Attached files
| file | filename |
|---|---|
| EX-32.2 - EX-32.2 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex322_8.htm |
| EX-32.1 - EX-32.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex321_9.htm |
| EX-31.2 - EX-31.2 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex312_10.htm |
| EX-31.1 - EX-31.1 - ALNYLAM PHARMACEUTICALS, INC. | alny-ex311_11.htm |
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
|
☒ |
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2017
OR
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission File Number 001-36407
ALNYLAM PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
|
Delaware |
|
77-0602661 |
|
(State or Other Jurisdiction of Incorporation or Organization) |
|
(I.R.S. Employer Identification No.) |
|
300 Third Street, Cambridge, MA |
|
02142 |
|
(Address of Principal Executive Offices) |
|
(Zip Code) |
(617) 551-8200
(Registrant’s Telephone Number, Including Area Code)
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer |
|
☒ |
|
Accelerated filer |
|
☐ |
|
|
|
|
|
|||
|
Non-accelerated filer |
|
☐ (Do not check if a smaller reporting company) |
|
Smaller reporting company |
|
☐ |
|
|
|
|
|
|
|
|
|
Emerging growth company |
|
☐ |
|
|
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
At April 28, 2017, the registrant had 86,190,192 shares of Common Stock, $0.01 par value per share, outstanding.
|
|
|
PAGE NUMBER |
|
|
|
|
|
PART I. FINANCIAL INFORMATION |
|
|
|
|
|
|
|
ITEM 1. FINANCIAL STATEMENTS (Unaudited) |
|
|
|
|
|
|
|
CONDENSED CONSOLIDATED BALANCE SHEETS AS OF MARCH 31, 2017 AND DECEMBER 31, 2016 |
|
2 |
|
|
3 |
|
|
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS FOR THE THREE MONTHS ENDED MARCH 31, 2017 AND 2016 |
|
4 |
|
|
5 |
|
|
|
|
|
|
ITEM 2. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
|
15 |
|
|
|
|
|
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
|
22 |
|
|
|
|
|
|
23 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
24 |
|
|
|
|
|
|
|
24 |
|
|
|
|
|
|
|
47 |
|
|
|
|
|
|
|
48 |
|
1
CONDENSED CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share amounts)
(Unaudited)
|
|
|
March 31, 2017 |
|
|
December 31, 2016 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
171,079 |
|
|
$ |
193,617 |
|
|
Marketable securities |
|
|
385,843 |
|
|
|
424,185 |
|
|
Investment in equity securities of Regulus Therapeutics Inc. |
|
|
2,682 |
|
|
|
8,997 |
|
|
Billed and unbilled collaboration receivables |
|
|
19,457 |
|
|
|
23,334 |
|
|
Prepaid expenses and other current assets |
|
|
18,127 |
|
|
|
21,744 |
|
|
Total current assets |
|
|
597,188 |
|
|
|
671,877 |
|
|
Marketable securities |
|
|
255,305 |
|
|
|
324,799 |
|
|
Property, plant and equipment, net |
|
|
129,962 |
|
|
|
114,572 |
|
|
Restricted investments |
|
|
150,000 |
|
|
|
150,000 |
|
|
Other assets |
|
|
1,471 |
|
|
|
1,562 |
|
|
Total assets |
|
$ |
1,133,926 |
|
|
$ |
1,262,810 |
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Accounts payable |
|
$ |
25,371 |
|
|
$ |
54,465 |
|
|
Accrued expenses |
|
|
31,842 |
|
|
|
42,118 |
|
|
Deferred rent |
|
|
1,818 |
|
|
|
1,576 |
|
|
Deferred revenue |
|
|
36,755 |
|
|
|
33,540 |
|
|
Total current liabilities |
|
|
95,786 |
|
|
|
131,699 |
|
|
Deferred rent, net of current portion |
|
|
8,006 |
|
|
|
8,431 |
|
|
Deferred revenue, net of current portion |
|
|
46,049 |
|
|
|
49,392 |
|
|
Long-term debt |
|
|
150,000 |
|
|
|
150,000 |
|
|
Other liabilities |
|
|
3,184 |
|
|
|
3,067 |
|
|
Total liabilities |
|
|
303,025 |
|
|
|
342,589 |
|
|
Commitments and contingencies (Note 5) |
|
|
|
|
|
|
|
|
|
Stockholders’ equity: |
|
|
|
|
|
|
|
|
|
Preferred stock, $0.01 par value per share, 5,000,000 shares authorized and no shares issued and outstanding at March 31, 2017 and December 31, 2016 |
|
|
— |
|
|
— |
|
|
|
Common stock, $0.01 par value per share, 125,000,000 shares authorized; 86,068,086 shares issued and outstanding at March 31, 2017; 85,941,344 shares issued and outstanding at December 31, 2016 |
|
|
861 |
|
|
|
859 |
|
|
Additional paid-in capital |
|
|
2,627,969 |
|
|
|
2,609,614 |
|
|
Accumulated other comprehensive loss |
|
|
(33,828 |
) |
|
|
(33,441 |
) |
|
Accumulated deficit |
|
|
(1,764,101 |
) |
|
|
(1,656,811 |
) |
|
Total stockholders’ equity |
|
|
830,901 |
|
|
|
920,221 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
1,133,926 |
|
|
$ |
1,262,810 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
2
CONDENSED CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands, except per share amounts)
(Unaudited)
|
|
|
Three Months Ended March 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net revenues from collaborators |
|
$ |
18,960 |
|
|
$ |
7,345 |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development (1) |
|
|
86,984 |
|
|
|
96,273 |
|
|
General and administrative (1) |
|
|
38,487 |
|
|
|
21,100 |
|
|
Total operating expenses |
|
|
125,471 |
|
|
|
117,373 |
|
|
Loss from operations |
|
|
(106,511 |
) |
|
|
(110,028 |
) |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
Interest income |
|
|
2,128 |
|
|
|
1,813 |
|
|
Other (expense) income |
|
|
(2,907 |
) |
|
|
5,241 |
|
|
Total other (expense) income |
|
|
(779 |
) |
|
|
7,054 |
|
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(102,974 |
) |
|
Net loss per common share - basic and diluted |
|
$ |
(1.25 |
) |
|
$ |
(1.21 |
) |
|
Weighted-average common shares used to compute basic and diluted net loss per common share |
|
|
86,027 |
|
|
|
85,277 |
|
|
Comprehensive loss: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(102,974 |
) |
|
Unrealized loss on marketable securities, net of tax |
|
|
(1,936 |
) |
|
|
(8,224 |
) |
|
Reclassification adjustment for realized loss (gain) on marketable securities included in net loss |
|
|
1,549 |
|
|
|
(5,156 |
) |
|
Comprehensive loss |
|
$ |
(107,677 |
) |
|
$ |
(116,354 |
) |
|
(1) |
Non-cash stock-based compensation expenses included in operating expenses are as follows: |
|
Research and development |
|
$ |
8,691 |
|
|
$ |
14,356 |
|
|
General and administrative |
|
|
7,026 |
|
|
|
9,124 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
3
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
(Unaudited)
|
|
|
Three Months Ended March 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(102,974 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
3,000 |
|
|
|
4,601 |
|
|
Non-cash stock-based compensation |
|
|
15,717 |
|
|
|
23,480 |
|
|
Charge for 401(k) company stock match |
|
|
551 |
|
|
|
331 |
|
|
Realized loss (gain) on sale of marketable equity securities |
|
|
1,549 |
|
|
|
(5,156 |
) |
|
Other |
|
|
608 |
|
|
|
— |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Billed and unbilled collaboration receivables |
|
|
3,877 |
|
|
|
(516 |
) |
|
Prepaid expenses and other assets |
|
|
3,708 |
|
|
|
(2,010 |
) |
|
Accounts payable |
|
|
(11,067 |
) |
|
|
347 |
|
|
Accrued expenses and other |
|
|
(10,855 |
) |
|
|
(4,305 |
) |
|
Deferred revenue |
|
|
(128 |
) |
|
|
2,579 |
|
|
Net cash used in operating activities |
|
|
(100,330 |
) |
|
|
(83,623 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
Purchases of property, plant and equipment |
|
|
(36,152 |
) |
|
|
(2,759 |
) |
|
Purchases of marketable securities |
|
|
(86,803 |
) |
|
|
(164,561 |
) |
|
Sales and maturities of marketable securities |
|
|
198,031 |
|
|
|
294,317 |
|
|
Deposit for manufacturing facility |
|
|
— |
|
|
|
(9,057 |
) |
|
Net cash provided by investing activities |
|
|
75,076 |
|
|
|
117,940 |
|
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
Proceeds from exercise of stock options and other types of equity |
|
|
2,769 |
|
|
|
1,824 |
|
|
Proceeds from issuance of common stock to Sanofi Genzyme |
|
|
— |
|
|
|
14,301 |
|
|
Payments for repurchase of common stock for employee tax withholding |
|
|
(53 |
) |
|
|
(59 |
) |
|
Net cash provided by financing activities |
|
|
2,716 |
|
|
|
16,066 |
|
|
Net (decrease) increase in cash and cash equivalents |
|
|
(22,538 |
) |
|
|
50,383 |
|
|
Cash and cash equivalents, beginning of period |
|
|
193,617 |
|
|
|
180,895 |
|
|
Cash and cash equivalents, end of period |
|
$ |
171,079 |
|
|
$ |
231,278 |
|
The accompanying notes are an integral part of these condensed consolidated financial statements.
4
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(Unaudited)
1. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Principles of Consolidation
The accompanying condensed consolidated financial statements of Alnylam Pharmaceuticals, Inc. are unaudited and have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP, applicable to interim periods and, in the opinion of management, include all normal and recurring adjustments that are necessary to state fairly the results of operations for the reported periods. Our condensed consolidated financial statements have also been prepared on a basis substantially consistent with, and should be read in conjunction with, our audited consolidated financial statements for the year ended December 31, 2016, which were included in our Annual Report on Form 10-K that was filed with the Securities and Exchange Commission, or SEC, on February 15, 2017. The year-end condensed consolidated balance sheet data was derived from our audited financial statements, but does not include all disclosures required by GAAP. The results of our operations for any interim period are not necessarily indicative of the results of our operations for any other interim period or for a full fiscal year.
The accompanying condensed consolidated financial statements reflect the operations of Alnylam and our wholly-owned subsidiaries. All intercompany accounts and transactions have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Liquidity
Based on our current operating plan, we believe that our cash, cash equivalents and fixed income marketable securities at March 31, 2017, together with the cash we expect to generate under our current alliances, will be sufficient to enable us to advance our Alnylam 2020 strategy for at least the next 12 months from the filing of this Quarterly Report on Form 10-Q.
Net Loss Per Common Share
We compute basic net loss per common share by dividing net loss by the weighted-average number of common shares outstanding. We compute diluted net loss per common share by dividing net loss by the weighted-average number of common shares and dilutive potential common share equivalents then outstanding. Potential common shares consist of shares issuable upon the exercise of stock options (the proceeds of which are then assumed to have been repurchased using the treasury stock method). Because the inclusion of potential common shares would be anti-dilutive for all periods presented, diluted net loss per common share is the same as basic net loss per common share.
The following table sets forth for the periods presented the potential common shares (prior to consideration of the treasury stock method) excluded from the calculation of net loss per common share because their inclusion would be anti-dilutive, in thousands:
|
|
|
At March 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Options to purchase common stock |
|
|
12,048 |
|
|
|
10,183 |
|
|
Unvested restricted common stock |
|
|
163 |
|
|
|
185 |
|
|
|
|
|
12,211 |
|
|
|
10,368 |
|
Equity
Total stockholders’ equity at March 31, 2017 decreased $89.3 million compared to December 31, 2016. This decrease was related primarily to our net loss, partially offset during the three months ended March 31, 2017 by increases to additional paid-in capital due to stock-based compensation.
5
The fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In general, fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities. Fair values determined by Level 2 inputs utilize data points that are observable, such as quoted prices (adjusted), interest rates and yield curves. Fair values determined by Level 3 inputs utilize unobservable data points for the asset or liability, and include situations where there is little, if any, market activity for the asset or liability. The fair value hierarchy level is determined by the lowest level of significant input.
Investments in Marketable Securities and Cash Equivalents
We invest our excess cash balances in short-term and long-term marketable debt and equity securities. We classify our investments in marketable debt securities as either held-to-maturity or available-for-sale based on facts and circumstances present at the time we purchased the securities. At each balance sheet date presented, we classified all of our investments in debt and equity securities as available-for-sale. We report available-for-sale investments at fair value at each balance sheet date and include any unrealized holding gains and losses (the adjustment to fair value) in accumulated other comprehensive income (loss), a component of stockholders’ equity. At March 31, 2017, the balance in our accumulated other comprehensive loss was composed solely of activity related to our available-for-sale marketable securities, including our investment in equity securities of Regulus Therapeutics Inc., or Regulus. Realized gains and losses are determined using the specific identification method and are included in other income (expense). We recognized $1.5 million of realized losses and $5.2 million of realized gains from sales of our Regulus available-for-sale securities as other income (expense) in our condensed consolidated statements of comprehensive loss during the three months ended March 31, 2017 and 2016, respectively. If any adjustment to arrive at fair value reflects a decline in the value of the investment, we consider all available evidence to evaluate the extent to which the decline is “other than temporary,” including our intention to sell and, if so, record a charge to our condensed consolidated statements of comprehensive loss. We did not record any impairment charges related to our fixed income marketable securities during the three months ended March 31, 2017 or 2016. Our marketable securities are classified as cash equivalents if the original maturity, from the date of purchase, is 90 days or less, and as marketable securities if the original maturity, from the date of purchase, is in excess of 90 days. Our cash equivalents are composed of commercial paper, corporate notes and money market funds.
We account for our investment in Regulus as an available-for-sale marketable security. Intraperiod tax allocation rules require us to allocate our provision for income taxes between continuing operations and other categories of earnings, such as other comprehensive income. In periods in which we have a year-to-date pre-tax loss from continuing operations and pre-tax income in other categories of earnings, such as other comprehensive income, we must allocate the tax provision to the other categories of earnings. We then record a related tax benefit in continuing operations. Upon sales of our available-for-sale marketable securities, we apply the aggregate portfolio approach to recognize the related tax provision or benefit into income (loss) from continuing operations. As a result, the disproportionate tax effect remains in accumulated other comprehensive income (loss) as long as we maintain an investment portfolio.
Recent Accounting Pronouncements
In May 2014, the Financial Accounting Standards Board, or FASB, issued a new revenue recognition standard which amends revenue recognition principles and provides a single, comprehensive set of criteria for revenue recognition within and across all industries. The new standard provides a five step framework whereby revenue is recognized when promised goods or services are transferred to a customer at an amount that reflects the consideration to which the entity expects to be entitled in exchange for those goods or services. The standard also requires enhanced disclosures pertaining to revenue recognition in both interim and annual periods. In August 2015, the FASB deferred the effective date of the new revenue standard from January 1, 2017 to January 1, 2018. In March 2016, the FASB issued amendments to clarify the implementation guidance on principal versus agent considerations. In April 2016, the FASB issued amendments to clarify the guidance on accounting for licenses of intellectual property and identifying performance obligations. In May 2016, the FASB issued amendments related to collectibility, non-cash consideration, the presentation of sales and other similar taxes collected from customers and transition. The standard allows for adoption using a full retrospective method or a modified retrospective method. We are currently evaluating the method of our adoption as well as the expected impact that the standard could have on our condensed consolidated financial statements and related disclosures.
In January 2016, the FASB issued new guidance on recognition and measurement of financial assets and financial liabilities. The new guidance will impact the accounting for equity investments, financial liabilities under the fair value option, and the presentation and disclosure requirements for financial instruments. All equity investments in unconsolidated entities (other than those accounted for under the equity method of accounting) will generally be measured at fair value with changes in fair value recognized through earnings. There will no longer be an available-for-sale classification (changes in fair value reported in other comprehensive income (loss)) for equity securities with readily determinable fair values. In addition, the FASB clarified the need for a valuation allowance on deferred tax assets resulting from unrealized losses on available-for-sale debt securities. In general, the new guidance
6
will require modified retrospective application to all outstanding instruments, with a cumulative effect adjustment recorded to opening retained earnings. This guidance will be effective for us on January 1, 2018. We are currently evaluating the expected impact that the standard could have on our condensed consolidated financial statements and related disclosures.
In February 2016, the FASB issued a new leasing standard that requires that all lessees recognize the assets and liabilities that arise from leases on the condensed consolidated balance sheet and disclose qualitative and quantitative information about its leasing arrangements. The new standard will be effective for us on January 1, 2019. Early adoption is permitted. We are currently evaluating the timing of our adoption and the expected impact that this standard could have on our consolidated financial statements and related disclosures.
In October 2016, the FASB issued guidance that an entity should recognize the income tax consequences of an intra-entity transfer of an asset other than inventory when the transfer occurs instead of deferring the income tax effects. The new standard will be effective for us on a modified retrospective basis on January 1, 2018. We are currently evaluating the expected impact that this standard could have on our condensed consolidated financial statements and related disclosures.
In November 2016, the FASB issued guidance that requires restricted cash and restricted cash equivalents be included with cash and cash equivalents when reconciling the total beginning and ending amounts for the periods shown on the statement of cash flows. The new standard will be effective for us on January 1, 2018 using a retrospective transition method to each period presented. Early adoption is permitted. We are currently evaluating the timing of our adoption and the expected impact that this standard could have on our condensed consolidated financial statements and related disclosures.
In March 2017, the FASB issued guidance that amends the amortization period for certain purchased callable debt securities held at a premium by shortening the amortization period for the premium to the earliest call date. The new standard will be effective for us on January 1, 2019. Early adoption is permitted. We are currently evaluating the timing of our adoption and the expected impact that this standard could have on our condensed consolidated financial statements and related disclosures.
2. COLLABORATION AGREEMENTS
The following table summarizes our total consolidated net revenues from collaborators, for the periods indicated, in thousands:
|
|
|
Three Months Ended March 31, |
|
|||||
|
Description |
|
2017 |
|
|
2016 |
|
||
|
Sanofi Genzyme |
|
$ |
12,277 |
|
|
$ |
4,415 |
|
|
The Medicines Company |
|
|
6,364 |
|
|
|
2,657 |
|
|
Other |
|
|
319 |
|
|
|
273 |
|
|
Total net revenues from collaborators |
|
$ |
18,960 |
|
|
$ |
7,345 |
|
Product Alliances
Sanofi Genzyme Collaboration
In January 2014, we entered into a global, strategic collaboration with Sanofi Genzyme, the specialty care global business unit of Sanofi, to discover, develop and commercialize RNA interference, or RNAi, therapeutics as Genetic Medicines to treat orphan diseases. The 2014 Sanofi Genzyme collaboration superseded and replaced the previous collaboration between us and Sanofi Genzyme entered into in October 2012 to develop and commercialize RNAi therapeutics targeting transthyretin, or TTR, for the treatment of hereditary ATTR amyloidosis, including patisiran and revusiran, in Japan and the Asia-Pacific region.
2012 Sanofi Genzyme Agreement
Under the 2012 Sanofi Genzyme agreement, Sanofi Genzyme paid us an upfront cash payment of $22.5 million. We were also entitled to receive certain milestone payments under the 2012 Sanofi Genzyme agreement. In the fourth quarter of 2013, we earned a milestone of $7.0 million based upon the completion of a successful patisiran Phase 2 clinical trial and a milestone of $4.0 million based upon the initiation of the APOLLO Phase 3 clinical trial for patisiran.
