1, 2017 through the separation date. These allocations were reflected as follows in the unaudited condensed consolidated financial statements:
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Research and development allocations |
|
$ |
6.0 |
|
$ |
19.8 |
|
Selling, general and administrative allocations |
|
|
5.6 |
|
|
17.0 |
|
Other (income) expense, net allocations |
|
|
(0.2) |
|
|
(0.6) |
|
Total corporate overhead and other allocations from Biogen |
|
$ |
11.4 |
|
$ |
36.2 |
12. Other Consolidated Financial Statement Detail
Other Income (Expense), Net
Components of other income (expense), net, are summarized as follows:
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Foreign exchange gains (losses), net |
|
$ |
(0.8) |
|
$ |
(0.4) |
|
Other, net |
|
|
0.4 |
|
|
— |
|
Total other income (expense), net |
|
$ |
(0.4) |
|
$ |
(0.4) |
Accrued Expenses and Other Current Liabilities
Accrued expenses and other consists of the following:
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Revenue-related rebates |
|
$ |
25.2 |
|
$ |
21.7 |
|
Employee compensation and benefits |
|
|
9.2 |
|
|
15.9 |
|
Clinical development expenses |
|
|
6.1 |
|
|
5.5 |
|
Royalty and collaboration expenses |
|
|
25.5 |
|
|
26.6 |
|
Taxes payable |
|
|
27.1 |
|
|
10.8 |
|
Other |
|
|
18.6 |
|
|
8.8 |
|
Total accrued expenses and other current liabilities |
|
$ |
111.7 |
|
$ |
89.3 |
20
Long‑Term Liabilities
Long‑term liabilities consist of the following:
|
|
|
As of |
|
As of |
||
|
|
|
March 31, |
|
December 31, |
||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Employee compensation and benefits |
|
$ |
18.3 |
|
$ |
18.7 |
|
Sobi payments |
|
|
53.9 |
|
|
45.0 |
|
Other |
|
|
0.9 |
|
|
— |
|
Total long term liabilities |
|
$ |
73.1 |
|
$ |
63.7 |
13. Litigation
From time to time, we may be involved in various claims and legal proceedings.
With respect to some loss contingencies, an estimate of the possible loss or range of loss cannot be made until management has further information, including, for example, (i) which claims, if any, will survive dispositive motion practice; (ii) information to be obtained through discovery; (iii) information as to the parties’ damages claims and supporting evidence; (iv) the parties’ legal theories; and (v) the parties’ settlement positions. For information as to our accounting policies relating to claims and legal proceedings, including use of estimates, and contingencies, please read Note 2, Summary of Significant Accounting Policies, of our 2016 Form 10-K.
Subject to the terms of the separation agreement we entered into with Biogen, the company may be responsible for certain liabilities relating to following legal matters involving Biogen prior to the separation, solely to the extent such liabilities relate to, arise out of or result from hemophilia business. The imposition of any such liability would be carefully evaluated by the company under the terms of the separation agreement.
Government Matters
On March 4, 2016, Biogen received a subpoena from the federal government for documents relating to Biogen’s relationship with non‑profit organizations that provide assistance to patients taking drugs sold by Biogen.
On July 1, 2016, Biogen received civil investigative demands from the federal government for documents and information relating to Biogen’s treatment of certain service agreements with wholesalers when calculating and reporting Average Manufacturer Prices in connection with the Medicaid Drug Rebate Program.
14. Commitments and Contingencies
Contingent Development, Regulatory and Commercial Milestone Payments
Based on our development plans primarily in gene editing and gene therapy for hemophilia and other blood disorders as of March 31, 2017, we could make potential future milestone payments to third party collaborators of up to approximately $440 million. Payments under these agreements generally become due and payable upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones had not occurred as of March 31, 2017, such contingencies have not been recorded in our financial statements. Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain development, regulatory approval and commercial milestones.
21
Item 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with our condensed consolidated financial statements and accompanying notes included elsewhere in this report on Form 10-Q and our audited consolidated financial statements and related notes included in our Annual Report on Form 10-K for the year ended December 31, 2016 (2016 Form 10-K). This Management’s Discussion and Analysis of Financial Condition and Results of Operations contains forward‑looking statements. The matters discussed in these forward‑looking statements are subject to risk, uncertainties and other factors that could cause actual results to differ materially from those made, projected or implied in the forward‑looking statements. Please see Item 1A, Risk Factors and “Note Regarding Forward‑Looking Statements” for a discussion of the uncertainties, risks and assumptions associated with these statements.
On May 3, 2016, Biogen announced its plans to separate into two independent, publicly traded companies. For purposes of the following discussion, Bioverativ refers to the hemophilia business of Biogen prior to the separation. To accomplish this separation, Biogen created a new company, Bioverativ Inc., to be the parent company for the hemophilia business. Bioverativ Inc. was incorporated in the State of Delaware on August 4, 2016. To effect the separation, Biogen made a 2‑1 pro rata distribution of Bioverativ Inc.’s common stock to Biogen’s stockholders. The distribution occurred on February 1, 2017 and since the distribution, Bioverativ Inc. has operated as an independent, publicly traded company.
Overview
We are a global biotechnology company focused on the discovery, research, development and commercialization of innovative therapies for the treatment of hemophilia and other blood disorders. We have two marketed products, ELOCTATE [Antihemophilic Factor (Recombinant), Fc Fusion Protein] and ALPROLIX [Coagulation Factor IX (Recombinant), Fc Fusion Protein], and an innovative product pipeline.
Our business strategy is aimed at improving treatment and standards of care for hemophilia and other blood disorder patients by further increasing sales and market share of our marketed products, advancing treatment attributes for our marketed products, leveraging our internal expertise to develop new products that meaningfully advance treatment and opportunistically pursuing strategic alliances and tactical acquisitions.
Our revenues are primarily derived from sales of ELOCTATE and ALPROLIX in the United States, Japan and Canada. We also earn revenue from the supply of ELOCTATE and ALPROLIX to Sobi and royalties on sales of ELOCTATE and ALPROLIX by Sobi in its commercialization territory, which is Europe, Russia and certain countries in Northern Africa and the Middle East.
Financial Results Overview—Three Months Ended March 31, 2017 and 2016
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Total revenues |
|
$ |
259.1 |
|
$ |
191.7 |
|
Net income |
|
$ |
69.3 |
|
$ |
66.7 |
Key Commercial Highlights
The United States, Japan and Canada are currently the principal markets outside of Sobi’s commercialization territory for our marketed products. We began selling ELOCTATE in the United States, Japan and Canada in the third quarter of 2014, the first quarter of 2015 and the first quarter of 2016, respectively. We began selling ALPROLIX in the United States, Japan and Canada in the second quarter of 2014, the fourth quarter of 2014 and the first quarter of 2016, respectively. We expect to continue to drive revenue growth and increased patient share of ELOCTATE and ALPROLIX by expanding into new geographies and continuing to penetrate our existing geographies. In addition, in 2016 we began earning royalties from Sobi on sales of ELOCTA and ALPROLIX following Sobi’s commercial launch of ELOCTA and ALPROLIX in the European Union.
22
Research and Development
We expect to make substantial investments in research and development in support of our ongoing proprietary research programs and through collaborations with third parties for the development of new products and therapies. Research and development expenses were $36.9 million, or approximately 14.2% of total revenue, during the first quarter of 2017.
Our overall research and development strategy includes the continued pursuit of collaborations and strategic relationships with third parties that are developing new products and therapies. These collaborations and relationships generally involve the granting or obtaining development and commercialization rights to or from third parties in exchange for an upfront payment upon execution of the agreement and potential future payments related to the achievement of development, regulatory approval or commercial milestones, as well as royalties. Our most significant collaboration is our relationship with Sobi for the development and commercialization of ELOCTATE and ALPROLIX. Please refer to Note 2, Collaborations, to the unaudited condensed consolidated financial statements included elsewhere in this report for additional details on our collaboration with Sobi.
Key Factors Affecting Results of Operations
Separation from Biogen on February 1, 2017
Bioverativ separated from Biogen on February 1, 2017 as a result of a special dividend distribution of all the outstanding shares of common stock of Bioverativ to Biogen stockholders. The distribution was made to Biogen stockholders of record as of the close of business on January 17, 2017, who received one share of Bioverativ common stock for every two shares of Biogen common stock held as of such date. As a result of the distribution, Bioverativ is now an independent public company.
Separation from Biogen
We did not operate as an independent, standalone company before the completion of the separation, but rather operated as a part of Biogen’s overall business. Accordingly, financial information for periods prior to the separation are shown on a carve-out basis for the hemophilia business as part of Biogen. There are limitations inherent in the preparation of all carve‑out financial statements due to the fact that the business was previously part of a larger organization. The basis of preparation included in our audited consolidated financial statements included in our 2016 Form 10-K provides a detailed description of the treatment of historical transactions prior to the separation. Our historical net income for periods prior to the separation may not be indicative of future results. Our historical net income for periods prior to the separation has been most notably impacted by the following consequences of carve‑out accounting and the separation:
|
· |
Biogen utilized a centralized treasury management system and cash or debt was not allocated to Bioverativ in the carve‑out financial statements. In connection with the separation, the capital structures of both companies were re‑aligned, resulting in Bioverativ having adequate cash to fund its operations. |
|
· |
The unaudited condensed consolidated statements of income include an allocation from Biogen to us for certain research and development and selling, general and administrative costs not directly attributable to the hemophilia business of Biogen. The research and development costs include depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. The selling, general and administrative costs include certain services provided by Biogen, which include executive oversight, treasury, finance, legal, human resources, tax planning, internal audit, financial reporting, information technology, investor relations, shared services, insurance, employee benefits and incentives and share‑based compensation. The amounts of these allocations may not necessarily be indicative of the similar costs we will incur subsequent to the separation. The total amount of research and development, selling, general and administrative costs and other income (expense) allocated to us from Biogen for the three months ended March 31, 2017 and 2016 were $11.4 million and $36.2 million, respectively. |
23
|
· |
We incurred certain one‑time separation costs, which are primarily associated with the design and establishment of us as a standalone public company. |
|
· |
Income tax expense (benefit) is computed on a “separate return basis”, as if operated as a standalone entity or a separate entity or a separate consolidated group in each material jurisdiction in which we operate. Income tax expense (benefit) prior to the separation included in the condensed consolidated financial statements may not be indicative of our future expected effective income tax rate. |
|
· |
Concurrent with the separation, we entered into a manufacturing and supply agreement with Biogen whereby Biogen will continue to produce ELOCTATE and ALPROLIX for us on terms on cost plus basis. As products were historically transferred at cost between Biogen and Bioverativ, these manufacturing and supply arrangements will result in changes to cost of goods sold in future periods. |
Results of Operations—Three Months Ended March 31, 2017 and 2016
Revenue
Total Revenues
|
|
|
For the Three Months Ended |
||||||||
|
|
|
March 31, |
||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
||||||
|
Product revenues |
|
|
|
|
|
|
|
|
|
|
|
United States |
|
$ |
204.5 |
|
78.9% |
|
$ |
163.4 |
|
85.2% |
|
All Other Markets |
|
|
37.4 |
|
14.4% |
|
|
19.4 |
|
10.1% |
|
Total product revenues |
|
|
241.9 |
|
93.4% |
|
|
182.8 |
|
95.4% |
|
Collaboration revenue |
|
|
17.2 |
|
6.6% |
|
|
8.9 |
|
4.6% |
|
Total revenues |
|
$ |
259.1 |
|
100.0% |
|
$ |
191.7 |
|
100.0% |
Revenues totaled $259.1 million for the three months ended March 31, 2017, an increase of 35.2% over the same period in 2016. In the three months ended March 31, 2017, product revenues in the United States totaled $204.5 million, an increase of 25.2% over the same period in 2016 and sales outside of the United States totaled $37.4 million, an increase of 92.8% over the same period in 2016. In the first quarter of 2016, Sobi had its first commercial sales of ELOCTATE. As a result, collaboration revenue, consisting of manufacturing and royalty revenues totaled $17.2 million, an increase of 93.3% over the same period in 2016.
Product Revenues
|
|
|
For the Three Months Ended |
||||||||
|
|
|
March 31, |
||||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
||||||
|
ELOCTATE |
|
$ |
155.9 |
|
64.4% |
|
$ |
107.8 |
|
59.0% |
|
ALPROLIX |
|
|
86.0 |
|
35.6% |
|
|
75.0 |
|
41.0% |
|
Total product revenues |
|
$ |
241.9 |
|
100.0% |
|
$ |
182.8 |
|
100.0% |
24
|
ELOCTATE |
ALPROLIX |
|
|
|
|
For the three months ended March 31, 2017 compared to the same period in 2016, the increase in U.S. ELOCTATE revenues was primarily due to an increase in unit sales volume of 35.3%. The increase in all other markets’ ELOCTATE revenues was due to an increase in unit sales volume in Japan of 108.5% compared to the same period in 2016, as well as an increase in unit sales volume in Canada of 261.3% compared to the same period in 2016. |
For the three months ended March 31, 2017 compared to the same period in 2016, the increase in U.S. ALPROLIX revenues was primarily due to an increase in unit sales volume of 7.2%. The increase in all other markets’ ALPROLIX revenues was due to an increase in unit sales volume in Japan of 30.3% compared to the same period in 2016, as well as an increase in unit sales volume in Canada of 20.0% compared to the same period in 2016. We expect that Alprolix will face an increasingly competitive environment. |
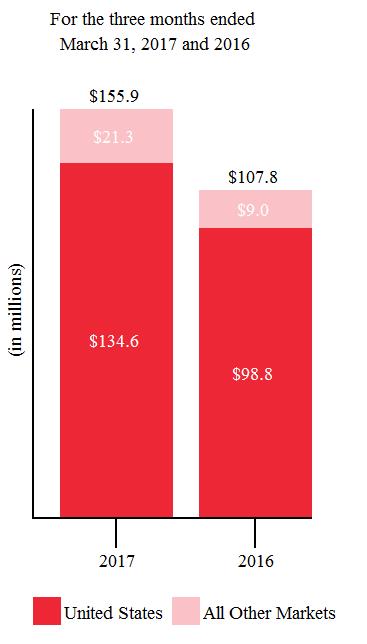
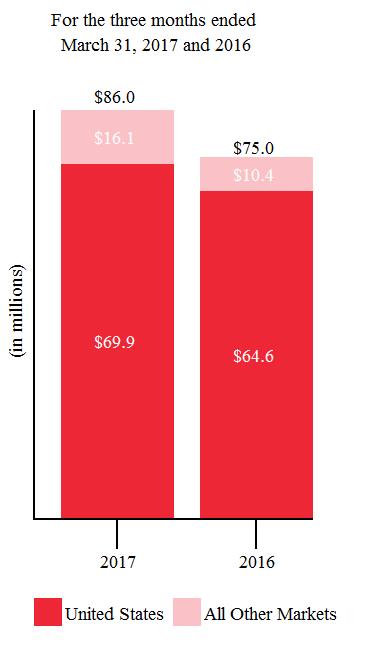
Discounts and allowances
Discounts and allowances for the three months ended March 31, 2017 and 2016 were approximately 30% and 27% of gross sales, respectively. The increase in discounts and allowances is due to increased Medicaid and government rebates.
Revenue from collaborative partners
For the three months ended March 31, 2017, compared to the same period in 2016, the increase in revenue from collaborative partners is attributable to a $4.3 million increase in contract manufacturing revenue and $4.1 million of royalty revenue both from Sobi. See Note 2, Collaborations, to the condensed consolidated financial statements included elsewhere in this report for additional information on our collaboration with Sobi.
Costs and Expenses
|
|
|
For the Three Months Ended |
||||||
|
|
|
March 31, |
||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
Change % |
||
|
Cost of sales |
|
$ |
63.3 |
|
$ |
32.5 |
|
94.8% |
|
Research and development |
|
|
36.9 |
|
|
54.3 |
|
(32.0%) |
|
Selling, general and administrative |
|
|
47.0 |
|
|
39.0 |
|
20.5% |
|
Total costs and expenses |
|
$ |
147.2 |
|
$ |
125.8 |
|
17.0% |
25
Cost of Sales
|
|
|
For the Three Months Ended |
||||||
|
|
|
March 31, |
||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
Change % |
||
|
Product |
|
$ |
33.8 |
|
$ |
17.0 |
|
98.8% |
|
Royalty |
|
|
28.1 |
|
|
14.8 |
|
89.9% |
|
Amortization of acquired intangibles |
|
|
1.4 |
|
|
0.7 |
|
100.0% |
|
Total cost of sales |
|
$ |
63.3 |
|
$ |
32.5 |
|
94.8% |
For the three months ended March 31, 2017 product cost of sales increased $16.8 million compared to the same period in 2016 as a result of increased volume of both ELOCTATE and ALPROLIX in our territory and manufacturing revenue in Sobi’s territory.
Royalty cost of sales consists mainly of our royalty to Sobi. As a result of Sobi’s first commercial sales of ELOCTATE and ALPROLIX in 2016, our royalty rate to Sobi for our sales of ELOCTATE and ALPROLIX increased in 2017 from approximately 7% to approximately 11%. Please refer to Note 2, Collaborations, to the condensed consolidated financial statements included elsewhere in this report for further information regarding our royalty structure with Sobi.
Inventory amounts written down as a result of excess, obsolescence, unmarketability or other reasons are charged to cost of sales. There were no write downs of inventory in either of the three months ended March 31, 2017 and 2016, respectively.
Research and Development Expenses
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Upfront and milestone payments |
|
$ |
(3.0) |
|
$ |
— |
|
Research and discovery |
|
|
15.5 |
|
|
10.8 |
|
Early stage programs |
|
|
— |
|
|
— |
|
Late stage programs |
|
|
— |
|
|
— |
|
Marketed programs |
|
|
16.7 |
|
|
23.9 |
|
Other research and development expenses |
|
|
7.7 |
|
|
19.6 |
|
Total research and development |
|
$ |
36.9 |
|
$ |
54.3 |
Research and discovery includes costs incurred to support our discovery research and translational science efforts up to the initiation of Phase 1 development. Early stage programs are programs in Phase 1 or Phase 2 development activities. Late stage programs are programs in Phase 3 development or in registration stage. Marketed programs are programs in support of our marketed products, including costs associated with product lifecycle management activities and, if applicable, costs associated with the development of new indications for existing products. Other research and development expenses consist mainly of allocations from Biogen prior to the separation and include costs not directly attributable to individual projects and depreciation and other facility‑based expenses, regulatory affairs function, pharmacovigilance, other infrastructure and management costs supporting multiple projects. Costs are reflected in the development stage based upon the program status when incurred. Therefore, the same program could be reflected in different development stages in the same year.
For the three months ended March 31, 2017, compared to the same period in 2016, the decrease in research and development was mainly due to a decrease in allocations from Biogen. However, we expect to further invest in research and development throughout 2017.
26
Selling, General and Administrative Expenses
|
|
|
For the Three Months Ended |
||||||
|
|
|
March 31, |
||||||
|
(In millions, except percentages) |
|
2017 |
|
2016 |
|
Change % |
||
|
Selling, general and administrative |
|
$ |
47.0 |
|
$ |
39.0 |
|
20.5% |
For the three months ended March 31, 2017, compared to the same period in 2016, the increase in selling, general and administrative is due to an increase in workforce partially offset by a decrease in allocations from Biogen. We expect to continue to invest in our infrastructure and people as we build capabilities to execute as a standalone company.
Income Taxes
We recorded income tax (benefit) expense of $42.2 million and $(1.2) million for the three months ended March 31, 2017 and 2016, respectively. Our effective income tax rate was 37.8% and (1.8)% of income before income taxes for the three months ended March 31, 2017 and 2016, respectively.
The effective income tax rate increased during the three months ended March 31, 2017 as compared to the prior year periods primarily due to the release of a valuation allowance associated with deferred tax assets that were available to us prior to separation. Substantially all of the deferred tax assets are no longer available to us as they have been utilized by Biogen. The effective rate in 2017 differs from the statutory rate mainly due to state taxes.
Liquidity and Capital Resources
Prior to the separation from Biogen, we participated in Biogen’s centralized treasury management, including centralized cash pooling and overall financing arrangements. Since the third quarter of 2015, we have generated and expect to continue to generate positive cash flow from operations on an annual basis. Net cash used for financing activities in the historical periods prior to the separation primarily reflects changes in Biogen’s investment in us. We had not reported cash or cash equivalents on our balance sheet as of December 31, 2016 due to our participation in Biogen’s centralized treasury management.