We determined that the deliverables under the 2012 Sanofi Genzyme agreement included the license, a joint steering committee and any additional TTR-specific RNAi therapeutic compounds that comprised the ALN-TTR program. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and undelivered joint steering committee and any additional TTR-specific RNAi therapeutic compounds did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme had the ability to grant sublicenses, it could not sublicense all or substantially all of its rights under the 2012 Sanofi Genzyme agreement. The uniqueness of our services and the
7
limited sublicense right were indicators that standalone value was not present in the arrangement. Therefore the deliverables were not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. We were unable to reasonably estimate the period of performance under the 2012 Sanofi Genzyme agreement, as we were unable to estimate the timeline of our deliverables related to the deliverable for any additional TTR-specific RNAi therapeutic compounds. Through December 31, 2013, we had deferred all revenue, or $33.5 million, under the 2012 Sanofi Genzyme agreement.
2014 Sanofi Genzyme Collaboration
In January 2014, we entered into the 2014 Sanofi Genzyme collaboration. As noted above, the 2014 Sanofi Genzyme collaboration superseded and replaced the 2012 Sanofi Genzyme agreement.
The 2014 Sanofi Genzyme collaboration is structured as an exclusive relationship for the worldwide development and commercialization of RNAi therapeutics in the field of Genetic Medicines, which includes our current and future Genetic Medicine programs that reach Human Proof-of-Principle Study Completion (as defined in the Sanofi Genzyme master agreement), or Human POP, by the end of 2019, subject to extension to the end of 2021 in various circumstances. We will retain product rights in the United States, Canada and Western Europe, referred to as the Alnylam Territory, while Sanofi Genzyme will obtain exclusive rights to develop and commercialize collaboration products in the rest of the world, referred to as the Sanofi Genzyme Territory, together with certain broader co-development/co-commercialize or worldwide rights for certain products. Sanofi Genzyme’s rights, described in detail below, are structured as an opt-in that is triggered upon achievement of Human POP. We maintain development control for all programs prior to Sanofi Genzyme’s opt-in and maintain development and commercialization control after Sanofi Genzyme’s opt-in for all programs in the Alnylam Territory. We will retain global rights to any RNAi therapeutic Genetic Medicine program that does not reach Human POP by the end of 2019, subject to certain limited exceptions. We retain full rights to all current and future RNAi therapeutic programs outside of the field of Genetic Medicines, including the right to form new collaborations.
Under the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme’s specific license rights and the programs into which Sanofi Genzyme has opted include the following:
|
|
• |
Regional license terms and programs — Upon opt-in, we will retain product rights in the Alnylam Territory, while Sanofi Genzyme will obtain exclusive rights to develop and commercialize the product in the Sanofi Genzyme Territory. Sanofi Genzyme can elect this license for any of our current and future Genetic Medicine programs that complete Human POP by the end of 2019, subject to limited extension. Development costs for products once Sanofi Genzyme exercises an option will be shared between Sanofi Genzyme and us, with Sanofi Genzyme responsible for twenty percent of the global development costs. Upon the effective date of the 2014 Sanofi Genzyme collaboration, Sanofi Genzyme expanded the scope of its regional license and collaboration for patisiran, an investigational RNAi therapeutic currently in Phase 3 clinical development, which was originally established under the 2012 Sanofi Genzyme agreement. In September 2015, Sanofi Genzyme elected to opt into our fitusiran clinical development program for the treatment of hemophilia and other rare bleeding disorders under the regional license terms. Cost-sharing for the fitusiran program began in January 2016 under the regional license terms. Sanofi Genzyme also had the right to elect to co-develop and co-commercialize fitusiran in the Alnylam Territory pursuant to the co-development/co-commercialize license terms described below. In November 2016, Sanofi Genzyme exercised this right and elected to co-develop and co-commercialize fitusiran in the Alnylam Territory. In addition, during 2016, Sanofi Genzyme elected not to opt into the development and commercialization of givosiran or ALN-CC5 in the Sanofi Genzyme Territory. Sanofi Genzyme will be required to make payments totaling up to $50.0 million upon the achievement of certain patisiran development milestones. We could potentially earn the next patisiran milestone payment, ranging between $5.0 million and $20.0 million based on the geographic region, upon the achievement of specified events in connection with a regulatory filing or approval. In addition, Sanofi Genzyme will be required to make payments totaling up to $75.0 million per regional product other than patisiran, consisting of up to $55.0 million in development milestones and $20.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for each regional product based on annual net sales, if any, of such regional product by Sanofi Genzyme, its affiliates and sublicensees. |
8
|
|
the exercise of its co-development/co-commercialize rights for fitusiran, Sanofi Genzyme paid us approximately $6.0 million in January 2017 for its incremental share of co-development costs incurred from January 2016 through September 2016. Sanofi Genzyme will be required to make payments totaling up to $75.0 million in development milestones for fitusiran, and, prior to the discontinuation of the revusiran program, was required to make certain milestone payments for revusiran. In December 2014, we earned a development milestone payment of $25.0 million based upon the initiation of the first global Phase 3 clinical trial for revusiran. We could potentially earn the first fitusiran milestone payment of $25.0 million upon the initiation of the first global Phase 3 clinical trial of fitusiran. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for each co-development/co-commercialize product based on annual net sales, if any, in the Sanofi Genzyme Territory for such co-development/co-commercialize product by Sanofi Genzyme, its affiliates and sublicensees. The parties will share profits equally and we expect to book product sales in the Alnylam Territory. |
|
|
• |
Global license terms and programs — Upon opt-in, Sanofi Genzyme will obtain a worldwide license to develop and commercialize the product. Sanofi Genzyme had the right to elect a global license for givosiran, but instead elected a license to co-develop and co-commercialize fitusiran, as described above. Sanofi Genzyme continues to have one right to a global license through 2019, subject to limited extension, for a future Genetic Medicine program that was not one of our defined Genetic Medicine programs as of the effective date of the 2014 Sanofi Genzyme collaboration. Sanofi Genzyme shall be responsible for one hundred percent of global development costs. Sanofi Genzyme will be required to make payments totaling up to $200.0 million per global product, including up to $60.0 million in development milestones and $140.0 million in commercial milestones. Sanofi Genzyme will also be required to pay tiered double-digit royalties up to twenty percent for each global product based on annual net sales, if any, of each global product by Sanofi Genzyme, its affiliates and sublicensees. |
Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development, we may not receive any additional milestone payments or any royalty payments from Sanofi Genzyme under the 2014 Sanofi Genzyme collaboration.
Under the master agreement, the parties will collaborate in the development of option products, with us leading development for all programs prior to Sanofi Genzyme’s opt-in and also leading development and commercialization for all programs in the Alnylam Territory after Sanofi Genzyme’s opt-in. If Sanofi Genzyme does not exercise its option to license rights to a particular program, we will retain the exclusive right to develop and commercialize such program throughout the world, including the right to sublicense to third parties.
The 2014 Sanofi Genzyme collaboration is governed by an alliance joint steering committee that is comprised of an equal number of representatives from each party. There are additional committees to manage various aspects of each regional, co-developed/co-commercialized and global program. We and Sanofi Genzyme intend to enter into supply agreements to provide for supply of collaboration products to Sanofi Genzyme for clinical studies, and, at Sanofi Genzyme’s request, commercial sales. Sanofi Genzyme also has certain rights to manufacture collaboration products. Additionally, Sanofi Genzyme has certain limited opt-out rights, as specified in the master agreement, upon which products revert fully back to us with no further obligations to Sanofi Genzyme.
Upon the closing of the equity transaction in February 2014, we sold to Sanofi Genzyme 8,766,338 shares of our common stock and Sanofi Genzyme paid $700.0 million in aggregate cash consideration to us. As a condition to the closing of the equity transaction, Sanofi Genzyme entered into an investor agreement with us containing provisions regarding Sanofi Genzyme’s holding and “standstill” obligations, additional purchase, voting and registration rights, as well as certain other rights and obligations of the parties.
We recorded the issuance of 8,766,338 shares of our common stock under the stock purchase agreement using the price of our common stock on the date the shares were issued to Sanofi Genzyme. Based on the common stock price of $85.72, the fair value of the shares issued was $751.5 million, which was $51.5 million in excess of the proceeds received from Sanofi Genzyme for the issuance of our common stock. This $51.5 million is being amortized on a straight-line basis over the performance period. In addition, due to intraperiod tax allocation rules, upon closing of the equity transaction we recorded a benefit from income taxes of $15.2 million due to the Sanofi Genzyme equity purchase being recorded in additional paid-in capital, net of tax.
In accordance with the investor agreement, as a result of our issuance of shares in connection with our acquisition of Sirna Therapeutics, Inc., or Sirna, in March 2014, Sanofi Genzyme exercised its right to purchase an additional 344,448 shares of our common stock for $23.0 million. In addition, in January 2015, in connection with our public offering, Sanofi Genzyme exercised its right to purchase directly from us, in concurrent private placements, 744,566 shares of common stock at the public offering price resulting in $70.7 million in proceeds to us. The sales of common stock to Sanofi Genzyme were not registered as part of the public offering, though they were consummated simultaneously with the public offering.
9
Sanofi Genzyme also has the right at the beginning of each year to purchase a number of shares of our common stock based on the number of shares we issued during the previous year for compensation-related purposes. Sanofi Genzyme exercised this right to purchase directly from us 196,251 shares of our common stock on January 22, 2015 for $18.3 million and 205,030 shares of our common stock on February 1, 2016 for $14.3 million. In January 2017, Sanofi Genzyme elected not to exercise its compensation-related right for 2016. The sales of these shares to Sanofi Genzyme were consummated as private placements.
In each instance, the purchase by Sanofi Genzyme described above allowed Sanofi Genzyme to maintain its ownership level of our common stock of approximately 12 percent.
We determined that the deliverables for the programs on which Sanofi Genzyme was collaborating with us upon initiation of the 2014 Sanofi Genzyme collaboration included the licenses to our patisiran and revusiran clinical programs, which licenses were delivered to Sanofi Genzyme upon the closing date of the transaction, and the associated development activities, joint steering committee participation and information exchange for these clinical programs. We also determined that, pursuant to the accounting guidance governing revenue recognition on multiple element arrangements, the license and associated undelivered development activities, joint steering committee participation and information exchange activities did not have standalone value due to the specialized nature of the services to be provided by us. In addition, while Sanofi Genzyme has the ability to grant sublicenses, it cannot sublicense all or substantially all of its rights under the 2014 Sanofi Genzyme collaboration. The uniqueness of our services and the limited sublicense rights are indicators that standalone value is not present in the arrangement. Therefore the deliverables are not separable and, accordingly, the license and undelivered services were treated as a single unit of accounting. When multiple deliverables are accounted for as a single unit of accounting, we base our revenue recognition model on the final deliverable. Under the 2014 Sanofi Genzyme collaboration, the last deliverables for patisiran and revusiran were expected to be completed within approximately six years from the closing date of the transaction and the last deliverables for fitusiran are expected to be completed within approximately five years from the date Sanofi Genzyme elected to opt into our fitusiran clinical development program under the regional license terms. Our estimate regarding the performance period under the 2014 Sanofi Genzyme collaboration related to the license to our patisiran and revusiran clinical programs was adjusted in October 2016 due to our decision to discontinue development of revusiran. As a result, with respect to these programs, we currently expect the last deliverables to be completed within approximately five years from the closing date of the transaction.
We determined that the total cash received from Sanofi Genzyme under the now superseded 2012 Sanofi Genzyme agreement reflects consideration for certain of the performance obligations for ALN-TTR programs included in the 2014 Sanofi Genzyme collaboration. Therefore we are recognizing the $33.5 million of deferred revenue under the 2012 Sanofi Genzyme agreement on a straight-line basis over the period of performance of the ALN-TTR programs. As consideration is achieved, including any milestones or reimbursement for development activities, we recognize as revenue a portion of these payments equal to the percentage of the performance period completed when the milestone or activities have been satisfied, multiplied by the amount of the payment. We recognize the remaining portion of consideration received over the remaining performance period on a straight-line basis.
The following table presents information related to the 2014 Sanofi Genzyme collaboration, in thousands:
|
Excess of fair value of our common stock issued to Sanofi Genzyme in February 2014 |
|
$ |
(51,450 |
) |
|
Deferred revenue remaining under the 2012 Sanofi Genzyme agreement upon execution of the 2014 Sanofi Genzyme collaboration |
|
|
33,500 |
|
|
Milestone payment received: |
|
|
|
|
|
Year-ended December 31, 2014 |
|
|
25,000 |
|
|
Expense reimbursement from Sanofi Genzyme: |
|
|
|
|
|
Year-ended December 31, 2015 |
|
|
33,949 |
|
|
Year-ended December 31, 2016 |
|
|
54,337 |
|
|
Quarter-ended March 31, 2017 |
|
|
13,012 |
|
|
Total consideration at March 31, 2017 |
|
$ |
108,348 |
|
|
|
|
|
|
|
|
Cumulative revenue recognized at March 31, 2017 |
|
$ |
55,666 |
|
|
Deferred revenue at March 31, 2017 |
|
$ |
52,682 |
|
We determined that the opt-in rights that Sanofi Genzyme has for future Genetic Medicine programs represent separate and additional deliverables that Sanofi Genzyme may receive from us in future periods. Upon each initial opt-in by Sanofi Genzyme, we have determined that each program and the related activities will represent a single unit of accounting and, consistent with our accounting policies, we will base our revenue recognition period on the final deliverable associated with each future opt-in.
10
The following tables present information about our assets that are measured at fair value on a recurring basis at March 31, 2017 and December 31, 2016, and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value, in thousands:
|
Description |
|
At March 31, 2017 |
|
|
Quoted Prices in Active Markets (Level 1) |
|
|
Significant Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
||||
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Commercial paper |
|
$ |
4,990 |
|
|
$ |
— |
|
|
$ |
4,990 |
|
|
$ |
— |
|
|
Corporate notes |
|
|
625 |
|
|
|
— |
|
|
|
625 |
|
|
|
— |
|
|
Money market funds |
|
|
88,835 |
|
|
|
88,835 |
|
|
|
— |
|
|
|
— |
|
|
Marketable securities (fixed income): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Certificates of deposit |
|
|
16,900 |
|
|
|
— |
|
|
|
16,900 |
|
|
|
— |
|
|
Commercial paper |
|
|
92,791 |
|
|
|
— |
|
|
|
92,791 |
|
|
|
— |
|
|
Corporate notes |
|
|
210,119 |
|
|
|
— |
|
|
|
210,119 |
|
|
|
— |
|
|
U.S. government-sponsored enterprise securities |
|
|
281,359 |
|
|
|
— |
|
|
|
281,359 |
|
|
|
— |
|
|
U.S. treasury securities |
|
|
39,979 |
|
|
|
— |
|
|
|
39,979 |
|
|
|
— |
|
|
Marketable securities (Regulus equity holdings) |
|
|
2,682 |
|
|
|
2,682 |
|
|
|
— |
|
|
|
— |
|
|
Restricted cash (money market funds) |
|
|
1,471 |
|
|
|
1,471 |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
739,751 |
|
|
$ |
92,988 |
|
|
$ |
646,763 |
|
|
$ |
— |
|
|
Description |
|
At December 31, 2016 |
|
|
Quoted Prices in Active Markets (Level 1) |
|
|
Significant Observable Inputs (Level 2) |
|
|
Significant Unobservable Inputs (Level 3) |
|
||||
|
Cash equivalents: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Commercial paper |
|
$ |
17,199 |
|
|
$ |
— |
|
|
$ |
17,199 |
|
|
$ |
— |
|
|
Money market funds |
|
|
151,479 |
|
|
|
151,479 |
|
|
|
— |
|
|
|
— |
|
|
Marketable securities (fixed income): |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Certificates of deposit |
|
|
17,999 |
|
|
|
— |
|
|
|
17,999 |
|
|
|
— |
|
|
Commercial paper |
|
|
59,340 |
|
|
|
— |
|
|
|
59,340 |
|
|
|
— |
|
|
Corporate notes |
|
|
333,872 |
|
|
|
— |
|
|
|
333,872 |
|
|
|
— |
|
|
U.S. government-sponsored enterprise securities |
|
|
297,773 |
|
|
|
— |
|
|
|
297,773 |
|
|
|
— |
|
|
U.S. treasury securities |
|
|
40,000 |
|
|
|
— |
|
|
|
40,000 |
|
|
|
— |
|
|
Marketable securities (Regulus equity holdings) |
|
|
8,997 |
|
|
|
8,997 |
|
|
|
— |
|
|
|
— |
|
|
Restricted cash (money market funds) |
|
|
1,471 |
|
|
|
1,471 |
|
|
|
— |
|
|
|
— |
|
|
Total |
|
$ |
928,130 |
|
|
$ |
161,947 |
|
|
$ |
766,183 |
|
|
$ |
— |
|
During the three months ended March 31, 2017, there were no transfers between Level 1 and Level 2 financial assets. The carrying amounts reflected in our condensed consolidated balance sheets for cash, billed and unbilled collaboration receivables, other current assets, accounts payable and accrued expenses approximate fair value due to their short-term maturities. The fair value of our long-term debt at March 31, 2017, computed pursuant to a discounted cash flow technique using a market interest rate, was $150.2 million and is considered a Level 3 fair value measurement. The effective interest rate reflects the current market rate.
11
The following tables summarize the fair value, accumulated other comprehensive income (loss) and intraperiod tax allocation regarding our investment in Regulus available-for-sale marketable securities at March 31, 2017 and 2016, and for the activity recorded for the three months ended March 31, 2017 and 2016, in thousands:
|
Description |
|
At December 31, 2016 |
|
|
Sales of Regulus Shares During Three Months Ended March 31, 2017 |
|
|
All Other Activity During Three Months Ended March 31, 2017 |
|
|
Balance at March 31, 2017 |
|
||||
|
Carrying value |
|
$ |
8,093 |
|
|
$ |
(4,803 |
) |
|
$ |
(608 |
) |
|
$ |
2,682 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
|
904 |
|
|
|
1,549 |
|
|
|
(2,453 |
) |
|
|
— |
|
|
Investment in equity securities of Regulus, as reported |
|
$ |
8,997 |
|
|
$ |
(3,254 |
) |
|
$ |
(3,061 |
) |
|
$ |
2,682 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
$ |
904 |
|
|
$ |
1,549 |
|
|
$ |
(2,453 |
) |
|
$ |
— |
|
|
Intraperiod tax allocation recorded as a benefit from income taxes |
|
|
(32,792 |
) |
|
|
— |
|
|
|
— |
|
|
|
(32,792 |
) |
|
Accumulated other comprehensive income (loss), net of tax |
|
$ |
(31,888 |
) |
|
$ |
1,549 |
|
|
$ |
(2,453 |
) |
|
$ |
(32,792 |
) |
|
Description |
|
At December 31, 2015 |
|
|
Sales of Regulus Shares During Three Months Ended March 31, 2016 |
|
|
All Other Activity During Three Months Ended March 31, 2016 |
|
|
Balance at March 31, 2016 |
|
||||
|
Carrying value |
|
$ |
11,935 |
|
|
$ |
(2,024 |
) |
|
$ |
— |
|
|
$ |
9,911 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
|
39,484 |
|
|
|
(5,156 |
) |
|
|
(10,305 |
) |
|
|
24,023 |
|
|
Investment in equity securities of Regulus, as reported |
|
$ |
51,419 |
|
|
$ |
(7,180 |
) |
|
$ |
(10,305 |
) |
|
$ |
33,934 |
|
|
Accumulated other comprehensive income (loss), before tax |
|
$ |
39,484 |
|
|
$ |
(5,156 |
) |
|
$ |
(10,305 |
) |
|
$ |
24,023 |
|
|
Intraperiod tax allocation recorded as a benefit from income taxes |
|
|
(32,792 |
) |
|
|
— |
|
|
|
— |
|
|
|
(32,792 |
) |
|
Accumulated other comprehensive income (loss), net of tax |
|
$ |
6,692 |
|
|
$ |
(5,156 |
) |
|
$ |
(10,305 |
) |
|
$ |
(8,769 |
) |
We obtain fair value measurement data for our marketable securities from independent pricing services. We perform validation procedures to ensure the reasonableness of this data. This includes meeting with the independent pricing services to understand the methods and data sources used. Additionally, we perform our own review of prices received from the independent pricing services by comparing these prices to other sources and confirming those securities are trading in active markets.