Subsequent to the separation from Biogen, we are no longer participating in cash management and funding arrangements with Biogen. Our ability to fund our operations and capital needs depends on our ongoing ability to generate cash from operations and access to capital markets, as further described under the “Debt and Capital” caption directly below. We anticipate that our principal uses of cash in the future will be primarily to fund our operations, working capital needs, capital expenditures and strategic investments.
Debt and Capital
We were capitalized by Biogen in connection with the distribution with $325.0 million in cash and we currently do not have any indebtedness for borrowed money. We expect that our initial cash capitalization, future cash from operations and access to capital markets will provide adequate resources to fund our ongoing cash flow obligations for the foreseeable future.
Cash Flows
|
|
|
For the Three Months Ended |
||||
|
|
|
March 31, |
||||
|
(In millions) |
|
2017 |
|
2016 |
||
|
Net cash flows provided by operating activities |
|
$ |
108.2 |
|
$ |
50.1 |
|
Net cash flows used in investing activities |
|
$ |
(6.3) |
|
$ |
(0.9) |
|
Net cash flows provided by (used) in financing activities |
|
$ |
256.3 |
|
$ |
(49.2) |
27
Net cash provided by operations increased $58.1 million for the three months ended March 31, 2017 as compared to the same period in 2016 driven primarily by increases in net income of $2.6 million and decreases in working capital of $69.0 million, which is primarily due to a change in inventory.
Net cash used for investing activities increased $5.4 million for the three months ended March 31, 2017 as compared to the same period in 2016 due to purchases of property, plant and equipment.
The $305.5 million increase in net cash provided for financing activities for the three months ended March 31, 2017 as compared to the same period in 2016 is mainly due to the initial capitalization from Biogen of $325.0 million offset by working capital payments between us and Biogen of $23.5 million.
Contractual Obligations
As of March 31, 2017, there have been no material changes in our contractual obligations since December 31, 2016.
For a further description of our contractual obligations, please read Part II, Item 7 Management’s Discussion and Analysis of Financial Condition and Results of Operations of our 2016 Form 10-K.
Critical Accounting Estimates
The preparation of our condensed consolidated financial statements, which have been prepared in accordance with U.S. GAAP requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses. On an ongoing basis we evaluate our estimates, judgments and methodologies. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable, the results of which form the basis for making judgments about the carrying value of assets, liabilities and equity and the amount of revenues and expenses. Actual results that differ from our estimates could have an unfavorable effect on our results of operations and financial position.
For a discussion of our critical accounting estimates, please read Part II, Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our 2016 Form 10-K. There have been no material changes to these critical accounting estimates since our 2016 Form 10-K.
New Accounting Standards
For a discussion of new accounting standards please read Note 1, Nature of Business and Basis of Presentation, to the condensed consolidated financial statements included elsewhere in this report.
Off‑Balance Sheet Arrangements
We do not have any relationships with entities often referred to as structured finance or special purpose entities that were established for the purpose of facilitating off‑balance sheet arrangements. As such, we are not exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in such relationships.
Item 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are subject to certain risks which may affect our results of operations, cash flows and fair values of assets and liabilities, including volatility in foreign currency exchange rates, interest rate movements, pricing pressures worldwide and weak economic conditions in the foreign markets in which we operate.
For a discussion of our quantitative and qualitative disclosures about market risk, please read Part II, Item 7A “Quantitative and Qualitative Disclosures About Market Risk” of our 2016 Form 10-K. In March 2017, we entered into an intercompany loan denominated in Japanese yen totaling approximately $71 million. In April 2017, we entered
28
into an undesignated hedge to mitigate any foreign exchange impact. The company believes that the intercompany loan and the corresponding hedge will not have a material impact on our results of operations.
Other than the item noted above, there has been no material changes to these market risks since our 2016 Form 10-K.
Item 4. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures and Internal Control over Financial Reporting
Bioverativ operated as part of Biogen through January 31, 2017.
Evaluation of Disclosure Controls and Procedures
Bioverativ has carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer (principal executive officer) and Chief Financial Officer (principal financial officer), of the effectiveness of the design and operation of the Bioverativ's disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended (the Exchange Act)) as of March 31, 2017. Based on that evaluation the Chief Executive Officer and Chief Financial Officer concluded that the company's disclosure controls and procedures were effective as of March 31, 2017.
Internal Control Over Financial Reporting
Under the rules and regulations of the Securities and Exchange Commission, Bioverativ is not required to comply with the requirements of Section 404 of the Sarbanes-Oxley Act of 2002 until its Annual Report on Form 10-K for the year ending December 31, 2017. In its Annual Report on Form 10-K for the year ending December 31, 2017, management and the company's independent registered public accounting firm may be required to provide an assessment as to the effectiveness of the company's internal control over financial reporting.
29
Please refer to Note 13, Litigation to the unaudited condensed consolidated financial statements included elsewhere in this report.
You should carefully consider the following risks and other information in this report in evaluating Bioverativ and Bioverativ’s common stock. Any of the following risks could materially and adversely affect our results of operations or financial condition and could adversely impact, or result in volatility to, our stock price following the distribution. The risk factors generally have been separated into three groups: risks related to our business, risks related to the separation from Biogen and risks related to our common stock.
Risks Related to Our Business
We are dependent on revenues from our products, ELOCTATE and ALPROLIX. If we or Sobi are unable to successfully commercialize ELOCTATE or ALPROLIX, our results of operations would be materially harmed.
Net sales of ELOCTATE and ALPROLIX represent substantially all of our revenues, and this concentration makes us dependent on these two products. Further, we currently have limited resources for commercializing ELOCTATE and ALPROLIX outside of the United States, Japan and Canada, and are dependent on the efforts of Sobi in its commercialization territory. If we were to experience difficulty with the commercialization of ELOCTATE or ALPROLIX, or if Sobi were to experience difficulty with the commercialization of ELOCTATE or ALPROLIX in its commercialization territory, we could experience a significant reduction in revenue and may not be profitable.
We expect that continued commercialization of ELOCTATE and ALPROLIX will depend on many factors, including the following:
|
· |
the effectiveness of our commercial strategy in and outside of the United States for the marketing of ELOCTATE and ALPROLIX; |
|
· |
our ability to maintain our development and commercialization arrangements with Sobi; |
|
· |
the success of our strategies for maintaining and enhancing a positive reputation among hemophilia patients and those in the hemophilia treatment community as to the efficacy and safety of ELOCTATE and ALPROLIX; and |
|
· |
other factors described below in this “Risk Factors” section. |
Many of these factors are beyond our control, and success in any one of these factors will not guarantee success in any of the others. Accordingly, we cannot assure you that we will be able to continue to generate revenue through the sale of ELOCTATE or ALPROLIX.
If our hemophilia products fail to compete effectively, our business and market position would suffer.
Due to our dependence on sales of our hemophilia products, our business may be harmed if our products are unable to successfully compete in the hemophilia treatment market. The hemophilia treatment market is highly competitive. We compete in the marketing and sale of our products, and in the development of, and acquisition of rights to, new products and technologies.
We compete with biotechnology and biopharmaceutical companies that have greater financial, technological and other resources than we do. One or more of our competitors may benefit from significantly greater sales and marketing capabilities, may develop products that are accepted more widely than ours or may receive patent protection that dominates, blocks or adversely affects our product development or business.
30
Our ability to successfully compete with other hemophilia treatments may be adversely affected if our therapies are not regarded by patients, healthcare providers or payors as offering significant benefits and value as compared to other current treatments. We are aware of a number of companies, including large biopharmaceutical companies, such as Bayer AG, Novo Nordisk, CSL Ltd., Pfizer Inc., Roche Holding AG and Shire Plc., that currently market or are pursuing the development of products for the treatment of hemophilia. We are also aware of other extended half‑life factor products as well as other new technologies, such as gene therapies and bi‑specific antibodies, that are in development and, if successfully developed and approved, would compete with ELOCTATE or ALPROLIX. New therapies and technologies have the potential to transform the standard of care for hemophilia patients, and our products may be unable to compete successfully with such new therapies and technologies that may be developed and marketed by other companies.
In addition, our relatively recent entrance into the hemophilia treatment market relative to certain of our competitors may impact our ability to develop relationships with the associated medical and scientific community that are necessary to properly inform these communities regarding the relative benefits that our products offer.
Our reliance on third parties for our manufacturing and distribution processes increases the risk that we will not have available sufficient quantities of ELOCTATE and ALPROLIX, or that such quantities may not be available at an acceptable cost, which could delay, prevent or impair our commercialization efforts and materially harm our business, results of operations and financial condition.
We rely, and expect to continue to rely, on third parties for the commercial manufacture and distribution of ELOCTATE and ALPROLIX. For example, in connection with the separation, we entered into a manufacturing and supply agreement with Biogen as our sole supplier of ELOCTATE and ALPROLIX for a specified period of time. Biogen is currently the sole manufacturer of ELOCTATE and ALPROLIX. Biogen also relies on third parties with respect to certain aspects of its manufacturing process, including certain sole sources of raw materials. We also rely, and expect to continue to rely, on third parties to distribute our products, including global, regional, and specialty distribution and logistics providers and, for a transition period, Biogen.
Biogen and other third party providers are independent entities subject to their own unique operational and financial risks that are outside of our control. Any of these third parties may not perform their obligations in a timely and cost‑effective manner or in compliance with applicable regulations. Biogen, for instance, may be unable or unwilling to increase production capacity commensurate with demand for our products. Finding alternative providers could take a significant amount of time and involve significant expense due to the specialized nature of these services.
In the event we change manufacturing partners or the third parties providing packaging, labeling and/or storage of our products, we may need to obtain approval from applicable regulatory authorities. Manufacturers are generally required to maintain compliance with current good manufacturing practices, or cGMPs, and other stringent requirements and are subject to inspections by the FDA and comparable agencies in other jurisdictions to confirm such compliance. These cGMP requirements and regulations are not prescriptive instructions on how to manufacture products, but rather a series of principles that must be observed during manufacturing; as a result, their implementation may not be clearly delineated and may present a challenging task as these regulatory requirements are complex, time‑consuming and expensive. Moreover, as our products are biologics, they require processing steps that are more difficult than those required for most chemical pharmaceuticals. Any delay, interruption or other issues that arise in the manufacture, fill‑finish, packaging or storage of our products as a result of a failure of our facilities or the facilities or operations of third parties to pass any regulatory agency inspection could result in administrative sanctions by the FDA or other U.S. or non‑U.S. regulatory agencies. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions.