The following tables summarize our marketable securities, other than our holdings in Regulus noted above, at March 31, 2017 and December 31, 2016, in thousands:
|
|
|
At March 31, 2017 |
|
|||||||||||||
|
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
|
Certificates of deposit |
|
$ |
16,900 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
16,900 |
|
|
Commercial paper |
|
|
92,791 |
|
|
|
— |
|
|
|
— |
|
|
|
92,791 |
|
|
Corporate notes |
|
|
210,363 |
|
|
|
42 |
|
|
|
(286 |
) |
|
|
210,119 |
|
|
U.S. government-sponsored enterprise securities |
|
|
282,117 |
|
|
|
— |
|
|
|
(758 |
) |
|
|
281,359 |
|
|
U.S. treasury securities |
|
|
40,013 |
|
|
|
— |
|
|
|
(34 |
) |
|
|
39,979 |
|
|
Total |
|
$ |
642,184 |
|
|
$ |
42 |
|
|
$ |
(1,078 |
) |
|
$ |
641,148 |
|
12
|
|
|
At December 31, 2016 |
|
|||||||||||||
|
|
|
Amortized Cost |
|
|
Gross Unrealized Gains |
|
|
Gross Unrealized Losses |
|
|
Fair Value |
|
||||
|
Certificates of deposit |
|
$ |
17,999 |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
17,999 |
|
|
Commercial paper |
|
|
59,340 |
|
|
|
— |
|
|
|
— |
|
|
|
59,340 |
|
|
Corporate notes |
|
|
334,266 |
|
|
|
47 |
|
|
|
(441 |
) |
|
|
333,872 |
|
|
U.S. government-sponsored enterprise securities |
|
|
298,910 |
|
|
|
9 |
|
|
|
(1,146 |
) |
|
|
297,773 |
|
|
U.S. treasury securities |
|
|
40,022 |
|
|
|
1 |
|
|
|
(23 |
) |
|
|
40,000 |
|
|
Total |
|
$ |
750,537 |
|
|
$ |
57 |
|
|
$ |
(1,610 |
) |
|
$ |
748,984 |
|
We classify our debt security investments based on their contractual maturity dates. The following table summarizes our available-for-sale debt securities by contractual maturity, at March 31, 2017, in thousands:
|
|
|
At March 31, 2017 |
|
|||||
|
|
|
Amortized Cost |
|
|
Fair Value |
|
||
|
Less than one year |
|
$ |
386,115 |
|
|
$ |
385,843 |
|
|
Greater than one year but less than two years |
|
|
256,069 |
|
|
|
255,305 |
|
|
Total |
|
$ |
642,184 |
|
|
$ |
641,148 |
|
5. COMMITMENTS AND CONTINGENCIES
Manufacturing Facility
In February 2016, we entered into an agreement with 20 Commerce LLC to purchase 12 acres of undeveloped land in Norton, Massachusetts. We completed the purchase and closed this transaction on April 4, 2016. We are constructing a manufacturing facility at this site for drug substance, including small interfering RNAs, or siRNAs, and siRNA conjugates, for clinical and commercial use. At March 31, 2017 and December 31, 2016, property, plant and equipment, net, on our condensed consolidated balance sheets reflects $88.6 million and $73.2 million, respectively, of land and associated costs related to the construction of our drug substance manufacturing facility.
Credit Agreements
On April 29, 2016, we entered into (i) a Credit Agreement, or the BOA Credit Agreement, with Alnylam U.S., Inc., our wholly-owned subsidiary, as the borrower, us, as a guarantor, and Bank of America N.A., or BOA, as the lender and (ii) a Credit Agreement, or the Wells Credit Agreement, together with the BOA Credit Agreement, the Credit Agreements, by and among Alnylam U.S., Inc., as the borrower, us, as a guarantor, and Wells Fargo Bank, National Association, or Wells, as the lender. The Credit Agreements were entered into in connection with the planned build out of our new drug substance manufacturing facility.
The BOA Credit Agreement and the Wells Credit Agreement provide for a $120.0 million and a $30.0 million term loan facility, respectively, and mature on April 29, 2021. The proceeds of the borrowing under each of the BOA Credit Agreement and the Wells Credit Agreement are to be used for working capital and general corporate purposes. Interest on borrowings under the Credit Agreements is calculated based on LIBOR plus 0.45 percent, except in the event of default. The borrower may prepay loans under each of the BOA Credit Agreement and the Wells Credit Agreement at any time, without premium or penalty, subject to certain notice requirements and LIBOR breakage costs.
The obligations of the borrower under each Credit Agreement are guaranteed by us. The obligations of the borrower and us under each Credit Agreement are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of all term loans outstanding under such Credit Agreement at such time. At each of March 31, 2017 and December 31, 2016, we have recorded $150.0 million of cash collateral in connection with the Credit Agreements as restricted investments on our condensed consolidated balance sheets.
Each Credit Agreement contains limited representations and warranties and limited affirmative and negative covenants, including quarterly reporting obligations, as well as certain customary events of default.
During the three months ended March 31, 2017, we recorded $0.4 million of interest expense related to the Credit Agreements that is reflected in other income (expense) on our condensed consolidated statements of comprehensive loss.
13
University of Utah Litigation
On March 22, 2011, The University of Utah, or Utah, filed a civil complaint in the United States District Court for the District of Massachusetts, or the MA District Court, against us, Max Planck Gesellschaft Zur Foerderung Der Wissenschaften e.V. and Max Planck Innovation GmbH, together, Max Planck, the Whitehead Institute for Biomedical Research, or Whitehead, the Massachusetts Institute of Technology, or MIT, and the University of Massachusetts, or UMass, claiming a professor at Utah is the sole inventor or, in the alternative, a joint inventor, of the Tuschl patents. Utah was seeking changes to the inventorship of the Tuschl patents, unspecified damages and other relief. After several years of court proceedings and discovery, on September 28, 2015, the MA District Court granted both of our motions for summary judgment, finding that there was no collaboration between Dr. Bass and Dr. Tuschl, which is a pre-requisite for co-inventorship, and dismissing Utah’s state law damages claims as well.
On October 28, 2015, Utah filed a notice of appeal to the United States Court of Appeals for the Federal Circuit, or the CAFC. On December 18, 2015, the CAFC entered an order dismissing Utah’s appeal following a joint motion filed by us and Utah seeking dismissal of the appeal with prejudice. This disposed of Utah’s inventorship claims and its state law claims for damages. On October 14, 2015, we filed a motion with the MA District Court seeking reimbursement of costs and fees associated with defending this action in the amount of approximately $8.0 million. On November 30, 2015, the MA District Court denied our motion and on December 15, 2015, we filed a notice of appeal of this ruling with the CAFC. Oral arguments on our appeal were heard at the CAFC on January 12, 2017. On March 23, 2017, the CAFC denied our appeal and we have decided not to appeal this ruling any further.
Dicerna Litigation
On June 10, 2015, we filed a trade secret misappropriation lawsuit against Dicerna Pharmaceuticals, Inc., or Dicerna, in the Superior Court of Middlesex County, Massachusetts, seeking to stop misappropriation by Dicerna of our confidential, proprietary and trade secret information related to the RNAi assets we purchased from Merck, including certain GalNAc conjugate technology. In addition to permanent injunctive relief, we are also seeking monetary damages from Dicerna. On July 10, 2015, Dicerna filed its answer to our complaint, in which it denied our claims, along with initial discovery requests, to which we responded in a timely fashion. On July 27, 2015, Dicerna filed a motion seeking removal of the case to the Business Litigation Session of the Superior Court of Suffolk County, which we opposed. On August 31, 2015, the Court denied Dicerna’s motion. We and Dicerna agreed to a protective order, which was entered by the Court on November 12, 2015. Discovery is ongoing and we now expect fact discovery to close in June 2017.
Although we believe we have meritorious claims in this matter, litigation is subject to inherent uncertainty and a court could ultimately rule against us. In addition, litigation and related matters are costly and may divert the attention of our management and other resources that would otherwise be engaged in other activities.
Our accounting policy for accrual of legal costs is to recognize such expenses as incurred.
14
ITEM 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS.
This Quarterly Report on Form 10-Q contains forward-looking statements that involve risks and uncertainties. The statements contained in this Quarterly Report on Form 10-Q that are not purely historical are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended and Section 21E of the Securities Exchange Act of 1934, as amended. Without limiting the foregoing, the words “may,” “will,” “should,” “could,” “expects,” “plans,” “intends,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue,” “target,” “goal” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these words. All forward-looking statements included in this Quarterly Report on Form 10-Q are based on information available to us up to, and including, the date of this document, and we expressly disclaim any obligation to update any such forward-looking statements to reflect events or circumstances that arise after the date hereof. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain important factors, including those set forth in this Item 2 — “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as under Part II, Item 1A — “Risk Factors” and elsewhere in this Quarterly Report on Form 10-Q. You should carefully review those factors and also carefully review the risks outlined in other documents that we file from time to time with the Securities and Exchange Commission, or SEC.
Overview
We are a biopharmaceutical company developing novel therapeutics based on RNA interference, or RNAi. RNAi is a naturally occurring biological pathway within cells for selectively silencing and regulating the expression of specific genes. Since many diseases are caused by the inappropriate activity of specific genes, the ability to silence genes selectively through RNAi could provide a new way to treat a wide range of human diseases. We believe that drugs that work through RNAi have the potential to become a broad new class of innovative medicines, and that this potential new drug class is similar to the opportunity created with other major biological discoveries such as recombinant DNA and monoclonal antibodies. Using our intellectual property and expertise, we are developing what we believe to be a reproducible and modular platform to develop RNAi therapeutics for a variety of human diseases.
Our research and development strategy is focused primarily on the use of our proprietary N-acetylgalactosamine, or GalNAc-conjugate platform for delivery of small interfering RNAs, or “siRNAs” — the molecules that mediate RNAi — toward genetically validated, liver-expressed target genes involved in the cause or pathway of human diseases. We are also focused on clinical indications where there are high unmet needs, early biomarkers for the assessment of clinical activity in Phase 1 clinical studies, and a definable path for drug development, regulatory approval, patient access and commercialization.
Specifically, our broad pipeline of investigational RNAi therapeutics is focused in three Strategic Therapeutic Areas, or “STArs:” Genetic Medicines, with multiple product candidates for the treatment of rare diseases; Cardio-Metabolic Diseases, with product candidates directed toward genetically validated, liver-expressed disease targets for unmet needs in cardiovascular and metabolic diseases; and Hepatic Infectious Diseases, with product candidates designed to address the major global health challenges of hepatic infectious diseases, beginning with hepatitis B and hepatitis D viral infections. We are focused on advancement of our Alnylam 2020 strategy for the development and commercialization of RNAi therapeutics as a potential new class of innovative medicines. Specifically, our goal is to achieve, by the end of 2020, a company profile with three marketed products and ten RNAi therapeutic clinical programs, including four in late stages of development, across our three STArs. Our most advanced investigational RNAi therapeutic in development, patisiran, targets the transthyretin, or TTR, gene for the treatment of patients with polyneuropathy due to hereditary TTR-mediated amyloidosis, or hATTR amyloidosis. We expect to report top-line data from our ongoing APOLLO Phase 3 study of patisiran in mid-2017. Assuming that the APOLLO data are positive, we plan to submit our first new drug application, or NDA, and marketing authorization application, or MAA, for patisiran by the end of 2017. We expect to advance additional investigational RNAi therapeutics into Phase 3 development during 2017, including fitusiran, for the treatment of hemophilia and rare bleeding disorders, and givosiran, for the treatment of acute hepatic porphyrias.
Based on our expertise in RNAi therapeutics and broad intellectual property estate, we have formed alliances with leading pharmaceutical and life sciences companies to support our development and commercialization efforts, including Sanofi Genzyme, the specialty care global business unit of Sanofi, and The Medicines Company, or MDCO. We have incurred significant losses since we commenced operations in 2002 and expect such losses to continue for the foreseeable future. At March 31, 2017, we had an accumulated deficit of $1.76 billion. Historically, we have generated losses principally from costs associated with research and development activities, acquiring, filing and expanding intellectual property rights and general administrative costs. As a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the establishment of late stage clinical and commercial capabilities, including European operations, the construction of our drug substance manufacturing facility, continued management and growth of our patent portfolio, collaborations and general corporate activities, we expect to incur additional operating losses for the foreseeable future. We also anticipate that our operating results will fluctuate for the foreseeable future. Therefore, period-to-period comparisons should not be relied upon as predictive of the results in future periods.
15
Although we currently have multiple clinical development programs, we are unable to predict when, if ever, we will successfully develop or be able to commence sales of any product. A substantial portion of our total net revenues in recent years has been derived from collaboration revenues from strategic alliances with Takeda, Monsanto, Sanofi Genzyme and MDCO. We expect our sources of potential funding for the next several years to be derived primarily from existing and new strategic alliances, which may include license and other fees, funded research and development and milestone payments, and proceeds from the sale of equity or debt.
Research and Development
Since our inception, we have focused on drug discovery and development programs. Research and development expenses represent a substantial percentage of our total operating expenses, as reflected by our broad pipeline of product development programs.
The following is a summary of our product development programs as of April 30, 2017, that identifies those programs in which we have achieved human proof of concept, or POC, by demonstrating target gene knockdown and/or additional evidence of activity in clinical studies, the development stage of our programs and our commercial rights to such programs:
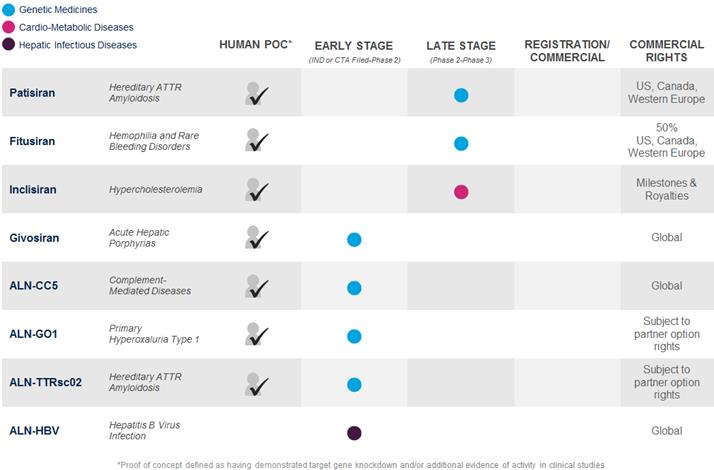
During the first quarter of 2017 and recent period, we reported the following updates from our late-stage clinical programs:
|
|
• |
We continued to advance patisiran, presenting final 24-month data from our Phase 2 open-label extension, or OLE, study in April 2017. We expect to report top-line results from our APOLLO Phase 3 clinical study of patisiran in mid-2017. |
|
|
• |
We continued to advance fitusiran, presenting positive new data from our Phase 1 clinical trial in February 2017. |
16
|
|
support for the development of medicines for diseases where there is an unmet medical need and where early clinical data show potential to benefit patients. |
|
|
• |
Our partner, MDCO, announced positive final results from its ORION-1 Phase 2 clinical study of inclisiran, an investigational RNAi therapeutic for the treatment of hypercholesterolemia, in March 2017. |
|
|
o |
MDCO also initiated its ORION-2 study of inclisiran in patients with Homozygous Familial Hypercholesterolemia, as well as its ORION-3 study, a Phase 2 open-label cross-over extension study for patients completing the ORION-1 study. |
|
|
o |
In addition, we and MDCO announced agreement with the United States Food and Drug Administration, or FDA, on the Phase 3 clinical program for inclisiran. |
There is a risk that any drug discovery or development program may not produce revenue for a variety of reasons, including the possibility that we will not be able to adequately demonstrate the safety and effectiveness of the product candidate. For example, in October 2016, we announced the discontinuation of our revusiran clinical development program due to safety concerns. Moreover, there are uncertainties specific to any new field of drug discovery, including RNAi. The success of any product candidate we develop is highly uncertain. Due to the numerous risks associated with developing drugs, we cannot reasonably estimate or know the nature, timing and estimated costs of the efforts necessary to complete the development of, or the period, if any, in which material net cash inflows will commence from, any potential product candidate.
Any failure to complete any stage of the development of any potential products in a timely manner could have a material adverse effect on our operations, financial position and liquidity. A discussion of some of the risks and uncertainties associated with completing our projects on schedule, or at all, and the potential consequences of failing to do so, are set forth in Part II, Item 1A below under the heading “Risk Factors.”
Strategic Alliances
Our business strategy is to develop and commercialize a broad pipeline of RNAi therapeutic products directed towards our three STArs. As part of this strategy, we have entered into, and expect to enter into additional, collaboration and licensing agreements as a means of obtaining resources, capabilities and funding to advance our investigational RNAi therapeutic programs.
Our collaboration strategy is to form alliances that create significant value for us and our collaborators in the advancement of RNAi therapeutics as a new class of innovative medicines. Specifically, with respect to our Genetic Medicine pipeline, we formed a broad strategic alliance with Sanofi Genzyme in 2014 pursuant to which we retain development and commercial rights for our current and future Genetic Medicine products in the United States, Canada and Western Europe, and Sanofi Genzyme will develop and commercialize our current and future Genetic Medicine products in the rest of the world, referred to as the Sanofi Genzyme Territory, subject to certain broader rights. In the case of patisiran, we are advancing the product in the United States, Canada and Western Europe, while Sanofi Genzyme will advance the product in the Sanofi Genzyme Territory. In the case of fitusiran, Sanofi Genzyme has elected to opt in to co-develop and co-commercialize fitusiran in the United States, Canada and Western Europe, in addition to developing and commercializing fitusiran in the Sanofi Genzyme Territory. With respect to our Cardio-Metabolic and Hepatic Infectious Disease pipelines, we intend to seek future strategic alliances for these programs, while retaining significant product development and commercialization rights. We currently have a global alliance with MDCO for the development and commercialization of our inclisiran program.
Intellectual Property
The strength of our intellectual property portfolio relating to the development and commercialization of siRNAs as therapeutics is essential to our business strategy. We own or license issued patents and pending patent applications in the United States and in key markets around the world claiming fundamental features of siRNAs and RNAi therapeutics as well as those claiming crucial chemical modifications and promising delivery technologies. Specifically, we have a portfolio of patents, patent applications and other intellectual property covering: fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms; chemical modifications to siRNAs that improve their suitability for therapeutic and other uses; siRNAs directed to specific targets as treatments for particular diseases; delivery technologies, such as in the fields of carbohydrate conjugates and cationic liposomes; and all aspects of our specific development candidates.
We believe that no other company possesses a portfolio of such broad and exclusive rights to the patents and patent applications required for the commercialization of RNAi therapeutics. Our intellectual property estate for RNAi therapeutics includes over 3,400 active cases and over 1,400 granted or issued patents, of which over 500 are issued or granted in the United States, the European Union, or EU, including by the European Patent Office, or EPO, and Japan. We continue to seek to grow our portfolio through the creation of new technology in this field. In addition, we are very active in our evaluation of third-party technologies.
17
Given the importance of our intellectual property portfolio to our business operations, we intend to vigorously enforce our rights and defend against challenges that have arisen or may arise in this area.
Critical Accounting Policies and Estimates
There have been no significant changes to our critical accounting policies since the beginning of this fiscal year. Our critical accounting policies are described in the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section of our Annual Report on Form 10-K for the year ended December 31, 2016, which we filed with the SEC on February 15, 2017.
Results of Operations
The following data summarizes the results of our operations for the periods indicated, in thousands:
|
|
|
Three Months Ended March 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net revenues from collaborators |
|
$ |
18,960 |
|
|
$ |
7,345 |
|
|
Operating expenses |
|
|
125,471 |
|
|
|
117,373 |
|
|
Loss from operations |
|
|
(106,511 |
) |
|
|
(110,028 |
) |
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(102,974 |
) |
Discussion of Results of Operations
Net revenues from collaborators
We generate revenues through research and development collaborations. The following table summarizes our total consolidated net revenues from collaborators, for the periods indicated, in thousands, together with the changes, in thousands:
|
|
|
Three Months Ended March 31, |
|
|
|
|
|
|||||
|
Description |
|
2017 |
|
|
2016 |
|
|
Dollar Change |
|
|||
|
Sanofi Genzyme |
|
$ |
12,277 |
|
|
$ |
4,415 |
|
|
$ |
7,862 |
|
|
MDCO |
|
|
6,364 |
|
|
|
2,657 |
|
|
|
3,707 |
|
|
Other |
|
|
319 |
|
|
|
273 |
|
|
|
46 |
|
|
Total net revenues from collaborators |
|
$ |
18,960 |
|
|
$ |
7,345 |
|
|
$ |
11,615 |
|
Net revenues from collaborators increased during the three months ended March 31, 2017 as compared to the three months ended March 31, 2016 due primarily to services performed by us in connection with our clinical development programs for which Sanofi Genzyme has opted-in. In addition, net revenues from collaborators increased during the first quarter of 2017 due to additional reimbursable activities under our MDCO agreement.
We expect net revenues from collaborators to continue to increase on a quarterly basis during the remainder of 2017 due primarily to an expected growth in revenues from Sanofi Genzyme related to increased reimbursement, as well as the potential receipt of milestone payments under our agreements with Sanofi Genzyme and MDCO. The receipt and timing of any milestone payments will be dependent on the progress of our and MDCO’s clinical development programs.
We had $82.8 million and $82.9 million of deferred revenue at March 31, 2017 and December 31, 2016, respectively, which consists primarily of payments we have received from collaborators, including Sanofi Genzyme, MDCO, Kyowa Hakko Kirin Co., Ltd. and Monsanto Company, but have not yet recognized pursuant to our revenue recognition policies.
For the foreseeable future, we expect our revenues to be derived primarily from our alliances with Sanofi Genzyme and MDCO, as well as other strategic alliances and potential new collaborations and licensing activities.
18
The following table summarizes our operating expenses for the periods indicated, in thousands and as a percentage of total operating expenses, together with the changes, in thousands:
|
|
|
Three Months Ended March 31, |
|
|
% of Total Operating |
|
|
Three Months Ended March 31, |
|
|
% of Total Operating |
|
|
Dollar |
|
|||||
|
Description |
|
2017 |
|
|
Expenses |
|
|
2016 |
|
|
Expenses |
|
|
Change |
|
|||||
|
Research and development |
|
$ |
86,984 |
|
|
|
69 |
% |
|
$ |
96,273 |
|
|
|
82 |
% |
|
$ |
(9,289 |
) |
|
General and administrative |
|
|
38,487 |
|
|
|
31 |
% |
|
|
21,100 |
|
|
|
18 |
% |
|
|
17,387 |
|
|
Total operating expenses |
|
$ |
125,471 |
|
|
|
100 |
% |
|
$ |
117,373 |
|
|
|
100 |
% |
|
$ |
8,098 |
|
Research and development. The following table summarizes the components of our research and development expenses for the periods indicated, in thousands and as a percentage of total research and development expenses, together with the changes, in thousands:
|
|
|
Three Months Ended March 31, |
|
|
% of Expense |
|
|
Three Months Ended March 31, |
|
|
% of Expense |
|
|
Dollar |
|
|||||
|
Description |
|
2017 |
|
|
Category |
|
|
2016 |
|
|
Category |
|
|
Change |
|
|||||
|
Research and development |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Compensation and related |
|
$ |
22,623 |
|
|
|
26 |
% |
|
$ |
21,070 |
|
|
|
22 |
% |
|
$ |
1,553 |
|
|
Manufacturing |
|
|
18,075 |
|
|
|
21 |
% |
|
|
15,387 |
|
|
|
16 |
% |
|
|
2,688 |
|
|
Clinical trial |
|
|
17,563 |
|
|
|
20 |
% |
|
|
20,257 |
|
|
|
21 |
% |
|
|
(2,694 |
) |
|
Non-cash stock-based compensation |
|
|
8,691 |
|
|
|
10 |
% |
|
|
14,356 |
|
|
|
15 |
% |
|
|
(5,665 |
) |
|
External services |
|
|
7,844 |
|
|
|
9 |
% |
|
|
12,992 |
|
|
|
13 |
% |
|
|
(5,148 |
) |
|
Facilities-related |
|
|
7,658 |
|
|
|
9 |
% |
|
|
7,049 |
|
|
|
7 |
% |
|
|
609 |
|
|
Lab supplies and materials |
|
|
2,146 |
|
|
|
2 |
% |
|
|
1,834 |
|
|
|
2 |
% |
|
|
312 |
|
|
Other |
|
|
2,384 |
|
|
|
3 |
% |
|
|
3,328 |
|
|
|
4 |
% |
|
|
(944 |
) |
|
Total research and development expenses |
|
$ |
86,984 |
|
|
|
100 |
% |
|
$ |
96,273 |
|
|
|
100 |
% |
|
$ |
(9,289 |
) |
Research and development expenses decreased during the three months ended March 31, 2017 as compared to the three months ended March 31, 2016 due in part to a decrease in non-cash stock-based compensation expense resulting from the vesting of certain performance-based stock option awards during the first quarter of 2016 upon completion of enrollment in our APOLLO Phase 3 clinical trial for patisiran. In addition, external services expenses decreased due to reduced pre-clinical activities. Clinical trial expenses also decreased as a result of our decision in October 2016 to discontinue development of revusiran. These decreases were partially offset by an increase in manufacturing expenses related to our late-stage clinical trials.
We expect to continue to devote a substantial portion of our resources to research and development expenses to support our goals for 2020. We expect that research and development expenses will remain relatively consistent with the first quarter during the remainder of 2017 as we continue to develop our pipeline and advance our product candidates into later-stage development, but expect that certain expenses will be variable depending on the timing of manufacturing batches, clinical trial enrollment and results, regulatory review of our product candidates and programs and non-cash stock-based compensation expenses due to the potential vesting of performance-based awards.
A significant portion of our research and development costs are not tracked by project as they benefit multiple projects or our technology platform. However, certain of our collaboration agreements contain cost-sharing arrangements pursuant to which certain costs incurred under the project are reimbursed. Costs reimbursed under these agreements typically include certain direct external costs and a negotiated full-time equivalent labor rate for the actual time worked on the project. As a result, although a significant portion of our research and development expenses are not tracked on a project-by-project basis, we do track direct external costs attributable to, and the actual time our employees worked on, our collaborations.
19
General and administrative. The following table summarizes the components of our general and administrative expenses for the periods indicated, in thousands and as a percentage of total general and administrative expenses, together with the changes, in thousands:
|
|
|
Three Months Ended March 31, |
|
|
% of Expense |
|
|
Three Months Ended March 31, |
|
|
% of Expense |
|
|
Dollar |
|
|||||
|
Description |
|
2017 |
|
|
Category |
|
|
2016 |
|
|
Category |
|
|
Change |
|
|||||
|
General and administrative |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Compensation and related |
|
$ |
13,245 |
|
|
|
34 |
% |
|
$ |
4,401 |
|
|
|
21 |
% |
|
$ |
8,844 |
|
|
Consulting and professional services |
|
|
13,132 |
|
|
|
34 |
% |
|
|
5,172 |
|
|
|
24 |
% |
|
|
7,960 |
|
|
Non-cash stock-based compensation |
|
|
7,026 |
|
|
|
18 |
% |
|
|
9,124 |
|
|
|
43 |
% |
|
|
(2,098 |
) |
|
Facilities-related |
|
|
2,213 |
|
|
|
6 |
% |
|
|
1,018 |
|
|
|
5 |
% |
|
|
1,195 |
|
|
Other |
|
|
2,871 |
|
|
|
8 |
% |
|
|
1,385 |
|
|
|
7 |
% |
|
|
1,486 |
|
|
Total general and administrative expenses |
|
$ |
38,487 |
|
|
|
100 |
% |
|
$ |
21,100 |
|
|
|
100 |
% |
|
$ |
17,387 |
|
General and administrative expenses increased during the three months ended March 31, 2017 as compared to the three months ended March 31, 2016 due primarily to an increase in commercial and medical affairs headcount to support corporate growth and to prepare for the potential launch of our first commercial product.
We expect that general and administrative expenses will increase on a quarterly basis during the remainder of 2017 as we continue to grow our operations, including the anticipated build-out of our commercial infrastructure, but expect that non-cash stock-based compensation expenses will be variable due to the potential vesting of performance-based awards.
Liquidity and Capital Resources
The following table summarizes our cash flow activities for the periods indicated, in thousands:
|
|
|
Three Months Ended March 31, |
|
|||||
|
|
|
2017 |
|
|
2016 |
|
||
|
Net loss |
|
$ |
(107,290 |
) |
|
$ |
(102,974 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities |
|
|
21,425 |
|
|
|
23,256 |
|
|
Changes in operating assets and liabilities |
|
|
(14,465 |
) |
|
|
(3,905 |
) |
|
Net cash used in operating activities |
|
|
(100,330 |
) |
|
|
(83,623 |
) |
|
Net cash provided by investing activities |
|
|
75,076 |
|
|
|
117,940 |
|
|
Net cash provided by financing activities |
|
|
2,716 |
|
|
|
16,066 |
|
|
Net (decrease) increase in cash and cash equivalents |
|
|
(22,538 |
) |
|
|
50,383 |
|
|
Cash and cash equivalents, beginning of period |
|
|
193,617 |
|
|
|
180,895 |
|
|
Cash and cash equivalents, end of period |
|
$ |
171,079 |
|
|
$ |
231,278 |
|
Since we commenced operations in 2002, we have generated significant losses. At March 31, 2017, we had an accumulated deficit of $1.76 billion. At March 31, 2017, we had cash, cash equivalents and fixed income marketable securities of $812.2 million, compared to $942.6 million at December 31, 2016, in each case excluding the $150.0 million of restricted investments related to our term loan agreements with Bank of America N.A., or BOA, and Wells Fargo Bank, National Association, or Wells, and our investment in equity securities of Regulus.
Sanofi Genzyme has certain rights to purchase additional shares from us under our investor agreement, including the right at the beginning of each year to purchase a number of shares of our common stock based on the number of shares we issued during the previous year for compensation-related purposes. Sanofi Genzyme exercised this right to purchase directly from us 205,030 shares of our common stock on February 1, 2016 for $14.3 million. This purchase allowed Sanofi Genzyme to maintain its ownership level of our outstanding common stock of approximately 12 percent. In January 2017, Sanofi Genzyme elected not to exercise its compensation-related right for 2016.
We invest primarily in money market funds, U.S. government-sponsored enterprise securities, U.S. treasury securities, high-grade corporate notes, certificates of deposit and commercial paper. Corporate notes may also include foreign bonds denominated in U.S. dollars. Our investment objectives are, primarily, to assure liquidity and preservation of capital and, secondarily, to obtain investment income. All of our investments in debt securities are recorded at fair value and are available-for-sale. Fair value is
20
determined based on quoted market prices and models using observable data inputs. We have not recorded any impairment charges to our fixed income marketable securities during the three months ended March 31, 2017 or 2016.
Operating activities
We have required significant amounts of cash to fund our operating activities as a result of net losses since our inception. Cash used in operating activities is adjusted for non-cash items to reconcile net loss to net cash used in operating activities. These non-cash adjustments have historically included stock-based compensation, intraperiod tax allocation, and depreciation and amortization.
We expect that we will require significant amounts of cash to fund our operating activities for the foreseeable future as we continue to execute on our Alnylam 2020 strategy through the advancement of our research, development, pre-commercial and potentially commercial initiatives. The actual amount of overall expenditures will depend on numerous factors, including the timing of expenses, the timing and terms of collaboration agreements or other strategic transactions, if any, and the timing and progress of our research, development and commercialization efforts.
The increase in net cash used in operating activities for the three months ended March 31, 2017 compared to the three months ended March 31, 2016 was primarily due to reductions in our operating assets and liabilities largely as a result of accounts payable and accrued expenses.
Investing activities
For the three months ended March 31, 2017 and 2016, net cash provided by investing activities was due primarily to activity related to our fixed income marketable securities in accordance with management of our liquidity needs. For the three months ended March 31, 2017, there were purchases of property, plant and equipment of $36.2 million primarily in connection with our construction of our drug substance manufacturing facility.
Financing activities
For the three months ended March 31, 2017, net cash of $2.7 million provided by financing activities was due primarily to the issuance of common stock in connection with stock option exercises. For the three months ended March 31, 2016, net cash of $16.1 million provided by financing activities was due primarily to proceeds of $14.3 million received from our issuance of common stock to Sanofi Genzyme in February 2016.
Operating Capital Requirements
We do not know when, if ever, we will successfully develop or be able to commence sales of any product. Therefore, we anticipate that we will continue to generate significant losses for the foreseeable future as a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the establishment of late stage clinical and commercial capabilities, including European operations, continued management and growth of our patent portfolio, collaborations and general corporate activities. In addition, we are expanding our manufacturing capabilities, including through construction of a drug substance manufacturing facility in Norton, Massachusetts. In April 2016, our subsidiary, Alnylam U.S., Inc., entered into an aggregate of $150.0 million in term loan agreements with BOA and Wells, for which we are the guarantor, related to the build out of our new drug substance manufacturing facility, that mature in April 2021. Interest on the borrowings is calculated based on LIBOR plus 0.45 percent. The obligations under the term loan agreements are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of all term loans outstanding under the agreements at such time.
Based on our current operating plan, we believe that our existing cash, cash equivalents and fixed income marketable securities, together with the cash we expect to generate under our current alliances, will be sufficient to enable us to advance our Alnylam 2020 strategy for at least the next several years. For reasons discussed below, we may require significant additional funds earlier than we currently expect in order to develop, conduct clinical trials for, manufacture and commercialize any product candidates.
In the future, we may seek additional funding through additional collaborative arrangements and public or private financings. Additional funding may not be available to us on acceptable terms or at all. For example, our decision in October 2016 to discontinue our revusiran clinical development program and the subsequent decline in our stock price may make it more difficult for us to obtain additional funding on acceptable terms. Moreover, the terms of any additional financing may further adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our existing stockholders will result. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. If we are unable to obtain funding on a timely basis, we may be required to significantly delay or curtail one or more of our research or development programs and our ability to achieve our goals for 2020 may
21
be delayed or diminished. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own.
Even if we are able to raise additional funds in a timely manner, our future capital requirements may vary from what we expect and will depend on many factors, including:
|
|
• |
our progress in demonstrating that siRNAs can be active as drugs and achieve desired clinical effects; |
|
|
• |
progress in our research and development programs, as well as what may be required by regulatory bodies to advance these programs; |
|
|
• |
the timing, receipt and amount of milestone and other payments, if any, from present and future collaborators, if any; |
|
|
• |
our ability to maintain and establish additional collaborative arrangements and/or new business initiatives; |
|
|
• |
the resources, time and costs required to successfully initiate and complete our pre-clinical and clinical trials, obtain regulatory approvals, prepare for commercialization of our product candidates and obtain and maintain licenses to third-party intellectual property; |
|
|
• |
our ability to establish, maintain and operate our own manufacturing facilities in a timely and cost effective manner; |
|
|
• |
our ability to manufacture, or contract with third-parties for the manufacture of, our product candidates for clinical testing and commercial sale; |
|
|
• |
the resources, time and cost required for the preparation, filing, prosecution, maintenance and enforcement of patent claims; |
|
|
• |
the costs associated with legal activities, including litigation, arising in the course of our business activities and our ability to prevail in any such legal disputes; and |
|
|
• |
the timing, receipt and amount of sales and royalties, if any, from our potential products. |
Contractual Obligations and Commitments
The disclosure of our contractual obligations and commitments is set forth under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Contractual Obligations” in our Annual Report on Form 10-K for the year ended December 31, 2016. There have been no material changes in our contractual obligations and commitments since December 31, 2016.
Recent Accounting Pronouncements
Please read Note 1 to our condensed consolidated financial statements included in Item 1, “Financial Statements (Unaudited),” of this Quarterly Report on Form 10-Q for a description of recent accounting pronouncements applicable to our business.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK.
As part of our investment portfolio, we own financial instruments that are sensitive to market risks. The investment portfolio is used to preserve our capital until it is required to fund operations, including our research and development activities. Our fixed income marketable securities consist primarily of U.S. government-sponsored enterprise securities, U.S. treasury securities, high-grade corporate notes, and commercial paper. Corporate notes may also include foreign bonds denominated in U.S. dollars. All of our investments in debt securities are classified as available-for-sale and are recorded at fair value. Our available-for-sale investments in debt securities are sensitive to changes in interest rates and changes in the credit ratings of the issuers. Interest rate changes would result in a change in the net fair value of these financial instruments due to the difference between the market interest rate and the market interest rate at the date of purchase of the financial instrument. If market interest rates were to increase immediately and uniformly by 50 basis points, or one-half of a percentage point, from levels at March 31, 2017, the net fair value of our interest-sensitive financial instruments would have resulted in a hypothetical decline of $2.2 million. We currently do not seek to hedge this exposure to fluctuations in interest rates. A downgrade in the credit rating of an issuer of a debt security or further deterioration of the credit markets could result in a decline in the fair value of the debt instruments. Our investment guidelines prohibit investment in auction rate securities and we do not believe we have any direct exposure to losses relating from mortgage-based securities or derivatives related thereto such as credit-default swaps. Historically, foreign currency fluctuations have not been material. We did not record any impairment charges to our fixed income marketable securities during the three months ended March 31, 2017.
22
ITEM 4. CONTROLS AND PROCEDURES.
Our management, with the participation of our chief executive officer (principal executive officer) and vice president of finance and treasurer (principal financial officer), evaluated the effectiveness of our disclosure controls and procedures as of March 31, 2017. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of March 31, 2017, our chief executive officer and vice president of finance and treasurer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
No change in our internal control over financial reporting (as defined in Rules 13a–15(f) and 15d–15(f) under the Exchange Act) occurred during the three months ended March 31, 2017 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
23
For a discussion of material pending legal proceedings, please read Note 5, Commitments and Contingencies – Litigation, to our condensed consolidated financial statements included in Part I, Item I, “Financial Statements (Unaudited),” of this quarterly report on Form 10-Q, which is incorporated into this item by reference.
Our business is subject to numerous risks. We caution you that the following important factors, among others, could cause our actual results to differ materially from those expressed in forward-looking statements made by us or on our behalf in filings with the SEC, press releases, communications with investors and oral statements. All statements other than statements relating to historical matters should be considered forward-looking statements. When used in this report, the words “believe,” “expect,” “plan,” “anticipate,” “estimate,” “predict,” “may,” “could,” “should,” “intend,” “will,” “target,” “goal” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these words. Any or all of our forward-looking statements in this quarterly report on Form 10-Q and in any other public statements we make may turn out to be wrong. They can be affected by inaccurate assumptions we might make or by known or unknown risks and uncertainties. Many factors mentioned in the discussion below will be important in determining future results. Consequently, no forward-looking statement can be guaranteed. Actual future results may vary materially from those anticipated in forward-looking statements. We explicitly disclaim any obligation to update any forward-looking statements to reflect events or circumstances that arise after the date hereof. You are advised, however, to consult any further disclosure we make in our reports filed with the SEC.
Risks Related to Our Business
Risks Related to Being a Clinical Stage Company
Although we have product candidates in late stage clinical development, there is limited information about our ability to successfully overcome many of the risks and uncertainties encountered by companies in the biopharmaceutical industry.