We cannot be certain that we could reach agreement with alternative providers or that the FDA or other regulatory authorities would approve our use of alternative manufacturers or providers on a timely basis. Any adverse developments affecting our supply chain may result in development delays, shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the commercial supply of our products. In addition, loss or damage to a manufacturing facility or storage site due to a natural disaster or otherwise could adversely affect our ability to manufacture sufficient quantities of our products or to deliver products to meet customer demand or contractual
31
requirements, any of which may result in a loss of revenue and other adverse business consequences. We may also have to take inventory write‑offs and incur other charges and expenses for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Such developments could increase our manufacturing or development costs, cause us to lose revenue or market share as patients and physicians turn to competing therapeutics, diminish our profitability or damage our reputation. Moreover, any failure of Biogen to supply ELOCTATE and ALPROLIX could cause us to breach our supply agreements to Sobi for these products, which may subject us to liability under those agreements and impair our relationship with Sobi.
Issues with product quality or safety, including the perception of such issues, could negatively affect our business, subject us to regulatory or other actions and cause a loss of confidence in us or our products.
Our success depends upon the quality and safety of our products. Even after a product is approved for marketing, new safety data may emerge from adverse event reports or post‑marketing studies. Previously unknown risks and adverse effects of our products may also be discovered in connection with unapproved or off‑label uses of our products. Additionally, global regulatory authorities, at their discretion, may review and raise new or additional questions related to approved product information. This could result in changes to the approved product prescribing information or to the product approval itself. A quality or safety issue, including the perception of such issues, may result in investigations by regulatory authorities, product liability, adverse inspection reports, warning letters, product recalls or seizures, monetary sanctions, injunctions to halt manufacture and distribution of products, civil or criminal sanctions, costly litigation, requirements for additional labeling or safety monitoring, refusal of a government to grant approvals and licenses, restrictions on operations or withdrawal of existing approvals and licenses. An inability to address any of these issues in an effective and timely manner may cause negative publicity, loss of physician and patient confidence in the company or its current or future products and may negatively impact physicians’ decisions to prescribe our products. These issues could also result in liabilities, loss of revenue, material write‑offs of inventory, withdrawal or voluntary recall of our products from the marketplace, delays or limitations in regulatory approvals, material impairments of intangible assets, goodwill and fixed assets, material restructuring charges, difficulty in successfully launching new products and other adverse impacts on our results of operations.
Our inability to maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business and results of operations.
Sales of ELOCTATE and ALPROLIX are dependent, in large part, on the availability and extent of coverage, pricing and reimbursement from government health administration authorities, private health insurers and other organizations. When a new biopharmaceutical product is approved, the availability of government and private insurance coverage for that product may be uncertain, as may be the pricing of the product and extent to which the product will be reimbursed. Failure to maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business and results of operations.
Pricing and reimbursement for our products may be adversely affected by a number of factors, including:
|
· |
changes in federal, state or foreign government regulations or private third party payors’ reimbursement policies; |
|
· |
pressure by employers on private health insurance plans to reduce costs; |
|
· |
consolidation and increasing assertiveness of payors, including managed care organizations, health insurers, pharmacy benefit managers, government health administration authorities, private health insurers and other organizations seeking price discounts or rebates in connection with the placement of our products on their formularies and, in some cases, imposing restrictions on access to, coverage of or pricing for particular drugs based on perceived value; and |
|
· |
the influence of third party organizations advocating for discounted pricing with respect to our products. |
Our ability to set the price for our products can vary significantly from country to country and, as a result, so can the price of our products. Pricing and acceptance of ELOCTATE and ALPROLIX in certain countries are also subject to risks due to the tendering process required in those countries, as well as the comparison of dose pricing of our products against conventional treatments. If we are unable to demonstrate to healthcare providers and payors the value of
32
prophylaxis treatment and reduced consumption of our products compared to conventional treatments, our products may not be accepted or we may not secure adequate prices in a particular country. Our inability to secure adequate prices in a particular country may limit the marketing of our products within that country, and may also adversely affect our ability to obtain acceptable prices in other markets. This may create the opportunity for third party cross‑border trade or influence our decision to sell or not to sell a product in a particular country, thus adversely affecting our geographic expansion plans and revenues.
Pricing for therapies and other health care costs are under significant scrutiny in the markets in which our products are prescribed and continue to be subject to intense political and societal pressures, which we anticipate will continue and escalate. As a result, our business and reputation may be harmed.
If we are unable to obtain and maintain adequate protection for our intellectual property and other proprietary rights, or if we are unable to avoid violation of the intellectual property or proprietary rights of others, we may be subject to liability, the operation of our business may be interrupted or our business or prospects may be otherwise harmed.
Our commercial success depends in part on our ability to obtain and defend patent and other intellectual property rights that are important to the development, manufacture and commercialization of our products and product candidates. The degree of patent protection that will be afforded to our products and processes in the United States and in other important non‑U.S. markets remains uncertain and is dependent upon the scope of protection decided upon by the patent offices, courts and lawmakers in those countries. We can provide no assurance that we will successfully obtain or preserve patent protection for the technologies incorporated into our products and processes, or that the scope of patent protection obtained will be sufficient to protect our commercial interests in all countries where we conduct business. If we cannot prevent others from exploiting our inventions, we will not derive the benefit from them that we currently expect.
We exclusively license, under an agreement with Amunix, the XTEN technology that is used in connection with certain of our pipeline product candidates. If that agreement were to be terminated or if we otherwise lost our rights to such technology, our ability to develop, manufacture and commercialize such product candidates could be adversely affected, and could materially harm our business prospects.
We also rely on regulatory exclusivity for protection of our products. Implementation and enforcement of regulatory exclusivity varies widely from country to country. Failure to qualify for regulatory exclusivity, or failure to obtain or maintain the extent or duration of such protections that we expect for our products in each of these markets due to challenges, changes of interpretations in the law or otherwise, could affect the revenue for our products, our decision on whether to market our products in a particular country or countries or could otherwise have an adverse impact on our results of operations.
Additionally, we rely in part on confidentiality and non‑use agreements with our employees, consultants, collaborators and other business partners to protect our proprietary technology and processes. If any of these individuals or entities breaches their confidentiality, non‑use or similar agreements with us, we may not have adequate remedies for that breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors and even patented by them. If that happens, the potential competitive advantages provided by our intellectual property may be adversely affected. We may then need to license such competing technologies, and we may not be able to obtain licenses on reasonable terms, if at all, which could cause material harm to our business. Moreover, to the extent that our employees, consultants, parties to collaboration agreements and other business partners use intellectual property owned by others in their work for the company, disputes may arise as to the rights in related or resulting know‑how and inventions.
Our success also depends in part on our, and on the people with whom we collaborate and do business, not infringing patents and proprietary rights of others, and not breaching any licenses or other agreements that we or they have entered into with regard to our technologies, products and business. We cannot be certain that patents have not or will not be issued to others that would block our ability to obtain patents or to operate our business as we would like or at all. There may be patents in some countries that, if valid and if we are unsuccessful in circumventing or acquiring rights to them, could block our ability to commercialize products in those countries. There also may be claims in patent applications filed in some countries that, if granted and valid, and if we are unable to circumvent or license them, could also block our ability to commercialize products or processes in those countries.
33
Litigation, interferences, oppositions, inter partes reviews or other proceedings are, and may in the future be, necessary in some instances to determine the validity and scope of certain of our proprietary rights, and in other instances to determine the validity, scope or non‑infringement of certain patent rights claimed by third parties to be pertinent to the manufacture, use or sale of our products. Patent‑related claims could include challenges to the scope and validity of our patents on products or processes as well as allegations that our products infringe patents held by competitors or others. We may also face challenges to regulatory or patent protections covering our products by manufacturers of biosimilars that may choose to launch or attempt to launch their products before the expiration of our regulatory or patent exclusivity.
The disposition of claims or proceedings is unpredictable and, regardless of the merits or the outcome, may be protracted, expensive and distracting to management. Moreover, the disposition and outcome of any such claims or proceedings could adversely affect the validity and scope of our patent or other proprietary rights; hinder our ability to manufacture, market and sell our products; lead to attempts on the part of other parties to pursue similar claims; force us to redesign those products or processes that use any allegedly infringing or misappropriated technology, which may result in significant cost or delay to us, or which the redesign of could be technically infeasible; require us to seek a license for the impacted product or technology and pay royalties; or result in the assessment of significant monetary damages against us that may exceed amounts, if any, accrued in our financial statements, including the possibility of treble damages in a patent case if a court finds us to have willfully infringed certain intellectual property rights. In addition, payments under any licenses that we are able to obtain would reduce our profits derived from the covered products and services. Furthermore, many of our collaboration agreements, including with Sobi, require us to indemnify the collaboration parties for third party intellectual property infringement claims, which would increase the cost to us of any such claim. Any of these adverse effects may be material and, consequently, may adversely impact our cash flow, financial position and results of operations.
Our sales and operations are subject to the risks of doing business in Japan and other international markets, which could adversely impact our business, results of operations and financial condition.