Although we have product candidates in late stage clinical development, we have limited experience and have not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical area. For example, to execute our business plan, we will need to successfully:
|
|
• |
execute product development activities using unproven technologies related to both RNAi and to the delivery of siRNAs to the relevant tissues and cells; |
|
|
• |
build and maintain a strong intellectual property portfolio; |
|
|
• |
gain regulatory acceptance for the development and commercialization of our product candidates and market success for any products we commercialize; |
|
|
• |
develop and maintain successful strategic alliances; and |
|
|
• |
manage our spending as costs and expenses increase due to clinical trials, regulatory approvals and commercialization. |
If we are unsuccessful in accomplishing these objectives, we may not be able to develop product candidates, commercialize products, raise capital, expand our business or continue our operations.
The approach we are taking to discover and develop novel RNAi therapeutics is unproven and may never lead to marketable products.
We have concentrated our efforts and therapeutic product research and development on RNAi technology and our future success depends on the successful development of this technology and products based on it. Neither we nor any other company has received regulatory approval to market therapeutics utilizing siRNAs, the class of molecule we are trying to develop into drugs. The scientific discoveries that form the basis for our efforts to discover and develop new drugs are relatively new. The scientific evidence to support the feasibility of developing drugs based on these discoveries is both early stage and limited. Skepticism as to the feasibility of developing RNAi therapeutics has been expressed in scientific literature. For example, there are potential challenges to achieving safe RNAi therapeutics based on the so-called off-target effects and activation of the interferon response. In addition, decisions by other companies with respect to their RNAi development efforts or their adoption of different or related technologies may increase skepticism in the marketplace regarding the potential for RNAi therapeutics.
24
Relatively few product candidates based on these discoveries have ever been tested in humans. siRNAs may not naturally possess the inherent properties typically required of drugs, such as the ability to be stable in the body, or the ability to enter cells within relevant tissues in order to exert their effects. We currently have limited data to suggest that we can introduce these properties into siRNAs. We have spent and expect to continue to spend large amounts of money trying to develop siRNAs that possess the properties typically required of drugs, and we may never succeed in doing so. In addition, these compounds may not demonstrate in patients the chemical and pharmacological properties ascribed to them in laboratory studies, and they may interact with human biological systems in unforeseen, ineffective or harmful ways. For example, in October 2016, we discontinued development of revusiran, an investigational RNAi therapeutic that was in development for the treatment of patients with cardiomyopathy due to hATTR amyloidosis, due to safety concerns, and are conducting a comprehensive evaluation of the revusiran data. We may never succeed in developing a marketable product, we may not become profitable and the value of our common stock will decline.
Further, our focus solely on RNAi technology for developing drugs, as opposed to multiple, more proven technologies for drug development, increases the risks associated with the ownership of our common stock. If we are not successful in developing a product candidate using RNAi technology, we may be required to change the scope and direction of our product development activities. In that case, we may not be able to identify and implement successfully an alternative product development strategy.
Risks Related to Our Financial Results and Need for Financing
We have a history of losses and may never become and remain consistently profitable.
We have experienced significant operating losses since our inception. At March 31, 2017, we had an accumulated deficit of $1.76 billion. To date, we have not received regulatory approval to market or sell any products nor generated any revenues from the sale of products. Further, we do not expect to generate any product revenues until at the earliest 2018, assuming we receive marketing approval for patisiran. We expect to continue to incur annual net operating losses over the next several years and will require substantial resources over the next several years as we expand our efforts to discover, develop and commercialize RNAi therapeutics. We anticipate that the majority of any revenues we generate over the next several years will be from alliances with pharmaceutical and biotechnology companies, but cannot be certain that we will be able to maintain our existing alliances or secure and maintain new alliances, or meet the obligations or achieve any milestones that we may be required to meet or achieve to receive payments. We anticipate that revenues derived from such sources will not be sufficient to make us consistently profitable.
We believe that to become and remain consistently profitable, we must succeed in discovering, developing and commercializing novel drugs with significant market potential. This will require us to be successful in a range of challenging activities, including pre-clinical testing and clinical trial stages of development, obtaining regulatory approval and reimbursement for these novel drugs and manufacturing, marketing and selling them. We may never succeed in these activities, and may never generate revenues that are significant enough to achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. If we cannot become and remain consistently profitable, the market price of our common stock could decline. In addition, we may be unable to raise capital, expand our business, develop additional product candidates or continue our operations.
We will require substantial additional funds to complete our research and development activities and if additional funds are not available, we may need to critically limit, significantly scale back or cease our operations.
We have used substantial funds to develop our RNAi technologies and will require substantial funds to conduct further research and development, including pre-clinical testing and clinical trials of our product candidates, and to manufacture, market and sell any products that are approved for commercial sale. Because we cannot be certain of the length of time or activities associated with successful development of our product candidates, we are unable to estimate the actual funds we will require to develop and commercialize them.
Our future capital requirements and the period for which we expect our existing resources to support our operations may vary from what we expect. We have based our expectations on a number of factors, many of which are difficult to predict or are outside of our control, including:
|
|
• |
our progress in demonstrating that siRNAs can be active as drugs and achieve desired clinical effects; |
|
|
• |
progress in our research and development programs, as well as what may be required by regulatory bodies to advance these programs; |
|
|
• |
the timing, receipt and amount of milestone and other payments, if any, from present and future collaborators, if any; |
|
|
• |
our ability to maintain and establish additional collaborative arrangements and/or new business initiatives; |
25
|
|
• |
our ability to establish, maintain and operate our own manufacturing facilities in a timely and cost effective manner; |
|
|
• |
our ability to manufacture, or contract with third parties for the manufacture of, our product candidates for clinical testing and commercial sale; |
|
|
• |
the resources, time and cost required for the preparation, filing, prosecution, maintenance and enforcement of patent claims; |
|
|
• |
the costs associated with legal activities, including litigation, arising in the course of our business activities and our ability to prevail in any such legal disputes; and |
|
|
• |
the timing, receipt and amount of sales and royalties, if any, from our potential products. |
If our estimates and predictions relating to these factors are incorrect, we may need to modify our operating plan.
Even if our estimates are correct, we will be required to seek additional funding in the future and intend to do so through either collaborative arrangements, public or private equity offerings or debt financings, or a combination of one or more of these funding sources. Additional funds may not be available to us on acceptable terms or at all. For example, our decision in October 2016 to discontinue development of revusiran and the subsequent decline in our stock price may make it more difficult for us to obtain additional funding on acceptable terms.
In April 2016, our subsidiary, Alnylam U.S., Inc., entered into an aggregate of $150.0 million in term loan agreements with BOA and Wells, for which we are the guarantor, related to the build out of our new drug substance manufacturing facility, that mature in April 2021. Interest on the borrowings is calculated based on LIBOR plus 0.45 percent. During an event of default under either agreement, the obligations under such agreement will bear interest at a rate per annum equal to the interest rate then in effect plus two percent. The obligations under the term loan agreements are secured by cash collateral in an amount equal to, at any given time, at least 100 percent of the principal amount of all term loans outstanding under the credit agreements at such time. The agreements include restrictive covenants that could limit our flexibility in conducting future business activities and further limit our ability to change the nature of our business and, in the event of insolvency, the lenders would be paid before holders of equity securities received any distribution of corporate assets. If an event of default occurs, the interest rate would increase and the lenders would be entitled to take various actions, including the acceleration of amounts due under the loan. Our ability to satisfy our obligations under these agreements and meet our debt service obligations will depend upon our future performance, which will be subject to financial, business and other factors affecting our operations, many of which are beyond our control.
In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities, further dilution to our existing stockholders will result. In addition, as a condition to providing additional funding to us, future investors may demand, and may be granted, rights superior to those of existing stockholders. Moreover, our investor agreement with Sanofi Genzyme provides Sanofi Genzyme with the right, subject to certain exceptions, generally to maintain its ownership position in us until Sanofi Genzyme owns less than 7.5 percent of our outstanding common stock, subject to certain additional limited rights of Sanofi Genzyme to maintain its ownership percentage. In accordance with the investor agreement, as a result of our issuance of shares in connection with our acquisition of Sirna in March 2014, Sanofi Genzyme exercised its right to purchase an additional 344,448 shares of our common stock. In January 2015, Sanofi Genzyme also exercised its right to purchase 196,251 shares based on its 2014 compensation-related right and its right to purchase 744,566 shares in connection with our public offering. In February 2016, Sanofi Genzyme purchased an additional 205,030 shares based on its 2015 compensation-related right. These purchases allowed Sanofi Genzyme to maintain its ownership level of approximately 12 percent of our outstanding common stock. While the exercise of these rights by Sanofi Genzyme has provided us with an additional $126.3 million in cash to date, and while any exercise of these rights by Sanofi Genzyme in the future will provide us with further additional cash, these exercises have caused, and any future exercise of these rights by Sanofi Genzyme will also cause further, dilution to our stockholders. In January 2017, Sanofi Genzyme elected not to exercise its compensation-related right for 2016. In November 2016, Sanofi Genzyme elected to expand its regional rights for fitusiran and opt-in to co-develop and co-commercialize fitusiran in the United States, Canada and Western Europe, in addition to developing and commercializing the product in the Sanofi Genzyme Territory. In connection with the exercise of this right, Sanofi Genzyme paid us in January 2017 for its incremental share of co-development costs incurred from January 2016 to September 2016, in accordance with the 2014 Sanofi Genzyme collaboration. Going forward, Sanofi Genzyme will share in 50 percent of certain development and sales and marketing costs for fitusiran, which will result in increased expense reimbursement to us.
If we are unable to obtain funding on a timely basis, we may be required to significantly delay or curtail one or more of our research or development programs or undergo future reductions in our workforce or other corporate restructuring activities, and our
26
ability to achieve our strategy for 2020 may be delayed or diminished. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own.
If the estimates we make, or the assumptions on which we rely, in preparing our condensed consolidated financial statements prove inaccurate, our actual results may vary from those reflected in our projections and accruals.
Our condensed consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these condensed consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
The investment of our cash, cash equivalents and fixed income marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
At March 31, 2017, we had $812.2 million in cash, cash equivalents and fixed income marketable securities, excluding our investment in equity securities of Regulus and the $150.0 million of restricted investments related to the term loan agreements with BOA and Wells. We historically have invested these amounts in high–grade corporate notes, commercial paper, securities issued or sponsored by the U.S. government, certificates of deposit and money market funds meeting the criteria of our investment policy, which is focused on the preservation of our capital. Corporate notes may also include foreign bonds denominated in U.S. dollars. These investments are subject to general credit, liquidity, market and interest rate risks. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our condensed consolidated financial statements. In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would suffer. The market risks associated with our investment portfolio may have an adverse effect on our results of operations, liquidity and financial condition.
Risks Related to Our Dependence on Third Parties
We may not be able to execute our business strategy if we are unable to enter into alliances with other companies that can provide business and scientific capabilities and funds for the development and commercialization of our product candidates. If we are unsuccessful in forming or maintaining these alliances on terms favorable to us, our business may not succeed.
We do not currently have any capability for sales or distribution and have early capability for marketing and market access, as well as limited capacity for drug development due to our growing pipeline of RNAi therapeutic opportunities. Accordingly, we have entered into alliances with other companies and collaborators that we believe can provide such capabilities in certain territories, and we intend to enter into additional such alliances in the future. Our collaboration strategy is to form alliances that create significant value for us and our collaborators in the advancement of RNAi therapeutics as a new class of innovative medicines. Specifically, with respect to our Genetic Medicine pipeline, we formed a broad strategic alliance with Sanofi Genzyme in 2014 pursuant to which we retain development and commercial rights for our current and future Genetic Medicine products in the United States, Canada and Western Europe, and Sanofi Genzyme has the right to develop and commercialize our current and future Genetic Medicine products principally in the rest of the world, subject to certain broader rights. With respect to our Cardio-Metabolic and Hepatic Infectious Disease pipelines, we intend to seek future strategic alliances for these programs, while retaining significant product development and commercialization rights. We currently have a global alliance with MDCO to advance inclisiran.
In such alliances, we expect our current, and may expect our future, collaborators to provide substantial capabilities in clinical development, regulatory affairs, and/or marketing, sales and distribution. Under certain of our alliances, we also may expect our collaborators to develop, market and/or sell certain of our product candidates. We may have limited or no control over the development, sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties. For example, we will rely entirely on (i) Sanofi Genzyme for the development and commercialization of patisiran, fitusiran and potentially other of our Genetic Medicine programs in territories outside of the United States, Canada and Western Europe, and (ii) MDCO for all future development and commercialization of inclisiran worldwide. If Sanofi Genzyme and/or MDCO are not successful in their commercialization efforts, our future revenues from RNAi therapeutics for these indications may be adversely affected. In addition, Sanofi Genzyme may elect not to opt into one or more of our Genetic Medicine programs. For example, during 2016, Sanofi Genzyme elected not to take a regional license for our givosiran and ALN-CC5 programs. While we intend to advance these programs independently, retaining global development and commercial rights, our ability to advance these programs and successfully develop and commercialize these product candidates may be adversely affected as a result of Sanofi Genzyme’s decision.
27
We may not be successful in entering into future alliances on terms favorable to us due to various factors, including our ability to successfully demonstrate proof of concept for our technology in humans, our ability to demonstrate the safety and efficacy of our specific drug candidates, our ability to manufacture or have third parties manufacture RNAi therapeutics, the strength of our intellectual property and/or concerns around challenges to our intellectual property. For example, our decision in October 2016 to discontinue development of revusiran could make it more difficult for us to attract collaborators due to concerns around the safety and/or efficacy of our technology platform or product candidates. Even if we do succeed in securing any such alliances, we may not be able to maintain them if, for example, development or approval of a product candidate is delayed, challenges are raised as to the validity or scope of our intellectual property, we are unable to secure adequate reimbursement from payors or sales of an approved drug are lower than we expected.
Furthermore, any delay in entering into collaboration agreements would likely either delay the development and commercialization of certain of our product candidates and reduce their competitiveness even if they reach the market, or prevent the development of certain product candidates. Any such delay related to our collaborations could adversely affect our business.
For certain product candidates that we may develop, we have formed collaborations to fund all or part of the costs of drug development and commercialization, such as our collaborations with Sanofi Genzyme and MDCO. We may not, however, be able to enter into additional collaborations for certain other programs, and the terms of any collaboration agreement we do secure may not be favorable to us. If we are not successful in our efforts to enter into future collaboration arrangements with respect to one or more of our product candidates, we may not have sufficient funds to develop that or other product candidates internally, or to bring our product candidates to market. If we do not have sufficient funds to develop and bring our product candidates to market, we will not be able to generate revenues from these product candidates, and this will substantially harm our business.
If any collaborator terminates or fails to perform its obligations under agreements with us, the development and commercialization of our product candidates could be delayed or terminated.
Our dependence on collaborators for capabilities and funding means that our business could be adversely affected if any collaborator terminates its collaboration agreement with us or fails to perform its obligations under that agreement. Our current or future collaborations, if any, may not be scientifically or commercially successful. Disputes may arise in the future with respect to the ownership of rights to technology or products developed with collaborators, which could have an adverse effect on our ability to develop and commercialize any affected product candidate.
Our current collaborations allow, and we expect that any future collaborations will allow, either party to terminate the collaboration for a material breach by the other party. In addition, our collaborators may have additional termination rights for convenience with respect to the collaboration or a particular program under the collaboration, under certain circumstances. Moreover, our agreement with MDCO relating to the development and commercialization of inclisiran worldwide may be terminated by MDCO at any time upon four months’ prior written notice. If we were to lose a commercialization collaborator, we would have to attract a new collaborator or develop expanded sales, distribution and marketing capabilities internally, which would require us to invest significant amounts of financial and management resources.
In addition, if we have a dispute with a collaborator over the ownership of technology or other matters, or if a collaborator terminates its collaboration with us, for breach or otherwise, or determines not to pursue the research, development and/or commercialization of RNAi therapeutics, it could delay our development of product candidates, result in the need for additional company resources to develop product candidates, require us to expend time and resources to develop expanded sales and marketing capabilities outside of the United States and EU, make it more difficult for us to attract new collaborators and could adversely affect how we are perceived in the business and financial communities. For example, in March 2011, Arbutus Biopharma Corporation, or ABC (formerly Tekmira Pharmaceuticals Corporation) and Protiva Biotherapeutics, Inc., a wholly owned subsidiary of ABC, and together with ABC, referred to as Arbutus, filed a civil complaint against us claiming, among other things, misappropriation of its confidential and proprietary information and trade secrets. As a result of the litigation, which was settled in November 2012, we were required to expend resources and management attention that would otherwise have been engaged in other activities. In addition, in August 2013, we initiated binding arbitration proceedings to resolve a disagreement with Arbutus regarding the achievement by Arbutus of a $5.0 million milestone payment under our cross-license agreement relating to the manufacture of ALN-VSP clinical trial material for use in China. The Arbutus arbitration hearing was held in May 2015. In March 2016, the arbitration panel ruled in our favor and as a result, no milestone payment is due to Arbutus at this time. The grounds on which Arbutus could appeal this ruling were limited and Arbutus did not appeal by the deadline.
28
Moreover, a collaborator, or in the event of a change in control of a collaborator or the assignment of a collaboration agreement to a third party, the successor entity or assignee, could determine that it is in its interests to:
|
|
• |
pursue alternative technologies or develop alternative products, either on its own or jointly with others, that may be competitive with the products on which it is collaborating with us or which could affect its commitment to the collaboration with us; |
|
|
• |
pursue higher-priority programs or change the focus of its development programs, which could affect the collaborator’s commitment to us; or |
|
|
• |
if it has marketing rights, choose to devote fewer resources to the marketing of our product candidates, if any are approved for marketing, than it does for product candidates developed without us. |
If any of these occur, the development and commercialization of one or more product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
We rely on third parties to conduct our clinical trials, and if they fail to fulfill their obligations, our development plans may be adversely affected.
We rely on independent clinical investigators, contract research organizations and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We have contracted, and we plan to continue to contract with, certain third parties to provide certain services, including site selection, enrollment, monitoring and data management services. Although we depend heavily on these parties, we control only certain aspects of their activity and therefore, we cannot be assured that these third parties will adequately perform all of their contractual obligations to us. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and our contract research organizations are required to comply with good clinical practice, or GCP, requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for all of our product candidates in clinical development. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of our contract research organizations fail to comply with applicable GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, the EMA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP regulations.
If our third-party service providers cannot adequately and timely fulfill their obligations to us, or if the quality and accuracy of our clinical trial data is compromised due to failure by such third party to adhere to our protocols or regulatory requirements or if such third parties otherwise fail to meet deadlines, our development plans and/or regulatory reviews for marketing approvals may be delayed or terminated. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase and our ability to generate revenues could be delayed.
We have limited manufacturing experience and resources and we must incur significant costs to develop this expertise and/or rely on third parties to manufacture our products.
We have limited manufacturing experience. In order to develop our product candidates, apply for regulatory approvals and commercialize our products, if approved, we will need to develop, contract for, or otherwise arrange for the necessary manufacturing capabilities. Historically, our internal manufacturing capabilities were limited to small-scale production of material for use in in vitro and in vivo experiments that is not required to be produced under current good manufacturing practices, or cGMP, standards. During 2012, we developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late stage clinical trial use and commercial supply. In addition, in April 2016, we completed our purchase of a parcel of land in Norton, Massachusetts, where we have commenced construction of a cGMP manufacturing facility for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use.