We are increasing our presence in Japan, Canada and other non‑U.S. markets, including in emerging markets, which subjects us to many risks that could adversely affect our business and revenues, such as:
|
· |
the inability to obtain necessary regulatory or pricing approvals of products in a timely manner; |
|
· |
differing local product preferences and product requirements; |
|
· |
changes in medical reimbursement policies and programs; |
|
· |
fluctuations in foreign currency exchange rates, in particular the recent strength of the U.S. dollar versus foreign currencies; |
|
· |
difficulties in staffing and managing non‑U.S. operations; |
|
· |
unexpected difficulties in establishing effective financial and other controls in non-US jurisdictions; |
|
· |
uncertainties regarding the collectability of accounts receivable; |
|
· |
differing and increased labor regulations, including non‑U.S. work councils; |
|
· |
the imposition of governmental controls; |
|
· |
less favorable intellectual property or other applicable laws; |
|
· |
increasingly complex standards for complying with non‑U.S. laws and regulations that may differ substantially from country to country and may conflict with corresponding U.S. laws and regulations; |
|
· |
government involvement in funding health care in major overseas markets; |
|
· |
the far‑reaching anti‑bribery and anti‑corruption legislation in the United Kingdom and elsewhere, including the U.K. Bribery Act 2010, and the escalation of investigations and prosecutions pursuant to such laws; |
|
· |
compliance with complex import and export control laws; |
|
· |
restrictions on direct investments by non‑U.S. entities and trade restrictions; |
|
· |
greater political or economic instability; and |
|
· |
changes in tax laws and tariffs. |
34
We cannot guarantee that our efforts to initiate or expand sales in these markets will succeed. Some non‑U.S. markets may be especially vulnerable to periods of financial instability or may have very limited resources to spend on health care. To successfully implement our strategy in non‑U.S. markets, we must attract and retain qualified personnel or may be required to increase our reliance on third party distributors within those markets. In addition, many of the countries in emerging markets have currencies that fluctuate substantially. If such currencies devalue and we cannot offset the devaluations, our financial performance within those countries could be adversely affected. In addition, price and currency exchange controls, limitations on participation in local enterprises, expropriation, nationalization and other governmental actions could affect our business and results of operations in these markets.
In addition, our non‑U.S. operations are subject to regulation under U.S. law. For example, the U.S. Foreign Corrupt Practices Act (FCPA) prohibits U.S. companies and their representatives from offering, promising, authorizing or making payments to foreign officials for the purpose of obtaining or retaining business abroad. In many countries, the health care professionals we regularly interact with may meet the definition of a foreign government official for purposes of the FCPA. Failure to comply with U.S. or non‑U.S. laws could result in various adverse consequences, including: possible delay in approval or refusal to approve a product; recalls, seizures or withdrawal of an approved product from the market; disruption in the supply or availability of our products or suspension of export or import privileges; the imposition of civil or criminal sanctions; the prosecution of executives overseeing our international operations; and damage to our reputation. Any significant impairment of our ability to sell products outside of the U.S. could adversely impact our business and financial results.
Development of our product candidates is expensive and uncertain. If we are unable to successfully develop and test our product candidates, our business, financial condition, results of operations and prospects will be harmed.
A part of our long‑term strategy is the continued development of marketed products and our product pipeline programs. The research and development of biological products is subject to numerous risks and uncertainties and requires significant capital expenditures and management resources. Only a small percentage of product candidates that enter the development process ever receive regulatory approval. The process of conducting the preclinical and clinical testing required to establish safety and efficacy and obtain regulatory approval is expensive and uncertain and takes many years. The FDA and non‑U.S. regulatory agencies generally require pre‑clinical (animal) testing as well as multiple stages of clinical (human) testing before a product gains regulatory approval, and failure may occur at any stage of testing. It is possible that positive results in a trial may not be replicated in subsequent or confirmatory trials, and success in preclinical work or early stage clinical trials does not ensure that later stage or larger scale clinical trials will be successful or that regulatory approval will be obtained. Furthermore, our ability to commence and complete clinical trials may be delayed, and our existing trials may be stopped, due to various factors, including: variability in the number and types of patients available for each study; difficulty in maintaining contact with patients after treatment, resulting in incomplete data, unforeseen safety issues or side effects; varying interpretations of clinical trial data; poor or unanticipated effectiveness of candidates during clinical trials; and government or regulatory delays.
These risks are enhanced by our reliance on third parties for aspects of the research and development process. We rely, and expect to continue to rely, on third parties to store and distribute drug supplies for our clinical trials as well as contract research organizations (CROs), clinical data management organizations, medical institutions and clinical investigators to conduct and manage our preclinical and clinical trials and to accurately report their results. Reduced control over these activities may impact our ability to control the timing, conduct, expense, reliability and quality of our clinical trials, but does not relieve us of our regulatory responsibility for trials that we sponsor. For example, we remain responsible for ensuring that each of our clinical trials is conducted in accordance with the general investigational plan and protocols for the trial as well as regulatory standards such as current cGCPs. Failure to fully comply with the study protocol or applicable regulations or regulatory standards could result in the clinical data generated in those studies being deemed unreliable. This failure may also result in the rejection of our product candidates by the FDA or a non‑U.S. regulatory agency, or may result in our having to conduct additional audits or require additional clinical studies, which would delay our development programs, require us to incur additional costs and could substantially harm our business and financial condition. If the third parties we rely on for research and development activities do not successfully carry out their contractual duties, do not meet expected deadlines, experience work stoppages, terminate their agreements with us, need to be replaced or do not conduct our clinical trials in accordance with regulatory requirements or our stated
35
protocols, we may need to enter into new arrangements with alternative third parties, which could be difficult, costly or impossible. As a result, our clinical trials may be extended, delayed, terminated or may need to be repeated.
Even if we are able to successfully develop new products or indications, sales of new products or products with additional indications may not meet expectations. Our products may not achieve an adequate level of acceptance in the medical community until longer‑term clinical data or other factors demonstrate their safety and efficacy as compared to other alternative treatments. We may also make a strategic decision to discontinue development of a product or indication if, for example, we believe commercialization will be difficult relative to the standard of care or other opportunities in our pipeline.
The occurrence of any of these events could result in significant costs and expenses and lost market opportunities.
If our business development activities are unsuccessful, our business could suffer and our financial performance could be adversely affected.
We intend to engage in business development activities, including evaluating potential acquisitions, strategic alliances, collaborations, technology licensing arrangements and other opportunities. These activities may require a substantial investment of our resources, with no certainty of success. Our success developing products or expanding our product portfolio from such business development activities will depend on a number of factors, including:
|
· |
our ability to find suitable opportunities for acquisition, investment or alliance; |
|
· |
whether we are able to complete an acquisition, investment or alliance on terms that are satisfactory to us; |
|
· |
the strength of the technology and products being licensed or acquired; |
|
· |
any intellectual property and litigation related to these products or technology; |
|
· |
our ability to successfully integrate the acquired company, business, product, technology or research into our existing operations, including the ability to adequately fund acquired in‑process research and development projects and to maintain adequate controls over the consolidated operations; and |
|
· |
our ability to access capital markets or incur indebtedness at terms that are satisfactory to us. |
If we are unsuccessful in our business development activities, we may be unable to grow or meet our financial targets and our business and financial performance could be adversely affected. Further, even if we are able to successfully identify and complete acquisitions and other strategic alliances, licenses and collaborations, we may face unanticipated costs or liabilities in connection with the transaction or we may not be able to integrate them or take full advantage of them or otherwise realize the benefits that we expect.
We depend on relationships with collaborators and other third parties for revenue, and for the development, regulatory approval, commercialization and marketing of certain products, which are outside our full control. If our collaborative efforts are unsuccessful, our commercialization strategies or product development may be delayed, which could have an adverse impact on our business, prospects and results of operations.
We rely, and expect to continue to rely, on a number of significant collaborative and other third party relationships for revenue, and for the development, regulatory approval, commercialization and marketing of our products and product candidates. These third parties may include other biotechnology and biopharmaceutical companies, academic and research institutions, governments and government agencies and other public and private research organizations. For example, in addition to our collaboration with Sobi, we are pursuing programs with other third parties in hemophilia A and hemophilia B using XTEN technology, gene therapy, gene editing and non‑factor bi‑specific antibodies, as well as other rare blood disorders.
Reliance on collaborative and other third party relationships subjects us to a number of risks, including:
|
· |
we may be unable to control the resources our collaborator devotes to our programs or product candidates; |
36
|
· |
disputes may arise with respect to ownership of rights to technology developed with our collaborator, and the underlying contract with our collaborator may fail to provide us with significant protection or may fail to be effectively enforced if the collaborator fails to perform; |
|
· |
our collaborators’ interests may not always be aligned with our interests and a collaborator may not pursue regulatory approvals or market a product in the same manner or to the same extent that we would, which could adversely affect our revenues; |
|
· |
the failure to effectively cooperate with our collaborators could adversely affect product sales or the clinical development or regulatory approvals of product candidates under joint control and could also result in termination of the research, development or commercialization of product candidates, litigation or arbitration; and |
|
· |
any failure on the part of our collaborator to comply with applicable laws and regulatory requirements in the marketing, sale and maintenance of the market authorization of our products or to fulfill any responsibilities our collaborator may have to protect and enforce any intellectual property rights underlying our products or product candidates could have an adverse effect on our revenues and involve us in legal proceedings. |
Given these risks, there is considerable uncertainty regarding the success of our current and future collaborative efforts. If these efforts fail, our product development or commercialization of new products could be delayed or revenues from products could decline.
If we or third parties with whom we do business fail to comply with the extensive legal and regulatory requirements affecting the health care industry, we could face increased costs, penalties and harm to our business.
Our activities, and the activities of our collaborators, distributors and other third party providers, are subject to extensive government regulation and oversight both in the U.S. and in non‑U.S. jurisdictions.
To be approved for marketing, a potential product must undergo lengthy and rigorous testing and other extensive, costly and time‑consuming procedures mandated by the FDA and non‑U.S. regulatory authorities. Satisfaction of these regulatory requirements typically takes many years. Moreover, regulatory oversight continues to apply after product marketing approval and covers, among other things, testing, manufacturing, distribution, quality control, labeling, advertising, promotion, risk mitigation and adverse event reporting requirements. Our facilities, or those of third parties on which we rely, must be licensed prior to production and remain subject to inspection from time to time thereafter. Separately, if previously unknown problems occur with regard to our marketed products, any of our products may have to be withdrawn from the market or subject to restrictions. Regulatory agencies may also require additional clinical trials or testing for our products, and our products may be recalled or may be subject to reformulation, changes in labeling, warnings to the public and negative publicity. We are relying on Biogen for a period following the effective time of the distribution to supply and distribute our products to customers while we obtain appropriate regulatory authorizations in certain jurisdictions, such as the United States and Canada. In the United States, we are in the process of securing a Department of Health and Human Services United States License Number, certain state authorizations and other regulatory and government authorizations. In Canada, we are in the process of applying for additional licenses, including a Drug Establishment License from Health Canada. We cannot guarantee that we will be able to obtain or maintain regulatory approval to market or engage in distribution or other activities regarding our products.