We may manufacture limited quantities of clinical trial materials ourselves, but otherwise we currently rely on third parties to manufacture the drug substance and, with the exception of patisiran, the finished product we will require for any clinical trials that we initiate and to support the commercial launch of our first several products. There are a limited number of manufacturers that supply synthetic siRNAs. We currently rely on a limited number of contract manufacturing organizations, or CMOs, for our supply of synthetic siRNAs. For example, in July 2015, we amended our manufacturing agreement with Agilent, to provide for Agilent to supply, subject to any conflicting obligations under our third-party agreements, a specified percentage of the active pharmaceutical ingredients required for certain of our products in clinical development, as well as other products the parties may agree upon in the future. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our CMOs, including Agilent, to meet
29
our delivery time requirements or provide adequate amounts of material to meet our needs. Included in these risks are potential synthesis and purification failures and/or contamination during the manufacturing process, as well as other issues with the CMO’s facility and ability to comply with the applicable manufacturing requirements, which could result in unusable product and cause delays in our manufacturing timelines and ultimately delay our clinical trials, as well as result in additional expense to us. To fulfill our siRNA requirements, we will likely need to secure alternative suppliers of synthetic siRNAs and such alternative suppliers are limited and may not be readily available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner. As noted above, in order to ensure long-term supply capabilities for our RNAi therapeutics, we are developing our own capabilities to manufacture drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use.
In addition to the manufacture of the synthetic siRNAs, we may have additional manufacturing requirements related to the technology required to deliver the siRNA to the relevant cell or tissue type, such as LNPs or conjugates. In some cases, the delivery technology we utilize is highly specialized or proprietary, and for technical and/or legal reasons, we may have access to only one or a limited number of potential manufacturers for such delivery technology. In addition, the scale-up of our delivery technologies could be very difficult and/or take significant time. We also have very limited experience in such scale-up and manufacturing, requiring us to depend on a limited number of third parties, who might not be able to deliver in a timely manner, or at all. Failure by manufacturers to properly manufacture our delivery technology and/or formulate our siRNAs for delivery could result in unusable product. Furthermore, competition for supply from our manufacturers from other companies, a breach by such manufacturers of their contractual obligations or a dispute with such manufacturers would cause delays in our discovery and development efforts, as well as additional expense to us.
Given the limited number of suppliers for our delivery technology and drug substance, we have developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late stage clinical use and commercial supply. During 2015, we scaled our cGMP manufacturing capacity for patisiran and believe we should have adequate resources to supply our commercial needs. In addition, as noted above, we are developing our own capabilities to manufacture drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use. In developing these manufacturing capabilities by building our own manufacturing facilities, we have incurred substantial expenditures, and expect to incur significant additional expenditures in the future. In addition, the construction and qualification of our drug substance facility is expected to take several years to complete and there are many risks inherent in the construction of a new facility that could result in delays and additional costs, including the need to obtain access to necessary equipment and third-party technology, if any. Also, we have had to, and will likely need to continue to, hire and train qualified employees to staff our facilities. We do not currently have a second source of supply for patisiran formulated bulk drug product. If we are unable to manufacture sufficient quantities of material or if we encounter problems with our facilities in the future, we may also need to secure alternative suppliers of patisiran formulated bulk drug product and drug substance, and such alternative suppliers may not be available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner.
The manufacturing process for any products that we may develop is subject to the FDA and foreign regulatory authority approval process and we will need to meet, and will need to contract with CMOs who can meet, all applicable FDA and foreign regulatory authority requirements on an ongoing basis. In addition, if we receive the necessary regulatory approval for any product candidate, we also expect to rely on third parties, including potentially our commercial collaborators, to produce materials required for commercial supply. We may experience difficulty in obtaining adequate manufacturing capacity for our needs and the needs of our collaborators, who we have, in some instances, the obligation to supply. If we are unable to obtain or maintain CMOs for these product candidates, or to do so on commercially reasonable terms, we may not be able to successfully develop and commercialize our products.
To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we depend, and will depend in the future, on these third parties, including Agilent, to perform their obligations in a timely manner and consistent with contractual and regulatory requirements, including those related to quality control and quality assurance. The failure of Agilent or any other CMO to perform its obligations as expected, or, to the extent we manufacture all or a portion of our product candidates ourselves, our failure to execute on our manufacturing requirements, could adversely affect our business in a number of ways, including:
|
|
• |
we or our current or future collaborators may not be able to initiate or continue clinical trials of product candidates that are under development; |
|
|
• |
we or our current or future collaborators may be delayed in submitting regulatory applications, or receiving regulatory approvals, for our product candidates; |
|
|
• |
we may lose the cooperation of our collaborators; |
|
|
• |
our facilities and those of our CMOs, and our products could be the subject of inspections by regulatory authorities that could have a negative outcome and result in delays in supply; |
30
|
|
• |
we may be required to cease distribution or recall some or all batches of our products or take action to recover clinical trial material from clinical trial sites; and |
|
|
• |
ultimately, we may not be able to meet commercial demands for our products. |
If any CMO with whom we contract, including Agilent, fails to perform its obligations, we may be forced to manufacture the materials ourselves, for which we may not have the capabilities or resources, or enter into an agreement with a different CMO, which we may not be able to do on reasonable terms, if at all. In either scenario, our clinical trials or commercial distribution could be delayed significantly as we establish alternative supply sources. In some cases, the technical skills required to manufacture our products or product candidates may be unique or proprietary to the original CMO and we may have difficulty, or there may be contractual restrictions prohibiting us from, transferring such skills to a back-up or alternate supplier, or we may be unable to transfer such skills at all. In addition, if we are required to change CMOs for any reason, we will be required to verify that the new CMO maintains facilities and procedures that comply with quality standards and with all applicable regulations and guidelines. We will also need to verify, such as through a manufacturing comparability study, that any new manufacturing process will produce our product according to the specifications previously submitted to or approved by the FDA or another regulatory authority. The delays associated with the verification of a new CMO could negatively affect our ability to develop product candidates in a timely manner or within budget. Furthermore, a CMO may possess technology related to the manufacture of our product candidate that such CMO owns independently. This would increase our reliance on such CMO or require us to obtain a license from such CMO in order to have another CMO manufacture our products or product candidates.
We have no sales or distribution experience and only early capabilities for marketing, sales and market access, and expect to invest significant financial and management resources to establish these capabilities and to establish infrastructure in the EU.
We have no sales or distribution experience and only early capabilities for marketing, sales and market access. We currently expect to rely heavily on third parties to launch and market certain of our product candidates in certain geographies, if approved. However, we intend to commercialize the majority of our products on our own in the United States and EU, as well as globally in the case of givosiran. Accordingly, we will need to develop internal sales, distribution and marketing capabilities as part of our core product strategy initially in the United States and the EU, and longer-term on a global basis, which will require significant financial and management resources. For the majority of our Genetic Medicine programs where we will perform sales, marketing and distribution functions ourselves in the United States, Canada and Western Europe, and for future Cardio-Metabolic and Hepatic Infectious Disease products we successfully develop where we intend to retain significant product development and commercialization rights, we could face a number of additional risks, including:
|
|
• |
we may not be able to attract and build a significant marketing or sales force; |
|
|
• |
we may not be able to establish our capabilities and infrastructure in the EU or in other territories in a timely manner; |
|
|
• |
the cost of establishing a marketing or sales force may not be justifiable in light of the revenues generated by any particular product; and |
|
|
• |
our direct sales and marketing efforts may not be successful. |
If we are unable to develop our own sales, marketing and distribution capabilities in the United States and the EU, as well as globally for certain products, we will not be able to successfully commercialize our products in our sales territories without reliance on third parties.
Credit and financial market conditions may exacerbate certain risks affecting our business from time to time.
Due to tightening of global credit, there may be a disruption or delay in the performance of our third-party contractors, suppliers or collaborators. We rely on third parties for several important aspects of our business, including significant portions of our manufacturing needs, development of product candidates and conduct of clinical trials. If such third parties are unable to satisfy their commitments to us, our business could be adversely affected.
Our ability to secure additional financing in addition to the term loan agreements with BOA and Wells and to satisfy our financial obligations under indebtedness outstanding from time to time will depend upon our future operating performance, which is subject to then prevailing general economic and credit market conditions, including interest rate levels and the availability of credit generally, and financial, business and other factors, many of which are beyond our control. In light of periodic uncertainty in the capital and credit markets, there can be no assurance that sufficient financing will be available on desirable or even any terms to fund investments, acquisitions, stock repurchases, dividends, debt refinancing or extraordinary actions.
31
Risks Related to Managing Our Operations
If we are unable to attract and retain qualified key management and scientists, development and commercial staff, consultants and advisors, our ability to implement our business plan may be adversely affected.
We are highly dependent upon our senior management and our scientific, clinical and medical staff. The loss of the service of any of the members of our senior management, including Dr. John Maraganore, our Chief Executive Officer, may significantly delay or prevent the achievement of product development and other business objectives. Our employment arrangements with our key personnel are terminable without notice. We do not carry key person life insurance on any of our employees.
We have grown our workforce significantly over the past year and anticipate continuing to add a significant number of additional employees as we focus on achieving our Alnylam 2020 strategy. We face intense competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities, governmental entities and other research institutions, many of which have substantially greater resources with which to reward qualified individuals than we do. In addition, due to our recent decision to discontinue our revusiran program, and the related decline in our stock price, we may face additional challenges in attracting and retaining employees. Accordingly, we may be unable to attract and retain suitably qualified individuals in order to support our growing research, development and commercialization efforts and initiatives, and our failure to do so could have an adverse effect on our ability to implement our future business plan.
We may have difficulty expanding our operations successfully as we evolve from a U.S.-based company primarily involved in discovery, pre-clinical testing and clinical development into a global company that develops and commercializes multiple drugs.
We expect that as we increase the number of product candidates we are developing we will also need to expand our operations in the United States and continue to build operations in the EU and eventually other territories. As noted above, we grew our workforce significantly during 2016 and anticipate continuing to hire additional employees, including employees in the EU, as we focus on achieving our Alnylam 2020 strategy. This expected growth is placing a strain on our administrative and operational infrastructure, and we will need to develop additional and/or new infrastructure and capabilities to support our growth and obtain additional space to conduct our operations in the United States and the EU. If we are unable to develop such additional infrastructure or obtain sufficient space to accommodate our growth in a timely manner and on commercially reasonable terms, our business could be negatively impacted. As product candidates we develop enter and advance through clinical trials, we will need to expand our development, regulatory, manufacturing, quality, compliance, and marketing and sales capabilities in the United States and the EU or contract with other organizations to provide these capabilities for us. In addition, as our operations expand due to our development progress, we expect that we will need to manage additional relationships with various collaborators, suppliers and other organizations. Our ability to manage our operations and future growth will require us to continue to improve our operational, financial and management controls and systems, reporting systems and infrastructure, and policies and procedures. We may not be able to implement improvements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
Our business and operations could suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war, and telecommunication and electrical failures. Such events could cause interruption of our operations. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our product candidates could result in delays in our regulatory filings and development efforts, as well as delays in the commercialization of our products, and significantly increase our costs. To the extent that any disruption or security breach were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the development and potential commercialization of our product candidates could be delayed.
The results of the United Kingdom’s referendum on withdrawal from the EU may have a negative effect on global economic conditions, financial markets and our business.
In June 2016, the United Kingdom, or UK, held a referendum in which voters approved an exit from the EU, commonly referred to as “Brexit.” This referendum has created political and economic uncertainty, particularly in the UK and the EU, and this uncertainty may persist for years. A withdrawal could, among other outcomes, disrupt the free movement of goods, services and people between the UK and the EU, and result in increased legal and regulatory complexities, as well as potential higher costs of conducting business in Europe. The UK's vote to exit the EU could also result in similar referendums or votes in other European countries in which we do business. Given the lack of comparable precedent, it is unclear what financial, trade and legal implications the withdrawal of the UK from the EU would have and how such withdrawal would affect us.
For example, Brexit could result in the UK or the EU significantly altering its regulations affecting the clearance or approval of our product candidates that are developed in the UK. Any new regulations could add time and expense to the conduct of our business,
32
as well as the process by which our products receive regulatory approval in the UK, the EU and elsewhere. In addition, the announcement of Brexit and the withdrawal of the UK from the EU have had and may continue to have a material adverse effect on global economic conditions and the stability of global financial markets, and may significantly reduce global market liquidity and restrict the ability of key market participants to operate in certain financial markets. Any of these effects of Brexit, among others, could adversely affect our business, our results of operations, liquidity and financial condition.
Risks Related to Our Industry
Risks Related to Development, Clinical Testing and Regulatory Approval of Our Product Candidates
Any product candidates we develop may fail in development or be delayed to a point where they do not become commercially viable.
Before obtaining regulatory approval for the commercial distribution of our product candidates, we must conduct, at our own expense, extensive nonclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. Nonclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome, and the historical failure rate for product candidates is high. In October 2016, we discontinued development of one of our product candidates, which included a Phase 3 clinical trial. We currently have multiple other programs in clinical development, including one program in a Phase 3 clinical trial, as well as several earlier stage clinical programs. However, we may not be able to further advance these or any other product candidate through clinical trials and regulatory approval.
If we enter into clinical trials, the results from nonclinical testing or early clinical trials of a product candidate may not predict the results that will be obtained in subsequent subjects or in subsequent human clinical trials of that product candidate or any other product candidate. For example, in July 2016, we announced updated results from our Phase 1 clinical trial of fitusiran, including initial clinical data on a small number of people with hemophilia with inhibitors. Although the initial clinical data from this trial are encouraging, the data are preliminary in nature, based on a limited number of people with hemophilia with inhibitors, and the fitusiran Phase 1 study is not complete. These data, or other positive data, may not continue for these people with hemophilia or occur for any future patients in this study, and may not be repeated or observed in any future studies. There can be no assurance that this study will ultimately be successful or support further clinical advancement of this product candidate. In addition, in June 2016, we reported initial data from PNH patients in our ALN-CC5 Phase 1/2 clinical trial, and we reiterated that we now plan to pursue a more focused development path in PNH where ALN-CC5 would be evaluated in eculizumab poor responders and for eculizumab sparing, as well as potentially in other indications. There is a high failure rate for drugs proceeding through clinical studies. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies, and any such setbacks in our clinical development could have a material adverse effect on our business and operating results. Moreover, patisiran, fitusiran and our other product candidates each employ novel delivery technologies that have yet to be extensively evaluated in human clinical trials and proven safe and effective.
In addition, we, the FDA or other applicable regulatory authorities, or an institutional review board, or IRB, or similar foreign review board or committee, may delay initiation of or suspend clinical trials of a product candidate at any time for various reasons, including if we or they believe the healthy volunteer subjects or patients participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a product candidate or related product on healthy volunteer subjects or patients in a clinical trial could result in our decision, or a decision by the FDA or foreign regulatory authorities, to suspend or terminate the trial, or, in the case of regulatory agencies, a refusal to approve a particular product candidate for any or all indications of use. For example, in October 2016, we announced our decision to discontinue development of revusiran, an investigational RNAi therapeutic that was being developed for the treatment of patients with cardiomyopathy due to hATTR amyloidosis. Our decision followed the recommendation of the revusiran ENDEAVOUR Phase 3 study DMC to suspend dosing and the observation of an imbalance in mortality in revusiran-treated patients as compared to those on placebo. Separately, the patisiran APOLLO DMC met at our request following our decision to discontinue development of revusiran, and recommended continuation of the APOLLO Phase 3 trial of patisiran, without modification. We are conducting a comprehensive evaluation of the revusiran data. While we believe that the decision to discontinue development of revusiran does not affect patisiran, which is in development for the treatment of hATTR amyloidosis, or any of our other investigational RNAi therapeutic programs in development, our comprehensive evaluation of the revusiran data is preliminary and ongoing. We expect this evaluation will take some time to complete and there remains uncertainty regarding the cause of the findings that led to the discontinuation of the revusiran program.
Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, including the size of the patient population, the age and condition of the patients, the stage and severity of disease, the availability of clinical trials for other investigational drugs for the same disease or condition, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, and the eligibility criteria for the clinical trial. For example, we may experience difficulty enrolling our clinical trials, including, but not limited to, our clinical trials for fitusiran, due to the availability of existing approved treatments, as well as other investigational treatments in development. Delays
33
or difficulties in patient enrollment or difficulties retaining trial participants, including as a result of the availability of existing or other investigational treatments, can result in increased costs, longer development times or termination of a clinical trial.
Although our investigational RNAi therapeutics have been generally well tolerated in our clinical trials to date, new safety findings may emerge. For example, as noted above, in October 2016, we made the decision to discontinue our revusiran program. Following reports in the revusiran Phase 2 OLE study of new onset or worsening peripheral neuropathy, the revusiran ENDEAVOUR Phase 3 study DMC assembled in early October 2016 at our request to review these reports and ENDEAVOUR safety data on an unblinded basis. The DMC did not find conclusive evidence for a drug-related neuropathy signal in the ENDEAVOUR trial, but informed us that the benefit-risk profile for revusiran no longer supported continued dosing. We subsequently reviewed unblinded ENDEAVOUR data which revealed an imbalance of mortality in the revusiran arm as compared to placebo. We had previously reported, in July 2016, preliminary data from our revusiran Phase 2 OLE study for 12 patients who had reached the 12-month endpoint as of the data transfer date of May 26, 2016. Serious adverse events, or SAEs, were observed in 14 patients, one of which, a case of lactic acidosis, was deemed possibly related to the study drug and the patient discontinued treatment. There were a total of seven deaths reported at that time in the revusiran OLE study, all of which were unrelated to study drug. The majority of the adverse events, or AEs, were mild or moderate in severity; injection site reactions, or ISRs, were reported in 12 patients. In August 2015, we reported that three patients had discontinued from the revusiran Phase 2 OLE study due to recurrent localized reactions at the injection site or a diffuse rash; no further discontinuations due to ISRs had occurred as of May 26, 2016. In our patisiran Phase 2 OLE study in patients with polyneuropathy due to hATTR amyloidosis, based on preliminary 24-month data reported from 27 patients as of the data cutoff on May 12, 2016, the most common drug-related or possibly drug-related AEs were flushing and infusion-related reactions, all of which were all mild in severity and did not result in any discontinuations. There were nine reports of SAEs in six patients, all of which were unrelated to study drug, including one discontinuation for gastroesophageal cancer at approximately 20 months in a patient who subsequently died and one death due to myocardial infarction in a 79 year-old patient who died after having completed the full 24 months of treatment. As noted above, the patisiran APOLLO Phase 3 study DMC met at our request following our decision to discontinue development of revusiran, and recommended continuation of the APOLLO Phase 3 trial without modification. In addition, in our ALN-VSP clinical trial, one patient with advanced pancreatic neuroendocrine cancer with extensive involvement of the liver developed hepatic failure five days following the second dose of ALN-VSP and subsequently died; this was deemed possibly related to the study drug. As demonstrated by the recent discontinuation of our revusiran program, the occurrence of AEs can result in the suspension or termination of clinical trials of a product candidate by us or the FDA or a foreign regulatory authority, or refusal to approve a particular product candidate for any or all indications of use.
Clinical trials also require the review, oversight and approval of IRBs or, outside of the United States, an independent ethics committee, which continually review clinical investigations and protect the rights and welfare of human subjects. Inability to obtain or delay in obtaining IRB or ethics committee approval can prevent or delay the initiation and completion of clinical trials, and the FDA or foreign regulatory authorities may decide not to consider any data or information derived from a clinical investigation not subject to initial and continuing IRB or ethics committee review and approval, as the case may be, in support of a marketing application.
Our product candidates that we develop may encounter problems during clinical trials that will cause us, an IRB, ethics committee or regulatory authorities to delay, suspend or terminate these trials, or that will delay or confound the analysis of data from these trials. If we experience any such problems, we may not have the financial resources to continue development of the product candidate that is affected, or development of any of our other product candidates. We may also lose, or be unable to enter into, collaborative arrangements for the affected product candidate and for other product candidates we are developing.
A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, nonclinical testing and the clinical trial process that could delay or prevent regulatory approval or our ability to commercialize our product candidates, including:
|
|
• |
our nonclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional nonclinical testing or clinical trials, or we may abandon projects that we expect to be promising; |
|
|
• |
delays in filing investigational new drug, or IND, applications or comparable foreign applications or delays or failure in obtaining the necessary approvals from regulators or IRBs/ethics committees in order to commence a clinical trial at a prospective trial site, or their suspension or termination of a clinical trial once commenced; |
|
|
• |
conditions imposed on us by an IRB or ethics committee, or the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
|
|
• |
problems in engaging IRBs or ethics committees to oversee clinical trials or problems in obtaining or maintaining IRB or ethics committee approval of trials; |
|
|
• |
delays in enrolling patients and volunteers into clinical trials, and variability in the number and types of patients and volunteers available for clinical trials; |
34
|
|
• |
negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours; |
|
|
• |
inadequate supply or quality of product candidate materials or other materials necessary for the conduct of our clinical trials; |
|
|
• |
greater than anticipated clinical trial costs; |
|
|
• |
serious and unexpected drug-related side effects experienced by participants in our clinical trials or by individuals using drugs similar to our product candidates; |
|
|
• |
poor or disappointing effectiveness of our product candidates during clinical trials; |
|
|
• |
unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or records of any clinical or nonclinical investigation; |
|
|
• |
failure of our third-party contractors or investigators to comply with regulatory requirements or otherwise meet their contractual obligations in a timely manner, or at all; |
|
|
• |
governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular; or |
|
|
• |
varying interpretations of data by the FDA and similar foreign regulatory agencies. |
Even if we successfully complete clinical trials of our product candidates, any given product candidate may not prove to be a safe and effective treatment for the disease for which it was being tested.
We may be unable to obtain United States or foreign regulatory approval and, as a result, unable to commercialize our product candidates.
Our product candidates are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, packaging, advertising and promotion, pricing, marketing and distribution of drugs. Rigorous nonclinical testing and clinical trials and an extensive regulatory approval process are required to be successfully completed in the United States and in many foreign jurisdictions before a new drug can be marketed. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. It is possible that none of the product candidates we may develop will obtain the regulatory approvals necessary for us or our collaborators to begin selling them.
We have limited experience in conducting and managing the clinical trials necessary to obtain regulatory approvals, including approval by the FDA. The time required to obtain FDA and other regulatory approvals is unpredictable but typically takes many years following the commencement of clinical trials, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when regulating us are not always applied predictably or uniformly and can change. Any analysis we perform of data from nonclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We may also encounter unexpected delays or increased costs due to new government regulations, for example, from future legislation or administrative action, or from changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be.
Because the drugs we are developing may represent a new class of drug, the FDA and its foreign counterparts have not yet established any definitive policies, practices or guidelines in relation to these drugs. The lack of policies, practices or guidelines may hinder or slow review by the FDA of any regulatory filings that we may submit. Moreover, the FDA may respond to these submissions by defining requirements we may not have anticipated. Such responses could lead to significant delays in the clinical development of our product candidates. In addition, because there may be approved treatments for some of the diseases for which we may seek approval, in order to receive regulatory approval, we may need to demonstrate through clinical trials that the product candidates we develop to treat these diseases, if any, are not only safe and effective, but safer or more effective than existing products. Furthermore, in recent years, there has been increased public and political pressure on the FDA with respect to the approval process for new drugs, and the FDA’s standards, especially regarding drug safety, appear to have become more stringent.
Assuming the data from our APOLLO Phase 3 clinical trial is positive, we expect to file our first NDA and MAA for patisiran at the end of 2017. Any delay or failure in obtaining required approvals could have a material adverse effect on our ability to generate
35
revenues from patisiran or any product candidate for which we may seek approval in the future. Furthermore, any regulatory approval to market patisiran or any other product may be subject to limitations on the approved uses for which we may market the product or the labeling or other restrictions. In addition, the FDA has the authority to require a Risk Evaluation and Mitigation Strategy, or REMS, plan as part of an NDA, or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. In the EU, we could be required to adopt a similar plan, known as a risk management plan, and our products could be subject to specific risk minimization measures, such as restrictions on prescription and supply, the conduct of post-marketing safety or efficacy studies, or the distribution of patient and/or prescriber educational materials. In either instance, these limitations and restrictions may limit the size of the market for the product and affect reimbursement by third-party payors.
We are also subject to numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process varies among countries and includes all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not ensure approval by regulatory authorities outside the United States and vice versa.
Even if we obtain regulatory approvals, our marketed drugs will be subject to ongoing regulatory oversight. If we fail to comply with continuing U.S. and foreign requirements, our approvals could be limited or withdrawn, we could be subject to other penalties, and our business would be seriously harmed.
Following any initial regulatory approval of any drugs we may develop, we will also be subject to continuing regulatory oversight, including the review of adverse drug experiences and clinical results that are reported after our drug products are made commercially available. This would include results from any post-marketing tests or surveillance to monitor the safety and efficacy of the drug product required as a condition of approval or agreed to by us. Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved uses for which the product may be marketed. Other ongoing regulatory requirements include, among other things, submissions of safety and other post-marketing information and reports, registration and listing, as well as continued compliance with cGMP requirements and GCP requirements for any clinical trials that we conduct post-approval. In addition, we are conducting, and intend to continue to conduct, clinical trials for our product candidates, and we intend to seek approval to market our product candidates, in jurisdictions outside of the United States, and therefore will be subject to, and must comply with, regulatory requirements in those jurisdictions.
The FDA has significant post-market authority, including, for example, the authority to require labeling changes based on new safety information and to require post-market studies or clinical trials to evaluate serious safety risks related to the use of a drug and to require withdrawal of the product from the market. The FDA also has the authority to require a REMS plan after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug.
The CMO and manufacturing facilities we use to make our product candidates, including our Cambridge facility, our future Norton facility, and Agilent and other CMOs, will also be subject to periodic review and inspection by the FDA and other regulatory agencies. To date, our Cambridge manufacturing facility has not been subject to an inspection by any regulatory authority. The discovery of any new or previously unknown problems with us or our CMOs, or our or their manufacturing processes or facilities, may result in restrictions on the drug or CMO or facility, including withdrawal of the drug from the market. We have developed cGMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for Phase 3 clinical and commercial use. In addition, in April 2016, we completed our purchase of a parcel of land in Norton, Massachusetts, where we have commenced construction of a cGMP manufacturing facility for drug substance, including siRNAs and siRNA conjugates, for clinical and commercial use. We may not have the ability or capacity to manufacture material at a broader commercial scale in the future. We may manufacture clinical trial materials or we may contract a third party to manufacture these materials for us. Reliance on CMOs entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the CMO for regulatory compliance. Our product promotion and advertising will also be subject to regulatory requirements and continuing regulatory review.
If we or our collaborators, CMOs or service providers fail to comply with applicable continuing regulatory requirements in the United States or foreign jurisdictions in which we may seek to market our products, we or they may be subject to, among other things, fines, warning letters, holds on clinical trials, refusal by the FDA or foreign regulatory authorities to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatory approval, product recalls and seizures, refusal to permit the import or export of products, operating restrictions, injunction, civil penalties and criminal prosecution.
36
Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our product candidates upon their commercial introduction, which will prevent us from becoming profitable.
The product candidates that we are developing are based upon new technologies or therapeutic approaches. Key participants in pharmaceutical marketplaces, such as physicians, third-party payors and consumers, may not accept a product intended to improve therapeutic results based on RNAi technology. As a result, it may be more difficult for us to convince the medical community and third-party payors to accept and use our product, or to provide favorable reimbursement.
Other factors that we believe will materially affect market acceptance of our product candidates include:
|
|
• |
the timing of our receipt of any marketing approvals, the terms of any approvals and the countries in which approvals are obtained; |
|
|
• |
the safety and efficacy of our product candidates, as demonstrated in clinical trials and as compared with alternative treatments, if any; |
|
|
• |
relative convenience and ease of administration of our product candidates; |
|
|
• |
the willingness of patients to accept potentially new routes of administration or new or different therapeutic approaches and mechanisms of action; |
|
|
• |
the success of our physician education programs; |
|
|
• |
the availability of adequate government and third-party payor reimbursement; |
|
|
• |
the pricing of our products, particularly as compared to alternative treatments; and |
|
|
• |
availability of alternative effective treatments for the diseases that product candidates we develop are intended to treat and the relative risks, benefits and costs of those treatments. |
For example, patisiran utilizes an intravenous mode of administration that physicians and/or patients may not readily adopt or which may not compete with other potentially available options. In addition, fitusiran represents a new approach to treating hemophilia which may not be readily accepted by patients and their caregivers.
In addition, our estimates regarding the potential market size may be materially different from what we currently expect at the time we commence commercialization, which could result in significant changes in our business plan and may have a material adverse effect on our results of operations and financial condition.
If we or our collaborators, CMOs or service providers fail to comply with healthcare laws and regulations, we or they could be subject to enforcement actions, which could affect our ability to develop, market and sell our products and may harm our reputation.
As a manufacturer of pharmaceuticals, we are subject to federal, state, and comparable foreign healthcare laws and regulations pertaining to fraud and abuse and patients’ rights. These laws and regulations include:
|
|
• |
the U.S. federal Anti-Kickback statute, which prohibits, among other things, persons from soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce either the referral of an individual for a healthcare item or service, or the purchasing or ordering of an item or service, for which payment may be made under a federal healthcare program such as Medicare or Medicaid; |
|
|
• |
the U.S. federal false claims laws, which prohibit, among other things, individuals or entities from knowingly presenting or causing to be presented, claims for payment by government-funded programs such as Medicare or Medicaid that are false or fraudulent, and which may apply to us by virtue of statements and representations made to customers or third parties; |
|
|
• |
the U.S. federal Health Insurance Portability and Accountability Act and Health Information Technology for Economic and Clinical Health Act, which impose requirements relating to the privacy, security, and transmission of individually identifiable health information; and require notification to affected individuals and regulatory authorities of certain breaches of security of individually identifiable health information; |
37
|
|
consulting fees, travel reimbursements, research grants, and other payments or gifts with values over $10 made to physicians and teaching hospitals; and |
|
|
• |
state and foreign laws comparable to each of the above federal laws, including in the EU laws prohibiting giving healthcare professionals any gift or benefit in kind as an inducement to prescribe our products, national transparency laws requiring the public disclosure of payments made to healthcare professionals and institutions, and data privacy laws, in addition to anti-kickback and false claims laws applicable to commercial insurers and other non-federal payors, requirements for mandatory corporate regulatory compliance programs, and laws relating to government reimbursement programs, patient data privacy and security. |
If our operations are found to be in violation of any such requirements, we may be subject to penalties, including civil or criminal penalties, criminal prosecution, monetary damages, the curtailment or restructuring of our operations, loss of eligibility to obtain approvals from the FDA, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, or the imposition of a corporate integrity agreement with the Office of Inspector General of the Department of Health and Human Services, any of which could adversely affect our financial results. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
If we or our collaborators, CMOs or service providers fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market and sell our products successfully and could harm our reputation and lead to reduced acceptance of our products by the market. These enforcement actions include, among others:
|
|
• |
adverse regulatory inspection findings; |
|
|
• |
warning letters; |
|
|
• |
voluntary or mandatory product recalls or public notification or medical product safety alerts to healthcare professionals; |
|
|
• |
restrictions on, or prohibitions against, marketing our products; |
|
|
• |
restrictions on, or prohibitions against, importation or exportation of our products; |
|
|
• |
suspension of review or refusal to approve pending applications or supplements to approved applications; |
|
|
• |
exclusion from participation in government-funded healthcare programs; |
|
|
• |
exclusion from eligibility for the award of government contracts for our products; |
|
|
• |
suspension or withdrawal of product approvals; |
|
|
• |
product seizures; |
|
|
• |
injunctions; and |
|
|
• |
civil and criminal penalties, up to and including criminal prosecution resulting in fines, exclusion from healthcare reimbursement programs and imprisonment. |
Moreover, federal, state or foreign laws or regulations are subject to change, and while we, our collaborators, CMOs and/or service providers currently may be compliant, that could change due to changes in interpretation, prevailing industry standards or the legal structure.
Any drugs we develop may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business.
The regulations that govern marketing approvals, pricing and reimbursement for new drugs vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. We are actively monitoring these regulations as several of our programs move into late stages of development, however, a number of our programs are currently in the earlier stages of development and we will not be able to assess the impact of price regulations for a number of years. As a result, we might obtain regulatory approval for a product in a particular country, but then be subject to price regulations that delay our commercial launch of
38
the product and negatively impact the revenues we are able to generate from the sale of the product in that country and potentially in other countries due to reference pricing.
Our ability to commercialize any products successfully also will depend in part on the extent to which reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Even if we succeed in bringing one or more products to the market, these products may not be considered cost-effective, and the amount reimbursed for any products may be insufficient to allow us to sell our products on a competitive basis. Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts reimbursed for drug products. If the price we are able to charge for any products we develop, or the reimbursement provided for such products, is inadequate in light of our development and other costs, or if reimbursement is denied, our return on investment could be adversely affected.
We currently expect that some of the drugs we develop may need to be administered under the supervision of a physician or other healthcare professional on an outpatient basis. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable drugs) may be eligible for coverage under the Medicare Part B program if:
|
|
• |
they are incident to a physician’s services; |
|
|
• |
they are reasonable and necessary for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standards of medical practice; and |
|
|
• |
they have been approved by the FDA and meet other requirements of the statute. |
There may be significant delays in obtaining coverage for newly-approved drugs, and coverage may be more limited than the purposes for which the drug is approved by the FDA or foreign regulatory authorities. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution or that covers a particular provider’s cost of acquiring the drug. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on payments allowed for lower-cost drugs that are already reimbursed, may be incorporated into existing payments for other services and may reflect budgetary constraints or imperfections in Medicare data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates. Our inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for new drugs that we develop and for which we obtain regulatory approval could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize products, and our overall financial condition.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies. A number of legislative and regulatory changes in the healthcare system in the United States and other major healthcare markets have been proposed in recent years, and such efforts have expanded substantially in recent years. These developments have included prescription drug benefit legislation that was enacted in 2003 and took effect in January 2006, healthcare reform legislation enacted by certain states, and major healthcare reform legislation that was passed by Congress and enacted into law in the United States in 2010. These developments could, directly or indirectly, affect our ability to sell our products, if approved, at a favorable price.
In particular, in March 2010, the PPACA was signed into law. This legislation changed the system of healthcare insurance and benefits intended to broaden coverage and control costs. The law also contains provisions that affect companies in the pharmaceutical industry and other healthcare related industries by imposing additional costs and changes to business practices. Provisions affecting pharmaceutical companies include the following:
|
|
• |
Mandatory rebates for drugs sold into the Medicaid program were increased, and the rebate requirement was extended to drugs used in risk-based Medicaid managed care plans. |
|
|
• |
The 340B Drug Pricing Program under the Public Health Service Act was extended to require mandatory discounts for drug products sold to certain critical access hospitals, cancer hospitals and other covered entities. |
|
|
• |
Pharmaceutical companies are required to offer discounts on brand-name drugs to patients who fall within the Medicare Part D coverage gap, commonly referred to as the “donut hole.” |
39
|
|
Medicare, Medicaid, Department of Veterans Affairs and Department of Defense. Since we expect our branded pharmaceutical sales to constitute a small portion of the total federal healthcare program pharmaceutical market, we do not expect this annual assessment to have a material impact on our financial condition. |
|
|
• |
The law provides that approval of an application for a follow-on biologic product may not become effective until 12 years after the date on which the reference innovator biologic product was first licensed by the FDA, with a possible six-month extension for pediatric products. After this exclusivity ends, it will be easier for generic manufacturers to enter the market, which is likely to reduce the pricing for such products and could affect our profitability. |
The full effects of the U.S. healthcare reform legislation cannot be known until the law is fully implemented through regulations or guidance issued by the CMS and other federal and state healthcare agencies. The financial impact of the U.S. healthcare reform legislation over the next few years will depend on a number of factors, including, but not limited, to the policies reflected in implementing regulations and guidance, and changes in sales volumes for products affected by the new system of rebates, discounts and fees. This legislation may also have a positive impact on our future net sales, if any, by increasing the aggregate number of persons with healthcare coverage in the United States.
As a result of the 2016 election in the United States, there is great political uncertainty concerning the fate of the PPACA and other healthcare laws. The United States Congress is expected to draft legislation to repeal parts of the PPACA, but it is uncertain when such legislation would be passed and whether Congress would replace the law and what any replacement law would encompass.
We cannot predict what healthcare reform initiatives may be adopted in the future. Further federal and state legislative and regulatory developments are likely, and we expect ongoing initiatives in the United States to increase pressure on drug pricing. Such reforms could have an adverse effect on anticipated revenues from product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our overall financial condition and ability to develop drug candidates.
Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending.
Under the Budget Control Act of 2011, the failure of Congress to enact deficit reduction measures of at least $1.2 trillion for the years 2013 through 2021 triggered automatic cuts to most federal programs. These cuts included aggregate reductions to Medicare payments to providers of up to 2 percent per fiscal year, starting in 2013. Certain of these automatic cuts have been implemented resulting in reductions in Medicare payments to physicians, hospitals, and other healthcare providers, among other things. The full impact on our business of these automatic cuts is uncertain.
If other federal spending is reduced, any budgetary shortfalls may also impact the ability of relevant agencies, such as the FDA or National Institutes of Health to continue to function. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell any products we may develop.
There is a substantial risk of product liability claims in our business. If we are unable to obtain sufficient insurance, a product liability claim against us could adversely affect our business.
Our business exposes us to significant potential product liability risks that are inherent in the development, testing, manufacturing and marketing of human therapeutic products. Product liability claims could delay or prevent completion of our clinical development programs. Following the decision to discontinue clinical development of revusiran, we have undertaken a comprehensive evaluation of available revusiran data, which is ongoing. Notwithstanding the risks undertaken by all persons who participate in clinical trials, and the information on risks provided to study investigators and patients participating in revusiran studies, it is possible that product liability claims will be asserted against us relating to the worsening of a patient’s condition alleged to have been caused by revusiran. Such claims might not be fully covered by product liability insurance. If we succeed in marketing products, product liability claims could result in an FDA investigation of the safety and effectiveness of our products, our manufacturing processes and facilities or our marketing programs, and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used, or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients and a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of development and may need to obtain higher levels prior to marketing any of our product candidates. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
40
If we do not comply with laws regulating the protection of the environment and health and human safety, our business could be adversely affected.
Our research, development and manufacturing involve the use of hazardous materials, chemicals and various radioactive compounds. We maintain quantities of various flammable and toxic chemicals in our facilities in Cambridge that are required for our research, development and manufacturing activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these materials in our Cambridge facilities comply with the relevant guidelines of the City of Cambridge, the Commonwealth of Massachusetts and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Risks Related to Patents, Licenses and Trade Secrets
If we are not able to obtain and enforce patent protection for our discoveries, our ability to develop and commercialize our product candidates will be harmed.
Our success depends, in part, on our ability to protect proprietary methods and technologies that we develop under the patent and other intellectual property laws of the United States and other countries, so that we can prevent others from unlawfully using our inventions and proprietary information. However, we may not hold proprietary rights to some patents required for us to manufacture and commercialize our proposed products. Because certain U.S. patent applications are confidential until the patents issue, such as applications filed prior to November 29, 2000, or applications filed after such date which will not be filed in foreign countries, third parties may have filed patent applications for technology covered by our pending patent applications without our being aware of those applications, and our patent applications may not have priority over those applications. For this and other reasons, we may be unable to secure desired patent rights, thereby losing desired exclusivity. Further, we may be required to obtain licenses under third-party patents to market our proposed products or conduct our research and development or other activities. If licenses are not available to us on acceptable terms, we may not be able to market the affected products or conduct the desired activities.