Further, even if we are successful in gaining regulatory approval of any of our product candidates, the extent to which we are able to commercialize the product may be less than we anticipate. Regulatory authorities may grant marketing approval that is more restricted than anticipated. These restrictions may include limiting indications to narrow patient populations and imposing safety monitoring, educational requirements and risk evaluation and mitigation strategies (REMS). In addition, if we seek to expand or change the use of any of our marketed products, those changes may be subject to vigorous review and include multiple regulatory submissions, and approvals are not certain.
In addition to FDA and related regulatory requirements, we are subject to health care “fraud and abuse” laws governing our interactions in the U.S. and non‑U.S. jurisdictions with physicians or other health care providers that prescribe or purchase our products. In the United States, these laws include the federal False Claims Act, the anti‑kickback provisions of the federal Social Security Act, the Physician Payment Sunshine provisions, and other state and federal laws and regulations. In both the United States and other jurisdictions, health care companies such as ours
37
are facing heightened scrutiny of their relationships with health care providers from anti‑corruption enforcement officials and private individuals. Many biotechnology and biopharmaceutical companies have been the target of lawsuits and investigations alleging violations of government regulation, including claims asserting submission of incorrect pricing information, impermissible off‑label promotion of biotechnology and biopharmaceutical products, payments intended to influence the referral of health care business, submission of false claims for government reimbursement, antitrust violations or violations related to environmental matters. There also recently has been enhanced scrutiny of company‑sponsored patient assistance programs, including insurance premium and co‑pay assistance programs and donations to third party charities that provide such assistance. If we, or our vendors or donation recipients, fail to comply with relevant laws, regulations or government guidance in the operation of these programs, we could be subject to significant fines or penalties. Our risks under health care fraud and abuse laws may be heightened as we continue to expand our global operations and if we enter new therapeutic areas with different patient populations, which may have product distribution methods distinct from those we currently utilize.
Violations of governmental regulation, such as a failed inspection or a failure in our adverse event reporting system, or any health care fraud and abuse law may be punishable by criminal, civil and administrative sanctions against us as well as against executives overseeing our business. These may include adverse inspection reports; refusal to grant approvals or licenses; warning letters; fines and civil monetary penalties; withdrawal of regulatory approval or licenses; interruption of production; operating restrictions; product recall or seizure; injunctions; criminal prosecution and exclusion from participation in government programs, including Medicare and Medicaid, as well as against executives overseeing our business. In addition to penalties for violation of laws and regulations, we could be required to repay amounts we received from government payors, or pay additional rebates and interest if we are found to have miscalculated the pricing information we have submitted to the government. We cannot ensure that our compliance controls, policies and procedures will, in every instance, protect us from acts committed by our employees, collaborators, partners or third party providers that would violate the laws or regulations of the jurisdictions in which we operate. Whether or not we have complied with the law, an investigation into alleged unlawful conduct could increase our expenses, damage our reputation, divert management time and attention and adversely affect our business. Any of these actions could cause a loss of confidence in us and our products, which could adversely affect our sales. Even if it is later determined that we are not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our position and have to divert significant management resources from other matters.
Our business and results of operations may be adversely affected by current and potential future health care reforms.
In the United States, federal and state legislatures, health agencies and third party payors continue to focus on containing the cost of health care. Legislative and regulatory proposals and enactments to reform health care insurance programs could significantly influence the manner in which our products are prescribed and purchased. For example, provisions of the Patient Protection and Affordable Care Act (PPACA) have resulted in changes in the way health care is paid for by both governmental and private insurers in the United States, including increased rebates owed by manufacturers under the Medicaid Drug Rebate Program, annual fees and taxes on manufacturers of certain branded prescription drugs, the requirement that manufacturers participate in a discount program for certain outpatient drugs under Medicare Part D and the expansion of the number of hospitals eligible for discounts under Section 340B of the Public Health Service Act. These changes have had and are expected to continue to have a significant impact on our business. In 2017, we continue to face uncertainties as a result of federal and administrative efforts to repeal, substantially modify or invalidate or replace some or all of the provisions of the PPACA. There is no assurance that the PPACA, as currently enacted or as amended or replaced in the future, will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform will affect our business.
There is also significant economic pressure on U.S. state budgets that may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for our drugs. In recent years, some states have considered legislation and ballot initiatives that would control the prices of drugs, including laws to allow importation of biotechnology and biopharmaceutical products from lower cost jurisdictions outside of the United States. and laws intended to impose price controls on state drug purchases. State Medicaid programs are increasingly requesting manufacturers pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not paid. Government efforts to reduce Medicaid expenses may lead to increased use of
38
managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription decisions for a larger segment of the population and a corresponding constraint on prices and reimbursement for our products.
In the European Union and some other non‑U.S. markets, the government provides health care at low cost to consumers and regulates biotechnology and biopharmaceutical prices, patient eligibility or reimbursement levels to control costs for the government‑sponsored health care system. Many countries have announced or implemented measures to reduce health care costs to constrain their overall level of government expenditures. These measures vary by country and may include, among other things, patient access restrictions, suspensions on price increases, prospective and possibly retroactive price reductions and other recoupments and increased mandatory discounts or rebates, recoveries of past price increases and greater importation of drugs from lower‑cost countries. These measures have negatively impacted our revenues and may continue to adversely affect our revenues and results of operations in the future.
If we fail to attract and retain key personnel, our business may suffer.
Our success depends in large part upon the leadership and performance of our management team and other key employees. Operating as an independent company demands a significant amount of time and effort from our management and other employees and may give rise to increased employee turnover. If we lose the services of members of our management team or other key employees, we may not be able to successfully manage our business or achieve our business objectives.
Our ability to attract, recruit and retain such talent will depend on a number of factors, including the hiring practices of our competitors, the performance of our commercial products and pipeline programs, our compensation and benefits, work location and work environment and economic conditions affecting our industry generally. If we cannot effectively hire and retain qualified employees, our business, results of operations and prospects could suffer.
A breakdown or breach of our technology systems could subject us to liability or interrupt the operation of our business.
We are increasingly dependent upon technology systems and data, many of which are new or unfamiliar systems. Our intellectual property, computer systems, other proprietary technology and other sensitive company data is potentially vulnerable to loss, damage or misappropriation from system malfunction, computer viruses, unauthorized access to data or misappropriation or misuse thereof by those with permitted access and other events. Likewise, data privacy or security breaches by individuals authorized to access our technology systems or others may pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to us, our patients, customers or other business partners, may be exposed to unauthorized persons or to the public. The increasing use and evolution of technology, including cloud‑based computing, creates additional opportunities for the unintentional dissemination of information and the intentional destruction of confidential information stored in the company’s systems or in non‑encrypted portable media or storage devices. Cyber attacks are increasing in their frequency, sophistication and intensity. While we continue to build and improve our systems and infrastructure and take appropriate security measures to reduce these risks to our intellectual property, data and information technology systems, there can be no assurance that our efforts will prevent breakdowns, breaches, cyber incidents or other events. Such events could have a negative effect on our reputation, business, financial condition or results of operations. Further, the misappropriation or other loss of our intellectual property from any of the foregoing could have an adverse effect on our competitive position and may cause us to incur substantial litigation costs.
Our business involves environmental risks, which include the cost of compliance and the risk of contamination or injury, which could harm our business.
Our business and the business of several of our third party contractors involve the controlled use of hazardous materials, chemicals, biologics and radioactive compounds. Although we believe that our and their safety procedures for the handling and disposing of such materials comply with state, federal and non‑U.S. laws and standards, there will always be the risk of accidental contamination or injury. If we were to become liable for an accident, or if a facility in which our products or product candidates were manufactured suffered an extended shutdown, we could incur significant
39
costs, damages or penalties that could harm our business. Manufacturing, distribution and disposal of our products and product candidates also requires compliance with environmental laws and may require permits from government agencies, including governmental authorizations or permits for water supply, wastewater discharge and waste disposal. If we or our contract parties do not obtain or comply with appropriate permits and other requirements of environmental laws, we or they could incur significant penalties and other costs and limits on manufacturing volumes that could harm our business.
Significant legal proceedings may adversely affect our results of operations or financial condition.
We are subject to the risk of litigation, derivative claims, securities class actions, regulatory and governmental investigations and other proceedings, including proceedings arising from investor dissatisfaction with us or our performance. If any claims were brought against us and resulted in a finding of substantial legal liability, the finding could materially adversely affect our business, financial condition or results of operations or cause significant reputational harm to us, which could seriously adversely impact our business. Allegations of improper conduct by private litigants or regulators, regardless of veracity, may harm our reputation and adversely impact our ability to grow our business.
Risks Related to the Separation
We may not achieve some or all of the expected benefits of the separation, and the separation could harm our business, results of operations and financial condition.
We may not be able to achieve some or all of the anticipated strategic, financial, operational, marketing or other benefits expected to result from the separation, or such benefits may be delayed or not occur at all. For example, we have undertaken and are undertaking strategic, structural and process realignment actions within our operations. These actions may not provide the benefits we currently expect, and could lead to disruption of our operations, loss of or inability to recruit, key personnel needed to operate and grow our businesses following the separation, weakening of our internal standards, controls or procedures and impairment of our key customer and supplier relationships.
By separating from Biogen, we may become more susceptible to market fluctuations and other adverse events than we would have been if we were still a part of the current Biogen organizational structure. As part of Biogen, we were able to enjoy certain benefits from Biogen’s operating diversity, purchasing power and opportunities to pursue integrated strategies with Biogen’s other businesses. As an independent, publicly traded company, we do not have similar diversity or integration opportunities and may not have similar purchasing power or access to capital markets. Additionally, as part of Biogen, we were able to leverage Biogen’s historical market reputation, performance and brand identity to recruit and retain key personnel to run our business. As an independent, publicly traded company, we do not have the same historical market reputation and performance or brand identity as Biogen. If we fail to achieve some or all of the benefits that we expect to achieve as an independent company, or do not achieve them in the time we expect, our business, operating results, financial condition or prospects may suffer.
We may be unable to make, on a timely or cost‑effective basis, the changes necessary to operate as an independent company, and we will be reliant on Biogen for the provision of certain services for a period of time.