Our strategy depends on our ability to rapidly identify and seek patent protection for our discoveries. In addition, we may rely on third-party collaborators to file patent applications relating to proprietary technology that we develop jointly during certain collaborations. The process of obtaining patent protection is expensive and time-consuming. If our present or future collaborators fail to file and prosecute all necessary and desirable patent applications at a reasonable cost and in a timely manner, our business may be adversely affected. Despite our efforts and the efforts of our collaborators to protect our proprietary rights, unauthorized parties may be able to obtain and use information that we regard as proprietary. While issued patents are presumed valid, this does not guarantee that the patent will survive a validity challenge or be held enforceable. Any patents we have obtained, or obtain in the future, may be challenged, invalidated, adjudged unenforceable or circumvented by parties attempting to design around our intellectual property. Moreover, third parties or the United States Patent and Trademark Office, or USPTO, may commence interference proceedings involving our patents or patent applications. Any challenge to, finding of unenforceability or invalidation or circumvention of, our patents or patent applications, would be costly, would require significant time and attention of our management and could have a material adverse effect on our business.
Our pending patent applications may not result in issued patents. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the USPTO and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. Similarly, the ultimate degree of protection that will be afforded to biotechnology inventions, including ours, in the United States and foreign countries, remains uncertain and is dependent upon the scope of the protection decided upon by patent offices, courts and lawmakers. Moreover, there are periodic discussions in the Congress of the United States and in international jurisdictions about modifying various aspects of patent law. For example, the America Invents Act included a number of changes to the patent laws of the United States. If any of the enacted changes do not provide adequate protection for discoveries, including our ability to pursue infringers of our patents for substantial damages, our business could be adversely affected. One major provision of the America Invents Act, which took effect in March 2013, changed United States patent practice from a first-to-invent to a first-to-file system. If we fail to file an
41
invention before a competitor files on the same invention, we no longer have the ability to provide proof that we were in possession of the invention prior to the competitor’s filing date, and thus would not be able to obtain patent protection for our invention. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents.
Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. We also rely to a certain extent on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business and financial condition could be materially adversely affected.
We license patent rights from third-party owners. If such owners do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, our competitive position and business prospects may be harmed.
We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained licenses from, among others, Cancer Research Technology Limited, Ionis Pharmaceuticals, Inc., or Ionis (formerly Isis Pharmaceuticals, Inc.), the Massachusetts Institute of Technology, or MIT, the Whitehead Institute for Biomedical Research, or Whitehead, Max Planck Innovation and Arbutus. We also intend to enter into additional licenses to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects. In addition, we sublicense our rights under various third-party licenses to our collaborators. Any impairment of these sublicensed rights could result in reduced revenues under our collaboration agreements or result in termination of an agreement by one or more of our collaborators.
Other companies or organizations may challenge our patent rights or may assert patent rights that prevent us from developing and commercializing our products.
RNAi is a relatively new scientific field, the commercial exploitation of which has resulted in many different patents and patent applications from organizations and individuals seeking to obtain patent protection in the field. We have obtained grants and issuances of RNAi patents and have licensed many of these patents from third parties on an exclusive basis. The issued patents and pending patent applications in the United States and in key markets around the world that we own or license claim many different methods, compositions and processes relating to the discovery, development, manufacture and commercialization of RNAi therapeutics.
Specifically, we have a portfolio of patents, patent applications and other intellectual property covering: fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms; chemical modifications to siRNAs that improve their suitability for therapeutic and other uses; siRNAs directed to specific targets as treatments for particular diseases; delivery technologies, such as in the fields of carbohydrate conjugates and cationic liposomes; and all aspects of our specific development candidates.
As the field of RNAi therapeutics is maturing, patent applications are being fully processed by national patent offices around the world. There is uncertainty about which patents will issue, and, if they do, as to when, to whom, and with what claims. It is likely that there will be significant litigation and other proceedings, such as interference, reexamination and opposition proceedings, as well as inter partes and post-grant review proceedings introduced by provisions of the America Invents Act, which became available to third party challengers on September 16, 2012, in various patent offices relating to patent rights in the RNAi field. For example, various third parties have initiated oppositions to patents in our McSwiggen, Kreutzer-Limmer and Tuschl II series in the EPO and in other jurisdictions. We expect that additional oppositions will be filed in the EPO and elsewhere, and other challenges will be raised relating to other patents and patent applications in our portfolio. In many cases, the possibility of appeal exists for either us or our opponents, and it may be years before final, unappealable rulings are made with respect to these patents in certain jurisdictions. The timing and outcome of these and other proceedings is uncertain and may adversely affect our business if we are not successful in defending the patentability and scope of our pending and issued patent claims. In addition, third parties may attempt to invalidate our intellectual property rights. Even if our rights are not directly challenged, disputes could lead to the weakening of our intellectual property rights. Our defense against any attempt by third parties to circumvent or invalidate our intellectual property rights could be costly to us, could require significant time and attention of our management and could have a material adverse effect on our business and our ability to successfully compete in the field of RNAi.
42
There are many issued and pending patents that claim aspects of oligonucleotide chemistry and modifications that we may need to apply to our siRNA therapeutic candidates. There are also many issued patents that claim targeting genes or portions of genes that may be relevant for siRNA drugs we wish to develop. Thus, it is possible that one or more organizations will hold patent rights to which we will need a license. If those organizations refuse to grant us a license to such patent rights on reasonable terms, we may not be able to market products or perform research and development or other activities covered by these patents.
If we become involved in patent litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, substantial liability for damages or be required to stop our product development and commercialization efforts.
Third parties may sue us for infringing their patent rights. Likewise, we may need to resort to litigation to enforce a patent issued or licensed to us or to determine the scope and validity of proprietary rights of others or protect our proprietary information and trade secrets. For example, during the second quarter of 2015, we filed a trade secret misappropriation lawsuit against Dicerna to protect our rights in the RNAi assets we purchased from Merck. A third party may also claim that we have improperly obtained or used its confidential or proprietary information. For example, in March 2011, Arbutus (formerly Tekmira) filed a civil complaint against us alleging, among other things, misappropriation of its confidential and proprietary information and trade secrets. In November 2012, we settled this litigation and restructured our contractual relationship with Arbutus. In connection with this restructuring, we incurred a $65.0 million charge to operating expenses during the quarter ended December 31, 2012. In addition, during the pendency of the litigation, we incurred significant costs, and the defense of this litigation diverted the attention of our management and other resources that would otherwise have been engaged in other activities.
Furthermore, third parties may challenge the inventorship of our patents or licensed patents. For example, in March 2011, The University of Utah, or Utah, filed a complaint in the United States District Court for the District of Massachusetts, or the MA District Court, against us, Max Planck Gesellschaft Zur Foerderung Der Wissenschaften e.V. and Max Planck Innovation, together, Max Planck, Whitehead, MIT and UMass, claiming that a professor of Utah is the sole inventor, or in the alternative, a joint inventor of certain of our in-licensed patents. Utah was seeking correction of inventorship of the Tuschl patents, unspecified damages and other relief. After several years of court proceedings and discovery, in September 2015, the MA District Court granted our motions for summary judgment, finding that there was no collaboration between Dr. Bass and Dr. Tuschl, which is a pre-requisite for co-inventorship, and dismissing Utah’s state law damages claims as well. On October 28, 2015, Utah filed a notice of appeal from this ruling to the United States Court of Appeals for the Federal Circuit, or CAFC. On December 18, 2015, the CAFC entered an order dismissing Utah’s appeal following a joint motion filed by us and Utah seeking dismissal of the appeal with prejudice. This disposed of Utah’s inventorship claims and its state law claims for damages. On October 14, 2015, we filed a motion with the MA District Court seeking reimbursement of costs and fees associated with defending this action in the amount of approximately $8.0 million. On November 30, 2015, the MA District Court denied our motion and on December 15, 2015 we filed a notice of appeal of this ruling with the CAFC. Oral arguments were heard on January 12, 2017. On March 23, 2017, the CAFC denied our appeal and we have decided not to appeal this ruling any further.
In addition, in connection with certain license and collaboration agreements, we have agreed to indemnify certain third parties for certain costs incurred in connection with litigation relating to intellectual property rights or the subject matter of the agreements. The cost to us of any litigation or other proceeding relating to intellectual property rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s efforts. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of any litigation could delay our research and development efforts and limit our ability to continue our operations.
If any parties successfully claim that our creation or use of proprietary technologies infringes upon or otherwise violates their intellectual property rights, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. In addition to any damages we might have to pay, a court could require us to stop the infringing activity or obtain a license. Any license required under any patent may not be made available on commercially acceptable terms, if at all. In addition, such licenses are likely to be non-exclusive and, therefore, our competitors may have access to the same technology licensed to us. If we fail to obtain a required license and are unable to design around a patent, we may be unable to effectively market some of our technology and products, which could limit our ability to generate revenues or achieve profitability and possibly prevent us from generating revenue sufficient to sustain our operations. Moreover, we expect that a number of our collaborations will provide that royalties payable to us for licenses to our intellectual property may be offset by amounts paid by our collaborators to third parties who have competing or superior intellectual property positions in the relevant fields, which could result in significant reductions in our revenues from products developed through collaborations.
43
If we fail to comply with our obligations under any licenses or related agreements, we may be required to pay damages and could lose license or other rights that are necessary for developing and protecting our RNAi technology and any related product candidates that we develop, or we could lose certain rights to grant sublicenses.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license or render the license non-exclusive, which could result in us being unable to develop, manufacture, market and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology. For example, in 2013, Arbutus (formerly Tekmira) notified us that it believed it had achieved a $5.0 million milestone payment under our cross-license agreement relating to the manufacture of ALN-VSP clinical trial material for use in China. We notified Arbutus that we did not believe that the milestone has been achieved under the terms of the cross-license agreement. In August 2013, we initiated binding arbitration proceedings seeking a declaratory judgment that Arbutus had not yet met the conditions of the milestone and was not entitled to payment at the time. The Arbutus arbitration hearing was held in May 2015. On March 9, 2016, the arbitration panel ruled in our favor and as a result, no milestone payment is due to Arbutus at this time. The grounds on which Arbutus could appeal this ruling were limited and Arbutus did not appeal by the deadline.
Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we will be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize products, we may be unable to achieve or maintain profitability.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, employees, consultants, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover trade secrets and proprietary information, and in such cases we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Risks Related to Competition
The pharmaceutical market is intensely competitive. If we are unable to compete effectively with existing drugs, new treatment methods and new technologies, we may be unable to commercialize successfully any drugs that we develop.
The pharmaceutical market is intensely competitive and rapidly changing. Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations are pursuing the development of novel drugs for the same diseases that we are targeting or expect to target. Many of our competitors have:
|
|
• |
much greater financial, technical and human resources than we have at every stage of the discovery, development, manufacture and commercialization of products; |
|
|
• |
more extensive experience in pre-clinical testing, conducting clinical trials, obtaining regulatory approvals, and in manufacturing, marketing and selling drug products; |
|
|
• |
product candidates that are based on previously tested or accepted technologies; |
|
|
• |
products that have been approved or are in late stages of development; and |
|
|
• |
collaborative arrangements in our target markets with leading companies and research institutions. |
We will face intense competition from drugs that have already been approved and accepted by the medical community for the treatment of the conditions for which we may develop drugs. We also expect to face competition from new drugs that enter the market. We believe a number of drugs are currently under development, and may become commercially available in the future, for the treatment of conditions for which we may try to develop drugs. These drugs may be more effective, safer, less expensive, or marketed and sold more effectively, than any products we develop. For example, we are developing patisiran for the treatment of hATTR amyloidosis. We have completed enrollment in our ongoing Phase 3 clinical trial and expect to report top-line data from our Phase 3 clinical trial in mid-2017. We are aware of other approved products used to treat this disease, including tafamidis, marketed by Pfizer,
44
as well as product candidates in various stages of clinical development, including an investigational drug being developed for which Ionis has completed enrollment in an ongoing Phase 3 clinical trial. Patisiran may not compete favorably with these products and product candidates, and even if approved, it may not achieve commercial success.
If we successfully develop product candidates, and obtain approval for them, we will face competition based on many different factors, including:
|
|
• |
the safety and effectiveness of our products relative to alternative therapies, if any; |
|
|
• |
the ease with which our products can be administered and the extent to which patients accept relatively new routes of administration; |
|
|
• |
the timing and scope of regulatory approvals for these products; |
|
|
• |
the availability and cost of manufacturing, marketing and sales capabilities; |
|
|
• |
price; |
|
|
• |
reimbursement coverage; and |
|
|
• |
patent position. |
Our competitors may develop or commercialize products with significant advantages over any products we develop based on any of the factors listed above or on other factors. Our competitors may therefore be more successful in commercializing their products than we are, which could adversely affect our competitive position and business. Competitive products may make any products we develop obsolete or noncompetitive before we can recover the expenses of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and the ability to execute on our business plan. Furthermore, we also face competition from existing and new treatment methods that reduce or eliminate the need for drugs, such as the use of advanced medical devices. The development of new medical devices or other treatment methods for the diseases we are targeting could make our product candidates noncompetitive, obsolete or uneconomical.
We face competition from other companies that are working to develop novel drugs and technology platforms using technology similar to ours. If these companies develop drugs more rapidly than we do or their technologies, including delivery technologies, are more effective, our ability to successfully commercialize drugs may be adversely affected.
In addition to the competition we face from competing drugs in general, we also face competition from other companies working to develop novel drugs using technology that competes more directly with our own. We are aware of multiple companies that are working in the field of RNAi. In addition, we granted licenses or options for licenses to Ionis (formerly Isis), Benitec Ltd., Arrowhead Research Corporation, or Arrowhead, and its subsidiary, Calando Pharmaceuticals, Inc., Arbutus, Quark Pharmaceuticals, Inc., Sylentis S.A. and others under which these companies may independently develop RNAi therapeutics against a limited number of targets. Any one of these companies may develop its RNAi technology more rapidly and more effectively than us.
In addition, as a result of agreements that we have entered into, Arrowhead, as the assignee of F. Hoffmann-La Roche Ltd, and Takeda Pharmaceutical Company Limited have obtained non-exclusive licenses, and Arrowhead, as the assignee of Novartis Pharma AG, has obtained specific exclusive licenses for 30 gene targets, to certain aspects of our technology that give them the right to compete with us in certain circumstances. We also compete with companies working to develop antisense-based drugs. Like RNAi therapeutics, antisense drugs target messenger RNAs, or mRNAs, in order to suppress the activity of specific genes. Ionis (formerly Isis) is currently marketing an antisense drug and has several antisense product candidates in clinical trials, including one for the treatment of hATTR amyloidosis. Ionis is also developing antisense drugs using ligand-conjugated GalNAc technology licensed from us, and these drugs have been shown to have increased potency at lower doses in clinical and pre-clinical studies, compared with antisense drugs that do not use such licensed GalNAc technology. The development of antisense drugs is more advanced than that of RNAi therapeutics, and antisense technology may become the preferred technology for drugs that target mRNAs to silence specific genes.
In addition to competition with respect to RNAi and with respect to specific products, we face substantial competition to discover and develop safe and effective means to deliver siRNAs to the relevant cell and tissue types. Safe and effective means to deliver siRNAs to the relevant cell and tissue types may be developed by our competitors, and our ability to successfully commercialize a competitive product would be adversely affected. In addition, substantial resources are being expended by third parties in the effort to discover and develop a safe and effective means of delivering siRNAs into the relevant cell and tissue types, both in academic laboratories and in the corporate sector. Some of our competitors have substantially greater resources than we do, and if our competitors are able to negotiate exclusive access to those delivery solutions developed by third parties, we may be unable to successfully commercialize our product candidates.
45
Risks Related to Our Common Stock
If our stock price fluctuates, purchasers of our common stock could incur substantial losses.
The market price of our common stock has fluctuated significantly and may continue to fluctuate significantly in response to factors that are beyond our control. The stock market in general has from time to time experienced extreme price and volume fluctuations, and the biotechnology in particular has very recently experienced extreme price and volume fluctuations. The market prices of securities of pharmaceutical and biotechnology companies have been extremely volatile, and have experienced fluctuations that often have been unrelated or disproportionate to the clinical development progress or operating performance of these companies, including as a result of adverse development events. These broad market and sector fluctuations have resulted and could in the future result in extreme fluctuations in the price of our common stock, which could cause purchasers of our common stock to incur substantial losses.
We may incur significant costs from class action litigation due to stock volatility.
Our stock price may fluctuate for many reasons, including as a result of public announcements regarding the progress of our development efforts or the development efforts of our collaborators and/or competitors, the addition or departure of our key personnel, variations in our quarterly operating results and changes in market valuations of pharmaceutical and biotechnology companies. For example, in October 2016, we announced that we were discontinuing the development of revusiran and our stock price declined significantly as a result. When the market price of a stock has been volatile as our stock price has been, holders of that stock have occasionally brought securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit is without merit, we could incur substantial costs defending the lawsuit. The lawsuit could also divert the time and attention of our management.
Sales of additional shares of our common stock, including by us or our directors and officers, could cause the price of our common stock to decline.
Sales of substantial amounts of our common stock in the public market, or the availability of such shares for sale, by us or others, including the issuance of common stock upon exercise of outstanding options, could adversely affect the price of our common stock.
Sanofi Genzyme’s ownership of our common stock could delay or prevent a change in corporate control.
Sanofi Genzyme currently holds approximately 12 percent of our outstanding common stock and has the right to increase its ownership up to 30 percent, as well as the right to maintain its ownership percentage through the term of our collaboration, subject to certain limitations. This concentration of ownership may harm the market price of our common stock by:
|
|
• |
delaying, deferring or preventing a change in control of our company; |
|
|
• |
impeding a merger, consolidation, takeover or other business combination involving our company; or |
|
|
• |
discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of our company. |
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation and our bylaws may delay or prevent an acquisition of us or a change in our management. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors. Because our board of directors is responsible for appointing the members of our management team, these provisions could in turn affect any attempt by our stockholders to replace current members of our management team. These provisions include:
|
|
• |
a classified board of directors; |
|
|
• |
a prohibition on actions by our stockholders by written consent; |
|
|
• |
limitations on the removal of directors; and |
|
|
• |
advance notice requirements for election to our board of directors and for proposing matters that can be acted upon at stockholder meetings. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15 percent of our outstanding voting stock from merging or
46
combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15 percent of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. These provisions would apply even if the proposed merger or acquisition could be considered beneficial by some stockholders.
|
|
|
|
|
31.1 |
|
Certification of principal executive officer pursuant to Rule 13a-14(a) promulgated under the Securities Exchange Act of 1934, as amended. |
|
|
|
|
|
31.2 |
|
Certification of principal financial officer pursuant to Rule 13a-14(a) promulgated under the Securities Exchange Act of 1934, as amended. |
|
|
|
|
|
32.1 |
|
Certification of principal executive officer pursuant to Rule 13a-14(b) promulgated under the Securities Exchange Act of 1934, as amended, and Section 1350 of Chapter 63 of Title 18 of the United States Code. |
|
|
|
|
|
32.2 |
|
Certification of principal financial officer pursuant to Rule 13a-14(b) promulgated under the Securities Exchange Act of 1934, as amended, and Section 1350 of Chapter 63 of Title 18 of the United States Code. |
|
|
|
|
|
101 |
|
The following materials from the Registrant’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2017, formatted in XBRL (Extensible Business Reporting Language): (i) the Condensed Consolidated Balance Sheets, (ii) the Condensed Consolidated Statements of Comprehensive Loss, (iii) the Condensed Consolidated Statements of Cash Flows, and (iv) Notes to Condensed Consolidated Financial Statements. |
47
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
ALNYLAM PHARMACEUTICALS, INC. |
|
|
|
|
|
Date: May 5, 2017 |
|
/s/ John M. Maraganore |
|
|
|
John M. Maraganore, Ph.D. |
|
|
|
Chief Executive Officer |
|
|
|
(Principal Executive Officer) |
|
|
|
|
|
Date: May 5, 2017 |
|
/s/ Michael P. Mason |
|
|
|
Michael P. Mason |
|
|
|
Vice President of Finance and Treasurer |
|
|
|
(Principal Financial and Accounting Officer) |
48