We have historically operated as part of Biogen’s corporate organization, and Biogen assisted us by providing various corporate and other business functions. Following the separation, Biogen has no obligation to provide us with assistance other than providing certain transition, manufacturing and supply services pursuant to certain agreements entered into in connection with the separation. If Biogen is unable or unwilling to satisfy its obligations under these agreements, we could incur operational difficulties or losses that could have a material and adverse effect on our business, operating results and financial condition.
The services to be provided by Biogen do not include every service or all of the information and technology systems that we have received from Biogen in the past, and Biogen is only obligated to provide these services for limited periods of time from the separation. Accordingly, following the separation, we will need to provide internally or obtain from unaffiliated third parties the systems and services we currently receive from Biogen.
40
If we do not have in place our own systems and services, including technology systems and services, or if we do not have agreements with other providers of these services in a timely manner or on terms and conditions as favorable as those we receive from Biogen, we may not be able to operate our business effectively and our profitability may decline. Furthermore, if we fail to obtain the quality of services necessary to operate effectively or incur greater costs in obtaining these services, our profitability, operating results and financial condition may be materially and adversely affected.
We have only operated as an independent company since February 1, 2017, and we expect to incur increased administrative and other costs following the separation by virtue of our status as an independent public company. Our historical financial information is not necessarily representative of the results that we would have achieved as a separate, publicly traded company and should not be relied upon as an indicator of our future results.
The historical information of Bioverativ included in this report for periods prior to our separation from Biogen refers to our business as operated by and integrated with Biogen and is derived from the consolidated financial statements and accounting records of Biogen. Accordingly, the historical financial information for periods prior to our separation from Biogen included in this report does not necessarily reflect the operating results, financial condition or cash flows that we would have achieved as a separate, publicly traded company during the periods presented or what we will achieve in the future primarily as a result of the following factors, among others:
|
· |
Prior to the separation, our business was operated by Biogen as part of its broader corporate organization, rather than as an independent company. Biogen, or one of its affiliates, performed various corporate functions for us, including executive oversight, treasury, finance, legal, human resources, tax planning, internal audit, financial reporting, information technology, investor relations, shared services, insurance, employee benefits and incentives and share‑based compensation. Following the separation, Biogen has continued to provide some of these functions to Bioverativ. Our historical financial results for periods prior to the separation reflect allocations of corporate expenses from Biogen for such functions, which are likely to be less than the expenses we would have incurred had we operated as a separate, publicly traded company. Following the separation, our costs related to such functions previously provided by Biogen will likely increase. |
|
· |
Historically, we shared economies of scope and scale in costs, employees, vendor relationships and customer relationships with Biogen. Although we have entered into certain agreements with Biogen in connection with the separation, these arrangements may not fully capture the benefits that we have enjoyed as a result of being integrated with Biogen and may result in us incurring higher costs than in the past. |
|
· |
Following the separation, we may lose certain synergies and benefits we enjoyed as a result of being a part of Biogen. As a part of Biogen, we benefited from, among other things, access to potential new customers from Biogen and capital to fund acquisitions and investments. In addition, being a part of Biogen enabled us to leverage Biogen’s technological capabilities, data and commerce platforms. |
|
· |
Generally, prior to the separation, our working capital requirements and capital for our general corporate purposes, including acquisitions and capital expenditures, were satisfied as part of the corporate‑wide cash management policies of Biogen. Following the separation, we may need to obtain additional financing from banks, through public offerings or private placements of debt or equity securities, or through strategic relationships or other arrangements, which may or may not be available and may be more costly. |
|
· |
After the separation, the cost of capital of our business may be higher than Biogen’s cost of capital prior to the separation. |
|
· |
Our historical financial information for periods prior to the separation does not reflect our obligations to purchase from Biogen certain operations and assets, and assumes the corresponding liabilities, of Biogen’s business after the separation date. |
Other significant changes may occur in our cost structure, management, financing and business operations as a result of operating as a company separate from Biogen.
41
The separation may result in disruptions to, and negatively impact our relationships with, our customers and other business partners.
Uncertainty related to the separation may lead customers and other parties with which we currently do business or may do business in the future to terminate or attempt to negotiate changes in our existing business relationships, or cause them to delay entering into business relationships with us or consider entering into business relationships with parties other than us. These disruptions could have a material and adverse effect on our business, operating results, financial condition and prospects. The effect of such disruptions could be exacerbated by any delays in the completion of the separation.
Our accounting and other management systems and resources may not be adequately prepared to meet the financial reporting and other requirements to which we are subject following the distribution.
Our financial results for periods prior to the separation historically were included within the consolidated results of Biogen, and until the distribution occurred, we were not directly subject to reporting and other requirements of the Securities and Exchange Act of 1934 (Exchange Act) and Section 404 of the Sarbanes‑Oxley Act of 2002. We currently qualify as an “emerging growth company” and for so long as we remain so qualified we will be exempt from Section 404(b) of the Sarbanes‑Oxley Act of 2002, which requires auditor attestation to the effectiveness of internal control over financial reporting. At such time as we no longer qualify as an emerging growth company, we will be broadly subject to reporting and other requirements under the Exchange Act and Sarbanes‑Oxley Act of 2002, which will require, among other things, annual management assessments of the effectiveness of our internal control over financial reporting and a report by our independent registered public accounting firm addressing these assessments. These and other obligations will place significant demands on our management, administrative and operational resources, including accounting and information technology resources. To comply with these requirements, we anticipate that we will need to further upgrade our systems, including duplicating computer hardware infrastructure, implement additional financial and management controls, reporting systems and procedures and hire additional accounting, finance and information technology staff. If we are unable to do this in a timely and effective fashion, our ability to comply with our financial reporting requirements and other rules that apply to reporting companies could be impaired and our business could be harmed.
If the distribution, together with certain related transactions, does not qualify as a transaction that is tax‑free for U.S. federal income tax purposes, Biogen and its stockholders could be subject to significant tax liabilities, and we could be required to indemnify Biogen for material taxes pursuant to indemnification obligations under the tax matters agreement.
Completion of the separation from Biogen was conditioned on the receipt by Biogen of an opinion from Biogen’s tax counsel or other third party advisor regarding the qualification of the distribution, together with certain related transactions, as a transaction that will qualify under Sections 368(a)(1)(D) and 355 of the Internal Revenue Code of 1986, as amended (the Code). Except as otherwise noted, it is expected that the distribution will qualify as a transaction that is tax‑free for U.S. federal income tax purposes to Biogen and the holders of Biogen common stock. The opinion was received and was based on and relied on, among other things, certain facts and assumptions, as well as certain representations, statements and undertakings of us and Biogen, including those relating to the past and future conduct of us and Biogen. If any of these facts, assumptions, representations, statements or undertakings are, or become, inaccurate or incomplete, or if we or Biogen breach any of our respective covenants in the separation documents, the opinion of counsel may be invalid and the conclusions reached therein could be jeopardized.
Notwithstanding the opinion of counsel, the Internal Revenue Service (IRS) could determine on audit that the distribution, together with certain related transactions, is taxable for U.S. federal income tax purposes if it determines that any of these facts, assumptions, representations, statements or undertakings are incorrect or have been violated or if it disagrees with the conclusions in the opinion of counsel. An opinion of counsel is not binding on the IRS or any court and there can be no assurance that the IRS will not challenge the conclusions reached in the opinion.
If the distribution, together with certain related transactions, is ultimately determined to be taxable, Biogen and its stockholders that are subject to U.S. federal income tax could incur significant tax liabilities. For example, if the
42
distribution fails to qualify for tax‑free treatment, Biogen would, for U.S. federal income tax purposes, be treated as if it had sold our common stock in a taxable sale for its fair market value, and those Biogen stockholders who are subject to U.S. federal income tax would be treated as receiving a taxable distribution in an amount equal to the fair market value of our common stock received in the distribution.
Under the tax matters agreement we entered into with Biogen, we would potentially be required to indemnify Biogen against taxes incurred by Biogen that arise as a result of our taking or failing to take, as the case may be, certain actions that result in the distribution failing to meet the requirements of a tax‑free distribution under Section 355 of the Code. If we are required to indemnify Biogen under the circumstances set forth in the tax matters agreement, we may be subject to substantial liabilities, which could materially adversely affect our financial condition.
We will be subject to numerous restrictions to preserve the tax‑free treatment of the transactions in the United States, which may reduce our strategic and operating flexibility.
Our ability to engage in significant transactions could be limited or restricted after the distribution in order to preserve, for U.S. federal income tax purposes, the tax‑free nature of the distribution by Biogen. Even if the distribution otherwise qualifies for tax‑free treatment, the distribution may result in corporate‑level taxable gain to Biogen under Section 355(e) of the Code if 50% or more, by vote or value, of shares of our stock or Biogen’s stock are acquired or issued as part of a plan or series of related transactions that includes the distribution. The process for determining whether an acquisition or issuance triggering these provisions has occurred is complex, inherently factual and subject to interpretation of the facts and circumstances of a particular case. Any acquisitions or issuances of our stock or Biogen’s stock within a two‑year period after the distribution generally are presumed to be part of such a plan, although we or Biogen, as applicable, may be able to rebut that presumption. Accordingly, under the tax matters agreement that we entered into with Biogen, for the two‑year period following the distribution, we will be prohibited, except in certain circumstances, from:
|
· |
entering into any transactions resulting in the acquisition of 40% or more of our stock or substantially all of our assets, whether by merger or otherwise; |
|
· |
merging, consolidating or liquidating; |
|
· |
issuing equity securities beyond certain thresholds; |
|
· |
repurchasing our capital stock; or |
|
· |
ceasing to actively conduct our business. |
These restrictions may limit our ability to pursue certain strategic transactions or other transactions that we may believe to otherwise be in the best interests of our stockholders or that might increase the value of our business. In addition, under the tax matters agreement, we will be required to indemnify Biogen against any such tax liabilities as a result of the acquisition of our stock or assets, even if we do not participate in or otherwise facilitate the acquisition.
Our agreements with Biogen may not reflect terms that would have resulted from negotiations with unaffiliated third parties.
The agreements we entered into with Biogen related to the separation, including, among others, the separation agreement, the tax matters agreement and the transition services agreement, were entered into in the context of the separation while we were still controlled by Biogen. Until the distribution occurred, Biogen effectively had the sole and absolute discretion to determine and change the terms of the separation, including the terms of any agreements between Biogen and us and the establishment of the record date and distribution date. As a result, the terms of such agreements may not reflect terms that would have resulted from negotiations between unaffiliated third parties.
We will be subject to continuing contingent tax related liabilities of Biogen following the distribution.
After the distribution, there are several significant areas where the liabilities of Biogen may become our obligations. For example, under the Code and the related rules and regulations, each corporation that was a member of Biogen’s consolidated tax reporting group during any taxable period or portion of any taxable period is jointly and severally liable for the U.S. federal income tax liability of the entire consolidated tax reporting group for such taxable
43
period. The tax matters agreement we entered into with Biogen allocates the responsibility for prior period taxes of Biogen’s consolidated tax reporting group between us and Biogen. If Biogen were unable to pay any prior period taxes for which it is responsible, however, under applicable law we could be required to pay the entire amount of such taxes, and such amounts could be significant. Other provisions of federal, state, local or foreign law may establish similar liability for other matters, including laws governing tax‑qualified pension plans, as well as other contingent liabilities.
In connection with the separation, we assumed and agreed to indemnify Biogen for certain liabilities. If we are required to make payments pursuant to these indemnities to Biogen, we may need to divert cash to meet those obligations and our financial results could be negatively impacted.
Pursuant to the separation agreement and certain other agreements we entered into with Biogen, we assumed and agreed to indemnify Biogen for certain liabilities for uncapped amounts, which may include, among other items, associated defense costs, settlement amounts and judgments Payments pursuant to these indemnities may be significant and could negatively impact our business, particularly indemnities relating to our actions that could impact the tax‑free nature of the distribution and certain related transactions. Third parties could also seek to hold us responsible for any of the liabilities of the Biogen business. Biogen agreed to indemnify us for liabilities of the Biogen business, but such indemnity from Biogen may not be sufficient to protect us against the full amount of such liabilities, and Biogen may not fully satisfy its indemnification obligations. Moreover, even if we ultimately succeed in recovering from Biogen any amounts for which we are held liable, we may be temporarily required to bear these losses ourselves. Each of these risks could negatively affect our business, operating results, financial condition and cash flows.
Risks Related to Our Common Stock
The market price for our common stock may fluctuate widely.
The market price of our shares of common stock may fluctuate widely, depending upon many factors, some of which are beyond our control, including the following:
|
· |
our quarterly or annual earnings, or those of other comparable companies; |
|
· |
actual or anticipated fluctuations in our operating results; |
|
· |
changes in accounting standards, policies, guidance, interpretations or principles; |
|
· |
announcements by us or our competitors of significant investments, acquisitions or dispositions; |
|
· |
the failure of securities analysts to cover our shares of common stock after the distribution; |
|
· |
changes in earnings estimates by securities analysts or our ability to meet those estimates; |
|
· |
the operating and stock price performance of other comparable companies; |
|
· |
overall market fluctuations; and |
|
· |
general economic conditions. |
Stock markets in general often experience volatility that is unrelated to the operating performance of a particular company. These broad market fluctuations may adversely affect the trading price of our shares of common stock. You may not be able to resell your shares of common stock following periods of volatility because of the market’s adverse reaction to volatility. When the market price of a company’s common stock drops significantly, stockholders often institute securities class action lawsuits against the company. This type of lawsuit against us could cause us to incur substantial costs and could divert the time and attention of management and other resources.
Future sales or distributions of our common stock could cause the market price of shares of our common stock to decline.
As a result of the separation, Biogen distributed approximately 108 million shares of our common stock to its stockholders, all of which are freely tradeable without restriction or further registration under the U.S. Securities Act of 1933, as amended (the Securities Act), unless the shares are owned by one of our affiliates, as that term is defined in Rule 405 of the Securities Act. The sale of significant amounts of our shares or the perception in the market that this
44
will occur may result in the lowering of the market price of our shares. We can offer no assurance that Biogen’s stockholders will continue to hold the shares they received in the distribution.
Your percentage ownership in the company may be diluted in the future.
In the future, your percentage ownership in the company may be diluted because of equity issuances for acquisitions, capital market transactions or otherwise, including equity awards that we grant to our directors, officers and employees. Such awards will have a dilutive effect on our earnings per share, which could adversely affect the market price of our common stock. From time to time, we issue stock options or other share‑based awards to employees under our employee benefits plans.
In addition, our amended and restated certificate of incorporation authorizes us to issue, without the approval of stockholders, one or more classes or series of preferred stock having such designation, powers, preferences and relative, participating, optional and other special rights, including preferences over the company’s common stock respecting dividends and distributions, as the board of directors generally may determine. The terms of one or more classes or series of preferred stock could dilute the voting power or reduce the value of our common stock. For example, we could grant the holders of preferred stock the right to elect some number of directors in all events or on the happening of specified events or the right to veto specified transactions. Similarly, the repurchase or redemption rights or liquidation preferences we could assign to holders of preferred stock could affect the residual value of the common stock.
The public announcement of data from clinical studies or news of any developments related to our or our competitors’ products or pipeline may cause significant volatility in our stock price.
As we continue to evolve as a standalone company, we will be focusing efforts and resources on further commercializing our existing products, as well as building a diversified pipeline of products into areas of unmet medical need. We expect that investors may place heightened scrutiny on some of our products in development when making investment decisions in the company compared to how such product developments relating to our business were previously viewed by investors when such programs part of the larger Biogen. The announcement of data from clinical studies by us or our collaborators or news of any developments related to our or our competitors’ products or pipeline may cause significant volatility in our stock price. Furthermore, the announcement of any negative or unexpected data or the discontinuation of development of any of our key pipeline product candidates, or any delay in anticipated timelines for filing for regulatory approval, could cause our stock price to decline significantly.
We do not expect to declare any dividends in the foreseeable future.
We do not anticipate declaring any cash dividends to holders of our common stock in the foreseeable future. Consequently, stockholders must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on the value of their shares of our common stock.
Provisions contained in our amended and restated certificate of incorporation and amended and restated bylaws, as well as provisions of Delaware law, could impair a takeover attempt.
Our amended and restated certificate of incorporation and amended and restated bylaws contain certain provisions that could have the effect of rendering more difficult or discouraging an acquisition deemed undesirable by our board of directors. For example, our corporate governance documents include provisions:
|
· |
authorizing blank check preferred stock, which could be issued with voting, liquidation, dividend and other rights superior to our common stock; |
|
· |
limiting the liability of, and providing indemnification to, our directors and officers; |
|
· |
limiting the ability of our stockholders to call and bring business before special meetings; |
|
· |
eliminating the ability of our stockholders to act by written consent in lieu of a meeting; |
|
· |
requiring advance notice of stockholder proposals for business to be conducted at meetings of our stockholders and for nominations of candidates for election to our board of directors; and |
45
|
· |
limiting the determination of the number of directors on our board of directors and the filling of newly created seats on the board to our board of directors then in office. |
These provisions, alone or together, could delay hostile takeovers and changes in control of our company or changes in our management.
As a Delaware corporation, we are also subject to provisions of Delaware law, including Section 203 of the Delaware General Corporation Law (DGCL), which prevents some stockholders holding more than 15% of our outstanding common stock from engaging in certain business combinations without approval of the holders of substantially all of our outstanding common stock. Any provision of our amended and restated certificate of incorporation or amended and restated bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock, and could also affect the price that some investors are willing to pay for our common stock.
In addition, an acquisition or further issuance of our stock could trigger the application of Section 355(e) of the Code. Under the tax matters agreement, we would be required to indemnify Biogen for any resulting taxes, and this indemnity obligation might discourage, delay or prevent a change of control that our stockholders may consider favorable.
Our amended and restated certificate of incorporation designates the state courts of the State of Delaware, or, if no state court located in the State of Delaware has jurisdiction, the federal court for the District of Delaware, as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could discourage lawsuits against us and our directors and officers.
Our amended and restated certificate of incorporation provides that unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware, to the fullest extent permitted by law, will be the sole and exclusive forum for:
|
· |
any derivative action or proceeding brought on behalf of us; |
|
· |
any action asserting a claim for breach of a fiduciary duty owed by any of our directors, officers or other employees of the company to us or our stockholders; |
|
· |
any action asserting a claim arising pursuant to any provision of the DGCL; or |
|
· |
any action asserting a claim governed by the internal affairs doctrine under Delaware state corporate law. |
This exclusive forum provision may limit the ability of our stockholders to bring a claim in a judicial forum that such stockholders find favorable for disputes with us or our directors or officers, which may discourage such lawsuits against the company and our directors and officers. Alternatively, if a court outside of Delaware were to find this exclusive forum provision inapplicable to, or unenforceable in respect of, one or more of the specified types of actions or proceedings described above, we may incur additional costs associated with resolving such matters in other jurisdictions, which could adversely affect our business, operating results or financial condition.
The exhibits listed on the Exhibit Index immediately preceding such exhibits, which is incorporated herein by reference, are filed or furnished as part of this Quarterly Report on Form 10-Q.
46
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
BIOVERATIV INC. |
|
|
|
|
|
|
Date: May 11, 2017 |
By: |
/s/ JOHN T. GREENE |
|
|
|
John T. Greene |
|
|
|
Executive Vice President, Chief Financial Officer (principal financial officer) |
EXHIBIT INDEX
|
Exhibit |
|
Exhibit Description |
|
|
|
|
| 31.1 |
+ |
Certification of the Chief Executive Officer pursuant to Section 302 of the Sarbanes‑Oxley Act of 2002 |
| 31.2 |
+ |
Certification of the Chief Financial Officer pursuant to Section 302 of the Sarbanes‑Oxley Act of 2002 |
| 32.1 |
++ |
Certification of the Chief Executive Officer and Chief Financial Officer pursuant to Section 906 of the Sarbanes‑Oxley Act of 2002 |
|
101.INS |
+ |
XBRL Instance Document |
|
101.SCH |
+ |
XBRL Taxonomy Extension Schema |
|
101.CAL |
+ |
XBRL Taxonomy Extension Calculation Linkbase |
|
101.DEF |
+ |
XBRL Taxonomy Extension Definition Linkbase |
|
101.LAB |
+ |
XBRL Taxonomy Extension Label Linkbase |
|
101.PRE |
+ |
XBRL Taxonomy Extension Presentation Linkbase |
+ Filed herewith
++ Furnished herewith
47